WO2023282245A1 - ヌクレオチド類の精製方法及びヌクレオチド類の精製装置並びに疎水性試薬及び疎水性基質 - Google Patents
ヌクレオチド類の精製方法及びヌクレオチド類の精製装置並びに疎水性試薬及び疎水性基質 Download PDFInfo
- Publication number
- WO2023282245A1 WO2023282245A1 PCT/JP2022/026665 JP2022026665W WO2023282245A1 WO 2023282245 A1 WO2023282245 A1 WO 2023282245A1 JP 2022026665 W JP2022026665 W JP 2022026665W WO 2023282245 A1 WO2023282245 A1 WO 2023282245A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydrophobic
- nucleotides
- group
- compound
- protecting group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images












































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1003—Extracting or separating nucleic acids from biological samples, e.g. pure separation or isolation methods; Conditions, buffers or apparatuses therefor
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H1/00—Processes for the preparation of sugar derivatives
- C07H1/06—Separation; Purification
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/7105—Natural ribonucleic acids, i.e. containing only riboses attached to adenine, guanine, cytosine or uracil and having 3'-5' phosphodiester links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/22—Amides of acids of phosphorus
- C07F9/24—Esteramides
- C07F9/2454—Esteramides the amide moiety containing a substituent or a structure which is considered as characteristic
- C07F9/2458—Esteramides the amide moiety containing a substituent or a structure which is considered as characteristic of aliphatic amines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/02—Heterocyclic radicals containing only nitrogen as ring hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/167—Purine radicals with ribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/20—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/26—Preparation of nitrogen-containing carbohydrates
- C12P19/28—N-glycosides
- C12P19/30—Nucleotides
- C12P19/34—Polynucleotides, e.g. nucleic acids, oligoribonucleotides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- the present invention relates to a method for purifying nucleotides, an apparatus for purifying nucleotides, a hydrophobic reagent, and a hydrophobic substrate.
- Nucleic acids such as DNA and RNA are important molecules in life science, and have been studied for many years in the medical field. In recent years, pharmaceutical applications of mRNA have attracted particular attention. Unlike antisense nucleic acids that suppress protein synthesis and siRNA (small interfering RNA) that causes RNA interference, mRNA drugs obtain therapeutic effects by synthesizing proteins within cells. Therefore, not only the suppression of abnormal proteins but also the replenishment of normal proteins is possible, and the realization of mRNA medicines will lead to the expansion of therapeutic strategies using nucleic acid medicines. In addition, since mRNA does not require transportation to the nucleus and is free from the risk of insertional mutation into the genome, it also has an aspect of excellent safety. Furthermore, in recent years, progress in the development of technologies related to nucleic acid delivery has made it possible to overcome the low cell membrane permeability of mRNA, and it has become widely recognized that mRNA medicine is feasible. .
- High performance liquid chromatography (HPLC) and denaturing polyacrylamide gel electrophoresis have been used for the isolation and purification of mRNA.
- HPLC high performance liquid chromatography
- denaturing polyacrylamide gel electrophoresis have been used for the isolation and purification of mRNA.
- methods for purifying RNA using hydrophobicity have been known. For example, a method has been reported in which a dinitrobenzene protecting group, which is a hydrophobic tag, is introduced to the ends of RNA synthesized on a solid phase based on the phosphoramidite method, and the RNA is purified by reversed-phase HPLC (e.g., non-patented See Document 1, and see c) in FIG. 5, which will be described later).
- the photoprotective groups are removed by ultraviolet light irradiation to obtain natural RNA.
- isolation and purification of 67-mer RNA were successful, and deprotection of the dinitrobenzene group was achieved with a high yield of 95% or more.
- This degradable protective group is composed of a nitrobenzyl group or the like, and is introduced into the 3'-position phosphate group of the intermediate nucleotide, not at the terminal position such as the 5'-end or 3'-end of the nucleotides constituting the nucleic acid. ing.
- Non-Patent Document 1 The method of Non-Patent Document 1 is only used to purify about 90 residues of chemically synthesized RNA.
- the dinitrobenzene protecting group of Non-Patent Document 1 has two nitrobenzyl groups, the carbon positioned between them is electrophilic and highly reactive. Therefore, the dinitrobenzene protective group is easily attacked by ammonium or the like and easily decomposed, resulting in poor stability after introduction of the protective group and low yield of the desired nucleotides.
- the dinitrobenzene protecting group of this document has two highly reactive nitrobenzyl groups in the molecule, and is therefore explosive, difficult to handle, and dangerous. Furthermore, this document does not describe purification of long RNAs of 100 bases or more and purification by transcription using a polymerase.
- An object of the present invention is to provide a method for purifying nucleotides and an apparatus for purifying nucleotides in which the nucleotides in which a protecting group is introduced are stable, the yield of the nucleotides after deprotection is high, and the safety is high. That's what it is.
- Another object of the present invention is to provide a hydrophobic reagent and a hydrophobic substrate for use in such a method and apparatus for purifying nucleotides.
- the inventors have conducted extensive research to solve the above problems. As a result, the inventors have found that the use of mononitrobenzene as a protecting group stabilizes the nucleotides into which the protecting group has been introduced, and that the yield of the nucleotides is improved by deprotection by light or reduction reaction, and thus completed the present invention. .
- the present invention is as follows.
- a method for purifying nucleotides having at least one nucleotide and/or derivative thereof as a structural unit a protecting group introduction step of introducing a hydrophobic protecting group represented by the following formula (P1) or (P2) into a nucleotide to generate a hydrophobic nucleotide; an isolation and purification step of isolating and purifying the hydrophobic nucleotides in a hydrophobic environment; and a deprotection step of deprotecting the hydrophobic protecting group from the hydrophobic nucleotides to produce the nucleotides.
- P1 hydrophobic protecting group represented by the following formula (P1) or (P2)
- R 1 represents a linear or branched alkyl group having 1 to 30 carbon atoms
- R 4 represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms
- R 2 , R 3 , R 5 and R 6 each represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms or a linear or branched alkoxy group having 1 to 10 carbon atoms
- R 2 , R 3 and R 5 and R 6 may be the same or different.
- an amidite reagent represented by the following formula (CR1) is introduced into a nucleotide to synthesize a protective group-introduced nucleotide, and a monovalent compound is added to the 3′ terminal side of the protective group-introduced nucleotide by solid-phase synthesis.
- a nucleic acid having a sequence complementary to the nucleotides is used as a template, and a hydrophobic substrate represented by the following formula (ER1) and a nucleoside triphosphate are used as substrates.
- ER1 hydrophobic substrate represented by the following formula (ER1) and a nucleoside triphosphate
- the step of introducing a protecting group includes synthesizing the hydrophobic nucleotides by reacting the 5' terminal phosphate group of the oligonucleotide containing the nucleotides with an adenylating reagent represented by the following formula (CA1):
- CA1 adenylating reagent represented by the following formula (CA1):
- CA1 The method for purifying nucleotides according to [1] above, characterized in that: (Here, Pro indicates the hydrophobic protecting group.)
- a nucleic acid having a sequence complementary to the nucleotides is used as a template, and a hydrophobic substrate represented by the following formula (EA1) and a nucleoside triphosphate are used as substrates.
- EA1 a hydrophobic substrate represented by the following formula
- the phosphate group at the 5' end of the oligonucleotide containing the nucleotides is reacted with a capping reagent selected from the group consisting of the following formulas (CC1) to (CC4).
- a capping reagent selected from the group consisting of the following formulas (CC1) to (CC4).
- the method for purifying nucleotides according to [1] above which comprises synthesizing the hydrophobic nucleotides. (Here, Pro represents the hydrophobic protecting group, and n represents an integer of 1 or 2.)
- a nucleic acid having a sequence complementary to the nucleotides is used as a template, a hydrophobic substrate having a structure of the following formula (EC0) and a nucleoside triphosphate are used as substrates, and an RNA polymerase is used to The method for purifying nucleotides according to [1] above, wherein the hydrophobic nucleotides are synthesized by transcribing the template.
- Pro1 to Pro4 represent the hydrophobic protecting group or hydrogen, at least one of which is a hydrophobic protecting group, and Pro1 to Pro4 may be the same or different.
- X is oxygen, selected from the group consisting of sulfur and selenium atoms
- Nuc denotes a natural or non-natural nucleoside or one or more natural or non-natural nucleotides at its 3' carbon.
- the hydrophobic substrate is a cap derivative represented by the formula (EC0);
- a device for purifying nucleotides comprising at least one nucleotide and/or derivative thereof as a structural unit, a protecting group introducing means for introducing a hydrophobic protecting group represented by the following formula (P1) or (P2) into a nucleotide to generate a hydrophobic nucleotide; an isolation and purification means for isolating and purifying the hydrophobic nucleotides in a hydrophobic environment; and deprotecting means for deprotecting the hydrophobic protecting group from the hydrophobic nucleotides to produce the nucleotides.
- a protecting group introducing means for introducing a hydrophobic protecting group represented by the following formula (P1) or (P2) into a nucleotide to generate a hydrophobic nucleotide
- an isolation and purification means for isolating and purifying the hydrophobic nucleotides in a hydrophobic environment
- deprotecting means for deprotecting the hydrophobic protecting group from the hydrophobic nucleot
- R 1 represents a linear or branched alkyl group having 1 to 30 carbon atoms
- R 4 represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms
- R 2 , R 3 , R 5 and R 6 each represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms or a linear or branched alkoxy group having 1 to 10 carbon atoms
- R 2 , R 3 and R 5 and R 6 may be the same or different.
- a hydrophobic reagent for synthesizing hydrophobic nucleotides by chemical synthesis which is selected from the group consisting of the following formula (CR1), formula (CA1) and formulas (CC1) to (CC4) Characterized Hydrophobic Reagents.
- Pro represents a hydrophobic protecting group represented by the following formula (P1) or (P2), and n represents an integer of 1 or 2.
- R 1 represents a linear or branched alkyl group having 1 to 30 carbon atoms
- R 4 represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms
- R 2 , R 3 , R 5 and R 6 each represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms or a linear or branched alkoxy group having 1 to 10 carbon atoms
- R 2 , R 3 and R 5 and R6 may be the same or different.
- * means a bond.
- a hydrophobic substrate for producing hydrophobic nucleotides by RNA polymerase characterized by being selected from the group consisting of the following formula (ER1), the following formula (EA1), and the following formula (EC0) hydrophobic substrate
- Pro represents a hydrophobic protecting group represented by the following formula (P1) or (P2).
- Pro represents the hydrophobic protective group represented by the following formula (P1) or (P2).
- Pro1 to Pro4 represent a hydrophobic protecting group or hydrogen represented by the following formula (P1) or (P2), at least one of which is a hydrophobic protecting group, and Pro1 to Pro4 are the same or may be different
- X is selected from the group consisting of oxygen, sulfur and selenium atoms
- Nuc represents a nucleoside, the 3'-carbon of which is linked to one or two natural or non-natural nucleotides; may be present.
- R 1 represents a linear or branched alkyl group having 1 to
- An mRNA drug comprising hydrophobic nucleotides in which a hydrophobic protecting group represented by the following formula (P1) or (P2) is introduced into the nucleotides having at least one nucleotide and/or derivative thereof as a structural unit.
- a hydrophobic protecting group represented by the following formula (P1) or (P2) is introduced into the nucleotides having at least one nucleotide and/or derivative thereof as a structural unit.
- R 1 represents a linear or branched alkyl group having 1 to 30 carbon atoms
- R 4 represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms
- R 2 , R 3 , R 5 and R 6 each represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms or a linear or branched alkoxy group having 1 to 10 carbon atoms
- R 2 , R 3 and R 5 and R 6 may be the same or different.
- RNA pharmaceutical of [20] which has a structure in which RNA is linked to the 3'-position carbon of the 3'-terminal nucleotide of the hydrophobic substrate, which includes the structure of the following formula (EC0).
- Pro1 to Pro4 represent the hydrophobic protecting group or hydrogen, at least one of which is a hydrophobic protecting group, and Pro1 to Pro4 may be the same or different.
- X is oxygen, selected from the group consisting of sulfur and selenium atoms
- Nuc denotes a natural or non-natural nucleoside or one or more natural or non-natural nucleotides at its 3' carbon).
- a method for producing the mRNA pharmaceutical according to [20] above comprising a protective group introduction step of introducing a hydrophobic protective group represented by the formula (P1) or (P2) into the nucleotides to produce hydrophobic nucleotides.
- a nucleic acid having a sequence complementary to the nucleotides is used as a template, a hydrophobic substrate having the structure of the formula (EC0) and a nucleoside triphosphate are used as substrates, and RNA polymerase The method for purifying an mRNA pharmaceutical according to [21] above, wherein the hydrophobic nucleotides are synthesized by transcribing the template.
- the hydrophobic substrate containing the structure of the formula (EC0) is selected from the group consisting of the formulas (EC1) to (EC12), and the hydrophobic protecting group is deprotected and the Pro is hydrogen.
- a method for purifying nucleotides and an apparatus for purifying nucleotides are provided in which the nucleotides in which a protecting group is introduced are stable, the yield of the nucleotides after deprotection is high, and the safety is high. becomes possible. Moreover, according to the present invention, it is possible to provide a hydrophobic reagent and a hydrophobic substrate for use in such a stable, high-yield, and safe method and apparatus for purifying nucleotides.
- FIG. 10 is a diagram showing the results of reverse-phase HPLC analysis of synthesis and purification of 5' phosphorylated oligonucleotide using amidite reagent_1, and post-purification deprotection reaction.
- FIG. 10 is a diagram showing the results of reverse-phase HPLC analysis of the synthesis and purification of 5′-phosphorylated oligonucleotides using amidite reagent_2, and the post-purification deprotection reaction.
- FIG. 10 is a diagram showing the results of reverse-phase HPLC analysis of the synthesis and purification of 5′-phosphorylated oligonucleotides using amidite reagent_2, and the post-purification deprotection reaction.
- FIG. 10 is a diagram showing the results of reverse-phase HPLC analysis of the synthesis and purification of 5′-phosphorylated oligonucleotides using amidite reagent_3, and the post-purification deprotection reaction.
- FIG. 10 is a diagram showing the results of synthesis yield comparison by reversed-phase HPLC analysis of 5′-phosphorylated oligonucleotides using amidite reagents_1 and 2.
- FIG. [ Fig. 10] Fig. 10 is a diagram showing the results of reverse-phase HPLC analysis of synthesis and purification of 5' phosphorylated oligonucleotide using amidite reagent_1, and post-purification deprotection reaction.
- FIG. 10 is a diagram showing the results of reverse-phase HPLC analysis of the synthesis and purification of 5' phosphorylated oligonucleotide using amidite reagent_1, and post-purification deprotection reaction.
- FIG. 10 is a diagram showing that 5'-terminal phosphorylated RNA synthesized using amidite reagent_1 was quantitatively deprotected by light irradiation.
- Fig. 3 is a diagram showing that 5'-terminal phosphorylated RNA synthesized using amidite reagent_1 can be isolated and purified by reversed-phase HPLC, and can be quantitatively deprotected by subsequent light irradiation.
- BRIEF DESCRIPTION OF THE DRAWINGS It is a figure which shows the outline of the method of transcribing and synthesizing RNA in this invention and a prior art.
- FIG. 2 shows the results of HPLC and R-pG reaction solutions analyzed.
- FIG. 1 shows the results of HPLC and R-pG reaction solutions analyzed.
- FIG. 4 shows the results of electrophoresis of RNA synthesized in a transcription reaction. It is a figure showing the analysis results of 34nt RNA by C18 column. It is a figure showing the analysis results of 34nt RNA by C4 column.
- FIG. 10 is a diagram showing analysis results of 100 nt RNA by C18 column. It is a figure showing the analysis results of 250 nt RNA by C18 column. It is a figure showing the analysis results of 250 nt RNA by C4 column. It is a diagram showing the analysis results of 650 nt RNA by C18 column. It is a figure showing the analysis results of 650 nt RNA by C4 column. It is a figure showing the analysis results of 1078 nt RNA by C18 column.
- FIG. 4 shows the results of synthesizing a branched cap analog compound (precursor).
- FIG. 10 shows the result of adding compound 11 to an in vitro transcription reaction using T7 RNA polymerase to obtain protective group-containing adenylated RNA.
- FIG. 2 shows the result that the protective group-containing adenylated RNA prepared in the transcription reaction was deprotected by light irradiation and converted into the desired adenylated RNA.
- FIG. 3 shows various analysis results of Cap Analog_1, which is a synthesized novel cap analog compound.
- FIG. 4 shows the results of various analyzes of cap analog_2, which is a synthesized novel cap analog compound.
- FIG. 4 shows that RNA introduced with novel cap analogs could be isolated and purified by reverse-phase HPLC.
- FIG. 10 shows translational activity evaluation of NanoLuc luciferase mRNA after reverse-phase HPLC isolation and purification.
- FIG. 10 is a diagram showing evaluation of translational activity of NanoLuc luciferase mRNA after isolation and purification by reversed-phase HPLC, in particular activity comparison with ARCA.
- FIG. 2 shows results of chemical capping reactions of RNA using hydrophobic chemical capping reagents.
- FIG. 10 shows the results of HPLC purification of co-transcription reaction product mRNA using the synthesized cap analogue.
- FIG. 10 shows the results of HPLC purification of co-transcription reaction product mRNA using the synthesized cap analogue.
- FIG. 10 shows the results of HPLC purification of co-transcription reaction product mRNA using the synthesized cap analogue.
- FIG. 10 shows the results of HPLC purification of co-transcription reaction product mRNA using the synthesized cap analogue.
- FIG. 10 shows the results of HPLC purification of co-transcription reaction product mRNA using the synthesized cap analogue.
- FIG. 2 is a diagram showing the results of translational activity evaluation in HeLa cells of RNA transcribed and synthesized using cap analogues.
- FIG. 10 is a diagram showing the results of translational activity evaluation in JAWS II cells of RNA transcribed and synthesized using cap analogues.
- FIG. 3 shows activity comparison.
- Fig. 2 shows a comparison of the translation activities of luciferase (Nluc) mRNA.
- FIG. 4 shows the results of analysis by reverse-phase HPLC of capped mRNA prepared by adding a hydrophobic protective group-containing cap analog (DiPure) during transcription of a 4247 base-long RNA strand.
- FIG. 10 is a diagram showing the result that PureCap-type mRNA exhibits a high amount of protein synthesis without the introduction of methylpseudouridine.
- FIG. 10 shows the results of evaluation of intracellular immune responses (HEK293 NF-kB cells).
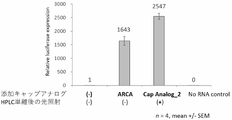
- FIG. 10 is a diagram showing results showing that highly pure PureCap-type mRNA has a higher ability to express protein than ARCA-type mRNA in individual animals.
- FIG. 10 shows the results of evaluation of intracellular immune responses (HEK293 NF-kB cells).
- the method for purifying nucleotides of the present invention is a method for purifying nucleotides to be purified, and comprises a protecting group introduction step, an isolation purification step, and a deprotection step. Each of these will be described below.
- “nucleotides” in the present invention are compounds having at least one nucleotide and/or a derivative thereof as a structural unit. Nucleotides are composed of sugars, bases and phosphates.
- Nucleotides include mononucleotides composed of one nucleotide, oligonucleotides and polynucleotides in which multiple nucleotides are linked together (both referred to as "nucleic acids"), and derivatives thereof.
- oligonucleotide means a nucleic acid (DNA, RNA) with 2 to 20 nucleotides and polynucleotide with 21 or more nucleotides.
- Nucleotides also include those having a branched structure in which the 5' end of the nucleotide is branched into two or more branches, and the branched structure includes a biantennary type, a triantennary type, and the like.
- Derivatives of at least one nucleotide include modified mononucleotides and nucleic acids (oligonucleotides, polynucleotides), for example, modified nucleotides such as methylation, 5' Terminally adenylated and capped are included.
- Sugars constituting the nucleotides of the present invention include ribose and deoxyribose.
- bases constituting the nucleotides of the present invention include adenine, guanine, cytosine, thymine, uracil, N-methyladenine, N-benzoyladenine, 2-methylthioadenine, 2-aminoadenine, 7-methylguanine, N -isobutyrylguanine, 5-fluorocytosine, 5-bromocytosine, 5-methylcytosine, 4-N-methylcytosine, 4-N,N-dimethylcytosine, 5-fluorouracil, 5-bromouracil, 5-chlorouracil , or 5,6-dihydrouracil.
- the phosphoric acid constituting the nucleotides of the present invention includes monophosphoric acid, diphosphoric acid, triphosphoric acid and the like.
- Protecting group introduction step In the protecting group introduction step, a hydrophobic protecting group represented by the following formula (P1) or (P2) (also referred to as “lipid-soluble protecting group” or “purification tag”) is introduced into nucleotides. to produce hydrophobic nucleotides.
- P1 or P2 also referred to as "lipid-soluble protecting group” or “purification tag
- R 1 represents a linear or branched alkyl group having 1 to 30 carbon atoms
- R 4 represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms
- R 2 , R 3 , R 5 and R 6 each represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms or a linear or branched alkoxy group having 1 to 10 carbon atoms
- R 2 , R 3 and R 5 and R 6 may be the same or different.
- R 1 is preferably a linear or branched alkyl group having 1 to 30 carbon atoms, and R 4 is preferably hydrogen. Furthermore, from the viewpoint of stabilizing the hydrophobic protecting group, R 1 is preferably a branched alkyl group, more preferably a secondary or tertiary alkyl group, and a tertiary alkyl group. It is particularly preferred to have On the other hand, the number of carbon atoms in R 1 is preferably 5 or more, more preferably 10 or more, from the viewpoint that the hydrophobicity of the hydrophobic protecting group becomes high and it becomes easy to separate from other compounds.
- R 4 is preferably hydrogen, and when the hydrophobic protective group is decomposable with a reducing agent, R4 is preferably an alkyl group.
- hydrophobic protecting group one having a tert-butyl group represented by the following formula (P3) is preferable. (Where R 2 and R 3 are as defined above.) In this case, it is particularly preferred that R 2 and R 3 are hydrogen.
- the hydrophobic protecting group can be introduced into any nucleotide, but is particularly introduced into the nucleotide located at the 5' end of the nucleotides. is preferred.
- the nucleotide positioned at the 5' end corresponds to the nucleotide positioned at the most 5' end in the case of DNA, RNA, and the like.
- AMP adenylation
- CAP capping
- the nucleotide located at the 5' end is adenosine or cap (7-methylguanylic acid) bound to the 5' terminal nucleotide of DNA or RNA.
- the hydrophobic protecting group can be introduced at any position of the nucleotide, but is particularly preferably introduced at the 5' phosphate group, 2' hydroxyl group or base of the sugar.
- the introduction position of the hydrophobic protective group is preferably the 5'-terminal nucleotide and a functional group that does not constitute a nucleic acid chain structure, such as a 5' phosphate group, a 2' hydroxyl group, and a base.
- the isolation and purification step is a step of isolating and purifying hydrophobic nucleotides in a hydrophobic environment.
- FIG. 1 shows a step (Step_1: Purification) of separating a nucleic acid introduced with a hydrophobic protecting group (Nucleic Acid).
- the isolation and purification step is not particularly limited as long as it is under hydrophobic conditions, and examples thereof include liquid chromatography, membrane separation, etc. Among them, reversed phase high performance liquid chromatography (reverse phase HPLC) is preferred.
- Reversed-phase HPLC is a method of separating objects to be separated according to the difference in hydrophobic interaction between a packing material in a chromatography column and a mobile phase.
- Silica gel or the like can be used as a stationary phase support for the packing material, and an alkyl group having about C4 to C18 carbon atoms can be used as the bonding phase on the surface of the packing material.
- water, organic solvents such as methanol, and acetonitrile can be used as mobile phases for reversed-phase HPLC, and gradient elution that continuously changes the composition of water/organic solvent (e.g., acetonitrile) makes the separation target hydrophobic.
- the temperature in the isolation and purification step is in the range of 10 to 40°C, preferably room temperature (25°C).
- the deprotection step is a step of deprotecting hydrophobic protecting groups from hydrophobic nucleotides to generate nucleotides.
- FIG. 1 shows a step of deprotecting the hydrophobic nucleotides purified in the isolation and purification step (Step_2: Deprotection).
- Examples of the method for deprotecting the hydrophobic protective group include light irradiation, reduction, and alkali decomposition, and light irradiation is particularly preferred. The conditions for light irradiation can be appropriately set according to the properties of the hydrophobic protective group, etc.
- the wavelength is 300 to 400 nm
- the light amount is 0.5 to 10 mW/cm 2
- the irradiation time is irradiation for 1 to 60 minutes.
- the conditions for the reduction reaction can be appropriately set according to the properties of the hydrophobic protecting group, etc.
- the reaction is performed in a 1 to 100 mM sodium dithionite aqueous solution at 37°C for 30 minutes, followed by reaction at 65°C for 10 minutes. React for 1 minute to deprotect.
- the protective group introduction step includes a method of synthesizing hydrophobic nucleotides by chemical synthesis using a hydrophobic reagent having a hydrophobic protective group (chemical synthesis), and a method of synthesizing hydrophobic nucleotides by an enzymatic reaction with a polymerase using a template and a substrate.
- a method for synthesizing nucleotides can be mentioned. Chemical synthesis and enzymatic synthesis of the above three purification methods will be described below.
- phosphorylation purification In phosphorylation purification, a hydrophobic protecting group (phosphorylation tag) is bound to the 5'-position phosphate group of the most terminal nucleotide on the 5' side of nucleotides, and this hydrophobicity is used to purify nucleotides. is a method of purifying
- a phosphorylated purification tag is a hydrophobic protecting group attached to the 5' phosphate group of the 5' most terminal nucleotide of the nucleotide group.
- Phosphorylation purification includes a method of introducing a hydrophobic protecting group by chemical synthesis and a method of introducing a hydrophobic protecting group by enzymatic synthesis.
- phosphorylation purification scheme as an example of a hydrophobic protecting group, in formula (P1), R 1 is a tert-butyl (t-Bu) group, R 2 to R 4 are hydrogen shows an example using "bNB". Note that bNB is just an example of the hydrophobic protecting group of the present invention, and the present invention is not limited thereto, and can be appropriately replaced with other compounds contained in formula (P1) or formula (P2).
- the amidite reagent represented by formula (CR1) can be obtained by reacting a phosphoramidite compound such as 2-cyanoethyl-N,N'-diisopropylchlorophosphoramidite with nitrobenzyl alcohol as a starting material.
- the amidite reagent in the protective group introduction step, is introduced into the nucleotide to synthesize the protective group-introduced nucleotide.
- mononucleotides are sequentially linked to the 3' terminal side of the protecting group-introduced nucleotide to synthesize hydrophobic nucleotides.
- Solid-phase synthesis can be performed using a known DNA synthesizer. Synthetic hydrophobic nucleotides can be isolated and purified from other nucleic acids by reversed-phase HPLC or the like using their hydrophobicity (fat solubility), and can be deprotected by light irradiation or the like.
- the lower part (b) of the phosphorylation purification scheme shows a scheme for enzymatically synthesizing hydrophobic nucleotides.
- the enzyme used is preferably RNA polymerase, particularly preferably T7 RNA polymerase.
- the above scheme shows an example of synthesizing RNA using T7 RNA polymerase.
- a nucleic acid (DNA) having a sequence complementary to nucleotides downstream of the T7 promoter sequence is used as a template.
- a hydrophobic substrate (guanosine monophosphate derivative) of the following formula (ER1) and nucleoside triphosphates (NTPs, namely ATP, GTP, CTP, UTP) are used.
- NTPs nucleoside triphosphates
- Pro denotes a hydrophobic protecting group as described above.
- RNA polymerase In the reaction of T7 RNA polymerase, transcription is initiated from GTP, so if guanosine monophosphate derivative and guanosine triphosphate (GTP) are used as substrates, two types of guanosine monophosphate derivative and guanosine triphosphate are produced at the 5' end. Produced in a mixed state of RNA. These two types of RNA can be separated according to their hydrophobicity by using reverse-phase HPLC or the like, and only the hydrophobic nucleotides derived from the guanosine monophosphate derivative can be isolated and purified.
- GTP guanosine monophosphate derivative and guanosine triphosphate
- T7 RNA polymerase Transcription of the template by T7 RNA polymerase can be carried out under appropriately set conditions, for example, the reaction temperature can be 30-45°C and the reaction time can be in the range of 1-5 hours.
- T7, T3, and SP6 RNA polymerases generally initiate transcription from GTP, and T7 RNA polymerase recognizes GTP and extends the RNA chain to its 2' hydroxyl group. Therefore, when using the hydrophobic substrate (guanosine monophosphate derivative) of formula (ER1) above, not only T7 RNA polymerase but also T3 and SP6 RNA polymerases can be used.
- E. coli RNA polymerase commercially available from New England Biolabs etc. initiates transcription also from ATP. Using this enzyme, an RNA chain can be extended from the 2' hydroxyl group using an adenosine monophosphate derivative in which ER1 guanine is replaced by adenine.
- Adenylation purification is a method of producing adenylated RNA in a transcription reaction using an adenylation purification tag and isolating it in high purity.
- An adenylation site is co-transcriptionally introduced into the 5′-end of the RNA transcript, and the hydrophobicity of the hydrophobic protective group (adenylation purification tag) introduced into the adenyl group site is used to detect co-existing RNA by reverse phase HPLC or the like. Separable. After that, the protective group is removed by light irradiation or the like to obtain the target adenylated RNA.
- Adenylation purification tags are hydrophobic tags for obtaining adenylated nucleic acids.
- An adenylated nucleic acid has a structure in which an adenosine monophosphate derivative introduced with a hydrophobic protective group is bound to the 3′-side nucleotide via two phosphate groups.
- Adenylation purification also includes a method of introducing a hydrophobic protecting group by chemical synthesis and a method of introducing a hydrophobic protecting group by enzymatic synthesis.
- the following synthesis scheme hereinafter, "adenylation purification scheme" also shows an example using "bNB". Also in this example, bNB can be replaced by other compounds of formula (P1) or formula (P2) as appropriate.
- Chemical synthesis (a) of the adenylation purification scheme shows a scheme for chemically synthesizing and purifying hydrophobic nucleotides.
- hydrophobic nucleotides are synthesized using an adenylating reagent represented by the following formula (CA1) as a hydrophobic reagent.
- CA1 adenylating reagent represented by the following formula (CA1) as a hydrophobic reagent.
- Pro represents the above hydrophobic protecting group (i.e. formula (P1) or (P2)).
- the adenylating reagent represented by formula (CA1) can be synthesized by the following procedure. First, iodonitrobenzene was used as a starting material to synthesize a nitrophenylimidazole compound, which was reacted with adenosine whose hydroxyl group was protected with a protective group such as tert-butyldimethylsilane, and then deprotected to bind a hydrophobic protective group. Synthesize adenosine derivatives. This adenosine derivative can be reacted with imidazole, dithiodipyridine, or the like to obtain an adenylating reagent represented by formula (CA1).
- the adenylation purification scheme (b) shows a scheme for synthesizing and purifying hydrophobic nucleotides by an enzymatic method.
- This scheme shows an example of RNA synthesis using T7 RNA polymerase.
- a nucleic acid (DNA) having a sequence complementary to nucleotides downstream of the T7 promoter sequence is used as a template.
- substrates for T7 RNA polymerase a hydrophobic substrate (dinucleotide derivative) of the following formula (EA1) and nucleoside triphosphates (NTPs, ie, ATP, GTP, CTP, UTP) are used.
- EA1 hydrophobic substrate
- NTPs nucleoside triphosphates
- the dinucleotide derivative represented by formula (EA1) can be obtained by reacting the adenosine derivative of "(1) chemical synthesis" above with guanosine 5'-phosphorimidazolide.
- RNA with an adenylated structure containing a protective group at the 5' end and triphosphate Two types of RNA are produced, structured RNA.
- the adenosine derivative at the 5' end and the nucleotide at the 3' end are linked via a diphosphate group.
- RNA having a protective group-containing adenylated structure can be isolated and purified.
- Transcription of the template by T7 RNA polymerase can be carried out under appropriately set conditions, for example, the reaction temperature can be 30-45°C and the reaction time can be in the range of 1-5 hours.
- Capped Purification Eukaryotic mRNAs have a cap (CAP) structure at the 5' end. Binding of ribosomes to the cap structure strongly promotes initiation of translation of mRNA. Therefore, when artificially producing an mRNA molecule, it is essential to impart a cap structure to its 5' end.
- An mRNA molecule having a length of several thousand bases is generally enzymatically synthesized by transcription from a template DNA using RNA polymerase. By adding a dinucleotide cap analog compound to the transcription reaction, a cap structure can be co-transcriptionally introduced to the 5' end of the mRNA.
- RNA transcripts of the product have a cap analogue at the desired 5' end (5' cap-RNA) and a triphosphate group. It becomes a mixture of things (5'ppp-RNA). RNAs having a triphosphate group at the 5' end are known to have undesirable immune response-inducing activity in vivo and must be removed.
- RNA with a triphosphate group at the end has degradative enzymes (RNA 5' polyphosphatase (epicentre, RP8092H), RNA 5' pyrophosphohydrolase (RppH) (New England Biolabs, M0356)), and can be converted to 5' monophosphorylated RNA using these enzymes. Furthermore, monophosphorylated RNA can be degraded and removed with an enzyme such as XRN-1 (New England Biolabs, M0338) (Reference: Chem. Sci., 2021, 12, 4383-4388). Until now, such isolation and purification of 5'cap-RNA has been troublesome.
- a novel cap analog compound having a photodegradable or reductively degradable hydrophobic protecting group is used to co-transcribe capped RNA, which is the target product, from the above-mentioned contaminants. It can be isolated by HPLC. This method is simpler and more pure than the enzymatic removal reaction, and can obtain the desired product with high purity.
- a capped purification tag is a hydrophobic tag for obtaining capped nucleic acids.
- a capped nucleic acid has a structure in which a cap structure introduced with a hydrophobic protecting group is bound to a 3′-side nucleotide via 3 or 4 phosphate groups.
- Capped purification also includes a method of introducing a hydrophobic protecting group by chemical synthesis and a method of introducing a hydrophobic protecting group by enzymatic synthesis.
- the following synthesis scheme hereinafter, “capped purification scheme” also shows an example using “bNB”. Also in this example, bNB can be replaced by other compounds of formula (P1) or formula (P2) as appropriate.
- capping purification scheme only the scheme using the capping reagent of formula (CC3) described later is exemplified. However, this scheme is not limited to this, and capping of nucleic acids and purification of hydrophobic nucleotides can be performed in a similar scheme using the capping reagents of formula (CC1), formula (CC2), and formula (CC4). It can be carried out.
- the capping reagents represented by formulas (CC1-CC4) can be synthesized by the following procedure. First, iodonitrobenzene was used as a starting material to synthesize a nitrophenylimidazole compound, which was reacted with guanosine whose hydroxyl group was protected with a protective group such as tert-butyldimethylsilane, and then deprotected to bind a hydrophobic protective group. Synthesize guanosine derivatives.
- This guanosine derivative is reacted with iodomethane, dimethyl sulfoxide (DMSO) or the like to methylate the 7' of guanosine, and reacted with imidazole, dithiodipyridine or the like to bind imidazole to the phosphate group, thereby obtaining formula (CC1- A capping reagent designated CC4) can be obtained.
- Various leaving groups other than imidazole can be used, and such leaving groups include, for example, 1-methylimidazole and 4-methylimidazole, and various imidazole derivatives described in Japanese Patent Application No. 2020-032889. and nitrogen-containing heteroaromatic compounds.
- the 5′-terminal monophosphate RNA is reacted with a diphosphate capping reagent (where n is an integer of 2 in the above formulas (CC1) to (CC4)). showing.
- the resulting hydrophobic nucleotides have a structure in which the 5'-terminal cap derivative and its 3'-terminal nucleoside are bonded via three phosphate groups.
- the 5′-terminal triphosphate RNA is reacted with a monophosphate capping reagent (where n is an integer of 1 in formulas (CC1) to (CC4) above). showing the scheme.
- the resulting hydrophobic nucleotides have a structure in which a 5'-terminal cap derivative and its 3'-terminal nucleoside are linked via four phosphate groups.
- Enzymatic synthesis (c) of the capped purification scheme shows a scheme for synthesizing and purifying hydrophobic nucleotides by an enzymatic method.
- This scheme shows an example of RNA synthesis using T7 RNA polymerase.
- a nucleic acid (DNA) having a sequence complementary to nucleotides downstream of the T7 promoter sequence is used as a template.
- substrates for T7 RNA polymerase a hydrophobic substrate (cap derivative) containing the structure of the following formula (EC0) and nucleoside triphosphates (NTPs, namely ATP, GTP, CTP, UTP are used.
- At least one of Pro1 to Pro4 is a hydrophobic protecting group represented by formula (P1) or formula (P2) above, and the rest are hydrogen.
- the number of hydrophobic protecting groups in Pro1 to Pro4 is preferably one or two.
- a nucleoside that constitutes Nuc has a structure in which a base is bound to the 1′-carbon of ribose, which is a sugar, or a derivative thereof. A hydroxyl group is bonded to the carbon at the 2'-position of this ribose.
- Ribose derivatives include those in which the hydroxyl group is substituted with an alkyl group, the oxygen bonded to the carbon at the 2'-position, and the Examples include those in which carbon atoms are bonded to carbon atoms to form heterocycles.
- the base is the same as Base, which will be described later, and may be a natural nucleic acid base or a non-natural base. Natural nucleobases include adenine, cytosine, thymine, uracil, and guanine.
- non-natural bases include N-methyladenine, N-benzoyladenine, 2-methylthioadenine, 2-aminoadenine, 7-methylguanine, N-isobutyrylguanine, 5-fluorocytosine, 5-bromocytosine, 5-methylcytosine, 4-N-methylcytosine, 4-N,N-dimethylcytosine, 5-fluorouracil, 5-bromouracil, 5-chlorouracil, or 5,6-dihydrouracil.
- nucleotides may be linked to the 3' carbon of the nucleoside that constitutes Nuc.
- the 3'-carbon of the nucleoside and the 5'-carbon of the nucleotide are preferably linked by a phosphodiester bond.
- a 5'-3' phosphodiester bond between the nucleotides is preferred.
- Nucleotides like the nucleosides constituting Nuc described above, are composed of sugars and bases, the sugars being ribose and derivatives thereof, and the bases being natural nucleobases and non-natural bases. Specific examples of Nuc include DNA, RNA, LNA, 2'OMe-RNA, 2'methoxyethyl RNA, acyclic nucleosides, heteroatom-containing linkers, and the like.
- hydrophobic substrates represented by the formula (EC0) examples include hydrophobic substrates selected from the group consisting of the following formulas (EC1) to (EC4) and the following formulas (EC5) to (EC12). can be done. (where Pro represents the above hydrophobic protecting group (i.e. formula (P1) or (P2)).)
- the capped derivatives represented by formulas (EC1) to (EC4) are the capping reagents of formulas (CA1) to (CA4) in the above "(1) Chemical synthesis", and guanosine 5'-phosphorimidazolide can be obtained by reacting
- R 1 represents a substituent selected from hydroxyl group (OH), methoxy group (OCH 3 ), methoxyethyl (-OCH 2 OCH 3 ), fluorine (F), formamide group (-NHCHO);
- R 2 represents hydrogen or a linear or branched alkyl group having 1 to 10 carbon atoms
- R 3 represents hydrogen or an amino group (—NH 2 ), where Base represents adenine, cytosine, thymine, A natural nucleobase selected from uracil, guanine, or a non-natural base, wherein X is selected from the group consisting of oxygen, sulfur and selenium atoms, where Pro is the above formula ( P1) or a hydrophobic protecting group represented by (P2).)
- natural nucleic acid bases and non-natural bases include adenine, guanine, cytosine, thymine, uracil, N-methyladenine, N-benzoyladenine, 2-methylthioadenine, 2-aminoadenine, 7-methylguanine, N -isobutyrylguanine, 5-fluorocytosine, 5-bromocytosine, 5-methylcytosine, 4-N-methylcytosine, 4-N,N-dimethylcytosine, 5-fluorouracil, 5-bromouracil, 5-chlorouracil , or 5,6-dihydrouracil.
- RNA having a protective group-containing capped structure at the 5′ end When the above-mentioned cap derivative and guanosine triphosphate (GTP) are used as substrates, two types of RNA, RNA having a protective group-containing capped structure at the 5′ end and RNA having a triphosphate group at the 5′ end, are produced. Generate. In an RNA having a protective group-containing capped structure at the 5' end, the cap structure at the 5' end and the nucleotide at the 3' end are linked via a triphosphate group. These two types of RNA can be separated according to their hydrophobicity by using reverse-phase HPLC or the like, and only the RNA having a protective group-containing capped structure can be isolated and purified. Transcription of the template by T7 RNA polymerase can be carried out under appropriately set conditions, for example, the reaction temperature can be 30-45°C and the reaction time can be in the range of 1-5 hours.
- mRNA bound with a hydrophobic protective group is bound with a hydrophobic protective group. It exhibits a high translational activity equal to or higher than that of mRNA in the free state. Therefore, when the cap derivative of formula (EC3) is used, translation can be performed using nucleotides to which a hydrophobic protecting group is attached prior to the deprotection step.
- the apparatus for purifying nucleotides is an apparatus for carrying out the above-described method for purifying nucleotides, and includes means for introducing protecting groups, means for isolating and purifying, and means for deprotecting.
- the protective group introduction means is a means for carrying out the protective group introduction step described above, in which a hydrophobic protective group represented by formula (P1) or (P2) is introduced into nucleotides to produce hydrophobic nucleotides.
- protecting group introduction means include the amidite reagent represented by the formula (CR1) and various reagents and reaction devices used for the phosphorylation purification in the above "chemical synthesis" of the phosphorylation purification.
- protective group introduction means include hydrophobic substrates represented by the formula (ER1), NTPs, various reagents used for phosphorylation purification, reaction devices, and the like. .
- the protecting group introducing means includes the adenylation reagents represented by formulas (CA1) to (CA4), and various reagents and reaction devices used for adenylation purification. can be mentioned.
- protective group introduction means include hydrophobic substrates represented by the formula (EA1), NTPs, various reagents used for adenylation purification, reaction devices, and the like. .
- the capping reagent represented by the formula (CA1) and various reagents and reaction devices used for the capping purification can be mentioned.
- the protecting group introducing means is a hydrophobic substrate having the structure of the formula (EC0), for example, the formulas (EC1) to (EC4), and the formulas (EC5) to Examples include hydrophobic substrates represented by (EC12), NTPs, various reagents used for capping purification, reaction devices, and the like.
- the isolation and purification means are means for carrying out the isolation and purification steps described above, and isolate and purify hydrophobic nucleotides in a hydrophobic environment.
- the isolation/purification device is not particularly limited as long as it is hydrophobic, and examples thereof include liquid chromatography and membrane separation devices, and examples of liquid chromatography include reversed-phase high-performance liquid chromatography.
- the deprotection means is a means for carrying out the above deprotection step, and deprotects the hydrophobic protecting group from the hydrophobic nucleotides to produce the nucleotides.
- Examples of deprotection means include devices for carrying out light irradiation treatment, reduction treatment, and the like.
- a light source apparatus for irradiating light with a wavelength of 300 to 400 nm for 1 to 30 minutes can be mentioned.
- a reducing agent such as sodium dithionite (Na 2 S 2 O 4 ) is used, and hydrophobic nucleotides are treated with this reducing agent at 25 to 80° C.
- reaction temperature is more preferably in the range of 25 to 50°C, particularly preferably 25°C or 37°C.
- Other treatments are similarly deprotected using a device for deprotecting a degradable protective group (deprotection device).
- the hydrophobic nucleotides of the present invention are also useful as an mRNA drug. That is, the mRNA pharmaceutical of the present invention is a hydrophobic nucleotide in which a hydrophobic protecting group represented by the following formula (P1) or (P2) is introduced into a nucleotide having at least one nucleotide and/or derivative thereof as a constituent unit.
- a hydrophobic protecting group represented by the following formula (P1) or (P2) is introduced into a nucleotide having at least one nucleotide and/or derivative thereof as a constituent unit.
- R 1 represents a linear or branched alkyl group having 1 to 30 carbon atoms
- R 4 represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms
- R 2 , R 3 , R 5 and R 6 each represents hydrogen, a linear or branched alkyl group having 1 to 10 carbon atoms or a linear or branched alkoxy group having 1 to 10 carbon atoms
- R 2 , R 3 and R 5 and R 6 may be the same or different.
- the mRNA drug is particularly preferably composed of mRNA having a cap structure (PureCap-type mRNA in Examples described later).
- some PureCap-type mRNAs exhibit higher intracellular translational activity than conventional methylpseudouridine-introduced mRNAs (formula (EC5) to formula (EC6) and formula (EC9) to formula (EC10), preferably using formula (EC10) and formula (EC9)).
- PureCap-type mRNAs such as ARCA (Anti-Reverse Cap Analog) exhibit lower immune responses than conventional mRNAs (formula (EC1) to formula (EC12 ) preferably using formula (EC3), formula (EC6), formula (EC5), formula (EC10), formula (EC9)). From these points of view, PureCap-type mRNA is particularly useful as a medicine compared to conventional mRNA.
- ARCA Anti-Reverse Cap Analog
- An mRNA drug having a cap structure has a structure in which RNA is linked to the 3'-position carbon of the 3'-terminal nucleotide of the hydrophobic substrate containing the structure of the above formula (EC0).
- Pro1 to Pro4 represent the above hydrophobic protecting groups or hydrogen, at least one of which is a hydrophobic protecting group, and Pro1 to Pro4 may be the same or different.
- X is oxygen , sulfur and selenium atoms
- Nuc denotes a natural or non-natural nucleoside or one or more natural or non-natural nucleotides at its 3' carbon).
- Nuc may be a natural or non-natural nucleoside or one or two natural or non-natural nucleotides linked to the 3'-carbon thereof. Since the details of the formula (EC0) are the same as those described above, the detailed description is omitted here.
- RNA has a sequence that encodes a protein or peptide that has therapeutic effects, and is translated by the ribosome to produce these useful proteins or peptides.
- hydrophobic substrates selected from the group consisting of formulas (EC1) to (EC12).
- Pro denotes a hydrophobic protecting group as described above.
- R 1 represents a substituent selected from hydroxyl group (OH), methoxy group (OCH 3 ), methoxyethyl (-OCH 2 OCH 3 ), fluorine (F), formamide group (-NHCHO);
- R 2 represents hydrogen or a linear or branched alkyl group having 1 to 10 carbon atoms
- R 3 represents hydrogen or an amino group (—NH 2 )
- Base represents adenine, cytosine, thymine, is a natural nucleobase selected from uracil, guanine, or represents a non-natural base
- X is selected from the group consisting of oxygen, sulfur and selenium atoms
- Pro is the above formula It represents a hydrophobic protecting group represented by (P1) or (P2).)
- the above-mentioned hydrophobic nucleotides to which the hydrophobic protecting group has been bound are also useful as medicines when the hydrophobic protecting group is deprotected.
- the mRNA pharmaceutical of the present invention includes both hydrophobic nucleotides and nucleotides from which the hydrophobic protecting group has been deprotected.
- the latter nucleotides are specifically nucleotides having at least one nucleotide and/or a derivative thereof as a structural unit, and are represented by the formula (P1), (P2) or (P3). is deprotected and is a nucleotide in which hydrogen or a substituent is bonded to * of the bond with the nucleotide.
- mRNA drugs having the cap structure described above are preferable as such nucleotides from which the hydrophobic protecting group has been deprotected.
- the hydrophobic protecting group is deprotected.
- all of Pro1 to Pro4 in formula (EC0) are hydrogen nucleotides.
- deprotected nucleotides of formulas (EC1) to (EC12) represented by formula (EC0) include nucleotides to which hydrophobic protecting groups of formulas (EC1) to (EC12) are bound. , the nucleotides in which Pro is hydrogen as a result of deprotection of this hydrophobic protecting group.
- the present invention includes the above-described method for producing an mRNA drug, which method comprises introducing a hydrophobic protecting group represented by the following formula (P1) or (P2) into a nucleotide to produce a hydrophobic nucleotide. Including a protective group introduction step to generate
- the protective group introduction step uses a nucleic acid having a sequence complementary to nucleotides as a template, and a hydrophobic substrate having the structure of the above formula (EC0) and a nucleoside triphosphate. is a substrate and a template is transcribed by RNA polymerase to synthesize hydrophobic nucleotides.
- a hydrophobic substrate having the structure of formula (EC0) include hydrophobic substrates selected from the group consisting of (EC1) to (EC12). Since the protective group introduction step has already been described in detail, detailed description thereof is omitted here.
- the mRNA pharmaceutical of the present invention can be administered to cells or tissues by encapsulating in carriers such as solid lipid nanoparticles (SNP) and lipid nanoparticles (LNP).
- Lipid nanoparticles are composed of cationic lipids, PEGylated lipids such as ALC-0159, DSPE-mPEG, DMG-mPEG, neutral phospholipids such as DSPC, DPPC, DOPE, and cholesterol.
- SNP solid lipid nanoparticles
- LNP lipid nanoparticles
- Lipid nanoparticles are composed of cationic lipids, PEGylated lipids such as ALC-0159, DSPE-mPEG, DMG-mPEG, neutral phospholipids such as DSPC, DPPC, DOPE, and cholesterol.
- uncapped RNA and transcriptional by-products may induce an immune response and decrease the amount of protein synthesis.
- the mRNA pharmaceutical of the present invention shows a high protein synthesis amount because by
- nucleotides in which the hydrophobic protecting group has been deprotected are also useful as pharmaceuticals. Also included are methods for producing such nucleotides.
- the hydrophobic protecting group represented by formula (P1), (P2) or (P3) is deprotected to generate nucleotides in which hydrogen or a substituent is bonded to * of the bond with the nucleotide.
- nucleotides in which all of Pro1 to Pro4 of the formula (EC0) are hydrogen are generated, and specific examples thereof are the deprotected nucleotides of the formulas (EC1) to (EC12).
- Pro are produced as nucleotides where Pro is hydrogen.
- oligonucleotides were redissolved in super-deionized water and concentrations were calculated by measuring absorbance at 260 nm.
- MALDI-TOF molecular weight determination of oligonucleotides was performed using 3-hydroxypicolinic acid as matrix and Ultrafle Xtreme (Bruker) in positive mode.
- FIG. 2 shows the synthesis and purification of 5'-phosphorylated oligonucleotides using amidite reagent_1, and the results of reverse-phase HPLC analysis of deprotection reaction after purification.
- amidite reagent_1 is used to synthesize a 19-base-long oligodeoxyribonucleotide 5'R-p-TAATACGACTCACTATAGG3' (SEQ ID NO: 2) having a phosphate group-protected body at the 5' end, and a reverse phase
- the desired product was isolated and purified by HPLC. The analysis results of the mixture before purification and the analysis results after isolation and purification are shown.
- (b) of the figure shows the results of MALDI-TOF molecular weight analysis of the oligodeoxynucleotide after isolation and purification. At the same time as the peak of the target product was confirmed, the main peak was confirmed as the protective group detached by laser light irradiation during the measurement.
- (c) of the figure shows the results of reverse-phase HPLC analysis of the oligodeoxynucleotides after isolation and purification by reverse-phase HPLC, and of the oligonucleotides after irradiation with 365 nm light. Quantitative progress of the deprotection reaction was confirmed.
- the HPLC analysis conditions for (a) and (c) of the figure are as follows.
- FIG. 3 shows the synthesis and purification of 5'-phosphorylated oligonucleotides using amidite reagent_2, and the reverse-phase HPLC analysis results of deprotection reaction after purification.
- amidite reagent_2 is used to synthesize a 19-base-long oligodeoxyribonucleotide 5'R-p-TAATACGACTCACTATAGG3' (SEQ ID NO: 2) having a phosphate group-protected body at the 5' end, and reverse phase
- the desired product was isolated and purified by HPLC. The analysis results of the mixture before purification and the analysis results after isolation and purification are shown.
- (b) of the figure shows the results of MALDI-TOF molecular weight analysis of the oligodeoxynucleotide after isolation and purification. At the same time as the peak of the target product was confirmed, the main peak was confirmed as the protective group detached by laser light irradiation during the measurement.
- (c) of the figure shows the results of reverse-phase HPLC analysis of the oligodeoxynucleotide after isolation and purification by reverse-phase HPLC, and of the oligonucleotide after irradiation with 365 nm light. Quantitative progress of the deprotection reaction was confirmed.
- the HPLC analysis conditions for (a) and (c) in the figure are as follows.
- FIG. 4 shows the synthesis and purification of 5'-phosphorylated oligonucleotides using amidite reagent_3, and the results of reverse-phase HPLC analysis of deprotection reaction after purification.
- amidite reagent_3 is used to synthesize a 19-base-long oligodeoxyribonucleotide 5'R-p-TAATACGACTCACTATAGG3' (SEQ ID NO: 2) having a phosphate group protected at the 5' end, and reverse phase
- the desired product was isolated and purified by HPLC. The analysis results of the mixture before purification and the analysis results after isolation and purification are shown.
- (b) of the figure shows the results of MALDI-TOF molecular weight analysis of the oligodeoxynucleotide after isolation and purification. At the same time as the peak of the target product was confirmed, the main peak was confirmed as the protective group detached by laser light irradiation during the measurement.
- the HPLC analysis conditions for (a) of the figure are as follows.
- the mixture before purification containing the phosphate-protected target compound was analyzed by reverse-phase HPLC to determine the content of the target compound.
- the HPLC analysis conditions are as follows. System used, Chromaster (Hitachi High Tech); column, YMC Hydrosphere C18 (250 ⁇ 4.6 mm ID, YMC) eluent A, 50 mM triethylammonium acetate (pH 7.0) containing 5% acetonitrile; eluent B, acetonitrile; gradient Conditions, 0-100% B (0-20 min); flow rate, 1.0 mL/min; column temperature, room temperature; detection wavelength, 260 nm.
- FIG. 1 a schematic diagram showing the chemical structure of the target substance, and its elution time and content % are described.
- (f) of the figure is a table summarizing the elution time and production yield (%) of the target 5′-terminal phosphate group-protected product from the results shown in (a) to (e). *Chem. Eur. J. 23, 5210 (2017). These results show that the amidite reagent_1 and the amidite reagent_2 of the present invention have a higher yield of the target nucleic acid than the conventional compounds.
- amidite reagent_1 when comparing amidite reagent_1 and amidite reagent_2, the yield of amidite reagent_1 in which R1 is a t-butyl group is higher than that of amidite reagent_2 in which R1 is a C19 linear alkyl group, and the yield of amidite reagent_2 It can be seen that amidite reagent_1 is more preferable than Amidite Reagent_1 in terms of yield.
- RNA synthesis Using an amidite reagent_1 and a commercially available phosphoramidite reagent (ChemGenes), an automatic nucleic acid synthesizer NR-2A 7MX (Nippon Techno Service) according to a conventional method oligoribonucleotides of 107-base length or 131-base length whose sequences are shown in the figure were synthesized. After completion of the synthesis, 1 mL of a 1:1 mixed solution of concentrated aqueous ammonia and 40% methylamine aqueous solution was added to the solid-phase carrier, and deprotected by heating at 65° C. for 15 minutes.
- the supernatant was filtered through a Millex LH filter (0.45 ⁇ m, Merck) and dried under reduced pressure using a centrifugal evaporator.
- the residue was dissolved by adding 1 mL of 1 M TBAF, THF solution and heated at 35° C. overnight.
- 1 mL of 1 M Tris-HCl (pH 7.5) buffer was added and mixed, concentrated with a centrifugal evaporator, and then desalted with a NAP-25 column (GE Healthcare).
- RNA concentration was calculated by redissolving the RNA in super-deionized water and measuring the absorbance at 260 nm.
- FIG. 6 shows the synthesis and purification of 5′-phosphorylated oligonucleotides using amidite reagent_1, and the results of reverse-phase HPLC analysis of deprotection reaction after purification.
- the 5'-end phosphorylated 107-nucleotide long RNA could be purified by reverse-phase HPLC after deprotection.
- (a) of the figure shows a chemically synthesized RNA sequence, and mG indicates that the hydroxyl group at the 2'-position is methylated.
- (b) of the figure shows the results of reverse-phase HPLC analysis of the mixture containing 107-ntRNA after deprotection.
- Peak(s)_1 contained the full-length 107-nt RNA of interest.
- Peak(s)_1 contained a synthetic by-product that was not extended to full length, and Peak(s)_3 contained an unidentified by-product.
- the polyacrylamide gel was stained with SYBR Green II nucleic acid staining reagent to visualize RNA.
- (d) of the figure shows the results of reversed-phase HPLC analysis of the target product (107-ntRNA) after purification using reversed-phase HPLC.
- the HPLC analysis conditions used in (b) and (d) of the figure are as follows. System used, LaChrom Elite (Hitachi High Tech): Column, YMC Triat Bio C4 (250 ⁇ 4.6 mm ID); Eluent A, 50 mM triethylammonium acetate (pH 7.0) containing 5% acetonitrile; Eluent B, Acetonitrile; Gradient condition, 5-20% B (0-20 min): Flow rate 1.0 mL/min: Column temperature, 50°C; Detection wavelength, 260 nm.
- FIG. 7 shows that the 5'-terminal phosphorylated 107-nucleotide long RNA synthesized using amidite reagent_1 was quantitatively deprotected by light irradiation.
- (a) of the figure shows a conceptual diagram of the deprotection reaction, in which 365 nm light is irradiated to remove the protecting group of the 5′ phosphate group.
- (b) and (c) of the figure show 107-ntRNA before light irradiation (before deprotection), (b) before light irradiation (before deprotection), and (c) light irradiation (deprotection). after protection) are shown, respectively. From this result, quantitative deprotection by light irradiation was confirmed.
- HPLC analysis conditions used in (b) and (c) of the figure are as follows.
- System used LaChrom Elite (Hitachi High Tech): Column, YMC Hydrosphere C18 (250 ⁇ 4.6 mm ID); Eluent A, 50 mM triethylammonium acetate (pH 7.0) containing 5% acetonitrile; Eluent B, acetonitrile; Gradient conditions, 5-20% B (0-20 min): flow rate 1.0 mL/min: column temperature, 50°C; detection wavelength, 260 nm.
- FIG. 8 shows that the 5′-end phosphorylated 131-nucleotide long RNA synthesized using amidite reagent_1 can be isolated and purified by reversed-phase HPLC after deprotection, and can be quantitatively deprotected by subsequent light irradiation. It is a figure which shows that there exists.
- (a) of the figure shows a chemically synthesized RNA sequence, and mG indicates that the hydroxyl group at the 2'-position is methylated.
- (b) of the figure shows the results of reverse-phase HPLC analysis of the mixture containing 131-ntRNA after deprotection. The eluate was divided into 3 groups and fractionated and purified (Peak(s)_1, 2, 3).
- Peak(s)_2 contained the full-length 131-nt RNA of interest.
- Peak(s)_1 contained synthetic by-products that were not extended to full length, and Peak(s)_3 contained unidentified by-products.
- the polyacrylamide gel was stained with SYBR Green II nucleic acid staining reagent to visualize RNA.
- (d) of the figure shows the results of reversed-phase HPLC analysis of the 5′ phosphate group-protected form (131-ntRNA) after purification using reversed-phase HPLC.
- (e) of the figure shows the results of reversed-phase HPLC analysis of the target product (131-ntRNA) deprotected by irradiation with 365-nm light after purification using reversed-phase HPLC.
- the HPLC analysis conditions used in (b) of the figure are as follows.
- Chromaster (Hitachi High Tech): Column, YMC Hydrosphere C18 (250 ⁇ 4.6 mm ID); Eluent A, 50 mM triethylammonium acetate (pH 7.0) containing 5% acetonitrile; Eluent B, acetonitrile; Conditions, 5-15% B (0-20 min): flow rate 1.0 mL/min; column temperature, 50°C; detection wavelength, 260 nm.
- FIG. 9 shows the outline of this experiment.
- RNA is transcribed and synthesized using T7 RNA polymerase.
- NTP ATP, UTP, GTP, CTP
- the transcription reaction is initiated by either GTP or GMP. Therefore, depending on the GTP/GMP mixing ratio, the 5′-terminal hydroxyl group of RNA becomes a mixture of triphosphate and monophosphate, but it was difficult to separate them by HPLC or the like (reaction scheme in the upper part of the figure).
- the present invention uses R-pG in which a protective group is attached to the 5' end of guanosine.
- the transcript becomes a mixture of 5'-terminal triphosphate and protected (R)-monophosphate depending on the mixing ratio of GTP/R-pG.
- the protecting groups it was possible to isolate the protected (R)-monophosphorylated RNA from the mixture, such as by reverse-phase HPLC.
- RNA transcripts 34, 100, 250, 650, and 1000 base lengths
- the reaction solution was electrophoresed on a denatured polyacrylamide gel to confirm the production of RNA.
- proteins were removed by phenol/chloroform extraction, and phenol was removed by chloroform extraction.
- transcription substrate R-pG was synthesized according to the conventional phosphoramidite method using an automated nucleic acid synthesizer. performed the synthesis. After synthesis, deprotection was performed, and then analysis and purification were performed by HPLC. The results are shown in FIG.
- RNAs synthesized by transcription have lengths of 34nt, 100nt, 250nt, 650nt and 1078nt, respectively.
- the reaction solution was analyzed by electrophoresis using a denatured polyacrylamide gel to confirm RNA of the desired chain length. The results are shown in FIG.
- RNA analysis is shown in Figures 12-20.
- the structure of RNA in each peak (peaks 1 to 3) of HPLC is as follows. Peak 1 is a mixture of triphosphate and monophosphate without protective groups, peak 2 is triphosphate without protective groups, and peak 3 is monophosphate with protective groups.
- FIG. 24 shows an experiment for synthesizing a branched cap analog compound using the novel phosphorylation reagent.
- (a) and (b) of the figure show synthetic schemes of 2- and 3-branched cap analog compounds. 1 and 4 were synthesized using an automated nucleic acid synthesizer and purified by reversed-phase HPLC utilizing the hydrophobic protecting groups of the novel phosphorylation reagents. The protective group of the phosphoric acid moiety was removed by light irradiation to obtain capped precursors 2 and 5.
- (c) and (d) of the figure show the results of reverse phase HPLC analysis of the mixture containing compounds 1 and 4 after deprotection and dialysis. Since the desired product was included in peaks with retention times of 12.17 minutes (c), 12.96 minutes and (d), it was isolated.
- (e) and (f) of the figure show the results of reverse phase HPLC analysis of compounds 2 and 5.
- Guanosine derivatives 1 and 4 were synthesized on a micromole scale using a nucleic acid synthesizer according to the phosphoramidite method.
- a commercially available branched amidite reagent (Symmetric Double Phosphoramidite, Glen Research, cat# 10-1920: Trebler Phosphoramidite, Glen Research, Inc.) was added to a CPG solid-phase carrier (ChemGenes, cat#N-3203-10) supporting a guanosine protector. #10-1922) were combined.
- the rG nucleotide and the novel chemical phosphorylation reagent (compound 1) were combined to obtain the target biantennary type compound (1) and triantennary type compound (4).
- An equal mixture of 1 mL of concentrated aqueous ammonia and 40% methylamine aqueous solution was added to the solid phase carrier and heated at 65° C. for 15 minutes. The supernatant was dried under reduced pressure, the residue was dissolved in 100 ⁇ L of dimethylsulfoxide, 125 ⁇ L of triethylamine trihydrofluoride was added, and the mixture was heated at 65°C for 2 hours.
- LaChrom Elite Hitachi High Tech
- Column YMC Triart Bio C4 (250 x 4.6 mm I.D.); Eluent A, 50 mM triethylammonium acetate (pH 7.0) containing 5% acetonitrile; Liquid B, acetonitrile; Gradient conditions, 5-80% B (0-20 minutes): Flow rate 1.0 mL/min: Column temperature, 25°C; Detection wavelength, 260 nm.
- the target product isolated by reversed-phase HPLC was concentrated and deprotected by irradiation with 365-nm light. Dialysis was performed overnight using a dialysis membrane (manufactured by Spectra/Pore, Biotech CE Tubing; MWCO 500-1000 Da) to obtain target compounds 2 and 5 in yields of 72 nmol and 96 nmol, respectively. The purity of the obtained target compounds 2 and 5 was confirmed by reverse phase HPLC. Analysis conditions are as follows. System used, LaChrom Elite (Hitachi High Tech): Column, YMC Hydroshere C18 250 ⁇ 4.6 mm I.V. D.
- eluent A 50 mM triethylammonium acetate (pH 7.0) containing 5% acetonitrile
- eluent B acetonitrile
- gradient conditions 0-20% B (0-20 min): flow rate, 1.0 mL/min: Column temperature, 25°C; detection wavelength, 260 nm.
- Imidazole (0.837 g, 12.3 mmol, 10 eq.) and 2,2′-dipyridyl disulfide (0.811 g, 3.68 mmol, 3.0 eq.) were added thereto and dissolved in DMSO (10.0 mL). Further, triethylamine (0.372 mL, 3.68 mmol, 3.0 eq.) and triphenylphosphine (0.965 g, 3.68 mmol, 3.0 eq.) were added and stirred overnight at room temperature.
- reaction tracking was performed by HPLC (column: CoresepSB, solvent: (A) 20 mM ammonium acetate 5% ACN, (B) 400 mM ammonium acetate 5% ACN, gradient: B conc. 2-22 min., 0-100%, flow rate : 1.0 mL/min., detection wavelength: 260 nm).
- a solution of sodium perchlorate (4.51 g, 36.8 mmol, 30 eq.) in dehydrated acetone (200 mL) was added thereto and ice-cooled in an ice bath. The deposited precipitate was collected by suction filtration to obtain white powdery compound 10 (0.530 g, 1.22 mmol, yield>99%).
- Various spectra showed good agreement with literature values [1] .
- reaction tracking was performed by HPLC (column: Hydrosphere C18, solvent: (A) 20 mM triethylammonium acetate (pH 7.0), (B) ACN, gradient: B conc. 0-30 min., 0-80%, flow rate: 1.0 mL/min., detection wavelength: 260 nm).
- HPLC columnumn: Hydrosphere C18, solvent: (A) 20 mM triethylammonium acetate (pH 7.0), (B) ACN, gradient: B conc.
- dsDNA transcription template (1105-bp PCR product containing T7 promoter sequence 5′TAATACGACTCACTATAG3′ (SEQ ID NO: 1)
- 2 mM ATP 2 mM UTP
- 2 mM CTP 0.5 mM GTP
- 2 mM compound 11 photolytic AppG dinucleotide compound with a hydrophobic protecting group
- 40 mM Tris-HCl pH 8.0
- 8 mM MgCl 2 2 mM spermidine
- 5 mM DTT 2.5 units/ ⁇ L T7 RNA polymerase
- the reaction mixture in both cases was mixed with an equal mixture of TE-saturated phenol and chloroform and vigorously mixed to remove any resulting proteinaceous insolubles. After the aqueous layer was extracted with chloroform, it was purified using an Amicon Ultra 10K ultrafiltration filter unit and analyzed by reverse phase HPLC.
- FIG. 25 shows that compound 11 can be added to an in vitro transcription reaction using T7 RNA polymerase to obtain protective group-containing adenylated RNA.
- (a) of the figure shows an outline of the experiment. A transcribed RNA is obtained as a mixture of one having a triphosphate structure at the 5'-end (ppp-RNA) and a target protecting group-containing adenylated structure (R-App-RNA). Since the latter has high hydrophobicity unlike the former, it can be isolated by reversed-phase HPLC using this property.
- (b) of the figure shows the results of reverse phase HPLC analysis of the 1078 base long RNA transcription reaction.
- RNA having a length of 250 bases was prepared by heating the reaction solution described below at 37° C. for 2 hours.
- 5 ng/ ⁇ L dsDNA transcription template (276-bp PCR product containing T7 promoter sequence 5′TAATACGACTCACTATAG3′ (SEQ ID NO: 1)
- 2 mM ATP 2 mM UTP
- 2 mM CTP 0.5 mM GTP
- 2 mM compound 11 photolytic AppG dinucleotide compound with a hydrophobic protecting group
- 8 mM MgCl 2 2 mM spermidine
- 5 mM DTT 2.5 units/ ⁇ L T7 RNA polymerase
- the reaction solution was mixed with an equal volume mixture of TE-saturated phenol and chloroform, and vigorously mixed to remove proteinaceous insoluble matter.
- the aqueous layer was extracted with chloroform and then purified using an Amicon Ultra 10K ultrafiltration filter unit. This was analyzed by reverse-phase HPLC, and photodegradable hydrophobic protective group-containing adenylated RNA was fractionated and purified. Eluates containing target RNA were desalted using Amicon Ultra 10K ultrafiltration filter units.
- the protective group-containing adenylated RNA obtained above was deprotected by light irradiation.
- RNA solution (16.5 ng RNA/ ⁇ L, 100 ⁇ L) was added to a transparent 96-well multiwell plate and irradiated with 365 nm light at a light intensity of 4 mW/cm 2 for 15 minutes using a MAX-305 light source device (Asahi Spectro).
- FIG. 26 shows that protective group-containing adenylated RNA prepared by transcription reaction can be deprotected by light irradiation and converted into the desired adenylated RNA.
- (a) of the figure shows an outline of the experiment.
- Figure (b) shows reversed-phase HPLC analysis of protective group-containing adenylated RNA (after isolation).
- Panel (c) shows reversed-phase HPLC analysis after irradiation of protective group-containing adenylated RNA (after isolation) with 365-nm light.
- reaction mixture was gradually warmed to room temperature. After stirring for 4 h, the reaction was quenched by adding saturated aqueous NH 4 Cl. The reaction was quenched by adding an aqueous solution of NH 4 Cl. The mixture was extracted three times with ethyl acetate. The organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography eluted with hexanes followed by 17 ⁇ 25% ethyl acetate/hexanes to give compound 2 as a dark brown solid (11 g, 65% yield). All spectral data of the compounds were in agreement with the literature.
- reaction mixture was treated in an ice bath with 0.5 M tetrabutylammonium dihydrogen phosphate/CH 3 CN (4.6 mL, 2.3 mmol) and tributylamine (1.6 mL, 1.3 g, 6.9 mmol) was added.
- the reaction mixture was warmed to room temperature and stirred at room temperature for 16 hours. Afterwards, 0.2 M TEAB buffer (pH 7.9, 10 mL) was added to quench the reaction mixture. After stirring for 1-2 hours at room temperature, the clear solution was diluted with water and washed 5 times with dichloromethane. The aqueous layer was concentrated on a rotary evaporator at 50°C.
- This crude product was passed through a YMC-Triart C8 column (250 ⁇ 10.0 mm ID, S-5 ⁇ m, 12 nm, flow rate 3 mL/min, temperature 50° C.) with 0.1 M triethylammonium carbonate buffer (pH 7.0). 9) was purified by reversed-phase HPLC using a linear gradient of 10-80% CH 3 CN over 25 minutes. Fractions containing product were pooled and acidified to pH 4.0 by adding a few drops of acetic acid.
- guanosine 5′-phosphorimidazolide monosodium salt (compound 10)
- the synthesis of guanosine-5'-phosphorimidazolide (compound 10) was performed according to the literature. Guanosine 5′-monophosphate (300 mg, 0.53 mmol), imidazole (290 mg, 4.2 mmol), 2,2′-dithiodipyridine (350 mg, 1.5 mmol) in N,N-dimethylformamide (7.5 mL). 6 mmol), triethylamine (150 ⁇ L, 110 mg, 1.1 mmol) was added followed by triphenylphosphine (420 mg, 1.6 mmol).
- Trifluoromethanesulfonic acid (1.12 g, 662 ⁇ L, 7.46 mmol) was added to the cooled suspension and stirred at ⁇ 40° C. for 19 hours.
- the reaction mixture was quenched by the addition of triethylamine (24.0 mL), diluted with ethyl acetate and washed with saturated aq. Washed twice with NaHCO3 .
- the organic layer was dried over Na2SO4 and concentrated. The residue was purified by silica gel column chromatography eluted with 0-3.2% methanol/dichloromethane to give compound 5 as a yellow foam (3.12 g, 76% yield).
- guanosine 5′-phosphorimidazolide monosodium salt (compound 10)
- the synthesis of guanosine-5'-phosphorimidazolide (compound 10) was performed according to the literature. Guanosine 5′-monophosphate (300 mg, 0.53 mmol), imidazole (290 mg, 4.2 mmol), 2,2′-dithiodipyridine (350 mg, 1 .6 mmol) was added triethylamine (150 ⁇ L, 110 mg, 1.1 mmol) followed by triphenylphosphine (420 mg, 1.6 mmol).
- This mixture was incubated at 37°C for 2 days.
- 38 mM aq. EDTA (1.6 mL, 60 ⁇ mol) was added to quench the reaction mixture and diluted with 0.2 M TEAB buffer (pH 7.9, 600 ⁇ L, pH adjusted to around 4.0).
- This mixture was passed through a YMC-Triart C8 column (250 ⁇ 4.6 mm ID, S-5 ⁇ m, 12 nm, flow rate 1 mL/min, temperature 50° C.) with 0.1 M triethylammonium carbonate buffer (pH 7.0). 9) was purified by reverse-phase HPLC running a 5-80% linear gradient of CH 3 CN in 25 minutes.
- FIG. 27 shows the results of various analyzes of the synthesized novel cap analog compound, cap analog_1. All of these analytical results indicated that the target compound was obtained with good purity.
- (a) of the figure shows the chemical structure of cap analog_1.
- (b) of the figure shows the results of reverse-phase HPLC analysis after isolation and purification of cap analog_1.
- the analysis conditions are as follows.
- FIG. 28 shows the results of various analyzes of cap analog_2, which is a synthesized novel cap analog compound. All of these analytical results indicated that the target compound was obtained with good purity.
- (a) of the figure shows the chemical structure of cap analog_2.
- (b) of the figure shows the results of reverse-phase HPLC analysis after isolation and purification. Analysis conditions are as follows.
- Fig. 29 shows that the novel cap analog-introduced RNA could be isolated and purified by reverse-phase HPLC.
- a cap analog compound was added, NanoLuc luciferase mRNA (650 base length) was transcribed and synthesized using T7 RNA polymerase, and the transcription reaction was performed on a denaturing polyacrylamide gel (5%, 7.5 M urea as a denaturant). including) was analyzed by electrophoresis.
- the composition of the transcription reaction solution and reaction conditions are shown below.
- dsDNA transcription template 15 ng/ ⁇ L dsDNA transcription template (a 676-bp PCR product containing the T7 promoter sequence 5′TAATACGACTCACTATAG3′ (SEQ ID NO: 1) containing the NanoLuc luciferase gene coding region from the pNL1.1TK vector (Promega)), 2 mM ATP , 2 mM UTP, 2 mM CTP, 2 mM GTP or [0.5 mM GTP + 2 mM cap analog], 40 mM Tris-HCl (pH 8.0), 8 mM MgCl 2 , 2 mM spermidine, 5 mM DTT, 10 units/ ⁇ L T7 RNA polymerase (Takara Bio) .
- (b) of the figure shows the results of re-analysis after fractionating the eluted RNA around 17.9 minutes shown in (a).
- (e) of the figure shows the results of re-analysis after fractionating the eluted RNA around 16.8 minutes shown in (d).
- (c) of the figure shows the results of RNA analysis after fractionating the eluted RNA around 17.9 minutes shown in (a) and then irradiating it with 365-nm light at 4 mW/cm 2 for 10 minutes. Since the elution time of RNA completely shifted from about 17.9 minutes to about 15 minutes, it can be judged that the hydrophobic protecting group of the cap analog site was completely deprotected.
- (f) of the figure shows the results of RNA analysis after fractionating the eluted RNA around 16.8 minutes shown in (d) and then irradiating 365-nm light at 4 mW/cm 2 for 10 minutes. Since the RNA elution time completely shifted from around 16.8 minutes to around 15 minutes, it could be determined that the hydrophobic protecting group of the cap analog site was completely deprotected. HPLC analysis conditions are shown below.
- FIG. 30 shows translational activity evaluation of NanoLuc luciferase mRNA after reverse-phase HPLC isolation and purification.
- the expression level of mRNA prepared with no cap analog added was shown as 1.
- HPLC-isolated mRNA (20 ng/well) was introduced into HeLa cells (1 ⁇ 10 4 cells/well seeded in a 96-well multiwell plate the day before) by lipofection (Lipofectamine Messenger MAX, 0.3 ⁇ L/well). After culturing for 7 hours, the cells were lysed, and the amount of expressed protein (NanoLuc luciferase) contained in the lysate was measured using the Nano-Glo Luciferase Assay System (Promega).
- mRNA capped with the novel cap analog compound exhibited high translational activity.
- mRNA modified with cap analog_1 did not show high translational activity without light irradiation and deprotection, but mRNA modified with cap analog_2 showed similar high translation activity before and after light irradiation and deprotection.
- Cap analog_1 is one in which the hydrogen bond of the base portion is protected, and translational activity can be completely suppressed in the protected state. It can be applied to mRNA that activates translation in vivo by irradiating with light.
- FIG. 31 shows translational activity evaluation of NanoLuc luciferase mRNA after reverse-phase HPLC isolation and purification.
- the expression level of mRNA prepared with no cap analog added was shown as 1.
- Translation activity was compared with similarly prepared mRNA using a commercially available ARCA cap analog (JENA BIOSCIENCE, cat#78862).
- HPLC-isolated mRNA (5 ng/well) was introduced into HeLa cells (1 ⁇ 10 4 cells/well seeded in a 96-well multiwell plate the day before) by lipofection (Lipofectamine Messenger MAX, 0.3 ⁇ L/well).
- mRNA capped with cap analog_2 showed higher translational activity compared to RNA capped with ARCA. It is presumed that this high translational activity is due to the fact that purification using a hydrophobic protecting group yielded mRNA with a capping rate of 100%.
- FIG. 32 shows chemical capping reactions of RNA using hydrophobic chemical capping reagents.
- (a) of the figure shows a conceptual diagram of the reaction.
- (b) of the figure shows the results of reverse-phase HPLC analysis of the reaction of 5'-monophosphorylated 19 base long RNA with a hydrophobic chemical capping reagent.
- (c) of the figure shows the results of denaturing PAGE analysis.
- a hydrophobic capping reagent (compound 8) was reacted with a 19 base-long 5′-end phosphorylated RNA (5′-p-GAACGUGCGAAAGUCCACA-3′: SEQ ID NO: 15) as follows. 38 ⁇ L of an aqueous solution containing 2 nmol of RNA and 500 nmol of calcium chloride was frozen and dried to dryness by a freeze-drying method. 18.5 ⁇ L of dimethyl sulfoxide (DMSO), 26.5 ⁇ L of hydrophobic capping reagent DMSO solution (19 mM) and 5 ⁇ L of 1-methylimidazole were added and mixed, and the mixture was heated at 55° C. for 5 hours.
- DMSO dimethyl sulfoxide
- 2-6.5 ⁇ L of hydrophobic capping reagent DMSO solution (19 mM) and 5 ⁇ L of 1-methylimidazole were added and mixed, and the mixture was heated at 55° C. for 5 hours.
- RNA was collected by alcohol precipitation and analyzed by reverse phase HPLC and denatured polyacrylamide gel electrophoresis (PAGE).
- HPLC analysis conditions shown in (b) of the figure are as follows.
- RNA was electrophoresed with 15% acrylamide (acrylamide:bis 19:1) containing 7.5 M urea as a denaturant, and the gel was stained with the nucleic acid staining reagent SYBR Green II. (Bio-Rad).
- reaction mixture was diluted with water and filtered through a membrane filter to remove insolubles.
- the resulting suspension was transferred to a centrifuge tube and centrifuged at 3,500 rpm for 10 minutes. The supernatant was removed and the precipitate was washed 5 times with acetone. The solid was dried in a desiccator over P2O5 under reduced pressure to give guanosine 5'-diphosphate derivative imidazolide disodium salt (5) as a white solid.
- the powdery molecular sieve 3 ⁇ was removed by filtering the resulting reaction solution through celite.
- the filtrate was diluted with ethyl acetate and washed with water.
- the organic layer was dried over anhydrous sodium sulfate and then concentrated.
- the residue was subjected to silica gel column chromatography eluting with 2.4-6.3% methanol/dichloromethane containing triethylamine to give the target compound (745 mg, quantitative) as a white amorphous solid.
- Trifluoroacetic acid (546 mg, 367 ⁇ L, 4.79 mmol) was added to a suspension of guanosine derivative 4 (56.9 mg, 63.9 ⁇ mol) in methanol (1.07 mL) and water (533 ⁇ L). added. After stirring for 19 hours at room temperature, the reaction mixture was diluted with a mixture of methanol/water (2:1, v/v, 5.00 mL). The mixture was concentrated and purified by silica gel column chromatography eluting with 1.9-9.1% methanol/dichloromethane to give the desired compound (24.3 mg, 71.5% yield) as a white solid.
- the mixture was diluted with water (14.0 mL) and purified by reverse-phase HPLC (instrument: Shimadzu Prep, column: YMC-Actus Triart C8 (Preparative, 250 ⁇ 20.0 mm I.D., solvent A: 50 mM TEAA buffer (pH 6.0)). 0, containing 0.5% CH 3 CN), solvent B: CH 3 CN, 5-80% B gradient (25 minutes), flow rate: 10 ml/min, detection: 254 nm).
- trifluoromethanesulfonic acid (756 mg, 448 ⁇ L, 5.04 mmol) was added to the suspension and stirred at ⁇ 40° C. for 16.5 hours.
- the reaction mixture was quenched by adding triethylamine (16.8 mL) and then diluted with ethyl acetate (100 mL).
- the mixture was saturated aqueous sodium bicarbonate solution (100 mL x 2). Washed with sodium thiosulfate (100 mL x 2), and brine (100 mL x 1).
- the organic layer was dried over Na2SO4 and concentrated.
- Fractions containing the target product were combined, concentrated and lyophilized to give the target compound as the triethylammonium form.
- the target triethylammonium salt was dissolved in methanol (1.00 mL).
- target compound 8 (280 mg, 410 ⁇ mol, 72% yield).
- solvent A water
- solvent B 1.5 MTEAB buffer (pH 7.7, with 10% CH 3 CN).
- N7-methyl-guanosine 5′-diphosphate imidazolide disodium salt 1 (65.0 mg, 89.4% yield) as a white solid.
- the following shows a synthetic scheme for nitrobenzyl-modified nucleotide phosphoroimidazolide compounds that are removable by reducing conditions.
- the coupling step involved 100 mM 2′-O-methyl-adenosine (n-benzoyl)-CE-phosphoramidite (ChemGenes), 100 mM 2′-O-methyl-N6-methyl-adenosine (n-benzoyl)-CE-phosphoramidite (ChemGenes) in CH 3 CN.
- the product-containing fractions were collected and concentrated to give the di-/tri-nucleotides.
- the extinction coefficient ( ⁇ 260) 40,100 or 33,200 M -1 cm -1 (for trinucleotide cap analogs) and 50,200, 46,800, or 43,300 M -1 cm -1 (for tetranucleotide cap analogues) was used.
- the mixture was diluted with 5.24 volumes of water and purified by reverse phase HPLC. Fractions containing the target compound were collected, concentrated and lyophilized to give the target tri/tetranucleotide cap analogs as triethylammonium salts.
- the product was redissolved in methanol (2.00 mL). A solution of 190 mM NaClO 4 (24.0 mL) in acetone was added to the mixture and the resulting suspension was centrifuged (4,000 rpm, 20 min). The supernatant was discarded and the precipitate was resuspended in acetone. The suspension centrifugation process was repeated 3-4 more times.
- the product was redissolved in methanol (2.0 mL).190 mM NaClO.sub.4 in acetone (12 mL) was added to the mixture and the resulting suspension The liquid was centrifuged (4,500 rpm, 20 min).The supernatant was discarded and the precipitate was resuspended in acetone.The suspension centrifugation process was repeated four more times. The precipitate was dried under reduced pressure.
- N7-methylguanosine diphosphate imidazolide disodium salt (67.4 mg, 87.2 ⁇ mol) and 2-nitroimidazole (14.8 mg, 131 ⁇ mol) were added.
- the mixture was incubated at 37° C. for 3 days and the reaction mixture was diluted with water (14.0 mL).
- the product was redissolved in methanol (2.0 mL). 190 mM NaClO 4 (12 mL) in acetone was added to the mixture and the resulting suspension was centrifuged (4,500 rpm, 20 min). The supernatant was discarded and the precipitate was resuspended in acetone. The suspension centrifugation process was repeated four more times.
- N7-methylguanosine diphosphate imidazolide disodium salt (11.7 mg, 15.1 ⁇ mol) and 2-nitroimidazole (2.57 mg, 22.7 ⁇ mol) were added.
- the mixture was incubated at 37° C. for 2.5 days and the reaction mixture was diluted with water (14.0 mL).
- the product was redissolved in methanol (1.0 mL).190 mM NaClO4 in acetone (12 mL) was added to the mixture to give The suspension was centrifuged (4,000 rpm, 20 min). The supernatant was discarded and the precipitate was resuspended in acetone.The suspension centrifugation process was repeated four more times. The precipitate was dried under reduced pressure.
- the target cap analog 4 was obtained as the sodium salt.
- the mixture was diluted with water (13.0 mL) and purified by reverse-phase HPLC (instrument: Shimadzu Prep, column: YMC-ActusTriart C8 (Preparative, 250 ⁇ 20.0 mm I.D.), solvent A: 50 mM TEAA buffer (pH 6.0). , 0.5% CH 3 CN), solvent B: CH 3 CN, linear gradient 5-80% B (25 min), flow rate: 10 ml/min, detection: 254 nm).
- N7-methylguanosine diphosphate imidazolide disodium salt (13.6 mg, 17.6 ⁇ mol) and 2-nitroimidazole (3.00 mg, 26.5 ⁇ mol) were added.
- the mixture was incubated at 37° C. for 2.5 days and the reaction mixture was diluted with water (10.0 mL).
- the product was redissolved in methanol (2.0 mL).190 mM NaClO4 in acetone (12 mL) was added to the mixture and the resulting suspension was Centrifuged (4,500 rpm, 20 min). The supernatant was discarded and the precipitate was resuspended in acetone.The suspension centrifugation process was repeated four more times. The precipitate was dried under vacuum to remove sodium
- the target cap analogue 9 (16.0 mg, 8.70 ⁇ mol, 42.2% yield) was obtained as a salt and the yield was calculated using the absorbance of the product at 260 nm measured by NanoDrop.
- N7-methylguanosine diphosphate imidazolide disodium salt (12.1 mg, 15.7 ⁇ mol) and 2-nitroimidazole (2.67 mg, 23.6 ⁇ mol) were added.
- the mixture was incubated at 37° C. for 2.5 days and the reaction mixture was diluted with water (14.0 mL).
- the product was redissolved in methanol (1.0 mL).190 mM NaClO.sub.4 in acetone (12 mL) was added to the mixture and the resulting The resulting suspension was centrifuged (4,000 rpm, 20 min).The supernatant was discarded and the precipitate was resuspended in acetone. The suspension centrifugation process was repeated four more times. The precipitate was removed under reduced pressure. Drying afforded the target cap analog 12 as the sodium salt.
- SEQ ID NO: 7 is the DNA sequence of the Luc2 mRNA transcription synthesis template.
- SEQ ID NO: 8 is the sequence of Luc2 mRNA transcribed using the template DNA of SEQ ID NO: 7.
- FIG. 33 is a diagram showing the results of HPLC purification of co-transcription reaction product mRNA using the synthesized cap analog.
- (a) of this figure shows the results of HPLC purification after co-transcription reaction using a trinucleotide cap analog (Nb-m7GpppApG). The uncapped compound was eluted at a retention time of 14 minutes, and the capped compound was eluted at 15 minutes.
- (b) of this figure is the result of HPLC purification after deprotection by light irradiation. The deprotection reaction by light irradiation shifted the peak to a retention time of 14 minutes, indicating that the photodegradable protective group was removed and capped mRNA was obtained with high purity.
- FIG. 34 is a diagram showing the results of HPLC purification of co-transcription reaction product mRNA using the synthesized cap analogue.
- (a) of this figure shows the results of HPLC purification after co-transcription reaction using a trinucleotide cap analog (Nb-m7GpppA(2'OCH3)pG). The uncapped compound was eluted at a retention time of 14 minutes, and the capped compound was eluted at 15 minutes.
- (b) of this figure is the result of HPLC purification after deprotection by light irradiation. The deprotection reaction by light irradiation shifted the peak to a retention time of 14 minutes, indicating that the photodegradable protective group was removed and capped mRNA was obtained with high purity.
- FIG. 35 is a diagram showing the results of HPLC purification of co-transcription reaction product mRNA using the synthesized cap analog.
- (a) of this figure shows the results of HPLC purification after co-transcription reaction using a tetranucleotide-type cap analog (Nb-m7GpppA(2'OCH3)pG(2'OCH3)pG). The uncapped compound was eluted at a retention time of 14 minutes, and the capped compound was eluted at 15 minutes.
- (b) of this figure is the result of HPLC purification after deprotection by light irradiation. The deprotection reaction by light irradiation shifted the peak to a retention time of 14 minutes, indicating that the photodegradable protective group was removed and capped mRNA was obtained with high purity.
- FIG. 36 is a diagram showing the results of HPLC purification of co-transcription reaction product mRNA using the synthesized cap analog.
- (a) of this figure shows the results of HPLC purification after co-transcription reaction using a tetranucleotide-type cap analog (Nb-m7Gppm6A(2'OCH3)pG(2'OCH3)pG). The uncapped compound was eluted at a retention time of 14 minutes, and the capped compound was eluted at 15 minutes.
- (b) of this figure is the result of HPLC purification after deprotection by light irradiation. The deprotection reaction by light irradiation shifted the peak to a retention time of 14 minutes, indicating that the photodegradable protective group was removed and capped mRNA was obtained with high purity.
- RNA was transcribed and synthesized with T7 RNA polymerase using the double-stranded DNA as a template.
- the template double-stranded DNA was pSP73-a-luc2 (sequence described at the end of the experimental section) encoding the Luc2 gene, KOD-Plus-Neo (TOYOBO), Fw primer (cgcgcgttggccgattcatt: SEQ ID NO: 3), Rev primer ( ttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttt
- Transcription reaction solution (template DNA 15 ng/ ⁇ L, 1 ⁇ T7 RNA Polymerase buffer (Takara) (40 mM Tris-HCl (pH 8.0), 8 mM MgCl 2 , 2 mM spermidine), 10 U/ ⁇ L T7 RNA polymerase (Takara), 5 mM DTT, 2 mM ATP, 2 mM UTP, 2 mM, CTP, 2 mM GTP, 2 mM Nb- m7GpppApG or Nb- m7GpppA ( 2'OCH3 )pG or Nb- m7GpppA ( 2'OCH3 )pG( 2'OCH3 ) pG or Nb- m7 Gppp m6 A(2'OCH 3 )pG(2'OCH 3 )pG) was prepared and incubated at 37° C.
- RNA solution was added to a transparent 96-well multiwell plate and irradiated with 365 nm light at a light intensity of 4 mW/cm 2 for 15 minutes using a MAX-305 light source device (Asahi Spectro). HPLC purification was performed.
- sequence information of pSP73-a-luc2 is as shown in SEQ ID NO:5.
- FIG. 37 shows the results of evaluation of translation activity in HeLa cells of RNA transcribed and synthesized using a cap analog. RNA was transfected into HeLa cells by the lipofection method. After 24 hours, cells were lysed and firefly luciferase luminescence was measured.
- TriPure_0 is Nb-m7GpppApG
- TriPure_1 is Nb-m7GpppA(2'OCH3)pG
- TetraPure_2 is Nb-m7GpppA(2'OCH3)pG(2'OCH3)pG
- m6A_TetraPurte_2 is Nb-m7Gppm6A(2'OCH3) pG(2'OCH3)pG
- Tri_1 are mRNAs synthesized by cotranscription of a compound having a structure equivalent to CleanCap (registered trademark) Reagent AG (Trilink). Translation activity was high in the order of Cap2>Cap1>Cap0.
- TriPure_1 purified by Nb tag exhibited about twice the activity as compared to Tri_1 using the existing CleanCap (registered trademark) Reagent AG.
- HeLa cells (RIKEN Cell Bank) were cultured in Dulbecco's modified Eagle's medium (DMEM; WAKO) supplemented with 10% fetal bovine serum (FBS; Invitrogen) (37° C., 5% CO 2 ).
- DMEM Dulbecco's modified Eagle's medium
- FBS fetal bovine serum
- FIG. 38 shows the results of translational activity evaluation in JAWS II cells of RNA transcribed and synthesized using cap analogues.
- RNA was transfected into JAWS II cells by the lipofection method. After 24 hours, cells were lysed and firefly luciferase luminescence was measured.
- TriPure_0 is Nb-m7GpppApG
- TriPure_1 is Nb-m7GpppA( 2'OCH3 )pG
- TetraPure_2 is Nb-m7GpppA( 2'OCH3 )pG( 2'OCH3 )pG
- m6A_TetraPurte_2 is Nb-m7Gppm6A(2 'OCH 3 )pG(2'OCH 3 )pG
- Tri — 1 is mRNA synthesized by cotranscription of a compound having a structure equivalent to CleanCap (registered trademark) Reagent AG (Trilink). Translation activity was high in the order of Cap_2>Cap_1>Cap_0.
- TriPure_1 purified by Nb tag exhibited about 24 times higher activity than Tri_1 using the existing CleanCap (registered trademark) Reagent AG.
- Evaluation of translational activity in cells JAWSII cells: JAWSII cells (ATCC) were prepared by adding 10% fetal bovine serum (FBS; Invitrogen) and GM-CSF (5 ng/mL) to Minimum Essential Medium ⁇ (MEM ⁇ ; Thermo Fisher SCIENTIFIC) (37° C., 5% CO 2 ). The day before transfection, JAWS II cells were seeded in a 96-well plate (3.0 ⁇ 10 4 cells/well).
- Opti-MEM registered trademark
- Thermo Fisher SCIENTIFIC 0.15 ⁇ L Lipofectamine® MessengerMAX®, 50 ng mRNA diluted in 10 ⁇ L Opti-MEM® and introduced into cells. After incubation at 37° C. for 5 hours, 20 ⁇ L/well 1 ⁇ Cell Lysis Buffer (Promega) was added to lyse the cells. Luminescence was measured using the ONE-Glo® luciferase assay® (Promega).
- the amino acid sequence information of Luc2 is as shown in SEQ ID NO:6.
- Figure 39 shows a 650-base long NanoLuc luciferase (Nluc ) It is a diagram showing a comparison of mRNA translation activity. These are experimental results showing that the translation activity of the former is high.
- a of this figure shows the chemical structures and abbreviations of the cap analogs used.
- B of this figure shows the results of 5'-end analysis of mRNA after purification using hydrophobic protecting groups. Full-length mRNA was cleaved with DNAzyme at the position 23 bases from the 5' end, and the cleaved products were analyzed by denaturing polyacrylamide electrophoresis. As a result, it was found that all the mRNAs purified using the hydrophobic protective group-containing capping derivatives contained only capped 5' ends.
- C of this figure shows the results of comparison of Nluc mRNA activity using HeLa cells.
- D in this figure shows the results of comparison of Nluc mRNA activity using JAWS II cells.
- the mRNAs prepared using the hydrophobic protective group-containing cap analogs DiPure, DiPure/3'ome, and DiPure/2'ome showed higher translation activity than the mRNA prepared using the conventional cap analog ARCA.
- ARCA/AP represents dephosphorylation of the 5' end of coexisting uncapped RNA by allowing Antarctic phosphatase to act on ARCA-containing mRNA.
- NLuc mRNA The template DNA sequence of NLuc mRNA is as shown in SEQ ID NO:9.
- sequence of NLuc mRNA (transcript) is as shown in SEQ ID NO:10.
- the capped mRNA used in FIG. 39 was transcribed and synthesized from a DNA template using T7 RNA polymerase in the following reaction solution.
- 15 ng/ ⁇ L dsDNA transcription template (a 676-bp PCR product containing the T7 promoter sequence 5′TAATACGACTCACTATAG3′ (SEQ ID NO: 1) containing the NanoLuc luciferase gene coding region from the pNL1.1TK vector (Promega)), 2 mM ATP , 2 mM UTP, 2 mM CTP, 2 mM GTP, 2 mM cap analog, 40 mM Tris-HCl (pH 8.0), 8 mM MgCl 2 , 2 mM spermidine, 5 mM DTT, 10 units/ ⁇ L T7 RNA polymerase (Takara Bio).
- the final concentration of ARCA was 2 mM and the final concentration of GTP was 0.5 mM.
- DNase Takara Bio
- An equal volume mixture of TE-saturated phenol and chloroform was added to the reaction mixture, vigorously stirred, and then centrifuged to separate the aqueous layer.
- An equal amount of chloroform was added thereto, and the mixture was vigorously stirred and then centrifuged to separate the aqueous layer.
- RNA product A 1/10 volume of 3M sodium acetate buffer (pH 5.2) and an equal volume of 2-propanol were added to the aqueous layer, cooled to minus 30°C for 1 hour, and centrifuged to obtain a crude RNA product. rice field.
- the obtained crude RNA was purified by reverse phase HPLC under the following conditions.
- Dephosphorylated ARCA-containing RNA was prepared as follows. Reaction solution (250 ng/ ⁇ L ARCA-mRNA, 50 mM Bis-Tris-Propane HCl, 1 mM MgCl 2 , 0.1 mM ZnCl 2 , pH 6.0 @ 25° C., 0.25 U/ ⁇ L Antarctic phosphatase (New England Biolabs), reaction solution 20 ⁇ L) was heated at 37° C.
- Terminal analysis experiment The purified Nluc mRNA was cleaved with DNAzyme (35-base-long DNA oligonucleotide), converted to a mixture containing a 23-base-long 5' terminal fragment, and analyzed by denaturing PAGE.
- the DNAzyme sequences used were as follows: 5' TTCGAGGCCAGGCTAGCTACAACGAACGCGTCACC 3' (SEQ ID NO: 11).
- the composition of the reaction solution for the cleavage reaction is as follows. 104 ng/ ⁇ L (0.5 ⁇ M) Nluc mRNA, 1 ⁇ M DNA, 5 mM Tris-HCl (pH 8.0), 5 mM magnesium chloride, reaction volume 35 ⁇ L.
- the resulting RNA was dissolved in 3.5 ⁇ L of water, and 2.5 ⁇ L of the solution was analyzed by electrophoresis on a 15% denaturing acrylamide gel containing 7.5 M urea as a denaturing agent.
- RNA bands were visualized.
- the molecular weight of the 5'-end RNA cleavage product was calculated using a MALDI-TOF mass spectrometer, ultrafleXtreme (Bruker), using 1 ⁇ L of the alcohol-precipitated RNA solution as a substrate. 3-Hydroxypicolinic acid was used as a matrix and the measurement was performed using a linear positive mode.
- HeLa cells HeLa cells (RIKEN Cell Bank) were cultured in Dulbecco's modified Eagle's medium (DMEM; WAKO) supplemented with 10% fetal bovine serum (FBS; Invitrogen) (37°C, 5 % CO2 ). The day before transfection, the medium was removed from HeLa cells (5 ⁇ 10 3 cells/well) seeded on a 96-well plate, and replaced with 40 ⁇ L/well Opti-MEM (registered trademark) (Thermo Fisher SCIENTIFIC), where mRNA (5 ng/well) was added by mixing Lipofectamine® MessengerMAX® (0.15 ⁇ L/well) with Opti-MEM® (10 ⁇ L/well).
- DMEM Dulbecco's modified Eagle's medium
- FBS fetal bovine serum
- DMEM medium containing 10% FBS was added at 100 ⁇ L/well and further cultured. After 12, 24 and 48 hours, cell lysates were prepared using the NanoGlo® Luciferase Assay System (Promega) and the amount of NanoLuc luciferase protein contained in the lysates was assessed by luminometric method.
- JAWSII cell line JAWSII cells (ATCC) were cultured in MEM ⁇ , nucleosides (Gibco) medium supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 5 ng/mL GM-CSF (37°C, 5% CO2 ).
- FBS fetal bovine serum
- GM-CSF GM-CSF
- the medium was removed from JAWS II cells (5 ⁇ 10 3 cells/well) seeded on a 96-well plate, replaced with 40 ⁇ L/well Opti-MEM (registered trademark) (Thermo Fisher SCIENTIFIC), and mRNA ( 5 ng/well) was mixed with Lipofectamine® MessengerMAX® (0.15 ⁇ L/well) and Opti-MEM® (10 ⁇ L) and added. After incubation at 37° C. for 3 hours, 100 ⁇ L/well of MEM ⁇ ,nucleosides medium containing 10% FBS and 5 ng/mL GM-CSF was added and cultured further.
- Opti-MEM registered trademark
- cell lysates were prepared using the NanoGlo® Luciferase Assay System (Promega) and the amount of NanoLuc luciferase protein contained in the lysates was assessed by luminometric method.
- amino acid sequence information of NLuc is as shown in SEQ ID NO: 12.
- FIG. 4 is a diagram showing a comparison of the translation activities of base-length NanoLuc luciferase (Nluc) mRNAs. These are experimental results showing that the translation activity of the former is high. A of this figure shows the chemical structures and abbreviations of the cap analogs used.
- FIG. 40C shows the results of comparison of Nluc mRNA activity using HeLa cells.
- D of this figure is the result of comparison of Nluc mRNA activity using JAWS II cells.
- mRNA prepared using hydrophobic protective group-containing cap analogs (TetraPure_2, TetraPure_2/m6A, TetraPure_2/G) is a conventional cap analog (Tri_1, CleanCap AG- sold by TriLink) A compound with the same structure and Tetra_2) exhibited higher translational activity than mRNA prepared using them.
- Tri — 1/AP and Tetra — 2/AP represent mRNAs prepared using Tri — 1 and Tetra — 2, respectively, and dephosphorylated at the 5′ end of the coexisting uncapped RNA by the action of Antarctic phosphatase. These dephosphorylated RNAs were prepared as follows.
- Reaction solution 250 ng/ ⁇ L mRNA, 50 mM Bis-Tris-Propane HCl, 1 mM MgCl 2 , 0.1 mM ZnCl 2 , pH 6.0@25° C., 0.25 U/ ⁇ L Antarctic phosphatase (New England Biolabs), reaction volume 20 ⁇ L
- reaction volume 20 ⁇ L
- 3M NaOAc pH 5.2
- the capped mRNA used in Figure 40 was prepared as follows. mRNA was transcribed and synthesized from the DNA template using T7 RNA polymerase reaction under the following conditions. 15 ng/ ⁇ L dsDNA transcription template (a 676-bp PCR product containing the T7 promoter sequence 5′TAATACGACTCACTATAG3′ (SEQ ID NO: 1) containing the NanoLuc luciferase gene coding region from the pNL1.1TK vector (Promega)), 2 mM ATP , 2 mM UTP, 2 mM CTP, 2 mM GTP or [0.5 mM GTP + 2 mM cap analog], 40 mM Tris-HCl (pH 8.0), 8 mM MgCl 2 , 2 mM spermidine, 5 mM DTT, 10 units/ ⁇ L T7 RNA polymerase (Takara Bio) .
- 15 ng/ ⁇ L dsDNA transcription template a 676-bp PCR product
- RNA was purified by reverse phase HPLC under the following conditions.
- RNA containing a cap analog containing a hydrophobic protecting group isolated by reversed-phase HPLC to a transparent 96-well multiwell plate and use a MAX-305 light source (Asahi Spectroscopy) at 4 mW/cm. 365 nm light was irradiated for 10 minutes at a light intensity of 2 . Subsequently, it was purified by reverse phase HPLC under the following conditions.
- Terminal analysis The purified Nluc mRNA was cleaved with DNAzyme (35-base-long DNA oligonucleotide), converted to a mixture containing a 23-base-long 5'-terminal fragment, and analyzed by denaturing PAGE.
- the DNAzyme sequences used were: 5'TTCGAGGCCAGGCTAGCTACAACGAACGCGTCACC 3' (SEQ ID NO: 11).
- the composition of the reaction solution for the cleavage reaction is as follows. 104 ng/ ⁇ L (0.5 ⁇ M) Nluc mRNA, 1 ⁇ M DNAzyme, 5 mM Tris-HCl (pH 8.0), 5 mM magnesium chloride, reaction volume 40 ⁇ L.
- RNA was recovered as a pellet by centrifugation. The obtained RNA was dissolved in 4 ⁇ L of water, and 3 ⁇ L of the solution was analyzed by electrophoresis on a 15% denaturing acrylamide gel containing 7.5 M urea as a denaturing agent. After electrophoresis, the gel was stained with a nucleic acid staining reagent SYBR Gold, photographed with a ChemiDoc MP gel imaging system (BioRad), and RNA bands were visualized.
- HeLa cell line HeLa cells (RIKEN Cell Bank) were cultured in Dulbecco's modified Eagle's medium (DMEM; WAKO) supplemented with 10% fetal bovine serum (FBS; Invitrogen) (37°C, 5% CO2 ). The day before transfection, HeLa cells seeded in a 96-well plate (5 ⁇ 10 3 cells / well), remove the medium, replace with 40 ⁇ L / well Opti-MEM (registered trademark) (Thermo Fisher SCIENTIFIC), mRNA here (5 ng/well) was added by mixing Lipofectamine® MessengerMAX® (0.15 ⁇ L/well) with Opti-MEM® (10 ⁇ L/well).
- DMEM Dulbecco's modified Eagle's medium
- FBS fetal bovine serum
- DMEM medium containing 10% FBS was added at 100 ⁇ L/well and further cultured. After 12, 24 and 48 hours, cell lysates were prepared using the NanoGlo® Luciferase Assay System (Promega) and the amount of NanoLuc luciferase protein contained in the lysates was assessed by luminometric method.
- JAWSII cells were cultured in MEM ⁇ , nucleosides (Gibco) medium supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 5 ng/mL GM-CSF (37°C, 5 % CO2 ).
- FBS fetal bovine serum
- GM-CSF GM-CSF
- the medium was removed from JAWS II cells (5 ⁇ 10 3 cells/well) seeded on a 96-well plate, replaced with 40 ⁇ L/well Opti-MEM (registered trademark) (Thermo Fisher SCIENTIFIC), and mRNA ( 5 ng/well) was mixed with Lipofectamine® MessengerMAX® (0.15 ⁇ L/well) and Opti-MEM® (10 ⁇ L) and added. After incubation at 37° C. for 3 hours, 100 ⁇ L/well of MEM ⁇ ,nucleosides medium containing 10% FBS and 5 ng/mL GM-CSF was added and cultured further.
- Opti-MEM registered trademark
- cell lysates were prepared using the NanoGlo® Luciferase Assay System (Promega) and the amount of NanoLuc luciferase protein contained in the lysates was assessed by luminometric method.
- the template DNA sequence (4375-bp) of Spike protein mRNA is shown in SEQ ID NO:13.
- sequence (transcript) 4247base of Spike protein mRNA is shown in SEQ ID NO: 14.
- the capped mRNA used in FIG. 41 was transcribed and synthesized from a DNA template using T7 RNA polymerase in the following reaction solution.
- 5 ng/ ⁇ L dsDNA transcription template (4375-bp PCR product containing T7 promoter sequence 5′TAATACGACTCACTATAG3′ (SEQ ID NO: 1)), 2 mM ATP, 2 mM UTP, 2 mM CTP, 2 mM GTP, 2 mM Pure cap analog (DiPure), 40 mM Tris-HCl (pH 8.0), 8 mM MgCl 2 , 2 mM spermidine, 5 mM DTT, 10 units/ ⁇ L T7 RNA polymerase (Takara Bio).
- RNA product After heating the reaction solution at 37° C. for 2 hours, DNase (Takara Bio) was added to a final concentration of 0.1 unit/ ⁇ L and heated at 37° C. for 20 minutes. An equal volume mixture of TE-saturated phenol and chloroform was added to the reaction mixture, vigorously stirred, and then centrifuged to separate the aqueous layer. An equal amount of chloroform was added thereto, and the mixture was vigorously stirred and then centrifuged to separate the aqueous layer. A 1/10 volume of 3M sodium acetate buffer (pH 5.2) and an equal volume of 2-propanol were added to the aqueous layer, cooled to -30°C for 1 hour, and centrifuged to obtain a crude RNA product.
- DNase Takara Bio
- RNA was analyzed by reverse-phase HPLC under the following conditions.
- Column Nacalai Tesque Cosmosil RNA RP-1 (200 mm x 4.6 mm ID); Solvent A, 100 mM triethylammonium acetate (pH 7.0), 5% acetonitrile; Solvent B, 100 mM triethylammonium acetate (pH 7.0) Linear gradient 10-25% Solvent B (0-60 min); flow rate: 1 mL/min; detection wavelength, 254 nm; column temperature, 50°C.
- the purified mRNA was cleaved with DNAzyme (39-base-long DNA oligonucleotide), converted to a mixture containing a 23-base-long 5'-terminal fragment, and analyzed by denaturing PAGE.
- the DNAzyme sequences used were: 5'TCTGTGGGGGAGGCTAGCTACAACGACAGAAGAATATACTAG 3' (SEQ ID NO: 19).
- the composition of the reaction solution for the cleavage reaction is as follows. 688 ng/ ⁇ L mRNA (DiPure), 1 ⁇ M DNAzyme, 5 mM Tris-HCl (pH 8.0), 5 mM magnesium chloride, reaction volume 9 ⁇ L.
- RNA was recovered as a pellet by centrifugation.
- the obtained RNA was dissolved in 3 ⁇ L of water, and 3 ⁇ L of the solution was analyzed by electrophoresis on a 15% denaturing acrylamide gel containing 7.5 M urea as a denaturing agent. After electrophoresis, the gel was stained with a nucleic acid staining reagent SYBR Gold, photographed with a ChemiDoc MP gel imaging system (BioRad), and the RNA band was visualized.
- FIG. 42 shows the result that PureCap-type mRNA shows a high protein synthesis amount without introduction of methylpseudouridine (see JP-A-2021-35979).
- Conventional ARCA (Cap_0) and CleanCap (Cap_1) increase the amount of protein synthesis by introducing methylpseudouridine.
- PureCap was shown to reduce the amount of protein synthesis by introducing methylpseudouridine. Overall, it was shown that the protein synthesis amount of PureCap-type mRNA was high.
- 1-Methyl pseudouridine-modified mRNA and unmodified mRNA were prepared as follows. mRNA was transcribed and synthesized from the DNA template using T7 RNA polymerase reaction under the following conditions. 15 ng/ ⁇ L dsDNA transcription template (a 676-bp PCR product containing the T7 promoter sequence 5′TAATACGACTCACTATAG3′ (SEQ ID NO: 1) containing the NanoLuc luciferase gene coding region from the pNL1.1TK vector (Promega)), 2 mM ATP , 2 mM 1-methyl pseudo UTP or UTP, 2 mM CTP, 2 mM GTP, 2 mM PureCap-type cap analog or [0.5 mM GTP + 2 mM ARCA cap analog (Jena Bioscience)], 40 mM Tris-HCl (pH 8.0), 8 mM MgCl 2 , 2 mM spermidine, 5 mM DTT, 10 units/ ⁇ L T7 RNA
- reaction solution was heated at 37°C for 2 hours, DNase (Takara Bio) was added to a final concentration of 0.1 unit/ ⁇ L and heated at 37°C for 20 minutes.
- DNase Tekara Bio
- To the reaction solution was added an equal volume mixture of TE-saturated phenol and chloroform in the same amount as the reaction solution, vigorously stirred, and then centrifuged to separate the aqueous layer.
- An equal amount of chloroform was added thereto, the mixture was vigorously stirred again, and then centrifuged to separate the aqueous layer.
- RNA product A 1/10 volume of 3M sodium acetate buffer (pH 5.2) and an equal volume of 2-propanol were added to the aqueous layer, cooled to minus 30°C for 1 hour, and centrifuged to obtain a crude RNA product. rice field.
- the obtained crude RNA was purified by reverse phase HPLC under the following conditions.
- RNA transcript containing the hydrophobic protecting group-containing cap analogue was 16.0 to 16.8 minutes, and the elution time of the RNA transcript without it was 13.4 to 14.0 minutes. was different. This made it possible to isolate and purify mRNA containing cap analogues containing hydrophobic protecting groups.
- JAWS II cells were cultured in MEM ⁇ ,nucleosides (Gibco) medium supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 5 ng/mL GM-CSF (37°C, 5% CO 2 ).
- FBS fetal bovine serum
- GM-CSF GM-CSF
- RNA 5 ng/well was mixed with Lipofectamine® MessengerMAX® (0.15 ⁇ L/well) and Opti-MEM® (10 ⁇ L) and added.
- Cell lysates were prepared after 24, 48, 72, 120 hours using the NanoGlo® Luciferase Assay System (Promega) and the amount of NanoLuc luciferase protein in the lysates was assessed by luminometric method.
- FIG. 43 shows the result of evaluation of immune response in cells (HEK293 NF-kB cells), and it was clarified that PureCap-type mRNA exhibits a lower immune response than conventional mRNA such as ARCA.
- NF- ⁇ B reporter (Luc)-HEK293 cells BPS Bioscience Inc.
- FBS fetal bovine serum
- KDM Dulbecco's modified Eagle's medium
- Opti-MEM® Thermo Fisher SCIENTIFIC
- 0.375 ⁇ L Lipofectamine® MessengerMAX®, 12.5 ng mRNA (DiPure) was diluted in 25 ⁇ L Opti-MEM® and introduced into the cells.
- 50 ⁇ L/well 1 ⁇ Cell Lysis Buffer 50 ⁇ L/well 1 ⁇ Cell Lysis Buffer (Promega) was added to lyse the cells.
- Luminescence was measured using the ONE-Glo® luciferase assay® (Promega). Protein levels were quantified using the Pierce® BCA Protein Assay Kit (Thermo Fisher SCIENTIFIC), and luminescence values were corrected for cell number.
- Fig. 44 shows the result that PureCap-type mRNA with high purity showed higher protein expression ability than ARCA-type mRNA in individual animals.
- PureCap-type mRNA has a high ability to express protein.
- As a method of administering mRNA to mice it was carried out by encapsulating in lipid nanoparticles (LNP).
- LNP lipid nanoparticles
- the prepared mRNA-encapsulating LNP was administered to balb/c female rats, 7w, through the tail vein at 2 ⁇ g mRNA/mouse. After 4 hours, the animals were sacrificed, and the organs were soaked in Passive Lysis Buffer (Promega) and homogenized. Luminescence was detected using NanoLuc® Luciferase Technology (Promega).
- FIG. 45 shows the results of evaluation of immune responses in cells (HEK293 NF-kB cells).
- Figure 45 (a) shows the results of immune response when 50 ng and 100 ng of mRNA were administered to HEK293 NF-kB cells
- Figure 45 (b) shows the results of immune response when 100 ng of mRNA was administered to HEK293 NF-kB cells. Shows the result of the response. It was revealed that PureCap-type mRNAs (DiPure, TriPure_0, TriPure_1, TetraPure_2, TetraPure_2/m6A) show a lower immune response than conventional mRNAs synthesized using ARCA.
- DiPure, TriPure_0, TriPure_1, TetraPure_2, TetraPure_2/m6A show a lower immune response than conventional mRNAs synthesized using ARCA.
- NF- ⁇ B reporter (Luc)-HEK293 cells BPS Bioscience Inc.
- FBS fetal bovine serum
- KEM Dulbecco's modified Eagle's medium
- NF- ⁇ B reporter (Luc)-HEK293 cells were seeded in a 48-well plate (1.0 ⁇ 10 5 cells/well). The next day, the medium was removed and replaced with 240 ⁇ L/well Opti-MEM® (Thermo Fisher SCIENTIFIC).
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Microbiology (AREA)
- Epidemiology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Plant Pathology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Crystallography & Structural Chemistry (AREA)
- Analytical Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Saccharide Compounds (AREA)
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP22837660.4A EP4368628A4 (en) | 2021-07-05 | 2022-07-05 | NUCLEOTIDE PURIFICATION PROCESS, NUCLEOTIDE PURIFICATION DEVICE, HYDROPHOBIC REAGENT AND HYDROPHOBIC SUBSTRATE |
| CN202280056963.6A CN117858885A (zh) | 2021-07-05 | 2022-07-05 | 核苷酸类的纯化方法和核苷酸类的纯化装置以及疏水性试剂和疏水性底物 |
| US18/576,587 US20240327444A1 (en) | 2021-07-05 | 2022-07-05 | Method for Purifying Nucleotides, Device for Purifying Nucleotides, Hy-Drophobic Reagent, and Hydrophobic Substrate |
| JP2023533132A JPWO2023282245A1 (https=) | 2021-07-05 | 2022-07-05 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021-111420 | 2021-07-05 | ||
| JP2021111420 | 2021-07-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2023282245A1 true WO2023282245A1 (ja) | 2023-01-12 |
Family
ID=84800657
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2022/026665 Ceased WO2023282245A1 (ja) | 2021-07-05 | 2022-07-05 | ヌクレオチド類の精製方法及びヌクレオチド類の精製装置並びに疎水性試薬及び疎水性基質 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20240327444A1 (https=) |
| EP (1) | EP4368628A4 (https=) |
| JP (1) | JPWO2023282245A1 (https=) |
| CN (1) | CN117858885A (https=) |
| WO (1) | WO2023282245A1 (https=) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN116478226A (zh) * | 2023-03-07 | 2023-07-25 | 江苏申基生物科技有限公司 | 一种锁核苷帽类似物和应用 |
| WO2024185697A1 (ja) * | 2023-03-03 | 2024-09-12 | 国立大学法人東海国立大学機構 | ポリヌクレオチド連結産物の製造方法 |
| WO2025050466A1 (zh) * | 2023-09-09 | 2025-03-13 | 南京鸿明生物科技有限公司 | 一种脱氧核酶及检测mRNA加帽率的方法 |
| WO2025054401A3 (en) * | 2023-09-06 | 2025-05-08 | Trilink Biotechnologies, Llc | Cap analogs and methods of use thereof |
| WO2025141025A3 (en) * | 2023-12-26 | 2025-08-07 | Eleven Therapeutics Ltd | Novel capping strategies for mrna 5'cap |
| WO2025187713A1 (en) * | 2024-03-07 | 2025-09-12 | Eisai R&D Management Co., Ltd. | Polynucleotide and producing method thereof |
| WO2026009964A1 (ja) * | 2024-07-05 | 2026-01-08 | 国立大学法人東海国立大学機構 | キャップ化ポリヌクレオチド |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4725949A1 (en) * | 2024-10-08 | 2026-04-15 | BioSpring Gesellschaft für Biotechnologie mbH | Improved solid-phase phosphoramidite-synthesis of long oligonucleotide molecules |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009152353A2 (en) | 2008-06-11 | 2009-12-17 | Lasergen, Inc. | Nucleotides and nucleosides and methods for their use in dna sequencing |
| CN102898464A (zh) * | 2012-05-23 | 2013-01-30 | 北京大学 | 光敏感基团保护的功能基团亚磷酰胺及其制备方法和应用 |
| WO2014150851A1 (en) * | 2013-03-15 | 2014-09-25 | Ibis Biosciences, Inc. | Nucleotide analogs for sequencing |
| JP2014532043A (ja) * | 2011-09-13 | 2014-12-04 | レーザーゲン インコーポレイテッド | 5−メトキシ3’−oh非遮断高速光開裂性末端ヌクレオチドおよび核酸配列決定のための方法 |
| WO2018162610A1 (en) | 2017-03-08 | 2018-09-13 | Eth Zurich | Novel phosphorylation reagents and uses thereof |
| US20180273576A1 (en) | 2015-09-21 | 2018-09-27 | Trilink Biotechnologies, Inc. | Compositions and methods for synthesizing 5'-capped rnas |
| JP2020032889A (ja) | 2018-08-30 | 2020-03-05 | 横浜ゴム株式会社 | 空気入りタイヤ |
| WO2021020562A1 (ja) | 2019-07-31 | 2021-02-04 | 国立研究開発法人科学技術振興機構 | プライマー及びこれを用いた二本鎖dnaの製造装置並びに二本鎖dnaの製造方法 |
| JP2021035979A (ja) | 2015-08-28 | 2021-03-04 | バイオンテック エールエヌアー ファーマシューティカルズ ゲーエムベーハー | Rnaの免疫原性を低減するための方法 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3433649A1 (de) * | 1984-09-13 | 1986-03-20 | Gesellschaft für Biotechnologische Forschung mbH (GBF), 3300 Braunschweig | Verfahren zur aufreinigung synthetischer oligonucleotide |
| US5998604A (en) * | 1997-09-15 | 1999-12-07 | The Perkin-Elmer Corporation | Polynucleotide purification method |
| US7345163B2 (en) * | 2002-08-28 | 2008-03-18 | Quiatech Ab | Process for separating and deprotecting oligonucleotides |
-
2022
- 2022-07-05 EP EP22837660.4A patent/EP4368628A4/en active Pending
- 2022-07-05 WO PCT/JP2022/026665 patent/WO2023282245A1/ja not_active Ceased
- 2022-07-05 US US18/576,587 patent/US20240327444A1/en active Pending
- 2022-07-05 CN CN202280056963.6A patent/CN117858885A/zh active Pending
- 2022-07-05 JP JP2023533132A patent/JPWO2023282245A1/ja active Pending
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009152353A2 (en) | 2008-06-11 | 2009-12-17 | Lasergen, Inc. | Nucleotides and nucleosides and methods for their use in dna sequencing |
| JP2011529449A (ja) * | 2008-06-11 | 2011-12-08 | レーザーゲン インコーポレイテッド | ヌクレオチドおよびヌクレオシドならびにdna配列決定におけるその使用の方法 |
| JP2014532043A (ja) * | 2011-09-13 | 2014-12-04 | レーザーゲン インコーポレイテッド | 5−メトキシ3’−oh非遮断高速光開裂性末端ヌクレオチドおよび核酸配列決定のための方法 |
| CN102898464A (zh) * | 2012-05-23 | 2013-01-30 | 北京大学 | 光敏感基团保护的功能基团亚磷酰胺及其制备方法和应用 |
| WO2014150851A1 (en) * | 2013-03-15 | 2014-09-25 | Ibis Biosciences, Inc. | Nucleotide analogs for sequencing |
| JP2021035979A (ja) | 2015-08-28 | 2021-03-04 | バイオンテック エールエヌアー ファーマシューティカルズ ゲーエムベーハー | Rnaの免疫原性を低減するための方法 |
| US20180273576A1 (en) | 2015-09-21 | 2018-09-27 | Trilink Biotechnologies, Inc. | Compositions and methods for synthesizing 5'-capped rnas |
| WO2018162610A1 (en) | 2017-03-08 | 2018-09-13 | Eth Zurich | Novel phosphorylation reagents and uses thereof |
| JP2020032889A (ja) | 2018-08-30 | 2020-03-05 | 横浜ゴム株式会社 | 空気入りタイヤ |
| WO2021020562A1 (ja) | 2019-07-31 | 2021-02-04 | 国立研究開発法人科学技術振興機構 | プライマー及びこれを用いた二本鎖dnaの製造装置並びに二本鎖dnaの製造方法 |
Non-Patent Citations (14)
| Title |
|---|
| BRIAN P. STUPI, HONG LI, JINCHUN WANG, WEIDONG WU, SIDNEY E. MORRIS, VLADISLAV A. LITOSH, JESSE MUNIZ, MEGAN N. HERSH, MICHAEL L. : "Stereochemistry of Benzylic Carbon Substitution Coupled with Ring Modification of 2-Nitrobenzyl Groups as Key Determinants for Fast-Cleaving Reversible Terminators", ANGEWANDTE CHEMIE INTERNATIONAL EDITION, ¬VERLAG CHEMIE| :, vol. 51, no. 7, 13 February 2012 (2012-02-13), pages 1724 - 1727, XP055043239, ISSN: 14337851, DOI: 10.1002/anie.201106516 * |
| CHEM. SCI., vol. 12, 2021, pages 4383 - 4388 |
| GRAY G. M.MACFARLANE M. G., BIOCHEM. J., vol. 81, 1961, pages 480 - 488 |
| LI WU, FEN PEI, JINHAO ZHANG, JUNZHOU WU, MENGKE FENG, YUAN WANG, HONGWEI JIN, LIANGREN ZHANG, XINJING TANG: "Synthesis of Site-Specifically Phosphate-Caged siRNAs and Evaluation of Their RNAi Activity and Stability", CHEMISTRY - A EUROPEAN JOURNAL, JOHN WILEY & SONS, INC, DE, vol. 20, no. 38, 15 September 2014 (2014-09-15), DE, pages 12114 - 12122, XP055410614, ISSN: 0947-6539, DOI: 10.1002/chem.201403430 * |
| MARCIN Z.MARIUSZ S.MACIEJ L.ANNA N.EDWARD D.ROBERT E. RHOADSZBIGNIEW W.JACEK J., RSC ADV, vol. 3, 2013, pages 20943 - 20958 |
| NUCLEIC ACID RES, vol. 48, no. 4, 2020, pages 1607 - 1626 |
| OGILVIE K. K.SCHIFMAN A. L.PENNEY C. L., CAN. J. CHEM., vol. 57, no. 17, 1979, pages 2230 - 2238 |
| PRADERE, U. ET AL., CHEM. EUR. J., vol. 23, 2017, pages 5210 - 5213 |
| See also references of EP4368628A4 |
| STUTZ ALFRED, CLAUDIA HOEBARTNER, STEFAN PITSCH: "Novel fluoride-labile nucleobase-protecting groups for the synthesis of 3'(2')-O-amino-acylated RNA sequences", HELVETICA CHIMICA ACTA, VERLAG HELVETICA CHIMICA ACTA., HOBOKEN, USA, vol. 83, no. 9, 1 January 2000 (2000-01-01), Hoboken, USA, pages 2477 - 2503, XP002979645, ISSN: 0018-019X, DOI: 10.1002/1522-2675(20000906)83:9<2477::AID-HLCA2477>3.0.CO;2-9 * |
| T. MUKOBATA ET AL., BIOORG. MED. CHEM. LETT., vol. 20, 2010, pages 129 - 131 |
| V. A. LITOSH ET AL., NUCLEIC ACIDS RES., vol. 29, no. 6, 2011, pages e39 |
| V. A. LITOSH, W. WU, B. P. STUPI, J. WANG, S. E. MORRIS, M. N. HERSH, M. L. METZKER: "Improved nucleotide selectivity and termination of 3'-OH unblocked reversible terminators by molecular tuning of 2-nitrobenzyl alkylated HOMedU triphosphates", NUCLEIC ACIDS RESEARCH, INFORMATION RETRIEVAL LTD., vol. 39, no. 6, 1 March 2011 (2011-03-01), pages e39 - e39, XP055043172, ISSN: 03051048, DOI: 10.1093/nar/gkq1293 * |
| YANGMING WANGSCOTT K. SILVERMAN: "Efficient RNA 5'-adenylation by T4 DNA ligase to facilitate practical applications", RNA, vol. 12, 2006, pages 1142 - 1146 |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024185697A1 (ja) * | 2023-03-03 | 2024-09-12 | 国立大学法人東海国立大学機構 | ポリヌクレオチド連結産物の製造方法 |
| CN116478226A (zh) * | 2023-03-07 | 2023-07-25 | 江苏申基生物科技有限公司 | 一种锁核苷帽类似物和应用 |
| WO2024183748A1 (zh) * | 2023-03-07 | 2024-09-12 | 江苏申基生物科技有限公司 | 一种锁核苷帽类似物和应用 |
| CN116478226B (zh) * | 2023-03-07 | 2025-08-19 | 江苏申基生物科技有限公司 | 一种锁核苷帽类似物和应用 |
| WO2025054401A3 (en) * | 2023-09-06 | 2025-05-08 | Trilink Biotechnologies, Llc | Cap analogs and methods of use thereof |
| WO2025050466A1 (zh) * | 2023-09-09 | 2025-03-13 | 南京鸿明生物科技有限公司 | 一种脱氧核酶及检测mRNA加帽率的方法 |
| WO2025141025A3 (en) * | 2023-12-26 | 2025-08-07 | Eleven Therapeutics Ltd | Novel capping strategies for mrna 5'cap |
| WO2025187713A1 (en) * | 2024-03-07 | 2025-09-12 | Eisai R&D Management Co., Ltd. | Polynucleotide and producing method thereof |
| WO2026009964A1 (ja) * | 2024-07-05 | 2026-01-08 | 国立大学法人東海国立大学機構 | キャップ化ポリヌクレオチド |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4368628A1 (en) | 2024-05-15 |
| JPWO2023282245A1 (https=) | 2023-01-12 |
| US20240327444A1 (en) | 2024-10-03 |
| CN117858885A (zh) | 2024-04-09 |
| EP4368628A4 (en) | 2025-11-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2023282245A1 (ja) | ヌクレオチド類の精製方法及びヌクレオチド類の精製装置並びに疎水性試薬及び疎水性基質 | |
| CN103068834B (zh) | 用于反向合成rna的亚磷酰胺 | |
| EP3409780B1 (en) | Nucleic acid complex | |
| US8309707B2 (en) | RNA synthesis-phosphoramidites for synthetic RNA in the reverse direction, and application in convenient introduction of ligands, chromophores and modifications of synthetic RNA at the 3′-end | |
| EP4234567A1 (en) | Oligonucleotide for 5'-capped rna synthesis | |
| EP3556764A1 (en) | Nucleoside derivative and use therefor | |
| WO2023277168A1 (ja) | ポリヌクレオチド及び医薬組成物 | |
| US8614312B2 (en) | Method for preparing nucleotides and related analogues by synthesis on soluble substrate, and biological tools thus prepared | |
| US11858953B2 (en) | Compositions and methods for synthesis of phosphorylated molecules | |
| US20040033967A1 (en) | Alkylated hexitol nucleoside analogues and oligomers thereof | |
| JP4802712B2 (ja) | リボ核酸化合物及びオリゴ核酸化合物の液相合成法 | |
| WO2004018494A1 (ja) | 4’−チオヌクレオチド | |
| KR20230123318A (ko) | 5'-캡핑된 rna 합성용 올리고뉴클레오티드 | |
| Mori et al. | Bridged nucleic acid conjugates at 6′-thiol: synthesis, hybridization properties and nuclease resistances | |
| WO2008025160A1 (en) | Oxepane nucleosides and oligonucleotides, uses thereof and methods of making the same | |
| Jabgunde et al. | Synthesis of 3′-fluoro-4′-amino-hexitol nucleosides with a pyrimidine nucleobase as building blocks for oligonucleotides | |
| Zatsepin et al. | Efficient conjugation and preferential DNA binding of oligonucleotides containing 2′-O-(2-oxoethyl) arabinouridine | |
| HK1184164B (en) | Phosphoramidites for synthetic rna in the reverse direction | |
| FR2612930A1 (fr) | Sondes oligonucleotidiques a | |
| HK1000191B (en) | Modified oligodeoxyribonucleotides, their preparation and their therapeutic use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 22837660 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023533132 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 18576587 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202417006053 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2022837660 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2022837660 Country of ref document: EP Effective date: 20240205 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202280056963.6 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11202400050X Country of ref document: SG |