WO2022070289A1 - ピリミジン含有含窒素二環化合物 - Google Patents
ピリミジン含有含窒素二環化合物 Download PDFInfo
- Publication number
- WO2022070289A1 WO2022070289A1 PCT/JP2020/037048 JP2020037048W WO2022070289A1 WO 2022070289 A1 WO2022070289 A1 WO 2022070289A1 JP 2020037048 W JP2020037048 W JP 2020037048W WO 2022070289 A1 WO2022070289 A1 WO 2022070289A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- salt
- mmol
- compound
- Prior art date
Links
- -1 Nitrogen-containing bicyclic compound Chemical class 0.000 title description 296
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 title description 23
- 150000001875 compounds Chemical class 0.000 claims abstract description 406
- 150000003839 salts Chemical class 0.000 claims abstract description 138
- 125000000217 alkyl group Chemical group 0.000 claims description 170
- 229920006395 saturated elastomer Polymers 0.000 claims description 151
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 105
- 125000001424 substituent group Chemical group 0.000 claims description 89
- 125000003545 alkoxy group Chemical group 0.000 claims description 76
- 125000005843 halogen group Chemical group 0.000 claims description 75
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 55
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 claims description 50
- 125000003003 spiro group Chemical group 0.000 claims description 49
- 125000001072 heteroaryl group Chemical group 0.000 claims description 38
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 37
- 125000005842 heteroatom Chemical group 0.000 claims description 30
- 229910052799 carbon Inorganic materials 0.000 claims description 29
- 229910052757 nitrogen Inorganic materials 0.000 claims description 28
- 125000003118 aryl group Chemical group 0.000 claims description 24
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 24
- 229910052739 hydrogen Inorganic materials 0.000 claims description 24
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 23
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 21
- 229910052760 oxygen Inorganic materials 0.000 claims description 16
- 125000004429 atom Chemical group 0.000 claims description 13
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 12
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 5
- 238000006467 substitution reaction Methods 0.000 claims description 4
- 239000003814 drug Substances 0.000 abstract description 55
- 239000004480 active ingredient Substances 0.000 abstract description 53
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 19
- 201000010099 disease Diseases 0.000 abstract description 18
- 230000005764 inhibitory process Effects 0.000 abstract description 18
- 230000002401 inhibitory effect Effects 0.000 abstract description 15
- 102100023533 Interleukin-1 receptor-associated kinase 4 Human genes 0.000 abstract description 3
- 101710199010 Interleukin-1 receptor-associated kinase 4 Proteins 0.000 abstract 2
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 194
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 191
- 239000000543 intermediate Substances 0.000 description 169
- 238000000034 method Methods 0.000 description 165
- 239000000203 mixture Substances 0.000 description 161
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 138
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 134
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 128
- 238000012360 testing method Methods 0.000 description 110
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 108
- 239000011541 reaction mixture Substances 0.000 description 105
- 238000006243 chemical reaction Methods 0.000 description 88
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 87
- 238000005160 1H NMR spectroscopy Methods 0.000 description 82
- 239000000243 solution Substances 0.000 description 70
- 239000012043 crude product Substances 0.000 description 67
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 60
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 58
- 239000012044 organic layer Substances 0.000 description 56
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 55
- 239000003480 eluent Substances 0.000 description 54
- 230000002829 reductive effect Effects 0.000 description 51
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 50
- 238000010898 silica gel chromatography Methods 0.000 description 50
- 229940079593 drug Drugs 0.000 description 47
- AIMMVWOEOZMVMS-UHFFFAOYSA-N cyclopropanecarboxamide Chemical compound NC(=O)C1CC1 AIMMVWOEOZMVMS-UHFFFAOYSA-N 0.000 description 46
- MFNYBOWJWGPXFM-UHFFFAOYSA-N cyclobutanecarboxamide Chemical compound NC(=O)C1CCC1 MFNYBOWJWGPXFM-UHFFFAOYSA-N 0.000 description 36
- YFPCLQKFNXUAAK-UHFFFAOYSA-N cyclopentyl acetate Chemical compound CC(=O)OC1CCCC1 YFPCLQKFNXUAAK-UHFFFAOYSA-N 0.000 description 33
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 32
- 238000004519 manufacturing process Methods 0.000 description 32
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 32
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 31
- 239000007810 chemical reaction solvent Substances 0.000 description 31
- 238000004128 high performance liquid chromatography Methods 0.000 description 31
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 30
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 30
- 235000017557 sodium bicarbonate Nutrition 0.000 description 30
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 29
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 28
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 28
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 28
- 239000002585 base Substances 0.000 description 26
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 25
- 210000004027 cell Anatomy 0.000 description 23
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 23
- 239000002904 solvent Substances 0.000 description 23
- 239000010410 layer Substances 0.000 description 22
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 21
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 21
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 21
- 150000002430 hydrocarbons Chemical group 0.000 description 21
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 20
- 125000000623 heterocyclic group Chemical group 0.000 description 20
- 238000005259 measurement Methods 0.000 description 20
- 238000007430 reference method Methods 0.000 description 20
- 229930195734 saturated hydrocarbon Natural products 0.000 description 19
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 18
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 18
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 17
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 16
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 16
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 16
- 238000007112 amidation reaction Methods 0.000 description 16
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 16
- 229910000024 caesium carbonate Inorganic materials 0.000 description 16
- 230000000694 effects Effects 0.000 description 16
- 229910052938 sodium sulfate Inorganic materials 0.000 description 16
- 235000011152 sodium sulphate Nutrition 0.000 description 16
- 125000005605 benzo group Chemical group 0.000 description 15
- 125000001246 bromo group Chemical group Br* 0.000 description 15
- 239000003153 chemical reaction reagent Substances 0.000 description 15
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 15
- 238000000746 purification Methods 0.000 description 15
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 14
- 239000003795 chemical substances by application Substances 0.000 description 14
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 14
- PAQZWJGSJMLPMG-UHFFFAOYSA-N propylphosphonic anhydride Substances CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 14
- NHGXDBSUJJNIRV-UHFFFAOYSA-M tetrabutylammonium chloride Chemical compound [Cl-].CCCC[N+](CCCC)(CCCC)CCCC NHGXDBSUJJNIRV-UHFFFAOYSA-M 0.000 description 14
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 13
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 13
- 238000005804 alkylation reaction Methods 0.000 description 13
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 13
- 238000002360 preparation method Methods 0.000 description 13
- 239000000047 product Substances 0.000 description 13
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 12
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 12
- 239000003054 catalyst Substances 0.000 description 12
- 230000002265 prevention Effects 0.000 description 12
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 12
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 12
- 102000004190 Enzymes Human genes 0.000 description 11
- 108090000790 Enzymes Proteins 0.000 description 11
- 230000029936 alkylation Effects 0.000 description 11
- 230000009435 amidation Effects 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 11
- 239000008280 blood Substances 0.000 description 11
- WORJEOGGNQDSOE-UHFFFAOYSA-N chloroform;methanol Chemical compound OC.ClC(Cl)Cl WORJEOGGNQDSOE-UHFFFAOYSA-N 0.000 description 11
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 11
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 11
- 241000282414 Homo sapiens Species 0.000 description 10
- 235000011054 acetic acid Nutrition 0.000 description 10
- 238000005859 coupling reaction Methods 0.000 description 10
- 238000010511 deprotection reaction Methods 0.000 description 10
- 231100000025 genetic toxicology Toxicity 0.000 description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 10
- 229910052751 metal Inorganic materials 0.000 description 10
- 239000002184 metal Substances 0.000 description 10
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 10
- 238000006722 reduction reaction Methods 0.000 description 10
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 9
- 102000002689 Toll-like receptor Human genes 0.000 description 9
- 108020000411 Toll-like receptor Proteins 0.000 description 9
- 239000007864 aqueous solution Substances 0.000 description 9
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 9
- 125000004432 carbon atom Chemical group C* 0.000 description 9
- 229910000027 potassium carbonate Inorganic materials 0.000 description 9
- 125000006239 protecting group Chemical group 0.000 description 9
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 8
- 241000699670 Mus sp. Species 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 8
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 8
- ZOCHARZZJNPSEU-UHFFFAOYSA-N diboron Chemical compound B#B ZOCHARZZJNPSEU-UHFFFAOYSA-N 0.000 description 8
- 230000001738 genotoxic effect Effects 0.000 description 8
- 125000002950 monocyclic group Chemical group 0.000 description 8
- 230000000144 pharmacologic effect Effects 0.000 description 8
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 8
- 230000009467 reduction Effects 0.000 description 8
- 238000006268 reductive amination reaction Methods 0.000 description 8
- 239000012312 sodium hydride Substances 0.000 description 8
- 229910000104 sodium hydride Inorganic materials 0.000 description 8
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 8
- 238000003756 stirring Methods 0.000 description 8
- 238000003786 synthesis reaction Methods 0.000 description 8
- 238000010998 test method Methods 0.000 description 8
- NAWXUBYGYWOOIX-SFHVURJKSA-N (2s)-2-[[4-[2-(2,4-diaminoquinazolin-6-yl)ethyl]benzoyl]amino]-4-methylidenepentanedioic acid Chemical compound C1=CC2=NC(N)=NC(N)=C2C=C1CCC1=CC=C(C(=O)N[C@@H](CC(=C)C(O)=O)C(O)=O)C=C1 NAWXUBYGYWOOIX-SFHVURJKSA-N 0.000 description 7
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 7
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 7
- 206010070835 Skin sensitisation Diseases 0.000 description 7
- 238000006161 Suzuki-Miyaura coupling reaction Methods 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 125000002947 alkylene group Chemical group 0.000 description 7
- 239000007809 chemical reaction catalyst Substances 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- 238000003682 fluorination reaction Methods 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 238000000691 measurement method Methods 0.000 description 7
- 229940002612 prodrug Drugs 0.000 description 7
- 239000000651 prodrug Substances 0.000 description 7
- 230000001850 reproductive effect Effects 0.000 description 7
- 231100000370 skin sensitisation Toxicity 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- 239000012453 solvate Substances 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 230000001629 suppression Effects 0.000 description 7
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 6
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 6
- 208000023275 Autoimmune disease Diseases 0.000 description 6
- 108010078791 Carrier Proteins Proteins 0.000 description 6
- 208000031404 Chromosome Aberrations Diseases 0.000 description 6
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 241001465754 Metazoa Species 0.000 description 6
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 6
- 241000283984 Rodentia Species 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 230000009471 action Effects 0.000 description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 6
- 229910000175 cerite Inorganic materials 0.000 description 6
- 230000007673 developmental toxicity Effects 0.000 description 6
- 231100000415 developmental toxicity Toxicity 0.000 description 6
- 238000000605 extraction Methods 0.000 description 6
- 239000002207 metabolite Substances 0.000 description 6
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 6
- 230000035699 permeability Effects 0.000 description 6
- 239000008194 pharmaceutical composition Substances 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 239000011734 sodium Substances 0.000 description 6
- 229910000029 sodium carbonate Inorganic materials 0.000 description 6
- 229910052717 sulfur Inorganic materials 0.000 description 6
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 6
- 230000007704 transition Effects 0.000 description 6
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 5
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 5
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 5
- QLRHRCMNDAZNLG-UHFFFAOYSA-N 3-oxocyclobutane-1-carboxamide Chemical compound NC(=O)C1CC(=O)C1 QLRHRCMNDAZNLG-UHFFFAOYSA-N 0.000 description 5
- IENOFRJPUPTEMI-UHFFFAOYSA-N 3-oxocyclobutane-1-carboxylic acid Chemical compound OC(=O)C1CC(=O)C1 IENOFRJPUPTEMI-UHFFFAOYSA-N 0.000 description 5
- 241000282693 Cercopithecidae Species 0.000 description 5
- 239000007821 HATU Substances 0.000 description 5
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 5
- 102000019223 Interleukin-1 receptor Human genes 0.000 description 5
- 108050006617 Interleukin-1 receptor Proteins 0.000 description 5
- 238000006751 Mitsunobu reaction Methods 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 238000005481 NMR spectroscopy Methods 0.000 description 5
- 108091000080 Phosphotransferase Proteins 0.000 description 5
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 5
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 5
- 238000005917 acylation reaction Methods 0.000 description 5
- 239000002671 adjuvant Substances 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 239000007806 chemical reaction intermediate Substances 0.000 description 5
- ZOOSILUVXHVRJE-UHFFFAOYSA-N cyclopropanecarbonyl chloride Chemical compound ClC(=O)C1CC1 ZOOSILUVXHVRJE-UHFFFAOYSA-N 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 238000011156 evaluation Methods 0.000 description 5
- 229910052736 halogen Inorganic materials 0.000 description 5
- 150000002367 halogens Chemical class 0.000 description 5
- 238000010438 heat treatment Methods 0.000 description 5
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 5
- 230000006698 induction Effects 0.000 description 5
- 230000002503 metabolic effect Effects 0.000 description 5
- 239000012038 nucleophile Substances 0.000 description 5
- 239000003960 organic solvent Substances 0.000 description 5
- 239000003208 petroleum Substances 0.000 description 5
- 239000000825 pharmaceutical preparation Substances 0.000 description 5
- 229940127557 pharmaceutical product Drugs 0.000 description 5
- 102000020233 phosphotransferase Human genes 0.000 description 5
- 235000011056 potassium acetate Nutrition 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- 229910052708 sodium Inorganic materials 0.000 description 5
- 241000894007 species Species 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- 230000006433 tumor necrosis factor production Effects 0.000 description 5
- PMDHIMMPXRSDML-UHFFFAOYSA-N 2-methylpyridine-4-carboxylic acid Chemical compound CC1=CC(C(O)=O)=CC=N1 PMDHIMMPXRSDML-UHFFFAOYSA-N 0.000 description 4
- DWOZNANUEDYIOF-UHFFFAOYSA-L 4-ditert-butylphosphanyl-n,n-dimethylaniline;dichloropalladium Chemical compound Cl[Pd]Cl.CN(C)C1=CC=C(P(C(C)(C)C)C(C)(C)C)C=C1.CN(C)C1=CC=C(P(C(C)(C)C)C(C)(C)C)C=C1 DWOZNANUEDYIOF-UHFFFAOYSA-L 0.000 description 4
- VZEBSJIOUMDNLY-UHFFFAOYSA-N 6-bromo-1,3-benzothiazol-2-amine Chemical compound C1=C(Br)C=C2SC(N)=NC2=C1 VZEBSJIOUMDNLY-UHFFFAOYSA-N 0.000 description 4
- 229910001369 Brass Inorganic materials 0.000 description 4
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 4
- 241000282472 Canis lupus familiaris Species 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 4
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 4
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 4
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 4
- VRFMCLZMWVACLN-KBPBESRZSA-N NC1=NC(C=CC(C2=CN=C(CO[C@@H](CCC3)[C@H]3O)N=C2)=C2)=C2S1 Chemical compound NC1=NC(C=CC(C2=CN=C(CO[C@@H](CCC3)[C@H]3O)N=C2)=C2)=C2S1 VRFMCLZMWVACLN-KBPBESRZSA-N 0.000 description 4
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- 206010003246 arthritis Diseases 0.000 description 4
- 238000006664 bond formation reaction Methods 0.000 description 4
- 239000010951 brass Substances 0.000 description 4
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 4
- 229910052794 bromium Inorganic materials 0.000 description 4
- 210000000748 cardiovascular system Anatomy 0.000 description 4
- 210000003169 central nervous system Anatomy 0.000 description 4
- 231100000005 chromosome aberration Toxicity 0.000 description 4
- 238000007796 conventional method Methods 0.000 description 4
- 150000001923 cyclic compounds Chemical class 0.000 description 4
- XCIXKGXIYUWCLL-UHFFFAOYSA-N cyclopentanol Chemical compound OC1CCCC1 XCIXKGXIYUWCLL-UHFFFAOYSA-N 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 150000002466 imines Chemical class 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 210000003141 lower extremity Anatomy 0.000 description 4
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 4
- 235000019341 magnesium sulphate Nutrition 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 150000007522 mineralic acids Chemical class 0.000 description 4
- AQNQGBUEVCAVML-UHFFFAOYSA-N oxazepane Chemical compound C1CCNOCC1 AQNQGBUEVCAVML-UHFFFAOYSA-N 0.000 description 4
- 230000001575 pathological effect Effects 0.000 description 4
- 125000000538 pentafluorophenyl group Chemical group FC1=C(F)C(F)=C(*)C(F)=C1F 0.000 description 4
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 4
- 210000002345 respiratory system Anatomy 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 208000024891 symptom Diseases 0.000 description 4
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 4
- 238000004809 thin layer chromatography Methods 0.000 description 4
- 231100000820 toxicity test Toxicity 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 239000003643 water by type Substances 0.000 description 4
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- MSFVEEFXECBJPG-UHFFFAOYSA-N 2-(chloromethyl)pyrimidine Chemical compound ClCC1=NC=CC=N1 MSFVEEFXECBJPG-UHFFFAOYSA-N 0.000 description 3
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 3
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 3
- YGZXZRBNENMLIE-UHFFFAOYSA-N 3-ethoxycyclobutane-1-carboxylic acid Chemical compound CCOC1CC(C1)C(O)=O YGZXZRBNENMLIE-UHFFFAOYSA-N 0.000 description 3
- 125000004938 5-pyridyl group Chemical group N1=CC=CC(=C1)* 0.000 description 3
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 3
- 108091006112 ATPases Proteins 0.000 description 3
- 102000057290 Adenosine Triphosphatases Human genes 0.000 description 3
- 238000010953 Ames test Methods 0.000 description 3
- 231100000039 Ames test Toxicity 0.000 description 3
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 3
- 239000005695 Ammonium acetate Substances 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- YVPIHUNTVUTOIV-UHFFFAOYSA-N C(CC)OCC1=NC=CC=N1 Chemical compound C(CC)OCC1=NC=CC=N1 YVPIHUNTVUTOIV-UHFFFAOYSA-N 0.000 description 3
- AJEKKRLHVNYEFU-UHFFFAOYSA-N COC(C1=C(C=C2)N=C(N(C(C3=CC=CC=C33)=O)C3=O)S1)=C2Br Chemical compound COC(C1=C(C=C2)N=C(N(C(C3=CC=CC=C33)=O)C3=O)S1)=C2Br AJEKKRLHVNYEFU-UHFFFAOYSA-N 0.000 description 3
- UFBBRWWGSVCTHF-UHFFFAOYSA-N COC(C1=C(C=C2)N=C(N)S1)=C2Br Chemical compound COC(C1=C(C=C2)N=C(N)S1)=C2Br UFBBRWWGSVCTHF-UHFFFAOYSA-N 0.000 description 3
- PLOMUEQWCQULJP-KBPBESRZSA-N COC(C1=C(C=C2)N=C(N)S1)=C2C1=CN=C(CO[C@@H](CCC2)[C@H]2O)N=C1 Chemical compound COC(C1=C(C=C2)N=C(N)S1)=C2C1=CN=C(CO[C@@H](CCC2)[C@H]2O)N=C1 PLOMUEQWCQULJP-KBPBESRZSA-N 0.000 description 3
- GCIQZENHSLKGEQ-UHFFFAOYSA-N COC(C1=C(C=C2)N=C(NC(C3CC3)=O)S1)=C2Br Chemical compound COC(C1=C(C=C2)N=C(NC(C3CC3)=O)S1)=C2Br GCIQZENHSLKGEQ-UHFFFAOYSA-N 0.000 description 3
- KUBVGFGFZBOAQB-UHFFFAOYSA-N COC(C1=CC(N)=NN1C=C1)=C1Cl Chemical compound COC(C1=CC(N)=NN1C=C1)=C1Cl KUBVGFGFZBOAQB-UHFFFAOYSA-N 0.000 description 3
- XJUZRXYOEPSWMB-UHFFFAOYSA-N Chloromethyl methyl ether Chemical compound COCCl XJUZRXYOEPSWMB-UHFFFAOYSA-N 0.000 description 3
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- 101000977771 Homo sapiens Interleukin-1 receptor-associated kinase 4 Proteins 0.000 description 3
- 101000932478 Homo sapiens Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 3
- 229940127590 IRAK4 inhibitor Drugs 0.000 description 3
- 101710199015 Interleukin-1 receptor-associated kinase 1 Proteins 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- AOIYYQFQSFMSSJ-UHFFFAOYSA-N NC1=NC(C=CC(Br)=C2O)=C2S1 Chemical compound NC1=NC(C=CC(Br)=C2O)=C2S1 AOIYYQFQSFMSSJ-UHFFFAOYSA-N 0.000 description 3
- 231100000220 OECD 429 (LLNA) Skin Sensitisation Toxicity 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 3
- 206010070834 Sensitisation Diseases 0.000 description 3
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 3
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 235000019257 ammonium acetate Nutrition 0.000 description 3
- 229940043376 ammonium acetate Drugs 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 150000004945 aromatic hydrocarbons Chemical group 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 230000003833 cell viability Effects 0.000 description 3
- 238000004296 chiral HPLC Methods 0.000 description 3
- 229940061627 chloromethyl methyl ether Drugs 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 210000004748 cultured cell Anatomy 0.000 description 3
- 238000012258 culturing Methods 0.000 description 3
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 3
- 231100000263 cytotoxicity test Toxicity 0.000 description 3
- 230000002950 deficient Effects 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Substances CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 238000001035 drying Methods 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- FUKUFMFMCZIRNT-UHFFFAOYSA-N hydron;methanol;chloride Chemical compound Cl.OC FUKUFMFMCZIRNT-UHFFFAOYSA-N 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- 229910017053 inorganic salt Inorganic materials 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 3
- 210000001853 liver microsome Anatomy 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 208000030159 metabolic disease Diseases 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- 239000012046 mixed solvent Substances 0.000 description 3
- 231100000286 mucous membrane, eye irritation or corrosion testing Toxicity 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- RFIOZSIHFNEKFF-UHFFFAOYSA-M piperazine-1-carboxylate Chemical compound [O-]C(=O)N1CCNCC1 RFIOZSIHFNEKFF-UHFFFAOYSA-M 0.000 description 3
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 230000008313 sensitization Effects 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 230000002194 synthesizing effect Effects 0.000 description 3
- FMLPQHJYUZTHQS-QMMMGPOBSA-N tert-butyl (3s)-3-methylpiperazine-1-carboxylate Chemical compound C[C@H]1CN(C(=O)OC(C)(C)C)CCN1 FMLPQHJYUZTHQS-QMMMGPOBSA-N 0.000 description 3
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 3
- 210000002438 upper gastrointestinal tract Anatomy 0.000 description 3
- AYEGPMGNMOIHDL-QWWZWVQMSA-N (1r,2r)-2-methylcyclopropane-1-carboxylic acid Chemical compound C[C@@H]1C[C@H]1C(O)=O AYEGPMGNMOIHDL-QWWZWVQMSA-N 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- VYXHVRARDIDEHS-UHFFFAOYSA-N 1,5-cyclooctadiene Chemical compound C1CC=CCCC=C1 VYXHVRARDIDEHS-UHFFFAOYSA-N 0.000 description 2
- 239000004912 1,5-cyclooctadiene Substances 0.000 description 2
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 2
- RRDRPVRKPDXOAZ-UHFFFAOYSA-N 2-(4-chloro-3-methoxypyridin-2-yl)acetonitrile Chemical compound COC1=C(Cl)C=CN=C1CC#N RRDRPVRKPDXOAZ-UHFFFAOYSA-N 0.000 description 2
- FOFSXFZRAOBLHA-UHFFFAOYSA-N 2-(bromomethyl)-4-chloro-3-methoxypyridine Chemical compound COC1=C(Cl)C=CN=C1CBr FOFSXFZRAOBLHA-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- UHGULLIUJBCTEF-UHFFFAOYSA-N 2-aminobenzothiazole Chemical compound C1=CC=C2SC(N)=NC2=C1 UHGULLIUJBCTEF-UHFFFAOYSA-N 0.000 description 2
- JBSLANHIBKBQGE-UHFFFAOYSA-N 2-cyclopropylacetamide Chemical compound NC(=O)CC1CC1 JBSLANHIBKBQGE-UHFFFAOYSA-N 0.000 description 2
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- QSWREPDNZURRPS-UHFFFAOYSA-N 5-bromo-2-(chloromethyl)pyrimidine Chemical compound ClCC1=NC=C(Br)C=N1 QSWREPDNZURRPS-UHFFFAOYSA-N 0.000 description 2
- ASNXCLBPISBIFU-UHFFFAOYSA-N 7-[(2-methylpropan-2-yl)oxycarbonyl]-7-azaspiro[3.5]nonane-2-carboxylic acid Chemical compound C1CN(C(=O)OC(C)(C)C)CCC11CC(C(O)=O)C1 ASNXCLBPISBIFU-UHFFFAOYSA-N 0.000 description 2
- AIUURUPEPJRTSD-UHFFFAOYSA-N 7-azaspiro[3.5]nonane-2-carboxylic acid Chemical compound C1C(C(=O)O)CC21CCNCC2 AIUURUPEPJRTSD-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- UJOBWOGCFQCDNV-UHFFFAOYSA-N 9H-carbazole Chemical compound C1=CC=C2C3=CC=CC=C3NC2=C1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical group [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- OKAQKFZXAQSKLA-UHFFFAOYSA-N BrC1=CN=C(COC2CCCC2)N=C1 Chemical compound BrC1=CN=C(COC2CCCC2)N=C1 OKAQKFZXAQSKLA-UHFFFAOYSA-N 0.000 description 2
- WLZPCEOEOLTFPX-VIFPVBQESA-N BrC1=CN=C(COC[C@H]2OCCC2)N=C1 Chemical compound BrC1=CN=C(COC[C@H]2OCCC2)N=C1 WLZPCEOEOLTFPX-VIFPVBQESA-N 0.000 description 2
- VPRKTOFFKOXERK-CHWSQXEVSA-N CC(C)(C)[Si](C)(C)O[C@H](CC(C1)(F)F)[C@@H]1OCC(N=C1)=NC=C1Br Chemical compound CC(C)(C)[Si](C)(C)O[C@H](CC(C1)(F)F)[C@@H]1OCC(N=C1)=NC=C1Br VPRKTOFFKOXERK-CHWSQXEVSA-N 0.000 description 2
- FKDOXAOMRDBQHP-UHFFFAOYSA-N CC(NC1=NC(C=CC(Br)=C2O)=C2S1)=O Chemical compound CC(NC1=NC(C=CC(Br)=C2O)=C2S1)=O FKDOXAOMRDBQHP-UHFFFAOYSA-N 0.000 description 2
- YGSRODCUFNGYFD-UHFFFAOYSA-N CC(NC1=NC(CCC(C2=O)(Br)Br)=C2S1)=O Chemical compound CC(NC1=NC(CCC(C2=O)(Br)Br)=C2S1)=O YGSRODCUFNGYFD-UHFFFAOYSA-N 0.000 description 2
- OYUOJVICUNODDU-ROUUACIJSA-N COC(C1=C(C=C2)N=C(NC(C(C3)CC3=O)=O)S1)=C2C1=CN=C(CO[C@@H](CCC2)[C@H]2O)N=C1 Chemical compound COC(C1=C(C=C2)N=C(NC(C(C3)CC3=O)=O)S1)=C2C1=CN=C(CO[C@@H](CCC2)[C@H]2O)N=C1 OYUOJVICUNODDU-ROUUACIJSA-N 0.000 description 2
- AXIAVMVRPUNOBS-JTQLQIEISA-N C[C@@H](CN(CC1)C2CC2)N1C(OC(C)(C)C)=O Chemical compound C[C@@H](CN(CC1)C2CC2)N1C(OC(C)(C)C)=O AXIAVMVRPUNOBS-JTQLQIEISA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- 102000000503 Collagen Type II Human genes 0.000 description 2
- 108010041390 Collagen Type II Proteins 0.000 description 2
- PMPVIKIVABFJJI-UHFFFAOYSA-N Cyclobutane Chemical compound C1CCC1 PMPVIKIVABFJJI-UHFFFAOYSA-N 0.000 description 2
- YVHAIVPPUIZFBA-UHFFFAOYSA-N Cyclopentylacetic acid Chemical compound OC(=O)CC1CCCC1 YVHAIVPPUIZFBA-UHFFFAOYSA-N 0.000 description 2
- 206010012442 Dermatitis contact Diseases 0.000 description 2
- 208000009386 Experimental Arthritis Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 201000005569 Gout Diseases 0.000 description 2
- 231100000938 Guinea Pig Maximization Test Toxicity 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- 101000831496 Homo sapiens Toll-like receptor 3 Proteins 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- 102000014150 Interferons Human genes 0.000 description 2
- 108010050904 Interferons Proteins 0.000 description 2
- 102100036342 Interleukin-1 receptor-associated kinase 1 Human genes 0.000 description 2
- 101710182491 Interleukin-1 receptor-associated kinase-like 2 Proteins 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 101000836282 Mus musculus Solute carrier organic anion transporter family member 1C1 Proteins 0.000 description 2
- 108010077432 Myeloid Differentiation Factor 88 Proteins 0.000 description 2
- 102000010168 Myeloid Differentiation Factor 88 Human genes 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- NBEFNTKXPKCQDC-UHFFFAOYSA-N N-[6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,3-benzothiazol-2-yl]cyclopropanecarboxamide Chemical compound C1=C(B2OC(C(C)(C)O2)(C)C)C=C2SC(=NC2=C1)NC(=O)C1CC1 NBEFNTKXPKCQDC-UHFFFAOYSA-N 0.000 description 2
- JWVANDDMKGDJPP-GJZGRUSLSA-N NC1=NN(C=CC(C2=CN=C(CO[C@@H](CCC3)[C@H]3O)N=C2)=C2)C2=C1 Chemical compound NC1=NN(C=CC(C2=CN=C(CO[C@@H](CCC3)[C@H]3O)N=C2)=C2)C2=C1 JWVANDDMKGDJPP-GJZGRUSLSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- NARYIABODNKKFI-UHFFFAOYSA-N OC(C1=C(C=C2)N=C(N(C(C3=CC=CC=C33)=O)C3=O)S1)=C2Br Chemical compound OC(C1=C(C=C2)N=C(N(C(C3=CC=CC=C33)=O)C3=O)S1)=C2Br NARYIABODNKKFI-UHFFFAOYSA-N 0.000 description 2
- NLXFWEBXSUXLMD-UHFFFAOYSA-N OC(C1=C(C=C2)N=C(NC(C3CC3)=O)S1)=C2Br Chemical compound OC(C1=C(C=C2)N=C(NC(C3CC3)=O)S1)=C2Br NLXFWEBXSUXLMD-UHFFFAOYSA-N 0.000 description 2
- JWWNTGGJQCMTAP-ROUUACIJSA-N O[C@@H](CCC1)[C@H]1OCC(N=C1)=NC=C1C(C=C1)=CC2=C1N=C(NC(C(C1)CC1=O)=O)S2 Chemical compound O[C@@H](CCC1)[C@H]1OCC(N=C1)=NC=C1C(C=C1)=CC2=C1N=C(NC(C(C1)CC1=O)=O)S2 JWWNTGGJQCMTAP-ROUUACIJSA-N 0.000 description 2
- DGQWCSICYJWRAR-HOTGVXAUSA-N O[C@@H](CCC1)[C@H]1OCC(N=C1)=NC=C1C(C=CC1=C2SC(NC(C3CC3)=O)=N1)=C2O Chemical compound O[C@@H](CCC1)[C@H]1OCC(N=C1)=NC=C1C(C=CC1=C2SC(NC(C3CC3)=O)=N1)=C2O DGQWCSICYJWRAR-HOTGVXAUSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 102000001253 Protein Kinase Human genes 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 102100027233 Solute carrier organic anion transporter family member 1B1 Human genes 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 102100024324 Toll-like receptor 3 Human genes 0.000 description 2
- JAZCEXBNIYKZDI-UHFFFAOYSA-N [Ir+] Chemical class [Ir+] JAZCEXBNIYKZDI-UHFFFAOYSA-N 0.000 description 2
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 2
- 239000012346 acetyl chloride Substances 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 239000012042 active reagent Substances 0.000 description 2
- 208000038016 acute inflammation Diseases 0.000 description 2
- 230000006022 acute inflammation Effects 0.000 description 2
- 125000002252 acyl group Chemical group 0.000 description 2
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 2
- UCTWMZQNUQWSLP-UHFFFAOYSA-N adrenaline Chemical compound CNCC(O)C1=CC=C(O)C(O)=C1 UCTWMZQNUQWSLP-UHFFFAOYSA-N 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- XSCHRSMBECNVNS-UHFFFAOYSA-N benzopyrazine Natural products N1=CC=NC2=CC=CC=C21 XSCHRSMBECNVNS-UHFFFAOYSA-N 0.000 description 2
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 2
- 230000036772 blood pressure Effects 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 235000013877 carbamide Nutrition 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 210000000170 cell membrane Anatomy 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 2
- 208000037976 chronic inflammation Diseases 0.000 description 2
- 230000006020 chronic inflammation Effects 0.000 description 2
- 229940000425 combination drug Drugs 0.000 description 2
- 208000010247 contact dermatitis Diseases 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- YMGUBTXCNDTFJI-UHFFFAOYSA-N cyclopropanecarboxylic acid Chemical compound OC(=O)C1CC1 YMGUBTXCNDTFJI-UHFFFAOYSA-N 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- WGLUMOCWFMKWIL-UHFFFAOYSA-N dichloromethane;methanol Chemical compound OC.ClCCl WGLUMOCWFMKWIL-UHFFFAOYSA-N 0.000 description 2
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- ASUTZQLVASHGKV-JDFRZJQESA-N galanthamine Chemical compound O1C(=C23)C(OC)=CC=C2CN(C)CC[C@]23[C@@H]1C[C@@H](O)C=C2 ASUTZQLVASHGKV-JDFRZJQESA-N 0.000 description 2
- 230000007674 genetic toxicity Effects 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 230000026030 halogenation Effects 0.000 description 2
- 238000005658 halogenation reaction Methods 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 2
- 102000046806 human IRAK4 Human genes 0.000 description 2
- DKAGJZJALZXOOV-UHFFFAOYSA-N hydrate;hydrochloride Chemical compound O.Cl DKAGJZJALZXOOV-UHFFFAOYSA-N 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 2
- 230000003053 immunization Effects 0.000 description 2
- 238000002649 immunization Methods 0.000 description 2
- 238000012606 in vitro cell culture Methods 0.000 description 2
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 229940079322 interferon Drugs 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 2
- 230000003907 kidney function Effects 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 210000005229 liver cell Anatomy 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 238000001819 mass spectrum Methods 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- 210000001589 microsome Anatomy 0.000 description 2
- DMRCQAFFSVEPQV-UHFFFAOYSA-N n-(6-bromo-1,3-benzothiazol-2-yl)cyclopropanecarboxamide Chemical compound S1C2=CC(Br)=CC=C2N=C1NC(=O)C1CC1 DMRCQAFFSVEPQV-UHFFFAOYSA-N 0.000 description 2
- VXXUKWLHWFFUNS-UHFFFAOYSA-N n-(7-oxo-5,6-dihydro-4h-1,3-benzothiazol-2-yl)acetamide Chemical compound C1CCC(=O)C2=C1N=C(NC(=O)C)S2 VXXUKWLHWFFUNS-UHFFFAOYSA-N 0.000 description 2
- PVWOIHVRPOBWPI-UHFFFAOYSA-N n-propyl iodide Chemical compound CCCI PVWOIHVRPOBWPI-UHFFFAOYSA-N 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 2
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 2
- 210000000287 oocyte Anatomy 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 description 2
- 239000008177 pharmaceutical agent Substances 0.000 description 2
- YNPNZTXNASCQKK-UHFFFAOYSA-N phenanthrene Chemical compound C1=CC=C2C3=CC=CC=C3C=CC2=C1 YNPNZTXNASCQKK-UHFFFAOYSA-N 0.000 description 2
- 208000017983 photosensitivity disease Diseases 0.000 description 2
- 231100000434 photosensitization Toxicity 0.000 description 2
- 238000012746 preparative thin layer chromatography Methods 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 125000005767 propoxymethyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])[#8]C([H])([H])* 0.000 description 2
- NSETWVJZUWGCKE-UHFFFAOYSA-N propylphosphonic acid Chemical compound CCCP(O)(O)=O NSETWVJZUWGCKE-UHFFFAOYSA-N 0.000 description 2
- 108060006633 protein kinase Proteins 0.000 description 2
- HZGCZRCZOMANHK-UHFFFAOYSA-N pyrimidin-2-ylmethanol Chemical compound OCC1=NC=CC=N1 HZGCZRCZOMANHK-UHFFFAOYSA-N 0.000 description 2
- 230000002285 radioactive effect Effects 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 239000011535 reaction buffer Substances 0.000 description 2
- 230000004202 respiratory function Effects 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 230000011664 signaling Effects 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 238000013268 sustained release Methods 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 208000011580 syndromic disease Diseases 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- 150000003952 β-lactams Chemical class 0.000 description 2
- BZMMRNKDONDVIB-UHFFFAOYSA-N (1-ethoxycyclopropyl)oxy-trimethylsilane Chemical compound CCOC1(O[Si](C)(C)C)CC1 BZMMRNKDONDVIB-UHFFFAOYSA-N 0.000 description 1
- RQJNPJXCMOLSSC-QWWZWVQMSA-N (1r,2r)-4,4-difluorocyclopentane-1,2-diol Chemical compound O[C@@H]1CC(F)(F)C[C@H]1O RQJNPJXCMOLSSC-QWWZWVQMSA-N 0.000 description 1
- VCVOSERVUCJNPR-RFZPGFLSSA-N (1r,2r)-cyclopentane-1,2-diol Chemical compound O[C@@H]1CCC[C@H]1O VCVOSERVUCJNPR-RFZPGFLSSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- MMIWDPMBWOTICQ-YFKPBYRVSA-N (2s)-2-methylpiperazine-1-carboxylic acid Chemical compound C[C@H]1CNCCN1C(O)=O MMIWDPMBWOTICQ-YFKPBYRVSA-N 0.000 description 1
- HSINOMROUCMIEA-FGVHQWLLSA-N (2s,4r)-4-[(3r,5s,6r,7r,8s,9s,10s,13r,14s,17r)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid Chemical compound C([C@@]12C)C[C@@H](O)C[C@H]1[C@@H](CC)[C@@H](O)[C@@H]1[C@@H]2CC[C@]2(C)[C@@H]([C@H](C)C[C@H](C)C(O)=O)CC[C@H]21 HSINOMROUCMIEA-FGVHQWLLSA-N 0.000 description 1
- LENYOXXELREKGZ-PGMHMLKASA-N (3r)-3-fluoropyrrolidine;hydrochloride Chemical compound Cl.F[C@@H]1CCNC1 LENYOXXELREKGZ-PGMHMLKASA-N 0.000 description 1
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- 125000006274 (C1-C3)alkoxy group Chemical group 0.000 description 1
- 125000003161 (C1-C6) alkylene group Chemical group 0.000 description 1
- PHIQHXFUZVPYII-ZCFIWIBFSA-N (R)-carnitine Chemical compound C[N+](C)(C)C[C@H](O)CC([O-])=O PHIQHXFUZVPYII-ZCFIWIBFSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- FQUYSHZXSKYCSY-UHFFFAOYSA-N 1,4-diazepane Chemical compound C1CNCCNC1 FQUYSHZXSKYCSY-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- 125000004818 1-methylbutylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- FUFLCEKSBBHCMO-UHFFFAOYSA-N 11-dehydrocorticosterone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 FUFLCEKSBBHCMO-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 1
- FZKCAHQKNJXICB-UHFFFAOYSA-N 2,1-benzoxazole Chemical compound C1=CC=CC2=CON=C21 FZKCAHQKNJXICB-UHFFFAOYSA-N 0.000 description 1
- RTMMSCJWQYWMNK-UHFFFAOYSA-N 2,2,2-trifluoroethyl trifluoromethanesulfonate Chemical compound FC(F)(F)COS(=O)(=O)C(F)(F)F RTMMSCJWQYWMNK-UHFFFAOYSA-N 0.000 description 1
- SURCGQGDUADKBL-UHFFFAOYSA-N 2-(2-hydroxyethylamino)-5-nitrobenzo[de]isoquinoline-1,3-dione Chemical compound [O-][N+](=O)C1=CC(C(N(NCCO)C2=O)=O)=C3C2=CC=CC3=C1 SURCGQGDUADKBL-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical group CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- JAZOMJIYYHHUBH-UHFFFAOYSA-N 2-amino-5,6-dihydro-4h-1,3-benzothiazol-7-one Chemical compound C1CCC(=O)C2=C1N=C(N)S2 JAZOMJIYYHHUBH-UHFFFAOYSA-N 0.000 description 1
- FDFCUHBHGYEOLQ-UHFFFAOYSA-N 2-chloro-3-iodo-1-methylpyridin-1-ium Chemical compound C[n+]1cccc(I)c1Cl FDFCUHBHGYEOLQ-UHFFFAOYSA-N 0.000 description 1
- DMUXSGAKEXSNGN-UHFFFAOYSA-N 2-ethyloctanoic acid Chemical compound CCCCCCC(CC)C(O)=O DMUXSGAKEXSNGN-UHFFFAOYSA-N 0.000 description 1
- DPNDWFVORIGXQO-UHFFFAOYSA-N 2-methoxypyridine-4-carboxylic acid Chemical compound COC1=CC(C(O)=O)=CC=N1 DPNDWFVORIGXQO-UHFFFAOYSA-N 0.000 description 1
- KUSYIGBGHPOWEL-UHFFFAOYSA-N 2-methyl nonaoic acid Chemical compound CCCCCCCC(C)C(O)=O KUSYIGBGHPOWEL-UHFFFAOYSA-N 0.000 description 1
- KCVKVMKMXZXHEN-UHFFFAOYSA-N 2-methylnonanamide Chemical compound CCCCCCCC(C)C(N)=O KCVKVMKMXZXHEN-UHFFFAOYSA-N 0.000 description 1
- VHMICKWLTGFITH-UHFFFAOYSA-N 2H-isoindole Chemical compound C1=CC=CC2=CNC=C21 VHMICKWLTGFITH-UHFFFAOYSA-N 0.000 description 1
- WADSJYLPJPTMLN-UHFFFAOYSA-N 3-(cycloundecen-1-yl)-1,2-diazacycloundec-2-ene Chemical compound C1CCCCCCCCC=C1C1=NNCCCCCCCC1 WADSJYLPJPTMLN-UHFFFAOYSA-N 0.000 description 1
- XXQQDDXBUSNVBQ-UHFFFAOYSA-N 3-[(2-methylpropan-2-yl)oxy]cyclobutane-1-carboxylic acid Chemical compound CC(C)(C)OC1CC(C(O)=O)C1 XXQQDDXBUSNVBQ-UHFFFAOYSA-N 0.000 description 1
- ZSHGVMYLGGANKU-UHFFFAOYSA-N 3-hydroxycyclobutane-1-carboxylic acid Chemical compound OC1CC(C(O)=O)C1 ZSHGVMYLGGANKU-UHFFFAOYSA-N 0.000 description 1
- RYGXNMUVOHCPBF-UHFFFAOYSA-N 4-chloro-3-methoxy-2-methylpyridine Chemical compound COC1=C(C)N=CC=C1Cl RYGXNMUVOHCPBF-UHFFFAOYSA-N 0.000 description 1
- BMXJFKLSEGBBSJ-UHFFFAOYSA-N 5-bromo-2-(bromomethyl)pyrimidine Chemical compound BrCC1=NC=C(Br)C=N1 BMXJFKLSEGBBSJ-UHFFFAOYSA-N 0.000 description 1
- DVEQCIBLXRSYPH-UHFFFAOYSA-N 5-butyl-1-cyclohexylbarbituric acid Chemical compound O=C1C(CCCC)C(=O)NC(=O)N1C1CCCCC1 DVEQCIBLXRSYPH-UHFFFAOYSA-N 0.000 description 1
- 125000006163 5-membered heteroaryl group Chemical group 0.000 description 1
- BRMIDWFYOMMSJW-UHFFFAOYSA-N 7-azaspiro[3.5]nonane-2-carboxamide Chemical compound C1C(CC11CCNCC1)C(=O)N BRMIDWFYOMMSJW-UHFFFAOYSA-N 0.000 description 1
- BKFMQUOAABZMKF-UHFFFAOYSA-N 7-azaspiro[3.5]nonane-7-carboxylic acid Chemical compound C1CN(C(=O)O)CCC11CCC1 BKFMQUOAABZMKF-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 102000043966 ABC-type transporter activity proteins Human genes 0.000 description 1
- 108010006533 ATP-Binding Cassette Transporters Proteins 0.000 description 1
- 206010000871 Acute monocytic leukaemia Diseases 0.000 description 1
- YCIPQJTZJGUXND-UHFFFAOYSA-N Aglaia odorata Alkaloid Natural products C1=CC(OC)=CC=C1C1(C(C=2C(=O)N3CCCC3=NC=22)C=3C=CC=CC=3)C2(O)C2=C(OC)C=C(OC)C=C2O1 YCIPQJTZJGUXND-UHFFFAOYSA-N 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical compound C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 239000004342 Benzoyl peroxide Substances 0.000 description 1
- OMPJBNCRMGITSC-UHFFFAOYSA-N Benzoylperoxide Chemical compound C=1C=CC=CC=1C(=O)OOC(=O)C1=CC=CC=C1 OMPJBNCRMGITSC-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- VDOHHYGCWBATEC-UHFFFAOYSA-N C1=CC2=C(C(=C1)OS(=O)(=O)C(F)(F)F)SC=N2 Chemical compound C1=CC2=C(C(=C1)OS(=O)(=O)C(F)(F)F)SC=N2 VDOHHYGCWBATEC-UHFFFAOYSA-N 0.000 description 1
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 description 1
- KKGUFBHCSOLJLG-KBPBESRZSA-N CC(C)(C)[Si](C)(C)O[C@@H](CCC1)[C@H]1OCC1=NC=CC=N1 Chemical compound CC(C)(C)[Si](C)(C)O[C@@H](CCC1)[C@H]1OCC1=NC=CC=N1 KKGUFBHCSOLJLG-KBPBESRZSA-N 0.000 description 1
- CDLUPRDBVODNPM-IRXDYDNUSA-N CC(O[C@@H](CCC1)[C@H]1OCC(N=C1)=NC=C1C1=CC2=CC(N)=NN2C=C1)=O Chemical compound CC(O[C@@H](CCC1)[C@H]1OCC(N=C1)=NC=C1C1=CC2=CC(N)=NN2C=C1)=O CDLUPRDBVODNPM-IRXDYDNUSA-N 0.000 description 1
- KRTHPWWFVGLCKQ-QWRGUYRKSA-N CC(O[C@@H](CCC1)[C@H]1OCC1=NC=CC=N1)=O Chemical compound CC(O[C@@H](CCC1)[C@H]1OCC1=NC=CC=N1)=O KRTHPWWFVGLCKQ-QWRGUYRKSA-N 0.000 description 1
- NMZCYJQAWBYMNB-GJZGRUSLSA-N CC1(C)OB(C2=CN=C(CO[C@@H](CCC3)[C@H]3OC(C)=O)N=C2)OC1(C)C Chemical compound CC1(C)OB(C2=CN=C(CO[C@@H](CCC3)[C@H]3OC(C)=O)N=C2)OC1(C)C NMZCYJQAWBYMNB-GJZGRUSLSA-N 0.000 description 1
- BRLROEBMSSDIMD-UHFFFAOYSA-N CCCOCC(N=C1)=NC=C1C(C=C1)=CC2=C1N=C(NC(C1CC1)=O)S2 Chemical compound CCCOCC(N=C1)=NC=C1C(C=C1)=CC2=C1N=C(NC(C1CC1)=O)S2 BRLROEBMSSDIMD-UHFFFAOYSA-N 0.000 description 1
- 108091054872 CK2 family Proteins 0.000 description 1
- MHXHNLNQRNPZOP-UHFFFAOYSA-N CO[Ir].C1CC=CCCC=C1 Chemical compound CO[Ir].C1CC=CCCC=C1 MHXHNLNQRNPZOP-UHFFFAOYSA-N 0.000 description 1
- RGDZMEMSCRKBCO-ZYUBPSHWSA-N C[C@H](C1)[C@@H]1C(NC1=NC(C=CC(C2=CN=C(CO[C@@H](CCC3)[C@H]3O)N=C2)=C2)=C2S1)=O Chemical compound C[C@H](C1)[C@@H]1C(NC1=NC(C=CC(C2=CN=C(CO[C@@H](CCC3)[C@H]3O)N=C2)=C2)=C2S1)=O RGDZMEMSCRKBCO-ZYUBPSHWSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- 206010008805 Chromosomal abnormalities Diseases 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- MFYSYFVPBJMHGN-ZPOLXVRWSA-N Cortisone Chemical compound O=C1CC[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 MFYSYFVPBJMHGN-ZPOLXVRWSA-N 0.000 description 1
- MFYSYFVPBJMHGN-UHFFFAOYSA-N Cortisone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)(O)C(=O)CO)C4C3CCC2=C1 MFYSYFVPBJMHGN-UHFFFAOYSA-N 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 208000030453 Drug-Related Side Effects and Adverse reaction Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- 229910052693 Europium Inorganic materials 0.000 description 1
- 206010015946 Eye irritation Diseases 0.000 description 1
- 206010016207 Familial Mediterranean fever Diseases 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 206010064571 Gene mutation Diseases 0.000 description 1
- 206010060891 General symptom Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101001050476 Homo sapiens Tyrosine-protein kinase ITK/TSK Proteins 0.000 description 1
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 1
- 101150057269 IKBKB gene Proteins 0.000 description 1
- 102000039996 IL-1 family Human genes 0.000 description 1
- 108091069196 IL-1 family Proteins 0.000 description 1
- HEFNNWSXXWATRW-UHFFFAOYSA-N Ibuprofen Chemical compound CC(C)CC1=CC=C(C(C)C(O)=O)C=C1 HEFNNWSXXWATRW-UHFFFAOYSA-N 0.000 description 1
- 231100000750 In vitro toxicology Toxicity 0.000 description 1
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000045959 Interleukin-1 Receptor-Like 1 Human genes 0.000 description 1
- 108700003107 Interleukin-1 Receptor-Like 1 Proteins 0.000 description 1
- 102100036433 Interleukin-1 receptor-associated kinase-like 2 Human genes 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- OWIKHYCFFJSOEH-UHFFFAOYSA-N Isocyanic acid Chemical compound N=C=O OWIKHYCFFJSOEH-UHFFFAOYSA-N 0.000 description 1
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 1
- ZCVMWBYGMWKGHF-UHFFFAOYSA-N Ketotifene Chemical compound C1CN(C)CCC1=C1C2=CC=CC=C2CC(=O)C2=C1C=CS2 ZCVMWBYGMWKGHF-UHFFFAOYSA-N 0.000 description 1
- PWKSKIMOESPYIA-BYPYZUCNSA-N L-N-acetyl-Cysteine Chemical compound CC(=O)N[C@@H](CS)C(O)=O PWKSKIMOESPYIA-BYPYZUCNSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 1
- 208000035489 Monocytic Acute Leukemia Diseases 0.000 description 1
- 102100022691 NACHT, LRR and PYD domains-containing protein 3 Human genes 0.000 description 1
- 101710126825 NACHT, LRR and PYD domains-containing protein 3 Proteins 0.000 description 1
- 102000003945 NF-kappa B Human genes 0.000 description 1
- 108010057466 NF-kappa B Proteins 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- JBHDOEWPFIVDCI-UHFFFAOYSA-N O=C(C1CC1)NC1=NC(C=CC(C2=CN=C(COC3CCCC3)N=C2)=C2)=C2S1 Chemical compound O=C(C1CC1)NC1=NC(C=CC(C2=CN=C(COC3CCCC3)N=C2)=C2)=C2S1 JBHDOEWPFIVDCI-UHFFFAOYSA-N 0.000 description 1
- AJKRANDMFQKKPG-ROUUACIJSA-N OCCOC(C1=C(C=C2)N=C(NC(C3CC3)=O)S1)=C2C1=CN=C(CO[C@@H](CCC2)[C@H]2O)N=C1 Chemical compound OCCOC(C1=C(C=C2)N=C(NC(C3CC3)=O)S1)=C2C1=CN=C(CO[C@@H](CCC2)[C@H]2O)N=C1 AJKRANDMFQKKPG-ROUUACIJSA-N 0.000 description 1
- NHAMRKJVGDFRFO-HOTGVXAUSA-N O[C@@H](CC(C1)(F)F)[C@H]1OCC(N=C1)=NC=C1C(C=C1)=CC2=C1N=C(NC(C1CC1)=O)S2 Chemical compound O[C@@H](CC(C1)(F)F)[C@H]1OCC(N=C1)=NC=C1C(C=C1)=CC2=C1N=C(NC(C1CC1)=O)S2 NHAMRKJVGDFRFO-HOTGVXAUSA-N 0.000 description 1
- MCKKBRBCLLYDMJ-OALUTQOASA-N O[C@@H](CCC1)[C@H]1OCC(N=C1)=NC=C1C1=CC2=CC(NC(C(C3)CC3=O)=O)=NN2C=C1 Chemical compound O[C@@H](CCC1)[C@H]1OCC(N=C1)=NC=C1C1=CC2=CC(NC(C(C3)CC3=O)=O)=NN2C=C1 MCKKBRBCLLYDMJ-OALUTQOASA-N 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 101150053185 P450 gene Proteins 0.000 description 1
- 102000016387 Pancreatic elastase Human genes 0.000 description 1
- 108010067372 Pancreatic elastase Proteins 0.000 description 1
- BYPFEZZEUUWMEJ-UHFFFAOYSA-N Pentoxifylline Chemical compound O=C1N(CCCCC(=O)C)C(=O)N(C)C2=C1N(C)C=N2 BYPFEZZEUUWMEJ-UHFFFAOYSA-N 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- CXOFVDLJLONNDW-UHFFFAOYSA-N Phenytoin Chemical compound N1C(=O)NC(=O)C1(C=1C=CC=CC=1)C1=CC=CC=C1 CXOFVDLJLONNDW-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 206010034972 Photosensitivity reaction Diseases 0.000 description 1
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 108091006207 SLC-Transporter Proteins 0.000 description 1
- 102000037054 SLC-Transporter Human genes 0.000 description 1
- 241000607142 Salmonella Species 0.000 description 1
- 108010087230 Sincalide Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 101150110875 Syk gene Proteins 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- YPWFISCTZQNZAU-UHFFFAOYSA-N Thiane Chemical compound C1CCSCC1 YPWFISCTZQNZAU-UHFFFAOYSA-N 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 206010070863 Toxicity to various agents Diseases 0.000 description 1
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 1
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 1
- LEHOTFFKMJEONL-UHFFFAOYSA-N Uric Acid Chemical compound N1C(=O)NC(=O)C2=C1NC(=O)N2 LEHOTFFKMJEONL-UHFFFAOYSA-N 0.000 description 1
- TVWHNULVHGKJHS-UHFFFAOYSA-N Uric acid Natural products N1C(=O)NC(=O)C2NC(=O)NC21 TVWHNULVHGKJHS-UHFFFAOYSA-N 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 241000269368 Xenopus laevis Species 0.000 description 1
- BSYVTEYKTMYBMK-YFKPBYRVSA-N [(2s)-oxolan-2-yl]methanol Chemical compound OC[C@@H]1CCCO1 BSYVTEYKTMYBMK-YFKPBYRVSA-N 0.000 description 1
- QJGAGWQFOSFZRE-JTQLQIEISA-N [(3S)-3-methylpiperazin-1-yl]-(2-methylpyridin-4-yl)methanone Chemical compound C[C@H]1CN(CCN1)C(=O)c1ccnc(C)c1 QJGAGWQFOSFZRE-JTQLQIEISA-N 0.000 description 1
- AYVGBNGTBQLJBG-UHFFFAOYSA-N [3-(hydroxymethyl)cyclopentyl]methanol Chemical compound OCC1CCC(CO)C1 AYVGBNGTBQLJBG-UHFFFAOYSA-N 0.000 description 1
- FRGBRXALGMTAID-UHFFFAOYSA-N [Si](C)(C)(C(C)(C)C)OCC1=NC=CC=N1 Chemical compound [Si](C)(C)(C(C)(C)C)OCC1=NC=CC=N1 FRGBRXALGMTAID-UHFFFAOYSA-N 0.000 description 1
- WLLIXJBWWFGEHT-UHFFFAOYSA-N [tert-butyl(dimethyl)silyl] trifluoromethanesulfonate Chemical compound CC(C)(C)[Si](C)(C)OS(=O)(=O)C(F)(F)F WLLIXJBWWFGEHT-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 229960004308 acetylcysteine Drugs 0.000 description 1
- 150000008065 acid anhydrides Chemical class 0.000 description 1
- 230000036982 action potential Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- NDAUXUAQIAJITI-UHFFFAOYSA-N albuterol Chemical compound CC(C)(C)NCC(O)C1=CC=C(O)C(CO)=C1 NDAUXUAQIAJITI-UHFFFAOYSA-N 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 125000004419 alkynylene group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- ZSBOMTDTBDDKMP-OAHLLOKOSA-N alogliptin Chemical compound C=1C=CC=C(C#N)C=1CN1C(=O)N(C)C(=O)C=C1N1CCC[C@@H](N)C1 ZSBOMTDTBDDKMP-OAHLLOKOSA-N 0.000 description 1
- 229960001667 alogliptin Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- CHKQALUEEULCPZ-UHFFFAOYSA-N amino 2,4,6-trimethylbenzenesulfonate Chemical compound CC1=CC(C)=C(S(=O)(=O)ON)C(C)=C1 CHKQALUEEULCPZ-UHFFFAOYSA-N 0.000 description 1
- 229940126575 aminoglycoside Drugs 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- XLJMAIOERFSOGZ-UHFFFAOYSA-N anhydrous cyanic acid Natural products OC#N XLJMAIOERFSOGZ-UHFFFAOYSA-N 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000001078 anti-cholinergic effect Effects 0.000 description 1
- 230000001315 anti-hyperlipaemic effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 239000000043 antiallergic agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 229940030600 antihypertensive agent Drugs 0.000 description 1
- 239000003430 antimalarial agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000003435 antirheumatic agent Substances 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- 201000003308 autosomal dominant familial periodic fever Diseases 0.000 description 1
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 239000003899 bactericide agent Substances 0.000 description 1
- HNYOPLTXPVRDBG-UHFFFAOYSA-N barbituric acid Chemical compound O=C1CC(=O)NC(=O)N1 HNYOPLTXPVRDBG-UHFFFAOYSA-N 0.000 description 1
- 230000003542 behavioural effect Effects 0.000 description 1
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 229940049706 benzodiazepine Drugs 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 description 1
- 239000012964 benzotriazole Substances 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 235000019400 benzoyl peroxide Nutrition 0.000 description 1
- FFBHFFJDDLITSX-UHFFFAOYSA-N benzyl N-[2-hydroxy-4-(3-oxomorpholin-4-yl)phenyl]carbamate Chemical compound OC1=C(NC(=O)OCC2=CC=CC=C2)C=CC(=C1)N1CCOCC1=O FFBHFFJDDLITSX-UHFFFAOYSA-N 0.000 description 1
- 229940124748 beta 2 agonist Drugs 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 229960002537 betamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-DVTGEIKXSA-N betamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-DVTGEIKXSA-N 0.000 description 1
- IIBYAHWJQTYFKB-UHFFFAOYSA-N bezafibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1CCNC(=O)C1=CC=C(Cl)C=C1 IIBYAHWJQTYFKB-UHFFFAOYSA-N 0.000 description 1
- MKCBRYIXFFGIKN-UHFFFAOYSA-N bicyclo[1.1.1]pentane Chemical compound C1C2CC1C2 MKCBRYIXFFGIKN-UHFFFAOYSA-N 0.000 description 1
- 239000003613 bile acid Substances 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- UORVGPXVDQYIDP-BJUDXGSMSA-N borane Chemical class [10BH3] UORVGPXVDQYIDP-BJUDXGSMSA-N 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 125000005997 bromomethyl group Chemical group 0.000 description 1
- 229950003872 bucolome Drugs 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- SGZAIDDFHDDFJU-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SGZAIDDFHDDFJU-UHFFFAOYSA-N 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 210000004413 cardiac myocyte Anatomy 0.000 description 1
- 229960004203 carnitine Drugs 0.000 description 1
- 238000010609 cell counting kit-8 assay Methods 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 229960005091 chloramphenicol Drugs 0.000 description 1
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 1
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Chemical compound ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 1
- SOYKEARSMXGVTM-UHFFFAOYSA-N chlorphenamine Chemical compound C=1C=CC=NC=1C(CCN(C)C)C1=CC=C(Cl)C=C1 SOYKEARSMXGVTM-UHFFFAOYSA-N 0.000 description 1
- 229960003291 chlorphenamine Drugs 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 229960004544 cortisone Drugs 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- IMZMKUWMOSJXDT-UHFFFAOYSA-N cromoglycic acid Chemical compound O1C(C(O)=O)=CC(=O)C2=C1C=CC=C2OCC(O)COC1=CC=CC2=C1C(=O)C=C(C(O)=O)O2 IMZMKUWMOSJXDT-UHFFFAOYSA-N 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 125000000000 cycloalkoxy group Chemical group 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 125000001352 cyclobutyloxy group Chemical group C1(CCC1)O* 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- USVZFSNDGFNNJT-UHFFFAOYSA-N cyclopenta-1,4-dien-1-yl(diphenyl)phosphane (2,3-dichlorocyclopenta-1,4-dien-1-yl)-diphenylphosphane iron(2+) Chemical compound [Fe++].c1cc[c-](c1)P(c1ccccc1)c1ccccc1.Clc1c(cc[c-]1Cl)P(c1ccccc1)c1ccccc1 USVZFSNDGFNNJT-UHFFFAOYSA-N 0.000 description 1
- 125000000058 cyclopentadienyl group Chemical group C1(=CC=CC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- JCWIWBWXCVGEAN-UHFFFAOYSA-L cyclopentyl(diphenyl)phosphane;dichloropalladium;iron Chemical compound [Fe].Cl[Pd]Cl.[CH]1[CH][CH][CH][C]1P(C=1C=CC=CC=1)C1=CC=CC=C1.[CH]1[CH][CH][CH][C]1P(C=1C=CC=CC=1)C1=CC=CC=C1 JCWIWBWXCVGEAN-UHFFFAOYSA-L 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000000131 cyclopropyloxy group Chemical group C1(CC1)O* 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 230000016396 cytokine production Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 125000001295 dansyl group Chemical group [H]C1=C([H])C(N(C([H])([H])[H])C([H])([H])[H])=C2C([H])=C([H])C([H])=C(C2=C1[H])S(*)(=O)=O 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- NTBIYBAYFBNTCD-UHFFFAOYSA-N dibenzoyl 2,3-dihydroxybutanedioate Chemical compound C=1C=CC=CC=1C(=O)OC(=O)C(O)C(O)C(=O)OC(=O)C1=CC=CC=C1 NTBIYBAYFBNTCD-UHFFFAOYSA-N 0.000 description 1
- 125000003963 dichloro group Chemical group Cl* 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 1
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 1
- 125000005057 dihydrothienyl group Chemical group S1C(CC=C1)* 0.000 description 1
- 125000005072 dihydrothiopyranyl group Chemical group S1C(CCC=C1)* 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 239000002988 disease modifying antirheumatic drug Substances 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 230000007783 downstream signaling Effects 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 238000002592 echocardiography Methods 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- ZSWFCLXCOIISFI-UHFFFAOYSA-N endo-cyclopentadiene Natural products C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 1
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- WHWZLSFABNNENI-UHFFFAOYSA-N epinastine Chemical compound C1C2=CC=CC=C2C2CN=C(N)N2C2=CC=CC=C21 WHWZLSFABNNENI-UHFFFAOYSA-N 0.000 description 1
- 229960003449 epinastine Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- UREBWPXBXRYXRJ-UHFFFAOYSA-N ethyl acetate;methanol Chemical compound OC.CCOC(C)=O UREBWPXBXRYXRJ-UHFFFAOYSA-N 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 description 1
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 1
- 208000012997 experimental autoimmune encephalomyelitis Diseases 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 231100000013 eye irritation Toxicity 0.000 description 1
- 230000009248 fertility and early embryonic development Effects 0.000 description 1
- 230000008175 fetal development Effects 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 229960002598 fumaric acid Drugs 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229960003980 galantamine Drugs 0.000 description 1
- ASUTZQLVASHGKV-UHFFFAOYSA-N galanthamine hydrochloride Natural products O1C(=C23)C(OC)=CC=C2CN(C)CCC23C1CC(O)C=C2 ASUTZQLVASHGKV-UHFFFAOYSA-N 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001156 gastric mucosa Anatomy 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 230000024924 glomerular filtration Effects 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 230000036732 histological change Effects 0.000 description 1
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 229940126904 hypoglycaemic agent Drugs 0.000 description 1
- 229960001680 ibuprofen Drugs 0.000 description 1
- KLZWOWYOHUKJIG-BPUTZDHNSA-N imidapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1C(N(C)C[C@H]1C(O)=O)=O)CC1=CC=CC=C1 KLZWOWYOHUKJIG-BPUTZDHNSA-N 0.000 description 1
- 229960001195 imidapril Drugs 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000003100 immobilizing effect Effects 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 229940125721 immunosuppressive agent Drugs 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 102000008625 interleukin-18 receptor activity proteins Human genes 0.000 description 1
- 108040002014 interleukin-18 receptor activity proteins Proteins 0.000 description 1
- 108040007659 interleukin-33 receptor activity proteins Proteins 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- OEXHQOGQTVQTAT-JRNQLAHRSA-N ipratropium Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)[N@@+]2(C)C(C)C)C(=O)C(CO)C1=CC=CC=C1 OEXHQOGQTVQTAT-JRNQLAHRSA-N 0.000 description 1
- 229960001888 ipratropium Drugs 0.000 description 1
- 229910052741 iridium Inorganic materials 0.000 description 1
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- IQPQWNKOIGAROB-UHFFFAOYSA-N isocyanate group Chemical group [N-]=C=O IQPQWNKOIGAROB-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 239000003199 leukotriene receptor blocking agent Substances 0.000 description 1
- 238000011694 lewis rat Methods 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 239000003120 macrolide antibiotic agent Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229940098895 maleic acid Drugs 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 210000001161 mammalian embryo Anatomy 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 230000008774 maternal effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 238000002779 membrane potential assay Methods 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000037323 metabolic rate Effects 0.000 description 1
- LVWZTYCIRDMTEY-UHFFFAOYSA-N metamizole Chemical compound O=C1C(N(CS(O)(=O)=O)C)=C(C)N(C)N1C1=CC=CC=C1 LVWZTYCIRDMTEY-UHFFFAOYSA-N 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- WSFSSNUMVMOOMR-BJUDXGSMSA-N methanone Chemical compound O=[11CH2] WSFSSNUMVMOOMR-BJUDXGSMSA-N 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- SJFNDMHZXCUXSA-UHFFFAOYSA-M methoxymethyl(triphenyl)phosphanium;chloride Chemical compound [Cl-].C=1C=CC=CC=1[P+](C=1C=CC=CC=1)(COC)C1=CC=CC=C1 SJFNDMHZXCUXSA-UHFFFAOYSA-M 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 239000003471 mutagenic agent Substances 0.000 description 1
- 231100000707 mutagenic chemical Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 229940014456 mycophenolate Drugs 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- DXASQZJWWGZNSF-UHFFFAOYSA-N n,n-dimethylmethanamine;sulfur trioxide Chemical group CN(C)C.O=S(=O)=O DXASQZJWWGZNSF-UHFFFAOYSA-N 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- NMUXPVMDAYGGGN-UHFFFAOYSA-N n-(1,3-benzothiazol-2-yl)-2-(4-chlorophenoxy)-2-methylpropanamide Chemical group N=1C2=CC=CC=C2SC=1NC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 NMUXPVMDAYGGGN-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- RIVIDPPYRINTTH-UHFFFAOYSA-N n-ethylpropan-2-amine Chemical compound CCNC(C)C RIVIDPPYRINTTH-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- OELFLUMRDSZNSF-BRWVUGGUSA-N nateglinide Chemical compound C1C[C@@H](C(C)C)CC[C@@H]1C(=O)N[C@@H](C(O)=O)CC1=CC=CC=C1 OELFLUMRDSZNSF-BRWVUGGUSA-N 0.000 description 1
- 229960000698 nateglinide Drugs 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 201000008383 nephritis Diseases 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 230000009437 off-target effect Effects 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- GNWXVOQHLPBSSR-UHFFFAOYSA-N oxolane;toluene Chemical compound C1CCOC1.CC1=CC=CC=C1 GNWXVOQHLPBSSR-UHFFFAOYSA-N 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 1
- 244000045947 parasite Species 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- HIANJWSAHKJQTH-UHFFFAOYSA-N pemirolast Chemical compound CC1=CC=CN(C2=O)C1=NC=C2C=1N=NNN=1 HIANJWSAHKJQTH-UHFFFAOYSA-N 0.000 description 1
- 229960004439 pemirolast Drugs 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 208000025487 periodic fever syndrome Diseases 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 125000001792 phenanthrenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C=CC12)* 0.000 description 1
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000036211 photosensitivity Effects 0.000 description 1
- LFSXCDWNBUNEEM-UHFFFAOYSA-N phthalazine Chemical compound C1=NN=CC2=CC=CC=C21 LFSXCDWNBUNEEM-UHFFFAOYSA-N 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 230000007542 postnatal development Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- NNFCIKHAZHQZJG-UHFFFAOYSA-N potassium cyanide Chemical compound [K+].N#[C-] NNFCIKHAZHQZJG-UHFFFAOYSA-N 0.000 description 1
- 239000012286 potassium permanganate Substances 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 230000009237 prenatal development Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- DBABZHXKTCFAPX-UHFFFAOYSA-N probenecid Chemical compound CCCN(CCC)S(=O)(=O)C1=CC=C(C(O)=O)C=C1 DBABZHXKTCFAPX-UHFFFAOYSA-N 0.000 description 1
- 229960003081 probenecid Drugs 0.000 description 1
- FYPMFJGVHOHGLL-UHFFFAOYSA-N probucol Chemical compound C=1C(C(C)(C)C)=C(O)C(C(C)(C)C)=CC=1SC(C)(C)SC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 FYPMFJGVHOHGLL-UHFFFAOYSA-N 0.000 description 1
- 229960003912 probucol Drugs 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- VTGOHKSTWXHQJK-UHFFFAOYSA-N pyrimidin-2-ol Chemical compound OC1=NC=CC=N1 VTGOHKSTWXHQJK-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000006085 pyrrolopyridyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical compound C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000008327 renal blood flow Effects 0.000 description 1
- 230000008085 renal dysfunction Effects 0.000 description 1
- 231100000205 reproductive and developmental toxicity Toxicity 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000036387 respiratory rate Effects 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229960002052 salbutamol Drugs 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- IZTQOLKUZKXIRV-YRVFCXMDSA-N sincalide Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(=O)NCC(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](N)CC(O)=O)C1=CC=C(OS(O)(=O)=O)C=C1 IZTQOLKUZKXIRV-YRVFCXMDSA-N 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- JUJBNYBVVQSIOU-UHFFFAOYSA-M sodium;4-[2-(4-iodophenyl)-3-(4-nitrophenyl)tetrazol-2-ium-5-yl]benzene-1,3-disulfonate Chemical compound [Na+].C1=CC([N+](=O)[O-])=CC=C1N1[N+](C=2C=CC(I)=CC=2)=NC(C=2C(=CC(=CC=2)S([O-])(=O)=O)S([O-])(=O)=O)=N1 JUJBNYBVVQSIOU-UHFFFAOYSA-M 0.000 description 1
- 238000003797 solvolysis reaction Methods 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000013112 stability test Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 1
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- NCEXYHBECQHGNR-QZQOTICOSA-N sulfasalazine Chemical compound C1=C(O)C(C(=O)O)=CC(\N=N\C=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-QZQOTICOSA-N 0.000 description 1
- 229960001940 sulfasalazine Drugs 0.000 description 1
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 238000004808 supercritical fluid chromatography Methods 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- DATRVIMZZZVHMP-QMMMGPOBSA-N tert-butyl (2s)-2-methylpiperazine-1-carboxylate Chemical compound C[C@H]1CNCCN1C(=O)OC(C)(C)C DATRVIMZZZVHMP-QMMMGPOBSA-N 0.000 description 1
- BONSKTIHNARTGJ-UHFFFAOYSA-N tert-butyl 3-fluoro-3-(hydroxymethyl)azetidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC(F)(CO)C1 BONSKTIHNARTGJ-UHFFFAOYSA-N 0.000 description 1
- TYVLAZGEMLWPQS-UHFFFAOYSA-N tert-butyl 3-hydroxycyclobutane-1-carboxylate Chemical compound CC(C)(C)OC(=O)C1CC(O)C1 TYVLAZGEMLWPQS-UHFFFAOYSA-N 0.000 description 1
- WMOVHXAZOJBABW-UHFFFAOYSA-N tert-butyl acetate Chemical compound CC(=O)OC(C)(C)C WMOVHXAZOJBABW-UHFFFAOYSA-N 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000005958 tetrahydrothienyl group Chemical group 0.000 description 1
- RAOIDOHSFRTOEL-UHFFFAOYSA-N tetrahydrothiophene Chemical compound C1CCSC1 RAOIDOHSFRTOEL-UHFFFAOYSA-N 0.000 description 1
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 1
- VXKWYPOMXBVZSJ-UHFFFAOYSA-N tetramethyltin Chemical compound C[Sn](C)(C)C VXKWYPOMXBVZSJ-UHFFFAOYSA-N 0.000 description 1
- 150000003536 tetrazoles Chemical group 0.000 description 1
- 229960000278 theophylline Drugs 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 125000000437 thiazol-2-yl group Chemical group [H]C1=C([H])N=C(*)S1 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 239000012929 tonicity agent Substances 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 229940116269 uric acid Drugs 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 238000009423 ventilation Methods 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Images
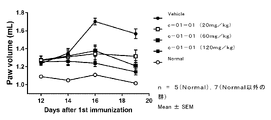
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/08—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/18—Bridged systems
Definitions
- the present invention relates to a novel nitrogen-containing bicyclic compound or a drug containing them as an active ingredient.
- Interleukin-1 receptor-related kinase 4 plays an important role in downstream signaling of Toll-like receptors (TLRs), interleukin-1 receptors (IL-1R), IL-18R and IL-33R. It is a protein kinase that plays a role (Non-Patent Document 1). Since the TLRs / IL-1 receptor family play important roles in inflammation and biological defense, this downstream signal is thought to play a major role in many diseases, including inflammatory and autoimmune diseases.
- TLRs use pathogen-associated molecular patterns (PAMPs) derived from infectious microorganisms such as bacteria, fungi, parasites, and viruses as ligands. In addition, it recognizes and activates damage-associated molecular patterns (DAMPs) released from damaged and apoptotic cells.
- PAMPs pathogen-associated molecular patterns
- DAMPs damage-associated molecular patterns
- MyD88 the adapter molecule MyD88 is recruited to a common intracellular region called the TIR (Toll / IL-1 receptor) region.
- IRAK-4 is recruited to the receptor by interacting with MyD88 and initiates downstream signal transduction (Non-Patent Document 2).
- IRAK-4 activates IRAK-1 and IRAK-2, and further regulates the production of inflammatory mediators such as cytokines and chemokines through the activation of signaling molecules such as NF-kB and MAPK downstream.
- Non-Patent Document 3 It has been reported that human-derived cells lacking the IRAK-4 gene do not respond to TLRs agonists other than TLR3 and IL-1 ⁇ and IL-18 (Non-Patent Document 3). In addition, IRAK-4 gene-deficient mice do not respond to IL-1 ⁇ and IL-18 with TLRs agonists other than TLR3 (Non-Patent Document 4). On the other hand, in IRAK-1 gene-deficient mice and IRAK-2 gene-deficient mice, these signals are only partially suppressed (Non-Patent Document 5). Therefore, it is considered that IRAK-4 plays a central role in these signal transductions in the IRAK family.
- Non-Patent Document 6 the kinase activity of IRAK-4 is essential for signal transduction involved in pathological conditions, and IRAK-4 inhibitors are acute and chronic inflammation, autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus, and metabolic diseases such as gout and diabetes. , May show high efficacy in the treatment of diseases such as tumors.
- the compound having IRAK-4 inhibitory activity for example, the compounds described in Patent Documents 1 to 6 are known.
- the problem to be solved by the present invention is to provide a novel compound having IRAK-4 inhibitory activity. Another challenge is to provide a novel compound useful as an active ingredient of a pharmaceutical for preventing and / or treating a disease associated with IRAK-4 inhibition. Yet another subject is to provide a pharmaceutical containing the compound.
- the compounds of the present invention represented by the following formula (1) have excellent IRAK-4 inhibitory activity, and these compounds have IRAK-. 4
- it is useful for the prevention and / or treatment of diseases related to inhibition, and have completed the present invention.
- the present invention includes the following.
- the following general formula (1) [In equation (1), Rg is the following general formula (1-1): Or the following general formula (1-2): (A and b indicate the bonding direction), R 1 is -H, -F, -Cl, methyl, or C 1-3 alkoxy, and the C 1-3 alkoxy is 1 to 3 identical or different substituents selected from the G1 group. May be replaced;
- the G1 group is a group consisting of -F, hydroxy, cyano, halogeno C 1-6 alkyl, C 1-4 alkoxy, phenyl, 5-6 member heteroaryl, and 3-7 member saturated ring group, and is a group consisting of G.
- Phenyl or 5-6 membered heteroaryls in group 1 may be substituted with 1 to 3 identical or different substituents selected from group G Ar ;
- the G Ar group is a group consisting of -F, -Cl, hydroxy, cyano, C 1-6 alkyl, halogeno C 1-6 alkyl, and -NH 2 ;
- R 2 is a C 1-6 alkyl, a halogeno C 1-6 alkyl, or a 3-7 member saturated ring group, wherein R 2 is 1 to 3 identical or different substituents selected from the G 2 group.
- the G2 group is a group consisting of -F, hydroxy, halogeno C 1-3 alkyl, and C 1-4 alkoxy; Cy is the following general formula (2-1): And; In equation (2-1), k is an integer of 0 or 1; R Cy1 and R Cy2 are independently -H, -F, hydroxy, cyano, C 1-6 alkyl, halogeno C 1-6 alkyl, C 1-6 alkoxy, -NR 11 R 12 , phenyl, 5 It is a -6-membered heteroaryl or a 3-7-membered saturated ring group, and the phenyl or 5-6-membered heteroaryl in RCy1 and RCy2 is one to three identical or different substitutions selected from the G Ar group.
- R Cy1 and R Cy2 may be substituted with 1 to 3 identical or different substituents selected from the G1 group;
- R 11 and R 12 are independently -H, C 1-6 alkyl, halogeno C 1-6 alkyl, hydroxy C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, 3-7 members, respectively.
- R 11 and R 12 may be substituted with 1 to 3 identical or different substituents selected from the G1 group;
- R 11 and R 12 are combined to form a 4-10-membered saturated ring or a 7-11-membered spiro ring, and the 4-10-membered saturated ring and the 7-11-membered spiro ring are added to N and O and It may have one or two identical or different heteroatoms or —S (O 2 ) — selected from the group consisting of N, and the 4-10-membered saturated ring and the 7-11-membered spiro ring may have.
- the G3 group includes -F , hydroxy, C 1-6 alkyl, halogeno C 1-6 alkyl, hydroxy C 1-6 alkyl, C 1-6 alkoxy, C 1-6 alkoxy-C 1-6 alkyl, and halogeno.
- R 13 and R 14 are combined to form a 4-7-membered saturated ring or a 7-11-membered spiro ring, and the 4-7-membered saturated ring and the 7-11-membered spiro ring are added to N and O and It may
- R 1 is -H; Cy is the following general formula (2-1-1): (R 11 and R 12 have the same meanings as described above) The compound according to any one of the above [2] to [5] or a salt thereof.
- R 11 and R 22 are independently -H, C 1-6 alkyl, halogeno C 1-6 alkyl, hydroxy C 1-6 alkyl, and C 1-6 alkoxy C 1-6 alkyl, respectively. , 3-7-membered saturated ring group, phenyl, or 5-6-membered heteroaryl, with R 11 and R 12 substituted with 1 to 3 identical or different substituents selected from the G1 group.
- R 11 and R 22 are independently ⁇ H, C 1-6 alkyl, halogeno C 1-6 alkyl, 3-7 member saturated ring group, or 5-6 member heteroaryl. , R 11 and R 12 may be substituted with 1 to 3 identical or different substituents selected from the G1 group; The compound according to any one of the above [2] to [5] or a salt thereof.
- R 11 and R 22 are independently ⁇ H, C 1-6 alkyl, halogeno C 1-6 alkyl, or 3-5 member saturated ring groups, and R 11 and R 12 are It may be substituted with 1 to 3 identical or different substituents selected from the G1 group; The compound according to any one of the above [2] to [5] or a salt thereof. [6-5] R 11 and R 12 are combined to form a 4-10-membered saturated ring or a 7-11-membered spiro ring, and the 4-7-membered saturated ring and the 7-11-membered spiro ring are added to N.
- the member spiro ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group;
- R 11 and R 12 are combined to form a 4-10-membered saturated ring or a 7-11-membered spiro ring, and the 4-7-membered saturated ring and the 7-11-membered spiro ring are added to N.
- the member spiro ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group; The compound according to any one of the above [2] to [5] or a salt thereof.
- R 11 and R 12 are combined into the following general formulas (2-1-1-a-1) to (2-1-1-a-7):
- the saturated ring of any of the above may be formed, and the saturated ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group;
- R 15 is -H, C 1-6 alkyl, halogeno C 1-6 alkyl, hydroxy C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, -C (O) R 16 , -S (O).
- R 16 is C 1-6 alkyl, halogeno C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, halogeno C 1-6 alkoxy C 1-6 alkyl, phenyl, 5-6 member heteroaryl, or It is a 3-7-membered saturated ring group, and the phenyl or 5-6-membered heteroaryl in the R16 may be substituted with 1 to 3 identical or different substituents selected from the GAr group; R 17 is -H, C 1-6 alkyl, halogeno C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, halogeno C 1-6 alkoxy C 1-6 alkyl, phenyl, 5-6
- Aryl, or a 3-7-membered saturated ring group, the phenyl or 5-6-membered heteroaryl in the R 17 is substituted with 1 to 3 identical or different substituents selected from the G Ar group.
- R 16 and R 17 are combined to form a 4-7-membered saturated ring or a 7-11-membered spiro ring, and the 4-7-membered saturated ring and the 7-11-membered spiro ring are added to N and O and It may have one or two identical or different heteroatoms or —S (O 2 ) — selected from the group consisting of N;
- R 11 and R 12 are combined to form the following general formula (2-1-1-b-1):
- the saturated ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group;
- X is the compound according to any one of the above [2] to [5], which is O or NR 15 (R 15 is synonymous with the above), or a salt thereof.
- R 11 and R 12 are combined into the following general formulas (2-1-1-c-1) to (2-1-1-c-3):
- the saturated ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group;
- X is the compound according to any one of the above [2] to [5], which is O or NR 15 (R 15 is synonymous with the above), or a salt thereof.
- a saturated ring having a crosslink of the above may be formed, and the saturated ring having the crosslink may be substituted with 1 to 3 identical or different substituents selected from the G3 group;
- X is the compound according to any one of the above [2] to [5], which is O or NR 15 (R 15 is synonymous with the above), or a salt thereof.
- R 11 and R 12 are combined to form the following general formula (2-1-1-e-1): A saturated ring having a condensed ring of No. 1 may be formed, and the saturated ring having the condensed ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group; X is the compound according to any one of the above [2] to [5], which is O or NR 15 (R 15 is synonymous with the above), or a salt thereof.
- Cy is the following general formula (2-1-2): And; [In formula (2-1-2), R Cy3 is C 1-4 alkyl, halogeno C 1-4 alkyl; X is O or NR 15 ; R 15 is -H, C 1-6 alkyl, halogeno C 1-6 alkyl, hydroxy C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, -C (O) R 16 , -S (O). 2 ) R 16 , -C (O) NR 16 R 17 , -C (O) OR 16 , or a 3-7-membered saturated ring group, wherein R 15 is 1 to 3 selected from the G1 group.
- R 16 is C 1-6 alkyl, halogeno C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, halogeno C 1-6 alkoxy C 1-6 alkyl, phenyl, 5-6 member heteroaryl, or It is a 3-7-membered saturated ring group, and the phenyl or 5-6-membered heteroaryl in the R16 may be substituted with 1 to 3 identical or different substituents selected from the GAr group;
- R 17 is -H, C 1-6 alkyl, halogeno C 1-6 alkyl, C 1-3 alkoxy C 1-6 alkyl, halogeno C 1-6 alkoxy C 1-6 alkyl, phenyl, 5-6 member hetero.
- Aryl, or a 3-7-membered saturated ring group, the phenyl or 5-6-membered heteroaryl in the R 17 is substituted with 1 to 3 identical or different substituents selected from the G Ar group.
- R 16 and R 17 are combined to form a 4-7-membered saturated ring or a 7-11-membered spiro ring, and the 4-7-membered saturated ring and the 7-11-membered spiro ring are added to N and O and May have one or two identical or different heteroatoms or -S (O 2 )- selected from the group consisting of N]
- the compound or salt thereof according to any one of [2] to [6-11] indicated by.
- R2 is the following equation (3-1): Alternatively, the compound according to any one of the above [2] to [9], which is normal propyl, or a salt thereof.
- R2 is the following equation (3-1): The compound according to any one of the above [2] to [9] or a salt thereof.
- Cy is the general formula (2-1); [In equation (2-1), k is an integer of 1; R Cy1 and R Cy2 are independently -H, -F, hydroxy, cyano, C 1-6 alkyl, halogeno C 1-6 alkyl, C 1-6 alkoxy, -NR 11 R 12 , phenyl, 5 It is a -6-membered heteroaryl or a 3-7-membered saturated ring group, and the phenyl or 5-6-membered heteroaryl in RCy1 and RCy2 is one to three identical or different substitutions selected from the G Ar group.
- R Cy1 and R Cy2 may be substituted with 1 to 3 identical or different substituents selected from the G1 group (R 11 and R 12 are the above. (Synonymous with)] The compound or salt thereof according to any one of [22] to [24] indicated by.
- Cy is the following general formula (2-1-1): (R 11 and R 12 have the same meanings as described above) The compound according to any one of the above [22] to [25] or a salt thereof.
- R 11 and R 22 are independently ⁇ H, C 1-6 alkyl, halogeno C 1-6 alkyl, hydroxy C 1-6 alkyl, and C 1-6 alkoxy C 1-6 alkyl, respectively. , 3-7-membered saturated ring group, phenyl, or 5-6-membered heteroaryl, with R 11 and R 12 substituted with 1 to 3 identical or different substituents selected from the G1 group. May; The compound according to any one of [22] to [25] or a salt thereof.
- R 11 and R 22 are independently ⁇ H, C 1-6 alkyl, halogeno C 1-6 alkyl, 3-7 member saturated ring group, or 5-6 member heteroaryl. , R 11 and R 12 may be substituted with 1 to 3 identical or different substituents selected from the G1 group; The compound according to any one of [22] to [25] or a salt thereof.
- R 11 and R 22 are independently ⁇ H, C 1-6 alkyl, halogeno C 1-6 alkyl, or 3-5 member saturated ring groups, and R 11 and R 12 are It may be substituted with 1 to 3 identical or different substituents selected from the G1 group; The compound according to any one of [22] to [25] or a salt thereof.
- R 11 and R 12 are combined to form a 4-10-membered saturated ring or a 7-11-membered spiro ring, and the 4-7-membered saturated ring and the 7-11-membered spiro ring are added to N.
- the member spiro ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group;
- R 11 and R 12 are combined to form a 4-10-membered saturated ring or a 7-11-membered spiro ring, and the 4-7-membered saturated ring and the 7-11-membered spiro ring are added to N.
- the member spiro ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group; The compound according to any one of [22] to [25] or a salt thereof.
- R 11 and R 12 are combined into the following general formulas (2-1-1-a-1) to (2-1-1-a-7): ( R15 is synonymous with the above)
- the saturated ring of any of the above may be formed, and the saturated ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group;
- R 11 and R 12 are combined to form the following general formula (2-1-1-b-1):
- the saturated ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group;
- X is the compound according to any one of the above [22] to [25], which is O or NR 15 (R 15 is synonymous with the above), or a salt thereof.
- R 11 and R 12 are combined into the following general formulas (2-1-1-c-1) to (2-1-1-c-3):
- the saturated ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group;
- X is the compound according to any one of the above [22] to [25], which is O or NR 15 (R 15 is synonymous with the above), or a salt thereof.
- R 11 and R 12 are combined to form the following general formulas (2-1-1-d-1) to (2-1-1-d-8):
- the spiro ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group;
- X is the compound according to any one of the above [22] to [25], which is O or NR 15 (R 15 is synonymous with the above), or a salt thereof.
- R 11 and R 12 are combined to form the following general formula (2-1-1-e-1): A saturated ring having a condensed ring of No. 1 may be formed, and the saturated ring having the condensed ring may be substituted with 1 to 3 identical or different substituents selected from the G3 group; X is the compound according to any one of the above [22] to [25], which is O or NR 15 (R 15 is synonymous with the above), or a salt thereof.
- Cy is the following general formula (2-1-2): ( RCy3 is synonymous with the above); X is the compound according to any one of the above [22] to [26-11], which is O or NR 15 (R 15 is synonymous with the above), or a salt thereof. [28] The compound or salt thereof according to any one of the above [22] to [27], wherein X is NR 15 (R 15 is synonymous with the above).
- Cy is the following general formula (2-1-3): ( RCy3 , X are synonymous with the above) The compound according to any one of the above [22] to [28] or a salt thereof.
- R 2 is the following equation (3-1): Alternatively, the compound according to any one of the above [22] to [29], which is normal propyl, or a salt thereof.
- R 2 is the following equation (3-1) :.
- a pharmaceutical composition for the prevention and / or treatment of rheumatism wherein the compound according to any one of the above [1] to [35] or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable salt thereof.
- a pharmaceutical composition comprising an acceptable carrier.
- a method for preventing and / or treating rheumatism in a mammal wherein an effective amount of the compound according to any one of the above [1] to [35] or a pharmaceutically acceptable salt thereof is used.
- a method comprising the step of administering to a mammal.
- the "compound represented by the formula (1) or a salt thereof” (hereinafter, may be simply referred to as “the compound of the present invention”) has excellent IRAK-4 inhibitory activity. Also, certain embodiments of the compounds of the invention show high selectivity for other kinases, especially FLT3. Moreover, certain embodiments of the compounds of the invention show low genotoxicity. In addition, certain embodiments of the compounds of the invention can be used as active ingredients in pharmaceuticals for the prevention and / or treatment of diseases associated with IRAK-4 inhibition, such as the prevention and / or treatment of autoimmune diseases. Furthermore, certain embodiments of the compounds of the invention can be used as reagents with IRAK-4 inhibitory activity.
- the carbon atom is simply “C”
- the hydrogen atom is “H”
- the oxygen atom is “O”
- the sulfur atom is “S”
- the nitrogen atom is “N”.
- the carbonyl group is simply “-C (O)-”
- the carboxyl group is “-COO-”
- the sulfinyl group is “-S (O)-”
- the sulfonyl group is "-S (O) 2- ".
- the ether bond may be represented by “-O-” and the thioether bond may be represented by "-S-” (in this case, "-” represents the bond).
- the alkyl may be a saturated hydrocarbon group that is linear, branched, cyclic, or a combination thereof.
- methyl, ethyl, propyl, butyl, their isomers [normal (n), iso (iso), secondary (sec), tertiary (t), etc.], or cycloalkyl such as cyclopropyl or cyclobutyl are exemplified.
- the alkyl include an alkyl having 1 to 6 carbon atoms.
- an alkyl having 1 to 3 carbon atoms is exemplified.
- An alkyl having 1 to 6 carbon atoms may be referred to as a C 1-6 alkyl.
- alkoxy may be linear, branched, cyclic, or a combination thereof, alkyloxy.
- alkoxy an alkoxy having 1 to 6 carbon atoms is exemplified.
- alkoxy having 1 to 3 carbon atoms is exemplified.
- Alkoxy having 1 to 6 carbon atoms may be referred to as C 1-6 alkoxy.
- Alkylene also includes linear or branched alkylene.
- alkylene examples include linear or branched alkylene.
- methylene, ethylene, propylene, butylene, methylmethylene, ethylmethylene, methylethylene, 1,2-dimethylethylene, 1,1,2,2-tetramethylethylene, or 1-methylbutylene are exemplified.
- the alkylene examples include an alkylene having 1 to 6 carbon atoms.
- an alkylene having 1 to 3 carbon atoms is exemplified.
- An alkylene having 1 to 6 carbon atoms may be referred to as a C 1-6 alkylene.
- the "halogen” is fluoro (-F), chloro (-Cl), bromo (-Br), or iodine (-I).
- -F or -Cl is exemplified.
- -F is exemplified.
- halogeno is meant substituted with the same or different 1-7 halogens. In another aspect, it means that they are substituted with the same or different 1-5 halogens. In yet another embodiment, it means that it is substituted with 1 to 3 halogens. In yet another embodiment, it means that it is substituted with one halogen. It is exemplified that it is substituted with ⁇ F.
- aromatic ring is not particularly limited as long as it is a ring having aromaticity, and examples thereof include monocyclic to tricyclic aromatic rings.
- aromatic ring include an aromatic hydrocarbon ring or an aromatic heterocycle. Specifically, benzene, naphthalene, phenanthrene, thiophene, furan, thiazole, isothiazole, oxazole, isoxazole, oxadiazole, pyrazole, pyrazole, imidazole, pyridine, pyrimidine, pyrazine, pyridazine, pyridone, pyrimidinone, indol, isoindole.
- Indazole quinoline, isoquinoline, benzimidazole, benzotriazole, benzothiophene, benzofuran, benzothiazole, phthalazine, quinoxalin, pyrolopyridine, or carbazole.
- aromatic ring group include a monovalent group formed by removing any one hydrogen atom from the aromatic ring. It may be a monocyclic to tricyclic aromatic ring group. For example, aryl or heteroaryl are exemplified.
- the "aryl” may be a monocyclic to tricyclic aromatic hydrocarbon ring group.
- Aryl also contains an aromatic hydrocarbon ring group condensed with a saturated hydrocarbon ring described later.
- 6-14 member aryl is exemplified.
- Another embodiment is exemplified by 6-10 member aryls.
- Yet another embodiment is exemplified by 6-membered aryl.
- phenyl, naphthyl, anthranil, phenanthrenyl, fluorenyl, indanyl, or 1,2,3,4-tetrahydronaphthalenyl are exemplified.
- Another embodiment is phenyl and yet another embodiment is naphthyl. Indanyl and 1,2,3,4-tetrahydronaphthalenyl are included in 6-10 member aryls.
- heteroaryl may be a monocyclic to tricyclic aromatic heterocyclic group containing 1 to 4 heteroatoms as ring-constituting atoms. Examples of the heteroatom include O, S, or N. 5-14 member heteroaryl is exemplified. As another embodiment, 5-10-membered heteroaryl is exemplified. Yet another embodiment is exemplified by 5-6 member heteroaryl.
- Benzoimidazolyl benzotriazolyl, benzothienyl, benzofuranyl, benzothiazolyl, phthalazinyl, quinoxalinyl, pyrrolopyridyl, or carbazolyl.
- saturated ring examples include a saturated hydrocarbon ring or a saturated heterocycle.
- the saturated ring may have a crosslink or may be condensed with the aromatic ring.
- the "saturated hydrocarbon ring” may be a monocyclic to tricyclic saturated hydrocarbon ring.
- a 3-10 member saturated hydrocarbon ring is exemplified.
- a 3-7-membered saturated hydrocarbon ring is exemplified.
- Yet another embodiment is exemplified by a 5- or 6-membered saturated hydrocarbon ring.
- the saturated hydrocarbon ring may have a crosslink or may be condensed with the aromatic ring. Specifically, cyclopropane, cyclobutane, cyclopentane, cyclohexane, or adamantane are exemplified.
- the "saturated heterocycle” may be a monocyclic to tricyclic saturated heterocycle containing 1 to 4 heteroatoms as ring-constituting atoms. Examples of the heteroatom include O, S, or N.
- a 3-10 member saturated heterocycle is exemplified.
- a 3-7 member saturated heterocycle is exemplified.
- Yet another embodiment is exemplified by a 5- or 6-membered saturated heterocycle.
- the saturated heterocycle may have a crosslink or may be condensed with the aromatic ring.
- tetrahydropyran tetrahydrofuran
- piperidine pyrrolidine
- azetidine oxetane
- aziridine oxylan
- tetrahydrothiopyran tetrahydrothiophene
- morpholine oxazepan, or piperazine.
- a “condensed ring” is a cyclic compound in which two or more rings share two or more atoms and are bonded to each other, and the two or more rings are independently 3-7-membered saturated rings. Is exemplified by the cyclic compound.
- the fused ring can have 1-3 heteroatoms selected from O, S, and N.
- a condensed ring a cyclic compound in which two rings share two adjacent atoms is exemplified.
- the "spiro ring” is exemplified by a cyclic compound containing one carbon atom common to the two rings, wherein the two rings are independently 3-7-membered saturated rings.
- the spiro ring can have 1-3 heteroatoms selected from O, S, and N. When the number of atoms constituting the spiro ring is 7 to 11, the spiro ring may be referred to as a 7-11-membered spiro ring. Examples of the spiro ring include a 7-13-membered spiro ring. As another embodiment, a 7-11-membered spiro ring is exemplified. Yet another embodiment is exemplified by a 7-9 member spiro ring.
- the "saturated ring group” is a monovalent group formed by removing any one hydrogen atom from the saturated ring, or a monovalent group formed by removing one hydrogen atom from each of two different ring-constituting atoms in the saturated ring.
- a divalent group can be mentioned.
- Saturated hydrocarbon ring groups or saturated heterocyclic groups are exemplified.
- a 3-10-membered saturated ring group is exemplified.
- a 3-7-membered saturated ring group is exemplified.
- Yet another embodiment is exemplified by a 5- or 6-membered saturated ring group.
- the "saturated hydrocarbon ring group” is a monovalent group formed by removing any one hydrogen atom from the saturated hydrocarbon ring, or one each from two different ring-constituting atoms in the saturated hydrocarbon ring.
- a divalent group formed by excluding a hydrogen atom can be mentioned. It may be a monocyclic to tricyclic saturated hydrocarbon ring group.
- the saturated hydrocarbon ring group may have a crosslink or may be condensed with the aromatic ring.
- a 3-10-membered saturated hydrocarbon ring group is exemplified.
- a 3-7-membered saturated hydrocarbon ring group is exemplified.
- Yet another embodiment is exemplified by a 5- or 6-membered saturated hydrocarbon ring group.
- the monovalent group examples include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and adamantyl.
- the divalent group specifically, among the specific examples of the above monovalent group, the divalent group obtained by further removing the hydrogen atom from the ring-constituting atom different from the ring-constituting atom excluding the hydrogen atom is used. Can be mentioned.
- the "saturated heterocyclic group” is a monovalent group formed by removing any one hydrogen atom from the saturated heterocycle, or one hydrogen atom from each of two different ring-constituting atoms in the saturated heterocycle.
- a divalent group that can be excluded can be mentioned. It may be a saturated heterocyclic group of monocyclic to tricyclic rings containing 1 to 4 heteroatoms as ring-constituting atoms.
- the saturated heterocyclic group may have a crosslink or may be condensed with the aromatic ring. Examples of the heteroatom include O, S, or N.
- a 3-10-membered heterocyclic group is exemplified. As another embodiment, a 3-7 member saturated heterocyclic group is exemplified.
- Yet another embodiment is exemplified by a 5- or 6-membered saturated heterocyclic group.
- the monovalent group include tetrahydropyranyl, tetrahydrofuranyl, piperidinyl, pyrrolidinyl, azetidinyl, oxetanyl, tetrahydrothiopyranyl, tetrahydrothienyl, morpholinyl, or piperazinyl.
- the "partially unsaturated ring group” may be any ring as long as a part of the saturated ring group is unsaturated, and is a partially unsaturated hydrocarbon ring group (partially unsaturated hydrocarbon ring group) or a partially unsaturated ring.
- Heterocyclic groups (partially unsaturated heterocyclic groups) are exemplified.
- a 3-10 member partially unsaturated ring group is exemplified.
- a 3-7 member partially unsaturated ring group is exemplified.
- Yet another embodiment is exemplified by a 5- or 6-membered partially unsaturated ring group.
- the “partially unsaturated hydrocarbon ring group” may be any group as long as a part of the saturated hydrocarbon ring group is unsaturated. Specific examples thereof include cyclopentenyl, cyclopentadienyl, cyclohexenyl, cyclohexadienyl, and bicyclooctatrienyl.
- the "partially unsaturated heterocyclic group” may be any group as long as a part of the saturated heterocyclic group is unsaturated. Specific examples thereof include dihydropyranyl, dihydrofuranyl, dihydrothiopyranyl, dihydrothienyl, 1,2-dihydroquinolyl, or 1,2,3,4-tetrahydroquinolyl.
- isomers include all of them unless otherwise specified.
- alkyl, alkenyl, alkynyl, alkoxy, alkylthio, alkylene, alkenylene, and alkynylene include linear and branched chains.
- isomers based on double bonds, rings, or fused rings (E or Z isomers, or cis or trans isomers), isomers based on the presence of asymmetric carbons (R- or S-isomers, ⁇ ).
- R 1 examples are ⁇ H, —F, —Cl, methyl, or C1-3 alkoxy.
- —H, or C 1-3 alkoxy is exemplified.
- Yet another embodiment is exemplified by —H, or methoxy.
- the R 1 may be substituted with 1 to 3 identical or different substituents selected from the G1 group.
- the G1 group include a group consisting of -F, hydroxy, cyano, halogeno C 1-6 alkyl, C 1-4 alkoxy, phenyl, 5-6 member heteroaryl, and 3-7 member saturated ring group.
- the G11 group consisting of —F, hydroxy, halogeno C 1-6 alkyl, C 1-4 alkoxy, 5-6 member heteroaryl, and 3-7 member saturated ring groups is exemplified.
- the G12 group consisting of —F, hydroxy, C 1-4 alkoxy, 5-membered heteroaryl, and 4-5 member saturated ring groups.
- the phenyl or 5-6 member heteroaryl in the G1 group may be substituted with 1 to 3 identical or different substituents selected from the GAr group.
- Examples of the G Ar group include a group consisting of -F, -Cl, hydroxy, cyano, C 1-6 alkyl, halogeno C 1-6 alkyl, and -NH 2 .
- the G Ar1 group consisting of —F, —Cl, cyano, C 1-6 alkyl, and halogeno C 1-6 alkyl is exemplified.
- R2 are C 1-6 alkyl, halogeno C 1-6 alkyl, and 3-7 member saturated ring groups.
- a 3-7-membered saturated ring group is exemplified.
- the R 2 may be substituted with 1 to 3 identical or different substituents selected from the G 2 group.
- the G2 group include a group consisting of -F, hydroxy, halogeno C 1-3 alkyl, and C 1-4 alkoxy.
- the G21 group consisting of ⁇ F and hydroxy is exemplified.
- R2 the following is exemplified as the R2 . Further, as another aspect of R2 , specifically, the following is exemplified.
- R2 As still another aspect of R2 , specifically, the following is exemplified. Further, as still another aspect of R2 , specifically, the following is exemplified.
- an integer of 0 or 1 is exemplified.
- an integer of 1 is exemplified.
- R Cy1 and R Cy2 respectively, -H, -F, hydroxy, cyano, C 1-6 alkyl, halogeno C 1-6 alkyl, C 1-6 alkoxy, -NR 11 R 12 , phenyl, 5-6-membered heteroaryl or 3-7-membered saturated ring groups are exemplified.
- —H, —F, C 1-6 alkyl, C 1-6 alkoxy, —NR 11 R 12 are exemplified.
- -NR 11 R 12 is exemplified.
- the phenyl or 5-6-membered heteroaryl in R Cy1 and R Cy2 may be substituted with 1 to 3 identical or different substituents selected from the G Ar group.
- the G Ar group in addition to the G Ar group, an embodiment of the G Ar 1 group is exemplified.
- the R Cy1 and R Cy2 may be substituted with 1 to 3 identical or different substituents selected from the G1 group.
- Examples of the G1 group include aspects of the G11 to G12 groups in addition to the G1 group.
- the R 11 and R 12 are independently -H, C 1-6 alkyl, halogeno C 1-6 alkyl, hydroxy C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, 3-.
- a 7-membered saturated ring group, phenyl, or a 5-6-membered heteroaryl is exemplified.
- —H, C 1-6 alkyl, halogeno C 1-6 alkyl, 3-7 member saturated ring group are exemplified.
- Examples of the 3-7-membered saturated ring group include cyclopropane, cyclobutane, cyclopentane, oxetane, and bicyclo [1.1.1] pentane.
- the R 11 and R 12 may be substituted with 1 to 3 identical or different substituents selected from the G1 group.
- Examples of the G1 group include aspects of the G11 to G12 groups in addition to the G1 group.
- the R 11 and R 12 can be combined to form a 4-10-membered saturated ring or a 7-11-membered spiro ring.
- the 4-10-membered saturated ring and the 7-11-membered spiro ring contain 1 to 2 identical or different heteroatoms or -S (O 2 )- selected from the group consisting of O and N. You may have.
- Examples of the 4-10-membered saturated ring include azetidine, pyrrolidine, piperidine, morpholine, oxazepan, piperazine, or homopiperazine.
- piperidine, morpholine, or piperazine is exemplified.
- Yet another embodiment is exemplified by morpholine or piperazine.
- the 4-10-membered saturated ring or 7-11-membered spiro ring formed by combining R 11 and R 12 is substituted with 1 to 3 identical or different substituents selected from the G3 group. May be.
- the G3 group include -F , hydroxy, C 1-6 alkyl, halogeno C 1-6 alkyl, hydroxy C 1-6 alkyl, C 1-6 alkoxy, and C 1-6 alkoxy-C 1-6 alkyl.
- —F, C 1-6 alkyl, halogeno C 1-6 alkyl, —C (O) R 14 , —C (O) NR 13 R 14 are exemplified.
- the phenyl or 5-6 member heteroaryl in the G3 group may be substituted with 1 to 3 identical or different substituents selected from the GAr group.
- G Ar group in addition to the G Ar group, an embodiment of the G Ar 1 group is exemplified.
- the R 13 includes —H, C 1-6 alkyl, halogeno C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, halogeno C 1-6 alkoxy C 1-6 alkyl, phenyl, 5- 6-membered heteroaryl or 3-7-membered saturated ring groups are exemplified.
- —H, C 1-6 alkyl, and halogeno C 1-6 alkyl are exemplified.
- —H is exemplified.
- R 14 examples include C 1-6 alkyl, halogeno C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, halogeno C 1-6 alkoxy C 1-6 alkyl, phenyl, and 5-6 member heteroaryl. , Or a 3-7 member saturated ring group is exemplified. As another embodiment, C 1-6 alkoxy C 1-6 alkyl, 5-6 member heteroaryl, or 3-7 member saturated ring group is exemplified. Yet another embodiment is exemplified by 5-6 member heteroaryl. Examples of the 5-6 member heteroaryl include pyridine, oxazole, isoxazole, and thiazole. As another embodiment, pyridine is exemplified. The phenyl or 5-6 member heteroaryl in the R 13 and R 14 may be substituted with 1 to 3 identical or different substituents selected from the G Ar group.
- the G Ar group in addition to the G Ar group, an embodiment of the G Ar 1 group is exemplified.
- the R 13 and R 14 can be combined to form a 4-7-membered saturated ring or a 7-11-membered spiro ring.
- the 4-7-membered saturated ring and the 7-11-membered spiro ring contain 1 to 2 identical or different heteroatoms or -S (O 2 )- selected from the group consisting of O and N. You may have.
- Examples of the 4-7-membered saturated ring include azetidine, pyrrolidine, piperidine, morpholine, oxazepan, and piperazine.
- azetidine and pyrrolidine are exemplified.
- Yet another embodiment is exemplified by azetidine.
- R 11 and R 12 together form a 4-10-membered saturated ring, and the 4-10-membered saturated ring is morpholine or piperazine.
- X O or NR 15 is exemplified.
- NR 15 is exemplified.
- Examples of RCy3 include C 1-4 alkyl and halogeno C 1-4 alkyl.
- methyl, ethyl, isopropyl are exemplified.
- Yet another embodiment is exemplified by methyl.
- the R15 includes —H, C 1-6 alkyl, halogeno C 1-6 alkyl, hydroxy C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, —C (O) R 16 , —S. (O 2 ) R 16 , -C (O) NR 16 R 17 , -C (O) OR 16 , or 3-7-membered saturated ring groups are exemplified.
- halogeno C 1-6 alkyl, —C (O) R 16 , —S (O 2 ) R 16 or —C (O) NR 16 R 17 are exemplified.
- —C (O) R 16 is exemplified.
- R 16 examples include C 1-6 alkyl, halogeno C 1-6 alkyl, C 1-6 alkoxy C 1-6 alkyl, halogeno C 1-6 alkoxy C 1-6 alkyl, phenyl, and 5-6 member heteroaryl. , Or a 3-7 member saturated ring group is exemplified. As another embodiment, C 1-6 alkoxy C 1-6 alkyl, 5-6 member heteroaryl, or 3-7 member saturated ring group is exemplified. Yet another embodiment is exemplified by 5-6 member heteroaryl. Examples of the 5-6 member heteroaryl include pyridine, oxazole, isoxazole, and thiazole. As another embodiment, pyridine is exemplified.
- R 17 examples include -H, C 1-3 alkyl, halogeno C 1-3 alkyl, C 1-6 alkoxy C 1-6 alkyl, halogeno C 1-6 alkoxy C 1-3 alkyl, phenyl, 5-6.
- a member heteroaryl or a 3-7 member saturated ring group is exemplified.
- —H, C 1-6 alkyl, and halogeno C 1-6 alkyl are exemplified.
- —H is exemplified.
- the phenyl or 5-6-membered heteroaryl in R 16 and R 17 may be substituted with 1 to 3 identical or different substituents selected from the G Ar group.
- the G Ar group in addition to the G Ar group, an embodiment of the G Ar 1 group is exemplified.
- the R 16 and R 17 can be combined to form a 4-7-membered saturated ring or a 7-11-membered spiro ring.
- the 4-7-membered saturated ring and the 7-11-membered spiro ring contain 1 to 2 identical or different heteroatoms or -S (O 2 )- selected from the group consisting of O and N. You may have.
- Examples of the 4-7-membered saturated ring include azetidine, pyrrolidine, piperidine, morpholine, oxazepan, and piperazine.
- azetidine and pyrrolidine are exemplified.
- azetidine is exemplified by azetidine.
- the R 15 may be substituted with 1 to 3 identical or different substituents selected from the G1 group.
- Examples of the G1 group include aspects of the G11 to G12 groups in addition to the G1 group.
- R 15 include the following.
- the RCy1 and RCy2 can also be combined to form a 4-7-membered saturated ring or a 7-11-membered spiro ring.
- the 4-7-membered saturated ring and the 7-11-membered spiro ring contain 1 to 2 identical or different heteroatoms or -S (O 2 )- selected from the group consisting of O and N. You may have.
- Examples of the 4-7-membered saturated ring formed by R Cy1 and R Cy2 together include azetidine, pyrrolidine, and piperidine. As another embodiment, piperidine is exemplified.
- the 4-7-membered saturated ring or 7-11-membered spiro ring formed by combining R Cy1 and R Cy2 is substituted with 1 to 3 identical or different substituents selected from the G1 group. May be.
- Examples of the G1 group include aspects of the G11 to G12 groups in addition to the G1 group.
- the "compound represented by the formula (1)” is generally understood as a free compound represented by the formula (1).
- the following salt can be mentioned as the salt. That is, the type of the salt of the compound represented by the formula (1) is not particularly limited, and may be either an acid addition salt or a base addition salt, or may be in the form of an intramolecular counterion. ..
- a pharmaceutically acceptable salt is preferable as the salt.
- the salt of the compound represented by the formula (1) is a pharmaceutically acceptable salt.
- the acid addition salt examples include acid addition salts with inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, and phosphoric acid, or formic acid, acetic acid, propionic acid, oxalic acid, and malonic acid. , Succinic acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, citric acid, malic acid, tartaric acid, dibenzoyl tartrate, mandelic acid, maleic acid, fumaric acid, aspartic acid, or acid addition salt with organic acids such as glutamate. Is included.
- the base addition salt examples include a base addition salt with an inorganic base such as sodium, potassium, magnesium, calcium and aluminum, and a base addition salt with an organic base such as methylamine, 2-aminoethanol, arginine, lysine or ornithine. Etc. can be exemplified. However, it is needless to say that the type of salt is not limited to these and can be appropriately selected by those skilled in the art.
- the compounds of the present invention include the form of hydrates.
- the compounds of the present invention also include the form of anhydrate.
- the compounds of the present invention include the form of solvates.
- the compound of the present invention also includes the form of a solvate.
- the compounds of the present invention include crystalline morphology.
- the compound of the present invention also includes an amorphous form.
- the compounds of the invention also include forms labeled with various radioactive or non-radioactive isotopes. More specifically, the compound of the present invention contains an anhydride and solvate of "the compound represented by the formula (1)", or a hydrate and / or a solvate thereof, or crystals thereof. including. Further, the compound of the present invention contains an anhydride and a solvate of "a salt of a compound represented by the formula (1)", or a hydrate and / or a solvate of the salt thereof, or further comprises crystals thereof. include.
- the compound of the present invention may also contain a pharmaceutically acceptable prodrug of "the compound represented by the formula (1)".
- a pharmaceutically acceptable prodrug is a compound having a group that can be converted into an amino group, a hydroxyl group, a carboxyl group, or the like by solvolysis or under physiological conditions.
- examples of the group forming a prodrug for a hydroxyl group and an amino group include an acyl group and an alkoxycarbonyl group.
- Examples of the group forming a prodrug for the carboxyl group include a methyl group, an ethyl group, an n-propyl group, an isopropyl group, an n-butyl group, an isobutyl group, an s-butyl group, a t-butyl group and an amino group. Examples thereof include a methylamino group, an ethylamino group, a dimethylamino group, or a diethylamino group.
- a group that appropriately forms a prodrug according to a conventional method into any one or more arbitrary groups selected from the hydroxyl group and the amino group in the compound of the present invention, for example, using a prodrug-forming reagent such as a corresponding halide. If desired, it can be produced by isolation and purification according to a conventional method as appropriate. Further, it is also possible to introduce a group that appropriately forms a prodrug according to a conventional method by using a prodrug-forming reagent such as an alcohol or an amine corresponding to the carboxyl group in the compound of the present invention.
- a prodrug-forming reagent such as an alcohol or an amine corresponding to the carboxyl group in the compound of the present invention.
- the compound represented by the formula (1) can be produced according to a known method, for example, a method shown below, a method according to these, or a method shown in Examples. It should be noted that the compound as a raw material in each of the following production methods is commercially available, or a known method described in, for example, "Compendium of Organic Synthesis Methods, Vol. I-XII (Wiley-Interscience)". Can be manufactured using.
- the reaction for compound synthesis of the present invention is carried out in a suitable solvent selected according to a known method. Suitable solvents do not react substantially with the starting material, intermediates, or products at the temperature at which the reaction takes place (eg, temperatures ranging from the melting point to the boiling point of the solvent).
- the reaction can be carried out in a single solvent or a mixed solvent. A solvent suitable for each reaction is used.
- the reaction can be followed by an appropriate method according to known methods.
- the product is a spectroscopic method, such as a nuclear magnetic resonance apparatus (NMR) such as 1H or 13C , an infrared spectrophotometer (IR), a mass spectrometer (MS), high performance liquid chromatography ( It can be tracked using HPLC), thin layer chromatography (TLC), and the like.
- NMR nuclear magnetic resonance apparatus
- IR infrared spectrophotometer
- MS mass spectrometer
- HPLC high performance liquid chromatography
- TLC thin layer chromatography
- the compound represented by the formula (1) and its intermediates can be produced by the synthetic method described below. Unless otherwise specified, the following reaction formulas and R 1 , R 2 , R 11 , R 12 , R 15 , R cy3 , and Cy in the description have the same meanings as described above.
- the compound of the present invention may be produced by a method other than the method described in the present specification by appropriately utilizing the method described in the present specification and the common general technical knowledge in the art.
- the reaction formulas and examples are intended for illustration purposes and do not limit the scope of the present invention.
- the compound of the present invention represented by the formula (1) can be produced, for example, according to the following reaction scheme.
- “STEP” means a process, and for example, “STEP 1" means a process 1.
- the compound represented by the formula (1) can form, for example, reaction scheme 1 (in the formula of each compound, L can form a leaving group such as -Cl, -Br, a pentafluorophenyl group, or an amide bond such as a hydroxyl group. Representing a substituent, hallo indicates -Cl, -Br, or -I, and M indicates a substituent that can be reacted by various couplings such as ZnI, MgBr, boronic acid, and boronic acid ester. ), It can be manufactured by the method described in.
- the compounds represented by the formulas (4)-(9) are commercially available or can be produced according to a known method, for example, the method shown below, or a method similar thereto.
- the compound represented by the formula (1) can be produced by an amidation reaction, an acylation reaction, or the like between the compound represented by the formula (4) and the compound represented by the formula (5).
- amidation condensing agent 1-propanephosphonic acid anhydride, 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide, HATU and the like can be used.
- nucleophile for amidation HOBt, HOAt and the like can be used.
- the base diisopropylethylamine or the like can be used.
- the reaction solvent for example, DMF, dichloromethane, THF and the like can be used.
- the reaction temperature can usually be 0 ° C to 150 ° C.
- Substituents Cy, R 1 , R 2 are represented by the compound represented by the formula (1) by further conversion, for example, deprotection, reduction, reductive amination, alkylation, fluorination and the like. Can be converted.
- the compound represented by the formula (5) can be produced by a coupling reaction with the compound represented by the formula (7) using a metal catalyst. More specifically, it can be produced by Suzuki-Miyaura coupling or the like between the compound represented by the formula (7) and the reagent represented by the formula (6).
- the reaction catalyst for example, Pd (dpppf) Cl 2 , PdAmphos, Pd (PPh 3 ) 4 , or the like can be used.
- the base cesium carbonate, cesium fluoride, sodium carbonate and the like can be used.
- THF, 1,4-dioxane, DMF, acetonitrile and the like can be used.
- the reaction temperature can usually be from room temperature to 180 ° C.
- the compound represented by the formula (5) can be produced by a coupling reaction with the compound represented by the formula (9) using a metal catalyst. More specifically, it can be produced by Suzuki-Miyaura coupling or the like between the compound represented by the formula (9) and the reagent represented by the formula (8).
- the reaction catalyst for example, Pd (dpppf) Cl 2 , PdAmphos, Pd (PPh 3 ) 4 , or the like can be used.
- the base cesium carbonate, cesium fluoride, sodium carbonate and the like can be used.
- THF, 1,4-dioxane, DMF, acetonitrile and the like can be used.
- the reaction temperature can usually be from room temperature to 180 ° C.
- the compound represented by the formula (9) can be produced, for example, by a coupling reaction using a metal catalyst with the compound represented by the formula (7) or a halogen-metal exchange reaction. More specifically, it can be produced by Suzuki-Miyaura coupling or the like between the compound represented by the formula (5) and bis (pinacolado) diboron or the like.
- the reaction catalyst for example, Pd (dpppf) Cl 2 or the like can be used.
- As the base potassium acetate or the like can be used.
- As the reaction solvent 1,4-dioxane or the like can be used.
- the reaction temperature can usually be 40 ° C to 150 ° C.
- the compound represented by the formula (1) can form, for example, reaction scheme 2 (in the formula of each compound, L can form a leaving group such as -Cl, -Br, pentafluorophenyl group, or an amide bond such as a hydroxyl group. Representing a substituent, hallo indicates -Cl, -Br, or -I, and M indicates a substituent that can be reacted by various couplings such as ZnI, MgBr, boronic acid, and boronic acid ester. ), It can be manufactured by the method described in.
- the compounds represented by the formulas (4), (6)-(8), (10), (11) are commercially available or according to a known method, for example, the method shown below, or a method similar thereto. Can be manufactured.
- the compound represented by the formula (1) can be produced by a coupling reaction with the compound represented by the formula (10) using a metal catalyst. More specifically, it can be produced by Suzuki-Miyaura coupling or the like between the compound represented by the formula (10) and the reagent represented by the formula (6).
- the reaction catalyst for example, Pd (dpppf) Cl 2 , PdAmphos, Pd (PPh 3 ) 4 , or the like can be used.
- the base cesium carbonate, cesium fluoride, sodium carbonate and the like can be used.
- THF, 1,4-dioxane, DMF, acetonitrile and the like can be used.
- the reaction temperature can usually be from room temperature to 180 ° C.
- Substituents Cy, R 1 , R 2 are represented by the compound represented by the formula (1) by further conversion, for example, deprotection, reduction, reductive amination, alkylation, fluorination and the like. Can be converted.
- the compound represented by the formula (10) can be produced by an amidation reaction, an acylation reaction, or the like between the compound represented by the formula (4) and the compound represented by the formula (7).
- amidation condensing agent 1-propanephosphonic acid anhydride, 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide, HATU and the like can be used.
- nucleophile for amidation HOBt, HOAt and the like can be used.
- the base diisopropylethylamine or the like can be used.
- the reaction solvent for example, DMF, dichloromethane, THF and the like can be used.
- the reaction temperature can usually be 0 ° C to 150 ° C.
- the compound represented by the formula (1) can be produced by a coupling reaction with the compound represented by the formula (11) using a metal catalyst. More specifically, it can be produced by Suzuki-Miyaura coupling or the like between the compound represented by the formula (11) and the reagent represented by the formula (8).
- the reaction catalyst for example, Pd (dpppf) Cl 2 , PdAmphos, Pd (PPh 3 ) 4 , or the like can be used.
- the base cesium carbonate, cesium fluoride, sodium carbonate and the like can be used.
- THF, 1,4-dioxane, DMF, acetonitrile and the like can be used.
- the reaction temperature can usually be from room temperature to 180 ° C.
- the compound represented by the formula (9) can be produced, for example, by a coupling reaction using a metal catalyst with the compound represented by the formula (10) or a halogen-metal exchange reaction. More specifically, it can be produced by Suzuki-Miyaura coupling or the like between the compound represented by the formula (10) and bis (pinacolad) diboron or the like.
- the reaction catalyst for example, Pd (dpppf) Cl 2 or the like can be used.
- As the base potassium acetate or the like can be used.
- As the reaction solvent 1,4-dioxane or the like can be used.
- the reaction temperature can usually be 40 ° C to 150 ° C.
- the compound (12) in which Cy is represented by the formula (2-1-1) is, for example, reaction scheme 3 (in the formula of each compound, L is ⁇ Cl, ⁇ Br, It can be produced by the method described in), which represents a leaving group such as a pentafluorophenyl group or a substituent capable of forming an amide bond such as a hydroxyl group.
- the compounds represented by the formulas (5), (13) and (14) are commercially available or can be produced according to a known method, for example, the method shown below, or a method similar thereto.
- the compound represented by the formula (10) can be produced by a reductive amination reaction of the compound represented by the formula (14).
- it can be produced by forming an imine in the presence of R 11 R 12 NH followed by a reduction reaction of the imine with sodium triacetoxyborohydride. THF and dichloromethane can be used as the reaction solvent.
- the reaction temperature can usually be 0 ° C to 80 ° C.
- Substituents R 11 R 12 , R 1 , R 2 transform the compound represented by formula (12) by further conversion, such as deprotection, reduction, alkylation, fluorination, etc. can do.
- the compound represented by the formula (14) can be produced by an amidation reaction, an acylation reaction, or the like between the compound represented by the formula (5) and the compound represented by the formula (15).
- amidation condensing agent 1-propanephosphonic acid anhydride, 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide, HATU and the like can be used.
- nucleophile for amidation HOBt, HOAt and the like can be used.
- the base diisopropylethylamine or the like can be used.
- the reaction solvent for example, DMF, dichloromethane, THF and the like can be used.
- the reaction temperature can usually be 0 ° C to 150 ° C.
- the compound (16) in which Cy is represented by the formula (1-1-2) and X in the formula is represented by NR 15 is, for example, reaction scheme 4 (Y CN in the formula of each compound).
- Y CN in the formula of each compound.
- the compounds represented by the formulas (14), (17)-(20) are commercially available or can be produced according to a known method, for example, the method shown below, or a method similar thereto.
- the compound represented by the formula (16) can be produced by a CN bond forming reaction such as amidation, acylation, alkylation and urea formation with the compound represented by the formula (18). More specifically, it can be produced by an amidation reaction between the compound represented by the formula (18) and the reagent represented by the formula (17).
- the amidation condensing agent 1-propanephosphonic acid anhydride, 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide, HATU and the like can be used.
- As the nucleophile for amidation HOBt, HOAt and the like can be used.
- As the base diisopropylethylamine or the like can be used.
- reaction solvent for example, DMF, dichloromethane, THF and the like can be used.
- the reaction temperature can usually be 0 ° C to 150 ° C.
- Substituents R 15 , R 1 , R 2 are represented by the formula (16) by performing further conversions such as deprotection, reduction, reductive amination, alkylation, fluorination and the like. Compounds can be converted.
- the compound represented by the formula (18) can be produced by deprotecting the compound represented by the formula (19).
- the deprotection reaction may be carried out according to a known method, for example, the method described in Greene's Protective Groups in Organic Synthesis, John Wiley and Sons (2014 edition), and the like.
- the compound represented by the formula (19) can be produced by a reductive amination reaction of the compound represented by the formula (14).
- it can be produced by forming an imine in the presence of the compound represented by the formula (20), followed by a reduction reaction of the imine with sodium triacetoxyborohydride.
- THF and dichloromethane can be used as the reaction solvent.
- the reaction temperature can usually be 0 ° C to 80 ° C.
- Rg is a general formula (1-1)
- hallo is a bromine
- R 1 is —OR al (R al is a substituted or unsubstituted C1-3 alkyl).
- the compound (21) shown is, for example, reaction scheme 5 (in the formula of each compound, YCO is a leaving group such as -Cl, -Br, -I, OTf, OMs , or OTs, or a C such as a hydroxyl group.
- -O represents a substituent capable of forming an O bond
- R ac represents an acyl group such as an acetyl group or a cyclopropyl group
- L represents a leaving of -Cl, -Br, a pentafluorophenyl group or the like.
- PG indicates a protective group published in Greene's Protective Groups in Organic Synthesis, John Wiley and Sons (2014 edition). ), It can be manufactured by the method described in.
- the compounds represented by the formulas (22)-(30) are commercially available or can be produced according to known methods, for example, the methods shown below, or methods similar thereto.
- the compound represented by the formula (21) can be produced by deprotecting the compound represented by the formula (19).
- the deprotection reaction may be carried out according to a known method, for example, the method described in Greene's Protective Groups in Organic Synthesis, John Wiley and Sons (2014 edition), and the like.
- the substituent of the substituent R al can convert the compound represented by the formula (21) by further conversion, for example, deprotection, reduction, reductive amination, alkylation, fluorination and the like. can.
- the compound represented by the formula (22) can be produced by a CO bond forming reaction such as an alkylation or a Mitsunobu reaction between the compound represented by the formula (24) and the compound represented by the formula (23). can. More specifically, it can be produced by an alkylation reaction between the compound represented by the formula (24) and the reagent represented by the formula (23).
- the alkylating agent iodomethane or the like can be used.
- the base diisopropylethylamine, potassium carbonate and the like can be used.
- the reaction solvent for example, DMF, dichloromethane, THF and the like can be used.
- the reaction temperature can usually be ⁇ 78 ° C. to 150 ° C.
- the compound represented by the formula (24) can be produced by protecting the compound represented by the formula (25).
- the protecting reaction may be carried out according to a known method, for example, the method described in Greene's Protective Groups in Organic Synthesis, John Wiley and Sons (2014 edition), and the like.
- the compound represented by the formula (25) can be produced by deacylated the compound represented by the formula (26). More specifically, it can be produced by hydrolyzing the compound represented by the formula (26) in the presence of an acid.
- an acid hydrochloric acid or the like can be used.
- the reaction solvent methanol, water, THF and the like can be used.
- the reaction temperature can usually be 100 ° C. from room temperature.
- the compound represented by the formula (26) can be produced by aromatizing the compound represented by the formula (27) in the presence of a base.
- a base DBU or the like can be used.
- the reaction solvent THF or the like can be used.
- the reaction temperature can usually be ⁇ 20 ° C. to 80 ° C.
- the compound represented by the formula (27) can be produced by brominating the compound represented by the formula (28) in the presence of a base.
- a base As the brominating agent, bromine, NBS and the like can be used.
- the acid hydrobromic acid or the like can be used.
- the reaction solvent acetic acid or the like can be used.
- the reaction temperature can usually be from room temperature to 60 ° C.
- the compound represented by the formula (28) can be produced by an amidation reaction, an acylation reaction, or the like between the compound represented by the formula (30) and the compound represented by the formula (29).
- an acid anhydride such as acetic anhydride can be used.
- an amidation condensing agent 1-propanephosphonic acid anhydride, 1- (3-dimethylaminopropyl) -3-ethylcarbodiimide, HATU and the like can be used.
- the nucleophile for amidation HOBt, HOAt and the like can be used.
- As the base diisopropylethylamine or the like can be used.
- the reaction solvent for example, DMF, dichloromethane, THF and the like can be used.
- the reaction temperature can usually be 0 ° C to 150 ° C.
- the compound represented by the general formula (1-1) in which Rg is represented by the general formula (1-1) and —OR al is, for example, reaction scheme 6 (in the formula of each compound, YCO has a leaving group such as -Cl, -Br, -I, OTf, OMs, or OTs, or a CO bond such as a hydroxyl group. It represents a formable substituent.) It can be produced by the method described in.
- the compound represented by the formula (32) is commercially available or can be produced according to a known method, for example, the method shown below, or a method similar thereto.
- the compound represented by the formula (31) can be produced by a CO bond forming reaction such as an alkylation or a Mitsunobu reaction between the compound represented by the formula (32) and the compound represented by the formula (23). can. More specifically, it can be produced by a Mitsunobu reaction between the compound represented by the formula (32) and the reagent represented by the hydroxyl group of YCO in the formula (23).
- a Mitsunobu reaction between the compound represented by the formula (32) and the reagent represented by the hydroxyl group of YCO in the formula (23).
- the reagent for the Mitsunobu reaction diethyl azocarboxylate, triphenylphosphine and the like can be used.
- the reaction solvent THF, toluene and the like can be used.
- the reaction temperature can usually be 0 ° C to 110 ° C.
- the compound represented by the general formula (1-1) in which Rg is represented by the general formula (1-1) and —OR al is, for example, reaction scheme 7 (in the formula of each compound, YCO has a leaving group such as -Cl, -Br, -I, OTf, OMs, or OTs, or a CO bond such as a hydroxyl group. Representing a formable substituent.
- PG is produced by the method described in Greene's Protective Groups in Organic Synthesis, John Wiley and Sons (2014 edition). Can be done.
- the compounds represented by the formula (23), the formula (34), and the formula (35) are commercially available or can be produced according to a known method, for example, the method shown below, or a method similar thereto.
- the compound represented by the formula (33) can be produced by deprotecting the compound represented by the formula (34).
- the deprotection reaction may be carried out according to a known method, for example, the method described in Greene's Protective Groups in Organic Synthesis, John Wiley and Sons (2014 edition), and the like.
- the substituent of the substituent R al can convert the compound represented by the formula (33) by further conversion, for example, deprotection, reduction, reductive amination, alkylation, fluorination and the like. can.
- the compound represented by the formula (34) can be produced by a CO bond forming reaction such as an alkylation or a Mitsunobu reaction between the compound represented by the formula (35) and the compound represented by the formula (23). can. More specifically, it can be produced by an alkylation reaction between the compound represented by the formula (35) and the reagent represented by the halogen group in which YCO is -Br, -I, etc. in the formula (23). ..
- As the base diisopropylethylamine, cesium carbonate and the like can be used.
- the reaction solvent for example, DMF, dichloromethane, THF and the like can be used.
- the reaction temperature can usually be 0 ° C to 150 ° C.
- the compound (36) in which Rg is represented by the general formula (1-2) can be produced, for example, by the method described in the reaction scheme 8.
- the compounds represented by the formulas (37)-(40) are commercially available or can be produced according to known methods, for example, the methods shown below, or methods similar thereto.
- the compound represented by the formula (36) can be produced by a cyclization reaction of the compound represented by the formula (37).
- As the base potassium carbonate, DBU and the like can be used.
- As the reaction solvent DMF or the like can be used.
- the reaction temperature can usually be 0 ° C to 180 ° C.
- the substituent of the substituent R 1 can convert the compound represented by the formula (36) by further conversion, for example, deprotection, reduction, reductive amination, alkylation, fluorination and the like. can.
- the compound represented by the formula (37) can be produced by reacting the compound represented by the formula (38) with O- (mesitylene sulfonyl) hydroxylamine. Chloroform and dichloromethane can be used as the reaction solvent. The reaction temperature can usually be 0 ° C to 50 ° C.
- the compound represented by the formula (38) can be produced by cyanating the compound represented by the formula (39).
- the cyanating agent sodium cyanide, potassium cyanide and the like can be used.
- the reaction solvent ethanol, water, DMF and the like can be used.
- the reaction temperature can usually be 0 ° C to 120 ° C.
- the compound represented by the formula (39) can be produced by brominating the compound represented by the formula (40).
- the brominating agent N-bromosuccinimide, bromine and the like can be used.
- the reaction solvent ethanol, water, DMF and the like can be used.
- the reaction temperature can usually be 0 ° C to 120 ° C.
- the compound (41) in which M is Z (Z represents a borane derivative such as boronic acid B (OH) 2 or a boronic acid ester) is, for example. It can be produced by the method described in Reaction Scheme 8 (in the formula of each compound, LG 1 indicates a desorbing group such as, for example, -Cl, -Br, -I, OTf, OMs, or OTs). ..
- the compounds represented by the formulas (42)-(46) are commercially available or can be produced according to known methods, for example, the methods shown below, or methods similar thereto.
- the compound represented by the formula (41) can be produced by CH borylizing the compound represented by the formula (42) using an iridium catalyst.
- the borane source for example, bis (pinacolato) diborone can be used.
- THF can be used as the reaction solvent.
- the reaction temperature can usually be 20 ° C to 80 ° C.
- the compound represented by the formula (42) can be produced by etherifying the compound represented by the formula (44) and the compound represented by the formula (43) under basic conditions.
- the base for example, sodium hydride or sodium hydroxide can be used.
- the catalyst for example, tetrabutylammonium chloride can be used.
- the reaction solvent THF, DMF, or dichloromethane can be used.
- the reaction temperature can usually be 0 ° C to 150 ° C.
- the compound represented by the formula (42) can be produced by etherifying the compound represented by the formula (46) and the compound represented by the formula (45) under basic conditions.
- the base for example, sodium hydride or sodium hydroxide can be used.
- the catalyst for example, tetrabutylammonium chloride can be used.
- the reaction solvent THF, DMF, or dichloromethane can be used.
- the reaction temperature can usually be 0 ° C to 150 ° C.
- the compound represented by the formula (46) can be produced by subjecting the compound represented by the formula (44) to halogenation including chlorolation.
- the chlorolytic agent for example, thionyl chloride can be used.
- Dichloromethane can be used as the reaction solvent.
- the reaction temperature can usually be 0 ° C to 40 ° C.
- the compound represented by the formula (6) represents, for example, a reaction scheme 10 (in the formula of each compound, LG 1 represents a leaving group such as, for example, -Cl, -Br, -I, OTf, OMs, or OTs. Further, hallo indicates -Cl, -Br, or -I, and M indicates a substituent that can be reacted by various couplings such as ZnI, MgBr, boronic acid, and boronic acid ester).
- Can be manufactured by The compounds represented by the formulas (8), (44), (45), (47), and (48) are commercially available or according to known methods, for example, the methods shown below, and methods similar thereto. Can be manufactured.
- the compound represented by the formula (6) can be produced, for example, by a coupling reaction using a metal catalyst with the compound represented by the formula (8) or a halogen-metal exchange reaction. More specifically, it can be produced by Suzuki-Miyaura coupling or the like between the compound represented by the formula (6) and bis (pinacolado) diboron or the like.
- the reaction catalyst for example, Pd (dpppf) Cl 2 or the like can be used.
- As the base potassium acetate or the like can be used.
- As the reaction solvent 1,4-dioxane or the like can be used.
- the reaction temperature can usually be 40 ° C to 150 ° C.
- the compound represented by the formula (8) can be produced by etherifying the compound represented by the formula (47) and the compound represented by the formula (44) under basic conditions.
- the base for example, sodium hydride or sodium hydroxide can be used.
- the catalyst for example, tetrabutylammonium chloride can be used.
- the reaction solvent THF, DMF, or dichloromethane can be used.
- the reaction temperature can usually be 0 ° C to 150 ° C.
- the compound represented by the formula (8) can be produced by etherifying the compound represented by the formula (48) and the compound represented by the formula (45) under basic conditions.
- the base for example, sodium hydride or sodium hydroxide can be used.
- the catalyst for example, tetrabutylammonium chloride can be used.
- the reaction solvent THF, DMF, or dichloromethane can be used.
- the reaction temperature can usually be 0 ° C to 150 ° C.
- the compound represented by the formula (48) can be produced by subjecting the compound represented by the formula (47) to halogenation including chlorolation.
- the chlorolytic agent for example, thionyl chloride can be used.
- Dichloromethane can be used as the reaction solvent.
- the reaction temperature can usually be 0 ° C to 40 ° C.
- the method for producing the compound of the present invention is not limited to the method described here.
- the compound of the present invention can be produced by modifying or converting a substituent of a compound that is a precursor thereof by combining one or a plurality of reactions described in ordinary chemical literature and the like.
- examples of the method for producing a compound containing asymmetric carbon include a production method by asymmetric reduction, and a commercially available (or known method or known method) in which the portion corresponding to the asymmetric carbon is optically active in advance.
- examples thereof include a method using a raw material compound (which can be prepared according to the above), a method of optically resolution by an enzyme, or a method of producing an optically active compound.
- the method is, for example, high performance liquid chromatography (HPLC) using an optically active column, supercritical fluid chromatography (SFC), or a salt formed with an optically active reagent and separated by fractional crystallization.
- HPLC high performance liquid chromatography
- SFC supercritical fluid chromatography
- a salt formed with an optically active reagent and separated by fractional crystallization there are a classical optical fractionated crystallization method for releasing the formation of the salt, or a method for separating and purifying the diastereomers produced by condensing with an optically active reagent and then decomposing them again.
- the optically active compound of the present invention can be produced by subsequently carrying out the production method shown above.
- a salt pharmaceutically acceptable by known means for example, an inorganic salt with sodium or the like or triethylamine. It is also possible to use an organic salt such as (organic salt).
- an inorganic salt when an inorganic salt is obtained, it is preferable to dissolve the compound of the present invention in water containing a hydroxide, a carbonate, a bicarbonate and the like corresponding to the desired inorganic salt.
- the reaction may be mixed with a water-miscible inert organic solvent such as methanol, ethanol, acetone or dioxane.
- a solution of sodium salts can be obtained by using sodium hydroxide, sodium carbonate or sodium bicarbonate.
- the compound when the compound contains an amino group contained in the compound or a basic functional group other than the compound, or when an aromatic ring having a basic property itself (for example, a pyridine ring) is contained.
- They can also be pharmaceutically acceptable salts (eg, salts with inorganic acids such as hydrochloric acid or salts with organic acids such as acetic acid) by known means.
- salts with inorganic acids such as hydrochloric acid or salts with organic acids such as acetic acid
- the reaction may be mixed with a water-miscible inert organic solvent such as methanol, ethanol, acetone or dioxane.
- a solution of hydrochloride can be obtained by using hydrochloric acid.
- the solution may be evaporated, or a water-miscible organic solvent having some polarity such as n-butanol, ethylmethylketone, etc. may be added to obtain the solid salt.
- a water-miscible organic solvent having some polarity such as n-butanol, ethylmethylketone, etc.
- the various compounds described in the present invention can be purified by known methods, for example, various chromatographies (columns, flash columns, thin layers, high performance liquids, supercritical fluids).
- a certain aspect of the compound of the present invention has IRAK-4 inhibitory activity and can be used as an IRAK-4 inhibitor. That is, certain embodiments of the compounds of the invention can be used as pharmaceuticals for the prevention and / or treatment of diseases associated with IRAK-4 inhibition. More specifically, the diseases associated with IRAK-4 inhibition are those that are successful with IRAK-4 inhibition, more specifically TLRs or IL-1 family signaling systems.
- the disease is not particularly limited as long as it is a disease that can be prevented and / or treated by suppressing the production of inflammatory mediators such as TNF ⁇ and IL-6 due to the inhibition of TNF ⁇ .
- the IRAK-4 inhibitory activity can be measured, for example, by the method shown in Test Example 1 or 2 described later.
- the disease related to IRAK-4 inhibition is not particularly limited as long as it is a disease that is successful by IRAK-4 inhibition, but specifically, for example, acute or chronic inflammation, autoimmune disease (rheumatoid arthritis, systemic erythematosus, lupus). Nephritis, etc.), autoimmune diseases (TNF receptor-related periodic syndrome (TRAPS), familial Mediterranean fever, cryopyrin-related periodic fever syndrome, high IgD syndrome, etc.), metabolic diseases (gout, etc.), etc. are exemplified. ..
- a certain aspect of the compound of the present invention has a TLR / IL-1 ⁇ signal inhibitory action as shown in a test example described later, and is useful as an active ingredient of a drug.
- certain embodiments of the compounds of the invention are preferably used for the prevention and / or treatment of diseases involving IRAK-4 signals.
- Some embodiments of the compounds of the invention show high selectivity for other kinases.
- other kinases include FLT3, ITK, CK2, IKKb, JAK1, Syk, PKC ⁇ , or p38.
- FLT3 is exemplified in particular. It is confirmed by, for example, a cytokine production suppression test using immune cells or a collagen-induced arthritis model that certain aspects of the pharmaceuticals of the present invention are useful for the prevention and / or treatment of diseases involving IRAK-4 signals. be able to. Specifically, the method described in Test Example 3 described later is exemplified.
- a certain aspect of the pharmaceutical of the present invention can be prepared as a pharmaceutical containing the compound represented by the formula (1) or a pharmaceutically acceptable salt thereof as an active ingredient, for example, a compound administered as a prodrug or a pharmaceutical product.
- a pharmaceutically acceptable salt undergoes metabolism in vivo to produce the compound represented by the formula (1) or the pharmaceutically acceptable salt thereof is also included in the scope of the pharmaceutical product of the present invention.
- the route of administration of certain aspects of the pharmaceutical of the present invention is not particularly limited, but for example, oral administration, subcutaneous administration, intradermal administration, intramuscular injection, intravenous administration, nasal administration, intravaginal administration, transrectal administration, or It can be appropriately selected from local administration to the affected area.
- the compound represented by the formula (1) or the pharmaceutically acceptable salt thereof may be used as it is, but the compound represented by the formula (1) or the pharmaceutically acceptable salt thereof may be used. It is preferred to add one or more pharmaceutically acceptable carriers to prepare and administer the pharmaceutical composition. Further, as the active ingredient of the pharmaceutical product of the present invention, a hydrate or solvate of the compound represented by the formula (1) or a pharmaceutically acceptable salt thereof may be used.
- Examples of the dosage form for formulating the pharmaceutical composition include tablets, powders, granules, syrups, suspensions, capsules, inhalants, injections and the like, and for the production thereof, Various carriers corresponding to these formulations are used.
- carriers for oral preparations may include excipients, binders, lubricants, fluidity promoters, or colorants.
- Examples of the inhalant include a method of inhaling the powder of the pharmaceutical composition or a chemical solution in which the pharmaceutical composition is dissolved or suspended in a solvent as it is, or a method of atomizing and inhaling using a sprayer called an atomizer or a nebulizer. Be done.
- an injection or the like generally distilled water for injection, physiological saline, aqueous glucose solution, vegetable oil for injection, propylene glycol, polyethylene glycol or the like can be used as the diluent. Further, if necessary, a bactericide, a preservative, a stabilizer, an tonicity agent, a painless agent, or the like may be added. An inclusion compound in which the compound of the present invention is included in cyclodextrin may be prepared and used as the pharmaceutical of the present invention.
- an appropriate dosage form may be appropriately selected and administered by an appropriate route.
- it can be orally administered in the form of tablets, powders, granules, syrups, suspensions, capsules and the like. It can also be administered trans-airway in the form of an inhalant. It can also be administered subcutaneously, intradermally, intravascularly, intramuscularly, or intraperitoneally in the form of an injection containing an infusion.
- it can be administered transmucosally in the form of a sublingual agent or a suppository, and can be administered transdermally in the form of a gel, lotion, ointment, cream, spray or the like. It can also be administered as a sustained-release preparation, such as a sustained-release injection or an implantable preparation (eg, a film preparation, etc.).
- the administration period of a certain aspect of the drug of the present invention is not particularly limited, but in principle, it is administered during the period when the clinical symptoms of the disease are judged to occur, and it is generally continued for several weeks to one year. be. However, it is possible to further extend the administration period depending on the pathological condition, or it is possible to continue administration even after the recovery of clinical symptoms. Furthermore, even if clinical symptoms are not manifested, it can be administered prophylactically at the discretion of the clinician.
- the dose of a certain aspect of the drug of the present invention is not particularly limited, but for example, when the drug of the present invention is orally administered, it is generally possible to administer 0.01 to 1000 mg of the active ingredient per adult. can. In that case, the administration frequency can be from once every 6 months to daily administration, preferably once / day.
- the daily and / or single dose, administration period, and frequency of administration are the patient's age, weight, physical health, type and severity of the disease to be treated, route of administration, and dosage form (active ingredient of the carrier). It may be increased or decreased as appropriate according to conditions such as sustained release of.
- an aspect of the drug of the present invention may be used at the same time as or at the same time as one or more selected drugs shown below. It can be changed and used together. Further, certain aspects of the pharmaceutical of the present invention can also be prepared and administered as a so-called combination drug together with the agents exemplified above.
- the combination is not only an administration form as a complete mixture of active ingredients as in a typical composition, but also an administration form in a non-mixed combination in which each active ingredient is separately administered from a plurality of containers arranged therein. It also includes kits and packaging.
- drugs examples include immunosuppressive agents (tachlorimus, cyclosporin, rapamycin, mofetyl mycophenolate, interferon preparations, cyclophosphamide, azathiopurine, methotrexate, etc.) and anti-inflammatory agents (steroids).
- NSAIDs non-steroidal anti-inflammatory drugs
- ibuprofen ibuprofen, selecoxib
- disease-modifying antirheumatic drugs gold preparation, methotrexate
- biopharmaceuticals used as therapeutic agents for autoimmune diseases, drugs for uric acid metabolism disorders (corhitin, probenecid, bucolome, etc.) , Benzbromalon, alloprinol, etc.), hypoglycemic agents (alogliptin, nateglinide, acarbose, metformin, pyoglitazone, insulin preparations, etc.), antihypertensive agents (imidapril, balsartan, candesartan, etc.) Bronchial dilators (adrenaline ⁇ 2 agonists such as salmetherol and salbutamol, anticholinergic drugs ipratropium, thiotropium, etc.), allergic disease therapeutic agents (theophylline, etc.), antiallergic drugs (fexoquinazine, epinastine, olopatazine, loratazine, cetilidine, bepot
- Ethyl icosapanthate, etc. Ethyl icosapanthate, etc.
- neurotransmitter regulators donevezil, galantamine, memantin, etc.
- antioxidants vitamin E, acetylcysteine, carnitine, betaine, pentoxyphyllin, etc.
- antibiotics ⁇ -lactam, ⁇ -lactam, etc.
- Various preparations such as macrolide, tetracycline, aminoglycoside, quinolone, chloramphenicol, etc. ). It can also be used in combination with various drugs created in the future. These combination drugs are not limited to any clinically significant combination.
- a certain aspect of the compound of the present invention contains a compound having excellent safety (various toxicity and safety pharmacology), pharmacokinetic performance, etc., and its usefulness as an active ingredient of a pharmaceutical can be confirmed by the following methods, for example. ..
- Safety-related tests include, for example, those listed below, but are not limited to this example. Cytotoxicity test (test using HL60 cells and hepatocytes, etc.), genotoxicity test (Ames test, mouse phosphorformer TK test, chromosomal abnormality test, micronucleus test, etc.), skin sensitization test (Buehler method, GPMT method, etc.) , APT method, LLNA test, etc.), Skin photosensitivity test (Adjuvant and Strip method, etc.), Eye irritation test (single instillation, short-term continuous instillation, repeated instillation, etc.), Safety pharmacology test for cardiovascular system (, etc.) Telemetry method, APD method, hERG inhibition evaluation method, etc.), safety pharmacology test for central nervous system (FOB method, modified Irwin method, etc.), safety pharmacology test for respiratory system (measurement method by respiratory function measuring device, blood gas) (Measurement method by analyzer, etc.), general toxicity
- the tests related to pharmacokinetic performance include, for example, those listed below, but are not limited to this example.
- Inhibition or induction test of chitochrome P450 enzyme, cell permeability test (test using CaCO-2 cells, MDCK cells, etc.), drug transporter ATPase assay, oral absorption test, blood concentration transition measurement test, metabolism test (stable) Includes sex tests, metabolic molecular species tests, reactivity tests, etc.), solubility tests (solubility test by turbidity method, etc.), etc.
- a certain aspect of the compound of the present invention is useful as an active ingredient of a pharmaceutical by conducting a cytotoxicity test.
- the cytotoxicity test include methods using various cultured cells such as HL-60 cells, which are pre-human leukemia cells, primary isolated cultured cells of liver cells, and neutrophil fractions prepared from human peripheral blood. This test can be performed by the method described below, but is not limited to this description. Prepare cells as a cell suspension of 105 to 107 cells / ml, and dispense 0.01 mL to 1 mL of the suspension into microtubes or microplates.
- a solution in which the compound was dissolved was added therein in an amount of 1/100 to 1 times the amount of the cell suspension, and the final concentration of the compound was, for example, 0.001 ⁇ M to 1000 ⁇ M in a cell culture medium at 37 ° C. Incubate under 5% CO 2 for 30 minutes to several days. After the culture is completed, the cell viability is evaluated using the MTT method or the WST-1 method (Ishiyama, M., et al., In Vitro Toxicology, 8, p.187, 1995). By measuring the cytotoxicity of a compound to cells, its usefulness as an active ingredient of a drug can be confirmed.
- genotoxicity tests include the Ames test, the mouse phosphorformer TK test, the chromosomal aberration test, and the micronucleus test.
- the Ames test is a method for determining sudden reversion mutations by culturing the bacteria on a culture dish mixed with a compound using Salmonella or Escherichia coli of the specified bacterial species (1999, Pharmaceutical Trial No. 1604). Refer to II-1. Genotoxicity test, etc. from the "Genotoxicity test guideline").
- the mouse lymphoma TK test is a gene mutation ability detection test targeting the thymidine kinase gene of mouse lymphoid L5178Y cells (1999, Pharmaceutical Cons No. 1604, "Genetic Toxicity Test Guidelines" II-3. Mouse phosphorus. Formal TK test; Clive, D. et al., Mutat. Res., 31, pp.17-29, 1975; Cole, J., et al., Mutat.Res., 111, pp.371-386, 1983 Etc.).
- the chromosomal aberration test is a method of co-culturing cultured mammalian cells and a compound, then immobilizing the cells, staining the chromosomes, and observing the cells to determine the activity that causes the chromosomal aberration (1999 Pharmaceutical Trial No. 1).
- the micronucleus test evaluates the ability to form micronuclei due to chromosomal abnormalities, and is based on the method using rodents (in vivo test) (1999, Pharmaceutical Cons No. 1604, "Genotoxicity Test Guidelines". II-4.
- a certain aspect of the compound of the present invention is useful as an active ingredient of a pharmaceutical.
- the Buehler method (Buehler, E.V. Arch.Dermatol., 91, pp.171-177, 1965) and GPMT method (maximization method) are used as skin sensitization tests using guinea pigs. (Magnusson, B. et al., J. Invest. Dermatol., 52, pp.268-276, 1969)) or APT method (adjuvant & patch method (Sato, Y.
- a skin photosensitization test As a skin light sensitization test, a skin light sensitization test using a guinea pig (see “Explanation of non-clinical test guidelines for pharmaceuticals 2002", Yakuji Nippo Co., Ltd. 2002, 1-9: Skin light sensitization test, etc.) Adjuvant and S Tori p method (Ichikawa, H. et al., J. Invest. Dermatol., 76, pp.498-501, 1981), Harber method (Harber, L.C., Arch).
- a certain aspect of the compound of the present invention is useful as an active ingredient of a pharmaceutical.
- eye irritation tests a single eye drop test method using rabbit eyes, monkey eyes, etc. (single instillation only once), a short-term continuous instillation test method (instillation in a short period of multiple times at regular intervals), and a repeated instillation test method (instillation at regular intervals) (Fukui, N. et al., Gendai no Rinsho, 4 (7), pp), which improves eye irritation symptoms for a certain period of time after instillation.
- There is a method of evaluation according to .277-289, 1970 By clarifying the ocular irritation of the compound by using any one or more of these methods, the usefulness as an active ingredient of a drug can be confirmed.
- a certain aspect of the compound of the present invention is useful as an active ingredient of a drug by conducting a safety pharmacological test on the cardiovascular system.
- a safety pharmacological test for the cardiovascular system the telemetry method (method for measuring the effects of compound administration without anesthesia on electrocardiogram, heart rate, blood pressure, blood flow, etc. (Shigeru Kanno, Hirokazu Bureau, Yoshiyoshi Nakata) Ed. Animal electrocardiogram, echocardiography, blood pressure, and pathological examination for basic and clinical use (2003 Maruzen Co., Ltd.), APD method (method for measuring cardiomyocyte action potential duration (Muraki, K. et al.). , AM. J.
- hERG inhibition evaluation method Patch clamp method (Chachin, M. et al., Nippon Yakurigaku Zasshi, 119, pp.345-351, 2002), Binding assay method (Gilbert, JD et al., J. Pharm. Tox. Methods, 50, pp) .187-199, 2004), Rb + efflex assay method (Cheng, CS et al., Drug Develop. Indust.
- a certain aspect of the compound of the present invention is useful as an active ingredient of a drug by conducting a safety pharmacological test on the central nervous system.
- the FOB method (Mattson, J. L. et al., J. American College of Technology, 15 (3), pp.239-254, 1996) is a safety pharmacological test for the central nervous system. ), Modified Irwin (method for evaluating general symptoms and behavioral observation (Irwin, S. Comprehensive Observational Assessment (Berl.) 13, pp.222-257, 1968), etc.
- a certain aspect of the compound of the present invention is useful as an active ingredient of a drug by conducting a safety pharmacological test on the respiratory system.
- Safety for the respiratory system As a pharmacological test, a measurement method using a respiratory function measuring device (measurement of respiratory rate, tidal volume, minute ventilation, etc.) (Drorbaugh, J.E. et al., Pediatrics, 16, pp.81- 87, 1955; Epstein, M.A.
- the general toxicity test is a single or repeated (multiple days) oral administration of a compound dissolved or suspended in an appropriate solvent using rodents such as rats and mice or non-rods such as monkeys and dogs. It is a method of observing the general condition of an administered animal and evaluating clinical chemical changes and pathological histological changes by intravenous administration or the like. By clarifying the general toxicity of the compound using these methods, the usefulness as an active ingredient of a pharmaceutical can be confirmed.
- the reproductive developmental toxicity test is a test that examines the induction of adverse effects on the reproductive development process of compounds using rodents such as rats and mice or non-rods such as monkeys and dogs ("Explanation of non-clinical test guidelines 2002"). See Yakuji Nippo, 2002, 1-6: Reproductive Developmental Toxicity Test, etc.). Reproductive developmental toxicity tests include tests on fertility and early embryonic development until implantation, tests on prenatal and postnatal development and maternal function, and tests on embryo / fetal development (2000, Pharmaceutical Cons No. 1834, Attachment). Please refer to [3] Reproductive developmental toxicity test) etc. from "Guidelines for Pharmaceutical Toxicity Test Method". By clarifying the reproductive developmental toxicity of the compound using these test methods, its usefulness as an active ingredient of a drug can be confirmed.
- cytochrome P450 enzymes Gomez-Lechon, MJ et al., Curr. Drug Metab. 5 (5), pp. It can be confirmed by performing 443-462, 2004.
- cytochrome P450 enzymes for example, a cytochrome P450 enzyme of each molecular species or a human P450 expression system microsome prepared from cells or prepared by using a recombinant is used, and the enzyme activity is compounded in vitro.
- the reactive metabolites are trapped as dGSH adducts by incubating the human liver microsome and the compound in the presence of NADPH and glutathione (dGSH) fluorescently labeled with a dansyl group, and the fluorescence intensity is increased.
- dGSH NADPH and glutathione
- a method for comprehensively detecting peaks caused by reactive metabolites from the amount of dGSH adduct produced Junping Gan. Et al., Chem. Res. Toxicol. 2005, 18, 896-903
- 14C labeled compound is incubated with human liver microsomes in the presence of NADPH to measure covalently bound activity to proteins (Baillie TA, Drug Metabolizing Enzymes.
- a cell permeability test for example, a method of measuring the cell membrane permeability of a compound in an in vitro cell culture system using Caco-2 cells (Delie, F. et al., Crit. Rev. Ther. Drug Carrier Syst., 14, pp. 221-286, 1997; Yamashita, S. et al., Eur. J. Pham. Sci., 10, pp.195-204, 2000; Ingels, F.M. et al., J. Pham. Sci.
- a certain aspect of the compound of the present invention is useful as an active ingredient of a drug, for example, by performing a drug transporter ATPase assay as an ATP-Binding Cassette (ABC) transporter.
- a drug transporter ATPase assay a method for investigating whether a compound is a substrate for P-gp using a P-glycoprotein (P-gp) baculovirus expression system (Germann, U.A., Methods Enzymol., 292, pp.427-41, 1998) and so on.
- a transport test using oocytes collected from Xenopus laevis as a Solute Carrier Transporter (SLC) transporter.
- SLC Solute Carrier Transporter
- Examples of the transport test include a method of investigating whether or not a compound is a substrate of OATP2 using OATP2-expressing Oocytes (Tamai I. et. Al., Pharm Res. 2001 Sep; 18 (9): 1262-1269). ..
- the usefulness as an active ingredient of a drug can be confirmed.
- a certain aspect of the compound of the present invention is useful as an active ingredient of a pharmaceutical.
- an oral absorption test using rodents, monkeys, dogs, etc., a certain amount of the compound is dissolved or suspended in an appropriate solvent, the blood concentration after oral administration is measured over time, and the compound is taken orally.
- Examples include a method for evaluating blood transferability by administration using the LC-MS / MS method (edited by Kenichi Harada et al., "Latest Mass Spectrometry for Life Sciences", Kodansha Scientific 2002, etc.). By clarifying the oral absorbability of the compound using these methods, the usefulness as an active ingredient of a drug can be confirmed.
- a certain aspect of the compound of the present invention is useful as an active ingredient of a pharmaceutical by conducting a blood concentration transition measurement test.
- a compound is orally or parenterally administered to rodents, monkeys, dogs, etc. (for example, intravenously, intramuscularly, intraperitoneally, subcutaneously, transdermally, instilled or nasally).
- the transition of the concentration of the compound in the blood after administration to is measured using the LC-MS / MS method (edited by Kenichi Harada et al., "Latest Mass Spectrometry for Life Science", Kodansha Scientific 2002, etc.). The method etc. can be mentioned.
- the usefulness as an active ingredient of a drug can be confirmed.
- a certain aspect of the compound of the present invention is useful as an active ingredient of a pharmaceutical.
- a blood stability test method (a method for predicting metabolic clearance in vivo from the metabolic rate of a compound in hepatic microsomes of humans or other animal species (Shou, W. Z. et al.,) J. Mass Spectrom., 40 (10), pp.1347-1356, 2005; Li, C. et al., Drug Metab. Dispos., 34 (6), 901-905, 2006) etc.) , Metabolism molecular species test method, reactive metabolite test method and the like.
- a certain aspect of the compound of the present invention is useful as an active ingredient of a pharmaceutical by conducting a solubility test.
- the evaluation of solubility in water exemplifies a method of confirming under acidic conditions, neutral conditions, or basic conditions, and further includes confirming a change in solubility depending on the presence or absence of bile acid.
- a solubility test a solubility test method by the turbidity method (Lipinski, C.A. et al., Adv. Drug Deliv. Rev., 23, pp.3-26, 1997; Bevan, C.D. et al., Anal. Chem. , 72, pp.1781-1787, 2000) and so on.
- a certain aspect of the compound of the present invention is useful as an active ingredient of a pharmaceutical by examining, for example, upper gastrointestinal tract disorder, renal dysfunction and the like.
- a pharmacological test on the upper gastrointestinal tract the effect on the gastric mucosa can be investigated using a fasted rat gastric mucosal injury model.
- Pharmacological tests for renal function include a method for measuring renal blood flow and glomerular filtration rate [Physiology, 18th Edition (Spectral Hall), 1986, Chapter 17].
- Examples and the like examples and test Examples
- All purchased reagents were used without further purification.
- the purchased anhydrous solvent was used without further drying.
- BIOTAGE Dalton was used as an MS detector connected to Yamazen's medium pressure preparative purification system SmartFlash or BIOTAGE's medium pressure preparative purification system Isolera ONE.
- As the column either SNAP Ultra manufactured by Biotage or DispoPack AT manufactured by YMC was used. In some cases, it was purified using BondElute SCX manufactured by Agilent as an ion exchange resin.
- the BondElute SCX may be abbreviated as SCX below.
- SCX As an example of the use of SCX, after washing the cartridge with methanol and dichloromethane, the crude product is dissolved in a minimum volume solvent (for example, a mixed solvent of dichloromethane-methanol) and adsorbed. Then, the impurities are washed away with methanol while applying pressure, and the product is eluted with 2.0 M ammonia-methanol.
- a minimum volume solvent for example, a mixed solvent of dichloromethane-methanol
- TLC thin layer chromatography
- Precoated silica gel 60 F254 manufactured by Merck, product number 5715-1M
- PTLC phosphomolybdic acid
- NMR nuclear magnetic resonance apparatus
- ppm parts per million
- 1H integral value
- multiplex term s, singlet; d, doublet; t, triplet; q, quartet; qui, quintet; m. , Multiplet; br, broad; dd, double doublet, etc.
- LCMS liquid chromatograph mass spectrometry spectrum
- LC-MS liquid chromatograph mass spectrometry spectrum
- ESI electrospray
- the liquid chromatography apparatus an Aquacity ⁇ Ltra Performance LC system manufactured by Waters was used.
- the separation column ACQUITY UPLC BEH C18 2.1 ⁇ 50 mm 1.7 ⁇ m (manufactured by Waters) was used.
- reaction solution was cooled to 0 ° C., imidazole (156 g, 2.29 mol) and TBS chloride (207 g, 1.38 mol) were sequentially added, and the mixture was stirred at room temperature for 1 and a half hours.
- the obtained crude reaction mixture was cooled to 0 ° C., water (530 mL) was added, the mixture was filtered through cerite, extracted with ethyl acetate, and then the organic layer was washed with water and saturated brine. The organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure.
- optical isomers were separated and analyzed under the following chiral HPLC conditions to obtain each optically active substance having an optical purity of 99.7% ee or more.
- Sorting conditions Column: CHIRALART Cellulose-SC (10um) 245x150mm I. D. , Eluent: heptane / 2-propanol (80/20) (v / v), flow rate: 518 mL / min, temperature: 24 ° C., detection: UV (245 nm), load: 180 mL (9 g) Analytical conditions; Column: CHIRALART Cellulose-SC (5um) 250x4.6mm I. D.
- Imidazole (330 mg, 4.82 mmol) and TBS chloride (545 mg, 3.62 mmol) were added thereto, and the mixture was stirred at room temperature for 6 hours. Then, imidazole (330 mg, 4.82 mmol) and TBS chloride (545 mg, 3.62 mmol) were added, and the mixture was stirred at room temperature for 52 hours. Further, imidazole (330 mg, 4.82 mmol) and TBS chloride (545 mg, 3.62 mmol) were added, and the mixture was stirred at room temperature for 24 hours.
- N- (6-bromo-1,3-benzothiazole-2-yl) cyclopropanecarbamide (intermediate A-2-3; 400 mg, 1.35 mmol) was dissolved in 1,4-dioxane (13 mL).
- Bis (pinacolado) diboron (513 mg, 2.03 mmol), [1,1'-bis (diphenylphosphino) ferrocene] dichloropalladium (II) (98 mg, 0.13 mmol), potassium acetate (400 mg, 2.69 mmol)
- the mixture was stirred at 80 ° C. for 3 hours, cooled to room temperature, and then the solvent was concentrated under reduced pressure.
- Example b-01-01 N- (6-(2-((((1S, 2S) -2-hydroxycyclopentyl) oxy) methyl) pyrimidine-5-yl) benzo [d] thiazole-2-yl) -3-Morpholine cyclobutane-1-carboxamide
- Example b-02-01 3-((R) -3-fluoropyrrolidine-1-yl) -N-6 ((((1S, 2S) -2-hydroxycyclopentyl) oxy) methyl) pyrimidine-5- Il) Benzo [d] Thiazole-2-yl) Cyclobutane-1-carboxamide
- 1,4-Dioxane 243.6 mL was added to 2-amino-6-bromobenzothiazole (20.3 mg, 88.61 mmol) to dissolve it, and cesium carbonate (88.61 g, 265.82 mmol), [1,1 '-Bis (diphenylphosphino) ferrocene] Dichloropalladium (II) (6.48 g, 8.86 mmol), [(1S, 2S) -2-[[5- (4,4,5,5-tetramethyl-] 1,3,2-dioxaborolan-2-yl) pyrimidin-2-yl] methoxy] cyclopentyl] acetate (88.15 g, 15,63 mmol) and water (60.9 mL) were added, and the mixture was heated and stirred at 80 ° C.
- Example b-04-01 tert-butyl (S) -4-((1S, 3R) -3-((6--2-((((1S, 2S) -2-hydroxycyclopentyl) oxy) methyl)) Pyrimidine-5-yl) benzo [d] thiazole-2-yl) carbamoyl) cyclobutyl) -3-methylpiperazin-1-carboxylate
- reaction mixture was purified by SCX and (1R, 3s) -N- (6-(((((1S, 2S) -2-hydroxycyclopentyl) oxy) methyl) pyrimidin-5-yl) benzo [ d] Thiazole-2-yl) -3-((S) -2-methylpiperazin-1-yl) cyclobutane-1-carboxamide (3.05 g, yield 74%) was obtained.
- Example b-04-02 (1R, 3s) -N- (6-((((1S, 2S) -2-hydroxycyclopentyl) oxy) methyl) pyrimidin-5-yl) benzo [d] Thiazole-2-yl) 3-((S) -2-methyl-4- (2-methylisonicotinoyle) piperazine-1-yl) cyclobutane-1-carboxamide
- Example b-05-01 (1R, 3s) -3-((S) -4-acetyl-2-methylpiperazin-1-yl) N- (6-(2-((((1S, 2S)) -2-Hydroxycyclopentyl) Oxy) Methyl) Pyrimidine-5-yl) Benzo [d] Thiazole-2-yl) Cyclobutane-1-carboxamide
- Example b-06-01 (S) -N-ethyl-4-((1s, 3R) -3-((6-(2-((((1S, 2S) -2-hydroxycyclopentyl) oxy)) Methyl) pyrimidine-5-yl) benzo [d] thiazole-2-yl) carbamoyl) -3-methylpiperazin-1-carboxamide
- Example b-07-01 N- (6-(((((1S, 2S) -2-hydroxycyclopentyl) oxy) methyl) pyrimidine-5-yl) benzo [d] thiazole-2-yl) -3-((S) -2-Methyl-4- (2,2,2-trifluoroethyl) piperazine-1-yl) cyclobutane-1-carboxamide
- Example b-08-01 3-((S) -4-cyclopropyl-2-methylpiperazin-1-yl) N-(6-(2-((((1S, 2S) -2-hydroxycyclopentyl) ) Oxy) Methyl) Pyrimidine-5-yl) Benzo [d] Thiazole-2-yl) Cyclobutane-1-carboxamide
- the crude reaction mixture was diluted with ethyl acetate (200 mL) and washed with water (200 mL). The aqueous layer was extracted again with ethyl acetate (200 mL), and the combined organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure.
- Example c-02-01 (1R, 2R) -N- (6-((((1S, 2S) -2-hydroxycyclopentyl) oxy) methyl) pyrimidin-5-yl) -7-methoxy Benzodiazepine [d] thiazole-2-yl) -2-methoxycyclopropane-1-carboxamide
- Example c-03-01 tert-butyl (S) -4-((1s, 3R) -3-((6-(2-((((1S, 2S) -2-hydroxycyclopentyl) oxy) methyl) ) Pyrimidine-5-yl) -7-methoxybenzo [d] thiazole-2-yl) carbamoyl) cyclobutyl) -3-methylpiperazin-1-carboxylate
- Example c-03-02 (1R, 3s) -N- (6-((((1S, 2S) -2-hydroxycyclopentyl) oxy) methyl) pyrimidin-5-yl) -7-methoxy Benzo [d] Thiazole-2-yl) -3-((S) -2-Methyl-4- (2-methylisonicotinoyle) piperazine-1-yl) Cyclobutane-1-carboxamide
- Example c-04-01 N-(6-(((((1S, 2S) -4,4-difluoro-2-hydroxycyclopentyl) oxy) methyl) pyrimidine-5-yl) -7-methoxy) Benzodiazepine [d] thiazole-2-yl) cyclopropanecarboxamide
- Trans-2-((5-bromopyrimidine-2-yl) methoxy) -4,4-difluorocyclopropane-1-ol (intermediate A-4-2; 1 g, 3.2 mmol) is 1,4-dioxane.
- Intermediate A-4-2 1 g, 3.2 mmol
- bis (pinacholado) diboron 1.1 g, 4.8 mmol
- Potassium acetate (0.95 g, 9.7 mmol) was added and stirred at 80 ° C. overnight.
- the obtained crude reaction mixture was concentrated under reduced pressure to obtain 1.50 g of a crude reaction intermediate.
- the obtained crude reaction intermediate was dissolved in acetonitrile (8 mL) and water (1 mL), and N- (6-bromo-7-methoxybenzo [d] thiazole-2-yl) cyclopropanecarboxamide (intermediate C-4) was dissolved.
- -1 400 mg, 1.22 mmol
- cesium fluoride (0.37 g, 2.4 mmol
- bis (di-tert-butyl (4-dimethylaminophenyl) phosphine) dichloropalladium (II) 43 mg, 61 ⁇ mol.
- the mixture was stirred at 130 ° C.
- Example c-05-01 N-(7-((3-fluoro-1-methylazetidine-3-yl) methoxy) -6-(2-((((1S, 2S) -2-hydroxycyclopentyl) ) Oxy) Methyl) Pyrimidine-5-yl) Benzo [d] Thiazole-2-yl) Cyclopropanecarboxamide
- the obtained crude reaction mixture was purified by silica gel chromatography to obtain a crude reaction intermediate (3 mg).
- the obtained crude reaction intermediate (30 mg) was dissolved in 3M hydrochloric acid-methanol (3 mL), and the mixture was stirred at room temperature for 3 hours.
- the obtained crude reaction mixture was concentrated under reduced pressure, dichloromethane and aqueous sodium hydrogen carbonate were added, and the organic layer was concentrated under reduced pressure.
- Fluoro-N-phenyl-N- (trifluoromethylsulfonyl) methanesulfonamide (629 mg, 1.76 mmol) was added, and the mixture was stirred at room temperature for 5.5 hours. Water is added to the reaction mixture solution and the mixture is extracted with ethyl acetate. The aqueous layer is extracted again with ethyl acetate. The organic layers are combined, washed with saturated brine, dried over sodium sulfate, and then filtered. It was concentrated under reduced pressure.
- reaction mixture was diluted with saturated brine, extracted with ethyl acetate, dried over anhydrous magnesium sulfate, concentrated under reduced pressure, and purified by HPLC to obtain N- [6- [2-[(1S, 2S). ) -2-Hydroxycyclopentoxy] pyrimidin-5-yl] -7-methyl-1,3-benzothiazole-2-yl] cyclopropanecasvoxamide (14.8 mg, yield 65%) was obtained. ..
- N-Ethyldiisopropylamine (921.1 ⁇ L, 5.29 mmol) and 1-propanephosphonic acid anhydride (1.24 mL, 4.17 mmol) were added, and the mixture was stirred at room temperature for 14 hours.
- 1- Propane phosphonic acid anhydride (1.24 mL, 4.17 mmol) was added, and the mixture was further stirred for 2 hours.
- tert-Butyl 3-hydroxycyclobutane carboxylate (0.30 g, 1.7 mmol) was dissolved in N, N-dimethylformamide (3.5 mL) and sodium hydride (55% oily, 5 mg, 2. 1 mmol) was added, and the mixture was stirred for 10 minutes.
- Ethyl iodide (0.28 mL, 3.5 mmol) was added to the reaction solution, and the mixture was stirred overnight at room temperature.
- a saturated aqueous solution of ammonium chloride was added to the reaction solution to stop the reaction, and the mixture was extracted with ethyl acetate.
- Example d-04-01 (1s, 3s) -3-ethoxy-N- (6-((((1S, 2S) -2-hydroxycyclopentyl) oxy) methyl) pyrimidin-5-yl) Benzodiazepine [d] thiazole-2-yl) cyclobutane-1-carboxamide
- N-ethyldiisopropylamine (5.17 mL, 29.7 mmol), 3-oxocyclobutane-1-carboxylic acid (1.36 g, 11.88 mmol), 1-propanephosphonic acid anhydride (8.74 mL, 14.85 mmol) was added, and the mixture was stirred at room temperature for 5 hours.
- Saturated aqueous sodium hydrogen carbonate and chloroform were added to the reaction mixture for extraction.
- the aqueous layer was extracted again with chloroform, the organic layers were combined and washed with saturated brine.
- the organic layer was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure.
- tert-butyl (S) -2-methylpiperazin-1-carboxylate 600 mg, 3.00 mmol was dissolved in dichloromethane (30.0 mL), 2-methylisonicotinic acid (821 mg, 6.00 mmol), N- Ethyldiisopropylamine (3.2 mL, 18.0 mmol) and propanephosphonic acid anhydride (6.0 mL, 1.7 M ethyl acetate solution) were added, and the mixture was stirred at room temperature for 1 hour. Saturated aqueous sodium hydrogen carbonate was added to the reaction mixture, the mixture was extracted with chloroform, and the organic layer was concentrated under reduced pressure.
- Example e-02-01 (1R, 3s) -N- (5-(2-(((((1S, 2S) -2-hydroxycyclopentyl) oxy) methyl) pyrimidin-5-yl) pyrazolo [1, 5-a] Pyridine-2-yl) -3-((S) -2-methyl-4- (2-methylisonicotinoyle) piperazine-1-yl) cyclobutane-1-carboxamide
- ⁇ Test Example 1 Measurement of human IRAK-4 inhibitory activity> (1) Measurement method The activity of human IRAK-4 (Invitrogen, Cat.PV3362) is the phosphorylation of the IRAK-4 peptide substrate (biotin-KKKKRFSFKKSFKKC) in the presence of 10 ⁇ M ATP (Sigma Aldrich, Cat.A7699). -Measured by the FRET method. The enzymatic reaction was performed in a reaction buffer consisting of 50 mM HEPES (pH 7.2), 1 mM DTT, 0.1 mM Na 3 VO 4 , 5 mM MgCl 2 , 1 mM MnCl 2 , and 0.1% bovine serum albumin.
- test compound was added to a reaction buffer containing 1 nM IRAK-4, 0.5 ⁇ M peptide substrate, and 10 ⁇ M ATP, and incubated at 23 ° C. for 30 minutes.
- Europium cryptotate-labeled antibody 0.3 ⁇ g / mL, antibody prepared using IRAK-4 peptide substrate as antigen
- streptavidin-XL665 2 ⁇ g / mL, CisBio, Cat.610SAXLB
- 50 mM HEPES pH 7.2.
- THP-1 LPS-stimulated TNF ⁇ production inhibition test using human acute monocytic leukemia-derived cell line THP-1>
- Measurement method in the THP-1 assay the effect of the test compound on TNF ⁇ production induced by LPS stimulation can be evaluated.
- THP-1 cells ATCC, Cat. TIB-202
- 20 ⁇ L of test compound was added, and 1 in a 5% CO 2 incubator at 37 ° C. Incubated for hours.
- 20 ⁇ L of LPS final concentration 2.5 ng / mL, Sigma, Cat. L2630
- the plates were centrifuged, and 100 ⁇ L of the supernatant was taken from each well and used for evaluation of the amount of TNF ⁇ by HTRF (Cisbio, Cat. 62TNFPEB).
- HTRF Transgene, Cat. 62TNFPEB
- To measure the amount of TNF ⁇ dilute the supernatant 2-fold with medium, add 10 ⁇ L to a 384-well plate, add anti-TNF ⁇ -criptate (5 ⁇ L) and anti-TNF ⁇ -XL665 (5 ⁇ L), and let stand overnight. bottom. The fluorescence intensity ratio at a wavelength of 620/665 nm was measured with a microplate reader, and the amount of TNF ⁇ in the supernatant was calculated by a calibration curve.
- the TNF ⁇ production suppression rate when LPS is not added is 100%
- the TNF ⁇ production suppression rate when the test compound is not added is 0%
- the IC50 of the test compound is used as 4 parameters of the data analysis software XLfit (ID Business Solutions Ltd.). It was determined using a compound model.
- Cell viability was measured using a 96-well plate after removing 100 ⁇ L of the supernatant, and the effect of the off-target effect by the test compound was evaluated. After adding 5 ⁇ L of CCK-8 (Dojindo, Cat.CK04-10) and incubating at 37 ° C. for 1 hour, the absorbance at 450 nm was measured with a microplate reader. The cell viability when no LPS was added was set to 100%, and the IC 50 of the test compound was determined using XLfit. It should be noted that each operation and condition during measurement can be appropriately changed within a range that can be understood by those skilled in the art and does not significantly affect the measurement.
- Rat collagen-induced arthritis model> (1) Measurement method An 8-week-old female Lewis rat (SLC Inc.) was immunized with bovine type II collagen (CII, Collagen Technology Workshop, product number K41) to induce arthritis. A one-to-one mixed emulsion of incomplete Freund's adjuvant (Difco, Cat. 263910) and CII 3 mg / mL solution was prepared, and 0.7 mL per rat was applied to 7 sites in the ridge and the skin of both anterior and posterior limbs. 1 mL each was injected.
- incomplete Freund's adjuvant Difco, Cat. 263910
- CII 3 mg / mL solution was prepared, and 0.7 mL per rat was applied to 7 sites in the ridge and the skin of both anterior and posterior limbs. 1 mL each was injected.
- the footpad volume of both hind limbs of the animal was measured using a Pletthysmometer every 1 or 2 days after the start of administration, and the effect of the test compound was evaluated. For each individual on each measurement day, the average footpad volume of both hind limbs was calculated using Excel2010 (Microsoft) and graphed using GraphPadPrism7.03 (GraphPad Software, Inc.). The Normal group was defined as a 100% inhibitory control, and the solvent-administered group was defined as a 0% inhibitory control. For these controls, the suppression rate for each test compound concentration was calculated using Excel2010 (Microsoft). It should be noted that each operation and condition during measurement can be appropriately changed within a range that can be understood by those skilled in the art and does not significantly affect the measurement.
- Figure 1 shows the transition results of the footpad capacity of each group.
- the vertical axis shows the sole capacity
- the horizontal axis shows the number of days after the first bovine type II collagen immunization.
- "n” indicates the number of examples of rats used.
- the suppression rates of Example c-01-01 at 20, 60, 120 mg / kg, twice daily administration group 19 days after the initial immunization were 45%, 65%, and 77%, respectively.
- one embodiment of the compound of the present invention (Example c-01-01) showed an excellent swelling-suppressing effect in rat arthritis.
- the compound of the general formula (1) or a salt thereof has excellent IRAK-4 inhibitory activity, and is useful as an active ingredient of a pharmaceutical agent for the prevention and / or treatment of diseases associated with IRAK-4 inhibition.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description
〔1〕下記一般式(1):

Rgは、下記一般式(1-1):


R1は、-H、-F、-Cl、メチル、又はC1-3アルコキシであり、前記C1-3アルコキシはG1群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
前記G1群は、-F、ヒドロキシ、シアノ、ハロゲノC1-6アルキル、C1-4アルコキシ、フェニル、5-6員ヘテロアリール、及び3-7員飽和環基からなる群であり、G1群におけるフェニル又は5-6員ヘテロアリールはGAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
前記GAr群は、-F、-Cl、ヒドロキシ、シアノ、C1-6アルキル、ハロゲノC1-6アルキル、及び-NH2からなる群であり;
R2は、C1-6アルキル、ハロゲノC1-6アルキル、又は3-7員飽和環基であり、前記R2はG2群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
前記G2群は、-F、ヒドロキシ、ハロゲノC1-3アルキル、及びC1-4アルコキシからなる群であり;
Cyは、下記一般式(2-1):

式(2-1)中、
kは、0又は1の整数であり;
RCy1及びRCy2は、各々独立して、-H、-F、ヒドロキシ、シアノ、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシ、-NR11R12、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、RCy1とRCy2におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく、前記RCy1、RCy2は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R11及びR12は、各々独立して、-H、C1-6アルキル、ハロゲノC1-6アルキル、ヒドロキシC1-6アルキル、C1-6アルコキシC1-6アルキル、3-7員飽和環基、フェニル、又は5-6員ヘテロアリールであり、R11及びR12はG1群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
又はR11とR12が一緒になって4-10員飽和環又は7-11員スピロ環を形成し、前記4-10員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよく、前記4-10員飽和環及び7-11員スピロ環はG3群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
前記G3群は、-F、ヒドロキシ、C1-6アルキル、ハロゲノC1-6アルキル、ヒドロキシC1-6アルキル、C1-6アルコキシ、C1-6アルコキシ-C1-6アルキル、ハロゲノC1-6アルコキシ、-C(O)R14、-NR13C(O)R14、-C(O)NR13R14、-C(O)NH2、-NR13S(O2)R14、-S(O2)NR13R14、-S(O2)NH2、-S(O2)R14、フェニル、5-6員ヘテロアリール、及び3-7員飽和環基からなる群であり、前記G3におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R13は、-H、C1-3アルキル、ハロゲノC1-3アルキル、C1-3アルコキシC1-3アルキル、ハロゲノC1-3アルコキシC1-3アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、前記R13におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R14は、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシC1-6アルキル、ハロゲノC1-6アルコキシC1-6アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、前記R14におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
又はR13とR14が一緒になって4-7員飽和環又は7-11員スピロ環を形成し、前記4-7員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよく;
又は、RCy1とRCy2が一緒になって4-7員飽和環を形成し、前記4-7員飽和環はO及びNからなる群より選択される1-2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよく、前記4-7員飽和環は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい]
で示される化合物又はその塩。
〔3〕R1が-F、-Cl、メチル、又はC1-3アルコキシである前記〔2〕に記載の化合物又はその塩。
〔4〕R1がC1-3アルコキシである前記〔2〕に記載の化合物又はその塩。
〔4-2〕R1がエトキシ、メトキシ、又はメトキシエトキシである前記〔2〕に記載の化合物又はその塩。
〔4-3〕R1がエトキシ、又はメトキシである前記〔2〕に記載の化合物又はその塩。
〔5〕R1がメトキシである請求項2に記載の化合物又はその塩。
Cyが、下記一般式(2-1-1):

〔6-2〕R11及びR22が、各々独立して、-H、C1-6アルキル、ハロゲノC1-6アルキル、ヒドロキシC1-6アルキル、C1-6アルコキシC1-6アルキル、3-7員飽和環基、フェニル、又は5-6員ヘテロアリールであり、R11及びR12は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい;
前記〔2〕~〔5〕のいずれかに記載の化合物又はその塩。
〔6-3〕R11及びR22が、各々独立して、-H、C1-6アルキル、ハロゲノC1-6アルキル、3-7員飽和環基、又は5-6員ヘテロアリールであり、R11及びR12は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい;
前記〔2〕~〔5〕のいずれかに記載の化合物又はその塩。
前記〔2〕~〔5〕のいずれかに記載の化合物又はその塩。
〔6-5〕R11とR12が一緒になって4-10員飽和環又は7-11員スピロ環を形成し、前記4-7員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよく、前記4-7員飽和環及び7-11員スピロ環は前記G3群より選択される1~3個の同一又は異なった置換基で置換されていてもよい;
前記〔2〕~〔5〕のいずれかに記載の化合物又はその塩。
〔6-6〕R11とR12が一緒になって4-10員飽和環又は7-11員スピロ環を形成し、前記4-7員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよく、前記4-7員飽和環及び7-11員スピロ環は前記G3群より選択される1~3個の同一又は異なった置換基で置換されていてもよい;
前記〔2〕~〔5〕のいずれかに記載の化合物又はその塩。

R15は、-H、C1-6アルキル、ハロゲノC1-6アルキル、ヒドロキシC1-6アルキル、C1-6アルコキシC1-6アルキル、-C(O)R16、-S(O2)R16、-C(O)NR16R17、-C(O)OR16、又は3-7員飽和環基であり、前記R15はG1群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R16は、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシC1-6アルキル、ハロゲノC1-6アルコキシC1-6アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、前記R16におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R17は、-H、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシC1-6アルキル、ハロゲノC1-6アルコキシC1-6アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、前記R17におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
又はR16とR17が一緒になって4-7員飽和環又は7-11員スピロ環を形成し、前記4-7員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよい;
前記〔2〕~〔5〕のいずれかに記載の化合物又はその塩。

Xは、O又はNR15(R15は前記と同義)である前記〔2〕~〔5〕のいずれかに記載の化合物又はその塩。
〔6-9〕R11とR12が一緒になって下記一般式(2-1-1-c-1)~(2-1-1-c-3):

Xは、O又はNR15(R15は前記と同義)である前記〔2〕~〔5〕のいずれかに記載の化合物又はその塩。

Xは、O又はNR15(R15は前記と同義)である前記〔2〕~〔5〕のいずれかに記載の化合物又はその塩。

Xは、O又はNR15(R15は前記と同義)である前記〔2〕~〔5〕のいずれかに記載の化合物又はその塩。

[式(2-1-2)中、RCy3は、C1-4アルキル、ハロゲノC1-4アルキルであり;
Xは、O又はNR15であり;
R15は、-H、C1-6アルキル、ハロゲノC1-6アルキル、ヒドロキシC1-6アルキル、C1-6アルコキシC1-6アルキル、-C(O)R16、-S(O2)R16、-C(O)NR16R17、-C(O)OR16、又は3-7員飽和環基であり、前記R15はG1群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R16は、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシC1-6アルキル、ハロゲノC1-6アルコキシC1-6アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、前記R16におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R17は、-H、C1-6アルキル、ハロゲノC1-6アルキル、C1-3アルコキシC1-6アルキル、ハロゲノC1-6アルコキシC1-6アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、前記R17におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
又はR16とR17が一緒になって4-7員飽和環又は7-11員スピロ環を形成し、前記4-7員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよい]
で示される〔2〕~〔6-11〕のいずれかに記載の化合物又はその塩。
〔9〕Cyが、下記一般式(2-1-3):

前記〔2〕~〔8〕のいずれかに記載の化合物又はその塩。
R2が、以下の式(3-1):

R2が、以下の式(3-1):











〔23〕R1が-H、又はメトキシである前記〔22〕に記載の化合物又はその塩。
〔24〕R1が-Hである前記〔22〕に記載の化合物又はその塩。
〔25〕Cyが、一般式(2-1)であり;
[式(2-1)中、
kが1の整数であり;
RCy1及びRCy2が、各々独立して、-H、-F、ヒドロキシ、シアノ、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシ、-NR11R12、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、RCy1とRCy2におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく、前記RCy1、RCy2は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい(R11及びR12は前記と同義)]
で示される〔22〕~〔24〕のいずれかに記載の化合物又はその塩。

前記〔22〕~〔25〕のいずれかに記載の化合物又はその塩。
〔26-2〕R11及びR22が、各々独立して、-H、C1-6アルキル、ハロゲノC1-6アルキル、ヒドロキシC1-6アルキル、C1-6アルコキシC1-6アルキル、3-7員飽和環基、フェニル、又は5-6員ヘテロアリールであり、R11及びR12は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい;
前記〔22〕~〔25〕のいずれかに記載の化合物又はその塩。
〔26-3〕R11及びR22が、各々独立して、-H、C1-6アルキル、ハロゲノC1-6アルキル、3-7員飽和環基、又は5-6員ヘテロアリールであり、R11及びR12は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい;
前記〔22〕~〔25〕のいずれかに記載の化合物又はその塩。
前記〔22〕~〔25〕のいずれかに記載の化合物又はその塩。
〔26-5〕R11とR12が一緒になって4-10員飽和環又は7-11員スピロ環を形成し、前記4-7員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよく、前記4-7員飽和環及び7-11員スピロ環は前記G3群より選択される1~3個の同一又は異なった置換基で置換されていてもよい;
前記〔22〕~〔25〕のいずれかに記載の化合物又はその塩。
〔26-6〕R11とR12が一緒になって4-10員飽和環又は7-11員スピロ環を形成し、前記4-7員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよく、前記4-7員飽和環及び7-11員スピロ環は前記G3群より選択される1~3個の同一又は異なった置換基で置換されていてもよい;
前記〔22〕~〔25〕のいずれかに記載の化合物又はその塩。

のいずれかの飽和環を形成し、前記飽和環はG3群より選択される1~3個の同一又は異なった置換基で置換されていてもよい;
前記〔22〕~〔25〕のいずれかに記載の化合物又はその塩。

Xは、O又はNR15(R15は前記と同義)である前記〔22〕~〔25〕のいずれかに記載の化合物又はその塩。

Xは、O又はNR15(R15は前記と同義)である前記〔22〕~〔25〕のいずれかに記載の化合物又はその塩。

Xは、O又はNR15(R15は前記と同義)である前記〔22〕~〔25〕のいずれかに記載の化合物又はその塩。

Xは、O又はNR15(R15は前記と同義)である前記〔22〕~〔25〕のいずれかに記載の化合物又はその塩。

Xは、O又はNR15(R15は前記と同義)である
前記〔22〕~〔26-11〕のいずれかに記載の化合物又はその塩。
〔28〕XがNR15(R15は前記と同義)である
前記〔22〕~〔27〕のいずれかに記載の化合物又はその塩。

前記〔22〕~〔28〕のいずれかに記載の化合物又はその塩。






〔35〕IRAK4阻害に関連する疾患の予防及び/又は治療のための前記〔36〕に記載の医薬。
〔38〕リウマチの予防及び/又は治療のための前記〔36〕に記載の医薬。
〔39〕前記〔1〕~〔35〕のいずれか1項に記載の化合物又は薬学的に許容されるその塩を有効成分として含むIRAK4阻害剤。
〔41〕リウマチの予防及び/又は治療のために使用される、前記〔1〕~〔35〕のいずれか1項に記載の化合物又は薬学的に許容されるその塩。
〔42〕哺乳動物におけるリウマチを予防及び/又は治療する方法であって、有効量の前記〔1〕~〔35〕のいずれか1項に記載の化合物又は薬学的に許容されるその塩を該哺乳動物に投与する工程を含む方法。
特に断らない限り、本明細書において、炭素原子を単に“C”で、水素原子を“H”で、酸素原子を“O”で、イオウ原子を“S”で、また窒素原子を“N”で表すことがある。またカルボニル基を単に“-C(O)-”で、カルボキシル基を“-COO-”で、スルフィニル基を“-S(O)-”で、スルホニル基を“-S(O)2-で、エーテル結合を“-O-”で、チオエーテル結合を“-S-”で表すことがある(この場合の“-”は結合を表す)。
「ハロゲン」は、フルオロ(-F)、クロロ(-Cl)、ブロモ(-Br)、又はヨード(-I)である。別の態様としては、-F又は-Clが例示される。さらに別の態様としては、-Fが例示される。「ハロゲノ」とは、同一又は異なった1~7個のハロゲンで置換されていることを意味する。別の態様としては、同一又は異なった1~5個のハロゲンで置換されていることを意味する。さらに別の態様としては、1~3個のハロゲンで置換されていることを意味する。さらに別の態様としては、1個のハロゲンで置換されていることを意味する。-Fで置換されていることが例示される。
「芳香環基」としては、芳香環から任意の1個の水素原子を除いてできる1価の基が挙げられる。単環から三環の芳香族環基であればよい。例えば、アリール又はヘテロアリールが例示される。
「飽和炭化水素環」としては、単環から三環の飽和炭化水素環であればよい。3-10員飽和炭化水素環が例示される。別の態様としては3-7員飽和炭化水素環が例示される。さらに別の態様としては5又は6員飽和炭化水素環が例示される。該飽和炭化水素環は架橋を有していてもよく、また、前記芳香環と縮合していてもよい。具体的には、シクロプロパン、シクロブタン、シクロペンタン、シクロヘキサン、又はアダマンタンが例示される。
「部分不飽和ヘテロ環基」は、飽和ヘテロ環基の一部が不飽和の基であればよい。具体的には、ジヒドロピラニル、ジヒドロフラニル、ジヒドロチオピラニル、ジヒドロチエニル、1,2-ジヒドロキノリル、又は1,2,3,4-テトラヒドロキノリルが例示される。






前記R1はG1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい。
前記G1群としては、-F、ヒドロキシ、シアノ、ハロゲノC1-6アルキル、C1-4アルコキシ、フェニル、5-6員ヘテロアリール、及び3-7員飽和環基からなる群が例示される。別の態様としては、-F、ヒドロキシ、ハロゲノC1-6アルキル、C1-4アルコキシ、5-6員ヘテロアリール、及び3-7員飽和環基からなるG11群が例示される。さらに別の態様としては、-F、ヒドロキシ、C1-4アルコキシ、5員ヘテロアリール、及び4-5員飽和環基からなるG12群が例示される。
前記G1群におけるフェニル又は5-6員ヘテロアリールはGAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよい。
前記GAr群としては、-F、-Cl、ヒドロキシ、シアノ、C1-6アルキル、ハロゲノC1-6アルキル、及び-NH2からなる群が例示される。別の態様としては、-F、-Cl、シアノ、C1-6アルキル、ハロゲノC1-6アルキルからなるGAr1群が例示される。
前記R2はG2群より選択される1~3個の同一又は異なった置換基で置換されていてもよい。
前記G2群としては、-F、ヒドロキシ、ハロゲノC1-3アルキル、及びC1-4アルコキシからなる群が例示される。別の態様としては、-F、ヒドロキシからなるG21群が例示される。





RCy1及びRCy2としては、各々独立して、-H、-F、ヒドロキシ、シアノ、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシ、-NR11R12、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基が例示される。別の態様としては、-H、-F、C1-6アルキル、C1-6アルコキシ、-NR11R12が例示される。さらに別の態様としては、-NR11R12が例示される。
前記RCy1とRCy2におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよい。
前記GAr群としては、前記GAr群の他、前記GAr1群の態様が例示される。
前記RCy1とRCy2は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい。
前記G1群としては、前記G1群の他、前記G11~G12群の態様が例示される。
前記R11及びR12は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい。
前記G1群としては、前記G1群の他、前記G11~G12群の態様が例示される。
前記4-10員飽和環としては、アゼチジン、ピロリジン、ピペリジン、モルホリン、オキサゼパン、ピペラジン、又はホモピペラジンが例示される。別の態様としては、ピペリジン、モルホリン、又はピペラジンが例示される。さらに別の態様としては、モルホリン、又はピペラジンが例示される。



前記G3群としては、-F、ヒドロキシ、C1-6アルキル、ハロゲノC1-6アルキル、ヒドロキシC1-6アルキル、C1-6アルコキシ、C1-6アルコキシ-C1-6アルキル、ハロゲノC1-6アルコキシ、-C(O)R14、-NR13C(O)R14、-C(O)NR13R14、-C(O)NH2、-NR13S(O2)R14、-S(O2)NR13R14、-S(O2)NH2、-S(O2)R14、フェニル、5-6員ヘテロアリール、及び3-7員飽和環基が例示される。別の態様としては、-F、C1-6アルキル、ハロゲノC1-6アルキル、-C(O)R14、-C(O)NR13R14が例示される。
前記GAr群としては、前記GAr群の他、前記GAr1群の態様が例示される。
前記R13はとしては、-H、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシC1-6アルキル、ハロゲノC1-6アルコキシC1-6アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基が例示される。別の態様としては、-H、C1-6アルキル、ハロゲノC1-6アルキルが例示される。さらに別の態様としては、-Hが例示される。
前記5-6員ヘテロアリールとしては、ピリジン、オキサゾール、イソオキサゾール、チアゾールが例示される。別の態様としては、ピリジンが例示される。
前記R13及びR14におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよい。
前記R13とR14は、一緒になって4-7員飽和環又は7-11員スピロ環を形成することもできる。前記4-7員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよい。
前記4-7員飽和環としては、アゼチジン、ピロリジン、ピペリジン、モルホリン、オキサゼパン、又はピペラジンが例示される。別の態様としては、アゼチジン、ピロリジンが例示される。さらに別の態様としては、アゼチジンが例示される。

RCy3としては、C1-4アルキル、ハロゲノC1-4アルキルが例示される。別の態様としては、メチル、エチル、イソプロピルが例示される。さらに別の態様としては、メチルが例示される。
前記R15としては、-H、C1-6アルキル、ハロゲノC1-6アルキル、ヒドロキシC1-6アルキル、C1-6アルコキシC1-6アルキル、-C(O)R16、-S(O2)R16、-C(O)NR16R17、-C(O)OR16、又は3-7員飽和環基が例示される。別の態様としては、ハロゲノC1-6アルキル、-C(O)R16、-S(O2)R16、又は-C(O)NR16R17が例示される。さらに別の態様としては、-C(O)R16が例示される。
前記5-6員ヘテロアリールとしては、ピリジン、オキサゾール、イソオキサゾール、チアゾールが例示される。別の態様としては、ピリジンが例示される。
前記R17としては、-H、C1-3アルキル、ハロゲノC1-3アルキル、C1-6アルコキシC1-6アルキル、ハロゲノC1-6アルコキシC1-3アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基が例示される。別の態様としては、-H、C1-6アルキル、ハロゲノC1-6アルキルが例示される。さらに別の態様としては、-Hが例示される。
前記R16とR17におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよい。
前記R16とR17は、一緒になって4-7員飽和環又は7-11員スピロ環を形成することもできる。前記4-7員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよい。
前記4-7員飽和環としては、アゼチジン、ピロリジン、ピペリジン、モルホリン、オキサゼパン、又はピペラジンが例示される。別の態様としては、アゼチジン、ピロリジンが例示される。さらに別の態様としては、アゼチジンが例示される。
前記R15は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい。
前記G1群としては、前記G1群の他、前記G11~G12群の態様が例示される。



前記RCy1とRCy2が一緒になって形成する4-7員飽和環としては、アゼチジン、ピロリジン、ピぺリジンが例示される。別の態様としては、ピぺリジンが例示される。
前記RCy1とRCy2が一緒になって形成される4-7員飽和環又は7-11員スピロ環は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい。
前記G1群としては、前記G1群の他、前記G11~G12群の態様が例示される。

すなわち、式(1)で示される化合物の塩としては、その種類は特に限定されず、酸付加塩又は塩基付加塩のいずれであってもよく、分子内対イオンの形態をとっていてもよい。特に医薬の有効成分とする際には、その塩としては薬学的に許容される塩が好ましい。本明細書において、医薬としての使用に関連して開示される場合には、式(1)で示される化合物の塩としては、薬学的に許容される塩であると通常は理解される。酸付加塩としては、例えば、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、硝酸、又はリン酸等の無機酸との酸付加塩、或いはギ酸、酢酸、プロピオン酸、シュウ酸、マロン酸、コハク酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、クエン酸、リンゴ酸、酒石酸、ジベンゾイル酒石酸、マンデル酸、マレイン酸、フマル酸、アスパラギン酸、又はグルタミン酸等の有機酸との酸付加塩が含まれる。塩基付加塩としては、例えば、ナトリウム、カリウム、マグネシウム、カルシウム、アルミニウム等の無機塩基との塩基付加塩、メチルアミン、2-アミノエタノール、アルギニン、リジン、又はオルニチン等の有機塩基との塩基付加塩などを例示することができる。もっとも、塩の種類はこれらに限定されることはなく、当業者が適宜選択可能であることは言うまでもない。
本発明の化合物は溶媒和物の形態を含む。また、本発明の化合物は無溶媒和物の形態も含む。
本発明の化合物は結晶の形態を含む。また、本発明の化合物は非晶質の形態も含む。
本発明の化合物は種々の放射性又は非放射性同位体でラベルされた形態も含む。
より具体的に記載すると、本発明の化合物は「式(1)で示される化合物」の無水物かつ無溶媒和物、又はその水和物及び/若しくは溶媒和物を含み、或いはさらにそれらの結晶を含む。
また、本発明の化合物は「式(1)で示される化合物の塩」の無水物かつ無溶媒和物、又はその塩の水和物及び/若しくは溶媒和物を含み、或いはさらにそれらの結晶を含む。
式(1)で表される化合物は、公知の方法、例えば以下に示す方法、これらに準ずる方法または実施例に示す方法に従って製造することができる。なお、以下の各製造方法における原料となる化合物は、商業的に入手可能であるか、例えば「Compendium of Organic Synthesis Methods, Vol. I-XII (Wiley-Interscience)」に記載された公知の方法を用いて製造することができる。

式(1)で示される化合物は、式(4)で示される化合物と、式(5)で示される化合物とのアミド化反応や、アシル化反応等により製造することができる。アミド化の縮合剤としては、1-プロパンホスホン酸無水物や、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド、HATUなどを用いることができる。アミド化の求核剤としては、HOBt、HOAtなどを用いることができる。塩基としては、ジイソプロピルエチルアミンなどを用いることができる。反応溶媒としては、例えば、DMF、ジクロロメタン、THFなどを用いることができる。反応温度は、通常、0℃から150℃で行うことができる。置換基Cy、R1、R2の置換基は、更なる変換、例えば、脱保護、還元、還元的アミノ化、アルキル化、フッ素化などを実施することで、式(1)で示される化合物を変換することができる。
式(5)で示される化合物は、式(7)で示される化合物との金属触媒を用いたカップリング反応により製造することができる。より具体的には、式(7)で示される化合物と、式(6)で示される試薬との鈴木-宮浦カップリング等によって製造することが出来る。反応触媒としては、例えば、Pd(dppf)Cl2やPdAmphos、Pd(PPh3)4などを用いることができる。塩基としては、炭酸セシウム、フッ化セシウム、炭酸ナトリウムなどを用いることができる。反応溶媒としては、THF、1,4―ジオキサン、DMF、アセトニトリルなどを用いることができる。反応温度は、通常、室温から180℃で行うことができる。
式(5)で示される化合物は、式(9)で示される化合物との金属触媒を用いたカップリング反応により製造することができる。より具体的には、式(9)で示される化合物と、式(8)で示される試薬との鈴木-宮浦カップリング等によって製造することが出来る。反応触媒としては、例えば、Pd(dppf)Cl2やPdAmphos、Pd(PPh3)4などを用いることができる。塩基としては、炭酸セシウム、フッ化セシウム、炭酸ナトリウムなどを用いることができる。反応溶媒としては、THF、1,4―ジオキサン、DMF、アセトニトリルなどを用いることができる。反応温度は、通常、室温から180℃で行うことができる。

式(1)で示される化合物は、式(10)で示される化合物との金属触媒を用いたカップリング反応により製造することができる。より具体的には、式(10)で示される化合物と、式(6)で示される試薬との鈴木-宮浦カップリング等によって製造することが出来る。反応触媒としては、例えば、Pd(dppf)Cl2やPdAmphos、Pd(PPh3)4などを用いることができる。塩基としては、炭酸セシウム、フッ化セシウム、炭酸ナトリウムなどを用いることができる。反応溶媒としては、THF、1,4―ジオキサン、DMF、アセトニトリルなどを用いることができる。反応温度は、通常、室温から180℃で行うことができる。置換基Cy、R1、R2の置換基は、更なる変換、例えば、脱保護、還元、還元的アミノ化、アルキル化、フッ素化などを実施することで、式(1)で示される化合物を変換することができる。
式(10)で示される化合物は、式(4)で示される化合物と、式(7)で示される化合物とのアミド化反応や、アシル化反応等により製造することができる。アミド化の縮合剤としては、1-プロパンホスホン酸無水物や、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド、HATUなどを用いることができる。アミド化の求核剤としては、HOBt、HOAtなどを用いることができる。塩基としては、ジイソプロピルエチルアミンなどを用いることができる。反応溶媒としては、例えば、DMF、ジクロロメタン、THFなどを用いることができる。反応温度は、通常、0℃から150℃で行うことができる。
式(1)で示される化合物は、式(11)で示される化合物との金属触媒を用いたカップリング反応により製造することができる。より具体的には、式(11)で示される化合物と、式(8)で示される試薬との鈴木-宮浦カップリング等によって製造することが出来る。反応触媒としては、例えば、Pd(dppf)Cl2やPdAmphos、Pd(PPh3)4などを用いることができる。塩基としては、炭酸セシウム、フッ化セシウム、炭酸ナトリウムなどを用いることができる。反応溶媒としては、THF、1,4―ジオキサン、DMF、アセトニトリルなどを用いることができる。反応温度は、通常、室温から180℃で行うことができる。
式(9)で示される化合物は、例えば、式(10)で示される化合物との金属触媒を用いたカップリング反応、ハロゲン―メタル交換反応により製造することができる。より具体的には、式(10)で示される化合物と、ビス(ピナコラド)ジボロンなどとの鈴木-宮浦カップリング等によって製造することが出来る。反応触媒としては、例えば、Pd(dppf)Cl2などを用いることができる。塩基としては、酢酸カリウムなどを用いることができる。反応溶媒としては、1,4―ジオキサンなどを用いることができる。反応温度は、通常、40℃から150℃で行うことができる。

式(10)で示される化合物は、式(14)で示される化合物の還元的アミノ化反応により製造することができる。例えば、R11R12NHの存在下でイミンを形成し、続くナトリウムトリアセトキシボロヒドリドを用いたイミンの還元反応によって製造することができる。反応溶媒としては、THF、ジクロロメタンを用いることができる。反応温度は、通常、0℃から80℃で行うことができる。置換基R11R12、R1、R2の置換基は、更なる変換、例えば、脱保護、還元、アルキル化、フッ素化などを実施することで、式(12)で示される化合物を変換することができる。
式(14)で示される化合物は、式(5)で示される化合物と、式(15)で示される化合物とのアミド化反応や、アシル化反応等により製造することができる。アミド化の縮合剤としては、1-プロパンホスホン酸無水物や、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド、HATUなどを用いることができる。アミド化の求核剤としては、HOBt、HOAtなどを用いることができる。塩基としては、ジイソプロピルエチルアミンなどを用いることができる。反応溶媒としては、例えば、DMF、ジクロロメタン、THFなどを用いることができる。反応温度は、通常、0℃から150℃で行うことができる。

式(16)で示される化合物は、式(18)で示される化合物との、アミド化、アシル化、アルキル化、ウレア化などのC-N結合形成反応により製造することができる。より具体的には、式(18)で示される化合物と、式(17)で示される試薬とのアミド化反応等によって製造することが出来る。アミド化の縮合剤としては、1-プロパンホスホン酸無水物や、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド、HATUなどを用いることができる。アミド化の求核剤としては、HOBt、HOAtなどを用いることができる。塩基としては、ジイソプロピルエチルアミンなどを用いることができる。反応溶媒としては、例えば、DMF、ジクロロメタン、THFなどを用いることができる。反応温度は、通常、0℃から150℃で行うことができる。置換基R15、R1、R2の置換基は、更なる変換、例えば、脱保護、還元、還元的アミノ化、アルキル化、フッ素化などを実施することで、式(16)で示される化合物を変換することができる。
式(18)で示される化合物は、式(19)で示される化合物を脱保護することにより製造することができる。脱保護反応は、公知の方法、例えばGreene’s Protective Groups in Organic Synthesis、John Wiley and Sons 刊(2014年版)に記載の方法などに準じて行えばよい。
式(19)で示される化合物は、式(14)で示される化合物の還元的アミノ化反応により製造することができる。例えば、式(20)で示される化合物の存在下でイミンを形成し、続くナトリウムトリアセトキシボロヒドリドを用いたイミンの還元反応によって製造することができる。反応溶媒としては、THF、ジクロロメタンを用いることができる。反応温度は、通常、0℃から80℃で行うことができる。

式(21)で示される化合物は、式(19)で示される化合物を脱保護することにより製造することができる。脱保護反応は、公知の方法、例えばGreene’s Protective Groups in Organic Synthesis、John Wiley and Sons 刊(2014年版)に記載の方法などに準じて行えばよい。置換基Ralの置換基は、更なる変換、例えば、脱保護、還元、還元的アミノ化、アルキル化、フッ素化などを実施することで、式(21)で示される化合物を変換することができる。
式(22)で示される化合物は、式(24)で示される化合物と、式(23)で示される化合物との、アルキル化、光延反応、などのC-O結合形成反応により製造することができる。より具体的には、式(24)で示される化合物と、式(23)で示される試薬とのアルキル化反応等によって製造することが出来る。アルキル化剤としては、ヨードメタンなどを用いることができる。塩基としては、ジイソプロピルエチルアミン、炭酸カリウムなどを用いることができる。反応溶媒としては、例えば、DMF、ジクロロメタン、THFなどを用いることができる。反応温度は、通常、-78℃から150℃で行うことができる。
式(24)で示される化合物は、式(25)で示される化合物を保護することにより製造することができる。保護反応は、公知の方法、例えばGreene’s Protective Groups in Organic Synthesis、John Wiley and Sons 刊(2014年版)に記載の方法などに準じて行えばよい。
式(25)で示される化合物は、式(26)で示される化合物を脱アシル化することにより製造することができる。より具体的には、式(26)で示される化合物を酸存在下、加水分解することにより製造できる。酸としては、塩酸などを用いることができる。反応溶媒としては、メタノール、水、THFなどを用いることができる。反応温度は、通常室温から100℃で行うことができる。
式(26)で示される化合物は、式(27)で示される化合物を、塩基存在下で芳香化することで製造できる。塩基としては、DBUなどを用いることができる。反応溶媒としては、THFなどを用いることができる。反応温度は、通常-20℃から80℃で行うことができる。
式(27)で示される化合物は、式(28)で示される化合物を、塩基存在下で臭素化することで製造できる。臭素化剤としては、臭素、NBSなどを用いることができる。酸としては、臭化水素酸などを用いることができる。反応溶媒としては、酢酸などを用いることができる。反応温度は、通常室温から60℃で行うことができる。
式(28)で示される化合物は、式(30)で示される化合物と、式(29)で示される化合物とのアミド化反応や、アシル化反応等により製造することができる。アシル化剤としては、無水酢酸のような酸無水物などを用いることができる。アミド化の縮合剤としては、1-プロパンホスホン酸無水物や、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド、HATUなどを用いることができる。アミド化の求核剤としては、HOBt、HOAtなどを用いることができる。塩基としては、ジイソプロピルエチルアミンなどを用いることができる。反応溶媒としては、例えば、DMF、ジクロロメタン、THFなどを用いることができる。反応温度は、通常、0℃から150℃で行うことができる。

式(31)で示される化合物は、式(32)で示される化合物と、式(23)で示される化合物との、アルキル化、光延反応、などのC-O結合形成反応により製造することができる。より具体的には、式(32)で示される化合物と、式(23)中、YCOが水酸基で示される試薬との光延反応等によって製造することが出来る。光延反応の試薬としては、ジエチルアゾカルボキシレート、トリフェニルホスフィンなどを用いることができる。反応溶媒としては、THF、トルエンなどを用いることができる。反応温度は、通常、0℃から110℃で行うことができる。

式(33)で示される化合物は、式(34)で示される化合物を脱保護することにより製造することができる。脱保護反応は、公知の方法、例えばGreene’s Protective Groups in Organic Synthesis、John Wiley and Sons 刊(2014年版)に記載の方法などに準じて行えばよい。置換基Ralの置換基は、更なる変換、例えば、脱保護、還元、還元的アミノ化、アルキル化、フッ素化などを実施することで、式(33)で示される化合物を変換することができる。
式(34)で示される化合物は、式(35)で示される化合物と、式(23)で示される化合物との、アルキル化、光延反応、などのC-O結合形成反応により製造することができる。より具体的には、式(35)で示される化合物と、式(23)中、YCOが-Br、-Iなどのハロゲン基で示される試薬とのアルキル化反応等によって製造することが出来る。塩基としては、ジイソプロピルエチルアミン、炭酸セシウムなどを用いることができる。反応溶媒としては、例えば、DMF、ジクロロメタン、THFなどを用いることができる。反応温度は、通常、0℃から150℃で行うことができる。

式(36)で示される化合物は、式(37)で示される化合物の環化反応により製造することができる。塩基としては、炭酸カリウム、DBUなどを用いることができる。反応溶媒としては、DMFなどを用いることができる。反応温度は、通常0℃から180℃で行うことができる。置換基R1の置換基は、更なる変換、例えば、脱保護、還元、還元的アミノ化、アルキル化、フッ素化などを実施することで、式(36)で示される化合物を変換することができる。
式(37)で示される化合物は、式(38)で示される化合物とO-(メシチレンスルホニル)ヒドロキシルアミンと反応させることにより製造することができる。反応溶媒としては、クロロホルム、ジクロロメタンを用いることができる。反応温度は、通常、0℃から50℃で行うことができる。
式(38)で示される化合物は、式(39)で示される化合物をシアノ化することにより製造することができる。シアノ化剤としては、シアン化ナトリウム、シアン化カリウムなどを用いることができる。反応溶媒としては、エタノール、水、DMFなどを用いることができる。反応温度は、通常、0℃から120℃で行うことができる。
式(39)で示される化合物は、式(40)で示される化合物を臭素化することにより製造することができる。臭素化剤としては、N-ブロモスクシンイミド、臭素などを用いることができる。反応溶媒としては、エタノール、水、DMFなどを用いることができる。反応温度は、通常、0℃から120℃で行うことができる。

式(41)で示される化合物は、式(42)で示される化合物を、イリジウム触媒を用いてC-Hボリル化することで製造することができる。ボラン源としては、例えばビス(ピナコラト)ジボロンを用いることができる。反応溶媒としては、THFを用いることができる。反応温度は、通常、20℃から80℃で行うことができる。
式(42)で示される化合物は、式(44)で示される化合物と、式(43)で示される化合物とを、塩基性条件下、エーテル化することにより製造することができる。塩基としては、例えば、水素化ナトリウム又は水酸化ナトリウムを用いることが出来る。触媒としては、例えば、テトラブチルアンモニウムクロライドを用いることができる。反応溶媒としては、THF、DMF、ジクロロメタンを用いることができる。反応温度は、通常、0℃から150℃で行うことができる。
式(42)で示される化合物は、式(46)で示される化合物と、式(45)で示される化合物とを、塩基性条件下、エーテル化することにより製造することができる。塩基としては、例えば、水素化ナトリウム又は水酸化ナトリウムを用いることが出来る。触媒としては、例えば、テトラブチルアンモニウムクロライドを用いることができる。反応溶媒としては、THF、DMF、ジクロロメタンを用いることができる。反応温度は、通常、0℃から150℃で行うことができる。
式(46)で示される化合物は、式(44)で示される化合物を、クロロ化を含むハロゲン化を行うことにより製造することができる。クロロ化剤としては、例えば、塩化チオニルを用いることができる。反応溶媒としては、ジクロロメタンを用いることができる。反応温度は、通常、0℃から40℃で行うことができる。

式(6)で示される化合物は、例えば、式(8)で示される化合物との金属触媒を用いたカップリング反応、ハロゲン―メタル交換反応により製造することができる。より具体的には、式(6)で示される化合物と、ビス(ピナコラド)ジボロンなどとの鈴木-宮浦カップリング等によって製造することが出来る。反応触媒としては、例えば、Pd(dppf)Cl2などを用いることができる。塩基としては、酢酸カリウムなどを用いることができる。反応溶媒としては、1,4―ジオキサンなどを用いることができる。反応温度は、通常、40℃から150℃で行うことができる。
式(8)で示される化合物は、式(47)で示される化合物と、式(44)で示される化合物とを、塩基性条件下、エーテル化することにより製造することができる。塩基としては、例えば、水素化ナトリウム又は水酸化ナトリウムを用いることが出来る。触媒としては、例えば、テトラブチルアンモニウムクロライドを用いることができる。反応溶媒としては、THF、DMF、ジクロロメタンを用いることができる。反応温度は、通常、0℃から150℃で行うことができる。
式(8)で示される化合物は、式(48)で示される化合物と、式(45)で示される化合物とを、塩基性条件下、エーテル化することにより製造することができる。塩基としては、例えば、水素化ナトリウム又は水酸化ナトリウムを用いることが出来る。触媒としては、例えば、テトラブチルアンモニウムクロライドを用いることができる。反応溶媒としては、THF、DMF、ジクロロメタンを用いることができる。反応温度は、通常、0℃から150℃で行うことができる。
式(48)で示される化合物は、式(47)で示される化合物を、クロロ化を含むハロゲン化を行うことにより製造することができる。クロロ化剤としては、例えば、塩化チオニルを用いることができる。反応溶媒としては、ジクロロメタンを用いることができる。反応温度は、通常、0℃から40℃で行うことができる。
本発明に記載の種々の化合物は、公知の方法、例えば、各種クロマトグラフィー(カラム、フラッシュカラム、薄層、高速液体、超臨界流体)により精製を行うことができる。
IRAK-4阻害に関連する疾患は、IRAK-4阻害により奏功する疾患であれば特に限定されないが、具体的には、例えば急性又は慢性炎症、自己免疫疾患(関節リウマチ、、全身性エリテマトーデス、ループス腎炎等)、自己炎症性疾患(TNF受容体関連周期性症候群(TRAPS) 、家族性地中海熱、クリオピリン関連周期性発熱症候群、高IgD症候群等)、代謝性疾患(痛風等)などが例示される。
本発明の医薬のある態様が、IRAK-4シグナルが関与する疾患の予防及び/又は治療に有用であることは、例えば免疫細胞を用いたサイトカイン産生抑制試験、または、コラーゲン誘発関節炎モデルにより確認することができる。具体的には後述する試験例3に記載の方法が例示される。
本発明の化合物のある態様が医薬の有効成分として有用であることは、例えば反応性代謝物生成確認試験を行うことにより確認できる。反応性代謝物生成確認試験としては、例えばヒト肝ミクロゾームと化合物をNADPHおよびダンシル基で蛍光標識したグルタチオン(dGSH)存在下でインキュベートすることで反応性代謝物をdGSH付加体としてトラップし、蛍光強度を指標にdGSH付加体の生成量から反応性代謝物に起因するピークを網羅的に検出する方法(Junping Gan. et al., Chem. Res. Toxicol. 2005, 18, 896-903)や、化合物の14C標識体をヒト肝ミクロゾームとともにNADPH存在下でインキュベートし、タンパク質に共有結合した放射能を測定する方法(Baillie T.A.,Drug Metabolizing Enzymes. Cytochrome P450 and Other Enzymes in Drug Discovery and Development, pp.147-154, 2003)などが挙げられる。これらのいずれか1つ又は2つ以上の方法を用いて、化合物の反応性代謝物生成によって生じる特異体質性薬物毒性の発生に対するリスクを明らかにすることにより、医薬の有効成分としての有用性を確認できる。
購入したすべての試薬は、更に精製することなく使用した。購入した無水溶媒は、更なる乾燥を行う事なく使用した。カラムクロマトグラフィーには、山善製中圧分取精製システムSmartFlash、又はBIOTAGE社製中圧分取精製システムIsolera ONEにMS検出器としてBIOTAGE Daltonを接続して用いた。カラムは、Biotage社製SNAP Ultra、またはYMC社製DispoPack ATのいずれかを用いた。いくつかの場合には、イオン交換樹脂としてAgilent社製BondElute SCXを用いて精製した。なお、BondElute SCXを以下SCXと略すことがある。SCXの使用例としては、メタノールおよびジクロロメタンでカートリッジを洗浄後、粗生成物を最小容量の溶媒(たとえばジクロロメタン-メタノールの混合溶媒など)に溶かして吸着させる。その後不純物を、圧力をかけながらメタノールで洗い流し、生成物を2.0Mアンモニア-メタノールで溶出させる方法が挙げられる。薄層ク口マトグラフィー (TLC) はPrecoated silica gel 60 F254(メルク社製、製品番号5715-1M) を使用した。クロロホルム:メタノール(1:0~1:1)、又は、酢酸エチル:ヘキサン(1:0~0:1)により展開後、UV(254nm又は又は365nm)照射、ヨウ素溶液、過マンガン酸カリウム水溶液、リンモリブデン酸(エタノール溶液)等による呈色により確認した。分取薄層クロマトグラフィー(以下、PTLC と呼ぶことがある。)はPLCプレートsilica gel 60 F254、20×20cm、層厚2mm、濃縮ゾーン(4cm)付(メルク社製、製品番号13793-1M)を試料の量に応じて1枚又は数枚使用して行った。有機溶媒の乾燥には無水硫酸マグネシウムあるいは無水硫酸ナトリウムを使用した。
1H(400MHx)核磁気共鳴装置(以下NMRと略すことがある)分析は、Bruker社製AVANCE III HD-400MHz又は、Bruker社製AVANCE III HD-600MHzを用いた。
内部標準は使用した溶媒または添加物の既知の値を使用した。1H NMRデータは化学シフト、parts per million(以下ppmと略す)、積分値(例えば1Hと記す)、多重項(s、シングレット;d、ダブレット;t、トリプレット;q、カルテット;qui、クインテット;m、マルチプレット;br、ブロード;dd、ダブルダブレット、など)を記した。

1H-NMR(CDCl3):δ(ppm)8.78(2H,d,J=4.9Hz),7.26(1H,t,J=4.9Hz),4.76(2H,s).


LCMS(LC-1);RT=2.10,m/z309[M+H]+
1H-NMR(CDCl3):δ(ppm)8.75(2H,d,J=4.9Hz),7.20(1H,t,J=4.9Hz),4.78(2H,d、7.3Hz)、4.26-4.23(1H,m), 3.86-3.83(1H,m)2.03-1.87(2H,m),1.75-1.67(3H,m),1.56-1.49(1H,m),0.86(9H,s),0.05(6H,s).
光学異性体は、次に示すキラルHPLCの条件で、分離、分析を行なうことで、各光学活性体を光学純度99.7%ee以上で得た。
分取条件;カラム:CHIRALART CelluLose-SC(10um)245x150mm I.D.、溶出液:ヘプタン/2-プロパノール(80/20)(v/v)、流速:518mL/min、温度:24℃、検出:UV(245nm)、ロード:180mL(9g)
分析条件;カラム:CHIRALART CelluLose-SC(5um)250x4.6mm I.D.、溶出液 ヘプタン/2-プロパノール(80/20)(v/v)、流速:0.5mL/min、温度:25℃、検出:UV(245nm)、インジェクション:10μL(0.5mg/mL)RT=11.6(1S,2S体;中間体A-6-2-a)、RT=16.5(1R,2R体;中間体A-6-2-b)

LCMS(LC-1);RT=1.80,m/z353[M+H]+(ボロン酸として検出)
1H-NMR(CDCl3):δ(ppm)9.00(2H,s), 4.78(2H,d、6.2Hz)、4.25-4.22(1H,m), 3.84-3.81(1H,m)1.97-1.87(2H,m),1.72-1.65(3H,m),1.55-1.48(1H,m),1.35(12H,s),0.85(9H,s),0.05(6H,s).

LCMS(LC-1);RT=2.07,m/z457[M+H]+
1H-NMR(CDCl3):δ(ppm)8.95(2H,s), 7.79(1H,J=2.0),7.76(1H,J=8.4),7.51(1H,J=8.4,2.0),7.26(2H,s),5.33(2H,s),4.83-4.79(2H,m), 4.29-4.26(1H,m),3.90-3.87(1H,m),2.05-1.89(2H,m),1.78-1.68(3H,m),1.58-1.50(1H,m).0.88(9H,s),0.07(6H,s).

LCMS(LC-1);RT=2.27,m/z525[M+H]+

LCMS(LC-1);RT=1.13,m/z411[M+H]+
1H-NMR(DMSOd6):δ(ppm)9.17(2H,s),8.45(1H,m),7.86(2H,m),4.71-4.69(3H,m),4.04-4.00(1H,m),3.83-3.80(1H,m),2.03-1.40(7H,m),0.98-0.96(4H,m).



1H-NMR(CDCl3):δ(ppm)8.76(2H,d,J=4.9Hz),7.21(1H,dd,J=4.9Hz),4.75(2H,s),3.60(2H,t,J=6.9Hz),1.73(2H,tt,J=6.9,7.4Hz),0.96(3H,t,J=7.4Hz).

LCMS(LC-1);RT=0.62,m/z197[M+H]+(ボロン酸として検出)

LCMS(LC-1);RT=1.58,m/z297[M+H]
1H-NMR(CDCl3):δ(ppm)7.95(1H,m),7.68(1H,m),7.62(2H,d,J=8.5Hz),7.53(2H,dd,J=8.5,2.0Hz),1.98-1.91(1H,m),1.27-1.23(4H,m).

LCMS(LC-1);RT=1.37,m/z369[M+H]+
1H-NMR(DMSOd6):δ(ppm)9.17(2H,s),8.38(1H,m),7.83-7.77(2H,m),4.65(2H,s),3.52(2H,J=6.7Hz),1.97-1.91(1H,m),1.62-1.53(2H,m),0.93-0.88(7H,m).

LCMS(LC-1);RT=2.25,m/z424[M+H]+
1H-NMR(CDCl3):δ(ppm)8.79(2H,s),4.77(2H,d,J=2.1Hz)4.36-4.31(1H,m),4.00-3.96(1H,m),2.62-2.44(2H,m),2.32-2.19(1H,m),2.13-2.00(1H,m),0.87(9H,s),0.07(6H,s).

LCMS(LC-1);RT=1.73,m/z346[M+2H]+
1H-NMR(CDCl3):δ(ppm)8.28(2H,s),7.86(1H,d,J=8.0Hz),7.71(1H,d,J=8.0Hz),1.80-1.74(1H,m),1.37(12H,s),1.26(4H,s).

LCMS(LC-1);RT=2.14,m/z561[M+H]+
1H-NMR(CDCl3):δ(ppm)9.95(1H,brs),9.00(2H,s),8.02(1H,d,J=2.0Hz),7.89(1H,d,J=8.4Hz),7.63(1H,dd,J=8.4,2.0Hz),4.92-4.84(2H,m),4.40-4.36(1H,m),4.06-4.02(1H,m),2.65-2.47(2H,m),2.37-2.25(1H,m),2.15-2.03(1H,m),1.73-1.66(1H,m),1.30-1.26(2H,m),1.08-1.04(2H,m),0.88(9H,s),0.09(6H,d,J=1.6Hz).

LCMS(LC-1);RT=1.23,m/z447[M+H]+
1H-NMR(DMSOd6):δ(ppm)12.76(1H,brs),9.19(2H,s),8.46(1H,m),7.89-7.85(2H,m),5.36-5.35(1H,m),4.76(2H,s),4.19(1H,m),4.03(1H,m),2.62-2.40(2H,m),2.26-2.14(1H,m),2.08-1.97(2H,m),0.99-0.97(4H,m).

LCMS(LC-1);RT=1.41,m/z258[M+H]+
1H-NMR(CDCl3):δ(ppm)8.79(2H,s),4.67(2H,s),4.15-4.09(1H,m),1.88-1.68(6H,m),1.58-1.50(2H,m).


LCMS(LC-1);RT=1.52,m/z395[M+H]+
1H-NMR(CDCl3):δ(ppm)9.00(2H,s),8.00(2H,s),7.89(2H,d,J=8.0Hz),7.63(2H,d,J=8.0Hz),4.79(2H,s),4.25-4.22(1H,m),4.21-4.13(1H,m),1.84-1.74(4H,m),1.72-1.70(1H,m),1.29-1.25(2H,m),1.09-1.05(2H,m).


LCMS(LC-1);RT=1.03,m/z273[M+H]+
1H-NMR(CDCl3):δ(ppm)8.79(2H,s),4.84-4.75(2H,m),4.19-4.13(1H,m),3.92-3.86(1H,m),3.80-3.75(1H,m),3.70-3.61(2H,m),2.03-1.95(1H,m),1.93-1.84(2H,m),1.70-1.61(1H,m).

LCMS(LC-1);RT=1.20,m/z411[M+H]+
1H-NMR(DMSOd6):δ(ppm)12.76(1H,s),9.18(2H,s),8.47(1H,s),7.90-7.85(2H,m),4.71(2H,s),4.03-3.97(1H,m),3.77-3.71(1H,m),3.65-3.60(1H,m),3.56(2H,d,J=5.3Hz),2.06-1.99(1H,m),1.95-1.74(3H,m),1.65-1.55(1H,m),0.99-0.97(4H,m).

LCMS(LC-1);RT=2.18,m/z553[M+H]+
1H-NMR(CDCl3):δ(ppm)10.4(1H,brs)9.02(2H,s),8.06(1H,d、J=1.5Hz),7.88(1H,d,J=8.0Hz),7.69-7.65(1H,m),7.59-7.55(1H,m),7.64-7.55(1H,m),4.98-4.74(2H,m),4.35-4.18(1H,m),3.98-3.80(1H,m),3.71-3.59(2H,m),3.42-3.28(3H,m),2.11-1.84(2H,m),1.80-1.71(3H,m),1.61-1.37(1H,m),0.88(9H,s),0.08(3H,s),0.07(3H,s).

LCMS(LC-1);RT=2.15,m/z624[M+H]+

LCMS(LC-1);RT=1.01,m/z510[M+H]+
1H-NMR(CDCl3):δ(ppm)9.00(2H,s),8.00(1H,d,J=1.5Hz),7.93-7.87(1H,m),7.65-7.58(1H,m),5.09-4.77(3H,m),4.30-4.19(1H,m),4.07-3.98(3H,m),3.97-3.91(1H,m),3.91-3.84(1H,m),3.30-3.15(2H,m),3.00-2.89(1H,m),2.76-2.63(2H,m),2.35-2.22(2H,m),2.14-1.99(2H,m),1.77-1.69(2H,m),1.29-1.23(2H,m).


LCMS(LC-1);RT=0.89,m/z343[M+H]+
1H-NMR(CDCl3):δ(ppm)9.91-9.65(2H,brs),9.12(2H,s),8.34(1H,d,J=2.0Hz),8.12(1H,d,J=2.0Hz),7.86(1H,dd,J=8.5,2.0Hz),7.65(1H,d,J=8.5Hz),7.60-7.50(1H,m),7.50-7.32(1H,m),4.70-4.69(2H,m),4.02(2H,m),3.99-3.72(3H,m),1.97-1.71(3H,m),1.68-1.52(5H,m),1.52-1.34(2H,m).

LCMS(LC-1);RT=1.03,m/z439[M+H]+
1H-NMR(CDCl3):δ(ppm)9.00(2H,s),8.06-8.00(1H,m),7.89-7.77(1H,m),7.66-7.56(1H,m),4.97(1H,d,J=14.4Hz),4.80(1H,d,J=14.4Hz),4.31-4.11(1H,m),3.92-3.83(1H,m),3.70-3.56(2H,m),3.52-3.30(3H,m),2.17-1.99(2H,m),1.81-1.50(4H,m).

LCMS(LC-1);RT=1.06,m/z512[M+H]+
1H-NMR(CDCl3):δ(ppm)9.00(2H,s),8.04-7.92(1H,m),7.83(1H,brd,J=7.1Hz),7.63-7.53(1H,m),5.53-5.18(2H,m),5.10-4.79(2H,m),4.30-4.20(1H,m),3.93-3.82(1H,m),3.28-3.14(3H,m),2.78(5H,brs),2.35-2.23(3H,m),2.16-1.99(3H,m),1.62-1.48(2H,m).











LCMS(LC-1);RT=1.31,m/z520[M+H]+
1H-NMR(CDCl3):δ(ppm)9.00(2H,s),8.06-7.99(1H,m),7.91-7.84(1H,m),7.67-7.58(1H,m),5.01-4.90(1H,m),4.84-4.75(1H,m),4.26-4.15(1H,m),3.90-3.79(1H,m),3.18-3.08(1H,m),3.08-2.97(1H,m),2.52-2.34(5H,m),2.17(3H,s),2.14-2.00(4H,m),1.78-1.50(5H,m).


LCMS(LC-1);RT=0.92,m/z238[M+H]+
1H-NMR(CDCl3):δ(ppm)8.73(2H,d,J=5.0Hz),7.19(1H,dd,J=5.0,5.0Hz),4.85-4.82(2H,m),4.36-4.24(1H、m),4.24-4.16(1H,m),4.19(1H,dd,J=4.0,4.0Hz,),2.12-1.95(2H,m),1.85-1.69(4H,m),1.75(3H,s).

LCMS(LC-1);RT=0.78,m/z281[M+H]+(ボロン酸として検出)
1H-NMR(CDCl3):δ(ppm)9.01(2H,s),4.89-4.77(2H,m),4.06-4.02(1H,m),2.02-2.01(3H,s),1.88-1.83(6H,m),1.36-1.35(12H,s).

LCMS(LC-1);RT=1.16,m/z385[M+H]+
1H-NMR(CDCl3):δ(ppm)8.96(2H,s),7.84-7.75(1H,m),7.70-7.62(1H,m),7.57-7.45(1H,m),5.34-5.24(2H,m),5.24-5.12(1H,m),4.94-4.80(2H,m),4.13-4.06(1H,m),2.23-2.11(1H,m),2.09-2.00(1H,m),2.04(3H,s),1.89-1.63(4H,m).

LCMS(LC-1);RT=1.36,m/z481[M+H]+
1H-NMR(CDCl3):δ(ppm)9.01(2H,s),8.04(1H,m),7.86(1H,m),7.68-7.61(1H,m),5.23-5.15(1H,m),4.89(2H,m),4.13-4.06(1H,m),3.69-3.58(1H,m),3.54-3.43(1H,m),3.43-3.31(2H,m),2.24-2.13(1H,m),2.06-2.02(3H,m),1.90-1.60(5H,m).

LCMS(LC-1);RT=1.69,m/z666[M+H]+

LCMS(LC-1);RT=1.44,m/z623[M+H]+

LCMS(LC-1);RT=0.88,m/z524[M+H]+
1H-NMR(CDCl3):δ(ppm)9.18(2H,s),8.52-8.45(1H,m),7.68-7.63(1H,m),7.48-7.34(1H,m),4.72-4.67(2H,m),4.06-3.98(1H,m),3.89-3.77(1H,m),3.13-3.01(4H,m),2.97-2.87(1H,m),2.87-2.78(1H,m),2.72-2.61(1H,m),2.43-2.22(3H,m),2.23-2.05(2H,m),1.94-1.75(2H,m),1.68-1.53(3H,m),1.52-1.38(1H,m),1.07(3H,d,J=5.5Hz).

LCMS(LC-1);RT=1.08,m/z642[M+H]+
1H-NMR(CD3OD):δ(ppm)9.01(s,2H),8.62-8.46(1H,m),8.10-8.00(1H,m),7.95-7.81(1H,m),7.71-7.57(1H,m),7.22-7.14(1H,m),7.14-7.05(1H,m),5.02-4.90(1H,m),4.90-4.71(1H,m),4.28-4.07(1H,m),3.99-3.81(2H,m),3.81-3.67(1H,m),3.57-3.30(2H,m),3.29-3.09(2H,m),3.09-2.94(1H,m),2.84-2.73(1H,m),2.73-2.63(2H,m),2.63-2.58(3H,m),2.49-2.32(3H,m),2.33-2.21(1H,m),2.18-1.96(2H,m),1.81-1.51(4H,m),1.18-1.09(1H,m),0.99-0.91(1H,m).







LCMS(LC-1);RT=0.99,m/z565[M+H]+
1H-NMR(CDCl3):δ(ppm)9.00-8.95(2H,m),8.02-7.98(1H,m),7.86-7.80(1H,m),7.63-7.57(1H,m),4.95-4.88(1H,m),4.81-4.71(1H,m),4.21-4.11(1H,m),3.85-3.78(1H,m),3.77-3.70(1H,m),3.59-3.52(1H,m),3.40-3.36(1H,m),3.35-3.25(1H,m),3.24-3.07(2H,m),3.06-2.96(1H,m),2.82-2.71(1H,m),2.69-2.57(2H,m),2.08(4H,m),2.03-1.96(1H,m),1.77-1.49(5H,m),1.06-0.99(3H,m).


LCMS(LC-1);RT=1.68,m/z665[M+H]+
1H-NMR(CDCl3):δ(ppm)9.00(s,2H),7.99(brs,1H),7.87(m,1H),7.61(brd,J=7.5Hz,2H),7.55-7.27(m,2H),7.00(s,2H),5.36-5.16(m,2H),4.98-4.77(m,2H),4.10(m,2H),3.82-3.31(m,7H),3.30-2.93(m,4H),2.67-2.48(m,3H),2.39-2.25(m,2H),2.13(s,2H),2.04(s,3H),1.82(m,3H),1.54-1.45(m,9H),1.00-0.90(m,3H).

LCMS(LC-1);RT=1.12,m/z565[M+H]+
1H-NMR(CDCl3):δ(ppm)9.00(s,2H),8.01(d,J=2.0Hz,1H),7.89(d,J=8.5Hz,1H),7.62(dd,J=8.5,2.0Hz,1H),5.20(d,J=6.5Hz,1H),4.98-4.77(m,2H),4.10(d,J=6.5Hz,1H),3.43-3.16(m,3H),3.15-3.00(m,2H),2.97(brs,1H),2.92-2.70(m,2H),2.68-2.44(m,3H),2.44-2.30(m,2H),2.22-1.98(m,5H),1.88-1.60(m,4H),1.43-1.19(m,1H),1.12(d,J=6.5Hz,3H).

LCMS(LC-1);RT=1.02,m/z594[M+H]+
1H-NMR(CDCl3):δ(ppm)8.98-8.95(2H,m),8.02-7.94(1H,m),7.86-7.78(1H,m),7.62-7.56(1H,m),4.94-4.86(1H,m),4.78-4.69(1H,m),4.20-4.09(1H,m),3.86-3.76(1H,m),3.50-3.40(2H,m),3.39-3.32(2H,m),3.25-3.17(2H,m),3.17-3.08(1H,m),3.07-2.95(2H,m),2.74-2.65(1H,m),2.53-2.45(1H,m),2.45-2.29(3H,m),2.29-2.19(1H,m),2.11-1.95(2H,m),1.75-1.49(4H,m),1.01(3H,d,J=6.6Hz).


LCMS(LC-1);RT=1.33,m/z605[M+H]+
1H-NMR(DMSOd6):δ(ppm)9.14(2H,s),8.29(1H,m),7.75-7.65(2H,m),4.71-4.67(3H,m),4.02(1H,brm),3.83-3.80(1H,m),3.17-3.07(2H,m),2.95-2.82(2H,m),2.67-2.62(3H,m),2.30-2.23(2H,m),2.54-2.43(2H,m),2.19-2.02(4H,m),1.92-1.76(2H,m),1.65-1.55(3H,m),1.47-1.40(1H,m),0.95(3H,d,J=6.4Hz).

1H-NMR(CDCl3):δ(ppm)4.18-4.16(1H,m),3.79-3.76(1H,m),2.99-2.92(1H,m),2.86-2.83(1H,m),2.71-2.68(1H,m),2.36-2.33(1H,m),2.19-2.12(1H,m),1.57-1.52(1H,m),1.46(9H,s),1.13(3H,d,J=6.7Hz),0.44-0.28(4H,m).

1H-NMR(CD3OD):δ(ppm)3.90-3.77(3H,m),3.74-3.70(1H,m),3.61-3.47(2H,m),3.36-3.30(1H,m),2.98-2.92(1H,m),1.44(3H,d、J=6.5Hz),1.23-1.19(2H,m),1.02-0.96(2H,m).

LCMS(LC-1);RT=1.13,m/z563[M+H]+
1H-NMR(CDCl3):δ(ppm)8.92(2H,s),7.93-7.88(1H,m),7.78-7.70(1H,m),7.55-7.48(1H,m),4.92-4.81(1H,m),4.80-4.67(1H,m),4.18-4.09(1H,m),3.84-3.76(1H,m),3.36-3.30(1H,m),2.97-2.88(1H,m),2.78-2.70(2H,m),2.59(4H,s),2.49-2.37(4H,m),2.35-2.25(2H,m),2.23-2.11(2H,m),2.08-1.92(3H,m),1.74-1.62(3H,m),1.61-1.48(3H,m),1.01(3H,brd,J=6.1Hz),0.45-0.33(4H,m).

LCMS(LC-1);RT=0.75,m/z211[M+H]+
1H-NMR(DMSOd6):δ(ppm)12.52(1H,s),2.85(2H,t,J=6.2Hz),2.51-2.47(2H,m),2.18(3H,s),2.11-2.05(2H,m).

LCMS(LC-1);RT=1.21,m/z366[M+H]+
1H-NMR(DMSOd6):δ(ppm)12.88(1H,s),3.15(2H,t,J=5.8Hz),2.97(2H,t,J=5.8Hz),2.22(3H,s).

LCMS(LC-1);RT=0.90,m/z286[M+H]+
1H-NMR(DMSOd6):δ(ppm)7.37(1H,d,J=8.4Hz),6.86(1H,d,J=8.4Hz),2.17(3H,s).


LCMS(LC-1);RT=0.84,m/z244[M+H]+(ボロン酸として検出)
1H-NMR(DMSOd6):δ(ppm)9.92(1H,s,br),7.52(2H,s),7.28(1H,d,J=8.4Hz),6.81(1H,d,J=8.4Hz).

LCMS(LC-1);RT=1.23,m/z374[M+H]+
1H-NMR(DMSOd6):δ(ppm)10.76(1H,s),8.08-8.04(2H,m),7.99-7.96(2H,m),7.65(1H,d,J=8.6),7.49(1H,d,J=8.6).

LCMS(LC-1);RT=1.74,m/z388[M+H]+
1H-NMR(DMSOd6):δ(ppm)8.09-8.06(2H,m),8.01-7.96(2H,m),7.76(2H,m),4.03(3H,s.)

LCMS(LC-1);RT=1.23,m/z258[M+H]+
1H-NMR(DMSOd6):δ(ppm)7.69(2H,s),7.40(1H,d,J=8.4Hz),7.05(1H,d,J=8.4Hz),3.85(3H,s).

LCMS(LC-1);RT=1.22,m/z415[M+H]+

LCMS(LC-1);RT=0.93,m/z373[M+H]+
1H-NMR(DMSOd6):δ(ppm)8.93(2H,s),7.73(2H,s),7.36(1H,d,J=8.3Hz),7.24(1H,d,J=8.3Hz),4.72(1H,d,J=4.0Hz),4.68(1H,m),4.05-4.01(1H,m),3.84-3.81(1H,m),3.70(3H,s),1.92-1.77(2H,m),1.69-1.53(3H,m),1.48-1.41(1H,m).

LCMS(LC-1);RT=1.19,m/z441[M+H]+
1H-NMR(CD3OD):δ(ppm)9.01(2H,s),7.64(1H,d,J=8.3Hz),7.53(1H,d,J=8.3Hz),4.81(2H,m),4.20-4.17(1H,m),3.91-3.88(1H,m),3.83(3H,s),2.06-1.93(3H,m),1.78-1.67(3H,m),1.60-1.52(1H,m),1.12-1.08(2H,m),1.07-1.01(2H,m).

LCMS(LC-1);RT=1.31,m/z455[M+H]+
1H-NMR(CD3OD):δ(ppm)9.01(2H,s),7.63(1H,d,J=8,4Hz)、7.53(1H,d,J=8.4Hz)、4.81(2H,m)、4.20-4.17(1H,m),3.91-3.88(1H,m),3.83(3H,s),2.06-1.94(2H,m),1.78-1.67(4H,m),1.60-1.48(2H,m),1.33-1.29(1H,m),1.20(3H,d,J=6.0Hz),0.90-0.85(1H,m).


LCMS(LC-1);RT=1.11,m/z469[M+H]+
1H-NMR(CD3OD):δ(ppm)0.92(2H,m),7.66-7.61(1H,m),7.54-7.51(1H,m),4.81(2H,s),4.21-4.17(1H,m),3.91-3.88(1H,m),3.85(3H,s),3.58-3.35(5H,m),2.62-2.41(1H,m),2.06-1.94(2H,m),1.78-1.67(3H,m),1.60-1.52(1H,m).

LCMS(LC-1);RT=1.48,m/z653[M+H]+
1H-NMR(CD3OD):δ(ppm)9.01(2H,s),7.63(1H,d,J=8.3Hz),7.52(1H,d,J=8.3Hz),4.81(2H,s),4.20-4.17(1H,m),3.91-3.88(1H,m),3.85(3H,s),3.51-3.35(4H,m),2.73-2.68(1H,m),2.60(1H,m),2.52-2.17(5H,m),2.06-1.94(2H,m),1.78-1.67(3H,m),1.61-1.52(1H,m),1.46(9H,s),1.03(3H,d,J=6.5Hz).

LCMS(LC-1);RT=0.92,m/z553[M+H]+
1H-NMR(CD3OD):δ(ppm)9.01(2H,s),7.62(1H,d,J=8.3Hz),7.52(1H,d,J=8.3Hz),4.81(2H,s),4.20-4.17(1H,m),3.91-3.88(1H,m),3.84(3H,s),3.19-3.02(2H,m),2.95-2.91(1H,m),2.85-2.73(3H,m),2.55-2.27(6H,m),2.18-2.11(1H,m),2.06-1.94(2H,m),1.78-1.67(3H,m),1.60-1.52(1H,m),1.06(3H,d,J=6.4Hz).

LCMS(LC-1);RT=1.11,m/z672[M+H]+
1H-NMR(DMSOd6):δ(ppm)12.52(1H,brs),8.99(2H,s),8.52(1H,d,J=5.0Hz),7.63(1H,d,J=8.4Hz),7.57(1H,d,J=8.4),7.24-7.21(1H,m),7.17-7.13(1H,m),4.73-4.72(1H,m),4.70(2H,m),3.85-3.83(1H,m),3.81(3H,s),3.70-3.52(2H,m),3.25(2H,m),3.10-2.94(3H,m),2.68-2.63(1H,m),2.38-2.32(1H,m),2.28-2.21(2H,m),2.18-2.06(3H,m),1.92-1.77(2H,m),1.66-1.55(3H,m),1.48-1.41(1H,m),1.25-1.24(1H,m),1.01-0.99(2H,m),0.88-0.82(2H,m).


LCMS(LC-1);RT=1.56,m/z326[M+H]+
1H-NMR(DMSOd6):δ(ppm)12.79(1H,brs),7.64(1H,d,J=8.4Hz),7.46(1H,d,J=8.4Hz),3.95(3H,s),2.01-1.99(1H,m),0.99-0.96(4H,m).

LCMS(LC-1);RT=1.30,m/z477[M+H]+
1H-NMR(DMSOd6):δ(ppm)9.00(2H,s),7.62(1H,d,J=8.4Hz),7.56(1H,d,J=8.4Hz),5.37-5.36(1H,m),4.77(2H,s),4.20(1H,m),4.05(1H,m),3.80(3H,s),2.58-2.44(2H,m),2.26-2.15(1H,m),2.09-1.97(2H,m),0.98-0.96(4H,m).


LCMS(LC-1);RT=1.86,m/z500[M+H]+

LCMS(LC-6);RT=1.57,m/z728[M+H]+

LCMS(LC-1);RT=0.98,m/z514[M+H]+

LCMS(LC-1);RT=1.16,m/z528[M+H]+
1H-NMR(CD3OD):δ(ppm)9.02(2H,s),7.67(1H,d,J=8.3Hz),7.55(1H,d,J=8.3Hz),4.81(2H,s),4.23(1H,s),4.20-4.16(2H,m),3.90-3.87(1H,m),3.50-3.44(2H,m),3.21-3.13(2H,m),2.37(3H,s),2.05-1.93(3H,m),1.78-1.67(3H,m),1.60-1.52(1H,m),1.12-1.01(4H,m).

LCMS(LC-1);RT=1.91,m/z401[M+H]+
1H-NMR(CDCl3):δ(ppm)7.59-7.56(1H,m),7.49-7.43(1H,m),5.94-5.91(2H,s),5.32-5.30(2H,s),3.72-3.69(3H,s),3.57-3.52(3H,s),1.30-1.25(2H,m),1.17-1.14(1H,m),1.11-1.05(2H,m).

LCMS(LC-1);RT=2.16,2.27,m/z629[M+H]+
1H-NMR(CDCl3):δ(ppm)8.97(2H,s),7.71(1H,d,J=8.0Hz),7.38(1H,d,J=8.0Hz),5.97(2H,s),5.04(2H,s),4.96(1H,s),4.36-4.24(1H,m),3.95-3.85(2H,m),3.57(3H,s),3.29(3H,s),2.31(1H,s),2.04-1.95(2H,m),1.76-1.73(2H,m),1.59-1.51(2H,m),1.32-1.28(2H,m),1.19-1.16(1H,m),1.13-1.06(2H,m),0.99-0.94(1H,m),0.88(9H,s),0.08(6H,s).

LCMS(LC-1);RT=0.98,m/z427[M+H]+
1H-NMR(DMSO):δ(ppm)12.7(1H,s),10.35-10.1(1H,m),8.96(2H,s),7.50-7.35(2H,m),4.78-4.70(1H,m),4.70-4.63(2H,m),4.08-3.96(1H,m),3.92-3.76(1H,m),2.10-1.98(1H,m),1.97-1.76(2H,m),1.70-1.52(3H,m),1.52-1.38(1H,m),1.00-0.96(2H,m).

LCMS(LC-6);RT=2.46,m/z655[M+H]+
1H-NMR(CDCl3):δ(ppm)10.19(1H,m),8.93(2H,m),7.55(1H,d,J=8.2Hz),7.34(1H,d,J=8.2Hz),4.88-4.80(2H,m),4.29-4.26(1H,m),3.89-3.85(1H,m),3.77-3.73(1H,m),2.05-1.89(2H,m),1.87-1.84(1H,m),1.76-1.68(2H,m),1.56-1.50(1H,m),1.30-1.26(2H,m),1.07-1.02(2H,m),0.96(9H,s),0.88(9H,s),0.07(6H,s),0.19(d,J=6.1Hz).

LCMS(LC-6);RT=2.72,m/z699[M+H]+

LCMS(LC-1);RT=2.26&2.35(2ピーク、メトキシメチル保護による位置異性体混合物),m/z585[M+H]+

LCMS(LC-1);RT=1.00,m/z471[M+H]+
1H-NMR(CDCl3):δ(ppm)9.04(2H,s),7.62(1H,d,J=8.4Hz),7.56(1H,d,J=8.4Hz),4.86-4.84(1H,m),4.73-4.72(1H,m),4.69(2H,m),4.05-4.01(1H,m),3.98-3.96(2H,m),3.85-3.82(1H,m),3.58-3.55(2H,m),2.04-1.98(1H,m),1.92-1.77(2H,m),1.66-1.56(3H,m),1.48-1.41(1H,m),0.99-0.96(4H,m).


LCMS(LC-1);RT=1.46,m/z559[M+H]+

LCMS(LC-1);RT=1.20,m/z425[M+H]+
1H-NMR(CDCl3):δ(ppm)8.78(2H,s),7.72(1H,d,J=8.3Hz),7.31(1H,d,J=8.3Hz),5.02(1H,d,J=15.0Hz),4.85(1H,d,J=15.0Hz),4.29-4.23(1H,m),3.93-3.88(1H,m),2.53(3H,s),2.13-2.03(1H,m),1.64-1.53(1H,m),1.30-1.26(2H,m),1.09-1.05(2H,m).

LCMS(LC-1);RT=1.24,m/z425[M+H]+
1H-NMR(DMSOd6):δ(ppm)12.68(1H,s),9.17(2H,s)、8.45(1H,s),7.86(2H,s),4.71-4.70(1H,m),4.69(2H,s),4.04-4.00(1H,m),3.83-3.81(1H,m),1.91-1.75(3H,m),1.65-1.56(3H,m),1.47-1.37(2H,m),1.22-1.16(1H,m),1.13(3H,d,J=6.0Hz).


LCMS(LC-1);RT=1.19,m/z483[M+H]+

LCMS(LC-1);RT=1.64,m/z539[M+H]+

LCMS(LC-1);RT=1.36,m/z497[M+H]+
1H-NMR(DMSOd6):δ(ppm)12.42(1H,m),9.17(2H,s),8.45(1H,s),7.87-7.81(2H,m),4.72-4.70(1H,m),4.69(2H,s),4.14-4.07(1H,m),4.04-4.00(1H,m),3.83-3.81(1H,m),2.94-2.84(1H,m),2.46-2.40(2H,m),2.13-2.06(2H,m),1.91-1.77(2H,m),1.66-1.56(3H,m),1.48-1.40(1H,m),1.13(9H,s).

LCMS(LC-1);RT=1.58,m/z594[M+H]+

LCMS(LC-1);RT=0.89,m/z494[M+H]+
1H-NMR(CDCl3):δ(ppm)8.93(2H,s),8.00-7.95(1H,m),7.81-7.74(1H,m),7.59-7.52(1H,m),4.87-4.66(2H,m),4.16-4.04(1H,m),3.80-3.74(1H,m),3.25-3.16(1H,m),3.25-3.16(1H,m),2.76-2.66(2H,m),2.16-2.06(4H,m),2.06-1.94(4H,m),1.72-1.52(4H,m),1.52-1.43(2H,m).

LCMS(LC-1);RT=1.08,m/z536[M+H]+
1H-NMR(CDCl3):δ(ppm)8.98(2H,s),8.01(1H,s),7.90-7.73(1H,m),7.65-7.58(1H,m),4.97-4.87(1H,m),4.80-4.72(1H,m),4.21-4.13(1H,m),3.86-3.78(1H,m),3.58-3.43(2H,m),3.43-3.26(3H,m),2.27-1.96(7H,m),2.06(3H,s),1.77-1.51(7H,m).

1H-NMR(CDCl3):δ(ppm)3.87-3.80(1H,m),3.40(2H,q,J=7.0Hz),2.57-2.42(3H,m),2.22-2.10(2H,m),1.44(9H,s),1.19(3H,t,J=7.0Hz).
1H-NMR(CDCl3):δ(ppm)3.93-3.86(1H,m),3.41(2H,q,J=7.0Hz),2.73-2.74(1H,m),2.58-2.50(2H,m),2.28-2.20(2H,m),1.19(3H,t,J=7.0Hz).

LCMS(LC-1);RT=1.19,m/z469[M+H]+
1H-NMR(DMSOd6):δ(ppm)12.45(1H,s),9.18(2H,s),8.48(1H,m),7.89-7.84(2H,m),4.72-4.71(1H,m),4.69(2H,s),4.04-4.00(1H,m),3.95-3.88(1H,m),3.84-3.81(1H,m),3.36(2H,q,J=7.0Hz),2.99-2.90(1H,m),2.48-2.44(2H,m),2.13-2.05(2H,m),1.91-1.77(2H,m),1.67-1.54(3H,m),1.48-1.40(1H,m),1.10(3H,t,J=7.0Hz).


LCMS(LC-1);RT=2.31,m/z581[M+H]+

LCMS(LC-1);RT=1.09,m/z453[M+H]+

LCMS(LC-1);RT=0.88,m/z482[M+H]+
1H-NMR(CDCl3):δ(ppm)8.98(2H,d,J=1.2Hz),8.00(1H,t,J=1.8Hz),7.86-7.78(1H,m),7.62-7.53(1H,m),4.96-4.88(1H,m),4.79-4.71(1H,m),4.21-4.12(1H,m),3.86-3.77(1H,m),3.40-3.35(1H,m),3.33-3.24(1H,m),3.25-3.14(1H,m),2.23-2.21(3H,s),2.21-2.20(3H,s),2.13-1.97(4H,m),1.77-1.62(3H,m),1.60-1.50(1H,m).


LCMS(LC-1);RT=2.00,m/z439[M+H]+

LCMS(LC-1);RT=0.99,m/z394[M+H]+
1H-NMR(CD3OD):δ(ppm)9.15(2H,s),8.51(1H,d,J=7.3Hz),7.95(1H,d,J=1.2Hz),7.18(1H,dd,J=7.2Hz,2.1Hz),6.96(1H,s),4.81(2H,s),4.19-4.15(1H,m),3.90-3.86(1H,m),2.05-1.94(2H,m),1.90-1.84(1H,m),1.77-1.66(3H,m),1.59-1.51(1H,m),1.02-0.98(2H,m),0.93-0.88(2H,m).


LCMS(LC-1);RT=1.09,m/z368[M+H]+
1H-NMR(CDCl3):δ(ppm)9.03-8.91(2H,m),8.48-8.09(1H,m),7.52-7.28(2H,m),6.97-6.47(1H,m),5.05-4.78(3H,m),4.21-4.01(3H,m),2.19-1.95(9H,m).

LCMS(LC-1);RT=0.81,m/z326[M+H]+
1H-NMR(CD3OD):δ(ppm)9.23-9.05(3H,m),8.27(1H,d,J=7.1Hz),7.67-7.63(1H,m),6.91(1H,dd,J=7.2,2.0Hz),5.88(1H,s),4.83-4.66(2H,m),4.22-4.12(2H,m),4.11-3.83(2H,m),2.10-1.82(2H,m),1.77-1.48(4H,m).

LCMS(LC-1);RT=0.92,m/z422[M+H]+
1H-NMR(CDCl3):δ(ppm)9.00(2H,s),8.36(1H,d,J=7Hz),8.30(1H,s),7.66(1H,d,J=1.3Hz),7.13(1H,s),6.91(1H,dd,J=7,1.8Hz),5.02-4.82(2H,m),4.27-4.22(1H,m),3.90-3.85(2H,m),3.66-3.60(2H,m),3.37-3.25(3H,m),2.13-2.02(2H,m),1.79-1.53(4H,m).

LCMS(LC-1);RT=1.21,m/z320[M+H]+
1H-NMR(CDCl3):δ(ppm)8.58(1H,d,J=4.9Hz),7.14(1H,brs),7.07(1H,brd,J=4.2Hz),4.58(0.5H,brd,J=13.1Hz),4.46(1H,brd,J=13.1Hz),4.22(0.5H,brs),3.96(0.5H,brd,J=13.7Hz),3.62-3.41(0.5H,m),3.34(1H,brs),3.21-2.96(2H,m),2.96-2.83(1H,m),2.60(3H,s),1.47(9H,s),1.33-1.03(3H,m).

LCMS(LC-1);RT=0.29,m/z220[M+H]+
1H-NMR(CD3OD):δ(ppm)8.51(1H,d,J=5.1Hz),7.31(1H,s),7.23(1H,d,J=5.1Hz),4.49(1H,brd,J=12.8Hz),3.56-3.39(1H,m),3.28-3.05(1H,m),2.95(1H,brd,J=12.8Hz),2.90-2.73(3H,m),2.65-2.56(4H,m),1.22-0.95(3H,m).

LCMS(LC-1);RT=0.96,m/z625[M+H]+
1H-NMR(DMSO-d6):δ(ppm)10.78(1H,s),9.23(2H,s),8.66(1H,d,J=7.2Hz),8.52(1H,d,J=5.0Hz),8.12(1H,s),7.30-7.12(3H,m),6.97(1H,s),5.76(1H,s),4.74-4.67(3H,m),4.02(1H,brd,J=3.1Hz),3.84-3.79(1H,m),3.62(1H,m),3.28-3.21(1H,m),3.10-2.89(3H,m),2.77-2.59(2H,m),2.27(1H,brs),2.24-2.00(5H,m),1.96-1.73(3H,m),1.68-1.51(4H,m),1.49-1.35(1H,m),0.99(1.5H,brd,J=6.2Hz),0.82(1.5H,brd,J=6.2Hz).




LCMS(LC-1);RT=2.09,m/z327[M+H]+
1H-NMR(CDCl3):δ(ppm)4.09-3.94(m,2H),2.70-2.61(m,2H),2.55-2.46(m,4H),2.30-2.21(m,4H),2.05(s,2H),1.19(s,18H).

LCMS(LC-1);RT=1.51,m/z522[M+H]+
1H-NMR(CD3OD):δ(ppm)9.15(s,2H),8.49(d,J=7.5 Hz,1H),7.99-7.93(m,1H),7.17(dd,J=7.5,2.0Hz,1H),7.01(s,1H),4.83-4.76(m,2H),4.28-4.01(m,2H),3.90-3.87(m,1H),2.94-2.72(m,1H),2.57-2.38(m,2H),2.35-2.13(m,2H),2.07-1.91(m,3H),1.81-1.63(m, 4H),1.60-1.51(m,2H),1.20(s,9H).

LCMS(LC-1);RT=1.35,m/z235[M+H]+
1H-NMR(CDCl3):δ(ppm)8.25(1H,d,J=5.1Hz),7.30(1H,d,J=5.1Hz),4.63(2H,s),4.04(3H,s).

LCMS(LC-1);RT=1.05,m/z183[M+H]+
1H-NMR(CDCl3):δ(ppm)8.25(1H,d,J=5.2Hz),7.35(1H,d,J=5.2Hz),4.01(3H,s),3.96(2H,s).

LCMS(LC-1);RT=1.03,m/z198[M+H]+
1H-NMR(CDCl3):δ(ppm)8.02(s,2H),7.91(d,J=7.3Hz,2H),6.50(d,J=7.3Hz,2H),5.86(s,2H),3.98(s,3H).

LCMS(LC-1);RT=1.11,m/z398[M+H]+
1H-NMR(CDCl3):δ(ppm)9.01-8.96(m,2H),8.12-8.05(m,1H),6.56-6.48(m,1H),6.01-5.93(m,1H),5.21-5.19(m,2H),4.95-4.82(m,4H),4.12-4.09(m,3H),3.86-3.77(m,3H)2.28-2.14(m,2H),2.04(s,3H),1.89-1.79(m,4H)1.32-1.22(m,2H).

LCMS(LC-1);RT=1.32,m/z466[M+H]+

LCMS(LC-1);RT=1.06,m/z424[M+H]+
1H-NMR(CDCl3):δ(ppm)8.99(s 2H),8.19(brs,1H),8.18-8.13(m,1H),7.25-7.19(m,1H),6.69(d,J=7.1Hz,1H),5.00(d,J=15Hz,1H),4.83(d,J=15Hz,1H),4.28-4.20(m,1H),3.93(s,3H),3.91-3.84(m,1H),2.17-2.00(m,3H),1.77-1.71(m,4H),1.19-1.12(m,2H),0.98-0.90(m,2H).
(1)測定方法
ヒトIRAK-4(Invitrogen,Cat.PV3362)の活性は、10μM ATP(Sigma Aldrich,Cat.A7699)存在下でのIRAK-4ペプチド基質(ビオチン-KKKKRFSFKKSFKC) のリン酸化を、TR-FRET法により測定した。酵素反応は、50mM HEPES (pH7.2)、1mM DTT、0.1mM Na3VO4、5mM MgCl2、1mM MnCl2、0.1% bovine serum albuminから成る反応バッファー中で行われた。IRAK-4阻害活性の測定のため、1nM IRAK-4、0.5μM ペプチド基質, 10μM ATPを含む反応バッファーに被検化合物を添加し、23℃で30分間インキュベートした。次に、ユーロピウムクリプテート標識した抗体(0.3μg/mL、抗体はIRAK-4ペプチド基質を抗原として作製)、streptavidin-XL665 (2μg/mL、CisBio, Cat.610SAXLB)、50mM HEPES (pH7.2)、0.1% BSA、120mM KF、66.7mM EDTAからなる検出液(各試薬とも終濃度)を添加して反応を停止後、さらに23℃で60分間インキュベートした。波長665nmと620nmの蛍光強度をマイクロプレートリーダーで測定し、酵素活性を665nm/620nmの蛍光強度の比として算出した。12.5μM Staurosporine(LC Laboratories,Cat.S-9300)添加時のIRAK-4抑制率を100%、被検化合物未添加時のIRAK-4抑制率を0%とし、被検化合物のIC50をデータ解析ソフトウェアXLfit (ID Business Solutions Ltd.)の4 Parameter Logistic Modelを用いて求めた。
なお、測定中の各操作、条件については、当業者が理解でき、測定に大きな影響を及ぼさない範囲で適宜変更することができる。
以下に示す通り、本発明の化合物のある態様は優れたIRAK-4阻害活性を示した。
なお、複数回測定した場合には、その平均値を示している。





(1) 測定方法
THP-1アッセイでは、LPS刺激により誘導されるTNFα産生に与える被検化合物の影響を評価することができる。THP-1細胞(ATCC, Cat.TIB-202)を1x105cells/160μL/wellで96wellプレートに播種し、20μLの被検化合物を添加して、5%CO2培養器にて37℃で1時間インキュベートした。次に、20μLのLPS(終濃度2.5ng/mL、Sigma, Cat.L2630)を添加して、さらに4時間インキュベートした。培養後にプレートを遠心し、各ウェルから100μLの上清を取り、HTRF(Cisbio, Cat.62TNFPEB)によるTNFα量の評価に使用した。TNFα量の測定では、上清を培地で2倍に希釈してから384wellプレートに10μL添加後、anti-TNFα-cryptate(5μL)とanti-TNFα-XL665(5μL)を加えて、一晩静置した。波長620/665nmの蛍光強度比をマイクロプレートリーダーで測定し、上清中のTNFα量を検量線により算出した。LPS未添加時のTNFα産生抑制率を100%、被検化合物未添加時のTNFα産生抑制率を0%とし、被検化合物のIC50をデータ解析ソフトウェアXLfit (ID Business Solutions Ltd.)の4 Parameter Logistic Modelを用いて求めた。
なお、測定中の各操作、条件については、当業者が理解でき、測定に大きな影響を及ぼさない範囲で適宜変更することができる。
以下に示す通り、本発明の化合物のある態様は優れたTNFα産生阻害活性を示した。
なお、複数回測定した場合には、その平均値を示している。小数点以下第4位を四捨五入した。





(1) 測定方法
8週齢の雌性Lewisラット(SLC Inc.)を、ウシII型コラーゲン(CII、コラーゲン技術研修会、製品番号K41)で免疫して関節炎を誘発した。不完全フロイントアジュバント(Difco,Cat.263910)とCII3mg/mL溶液との1対1混合エマルジョンを作製し、ラット1匹あたり0.7mLを尾根部および両前後肢根部皮内の7箇所に0.1mLずつ注入した。7日後に、初日と同様に不完全フロイントアジュバントとCIIを1対1で混合したエマルジョン0.2mLを、尾根部2箇所に0.1mLずつ追加免疫した。12日後に、Plethysmometer(UGO BASILE,Cat.37140)を用いて、動物の両後肢の足蹠容量を測定し、Normal群に対する足蹠容量比および体重で群分けを行った。群分け後、被検化合物または溶媒(Vehicle:0.5%メチルセルロース)の投与を開始し、1日2回、7日間経口投与した(群分け日は、群分け後1回のみ)。投薬開始から1日あるいは2日おきにPlethysmometerを用いて、動物の両後肢の足蹠容量を測定し、被検化合物による影響を評価した。
各測定日における各個体について、両後肢の足蹠容量平均値を、Excel2010(Microsoft)を用いて算出し、GraphPadPrism7.03(GraphPad Software,Inc.)を用いてグラフ化した。Normal群を100%、溶媒投与群を0%抑制対照と規定した。これらの対照に対して、各被検化合物濃度についての抑制率を、Excel2010(Microsoft)を用いて計算した。
なお、測定中の各操作、条件については、当業者が理解でき、測定に大きな影響を及ぼさない範囲で適宜変更することができる。
各群の足蹠容量の推移結果を図1に示す。縦軸は足蹠容量、横軸は初回のウシII型コラーゲン免疫後の日数を示す。また、「n」は使用したラットの例数を示す。
実施例c-01-01の20、60、120mg/kg,1日2回投与群の初回免疫から19日後における抑制率はそれぞれ45%、65%、77%であった。
このように本発明の化合物のある態様(実施例c-01-01)はラット関節炎において優れた腫脹抑制効果を示した。
Claims (35)
- 下記一般式(1):

Rgは、下記一般式(1-1):


R1は、-H、-F、-Cl、メチル、又はC1-3アルコキシであり、前記C1-3アルコキシはG1群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
前記G1群は、-F、ヒドロキシ、シアノ、ハロゲノC1-6アルキル、C1-4アルコキシ、フェニル、5-6員ヘテロアリール、及び3-7員飽和環基からなる群であり、G1群におけるフェニル又は5-6員ヘテロアリールはGAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
前記GAr群は、-F、-Cl、ヒドロキシ、シアノ、C1-6アルキル、ハロゲノC1-6アルキル、及び-NH2からなる群であり;
R2は、C1-6アルキル、ハロゲノC1-6アルキル、又は3-7員飽和環基であり、前記R2はG2群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
前記G2群は、-F、ヒドロキシ、ハロゲノC1-3アルキル、及びC1-4アルコキシからなる群であり;
Cyは、下記一般式(2-1):

式(2-1)中、
kは、0又は1の整数であり;
RCy1及びRCy2は、各々独立して、-H、-F、ヒドロキシ、シアノ、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシ、-NR11R12、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、RCy1とRCy2におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく、前記RCy1、RCy2は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R11及びR12は、各々独立して、-H、C1-6アルキル、ハロゲノC1-6アルキル、ヒドロキシC1-6アルキル、C1-6アルコキシC1-6アルキル、3-7員飽和環基、フェニル、又は5-6員ヘテロアリールであり、R11及びR12はG1群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
又はR11とR12が一緒になって4-10員飽和環又は7-11員スピロ環を形成し、前記4-10員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよく、前記4-10員飽和環及び7-11員スピロ環はG3群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
前記G3群は、-F、ヒドロキシ、C1-6アルキル、ハロゲノC1-6アルキル、ヒドロキシC1-6アルキル、C1-6アルコキシ、C1-6アルコキシ-C1-6アルキル、ハロゲノC1-6アルコキシ、-C(O)R14、-NR13C(O)R14、-C(O)NR13R14、-C(O)NH2、-NR13S(O2)R14、-S(O2)NR13R14、-S(O2)NH2、-S(O2)R14、フェニル、5-6員ヘテロアリール、及び3-7員飽和環基からなる群であり、前記G3におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R13は、-H、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシC1-6アルキル、ハロゲノC1-6アルコキシC1-6アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、前記R13におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R14は、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシC1-6アルキル、ハロゲノC1-6アルコキシC1-6アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、前記R14におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
又はR13とR14が一緒になって4-7員飽和環又は7-11員スピロ環を形成し、前記4-7員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよく;
又は、RCy1とRCy2が一緒になって4-7員飽和環を形成し、前記4-7員飽和環はO及びNからなる群より選択される1-2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよく、前記4-7員飽和環は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい]
で示される化合物又はその塩。 - Rgが一般式(1-1)
である請求項1に記載の化合物又はその塩。 - R1が-F、-Cl、メチル、又はC1-3アルコキシである請求項2に記載の化合物又はその塩。
- R1がC1-3アルコキシである請求項2に記載の化合物又はその塩。
- R1がメトキシである請求項2に記載の化合物又はその塩。
- R1が-Hであり;
Cyが、下記一般式(2-1-1):

- Cyが、下記一般式(2-1-2):

[式(2-1-2)中、RCy3は、C1-4アルキル、ハロゲノC1-4アルキルであり;
Xは、O又はNR15であり;
R15は、-H、C1-6アルキル、ハロゲノC1-6アルキル、ヒドロキシC1-6アルキル、C1-6アルコキシC1-6アルキル、-C(O)R16、-S(O2)R16、-C(O)NR16R17、-C(O)OR16、又は3-7員飽和環基であり、前記R15はG1群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R16は、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシC1-6アルキル、ハロゲノC1-6アルコキシC1-6アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、前記R16におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
R17は、-H、C1-6アルキル、ハロゲノC1-6アルキル、C1-3アルコキシC1-6アルキル、ハロゲノC1-6アルコキシC1-6アルキル、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、前記R17におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく;
又はR16とR17が一緒になって4-7員飽和環又は7-11員スピロ環を形成し、前記4-7員飽和環及び7-11員スピロ環はNに加えて、O及びNからなる群より選択される1~2個の同一又は異なったヘテロ原子又は-S(O2)-を有していてもよい]
で示される請求項2~6のいずれかに記載の化合物又はその塩。 - XがNR15(R15は前記と同義)である請求項2~7のいずれかに記載の化合物又はその塩。
- Cyが、下記一般式(2-1-3):

- R2が、以下の式(3-1):

- R2が、以下の式(3-1):

- 以下の式:

- 以下の式:

- 以下の式:

- 以下の式:

- 以下の式:

- 以下の式:

- 以下の式:

- 以下の式:

- 以下の式:

- 以下の式:

- Rgが一般式(1-2)である請求項1に記載の化合物又はその塩。
- R1が-H、又はメトキシである請求項22に記載の化合物又はその塩。
- R1が-Hである請求項22に記載の化合物又はその塩。
- Cyが、一般式(2-1)であり;
[式(2-1)中、
kが1の整数であり;
RCy1及びRCy2が、各々独立して、-H、-F、ヒドロキシ、シアノ、C1-6アルキル、ハロゲノC1-6アルキル、C1-6アルコキシ、-NR11R12、フェニル、5-6員ヘテロアリール、又は3-7員飽和環基であり、RCy1とRCy2におけるフェニル又は5-6員ヘテロアリールは前記GAr群より選択される1~3個の同一又は異なった置換基で置換されていてもよく、前記RCy1、RCy2は前記G1群より選択される1~3個の同一又は異なった置換基で置換されていてもよい(R11及びR12は前記と同義)]
で示される請求項22~24のいずれかに記載の化合物又はその塩。 - Cyが、下記一般式(2-1-1):

- Cyが、下記一般式(2-1-2):

Xは、O又はNR15(R15は前記と同義)である請求項22~26のいずれかに記載の化合物又はその塩。 - XがNR15(R15は前記と同義)である請求項22~27のいずれかに記載の化合物又はその塩。
- Cyが、下記一般式(2-1-3):

- R2が、以下の式(3-1):

- R2が、以下の式(3-1):

- 以下の式:

- 以下の式:

- 以下の式:

- 以下の式:

Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2020471055A AU2020471055B8 (en) | 2020-09-30 | 2020-09-30 | Pyrimidine- and nitrogen-containing bicyclic compound |
| CA3194164A CA3194164A1 (en) | 2020-09-30 | 2020-09-30 | Pyrimidine- and nitrogen-containing bicyclic compound |
| US18/029,289 US20240025893A1 (en) | 2020-09-30 | 2020-09-30 | Pyrimidine-and nitrogen-containing bicyclic compound |
| JP2022553287A JP7433463B2 (ja) | 2020-09-30 | 2020-09-30 | ピリミジン含有含窒素二環化合物 |
| KR1020237010766A KR20230084145A (ko) | 2020-09-30 | 2020-09-30 | 피리미딘 함유 함질소 2환 화합물 |
| EP20956217.2A EP4223750A4 (en) | 2020-09-30 | 2020-09-30 | NITROGEN-CONTAINING BICYCLIC COMPOUND WITH PYRIMIDINE |
| MX2023003698A MX2023003698A (es) | 2020-09-30 | 2020-09-30 | Compuesto biciclico que contiene nitrogeno y pirimidina. |
| PCT/JP2020/037048 WO2022070289A1 (ja) | 2020-09-30 | 2020-09-30 | ピリミジン含有含窒素二環化合物 |
| CN202080105488.8A CN116194454A (zh) | 2020-09-30 | 2020-09-30 | 含有嘧啶的含氮双环化合物 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2020/037048 WO2022070289A1 (ja) | 2020-09-30 | 2020-09-30 | ピリミジン含有含窒素二環化合物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022070289A1 true WO2022070289A1 (ja) | 2022-04-07 |
Family
ID=80951519
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/037048 WO2022070289A1 (ja) | 2020-09-30 | 2020-09-30 | ピリミジン含有含窒素二環化合物 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20240025893A1 (ja) |
| EP (1) | EP4223750A4 (ja) |
| JP (1) | JP7433463B2 (ja) |
| KR (1) | KR20230084145A (ja) |
| CN (1) | CN116194454A (ja) |
| AU (1) | AU2020471055B8 (ja) |
| CA (1) | CA3194164A1 (ja) |
| MX (1) | MX2023003698A (ja) |
| WO (1) | WO2022070289A1 (ja) |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012068546A1 (en) | 2010-11-19 | 2012-05-24 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| WO2013042137A1 (en) | 2011-09-19 | 2013-03-28 | Aurigene Discovery Technologies Limited | Bicyclic heterocycles as irak4 inhibitors |
| WO2013106614A1 (en) * | 2012-01-13 | 2013-07-18 | Bristol-Myers Squibb Company | Triazolyl-substituted pyridyl compounds useful as kinase inhibitors |
| WO2015048281A1 (en) | 2013-09-27 | 2015-04-02 | Nimbus Iris, Inc. | Irak inhibitors and uses thereof |
| WO2015150995A1 (en) | 2014-04-04 | 2015-10-08 | Pfizer Inc. | Bicyclic-fused heteroaryl or aryl compounds and their use as irak4 inhibitors |
| WO2016053771A1 (en) | 2014-09-30 | 2016-04-07 | Merck Sharp & Dohme Corp. | Inhibitors of irak4 activity |
| WO2016144846A1 (en) | 2015-03-12 | 2016-09-15 | Merck Sharp & Dohme Corp. | Pyrazolopyrimidine inhibitors of irak4 activity |
| WO2019070093A1 (en) * | 2017-10-02 | 2019-04-11 | 1ST Biotherapeutics, Inc. | BENZOTHIAZOL COMPOUNDS AND METHODS OF USING THE SAME FOR TREATING NEURODEGENERATIVE DISORDERS |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2593457B1 (en) * | 2010-07-13 | 2017-08-23 | F. Hoffmann-La Roche AG | Pyrazolo[1,5a]pyrimidine and thieno[3,2b]pyrimidine derivatives as irak4 modulators |
| WO2012129258A1 (en) * | 2011-03-22 | 2012-09-27 | Merck Sharp & Dohme Corp. | Amidopyrazole inhibitors of interleukin receptor-associated kinases |
| JP6895439B2 (ja) * | 2015-12-22 | 2021-06-30 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | IRAK4調節因子としてのピラゾロ[1,5a]ピリミジン誘導体 |
| CN106946890A (zh) * | 2017-04-26 | 2017-07-14 | 中国药科大学 | 吡啶类irak4抑制剂、其制备方法及应用 |
-
2020
- 2020-09-30 CA CA3194164A patent/CA3194164A1/en active Pending
- 2020-09-30 MX MX2023003698A patent/MX2023003698A/es unknown
- 2020-09-30 AU AU2020471055A patent/AU2020471055B8/en active Active
- 2020-09-30 US US18/029,289 patent/US20240025893A1/en active Pending
- 2020-09-30 CN CN202080105488.8A patent/CN116194454A/zh active Pending
- 2020-09-30 JP JP2022553287A patent/JP7433463B2/ja active Active
- 2020-09-30 EP EP20956217.2A patent/EP4223750A4/en active Pending
- 2020-09-30 WO PCT/JP2020/037048 patent/WO2022070289A1/ja active Application Filing
- 2020-09-30 KR KR1020237010766A patent/KR20230084145A/ko unknown
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012068546A1 (en) | 2010-11-19 | 2012-05-24 | Ligand Pharmaceuticals Incorporated | Heterocycle amines and uses thereof |
| WO2013042137A1 (en) | 2011-09-19 | 2013-03-28 | Aurigene Discovery Technologies Limited | Bicyclic heterocycles as irak4 inhibitors |
| WO2013106614A1 (en) * | 2012-01-13 | 2013-07-18 | Bristol-Myers Squibb Company | Triazolyl-substituted pyridyl compounds useful as kinase inhibitors |
| WO2015048281A1 (en) | 2013-09-27 | 2015-04-02 | Nimbus Iris, Inc. | Irak inhibitors and uses thereof |
| WO2015150995A1 (en) | 2014-04-04 | 2015-10-08 | Pfizer Inc. | Bicyclic-fused heteroaryl or aryl compounds and their use as irak4 inhibitors |
| WO2016053771A1 (en) | 2014-09-30 | 2016-04-07 | Merck Sharp & Dohme Corp. | Inhibitors of irak4 activity |
| WO2016144846A1 (en) | 2015-03-12 | 2016-09-15 | Merck Sharp & Dohme Corp. | Pyrazolopyrimidine inhibitors of irak4 activity |
| WO2019070093A1 (en) * | 2017-10-02 | 2019-04-11 | 1ST Biotherapeutics, Inc. | BENZOTHIAZOL COMPOUNDS AND METHODS OF USING THE SAME FOR TREATING NEURODEGENERATIVE DISORDERS |
Non-Patent Citations (63)
| Title |
|---|
| "Newest Mass Spectrometry for Life Science", 2002, KODANSHA SCIENTIFIC |
| "Physiology", 1986, BUNKODO |
| BAILLIE T.A.: "Cytochrome P450 and Other Enzymes in Drug Discovery and Development", DRUG METABOLIZING ENZYMES, 2003, pages 147 - 154 |
| BEVAN, C.D ET AL., ANAL. CHEM., vol. 72, 2000, pages 1781 - 1787 |
| BUEHLER, E.V., ARCH. DERMATOL., vol. 91, 1965, pages 171 - 177 |
| CHACHIN, M ET AL., NIPPON YAKURIGAKU ZASSHI, vol. 119, 2002, pages 345 - 351 |
| CHENG, C.S ET AL., DRUG DEVELOP. INDUST. PHARM., vol. 28, 2002, pages 177 - 191 |
| CLIVE, D ET AL., MUTAT. RES., vol. 31, 1975, pages 17 - 29 |
| COLE, J. ET AL., MUTAT. RES., vol. 111, 1983, pages 371 - 386 |
| DELIE, F ET AL., CRIT. REV. THER. DRUG CARRIER SYST., vol. 14, 1997, pages 221 - 286 |
| DOM, A ET AL., J. BIOMOL. SCREEN., vol. 10, 2005, pages 339 - 347 |
| DRORBAUGH, J.E ET AL., PEDIATRICS, vol. 16, 1955, pages 81 - 87 |
| DUCIC ET AL., J. CARDIOVASC. PHARMACOL., vol. 30, no. 1, 1997, pages 42 - 54 |
| EPSTEIN, M.A ET AL., RESPIR. PHYSIOL., vol. 32, 1978, pages 105 - 120 |
| FENECH M. ET AL., MUTAT. RES., vol. 147, 1985, pages 29 - 36 |
| FLANNERY SBOWIE A.G., BIOCHEMICAL PHARMACOLOGY, vol. 80, 2010, pages 1981 - 1991 |
| FUKUI, N ET AL., GENDAI NO RINSHO, vol. 4, no. 7, 1970, pages 277 - 289 |
| GERMANN, U.A., METHODS ENZYMOL., vol. 292, 1998, pages 427 - 41 |
| GILBERT, J.D ET AL., J. PHARM. TOX. METHODS, vol. 50, 2004, pages 187 - 199 |
| GOMEZ-LECHON, M.J ET AL., CURR. DRUG METAB., vol. 5, no. 5, 2004, pages 443 - 462 |
| HARBER, L.C., ARCH. DERMATOL., vol. 96, 1967, pages 646 - 653 |
| HAYASHI M ET AL., MUTAT. RES., vol. 312, 1994, pages 293 - 304 |
| HAYASHI, M ET AL., ENVIRON. MOL. MUTAGEN., vol. 35, 2000, pages 234 - 252 |
| HEIFETZ, A. ET AL.: "Fragment Molecular Orbital Method Applied to Lead Optimization of Novel Interleukin-2 Inducible T- Cell Kinase (ITK) Inhibitors", JOURNAL OF MEDICINAL CHEMISTRY, vol. 59, no. 9, 2016, pages 4352 - 4363, XP055320239, DOI: 10.1021/acs.jmedchem.6b00045 entire text * |
| HENGSTLER, J.G ET AL., DRUG METAB. REV., vol. 32, 2000, pages 81 - 118 |
| HORIO, T., J. INVEST. DERMATOL., vol. 67, 1976, pages 591 - 593 |
| ICHIKAWA, H ET AL., J. INVEST. DERMATOL., vol. 76, 1981, pages 498 - 501 |
| INGELS, F.M ET AL., J. PHAM. SCI., vol. 92, 2003, pages 1545 - 1558 |
| IRVINE, J.D ET AL., J. PHAM. SCI., vol. 88, 1999, pages 28 - 33 |
| IRWIN, S., COMPREHENSIVE OBSERVATIONAL ASSESSMENT, vol. 13, 1968, pages 222 - 257 |
| ISHIYAMA, M. ET AL., IN VITRO TOXICOLOGY, vol. 8, 1995, pages 187 |
| IYAKUSHIN, APPENDIX, ''GUIDELINE FOR DRUG TOXICITY TEST, no. 1834, 2000 |
| IYAKUSHIN, GUIDELINE FOR GENOTOXICITY TEST, no. 1604, 1999, pages II-4 - 3 |
| JAIN A ET AL., FRONITERS IN IMMUNOLOGY, vol. 5, 2014 |
| JORDAN, W.P., CONTACT DERMATITIS, vol. 8, 1982, pages 109 - 116 |
| JUNPING GAN ET AL., CHEM. RES. TOXICOL., vol. 18, 2005, pages 896 - 903 |
| KATO, M ET AL., DRUG METAB. PHARMACOKINET., vol. 20, no. 4, 2005, pages 236 - 243 |
| KOCHEVER, L.E ET AL., J. INVEST. DERMATOL., vol. 73, 1979, pages 144 - 146 |
| KOZICZAK-HOLBRO M ET AL., ARTHRITIS & RHEUMATISM, vol. 60, 2009, pages 1661 - 1671 |
| LI, C ET AL., DRUG METAB. DISPOS., vol. 34, no. 6, 2006, pages 901 - 905 |
| LIPINSKI, C.A ET AL., ADV. DRUG DELIV. REV., vol. 23, 1997, pages 3 - 26 |
| MAGNUSSON B. ET AL., J. INVEST. DERMATOL., vol. 52, 1969, pages 268 - 276 |
| MATTSON, J.L ET AL.: "Functional Observational Battery", J. AMERICAN COLLEGE OF TECHNOLOGY, vol. 15, no. 3, 1996, pages 239 - 254 |
| MAURER, T ET AL., BR. J. DERMATOL., vol. 63, 1980, pages 593 - 605 |
| MILLER, B. ET AL., MUTAT. RES., vol. 392, 1997, pages 45 - 59 |
| MILLER, V.P ET AL., ANN. N.Y. ACAD. SCI., vol. 919, 2000, pages 26 - 32 |
| MORIKAWA, F ET AL.: "Sunlight and Man", 1974, TOKYO UNIV. PRESS, pages: 529 - 557 |
| MURAKI, K ET AL., AM. J. PHYSIOL., vol. 269, 1995, pages 524 - 532 |
| NAIR, S. ET AL.: "Optimization of Nicotinamides as Potent and Selective IRAK 4 Inhibitors with Efficacy in a Murine Model of Psoriasis", ACS MEDICINAL CHEMISTRY LETTERS, vol. 11, no. 7, 10 June 2020 (2020-06-10), pages 1402 - 1409, XP055923635, DOI: 10.1021/acsmedchemlett.0c00082 entire text * |
| PETER G.M.WUTS: "Greene's Protective Groups in Organic Synthesis", vol. 1-12, 2014, JOHN WILEY AND SONS |
| PICAD C ET AL., SCIENCE, vol. 299, 2003, pages 2076 - 2079 |
| SATO, Y ET AL., CONTACT DERMATITIS, vol. 7, 1981, pages 225 - 237 |
| See also references of EP4223750A4 |
| SHOU, W.Z ET AL., J. MASS SPECTROM., vol. 40, no. 10, 2005, pages 1347 - 1356 |
| SUGANO S.TSUBONE H.NAKADA Y, MARUZEN, 2003 |
| SUZUKI N ET AL., NATURE, vol. 416, 2002, pages 750 - 754 |
| TAKEYOSHI, M ET AL., J. APPL. TOXICOL., vol. 25, no. 2, 2005, pages 129 - 34 |
| TAKEYOSHI, M, TOXICOL. LETT., vol. 119, no. 3, 2001, pages 203 - 8 |
| TAMAI I ET AL., PHARM. RES., vol. 18, no. 9, September 2001 (2001-09-01), pages 1262 - 1269 |
| VINSON, L.J., J. SOC. COSM. CHEM., vol. 17, 1966, pages 123 - 130 |
| WAN Y ET AL., J. BIOL. CHEM., vol. 284, 2009, pages 10367 - 10375 |
| YAKUJI NIPPO, DRUG NONCLINICAL TEST GUIDELINE COMMENTARY 2002, 2002, pages 1 - 6 |
| YAMASHITA, S ET AL., EUR. J. PHAM. SCI., vol. 10, 2000, pages 195 - 204 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2022070289A1 (ja) | 2022-04-07 |
| US20240025893A1 (en) | 2024-01-25 |
| CN116194454A (zh) | 2023-05-30 |
| AU2020471055A1 (en) | 2023-05-04 |
| KR20230084145A (ko) | 2023-06-12 |
| CA3194164A1 (en) | 2022-04-07 |
| JP7433463B2 (ja) | 2024-02-19 |
| AU2020471055B2 (en) | 2023-12-14 |
| EP4223750A4 (en) | 2024-06-19 |
| MX2023003698A (es) | 2023-06-15 |
| EP4223750A1 (en) | 2023-08-09 |
| AU2020471055B8 (en) | 2023-12-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI788655B (zh) | 酪胺酸蛋白質激酶2(tyk2)抑制劑及其用途 | |
| JP6800885B2 (ja) | イミダゾピラジン及びピラゾロピリミジン、並びにampa受容体調節物質としてのこれらの使用 | |
| KR20140096100A (ko) | 이환 피페라진 화합물 | |
| WO2011028685A1 (en) | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors | |
| US20090105261A1 (en) | Novel substituted pyrazolo[1,5<I>A</I>]-1,3,5-Triazine derivatives and their analogues, pharmaceutical compositions containing same, use thereof as medicine and methods for preparing same | |
| US9663525B2 (en) | Pyrazolopyrimidinyl inhibitors of ubiquitin-activating enzyme | |
| JP6192741B2 (ja) | 新規ピロロピリミジン化合物又はその塩、及びこれを含有する医薬組成物、特にnae阻害作用に基づく腫瘍等の予防剤及び/又は治療剤 | |
| JP6322275B2 (ja) | ヤヌスキナーゼ阻害剤としてのn−(2−シアノヘテロシクリル)ピラゾロピリドン | |
| BR112019015273A2 (pt) | compostos | |
| CN117881683A (zh) | PI3Kα抑制剂及其使用方法 | |
| JP2023538304A (ja) | 化合物、組成物及び方法 | |
| JP2024535867A (ja) | 治療薬としてn-アリールピリミジン-2-アミン誘導体を有する化合物 | |
| WO2022070289A1 (ja) | ピリミジン含有含窒素二環化合物 | |
| EP4408845A1 (en) | Novel compounds | |
| RU2827867C1 (ru) | Пиримидин- и азотсодержащее бициклическое соединение | |
| TW202328123A (zh) | 含有嘧啶之含氮雙環化合物 | |
| JP7422242B2 (ja) | マクロサイクル化合物 | |
| WO2022070288A1 (ja) | 含窒素二環化合物 | |
| TW202327581A (zh) | 含氮雙環化合物 | |
| TW202328149A (zh) | 巨環化合物 | |
| WO2023086800A1 (en) | Heterocyclic compounds as triggering receptor expressed on myeloid cells 2 agonists and methods of use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20956217 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2022553287 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202317021013 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 3194164 Country of ref document: CA |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112023005856 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2023107549 Country of ref document: RU |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020471055 Country of ref document: AU Date of ref document: 20200930 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2020956217 Country of ref document: EP Effective date: 20230502 |
|
| ENP | Entry into the national phase |
Ref document number: 112023005856 Country of ref document: BR Kind code of ref document: A2 Effective date: 20230329 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 523440074 Country of ref document: SA |