WO2020171092A1 - 相補部分を含む修飾オリゴヌクレオチドの製造方法 - Google Patents
相補部分を含む修飾オリゴヌクレオチドの製造方法 Download PDFInfo
- Publication number
- WO2020171092A1 WO2020171092A1 PCT/JP2020/006366 JP2020006366W WO2020171092A1 WO 2020171092 A1 WO2020171092 A1 WO 2020171092A1 JP 2020006366 W JP2020006366 W JP 2020006366W WO 2020171092 A1 WO2020171092 A1 WO 2020171092A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- oligonucleotide
- modified
- raw material
- fragment
- fragments
- Prior art date
Links
Images

























Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/102—Mutagenizing nucleic acids
- C12N15/1031—Mutagenizing nucleic acids mutagenesis by gene assembly, e.g. assembly by oligonucleotide extension PCR
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/04—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with deoxyribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H21/00—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids
- C07H21/02—Compounds containing two or more mononucleotide units having separate phosphate or polyphosphate groups linked by saccharide radicals of nucleoside groups, e.g. nucleic acids with ribosyl as saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/93—Ligases (6)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/26—Preparation of nitrogen-containing carbohydrates
- C12P19/28—N-glycosides
- C12P19/30—Nucleotides
- C12P19/34—Polynucleotides, e.g. nucleic acids, oligoribonucleotides
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/70—Vectors or expression systems specially adapted for E. coli
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y605/00—Ligases forming phosphoric ester bonds (6.5)
- C12Y605/01—Ligases forming phosphoric ester bonds (6.5) forming phosphoric ester bonds (6.5.1)
- C12Y605/01001—DNA ligase (ATP) (6.5.1.1)
Definitions
- the present invention relates to a method for producing a modified oligonucleotide containing a complementary portion.
- Oligonucleotides such as siRNA and antisense have been shown to be useful as nucleic acid drugs, and their development has been activated in recent years. Oligonucleotides are mainly produced by a synthetic method, and can be produced, for example, by sequentially extending and synthesizing nucleotide residues one by one in series by solid phase synthesis such as the phosphoramidite method. However, this method has problems that the purity and yield of the product decrease as the chain length of the oligonucleotide increases, and the production efficiency is low. Therefore, there is a demand for a parallel synthesis method in which oligonucleotides are synthesized as short-chain fragments and condensed to obtain the desired oligonucleotide.
- Patent Document 1 a plurality of oligonucleotide raw material fragments corresponding to fragments obtained by dividing the target oligonucleotide are annealed with a template oligonucleotide complementary to the target oligonucleotide, and the annealed oligonucleotide raw material fragments are enzymatically bound to each other. It is described that a single-stranded oligonucleotide is produced by condensing the obtained target oligonucleotide chain from the template oligonucleotide.
- Non-Patent Document 1 describes PEGylation of oligo DNA by ligating the oligo DNA fragment and the PEGylated oligo DNA fragment with a DNA ligase at the cohesive ends.
- an oligonucleotide containing a complementary portion which is used as a raw material, is an oligonucleotide containing a short base and a modified base, which is suggested to have a reduced annealing ability. It is unclear whether the enzymatic condensation of is possible.
- Non-Patent Documents 2 and 3 describe that a nick formed by annealing one oligonucleotide chain and two oligonucleotide raw material fragments complementary thereto is ligated.
- Non-Patent Document 4 describes that a 24-mer double-stranded oligo RNA having sticky ends is ligated with an RNA ligase to form a double-stranded RNA having a size of 48 mer or more.
- the substrate since the substrate has a long length of 24 bases and the product has a long length of 48 bases, the annealing ability of the substrate is significantly reduced because the annealing is performed only under the condition that the substrate has a high annealing ability.
- Non-Patent Document 5 describes that siRNA was synthetically prepared.
- Non-Patent Document 6 describes that the RNA ligase DraRnl is included in the Rn15 family.
- RNA substratecity and structure-guided mutational analysis of bacteriophage T4 RNA ligase J Biol Chem 279:31337-31347. Jayaprakash K. Nair, et al. (2014), Multivalent N-Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Eliminates Robust RNAi-Mediated Silent. J. Am. Chem. Soc. , 136, 16958-16961. MIHAELA-CARMEN UNCULUREAC and STEWART SHUMAN (2019), RNA 21:824-832
- An object of the present invention is to provide an efficient method for producing an oligonucleotide containing a complementary portion such as siRNA or heteroduplex oligonucleotide.
- the inventors of the present invention treated four or more oligonucleotide raw material fragments corresponding to fragments obtained by dividing both complementary parts of an oligonucleotide containing a complementary part of interest with an oligonucleotide ligase to give two fragments.
- the present invention has been completed by finding out directly constructing an oligonucleotide containing a complementary portion such as a chain and finding out that it can be produced with high production efficiency and high purity as compared with a serial synthesis method such as solid phase synthesis.
- oligonucleotides such as siRNA have been produced by chemical synthesis because chemical synthesis is easy and convenient. Specifically, it was produced by chemically synthesizing (for example, solid phase synthesis) each of two constituent oligonucleotide chains, purifying them, and then annealing both chains. Therefore, a method using enzymatic condensation has not been reported so far, and among them, a method of enzymatically synthesizing from 4 or more oligonucleotide raw material fragments is a relatively long-chain double-stranded oligonucleotide having a length of 28 bases or more. Only the method of manufacturing was reported.
- a short oligonucleotide naturally shortens the base length of the oligonucleotide raw material fragment used for its production, but it is considered that the oligonucleotide raw material fragment having a short base length has a reduced annealing ability. It was also considered that if the constituent nucleotides were modified, the annealing ability would be further reduced. Therefore, in the enzymatic oligonucleotide synthesis method using ligase, since the annealing ability of the oligonucleotide raw material fragment was considered to be important, a length of less than 28 bases such as 4 or more oligonucleotide raw material fragments using ligase was used.
- the method for producing the shorter-chain oligonucleotides has not been tried.
- the present inventors have unexpectedly found that when a ligase and four or more oligonucleotide raw material fragments are used, a double strand having a length of less than 28 bases is not affected by the annealing ability. It has been found that oligonucleotides can be successfully produced. Further, in the case of a short-chain oligonucleotide, it was found that the purity of the produced oligonucleotide is also improved, and the present invention has been completed.
- a method for producing a modified oligonucleotide containing a complementary portion having a length of 11 to 27 bases comprising: The method comprises treating a total of four or more oligonucleotide source fragments in the presence of oligonucleotide ligase, The method in which the total of 4 or more oligonucleotide raw material fragments corresponds to the oligonucleotide raw material fragment obtained when the modified oligonucleotide is divided at a fragment ligation part satisfying the following conditions (i) to (v): (I) one or more fragment ligation sites are present in each strand of the complementary portion, and two or more fragment ligation sites are present in the modified oligonucleotide in total; (Ii) when the modified oligonucleotide is separated at a fragment junction, a protruding end is formed in a complementary portion, and the protruding end has a length of 1
- All of the base lengths corresponding to are 11 to 27 base lengths.
- [2] The method of [1], wherein the protruding end in (ii) has a length of 2 to 6 bases.
- [3] The method according to [1] or [2], wherein the portion other than the protruding end in the complementary portion of the four oligonucleotide raw material fragments defined in (iv) has a length of 4 to 16 bases.
- [5] The method of [4], wherein the oligonucleotide ligase is a double-stranded RNA ligase.
- RNA ligase is Rnl2 family or Rnl5 family RNA ligase.
- the modified nucleotide residue is a 1′, 2′, 3′, or 4′ chemically modified nucleotide residue, a 5′- or 3′-phosphate group modified nucleotide residue, a crosslinked modified nucleotide
- the method according to [7] which is a residue, a carrier addition modified nucleotide residue, or a sugar skeleton substitution nucleotide residue.
- the modified nucleotide residue is i) 1 ', 2', 3 ', or 4' site, C 1 ⁇ 6 alkyloxy C 1 ⁇ 6 alkylene, -O-C 1 ⁇ 6 alkyl, -O-C 6 ⁇ 14 aryl, -C- aryl, halogen atom, -O-C 1 ⁇ 6 alkyl N- amido C 1 ⁇ 6 alkylene, -O-C 1 ⁇ 6 alkyl - (C 1 ⁇ 6 alkyl -) amino -C 1 ⁇ 6 alkylene or -O, A 1′, 2′, 3′, or 4′ chemically modified nucleotide residue substituted with an amino C 1-6 alkyl (eg, —O-aminopropyl, —O-AP); ii) -O- ⁇ (S)(OH) 2 , -NH- ⁇ (O)(OH) 2 , or -NH- ⁇ (S)(OH) in
- the 2'-site and the 4'-site are 2'-OC 1-6 alkylene-4', 2'-O-ethylene-4', 2'-O-methyl substituted methylene-4', 2'-.
- modified oligonucleotides such as siRNA and heteroduplex oligonucleotides can be efficiently produced with high purity.
- FIG. 1 is a schematic view showing an example of the mechanism of the present invention.
- FIGS. 2-1 to 2-6 show that, in Example 1, a combination of 6 fragments (combination numbers 1 to 6) of 4 short-chain naturally occurring RNAs for producing the same siRNA was reacted with T4 RNA ligase 2.
- 6 is a graph showing the amount of siRNA produced depending on the concentration of T4 RNA ligase 2 in the case of allowing the reaction.
- 2-1 to 2-6 are as described above.
- 2-1 to 2-6 are as described above.
- 2-1 to 2-6 are as described above.
- 2-1 to 2-6 are as described above.
- 2-1 to 2-6 are as described above.
- 2-1 to 2-6 are as described above.
- 3-1 to 3-4 show changes with time in the production amount of siRNA at each reaction temperature when a combination of four short-chain natural RNAs was reacted with T4 RNA ligase 2 in Example 2. It is a graph shown. 3-1 to 3-4 are as described above. 3-1 to 3-4 are as described above. 3-1 to 3-4 are as described above. FIG. 4 is a diagram showing confirmation by HPLC analysis of siRNA and oligonucleotide preparations produced from modified RNA at each T4 RNA ligase 2 concentration in Example 3.
- FIG. 4 is a diagram showing confirmation by HPLC analysis of siRNA and oligonucleotide preparations produced from modified RNA at each T4 RNA ligase 2 concentration in Example 3.
- FIG. 5 is a diagram showing a HPLC analysis chart of a reaction product in the absence of enzyme and in the presence of Deinococcus radiodurans-derived RNA ligase (DraRnl), and an RNA preparation (sense strand and antisense strand) in Example 5.
- DraRnl Deinococcus radiodurans-derived RNA ligase
- FIG. 6 is a schematic diagram of a reaction product and an HPLC analysis chart of the reaction product in the absence of enzyme and in the presence of T4 RNA ligase 2 in Example 6.
- FIG. 7 is a diagram showing a relationship between four oligonucleotide raw material fragments (including an oligonucleotide raw material fragment containing a mismatch base pair) and a double-stranded modified oligonucleotide generated from the oligonucleotide raw material fragment.
- FIG. 8 is a diagram showing the relationship between 5 or 6 oligonucleotide raw material fragments and double-stranded modified oligonucleotides produced therefrom.
- FIG. 9 is a diagram showing confirmation by HPLC analysis of generation of a double-stranded modified oligonucleotide in a reaction using 5 or 6 oligonucleotide raw material fragments.
- FIG. 10 is a diagram showing a relationship between four oligonucleotide raw material fragments including an oligonucleotide raw material fragment having a DMTr group added to the 5'end and a double-stranded modified oligonucleotide produced from the four oligonucleotide raw material fragments.
- FIG. 11 is a diagram showing a relationship between four oligonucleotide raw material fragments containing an oligonucleotide raw material fragment having a carrier added to the 5'end and a double-stranded modified oligonucleotide produced from the four oligonucleotide raw material fragments.
- FIG. 11 is a diagram showing a relationship between four oligonucleotide raw material fragments containing an oligonucleotide raw material fragment having a carrier added to the 5'end and a double-stranded modified oligonucleotide produced from the four oligonucleotide raw material fragments
- FIG. 12 is a diagram showing the relationship between four oligonucleotide raw material fragments in which the phosphate group at the linking portion of the nucleotide residue is replaced with a thiophosphate group and the double-stranded modified oligonucleotide produced therefrom.
- FIG. 13 is a diagram showing the relationship between four oligonucleotide raw material fragments and the hairpin-type modified oligonucleotide produced from them.
- FIG. 14 shows the relationship between the four oligonucleotide raw material fragments and the double-stranded modified oligonucleotide produced from the four oligonucleotide raw material fragments, which were used to compare the influence of the base length of the protruding end on the reactivity in the oligonucleotide raw material fragment.
- FIG. 15 is a diagram showing the relationship between the four oligonucleotide raw material fragments used for comparison of the influence of the base length of the product on the reactivity, and the double-stranded modified oligonucleotide produced from the four fragments. .. FIG.
- FIG. 16 is a diagram showing the relationship between the four oligonucleotide raw material fragments used for studying the initial reaction rate with a high-concentration substrate and the double-stranded modified oligonucleotide produced therefrom.
- FIG. 17 is a diagram showing the relationship between the four oligonucleotide raw material fragments used for comparison of the base lengths of the products and the double-stranded modified oligonucleotides produced therefrom.
- the present invention provides a method for producing a modified oligonucleotide containing a complementary portion having a length of 11 to 27 bases (hereinafter, also referred to as “target modified oligonucleotide” or the like).
- the method of the present invention includes treating a total of four or more oligonucleotide raw material fragments as raw materials in the presence of an oligonucleotide ligase to produce a modified oligonucleotide of interest.
- modified oligonucleotide of interest produced by the method of the present invention the oligonucleotide raw material fragment and the oligonucleotide ligase used in the method of the present invention, and the details of each treatment condition for carrying out the method of the present invention will be described. To do.
- the modified oligonucleotide of interest produced by the method of the present invention is a modified oligonucleotide containing a complementary portion of 11 to 27 bases in length.
- Oligonucleotide refers to an oligomer containing nucleotide residues as monomer units.
- examples of the “oligonucleotide” include oligo RNA, oligo DNA, and RNA-DNA hybrid oligonucleotide.
- Oligonucleotides can be classified into “natural oligonucleotides” and “modified oligonucleotides”. “Natural type oligonucleotide” refers to nucleotide residues (adenosine (A), guanosine (G), cytidine (C), uridine (U), deoxyadenosine (which constitute polynucleotides (RNA and DNA) contained in cells. dA), deoxyguanosine (dG), deoxycytidine (dC), thymidine (dT) (hereinafter referred to as "natural nucleotide residue").
- modified oligonucleotide refers to an oligonucleotide other than the "natural oligonucleotide", and is an oligonucleotide containing a constituent element (hereinafter referred to as "modified residue") other than the natural nucleotide residue.
- modified residues include modified nucleotide residues, amino acid residues, and linkers.
- modified nucleotide residue include a nucleotide residue containing a modification described below.
- Amino acids include amino acid derivatives.
- amino acid for example, glycine, alanine, valine, leucine, isoleucine, proline, methionine, phenylalanine, tryptophan, serine, threonine, asparagine, glutamine, tyrosine, cysteine, aspartic acid, glutamic acid, histidine, lysine, arginine, and these Examples include derivatives.
- the amino acid derivative refers to an amino acid in which any atom or group in the amino acid is substituted with another atom or group, for example, a hydrogen atom in an amino group, a hydrogen atom in a carboxyl group, an oxygen atom, a hydroxyl group, Any atom or group in the side chain or a hydrogen atom bonded to a skeletal carbon atom (eg, ⁇ -, ⁇ -, ⁇ -, ⁇ -carbon atom) is replaced with another atom (eg, fluorine atom, chlorine atom, Examples thereof include a halogen atom such as a bromine atom and an iodine atom) or an amino acid substituted with a group (eg, a substituent after substitution in the chemical modification described later).
- a hydrogen atom in an amino group e.g, a hydrogen atom in a carboxyl group, an oxygen atom, a hydroxyl group
- Modification in “modified nucleotide residue” includes substitution of a substituent of a sugar moiety (ribose or deoxyribose) of a nucleotide residue, replacement of a sugar moiety of a nucleotide residue (sugar backbone), and nucleotide residue Modification of the nucleobase moiety (eg, substitution of a substituent on the nucleobase moiety) of the.
- substitution of the sugar moiety of the nucleotide residue is, for example, 1′-H, 2′-OH (ribose only), 2′-H, 3′-OH, 3′-NH 2 , 3′.
- the “phosphate group” includes not only —OP(O)(OH) 2 but also a group in which an oxygen atom is replaced by a sulfur atom or ⁇ H (eg, —O— ⁇ (S)(OH) 2 , -NH- ⁇ (O)(OH) 2 , -NH- ⁇ (S)(OH) 2 ) are also included.
- a group in which a hydroxyl group (—OH) in a phosphoric acid group is replaced with an OR * (in the formula, R * represents an organic group such as a protecting group for a phosphoric acid group) (for example, a protected phosphoric acid group) Also included in the above "phosphate group".
- R * represents an organic group such as a protecting group for a phosphoric acid group
- Such substitution includes, for example, 1′, 2′, 3′, or 4′-chemical modification (substitution with another substituent at 1′, 2′, 3′, or 4′ site), 5′.
- Chemical modifications may be introduced, for example, to improve the resistance of the oligonucleotide to degradation.
- the substituent of the following substitutions in the chemical modification for example, C 1 ⁇ 6 alkyloxy C 1 ⁇ 6 alkylene (e.g., methoxyethyl: MOE), - O-C 1 ⁇ 6 alkyl (e.g., -O-Me), -O-C 6 ⁇ 14 aryl (e.g., -O- phenyl), - C-aryl (e.g., -C- phenyl), halogen atom (e.g., fluorine atom), - O-C 1 ⁇ 6 alkyl N- amide C 1 ⁇ 6 alkylene (e.g., -O-N-methylacetamide, -O-NMA), - O -C 1 ⁇ 6 alkyl - (C 1 ⁇ 6 alkyl -) amino -C 1 ⁇ 6 alkylene (
- the chemical modification is preferably 2'-chemical modification (substitution of 2'site), 3'-chemical modification (substitution of 3'site), and more preferably 2'-chemical modification (substitution of 2'site).
- the substituent after the replacement of 2'chemical modification e.g., 2'-C 1 ⁇ 6 alkyloxy C 1 ⁇ 6 alkylene (e.g., 2'-methoxyethyl), 2'-O-C 1 ⁇ 6 alkyl (eg, 2'-O-Me), 2'-O-C 6 ⁇ 14 aryl (e.g., 2'-O-phenyl), 2'-C-aryl (e.g., 2'-C-phenyl), 2 '-Halogen atom (eg 2'-F), 2'-O-C 1-6 alkyl N-amide C 1-6 alkylene (eg 2'-O-N-methylacetamide, 2'-O-NMA ), 2'-O-C 1 ⁇ 6 alkyl
- Examples of the substituent after the substitution in the 3′-chemical modification include 3′-OP(O)(OH) 2, 3′-OP(S)(OH) 2 , 3′-NH- ⁇ . (O)(OH) 2 , 3'-NH- ⁇ (S)(OH) 2 ) and the hydroxyl group (-OH) in the phosphoric acid group is OR * (in the formula, R * is phosphoric acid described later). Groups which represent an organic group such as a protecting group for the group).
- 5'- or 3'-phosphate group modifications may be introduced, for example to improve the resistance of the oligonucleotide to degradation.
- the modification of the 5'- or 3'-phosphate group is, for example, from a phosphate group (-OP(O)(OH) 2 ) to a group in which an oxygen atom in a phosphate group is replaced with a sulfur atom or ⁇ H.
- a phosphate group phosphate group
- Examples of such groups include —O— ⁇ (S)(OH) 2 (thiophosphoric acid group: phosphorothioate type modification), —NH— ⁇ (O)(OH) 2 , —NH- ⁇ (S)( OH) 2 .
- the hydroxyl group (—OH) in the phosphoric acid group represents OR * (in the formula, R * represents an organic group such as a phosphoric acid group-protecting group). ) Is also included (for example, a protected phosphate group).
- the protective group for a phosphoric acid group include a trityl (Tr) group, a p-methoxyphenyldiphenylmethyl (MMTr) group, a di(p-methoxyphenyl)phenylmethyl (DMTr) group, a cyanoethyl group (CN-C 2 H). 4-- ).
- Cross-linking modifications may be introduced, for example, to improve the conformational stability of nucleotide residues.
- Examples of the cross-linking modification include 2'4'-crosslinking modification (substitution that crosslinks 2'-OH and 4'-H), 3'5'-crosslinking modification (3'-H and 5'-H. And the like).
- Examples of the 2'4'-crosslinking type modification include substitution of 2'-OH and 4'-H with 2'-O-C 1-6 alkylene-4' (eg, 2'-O-methylene-4).
- the carrier in the carrier addition modification may be a carrier for improving or imparting performance such as stability, targeting property, and drug efficacy to the target modified oligonucleotide.
- a carrier can be appropriately selected from known carriers according to the purpose of use.
- the carrier include N-acetylgalactosamine (GalNAc), peptide, phosphoric acid, cholesterol, tocopherol, fatty chain, and folic acid.
- the addition site in the carrier addition modification is preferably the 3'or 5'site corresponding to the end of the modified oligonucleotide of interest.
- modified nucleotide residue sucgar backbone-substituted nucleotide residue
- substitution of sugar moiety of nucleotide residue include, for example, hexitol nucleic acid (HNA), cyclohexenyl nucleic acid (CeNA), and other oligonucleotides.
- modified nucleotide residue containing “substitution of the sugar moiety itself of the nucleotide residue” further includes a morpholino ring structure which is not decomposed by an in vivo enzyme (eg, nuclease such as RNase) and does not induce an immune response.
- morpholino nucleic acid (PMO) residues are artificial compounds that mimic nucleotides.
- the modification of the nucleobase moiety of the nucleotide residue includes, for example, alkyl substitution of the nucleobase moiety of the nucleotide residue (eg, substitution of a methyl group at the 5-position of the cytosyl group).
- the oligonucleotide containing a complementary portion refers to an oligonucleotide containing a structure in which complementary nucleotide sequences are paired with each other.
- Examples of the “oligonucleotide containing a complementary portion” include, for example, double-stranded oligonucleotide, double-stranded structure-containing single-stranded oligonucleotide (eg, hairpin-shaped oligonucleotide, loop-shaped oligonucleotide such as dumbbell-shaped oligonucleotide). Can be mentioned.
- the double-stranded oligonucleotide may be a double-stranded oligonucleotide in which each strand is the above-mentioned oligonucleotide, for example, double-stranded oligo RNA, double-stranded oligo DNA, hetero RNA consisting of oligo RNA and oligo DNA.
- Examples include double-stranded oligonucleotides.
- double-stranded oligonucleotides include siRNA and heteroduplex oligonucleotides.
- a portion in which complementary nucleotide sequences are paired with each other is called a “complementary portion”.
- complementary portion means not only a complementary portion in an oligonucleotide containing a complementary portion but also a complementary portion in an oligonucleotide containing a complementary portion when the oligonucleotide containing a complementary portion is divided into oligonucleotide raw material fragments. It also refers to the part in the corresponding oligonucleotide material fragment.
- any one complementary nucleotide sequence in the complementary portion may be referred to as a "sense strand", and the other complementary nucleotide sequence may be referred to as an "antisense strand".
- the terms “sense” and “antisense” are merely convenient names referring to any one and the other of complementary portions, and are not intended to have biological meaning (in particular, RNAi).
- Oligonucleotides containing complementary portions may or may not include loop portions.
- the “loop part” refers to a linker that connects the sense side and the antisense side of the complementary part at the same end side (for example, the 5′ end and the 3′ end).
- Oligonucleotides containing complementary portions are used, inter alia, for post-transcriptional gene silencing (eg, RNA interference (RNAi)) effects.
- RNAi RNA interference
- the target modified oligonucleotide contains the above-mentioned modified residue in the complementary portion.
- modified oligonucleotide of interest include, for example, a double-stranded oligonucleotide containing a modified nucleotide residue or a loop-type oligonucleotide (e.g., a double-stranded oligonucleotide containing a modified nucleotide residue in a complementary portion or a loop-type oligonucleotide).
- a loop-type oligonucleotide containing a modified nucleotide residue or a residue other than the nucleotide residue (eg, an amino acid residue, a linker, etc.) in the loop portion eg, WO 2012/005368.
- the target modified oligonucleotide some nucleotide residues may be modified nucleotide residues, and all nucleotide residues may be modified nucleotide residues, but "modified nucleotide residues" Is a morpholino nucleic acid (PMO) residue, it is preferable that some nucleotide residues in the target modified oligonucleotide are morpholino nucleic acid (PMO) residues.
- the modified oligonucleotide of interest includes a gapmer which is an oligonucleotide having modified nucleotide residues at both ends of the sequence and having a gap region for recognition of RNase in the center of the sequence.
- the complementary portion in the modified oligonucleotide of interest has a length of 11 to 27 bases (eg, 12 to 27 bases, 15 to 27 bases, or 18 to 27 bases).
- the modified oligonucleotide of interest may be 11 to 27 bases in length when it is a double-stranded modified oligonucleotide consisting of a complementary portion only.
- the modified oligonucleotide of interest may have a non-complementary portion in addition to the complementary portion.
- the non-complementary portion may be 1 to 16 bases long, for example 1 to 10 bases long, preferably 1 to 5 bases long, more preferably 1, 2 or 3 bases long.
- the complementary portion of 11 to 27 bases may be in a continuous form, but mismatch base pairs as the non-complementary portion. It may be a non-continuous form divided by.
- the total number of residues of the modified oligonucleotide of interest may be appropriately selected depending on the function of the modified oligonucleotide of interest and each condition in the method of the present invention.
- the total number of residues of the modified oligonucleotide of interest may be, for example, 24-74.
- the modified oligonucleotide of interest is divided at the fragment junctions (also referred to as “cleavage sites”) that satisfy the following conditions (i) to (v).
- oligonucleotide raw material fragment (I) one or more fragment ligation sites are present in each strand of the complementary portion, and two or more fragment ligation sites are present in the modified oligonucleotide in total; (Ii) a protruding end (also referred to as a "sticky end") is formed in the complementary portion when the modified oligonucleotide is separated at the fragment junction, and the protruding end is 1-10 bases in length; (Iii) at least one oligonucleotide material fragment comprises a modified nucleotide, (Iv) Of the total four or more oligonucleotide raw material fragments, four oligonucleotide raw material fragments include a complementary portion having a length of 5 to 25 bases, and (v) each strand side of the complementary portion of the oligonucleotide raw material fragment. All of the base lengths corresponding to are 11 to 27 base lengths.
- the number of oligonucleotide raw material fragments is 4 or more, preferably 4 to 6 (4,5, 6).
- the number of oligonucleotide raw material fragments can also be characterized in terms of the number mainly corresponding to the sense strand and antisense strand constituting the target modified oligonucleotide (mainly double-stranded nucleic acid). From the above condition (i), it is understood that the number of oligonucleotide raw material fragments mainly corresponding to such sense strand and antisense strand is 2 or more, respectively.
- the number of oligonucleotide raw material fragments corresponding to such a sense strand and an antisense strand may be 3 or 4 fragments, and is preferably 2 or 3 fragments.
- the overhanging end in the above condition (ii) may be either a 5'overhanging end or a 3'overhanging end.
- the “complementary portion” in the above condition (iv) refers to a portion in the oligonucleotide raw material fragment corresponding to the complementary portion in the target modified oligonucleotide.
- fragment junction and “cleavage site” have the same meaning.
- “Fragment junction” (“cleavage site”) means a site set for the purpose of designing a combination of oligonucleotide raw material fragments, and does not mean a site actually cleaved in the method of the present invention.
- the four oligonucleotide raw material fragments in the above (iv) are preferably 5 to 25 bases long, more preferably 5 to 20 bases long, and even more preferably 5 to 17 bases long. It may be designed to include.
- the base length of the “complementary portion” in the above condition (iv) may be any base length that can form a pair, and may be one base length or more.
- four oligonucleotide raw material fragments out of all the oligonucleotide raw material fragments have a complementary portion of 17 base length or less. It is preferably designed to be
- the two strands constituting the complementary portion have a length of 5 to 25 bases (eg, 5 to 22 bases, 5 to 20 bases, 5 to 17 bases, 8 to 25 bases, 8 to 22 bases, 8 The length is preferably 20 to 20 bases, 8 to 17 bases).
- the protruding end is, for example, 1 to 10 bases long, preferably 1 to 8 bases long, more preferably 1 to 6 bases long, and even more preferably 2 to 6 bases long, 3 to 6 bases long or It is 4 to 6 bases long.
- the numerical value of the base length of the "complementary portion” and the numerical value of the base length of the protruding end under the above condition (iv) are set to values satisfying the above-mentioned range and mutually matching.
- the complementary portion may be 6-25 bases long to form the overhanging end, eg, if the overhanging end is 6 bases long, the complementary portion may be It may be 7-25 bases long to form overhanging ends.
- the portion other than the protruding end in the “complementary portion” of the four oligonucleotide raw material fragments defined by the above condition (iv) has a length of 4 to 24 bases, 4 to 21 bases, 4 to 19 bases, or 4 to It is preferably 16 bases long.
- each of the four oligonucleotide raw material fragments defined by the above condition (iv) has a length of 5 bases or more, preferably 6 bases or more, more preferably 7 bases or more, and even more preferably It may be 8 bases or more, particularly preferably 9 bases or more.
- Such four oligonucleotide raw material fragments also have a length of 19 bases or less, preferably 18 bases or less, more preferably 17 bases or less, still more preferably 16 bases or less, and particularly preferably 15 bases or less. It may be.
- Such four oligonucleotide raw material fragments also have a length of 5 to 19 bases, preferably 6 to 18 bases, more preferably 7 to 17 bases, still more preferably 8 to 16 bases, particularly preferably 9 bases. It may be up to 15 bases long.
- the 5'end of the oligonucleotide raw material fragment corresponding to the 5'end of the target modified oligonucleotide may remain the 5'-phosphate group, or may be substituted with 5'-OH.
- -A phosphate group modification may be introduced, or it may have the same structure as the 5'end of the modified oligonucleotide of interest. Examples of the 5'-phosphate group modification include those mentioned above. From the viewpoint of the ligation reaction with an oligonucleotide ligase, it is preferable that the 5'-ends of the other oligonucleotide raw material fragments remain as 5'-phosphate groups.
- the 3'end of the oligonucleotide raw material fragment corresponding to the 3'end of the modified oligonucleotide of interest may remain 3'-OH or may have a 3'-phosphate modification introduced, or
- the modified oligonucleotide of interest may have the same structure as the 3'end.
- Examples of the 3'-phosphate group modification include those mentioned above. From the viewpoint of the ligation reaction with an oligonucleotide ligase, it is preferable that the 3'end of the other oligonucleotide raw material fragment remains 3'-OH.
- the oligonucleotide raw material fragment may be in a free form, complexed, or immobilized.
- the oligonucleotide raw material fragment may be produced by a known chemical synthesis method or enzymatic synthesis method.
- Known chemical synthesis methods include solid-phase synthesis methods and liquid-phase synthesis methods, for example, methods described in WO 2012/157723 and WO 2005/070859.
- the oligonucleotide raw material fragment may have a functional part added at its corresponding part.
- Oligonucleotide ligase is an enzyme that links oligonucleotide raw material fragments together.
- the oligonucleotide raw material fragments are ligated at the “cleavage site” (also referred to as the “ligation site”) by the catalytic action of the oligonucleotide ligase to produce the modified oligonucleotide of interest.
- the oligonucleotide ligase include RNA ligase and DNA ligase.
- the RNA ligase may be either single-stranded RNA ligase or double-stranded RNA ligase, and double-stranded RNA ligase is preferable.
- double-stranded RNA ligase examples include Rnl2 family RNA ligase (sometimes referred to as "RNA ligase 2") and Rnl5 family RNA ligase.
- RNA ligase RNA ligase derived from any biological species or virus species may be used as long as the object of the present invention is achieved.
- T4 phage-derived RNA ligase T4 phage-derived RNA ligase 1, T4 RNA ligase 2 is used.
- T4 phage-derived DNA ligase DNA ligase derived from any biological species or virus species may be used as long as the object of the present invention is achieved, and for example, T4 phage-derived DNA ligase may be used.
- ligase treatment is a reaction in which oligonucleotide raw material fragments are ligated by the catalytic action of oligonucleotide ligase.
- the operation of ligase treatment is to mix the oligonucleotide raw material fragment and the oligonucleotide ligase.
- all the oligonucleotide raw material fragments and the oligonucleotide ligase may be mixed and the ligation reaction may be performed in one step.
- ligase treatment as a multi-step ligation reaction, a part of the oligonucleotide raw material fragments and the oligonucleotide ligase are mixed and the ligation reaction is performed, and then the remaining oligonucleotide raw material fragments and the reaction product are mixed.
- a ligation reaction may be performed.
- Mixing is done by adding the oligonucleotide ligase to the oligonucleotide starting material fragment, adding the oligonucleotide starting material fragment to the system containing the oligonucleotide ligase, or adding the oligonucleotide starting material fragment and the oligonucleotide ligase to the system for the reaction. May be that.
- An aqueous solution can be used as the system for ligase treatment.
- a buffer solution is preferable.
- the buffer solution include a phosphate buffer solution, a Tris buffer solution, a carbonate buffer solution, an acetate buffer solution, and a citrate buffer solution.
- the pH may be, for example, about 5-9.
- the pH may be 7.5 to 9.0 (for example, 8.0 to 8.5).
- the concentration of each oligonucleotide raw material fragment in the ligase treatment may be a concentration sufficient to dissolve the oligonucleotide raw material fragment and generate the target modified oligonucleotide.
- the concentration of each oligonucleotide raw material fragment may be, for example, 1 ⁇ M or more, 10 ⁇ M or more, 50 ⁇ M or more, 100 ⁇ M or more, 300 ⁇ M or more, 500 ⁇ M or more, or 1000 ⁇ M or more.
- the concentration of each oligonucleotide source fragment may also be, for example, 1 M, 100 mM, or 10 mM or less.
- each oligonucleotide raw material fragment is used at a concentration of 100 ⁇ M or more in the above concentration and a pH range of 7.5 to 9.0 in the above pH range. Is preferably used.
- the number of moles of all oligonucleotide raw material fragments in the ligase treatment is preferably approximately equal from the viewpoint of improving production efficiency by reducing the amount of unreacted oligonucleotide raw material fragments.
- the total molar ratio of any two oligonucleotide raw material fragments selected from a total of four or more oligonucleotide raw material fragments is, for example, 0.5 to 2, preferably 1/1.8 to 1.8, more preferably 1/1.5 to 1.5, even more preferably 1/1.2 to 1.2, particularly preferably 1/1.1 to It may be in the range of 1.1.
- the concentration of the oligonucleotide ligase in the ligase treatment may be a concentration sufficient to generate the modified oligonucleotide of interest.
- the concentration of the oligonucleotide ligase may be, for example, 0.01 U/ ⁇ L or higher, preferably 0.02 U/ ⁇ L or higher, more preferably 0.03 U/ ⁇ L or higher, even more preferably 0.04 U/ ⁇ L or higher.
- the concentration of the oligonucleotide ligase may be, for example, 1 U/ ⁇ L or less, preferably 0.5 U/ ⁇ L or less, more preferably 0.2 U/ ⁇ L or less, and even more preferably 0.1 U/ ⁇ L or less.
- the concentration of the oligonucleotide ligase is, for example, 0.01 to 1 U/ ⁇ L, preferably 0.02 to 0.5 U/ ⁇ L, more preferably 0.03 to 0.2 U/ ⁇ L, and even more preferably May be 0.04 to 0.1 U/ ⁇ L.
- the system for ligase treatment may contain a cofactor for oligonucleotide ligase.
- oligonucleotide ligase cofactors include ATP and divalent metal salts (eg, magnesium salts such as magnesium chloride).
- the treatment system may include a stabilizer for the oligonucleotide ligase.
- the stabilizer for oligonucleotide ligase include antioxidants (eg, reducing agents such as dithiothreitol and mercaptoethanol).
- the system for carrying out the ligase treatment may contain a surfactant in order to stably maintain the enzyme and improve the reaction rate.
- the surfactant examples include nonionic surfactants (eg, Triton series surfactants such as Triton X-100), and ionic surfactants.
- examples of the ionic surfactant include a cationic surfactant, an anionic surfactant, and an amphoteric surfactant.
- the system for ligase treatment may contain polyethylene glycol in order to improve the reaction rate.
- the ligase treatment system may have a low concentration of monovalent cation salt or be substantially free of monovalent cation salt.
- the monovalent cation salt concentration of the system to be treated may be, for example, 10 mM or less, preferably 1 mM or less, more preferably 0.1 mM or less, and even more preferably 0.01 mM or less.
- the treatment system may be substantially free of monovalent cation salts.
- Examples of the monovalent cation salt include monovalent cations such as lithium ion, sodium ion, potassium ion, rubidium ion, cesium ion, and ammonium ion, and anions such as fluoride ion, chloride ion, bromide ion, and iodide ion. And salt.
- the temperature in the ligase treatment may be a temperature sufficient to activate the oligonucleotide ligase.
- a temperature may be, for example, 2 to 50° C., preferably 16 to 50° C., more preferably 25 to 50° C.
- the ligase treatment may be performed for a time sufficient to generate the modified oligonucleotide of interest. Such time may be, for example, 1 to 72 hours.
- the modified oligonucleotide of interest is produced as a single double-stranded nucleic acid (eg, siRNA, heteroduplex oligonucleotide).
- the sense strand and the antisense strand constituting such a single double-stranded nucleic acid have N and M base lengths, respectively.
- the N and M base lengths are each independently 11 to 30 base length (eg, 18 to 30 base length).
- the N and M base lengths may each independently be 11 to 27 base lengths (eg, 18 to 27 base lengths).
- the impurities of the intended modified oligonucleotide are intended to be nucleic acid contaminants other than the above-mentioned intended modified oligonucleotide).
- the sense strand and antisense strand constituting such a nucleic acid contaminant are not N and M bases in length, respectively, but (N ⁇ ) and M bases in length, N and (M ⁇ in length, respectively. ) Base lengths or (N ⁇ ) and (M ⁇ ) base lengths.
- N and M are the same as above, and ⁇ and ⁇ are, for example, 1, 2, or 3.
- the method of the present invention may include a step of synthesizing an oligonucleotide raw material fragment (eg, chemical synthesis such as solid phase synthesis). Since the method of the present invention can suppress the production of nucleic acid contaminants other than the desired modified oligonucleotide, the purification of the desired modified oligonucleotide from the sample of the synthesized oligonucleotide raw material fragment can be omitted.
- an oligonucleotide raw material fragment eg, chemical synthesis such as solid phase synthesis
- the method of the present invention is a double-stranded nucleic acid contaminant caused by a small amount of a non-target oligonucleotide raw material fragment that can remain after the purification even when the target modified oligonucleotide is purified ( Since the production of impurities) can be suppressed, the target modified oligonucleotide may be purified.
- Such purification can be performed, for example, by a method such as chromatography (eg, HPLC, IEX), gel filtration and the like.
- the method of the present invention may include a reaction termination step after the ligase treatment step.
- the reaction stopping step include high temperature treatment (eg, 80° C.), oligonucleotide/ligase deactivation treatment by addition of acid/alkali, organic solvent, and removal of cofactor metal ion by addition of chelating agent such as EDTA. Can be mentioned. Further, a method in which an enzyme is immobilized on a carrier to carry out a reaction, and the enzyme is removed from the reaction solution by membrane separation is also mentioned.
- the method of the present invention may include a step of purifying the modified oligonucleotide of interest after the ligase treatment step.
- this step can be performed by any appropriate method such as chromatography (eg, HPLC), gel filtration, or the like.
- the oligonucleotide raw material fragments are annealed, generally, in order to bring the oligonucleotide raw material fragments into a denatured state (non-pairing state), the oligonucleotide raw material fragment mixed solution is heated to a high temperature, and then the complementary nucleotide sequences are In order to form the pairing, an operation of gradually cooling the high-temperature oligonucleotide raw material fragment mixed solution by air cooling or the like is often performed.
- the ligase treatment step can be simplified without performing a heating operation at a high temperature for such denaturation and a cooling treatment for heat treatment (heating-cooling treatment).
- the desired modified oligonucleotide can be produced by the procedure described above.
- the high temperature for example, the oligonucleotide raw material fragment mixed solution is kept at 65°C or higher, 70°C or higher, 75°C or higher, 80°C or higher, 85°C or higher, 90°C or higher, 95°C or higher, or 100°C or higher. (Eg, 5 minutes or more, or 10 minutes or more).
- the oligonucleotide raw material fragment mixed solution is allowed to stand at room temperature (eg, 15 to 25° C., or 20 to 25° C.) or a predetermined temperature (eg, 37° C.) (eg, 5 hours or more). ) And maintaining the oligonucleotide raw material fragment mixed solution at a predetermined temperature (eg, 37° C.) (eg, 15 minutes or more).
- the time for which the oligonucleotide raw material fragment mixed solution is kept at a high temperature before the ligase treatment step is, for example, less than 5 minutes, 4.5 minutes or less, 4 minutes or less, 3.5 minutes. It may be controlled to be 3 minutes or less, 2.5 minutes or less, 2 minutes or less, 1.5 minutes or less, 1 minute or less, or 0.5 minutes or less.
- the solution containing the oligonucleotide raw material fragments is kept at 2 to 50° C. until the oligonucleotide raw material fragments are mixed in the solution and the ligase treatment step is performed. It may be held. That is, in the method of the present invention, any mixing of all the oligonucleotide raw material fragments and oligonucleotide ligases contained in the above combination, any interval between mixings, and the ligase reaction may be carried out under the condition of 2 to 50°C. Good.
- the method of the invention comprises: (1) All the oligonucleotide raw material fragments and the oligonucleotide ligases contained in the above combination present in separate systems are mixed at 2 to 50° C. and the mixture is mixed at 2 to 50° C. Mixing under conditions maintained at 50° C. to obtain a mixed solution; and (2) reacting while maintaining the mixed solution at 2 to 50° C. to obtain a solution containing the modified oligonucleotide of interest ..
- all oligonucleotide raw material fragments included in the above combination are obtained in separate systems.
- the present invention provides, for example, mixing the oligonucleotide raw material fragments at 2 to 50°C to obtain an oligonucleotide raw material fragment mixture, and mixing the oligonucleotide raw material fragment mixture and the oligonucleotide ligase at 2 to 50°C. It may be performed by mixing.
- the present invention may be carried out, for example, by sequentially adding the oligonucleotide raw material fragments to a solution containing the oligonucleotide ligase at 2 to 50°C.
- the method of the present invention can be used, for example, in industrial production of a modified oligonucleotide of interest on a large scale.
- Example 1 Comparison of patterns of combination of fragments using natural RNA 1) Synthesis of substrate and product preparation In a reaction for enzymatically synthesizing siRNA from four fragments of short natural RNA, The effect of base length was evaluated.
- the target siRNA was a double strand consisting of RNA1-S (21mer) and RNA1-A (23mer) in Table 1 (hereinafter, referred to as sense strand and antisense strand, respectively).
- 18 kinds of RNA fragments shown in Table 1 were synthesized, and using these, the combinations of 6 patterns of fragments shown in Table 2 were evaluated.
- T4 RNA ligase 2 Reaction was performed with T4 RNA ligase 2 (New England Biolabs) using the oligonucleotides of 4 fragments.
- the composition of the reaction solution was 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5, and each RNA fragment was 10 ⁇ M, and the reaction solution volume was 10 ⁇ L.
- the concentrations of the products were compared by setting the added concentrations of the enzyme to 0.025, 0.05, 0.1 and 0.2 U/ ⁇ L. After reacting at 25°C for 1 hour using a thermal cycler, the reaction was stopped by heating at 80°C for 5 minutes.
- Example 2 Evaluation of influence of reaction temperature on ligation reaction of natural RNA The influence of reaction temperature on the ligation reaction of a short chain was examined.
- the oligonucleotide of 1 in Table 2 was used as a substrate and reacted with T4 RNA ligase 2.
- the composition of the reaction solution was 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5, enzyme concentration 0.2 U/ ⁇ L, RNA fragment was 10 ⁇ M each, and the reaction solution volume was 10 ⁇ L.
- the reaction temperature was set to 16° C., 25° C., 30° C. and 37° C., and the reaction was stopped by heating at 80° C. for 5 minutes after reacting for 1, 2 and 4 hours, respectively.
- the concentration of the ligation product contained in the reaction solution was analyzed by HPLC under the conditions described in Example 1.
- Example 3 Confirmation of reaction progress using modified RNA 1) Synthesis of substrate and product preparation The progress of enzymatic ligation reaction of siRNA from 4 fragments in modified oligonucleotide was evaluated.
- the target siRNA was a double strand consisting of a sense strand (MOD1-S) and an antisense strand (MOD1-A) shown in Table 3.
- This siRNA has the same nucleotide sequence as the natural RNA used in Examples 1 and 2, but all residues are modified with 2'-F or 2'-O-methyl, and some of the phosphates are The group is substituted with a thiophosphate group.
- 4 fragments shown in Table 3 were synthesized as the respective fragments. The nucleotide sequences of these 4 fragments are the same as the combination of No. 1 in Table 2.
- T4 RNA ligase 2 Reaction was performed with T4 RNA ligase 2 using the modified RNA of 4 fragments.
- the composition of the reaction solution was 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5, and each modified RNA fragment was 10 ⁇ M, and the reaction solution volume was 50 ⁇ L.
- the enzyme was added at a concentration of 0.2 or 1.0 U/ ⁇ L, and as a negative control, the reaction was performed without addition of the enzyme. After reacting at 25°C for 1 hour using a thermal cycler, the reaction was stopped by heating at 80°C for 5 minutes.
- the composition of the reaction solution was DNA ligase 50 mM Tris-HCl, 10 mM MgCl 2 , 10 mM dithiothreitol, 1 mM ATP, pH 7.5.
- the enzyme concentration was 470 nM
- each oligonucleotide fragment was 10 ⁇ M
- the reaction solution volume was 30 ⁇ L.
- the combinations shown in Table 3 were used.
- the reaction was performed at 25° C. using a thermal cycler, 10 ⁇ L was sampled after 4 hours, and the reaction was stopped by heating at 80° C. for 5 minutes.
- the concentration of the ligation product contained in the reaction solution was analyzed by HPLC under the conditions described in Example 3. The standard product was also analyzed in the same manner to quantify the concentration of the ligation product.
- RNA ligase and ligation reaction (1) Construction of Recombinant Expression Strain by E. coli
- the RNA ligase DraRnl belonging to the Rn15 family derived from Deinococcus radiodurans was transformed into E. coli.
- a strain expressing E. coli was constructed and purified enzyme was prepared.
- the amino acid sequence of DraRnl (SEQ ID NO: 17) was transformed into E.
- a plasmid having a base sequence optimized for the E. coli codon was prepared by total gene synthesis, and then this sequence was subcloned into the NdeI/BamHI site of the pET16b vector.
- This expression plasmid was transformed into E. E. coli BL21(DE3) was transformed to obtain an expression strain of DraRnl. In this expression strain, DraRnl having His-tag added to the N-terminus is expressed.
- the obtained soluble fraction was applied to a His-TAG protein purification column HisTALON Superflow Cartridge (Takara Bio) equilibrated with the above-mentioned buffer and adsorbed on a carrier. Proteins that were not adsorbed on the carrier (non-adsorbed proteins) were washed off with a buffer solution consisting of 50 mM Tris-HCl (pH 7.6), 250 mM NaCl, 10% sucrose, and 15 mM imidazole, and then 50 mM Tris-HCl (pH 8. 0), 250 mM NaCl, 10% glycerol, and 200 mM imidazole were used to elute the adsorbed protein.
- a buffer solution consisting of 50 mM Tris-HCl (pH 7.6), 250 mM NaCl, 10% sucrose, and 15 mM imidazole, and then 50 mM Tris-HCl (pH 8. 0), 250 mM NaCl, 10%
- the elution fractions containing the enzyme were collected, and using Amicon Ultra-15 10 kDa (Merck Millipore), 50 mM Tris-HCl (pH 8.0), 200 mM NaCl, 2 mM DTT, 2 mM EDTA, 10% glycerol, 0.1
- the buffer was exchanged with a buffer consisting of% Triton X-10 to obtain a purified enzyme solution.
- the reaction solution was analyzed by HPLC using an ACQUITY HPLC Oligonucleotide BEH C18 Column (Waters, 2.1 ⁇ 100 mm, 1.7 ⁇ m).
- the analysis conditions were a column temperature of 60° C., a detection wavelength of 260 nm, an injection amount of 10 ⁇ L, and a flow rate of 0.4 mL/min.
- the mobile phase was analyzed by a linear gradient using solution A hexafluoroisopropanol-triethylamine and solution B (methanol).
- the production of the ligation product was confirmed by analyzing the preparations of the sense strand and the antisense strand in the same manner.
- Example 6 Production of modified oligonucleotide having loop structure The progress of the reaction for producing a modified oligonucleotide having a loop structure was evaluated by enzymatic ligation from oligonucleotides of 4 fragments. The sequences of the desired product and the synthesized substrate fragment are shown in Table 4.
- Reaction was performed with T4 RNA ligase 2 (New England Biolabs) using the substrate oligonucleotides of the four fragments in Table 4.
- the composition of the reaction solution was 50 mM Tris-HCl, 2 mM MgCl2, 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5, and each oligonucleotide fragment was 10 ⁇ M, and the reaction solution volume was 100 ⁇ L.
- the concentrations of the products were compared at an enzyme addition concentration of 17.8 ⁇ g/ ⁇ L.
- As a negative control the reaction was performed under the condition that no enzyme was added. After reacting at 25° C. for 3 hours using a thermal cycler, the reaction was stopped by heating at 80° C. for 5 minutes.
- the reaction solution was analyzed by HPLC and LC-TOF/MS under the conditions of Example 5.
- the target modified oligonucleotide having a loop structure can be produced from 4 fragments by T4 RNA ligase 2.
- Example 7 Generation of heteroduplex composed of DNA chain and RNA chain A heteroduplex composed of a modified DNA chain and a modified RNA chain was produced by a double-stranded RNA ligase. T4 RNA ligase 2 (New England Biolabs) was used as the double-stranded RNA ligase.
- the composition of the reaction solution was 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5.
- the enzyme concentration was 3.56 ⁇ g/mL
- the substrate was 10 ⁇ M each of the four fragment oligonucleotides shown in Table 5, and the reaction volume was 40 ⁇ L.
- the reaction was carried out at 37° C. using a thermal cycler, and 18 hours later, the reaction was stopped by adding EDTA to a final concentration of 10 mM. As a negative control, the reaction was performed under the condition that no enzyme was added.
- the concentration of the ligation product contained in the reaction solution was analyzed by HPLC and LC-TOF/MS under the conditions described in Example 5.
- T4 RNA ligase 2 can generate a heteroduplex composed of a modified DNA chain and a modified RNA chain.
- Example 8 Reaction Using Oligonucleotide Fragments Containing Mismatched Base Pairs
- the progress of the reaction for producing a double-stranded modified oligonucleotide having a mismatched portion in the base pair is evaluated by enzymatic ligation from 4 fragments of the oligonucleotide. did.
- the respective chains of the desired product were designated as A chain and B chain.
- the sequences of the target product and the synthesized substrate fragment are shown in Table 6, and the combination of 4 fragments is shown in FIG.
- the composition of the reaction solution was 1.78 ⁇ g/mL T4 RNA ligase 2 (New England Biolabs), 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5.
- As a substrate 4 oligonucleotide fragments were added to each so that the final concentration was 10 ⁇ M, and the reaction was carried out at a volume of 40 ⁇ L.
- the reaction was performed at 25° C. using a thermal cycler, and 4 hours later, the reaction was stopped by adding EDTA to a final concentration of 10 mM.
- As a negative control the reaction was performed under the condition that no enzyme was added.
- the concentration of the ligation product contained in the reaction solution was analyzed by HPLC and LC-TOF/MS under the conditions described in Example 5.
- T4 RNA ligase 2 can generate a double-stranded modified oligonucleotide having a mismatched sequence.
- Example 9 Reaction using oligonucleotides of 5 and 6 fragments The progress of the reaction for producing a double-stranded modified oligonucleotide was evaluated from the oligonucleotides of 5 and 6 fragments by enzymatic ligation. The respective chains of the desired product were designated as A chain and B chain. The sequences of the target product and the synthesized substrate fragment are shown in Table 7, and the combination of 4 fragments is shown in FIG.
- the composition of the reaction solution was 1.78 ⁇ g/mL T4 RNA ligase 2 (New England Biolabs), 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5. Oligonucleotide fragments were added as substrates to a final concentration of 10 ⁇ M, and reacted in a volume of 30 ⁇ L. The reaction was carried out at 37° C. using a thermal cycler, and 16 hours later, the reaction was stopped by adding EDTA to a final concentration of 10 mM. As a negative control, the reaction was performed under the condition that no enzyme was added. The concentration of the ligation product contained in the reaction solution was analyzed by HPLC under the conditions described in Example 5.
- T4 RNA ligase 2 can generate a double-stranded modified oligonucleotide from 5 and 6 fragment substrate oligonucleotides.
- Example 10 Reaction using an oligonucleotide having a DMTr group added to the 5'end A two-fragment oligonucleotide containing two fragments having a dimethoxytrityl (DMTr) group added to the 5'end was subjected to enzymatic ligation to obtain two fragments. The progress of the reaction for producing the double-stranded modified oligonucleotide was evaluated. The respective chains of the desired product were designated as A chain and B chain. The sequences of the target product and the synthesized substrate fragment are shown in Table 8, and the combination of 4 fragments is shown in FIG.
- DMTr dimethoxytrityl
- the composition of the reaction solution was 1.78 ⁇ g/mL T4 RNA ligase 2 (New England Biolabs), 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5.
- As a substrate four oligonucleotide fragments were added to each to give a final concentration of 10 ⁇ M, and reacted at a volume of 40 ⁇ L.
- the reaction was performed at 25° C. using a thermal cycler, and 4 hours later, the reaction was stopped by adding EDTA to a final concentration of 10 mM.
- As a negative control the reaction was performed under the condition that no enzyme was added.
- the concentration of the ligation product contained in the reaction solution was analyzed by HPLC and LC-TOF/MS under the conditions described in Example 5.
- T4 RNA ligase 2 can generate a double-stranded modified oligonucleotide from a substrate fragment having a DMTr group.
- Example 11 Reaction using a carrier-added oligonucleotide fragment A double-stranded modified oligo was enzymatically ligated from a 4-fragment oligonucleotide containing 1 fragment having N-acetylgalactosamine (GalNAc) added to the 5'end. The progress of the reaction producing nucleotides was evaluated. The respective chains of the desired product were designated as A chain and B chain. The sequences of the target product and the synthesized substrate fragment are shown in Table 9, and the combination of 4 fragments is shown in FIG.
- the fragment modified with GalNAc was synthesized by linking Trivalent ⁇ -D-GalNAc with carboxyl-functionalized PEG5 Linker (manufactured by Wales) to the 5′ end of the oligonucleotide via an amino C6 linker.
- the composition of the reaction solution was 1.78 ⁇ g/mL T4 RNA ligase 2 (New England Biolabs), 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5.
- As a substrate four oligonucleotide fragments were added to each to give a final concentration of 10 ⁇ M, and reacted at a volume of 40 ⁇ L.
- the reaction was performed at 25° C. using a thermal cycler, and 4 hours later, the reaction was stopped by adding EDTA to a final concentration of 10 mM.
- As a negative control the reaction was performed under the condition that no enzyme was added.
- the ligation product contained in the reaction solution was analyzed by HPLC and LC-TOF/MS under the conditions described in Example 5.
- a double-stranded modified oligonucleotide having the terminal modified with N-acetylgalactosamine can be produced by the reaction from four fragments using T4 RNA ligase 2.
- Example 12 Generation reaction of double-stranded modified oligonucleotide having thiophosphoric acid diester bond at the connecting portion
- a double-stranded modified oligonucleotide was enzymatically ligated from an oligonucleotide of 4 fragments in which a phosphoric acid group was replaced with a thiophosphoric acid group.
- the progress of the reaction for producing was evaluated.
- the respective chains of the desired product were designated as A chain and B chain.
- the sequences of the target product and the synthesized substrate fragment are shown in Table 10, and the combination of 4 fragments is shown in FIG.
- the composition of the reaction solution was 1.78 ⁇ g/mL T4 RNA ligase 2 (New England Biolabs), 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5.
- As a substrate four oligonucleotide fragments were added to each to give a final concentration of 10 ⁇ M, and reacted at a volume of 40 ⁇ L.
- the reaction was performed at 25° C. using a thermal cycler, and 4 hours later, the reaction was stopped by adding EDTA to a final concentration of 10 mM.
- As a negative control the reaction was performed under the condition that no enzyme was added.
- the ligation product contained in the reaction solution was analyzed by HPLC and LC-TOF/MS under the conditions described in Example 5.
- a double-stranded modified oligonucleotide having a thiophosphate bond at the junction can be produced by the reaction from 4 fragments using T4 RNA ligase 2.
- Example 13 Production reaction of hairpin-type oligonucleotides The progress of the reaction for producing hairpin-type oligonucleotides by enzymatic ligation from the oligonucleotides of four fragments was evaluated. The sequences of the target product and the synthesized substrate fragment are shown in Table 11, and the combination of 4 fragments is shown in FIG. As a linker (Hamasaki T, Suzuki H, Shirohzu H, et al. Efficacy of a novel class of RNA interference therapeutic agents. 55).
- the composition of the reaction solution was 1.78 ⁇ g/mL T4 RNA ligase 2 (New England Biolabs), 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5.
- As a substrate four oligonucleotide fragments were added to each to give a final concentration of 10 ⁇ M, and reacted at a volume of 40 ⁇ L.
- the reaction was performed at 25° C. using a thermal cycler, and 4 hours later, the reaction was stopped by adding EDTA to a final concentration of 10 mM.
- As a negative control the reaction was performed under the condition that no enzyme was added.
- the ligation product contained in the reaction solution was analyzed by HPLC and LC-TOF/MS under the conditions described in Example 5.
- a hairpin type oligonucleotide can be produced by the reaction from the four fragments using T4 RNA ligase 2.
- Example 14 Effect of Base Length of Overhanging End on Reactivity
- the substrate oligonucleic acid was designed so as to form a protruding end having a length of 1 to 6 bases, and the difference in the base length of the overhanging end was designed.
- the respective chains of the desired product were designated as A chain and B chain.
- the substrates constituting the B chain had a common sequence, and the cleavage positions of the A chain were different positions.
- the sequences of the synthesized target product preparation and the substrate fragment are shown in Table 12, and the combination of 4 fragments is shown in FIG.
- the composition of the reaction solution was 1.78 ⁇ g/mL T4 RNA ligase 2 (New England Biolabs), 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5.
- As a substrate four oligonucleotide fragments were added to each to give a final concentration of 10 ⁇ M, and reacted at a volume of 40 ⁇ L.
- the reaction was carried out at 25° C. using a thermal cycler, and after 15 minutes, 30 minutes, 1 hour, 2 hours, and 4 hours, 5 ⁇ L was collected and EDTA was added to a final concentration of 10 mM to stop the reaction.
- As a negative control the reaction was performed under the condition that no enzyme was added.
- the concentrations of the ligation product and the product preparation contained in the reaction solution were analyzed by HPLC under the conditions described in Example 5, and the concentration of the ligation product was calculated.
- Example 15 Effect of base length of product on reactivity Differences in reactivity when a short target product consisting of a complementary portion of 11 to 14 base length was produced were compared.
- the respective chains of the desired product were designated as A chain and B chain.
- the sequences of the target product and the synthesized substrate fragment are shown in Table 13, and the combination of 4 fragments is shown in FIG.
- the composition of the reaction solution was 1.78 ⁇ g/mL T4 RNA ligase 2 (New England Biolabs), 50 mM Tris-HCl, 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5.
- As a substrate four oligonucleotide fragments were added to each to give a final concentration of 10 ⁇ M, and reacted at a volume of 40 ⁇ L.
- the reaction was performed at 25° C. using a thermal cycler, and 4 hours later, the reaction was stopped by adding EDTA to a final concentration of 10 mM.
- As a negative control the reaction was performed under the condition that no enzyme was added.
- the ligation product contained in the reaction solution was analyzed by HPLC and LC-TOF/MS under the conditions described in Example 5.
- Residual rate (%) (sum of substrate peak areas under enzyme addition conditions) /Sum of substrate peak areas in negative control) x 100
- the product consisting of the complementary portion having a length of 11 bases tended to have a higher residual ratio of the substrate, compared with the product consisting of the complementary portion having a length of 12 bases or more.
- Example 16 Reaction with High Concentration Substrate
- the reaction rates of modified oligonucleotide production in the presence of a higher concentration of substrate were compared at each pH.
- the respective chains of the desired product of the reaction were designated as A chain and B chain.
- the sequences of the target product and the synthesized substrate fragment are shown in Table 15, and the combination of 4 fragments is shown in FIG.
- the composition of the reaction solution was 1.78 ⁇ g/mL T4 RNA ligase 2 (New England Biolabs), 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, and 50 mM Tris-HCl (pH 7.0 to 9.0) as a buffer. Using. Oligonucleotide fragments were added as substrates so that the final concentration was 10 ⁇ M, 300 ⁇ M, 500 ⁇ M, or 1000 ⁇ M, and the reaction was carried out at a volume of 40 ⁇ L. The reaction was carried out at 25° C.
- the ligation product contained in the reaction solution was analyzed by HPLC under the conditions described in Example 5, and the production rate was calculated from the total concentration of A chain and B chain. The progress of the reaction was confirmed even at a substrate concentration of 300 ⁇ M or higher, and at these concentrations, a high reaction rate was exhibited at pH 8.0 and 8.5.
- Example 17 Effect of Addition of Surfactant
- the reaction rate of modified oligonucleotide formation by addition of a surfactant was evaluated.
- the sequences of the target product and the synthesized substrate fragment are shown in Table 15, and the combination of 4 fragments is shown in FIG.
- the composition of the reaction solution was 1.78 ⁇ g/mL T4 RNA ligase 2 (New England Biolabs), 2 mM MgCl 2 , 1 mM dithiothreitol, 400 ⁇ M ATP, 50 mM Tris-HCl (pH 7.5), and a final concentration as a surfactant. 0.1% Triton X-100 was used.
- Oligonucleotide fragments were added as substrates so that the final concentration was 20 ⁇ M, and the reaction was carried out at a volume of 40 ⁇ L.
- the enzyme solution was adjusted to 17.8 ⁇ g/mL with storage buffer (10 mM Tris-HCl, 50 mM KCl, 35 mM ammonium sulfate, 0.1 mM dithiothreitol, 0.1 mM EDTA, 50% glycerol, pH 7.5).
- test condition 1 The condition that 1/10 amount is added to the reaction solution after dilution (control condition), the reaction solution containing Triton X-100 with a final concentration of 0.1% after diluting the enzyme solution with the storage buffer to 17.8 ⁇ g/mL 1/10 amount (test condition 1), the enzyme solution was diluted to 17.8 ⁇ g/mL with a storage buffer containing 0.1% Triton X-100, and then 0.09% Triton X-100 was added. Three conditions were compared: a condition of adding 1/10 amount to the reaction solution (test condition 2, Triton X-100 final concentration 0.1%). The reaction was carried out at 25° C.
- Example 18 Comparison of Selectivity of Impurities Due to Difference in Base Length of Product Substrates and products were subjected to reactions having different base lengths, and impurities contained in the substrate oligonucleic acid fragment and the solution after the enzymatic reaction were analyzed. ..
- the respective chains of the desired product of the reaction were designated as A chain and B chain.
- the sequences of the target product and the synthesized substrate fragment are shown in Table 16, and the combination of 4 fragments is shown in FIG.
- the composition of the reaction solution was 8.9 ⁇ g/mL T4 RNA ligase 2 (New England Biolabs), 50 mM Tris-HCl, 2 mM MgCl 2, 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5 in the reaction using the modified oligonucleotide. ..
- T4 RNA ligase 2 New England Biolabs
- 50 mM Tris-HCl, 2 mM MgCl 2, 1 mM dithiothreitol, 400 ⁇ M ATP, pH 7.5 was used.
- oligonucleotide fragments were added to each to give a final concentration of 50 ⁇ M, and the reaction was carried out in a volume of 30 ⁇ L. Reaction was carried out at 25° C. using a thermal cycler, and after 8 hours, EDTA was added so that the final concentration was 12.5 mM to stop the reaction.
- the reaction solution was analyzed by HPLC and LC-TOF/MS under the conditions described in Example 5 to confirm the intended reaction progress.
- the content of impurities (N ⁇ 1 mer) in the product of the target structure is based on the method of the prior literature (Roussis et al., Journal of Chromatogr A. 2019; 1584:106-114.). Was calculated.
- the substrate oligonucleotide solution used in the reaction was also analyzed by LC-TOF/MS to calculate the content rate of impurities (N ⁇ 1 mer) with respect to the substrate.
- the results are shown in Table 17.
- the residual rate of impurities (N ⁇ 1 mer) is 86% or more in the reaction that produces an oligonucleic acid having a length of 28 bases, whereas it is 23 to 59% in the reaction that produces an oligonucleic acid having a length of 25 bases or less. It was
- the present invention is useful for producing modified oligonucleotides (eg, siRNA, heteroduplex oligonucleotides, etc.) that can be used in products such as nucleic acid drugs.
- modified oligonucleotides eg, siRNA, heteroduplex oligonucleotides, etc.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Biomedical Technology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
- Plant Pathology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
本発明は、siRNAやヘテロ二本鎖オリゴヌクレオチド等の相補部分を含むオリゴヌクレオチドの効率的な製造方法を提供する。より具体的には、11~27塩基長の相補部分を含む修飾オリゴヌクレオチドの製造方法であって、 該方法は、合計4本以上のオリゴヌクレオチド原料断片をオリゴヌクレオチドリガーゼの存在下で処理して該修飾オリゴヌクレオチドを生成することを含み、 該合計4本以上のオリゴヌクレオチド原料断片は、該修飾オリゴヌクレオチドを、下記条件(i)~(v)を満たす断片連結部で分けた場合に得られるオリゴヌクレオチド原料断片に相当する、方法: (i)断片連結部は、相補部分の各鎖側中のそれぞれに1以上存在し、かつ断片連結部は、該修飾オリゴヌクレオチド中に合計2つ以上存在し; (ii)該修飾オリゴヌクレオチドを断片連結部で分けた場合に、突出末端が相補部分中に形成され、かつ該突出末端が1~10塩基長であり; (iii)少なくとも1本のオリゴヌクレオチド原料断片が、修飾ヌクレオチドを含み、 (iv)該合計4本以上のオリゴヌクレオチド原料断片のうち4本のオリゴヌクレオチド原料断片が、5~25塩基長の相補部分を含み、かつ (v)オリゴヌクレオチド原料断片の相補部分の各鎖側に相当する塩基長の合計が、いずれも11~27塩基長である、方法を提供する。
Description
本発明は、相補部分を含む修飾オリゴヌクレオチドの製造方法に関する。
siRNA、アンチセンス等のオリゴヌクレオチドは、核酸医薬としての有用性が示され、近年、開発が活発化している。オリゴヌクレオチドは、主に合成法にて製造されており、例えば、ホスホロアミダイト法など固相合成にてヌクレオチド残基を1塩基ずつ順次直列的に伸長合成することにより製造することができる。しかしながら、この手法は、オリゴヌクレオチドの鎖長が長くなるにつれて産物の純度および収量が低下したり、製造効率が低いなどの課題を有している。そこで、オリゴヌクレオチドを各短鎖断片として合成し、それらを縮合して目的のオリゴヌクレオチドを得る並列的合成手法が求められている。
特許文献1には、目的のオリゴヌクレオチドを分割した断片に相当する複数のオリゴヌクレオチド原料断片を、目的のオリゴヌクレオチドに相補的な鋳型オリゴヌクレオチドとアニールさせ、アニールしたオリゴヌクレオチド原料断片同士を酵素的に縮合させ、得られた目的のオリゴヌクレオチド鎖を鋳型オリゴヌクレオチドから分離することにより、一本鎖オリゴヌクレオチドを製造することが記載されている。
非特許文献1には、オリゴDNA断片とPEG化オリゴDNA断片を、付着性末端においてDNAリガーゼで連結させることによる、オリゴDNAのPEG化が記載されている。しかし、非特許文献1では天然型オリゴDNA断片でしか実施していないため、アニーリング能の低下が示唆される短鎖かつ修飾型塩基を含むオリゴヌクレオチドを原料として用いた、相補部分を含むオリゴヌクレオチドの酵素的縮合が可能であるかどうかは不明である。
非特許文献2および3には、1本のオリゴヌクレオチド鎖とこれに相補的な2本のオリゴヌクレオチド原料断片をアニールさせることにより形成されるニックを、リガーゼで連結させることが記載されている。
非特許文献4には、付着性末端を有する24mer二本鎖オリゴRNAをRNAリガーゼで連結させて、48mer以上の大きさを有する二本鎖RNAを形成させることが記載されている。しかしながら、非特許文献4では、基質は24塩基長、生成物も48塩基長といずれも長いことから、基質のアニーリング能が高い条件でしか実施していないため、アニーリング能が著しく低下することが示唆される短鎖かつ修飾型塩基の導入による酵素の連結活性の低下が予想されるオリゴヌクレオチド基質を用いた場合に2箇所以上の縮合点を有する酵素反応が進行するかは知られていなかった。
非特許文献5には、siRNAを合成的に作成したことが記載されている。
非特許文献6には、RNAリガーゼDraRnlがRnl5ファミリーに含まれることが記載されている。
非特許文献1には、オリゴDNA断片とPEG化オリゴDNA断片を、付着性末端においてDNAリガーゼで連結させることによる、オリゴDNAのPEG化が記載されている。しかし、非特許文献1では天然型オリゴDNA断片でしか実施していないため、アニーリング能の低下が示唆される短鎖かつ修飾型塩基を含むオリゴヌクレオチドを原料として用いた、相補部分を含むオリゴヌクレオチドの酵素的縮合が可能であるかどうかは不明である。
非特許文献2および3には、1本のオリゴヌクレオチド鎖とこれに相補的な2本のオリゴヌクレオチド原料断片をアニールさせることにより形成されるニックを、リガーゼで連結させることが記載されている。
非特許文献4には、付着性末端を有する24mer二本鎖オリゴRNAをRNAリガーゼで連結させて、48mer以上の大きさを有する二本鎖RNAを形成させることが記載されている。しかしながら、非特許文献4では、基質は24塩基長、生成物も48塩基長といずれも長いことから、基質のアニーリング能が高い条件でしか実施していないため、アニーリング能が著しく低下することが示唆される短鎖かつ修飾型塩基の導入による酵素の連結活性の低下が予想されるオリゴヌクレオチド基質を用いた場合に2箇所以上の縮合点を有する酵素反応が進行するかは知られていなかった。
非特許文献5には、siRNAを合成的に作成したことが記載されている。
非特許文献6には、RNAリガーゼDraRnlがRnl5ファミリーに含まれることが記載されている。
Sosic A, Pasqualin M, Pasut G, Gatto B. 2014. Enzymatic formation of PEGylated oligonucleotides. Bioconjug Chem 25:433-441.
Bullard, D. R., & Bowater, R. P. (2006). Direct comparison of nick-joining activity of the nucleic acid ligases from bacteriophage T4. Biochem. J, 398, 135-144.
Nandakumar, J., & Shuman, S. (2004). How an RNA Ligase Discriminates RNA versus DNA Damage. Molecular Cell, 16(2), 211-221.
Nandakumar J, Ho CK, Lima CD, Shuman S. 2004. RNA substrate specificity and structure-guided mutational analysis of bacteriophage T4 RNA ligase 2. J Biol Chem 279:31337-31347.
Jayaprakash K. Nair, et al. (2014), Multivalent N-Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J. Am. Chem. Soc., 136, 16958-16961.
MIHAELA-CARMEN UNCIULEAC and STEWART SHUMAN (2019),RNA 21:824-832
本発明の目的は、siRNAやヘテロ二本鎖オリゴヌクレオチド等の相補部分を含むオリゴヌクレオチドの効率的な製造方法を提供することである。
本発明者らは、鋭意検討した結果、目的の相補部分を含むオリゴヌクレオチドの両相補部分を分割した断片に相当する4本以上のオリゴヌクレオチド原料断片をオリゴヌクレオチドリガーゼで処理することにより、二本鎖などの相補部分を含むオリゴヌクレオチドを直接構築することを見出し、固相合成などの直列合成法に比し高い製造効率、高い純度で製造できることなどを見出し、本発明を完成するに至った。
従来、siRNAなど、比較的短い二本鎖オリゴヌクレオチドは、化学合成が容易かつ簡便であったことから、化学合成により製造されていた。具体的には、構成する2本のオリゴヌクレオチド鎖をそれぞれ化学合成(例、固相合成)し、それらを精製後、両鎖をアニーリングさせることによって製造されていた。そのため、酵素的縮合を用いる方法はさほど報告されておらず、その中でも、4本以上のオリゴヌクレオチド原料断片から酵素的合成する方法は、28塩基長以上の比較的長鎖の二本鎖オリゴヌクレオチドを製造する方法しか報告されていなかった。短いオリゴヌクレオチドは、その製造に用いられるオリゴヌクレオチド原料断片の塩基長も自ずと短くなるが、短い塩基長のオリゴヌクレオチド原料断片は、アニーリング能も低下すると考えられる。構成するヌクレオチドが修飾されているとさらにアニーリング能が低下してしまうことも考えられた。したがって、リガーゼを用いる酵素的なオリゴヌクレオチド合成手法では、オリゴヌクレオチド原料断片のアニーリング能が重要と考えられていたことから、リガーゼを用いて4本以上のオリゴヌクレオチド原料断片とした28塩基長未満などのより短鎖のオリゴヌクレオチドを製造する方法は試みられていなかった。
しかし、本発明者らは、鋭意検討した結果、予想に反して、リガーゼおよび4本以上のオリゴヌクレオチド原料断片を用いた場合、アニーリング能に影響されることなく、28塩基長未満の二本鎖オリゴヌクレオチドを首尾よく製造できることを見出した。また、短鎖のオリゴヌクレオチドの場合、製造されるオリゴヌクレオチドの純度も向上することなどを見出し、本発明を完成するに至った。
しかし、本発明者らは、鋭意検討した結果、予想に反して、リガーゼおよび4本以上のオリゴヌクレオチド原料断片を用いた場合、アニーリング能に影響されることなく、28塩基長未満の二本鎖オリゴヌクレオチドを首尾よく製造できることを見出した。また、短鎖のオリゴヌクレオチドの場合、製造されるオリゴヌクレオチドの純度も向上することなどを見出し、本発明を完成するに至った。
すなわち、本発明は、以下のとおりである。
〔1〕11~27塩基長の相補部分を含む修飾オリゴヌクレオチドの製造方法であって、
該方法は、合計4本以上のオリゴヌクレオチド原料断片をオリゴヌクレオチドリガーゼの存在下で処理することを含み、
該合計4本以上のオリゴヌクレオチド原料断片は、該修飾オリゴヌクレオチドを、下記条件(i)~(v)を満たす断片連結部で分けた場合に得られるオリゴヌクレオチド原料断片に相当する、方法:
(i)断片連結部は、相補部分の各鎖側中のそれぞれに1以上存在し、かつ断片連結部は、該修飾オリゴヌクレオチド中に合計2つ以上存在し;
(ii)該修飾オリゴヌクレオチドを断片連結部で分けた場合に、突出末端が相補部分中に形成され、かつ該突出末端が1~10塩基長であり;
(iii)少なくとも1本のオリゴヌクレオチド原料断片が、修飾ヌクレオチドを含み、
(iv)該合計4本以上のオリゴヌクレオチド原料断片のうち4本のオリゴヌクレオチド原料断片が、5~25塩基長の相補部分を含み、かつ
(v)オリゴヌクレオチド原料断片の相補部分の各鎖側に相当する塩基長の合計が、いずれも11~27塩基長である。
〔2〕(ii)における突出末端が、2~6塩基長である、〔1〕の方法。
〔3〕(iv)で規定される4本のオリゴヌクレオチド原料断片の相補部分における突出末端以外の部分が、4~16塩基長である、〔1〕または〔2〕の方法。
〔4〕オリゴヌクレオチドリガーゼが、RNAリガーゼである、〔1〕~〔3〕のいずれかの方法。
〔5〕オリゴヌクレオチドリガーゼが、二本鎖RNAリガーゼである、〔4〕の方法。
〔6〕二本鎖RNAリガーゼが、Rnl2ファミリーまたはRnl5ファミリーのRNAリガーゼである、〔5〕の方法。
〔7〕修飾オリゴヌクレオチドが、修飾型ヌクレオチド残基を含む、〔1〕~〔6〕のいずれかの方法。
〔8〕修飾型ヌクレオチド残基が、1’、2’、3’、もしくは4’化学修飾型ヌクレオチド残基、5’-または3’-リン酸基修飾型ヌクレオチド残基、架橋型修飾型ヌクレオチド残基、キャリア付加修飾型ヌクレオチド残基、または糖骨格置換型ヌクレオチド残基である、〔7〕の方法。
〔9〕修飾型ヌクレオチド残基が、
i)1’、2’、3’、もしくは4’部位が、C1~6アルキルオキシC1~6アルキレン、-O-C1~6アルキル、-O-C6~14アリール、-C-アリール、ハロゲン原子、-O-C1~6アルキルN-アミドC1~6アルキレン、-O-C1~6アルキル-(C1~6アルキル-)アミノ-C1~6アルキレン、または-O-アミノC1~6アルキル(例、-O-アミノプロピル、-O-AP)で置換された、1’、2’、3’、もしくは4’化学修飾型ヌクレオチド残基;
ii)水酸基が、保護基で置換されていてもよい、-O-Ρ(S)(OH)2、-NH-Ρ(O)(OH)2、もしくは-NH-Ρ(S)(OH)2で置換された、5’-もしくは3’-リン酸基修飾型ヌクレオチド残基;
iii)2’部位と4’部位が、2’-O-C1~6アルキレン-4’、2’-O-エチレン-4’、2’-O-メチル置換メチレン-4’、2’-O-C1~6アルキレン-O-C1~6アルキレン-4’、2’-O-N(R)-C1~6アルキレン-4’(ここで、Rはメチル、水素原子またはベンジルを示す)、2’-N(R)-C(O)-4’、2’-NH-C1~6アルキレン-4’、もしくは、2’-C1~6アルキレン-4’に置換された、または、3’部位と5’部位が、3’-C1~6アルキレン-5’に置換された、架橋型修飾型ヌクレオチド残基;または
iv)ヘキシトール核酸(HNA)残基、シクロヘキセニル核酸(CeNA)残基、もしくはモルフォリノ核酸(PMO)残基
である、〔8〕の方法。
〔10〕合計4本以上のオリゴヌクレオチド原料断片より選ばれる任意の2本のオリゴヌクレオチド原料断片の全モル比が、0.5~2である、〔1〕~〔9〕のいずれかの方法。
〔11〕オリゴヌクレオチド原料断片が、10mM以下の一価カチオン塩濃度下で処理される、〔1〕~〔10〕のいずれかの方法。
〔12〕前記処理することの前に、オリゴヌクレオチド原料断片混合溶液を高温に置きその後クールダウンすることが行われない、〔1〕~〔11〕のいずれかの方法。
〔13〕前記修飾オリゴヌクレオチドの不純物の生成が抑制される、〔1〕~〔12〕のいずれかの方法。
〔14〕前記修飾オリゴヌクレオチドを精製することをさらに含む、〔1〕~〔13〕のいずれかの方法。
〔1〕11~27塩基長の相補部分を含む修飾オリゴヌクレオチドの製造方法であって、
該方法は、合計4本以上のオリゴヌクレオチド原料断片をオリゴヌクレオチドリガーゼの存在下で処理することを含み、
該合計4本以上のオリゴヌクレオチド原料断片は、該修飾オリゴヌクレオチドを、下記条件(i)~(v)を満たす断片連結部で分けた場合に得られるオリゴヌクレオチド原料断片に相当する、方法:
(i)断片連結部は、相補部分の各鎖側中のそれぞれに1以上存在し、かつ断片連結部は、該修飾オリゴヌクレオチド中に合計2つ以上存在し;
(ii)該修飾オリゴヌクレオチドを断片連結部で分けた場合に、突出末端が相補部分中に形成され、かつ該突出末端が1~10塩基長であり;
(iii)少なくとも1本のオリゴヌクレオチド原料断片が、修飾ヌクレオチドを含み、
(iv)該合計4本以上のオリゴヌクレオチド原料断片のうち4本のオリゴヌクレオチド原料断片が、5~25塩基長の相補部分を含み、かつ
(v)オリゴヌクレオチド原料断片の相補部分の各鎖側に相当する塩基長の合計が、いずれも11~27塩基長である。
〔2〕(ii)における突出末端が、2~6塩基長である、〔1〕の方法。
〔3〕(iv)で規定される4本のオリゴヌクレオチド原料断片の相補部分における突出末端以外の部分が、4~16塩基長である、〔1〕または〔2〕の方法。
〔4〕オリゴヌクレオチドリガーゼが、RNAリガーゼである、〔1〕~〔3〕のいずれかの方法。
〔5〕オリゴヌクレオチドリガーゼが、二本鎖RNAリガーゼである、〔4〕の方法。
〔6〕二本鎖RNAリガーゼが、Rnl2ファミリーまたはRnl5ファミリーのRNAリガーゼである、〔5〕の方法。
〔7〕修飾オリゴヌクレオチドが、修飾型ヌクレオチド残基を含む、〔1〕~〔6〕のいずれかの方法。
〔8〕修飾型ヌクレオチド残基が、1’、2’、3’、もしくは4’化学修飾型ヌクレオチド残基、5’-または3’-リン酸基修飾型ヌクレオチド残基、架橋型修飾型ヌクレオチド残基、キャリア付加修飾型ヌクレオチド残基、または糖骨格置換型ヌクレオチド残基である、〔7〕の方法。
〔9〕修飾型ヌクレオチド残基が、
i)1’、2’、3’、もしくは4’部位が、C1~6アルキルオキシC1~6アルキレン、-O-C1~6アルキル、-O-C6~14アリール、-C-アリール、ハロゲン原子、-O-C1~6アルキルN-アミドC1~6アルキレン、-O-C1~6アルキル-(C1~6アルキル-)アミノ-C1~6アルキレン、または-O-アミノC1~6アルキル(例、-O-アミノプロピル、-O-AP)で置換された、1’、2’、3’、もしくは4’化学修飾型ヌクレオチド残基;
ii)水酸基が、保護基で置換されていてもよい、-O-Ρ(S)(OH)2、-NH-Ρ(O)(OH)2、もしくは-NH-Ρ(S)(OH)2で置換された、5’-もしくは3’-リン酸基修飾型ヌクレオチド残基;
iii)2’部位と4’部位が、2’-O-C1~6アルキレン-4’、2’-O-エチレン-4’、2’-O-メチル置換メチレン-4’、2’-O-C1~6アルキレン-O-C1~6アルキレン-4’、2’-O-N(R)-C1~6アルキレン-4’(ここで、Rはメチル、水素原子またはベンジルを示す)、2’-N(R)-C(O)-4’、2’-NH-C1~6アルキレン-4’、もしくは、2’-C1~6アルキレン-4’に置換された、または、3’部位と5’部位が、3’-C1~6アルキレン-5’に置換された、架橋型修飾型ヌクレオチド残基;または
iv)ヘキシトール核酸(HNA)残基、シクロヘキセニル核酸(CeNA)残基、もしくはモルフォリノ核酸(PMO)残基
である、〔8〕の方法。
〔10〕合計4本以上のオリゴヌクレオチド原料断片より選ばれる任意の2本のオリゴヌクレオチド原料断片の全モル比が、0.5~2である、〔1〕~〔9〕のいずれかの方法。
〔11〕オリゴヌクレオチド原料断片が、10mM以下の一価カチオン塩濃度下で処理される、〔1〕~〔10〕のいずれかの方法。
〔12〕前記処理することの前に、オリゴヌクレオチド原料断片混合溶液を高温に置きその後クールダウンすることが行われない、〔1〕~〔11〕のいずれかの方法。
〔13〕前記修飾オリゴヌクレオチドの不純物の生成が抑制される、〔1〕~〔12〕のいずれかの方法。
〔14〕前記修飾オリゴヌクレオチドを精製することをさらに含む、〔1〕~〔13〕のいずれかの方法。
本発明の方法によれば、siRNAやヘテロ二本鎖オリゴヌクレオチド等の修飾オリゴヌクレオチドを効率的に高純度で製造することができる。
(本発明の概要)
以下、本発明を説明する。本発明の説明を容易にするため、本発明の機構の一例を図1の模式図に示す。ただし、この模式図は、本発明を説明するための例示にすぎず、本発明を限定するものではない。
以下、本発明を説明する。本発明の説明を容易にするため、本発明の機構の一例を図1の模式図に示す。ただし、この模式図は、本発明を説明するための例示にすぎず、本発明を限定するものではない。
本発明は、11~27塩基長の相補部分を含む修飾オリゴヌクレオチド(以下、「目的の修飾オリゴヌクレオチド」等と呼ぶこともある)の製造方法を提供する。本発明の方法は、原料である合計4以上のオリゴヌクレオチド原料断片を、オリゴヌクレオチドリガーゼの存在下で処理して、目的の修飾オリゴヌクレオチドを生成することを含む。以下、本発明の方法で製造される目的の修飾オリゴヌクレオチド、本発明の方法に用いるオリゴヌクレオチド原料断片およびオリゴヌクレオチドリガーゼ、ならびに、本発明の方法を行なうための処理の各条件等の詳細を説明する。
(目的の修飾オリゴヌクレオチド)
本発明の方法で製造される目的の修飾オリゴヌクレオチドは、11~27塩基長の相補部分を含む修飾オリゴヌクレオチドである。
本発明の方法で製造される目的の修飾オリゴヌクレオチドは、11~27塩基長の相補部分を含む修飾オリゴヌクレオチドである。
「オリゴヌクレオチド」とは、ヌクレオチド残基をモノマー単位として含むオリゴマーをいう。「オリゴヌクレオチド」としては、例えば、オリゴRNA、オリゴDNA、およびRNA-DNA混成オリゴヌクレオチドが挙げられる。
オリゴヌクレオチドは、「天然型オリゴヌクレオチド」および「修飾オリゴヌクレオチド」に分類することができる。「天然型オリゴヌクレオチド」とは、細胞に含まれるポリヌクレオチド(RNAおよびDNA)を構成するヌクレオチド残基(アデノシン(A)、グアノシン(G)、シチジン(C)、ウリジン(U)、デオキシアデノシン(dA)、デオキシグアノシン(dG)、デオキシシチジン(dC)、チミジン(dT)。以下、「天然型ヌクレオチド残基」と呼ぶ。)から構成されるオリゴヌクレオチドをいう。「修飾オリゴヌクレオチド」とは、「天然型オリゴヌクレオチド」以外のオリゴヌクレオチドをいい、天然型ヌクレオチド残基以外の構成要素(以下、「修飾残基」と呼ぶ。)を含むオリゴヌクレオチドである。修飾残基としては、例えば、修飾型ヌクレオチド残基、アミノ酸残基、リンカーが挙げられる。修飾型ヌクレオチド残基としては、例えば、後述する修飾を含むヌクレオチド残基が挙げられる。アミノ酸には、アミノ酸の誘導体を含む。アミノ酸としては、例えば、グリシン、アラニン、バリン、ロイシン、イソロイシン、プロリン、メチオニン、フェニルアラニン、トリプトファン、セリン、スレオニン、アスパラギン、グルタミン、チロシン、システイン、アスパラギン酸、グルタミン酸、ヒスチジン、リジン、アルギニン、およびこれらの誘導体が挙げられる。アミノ酸の誘導体とは、アミノ酸中の任意の原子または基が、別の原子または基で置換されたアミノ酸をいい、例えば、アミノ基中の水素原子、カルボキシル基中の水素原子、酸素原子、水酸基、側鎖中の任意の原子もしくは基、または骨格炭素原子(例、α-、β-、γ-、δ-炭素原子)に結合した水素原子が、別の原子(例、フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子)または基(例、後述する化学修飾における置換後の置換基)で置換されたアミノ酸が挙げられる。
「修飾型ヌクレオチド残基」における「修飾」には、ヌクレオチド残基の糖部分(リボースまたはデオキシリボース)の置換基の置換、ヌクレオチド残基の糖部分自体(糖骨格)の置換、およびヌクレオチド残基の核酸塩基部分の修飾(例、核酸塩基部分の置換基の置換)が含まれる。
「ヌクレオチド残基の糖部分の置換基の置換」としては、例えば、1’-H、2’-OH(リボースのみ)、2’-H、3’-OH、3’-NH2、3’-H、3’-リン酸基、4’-H、5’-リン酸基、またはこれらの組合せの置換が挙げられる。ここで、「リン酸基」は、-O-P(O)(OH)2だけでなく、酸素原子が硫黄原子またはΝHに置き換わった基(例えば、-O-Ρ(S)(OH)2、-NH-Ρ(O)(OH)2、-NH-Ρ(S)(OH)2)も包含する。また、リン酸基中の水酸基(-OH)がOR*(式中、R*は、リン酸基の保護基などの有機基を示す)に置き換わった基(例えば、保護されたリン酸基)も、上記「リン酸基」に包含される。このような置換としては、例えば、1’、2’、3’、もしくは4’-化学修飾(1’、2’、3’、または4’部位における他の置換基への置換)、5’-または3’-リン酸基修飾(5’-または3’-リン酸基の他の置換基への置換)、架橋型修飾(1’、2’、3’、または4’部位の2つ同士を架橋する置換)、およびキャリア付加修飾(1’、2’、3’、4’、または5’部位におけるキャリアへの置換)が挙げられる。
化学修飾は、例えば、オリゴヌクレオチドの分解耐性を向上させるために導入されてもよい。化学修飾における置換後の置換基としては、例えば、C1~6アルキルオキシC1~6アルキレン(例、メトキシエチル:MOE)、-O-C1~6アルキル(例、-O-Me)、-O-C6~14アリール(例、-O-フェニル)、-C-アリール(例、-C-フェニル)、ハロゲン原子(例、フッ素原子)、-O-C1~6アルキルN-アミドC1~6アルキレン(例、-O-N-メチルアセトアミド、-O-NMA)、-O-C1~6アルキル-(C1~6アルキル-)アミノ-C1~6アルキレン(例、-O-ジメチルアミノエトキシエチル、-O-DMAEOE)、および-O-アミノC1~6アルキル(例、-O-アミノプロピル、-O-AP)が挙げられる。化学修飾は、2’-化学修飾(2’部位の置換)、3’-化学修飾(3’部位の置換)が好ましく、中でも、2’-化学修飾(2’部位の置換)がより好ましい。2’-化学修飾における置換後の置換基としては、例えば、2’-C1~6アルキルオキシC1~6アルキレン(例、2’-メトキシエチル)、2’-O-C1~6アルキル(例、2’-O-Me)、2’-O-C6~14アリール(例、2’-O-フェニル)、2’-C-アリール(例、2’-C-フェニル)、2’-ハロゲン原子(例、2’-F)、2’-O-C1~6アルキルN-アミドC1~6アルキレン(例、2’-O-N-メチルアセトアミド、2’-O-NMA)、2’-O-C1~6アルキル-(C1~6アルキル-)アミノ-C1~6アルキレン(例、2’-O-ジメチルアミノエトキシエチル、2’-O-DMAEOE)、および2’-O-アミノC1~6アルキル(例、2’-O-アミノプロピル、2’-O-AP)が挙げられる。3’-化学修飾における置換後の置換基としては、例えば、3’-O-P(O)(OH)2、3’-O-Ρ(S)(OH)2、3’-NH-Ρ(O)(OH)2、3’-NH-Ρ(S)(OH)2)、および、リン酸基中の水酸基(-OH)がOR*(式中、R*は、後述するリン酸基の保護基などの有機基を示す)に置き換わった基が挙げられる。
5’-または3’-リン酸基修飾は、例えば、オリゴヌクレオチドの分解耐性を向上させるために導入されてもよい。5’-または3’-リン酸基修飾としては、例えば、リン酸基(-O-P(O)(OH)2)から、リン酸基において酸素原子が硫黄原子またはΝHに置き換わった基への置換が挙げられる。このような基としては、例えば、-O-Ρ(S)(OH)2(チオリン酸基:ホスホロチオエート型修飾)、-NH-Ρ(O)(OH)2、-NH-Ρ(S)(OH)2が挙げられる。また、5’-または3’-リン酸基修飾には、リン酸基中の水酸基(-OH)がOR*(式中、R*は、リン酸基の保護基などの有機基、を示す)に置き換わった基(例えば、保護されたリン酸基)も含まれる。リン酸基の保護基としては、例えば、トリチル(Tr)基、p-メトキシフェニルジフェニルメチル(MMTr)基、ジ(p-メトキシフェニル)フェニルメチル(DMTr)基、シアノエチル基(CN-C2H4-)が挙げられる。
架橋型修飾は、例えば、ヌクレオチド残基の立体構造安定性を向上させるために導入されてもよい。架橋型修飾としては、例えば、2’4’-架橋型修飾(2’-OHと4’-Hを架橋する置換)、3’5’-架橋型修飾(3’-Hと5’-Hを架橋する置換)等が挙げられる。2’4’-架橋型修飾としては、例えば、2’-OHと4’-Hの2’-O-C1~6アルキレン-4’への置換(例、2’-O-メチレン-4’(ロック核酸:LNA)、2’-O-エチレン-4’(エチレン架橋核酸:ENA)、2’-O-メチル置換メチレン-4’への置換(コンストレインドエチル架橋化核酸:BNAの一種(cEt-BNA))、2’-OHと4’-Hの2’-O-C1~6アルキレン-O-C1~6アルキレン-4’への置換(例、2’-O-メチレン-O-メチレン-4’(架橋化核酸:BNAの一種(BNACOC))、2’-OHと4’-Hの2’-O-N(R)-C1~6アルキレン-4’への置換(例、2’-O-N(R)-メチレン-4’(架橋化核酸:BNAの一種(BNANC)、ここで、Rはメチル、水素原子またはベンジルを示す)、2’-NH2と4’-Hの2’-N(R)-C(O)-4’への置換(例、2’-N(メチル)-C(O)-4’(アミド架橋核酸:AmNA))、2’-NH2と4’-Hの2’-NH-C1~6アルキレン-4’への置換(例、2’-NH-メチレン-4’)、2’-Hと4’-Hの2’-C1~6アルキレン-4’への置換(例、2’-メチル置換エチレン-4’)が挙げられる。また、3’5’-架橋型修飾としては、例えば、3’-Hと5’-Hの3’ -C1~6アルキレン-5’への置換(例、3’-エチレン-5’(ビシクロ核酸:Bc核酸)、Bc核酸の一種:tc核酸等))が挙げられる。
キャリア付加修飾におけるキャリアは、目的の修飾オリゴヌクレオチドに安定性、標的指向性、薬効等の性能を向上または付与するためのキャリアであってもよい。このようなキャリアは、使用目的に応じて、公知のキャリアから適宜選択することができる。キャリアとしては、例えば、N-アセチルガラクトサミン(GalNAc)、ペプチド、リン酸、コレステロール、トコフェロール、脂肪鎖、葉酸が挙げられる。キャリア付加修飾における付加部位は、目的の修飾オリゴヌクレオチドの末端に相当する3’または5’部位が好ましい。
「ヌクレオチド残基の糖部分自体の置換」を含む修飾型ヌクレオチド残基(糖骨格置換型ヌクレオチド残基)としては、例えば、ヘキシトール核酸(HNA)、シクロヘキセニル核酸(CeNA)等のオリゴヌクレオチド中の5員環の糖から6員環の擬似糖への置換を含むヌクレオチド残基が挙げられる。また、「ヌクレオチド残基の糖部分自体の置換」を含む修飾型ヌクレオチド残基としてはさらに、生体内の酵素(例、RNase等のヌクレアーゼ)により分解を受けずかつ免疫応答を誘導しないモルフォリノ環構造を持つヌクレオチド類似人工化合物であるモルフォリノ核酸(PMO)残基も挙げられる。
「ヌクレオチド残基の核酸塩基部分の修飾」としては、例えば、ヌクレオチド残基の核酸塩基部分がアルキル置換したもの(例えば、シトシル基の5位にメチル基が置換したもの)が挙げられる。
「相補部分を含むオリゴヌクレオチド」とは、相補的ヌクレオチド配列同士が対合した構造を含むオリゴヌクレオチドをいう。「相補部分を含むオリゴヌクレオチド」としては、例えば、二本鎖オリゴヌクレオチド、二本鎖様構造含有一本鎖オリゴヌクレオチド(例、ヘアピン型オリゴヌクレオチド、ダンベル型オリゴヌクレオチド等のループ型オリゴヌクレオチド)が挙げられる。二本鎖オリゴヌクレオチドは、各鎖が上述したオリゴヌクレオチドである二本鎖オリゴヌクレオチドであってもよく、例えば、二本鎖オリゴRNA、二本鎖オリゴDNA、オリゴRNAおよびオリゴDNAからなるヘテロ二本鎖オリゴヌクレオチド、オリゴRNAおよびRNA-DNA混成オリゴヌクレオチドからなる二本鎖オリゴヌクレオチド、オリゴDNAおよびRNA-DNA混成オリゴヌクレオチドからなる二本鎖オリゴヌクレオチド、およびRNA-DNA混成オリゴヌクレオチド同士からなる二本鎖オリゴヌクレオチドが挙げられる。二本鎖オリゴヌクレオチドとしては、例えば、siRNAやヘテロ二本鎖オリゴヌクレオチドが挙げられる。相補部分を含むオリゴヌクレオチドにおいて、相補的ヌクレオチド配列同士が対合した部分を「相補部分」と呼ぶ。用語「相補部分」とは、相補部分を含むオリゴヌクレオチド中の相補部分のみならず、相補部分を含むオリゴヌクレオチドをオリゴヌクレオチド原料断片に分けた場合における、相補部分を含むオリゴヌクレオチド中の相補部分に相当するオリゴヌクレオチド原料断片中の部分も指す。便宜上、相補部分における任意の片方の相補的ヌクレオチド配列を「センス鎖」と呼び、他方の相補的ヌクレオチド配列を「アンチセンス鎖」と呼ぶことがある。本発明において、用語「センス」および「アンチセンス」は、相補部分の任意の一方および他方を指す便宜上の呼称にすぎず、生物学的意義(特に、RNAiにおける意義)を意図するものではない。相補部分を含むオリゴヌクレオチドは、ループ部分を含んでも含まなくてもよい。「ループ部分」とは、相補部分のセンス側とアンチセンス側とを同一末端側で(例えば、5’末端と3’末端とを)連結するリンカーをいう。相補部分を含むオリゴヌクレオチドは、特に、転写後遺伝子サイレンシング(例、RNA干渉(RNAi))作用のために用いられる。
目的の修飾オリゴヌクレオチドは、上述した修飾残基を相補部分に含む。目的の修飾オリゴヌクレオチドとしては、例えば、修飾型ヌクレオチド残基を含む二本鎖オリゴヌクレオチドまたはループ型オリゴヌクレオチド(例、相補部分に修飾型ヌクレオチド残基を含む二本鎖オリゴヌクレオチドまたはループ型オリゴヌクレオチド)、ループ部分に修飾型ヌクレオチド残基またはヌクレオチド残基以外の残基(例、アミノ酸残基やリンカー等)を含むループ型オリゴヌクレオチドが挙げられる(例、国際公開第2012/005368号)。目的の修飾オリゴヌクレオチドにおいて、一部のヌクレオチド残基が修飾型ヌクレオチド残基であってもよく、全てのヌクレオチド残基が修飾型ヌクレオチド残基であってもよいが、「修飾型ヌクレオチド残基」がモルフォリノ核酸(PMO)残基の場合には、目的の修飾オリゴヌクレオチドにおいて、一部のヌクレオチド残基がモルフォリノ核酸(PMO)残基であるのが好ましい。また、目的の修飾オリゴヌクレオチドには、その配列の両端に修飾型ヌクレオチド残基を有し、その配列の中央部にRNaseの認識を受けるギャップ領域を有するオリゴヌクレオチドであるギャップマーが含まれ、さらに、その配列中に修飾型ヌクレオチド残基が混在しているオリゴヌクレオチドであるミックスマー、その配列中の全てのヌクレオチド残基が修飾型ヌクレオチド残基であるオリゴヌクレオチドである全修飾型オリゴヌクレオチド等のRNase活性を誘導しないオリゴヌクレオチドも含まれる。
本発明では、目的の修飾オリゴヌクレオチド中の相補部分は、11~27塩基長(例、12~27塩基長、15~27塩基長、または18~27塩基長)である。例えば、目的の修飾オリゴヌクレオチドは、相補部分のみからなる二本鎖修飾オリゴヌクレオチドである場合、11~27塩基長であってもよい。あるいは、目的の修飾オリゴヌクレオチドは、相補部分に加えて非相補部分を有していてもよい。この場合、非相補部分は、1~16塩基長、例えば1~10塩基長、好ましくは1~5塩基長、より好ましくは1、2または3塩基長であってもよい。11~27塩基長の相補部分に加えて非相補部分を有する目的の修飾オリゴヌクレオチドにおいて、11~27塩基長の相補部分は、連続形態であってもよいが、非相補部分としてのミスマッチ塩基対により分断された非連続形態であってもよい。
目的の修飾オリゴヌクレオチドの合計残基数は、目的の修飾オリゴヌクレオチドの機能、本発明の方法における各条件から適宜選択してもよい。目的の修飾オリゴヌクレオチドの合計残基数は、例えば、24~74であってもよい。
(オリゴヌクレオチド原料断片)
本発明の方法で原料として用いられる合計4以上のオリゴヌクレオチド原料断片は、目的の修飾オリゴヌクレオチドを、下記条件(i)~(v)を満たす断片連結部(「切断部位」とも呼ぶ)で分けた場合に得られるオリゴヌクレオチド原料断片に相当するように設計することができる:
(i)断片連結部は、相補部分の各鎖側中のそれぞれに1以上存在し、かつ断片連結部は、該修飾オリゴヌクレオチド中に合計2つ以上存在し;
(ii)該修飾オリゴヌクレオチドを断片連結部で分けた場合に、突出末端(「付着性末端」とも呼ぶ)が相補部分中に形成され、かつ該突出末端が1~10塩基長であり;
(iii)少なくとも1本のオリゴヌクレオチド原料断片が、修飾ヌクレオチドを含み、
(iv)該合計4本以上のオリゴヌクレオチド原料断片のうち4本のオリゴヌクレオチド原料断片が、5~25塩基長の相補部分を含み、かつ
(v)オリゴヌクレオチド原料断片の相補部分の各鎖側に相当する塩基長の合計が、いずれも11~27塩基長である。
本発明の方法で原料として用いられる合計4以上のオリゴヌクレオチド原料断片は、目的の修飾オリゴヌクレオチドを、下記条件(i)~(v)を満たす断片連結部(「切断部位」とも呼ぶ)で分けた場合に得られるオリゴヌクレオチド原料断片に相当するように設計することができる:
(i)断片連結部は、相補部分の各鎖側中のそれぞれに1以上存在し、かつ断片連結部は、該修飾オリゴヌクレオチド中に合計2つ以上存在し;
(ii)該修飾オリゴヌクレオチドを断片連結部で分けた場合に、突出末端(「付着性末端」とも呼ぶ)が相補部分中に形成され、かつ該突出末端が1~10塩基長であり;
(iii)少なくとも1本のオリゴヌクレオチド原料断片が、修飾ヌクレオチドを含み、
(iv)該合計4本以上のオリゴヌクレオチド原料断片のうち4本のオリゴヌクレオチド原料断片が、5~25塩基長の相補部分を含み、かつ
(v)オリゴヌクレオチド原料断片の相補部分の各鎖側に相当する塩基長の合計が、いずれも11~27塩基長である。
オリゴヌクレオチド原料断片の数は、4本以上であり、4~6本(4、5、6本)が好ましい。オリゴヌクレオチド原料断片の数は、目的の修飾オリゴヌクレオチド(主に2本鎖核酸)を構成するセンス鎖及びアンチセンス鎖に主に対応する数の観点から特徴付けることもできる。上記条件(i)より、このようなセンス鎖及びアンチセンス鎖に主に対応するオリゴヌクレオチド原料断片の数は、それぞれ、2本以上であることが理解される。このようなセンス鎖及びアンチセンス鎖に対応するオリゴヌクレオチド原料断片の数は、それぞれ、3断片または4断片であってもよく、2断片又は3断片が好ましい。上記条件(ii)における突出末端は、5’突出末端または3’突出末端のいずれであってもよい。上記条件(iv)における「相補部分」とは、目的の修飾オリゴヌクレオチド中の相補部分に相当するオリゴヌクレオチド原料断片中の部分を指す。本発明において、用語「断片連結部」と「切断部位」は同じ意味を有する。「断片連結部」(「切断部位」)は、オリゴヌクレオチド原料断片の組合せを設計するために便宜上設定される部位を意味し、本発明の方法において実際に切断される部位を意味しない。上記(iv)における4本のオリゴヌクレオチド原料断片は、好ましくは5~25塩基長、より好ましくは5~20塩基長、さらにより好ましくは5~17塩基長の相補部分を目的の修飾オリゴヌクレオチドが含むように設計されたものであってもよい。
上記条件(iv)における「相補部分」の塩基長は、対合を形成することができる塩基長であればよく、1塩基長以上であればよい。また、オリゴヌクレオチド合成において塩基長が長くなるにつれて産物の純度、収量、製造効率が低下し得るため、全オリゴヌクレオチド原料断片のうち4本のオリゴヌクレオチド原料断片は、相補部分が17塩基長以下となるように設計されるのが好ましい。相補部分を構成する2本の鎖が、5~25塩基長(例、5~22塩基長、5~20塩基長、5~17塩基長、8~25塩基長、8~22塩基長、8~20塩基長、8~17塩基長)であることが好ましい。
突出末端は、例えば1~10塩基長であり、好ましくは1~8塩基長であり、より好ましくは1~6塩基長であり、さらにより好ましくは2~6塩基長、3~6塩基長または4~6塩基長である。
上記条件(iv)における「相補部分」の塩基長の数値と、突出末端の塩基長の数値は、上述した範囲を満たしかつ互いに整合する値が設定される。例えば、突出末端が5塩基長である場合、相補部分は、突出末端を形成するために6~25塩基長であってもよく、例えば、突出末端が6塩基長である場合、相補部分は、突出末端を形成するために7~25塩基長であってもよい。
上記条件(iv)で規定される4本のオリゴヌクレオチド原料断片の「相補部分」における突出末端以外の部分は、4~24塩基長、4~21塩基長、4~19塩基長、または4~16塩基長であることが好ましい。
特定の実施形態では、上記条件(iv)で規定される4本のオリゴヌクレオチド原料断片は、それぞれ、5塩基長以上、好ましくは6塩基長以上、より好ましくは7塩基長以上、さらにより好ましくは8塩基長以上、特に好ましくは9塩基長以上であってもよい。このような4本のオリゴヌクレオチド原料断片はまた、19塩基長以下、好ましくは18塩基長以下、より好ましくは17塩基長以下、さらにより好ましくは16塩基長以下、特に好ましくは15塩基長以下であってもよい。このような4本のオリゴヌクレオチド原料断片はまた、5~19塩基長、好ましくは6~18塩基長、より好ましくは7~17塩基長、さらにより好ましくは8~16塩基長、特に好ましくは9~15塩基長であってもよい。
目的の修飾オリゴヌクレオチドの5’末端に相当するオリゴヌクレオチド原料断片の5’末端は、5’-リン酸基のままであってもよく、5’-OHに置換されていてもよく、5’-リン酸基修飾が導入されていてもよく、あるいは目的の修飾オリゴヌクレオチドの5’末端と同じ構造を有していてもよい。5’-リン酸基修飾としては、例えば上述のものが挙げられる。それ以外のオリゴヌクレオチド原料断片の5’末端は、オリゴヌクレオチドリガーゼによる連結反応の観点から、5’-リン酸基のままであることが好ましい。目的の修飾オリゴヌクレオチドの3’末端に相当するオリゴヌクレオチド原料断片の3’末端は、3’-OHのままであってもよく、3’-リン酸基修飾が導入されていてもよく、あるいは目的の修飾オリゴヌクレオチドの3’末端と同じ構造を有していてもよい。3’-リン酸基修飾としては、例えば上述のものが挙げられる。それ以外のオリゴヌクレオチド原料断片の3’末端は、オリゴヌクレオチドリガーゼによる連結反応の観点から、3’-OHのままであることが好ましい。
オリゴヌクレオチド原料断片は、遊離の形態であっても、複合体化されていても、固定化されていてもよい。
オリゴヌクレオチド原料断片は、公知の化学合成法または酵素的合成法で製造されてもよい。公知の化学合成法としては、固相合成法や液相合成法、例えば、国際公開第2012/157723号、国際公開第2005/070859号に記載の方法などが挙げられる。
目的の修飾オリゴヌクレオチドへの機能性部分の付加が所望される場合、オリゴヌクレオチド原料断片は、その対応部分において機能性部分が付加されていてもよい。
(リガーゼ処理)
オリゴヌクレオチドリガーゼは、オリゴヌクレオチド原料断片同士を連結させる酵素である。本発明の方法において、オリゴヌクレオチドリガーゼの触媒作用により、オリゴヌクレオチド原料断片同士が「切断部位」(「連結部位」とも呼ぶ)において連結されて、目的の修飾オリゴヌクレオチドが生成される。オリゴヌクレオチドリガーゼとしては、例えば、RNAリガーゼおよびDNAリガーゼが挙げられる。RNAリガーゼは、一本鎖RNAリガーゼまたは二本鎖RNAリガーゼのいずれであってもよく、二本鎖RNAリガーゼが好ましい。二本鎖RNAリガーゼとしては、例えばRnl2ファミリーのRNAリガーゼ(「RNAリガーゼ2」と呼ぶこともある)、Rnl5ファミリーのRNAリガーゼが挙げられる。RNAリガーゼとしては、本発明の目的を達成する限りにおいて如何なる生物種またはウイルス種に由来するRNAリガーゼを用いてもよく、例えば、T4ファージ由来RNAリガーゼ(T4RNAリガーゼ1、T4RNAリガーゼ2)を用いてもよい。DNAリガーゼとしては、本発明の目的を達成する限りにおいて如何なる生物種またはウイルス種に由来するDNAリガーゼを用いてもよく、例えば、T4ファージ由来DNAリガーゼを用いてもよい。
オリゴヌクレオチドリガーゼは、オリゴヌクレオチド原料断片同士を連結させる酵素である。本発明の方法において、オリゴヌクレオチドリガーゼの触媒作用により、オリゴヌクレオチド原料断片同士が「切断部位」(「連結部位」とも呼ぶ)において連結されて、目的の修飾オリゴヌクレオチドが生成される。オリゴヌクレオチドリガーゼとしては、例えば、RNAリガーゼおよびDNAリガーゼが挙げられる。RNAリガーゼは、一本鎖RNAリガーゼまたは二本鎖RNAリガーゼのいずれであってもよく、二本鎖RNAリガーゼが好ましい。二本鎖RNAリガーゼとしては、例えばRnl2ファミリーのRNAリガーゼ(「RNAリガーゼ2」と呼ぶこともある)、Rnl5ファミリーのRNAリガーゼが挙げられる。RNAリガーゼとしては、本発明の目的を達成する限りにおいて如何なる生物種またはウイルス種に由来するRNAリガーゼを用いてもよく、例えば、T4ファージ由来RNAリガーゼ(T4RNAリガーゼ1、T4RNAリガーゼ2)を用いてもよい。DNAリガーゼとしては、本発明の目的を達成する限りにおいて如何なる生物種またはウイルス種に由来するDNAリガーゼを用いてもよく、例えば、T4ファージ由来DNAリガーゼを用いてもよい。
オリゴヌクレオチドリガーゼの存在下での処理(以下、「リガーゼ処理」と呼ぶ)は、オリゴヌクレオチドリガーゼの触媒作用によりオリゴヌクレオチド原料断片を連結させる反応である。リガーゼ処理の操作は、オリゴヌクレオチド原料断片とオリゴヌクレオチドリガーゼを混合することである。リガーゼ処理においては、全てのオリゴヌクレオチド原料断片とオリゴヌクレオチドリガーゼを混合して、連結反応を一段階で行なってもよい。また、リガーゼ処理においては、多段階連結反応として、一部のオリゴヌクレオチド原料断片とオリゴヌクレオチドリガーゼを混合して連結反応を行なった後で残りのオリゴヌクレオチド原料断片と反応物を混合して次なる連結反応を行なってもよい。混合は、オリゴヌクレオチドリガーゼをオリゴヌクレオチド原料断片に添加すること、オリゴヌクレオチド原料断片をオリゴヌクレオチドリガーゼを含む系に添加すること、またはオリゴヌクレオチド原料断片およびオリゴヌクレオチドリガーゼを反応のための系に添加することであってもよい。
リガーゼ処理を行う系としては、水溶液を用いることができる。水溶液としては、緩衝液が好ましい。緩衝液としては、例えば、リン酸緩衝液、Tris緩衝液、炭酸緩衝液、酢酸緩衝液、クエン酸緩衝液が挙げられる。pHは、例えば約5~9であってもよい。例えば、リガーゼ処理におけるオリゴヌクレオチド原料断片の濃度が高い場合、pHは、7.5~9.0(例えば、8.0~8.5)であってもよい。
リガーゼ処理における各オリゴヌクレオチド原料断片の濃度は、オリゴヌクレオチド原料断片が溶解し、かつ目的の修飾オリゴヌクレオチドを生成するために十分な濃度であればよい。各オリゴヌクレオチド原料断片の濃度は、例えば1μM以上、10μM以上、50μM以上、100μM以上、300μM以上、500μM以上または1000μM以上であってもよい。各オリゴヌクレオチド原料断片の濃度はまた、例えば1M、100mM、または10mM以下であってもよい。目的の修飾オリゴヌクレオチドの効率的な大量生産が特に所望される場合には、上記濃度のうち100μM以上の濃度および上記pH範囲のうち7.5~9.0のpH範囲で各オリゴヌクレオチド原料断片を用いることが好ましい。
リガーゼ処理における全オリゴヌクレオチド原料断片のモル数は、未反応のオリゴヌクレオチド原料断片量の低減により製造効率を向上させる観点から、ほぼ等量であることが好ましい。全オリゴヌクレオチド原料断片のモル数がほぼ等量であるためには、合計4本以上のオリゴヌクレオチド原料断片より選ばれる任意の2本のオリゴヌクレオチド原料断片の全モル比が、例えば0.5~2、好ましくは1/1.8~1.8、より好ましくは1/1.5~1.5、さらにより好ましくは1/1.2~1.2、特に好ましくは1/1.1~1.1の範囲内であってもよい。
リガーゼ処理におけるオリゴヌクレオチドリガーゼの濃度は、目的の修飾オリゴヌクレオチドを生成するために十分な濃度であればよい。オリゴヌクレオチドリガーゼの濃度は、例えば0.01U/μL以上、好ましくは0.02U/μL以上、より好ましくは0.03U/μL以上、さらにより好ましくは0.04U/μL以上であってもよい。オリゴヌクレオチドリガーゼの濃度は、例えば1U/μL以下、好ましくは0.5U/μL以下、より好ましくは0.2U/μL以下、さらにより好ましくは0.1U/μL以下であってもよい。より具体的には、オリゴヌクレオチドリガーゼの濃度は、例えば0.01~1U/μL、好ましくは0.02~0.5U/μL、より好ましくは0.03~0.2U/μL、さらにより好ましくは0.04~0.1U/μLであってもよい。
リガーゼ処理を行う系は、オリゴヌクレオチドリガーゼの補因子を含んでいてもよい。オリゴヌクレオチドリガーゼの補因子としては、例えば、ATP、二価金属塩(例、塩化マグネシウム等のマグネシウム塩)が挙げられる。処理を行う系は、オリゴヌクレオチドリガーゼの安定化剤を含んでいてもよい。オリゴヌクレオチドリガーゼの安定化剤としては、例えば、酸化防止剤(例、ジチオスレイトール、メルカプトエタノール等の還元剤)が挙げられる。リガーゼ処理を行う系は、酵素の安定保持および反応速度向上のため、界面活性剤を含んでいてもよい。界面活性剤としては、例えば、非イオン性界面活性剤(例、Triton X-100等のTritonシリーズの界面活性剤)、およびイオン性界面活性剤が挙げられる。イオン性界面活性剤としては、例えば、カチオン性界面活性剤、アニオン性界面活性剤、両イオン性界面活性剤)が挙げられる。また、リガーゼ処理を行う系は、反応速度向上のため、ポリエチレングリコールを含んでいてもよい。
リガーゼ処理を行う系は、低濃度の一価カチオン塩濃度を有するか、または一価カチオン塩を実質的に含まなくてもよい。処理を行う系の一価カチオン塩濃度は、例えば10mM以下、好ましくは1mM以下、より好ましくは0.1mM以下、さらにより好ましくは0.01mM以下であってもよい。特に好ましくは、処理を行う系は、一価カチオン塩を実質的に含まなくてよい。一価カチオン塩としては、例えば、リチウムイオン、ナトリウムイオン、カリウムイオン、ルビジウムイオン、セシウムイオン、アンモニウムイオン等の一価カチオンと、フッ化物イオン、塩化物イオン、臭化物イオン、ヨウ化物イオン等のアニオンとの塩が挙げられる。
リガーゼ処理における温度は、オリゴヌクレオチドリガーゼを活性化するために十分な温度であればよい。このような温度は、例えば2~50℃、好ましくは16~50℃、より好ましくは25~50℃であってもよい。
リガーゼ処理が行われる時間は、目的の修飾オリゴヌクレオチドを生成するために十分な時間であればよい。このような時間は、例えば1~72時間であってもよい。
本発明によれば、目的の修飾オリゴヌクレオチドについて、N-1merやN+1merなどの目的塩基長以外の塩基長を有する不純物の生成および混入を抑制することができる。例えば、本発明では、目的の修飾オリゴヌクレオチドが、単一の二本鎖核酸(例、siRNA、ヘテロ二本鎖オリゴヌクレオチド)として製造される。このような単一の二本鎖核酸を構成するセンス鎖及びアンチセンス鎖は、それぞれ、N個及びM個の塩基長である。N個及びM個の塩基長は、それぞれ独立して、11~30塩基長(例、18~30塩基長)である。N個及びM個の塩基長はまた、それぞれ独立して、11~27塩基長(例、18~27塩基長)であってもよい。本発明において、目的の修飾オリゴヌクレオチドの不純物は、上記目的の修飾オリゴヌクレオチド)以外の核酸夾雑物が意図される。このような核酸夾雑物を構成するセンス鎖及びアンチセンス鎖は、それぞれ、N個及びM個の塩基長ではなく、(N±α)個及びM個の塩基長、N個及び(M±β)個の塩基長、又は(N±α)個及び(M±β)個の塩基長からなる。ここで、N個及びM個は上記と同様であり、α個及びβ個は、例えば1個、2個又は3個である。
(その他の任意工程)
本発明の方法は、オリゴヌクレオチド原料断片を合成する工程(例、固相合成等の化学合成)を含んでいてもよい。本発明の方法は、目的の修飾オリゴヌクレオチド以外の核酸夾雑物の生成を抑制できることから、合成されたオリゴヌクレオチド原料断片の試料からの目的の修飾オリゴヌクレオチドの精製を省略することができる。しかし、本発明の方法は、目的の修飾オリゴヌクレオチドの精製が行われた場合であっても、当該精製後に残存し得る少量の目的外のオリゴヌクレオチド原料断片に起因する二本鎖核酸夾雑物(不純物)の生成を抑制できるため、目的の修飾オリゴヌクレオチドの精製が行われてもよい。このような精製は、例えば、クロマトグラフィ(例、HPLC、IEX)、ゲル濾過等の方法により行うことができる。
本発明の方法は、オリゴヌクレオチド原料断片を合成する工程(例、固相合成等の化学合成)を含んでいてもよい。本発明の方法は、目的の修飾オリゴヌクレオチド以外の核酸夾雑物の生成を抑制できることから、合成されたオリゴヌクレオチド原料断片の試料からの目的の修飾オリゴヌクレオチドの精製を省略することができる。しかし、本発明の方法は、目的の修飾オリゴヌクレオチドの精製が行われた場合であっても、当該精製後に残存し得る少量の目的外のオリゴヌクレオチド原料断片に起因する二本鎖核酸夾雑物(不純物)の生成を抑制できるため、目的の修飾オリゴヌクレオチドの精製が行われてもよい。このような精製は、例えば、クロマトグラフィ(例、HPLC、IEX)、ゲル濾過等の方法により行うことができる。
本発明の方法は、リガーゼ処理工程の後に反応停止工程を含んでもよい。反応停止工程としては、例えば、高温処理(例、80℃)や酸・アルカリ、有機溶剤の添加によるオリゴヌクレオチドリガーゼ失活処理、EDTAなどのキレート剤の添加による補因子の金属イオンの除去、が挙げられる。また、担体に酵素を固定化して反応を行い、膜分離によって酵素を反応液から除去する方法も挙げられる。
本発明の方法は、リガーゼ処理工程の後に目的の修飾オリゴヌクレオチドを精製する工程を含んでもよい。例えば、本工程は、クロマトグラフィ(例、HPLC)、ゲル濾過等の任意の適切な方法により行うことができる。
オリゴヌクレオチド原料断片をアニールさせる場合において、一般に、オリゴヌクレオチド原料断片を変性状態(非対合状態)にするために、オリゴヌクレオチド原料断片混合溶液を高温に加熱し、その後、相補的ヌクレオチド配列同士の対合を形成させるために、高温のオリゴヌクレオチド原料断片混合溶液を空冷等により徐々に冷却する操作が行われることが多い。しかし、本発明の方法では、リガーゼ処理工程の前に、そのような変性のために高温に加熱する操作および対合形成のために冷却する処理(加熱-冷却処理)を行わずに、簡略化された操作で、目的の修飾オリゴヌクレオチドを製造することができる。高温に置くこととしては、例えば、オリゴヌクレオチド原料断片混合溶液を65℃以上、70℃以上、75℃以上、80℃以上、85℃以上、90℃以上、95℃以上、または100℃以上で保持(例、5分以上、または10分以上)することが挙げられる。冷却することとしては、例えば、オリゴヌクレオチド原料断片混合溶液を室温(例、15~25℃、または20~25℃)または所定の温度(例、37℃)下で静置(例、5時間以上)すること、オリゴヌクレオチド原料断片混合溶液を所定の温度(例、37℃)に保持(例、15分以上)することが挙げられる。
加熱-冷却処理を省略するために、リガーゼ処理工程の前にオリゴヌクレオチド原料断片混合溶液が高温に置かれる時間は、例えば、5分未満、4.5分以下、4分以下、3.5分以下、3分以下、2.5分以下、2分以下、1.5分以下、1分以下、または0.5分以下となるように制御されてもよい。
加熱-冷却処理を省略するために、本発明の方法において、オリゴヌクレオチド原料断片同士を溶液中で混合してからリガーゼ処理工程を行なうまで、オリゴヌクレオチド原料断片を含む溶液は、2~50℃に保持されていてもよい。すなわち、本発明の方法において、上記組合せに含まれる全てのオリゴヌクレオチド原料断片およびオリゴヌクレオチドリガーゼのいかなる混合およびいかなる混合間のインターバル、ならびにリガーゼ反応は、2~50℃の条件下で行なわれてもよい。このような実施形態において、本発明の方法は、以下を含む:
(1)別個の系に存在する上記組合せに含まれる全てのオリゴヌクレオチド原料断片、およびオリゴヌクレオチドリガーゼを、全ての混合が2~50℃で行なわれかつ全ての混合間のインターバルにおいて混合物が2~50℃に保持される条件下で混合して、混合溶液を得ること;および
(2)混合溶液を2~50℃で保持したままで反応させて、目的の修飾オリゴヌクレオチドを含む溶液を得ること。
(1)別個の系に存在する上記組合せに含まれる全てのオリゴヌクレオチド原料断片、およびオリゴヌクレオチドリガーゼを、全ての混合が2~50℃で行なわれかつ全ての混合間のインターバルにおいて混合物が2~50℃に保持される条件下で混合して、混合溶液を得ること;および
(2)混合溶液を2~50℃で保持したままで反応させて、目的の修飾オリゴヌクレオチドを含む溶液を得ること。
この実施形態において、上記組合せに含まれる全てのオリゴヌクレオチド原料断片は、別個の系で得られる。この実施形態において、本発明は、例えば、当該オリゴヌクレオチド原料断片を2~50℃で混合してオリゴヌクレオチド原料断片混合物を得て、当該オリゴヌクレオチド原料断片混合物とオリゴヌクレオチドリガーゼを2~50℃で混合することにより行われてもよい。この実施形態において、本発明は、例えば、オリゴヌクレオチドリガーゼを含む溶液に当該オリゴヌクレオチド原料断片を順次2~50℃で添加することにより行われてもよい。
本発明の方法は、例えばラージスケールでの目的の修飾オリゴヌクレオチドの工業的製造に用いることができる。
次に実施例を示して本発明をさらに詳細に説明するが、本発明は以下の実施例に限定されるものではない。
〔実施例1〕天然型RNAを用いた断片の組み合わせのパターンの比較
1)基質および生成物標品の合成
4断片の短鎖天然型RNAからsiRNAを酵素的に合成する反応において、当該断片の塩基長の影響を評価した。目的とするsiRNAを表1のRNA1-S(21mer)およびRNA1-A(23mer)からなる二本鎖とした(以下、それぞれをセンス鎖、アンチセンス鎖と呼ぶ)。表1に示す18種のRNA断片を合成し、これらを用いて表2に示す6パターンの断片の組合せを評価した。
1)基質および生成物標品の合成
4断片の短鎖天然型RNAからsiRNAを酵素的に合成する反応において、当該断片の塩基長の影響を評価した。目的とするsiRNAを表1のRNA1-S(21mer)およびRNA1-A(23mer)からなる二本鎖とした(以下、それぞれをセンス鎖、アンチセンス鎖と呼ぶ)。表1に示す18種のRNA断片を合成し、これらを用いて表2に示す6パターンの断片の組合せを評価した。


2)T4 RNAリガーゼ2によるライゲーション反応
4断片のオリゴヌクレオチドを用いてT4 RNAリガーゼ2(New England Biolabs)により反応した。反応液の組成は50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5、RNA断片をそれぞれ10μMとし、反応液量は10μLとした。酵素の添加濃度を0.025、0.05、0.1、0.2U/μLとして生成物の濃度を比較した。サーマルサイクラーを用いて25℃で1時間反応した後、80℃で5分間加熱することにより反応を停止した。
4断片のオリゴヌクレオチドを用いてT4 RNAリガーゼ2(New England Biolabs)により反応した。反応液の組成は50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5、RNA断片をそれぞれ10μMとし、反応液量は10μLとした。酵素の添加濃度を0.025、0.05、0.1、0.2U/μLとして生成物の濃度を比較した。サーマルサイクラーを用いて25℃で1時間反応した後、80℃で5分間加熱することにより反応を停止した。
3)HPLCによる分析
反応液をXbridge Oligonucleotide BEH C18 column(Waters、2.5μm,4.6mm×50mm)を用いたHPLCにより分析した。分析条件は、カラム温度60℃、検出波長254nm、インジェクション量10μL、流速0.4mL/minとした。移動相は溶離液A(ヘキサフルオロイソプロパノール-トリエチルアミン)および溶離液B(メタノール)を用いたリニアグラジエントにより分析した。センス鎖およびアンチセンス鎖の標品も同様に分析することにより、ライゲーション産物の濃度を定量した。
反応液をXbridge Oligonucleotide BEH C18 column(Waters、2.5μm,4.6mm×50mm)を用いたHPLCにより分析した。分析条件は、カラム温度60℃、検出波長254nm、インジェクション量10μL、流速0.4mL/minとした。移動相は溶離液A(ヘキサフルオロイソプロパノール-トリエチルアミン)および溶離液B(メタノール)を用いたリニアグラジエントにより分析した。センス鎖およびアンチセンス鎖の標品も同様に分析することにより、ライゲーション産物の濃度を定量した。
4)結果
各断片の塩基長の組み合わせにおけるセンス鎖およびアンチセンス鎖の蓄積を図2に示す。組み合わせ3ではライゲーション産物がほとんど蓄積しなかったが、それ以外の組合せでは酵素濃度の増加に応じてライゲーション産物の蓄積が増大することが観察された。
各断片の塩基長の組み合わせにおけるセンス鎖およびアンチセンス鎖の蓄積を図2に示す。組み合わせ3ではライゲーション産物がほとんど蓄積しなかったが、それ以外の組合せでは酵素濃度の増加に応じてライゲーション産物の蓄積が増大することが観察された。
〔実施例2〕天然型RNAのライゲーション反応における反応温度の影響の評価
短鎖のライゲーション反応における反応温度による影響を検討した。基質として表2の1のオリゴヌクレオチドを用い、T4 RNAリガーゼ2により反応した。反応液の組成は50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5、酵素濃度0.2U/μL、RNA断片をそれぞれ10μMとし、反応液量は10μLとした。反応温度を16℃、25℃、30℃および37℃として、それぞれ1、2、および4時間反応後に、80℃で5分間加熱することにより反応を停止した。反応液に含まれるライゲーション産物の濃度を実施例1に記載の条件にてHPLCにより分析した。
短鎖のライゲーション反応における反応温度による影響を検討した。基質として表2の1のオリゴヌクレオチドを用い、T4 RNAリガーゼ2により反応した。反応液の組成は50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5、酵素濃度0.2U/μL、RNA断片をそれぞれ10μMとし、反応液量は10μLとした。反応温度を16℃、25℃、30℃および37℃として、それぞれ1、2、および4時間反応後に、80℃で5分間加熱することにより反応を停止した。反応液に含まれるライゲーション産物の濃度を実施例1に記載の条件にてHPLCにより分析した。
結果を図3に示す。反応温度25℃以上では反応1時間後にセンス鎖、アンチセンス鎖ともに約10μMのライゲーション産物が蓄積した。一方、25℃以上に比べ16℃では特にセンス鎖のライゲーション差物の生成速度が低かった。
〔実施例3〕修飾型RNAを用いた反応進行の確認
1)基質および生成物標品の合成
修飾型オリゴヌクレオチドにおける4断片からのsiRNAの酵素的ライゲーション反応の進行を評価した。目的のsiRNAを表3に示す、センス鎖(MOD1-S)およびアンチセンス鎖(MOD1-A)からなる二本鎖とした。このsiRNAは実施例1および2で用いた天然型RNAと塩基配列は同一であるが、すべての残基が2’-Fまたは2’-O-メチルにより修飾されており、一部のリン酸基がチオリン酸基に置換されている。また、それぞれの断片として表3に示す4断片を合成した。これら4断片の塩基配列は表2の番号1の組合せと同一である。
1)基質および生成物標品の合成
修飾型オリゴヌクレオチドにおける4断片からのsiRNAの酵素的ライゲーション反応の進行を評価した。目的のsiRNAを表3に示す、センス鎖(MOD1-S)およびアンチセンス鎖(MOD1-A)からなる二本鎖とした。このsiRNAは実施例1および2で用いた天然型RNAと塩基配列は同一であるが、すべての残基が2’-Fまたは2’-O-メチルにより修飾されており、一部のリン酸基がチオリン酸基に置換されている。また、それぞれの断片として表3に示す4断片を合成した。これら4断片の塩基配列は表2の番号1の組合せと同一である。

2)T4RNAリガーゼ2によるライゲーション反応
4断片の修飾型RNAを用いてT4 RNAリガーゼ2により反応した。反応液の組成は50mM Tris-HCl、2mM MgCl2、1mM ジチオスレイトール、400μM ATP、pH7.5、修飾型RNA断片をそれぞれ10μMとし、反応液量は50μLとした。酵素の添加濃度を0.2または1.0U/μLとし、陰性対照として酵素非添加の条件でも反応した。サーマルサイクラーを用いて25℃で1時間反応した後、80℃で5分間加熱することにより反応を停止した。
4断片の修飾型RNAを用いてT4 RNAリガーゼ2により反応した。反応液の組成は50mM Tris-HCl、2mM MgCl2、1mM ジチオスレイトール、400μM ATP、pH7.5、修飾型RNA断片をそれぞれ10μMとし、反応液量は50μLとした。酵素の添加濃度を0.2または1.0U/μLとし、陰性対照として酵素非添加の条件でも反応した。サーマルサイクラーを用いて25℃で1時間反応した後、80℃で5分間加熱することにより反応を停止した。
3)HPLCおよびLC-TOF/MSによる分析
反応液をACQUITY UPLC Oligonucleotide BEH C18 Column (Waters、2.1×100mm、1.7μm)を用いたHPLCにより分析した。分析条件は、カラム温度80℃、検出波長260nm、インジェクション量10μL、流速0.4mL/minとした。移動相はA液(ヘキサフルオロイソプロパノール-トリエチルアミン)およびB液(メタノール)を用いたリニアグラジエントにより分析した。センス鎖およびアンチセンス鎖の標品も同様に分析することにより、ライゲーション産物の生成を確認した。また、Agilent 6230 TOF LC/MSシステム(Agilent technologies)によりライゲーション産物の質量分析を行った。
反応液をACQUITY UPLC Oligonucleotide BEH C18 Column (Waters、2.1×100mm、1.7μm)を用いたHPLCにより分析した。分析条件は、カラム温度80℃、検出波長260nm、インジェクション量10μL、流速0.4mL/minとした。移動相はA液(ヘキサフルオロイソプロパノール-トリエチルアミン)およびB液(メタノール)を用いたリニアグラジエントにより分析した。センス鎖およびアンチセンス鎖の標品も同様に分析することにより、ライゲーション産物の生成を確認した。また、Agilent 6230 TOF LC/MSシステム(Agilent technologies)によりライゲーション産物の質量分析を行った。
4)結果
HPLCによる分析の結果を図4に示す。HPLCによる分析では酵素の添加により、標品と一致する保持時間にライゲーション産物のピークが得られるとともに、陰性対照と比較して基質の修飾型RNAのピーク面積が減少した。また、酵素添加量の増加に伴いライゲーション産物のピーク面積も増加した。さらに反応液のLC-TOF/MSによる分析では酵素添加条件においてセンス鎖およびアンチセンス鎖の生成が観察された。
センス鎖LC/MS m/z: calcd 2266.13, found 2265.9703[M-3H]3-
アンチセンス鎖LC/MS m/z: calcd 2531.04, found 2531.0199[M-3H]3-
HPLCによる分析の結果を図4に示す。HPLCによる分析では酵素の添加により、標品と一致する保持時間にライゲーション産物のピークが得られるとともに、陰性対照と比較して基質の修飾型RNAのピーク面積が減少した。また、酵素添加量の増加に伴いライゲーション産物のピーク面積も増加した。さらに反応液のLC-TOF/MSによる分析では酵素添加条件においてセンス鎖およびアンチセンス鎖の生成が観察された。
センス鎖LC/MS m/z: calcd 2266.13, found 2265.9703[M-3H]3-
アンチセンス鎖LC/MS m/z: calcd 2531.04, found 2531.0199[M-3H]3-
以上から、T4 RNAリガーゼ2による修飾型RNAの4断片からのsiRNAの生成が可能であることが示された。
〔実施例4〕DNAリガーゼによる反応進行の確認
修飾型オリゴヌクレオチドにおける4断片からのsiRNAの酵素的ライゲーション反応の進行を、DNAリガーゼを用いて評価した。DNAリガーゼとしてT4 DNAリガーゼ(New England Biolabs)を用いた。
修飾型オリゴヌクレオチドにおける4断片からのsiRNAの酵素的ライゲーション反応の進行を、DNAリガーゼを用いて評価した。DNAリガーゼとしてT4 DNAリガーゼ(New England Biolabs)を用いた。
反応液の組成は、DNAリガーゼ50mM Tris-HCl、10mM MgCl2、10mMジチオスレイトール、1mM ATP、pH7.5とした。酵素濃度は470nM、オリゴヌクレオチド断片をそれぞれ10μMとし、反応液量は30μLとした。オリゴヌクレオチド断片は表3に示す組み合わせを用いた。サーマルサイクラーを用いて25℃で反応し、4時間後に10μLを採取し、80℃で5分間加熱することにより反応を停止した。反応液に含まれるライゲーション産物の濃度を実施例3に記載の条件にてHPLCにより分析した。標品も同様に分析することにより、ライゲーション産物の濃度を定量した。
HPLCによる分析の結果、ライゲーション産物の蓄積が確認され、反応4時間後の蓄積はセンス鎖が0.58μM、アンチセンス鎖が5.1μMであった。以上のように、DNAリガーゼにおいても4断片の修飾型RNAからのsiRNAの生成反応が進行した。
〔実施例5〕デイノコッカス・ラディオデュランス(Deinococcus radiodurans)RNAリガーゼの調製とライゲーション反応
(1)E. coliによる組換え発現株の構築
Deinococcus radiodurans由来のRnl5ファミリーに属するRNAリガーゼDraRnlをE. coliで発現する株を構築し、精製酵素を調製した。まず、DraRnlのアミノ酸配列(配列番号17)をE. coliコドンに最適化した塩基配列を有するプラスミドを遺伝子全合成により作成し、次にこの配列をpET16bベクターのNdeI/BamHIサイトにサブクローニングした。本発現プラスミドをE. coli BL21(DE3)を形質転換し、DraRnlの発現株を得た。この発現株では、N末端にHis-tagが付与されたDraRnlが発現される。
(1)E. coliによる組換え発現株の構築
Deinococcus radiodurans由来のRnl5ファミリーに属するRNAリガーゼDraRnlをE. coliで発現する株を構築し、精製酵素を調製した。まず、DraRnlのアミノ酸配列(配列番号17)をE. coliコドンに最適化した塩基配列を有するプラスミドを遺伝子全合成により作成し、次にこの配列をpET16bベクターのNdeI/BamHIサイトにサブクローニングした。本発現プラスミドをE. coli BL21(DE3)を形質転換し、DraRnlの発現株を得た。この発現株では、N末端にHis-tagが付与されたDraRnlが発現される。
(2)組換え酵素の調製
各発現株をアンピシリン100mg/Lを含むLB寒天培地で37℃、一晩生育させた。得られたコロニーをアンピシリン100mg/Lを含むLB培地100mLに植菌し、坂口フラスコを用いて振とう培養を行った。37℃で2時間培養後、終濃度でそれぞれ0.1mMまたは2%になるようにIPTGおよびエタノールを添加した。さらに17℃で16時間培養した。
各発現株をアンピシリン100mg/Lを含むLB寒天培地で37℃、一晩生育させた。得られたコロニーをアンピシリン100mg/Lを含むLB培地100mLに植菌し、坂口フラスコを用いて振とう培養を行った。37℃で2時間培養後、終濃度でそれぞれ0.1mMまたは2%になるようにIPTGおよびエタノールを添加した。さらに17℃で16時間培養した。
培養終了後、得られた培養液より菌体を遠心分離により集め、50mM Tris-HCl(pH7.6)、250mM NaCl、10% sucrose、15mM imidazole、1% Lysozyme、0.1% Triton-X100からなる緩衝液に懸濁し、超音波破砕を行った。遠心分離により破砕液から菌体残渣を除き、得られた上清を可溶性画分とした。
得られた可溶性画分を、前記緩衝液で平衡化したHis-tagタンパク質精製カラムHisTALON Superflow Cartridge(タカラバイオ)に供して、担体に吸着させた。担体に吸着しなかったタンパク質(非吸着タンパク質)を50mM Tris-HCl(pH7.6)、250mM NaCl、10% sucrose、15mM imidazoleからなる緩衝液を用いて洗い流した後、50mM Tris-HCl(pH8.0)、250mM NaCl、10% glycerol、200mM imidazoleからなる緩衝液で吸着したタンパク質の溶出を行った。
酵素が含まれる溶出画分を集めて、Amicon Ultra-15 10kDa(メルクミリポア)を用いて、50mM Tris-HCl(pH8.0)、200mM NaCl、2mM DTT、2mM EDTA、10% glycerol、0.1% Triton X-10からなる緩衝液にバッファー交換し、精製酵素溶液とした。
(3)DraRnlによるライゲーション反応
4断片の修飾型RNAを用いてDraRnlにより反応した。反応液の組成は50mM Tris-HCl(pH7.5)、10mM MnSO4、1mM ジチオスレイトール、400μM ATP、修飾型RNA断片をそれぞれ10μMとし、反応液量は25μLとした。修飾型RNA断片は表3の組み合わせを用いた。酵素の添加濃度を72μg/mLとし、陰性対照として酵素非添加の条件でも反応した。サーマルサイクラーを用いて25℃で3時間反応した後、終濃度1mMになるようにEDTAを加えることで反応を停止した。
4断片の修飾型RNAを用いてDraRnlにより反応した。反応液の組成は50mM Tris-HCl(pH7.5)、10mM MnSO4、1mM ジチオスレイトール、400μM ATP、修飾型RNA断片をそれぞれ10μMとし、反応液量は25μLとした。修飾型RNA断片は表3の組み合わせを用いた。酵素の添加濃度を72μg/mLとし、陰性対照として酵素非添加の条件でも反応した。サーマルサイクラーを用いて25℃で3時間反応した後、終濃度1mMになるようにEDTAを加えることで反応を停止した。
(4)HPLCによる分析
反応液をACQUITY HPLC Oligonucleotide BEH C18 Column (Waters、2.1×100mm、1.7μm)を用いたHPLCにより分析した。分析条件は、カラム温度60℃、検出波長260nm、インジェクション量10μL、流速0.4mL/minとした。移動相はA液ヘキサフルオロイソプロパノール-トリエチルアミンおよびB液(メタノール)を用いたリニアグラジエントにより分析した。センス鎖およびアンチセンス鎖の標品も同様に分析することにより、ライゲーション産物の生成を確認した。
反応液をACQUITY HPLC Oligonucleotide BEH C18 Column (Waters、2.1×100mm、1.7μm)を用いたHPLCにより分析した。分析条件は、カラム温度60℃、検出波長260nm、インジェクション量10μL、流速0.4mL/minとした。移動相はA液ヘキサフルオロイソプロパノール-トリエチルアミンおよびB液(メタノール)を用いたリニアグラジエントにより分析した。センス鎖およびアンチセンス鎖の標品も同様に分析することにより、ライゲーション産物の生成を確認した。
HPLCによる分析の結果を図5に示す。HPLCによる分析では酵素の添加により、標品と一致する保持時間にライゲーション産物のピークが得られるとともに、陰性対照と比較して基質の修飾型RNAのピーク面積が減少した。以上から、DraRnlによる修飾型RNAの4断片からの目的の修飾オリゴヌクレオチドの生成が可能であることが示された。
〔実施例6〕ループ構造を持つ修飾オリゴヌクレオチドの生成
4断片のオリゴヌクレオチドから酵素的ライゲーションにより、ループ構造を持つ修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物および合成した基質断片の配列を表4に示す。
4断片のオリゴヌクレオチドから酵素的ライゲーションにより、ループ構造を持つ修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物および合成した基質断片の配列を表4に示す。

表4の4断片の基質オリゴヌクレオチドを用いてT4 RNAリガーゼ2(New England Biolabs)により反応した。反応液の組成は50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5、オリゴヌクレオチド断片をそれぞれ10μMとし、反応液量は100μLとした。酵素の添加濃度17.8μg/μLとして生成物の濃度を比較した。陰性対照として酵素非添加の条件でも反応した。サーマルサイクラーを用いて25℃で3時間反応した後、80℃で5分間加熱することにより反応を停止した。実施例5の条件でHPLCおよびLC-TOF/MSにより反応液を分析した。
HPLCによる分析では図6に示すように、酵素の添加により、陰性対照と比較して、基質の修飾型RNAのピーク面積が減少し、基質と比較して保持時間の長い位置にピーク(5.6分付近)を検出することができた。さらに反応液のLC-TOF/MSによる分析では酵素添加条件において目的生成物が観察された。
LC/MS m/z: calcd 2727.33, found 2727.21[M-6H]6-
LC/MS m/z: calcd 2727.33, found 2727.21[M-6H]6-
以上から、T4 RNAリガーゼ2により、ループ構造を持つ目的の修飾オリゴヌクレオチドを4断片から生成可能であることが示された。
〔実施例7〕DNA鎖およびRNA鎖からなるヘテロ二本鎖の生成
二本鎖RNAリガーゼにより、修飾型DNA鎖および修飾型RNA鎖からなるヘテロ二本鎖を生成した。二本鎖RNAリガーゼとしてT4 RNAリガーゼ2(New England Biolabs)を用いた。
二本鎖RNAリガーゼにより、修飾型DNA鎖および修飾型RNA鎖からなるヘテロ二本鎖を生成した。二本鎖RNAリガーゼとしてT4 RNAリガーゼ2(New England Biolabs)を用いた。
反応液の組成は、50mM Tris-HCl、2mM MgCl2、1mM ジチオスレイトール、400μM ATP、pH7.5とした。酵素濃度は3.56μg/mL、基質は表5に示す4断片のオリゴヌクレオチドをそれぞれ10μMとし、反応液量は40μLとした。サーマルサイクラーを用いて37℃で反応し、18時間後に終濃度10mMになるようにEDTAを加えることで反応を停止した。陰性対照として酵素非添加の条件でも反応した。反応液に含まれるライゲーション産物の濃度を実施例5に記載の条件にてHPLCおよびLC-TOF/MSにより分析した。
HPLCによる分析では、酵素の添加により、陰性対照と比較して、各基質のピーク面積が減少し、新たに2本のピークを検出することができた。さらに反応液のLC-TOF/MSによる分析では、酵素添加条件において修飾型DNA鎖および修飾型RNA鎖の生成が観察された。
修飾型DNA鎖LC/MS m/z: calcd 2119.50, found 2119.34[M-2H]2-
修飾型RNA鎖LC/MS m/z: calcd 2126.89, found 2126.36[M-2H]2-
修飾型DNA鎖LC/MS m/z: calcd 2119.50, found 2119.34[M-2H]2-
修飾型RNA鎖LC/MS m/z: calcd 2126.89, found 2126.36[M-2H]2-
以上から、T4 RNAリガーゼ2により、修飾型DNA鎖および修飾型RNA鎖からなるヘテロ二本鎖の生成が可能であることが示された。

〔実施例8〕ミスマッチ塩基対を含むオリゴヌクレオチド断片を用いた反応
4断片のオリゴヌクレオチドから酵素的ライゲーションにより、塩基対にミスマッチ部分を持った二本鎖修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表6に、4断片の組み合わせを図7に示す。
4断片のオリゴヌクレオチドから酵素的ライゲーションにより、塩基対にミスマッチ部分を持った二本鎖修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表6に、4断片の組み合わせを図7に示す。
反応液の組成は、1.78μg/mL T4 RNAリガーゼ2(New England Biolabs)、50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5とした。基質として4断片のオリゴヌクレオチド断片をそれぞれ終濃度10μMとなるように添加し、40μLの液量で反応した。サーマルサイクラーを用いて25℃で反応し、4時間後に終濃度10mMになるようにEDTAを加えることで反応を停止した。陰性対照として酵素非添加の条件でも反応した。反応液に含まれるライゲーション産物の濃度を実施例5に記載の条件にてHPLCおよびLC-TOF/MSにより分析した。
HPLCによる分析では、酵素の添加により、陰性対照と比較して、各基質のピーク面積が減少し、新たに2本のピークを検出することができた。さらに反応液のLC-TOF/MSによる分析では、酵素添加条件においてA鎖およびB鎖の生成を確認できた。
A鎖 LC/MS m/z: calcd 1361.77, found 1361.75[M-5H]5-
B鎖 LC/MS m/z: calcd 1517.41, found 1517.35[M-5H]5-
A鎖 LC/MS m/z: calcd 1361.77, found 1361.75[M-5H]5-
B鎖 LC/MS m/z: calcd 1517.41, found 1517.35[M-5H]5-
以上から、T4 RNAリガーゼ2により、ミスマッチ配列を有する二本鎖修飾オリゴヌクレオチドが生成可能であることが示された。

〔実施例9〕5断片および6断片のオリゴヌクレオチドを用いた反応
5断片および6断片のオリゴヌクレオチドから酵素的ライゲーションにより、二本鎖修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表7に、4断片の組み合わせを図8に示す。
5断片および6断片のオリゴヌクレオチドから酵素的ライゲーションにより、二本鎖修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表7に、4断片の組み合わせを図8に示す。
反応液の組成は、1.78μg/mL T4 RNAリガーゼ2(New England Biolabs)、50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5とした。基質としてオリゴヌクレオチド断片をそれぞれ終濃度10μMとなるように添加し、30μLの液量で反応した。サーマルサイクラーを用いて37℃で反応し、16時間後に終濃度10mMになるようにEDTAを加えることで反応を停止した。陰性対照として酵素非添加の条件でも反応した。反応液に含まれるライゲーション産物の濃度を実施例5に記載の条件にてHPLCにより分析した。
HPLCによる分析では、図9に示すように酵素の添加により、陰性対照と比較して、各基質のピーク面積が減少し、新たに2本のピークを検出することができた。これらのピークの保持時間は生成物の標品と一致した。
以上から、T4 RNAリガーゼ2により、5断片および6断片の基質オリゴヌクレオチドから二本鎖修飾オリゴヌクレオチドが生成可能であることが示された。

〔実施例10〕5’末端にDMTr基が付加されたオリゴヌクレオチドを用いた反応
5’末端にジメトキシトリチル(DMTr)基が付加された2断片を含む4断片のオリゴヌクレオチドから酵素的ライゲーションにより二本鎖修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表8に、4断片の組み合わせを図10に示す。
5’末端にジメトキシトリチル(DMTr)基が付加された2断片を含む4断片のオリゴヌクレオチドから酵素的ライゲーションにより二本鎖修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表8に、4断片の組み合わせを図10に示す。
反応液の組成は、1.78μg/mL T4 RNAリガーゼ2(New England Biolabs)、50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5とした。基質として4断片のオリゴヌクレオチド断片をそれぞれ終濃度10μMとなるように添加し、40μLの液量で反応した。サーマルサイクラーを用いて25℃で反応し、4時間後に終濃度10mMになるようにEDTAを加えることで反応を停止した。陰性対照として酵素非添加の条件でも反応した。反応液に含まれるライゲーション産物の濃度を実施例5に記載の条件にてHPLCおよびLC-TOF/MSにより分析した。
HPLCによる分析では、酵素の添加により、陰性対照と比較して、各基質のピーク面積が減少し、新たに2本のピークを検出することができた。さらに反応液のLC-TOF/MSによる分析では、酵素添加条件においてA鎖およびB鎖の生成を確認できた。
A鎖 LC/MS m/z: calcd 1764.80, found 1764.79[M-4H]4-
B鎖 LC/MS m/z: calcd 1738.76, found 1738.75[M-4H]4-
A鎖 LC/MS m/z: calcd 1764.80, found 1764.79[M-4H]4-
B鎖 LC/MS m/z: calcd 1738.76, found 1738.75[M-4H]4-
以上から、T4 RNAリガーゼ2により、DMTr基を有する基質断片から二本鎖修飾オリゴヌクレオチドが生成可能であることが示された。

〔実施例11〕キャリア付加型オリゴヌクレオチド断片を用いた反応
5’末端にN-アセチルガラクトサミン(GalNAc)が付加された1断片を含む4断片のオリゴヌクレオチドから、酵素的ライゲーションにより二本鎖修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表9に、4断片の組み合わせを図11に示す。GalNAcで修飾した断片は、Trivalent β-D-GalNAc with carboxyl-functionalized PEG5 Linker (Sussex社製)をアミノC6リンカーを介してオリゴヌクレオチドの5’末端に連結して合成した。
5’末端にN-アセチルガラクトサミン(GalNAc)が付加された1断片を含む4断片のオリゴヌクレオチドから、酵素的ライゲーションにより二本鎖修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表9に、4断片の組み合わせを図11に示す。GalNAcで修飾した断片は、Trivalent β-D-GalNAc with carboxyl-functionalized PEG5 Linker (Sussex社製)をアミノC6リンカーを介してオリゴヌクレオチドの5’末端に連結して合成した。
反応液の組成は、1.78μg/mL T4 RNAリガーゼ2(New England Biolabs)、50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5とした。基質として4断片のオリゴヌクレオチド断片をそれぞれ終濃度10μMとなるように添加し、40μLの液量で反応した。サーマルサイクラーを用いて25℃で反応し、4時間後に終濃度10mMになるようにEDTAを加えることで反応を停止した。陰性対照として酵素非添加の条件でも反応した。反応液に含まれるライゲーション産物を実施例5に記載の条件にてHPLCおよびLC-TOF/MSにより分析した。
HPLCによる分析では、酵素の添加により、陰性対照と比較して、各基質のピーク面積が減少し、新たに2本のピークを検出することができた。さらに反応液のLC-TOF/MSによる分析では、酵素添加条件においてA鎖およびB鎖が検出され、生成を確認できた。
A鎖 LC/MS m/z: calcd 1730.40, found 1730.37[M-5H]5-
B鎖 LC/MS m/z: calcd 1330.38 found 1330.36[M-5H]5-
A鎖 LC/MS m/z: calcd 1730.40, found 1730.37[M-5H]5-
B鎖 LC/MS m/z: calcd 1330.38 found 1330.36[M-5H]5-
以上から、T4 RNAリガーゼ2を用いた4断片からの反応により、末端をN-アセチルガラクトサミンで修飾した二本鎖修飾オリゴヌクレオチドが生成可能であることが示された。
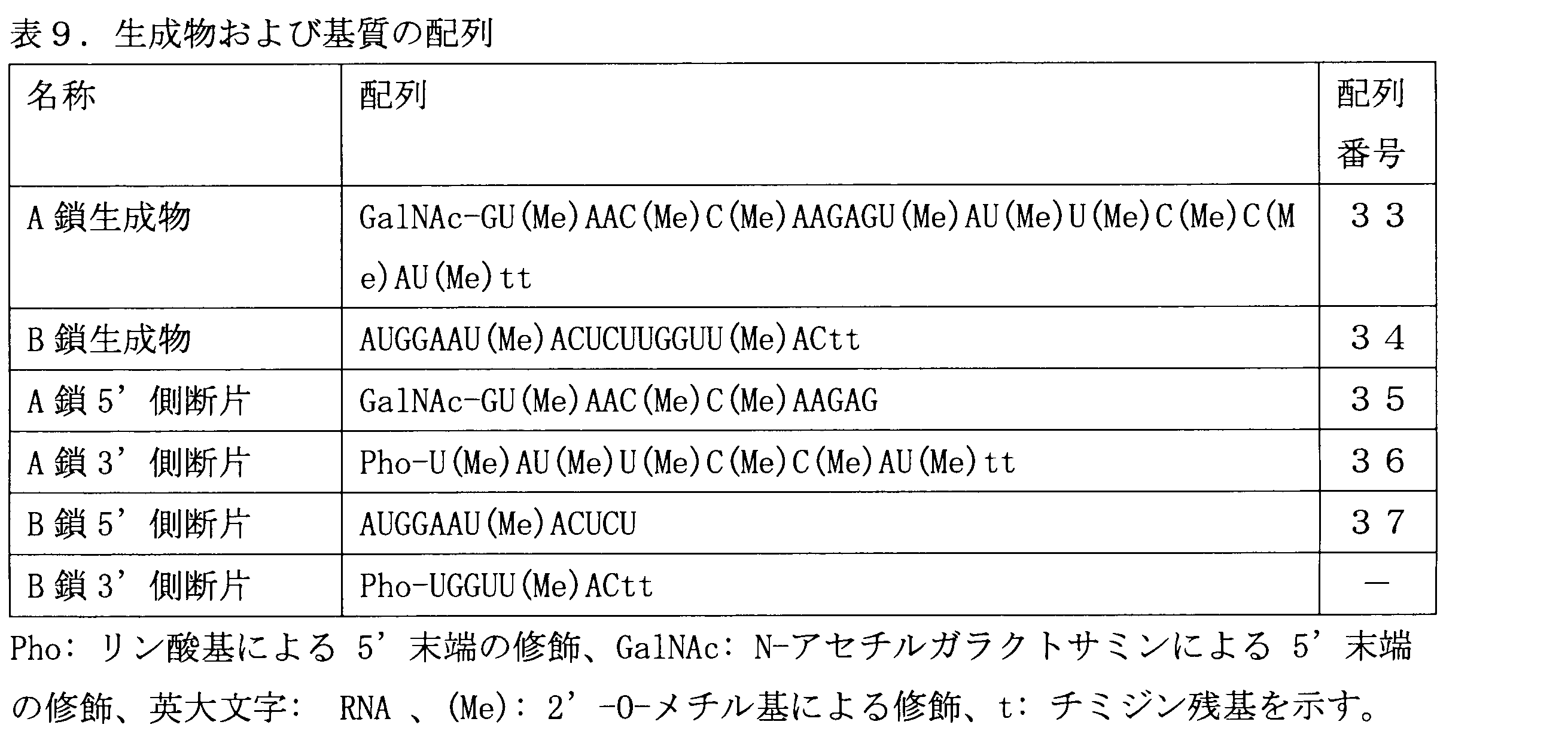
〔実施例12〕連結部にチオリン酸ジエステル結合を有する二本鎖修飾オリゴヌクレオチドの生成反応
リン酸基をチオリン酸基に置き換えた4断片のオリゴヌクレオチドから、酵素的ライゲーションにより二本鎖修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表10に、4断片の組み合わせを図12に示す。
リン酸基をチオリン酸基に置き換えた4断片のオリゴヌクレオチドから、酵素的ライゲーションにより二本鎖修飾オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表10に、4断片の組み合わせを図12に示す。
反応液の組成は、1.78μg/mL T4 RNAリガーゼ2(New England Biolabs)、50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5とした。基質として4断片のオリゴヌクレオチド断片をそれぞれ終濃度10μMとなるように添加し、40μLの液量で反応した。サーマルサイクラーを用いて25℃で反応し、4時間後に終濃度10mMになるようにEDTAを加えることで反応を停止した。陰性対照として酵素非添加の条件でも反応した。反応液に含まれるライゲーション産物を実施例5に記載の条件にてHPLCおよびLC-TOF/MSにより分析した。
HPLCによる分析では、酵素の添加により、陰性対照と比較して、各基質のピーク面積が減少し、新たに2本のピークを検出することができた。さらに反応液のLC-TOF/MSによる分析では、酵素添加条件においてA鎖およびB鎖が検出され、生成を確認できた。
A鎖 LC/MS m/z: calcd 1769.15, found 1769.13[M-4H]4-
B鎖 LC/MS m/z: calcd 1743.11 found 1743.10[M-4H]4-
A鎖 LC/MS m/z: calcd 1769.15, found 1769.13[M-4H]4-
B鎖 LC/MS m/z: calcd 1743.11 found 1743.10[M-4H]4-
以上から、T4 RNAリガーゼ2を用いた4断片からの反応により、連結部にチオリン酸結合を有する二本鎖修飾オリゴヌクレオチドが生成可能であることが示された。

〔実施例13〕ヘアピン型オリゴヌクレオチドの生成反応
4断片のオリゴヌクレオチドから、酵素的ライゲーションによりヘアピン型オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物および合成した基質断片の配列を表11に、4断片の組み合わせを図13に示す。リンカーとして文献(Hamasaki T, Suzuki H, Shirohzu H, et al. Efficacy of a novel class of RNA interference therapeutic agents. PLoS ONE. 2012;7(8):e42655.)に記載のプロリン誘導体を用いた。
4断片のオリゴヌクレオチドから、酵素的ライゲーションによりヘアピン型オリゴヌクレオチドを生成する反応の進行を評価した。目的生成物および合成した基質断片の配列を表11に、4断片の組み合わせを図13に示す。リンカーとして文献(Hamasaki T, Suzuki H, Shirohzu H, et al. Efficacy of a novel class of RNA interference therapeutic agents. PLoS ONE. 2012;7(8):e42655.)に記載のプロリン誘導体を用いた。
反応液の組成は、1.78μg/mL T4 RNAリガーゼ2(New England Biolabs)、50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5とした。基質として4断片のオリゴヌクレオチド断片をそれぞれ終濃度10μMとなるように添加し、40μLの液量で反応した。サーマルサイクラーを用いて25℃で反応し、4時間後に終濃度10mMになるようにEDTAを加えることで反応を停止した。陰性対照として酵素非添加の条件でも反応した。反応液に含まれるライゲーション産物を実施例5に記載の条件にてHPLCおよびLC-TOF/MSにより分析した。
HPLCによる分析では、酵素の添加により、陰性対照と比較して、各基質のピーク面積が減少し、新たに1本のピークを検出することができた。さらに反応液のLC-TOF/MSによる分析では、酵素添加条件において目的生成物が検出され、生成を確認できた。
LC/MS m/z: calcd 1891.61 found 1891.60[M-9H]9-
LC/MS m/z: calcd 1891.61 found 1891.60[M-9H]9-
以上から、T4 RNAリガーゼ2を用いた4断片からの反応により、ヘアピン型オリゴヌクレオチドが生成可能であることが示された。

〔実施例14〕突出末端の塩基長の反応性への影響
生成物は同一で、1~6塩基長の突出末端を形成するように基質のオリゴ核酸を設計し、突出末端の塩基長の差による反応性の違いを比較した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。それぞれの組み合わせにおいて、B鎖を構成する基質は共通する配列とし、A鎖の切断位置を異なる位置とした。合成した目的生成物標品および基質断片の配列を表12に、4断片の組み合わせを図14に示す。
生成物は同一で、1~6塩基長の突出末端を形成するように基質のオリゴ核酸を設計し、突出末端の塩基長の差による反応性の違いを比較した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。それぞれの組み合わせにおいて、B鎖を構成する基質は共通する配列とし、A鎖の切断位置を異なる位置とした。合成した目的生成物標品および基質断片の配列を表12に、4断片の組み合わせを図14に示す。
反応液の組成は、1.78μg/mL T4 RNAリガーゼ2(New England Biolabs)、50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5とした。基質として4断片のオリゴヌクレオチド断片をそれぞれ終濃度10μMとなるように添加し、40μLの液量で反応した。サーマルサイクラーを用いて25℃で反応し、15分、30分、1時間、2時間および4時間後に5μLを回収し終濃度10mMになるようにEDTAを加えることで反応を停止した。陰性対照として酵素非添加の条件でも反応した。反応液に含まれるライゲーション産物および生成物標品の濃度を実施例5に記載の条件にてHPLCにより分析し、ライゲーション産物の濃度を算出した。
HPLC分析により確認した結果、いずれの塩基長の突出末端においても、A鎖およびB鎖の生成が観察された。1塩基長の突出末端では反応速度が低い傾向があった。

〔実施例15〕生成物の塩基長の反応性への影響
11~14塩基長の相補部分からなる短い目的生成物を製造した場合の反応性の違いを比較した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表13に、4断片の組み合わせを図15に示す。
11~14塩基長の相補部分からなる短い目的生成物を製造した場合の反応性の違いを比較した。目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表13に、4断片の組み合わせを図15に示す。
反応液の組成は、1.78μg/mL T4 RNAリガーゼ2(New England Biolabs)、50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5とした。基質として4断片のオリゴヌクレオチド断片をそれぞれ終濃度10μMとなるように添加し、40μLの液量で反応した。サーマルサイクラーを用いて25℃で反応し、4時間後に終濃度10mMになるようにEDTAを加えることで反応を停止した。陰性対照として酵素非添加の条件でも反応した。反応液に含まれるライゲーション産物を実施例5に記載の条件にてHPLCおよびLC-TOF/MSにより分析した。
HPLCによる分析では、酵素の添加により、陰性対照と比較して、各基質のピーク面積が減少し、保持時間の遅い位置に新たなピークを検出することができた。さらに反応液のLC-TOF/MSによる分析では、酵素添加条件において表14に示すように、いずれの塩基長の生成物においてもA鎖およびB鎖の2価または3価のイオンが検出され、反応の進行が確認された。
HPLCにより各反応条件における基質のピーク面積値を算出し、基質の残存率を以下の式で算出した。
残存率(%)=(酵素添加条件における基質のピーク面積の合計)
/陰性対照における基質のピーク面積の合計)×100
残存率(%)=(酵素添加条件における基質のピーク面積の合計)
/陰性対照における基質のピーク面積の合計)×100
11塩基長の相補部分からなる生成物では、12塩基長以上の相補部分からなる生成物と比べて、基質の残存率が高い傾向があった。


〔実施例16〕高濃度基質での反応
より高濃度の基質存在下での修飾オリゴヌクレオチドの生成反応速度を、各pHで比較した。反応の目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表15に、4断片の組み合わせを図16に示す。
より高濃度の基質存在下での修飾オリゴヌクレオチドの生成反応速度を、各pHで比較した。反応の目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表15に、4断片の組み合わせを図16に示す。
反応液の組成は、1.78μg/mL T4 RNAリガーゼ2(New England Biolabs)、2mM MgCl2、1mMジチオスレイトール、400μM ATPとし、バッファーとして50mM Tris-HCl(pH7.0~9.0)を用いた。基質としてオリゴヌクレオチド断片をそれぞれ終濃度10μM、300μM、500μMまたは1000μMとなるように添加し、40μLの液量で反応した。サーマルサイクラーを用いて25℃で反応し、15分または1時間後にサンプリングし終濃度10mMになるようにEDTAを加えることで反応を停止した。反応液に含まれるライゲーション産物を実施例5に記載の条件にてHPLCにより分析し、A鎖およびB鎖の合計の濃度から生成速度を算出した。基質濃度300μM以上においても反応進行が確認され、これらの濃度ではpH8.0および8.5で高い反応速度を示した。

〔実施例17〕界面活性剤添加の効果
界面活性剤の添加による修飾オリゴヌクレオチドの生成反応速度を評価した。目的生成物および合成した基質断片の配列を表15に、4断片の組み合わせを図16に示す。
反応液の組成は、1.78μg/mL T4 RNAリガーゼ2(New England Biolabs)、2mM MgCl2、1mMジチオスレイトール、400μM ATP、50mM Tris-HCl(pH7.5)とし、界面活性剤として終濃度0.1%のTritonX-100を使用した。基質としてオリゴヌクレオチド断片をそれぞれ終濃度20μMとなるように添加し、40μLの液量で反応した。
評価では、酵素溶液を保存バッファー(10 mM Tris-HCl、50 mM KCl、35 mM 硫酸アンモニウム、0.1mMジチオスレイトール、0.1mM EDTA、50% グリセロール、pH7.5)で17.8μg/mLに希釈したのちに反応液中に1/10量添加する条件(対照条件)、酵素溶液を保存バッファーで17.8μg/mLに希釈したのちに終濃度0.1%の TritonX-100を含む反応液中に1/10量添加する条件(試験条件1)、酵素溶液を0.1% TritonX-100を含む保存バッファーで17.8μg/mLに希釈したのちに0.09%のTritonX-100を含む反応液中に1/10量添加する条件(試験条件2、TritonX-100終濃度0.1%)、の3条件を比較した。
サーマルサイクラーを用いて25℃で反応し、4時間後にサンプリングし終濃度10mMになるようにEDTAを加えることで反応を停止した。反応液に含まれるライゲーション産物を実施例5に記載の条件にてHPLCにより分析し、A鎖およびB鎖の濃度を算出した。
試験条件1および試験条件2では、対照条件に比べて、より高濃度のA鎖およびB鎖の生成物が観察された。
界面活性剤の添加による修飾オリゴヌクレオチドの生成反応速度を評価した。目的生成物および合成した基質断片の配列を表15に、4断片の組み合わせを図16に示す。
反応液の組成は、1.78μg/mL T4 RNAリガーゼ2(New England Biolabs)、2mM MgCl2、1mMジチオスレイトール、400μM ATP、50mM Tris-HCl(pH7.5)とし、界面活性剤として終濃度0.1%のTritonX-100を使用した。基質としてオリゴヌクレオチド断片をそれぞれ終濃度20μMとなるように添加し、40μLの液量で反応した。
評価では、酵素溶液を保存バッファー(10 mM Tris-HCl、50 mM KCl、35 mM 硫酸アンモニウム、0.1mMジチオスレイトール、0.1mM EDTA、50% グリセロール、pH7.5)で17.8μg/mLに希釈したのちに反応液中に1/10量添加する条件(対照条件)、酵素溶液を保存バッファーで17.8μg/mLに希釈したのちに終濃度0.1%の TritonX-100を含む反応液中に1/10量添加する条件(試験条件1)、酵素溶液を0.1% TritonX-100を含む保存バッファーで17.8μg/mLに希釈したのちに0.09%のTritonX-100を含む反応液中に1/10量添加する条件(試験条件2、TritonX-100終濃度0.1%)、の3条件を比較した。
サーマルサイクラーを用いて25℃で反応し、4時間後にサンプリングし終濃度10mMになるようにEDTAを加えることで反応を停止した。反応液に含まれるライゲーション産物を実施例5に記載の条件にてHPLCにより分析し、A鎖およびB鎖の濃度を算出した。
試験条件1および試験条件2では、対照条件に比べて、より高濃度のA鎖およびB鎖の生成物が観察された。
〔実施例18〕生成物の塩基長の違いによる不純物の淘汰性の比較
基質および生成物について塩基長の異なる反応を実施し、基質オリゴ核酸断片および酵素反応後の溶液に含まれる不純物を分析した。反応の目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表16に、4断片の組み合わせを図17に示す。
基質および生成物について塩基長の異なる反応を実施し、基質オリゴ核酸断片および酵素反応後の溶液に含まれる不純物を分析した。反応の目的生成物のそれぞれの鎖をA鎖及びB鎖とした。目的生成物および合成した基質断片の配列を表16に、4断片の組み合わせを図17に示す。
反応液の組成は、修飾オリゴヌクレオチドを用いた反応では8.9μg/mL T4 RNAリガーゼ2(New England Biolabs)、50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5とした。DNAを用いた反応では22.0μg/mL T7 DNAリガーゼ(New England Biolabs)、50mM Tris-HCl、2mM MgCl2、1mMジチオスレイトール、400μM ATP、pH7.5とした。基質として4断片のオリゴヌクレオチド断片をそれぞれ終濃度50μMとなるように添加し、30μLの液量で反応した。サーマルサイクラーを用いて25℃で反応し、8時間後に終濃度12.5mMになるようにEDTAを加えることで反応を停止した。反応液を実施例5に記載の条件にてHPLCおよびLC-TOF/MSにより分析し、目的の反応進行を確認した。
得られた質量分析結果から先行文献(Roussis et al., Journal of Chromatogr A. 2019;1584:106-114.)の方法に基づき、目的の構造の生成物に対する不純物(N±1mer)の含有率を算出した。また、反応に供した基質オリゴヌクレオチド溶液も同様にLC-TOF/MSにより分析し、基質に対する不純物(N±1mer)の含有率を算出した。
上記で算出した値から、各反応のA鎖およびB鎖のそれぞれについて、以下の式で不純物の残存率(%)を算出した。
不純物の残存率(%)=(反応液中の目的生成物に対するN±1merの比率)/(基質溶液中の基質に対するN±1merの比率)×100
不純物の残存率(%)=(反応液中の目的生成物に対するN±1merの比率)/(基質溶液中の基質に対するN±1merの比率)×100
結果を表17に示す。28塩基長のオリゴ核酸を生成する反応では、不純物(N±1mer)の残存率は86%以上であるのに対し、25塩基長以下のオリゴ核酸を生成する反応では、23~59%であった。


本発明は、核酸医薬等の製品に利用可能な修飾オリゴヌクレオチド(例、siRNAやヘテロ二本鎖オリゴヌクレオチド等)の製造のために有用である。
Claims (14)
- 11~27塩基長の相補部分を含む修飾オリゴヌクレオチドの製造方法であって、
該方法は、合計4本以上のオリゴヌクレオチド原料断片をオリゴヌクレオチドリガーゼの存在下で処理して該修飾オリゴヌクレオチドを生成することを含み、
合計4本以上のオリゴヌクレオチド原料断片は、該修飾オリゴヌクレオチドを、下記条件(i)~(v)を満たす断片連結部で分けた場合に得られるオリゴヌクレオチド原料断片に相当する、方法:
(i)断片連結部は、相補部分の各鎖側中のそれぞれに1以上存在し、かつ断片連結部は、該修飾オリゴヌクレオチド中に合計2つ以上存在し;
(ii)該修飾オリゴヌクレオチドを断片連結部で分けた場合に、突出末端が相補部分中に形成され、かつ該突出末端が1~10塩基長であり;
(iii)少なくとも1本のオリゴヌクレオチド原料断片が、修飾ヌクレオチドを含み、
(iv)該合計4本以上のオリゴヌクレオチド原料断片のうち4本のオリゴヌクレオチド原料断片が、5~25塩基長の相補部分を含み、かつ
(v)オリゴヌクレオチド原料断片の相補部分の各鎖側に相当する塩基長の合計が、いずれも11~27塩基長である。 - (ii)における突出末端が、2~6塩基長である、請求項1記載の方法。
- (iv)で規定される4本のオリゴヌクレオチド原料断片の相補部分における突出末端以外の部分が、4~16塩基長である、請求項1または2記載の方法。
- オリゴヌクレオチドリガーゼが、RNAリガーゼである、請求項1~3のいずれか一項記載の方法。
- オリゴヌクレオチドリガーゼが、二本鎖RNAリガーゼである、請求項4記載の方法。
- 二本鎖RNAリガーゼが、Rnl2ファミリーまたはRnl5ファミリーのRNAリガーゼである、請求項5記載の方法。
- 修飾オリゴヌクレオチドが、修飾型ヌクレオチド残基を含む、請求項1~6のいずれか一項記載の方法。
- 修飾型ヌクレオチド残基が、1’、2’、3’、もしくは4’化学修飾型ヌクレオチド残基、5’-または3’-リン酸基修飾型ヌクレオチド残基、架橋型修飾型ヌクレオチド残基、キャリア付加修飾型ヌクレオチド残基、または糖骨格置換型ヌクレオチド残基である、請求項7記載の方法。
- 修飾型ヌクレオチド残基が、
i)1’、2’、3’、もしくは4’部位が、C1~6アルキルオキシC1~6アルキレン、-O-C1~6アルキル、-O-C6~14アリール、-C-アリール、ハロゲン原子、-O-C1~6アルキルN-アミドC1~6アルキレン、-O-C1~6アルキル-(C1~6アルキル-)アミノ-C1~6アルキレン、または-O-アミノC1~6アルキル(例、-O-アミノプロピル、-O-AP)で置換された、1’、2’、3’、もしくは4’化学修飾型ヌクレオチド残基;
ii)水酸基が、保護基で置換されていてもよい、-O-Ρ(S)(OH)2、-NH-Ρ(O)(OH)2、もしくは-NH-Ρ(S)(OH)2で置換された、5’-もしくは3’-リン酸基修飾型ヌクレオチド残基;
iii)2’部位と4’部位が、2’-O-C1~6アルキレン-4’、2’-O-エチレン-4’、2’-O-メチル置換メチレン-4’、2’-O-C1~6アルキレン-O-C1~6アルキレン-4’、2’-O-N(R)-C1~6アルキレン-4’(ここで、Rはメチル、水素原子またはベンジルを示す)、2’-N(R)-C(O)-4’、2’-NH-C1~6アルキレン-4’、もしくは、2’-C1~6アルキレン-4’に置換された、または、3’部位と5’部位が、3’-C1~6アルキレン-5’に置換された、架橋型修飾型ヌクレオチド残基;または
iv)ヘキシトール核酸(HNA)残基、シクロヘキセニル核酸(CeNA)残基、もしくはモルフォリノ核酸(PMO)残基
である、請求項8記載の方法。 - オリゴヌクレオチド原料断片より選ばれる任意の2本のオリゴヌクレオチド原料断片の全モル比が、0.5~2である、請求項1~9のいずれか一項記載の方法。
- オリゴヌクレオチド原料断片が、10mM以下の一価カチオン塩濃度下で処理される、請求項1~10のいずれか一項記載の方法。
- 前記処理することの前に、オリゴヌクレオチド原料断片混合溶液を高温に置きその後クールダウンすることが行われない、請求項1~11のいずれか一項記載の方法。
- 前記修飾オリゴヌクレオチドの不純物の生成が抑制される、請求項1~12のいずれか一項記載の方法。
- 前記修飾オリゴヌクレオチドを精製することをさらに含む、請求項1~13のいずれか一項記載の方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20759682.6A EP3929205A4 (en) | 2019-02-18 | 2020-02-18 | METHOD FOR PRODUCING A MODIFIED OLIGONUCLEOTIDE COMPRISING A COMPLEMENTARY SEQUENCE |
| JP2021502052A JPWO2020171092A1 (ja) | 2019-02-18 | 2020-02-18 | 相補部分を含む修飾オリゴヌクレオチドの製造方法 |
| CN202080014985.7A CN113439085A (zh) | 2019-02-18 | 2020-02-18 | 包含互补部分的修饰寡核苷酸的制造方法 |
| US17/403,317 US20220041645A1 (en) | 2019-02-18 | 2021-08-16 | Method for producing modified oligonucleotide comprising complementary portion |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019026868 | 2019-02-18 | ||
| JP2019-026868 | 2019-02-18 | ||
| JP2019174557 | 2019-09-25 | ||
| JP2019-174557 | 2019-09-25 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US17/403,317 Continuation US20220041645A1 (en) | 2019-02-18 | 2021-08-16 | Method for producing modified oligonucleotide comprising complementary portion |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020171092A1 true WO2020171092A1 (ja) | 2020-08-27 |
Family
ID=72143373
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2020/006366 WO2020171092A1 (ja) | 2019-02-18 | 2020-02-18 | 相補部分を含む修飾オリゴヌクレオチドの製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20220041645A1 (ja) |
| EP (1) | EP3929205A4 (ja) |
| JP (1) | JPWO2020171092A1 (ja) |
| CN (1) | CN113439085A (ja) |
| WO (1) | WO2020171092A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024029547A1 (ja) * | 2022-08-02 | 2024-02-08 | 味の素株式会社 | オリゴ核酸の製造法 |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117660374A (zh) * | 2022-09-08 | 2024-03-08 | 凯莱英医药集团(天津)股份有限公司 | Rna连接酶在寡核苷酸制备上的应用 |
| CN117070513B (zh) * | 2023-10-16 | 2024-04-26 | 吉林凯莱英医药化学有限公司 | 含有硫代磷酸酯键的rna及其制备方法 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20180023122A1 (en) | 2016-07-11 | 2018-01-25 | Glaxosmithkline Intellectual Property Development Limited | Novel processes for the production of oligonucleotides |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2780572B2 (ja) * | 1991-09-13 | 1998-07-30 | 株式会社島津製作所 | オリゴヌクレオチドの酵素的合成法及びオリゴヌクレオチドのプライマーとしての使用 |
| JPH08268A (ja) * | 1994-06-16 | 1996-01-09 | Koichi Nishigaki | 連結した2本鎖dnaの製造方法 |
| KR102242879B1 (ko) * | 2011-09-07 | 2021-04-20 | 엑스-켐, 인크. | Dna-코딩된 라이브러리에 태그부착하는 방법 |
| CA3066744A1 (en) * | 2017-06-12 | 2018-12-20 | Twist Bioscience Corporation | Methods for seamless nucleic acid assembly |
| GB201721307D0 (en) * | 2017-12-19 | 2018-01-31 | Glaxosmithkline Ip Dev Ltd | Novel processes for the production of oligonucleotides |
-
2020
- 2020-02-18 WO PCT/JP2020/006366 patent/WO2020171092A1/ja unknown
- 2020-02-18 JP JP2021502052A patent/JPWO2020171092A1/ja active Pending
- 2020-02-18 CN CN202080014985.7A patent/CN113439085A/zh active Pending
- 2020-02-18 EP EP20759682.6A patent/EP3929205A4/en active Pending
-
2021
- 2021-08-16 US US17/403,317 patent/US20220041645A1/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20180023122A1 (en) | 2016-07-11 | 2018-01-25 | Glaxosmithkline Intellectual Property Development Limited | Novel processes for the production of oligonucleotides |
Non-Patent Citations (16)
| Title |
|---|
| "How an RNA Ligase Discriminates RNA versus DNA Damage", MOLECULAR CELL, vol. 16, 2004, pages 211 - 221, XP055732818 |
| ALICE SOSIC, MATTEO PASQUALIN, GIANFRANCO PASUT, BARBARA GATTO: "Enzymatic Formation of PEGylated Oligonucleotides", BIOCONJUGATE CHEMISTRY, vol. 25, no. 2, 19 February 2014 (2014-02-19), US , pages 433 - 441, XP055732817, ISSN: 1043-1802, DOI: 10.1021/bc400569z |
| BULLARD DESMOND R, BOWATER RICHARD P: "Direct comparison of nick-joining activity of the nucleic acid ligases from bacteriophage T4.", BIOCHEMICAL JOURNAL, vol. 398, no. 1, 15 August 2006 (2006-08-15), GB , pages 135 - 144, XP002467857, ISSN: 0264-6021, DOI: 10.1042/BJ20060313 |
| BULLARD, D. R. ET AL.: "Direct comparison of nick-joining activity of the nucleic acid ligases from bacteriophage T4", BIOCHEM. J., vol. 398, 2006, pages 135 - 144, XP002467857, DOI: 10.1042/BJ20060313 * |
| BULLARD, D. R.BOWATER R. P.: "Direct comparison of nick-joining activity of the nucleic acid ligases from bacteriophage T4.", BIOCHEM. J, vol. 398, 2006, pages 135 - 144, XP002467857, DOI: 10.1042/BJ20060313 |
| HSIUNG HANSEN M., BROUSSEAU ROLAND, MICHNIEWICZ JOSEPH, NARANG SARAN A.: "Synthesis of human insulin gene. Part I. Development of reversed-phase chromatography in the modified triester method. Its application in the rapid and efficient synthesis of eight deoxyribo-oligonucleotides fragments constituting human insulin A DNA", NUCLEIC ACIDS RESEARCH, vol. 6, no. 4, 1 January 1979 (1979-01-01), GB , pages 1371 - 1385, XP093034576, ISSN: 0305-1048, DOI: 10.1093/nar/6.4.1371 |
| JAYAPRAKASH K. NAIR ET AL.: "Multivalent N-Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing.", J. AM. CHEM. SOC., vol. 136, 2014, pages 16958 - 16961, XP055181463, DOI: 10.1021/ja505986a |
| MIHAELA-CARMEN UNCIULEACSTEWART SHUMAN, RNA, vol. 21, 2019, pages 824 - 832 |
| NANDAKUMAR, J. ET AL.: "How an RNA ligase discriminates RNA versus DNA damage", MOL. CELL, vol. 16, 2004, pages 211 - 221, XP055732818 * |
| NANDAKUMAR, J.SHUMAN, S.: "How an RNA Ligase Discriminates RNA versus DNA Damage.", MOLECULAR CELL, vol. 16, no. 2, 2004, pages 211 - 221, XP055732818 |
| NANDAKUMAR, JHO CKLIMA CDSHUMAN S: "RNA substrate specificity and structure-guided mutational analysis of bacteriophage T4 RNA ligase 2.", J BIOL CHEM, vol. 279, 2004, pages 31337 - 31347, XP002468194, DOI: 10.1074/jbc.M402394200 |
| PASCAL: "DNA and RNA ligases: structural variations and shared mechanisms", CURRENT OPINION IN STRUCTURAL BIOLOGY, vol. 18, 2008, pages 96 - 105, XP022485997, DOI: 10.1016/j.sbi.2007.12.008 |
| ROUSSIS ET AL., JOURNAL OF CHROMATOGR A., vol. 1584, 2019, pages 106 - 114 |
| See also references of EP3929205A4 |
| SOSIC APASQUALIN MPASUT GGATTO B.: "Enzymatic formation of PEGylated oligonucleotides.", BIOCONJUG CHEM, vol. 25, 2014, pages 433 - 441, XP055732817, DOI: 10.1021/bc400569z |
| SOSIC, A. ET AL.: "Enzymatic formation of PEGylated oligonucleotides", BIOCONJUGATE CHEM., vol. 25, 2014, pages 433 - 441, XP055732817 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024029547A1 (ja) * | 2022-08-02 | 2024-02-08 | 味の素株式会社 | オリゴ核酸の製造法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20220041645A1 (en) | 2022-02-10 |
| CN113439085A (zh) | 2021-09-24 |
| EP3929205A1 (en) | 2021-12-29 |
| JPWO2020171092A1 (ja) | 2021-12-16 |
| EP3929205A4 (en) | 2022-11-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2020171092A1 (ja) | 相補部分を含む修飾オリゴヌクレオチドの製造方法 | |
| AU2020256391B2 (en) | A method for the site-specific enzymatic labelling of nucleic acids in vitro by incorporation of unnatural nucleotides | |
| US20200277659A1 (en) | Novel processes for the production of oligonucleotides | |
| JP2022521094A (ja) | 共転写キャッピング用rnaポリメラーゼバリアント | |
| US11879145B2 (en) | Reagents and methods for replication, transcription, and translation in semi-synthetic organisms | |
| JP7065970B2 (ja) | オリゴヌクレオチドの産生のための新規プロセス | |
| JP6522496B2 (ja) | L−核酸の酵素性合成 | |
| CN113444698A (zh) | 连接酶突变体 | |
| JP2016537981A (ja) | 酵素によるl−核酸の合成 | |
| McManus et al. | O6‐Alkylguanine‐DNA Alkyltransferase‐Mediated Repair of O4‐Alkylated 2′‐Deoxyuridines | |
| JP2021153573A (ja) | リガーゼ変異体 | |
| Carnero Martín et al. | The impact of an extended nucleobase-2'-deoxyribose linker in the biophysical and biological properties of oligonucleotides | |
| Bley | Mapping the RNA-protein interface in telomerase RNP | |
| Veliath | Synthesis and characterization of c8 analogs of c-di-GMP; new synthetic method for 5'-capped oligoribonucleotides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 20759682 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021502052 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2020759682 Country of ref document: EP Effective date: 20210920 |