WO2020066976A1 - Resin composition, cured film, multilayer body, method for producing cured film and semiconductor device - Google Patents
Resin composition, cured film, multilayer body, method for producing cured film and semiconductor device Download PDFInfo
- Publication number
- WO2020066976A1 WO2020066976A1 PCT/JP2019/037188 JP2019037188W WO2020066976A1 WO 2020066976 A1 WO2020066976 A1 WO 2020066976A1 JP 2019037188 W JP2019037188 W JP 2019037188W WO 2020066976 A1 WO2020066976 A1 WO 2020066976A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- resin composition
- compound
- mass
- composition according
- Prior art date
Links
- NWVVVBRKAWDGAB-UHFFFAOYSA-N COc(cc1)ccc1O Chemical compound COc(cc1)ccc1O NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 1
- POTQBGGWSWSMCX-UHFFFAOYSA-N NCCCOCCOCCCN Chemical compound NCCCOCCOCCCN POTQBGGWSWSMCX-UHFFFAOYSA-N 0.000 description 1
- IWBOPFCKHIJFMS-UHFFFAOYSA-N NCCOCCOCCN Chemical compound NCCOCCOCCN IWBOPFCKHIJFMS-UHFFFAOYSA-N 0.000 description 1
- AZQWKYJCGOJGHM-UHFFFAOYSA-N O=C(C=C1)C=CC1=O Chemical compound O=C(C=C1)C=CC1=O AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 1
- YXAOOTNFFAQIPZ-UHFFFAOYSA-N Oc(ccc1c2cccc1)c2N=O Chemical compound Oc(ccc1c2cccc1)c2N=O YXAOOTNFFAQIPZ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B15/00—Layered products comprising a layer of metal
- B32B15/04—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material
- B32B15/08—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin
- B32B15/088—Layered products comprising a layer of metal comprising metal as the main or only constituent of a layer, which is next to another layer of the same or of a different material of synthetic resin comprising polyamides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B32—LAYERED PRODUCTS
- B32B—LAYERED PRODUCTS, i.e. PRODUCTS BUILT-UP OF STRATA OF FLAT OR NON-FLAT, e.g. CELLULAR OR HONEYCOMB, FORM
- B32B27/00—Layered products comprising a layer of synthetic resin
- B32B27/34—Layered products comprising a layer of synthetic resin comprising polyamides
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/32—Epoxy compounds containing three or more epoxy groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/10—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
- C08G73/12—Unsaturated polyimide precursors
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/01—Use of inorganic substances as compounding ingredients characterized by their specific function
- C08K3/011—Crosslinking or vulcanising agents, e.g. accelerators
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/0008—Organic ingredients according to more than one of the "one dot" groups of C08K5/01 - C08K5/59
- C08K5/0025—Crosslinking or vulcanising agents; including accelerators
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L79/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing nitrogen with or without oxygen or carbon only, not provided for in groups C08L61/00 - C08L77/00
- C08L79/04—Polycondensates having nitrogen-containing heterocyclic rings in the main chain; Polyhydrazides; Polyamide acids or similar polyimide precursors
- C08L79/08—Polyimides; Polyester-imides; Polyamide-imides; Polyamide acids or similar polyimide precursors
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/027—Non-macromolecular photopolymerisable compounds having carbon-to-carbon double bonds, e.g. ethylenic compounds
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/004—Photosensitive materials
- G03F7/09—Photosensitive materials characterised by structural details, e.g. supports, auxiliary layers
- G03F7/094—Multilayer resist systems, e.g. planarising layers
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/16—Coating processes; Apparatus therefor
- G03F7/168—Finishing the coated layer, e.g. drying, baking, soaking
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/20—Exposure; Apparatus therefor
-
- G—PHYSICS
- G03—PHOTOGRAPHY; CINEMATOGRAPHY; ANALOGOUS TECHNIQUES USING WAVES OTHER THAN OPTICAL WAVES; ELECTROGRAPHY; HOLOGRAPHY
- G03F—PHOTOMECHANICAL PRODUCTION OF TEXTURED OR PATTERNED SURFACES, e.g. FOR PRINTING, FOR PROCESSING OF SEMICONDUCTOR DEVICES; MATERIALS THEREFOR; ORIGINALS THEREFOR; APPARATUS SPECIALLY ADAPTED THEREFOR
- G03F7/00—Photomechanical, e.g. photolithographic, production of textured or patterned surfaces, e.g. printing surfaces; Materials therefor, e.g. comprising photoresists; Apparatus specially adapted therefor
- G03F7/26—Processing photosensitive materials; Apparatus therefor
Definitions
- the present invention relates to a resin composition containing a polyimide precursor.
- the present invention also relates to a cured film, a laminate, a method for producing a cured film, and a semiconductor device using the resin composition containing the polymer precursor described above.
- Polyimide resins have been applied to various uses because of their excellent heat resistance and insulation properties.
- the application of the semiconductor device is not particularly limited.
- a semiconductor device for mounting may be used as a material for an insulating film or a sealing material, or as a protective film (see Non-Patent Documents 1 and 2). Further, it is also used as a base film or coverlay of a flexible substrate.
- polyimide resins generally have low solubility in solvents. Therefore, a method of dissolving in a solvent in a state of a polymer precursor before the cyclization reaction, specifically, a polyimide precursor is often used.
- the polyimide precursor can be cyclized by heating to form a cured product.
- industrial adaptability is increasingly expected from the viewpoint of excellent manufacturing adaptability.
- Patent Document 1 discloses that (A) a polyamide having a photopolymerizable unsaturated bond: 100 parts by mass, (B) a monomer having a photopolymerizable unsaturated double bond: 1 to 50 parts by mass, (C) light A photosensitive resin composition containing a polymerization initiator: 1 to 20 parts by mass and (D) a thermal crosslinking agent: 5 to 30 parts by mass is described.
- Patent Document 2 discloses (A) a polyimide precursor having a polymerizable unsaturated bond, (B) a polymerizable monomer having an aliphatic cyclic skeleton, (C) a photopolymerization initiator, and (D) a thermal crosslinking agent.
- a photosensitive resin composition is described.
- an object of the present invention is to provide a resin composition, a cured film, a laminate, a method for producing a cured film, and a semiconductor device capable of forming a cured film having excellent elongation at break and chemical resistance. .
- the present inventors have conducted intensive studies on a resin composition containing at least one polymer precursor selected from the group consisting of a polyimide precursor and a polybenzoxazole precursor. And found that a cured film having excellent chemical resistance can be formed, and completed the present invention.
- the present invention provides the following.
- thermosetting compound having a plurality of functional groups selected from the group consisting of an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group and a blocked isocyanate group, A resin composition comprising: ⁇ 2> the resin composition according to ⁇ 1>, wherein the thermosetting compound is a compound represented by the following formula (TC1); X 1- (Y 1 ) n ...
- X 1 represents an n-valent linking group
- Y 1 represents an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group or a blocked isocyanate group
- n is Represents an integer of 2 or more.
- X 1 in the formula (TC1) includes a cyclic structure.
- thermosetting compound is a compound having a plurality of alkoxymethyl groups.
- thermosetting compound is a compound having a plurality of methoxymethyl groups.
- thermosetting compound is a compound having a plurality of methoxymethyl groups.
- base generation temperature of the thermobase generator is lower than the curing start temperature of the thermosetting compound.
- ⁇ 7> The resin composition according to any one of ⁇ 1> to ⁇ 6>, wherein the content of the polymerizable monomer having a plurality of (meth) acryloyl groups is 20% by mass or less based on the total solid content of the resin composition. Stuff.
- ⁇ 11> The resin composition according to ⁇ 8> or ⁇ 10>, further including a photopolymerization initiator.
- ⁇ 12> The resin composition according to any one of ⁇ 1> to ⁇ 11>, which is used for forming an interlayer insulating film for a redistribution layer.
- ⁇ 13> A cured film obtained by curing the resin composition according to any one of ⁇ 1> to ⁇ 12>.
- ⁇ 14> A laminate having at least two cured films according to ⁇ 13> and having a metal layer between the two cured films.
- ⁇ 15> A method for producing a cured film, comprising a film formation step of forming a film by applying the resin composition according to any one of ⁇ 1> to ⁇ 12> to a substrate.
- ⁇ 16> The method for producing a cured film according to ⁇ 15>, comprising an exposure step of exposing the film and a development step of developing the film.
- ⁇ 17> The method for producing a cured film according to ⁇ 16>, comprising a step of heating the film at 80 to 450 ° C.
- ⁇ 18> A semiconductor device having the cured film according to ⁇ 13> or the laminate according to ⁇ 14>.
- a resin composition a cured film, a laminate, a method for producing a cured film, and a semiconductor device capable of forming a cured film excellent in elongation at break and chemical resistance can be provided.
- the description of the components of the present invention described below may be made based on typical embodiments of the present invention, but the present invention is not limited to such embodiments.
- the notation of not indicating substituted or unsubstituted includes not only a group having no substituent but also a group having a substituent.
- the “alkyl group” includes not only an alkyl group having no substituent (unsubstituted alkyl group) but also an alkyl group having a substituent (substituted alkyl group).
- exposure includes not only exposure using light but also drawing using particle beams such as electron beams and ion beams, unless otherwise specified.
- the light used for exposure generally includes an active ray or radiation such as a bright line spectrum of a mercury lamp, far ultraviolet represented by excimer laser, extreme ultraviolet (EUV light), X-ray, and electron beam.
- active ray or radiation such as a bright line spectrum of a mercury lamp, far ultraviolet represented by excimer laser, extreme ultraviolet (EUV light), X-ray, and electron beam.
- active ray or radiation such as a bright line spectrum of a mercury lamp, far ultraviolet represented by excimer laser, extreme ultraviolet (EUV light), X-ray, and electron beam.
- active ray or radiation such as a bright line spectrum of a mercury lamp, far ultraviolet represented by excimer laser, extreme ultraviolet (EUV light), X-ray, and electron beam.
- EUV light extreme ultraviolet
- X-ray extreme ultraviolet
- the physical property values in the present invention are values at a temperature of 23 ° C. and a pressure of 101325 Pa unless otherwise specified.
- the weight average molecular weight (Mw) and the number average molecular weight (Mn) are measured by gel permeation chromatography (GPC measurement) and are defined as polystyrene equivalent values, unless otherwise specified.
- the weight average molecular weight (Mw) and the number average molecular weight (Mn) are determined, for example, using HLC-8220 (manufactured by Tosoh Corporation), and using guard columns HZ-L, TSKgel Super HZM-M, and TSKgel as columns.
- THF tetrahydrofuran
- detection is performed using a detector having a wavelength of 254 nm of UV rays (ultraviolet rays).
- the resin composition of the present invention a polyimide precursor, A thermal base generator, A thermosetting compound having a plurality of functional groups selected from the group consisting of an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group and a blocked isocyanate; including.
- the resin composition of the present invention can form a cured film excellent in elongation at break and chemical resistance.
- a cured film excellent in elongation at break and chemical resistance can be formed even when the curing treatment is performed at a low temperature of 200 ° C. or lower.
- the base generation temperature of the thermal base generator is preferably lower than the curing start temperature of the thermosetting compound.
- the content of the polymerizable monomer having a plurality of (meth) acryloyl groups is preferably 20% by mass or less based on the total solid content of the resin composition. Also according to this aspect, the effects of the present invention can be more remarkably obtained.
- the lower limit can be more than 0% by mass.
- the resin composition of the present invention contains a polyimide precursor.
- the polyimide precursor used in the present invention preferably contains a radically polymerizable group.
- the radical polymerizable group is a group capable of undergoing a cross-linking reaction by the action of a radical, and a preferable example is a group having an ethylenically unsaturated bond. Examples of the group having an ethylenically unsaturated bond include a vinyl group, an allyl group, a (meth) acryloyl group, and a group represented by the following formula (III).
- a polyimide precursor containing a radical polymerizable group is used, a cured film having more excellent properties is easily obtained.
- the resin composition of the present invention contains a photoradical polymerization initiator, it can be a resin composition having excellent pattern formability by a photolithography method.
- the polyimide precursor preferably contains a structural unit represented by the following formula (1).
- a 1 and A 2 each independently represent an oxygen atom or NH
- R 111 represents a divalent organic group
- R 115 represents a tetravalent organic group
- R 113 and R 114 are each independently Represents a hydrogen atom or a monovalent organic group.
- a 1 and A 2 are each independently an oxygen atom or NH, and an oxygen atom is preferable.
- R 111 represents a divalent organic group.
- the divalent organic group include a linear or branched aliphatic group, a cyclic aliphatic group, and an aromatic group, a heteroaromatic group, or a group composed of a combination thereof.
- an aromatic group having 6 to 20 carbon atoms is more preferable.
- R 111 is derived from a diamine.
- diamine used in the production of the polyimide precursor examples include linear or branched aliphatic, cyclic aliphatic or aromatic diamines.
- Diamines may be used alone or in combination of two or more.
- the diamine may be a linear aliphatic group having 2 to 20 carbon atoms, a branched or cyclic aliphatic group having 3 to 20 carbon atoms, an aromatic group having 6 to 20 carbon atoms, or a combination thereof.
- the diamine is a diamine containing an aromatic group having 6 to 20 carbon atoms. Examples of the aromatic group include the following.
- diamine examples include 1,2-diaminoethane, 1,2-diaminopropane, 1,3-diaminopropane, 1,4-diaminobutane, and 1,6-diaminohexane; 1,3-diaminocyclopentane, 1,2-, 1,3- or 1,4-diaminocyclohexane, 1,2-, 1,3- or 1,4-bis (aminomethyl) cyclohexane, bis- (4- Aminocyclohexyl) methane, bis- (3-aminocyclohexyl) methane, 4,4'-diamino-3,3'-dimethylcyclohexylmethane and isophoronediamine; meta and paraphenylenediamine, diaminotoluene, 4,4'- and 3 3,3'-diaminobiphenyl, 4,4'-diaminodiphenyl ether, 3,3-
- Diamines having two or more alkylene glycol units in the main chain are also preferred examples.
- it is a diamine containing one or both of an ethylene glycol chain and a propylene glycol chain in one molecule, and more preferably a diamine containing no aromatic ring.
- x, y, and z are average values.
- R 111 is preferably represented by -Ar 0 -L 0 -Ar 0- from the viewpoint of the flexibility of the obtained cured film.
- Ar 0 is each independently an aromatic hydrocarbon group (preferably having 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, and particularly preferably 6 to 10 carbon atoms), and is preferably a phenylene group.
- the preferred range is the same as that of A described above.
- R 111 is preferably a divalent organic group represented by the following formula (51) or (61) from the viewpoint of i-ray transmittance.
- a divalent organic group represented by the formula (61) is more preferable from the viewpoints of i-line transmittance and availability.
- R 50 to R 57 are each independently a hydrogen atom, a fluorine atom or a monovalent organic group, and at least one of R 50 to R 57 is a fluorine atom, a methyl group, a fluoromethyl group, a difluoromethyl group, or It is a trifluoromethyl group.
- Examples of the monovalent organic group represented by R 50 to R 57 include an unsubstituted alkyl group having 1 to 10 (preferably 1 to 6) carbon atoms and a fluorine atom having 1 to 10 (preferably 1 to 6) carbon atoms. Alkyl group and the like.
- R 58 and R 59 are each independently a fluorine atom, a fluoromethyl group, a difluoromethyl group, or a trifluoromethyl group.
- Diamine compounds giving the structure of formula (51) or (61) include dimethyl-4,4′-diaminobiphenyl, 2,2′-bis (trifluoromethyl) -4,4′-diaminobiphenyl, 2,2 '-Bis (fluoro) -4,4'-diaminobiphenyl, 4,4'-diaminooctafluorobiphenyl and the like. One of these may be used, or two or more may be used in combination.
- R 115 in the formula (1) represents a tetravalent organic group.
- the tetravalent organic group is preferably a group containing an aromatic ring, and more preferably a group represented by the following formula (5) or (6).
- R 112 has the same meaning as A, and the preferred range is also the same.
- tetravalent organic group represented by R 115 in the formula (1) include a tetracarboxylic acid residue remaining after removing the acid dianhydride group from the tetracarboxylic dianhydride.
- the tetracarboxylic dianhydride may be used alone or in combination of two or more.
- the compound represented by the following formula (7) is preferable as the tetracarboxylic dianhydride.
- R 115 represents a tetravalent organic group.
- R 115 has the same meaning as R 115 in formula (1).
- tetracarboxylic dianhydride examples include pyromellitic acid, pyromellitic dianhydride (PMDA), 3,3 ′, 4,4′-biphenyltetracarboxylic dianhydride, 3,3 ′, 4 4,4'-diphenylsulfidetetracarboxylic dianhydride, 3,3 ', 4,4'-diphenylsulfonetetracarboxylic dianhydride, 3,3', 4,4'-benzophenonetetracarboxylic dianhydride, 3,3 ', 4,4'-diphenylmethanetetracarboxylic dianhydride, 2,2', 3,3'-diphenylmethanetetracarboxylic dianhydride, 2,3,3 ', 4'-biphenyltetracarboxylic acid Dianhydride, 2,3,3 ', 4'-benzophenonetetracarboxylic dianhydride,
- DAA-1 tetracarboxylic dianhydrides
- DAA-5 tetracarboxylic dianhydrides
- R 113 and R 114 each independently represent a hydrogen atom or a monovalent organic group.
- at least one of R 113 and R 114 contains a radically polymerizable group, and more preferably both contain a radically polymerizable group.
- the radical polymerizable group is a group capable of undergoing a cross-linking reaction by the action of a radical, and a preferable example is a group having an ethylenically unsaturated bond. Examples of the group having an ethylenically unsaturated bond include a vinyl group, an allyl group, a (meth) acryloyl group, and a group represented by the following formula (III).
- R 200 represents a hydrogen atom or a methyl group, and a methyl group is more preferable.
- R 201 is an alkylene group having 2 to 12 carbon atoms, —CH 2 CH (OH) CH 2 — or a (poly) oxyalkylene group having 4 to 30 carbon atoms (an alkylene group having 1 carbon atom To 12, preferably 1 to 6, more preferably 1 to 3, and the number of repetitions is preferably 1 to 12, more preferably 1 to 6, and particularly preferably 1 to 3.)
- a (poly) oxyalkylene group means an oxyalkylene group or a polyoxyalkylene group.
- R201 examples include ethylene, propylene, trimethylene, tetramethylene, 1,2-butanediyl, 1,3-butanediyl, pentamethylene, hexamethylene, octamethylene, dodecamethylene. , —CH 2 CH (OH) CH 2 —, and more preferably an ethylene group, a propylene group, a trimethylene group, and —CH 2 CH (OH) CH 2 —.
- R 200 is a methyl group and R 201 is an ethylene group.
- an aliphatic group, an aromatic group, and an aryl group having one, two or three, preferably one acid group And an alkyl group include an aromatic group having 6 to 20 carbon atoms having an acid group and an arylalkyl group having 7 to 25 carbon atoms having an acid group. More specifically, a phenyl group having an acid group and a benzyl group having an acid group are exemplified.
- the acid group is preferably a hydroxyl group. That is, R 113 or R 114 is preferably a group having a hydroxyl group.
- R 113 or R 114 As the monovalent organic group represented by R 113 or R 114, a substituent that improves the solubility of a developer is preferably used.
- R 113 or R 114 is more preferably a hydrogen atom, 2-hydroxybenzyl, 3-hydroxybenzyl or 4-hydroxybenzyl from the viewpoint of solubility in an aqueous developer.
- R 113 or R 114 is preferably a monovalent organic group.
- the monovalent organic group preferably contains a linear or branched alkyl group, a cyclic alkyl group, or an aromatic group, and more preferably an alkyl group substituted with an aromatic group.
- the alkyl group preferably has 1 to 30 carbon atoms (3 or more in the case of a cyclic group).
- the alkyl group may be linear, branched or cyclic.
- linear or branched alkyl group examples include a methyl group, an ethyl group, a propyl group, a butyl group, a pentyl group, a hexyl group, a heptyl group, an octyl group, a nonyl group, a decyl group, a dodecyl group, a tetradecyl group, and an octadecyl group.
- the cyclic alkyl group may be a monocyclic alkyl group or a polycyclic alkyl group.
- Examples of the monocyclic alkyl group include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, a cycloheptyl group, and a cyclooctyl group.
- polycyclic alkyl group examples include, for example, an adamantyl group, a norbornyl group, a bornyl group, a camphenyl group, a decahydronaphthyl group, a tricyclodecanyl group, a tetracyclodecanyl group, a camphoroyl group, a dicyclohexyl group, and a pinenyl group.
- an alkyl group substituted with an aromatic group a linear alkyl group substituted with an aromatic group described below is preferable.
- a substituted or unsubstituted aromatic hydrocarbon group (the cyclic structure constituting the group includes a benzene ring, a naphthalene ring, a biphenyl ring, a fluorene ring, a pentalene ring, an indene ring, an azulene ring) Ring, heptarene ring, indacene ring, perylene ring, pentacene ring, acenaphthene ring, phenanthrene ring, anthracene ring, naphthacene ring, chrysene ring, triphenylene ring, etc.) or a substituted or unsubstituted aromatic heterocyclic group (group As the constituting cyclic structure, fluorene ring, pyrrole ring, furan ring, thiophene ring, imidazole ring, oxazole ring, a substituted or unsub
- the polyimide precursor has a fluorine atom in the structural unit.
- the content of fluorine atoms in the polyimide precursor is preferably 10% by mass or more, more preferably 20% by mass or less. There is no particular upper limit, but 50% by mass or less is practical.
- an aliphatic group having a siloxane structure may be copolymerized with the structural unit represented by the formula (1). Specifically, bis (3-aminopropyl) tetramethyldisiloxane, bis (paraaminophenyl) octamethylpentasiloxane, and the like can be given as the diamine component.
- the structural unit represented by the formula (1) is preferably a structural unit represented by the formula (1-A) or (1-B).
- a 11 and A 12 represent an oxygen atom or NH
- R 111 and R 112 each independently represent a divalent organic group
- R 113 and R 114 each independently represent a hydrogen atom or a monovalent It represents an organic group
- at least one of R 113 and R 114 is preferably a group containing a radical polymerizable group, and more preferably a radical polymerizable group.
- a preferred ranges of A 11 , A 12 , R 111 , R 113 and R 114 are each independently the same as the preferred ranges of A 1 , A 2 , R 111 , R 113 and R 114 in the formula (1).
- a preferred range of R 112 has the same meaning as R 112 in formula (5), and more preferably among others oxygen atoms.
- the bonding position of the carbonyl group in the formula to the benzene ring is preferably 4, 5, 3 ', 4' in formula (1-A). In the formula (1-B), 1,2,4,5 is preferable.
- the structural unit represented by the formula (1) may be one type, or may be two or more types. Further, it may contain a structural isomer of the structural unit represented by the formula (1). Further, the polyimide precursor may include other types of structural units in addition to the structural units of the above formula (1).
- a polyimide precursor in which 50 mol% or more, more preferably 70 mol% or more, and particularly 90 mol% or more of all the structural units is a structural unit represented by the formula (1).
- the upper limit is practically 100 mol% or less.
- the weight average molecular weight (Mw) of the polyimide precursor is preferably from 2,000 to 500,000, more preferably from 5,000 to 100,000, and further preferably from 10,000 to 50,000.
- the number average molecular weight (Mn) is preferably from 800 to 250,000, more preferably from 2,000 to 50,000, and still more preferably from 4,000 to 25,000.
- the degree of dispersion of the molecular weight of the polyimide precursor is preferably from 1.5 to 3.5, more preferably from 2 to 3.
- the polyimide precursor can be obtained by reacting a dicarboxylic acid or a dicarboxylic acid derivative with a diamine. Preferably, it is obtained by halogenating a dicarboxylic acid or a dicarboxylic acid derivative with a halogenating agent and then reacting the dicarboxylic acid or dicarboxylic acid derivative with a diamine.
- an organic solvent may be one type or two or more types.
- the organic solvent can be appropriately determined according to the raw material, and examples thereof include pyridine, diethylene glycol dimethyl ether (diglyme), N-methylpyrrolidone and N-ethylpyrrolidone.
- the production of the polyimide precursor includes a step of depositing a solid.
- the polyimide precursor in the reaction solution can be precipitated in water and dissolved in a solvent in which the polyimide precursor is soluble, such as tetrahydrofuran, to perform solid deposition.
- the content of the polyimide precursor in the resin composition of the present invention is preferably 20% by mass or more, more preferably 30% by mass or more, and preferably 40% by mass or more based on the total solid content of the resin composition. Is more preferably 50% by mass or more, still more preferably 60% by mass or more, and even more preferably 70% by mass or more. Further, the content of the polyimide precursor in the resin composition of the present invention is preferably 99.5% by mass or less, more preferably 99% by mass or less, based on the total solid content of the resin composition. , 98% by mass or less, more preferably 95% by mass or less.
- the resin composition of the present invention may include only one kind of the polyimide precursor, or may include two or more kinds of the polyimide precursor. When two or more kinds are included, the total amount is preferably in the above range.
- the resin composition of the present invention contains a thermal base generator.
- the type of the thermal base generator is not particularly limited, but is selected from an acidic compound which generates a base when heated to 40 ° C. or more, and an ammonium salt having an anion having an pKa of 0 to 4 and an ammonium cation. It is preferable to include a thermal base generator containing at least one of these.
- pKa1 represents the logarithm ( ⁇ Log 10 Ka) of the dissociation constant (Ka) of the first proton of the acid, which will be described in detail later.
- thermal base generator examples include an acidic compound (A1) that generates a base when heated to 40 ° C. or higher, an ammonium salt (A2) having an anion having a pKa of 0 to 4 and an ammonium cation, and a nonionic thermal base generator (A3). ), And more preferably a nonionic thermal base generator (A3). Since these compounds generate bases when heated, the bases generated from these compounds can promote the cyclization reaction of the polyimide precursor, and the cyclization of the polyimide precursor can be performed at a low temperature.
- A1 acidic compound
- A2 ammonium salt
- A3 nonionic thermal base generator
- the solution obtained by stirring the solution is a compound having a value of less than 7 when measured at 20 ° C. using a pH (power of hydrogen) meter.
- the base generation temperature of the thermal base generator used in the present invention is preferably 40 ° C or higher, more preferably 120 to 200 ° C.
- the upper limit of the base generation temperature is preferably 190 ° C. or lower, more preferably 180 ° C. or lower, and further preferably 165 ° C. or lower.
- the base generation temperature is measured, for example, by differential scanning calorimetry, by heating the compound in a pressure-resistant capsule at 250C at 5C / min, reading the peak temperature of the lowest exothermic peak, and measuring the peak temperature as the base generation temperature. can do.
- the base generation temperature of the thermal base generator used in the present invention is preferably lower than the curing start temperature of the thermosetting compound described above.
- the base generated by the thermal base generator is preferably a secondary amine or a tertiary amine, and more preferably a tertiary amine. Since the tertiary amine has a high basicity, the cyclization temperature of the polyimide precursor can be lowered. Further, the boiling point of the base generated by the thermal base generator is preferably 80 ° C. or higher, more preferably 100 ° C. or higher, and even more preferably 140 ° C. or higher. Further, the molecular weight of the generated base is preferably from 80 to 2,000. The lower limit is more preferably 100 or more. The upper limit is more preferably 500 or less. In addition, the value of molecular weight is a theoretical value obtained from the structural formula.
- the acidic compound (A1) preferably contains at least one selected from an ammonium salt and a compound represented by the following formula (101) or (102).
- the ammonium salt (A2) is preferably an acidic compound.
- the ammonium salt (A2) may be a compound containing an acidic compound that generates a base when heated to 40 ° C. or higher (preferably 120 to 200 ° C.) or 40 ° C. or higher (preferably 120 to 200 ° C.)
- the compound may be a compound excluding an acidic compound which generates a base when heated to the step (1).
- the ammonium salt means a salt of an ammonium cation represented by the following formula (101) or (102) and an anion.
- the anion may be bonded to any part of the ammonium cation through a covalent bond and may be present outside the ammonium cation molecule, but may be present outside the ammonium cation molecule. preferable.
- numerator means the case where an ammonium cation and an anion are not couple
- an anion outside the cation moiety is also referred to as a counter anion.
- Equation (101) Equation (102) R 1 to R 6 each independently represent a hydrogen atom or a hydrocarbon group, and R 7 represents a hydrocarbon group.
- R 1 and R 2 , R 3 and R 4 , R 5 and R 6 , and R 5 and R 7 in the formulas (101) and (102) may be respectively bonded to form a ring.
- the ammonium cation is preferably represented by any of the following formulas (Y1-1) to (Y1-5).
- R 101 represents an n-valent organic group
- R 1 and R 7 have the same meaning as in Formula (101) or Formula (102).
- Ar 101 and Ar 102 each independently represent an aryl group
- n represents an integer of 1 or more
- m represents an integer of 0 to 5.
- the ammonium salt preferably has an anion having a pKa of 0 to 4 and an ammonium cation.
- the upper limit of the pKa1 of the anion is more preferably 3.5 or less, even more preferably 3.2 or less.
- the lower limit is preferably 0.5 or more, and more preferably 1.0 or more.
- the type of the anion is preferably one selected from a carboxylate anion, a phenol anion, a phosphate anion and a sulfate anion, and more preferably a carboxylate anion because both salt stability and thermal decomposability can be achieved. That is, the ammonium salt is more preferably a salt of an ammonium cation and a carboxylate anion.
- the carboxylate anion is preferably a divalent or higher carboxylic acid anion having two or more carboxyl groups, and more preferably a divalent carboxylic acid anion.
- the thermal base generator that can further improve the stability, curability, and developability of the resin composition.
- the stability, curability and developability of the resin composition can be further improved.
- the carboxylate anion is preferably a carboxylate anion having a pKa of 4 or less.
- pKa1 is more preferably 3.5 or less, even more preferably 3.2 or less.
- the stability of the resin composition can be further improved.
- pKa1 represents the logarithm of the reciprocal of the dissociation constant of the first proton of the acid and is determined by Organic Structures by Physical Methods (author: Brown, HC, McDaniel, DH, Hafliger, Hafliger). Compiled by: Braude, EA, Nachod, FC; Academic Press, New York, 1955), and Data for Biochemical Research (author: Dawson, R., R.M. al; Oxford, Clarendon Press, 1959). For compounds not described in these documents, values calculated from the structural formula using ACD / pKa (manufactured by ACD / Labs) software will be used.
- the carboxylate anion is preferably represented by the following formula (X1).
- EWG represents an electron-withdrawing group.
- the electron withdrawing group means a group having a positive Hammett's substituent constant ⁇ m.
- ⁇ m is described by Yuno Tsuno, Synthetic Organic Chemistry Society, Vol. 631-642. Note that the electron-withdrawing group in the present embodiment is not limited to the substituents described in the above documents.
- Me represents a methyl group
- Ac represents an acetyl group
- Ph represents a phenyl group.
- EWG is preferably a group represented by the following formulas (EWG-1) to (EWG-6).
- R x1 to R x3 each independently represent a hydrogen atom, an alkyl group, an alkenyl group, an aryl group, a hydroxyl group or a carboxyl group, and Ar represents an aromatic group.
- the carboxylate anion is preferably represented by the following formula (XA).
- Formula (XA) In the formula (XA), L 10 represents a single bond or a divalent linking group selected from an alkylene group, an alkenylene group, an aromatic group, —NR X — and a combination thereof, and R X represents a hydrogen atom , An alkyl group, an alkenyl group or an aryl group.
- carboxylate anion examples include maleate anion, phthalate anion, N-phenyliminodiacetic acid anion and oxalate anion. These can be preferably used.
- Examples of the nonionic thermal base generator (A3) include a compound represented by the formula (B1) or the formula (B2).
- Rb 1 , Rb 2 and Rb 3 are each independently an organic group having no tertiary amine structure, a halogen atom or a hydrogen atom. However, Rb 1 and Rb 2 are not simultaneously hydrogen atoms. Further, Rb 1 , Rb 2 and Rb 3 do not have a carboxyl group.
- a tertiary amine structure refers to a structure in which all three bonds of a trivalent nitrogen atom are covalently bonded to a hydrocarbon-based carbon atom. Therefore, this does not apply when the bonded carbon atom is a carbon atom forming a carbonyl group, that is, when forming an amide group together with a nitrogen atom.
- Rb 1 , Rb 2 and Rb 3 preferably has a cyclic structure, and more preferably at least two of them have a cyclic structure.
- the cyclic structure may be either a single ring or a condensed ring, and is preferably a single ring or a condensed ring in which two single rings are condensed.
- the monocyclic ring is preferably a 5- or 6-membered ring, and more preferably a 6-membered ring.
- the single ring is preferably a cyclohexane ring and a benzene ring, and more preferably a cyclohexane ring.
- Rb 1 and Rb 2 each represent a hydrogen atom, an alkyl group (preferably having 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, still more preferably 3 to 12 carbon atoms), and an alkenyl group (preferably having 2 to 24 carbon atoms).
- 2 to 18 are more preferable
- 3 to 12 are more preferable, an aryl group (preferably having 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, still more preferably 6 to 10 carbon atoms), or an arylalkyl group (having 7 carbon atoms) To 25, more preferably 7 to 19, and even more preferably 7 to 12).
- These groups may have a substituent as long as the effects of the present invention are exhibited.
- Rb 1 and Rb 2 may combine with each other to form a ring.
- the formed ring is preferably a 4- to 7-membered nitrogen-containing heterocyclic ring.
- Rb 1 and Rb 2 are particularly an optionally substituted linear, branched or cyclic alkyl group (preferably having 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, and still more preferably 3 to 12 carbon atoms). More preferably, it is a cycloalkyl group which may have a substituent (preferably having 3 to 24 carbon atoms, more preferably 3 to 18 carbon atoms, and still more preferably 3 to 12 carbon atoms). A cyclohexyl group which may be further preferred.
- an alkyl group preferably having 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, still more preferably 3 to 12 carbon atoms
- an aryl group preferably having 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, To 10
- an alkenyl group preferably having 2 to 24 carbon atoms, more preferably 2 to 12 carbon atoms
- an arylalkyl group preferably having 7 to 23 carbon atoms, more preferably 7 to 19 carbon atoms.
- 7 to 12 are more preferable, an arylalkenyl group (preferably having 8 to 24 carbon atoms, more preferably 8 to 20, more preferably 8 to 16), and an alkoxyl group (preferably having 1 to 24 carbon atoms, 2 to 2) 18 is more preferable, and 3 to 12 is more preferable.
- the aryloxy group (6 to 22 carbon atoms is preferable, 6 to 18 is more preferable, and 6 to 12 is more preferable.
- arylalkyloxy group having 7 to 23 carbon atoms, more preferably 7 to 19, more preferably 7 to 12 are exemplified.
- a cycloalkyl group (preferably having 3 to 24 carbon atoms, more preferably 3 to 18 carbon atoms, and still more preferably 3 to 12 carbon atoms), an arylalkenyl group and an arylalkyloxy group are preferred.
- Rb 3 may further have a substituent within a range in which the effects of the present invention can be obtained.
- the compound represented by the formula (B1) is preferably a compound represented by the following formula (B1-1) or the following formula (B1-2).
- Rb 11 and Rb 12 , and Rb 31 and Rb 32 are the same as Rb 1 and Rb 2 in formula (B1), respectively.
- Rb 13 is an alkyl group (preferably having 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, still more preferably 3 to 12 carbon atoms), and an alkenyl group (preferably having 2 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, and 3 to 12 carbon atoms.
- Rb 13 is preferably an arylalkyl group.
- Rb 33 and Rb 34 each independently represent a hydrogen atom, an alkyl group (preferably having 1 to 12 carbon atoms, more preferably 1 to 8 and still more preferably 1 to 3), and an alkenyl group (preferably having 2 to 12 carbon atoms). , 2 to 8, more preferably 2 to 3, an aryl group (preferably having 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, and still more preferably 6 to 10 carbon atoms), and an arylalkyl group (having 7 to 7 carbon atoms). 23 is preferable, 7 to 19 are more preferable, and 7 to 11 are more preferable), and a hydrogen atom is preferable.
- Rb 35 is an alkyl group (preferably having 1 to 24 carbon atoms, more preferably 1 to 12 carbon atoms, still more preferably 3 to 8), an alkenyl group (preferably having 2 to 12 carbon atoms and more preferably 3 to 8), and aryl. Groups (preferably having 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, and still more preferably 6 to 12 carbon atoms), and arylalkyl groups (preferably having 7 to 23 carbon atoms, more preferably 7 to 19 carbon atoms, and still more preferably 7 to 12 carbon atoms). ), And an aryl group is preferred.
- the compound represented by the formula (B1-1) is also preferably a compound represented by the formula (B1-1a).
- Rb 11 and Rb 12 have the same meanings as Rb 11 and Rb 12 in the formula (B1-1).
- Rb 15 and Rb 16 each represent a hydrogen atom, an alkyl group (preferably having 1 to 12 carbon atoms, more preferably 1 to 6), and an alkenyl group (preferably having 2 to 12 carbon atoms, preferably having 2 to 6 carbon atoms).
- Rb 17 is an alkyl group (preferably having 1 to 24 carbon atoms, more preferably 1 to 12 carbon atoms, still more preferably 3 to 8), an alkenyl group (preferably having 2 to 12 carbon atoms, more preferably 3 to 8), and an aryl group.
- an arylalkyl group (preferably having 7 to 23 carbon atoms, more preferably 7 to 19, and still more preferably 7 to 12). And among them, an aryl group is preferable.
- the molecular weight of the nonionic thermal base generator (A3) is preferably 800 or less, more preferably 600 or less, and even more preferably 500 or less.
- the lower limit is preferably 100 or more, more preferably 200 or more, and even more preferably 300 or more.
- thermal base generator examples include the following compounds.
- the content of the thermal base generator is preferably 0.1 to 50% by mass based on the total solid content of the resin composition of the present invention.
- the lower limit is more preferably 0.5% by mass or more, and still more preferably 1% by mass or more.
- the upper limit is more preferably 30% by mass or less, and even more preferably 20% by mass or less.
- One or more thermal base generators can be used. When two or more kinds are used, the total amount is preferably in the above range.
- the resin composition of the present invention is a thermosetting compound having a plurality of functional groups selected from the group consisting of an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group and a blocked isocyanate group. contains.
- the thermosetting compound means a compound which is cured by heating.
- the blocked isocyanate group is a group capable of generating an isocyanate group by heat, and for example, a group obtained by reacting a blocking agent with an isocyanate group to protect the isocyanate group can be preferably exemplified.
- the molecular weight of the thermosetting compound (weight average molecular weight in the case of a polymer) is preferably 100 to 10,000.
- the lower limit is preferably 150 or more, and more preferably 200 or more.
- the upper limit is preferably 1000 or less, more preferably 500 or less.
- the curing start temperature of the thermosetting compound is preferably 100 to 350 ° C.
- the lower limit is preferably 110 ° C. or higher, more preferably 120 ° C. or higher.
- the upper limit is preferably 200 ° C. or lower, more preferably 180 ° C. or lower.
- the curing start temperature of the thermosetting compound is defined as the difference between the temperature of 1 mg of a sample (thermosetting compound) and the temperature of 25 ° C. at a rate of 5 ° C./min. And the temperature at which the exothermic reaction of the thermosetting compound starts.
- the temperature at which the exothermic reaction of the thermosetting compound starts means the temperature at which the peak of the exothermic reaction appears in the differential scanning calorimetry curve in which the vertical axis represents heat flow (mW) and the horizontal axis represents temperature (° C.). I do.
- the exothermic reaction peak at a low temperature is defined as "the temperature at which the exothermic reaction of the thermosetting compound starts (curing start temperature)" in the present invention.
- thermosetting compound used in the present invention is preferably a compound having a plurality of functional groups selected from an epoxy group, an oxetanyl group, a methylol group and an alkoxymethyl group, and a functional group selected from a methylol group and an alkoxymethyl group. It is more preferable that the compound has a plurality of compounds, and it is further preferable that the compound has a plurality of alkoxymethyl groups because the effect of the present invention is more easily obtained, and it is particularly preferable that the compound has a plurality of methoxymethyl groups. preferable.
- the number of the above-mentioned functional groups contained in the thermosetting compound may be two or more, and is preferably three or more.
- the upper limit is preferably 10 or less, more preferably 6 or less.
- thermosetting compound used in the present invention is preferably a compound represented by the formula (TC1).
- X 1- (Y 1 ) n ... (TC1) X 1 represents an n-valent linking group
- Y 1 represents an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group or a blocked isocyanate group
- n is Represents an integer of 2 or more.
- Examples of the n-valent linking group represented by X 1 in the formula (TC1) include an aliphatic hydrocarbon group, an aromatic hydrocarbon group, a heterocyclic group, —O—, —NH—, —NHCO—, —CONH—, and —. OCO—, —COO—, —CO—, —SO 2 NH—, —SO 2 — and combinations thereof.
- the number of carbon atoms of the aliphatic hydrocarbon group is preferably 1 to 30, more preferably 1 to 15, still more preferably 1 to 8, and particularly preferably 1 to 5.
- the aliphatic hydrocarbon group may be linear, branched or cyclic, preferably linear or branched, and particularly preferably linear.
- the aromatic hydrocarbon group preferably has 6 to 30 carbon atoms, more preferably 6 to 15 carbon atoms.
- the aromatic hydrocarbon group may be a single tube or a condensed ring.
- the heterocyclic group may be a single ring or a condensed ring.
- the heterocyclic group is preferably a single ring or a condensed ring having 2 to 4 condensed numbers.
- the number of hetero atoms constituting the ring of the heterocyclic group is preferably from 1 to 3.
- the hetero atom constituting the ring of the heterocyclic group is preferably a nitrogen atom, an oxygen atom or a sulfur atom.
- the number of carbon atoms constituting the ring of the heterocyclic group is preferably from 3 to 30, more preferably from 3 to 18, and even more preferably from 3 to 12.
- X 1 in the formula (TC1) is preferably a group containing a cyclic structure.
- the cyclic structure include an aliphatic ring, an aromatic hydrocarbon ring, and a heterocyclic ring, and are preferably an aromatic hydrocarbon ring or a heterocyclic ring, and more preferably a heterocyclic ring.
- the heterocycle is preferably a nitrogen-containing heterocycle. Examples of the nitrogen-containing heterocyclic ring include a pyridine ring, a triazine ring, an imidazolidinone ring and the like, and an imidazolidinone ring is preferable.
- Y 1 in the formula (TC1) represents an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group or a blocked isocyanate group, and is an epoxy group, an oxetanyl group, a methylol group or an alkoxymethyl group.
- it is a methylol group or an alkoxymethyl group, more preferably an alkoxymethyl group.
- An alkoxymethyl group is a group represented by —CH 2 —ORy 1 .
- Ry 1 is an alkyl group, preferably an alkyl group having 1 to 30 carbon atoms, more preferably an alkyl group having 1 to 15 carbon atoms, and further preferably an alkyl group having 1 to 8 carbon atoms. It is particularly preferably an alkyl group having 1 to 3 carbon atoms, and most preferably an alkyl group having 1 carbon atom (methyl group). That is, the alkoxymethyl group is most preferably a methoxymethyl group.
- thermosetting compound used in the present invention is also preferably a compound in which a methylol group or an alkoxymethyl group is bonded to a nitrogen atom or a carbon atom forming an aromatic ring.
- alkoxymethylated melamine, methylolated melamine, alkoxymethylated benzoguanamine, methylolated benzoguanamine, alkoxymethylated glycoluril, methylolated glycoluril, alkoxymethylated urea, and methylolated urea are preferred.
- compounds described in paragraphs 0056 to 0065 of JP-A-2003-287889 and paragraphs 0058 to 0060 of JP-A-2018-084626 can also be used.
- Preferred structures of the compound having an alkoxymethyl group or a methylol group bonded to a nitrogen atom include compounds represented by the following formulas (AM-101) to (AM-105).
- Rm 1 to Rm 4 each independently represent a hydrogen atom or a group represented by Formula (Rm). However, two or more of Rm 1 to Rm 4 are groups represented by the formula (Rm).
- Rm 5 to Rm 8 each independently represent a hydrogen atom or a group represented by Formula (Rm). However, two or more of Rm 5 to Rm 8 are groups represented by the formula (Rm).
- Rm 9 and Rm 10 each independently represent a group represented by Formula (Rm).
- Rm 11 to Rm 16 each independently represent a hydrogen atom or a group represented by Formula (Rm).
- Rm 11 to Rm 16 are groups represented by the formula (Rm).
- Rm 17 to Rm 20 each independently represent a hydrogen atom or a group represented by Formula (Rm).
- Rm 17 to Rm 20 are groups represented by the formula (Rm).
- Rm 100 represents a hydrogen atom or an alkyl group.
- the number of carbon atoms of the alkyl group represented by Rm 100 is preferably 1 to 30, more preferably 1 to 15, still more preferably 1 to 8, and particularly preferably 1 to 3, Most preferably, it is 1. That is, the alkoxymethyl group is most preferably a methoxymethyl group.
- Examples of compounds in which an alkoxymethyl group or a methylol group is bonded to a carbon atom forming an aromatic ring include, for example, a compound represented by the following formula (AM-110).
- Formula (AM-110) In the formula (AM-110), X represents a single bond or a monovalent to tetravalent organic group, R 11 to R 13 each independently represent a hydrogen atom or an alkyl group, and R 15 represents a hydrogen atom, a hydroxyl group or an alkyl group. Represents a group, n, p and r are each independently an integer of 1-4, and q is an integer of 0-4.
- thermosetting compound having a methylol group or an alkoxymethyl group examples include a compound having the following structure.
- Commercial products include 46DMOC, 46DMOP, TM-BIP-A (manufactured by Asahi Organic Materials Co., Ltd.), DML-MBPC, DML-MBOC, DML-OCHP, DML-PCHP, DML-PC, DML-PTBP, DML.
- thermosetting compound having an epoxy group (hereinafter, also referred to as an epoxy compound) can be used as the thermosetting compound.
- the epoxy compound may be a compound having two or more epoxy groups, and is preferably a compound having 2 to 100 epoxy groups.
- the upper limit of the number of epoxy groups can be, for example, 10 or less, or 5 or less.
- epoxy compounds include bisphenol A type epoxy resin; bisphenol F type epoxy resin; alkylene glycol type epoxy resin such as propylene glycol diglycidyl ether; polyalkylene glycol type epoxy resin such as polypropylene glycol diglycidyl ether; polymethyl (glycidyl)
- examples thereof include, but are not limited to, epoxy group-containing silicones such as (roxypropyl) siloxane.
- Epicron (registered trademark) 850-S Epicron (registered trademark) HP-4032, Epicron (registered trademark) HP-7200, Epicron (registered trademark) HP-820, Epicron (registered trademark) HP-4700, Epicron® EXA-4710, Epicron® HP-4770, Epicron® EXA-859CRP, Epicron® EXA-1514, Epicron® EXA-4880, Epicron® EXA-4850-150, EPICLON EXA-4850-1000, EPICLON (registered trademark) EXA-4816, EPICLON (registered trademark) EXA-4822 (manufactured by DIC Corporation), Rica Resin (registered trademark) BEO-60E (trade name, Shin-Nippon Rika Co., Ltd., EP-4003S, Such as P-4000S ((Ltd.) ADEKA), and the like. Further, a compound having the following structure can also be used.
- thermosetting compound having an oxetanyl group (hereinafter, also referred to as an oxetane compound) may be used as the thermosetting compound.
- oxetane compounds include 3-ethyl-3-hydroxymethyloxetane, 1,4-bis ⁇ [(3-ethyl-3-oxetanyl) methoxy] methyl ⁇ benzene, and 3-ethyl-3- (2-ethylhexylmethyl) oxetane And 1,4-benzenedicarboxylic acid-bis [(3-ethyl-3-oxetanyl) methyl] ester.
- examples of commercially available products include Aron Oxetane Series (eg, OXT-121, OXT-221, OXT-191, and OXT-223) manufactured by Toagosei Co., Ltd.
- thermosetting compound having a blocked isocyanate group (hereinafter, also referred to as a blocked isocyanate compound) may be used as the thermosetting compound.
- the skeleton of the blocked isocyanate compound is not particularly limited, and may be an aliphatic, alicyclic or aromatic polyisocyanate. Regarding specific examples of the skeleton, the description in paragraph No. 0144 of JP-A-2014-238438 can be referred to, and the contents thereof are incorporated herein.
- Examples of the parent structure of the blocked isocyanate compound include a biuret type, an isocyanurate type, an adduct type, and a bifunctional prepolymer type.
- Examples of the blocking agent forming the block structure of the blocked isocyanate compound include oxime compounds, lactam compounds, phenol compounds, alcohol compounds, amine compounds, active methylene compounds, pyrazole compounds, mercaptan compounds, imidazole compounds, imide compounds, and the like. Can be. Among these, a blocking agent selected from oxime compounds, lactam compounds, phenol compounds, alcohol compounds, amine compounds, active methylene compounds, and pyrazole compounds is particularly preferred. As specific examples of the blocking agent, the description in paragraph No. 0146 of JP-A-2014-238438 can be referred to, and the contents thereof are incorporated in the present specification.
- the blocked isocyanate compound is available as a commercial product, for example, Coronate AP Stable M, Coronate 2503, 2515, 2507, 2513, 2555, Millionate MS-50 (all manufactured by Nippon Polyurethane Industry Co., Ltd.), Takenate B -830, B-815N, B-820NSU, B-842N, B-846N, B-870N, B-874N, B-882N (all manufactured by Mitsui Chemicals, Inc.), Duranate 17B-60PX, 17B-60P, TPA-B80X, TPA-B80E, MF-B60X, MF-B60B, MF-K60X, MF-K60B, E402-B80B, SBN-70D, SBB-70P, K6000 (all manufactured by Asahi Kasei Corporation), Desmodur BL1100 , BL1265 @ MPA / X, BL357 / 1, BL3272MPA, BL3370MPA
- thermosetting compound having a group selected from a phenol group, a maleimide group and a cyanate group can also be used as the thermosetting compound.
- the content of the thermosetting compound is preferably 1 to 20% by mass based on the total solid content of the resin composition of the present invention.
- the lower limit is preferably 3% by mass or more, more preferably 5% by mass or more.
- the upper limit is preferably at most 18% by mass, more preferably at most 15% by mass.
- the content of the thermosetting compound is preferably 1 to 25 parts by mass with respect to 100 parts by mass of the polyimide precursor.
- the upper limit is preferably at most 23 parts by mass, more preferably at most 20 parts by mass.
- the lower limit is preferably 4 parts by mass or more, more preferably 6 parts by mass or more.
- thermosetting compound When the content of the thermosetting compound is 6 parts by mass or more with respect to 100 parts by mass of the polyimide precursor, a cured film having excellent chemical resistance is easily obtained. When the content of the thermosetting compound is 20 parts by mass or less based on 100 parts by mass of the polyimide precursor, a cured film having excellent substrate adhesion is easily obtained. Further, the content of the thermosetting compound is preferably 10 to 1000 parts by mass with respect to 100 parts by mass of the thermal base generator. The upper limit is preferably 800 parts by mass or less, more preferably 600 parts by mass or less. The lower limit is preferably at least 20 parts by mass, more preferably at least 50 parts by mass. One thermosetting compound may be used alone, or two or more thermosetting compounds may be used in combination. When two or more kinds are used in combination, the total amount is preferably within the above range.
- the resin composition of the present invention preferably contains a photopolymerization initiator. It is preferably a photo-radical polymerization initiator.
- the photo-radical polymerization initiator is not particularly limited, and can be appropriately selected from known photo-radical polymerization initiators. For example, a photo-radical polymerization initiator having photosensitivity to light in the ultraviolet region to the visible region is preferable. In addition, an activator that produces some action with the photoexcited sensitizer and generates an active radical may be used.
- the photo-radical polymerization initiator preferably contains at least one compound having a molar extinction coefficient of at least about 50 in the range of about 300 to 800 nm (preferably 330 to 500 nm).
- the molar extinction coefficient of a compound can be measured using a known method. For example, it is preferable to measure with an ultraviolet-visible spectrophotometer (Cary-5 spectrophotometer manufactured by Varian) using an ethyl acetate solvent at a concentration of 0.01 g / L.
- a known compound can be arbitrarily used as the photoradical polymerization initiator.
- halogenated hydrocarbon derivatives eg, compounds having a triazine skeleton, compounds having an oxadiazole skeleton, compounds having a trihalomethyl group, etc.
- acylphosphine compounds such as acylphosphine oxide, hexaarylbiimidazole, oxime derivatives, etc.
- the descriptions in paragraphs 0165 to 0182 of JP-A-2016-027357 and paragraphs 0138 to 0151 of WO 2015/199219 can be referred to, and the contents are incorporated herein.
- ketone compound examples include the compounds described in paragraph 0087 of JP-A-2015-087611, the contents of which are incorporated herein.
- Kayacure DETX manufactured by Nippon Kayaku Co., Ltd.
- Nippon Kayaku Co., Ltd. is also suitably used.
- a hydroxyacetophenone compound, an aminoacetophenone compound, and an acylphosphine compound can also be suitably used. More specifically, for example, an aminoacetophenone-based initiator described in JP-A-10-291969 and an acylphosphine oxide-based initiator described in Patent No. 4225988 can also be used.
- a hydroxyacetophenone-based initiator IRGACURE 184 (IRGACURE is a registered trademark), DAROCUR 1173, IRGACURE 500, IRGACURE-2959, IRGACURE 127 (trade name: all manufactured by BASF) can be used.
- aminoacetophenone-based initiator commercially available products IRGACURE 907, IRGACURE 369, and IRGACURE 379 (trade names: all manufactured by BASF) can be used.
- aminoacetophenone-based initiator a compound described in JP-A-2009-191179 in which the absorption maximum wavelength is matched to a light source having a wavelength of 365 nm or 405 nm can also be used.
- the acylphosphine initiator include 2,4,6-trimethylbenzoyl-diphenyl-phosphine oxide.
- IRGACURE-819 and IRGACURE-TPO commercially available products such as IRGACURE-819 and IRGACURE-TPO (trade names, both manufactured by BASF) can be used.
- metallocene compound include IRGACURE-784 (manufactured by BASF).
- An oxime compound is more preferably used as the photoradical polymerization initiator.
- the exposure latitude can be more effectively improved.
- Oxime compounds are particularly preferred because they have a wide exposure latitude (exposure margin) and also act as photocuring accelerators.
- compounds described in JP-A-2001-233842 compounds described in JP-A-2000-080068, and compounds described in JP-A-2006-342166 can be used.
- Preferred oxime compounds include, for example, compounds having the following structures, 3-benzooxyiminobutan-2-one, 3-acetoxyiminobutan-2-one, 3-propionyloxyimiminobtan-2-one, 2-acetoxy Iminopentan-3-one, 2-acetoxyimino-1-phenylpropan-1-one, 2-benzoyloxyimino-1-phenylpropan-1-one, 3- (4-toluenesulfonyloxy) iminobutan-2-one And 2-ethoxycarbonyloxyimino-1-phenylpropan-1-one.
- an oxime compound (oxime-based photopolymerization initiator) as the photoradical polymerization initiator.
- Commercially available products include IRGACURE OXE 01, IRGACURE OXE 02, IRGACURE OXE 03, IRGACURE OXE 04 (both manufactured by BASF) and Adeka Optomer N-1919 (manufactured by ADEKA CORPORATION, JP 2012-014052A).
- a radical polymerization initiator 2) is also preferably used.
- TR-PBG-304 manufactured by Changzhou Strong Electronics New Materials Co., Ltd.
- Adeka Aquel's NCI-831 and Adeka Aquel's NCI-930 manufactured by ADEKA Corporation
- DFI-091 manufactured by Daito Mix
- Specific examples of such oxime compounds include compounds described in JP-A-2010-262028, compounds 24 and 36 to 40 described in paragraph 0345 of JP-A-2014-500852, and JP-A-2013-2013.
- the most preferred oxime compounds include oxime compounds having a specific substituent described in JP-A-2007-269779, oxime compounds having a thioaryl group described in JP-A-2009-191061, and the like.
- the photoradical polymerization initiator may be a trihalomethyltriazine compound, a benzyldimethylketal compound, an ⁇ -hydroxyketone compound, an ⁇ -aminoketone compound, an acylphosphine compound, a phosphine oxide compound, a metallocene compound, an oxime compound, a triaryl Selected from the group consisting of imidazole dimers, onium salt compounds, benzothiazole compounds, benzophenone compounds, acetophenone compounds and derivatives thereof, cyclopentadiene-benzene-iron complexes and salts thereof, halomethyloxadiazole compounds, and 3-aryl-substituted coumarin compounds.
- More preferred photoradical polymerization initiators are trihalomethyltriazine compounds, ⁇ -aminoketone compounds, acylphosphine compounds, phosphine oxide compounds, metallocene compounds, oxime compounds, triarylimidazole dimers, onium salt compounds, benzophenone compounds, and acetophenone compounds.
- At least one compound selected from the group consisting of a trihalomethyltriazine compound, an ⁇ -aminoketone compound, an oxime compound, a triarylimidazole dimer, and a benzophenone compound is more preferable, and a metallocene compound or an oxime compound is more preferably used. Is even more preferred.
- the photo-radical polymerization initiators include N, N'-tetraalkyl-4,4'-diaminobenzophenone such as benzophenone, N, N'-tetramethyl-4,4'-diaminobenzophenone (Michler's ketone), 2-benzyl Aromatic ketones such as -2-dimethylamino-1- (4-morpholinophenyl) -butanone-1,2-methyl-1- [4- (methylthio) phenyl] -2-morpholino-propanone-1, alkylanthraquinone, etc.
- benzoin ether compounds such as benzoin alkyl ethers, benzoin compounds such as benzoin and alkyl benzoin, and benzyl derivatives such as benzyl dimethyl ketal.
- a compound represented by the following formula (I) can also be used.
- R I00 is an alkyl group having 1 to 20 carbon atoms, an alkyl group having 2 to 20 carbon atoms interrupted by at least one oxygen atom, an alkoxyl group having 1 to 12 carbon atoms, a phenyl group, An alkyl group having 1 to 20 carbon atoms, an alkoxyl group having 1 to 12 carbon atoms, a halogen atom, a cyclopentyl group, a cyclohexyl group, an alkenyl group having 2 to 12 carbon atoms, and 2 to 2 carbon atoms interrupted by at least one oxygen atom 18 alkyl group and at least one substituted phenyl group of the alkyl group having 1 to 4 carbon atoms or a biphenyl,
- R I01 is a group represented by formula (II), the same as R I00 And R I02 to R I04 are each independently alkyl having 1 to 12 carbons, alkoxy or halogen having 1 to 12 carbons.
- a photopolymerization initiator When a photopolymerization initiator is contained, its content is preferably from 0.1 to 30% by mass, more preferably from 0.1 to 20% by mass, based on the total solid content of the resin composition of the present invention. More preferably, it is from 0.5 to 15% by mass, more preferably from 1.0 to 10% by mass.
- the photopolymerization initiator may contain only one kind or two or more kinds. When two or more photopolymerization initiators are contained, the total is preferably within the above range.
- the resin composition of the present invention may contain a thermal radical polymerization initiator.
- a thermal radical polymerization initiator is a compound that generates a radical by the energy of heat and initiates or promotes a polymerization reaction of a compound having a radical polymerizable group.
- the thermal radical polymerization initiator By adding a thermal radical polymerization initiator, the cyclization of the polyimide precursor and, when the polyimide precursor has a radical polymerizable group, the polymerization reaction of the polyimide precursor can be advanced, so that higher heat resistance can be achieved. Can be achieved.
- Specific examples of the thermal radical polymerization initiator include compounds described in paragraphs 0074 to 0118 of JP-A-2008-063554.
- thermal radical polymerization initiator When a thermal radical polymerization initiator is contained, its content is preferably from 0.1 to 30% by mass, more preferably from 0.1 to 20% by mass, based on the total solid content of the resin composition of the present invention. And more preferably 5 to 15% by mass.
- the thermal radical polymerization initiator may contain only one kind or two or more kinds. When two or more thermal radical polymerization initiators are contained, the total is preferably within the above range.
- the resin composition of the present invention may contain a polymerizable monomer having a plurality of (meth) acryloyl groups.
- the number of (meth) acryloyl groups contained in the polymerizable monomer may be two or more, and is preferably three or more.
- the upper limit is preferably 15 or less, more preferably 10 or less, and still more preferably 8 or less.
- the molecular weight of the polymerizable monomer is preferably 2000 or less, more preferably 1500 or less, and even more preferably 900 or less.
- the lower limit of the molecular weight of the polymerizable monomer is preferably 100 or more, and more preferably 150 or more.
- examples of the polymerizable monomer include compounds having a boiling point of 100 ° C. or more under normal pressure.
- examples include polyethylene glycol di (meth) acrylate, trimethylolethane tri (meth) acrylate, neopentyl glycol di (meth) acrylate, pentaerythritol tri (meth) acrylate, pentaerythritol tetra (meth) acrylate, dipentaerythritol Penta (meth) acrylate, dipentaerythritol hexa (meth) acrylate, hexanediol (meth) acrylate, trimethylolpropane tri (acryloyloxypropyl) ether, tri (acryloyloxyethyl) isocyanurate, glycerin and trimethylolethane A compound obtained by adding ethylene oxide or propylene oxide to a functional alcohol and then (meth) acrylated,
- JP-B-50-006034 and urethane (meth) acrylates as described in JP-A-51-037193, JP-A-48-064183, and JP-A-49-043191.
- polyfunctional acrylates and methacrylates such as polyester acrylates described in JP-B-52-030490, epoxy acrylates which are reaction products of epoxy resins with (meth) acrylic acid, and mixtures thereof. Can be. Further, compounds described in paragraphs 0254 to 0257 of JP-A-2008-292970 are also suitable.
- a polyfunctional (meth) acrylate obtained by reacting a compound having an ethylenically unsaturated bond with a cyclic ether group such as glycidyl (meth) acrylate with a polyfunctional carboxylic acid can also be mentioned.
- a polymerizable monomer other than those described above it has a fluorene ring and an ethylenically unsaturated bond described in JP-A-2010-160418, JP-A-2010-129825, and JP-A-4364216. Compounds having two or more groups and cardo resins are also included.
- Examples of the polymerizable monomer include dipentaerythritol triacrylate (a commercially available product, KAYARAD D-330; manufactured by Nippon Kayaku Co., Ltd.), and dipentaerythritol tetraacrylate (a commercially available product, KAYARAD D-320; Nippon Kayaku Co., Ltd.) ), A-TMMT: Shin-Nakamura Chemical Co., Ltd.), dipentaerythritol penta (meth) acrylate (commercially available: KAYARAD D-310; manufactured by Nippon Kayaku Co., Ltd.), dipentaerythritol hexa (meth) acrylate (Commercially available products are KAYARAD DPHA; manufactured by Nippon Kayaku Co., Ltd., A-DPH; manufactured by Shin-Nakamura Chemical Co., Ltd.) Compounds having the following structures:
- polymerizable monomers include, for example, SR-494, a tetrafunctional acrylate having four ethyleneoxy chains, manufactured by Sartomer, SR-209, manufactured by Sartomer, which is a bifunctional methacrylate having four ethyleneoxy chains, 231, 239; DPCA-60, a hexafunctional acrylate having six pentyleneoxy chains, TPA-330, a trifunctional acrylate having three isobutyleneoxy chains, and urethane oligomer UAS-10, manufactured by Nippon Kayaku Co., Ltd.
- Examples of the polymerizable monomer include urethane acrylates described in JP-B-48-041708, JP-A-51-037193, JP-B-02-032293, and JP-B-02-016765.
- Urethane compounds having an ethylene oxide skeleton described in JP-B-58-49860, JP-B-56-017654, JP-B-62-039417, and JP-B-62-039418 are also suitable.
- compounds having an amino structure or a sulfide structure in the molecule described in JP-A-63-277563, JP-A-63-260909, and JP-A-01-105238 may be used as radical polymerizable compounds. It can also be used.
- the polymerizable monomer may be a compound having an acid group such as a carboxyl group and a phosphoric acid group.
- the polymerizable monomer having an acid group is preferably an ester of an aliphatic polyhydroxy compound and an unsaturated carboxylic acid, and the unreacted hydroxyl group of the aliphatic polyhydroxy compound is reacted with a non-aromatic carboxylic anhydride to form an acid group. Is more preferable.
- the aliphatic polyhydroxy compound is preferably pentaerythritol or dipentaerythritol. Is a compound.
- examples of commercially available products include M-510 and M-520 as polybasic acid-modified acrylic oligomers manufactured by Toagosei Co., Ltd.
- the preferred acid value of the polymerizable monomer having an acid group is 0.1 to 40 mgKOH / g, particularly preferably 5 to 30 mgKOH / g.
- the content of the polymerizable monomer is preferably 20% by mass or less, more preferably 18% by mass or less, and preferably 15% by mass or less based on the total solid content of the resin composition of the present invention. Is more preferred.
- the lower limit may be more than 0% by mass, 1% by mass or more, or 3% by mass or more.
- the resin composition of the present invention does not substantially contain a polymerizable monomer. According to this aspect, the elongation at break and the chemical resistance of the obtained cured film can be further improved.
- the case where the polymerizable monomer is not substantially contained means that the content of the polymerizable monomer is 0.01% by mass or less based on the total solid content of the resin composition.
- the resin composition of the present invention preferably contains a solvent.
- a solvent a known solvent can be arbitrarily used.
- the solvent is preferably an organic solvent.
- the organic solvent include compounds such as esters, ethers, ketones, aromatic hydrocarbons, sulfoxides, and amides.
- esters for example, ethyl acetate, n-butyl acetate, isobutyl acetate, amyl formate, isoamyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, butyl butyrate, methyl lactate, ethyl lactate, ⁇ -butyrolactone, ⁇ -caprolactone , ⁇ -valerolactone, alkyl alkyloxyacetates (eg, methyl alkyloxyacetate, ethylalkyloxyacetate, butylalkyloxyacetate (eg, methyl methoxyacetate, ethyl methoxyacetate, butyl methoxyacetate, methyl ethoxyacetate, methyl ethoxyacetate, etc.) )), Alkyl 3-alkyloxypropionates (eg, methyl 3-alkyloxypropionate, ethyl 3-
- alkyl 2-alkyloxypropionates eg, methyl 2-alkyloxypropionate, ethyl 2-alkyloxypropionate, 2 Propyl alkyloxypropionate and the like (eg, methyl 2-methoxypropionate, ethyl 2-methoxypropionate, propyl 2-methoxypropionate, methyl 2-ethoxypropionate, ethyl 2-ethoxypropionate), 2-alkyl Methyl oxy-2-methylpropionate and ethyl 2-alkyloxy-2-methylpropionate (eg, methyl 2-methoxy-2-methylpropionate, ethyl 2-ethoxy-2-methylpropionate), methyl pyruvate , Pyruvate Chill, propyl
- ethers for example, diethylene glycol dimethyl ether, tetrahydrofuran, ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, methyl cellosolve acetate, ethyl cellosolve acetate, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, diethylene glycol monobutyl ether, propylene glycol monomethyl ether, propylene glycol Suitable examples include monomethyl ether acetate, propylene glycol monoethyl ether acetate, and propylene glycol monopropyl ether acetate.
- Suitable ketones include, for example, methyl ethyl ketone, cyclohexanone, cyclopentanone, 2-heptanone, 3-heptanone and the like.
- Suitable aromatic hydrocarbons include, for example, toluene, xylene, anisole, limonene and the like.
- Suitable sulfoxides include, for example, dimethyl sulfoxide.
- Suitable amides include N-methyl-2-pyrrolidone, N-ethyl-2-pyrrolidone, N, N-dimethylacetamide, N, N-dimethylformamide and the like.
- a form in which two or more solvents are mixed is also preferable.
- solvents selected from butyrolactone, dimethylsulfoxide, ethyl carbitol acetate, butyl carbitol acetate, N-methyl-2-pyrrolidone, propylene glycol methyl ether, and propylene glycol methyl ether acetate, or two or more solvents is preferred. Particularly preferred is a combination of dimethyl sulfoxide and ⁇ -butyrolactone.
- the content of the solvent is preferably such that the total solid content of the resin composition of the present invention is 5 to 80% by mass, and more preferably 5 to 75% by mass. More preferably, the amount is 10 to 70% by mass, even more preferably 40 to 70% by mass.
- the solvent content may be adjusted depending on the desired thickness and the coating method.
- the solvent may contain only one kind, or may contain two or more kinds. When two or more solvents are contained, the total is preferably within the above range.
- the resin composition of the present invention further contains a migration inhibitor.
- the migration inhibitor is not particularly limited, but may be a heterocycle (pyrrole ring, furan ring, thiophene ring, imidazole ring, oxazole ring, thiazole ring, pyrazole ring, isoxazole ring, isothiazole ring, tetrazole ring, pyridine ring, Compounds having pyridazine ring, pyrimidine ring, pyrazine ring, piperidine ring, piperazine ring, morpholine ring, 2H-pyran ring and 6H-pyran ring, triazine ring), compounds having thioureas and sulfanyl groups, hindered phenol compounds , Salicylic
- an ion trapping agent for trapping anions such as halogen ions can be used.
- the migration inhibitor examples include rust preventives described in paragraph 0094 of JP-A-2013-015701, compounds described in paragraphs 0073 to 0076 of JP-A-2009-283711, and JP-A-2011-059656.
- Compounds described in paragraph 0052, compounds described in paragraphs 0114, 0116 and 0118 of JP-A-2012-194520, compounds described in paragraph 0166 of WO 2015/199219, and the like can be used.
- the migration inhibitor include the following compounds.
- the content of the migration inhibitor is preferably from 0.01 to 5.0% by mass, and more preferably from 0.05 to 2% by mass, based on the total solid content of the resin composition.
- the content is more preferably 0.0% by mass, and further preferably 0.1 to 1.0% by mass.
- the migration inhibitor may be only one kind or two or more kinds. When two or more types of migration inhibitors are used, the total is preferably within the above range.
- the resin composition of the present invention preferably contains a polymerization inhibitor.
- the polymerization inhibitor include hydroquinone, p-methoxyphenol, di-tert-butyl-p-cresol, pyrogallol, p-tert-butylcatechol, 1,4-benzoquinone, diphenyl-p-benzoquinone, and 4,4 ′.
- -Thiobis (3-methyl-6-tert-butylphenol), 2,2'-methylenebis (4-methyl-6-tert-butylphenol), N-nitroso-N-phenylhydroxyamine aluminum salt, phenothiazine, N-nitrosodiphenylamine , N-phenylnaphthylamine, ethylenediaminetetraacetic acid, 1,2-cyclohexanediaminetetraacetic acid, glycol etherdiaminetetraacetic acid, 2,6-di-tert-butyl-4-methylphenol, 5-nitroso-8-hydroxyquinoline, -Nitroso 2-naphthol, 2-nitroso-1-naphthol, 2-nitroso-5- (N-ethyl-N-sulfopropylamino) phenol, N-nitroso-N- (1-naphthyl) hydroxyamine ammonium salt, bis (4 -Hydroxy-3,5-ter
- polymerization inhibitors described in paragraph 0060 of JP-A-2015-127817 and the compounds described in paragraphs 0031 to 0046 of WO2015 / 125469 can also be used. Further, the following compounds can be used (Me is a methyl group).
- the content of the polymerization inhibitor is preferably from 0.01 to 5% by mass based on the total solid content of the resin composition of the present invention. It is more preferably 0.02 to 3% by mass, and further preferably 0.05 to 2.5% by mass.
- the polymerization inhibitor may be only one kind or two or more kinds. When there are two or more polymerization inhibitors, the total is preferably within the above range.
- the resin composition of the present invention preferably contains a metal adhesion improver for improving the adhesion to a metal material used for an electrode or a wiring.
- a metal adhesion improver for improving the adhesion to a metal material used for an electrode or a wiring.
- the metal adhesion improver include a silane coupling agent.
- silane coupling agent examples include the compounds described in paragraph 0167 of WO 2015/199219, the compounds described in paragraphs 0062 to 0073 of JP-A-2014-191002, and the paragraphs of WO 2011/080992.
- the compounds described in paragraph 0055 are mentioned. It is also preferable to use two or more different silane coupling agents as described in paragraphs 0050 to 0058 of JP-A-2011-128358. It is also preferable to use the following compound as the silane coupling agent.
- Et represents an ethyl group.
- the content of the metal adhesion improver is preferably from 0.1 to 30 parts by mass, more preferably from 0.5 to 15 parts by mass, and still more preferably from 0 to 15 parts by mass, per 100 parts by mass of the polymer precursor. It is in the range of 5 to 5 parts by mass.
- the metal adhesion improver may be only one kind or two or more kinds. When two or more types are used, the total is preferably within the above range.
- the resin composition of the present invention may contain various additives, for example, a thermal acid generator, a sensitizing dye, a chain transfer agent, a surfactant, and a higher fatty acid derivative, as long as the effects of the present invention are not impaired. , Inorganic particles, a curing agent, a curing catalyst, a filler, an antioxidant, an ultraviolet absorber, an anti-agglomeration agent, and the like. When these additives are blended, the total blending amount is preferably 3% by mass or less of the solid content of the composition.
- the resin composition of the present invention may contain a thermal acid generator.
- the thermal acid generator is used for elimination of the protective group.
- the content of the thermal acid generator is preferably at least 0.01 part by mass, more preferably at least 0.1 part by mass, based on 100 parts by mass of the polymer precursor. Since the crosslinking reaction and the cyclization of the polymer precursor are promoted by containing 0.01 parts by mass or more of the thermal acid generator, the mechanical properties and chemical resistance of the cured film can be further improved. Further, the content of the thermal acid generator is preferably 20 parts by mass or less, more preferably 15 parts by mass or less, and still more preferably 10 parts by mass or less, from the viewpoint of the electrical insulation of the cured film. Only one thermal acid generator may be used, or two or more thermal acid generators may be used. When two or more kinds are used, the total amount is preferably within the above range.
- the resin composition of the present invention may contain a sensitizing dye.
- the sensitizing dye absorbs a specific actinic radiation and enters an electronically excited state.
- the sensitizing dye in the electronically excited state comes into contact with a thermosetting accelerator, a thermal radical polymerization initiator, a photoradical polymerization initiator, or the like, and causes effects such as electron transfer, energy transfer, and heat generation.
- the thermal curing accelerator, the thermal radical polymerization initiator, and the photoradical polymerization initiator undergo chemical changes and are decomposed to generate radicals, acids, or bases.
- the description in paragraphs 0161 to 0163 of JP-A-2016-027357 can be referred to, and the contents thereof are incorporated herein.
- the content of the sensitizing dye is preferably 0.01 to 20% by mass relative to the total solid content of the resin composition of the present invention.
- the content is more preferably 1 to 15% by mass, and further preferably 0.5 to 10% by mass.
- the sensitizing dyes may be used alone or in combination of two or more.
- the resin composition of the present invention may contain a chain transfer agent.
- the chain transfer agent is defined, for example, in Polymer Dictionary, Third Edition (edited by The Society of Polymer Science, 2005), pages 683-684.
- As the chain transfer agent for example, a compound group having SH, PH, SiH, and GeH in the molecule is used. These can generate a radical by donating hydrogen to a low activity radical, or can generate a radical by being oxidized and then deprotonated.
- a thiol compound can be preferably used.
- compounds described in paragraphs 0152 to 0153 of WO 2015/199219 can also be used.
- the content of the chain transfer agent is preferably 0.01 to 20 parts by mass, preferably 1 to 20 parts by mass, based on 100 parts by mass of the total solid content of the resin composition of the present invention. 10 parts by mass is more preferable, and 1 to 5 parts by mass is further preferable. Only one type of chain transfer agent may be used, or two or more types may be used. When there are two or more chain transfer agents, the total is preferably within the above range.
- Each type of surfactant may be added to the resin composition of the present invention from the viewpoint of further improving coatability.
- various types of surfactants such as a fluorine-based surfactant, a nonionic surfactant, a cationic surfactant, an anionic surfactant, and a silicone-based surfactant can be used. Further, the following surfactants are also preferable.
- the content of the surfactant is preferably 0.001 to 2.0% by mass based on the total solid content of the resin composition of the present invention. , More preferably 0.005 to 1.0% by mass.
- the surfactant may be only one kind or two or more kinds. When two or more surfactants are used, the total is preferably within the above range.
- the resin composition of the present invention is added with a higher fatty acid derivative such as behenic acid or behenic acid amide, and is unevenly distributed on the surface of the composition in a drying process after application. May be. Further, as the higher fatty acid derivative, a compound described in paragraph 0155 of WO 2015/199219 can be used.
- the content of the higher fatty acid derivative is preferably 0.1 to 10% by mass based on the total solid content of the resin composition of the present invention.
- the higher fatty acid derivative may be only one kind or two or more kinds. When there are two or more higher fatty acid derivatives, the total is preferably within the above range.
- the water content of the resin composition of the present invention is preferably less than 5% by mass, more preferably less than 1% by mass, even more preferably less than 0.6% by mass from the viewpoint of the properties of the coated surface.
- the metal content of the resin composition of the present invention is preferably less than 5 ppm by mass (parts per million), more preferably less than 1 ppm by mass, and even more preferably less than 0.5 ppm by mass.
- the metal include sodium, potassium, magnesium, calcium, iron, chromium, nickel and the like. When a plurality of metals are included, the total of these metals is preferably within the above range.
- a material having a low metal content is selected as a raw material constituting the resin composition of the present invention. Examples of the method include filtering the raw material constituting the product with a filter, lining the inside of the apparatus with polytetrafluoroethylene or the like, and performing distillation under the condition that contamination is suppressed as much as possible.
- the resin composition of the present invention has a halogen atom content of preferably less than 500 ppm by mass, more preferably less than 300 ppm by mass, and less than 200 ppm by mass, from the viewpoint of wiring corrosiveness, when considering the use as a semiconductor material. Is more preferred. Above all, those present in the form of halogen ions are preferably less than 5 ppm by mass, more preferably less than 1 ppm by mass, even more preferably less than 0.5 ppm by mass.
- the halogen atom include a chlorine atom and a bromine atom. It is preferable that each of the chlorine atom and the bromine atom, or the total of the chlorine ion and the bromine ion is within the above range.
- the container for storing the resin composition of the present invention a conventionally known container can be used.
- the inner wall of the container is formed into a multi-layer bottle composed of six types and six layers of resin, or six types of resin is formed into a seven-layer structure. It is also preferred to use a bottle that has been used. Examples of such a container include a container described in JP-A-2015-123351.
- the resin composition of the present invention can be prepared by mixing the above components.
- the mixing method is not particularly limited, and can be performed by a conventionally known method.
- the filter pore size is preferably 1 ⁇ m or less, more preferably 0.5 ⁇ m or less, and even more preferably 0.1 ⁇ m or less.
- the material of the filter is preferably polytetrafluoroethylene, polyethylene or nylon.
- the filter may be one that has been washed in advance with an organic solvent. In the filter filtration step, a plurality of types of filters may be connected in series or in parallel.
- filters having different pore sizes or materials may be used in combination. Further, various materials may be filtered plural times. When filtration is performed a plurality of times, circulation filtration may be used. Also, filtration may be performed under pressure. In the case of performing filtration by applying pressure, the pressure to be applied is preferably 0.05 MPa or more and 0.3 MPa or less.
- a treatment for removing impurities using an adsorbent may be performed. Filter filtration and impurity removal treatment using an adsorbent may be combined.
- the adsorbent a known adsorbent can be used. For example, inorganic adsorbents such as silica gel and zeolite, and organic adsorbents such as activated carbon can be used.
- the cured film of the present invention is obtained by curing the resin composition of the present invention.
- the thickness of the cured film of the present invention can be, for example, 0.5 ⁇ m or more, and can be 1 ⁇ m or more. Further, the upper limit can be set to 100 ⁇ m or less, and can be set to 30 ⁇ m or less.
- the cured film of the present invention may be formed into a laminate by laminating two or more, more preferably three to seven layers.
- the laminate having two or more cured films of the present invention preferably has a metal layer between the cured films.
- Such a metal layer is preferably used as a metal wiring such as a rewiring layer.
- Fields to which the cured film of the present invention can be applied include an insulating film of a semiconductor device, an interlayer insulating film for a rewiring layer, a stress buffer film, and the like.
- Other examples include patterning a sealing film, a substrate material (a base film or a coverlay of a flexible printed board, an interlayer insulating film), or an insulating film for mounting as described above by etching. These applications are described in, for example, Science & Technology Co., Ltd.
- the cured film according to the present invention can also be used for producing plate surfaces such as offset plate surfaces or screen plate surfaces, for use in etching molded parts, and for producing protective lacquers and dielectric layers in electronics, particularly microelectronics.
- the method for producing a cured film of the present invention includes using the resin composition of the present invention. Specifically, it is preferable to include the following steps (a) to (d). (A) a film forming step of forming a film by applying the resin composition to a substrate; (b) an exposing step of exposing the film after the film forming step; and (c) a developing treatment of the exposed resin composition layer. (D) heating step of heating the developed resin composition at 80 to 450 ° C. As in this embodiment, the exposed resin layer can be further cured by heating after development. . In this heating step, the thermobase generator and the thermosetting compound act to obtain sufficient curability.
- the method for producing a laminate according to a preferred embodiment of the present invention includes the method for producing a cured film of the present invention.
- the step (a), the steps (a) to (c), or the step (a) is performed again.
- ) To (d) are performed.
- a metal layer on the portion where the cured film is provided, between the cured films, or both.
- the manufacturing method includes a film forming step (layer forming step) of applying a resin composition to a substrate to form a film (layered).
- the type of the substrate can be appropriately determined according to the application, but a semiconductor production substrate such as silicon, silicon nitride, polysilicon, silicon oxide, and amorphous silicon, quartz, glass, an optical film, a ceramic material, a vapor-deposited film, and a magnetic film , A reflective film, a metal substrate of Ni, Cu, Cr, Fe, etc., paper, SOG (Spin On Glass), TFT (thin film transistor) array substrate, and an electrode plate of a plasma display panel (PDP).
- a semiconductor production substrate such as silicon, silicon nitride, polysilicon, silicon oxide, and amorphous silicon, quartz, glass, an optical film, a ceramic material, a vapor-deposited film, and a magnetic film
- a semiconductor fabrication substrate is particularly preferable, and a silicon substrate is more preferable.
- the resin composition layer is formed on the surface of the resin layer or the surface of the metal layer, the resin layer or the metal layer serves as a substrate.
- coating is preferable. Specifically, as means to be applied, dip coating, air knife coating, curtain coating, wire bar coating, gravure coating, extrusion coating, spray coating, spin coating, slit coating, slit coating, And an inkjet method. From the viewpoint of the uniformity of the thickness of the resin composition layer, more preferred are a spin coating method, a slit coating method, a spray coating method and an ink jet method.
- a resin layer having a desired thickness can be obtained by adjusting an appropriate solid content concentration and application conditions according to the method.
- the coating method can also be appropriately selected depending on the shape of the substrate.
- a spin coating method, a spray coating method, or an inkjet method is preferable.
- a slit coating method, a spray coating method, or an inkjet method is used. Method is preferred.
- a rotation speed of 500 to 2000 rpm can be applied for about 10 seconds to 1 minute.
- the production method of the present invention may include a step of drying after removing the solvent after forming the resin composition layer and after the film forming step (layer forming step).
- the preferred drying temperature is 50 to 150 ° C., more preferably 70 to 130 ° C., and even more preferably 90 to 110 ° C.
- the drying time is, for example, 30 seconds to 20 minutes, preferably 1 minute to 10 minutes, and more preferably 3 minutes to 7 minutes.
- the production method of the present invention may include an exposure step of exposing the resin composition layer.
- the amount of exposure is not particularly limited as long as the resin composition can be cured.
- irradiation is preferably 100 to 10,000 mJ / cm 2 , and more preferably 200 to 8000 mJ / cm 2 in terms of exposure energy at a wavelength of 365 nm. More preferred.
- the exposure wavelength can be appropriately determined within the range of 190 to 1000 nm, and preferably 240 to 550 nm.
- the exposure wavelength is, in terms of the light source, (1) semiconductor laser (wavelength 830 nm, 532 nm, 488 nm, 405 nm etc.), (2) metal halide lamp, (3) high-pressure mercury lamp, g-line (wavelength 436 nm), h Line (wavelength 405 nm), i-line (wavelength 365 nm), broad (three wavelengths of g, h, i-line), (4) excimer laser, KrF excimer laser (wavelength 248 nm), ArF excimer laser (wavelength 193 nm), F2 excimer Laser (wavelength: 157 nm); (5) extreme ultraviolet; EUV (wavelength: 13.6 nm); (6) electron beam.
- exposure with a high-pressure mercury lamp is particularly preferred, and exposure with i-line is particularly preferred. Thereby, a particularly high exposure sensitivity can be obtained.
- the production method of the present invention may include a development processing step of performing development processing on the exposed resin composition layer. By performing the development, an unexposed portion (non-exposed portion) is removed.
- the developing method is not particularly limited as long as a desired pattern can be formed. For example, a developing method such as paddle, spray, immersion, or ultrasonic wave can be adopted. Development is performed using a developer.
- the developer can be used without particular limitation as long as the unexposed portions (non-exposed portions) are removed.
- the developer preferably contains an organic solvent, and more preferably the developer contains 90% or more of the organic solvent.
- the developer preferably contains an organic solvent having a ClogP value of -1 to 5, more preferably an organic solvent having a ClogP value of 0 to 3.
- the ClogP value can be obtained as a calculated value by inputting a structural formula in ChemBioDraw.
- the organic solvent include esters such as ethyl acetate, n-butyl acetate, amyl formate, isoamyl acetate, isobutyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, butyl butyrate, methyl lactate, ethyl lactate, and ⁇ -butyrolactone.
- alkyl alkyl oxyacetates eg, methyl alkyl oxyacetate, ethyl oxyacetate, alkyl butyl oxyacetate (eg, methyl methoxy acetate, ethyl methoxy acetate, butyl methoxy acetate, methyl ethoxy acetate, Ethyl ethoxyacetate, etc.
- alkyl 3-alkyloxypropionates eg, methyl 3-alkyloxypropionate, ethyl 3-alkyloxypropionate, etc.
- alkyl 2-alkyloxypropionates eg, methyl 2-alkyloxypropionate, ethyl 2-alkyloxypropionate, 2 Propyl alkyloxypropionate and the like (eg, methyl 2-methoxypropionate, ethyl 2-methoxypropionate, propyl 2-methoxypropionate, methyl 2-ethoxypropionate, ethyl 2-ethoxypropionate), 2-alkyl Methyl oxy-2-methylpropionate and ethyl 2-alkyloxy-2-methylpropionate (eg, methyl 2-methoxy-2-methylpropionate, ethyl 2-ethoxy-2-methylpropionate), methyl pyruvate , Pyruvate Tyl, propyl pionate, methyl 3-ethoxypropionate, ethyl 3-ethoxypropionate)), alkyl 2-alkyloxypropionates (eg, methyl 2-alkyloxyprop
- dimethyl sulfoxide is preferably exemplified.
- cyclopentanone and ⁇ -butyrolactone are particularly preferred, and cyclopentanone is more preferred.
- the developer preferably contains 50% by mass or more of an organic solvent, more preferably 70% by mass or more of an organic solvent, and even more preferably 90% by mass or more of an organic solvent. Further, 100% by mass of the developer may be an organic solvent.
- the development time is preferably from 10 seconds to 5 minutes.
- the temperature of the developing solution at the time of development is not particularly limited, but it can be usually 20 to 40 ° C.
- rinsing may be further performed. Rinsing is preferably performed with a solvent different from the developer. For example, rinsing can be performed using a solvent contained in the resin composition.
- the rinsing time is preferably from 5 seconds to 1 minute.
- the manufacturing method of the present invention preferably includes a heating step after the film forming step (layer forming step), the drying step, or the developing step.
- a heating step a cyclization reaction of the polymer precursor and a curing reaction of the thermosetting compound proceed.
- the heating temperature (maximum heating temperature) of the layer in the heating step is preferably 50 ° C. or higher, more preferably 80 ° C. or higher, further preferably 140 ° C. or higher, and more preferably 150 ° C. or higher. Is more preferably 160 ° C. or higher, and even more preferably 170 ° C. or higher.
- the upper limit is preferably 500 ° C. or lower, more preferably 450 ° C.
- Heating is preferably performed at a rate of 1 to 12 ° C./min from the temperature at the start of heating to the maximum heating temperature, more preferably 2 to 10 ° C./min, even more preferably 3 to 10 ° C./min.
- the heating rate By setting the heating rate to 1 ° C./min or more, the amine can be prevented from being excessively volatilized while securing the productivity.
- the heating rate By setting the heating rate to 12 ° C./min or less, the cured film can be cured. The residual stress can be reduced.
- the temperature at the start of heating is preferably from 20 ° C to 150 ° C, more preferably from 20 ° C to 130 ° C, even more preferably from 25 ° C to 120 ° C.
- the temperature at the start of heating refers to the temperature at which the step of heating to the maximum heating temperature is started.
- the heating time is preferably from 10 to 360 minutes, more preferably from 20 to 300 minutes, even more preferably from 30 to 240 minutes.
- the heating temperature is preferably from 180 ° C. to 320 ° C., and more preferably from 180 ° C. to 260 ° C., from the viewpoint of adhesion between the cured films.
- the reason is not clear, it is considered that the ethynyl groups of the polymer precursor between the layers are undergoing a crosslinking reaction at this temperature.
- the heating may be performed stepwise. As an example, the temperature is raised from 25 ° C. to 180 ° C. at 3 ° C./min, maintained at 180 ° C. for 60 minutes, raised from 180 ° C. to 200 ° C. at 2 ° C./min, and maintained at 200 ° C. for 120 minutes. , Etc., may be performed.
- the heating temperature in the pretreatment step is preferably from 100 to 200 ° C, more preferably from 110 to 190 ° C, and even more preferably from 120 to 185 ° C. In this pretreatment step, it is also preferable to perform the treatment while irradiating ultraviolet rays as described in US Pat. No. 9,159,547.
- the pretreatment step may be performed in a short time of about 10 seconds to 2 hours, more preferably 15 seconds to 30 minutes.
- the pretreatment may include two or more steps. For example, pretreatment step 1 may be performed at a temperature in the range of 100 to 150 ° C., and then pretreatment step 2 may be performed at a temperature in the range of 150 to 200 ° C. Furthermore, cooling may be performed after heating, and the cooling rate in this case is preferably 1 to 5 ° C./min.
- the heating step is preferably performed in an atmosphere having a low oxygen concentration by flowing an inert gas such as nitrogen, helium, or argon from the viewpoint of preventing decomposition of the polymer precursor.
- the oxygen concentration is preferably 50 ppm (volume ratio) or less, more preferably 20 ppm (volume ratio) or less.
- the production method of the present invention preferably includes a metal layer forming step of forming a metal layer on the surface of the resin composition layer after the development processing.
- the metal layer is not particularly limited, and any existing metal species can be used.Examples include copper, aluminum, nickel, vanadium, titanium, chromium, cobalt, gold, and tungsten, and copper and aluminum are more preferable, and copper is preferable. More preferred.
- the method for forming the metal layer is not particularly limited, and an existing method can be applied. For example, the methods described in JP-A-2007-157879, JP-T-2001-521288, JP-A-2004-214501, and JP-A-2004-101850 can be used.
- the thickness of the metal layer at the thickest part is preferably 0.1 to 50 ⁇ m, more preferably 1 to 10 ⁇ m.
- the manufacturing method of the present invention further includes a lamination step.
- the laminating step means (a) a film forming step (layer forming step), (b) an exposing step, (c) a developing step, and (d) a heating step on the surface of the cured film (resin layer) or the metal layer. Are performed in this order. However, an embodiment in which only the film forming step (a) is repeated may be employed. Further, the heating step (d) may be performed at the end of or in the middle of the lamination. That is, the steps (a) to (c) may be repeated a predetermined number of times, and then the heating of (d) may be performed to cure the laminated resin composition layers at once.
- a metal layer forming step may be included after the (c) developing step.
- the laminating step may further include the above-mentioned drying step, heating step and the like as appropriate.
- a surface activation treatment step may be further performed after the heating step, after the exposing step, or after the metal layer forming step.
- a plasma treatment is exemplified.
- the above lamination step is preferably performed 2 to 5 times, more preferably 3 to 5 times.
- a configuration of three to seven resin layers is preferable, such as a resin layer / metal layer / resin layer / metal layer / resin layer / metal layer, and more preferably three to five layers.
- a cured film (resin layer) of the resin composition is further formed so as to cover the metal layer.
- the present invention also discloses a semiconductor device having the cured film or the laminate of the present invention.
- the semiconductor device using the resin composition of the present invention for forming an interlayer insulating film for a rewiring layer the description of paragraphs 0213 to 0218 of JP-A-2016-027357 and the description of FIG. 1 can be referred to. These contents are incorporated herein.
- reaction mixture was cooled to room temperature and 21.43 g (270.9 mmol) of pyridine and 90 mL of N-methylpyrrolidone were added. The reaction mixture was then cooled to ⁇ 10 ° C. and 16.12 g (135.5 mmol) SOCl 2 was added over 10 minutes keeping the temperature at ⁇ 10 ⁇ 4 ° C. The viscosity increased during the addition of SOCl 2 . After dilution with 50 mL of N-methylpyrrolidone, the reaction mixture was stirred at room temperature for 2 hours.
- Examples and Comparative Examples> The components shown in the following table were mixed to obtain each resin composition.
- the obtained resin composition was subjected to pressure filtration through a filter having a pore width of 0.8 ⁇ m.
- Each resin composition was applied on a silicon wafer by a spin coating method to form a resin composition layer.
- the silicon wafer to which the obtained resin composition layer was applied was dried on a hot plate at 100 ° C. for 4 minutes to form a uniform resin composition layer having a thickness of 20 ⁇ m on the silicon wafer.
- the resin composition layer on the silicon wafer was exposed to light at an exposure energy of 400 mJ / cm 2 using a broadband exposure machine (UX-1000SN-EH01 manufactured by Ushio Inc.), and the exposed photosensitive resin composition layer ( The resin layer) was heated at a rate of 5 ° C./min in a nitrogen atmosphere, and after reaching 180 ° C., was heated for 2 hours.
- the cured resin layer was immersed in a 3% hydrofluoric acid solution, and the resin layer was separated from the silicon wafer to obtain a cured film.
- Elongation at break was measured 10 times each, and the average value was used. The results were classified and evaluated as follows. A: Elongation at break is 60% or more B: Elongation at break is 50% or more and less than 60% C: Elongation at break is less than 50%
- Each resin composition was spin-coated on a silicon wafer.
- the silicon wafer to which the resin composition was applied was dried on a hot plate at 100 ° C. for 4 minutes to form a uniform resin composition layer having a thickness of 20 ⁇ m on the silicon wafer.
- the resin composition layer on the silicon wafer was exposed using a stepper (Nikon NSR 2005 i9C). Exposure is performed with i-line, at a wavelength of 365 nm, using a line and space photomask in increments of 1 ⁇ m from 5 ⁇ m to 25 ⁇ m at exposure energy of 200, 300, 400, 500, 600, 700, 800 mJ / cm 2. Exposure was performed to obtain a resin layer.
- This resin layer was developed with cyclopentanone for 60 seconds.
- the minimum line width that can be formed hardly changes with respect to the fluctuation of the exposure amount, the arbitrariness of the exposure amount in the formation of the fine pattern increases, and a preferable result is obtained.
- B The line width is 10 ⁇ m or more and less than 20 ⁇ m.
Abstract
Description
一方で、ポリイミド樹脂は、一般に、溶剤への溶解性が低い。そのため、環化反応前のポリマー前駆体、具体的には、ポリイミド前駆体の状態で溶剤に溶解する方法がよく用いられる。これにより、優れた取り扱い性を実現することができ、上述のような各製品を製造する際に基板などに多様な形態で塗布して加工することができる。その後、加熱してポリイミド前駆体を環化し、硬化した製品を形成することができる。ポリイミド樹脂等がもつ高い性能に加え、このような製造上の適応性に優れる観点から、その産業上の応用展開がますます期待されている。 Polyimide resins have been applied to various uses because of their excellent heat resistance and insulation properties. The application of the semiconductor device is not particularly limited. For example, a semiconductor device for mounting may be used as a material for an insulating film or a sealing material, or as a protective film (see Non-Patent Documents 1 and 2). Further, it is also used as a base film or coverlay of a flexible substrate.
On the other hand, polyimide resins generally have low solubility in solvents. Therefore, a method of dissolving in a solvent in a state of a polymer precursor before the cyclization reaction, specifically, a polyimide precursor is often used. Thereby, excellent handling properties can be realized, and when each of the above products is manufactured, it can be applied and processed in various forms on a substrate or the like. Thereafter, the polyimide precursor can be cyclized by heating to form a cured product. In addition to the high performance of polyimide resins and the like, such industrial adaptability is increasingly expected from the viewpoint of excellent manufacturing adaptability.
熱塩基発生剤と、
エポキシ基、オキセタニル基、メチロール基、アルコキシメチル基、フェノール基、マレイミド基、シアネート基及びブロックイソシアネート基からなる群から選択される官能基を複数有する熱硬化性化合物と、
を含む樹脂組成物。
<2> 熱硬化性化合物は、下記式(TC1)で表される化合物である、<1>に記載の樹脂組成物;
X1-(Y1)n ・・・(TC1)
式(TC1)中、X1はn価の連結基を表し、Y1はエポキシ基、オキセタニル基、メチロール基、アルコキシメチル基、フェノール基、マレイミド基、シアネート基またはブロックイソシアネート基を表し、nは2以上の整数を表す。
<3> 式(TC1)のX1は環状構造を含む、<2>に記載の樹脂組成物。
<4> 熱硬化性化合物は、アルコキシメチル基を複数有する化合物である、<1>~<3>のいずれか1つに記載の樹脂組成物。
<5> 熱硬化性化合物は、メトキシメチル基を複数有する化合物である、<1>~<3>のいずれか1つに記載の樹脂組成物。
<6> 熱塩基発生剤の塩基発生温度は、熱硬化性化合物の硬化開始温度よりも低い<1>~<5>のいずれか1つに記載の樹脂組成物。
<7> (メタ)アクリロイル基を複数有する重合性モノマーの含有量が樹脂組成物の全固形分中20質量%以下である、<1>~<6>のいずれか1つに記載の樹脂組成物。
<8> ポリイミド前駆体がラジカル重合性基を含む、<1>~<7>のいずれか1つに記載の樹脂組成物。
<9> ポリイミド前駆体が下記式(1)で表される構成単位を有する、<1>~<8>のいずれか1つに記載の樹脂組成物;

<10> 式(1)におけるR113およびR114の少なくとも一方がラジカル重合性基を含む、<9>に記載の樹脂組成物。
<11> 更に、光重合開始剤を含む<8>または<10>に記載の樹脂組成物。
<12> 再配線層用層間絶縁膜の形成に用いられる、<1>~<11>のいずれか1つに記載の樹脂組成物。
<13> <1>~<12>のいずれか1つに記載の樹脂組成物を硬化して得られる硬化膜。
<14> <13>に記載の硬化膜を2層以上有し、2層の硬化膜の間に金属層を有する、積層体。
<15> <1>~<12>のいずれか1つに記載の樹脂組成物を基板に適用して膜を形成する膜形成工程を含む、硬化膜の製造方法。
<16> 膜を露光する露光工程および膜を現像する現像工程を有する、<15>に記載の硬化膜の製造方法。
<17> 膜を80~450℃で加熱する工程を含む、<16>に記載の硬化膜の製造方法。
<18> <13>に記載の硬化膜または<14>に記載の積層体を有する、半導体デバイス。 <1> a polyimide precursor,
A thermal base generator,
A thermosetting compound having a plurality of functional groups selected from the group consisting of an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group and a blocked isocyanate group,
A resin composition comprising:
<2> the resin composition according to <1>, wherein the thermosetting compound is a compound represented by the following formula (TC1);
X 1- (Y 1 ) n ... (TC1)
In the formula (TC1), X 1 represents an n-valent linking group, Y 1 represents an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group or a blocked isocyanate group, and n is Represents an integer of 2 or more.
<3> The resin composition according to <2>, wherein X 1 in the formula (TC1) includes a cyclic structure.
<4> The resin composition according to any one of <1> to <3>, wherein the thermosetting compound is a compound having a plurality of alkoxymethyl groups.
<5> The resin composition according to any one of <1> to <3>, wherein the thermosetting compound is a compound having a plurality of methoxymethyl groups.
<6> The resin composition according to any one of <1> to <5>, wherein the base generation temperature of the thermobase generator is lower than the curing start temperature of the thermosetting compound.
<7> The resin composition according to any one of <1> to <6>, wherein the content of the polymerizable monomer having a plurality of (meth) acryloyl groups is 20% by mass or less based on the total solid content of the resin composition. Stuff.
<8> The resin composition according to any one of <1> to <7>, wherein the polyimide precursor contains a radical polymerizable group.
<9> The resin composition according to any one of <1> to <8>, wherein the polyimide precursor has a structural unit represented by the following formula (1);
<10> The resin composition according to <9>, wherein at least one of R 113 and R 114 in the formula (1) contains a radically polymerizable group.
<11> The resin composition according to <8> or <10>, further including a photopolymerization initiator.
<12> The resin composition according to any one of <1> to <11>, which is used for forming an interlayer insulating film for a redistribution layer.
<13> A cured film obtained by curing the resin composition according to any one of <1> to <12>.
<14> A laminate having at least two cured films according to <13> and having a metal layer between the two cured films.
<15> A method for producing a cured film, comprising a film formation step of forming a film by applying the resin composition according to any one of <1> to <12> to a substrate.
<16> The method for producing a cured film according to <15>, comprising an exposure step of exposing the film and a development step of developing the film.
<17> The method for producing a cured film according to <16>, comprising a step of heating the film at 80 to 450 ° C.
<18> A semiconductor device having the cured film according to <13> or the laminate according to <14>.
本明細書における基(原子団)の表記に於いて、置換および無置換を記していない表記は、置換基を有さないものと共に置換基を有するものをも包含するものである。例えば、「アルキル基」とは、置換基を有さないアルキル基(無置換アルキル基)のみならず、置換基を有するアルキル基(置換アルキル基)をも包含するものである。
本明細書において「露光」とは、特に断らない限り、光を用いた露光のみならず、電子線、イオンビーム等の粒子線を用いた描画も露光に含める。また、露光に用いられる光としては、一般的に、水銀灯の輝線スペクトル、エキシマレーザーに代表される遠紫外線、極紫外線(EUV光)、X線、電子線等の活性光線または放射線が挙げられる。
本明細書において、「(メタ)アクリレート」は、「アクリレート」および「メタクリレート」の双方、または、いずれかを表し、「(メタ)アクリル」は、「アクリル」および「メタクリル」の双方、または、いずれかを表し、「(メタ)アクリロイル」は、「アクリロイル」および「メタクリロイル」の双方、または、いずれかを表す。
本明細書において「工程」との語は、独立した工程だけではなく、他の工程と明確に区別できない場合であってもその工程の所期の作用が達成されれば、本用語に含まれる。
本発明における物性値は特に述べない限り、温度23℃、気圧101325Paの下での値とする。
本明細書において、重量平均分子量(Mw)および数平均分子量(Mn)は、特に述べない限り、ゲル浸透クロマトグラフィ(GPC測定)によって測定されたものであり、ポリスチレン換算値として定義される。本明細書において、重量平均分子量(Mw)および数平均分子量(Mn)は、例えば、HLC-8220(東ソー(株)製)を用い、カラムとしてガードカラムHZ-L、TSKgel Super HZM-M、TSKgel Super HZ4000、TSKgel Super HZ3000およびTSKgel Super HZ2000(東ソー(株)製)を用いることによって求めることができる。この測定において、溶離液は特に述べない限り、THF(テトラヒドロフラン)を用いる。また、検出は特に述べない限り、UV線(紫外線)の波長254nm検出器を使用したものとする。 The description of the components of the present invention described below may be made based on typical embodiments of the present invention, but the present invention is not limited to such embodiments.
In the description of the group (atomic group) in the present specification, the notation of not indicating substituted or unsubstituted includes not only a group having no substituent but also a group having a substituent. For example, the “alkyl group” includes not only an alkyl group having no substituent (unsubstituted alkyl group) but also an alkyl group having a substituent (substituted alkyl group).
In this specification, “exposure” includes not only exposure using light but also drawing using particle beams such as electron beams and ion beams, unless otherwise specified. The light used for exposure generally includes an active ray or radiation such as a bright line spectrum of a mercury lamp, far ultraviolet represented by excimer laser, extreme ultraviolet (EUV light), X-ray, and electron beam.
In the present specification, “(meth) acrylate” represents both or “acrylate” and “methacrylate”, and “(meth) acryl” represents both “acryl” and “methacryl”, or The term “(meth) acryloyl” represents either “acryloyl” or “methacryloyl”, or any one of them.
In the present specification, the term "step" is included not only in an independent step but also in the case where the intended action of the step is achieved even if it cannot be clearly distinguished from other steps. .
The physical property values in the present invention are values at a temperature of 23 ° C. and a pressure of 101325 Pa unless otherwise specified.
In this specification, the weight average molecular weight (Mw) and the number average molecular weight (Mn) are measured by gel permeation chromatography (GPC measurement) and are defined as polystyrene equivalent values, unless otherwise specified. In the present specification, the weight average molecular weight (Mw) and the number average molecular weight (Mn) are determined, for example, using HLC-8220 (manufactured by Tosoh Corporation), and using guard columns HZ-L, TSKgel Super HZM-M, and TSKgel as columns. It can be determined by using Super HZ4000, TSKgel Super HZ3000 and TSKgel Super HZ2000 (manufactured by Tosoh Corporation). In this measurement, THF (tetrahydrofuran) is used as an eluent unless otherwise specified. Unless otherwise specified, detection is performed using a detector having a wavelength of 254 nm of UV rays (ultraviolet rays).
本発明の樹脂組成物は、ポリイミド前駆体と、
熱塩基発生剤と、
エポキシ基、オキセタニル基、メチロール基、アルコキシメチル基、フェノール基、マレイミド基、シアネート基及びブロックイソシアネートからなる群から選択される官能基を複数有する熱硬化性化合物と、
を含む。 [Resin composition]
The resin composition of the present invention, a polyimide precursor,
A thermal base generator,
A thermosetting compound having a plurality of functional groups selected from the group consisting of an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group and a blocked isocyanate;
including.
本発明の樹脂組成物は、ポリイミド前駆体を含む。 <Polyimide precursor>
The resin composition of the present invention contains a polyimide precursor.

R111は2価の有機基を表す。2価の有機基としては、直鎖または分岐の脂肪族基、環状の脂肪族基、および芳香族基、複素芳香族基、またはこれらの組み合わせからなる基が例示され、炭素数2~20の直鎖の脂肪族基、炭素数3~20の分岐の脂肪族基、炭素数3~20の環状の脂肪族基、炭素数6~20の芳香族基、または、これらの組み合わせからなる基が好ましく、炭素数6~20の芳香族基がより好ましい。
R111は、ジアミンから誘導されることが好ましい。ポリイミド前駆体の製造に用いられるジアミンとしては、直鎖または分岐の脂肪族、環状の脂肪族または芳香族ジアミンなどが挙げられる。ジアミンは、1種のみ用いてもよいし、2種以上用いてもよい。
具体的には、ジアミンは、炭素数2~20の直鎖脂肪族基、炭素数3~20の分岐または環状の脂肪族基、炭素数6~20の芳香族基、または、これらの組み合わせからなる基を含むものであることが好ましく、炭素数6~20の芳香族基を含むジアミンであることがより好ましい。芳香族基の例としては、下記が挙げられる。 <<<< R 111 >>>
R 111 represents a divalent organic group. Examples of the divalent organic group include a linear or branched aliphatic group, a cyclic aliphatic group, and an aromatic group, a heteroaromatic group, or a group composed of a combination thereof. A linear aliphatic group, a branched aliphatic group having 3 to 20 carbon atoms, a cyclic aliphatic group having 3 to 20 carbon atoms, an aromatic group having 6 to 20 carbon atoms, or a group composed of a combination thereof. Preferably, an aromatic group having 6 to 20 carbon atoms is more preferable.
Preferably, R 111 is derived from a diamine. Examples of the diamine used in the production of the polyimide precursor include linear or branched aliphatic, cyclic aliphatic or aromatic diamines. Diamines may be used alone or in combination of two or more.
Specifically, the diamine may be a linear aliphatic group having 2 to 20 carbon atoms, a branched or cyclic aliphatic group having 3 to 20 carbon atoms, an aromatic group having 6 to 20 carbon atoms, or a combination thereof. Preferably, the diamine is a diamine containing an aromatic group having 6 to 20 carbon atoms. Examples of the aromatic group include the following.


ジェファーミン(登録商標)KH-511、ジェファーミン(登録商標)ED-600、ジェファーミン(登録商標)ED-900、ジェファーミン(登録商標)ED-2003、ジェファーミン(登録商標)EDR-148、ジェファーミン(登録商標)EDR-176の構造を以下に示す。 Diamines having two or more alkylene glycol units in the main chain are also preferred examples. Preferably, it is a diamine containing one or both of an ethylene glycol chain and a propylene glycol chain in one molecule, and more preferably a diamine containing no aromatic ring. Specific examples include Jeffamine (registered trademark) KH-511, Jeffamine (registered trademark) ED-600, Jeffamine (registered trademark) ED-900, Jeffamine (registered trademark) ED-2003, Jeffamine (registered trademark) ) EDR-148, Jeffamine (registered trademark) EDR-176, D-200, D-400, D-2000, D-4000 (manufactured by HUNTSMAN), 1- (2- (2- (2-aminopropoxy)) Examples include, but are not limited to, ethoxy) propoxy) propan-2-amine, 1- (1- (1- (2-aminopropoxy) propan-2-yl) oxy) propan-2-amine.
Jeffamine (registered trademark) KH-511, Jeffamine (registered trademark) ED-600, Jeffamine (registered trademark) ED-900, Jeffamine (registered trademark) ED-2003, Jeffamine (registered trademark) EDR-148, The structure of Jeffamine® EDR-176 is shown below.


R50~R57の1価の有機基として、炭素数1~10(好ましくは炭素数1~6)の無置換のアルキル基、炭素数1~10(好ましくは炭素数1~6)のフッ化アルキル基等が挙げられる。

Examples of the monovalent organic group represented by R 50 to R 57 include an unsubstituted alkyl group having 1 to 10 (preferably 1 to 6) carbon atoms and a fluorine atom having 1 to 10 (preferably 1 to 6) carbon atoms. Alkyl group and the like.
式(1)におけるR115は、4価の有機基を表す。4価の有機基としては、芳香環を含む基であることが好ましく、下記式(5)または式(6)で表される基がより好ましい。

R 115 in the formula (1) represents a tetravalent organic group. The tetravalent organic group is preferably a group containing an aromatic ring, and more preferably a group represented by the following formula (5) or (6).


式(1)におけるR113およびR114は、それぞれ独立に、水素原子または1価の有機基を表す。R113およびR114の少なくとも一方がラジカル重合性基を含むことが好ましく、両方がラジカル重合性基を含むことがより好ましい。ラジカル重合性基としては、ラジカルの作用により、架橋反応することが可能な基であって、好ましい例として、エチレン性不飽和結合を有する基が挙げられる。エチレン性不飽和結合を有する基としては、ビニル基、アリル基、(メタ)アクリロイル基、下記式(III)で表される基などが挙げられる。 <<<< R 113 and R 114 >>>
R 113 and R 114 in the formula (1) each independently represent a hydrogen atom or a monovalent organic group. Preferably, at least one of R 113 and R 114 contains a radically polymerizable group, and more preferably both contain a radically polymerizable group. The radical polymerizable group is a group capable of undergoing a cross-linking reaction by the action of a radical, and a preferable example is a group having an ethylenically unsaturated bond. Examples of the group having an ethylenically unsaturated bond include a vinyl group, an allyl group, a (meth) acryloyl group, and a group represented by the following formula (III).

式(III)において、R201は、炭素数2~12のアルキレン基、-CH2CH(OH)CH2-または炭素数4~30の(ポリ)オキシアルキレン基(アルキレン基としては炭素数1~12が好ましく、1~6がより好ましく、1~3が特に好ましい;繰り返し数は1~12が好ましく、1~6がより好ましく、1~3が特に好ましい)を表す。なお、(ポリ)オキシアルキレン基とは、オキシアルキレン基またはポリオキシアルキレン基を意味する。
好適なR201の例は、エチレン基、プロピレン基、トリメチレン基、テトラメチレン基、1,2-ブタンジイル基、1,3-ブタンジイル基、ペンタメチレン基、ヘキサメチレン基、オクタメチレン基、ドデカメチレン基、-CH2CH(OH)CH2-が挙げられ、エチレン基、プロピレン基、トリメチレン基、-CH2CH(OH)CH2-がより好ましい。
特に好ましくは、R200がメチル基で、R201がエチレン基である。 In the formula (III), R 200 represents a hydrogen atom or a methyl group, and a methyl group is more preferable.
In the formula (III), R 201 is an alkylene group having 2 to 12 carbon atoms, —CH 2 CH (OH) CH 2 — or a (poly) oxyalkylene group having 4 to 30 carbon atoms (an alkylene group having 1 carbon atom To 12, preferably 1 to 6, more preferably 1 to 3, and the number of repetitions is preferably 1 to 12, more preferably 1 to 6, and particularly preferably 1 to 3.) In addition, a (poly) oxyalkylene group means an oxyalkylene group or a polyoxyalkylene group.
Examples of suitable R201 include ethylene, propylene, trimethylene, tetramethylene, 1,2-butanediyl, 1,3-butanediyl, pentamethylene, hexamethylene, octamethylene, dodecamethylene. , —CH 2 CH (OH) CH 2 —, and more preferably an ethylene group, a propylene group, a trimethylene group, and —CH 2 CH (OH) CH 2 —.
Particularly preferably, R 200 is a methyl group and R 201 is an ethylene group.
R113またはR114が表す1価の有機基としては、現像液の溶解度を向上させる置換基が好ましく用いられる。
R113またはR114が、水素原子、2-ヒドロキシベンジル、3-ヒドロキシベンジルおよび4-ヒドロキシベンジルであることが、水性現像液に対する溶解性の点からは、より好ましい。 As a preferred embodiment of the polyimide precursor in the present invention, as a monovalent organic group of R 113 or R 114 , an aliphatic group, an aromatic group, and an aryl group having one, two or three, preferably one acid group And an alkyl group. Specific examples include an aromatic group having 6 to 20 carbon atoms having an acid group and an arylalkyl group having 7 to 25 carbon atoms having an acid group. More specifically, a phenyl group having an acid group and a benzyl group having an acid group are exemplified. The acid group is preferably a hydroxyl group. That is, R 113 or R 114 is preferably a group having a hydroxyl group.
As the monovalent organic group represented by R 113 or R 114, a substituent that improves the solubility of a developer is preferably used.
R 113 or R 114 is more preferably a hydrogen atom, 2-hydroxybenzyl, 3-hydroxybenzyl or 4-hydroxybenzyl from the viewpoint of solubility in an aqueous developer.
アルキル基の炭素数は1~30が好ましい(環状の場合は3以上)。アルキル基は直鎖、分岐、環状のいずれであってもよい。直鎖または分岐のアルキル基としては、例えば、メチル基、エチル基、プロピル基、ブチル基、ペンチル基、ヘキシル基、ヘプチル基、オクチル基、ノニル基、デシル基、ドデシル基、テトラデシル基、オクタデシル基、イソプロピル基、イソブチル基、sec-ブチル基、t-ブチル基、1-エチルペンチル基、および2-エチルヘキシル基が挙げられる。環状のアルキル基は、単環の環状のアルキル基であってもよく、多環の環状のアルキル基であってもよい。単環の環状のアルキル基としては、例えば、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基およびシクロオクチル基が挙げられる。多環の環状のアルキル基としては、例えば、アダマンチル基、ノルボルニル基、ボルニル基、カンフェニル基、デカヒドロナフチル基、トリシクロデカニル基、テトラシクロデカニル基、カンホロイル基、ジシクロヘキシル基およびピネニル基が挙げられる。また、芳香族基で置換されたアルキル基としては、次に述べる芳香族基で置換された直鎖アルキル基が好ましい。 From the viewpoint of solubility in an organic solvent, R 113 or R 114 is preferably a monovalent organic group. The monovalent organic group preferably contains a linear or branched alkyl group, a cyclic alkyl group, or an aromatic group, and more preferably an alkyl group substituted with an aromatic group.
The alkyl group preferably has 1 to 30 carbon atoms (3 or more in the case of a cyclic group). The alkyl group may be linear, branched or cyclic. Examples of the linear or branched alkyl group include a methyl group, an ethyl group, a propyl group, a butyl group, a pentyl group, a hexyl group, a heptyl group, an octyl group, a nonyl group, a decyl group, a dodecyl group, a tetradecyl group, and an octadecyl group. Isopropyl group, isobutyl group, sec-butyl group, t-butyl group, 1-ethylpentyl group, and 2-ethylhexyl group. The cyclic alkyl group may be a monocyclic alkyl group or a polycyclic alkyl group. Examples of the monocyclic alkyl group include a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, a cycloheptyl group, and a cyclooctyl group. Examples of the polycyclic alkyl group include, for example, an adamantyl group, a norbornyl group, a bornyl group, a camphenyl group, a decahydronaphthyl group, a tricyclodecanyl group, a tetracyclodecanyl group, a camphoroyl group, a dicyclohexyl group, and a pinenyl group. Is mentioned. Further, as the alkyl group substituted with an aromatic group, a linear alkyl group substituted with an aromatic group described below is preferable.

R112の好ましい範囲は、式(5)におけるR112と同義であり、中でも酸素原子であることがより好ましい。
式中のカルボニル基のベンゼン環への結合位置は、式(1-A)において、4,5,3’,4’であることが好ましい。式(1-B)においては、1,2,4,5であることが好ましい。 The preferred ranges of A 11 , A 12 , R 111 , R 113 and R 114 are each independently the same as the preferred ranges of A 1 , A 2 , R 111 , R 113 and R 114 in the formula (1). .
A preferred range of R 112 has the same meaning as R 112 in formula (5), and more preferably among others oxygen atoms.
The bonding position of the carbonyl group in the formula to the benzene ring is preferably 4, 5, 3 ', 4' in formula (1-A). In the formula (1-B), 1,2,4,5 is preferable.
ポリイミド前駆体の分子量の分散度は、1.5~3.5が好ましく、2~3がより好ましい。 The weight average molecular weight (Mw) of the polyimide precursor is preferably from 2,000 to 500,000, more preferably from 5,000 to 100,000, and further preferably from 10,000 to 50,000. The number average molecular weight (Mn) is preferably from 800 to 250,000, more preferably from 2,000 to 50,000, and still more preferably from 4,000 to 25,000.
The degree of dispersion of the molecular weight of the polyimide precursor is preferably from 1.5 to 3.5, more preferably from 2 to 3.
ポリイミド前駆体の製造方法では、反応に際し、有機溶剤を用いることが好ましい。有機溶剤は1種でもよいし、2種以上でもよい。
有機溶剤としては、原料に応じて適宜定めることができるが、ピリジン、ジエチレングリコールジメチルエーテル(ジグリム)、N-メチルピロリドンおよびN-エチルピロリドンが例示される。 The polyimide precursor can be obtained by reacting a dicarboxylic acid or a dicarboxylic acid derivative with a diamine. Preferably, it is obtained by halogenating a dicarboxylic acid or a dicarboxylic acid derivative with a halogenating agent and then reacting the dicarboxylic acid or dicarboxylic acid derivative with a diamine.
In the method for producing a polyimide precursor, it is preferable to use an organic solvent during the reaction. The organic solvent may be one type or two or more types.
The organic solvent can be appropriately determined according to the raw material, and examples thereof include pyridine, diethylene glycol dimethyl ether (diglyme), N-methylpyrrolidone and N-ethylpyrrolidone.
本発明の樹脂組成物は、ポリイミド前駆体を1種のみ含んでいてもよいし、2種以上含んでいてもよい。2種以上含む場合、合計量が上記範囲となることが好ましい。 The content of the polyimide precursor in the resin composition of the present invention is preferably 20% by mass or more, more preferably 30% by mass or more, and preferably 40% by mass or more based on the total solid content of the resin composition. Is more preferably 50% by mass or more, still more preferably 60% by mass or more, and even more preferably 70% by mass or more. Further, the content of the polyimide precursor in the resin composition of the present invention is preferably 99.5% by mass or less, more preferably 99% by mass or less, based on the total solid content of the resin composition. , 98% by mass or less, more preferably 95% by mass or less.
The resin composition of the present invention may include only one kind of the polyimide precursor, or may include two or more kinds of the polyimide precursor. When two or more kinds are included, the total amount is preferably in the above range.
本発明の樹脂組成物は、熱塩基発生剤を含む。熱塩基発生剤としては、その種類等は特に定めるものではないが、40℃以上に加熱すると塩基を発生する酸性化合物、および、pKa1が0~4のアニオンとアンモニウムカチオンとを有するアンモニウム塩から選ばれる少なくとも1種を含む熱塩基発生剤を含むことが好ましい。ここで、pKa1とは、酸の第一のプロトンの解離定数(Ka)の対数(-Log10Ka)を表し、詳細は後述する。
このような化合物を配合することにより、ポリイミド前駆体の環化反応を低温で行うことができる。また、熱塩基発生剤は、加熱しなければ塩基を発生しないので、ポリマー前駆体と共存させても、保存中におけるポリマー前駆体の環化を抑制でき、保存安定性に優れている。 <Heat base generator>
The resin composition of the present invention contains a thermal base generator. The type of the thermal base generator is not particularly limited, but is selected from an acidic compound which generates a base when heated to 40 ° C. or more, and an ammonium salt having an anion having an pKa of 0 to 4 and an ammonium cation. It is preferable to include a thermal base generator containing at least one of these. Here, pKa1 represents the logarithm (−Log 10 Ka) of the dissociation constant (Ka) of the first proton of the acid, which will be described in detail later.
By compounding such a compound, the cyclization reaction of the polyimide precursor can be performed at a low temperature. Further, since the thermal base generator does not generate a base unless heated, it can suppress cyclization of the polymer precursor during storage even when it is present together with the polymer precursor, and is excellent in storage stability.
式(101) 式(102)

Equation (101) Equation (102)

式(Y1-1)~(Y1-5)において、Ar101およびAr102は、それぞれ独立に、アリール基を表し、nは、1以上の整数を表し、mは、0~5の整数を表す。 In Formulas (Y1-1) to (Y1-5), R 101 represents an n-valent organic group, and R 1 and R 7 have the same meaning as in Formula (101) or Formula (102).
In the formulas (Y1-1) to (Y1-5), Ar 101 and Ar 102 each independently represent an aryl group, n represents an integer of 1 or more, and m represents an integer of 0 to 5. .
アニオンの種類は、カルボン酸アニオン、フェノールアニオン、リン酸アニオンおよび硫酸アニオンから選ばれる1種が好ましく、塩の安定性と熱分解性を両立させられるという理由からカルボン酸アニオンがより好ましい。すなわち、アンモニウム塩は、アンモニウムカチオンとカルボン酸アニオンとの塩がより好ましい。
カルボン酸アニオンは、2個以上のカルボキシル基を持つ2価以上のカルボン酸のアニオンが好ましく、2価のカルボン酸のアニオンがより好ましい。この態様によれば、樹脂組成物の安定性、硬化性および現像性をより向上できる熱塩基発生剤とすることができる。特に、2価のカルボン酸のアニオンを用いることで、樹脂組成物の安定性、硬化性および現像性をさらに向上できる。
本実施形態において、カルボン酸アニオンは、pKa1が4以下のカルボン酸のアニオンであることが好ましい。pKa1は、3.5以下がより好ましく、3.2以下が一層好ましい。この態様によれば、樹脂組成物の安定性をより向上できる。
ここでpKa1とは、酸の第一のプロトンの解離定数の逆数の対数を表し、Determination of Organic Structures by Physical Methods(著者:Brown, H. C., McDaniel, D. H., Hafliger, O., Nachod, F. C.; 編纂:Braude, E. A., Nachod, F. C.; Academic Press, New York, 1955)や、Data for Biochemical Research(著者:Dawson, R.M.C.et al; Oxford, Clarendon Press, 1959)に記載の値を参照することができる。これらの文献に記載の無い化合物については、ACD/pKa(ACD/Labs製)のソフトを用いて構造式より算出した値を用いることとする。 In the present embodiment, the ammonium salt preferably has an anion having a pKa of 0 to 4 and an ammonium cation. The upper limit of the pKa1 of the anion is more preferably 3.5 or less, even more preferably 3.2 or less. The lower limit is preferably 0.5 or more, and more preferably 1.0 or more. When the pKa1 of the anion is in the above range, the polymer precursor can be cyclized at a lower temperature, and further, the stability of the resin composition can be improved. When the pKa1 is 4 or less, the stability of the thermal base generator is good, the generation of a base without heating can be suppressed, and the stability of the resin composition is good. When pKa1 is 0 or more, the generated base is difficult to be neutralized, and the cyclization efficiency of the polymer precursor is good.
The type of the anion is preferably one selected from a carboxylate anion, a phenol anion, a phosphate anion and a sulfate anion, and more preferably a carboxylate anion because both salt stability and thermal decomposability can be achieved. That is, the ammonium salt is more preferably a salt of an ammonium cation and a carboxylate anion.
The carboxylate anion is preferably a divalent or higher carboxylic acid anion having two or more carboxyl groups, and more preferably a divalent carboxylic acid anion. According to this aspect, it is possible to provide a thermal base generator that can further improve the stability, curability, and developability of the resin composition. In particular, by using an anion of a divalent carboxylic acid, the stability, curability and developability of the resin composition can be further improved.
In the present embodiment, the carboxylate anion is preferably a carboxylate anion having a pKa of 4 or less. pKa1 is more preferably 3.5 or less, even more preferably 3.2 or less. According to this aspect, the stability of the resin composition can be further improved.
Here, pKa1 represents the logarithm of the reciprocal of the dissociation constant of the first proton of the acid and is determined by Organic Structures by Physical Methods (author: Brown, HC, McDaniel, DH, Hafliger, Hafliger). Compiled by: Braude, EA, Nachod, FC; Academic Press, New York, 1955), and Data for Biochemical Research (author: Dawson, R., R.M. al; Oxford, Clarendon Press, 1959). For compounds not described in these documents, values calculated from the structural formula using ACD / pKa (manufactured by ACD / Labs) software will be used.

σmが正の値を示す置換基の例としては、CF3基(σm=0.43)、CF3CO基(σm=0.63)、HC≡C基(σm=0.21)、CH2=CH基(σm=0.06)、Ac基(σm=0.38)、MeOCO基(σm=0.37)、MeCOCH=CH基(σm=0.21)、PhCO基(σm=0.34)、H2NCOCH2基(σm=0.06)などが挙げられる。なお、Meはメチル基を表し、Acはアセチル基を表し、Phはフェニル基を表す。 In the present embodiment, the electron withdrawing group means a group having a positive Hammett's substituent constant σm. Here, σm is described by Yuno Tsuno, Synthetic Organic Chemistry Society, Vol. 631-642. Note that the electron-withdrawing group in the present embodiment is not limited to the substituents described in the above documents.
Examples of the substituent having a positive value of σm include a CF 3 group (σm = 0.43), a CF 3 CO group (σm = 0.63), an HC≡C group (σm = 0.21), and CH 2 = CH group (σm = 0.06), Ac group (σm = 0.38), MeOCO group (σm = 0.37), MeCOCH = CH group (σm = 0.21), PhCO group (σm = 0 .34), H 2 NCOCH 2 group (σm = 0.06) and the like. Note that Me represents a methyl group, Ac represents an acetyl group, and Ph represents a phenyl group.

式(XA)

Formula (XA)
ノニオン系熱塩基発生剤(A3)としては、式(B1)または式(B2)で表される化合物が挙げられる。

Examples of the nonionic thermal base generator (A3) include a compound represented by the formula (B1) or the formula (B2).

Rb13はアルキル基(炭素数1~24が好ましく、2~18がより好ましく、3~12がさらに好ましい)、アルケニル基(炭素数2~24が好ましく、2~18がより好ましく、3~12がさらに好ましい)、アリール基(炭素数6~22が好ましく、6~18がより好ましく、6~12がさらに好ましい)、アリールアルキル基(炭素数7~23が好ましく、7~19がより好ましく、7~12がさらに好ましい)であり、本発明の効果を奏する範囲で置換基を有していてもよい。なかでも、Rb13はアリールアルキル基が好ましい。 In the formula, Rb 11 and Rb 12 , and Rb 31 and Rb 32 are the same as Rb 1 and Rb 2 in formula (B1), respectively.
Rb 13 is an alkyl group (preferably having 1 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, still more preferably 3 to 12 carbon atoms), and an alkenyl group (preferably having 2 to 24 carbon atoms, more preferably 2 to 18 carbon atoms, and 3 to 12 carbon atoms. ), An aryl group (preferably having 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, still more preferably 6 to 12), and an arylalkyl group (preferably having 7 to 23 carbon atoms, more preferably 7 to 19, 7 to 12 are more preferable), and may have a substituent within a range in which the effects of the present invention are exhibited. Above all, Rb 13 is preferably an arylalkyl group.

Rb15およびRb16は水素原子、アルキル基(炭素数1~12が好ましく、1~6がより好ましく、1~3がさらに好ましい)、アルケニル基(炭素数2~12が好ましく、2~6がより好ましく、2~3がさらに好ましい)、アリール基(炭素数6~22が好ましく、6~18がより好ましく、6~10がさらに好ましい)、アリールアルキル基(炭素数7~23が好ましく、7~19がより好ましく、7~11がさらに好ましい)であり、水素原子またはメチル基が好ましい。
Rb17はアルキル基(炭素数1~24が好ましく、1~12がより好ましく、3~8がさらに好ましい)、アルケニル基(炭素数2~12が好ましく、3~8がより好ましい)、アリール基(炭素数6~22が好ましく、6~18がより好ましく、6~12がさらに好ましい)、アリールアルキル基(炭素数7~23が好ましく、7~19がより好ましく、7~12がさらに好ましい)であり、なかでもアリール基が好ましい。 Rb 11 and Rb 12 have the same meanings as Rb 11 and Rb 12 in the formula (B1-1).
Rb 15 and Rb 16 each represent a hydrogen atom, an alkyl group (preferably having 1 to 12 carbon atoms, more preferably 1 to 6), and an alkenyl group (preferably having 2 to 12 carbon atoms, preferably having 2 to 6 carbon atoms). More preferably, 2 to 3 are more preferable, an aryl group (preferably having 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, still more preferably 6 to 10), and an arylalkyl group (preferably having 7 to 23 carbon atoms, and 7 To 19 are more preferable, and 7 to 11 are more preferable), and a hydrogen atom or a methyl group is preferable.
Rb 17 is an alkyl group (preferably having 1 to 24 carbon atoms, more preferably 1 to 12 carbon atoms, still more preferably 3 to 8), an alkenyl group (preferably having 2 to 12 carbon atoms, more preferably 3 to 8), and an aryl group. (Preferably having 6 to 22 carbon atoms, more preferably 6 to 18 carbon atoms, and still more preferably 6 to 12), and an arylalkyl group (preferably having 7 to 23 carbon atoms, more preferably 7 to 19, and still more preferably 7 to 12). And among them, an aryl group is preferable.




本発明の樹脂組成物は、エポキシ基、オキセタニル基、メチロール基、アルコキシメチル基、フェノール基、マレイミド基、シアネート基及びブロックイソシアネート基からなる群から選択される官能基を複数有する熱硬化性化合物を含有する。本発明において、熱硬化性化合物とは、加熱によって硬化する化合物を意味する。また、ブロックイソシアネート基とは、熱によりイソシアネート基を生成することが可能な基のことであり、例えば、ブロック剤とイソシアネート基とを反応させイソシアネート基を保護した基が好ましく例示できる。 <Thermosetting compound>
The resin composition of the present invention is a thermosetting compound having a plurality of functional groups selected from the group consisting of an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group and a blocked isocyanate group. contains. In the present invention, the thermosetting compound means a compound which is cured by heating. The blocked isocyanate group is a group capable of generating an isocyanate group by heat, and for example, a group obtained by reacting a blocking agent with an isocyanate group to protect the isocyanate group can be preferably exemplified.
X1-(Y1)n ・・・(TC1)
式(TC1)中、X1はn価の連結基を表し、Y1はエポキシ基、オキセタニル基、メチロール基、アルコキシメチル基、フェノール基、マレイミド基、シアネート基またはブロックイソシアネート基を表し、nは2以上の整数を表す。 The thermosetting compound used in the present invention is preferably a compound represented by the formula (TC1).
X 1- (Y 1 ) n ... (TC1)
In the formula (TC1), X 1 represents an n-valent linking group, Y 1 represents an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group or a blocked isocyanate group, and n is Represents an integer of 2 or more.

式(AM-102)中、Rm5~Rm8は、それぞれ独立して水素原子、または、式(Rm)で表される基を表す。ただし、Rm5~Rm8のうち2以上は式(Rm)で表される基である。
式(AM-103)中、Rm9およびRm10は、それぞれ独立して式(Rm)で表される基を表す。
式(AM-104)中、Rm11~Rm16は、それぞれ独立して水素原子、または、式(Rm)で表される基を表す。ただし、Rm11~Rm16のうち2以上は式(Rm)で表される基である。
式(AM-105)中、Rm17~Rm20は、それぞれ独立して水素原子、または、式(Rm)で表される基を表す。ただし、Rm17~Rm20のうち2以上は式(Rm)で表される基である。 In Formula (AM-101), Rm 1 to Rm 4 each independently represent a hydrogen atom or a group represented by Formula (Rm). However, two or more of Rm 1 to Rm 4 are groups represented by the formula (Rm).
In Formula (AM-102), Rm 5 to Rm 8 each independently represent a hydrogen atom or a group represented by Formula (Rm). However, two or more of Rm 5 to Rm 8 are groups represented by the formula (Rm).
In Formula (AM-103), Rm 9 and Rm 10 each independently represent a group represented by Formula (Rm).
In Formula (AM-104), Rm 11 to Rm 16 each independently represent a hydrogen atom or a group represented by Formula (Rm). However, two or more of Rm 11 to Rm 16 are groups represented by the formula (Rm).
In Formula (AM-105), Rm 17 to Rm 20 each independently represent a hydrogen atom or a group represented by Formula (Rm). However, two or more of Rm 17 to Rm 20 are groups represented by the formula (Rm).
-CH2-O-Rm100
式(Rm)中、Rm100は、水素原子またはアルキル基を表す。Rm100が表すアルキル基の炭素数は、1~30であることが好ましく、1~15であることがより好ましく、1~8であることが更に好ましく、1~3であることが特に好ましく、1であることが最も好ましい。すなわち、アルコキシメチル基は、メトキシメチル基であることが最も好ましい。 (Formula (Rm))
—CH 2 —O—Rm 100
In the formula (Rm), Rm 100 represents a hydrogen atom or an alkyl group. The number of carbon atoms of the alkyl group represented by Rm 100 is preferably 1 to 30, more preferably 1 to 15, still more preferably 1 to 8, and particularly preferably 1 to 3, Most preferably, it is 1. That is, the alkoxymethyl group is most preferably a methoxymethyl group.

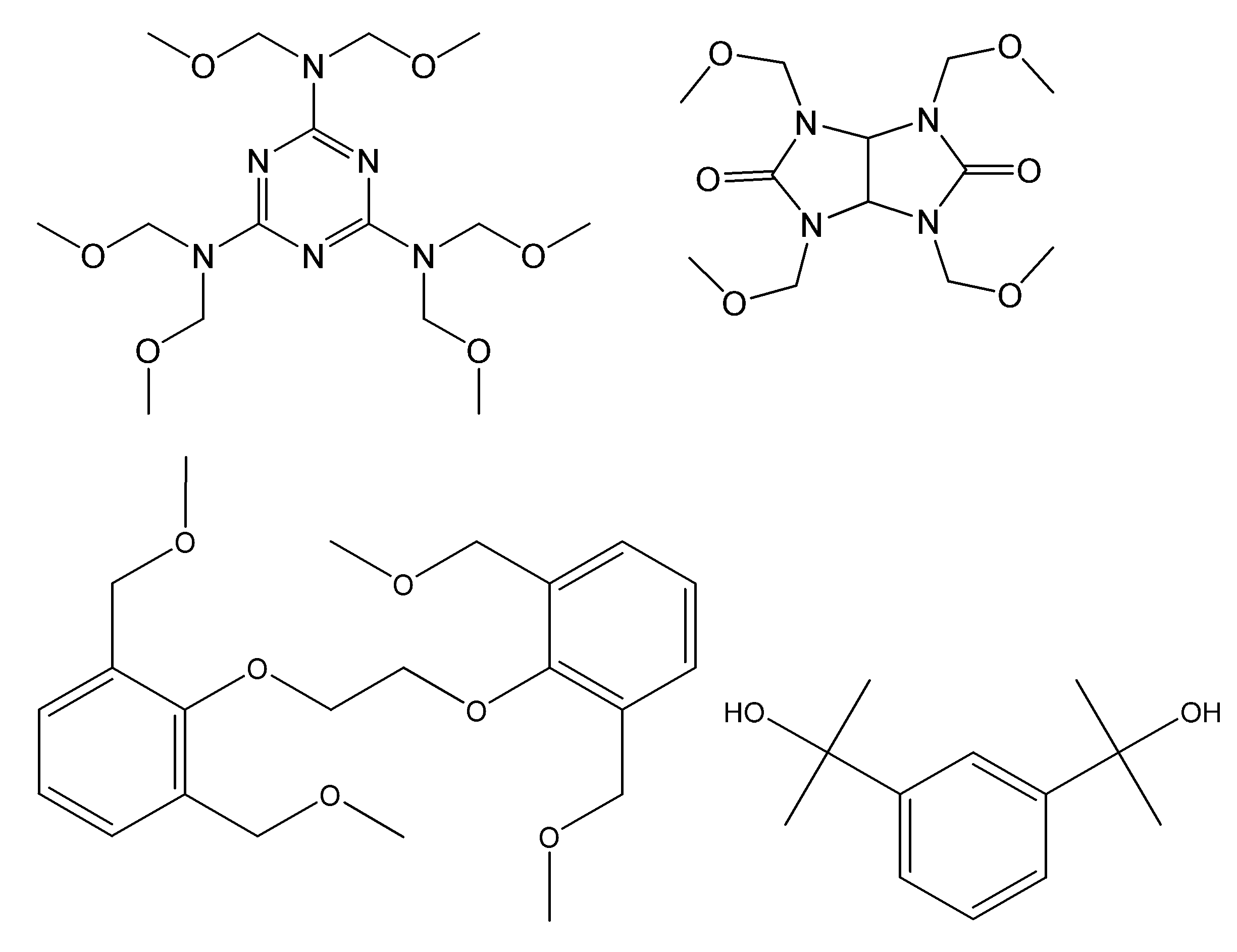

また、熱硬化性化合物の含有量は、ポリイミド前駆体の100質量部に対して1~25質量部であることが好ましい。上限は23質量部以下が好ましく、20質量部以下がより好ましい。下限は、4質量部以上が好ましく、6質量部以上がより好ましい。熱硬化性化合物の含有量がポリイミド前駆体の100質量部に対して6質量部以上であれば薬品耐性に優れた硬化膜が得られやすい。また、熱硬化性化合物の含有量がポリイミド前駆体の100質量部に対して20質量部以下であれば基板密着性に優れた硬化膜が得られやすい。
また、熱硬化性化合物の含有量は、熱塩基発生剤の100質量部に対して10~1000質量部であることが好ましい。上限は800質量部以下が好ましく、600質量部以下がより好ましい。下限は、20質量部以上が好ましく、50質量部以上がより好ましい。
熱硬化性化合物は1種を単独で用いてもよいが、2種以上を混合して用いてもよい。2種以上を併用する場合にはその合計量が上記の範囲となることが好ましい。 The content of the thermosetting compound is preferably 1 to 20% by mass based on the total solid content of the resin composition of the present invention. The lower limit is preferably 3% by mass or more, more preferably 5% by mass or more. The upper limit is preferably at most 18% by mass, more preferably at most 15% by mass.
Further, the content of the thermosetting compound is preferably 1 to 25 parts by mass with respect to 100 parts by mass of the polyimide precursor. The upper limit is preferably at most 23 parts by mass, more preferably at most 20 parts by mass. The lower limit is preferably 4 parts by mass or more, more preferably 6 parts by mass or more. When the content of the thermosetting compound is 6 parts by mass or more with respect to 100 parts by mass of the polyimide precursor, a cured film having excellent chemical resistance is easily obtained. When the content of the thermosetting compound is 20 parts by mass or less based on 100 parts by mass of the polyimide precursor, a cured film having excellent substrate adhesion is easily obtained.
Further, the content of the thermosetting compound is preferably 10 to 1000 parts by mass with respect to 100 parts by mass of the thermal base generator. The upper limit is preferably 800 parts by mass or less, more preferably 600 parts by mass or less. The lower limit is preferably at least 20 parts by mass, more preferably at least 50 parts by mass.
One thermosetting compound may be used alone, or two or more thermosetting compounds may be used in combination. When two or more kinds are used in combination, the total amount is preferably within the above range.
本発明の樹脂組成物は、光重合開始剤を含有することが好ましい。光ラジカル重合開始剤であることが好ましい。光ラジカル重合開始剤としては、特に制限はなく、公知の光ラジカル重合開始剤の中から適宜選択することができる。例えば、紫外線領域から可視領域の光線に対して感光性を有する光ラジカル重合開始剤が好ましい。また、光励起された増感剤と何らかの作用を生じ、活性ラジカルを生成する活性剤であってもよい。
光ラジカル重合開始剤は、約300~800nm(好ましくは330~500nm)の範囲内で少なくとも約50のモル吸光係数を有する化合物を、少なくとも1種含有していることが好ましい。化合物のモル吸光係数は、公知の方法を用いて測定することができる。例えば、紫外可視分光光度計(Varian社製Cary-5 spectrophotometer)にて、酢酸エチル溶剤を用い、0.01g/Lの濃度で測定することが好ましい。 <Photopolymerization initiator>
The resin composition of the present invention preferably contains a photopolymerization initiator. It is preferably a photo-radical polymerization initiator. The photo-radical polymerization initiator is not particularly limited, and can be appropriately selected from known photo-radical polymerization initiators. For example, a photo-radical polymerization initiator having photosensitivity to light in the ultraviolet region to the visible region is preferable. In addition, an activator that produces some action with the photoexcited sensitizer and generates an active radical may be used.
The photo-radical polymerization initiator preferably contains at least one compound having a molar extinction coefficient of at least about 50 in the range of about 300 to 800 nm (preferably 330 to 500 nm). The molar extinction coefficient of a compound can be measured using a known method. For example, it is preferable to measure with an ultraviolet-visible spectrophotometer (Cary-5 spectrophotometer manufactured by Varian) using an ethyl acetate solvent at a concentration of 0.01 g / L.
ヒドロキシアセトフェノン系開始剤としては、IRGACURE 184(IRGACUREは登録商標)、DAROCUR 1173、IRGACURE 500、IRGACURE-2959、IRGACURE 127(商品名:いずれもBASF社製)を用いることができる。
アミノアセトフェノン系開始剤としては、市販品であるIRGACURE 907、IRGACURE 369、および、IRGACURE 379(商品名:いずれもBASF社製)を用いることができる。
アミノアセトフェノン系開始剤として、365nmまたは405nm等の波長光源に吸収極大波長がマッチングされた特開2009-191179号公報に記載の化合物も用いることができる。
アシルホスフィン系開始剤としては、2,4,6-トリメチルベンゾイル-ジフェニル-ホスフィンオキサイドなどが挙げられる。また、市販品であるIRGACURE-819やIRGACURE-TPO(商品名:いずれもBASF社製)を用いることができる。
メタロセン化合物としては、IRGACURE-784(BASF社製)などが例示される。 As the photoradical polymerization initiator, a hydroxyacetophenone compound, an aminoacetophenone compound, and an acylphosphine compound can also be suitably used. More specifically, for example, an aminoacetophenone-based initiator described in JP-A-10-291969 and an acylphosphine oxide-based initiator described in Patent No. 4225988 can also be used.
As the hydroxyacetophenone-based initiator, IRGACURE 184 (IRGACURE is a registered trademark), DAROCUR 1173, IRGACURE 500, IRGACURE-2959, IRGACURE 127 (trade name: all manufactured by BASF) can be used.
As the aminoacetophenone-based initiator, commercially available products IRGACURE 907, IRGACURE 369, and IRGACURE 379 (trade names: all manufactured by BASF) can be used.
As the aminoacetophenone-based initiator, a compound described in JP-A-2009-191179 in which the absorption maximum wavelength is matched to a light source having a wavelength of 365 nm or 405 nm can also be used.
Examples of the acylphosphine initiator include 2,4,6-trimethylbenzoyl-diphenyl-phosphine oxide. Further, commercially available products such as IRGACURE-819 and IRGACURE-TPO (trade names, both manufactured by BASF) can be used.
Examples of the metallocene compound include IRGACURE-784 (manufactured by BASF).
オキシム化合物の具体例としては、特開2001-233842号公報に記載の化合物、特開2000-080068号公報に記載の化合物、特開2006-342166号公報に記載の化合物を用いることができる。
好ましいオキシム化合物としては、例えば、下記の構造の化合物や、3-ベンゾオキシイミノブタン-2-オン、3-アセトキシイミノブタン-2-オン、3-プロピオニルオキシイミノブタン-2-オン、2-アセトキシイミノペンタン-3-オン、2-アセトキシイミノ-1-フェニルプロパン-1-オン、2-ベンゾイルオキシイミノ-1-フェニルプロパン-1-オン、3-(4-トルエンスルホニルオキシ)イミノブタン-2-オン、および2-エトキシカルボニルオキシイミノ-1-フェニルプロパン-1-オンなどが挙げられる。本発明の樹脂組成物においては、特に光ラジカル重合開始剤としてオキシム化合物(オキシム系の光重合開始剤)を用いることが好ましい。オキシム系の光重合開始剤は、分子内に >C=N-O-C(=O)- の連結基を有する。

さらに、また、フッ素原子を有するオキシム化合物を用いることも可能である。そのようなオキシム化合物の具体例としては、特開2010-262028号公報に記載されている化合物、特表2014-500852号公報の段落0345に記載されている化合物24、36~40、特開2013-164471号公報の段落0101に記載されている化合物(C-3)などが挙げられる。
最も好ましいオキシム化合物としては、特開2007-269779号公報に示される特定置換基を有するオキシム化合物や、特開2009-191061号公報に示されるチオアリール基を有するオキシム化合物などが挙げられる。 An oxime compound is more preferably used as the photoradical polymerization initiator. By using the oxime compound, the exposure latitude can be more effectively improved. Oxime compounds are particularly preferred because they have a wide exposure latitude (exposure margin) and also act as photocuring accelerators.
As specific examples of the oxime compound, compounds described in JP-A-2001-233842, compounds described in JP-A-2000-080068, and compounds described in JP-A-2006-342166 can be used.
Preferred oxime compounds include, for example, compounds having the following structures, 3-benzooxyiminobutan-2-one, 3-acetoxyiminobutan-2-one, 3-propionyloxyimiminobtan-2-one, 2-acetoxy Iminopentan-3-one, 2-acetoxyimino-1-phenylpropan-1-one, 2-benzoyloxyimino-1-phenylpropan-1-one, 3- (4-toluenesulfonyloxy) iminobutan-2-one And 2-ethoxycarbonyloxyimino-1-phenylpropan-1-one. In the resin composition of the present invention, it is particularly preferable to use an oxime compound (oxime-based photopolymerization initiator) as the photoradical polymerization initiator. The oxime-based photopolymerization initiator has a> C = NOC (= O)-linking group in the molecule.
Furthermore, it is also possible to use an oxime compound having a fluorine atom. Specific examples of such oxime compounds include compounds described in JP-A-2010-262028, compounds 24 and 36 to 40 described in paragraph 0345 of JP-A-2014-500852, and JP-A-2013-2013. Compound (C-3) described in paragraph 0101 of JP-A-164471.
The most preferred oxime compounds include oxime compounds having a specific substituent described in JP-A-2007-269779, oxime compounds having a thioaryl group described in JP-A-2009-191061, and the like.
さらに好ましい光ラジカル重合開始剤は、トリハロメチルトリアジン化合物、α-アミノケトン化合物、アシルホスフィン化合物、ホスフィンオキサイド化合物、メタロセン化合物、オキシム化合物、トリアリールイミダゾールダイマー、オニウム塩化合物、ベンゾフェノン化合物、アセトフェノン化合物であり、トリハロメチルトリアジン化合物、α-アミノケトン化合物、オキシム化合物、トリアリールイミダゾールダイマー、ベンゾフェノン化合物からなる群より選ばれる少なくとも1種の化合物が一層好ましく、メタロセン化合物またはオキシム化合物を用いるのがより一層好ましく、オキシム化合物がさらに一層好ましい。
また、光ラジカル重合開始剤は、ベンゾフェノン、N,N’-テトラメチル-4,4’-ジアミノベンゾフェノン(ミヒラーケトン)等のN,N’-テトラアルキル-4,4’-ジアミノベンゾフェノン、2-ベンジル-2-ジメチルアミノ-1-(4-モルホリノフェニル)-ブタノン-1,2-メチル-1-[4-(メチルチオ)フェニル]-2-モルホリノ-プロパノン-1等の芳香族ケトン、アルキルアントラキノン等の芳香環と縮環したキノン類、ベンゾインアルキルエーテル等のベンゾインエーテル化合物、ベンゾイン、アルキルベンゾイン等のベンゾイン化合物、ベンジルジメチルケタール等のベンジル誘導体などを用いることもできる。また、下記式(I)で表される化合物を用いることもできる。


More preferred photoradical polymerization initiators are trihalomethyltriazine compounds, α-aminoketone compounds, acylphosphine compounds, phosphine oxide compounds, metallocene compounds, oxime compounds, triarylimidazole dimers, onium salt compounds, benzophenone compounds, and acetophenone compounds. At least one compound selected from the group consisting of a trihalomethyltriazine compound, an α-aminoketone compound, an oxime compound, a triarylimidazole dimer, and a benzophenone compound is more preferable, and a metallocene compound or an oxime compound is more preferably used. Is even more preferred.
The photo-radical polymerization initiators include N, N'-tetraalkyl-4,4'-diaminobenzophenone such as benzophenone, N, N'-tetramethyl-4,4'-diaminobenzophenone (Michler's ketone), 2-benzyl Aromatic ketones such as -2-dimethylamino-1- (4-morpholinophenyl) -butanone-1,2-methyl-1- [4- (methylthio) phenyl] -2-morpholino-propanone-1, alkylanthraquinone, etc. And benzoin ether compounds such as benzoin alkyl ethers, benzoin compounds such as benzoin and alkyl benzoin, and benzyl derivatives such as benzyl dimethyl ketal. Further, a compound represented by the following formula (I) can also be used.
本発明の樹脂組成物は、熱ラジカル重合開始剤を含有してもよい。熱ラジカル重合開始剤は、熱のエネルギーによってラジカルを発生し、ラジカル重合性基を有する化合物の重合反応を開始または促進させる化合物である。熱ラジカル重合開始剤を添加することによって、ポリイミド前駆体の環化と共に、ポリイミド前駆体がラジカル重合性基を有する場合はポリイミド前駆体の重合反応を進行させることもできるので、より高度な耐熱化が達成できることとなる。
熱ラジカル重合開始剤として、具体的には、特開2008-063554号公報の段落0074~0118に記載されている化合物が挙げられる。 <Thermal radical polymerization initiator>
The resin composition of the present invention may contain a thermal radical polymerization initiator. A thermal radical polymerization initiator is a compound that generates a radical by the energy of heat and initiates or promotes a polymerization reaction of a compound having a radical polymerizable group. By adding a thermal radical polymerization initiator, the cyclization of the polyimide precursor and, when the polyimide precursor has a radical polymerizable group, the polymerization reaction of the polyimide precursor can be advanced, so that higher heat resistance can be achieved. Can be achieved.
Specific examples of the thermal radical polymerization initiator include compounds described in paragraphs 0074 to 0118 of JP-A-2008-063554.
本発明の樹脂組成物は(メタ)アクリロイル基を複数有する重合性モノマーを含有してもよい。重合性モノマーが有する(メタ)アクリロイル基の数は、2個以上でもよく、3個以上であることが好ましい。上限は、15個以下が好ましく、10個以下がより好ましく、8個以下がさらに好ましい。 <Polymerizable monomer>
The resin composition of the present invention may contain a polymerizable monomer having a plurality of (meth) acryloyl groups. The number of (meth) acryloyl groups contained in the polymerizable monomer may be two or more, and is preferably three or more. The upper limit is preferably 15 or less, more preferably 10 or less, and still more preferably 8 or less.
酸基を有する重合性モノマーの好ましい酸価は、0.1~40mgKOH/gであり、特に好ましくは5~30mgKOH/gである。重合性モノマーの酸価が上記範囲であれば、製造や取扱性に優れ、さらには、現像性に優れる。また、重合性が良好である。 The polymerizable monomer may be a compound having an acid group such as a carboxyl group and a phosphoric acid group. The polymerizable monomer having an acid group is preferably an ester of an aliphatic polyhydroxy compound and an unsaturated carboxylic acid, and the unreacted hydroxyl group of the aliphatic polyhydroxy compound is reacted with a non-aromatic carboxylic anhydride to form an acid group. Is more preferable. Particularly preferably, in a polymerizable monomer obtained by reacting a non-aromatic carboxylic anhydride with an unreacted hydroxyl group of an aliphatic polyhydroxy compound to have an acid group, the aliphatic polyhydroxy compound is preferably pentaerythritol or dipentaerythritol. Is a compound. Examples of commercially available products include M-510 and M-520 as polybasic acid-modified acrylic oligomers manufactured by Toagosei Co., Ltd.
The preferred acid value of the polymerizable monomer having an acid group is 0.1 to 40 mgKOH / g, particularly preferably 5 to 30 mgKOH / g. When the acid value of the polymerizable monomer is in the above range, it is excellent in production and handleability, and further excellent in developability. Further, the polymerizability is good.
本発明の樹脂組成物は重合性モノマーを実質的に含有しないことも好ましい。この態様によれば、得られる硬化膜の破断伸びや耐薬品性をより向上させることができる。なお、本明細書において、重合性モノマーを実質的に含有しない場合とは、重合性モノマーの含有量が樹脂組成物の全固形分に対して、0.01質量%以下であることを意味し、0.005質量%以下であることが好ましく、含有しないことがより好ましい。このような効果が得られる詳細な理由は不明であるが、重合性モノマーの重合反応は、ポリイミド前駆体の環化反応よりも先に進行しやすいと推測される。このため、ポリイミド前駆体の環化反応時に、重合性モノマーが存在していると、重合性モノマーの重合反応が先に進み、ポリイミド前駆体の環化が進行しにくくなると推測される。樹脂組成物が重合性モノマーを実質的に含有しないことでポリイミド前駆体の環化が進行しやすくなり、その結果得られる硬化膜の破断伸びや耐薬品性をより向上させることができたと推測される。 The content of the polymerizable monomer is preferably 20% by mass or less, more preferably 18% by mass or less, and preferably 15% by mass or less based on the total solid content of the resin composition of the present invention. Is more preferred. The lower limit may be more than 0% by mass, 1% by mass or more, or 3% by mass or more.
It is also preferable that the resin composition of the present invention does not substantially contain a polymerizable monomer. According to this aspect, the elongation at break and the chemical resistance of the obtained cured film can be further improved. In this specification, the case where the polymerizable monomer is not substantially contained means that the content of the polymerizable monomer is 0.01% by mass or less based on the total solid content of the resin composition. , 0.005% by mass or less, and more preferably not contained. Although the detailed reason for obtaining such an effect is unknown, it is presumed that the polymerization reaction of the polymerizable monomer tends to proceed earlier than the cyclization reaction of the polyimide precursor. For this reason, when the polymerizable monomer is present during the cyclization reaction of the polyimide precursor, it is presumed that the polymerization reaction of the polymerizable monomer proceeds first and the cyclization of the polyimide precursor does not easily progress. It is presumed that the cyclization of the polyimide precursor easily progressed because the resin composition did not substantially contain a polymerizable monomer, and the resulting cured film could have further improved elongation at break and chemical resistance. You.
本発明の樹脂組成物は、溶剤を含有することが好ましい。溶剤は、公知の溶剤を任意に使用できる。溶剤は有機溶剤が好ましい。有機溶剤としては、エステル類、エーテル類、ケトン類、芳香族炭化水素類、スルホキシド類、アミド類などの化合物が挙げられる。
エステル類として、例えば、酢酸エチル、酢酸-n-ブチル、酢酸イソブチル、ギ酸アミル、酢酸イソアミル、プロピオン酸ブチル、酪酸イソプロピル、酪酸エチル、酪酸ブチル、乳酸メチル、乳酸エチル、γ-ブチロラクトン、ε-カプロラクトン、δ-バレロラクトン、アルキルオキシ酢酸アルキル(例えば、アルキルオキシ酢酸メチル、アルキルオキシ酢酸エチル、アルキルオキシ酢酸ブチル(例えば、メトキシ酢酸メチル、メトキシ酢酸エチル、メトキシ酢酸ブチル、エトキシ酢酸メチル、エトキシ酢酸エチル等))、3-アルキルオキシプロピオン酸アルキルエステル類(例えば、3-アルキルオキシプロピオン酸メチル、3-アルキルオキシプロピオン酸エチル等(例えば、3-メトキシプロピオン酸メチル、3-メトキシプロピオン酸エチル、3-エトキシプロピオン酸メチル、3-エトキシプロピオン酸エチル等))、2-アルキルオキシプロピオン酸アルキルエステル類(例えば、2-アルキルオキシプロピオン酸メチル、2-アルキルオキシプロピオン酸エチル、2-アルキルオキシプロピオン酸プロピル等(例えば、2-メトキシプロピオン酸メチル、2-メトキシプロピオン酸エチル、2-メトキシプロピオン酸プロピル、2-エトキシプロピオン酸メチル、2-エトキシプロピオン酸エチル))、2-アルキルオキシ-2-メチルプロピオン酸メチルおよび2-アルキルオキシ-2-メチルプロピオン酸エチル(例えば、2-メトキシ-2-メチルプロピオン酸メチル、2-エトキシ-2-メチルプロピオン酸エチル等)、ピルビン酸メチル、ピルビン酸エチル、ピルビン酸プロピル、アセト酢酸メチル、アセト酢酸エチル、2-オキソブタン酸メチル、2-オキソブタン酸エチル等が好適なものとして挙げられる。
エーテル類として、例えば、ジエチレングリコールジメチルエーテル、テトラヒドロフラン、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、メチルセロソルブアセテート、エチルセロソルブアセテート、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールモノブチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノエチルエーテルアセテート、プロピレングリコールモノプロピルエーテルアセテート等が好適なものとして挙げられる。
ケトン類として、例えば、メチルエチルケトン、シクロヘキサノン、シクロペンタノン、2-ヘプタノン、3-ヘプタノン等が好適なものとして挙げられる。
芳香族炭化水素類として、例えば、トルエン、キシレン、アニソール、リモネン等が好適なものとして挙げられる。
スルホキシド類として、例えば、ジメチルスルホキシドが好適なものとして挙げられる。
アミド類として、N-メチル-2-ピロリドン、N -エチル-2-ピロリドン、N,N-ジメチルアセトアミド、N,N-ジメチルホルムアミド等が好適なものとして挙げられる。 <Solvent>
The resin composition of the present invention preferably contains a solvent. As the solvent, a known solvent can be arbitrarily used. The solvent is preferably an organic solvent. Examples of the organic solvent include compounds such as esters, ethers, ketones, aromatic hydrocarbons, sulfoxides, and amides.
As esters, for example, ethyl acetate, n-butyl acetate, isobutyl acetate, amyl formate, isoamyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, butyl butyrate, methyl lactate, ethyl lactate, γ-butyrolactone, ε-caprolactone , Δ-valerolactone, alkyl alkyloxyacetates (eg, methyl alkyloxyacetate, ethylalkyloxyacetate, butylalkyloxyacetate (eg, methyl methoxyacetate, ethyl methoxyacetate, butyl methoxyacetate, methyl ethoxyacetate, methyl ethoxyacetate, etc.) )), Alkyl 3-alkyloxypropionates (eg, methyl 3-alkyloxypropionate, ethyl 3-alkyloxypropionate, etc. (eg, methyl 3-methoxypropionate, 3-methoxypropionate) Ethyl ester, methyl 3-ethoxypropionate, ethyl 3-ethoxypropionate)), alkyl 2-alkyloxypropionates (eg, methyl 2-alkyloxypropionate, ethyl 2-alkyloxypropionate, 2 Propyl alkyloxypropionate and the like (eg, methyl 2-methoxypropionate, ethyl 2-methoxypropionate, propyl 2-methoxypropionate, methyl 2-ethoxypropionate, ethyl 2-ethoxypropionate), 2-alkyl Methyl oxy-2-methylpropionate and ethyl 2-alkyloxy-2-methylpropionate (eg, methyl 2-methoxy-2-methylpropionate, ethyl 2-ethoxy-2-methylpropionate), methyl pyruvate , Pyruvate Chill, propyl pyruvate, methyl acetoacetate, ethyl acetoacetate, methyl 2-oxobutanoate, ethyl 2-oxobutanoate, and the like as preferred.
As ethers, for example, diethylene glycol dimethyl ether, tetrahydrofuran, ethylene glycol monomethyl ether, ethylene glycol monoethyl ether, methyl cellosolve acetate, ethyl cellosolve acetate, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, diethylene glycol monobutyl ether, propylene glycol monomethyl ether, propylene glycol Suitable examples include monomethyl ether acetate, propylene glycol monoethyl ether acetate, and propylene glycol monopropyl ether acetate.
Suitable ketones include, for example, methyl ethyl ketone, cyclohexanone, cyclopentanone, 2-heptanone, 3-heptanone and the like.
Suitable aromatic hydrocarbons include, for example, toluene, xylene, anisole, limonene and the like.
Suitable sulfoxides include, for example, dimethyl sulfoxide.
Suitable amides include N-methyl-2-pyrrolidone, N-ethyl-2-pyrrolidone, N, N-dimethylacetamide, N, N-dimethylformamide and the like.
本発明では、3-エトキシプロピオン酸メチル、3-エトキシプロピオン酸エチル、エチルセロソルブアセテート、乳酸エチル、ジエチレングリコールジメチルエーテル、酢酸ブチル、3-メトキシプロピオン酸メチル、2-ヘプタノン、シクロヘキサノン、シクロペンタノン、γ-ブチロラクトン、ジメチルスルホキシド、エチルカルビトールアセテート、ブチルカルビトールアセテート、N-メチル-2-ピロリドン、プロピレングリコールメチルエーテル、およびプロピレングリコールメチルエーテルアセテートから選択される1種の溶剤、または、2種以上で構成される混合溶剤が好ましい。ジメチルスルホキシドとγ-ブチロラクトンとの併用が特に好ましい。 From the viewpoint of improving the properties of the coated surface, a form in which two or more solvents are mixed is also preferable.
In the present invention, methyl 3-ethoxypropionate, ethyl 3-ethoxypropionate, ethyl cellosolve acetate, ethyl lactate, diethylene glycol dimethyl ether, butyl acetate, methyl 3-methoxypropionate, 2-heptanone, cyclohexanone, cyclopentanone, γ- One or more solvents selected from butyrolactone, dimethylsulfoxide, ethyl carbitol acetate, butyl carbitol acetate, N-methyl-2-pyrrolidone, propylene glycol methyl ether, and propylene glycol methyl ether acetate, or two or more solvents Is preferred. Particularly preferred is a combination of dimethyl sulfoxide and γ-butyrolactone.
溶剤は1種のみ含有していてもよいし、2種以上含有していてもよい。溶剤を2種以上含有する場合は、その合計が上記範囲であることが好ましい。 From the viewpoint of applicability, the content of the solvent is preferably such that the total solid content of the resin composition of the present invention is 5 to 80% by mass, and more preferably 5 to 75% by mass. More preferably, the amount is 10 to 70% by mass, even more preferably 40 to 70% by mass. The solvent content may be adjusted depending on the desired thickness and the coating method.
The solvent may contain only one kind, or may contain two or more kinds. When two or more solvents are contained, the total is preferably within the above range.
本発明の樹脂組成物は、さらにマイグレーション抑制剤を含むことが好ましい。マイグレーション抑制剤を含むことにより、金属層(金属配線)由来の金属イオンが樹脂組成物層内へ移動することを効果的に抑制可能となる。
マイグレーション抑制剤としては、特に制限はないが、複素環(ピロール環、フラン環、チオフェン環、イミダゾール環、オキサゾール環、チアゾール環、ピラゾール環、イソオキサゾール環、イソチアゾール環、テトラゾール環、ピリジン環、ピリダジン環、ピリミジン環、ピラジン環、ピペリジン環、ピペラジン環、モルホリン環、2H-ピラン環および6H-ピラン環、トリアジン環)を有する化合物、チオ尿素類およびスルファニル基を有する化合物、ヒンダードフェノール系化合物、サリチル酸誘導体系化合物、ヒドラジド誘導体系化合物が挙げられる。特に、1,2,4-トリアゾール、ベンゾトリアゾール等のトリアゾール系化合物、1H-テトラゾール、5-フェニルテトラゾール等のテトラゾール系化合物が好ましく使用できる。 <Migration inhibitor>
It is preferable that the resin composition of the present invention further contains a migration inhibitor. By including the migration inhibitor, it is possible to effectively suppress migration of metal ions derived from the metal layer (metal wiring) into the resin composition layer.
The migration inhibitor is not particularly limited, but may be a heterocycle (pyrrole ring, furan ring, thiophene ring, imidazole ring, oxazole ring, thiazole ring, pyrazole ring, isoxazole ring, isothiazole ring, tetrazole ring, pyridine ring, Compounds having pyridazine ring, pyrimidine ring, pyrazine ring, piperidine ring, piperazine ring, morpholine ring, 2H-pyran ring and 6H-pyran ring, triazine ring), compounds having thioureas and sulfanyl groups, hindered phenol compounds , Salicylic acid derivative compounds, and hydrazide derivative compounds. In particular, triazole compounds such as 1,2,4-triazole and benzotriazole, and tetrazole compounds such as 1H-tetrazole and 5-phenyltetrazole can be preferably used.

本発明の樹脂組成物は、重合禁止剤を含むことが好ましい。重合禁止剤としては、例えば、ヒドロキノン、p-メトキシフェノール、ジ-tert-ブチル-p-クレゾール、ピロガロール、p-tert-ブチルカテコール、1,4-ベンゾキノン、ジフェニル-p-ベンゾキノン、4,4’-チオビス(3-メチル-6-tert-ブチルフェノール)、2,2’-メチレンビス(4-メチル-6-tert-ブチルフェノール)、N-ニトロソ-N-フェニルヒドロキシアミンアルミニウム塩、フェノチアジン、N-ニトロソジフェニルアミン、N-フェニルナフチルアミン、エチレンジアミン四酢酸、1,2-シクロヘキサンジアミン四酢酸、グリコールエーテルジアミン四酢酸、2,6-ジ-tert-ブチル-4-メチルフェノール、5-ニトロソ-8-ヒドロキシキノリン、1-ニトロソ-2-ナフトール、2-ニトロソ-1-ナフトール、2-ニトロソ-5-(N-エチル-N-スルホプロピルアミノ)フェノール、N-ニトロソ-N-(1-ナフチル)ヒドロキシアミンアンモニウム塩、ビス(4-ヒドロキシ-3,5-tert-ブチル)フェニルメタンなどが好適に用いられる。また、特開2015-127817号公報の段落0060に記載の重合禁止剤、および、国際公開第2015/125469号の段落0031~0046に記載の化合物を用いることもできる。また、下記化合物を用いることができる(Meはメチル基である)。
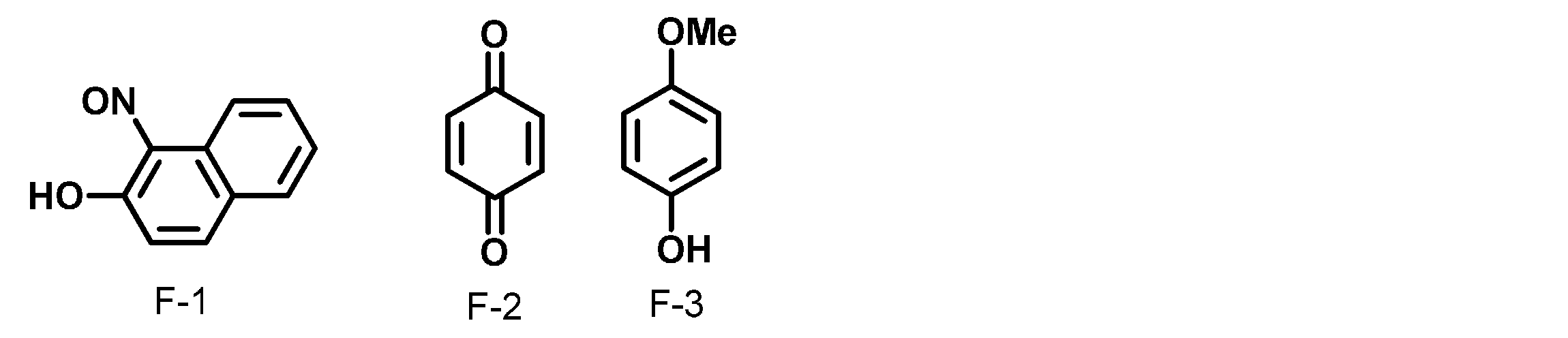
The resin composition of the present invention preferably contains a polymerization inhibitor. Examples of the polymerization inhibitor include hydroquinone, p-methoxyphenol, di-tert-butyl-p-cresol, pyrogallol, p-tert-butylcatechol, 1,4-benzoquinone, diphenyl-p-benzoquinone, and 4,4 ′. -Thiobis (3-methyl-6-tert-butylphenol), 2,2'-methylenebis (4-methyl-6-tert-butylphenol), N-nitroso-N-phenylhydroxyamine aluminum salt, phenothiazine, N-nitrosodiphenylamine , N-phenylnaphthylamine, ethylenediaminetetraacetic acid, 1,2-cyclohexanediaminetetraacetic acid, glycol etherdiaminetetraacetic acid, 2,6-di-tert-butyl-4-methylphenol, 5-nitroso-8-hydroxyquinoline, -Nitroso 2-naphthol, 2-nitroso-1-naphthol, 2-nitroso-5- (N-ethyl-N-sulfopropylamino) phenol, N-nitroso-N- (1-naphthyl) hydroxyamine ammonium salt, bis (4 -Hydroxy-3,5-tert-butyl) phenylmethane and the like are preferably used. Further, the polymerization inhibitors described in paragraph 0060 of JP-A-2015-127817 and the compounds described in paragraphs 0031 to 0046 of WO2015 / 125469 can also be used. Further, the following compounds can be used (Me is a methyl group).
本発明の樹脂組成物は、電極や配線などに用いられる金属材料との接着性を向上させるための金属接着性改良剤を含んでいることが好ましい。金属接着性改良剤としては、シランカップリング剤などが挙げられる。 <Metal adhesion improver>
The resin composition of the present invention preferably contains a metal adhesion improver for improving the adhesion to a metal material used for an electrode or a wiring. Examples of the metal adhesion improver include a silane coupling agent.

本発明の樹脂組成物は、本発明の効果を損なわない範囲で、必要に応じて、各種の添加物、例えば、熱酸発生剤、増感色素、連鎖移動剤、界面活性剤、高級脂肪酸誘導体、無機粒子、硬化剤、硬化触媒、充填剤、酸化防止剤、紫外線吸収剤、凝集防止剤等を配合することができる。これらの添加剤を配合する場合、その合計配合量は組成物の固形分の3質量%以下とすることが好ましい。 <Other additives>
The resin composition of the present invention may contain various additives, for example, a thermal acid generator, a sensitizing dye, a chain transfer agent, a surfactant, and a higher fatty acid derivative, as long as the effects of the present invention are not impaired. , Inorganic particles, a curing agent, a curing catalyst, a filler, an antioxidant, an ultraviolet absorber, an anti-agglomeration agent, and the like. When these additives are blended, the total blending amount is preferably 3% by mass or less of the solid content of the composition.
本発明の樹脂組成物は、熱酸発生剤を含んでいてもよい。熱酸発生剤は、特定熱塩基発生剤が保護基を有する場合、保護基の脱離に用いられる。 << thermal acid generator >>
The resin composition of the present invention may contain a thermal acid generator. When the specific thermal base generator has a protective group, the thermal acid generator is used for elimination of the protective group.
熱酸発生剤は、1種のみ用いても、2種以上用いてもよい。2種以上用いる場合は、合計量が上記範囲となることが好ましい。 The content of the thermal acid generator is preferably at least 0.01 part by mass, more preferably at least 0.1 part by mass, based on 100 parts by mass of the polymer precursor. Since the crosslinking reaction and the cyclization of the polymer precursor are promoted by containing 0.01 parts by mass or more of the thermal acid generator, the mechanical properties and chemical resistance of the cured film can be further improved. Further, the content of the thermal acid generator is preferably 20 parts by mass or less, more preferably 15 parts by mass or less, and still more preferably 10 parts by mass or less, from the viewpoint of the electrical insulation of the cured film.
Only one thermal acid generator may be used, or two or more thermal acid generators may be used. When two or more kinds are used, the total amount is preferably within the above range.
本発明の樹脂組成物は、増感色素を含んでいてもよい。増感色素は、特定の活性放射線を吸収して電子励起状態となる。電子励起状態となった増感色素は、熱硬化促進剤、熱ラジカル重合開始剤、光ラジカル重合開始剤などと接触して、電子移動、エネルギー移動、発熱などの作用が生じる。これにより、熱硬化促進剤、熱ラジカル重合開始剤、光ラジカル重合開始剤は化学変化を起こして分解し、ラジカル、酸あるいは塩基を生成する。増感色素の詳細については、特開2016-027357号公報の段落0161~0163の記載を参酌でき、この内容は本明細書に組み込まれる。 << Sensitizing dye >>
The resin composition of the present invention may contain a sensitizing dye. The sensitizing dye absorbs a specific actinic radiation and enters an electronically excited state. The sensitizing dye in the electronically excited state comes into contact with a thermosetting accelerator, a thermal radical polymerization initiator, a photoradical polymerization initiator, or the like, and causes effects such as electron transfer, energy transfer, and heat generation. As a result, the thermal curing accelerator, the thermal radical polymerization initiator, and the photoradical polymerization initiator undergo chemical changes and are decomposed to generate radicals, acids, or bases. For the details of the sensitizing dye, the description in paragraphs 0161 to 0163 of JP-A-2016-027357 can be referred to, and the contents thereof are incorporated herein.
本発明の樹脂組成物は、連鎖移動剤を含有してもよい。連鎖移動剤は、例えば高分子辞典第三版(高分子学会編、2005年)683-684頁に定義されている。連鎖移動剤としては、例えば、分子内にSH、PH、SiH、およびGeHを有する化合物群が用いられる。これらは、低活性のラジカルに水素を供与して、ラジカルを生成するか、もしくは、酸化された後、脱プロトンすることによりラジカルを生成しうる。特に、チオール化合物を好ましく用いることができる。
また、連鎖移動剤は、国際公開第2015/199219号の段落0152~0153に記載の化合物を用いることもできる。 << Chain transfer agent >>
The resin composition of the present invention may contain a chain transfer agent. The chain transfer agent is defined, for example, in Polymer Dictionary, Third Edition (edited by The Society of Polymer Science, 2005), pages 683-684. As the chain transfer agent, for example, a compound group having SH, PH, SiH, and GeH in the molecule is used. These can generate a radical by donating hydrogen to a low activity radical, or can generate a radical by being oxidized and then deprotonated. In particular, a thiol compound can be preferably used.
Further, as the chain transfer agent, compounds described in paragraphs 0152 to 0153 of WO 2015/199219 can also be used.
本発明の樹脂組成物には、塗布性をより向上させる観点から、各種類の界面活性剤を添加してもよい。界面活性剤としては、フッ素系界面活性剤、ノニオン系界面活性剤、カチオン系界面活性剤、アニオン系界面活性剤、シリコーン系界面活性剤などの各種類の界面活性剤を使用できる。また、下記界面活性剤も好ましい。

Each type of surfactant may be added to the resin composition of the present invention from the viewpoint of further improving coatability. As the surfactant, various types of surfactants such as a fluorine-based surfactant, a nonionic surfactant, a cationic surfactant, an anionic surfactant, and a silicone-based surfactant can be used. Further, the following surfactants are also preferable.
本発明の樹脂組成物は、酸素に起因する重合阻害を防止するために、ベヘン酸やベヘン酸アミドのような高級脂肪酸誘導体を添加して、塗布後の乾燥の過程で組成物の表面に偏在させてもよい。
また、高級脂肪酸誘導体は、国際公開第2015/199219号の段落0155に記載の化合物を用いることもできる。
本発明の樹脂組成物が高級脂肪酸誘導体を有する場合、高級脂肪酸誘導体の含有量は、本発明の樹脂組成物の全固形分に対して、0.1~10質量%であることが好ましい。高級脂肪酸誘導体は1種のみでもよいし、2種以上であってもよい。高級脂肪酸誘導体が2種以上の場合は、その合計が上記範囲であることが好ましい。 << Higher fatty acid derivative >>
In order to prevent polymerization inhibition caused by oxygen, the resin composition of the present invention is added with a higher fatty acid derivative such as behenic acid or behenic acid amide, and is unevenly distributed on the surface of the composition in a drying process after application. May be.
Further, as the higher fatty acid derivative, a compound described in paragraph 0155 of WO 2015/199219 can be used.
When the resin composition of the present invention has a higher fatty acid derivative, the content of the higher fatty acid derivative is preferably 0.1 to 10% by mass based on the total solid content of the resin composition of the present invention. The higher fatty acid derivative may be only one kind or two or more kinds. When there are two or more higher fatty acid derivatives, the total is preferably within the above range.
本発明の樹脂組成物の水分含有量は、塗布面性状の観点から、5質量%未満が好ましく、1質量%未満がより好ましく、0.6質量%未満がさらに好ましい。 <Restrictions on other contained substances>
The water content of the resin composition of the present invention is preferably less than 5% by mass, more preferably less than 1% by mass, even more preferably less than 0.6% by mass from the viewpoint of the properties of the coated surface.
また、本発明の樹脂組成物に意図せずに含まれる金属不純物を低減する方法としては、本発明の樹脂組成物を構成する原料として金属含有量が少ない原料を選択する、本発明の樹脂組成物を構成する原料に対してフィルターろ過を行う、装置内をポリテトラフロロエチレン等でライニングしてコンタミネーションを可能な限り抑制した条件下で蒸留を行う等の方法を挙げることができる。 From the viewpoint of insulating properties, the metal content of the resin composition of the present invention is preferably less than 5 ppm by mass (parts per million), more preferably less than 1 ppm by mass, and even more preferably less than 0.5 ppm by mass. Examples of the metal include sodium, potassium, magnesium, calcium, iron, chromium, nickel and the like. When a plurality of metals are included, the total of these metals is preferably within the above range.
In addition, as a method for reducing metal impurities unintentionally contained in the resin composition of the present invention, a material having a low metal content is selected as a raw material constituting the resin composition of the present invention. Examples of the method include filtering the raw material constituting the product with a filter, lining the inside of the apparatus with polytetrafluoroethylene or the like, and performing distillation under the condition that contamination is suppressed as much as possible.
本発明の樹脂組成物は、上記各成分を混合して調製することができる。混合方法は特に限定はなく、従来公知の方法で行うことができる。
また、組成物中のゴミや微粒子等の異物を除去する目的で、フィルターを用いたろ過を行うことが好ましい。フィルター孔径は、1μm以下が好ましく、0.5μm以下がより好ましく、0.1μm以下がさらに好ましい。フィルターの材質は、ポリテトラフロロエチレン、ポリエチレンまたはナイロンが好ましい。フィルターは、有機溶剤であらかじめ洗浄したものを用いてもよい。フィルターろ過工程では、複数種のフィルターを直列または並列に接続して用いてもよい。複数種のフィルターを使用する場合は、孔径または材質が異なるフィルターを組み合わせて使用してもよい。また、各種材料を複数回ろ過してもよい。複数回ろ過する場合は、循環ろ過であってもよい。また、加圧してろ過を行ってもよい。加圧してろ過を行う場合、加圧する圧力は0.05MPa以上0.3MPa以下が好ましい。
フィルターを用いたろ過の他、吸着材を用いた不純物の除去処理を行ってもよい。フィルターろ過と吸着材を用いた不純物除去処理とを組み合わせてもよい。吸着材としては、公知の吸着材を用いることができる。例えば、シリカゲル、ゼオライトなどの無機系吸着材、活性炭などの有機系吸着材が挙げられる。 [Preparation of resin composition]
The resin composition of the present invention can be prepared by mixing the above components. The mixing method is not particularly limited, and can be performed by a conventionally known method.
In addition, it is preferable to perform filtration using a filter for the purpose of removing foreign substances such as dust and fine particles in the composition. The filter pore size is preferably 1 μm or less, more preferably 0.5 μm or less, and even more preferably 0.1 μm or less. The material of the filter is preferably polytetrafluoroethylene, polyethylene or nylon. The filter may be one that has been washed in advance with an organic solvent. In the filter filtration step, a plurality of types of filters may be connected in series or in parallel. When a plurality of types of filters are used, filters having different pore sizes or materials may be used in combination. Further, various materials may be filtered plural times. When filtration is performed a plurality of times, circulation filtration may be used. Also, filtration may be performed under pressure. In the case of performing filtration by applying pressure, the pressure to be applied is preferably 0.05 MPa or more and 0.3 MPa or less.
In addition to the filtration using a filter, a treatment for removing impurities using an adsorbent may be performed. Filter filtration and impurity removal treatment using an adsorbent may be combined. As the adsorbent, a known adsorbent can be used. For example, inorganic adsorbents such as silica gel and zeolite, and organic adsorbents such as activated carbon can be used.
次に、硬化膜、積層体、半導体デバイス、およびそれらの製造方法について説明する。
本発明の硬化膜は、本発明の樹脂組成物を硬化して得られるものである。本発明の硬化膜の膜厚は、例えば、0.5μm以上とすることができ、1μm以上とすることができる。また、上限値としては、100μm以下とすることができ、30μm以下とすることもできる。 [Cured film, laminate, semiconductor device, and manufacturing method thereof]
Next, a cured film, a laminate, a semiconductor device, and a method for manufacturing the same will be described.
The cured film of the present invention is obtained by curing the resin composition of the present invention. The thickness of the cured film of the present invention can be, for example, 0.5 μm or more, and can be 1 μm or more. Further, the upper limit can be set to 100 μm or less, and can be set to 30 μm or less.
(a)樹脂組成物を基板に適用して膜を形成する膜形成工程
(b)膜形成工程の後、膜を露光する露光工程
(c)露光された樹脂組成物層に対して、現像処理を行う現像工程
(d)現像された樹脂組成物を80~450℃で加熱する加熱工程
この実施形態のように、現像の後、加熱することで露光された樹脂層をさらに硬化させることができる。この加熱工程で熱塩基発生剤および熱硬化性化合物が作用し十分な硬化性が得られる。 The method for producing a cured film of the present invention includes using the resin composition of the present invention. Specifically, it is preferable to include the following steps (a) to (d).
(A) a film forming step of forming a film by applying the resin composition to a substrate; (b) an exposing step of exposing the film after the film forming step; and (c) a developing treatment of the exposed resin composition layer. (D) heating step of heating the developed resin composition at 80 to 450 ° C. As in this embodiment, the exposed resin layer can be further cured by heating after development. . In this heating step, the thermobase generator and the thermosetting compound act to obtain sufficient curability.
本発明の好ましい実施形態に係る製造方法は、樹脂組成物を基板に適用して膜(層状)にする、膜形成工程(層形成工程)を含む。
基板の種類は、用途に応じて適宜定めることができるが、シリコン、窒化シリコン、ポリシリコン、酸化シリコン、アモルファスシリコンなどの半導体作製基板、石英、ガラス、光学フィルム、セラミック材料、蒸着膜、磁性膜、反射膜、Ni、Cu、Cr、Feなどの金属基板、紙、SOG(Spin On Glass)、TFT(薄膜トランジスタ)アレイ基板、プラズマディスプレイパネル(PDP)の電極板など特に制約されない。本発明では、特に、半導体作製基板が好ましく、シリコン基板がより好ましい。
また、樹脂層の表面や金属層の表面に樹脂組成物層を形成する場合は、樹脂層や金属層が基板となる。
樹脂組成物を基板に適用する手段としては、塗布が好ましい。
具体的には、適用する手段としては、ディップコート法、エアーナイフコート法、カーテンコート法、ワイヤーバーコート法、グラビアコート法、エクストルージョンコート法、スプレーコート法、スピンコート法、スリットコート法、およびインクジェット法などが例示される。樹脂組成物層の厚さの均一性の観点から、より好ましくはスピンコート法、スリットコート法、スプレーコート法、インクジェット法である。方法に応じて適切な固形分濃度や塗布条件を調整することで、所望の厚さの樹脂層を得ることができる。また、基板の形状によっても塗布方法を適宜選択でき、ウェハ等の円形基板であればスピンコート法やスプレーコート法、インクジェット法等が好ましく、矩形基板であればスリットコート法やスプレーコート法、インクジェット法等が好ましい。スピンコート法の場合は、例えば、500~2000rpmの回転数で、10秒~1分程度適用することができる。 <Film forming step (layer forming step)>
The manufacturing method according to a preferred embodiment of the present invention includes a film forming step (layer forming step) of applying a resin composition to a substrate to form a film (layered).
The type of the substrate can be appropriately determined according to the application, but a semiconductor production substrate such as silicon, silicon nitride, polysilicon, silicon oxide, and amorphous silicon, quartz, glass, an optical film, a ceramic material, a vapor-deposited film, and a magnetic film , A reflective film, a metal substrate of Ni, Cu, Cr, Fe, etc., paper, SOG (Spin On Glass), TFT (thin film transistor) array substrate, and an electrode plate of a plasma display panel (PDP). In the present invention, a semiconductor fabrication substrate is particularly preferable, and a silicon substrate is more preferable.
When the resin composition layer is formed on the surface of the resin layer or the surface of the metal layer, the resin layer or the metal layer serves as a substrate.
As a means for applying the resin composition to the substrate, coating is preferable.
Specifically, as means to be applied, dip coating, air knife coating, curtain coating, wire bar coating, gravure coating, extrusion coating, spray coating, spin coating, slit coating, slit coating, And an inkjet method. From the viewpoint of the uniformity of the thickness of the resin composition layer, more preferred are a spin coating method, a slit coating method, a spray coating method and an ink jet method. A resin layer having a desired thickness can be obtained by adjusting an appropriate solid content concentration and application conditions according to the method. The coating method can also be appropriately selected depending on the shape of the substrate. For a circular substrate such as a wafer, a spin coating method, a spray coating method, or an inkjet method is preferable. For a rectangular substrate, a slit coating method, a spray coating method, or an inkjet method is used. Method is preferred. In the case of the spin coating method, for example, a rotation speed of 500 to 2000 rpm can be applied for about 10 seconds to 1 minute.
本発明の製造方法は、樹脂組成物層を形成後、膜形成工程(層形成工程)の後に、溶剤を除去するために乾燥する工程を含んでいてもよい。好ましい乾燥温度は50~150℃で、70℃~130℃がより好ましく、90℃~110℃がさらに好ましい。乾燥時間としては、30秒~20分が例示され、1分~10分が好ましく、3分~7分がより好ましい。 <Drying process>
The production method of the present invention may include a step of drying after removing the solvent after forming the resin composition layer and after the film forming step (layer forming step). The preferred drying temperature is 50 to 150 ° C., more preferably 70 to 130 ° C., and even more preferably 90 to 110 ° C. The drying time is, for example, 30 seconds to 20 minutes, preferably 1 minute to 10 minutes, and more preferably 3 minutes to 7 minutes.
本発明の製造方法は、上記樹脂組成物層を露光する露光工程を含んでもよい。露光量は、樹脂組成物を硬化できる限り特に定めるものではないが、例えば、波長365nmでの露光エネルギー換算で100~10000mJ/cm2照射することが好ましく、200~8000mJ/cm2照射することがより好ましい。
露光波長は、190~1000nmの範囲で適宜定めることができ、240~550nmが好ましい。
露光波長は、光源との関係でいうと、(1)半導体レーザー(波長 830nm、532nm、488nm、405nm etc.)、(2)メタルハライドランプ、(3)高圧水銀灯、g線(波長 436nm)、h線(波長 405nm)、i線(波長 365nm)、ブロード(g,h,i線の3波長)、(4)エキシマレーザー、KrFエキシマレーザー(波長 248nm)、ArFエキシマレーザー(波長 193nm)、F2エキシマレーザー(波長 157nm)、(5)極端紫外線;EUV(波長 13.6nm)、(6)電子線等が挙げられる。本発明の樹脂組成物については、特に高圧水銀灯による露光が好ましく、なかでも、i線による露光が好ましい。これにより、特に高い露光感度が得られうる。 <Exposure process>
The production method of the present invention may include an exposure step of exposing the resin composition layer. The amount of exposure is not particularly limited as long as the resin composition can be cured. For example, irradiation is preferably 100 to 10,000 mJ / cm 2 , and more preferably 200 to 8000 mJ / cm 2 in terms of exposure energy at a wavelength of 365 nm. More preferred.
The exposure wavelength can be appropriately determined within the range of 190 to 1000 nm, and preferably 240 to 550 nm.
The exposure wavelength is, in terms of the light source, (1) semiconductor laser (wavelength 830 nm, 532 nm, 488 nm, 405 nm etc.), (2) metal halide lamp, (3) high-pressure mercury lamp, g-line (wavelength 436 nm), h Line (wavelength 405 nm), i-line (wavelength 365 nm), broad (three wavelengths of g, h, i-line), (4) excimer laser, KrF excimer laser (wavelength 248 nm), ArF excimer laser (wavelength 193 nm), F2 excimer Laser (wavelength: 157 nm); (5) extreme ultraviolet; EUV (wavelength: 13.6 nm); (6) electron beam. With respect to the resin composition of the present invention, exposure with a high-pressure mercury lamp is particularly preferred, and exposure with i-line is particularly preferred. Thereby, a particularly high exposure sensitivity can be obtained.
本発明の製造方法は、露光された樹脂組成物層に対して、現像処理を行う現像処理工程を含んでもよい。現像を行うことにより、露光されていない部分(非露光部)が除去される。現像方法は、所望のパターンを形成できれば特に制限は無く、例えば、パドル、スプレー、浸漬、超音波等の現像方法が採用可能である。
現像は現像液を用いて行う。現像液は、露光されていない部分(非露光部)が除去されるのであれば、特に制限なく使用できる。現像液は、有機溶剤を含むことが好ましく、現像液が有機溶剤を90%以上含むことがより好ましい。本発明では、現像液は、ClogP値が-1~5の有機溶剤を含むことが好ましく、ClogP値が0~3の有機溶剤を含むことがより好ましい。ClogP値は、ChemBioDrawにて構造式を入力して計算値として求めることができる。
有機溶剤は、エステル類として、例えば、酢酸エチル、酢酸-n-ブチル、ギ酸アミル、酢酸イソアミル、酢酸イソブチル、プロピオン酸ブチル、酪酸イソプロピル、酪酸エチル、酪酸ブチル、乳酸メチル、乳酸エチル、γ-ブチロラクトン、ε-カプロラクトン、δ-バレロラクトン、アルキルオキシ酢酸アルキル(例:アルキルオキシ酢酸メチル、アルキルオキシ酢酸エチル、アルキルオキシ酢酸ブチル(例えば、メトキシ酢酸メチル、メトキシ酢酸エチル、メトキシ酢酸ブチル、エトキシ酢酸メチル、エトキシ酢酸エチル等))、3-アルキルオキシプロピオン酸アルキルエステル類(例:3-アルキルオキシプロピオン酸メチル、3-アルキルオキシプロピオン酸エチル等(例えば、3-メトキシプロピオン酸メチル、3-メトキシプロピオン酸エチル、3-エトキシプロピオン酸メチル、3-エトキシプロピオン酸エチル等))、2-アルキルオキシプロピオン酸アルキルエステル類(例:2-アルキルオキシプロピオン酸メチル、2-アルキルオキシプロピオン酸エチル、2-アルキルオキシプロピオン酸プロピル等(例えば、2-メトキシプロピオン酸メチル、2-メトキシプロピオン酸エチル、2-メトキシプロピオン酸プロピル、2-エトキシプロピオン酸メチル、2-エトキシプロピオン酸エチル))、2-アルキルオキシ-2-メチルプロピオン酸メチルおよび2-アルキルオキシ-2-メチルプロピオン酸エチル(例えば、2-メトキシ-2-メチルプロピオン酸メチル、2-エトキシ-2-メチルプロピオン酸エチル等)、ピルビン酸メチル、ピルビン酸エチル、ピルビン酸プロピル、アセト酢酸メチル、アセト酢酸エチル、2-オキソブタン酸メチル、2-オキソブタン酸エチル等、ならびに、エーテル類として、例えば、ジエチレングリコールジメチルエーテル、テトラヒドロフラン、エチレングリコールモノメチルエーテル、エチレングリコールモノエチルエーテル、メチルセロソルブアセテート、エチルセロソルブアセテート、ジエチレングリコールモノメチルエーテル、ジエチレングリコールモノエチルエーテル、ジエチレングリコールモノブチルエーテル、プロピレングリコールモノメチルエーテル、プロピレングリコールモノメチルエーテルアセテート、プロピレングリコールモノエチルエーテルアセテート、プロピレングリコールモノプロピルエーテルアセテート等、ならびに、ケトン類として、例えば、メチルエチルケトン、シクロヘキサノン、シクロペンタノン、2-ヘプタノン、3-ヘプタノン、N-メチル-2-ピロリドン等、ならびに、芳香族炭化水素類として、例えば、トルエン、キシレン、アニソール、リモネン等、スルホキシド類としてジメチルスルホキシドが好適に挙げられる。
本発明では、特にシクロペンタノン、γ-ブチロラクトンが好ましく、シクロペンタノンがより好ましい。
現像液は、50質量%以上が有機溶剤であることが好ましく、70質量%以上が有機溶剤であることがより好ましく、90質量%以上が有機溶剤であることがさらに好ましい。また、現像液は、100質量%が有機溶剤であってもよい。 <Development process>
The production method of the present invention may include a development processing step of performing development processing on the exposed resin composition layer. By performing the development, an unexposed portion (non-exposed portion) is removed. The developing method is not particularly limited as long as a desired pattern can be formed. For example, a developing method such as paddle, spray, immersion, or ultrasonic wave can be adopted.
Development is performed using a developer. The developer can be used without particular limitation as long as the unexposed portions (non-exposed portions) are removed. The developer preferably contains an organic solvent, and more preferably the developer contains 90% or more of the organic solvent. In the present invention, the developer preferably contains an organic solvent having a ClogP value of -1 to 5, more preferably an organic solvent having a ClogP value of 0 to 3. The ClogP value can be obtained as a calculated value by inputting a structural formula in ChemBioDraw.
Examples of the organic solvent include esters such as ethyl acetate, n-butyl acetate, amyl formate, isoamyl acetate, isobutyl acetate, butyl propionate, isopropyl butyrate, ethyl butyrate, butyl butyrate, methyl lactate, ethyl lactate, and γ-butyrolactone. , Ε-caprolactone, δ-valerolactone, alkyl alkyl oxyacetates (eg, methyl alkyl oxyacetate, ethyl oxyacetate, alkyl butyl oxyacetate (eg, methyl methoxy acetate, ethyl methoxy acetate, butyl methoxy acetate, methyl ethoxy acetate, Ethyl ethoxyacetate, etc.), alkyl 3-alkyloxypropionates (eg, methyl 3-alkyloxypropionate, ethyl 3-alkyloxypropionate, etc. (eg, methyl 3-methoxypropionate, 3-methoxypropionate) Ethyl pionate, methyl 3-ethoxypropionate, ethyl 3-ethoxypropionate)), alkyl 2-alkyloxypropionates (eg, methyl 2-alkyloxypropionate, ethyl 2-alkyloxypropionate, 2 Propyl alkyloxypropionate and the like (eg, methyl 2-methoxypropionate, ethyl 2-methoxypropionate, propyl 2-methoxypropionate, methyl 2-ethoxypropionate, ethyl 2-ethoxypropionate), 2-alkyl Methyl oxy-2-methylpropionate and ethyl 2-alkyloxy-2-methylpropionate (eg, methyl 2-methoxy-2-methylpropionate, ethyl 2-ethoxy-2-methylpropionate), methyl pyruvate , Pyruvate Tyl, propyl pyruvate, methyl acetoacetate, ethyl acetoacetate, methyl 2-oxobutanoate, ethyl 2-oxobutanoate, and ethers such as diethylene glycol dimethyl ether, tetrahydrofuran, ethylene glycol monomethyl ether, ethylene glycol monoethyl ether , Methyl cellosolve acetate, ethyl cellosolve acetate, diethylene glycol monomethyl ether, diethylene glycol monoethyl ether, diethylene glycol monobutyl ether, propylene glycol monomethyl ether, propylene glycol monomethyl ether acetate, propylene glycol monoethyl ether acetate, propylene glycol monopropyl ether acetate, and the like, and Examples of ketones include methyl ethyl ketone, cyclohexanone, cyclopentanone, 2-heptanone, 3-heptanone, N-methyl-2-pyrrolidone, and the like, and aromatic hydrocarbons such as toluene, xylene, anisole, limonene, and the like. As the sulfoxides, dimethyl sulfoxide is preferably exemplified.
In the present invention, cyclopentanone and γ-butyrolactone are particularly preferred, and cyclopentanone is more preferred.
The developer preferably contains 50% by mass or more of an organic solvent, more preferably 70% by mass or more of an organic solvent, and even more preferably 90% by mass or more of an organic solvent. Further, 100% by mass of the developer may be an organic solvent.
現像液を用いた処理の後、さらに、リンスを行ってもよい。リンスは、現像液とは異なる溶剤で行うことが好ましい。例えば、樹脂組成物に含まれる溶剤を用いてリンスすることができる。リンス時間は、5秒~1分が好ましい。 The development time is preferably from 10 seconds to 5 minutes. The temperature of the developing solution at the time of development is not particularly limited, but it can be usually 20 to 40 ° C.
After the treatment using the developer, rinsing may be further performed. Rinsing is preferably performed with a solvent different from the developer. For example, rinsing can be performed using a solvent contained in the resin composition. The rinsing time is preferably from 5 seconds to 1 minute.
本発明の製造方法は、膜形成工程(層形成工程)、乾燥工程、または現像工程の後に加熱する工程を含むことが好ましい。加熱工程では、ポリマー前駆体の環化反応および熱硬化性化合物の硬化反応が進行する。加熱工程における層の加熱温度(最高加熱温度)としては、50℃以上であることが好ましく、80℃以上であることがより好ましく、140℃以上であることがさらに好ましく、150℃以上であることが一層好ましく、160℃以上であることがより一層好ましく、170℃以上であることがさらに一層好ましい。上限としては、500℃以下であることが好ましく、450℃以下であることがより好ましく、350℃以下であることがさらに好ましく、250℃以下であることが一層好ましく、220℃以下であることがより一層好ましい。
加熱は、加熱開始時の温度から最高加熱温度まで1~12℃/分の昇温速度で行うことが好ましく、2~10℃/分がより好ましく、3~10℃/分がさらに好ましい。昇温速度を1℃/分以上とすることにより、生産性を確保しつつ、アミンの過剰な揮発を防止することができ、昇温速度を12℃/分以下とすることにより、硬化膜の残存応力を緩和することができる。
加熱開始時の温度は、20℃~150℃が好ましく、20℃~130℃がより好ましく、25℃~120℃がさらに好ましい。加熱開始時の温度は、最高加熱温度まで加熱する工程を開始する際の温度のことをいう。例えば、樹脂組成物を基板の上に適用した後、乾燥させる場合、この乾燥後の膜(層)の温度であり、例えば、樹脂組成物に含まれる溶剤の沸点よりも、30~200℃低い温度から徐々に昇温させることが好ましい。
加熱時間(最高加熱温度での加熱時間)は、10~360分であることが好ましく、20~300分であることがより好ましく、30~240分であることがさらに好ましい。
特に多層の積層体を形成する場合、硬化膜の層間の密着性の観点から、加熱温度は180℃~320℃で加熱することが好ましく、180℃~260℃で加熱することがより好ましい。その理由は定かではないが、この温度とすることで、層間のポリマー前駆体のエチニル基同士が架橋反応を進行しているためと考えられる。 <Heating process>
The manufacturing method of the present invention preferably includes a heating step after the film forming step (layer forming step), the drying step, or the developing step. In the heating step, a cyclization reaction of the polymer precursor and a curing reaction of the thermosetting compound proceed. The heating temperature (maximum heating temperature) of the layer in the heating step is preferably 50 ° C. or higher, more preferably 80 ° C. or higher, further preferably 140 ° C. or higher, and more preferably 150 ° C. or higher. Is more preferably 160 ° C. or higher, and even more preferably 170 ° C. or higher. The upper limit is preferably 500 ° C. or lower, more preferably 450 ° C. or lower, even more preferably 350 ° C. or lower, further preferably 250 ° C. or lower, and more preferably 220 ° C. or lower. Even more preferred.
Heating is preferably performed at a rate of 1 to 12 ° C./min from the temperature at the start of heating to the maximum heating temperature, more preferably 2 to 10 ° C./min, even more preferably 3 to 10 ° C./min. By setting the heating rate to 1 ° C./min or more, the amine can be prevented from being excessively volatilized while securing the productivity. By setting the heating rate to 12 ° C./min or less, the cured film can be cured. The residual stress can be reduced.
The temperature at the start of heating is preferably from 20 ° C to 150 ° C, more preferably from 20 ° C to 130 ° C, even more preferably from 25 ° C to 120 ° C. The temperature at the start of heating refers to the temperature at which the step of heating to the maximum heating temperature is started. For example, when the resin composition is applied on a substrate and then dried, it is the temperature of the film (layer) after the drying, for example, 30 to 200 ° C. lower than the boiling point of the solvent contained in the resin composition. It is preferable to gradually raise the temperature from the temperature.
The heating time (heating time at the maximum heating temperature) is preferably from 10 to 360 minutes, more preferably from 20 to 300 minutes, even more preferably from 30 to 240 minutes.
In particular, when a multilayer laminate is formed, the heating temperature is preferably from 180 ° C. to 320 ° C., and more preferably from 180 ° C. to 260 ° C., from the viewpoint of adhesion between the cured films. Although the reason is not clear, it is considered that the ethynyl groups of the polymer precursor between the layers are undergoing a crosslinking reaction at this temperature.
さらに、加熱後冷却してもよく、この場合の冷却速度としては、1~5℃/分であることが好ましい。 The heating may be performed stepwise. As an example, the temperature is raised from 25 ° C. to 180 ° C. at 3 ° C./min, maintained at 180 ° C. for 60 minutes, raised from 180 ° C. to 200 ° C. at 2 ° C./min, and maintained at 200 ° C. for 120 minutes. , Etc., may be performed. The heating temperature in the pretreatment step is preferably from 100 to 200 ° C, more preferably from 110 to 190 ° C, and even more preferably from 120 to 185 ° C. In this pretreatment step, it is also preferable to perform the treatment while irradiating ultraviolet rays as described in US Pat. No. 9,159,547. By such a pretreatment step, the characteristics of the film can be improved. The pretreatment step may be performed in a short time of about 10 seconds to 2 hours, more preferably 15 seconds to 30 minutes. The pretreatment may include two or more steps. For example, pretreatment step 1 may be performed at a temperature in the range of 100 to 150 ° C., and then pretreatment step 2 may be performed at a temperature in the range of 150 to 200 ° C.
Furthermore, cooling may be performed after heating, and the cooling rate in this case is preferably 1 to 5 ° C./min.
本発明の製造方法は、現像処理後の樹脂組成物層の表面に金属層を形成する金属層形成工程を含んでいることが好ましい。
金属層としては、特に限定なく、既存の金属種を使用することができ、銅、アルミニウム、ニッケル、バナジウム、チタン、クロム、コバルト、金およびタングステンが例示され、銅およびアルミニウムがより好ましく、銅がさらに好ましい。
金属層の形成方法は、特に限定なく、既存の方法を適用することができる。例えば、特開2007-157879号公報、特表2001-521288号公報、特開2004-214501号公報、特開2004-101850号公報に記載された方法を使用することができる。例えば、フォトリソグラフィ、リフトオフ、電解メッキ、無電解メッキ、エッチング、印刷、およびこれらを組み合わせた方法などが考えられる。より具体的には、スパッタリング、フォトリソグラフィおよびエッチングを組み合わせたパターニング方法、フォトリソグラフィと電解メッキを組み合わせたパターニング方法が挙げられる。
金属層の厚さとしては、最も厚肉部で、0.1~50μmが好ましく、1~10μmがより好ましい。 <Metal layer forming step>
The production method of the present invention preferably includes a metal layer forming step of forming a metal layer on the surface of the resin composition layer after the development processing.
The metal layer is not particularly limited, and any existing metal species can be used.Examples include copper, aluminum, nickel, vanadium, titanium, chromium, cobalt, gold, and tungsten, and copper and aluminum are more preferable, and copper is preferable. More preferred.
The method for forming the metal layer is not particularly limited, and an existing method can be applied. For example, the methods described in JP-A-2007-157879, JP-T-2001-521288, JP-A-2004-214501, and JP-A-2004-101850 can be used. For example, photolithography, lift-off, electrolytic plating, electroless plating, etching, printing, and a method combining these are conceivable. More specifically, a patterning method that combines sputtering, photolithography, and etching, and a patterning method that combines photolithography and electrolytic plating can be given.
The thickness of the metal layer at the thickest part is preferably 0.1 to 50 μm, more preferably 1 to 10 μm.
本発明の製造方法は、さらに、積層工程を含むことが好ましい。
積層工程とは、硬化膜(樹脂層)または金属層の表面に、再度、(a)膜形成工程(層形成工程)、(b)露光工程、(c)現像処理工程、(d)加熱工程を、この順に行うことを含む一連の工程である。ただし、(a)の膜形成工程のみを繰り返す態様であってもよい。また、(d)加熱工程は積層の最後または中間に一括して行う態様としてもよい。すなわち、(a)~(c)の工程を所定の回数繰り返し行い、その後に(d)の加熱をすることで、積層された樹脂組成物層を一括で硬化する態様としてもよい。また、(c)現像工程の後には(e)金属層形成工程を含んでもよく、このときにも都度(d)の加熱を行っても、所定回数積層させた後に一括して(d)の加熱を行ってもよい。積層工程には、さらに、上記乾燥工程や加熱工程等を適宜含んでいてもよいことは言うまでもない。
積層工程後、さらに積層工程を行う場合には、上記加熱工程後、上記露光工程後、または、上記金属層形成工程後に、さらに、表面活性化処理工程を行ってもよい。表面活性化処理としては、プラズマ処理が例示される。
上記積層工程は、2~5回行うことが好ましく、3~5回行うことがより好ましい。
例えば、樹脂層/金属層/樹脂層/金属層/樹脂層/金属層のような、樹脂層が3層以上7層以下の構成が好ましく、3層以上5層以下がさらに好ましい。
本発明では特に、金属層を設けた後、さらに、上記金属層を覆うように、上記樹脂組成物の硬化膜(樹脂層)を形成する態様が好ましい。具体的には、(a)膜形成工程、(b)露光工程、(c)現像工程、(e)金属層形成工程、(d)加熱工程の順序で繰り返す態様、あるいは、(a)膜形成工程、(b)露光工程、(c)現像工程、(e)金属層形成工程の順序で繰り返し、最後または中間に一括して(d)加熱工程を設ける態様が挙げられる。樹脂組成物層(樹脂)を積層する積層工程と、金属層形成工程を交互に行うことにより、樹脂組成物層(樹脂層)と金属層を交互に積層することができる。 <Lamination process>
It is preferable that the manufacturing method of the present invention further includes a lamination step.
The laminating step means (a) a film forming step (layer forming step), (b) an exposing step, (c) a developing step, and (d) a heating step on the surface of the cured film (resin layer) or the metal layer. Are performed in this order. However, an embodiment in which only the film forming step (a) is repeated may be employed. Further, the heating step (d) may be performed at the end of or in the middle of the lamination. That is, the steps (a) to (c) may be repeated a predetermined number of times, and then the heating of (d) may be performed to cure the laminated resin composition layers at once. In addition, (e) a metal layer forming step may be included after the (c) developing step. In this case, even if heating (d) is performed each time, after laminating a predetermined number of times, (d) Heating may be performed. Needless to say, the laminating step may further include the above-mentioned drying step, heating step and the like as appropriate.
When the laminating step is further performed after the laminating step, a surface activation treatment step may be further performed after the heating step, after the exposing step, or after the metal layer forming step. As the surface activation treatment, a plasma treatment is exemplified.
The above lamination step is preferably performed 2 to 5 times, more preferably 3 to 5 times.
For example, a configuration of three to seven resin layers is preferable, such as a resin layer / metal layer / resin layer / metal layer / resin layer / metal layer, and more preferably three to five layers.
In the present invention, it is particularly preferable that after the metal layer is provided, a cured film (resin layer) of the resin composition is further formed so as to cover the metal layer. Specifically, a mode in which (a) a film forming step, (b) an exposing step, (c) a developing step, (e) a metal layer forming step, and (d) a heating step are repeated in this order, or (a) a film forming step Step, (b) exposure step, (c) development step, and (e) repetition in the order of the metal layer forming step, and (d) heating step is provided at the end or in the middle in a lump. By alternately performing the laminating step of laminating the resin composition layer (resin) and the metal layer forming step, the resin composition layer (resin layer) and the metal layer can be laminated alternately.
[ピロメリット酸二無水物、4,4’-ジアミノジフェニルエーテルおよびベンジルアルコールからのポリイミド前駆体(A-1:ラジカル重合性基を有さないポリイミド前駆体)の合成]
14.06g(64.5ミリモル)のピロメリット酸二無水物(140℃で12時間乾燥)と、14.22g(131.58ミリモル)のベンジルアルコールを、50mLのN-メチルピロリドンに懸濁させ、モレキュラーシーブで乾燥させた。懸濁液を100℃で3時間加熱した。反応混合物を室温に冷却し、21.43g(270.9ミリモル)のピリジンおよび90mLのN-メチルピロリドンを加えた。次いで、反応混合物を-10℃に冷却し、温度を-10±4℃に保ちながら16.12g(135.5ミリモル)のSOCl2を10分かけて加えた。SOCl2を加えている間、粘度が増加した。50mLのN-メチルピロリドンで希釈した後、反応混合物を室温で2時間撹拌した。次いで、100mLのN-メチルピロリドンに11.08g(58.7ミリモル)の4,4’-ジアミノジフェニルエーテルを溶解させた溶液を、-5~0℃で20分かけて反応混合物に滴下した。次いで、反応混合物を0℃で1時間反応させたのち、エタノールを70g加えて、室温で1晩撹拌した。次いで、5リットルの水の中でポリイミド前駆体を沈殿させ、水-ポリイミド前駆体混合物を5000rpmの速度で15分間撹拌した。ポリイミド前駆体をろ過して除き、4リットルの水の中で再度30分間撹拌し再びろ過した。次いで、得られたポリイミド前駆体を減圧下で、45℃で3日間乾燥した。このポリイミド前駆体の重量平均分子量は、18,000であった。 <Synthesis example 1>
[Synthesis of polyimide precursor (A-1: polyimide precursor having no radical polymerizable group) from pyromellitic dianhydride, 4,4'-diaminodiphenyl ether and benzyl alcohol]
14.06 g (64.5 mmol) of pyromellitic dianhydride (dried at 140 ° C. for 12 hours) and 14.22 g (131.58 mmol) of benzyl alcohol were suspended in 50 mL of N-methylpyrrolidone. , Dried over molecular sieves. The suspension was heated at 100 ° C. for 3 hours. The reaction mixture was cooled to room temperature and 21.43 g (270.9 mmol) of pyridine and 90 mL of N-methylpyrrolidone were added. The reaction mixture was then cooled to −10 ° C. and 16.12 g (135.5 mmol) SOCl 2 was added over 10 minutes keeping the temperature at −10 ± 4 ° C. The viscosity increased during the addition of SOCl 2 . After dilution with 50 mL of N-methylpyrrolidone, the reaction mixture was stirred at room temperature for 2 hours. Next, a solution of 11.08 g (58.7 mmol) of 4,4′-diaminodiphenyl ether dissolved in 100 mL of N-methylpyrrolidone was added dropwise to the reaction mixture at −5 to 0 ° C. over 20 minutes. Next, the reaction mixture was reacted at 0 ° C. for 1 hour, and then 70 g of ethanol was added, followed by stirring at room temperature overnight. The polyimide precursor was then precipitated in 5 liters of water, and the water-polyimide precursor mixture was stirred at 5000 rpm for 15 minutes. The polyimide precursor was removed by filtration, stirred again for 30 minutes in 4 liters of water, and filtered again. Next, the obtained polyimide precursor was dried under reduced pressure at 45 ° C. for 3 days. The weight average molecular weight of this polyimide precursor was 18,000.

[ピロメリット酸二無水物、4,4’-ジアミノジフェニルエーテルおよび2-ヒドロキシエチルメタクリレートからのポリイミド前駆体(A-2:ラジカル重合性基を有するポリイミド前駆体)の合成]
14.06g(64.5ミリモル)のピロメリット酸二無水物(140℃で12時間乾燥した)と、16.8g(129ミリモル)の2-ヒドロキシエチルメタクリレートと、0.05gのハイドロキノンと、20.4gのピリジン(258ミリモル)と、100gのダイグライム(ジエチレングリコールジメチルエーテル)を混合し、60℃の温度で18時間撹拌して、ピロメリット酸と2-ヒドロキシエチルメタクリレートのジエステルを製造した。次いで、得られたジエステルをSOCl2により塩素化した後、合成例1と同様の方法で4,4’-ジアミノジフェニルエーテルでポリイミド前駆体に変換し、合成例1と同様の方法でポリイミド前駆体を得た。このポリイミド前駆体の重量平均分子量は、19,000であった。 <Synthesis Example 2>
[Synthesis of a polyimide precursor (A-2: a polyimide precursor having a radical polymerizable group) from pyromellitic dianhydride, 4,4'-diaminodiphenyl ether and 2-hydroxyethyl methacrylate]
14.06 g (64.5 mmol) of pyromellitic dianhydride (dried at 140 ° C. for 12 hours), 16.8 g (129 mmol) of 2-hydroxyethyl methacrylate, 0.05 g of hydroquinone and 20 0.4 g of pyridine (258 mmol) and 100 g of diglyme (diethylene glycol dimethyl ether) were mixed and stirred at a temperature of 60 ° C. for 18 hours to produce a diester of pyromellitic acid and 2-hydroxyethyl methacrylate. Next, the obtained diester is chlorinated with SOCl 2 , and then converted into a polyimide precursor with 4,4′-diaminodiphenyl ether in the same manner as in Synthesis Example 1, and the polyimide precursor is converted in the same manner as in Synthesis Example 1. Obtained. The weight average molecular weight of this polyimide precursor was 19,000.

[4,4’-オキシジフタル酸無水物、4,4’-ジアミノジフェニルエーテルおよび2-ヒドロキシエチルメタクリレートからのポリイミド前駆体(A-3:ラジカル重合性基を有するポリイミド前駆体)の合成]
20.0g(64.5ミリモル)の4,4’-オキシジフタル酸無水物(140℃で12時間乾燥した)と、16.8g(129ミリモル)の2-ヒドロキシエチルメタクリレートと、0.05gのハイドロキノンと、20.4gのピリジン(258ミリモル)と、100gのダイグライムとを混合し、60℃の温度で18時間撹拌して、4,4’-オキシジフタル酸と2-ヒドロキシエチルメタクリレートのジエステルを製造した。次いで、得られたジエステルをSOCl2により塩素化した後、合成例1と同様の方法で4,4’-ジアミノジフェニルエーテルでポリイミド前駆体に変換し、合成例1と同様の方法でポリイミド前駆体を得た。このポリイミド前駆体の重量平均分子量は、18,000であった。 <Synthesis Example 3>
[Synthesis of polyimide precursor (A-3: polyimide precursor having radically polymerizable group) from 4,4'-oxydiphthalic anhydride, 4,4'-diaminodiphenyl ether and 2-hydroxyethyl methacrylate]
20.0 g (64.5 mmol) of 4,4′-oxydiphthalic anhydride (dried at 140 ° C. for 12 hours), 16.8 g (129 mmol) of 2-hydroxyethyl methacrylate and 0.05 g of hydroquinone And 20.4 g of pyridine (258 mmol) and 100 g of diglyme were stirred at a temperature of 60 ° C. for 18 hours to produce a diester of 4,4′-oxydiphthalic acid and 2-hydroxyethyl methacrylate. . Next, the obtained diester is chlorinated with SOCl 2 , and then converted into a polyimide precursor with 4,4′-diaminodiphenyl ether in the same manner as in Synthesis Example 1, and the polyimide precursor is converted in the same manner as in Synthesis Example 1. Obtained. The weight average molecular weight of this polyimide precursor was 18,000.

[4,4’-オキシジフタル酸無水物、4,4’-ジアミノ-2,2’-ジメチルビフェニル(オルトトリジン)および2-ヒドロキシエチルメタクリレートからのポリイミド前駆体(A-4:ラジカル重合性基を有するポリイミド前駆体)の合成]
20.0g(64.5ミリモル)の4,4’-オキシジフタル酸無水物(140℃で12時間乾燥した)と、16.8g(129ミリモル)の2-ヒドロキシエチルメタクリレートと、0.05gのハイドロキノンと、20.4gのピリジン(258ミリモル)と、100gのダイグライムとを混合し、60℃の温度で18時間撹拌して、4,4’-オキシジフタル酸と2-ヒドロキシエチルメタクリレートのジエステルを製造した。次いで、得られたジエステルをSOCl2により塩素化した後、合成例1と同様の方法で4,4’-ジアミノ-2,2’-ジメチルビフェニルでポリイミド前駆体に変換し、合成例1と同様の方法でポリイミド前駆体を得た。このポリイミド前駆体の重量平均分子量は、19,000であった。 <Synthesis example 4>
[Polyimide precursor from 4,4'-oxydiphthalic anhydride, 4,4'-diamino-2,2'-dimethylbiphenyl (orthotrizine) and 2-hydroxyethyl methacrylate (A-4: having a radical polymerizable group Synthesis of Polyimide Precursor)
20.0 g (64.5 mmol) of 4,4′-oxydiphthalic anhydride (dried at 140 ° C. for 12 hours), 16.8 g (129 mmol) of 2-hydroxyethyl methacrylate and 0.05 g of hydroquinone And 20.4 g of pyridine (258 mmol) and 100 g of diglyme were stirred at a temperature of 60 ° C. for 18 hours to produce a diester of 4,4′-oxydiphthalic acid and 2-hydroxyethyl methacrylate. . Next, the obtained diester was chlorinated with SOCl 2 , and then converted to a polyimide precursor with 4,4′-diamino-2,2′-dimethylbiphenyl in the same manner as in Synthesis Example 1. Thus, a polyimide precursor was obtained. The weight average molecular weight of this polyimide precursor was 19,000.

下記表に記載の成分を混合し、各樹脂組成物を得た。得られた樹脂組成物を、細孔の幅が0.8μmのフィルターを通して加圧ろ過した。 <Examples and Comparative Examples>
The components shown in the following table were mixed to obtain each resin composition. The obtained resin composition was subjected to pressure filtration through a filter having a pore width of 0.8 μm.



(A)ポリイミド前駆体
A-1~A-4:上記で合成したポリイミド前駆体A-1~A-4
(B)重合性モノマー:
B-1、B-2:下記構造の化合物

C-1~C-3:下記構造の化合物

D-1、D-2:下記構造の化合物

E-1~E-3:下記構造の化合物

F-1、F-2:下記構造の化合物

G-1、G-2:下記構造の化合物

H-1、H-2:下記構造の化合物

I-1:γ-ブチロラクトン
I-2:ジメチルスルホキシド The raw materials described in the above table are as follows.
(A) Polyimide precursors A-1 to A-4: polyimide precursors A-1 to A-4 synthesized above
(B) Polymerizable monomer:
B-1, B-2: compounds having the following structure
各樹脂組成物をシリコンウェハ上にスピンコート法により適用し、樹脂組成物層を形成した。得られた樹脂組成物層を適用したシリコンウェハをホットプレート上で、100℃で4分間乾燥し、シリコンウェハ上に20μmの厚さの均一な樹脂組成物層とした。シリコンウェハ上の樹脂組成物層を、ブロードバンド露光機(ウシオ電機株式会社製:UX-1000SN-EH01)を用いて、400mJ/cm2の露光エネルギーで露光し、露光した感光性樹脂組成物層(樹脂層)を、窒素雰囲気下で、5℃/分の昇温速度で昇温し、180℃に達した後、2時間加熱した。硬化後の樹脂層を3%フッ化水素酸溶液に浸漬し、シリコンウェハから樹脂層を剥離し、硬化膜を得た。 <Production of cured film>
Each resin composition was applied on a silicon wafer by a spin coating method to form a resin composition layer. The silicon wafer to which the obtained resin composition layer was applied was dried on a hot plate at 100 ° C. for 4 minutes to form a uniform resin composition layer having a thickness of 20 μm on the silicon wafer. The resin composition layer on the silicon wafer was exposed to light at an exposure energy of 400 mJ / cm 2 using a broadband exposure machine (UX-1000SN-EH01 manufactured by Ushio Inc.), and the exposed photosensitive resin composition layer ( The resin layer) was heated at a rate of 5 ° C./min in a nitrogen atmosphere, and after reaching 180 ° C., was heated for 2 hours. The cured resin layer was immersed in a 3% hydrofluoric acid solution, and the resin layer was separated from the silicon wafer to obtain a cured film.
得られた硬化膜の破断伸びを測定した。硬化膜の破断伸びは、引張り試験機(テンシロン)を用い、クロスヘッドスピード300mm/分、幅10mm、試料長50mmとして、フィルムの長手方向および幅方向について、25℃、65%RH(相対湿度)の環境下にて、JIS-K6251(日本工業規格)に準拠して測定した。破断伸びは、Eb=(Lb-L0)/L0(Eb:破断伸び、L0:試験前の試験片の長さ、Lb:試験片が切断した時の試験片の長さ)で算出した。評価は破断伸びを各10回ずつ測定し、平均値を用いた。結果は下記のとおり区分して評価した。
A:破断伸びが60%以上である
B:破断伸びが50%以上60%未満である
C:破断伸びが50%未満である <Elongation at break>
The breaking elongation of the obtained cured film was measured. The elongation at break of the cured film was measured using a tensile tester (Tensilon) at a crosshead speed of 300 mm / min, a width of 10 mm, and a sample length of 50 mm. The measurement was carried out in accordance with JIS-K6251 (Japanese Industrial Standard) under the following environment. Elongation at break, Eb = (L b -L 0 ) / L 0 (E b: elongation at break, L 0: length of the test piece before the test, L b: the length of the specimen when the specimen was cut ). For evaluation, the elongation at break was measured 10 times each, and the average value was used. The results were classified and evaluated as follows.
A: Elongation at break is 60% or more B: Elongation at break is 50% or more and less than 60% C: Elongation at break is less than 50%
得られた硬化膜について下記の薬液に下記の条件で浸漬し、溶解速度を算定した。
薬品:ジメチルスルホキシド(DMSO)と25質量%のテトラメチルアンモニウムヒドロキシド(TMAH)溶液の90:10(質量比)の混合物
評価条件:溶液中で樹脂層を75℃で15分間浸漬して前後の膜厚を比較し、溶解速度(nm/分)を算出した。
A:250nm/分未満
B:250nm/分以上500nm/分未満
C:500nm/分以上 <Chemical resistance>
The obtained cured film was immersed in the following chemical solution under the following conditions, and the dissolution rate was calculated.
Chemicals: 90:10 (mass ratio) mixture of dimethyl sulfoxide (DMSO) and 25% by mass of tetramethylammonium hydroxide (TMAH) solution Evaluation conditions: Before and after immersing the resin layer in the solution at 75 ° C. for 15 minutes The film thickness was compared and the dissolution rate (nm / min) was calculated.
A: less than 250 nm / min B: 250 nm / min or more and less than 500 nm / min C: 500 nm / min or more
各樹脂組成物を、シリコンウェハ上にスピンコートした。樹脂組成物を適用したシリコンウェハをホットプレート上で、100℃で4分間乾燥し、シリコンウェハ上に20μmの膜厚の均一な樹脂組成物層を形成した。シリコンウェハ上の樹脂組成物層を、ステッパー(Nikon NSR 2005 i9C)を用いて露光した。露光はi線で行い、波長365nmにおいて、200、300、400、500、600、700、800mJ/cm2の各露光エネルギーで、5μmから25μmまで1μm刻みのラインアンドスペースのフォトマスクを使用して、露光を行って、樹脂層を得た。この樹脂層を、シクロペンタノンで60秒間現像した。得られた樹脂層(ラインパターン)の線幅が小さければ小さいほど微細なパターンを形成可能であることを表し、好ましい結果となる。また、形成可能な最小線幅が露光量の変動に対して変化しにくいほど、微細パターン形成における露光量の任意性が増大し、好ましい結果となる。
A:線幅が10μm未満である
B:線幅が10μm以上20μm未満である
C:線幅が20μm以上であるか、または、エッジの鋭さを持つ線幅を有するパターンが得られなかった。 <Lithography evaluation>
Each resin composition was spin-coated on a silicon wafer. The silicon wafer to which the resin composition was applied was dried on a hot plate at 100 ° C. for 4 minutes to form a uniform resin composition layer having a thickness of 20 μm on the silicon wafer. The resin composition layer on the silicon wafer was exposed using a stepper (Nikon NSR 2005 i9C). Exposure is performed with i-line, at a wavelength of 365 nm, using a line and space photomask in increments of 1 μm from 5 μm to 25 μm at exposure energy of 200, 300, 400, 500, 600, 700, 800 mJ / cm 2. Exposure was performed to obtain a resin layer. This resin layer was developed with cyclopentanone for 60 seconds. The smaller the line width of the obtained resin layer (line pattern), the finer the pattern can be formed, which is a preferable result. In addition, as the minimum line width that can be formed hardly changes with respect to the fluctuation of the exposure amount, the arbitrariness of the exposure amount in the formation of the fine pattern increases, and a preferable result is obtained.
A: The line width is less than 10 μm. B: The line width is 10 μm or more and less than 20 μm.

Claims (18)
- ポリイミド前駆体と、
熱塩基発生剤と、
エポキシ基、オキセタニル基、メチロール基、アルコキシメチル基、フェノール基、マレイミド基、シアネート基及びブロックイソシアネート基からなる群から選択される官能基を複数有する熱硬化性化合物と、
を含む樹脂組成物。 A polyimide precursor,
A thermal base generator,
A thermosetting compound having a plurality of functional groups selected from the group consisting of an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group and a blocked isocyanate group,
A resin composition comprising: - 前記熱硬化性化合物は、下記式(TC1)で表される化合物である、請求項1に記載の樹脂組成物;
X1-(Y1)n ・・・(TC1)
式(TC1)中、X1はn価の連結基を表し、Y1はエポキシ基、オキセタニル基、メチロール基、アルコキシメチル基、フェノール基、マレイミド基、シアネート基またはブロックイソシアネート基を表し、nは2以上の整数を表す。 The resin composition according to claim 1, wherein the thermosetting compound is a compound represented by the following formula (TC1);
X 1- (Y 1 ) n ... (TC1)
In the formula (TC1), X 1 represents an n-valent linking group, Y 1 represents an epoxy group, an oxetanyl group, a methylol group, an alkoxymethyl group, a phenol group, a maleimide group, a cyanate group or a blocked isocyanate group, and n is Represents an integer of 2 or more. - 前記式(TC1)のX1は環状構造を含む、請求項2に記載の樹脂組成物。 X 1 comprises a cyclic structure, a resin composition according to claim 2 of the formula (TC1).
- 前記熱硬化性化合物は、アルコキシメチル基を複数有する化合物である、請求項1~3のいずれか1項に記載の樹脂組成物。 The resin composition according to any one of claims 1 to 3, wherein the thermosetting compound is a compound having a plurality of alkoxymethyl groups.
- 前記熱硬化性化合物は、メトキシメチル基を複数有する化合物である、請求項1~3のいずれか1項に記載の樹脂組成物。 樹脂 The resin composition according to any one of claims 1 to 3, wherein the thermosetting compound is a compound having a plurality of methoxymethyl groups.
- 前記熱塩基発生剤の塩基発生温度は、前記熱硬化性化合物の硬化開始温度よりも低い請求項1~5のいずれか1項に記載の樹脂組成物。 The resin composition according to any one of claims 1 to 5, wherein the base generation temperature of the thermal base generator is lower than the curing start temperature of the thermosetting compound.
- (メタ)アクリロイル基を複数有する重合性モノマーの含有量が樹脂組成物の全固形分中20質量%以下である、請求項1~6のいずれか1項に記載の樹脂組成物。 The resin composition according to any one of claims 1 to 6, wherein the content of the polymerizable monomer having a plurality of (meth) acryloyl groups is 20% by mass or less based on the total solid content of the resin composition.
- 前記ポリイミド前駆体がラジカル重合性基を含む、請求項1~7のいずれか1項に記載の樹脂組成物。 (8) The resin composition according to any one of (1) to (7), wherein the polyimide precursor contains a radical polymerizable group.
- 前記ポリイミド前駆体が下記式(1)で表される構成単位を有する、請求項1~8のいずれか1項に記載の樹脂組成物;

- 前記式(1)におけるR113およびR114の少なくとも一方がラジカル重合性基を含む、請求項9に記載の樹脂組成物。 At least one comprises a radical polymerizable group, a resin composition according to claim 9 of R 113 and R 114 in the formula (1).
- 更に、光重合開始剤を含む請求項8または10に記載の樹脂組成物。 (11) The resin composition according to (8) or (10), further comprising a photopolymerization initiator.
- 再配線層用層間絶縁膜の形成に用いられる、請求項1~11のいずれか1項に記載の樹脂組成物。 The resin composition according to any one of claims 1 to 11, which is used for forming an interlayer insulating film for a redistribution layer.
- 請求項1~12のいずれか1項に記載の樹脂組成物を硬化して得られる硬化膜。 A cured film obtained by curing the resin composition according to any one of claims 1 to 12.
- 請求項13に記載の硬化膜を2層以上有し、前記2層の硬化膜の間に金属層を有する、積層体。 A laminate having two or more cured films according to claim 13, and a metal layer between the two cured films.
- 請求項1~12のいずれか1項に記載の樹脂組成物を基板に適用して膜を形成する膜形成工程を含む、硬化膜の製造方法。 A method for producing a cured film, comprising a film forming step of forming a film by applying the resin composition according to any one of claims 1 to 12 to a substrate.
- 前記膜を露光する露光工程および前記膜を現像する現像工程を有する、請求項15に記載の硬化膜の製造方法。 The method for producing a cured film according to claim 15, further comprising an exposure step of exposing the film and a developing step of developing the film.
- 前記膜を80~450℃で加熱する工程を含む、請求項16に記載の硬化膜の製造方法。 17. The method for producing a cured film according to claim 16, comprising a step of heating the film at 80 to 450 ° C.
- 請求項13に記載の硬化膜または請求項14に記載の積層体を有する、半導体デバイス。 A semiconductor device having the cured film according to claim 13 or the laminate according to claim 14.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201980062870.2A CN112752798A (en) | 2018-09-27 | 2019-09-24 | Resin composition, cured film, laminate, method for producing cured film, and semiconductor device |
| JP2020549195A JP7237978B2 (en) | 2018-09-27 | 2019-09-24 | Resin composition, cured film, laminate, method for producing cured film, and semiconductor device |
| KR1020217008567A KR20210048519A (en) | 2018-09-27 | 2019-09-24 | Resin composition, cured film, laminate, method for producing cured film, and semiconductor device |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018181637 | 2018-09-27 | ||
| JP2018-181637 | 2018-09-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020066976A1 true WO2020066976A1 (en) | 2020-04-02 |
Family
ID=69952378
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2019/037188 WO2020066976A1 (en) | 2018-09-27 | 2019-09-24 | Resin composition, cured film, multilayer body, method for producing cured film and semiconductor device |
Country Status (5)
| Country | Link |
|---|---|
| JP (1) | JP7237978B2 (en) |
| KR (1) | KR20210048519A (en) |
| CN (1) | CN112752798A (en) |
| TW (1) | TW202024187A (en) |
| WO (1) | WO2020066976A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3896521A1 (en) * | 2020-04-14 | 2021-10-20 | Shin-Etsu Chemical Co., Ltd. | Polymer, photosensitive resin composition, patterning method, method of forming cured film, interlayer insulating film, surface protective film, and electronic component |
| JPWO2022059622A1 (en) * | 2020-09-16 | 2022-03-24 | ||
| WO2022059621A1 (en) * | 2020-09-16 | 2022-03-24 | 富士フイルム株式会社 | Resin composition, cured object, layered product, method for producing cured object, and semiconductor device |
| US11572442B2 (en) | 2020-04-14 | 2023-02-07 | International Business Machines Corporation | Compound, polyimide resin and method of producing the same, photosensitive resin composition, patterning method and method of forming cured film, interlayer insulating film, surface protective film, and electronic component |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014066764A (en) * | 2012-09-24 | 2014-04-17 | Jsr Corp | Radiation-sensitive resin composition, polyimide film, semiconductor element, and organic el element |
| WO2017038664A1 (en) * | 2015-08-31 | 2017-03-09 | 富士フイルム株式会社 | Composition, cured film, method for manufacturing cured film, method for manufacturing semiconductor device, and semiconductor device |
| WO2018025738A1 (en) * | 2016-08-01 | 2018-02-08 | 富士フイルム株式会社 | Photosensitive resin composition, cured film, laminate, method for producing cured film, method for producing laminate, and semiconductor device |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH08257497A (en) * | 1995-03-23 | 1996-10-08 | Nippon Steel Corp | Formation of high-hardness colorful-pattern coating film and coated metallic sheet |
| JP4046563B2 (en) | 2002-01-25 | 2008-02-13 | 旭化成エレクトロニクス株式会社 | High heat-resistant photosensitive resin composition |
| JP2007056196A (en) * | 2005-08-26 | 2007-03-08 | Tokyo Institute Of Technology | Polyimide precursor composition, method for producing polyimide film and semiconductor device |
| JP6136486B2 (en) | 2013-04-08 | 2017-05-31 | 日立化成デュポンマイクロシステムズ株式会社 | Resin composition and pattern forming method using the same |
| TWI671343B (en) * | 2014-06-27 | 2019-09-11 | 日商富士軟片股份有限公司 | Thermosetting resin composition, cured film, method for producing cured film, and semiconductor device |
| WO2016194769A1 (en) * | 2015-05-29 | 2016-12-08 | 富士フイルム株式会社 | Polyimide precursor composition, photosensitive resin composition, cured film, method for producing cured film, semiconductor device, and method for producing polyimide precursor composition |
| TWI634135B (en) * | 2015-12-25 | 2018-09-01 | 日商富士軟片股份有限公司 | Resin, composition, cured film, method for producing cured film, and semiconductor element |
| JP6704048B2 (en) * | 2016-06-29 | 2020-06-03 | 富士フイルム株式会社 | Negative-type photosensitive resin composition, cured film, cured film manufacturing method, semiconductor device, laminated body manufacturing method, semiconductor device manufacturing method, and polyimide precursor |
-
2019
- 2019-09-24 WO PCT/JP2019/037188 patent/WO2020066976A1/en active Application Filing
- 2019-09-24 CN CN201980062870.2A patent/CN112752798A/en active Pending
- 2019-09-24 JP JP2020549195A patent/JP7237978B2/en active Active
- 2019-09-24 KR KR1020217008567A patent/KR20210048519A/en not_active Application Discontinuation
- 2019-09-26 TW TW108134755A patent/TW202024187A/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2014066764A (en) * | 2012-09-24 | 2014-04-17 | Jsr Corp | Radiation-sensitive resin composition, polyimide film, semiconductor element, and organic el element |
| WO2017038664A1 (en) * | 2015-08-31 | 2017-03-09 | 富士フイルム株式会社 | Composition, cured film, method for manufacturing cured film, method for manufacturing semiconductor device, and semiconductor device |
| WO2018025738A1 (en) * | 2016-08-01 | 2018-02-08 | 富士フイルム株式会社 | Photosensitive resin composition, cured film, laminate, method for producing cured film, method for producing laminate, and semiconductor device |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3896521A1 (en) * | 2020-04-14 | 2021-10-20 | Shin-Etsu Chemical Co., Ltd. | Polymer, photosensitive resin composition, patterning method, method of forming cured film, interlayer insulating film, surface protective film, and electronic component |
| CN113527680A (en) * | 2020-04-14 | 2021-10-22 | 信越化学工业株式会社 | Polymer, photosensitive resin composition, pattern forming method, cured film, and electronic component |
| US11333975B2 (en) | 2020-04-14 | 2022-05-17 | International Business Machines Corporation | Polymer, photosensitive resin composition, patterning method, method of forming cured film, interlayer insulating film, surface protective film, and electronic component |
| US11572442B2 (en) | 2020-04-14 | 2023-02-07 | International Business Machines Corporation | Compound, polyimide resin and method of producing the same, photosensitive resin composition, patterning method and method of forming cured film, interlayer insulating film, surface protective film, and electronic component |
| CN113527680B (en) * | 2020-04-14 | 2023-04-28 | 信越化学工业株式会社 | Polymer, photosensitive resin composition, pattern forming method, cured film, and electronic component |
| JPWO2022059622A1 (en) * | 2020-09-16 | 2022-03-24 | ||
| WO2022059621A1 (en) * | 2020-09-16 | 2022-03-24 | 富士フイルム株式会社 | Resin composition, cured object, layered product, method for producing cured object, and semiconductor device |
| JPWO2022059621A1 (en) * | 2020-09-16 | 2022-03-24 | ||
| WO2022059622A1 (en) * | 2020-09-16 | 2022-03-24 | 富士フイルム株式会社 | Resin composition, cured object, layered product, method for producing cured object, and semiconductor device |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2020066976A1 (en) | 2021-09-24 |
| TW202024187A (en) | 2020-07-01 |
| KR20210048519A (en) | 2021-05-03 |
| CN112752798A (en) | 2021-05-04 |
| JP7237978B2 (en) | 2023-03-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6813602B2 (en) | Photosensitive resin compositions, heterocyclic-containing polymer precursors, cured films, laminates, methods for producing cured films, and semiconductor devices. | |
| JP6808831B2 (en) | Photosensitive resin compositions, cured films, laminates, methods for producing cured films, semiconductor devices and compounds | |
| JP7237978B2 (en) | Resin composition, cured film, laminate, method for producing cured film, and semiconductor device | |
| JP7333383B2 (en) | Curable resin composition, cured film, laminate, method for producing cured film, and semiconductor device | |
| CN112639616A (en) | Photosensitive resin composition, cured film, laminate, method for producing cured film, and semiconductor device | |
| WO2020066315A1 (en) | Photosensitive resin composition, cured product, layered product, production method for cured product, and semiconductor device | |
| JP7177249B2 (en) | Curable resin composition, cured film, laminate, method for producing cured film, and semiconductor device | |
| JP7023379B2 (en) | Resin composition, cured film, laminate, method for manufacturing cured film, and semiconductor device | |
| JP2023027046A (en) | Curable resin composition, cured film, laminate, production method of cured film, semiconductor device, and polymer precursor | |
| WO2020189481A1 (en) | Curable resin composition, cured film, multilayer body, method for producing cured film, and semiconductor device | |
| WO2020054226A1 (en) | Photosensitive resin composition, cured film, laminate, method for producing cured film, and semiconductor device | |
| JPWO2019146611A1 (en) | Photosensitive resin composition, resin, cured film, laminate, method for manufacturing cured film, semiconductor device | |
| JP7351896B2 (en) | Curable resin composition, cured film, laminate, method for producing cured film, semiconductor device, and thermal base generator | |
| JP7086882B2 (en) | Curable resin composition, cured film, laminate, method for manufacturing cured film, and semiconductor device | |
| JPWO2019189327A1 (en) | Photosensitive resin compositions, cured films, laminates and their applications | |
| JP7078744B2 (en) | Resin composition, cured film, laminate, method for manufacturing cured film, and semiconductor device | |
| JP7037664B2 (en) | Photosensitive resin compositions, cured films, laminates, methods for manufacturing cured films, semiconductor devices, and thermal base generators. | |
| JP7083392B2 (en) | Photosensitive resin compositions, cured films, laminates, methods for producing these, semiconductor devices, thermal base generators used in these. | |
| WO2019189110A1 (en) | Photosensitive resin composition, cured film, laminate, method for manufacturing cured film, and semiconductor device | |
| JP7426375B2 (en) | Curable resin composition, cured film, laminate, method for producing cured film, semiconductor device, and thermal base generator | |
| WO2020066244A1 (en) | Photosensitive resin composition, cured film, layered product, production method for cured film, semiconductor device, and thermal base generator |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19864832 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2020549195 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20217008567 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 19864832 Country of ref document: EP Kind code of ref document: A1 |