WO2019117136A1 - 動物細胞、動物細胞の製造方法および目的タンパク質の製造方法 - Google Patents
動物細胞、動物細胞の製造方法および目的タンパク質の製造方法 Download PDFInfo
- Publication number
- WO2019117136A1 WO2019117136A1 PCT/JP2018/045479 JP2018045479W WO2019117136A1 WO 2019117136 A1 WO2019117136 A1 WO 2019117136A1 JP 2018045479 W JP2018045479 W JP 2018045479W WO 2019117136 A1 WO2019117136 A1 WO 2019117136A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- culture
- cells
- cell
- gene encoding
- coa dehydrogenase
- Prior art date
Links
Images


















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P21/00—Preparation of peptides or proteins
- C12P21/02—Preparation of peptides or proteins having a known sequence of two or more amino acids, e.g. glutathione
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0681—Cells of the genital tract; Non-germinal cells from gonads
- C12N5/0682—Cells of the female genital tract, e.g. endometrium; Non-germinal cells from ovaries, e.g. ovarian follicle cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/0004—Oxidoreductases (1.)
- C12N9/001—Oxidoreductases (1.) acting on the CH-CH group of donors (1.3)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y103/00—Oxidoreductases acting on the CH-CH group of donors (1.3)
- C12Y103/08—Oxidoreductases acting on the CH-CH group of donors (1.3) with flavin as acceptor (1.3.8)
- C12Y103/08004—Isovaleryl-CoA dehydrogenase (1.3.8.4)
Abstract
本発明の課題は、増殖能および生存率が向上した動物細胞、上記動物細胞の製造方法、および上記動物細胞を使用した目的タンパク質の製造方法を提供することである。本発明によれば、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを有し、イソバレリルCoAデヒドロゲナーゼが過剰発現している、動物細胞が提供される。
Description
本発明は、目的タンパク質を発現する動物細胞に関する。本発明は、上記動物細胞の製造方法、および上記動物細胞を使用した目的タンパク質の製造方法に関する。
抗体などのバイオ医薬品の製造においては、抗体の生産性を向上するため、培養液に栄養を追添加することで細胞の状態を良化させるフェドバッチ培養がよく用いられている。また、抗体などのバイオ医薬品の製造においては、抗体の生産性を向上するため、培養液を連続的に濾過および排出し、一方で栄養成分を含むフレッシュな培地を連続的に培養槽に供給する、灌流培養法がよく用いられている。
細胞の増殖性、生存性を低下させる老廃物として、有機酸があげられる。その中でも特に阻害性の高いイソ吉草酸はロイシンの代謝による中間生成物として生じる有機酸であり、一般的に悪臭を生じる物質として知られている。ロイシンの代謝経路において、ロイシンは細胞内でイソバレリルCoA(3-メチルブタノイルCoA)に酵素的に変換される。イソバレリルCoAは、その下流酵素であるイソバレリルCoAデヒドロゲナーゼ(IVDとも略記する)によって3-メチルブタ-2-エノイルCoAへ変換される。IVD活性が低下または欠損すると中間体であるイソバレリルCoAが非酵素的に分解し、イソ吉草酸が生じる(図1)。遺伝的にIVD遺伝子が変異または欠損している患者はイソ吉草酸が蓄積し、脳神経障害が起こる(イソ吉草酸血症)。
非特許文献1においては、チャイニーズハムスター卵巣(CHO)細胞において、細胞から分泌されるイソ吉草酸が増殖阻害に働くことが記載されている。非特許文献1においては、低ロイシン培地を用いたフェドバッチ培養を実施することでイソ吉草酸の分泌量を低減し、増殖性を向上させている。
特許文献1には、微生物にイソバレリルCoAデヒドロゲナーゼ遺伝子を導入することにより、イソ吉草酸生産量を低減し、不精臭の少ない食品を生産することが記載されている。
特許文献2には、CHO細胞をロイシンを低減した培地で培養することについて記載されている。
Biotechnology and bioengineering, vol 114,2017,1779-1790
フェドバッチ培養においては、栄養を追加して添加しない場合に比べて長期間かつ高濃度に細胞培養が可能になるが、細胞から分泌される老廃物が蓄積することにより培養後半で生存性が低下するという問題がある。また、灌流培養法において高密度で培養するためには、栄養の供給のほか、老廃物を系外に排出するために培地を1日当たり、培養体積の1~3倍量を交換しながら培養するためコストが高くなることが問題となっている。
本発明は、増殖能および生存率が向上した動物細胞を提供することを解決すべき課題とする。本発明はさらに、上記動物細胞の製造方法、および上記動物細胞を使用した目的タンパク質の製造方法を提供することを解決すべき課題とする。
本発明者は上記課題を解決するために鋭意検討した結果、イソ吉草酸代謝経路の下流反応を促進する酵素イソバレリルCoAデヒドロゲナーゼをコードする遺伝子をCHO細胞に強制発現させることでイソ吉草酸および酪酸の生産を低減できることを見出した。また、本発明者らは、上記の結果として細胞の状態が向上し、増殖性および生存性が向上することを実証した。本発明は、上記の知見に基づいて完成したものである。
即ち、本発明によれば、以下の発明が提供される。
<1> 目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを有し、イソバレリルCoAデヒドロゲナーゼが過剰発現している、動物細胞。
<2> イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子がプロモーターに連結されている、<1>に記載の動物細胞。
<3> 動物細胞が、CHO細胞である、<1>または<2>に記載の動物細胞。
<4> イソバレリルCoAデヒドロゲナーゼの発現量が、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子を有さない上記動物細胞に対して、3倍以上である、<1>から<3>の何れか一に記載の動物細胞。
<5> 目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とが、同一の発現ベクター上に存在している、<1>から<4>の何れか一に記載の動物細胞。
<1> 目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを有し、イソバレリルCoAデヒドロゲナーゼが過剰発現している、動物細胞。
<2> イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子がプロモーターに連結されている、<1>に記載の動物細胞。
<3> 動物細胞が、CHO細胞である、<1>または<2>に記載の動物細胞。
<4> イソバレリルCoAデヒドロゲナーゼの発現量が、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子を有さない上記動物細胞に対して、3倍以上である、<1>から<3>の何れか一に記載の動物細胞。
<5> 目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とが、同一の発現ベクター上に存在している、<1>から<4>の何れか一に記載の動物細胞。
<6> 動物細胞に対して、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを導入する工程を含む、<1>から<5>の何れか一に記載の動物細胞の製造方法。
<7> イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子を導入する工程が、エレクトロポレーションにより行われる、<6>に記載の方法。
<8> 目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを含む同一の発現ベクターを用いて、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを導入する、<6>または<7>に記載の方法。
<7> イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子を導入する工程が、エレクトロポレーションにより行われる、<6>に記載の方法。
<8> 目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを含む同一の発現ベクターを用いて、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを導入する、<6>または<7>に記載の方法。
<9> <1>から<5>の何れか一に記載の動物細胞を培養することを含む、目的タンパク質の製造方法。
<10> 動物細胞の培養がフェドバッチ培養またはバッチ培養である、<9>に記載の方法。
<11> 細胞培養の播種細胞密度が0.2×106cells/mL以上5×106cells/mL以下である、<10>に記載の方法。
<12> 培養期間中の生細胞率が全期間において60%以上である、<10>または<11>に記載の方法。
<13> 培養期間を通した培養液中のイソ吉草酸の最大濃度が3000μmol/L以下である、<10>から<12>の何れか一に記載の方法。
<14> 培養期間を通した細胞あたりのイソ吉草酸分泌量が30fmol/細胞/日以下である、<10>から<13>の何れか一に記載の方法。
<15> 培養期間を通した培養液中の酪酸の最大濃度が3000μmol/L以下である、<10>から<14>の何れか一に記載の方法。
<16> 培養期間を通した細胞あたりの酪酸分泌量が30fmol/細胞/日以下である、<10>から<15>の何れか一に記載の方法。
<10> 動物細胞の培養がフェドバッチ培養またはバッチ培養である、<9>に記載の方法。
<11> 細胞培養の播種細胞密度が0.2×106cells/mL以上5×106cells/mL以下である、<10>に記載の方法。
<12> 培養期間中の生細胞率が全期間において60%以上である、<10>または<11>に記載の方法。
<13> 培養期間を通した培養液中のイソ吉草酸の最大濃度が3000μmol/L以下である、<10>から<12>の何れか一に記載の方法。
<14> 培養期間を通した細胞あたりのイソ吉草酸分泌量が30fmol/細胞/日以下である、<10>から<13>の何れか一に記載の方法。
<15> 培養期間を通した培養液中の酪酸の最大濃度が3000μmol/L以下である、<10>から<14>の何れか一に記載の方法。
<16> 培養期間を通した細胞あたりの酪酸分泌量が30fmol/細胞/日以下である、<10>から<15>の何れか一に記載の方法。
<17> 動物細胞の培養が、灌流培養である、<9>に記載の方法。
<18> 細胞培養の播種細胞密度が0.2×106cells/mL以上3×107cells/mL以下である、<17>に記載の方法。
<19> 培養期間中の生細胞率が全期間において90%以上である、<17>または<18>に記載の方法。
<20> 培養液中のイソ吉草酸の濃度が3000μmol/L以下である、<17>から<19>の何れか一に記載の方法。
<21> 細胞あたりのイソ吉草酸分泌量が30fmol/細胞/日以下である、<17>から<20>の何れか一に記載の方法。
<22> 培養期間を通した培養液中の酪酸の濃度が3000μmol/L以下である、<17>から<21>の何れか一に記載の方法。
<23> 培養期間を通した細胞あたりの酪酸分泌量が30fmol/細胞/日以下である、<17>から<22>の何れか一に記載の方法。
<18> 細胞培養の播種細胞密度が0.2×106cells/mL以上3×107cells/mL以下である、<17>に記載の方法。
<19> 培養期間中の生細胞率が全期間において90%以上である、<17>または<18>に記載の方法。
<20> 培養液中のイソ吉草酸の濃度が3000μmol/L以下である、<17>から<19>の何れか一に記載の方法。
<21> 細胞あたりのイソ吉草酸分泌量が30fmol/細胞/日以下である、<17>から<20>の何れか一に記載の方法。
<22> 培養期間を通した培養液中の酪酸の濃度が3000μmol/L以下である、<17>から<21>の何れか一に記載の方法。
<23> 培養期間を通した細胞あたりの酪酸分泌量が30fmol/細胞/日以下である、<17>から<22>の何れか一に記載の方法。
本発明の動物細胞は、高い増殖能および高い生存率を有している。本発明の動物細胞によれば、目的タンパク質を高い生産性で製造することができる。
以下、本発明を実施するための形態を、詳細に説明する。
本明細書において「~」を用いて示された数値範囲は、「~」の前後に記載される数値をそれぞれ最小値および最大値として含む範囲を意味する。
本明細書において「~」を用いて示された数値範囲は、「~」の前後に記載される数値をそれぞれ最小値および最大値として含む範囲を意味する。
[動物細胞]
本発明の動物細胞は、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを有し、イソバレリルCoAデヒドロゲナーゼが過剰発現している、動物細胞である。
本発明の動物細胞は、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを有し、イソバレリルCoAデヒドロゲナーゼが過剰発現している、動物細胞である。
イソ吉草酸は、多くの低分子有機酸と同様に、ヒストン脱アセチル化酵素を阻害することで細胞周期を止め、増殖を阻害することが分かっている。CHO細胞を培養すると上記理由によりイソ吉草酸が蓄積することにより培養後期において、細胞の増殖が停止し、生存率が低下する。生存率の低下により、細胞あたりのタンパク質生産量が低下する。
非特許文献1においては、低ロイシン培地を用いたフェドバッチ培養を実施することによりイソ吉草酸の分泌量を低減している。しかし、市販培地ではロイシンの低減処方を用いることができないことから非特許文献1に記載の方法は汎用性に欠ける。また、非特許文献1においては、細胞の増殖性は向上しているが、培養後半の生存性を保つことができず、また細胞あたりのIgG生産量(Qp)が低下しているために、総抗体生産量には変化がない。これに対し、本発明では細胞自体を制御することで、培地種によらずイソ吉草酸の分泌を抑えることが可能である。また本発明の細胞は培養後半の生存性を高く保つことができ、細胞あたりの抗体生産量を保ちながら増殖性を向上させることで抗体の総生産量を向上させることができる。
特許文献1においては、イソ吉草酸の低減は食品生産物の悪臭を低減することを目的としており、イソ吉草酸自体の細胞増殖阻害性に関しては言及していない。また、特許文献1の対象は微生物であり、動物細胞は対象外であり、目的タンパク質の生産に関するものでもない。これに対し、本発明ではイソ吉草酸の生産を低減することでCHO細胞の増殖を増大させることができ、目的タンパク質を効率的に製造することができる。
本発明においては、ロイシンの添加量を減らすのではなく、ロイシンの代謝経路を活性化し、中間体であるイソバレリルCoAの蓄積を解消することを目的として、動物細胞にイソバレリルCoAデヒドロゲナーゼ遺伝子を導入する。イソバレリルCoAデヒドロゲナーゼを高発現する細胞は、イソバレリルCoAを速やかに3-メチルブタ-2-エノイルCoAに変換することにより、培地種によらずイソ吉草酸の分泌量を抑えることができる。上記の通り、培地種によらずイソ吉草酸の分泌の少ない細胞を提供することにより、本発明の細胞を用いて増殖性および生存性の高い培養を実施することができる。特に、老廃物が溜まりやすい培養後期(培養開始10日目以降)において、細胞の生存性を高く保つことができる。本発明においては、高い生存性を保つことにより、細胞あたりのタンパク質生産性を高めることができ、通常の培養法よりもタンパク質の総生産量を向上させることができる。さらに、副次的効果として、死滅細胞が減ることで下流工程であるタンパク質精製時に問題となる宿主由来タンパク質およびDNA混入量を減らすことが期待できる。さらに、本発明の細胞においては、イソ吉草酸と同様に増殖阻害活性を有することが知られている酪酸の分泌量を抑えられている。イソバレリルCoAデヒドロゲナーゼは酪酸生成の代謝経路には直接かかわっていないことから、本発明の動物細胞において酪酸の分泌量を低減できたことは予想外の結果である。
また、イソ吉草酸および酪酸はヒストン脱アセチル化酵素(HDAC)を阻害することにより細胞分裂を抑制する一方で、遺伝子の発現を活性化させることから目的タンパク質の生産性を向上させること、即ち、細胞あたりの抗体産生量(Qp)を増加させることが知られている。そのため、イソ吉草酸および酪酸の分泌量が低下すると、細胞あたりの抗体産生量(Qp)も低下することが予想された。しかし、本発明では、イソバレリルCoAデヒドロゲナーゼの高発現によってイソ吉草酸、酪酸を低減したにも関わらず、目的タンパク質の生産性(細胞あたりの抗体産生量(Qp))を低下させることはなく、細胞の増殖率が向上したことにより、目的タンパク質の生産量を向上させることができた。
また、本発明の効果は、フェドバッチ培養、バッチ培養および灌流培養の両方において発揮することができる。灌流培養では、低灌流比培養時には老廃物の蓄積が問題となるが、本発明の動物細胞を用いることで増殖性および生存性を保ちながら培養が可能である。さらに、老廃物の分泌・蓄積を抑えることができるため、灌流培養を用いた高密度培養時でも高い灌流比が不要となる。 そのため、低灌流比状態でも品質を保ったまま培養することが可能となる。また、高い生存性により細胞デブリを生じさせずに培養可能なため、灌流培養時に用いる分離膜の詰まりを回避可能となる。
<目的タンパク質>
本発明において、目的タンパク質の種類は特に限定されず、例えば、組み換えポリペプチド鎖、組み換え分泌ポリペプチド鎖、抗原結合タンパク質、ヒト抗体、ヒト化抗体、キメラ抗体、マウス抗体、バイスペシフィック抗体、Fc融合タンパク質、断片化免疫イムノグロブリン、一本鎖抗体(scFv)である。目的タンパク質は、好ましくはヒト抗体、ヒト化抗体、キメラ抗体、またはマウス抗体である。断片化免疫イムノグロブリンとしては、Fab、F(ab’)2、Fvなどが挙げられる。抗体のクラスも特に限定されるものではなく、IgG1、IgG2、IgG3、IgG4などのIgG、IgA、IgD、IgE、IgMなどいずれのクラスでもよいが、医薬として用いる場合はIgGおよびIgMが好ましい。
本発明において、目的タンパク質の種類は特に限定されず、例えば、組み換えポリペプチド鎖、組み換え分泌ポリペプチド鎖、抗原結合タンパク質、ヒト抗体、ヒト化抗体、キメラ抗体、マウス抗体、バイスペシフィック抗体、Fc融合タンパク質、断片化免疫イムノグロブリン、一本鎖抗体(scFv)である。目的タンパク質は、好ましくはヒト抗体、ヒト化抗体、キメラ抗体、またはマウス抗体である。断片化免疫イムノグロブリンとしては、Fab、F(ab’)2、Fvなどが挙げられる。抗体のクラスも特に限定されるものではなく、IgG1、IgG2、IgG3、IgG4などのIgG、IgA、IgD、IgE、IgMなどいずれのクラスでもよいが、医薬として用いる場合はIgGおよびIgMが好ましい。
ヒト抗体は、ヒト免疫グロブリン配列から誘導される1つまたは複数の可変および定常領域を有する全ての抗体を含む。一実施形態では、可変および定常ドメインの全てが、ヒト免疫グロブリン配列から誘導される(完全ヒト抗体)。
ヒト化抗体は、ヒト対象に投与されたときに、非ヒト種抗体と比較して、ヒト化抗体が免疫反応を誘発する可能性が低くなるように、および/または重篤な免疫反応の誘発がより少なくなるように、1つまたは複数のアミノ酸置換、欠失、および/または付加により非ヒト種から誘導された抗体の配列と異なる配列を有する。一実施形態では、非ヒト種抗体の重鎖および/または軽鎖のフレームワークおよび定常ドメイン内のある特定のアミノ酸は、ヒト化抗体を産生するように変異している。別の実施形態では、ヒト抗体からの定常ドメインは、非ヒト種の可変ドメインに融合される。
ヒト化抗体は、ヒト対象に投与されたときに、非ヒト種抗体と比較して、ヒト化抗体が免疫反応を誘発する可能性が低くなるように、および/または重篤な免疫反応の誘発がより少なくなるように、1つまたは複数のアミノ酸置換、欠失、および/または付加により非ヒト種から誘導された抗体の配列と異なる配列を有する。一実施形態では、非ヒト種抗体の重鎖および/または軽鎖のフレームワークおよび定常ドメイン内のある特定のアミノ酸は、ヒト化抗体を産生するように変異している。別の実施形態では、ヒト抗体からの定常ドメインは、非ヒト種の可変ドメインに融合される。
キメラ抗体とは、互いに由来の異なる可変領域と定常領域を連結した抗体である。例えば、マウス抗体の重鎖および軽鎖の可変領域と、ヒト抗体の重鎖および軽鎖の定常領域からなる抗体は、マウス・ヒト異種キメラ抗体である。マウス抗体の可変領域をコードするDNAをヒト抗体の定常領域をコードするDNAと連結させ、これを発現ベクターに組み込むことによって、キメラ抗体を発現する組換えベクターが作製できる。上記ベクターにより形質転換された組換え細胞を培養し、組み込まれたDNAを発現させることによって、培養中に生産されるキメラ抗体を取得できる。
バイスペシフィック抗体とは、2つの異なる抗原特異性を認識する、化学的方法または細胞融合によって作製された抗体である。バイスペシフィック抗体を作製する方法としては、2つのイムノグロブリン分子をN-サクシンイミジル 3-(2-ピリジルジチオール) プロピオネートまたはS-アセチルメルカプトサクシニックアシッドアンハイドライドなどの架橋剤を用いて結合して作製する方法、イムノグロブリン分子のFabフラグメントどうしを結合して作製する方法などが報告されている。
Fc融合タンパク質とは、Fc領域を有するタンパク質を示し、抗体を含む。
Fabは、VL、VH、CLおよびCH1ドメインを有する一価断片である。
F(ab’)2は、ヒンジ領域でジスルフィド架橋により結合された2つのFab断片を有する二価断片である。
Fv断片は、抗体のシングルアームのVLおよびVHドメインを有する。
一本鎖抗体(scFv)は、VLおよびVH領域がリンカー(例えば、アミノ酸残基の合成配列)を介して接合して、連続したタンパク質鎖を形成する抗体であり、ここでリンカーは、タンパク質鎖をそれ自身に折り重ね、一価抗原結合部位を形成させるのに十分な長さである。
Fabは、VL、VH、CLおよびCH1ドメインを有する一価断片である。
F(ab’)2は、ヒンジ領域でジスルフィド架橋により結合された2つのFab断片を有する二価断片である。
Fv断片は、抗体のシングルアームのVLおよびVHドメインを有する。
一本鎖抗体(scFv)は、VLおよびVH領域がリンカー(例えば、アミノ酸残基の合成配列)を介して接合して、連続したタンパク質鎖を形成する抗体であり、ここでリンカーは、タンパク質鎖をそれ自身に折り重ね、一価抗原結合部位を形成させるのに十分な長さである。
目的タンパク質をコードする遺伝子は、当業者に公知の方法により入手することができる。目的タンパク質が抗体である場合には、抗体のL鎖をコードするDNAおよびH鎖をコードするDNAを使用することができる。
抗体のL鎖をコードするDNAおよびH鎖をコードするDNAは、以下のようにして調製することができる。抗体を発現する遺伝子を持つハイブリドーマ、細胞、ファージ、リボソームなどからmRNAを抽出する。このmRNAより逆転写酵素を用いる逆転写反応によりcDNAを作製する。L鎖遺伝子またはH鎖遺伝子と相補塩基配列を持つプライマーとcDNAを用いるPCRによりL鎖遺伝子またはH鎖遺伝子を増幅し、クローニング用プラスミドと結合することにより各遺伝子を取得する。
抗体のL鎖の断片をコードするDNAおよびH鎖の断片をコードするDNAは、以下のようにして調製することができる。抗体を発現する遺伝子を持つハイブリドーマ、細胞、ファージ、リボソームなどからmRNAを抽出する。このmRNAより逆転写酵素を用いる逆転写反応によりcDNAを作製する。L鎖遺伝子断片またはH鎖遺伝子断片と相補塩基配列を持つプライマーとcDNAを用いるPCRによりL鎖遺伝子断片またはH鎖遺伝子断片を増幅し、クローニング用プラスミドと結合することにより各遺伝子断片を取得する。
<イソバレリルCoAデヒドロゲナーゼおよびイソバレリルCoAデヒドロゲナーゼ遺伝子>
本発明において、イソバレリルCoAデヒドロゲナーゼの由来は特に限定されず、ヒト、サル、マウス、ラット、ハムスターなどの哺乳動物由来のイソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子を使用することができる。本発明において、外来遺伝子とは、動物細胞に外から導入された遺伝子のことを言う。また、内在遺伝子を増幅させて遺伝子導入した場合でも外来遺伝子とみなす。
本発明において、イソバレリルCoAデヒドロゲナーゼの由来は特に限定されず、ヒト、サル、マウス、ラット、ハムスターなどの哺乳動物由来のイソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子を使用することができる。本発明において、外来遺伝子とは、動物細胞に外から導入された遺伝子のことを言う。また、内在遺伝子を増幅させて遺伝子導入した場合でも外来遺伝子とみなす。
ヒトイソバレリルCoAデヒドロゲナーゼの塩基配列とアミノ酸配列を配列表の配列番号1および2に示す。
イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子としては、
(1)配列番号2に記載のアミノ酸配列からなるタンパク質をコードする遺伝子;
(2)配列番号2に記載のアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;または
(3)配列番号2に記載のアミノ酸配列と85%以上(さらに好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上)の配列同一性を有するアミノ酸配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;
を使用することができる。
イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子としては、
(1)配列番号2に記載のアミノ酸配列からなるタンパク質をコードする遺伝子;
(2)配列番号2に記載のアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;または
(3)配列番号2に記載のアミノ酸配列と85%以上(さらに好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上)の配列同一性を有するアミノ酸配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;
を使用することができる。
イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子としてはさらに、
(4)配列番号1に記載の塩基配列からなる遺伝子;
(5)配列番号1に記載の塩基配列において1若しくは数個の塩基が欠失、置換若しくは付加された塩基配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;または
(6)配列番号1に記載の塩基配列の相補配列に対してストリンジェントな条件下でハイブリダイズする塩基配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;
を使用することもできる。
(4)配列番号1に記載の塩基配列からなる遺伝子;
(5)配列番号1に記載の塩基配列において1若しくは数個の塩基が欠失、置換若しくは付加された塩基配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;または
(6)配列番号1に記載の塩基配列の相補配列に対してストリンジェントな条件下でハイブリダイズする塩基配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;
を使用することもできる。
「1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列」における「1若しくは数個」とは、好ましくは1~20個、より好ましくは1~10個、さらに好ましくは1~5個、特に好ましくは1~3個を意味する。
本発明における配列同一性は、以下の式で計算される値を指す。
%配列同一性=[(同一残基数)/(長い方のアラインメント長)]×100
2つのアミノ酸配列における配列同一性は当業者に公知の任意の方法で決定することができ、BLAST((Basic Local Alignment Search Tool))プログラム(J.Mol.Biol.215:403-410,1990)等を使用して決定することができる。分母の「長い方のアラインメント長」とは、二つのアラインメントを比較した場合に、分母には長い方のアラインメント長を用いることを意味する。
%配列同一性=[(同一残基数)/(長い方のアラインメント長)]×100
2つのアミノ酸配列における配列同一性は当業者に公知の任意の方法で決定することができ、BLAST((Basic Local Alignment Search Tool))プログラム(J.Mol.Biol.215:403-410,1990)等を使用して決定することができる。分母の「長い方のアラインメント長」とは、二つのアラインメントを比較した場合に、分母には長い方のアラインメント長を用いることを意味する。
「1若しくは数個の塩基が欠失、置換若しくは付加された塩基配列」における「1若しくは数個」とは、好ましくは1~20個、より好ましくは1~10個、さらに好ましくは1~5個、特に好ましくは1~3個を意味する。
「ストリンジェントな条件下でハイブリダイズする」における「ストリンジェントな条件下」とは、中程度または高程度のストリンジェント条件下でハイブリダイズすることを意味し、これらは当業者であれば認識することができる。中程度のストリンジェントな条件としては、Sambrookら、Molecular Cloning: A Laboratory Manual、第3版、Vol.1、7.42-7.45 Cold Spring Harbor Laboratory Press, 2001に記載されている条件を挙げることができる。中程度のストリンジェントな条件は、例えば、ニトロセルロースフィルターにおいて5×SSC、0.5%SDS、1.0mmol/L EDTA(pH8.0)の前洗浄溶液、約40~50℃での約50%ホルムアミド、2×SSC~6×SSC(または約42℃での約50%ホルムアミド中の、スターク溶液(Stark’s solution)などの他の同様のハイブリダイゼーション溶液)のハイブリダイゼーション条件、および約60℃、0.5×SSC、0.1%SDSの洗浄条件を挙げることができる。高ストリンジェントな条件もまた当業者により容易に決定することができ、例えば、上記した中程度にストリンジェントな条件よりも高い温度および/または低い塩濃度でのハイブリダイゼーションおよび/または洗浄を含む。例えば、上記のようなハイブリダイゼーション条件、および68℃、0.2×SSC、0.1%SDSでの洗浄を挙げることができる。ここで、1×SSCの組成は、150mmol/L NaCl、15mmol/Lクエン酸ナトリウム、pH7.4である。SDSはドデシル硫酸ナトリウムであり、EDTAはエチレンジアミン四酢酸である。
ヒト以外のイソバレリルCoAデヒドロゲナーゼのアミノ酸配列と塩基配列の情報を以下に示す。以下の番号はNCBI (National Center for Biotechnology Information)のGene No.を示す。
ID: 24513 ラット
ID: 56357 マウス
ID: 368775 ゼブラフィッシュ
ID: 510440 ウシ
ID: 423011 ニワトリ
ID: 702867 アカゲサル
ID: 100856316 イヌ
ID: 100156047 ブタ
ID: 100759847 チャイニーズハムスター
ID: 24513 ラット
ID: 56357 マウス
ID: 368775 ゼブラフィッシュ
ID: 510440 ウシ
ID: 423011 ニワトリ
ID: 702867 アカゲサル
ID: 100856316 イヌ
ID: 100156047 ブタ
ID: 100759847 チャイニーズハムスター
ヒト以外のイソバレリルCoAデヒドロゲナーゼについても、ヒトイソバレリルCoAデヒドロゲナーゼの場合と同様に、
(2A)所定のアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;
(3A)所定のアミノ酸配列と85%以上(さらに好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上)の配列同一性を有するアミノ酸配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;
(5A)所定の塩基配列において1若しくは数個の塩基が欠失、置換若しくは付加された塩基配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;または
(6A)所定の塩基配列の相補配列に対してストリンジェントな条件下でハイブリダイズする塩基配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;
を使用してもよい。
(2A)所定のアミノ酸配列において1若しくは数個のアミノ酸が欠失、置換若しくは付加されたアミノ酸配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;
(3A)所定のアミノ酸配列と85%以上(さらに好ましくは90%以上、特に好ましくは95%以上、最も好ましくは98%以上)の配列同一性を有するアミノ酸配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;
(5A)所定の塩基配列において1若しくは数個の塩基が欠失、置換若しくは付加された塩基配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;または
(6A)所定の塩基配列の相補配列に対してストリンジェントな条件下でハイブリダイズする塩基配列からなり、イソバレリルCoAデヒドロゲナーゼ活性を有するタンパク質をコードする遺伝子;
を使用してもよい。
イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子はプロモーターに連結されていることが好ましい。
プロモーターとしては宿主の動物細胞において機能してイソバレリルCoAデヒドロゲナーゼを発現させることができるものであれば特に限定されない。プロモーターとしては、CMVプロモーター(サイトメガロウィルスプロモーター)、EF1αプロモーター(ヒトポリペプチド鎖伸長因子遺伝子のプロモーター)、SV40プロモーター(シアミンウイルス40プロモーター)、β-actinプロモーター、MMLV-LTRプロモーター(モロニーマウス白血病ウイルスの長い末端反復のプロモーター)、またはマウスβグロビンプロモーターが好ましく、CMVプロモーターがより好ましい。
プロモーターとしては宿主の動物細胞において機能してイソバレリルCoAデヒドロゲナーゼを発現させることができるものであれば特に限定されない。プロモーターとしては、CMVプロモーター(サイトメガロウィルスプロモーター)、EF1αプロモーター(ヒトポリペプチド鎖伸長因子遺伝子のプロモーター)、SV40プロモーター(シアミンウイルス40プロモーター)、β-actinプロモーター、MMLV-LTRプロモーター(モロニーマウス白血病ウイルスの長い末端反復のプロモーター)、またはマウスβグロビンプロモーターが好ましく、CMVプロモーターがより好ましい。
本発明の動物細胞においては、イソバレリルCoAデヒドロゲナーゼが過剰発現している。過剰発現とは、ある遺伝子の発現が、宿主における通常の発現量を超えていることを意味する。イソバレリルCoAデヒドロゲナーゼの外来遺伝子を宿主に導入し、上記外来遺伝子を宿主において発現させることにより、イソバレリルCoAデヒドロゲナーゼを過剰発現している動物細胞を得ることができる。
本発明の動物細胞におけるイソバレリルCoAデヒドロゲナーゼの発現量は、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子を有さない動物細胞に対して、3倍以上であることが好ましく、3.5倍以上であることがより好ましく、4倍以上であることがより一層好ましく、4.5倍以上であることがさらに好ましく、5倍以上であることがさらに一層好ましく、5.5倍以上であることが特に好ましい。上限はなくてもよいが、30000倍以下でもよくが、10000倍以下でもよい。
イソバレリルCoAデヒドロゲナーゼの発現量は、RT-PCR法(逆転写-ポリメラーゼ連鎖反応)などによって調べることができる。イソバレリルCoAデヒドロゲナーゼの発現量は、mRNAの逆転写とリアルタイムPCRにより実施することが好ましい。イソバレリルCoAデヒドロゲナーゼの発現量は、ノーマライゼーションによって算出される相対的発現量であることが好ましい。ノーマライゼーションは、たとえば、β―アクチンやHPRT1などのハウスキーピング遺伝子の発現量を内在性コントロールとした比較定量によって行える。
<動物細胞>
本発明における細胞は、動物細胞であれば特に限定されない。動物細胞としては、チャイニーズハムスター卵巣(CHO)細胞、BHK細胞、293細胞、ミエローマ細胞(NS0細胞など)、PerC6細胞、SP2/0細胞、ハイブリドーマ細胞、COS細胞、3T3細胞、HeLa細胞、Vero細胞、MDCK細胞、PC12細胞、WI38細胞などを挙げることができる。上記の中でも、特にCHO細胞、BHK細胞、293細胞、ミエローマ細胞(NS0細胞など)、PerC6細胞、SP2/0細胞、ハイブリドーマ細胞が好ましく、さらにCHO細胞が好ましい。CHO細胞は、組換えタンパク質、例えば、サイトカイン、凝固因子、および抗体の産生に広く使用されている。ジヒドロ葉酸還元酵素(DHFR)を欠損したCHO細胞を使用することが好ましく、DHFR欠損CHO細胞としては、例えば、CHO-DG44を使用することができる。
本発明における細胞は、動物細胞であれば特に限定されない。動物細胞としては、チャイニーズハムスター卵巣(CHO)細胞、BHK細胞、293細胞、ミエローマ細胞(NS0細胞など)、PerC6細胞、SP2/0細胞、ハイブリドーマ細胞、COS細胞、3T3細胞、HeLa細胞、Vero細胞、MDCK細胞、PC12細胞、WI38細胞などを挙げることができる。上記の中でも、特にCHO細胞、BHK細胞、293細胞、ミエローマ細胞(NS0細胞など)、PerC6細胞、SP2/0細胞、ハイブリドーマ細胞が好ましく、さらにCHO細胞が好ましい。CHO細胞は、組換えタンパク質、例えば、サイトカイン、凝固因子、および抗体の産生に広く使用されている。ジヒドロ葉酸還元酵素(DHFR)を欠損したCHO細胞を使用することが好ましく、DHFR欠損CHO細胞としては、例えば、CHO-DG44を使用することができる。
本発明の動物細胞においては、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とが、同一の発現ベクター上に存在していてもよいし、異なる発現ベクターに存在していてもよい。好ましくは、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とが、同一の発現ベクター上に存在している。目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とが、同一の発現ベクター上に存在していることにより、イソバレリルCoAデヒドロゲナーゼはイソ吉草酸という増殖阻害物質を抑制することにより増殖作用を発揮し、イソバレリルCoAデヒドロゲナーゼ遺伝子を保有する細胞は増殖しやすいと考えられる。イソバレリルCoAデヒドロゲナーゼ遺伝子を有する発現ベクター上に、発現させたい目的タンパク質をコードする遺伝子を存在させることにより、相乗効果(正のセレクション)が得られるものと考えられる。また、同一の発現ベクター上に、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子を載せることにより、上記の遺伝子の数の比率は1:1の関係になる。イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子の導入比率と、目的タンパク質の発現効率とが相関することにより、本発明の効果が発揮される可能性がある。イソバレリルCoAデヒドロゲナーゼの発現が高くなれば、それに応じて目的タンパク質の発現も高くなることが予想される。
[動物細胞の製造方法]
本発明によれば、動物細胞に対して、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを導入する工程を含む、本発明の動物細胞の製造方法が提供される。
本発明によれば、動物細胞に対して、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを導入する工程を含む、本発明の動物細胞の製造方法が提供される。
目的タンパク質をコードする遺伝子、およびイソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子はそれぞれベクターに組み込まれた形で、宿主である動物細胞に導入されることが好ましい。
遺伝子を宿主に導入するために使用できるベクターとしては、例えば、哺乳動物由来の発現ベクターを使用することができ、例えば、pCMV6-Entry(OriGene社製)、pcDNA3(Invitrogen社製)、pEGF-BOS(Nucleic Acids.Res.1990,18(17),p5322)、pEF、pCDM8(フナコシ社製)、INPEP4(Biogen-IDEC社製))などが挙げられるが、特に限定されない。
また、ポリAを持つmRNAは細胞内で安定することが知られている。目的タンパク質をコードする遺伝子、およびイソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子は、ポリAを遺伝子に付加させるために必要なポリAシグナル、例えばマウスβグロビンポリAシグナル、ウシ成長ホルモンポリAシグナル、SV40ポリAシグナルなどを持っていてもよい。
動物細胞への、目的タンパク質をコードする遺伝子、およびイソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子の導入方法は特に限定されず、当業者に公知の方法により行うことができる。例えば、エレクトロポレーション、リポフェクション、リン酸カルシウム法、DEAEデキストラン法、カチオニックリポソームDOTAP(ロシュ・ライフサイエンス社製)を用いた方法、またはウイルスベクターを用いた方法で行うことが可能である。上記の中でも好ましくは、エレクトロポレーションである。
動物細胞に遺伝子を導入する場合、使用する発現ベクターの種類および遺伝子導入法に応じて、遺伝子は、遺伝子導入に供された細胞のうちの一部の細胞のみに導入される。遺伝子を導入した細胞を同定および選択するために、例えば抗生物質に対する耐性について選択可能なマーカーをコードする遺伝子を、目的の遺伝子とともに宿主細胞内に導入してもよい。好ましい選択可能マーカーには、G418、ハイグロマイシンおよびメトトレキサートなどの、薬物に耐性を与えるものが含まれる。
本明細書中において上記した通り、本発明においては、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを含む同一の発現ベクターを用いて、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを導入することが好ましい。
本発明の細胞においては、目的タンパク質をコードする遺伝子は、一過性発現系で発現していてもよいし、恒常的発現系で発現していてもよいが、恒常的発現系で発現しているものが好ましい。
本発明の細胞においては、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子は、一過性発現系で発現していてもよいし、恒常的発現系で発現していてもよいが、恒常的発現系で発現しているものが好ましい。
一過性発現系とは、環状プラスミドをリン酸カルシウム法,エレクトロポレーション法,リポフェクション法などにより細胞内に取り込ませ発現させる方法である。目的タンパク質をコードする遺伝子、およびイソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子は染色体外に存在することが多い。
恒常的発現系とは、環状プラスミドまたは制限酵素処理などにより作成した直鎖上プラスミドをリン酸カルシウム法、エレクトロポレーション法、リポフェクション法などにより細胞内に取り込ませ、一部が細胞のゲノム中に挿入されることで目的タンパク質を発現させる方法である。目的タンパク質をコードする遺伝子、およびイソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子の発現を長期間維持することが可能である。またプラスミドへの薬剤耐性遺伝子の導入を行えば薬剤選抜が可能となり、目的タンパク質をコードする遺伝子、およびイソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子が染色体上に維持された細胞を効率的に選択することができる。
[目的タンパク質の製造方法]
本発明によれば、本発明の動物細胞を培養することを含む、目的タンパク質の製造方法が提供される。
本発明によれば、本発明の動物細胞を培養することを含む、目的タンパク質の製造方法が提供される。
本発明の動物細胞を培養することにより、目的タンパク質を製造することができる。培養は、公知の方法に従い行うことができる。
本発明の動物細胞の培養に用いる培地としては、通常の動物細胞の培養で使用されている培地を用いることができる。例えば、OptiCHO(Lifetechnologies社、12681011)培地、ダルベッコ変法イーグル培地(DMEM)、イーグル最小必須培地(MEM)、RPMI-1640培地、RPMI-1641培地、F-12K培地、ハムF12培地、イスコブ変法ダルベッコ培地(IMDM)、マッコイ5A培地、ライボビッツL-15培地、およびEX-CELL(商標)300シリーズ(JRH Biosciences社)、CHO-S-SFMII(Invitrogen社)、CHO-SF(Sigma-Aldrich社)、CD-CHO(Invitrogen社)、 IS CHO-V(Irvine Scientific社)、PF-ACF-CHO (Sigma-Aldrich社)などを使用することができる。
本発明の動物細胞の培養に用いる培地としては、通常の動物細胞の培養で使用されている培地を用いることができる。例えば、OptiCHO(Lifetechnologies社、12681011)培地、ダルベッコ変法イーグル培地(DMEM)、イーグル最小必須培地(MEM)、RPMI-1640培地、RPMI-1641培地、F-12K培地、ハムF12培地、イスコブ変法ダルベッコ培地(IMDM)、マッコイ5A培地、ライボビッツL-15培地、およびEX-CELL(商標)300シリーズ(JRH Biosciences社)、CHO-S-SFMII(Invitrogen社)、CHO-SF(Sigma-Aldrich社)、CD-CHO(Invitrogen社)、 IS CHO-V(Irvine Scientific社)、PF-ACF-CHO (Sigma-Aldrich社)などを使用することができる。
培地には牛胎児血清(FCS)等の血清を添加してもよく、より好ましくは無血清培地で培養してもよく、最も好ましくは完全合成培地がよい。
培地には、アミノ酸、塩、糖類、ビタミン、ホルモン、増殖因子、緩衝液、抗生物質、脂質、微量元素、植物タンパク質の加水分解物などの追加成分を補充してもよい。
培地には、アミノ酸、塩、糖類、ビタミン、ホルモン、増殖因子、緩衝液、抗生物質、脂質、微量元素、植物タンパク質の加水分解物などの追加成分を補充してもよい。
培地のpHは培養する細胞により異なるが、一般的にはpH6.0~8.0であり、好ましくはpH6.8~7.6であり、より好ましくはpH7.0~7.4である。
培養温度は、一般的には30℃~40℃であり、好ましくは32℃~37℃であり、より好ましくは36℃~37℃であり、培養中に培養温度を変更してもよい。
培養温度は、一般的には30℃~40℃であり、好ましくは32℃~37℃であり、より好ましくは36℃~37℃であり、培養中に培養温度を変更してもよい。
培養は、CO2濃度が0~40%、好ましくは2~10%の雰囲気下で行うことが好ましい。
培養時間は特に限定されないが、一般的には12時間~90日間であり、好ましくは24時間~60日間であり、より好ましくは24時間~30日間である。
培養においては、必要に応じて培地の交換、通気、攪拌を加えることができる。
培養時間は特に限定されないが、一般的には12時間~90日間であり、好ましくは24時間~60日間であり、より好ましくは24時間~30日間である。
培養においては、必要に応じて培地の交換、通気、攪拌を加えることができる。
本発明の動物細胞の培養は、培養装置(バイオリアクターとも言う)、またはそれ以外の好適な容器内で行うことができる。培養装置としては、発酵槽型タンク培養装置、エアーリフト型培養装置、カルチャーフラスコ型培養装置、スピンナーフラスコ型培養装置、マイクロキャリアー型培養装置、流動層型培養装置、ホロファイバー型培養装置、ローラーボトル型培養装置、充填槽型培養装置等を用いて培養することができる。
培養スケールは、一般的には1L~20000Lであり、好ましくは200L~2000Lであり、より好ましくは500L~2000Lである。
培養は、バッチ培養(batch culture)、フェドバッチ培養 (fed-batch culture;流加培養とも言う)、灌流培養(perfusion culture)などのいずれの方法を用いてもよいが、フェドバッチ培養または灌流培養が好ましい。
バッチ培養とは、細胞を固定体積の培養培地中で短期間増殖させ、その後、完全に回収する不連続な方法である。バッチ法を用いて増殖させた培養物は、最大細胞密度に達するまで細胞密度の増加を経験し、その後、培地成分が消費され、代謝副産物(乳酸塩およびアンモニア等)のレベルが蓄積するにつれて生存細胞密度が減退する。回収は、典型的には、最大細胞密度(典型的には、5~10×106細胞/mL)が達成された時点で生じる。バッチプロセスは、最も単純な培養方法であるが、しかしながら生存細胞密度は、栄養分利用能によって制限され、細胞が一度最大密度になると、培養は減退し、目的タンパク質の産生が低減する。廃棄産物の蓄積および栄養枯渇が培養減退に迅速につながるため(典型的には、約3~7日)、目的タンパク質の産生期間を延長することはできない。
フェドバッチ培養は、ボーラスまたは連続的に培地を供給して、消費された培地成分を補給することによって、バッチプロセスを改善する培養方法である。即ち、フェドバッチ培養においては、培養形態は懸濁培養であり、培養プロセスの開始後の一以上の時点において培養に追加成分を提供する。追加成分としては、培養プロセス中に枯渇している細胞のための栄養補助成分が挙げられ、その他の補助成分(例えば、細胞周期阻害化合物)を含めてもよい。
フェドバッチ培養では、培養期間を通して追加の栄養分を添加するため、バッチ培養と比較して、より高い細胞密度、および目的タンパク質の高い産生量を達成できる可能性がある。フェドバッチ培養では、バッチ培養とは異なり、所望の細胞密度を達成するための細胞増殖の期間(増殖期)を、中止したまたは遅い細胞増殖の期間(産生期)から区別するように供給スケジュールおよび培地成分を操作することにより二相培養を作製および持続することができる。これにより、フェドバッチ培養は、バッチ培養と比較して、目的タンパク質のより高い産生量を達成できる可能性がある。
フェドバッチ培養またはバッチ培養において、細胞培養の播種細胞密度は、一般的には0.2×106cells/mL以上1×107cells/mL以下であり、好ましくは0.2×106cells/mL以上5×106cells/mL以下であり、より好ましくは0.5×106cells/mL以上2.5×106cells/mL以下であり、さらに好ましくは0.5×106cells/mL以上1.5×106cells/mL以下である。
フェドバッチ培養またはバッチ培養において、培養期間中の生細胞率は全期間において、好ましくは60%以上100%以下であり、より好ましくは70%以上100%以下であり、さらに好ましくは75%以上100%以下である。
フェドバッチ培養またはバッチ培養において、培養期間中の生細胞率は全期間において、好ましくは60%以上100%以下であり、より好ましくは70%以上100%以下であり、さらに好ましくは75%以上100%以下である。
フェドバッチ培養またはバッチ培養において、培養期間を通した培養液中のイソ吉草酸の最大濃度は、好ましくは3000μmol/L以下であり、より好ましくは1500μmol/L以下であり、さらに好ましくは1000μmol/L以下であり、特に好ましくは500μmol/L以下であり、最も好ましくは300μmol/L以下である。培養期間を通した培養液中のイソ吉草酸の最大濃度の下限は特に限定されないが、一般的には1μmol/L以上である。
フェドバッチ培養またはバッチ培養において、培養期間を通した細胞あたりのイソ吉草酸分泌量は、好ましくは30fmol/細胞/日以下であり、より好ましくは15fmol/細胞/日以下であり、さらに好ましくは5fmol/細胞/日以下である。培養期間を通した細胞あたりのイソ吉草酸分泌量の下限は特に限定されないが、一般的には0.1fmol/細胞/日以上である。
フェドバッチ培養またはバッチ培養において、培養期間を通した培養液中の酪酸の最大濃度は、好ましくは3000μmol/L以下であり、より好ましくは1500μmol/L以下であり、さらに好ましくは1000μmol/L以下であり、特に好ましくは500μmol/L以下であり、最も好ましくは300μmol/L以下である。培養期間を通した培養液中の酪酸の最大濃度の下限は特に限定されないが、一般的には1μmol/L以上である。
フェドバッチ培養またはバッチ培養において、培養期間を通した細胞あたりの酪酸分泌量は、好ましくは30fmol/細胞/日以下であり、より好ましくは15fmol/細胞/日以下であり、さらに好ましくは5fmol/細胞/日以下である。培養期間を通した細胞あたりの酪酸分泌量の下限は特に限定されないが、一般的には0.1fmol/細胞/日以上である。
灌流培養は、新鮮な培地を添加し、同時に使用済み培地を除去する培養法であり、バッチ培養およびフェドバッチ培養をさらに改善できる可能性がある。灌流培養によれば、1×108細胞/mLを超える高い細胞密度を達成することが可能である。典型的な灌流培養は、1日間または2日間続くバッチ培養スタートアップで始まり、その後、培養物に新鮮な供給培地を連続的、段階的、および/または断続的に添加し、使用済み培地を同時に除去する。灌流培養においては、沈降、遠心分離または濾過などの方法を用いて、細胞密度を維持しながら使用済み培地を除去することができる。
灌流培養の利点は、目的タンパク質が生産される培養が、バッチ培養法またはフェドバット培養よりも長期間維持されることである。しかし、長期の灌流培養、特に高細胞密度での灌流培養を維持するためには、培地の調製、使用、保存および廃棄が必要である。灌流培養では、多くの栄養分が必要であり、バッチ培養およびフェドバッチ培養と比較して、目的タンパク質の生産コストが高くなる傾向にある。また、膜孔径の選択により、抗体を系外へ回収しながら培養を継続することが可能であるため、抗体の培養液中での滞留時間を短くし、化学的変化を減らすことで抗体の品質を高く保つことが可能である。
上記の通り、灌流培養は老廃物の蓄積を抑制するために培地を引き抜きながら新鮮な培地を足して培養を行うことにより高細胞密度培養が可能であるが、大量の培地を使用することから高コストになることが問題であった。しかし、本発明の動物細胞によれば、増殖阻害の原因物質であるイソ吉草酸および酪酸を低減させることで低灌流比であっても高細胞密度培養が可能であり、目的タンパク質の生産量及び生産性は低下しないことから、コスト低減に寄与することができる。
フェドバッチ培養および灌流培養を組み合わせた培養を行うことも可能である。 一例としては、ボーラス供給を伴うフェドバッチ培養を、増殖期の細胞の培養を維持するために使用し、次いで、灌流培養を目的タンパク質の産生のために使用することができる。
灌流は、連続的、段階的、断続的またはこれらの組み合わせの何れの形態でもよい。動物細胞は、培養物中に保持され、除去される使用済みの培地は、細胞を実質的に含まないか、または培養物よりもはるかに少ない細胞を有していてもよい。細胞培養によって発現される目的タンパク質は膜孔径の選択により、培養物中に保持または回収することができる。培養中の細胞密度が過剰にならないよう、培養液の一部を細胞ごと抜き取り、新鮮な培地を同量加えることにより細胞密度を減らす(セルブリーディング)ことを行っても良い。
灌流培養において、細胞培養の播種細胞密度は、一般的には0.2×106cells/mL以上3×107cells/mL以下であり、好ましくは0.5×106cells/mL以上1×107cells/mL以下である。
灌流培養において、培養期間中の生細胞率は全期間において、好ましくは80%以上であり、より好ましくは85%以上であり、さらに好ましくは90%以上である。
灌流培養において、培養期間中の生細胞率は全期間において、好ましくは80%以上であり、より好ましくは85%以上であり、さらに好ましくは90%以上である。
灌流培養において、最高到達細胞密度は、好ましくは2×108cells/mL以下であり、より好ましくは1.5×108cells/mL以下であり、さらに好ましくは1.0×108cells/mL以下である。
灌流培養において、培養期間を通した培養液中のイソ吉草酸の最大濃度は、好ましくは3000μmol/L以下であり、より好ましくは1500μmol/L以下であり、さらに好ましくは1000μmol/L以下であり、特に好ましくは500μmol/L以下であり、最も好ましくは300μmol/L以下である。培養期間を通した培養液中のイソ吉草酸の最大濃度の下限は特に限定されないが、一般的には1μmol/L以上である。
灌流培養において、培養期間を通した細胞あたりのイソ吉草酸分泌量は、好ましくは30fmol/細胞/日以下であり、より好ましくは15fmol/細胞/日以下であり、さらに好ましくは5fmol/細胞/日以下である。培養期間を通した細胞あたりのイソ吉草酸分泌量の下限は特に限定されないが、一般的には0.1fmol/細胞/日以上である。
灌流培養において、培養期間を通した培養液中の酪酸の最大濃度は、好ましくは3000μmol/L以下であり、より好ましくは1500μmol/L以下であり、さらに好ましくは1000μmol/L以下であり、特に好ましくは500μmol/L以下であり、最も好ましくは300μmol/L以下である。培養期間を通した培養液中の酪酸の最大濃度の下限は特に限定されないが、一般的には1μmol/L以上である。
灌流培養において、培養期間を通した細胞あたりの酪酸分泌量は、好ましくは30fmol/細胞/日以下であり、より好ましくは15fmol/細胞/日以下であり、さらに好ましくは5fmol/細胞/日以下である。培養期間を通した細胞あたりの酪酸分泌量の下限は特に限定されないが、一般的には0.1fmol/細胞/日以上である。
灌流培養における灌流比としては、好ましくは0.3vvd以上5.0vvd以下であり、より好ましくは0.3vvd以上1.5vvd以下である。但し、vvdは以下を表す。
vvd=(volume of fresh medium/working volume of reactor/day, 1日当たりの供給培養液量/培養液量)
vvd=(volume of fresh medium/working volume of reactor/day, 1日当たりの供給培養液量/培養液量)
細胞培養の播種細胞密度、および培養における最高到達細胞密度は、細胞数を常法により測定し、細胞数を培養液量で割ることにより求めることができる。
培養期間中の生細胞率(生存率)は、生細胞数を(生細胞数+死細胞数)で除算することで求められる。たとえば、細胞数の測定はVi-CELL XR(Beckman Coulter社)を用いて測定することができる。 培養液中のイソ吉草酸の濃度は、クロマト分離(例えば、DIONEX製ICS-2100および Ion Pac AS11-HCカラムを用いたクロマト分離)により測定することができる。
細胞あたりのイソ吉草酸分泌量および酪酸分泌量は、後記の実施例に記載の数式から求めることができる。
培養期間中の生細胞率(生存率)は、生細胞数を(生細胞数+死細胞数)で除算することで求められる。たとえば、細胞数の測定はVi-CELL XR(Beckman Coulter社)を用いて測定することができる。 培養液中のイソ吉草酸の濃度は、クロマト分離(例えば、DIONEX製ICS-2100および Ion Pac AS11-HCカラムを用いたクロマト分離)により測定することができる。
細胞あたりのイソ吉草酸分泌量および酪酸分泌量は、後記の実施例に記載の数式から求めることができる。
上記した培養により製造される目的タンパク質は精製することができる。目的タンパク質の分離および精製は通常のタンパク質で使用されている分離および精製方法を使用すればよい。例えば、アフィニティークロマトグラフィー等のクロマトグラフィーカラム、フィルター、限外濾過、塩析、透析、ドデシル硫酸ナトリウム(SDS)ポリアクリルアミドゲル電気泳動、等電点電気泳動等を適宜選択および組み合わせることにより、目的タンパク質を分離および精製することができるが、これらに限定されるものではない。上記で得られた目的タンパク質の濃度測定は、吸光度測定または酵素結合免疫吸着検定法(Enzyme-linked immunosorbent assay;ELISA)等により行うことができる。
アフィニティークロマトグラフィーに用いるカラムとしては、プロテインAカラム、プロテインGカラムが挙げられる。アフィニティークロマトグラフィー以外のクロマトグラフィーとしては、例えば、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲル濾過、逆相クロマトグラフィー、吸着クロマトグラフィー等が挙げられる。これらのクロマトグラフィーはHPLC(high performance liquid chromatography;高速液体クロマトグラフィー)またはFPLC(fast protein liquid chromatography)等の液相クロマトグラフィーを用いて行うことができる。
なお、目的タンパク質は、精製前または精製後に適当なポリペプチド修飾酵素を作用させることにより、目的タンパク質を修飾したり、部分的にペプチドを除去することもできる。ポリペプチド修飾酵素としては、例えば、トリプシン、キモトリプシン、リシルエンドペプチダーゼ、プロテインキナーゼ、グルコシダーゼなどが用いられる。
[目的タンパク質の利用]
本発明の方法により製造された目的タンパク質が、医薬品として有用な生物学的活性を有する場合には、目的タンパク質を、薬学的に許容される担体または添加剤と混合して製剤化することにより、医薬品を製造することができる。
本発明の方法により製造された目的タンパク質が、医薬品として有用な生物学的活性を有する場合には、目的タンパク質を、薬学的に許容される担体または添加剤と混合して製剤化することにより、医薬品を製造することができる。
薬学的に許容される担体および添加剤の例として、水、薬学的に許容される有機溶剤、コラーゲン、ポリビニルアルコール、ポリビニルピロリドン、カルボキシビニルポリマー、カルボキシメチルセルロースナトリウム、ポリアクリル酸ナトリウム、アルギン酸ナトリウム、水溶性デキストラン、カルボキシメチルスターチナトリウム、ペクチン、メチルセルロース、エチルセルロース、キサンタンガム、アラビアゴム、カゼイン、寒天、ポリエチレングリコール、ジグリセリン、グリセリン、プロピレングリコール、ワセリン、パラフィン、ステアリルアルコール、ステアリン酸、ヒト血清アルブミン(HSA)、マンニトール、ソルビトール、ラクトース、薬学的に許容される界面活性剤等が挙げられる。
例えば、注射用製剤として使用する場合、精製された目的タンパク質を溶剤、例えば生理食塩水、緩衝液、ブドウ糖溶液等に溶解し、これに吸着防止剤、例えばTween80、Tween20、ゼラチン、ヒト血清アルブミン等を加えたものを使用することができる。あるいは、使用前に溶解再構成する剤形とするために凍結乾燥したものであってもよく、凍結乾燥のための賦形剤としては、例えば、マンニトール、ブドウ糖等の糖アルコールや糖類を使用することができる。
目的タンパク質の投与方法は、経口投与または非経口投与のいずれでもよいが、好ましくは非経口投与である。例えば、注射(例えば、静脈内注射、筋肉内注射、腹腔内注射、皮下注射などによる全身または局所投与)、経鼻投与、経肺投与、または経皮投与などが挙げられる。
目的タンパク質の投与量は、目的タンパク質の種類、治療や予防の対象とする疾患の種類、患者の年齢、疾患の重篤度などにより適宜選択される。一般的には、一回につき体重1kgあたり0.001mgから1000mgの範囲であるが、特に限定されない。
以下の実施例により本発明をさらに具体的に説明するが、本発明は実施例によって限定されるものではない。
<実施例1>動物細胞の作製
キメラIgG1(リツキシマブ)をコードする核酸配列を含むベクターを構築し、構築したベクターをCHO-DG44細胞へ導入することにより、IgG1を発現させたCHO-DG44細胞(IgG1細胞)を作製した。ベクターの構築および細胞への導入は、特表2016-517691号公報の実施例2に準じて行った。
キメラIgG1(リツキシマブ)をコードする核酸配列を含むベクターを構築し、構築したベクターをCHO-DG44細胞へ導入することにより、IgG1を発現させたCHO-DG44細胞(IgG1細胞)を作製した。ベクターの構築および細胞への導入は、特表2016-517691号公報の実施例2に準じて行った。
緑色蛍光タンパク質(GFP)をコンジュゲートしたヒトイソバレリルCoAデヒドロゲナーゼをコードする遺伝子IVD-GFPを含むベクター(pCMV6-AC-GFP:図2)を購入した(OriGene社)。
また、pCMV6-Entryベクター(pCMV6-Entry Vector:図3)(OriGene社)を購入し、IVD-GFPを発現しない対照群として使用した。
また、pCMV6-Entryベクター(pCMV6-Entry Vector:図3)(OriGene社)を購入し、IVD-GFPを発現しない対照群として使用した。
IgG1を発現させたCHO-DG44細胞(IgG1細胞)はすべて37℃、5%CO2雰囲気下のインキュベーターで培養した。IgG1細胞1×106cellsにエレクトロポレーション法(Lonza社、4D-Nucleofector)を用いて上記ベクターを導入し、2mLのOptiCHO(Lifetechnologies社、12681011)培地に播種した。1日後、終濃度200μg/mLとなるようにG418(Lifetechnologies社)を添加した。遺伝子導入後7日目に96ウェルプレート(IWAKI,3860-096)1枚分に5000cells/ウェルとなるように200μLの培地に播種し、終濃度200μg/mLとなるようにG418(Lifetechnologies社)を添加した。
遺伝子導入後18日目に培養液量を1mLに拡大し、24ウェルプレートへ移した。フローサイトメーター(BD FACSCalibur,BDbiosciences社)を用いてIVD-GFP発現細胞の蛍光を測定し、蛍光シグナルを発する細胞を選別した。蛍光シグナルを発する細胞(GFP+cells)の割合を測定した結果を図4に示す。
また、細胞をRNeasy plus mini kit(Qiagen)を用いて処理し、Total RNAを回収した。得られたTotal RNAをPrimeScriptTM RT Master Mix(Perfect Real Time, Takara)を用いて逆転写を実施した。内在性IVDおよび外来性IVDの両方に相同性のあるプライマー(配列番号3および4)および内部標準としてβ-actin遺伝子に対するプライマー(配列番号5および6)を設計し、SYBR(登録商標)Premix Ex TaqTM(Tli RNaseH Plus,Takara)を用いたリアルタイムPCR(ポリメラーゼ連鎖反応)を実施した。β-actinを標準化に用いてIVD遺伝子の発現を確認した結果を図5に示す。IVD遺伝子を導入した細胞ではIVDの発現が4~6倍程度に上昇していた(図5)。
図4および図5において 、Vectorは、pCMV6-Entry Vectorを導入した細胞を示し、IVD-GFPは、IVD-GFPを含むベクターを導入した細胞を示す。Pool1及びPool2は、1回目の実験と2回目の実験を示す。
図4および図5において 、Vectorは、pCMV6-Entry Vectorを導入した細胞を示し、IVD-GFPは、IVD-GFPを含むベクターを導入した細胞を示す。Pool1及びPool2は、1回目の実験と2回目の実験を示す。
チャイニーズハムスターおよびヒトIVDに対する特異的プライマー:
フォワードプライマー : AGTTGATGCAGGGGAAGATG(配列番号3)
リバースプライマー : TCATACAGCTTGGCATCTCG(配列番号4)
フォワードプライマー : AGTTGATGCAGGGGAAGATG(配列番号3)
リバースプライマー : TCATACAGCTTGGCATCTCG(配列番号4)
チャイニーズハムスターβactinに対する特異的プライマー:
フォワードプライマー : AGCTGAGAGGGAAATTGTGCG(配列番号5)
リバースプライマー : GCAACGGAACCGCTCATT(配列番号6)
フォワードプライマー : AGCTGAGAGGGAAATTGTGCG(配列番号5)
リバースプライマー : GCAACGGAACCGCTCATT(配列番号6)
図4および図5に示す結果から、IgG1-hIVD-GFP(CHO-DG44)細胞におけるIVDの発現が確認された。Pool1およびPool2ともにIVD-GFP発現細胞の割合は50%以上存在していた。Pool2の方が、存在割合が高いものの、qPCRによる発現量は低かった。GFP発現細胞×発現量で比較すると、両細胞とも同程度であり、イソ吉草酸低減効果は同程度である。
遺伝子導入後22日目に培養体積を5mLへ拡大し、Tubespin(SIGMA社)を用いて180rpm、37℃で振とう培養を実施した。
遺伝子導入後26日目に培養体積を20mLへ拡大し、125mL容振とうフラスコ(Corning)へ播種し、140rpmで振とう培養した。
上記の方法でヒトイソバレリルCoAデヒドロゲナーゼを強制発現するIgG1-hIVD-GFP(CHO-DG44)細胞を樹立した。また、pCMV6-Entryベクターについても、IVD-GFPを発現しない対照群として遺伝子導入を実施し、IgG1-vector(CHO-DG44)細胞を構築した。各遺伝子導入細胞はそれぞれ2種類のプール細胞として樹立した。
遺伝子導入後26日目に培養体積を20mLへ拡大し、125mL容振とうフラスコ(Corning)へ播種し、140rpmで振とう培養した。
上記の方法でヒトイソバレリルCoAデヒドロゲナーゼを強制発現するIgG1-hIVD-GFP(CHO-DG44)細胞を樹立した。また、pCMV6-Entryベクターについても、IVD-GFPを発現しない対照群として遺伝子導入を実施し、IgG1-vector(CHO-DG44)細胞を構築した。各遺伝子導入細胞はそれぞれ2種類のプール細胞として樹立した。
<実施例2>フェドバッチ培養実験
IgG1-vector(CHO-DG44)細胞およびIgG1-hIVD-GFP(CHO-DG44)細胞をそれぞれ2水準ずつ用いてフェドバッチ培養実験を実施した。測定方法は、以下の通りである。
細胞数測定方法:Vi-CELL (BeckmanCoulter)
イソ吉草酸測定方法:DIONEX製ICS-2100および Ion Pac AS11-HCカラムを用いたクロマト分離
抗体濃度測定方法:Prominence(島津) HPLCおよびPOROS 50A 4.6×50mm(applied biosystems )を用いたクロマト分離
IgG1-vector(CHO-DG44)細胞およびIgG1-hIVD-GFP(CHO-DG44)細胞をそれぞれ2水準ずつ用いてフェドバッチ培養実験を実施した。測定方法は、以下の通りである。
細胞数測定方法:Vi-CELL (BeckmanCoulter)
イソ吉草酸測定方法:DIONEX製ICS-2100および Ion Pac AS11-HCカラムを用いたクロマト分離
抗体濃度測定方法:Prominence(島津) HPLCおよびPOROS 50A 4.6×50mm(applied biosystems )を用いたクロマト分離
細胞を5×105cells/mLの細胞密度でOptiCHO培地40mLに懸濁し、125mL容フラスコに播種し、37℃、5%CO2雰囲気下、140rpmの速度で振とう培養を行った。培養開始3日目から13日目まで、毎日フィード培地(Cellboost7a,7b,GE healthcare社)を初期培養体積比で2%ずつ加えた。経時的に細胞密度、培養液成分および抗体濃度を測定するため、1~3日おきにサンプリングを実施した。IgG1-vector(CHO-DG44)細胞およびIgG1-hIVD-GFP(CHO-DG44)細胞はそれぞれ培養開始10日目、および11日目でピーク細胞密度を観測し、IgG1-hIVD-GFP(CHO-DG44)細胞では20%高い細胞密度で培養できた(図6)。さらに、両細胞とも細胞生存率が徐々に低下する傾向があったが、培養開始14日目においてIgG1-hIVD-GFP(CHO-DG44)細胞が平均15%高い生存率を示した(図7)。培養開始14日目に培養液を回収し、0.22μmのデプスフィルター(メルクミリポア社)を用いて細胞および細胞デブリを除いた。上清中の抗体濃度および細胞あたりのIgG生産量(Qp)を液体クロマトグラフィーによって測定したところ、IgG生産量は平均20%増加していた(図8)。一般的に、増殖性を向上させた場合はQpが低下することが知られているが、IgG1-hIVD-GFP(CHO-DG44)細胞ではQpの低下は見られなかった(図9)。
培養開始12日目の培養上清中のイソ吉草酸の濃度を液体クロマトグラフィーで測定したところ、IgG1-hIVD-GFP(CHO-DG44)細胞では46.9%低い値を示していた(図10)。イソ吉草酸は対数増殖期に多く分泌されるが、培養開始10日目までのイソ吉草酸量から細胞あたりのイソ吉草酸生産量(QIVA)を計算したところ、49%低減できていることが分かった(図11)。
図6、図7および図10において 、Vector-1およびVector-2は、pCMV6-Entry Vectorを導入した細胞を示し、IVD-GFP-1およびIVD-GFP-2は、IVD-GFPを含むベクターを導入した細胞を示す。「-1」および「-2」はそれぞれ異なるプールの細胞を示す。
図8、図9および図11において 、Vectorは、pCMV6-Entry Vectorを導入した細胞を示し、IVD-GFPおよびIVDは、IVD-GFPを含むベクターを導入した細胞を示す。Pool1及びPool2は、それぞれ異なるプールの細胞を示す。
図8、図9および図11において 、Vectorは、pCMV6-Entry Vectorを導入した細胞を示し、IVD-GFPおよびIVDは、IVD-GFPを含むベクターを導入した細胞を示す。Pool1及びPool2は、それぞれ異なるプールの細胞を示す。
実施例において、細胞あたりの抗体生産量Qp[pg/cell/day]、細胞あたりのイソ吉草酸生産量QIVA [fmol/cell/day]および細胞あたりの酪酸生産性QBA[fmol/cell/day]は、以下の式で求めた。

Pti[g/L] = 培養日tiにおける精製物の濃度
Ati[μmol/L] = 培養日tiにおけるイソ吉草酸の濃度
Bti[μmol/L] = 培養日tiにおける酪酸の濃度
Xti[cell/day]= 培養日tiにおける生細胞密度
Ati[μmol/L] = 培養日tiにおけるイソ吉草酸の濃度
Bti[μmol/L] = 培養日tiにおける酪酸の濃度
Xti[cell/day]= 培養日tiにおける生細胞密度
積分の近似値は、時間t1からt2までの増殖曲線下面積を台形の面積として求めることで算定した。

イソ吉草酸を低減させることにより、Qpを低下させずに細胞濃度および細胞生存率を向上できたため、IgG生産量の向上をもたらしたと考えられる。細胞あたりのQpが下がることなく、増殖能が向上したことは予想外な効果である。
<実施例3>動物細胞の作製
Mab2(抗MUC-1抗体)をコードする核酸配列を含むベクターを構築し(pmab2)、pmab2上にさらにIVD遺伝子をコードする核酸配列を追加したベクター(pmab2/IVD)を構築した。構築したベクターをCHO-DG44細胞へ導入することにより、mab2を発現させたCHO-DG44細胞(mab2細胞)、並びにmab2とIVDを共発現させたCHO-DG44細胞(mab2/IVD細胞)を作製した。ベクターの構築および細胞への導入は、特表2016-517691号公報の実施例2に準じて行った。
Mab2(抗MUC-1抗体)をコードする核酸配列を含むベクターを構築し(pmab2)、pmab2上にさらにIVD遺伝子をコードする核酸配列を追加したベクター(pmab2/IVD)を構築した。構築したベクターをCHO-DG44細胞へ導入することにより、mab2を発現させたCHO-DG44細胞(mab2細胞)、並びにmab2とIVDを共発現させたCHO-DG44細胞(mab2/IVD細胞)を作製した。ベクターの構築および細胞への導入は、特表2016-517691号公報の実施例2に準じて行った。
mab2細胞およびmab2/IVD細胞はすべて37℃、5%CO2雰囲気下のインキュベーターで培養した。mab2細胞およびmab2/IVD細胞5×106cellsにエレクトロポレーション法(Lonza社、4D-Nucleofector)を用いて上記ベクターを導入し、100倍希釈したHT Supplement(x100)(Lifetechnologies社、11067-030)を含む20mLのOptiCHO(Lifetechnologies社、12681011)培地に播種した。トランスフェクションは独立に8回実施し、1日後、HT Supplementを除去するために培地をOpti-CHO培地に交換し、終濃度175nmol/Lとなるようにメトトレキサート(和光純薬工業、139-13571)を添加した。遺伝子導入後14日目に培養体積を20mLへ拡大し、125mL容振とうフラスコ(Corning)へ播種し、140rpmで振とう培養した。
また、細胞をRNeasy plus mini kit(Qiagen)を用いて処理し、Total RNAを回収した。得られたTotal RNAをPrimeScriptTM RT Master Mix(Perfect Real Time, Takara)を用いて逆転写を実施した。内在性IVDおよび外来性IVDの両方に相同性のあるプライマー(配列番号3および4)および内部標準としてHprt1遺伝子に対するプライマー(配列番号7および8)を設計し、SYBR(登録商標)Premix Ex TaqTM(Tli RNaseH Plus,Takara)を用いたリアルタイムPCR(ポリメラーゼ連鎖反応)を実施した。Hprt1を標準化に用いてIVD遺伝子の発現を確認した結果を(図12)に示す。IVD遺伝子を導入した細胞ではIVDの発現が平均136倍程度に上昇していた(図12)。実施例1と比較して、目的たんぱく質と同一ベクターにIVD遺伝子を導入したほうがよりIVD遺伝子発現の高い細胞を取得できた。
チャイニーズハムスターHprt1に対する特異的プライマー:
フォワードプライマー : TCGAGGATTTGGAAAAGGTG(配列番号7)
リバースプライマー : AATCCAGCAGGTCAGCAAAG(配列番号8)
フォワードプライマー : TCGAGGATTTGGAAAAGGTG(配列番号7)
リバースプライマー : AATCCAGCAGGTCAGCAAAG(配列番号8)
<実施例4>バッチ培養試験
mab2細胞(8種類)およびmab2/IVD細胞(8種類)をそれぞれ用いてバッチ培養試験を実施した。細胞を4×105cells/mLの細胞密度でHT Supplement(x100)(Lifetechnologies社、11067-030)を含む20mLのOptiCHO(Lifetechnologies社、12681011)培地30mLに懸濁し、125mL容フラスコに播種し、37℃、5%CO2雰囲気下、140rpmの速度で振とう培養を行った。経時的に細胞密度、培養液成分および抗体濃度を測定するため、培養開始0、2、3、6、7日にサンプリングを実施した。
mab2細胞(8種類)およびmab2/IVD細胞(8種類)をそれぞれ用いてバッチ培養試験を実施した。細胞を4×105cells/mLの細胞密度でHT Supplement(x100)(Lifetechnologies社、11067-030)を含む20mLのOptiCHO(Lifetechnologies社、12681011)培地30mLに懸濁し、125mL容フラスコに播種し、37℃、5%CO2雰囲気下、140rpmの速度で振とう培養を行った。経時的に細胞密度、培養液成分および抗体濃度を測定するため、培養開始0、2、3、6、7日にサンプリングを実施した。
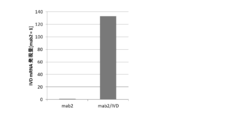
mab2(CHO-DG44)細胞およびmab2/IVD(CHO-DG44)細胞はそれぞれ7日間培養し、mab2/IVD(CHO-DG44)細胞では培養開始7日目時点で19%高い細胞密度で培養できた(図13)。上清中のmab2濃度をCedexBio(ロシュダイアグノスティクス)によって測定したところ、mab2生産量は培養開始7日目時点で平均31%増加していた(図14)。一般的に、増殖性を向上させた場合はQpが低下することが知られているが、mab2/IVD(CHO-DG44)細胞ではQpの低下は見られず、逆に平均11%向上する結果であった(図15)。
培養開始7日目の培養上清中のイソ吉草酸および酪酸の濃度を液体クロマトグラフィーで測定したところ、mab2/IVD(CHO-DG44)細胞ではそれぞれ59%、45%低い値を示していた(図16)。イソ吉草酸、酪酸は対数増殖期に多く分泌されるが、培養開始10日目までのイソ吉草酸量から細胞あたりのイソ吉草酸生産量(QIVA)および酪酸生成量(QBA)を計算したところ、それぞれ70%、61%低減できていることが分かった(図17)。
酪酸はイソ吉草酸と同様に増殖阻害活性を有することが知られているが、IVDは酪酸生成の代謝経路には直接かかわっていないにもかかわらず分泌量を低減できたことは予想外の結果であった。
<実施例5>灌流培養実験
Mab2(CHO-DG44)細胞およびmab2-IVD(CHO-DG44)細胞を用いて灌流培養実験を実施した。
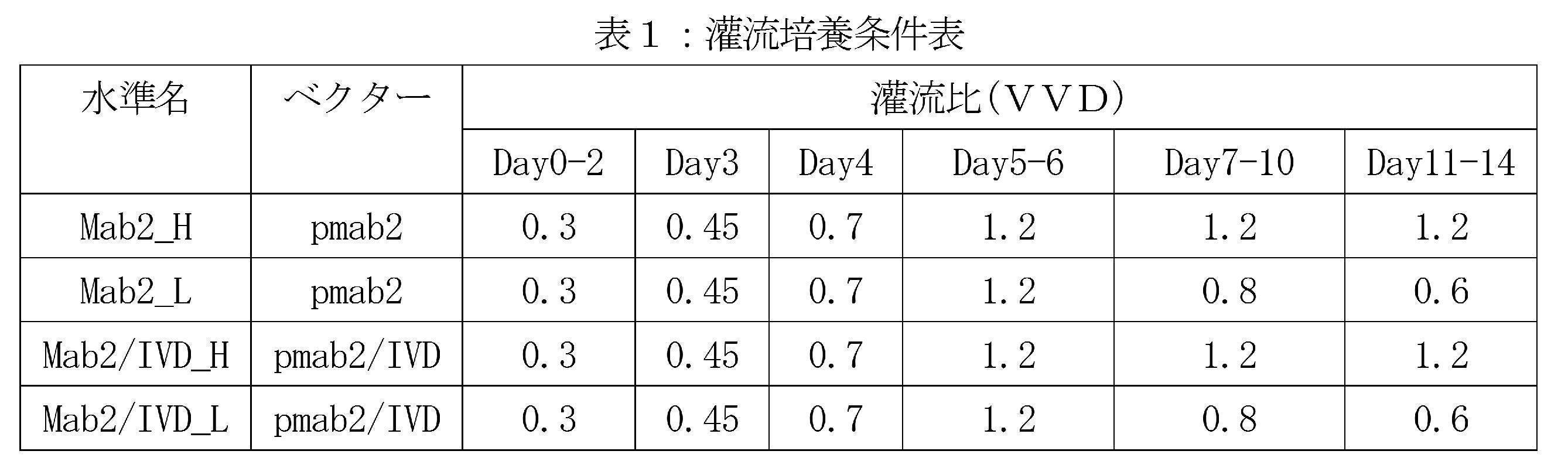
細胞を25×105cells/mLの細胞密度でバイオリアクターに播種し、37℃、5%CO2雰囲気下、200rpmの速度で攪拌培養を行った。培養槽内に細胞をとどめるための分離用に中空糸膜とATFポンプを用いた灌流により、灌流比を0.3VVDから1.2VVDまで徐々に増加させ、細胞数が8×107cells/mLを超えた時点で一部の水準において灌流比を段階的に低下させて培養した(表1)。細胞数が8x107cells/mLを超えた時点で、7.3x107cells/mLになるように培養液を引き抜き(セルブリーディング)、初期培地を引き抜いた分補てんすることにより、細胞密度が増えすぎないように調整しながら培養した。経時的に細胞密度、培養液成分および抗体濃度を測定するため、毎日培養槽内の培養液と透過側の培養液のサンプリングを実施した。
Mab2(CHO-DG44)細胞およびmab2-IVD(CHO-DG44)細胞を用いて灌流培養実験を実施した。
細胞を25×105cells/mLの細胞密度でバイオリアクターに播種し、37℃、5%CO2雰囲気下、200rpmの速度で攪拌培養を行った。培養槽内に細胞をとどめるための分離用に中空糸膜とATFポンプを用いた灌流により、灌流比を0.3VVDから1.2VVDまで徐々に増加させ、細胞数が8×107cells/mLを超えた時点で一部の水準において灌流比を段階的に低下させて培養した(表1)。細胞数が8x107cells/mLを超えた時点で、7.3x107cells/mLになるように培養液を引き抜き(セルブリーディング)、初期培地を引き抜いた分補てんすることにより、細胞密度が増えすぎないように調整しながら培養した。経時的に細胞密度、培養液成分および抗体濃度を測定するため、毎日培養槽内の培養液と透過側の培養液のサンプリングを実施した。
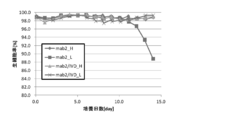
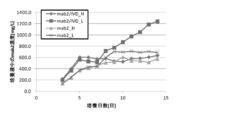
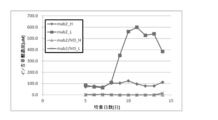
mab2/IVD(CHO-DG44)細胞は灌流培養において、灌流比を低減させた場合でも生細胞数、高生細胞率を保つことができた一方、mab2(CHO-DG44)細胞では生細胞数、生細胞率が徐々に低下することが観察された(図18および19)。灌流培養における、1日当たりに細胞が生産したmab2量を計算したところ、mab2/IVD細胞は灌流比が低下した場合でもmab2生産性を低下させることがなかった(図20および21)。mab2細胞は灌流比を低減させたday6付近から培養上清中のイソ吉草酸、酪酸が蓄積しているのに対し、mab2/IVD細胞は14日間の培養期間を通してイソ吉草酸および酪酸が培養液中に蓄積が見られなかった(図22および23)。mab2/IVD細胞は増殖阻害物質を培養槽内に蓄積させないことにより増殖性を低灌流比でも保つことができ、高密度培養が可能にできた。また。2000Lスケールでmab2/IVD細胞を低灌流比で培養した場合の培地使用量を見積ると、24日間の培養で28800Lもの培地を節約できることがわかった(表2)。
一日当たり細胞が生産した総mab2量Ymab2は以下の式で求めた
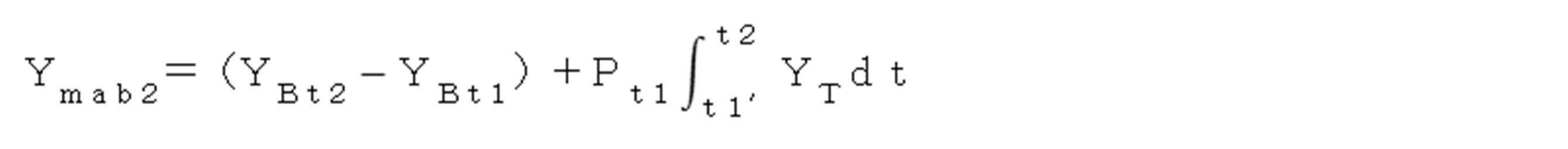
YTti [g/L] = 培養日tiにおける透過側の抗体濃度
YTti’ [g/L] = 培養日tiにおけるブリーディング後の透過側の抗体濃度
YBti [g/L] = 培養日tiにおける培養槽側の抗体濃度
Pti[VVD] = 培養日tiからt(i+1)までの灌流比
YTti’ [g/L] = 培養日tiにおけるブリーディング後の透過側の抗体濃度
YBti [g/L] = 培養日tiにおける培養槽側の抗体濃度
Pti[VVD] = 培養日tiからt(i+1)までの灌流比
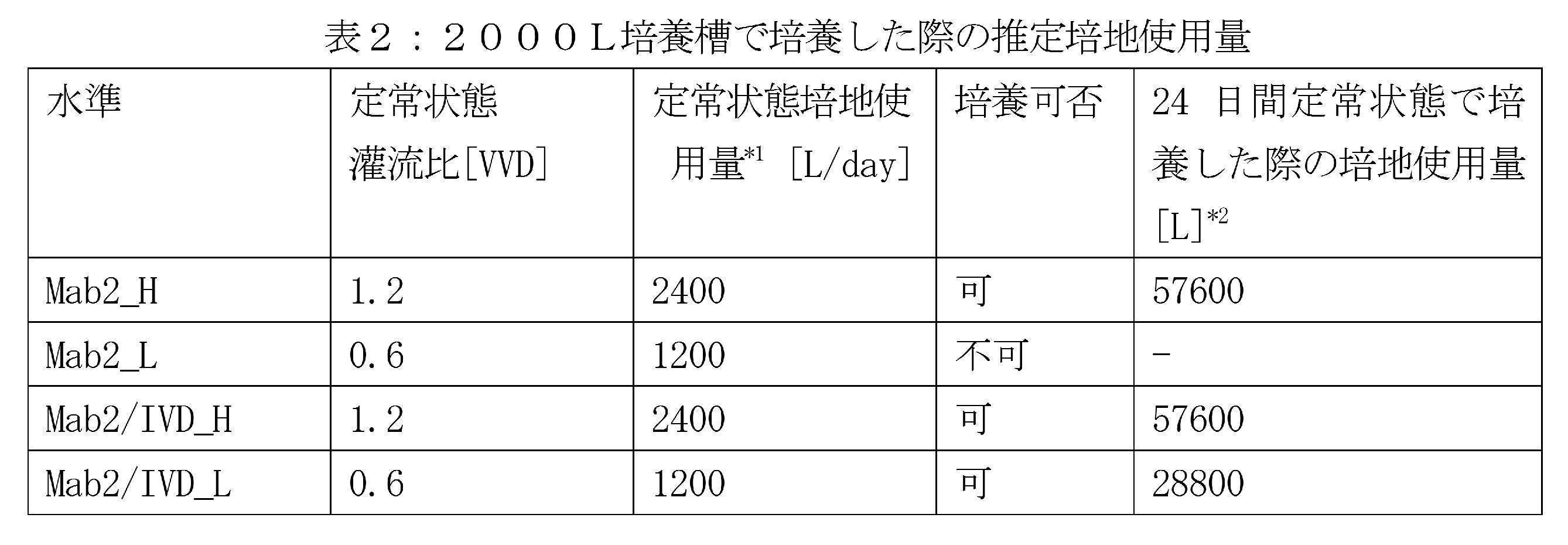
*1:セルブリーディング時に補填する培地量は除く
*2:24日間、定常状態が続いたと仮定
*2:24日間、定常状態が続いたと仮定
Claims (23)
- 目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを有し、イソバレリルCoAデヒドロゲナーゼが過剰発現している、動物細胞。
- イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子がプロモーターに連結されている、請求項1に記載の動物細胞。
- 動物細胞が、CHO細胞である、請求項1または2に記載の動物細胞。
- イソバレリルCoAデヒドロゲナーゼの発現量が、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子を有さない前記動物細胞に対して、3倍以上である、請求項1から3の何れか一項に記載の動物細胞。
- 目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とが、同一の発現ベクター上に存在している、請求項1から4の何れか一項に記載の動物細胞。
- 動物細胞に対して、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを導入する工程を含む、請求項1から5の何れか一項に記載の動物細胞の製造方法。
- イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子を導入する工程が、エレクトロポレーションにより行われる、請求項6に記載の方法。
- 目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを含む同一の発現ベクターを用いて、目的タンパク質をコードする遺伝子と、イソバレリルCoAデヒドロゲナーゼをコードする外来遺伝子とを導入する、請求項6または7に記載の方法。
- 請求項1から5の何れか一項に記載の動物細胞を培養することを含む、目的タンパク質の製造方法。
- 動物細胞の培養がフェドバッチ培養またはバッチ培養である、請求項9に記載の方法。
- 細胞培養の播種細胞密度が0.2×106cells/mL以上5×106cells/mL以下である、請求項10に記載の方法。
- 培養期間中の生細胞率が全期間において60%以上である、請求項10または11に記載の方法。
- 培養期間を通した培養液中のイソ吉草酸の最大濃度が3000μmol/L以下である、請求項10から12の何れか一項に記載の方法。
- 培養期間を通した細胞あたりのイソ吉草酸分泌量が30fmol/細胞/日以下である、請求項10から13の何れか一項に記載の方法。
- 培養期間を通した培養液中の酪酸の最大濃度が3000μmol/L以下である、請求項10から14の何れか一項に記載の方法。
- 培養期間を通した細胞あたりの酪酸分泌量が30fmol/細胞/日以下である、請求項10から15の何れか一項に記載の方法。
- 動物細胞の培養が、灌流培養である、請求項9に記載の方法。
- 細胞培養の播種細胞密度が0.2×106cells/mL以上3×107cells/mL以下である、請求項17に記載の方法。
- 培養期間中の生細胞率が全期間において90%以上である、請求項17または18に記載の方法。
- 培養液中のイソ吉草酸の濃度が3000μmol/L以下である、請求項17から19の何れか一項に記載の方法。
- 細胞あたりのイソ吉草酸分泌量が30fmol/細胞/日以下である、請求項17から20の何れか一項に記載の方法。
- 培養期間を通した培養液中の酪酸の濃度が3000μmol/L以下である、請求項17から21の何れか一項に記載の方法。
- 培養期間を通した細胞あたりの酪酸分泌量が30fmol/細胞/日以下である、請求項17から22の何れか一項に記載の方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18887753.4A EP3725877A4 (en) | 2017-12-11 | 2018-12-11 | ANIMAL CELLS, METHOD FOR MANUFACTURING ANIMAL CELLS AND METHOD FOR MANUFACTURING TARGET PROTEIN |
| CN201880079952.3A CN111465686B (zh) | 2017-12-11 | 2018-12-11 | 动物细胞、动物细胞的制造方法及靶蛋白的制造方法 |
| JP2019559655A JP7032438B2 (ja) | 2017-12-11 | 2018-12-11 | 動物細胞、動物細胞の製造方法および目的タンパク質の製造方法 |
| US16/897,860 US11542523B2 (en) | 2017-12-11 | 2020-06-10 | Animal cell, method for producing animal cell, and method for producing target protein |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017-236641 | 2017-12-11 | ||
| JP2017236641 | 2017-12-11 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US16/897,860 Continuation US11542523B2 (en) | 2017-12-11 | 2020-06-10 | Animal cell, method for producing animal cell, and method for producing target protein |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019117136A1 true WO2019117136A1 (ja) | 2019-06-20 |
Family
ID=66820862
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2018/045479 WO2019117136A1 (ja) | 2017-12-11 | 2018-12-11 | 動物細胞、動物細胞の製造方法および目的タンパク質の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11542523B2 (ja) |
| EP (1) | EP3725877A4 (ja) |
| JP (1) | JP7032438B2 (ja) |
| CN (1) | CN111465686B (ja) |
| WO (1) | WO2019117136A1 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2021107123A1 (ja) * | 2019-11-29 | 2021-06-03 | ||
| WO2023053585A1 (ja) | 2021-09-30 | 2023-04-06 | 富士フイルム株式会社 | 学習用データの取得方法、学習用データ取得システム、ソフトセンサの構築方法、ソフトセンサ、学習用データ |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024040519A1 (en) * | 2022-08-25 | 2024-02-29 | Wuxi Biologics Co., Ltd. | Intermittent perfusion fed-batch culture |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006174786A (ja) | 2004-12-24 | 2006-07-06 | Kikkoman Corp | 不精臭が低減した飲食品 |
| JP2016517691A (ja) | 2013-05-03 | 2016-06-20 | フジフィルム・ダイオシンス・バイオテクノロジーズ ・ユーケイ・リミテッド | 発現方法 |
| WO2017051347A2 (en) | 2015-09-23 | 2017-03-30 | Pfizer Inc. | Cells and method of cell culture |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009086215A2 (en) * | 2007-12-21 | 2009-07-09 | Wyeth | Pathway analysis of cell culture phenotypes and uses thereof |
| EP2296701A1 (en) * | 2008-06-11 | 2011-03-23 | Flugen, Inc. | Cell-based systems for producing influenza vaccines |
| WO2014022102A1 (en) * | 2012-08-01 | 2014-02-06 | Amgen Inc. | Methods of using anti-apoptotic compounds to modulate one or more properties of a cell culture |
-
2018
- 2018-12-11 EP EP18887753.4A patent/EP3725877A4/en active Pending
- 2018-12-11 CN CN201880079952.3A patent/CN111465686B/zh active Active
- 2018-12-11 WO PCT/JP2018/045479 patent/WO2019117136A1/ja unknown
- 2018-12-11 JP JP2019559655A patent/JP7032438B2/ja active Active
-
2020
- 2020-06-10 US US16/897,860 patent/US11542523B2/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006174786A (ja) | 2004-12-24 | 2006-07-06 | Kikkoman Corp | 不精臭が低減した飲食品 |
| JP2016517691A (ja) | 2013-05-03 | 2016-06-20 | フジフィルム・ダイオシンス・バイオテクノロジーズ ・ユーケイ・リミテッド | 発現方法 |
| WO2017051347A2 (en) | 2015-09-23 | 2017-03-30 | Pfizer Inc. | Cells and method of cell culture |
Non-Patent Citations (7)
| Title |
|---|
| BIOTECHNOLOGY AND BIOENGINEERING, vol. 114, 2017, pages 1779 - 1790 |
| J. MOL. BIOL., vol. 215, 1990, pages 403 - 410 |
| MOHSEN, A.-W. A. ET AL.: "High-level expression of an altered cDNA encoding human isovaleryl-CoA dehydrogenase in Escherichia coli.", GENE, vol. 160, 1995, pages 263 - 267, XP004042156 * |
| MULUKUTLA, B. C. ET AL.: "Identification and control of novel growth inhibitors in fed-batch cultures of chinese hamster ovary cells", BIOTECHNOL. BIOENG ., vol. 114, no. 8, 2017, pages 1779 - 1790, XP055617764 * |
| SAMBROOK ET AL.: "Molecular Cloning: A Laboratory Manual", vol. 1, 2001, COLD SPRING HARBOR LABORATORY PRESS |
| See also references of EP3725877A4 |
| URANO, K. ET AL.: "Molecular defect of isovaleryl-CoA dehydrogenase in the skunk mutant of silkworm , Bombyx mori.", FEBS J., vol. 277, no. 21, 2010, pages 4452 - 4463, XP055617756 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2021107123A1 (ja) * | 2019-11-29 | 2021-06-03 | ||
| WO2021107123A1 (ja) * | 2019-11-29 | 2021-06-03 | 富士フイルム株式会社 | 細胞培養方法、抗体製造方法、有機酸除去方法、及び、抗体 |
| JP7352650B2 (ja) | 2019-11-29 | 2023-09-28 | 富士フイルム株式会社 | 細胞培養方法、抗体製造方法、有機酸除去方法、及び、抗体 |
| WO2023053585A1 (ja) | 2021-09-30 | 2023-04-06 | 富士フイルム株式会社 | 学習用データの取得方法、学習用データ取得システム、ソフトセンサの構築方法、ソフトセンサ、学習用データ |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3725877A4 (en) | 2021-02-17 |
| US20200299723A1 (en) | 2020-09-24 |
| JP7032438B2 (ja) | 2022-03-08 |
| EP3725877A1 (en) | 2020-10-21 |
| CN111465686B (zh) | 2024-03-22 |
| CN111465686A (zh) | 2020-07-28 |
| JPWO2019117136A1 (ja) | 2020-12-17 |
| US11542523B2 (en) | 2023-01-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2020281075B2 (en) | Mammalian cell culture | |
| JP6943972B2 (ja) | 灌流培地 | |
| US11542523B2 (en) | Animal cell, method for producing animal cell, and method for producing target protein | |
| JPWO2020091041A1 (ja) | 液体培地の調製方法 | |
| JP7123168B2 (ja) | 動物細胞、動物細胞の製造方法および目的タンパク質の製造方法 | |
| WO2020122048A1 (ja) | 動物細胞、動物細胞の製造方法および目的タンパク質の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18887753 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019559655 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2018887753 Country of ref document: EP Effective date: 20200713 |