WO2015072537A1 - Light-emitting material, organic light-emitting element, and compound - Google Patents
Light-emitting material, organic light-emitting element, and compound Download PDFInfo
- Publication number
- WO2015072537A1 WO2015072537A1 PCT/JP2014/080162 JP2014080162W WO2015072537A1 WO 2015072537 A1 WO2015072537 A1 WO 2015072537A1 JP 2014080162 W JP2014080162 W JP 2014080162W WO 2015072537 A1 WO2015072537 A1 WO 2015072537A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- general formula
- compound
- atom
- group
- substituent
- Prior art date
Links
- 0 CCC(*)c1ccc(*)c(Oc2c(*)ccc(*)c2)c1 Chemical compound CCC(*)c1ccc(*)c(Oc2c(*)ccc(*)c2)c1 0.000 description 2
- VMYBTZYBKMKNIO-UHFFFAOYSA-N Bc1cc(C)ccc1Nc1c(C)cc(C)cc1 Chemical compound Bc1cc(C)ccc1Nc1c(C)cc(C)cc1 VMYBTZYBKMKNIO-UHFFFAOYSA-N 0.000 description 1
- UYZSPNQAUQZJNH-UHFFFAOYSA-N Cc(cc1)cc([BrH]C)c1N Chemical compound Cc(cc1)cc([BrH]C)c1N UYZSPNQAUQZJNH-UHFFFAOYSA-N 0.000 description 1
- URLKBWYHVLBVBO-UHFFFAOYSA-N Cc1ccc(C)cc1 Chemical compound Cc1ccc(C)cc1 URLKBWYHVLBVBO-UHFFFAOYSA-N 0.000 description 1
Images
























Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K11/00—Luminescent, e.g. electroluminescent, chemiluminescent materials
- C09K11/06—Luminescent, e.g. electroluminescent, chemiluminescent materials containing organic luminescent materials
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic System
- C07F5/02—Boron compounds
- C07F5/027—Organoboranes and organoborohydrides
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/30—Coordination compounds
- H10K85/321—Metal complexes comprising a group IIIA element, e.g. Tris (8-hydroxyquinoline) gallium [Gaq3]
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/631—Amine compounds having at least two aryl rest on at least one amine-nitrogen atom, e.g. triphenylamine
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K85/00—Organic materials used in the body or electrodes of devices covered by this subclass
- H10K85/60—Organic compounds having low molecular weight
- H10K85/649—Aromatic compounds comprising a hetero atom
- H10K85/657—Polycyclic condensed heteroaromatic hydrocarbons
- H10K85/6572—Polycyclic condensed heteroaromatic hydrocarbons comprising only nitrogen in the heteroaromatic polycondensed ring system, e.g. phenanthroline or carbazole
-
- H—ELECTRICITY
- H10—SEMICONDUCTOR DEVICES; ELECTRIC SOLID-STATE DEVICES NOT OTHERWISE PROVIDED FOR
- H10K—ORGANIC ELECTRIC SOLID-STATE DEVICES
- H10K50/00—Organic light-emitting devices
- H10K50/10—OLEDs or polymer light-emitting diodes [PLED]
- H10K50/11—OLEDs or polymer light-emitting diodes [PLED] characterised by the electroluminescent [EL] layers
Definitions
- the present invention relates to a compound useful as a light emitting material and an organic light emitting device using the compound.
- organic light emitting devices such as organic electroluminescence devices (organic EL devices)
- organic electroluminescence devices organic electroluminescence devices
- various efforts have been made to increase the light emission efficiency by newly developing and combining electron transport materials, hole transport materials, light emitting materials, and the like constituting the organic electroluminescence element.
- research on organic electroluminescence devices using a compound having a structure in which a substituent such as a carbazolyl group is bonded to a tricyclic structure having a heteraborine ring containing a boron atom and another heteroatom can be seen.
- Non-Patent Document 1 describes the results of studying luminescence characteristics of a compound having a heteraboline ring containing a boron atom and a nitrogen atom. According to the document, it was described that when a cyclohexane solution or a solid film of a compound having the following structure was irradiated with excitation light, light emission was observed in the visible region (Hex n represents an n-hexyl group, and Tip represents Represents a 2,4,6-triisopropylphenyl group). However, Non-Patent Document 1 does not describe a compound in which a heteroatom of a heteraboline ring is changed to a heteroatom other than a nitrogen atom such as an oxygen atom or a sulfur atom.
- Patent Document 1 discloses a compound having a tricyclic structure having a heterolabrin ring represented by the following general formula.
- R 1 in the following general formula is a group containing two or more aryl groups or heteroaryl groups
- R 3 and R 4 represent 1 to 5 substituents
- R 2 to R 4 represent a hydrogen atom, an alkyl group
- R 1 in the following general formula is a 2,4,6-triphenylphenyl group
- R 2 is a phenyl group
- R 3 and R 4 are hydrogen atoms
- Patent Document 1 does not describe the light emission characteristics of the compound represented by this general formula, nor does it describe a compound in which the heteroatom of the heterolabrin ring is changed to a heteroatom other than a nitrogen atom. .
- Non-Patent Document 2 describes the results of studying the emission characteristics of a compound having a heteraboline ring containing a boron atom and other heteroatoms, and light emission in the visible region was observed in a cyclohexane solution of a compound having the following structure. It is described.
- R represents a hydrogen atom or a methyl group
- E represents an N-methyl group or a sulfur atom
- Mes represents a mesityl group.
- Non-Patent Document 1 does not describe a compound in which R is a substituted amino group such as a carbazolyl group.
- Non-Patent Document 1 discloses that a compound in which a carbazolyl group is introduced into a tricyclic structure having a heteraboline ring (azaborin ring) containing a boron atom and a nitrogen atom emits light in the visible region.
- azaborin ring a heteraboline ring
- the inventors actually evaluated the light emission characteristics of a compound in which a carbazolyl group substituted with a carbazolyl group, a methyl group, or the like was introduced into a tricyclic structure having an azaborin ring, the light emission characteristics can be sufficiently satisfied. It was proved that it was not (see Comparative Examples 1 to 3 below).
- Non-Patent Document 2 it is described in Non-Patent Document 2 that light emission was observed in the visible region of a compound having a tricyclic structure having a heteraboline ring containing a boron atom and a sulfur atom.
- Non-Patent Document 2 does not describe a compound in which a substituted amino group such as a carbazole group is bonded to a tricyclic structure having a heterolabrin ring. For this reason, the usefulness as a light-emitting material of a compound in which a substituted amino group such as a carbazolyl group is bonded to a tricyclic structure having a heteraboline ring containing a boron atom and an oxygen atom or a sulfur atom cannot be predicted.
- the present inventors have further investigated the usefulness of a compound having a heteraboline ring containing a boron atom and an oxygen atom or sulfur atom as a luminescent material, and found a compound having excellent luminescent properties. Aimed at research. And the general formula of the compound useful as a luminescent material was derived, and the earnest examination was advanced for the purpose of generalizing the structure of the organic light emitting element with high luminous efficiency.
- the present inventors have found that compounds having a specific structure among the compounds having a heteraboline ring containing a boron atom and an oxygen atom or a sulfur atom have excellent properties as a luminescent material.
- a group of compounds is useful as a delayed fluorescent material, and it has been clarified that an organic light-emitting device having high emission efficiency can be provided at low cost. Based on these findings, the present inventors have provided the following present invention as means for solving the above problems.
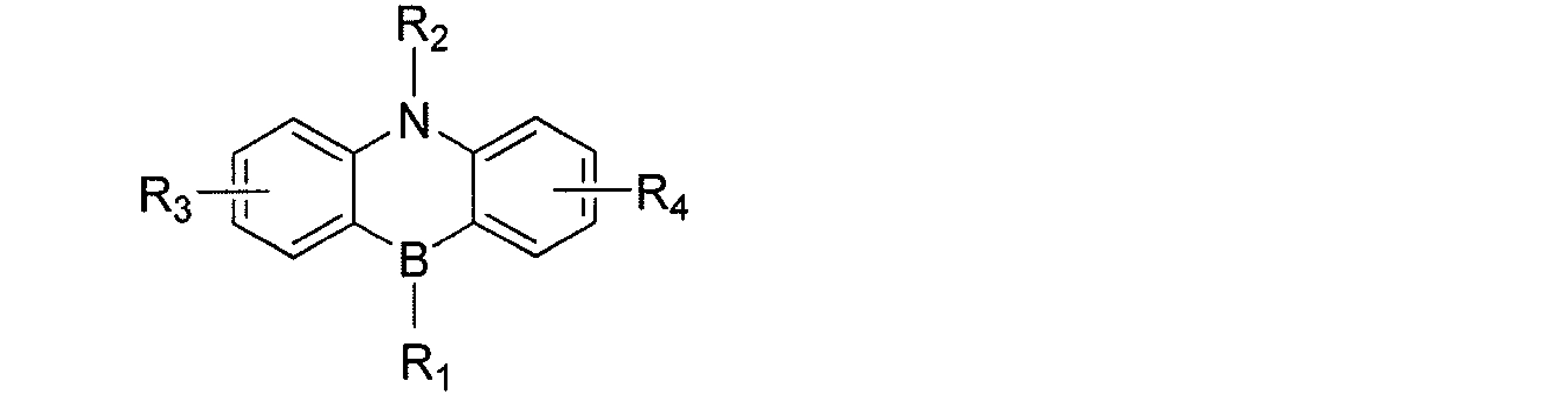
- a light emitting material comprising a compound represented by the following general formula (1).
- X represents an oxygen atom or a sulfur atom.
- R 1 ⁇ R 8 each independently represent a hydrogen atom or a substituent. However, at least one of R 1 to R 8 is each independently a group represented by any one of the following general formulas (2) to (7).
- R 1 and R 2 , R 2 and R 3 , R 3 and R 4 , R 5 and R 6 , R 6 and R 7 , R 7 and R 8 , R 8 and R 9 , R 9 and R 1 are bonded to each other Thus, a ring structure may be formed.
- R 9 represents a substituent.
- R 9 contains an atom having a lone pair of electrons that does not form a single bond with a boron atom
- the atom may be coordinated with the boron atom to form a cyclic structure.
- L 12 to L 17 each independently represents a single bond or a divalent linking group
- * represents a bonding site to the benzene ring in the general formula (1).
- R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a , R 3b , R 41 to R 48 , R 4a , R 51 to R 58 , R 61 to R 68 are each independently hydrogen Represents an atom or substituent.
- R 9 in the general formula (1) is a group represented by the following general formula (a).
- * represents the bonding site to the boron atom in the general formula (1).
- R 9a , R 9b , R 9c , R 9d and R 9e each independently represent a hydrogen atom or a substituent.
- R 9a and R 9b , R 9b and R 9c , R 9c and R 9d , and R 9d and R 9e may be bonded to each other to form a cyclic structure.
- X ′ represents an oxygen atom or a sulfur atom.
- R 1 ′ to R 8 ′ each independently represents a hydrogen atom or a substituent. However, at least one of R 1 ′ to R 8 ′ is a group independently represented by any one of the following general formulas (2 ′) to (7 ′).
- R 1 'and R 2', R 2 'and R 3', R 3 'and R 4', R 5 'and R 6', R 6 'and R 7', R 7 'and R 8', R 8 'And R 9 ' and R 9 'and R 1 ' may be bonded to each other to form a cyclic structure.
- R 9 ′ represents a substituent. When R 9 ′ includes an atom having a lone pair of electrons that does not form a single bond with a boron atom, the atom may be coordinated with the boron atom to form a cyclic structure.
- L 12 ′ to L 17 ′ each independently represents a single bond or a divalent linking group, and * represents a bond to the benzene ring in the general formula (1 ′).
- the compound of the present invention is useful as a light emitting material.
- the compounds of the present invention include those that emit delayed fluorescence.
- An organic light emitting device using the compound of the present invention as a light emitting material can realize high luminous efficiency.
- 2 is an emission spectrum of a solution of compound 5 in Example 1.
- 2 is an emission spectrum of a thin film type organic photoluminescence device of Compound 5 of Example 1.
- 2 is a transient decay curve of a solution of compound 5 of Example 1. It is the emission spectrum of the solution of the compound 6 of Example 2, and a thin film type organic photoluminescent element.
- 2 is an emission spectrum of a thin film type organic photoluminescence device of Compound 6 of Example 2.
- 4 is a transient decay curve of a solution of compound 6 of Example 2.
- 2 is an emission spectrum of a solution of the compound 18 of Example 3.
- 3 is a transient decay curve of a solution of the compound 18 of Example 3.
- 4 is an emission spectrum of a solution of the compound 59 of Example 4.
- 7 is an emission spectrum of a solution of the compound 65 of Example 5.
- 7 is an emission spectrum of a solution of the compound 67 of Example 6.
- 4 is an emission spectrum of a solution of compound 156 of Example 7.
- 4 is a transient decay curve of a solution of compound 156 of Example 7.
- 2 is an emission spectrum of a solution of Comparative Compound A of Comparative Example 1.
- 2 is a transient decay curve of a solution of Comparative Compound A of Comparative Example 1.
- 3 is an emission spectrum of a solution of Comparative Compound B of Comparative Example 2.
- 4 is a transient decay curve of a solution of Comparative Compound B of Comparative Example 2.
- 2 is an emission spectrum of a solution of Comparative Compound C of Comparative Example 3.
- 4 is a transient decay curve of a solution of Comparative Compound C of Comparative Example 3.
- 2 is an emission spectrum of an organic electroluminescent element of the compound 5 of Example 11.
- 6 is a graph showing current density-external quantum efficiency characteristics of an organic electroluminescent device of Compound 5 of Example 11.
- 7 is an emission spectrum of an organic electroluminescent element of the compound 6 of Example 12.
- 10 is a graph showing current density-external quantum efficiency characteristics of an organic electroluminescence device of Compound 6 of Example 12.
- a numerical range represented by using “to” means a range including numerical values described before and after “to” as a lower limit value and an upper limit value.
- the isotope species of the hydrogen atom present in the molecule of the compound used in the present invention is not particularly limited. For example, all the hydrogen atoms in the molecule may be 1 H, or a part or all of them are 2 H. (Deuterium D) may be used.
- the luminescent material of the present invention is characterized by comprising a compound represented by the following general formula (1).
- X represents an oxygen atom or a sulfur atom, and is preferably an oxygen atom.
- R 1 to R 8 each independently represents a hydrogen atom or a substituent. However, at least one of R 1 to R 8 is each independently a group represented by any one of the following general formulas (2) to (7). The group represented by any of the following general formulas (2) to (7) may be only one of R 1 to R 8 , or may be two or more.
- any one of R 2 , R 3 , R 6 and R 7 Is preferably a group represented by any one of the following general formulas (2) to (7)
- R 3 or R 6 is a group represented by any one of the following general formulas (2) to (7). More preferably.
- any one of the following general formulas (2) to (7) Is preferably at least one of R 1 to R 4 and at least one of R 5 to R 8 .
- the groups represented by any of the following general formulas (2) to (7) are 1 to 3 of R 1 to R 4 and 1 to 3 of R 5 to R 8. It is preferably 1 or 2 of R 1 to R 4 , and more preferably 1 or 2 of R 5 to R 8 .
- R 1 to R 4 are represented by any of the general formulas (2) to (7), and R 5 to R 8 are represented by any of the general formulas (2) to (7).
- the number of groups may be the same or different, but is preferably the same.
- R 1 to R 4 at least one of R 2 or R 3 is preferably a group represented by any one of the general formulas (2) to (7), and at least R 3 is a group represented by the general formula (2) A group represented by any one of (7) to (7) is more preferable.
- R 5 to R 8 at least one of R 6 or R 7 is preferably a group represented by any one of the general formulas (2) to (7), and at least R 6 is a group represented by the general formula ( A group represented by any one of 2) to (7) is more preferable.
- Preferred compounds are those in which R 3 and R 6 in the general formula (1) are groups represented by any one of the general formulas (2) to (7), and R 2 and R 7 in the general formula (1) are general.
- a more preferred compound is a compound in which R 3 and R 6 are groups represented by any one of the general formulas (2) to (7).
- the groups represented by any of the plurality of general formulas (2) to (7) present in the general formula (1) may be the same or different, but are preferably the same.
- the group represented by the general formula (1) has a symmetrical structure. That is, R 1 and R 8 , R 2 and R 7 , R 3 and R 6 , and R 4 and R 5 are preferably the same.
- L 12 to L 17 each independently represent a single bond or a divalent linking group, and preferably a single bond.
- examples of the divalent linking group include an aromatic hydrocarbon ring group having 6 to 18 carbon atoms and a heteroaromatic hydrocarbon ring group having 6 to 18 ring members. Can be mentioned.
- Preferred linking groups are a phenylene group, a biphenylene group, a fluorenylene group, a triphenylenylene group, and a thiophenylene group, a more preferred linking group is a phenylene group, and a more preferred linking group is a 1,4-phenylene group. .
- These linking groups may have a substituent introduced therein.
- the explanation and preferred range of the substituent that can be introduced into the linking group the explanation and preferred range of the substituent that can be taken by the following R 1 to R 8 and the like can be referred to.
- R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a , R 3b , R 41 to R 48 , R 4a , R 51 to R 58 , R 61 to R 68 each independently represents a hydrogen atom or a substituent.
- R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a , R 3b , R 41 to R 48 , R 4a , R 51 to R 58 , R All of 61 to R 68 may be unsubstituted (that is, a hydrogen atom), but R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a and R 3b , R 41 to R 48 , R 51 to R 58 , and R 61 to R 68 are preferably a substituent, and R 13 and R 18 , R 23 and R 26 , R 33 and R 36 , R 3a and More preferably, at least one of the respective groups of R 3b , R 43 and R 46 , R 53 and R 56 , R 63 and R 66 is a substituent. In each of the general formulas (2) to (7), when there are two or more substituents, these substituents may be the same or different.
- R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a , R 3b , R 41 to R 48 , R 4a , R 51 to R 58 , R 61 to R 68 , R 1 to R 8 can be substituted with a substituent that can be introduced into L 12 to L 17 , for example, a hydroxy group, a halogen atom, a cyano group, an alkyl group having 1 to 20 carbon atoms, 20 alkoxy groups, alkylthio groups having 1 to 20 carbon atoms, alkyl-substituted amino groups having 1 to 20 carbon atoms, acyl groups having 2 to 20 carbon atoms, aryl groups having 6 to 40 carbon atoms, heterocycles having 3 to 40 carbon atoms An aryl group, an alkenyl group having 2 to 10 carbon atoms, an alkynyl group having 2 to 10 carbon atoms, an alkoxycarbonyl group having 2 to 10 carbon atom
- substituents are a halogen atom, a cyano group, a substituted or unsubstituted alkyl group having 1 to 20 carbon atoms, an alkoxy group having 1 to 20 carbon atoms, a substituted or unsubstituted aryl group having 6 to 40 carbon atoms, carbon A substituted or unsubstituted heteroaryl group having 3 to 40 carbon atoms, and a dialkyl-substituted amino group having 1 to 20 carbon atoms.
- substituents are a halogen atom, a cyano group, a substituted or unsubstituted alkyl group having 1 to 20 carbon atoms, an alkoxy group having 1 to 20 carbon atoms, a substituted or unsubstituted aryl group having 6 to 40 carbon atoms, carbon A substituted or unsubstituted heteroaryl group having 3 to 40 carbon atoms, and a dialkyl-substituted amino group having 1 to 20 carbon
- substituents are a fluorine atom, a chlorine atom, a cyano group, a substituted or unsubstituted alkyl group having 1 to 10 carbon atoms, a substituted or unsubstituted alkoxy group having 1 to 10 carbon atoms, and a substituted group having 6 to 15 carbon atoms.
- it is an unsubstituted aryl group or a substituted or unsubstituted heteroaryl group having 3 to 12 carbon atoms.
- the cyclic structure may be an aromatic ring or an alicyclic ring, may contain a hetero atom, and the cyclic structure may be a condensed ring of two or more rings.
- the hetero atom here is preferably selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom.
- Examples of cyclic structures formed include benzene ring, naphthalene ring, pyridine ring, pyridazine ring, pyrimidine ring, pyrazine ring, pyrrole ring, imidazole ring, pyrazole ring, triazole ring, imidazoline ring, oxazole ring, isoxazole ring, thiazole And a ring, an isothiazole ring, a cyclohexadiene ring, a cyclohexene ring, a cyclopentaene ring, a cycloheptatriene ring, a cycloheptadiene ring, and a cycloheptaene ring.
- R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a , R 3b , R 41 to R 48 , R 4a , R 51 to R 58 , and R 61 to R 68 are each independently A group represented by any one of the general formulas (2) to (7) is also preferable.
- R 1 to R 8 may be, for example, at least one selected from the groups represented by any of the general formulas (3) to (7), and at least one of the general formulas (4) to (7 ) May be selected.
- R 1 ⁇ R 8 is a group represented by the general formula (4)
- R 3a and R 3b are preferably bonded to each other to form a cyclic structure.
- the cyclic structure is preferably a tricyclic structure such as a fluorene ring, a xanthene ring, or a thioxanthene ring.
- R 9 represents a substituent.
- R 9 is preferably a substituted or unsubstituted phenyl group represented by the following general formula (8).
- R 9a , R 9b , R 9c , R 9d and R 9e each independently represent a hydrogen atom or a substituent.
- the number of substituents is not particularly limited, and all of R 9a , R 9b , R 9c , R 9d , and R 9e may be unsubstituted (that is, a hydrogen atom), but R 9a , R 9c , and R 9e At least one of them is preferably a substituent, and R 9a and R 9e are more preferably substituents.
- these substituents may be the same or different.
- R 9a and R 9b , R 9b and R 9c , R 9c and R 9d , and R 9d and R 9e may be bonded to each other to form a cyclic structure.
- R 9a , R 9b , R 9c , R 9d , and R 9e may take, see the explanations and preferred ranges of the substituents that can be taken by R 1 to R 8 below.
- R 1 to R 8 For the explanation and preferred examples of the cyclic structure, reference can be made to the explanation and preferred examples of the cyclic structure formed by combining R 1 and R 2 in the general formula (1).
- R 9 is a substituent that includes an atom that does not form a single bond with a boron atom and has a lone pair
- the atom coordinates with the boron atom to form a cyclic structure. It may be.
- the atoms that do not form a single bond with the boron atom and have a lone pair include oxygen atoms and nitrogen atoms.
- the cyclic structure formed by coordination bonding of the atom with a boron atom is preferably a 5- to 7-membered ring, and more preferably a 5- or 6-membered ring.
- the following compounds 177 to 186 can be referred to. In these compounds, an oxygen atom or a nitrogen atom that does not form a single bond with a boron atom is coordinated with the boron atom to form a 5- or 6-membered ring.
- the molecular weight of the compound represented by the general formula (1) is, for example, 1500 or less when the organic layer containing the compound represented by the general formula (1) is intended to be formed by vapor deposition. Preferably, it is preferably 1200 or less, more preferably 1000 or less, and even more preferably 800 or less.
- the lower limit of the molecular weight is the molecular weight of the minimum compound represented by the general formula (1).
- the compound represented by the general formula (1) may be formed by a coating method regardless of the molecular weight. If a coating method is used, a film can be formed even with a compound having a relatively large molecular weight.
- a compound containing a plurality of structures represented by the general formula (1) in the molecule as a light emitting material.
- a polymer obtained by previously polymerizing a polymerizable group in the structure represented by the general formula (1) and polymerizing the polymerizable group as a light emitting material.
- a monomer containing a polymerizable functional group in any of R 1 to R 9 in the general formula (1) and polymerizing it alone or copolymerizing with other monomers, It is conceivable to obtain a polymer having a repeating unit and use the polymer as a light emitting material.
- dimers and trimers are obtained by reacting compounds having a structure represented by the general formula (1) and used as a luminescent material.
- Examples of the polymer having a repeating unit containing a structure represented by the general formula (1) include a polymer containing a structure represented by the following general formula (9) or (10).
- Q represents a group including the structure represented by General Formula (1)
- L 1 and L 2 represent a linking group.
- the linking group preferably has 0 to 20 carbon atoms, more preferably 1 to 15 carbon atoms, and still more preferably 2 to 10 carbon atoms. And preferably has a structure represented by - linking group -X 11 -L 11.
- X 11 represents an oxygen atom or a sulfur atom, and is preferably an oxygen atom.
- L 11 represents a linking group, preferably a substituted or unsubstituted alkylene group, or a substituted or unsubstituted arylene group, and a substituted or unsubstituted alkylene group having 1 to 10 carbon atoms, or a substituted or unsubstituted group A phenylene group is more preferable.
- R 101 , R 102 , R 103 and R 104 each independently represent a substituent.
- it is a substituted or unsubstituted alkyl group having 1 to 6 carbon atoms, a substituted or unsubstituted alkoxy group having 1 to 6 carbon atoms, or a halogen atom, more preferably an unsubstituted alkyl group having 1 to 3 carbon atoms.
- An unsubstituted alkoxy group having 1 to 3 carbon atoms, a fluorine atom, and a chlorine atom and more preferably an unsubstituted alkyl group having 1 to 3 carbon atoms and an unsubstituted alkoxy group having 1 to 3 carbon atoms.
- the linking group represented by L 1 and L 2 is any one of R 1 to R 8 in the structure of the general formula (1) constituting Q, any one of R 11 to R 20 in the general formula (2), Any one of R 21 to R 28 in the structure of formula (3), any of R 31 to R 38 in the structure of general formula (4), any of R 41 to R 48 in the structure of general formula (5), It can be bonded to any one of R 51 to R 58 of the structure of the general formula (6) and any of R 61 to R 68 of the structure of the general formula (7).
- Two or more linking groups may be linked to one Q to form a crosslinked structure or a network structure.
- repeating unit examples include structures represented by the following formulas (11) to (14).
- a hydroxy group is introduced into any one of R 1 to R 9 of the structure of the general formula (1), and this is used as a linker as described below. It can be synthesized by reacting a compound to introduce a polymerizable group and polymerizing the polymerizable group.
- the polymer containing the structure represented by the general formula (1) in the molecule may be a polymer composed only of repeating units having the structure represented by the general formula (1), or other structures may be used. It may be a polymer containing repeating units.
- the repeating unit having a structure represented by the general formula (1) contained in the polymer may be a single type or two or more types. Examples of the repeating unit not having the structure represented by the general formula (1) include those derived from monomers used in ordinary copolymerization. Examples thereof include a repeating unit derived from a monomer having an ethylenically unsaturated bond such as ethylene and styrene.
- X ′ represents an oxygen atom or a sulfur atom.
- R 1 ′ to R 8 ′ each independently represents a hydrogen atom or a substituent. However, at least one of R 1 ′ to R 8 ′ is a group independently represented by the following general formulas (2 ′) to (7 ′).
- R 1 'and R 2', R 2 'and R 3', R 3 'and R 4', R 5 'and R 6', R 6 'and R 7', R 7 'and R 8', R 8 'And R 9 ' and R 9 'and R 1 ' may be bonded to each other to form a cyclic structure.
- R 9 ′ represents a substituent. When R 9 ′ includes an atom having a lone pair of electrons that does not form a single bond with a boron atom, the atom may be coordinated with the boron atom to form a cyclic structure.
- L 12 ′ to L 17 ′ each independently represents a single bond or a divalent linking group
- * represents a binding site to the benzene ring in the general formula (1 ′).
- R 1 ′ to R 8 ′ in general formula (1 ′) and R 11 ′ to R 20 ′, R 21 ′ to R 28 ′, R 31 ′ to R 38 in general formulas (2 ′) to (7 ′) R, R 3a , R 3b ′, R 41 ′ to R 48 ′, R 4a ′, R 51 ′ to R 58 ′, R 61 ′ to R 68 ′ are explained in general formula (1).
- the description of the compound represented by can be referred to.
- A represents a halogen atom, and examples include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom, and a chlorine atom, a bromine atom, and an iodine atom are preferable.
- the above reaction is an application of a known reaction, and known reaction conditions can be appropriately selected and used. The details of the above reaction can be referred to the synthesis examples described below.
- the compound represented by the general formula (1 ′) can also be synthesized by combining other known synthesis reactions.
- the compound represented by the general formula (1) of the present invention is useful as a light emitting material of an organic light emitting device. For this reason, the compound represented by General formula (1) of this invention can be effectively used as a luminescent material for the light emitting layer of an organic light emitting element.
- the compound represented by the general formula (1) includes a delayed fluorescent material (delayed phosphor) that emits delayed fluorescence. That is, the present invention relates to a delayed phosphor having a structure represented by the general formula (1), an invention using a compound represented by the general formula (1) as a delayed phosphor, and a general formula (1).
- An invention of a method for emitting delayed fluorescence using the represented compound is also provided.
- An organic light emitting device using such a compound as a light emitting material emits delayed fluorescence and has a feature of high luminous efficiency. The principle will be described below by taking an organic electroluminescence element as an example.
- the organic electroluminescence element carriers are injected into the light emitting material from both positive and negative electrodes to generate an excited light emitting material and emit light.
- 25% of the generated excitons are excited to the excited singlet state, and the remaining 75% are excited to the excited triplet state. Therefore, the use efficiency of energy is higher when phosphorescence, which is light emission from an excited triplet state, is used.
- the excited triplet state has a long lifetime, energy saturation occurs due to saturation of the excited state and interaction with excitons in the excited triplet state, and in general, the quantum yield of phosphorescence is often not high.
- delayed fluorescent materials after energy transition to an excited triplet state due to intersystem crossing, etc., are then crossed back to an excited singlet state due to triplet-triplet annihilation or absorption of thermal energy, and emit fluorescence.
- a thermally activated delayed fluorescent material by absorption of thermal energy is particularly useful.
- excitons in the excited singlet state emit fluorescence as usual.
- excitons in the excited triplet state absorb heat generated by the device and cross between the excited singlets to emit fluorescence.
- the light is emitted from the excited singlet, the light is emitted at the same wavelength as the fluorescence, but the light lifetime (luminescence lifetime) generated by the reverse intersystem crossing from the excited triplet state to the excited singlet state is normal. Since the fluorescence becomes longer than the fluorescence and phosphorescence, it is observed as fluorescence delayed from these. This can be defined as delayed fluorescence. If such a heat-activated exciton transfer mechanism is used, the ratio of the compound in an excited singlet state, which normally generated only 25%, is increased to 25% or more by absorbing thermal energy after carrier injection. It can be raised.
- the heat of the device will sufficiently cause intersystem crossing from the excited triplet state to the excited singlet state and emit delayed fluorescence. Efficiency can be improved dramatically.
- the compound represented by the general formula (1) of the present invention as a light-emitting material of a light-emitting layer, excellent organic light-emitting devices such as an organic photoluminescence device (organic PL device) and an organic electroluminescence device (organic EL device) Can be provided.
- the compound represented by the general formula (1) of the present invention may have a function of assisting light emission of another light emitting material included in the light emitting layer as a so-called assist dopant. That is, the compound represented by the general formula (1) of the present invention contained in the light emitting layer includes the lowest excitation singlet energy level of the host material contained in the light emitting layer and the lowest excitation of other light emitting materials contained in the light emitting layer.
- the organic photoluminescence element has a structure in which at least a light emitting layer is formed on a substrate.
- the organic electroluminescence element has a structure in which an organic layer is formed at least between an anode, a cathode, and an anode and a cathode.
- the organic layer includes at least a light emitting layer, and may consist of only the light emitting layer, or may have one or more organic layers in addition to the light emitting layer. Examples of such other organic layers include a hole transport layer, a hole injection layer, an electron blocking layer, a hole blocking layer, an electron injection layer, an electron transport layer, and an exciton blocking layer.
- the hole transport layer may be a hole injection / transport layer having a hole injection function
- the electron transport layer may be an electron injection / transport layer having an electron injection function.
- FIG. 1 A specific example of the structure of an organic electroluminescence element is shown in FIG.
- 1 is a substrate
- 2 is an anode
- 3 is a hole injection layer
- 4 is a hole transport layer
- 5 is a light emitting layer
- 6 is an electron transport layer
- 7 is a cathode.
- each member and each layer of an organic electroluminescent element are demonstrated.
- substrate and a light emitting layer corresponds also to the board
- the organic electroluminescence device of the present invention is preferably supported on a substrate.
- the substrate is not particularly limited and may be any substrate conventionally used for organic electroluminescence elements.
- a substrate made of glass, transparent plastic, quartz, silicon, or the like can be used.
- an electrode material made of a metal, an alloy, an electrically conductive compound, or a mixture thereof having a high work function (4 eV or more) is preferably used.
- electrode materials include metals such as Au, and conductive transparent materials such as CuI, indium tin oxide (ITO), SnO 2 , and ZnO.
- conductive transparent materials such as CuI, indium tin oxide (ITO), SnO 2 , and ZnO.
- an amorphous material such as IDIXO (In 2 O 3 —ZnO) that can form a transparent conductive film may be used.
- a thin film may be formed by vapor deposition or sputtering of these electrode materials, and a pattern of a desired shape may be formed by photolithography, or when pattern accuracy is not so high (about 100 ⁇ m or more) ), A pattern may be formed through a mask having a desired shape at the time of vapor deposition or sputtering of the electrode material.
- wet film-forming methods such as a printing system and a coating system, can also be used.
- the transmittance be greater than 10%, and the sheet resistance as the anode is preferably several hundred ⁇ / ⁇ or less.
- the film thickness depends on the material, it is usually selected in the range of 10 to 1000 nm, preferably 10 to 200 nm.
- cathode a material having a low work function (4 eV or less) metal (referred to as an electron injecting metal), an alloy, an electrically conductive compound, and a mixture thereof as an electrode material is used.
- electrode materials include sodium, sodium-potassium alloy, magnesium, lithium, magnesium / copper mixture, magnesium / silver mixture, magnesium / aluminum mixture, magnesium / indium mixture, aluminum / aluminum oxide (Al 2 O 3 ) Mixtures, indium, lithium / aluminum mixtures, rare earth metals and the like.
- a mixture of an electron injecting metal and a second metal which is a stable metal having a larger work function value than this for example, a magnesium / silver mixture
- Suitable are a magnesium / aluminum mixture, a magnesium / indium mixture, an aluminum / aluminum oxide (Al 2 O 3 ) mixture, a lithium / aluminum mixture, aluminum and the like.
- the cathode can be produced by forming a thin film of these electrode materials by a method such as vapor deposition or sputtering.
- the sheet resistance as the cathode is preferably several hundred ⁇ / ⁇ or less, and the film thickness is usually selected in the range of 10 nm to 5 ⁇ m, preferably 50 to 200 nm.
- the emission luminance is advantageously improved.
- a transparent or semi-transparent cathode can be produced. By applying this, an element in which both the anode and the cathode are transparent is used. Can be produced.
- the light emitting layer is a layer that emits light after excitons are generated by recombination of holes and electrons injected from each of the anode and the cathode, and the light emitting material may be used alone for the light emitting layer. , Preferably including a luminescent material and a host material. As a luminescent material, the 1 type (s) or 2 or more types chosen from the compound group of this invention represented by General formula (1) can be used. In order for the organic electroluminescence device and the organic photoluminescence device of the present invention to exhibit high luminous efficiency, it is important to confine singlet excitons and triplet excitons generated in the light emitting material in the light emitting material.
- a host material in addition to the light emitting material in the light emitting layer.
- the host material an organic compound having at least one of excited singlet energy and excited triplet energy higher than that of the light emitting material of the present invention can be used.
- singlet excitons and triplet excitons generated in the light emitting material of the present invention can be confined in the molecules of the light emitting material of the present invention, and the light emission efficiency can be sufficiently extracted.
- high luminous efficiency can be obtained, so that host materials that can achieve high luminous efficiency are particularly limited. And can be used in the present invention.
- the organic light emitting device or organic electroluminescent device of the present invention light emission is generated from the light emitting material of the present invention contained in the light emitting layer. This emission includes both fluorescence and delayed fluorescence. However, light emission from the host material may be partly or partly emitted.
- the amount of the compound of the present invention, which is a light emitting material is preferably 0.1% by weight or more, more preferably 1% by weight or more, and 50% or more. It is preferably no greater than wt%, more preferably no greater than 20 wt%, and even more preferably no greater than 10 wt%.
- the host material in the light-emitting layer is preferably an organic compound that has a hole transporting ability and an electron transporting ability, prevents the emission of longer wavelengths, and has a high glass transition temperature.
- the injection layer is a layer provided between the electrode and the organic layer for lowering the driving voltage and improving the luminance of light emission, and includes a hole injection layer and an electron injection layer, Further, it may be present between the cathode and the light emitting layer or the electron transport layer.
- the injection layer can be provided as necessary.
- the blocking layer is a layer that can prevent diffusion of charges (electrons or holes) and / or excitons existing in the light emitting layer to the outside of the light emitting layer.
- the electron blocking layer can be disposed between the light emitting layer and the hole transport layer and blocks electrons from passing through the light emitting layer toward the hole transport layer.
- a hole blocking layer can be disposed between the light emitting layer and the electron transporting layer to prevent holes from passing through the light emitting layer toward the electron transporting layer.
- the blocking layer can also be used to block excitons from diffusing outside the light emitting layer. That is, each of the electron blocking layer and the hole blocking layer can also function as an exciton blocking layer.
- the term “electron blocking layer” or “exciton blocking layer” as used herein is used in the sense of including a layer having the functions of an electron blocking layer and an exciton blocking layer in one layer.
- the hole blocking layer has a function of an electron transport layer in a broad sense.
- the hole blocking layer has a role of blocking holes from reaching the electron transport layer while transporting electrons, thereby improving the recombination probability of electrons and holes in the light emitting layer.
- the material for the hole blocking layer the material for the electron transport layer described later can be used as necessary.
- the electron blocking layer has a function of transporting holes in a broad sense.
- the electron blocking layer has a role to block electrons from reaching the hole transport layer while transporting holes, thereby improving the probability of recombination of electrons and holes in the light emitting layer. .
- the exciton blocking layer is a layer for preventing excitons generated by recombination of holes and electrons in the light emitting layer from diffusing into the charge transport layer. It becomes possible to efficiently confine in the light emitting layer, and the light emission efficiency of the device can be improved.
- the exciton blocking layer can be inserted on either the anode side or the cathode side adjacent to the light emitting layer, or both can be inserted simultaneously.
- the layer when the exciton blocking layer is provided on the anode side, the layer can be inserted adjacent to the light emitting layer between the hole transport layer and the light emitting layer, and when inserted on the cathode side, the light emitting layer and the cathode Between the luminescent layer and the light-emitting layer.
- a hole injection layer, an electron blocking layer, or the like can be provided between the anode and the exciton blocking layer adjacent to the anode side of the light emitting layer, and the excitation adjacent to the cathode and the cathode side of the light emitting layer can be provided.
- an electron injection layer, an electron transport layer, a hole blocking layer, and the like can be provided.
- the blocking layer is disposed, at least one of the excited singlet energy and the excited triplet energy of the material used as the blocking layer is preferably higher than the excited singlet energy and the excited triplet energy of the light emitting material.
- the hole transport layer is made of a hole transport material having a function of transporting holes, and the hole transport layer can be provided as a single layer or a plurality of layers.
- the hole transport material has any one of hole injection or transport and electron barrier properties, and may be either organic or inorganic.
- hole transport materials that can be used include, for example, triazole derivatives, oxadiazole derivatives, imidazole derivatives, carbazole derivatives, indolocarbazole derivatives, polyarylalkane derivatives, pyrazoline derivatives and pyrazolone derivatives, phenylenediamine derivatives, arylamine derivatives, Examples include amino-substituted chalcone derivatives, oxazole derivatives, styrylanthracene derivatives, fluorenone derivatives, hydrazone derivatives, stilbene derivatives, silazane derivatives, aniline copolymers, and conductive polymer oligomers, particularly thiophene oligomers.
- An aromatic tertiary amine compound and an styrylamine compound are preferably used, and an aromatic tertiary amine compound is more preferably used.
- the electron transport layer is made of a material having a function of transporting electrons, and the electron transport layer can be provided as a single layer or a plurality of layers.
- the electron transport material (which may also serve as a hole blocking material) may have a function of transmitting electrons injected from the cathode to the light emitting layer.
- Examples of the electron transport layer that can be used include nitro-substituted fluorene derivatives, diphenylquinone derivatives, thiopyrandioxide derivatives, carbodiimides, fluorenylidenemethane derivatives, anthraquinodimethane and anthrone derivatives, oxadiazole derivatives, and the like.
- a thiadiazole derivative in which the oxygen atom of the oxadiazole ring is substituted with a sulfur atom, and a quinoxaline derivative having a quinoxaline ring known as an electron withdrawing group can also be used as an electron transport material.
- a polymer material in which these materials are introduced into a polymer chain or these materials are used as a polymer main chain can also be used.
- the compound represented by the general formula (1) may be used not only for the light emitting layer but also for layers other than the light emitting layer.
- the compound represented by General formula (1) used for a light emitting layer and the compound represented by General formula (1) used for layers other than a light emitting layer may be same or different.
- the compound represented by the general formula (1) may be used for the injection layer, blocking layer, hole blocking layer, electron blocking layer, exciton blocking layer, hole transporting layer, electron transporting layer, and the like. .
- the method for forming these layers is not particularly limited, and the layer may be formed by either a dry process or a wet process.
- the preferable material which can be used for an organic electroluminescent element is illustrated concretely.
- the material that can be used in the present invention is not limited to the following exemplary compounds.
- R and R 1 to R 10 in the structural formulas of the following exemplary compounds each independently represent a hydrogen atom or a substituent.
- n represents an integer of 3 to 5.
- the organic electroluminescent device produced by the above-described method emits light by applying an electric field between the anode and the cathode of the obtained device. At this time, if the light is emitted by excited singlet energy, light having a wavelength corresponding to the energy level is confirmed as fluorescence emission and delayed fluorescence emission. In addition, in the case of light emission by excited triplet energy, a wavelength corresponding to the energy level is confirmed as phosphorescence. Since normal fluorescence has a shorter fluorescence lifetime than delayed fluorescence, the emission lifetime can be distinguished from fluorescence and delayed fluorescence.
- the excited triplet energy is unstable and is converted into heat and the like, and the lifetime is short and it is immediately deactivated.
- the excited triplet energy of a normal organic compound it can be measured by observing light emission under extremely low temperature conditions.
- the organic electroluminescence element of the present invention can be applied to any of a single element, an element having a structure arranged in an array, and a structure in which an anode and a cathode are arranged in an XY matrix. According to the present invention, an organic light emitting device with greatly improved light emission efficiency can be obtained by containing the compound represented by the general formula (1) in the light emitting layer.
- the organic light emitting device such as the organic electroluminescence device of the present invention can be further applied to various uses. For example, it is possible to produce an organic electroluminescence display device using the organic electroluminescence element of the present invention.
- organic electroluminescence device of the present invention can be applied to organic electroluminescence illumination and backlights that are in great demand.
- Methyl anthranilate (349.2 g, 2310 mmol), iodobenzene (428.4 g, 2100 mmol), potassium carbonate (348.3 g, 2520 mmol), and copper iodide (20.0 g, 105 mmol) were placed in a three-necked flask under a nitrogen atmosphere. The mixture was heated and stirred at 180 ° C. After completion of the reaction, the mixture was cooled to room temperature, diluted with 500 ml of hexane, and filtered using celite.
- the target pale yellow oily liquid (intermediate 1a) was obtained in a yield of 427.5 g and a yield of 90%.
- This solution was washed with water using a separatory funnel and dried over anhydrous magnesium sulfate. Thereafter, the solution was filtered, concentrated, and vacuum dried (60 ° C., 6 h) to obtain an oily liquid.
- This oily liquid was put into a three-necked flask, 960 ml of chloroform and 62.3 ml of methanesulfonic acid were added, and the mixture was refluxed for 3 hours in a nitrogen atmosphere. After cooling this mixture to room temperature, it neutralized carefully using sodium hydrogencarbonate aqueous solution, and extracted with chloroform using the separatory funnel. The obtained extract was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated.
- the concentrate obtained by concentrating the obtained fraction was recrystallized with a mixed solvent of toluene and hexane (1: 5 ml / g).
- the target intermediate 1b was obtained in a yield of 47.9 g and a yield of 48%.
- This solution was washed with water using a separatory funnel and dried over anhydrous magnesium sulfate. Thereafter, the solution was filtered, concentrated, and vacuum dried (60 ° C., 6 h) to obtain an oily substance.
- This oily substance was put into a three-necked flask, 672 ml of chloroform and 21.8 ml of methanesulfonic acid were added, and the mixture was refluxed for 3 hours in a nitrogen atmosphere. The mixture was cooled to room temperature, then carefully neutralized with an aqueous sodium hydrogen carbonate solution, and extracted with dichloromethane using a separatory funnel. The extract was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated.
- This concentrate was recrystallized with a mixed solvent of toluene and hexane (5:10 ml / g). As a result, the target intermediate 2a was obtained in a yield of 27.9 g and a yield of 50%.
- the resulting filtrate was filtered through a silica gel pad and concentrated.
- the concentrate obtained by concentrating the obtained fraction was recrystallized from hexane (5 ml / g, 5 ° C.). As a result, the target product Intermediate 3a was obtained in a yield of 19.68 g and a yield of 61%.
- 2-Bromo-1,3,5-triisopropylbenzene (43.9 g, 155 mmol) and dehydrated tetrahydrofuran 155 ml were placed in a three-necked flask and cooled to -78 ° C.
- a 1.60 M hexane solution of n-butyllithium (106.6 ml, 170.5 mmol) was added with a syringe and stirred at ⁇ 78 ° C. for 10 minutes, and then trimethoxyborane (34.5 ml, 310 mmol) was added for 2 minutes. It was dripped over. The mixture was then stirred at ⁇ 78 ° C. for 30 minutes and then at room temperature overnight.
- the mixture was concentrated to about half by an evaporator, 300 ml of 1N hydrochloric acid aqueous solution was added, and the mixture was stirred at room temperature for 4 hours.
- the resulting solution was placed in a separatory funnel and extracted with dichloromethane.
- the extract was washed twice with water, dried over anhydrous magnesium sulfate, filtered and concentrated.
- This concentrate was put into a three-necked flask equipped with a Soxhlet extractor charged with molecular sieves, and 200 ml of toluene and 30 ml of methanol were added and refluxed for 12 hours. After completion of the reaction, the mixture was cooled to room temperature and concentrated with an evaporator.
- the solvent of the obtained fraction was removed by vacuum drying, and the target colorless oily substance (intermediate 3c) was obtained in a yield of 31.88 g and a yield of 74%.
- 1,4-dibromo-2-fluorobenzene 50.78 g, 200 mmol
- potassium hydroxide (6.73 g, 120 mmol)
- potassium carbonate 27.64 g, 200 mmol
- 20 ml of triethylene glycol dimethyl ether 20 ml
- the mixture was heated and stirred at 180 ° C. for 96 hours in a nitrogen atmosphere.
- the mixture was cooled to room temperature, diluted by adding 500 ml of toluene, and further subjected to sonication for 10 minutes by adding 500 ml of water.
- the mixture was filtered using celite, and the filtrate was collected.
- the filtrate was transferred to a separatory funnel and washed twice with water to recover the organic phase.
- the obtained organic phase was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated.
- the intermediate product 4a was obtained in a yield of 8.51 g and a yield of 18%.
- the obtained organic phase was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated.
- This white solid was rinsed with hexane and then vacuum-dried to obtain 9.40 g of the target intermediate 5a in a yield of 88%.
- 2,5-dibromofluorobenzene (15.23 g, 60 mmol), 2-bromobenzenethiol (9.45 g, 50 mmol), potassium carbonate (13.82 g, 100 mmol), and 10 ml of triethylene glycol dimethyl ether were placed in a three-necked flask, and nitrogen was added. The mixture was heated and stirred at 180 ° C. for 24 hours in an atmosphere. After completion of the reaction, the mixture was cooled to room temperature, diluted with 100 ml of toluene, and further sonicated with 100 ml of water for 10 minutes. The mixture was filtered using celite, and the filtrate was collected.
- reaction solution was deactivated by adding a small amount of water, and extracted with dichloromethane using a separatory funnel.
- the obtained extract was dried over anhydrous magnesium sulfate, filtered, and concentrated to obtain a reaction crude product.
- This crude reaction product was placed in a three-necked flask, 600 ml of chloroform and 3.9 ml of methanesulfonic acid were added, and the mixture was refluxed for 3 hours in a nitrogen atmosphere.
- the mixture was cooled to room temperature and washed with water using a separatory funnel.
- the washed mixture was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated to obtain a precipitate.
- This precipitate was recrystallized with ethanol to obtain 10.2 g of a target intermediate 8b in a yield of 45%.
- the resulting solution was washed with a separatory funnel, and then residual bromine was deactivated with a sodium thiosulfate / sodium hydroxide aqueous solution.
- the solution was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated.
- the obtained concentrate was purified by silica gel chromatography using hexane as a developing solvent.
- the resulting fraction concentrate was recrystallized from hexane.
- the target intermediate 11a was obtained in a yield of 38.29 g and a yield of 79%.
- Intermediate 11b was synthesized in the same manner as Compound 5 Synthesis Method A described later, except that Intermediate 11a (14.57 g, 30 mmol) was used as a raw material. The obtained intermediate 11b was 6.27 g in yield and 39% in yield.
- 2,4,6-Triphenylbromobenzene (25.5 g, 66 mmol) and 264 ml of dehydrated tetrahydrofuran were placed in a three-necked flask, cooled to ⁇ 80 ° C., and 1.60 M hexane solution (45. 4 ml, 72.6 mmol) was added via syringe. Thereafter, the reaction solution was stirred at ⁇ 80 ° C. for 15 minutes, and then trimethoxyborane (14.7 ml, 132 mmol) was added by syringe and stirred overnight at room temperature.
- the resulting solution was filtered through a silica gel pad and the filtrate was concentrated.
- the concentrate of the obtained fraction was recrystallized with hexane to obtain 2.20 g of the target compound 5 in a yield of 37%.
- the resulting solution was filtered using celite and concentrated.
- Synthesis Example 11 Synthesis of Compound 176
- Compound 176 was synthesized in the same manner as the compound 59 synthesis method, except that intermediate 7b (2.16 mmol) and intermediate 2a (2.59 mmol) were used as raw materials.
- the compound 176 obtained was 0.85 g in yield and 54% in yield.
- an organic photoluminescence element and an organic electroluminescence element were produced and evaluated. Emission characteristics are evaluated by source meter (Keithley: 2400 series), semiconductor parameter analyzer (Agilent Technology: E5273A), optical power meter measuring device (Newport: 1930C), optical spectrometer (Ocean) Optics: USB2000), a spectroradiometer (Topcon: SR-3) and a streak camera (Hamamatsu Photonics C4334) were used.
- Singlet energy E S1 A sample having a thickness of 100 nm was prepared on a Si substrate by co-evaporating the measurement target compound and mCP so that the measurement target compound had a concentration of 6% by weight. The fluorescence spectrum of this sample was measured at room temperature (300K). By integrating the luminescence from immediately after the excitation light incidence to 100 nanoseconds after the incidence, a fluorescence spectrum having a luminescence intensity on the vertical axis and a wavelength on the horizontal axis was obtained.
- the vertical axis represents light emission and the horizontal axis represents wavelength.
- a tangent line was drawn with respect to the short-wave rise of the emission spectrum, and the wavelength value ⁇ edge [nm] at the intersection of the tangent line and the horizontal axis was obtained.
- a value obtained by converting this wavelength value into an energy value by the following conversion formula was defined as E S1 .
- Conversion formula: E S1 [eV] 1239.85 / ⁇ edge
- the maximum point having a peak intensity of 10% or less of the maximum peak intensity of the spectrum is not included in the above-mentioned maximum value on the shortest wavelength side, and has the maximum slope value closest to the maximum value on the shortest wavelength side.
- the tangent drawn at the point where the value was taken was taken as the tangent to the rising edge of the phosphorescence spectrum on the short wavelength side.
- Example 1 Preparation and Evaluation of Organic Photoluminescence Device Using Compound 5
- a toluene solution (concentration 10 ⁇ 5 mol / L) and a hexane solution (concentration 10 ⁇ 5 mol / L) of compound 5 were prepared in the atmosphere. Then, after deoxygenation by bubbling with nitrogen for 15 minutes, it was used for measurement of light emission characteristics.
- a thin film of compound 5 was formed to a thickness of 100 nm on a quartz substrate by a vacuum deposition method under a vacuum degree of 10 ⁇ 4 Pa or less to obtain an organic photoluminescence device.
- the result of measuring the emission spectrum of the toluene solution and the hexane solution of Compound 5 is shown in FIG.
- the photoluminescence quantum yield ⁇ was 0.97 for the toluene solution, 0.54 for the hexane solution, and 0.83 for the organic photoluminescence device having a thin film of Compound 5.
- Energy difference Delta] E ST of the singlet excited state and a triplet excited state as determined from the fluorescence spectrum and phosphorescence spectrum was 0.05 eV.
- the transient decay curve of the toluene solution of compound 5 and a hexane solution is shown in FIG.
- This transient decay curve shows the result of measuring the luminescence lifetime obtained by measuring the process in which the emission intensity is deactivated by applying excitation light to the compound.
- the light emission intensity decays in a single exponential manner. This means that if the vertical axis of the graph is semi-log, it will decay linearly.
- the transient decay curve of Compound 5 shown in FIG. 4 such a linear component (fluorescence) is observed at the beginning of observation, but a component deviating from linearity appears after several ⁇ sec. This is light emission of the delay component, and the signal added to the initial component becomes a loose curve with a tail on the long time side.
- Compound 5 is a luminescent material containing a delay component in addition to the fluorescent component.
- the emission lifetime ⁇ 1 of the immediate fluorescence component in the toluene solution is 35 ns
- the emission lifetime ⁇ 2 of the delayed fluorescence component is 1.35 ⁇ s
- the emission lifetime ⁇ 1 of the immediate fluorescence component in the hexane solution is 21 ns
- the emission lifetime ⁇ 2 of the delay fluorescence component is 2.09 ⁇ s.
- Example 2 Production and evaluation of organic photoluminescence device using compound 6
- a toluene solution and a hexane solution of compound 6 by the same method as in Example 1, An organic photoluminescence device having a thin film of Compound 6 was produced. Further, a thin film in which the concentration of compound 6 is 6.0% by weight is deposited on a quartz substrate by a vacuum deposition method under the condition of a vacuum degree of 10 ⁇ 4 Pa or less from different deposition sources. An organic photoluminescence device was formed with a thickness of 100 nm.
- FIG. 5 shows the results of measuring the emission spectrum of an organic photoluminescence device having a toluene solution and a hexane solution of compound 6 and a thin film of compound 6 and mCP. The results are shown in FIG.
- the photoluminescence quantum yield ⁇ was 1.00 for the toluene solution, 0.54 for the hexane solution, and 0.88 for the organic photoluminescence device having a thin film of compound 6 alone.
- Energy difference Delta] E ST of the singlet excited state and a triplet excited state as determined from the fluorescence spectrum and phosphorescence spectrum was 0.08 eV.
- the transient decay curve of the toluene solution of compound 6 and a hexane solution is shown in FIG.
- the emission lifetime ⁇ 1 of the immediate fluorescence component in the toluene solution is 33 ns
- the emission lifetime ⁇ 2 of the delayed fluorescence component is 1.60 ⁇ s
- the emission lifetime ⁇ 1 of the immediate fluorescence component in the hexane solution is 20 ns
- the emission lifetime ⁇ 2 of the delay fluorescence component is 1.29 ⁇ s.
- Example 3 Preparation and Evaluation of Organic Photoluminescence Device Using Compound 18
- a toluene solution of Compound 18 was prepared in the same manner as in Example 1 except that Compound 18 was used instead of Compound 5.
- the emission spectrum and transient decay curve of this toluene solution were measured.
- the measured emission spectrum is shown in FIG. 8, and the transient decay curve is shown in FIG.
- the photoluminescence quantum yield ⁇ of the toluene solution of compound 18 was 0.81.
- the emission lifetime ⁇ 1 of the immediate fluorescence component was 89.3 ns
- the emission lifetime ⁇ 2 of the delayed fluorescence component was 39.5 ⁇ s.
- Example 4 Production and Evaluation of Organic Photoluminescence Device Using Compound 59
- a toluene solution of compound 59 was prepared in the same manner as in Example 1, except that compound 59 was used instead of compound 5.
- the emission spectrum and transient decay curve of this toluene solution were measured.
- the measured emission spectrum is shown in FIG.
- the photoluminescence quantum yield ⁇ of the toluene solution of compound 59 was 0.76. Further, the emission lifetime ⁇ 1 of the immediate fluorescence component was 27.7 ns, and the emission lifetime ⁇ 2 of the delayed fluorescence component was 4.02 ⁇ s.
- Example 5 Production and Evaluation of Organic Photoluminescence Device Using Compound 65
- a toluene solution of compound 65 was prepared by the same method as in Example 1 except that compound 65 was used instead of compound 5.
- the emission spectrum and transient decay curve of this toluene solution were measured.
- the measured emission spectrum is shown in FIG.
- the photoluminescence quantum yield ⁇ of the toluene solution of compound 65 was 0.56. Further, the emission lifetime ⁇ 1 of the immediate fluorescence component was 25.9 ns, and the emission lifetime ⁇ 2 of the delayed fluorescence component was 2.06 ⁇ s.
- Example 6 Production and Evaluation of Organic Photoluminescence Device Using Compound 67
- a toluene solution of compound 67 was prepared in the same manner as in Example 1 except that compound 67 was used instead of compound 5.
- the emission spectrum and transient decay curve of this toluene solution were measured.
- the measured emission spectrum is shown in FIG.
- the photoluminescence quantum yield ⁇ of the toluene solution of compound 67 was 1.00. Further, the emission lifetime ⁇ 1 of the immediate fluorescence component was 35 ns, and the emission lifetime ⁇ 2 of the delayed fluorescence component was 2.33 ⁇ s.
- Example 7 Production and Evaluation of Organic Photoluminescence Device Using Compound 156
- a toluene solution of compound 156 was prepared in the same manner as in Example 1 except that compound 156 was used instead of compound 5.
- the emission spectrum and transient decay curve of this toluene solution were measured.
- the measured emission spectrum is shown in FIG. 13, and the transient decay curve is shown in FIG.
- the photoluminescence quantum yield ⁇ of the toluene solution of compound 156 was 1.00.
- the emission lifetime ⁇ 1 of the immediate fluorescence component was 3.2 ns
- the emission lifetime ⁇ 2 of the delayed fluorescence component was 1.61 ⁇ s.
- Example 8 A toluene solution was prepared in the same manner as in Example 1, except that the compounds 88, 116, and 176 were used instead of the compound 5. The emission spectrum and transient decay curve of this toluene solution were measured. Table 1 shows the photoluminescence quantum yield ⁇ and the emission lifetime determined from the measured emission spectrum and transient decay curve. As shown in Table 1, all the toluene solutions of the compounds 88, 116, and 176 obtained a sufficient photoluminescence quantum yield ⁇ , and confirmed delayed fluorescence.
- Comparative Example 1 Preparation and Evaluation of Organic Photoluminescence Device Using Comparative Compound A Using Comparative Compound A having the following structure instead of Compound 1, a toluene solution of Comparative Compound A was prepared in the same manner as Example 1. Prepared. The emission spectrum and transient decay curve of this toluene solution were measured. The measured emission spectrum is shown in FIG. 15, and the transient decay curve is shown in FIG. The emission lifetime was 16.0 ns, and no delayed fluorescence was observed.
- Comparative Example 2 Preparation and Evaluation of Organic Photoluminescence Element Using Comparative Compound B Using Comparative Compound B having the following structure instead of Compound 1, a toluene solution of Comparative Compound A was prepared in the same manner as Example 1. Prepared. The emission spectrum and transient decay curve of this toluene solution were measured. The measured emission spectrum is shown in FIG. 17, and the transient decay curve is shown in FIG. The photoluminescence quantum yield ⁇ of the toluene solution of Comparative Compound B was 0.27. Moreover, the light emission lifetime was 8.1 ns, and no delayed fluorescence was observed.
- Comparative Example 3 Preparation and Evaluation of Organic Photoluminescence Device Using Comparative Compound C having the following structure instead of Compound 1, a toluene solution of Comparative Compound C was prepared in the same manner as Example 1. Prepared. The emission spectrum and transient decay curve of this toluene solution were measured. The measured emission spectrum is shown in FIG. 19, and the transient decay curve is shown in FIG. The photoluminescence quantum yield ⁇ of the toluene solution of Comparative Compound C was 0.26. The emission lifetime was 12.7 ns, and no delayed fluorescence was observed.
- Example 11 Production and evaluation of organic electroluminescence device using compound 5 Each thin film was formed by vacuum deposition on a glass substrate on which an anode made of indium tin oxide (ITO) having a thickness of 100 nm was formed. And a degree of vacuum of 5.0 ⁇ 10 ⁇ 4 Pa or less. First, ⁇ -NPD was formed on ITO with a thickness of 30 nm, and mCP was formed thereon with a thickness of 10 nm. Next, Compound 5 and (Cz) 2 DBF were co-evaporated from different vapor deposition sources to form a layer having a thickness of 15 nm as a light emitting layer. At this time, the concentration of Compound 5 was 6.0% by weight.
- ITO indium tin oxide
- PPT was formed to a thickness of 10 nm, and TPBi was formed thereon to a thickness of 40 nm. Further, lithium fluoride (LiF) was vacuum-deposited at 0.8 nm, and then aluminum (Al) was evaporated at a thickness of 100 nm to form a cathode, thereby obtaining an organic electroluminescence device.
- LiF lithium fluoride
- Al aluminum
- the emission spectrum of the produced organic electroluminescence device is shown in FIG. 21, and the current density-external quantum efficiency characteristic is shown in FIG.
- the organic electroluminescence device using Compound 5 as the light emitting material achieved a high external quantum efficiency of 15.3%.
- Example 12 Production and evaluation of organic electroluminescence device using compound 6 An organic electroluminescence device was produced in the same manner as in Example 4 using compound 6 instead of compound 5. The emission spectrum of the produced organic electroluminescence element is shown in FIG. 23, and the current density-external quantum efficiency characteristic is shown in FIG. The organic electroluminescence device using Compound 5 as the light emitting material achieved a high external quantum efficiency of 8.9%.
- the compound of the present invention is useful as a luminescent material. For this reason, the compound of this invention is effectively used as a luminescent material for organic light emitting elements, such as an organic electroluminescent element. Since the compounds of the present invention include those that emit delayed fluorescence, it is also possible to provide an organic light-emitting device with high luminous efficiency. For this reason, this invention has high industrial applicability.
Abstract
A compound represented by general formula (1) is useful as a light-emitting material. R1 to R8 independently represent a hydrogen atom or a substituent, and R9 represents a substituent, wherein at least one of R1 to R8 represents a group represented by general formula (2) or the like. L12 represents a single bond or a bivalent linking group, and * represents a bonding site to a benzene ring in general formula (1). R11 to R20 independently represent a hydrogen atom or a substituent.
Description
本発明は、発光材料として有用な化合物とそれを用いた有機発光素子に関する。
The present invention relates to a compound useful as a light emitting material and an organic light emitting device using the compound.
有機エレクトロルミネッセンス素子(有機EL素子)などの有機発光素子の発光効率を高める研究が盛んに行われている。特に、有機エレクトロルミネッセンス素子を構成する電子輸送材料、正孔輸送材料、発光材料などを新たに開発して組み合わせることにより、発光効率を高める工夫が種々なされてきている。その中には、ホウ素原子と他のヘテロ原子を含むヘテラボリン環を有する三環構造にカルバゾリル基等の置換基が結合した構造を有する化合物を利用した、有機エレクトロルミネッセンス素子に関する研究も見受けられる。
Researches for increasing the light emission efficiency of organic light emitting devices such as organic electroluminescence devices (organic EL devices) are being actively conducted. In particular, various efforts have been made to increase the light emission efficiency by newly developing and combining electron transport materials, hole transport materials, light emitting materials, and the like constituting the organic electroluminescence element. Among them, research on organic electroluminescence devices using a compound having a structure in which a substituent such as a carbazolyl group is bonded to a tricyclic structure having a heteraborine ring containing a boron atom and another heteroatom can be seen.
非特許文献1には、ホウ素原子と窒素原子を含むヘテラボリン環を有する化合物について発光特性を検討した結果が記載されている。それによると、下記の構造を有する化合物のシクロヘキサン溶液または固体膜に励起光を照射したところ可視領域に発光が認められたことが記載されている(Hexnはn-ヘキシル基を表し、Tipは2,4,6-トリイソプロピルフェニル基を表す)。
しかしながら、非特許文献1には、ヘテラボリン環のヘテロ原子を、酸素原子や硫黄原子などの窒素原子以外のヘテロ原子に変えた化合物については記載されていない。

Non-Patent Document 1 describes the results of studying luminescence characteristics of a compound having a heteraboline ring containing a boron atom and a nitrogen atom. According to the document, it was described that when a cyclohexane solution or a solid film of a compound having the following structure was irradiated with excitation light, light emission was observed in the visible region (Hex n represents an n-hexyl group, and Tip represents Represents a 2,4,6-triisopropylphenyl group).
However, Non-PatentDocument 1 does not describe a compound in which a heteroatom of a heteraboline ring is changed to a heteroatom other than a nitrogen atom such as an oxygen atom or a sulfur atom.
しかしながら、非特許文献1には、ヘテラボリン環のヘテロ原子を、酸素原子や硫黄原子などの窒素原子以外のヘテロ原子に変えた化合物については記載されていない。
However, Non-Patent
また、特許文献1には、下記一般式で表されるヘテラボリン環を有する三環構造の化合物が開示されている。下記の一般式におけるR1は2以上のアリール基またはヘテロアリール基を含む基であり、R3、R4は1~5つの置換基を表し、R2~R4は水素原子、アルキル基、アルコキシ基、アミノ基、アルケニル基、アルキニル基、アリール基、ヘテロアリール基を表すものと規定されている。ここでは、下記一般式のR1が2,4,6-トリフェニルフェニル基、R2がフェニル基、R3、R4が水素原子である化合物を、有機エレクトロルミネッセンス素子を構成する一対の電極間に存在する発光層の中にホスト材料として用いた例や、電子輸送層の中に用いた例が記載されている。しかしながら、特許文献1には、この一般式で表される化合物の発光特性については記載されておらず、ヘテラボリン環のヘテロ原子を、窒素原子以外のヘテロ原子に変えた化合物についても記載されていない。
Further, Patent Document 1 discloses a compound having a tricyclic structure having a heterolabrin ring represented by the following general formula. R 1 in the following general formula is a group containing two or more aryl groups or heteroaryl groups, R 3 and R 4 represent 1 to 5 substituents, R 2 to R 4 represent a hydrogen atom, an alkyl group, It is defined to represent an alkoxy group, amino group, alkenyl group, alkynyl group, aryl group, heteroaryl group. Here, a compound in which R 1 in the following general formula is a 2,4,6-triphenylphenyl group, R 2 is a phenyl group, and R 3 and R 4 are hydrogen atoms is used as a pair of electrodes constituting an organic electroluminescence device. An example of using it as a host material in a light emitting layer existing between them and an example of using it in an electron transport layer are described. However, Patent Document 1 does not describe the light emission characteristics of the compound represented by this general formula, nor does it describe a compound in which the heteroatom of the heterolabrin ring is changed to a heteroatom other than a nitrogen atom. .
非特許文献2には、ホウ素原子と他のヘテロ原子を含むヘテラボリン環を有する化合物について発光特性を検討した結果が記載され、下記の構造を有する化合物のシクロヘキサン溶液に可視領域の発光が認められたことが記載されている。下記式におけるRは水素原子またはメチル基を表し、EはN-メチル基または硫黄原子を表し、Mesはメシチル基を表す。しかしながら、非特許文献1には、Rがカルバゾリル基などの置換アミノ基である化合物については記載されていない。

Non-Patent Document 2 describes the results of studying the emission characteristics of a compound having a heteraboline ring containing a boron atom and other heteroatoms, and light emission in the visible region was observed in a cyclohexane solution of a compound having the following structure. It is described. In the following formula, R represents a hydrogen atom or a methyl group, E represents an N-methyl group or a sulfur atom, and Mes represents a mesityl group. However, Non-Patent Document 1 does not describe a compound in which R is a substituted amino group such as a carbazolyl group.
上記のように、非特許文献1には、ホウ素原子と窒素原子を含むヘテラボリン環(アザボリン環)を有する三環構造にカルバゾリル基を導入した化合物が可視領域で発光することが開示されている。しかしながら、本発明者らが、カルバゾリル基やメチル基等で置換したカルバゾリル基を、アザボリン環を有する三環構造に導入した化合物について発光特性を実際に評価したところ、発光特性は十分に満足しうるものではないことが判明した(後掲の比較例1~3参照)。
そこで本発明者らは、ヘテラボリン環を有する化合物群について種々の検討を始め、多数の類似骨格の中からホウ素原子と酸素原子または硫黄原子とを含むヘテラボリン環を有する化合物群に発光材料として有用性があることを初めて見出し、さらに検討を進めることにした。上記のように、ホウ素原子と硫黄原子を含むヘテラボリン環を有する三環構造の化合物については、非特許文献2に可視領域で発光が認められたことが記載されている。しかしながら、非特許文献2には、ヘテラボリン環を有する三環構造にカルバゾール基等の置換アミノ基が結合した化合物については記載されていない。このため、ホウ素原子と酸素原子または硫黄原子を含むヘテラボリン環を有する三環構造にカルバゾリル基等の置換アミノ基が結合した化合物の発光材料としての有用性は予測がつかない。 As described above, Non-PatentDocument 1 discloses that a compound in which a carbazolyl group is introduced into a tricyclic structure having a heteraboline ring (azaborin ring) containing a boron atom and a nitrogen atom emits light in the visible region. However, when the inventors actually evaluated the light emission characteristics of a compound in which a carbazolyl group substituted with a carbazolyl group, a methyl group, or the like was introduced into a tricyclic structure having an azaborin ring, the light emission characteristics can be sufficiently satisfied. It was proved that it was not (see Comparative Examples 1 to 3 below).
Therefore, the present inventors have started various studies on a compound group having a heteraboline ring, and are useful as a luminescent material for a compound group having a heteraboline ring containing a boron atom and an oxygen atom or a sulfur atom from many similar skeletons. I found that there was for the first time and decided to proceed with further study. As described above, it is described inNon-Patent Document 2 that light emission was observed in the visible region of a compound having a tricyclic structure having a heteraboline ring containing a boron atom and a sulfur atom. However, Non-Patent Document 2 does not describe a compound in which a substituted amino group such as a carbazole group is bonded to a tricyclic structure having a heterolabrin ring. For this reason, the usefulness as a light-emitting material of a compound in which a substituted amino group such as a carbazolyl group is bonded to a tricyclic structure having a heteraboline ring containing a boron atom and an oxygen atom or a sulfur atom cannot be predicted.
そこで本発明者らは、ヘテラボリン環を有する化合物群について種々の検討を始め、多数の類似骨格の中からホウ素原子と酸素原子または硫黄原子とを含むヘテラボリン環を有する化合物群に発光材料として有用性があることを初めて見出し、さらに検討を進めることにした。上記のように、ホウ素原子と硫黄原子を含むヘテラボリン環を有する三環構造の化合物については、非特許文献2に可視領域で発光が認められたことが記載されている。しかしながら、非特許文献2には、ヘテラボリン環を有する三環構造にカルバゾール基等の置換アミノ基が結合した化合物については記載されていない。このため、ホウ素原子と酸素原子または硫黄原子を含むヘテラボリン環を有する三環構造にカルバゾリル基等の置換アミノ基が結合した化合物の発光材料としての有用性は予測がつかない。 As described above, Non-Patent
Therefore, the present inventors have started various studies on a compound group having a heteraboline ring, and are useful as a luminescent material for a compound group having a heteraboline ring containing a boron atom and an oxygen atom or a sulfur atom from many similar skeletons. I found that there was for the first time and decided to proceed with further study. As described above, it is described in
このような状況下において本発明者らは、ホウ素原子と酸素原子または硫黄原子を含むヘテラボリン環を有する化合物の発光材料としての有用性についてさらに検討を進め、発光特性が優れた化合物を見出すことを目指して研究を重ねた。そして、発光材料として有用な化合物の一般式を導きだし、発光効率が高い有機発光素子の構成を一般化することを目的として鋭意検討を進めた。
Under such circumstances, the present inventors have further investigated the usefulness of a compound having a heteraboline ring containing a boron atom and an oxygen atom or sulfur atom as a luminescent material, and found a compound having excellent luminescent properties. Aimed at research. And the general formula of the compound useful as a luminescent material was derived, and the earnest examination was advanced for the purpose of generalizing the structure of the organic light emitting element with high luminous efficiency.
鋭意検討を進めた結果、本発明者らは、ホウ素原子と酸素原子または硫黄原子を含むヘテラボリン環を有する化合物のうち特定の構造を有するものが発光材料として優れた性質を有することを見出した。また、そのような化合物群の中に、遅延蛍光材料として有用なものがあることを見出し、発光効率が高い有機発光素子を安価に提供しうることを明らかにした。本発明者らは、これらの知見に基づいて、上記の課題を解決する手段として、以下の本発明を提供するに至った。
As a result of intensive investigations, the present inventors have found that compounds having a specific structure among the compounds having a heteraboline ring containing a boron atom and an oxygen atom or a sulfur atom have excellent properties as a luminescent material. In addition, it has been found that such a group of compounds is useful as a delayed fluorescent material, and it has been clarified that an organic light-emitting device having high emission efficiency can be provided at low cost. Based on these findings, the present inventors have provided the following present invention as means for solving the above problems.
[1] 下記一般式(1)で表される化合物からなる発光材料。

[一般式(1)において、Xは酸素原子または硫黄原子を表す。R1~R8は各々独立に水素原子または置換基を表す。ただし、R1~R8の少なくとも1つは、各々独立に下記一般式(2)~(7)のいずれかで表される基である。R1とR2、R2とR3、R3とR4、R5とR6、R6とR7、R7とR8、R8とR9、R9とR1は互いに結合して環状構造を形成していてもよい。R9は置換基を表す。R9がホウ素原子と単結合を形成していない孤立電子対を有する原子を含むとき、該原子はホウ素原子と配位結合して環状構造を形成していてもよい。]
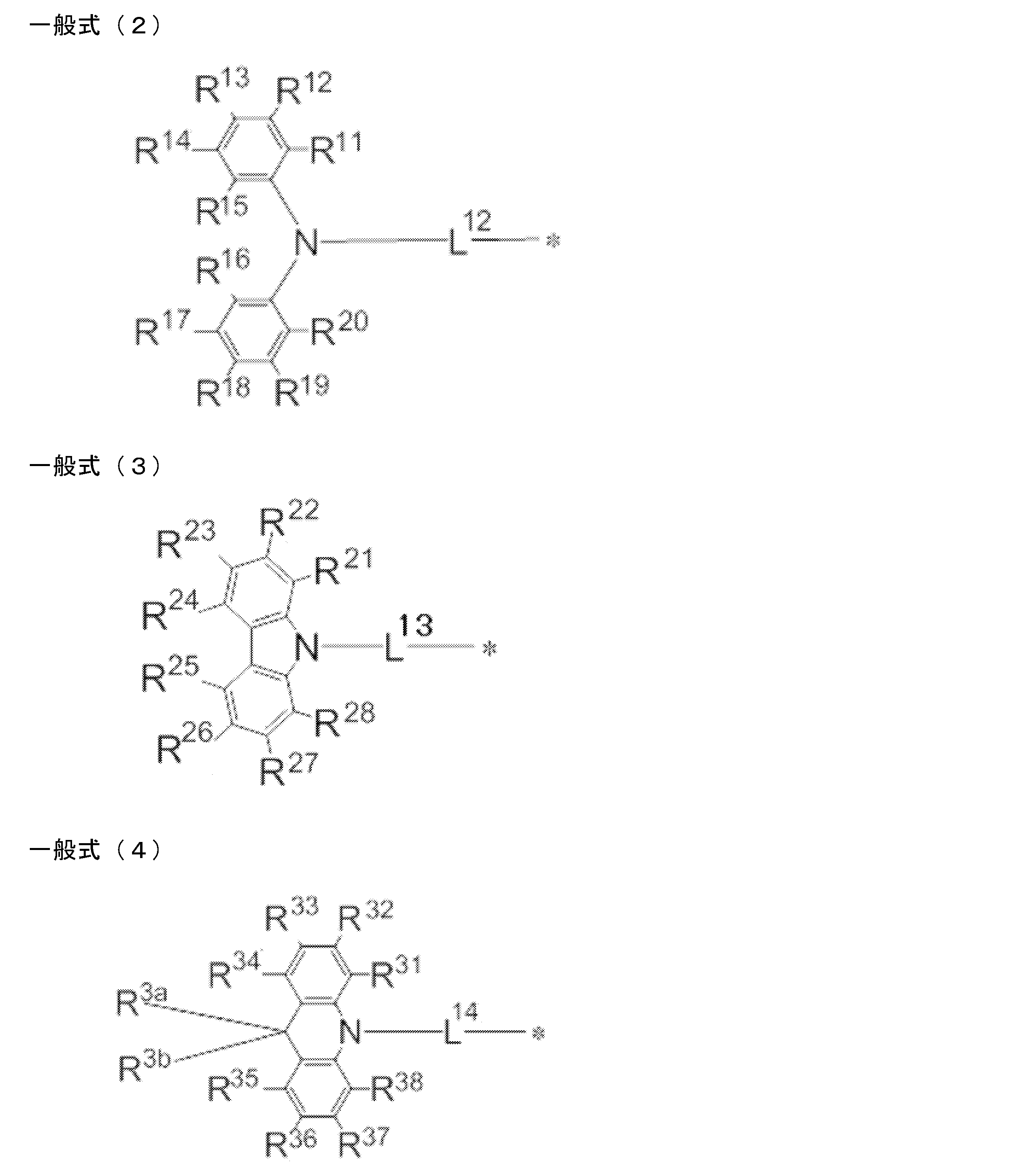

[一般式(2)~(7)において、L12~L17は各々独立に単結合または二価の連結基を表し、*は一般式(1)におけるベンゼン環への結合部位を表す。R11~R20、R21~R28、R31~R38、R3a、R3b、R41~R48、R4a、R51~R58、R61~R68は、各々独立に水素原子または置換基を表す。R11とR12、R12とR13、R13とR14、R14とR15、R16とR17、R17とR18、R18とR19、R19とR20、R21とR22、R22とR23、R23とR24、R24とR25、R25とR26、R26とR27、R27とR28、R31とR32、R32とR33、R33とR34、R35とR36、R36とR37、R37とR38、R3aとR3b、R41とR42、R42とR43、R43とR44、R45とR46、R46とR47、R47とR48、R51とR52、R52とR53、R53とR54、R55とR56、R56とR57、R57とR58、R61とR62、R62とR63、R63とR64、R65とR66、R66とR67、R67とR68はそれぞれ互いに結合して環状構造を形成していてもよい。]
[2] 一般式(1)のR1~R8の少なくとも1つが前記一般式(3)~(7)のいずれかで表される基であることを特徴とする[1]に記載の発光材料。
[3] 一般式(1)のR1~R8の少なくとも1つが前記一般式(3)で表される基である場合に、前記一般式(3)のR21~R28のうち少なくとも1つは置換基であることを特徴とする[1]または[2]に記載の発光材料。
[4] 一般式(1)のR2、R3、R6、およびR7の少なくとも1つが前記一般式(2)~(7)のいずれかで表される基であることを特徴とする[1]~[3]のいずれか1項に記載の発光材料。
[5] 一般式(1)のR3およびR6の少なくとも1つが前記一般式(2)~(7)のいずれかで表される基であることを特徴とする[4]に記載の発光材料。
[6] 一般式(1)のR3とR6が、各々独立に前記一般式(2)~(7)のいずれかで表される基であることを特徴とする[5]に記載の発光材料。
[7] 前記一般式(2)のR11~R20の少なくとも1つ、前記一般式(3)のR21~R28の少なくとも1つ、前記一般式(4)のR31~R38の少なくとも1つと、R3aおよびR3bの少なくとも1つ、前記一般式(5)のR41~R48の少なくとも1つ、前記一般式(6)のR51~R58の少なくとも1つ、および前記一般式(7)のR61~R68の少なくとも1つが置換基であることを特徴とする[1]~[6]のいずれか1項に記載の発光材料。
[8] 前記一般式(2)のR13およびR18の少なくとも一方、前記一般式(3)のR23およびR26の少なくとも一方、前記一般式(4)のR33およびR36の少なくとも一方と、R3aおよびR3bの少なくとも一方、前記一般式(5)のR43およびR46の少なくとも一方、前記一般式(6)のR53およびR56の少なくとも一方、並びに前記一般式(7)のR63およびR66の少なくとも一方が置換基であることを特徴とする[7]に記載の発光材料。
[9] 前記一般式(2)のR13およびR18の少なくとも一方、前記一般式(3)のR23およびR26の少なくとも一方、前記一般式(4)のR33およびR36の少なくとも一方と、R3aおよびR3bの少なくとも一方、前記一般式(5)のR43およびR46の少なくとも一方、前記一般式(6)のR53およびR56の少なくとも一方、並びに前記一般式(7)のR63およびR66の少なくとも一方が、前記一般式(2)~(7)のいずれかで表される基であることを特徴とする[8]に記載の発光材料。
[10] 前記一般式(2)~(7)のL12~L17が、単結合であることを特徴とする[1]~[9]のいずれか1項に記載の発光材料。
[11] 一般式(1)のXが、酸素原子であることを特徴とする[1]~[10]のいずれか1項に記載の発光材料。
[12] 一般式(1)のR9が、下記一般式(a)で表される基であることを特徴とする[1]~[11]のいずれか1項に記載の発光材料。

[式(a)において、*は前記一般式(1)におけるホウ素原子への結合部位を表す。R9a、R9b、R9c、R9d、R9eは、各々独立に水素原子または置換基を表す。R9aとR9b、R9bとR9c、R9cとR9d、R9dとR9eはそれぞれ互いに結合して環状構造を形成していてもよい。]
[13] 前記一般式(a)のR9aとR9eが置換基であることを特徴とする[12]に記載の発光材料。
[14] 一般式(1)のR1~R8の少なくとも1つが前記一般式(4)で表される基であることを特徴とする[1]~[13]のいずれか1項に記載の発光材料。
[15] 一般式(1)のR3とR6、またはR2とR7が、前記一般式(4)で表される基であることを特徴とする[1]~[4]、[7]~[14]のいずれか1項に記載の発光材料。
[16] 前記一般式(4)のR3aとR3bが、置換基であることを特徴とする[14]または[15]に記載の発光材料。
[17] 前記置換基が、炭素数1~15のアルキル基またはフェニル基であることを特徴とする[14]~[16]のいずれか1項に記載の発光材料。
[18] 前記一般式(4)のR3aとR3bが互いに結合して環状構造を形成していることを特徴とする[14]~[16]のいずれか1項に記載の発光材料。
[19] 前記一般式(1)で表される化合物からなる遅延蛍光体。
[20] [1]~[18]のいずれか1項に記載の発光材料を含むことを特徴とする有機発光素子。
[21] 遅延蛍光を放射することを特徴とする[20]に記載の有機発光素子。
[22] 有機エレクトロルミネッセンス素子であることを特徴とする[20]または[21]に記載の有機発光素子。
[23] 下記一般式(1’)で表される化合物。

[一般式(1’)において、X’は酸素原子または硫黄原子を表す。R1’~R8’は各々独立に水素原子または置換基を表す。ただし、R1’~R8’の少なくとも1つは、各々独立に下記一般式(2’)~(7’)のいずれかで表される基である。R1’とR2’、R2’とR3’、R3’とR4’、R5’とR6’、R6’とR7’、R7’とR8’、R8’とR9’、R9’とR1’は互いに結合して環状構造を形成していてもよい。R9’は置換基を表す。R9’がホウ素原子と単結合を形成していない孤立電子対を有する原子を含むとき、該原子はホウ素原子と配位結合して環状構造を形成していてもよい。]
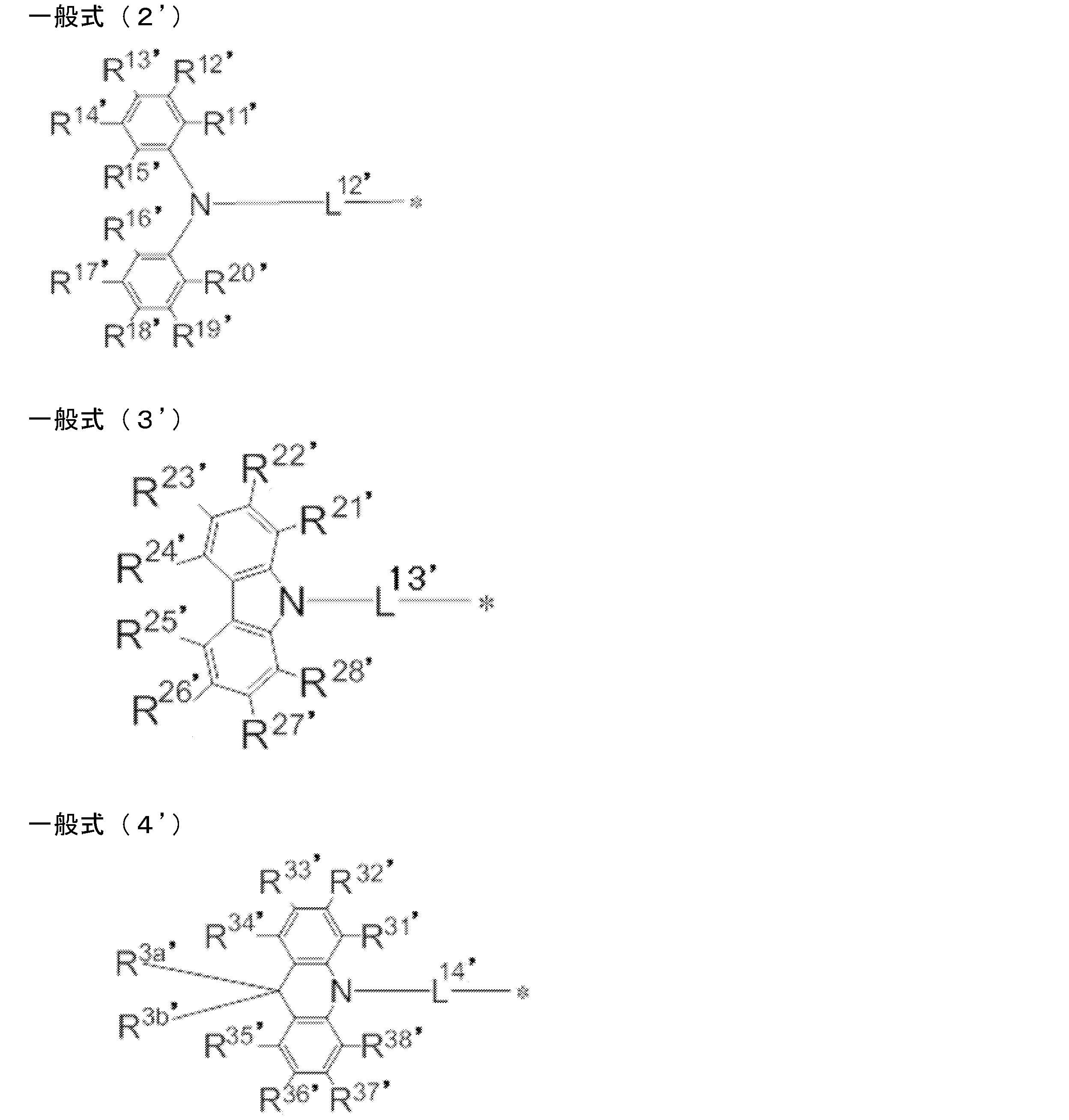
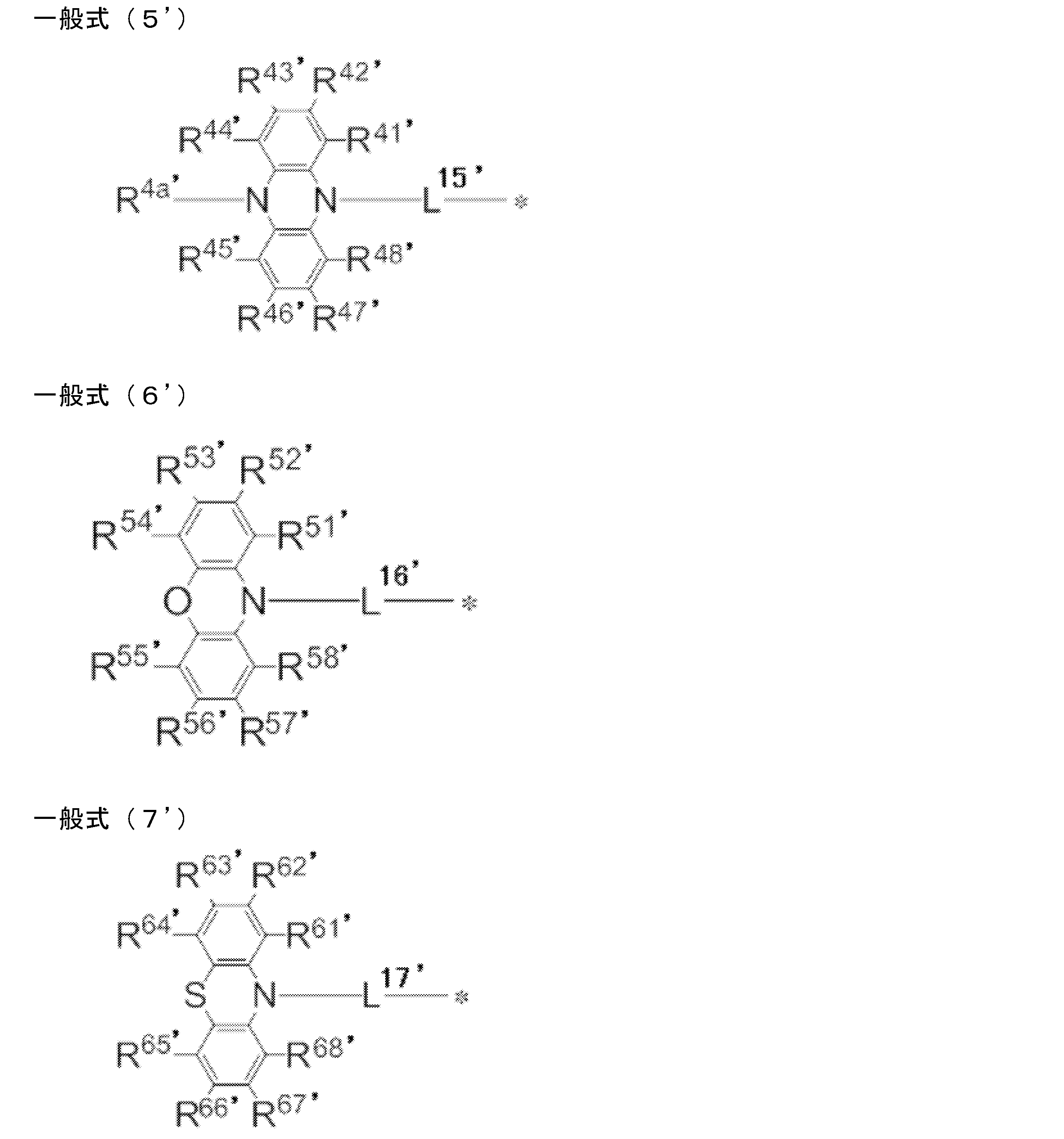
[一般式(2’)~(7’)において、L12’~L17’は各々独立に単結合または二価の連結基を表し、*は一般式(1’)におけるベンゼン環への結合部位を表す。R11’~R20’、R21’~R28’、R31’~R38’、R3a’、R3b’、R41’~R48’、R4a’、R51’~R58’、R61’~R68’は、各々独立に水素原子または置換基を表す。R11’とR12’、R12’とR13’、R13’とR14’、R14’とR15’、R16’とR17’、R17’とR18’、R18’とR19’、R19’とR20’、R21’とR22’、R22’とR23’、R23’とR24’、R24’とR25’、R25’とR26’、R26’とR27’、R27’とR28’、R31’とR32’、R32’とR33’、R33’とR34’、R35’とR36’、R36’とR37’、R37’とR38’、R3a’とR3b’、R41’とR42’、R42’とR43’、R43’とR44’、R45’とR46’、R46’とR47’、R47’とR48’、R51’とR52’、R52’とR53’、R53’とR54’、R55’とR56’、R56’とR57’、R57’とR58’、R61’とR62’、R62’とR63’、R63’とR64’、R65’とR66’、R66’とR67’、R67’とR68’はそれぞれ互いに結合して環状構造を形成していてもよい。]
[1] A light emitting material comprising a compound represented by the following general formula (1).
[In General Formula (1), X represents an oxygen atom or a sulfur atom. R 1 ~ R 8 each independently represent a hydrogen atom or a substituent. However, at least one of R 1 to R 8 is each independently a group represented by any one of the following general formulas (2) to (7). R 1 and R 2 , R 2 and R 3 , R 3 and R 4 , R 5 and R 6 , R 6 and R 7 , R 7 and R 8 , R 8 and R 9 , R 9 and R 1 are bonded to each other Thus, a ring structure may be formed. R 9 represents a substituent. When R 9 contains an atom having a lone pair of electrons that does not form a single bond with a boron atom, the atom may be coordinated with the boron atom to form a cyclic structure. ]
[In the general formulas (2) to (7), L 12 to L 17 each independently represents a single bond or a divalent linking group, and * represents a bonding site to the benzene ring in the general formula (1). R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a , R 3b , R 41 to R 48 , R 4a , R 51 to R 58 , R 61 to R 68 are each independently hydrogen Represents an atom or substituent. R 11 and R 12 , R 12 and R 13 , R 13 and R 14 , R 14 and R 15 , R 16 and R 17 , R 17 and R 18 , R 18 and R 19 , R 19 and R 20 , R 21 And R 22 , R 22 and R 23 , R 23 and R 24 , R 24 and R 25 , R 25 and R 26 , R 26 and R 27 , R 27 and R 28 , R 31 and R 32 , R 32 and R 33, R 33 and R 34, R 35 and R 36, R 36 and R 37, R 37 and R 38, R 3a and R 3b, R 41 and R 42, R 42 and R 43, R 43 and R 44, R45 and R46 , R46 and R47 , R47 and R48 , R51 and R52 , R52 and R53 , R53 and R54 , R55 and R56 , R56 and R57 , R57 And R 58 , R 61 and R 62 , R 62 and R 63 , R 63 and R 64 , R 65 and R 66 , R 66 and R 67 , R 67 and R 68 are bonded to each other to form a cyclic structure. It may be. ]
[2] The luminescence according to [1], wherein at least one of R 1 to R 8 in the general formula (1) is a group represented by any one of the general formulas (3) to (7) material.
[3] When at least one of R 1 to R 8 in the general formula (1) is a group represented by the general formula (3), at least one of R 21 to R 28 in the general formula (3) One is a substituent, The luminescent material as described in [1] or [2] characterized by the above-mentioned.
[4] It is characterized in that at least one of R 2 , R 3 , R 6 and R 7 in the general formula (1) is a group represented by any one of the general formulas (2) to (7). The light emitting material according to any one of [1] to [3].
[5] The luminescence according to [4], wherein at least one of R 3 and R 6 in the general formula (1) is a group represented by any one of the general formulas (2) to (7) material.
[6] R 3 and R 6 in formula (1) is, according to each, characterized in that a group represented by any one of formulas independently (2) to (7) [5] Luminescent material.
[7] At least one of R 11 to R 20 in the general formula (2), at least one of R 21 to R 28 in the general formula (3), and R 31 to R 38 in the general formula (4) At least one, at least one of R 3a and R 3b , at least one of R 41 to R 48 of the general formula (5), at least one of R 51 to R 58 of the general formula (6), and The light emitting material according to any one of [1] to [6], wherein at least one of R 61 to R 68 in the general formula (7) is a substituent.
[8] At least one of R 13 and R 18 in the general formula (2), at least one of R 23 and R 26 in the general formula (3), and at least one of R 33 and R 36 in the general formula (4) And at least one of R 3a and R 3b , at least one of R 43 and R 46 in the general formula (5), at least one of R 53 and R 56 in the general formula (6), and the general formula (7) The luminescent material according to [7], wherein at least one of R 63 and R 66 is a substituent.
[9] At least one of R 13 and R 18 in the general formula (2), at least one of R 23 and R 26 in the general formula (3), and at least one of R 33 and R 36 in the general formula (4) And at least one of R 3a and R 3b , at least one of R 43 and R 46 in the general formula (5), at least one of R 53 and R 56 in the general formula (6), and the general formula (7) The light emitting material according to [8], wherein at least one of R 63 and R 66 is a group represented by any one of the general formulas (2) to (7).
[10] The light emitting material according to any one of [1] to [9], wherein L 12 to L 17 in the general formulas (2) to (7) are single bonds.
[11] The light emitting material according to any one of [1] to [10], wherein X in the general formula (1) is an oxygen atom.
[12] The light-emitting material according to any one of [1] to [11], wherein R 9 in the general formula (1) is a group represented by the following general formula (a).
[In formula (a), * represents the bonding site to the boron atom in the general formula (1). R 9a , R 9b , R 9c , R 9d and R 9e each independently represent a hydrogen atom or a substituent. R 9a and R 9b , R 9b and R 9c , R 9c and R 9d , and R 9d and R 9e may be bonded to each other to form a cyclic structure. ]
[13] The luminescent material according to [12], wherein R 9a and R 9e in the general formula (a) are substituents.
[14] Any one of [1] to [13], wherein at least one of R 1 to R 8 in the general formula (1) is a group represented by the general formula (4). Luminescent material.
[15] R 3 and R 6 in formula (1) or R 2 and R 7, is characterized in that it is a group represented by the general formula (4) [1] to [4], [ [7] The luminescent material according to any one of [14] to [14].
[16] The light-emitting material according to [14] or [15], wherein R 3a and R 3b in the general formula (4) are substituents.
[17] The light-emitting material according to any one of [14] to [16], wherein the substituent is an alkyl group having 1 to 15 carbon atoms or a phenyl group.
[18] The luminescent material according to any one of [14] to [16], wherein R 3a and R 3b in the general formula (4) are bonded to each other to form a cyclic structure.
[19] A delayed phosphor comprising the compound represented by the general formula (1).
[20] An organic light-emitting device comprising the light-emitting material according to any one of [1] to [18].
[21] The organic light-emitting device according to [20], which emits delayed fluorescence.
[22] The organic light-emitting device according to [20] or [21], which is an organic electroluminescence device.
[23] A compound represented by the following general formula (1 ′).
[In General Formula (1 ′), X ′ represents an oxygen atom or a sulfur atom. R 1 ′ to R 8 ′ each independently represents a hydrogen atom or a substituent. However, at least one of R 1 ′ to R 8 ′ is a group independently represented by any one of the following general formulas (2 ′) to (7 ′). R 1 'and R 2', R 2 'and R 3', R 3 'and R 4', R 5 'and R 6', R 6 'and R 7', R 7 'and R 8', R 8 'And R 9 ' and R 9 'and R 1 ' may be bonded to each other to form a cyclic structure. R 9 ′ represents a substituent. When R 9 ′ includes an atom having a lone pair of electrons that does not form a single bond with a boron atom, the atom may be coordinated with the boron atom to form a cyclic structure. ]
[In the general formulas (2 ′) to (7 ′), L 12 ′ to L 17 ′ each independently represents a single bond or a divalent linking group, and * represents a bond to the benzene ring in the general formula (1 ′). Represents a site. R 11 '~ R 20', R 21 '~ R 28', R 31 '~ R 38', R 3a ', R 3b', R 41 '~ R 48', R 4a ', R 51' ~ R 58 ', R 61 ' to R 68 'each independently represents a hydrogen atom or a substituent. R 11 'and R 12', R 12 'and R 13', R 13 'and R 14', R 14 'and R 15', 'R 17 and' R 16, R 17 'and R 18', R 18 'And R 19 ', R 19 'and R 20 ', R 21 'and R 22 ', R 22 'and R 23 ', R 23 'and R 24 ', R 24 'and R 25 ', R 25 'and R 26 ', R 26' and R 27 ', R 27' and R 28 ', R 31' and R 32 ', R 32' and R 33 ', R 33' and R 34 ', R 35' and R 36 ', R 36 ' and R 37 ', R 37 ' and R 38 ', R 3a ' and R 3b ', R 41 ' and R 42 ', R 42 ' and R 43 ', R 43 ' and R 44 ', R45 'and R46 ', R46 'and R47 ', R47 'and R48 ', R51 'and R52 ', R52 'and R53 ', R53 'and R54 ', R55 'And R 56 ', R 56 'and R 57 ', R 57 'and R 58 ', R 61 'and R 62 ', R 62 'and R 63 ', R 63 'and R 64 ', R 65 'and R 66 ′, R 66 ′ and R 67 ′, R 67 ′ and R 68 ′ may be bonded to each other to form a cyclic structure. ]
[2] 一般式(1)のR1~R8の少なくとも1つが前記一般式(3)~(7)のいずれかで表される基であることを特徴とする[1]に記載の発光材料。
[3] 一般式(1)のR1~R8の少なくとも1つが前記一般式(3)で表される基である場合に、前記一般式(3)のR21~R28のうち少なくとも1つは置換基であることを特徴とする[1]または[2]に記載の発光材料。
[4] 一般式(1)のR2、R3、R6、およびR7の少なくとも1つが前記一般式(2)~(7)のいずれかで表される基であることを特徴とする[1]~[3]のいずれか1項に記載の発光材料。
[5] 一般式(1)のR3およびR6の少なくとも1つが前記一般式(2)~(7)のいずれかで表される基であることを特徴とする[4]に記載の発光材料。
[6] 一般式(1)のR3とR6が、各々独立に前記一般式(2)~(7)のいずれかで表される基であることを特徴とする[5]に記載の発光材料。
[7] 前記一般式(2)のR11~R20の少なくとも1つ、前記一般式(3)のR21~R28の少なくとも1つ、前記一般式(4)のR31~R38の少なくとも1つと、R3aおよびR3bの少なくとも1つ、前記一般式(5)のR41~R48の少なくとも1つ、前記一般式(6)のR51~R58の少なくとも1つ、および前記一般式(7)のR61~R68の少なくとも1つが置換基であることを特徴とする[1]~[6]のいずれか1項に記載の発光材料。
[8] 前記一般式(2)のR13およびR18の少なくとも一方、前記一般式(3)のR23およびR26の少なくとも一方、前記一般式(4)のR33およびR36の少なくとも一方と、R3aおよびR3bの少なくとも一方、前記一般式(5)のR43およびR46の少なくとも一方、前記一般式(6)のR53およびR56の少なくとも一方、並びに前記一般式(7)のR63およびR66の少なくとも一方が置換基であることを特徴とする[7]に記載の発光材料。
[9] 前記一般式(2)のR13およびR18の少なくとも一方、前記一般式(3)のR23およびR26の少なくとも一方、前記一般式(4)のR33およびR36の少なくとも一方と、R3aおよびR3bの少なくとも一方、前記一般式(5)のR43およびR46の少なくとも一方、前記一般式(6)のR53およびR56の少なくとも一方、並びに前記一般式(7)のR63およびR66の少なくとも一方が、前記一般式(2)~(7)のいずれかで表される基であることを特徴とする[8]に記載の発光材料。
[10] 前記一般式(2)~(7)のL12~L17が、単結合であることを特徴とする[1]~[9]のいずれか1項に記載の発光材料。
[11] 一般式(1)のXが、酸素原子であることを特徴とする[1]~[10]のいずれか1項に記載の発光材料。
[12] 一般式(1)のR9が、下記一般式(a)で表される基であることを特徴とする[1]~[11]のいずれか1項に記載の発光材料。
[13] 前記一般式(a)のR9aとR9eが置換基であることを特徴とする[12]に記載の発光材料。
[14] 一般式(1)のR1~R8の少なくとも1つが前記一般式(4)で表される基であることを特徴とする[1]~[13]のいずれか1項に記載の発光材料。
[15] 一般式(1)のR3とR6、またはR2とR7が、前記一般式(4)で表される基であることを特徴とする[1]~[4]、[7]~[14]のいずれか1項に記載の発光材料。
[16] 前記一般式(4)のR3aとR3bが、置換基であることを特徴とする[14]または[15]に記載の発光材料。
[17] 前記置換基が、炭素数1~15のアルキル基またはフェニル基であることを特徴とする[14]~[16]のいずれか1項に記載の発光材料。
[18] 前記一般式(4)のR3aとR3bが互いに結合して環状構造を形成していることを特徴とする[14]~[16]のいずれか1項に記載の発光材料。
[19] 前記一般式(1)で表される化合物からなる遅延蛍光体。
[20] [1]~[18]のいずれか1項に記載の発光材料を含むことを特徴とする有機発光素子。
[21] 遅延蛍光を放射することを特徴とする[20]に記載の有機発光素子。
[22] 有機エレクトロルミネッセンス素子であることを特徴とする[20]または[21]に記載の有機発光素子。
[23] 下記一般式(1’)で表される化合物。
[2] The luminescence according to [1], wherein at least one of R 1 to R 8 in the general formula (1) is a group represented by any one of the general formulas (3) to (7) material.
[3] When at least one of R 1 to R 8 in the general formula (1) is a group represented by the general formula (3), at least one of R 21 to R 28 in the general formula (3) One is a substituent, The luminescent material as described in [1] or [2] characterized by the above-mentioned.
[4] It is characterized in that at least one of R 2 , R 3 , R 6 and R 7 in the general formula (1) is a group represented by any one of the general formulas (2) to (7). The light emitting material according to any one of [1] to [3].
[5] The luminescence according to [4], wherein at least one of R 3 and R 6 in the general formula (1) is a group represented by any one of the general formulas (2) to (7) material.
[6] R 3 and R 6 in formula (1) is, according to each, characterized in that a group represented by any one of formulas independently (2) to (7) [5] Luminescent material.
[7] At least one of R 11 to R 20 in the general formula (2), at least one of R 21 to R 28 in the general formula (3), and R 31 to R 38 in the general formula (4) At least one, at least one of R 3a and R 3b , at least one of R 41 to R 48 of the general formula (5), at least one of R 51 to R 58 of the general formula (6), and The light emitting material according to any one of [1] to [6], wherein at least one of R 61 to R 68 in the general formula (7) is a substituent.
[8] At least one of R 13 and R 18 in the general formula (2), at least one of R 23 and R 26 in the general formula (3), and at least one of R 33 and R 36 in the general formula (4) And at least one of R 3a and R 3b , at least one of R 43 and R 46 in the general formula (5), at least one of R 53 and R 56 in the general formula (6), and the general formula (7) The luminescent material according to [7], wherein at least one of R 63 and R 66 is a substituent.
[9] At least one of R 13 and R 18 in the general formula (2), at least one of R 23 and R 26 in the general formula (3), and at least one of R 33 and R 36 in the general formula (4) And at least one of R 3a and R 3b , at least one of R 43 and R 46 in the general formula (5), at least one of R 53 and R 56 in the general formula (6), and the general formula (7) The light emitting material according to [8], wherein at least one of R 63 and R 66 is a group represented by any one of the general formulas (2) to (7).
[10] The light emitting material according to any one of [1] to [9], wherein L 12 to L 17 in the general formulas (2) to (7) are single bonds.
[11] The light emitting material according to any one of [1] to [10], wherein X in the general formula (1) is an oxygen atom.
[12] The light-emitting material according to any one of [1] to [11], wherein R 9 in the general formula (1) is a group represented by the following general formula (a).
[13] The luminescent material according to [12], wherein R 9a and R 9e in the general formula (a) are substituents.
[14] Any one of [1] to [13], wherein at least one of R 1 to R 8 in the general formula (1) is a group represented by the general formula (4). Luminescent material.
[15] R 3 and R 6 in formula (1) or R 2 and R 7, is characterized in that it is a group represented by the general formula (4) [1] to [4], [ [7] The luminescent material according to any one of [14] to [14].
[16] The light-emitting material according to [14] or [15], wherein R 3a and R 3b in the general formula (4) are substituents.
[17] The light-emitting material according to any one of [14] to [16], wherein the substituent is an alkyl group having 1 to 15 carbon atoms or a phenyl group.
[18] The luminescent material according to any one of [14] to [16], wherein R 3a and R 3b in the general formula (4) are bonded to each other to form a cyclic structure.
[19] A delayed phosphor comprising the compound represented by the general formula (1).
[20] An organic light-emitting device comprising the light-emitting material according to any one of [1] to [18].
[21] The organic light-emitting device according to [20], which emits delayed fluorescence.
[22] The organic light-emitting device according to [20] or [21], which is an organic electroluminescence device.
[23] A compound represented by the following general formula (1 ′).
本発明の化合物は、発光材料として有用である。また、本発明の化合物の中には遅延蛍光を放射するものが含まれている。本発明の化合物を発光材料として用いた有機発光素子は、高い発光効率を実現しうる。
The compound of the present invention is useful as a light emitting material. The compounds of the present invention include those that emit delayed fluorescence. An organic light emitting device using the compound of the present invention as a light emitting material can realize high luminous efficiency.
以下において、本発明の内容について詳細に説明する。以下に記載する構成要件の説明は、本発明の代表的な実施態様や具体例に基づいてなされることがあるが、本発明はそのような実施態様や具体例に限定されるものではない。なお、本明細書において「~」を用いて表される数値範囲は、「~」の前後に記載される数値を下限値および上限値として含む範囲を意味する。また、本発明に用いられる化合物の分子内に存在する水素原子の同位体種は特に限定されず、例えば分子内の水素原子がすべて1Hであってもよいし、一部または全部が2H(デューテリウムD)であってもよい。
Hereinafter, the contents of the present invention will be described in detail. The description of the constituent elements described below may be made based on typical embodiments and specific examples of the present invention, but the present invention is not limited to such embodiments and specific examples. In the present specification, a numerical range represented by using “to” means a range including numerical values described before and after “to” as a lower limit value and an upper limit value. In addition, the isotope species of the hydrogen atom present in the molecule of the compound used in the present invention is not particularly limited. For example, all the hydrogen atoms in the molecule may be 1 H, or a part or all of them are 2 H. (Deuterium D) may be used.
[一般式(1)で表される化合物]
本発明の発光材料は、下記一般式(1)で表される化合物からなることを特徴とする。

[Compound represented by general formula (1)]
The luminescent material of the present invention is characterized by comprising a compound represented by the following general formula (1).
本発明の発光材料は、下記一般式(1)で表される化合物からなることを特徴とする。
The luminescent material of the present invention is characterized by comprising a compound represented by the following general formula (1).
一般式(1)において、Xは酸素原子または硫黄原子を表し、酸素原子であることが好ましい。
一般式(1)において、R1~R8は各々独立に水素原子または置換基を表す。ただし、R1~R8の少なくとも1つは、各々独立に下記一般式(2)~(7)のいずれかで表される基である。
下記一般式(2)~(7)のいずれかで表される基は、R1~R8のうちの1つのみであってもよいし、2つ以上であってもよい。
下記一般式(2)~(7)のいずれかで表される基がR1~R8のうちの1つのみであるときは、R2、R3、R6およびR7のいずれか1つが下記一般式(2)~(7)のいずれかで表される基であることが好ましく、R3またはR6が下記一般式(2)~(7)のいずれかで表される基であることがより好ましい。
一方、R1~R8のうちの2つ以上が下記一般式(2)~(7)のいずれかで表される基であるときは、下記一般式(2)~(7)のいずれかで表される基は、R1~R4の少なくとも1つと、R5~R8の少なくとも1つであることが好ましい。このとき、下記一般式(2)~(7)のいずれかで表される基は、R1~R4のうちの1~3つ、R5~R8のうちの1~3つであることが好ましく、R1~R4のうちの1または2つ、R5~R8のうちの1または2つであることがより好ましい。R1~R4のうち一般式(2)~(7)のいずれかで表される基の数と、R5~R8のうち一般式(2)~(7)のいずれかで表される基の数は同じであっても異なっていてもよいが、同じであることが好ましい。R1~R4のうちでは、R2またはR3の少なくとも1つが一般式(2)~(7)のいずれかで表される基であることが好ましく、少なくともR3が一般式(2)~(7)のいずれかで表される基であることがより好ましい。また、R5~R8のうちでは、R6またはR7の少なくとも1つが一般式(2)~(7)のいずれかで表される基であることが好ましく、少なくともR6が一般式(2)~(7)のいずれかで表される基であることがより好ましい。好ましい化合物は、一般式(1)のR3とR6が一般式(2)~(7)のいずれかで表される基である化合物、一般式(1)のR2とR7が一般式(2)~(7)のいずれかで表される基である化合物、一般式(1)のR2、R3、R6、R7が一般式(2)~(7)のいずれかで表される基である化合物であり、さらに好ましい化合物はR3とR6が一般式(2)~(7)のいずれかで表される基である化合物である。一般式(1)中に存在する複数の一般式(2)~(7)のいずれかで表される基は、同一であっても異なっていてもよいが、同一であることが好ましい。また、一般式(1)で表される基は対称構造をとっていることも好ましい。すなわち、R1とR8、R2とR7、R3とR6、R4とR5は、それぞれ同一であることが好ましい。 In the general formula (1), X represents an oxygen atom or a sulfur atom, and is preferably an oxygen atom.
In the general formula (1), R 1 to R 8 each independently represents a hydrogen atom or a substituent. However, at least one of R 1 to R 8 is each independently a group represented by any one of the following general formulas (2) to (7).
The group represented by any of the following general formulas (2) to (7) may be only one of R 1 to R 8 , or may be two or more.
When the group represented by any one of the following general formulas (2) to (7) is only one of R 1 to R 8, any one of R 2 , R 3 , R 6 and R 7 Is preferably a group represented by any one of the following general formulas (2) to (7), and R 3 or R 6 is a group represented by any one of the following general formulas (2) to (7). More preferably.
On the other hand, when two or more of R 1 to R 8 are groups represented by any of the following general formulas (2) to (7), any one of the following general formulas (2) to (7) Is preferably at least one of R 1 to R 4 and at least one of R 5 to R 8 . At this time, the groups represented by any of the following general formulas (2) to (7) are 1 to 3 of R 1 to R 4 and 1 to 3 of R 5 to R 8. It is preferably 1 or 2 of R 1 to R 4 , and more preferably 1 or 2 of R 5 to R 8 . R 1 to R 4 are represented by any of the general formulas (2) to (7), and R 5 to R 8 are represented by any of the general formulas (2) to (7). The number of groups may be the same or different, but is preferably the same. Among R 1 to R 4 , at least one of R 2 or R 3 is preferably a group represented by any one of the general formulas (2) to (7), and at least R 3 is a group represented by the general formula (2) A group represented by any one of (7) to (7) is more preferable. Of R 5 to R 8 , at least one of R 6 or R 7 is preferably a group represented by any one of the general formulas (2) to (7), and at least R 6 is a group represented by the general formula ( A group represented by any one of 2) to (7) is more preferable. Preferred compounds are those in which R 3 and R 6 in the general formula (1) are groups represented by any one of the general formulas (2) to (7), and R 2 and R 7 in the general formula (1) are general. A compound which is a group represented by any one of formulas (2) to (7), wherein R 2 , R 3 , R 6 and R 7 in formula (1) are any of formulas (2) to (7) A more preferred compound is a compound in which R 3 and R 6 are groups represented by any one of the general formulas (2) to (7). The groups represented by any of the plurality of general formulas (2) to (7) present in the general formula (1) may be the same or different, but are preferably the same. Moreover, it is also preferable that the group represented by the general formula (1) has a symmetrical structure. That is, R 1 and R 8 , R 2 and R 7 , R 3 and R 6 , and R 4 and R 5 are preferably the same.
一般式(1)において、R1~R8は各々独立に水素原子または置換基を表す。ただし、R1~R8の少なくとも1つは、各々独立に下記一般式(2)~(7)のいずれかで表される基である。
下記一般式(2)~(7)のいずれかで表される基は、R1~R8のうちの1つのみであってもよいし、2つ以上であってもよい。
下記一般式(2)~(7)のいずれかで表される基がR1~R8のうちの1つのみであるときは、R2、R3、R6およびR7のいずれか1つが下記一般式(2)~(7)のいずれかで表される基であることが好ましく、R3またはR6が下記一般式(2)~(7)のいずれかで表される基であることがより好ましい。
一方、R1~R8のうちの2つ以上が下記一般式(2)~(7)のいずれかで表される基であるときは、下記一般式(2)~(7)のいずれかで表される基は、R1~R4の少なくとも1つと、R5~R8の少なくとも1つであることが好ましい。このとき、下記一般式(2)~(7)のいずれかで表される基は、R1~R4のうちの1~3つ、R5~R8のうちの1~3つであることが好ましく、R1~R4のうちの1または2つ、R5~R8のうちの1または2つであることがより好ましい。R1~R4のうち一般式(2)~(7)のいずれかで表される基の数と、R5~R8のうち一般式(2)~(7)のいずれかで表される基の数は同じであっても異なっていてもよいが、同じであることが好ましい。R1~R4のうちでは、R2またはR3の少なくとも1つが一般式(2)~(7)のいずれかで表される基であることが好ましく、少なくともR3が一般式(2)~(7)のいずれかで表される基であることがより好ましい。また、R5~R8のうちでは、R6またはR7の少なくとも1つが一般式(2)~(7)のいずれかで表される基であることが好ましく、少なくともR6が一般式(2)~(7)のいずれかで表される基であることがより好ましい。好ましい化合物は、一般式(1)のR3とR6が一般式(2)~(7)のいずれかで表される基である化合物、一般式(1)のR2とR7が一般式(2)~(7)のいずれかで表される基である化合物、一般式(1)のR2、R3、R6、R7が一般式(2)~(7)のいずれかで表される基である化合物であり、さらに好ましい化合物はR3とR6が一般式(2)~(7)のいずれかで表される基である化合物である。一般式(1)中に存在する複数の一般式(2)~(7)のいずれかで表される基は、同一であっても異なっていてもよいが、同一であることが好ましい。また、一般式(1)で表される基は対称構造をとっていることも好ましい。すなわち、R1とR8、R2とR7、R3とR6、R4とR5は、それぞれ同一であることが好ましい。 In the general formula (1), X represents an oxygen atom or a sulfur atom, and is preferably an oxygen atom.
In the general formula (1), R 1 to R 8 each independently represents a hydrogen atom or a substituent. However, at least one of R 1 to R 8 is each independently a group represented by any one of the following general formulas (2) to (7).
The group represented by any of the following general formulas (2) to (7) may be only one of R 1 to R 8 , or may be two or more.
When the group represented by any one of the following general formulas (2) to (7) is only one of R 1 to R 8, any one of R 2 , R 3 , R 6 and R 7 Is preferably a group represented by any one of the following general formulas (2) to (7), and R 3 or R 6 is a group represented by any one of the following general formulas (2) to (7). More preferably.
On the other hand, when two or more of R 1 to R 8 are groups represented by any of the following general formulas (2) to (7), any one of the following general formulas (2) to (7) Is preferably at least one of R 1 to R 4 and at least one of R 5 to R 8 . At this time, the groups represented by any of the following general formulas (2) to (7) are 1 to 3 of R 1 to R 4 and 1 to 3 of R 5 to R 8. It is preferably 1 or 2 of R 1 to R 4 , and more preferably 1 or 2 of R 5 to R 8 . R 1 to R 4 are represented by any of the general formulas (2) to (7), and R 5 to R 8 are represented by any of the general formulas (2) to (7). The number of groups may be the same or different, but is preferably the same. Among R 1 to R 4 , at least one of R 2 or R 3 is preferably a group represented by any one of the general formulas (2) to (7), and at least R 3 is a group represented by the general formula (2) A group represented by any one of (7) to (7) is more preferable. Of R 5 to R 8 , at least one of R 6 or R 7 is preferably a group represented by any one of the general formulas (2) to (7), and at least R 6 is a group represented by the general formula ( A group represented by any one of 2) to (7) is more preferable. Preferred compounds are those in which R 3 and R 6 in the general formula (1) are groups represented by any one of the general formulas (2) to (7), and R 2 and R 7 in the general formula (1) are general. A compound which is a group represented by any one of formulas (2) to (7), wherein R 2 , R 3 , R 6 and R 7 in formula (1) are any of formulas (2) to (7) A more preferred compound is a compound in which R 3 and R 6 are groups represented by any one of the general formulas (2) to (7). The groups represented by any of the plurality of general formulas (2) to (7) present in the general formula (1) may be the same or different, but are preferably the same. Moreover, it is also preferable that the group represented by the general formula (1) has a symmetrical structure. That is, R 1 and R 8 , R 2 and R 7 , R 3 and R 6 , and R 4 and R 5 are preferably the same.

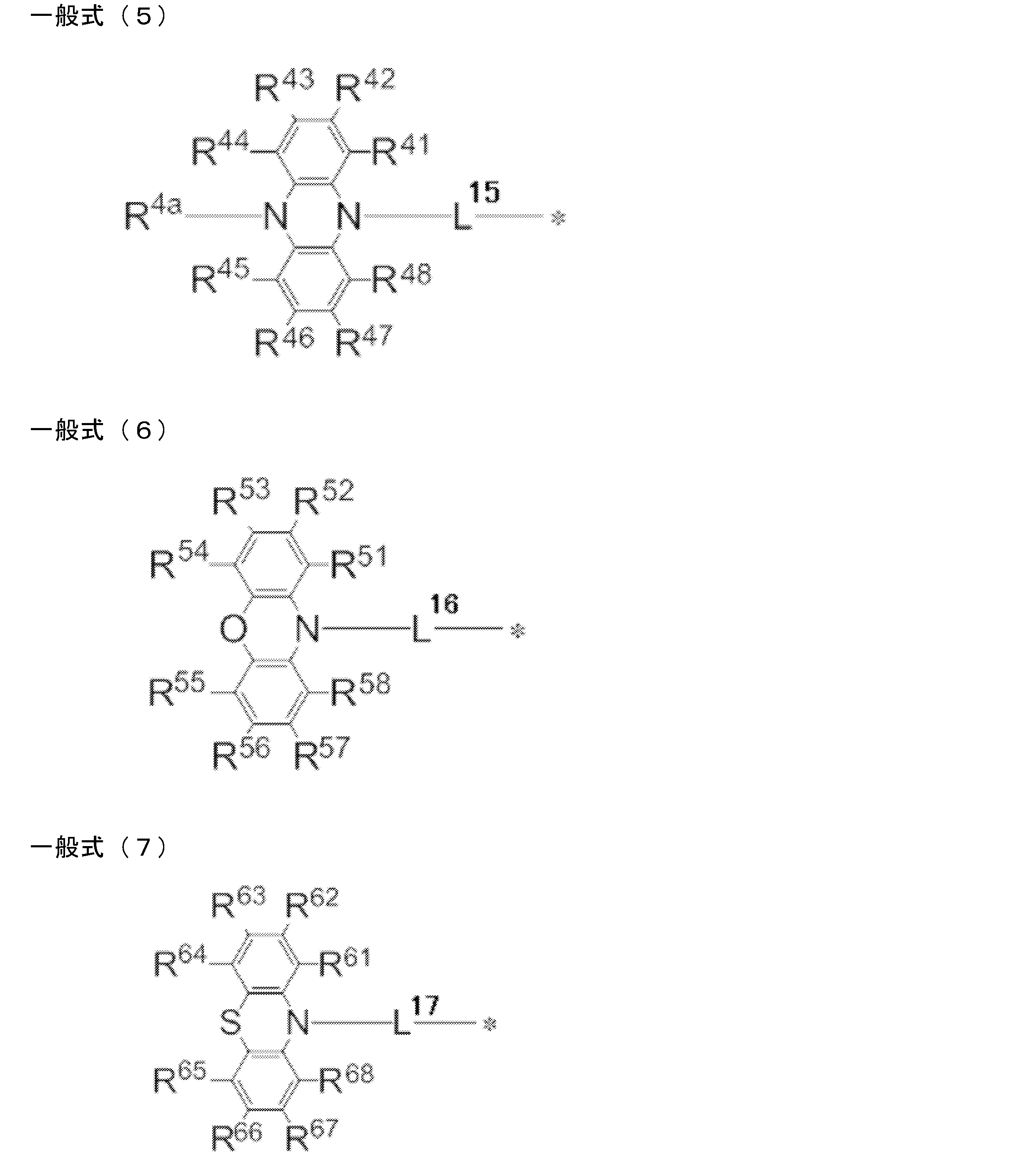
一般式(2)~(7)において、*は一般式(1)におけるベンゼン環への結合部位を表す。
L12~L17は、各々独立に単結合または二価の連結基を表し、単結合であることが好ましい。L12~L17が二価の連結基である場合、二価の連結基としては、炭素数6~18の芳香族炭化水素環基、環員数6~18の複素芳香族炭化水素環基などを挙げることができる。好ましい連結基は、フェニレン基、ビフェニレン基、フルオレニレン基、トリフェニレニレン基、チオフェニレン基であり、より好ましくい連結基はフェニレン基であり、さらに好ましい連結基は、1,4-フェニレン基である。これらの連結基は置換基が導入されていてもよい。連結基に導入しうる置換基の説明と好ましい範囲については、下記のR1~R8等がとりうる置換基の説明と好ましい範囲を参照することができる。 In the general formulas (2) to (7), * represents a bonding site to the benzene ring in the general formula (1).
L 12 to L 17 each independently represent a single bond or a divalent linking group, and preferably a single bond. When L 12 to L 17 are a divalent linking group, examples of the divalent linking group include an aromatic hydrocarbon ring group having 6 to 18 carbon atoms and a heteroaromatic hydrocarbon ring group having 6 to 18 ring members. Can be mentioned. Preferred linking groups are a phenylene group, a biphenylene group, a fluorenylene group, a triphenylenylene group, and a thiophenylene group, a more preferred linking group is a phenylene group, and a more preferred linking group is a 1,4-phenylene group. . These linking groups may have a substituent introduced therein. For the explanation and preferred range of the substituent that can be introduced into the linking group, the explanation and preferred range of the substituent that can be taken by the following R 1 to R 8 and the like can be referred to.
L12~L17は、各々独立に単結合または二価の連結基を表し、単結合であることが好ましい。L12~L17が二価の連結基である場合、二価の連結基としては、炭素数6~18の芳香族炭化水素環基、環員数6~18の複素芳香族炭化水素環基などを挙げることができる。好ましい連結基は、フェニレン基、ビフェニレン基、フルオレニレン基、トリフェニレニレン基、チオフェニレン基であり、より好ましくい連結基はフェニレン基であり、さらに好ましい連結基は、1,4-フェニレン基である。これらの連結基は置換基が導入されていてもよい。連結基に導入しうる置換基の説明と好ましい範囲については、下記のR1~R8等がとりうる置換基の説明と好ましい範囲を参照することができる。 In the general formulas (2) to (7), * represents a bonding site to the benzene ring in the general formula (1).
L 12 to L 17 each independently represent a single bond or a divalent linking group, and preferably a single bond. When L 12 to L 17 are a divalent linking group, examples of the divalent linking group include an aromatic hydrocarbon ring group having 6 to 18 carbon atoms and a heteroaromatic hydrocarbon ring group having 6 to 18 ring members. Can be mentioned. Preferred linking groups are a phenylene group, a biphenylene group, a fluorenylene group, a triphenylenylene group, and a thiophenylene group, a more preferred linking group is a phenylene group, and a more preferred linking group is a 1,4-phenylene group. . These linking groups may have a substituent introduced therein. For the explanation and preferred range of the substituent that can be introduced into the linking group, the explanation and preferred range of the substituent that can be taken by the following R 1 to R 8 and the like can be referred to.
一般式(2)~(7)において、R11~R20、R21~R28、R31~R38、R3a、R3b、R41~R48、R4a、R51~R58、R61~R68は、各々独立に水素原子または置換基を表す。置換基の数は特に制限されず、R11~R20、R21~R28、R31~R38、R3a、R3b、R41~R48、R4a、R51~R58、R61~R68のすべてが無置換(すなわち水素原子)であってもよいが、R11~R20、R21~R28、R31~R38、R3aおよびR3b、R41~R48、R51~R58、R61~R68のそれぞれの組の少なくとも1つは置換基であることが好ましく、R13およびR18、R23およびR26、R33およびR36、R3aおよびR3b、R43およびR46、R53およびR56、R63およびR66のそれぞれの組の少なくとも1つが置換基であることがより好ましい。一般式(2)~(7)のそれぞれにおいて置換基が2つ以上ある場合、それらの置換基は同一であっても異なっていてもよい。
In the general formulas (2) to (7), R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a , R 3b , R 41 to R 48 , R 4a , R 51 to R 58 , R 61 to R 68 each independently represents a hydrogen atom or a substituent. The number of substituents is not particularly limited, and R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a , R 3b , R 41 to R 48 , R 4a , R 51 to R 58 , R All of 61 to R 68 may be unsubstituted (that is, a hydrogen atom), but R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a and R 3b , R 41 to R 48 , R 51 to R 58 , and R 61 to R 68 are preferably a substituent, and R 13 and R 18 , R 23 and R 26 , R 33 and R 36 , R 3a and More preferably, at least one of the respective groups of R 3b , R 43 and R 46 , R 53 and R 56 , R 63 and R 66 is a substituent. In each of the general formulas (2) to (7), when there are two or more substituents, these substituents may be the same or different.
R11~R20、R21~R28、R31~R38、R3a、R3b、R41~R48、R4a、R51~R58、R61~R68がとりうる置換基と、R1~R8がとりうる置換基と、L12~L17に導入しうる置換基して、例えばヒドロキシ基、ハロゲン原子、シアノ基、炭素数1~20のアルキル基、炭素数1~20のアルコキシ基、炭素数1~20のアルキルチオ基、炭素数1~20のアルキル置換アミノ基、炭素数2~20のアシル基、炭素数6~40のアリール基、炭素数3~40のヘテロアリール基、炭素数2~10のアルケニル基、炭素数2~10のアルキニル基、炭素数2~10のアルコキシカルボニル基、炭素数1~10のアルキルスルホニル基、炭素数1~10のハロアルキル基、アミド基、炭素数2~10のアルキルアミド基、炭素数3~20のトリアルキルシリル基、炭素数4~20のトリアルキルシリルアルキル基、炭素数5~20のトリアルキルシリルアルケニル基、炭素数5~20のトリアルキルシリルアルキニル基およびニトロ基等が挙げられる。これらの具体例のうち、さらに置換基により置換可能なものは置換されていてもよい。より好ましい置換基は、ハロゲン原子、シアノ基、炭素数1~20の置換もしくは無置換のアルキル基、炭素数1~20のアルコキシ基、炭素数6~40の置換もしくは無置換のアリール基、炭素数3~40の置換もしくは無置換のヘテロアリール基、炭素数1~20のジアルキル置換アミノ基である。さらに好ましい置換基は、フッ素原子、塩素原子、シアノ基、炭素数1~10の置換もしくは無置換のアルキル基、炭素数1~10の置換もしくは無置換のアルコキシ基、炭素数6~15の置換もしくは無置換のアリール基、炭素数3~12の置換もしくは無置換のヘテロアリール基である。
R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a , R 3b , R 41 to R 48 , R 4a , R 51 to R 58 , R 61 to R 68 , R 1 to R 8 can be substituted with a substituent that can be introduced into L 12 to L 17 , for example, a hydroxy group, a halogen atom, a cyano group, an alkyl group having 1 to 20 carbon atoms, 20 alkoxy groups, alkylthio groups having 1 to 20 carbon atoms, alkyl-substituted amino groups having 1 to 20 carbon atoms, acyl groups having 2 to 20 carbon atoms, aryl groups having 6 to 40 carbon atoms, heterocycles having 3 to 40 carbon atoms An aryl group, an alkenyl group having 2 to 10 carbon atoms, an alkynyl group having 2 to 10 carbon atoms, an alkoxycarbonyl group having 2 to 10 carbon atoms, an alkylsulfonyl group having 1 to 10 carbon atoms, a haloalkyl group having 1 to 10 carbon atoms, Amido group, C2-C10 alkylamide group, C3-C3 0 trialkylsilyl group, trialkylsilyl group having 4 to 20 carbon atoms, trialkylsilyl alkenyl group having 5 to 20 carbon atoms, and the like trialkylsilyl alkynyl group and a nitro group having 5 to 20 carbon atoms. Among these specific examples, those that can be substituted with a substituent may be further substituted. More preferred substituents are a halogen atom, a cyano group, a substituted or unsubstituted alkyl group having 1 to 20 carbon atoms, an alkoxy group having 1 to 20 carbon atoms, a substituted or unsubstituted aryl group having 6 to 40 carbon atoms, carbon A substituted or unsubstituted heteroaryl group having 3 to 40 carbon atoms, and a dialkyl-substituted amino group having 1 to 20 carbon atoms. More preferable substituents are a fluorine atom, a chlorine atom, a cyano group, a substituted or unsubstituted alkyl group having 1 to 10 carbon atoms, a substituted or unsubstituted alkoxy group having 1 to 10 carbon atoms, and a substituted group having 6 to 15 carbon atoms. Alternatively, it is an unsubstituted aryl group or a substituted or unsubstituted heteroaryl group having 3 to 12 carbon atoms.
R1とR2、R2とR3、R3とR4、R5とR6、R6とR7、R7とR8、R8とR9、R9とR1、R11とR12、R12とR13、R13とR14、R14とR15、R16とR17、R17とR18、R18とR19、R19とR20、R21とR22、R22とR23、R23とR24、R24とR25、R25とR26、R26とR27、R27とR28、R31とR32、R32とR33、R33とR34、R35とR36、R36とR37、R37とR38、R3aとR3b、R41とR42、R42とR43、R43とR44、R45とR46、R46とR47、R47とR48、R51とR52、R52とR53、R53とR54、R55とR56、R56とR57、R57とR58、R61とR62、R62とR63、R63とR64、R65とR66、R66とR67、R67とR68は互いに結合して環状構造を形成していてもよい。環状構造は芳香環であっても脂肪環であってもよく、またヘテロ原子を含むものであってもよく、さらに環状構造は2環以上の縮合環であってもよい。ここでいうヘテロ原子としては、窒素原子、酸素原子および硫黄原子からなる群より選択されるものであることが好ましい。形成される環状構造の例として、ベンゼン環、ナフタレン環、ピリジン環、ピリダジン環、ピリミジン環、ピラジン環、ピロール環、イミダゾール環、ピラゾール環、トリアゾール環、イミダゾリン環、オキサゾール環、イソオキサゾール環、チアゾール環、イソチアゾール環、シクロヘキサジエン環、シクロヘキセン環、シクロペンタエン環、シクロヘプタトリエン環、シクロヘプタジエン環、シクロヘプタエン環などを挙げることができる。
R11~R20、R21~R28、R31~R38、R3a、R3b、R41~R48、R4a、R51~R58、R61~R68は、各々独立に上記一般式(2)~(7)のいずれかで表される基であることも好ましい。 R 1 and R 2 , R 2 and R 3 , R 3 and R 4 , R 5 and R 6 , R 6 and R 7 , R 7 and R 8 , R 8 and R 9 , R 9 and R 1 , R 11 And R 12 , R 12 and R 13 , R 13 and R 14 , R 14 and R 15 , R 16 and R 17 , R 17 and R 18 , R 18 and R 19 , R 19 and R 20 , R 21 and R 22 , R 22 and R 23 , R 23 and R 24 , R 24 and R 25 , R 25 and R 26 , R 26 and R 27 , R 27 and R 28 , R 31 and R 32 , R 32 and R 33 , R 33 and R 34, R 35 and R 36, R 36 and R 37, R 37 and R 38, R 3a and R 3b, R 41 and R 42, R 42 and R 43, R 43 and R 44, R 45 And R 46 , R 46 and R 47 , R 47 and R 48 , R 51 and R 52 , R 52 and R 53 , R 53 and R 54 , R 55 and R 56 , R 56 and R 57 , R 57 and R 58 , R 61 and R 62 , R 62 and R 63 , R 63 and R 64 , R 65 and R 66 , R 66 and R 67 , R 67 and R 68 may be bonded to each other to form a cyclic structure. Good. The cyclic structure may be an aromatic ring or an alicyclic ring, may contain a hetero atom, and the cyclic structure may be a condensed ring of two or more rings. The hetero atom here is preferably selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom. Examples of cyclic structures formed include benzene ring, naphthalene ring, pyridine ring, pyridazine ring, pyrimidine ring, pyrazine ring, pyrrole ring, imidazole ring, pyrazole ring, triazole ring, imidazoline ring, oxazole ring, isoxazole ring, thiazole And a ring, an isothiazole ring, a cyclohexadiene ring, a cyclohexene ring, a cyclopentaene ring, a cycloheptatriene ring, a cycloheptadiene ring, and a cycloheptaene ring.
R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a , R 3b , R 41 to R 48 , R 4a , R 51 to R 58 , and R 61 to R 68 are each independently A group represented by any one of the general formulas (2) to (7) is also preferable.
R11~R20、R21~R28、R31~R38、R3a、R3b、R41~R48、R4a、R51~R58、R61~R68は、各々独立に上記一般式(2)~(7)のいずれかで表される基であることも好ましい。 R 1 and R 2 , R 2 and R 3 , R 3 and R 4 , R 5 and R 6 , R 6 and R 7 , R 7 and R 8 , R 8 and R 9 , R 9 and R 1 , R 11 And R 12 , R 12 and R 13 , R 13 and R 14 , R 14 and R 15 , R 16 and R 17 , R 17 and R 18 , R 18 and R 19 , R 19 and R 20 , R 21 and R 22 , R 22 and R 23 , R 23 and R 24 , R 24 and R 25 , R 25 and R 26 , R 26 and R 27 , R 27 and R 28 , R 31 and R 32 , R 32 and R 33 , R 33 and R 34, R 35 and R 36, R 36 and R 37, R 37 and R 38, R 3a and R 3b, R 41 and R 42, R 42 and R 43, R 43 and R 44, R 45 And R 46 , R 46 and R 47 , R 47 and R 48 , R 51 and R 52 , R 52 and R 53 , R 53 and R 54 , R 55 and R 56 , R 56 and R 57 , R 57 and R 58 , R 61 and R 62 , R 62 and R 63 , R 63 and R 64 , R 65 and R 66 , R 66 and R 67 , R 67 and R 68 may be bonded to each other to form a cyclic structure. Good. The cyclic structure may be an aromatic ring or an alicyclic ring, may contain a hetero atom, and the cyclic structure may be a condensed ring of two or more rings. The hetero atom here is preferably selected from the group consisting of a nitrogen atom, an oxygen atom and a sulfur atom. Examples of cyclic structures formed include benzene ring, naphthalene ring, pyridine ring, pyridazine ring, pyrimidine ring, pyrazine ring, pyrrole ring, imidazole ring, pyrazole ring, triazole ring, imidazoline ring, oxazole ring, isoxazole ring, thiazole And a ring, an isothiazole ring, a cyclohexadiene ring, a cyclohexene ring, a cyclopentaene ring, a cycloheptatriene ring, a cycloheptadiene ring, and a cycloheptaene ring.
R 11 to R 20 , R 21 to R 28 , R 31 to R 38 , R 3a , R 3b , R 41 to R 48 , R 4a , R 51 to R 58 , and R 61 to R 68 are each independently A group represented by any one of the general formulas (2) to (7) is also preferable.
また、R1~R8は、例えば少なくとも1つとして一般式(3)~(7)のいずれかで表される基を選択してもよく、少なくとも1つとして一般式(4)~(7)のいずれかで表される基を選択してもよい。R1~R8の少なくとも1つが一般式(4)で表される基であるとき、一般式(4)のR3aおよびR3bの少なくともいずれかが置換基であることが好ましく、R3aとR3bがともに置換基であることがより好ましい。置換基としては、炭素数1~15のアルキル基、フェニル基であることが好ましく、炭素数1~15のアルキル基であることがより好ましい。また、R3aおよびR3bは互いに結合して環状構造を形成していることも好ましい。この環状構造としては、フルオレン環、キサンテン環、チオキサンテン環等の三環構造であることが好ましい。
一般式(1)において、R9は置換基を表す。R9が表す置換基の説明と好ましい範囲については、
上記のR1~R8等がとりうる置換基の説明と好ましい範囲を参照することができる。このうち、R9は下記一般式(8)で表される置換もしくは無置換のフェニル基であることが好ましい。

R 1 to R 8 may be, for example, at least one selected from the groups represented by any of the general formulas (3) to (7), and at least one of the general formulas (4) to (7 ) May be selected. When at least one of R 1 ~ R 8 is a group represented by the general formula (4), it is preferable that at least one of R 3a and R 3b in the formula (4) is a substituent, and R 3a More preferably, both R 3b are substituents. As the substituent, an alkyl group having 1 to 15 carbon atoms and a phenyl group are preferable, and an alkyl group having 1 to 15 carbon atoms is more preferable. R 3a and R 3b are preferably bonded to each other to form a cyclic structure. The cyclic structure is preferably a tricyclic structure such as a fluorene ring, a xanthene ring, or a thioxanthene ring.
In the general formula (1), R 9 represents a substituent. For the explanation and preferred range of the substituent represented by R 9 ,
Reference can be made to the description and preferred ranges of the substituents that can be taken by the above R 1 to R 8 and the like. Among these, R 9 is preferably a substituted or unsubstituted phenyl group represented by the following general formula (8).
一般式(1)において、R9は置換基を表す。R9が表す置換基の説明と好ましい範囲については、
上記のR1~R8等がとりうる置換基の説明と好ましい範囲を参照することができる。このうち、R9は下記一般式(8)で表される置換もしくは無置換のフェニル基であることが好ましい。
In the general formula (1), R 9 represents a substituent. For the explanation and preferred range of the substituent represented by R 9 ,
Reference can be made to the description and preferred ranges of the substituents that can be taken by the above R 1 to R 8 and the like. Among these, R 9 is preferably a substituted or unsubstituted phenyl group represented by the following general formula (8).
一般式(8)において、*は一般式(1)におけるホウ素原子への結合部位を表す。R9a、R9b、R9c、R9d、R9eは、各々独立に水素原子または置換基を表す。置換基の数は特に制限されず、R9a、R9b、R9c、R9d、R9eのすべてが無置換(すなわち水素原子)であってもよいが、R9a、R9c、R9eの少なくともいずれかは置換基であることが好ましく、R9aとR9eが置換基であることがより好ましい。一般式(8)において置換基が2つ以上ある場合、それらの置換基は同一であっても異なっていてもよい。R9aとR9b、R9bとR9c、R9cとR9d、R9dとR9eはそれぞれ互いに結合して環状構造を形成していてもよい。R9a、R9b、R9c、R9d、R9eがとりうる置換基の説明と好ましい範囲については、下記のR1~R8等がとりうる置換基の説明と好ましい範囲を参照することができ、環状構造の説明と好ましい例については、上記の一般式(1)において、R1とR2等が互いに結合して形成する環状構造の説明と好ましい例を参照することができる。
In the general formula (8), * represents a bonding site to the boron atom in the general formula (1). R 9a , R 9b , R 9c , R 9d and R 9e each independently represent a hydrogen atom or a substituent. The number of substituents is not particularly limited, and all of R 9a , R 9b , R 9c , R 9d , and R 9e may be unsubstituted (that is, a hydrogen atom), but R 9a , R 9c , and R 9e At least one of them is preferably a substituent, and R 9a and R 9e are more preferably substituents. In the general formula (8), when there are two or more substituents, these substituents may be the same or different. R 9a and R 9b , R 9b and R 9c , R 9c and R 9d , and R 9d and R 9e may be bonded to each other to form a cyclic structure. For the explanation and preferred ranges of the substituents that R 9a , R 9b , R 9c , R 9d , and R 9e can take, see the explanations and preferred ranges of the substituents that can be taken by R 1 to R 8 below. For the explanation and preferred examples of the cyclic structure, reference can be made to the explanation and preferred examples of the cyclic structure formed by combining R 1 and R 2 in the general formula (1).
ここで、R9がホウ素原子と単結合を形成していない原子であって孤立電子対を有するものを含む置換基であるとき、該原子はホウ素原子と配位結合して環状構造を形成していてもよい。ここでいうホウ素原子と単結合を形成していない原子であって孤立電子対を有するものとして、酸素原子や窒素原子を挙げることができる。また、該原子がホウ素原子と配位結合することによって形成される環状構造は、5~7員環であることが好ましく、5または6員環であることがより好ましい。具体的な化合物例として、下記の化合物177~186を参照することができる。これらの化合物では、ホウ素原子と単結合を形成していない酸素原子または窒素原子がホウ素原子と配位結合して5または6員環を形成している。
Here, when R 9 is a substituent that includes an atom that does not form a single bond with a boron atom and has a lone pair, the atom coordinates with the boron atom to form a cyclic structure. It may be. Examples of the atoms that do not form a single bond with the boron atom and have a lone pair include oxygen atoms and nitrogen atoms. Further, the cyclic structure formed by coordination bonding of the atom with a boron atom is preferably a 5- to 7-membered ring, and more preferably a 5- or 6-membered ring. As specific compound examples, the following compounds 177 to 186 can be referred to. In these compounds, an oxygen atom or a nitrogen atom that does not form a single bond with a boron atom is coordinated with the boron atom to form a 5- or 6-membered ring.
以下において、一般式(1)で表される化合物の具体例を例示する。ただし、本発明において用いることができる一般式(1)で表される化合物はこれらの具体例によって限定的に解釈されるべきものではない。
Below, the specific example of a compound represented by General formula (1) is illustrated. However, the compound represented by the general formula (1) that can be used in the present invention should not be limitedly interpreted by these specific examples.
Below, the specific example of a compound represented by General formula (1) is illustrated. However, the compound represented by the general formula (1) that can be used in the present invention should not be limitedly interpreted by these specific examples.













一般式(1)で表される化合物の分子量は、例えば一般式(1)で表される化合物を含む有機層を蒸着法により製膜して利用することを意図する場合には、1500以下であることが好ましく、1200以下であることがより好ましく、1000以下であることがさらに好ましく、800以下であることがさらにより好ましい。分子量の下限値は、一般式(1)で表される最小化合物の分子量である。
一般式(1)で表される化合物は、分子量にかかわらず塗布法で成膜してもよい。塗布法を用いれば、分子量が比較的大きな化合物であっても成膜することが可能である。 The molecular weight of the compound represented by the general formula (1) is, for example, 1500 or less when the organic layer containing the compound represented by the general formula (1) is intended to be formed by vapor deposition. Preferably, it is preferably 1200 or less, more preferably 1000 or less, and even more preferably 800 or less. The lower limit of the molecular weight is the molecular weight of the minimum compound represented by the general formula (1).
The compound represented by the general formula (1) may be formed by a coating method regardless of the molecular weight. If a coating method is used, a film can be formed even with a compound having a relatively large molecular weight.
一般式(1)で表される化合物は、分子量にかかわらず塗布法で成膜してもよい。塗布法を用いれば、分子量が比較的大きな化合物であっても成膜することが可能である。 The molecular weight of the compound represented by the general formula (1) is, for example, 1500 or less when the organic layer containing the compound represented by the general formula (1) is intended to be formed by vapor deposition. Preferably, it is preferably 1200 or less, more preferably 1000 or less, and even more preferably 800 or less. The lower limit of the molecular weight is the molecular weight of the minimum compound represented by the general formula (1).
The compound represented by the general formula (1) may be formed by a coating method regardless of the molecular weight. If a coating method is used, a film can be formed even with a compound having a relatively large molecular weight.
本発明を応用して、分子内に一般式(1)で表される構造を複数個含む化合物を、発光材料として用いることも考えられる。
例えば、一般式(1)で表される構造中にあらかじめ重合性基を存在させておいて、その重合性基を重合させることによって得られる重合体を、発光材料として用いることが考えられる。具体的には、一般式(1)のR1~R9のいずれかに重合性官能基を含むモノマーを用意して、これを単独で重合させるか、他のモノマーとともに共重合させることにより、繰り返し単位を有する重合体を得て、その重合体を発光材料として用いることが考えられる。あるいは、一般式(1)で表される構造を有する化合物どうしを反応させることにより、二量体や三量体を得て、それらを発光材料として用いることも考えられる。 By applying the present invention, it is also conceivable to use a compound containing a plurality of structures represented by the general formula (1) in the molecule as a light emitting material.
For example, it is conceivable to use a polymer obtained by previously polymerizing a polymerizable group in the structure represented by the general formula (1) and polymerizing the polymerizable group as a light emitting material. Specifically, by preparing a monomer containing a polymerizable functional group in any of R 1 to R 9 in the general formula (1) and polymerizing it alone or copolymerizing with other monomers, It is conceivable to obtain a polymer having a repeating unit and use the polymer as a light emitting material. Alternatively, it is also conceivable that dimers and trimers are obtained by reacting compounds having a structure represented by the general formula (1) and used as a luminescent material.
例えば、一般式(1)で表される構造中にあらかじめ重合性基を存在させておいて、その重合性基を重合させることによって得られる重合体を、発光材料として用いることが考えられる。具体的には、一般式(1)のR1~R9のいずれかに重合性官能基を含むモノマーを用意して、これを単独で重合させるか、他のモノマーとともに共重合させることにより、繰り返し単位を有する重合体を得て、その重合体を発光材料として用いることが考えられる。あるいは、一般式(1)で表される構造を有する化合物どうしを反応させることにより、二量体や三量体を得て、それらを発光材料として用いることも考えられる。 By applying the present invention, it is also conceivable to use a compound containing a plurality of structures represented by the general formula (1) in the molecule as a light emitting material.
For example, it is conceivable to use a polymer obtained by previously polymerizing a polymerizable group in the structure represented by the general formula (1) and polymerizing the polymerizable group as a light emitting material. Specifically, by preparing a monomer containing a polymerizable functional group in any of R 1 to R 9 in the general formula (1) and polymerizing it alone or copolymerizing with other monomers, It is conceivable to obtain a polymer having a repeating unit and use the polymer as a light emitting material. Alternatively, it is also conceivable that dimers and trimers are obtained by reacting compounds having a structure represented by the general formula (1) and used as a luminescent material.
一般式(1)で表される構造を含む繰り返し単位を有する重合体の例として、下記一般式(9)または(10)で表される構造を含む重合体を挙げることができる。

Examples of the polymer having a repeating unit containing a structure represented by the general formula (1) include a polymer containing a structure represented by the following general formula (9) or (10).
一般式(9)または(10)において、Qは一般式(1)で表される構造を含む基を表し、L1およびL2は連結基を表す。連結基の炭素数は、好ましくは0~20であり、より好ましくは1~15であり、さらに好ましくは2~10である。連結基は-X11-L11-で表される構造を有するものであることが好ましい。ここで、X11は酸素原子または硫黄原子を表し、酸素原子であることが好ましい。L11は連結基を表し、置換もしくは無置換のアルキレン基、または置換もしくは無置換のアリーレン基であることが好ましく、炭素数1~10の置換もしくは無置換のアルキレン基、または置換もしくは無置換のフェニレン基であることがより好ましい。
一般式(9)または(10)において、R101、R102、R103およびR104は、各々独立に置換基を表す。好ましくは、炭素数1~6の置換もしくは無置換のアルキル基、炭素数1~6の置換もしくは無置換のアルコキシ基、ハロゲン原子であり、より好ましくは炭素数1~3の無置換のアルキル基、炭素数1~3の無置換のアルコキシ基、フッ素原子、塩素原子であり、さらに好ましくは炭素数1~3の無置換のアルキル基、炭素数1~3の無置換のアルコキシ基である。
L1およびL2で表される連結基は、Qを構成する一般式(1)の構造のR1~R8のいずれか、一般式(2)のR11~R20のいずれか、一般式(3)の構造のR21~R28のいずれか、一般式(4)の構造のR31~R38のいずれか、一般式(5)の構造のR41~R48のいずれか、一般式(6)の構造のR51~R58のいずれか、一般式(7)の構造のR61~R68のいずれかに結合することができる。1つのQに対して連結基が2つ以上連結して架橋構造や網目構造を形成していてもよい。 In General Formula (9) or (10), Q represents a group including the structure represented by General Formula (1), and L 1 and L 2 represent a linking group. The linking group preferably has 0 to 20 carbon atoms, more preferably 1 to 15 carbon atoms, and still more preferably 2 to 10 carbon atoms. And preferably has a structure represented by - linking group -X 11 -L 11. Here, X 11 represents an oxygen atom or a sulfur atom, and is preferably an oxygen atom. L 11 represents a linking group, preferably a substituted or unsubstituted alkylene group, or a substituted or unsubstituted arylene group, and a substituted or unsubstituted alkylene group having 1 to 10 carbon atoms, or a substituted or unsubstituted group A phenylene group is more preferable.
In general formula (9) or (10), R 101 , R 102 , R 103 and R 104 each independently represent a substituent. Preferably, it is a substituted or unsubstituted alkyl group having 1 to 6 carbon atoms, a substituted or unsubstituted alkoxy group having 1 to 6 carbon atoms, or a halogen atom, more preferably an unsubstituted alkyl group having 1 to 3 carbon atoms. An unsubstituted alkoxy group having 1 to 3 carbon atoms, a fluorine atom, and a chlorine atom, and more preferably an unsubstituted alkyl group having 1 to 3 carbon atoms and an unsubstituted alkoxy group having 1 to 3 carbon atoms.
The linking group represented by L 1 and L 2 is any one of R 1 to R 8 in the structure of the general formula (1) constituting Q, any one of R 11 to R 20 in the general formula (2), Any one of R 21 to R 28 in the structure of formula (3), any of R 31 to R 38 in the structure of general formula (4), any of R 41 to R 48 in the structure of general formula (5), It can be bonded to any one of R 51 to R 58 of the structure of the general formula (6) and any of R 61 to R 68 of the structure of the general formula (7). Two or more linking groups may be linked to one Q to form a crosslinked structure or a network structure.
一般式(9)または(10)において、R101、R102、R103およびR104は、各々独立に置換基を表す。好ましくは、炭素数1~6の置換もしくは無置換のアルキル基、炭素数1~6の置換もしくは無置換のアルコキシ基、ハロゲン原子であり、より好ましくは炭素数1~3の無置換のアルキル基、炭素数1~3の無置換のアルコキシ基、フッ素原子、塩素原子であり、さらに好ましくは炭素数1~3の無置換のアルキル基、炭素数1~3の無置換のアルコキシ基である。
L1およびL2で表される連結基は、Qを構成する一般式(1)の構造のR1~R8のいずれか、一般式(2)のR11~R20のいずれか、一般式(3)の構造のR21~R28のいずれか、一般式(4)の構造のR31~R38のいずれか、一般式(5)の構造のR41~R48のいずれか、一般式(6)の構造のR51~R58のいずれか、一般式(7)の構造のR61~R68のいずれかに結合することができる。1つのQに対して連結基が2つ以上連結して架橋構造や網目構造を形成していてもよい。 In General Formula (9) or (10), Q represents a group including the structure represented by General Formula (1), and L 1 and L 2 represent a linking group. The linking group preferably has 0 to 20 carbon atoms, more preferably 1 to 15 carbon atoms, and still more preferably 2 to 10 carbon atoms. And preferably has a structure represented by - linking group -X 11 -L 11. Here, X 11 represents an oxygen atom or a sulfur atom, and is preferably an oxygen atom. L 11 represents a linking group, preferably a substituted or unsubstituted alkylene group, or a substituted or unsubstituted arylene group, and a substituted or unsubstituted alkylene group having 1 to 10 carbon atoms, or a substituted or unsubstituted group A phenylene group is more preferable.
In general formula (9) or (10), R 101 , R 102 , R 103 and R 104 each independently represent a substituent. Preferably, it is a substituted or unsubstituted alkyl group having 1 to 6 carbon atoms, a substituted or unsubstituted alkoxy group having 1 to 6 carbon atoms, or a halogen atom, more preferably an unsubstituted alkyl group having 1 to 3 carbon atoms. An unsubstituted alkoxy group having 1 to 3 carbon atoms, a fluorine atom, and a chlorine atom, and more preferably an unsubstituted alkyl group having 1 to 3 carbon atoms and an unsubstituted alkoxy group having 1 to 3 carbon atoms.
The linking group represented by L 1 and L 2 is any one of R 1 to R 8 in the structure of the general formula (1) constituting Q, any one of R 11 to R 20 in the general formula (2), Any one of R 21 to R 28 in the structure of formula (3), any of R 31 to R 38 in the structure of general formula (4), any of R 41 to R 48 in the structure of general formula (5), It can be bonded to any one of R 51 to R 58 of the structure of the general formula (6) and any of R 61 to R 68 of the structure of the general formula (7). Two or more linking groups may be linked to one Q to form a crosslinked structure or a network structure.
繰り返し単位の具体的な構造例として、下記式(11)~(14)で表される構造を挙げることができる。

Specific examples of the structure of the repeating unit include structures represented by the following formulas (11) to (14).
これらの式(11)~(14)を含む繰り返し単位を有する重合体は、一般式(1)の構造のR1~R9のいずれかにヒドロキシ基を導入しておき、それをリンカーとして下記化合物を反応させて重合性基を導入し、その重合性基を重合させることにより合成することができる。
In the polymer having a repeating unit containing these formulas (11) to (14), a hydroxy group is introduced into any one of R 1 to R 9 of the structure of the general formula (1), and this is used as a linker as described below. It can be synthesized by reacting a compound to introduce a polymerizable group and polymerizing the polymerizable group.
分子内に一般式(1)で表される構造を含む重合体は、一般式(1)で表される構造を有する繰り返し単位のみからなる重合体であってもよいし、それ以外の構造を有する繰り返し単位を含む重合体であってもよい。また、重合体の中に含まれる一般式(1)で表される構造を有する繰り返し単位は、単一種であってもよいし、2種以上であってもよい。一般式(1)で表される構造を有さない繰り返し単位としては、通常の共重合に用いられるモノマーから誘導されるものを挙げることができる。例えば、エチレン、スチレンなどのエチレン性不飽和結合を有するモノマーから誘導される繰り返し単位を挙げることができる。
The polymer containing the structure represented by the general formula (1) in the molecule may be a polymer composed only of repeating units having the structure represented by the general formula (1), or other structures may be used. It may be a polymer containing repeating units. The repeating unit having a structure represented by the general formula (1) contained in the polymer may be a single type or two or more types. Examples of the repeating unit not having the structure represented by the general formula (1) include those derived from monomers used in ordinary copolymerization. Examples thereof include a repeating unit derived from a monomer having an ethylenically unsaturated bond such as ethylene and styrene.
[一般式(1’)で表される化合物の合成方法]
一般式(1)で表される化合物は新規化合物である。

[Synthesis Method of Compound Represented by General Formula (1 ′)]
The compound represented by the general formula (1) is a novel compound.
一般式(1)で表される化合物は新規化合物である。
The compound represented by the general formula (1) is a novel compound.
一般式(1’)において、X’は酸素原子または硫黄原子を表す。R1’~R8’は各々独立に水素原子または置換基を表す。ただし、R1’~R8’の少なくとも1つは、各々独立に下記一般式(2’)~(7’)で表される基である。R1’とR2’、R2’とR3’、R3’とR4’、R5’とR6’、R6’とR7’、R7’とR8’、R8’とR9’、R9’とR1’は互いに結合して環状構造を形成していてもよい。R9’は置換基を表す。R9’がホウ素原子と単結合を形成していない孤立電子対を有する原子を含むとき、該原子はホウ素原子と配位結合して環状構造を形成していてもよい。


In the general formula (1 ′), X ′ represents an oxygen atom or a sulfur atom. R 1 ′ to R 8 ′ each independently represents a hydrogen atom or a substituent. However, at least one of R 1 ′ to R 8 ′ is a group independently represented by the following general formulas (2 ′) to (7 ′). R 1 'and R 2', R 2 'and R 3', R 3 'and R 4', R 5 'and R 6', R 6 'and R 7', R 7 'and R 8', R 8 'And R 9 ' and R 9 'and R 1 ' may be bonded to each other to form a cyclic structure. R 9 ′ represents a substituent. When R 9 ′ includes an atom having a lone pair of electrons that does not form a single bond with a boron atom, the atom may be coordinated with the boron atom to form a cyclic structure.
一般式(2’)~(7’)において、L12’~L17’は各々独立に単結合または二価の連結基を表し、*は一般式(1’)におけるベンゼン環への結合部位を表す。R11’~R20’、R21’~R28’、R31’~R38 ’、R3a’、R3b’、R41’~R48’、R4a’、R51’~R58’、R61’~R68 ’は、各々独立に水素原子または置換基を表す。R11’とR12’、R12’とR13’、R13’とR14’、R14’とR15’、R16’とR17’、R17’とR18’、R18’とR19’、R19’とR20’、R21’とR22’、R22’とR23’、R23’とR24’、R24’とR25’、R25’とR26’、R26’とR27’、R27’とR28’、R31’とR32’、R32’とR33’、R33’とR34’、R35’とR36’、R36’とR37’、R37’とR38’、R3a’とR3b’、R41’とR42’、R42’とR43’、R43’とR44’、R45’とR46’、R46’とR47’、R47’とR48’、R51’とR52’、R52’とR53’、R53’とR54’、R55’とR56’、R56’とR57’、R57’とR58’、R61’とR62’、R62’とR63’、R63’とR64’、R65’とR66’、R66’とR67’、R67’とR68’はそれぞれ互いに結合して環状構造を形成していてもよい。
一般式(1’)におけるR1'~R8’と、一般式(2’)~(7’)におけるR11’~R20 ’、R21’~R28 ’、R31’~R38 ’、R3a’、R3b’、R41’~R48 ’、R4a’、R51’~R58 ’、R61’~R68 ’の説明と好ましい範囲については、一般式(1)で表される化合物の説明を参照することができる。 In the general formulas (2 ′) to (7 ′), L 12 ′ to L 17 ′ each independently represents a single bond or a divalent linking group, and * represents a binding site to the benzene ring in the general formula (1 ′). Represents. R 11 '~ R 20', R 21 '~ R 28', R 31 '~ R 38', R 3a ', R 3b', R 41 '~ R 48', R 4a ', R 51' ~ R 58 ', R 61 ' to R 68 'each independently represents a hydrogen atom or a substituent. R 11 'and R 12', R 12 'and R 13', R 13 'and R 14', R 14 'and R 15', 'R 17 and' R 16, R 17 'and R 18', R 18 'And R 19 ', R 19 'and R 20 ', R 21 'and R 22 ', R 22 'and R 23 ', R 23 'and R 24 ', R 24 'and R 25 ', R 25 'and R 26 ', R 26' and R 27 ', R 27' and R 28 ', R 31' and R 32 ', R 32' and R 33 ', R 33' and R 34 ', R 35' and R 36 ', R 36 ' and R 37 ', R 37 ' and R 38 ', R 3a ' and R 3b ', R 41 ' and R 42 ', R 42 ' and R 43 ', R 43 ' and R 44 ', R45 'and R46 ', R46 'and R47 ', R47 'and R48 ', R51 'and R52 ', R52 'and R53 ', R53 'and R54 ', R55 'And R 56 ', R 56 'and R 57 ', R 57 'and R 58 ', R 61 'and R 62 ', R 62 'and R 63 ', R 63 'and R 64 ', R 65 'and R 66 ′, R 66 ′ and R 67 ′, R 67 ′ and R 68 ′ may be bonded to each other to form a cyclic structure.
R 1 ′ to R 8 ′ in general formula (1 ′) and R 11 ′ to R 20 ′, R 21 ′ to R 28 ′, R 31 ′ to R 38 in general formulas (2 ′) to (7 ′) R, R 3a , R 3b ′, R 41 ′ to R 48 ′, R 4a ′, R 51 ′ to R 58 ′, R 61 ′ to R 68 ′ are explained in general formula (1). The description of the compound represented by can be referred to.
一般式(1’)におけるR1'~R8’と、一般式(2’)~(7’)におけるR11’~R20 ’、R21’~R28 ’、R31’~R38 ’、R3a’、R3b’、R41’~R48 ’、R4a’、R51’~R58 ’、R61’~R68 ’の説明と好ましい範囲については、一般式(1)で表される化合物の説明を参照することができる。 In the general formulas (2 ′) to (7 ′), L 12 ′ to L 17 ′ each independently represents a single bond or a divalent linking group, and * represents a binding site to the benzene ring in the general formula (1 ′). Represents. R 11 '~ R 20', R 21 '~ R 28', R 31 '~ R 38', R 3a ', R 3b', R 41 '~ R 48', R 4a ', R 51' ~ R 58 ', R 61 ' to R 68 'each independently represents a hydrogen atom or a substituent. R 11 'and R 12', R 12 'and R 13', R 13 'and R 14', R 14 'and R 15', 'R 17 and' R 16, R 17 'and R 18', R 18 'And R 19 ', R 19 'and R 20 ', R 21 'and R 22 ', R 22 'and R 23 ', R 23 'and R 24 ', R 24 'and R 25 ', R 25 'and R 26 ', R 26' and R 27 ', R 27' and R 28 ', R 31' and R 32 ', R 32' and R 33 ', R 33' and R 34 ', R 35' and R 36 ', R 36 ' and R 37 ', R 37 ' and R 38 ', R 3a ' and R 3b ', R 41 ' and R 42 ', R 42 ' and R 43 ', R 43 ' and R 44 ', R45 'and R46 ', R46 'and R47 ', R47 'and R48 ', R51 'and R52 ', R52 'and R53 ', R53 'and R54 ', R55 'And R 56 ', R 56 'and R 57 ', R 57 'and R 58 ', R 61 'and R 62 ', R 62 'and R 63 ', R 63 'and R 64 ', R 65 'and R 66 ′, R 66 ′ and R 67 ′, R 67 ′ and R 68 ′ may be bonded to each other to form a cyclic structure.
R 1 ′ to R 8 ′ in general formula (1 ′) and R 11 ′ to R 20 ′, R 21 ′ to R 28 ′, R 31 ′ to R 38 in general formulas (2 ′) to (7 ′) R, R 3a , R 3b ′, R 41 ′ to R 48 ′, R 4a ′, R 51 ′ to R 58 ′, R 61 ′ to R 68 ′ are explained in general formula (1). The description of the compound represented by can be referred to.
[一般式(1’)で表される化合物の合成方法]
一般式(1’)で表される化合物は、既知の反応を組み合わせることによって合成することができる。例えば、一般式(1’)のR3’とR6’が一般式(4’)で表される基である化合物は、以下の2つの化合物を反応させることにより合成することが可能である。

[Synthesis Method of Compound Represented by General Formula (1 ′)]
The compound represented by the general formula (1 ′) can be synthesized by combining known reactions. For example, a compound in which R 3 ′ and R 6 ′ in general formula (1 ′) are a group represented by general formula (4 ′) can be synthesized by reacting the following two compounds. .
一般式(1’)で表される化合物は、既知の反応を組み合わせることによって合成することができる。例えば、一般式(1’)のR3’とR6’が一般式(4’)で表される基である化合物は、以下の2つの化合物を反応させることにより合成することが可能である。
The compound represented by the general formula (1 ′) can be synthesized by combining known reactions. For example, a compound in which R 3 ′ and R 6 ′ in general formula (1 ′) are a group represented by general formula (4 ′) can be synthesized by reacting the following two compounds. .
上記の反応式におけるR1’、R2’、R4’、R5’、R7’~R9’、R31’~R38’、R3a’、R3b’の説明については、一般式(1’)における対応する記載を参照することができる。Aはハロゲン原子を表し、フッ素原子、塩素原子、臭素原子、ヨウ素原子を挙げることができ、塩素原子、臭素原子、ヨウ素原子が好ましい。
上記の反応は、公知の反応を応用したものであり、公知の反応条件を適宜選択して用いることができる。上記の反応の詳細については、後述の合成例を参考にすることができる。また、一般式(1’)で表される化合物は、その他の公知の合成反応を組み合わせることによっても合成することができる。 For the explanation of R 1 ′, R 2 ′, R 4 ′, R 5 ′, R 7 ′ to R 9 ′, R 31 ′ to R 38 ′, R 3a ′, R 3b ′ in the above reaction formula, Reference may be made to the corresponding description in formula (1 ′). A represents a halogen atom, and examples include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom, and a chlorine atom, a bromine atom, and an iodine atom are preferable.
The above reaction is an application of a known reaction, and known reaction conditions can be appropriately selected and used. The details of the above reaction can be referred to the synthesis examples described below. The compound represented by the general formula (1 ′) can also be synthesized by combining other known synthesis reactions.
上記の反応は、公知の反応を応用したものであり、公知の反応条件を適宜選択して用いることができる。上記の反応の詳細については、後述の合成例を参考にすることができる。また、一般式(1’)で表される化合物は、その他の公知の合成反応を組み合わせることによっても合成することができる。 For the explanation of R 1 ′, R 2 ′, R 4 ′, R 5 ′, R 7 ′ to R 9 ′, R 31 ′ to R 38 ′, R 3a ′, R 3b ′ in the above reaction formula, Reference may be made to the corresponding description in formula (1 ′). A represents a halogen atom, and examples include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom, and a chlorine atom, a bromine atom, and an iodine atom are preferable.
The above reaction is an application of a known reaction, and known reaction conditions can be appropriately selected and used. The details of the above reaction can be referred to the synthesis examples described below. The compound represented by the general formula (1 ′) can also be synthesized by combining other known synthesis reactions.
[有機発光素子]
本発明の一般式(1)で表される化合物は、有機発光素子の発光材料として有用である。このため、本発明の一般式(1)で表される化合物は、有機発光素子の発光層に発光材料として効果的に用いることができる。一般式(1)で表される化合物の中には、遅延蛍光を放射する遅延蛍光材料(遅延蛍光体)が含まれている。すなわち本発明は、一般式(1)で表される構造を有する遅延蛍光体の発明と、一般式(1)で表される化合物を遅延蛍光体として使用する発明と、一般式(1)で表される化合物を用いて遅延蛍光を発光させる方法の発明も提供する。そのような化合物を発光材料として用いた有機発光素子は、遅延蛍光を放射し、発光効率が高いという特徴を有する。その原理を、有機エレクトロルミネッセンス素子を例にとって説明すると以下のようになる。 [Organic light emitting device]
The compound represented by the general formula (1) of the present invention is useful as a light emitting material of an organic light emitting device. For this reason, the compound represented by General formula (1) of this invention can be effectively used as a luminescent material for the light emitting layer of an organic light emitting element. The compound represented by the general formula (1) includes a delayed fluorescent material (delayed phosphor) that emits delayed fluorescence. That is, the present invention relates to a delayed phosphor having a structure represented by the general formula (1), an invention using a compound represented by the general formula (1) as a delayed phosphor, and a general formula (1). An invention of a method for emitting delayed fluorescence using the represented compound is also provided. An organic light emitting device using such a compound as a light emitting material emits delayed fluorescence and has a feature of high luminous efficiency. The principle will be described below by taking an organic electroluminescence element as an example.
本発明の一般式(1)で表される化合物は、有機発光素子の発光材料として有用である。このため、本発明の一般式(1)で表される化合物は、有機発光素子の発光層に発光材料として効果的に用いることができる。一般式(1)で表される化合物の中には、遅延蛍光を放射する遅延蛍光材料(遅延蛍光体)が含まれている。すなわち本発明は、一般式(1)で表される構造を有する遅延蛍光体の発明と、一般式(1)で表される化合物を遅延蛍光体として使用する発明と、一般式(1)で表される化合物を用いて遅延蛍光を発光させる方法の発明も提供する。そのような化合物を発光材料として用いた有機発光素子は、遅延蛍光を放射し、発光効率が高いという特徴を有する。その原理を、有機エレクトロルミネッセンス素子を例にとって説明すると以下のようになる。 [Organic light emitting device]
The compound represented by the general formula (1) of the present invention is useful as a light emitting material of an organic light emitting device. For this reason, the compound represented by General formula (1) of this invention can be effectively used as a luminescent material for the light emitting layer of an organic light emitting element. The compound represented by the general formula (1) includes a delayed fluorescent material (delayed phosphor) that emits delayed fluorescence. That is, the present invention relates to a delayed phosphor having a structure represented by the general formula (1), an invention using a compound represented by the general formula (1) as a delayed phosphor, and a general formula (1). An invention of a method for emitting delayed fluorescence using the represented compound is also provided. An organic light emitting device using such a compound as a light emitting material emits delayed fluorescence and has a feature of high luminous efficiency. The principle will be described below by taking an organic electroluminescence element as an example.
有機エレクトロルミネッセンス素子においては、正負の両電極より発光材料にキャリアを注入し、励起状態の発光材料を生成し、発光させる。通常、キャリア注入型の有機エレクトロルミネッセンス素子の場合、生成した励起子のうち、励起一重項状態に励起されるのは25%であり、残り75%は励起三重項状態に励起される。従って、励起三重項状態からの発光であるリン光を利用するほうが、エネルギーの利用効率が高い。しかしながら、励起三重項状態は寿命が長いため、励起状態の飽和や励起三重項状態の励起子との相互作用によるエネルギーの失活が起こり、一般にリン光の量子収率が高くないことが多い。一方、遅延蛍光材料は、項間交差等により励起三重項状態へとエネルギーが遷移した後、三重項-三重項消滅あるいは熱エネルギーの吸収により、励起一重項状態に逆項間交差され蛍光を放射する。有機エレクトロルミネッセンス素子においては、なかでも熱エネルギーの吸収による熱活性化型の遅延蛍光材料が特に有用であると考えられる。有機エレクトロルミネッセンス素子に遅延蛍光材料を利用した場合、励起一重項状態の励起子は通常通り蛍光を放射する。一方、励起三重項状態の励起子は、デバイスが発する熱を吸収して励起一重項へ項間交差され蛍光を放射する。このとき、励起一重項からの発光であるため蛍光と同波長での発光でありながら、励起三重項状態から励起一重項状態への逆項間交差により、生じる光の寿命(発光寿命)は通常の蛍光やりん光よりも長くなるため、これらよりも遅延した蛍光として観察される。これを遅延蛍光として定義できる。このような熱活性化型の励起子移動機構を用いれば、キャリア注入後に熱エネルギーの吸収を経ることにより、通常は25%しか生成しなかった励起一重項状態の化合物の比率を25%以上に引き上げることが可能となる。100℃未満の低い温度でも強い蛍光および遅延蛍光を発する化合物を用いれば、デバイスの熱で充分に励起三重項状態から励起一重項状態への項間交差が生じて遅延蛍光を放射するため、発光効率を飛躍的に向上させることができる。
In the organic electroluminescence element, carriers are injected into the light emitting material from both positive and negative electrodes to generate an excited light emitting material and emit light. In general, in the case of a carrier injection type organic electroluminescence element, 25% of the generated excitons are excited to the excited singlet state, and the remaining 75% are excited to the excited triplet state. Therefore, the use efficiency of energy is higher when phosphorescence, which is light emission from an excited triplet state, is used. However, since the excited triplet state has a long lifetime, energy saturation occurs due to saturation of the excited state and interaction with excitons in the excited triplet state, and in general, the quantum yield of phosphorescence is often not high. On the other hand, delayed fluorescent materials, after energy transition to an excited triplet state due to intersystem crossing, etc., are then crossed back to an excited singlet state due to triplet-triplet annihilation or absorption of thermal energy, and emit fluorescence. To do. In the organic electroluminescence device, it is considered that a thermally activated delayed fluorescent material by absorption of thermal energy is particularly useful. When a delayed fluorescent material is used for the organic electroluminescence element, excitons in the excited singlet state emit fluorescence as usual. On the other hand, excitons in the excited triplet state absorb heat generated by the device and cross between the excited singlets to emit fluorescence. At this time, since the light is emitted from the excited singlet, the light is emitted at the same wavelength as the fluorescence, but the light lifetime (luminescence lifetime) generated by the reverse intersystem crossing from the excited triplet state to the excited singlet state is normal. Since the fluorescence becomes longer than the fluorescence and phosphorescence, it is observed as fluorescence delayed from these. This can be defined as delayed fluorescence. If such a heat-activated exciton transfer mechanism is used, the ratio of the compound in an excited singlet state, which normally generated only 25%, is increased to 25% or more by absorbing thermal energy after carrier injection. It can be raised. If a compound that emits strong fluorescence and delayed fluorescence even at a low temperature of less than 100 ° C is used, the heat of the device will sufficiently cause intersystem crossing from the excited triplet state to the excited singlet state and emit delayed fluorescence. Efficiency can be improved dramatically.
本発明の一般式(1)で表される化合物を発光層の発光材料として用いることにより、有機フォトルミネッセンス素子(有機PL素子)や有機エレクトロルミネッセンス素子(有機EL素子)などの優れた有機発光素子を提供することができる。このとき、本発明の一般式(1)で表される化合物は、いわゆるアシストドーパントとして、発光層に含まれる他の発光材料の発光をアシストする機能を有するものであってもよい。すなわち、発光層に含まれる本発明の一般式(1)で表される化合物は、発光層に含まれるホスト材料の最低励起一重項エネルギー準位と発光層に含まれる他の発光材料の最低励起一重項エネルギー準位の間の最低励起一重項エネルギー準位を有するものであってもよい。
有機フォトルミネッセンス素子は、基板上に少なくとも発光層を形成した構造を有する。また、有機エレクトロルミネッセンス素子は、少なくとも陽極、陰極、および陽極と陰極の間に有機層を形成した構造を有する。有機層は、少なくとも発光層を含むものであり、発光層のみからなるものであってもよいし、発光層の他に1層以上の有機層を有するものであってもよい。そのような他の有機層として、正孔輸送層、正孔注入層、電子阻止層、正孔阻止層、電子注入層、電子輸送層、励起子阻止層などを挙げることができる。正孔輸送層は正孔注入機能を有した正孔注入輸送層でもよく、電子輸送層は電子注入機能を有した電子注入輸送層でもよい。具体的な有機エレクトロルミネッセンス素子の構造例を図1に示す。図1において、1は基板、2は陽極、3は正孔注入層、4は正孔輸送層、5は発光層、6は電子輸送層、7は陰極を表わす。
以下において、有機エレクトロルミネッセンス素子の各部材および各層について説明する。なお、基板と発光層の説明は有機フォトルミネッセンス素子の基板と発光層にも該当する。 By using the compound represented by the general formula (1) of the present invention as a light-emitting material of a light-emitting layer, excellent organic light-emitting devices such as an organic photoluminescence device (organic PL device) and an organic electroluminescence device (organic EL device) Can be provided. At this time, the compound represented by the general formula (1) of the present invention may have a function of assisting light emission of another light emitting material included in the light emitting layer as a so-called assist dopant. That is, the compound represented by the general formula (1) of the present invention contained in the light emitting layer includes the lowest excitation singlet energy level of the host material contained in the light emitting layer and the lowest excitation of other light emitting materials contained in the light emitting layer. It may have the lowest excited singlet energy level between singlet energy levels.
The organic photoluminescence element has a structure in which at least a light emitting layer is formed on a substrate. The organic electroluminescence element has a structure in which an organic layer is formed at least between an anode, a cathode, and an anode and a cathode. The organic layer includes at least a light emitting layer, and may consist of only the light emitting layer, or may have one or more organic layers in addition to the light emitting layer. Examples of such other organic layers include a hole transport layer, a hole injection layer, an electron blocking layer, a hole blocking layer, an electron injection layer, an electron transport layer, and an exciton blocking layer. The hole transport layer may be a hole injection / transport layer having a hole injection function, and the electron transport layer may be an electron injection / transport layer having an electron injection function. A specific example of the structure of an organic electroluminescence element is shown in FIG. In FIG. 1, 1 is a substrate, 2 is an anode, 3 is a hole injection layer, 4 is a hole transport layer, 5 is a light emitting layer, 6 is an electron transport layer, and 7 is a cathode.
Below, each member and each layer of an organic electroluminescent element are demonstrated. In addition, description of a board | substrate and a light emitting layer corresponds also to the board | substrate and light emitting layer of an organic photo-luminescence element.
有機フォトルミネッセンス素子は、基板上に少なくとも発光層を形成した構造を有する。また、有機エレクトロルミネッセンス素子は、少なくとも陽極、陰極、および陽極と陰極の間に有機層を形成した構造を有する。有機層は、少なくとも発光層を含むものであり、発光層のみからなるものであってもよいし、発光層の他に1層以上の有機層を有するものであってもよい。そのような他の有機層として、正孔輸送層、正孔注入層、電子阻止層、正孔阻止層、電子注入層、電子輸送層、励起子阻止層などを挙げることができる。正孔輸送層は正孔注入機能を有した正孔注入輸送層でもよく、電子輸送層は電子注入機能を有した電子注入輸送層でもよい。具体的な有機エレクトロルミネッセンス素子の構造例を図1に示す。図1において、1は基板、2は陽極、3は正孔注入層、4は正孔輸送層、5は発光層、6は電子輸送層、7は陰極を表わす。
以下において、有機エレクトロルミネッセンス素子の各部材および各層について説明する。なお、基板と発光層の説明は有機フォトルミネッセンス素子の基板と発光層にも該当する。 By using the compound represented by the general formula (1) of the present invention as a light-emitting material of a light-emitting layer, excellent organic light-emitting devices such as an organic photoluminescence device (organic PL device) and an organic electroluminescence device (organic EL device) Can be provided. At this time, the compound represented by the general formula (1) of the present invention may have a function of assisting light emission of another light emitting material included in the light emitting layer as a so-called assist dopant. That is, the compound represented by the general formula (1) of the present invention contained in the light emitting layer includes the lowest excitation singlet energy level of the host material contained in the light emitting layer and the lowest excitation of other light emitting materials contained in the light emitting layer. It may have the lowest excited singlet energy level between singlet energy levels.
The organic photoluminescence element has a structure in which at least a light emitting layer is formed on a substrate. The organic electroluminescence element has a structure in which an organic layer is formed at least between an anode, a cathode, and an anode and a cathode. The organic layer includes at least a light emitting layer, and may consist of only the light emitting layer, or may have one or more organic layers in addition to the light emitting layer. Examples of such other organic layers include a hole transport layer, a hole injection layer, an electron blocking layer, a hole blocking layer, an electron injection layer, an electron transport layer, and an exciton blocking layer. The hole transport layer may be a hole injection / transport layer having a hole injection function, and the electron transport layer may be an electron injection / transport layer having an electron injection function. A specific example of the structure of an organic electroluminescence element is shown in FIG. In FIG. 1, 1 is a substrate, 2 is an anode, 3 is a hole injection layer, 4 is a hole transport layer, 5 is a light emitting layer, 6 is an electron transport layer, and 7 is a cathode.
Below, each member and each layer of an organic electroluminescent element are demonstrated. In addition, description of a board | substrate and a light emitting layer corresponds also to the board | substrate and light emitting layer of an organic photo-luminescence element.
(基板)
本発明の有機エレクトロルミネッセンス素子は、基板に支持されていることが好ましい。この基板については、特に制限はなく、従来から有機エレクトロルミネッセンス素子に慣用されているものであればよく、例えば、ガラス、透明プラスチック、石英、シリコンなどからなるものを用いることができる。 (substrate)
The organic electroluminescence device of the present invention is preferably supported on a substrate. The substrate is not particularly limited and may be any substrate conventionally used for organic electroluminescence elements. For example, a substrate made of glass, transparent plastic, quartz, silicon, or the like can be used.
本発明の有機エレクトロルミネッセンス素子は、基板に支持されていることが好ましい。この基板については、特に制限はなく、従来から有機エレクトロルミネッセンス素子に慣用されているものであればよく、例えば、ガラス、透明プラスチック、石英、シリコンなどからなるものを用いることができる。 (substrate)
The organic electroluminescence device of the present invention is preferably supported on a substrate. The substrate is not particularly limited and may be any substrate conventionally used for organic electroluminescence elements. For example, a substrate made of glass, transparent plastic, quartz, silicon, or the like can be used.
(陽極)
有機エレクトロルミネッセンス素子における陽極としては、仕事関数の大きい(4eV以上)金属、合金、電気伝導性化合物およびこれらの混合物を電極材料とするものが好ましく用いられる。このような電極材料の具体例としてはAu等の金属、CuI、インジウムチンオキシド(ITO)、SnO2、ZnO等の導電性透明材料が挙げられる。また、IDIXO(In2O3-ZnO)等非晶質で透明導電膜を作製可能な材料を用いてもよい。陽極はこれらの電極材料を蒸着やスパッタリング等の方法により、薄膜を形成させ、フォトリソグラフィー法で所望の形状のパターンを形成してもよく、あるいはパターン精度をあまり必要としない場合は(100μm以上程度)、上記電極材料の蒸着やスパッタリング時に所望の形状のマスクを介してパターンを形成してもよい。あるいは、有機導電性化合物のように塗布可能な材料を用いる場合には、印刷方式、コーティング方式等湿式成膜法を用いることもできる。この陽極より発光を取り出す場合には、透過率を10%より大きくすることが望ましく、また陽極としてのシート抵抗は数百Ω/□以下が好ましい。さらに膜厚は材料にもよるが、通常10~1000nm、好ましくは10~200nmの範囲で選ばれる。 (anode)
As the anode in the organic electroluminescence element, an electrode material made of a metal, an alloy, an electrically conductive compound, or a mixture thereof having a high work function (4 eV or more) is preferably used. Specific examples of such electrode materials include metals such as Au, and conductive transparent materials such as CuI, indium tin oxide (ITO), SnO 2 , and ZnO. Alternatively, an amorphous material such as IDIXO (In 2 O 3 —ZnO) that can form a transparent conductive film may be used. For the anode, a thin film may be formed by vapor deposition or sputtering of these electrode materials, and a pattern of a desired shape may be formed by photolithography, or when pattern accuracy is not so high (about 100 μm or more) ), A pattern may be formed through a mask having a desired shape at the time of vapor deposition or sputtering of the electrode material. Or when using the material which can be apply | coated like an organic electroconductivity compound, wet film-forming methods, such as a printing system and a coating system, can also be used. When light emission is extracted from the anode, it is desirable that the transmittance be greater than 10%, and the sheet resistance as the anode is preferably several hundred Ω / □ or less. Further, although the film thickness depends on the material, it is usually selected in the range of 10 to 1000 nm, preferably 10 to 200 nm.
有機エレクトロルミネッセンス素子における陽極としては、仕事関数の大きい(4eV以上)金属、合金、電気伝導性化合物およびこれらの混合物を電極材料とするものが好ましく用いられる。このような電極材料の具体例としてはAu等の金属、CuI、インジウムチンオキシド(ITO)、SnO2、ZnO等の導電性透明材料が挙げられる。また、IDIXO(In2O3-ZnO)等非晶質で透明導電膜を作製可能な材料を用いてもよい。陽極はこれらの電極材料を蒸着やスパッタリング等の方法により、薄膜を形成させ、フォトリソグラフィー法で所望の形状のパターンを形成してもよく、あるいはパターン精度をあまり必要としない場合は(100μm以上程度)、上記電極材料の蒸着やスパッタリング時に所望の形状のマスクを介してパターンを形成してもよい。あるいは、有機導電性化合物のように塗布可能な材料を用いる場合には、印刷方式、コーティング方式等湿式成膜法を用いることもできる。この陽極より発光を取り出す場合には、透過率を10%より大きくすることが望ましく、また陽極としてのシート抵抗は数百Ω/□以下が好ましい。さらに膜厚は材料にもよるが、通常10~1000nm、好ましくは10~200nmの範囲で選ばれる。 (anode)
As the anode in the organic electroluminescence element, an electrode material made of a metal, an alloy, an electrically conductive compound, or a mixture thereof having a high work function (4 eV or more) is preferably used. Specific examples of such electrode materials include metals such as Au, and conductive transparent materials such as CuI, indium tin oxide (ITO), SnO 2 , and ZnO. Alternatively, an amorphous material such as IDIXO (In 2 O 3 —ZnO) that can form a transparent conductive film may be used. For the anode, a thin film may be formed by vapor deposition or sputtering of these electrode materials, and a pattern of a desired shape may be formed by photolithography, or when pattern accuracy is not so high (about 100 μm or more) ), A pattern may be formed through a mask having a desired shape at the time of vapor deposition or sputtering of the electrode material. Or when using the material which can be apply | coated like an organic electroconductivity compound, wet film-forming methods, such as a printing system and a coating system, can also be used. When light emission is extracted from the anode, it is desirable that the transmittance be greater than 10%, and the sheet resistance as the anode is preferably several hundred Ω / □ or less. Further, although the film thickness depends on the material, it is usually selected in the range of 10 to 1000 nm, preferably 10 to 200 nm.
(陰極)
一方、陰極としては、仕事関数の小さい(4eV以下)金属(電子注入性金属と称する)、合金、電気伝導性化合物およびこれらの混合物を電極材料とするものが用いられる。このような電極材料の具体例としては、ナトリウム、ナトリウム-カリウム合金、マグネシウム、リチウム、マグネシウム/銅混合物、マグネシウム/銀混合物、マグネシウム/アルミニウム混合物、マグネシウム/インジウム混合物、アルミニウム/酸化アルミニウム(Al2O3)混合物、インジウム、リチウム/アルミニウム混合物、希土類金属等が挙げられる。これらの中で、電子注入性および酸化等に対する耐久性の点から、電子注入性金属とこれより仕事関数の値が大きく安定な金属である第二金属との混合物、例えば、マグネシウム/銀混合物、マグネシウム/アルミニウム混合物、マグネシウム/インジウム混合物、アルミニウム/酸化アルミニウム(Al2O3)混合物、リチウム/アルミニウム混合物、アルミニウム等が好適である。陰極はこれらの電極材料を蒸着やスパッタリング等の方法により薄膜を形成させることにより、作製することができる。また、陰極としてのシート抵抗は数百Ω/□以下が好ましく、膜厚は通常10nm~5μm、好ましくは50~200nmの範囲で選ばれる。なお、発光した光を透過させるため、有機エレクトロルミネッセンス素子の陽極または陰極のいずれか一方が、透明または半透明であれば発光輝度が向上し好都合である。
また、陽極の説明で挙げた導電性透明材料を陰極に用いることで、透明または半透明の陰極を作製することができ、これを応用することで陽極と陰極の両方が透過性を有する素子を作製することができる。 (cathode)
On the other hand, as the cathode, a material having a low work function (4 eV or less) metal (referred to as an electron injecting metal), an alloy, an electrically conductive compound, and a mixture thereof as an electrode material is used. Specific examples of such electrode materials include sodium, sodium-potassium alloy, magnesium, lithium, magnesium / copper mixture, magnesium / silver mixture, magnesium / aluminum mixture, magnesium / indium mixture, aluminum / aluminum oxide (Al 2 O 3 ) Mixtures, indium, lithium / aluminum mixtures, rare earth metals and the like. Among these, from the point of durability against electron injection and oxidation, a mixture of an electron injecting metal and a second metal which is a stable metal having a larger work function value than this, for example, a magnesium / silver mixture, Suitable are a magnesium / aluminum mixture, a magnesium / indium mixture, an aluminum / aluminum oxide (Al 2 O 3 ) mixture, a lithium / aluminum mixture, aluminum and the like. The cathode can be produced by forming a thin film of these electrode materials by a method such as vapor deposition or sputtering. The sheet resistance as the cathode is preferably several hundred Ω / □ or less, and the film thickness is usually selected in the range of 10 nm to 5 μm, preferably 50 to 200 nm. In order to transmit the emitted light, if either one of the anode or the cathode of the organic electroluminescence element is transparent or translucent, the emission luminance is advantageously improved.
In addition, by using the conductive transparent material mentioned in the description of the anode as a cathode, a transparent or semi-transparent cathode can be produced. By applying this, an element in which both the anode and the cathode are transparent is used. Can be produced.
一方、陰極としては、仕事関数の小さい(4eV以下)金属(電子注入性金属と称する)、合金、電気伝導性化合物およびこれらの混合物を電極材料とするものが用いられる。このような電極材料の具体例としては、ナトリウム、ナトリウム-カリウム合金、マグネシウム、リチウム、マグネシウム/銅混合物、マグネシウム/銀混合物、マグネシウム/アルミニウム混合物、マグネシウム/インジウム混合物、アルミニウム/酸化アルミニウム(Al2O3)混合物、インジウム、リチウム/アルミニウム混合物、希土類金属等が挙げられる。これらの中で、電子注入性および酸化等に対する耐久性の点から、電子注入性金属とこれより仕事関数の値が大きく安定な金属である第二金属との混合物、例えば、マグネシウム/銀混合物、マグネシウム/アルミニウム混合物、マグネシウム/インジウム混合物、アルミニウム/酸化アルミニウム(Al2O3)混合物、リチウム/アルミニウム混合物、アルミニウム等が好適である。陰極はこれらの電極材料を蒸着やスパッタリング等の方法により薄膜を形成させることにより、作製することができる。また、陰極としてのシート抵抗は数百Ω/□以下が好ましく、膜厚は通常10nm~5μm、好ましくは50~200nmの範囲で選ばれる。なお、発光した光を透過させるため、有機エレクトロルミネッセンス素子の陽極または陰極のいずれか一方が、透明または半透明であれば発光輝度が向上し好都合である。
また、陽極の説明で挙げた導電性透明材料を陰極に用いることで、透明または半透明の陰極を作製することができ、これを応用することで陽極と陰極の両方が透過性を有する素子を作製することができる。 (cathode)
On the other hand, as the cathode, a material having a low work function (4 eV or less) metal (referred to as an electron injecting metal), an alloy, an electrically conductive compound, and a mixture thereof as an electrode material is used. Specific examples of such electrode materials include sodium, sodium-potassium alloy, magnesium, lithium, magnesium / copper mixture, magnesium / silver mixture, magnesium / aluminum mixture, magnesium / indium mixture, aluminum / aluminum oxide (Al 2 O 3 ) Mixtures, indium, lithium / aluminum mixtures, rare earth metals and the like. Among these, from the point of durability against electron injection and oxidation, a mixture of an electron injecting metal and a second metal which is a stable metal having a larger work function value than this, for example, a magnesium / silver mixture, Suitable are a magnesium / aluminum mixture, a magnesium / indium mixture, an aluminum / aluminum oxide (Al 2 O 3 ) mixture, a lithium / aluminum mixture, aluminum and the like. The cathode can be produced by forming a thin film of these electrode materials by a method such as vapor deposition or sputtering. The sheet resistance as the cathode is preferably several hundred Ω / □ or less, and the film thickness is usually selected in the range of 10 nm to 5 μm, preferably 50 to 200 nm. In order to transmit the emitted light, if either one of the anode or the cathode of the organic electroluminescence element is transparent or translucent, the emission luminance is advantageously improved.
In addition, by using the conductive transparent material mentioned in the description of the anode as a cathode, a transparent or semi-transparent cathode can be produced. By applying this, an element in which both the anode and the cathode are transparent is used. Can be produced.
(発光層)
発光層は、陽極および陰極のそれぞれから注入された正孔および電子が再結合することにより励起子が生成した後、発光する層であり、発光材料を単独で発光層に使用しても良いが、好ましくは発光材料とホスト材料を含む。発光材料としては、一般式(1)で表される本発明の化合物群から選ばれる1種または2種以上を用いることができる。本発明の有機エレクトロルミネッセンス素子および有機フォトルミネッセンス素子が高い発光効率を発現するためには、発光材料に生成した一重項励起子および三重項励起子を、発光材料中に閉じ込めることが重要である。従って、発光層中に発光材料に加えてホスト材料を用いることが好ましい。ホスト材料としては、励起一重項エネルギー、励起三重項エネルギーの少なくとも何れか一方が本発明の発光材料よりも高い値を有する有機化合物を用いることができる。その結果、本発明の発光材料に生成した一重項励起子および三重項励起子を、本発明の発光材料の分子中に閉じ込めることが可能となり、その発光効率を十分に引き出すことが可能となる。もっとも、一重項励起子および三重項励起子を十分に閉じ込めることができなくても、高い発光効率を得ることが可能な場合もあるため、高い発光効率を実現しうるホスト材料であれば特に制約なく本発明に用いることができる。本発明の有機発光素子または有機エレクトロルミネッセンス素子において、発光は発光層に含まれる本発明の発光材料から生じる。この発光は蛍光発光および遅延蛍光発光の両方を含む。但し、発光の一部或いは部分的にホスト材料からの発光があってもかまわない。
ホスト材料を用いる場合、発光材料である本発明の化合物が発光層中に含有される量は0.1重量%以上であることが好ましく、1重量%以上であることがより好ましく、また、50重量%以下であることが好ましく、20重量%以下であることがより好ましく、10重量%以下であることがさらに好ましい。
発光層におけるホスト材料としては、正孔輸送能、電子輸送能を有し、かつ発光の長波長化を防ぎ、なおかつ高いガラス転移温度を有する有機化合物であることが好ましい。 (Light emitting layer)
The light emitting layer is a layer that emits light after excitons are generated by recombination of holes and electrons injected from each of the anode and the cathode, and the light emitting material may be used alone for the light emitting layer. , Preferably including a luminescent material and a host material. As a luminescent material, the 1 type (s) or 2 or more types chosen from the compound group of this invention represented by General formula (1) can be used. In order for the organic electroluminescence device and the organic photoluminescence device of the present invention to exhibit high luminous efficiency, it is important to confine singlet excitons and triplet excitons generated in the light emitting material in the light emitting material. Therefore, it is preferable to use a host material in addition to the light emitting material in the light emitting layer. As the host material, an organic compound having at least one of excited singlet energy and excited triplet energy higher than that of the light emitting material of the present invention can be used. As a result, singlet excitons and triplet excitons generated in the light emitting material of the present invention can be confined in the molecules of the light emitting material of the present invention, and the light emission efficiency can be sufficiently extracted. However, even if singlet excitons and triplet excitons cannot be sufficiently confined, there are cases where high luminous efficiency can be obtained, so that host materials that can achieve high luminous efficiency are particularly limited. And can be used in the present invention. In the organic light emitting device or organic electroluminescent device of the present invention, light emission is generated from the light emitting material of the present invention contained in the light emitting layer. This emission includes both fluorescence and delayed fluorescence. However, light emission from the host material may be partly or partly emitted.
When the host material is used, the amount of the compound of the present invention, which is a light emitting material, is preferably 0.1% by weight or more, more preferably 1% by weight or more, and 50% or more. It is preferably no greater than wt%, more preferably no greater than 20 wt%, and even more preferably no greater than 10 wt%.
The host material in the light-emitting layer is preferably an organic compound that has a hole transporting ability and an electron transporting ability, prevents the emission of longer wavelengths, and has a high glass transition temperature.
発光層は、陽極および陰極のそれぞれから注入された正孔および電子が再結合することにより励起子が生成した後、発光する層であり、発光材料を単独で発光層に使用しても良いが、好ましくは発光材料とホスト材料を含む。発光材料としては、一般式(1)で表される本発明の化合物群から選ばれる1種または2種以上を用いることができる。本発明の有機エレクトロルミネッセンス素子および有機フォトルミネッセンス素子が高い発光効率を発現するためには、発光材料に生成した一重項励起子および三重項励起子を、発光材料中に閉じ込めることが重要である。従って、発光層中に発光材料に加えてホスト材料を用いることが好ましい。ホスト材料としては、励起一重項エネルギー、励起三重項エネルギーの少なくとも何れか一方が本発明の発光材料よりも高い値を有する有機化合物を用いることができる。その結果、本発明の発光材料に生成した一重項励起子および三重項励起子を、本発明の発光材料の分子中に閉じ込めることが可能となり、その発光効率を十分に引き出すことが可能となる。もっとも、一重項励起子および三重項励起子を十分に閉じ込めることができなくても、高い発光効率を得ることが可能な場合もあるため、高い発光効率を実現しうるホスト材料であれば特に制約なく本発明に用いることができる。本発明の有機発光素子または有機エレクトロルミネッセンス素子において、発光は発光層に含まれる本発明の発光材料から生じる。この発光は蛍光発光および遅延蛍光発光の両方を含む。但し、発光の一部或いは部分的にホスト材料からの発光があってもかまわない。
ホスト材料を用いる場合、発光材料である本発明の化合物が発光層中に含有される量は0.1重量%以上であることが好ましく、1重量%以上であることがより好ましく、また、50重量%以下であることが好ましく、20重量%以下であることがより好ましく、10重量%以下であることがさらに好ましい。
発光層におけるホスト材料としては、正孔輸送能、電子輸送能を有し、かつ発光の長波長化を防ぎ、なおかつ高いガラス転移温度を有する有機化合物であることが好ましい。 (Light emitting layer)
The light emitting layer is a layer that emits light after excitons are generated by recombination of holes and electrons injected from each of the anode and the cathode, and the light emitting material may be used alone for the light emitting layer. , Preferably including a luminescent material and a host material. As a luminescent material, the 1 type (s) or 2 or more types chosen from the compound group of this invention represented by General formula (1) can be used. In order for the organic electroluminescence device and the organic photoluminescence device of the present invention to exhibit high luminous efficiency, it is important to confine singlet excitons and triplet excitons generated in the light emitting material in the light emitting material. Therefore, it is preferable to use a host material in addition to the light emitting material in the light emitting layer. As the host material, an organic compound having at least one of excited singlet energy and excited triplet energy higher than that of the light emitting material of the present invention can be used. As a result, singlet excitons and triplet excitons generated in the light emitting material of the present invention can be confined in the molecules of the light emitting material of the present invention, and the light emission efficiency can be sufficiently extracted. However, even if singlet excitons and triplet excitons cannot be sufficiently confined, there are cases where high luminous efficiency can be obtained, so that host materials that can achieve high luminous efficiency are particularly limited. And can be used in the present invention. In the organic light emitting device or organic electroluminescent device of the present invention, light emission is generated from the light emitting material of the present invention contained in the light emitting layer. This emission includes both fluorescence and delayed fluorescence. However, light emission from the host material may be partly or partly emitted.
When the host material is used, the amount of the compound of the present invention, which is a light emitting material, is preferably 0.1% by weight or more, more preferably 1% by weight or more, and 50% or more. It is preferably no greater than wt%, more preferably no greater than 20 wt%, and even more preferably no greater than 10 wt%.
The host material in the light-emitting layer is preferably an organic compound that has a hole transporting ability and an electron transporting ability, prevents the emission of longer wavelengths, and has a high glass transition temperature.
(注入層)
注入層とは、駆動電圧低下や発光輝度向上のために電極と有機層間に設けられる層のことで、正孔注入層と電子注入層があり、陽極と発光層または正孔輸送層の間、および陰極と発光層または電子輸送層との間に存在させてもよい。注入層は必要に応じて設けることができる。 (Injection layer)
The injection layer is a layer provided between the electrode and the organic layer for lowering the driving voltage and improving the luminance of light emission, and includes a hole injection layer and an electron injection layer, Further, it may be present between the cathode and the light emitting layer or the electron transport layer. The injection layer can be provided as necessary.
注入層とは、駆動電圧低下や発光輝度向上のために電極と有機層間に設けられる層のことで、正孔注入層と電子注入層があり、陽極と発光層または正孔輸送層の間、および陰極と発光層または電子輸送層との間に存在させてもよい。注入層は必要に応じて設けることができる。 (Injection layer)
The injection layer is a layer provided between the electrode and the organic layer for lowering the driving voltage and improving the luminance of light emission, and includes a hole injection layer and an electron injection layer, Further, it may be present between the cathode and the light emitting layer or the electron transport layer. The injection layer can be provided as necessary.
(阻止層)
阻止層は、発光層中に存在する電荷(電子もしくは正孔)および/または励起子の発光層外への拡散を阻止することができる層である。電子阻止層は、発光層および正孔輸送層の間に配置されることができ、電子が正孔輸送層の方に向かって発光層を通過することを阻止する。同様に、正孔阻止層は発光層および電子輸送層の間に配置されることができ、正孔が電子輸送層の方に向かって発光層を通過することを阻止する。阻止層はまた、励起子が発光層の外側に拡散することを阻止するために用いることができる。すなわち電子阻止層、正孔阻止層はそれぞれ励起子阻止層としての機能も兼ね備えることができる。本明細書でいう電子阻止層または励起子阻止層は、一つの層で電子阻止層および励起子阻止層の機能を有する層を含む意味で使用される。 (Blocking layer)
The blocking layer is a layer that can prevent diffusion of charges (electrons or holes) and / or excitons existing in the light emitting layer to the outside of the light emitting layer. The electron blocking layer can be disposed between the light emitting layer and the hole transport layer and blocks electrons from passing through the light emitting layer toward the hole transport layer. Similarly, a hole blocking layer can be disposed between the light emitting layer and the electron transporting layer to prevent holes from passing through the light emitting layer toward the electron transporting layer. The blocking layer can also be used to block excitons from diffusing outside the light emitting layer. That is, each of the electron blocking layer and the hole blocking layer can also function as an exciton blocking layer. The term “electron blocking layer” or “exciton blocking layer” as used herein is used in the sense of including a layer having the functions of an electron blocking layer and an exciton blocking layer in one layer.
阻止層は、発光層中に存在する電荷(電子もしくは正孔)および/または励起子の発光層外への拡散を阻止することができる層である。電子阻止層は、発光層および正孔輸送層の間に配置されることができ、電子が正孔輸送層の方に向かって発光層を通過することを阻止する。同様に、正孔阻止層は発光層および電子輸送層の間に配置されることができ、正孔が電子輸送層の方に向かって発光層を通過することを阻止する。阻止層はまた、励起子が発光層の外側に拡散することを阻止するために用いることができる。すなわち電子阻止層、正孔阻止層はそれぞれ励起子阻止層としての機能も兼ね備えることができる。本明細書でいう電子阻止層または励起子阻止層は、一つの層で電子阻止層および励起子阻止層の機能を有する層を含む意味で使用される。 (Blocking layer)
The blocking layer is a layer that can prevent diffusion of charges (electrons or holes) and / or excitons existing in the light emitting layer to the outside of the light emitting layer. The electron blocking layer can be disposed between the light emitting layer and the hole transport layer and blocks electrons from passing through the light emitting layer toward the hole transport layer. Similarly, a hole blocking layer can be disposed between the light emitting layer and the electron transporting layer to prevent holes from passing through the light emitting layer toward the electron transporting layer. The blocking layer can also be used to block excitons from diffusing outside the light emitting layer. That is, each of the electron blocking layer and the hole blocking layer can also function as an exciton blocking layer. The term “electron blocking layer” or “exciton blocking layer” as used herein is used in the sense of including a layer having the functions of an electron blocking layer and an exciton blocking layer in one layer.
(正孔阻止層)
正孔阻止層とは広い意味では電子輸送層の機能を有する。正孔阻止層は電子を輸送しつつ、正孔が電子輸送層へ到達することを阻止する役割があり、これにより発光層中での電子と正孔の再結合確率を向上させることができる。正孔阻止層の材料としては、後述する電子輸送層の材料を必要に応じて用いることができる。 (Hole blocking layer)
The hole blocking layer has a function of an electron transport layer in a broad sense. The hole blocking layer has a role of blocking holes from reaching the electron transport layer while transporting electrons, thereby improving the recombination probability of electrons and holes in the light emitting layer. As the material for the hole blocking layer, the material for the electron transport layer described later can be used as necessary.
正孔阻止層とは広い意味では電子輸送層の機能を有する。正孔阻止層は電子を輸送しつつ、正孔が電子輸送層へ到達することを阻止する役割があり、これにより発光層中での電子と正孔の再結合確率を向上させることができる。正孔阻止層の材料としては、後述する電子輸送層の材料を必要に応じて用いることができる。 (Hole blocking layer)
The hole blocking layer has a function of an electron transport layer in a broad sense. The hole blocking layer has a role of blocking holes from reaching the electron transport layer while transporting electrons, thereby improving the recombination probability of electrons and holes in the light emitting layer. As the material for the hole blocking layer, the material for the electron transport layer described later can be used as necessary.
(電子阻止層)
電子阻止層とは、広い意味では正孔を輸送する機能を有する。電子阻止層は正孔を輸送しつつ、電子が正孔輸送層へ到達することを阻止する役割があり、これにより発光層中での電子と正孔が再結合する確率を向上させることができる。 (Electron blocking layer)
The electron blocking layer has a function of transporting holes in a broad sense. The electron blocking layer has a role to block electrons from reaching the hole transport layer while transporting holes, thereby improving the probability of recombination of electrons and holes in the light emitting layer. .
電子阻止層とは、広い意味では正孔を輸送する機能を有する。電子阻止層は正孔を輸送しつつ、電子が正孔輸送層へ到達することを阻止する役割があり、これにより発光層中での電子と正孔が再結合する確率を向上させることができる。 (Electron blocking layer)
The electron blocking layer has a function of transporting holes in a broad sense. The electron blocking layer has a role to block electrons from reaching the hole transport layer while transporting holes, thereby improving the probability of recombination of electrons and holes in the light emitting layer. .
(励起子阻止層)
励起子阻止層とは、発光層内で正孔と電子が再結合することにより生じた励起子が電荷輸送層に拡散することを阻止するための層であり、本層の挿入により励起子を効率的に発光層内に閉じ込めることが可能となり、素子の発光効率を向上させることができる。励起子阻止層は発光層に隣接して陽極側、陰極側のいずれにも挿入することができ、両方同時に挿入することも可能である。すなわち、励起子阻止層を陽極側に有する場合、正孔輸送層と発光層の間に、発光層に隣接して該層を挿入することができ、陰極側に挿入する場合、発光層と陰極との間に、発光層に隣接して該層を挿入することができる。また、陽極と、発光層の陽極側に隣接する励起子阻止層との間には、正孔注入層や電子阻止層などを有することができ、陰極と、発光層の陰極側に隣接する励起子阻止層との間には、電子注入層、電子輸送層、正孔阻止層などを有することができる。阻止層を配置する場合、阻止層として用いる材料の励起一重項エネルギーおよび励起三重項エネルギーの少なくともいずれか一方は、発光材料の励起一重項エネルギーおよび励起三重項エネルギーよりも高いことが好ましい。 (Exciton blocking layer)
The exciton blocking layer is a layer for preventing excitons generated by recombination of holes and electrons in the light emitting layer from diffusing into the charge transport layer. It becomes possible to efficiently confine in the light emitting layer, and the light emission efficiency of the device can be improved. The exciton blocking layer can be inserted on either the anode side or the cathode side adjacent to the light emitting layer, or both can be inserted simultaneously. That is, when the exciton blocking layer is provided on the anode side, the layer can be inserted adjacent to the light emitting layer between the hole transport layer and the light emitting layer, and when inserted on the cathode side, the light emitting layer and the cathode Between the luminescent layer and the light-emitting layer. Further, a hole injection layer, an electron blocking layer, or the like can be provided between the anode and the exciton blocking layer adjacent to the anode side of the light emitting layer, and the excitation adjacent to the cathode and the cathode side of the light emitting layer can be provided. Between the child blocking layer, an electron injection layer, an electron transport layer, a hole blocking layer, and the like can be provided. When the blocking layer is disposed, at least one of the excited singlet energy and the excited triplet energy of the material used as the blocking layer is preferably higher than the excited singlet energy and the excited triplet energy of the light emitting material.
励起子阻止層とは、発光層内で正孔と電子が再結合することにより生じた励起子が電荷輸送層に拡散することを阻止するための層であり、本層の挿入により励起子を効率的に発光層内に閉じ込めることが可能となり、素子の発光効率を向上させることができる。励起子阻止層は発光層に隣接して陽極側、陰極側のいずれにも挿入することができ、両方同時に挿入することも可能である。すなわち、励起子阻止層を陽極側に有する場合、正孔輸送層と発光層の間に、発光層に隣接して該層を挿入することができ、陰極側に挿入する場合、発光層と陰極との間に、発光層に隣接して該層を挿入することができる。また、陽極と、発光層の陽極側に隣接する励起子阻止層との間には、正孔注入層や電子阻止層などを有することができ、陰極と、発光層の陰極側に隣接する励起子阻止層との間には、電子注入層、電子輸送層、正孔阻止層などを有することができる。阻止層を配置する場合、阻止層として用いる材料の励起一重項エネルギーおよび励起三重項エネルギーの少なくともいずれか一方は、発光材料の励起一重項エネルギーおよび励起三重項エネルギーよりも高いことが好ましい。 (Exciton blocking layer)
The exciton blocking layer is a layer for preventing excitons generated by recombination of holes and electrons in the light emitting layer from diffusing into the charge transport layer. It becomes possible to efficiently confine in the light emitting layer, and the light emission efficiency of the device can be improved. The exciton blocking layer can be inserted on either the anode side or the cathode side adjacent to the light emitting layer, or both can be inserted simultaneously. That is, when the exciton blocking layer is provided on the anode side, the layer can be inserted adjacent to the light emitting layer between the hole transport layer and the light emitting layer, and when inserted on the cathode side, the light emitting layer and the cathode Between the luminescent layer and the light-emitting layer. Further, a hole injection layer, an electron blocking layer, or the like can be provided between the anode and the exciton blocking layer adjacent to the anode side of the light emitting layer, and the excitation adjacent to the cathode and the cathode side of the light emitting layer can be provided. Between the child blocking layer, an electron injection layer, an electron transport layer, a hole blocking layer, and the like can be provided. When the blocking layer is disposed, at least one of the excited singlet energy and the excited triplet energy of the material used as the blocking layer is preferably higher than the excited singlet energy and the excited triplet energy of the light emitting material.
(正孔輸送層)
正孔輸送層とは正孔を輸送する機能を有する正孔輸送材料からなり、正孔輸送層は単層または複数層設けることができる。
正孔輸送材料としては、正孔の注入または輸送、電子の障壁性のいずれかを有するものであり、有機物、無機物のいずれであってもよい。使用できる公知の正孔輸送材料としては例えば、トリアゾール誘導体、オキサジアゾール誘導体、イミダゾール誘導体、カルバゾール誘導体、インドロカルバゾール誘導体、ポリアリールアルカン誘導体、ピラゾリン誘導体およびピラゾロン誘導体、フェニレンジアミン誘導体、アリールアミン誘導体、アミノ置換カルコン誘導体、オキサゾール誘導体、スチリルアントラセン誘導体、フルオレノン誘導体、ヒドラゾン誘導体、スチルベン誘導体、シラザン誘導体、アニリン系共重合体、また導電性高分子オリゴマー、特にチオフェンオリゴマー等が挙げられるが、ポルフィリン化合物、芳香族第3級アミン化合物およびスチリルアミン化合物を用いることが好ましく、芳香族第3級アミン化合物を用いることがより好ましい。 (Hole transport layer)
The hole transport layer is made of a hole transport material having a function of transporting holes, and the hole transport layer can be provided as a single layer or a plurality of layers.
The hole transport material has any one of hole injection or transport and electron barrier properties, and may be either organic or inorganic. Known hole transport materials that can be used include, for example, triazole derivatives, oxadiazole derivatives, imidazole derivatives, carbazole derivatives, indolocarbazole derivatives, polyarylalkane derivatives, pyrazoline derivatives and pyrazolone derivatives, phenylenediamine derivatives, arylamine derivatives, Examples include amino-substituted chalcone derivatives, oxazole derivatives, styrylanthracene derivatives, fluorenone derivatives, hydrazone derivatives, stilbene derivatives, silazane derivatives, aniline copolymers, and conductive polymer oligomers, particularly thiophene oligomers. An aromatic tertiary amine compound and an styrylamine compound are preferably used, and an aromatic tertiary amine compound is more preferably used.
正孔輸送層とは正孔を輸送する機能を有する正孔輸送材料からなり、正孔輸送層は単層または複数層設けることができる。
正孔輸送材料としては、正孔の注入または輸送、電子の障壁性のいずれかを有するものであり、有機物、無機物のいずれであってもよい。使用できる公知の正孔輸送材料としては例えば、トリアゾール誘導体、オキサジアゾール誘導体、イミダゾール誘導体、カルバゾール誘導体、インドロカルバゾール誘導体、ポリアリールアルカン誘導体、ピラゾリン誘導体およびピラゾロン誘導体、フェニレンジアミン誘導体、アリールアミン誘導体、アミノ置換カルコン誘導体、オキサゾール誘導体、スチリルアントラセン誘導体、フルオレノン誘導体、ヒドラゾン誘導体、スチルベン誘導体、シラザン誘導体、アニリン系共重合体、また導電性高分子オリゴマー、特にチオフェンオリゴマー等が挙げられるが、ポルフィリン化合物、芳香族第3級アミン化合物およびスチリルアミン化合物を用いることが好ましく、芳香族第3級アミン化合物を用いることがより好ましい。 (Hole transport layer)
The hole transport layer is made of a hole transport material having a function of transporting holes, and the hole transport layer can be provided as a single layer or a plurality of layers.
The hole transport material has any one of hole injection or transport and electron barrier properties, and may be either organic or inorganic. Known hole transport materials that can be used include, for example, triazole derivatives, oxadiazole derivatives, imidazole derivatives, carbazole derivatives, indolocarbazole derivatives, polyarylalkane derivatives, pyrazoline derivatives and pyrazolone derivatives, phenylenediamine derivatives, arylamine derivatives, Examples include amino-substituted chalcone derivatives, oxazole derivatives, styrylanthracene derivatives, fluorenone derivatives, hydrazone derivatives, stilbene derivatives, silazane derivatives, aniline copolymers, and conductive polymer oligomers, particularly thiophene oligomers. An aromatic tertiary amine compound and an styrylamine compound are preferably used, and an aromatic tertiary amine compound is more preferably used.
(電子輸送層)
電子輸送層とは電子を輸送する機能を有する材料からなり、電子輸送層は単層または複数層設けることができる。
電子輸送材料(正孔阻止材料を兼ねる場合もある)としては、陰極より注入された電子を発光層に伝達する機能を有していればよい。使用できる電子輸送層としては例えば、ニトロ置換フルオレン誘導体、ジフェニルキノン誘導体、チオピランジオキシド誘導体、カルボジイミド、フレオレニリデンメタン誘導体、アントラキノジメタンおよびアントロン誘導体、オキサジアゾール誘導体等が挙げられる。さらに、上記オキサジアゾール誘導体において、オキサジアゾール環の酸素原子を硫黄原子に置換したチアジアゾール誘導体、電子吸引基として知られているキノキサリン環を有するキノキサリン誘導体も、電子輸送材料として用いることができる。さらにこれらの材料を高分子鎖に導入した、またはこれらの材料を高分子の主鎖とした高分子材料を用いることもできる。 (Electron transport layer)
The electron transport layer is made of a material having a function of transporting electrons, and the electron transport layer can be provided as a single layer or a plurality of layers.
The electron transport material (which may also serve as a hole blocking material) may have a function of transmitting electrons injected from the cathode to the light emitting layer. Examples of the electron transport layer that can be used include nitro-substituted fluorene derivatives, diphenylquinone derivatives, thiopyrandioxide derivatives, carbodiimides, fluorenylidenemethane derivatives, anthraquinodimethane and anthrone derivatives, oxadiazole derivatives, and the like. Furthermore, in the above oxadiazole derivative, a thiadiazole derivative in which the oxygen atom of the oxadiazole ring is substituted with a sulfur atom, and a quinoxaline derivative having a quinoxaline ring known as an electron withdrawing group can also be used as an electron transport material. Furthermore, a polymer material in which these materials are introduced into a polymer chain or these materials are used as a polymer main chain can also be used.
電子輸送層とは電子を輸送する機能を有する材料からなり、電子輸送層は単層または複数層設けることができる。
電子輸送材料(正孔阻止材料を兼ねる場合もある)としては、陰極より注入された電子を発光層に伝達する機能を有していればよい。使用できる電子輸送層としては例えば、ニトロ置換フルオレン誘導体、ジフェニルキノン誘導体、チオピランジオキシド誘導体、カルボジイミド、フレオレニリデンメタン誘導体、アントラキノジメタンおよびアントロン誘導体、オキサジアゾール誘導体等が挙げられる。さらに、上記オキサジアゾール誘導体において、オキサジアゾール環の酸素原子を硫黄原子に置換したチアジアゾール誘導体、電子吸引基として知られているキノキサリン環を有するキノキサリン誘導体も、電子輸送材料として用いることができる。さらにこれらの材料を高分子鎖に導入した、またはこれらの材料を高分子の主鎖とした高分子材料を用いることもできる。 (Electron transport layer)
The electron transport layer is made of a material having a function of transporting electrons, and the electron transport layer can be provided as a single layer or a plurality of layers.
The electron transport material (which may also serve as a hole blocking material) may have a function of transmitting electrons injected from the cathode to the light emitting layer. Examples of the electron transport layer that can be used include nitro-substituted fluorene derivatives, diphenylquinone derivatives, thiopyrandioxide derivatives, carbodiimides, fluorenylidenemethane derivatives, anthraquinodimethane and anthrone derivatives, oxadiazole derivatives, and the like. Furthermore, in the above oxadiazole derivative, a thiadiazole derivative in which the oxygen atom of the oxadiazole ring is substituted with a sulfur atom, and a quinoxaline derivative having a quinoxaline ring known as an electron withdrawing group can also be used as an electron transport material. Furthermore, a polymer material in which these materials are introduced into a polymer chain or these materials are used as a polymer main chain can also be used.
有機エレクトロルミネッセンス素子を作製する際には、一般式(1)で表される化合物を発光層に用いるだけでなく、発光層以外の層にも用いてもよい。その際、発光層に用いる一般式(1)で表される化合物と、発光層以外の層に用いる一般式(1)で表される化合物は、同一であっても異なっていてもよい。例えば、上記の注入層、阻止層、正孔阻止層、電子阻止層、励起子阻止層、正孔輸送層、電子輸送層などにも一般式(1)で表される化合物を用いてもよい。これらの層の製膜方法は特に限定されず、ドライプロセス、ウェットプロセスのどちらで作製してもよい。
When producing an organic electroluminescence device, the compound represented by the general formula (1) may be used not only for the light emitting layer but also for layers other than the light emitting layer. In that case, the compound represented by General formula (1) used for a light emitting layer and the compound represented by General formula (1) used for layers other than a light emitting layer may be same or different. For example, the compound represented by the general formula (1) may be used for the injection layer, blocking layer, hole blocking layer, electron blocking layer, exciton blocking layer, hole transporting layer, electron transporting layer, and the like. . The method for forming these layers is not particularly limited, and the layer may be formed by either a dry process or a wet process.
以下に、有機エレクトロルミネッセンス素子に用いることができる好ましい材料を具体的に例示する。ただし、本発明において用いることができる材料は、以下の例示化合物によって限定的に解釈されることはない。また、特定の機能を有する材料として例示した化合物であっても、その他の機能を有する材料として転用することも可能である。なお、以下の例示化合物の構造式におけるR、R1~R10は、各々独立に水素原子または置換基を表す。nは3~5の整数を表す。
Below, the preferable material which can be used for an organic electroluminescent element is illustrated concretely. However, the material that can be used in the present invention is not limited to the following exemplary compounds. Moreover, even if it is a compound illustrated as a material which has a specific function, it can also be diverted as a material which has another function. Note that R and R 1 to R 10 in the structural formulas of the following exemplary compounds each independently represent a hydrogen atom or a substituent. n represents an integer of 3 to 5.
まず、発光層のホスト材料としても用いることができる好ましい化合物を挙げる。
First, preferred compounds that can also be used as a host material for the light emitting layer are listed.



次に、正孔注入材料として用いることができる好ましい化合物例を挙げる。
Next, preferred examples of compounds that can be used as the hole injection material are given.
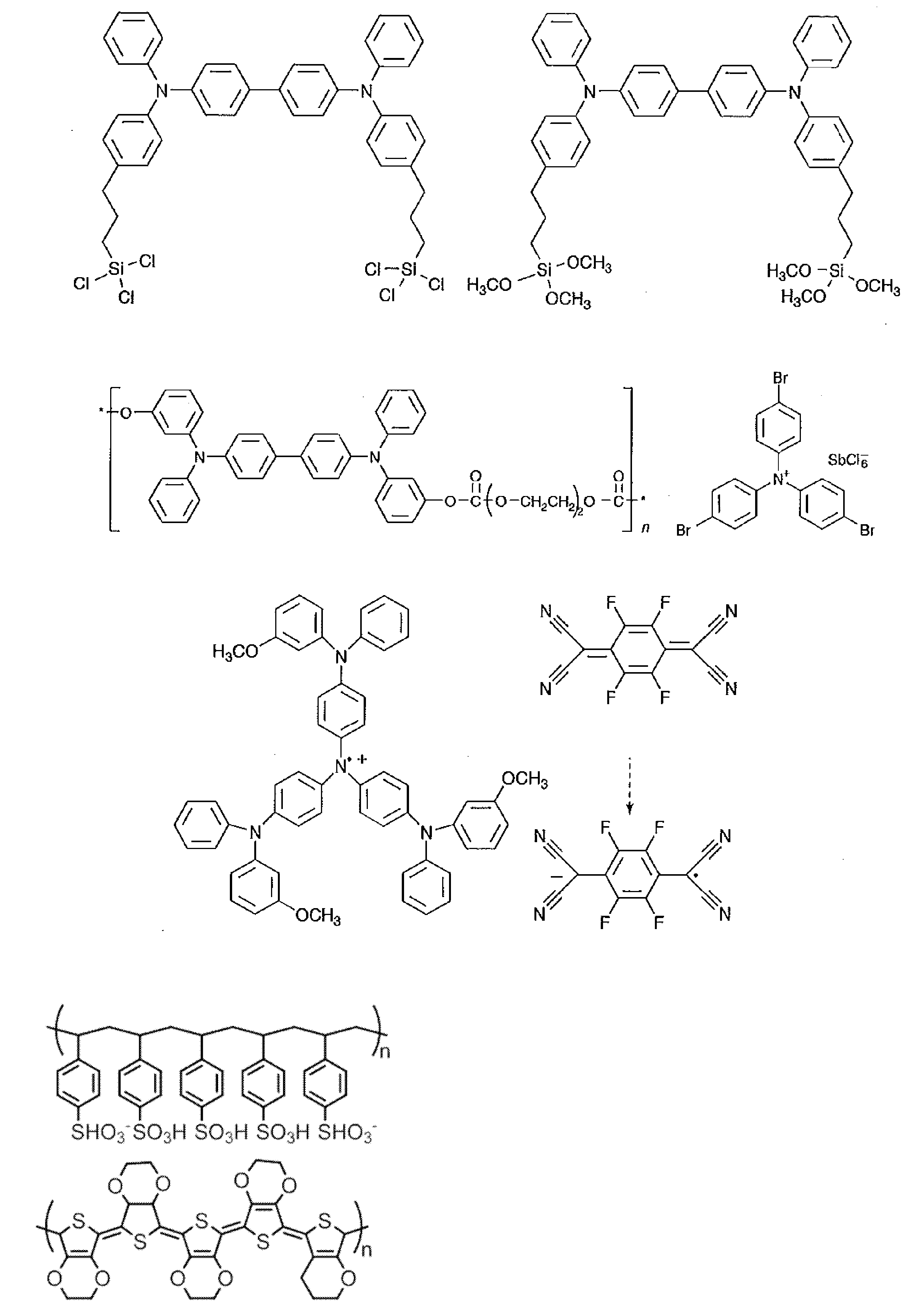
次に、正孔輸送材料として用いることができる好ましい化合物例を挙げる。
Next, preferred examples of compounds that can be used as a hole transport material are given.



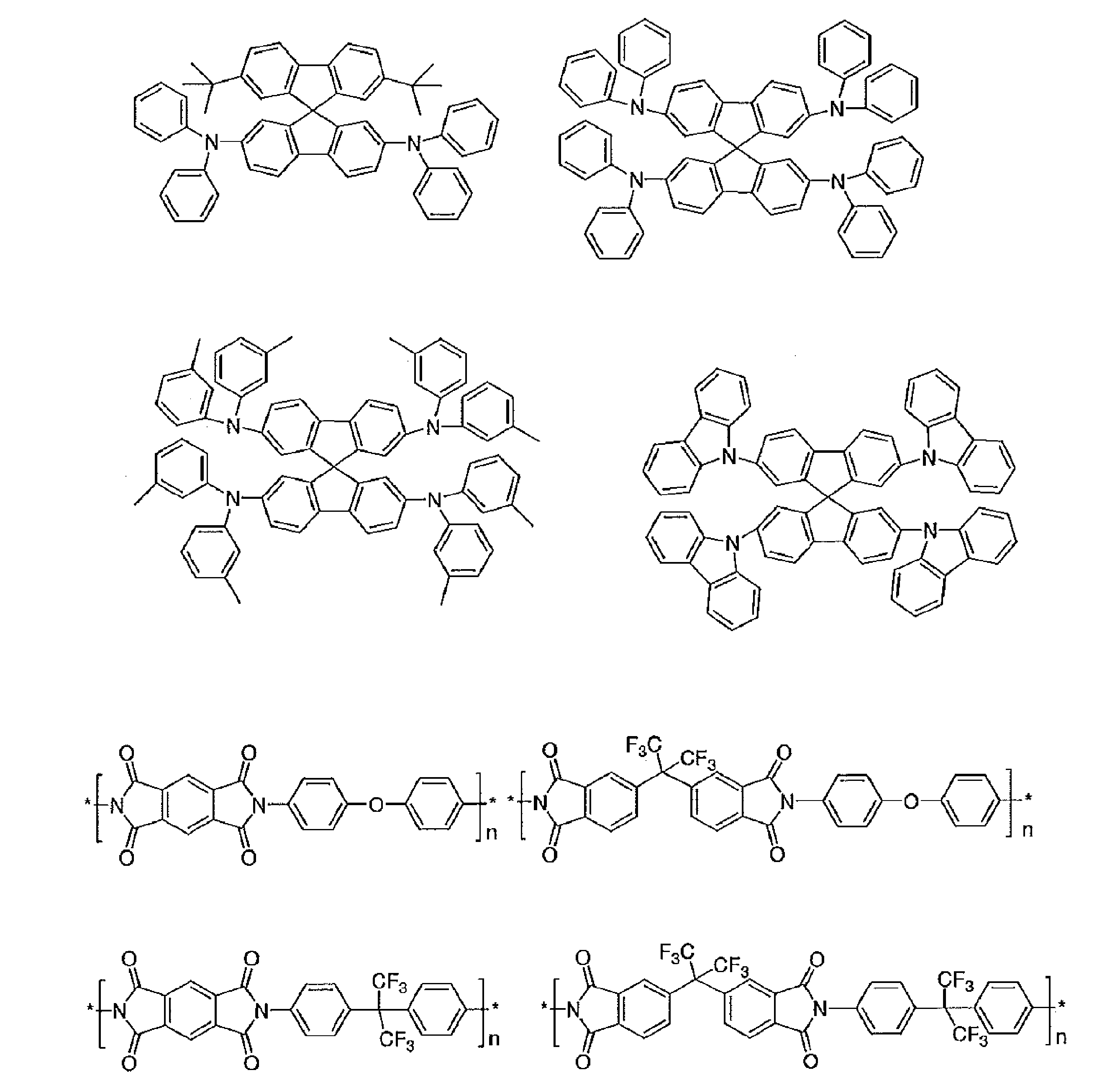
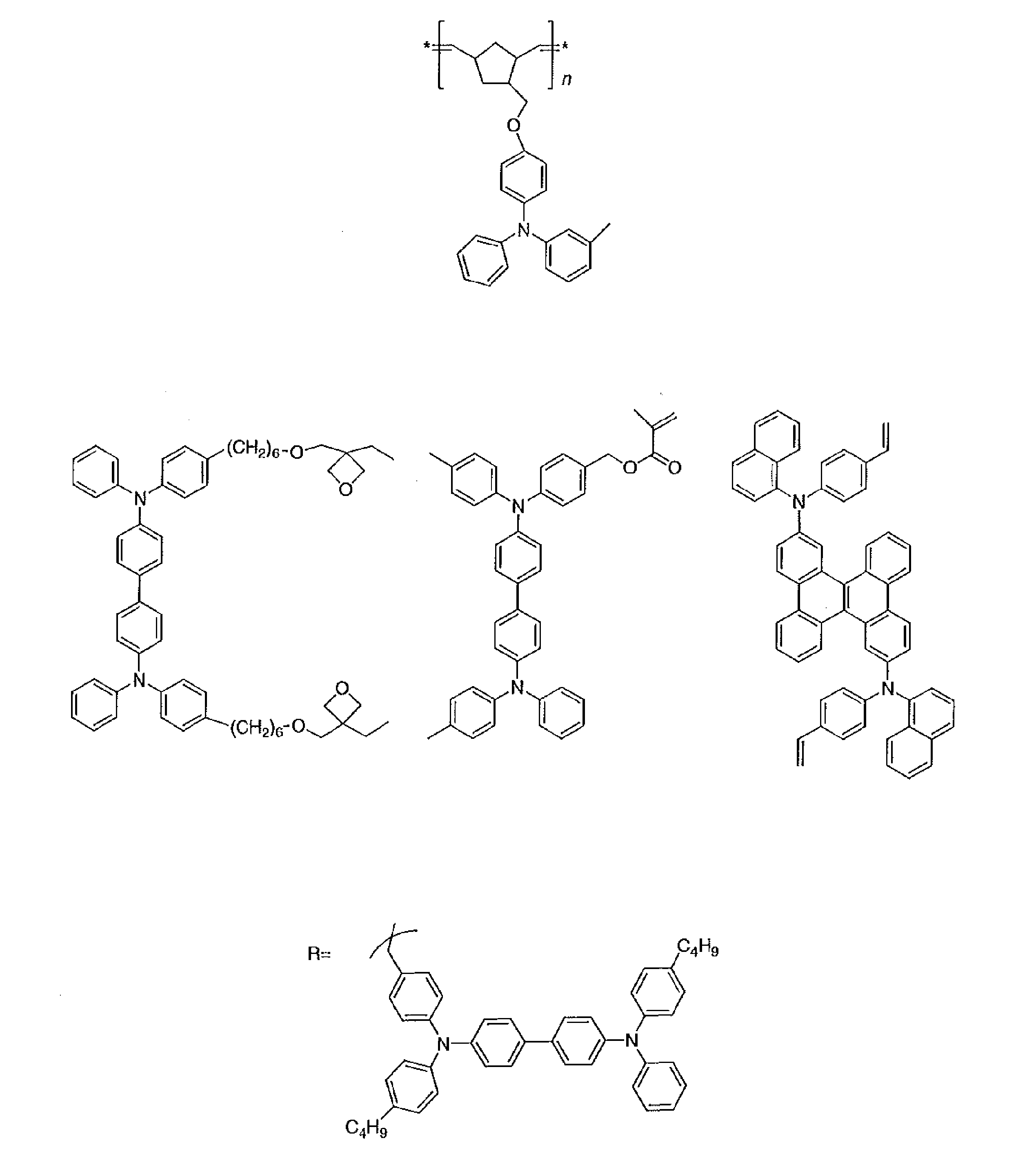

次に、電子阻止材料として用いることができる好ましい化合物例を挙げる。
Next, preferred examples of compounds that can be used as an electron blocking material are given.

次に、正孔阻止材料として用いることができる好ましい化合物例を挙げる。
Next, preferred examples of compounds that can be used as hole blocking materials are given.

次に、電子輸送材料として用いることができる好ましい化合物例を挙げる。
Next, preferred compound examples that can be used as an electron transporting material are listed.


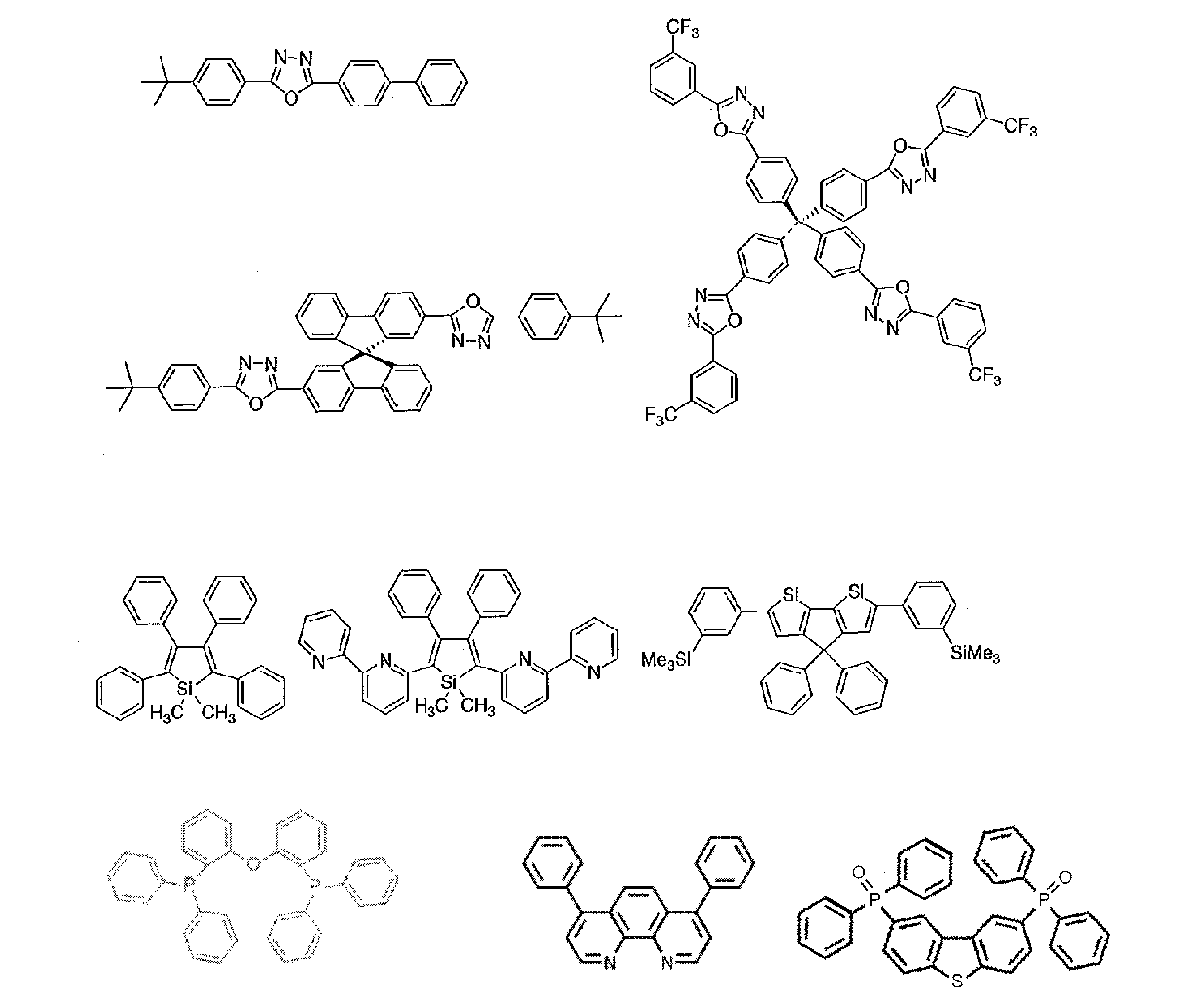
次に、電子注入材料として用いることができる好ましい化合物例を挙げる。
Next, preferred examples of compounds that can be used as an electron injection material will be given.

さらに添加可能な材料として好ましい化合物例を挙げる。例えば、安定化材料として添加すること等が考えられる。
Further preferred compound examples are given as materials that can be added. For example, adding as a stabilizing material can be considered.
上述の方法により作製された有機エレクトロルミネッセンス素子は、得られた素子の陽極と陰極の間に電界を印加することにより発光する。このとき、励起一重項エネルギーによる発光であれば、そのエネルギーレベルに応じた波長の光が、蛍光発光および遅延蛍光発光として確認される。また、励起三重項エネルギーによる発光であれば、そのエネルギーレベルに応じた波長が、りん光として確認される。通常の蛍光は、遅延蛍光発光よりも蛍光寿命が短いため、発光寿命は蛍光と遅延蛍光で区別できる。
一方、りん光については、本発明の化合物のような通常の有機化合物では、励起三重項エネルギーは不安定で熱等に変換され、寿命が短く直ちに失活するため、室温では殆ど観測できない。通常の有機化合物の励起三重項エネルギーを測定するためには、極低温の条件での発光を観測することにより測定可能である。 The organic electroluminescent device produced by the above-described method emits light by applying an electric field between the anode and the cathode of the obtained device. At this time, if the light is emitted by excited singlet energy, light having a wavelength corresponding to the energy level is confirmed as fluorescence emission and delayed fluorescence emission. In addition, in the case of light emission by excited triplet energy, a wavelength corresponding to the energy level is confirmed as phosphorescence. Since normal fluorescence has a shorter fluorescence lifetime than delayed fluorescence, the emission lifetime can be distinguished from fluorescence and delayed fluorescence.
On the other hand, with respect to phosphorescence, in ordinary organic compounds such as the compounds of the present invention, the excited triplet energy is unstable and is converted into heat and the like, and the lifetime is short and it is immediately deactivated. In order to measure the excited triplet energy of a normal organic compound, it can be measured by observing light emission under extremely low temperature conditions.
一方、りん光については、本発明の化合物のような通常の有機化合物では、励起三重項エネルギーは不安定で熱等に変換され、寿命が短く直ちに失活するため、室温では殆ど観測できない。通常の有機化合物の励起三重項エネルギーを測定するためには、極低温の条件での発光を観測することにより測定可能である。 The organic electroluminescent device produced by the above-described method emits light by applying an electric field between the anode and the cathode of the obtained device. At this time, if the light is emitted by excited singlet energy, light having a wavelength corresponding to the energy level is confirmed as fluorescence emission and delayed fluorescence emission. In addition, in the case of light emission by excited triplet energy, a wavelength corresponding to the energy level is confirmed as phosphorescence. Since normal fluorescence has a shorter fluorescence lifetime than delayed fluorescence, the emission lifetime can be distinguished from fluorescence and delayed fluorescence.
On the other hand, with respect to phosphorescence, in ordinary organic compounds such as the compounds of the present invention, the excited triplet energy is unstable and is converted into heat and the like, and the lifetime is short and it is immediately deactivated. In order to measure the excited triplet energy of a normal organic compound, it can be measured by observing light emission under extremely low temperature conditions.
本発明の有機エレクトロルミネッセンス素子は、単一の素子、アレイ状に配置された構造からなる素子、陽極と陰極がX-Yマトリックス状に配置された構造のいずれにおいても適用することができる。本発明によれば、発光層に一般式(1)で表される化合物を含有させることにより、発光効率が大きく改善された有機発光素子が得られる。本発明の有機エレクトロルミネッセンス素子などの有機発光素子は、さらに様々な用途へ応用することが可能である。例えば、本発明の有機エレクトロルミネッセンス素子を用いて、有機エレクトロルミネッセンス表示装置を製造することが可能であり、詳細については、時任静士、安達千波矢、村田英幸共著「有機ELディスプレイ」(オーム社)を参照することができる。また、特に本発明の有機エレクトロルミネッセンス素子は、需要が大きい有機エレクトロルミネッセンス照明やバックライトに応用することもできる。
The organic electroluminescence element of the present invention can be applied to any of a single element, an element having a structure arranged in an array, and a structure in which an anode and a cathode are arranged in an XY matrix. According to the present invention, an organic light emitting device with greatly improved light emission efficiency can be obtained by containing the compound represented by the general formula (1) in the light emitting layer. The organic light emitting device such as the organic electroluminescence device of the present invention can be further applied to various uses. For example, it is possible to produce an organic electroluminescence display device using the organic electroluminescence element of the present invention. For details, see “Organic EL Display” (Ohm Co., Ltd.) written by Shizushi Tokito, Chiba Adachi and Hideyuki Murata. ) Can be referred to. In particular, the organic electroluminescence device of the present invention can be applied to organic electroluminescence illumination and backlights that are in great demand.
以下に合成例および実施例を挙げて本発明の特徴をさらに具体的に説明する。以下に示す材料、処理内容、処理手順等は、本発明の趣旨を逸脱しない限り適宜変更することができる。したがって、本発明の範囲は以下に示す具体例により限定的に解釈されるべきものではない。
Hereinafter, the features of the present invention will be described more specifically with reference to synthesis examples and examples. The following materials, processing details, processing procedures, and the like can be changed as appropriate without departing from the spirit of the present invention. Therefore, the scope of the present invention should not be construed as being limited by the specific examples shown below.
[中間体の合成]
実施例の各化合物を合成するための中間体を以下のようにして合成した。
(中間体1aの合成)

[Synthesis of Intermediate]
Intermediates for synthesizing each compound of the examples were synthesized as follows.
(Synthesis of Intermediate 1a)
実施例の各化合物を合成するための中間体を以下のようにして合成した。
(中間体1aの合成)
Intermediates for synthesizing each compound of the examples were synthesized as follows.
(Synthesis of Intermediate 1a)
アントラニル酸メチル(349.2g,2310mmol)、ヨードベンゼン(428.4g,2100mmol)、炭酸カリウム(348.3g,2520mmol)、ヨウ化銅(20.0g,105mmol)を三口フラスコに入れ、窒素雰囲気下、180℃で加熱撹拌した。
反応終了後、この混合物を室温まで冷却し、ヘキサン500mlで希釈し、セライトを用いて濾過した。得られたろ液を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=8:2の混合溶媒を展開溶媒に用いて精製し、得られたフラクションを真空乾燥した。これにより、目的物の淡黄色オイル状液体(中間体1a)を収量427.5g、収率90%で得た。 Methyl anthranilate (349.2 g, 2310 mmol), iodobenzene (428.4 g, 2100 mmol), potassium carbonate (348.3 g, 2520 mmol), and copper iodide (20.0 g, 105 mmol) were placed in a three-necked flask under a nitrogen atmosphere. The mixture was heated and stirred at 180 ° C.
After completion of the reaction, the mixture was cooled to room temperature, diluted with 500 ml of hexane, and filtered using celite. The obtained filtrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 8: 2 as a developing solvent, and the obtained fraction was vacuum-dried. As a result, the target pale yellow oily liquid (intermediate 1a) was obtained in a yield of 427.5 g and a yield of 90%.
反応終了後、この混合物を室温まで冷却し、ヘキサン500mlで希釈し、セライトを用いて濾過した。得られたろ液を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=8:2の混合溶媒を展開溶媒に用いて精製し、得られたフラクションを真空乾燥した。これにより、目的物の淡黄色オイル状液体(中間体1a)を収量427.5g、収率90%で得た。 Methyl anthranilate (349.2 g, 2310 mmol), iodobenzene (428.4 g, 2100 mmol), potassium carbonate (348.3 g, 2520 mmol), and copper iodide (20.0 g, 105 mmol) were placed in a three-necked flask under a nitrogen atmosphere. The mixture was heated and stirred at 180 ° C.
After completion of the reaction, the mixture was cooled to room temperature, diluted with 500 ml of hexane, and filtered using celite. The obtained filtrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 8: 2 as a developing solvent, and the obtained fraction was vacuum-dried. As a result, the target pale yellow oily liquid (intermediate 1a) was obtained in a yield of 427.5 g and a yield of 90%.
(中間体1bの合成)

(Synthesis of Intermediate 1b)
中間体1a(109.08g,480mmol)、脱水テトラヒドロフラン960mlを三口フラスコに入れ、-40℃に冷却した。この溶液に、メタンガスの発生に注意しつつ、メチルリチウムの3.0Mジエトキメタン溶液(504ml,1512mmol)を30分かけて滴下した。その後、この混合物を0℃で1時間撹拌し、次いで室温で一晩撹拌した。その後、0℃に冷却した混合物に、慎重にメタノールを滴下して残存メチルリチウムを失活させた。
この混合物を濃縮した後、ジクロロメタンで希釈して溶液を得た。この溶液を、分液ロートを用いて水洗し、無水硫酸マグネシウムで乾燥した。その後、溶液を濾過した後、濃縮し、真空乾燥(60℃、6h)を行ってオイル状液体を得た。このオイル状液体を三口フラスコに入れ、クロロホルム960ml、メタンスルホン酸62.3mlを加えて、窒素雰囲気下、3時間還流させた。
この混合物を室温まで冷却した後、炭酸水素ナトリウム水溶液を用いて慎重に中和し、分液ロートを用いてクロロホルムによる抽出を行った。得られた抽出物を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=1:1の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを濃縮して得た濃縮物をトルエンとヘキサンの混合溶媒(1:5ml/g)で再結晶した。これにより、目的物の中間体1bを収量47.9g、収率48%で得た。 Intermediate 1a (109.08 g, 480 mmol) and 960 ml of dehydrated tetrahydrofuran were placed in a three-necked flask and cooled to −40 ° C. To this solution, a 3.0M diethyloxymethane solution (504 ml, 1512 mmol) of methyllithium was added dropwise over 30 minutes while paying attention to the generation of methane gas. The mixture was then stirred at 0 ° C. for 1 hour and then at room temperature overnight. Thereafter, methanol was carefully added dropwise to the mixture cooled to 0 ° C. to deactivate the remaining methyllithium.
The mixture was concentrated and then diluted with dichloromethane to obtain a solution. This solution was washed with water using a separatory funnel and dried over anhydrous magnesium sulfate. Thereafter, the solution was filtered, concentrated, and vacuum dried (60 ° C., 6 h) to obtain an oily liquid. This oily liquid was put into a three-necked flask, 960 ml of chloroform and 62.3 ml of methanesulfonic acid were added, and the mixture was refluxed for 3 hours in a nitrogen atmosphere.
After cooling this mixture to room temperature, it neutralized carefully using sodium hydrogencarbonate aqueous solution, and extracted with chloroform using the separatory funnel. The obtained extract was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 1: 1 as a developing solvent. The concentrate obtained by concentrating the obtained fraction was recrystallized with a mixed solvent of toluene and hexane (1: 5 ml / g). As a result, the target intermediate 1b was obtained in a yield of 47.9 g and a yield of 48%.
この混合物を濃縮した後、ジクロロメタンで希釈して溶液を得た。この溶液を、分液ロートを用いて水洗し、無水硫酸マグネシウムで乾燥した。その後、溶液を濾過した後、濃縮し、真空乾燥(60℃、6h)を行ってオイル状液体を得た。このオイル状液体を三口フラスコに入れ、クロロホルム960ml、メタンスルホン酸62.3mlを加えて、窒素雰囲気下、3時間還流させた。
この混合物を室温まで冷却した後、炭酸水素ナトリウム水溶液を用いて慎重に中和し、分液ロートを用いてクロロホルムによる抽出を行った。得られた抽出物を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=1:1の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを濃縮して得た濃縮物をトルエンとヘキサンの混合溶媒(1:5ml/g)で再結晶した。これにより、目的物の中間体1bを収量47.9g、収率48%で得た。 Intermediate 1a (109.08 g, 480 mmol) and 960 ml of dehydrated tetrahydrofuran were placed in a three-necked flask and cooled to −40 ° C. To this solution, a 3.0M diethyloxymethane solution (504 ml, 1512 mmol) of methyllithium was added dropwise over 30 minutes while paying attention to the generation of methane gas. The mixture was then stirred at 0 ° C. for 1 hour and then at room temperature overnight. Thereafter, methanol was carefully added dropwise to the mixture cooled to 0 ° C. to deactivate the remaining methyllithium.
The mixture was concentrated and then diluted with dichloromethane to obtain a solution. This solution was washed with water using a separatory funnel and dried over anhydrous magnesium sulfate. Thereafter, the solution was filtered, concentrated, and vacuum dried (60 ° C., 6 h) to obtain an oily liquid. This oily liquid was put into a three-necked flask, 960 ml of chloroform and 62.3 ml of methanesulfonic acid were added, and the mixture was refluxed for 3 hours in a nitrogen atmosphere.
After cooling this mixture to room temperature, it neutralized carefully using sodium hydrogencarbonate aqueous solution, and extracted with chloroform using the separatory funnel. The obtained extract was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 1: 1 as a developing solvent. The concentrate obtained by concentrating the obtained fraction was recrystallized with a mixed solvent of toluene and hexane (1: 5 ml / g). As a result, the target intermediate 1b was obtained in a yield of 47.9 g and a yield of 48%.
(中間体2aの合成)

(Synthesis of Intermediate 2a)
中間体1a(38.18g,168mmol)、脱水テトラヒドロフラン336mlを三口フラスコに入れて0℃に冷却し、この溶液にフェニルリチウムの1.06Mシクロヘキサン-ジエチルエーテル溶液(500ml,529mmol)を30分かけて滴下した。その後、この混合物を0℃で2時間撹拌し、次いで室温で一晩撹拌した。その後、0℃に冷却した混合物に、慎重にメタノールを滴下して残存フェニルリチウムを失活させた。
この混合物を濃縮した後、ジクロロメタンで希釈して溶液を得た。この溶液を、分液ロートで水洗し、無水硫酸マグネシウムで乾燥した。その後、溶液を濾過した後、濃縮し、真空乾燥(60℃、6h)を行ってオイル状物質を得た。このオイル状物質を三口フラスコに入れ、クロロホルム672ml、メタンスルホン酸21.8mlを加えて、窒素雰囲気下、3時間還流させた。
この混合物を室温まで冷却した後、炭酸水素ナトリウム水溶液を用いて慎重に中和し、分液ロートを用いてジクロロメタンによる抽出を行った。この抽出物を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物をトルエンとヘキサンの混合溶媒(5:10ml/g)で再結晶した。これにより、目的物の中間体2aを収量27.9g、収率50%で得た。 Intermediate 1a (38.18 g, 168 mmol) and 336 ml of dehydrated tetrahydrofuran were placed in a three-necked flask and cooled to 0 ° C., and 1.06 M cyclohexane-diethyl ether solution of phenyl lithium (500 ml, 529 mmol) was added to this solution over 30 minutes. It was dripped. The mixture was then stirred at 0 ° C. for 2 hours and then at room temperature overnight. Thereafter, methanol was carefully added dropwise to the mixture cooled to 0 ° C. to deactivate the residual phenyllithium.
The mixture was concentrated and then diluted with dichloromethane to obtain a solution. This solution was washed with water using a separatory funnel and dried over anhydrous magnesium sulfate. Thereafter, the solution was filtered, concentrated, and vacuum dried (60 ° C., 6 h) to obtain an oily substance. This oily substance was put into a three-necked flask, 672 ml of chloroform and 21.8 ml of methanesulfonic acid were added, and the mixture was refluxed for 3 hours in a nitrogen atmosphere.
The mixture was cooled to room temperature, then carefully neutralized with an aqueous sodium hydrogen carbonate solution, and extracted with dichloromethane using a separatory funnel. The extract was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. This concentrate was recrystallized with a mixed solvent of toluene and hexane (5:10 ml / g). As a result, the target intermediate 2a was obtained in a yield of 27.9 g and a yield of 50%.
この混合物を濃縮した後、ジクロロメタンで希釈して溶液を得た。この溶液を、分液ロートで水洗し、無水硫酸マグネシウムで乾燥した。その後、溶液を濾過した後、濃縮し、真空乾燥(60℃、6h)を行ってオイル状物質を得た。このオイル状物質を三口フラスコに入れ、クロロホルム672ml、メタンスルホン酸21.8mlを加えて、窒素雰囲気下、3時間還流させた。
この混合物を室温まで冷却した後、炭酸水素ナトリウム水溶液を用いて慎重に中和し、分液ロートを用いてジクロロメタンによる抽出を行った。この抽出物を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物をトルエンとヘキサンの混合溶媒(5:10ml/g)で再結晶した。これにより、目的物の中間体2aを収量27.9g、収率50%で得た。 Intermediate 1a (38.18 g, 168 mmol) and 336 ml of dehydrated tetrahydrofuran were placed in a three-necked flask and cooled to 0 ° C., and 1.06 M cyclohexane-diethyl ether solution of phenyl lithium (500 ml, 529 mmol) was added to this solution over 30 minutes. It was dripped. The mixture was then stirred at 0 ° C. for 2 hours and then at room temperature overnight. Thereafter, methanol was carefully added dropwise to the mixture cooled to 0 ° C. to deactivate the residual phenyllithium.
The mixture was concentrated and then diluted with dichloromethane to obtain a solution. This solution was washed with water using a separatory funnel and dried over anhydrous magnesium sulfate. Thereafter, the solution was filtered, concentrated, and vacuum dried (60 ° C., 6 h) to obtain an oily substance. This oily substance was put into a three-necked flask, 672 ml of chloroform and 21.8 ml of methanesulfonic acid were added, and the mixture was refluxed for 3 hours in a nitrogen atmosphere.
The mixture was cooled to room temperature, then carefully neutralized with an aqueous sodium hydrogen carbonate solution, and extracted with dichloromethane using a separatory funnel. The extract was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. This concentrate was recrystallized with a mixed solvent of toluene and hexane (5:10 ml / g). As a result, the target intermediate 2a was obtained in a yield of 27.9 g and a yield of 50%.
(中間体3aの合成)

(Synthesis of Intermediate 3a)
1-ブロモ-2-フルオロ-4-ヨードベンゼン(25.3g,84mmol)、中間体1b(17.58g,84mmol)、ナトリウム-tert-ブトキシド(12.11g,126mmol)、ヨウ化銅(0.32g,1.68mmol)、trans-1,2-ジアミノシクロヘキサン(1.01ml,8.4mmol)、1,4-ジオキサン84mlを三口フラスコに入れ、窒素雰囲気下、24時間還流させた。
反応終了後、この混合物をトルエン200mlで希釈し、セライトで濾過した。得られたろ液をシリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:トルエン=6:4の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを濃縮して得た濃縮物をヘキサン(5ml/g,5℃)で再結晶した。これにより、目的物の中間体3aを収量19.68g、収率61%で得た。 1-bromo-2-fluoro-4-iodobenzene (25.3 g, 84 mmol), intermediate 1b (17.58 g, 84 mmol), sodium-tert-butoxide (12.11 g, 126 mmol), copper iodide (0. 32 g, 1.68 mmol), trans-1,2-diaminocyclohexane (1.01 ml, 8.4 mmol) and 84 ml of 1,4-dioxane were placed in a three-necked flask and refluxed for 24 hours under a nitrogen atmosphere.
After completion of the reaction, the mixture was diluted with 200 ml of toluene and filtered through celite. The resulting filtrate was filtered through a silica gel pad and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 6: 4 as a developing solvent. The concentrate obtained by concentrating the obtained fraction was recrystallized from hexane (5 ml / g, 5 ° C.). As a result, the target product Intermediate 3a was obtained in a yield of 19.68 g and a yield of 61%.
反応終了後、この混合物をトルエン200mlで希釈し、セライトで濾過した。得られたろ液をシリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:トルエン=6:4の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを濃縮して得た濃縮物をヘキサン(5ml/g,5℃)で再結晶した。これにより、目的物の中間体3aを収量19.68g、収率61%で得た。 1-bromo-2-fluoro-4-iodobenzene (25.3 g, 84 mmol), intermediate 1b (17.58 g, 84 mmol), sodium-tert-butoxide (12.11 g, 126 mmol), copper iodide (0. 32 g, 1.68 mmol), trans-1,2-diaminocyclohexane (1.01 ml, 8.4 mmol) and 84 ml of 1,4-dioxane were placed in a three-necked flask and refluxed for 24 hours under a nitrogen atmosphere.
After completion of the reaction, the mixture was diluted with 200 ml of toluene and filtered through celite. The resulting filtrate was filtered through a silica gel pad and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 6: 4 as a developing solvent. The concentrate obtained by concentrating the obtained fraction was recrystallized from hexane (5 ml / g, 5 ° C.). As a result, the target product Intermediate 3a was obtained in a yield of 19.68 g and a yield of 61%.
(中間体3bの合成)

(Synthesis of Intermediate 3b)
中間体3a(7.65g,20mmol)、水酸化カリウム(1.12g,20mmol)、炭酸カリウム(2.76g,20mmol)、トリエチレングリコールジメチルエーテル5mlを三口フラスコに入れ、窒素雰囲気下、190℃で48時間加熱撹拌した。反応終了後、この混合物を室温まで冷却した後、トルエン200mlを加えて希釈し、さらに水200mlを加えて超音波処理を10分行った。
この混合物を、セライトを用いて濾過し、ろ液を回収した。このろ液を分液ロートに移し、水洗を2回行い、有機相を回収した。得られた有機相を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:トルエン=6:4の混合溶媒を展開溶媒に用いて精製し、さらに得られたフラクションを濃縮して得た濃縮物をヘキサンで再結晶した。これにより、目的物の中間体3bを収量4.16g、収率56%で得た。 Intermediate 3a (7.65 g, 20 mmol), potassium hydroxide (1.12 g, 20 mmol), potassium carbonate (2.76 g, 20 mmol), and 5 ml of triethylene glycol dimethyl ether were placed in a three-necked flask at 190 ° C. under a nitrogen atmosphere. The mixture was heated and stirred for 48 hours. After completion of the reaction, the mixture was cooled to room temperature, diluted with 200 ml of toluene, and further sonicated with 200 ml of water for 10 minutes.
The mixture was filtered using celite, and the filtrate was collected. The filtrate was transferred to a separatory funnel and washed twice with water to recover the organic phase. The obtained organic phase was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 6: 4 as a developing solvent, and the resulting fraction was concentrated to recrystallize with hexane. As a result, 4.16 g of a target intermediate 3b was obtained in a yield of 56%.
この混合物を、セライトを用いて濾過し、ろ液を回収した。このろ液を分液ロートに移し、水洗を2回行い、有機相を回収した。得られた有機相を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:トルエン=6:4の混合溶媒を展開溶媒に用いて精製し、さらに得られたフラクションを濃縮して得た濃縮物をヘキサンで再結晶した。これにより、目的物の中間体3bを収量4.16g、収率56%で得た。 Intermediate 3a (7.65 g, 20 mmol), potassium hydroxide (1.12 g, 20 mmol), potassium carbonate (2.76 g, 20 mmol), and 5 ml of triethylene glycol dimethyl ether were placed in a three-necked flask at 190 ° C. under a nitrogen atmosphere. The mixture was heated and stirred for 48 hours. After completion of the reaction, the mixture was cooled to room temperature, diluted with 200 ml of toluene, and further sonicated with 200 ml of water for 10 minutes.
The mixture was filtered using celite, and the filtrate was collected. The filtrate was transferred to a separatory funnel and washed twice with water to recover the organic phase. The obtained organic phase was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 6: 4 as a developing solvent, and the resulting fraction was concentrated to recrystallize with hexane. As a result, 4.16 g of a target intermediate 3b was obtained in a yield of 56%.
(中間体3cの合成)

(Synthesis of Intermediate 3c)
2-ブロモ-1,3,5-トリイソプロピルベンゼン(43.9g,155mmol)、脱水テトラヒドロフラン155mlを三口フラスコに入れ、-78℃に冷却した。この溶液にn-ブチルリチウムの1.60Mヘキサン溶液(106.6ml,170.5mmol)をシリンジで加え、-78℃で10分撹拌した後、トリメトキシボラン(34.5ml,310mmol)を2分かけて滴下した。その後、この混合物を-78℃で30分撹拌した後、室温で一晩撹拌した。
この混合物をエバポレータで半分程度に濃縮し、1N塩酸水溶液300mlを加え、室温で4時間撹拌した。得られた溶液を分液ロートに入れ、ジクロロメタンによる抽出を行った。抽出物を、2回水洗した後、無水硫酸マグネシウムで乾燥し、濾過、濃縮した。この濃縮物を、モレキュラーシーブスを中に仕込んだソックスレー抽出器を取り付けた三口フラスコに入れ、トルエン200ml、メタノール30mlを加えて12時間還流させた。反応終了後、混合物を室温まで冷却し、エバポレータで濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=8:2の混合溶媒を展開溶媒に用いて精製した。得られたフラクションの溶媒を真空乾燥で除去し、目的物の無色オイル状物質(中間体3c)を収量31.88g、収率74%で得た。 2-Bromo-1,3,5-triisopropylbenzene (43.9 g, 155 mmol) and dehydrated tetrahydrofuran 155 ml were placed in a three-necked flask and cooled to -78 ° C. To this solution, a 1.60 M hexane solution of n-butyllithium (106.6 ml, 170.5 mmol) was added with a syringe and stirred at −78 ° C. for 10 minutes, and then trimethoxyborane (34.5 ml, 310 mmol) was added for 2 minutes. It was dripped over. The mixture was then stirred at −78 ° C. for 30 minutes and then at room temperature overnight.
The mixture was concentrated to about half by an evaporator, 300 ml of 1N hydrochloric acid aqueous solution was added, and the mixture was stirred at room temperature for 4 hours. The resulting solution was placed in a separatory funnel and extracted with dichloromethane. The extract was washed twice with water, dried over anhydrous magnesium sulfate, filtered and concentrated. This concentrate was put into a three-necked flask equipped with a Soxhlet extractor charged with molecular sieves, and 200 ml of toluene and 30 ml of methanol were added and refluxed for 12 hours. After completion of the reaction, the mixture was cooled to room temperature and concentrated with an evaporator. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 8: 2 as a developing solvent. The solvent of the obtained fraction was removed by vacuum drying, and the target colorless oily substance (intermediate 3c) was obtained in a yield of 31.88 g and a yield of 74%.
この混合物をエバポレータで半分程度に濃縮し、1N塩酸水溶液300mlを加え、室温で4時間撹拌した。得られた溶液を分液ロートに入れ、ジクロロメタンによる抽出を行った。抽出物を、2回水洗した後、無水硫酸マグネシウムで乾燥し、濾過、濃縮した。この濃縮物を、モレキュラーシーブスを中に仕込んだソックスレー抽出器を取り付けた三口フラスコに入れ、トルエン200ml、メタノール30mlを加えて12時間還流させた。反応終了後、混合物を室温まで冷却し、エバポレータで濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=8:2の混合溶媒を展開溶媒に用いて精製した。得られたフラクションの溶媒を真空乾燥で除去し、目的物の無色オイル状物質(中間体3c)を収量31.88g、収率74%で得た。 2-Bromo-1,3,5-triisopropylbenzene (43.9 g, 155 mmol) and dehydrated tetrahydrofuran 155 ml were placed in a three-necked flask and cooled to -78 ° C. To this solution, a 1.60 M hexane solution of n-butyllithium (106.6 ml, 170.5 mmol) was added with a syringe and stirred at −78 ° C. for 10 minutes, and then trimethoxyborane (34.5 ml, 310 mmol) was added for 2 minutes. It was dripped over. The mixture was then stirred at −78 ° C. for 30 minutes and then at room temperature overnight.
The mixture was concentrated to about half by an evaporator, 300 ml of 1N hydrochloric acid aqueous solution was added, and the mixture was stirred at room temperature for 4 hours. The resulting solution was placed in a separatory funnel and extracted with dichloromethane. The extract was washed twice with water, dried over anhydrous magnesium sulfate, filtered and concentrated. This concentrate was put into a three-necked flask equipped with a Soxhlet extractor charged with molecular sieves, and 200 ml of toluene and 30 ml of methanol were added and refluxed for 12 hours. After completion of the reaction, the mixture was cooled to room temperature and concentrated with an evaporator. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 8: 2 as a developing solvent. The solvent of the obtained fraction was removed by vacuum drying, and the target colorless oily substance (intermediate 3c) was obtained in a yield of 31.88 g and a yield of 74%.
(中間体4aの合成)
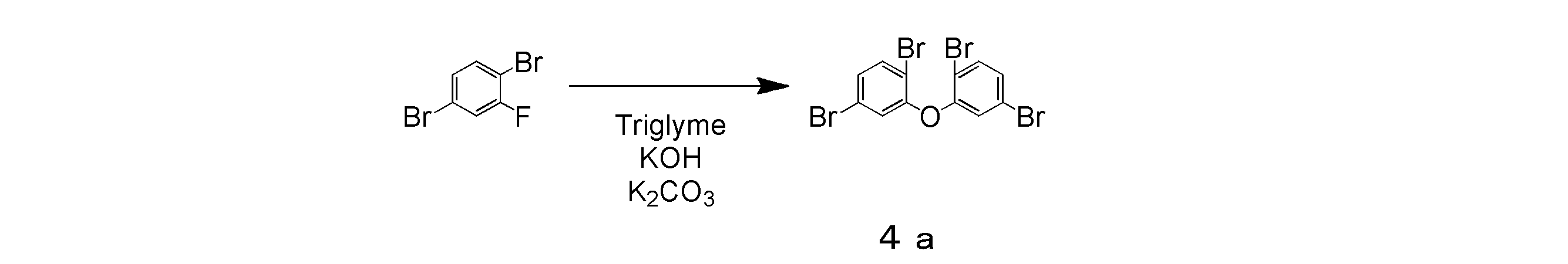
(Synthesis of Intermediate 4a)
1,4-ジブロモ-2-フルオロベンゼン(50.78g,200mmol)、水酸化カリウム(6.73g,120mmol)、炭酸カリウム(27.64g,200mmol)、トリエチレングリコールジメチルエーテル20mlを三口フラスコに入れ、窒素雰囲気下、180℃で96時間加熱撹拌した。反応終了後、混合物を室温まで冷却し、トルエン500mlを加えて希釈し、さらに水500mlを加えて超音波処理を10分行った。
この混合物を、セライトを用いて濾過し、ろ液を回収した。このろ液を分液ロートに移し、水洗を2回行い、有機相を回収した。得られた有機相を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=7:3の混合溶媒を展開溶媒に用いて精製し、得られたフラクションの濃縮物をヘキサンで再結晶した。これにより、目的物の中間体4aを収量8.51g、収率18%で得た。 1,4-dibromo-2-fluorobenzene (50.78 g, 200 mmol), potassium hydroxide (6.73 g, 120 mmol), potassium carbonate (27.64 g, 200 mmol), and 20 ml of triethylene glycol dimethyl ether were placed in a three-necked flask. The mixture was heated and stirred at 180 ° C. for 96 hours in a nitrogen atmosphere. After completion of the reaction, the mixture was cooled to room temperature, diluted by adding 500 ml of toluene, and further subjected to sonication for 10 minutes by adding 500 ml of water.
The mixture was filtered using celite, and the filtrate was collected. The filtrate was transferred to a separatory funnel and washed twice with water to recover the organic phase. The obtained organic phase was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. The concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 7: 3 as a developing solvent, and the resulting fraction concentrate was recrystallized from hexane. As a result, the intermediate product 4a was obtained in a yield of 8.51 g and a yield of 18%.
この混合物を、セライトを用いて濾過し、ろ液を回収した。このろ液を分液ロートに移し、水洗を2回行い、有機相を回収した。得られた有機相を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=7:3の混合溶媒を展開溶媒に用いて精製し、得られたフラクションの濃縮物をヘキサンで再結晶した。これにより、目的物の中間体4aを収量8.51g、収率18%で得た。 1,4-dibromo-2-fluorobenzene (50.78 g, 200 mmol), potassium hydroxide (6.73 g, 120 mmol), potassium carbonate (27.64 g, 200 mmol), and 20 ml of triethylene glycol dimethyl ether were placed in a three-necked flask. The mixture was heated and stirred at 180 ° C. for 96 hours in a nitrogen atmosphere. After completion of the reaction, the mixture was cooled to room temperature, diluted by adding 500 ml of toluene, and further subjected to sonication for 10 minutes by adding 500 ml of water.
The mixture was filtered using celite, and the filtrate was collected. The filtrate was transferred to a separatory funnel and washed twice with water to recover the organic phase. The obtained organic phase was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. The concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 7: 3 as a developing solvent, and the resulting fraction concentrate was recrystallized from hexane. As a result, the intermediate product 4a was obtained in a yield of 8.51 g and a yield of 18%.
(中間体4bの合成)
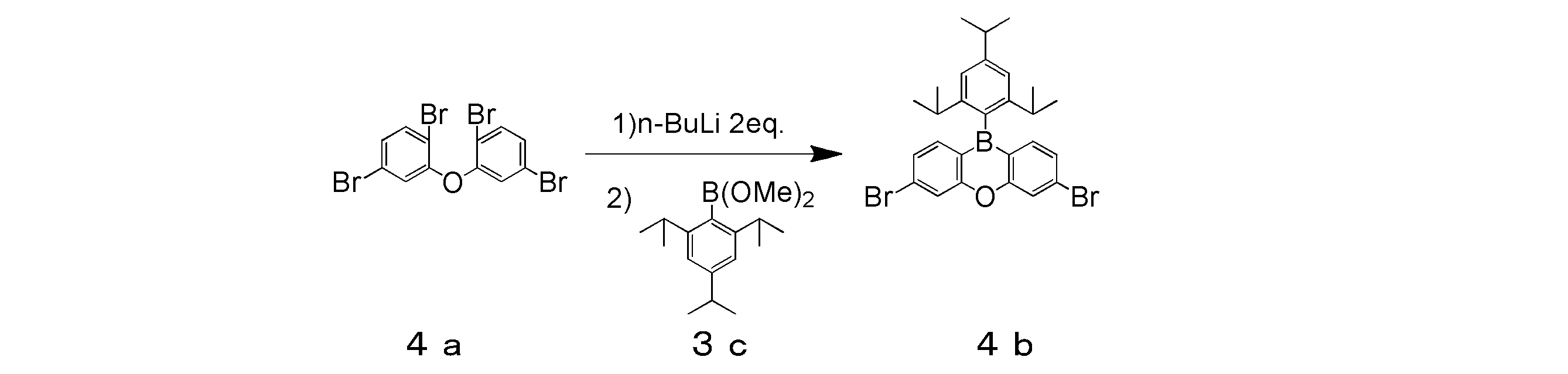
(Synthesis of Intermediate 4b)
中間体4a(2.91g,6mmol)、脱水ジエチルエーテル60mlを三口フラスコに入れて懸濁させ、0℃に冷却した。この溶液にn-ブチルリチウムの1.58Mヘキサン溶液(7.6ml,12mmol)をシリンジで加え、0℃で10分撹拌した後、中間体3c(1.66g,6mmol)をジエチルエーテル6mlに溶解した溶液を2分かけて滴下した。その後、この混合物を0℃で30分撹拌した後、室温で一晩撹拌した。
この混合物をエバポレータで濃縮し、得られた濃縮物をジクロロメタン100mlで希釈して溶液を得た。得られた溶液をシリカゲルパッドで濾過し、ろ液を濃縮した。濃縮によって析出した試料をヘキサンでリンスしてろ取し、真空乾燥(50℃、8時間)した。これにより、目的物の中間体4bを収量1.58g、収率49%で得た。 Intermediate 4a (2.91 g, 6 mmol) and 60 ml of dehydrated diethyl ether were suspended in a three-necked flask and cooled to 0 ° C. To this solution was added a 1.58M hexane solution (7.6 ml, 12 mmol) of n-butyllithium with a syringe, and the mixture was stirred at 0 ° C. for 10 minutes. Then, Intermediate 3c (1.66 g, 6 mmol) was dissolved in 6 ml of diethyl ether. The solution was added dropwise over 2 minutes. The mixture was then stirred at 0 ° C. for 30 minutes and then at room temperature overnight.
This mixture was concentrated by an evaporator, and the obtained concentrate was diluted with 100 ml of dichloromethane to obtain a solution. The resulting solution was filtered through a silica gel pad and the filtrate was concentrated. A sample precipitated by concentration was rinsed with hexane, collected by filtration, and vacuum-dried (50 ° C., 8 hours). As a result, the intermediate 4b as a target product was obtained in a yield of 1.58 g and a yield of 49%.
この混合物をエバポレータで濃縮し、得られた濃縮物をジクロロメタン100mlで希釈して溶液を得た。得られた溶液をシリカゲルパッドで濾過し、ろ液を濃縮した。濃縮によって析出した試料をヘキサンでリンスしてろ取し、真空乾燥(50℃、8時間)した。これにより、目的物の中間体4bを収量1.58g、収率49%で得た。 Intermediate 4a (2.91 g, 6 mmol) and 60 ml of dehydrated diethyl ether were suspended in a three-necked flask and cooled to 0 ° C. To this solution was added a 1.58M hexane solution (7.6 ml, 12 mmol) of n-butyllithium with a syringe, and the mixture was stirred at 0 ° C. for 10 minutes. Then, Intermediate 3c (1.66 g, 6 mmol) was dissolved in 6 ml of diethyl ether. The solution was added dropwise over 2 minutes. The mixture was then stirred at 0 ° C. for 30 minutes and then at room temperature overnight.
This mixture was concentrated by an evaporator, and the obtained concentrate was diluted with 100 ml of dichloromethane to obtain a solution. The resulting solution was filtered through a silica gel pad and the filtrate was concentrated. A sample precipitated by concentration was rinsed with hexane, collected by filtration, and vacuum-dried (50 ° C., 8 hours). As a result, the intermediate 4b as a target product was obtained in a yield of 1.58 g and a yield of 49%.
(中間体5aの合成)
(Synthesis of Intermediate 5a)
中間体3a(7.65g,20mmol)、2-ブロモフェノール(13.84g,80mmol)、炭酸カリウム(11.06g,80mmol)、トリエチレングリコールジメチルエーテル10mlを三口フラスコに入れ、窒素雰囲気下、190℃で72時間加熱撹拌した。反応終了後、混合物を室温まで冷却した後、トルエン200mlを加えて希釈し、さらに水200mlを加えて超音波処理を10分行った。
この混合物を、セライトを用いて濾過し、ろ液を回収した。このろ液を分液ロートに移し、水洗を2回行い、有機相を回収した。得られた有機相を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:トルエン=6:4の混合溶媒を展開溶媒に用いて精製し、得られたフラクションを濃縮して白色固体を得た。この白色固体をヘキサンでリンスした後、真空乾燥して目的物の中間体5aを収量9.40g、収率88%で得た。 Intermediate 3a (7.65 g, 20 mmol), 2-bromophenol (13.84 g, 80 mmol), potassium carbonate (11.06 g, 80 mmol), and 10 ml of triethylene glycol dimethyl ether were placed in a three-necked flask and treated at 190 ° C. under a nitrogen atmosphere. And stirred for 72 hours. After completion of the reaction, the mixture was cooled to room temperature, diluted with 200 ml of toluene, and further 200 ml of water was added and sonication was performed for 10 minutes.
The mixture was filtered using celite, and the filtrate was collected. The filtrate was transferred to a separatory funnel and washed twice with water to recover the organic phase. The obtained organic phase was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 6: 4 as a developing solvent, and the obtained fraction was concentrated to obtain a white solid. This white solid was rinsed with hexane and then vacuum-dried to obtain 9.40 g of the target intermediate 5a in a yield of 88%.
この混合物を、セライトを用いて濾過し、ろ液を回収した。このろ液を分液ロートに移し、水洗を2回行い、有機相を回収した。得られた有機相を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:トルエン=6:4の混合溶媒を展開溶媒に用いて精製し、得られたフラクションを濃縮して白色固体を得た。この白色固体をヘキサンでリンスした後、真空乾燥して目的物の中間体5aを収量9.40g、収率88%で得た。 Intermediate 3a (7.65 g, 20 mmol), 2-bromophenol (13.84 g, 80 mmol), potassium carbonate (11.06 g, 80 mmol), and 10 ml of triethylene glycol dimethyl ether were placed in a three-necked flask and treated at 190 ° C. under a nitrogen atmosphere. And stirred for 72 hours. After completion of the reaction, the mixture was cooled to room temperature, diluted with 200 ml of toluene, and further 200 ml of water was added and sonication was performed for 10 minutes.
The mixture was filtered using celite, and the filtrate was collected. The filtrate was transferred to a separatory funnel and washed twice with water to recover the organic phase. The obtained organic phase was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 6: 4 as a developing solvent, and the obtained fraction was concentrated to obtain a white solid. This white solid was rinsed with hexane and then vacuum-dried to obtain 9.40 g of the target intermediate 5a in a yield of 88%.
(中間体6aの合成)
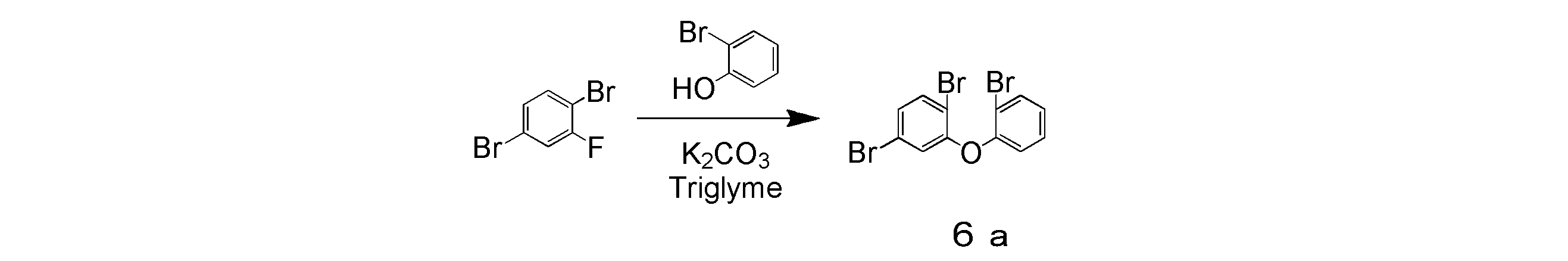
(Synthesis of Intermediate 6a)
2,5-ジブロモフルオロベンゼン(25.39g,100mmol)、2-ブロモフェノール(25.95g,150mmol)、炭酸カリウム(27.64g,200mmol)、トリエチレングリコールジメチルエーテル10mlを三口フラスコに入れ、窒素雰囲気下、190℃で72時間加熱撹拌した。反応終了後、この混合物を室温まで冷却した後、トルエン200mlを加えて希釈し、さらに水200mlを加えて超音波処理を10分行った。
この混合物を、セライトを用いて濾過し、ろ液を回収した。このろ液を分液ロートに移し、水洗を2回行い、有機相を回収した。得られた有機相を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=9:1の混合溶媒を展開溶媒に用いて精製し、濃縮した。これにより、目的物の白色固体(中間体6a)を収量50.53g、収率89%で得た。 2,5-dibromofluorobenzene (25.39 g, 100 mmol), 2-bromophenol (25.95 g, 150 mmol), potassium carbonate (27.64 g, 200 mmol), and 10 ml of triethylene glycol dimethyl ether were placed in a three-necked flask and a nitrogen atmosphere. Under stirring at 190 ° C. for 72 hours. After completion of the reaction, the mixture was cooled to room temperature, diluted with 200 ml of toluene, and further sonicated with 200 ml of water for 10 minutes.
The mixture was filtered using celite, and the filtrate was collected. The filtrate was transferred to a separatory funnel and washed twice with water to recover the organic phase. The obtained organic phase was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 9: 1 as a developing solvent, and concentrated. As a result, the target white solid (intermediate 6a) was obtained in a yield of 50.53 g and a yield of 89%.
この混合物を、セライトを用いて濾過し、ろ液を回収した。このろ液を分液ロートに移し、水洗を2回行い、有機相を回収した。得られた有機相を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=9:1の混合溶媒を展開溶媒に用いて精製し、濃縮した。これにより、目的物の白色固体(中間体6a)を収量50.53g、収率89%で得た。 2,5-dibromofluorobenzene (25.39 g, 100 mmol), 2-bromophenol (25.95 g, 150 mmol), potassium carbonate (27.64 g, 200 mmol), and 10 ml of triethylene glycol dimethyl ether were placed in a three-necked flask and a nitrogen atmosphere. Under stirring at 190 ° C. for 72 hours. After completion of the reaction, the mixture was cooled to room temperature, diluted with 200 ml of toluene, and further sonicated with 200 ml of water for 10 minutes.
The mixture was filtered using celite, and the filtrate was collected. The filtrate was transferred to a separatory funnel and washed twice with water to recover the organic phase. The obtained organic phase was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 9: 1 as a developing solvent, and concentrated. As a result, the target white solid (intermediate 6a) was obtained in a yield of 50.53 g and a yield of 89%.
(中間体6bの合成)

(Synthesis of Intermediate 6b)
中間体6a(32.55g,80mmol)、脱水ジエチルエーテル400mlを三口フラスコに入れて溶解させ、0℃に冷却した。この溶液にn-ブチルリチウムの1.60Mヘキサン溶液(102.5ml,164mmol)をシリンジで加え、0℃で10分撹拌した。その後、この混合物に、中間体3c(22.10g,80mmol)をジエチルエーテル40mlに溶解した溶液を5分かけて滴下し、0℃で30分撹拌した後、室温で一晩撹拌した。
この混合物をエバポレータで濃縮し、トルエン500mlで希釈して溶液を得た。この溶液を、分液ロートで2回水洗し、無水硫酸マグネシウムで乾燥、濾過した後、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=9:1の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを、ヘキサンを用いて5℃で再結晶した。これにより、目的物の中間体6bを収量16.51g、収率45%で得た。
(中間体7aの合成)

Intermediate 6a (32.55 g, 80 mmol) and dehydrated diethyl ether 400 ml were dissolved in a three-necked flask and cooled to 0 ° C. To this solution, a 1.60 M hexane solution of n-butyllithium (102.5 ml, 164 mmol) was added by syringe, and the mixture was stirred at 0 ° C. for 10 minutes. Thereafter, a solution of Intermediate 3c (22.10 g, 80 mmol) dissolved in 40 ml of diethyl ether was added dropwise to this mixture over 5 minutes, stirred at 0 ° C. for 30 minutes, and then stirred overnight at room temperature.
This mixture was concentrated by an evaporator and diluted with 500 ml of toluene to obtain a solution. This solution was washed twice with a separatory funnel, dried over anhydrous magnesium sulfate, filtered, and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 9: 1 as a developing solvent. The obtained fraction was recrystallized at 5 ° C. using hexane. As a result, the intermediate product 6b was obtained in a yield of 16.51 g and a yield of 45%.
(Synthesis of Intermediate 7a)
この混合物をエバポレータで濃縮し、トルエン500mlで希釈して溶液を得た。この溶液を、分液ロートで2回水洗し、無水硫酸マグネシウムで乾燥、濾過した後、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=9:1の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを、ヘキサンを用いて5℃で再結晶した。これにより、目的物の中間体6bを収量16.51g、収率45%で得た。
(中間体7aの合成)
This mixture was concentrated by an evaporator and diluted with 500 ml of toluene to obtain a solution. This solution was washed twice with a separatory funnel, dried over anhydrous magnesium sulfate, filtered, and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 9: 1 as a developing solvent. The obtained fraction was recrystallized at 5 ° C. using hexane. As a result, the intermediate product 6b was obtained in a yield of 16.51 g and a yield of 45%.
(Synthesis of Intermediate 7a)
2,5-ジブロモフルオロベンゼン(15.23g,60mmol)、2-ブロモベンゼンチオール(9.45g,50mmol)、炭酸カリウム(13.82g,100mmol)、トリエチレングリコールジメチルエーテル10mlを三口フラスコに入れ、窒素雰囲気下、180℃で24時間加熱撹拌した。反応終了後、この混合物を室温まで冷却した後、トルエン100mlを加えて希釈し、さらに水100mlを加えて超音波処理を10分行った。
この混合物を、セライトを用いて濾過し、ろ液を回収した。このろ液を分液ロートに移し、水洗を2回行い、有機相を回収した。得られた有機相を無水硫酸マグネシウムで乾燥した後、濾過、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサンを展開溶媒に用いて精製した。得られたフラクションを濃縮、真空乾燥して目的物の無色オイル状物質(中間体7a)を収量14.01g、収率66%で得た。 2,5-dibromofluorobenzene (15.23 g, 60 mmol), 2-bromobenzenethiol (9.45 g, 50 mmol), potassium carbonate (13.82 g, 100 mmol), and 10 ml of triethylene glycol dimethyl ether were placed in a three-necked flask, and nitrogen was added. The mixture was heated and stirred at 180 ° C. for 24 hours in an atmosphere. After completion of the reaction, the mixture was cooled to room temperature, diluted with 100 ml of toluene, and further sonicated with 100 ml of water for 10 minutes.
The mixture was filtered using celite, and the filtrate was collected. The filtrate was transferred to a separatory funnel and washed twice with water to recover the organic phase. The obtained organic phase was dried over anhydrous magnesium sulfate, filtered and concentrated. This concentrate was purified by silica gel chromatography using hexane as a developing solvent. The obtained fraction was concentrated and vacuum-dried to obtain the desired colorless oily substance (intermediate 7a) in a yield of 14.01 g and a yield of 66%.
この混合物を、セライトを用いて濾過し、ろ液を回収した。このろ液を分液ロートに移し、水洗を2回行い、有機相を回収した。得られた有機相を無水硫酸マグネシウムで乾燥した後、濾過、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサンを展開溶媒に用いて精製した。得られたフラクションを濃縮、真空乾燥して目的物の無色オイル状物質(中間体7a)を収量14.01g、収率66%で得た。 2,5-dibromofluorobenzene (15.23 g, 60 mmol), 2-bromobenzenethiol (9.45 g, 50 mmol), potassium carbonate (13.82 g, 100 mmol), and 10 ml of triethylene glycol dimethyl ether were placed in a three-necked flask, and nitrogen was added. The mixture was heated and stirred at 180 ° C. for 24 hours in an atmosphere. After completion of the reaction, the mixture was cooled to room temperature, diluted with 100 ml of toluene, and further sonicated with 100 ml of water for 10 minutes.
The mixture was filtered using celite, and the filtrate was collected. The filtrate was transferred to a separatory funnel and washed twice with water to recover the organic phase. The obtained organic phase was dried over anhydrous magnesium sulfate, filtered and concentrated. This concentrate was purified by silica gel chromatography using hexane as a developing solvent. The obtained fraction was concentrated and vacuum-dried to obtain the desired colorless oily substance (intermediate 7a) in a yield of 14.01 g and a yield of 66%.
(中間体7bの合成)

中間体6aの代わりに中間体7aを用い、各原料の量をモル換算で0.41倍にした以外は中間体6bの合成方法と同様にして中間体7bを合成した。得られた中間体7bは収量2.49g、収率16%であった。
(Synthesis of Intermediate 7b)
Intermediate 7b was synthesized in the same manner as intermediate 6b except that intermediate 7a was used instead of intermediate 6a and the amount of each raw material was 0.41 times in terms of mole. The obtained intermediate 7b was 2.49 g in yield and 16% in yield.
(中間体8aの合成)
(Synthesis of Intermediate 8a)
2-ブロモ-4-メチルアニリン(50.23g,270mmol)、4-ヨードトルエン(64.75g,297mmol)、ナトリウム-tert-ブトキシド(38.92g,405mmol)、酢酸パラジウム(606mg,2.7mmol)、1,1‘-ビス(ジフェニルホスフィノ)フェロセン(1.50g,2.7mmol)、脱水トルエン540mlを三口フラスコに入れ、窒素雰囲気下で8時間還流撹拌した。
反応終了後、混合物を室温まで冷却した後、トルエン500mlを加えて希釈し、得られた溶液を、セライトを用いて濾過し濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=9:1の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを濃縮し、真空乾燥して目的物の無色オイル状液体(中間体8a)を収量54.85g、収率74%で得た。 2-Bromo-4-methylaniline (50.23 g, 270 mmol), 4-iodotoluene (64.75 g, 297 mmol), sodium tert-butoxide (38.92 g, 405 mmol), palladium acetate (606 mg, 2.7 mmol) 1,1′-bis (diphenylphosphino) ferrocene (1.50 g, 2.7 mmol) and 540 ml of dehydrated toluene were placed in a three-necked flask and stirred at reflux under a nitrogen atmosphere for 8 hours.
After completion of the reaction, the mixture was cooled to room temperature, diluted with 500 ml of toluene, and the resulting solution was filtered using celite and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 9: 1 as a developing solvent. The obtained fraction was concentrated and vacuum-dried to obtain the desired colorless oily liquid (intermediate 8a) in a yield of 54.85 g and a yield of 74%.
反応終了後、混合物を室温まで冷却した後、トルエン500mlを加えて希釈し、得られた溶液を、セライトを用いて濾過し濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサン:ジクロロメタン=9:1の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを濃縮し、真空乾燥して目的物の無色オイル状液体(中間体8a)を収量54.85g、収率74%で得た。 2-Bromo-4-methylaniline (50.23 g, 270 mmol), 4-iodotoluene (64.75 g, 297 mmol), sodium tert-butoxide (38.92 g, 405 mmol), palladium acetate (606 mg, 2.7 mmol) 1,1′-bis (diphenylphosphino) ferrocene (1.50 g, 2.7 mmol) and 540 ml of dehydrated toluene were placed in a three-necked flask and stirred at reflux under a nitrogen atmosphere for 8 hours.
After completion of the reaction, the mixture was cooled to room temperature, diluted with 500 ml of toluene, and the resulting solution was filtered using celite and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: dichloromethane = 9: 1 as a developing solvent. The obtained fraction was concentrated and vacuum-dried to obtain the desired colorless oily liquid (intermediate 8a) in a yield of 54.85 g and a yield of 74%.
(中間体8bの合成)

(Synthesis of Intermediate 8b)
中間体8a(16.57g,60mmol)、脱水テトラヒドロフラン240mlを三口フラスコに入れて-78℃に冷却し、この溶液にn-ブチルリチウムの2.65Mヘキサン溶液(47.5ml,126mmol)をシリンジで加えた。その後、反応溶液を0℃で50分撹拌した。その後、この反応溶液に、キサントンの粉末(12.95g,66mmol)を加え、0℃で30分撹拌した後、室温で2時間撹拌した。
反応終了後、反応溶液に少量の水を加えて失活させ、分液ロートを用いてジクロロメタンによる抽出を行った。得られた抽出物を、無水硫酸マグネシウムで乾燥、濾過した後、濃縮して反応粗物を得た。
この反応粗物を三口フラスコに入れ、クロロホルム600ml、メタンスルホン酸3.9mlを加えて、窒素雰囲気下、3時間還流させた。この混合物を、室温まで冷却した後、分液ロートを用いて水洗した。水洗した混合物を、無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮して析出物を得た。この析出物をエタノールで再結晶して目的物の中間体8bを収量10.2g、収率45%で得た。 Intermediate 8a (16.57 g, 60 mmol) and 240 ml of dehydrated tetrahydrofuran were placed in a three-necked flask and cooled to −78 ° C., and a 2.65 M hexane solution (47.5 ml, 126 mmol) of n-butyllithium was added to this solution with a syringe. added. Thereafter, the reaction solution was stirred at 0 ° C. for 50 minutes. Thereafter, xanthone powder (12.95 g, 66 mmol) was added to the reaction solution, stirred at 0 ° C. for 30 minutes, and then stirred at room temperature for 2 hours.
After completion of the reaction, the reaction solution was deactivated by adding a small amount of water, and extracted with dichloromethane using a separatory funnel. The obtained extract was dried over anhydrous magnesium sulfate, filtered, and concentrated to obtain a reaction crude product.
This crude reaction product was placed in a three-necked flask, 600 ml of chloroform and 3.9 ml of methanesulfonic acid were added, and the mixture was refluxed for 3 hours in a nitrogen atmosphere. The mixture was cooled to room temperature and washed with water using a separatory funnel. The washed mixture was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated to obtain a precipitate. This precipitate was recrystallized with ethanol to obtain 10.2 g of a target intermediate 8b in a yield of 45%.
反応終了後、反応溶液に少量の水を加えて失活させ、分液ロートを用いてジクロロメタンによる抽出を行った。得られた抽出物を、無水硫酸マグネシウムで乾燥、濾過した後、濃縮して反応粗物を得た。
この反応粗物を三口フラスコに入れ、クロロホルム600ml、メタンスルホン酸3.9mlを加えて、窒素雰囲気下、3時間還流させた。この混合物を、室温まで冷却した後、分液ロートを用いて水洗した。水洗した混合物を、無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮して析出物を得た。この析出物をエタノールで再結晶して目的物の中間体8bを収量10.2g、収率45%で得た。 Intermediate 8a (16.57 g, 60 mmol) and 240 ml of dehydrated tetrahydrofuran were placed in a three-necked flask and cooled to −78 ° C., and a 2.65 M hexane solution (47.5 ml, 126 mmol) of n-butyllithium was added to this solution with a syringe. added. Thereafter, the reaction solution was stirred at 0 ° C. for 50 minutes. Thereafter, xanthone powder (12.95 g, 66 mmol) was added to the reaction solution, stirred at 0 ° C. for 30 minutes, and then stirred at room temperature for 2 hours.
After completion of the reaction, the reaction solution was deactivated by adding a small amount of water, and extracted with dichloromethane using a separatory funnel. The obtained extract was dried over anhydrous magnesium sulfate, filtered, and concentrated to obtain a reaction crude product.
This crude reaction product was placed in a three-necked flask, 600 ml of chloroform and 3.9 ml of methanesulfonic acid were added, and the mixture was refluxed for 3 hours in a nitrogen atmosphere. The mixture was cooled to room temperature and washed with water using a separatory funnel. The washed mixture was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated to obtain a precipitate. This precipitate was recrystallized with ethanol to obtain 10.2 g of a target intermediate 8b in a yield of 45%.
(中間体9aの合成)

原料として2-ブロモアニリン(50.06g,291mmol)と4-ヨードベンゼン(62.43g,306mmol)を用いた以外は、中間体8aの合成方法と同様にして中間体9aを合成した。得られた中間体9aは、収量71.38g、収率99%であった。
(Synthesis of Intermediate 9a)
Intermediate 9a was synthesized in the same manner as Intermediate 8a except that 2-bromoaniline (50.06 g, 291 mmol) and 4-iodobenzene (62.43 g, 306 mmol) were used as raw materials. The obtained intermediate 9a was 71.38 g in yield and 99% in yield.
(中間体9bの合成)

原料として中間体9a(24.81g,100mmol)、フルオレノン(19.82g,110mmol)を用いた以外は中間体8bの合成方法と同様にして中間体9bを合成した。得られた中間体9bは、収量13.74g、収率41%であった。
(Synthesis of Intermediate 9b)
Intermediate 9b was synthesized in the same manner as intermediate 8b except that intermediate 9a (24.81 g, 100 mmol) and fluorenone (19.82 g, 110 mmol) were used as raw materials. The obtained intermediate 9b was 13.74 g in yield and 41% in yield.
(中間体10aの合成)

原料として中間体9a(12.9g,52mmol)を用い、フルオレノンの代わりにキサントン(11.22g,57.2mmol)を用いた以外は、中間体9bの合成方法と同様にして中間体10aを合成した。得られた中間体10aは、収量13.45g、収率74%であった。
(Synthesis of Intermediate 10a)
Intermediate 10a was synthesized in the same manner as Intermediate 9b, except that intermediate 9a (12.9 g, 52 mmol) was used as a raw material, and xanthone (11.22 g, 57.2 mmol) was used instead of fluorenone. did. The obtained intermediate 10a was 13.45 g in yield and 74% in yield.
(中間体11aの合成)

(Synthesis of Intermediate 11a)
ジフェニルエーテル(17.02g,100mmol)、FeCl3(0.81g,5mmol)、ニトロベンゼン100mlを三口フラスコに入れ、0℃に冷却した後、この混合物に臭素(21.5ml,420mmol)を15分かけて滴下した。この混合物を、0℃で30分撹拌した後、室温で1時間撹拌し、さらに120℃で24時間加熱撹拌した。反応終了後、この混合物を室温まで冷却し、ジクロロメタン200mlで希釈した。得られた溶液を分液ロートで水洗した後、チオ硫酸ナトリウム/水酸化ナトリウム水溶液で残存臭素を失活させた。この溶液を無水硫酸マグネシウムで乾燥した後、シリカゲルパッドで濾過し、濃縮した。得られた濃縮物を、シリカゲルクロマトグラフィーにより、ヘキサンを展開溶媒に用いて精製した。得られたフラクションの濃縮物をヘキサンで再結晶した。これにより、目的物の中間体11aを収量38.29g、収率79%で得た。
Diphenyl ether (17.02 g, 100 mmol), FeCl 3 (0.81 g, 5 mmol), and 100 ml of nitrobenzene were placed in a three-necked flask, cooled to 0 ° C., and bromine (21.5 ml, 420 mmol) was added to the mixture over 15 minutes. It was dripped. The mixture was stirred at 0 ° C. for 30 minutes, then stirred at room temperature for 1 hour, and further heated and stirred at 120 ° C. for 24 hours. After completion of the reaction, the mixture was cooled to room temperature and diluted with 200 ml of dichloromethane. The resulting solution was washed with a separatory funnel, and then residual bromine was deactivated with a sodium thiosulfate / sodium hydroxide aqueous solution. The solution was dried over anhydrous magnesium sulfate, filtered through a silica gel pad, and concentrated. The obtained concentrate was purified by silica gel chromatography using hexane as a developing solvent. The resulting fraction concentrate was recrystallized from hexane. As a result, the target intermediate 11a was obtained in a yield of 38.29 g and a yield of 79%.
(中間体11bの合成)

(Synthesis of Intermediate 11b)
原料として中間体11a(14.57g,30mmol)を用いた以外は、後述する化合物5の合成方法Aと同様にして中間体11bを合成した。得られた中間体11bは、収量6.27g、収率39%であった。
Intermediate 11b was synthesized in the same manner as Compound 5 Synthesis Method A described later, except that Intermediate 11a (14.57 g, 30 mmol) was used as a raw material. The obtained intermediate 11b was 6.27 g in yield and 39% in yield.
(中間体12aの合成)
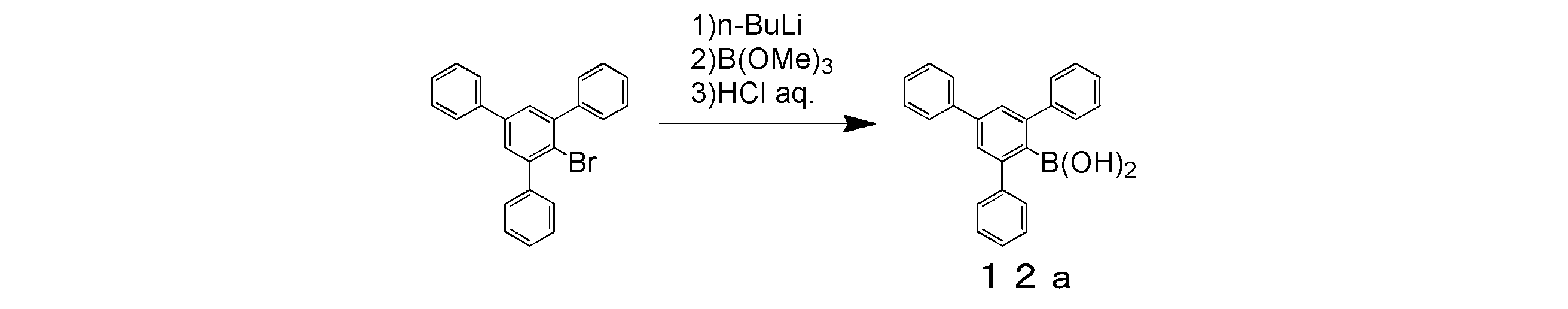
(Synthesis of Intermediate 12a)
2,4,6-トリフェニルブロモベンゼン(25.5g,66mmol)、脱水テトラヒドロフラン264mlを三口フラスコに入れ、-80℃に冷却し、この溶液にn-ブチルリチウムの1.60Mヘキサン溶液(45.4ml,72.6mmol)をシリンジで加えた。その後、反応溶液を-80℃で15分撹拌し、その後、トリメトキシボラン(14.7ml,132mmol)をシリンジで加え、室温で一晩撹拌した。この反応溶液に、少量の水を加えて反応系を失活させた後、1N塩酸水溶液400mlを加え、一晩撹拌した。この反応溶液を、分液ロートを用いてジクロロメタンで抽出し、水洗した。水洗した反応溶液を、無水硫酸マグネシウムで乾燥した後、濾過、濃縮した。この濃縮物を、シリカゲルクロマトグラフィーにより、ジクロロメタン:酢酸エチル=4:1の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを濃縮して得た濃縮物にヘキサン200mlを加えて析出物を得た。得られた析出物を、ろ取、真空乾燥(50℃、6時間)して目的物の中間体12aを収量15.9g、収率69%で得た。
2,4,6-Triphenylbromobenzene (25.5 g, 66 mmol) and 264 ml of dehydrated tetrahydrofuran were placed in a three-necked flask, cooled to −80 ° C., and 1.60 M hexane solution (45. 4 ml, 72.6 mmol) was added via syringe. Thereafter, the reaction solution was stirred at −80 ° C. for 15 minutes, and then trimethoxyborane (14.7 ml, 132 mmol) was added by syringe and stirred overnight at room temperature. A small amount of water was added to the reaction solution to inactivate the reaction system, and then 400 ml of a 1N hydrochloric acid aqueous solution was added and stirred overnight. The reaction solution was extracted with dichloromethane using a separatory funnel and washed with water. The reaction solution washed with water was dried over anhydrous magnesium sulfate, filtered and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of dichloromethane: ethyl acetate = 4: 1 as a developing solvent. 200 ml of hexane was added to the concentrate obtained by concentrating the obtained fraction to obtain a precipitate. The obtained precipitate was collected by filtration and dried in vacuo (50 ° C., 6 hours) to obtain the target intermediate 12a in a yield of 15.9 g and a yield of 69%.
(中間体12bの合成)

中間体12a(15.76g,45mmol)を、モレキュラーシーブスを中に仕込んだソックスレー抽出器を取り付けた三口フラスコに入れ、トルエン200ml、メタノール30mlを加えて12時間還流させた。反応終了後、この混合物を室温まで冷却した後、エバポレータで濃縮し、さらに真空乾燥した。これにより、目的物の無色オイル状物質(中間体12b)を得た。
(Synthesis of Intermediate 12b)
Intermediate 12a (15.76 g, 45 mmol) was placed in a three-necked flask equipped with a Soxhlet extractor charged with molecular sieves, and 200 ml of toluene and 30 ml of methanol were added and refluxed for 12 hours. After completion of the reaction, the mixture was cooled to room temperature, concentrated with an evaporator, and further dried under vacuum. As a result, a colorless oily substance (intermediate 12b) as a target product was obtained.
(中間体12cの合成)

原料として中間体6a(10.74g,26.4mmol)、中間体12b(10.0g,26.4mmol)を用いた以外は、中間体6bの合成方法と同様にして中間体12cを合成した。得られた中間体12cは、収量3.53g、収率24%であった。
(Synthesis of Intermediate 12c)
Intermediate 12c was synthesized in the same manner as Intermediate 6b, except that Intermediate 6a (10.74 g, 26.4 mmol) and Intermediate 12b (10.0 g, 26.4 mmol) were used as raw materials. The obtained intermediate 12c was 3.53 g in yield and 24% in yield.
[化合物の合成例]
(合成例1) 化合物5の合成(合成方法A)
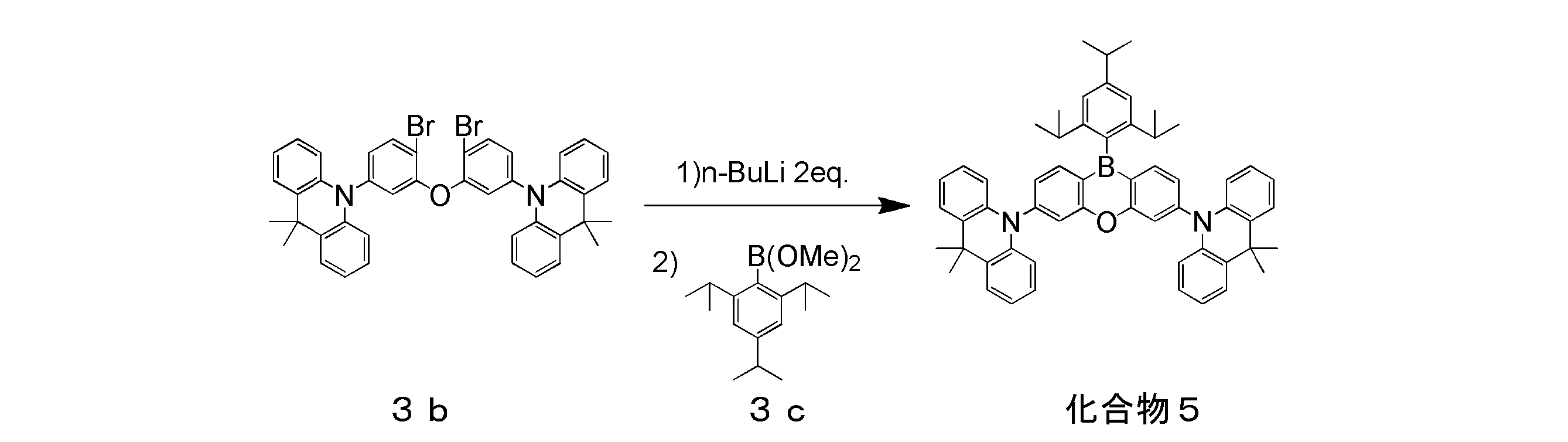
[Examples of compound synthesis]
Synthesis Example 1 Synthesis of Compound 5 (Synthesis Method A)
(合成例1) 化合物5の合成(合成方法A)
Synthesis Example 1 Synthesis of Compound 5 (Synthesis Method A)
中間体3b(5.56g,7.5mmol)、脱水ジエチルエーテル150mlを三口フラスコに入れて懸濁させ、0℃に冷却した。この溶液にn-ブチルリチウムの1.58Mヘキサン溶液(9.5ml,15mmol)をシリンジで加えた。この混合物を0℃で10分撹拌した後、中間体3c(2.07g,7.5mmol)をジエチルエーテル15mlに溶解した溶液を10分かけて滴下した。その後、この混合物を0℃で30分撹拌した後、室温で一晩撹拌した。
この混合物を、エバポレータで濃縮し、トルエン:ヘキサン=1:1の混合溶媒200mlで希釈して溶液を得た。得られた溶液をシリカゲルパッドで濾過し、ろ液を濃縮した。このろ液をシリカゲルクロマトグラフィーにより、ヘキサン:トルエン=6:4の混合溶媒を展開溶媒に用いて精製した。得られたフラクションの濃縮物をヘキサンで再結晶して目的物の化合物5を収量2.20g、収率37%で得た。 Intermediate 3b (5.56 g, 7.5 mmol) and 150 ml of dehydrated diethyl ether were suspended in a three-necked flask and cooled to 0 ° C. To this solution, a 1.58 M hexane solution of n-butyllithium (9.5 ml, 15 mmol) was added by syringe. The mixture was stirred at 0 ° C. for 10 minutes, and then a solution of intermediate 3c (2.07 g, 7.5 mmol) dissolved in 15 ml of diethyl ether was added dropwise over 10 minutes. The mixture was then stirred at 0 ° C. for 30 minutes and then at room temperature overnight.
This mixture was concentrated by an evaporator and diluted with 200 ml of a mixed solvent of toluene: hexane = 1: 1 to obtain a solution. The resulting solution was filtered through a silica gel pad and the filtrate was concentrated. This filtrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 6: 4 as a developing solvent. The concentrate of the obtained fraction was recrystallized with hexane to obtain 2.20 g of thetarget compound 5 in a yield of 37%.
この混合物を、エバポレータで濃縮し、トルエン:ヘキサン=1:1の混合溶媒200mlで希釈して溶液を得た。得られた溶液をシリカゲルパッドで濾過し、ろ液を濃縮した。このろ液をシリカゲルクロマトグラフィーにより、ヘキサン:トルエン=6:4の混合溶媒を展開溶媒に用いて精製した。得られたフラクションの濃縮物をヘキサンで再結晶して目的物の化合物5を収量2.20g、収率37%で得た。 Intermediate 3b (5.56 g, 7.5 mmol) and 150 ml of dehydrated diethyl ether were suspended in a three-necked flask and cooled to 0 ° C. To this solution, a 1.58 M hexane solution of n-butyllithium (9.5 ml, 15 mmol) was added by syringe. The mixture was stirred at 0 ° C. for 10 minutes, and then a solution of intermediate 3c (2.07 g, 7.5 mmol) dissolved in 15 ml of diethyl ether was added dropwise over 10 minutes. The mixture was then stirred at 0 ° C. for 30 minutes and then at room temperature overnight.
This mixture was concentrated by an evaporator and diluted with 200 ml of a mixed solvent of toluene: hexane = 1: 1 to obtain a solution. The resulting solution was filtered through a silica gel pad and the filtrate was concentrated. This filtrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 6: 4 as a developing solvent. The concentrate of the obtained fraction was recrystallized with hexane to obtain 2.20 g of the
(合成例2) 化合物5の合成(合成方法B)
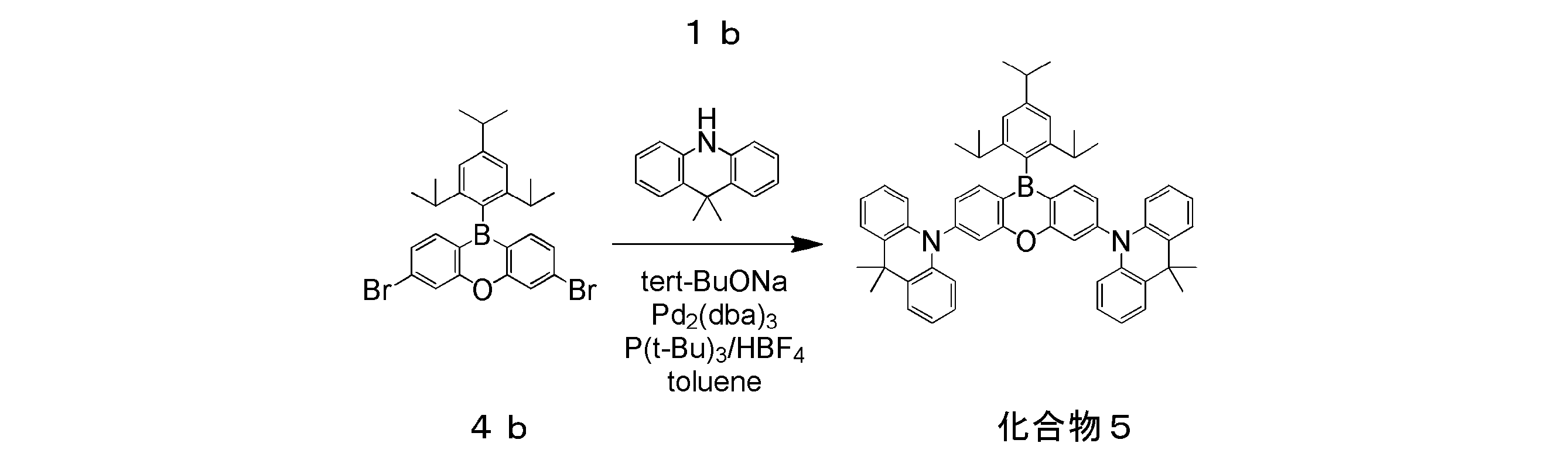
Synthesis Example 2 Synthesis of Compound 5 (Synthesis Method B)
中間体4b(1.58g,2.93mmol)、中間体1b(1.47g,7.03mmol)、ナトリウム-tert-ブトキシド(0.84g,8.79mmol)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(54mg,59μmol)、トリ-tert-ブチルホスホニウムテトラフルオロボラート(68mg,234μmol)、脱水トルエン15mlを三口フラスコに入れ、窒素雰囲気下で4時間還流撹拌した。
反応終了後、この混合物を室温まで冷却した後、トルエン50mlを加えて希釈した。得られた溶液を、セライトを用いて濾過し濃縮した。この濃縮物をシリカゲルクロマトグラフィーにより、ヘキサン:トルエン=6:4の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを濃縮して得た濃縮物にアセトニトリル30mlを加え、超音波処理30分を行った。この混合物から析出物をろ取し、アセトニトリルでリンスし、真空乾燥(50℃、6時間)して目的物の化合物6を収量2.14g、収率92%で得た。 Intermediate 4b (1.58 g, 2.93 mmol), Intermediate 1b (1.47 g, 7.03 mmol), Sodium tert-butoxide (0.84 g, 8.79 mmol), Tris (dibenzylideneacetone) dipalladium ( 0) (54 mg, 59 μmol), tri-tert-butylphosphonium tetrafluoroborate (68 mg, 234 μmol), and 15 ml of dehydrated toluene were placed in a three-necked flask and stirred at reflux under a nitrogen atmosphere for 4 hours.
After completion of the reaction, the mixture was cooled to room temperature and diluted with 50 ml of toluene. The resulting solution was filtered using celite and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 6: 4 as a developing solvent. 30 ml of acetonitrile was added to the concentrate obtained by concentrating the obtained fraction, and sonication was performed for 30 minutes. A precipitate was collected from this mixture by filtration, rinsed with acetonitrile, and vacuum-dried (50 ° C., 6 hours) to obtain 2.14 g of the target compound in a yield of 92%.
反応終了後、この混合物を室温まで冷却した後、トルエン50mlを加えて希釈した。得られた溶液を、セライトを用いて濾過し濃縮した。この濃縮物をシリカゲルクロマトグラフィーにより、ヘキサン:トルエン=6:4の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを濃縮して得た濃縮物にアセトニトリル30mlを加え、超音波処理30分を行った。この混合物から析出物をろ取し、アセトニトリルでリンスし、真空乾燥(50℃、6時間)して目的物の化合物6を収量2.14g、収率92%で得た。 Intermediate 4b (1.58 g, 2.93 mmol), Intermediate 1b (1.47 g, 7.03 mmol), Sodium tert-butoxide (0.84 g, 8.79 mmol), Tris (dibenzylideneacetone) dipalladium ( 0) (54 mg, 59 μmol), tri-tert-butylphosphonium tetrafluoroborate (68 mg, 234 μmol), and 15 ml of dehydrated toluene were placed in a three-necked flask and stirred at reflux under a nitrogen atmosphere for 4 hours.
After completion of the reaction, the mixture was cooled to room temperature and diluted with 50 ml of toluene. The resulting solution was filtered using celite and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 6: 4 as a developing solvent. 30 ml of acetonitrile was added to the concentrate obtained by concentrating the obtained fraction, and sonication was performed for 30 minutes. A precipitate was collected from this mixture by filtration, rinsed with acetonitrile, and vacuum-dried (50 ° C., 6 hours) to obtain 2.14 g of the target compound in a yield of 92%.
(合成例3) 化合物6の合成

中間体3bの代わりに中間体5aを用い、原料の量をモル換算で2.33倍に変更した以外は、化合物5の合成方法Aと同様にして化合物6を合成した。得られた化合物6は、収量5.82g、収率56%であった。
(Synthesis Example 3) Synthesis of Compound 6
Compound 6 was synthesized in the same manner as Compound 5 Synthesis Method A, except that Intermediate 5a was used instead of Intermediate 3b, and the amount of raw material was changed to 2.33 times in terms of mole. The obtained compound 6 was 5.82 g in yield and 56% in yield.
(合成例4) 化合物18の合成
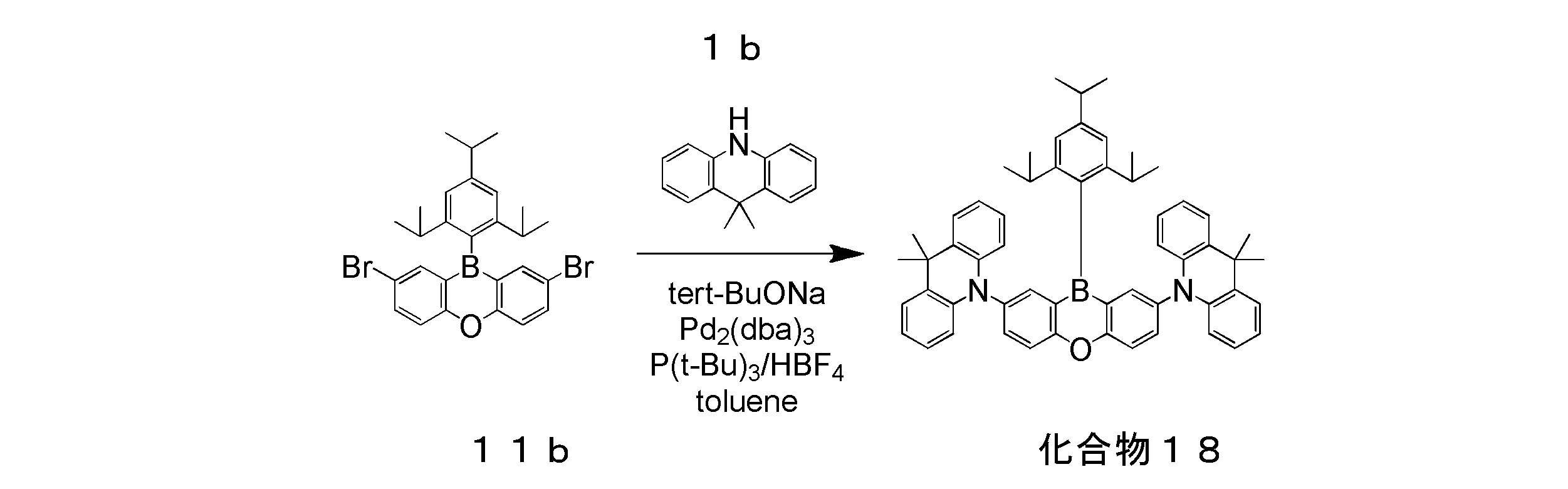
原料として中間体11b(2.70g,5mmol)を用いた以外は、化合物5の合成方法Bと同様にして化合物18を合成した。得られた化合物18は、収量3.65g、収率92%であった。
Synthesis Example 4 Synthesis of Compound 18
Compound 18 was synthesized in the same manner as Compound 5 Synthesis Method B except that Intermediate 11b (2.70 g, 5 mmol) was used as a raw material. The obtained compound 18 was 3.65 g in yield and 92% in yield.
(合成例5) 化合物59の合成

Synthesis Example 5 Synthesis of Compound 59
中間体6b(2.31g,5mmol)、中間体9b(1.99g,6mmol)、ナトリウム-tert-ブトキシド(1.15g,12mmol)、トリス(ジベンジリデンアセトン)ジパラジウム(0)(110mg,120μmol)、トリ-tert-ブチルホスホニウムテトラフルオロボラート(139mg,(480μmol)、脱水トルエン30mlを三口フラスコに入れ、窒素雰囲気下で8時間還流撹拌した。
反応終了後、混合物を室温まで冷却した後、トルエン50mlを加えて希釈して溶液を得た。この溶液を、セライトを用いて濾過し、濃縮した。この濃縮物をシリカゲルクロマトグラフィーにより、ヘキサン:トルエン=8:2の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを濃縮して得た濃縮物を、酢酸エチル:ヘキサン(v/v)=2:8の混合溶媒に10ml/gで溶解して再結晶し、真空乾燥(50℃、6時間)した。これにより、目的物の化合物59を収量3.17g、収率89%で得た。 Intermediate 6b (2.31 g, 5 mmol), Intermediate 9b (1.99 g, 6 mmol), Sodium-tert-butoxide (1.15 g, 12 mmol), Tris (dibenzylideneacetone) dipalladium (0) (110 mg, 120 μmol) ), Tri-tert-butylphosphonium tetrafluoroborate (139 mg, (480 μmol)) and 30 ml of dehydrated toluene were placed in a three-necked flask and stirred at reflux under a nitrogen atmosphere for 8 hours.
After completion of the reaction, the mixture was cooled to room temperature and diluted by adding 50 ml of toluene to obtain a solution. The solution was filtered using celite and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 8: 2 as a developing solvent. The concentrate obtained by concentrating the obtained fraction was dissolved in a mixed solvent of ethyl acetate: hexane (v / v) = 2: 8 at 10 ml / g, recrystallized, and dried under vacuum (50 ° C., 6 hours). )did. As a result, 3.59 g of the target compound 59 was obtained in a yield of 89%.
反応終了後、混合物を室温まで冷却した後、トルエン50mlを加えて希釈して溶液を得た。この溶液を、セライトを用いて濾過し、濃縮した。この濃縮物をシリカゲルクロマトグラフィーにより、ヘキサン:トルエン=8:2の混合溶媒を展開溶媒に用いて精製した。得られたフラクションを濃縮して得た濃縮物を、酢酸エチル:ヘキサン(v/v)=2:8の混合溶媒に10ml/gで溶解して再結晶し、真空乾燥(50℃、6時間)した。これにより、目的物の化合物59を収量3.17g、収率89%で得た。 Intermediate 6b (2.31 g, 5 mmol), Intermediate 9b (1.99 g, 6 mmol), Sodium-tert-butoxide (1.15 g, 12 mmol), Tris (dibenzylideneacetone) dipalladium (0) (110 mg, 120 μmol) ), Tri-tert-butylphosphonium tetrafluoroborate (139 mg, (480 μmol)) and 30 ml of dehydrated toluene were placed in a three-necked flask and stirred at reflux under a nitrogen atmosphere for 8 hours.
After completion of the reaction, the mixture was cooled to room temperature and diluted by adding 50 ml of toluene to obtain a solution. The solution was filtered using celite and concentrated. This concentrate was purified by silica gel chromatography using a mixed solvent of hexane: toluene = 8: 2 as a developing solvent. The concentrate obtained by concentrating the obtained fraction was dissolved in a mixed solvent of ethyl acetate: hexane (v / v) = 2: 8 at 10 ml / g, recrystallized, and dried under vacuum (50 ° C., 6 hours). )did. As a result, 3.59 g of the target compound 59 was obtained in a yield of 89%.
(合成例6) 化合物65の合成

原料として中間体6b(2.31g,5mmol)、中間体10b(1.91g,5.5mmol)を用いた以外は、化合物59の合成方法と同様にして化合物65を合成した。得られた化合物65は、収量3.37g,収率93%であった。
Synthesis Example 6 Synthesis of Compound 65
Compound 65 was synthesized in the same manner as Compound 59 except that Intermediate 6b (2.31 g, 5 mmol) and Intermediate 10b (1.91 g, 5.5 mmol) were used as raw materials. The obtained compound 65 was 3.37 g in yield, 93% in yield.
(合成例7) 化合物67の合成

原料として中間体6b(3.69g,8mmol)、中間体8b(3.30g,8.8mmol)を用いた以外は、化合物59の合成方法と同様にして化合物67を合成した。得られた化合物67は、収量4.66g、収率77%であった。
Synthesis Example 7 Synthesis of Compound 67
Compound 67 was synthesized in the same manner as Compound 59 except that Intermediate 6b (3.69 g, 8 mmol) and Intermediate 8b (3.30 g, 8.8 mmol) were used as raw materials. The obtained compound 67 was 4.66 g in yield and 77% in yield.
(合成例8) 化合物88の合成
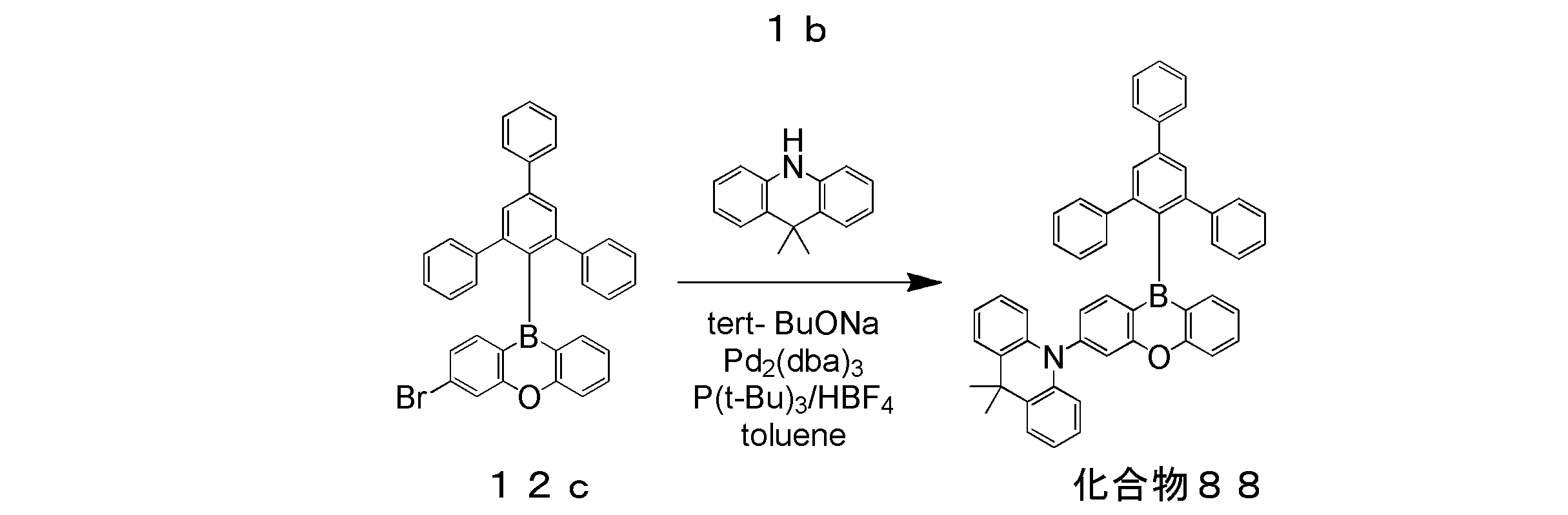
原料として中間体12c(563mg,1mmol)、中間体1b(319mg,1.1mmol)を用いた以外は、化合物59の合成方法と同様にして化合物88を合成した。得られた化合物88は、収量0.49g、収率71%であった。
Synthesis Example 8 Synthesis of Compound 88
Compound 88 was synthesized in the same manner as Compound 59 except that Intermediate 12c (563 mg, 1 mmol) and Intermediate 1b (319 mg, 1.1 mmol) were used as raw materials. The obtained compound 88 was 0.49 g in yield and 71% in yield.
(合成例9) 化合物116の合成

原料として中間体6b(2.77g,6mmol)、中間体2a(2.40g,7.2mmol)を用いた以外は、化合物59の合成方法と同様にして化合物116を合成した。得られた化合物116は、収量3.56g、収率83%であった。
Synthesis Example 9 Synthesis of Compound 116
Compound 116 was synthesized in the same manner as Compound 59 except that Intermediate 6b (2.77 g, 6 mmol) and Intermediate 2a (2.40 g, 7.2 mmol) were used as raw materials. The obtained compound 116 was 3.56 g in yield and 83% in yield.
(合成例10) 化合物156の合成
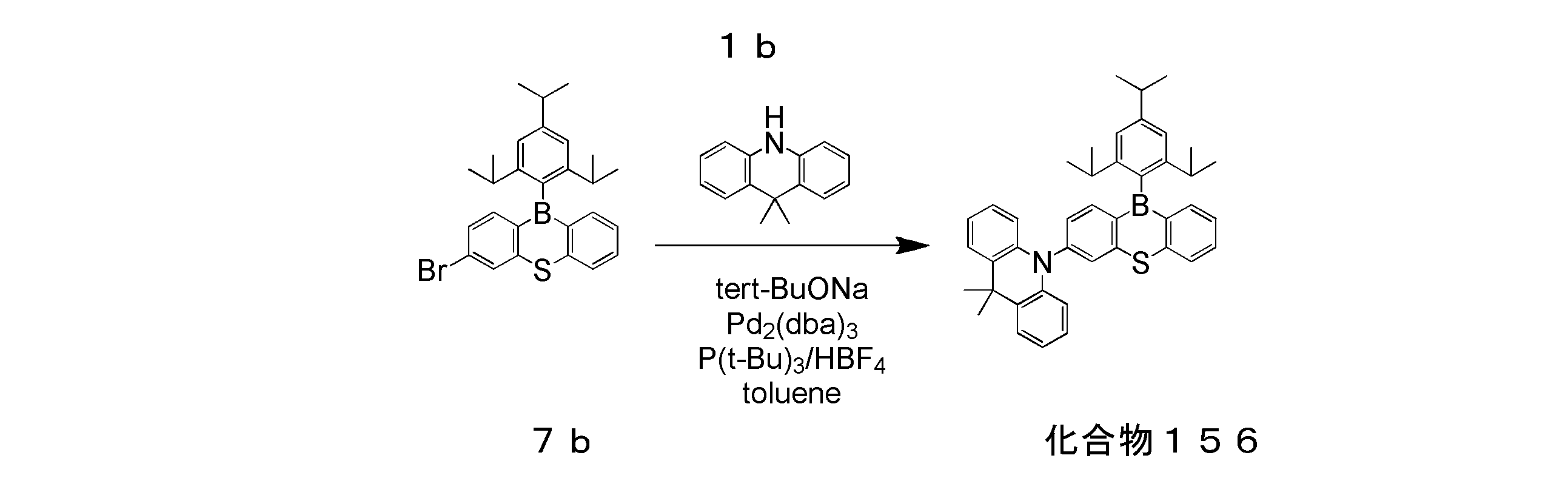
原料として中間体7b(3mmol)、中間体1b(3.6mmol)を用いた以外は、化合物59の合成方法と同様にして化合物156を合成した。得られた化合物156は、収量1.36g、収率75%であった。
Synthesis Example 10 Synthesis of Compound 156
Compound 156 was synthesized in the same manner as Compound 59 except that Intermediate 7b (3 mmol) and Intermediate 1b (3.6 mmol) were used as raw materials. The obtained compound 156 was 1.36 g in yield and 75% in yield.
(合成例11) 化合物176の合成

原料として中間体7b(2.16mmol)、中間体2a(2.59mmol)を用いた以外は、化合物59の合成方法と同様にして化合物176を合成した。得られた化合物176は、収量0.85g、収率54%であった。
Synthesis Example 11 Synthesis of Compound 176
Compound 176 was synthesized in the same manner as the compound 59 synthesis method, except that intermediate 7b (2.16 mmol) and intermediate 2a (2.59 mmol) were used as raw materials. The compound 176 obtained was 0.85 g in yield and 54% in yield.
[素子の作製と評価]
以下において、有機フォトルミネッセンス素子と有機エレクトロルミネッセンス素子を作製して、評価した。
発光特性の評価は、ソースメータ(ケースレー社製:2400シリーズ)、半導体パラメータ・アナライザ(アジレント・テクノロジー社製:E5273A)、光パワーメータ測定装置(ニューポート社製:1930C)、光学分光器(オーシャンオプティクス社製:USB2000)、分光放射計(トプコン社製:SR-3)およびストリークカメラ(浜松ホトニクス(株)製C4334型)を用いて行った。 [Production and evaluation of elements]
In the following, an organic photoluminescence element and an organic electroluminescence element were produced and evaluated.
Emission characteristics are evaluated by source meter (Keithley: 2400 series), semiconductor parameter analyzer (Agilent Technology: E5273A), optical power meter measuring device (Newport: 1930C), optical spectrometer (Ocean) Optics: USB2000), a spectroradiometer (Topcon: SR-3) and a streak camera (Hamamatsu Photonics C4334) were used.
以下において、有機フォトルミネッセンス素子と有機エレクトロルミネッセンス素子を作製して、評価した。
発光特性の評価は、ソースメータ(ケースレー社製:2400シリーズ)、半導体パラメータ・アナライザ(アジレント・テクノロジー社製:E5273A)、光パワーメータ測定装置(ニューポート社製:1930C)、光学分光器(オーシャンオプティクス社製:USB2000)、分光放射計(トプコン社製:SR-3)およびストリークカメラ(浜松ホトニクス(株)製C4334型)を用いて行った。 [Production and evaluation of elements]
In the following, an organic photoluminescence element and an organic electroluminescence element were produced and evaluated.
Emission characteristics are evaluated by source meter (Keithley: 2400 series), semiconductor parameter analyzer (Agilent Technology: E5273A), optical power meter measuring device (Newport: 1930C), optical spectrometer (Ocean) Optics: USB2000), a spectroradiometer (Topcon: SR-3) and a streak camera (Hamamatsu Photonics C4334) were used.
また、各材料の一重項エネルギー(ES1)と三重項エネルギー(ET1)の差(ΔEST)は、一重項エネルギー(ES1)と三重項エネルギーを以下の方法で算出し、ΔEST=ES1-ET1により求めた。
(1)一重項エネルギーES1
測定対象化合物とmCPとを、測定対象化合物が濃度6重量%となるように共蒸着することでSi基板上に厚さ100nmの試料を作製した。常温(300K)でこの試料の蛍光スペクトルを測定した。励起光入射直後から入射後100ナノ秒までの発光を積算することで、縦軸を発光強度、横軸を波長の蛍光スペクトルを得た。蛍光スペクトルは、縦軸を発光、横軸を波長とした。この発光スペクトルの短波側の立ち上がりに対して接線を引き、その接線と横軸との交点の波長値 λedge[nm]を求めた。この波長値を次に示す換算式でエネルギー値に換算した値をES1とした。
換算式:ES1[eV]=1239.85/λedge
発光スペクトルの測定には、励起光源に窒素レーザー(Lasertechnik Berlin社製、MNL200)を検出器には、ストリークカメラ(浜松ホトニクス社製、C4334)を用いた。
(2) 三重項エネルギーET1
一重項エネルギーES1と同じ試料を5[K]に冷却し、励起光(337nm)を燐光測定用試料に照射し、ストリークカメラを用いて、燐光強度を測定した。励起光入射後1ミリ秒から入射後10ミリ秒の発光を積算することで、縦軸を発光強度、横軸を波長の燐光スペクトルを得た。この燐光スペクトルの短波長側の立ち上がりに対して接線を引き、その接線と横軸との交点の波長値λedge[nm]を求めた。この波長値を次に示す換算式でエネルギー値に換算した値をET1とした。
換算式:ET1[eV]=1239.85/λedge
燐光スペクトルの短波長側の立ち上がりに対する接線は以下のように引いた。燐光スペクトルの短波長側から、スペクトルの極大値のうち、最も短波長側の極大値までスペクトル曲線上を移動する際に、長波長側に向けて曲線上の各点における接線を考える。この接線は、曲線が立ち上がるにつれ(つまり縦軸が増加するにつれ)、傾きが増加する。この傾きの値が極大値をとる点において引いた接線を、当該燐光スペクトルの短波長側の立ち上がりに対する接線とした。
なお、スペクトルの最大ピーク強度の10%以下のピーク強度をもつ極大点は、上述の最も短波長側の極大値には含めず、最も短波長側の極大値に最も近い、傾きの値が極大値をとる点において引いた接線を当該燐光スペクトルの短波長側の立ち上がりに対する接線とした。 The difference between the singlet energy of each material (E S1) and triplet energy (E T1) (ΔE ST) calculates the triplet energy singlet energy (E S1) in the following manner, Delta] E ST = It was determined by E S1 -E T1 .
(1) Singlet energy E S1
A sample having a thickness of 100 nm was prepared on a Si substrate by co-evaporating the measurement target compound and mCP so that the measurement target compound had a concentration of 6% by weight. The fluorescence spectrum of this sample was measured at room temperature (300K). By integrating the luminescence from immediately after the excitation light incidence to 100 nanoseconds after the incidence, a fluorescence spectrum having a luminescence intensity on the vertical axis and a wavelength on the horizontal axis was obtained. In the fluorescence spectrum, the vertical axis represents light emission and the horizontal axis represents wavelength. A tangent line was drawn with respect to the short-wave rise of the emission spectrum, and the wavelength value λedge [nm] at the intersection of the tangent line and the horizontal axis was obtained. A value obtained by converting this wavelength value into an energy value by the following conversion formula was defined as E S1 .
Conversion formula: E S1 [eV] = 1239.85 / λedge
For the measurement of the emission spectrum, a nitrogen laser (Lasertechnik Berlin, MNL200) was used as an excitation light source, and a streak camera (Hamamatsu Photonics, C4334) was used as a detector.
(2) Triplet energy E T1
The same sample as the singlet energy E S1 was cooled to 5 [K], the sample for phosphorescence measurement was irradiated with excitation light (337 nm), and the phosphorescence intensity was measured using a streak camera. By integrating the luminescence from 1 millisecond after the excitation light incidence to 10 milliseconds after the incidence, a phosphorescence spectrum having the luminescence intensity on the vertical axis and the wavelength on the horizontal axis was obtained. A tangent line was drawn with respect to the rising edge of the phosphorescence spectrum on the short wavelength side, and a wavelength value λ edge [nm] at the intersection of the tangent line and the horizontal axis was obtained. A value obtained by converting this wavelength value into an energy value by the following conversion formula was defined as ET1 .
Conversion formula: E T1 [eV] = 1239.85 / λedge
The tangent to the short wavelength rising edge of the phosphorescence spectrum was drawn as follows. When moving on the spectrum curve from the short wavelength side of the phosphorescence spectrum to the maximum value on the shortest wavelength side among the maximum values of the spectrum, tangents at each point on the curve are considered toward the long wavelength side. The slope of this tangent line increases as the curve rises (that is, as the vertical axis increases). The tangent drawn at the point where the value of the slope takes the maximum value was taken as the tangent to the rising edge of the phosphorescence spectrum on the short wavelength side.
In addition, the maximum point having a peak intensity of 10% or less of the maximum peak intensity of the spectrum is not included in the above-mentioned maximum value on the shortest wavelength side, and has the maximum slope value closest to the maximum value on the shortest wavelength side. The tangent drawn at the point where the value was taken was taken as the tangent to the rising edge of the phosphorescence spectrum on the short wavelength side.
(1)一重項エネルギーES1
測定対象化合物とmCPとを、測定対象化合物が濃度6重量%となるように共蒸着することでSi基板上に厚さ100nmの試料を作製した。常温(300K)でこの試料の蛍光スペクトルを測定した。励起光入射直後から入射後100ナノ秒までの発光を積算することで、縦軸を発光強度、横軸を波長の蛍光スペクトルを得た。蛍光スペクトルは、縦軸を発光、横軸を波長とした。この発光スペクトルの短波側の立ち上がりに対して接線を引き、その接線と横軸との交点の波長値 λedge[nm]を求めた。この波長値を次に示す換算式でエネルギー値に換算した値をES1とした。
換算式:ES1[eV]=1239.85/λedge
発光スペクトルの測定には、励起光源に窒素レーザー(Lasertechnik Berlin社製、MNL200)を検出器には、ストリークカメラ(浜松ホトニクス社製、C4334)を用いた。
(2) 三重項エネルギーET1
一重項エネルギーES1と同じ試料を5[K]に冷却し、励起光(337nm)を燐光測定用試料に照射し、ストリークカメラを用いて、燐光強度を測定した。励起光入射後1ミリ秒から入射後10ミリ秒の発光を積算することで、縦軸を発光強度、横軸を波長の燐光スペクトルを得た。この燐光スペクトルの短波長側の立ち上がりに対して接線を引き、その接線と横軸との交点の波長値λedge[nm]を求めた。この波長値を次に示す換算式でエネルギー値に換算した値をET1とした。
換算式:ET1[eV]=1239.85/λedge
燐光スペクトルの短波長側の立ち上がりに対する接線は以下のように引いた。燐光スペクトルの短波長側から、スペクトルの極大値のうち、最も短波長側の極大値までスペクトル曲線上を移動する際に、長波長側に向けて曲線上の各点における接線を考える。この接線は、曲線が立ち上がるにつれ(つまり縦軸が増加するにつれ)、傾きが増加する。この傾きの値が極大値をとる点において引いた接線を、当該燐光スペクトルの短波長側の立ち上がりに対する接線とした。
なお、スペクトルの最大ピーク強度の10%以下のピーク強度をもつ極大点は、上述の最も短波長側の極大値には含めず、最も短波長側の極大値に最も近い、傾きの値が極大値をとる点において引いた接線を当該燐光スペクトルの短波長側の立ち上がりに対する接線とした。 The difference between the singlet energy of each material (E S1) and triplet energy (E T1) (ΔE ST) calculates the triplet energy singlet energy (E S1) in the following manner, Delta] E ST = It was determined by E S1 -E T1 .
(1) Singlet energy E S1
A sample having a thickness of 100 nm was prepared on a Si substrate by co-evaporating the measurement target compound and mCP so that the measurement target compound had a concentration of 6% by weight. The fluorescence spectrum of this sample was measured at room temperature (300K). By integrating the luminescence from immediately after the excitation light incidence to 100 nanoseconds after the incidence, a fluorescence spectrum having a luminescence intensity on the vertical axis and a wavelength on the horizontal axis was obtained. In the fluorescence spectrum, the vertical axis represents light emission and the horizontal axis represents wavelength. A tangent line was drawn with respect to the short-wave rise of the emission spectrum, and the wavelength value λedge [nm] at the intersection of the tangent line and the horizontal axis was obtained. A value obtained by converting this wavelength value into an energy value by the following conversion formula was defined as E S1 .
Conversion formula: E S1 [eV] = 1239.85 / λedge
For the measurement of the emission spectrum, a nitrogen laser (Lasertechnik Berlin, MNL200) was used as an excitation light source, and a streak camera (Hamamatsu Photonics, C4334) was used as a detector.
(2) Triplet energy E T1
The same sample as the singlet energy E S1 was cooled to 5 [K], the sample for phosphorescence measurement was irradiated with excitation light (337 nm), and the phosphorescence intensity was measured using a streak camera. By integrating the luminescence from 1 millisecond after the excitation light incidence to 10 milliseconds after the incidence, a phosphorescence spectrum having the luminescence intensity on the vertical axis and the wavelength on the horizontal axis was obtained. A tangent line was drawn with respect to the rising edge of the phosphorescence spectrum on the short wavelength side, and a wavelength value λ edge [nm] at the intersection of the tangent line and the horizontal axis was obtained. A value obtained by converting this wavelength value into an energy value by the following conversion formula was defined as ET1 .
Conversion formula: E T1 [eV] = 1239.85 / λedge
The tangent to the short wavelength rising edge of the phosphorescence spectrum was drawn as follows. When moving on the spectrum curve from the short wavelength side of the phosphorescence spectrum to the maximum value on the shortest wavelength side among the maximum values of the spectrum, tangents at each point on the curve are considered toward the long wavelength side. The slope of this tangent line increases as the curve rises (that is, as the vertical axis increases). The tangent drawn at the point where the value of the slope takes the maximum value was taken as the tangent to the rising edge of the phosphorescence spectrum on the short wavelength side.
In addition, the maximum point having a peak intensity of 10% or less of the maximum peak intensity of the spectrum is not included in the above-mentioned maximum value on the shortest wavelength side, and has the maximum slope value closest to the maximum value on the shortest wavelength side. The tangent drawn at the point where the value was taken was taken as the tangent to the rising edge of the phosphorescence spectrum on the short wavelength side.
(実施例1) 化合物5を用いた有機フォトルミネッセンス素子の作製と評価
大気下にて化合物5のトルエン溶液(濃度10-5mol/L)およびヘキサン溶液(濃度10-5mol/L)を調製し、窒素で15分間のバブリングによる脱酸素処理を行った後、発光特性測定に用いた。
また、石英基板上に真空蒸着法にて、真空度10-4Pa以下の条件にて化合物5の薄膜を100nmの厚さで形成して有機フォトルミネッセンス素子とした。
化合物5のトルエン溶液およびヘキサン溶液について発光スペクトルを測定した結果を図2に示し、化合物5の薄膜を有する有機フォトルミネッセンス素子について発光スペクトルを測定した結果を図3に示す。
フォトルミネッセンス量子収率φは、トルエン溶液で0.97、ヘキサン溶液で0.54、化合物5の薄膜を有する有機フォトルミネッセンス素子で0.83であった。蛍光スペクトルとリン光スペクトルから求めた一重項励起状態と三重項励起状態とのエネルギー差ΔESTは0.05eVであった。
また、化合物5のトルエン溶液およびヘキサン溶液の過渡減衰曲線を図4に示す。この過渡減衰曲線は、化合物に励起光を当てて発光強度が失活してゆく過程を測定した発光寿命測定結果を示すものである。通常の一成分の発光(蛍光もしくはリン光)では発光強度は単一指数関数的に減衰する。これは、グラフの縦軸がセミlog である場合には、直線的に減衰することを意味している。図4に示す化合物5の過渡減衰曲線では、観測初期にこのような直線的成分(蛍光)が観測されているが、数μ秒以降には直線性から外れる成分が現れている。これは遅延成分の発光であり、初期の成分と加算される信号は、長時間側に裾をひくゆるい曲線になる。このように発光寿命を測定することによって、化合物5は蛍光成分のほかに遅延成分を含む発光体であることが確認された。トルエン溶液での即時蛍光成分の発光寿命τ1は35ns、遅延蛍光成分の発光寿命τ2は1.35μsであり、ヘキサン溶液での即時蛍光成分の発光寿命τ1は21ns、遅延蛍光成分の発光寿命τ2は2.09μsであった。 Example 1 Preparation and Evaluation of Organic Photoluminescence Device Using Compound 5 A toluene solution (concentration 10 −5 mol / L) and a hexane solution (concentration 10 −5 mol / L) of compound 5 were prepared in the atmosphere. Then, after deoxygenation by bubbling with nitrogen for 15 minutes, it was used for measurement of light emission characteristics.
In addition, a thin film ofcompound 5 was formed to a thickness of 100 nm on a quartz substrate by a vacuum deposition method under a vacuum degree of 10 −4 Pa or less to obtain an organic photoluminescence device.
The result of measuring the emission spectrum of the toluene solution and the hexane solution ofCompound 5 is shown in FIG. 2, and the result of measuring the emission spectrum of the organic photoluminescence device having a thin film of Compound 5 is shown in FIG.
The photoluminescence quantum yield φ was 0.97 for the toluene solution, 0.54 for the hexane solution, and 0.83 for the organic photoluminescence device having a thin film ofCompound 5. Energy difference Delta] E ST of the singlet excited state and a triplet excited state as determined from the fluorescence spectrum and phosphorescence spectrum was 0.05 eV.
Moreover, the transient decay curve of the toluene solution ofcompound 5 and a hexane solution is shown in FIG. This transient decay curve shows the result of measuring the luminescence lifetime obtained by measuring the process in which the emission intensity is deactivated by applying excitation light to the compound. In the case of normal single component light emission (fluorescence or phosphorescence), the light emission intensity decays in a single exponential manner. This means that if the vertical axis of the graph is semi-log, it will decay linearly. In the transient decay curve of Compound 5 shown in FIG. 4, such a linear component (fluorescence) is observed at the beginning of observation, but a component deviating from linearity appears after several μsec. This is light emission of the delay component, and the signal added to the initial component becomes a loose curve with a tail on the long time side. By measuring the luminescence lifetime in this way, it was confirmed that Compound 5 is a luminescent material containing a delay component in addition to the fluorescent component. The emission lifetime τ1 of the immediate fluorescence component in the toluene solution is 35 ns, the emission lifetime τ2 of the delayed fluorescence component is 1.35 μs, the emission lifetime τ1 of the immediate fluorescence component in the hexane solution is 21 ns, and the emission lifetime τ2 of the delay fluorescence component is 2.09 μs.
大気下にて化合物5のトルエン溶液(濃度10-5mol/L)およびヘキサン溶液(濃度10-5mol/L)を調製し、窒素で15分間のバブリングによる脱酸素処理を行った後、発光特性測定に用いた。
また、石英基板上に真空蒸着法にて、真空度10-4Pa以下の条件にて化合物5の薄膜を100nmの厚さで形成して有機フォトルミネッセンス素子とした。
化合物5のトルエン溶液およびヘキサン溶液について発光スペクトルを測定した結果を図2に示し、化合物5の薄膜を有する有機フォトルミネッセンス素子について発光スペクトルを測定した結果を図3に示す。
フォトルミネッセンス量子収率φは、トルエン溶液で0.97、ヘキサン溶液で0.54、化合物5の薄膜を有する有機フォトルミネッセンス素子で0.83であった。蛍光スペクトルとリン光スペクトルから求めた一重項励起状態と三重項励起状態とのエネルギー差ΔESTは0.05eVであった。
また、化合物5のトルエン溶液およびヘキサン溶液の過渡減衰曲線を図4に示す。この過渡減衰曲線は、化合物に励起光を当てて発光強度が失活してゆく過程を測定した発光寿命測定結果を示すものである。通常の一成分の発光(蛍光もしくはリン光)では発光強度は単一指数関数的に減衰する。これは、グラフの縦軸がセミlog である場合には、直線的に減衰することを意味している。図4に示す化合物5の過渡減衰曲線では、観測初期にこのような直線的成分(蛍光)が観測されているが、数μ秒以降には直線性から外れる成分が現れている。これは遅延成分の発光であり、初期の成分と加算される信号は、長時間側に裾をひくゆるい曲線になる。このように発光寿命を測定することによって、化合物5は蛍光成分のほかに遅延成分を含む発光体であることが確認された。トルエン溶液での即時蛍光成分の発光寿命τ1は35ns、遅延蛍光成分の発光寿命τ2は1.35μsであり、ヘキサン溶液での即時蛍光成分の発光寿命τ1は21ns、遅延蛍光成分の発光寿命τ2は2.09μsであった。 Example 1 Preparation and Evaluation of Organic Photoluminescence Device Using Compound 5 A toluene solution (
In addition, a thin film of
The result of measuring the emission spectrum of the toluene solution and the hexane solution of
The photoluminescence quantum yield φ was 0.97 for the toluene solution, 0.54 for the hexane solution, and 0.83 for the organic photoluminescence device having a thin film of
Moreover, the transient decay curve of the toluene solution of
(実施例2) 化合物6を用いた有機フォトルミネッセンス素子の作製と評価
化合物5のかわりに化合物6を用いた点を変更して、実施例1と同じ方法により化合物6のトルエン溶液およびヘキサン溶液、化合物6の薄膜を有する有機フォトルミネッセンス素子を作製した。
また、石英基板上に真空蒸着法にて、真空度10-4Pa以下の条件にて化合物6とmCPとを異なる蒸着源から蒸着し、化合物6の濃度が6.0重量%である薄膜を100nmの厚さで形成して有機フォトルミネッセンス素子とした。
化合物6のトルエン溶液およびヘキサン溶液、化合物6とmCPの薄膜を有する有機フォトルミネッセンス素子について発光スペクトルを測定した結果を図5に示し、化合物6のみの薄膜を有する有機フォトルミネッセンス素子について発光スペクトルを測定した結果を図6に示す。
フォトルミネッセンス量子収率φは、トルエン溶液で1.00、ヘキサン溶液で0.54、化合物6のみの薄膜を有する有機フォトルミネッセンス素子で0.88であった。蛍光スペクトルとリン光スペクトルから求めた一重項励起状態と三重項励起状態とのエネルギー差ΔESTは0.08eVであった。
また、化合物6のトルエン溶液およびヘキサン溶液の過渡減衰曲線を図7に示す。トルエン溶液での即時蛍光成分の発光寿命τ1は33ns、遅延蛍光成分の発光寿命τ2は1.60μsであり、ヘキサン溶液での即時蛍光成分の発光寿命τ1は20ns、遅延蛍光成分の発光寿命τ2は1.29μsであった。 (Example 2) Production and evaluation of organic photoluminescencedevice using compound 6 By changing the point of using compound 6 instead of compound 5, a toluene solution and a hexane solution of compound 6 by the same method as in Example 1, An organic photoluminescence device having a thin film of Compound 6 was produced.
Further, a thin film in which the concentration ofcompound 6 is 6.0% by weight is deposited on a quartz substrate by a vacuum deposition method under the condition of a vacuum degree of 10 −4 Pa or less from different deposition sources. An organic photoluminescence device was formed with a thickness of 100 nm.
FIG. 5 shows the results of measuring the emission spectrum of an organic photoluminescence device having a toluene solution and a hexane solution ofcompound 6 and a thin film of compound 6 and mCP. The results are shown in FIG.
The photoluminescence quantum yield φ was 1.00 for the toluene solution, 0.54 for the hexane solution, and 0.88 for the organic photoluminescence device having a thin film ofcompound 6 alone. Energy difference Delta] E ST of the singlet excited state and a triplet excited state as determined from the fluorescence spectrum and phosphorescence spectrum was 0.08 eV.
Moreover, the transient decay curve of the toluene solution ofcompound 6 and a hexane solution is shown in FIG. The emission lifetime τ1 of the immediate fluorescence component in the toluene solution is 33 ns, the emission lifetime τ2 of the delayed fluorescence component is 1.60 μs, the emission lifetime τ1 of the immediate fluorescence component in the hexane solution is 20 ns, and the emission lifetime τ2 of the delay fluorescence component is 1.29 μs.
化合物5のかわりに化合物6を用いた点を変更して、実施例1と同じ方法により化合物6のトルエン溶液およびヘキサン溶液、化合物6の薄膜を有する有機フォトルミネッセンス素子を作製した。
また、石英基板上に真空蒸着法にて、真空度10-4Pa以下の条件にて化合物6とmCPとを異なる蒸着源から蒸着し、化合物6の濃度が6.0重量%である薄膜を100nmの厚さで形成して有機フォトルミネッセンス素子とした。
化合物6のトルエン溶液およびヘキサン溶液、化合物6とmCPの薄膜を有する有機フォトルミネッセンス素子について発光スペクトルを測定した結果を図5に示し、化合物6のみの薄膜を有する有機フォトルミネッセンス素子について発光スペクトルを測定した結果を図6に示す。
フォトルミネッセンス量子収率φは、トルエン溶液で1.00、ヘキサン溶液で0.54、化合物6のみの薄膜を有する有機フォトルミネッセンス素子で0.88であった。蛍光スペクトルとリン光スペクトルから求めた一重項励起状態と三重項励起状態とのエネルギー差ΔESTは0.08eVであった。
また、化合物6のトルエン溶液およびヘキサン溶液の過渡減衰曲線を図7に示す。トルエン溶液での即時蛍光成分の発光寿命τ1は33ns、遅延蛍光成分の発光寿命τ2は1.60μsであり、ヘキサン溶液での即時蛍光成分の発光寿命τ1は20ns、遅延蛍光成分の発光寿命τ2は1.29μsであった。 (Example 2) Production and evaluation of organic photoluminescence
Further, a thin film in which the concentration of
FIG. 5 shows the results of measuring the emission spectrum of an organic photoluminescence device having a toluene solution and a hexane solution of
The photoluminescence quantum yield φ was 1.00 for the toluene solution, 0.54 for the hexane solution, and 0.88 for the organic photoluminescence device having a thin film of
Moreover, the transient decay curve of the toluene solution of
(実施例3) 化合物18を用いた有機フォトルミネッセンス素子の作製と評価
化合物5のかわりに化合物18を用いた点を変更して、実施例1と同じ方法により化合物18のトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図8に示し、過渡減衰曲線を図9に示す。化合物18のトルエン溶液のフォトルミネッセンス量子収率φは0.81であった。また、即時蛍光成分の発光寿命τ1は89.3ns、遅延蛍光成分の発光寿命τ2は39.5μsであった。 Example 3 Preparation and Evaluation of Organic Photoluminescence Device Using Compound 18 A toluene solution of Compound 18 was prepared in the same manner as in Example 1 except that Compound 18 was used instead ofCompound 5. The emission spectrum and transient decay curve of this toluene solution were measured. The measured emission spectrum is shown in FIG. 8, and the transient decay curve is shown in FIG. The photoluminescence quantum yield φ of the toluene solution of compound 18 was 0.81. Further, the emission lifetime τ1 of the immediate fluorescence component was 89.3 ns, and the emission lifetime τ2 of the delayed fluorescence component was 39.5 μs.
化合物5のかわりに化合物18を用いた点を変更して、実施例1と同じ方法により化合物18のトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図8に示し、過渡減衰曲線を図9に示す。化合物18のトルエン溶液のフォトルミネッセンス量子収率φは0.81であった。また、即時蛍光成分の発光寿命τ1は89.3ns、遅延蛍光成分の発光寿命τ2は39.5μsであった。 Example 3 Preparation and Evaluation of Organic Photoluminescence Device Using Compound 18 A toluene solution of Compound 18 was prepared in the same manner as in Example 1 except that Compound 18 was used instead of
(実施例4) 化合物59を用いた有機フォトルミネッセンス素子の作製と評価
化合物5のかわりに化合物59を用いた点を変更して、実施例1と同じ方法により化合物59のトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図10に示す。化合物59のトルエン溶液のフォトルミネッセンス量子収率φは0.76であった。また、即時蛍光成分の発光寿命τ1は27.7ns、遅延蛍光成分の発光寿命τ2は4.02μsであった。 Example 4 Production and Evaluation of Organic Photoluminescence Device Using Compound 59 A toluene solution of compound 59 was prepared in the same manner as in Example 1, except that compound 59 was used instead ofcompound 5. The emission spectrum and transient decay curve of this toluene solution were measured. The measured emission spectrum is shown in FIG. The photoluminescence quantum yield φ of the toluene solution of compound 59 was 0.76. Further, the emission lifetime τ1 of the immediate fluorescence component was 27.7 ns, and the emission lifetime τ2 of the delayed fluorescence component was 4.02 μs.
化合物5のかわりに化合物59を用いた点を変更して、実施例1と同じ方法により化合物59のトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図10に示す。化合物59のトルエン溶液のフォトルミネッセンス量子収率φは0.76であった。また、即時蛍光成分の発光寿命τ1は27.7ns、遅延蛍光成分の発光寿命τ2は4.02μsであった。 Example 4 Production and Evaluation of Organic Photoluminescence Device Using Compound 59 A toluene solution of compound 59 was prepared in the same manner as in Example 1, except that compound 59 was used instead of
(実施例5) 化合物65を用いた有機フォトルミネッセンス素子の作製と評価
化合物5のかわりに化合物65を用いた点を変更して、実施例1と同じ方法により化合物65のトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図11に示す。化合物65のトルエン溶液のフォトルミネッセンス量子収率φは0.56であった。また、即時蛍光成分の発光寿命τ1は25.9ns、遅延蛍光成分の発光寿命τ2は2.06μsであった。 Example 5 Production and Evaluation of Organic Photoluminescence Device Using Compound 65 A toluene solution of compound 65 was prepared by the same method as in Example 1 except that compound 65 was used instead ofcompound 5. The emission spectrum and transient decay curve of this toluene solution were measured. The measured emission spectrum is shown in FIG. The photoluminescence quantum yield φ of the toluene solution of compound 65 was 0.56. Further, the emission lifetime τ1 of the immediate fluorescence component was 25.9 ns, and the emission lifetime τ2 of the delayed fluorescence component was 2.06 μs.
化合物5のかわりに化合物65を用いた点を変更して、実施例1と同じ方法により化合物65のトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図11に示す。化合物65のトルエン溶液のフォトルミネッセンス量子収率φは0.56であった。また、即時蛍光成分の発光寿命τ1は25.9ns、遅延蛍光成分の発光寿命τ2は2.06μsであった。 Example 5 Production and Evaluation of Organic Photoluminescence Device Using Compound 65 A toluene solution of compound 65 was prepared by the same method as in Example 1 except that compound 65 was used instead of
(実施例6) 化合物67を用いた有機フォトルミネッセンス素子の作製と評価
化合物5のかわりに化合物67を用いた点を変更して、実施例1と同じ方法により化合物67のトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図12に示す。化合物67のトルエン溶液のフォトルミネッセンス量子収率φは1.00であった。また、即時蛍光成分の発光寿命τ1は35ns、遅延蛍光成分の発光寿命τ2は2.33μsであった。 Example 6 Production and Evaluation of Organic Photoluminescence Device Using Compound 67 A toluene solution of compound 67 was prepared in the same manner as in Example 1 except that compound 67 was used instead ofcompound 5. The emission spectrum and transient decay curve of this toluene solution were measured. The measured emission spectrum is shown in FIG. The photoluminescence quantum yield φ of the toluene solution of compound 67 was 1.00. Further, the emission lifetime τ1 of the immediate fluorescence component was 35 ns, and the emission lifetime τ2 of the delayed fluorescence component was 2.33 μs.
化合物5のかわりに化合物67を用いた点を変更して、実施例1と同じ方法により化合物67のトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図12に示す。化合物67のトルエン溶液のフォトルミネッセンス量子収率φは1.00であった。また、即時蛍光成分の発光寿命τ1は35ns、遅延蛍光成分の発光寿命τ2は2.33μsであった。 Example 6 Production and Evaluation of Organic Photoluminescence Device Using Compound 67 A toluene solution of compound 67 was prepared in the same manner as in Example 1 except that compound 67 was used instead of
(実施例7) 化合物156を用いた有機フォトルミネッセンス素子の作製と評価
化合物5のかわりに化合物156を用いた点を変更して、実施例1と同じ方法により化合物156のトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図13に示し、過渡減衰曲線を図14に示す。化合物156のトルエン溶液のフォトルミネッセンス量子収率φは1.00であった。また、即時蛍光成分の発光寿命τ1は3.2ns、遅延蛍光成分の発光寿命τ2は1.61μsであった。 Example 7 Production and Evaluation of Organic Photoluminescence Device Using Compound 156 A toluene solution of compound 156 was prepared in the same manner as in Example 1 except that compound 156 was used instead ofcompound 5. The emission spectrum and transient decay curve of this toluene solution were measured. The measured emission spectrum is shown in FIG. 13, and the transient decay curve is shown in FIG. The photoluminescence quantum yield φ of the toluene solution of compound 156 was 1.00. Further, the emission lifetime τ1 of the immediate fluorescence component was 3.2 ns, and the emission lifetime τ2 of the delayed fluorescence component was 1.61 μs.
化合物5のかわりに化合物156を用いた点を変更して、実施例1と同じ方法により化合物156のトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図13に示し、過渡減衰曲線を図14に示す。化合物156のトルエン溶液のフォトルミネッセンス量子収率φは1.00であった。また、即時蛍光成分の発光寿命τ1は3.2ns、遅延蛍光成分の発光寿命τ2は1.61μsであった。 Example 7 Production and Evaluation of Organic Photoluminescence Device Using Compound 156 A toluene solution of compound 156 was prepared in the same manner as in Example 1 except that compound 156 was used instead of
(実施例8~10)
化合物5のかわりに化合物88、116、176を用いた点を変更して、実施例1と同じ方法によりトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルおよび過渡減衰曲線から求めたフォトルミネッセンス量子収率φおよび発光寿命を表1に示す。表1に示すように、化合物88、116、176のトルエン溶液は、いずれも十分なフォトルミネッセンス量子収率φが得られ、また遅延蛍光を確認することができた。 (Examples 8 to 10)
A toluene solution was prepared in the same manner as in Example 1, except that the compounds 88, 116, and 176 were used instead of thecompound 5. The emission spectrum and transient decay curve of this toluene solution were measured. Table 1 shows the photoluminescence quantum yield φ and the emission lifetime determined from the measured emission spectrum and transient decay curve. As shown in Table 1, all the toluene solutions of the compounds 88, 116, and 176 obtained a sufficient photoluminescence quantum yield φ, and confirmed delayed fluorescence.
化合物5のかわりに化合物88、116、176を用いた点を変更して、実施例1と同じ方法によりトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルおよび過渡減衰曲線から求めたフォトルミネッセンス量子収率φおよび発光寿命を表1に示す。表1に示すように、化合物88、116、176のトルエン溶液は、いずれも十分なフォトルミネッセンス量子収率φが得られ、また遅延蛍光を確認することができた。 (Examples 8 to 10)
A toluene solution was prepared in the same manner as in Example 1, except that the compounds 88, 116, and 176 were used instead of the

(比較例1) 比較化合物Aを用いた有機フォトルミネッセンス素子の作製と評価
化合物1のかわりに下記の構造を有する比較化合物Aを用いて、実施例1と同じ方法により比較化合物Aのトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図15に示し、過渡減衰曲線を図16に示す。発光寿命は16.0nsであり、遅延蛍光は認められなかった。

(Comparative Example 1) Preparation and Evaluation of Organic Photoluminescence Device Using Comparative Compound A Using Comparative Compound A having the following structure instead of Compound 1, a toluene solution of Comparative Compound A was prepared in the same manner as Example 1. Prepared. The emission spectrum and transient decay curve of this toluene solution were measured. The measured emission spectrum is shown in FIG. 15, and the transient decay curve is shown in FIG. The emission lifetime was 16.0 ns, and no delayed fluorescence was observed.
化合物1のかわりに下記の構造を有する比較化合物Aを用いて、実施例1と同じ方法により比較化合物Aのトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図15に示し、過渡減衰曲線を図16に示す。発光寿命は16.0nsであり、遅延蛍光は認められなかった。
(比較例2) 比較化合物Bを用いた有機フォトルミネッセンス素子の作製と評価
化合物1のかわりに下記の構造を有する比較化合物Bを用いて、実施例1と同じ方法により比較化合物Aのトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図17に示し、過渡減衰曲線を図18に示す。比較化合物Bのトルエン溶液のフォトルミネッセンス量子収率φは0.27であった。また、発光寿命は8.1nsであり、遅延蛍光は認められなかった。

(Comparative Example 2) Preparation and Evaluation of Organic Photoluminescence Element Using Comparative Compound B Using Comparative Compound B having the following structure instead of Compound 1, a toluene solution of Comparative Compound A was prepared in the same manner as Example 1. Prepared. The emission spectrum and transient decay curve of this toluene solution were measured. The measured emission spectrum is shown in FIG. 17, and the transient decay curve is shown in FIG. The photoluminescence quantum yield φ of the toluene solution of Comparative Compound B was 0.27. Moreover, the light emission lifetime was 8.1 ns, and no delayed fluorescence was observed.
化合物1のかわりに下記の構造を有する比較化合物Bを用いて、実施例1と同じ方法により比較化合物Aのトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図17に示し、過渡減衰曲線を図18に示す。比較化合物Bのトルエン溶液のフォトルミネッセンス量子収率φは0.27であった。また、発光寿命は8.1nsであり、遅延蛍光は認められなかった。
(比較例3) 比較化合物Cを用いた有機フォトルミネッセンス素子の作製と評価
化合物1のかわりに下記の構造を有する比較化合物Cを用いて、実施例1と同じ方法により比較化合物Cのトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図19に示し、過渡減衰曲線を図20に示す。比較化合物Cのトルエン溶液のフォトルミネッセンス量子収率φは0.26であった。また、発光寿命は12.7nsであり、遅延蛍光は認められなかった。

(Comparative Example 3) Preparation and Evaluation of Organic Photoluminescence Device Using Comparative Compound C Using Comparative Compound C having the following structure instead of Compound 1, a toluene solution of Comparative Compound C was prepared in the same manner as Example 1. Prepared. The emission spectrum and transient decay curve of this toluene solution were measured. The measured emission spectrum is shown in FIG. 19, and the transient decay curve is shown in FIG. The photoluminescence quantum yield φ of the toluene solution of Comparative Compound C was 0.26. The emission lifetime was 12.7 ns, and no delayed fluorescence was observed.
化合物1のかわりに下記の構造を有する比較化合物Cを用いて、実施例1と同じ方法により比較化合物Cのトルエン溶液を調製した。このトルエン溶液について発光スペクトルおよび過渡減衰曲線を測定した。測定された発光スペクトルを図19に示し、過渡減衰曲線を図20に示す。比較化合物Cのトルエン溶液のフォトルミネッセンス量子収率φは0.26であった。また、発光寿命は12.7nsであり、遅延蛍光は認められなかった。
(実施例11) 化合物5を用いた有機エレクトロルミネッセンス素子の作製と評価
膜厚100nmのインジウム・スズ酸化物(ITO)からなる陽極が形成されたガラス基板上に、各薄膜を真空蒸着法にて、真空度5.0×10-4Pa以下で積層した。まず、ITO上にα-NPDを30nmの厚さに形成し、この上に、mCPを10nmの厚さに形成した。次に、化合物5と(Cz)2DBFを異なる蒸着源から共蒸着し、15nmの厚さの層を形成して発光層とした。この時、化合物5の濃度は6.0重量%とした。次に、PPTを10nmの厚さに形成し、この上に、TPBiを40nmの厚さに形成した。さらにフッ化リチウム(LiF)を0.8nm真空蒸着し、次いでアルミニウム(Al)を100nmの厚さに蒸着することにより陰極を形成し、有機エレクトロルミネッセンス素子とした。
作製した有機エレクトロルミネッセンス素子の発光スペクトルを図21に示し、電流密度-外部量子効率特性を図22に示す。化合物5を発光材料として用いた有機エレクトロルミネッセンス素子は15.3%の高い外部量子効率を達成した。仮に発光量子効率が100%の蛍光材料を用いてバランスの取れた理想的な有機エレクトロルミネッセンス素子を試作したとすると、光取り出し効率が20~30%であれば、蛍光発光の外部量子効率は5~7.5%となる。この値が一般に、蛍光材料を用いた有機エレクトロルミネッセンス素子の外部量子効率の理論限界値とされている。したがって、化合物5を用いた本発明の有機エレクトロルミネッセンス素子は、理論限界値を超える高い外部量子効率を実現している点で極めて優れている。 (Example 11) Production and evaluation of organic electroluminescencedevice using compound 5 Each thin film was formed by vacuum deposition on a glass substrate on which an anode made of indium tin oxide (ITO) having a thickness of 100 nm was formed. And a degree of vacuum of 5.0 × 10 −4 Pa or less. First, α-NPD was formed on ITO with a thickness of 30 nm, and mCP was formed thereon with a thickness of 10 nm. Next, Compound 5 and (Cz) 2 DBF were co-evaporated from different vapor deposition sources to form a layer having a thickness of 15 nm as a light emitting layer. At this time, the concentration of Compound 5 was 6.0% by weight. Next, PPT was formed to a thickness of 10 nm, and TPBi was formed thereon to a thickness of 40 nm. Further, lithium fluoride (LiF) was vacuum-deposited at 0.8 nm, and then aluminum (Al) was evaporated at a thickness of 100 nm to form a cathode, thereby obtaining an organic electroluminescence device.
The emission spectrum of the produced organic electroluminescence device is shown in FIG. 21, and the current density-external quantum efficiency characteristic is shown in FIG. The organic electroluminescencedevice using Compound 5 as the light emitting material achieved a high external quantum efficiency of 15.3%. Assuming that an ideal organic electroluminescence device balanced using a fluorescent material having a light emission quantum efficiency of 100% is prototyped, if the light extraction efficiency is 20 to 30%, the external quantum efficiency of fluorescence emission is 5%. 7.5%. This value is generally regarded as a theoretical limit value of the external quantum efficiency of an organic electroluminescence device using a fluorescent material. Therefore, the organic electroluminescence device of the present invention using Compound 5 is extremely excellent in that high external quantum efficiency exceeding the theoretical limit value is realized.
膜厚100nmのインジウム・スズ酸化物(ITO)からなる陽極が形成されたガラス基板上に、各薄膜を真空蒸着法にて、真空度5.0×10-4Pa以下で積層した。まず、ITO上にα-NPDを30nmの厚さに形成し、この上に、mCPを10nmの厚さに形成した。次に、化合物5と(Cz)2DBFを異なる蒸着源から共蒸着し、15nmの厚さの層を形成して発光層とした。この時、化合物5の濃度は6.0重量%とした。次に、PPTを10nmの厚さに形成し、この上に、TPBiを40nmの厚さに形成した。さらにフッ化リチウム(LiF)を0.8nm真空蒸着し、次いでアルミニウム(Al)を100nmの厚さに蒸着することにより陰極を形成し、有機エレクトロルミネッセンス素子とした。
作製した有機エレクトロルミネッセンス素子の発光スペクトルを図21に示し、電流密度-外部量子効率特性を図22に示す。化合物5を発光材料として用いた有機エレクトロルミネッセンス素子は15.3%の高い外部量子効率を達成した。仮に発光量子効率が100%の蛍光材料を用いてバランスの取れた理想的な有機エレクトロルミネッセンス素子を試作したとすると、光取り出し効率が20~30%であれば、蛍光発光の外部量子効率は5~7.5%となる。この値が一般に、蛍光材料を用いた有機エレクトロルミネッセンス素子の外部量子効率の理論限界値とされている。したがって、化合物5を用いた本発明の有機エレクトロルミネッセンス素子は、理論限界値を超える高い外部量子効率を実現している点で極めて優れている。 (Example 11) Production and evaluation of organic electroluminescence
The emission spectrum of the produced organic electroluminescence device is shown in FIG. 21, and the current density-external quantum efficiency characteristic is shown in FIG. The organic electroluminescence
(実施例12) 化合物6を用いた有機エレクトロルミネッセンス素子の作製と評価
化合物5のかわりに化合物6を用いて、実施例4と同じ方法により有機エレクトロルミネッセンス素子を作製した。
作製した有機エレクトロルミネッセンス素子の発光スペクトルを図23に示し、電流密度-外部量子効率特性を図24に示す。化合物5を発光材料として用いた有機エレクトロルミネッセンス素子は8.9%の高い外部量子効率を達成した。 (Example 12) Production and evaluation of organic electroluminescencedevice using compound 6 An organic electroluminescence device was produced in the same manner as in Example 4 using compound 6 instead of compound 5.
The emission spectrum of the produced organic electroluminescence element is shown in FIG. 23, and the current density-external quantum efficiency characteristic is shown in FIG. The organic electroluminescencedevice using Compound 5 as the light emitting material achieved a high external quantum efficiency of 8.9%.
化合物5のかわりに化合物6を用いて、実施例4と同じ方法により有機エレクトロルミネッセンス素子を作製した。
作製した有機エレクトロルミネッセンス素子の発光スペクトルを図23に示し、電流密度-外部量子効率特性を図24に示す。化合物5を発光材料として用いた有機エレクトロルミネッセンス素子は8.9%の高い外部量子効率を達成した。 (Example 12) Production and evaluation of organic electroluminescence
The emission spectrum of the produced organic electroluminescence element is shown in FIG. 23, and the current density-external quantum efficiency characteristic is shown in FIG. The organic electroluminescence

本発明の化合物は発光材料として有用である。このため本発明の化合物は、有機エレクトロルミネッセンス素子などの有機発光素子用の発光材料として効果的に用いられる。本発明の化合物の中には、遅延蛍光が放射するものも含まれているため、発光効率が高い有機発光素子を提供することも可能である。このため、本発明は産業上の利用可能性が高い。
The compound of the present invention is useful as a luminescent material. For this reason, the compound of this invention is effectively used as a luminescent material for organic light emitting elements, such as an organic electroluminescent element. Since the compounds of the present invention include those that emit delayed fluorescence, it is also possible to provide an organic light-emitting device with high luminous efficiency. For this reason, this invention has high industrial applicability.
1 基板
2 陽極
3 正孔注入層
4 正孔輸送層
5 発光層
6 電子輸送層
7 陰極 DESCRIPTION OFSYMBOLS 1 Substrate 2 Anode 3 Hole injection layer 4 Hole transport layer 5 Light emitting layer 6 Electron transport layer 7 Cathode
2 陽極
3 正孔注入層
4 正孔輸送層
5 発光層
6 電子輸送層
7 陰極 DESCRIPTION OF
Claims (23)
- 下記一般式(1)で表される化合物からなる発光材料。

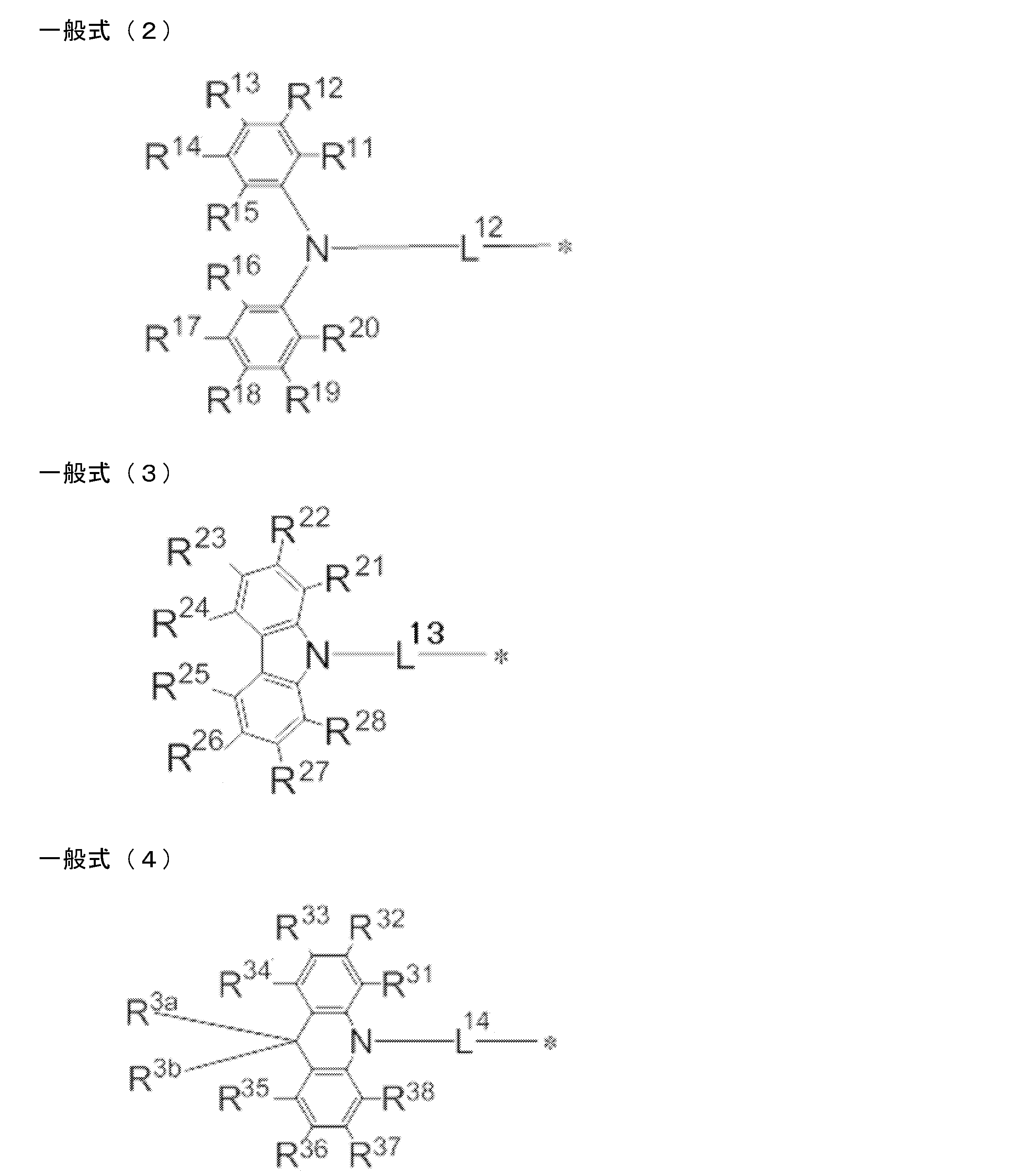

- 一般式(1)のR1~R8の少なくとも1つが前記一般式(3)~(7)のいずれかで表される基であることを特徴とする請求項1に記載の発光材料。 2. The luminescent material according to claim 1, wherein at least one of R 1 to R 8 in the general formula (1) is a group represented by any one of the general formulas (3) to (7).
- 一般式(1)のR1~R8の少なくとも1つが前記一般式(3)で表される基である場合に、前記一般式(3)のR21~R28のうち少なくとも1つは置換基であることを特徴とする請求項1または2に記載の発光材料。 When at least one of R 1 to R 8 in the general formula (1) is a group represented by the general formula (3), at least one of R 21 to R 28 in the general formula (3) is substituted. The luminescent material according to claim 1, wherein the luminescent material is a group.
- 一般式(1)のR2、R3、R6、およびR7の少なくとも1つが前記一般式(2)~(7)のいずれかで表される基であることを特徴とする請求項1~3のいずれか1項に記載の発光材料。 2. At least one of R 2 , R 3 , R 6 , and R 7 in the general formula (1) is a group represented by any one of the general formulas (2) to (7). 4. The light emitting material according to any one of items 1 to 3.
- 一般式(1)のR3およびR6の少なくとも1つが前記一般式(2)~(7)のいずれかで表される基であることを特徴とする請求項4に記載の発光材料。 The luminescent material according to claim 4, wherein at least one of R 3 and R 6 in the general formula (1) is a group represented by any one of the general formulas (2) to (7).
- 一般式(1)のR3とR6が、各々独立に前記一般式(2)~(7)のいずれかで表される基であることを特徴とする請求項5に記載の発光材料。 6. The luminescent material according to claim 5, wherein R 3 and R 6 in the general formula (1) are each independently a group represented by any one of the general formulas (2) to (7).
- 前記一般式(2)のR11~R20の少なくとも1つ、前記一般式(3)のR21~R28の少なくとも1つ、前記一般式(4)のR31~R38の少なくとも1つと、R3aおよびR3bの少なくとも1つ、前記一般式(5)のR41~R48の少なくとも1つ、前記一般式(6)のR51~R58の少なくとも1つ、および前記一般式(7)のR61~R68の少なくとも1つが置換基であることを特徴とする請求項1~6のいずれか1項に記載の発光材料。 At least one of R 11 to R 20 in the general formula (2), at least one of R 21 to R 28 in the general formula (3), and at least one of R 31 to R 38 in the general formula (4); , R 3a and R 3b , at least one of R 41 to R 48 in the general formula (5), at least one of R 51 to R 58 in the general formula (6), and the general formula ( The luminescent material according to any one of claims 1 to 6, wherein at least one of R 61 to R 68 in 7) is a substituent.
- 前記一般式(2)のR13およびR18の少なくとも一方、前記一般式(3)のR23およびR26の少なくとも一方、前記一般式(4)のR33およびR36の少なくとも一方と、R3aおよびR3bの少なくとも一方、前記一般式(5)のR43およびR46の少なくとも一方、前記一般式(6)のR53およびR56の少なくとも一方、並びに前記一般式(7)のR63およびR66の少なくとも一方が置換基であることを特徴とする請求項7に記載の発光材料。 At least one of R 13 and R 18 in the general formula (2), at least one of R 23 and R 26 in the general formula (3), at least one of R 33 and R 36 in the general formula (4), and R At least one of 3a and R 3b , at least one of R 43 and R 46 in the general formula (5), at least one of R 53 and R 56 in the general formula (6), and R 63 in the general formula (7). The luminescent material according to claim 7, wherein at least one of R 66 and R 66 is a substituent.
- 前記一般式(2)のR13およびR18の少なくとも一方、前記一般式(3)のR23およびR26の少なくとも一方、前記一般式(4)のR33およびR36の少なくとも一方と、R3aおよびR3bの少なくとも一方、前記一般式(5)のR43およびR46の少なくとも一方、前記一般式(6)のR53およびR56の少なくとも一方、並びに前記一般式(7)のR63およびR66の少なくとも一方が、前記一般式(2)~(7)のいずれかで表される基であることを特徴とする請求項8に記載の発光材料。 At least one of R 13 and R 18 in the general formula (2), at least one of R 23 and R 26 in the general formula (3), at least one of R 33 and R 36 in the general formula (4), and R At least one of 3a and R 3b , at least one of R 43 and R 46 in the general formula (5), at least one of R 53 and R 56 in the general formula (6), and R 63 in the general formula (7). 9. The luminescent material according to claim 8, wherein at least one of R 66 and R 66 is a group represented by any one of the general formulas (2) to (7).
- 前記一般式(2)~(7)のL12~L17が、単結合であることを特徴とする請求項1~9のいずれか1項に記載の発光材料。 10. The luminescent material according to claim 1, wherein L 12 to L 17 in the general formulas (2) to (7) are single bonds.
- 一般式(1)のXが、酸素原子であることを特徴とする請求項1~10のいずれか1項に記載の発光材料。 11. The luminescent material according to claim 1, wherein X in the general formula (1) is an oxygen atom.
- 一般式(1)のR9が、下記一般式(a)で表される基であることを特徴とする請求項1~11のいずれか1項に記載の発光材料。

- 前記一般式(a)のR9aとR9eが置換基であることを特徴とする請求項12に記載の発光材料。 The luminescent material according to claim 12, wherein R 9a and R 9e in the general formula (a) are substituents.
- 一般式(1)のR1~R8の少なくとも1つが前記一般式(4)で表される基であることを特徴とする請求項1~13のいずれか1項に記載の発光材料。 The luminescent material according to any one of claims 1 to 13, wherein at least one of R 1 to R 8 in the general formula (1) is a group represented by the general formula (4).
- 一般式(1)のR3とR6、またはR2とR7が、前記一般式(4)で表される基であることを特徴とする請求項1~4、7~14のいずれか1項に記載の発光材料。 R 3 and R 6 or R 2 and R 7, the general formula (1) is any one of claims 1 to 4 and 7 to 14, which is a group represented by the general formula (4) 2. The light emitting material according to item 1.
- 前記一般式(4)のR3aとR3bが、置換基であることを特徴とする請求項14または15に記載の発光材料。 16. The light emitting material according to claim 14, wherein R 3a and R 3b in the general formula (4) are substituents.
- 前記置換基が、炭素数1~15のアルキル基またはフェニル基であることを特徴とする請求項14~16のいずれか1項に記載の発光材料。 The luminescent material according to any one of claims 14 to 16, wherein the substituent is an alkyl group having 1 to 15 carbon atoms or a phenyl group.
- 前記一般式(4)のR3aとR3bが互いに結合して環状構造を形成していることを特徴とする請求項14~16のいずれか1項に記載の発光材料。 The luminescent material according to any one of claims 14 to 16, wherein R 3a and R 3b in the general formula (4) are bonded to each other to form a cyclic structure.
- 下記一般式(1)で表される化合物からなる遅延蛍光体。

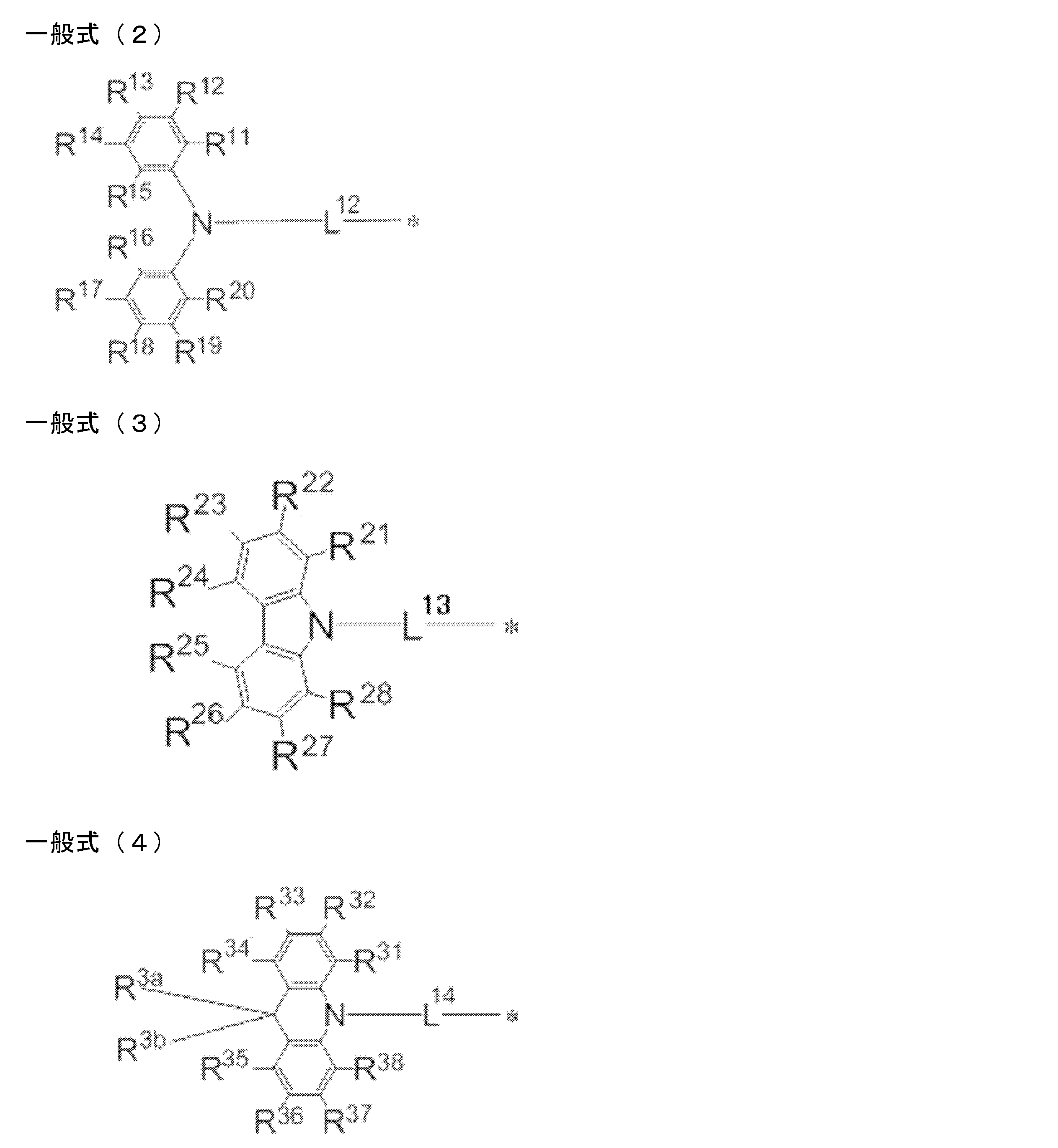
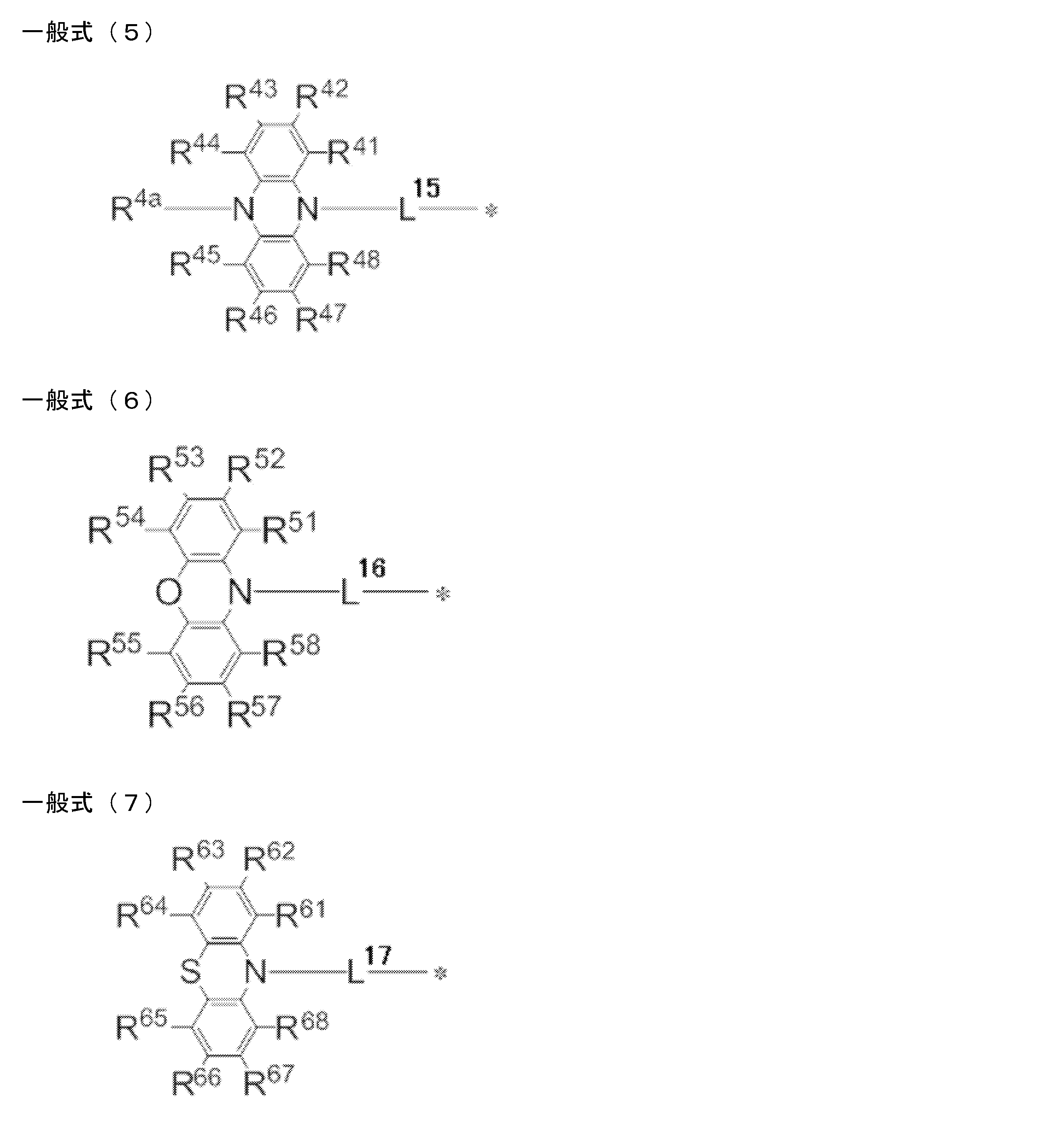
- 請求項1~18のいずれか1項に記載の発光材料を含むことを特徴とする有機発光素子。 An organic light emitting device comprising the light emitting material according to any one of claims 1 to 18.
- 遅延蛍光を放射することを特徴とする請求項20に記載の有機発光素子。 The organic light-emitting device according to claim 20, which emits delayed fluorescence.
- 有機エレクトロルミネッセンス素子であることを特徴とする請求項20または21に記載の有機発光素子。 The organic light-emitting device according to claim 20 or 21, wherein the organic light-emitting device is an organic electroluminescence device.
- 下記一般式(1’)で表される化合物。



Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015547802A JP6392240B2 (en) | 2013-11-18 | 2014-11-14 | Luminescent materials, organic light emitting devices and compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-237824 | 2013-11-18 | ||
| JP2013237824 | 2013-11-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015072537A1 true WO2015072537A1 (en) | 2015-05-21 |
Family
ID=53057471
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/080162 WO2015072537A1 (en) | 2013-11-18 | 2014-11-14 | Light-emitting material, organic light-emitting element, and compound |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP6392240B2 (en) |
| TW (1) | TW201527485A (en) |
| WO (1) | WO2015072537A1 (en) |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017126606A (en) * | 2016-01-12 | 2017-07-20 | コニカミノルタ株式会社 | Electronic device material, organic electroluminescent element, display device, and illuminating device |
| WO2018159662A1 (en) * | 2017-02-28 | 2018-09-07 | 国立大学法人九州大学 | Compound, light emitting material and organic light emitting element |
| WO2018212169A1 (en) * | 2017-05-16 | 2018-11-22 | 学校法人関西学院 | Polycyclic aromatic compound |
| US10333085B2 (en) | 2016-07-13 | 2019-06-25 | Samsung Display Co., Ltd. | Polycyclic compound and organic electroluminescence device including the same |
| WO2019131079A1 (en) * | 2017-12-25 | 2019-07-04 | 学校法人関西学院 | Compound including boron as spiro atom and macromolecular compound thereof |
| EP3508490A1 (en) * | 2018-01-08 | 2019-07-10 | Samsung Display Co., Ltd. | Organic electroluminescence device and polycyclic compound for organic electroluminescence device |
| CN110272440A (en) * | 2018-03-15 | 2019-09-24 | 江苏三月光电科技有限公司 | A kind of boracic 6-membered heterocyclic compound and its application in organic electroluminescence device |
| WO2020076796A1 (en) | 2018-10-09 | 2020-04-16 | Kyulux, Inc. | Novel composition of matter for use in organic light-emitting diodes |
| CN112661778A (en) * | 2020-12-30 | 2021-04-16 | 中节能万润股份有限公司 | Boron-containing aryl imide organic electroluminescent material and application thereof |
| WO2021157642A1 (en) | 2020-02-04 | 2021-08-12 | 株式会社Kyulux | Host material, composition, and organic electroluminescent element |
| US11101440B2 (en) | 2015-07-01 | 2021-08-24 | Kyushu University, National University Corporation | Organic electroluminescent device |
| WO2021235549A1 (en) | 2020-05-22 | 2021-11-25 | 株式会社Kyulux | Compound, light-emitting material, and light-emitting element |
| WO2022009883A1 (en) | 2020-07-08 | 2022-01-13 | 日鉄ケミカル&マテリアル株式会社 | Heat-activated delayed fluorescence material and organic electroluminescent element |
| WO2022025248A1 (en) | 2020-07-31 | 2022-02-03 | 株式会社Kyulux | Compound, light-emitting material, and light-emitting element |
| US11335872B2 (en) | 2016-09-06 | 2022-05-17 | Kyulux, Inc. | Organic light-emitting device |
| JP2022077011A (en) * | 2020-11-10 | 2022-05-20 | エルジー ディスプレイ カンパニー リミテッド | Organic light-emitting diode and organic light-emitting device |
| WO2022168956A1 (en) | 2021-02-04 | 2022-08-11 | 株式会社Kyulux | Compound, light-emitting material, and organic light-emitting element |
| US11462690B2 (en) | 2018-11-29 | 2022-10-04 | Samsung Display Co., Ltd. | Organic electroluminescence device and polycyclic compound for organic electroluminescence device |
| US11476435B2 (en) | 2017-08-24 | 2022-10-18 | Kyushu University, National University Corporation | Film and organic light-emitting device containing perovskite-type compound and organic light-emitting material |
| US11482679B2 (en) | 2017-05-23 | 2022-10-25 | Kyushu University, National University Corporation | Compound, light-emitting lifetime lengthening agent, use of n-type compound, film and light-emitting device |
| WO2022244503A1 (en) | 2021-05-20 | 2022-11-24 | 株式会社Kyulux | Organic light emitting element |
| WO2022270602A1 (en) | 2021-06-23 | 2022-12-29 | 株式会社Kyulux | Organic light-emitting element and film |
| WO2022270113A1 (en) | 2021-06-23 | 2022-12-29 | 株式会社Kyulux | Organic electroluminescent element |
| WO2022270354A1 (en) | 2021-06-23 | 2022-12-29 | 株式会社Kyulux | Compound, light-emitting material, and organic light-emitting element |
| WO2023282224A1 (en) | 2021-07-06 | 2023-01-12 | 株式会社Kyulux | Organic light-emitting element and design method therefor |
| US11634438B2 (en) | 2018-04-12 | 2023-04-25 | Samsung Display Co., Ltd. | Heterocyclic compound and organic light-emitting device including the same |
| US11785837B2 (en) | 2019-11-08 | 2023-10-10 | Samsung Display Co., Ltd. | Organic electroluminescence device and polycyclic compound for organic electroluminescence device |
| US11930654B2 (en) | 2017-07-06 | 2024-03-12 | Kyulux, Inc. | Organic light-emitting element |
| US11950492B2 (en) | 2019-01-29 | 2024-04-02 | Samsung Display Co., Ltd. | Organic electroluminescence device and polycyclic compound for organic electroluminescence device |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109575056A (en) * | 2017-09-29 | 2019-04-05 | 江苏三月光电科技有限公司 | A kind of boracic organic compound and its application in organic electroluminescence device |
| WO2019062684A1 (en) * | 2017-09-29 | 2019-04-04 | 江苏三月光电科技有限公司 | Boron-containing organic compound and application thereof in organic electroluminescent device |
| CN113980054B (en) * | 2021-11-18 | 2023-12-15 | 广东工业大学 | Phosphine oxide-containing compound and preparation and application thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013526548A (en) * | 2010-05-14 | 2013-06-24 | ユニバーサル ディスプレイ コーポレイション | Azaborin compounds as host materials and dopants for PHOLEDs |
-
2014
- 2014-11-14 TW TW103139668A patent/TW201527485A/en unknown
- 2014-11-14 JP JP2015547802A patent/JP6392240B2/en active Active
- 2014-11-14 WO PCT/JP2014/080162 patent/WO2015072537A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013526548A (en) * | 2010-05-14 | 2013-06-24 | ユニバーサル ディスプレイ コーポレイション | Azaborin compounds as host materials and dopants for PHOLEDs |
Non-Patent Citations (4)
| Title |
|---|
| A. K. C. MENGEL ET AL.: "A Triarylamine- Triarylborane Dyad with a Photochromic Dithienylethene Bridge", THE JOURNAL OF ORGANIC CHEMISTRY, vol. 77, 16 July 2012 (2012-07-16), pages 6545 - 6552 * |
| J. KOBAYASHI ET AL.: "Synthesis of Dibenzochalcogenaborins and Systematic Comparisons of Their Optical Properties by Changing a Bridging Chalcogen Atom", CHEMISTRY - AN ASIAN JOURNAL, vol. 4, 19 November 2008 (2008-11-19), pages 42 - 49 * |
| P. CHEN ET AL.: "n-Expanded Borazine: An Ambipolar Conjugated B-n-N Macrocycle", ANGEWANDTE CHEMIE INTERNATIONAL EDITION, vol. 51, 5 July 2012 (2012-07-05), pages 7994 - 7998 * |
| T. AGOU ET AL.: "Development of a general route to periphery-functionalized azaborines and ladder-type azaborines by using common intermediates", CHEMICAL COMMUNICATIONS, pages 3204 - 3206 * |
Cited By (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11101440B2 (en) | 2015-07-01 | 2021-08-24 | Kyushu University, National University Corporation | Organic electroluminescent device |
| JP2017126606A (en) * | 2016-01-12 | 2017-07-20 | コニカミノルタ株式会社 | Electronic device material, organic electroluminescent element, display device, and illuminating device |
| US10333085B2 (en) | 2016-07-13 | 2019-06-25 | Samsung Display Co., Ltd. | Polycyclic compound and organic electroluminescence device including the same |
| US11335872B2 (en) | 2016-09-06 | 2022-05-17 | Kyulux, Inc. | Organic light-emitting device |
| WO2018159662A1 (en) * | 2017-02-28 | 2018-09-07 | 国立大学法人九州大学 | Compound, light emitting material and organic light emitting element |
| CN110520421A (en) * | 2017-02-28 | 2019-11-29 | 国立大学法人九州大学 | Compound, luminescent material and organic illuminating element |
| WO2018212169A1 (en) * | 2017-05-16 | 2018-11-22 | 学校法人関西学院 | Polycyclic aromatic compound |
| JP7202572B2 (en) | 2017-05-16 | 2023-01-12 | 学校法人関西学院 | polycyclic aromatic compound |
| CN110612304A (en) * | 2017-05-16 | 2019-12-24 | 学校法人关西学院 | Polycyclic aromatic compound |
| JPWO2018212169A1 (en) * | 2017-05-16 | 2020-03-19 | 学校法人関西学院 | Polycyclic aromatic compounds |
| US11482679B2 (en) | 2017-05-23 | 2022-10-25 | Kyushu University, National University Corporation | Compound, light-emitting lifetime lengthening agent, use of n-type compound, film and light-emitting device |
| US11930654B2 (en) | 2017-07-06 | 2024-03-12 | Kyulux, Inc. | Organic light-emitting element |
| US11476435B2 (en) | 2017-08-24 | 2022-10-18 | Kyushu University, National University Corporation | Film and organic light-emitting device containing perovskite-type compound and organic light-emitting material |
| WO2019131079A1 (en) * | 2017-12-25 | 2019-07-04 | 学校法人関西学院 | Compound including boron as spiro atom and macromolecular compound thereof |
| JP7341412B2 (en) | 2017-12-25 | 2023-09-11 | 学校法人関西学院 | Compounds with boron as a spiro atom and their polymer compounds |
| KR20200101969A (en) | 2017-12-25 | 2020-08-28 | 가꼬우 호징 관세이 가쿠잉 | Compounds containing boron as a spiro atom, and polymer compounds thereof |
| JPWO2019131079A1 (en) * | 2017-12-25 | 2021-01-28 | 学校法人関西学院 | Compounds with boron as a spiro atom and its polymer compounds |
| CN111527094A (en) * | 2017-12-25 | 2020-08-11 | 学校法人关西学院 | Compound having boron as spiro atom and polymer compound thereof |
| CN111527094B (en) * | 2017-12-25 | 2024-03-26 | 学校法人关西学院 | Compound, polymer compound, material for organic element, organic electroluminescent element, display device, and lighting device |
| EP3508490A1 (en) * | 2018-01-08 | 2019-07-10 | Samsung Display Co., Ltd. | Organic electroluminescence device and polycyclic compound for organic electroluminescence device |
| US11251380B2 (en) | 2018-01-08 | 2022-02-15 | Samsung Display Co., Ltd. | Organic electroluminescence device and polycyclic compound for organic electroluminescence device |
| CN110272440A (en) * | 2018-03-15 | 2019-09-24 | 江苏三月光电科技有限公司 | A kind of boracic 6-membered heterocyclic compound and its application in organic electroluminescence device |
| US11634438B2 (en) | 2018-04-12 | 2023-04-25 | Samsung Display Co., Ltd. | Heterocyclic compound and organic light-emitting device including the same |
| WO2020076796A1 (en) | 2018-10-09 | 2020-04-16 | Kyulux, Inc. | Novel composition of matter for use in organic light-emitting diodes |
| US11462690B2 (en) | 2018-11-29 | 2022-10-04 | Samsung Display Co., Ltd. | Organic electroluminescence device and polycyclic compound for organic electroluminescence device |
| US11950492B2 (en) | 2019-01-29 | 2024-04-02 | Samsung Display Co., Ltd. | Organic electroluminescence device and polycyclic compound for organic electroluminescence device |
| US11785837B2 (en) | 2019-11-08 | 2023-10-10 | Samsung Display Co., Ltd. | Organic electroluminescence device and polycyclic compound for organic electroluminescence device |
| WO2021157642A1 (en) | 2020-02-04 | 2021-08-12 | 株式会社Kyulux | Host material, composition, and organic electroluminescent element |
| WO2021157593A1 (en) | 2020-02-04 | 2021-08-12 | 株式会社Kyulux | Composition, film, organic light emitting element, method for providing light emitting composition, and program |
| WO2021235549A1 (en) | 2020-05-22 | 2021-11-25 | 株式会社Kyulux | Compound, light-emitting material, and light-emitting element |
| KR20230035528A (en) | 2020-07-08 | 2023-03-14 | 닛테츠 케미컬 앤드 머티리얼 가부시키가이샤 | Thermally activated delayed fluorescent materials, and organic electroluminescent devices |
| WO2022009883A1 (en) | 2020-07-08 | 2022-01-13 | 日鉄ケミカル&マテリアル株式会社 | Heat-activated delayed fluorescence material and organic electroluminescent element |
| WO2022025248A1 (en) | 2020-07-31 | 2022-02-03 | 株式会社Kyulux | Compound, light-emitting material, and light-emitting element |
| JP7246453B2 (en) | 2020-11-10 | 2023-03-27 | エルジー ディスプレイ カンパニー リミテッド | Organic Light Emitting Diodes and Organic Light Emitting Devices |
| JP2022077011A (en) * | 2020-11-10 | 2022-05-20 | エルジー ディスプレイ カンパニー リミテッド | Organic light-emitting diode and organic light-emitting device |
| CN112661778A (en) * | 2020-12-30 | 2021-04-16 | 中节能万润股份有限公司 | Boron-containing aryl imide organic electroluminescent material and application thereof |
| WO2022168956A1 (en) | 2021-02-04 | 2022-08-11 | 株式会社Kyulux | Compound, light-emitting material, and organic light-emitting element |
| WO2022244503A1 (en) | 2021-05-20 | 2022-11-24 | 株式会社Kyulux | Organic light emitting element |
| WO2022270354A1 (en) | 2021-06-23 | 2022-12-29 | 株式会社Kyulux | Compound, light-emitting material, and organic light-emitting element |
| WO2022270113A1 (en) | 2021-06-23 | 2022-12-29 | 株式会社Kyulux | Organic electroluminescent element |
| WO2022270602A1 (en) | 2021-06-23 | 2022-12-29 | 株式会社Kyulux | Organic light-emitting element and film |
| WO2023282224A1 (en) | 2021-07-06 | 2023-01-12 | 株式会社Kyulux | Organic light-emitting element and design method therefor |
Also Published As
| Publication number | Publication date |
|---|---|
| TW201527485A (en) | 2015-07-16 |
| JPWO2015072537A1 (en) | 2017-03-16 |
| JP6392240B2 (en) | 2018-09-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6392240B2 (en) | Luminescent materials, organic light emitting devices and compounds | |
| JP6318155B2 (en) | COMPOUND, LIGHT EMITTING MATERIAL AND ORGANIC LIGHT EMITTING DEVICE | |
| WO2015080182A1 (en) | Light-emitting material, organic light-emitting element, and compound | |
| JP6430370B2 (en) | Luminescent materials, organic light emitting devices and compounds | |
| JP6284370B2 (en) | Luminescent materials, organic light emitting devices and compounds | |
| WO2015080183A1 (en) | Light-emitting material, organic light-emitting element, and compound | |
| JP6225111B2 (en) | Luminescent material, compound, and organic light emitting device using the same | |
| JP6466913B2 (en) | Luminescent materials, organic light emitting devices and compounds | |
| JP6293417B2 (en) | COMPOUND, LIGHT EMITTING MATERIAL AND ORGANIC LIGHT EMITTING DEVICE | |
| JP6472383B2 (en) | Luminescent material, delayed phosphor and organic light emitting device using the same | |
| JP6367189B2 (en) | Luminescent materials, organic light emitting devices and compounds | |
| JP6326050B2 (en) | COMPOUND, LIGHT EMITTING MATERIAL AND ORGANIC LIGHT EMITTING DEVICE | |
| JP6383538B2 (en) | Luminescent materials, organic light emitting devices and compounds | |
| WO2015002213A1 (en) | Light-emitting material, long-persistent phosphor, organic light-emitting element and compound | |
| WO2015129715A1 (en) | Luminescent material, organic luminescent element, and compound | |
| WO2015133501A1 (en) | Light-emitting material, organic light-emitting element, and compound | |
| WO2014133121A1 (en) | Compound, light-emitting material, and organic light-emitting element | |
| WO2015129714A1 (en) | Light-emitting material, organic light-emitting element, and compound | |
| JPWO2013161437A1 (en) | Luminescent material and organic light emitting device | |
| JP6647514B2 (en) | Organic light emitting device and light emitting material and compound used therefor | |
| WO2019004254A1 (en) | Light-emitting material, compound, long-persistent phosphor and light-emitting element | |
| WO2018043241A1 (en) | Organic light-emitting element, and luminescent material and compound used therefor | |
| JP6622484B2 (en) | Luminescent materials, organic light emitting devices and compounds | |
| WO2016111196A1 (en) | Compound, mixture, light-emitting layer, organic light-emitting element, and assist dopant | |
| JP2019006988A (en) | Light-emitting material, compound, delayed fluorescent material, and light-emitting element |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14862491 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015547802 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14862491 Country of ref document: EP Kind code of ref document: A1 |