WO2014201972A1 - 苯并咪唑-2-哌嗪杂环类化合物、其药物组合物及其制备方法和用途 - Google Patents
苯并咪唑-2-哌嗪杂环类化合物、其药物组合物及其制备方法和用途 Download PDFInfo
- Publication number
- WO2014201972A1 WO2014201972A1 PCT/CN2014/079827 CN2014079827W WO2014201972A1 WO 2014201972 A1 WO2014201972 A1 WO 2014201972A1 CN 2014079827 W CN2014079827 W CN 2014079827W WO 2014201972 A1 WO2014201972 A1 WO 2014201972A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cancer
- compound
- pharmaceutical composition
- formula
- medicament
- Prior art date
Links
- 238000002360 preparation method Methods 0.000 title claims abstract description 89
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 73
- 239000003814 drug Substances 0.000 claims abstract description 68
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 claims abstract description 65
- 150000001875 compounds Chemical class 0.000 claims description 260
- 206010028980 Neoplasm Diseases 0.000 claims description 116
- 201000011510 cancer Diseases 0.000 claims description 74
- 238000010534 nucleophilic substitution reaction Methods 0.000 claims description 43
- 238000004519 manufacturing process Methods 0.000 claims description 38
- 230000006801 homologous recombination Effects 0.000 claims description 36
- 238000002744 homologous recombination Methods 0.000 claims description 36
- 238000011282 treatment Methods 0.000 claims description 36
- 230000000694 effects Effects 0.000 claims description 33
- 230000005782 double-strand break Effects 0.000 claims description 30
- 230000008439 repair process Effects 0.000 claims description 29
- 102100023712 Poly [ADP-ribose] polymerase 1 Human genes 0.000 claims description 27
- 150000003839 salts Chemical class 0.000 claims description 27
- 201000010099 disease Diseases 0.000 claims description 26
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 26
- 230000005764 inhibitory process Effects 0.000 claims description 25
- -1 pyrimidoimidazole compound Chemical class 0.000 claims description 24
- 239000000243 solution Substances 0.000 claims description 22
- 230000001668 ameliorated effect Effects 0.000 claims description 21
- 101710179684 Poly [ADP-ribose] polymerase Proteins 0.000 claims description 20
- 229910052739 hydrogen Inorganic materials 0.000 claims description 19
- 239000001257 hydrogen Substances 0.000 claims description 19
- 238000000034 method Methods 0.000 claims description 19
- 238000006243 chemical reaction Methods 0.000 claims description 16
- 230000001419 dependent effect Effects 0.000 claims description 15
- 239000003937 drug carrier Substances 0.000 claims description 15
- 238000001959 radiotherapy Methods 0.000 claims description 14
- 239000004480 active ingredient Substances 0.000 claims description 13
- 238000002512 chemotherapy Methods 0.000 claims description 13
- 239000003085 diluting agent Substances 0.000 claims description 12
- 229940079593 drug Drugs 0.000 claims description 12
- 208000026310 Breast neoplasm Diseases 0.000 claims description 11
- 206010033128 Ovarian cancer Diseases 0.000 claims description 11
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 11
- 206010040070 Septic Shock Diseases 0.000 claims description 11
- 208000027866 inflammatory disease Diseases 0.000 claims description 11
- 230000036303 septic shock Effects 0.000 claims description 11
- 206010006187 Breast cancer Diseases 0.000 claims description 10
- 208000032456 Hemorrhagic Shock Diseases 0.000 claims description 10
- 206010029350 Neurotoxicity Diseases 0.000 claims description 10
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 10
- 206010060862 Prostate cancer Diseases 0.000 claims description 10
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 10
- 206010049771 Shock haemorrhagic Diseases 0.000 claims description 10
- 206010044221 Toxic encephalopathy Diseases 0.000 claims description 10
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 10
- 208000037906 ischaemic injury Diseases 0.000 claims description 10
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 10
- 201000006417 multiple sclerosis Diseases 0.000 claims description 10
- 230000007135 neurotoxicity Effects 0.000 claims description 10
- 231100000228 neurotoxicity Toxicity 0.000 claims description 10
- 201000002528 pancreatic cancer Diseases 0.000 claims description 10
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 10
- 208000019553 vascular disease Diseases 0.000 claims description 10
- 230000008569 process Effects 0.000 claims description 9
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 claims description 8
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 claims description 8
- 229930195733 hydrocarbon Natural products 0.000 claims description 8
- 239000007900 aqueous suspension Substances 0.000 claims description 7
- 239000002775 capsule Substances 0.000 claims description 7
- 239000006071 cream Substances 0.000 claims description 7
- 239000000839 emulsion Substances 0.000 claims description 7
- 239000008187 granular material Substances 0.000 claims description 7
- 239000007924 injection Substances 0.000 claims description 7
- 238000002347 injection Methods 0.000 claims description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 7
- 239000002674 ointment Substances 0.000 claims description 7
- 239000000843 powder Substances 0.000 claims description 7
- 230000002829 reductive effect Effects 0.000 claims description 7
- 239000000829 suppository Substances 0.000 claims description 7
- 239000000725 suspension Substances 0.000 claims description 7
- 239000006188 syrup Substances 0.000 claims description 7
- 235000020357 syrup Nutrition 0.000 claims description 7
- 239000003826 tablet Substances 0.000 claims description 7
- 230000007812 deficiency Effects 0.000 claims description 6
- 229910052736 halogen Chemical group 0.000 claims description 6
- 150000002367 halogens Chemical group 0.000 claims description 6
- 239000000463 material Substances 0.000 claims description 6
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 6
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 claims description 6
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 claims description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 5
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 claims description 5
- 125000001183 hydrocarbyl group Chemical group 0.000 claims description 5
- 239000000203 mixture Substances 0.000 claims description 5
- 239000004215 Carbon black (E152) Substances 0.000 claims description 4
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 claims description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical group Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 4
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 claims description 4
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 4
- 230000002950 deficient Effects 0.000 claims description 4
- 150000002430 hydrocarbons Chemical class 0.000 claims description 4
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 4
- BLJHLOLVEXWHFS-UHFFFAOYSA-N methyl 2,3-diaminobenzoate Chemical class COC(=O)C1=CC=CC(N)=C1N BLJHLOLVEXWHFS-UHFFFAOYSA-N 0.000 claims description 4
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 4
- 229910052757 nitrogen Inorganic materials 0.000 claims description 4
- 125000003554 tetrahydropyrrolyl group Chemical group 0.000 claims description 4
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 4
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 claims description 3
- 239000002253 acid Substances 0.000 claims description 3
- 239000002671 adjuvant Substances 0.000 claims description 3
- 150000001412 amines Chemical class 0.000 claims description 3
- 229910052731 fluorine Chemical group 0.000 claims description 3
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 claims description 3
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 claims description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 3
- 238000006467 substitution reaction Methods 0.000 claims description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 claims description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical group FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 2
- 229910019142 PO4 Inorganic materials 0.000 claims description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 2
- CBHOOMGKXCMKIR-UHFFFAOYSA-N azane;methanol Chemical compound N.OC CBHOOMGKXCMKIR-UHFFFAOYSA-N 0.000 claims description 2
- 230000008878 coupling Effects 0.000 claims description 2
- 238000010168 coupling process Methods 0.000 claims description 2
- 238000005859 coupling reaction Methods 0.000 claims description 2
- 230000007547 defect Effects 0.000 claims description 2
- 239000011737 fluorine Chemical group 0.000 claims description 2
- 125000001153 fluoro group Chemical group F* 0.000 claims description 2
- 239000007937 lozenge Substances 0.000 claims description 2
- 229940049920 malate Drugs 0.000 claims description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 2
- 239000010452 phosphate Substances 0.000 claims description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 claims description 2
- 229940095064 tartrate Drugs 0.000 claims description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 claims description 2
- 125000004185 ester group Chemical group 0.000 claims 1
- QEWYKACRFQMRMB-UHFFFAOYSA-N fluoroacetic acid Chemical compound OC(=O)CF QEWYKACRFQMRMB-UHFFFAOYSA-N 0.000 claims 1
- 125000002485 formyl group Chemical class [H]C(*)=O 0.000 claims 1
- 238000006460 hydrolysis reaction Methods 0.000 claims 1
- 230000035772 mutation Effects 0.000 claims 1
- 125000003386 piperidinyl group Chemical group 0.000 claims 1
- 238000002560 therapeutic procedure Methods 0.000 claims 1
- 102000012338 Poly(ADP-ribose) Polymerases Human genes 0.000 abstract description 50
- 108010061844 Poly(ADP-ribose) Polymerases Proteins 0.000 abstract description 50
- 239000003112 inhibitor Substances 0.000 abstract description 6
- 229940124597 therapeutic agent Drugs 0.000 abstract description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 35
- 229940125904 compound 1 Drugs 0.000 description 26
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 230000005778 DNA damage Effects 0.000 description 12
- 231100000277 DNA damage Toxicity 0.000 description 12
- OPGUZRRLMQSMAQ-UHFFFAOYSA-N 5-(4-methoxyphenyl)-1-phenylbenzimidazole Chemical compound C1=CC(OC)=CC=C1C1=CC=C(N(C=N2)C=3C=CC=CC=3)C2=C1 OPGUZRRLMQSMAQ-UHFFFAOYSA-N 0.000 description 10
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 9
- ZHNUHDYFZUAESO-UHFFFAOYSA-N formamide Substances NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 108010064218 Poly (ADP-Ribose) Polymerase-1 Proteins 0.000 description 8
- 210000004881 tumor cell Anatomy 0.000 description 8
- 210000004027 cell Anatomy 0.000 description 7
- 239000013642 negative control Substances 0.000 description 7
- 102000004190 Enzymes Human genes 0.000 description 6
- 108090000790 Enzymes Proteins 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 6
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 6
- 150000002431 hydrogen Chemical class 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- 108010033040 Histones Proteins 0.000 description 5
- 239000012661 PARP inhibitor Substances 0.000 description 5
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 230000003197 catalytic effect Effects 0.000 description 5
- 239000013641 positive control Substances 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 5
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- DBPKMSBWOKAKLA-UHFFFAOYSA-N 4-chloropyrimidine Chemical compound ClC1=CC=NC=N1 DBPKMSBWOKAKLA-UHFFFAOYSA-N 0.000 description 4
- 108091026813 Poly(ADPribose) Proteins 0.000 description 4
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 238000003818 flash chromatography Methods 0.000 description 4
- 230000007246 mechanism Effects 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 239000005711 Benzoic acid Substances 0.000 description 3
- 230000033616 DNA repair Effects 0.000 description 3
- 230000008265 DNA repair mechanism Effects 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 102000007999 Nuclear Proteins Human genes 0.000 description 3
- 108010089610 Nuclear Proteins Proteins 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- 239000013504 Triton X-100 Substances 0.000 description 3
- 229920004890 Triton X-100 Polymers 0.000 description 3
- 235000010233 benzoic acid Nutrition 0.000 description 3
- 239000004359 castor oil Substances 0.000 description 3
- 235000019438 castor oil Nutrition 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 229940044683 chemotherapy drug Drugs 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 231100000433 cytotoxic Toxicity 0.000 description 3
- 230000001472 cytotoxic effect Effects 0.000 description 3
- 235000019439 ethyl acetate Nutrition 0.000 description 3
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 3
- 230000005855 radiation Effects 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 229940126585 therapeutic drug Drugs 0.000 description 3
- ZBNZAJFNDPPMDT-UHFFFAOYSA-N 1h-imidazole-5-carboxamide Chemical compound NC(=O)C1=CNC=N1 ZBNZAJFNDPPMDT-UHFFFAOYSA-N 0.000 description 2
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 2
- PJWWMVQRAZJKGT-UHFFFAOYSA-N 2-amino-5-fluoro-3-nitrobenzoic acid Chemical compound NC1=C(C(O)=O)C=C(F)C=C1[N+]([O-])=O PJWWMVQRAZJKGT-UHFFFAOYSA-N 0.000 description 2
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- SRNWOUGRCWSEMX-KEOHHSTQSA-N ADP-beta-D-ribose Chemical group C([C@H]1O[C@H]([C@@H]([C@@H]1O)O)N1C=2N=CN=C(C=2N=C1)N)OP(O)(=O)OP(O)(=O)OC[C@H]1O[C@@H](O)[C@H](O)[C@@H]1O SRNWOUGRCWSEMX-KEOHHSTQSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 230000005971 DNA damage repair Effects 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- 206010028851 Necrosis Diseases 0.000 description 2
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 102100023652 Poly [ADP-ribose] polymerase 2 Human genes 0.000 description 2
- 101710144590 Poly [ADP-ribose] polymerase 2 Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 238000003149 assay kit Methods 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 230000022131 cell cycle Effects 0.000 description 2
- 231100000244 chromosomal damage Toxicity 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 230000005865 ionizing radiation Effects 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000002207 metabolite Substances 0.000 description 2
- ADEDGAKMZOCLDK-UHFFFAOYSA-N methyl 2,3-diamino-5-fluorobenzoate Chemical compound COC(=O)C1=CC(F)=CC(N)=C1N ADEDGAKMZOCLDK-UHFFFAOYSA-N 0.000 description 2
- DQKJPBJOMJODBX-UHFFFAOYSA-N methyl 2-amino-5-fluoro-3-nitrobenzoate Chemical compound COC(=O)C1=CC(F)=CC([N+]([O-])=O)=C1N DQKJPBJOMJODBX-UHFFFAOYSA-N 0.000 description 2
- TZIHFWKZFHZASV-UHFFFAOYSA-N methyl formate Chemical compound COC=O TZIHFWKZFHZASV-UHFFFAOYSA-N 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000017074 necrotic cell death Effects 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- 0 *c1cnc(N(CC2)CCN2c([n]c2ccc3)nc2c3C(N)=O)nc1 Chemical compound *c1cnc(N(CC2)CCN2c([n]c2ccc3)nc2c3C(N)=O)nc1 0.000 description 1
- AGRIQBHIKABLPJ-UHFFFAOYSA-N 1-Pyrrolidinecarboxaldehyde Chemical compound O=CN1CCCC1 AGRIQBHIKABLPJ-UHFFFAOYSA-N 0.000 description 1
- SMZOUWXMTYCWNB-UHFFFAOYSA-N 2-(2-methoxy-5-methylphenyl)ethanamine Chemical compound COC1=CC=C(C)C=C1CCN SMZOUWXMTYCWNB-UHFFFAOYSA-N 0.000 description 1
- CEAMSISEJZMQEP-UHFFFAOYSA-N 2-(trifluoromethyl)pyrazine Chemical compound FC(F)(F)C1=CN=CC=N1 CEAMSISEJZMQEP-UHFFFAOYSA-N 0.000 description 1
- ATRQECRSCHYSNP-UHFFFAOYSA-N 2-(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CC=CC=N1 ATRQECRSCHYSNP-UHFFFAOYSA-N 0.000 description 1
- BQDJLAWUTBCDHK-UHFFFAOYSA-N 2-(trifluoromethyl)pyrimidine Chemical compound FC(F)(F)C1=NC=CC=N1 BQDJLAWUTBCDHK-UHFFFAOYSA-N 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-N 2-Propenoic acid Natural products OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 1
- FZRBTBCCMVNZBD-UHFFFAOYSA-N 2-chloro-4-(trifluoromethyl)pyrimidine Chemical compound FC(F)(F)C1=CC=NC(Cl)=N1 FZRBTBCCMVNZBD-UHFFFAOYSA-N 0.000 description 1
- ZOCBWRJIEHXNMD-UHFFFAOYSA-N 2-chloro-n-ethylpyridin-3-amine Chemical compound CCNC1=CC=CN=C1Cl ZOCBWRJIEHXNMD-UHFFFAOYSA-N 0.000 description 1
- UNCQVRBWJWWJBF-UHFFFAOYSA-N 2-chloropyrimidine Chemical compound ClC1=NC=CC=N1 UNCQVRBWJWWJBF-UHFFFAOYSA-N 0.000 description 1
- VVQHIHPZHJQYEY-UHFFFAOYSA-N 3-(trifluoromethyl)pyridazine Chemical compound FC(F)(F)C1=CC=CN=N1 VVQHIHPZHJQYEY-UHFFFAOYSA-N 0.000 description 1
- JTZSFNHHVULOGJ-UHFFFAOYSA-N 3-(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CC=CN=C1 JTZSFNHHVULOGJ-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- FZINIBTZNSPWQR-UHFFFAOYSA-N 4-chloro-2-(trifluoromethyl)pyridine Chemical compound FC(F)(F)C1=CC(Cl)=CC=N1 FZINIBTZNSPWQR-UHFFFAOYSA-N 0.000 description 1
- TYSPDLZOMUDHQZ-UHFFFAOYSA-N 4-chloro-6-(trifluoromethyl)pyrimidine Chemical compound FC(F)(F)C1=CC(Cl)=NC=N1 TYSPDLZOMUDHQZ-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-M 4-hydroxybenzoate Chemical compound OC1=CC=C(C([O-])=O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-M 0.000 description 1
- SJZRECIVHVDYJC-UHFFFAOYSA-N 4-hydroxybutyric acid Chemical compound OCCCC(O)=O SJZRECIVHVDYJC-UHFFFAOYSA-N 0.000 description 1
- ZEYHEAKUIGZSGI-UHFFFAOYSA-M 4-methoxybenzoate Chemical compound COC1=CC=C(C([O-])=O)C=C1 ZEYHEAKUIGZSGI-UHFFFAOYSA-M 0.000 description 1
- CPCHRGFQWZMVNX-UHFFFAOYSA-N 5-(trifluoromethyl)pyrimidine Chemical compound FC(F)(F)C1=CN=CN=C1 CPCHRGFQWZMVNX-UHFFFAOYSA-N 0.000 description 1
- KSPDSMOWMQFPBL-UHFFFAOYSA-N 5-fluoropyrimidine Chemical compound FC1=CN=CN=C1 KSPDSMOWMQFPBL-UHFFFAOYSA-N 0.000 description 1
- BPMBBXCPGAFTCX-UHFFFAOYSA-N 6-chloro-n,n-dimethylpyridine-3-carboxamide Chemical compound CN(C)C(=O)C1=CC=C(Cl)N=C1 BPMBBXCPGAFTCX-UHFFFAOYSA-N 0.000 description 1
- DEYOMDZKXMQLCF-UHFFFAOYSA-N 6-chloro-n-methylpyridine-3-carboxamide Chemical compound CNC(=O)C1=CC=C(Cl)N=C1 DEYOMDZKXMQLCF-UHFFFAOYSA-N 0.000 description 1
- ZIJAZUBWHAZHPL-UHFFFAOYSA-N 6-chloropyridine-3-carboxamide Chemical compound NC(=O)C1=CC=C(Cl)N=C1 ZIJAZUBWHAZHPL-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- PWJFNRJRHXWEPT-UHFFFAOYSA-N ADP ribose Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OCC(O)C(O)C(O)C=O)C(O)C1O PWJFNRJRHXWEPT-UHFFFAOYSA-N 0.000 description 1
- 230000005730 ADP ribosylation Effects 0.000 description 1
- 208000009304 Acute Kidney Injury Diseases 0.000 description 1
- 206010067484 Adverse reaction Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- YKDJCJFTWASNNK-UHFFFAOYSA-N CC=1N=C(C(=NC1)C(=O)N)C.NN Chemical compound CC=1N=C(C(=NC1)C(=O)N)C.NN YKDJCJFTWASNNK-UHFFFAOYSA-N 0.000 description 1
- BOBBLKRZGGRAOC-UHFFFAOYSA-N CN(CC1)CCN1c1nc(c(C(N)=O)ccc2)c2[nH]1 Chemical compound CN(CC1)CCN1c1nc(c(C(N)=O)ccc2)c2[nH]1 BOBBLKRZGGRAOC-UHFFFAOYSA-N 0.000 description 1
- ILEUCNCKSCQHGO-UHFFFAOYSA-N COC(c1cccc([nH]2)c1nc2Cl)=O Chemical compound COC(c1cccc([nH]2)c1nc2Cl)=O ILEUCNCKSCQHGO-UHFFFAOYSA-N 0.000 description 1
- VNYMKKRTIQDAIY-UHFFFAOYSA-N COC(c1cccc([nH]2)c1nc2N1CCNCC1)=O Chemical compound COC(c1cccc([nH]2)c1nc2N1CCNCC1)=O VNYMKKRTIQDAIY-UHFFFAOYSA-N 0.000 description 1
- FOZGFQHZXQLOSG-UHFFFAOYSA-N C[N]C(c(nc1)cnc1N(CC1)CCN1c([nH]c1cc(F)c2)nc1c2C(N)=O)=O Chemical compound C[N]C(c(nc1)cnc1N(CC1)CCN1c([nH]c1cc(F)c2)nc1c2C(N)=O)=O FOZGFQHZXQLOSG-UHFFFAOYSA-N 0.000 description 1
- WKWNPVUAOVEGFQ-UHFFFAOYSA-N C[N]C(c(nc1)ncc1N(CC1)CCN1c([nH]c1cc(F)c2)nc1c2C(N)=O)=O Chemical compound C[N]C(c(nc1)ncc1N(CC1)CCN1c([nH]c1cc(F)c2)nc1c2C(N)=O)=O WKWNPVUAOVEGFQ-UHFFFAOYSA-N 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- KMLJVJRQAFXKKA-UHFFFAOYSA-N Cl[P](Cl)(Cl)Cl Chemical compound Cl[P](Cl)(Cl)Cl KMLJVJRQAFXKKA-UHFFFAOYSA-N 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 208000031448 Genomic Instability Diseases 0.000 description 1
- 102000006947 Histones Human genes 0.000 description 1
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 1
- 208000023105 Huntington disease Diseases 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-N Metaphosphoric acid Chemical compound OP(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-N 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 description 1
- BXDZOYLPNAIDOC-UHFFFAOYSA-N N-[5-[(5-tert-butyl-1,3-oxazol-2-yl)methylsulfanyl]-1,3-thiazol-2-yl]-1-[2-[2-[2-[2-[2-[[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxoisoindol-4-yl]amino]ethoxy]ethoxy]ethoxy]ethylamino]-2-oxoethyl]piperidine-4-carboxamide Chemical compound CC(C)(C)c1cnc(CSc2cnc(NC(=O)C3CCN(CC(=O)NCCOCCOCCOCCNc4cccc5C(=O)N(C6CCC(=O)NC6=O)C(=O)c45)CC3)s2)o1 BXDZOYLPNAIDOC-UHFFFAOYSA-N 0.000 description 1
- ZYVXHFWBYUDDBM-UHFFFAOYSA-N N-methylnicotinamide Chemical compound CNC(=O)C1=CC=CN=C1 ZYVXHFWBYUDDBM-UHFFFAOYSA-N 0.000 description 1
- ADHVDIDMAQCSQF-UHFFFAOYSA-N NC(c(cccc1[nH]2)c1nc2N(CC1)CCN1c(nc1)ncc1F)=O Chemical compound NC(c(cccc1[nH]2)c1nc2N(CC1)CCN1c(nc1)ncc1F)=O ADHVDIDMAQCSQF-UHFFFAOYSA-N 0.000 description 1
- GEGQNLXASRPTBN-UHFFFAOYSA-N NC(c(cccc1[nH]2)c1nc2N(CC1)CCN1c1ncncc1)=O Chemical compound NC(c(cccc1[nH]2)c1nc2N(CC1)CCN1c1ncncc1)=O GEGQNLXASRPTBN-UHFFFAOYSA-N 0.000 description 1
- XJKKXWQIBNHZOK-UHFFFAOYSA-N NC(c1cccc([nH]2)c1nc2N1CCNCC1)=O Chemical compound NC(c1cccc([nH]2)c1nc2N1CCNCC1)=O XJKKXWQIBNHZOK-UHFFFAOYSA-N 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229940123066 Polymerase inhibitor Drugs 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 102000018120 Recombinases Human genes 0.000 description 1
- 108010091086 Recombinases Proteins 0.000 description 1
- 208000033626 Renal failure acute Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 201000011040 acute kidney failure Diseases 0.000 description 1
- 208000012998 acute renal failure Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000006838 adverse reaction Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 229910001413 alkali metal ion Inorganic materials 0.000 description 1
- 229910001420 alkaline earth metal ion Inorganic materials 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- UHOVQNZJYSORNB-UHFFFAOYSA-N benzene Substances C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 1
- 238000012925 biological evaluation Methods 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 230000005779 cell damage Effects 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 208000037887 cell injury Diseases 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- CIPMQPTWGWCBLF-UHFFFAOYSA-N chembl382387 Chemical compound C=1C(CC(=O)O)=CC(C=2NC3=CC=CC=C3N=2)=C(O)C=1C1=CC=CC([N+]([O-])=O)=C1 CIPMQPTWGWCBLF-UHFFFAOYSA-N 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000000536 complexating effect Effects 0.000 description 1
- 239000008139 complexing agent Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- PMSVVUSIPKHUMT-UHFFFAOYSA-N cyanopyrazine Chemical compound N#CC1=CN=CC=N1 PMSVVUSIPKHUMT-UHFFFAOYSA-N 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000002498 deadly effect Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- MVZXIDCKTRPHIU-UHFFFAOYSA-N formic acid;phthalic acid Chemical compound OC=O.OC(=O)C1=CC=CC=C1C(O)=O MVZXIDCKTRPHIU-UHFFFAOYSA-N 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000012224 gene deletion Methods 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 239000005414 inactive ingredient Substances 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 description 1
- 239000001095 magnesium carbonate Substances 0.000 description 1
- 229910000021 magnesium carbonate Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- CKJNUZNMWOVDFN-UHFFFAOYSA-N methanone Chemical compound O=[CH-] CKJNUZNMWOVDFN-UHFFFAOYSA-N 0.000 description 1
- DVLGIQNHKLWSRU-UHFFFAOYSA-N methyl 1h-imidazole-5-carboxylate Chemical compound COC(=O)C1=CN=CN1 DVLGIQNHKLWSRU-UHFFFAOYSA-N 0.000 description 1
- AXLYJLKKPUICKV-UHFFFAOYSA-N methyl 3-nitrobenzoate Chemical compound COC(=O)C1=CC=CC([N+]([O-])=O)=C1 AXLYJLKKPUICKV-UHFFFAOYSA-N 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 230000011278 mitosis Effects 0.000 description 1
- 239000010413 mother solution Substances 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- FJEVKYZLIRAAKE-UHFFFAOYSA-N n,n-dimethylpyridine-3-carboxamide Chemical compound CN(C)C(=O)C1=CC=CN=C1 FJEVKYZLIRAAKE-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- JJNNWWOHMWHRAB-UHFFFAOYSA-N n-ethylpyrazine-2-carboxamide Chemical compound CCNC(=O)C1=CN=CC=N1 JJNNWWOHMWHRAB-UHFFFAOYSA-N 0.000 description 1
- OPVFDBZMQPCJEB-UHFFFAOYSA-N n-methylpyrazine-2-carboxamide Chemical compound CNC(=O)C1=CN=CC=N1 OPVFDBZMQPCJEB-UHFFFAOYSA-N 0.000 description 1
- FJMKLLGSSGMGBM-UHFFFAOYSA-N n-methylpyrimidine-2-carboxamide Chemical compound CNC(=O)C1=NC=CC=N1 FJMKLLGSSGMGBM-UHFFFAOYSA-N 0.000 description 1
- OLJJTYPPPKTNCJ-UHFFFAOYSA-N n-methylpyrimidine-5-carboxamide Chemical compound CNC(=O)C1=CN=CN=C1 OLJJTYPPPKTNCJ-UHFFFAOYSA-N 0.000 description 1
- DLFPZLKEZOVQNJ-UHFFFAOYSA-N n-propan-2-ylpyrazine-2-carboxamide Chemical compound CC(C)NC(=O)C1=CN=CC=N1 DLFPZLKEZOVQNJ-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 230000004770 neurodegeneration Effects 0.000 description 1
- 208000015122 neurodegenerative disease Diseases 0.000 description 1
- 229960003966 nicotinamide Drugs 0.000 description 1
- 235000005152 nicotinamide Nutrition 0.000 description 1
- 239000011570 nicotinamide Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- ILUJQPXNXACGAN-UHFFFAOYSA-N ortho-methoxybenzoic acid Natural products COC1=CC=CC=C1C(O)=O ILUJQPXNXACGAN-UHFFFAOYSA-N 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 230000005731 poly ADP ribosylation Effects 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 230000001124 posttranscriptional effect Effects 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000006916 protein interaction Effects 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- IIHQNAXFIODVDU-UHFFFAOYSA-N pyrimidine-2-carbonitrile Chemical compound N#CC1=NC=CC=N1 IIHQNAXFIODVDU-UHFFFAOYSA-N 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 230000008263 repair mechanism Effects 0.000 description 1
- 230000028617 response to DNA damage stimulus Effects 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 102000034285 signal transducing proteins Human genes 0.000 description 1
- 108091006024 signal transducing proteins Proteins 0.000 description 1
- 230000005783 single-strand break Effects 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical class [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 239000011573 trace mineral Substances 0.000 description 1
- 235000013619 trace mineral Nutrition 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 208000022679 triple-negative breast carcinoma Diseases 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/08—Plasma substitutes; Perfusion solutions; Dialytics or haemodialytics; Drugs for electrolytic or acid-base disorders, e.g. hypovolemic shock
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Definitions
- the present invention relates to a benzimidazole-2-piperazine heterocyclic compound, a process for the preparation thereof, and a pharmaceutical composition containing the same, and use thereof as a therapeutic agent and as a poly(ADP-ribose) polymerase (PARP) inhibitor .
- Background technique a benzimidazole-2-piperazine heterocyclic compound, a process for the preparation thereof, and a pharmaceutical composition containing the same, and use thereof as a therapeutic agent and as a poly(ADP-ribose) polymerase (PARP) inhibitor.
- PARP poly(ADP-ribose) polymerase
- Chemotherapeutic drugs and ionizing radiation therapy are two common methods of treating cancer. Both treatments induce DNA single-strand and/or double-strand breaks to produce cytotoxic effects, and the target tumor cells die due to chromosome damage.
- An important consequence of responding to DNA damage signals is that the cell cycle regulatory site signal is activated in order to protect cells from mitosis in the event of DNA damage and to avoid cell damage.
- tumor cells have a high rate of proliferation while exhibiting signal defects at the cell cycle regulatory site. Therefore, it can be inferred that there is a specific DNA repair mechanism in tumor cells, which can quickly respond to and repair the chromosomal damage associated with proliferation regulation, thereby freeing itself from the cytotoxic effects of some therapeutic drugs and maintaining continued survival.
- the effective concentration of the chemotherapeutic drug or the therapeutic radiation intensity can counteract these DNA repair mechanisms and ensure the killing effect on the target tumor cells.
- tumor cells can be tolerant to treatment by enhancing their DNA damage repair mechanisms, allowing them to survive deadly DNA damage.
- it is usually necessary to increase the dose of the therapeutic drug or increase the radiation intensity, which will adversely affect the normal tissues in the vicinity of the lesion, thus causing serious adverse reactions in the treatment process, thereby increasing the treatment. risk.
- increasing tolerance will reduce the therapeutic effect, so it can be inferred that by modulating the DNA damage signal repair mechanism, the cytotoxicity of the DNA damage agent can be improved in a tumor cell-specific manner.
- PARPs Poly (ADP-ribose) polymerases characterized by polyadenosine diphosphate-ribosylation activity constitute a superfamily of 18 nuclear ribozymes. This polyadenosine diphosphate-ribosylation regulates the catalytic activity and protein interactions of the target protein and regulates many basic biological processes, including DNA repair, cell death, and genomic stability.
- PARP-1 activity accounts for approximately 80% of the total cellular PARP activity, and together with its closest counterpart, PARP-2, becomes a member of the PARP family with the ability to repair DNA damage.
- PARP-1 can rapidly detect and directly bind to DNA damage sites, and then induce a variety of proteins required for DNA repair, thereby repairing DNA damage.
- PARP-2 can replace PARP-1 to repair DNA damage.
- tumors for DNA repair-related gene deletions (such as BRCA-1 or BRCA-2) (such as breast tumors and ovarian cancer) show extreme sensitivity to PARP-1 inhibitors, indicating that PARP inhibitors are used as single agents. A potential use in treating this type of triple-negative breast cancer.
- PARP-1 is considered to be an effective target for exploring new cancer treatment methods.
- a PARP inhibitor was developed using a nicotinamide of NAD as a PARP catalytic substrate as a template. These inhibitors, as competitive inhibitors of NAD, compete with NAD for the catalytic site of PARP, thereby preventing the synthesis of poly(ADP-ribose) chains.
- PARP without poly(ADP-ribosylation) modification cannot be dissociated from the DNA damage site, which will cause other proteins involved in the repair to enter the injury site, and thus the repair process cannot be performed. Therefore, under the action of cytotoxic drugs or radiation, the presence of a PARP inhibitor ultimately causes the DNA-damaged tumor cells to eventually die.
- NAD which is consumed as a PARP catalytic substrate, is essential for the synthesis of ATP by cells.
- NAD which is consumed as a PARP catalytic substrate, is essential for the synthesis of ATP by cells.
- intracellular NAD levels are significantly reduced, which in turn affects intracellular ATP levels.
- Due to insufficient intracellular ATP content cells are unable to achieve an ATP-dependent programmed death process and can only turn to a special apoptotic process of necrosis. During the process of necrosis, a large number of inflammatory factors are released, which can cause toxic effects on other organs and tissues.
- PARP inhibitors can also be used to treat a variety of diseases associated with this mechanism, including neurodegenerative diseases (such as Alzheimer's disease, Huntington's disease, Parkinson's disease), diabetes, ischemia or ischemia-reperfusion Concurrent diseases such as myocardial infarction and acute renal failure, circulatory diseases such as septic shock, and inflammatory diseases such as chronic rheumatism.
- neurodegenerative diseases such as Alzheimer's disease, Huntington's disease, Parkinson's disease
- diabetes ischemia or ischemia-reperfusion
- Concurrent diseases such as myocardial infarction and acute renal failure
- circulatory diseases such as septic shock
- inflammatory diseases such as chronic rheumatism.
- One of the objects of the present invention is to provide a novel benzimidazole-2-piperazine heterocyclic compound and derivatives thereof, and their tautomers, enantiomers, diastereomers, racemates And pharmaceutically acceptable salts, as well as metabolites and metabolite precursors or prodrugs.
- Another object of the present invention is to provide a pharmaceutical composition comprising the benzimidazole-2-piperazine heterocyclic compound as an active ingredient.
- a third object of the present invention is to provide a process for producing the above benzimidazole-2-piperazine heterocyclic compound.
- a fourth object of the present invention is to provide the use of the above benzimidazole-2-piperazine heterocyclic compound in medicine.
- the benzimidazole-2-piperazine heterocyclic compound of the first aspect of the invention is a compound represented by the formula (I):
- R is hydrogen or halogen
- X, ⁇ , ⁇ one of them is nitrogen, the rest is hydrocarbon or X, ⁇ , ⁇ are all hydrocarbons;
- R2 is hydrogen, c plant c 6 burning base
- X, ⁇ , ⁇ one of them is nitrogen, the rest is hydrocarbon or X, ⁇ , ⁇ are all hydrocarbons;
- the compound of the formula (I) is any one of an enantiomer, a diastereomer and a conformational isomer or a mixture of any two or three.
- the compound of the formula (I) is a pharmaceutically acceptable derivative.
- the compound of the formula (I) of the present invention may exist in the form of a pharmaceutically acceptable salt.
- the pharmaceutically acceptable salt of the present invention is a hydrochloride, a sulfate, a phosphate, an acetate, a trifluoroacetate, a methanesulfonate or a triflate of the compound of the formula (I). P-toluenesulfonate, tartrate, maleate, fumarate, succinate or malate.
- the benzimidazole-2-piperazine heterocyclic compound of the formula (I) is 2-(piperazin-1-yl)-1 hydrogen-benzimidazole-4 a formamide compound and a pharmaceutically acceptable salt thereof.
- Step 1) The substituted methyl 2,3-diaminobenzoate is cyclized with carbonyldiimidazole to give a substituted 2-oxo-2,3-dihydro-1hydro-benzimidazole-4-carboxylic acid Methyl ester ( II );
- Step 2) Step 1) The obtained substituted 2-oxo-2,3-dihydro-1hydro-benzimidazole-4-carboxylic acid methyl ester (II) is chlorinated with phosphorus oxychloride to obtain a substitution.
- Step 3) nucleophilic substitution reaction of the substituted 2-chloro-1 hydrogen-benzimidazole-4-carboxylic acid methyl ester (III) obtained in the step 2) with piperazine in the presence of a base to obtain a substituted 2- (piperazin-1-yl)-1 hydrogen-benzimidazole-4-carboxylic acid methyl ester (IV);
- Step 4) Amino acid hydrolysis of the substituted 2-(piperazin-1-yl)-1 -hydrogen-benzimidazole-4-carboxylic acid methyl ester (IV) obtained in the step 3) in an ammonia methanol solution , a substituted 2-(piperazin-1-yl)-1 hydrogen-benzimidazole-4-carboxamide (V);
- Step 5) coupling the substituted 2-(piperazin-1-yl)-1 hydrogen-benzimidazole-4-carboxamide (V) obtained in the step 4) with an acid or reducing amine with an aldehyde
- the reaction is carried out to give a compound of the formula (I).
- the pharmaceutical composition of the third aspect of the invention comprises a therapeutically effective amount of a compound of the formula (I) and one or more pharmaceutically acceptable carrier materials and/or diluents constituting the active ingredient. Or a therapeutically effective amount of a compound of formula (I) constituting the active ingredient and a pharmaceutically acceptable carrier, excipient or diluent.
- the pharmaceutical composition of the third aspect of the invention comprises a therapeutically effective amount of a pharmaceutically acceptable derivative of the compound of the formula (I) and one or more pharmaceutically acceptable carrier materials and/or diluents constituting the active ingredient. Or a pharmaceutically acceptable derivative of a compound of formula (I) constituting the active ingredient, and a pharmaceutically acceptable carrier, excipient or diluent.
- the pharmaceutical composition of the third aspect of the present invention comprises a therapeutically effective amount of a pharmaceutically acceptable salt of the compound of the formula (I) and one or more pharmaceutically acceptable carrier materials and/or diluents constituting the active ingredient. Or a pharmaceutically acceptable salt of a compound of formula (I) constituting the active ingredient, and a pharmaceutically acceptable carrier, Excipient or diluent.
- the pharmaceutical composition is formulated into a tablet, a capsule, an aqueous suspension, an oily suspension, a dispersible powder, a granule, a lozenge, an emulsion, a syrup, a cream, an ointment, a suppository or an injection.
- the compound of the formula (I) is present in free form.
- the pharmaceutical composition for the preparation of a medicament for treating a disease which is ameliorated by inhibition of PARP activity.
- the disease which is ameliorated by inhibition of PARP activity is vascular disease, septic shock, ischemic injury, neurotoxicity, hemorrhagic shock, inflammatory disease or multiple sclerosis.
- the pharmaceutical composition for the preparation of an adjuvant for the treatment of tumors.
- the pharmaceutical composition for the preparation of a medicament for tumor intensive radiation therapy.
- the compound of the formula (I) for the preparation of a medicament for tumor chemotherapy.
- the pharmaceutical composition for the preparation of a medicament for tumor chemotherapy.
- Use as a fourth aspect of the invention is the use of the compound of formula (I) for the manufacture of a medicament for the treatment of individualized cancer which lacks homologous recombination (HR)-dependent DNA double-strand break (DSB) repair.
- HR homologous recombination
- DFB DNA double-strand break
- the pharmaceutical composition for the preparation of a medicament for the treatment of individualized cancer which lacks homologous recombination (HR)-dependent DNA double-strand break (DSB) repair.
- the cancer is a cancer of a cancer cell that contains one or more of the ability to repair the DSB of DNA by HR relative to normal cells.
- the cancer is a cancer having a BRCA-1 or BRCA-2 deficiency, a mutant phenotype.
- the cancer is breast cancer, ovarian cancer, pancreatic cancer or prostate cancer.
- biochemical level enzyme activity assays were employed to determine the activity of various compounds of the invention on PARP enzymes.
- PARP is a post-transcriptional modification enzyme. DNA damage can activate the enzyme.
- the catalytic process of PARP in vivo is mainly an NAD-dependent poly (ADP-ribose) process.
- the substrate is mainly some nuclear proteins including PARP. , histone is one of them, the present invention determines PARP activity by measuring the degree of Histone poly (ADP-ribose) coated with a PARP in a 96-well plate under the action of NAD, and accordingly determines the PARP activity after the action of the PARP inhibitor, thereby evaluating The extent to which this class of compounds inhibits PARP activity.
- CfCe fluorenyl group means a saturated monovalent hydrocarbon group having a linear or branched portion and having 1 to 6 carbon atoms.
- examples of such groups include, but are not limited to, methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl and t-butyl.
- halogen and halogen refer to F, Cl, Br, I.
- “Pharmaceutically acceptable salt” means those salts which retain the biological effectiveness and properties of the parent compound. Such salts include -
- Forming a salt with an acid which is obtained by a reaction of a free base of a parent compound with an inorganic acid or an organic acid, and the inorganic acid includes hydrochloric acid, hydrobromic acid, nitric acid, phosphoric acid, metaphosphoric acid, sulfuric acid, sulfurous acid, perchloric acid, and the like.
- organic acids including acetic acid, propionic acid, acrylic acid, oxalic acid, (D) or (L) malic acid, fumaric acid, maleic acid, hydroxybenzoic acid, ⁇ -hydroxybutyric acid, methoxybenzoic acid, phthalic acid
- malic acid fumaric acid, maleic acid, hydroxybenzoic acid, ⁇ -hydroxybutyric acid, methoxybenzoic acid, phthalic acid
- an organic base such as ethanolamine, diethanolamine, or the like.
- “Pharmaceutical composition” means that one or more of the compounds of the present invention, or a pharmaceutically acceptable salt, solvate, hydrate or prodrug thereof, is mixed with another chemical component, such as a pharmaceutically acceptable carrier. .
- the purpose of the pharmaceutical composition is to facilitate the administration to the animal.
- “Pharmaceutically acceptable carrier” refers to an inactive ingredient in a pharmaceutical composition that does not cause significant irritation to the organism and does not interfere with the biological activity and properties of the administered compound, such as, but not limited to, calcium carbonate, calcium phosphate, each Sugar (eg lactose, mannitol, etc.), starch, cyclodextrin, magnesium stearate, cellulose, magnesium carbonate, acrylic polymer or methacrylic acid polymer, gel, water, polyethylene glycol, propylene glycol, Ethylene glycol, castor oil or hydrogenated castor oil or polyethoxy hydrogenated castor oil, sesame oil, corn oil, peanut oil, and the like.
- the aforementioned pharmaceutical composition may include, in addition to a pharmaceutically acceptable carrier, an adjuvant commonly used in medicine, for example: an antibacterial agent, an antifungal agent, an antimicrobial agent, a shelf life agent, a tone.
- an adjuvant commonly used in medicine for example: an antibacterial agent, an antifungal agent, an antimicrobial agent, a shelf life agent, a tone.
- the present invention discloses a compound and the use of the compound as a poly(ADP-ribose) polymerase inhibitor, and those skilled in the art can learn from the contents of the present invention and appropriately improve the process parameters. In particular, all classes are Such substitutions and modifications will be apparent to those skilled in the art and are considered to be included in the present invention.
- the method and the application of the present invention have been described by the preferred embodiments, and it is obvious that the method and application described herein may be modified or appropriately modified and combined without departing from the scope of the present invention. The technique of the present invention is applied.
- Step 1 Preparation of methyl 2-oxo-2,3-dihydro-1hydro-benzimidazole-4-carboxylate
- Step 5 Preparation of 2-(4-(pyrimidin-2-yl)piperazin-1-yl)-1hydro-benzimidazole-4-carboxamide will dissolve compound d: 2- (piperazin-1- -1 Hydrogen-benzimidazole-4-carboxamide (74 mg, 0.3 mmol) in dimethylformamide (5 mL), 2-chloropyrimidine (34 mg, 0.3 ol) and triethylamine (30 mg, 0.3 ol) The temperature was raised to 100 ° C, the reaction was allowed to cool for 8 hours, and the solvent was removed under reduced pressure.
- the compound (1) was prepared in a similar manner as in Step 5 of Example 1, by the compound d: 2-(piperazin-1-yl)-1 hydrogen-benzimidazole-4-carboxamide and 2-chloro-5-fluoropyrimidine Aromatic nucleophilic substitution reaction occurs to give compound (2): 2-(4-(5-fluoropyrimidin-2-yl)piperazin-1-yl)-1 -hydro-benzimidazole-4-carboxamide (30 mg, Yield 72 LC-MS (ESI): m/z 342 (M+l) + .
- Example 3 Compound (3): Preparation of 2-(4-(5-ethylaminopyrimidin-2-yl)piperazin-1-yl)-1hydro-benzimidazole-4-carboxamide: The compound (1) was prepared in a similar manner as in Step 5 of Example 1, by the compound d: 2-(piperazin-1-yl)-1 hydrogen-benzimidazole-4-carboxamide and 2-chloro-5-ethylamine Aromatic nucleophilic substitution reaction of a pyrimidine to give compound (3): 2-(4-(5-ethylaminopyrimidin-2-yl)piperazin-1-yl)-1 hydrogen-benzimidazole-4-methyl Amide (23 mg, yield 42%).
- the compound (1) was prepared in a similar manner as in Step 5 of Example 1, by the compound d: 2-(piperazin-1-yl)-1 hydrogen-benzimidazole-4-carboxamide and 2-chloro-5-amino group Aromatic nucleophilic substitution reaction of pyrimidine to give compound (6): 2-(4-(5-aminopyrimidin-2-yl)piperazin-1-yl)-1hydro-benzimidazole-4-carboxamide ( 190 mg, yield 83%).
- the compound (1) was prepared in the same manner as in Example 1 except that the compound d: 2-(4-(pyrimidin-4-yl)piperazin-1-yl)-1 -hydro-benzimidazole-4-carboxamide Aromatic nucleophilic substitution reaction with 4-chloropyrimidine (7): 2-(4-(Pyrimidin-4-yl)piperazin-1-yl)-hydrogen-benzimidazole-4-carboxamide (25 mg, yield: 65%). LC-MS (ESI): m/z 324 (M+l) + .
- the compound (1) was prepared in the same manner as in Example 1 except that the compound d: 2-(4-(pyrimidin-4-yl)piperazin-1-yl)-1 -hydro-benzimidazole-4-carboxamide Aromatic nucleophilic substitution reaction with 2-chloro-5-cyanoimidate gives compound (14): 2-(4-(5-cyanopyrimidin-2-yl)piperazin-1-yl)-1 hydrogen -benzimidazole-4-carboxamide (40 mg, yield 71%).
- the compound (1) was prepared in the same manner as in Example 1 except that the compound d: 2-(4-(pyrimidin-4-yl)piperazin-1-yl)-1 -hydro-benzimidazole-4-carboxamide Aromatic nucleophilic substitution reaction with 6-chloro-3-dimethylcarbamoylpyridine to give compound (15): 2-(4-(5-dimethylaminoformylpyridin-2-yl)piperazine- 1-yl)-hydrogen-benzimidazole-4-carboxamide (35 mg, yield 44%).
- Step 5 Preparation of 6-fluoro-2-oxo-2,3-dihydro-1hydrogen-benzimidazole-4-carboxylic acid methyl ester
- the compound h 2, 3-diamino-5-fluorobenzoic acid methyl ester and carbonyldiimidazole (CDI) are cyclized to obtain a compound i: 6 -Fluoro-2-oxo-2,3-dihydro-1hydrogen-benzimidazole-4-carboxylic acid methyl ester (711 mg, yield 37%).
- MS (ESI) m/z: [M+H] + 2U.
- Step 6 Preparation of methyl 2-chloro-6-fluoro-1 hydrogen-benzimidazole-4-carboxylate
- the compound (1) was prepared in a similar manner as in Step 2 of Example 1, by the compound i: 6-fluoro-2-oxo-2,3-dihydro-1hydro-benzimidazole-4-carboxylic acid methyl ester and trichloro Chlorophosphorus chlorination gave compound j: 2-chloro-6-fluoro-1 hydrogen-benzimidazole-4-carboxylic acid methyl ester (681 mg, yield 94%).
- Step 7 Preparation of 6-fluoro-2-(piperazin-1-yl)-1 -hydrogen-benzimidazole-4-carboxylic acid methyl ester:
- Step 8 Preparation of 6-fluoro-2-(piperazin-1-yl)-1hydro-benzimidazole-4-carboxamide:
- Step 9 Preparation of 6-fluoro-2-(4-(pyrimidin-4-yl)piperazin-1-yl)-1 -hydrogen-benzimidazole-4-carboxamide Using the procedure of Example 1 for the preparation of compound (1) 5, a similar method, by compound 1: 6-fluoro-2-(piperazin-1-yl)-1 hydrogen-benzimidazole-4-carboxamide and 4-chloropyrimidine aromatic nucleophilic substitution reaction to prepare a compound ( 16): 6-Fluoro-2-(4-(pyrimidin-4-yl)piperazin-1-yl)-hydrogen-benzimidazole-4-carboxamide (21 mg, yield 48%).
- the compound (1) was prepared in the same manner as in Example 1 except that the compound 1: 6-fluoro-2-(piperazin-1-yl)-1 hydrogen-benzimidazole-4-carboxamide and 6-chloro- Aromatic nucleophilic substitution reaction of 3-dimethylcarbamoylpyridine to give compound (18): 2-(4-(5-(dimethylformamide)pyridin-2-yl)piperazin-1-yl)-6- Fluorine-1 hydrogen-benzimidazole-4-carboxamide (14 mg, yield 18%).
- the compound (1) was prepared in the same manner as in Example 1 except that the compound 1: 6-fluoro-2-(piperazin-1-yl)-1 -hydrogen-benzimidazole-4-carboxamide and 6-chloro- Aromatic nucleophilic substitution reaction of hydrazine-methylpyridazine-3-carboxamide to give compound (23): 6-fluoro-2-(4-(6-methylamidopyridazin-3-yl)piperazine- 1-yl)-hydrogen-benzimidazole-4-carboxamide (20 mg, yield 27%) LC-MS (ESI): m/z 399 (M+l) + .
- the compound (1) was prepared in the same manner as in Example 1 except that the compound 1: 6-fluoro-2-(piperazin-1-yl)-1 hydrogen-benzimidazole-4-carboxamide and 6-chloro- Aromatic nucleophilic substitution reaction of hydrazine-methylnicotinamide to give compound (24): 6-fluoro-2-(4-(5-methylamidopyridin-2-yl)piperazin-1-yl)-1 Hydrogen-benzimidazole _4-formamide (6 mg, yield 13%).
- the compound (1) was prepared in the same manner as in Example 1 except that the compound 1: 6-fluoro-2-(piperazin-1-yl)-1 -hydrogen-benzimidazole-4-carboxamide and 5-bromo- Aromatic nucleophilic substitution reaction of N-methylpyrimidine-2-carboxamide to give compound (39): 6-fluoro-2-(4-(2-methylamidopyrimidin-2-yl)piperazine-5- Base) -1 Hydrogen-benzimidazole-4-carboxamide (16 mg, yield 29%).
- the compound (1) was prepared in the same manner as in Example 1 except that the compound 1: 6-fluoro-2-(piperazin-1-yl)-1 -hydrogen-benzimidazole-4-carboxamide and 5-bromo- Aromatic nucleophilic substitution reaction of hydrazine-ethylpyrimidine-2-carboxamide to give compound (40): 2-(4-(2-ethylamidopyrimidin-5-yl)piperazin-1-yl)-6 -Fluoro-1 Hydrogen-benzimidazole-4-carboxamide (17 mg, yield 23%).
- the compound (1) was prepared in the same manner as in Example 1 except that the compound 1: 6-fluoro-2-(piperazin-1-yl)-1 -hydrogen-benzimidazole-4-carboxamide and 5-bromo- Aromatic nucleophilic substitution reaction of fluorene-dimethylpyrimidine-2-carboxamide to give compound (41): 2-(4-(2-dimethylamidopyrimidin-5-yl)piperazin-1-yl) Preparation of -6-fluoro-1 hydrogen-benzimidazole-4-carboxamide (19 mg, yield 26%).
- Poly ADP ribosylation of nuclear proteins is post-translational in response to DNA damage.
- PARP the full name of poly ADP-ribose polymerase, catalyzes the binding of poly(ADP-ribose) to adjacent nuclear proteins in the presence of NAD, thereby triggering a DNA repair mechanism via the base-clearing repair pathway.
- Trevigen's HT Universal Chemi luminescent PARP Assay Kit measures the level of binding of this biotinylated ADP-ribose to histones.
- Strep-HRP Dilute Strep-HRP 500 times to obtain IX solution only with IX Strep dilution before use.
- the chemiluminescent substrate is mixed with the same volume of PeroxyGlow A and B solution to obtain a horseradish peroxidase substrate only before use.
- the readings in each well need to be converted to inhibition rates.
- the inhibition rate of the compound can be calculated using the following formula: Qing Library (.%) ⁇ ' ⁇ t ⁇ TL reading - X X ⁇
- Positive control hole reading is positive hole reading, meaning 100% activity of enzyme; negative control hole reading is negative hole reading, meaning enzyme 0%; active X is each sample The reading of the concentration.
- the activity data listed in Table 1 sufficiently shows that the compounds of the present invention are all inhibitors of PARP-1, in which, in the examples, the compounds (1), (2), (5), (6), (7), (10), (11), (12), (13), (14), (15), (16), (17), (18), (19), (20), (21), (22 ), (23), (24), (25), (26), (27), (28), (29), (30), (31), (32), (33), (34), (35), (36), (37), (38), (39), (40), (41) IC 5 .
- the value is not more than ⁇ , compound (16), (19), (25), (26), (30), (31), (33), (35), (36), (37), (38), (39), (40), (41) IC 5 .
- the value is not more than 10nM.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Heart & Thoracic Surgery (AREA)
- Immunology (AREA)
- Diabetes (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Rheumatology (AREA)
- Urology & Nephrology (AREA)
- Psychiatry (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
Abstract
Description











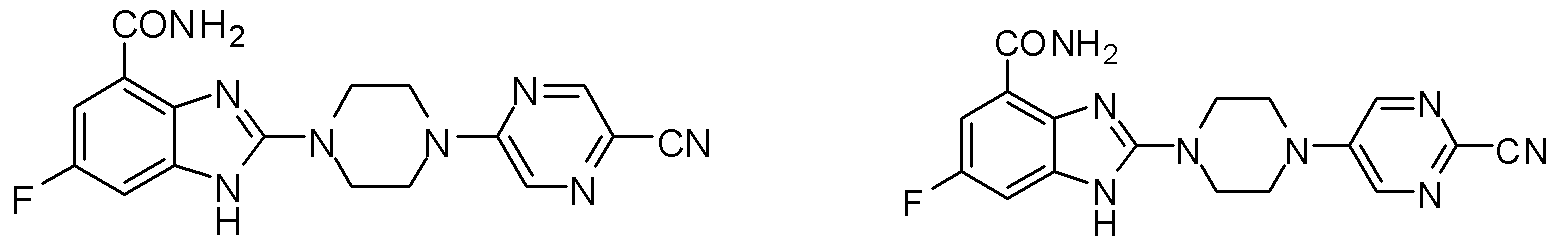









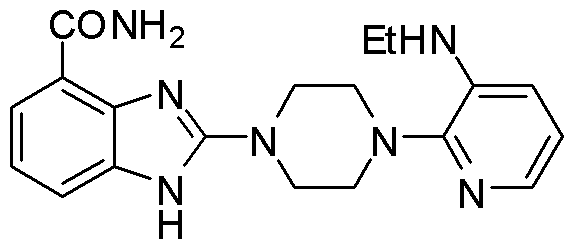












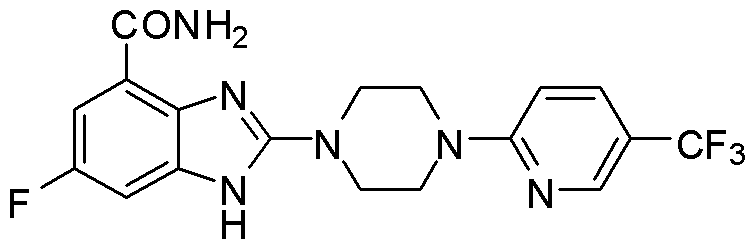

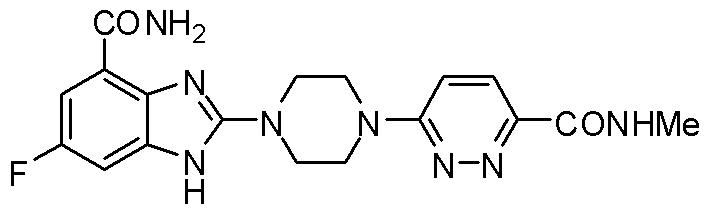



















Claims






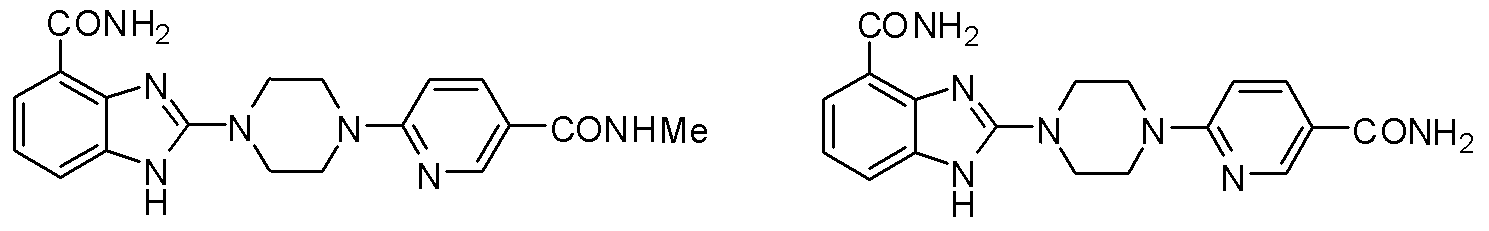









Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/898,658 US10196381B2 (en) | 2013-06-17 | 2014-06-13 | Benzimidazole-2-piperazine heterocyclic compound, pharmaceutical composition containing the same, preparation method and use thereof |
| CA2915319A CA2915319C (en) | 2013-06-17 | 2014-06-13 | Benzimidazole-2-piperazine heterocyclic compound, pharmaceutical composition containing the same, preparation method and use thereof |
| EP14814343.1A EP3012256B1 (en) | 2013-06-17 | 2014-06-13 | Benzimidazole-2-piperazine heterocyclic compound, pharmaceutical composition thereof, preparation method and use thereof |
| CN201480007034.1A CN104981468B (zh) | 2013-06-17 | 2014-06-13 | 苯并咪唑‑2‑哌嗪杂环类化合物、其药物组合物及其制备方法和用途 |
| KR1020217041080A KR102443509B1 (ko) | 2013-06-17 | 2014-06-13 | 벤조이미다졸-2-피페라진 헤테로고리 화합물, 및 그 약물 조성물 |
| ES14814343.1T ES2688191T3 (es) | 2013-06-17 | 2014-06-13 | Compuesto heterocíclico de bencimidazol-2-piperazina, composición farmacéutica del mismo, método de preparación y uso del mismo |
| AU2014283879A AU2014283879B2 (en) | 2013-06-17 | 2014-06-13 | Benzimidazole-2-piperazine heterocyclic compound, pharmaceutical composition thereof, preparation method and use thereof |
| KR1020167001349A KR20160021845A (ko) | 2013-06-17 | 2014-06-13 | 벤조이미다졸-2-피페라진 헤테로고리 화합물, 그 약물 조성물 및 그 제조방법과 용도 |
| RU2015153319A RU2649002C2 (ru) | 2013-06-17 | 2014-06-13 | Производные бензимидазол-2-пиперазина, полезные в качестве ингибитора поли(АДФ-рибоза)-полимеразы (PARP) |
| JP2016520260A JP6263614B2 (ja) | 2013-06-17 | 2014-06-13 | ベンゾイミダゾール−2−ピペラジン複素環系化合物、その医薬組成物及びその調製方法と用途 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201310240069.7 | 2013-06-17 | ||
| CN201310240069.7A CN104230896A (zh) | 2013-06-17 | 2013-06-17 | 苯并咪唑-2-哌嗪杂环类化合物、其药物组合物及其制备方法和用途 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014201972A1 true WO2014201972A1 (zh) | 2014-12-24 |
Family
ID=52103947
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2014/079827 WO2014201972A1 (zh) | 2013-06-17 | 2014-06-13 | 苯并咪唑-2-哌嗪杂环类化合物、其药物组合物及其制备方法和用途 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US10196381B2 (zh) |
| EP (1) | EP3012256B1 (zh) |
| JP (1) | JP6263614B2 (zh) |
| KR (2) | KR20160021845A (zh) |
| CN (2) | CN104230896A (zh) |
| AU (1) | AU2014283879B2 (zh) |
| CA (1) | CA2915319C (zh) |
| ES (1) | ES2688191T3 (zh) |
| RU (1) | RU2649002C2 (zh) |
| WO (1) | WO2014201972A1 (zh) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017174879A1 (en) | 2016-04-06 | 2017-10-12 | University Of Oulu | Compounds for use in the treatment of cancer |
| WO2018162439A1 (en) | 2017-03-08 | 2018-09-13 | Onxeo | New predictive biomarker for the sensitivity to a treatment of cancer with a dbait molecule |
| WO2019175132A1 (en) | 2018-03-13 | 2019-09-19 | Onxeo | A dbait molecule against acquired resistance in the treatment of cancer |
| EP3594343A1 (en) | 2015-07-23 | 2020-01-15 | Institut Curie | Use of a combination of dbait molecule and parp inhibitors to treat cancer |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN109810100B (zh) * | 2017-11-21 | 2022-03-11 | 中国药科大学 | 含有苯并呋喃的parp-1和pi3k双靶点抑制剂 |
| CN111386114A (zh) | 2017-11-25 | 2020-07-07 | 百济神州有限公司 | 作为吲哚胺2,3-双加氧酶的选择性抑制剂的新颖苯并咪唑 |
| US20220227762A1 (en) * | 2019-05-22 | 2022-07-21 | Beigene, Ltd. | Amide-substituted imidazo compounds as selective inhibitors of indoleamine 2,3-dioxygenases |
| US20240238283A1 (en) * | 2021-05-18 | 2024-07-18 | Onconic Therapeutics Inc. | Parp inhibitor-resistant cancer therapeutic agent |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1195985A (zh) * | 1995-08-02 | 1998-10-14 | 钮卡斯尔大学风险投资有限公司 | 苯并咪唑化合物 |
| DE19920936A1 (de) * | 1999-05-07 | 2000-11-09 | Basf Ag | Heterozyklisch substituierte Benzimidazole, deren Herstellung und Anwendung |
| CN1425665A (zh) * | 1997-07-24 | 2003-06-25 | 拜尔公司 | 2-氯苯并咪唑衍生物的制备方法 |
| CN1446218A (zh) * | 2000-08-08 | 2003-10-01 | 圣诺菲-合成实验室公司 | 苯并咪唑衍生物,它们的制备方法以及它们的治疗应用 |
| WO2003106430A1 (en) * | 2002-06-14 | 2003-12-24 | Pfizer Inc. | Benzimidazole inhibitors of poly(adp-ribosyl) polymerase |
| CN1486984A (zh) * | 2002-09-30 | 2004-04-07 | 天津天士力集团有限公司 | 一种哌啶酸酯制备方法 |
| CN101754967A (zh) * | 2007-07-16 | 2010-06-23 | 雅培制药有限公司 | 苯并咪唑聚(adp-核糖)聚合酶抑制剂 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR9915381A (pt) * | 1998-11-17 | 2001-08-14 | Basf Ag | Uso de um composto, e, composto |
| FR2833948B1 (fr) * | 2001-12-21 | 2004-02-06 | Sod Conseils Rech Applic | Nouveaux derives de benzimidazole et leur utilisation en tant que medicament |
| WO2007084532A2 (en) * | 2006-01-17 | 2007-07-26 | Abbott Laboratories | Combination therapy with parp inhibitors |
| ATE553104T1 (de) * | 2006-05-02 | 2012-04-15 | Abbott Lab | Substituierte 1h-benzimidazol-4-carboxamide als potente parp-hemmer |
| WO2013014038A1 (en) * | 2011-07-26 | 2013-01-31 | Nerviano Medical Sciences S.R.L. | 3-oxo-2,3-dihydro-1h-indazole-4-carboxamide derivatives as parp-1 inhibitors |
-
2013
- 2013-06-17 CN CN201310240069.7A patent/CN104230896A/zh active Pending
-
2014
- 2014-06-13 JP JP2016520260A patent/JP6263614B2/ja active Active
- 2014-06-13 KR KR1020167001349A patent/KR20160021845A/ko not_active IP Right Cessation
- 2014-06-13 KR KR1020217041080A patent/KR102443509B1/ko active IP Right Grant
- 2014-06-13 CA CA2915319A patent/CA2915319C/en active Active
- 2014-06-13 WO PCT/CN2014/079827 patent/WO2014201972A1/zh active Application Filing
- 2014-06-13 AU AU2014283879A patent/AU2014283879B2/en active Active
- 2014-06-13 ES ES14814343.1T patent/ES2688191T3/es active Active
- 2014-06-13 US US14/898,658 patent/US10196381B2/en active Active
- 2014-06-13 CN CN201480007034.1A patent/CN104981468B/zh active Active
- 2014-06-13 RU RU2015153319A patent/RU2649002C2/ru active
- 2014-06-13 EP EP14814343.1A patent/EP3012256B1/en active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1195985A (zh) * | 1995-08-02 | 1998-10-14 | 钮卡斯尔大学风险投资有限公司 | 苯并咪唑化合物 |
| CN1425665A (zh) * | 1997-07-24 | 2003-06-25 | 拜尔公司 | 2-氯苯并咪唑衍生物的制备方法 |
| DE19920936A1 (de) * | 1999-05-07 | 2000-11-09 | Basf Ag | Heterozyklisch substituierte Benzimidazole, deren Herstellung und Anwendung |
| CN1353695A (zh) * | 1999-05-07 | 2002-06-12 | 巴斯福股份公司 | 杂环取代的苯并咪唑、它们的制备及其用途 |
| CN1446218A (zh) * | 2000-08-08 | 2003-10-01 | 圣诺菲-合成实验室公司 | 苯并咪唑衍生物,它们的制备方法以及它们的治疗应用 |
| WO2003106430A1 (en) * | 2002-06-14 | 2003-12-24 | Pfizer Inc. | Benzimidazole inhibitors of poly(adp-ribosyl) polymerase |
| CN1486984A (zh) * | 2002-09-30 | 2004-04-07 | 天津天士力集团有限公司 | 一种哌啶酸酯制备方法 |
| CN101754967A (zh) * | 2007-07-16 | 2010-06-23 | 雅培制药有限公司 | 苯并咪唑聚(adp-核糖)聚合酶抑制剂 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3012256A4 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3594343A1 (en) | 2015-07-23 | 2020-01-15 | Institut Curie | Use of a combination of dbait molecule and parp inhibitors to treat cancer |
| WO2017174879A1 (en) | 2016-04-06 | 2017-10-12 | University Of Oulu | Compounds for use in the treatment of cancer |
| WO2018162439A1 (en) | 2017-03-08 | 2018-09-13 | Onxeo | New predictive biomarker for the sensitivity to a treatment of cancer with a dbait molecule |
| WO2019175132A1 (en) | 2018-03-13 | 2019-09-19 | Onxeo | A dbait molecule against acquired resistance in the treatment of cancer |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
Also Published As
| Publication number | Publication date |
|---|---|
| RU2015153319A (ru) | 2017-07-24 |
| KR20210155826A (ko) | 2021-12-23 |
| US20160159776A1 (en) | 2016-06-09 |
| ES2688191T3 (es) | 2018-10-31 |
| EP3012256A1 (en) | 2016-04-27 |
| JP6263614B2 (ja) | 2018-01-17 |
| EP3012256B1 (en) | 2018-08-08 |
| CN104981468B (zh) | 2018-01-05 |
| CN104230896A (zh) | 2014-12-24 |
| AU2014283879A1 (en) | 2016-01-21 |
| KR20160021845A (ko) | 2016-02-26 |
| CA2915319A1 (en) | 2014-12-24 |
| RU2649002C2 (ru) | 2018-03-29 |
| AU2014283879B2 (en) | 2017-06-08 |
| CN104981468A (zh) | 2015-10-14 |
| KR102443509B1 (ko) | 2022-09-15 |
| US10196381B2 (en) | 2019-02-05 |
| EP3012256A4 (en) | 2016-05-04 |
| JP2016521758A (ja) | 2016-07-25 |
| CA2915319C (en) | 2017-10-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2014201972A1 (zh) | 苯并咪唑-2-哌嗪杂环类化合物、其药物组合物及其制备方法和用途 | |
| US20100048561A1 (en) | Quinazolines for pdk1 inhibition | |
| CN104140426B (zh) | 嘧啶并咪唑类化合物、其药物组合物及其制备方法和用途 | |
| TW200848049A (en) | Pyrimidine derivatives | |
| WO2013097280A1 (zh) | 喹啉类及噌啉类化合物及其应用 | |
| JP5301456B2 (ja) | ヘテロアリールオキシキナゾリン誘導体 | |
| WO2012020780A1 (ja) | 複素環化合物およびその用途 | |
| AU2017289021B2 (en) | Novel pyrazole derivative as ALK5 inhibitor and uses thereof | |
| KR102286526B1 (ko) | 헤테로시클릭-이미다졸계 화합물, 그 약물 조성물 및 그 제조방법과 용도 | |
| CN106146504B (zh) | 一种杂环并咪唑类化合物、其药物组合物及其制备方法和用途 | |
| KR102456432B1 (ko) | 항종양을 위한 복소환 이미다졸계 화합물의 약용염 | |
| CN104230897B (zh) | 苯并咪唑-2-哌嗪杂环类化合物、其药物组合物及其制备方法和用途 | |
| CN104230898B (zh) | 苯并咪唑-2-哌嗪杂环类化合物、其药物组合物及其制备方法和用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14814343 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2915319 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14898658 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2016520260 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014814343 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 20167001349 Country of ref document: KR Kind code of ref document: A Ref document number: 2015153319 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2014283879 Country of ref document: AU Date of ref document: 20140613 Kind code of ref document: A |