WO2014119600A1 - Flexible Display法 - Google Patents
Flexible Display法 Download PDFInfo
- Publication number
- WO2014119600A1 WO2014119600A1 PCT/JP2014/051907 JP2014051907W WO2014119600A1 WO 2014119600 A1 WO2014119600 A1 WO 2014119600A1 JP 2014051907 W JP2014051907 W JP 2014051907W WO 2014119600 A1 WO2014119600 A1 WO 2014119600A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mrna
- linker
- translation
- peptide
- library
- Prior art date
Links
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1062—Isolating an individual clone by screening libraries mRNA-Display, e.g. polypeptide and encoding template are connected covalently
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1058—Directional evolution of libraries, e.g. evolution of libraries is achieved by mutagenesis and screening or selection of mixed population of organisms
Definitions
- the present invention is used to select a functional protein, peptide, or peptide analog by preparing a conjugate of mRNA and its translation product using a genotype-phenotype association technique (display method). It relates to an improved method.
- RNA display Various functional proteins and peptides have been created by using evolutionary molecular engineering techniques such as phage display (phage display) and mRNA display (mRNA display).
- mRNA display method (“In vitro virus,” Nemoto N, et al. FEBS Lett. 414, 405-408 (1997), International Publication WO 98/16636; or “RNA-peptide fusions,” Roberts, RW & Szostak, JW, Proc. Natl. Acad. Sci. USA., 94, 12297-12302 (1997), International Publication WO98 / 31700) is a useful method for able to produce peptide libraries with a high diversity of about 10 12-13 It is believed that there is.
- the mRNA display method has a major drawback. Creating a peptide library requires a number of laborious steps.
- the “one-to-one correspondence” between the amino acid sequence of the peptide and the base sequence of the mRNA is maintained by a covalent bond via a linker DNA.
- a covalent bond is formed between linker DNA and mRNA by enzymatic reaction or photocrosslinking, and (2) this is added to a cell-free translation system to form linker DNA. It is necessary to link the mRNA and the translation product via terminal puromycin.
- puromycin which is an analog of the tyrosyl tRNA 3 ′ end, is linked to the 3 ′ end of the mRNA via a linker DNA.
- this Pu-linker DNA / mRNA complex is introduced into a cell-free translation system and a peptide is synthesized from mRNA, puromycin attacks the C-terminus of the growing peptide chain as a substrate for the peptide transfer reaction in the ribosome, and the translation product The peptide molecule is linked to mRNA via puromycin.
- Non-patent Documents 1-4 As a method for binding linker DNA and mRNA, known are ligation reaction, photocrosslinking reaction, or hybridization using 2′-O-methyl RNA (Non-patent Documents 1-4). In this way, since the reaction of linking the linker to the mRNA is performed outside the cell-free translation system as an independent process, the mRNA display method performs a transcription reaction from the pool DNA, and purifies this to link the linker. It is necessary to perform a complicated operation of further purifying this and adding it to the translation system.
- the mRNA presentation method is a very complicated method requiring one to two days for one selection operation. Furthermore, in the creation of an actual peptide, it is necessary to repeat the selection operation about 5 to 10 times, and when a useful peptide cannot be obtained, it is necessary to change the selection conditions and repeat all the operations. Therefore, it takes a lot of time to select useful peptides using the mRNA display method, and development of a more rapid selection method is desired.
- Patent No. 3683282 Publication WO98 / 16636
- Patent No. 3683902 Japanese Patent No. 3692542 (International Publication WO98 / 31700)
- the present invention provides a method for selecting a target peptide, protein, or peptide analog quickly and easily.
- the present invention provides a novel reaction system that enables formation of a translation product / linker / mRNA complex in a single step.
- a series of reactions such as transcription, binding of mRNA and linker, translation on ribosome, and complex formation between translation product and linker / mRNA automatically proceed in a cell-free translation system.
- a library of translation products is generated in substantially one step. Therefore, the time required for the preparation of the translation product presented to the mRNA from the transcription of the template DNA is greatly shortened.
- a translation product / linker / mRNA complex can also be obtained by adding an mRNA library and a linker to a reaction system consisting only of a translation system (that is, containing no factors necessary for transcription) and carrying out a similar reaction. The same effect that it can be formed quickly and easily in one step can be obtained.
- this new reaction system was recovered from the previous round and added as it was without purification to the second and subsequent round DNA libraries obtained by PCR.
- the series of reactions described above up to the formation of the mRNA complex can also be performed. Thereby, the time required for the selection operation of useful translation products is further reduced.
- FIG. 6-2 is a schematic diagram of a linker.
- (1) By adding a template DNA library, translation of the template DNA library to mRNA, binding of mRNA and linker, translation of mRNA, binding of translation product, translation product / linker / mRNA complex
- a transcription-linker bond-translation reaction coupling reaction system that enables the step of forming a reaction to be performed automatically in the reaction system, and this reaction system includes factors necessary for transcription, factors necessary for translation, and Includes a linker.
- DNA contains a promoter sequence of a transcriptase, and an mRNA library can be rapidly generated from a template DNA library by factors necessary for transcription existing in the reaction system.
- a factor necessary for transcription promotes transcription from DNA to mRNA.
- the factor required for transcription may contain 0.1 ⁇ M or more of transcriptase.
- a factor necessary for translation existing in the reaction system promotes translation.
- EF-P may be included in factors necessary for translation.
- factors necessary for transcription or translation include factors such as Escherichia coli extract, wheat germ extract, rabbit reticulocyte, insect cell extract, and human cell extract. Can be derived from.
- Factors necessary for translation may include 20 types of aminoacyl-tRNA synthetases (ARS).
- ARS aminoacyl-tRNA synthetases
- (1-6) It is possible to stabilize the translation product / linker / mRNA complex in the translation system by introducing an empty codon with low translation efficiency or not translated into the DNA sequence.
- the location of the vacant codon can desirably overlap the sequence that the linker anneals or be within 20 bases upstream.
- (1-7) 1 hour is a process of forming a translation product / linker / mRNA complex through transcription of a template DNA library to mRNA, binding of mRNA and linker, translation of mRNA, binding of translation product Can be completed within. (2) mRNA and linker are bound in the translation system.
- the linker has a part that forms a complex with mRNA in the translation system, and examples thereof include nucleic acids such as DNA and RNA, and nucleic acid analogs, but are not limited to these examples.
- the base sequence of the linker in this part anneals with the sequence of the 3 ′ end untranslated region of the mRNA, and the mRNA and the linker are bound via a covalent bond and / or a non-covalent bond. (2-2) It is preferable that the linker and each mRNA are bonded without a covalent bond by annealing.
- the complementarity of the first 10 bases on the 3 ′ side of the linker base sequence of the part forming a complex with mRNA and the sequence of the 3 ′ end untranslated region of the mRNA is preferably 50% or more.
- the first 13 bases on the 3 ′ side of the linker base sequence of the part that forms a complex with mRNA have the same base sequence as the An13-type Pu-linker described in FIG. 1c. Also good.
- the first 21 bases on the 3 ′ side of the linker base sequence of the part that forms a complex with mRNA have the same base sequence as the An21-type Pu-linker described in FIG. Also good.
- the linker contains a nucleophile that can bind to the translation product, which includes aminoacyl RNA, aminoacyl RNA analog, amino acid, amino acid analog, puromycin, puromycin analog, etc. that can bind to the translation product. However, it is not limited to these examples.
- mRNA is used instead of DNA, mRNA and a linker can be combined by the above method, and in this case, a reaction system consisting only of a translation system can also be used.
- a method for producing a library of translation products by forming a translation product / linker / mRNA complex in a single step.
- This step includes adding a template DNA library to a transcription-linker binding-translation reaction coupling reaction system, which comprises the factors necessary for transcription described in (1) to (4) and translation. It is a reaction system including necessary factors and a linker.
- a translation product having a useful desired function is selected from the translation product library prepared by the above method, and desired mRNA or DNA is amplified by binding to the selected translation product.
- a nucleic acid library encoding a translation product having a function can be prepared. Using this nucleic acid library as a template, a library of translation products was obtained again by adding to the above reaction system, and the selection-amplification step was repeated several times to enrich the nucleic acids encoding useful translation products. A nucleic acid library can be obtained.
- a translation product having a desired function can be a translation product that binds to a target.
- the above selection-amplification operation can be completed within 3 hours. (6-5) The above selection-amplification operation can be repeated twice or more in one day. (6-6) The above selection-amplification operation can be automated by machine operation. (6-7) Examples of the target include, but are not limited to, a purified target, a target inserted into a membrane, a target displayed on a phage, a target displayed on a baculovirus, and a target displayed on a cell.
- the translation product can be a peptide, protein, or peptide analog, but is not limited thereto. (6-9) The translation product can be modified by post-translational modification after complex formation. (6-10) The method according to (6-3), wherein the target substance is human serum albumin. (7) Compounds p1 and p2 of Example 4 obtained by the method of (6-10) above.
- a stable complex of a translation product and mRNA is formed simply by adding a template DNA to a cell-free translation system, and a selection operation that takes 1 to 2 days by a conventional mRNA display method is completed in about 3 hours.
- Cyclic peptides containing non-protein amino acids in the skeleton are promising as drug candidates and bioprobes because they are less susceptible to degradation by peptidase.
- a selection operation using a reaction system (TRAPTRA system: Transcription-translation coupled with Association of Puromycin-linker) that couples transcription and translation reactions and linker binding.
- TRAPTRA system Transcription-translation coupled with Association of Puromycin-linker
- RT reverse transcription
- selection selection of beads with immobilized target
- Selection selection
- PCR Pattern diagram PCR Pattern diagram.
- b Complex of mRNA random pool and Pu-linker (An21) used in Examples.
- c Structure of mRNA / Pu-linker via (Li13) ⁇ with ligation or (An13) 13 base pair duplex without ligation.
- Figure 3 shows the stability study of peptide / linker / mRNA complex using T7-peptide pull-down assay.
- Figure 7 shows an analysis of the efficiency of random peptide / linker / mRNA complex formation in the TRAP system.
- X and C represent ClAB-L-Phe and Cys, respectively.
- the first aspect of the present invention is a cell-free transcription-translation coupling system containing a linker.
- This reaction system includes factors necessary for transcription / translation and a linker with an acceptor of translation products.
- transcription of the template DNA library to mRNA, mRNA and linker By adding a template DNA library to the reaction system, transcription of the template DNA library to mRNA, mRNA and linker
- the step of forming a translation product / linker / mRNA complex can be automatically performed in the reaction system through the binding of mRNA, translation of mRNA on a ribosome, and binding with a translation product.
- FIG. 1 A reaction system comprising T7 RNA polymerase for transcription and a linker whose translation product acceptor is puromycin (Pu) is shown.
- a reaction system comprising T7 RNA polymerase for transcription and a linker whose translation product acceptor is puromycin (Pu) is shown.
- the transcription-linker bond-translation coupling reaction system of the present invention is referred to as a TRAP ⁇ system.
- pre-transcribed mRNA linker binding-translation coupling reaction system
- the term cell-free translation system sometimes refers to a reaction system (transcription / translation system) containing factors necessary for transcription. Therefore, the cell-free translation system and the cell-free transcription / translation system are used as interchangeable terms.
- Fig. 1a shows the selection process consisting of four steps: translation product / linker / mRNA complex formation in TRAP system, reverse transcription (RT), selection of target immobilized beads (selection), and PCR.
- FIG. The reaction in the TRAP system depicted in the balloon on the right in Fig. 1a is a step in which transcription, binding of Puromycin-DNA linker, translation, and complexation of translation products (peptides) and peptide acceptors are coupled. ing.
- any cell-free translation system can be used.
- Such systems generally include ribosomal proteins, ribosomal RNA, amino acids, tRNA, GTP, ATP, translation initiation and elongation factors, and other factors required for translation.
- Known cell-free translation systems that can be used in the TRAP system include Escherichia coli extract, wheat germ extract, rabbit reticulocyte extract, insect cell extract, animal cell extract and the like. Since the cell-free translation system is a system in a test tube that does not use a living organism, conditions such as the composition of the reaction solution and the reaction temperature can be arbitrarily changed.
- the modified system is suitably used by means such as removing specific components or adding necessary components.
- a translation system recombined by combining components necessary for transcription and / or translation isolated from one or more arbitrary organisms can also be used.
- a component isolated from one or more arbitrary organisms can be used in appropriate combination with a component synthesized in vitro.
- Such isolated components can be separated and purified from biological sources, but can also be produced by genetic engineering techniques, chemical synthesis, or a combination thereof.
- the description is made using a prokaryotic-derived system.
- the reaction using a eukaryotic organism as a transcription / translation enzyme source It is not intended to exclude the use of the system.
- a transcription / translation system derived from plant seeds can be suitably used.
- a system derived from wheat germ extract is preferable because there is a known high-efficiency system.
- the difference between the conventional cell-free translation system and the TRAP® system is that the latter contains a linker as an essential component.
- the present inventors have found that the stability and translation of the translation product / linker / mRNA when using the TRAP® system and a known linker for mRNA display in a conventional cell-free translation system. The efficiency of product library formation was compared.
- a cell-free translation system using purified E. coli-derived components was used in the examples.
- This is a translation system in which factors involved in translation reactions such as Escherichia coli ribosome, transfer RNA (tRNA), aminoacyl tRNA synthetase (ARS), etc. are isolated and purified, and then arbitrarily mixed and reconstituted.
- factors involved in translation reactions such as Escherichia coli ribosome, transfer RNA (tRNA), aminoacyl tRNA synthetase (ARS), etc. are isolated and purified, and then arbitrarily mixed and reconstituted.
- tRNA transfer RNA
- ARS aminoacyl tRNA synthetase
- the techniques described in the following literature are known: H. F. Kung, B. Redfield, B. V. Treadwell, B. Eskin, C. Spears and H. Weissbach (1977) “DNA-directed in vitro synthesis of beta-galactosidase. Studies with purified
- Typical components of a cell-free translation system that uses E. coli-derived components include (i) T7 RNA polymerase (when transcription is also performed from DNA), (ii) E. coli initiation factor as a translation factor (for example, , IF1, IF2, IF3)-Elongation factors (eg EF-Tu, EF-Ts, EF-G)-Termination factors and ribosome recycling factors (eg RF1, RF2, RF3, RRF), (iii) 20 types Aminoacyl tRNA synthetase (ARS), methionyl tRNA formyltransferase (MTF), (iv) E. coli 70S ribosome, (v) E.
- T7 RNA polymerase when transcription is also performed from DNA
- E. coli initiation factor as a translation factor (for example, , IF1, IF2, IF3)-Elongation factors (eg EF-Tu, EF-Ts, EF
- coli tRNA isolated from E. coli
- various amino acids NTP, energy regeneration system (eg creatine Enzymes for regeneration of energy sources such as kinase, myokinase, pyrophosphatase, nucleotide-diphosphatase kinase, etc.
- Translation systems with artificially synthesized aminoacyl-tRNA are also known, mainly non-protein proteins. Used to introduce acid and hydroxy acids into translation products (A. C. Forster, Z. Tan, M. N. L. Nalam et al., “Programming peptidomimetic syntheses by translating genetic codes designed de novo, “PNAS 2003, vol. 100, no. 11, 6353-6357; T. Kawakami and H. Murakami“ Genetically Encoded Libraries of Nonstandard Peptides, ”Journal of Nucleic Acids Volume 2012 (2012), Article ID 713
- the TRAP® system (including variations thereof) of the present invention is characterized in that it includes a linker having an acceptor of a translation product in addition to factors involved in the translation reaction. Due to this feature, binding of mRNA and linker, translation of mRNA on ribosome, and binding of mRNA and translation product via the linker all occur in a cell-free translation system. Therefore, the linker molecule in the TRAP system has a structure for complex formation with mRNA to occur in a cell-free translation system. On the other hand, in a known linker for mRNA presentation, the binding of mRNA and linker is due to a ligation reaction outside the translation system. The details of the linker structure used in the TRAP® system will be described later.
- the components of the reaction system are changed from the known cell-free translation system and optimized.
- An example of a modified cell-free translation system is a translation system that does not contain amino acids, ARS, MTF, termination factor, tRNA, and the like.
- Terminating factor is a proteinaceous factor that recognizes the termination codon on mRNA and releases the completed peptide chain from the ribosome. Due to the absence of termination factors, translation can be paused rather than terminated. Terminating factors, also called peptide chain dissociation factors, are classified into two classes, called class I and class II. Class I dissociation factors are thought to promote peptide chain dissociation by recognizing a stop codon and activating the peptide chain transfer reaction center of the ribosome. In Escherichia coli, two types of class I dissociation factors named RF1RF and RF2 are known.
- RF1 recognizes UAA and UAG among termination codons
- RF-2 recognizes UAA and UGA.
- Class II dissociation factors are thought to promote the removal of class I dissociation factors that remain on the mRNA ⁇ in the ribosome after the peptide chain dissociation reaction by class I dissociation factors.
- An example of a translation system that can be used in the TRAP system is one that lacks a class I dissociation factor. For systems utilizing E. coli ribosomes, both RF1 and RF2, or either class I dissociation factor can be lacking.
- factors necessary for translation may further include EF-P.
- EF-P is a bacterial elongation factor P, a protein that is highly conserved across species. EF-P binds to ribosomes, optimizes interaction with tRNA and dissociation factors, and promotes translation. The binding site of EF-P on the ribosome is located between the P and E sites. The N-terminal domain of EF-P interacts with the acceptor stem of tRNA bound to the P site, promotes peptide bond formation, and is thought to stabilize peptidyl tRNA at the catalytic center of ribosomal peptidyltransferase. Yes.
- EF-P may be a modified EF-P protein, and the modification preferably improves the affinity of EF-P for ribosomes.
- the modification of EF-P can be in the form of a post-translational modification (eg on a lysine residue).
- the TRAP® system can be a system given only limited amino acids.
- the codon of the proteinaceous amino acid that is not added corresponds to the anticodon of tRNA acylated with the special amino acid, so that the peptide containing the special amino acid is inherited from the mRNA.
- tRNA acylated with any amino acid regardless of whether it is a proteinaceous amino acid.
- the tRNA acylated with any amino acid outside the system can be an orthogonal tRNA that is orthogonal to the ARS present in the translation system.
- Orthogonal tRNA is not recognized by naturally-occurring ARS (for example, ARS protein enzyme derived from E. coli) inherent in the translation system, so that it is not aminoacylated in the translation system, but peptide synthesis on the ribosome In the reaction, it is a tRNA that can efficiently express a designated amino acid by pairing with a codon of mRNA.
- the orthogonal tRNA for example, a natural suppressor tRNA derived from a different species or an artificially constructed tRNA is used.
- the acylated tRNA can be replaced by adding tRNA, ARS and its substrate to the reaction solution.
- the TRAP system may include 20 types of ARS.
- the TRAP® system When performing transcription from template DNA, the TRAP® system includes factors necessary for performing transcription from template DNA.
- factors necessary for transcription is a suitable DNA-dependent RNA synthetase or transcriptase and its substrate, such as T7 RNA polymerase, T3 RNA polymerase, SP6 RNA polymerase.
- Each DNA of the template DNA library contains a promoter sequence corresponding to the transcriptase in the reaction system, and mRNA is transcribed.
- Factors necessary for performing transcription further include a reducing agent such as nucleoside triphosphate (NTP), divalent magnesium ion, and DTT.
- NTP nucleoside triphosphate
- polyamines such as spermidine, Triton® X-100, which is a nonionic surfactant, and the like may be added as reagents that aid in stabilization and activation of the polymerase.
- the present invention provides an improved method used in selecting functional proteins, peptides, and peptide analogs from a random DNA pool that is a template for a translation product, the method comprising a TRAP system, A round consisting of four steps of reverse transcription (RT), selection (selection), and PCR is repeated.
- the translation product / linker / mRNA complex (display library) is automatically formed in the reaction system by adding the template DNA library to the TRAP IV system.
- TRAP system by devising factors necessary for transcription, DNA amplified by the previous round of PCR can be added as it is as a template DNA library without being purified. This eliminates the need for purification of the template DNA library required in the conventional mRNA display method, and speeds up the stage of generating the mRNA library from the template DNA library.
- a factor necessary for transcription existing in the TRAP system promotes transcription from DNA to mRNA.
- the factors necessary for transcription are preferably present in such an amount that an mRNA library can be rapidly generated from a template DNA library.
- the factor required for transcription may contain 0.1 ⁇ M or more of transcriptase. It is also possible to increase the yield of translation product / linker / mRNA complex by increasing the concentration of the transcriptase.
- the transcriptase is about 0.2 ⁇ M or more, 0.3 ⁇ M or more, 0.4 ⁇ M or more, or 0.5 ⁇ M or more, 0.6 ⁇ M or more, 0.7 ⁇ M or more, 0.8 ⁇ M or more, or 0.9 ⁇ M or more may be contained.
- T7 RNA polymerase for example, 0.05 ⁇ M to 20 ⁇ M, preferably 0.1 ⁇ M to 20 ⁇ M, alternatively 0.2 ⁇ M to 10 ⁇ M, more preferably about 0.5 ⁇ M to about 5 ⁇ M, even more preferably about 0.8 to about 1.2 ⁇ M, Particularly preferably, it can contain about 0.9 ⁇ M to about 1.1 ⁇ M, or about 1.0 ⁇ M.
- the cell-free translation system (TRAP system) of the present invention is a reaction system containing a linker, and is capable of forming a complex between the linker and mRNA. Occurs in the reaction system.
- a template DNA library is transcribed into mRNA.
- the mRNA is captured by a linker (shown as Puromycin-DNA linker in the figure) by hybridization.
- a GC-rich sequence complementary to the linker DNA is placed in the 3'-untranslated region (3'-UTR) of the mRNA, and after annealing these base sequences, the linker DNA molecule and the mRNA molecule A stable duplex is formed between them.
- the ribosome moves along the mRNA chain from the start codon to the end of the open reading frame (ORF) and stops just before the linker / mRNA duplex site.
- the peptide acceptor on the linker attacks the nascent polypeptide chain (specifically, the junction between the peptide and tRNA in the peptidyl tRNA).
- An mRNA complex is formed.
- the peptide-linker / mRNA complex thus formed is stably maintained even after the peptide chain is dissociated from the ribosome.
- FIG. 1b shows how the linker available in the TRAP system is complexed with the mRNA random pool.
- the structure in which the DNA base sequence portion of the linker is annealed to the 3'-UTR of mRNA to form a 21 base pair DNA / RNA duplex is depicted.
- the base sequence and UAG codon corresponding to the cysteine residue and spacer are arranged downstream of the (NNK) 12 random sequence in the mRNA
- the 21-base GC-rich sequence including the last G is the sequence for annealing with the Pu-linker.
- N is a ribonucleotide of either A, U, C or G
- K is a ribonucleotide of either U or G.
- An13 in FIG. 1c shows the structure of the Pu-linker / mRNA complex formed by 13 base pair duplex formation.
- Lic in FIG. 1c is an example of a linker / mRNA linking structure using a linker of a known mRNA display method. This is a comparative example to the present invention, and the 3 ′ end of mRNA is ligated to 5 of the Pu-linker. It is covalently bonded to the terminus.
- the end codon UAG is located at the end of the mRNA open reading frame.
- the UAG codon is an empty codon (blank). By placing this UAG codon, the UAG codon is placed before the linker / mRNA duplex site at the time of translation termination. The action of stopping the ribosome is enhanced.
- the double-stranded site formed by hydrogen bonding is dissociated relatively easily when it comes into contact with a ribosome having RNA helicase (double-strand cleavage enzyme) activity.
- stopping the ribosome on mRNA by an empty codon is considered to have the following two significances: (1) Stabilizing the linker-mRNA binding and the mRNA of the peptide-linker / mRNA complex Maintaining the correct one-to-one correspondence between the translation product and the template nucleic acid by preventing the peptide-linker moiety from dissociating from and recombining with another mRNA; Increase the efficiency of peptide-linker / mRNA complex formation by providing a delay in attacking the peptide chain.
- a codon with low translation efficiency or an untranslated codon can be introduced into the sequence of the template nucleic acid.
- the location of codons with low translation efficiency or empty codons can be preferably in the sequence where the linker anneals to mRNA (3′-UTR annealing sequence) or within 20 bases upstream thereof.
- a vacant codon or a codon having low translation efficiency can be introduced at the end point of the ORF.
- Vacant codons are codons that are not translated. Unoccupied codons can be created from cell-free translation systems by removing amino acids, aminoacyl synthases, or proteinaceous factors such as tRNA or termination factors. For example, by removing a specific proteinous amino acid and ARS corresponding to the amino acid, a codon in which the amino acid is defined in the genetic code table can be made “empty (blank)”.
- Minor codons are codons with a low usage frequency (codon usage), and differ depending on the species. Such minor codons can be examined in public databases such as http://www.kazusa.or.jp/codon/. In the present invention, an appropriate minor codon suitable for the translation system to be used can be selected and used. Many codons with low usage frequency have been clarified from studies of codon usage with genes expressed in E. coli. Examples of such codons include AGA / AGG / CGA (Arg), AUA (Ile), CUA (Leu) GGA (Gly), CGG (Arg), and CCC (Pro) codons (Ikemura, T., (1981) J. Mol. Biol. 146, 1-21; Dong, H. (et Jal., (1996) J. Mol. Biol. 260, 649-663).
- empty codons and minor codons are not necessarily required for the formation of peptide-linker / mRNA complexes.
- the linker links the mRNA and its translation product by binding to the 3 ′ end of the mRNA at one end and the C-terminal side of the peptide at the other end.
- the function of such a linker itself is the same whether it is a TRAP system or a linker / mRNA conjugate is added to a cell-free translation system by a conventional mRNA display method.
- mRNA and a linker are bound via a covalent bond and / or a non-covalent bond.
- the mRNA is preferably 3 'terminal sequence outside the coding region (3'-UTR), annealed to the linker, and linked to the linker via a covalent bond or non-covalently.
- mRNA has a predetermined sequence near the 3 ′ end (referred to as 3′-UTR annealing sequence), and the linker has a part that forms a complex with mRNA in the translation system.
- the structure of the linker that forms a complex with mRNA is a structure having a nucleobase in the side chain and can be a nucleic acid such as DNA or RNA, or a nucleic acid analog, but is not limited to these examples.
- the linker and each mRNA do not undergo a covalent bond by hybridization (ie, hydrogen bonding) based on base pairing between the 5 ′ end of the nucleobase portion of the linker and the 3 ′ end of the mRNA molecule.
- hybridization ie, hydrogen bonding
- the 5 ′ end side of the linker and the 3 ′ end vicinity sequence of the mRNA are sequences that can be linked to each other by base pair formation over several bases to several tens of bases.
- the sequences that can bind by base pairing are not limited to those that perfectly pair (that is, 100% complementary), and the presence of unpaired bases is allowed to the extent that does not hinder the function.
- the complementarity of the sequence on the other side is preferably 50% or more, but the downstream sequence may not have any base to be paired.
- linker / mRNA duplex site a site where the base sequence of the linker and the 3′-UTR annealing sequence of the mRNA molecule form a duplex by hybridization is referred to as a linker / mRNA duplex site.
- the mRNA and linker are linked via a covalent bond after annealing.
- a reaction that crosslinks with light or a non-natural crosslinkable base is added, a covalent bond can be formed after annealing in the reaction system.
- the structure of a base capable of such a cross-linking reaction is known.
- the other end of the linker has a structure capable of binding to the translation product.
- this part is referred to as a “peptide acceptor” for convenience, and the translation product is referred to as “peptide” or It should be noted that the translation product actually synthesized on the ribosome includes polymers other than those due to typical peptide bonds, although sometimes referred to as a “peptide chain”.
- a peptide acceptor means a molecule having a structure capable of receiving a C-terminus of a growing peptide (peptidyl tRNA) by a peptide transfer reaction in a ribosome and covalently binding to the peptide.
- peptide transfer reaction a reaction in which a peptide acceptor is a peptide transfer reaction is performed.
- Many molecules that perform such a function are known, and any peptide acceptor can be used in the present invention.
- peptide acceptors include nucleophilic compounds such as aminoacyl RNAs, aminoacyl RNA analogs, amino acids, amino acid analogs, puromycins, and puromycin analogs that can bind to translation products, but are not limited to these examples.
- a bond between a typical peptide acceptor and the peptide C-terminus is incorporated into the A-site by an ester bond at the C-terminal of the peptide attached to the peptidyl tRNA at the P-site, similar to the normal peptide transfer reaction that occurs on the ribosome. This is caused by the proximity of the amino group of the peptide acceptor. Therefore, the covalent bond formed between the peptide chain C-terminal is typically an amide bond. Similar to known mRNA display methods, a specific example of a peptide acceptor is puromycin (Pu).
- nucleophile In addition to amino groups, compounds having various nucleophilic functional groups such as hydroxyl groups and thiol groups can also be used to form covalent bonds with translation products. In the present specification, any compound that functions as a peptide acceptor at the linker end is collectively referred to as a “nucleophile”.
- the portions other than both ends of the linker are the same as the structure of the linker used in the known mRNA display method.
- oligonucleotides such as single-stranded or double-stranded DNA and RNA
- polyalkylenes such as polyethylene
- polyalkylene glycols such as polyethylene glycol
- linear substances such as polystyrene and polysaccharides, or combinations thereof are appropriately selected.
- Non-limiting specific examples of the linker that can be used in the present invention include a portion that forms a complex with mRNA is a single-stranded DNA, and a polyethylene glycol between the 3'-terminal dCdC-Pu-3 'sequence.
- the Pu-DNA linker linked in is raised.
- the linker used in the Examples is a typical example.
- a template nucleic acid having a necessary sequence is provided to a cell-free transcription / translation reaction system comprising constituent factors optimized for the purpose.
- a sequence in which cDNA is transcribed into mRNA in the TRAP system is required.
- Such sequences include promoter sequences corresponding to the RNA polymerase used.
- an appropriate sequence upstream of the start codon is required.
- sequences include, for example, SD sequences when using E. coli-derived ribosomes.
- the start codon is a codon that specifies the start of translation, and encodes the start amino acid that becomes the N-terminus of the translation product on the mRNA. Since the initiation codon on mRNA is generally AUG, which is a methionine codon, the initiation tRNA has an anticodon corresponding to methionine, and the initiation tRNA carries methionine (formylmethionine in prokaryotes). However, reprogramming of the genetic code can be used to initiate translation by binding any amino acid other than methionine to the start tRNA. Furthermore, by changing the anticodon sequence of the start tRNA, sequences other than AUG can be used as the start codon.
- a spacer sequence consisting of a peptide for flexibility is linked to the C-terminal side of a portion encoding a random amino acid sequence for a peptide library, and immediately after that a stop codon or a vacant codon. Or it is desirable that minor codons come.
- the amino acid sequence Gly-Gly-Gly-Gly-Gly-Gly-Ser and the empty codon UAG are encoded immediately after cysteine.
- the term “immediately after the spacer sequence” does not mean that it must be in contact with the spacer sequence, and a non-coding sequence or other linear structure may be further included downstream of the spacer sequence.
- the location of the empty codon or minor codon can be in the 3′-UTR annealing sequence of the mRNA or upstream of it.
- the sense codon can be made a vacant codon by removing a specific proteinaceous amino acid and / or ARS corresponding to the amino acid.
- the random sequence is composed of codon repeats consisting of an arbitrary base sequence.
- NNU codon or NNK codon ⁇ N is any ribonucleotide of A, U, C or G, and K is either U or G which is a ribonucleotide of
- a stop codon defined by UAA, UAG, and UGA does not appear in a random region, a highly accurate library can be constructed.
- the codon not only a normal three-base (triplet) codon but also a four-base codon can be used.
- a sequence (3′-UTR annealing sequence) capable of binding to the linker by base pairing is arranged.
- a DNA library is first prepared, an RNA library is obtained as an in vitro transcript, and a peptide library is obtained as an in vitro translation product. From this peptide library, one having a desired function or property is selected by some screening system. For example, if you want to obtain peptide molecules that bind to a specific protein, mix the magnetic beads with the target protein immobilized on the peptide library and collect the mixture of peptide molecules bound to the magnetic beads using a magnet. Can do. At this time, since the template mRNA is added to each peptide molecule like a tag, the mRNA is returned to DNA from the collected peptide-mRNA complex library with reverse transcriptase and amplified by PCR.
- a similar selection experiment is performed again.
- a reverse transcription reaction can be performed before selection for the purpose of making the nucleic acid portion into a double strand. By repeating this operation, clones having a desired phenotype are enriched in the library as the generation progresses.
- a nucleic acid library is prepared from the nucleic acid portion of the selected corresponding molecule by PCR and its base sequence is determined.
- the sequence information of the peptide aptamer that binds to the target substance is obtained by translating according to the genetic code using the base sequence.
- the target substance In order to separate the complex, which is an active species that binds to the target substance, from other complexes, it is convenient that the target substance be modified so that it can be recovered by binding to the solid phase.
- the target substance is modified with biotin and recovered using specific binding to a biotin-binding protein immobilized on magnetic beads.
- biotin binding protein avidin, streptavidin, etc.
- Biotin combinations such specific binding includes maltose binding protein / maltose, polyhistidine peptide / metal ions (nickel, cobalt, etc.), glutathione- S-transferase / glutathione, antibody / antigen (epitope) and the like can be used, but are not limited thereto.
- avidin, streptavidin, etc. avidin, streptavidin, etc.
- Biotin combinations such specific binding includes maltose binding protein / maltose, polyhistidine peptide / metal ions (nickel, cobalt, etc.), glutathione- S-transferase / glutathione, antibody / antigen (epitope) and the like can be used, but are not limited thereto.
- As a method for immobilizing a target substance nonspecific adsorption onto a solid phase or random covalent bond formation with a
- the translation product can be a peptide consisting of proteinaceous amino acids, a protein, or a peptide analog containing a special amino acid, but is not limited thereto.
- an active species presenting a translation product having a desired binding property is selected from a library of complexes of all translation products and mRNA (or cDNA) that can be synthesized in a template-dependent manner.
- the flow of the Rapid system taking as an example the application to a special peptide library containing a thioether cyclic peptide, is as follows. (Hereafter, Satoshi Yamaguchi, Takayuki Kato, Hiroaki Tsuji, “Drug ⁇ discovery of non-standard peptide with genetic code reprogramming” Drug Delivery System 26-6: 584-592, (Quoted from 2011)
- a library of NA-special peptide complexes can be constructed, after reverse-transcription of the mRNA site of this complex and mixing with a target protein immobilized on a solid support, non-specifically bound complexes or unbound The complex complex is washed away, and only the special peptide-mRNA-cDNA complex bound to the target protein is selected, the cDNA of the complex recovered from this is amplified by PCR, and then transcribed again to be converted into an mRNA library. , Ligation, translation synthesis, etc.
- peptides that bind to the target protein can be concentrated in the library, and finally the cDNA of the peptide complex obtained is collected and analyzed for its base sequence This makes it possible to analyze the amino acid sequence of a specific special peptide that binds to the target protein.
- the difference between the selection system using the Rapid system that uses the conventional mRNA display method and the transcription-linker binding-translation coupling reaction system (TRAP system) of the present invention is mainly the following two points: (1 The former uses a linker that binds to mRNA by ligation, while the latter uses a linker that captures mRNA by annealing; (2) Uses a transcription-translation reaction system (TRAP system) containing such a linker. Thus, only by adding the cDNA library to the reaction system, a translation product / linker / mRNA complex is automatically formed.
- FIG. 1a is a schematic diagram of a selection operation realized by the present invention.
- a template DNA library (Template DNA) is prepared and added to the TRAP system to form a translation product / linker / mRNA complex library (Transcription ⁇ ⁇ Association ⁇ Translation ⁇ Conjugation).
- RT reverse transcription
- PCR amplified
- the PCR reaction solution containing the amplified cDNA can be added to the TRAP® system without purification. Therefore, the selection operation using the TRAP system) includes (1) translation product / linker / mRNA complex formation, (2) reverse transcription (RT), (3) selection (4), and (4) PCR 4 A series of operations consisting of processes is defined as one round, and this is repeated a plurality of times. That is, the process can proceed to the process 1 immediately after the process 4.
- the selection operation using the conventional mRNA display method the cDNA after PCR is purified, and then the transcription reaction is performed. Then, this is purified, the linker is ligated, and this is further purified and translated. An additional step of adding to the system is required.
- a linker bond-translation coupling system (TRAP system) can also be used.
- RNA is transcribed separately from cDNA in each round, and the transcription reaction solution is directly added to a translation reaction system containing a linker without purification. This method appears to be useful when using mRNA that is difficult to transcribe.
- the selection operation which takes 1 to 2 days by the conventional mRNA presentation method is completed in about 3 hours. That is, a series of operations (one round) can be completed within 3 hours. It is also easy to do 2 rounds in a day. In the case of a gene that is easily transcribed, one round can be completed within 2.5 hours.
- the above selection-amplification operation can also be automated (automated) by machine operation.
- the translation product can be modified by post-translational modification after complex formation.
- Post-translational modification is a reaction that changes the chemical structure of a protein synthesized based on a gene sequence, and examples thereof include phosphorylation, sugar chain addition, lipid addition, methylation, and acetylation. Modifications with polypeptides such as ubiquitin and SUMO are also included in post-translational modifications.
- Target substance can be any compound that may be useful to interact with a translation product.
- the target substance may be a protein, nucleic acid, carbohydrate, lipid, or any other compound.
- an example of a preferable target is a substance that can be a target for drug development.
- Targets include, but are not limited to, a purified target (which may be partially purified), a target inserted into a membrane, a target displayed on a phage, a target displayed on a baculovirus, a target displayed on a cell, and the like.
- Serum albumin is the most abundant protein in serum and has many actions. For example, it acts as a carrier protein for many hydrophobic molecules such as fatty acids and drugs, or as an energy source for tumor cells (ElenaElNeumann, mannEva Frei, Dorothee Funk et al., Expert Opin. Drug Deliv. 7 (8), 915 (2010)). Furthermore, albumin degradation is inhibited by the Fc receptor-mediated recycling system, which increases its half-life in serum (Jan Terje Andersen and Inger Sandlie, Drug Metab. Pharmacokinet. 24 (4), 318 (2009)). Thus, an agent conjugated to an albumin binding molecule can be efficiently delivered to tumor cells and effective for a long time.
- albumin-binding biomolecules such as bacterial protein domains and antibodies
- Such an albumin-binding molecule is considered to be effective in extending the blood half-life of the drug.
- Peptide, protein, peptide analog, special peptide Peptide is a general term for biopolymer compounds formed by connecting various amino acids by amide bonds (peptide bonds).
- a relatively short chain having 50 amino acid residues or less in the chain is called a peptide, and a longer chain is often called a protein or polypeptide. Those having 30 residues or less are sometimes called short-chain peptides.
- the terms short peptide, peptide, protein, and polypeptide are not particularly distinguished and are used as terms that can be read each other.
- Normal (natural) peptides are composed of 20 kinds of proteinaceous amino acids.
- a peptide containing a partial structure that is not usually present in a peptide is called a special peptide (abnormal peptide), a non-natural peptide, a peptidomimetic, a peptide analog, or the like.
- antibiotics such as vancomycin and cyclosporin A used as an immunosuppressant are known.
- any molecule containing amino acids other than 20 kinds of proteinous amino acids and hydroxy acids, which are translationally synthesized by ribosomes is called a special peptide, a non-natural peptide or a peptide analog.
- Proteinaceous amino ⁇ br/> proteinaceous amino acids or naturally occurring amino acids are typically used in translation ⁇ - amino acid (or substituted ⁇ - amino acids), alanine (Ala), valine (Val) , Leucine (Leu), isoleucine (Ile), proline (Pro), tryptophan (Trp), phenylalanine (Phe), methionine (Met), glycine (Gly), serine (Ser), threonine (Thr), tyrosine (Tyr) Cysteine (Cys), glutamine (Gln), asparagine (Asn), lysine (Lys), arginine (Arg), histidine (His), aspartic acid (Asp), and glutamic acid (Glu).
- amino acids include both proteinaceous amino acids and special amino acids
- peptides include special peptides (or non-natural peptides or peptide analogs).
- “Special amino acid” refers to all amino acids having a structure different from that of 20 proteinic amino acids used in natural translation, and includes some hydroxy acids. In other words, some non-protein amino acids, artificial amino acids, D-form amino acids, N-methyl amino acids, N-acyl amino acids, ⁇ (beta) -amino acids, in which part of the side chain structure of protein amino acids has been chemically modified or modified, ⁇ (gamma) -amino acid, ⁇ (delta) -amino acid, derivatives having a structure in which an amino group or a carboxyl group on the amino acid skeleton is substituted are all included.
- Peptides into which special amino acids have been introduced, or the above-mentioned “special peptides”, include polymers having these various special amino acids as constituent elements.
- a special peptide can have some or all of its constituent elements special amino acids. Therefore, a special peptide can also have a structure different from a normal amide bond as a main chain skeleton.
- depsipeptides composed of amino acids and hydroxy acids, polyesters in which hydroxy acids are continuously condensed, N-methyl peptides, peptides having various acyl groups at the N-terminus (acetyl groups, pyroglutamic acids, fatty acids, etc.) Included in special peptides.
- the special peptide also includes a cyclic peptide.
- a method for cyclizing linear peptides synthesized by translation by intramolecular reaction is known (Goto et al., ACS Chem. Biol., 2008, 3, 120-129, WO2008 / 117833 " Synthesis method ").
- a cyclic peptide cyclized with a thioether bond obtained by translating a peptide sequence in which a special amino acid having a chloroacetyl group at the N-terminus and a cysteine at the C-terminus is located in the peptide chain is an example.
- it can be cyclized with various structures according to various combinations of functional groups capable of bond formation.
- Translation generally refers to reading mRNA information consisting of a base sequence and converting it into an amino acid sequence on a ribosome, which is substantially synonymous with protein (peptide) biosynthesis. Since the translation reaction is a reaction for synthesizing a peptide using a nucleic acid as a template, peptides having various sequences can be synthesized by changing the sequence of the cDNA or mRNA used as the template. Translation products are peptides (including peptides, proteins, peptide analogs, and special peptides) synthesized by ribosomes.
- Cell-free translation is a technique for artificially performing a translation reaction in a test tube and is also called in-vitro protein synthesis.
- a cell-free translation system is a concept that includes both a method and a product (solution mixture or kit) for translation synthesis, but if this term is used in the claims, it is translated It shall mean the invention of “things” used in A transcription-linker bond-translation coupling reaction system and a linker bond-translation coupling reaction system are also described as product inventions.
- Annealing and hybridization Annealing generally means that a denatured single-stranded DNA becomes a double-stranded molecule through complementary base pairing under appropriate conditions.
- Complementary base pairing also referred to as hybridization or complementary base pairing, is the pairing of complementary combinations of nucleobases by hydrogen bonding.
- Hybridization means that single-stranded DNA or RNA forms a hybrid (hybrid double-stranded nucleic acid molecule) by complementary base pairing.
- the linker base sequence and the complementary sequence portion of mRNA form a double strand by annealing.
- Complementary base pairing is the natural binding of a polynucleotide (RNA or DNA, or other linear molecule with a nucleobase in the side chain) under salt and temperature tolerance conditions, and is a single strand of two types. Intermolecular complementarity may be “partial”, in which case only some of the bases of the nucleic acid sequence bind and the ratio of complementary base pairs can be indicated numerically. When perfect complementarity exists between two single stranded molecules, the complementarity is “perfect” (100%). The degree of complementarity significantly affects the efficiency and intensity of hybridization. Various methods known to those skilled in the art can be used to determine whether two nucleic acid molecules hybridize.
- nucleic acid base combinations that form complementary base pairs are adenine (A) and thymine (T) or uracil (U), and guanine (G) and cytosine (C).
- Thymine (T) and uracil (U) can be used interchangeably depending on the respective type of polynucleotide (DNA or RNA).
- non-Watson-Crick base pairs such as GU also exist as thermodynamically stable base pairs, such combinations may also be complementary.
- a library refers to a population of multiple molecules (e.g., multiple nucleic acids, translation products, translation product / linker / mRNA complex, translation product / linker / mRNA-cDNA complex molecule).
- the library according to the present invention has a phenotype aimed at a large-scale preparation of potential genes for the purpose of creating peptides, proteins and peptide analogs having desired functions and properties. It is preferable to use a library containing a large number of candidate molecules.
- the diversity of the library can be for example 10 11 , typically 10 12 to 10 13 , preferably 10 15 different gene sequences.
- Selection means that one molecule is substantially separated from other molecules in the library.
- a peptide library (a group of molecules in which peptides and nucleic acids are associated) is obtained as a translation product of a gene library, and appropriate screening of those having desired functions and properties from this peptide library Select by system.
- a nucleic acid that is the gene is added like a tag.
- the stability of the translation product-Pu-linker / mRNA complex obtained by ligation used in the conventional mRNA display method and the translation product-Pu-linker / mRNA complex obtained by annealing were compared.
- the efficiency of library formation using random peptides as translation products was also compared.
- T7-tag peptide (T7-peptide) as a model translation product can be selectively concentrated from a random DNA pool.
- Example 1 Stability of peptide-Pu-linker / mRNA complex
- the linker and mRNA are bound by non-covalent bonds.
- T7-peptide pull-down is used to confirm that the peptide-mRNA bond via this linker is dissociated and replaced with other unrelated mRNAs. The assay was performed.
- the assay scheme is shown in FIG. 2a.
- Pu-linker / mRNA complex of mRNA encoding T7- peptide and mRNA encoding a peptide of random sequence (these mRNAs have the same Pu-linker annealing sequence in 3′-UTR) at a ratio of 1:10
- RT translation and reverse transcription
- Fig. 2b shows the result of amplifying the cDNA of the selected complex and analyzing it using electrophoresis.
- Lanes 1-4 contain markers synthesized from DNA T7 mRNA, random mRNA, and a mixture of 1: 1 or 1:10 of T7- mRNA and random mRNAm.
- lanes 5-10 a 1:10 mixture of T7-RNA and random mRNA was added to the translation solution.
- the reaction was carried out in a translation reaction solution (lanes 5-7) or Ulanes (lanes 8-10) in which the UAG codon was assigned to Phe.
- the following types of mRNA templates were used in the reaction: lanes and 8, Li13-type mRNA; lanes 6 and 9, An13-type mRNA; lanes 7 and 10, An21-type mRNA.
- T7-cDNA / (T7-cDNA + random cDNA) only changed from 34% to 40% (FIG. 2b, lane). 6 and 7).
- This result indicates that more than half of the peptide-Pu-linker was dissociated from the mRNA during either the translation reaction, RT, or T7 peptide pull-down process.
- the Pu-linker DNA / mRNA duplex was stable during the RT and T7 peptide pulldown process, so dissociation was expected to occur during the translation reaction.
- T7-cDNA / (T7-cDNA + random cDNA) was 34% to 63% for the An13-type complex, and the An21-type complex. Increased from 40% to 73% ( Figure 2b, lanes 6 and 9, and lanes 7 and 10, respectively).
- This result suggests that the main cause of the dissociation of T7-peptide-Pu-linker from T7-mRNA is ribosomal helicase activity.
- the T7-peptide-Pu-linker / mRNA complex is stabilized by extending the length of the Pu-linker DNA / mRNA duplex and inserting a blank codon immediately before the double-stranded site. I found out that
- Example 2 Efficiency of formation of a random sequence peptide-Pu-linker / mRNA complex
- the efficiency of formation of a complex in which the peptide and mRNA are linked depends on the diversity of the library. decide.
- biotinylated Phe (Fig. 3a) bound to the starting tRNA fMet CAU was added to the translation system excluding Met, and a peptide labeled with biotin at the N-terminus was synthesized (Fig. 3b).
- Figure 3c shows the result of quantifying the recovered complex.
- the recovery of the cDNA complex was calculated by dividing the amount of the recovered cDNA by the theoretical value of the amount of mRNA / Pu-linker (1 ⁇ M) in the reaction solution. Error bars indicate the standard deviation calculated from 3 experiments.
- Reactions were performed in the translation solution where the UAG codon was assigned to Phe (first and third columns) or empty (second and 4-6 rows).
- the following types of templates were used in the reaction: 1-2 rows were Li13-type mRNA; 3-4 rows were An21-type mRNA; 5 rows were An21-type DNA; 6 rows were 37 An21-type DNA template without incubation at ° C.
- the Pu13 of Li13 type or An21 type The linker / mRNA complex was added to the translation system as a template.
- CDNA recovered by biotin pull-down assay was quantified by real-time PCR.
- the recovery rate did not change at 7% in the reaction system in which the UAG codon was assigned to Phe or in the reaction system of an empty codon (FIG. 3c, first and second columns).
- the PCR mixture containing the reverse transcribed and amplified DNA was directly added to the transcription / translation reaction solution without purification, and the obtained mRNA was quantified by RT real-time PCR.
- the transcription / translation reaction solution was prepared by mixing solA and solC (see Tables 2 and 3 below). After the RT-PCR reaction, 5% (v / v) of a crude solution containing random-DNA was added, and a transcription / translation reaction (1 ⁇ L) was performed at 37 ° C. for 20 minutes. The amount of mRNA at each time point is shown in FIG. Production of a sufficient amount of mRNA ( ⁇ 1 pmol / ⁇ L) was confirmed after 5-10 minutes incubation in a reaction containing 1 ⁇ M T7 RNA polymerase.
- Example 3 Selective enrichment of T7-peptide-Pu-linker / mRNA complex
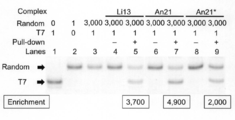
- the T7 peptide pull-down assay described above was performed using an An21 type linker and a conventional mRNA display method (Li13 type linker) to compare the T7-DNA enrichment efficiency.
- the Pu-linker / mRNA complex of T7-mRNA and random mRNA was mixed at a ratio of 1: 3000 and added to the translation reaction solution. After the pull-down assay, RT-PCR products were analyzed.
- Lanes 1-3 contain T7-mRNA, random mRNA, and DNA markers synthesized from a 1: 3000 mixture of T7-mRNA and random mRNA.
- T7-mRNA and random mRNA or DNA were mixed at a ratio of 1: 3000.
- the following types of templates were used in the reaction: lanes 4-5 are Li13-type mRNA; lanes 6-7 are An21-type mRNA; lanes 8-9 are An21-type DNA.
- T7-mRNA was concentrated 3700 times (FIG. 4, lanes 4 and 5), and 4900 times (FIG. 4, lanes 6 and 7) in the system using the An21 type linker. From this result, translation system using mRNA template and adding linker that captures by annealing selects specific peptide DNA (T7-DNA) comparable to or higher than conventional mRNA display method. It was revealed that it can be concentrated.
- T7-DNA and random-DNA were added to the transcription / translation reaction system (TRAP system) at a ratio of 1: 3000. From the results of the T7 peptide pull-down assay, it was found that T7-DNA was concentrated 2000 times (FIG. 4, lanes 8 and 9). Therefore, in a system using a linker that captures mRNA by annealing, the polypeptide-Pu-linker / mRNA can be generated not only from the mRNA template but also from the DNA template, and the target is immobilized. Pull-down experiments with beads showed that specific polypeptides can also be concentrated.
- Example 4 In vitro selection of HSA binding peptides Using the Flexible Display method, peptidaptamers that bind to human serum-derived albumin (HSA) were selected from a library of cyclic peptides.
- HSA human serum-derived albumin
- mRNA library was prepared for peptides consisting of 8-12 random amino acid sequences having ClAB-L-Phe and Cys at both ends.
- the encoded peptide cyclizes spontaneously by an intramolecular thioether bond formed between the chloroacetyl of ClAB-L-Phe and the thiol group of Cys (FIG. 5b).
- the same HSA-binding cyclic peptide was selected using the conventional mRNA display method (Fig. 5c), and the results obtained by comparing the sequences selected by the mRNA display method and the Flexible-display method were the two most frequently obtained.
- the two sequences were identical (Fig. 5d, p1: XTYNERLFWC and p2: XSQWDPWAIFWC, where X represents ClAB-L-Phe). Therefore, the flexible display method demonstrated the same performance as the mRNA display method.
- Binding assays were performed at 30 ° C. under conditions of 50 mM Hepes-KOH pH 7.5, 300 mM NaCl, 0.05% Tween 20 and 0.001% DMSO. Each step of the assay consists of 500 ⁇ s for the binding reaction of the peptide to HSA and 500 ⁇ s for the dissociation reaction. Peptide solutions were prepared in the following dilution series (p1: 16, 12, 8, 2, 1 nM, and p 2: 16, 12, 8, 4 nM).
- Example 5 Confirmation of the amount of associated molecules produced in the presence / absence of EF-P mRNA was prepared from DNA of the following sequence by a conventional method.
- N represents A / T / G / C and W represents A / T.
- the amino acid sequence when this mRNA is translated by introducing N- ⁇ -biotinyl-L-phenylalanine at the start ATG codon and tryptophan at the extended ATG codon by flexizyme is as follows if NNW is not TAA or TGA, which is the stop codon It is expected that Biotin-Phe-Xaa-Xaa-Xaa-Xaa-Xaa-Xaa-Xaa-Xaa-Xaa-Trp-Gly-Gly-Gly-Ser-Gly-Ser
- Xaa corresponds to 18 kinds of natural amino acids other than methionine and tryptophan.
- This mRNA was mixed with 1.1 equivalent of Puromycin-linker.an21, added to the translation solution to a final concentration of 2 ⁇ M, and reacted at 37 ° C. for 25 minutes.
- N- ⁇ -biotinyl-L-phenylalanine-tRNA (starting CAU) and L-tryptophan-tRNA (extended CAU) prepared with 19 kinds of natural amino acids other than methionine, corresponding ARS and flexizyme was translated under two conditions: a system containing 3 ⁇ M EF-P and a system containing no EF-P at all.
- EDTA was added and reverse transcription was performed by a conventional method, and then a part was mixed with the streptavidin bead suspension and stirred for 30 minutes.
- CME cyanomethyl ester
- MgSO 4 magnesium sulfate
- MgCl 2 magnesium chloride
- DMSO dimethyl sufoxide
- Tris tris (hydroxymethyl) aminoethane (Tris ( Hexy, 2-ethansufonic acid
- EDTA ethylene diaminetetraacetic acid
- DTT dithiothreitol
- KOH potassium hydroxide
- T7-DNA, Random-DNA, T7-mRNA, and Random-mRNA Template DNA was synthesized by extension reaction and PCR using the oligonucleotides shown in Table 1.
- N represents any one of A, T, G and C
- M represents any one of A and C.
- oligonucleotides were used as primers for PCR. T7SD8M2.F44 and G5S-4an13.R36 (Li13, An13) or G5S-4an21.R41 (An21)
- MRNA was synthesized from template DNA using T7 RNA polymerase.
- T7 RNA polymerase For selection of HSA-binding peptides, mRNA purified by isopropanol precipitation was used, and for other experiments, mRNA purified by electrophoresis was used.
- Table 2 shows the concentration of proteinaceous factor and ribosome in the stock solution B (solB) and the final concentration in the reaction solution.
- Table 3 shows the concentrations of tRNA and other small molecule factors in stock solution A (solA) and the final concentration in the reaction solution.
- SolC was prepared by mixing 100 ⁇ L of solB and 10 ⁇ L of 90 ⁇ M T7 RNA polymerase.
- a reaction solution of a conventional cell-free translation system was prepared by mixing solA (11%, v / v) and solB (10%, v / v) with other solutions.
- the reaction solution of TRAP system ⁇ ⁇ was prepared by mixing solA (11%, v / v), solC (11%, v / v), and Pu-linker (final concentration 1.1 ⁇ M) with other solutions.
- Li13-type Pu-linker / mRNA complex was prepared by mixing Puromycin-linker.an13 and mRNA in Table 1 and forming a covalent bond with T4 RNA ligase.
- An13-type and An21-type Pu-linker / mRNA complexes were prepared by mixing mRNA and 1.1 equivalents of Puromycin-linker.an13 or Puromycin-linker.an21 in Table 1.
- the concentration of Pu-linker / mRNA complex described below is the mRNA concentration.
- T7-peptide-Pu-linker / mRNA / cDNA complex ⁇ br/> 20 proteinaceous amino acids of 0.25 mM each, 0.1 ⁇ M Li13-, An13- or An21-type Pu-linker /
- a translation reaction containing the T7-mRNA complex and the corresponding 1 ⁇ M Pu-linker / random-mRNA complex was used.
- 25 ⁇ M Phe-tRNA Asn-E2 CUA was added to the reaction to reassign the UAG codon from blank to Phe.
- Translation and reverse transcription were performed as follows.
- the translation reaction (2 ⁇ L) was incubated at 37 ° C. for 15 minutes.
- the reaction solution (3 ⁇ L) was added to 1 ⁇ L of 4 ⁇ RT mix: 200 mM Tris-HCl (pH 8.3), 300 mM KCl, 75 mM MgCl 2 , 4 mM DTT, 2 mM each dNTP (dATP, dTTP, dGTP, dCTP), 10 ⁇ M G5S-4.R20, 6 U ReverTraAce (TOYOBO).
- the final concentrations of mRNA template and primer are: 0.05 ⁇ M T7-mRNA, 0.5 ⁇ M random-mRNA, 2.5 ⁇ M G5S-4.R20.
- the reaction was incubated at 42 ° C for 30 minutes.
- T7-peptide pull-down was performed as follows.
- the reverse transcription reaction solution was diluted 10-fold with HBST (50 mM Hepes-KOH pH7.5, 300 mM NaCl, 0.05% Tween20), and the diluted reaction solution (10 ⁇ L) was immobilized on Dynabeads Protein G (VERITAS).
- the anti-T7 peptide antibody (MBL) was mixed and incubated for 15 minutes at 4 ° C.
- the beads were washed 3 times with 10 ⁇ L HBST and suspended in 25 ⁇ L 0.5 ⁇ PCR buffer [5 mM Tris-HCl pH 8.4, 25 mM KCl, 0.05% (v / v) Triton X-100].
- TRAP system reaction containing 0.25 mM each of 19 proteinaceous amino acids (excluding methionine), 25 ⁇ M biotin-Phe-tRNA fMet CAU and 5% (v / v) random-DNA RT-PCR solution Also used.
- the beads were washed 3 times with 60 ⁇ L HBST and 5 ⁇ L of 1 ⁇ RT mix [1 ⁇ M G5S-4.R20, 50 mM Tris-HCl pH 8.3, 75 mM KCl, 3 mM MgCl 2 , 10 mM DTT, 0.5 Suspended in mM dNTPs, 5 U M-MLV (+) (Promega)].
- the TRAP system contains 19 proteinaceous amino acids of 0.25 mM each (except Met), 25 ⁇ M ClAB-L-Phe-tRNA fMet CAU and 5% (v / v) PCR crude solution.
- the reaction mixture (20 ⁇ L) was incubated at 37 ° C. for 15 minutes.
- reaction mixture After adding 5 ⁇ L of 100 mM EDTA (pH 7.5), the reaction mixture is mixed with 8.5 ⁇ L of 4 ⁇ RT mix [200 mM Tris-HCl (pH 8.3), 300 mM KCl, 75 mM MgCl 2 , 4 mM DTT , 2 mM of each dNTP, 10 ⁇ M G5S-4.R20, 6 U ReverTraAce] and incubated at 42 ° C. for 30 minutes.
- 4 ⁇ RT mix 200 mM Tris-HCl (pH 8.3), 300 mM KCl, 75 mM MgCl 2 , 4 mM DTT , 2 mM of each dNTP, 10 ⁇ M G5S-4.R20, 6 U ReverTraAce
- the reaction solution was reduced to 1/4 scale.
- negative selection using streptavidin beads was performed three times before positive selection.
- a more stringent wash with 200 ⁇ L HBST was performed at 37 ° C for 30 minutes with a second wash.
- the amplified cDNA was cloned and sequenced.
- Binding of cloned peptides and their derivatives to HSA in display form p1 and p2 mRNAs were synthesized from colony PCR products.
- the reverse or shuffled sequence of each mRNA of p1 and p2 peptides was prepared as follows.
- a DNA template was prepared using T7SD8M2.F44 as a forward primer and SD8No38revG5S4.R69, SD8No38ranG5S4.R69, SD8No41revG5S4.R75 or SD8No41ranG5S4.R75 as a reverse primer.
- Extension PCR and transcription were performed in a manner similar to that used to prepare T7-mRNA.
- MRNA was purified by phenol / chloroform extraction and 2-propanol precipitation and dissolved in ultrapure water.
- HSA-immobilized SA beads 0.3 pmol HSA
- SA-beads After 10 minutes incubation at room temperature, the beads were washed 3 times with 10 ⁇ L of HBST. In the second washing, stringent washing was performed for 30 minutes at 37 ° C. using 100 ⁇ L of HBST.
- the recovered cDNA was quantified by real-time PCR.
- ⁇ 210> is the sequence number, ⁇ 223> is other information ⁇ 210> 1 ⁇ 223> T7SD8M2.F44 ⁇ 210> 2 ⁇ 223> G5S-4an21.R41 ⁇ 210> 3 ⁇ 223> G5S-4an13.R36 ⁇ 210> 4 ⁇ 223> G5S-4.R20 ⁇ 210> 5 ⁇ 223> Biotin-DNA ⁇ 210> 6 ⁇ 223> Competitor DNA ⁇ 210> 7 ⁇ 223> Puromycin-linker.an13 ⁇ 210> 8 ⁇ 223> Puromycin-linker.an21 ⁇ 210> 9 ⁇ 223> SD8-NNK ⁇ 210> 10 ⁇ 223> NNK-CG5S4 ⁇ 210> 11 ⁇ 223> SD8T7G5S4.R72 ⁇ 210> 12 ⁇ 223> SD8No38revG5S4.R69 ⁇ 210> 13 ⁇ 223> SD8No38ranG5S4.R69 ⁇
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Organic Chemistry (AREA)
- Biotechnology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Plant Pathology (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Bioinformatics & Computational Biology (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biophysics (AREA)
- Ecology (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description
(1)鋳型DNAライブラリーを加えることで、鋳型DNAライブラリーのmRNAへの転写、mRNAとリンカーとの結合、mRNAの翻訳、翻訳産物との結合を介して、翻訳産物/リンカー/mRNA複合体を形成させる工程を反応系中で自動的に行うことを可能にする、転写-リンカー結合-翻訳反応カップリング反応系であり、本反応系は転写に必要な因子、翻訳に必要な因子、及びリンカーを含む。
(1-2)DNAは転写酵素のプロモータ配列を含み、反応系中に存在する転写に必要な因子により、鋳型DNAライブラリーからmRNAライブラリーが迅速に生成可能となる。転写に必要な因子が、DNAからmRNAへの転写を促進することが好ましい。例えば、転写に必要な因子には、転写酵素が0.1μM以上含まれていてもよい。
(1-3)反応系中に存在する翻訳に必要な因子が、翻訳を促進することが好ましい。例えば、翻訳に必要な因子には、EF-Pが含まれていてもよい。
(1-4)無細胞転写・翻訳系において、転写に必要な因子または翻訳に必要な因子は大腸菌抽出液、小麦胚芽抽出液、ウサギの網状赤血球、昆虫細胞抽出液、ヒト細胞抽出液等に由来することができる。
(1-5)翻訳に必要な因子には、20種類のアミノアシルtRNA合成酵素(ARS)が含まれていてもよい。
(1-6)DNA配列中に、翻訳効率の低いもしくは翻訳されない空きコドンを導入することで、翻訳系中で、翻訳産物/リンカー/mRNA複合体を安定化することも可能である。空きコドンの場所は、望ましくはリンカーがアニールする配列と重なるか、その上流20塩基以内であることができる。
(1-7)鋳型DNAライブラリーのmRNAへの転写、mRNAとリンカーとの結合、mRNAの翻訳、翻訳産物との結合を介して、翻訳産物/リンカー/mRNA複合体を形成させる工程を1時間以内で完了することが可能である。
(2)mRNAとリンカーは、翻訳系中で結合する。リンカーは、翻訳系中でmRNAと複合体を形成する部分を持ち、これにはDNA、RNAなどの核酸や、核酸アナログなどがあるが、これらの例に限らない。当該部分のリンカーの塩基配列がmRNAの3´末端非翻訳領域の配列とアニーリングし、mRNAとリンカーは、共有結合および/または非共有結合を介して結合する。
(2-2)アニーリングにより、リンカーと各mRNAが共有結合を介さずに結合することが好ましい。
(2-3)mRNAと複合体を形成する部分のリンカー塩基配列の3´側の最初の10塩基とmRNAの3´末端非翻訳領域の配列の相補性が50%以上であることが好ましい。
(2-4)mRNAと複合体を形成する部分のリンカー塩基配列の3´側の最初の13塩基は、図1cに記載されたAn13-タイプの Pu-linkerと同じ塩基配列を有していてもよい。
(2-5)mRNAと複合体を形成する部分のリンカー塩基配列の3´側の最初の21塩基は、図1bに記載されたAn21-タイプの Pu-linkerと同じ塩基配列を有していてもよい。
(3)リンカーは翻訳産物と結合可能な求核剤(Nucleophile)を含み、これには翻訳産物と結合可能なアミノアシルRNA、アミノアシルRNAアナログ、アミノ酸、アミノ酸アナログ、ピューロマイシン、ピューロマイシンアナログなどがあるが、これらの例に限らない。
(4)mRNAをDNAの代わりに使用する場合は、mRNAとリンカーを上記の方法により結合させて用いることができ、この場合は翻訳系のみからなる反応系も使用できる。
(5)翻訳産物/リンカー/mRNA複合体の形成を単一の工程で実施して、翻訳産物のライブラリーを作製する方法。当該工程は転写-リンカー結合-翻訳反応カップリング反応系に鋳型DNAライブラリーを加えることを含み、当該反応系は、(1)~(4)に記載された、転写に必要な因子、翻訳に必要な因子、及びリンカーを含む反応系である。
(6)上記の方法で作成した翻訳産物のライブラリーから、有用な所望の機能を有する翻訳産物を選択し、選択された翻訳産物に結合しているmRNAまたはDNAを増幅することで、所望の機能を有する翻訳産物をコードする核酸ライブラリーを作製できる。この核酸ライブラリーを鋳型として再び上記反応系に加えて翻訳産物のライブラリーを得て、さらに選択-増幅の工程を実施することを複数回繰り返し、有用な翻訳産物をコードする核酸が濃縮された核酸ライブラリーを得ることができる。
(6-2)上記の方法を、第二ラウンド以降で使用することが好ましい。その場合、先のラウンドからのPCR産物を精製しないでそのまま、鋳型DNAライブラリーとして添加することができる。
(6-3)所望の機能を持つ翻訳産物は、標的に結合する翻訳産物であることができる。
(6-4)上記の選択-増幅操作を3時間以内に完了させることが可能である。
(6-5)上記の選択-増幅操作を1日で2回以上繰り返すことが可能である。
(6-6)上記の選択-増幅操作は機械操作により自動化することも可能である。
(6-7)標的は、精製した標的、膜に挿入した標的、ファージに提示した標的、バキュロウイルスに提示した標的、細胞に提示した標的などがあるが、これに限定しない。
(6-8)翻訳産物はペプチド、タンパク質や、ペプチドアナログとすることもできるが、これに限らない。
(6-9)翻訳産物は複合体形成後に、翻訳後修飾により修飾することもできる。
(6-10)標的物質が、ヒト血清アルブミンである、(6-3)の方法。
(7)前記(6-10)の方法により得られた、実施例4の化合物p1とp2。
本発明の第一の側面は、リンカーを含有する無細胞転写・翻訳反応系(cell-free transcription-translation coupling system)である。この反応系は、転写・翻訳に必要な因子と、翻訳産物のアクセプターをもつリンカーを含み、反応系に鋳型DNAライブラリーを加えることで、鋳型DNAライブラリーのmRNAへの転写、mRNAとリンカーとの結合、リボソーム上でのmRNAの翻訳、及び翻訳産物との結合を介して、翻訳産物/リンカー/mRNA複合体を形成させる工程を反応系中で自動的に行うことを可能にする。
上述したように、本発明の無細胞翻訳系(TRAP システム)は、リンカーを含有する反応系であり、リンカーとmRNAとの複合体形成が反応系内で起こる。
本発明のTRAPシステムで作成された「翻訳産物-リンカー/mRNA複合体」のライブラリーから、標的に結合する翻訳産物を選択し、これに結合しているmRNAまたはDNAを増幅して、さらにTRAPシステムで翻訳産物ライブラリーを作ることで、有用な翻訳産物を増幅できる。これが、本発明の第二の側面である。
標的物質は、翻訳産物と相互作用することが有用であり得る任意の化合物であることができる。標的物質としては、タンパク質、核酸、糖質、脂質、その他どのような化合物でもよい。
p1:XTYNERLFWC
p2:XSQWDPWAIFWC
(X は ClAB-L-Pheを示す)
これらの環状ペプチドも、本発明の範囲に含まれる。
これまでの説明および後述の実施例の内容に関し、分子生物学的手法の詳細は、例えば、Sambrook, Molecular Cloning: A Laboratory Manual, 3rd edition, Cold Spring Harbor Laboratory Press, 2001; Golemis, Protein-Protein Interactions: A Molecular Cloning Manual, 2nd edition, Cold Spring Laboratory Press, 2005などを参照されたい。
ペプチドとは、様々なアミノ酸がアミド結合(ペプチド結合)によって連結してできた生体高分子化合物の総称である。一般に、鎖中が50アミノ酸残基以下の比較的短いものをペプチドと呼び、それ以上の長さを持つものはタンパク質あるいはポリペプチドと呼ばれることが多い。30残基以下のものは短鎖ペプチドと呼ばれることもある。本明細書では、短鎖ペプチド、ペプチド、タンパク質、及びポリペプチドの用語を特に区別せず、相互に読み替えることができる語として用いる。
タンパク質性アミノ酸または天然アミノ酸とは、通常の翻訳で使用されるα-アミノカルボン酸(または置換型α-アミノカルボン酸)である、アラニン(Ala)、バリン(Val)、ロイシン(Leu)、イソロイシン(Ile)、プロリン(Pro)、トリプトファン(Trp)、フェニルアラニン(Phe)、メチオニン(Met)、グリシン(Gly)、セリン(Ser)、トレオニン(Thr)、チロシン(Tyr)、システイン(Cys)、グルタミン(Gln)、アスパラギン(Asn)、リジン(Lys)、アルギニン(Arg)、ヒスチジン(His)、アスパラギン酸(Asp)、及びグルタミン酸(Glu)の20種類のアミノ酸を指す。
本明細書においてアミノ酸にはタンパク質性アミノ酸と特殊アミノ酸の両方が含まれ、ペプチドには特殊ペプチド(または非天然ペプチドまたはペプチドアナログ)も含まれる。
翻訳とは、一般的には塩基配列からなるmRNAの情報を読み取ってリボソーム上でアミノ酸配列に変換することをいい、実質的にはタンパク質(ペプチド)生合成と同義である。翻訳反応は核酸を鋳型としてペプチドを合成する反応であるため、鋳型となるcDNAまたはmRNAの配列を変えることで様々な配列を持つペプチドを合成できる。翻訳産物は、リボソームにより合成されるペプチド(ペプチド、タンパク質、ペプチドアナログ、特殊ペプチドを含む)である。
アニーリングとは、一般的には、変性して1本鎖になったDNAが適当な条件下で相補的塩基対形成により二本鎖分子になることである。本明細書では、DNAに限らず、核酸塩基を側鎖に持つ構造の1本鎖が相補的塩基対同士で会合して二本鎖となることをいう。相補的塩基対形成は、ハイブリダイゼーションまたは相補的塩基対合ともいい、相補的な組み合わせの核酸塩基が水素結合により対合することである。ハイブリダイゼーションは1本鎖DNAまたはRNAが相補的塩基対形成によってハイブリッド(雑種二本鎖核酸分子)を形成することをいう。本明細書では、リンカー塩基配列とmRNAの相補的配列部分がアニーリングにより二本鎖を形成する。相補的塩基対合は、塩と温度の許容条件下でのポリヌクレオチド(RNAまたはDNA、または核酸塩基を側鎖に持つ他の直鎖分子)の自然な結合であり、2種の一本鎖分子間での相補性は、「部分的」であってもよく、その場合、核酸配列のうちのいくつかの塩基のみが結合し、相補的塩基対の比率を数値で示すことができる。2種の一本鎖分子間に完全な相補性が存在する場合、その相補性は「完全」(100%)である。相補性の度合いは、ハイブリダイゼーションの効率と強度に有意な影響を及ぼす。2種の核酸分子がハイブリダイズするか否かを決定するためには、当業者に周知の様々な方法を利用することができる。典型的には、相補的塩基対となる核酸の塩基の組合わせは、アデニン(A)とチミン(T)またはウラシル(U)、及び、グアニン(G)とシトシン(C)である。チミン(T)とウラシル(U)は、ポリヌクレオチドのそれぞれの型(DNAあるいはRNA)により、交換可能に使用され得る。さらに、G-Uなどのいわゆる非ワトソン-クリック塩基対も熱力学的に安定な塩基対として存在するので、そのような組合せも相補的という場合がある。
ライブラリーは、複数の分子(例えば、複数の核酸、翻訳産物、翻訳産物/リンカー/mRNA複合体、翻訳産物/リンカー/mRNA-cDNA複合体分子)の集団を意味する。本発明におけるライブラリーは、所望の機能や性質を持つペプチド、タンパク質、ペプチドアナログを創製することを目的として、可能性のある遺伝子を大規模に準備し、その中から狙った表現型を有するものを選択するためのものであるから、多数の候補分子を含むライブラリーを用いることが好ましい。ライブラリーの多様性は、例えば1011個、典型的には1012~1013個、好ましくは1015個の異なる遺伝子配列であることができる。
選択(セレクション)とは、ある分子をライブラリー中の他の分子から実質的に分離することを意味する。進化分子工学では、遺伝子ライブラリーの翻訳産物としてペプチドライブラリー(ペプチドと核酸が対応付けされた分子の集団)を得て、このペプチドライブラリーから、所望の機能や性質を持つものを適切なスクリーニング系で選択する。選択された各ペプチド分子には、その遺伝子である核酸がタグのように付加されている。選択された対応付け分子の核酸部分のPCR増幅により核酸ライブラリーを調製して、狙った表現型を有するクローンが多く含まれるバイアスのかかったライブラリーを得た後に、再度同じような選択実験を行う。この選択-増幅操作を繰返すことで、世代の経過とともに所望の表現型を有する分子がライブラリー中で濃縮(富化:enrich)されていく。
本実施例の方法では、リンカーとmRNAの結合は非共有結合によるものである。ペプチド-リンカー/mRNA複合体の形成とペプチドアプタマー選択過程の間に、このリンカーを介するペプチドとmRNAの結合が解離して他の無関係なmRNAと入れ替わってしまわないか確認するため、T7-ペプチドプルダウンアッセイを実施した。
ランダムライブラリーからのペプチド選択では、ペプチドとmRNAが連結した複合体の形成効率がライブラリーの多様性を決定する。複合体形成の効率を評価するために、ビオチン化Phe(図3a)が結合した開始tRNAfMet CAUを、Metを除いた翻訳系に加えて、N末端がビオチン標識されたペプチドを合成した(図3b)。これにより、ストレプトアビジンを固定化したビーズ(SA-bead)を用いて、ビオチン化ペプチドを提示するmRNAのみを選択的に回収できる。
An21タイプのリンカーを使用する系と従来のmRNA提示法(Li13タイプのリンカー)で、上述のT7ペプチドプルダウンアッセイを行って、T7-DNAの濃縮効率を比較した。
Flexible Display法を使用して、環状ペプチドのライブラリーから、ヒト血清由来アルブミン(HSA)に結合するペプチアプタマーの選択をおこなった。
以下の配列のDNAからmRNAを常法により調製した。
CTAGTAATACGACTCACTATAGGGTTAACTTTAAGAAGGAGATATACATATGNNWNNWNNWNNWNNWNNWNNWNNWATGGGAGGTGGTTCAGGAAGTTAGGACGGGGGGCGGGAGGCGGG
NはA/T/G/C、WはA/Tを表す。フレキシザイムにより開始ATGコドンにN-α-ビオチニル-L-フェニルアラニン、伸長ATGコドンにトリプトファンを導入してこのmRNAを翻訳した時のアミノ酸配列はNNWが終止コドンであるTAAまたはTGAでない場合は以下のようになると期待される。
Biotin-Phe-Xaa-Xaa-Xaa-Xaa-Xaa-Xaa-Xaa-Xaa-Trp-Gly-Gly-Gly-Ser-Gly-Ser
ここでXaaはメチオニン、トリプトファン以外の18種類の天然型アミノ酸に相当する。
略語
CME, シアノメチルエステル(cyanomethyl ester); MgSO4, 硫酸マグネシウム(magnesium sulfate); MgCl2, 塩化マグネシウム(magnesium chloride); DMSO, ジメチルスルホキシド(dimethyl sufoxide); Tris, トリス(ヒドロキシメチル)アミノエタン(Tris(hydroxymethyl)aminomethane); Hepes, 2-エタンスルホン酸(2- ethansufonic acid); EDTA, エチレンジアミン四酢酸(ethylene diaminetetraacetic acid); DTT, ジチオスレイトール(dithiothreitol); KOH, 水酸化カリウム
L-Phe-tRNAAsn-E2 CUA及びビオチン-L-Phe-tRNAfMet CAUは以前記載した方法で調製した。N-[3-(2-chloroacetamide)benzoyl]-L-Phe-CME (ClAB-L-Phe-CME)は、L-Pheに3-(2-chloroacetamide)benzoyl chlorideをカップリングし、以前記載した方法と同様の方法でシアノメチルエステル化を行い合成した。ClAB-L-Phe-tRNAfMet CAUは以前記載した方法と同様の方法で調製した。(H. Murakami, A. Ohta, H. Ashigai et al., Nat. Methods 3 (5), 357 (2006); Y. Goto, T. Katoh, and H. Suga, Nat. Protoc. 6 (6), 779 (2011).)
鋳型DNAは、表1に示したオリゴヌクレオチドを用いて、伸長反応とPCRにより合成した。

ランダム-DNA :T7SD8M2.F44と、SD8NNK8CG5S4.R69 ~SD8NNK12CG5S4.R81のいずれか
T7-DNA :T7SD8M2.F44と、SD8T7G5S4.R72
T7SD8M2.F44とG5S-4an13.R36 (Li13, An13) 又はG5S-4an21.R41 (An21)
クレアチンキナーゼ(creatine kinase)、クレアチンリン酸(creatine phosphate)及び 大腸菌tRNA類は Roche Diagnostics (Tokyo, Japan)から購入した。ミオキナーゼ(myokinase)はSigma-Aldrich Japan (Tokyo, Japan)から購入した。翻訳のための化学物質、タンパク質類、及びリボソームは以前報告された論文と同様の方法で調製した(K. Josephson, M. C. T. Hartman, and J. W. Szostak, J. Am. Chem. Soc. 127 (33), 11727 (2005); Y. Shimizu, A. Inoue, Y. Tomari et al., Nat. Biotechnol. 19 (8), 751 (2001); Hiroyuki Ohashi, Yoshihiro Shimizu, Bei-Wen Ying et al., Biochem. Biophys. Res. Commun. 352 (1), 270 (2007). Patrick C. Reid, Yuki Goto, Takayuki Katoh et al., Methods Mol. Biol. 805, 335 (2012).)。
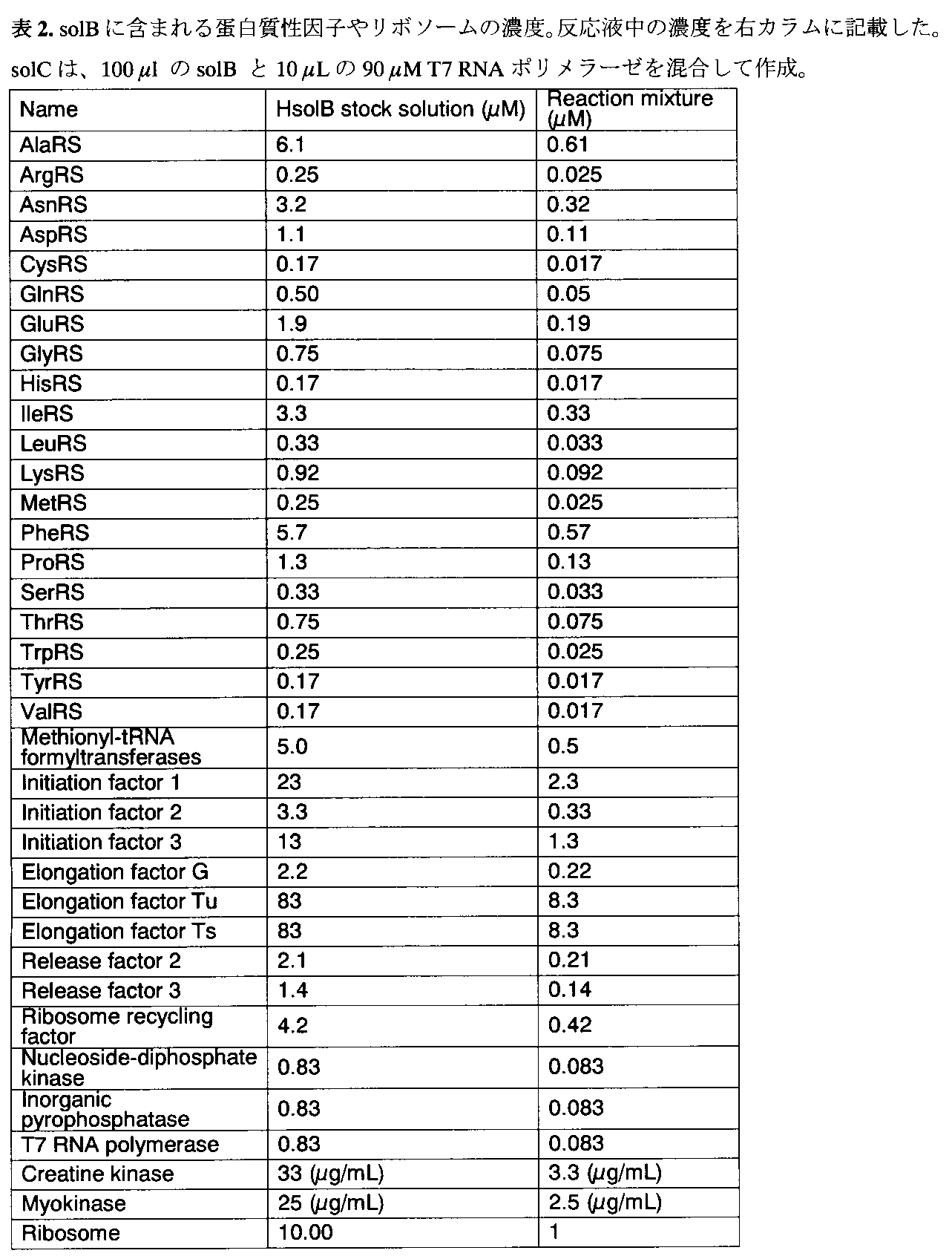
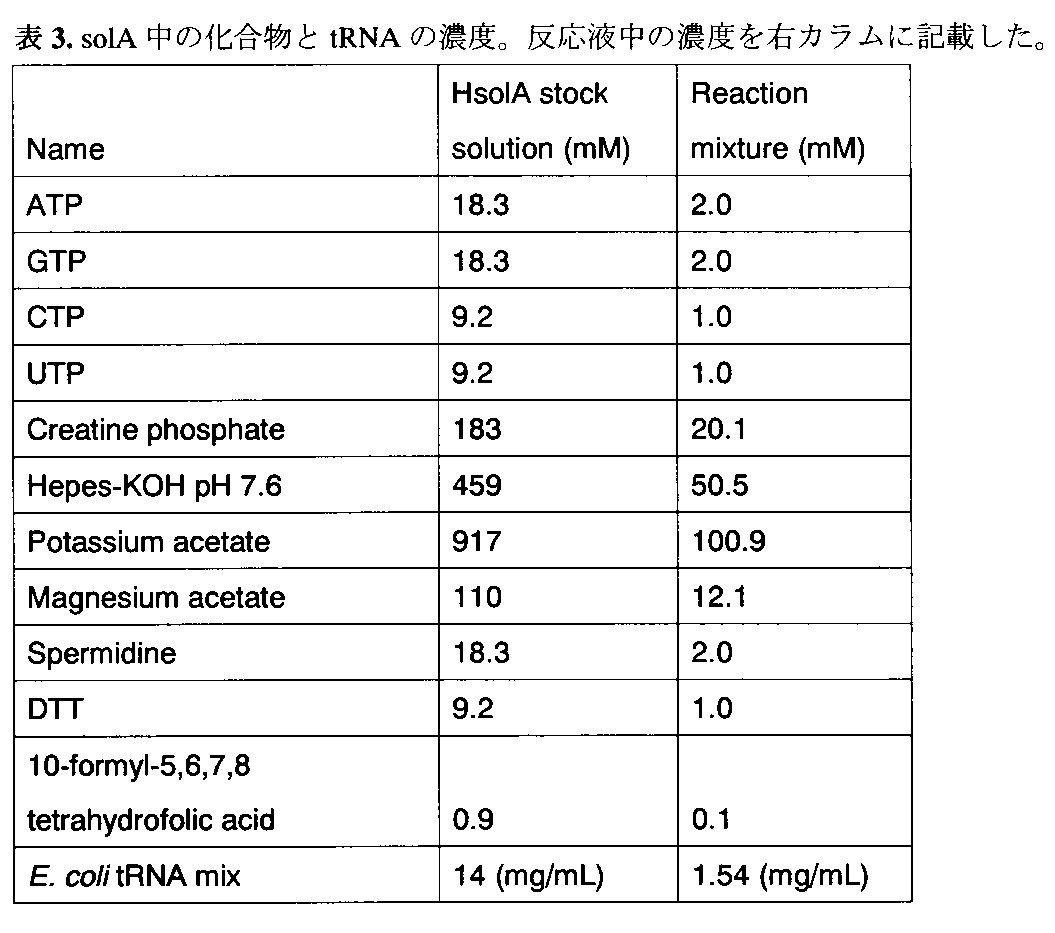
Li13-タイプの Pu-リンカー/mRNA 複合体は、表1のPuromycin-linker.an13とmRNAを混合し、T4 RNA リガーゼで共有結合を形成させて調製した。An13-タイプ及びAn21-タイプの Pu-linker/mRNA 複合体は、mRNA と 1.1 当量の表1のPuromycin-linker.an13または、Puromycin-linker.an21を混合して調製した。以下に記載したPu-リンカー/mRNA 複合体の濃度はmRNA濃度である。
各0.25 mMの20 種のタンパク質性アミノ酸、0.1 μMのLi13-, An13- または An21-タイプの Pu-リンカー/T7-mRNA 複合体、及び対応する1 μMのPu-リンカー/ランダム-mRNA 複合体を含む翻訳反応液を使用した。別法として、UAG コドンをブランクから Pheに再割り当てするために、25 μMのPhe-tRNAAsn-E2 CUA を反応液に加えた。
各 0.25 mMの19種の(メチオニンを除いた)タンパク質性アミノ酸、 25 μMのビオチン-Phe-tRNAfMet CAU及び Li13-タイプ または An21-タイプのPu-リンカー/ランダム-mRNA 複合体 1 μMを含有する翻訳反応液を使用した。別法として、反応液に25 μM のPhe-tRNAAsn-E2 CUA を加えて、UAG コドンをブランクから Pheに割り当てし直した。各 0.25 mMの19種の(メチオニンを除いた)タンパク質性アミノ酸、 25 μMのビオチン-Phe-tRNAfMet CAU及び 5% (v/v) ランダム-DNA RT-PCR 溶液を含有するTRAP システムの反応液も使用した。
各 0.25 mMの20種のタンパク質性アミノ酸、0.3 nMのLi13タイプまたはAn21タイプの Pu-リンカー/T7-mRNA 複合体、及び対応する1 μMのPu-リンカー/ランダム-mRNA複合体を含有する翻訳反応液を使用した。従来型のmRNA提示法 (Li13)のために, 25 μMのPhe-tRNAAsn-E2 CUA を加えて、UAGコドンをブランクからPheに割り当てし直した。各0.25 mMの20種のタンパク質性アミノ酸と、T7-cDNAとランダム-cDNAを1:3000 の比率で混合した5% (v/v) RT-PCR 溶液を含有するTRAPシステムの反応液も使用した。
第1ラウンドでは、各 0.25 mMの19種のタンパク質性アミノ酸(-Met)、10 μMのClAB-L-Phe-tRNAfMet CAU、1 μMのmRNA ライブラリー、及び1.1 μM Pu-リンカー(an21)を含有する翻訳反応液 (50 μL) を37℃で 15分間インキュベートした。12.5 μL の100 mM EDTA (pH7.5)を加えた後、反応混合液をHSA 固定化 SA-ビーズ (1.6 pmol HSA)と、室温で20分間インキュベートした。ビーズを 60 μLのHBSTで3回洗浄し、5 μL の 1×RT ミックス [1 μM G5S-4.R20, 50 mM Tris-HCl pH 8.3, 75 mM KCl, 3 mM MgCl2, 10 mM DTT, 0.5 mM の各 dNTP, 5 U M-MLV (+)(Promega)] 中に懸濁した。42°Cで30分間インキュベーションした後、混合物を100 μLの1×PCR ミックス (1×PCRバッファー, 2 mM MgCl2, 0.25 mMの各dNTP, 0.25 μM T7SDM2.F44, 0.25 μM G5S-4.R41,及び Taq DNA ポリメラーゼ)に添加し、95°Cで5 分間加熱した。1 μL の混合物を回収されたcDNAの定量に使用し、残りをTaq DNA ポリメラーゼの添加後、PCR (94°C で20 s, 55°Cで20 s, 72°Cで30 s)に供した。
p1 及び p2 のmRNA をcolony PCR産物 から合成した。p1 及び p2ペプチドの各 mRNAのreverse またはshuffled配列を、以下のように調製した。T7SD8M2.F44 を forwardプライマーとして、SD8No38revG5S4.R69, SD8No38ranG5S4.R69, SD8No41revG5S4.R75 またはSD8No41ranG5S4.R75 を reverseプライマーとして使用してDNA鋳型を調製した。伸長PCR 及び転写を、T7-mRNAの調製に用いた手法と同様の手法で実施した。フェノール/クロロホルム抽出と 2-プロパノール沈殿で mRNAを精製し、超純水に溶解した。
<210> 1
<223> T7SD8M2.F44
<210> 2
<223> G5S-4an21.R41
<210> 3
<223> G5S-4an13.R36
<210> 4
<223> G5S-4.R20
<210> 5
<223> Biotin-DNA
<210> 6
<223> Competitor DNA
<210> 7
<223> Puromycin-linker.an13
<210> 8
<223> Puromycin-linker.an21
<210> 9
<223> SD8-NNK
<210> 10
<223> NNK-CG5S4
<210> 11
<223> SD8T7G5S4.R72
<210> 12
<223> SD8No38revG5S4.R69
<210> 13
<223> SD8No38ranG5S4.R69
<210> 14
<223> SD8No41revG5S4.R75
<210> 15
<223> SD8No41ranG5S4.R75
<210> 16
<223> C+GGGGGS spacer
<210> 17
<223> C + spacer pepide
<210> 18
<223> An21 mRNA annealing sequence
<210> 19
<223> spacer and annealing sequence
<210> 20
<223> stop and annealing sequence to An13
<210> 21
<223> p1
<210> 22
<223> p2
<210> 23
<223> p3
<210> 24
<223> p4
<210> 25
<223> p5
<210> 26
<223> p6
<210> 27
<223> p7
<210> 28
<223> p8
<210> 29
<223> p9
<210> 30
<223> Reversed p1
<210> 31
<223> Shuffled p1
<210> 32
<223> Reversed p2
<210> 33
<223> Shuffled p2
<210> 34
<223> 5'-SD-AUG (Ex.5)
<210> 35
<223> AUG-spacer-an21 (Ex.5)
<210> 36
<223> spacer (Ex.5)
Claims (17)
- 鋳型DNAライブラリーを加えることで、鋳型DNAライブラリーのmRNAへの転写、mRNAとリンカーとの結合、mRNAの翻訳、翻訳産物との結合を介して、翻訳産物/リンカー/mRNA複合体を形成させる工程を反応系中で自動的に行うことを可能にする、転写-リンカー結合-翻訳反応カップリング反応系であって、転写に必要な因子、翻訳に必要な因子、及びリンカーを含む、前記反応系。
- 鋳型DNAライブラリーのmRNAへの転写、mRNAとリンカーとの結合、mRNAの翻訳、翻訳産物との結合を介して、翻訳産物/リンカー/mRNA複合体を形成させる工程が1時間以内で完了することができる、請求項1の反応系。
- 反応系がEF-Pを含む、請求項1または2の反応系。
- リンカーは、翻訳系中でmRNAと複合体を形成する部分を持ち、当該部分のリンカーの塩基配列がmRNAの3´末端非翻訳領域の配列とアニーリングし、mRNAとリンカーは、共有結合および/または非共有結合を介して結合する、請求項1~3のいずれかに記載の反応系。
- リンカーと各mRNAが非共有結合により結合する、請求項4の反応系。
- mRNAと複合体を形成する部分のリンカー塩基配列の3´側の最初の10塩基とmRNAの3´末端非翻訳領域の配列の相補性が50%以上である、請求項4または5の反応系。
- リンカーは翻訳産物と結合可能な求核剤を含む、請求項1~6のいずれかに記載の反応系。
- ライブラリー中の各DNAが転写酵素のプロモータ配列を含み、反応系が転写酵素を0.1μM以上含む、請求項1~7のいずれかに記載の反応系。
- 鋳型DNAライブラリーを請求項1~8のいずれかに記載された反応系に加えることにより、翻訳産物のライブラリーを作製する方法。
- 所望の機能を持つ翻訳産物をコードする核酸ライブラリーを作製する方法であって、
(ア)鋳型核酸ライブラリーを請求項1~8のいずれかに記載された反応系に加えることにより、翻訳産物/リンカー/mRNA複合体のライブラリーを作製し、
(イ)翻訳産物/リンカー/mRNA複合体のライブラリーから、所望の機能を持つ翻訳産物を選択し、
(ウ)選択された所望の機能を持つ翻訳産物に結合しているmRNAまたはcDNAを増幅して核酸ライブラリーを得ること
を含む、前記方法。 - 所望の機能を持つ翻訳産物は、標的に結合する翻訳産物である、請求項10の方法。
- 標的に結合する翻訳産物をコードするDNAを得る方法であって、
(ア)鋳型DNAライブラリーを請求項1~8のいずれかに記載された反応系に加えることにより、翻訳産物/リンカー/mRNA複合体のライブラリーを作製し、
(イ)翻訳産物/リンカー/mRNA複合体のmRNA部分の逆転写を行って翻訳産物/リンカー/mRNA-cDNA複合体のライブラリーを得て、
(ウ)翻訳産物/リンカー/mRNA-cDNA複合体のライブラリーから、標的に結合する翻訳産物を選択し、
(エ)選択された翻訳産物に結合しているcDNAを増幅すること、
を含む、前記方法。 - 前記(エ)の工程が、翻訳産物/リンカー/mRNA-cDNA複合体からcDNAを回収し、回収されたcDNAをPCRにより増幅することを含み、
得られたPCR産物を、精製しないでそのまま鋳型DNAライブラリーとして使用してさらに(ア)~(エ)の工程が実施される、請求項12の方法。 - (ア)~(エ)の工程が3時間以内で実行できる、請求項12または13に記載の方法。
- (ア)~(エ)の工程を複数回繰り返すことを含む、請求項12または13に記載の方法。
- (ア)~(エ)の工程を1日で2回またはそれ以上の回数繰り返すことのできる、請求項15の方法。
- 機械操作により自動化される、請求項10~16のいずれかに記載の方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014559708A JPWO2014119600A1 (ja) | 2013-01-30 | 2014-01-29 | FlexibleDisplay法 |
| EP14746442.4A EP2952582A4 (en) | 2013-01-30 | 2014-01-29 | FLEXIBLE DISPLAY METHOD |
| US14/764,475 US20160068835A1 (en) | 2013-01-30 | 2014-01-29 | Flexible display method |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013015423 | 2013-01-30 | ||
| JP2013-015423 | 2013-07-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014119600A1 true WO2014119600A1 (ja) | 2014-08-07 |
Family
ID=51262313
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2014/051907 WO2014119600A1 (ja) | 2013-01-30 | 2014-01-29 | Flexible Display法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20160068835A1 (ja) |
| EP (1) | EP2952582A4 (ja) |
| JP (1) | JPWO2014119600A1 (ja) |
| WO (1) | WO2014119600A1 (ja) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017195901A1 (ja) | 2016-05-13 | 2017-11-16 | 国立大学法人 東京大学 | 肝分泌型代謝制御因子阻害作用による肥満関連疾患治療剤 |
| WO2021167107A1 (ja) | 2020-02-22 | 2021-08-26 | Jcrファーマ株式会社 | ヒトトランスフェリンレセプター結合ペプチド |
| WO2022202761A1 (ja) | 2021-03-22 | 2022-09-29 | ペプチドリーム株式会社 | c-Metタンパク質結合ペプチド複合体 |
| WO2022202816A1 (ja) | 2021-03-22 | 2022-09-29 | ペプチエイド株式会社 | ペプチド及びペプチドを含む組成物 |
| WO2022265109A1 (en) | 2021-06-18 | 2022-12-22 | Peptidream Inc. | Ghr-binding pending peptide and composition comprising same |
| JP7573304B2 (ja) | 2019-07-03 | 2024-10-25 | ペプチドリーム株式会社 | Cd38結合剤およびその使用 |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG11201908579PA (en) * | 2017-03-17 | 2019-10-30 | Cubicstars Inc | Method for producing complex of rna molecule and peptide, and utilization thereof |
| US11814621B2 (en) | 2018-06-01 | 2023-11-14 | Northwestern University | Expanding the chemical substrates for genetic code reprogramming |
| EP3993874A4 (en) | 2019-07-03 | 2023-06-28 | PeptiDream Inc. | Cd38-binding agents and uses thereof |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998016636A1 (fr) | 1996-10-17 | 1998-04-23 | Mitsubishi Chemical Corporation | Molecule permettant d'homologuer un genotype et un phenotype, et utilisation de celle-ci |
| WO1998031700A1 (en) | 1997-01-21 | 1998-07-23 | The General Hospital Corporation | Selection of proteins using rna-protein fusions |
| JP3683902B2 (ja) | 1996-10-17 | 2005-08-17 | 三菱化学株式会社 | 遺伝子型と表現型の対応付け分子及びその利用 |
| WO2008117833A1 (ja) | 2007-03-26 | 2008-10-02 | The University Of Tokyo | 環状ペプチド化合物の合成方法 |
| JP2008253176A (ja) * | 2007-04-03 | 2008-10-23 | Janusys Kk | 高親和性分子取得のためのリンカー |
| WO2010011944A2 (en) * | 2008-07-25 | 2010-01-28 | Wagner Richard W | Protein screeing methods |
| WO2011049157A1 (ja) * | 2009-10-22 | 2011-04-28 | ペプチドリーム株式会社 | ペプチド翻訳合成におけるrapidディスプレイ法 |
| WO2012074130A1 (ja) * | 2010-12-03 | 2012-06-07 | 国立大学法人東京大学 | ペプチドライブラリーの製造方法、ペプチドライブラリー、及びスクリーニング方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6261804B1 (en) * | 1997-01-21 | 2001-07-17 | The General Hospital Corporation | Selection of proteins using RNA-protein fusions |
-
2014
- 2014-01-29 EP EP14746442.4A patent/EP2952582A4/en not_active Withdrawn
- 2014-01-29 US US14/764,475 patent/US20160068835A1/en not_active Abandoned
- 2014-01-29 WO PCT/JP2014/051907 patent/WO2014119600A1/ja active Application Filing
- 2014-01-29 JP JP2014559708A patent/JPWO2014119600A1/ja active Pending
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998016636A1 (fr) | 1996-10-17 | 1998-04-23 | Mitsubishi Chemical Corporation | Molecule permettant d'homologuer un genotype et un phenotype, et utilisation de celle-ci |
| JP3683282B2 (ja) | 1996-10-17 | 2005-08-17 | 三菱化学株式会社 | 遺伝子型と表現型の対応付け分子及びその利用 |
| JP3683902B2 (ja) | 1996-10-17 | 2005-08-17 | 三菱化学株式会社 | 遺伝子型と表現型の対応付け分子及びその利用 |
| WO1998031700A1 (en) | 1997-01-21 | 1998-07-23 | The General Hospital Corporation | Selection of proteins using rna-protein fusions |
| JP3692542B2 (ja) | 1997-01-21 | 2005-09-07 | ザ ジェネラル ホスピタル コーポレーション | Rna−蛋白質の融合体を用いた蛋白質の選抜 |
| WO2008117833A1 (ja) | 2007-03-26 | 2008-10-02 | The University Of Tokyo | 環状ペプチド化合物の合成方法 |
| JP2008253176A (ja) * | 2007-04-03 | 2008-10-23 | Janusys Kk | 高親和性分子取得のためのリンカー |
| WO2010011944A2 (en) * | 2008-07-25 | 2010-01-28 | Wagner Richard W | Protein screeing methods |
| WO2011049157A1 (ja) * | 2009-10-22 | 2011-04-28 | ペプチドリーム株式会社 | ペプチド翻訳合成におけるrapidディスプレイ法 |
| WO2012074130A1 (ja) * | 2010-12-03 | 2012-06-07 | 国立大学法人東京大学 | ペプチドライブラリーの製造方法、ペプチドライブラリー、及びスクリーニング方法 |
Non-Patent Citations (36)
| Title |
|---|
| A. C. FORSTER; Z. TAN; M. N. L. NALAM ET AL.: "Programming peptidomimetic syntheses by translating genetic codes designed de novo", PNAS, vol. 100, no. 11, 2003, pages 6353 - 6357, XP002316170, DOI: doi:10.1073/pnas.1132122100 |
| ATSUSHI YAMAGUCHI; TAKAYUKI KATOH; HIROAKI SUGA, DRUG DELIVERY SYSTEM, vol. 26-6, 2011, pages 584 - 592 |
| CHRISTOPHER J HIPOLITO; HIROAKI SUGA, CURRENT OPINION IN CHEMICAL BIOLOGY, vol. 16, no. 1-2, 2012, pages 196 - 203 |
| DONG, H. ET AL., J. MOL. BIOL., vol. 260, 1996, pages 649 - 663 |
| ELENA NEUMANN; EVA FREI; DOROTHEE FUNK ET AL., EXPERT OPIN. DRUG DELIV., vol. 7, no. 8, 2010, pages 915 |
| GOLEMIS: "Protein-Protein Interactions: A Molecular Cloning Manual, 2nd ed.", 2005, COLD SPRING LABORATORY PRESS |
| GOTO ET AL., ACS CHEM. BIOL., vol. 3, 2008, pages 120 - 129 |
| H. F. KUNG; B. REDFIELD; B. V. TREADWELL; B. ESKIN; C. SPEARS; H. WEISSBACH: "DNA-directed in vitro synthesis of beta-galactosidase. Studies with purified factors", THE JOURNAL OF BIOLOGICAL CHEMISTRY, vol. 252, no. 19, 1977, pages 6889 - 6894 |
| H. MURAKAMI; A. OHTA; H. ASHIGAI ET AL., NAT. METHODS, vol. 3, no. 5, 2006, pages 357 |
| H. OHASHI; Y. SHIMIZU; B. W. YING; T. UEDA: "Efficient protein selection based on ribosome display system with purified components", BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, vol. 352, no. 1, 2007, pages 270 - 276, XP005727956, DOI: doi:10.1016/j.bbrc.2006.11.017 |
| HIROYUKI OHASHI; YOSHIHIRO SHIMIZU; BEI-WEN YING ET AL., BIOCHEM. BIOPHYS. RES. COMMUN., vol. 352, no. 1, 2007, pages 270 |
| I. TABUCHI; S. SORAMOTO; N. NEMOTO ET AL., FEBS LETT., vol. 508, no. 3, 2001, pages 309 |
| IKEMURA, T., J. MOL. BIOL., vol. 146, 1981, pages 1 - 21 |
| JAN TERJE ANDERSEN; INGER SANDLIE, DRUG METAB. PHARMACOKINET., vol. 24, no. 4, 2009, pages 318 |
| JUN YAMAGUCHI; TAKAYUKI KATOH; HIROAKI SUGA: "Drug discovery of non-standard peptide with genetic code reprogramming", DRUG DELIVERY SYSTEM, vol. 26-6, 2011, pages 584 - 592 |
| K. JOSEPHSON; M. C. T. HARTMAN; J. W. SZOSTAK, J. AM. CHEM. SOC., vol. 127, no. 33, 2005, pages 11727 |
| LUCY J. HOLT; AMRIK BASRAN; KATE JONES ET AL., PROTEIN ENG. DES. SEL., vol. 21, no. 5, 2008, pages 283 |
| M KURZ; K GU; P. A. LOHSE, NUCLEIC ACIDS RES., vol. 28, no. E83, 2000 |
| M. C. GONZA; C. CUNNINGHAM; R. M. GREEN: "Isolation and point of action of a factor from Escherichia coli required to reconstruct translation", PROCEEDING OF NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA, vol. 82, 1985, pages 1648 - 1652, XP055124549, DOI: doi:10.1073/pnas.82.6.1648 |
| M. Y. PAVLOV; M. EHRENBERG: "Rate of translation of natural mRNAs in an optimized in Vitro system", ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS, vol. 328, no. 1, 1996, pages 9 - 16, XP001066398, DOI: doi:10.1006/abbi.1996.0136 |
| MOCHIZUKI Y. ET AL.: "One-pot preparation of mRNA/cDNA display by a novel and versatile puromycin-linker DNA", ACS COMB SCI., vol. 13, 2011, pages 478 - 485, XP055271124 * |
| NEMOTO N ET AL., FEBS LETT, vol. 414, 1997, pages 405 - 408 |
| NEMOTO N ET AL.: "In vitro virus", FEBS LETT., vol. 414, 1997, pages 405 - 408, XP004366358, DOI: doi:10.1016/S0014-5793(97)01026-0 |
| PATRICK C. REID; YUKI GOTO; TAKAYUKI KATOH ET AL., METHODS MOL. BIOL., vol. 805, 2012, pages 335 |
| ROBERTS, R. W.; SZOSTAK, J. W., PROC. NATL. ACAD. SCI. USA., vol. 94, 1997, pages 12297 - 12302 |
| ROBERTS, R. W.; SZOSTAK, J. W.: "RNA-peptide fusions", PROC. NATL. ACAD. SCI. USA., vol. 94, 1997, pages 12297 - 12302, XP002913434, DOI: doi:10.1073/pnas.94.23.12297 |
| S. C. MAKRIDES; P. A. NYGREN; B. ANDREWS ET AL., J. PHARMACOL. EXP. THER., vol. 277, no. 1, 1996, pages 534 |
| SAMBROOK: "Molecular Cloning: A Laboratory Manual, 3rd ed.", 2001, COLD SPRING HARBOR LABORATORY PRESS |
| See also references of EP2952582A4 * |
| T. KAWAKAMI; H. MURAKAMI: "Genetically Encoded Libraries of Nonstandard Peptides", JOURNAL OF NUCLEIC ACIDS, vol. 2012, pages 15 |
| TAKAHIRO ISHIZAWA ET AL.: "Hyoteki Ketsugo Peptide no Jinsoku na Sentakuho no Kaihatsu", CSJ: THE CHEMICAL SOCIETY OF JAPAN KOEN YOKOSHU, vol. 92, no. 3, 9 March 2012 (2012-03-09), pages 782, XP008180182 * |
| Y. GOTO; T. KATOH; H. SUGA, NAT. PROTOC., vol. 6, no. 6, 2011, pages 779 |
| Y. SHIMIZU; A. INOUE; Y. TOMARI ET AL., NAT. BIOTECHNOL., vol. 19, no. 8, 2001, pages 751 |
| Y. SHIMIZU; A. INOUE; Y. TOMARI; T. SUZUKI; T. YOKOGAWA; K. NISHIKAWA; T. UEDA: "Cell-free translation reconstituted with purified components", NATURE BIOTECHNOLOGY, vol. 19, no. 8, 2001, pages 751 - 755, XP002677144, DOI: doi:10.1038/90802 |
| YAMAGUCHI J. ET AL.: "cDNA display: a novel screening method for functional disulfide-rich peptides by solid-phase synthesis and stabilization of mRNA-protein fusions. art e108", NUCLEIC ACIDS RES., vol. 37, no. 16, 2009, pages 1 - 13, XP008154704 * |
| YUKI GOTO; TAKAYUKI KATOH; HIROAKI SUGA, NATURE PROTOCOLS, vol. 6, 2011, pages 779 - 790 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017195901A1 (ja) | 2016-05-13 | 2017-11-16 | 国立大学法人 東京大学 | 肝分泌型代謝制御因子阻害作用による肥満関連疾患治療剤 |
| JP7573304B2 (ja) | 2019-07-03 | 2024-10-25 | ペプチドリーム株式会社 | Cd38結合剤およびその使用 |
| WO2021167107A1 (ja) | 2020-02-22 | 2021-08-26 | Jcrファーマ株式会社 | ヒトトランスフェリンレセプター結合ペプチド |
| WO2022202761A1 (ja) | 2021-03-22 | 2022-09-29 | ペプチドリーム株式会社 | c-Metタンパク質結合ペプチド複合体 |
| WO2022202816A1 (ja) | 2021-03-22 | 2022-09-29 | ペプチエイド株式会社 | ペプチド及びペプチドを含む組成物 |
| WO2022265109A1 (en) | 2021-06-18 | 2022-12-22 | Peptidream Inc. | Ghr-binding pending peptide and composition comprising same |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2014119600A1 (ja) | 2017-01-26 |
| EP2952582A4 (en) | 2016-11-16 |
| US20160068835A1 (en) | 2016-03-10 |
| EP2952582A1 (en) | 2015-12-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2014119600A1 (ja) | Flexible Display法 | |
| US10195578B2 (en) | Peptide library production method, peptide library, and screening method | |
| CN110637086B (zh) | Rna分子与肽的复合体的制造方法及其利用 | |
| WO2016154675A1 (en) | Platform for non-natural amino acid incorporation into proteins | |
| JP6440055B2 (ja) | ペプチドライブラリの製造方法、ペプチドライブラリ、及びスクリーニング方法 | |
| US7622248B2 (en) | Ribozymes with broad tRNA aminoacylation activity | |
| US7846694B2 (en) | Process for producing template DNA and process for producing protein in cell-free protein synthesis system with the use of the same | |
| JP6357154B2 (ja) | 荷電性非タンパク質性アミノ酸含有ペプチドの製造方法 | |
| EP3487998B1 (en) | Compositions and methods for identifying rna binding polypeptide targets | |
| KR20200036925A (ko) | 이중-가닥 콘카테머 dna를 사용하는 무세포 단백질 발현 | |
| WO2003062417A1 (fr) | Produit de ligation arn-adn, et utilisation correspondante | |
| US9783800B2 (en) | Method for producing peptides having azole-derived skeleton | |
| JP2017216961A (ja) | 非天然アミノ酸含有ペプチドライブラリ | |
| Fujino et al. | In Vitro Selection Combined with Ribosomal Translation Containing Non‐proteinogenic Amino Acids | |
| US20220002719A1 (en) | Oligonucleotide-mediated sense codon reassignment | |
| WO2023048291A1 (ja) | tRNA、アミノアシルtRNA、ポリペプチド合成用試薬、非天然アミノ酸の導入方法、ポリペプチドの作製方法、核酸ディスプレイライブラリの作製方法、核酸-ポリペプチド連結体及びスクリーニング方法 | |
| EP4410982A1 (en) | Method for producing polypeptide, tag, expression vector, method for evaluating polypeptide, method for producing nucleic acid display library, and screening method | |
| JP2016123343A (ja) | 核酸リンカー | |
| WO2024172058A1 (ja) | EF-P応答性人工開始tRNAを用いた外来アミノ酸による翻訳開始 | |
| EP4063377A1 (en) | Modification of trna t-stem for enhancing n-methyl amino acid incorporation | |
| JP2017035063A (ja) | 任意の標的物質と共有結合可能な(ポリ)ペプチド/タンパク質タグの選択方法及び選択されて得られた(ポリ)ペプチド/タンパク質タグ | |
| JP4198387B2 (ja) | 無細胞タンパク質合成系におけるタンパク質またはペプチドの製造方法、およびこれを用いて製造したタンパク質またはペプチド | |
| JP2012139197A (ja) | mRNA/cDNA−タンパク質連結体作製用リンカーとそれを用いたヌクレオチド−タンパク質連結体の精製方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14746442 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014559708 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14764475 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014746442 Country of ref document: EP |