WO2013101968A1 - Process for the production of hexanediols - Google Patents
Process for the production of hexanediols Download PDFInfo
- Publication number
- WO2013101968A1 WO2013101968A1 PCT/US2012/071891 US2012071891W WO2013101968A1 WO 2013101968 A1 WO2013101968 A1 WO 2013101968A1 US 2012071891 W US2012071891 W US 2012071891W WO 2013101968 A1 WO2013101968 A1 WO 2013101968A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cuo
- oxide
- catalyst
- range
- psi
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C29/00—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring
- C07C29/60—Preparation of compounds having hydroxy or O-metal groups bound to a carbon atom not belonging to a six-membered aromatic ring by elimination of -OH groups, e.g. by dehydration
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/04—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D309/06—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
Definitions
- Processes for preparing 1 ,2-cyclohexanediol and mixtures of 1 ,2- cyclohexanediol and 1 ,6-hexanediol are provided.
- Industrial chemicals obtained from inexpensive sources are desirable for use in industrial processes, for example as raw materials, solvents, or starting materials. It has become increasingly desirable to obtain industrial chemicals or their precursors from materials that are not only inexpensive but also benign in the environment.
- materials which can be obtained from renewable sources that is, materials that are produced by a biological activity such as planting, farming, or harvesting.
- the terms “renewable” and “biosourced” can be used interchangeably.
- 1 ,2-Cyclohexanediol and related compounds such as 1 ,6-hexanediol are useful precursors in the synthesis of industrially useful chemicals such as pharmaceuticals, herbicides, stabilizers, and polymers.
- 1 ,2- cyclohexanediol can be converted to adipic acid, o-phenylenediamine, catechol, phenol, benzoquinone, and hydroquinone.
- 1 ,6-Hexanediol is used in the production of polyesters for polyurethane elastomers, coatings, adhesives and polymeric plasticizers.
- 1 ,6-Hexanediol can also be converted to 1 ,6- hexamethylenediamine, a useful monomer in nylon production. Partial oxidation of the petrochemicals cyclohexane and cyclohexene has been used to synthesize 1 ,2-cyclohexanediol.
- renewable sources for materials such as 1 ,2-cyclohexanediol and 1 ,6-hexanediol are desired, in particular renewable sources which are economically attractive in comparison to petroleum-based sources.
- a process comprising: contacting 1 ,2,6-hexanetriol with hydrogen in the presence of a hydrogenation catalyst at a temperature in the range of from about 120 °C to about 300 °C and at a pressure in the range of from about 200 psi to about 3000 psi to form a product mixture comprising
- the product mixture further comprises 1 ,6-hexanediol.
- indefinite article “a” or “an” is used with respect to a statement or description of the presence of a step in a process disclosed herein, unless the statement or description explicitly provides to the contrary, the use of such indefinite article does not limit the presence of the step in the process to one in number.
- the terms “comprises,” “comprising,” “includes,” “including,” “has,” “having,” “contains” or “containing,” or any other variation thereof, are intended to cover a non-exclusive inclusion.
- a composition, a mixture, process, method, article, or apparatus that comprises a list of elements is not necessarily limited to only those elements but can include other elements not expressly listed or inherent to such composition, mixture, process, method, article, or apparatus.
- “or” refers to an inclusive or and not to an exclusive or. For example, a condition A or B is satisfied by any one of the following: A is true (or present) and B is false (or not present), A is false (or not present) and B is true (or present), and both A and B are true (or present).
- the term "about" modifying the quantity of an ingredient or reactant employed refers to variation in the numerical quantity that can occur, for example, through typical measuring and liquid handling procedures used for making concentrates or use solutions in the real world; through inadvertent error in these procedures; through differences in the manufacture, source, or purity of the ingredients employed to make the compositions or carry out the methods; and the like.
- the term “about” also encompasses amounts that differ due to different equilibrium conditions for a composition resulting from a particular initial mixture. Whether or not modified by the term “about,” the claims include equivalents to the quantities.
- the term “about” can mean within 10% of the reported numerical value, preferably within 5% of the reported numerical value.
- biomass refers to any hemicellulosic or lignocellulosic material and includes materials comprising hemicellulose, and optionally further comprising cellulose, lignin, starch, oligosaccharides and/or monosaccharides.
- lignocellulosic means comprising both lignin and cellulose.
- Lignocellulosic material can also comprise hemicellulose.
- lignocellulosic material contains glucan and xylan.
- Hemicellulose is a non-cellulosic polysaccharide found in lignocellulosic biomass. Hemicellulose is a branched heteropolymer consisting of different sugar monomers. It typically comprises from 500 to 3000 sugar monomeric units.
- Lignin is a complex high molecular weight polymer and can comprise guaiacyl units as in softwood lignin, or a mixture of guaiacyl and syringyl units as in hardwood lignin.
- 126HT refers to 1 ,2,6-hexanetriol and includes a racemic mixture of isomers.
- the chemical structure of 1 ,2,6- hexanetriol is represented by Formula (I).
- THPM tetrahydro-2H-pyran- 2-methanol, also known as 2-hydroxymethyltetrahydropyran, and includes a racemic mixture of isomers.
- the chemical structure of tetrahydro-2H-pyran-2- methanol is represented by Formula (II).
- the abbreviation "16HD” refers to 1 ,6-hexanediol.
- the chemical structure of 1 ,6-hexanediol is represented by Formula (III).
- the abbreviation “12CHD” refers to 1 ,2-cyclohexanediol and includes a mixture of stereoisomers (cis and racemic trans isomers).
- the abbreviation “c12CHD” refers to cis-1 ,2-cyclohexanediol.
- the abbreviation “t12CHD” refers to trans-1 ,2-cyclohexanediol.
- the chemical structure of 1 ,2-cyclohexanediol is represented by Formula (IV).
- 15HD refers to 1 ,5-hexanediol and includes a racemic mixture of isomers.
- the chemical structure of 1 ,5- hexanediol is represented by Formula (V).
- 15PD refers to 1 ,5-pentanediol.
- the chemical structure of 1 ,5-pentanediol is represented by Formula (VI).
- the term "renewable biosource” includes biomass and animal or vegetable fats or oils.
- a renewable biosource can be pyrolyzed under high temperature conditions in the presence of an acid catalyst to provide useful chemical intermediates.
- pyrolysis of wood, starch, glucose or cellulose can produce levoglucosenone by known and conventional methods (see, for example, Ponder (Applied Biochemistry and Biotechnology, Vol 24/25, 41 -47 (1990)) or Shafizadeh (Carbohydrate Research, 71 , 169-191 (1979)).
- Glycerol can be obtained from a renewable biosource, for example from hydrolysis of vegetable and animal fats and oils (that is, triacylglycerides comprising ester functionality resulting from the combination of glycerol with Ci2 or greater fatty acids).
- 1 ,2,6-Hexanetriol can be obtained from materials such as glucose, cellulose or glycerol which can be derived from a renewable biosource.
- 1 ,2,6-hexanetriol can be obtained by a process comprising the steps of contacting glycerol with a catalyst to prepare acrolein, heating acrolein optionally in the presence of a catalyst to prepare 2-formyl-3,4-dihydro- 2H-pyran, contacting 2-formyl-3,4-dihydro-2H-pyran with water to prepare 2-hydroxyadipic aldehyde and contacting 2-hydroxyadipic aldehyde with hydrogen and a catalyst to produce a product mixture comprising
- 1 ,2,6-hexanetriol is contacted with hydrogen in the presence of a hydrogenation catalyst comprising a transition metal under suitable temperature and temperature conditions to form a product mixture comprising 1 ,2-cyclohexanediol.
- the product mixture further comprises 1 ,6-hexanediol.
- the product mixture further comprises one or more of tetrahydro-2H-pyran- 2-methanol, 1 ,5-hexanediol, and 1 ,5-pentanediol.
- the hydrogenation catalyst comprises a transition metal selected from the group consisting of platinum, nickel, cobalt, silver, copper, ruthenium, rhodium, iron, palladium, and mixtures thereof. In some embodiments, the catalyst comprises a transition metal selected from platinum, palladium, copper, nickel, or mixtures thereof. In some embodiments, the catalyst comprises copper.
- the hydrogenation catalyst comprises CuO.
- the catalyst comprises from 2 wt% to 98 wt% CuO and further comprises from 98 wt% to 2 wt% of at least one oxide selected from the group consisting of zinc oxide (ZnO), magnesium oxide (MgO), barium oxide (BaO), chromium oxide (Cr 2 O3), silica (S1O2), alumina (AI2O3), zirconium dioxide (ZrO2), nickel oxide (NiO), manganese oxide (MnO2), sodium oxide (Na 2 O), potassium oxide (K 2 O), cerium oxide (CeO 2 ), lanthanum oxide (La2O3), iron oxide (Fe2Os), silver oxide (Ag 2 O) and cobalt oxide (C02O3), based on the total weight of the catalyst.
- the catalyst further comprises ZnO.
- the catalyst further comprises MgO.
- the catalyst further comprises carbon.
- Suitable commercially available catalysts include but are not limited to the following: CuO/ZnO, BaO/CuO/Cr 2 O 3 /SiO 2 , BaO/CuO/Cr 2 O 3 , BaO/CuO/MnO 2 /Cr 2 O 3 , CuO/SiO 2 , CuO/AI 2 O 3 , CuO/NiO/AI 2 O 3 ,
- the catalyst comprises CuO/ZnO, CuO/ZnO/AI 2 O 3 , or CuO/ZnO/CeO 2 /AI 2 O3/Na2O/C.
- catalysts comprising CuO can further comprise a support.
- suitable supports include aluminas, zeolites, CeO 2 , ZrO2, MgO, MgAI 2 O 4 , and ⁇ 2.
- the supports are impregnated with promoters, such as Ba, La, Mg, Ca, Na, and K.
- suitable supported copper catalysts include CuO La2Os ZrO2,
- Catalysts comprising CuO and at least one oxide as described above can be prepared by forming a co-precipitated catalyst comprising compounds which are thermally decomposable to oxides or mixed oxides.
- the precipitated catalyst can be formed by admixing solutions of the elements and heating the resultant mixture to its precipitation temperature; separately heating a solution of a precipitant in water; and thereafter adding both solutions to preheated demineralized water with vigorous stirring and strict pH control, for example in a precipitation reactor.
- the precipitate can be formed by admixing solutions of the elements and heating the resultant mixture to its precipitation temperature; then adding the preheated mixture or solution of elements rapidly to a predetermined volume of a preheated solution of a precipitant in water.
- the precipitate can be formed by admixing solutions of the elements and heating the resultant mixture to its precipitation temperature; then adding a preheated solution of precipitant in water
- the precipitant can be a solution of sodium, potassium and/or ammonium carbonate or bicarbonate in water.
- the precipitation can be carried out at high temperature, for example between about 75 °C and 100 °C. Lower temperatures, for example between about 50 °C and 60 ° C can also be used, but the crystallite size of the catalyst precursor so formed is larger, and the activity of such a catalyst may be lower.
- the precipitation can be effected at a pH in the range of 6.5-9.5.
- the precipitate can then be separated from the residual liquid.
- the separation can be effected by filtration.
- the precipitate can be re-suspended at least once, but typically a few times, in demineralized water, then separated from the water by filtration, and finally washed thoroughly on the filter.
- the washed precipitate comprising a homogeneous hydrated catalyst precursor can then be dried by any known drying process, for example in an oven at temperatures between 50 °C and 130 °C, under vacuum or at normal pressure. Alternatively, spray drying can be employed.
- the dried precipitate also referred to herein as a precursor, comprises an essentially homogeneous association of carbonates and
- the elements may initially be in soluble nitrate form or optionally in the form of a thermally decomposable ammonium salt.
- the dried precipitate can be calcined to provide a catalyst.
- the calcination can comprise treating the dried precipitate at a temperature of between 200 °C and 450 °C, for example between 250 °C and 350 °C, for between 3 and 10 hours, to obtain a homogeneous catalyst.
- the homogeneous catalyst can be densified and pelletized after addition of 1 -3 wt%, for example about 2 wt%, graphite. It can also be made into extrudates using, for example, methyl cellulose as a binder. The homogeneous catalyst can also be sieved to a desired particle size distribution to be used in batch or continuous stirred tank reactors.
- the copper component of the active catalyst contains the copper in a dispersed form, and after activation acts primarily as the active constituent of the catalyst, while the additional oxide component(s) acts primarily but not exclusively as a structural support.
- the active catalyst can be reduced by thermal activation to produce an active catalyst in which at least a portion of the copper, and other element(s) present in the catalyst, are in metallic form.
- the thermal activation can comprise reduction treatment of the calcined catalyst in a reactor, using a mixture of an inert gas, preferably nitrogen, and at least one reducing gas, such as hydrogen, carbon monoxide or a mixture thereof.
- the molar ratio between reducing gas and inert gas should be between 1 :30 and 1 :100.
- the reduction temperature can be between 100 °C to 280 °C, preferably between 130 °C and 240 °C, and the pressure can be 0.1 to 1 MPa.
- the catalyst is preferably first slowly heated at a rate of 30-50 °C/hour under the inert gas at a pressure between 0.6-0.9 MPa, until a temperature between 120 °C and 150 °C has been reached. Thereafter the reduction takes place by adding the reducing gas to the inert gas in a molar ratio as described above, but preferably between 1 :50 and 1 :40. The temperature is then slowly further increased at a rate of 15-25 °C/h to reach a temperature between 190 °C and 210 °C. The thermal reductive activation is continued at this
- the temperature for a time period of between 10 and 24 hours. Thereafter, in a final step, the temperature can be increased to between 230 °C and 250 °C and the molar ratio of reducing gas to inert gas adjusted to between 1 :10 and 1 :6 for a time period of 1 -3 hours, in order to complete activation.
- the reduced catalyst can then be stabilized by passivating the catalyst in a mixture of nitrogen and oxygen to prevent complete oxidation of the catalyst when exposed to air.
- a wide range of commercially available catalyst supports comprising metal oxides, mixed metal oxides or metal-incorporated metal oxides (such as gamma-alumina, La-doped alumina, Ce-doped zirconia, magnesium oxide, and USY zeolite) can be used as supports with the CuO catalyst.
- the metals so incorporated in the metal oxide or mixed metal oxide support can be an alkali, an alkaline earth metal, a rare earth metal, or a mixture of one or more such metals. Incorporation of the specified metal or metals onto the metal oxide or mixed metal oxide support can be
- the calcining step at 250 °C to 600 °C prior to depositing the copper on the support is necessary.
- the time of calcining should be sufficient to decompose the metal salt(s) to the metal oxide(s).
- the total amount of added metal(s) in the support is in the range of 0.5% to 20% by weight based upon the weight of the support.
- copper preferably as copper nitrate
- the metal-modified metal oxide or mixed metal oxide support is impregnated on the metal-modified metal oxide or mixed metal oxide support in any manner known to those skilled in the art.
- the amount of copper deposited will depend on the desired activity of the catalyst, and can be as little as 2% by weight to as much as 20% by weight.
- the final catalyst composition containing the copper catalyst on the modified support can be in the form of powder, granules, extrudates or tablets, but certain specific characteristics such as surface area and pore volume, for example, are modified by reason of the deposit of copper.
- the catalyst comprising active metal(s) either in the co-precipitated form with other elements, or active metal(s) dispersed on a first oxide, mixed metal oxides or metal-modified metal oxide support, as described herein above can be either physically mixed and sieved to appropriate size, or intimately mixed and optionally co-extruded or pelletized with a second metal oxide, mixed metal oxides or metal-modified metal oxide support.
- the pelletized or co-extruded catalyst can be optionally crushed and sieved to appropriate size for use in slurry batch, continuous stirred tank, or fixed bed reactors.
- the 1 ,2,6-hexanetriol, catalyst, and hydrogen are contacted at a reaction temperature within the range from about 120 °C and 300 °C and at a pressure within the range from about 200 psi to about 3000 psi for a time sufficient to form a product mixture comprising 1 ,2-cyclohexanediol as a mixture of cis and trans isomers.
- the product mixture can further comprise 1 ,6-hexanediol.
- 1 ,2,6-hexanetriol, catalyst, and hydrogen are contacted at a temperature between and optionally including any two of the following values: 120 °C, 130 °C, 140 °C, 150 °C, 160 °C, 170 °C, 180 °C, 190 °C, 200 °C, 210 °C, 220 °C, 230 °C, 240 °C, 250 °C, 260 °C, 270 °C, 280 °C, 290 °C, and 300 °C.
- the temperature is within the range from about 200 °C to about 290 °C, for example between and optionally including any two of the following values: 200 °C, 210 °C, 220 °C, 230 °C, 240 °C, 250 °C, 260 °C, 270 °C, 280 °C, and 290 °C.
- the period of time for contacting is within the range of about 1 minute to about 10 hours.
- the 1 ,2,6-hexanetriol, catalyst, and hydrogen are contacted at a pressure between 200 and 3000 psi.
- the contacting is at a pressure between and optionally including any two of the following values: 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1 100, 1200, 1300, 1400, 1500, 1600, 1700, 1800, 1900, 2000, 2100, 2200, 2300, 2400, 2500, 2600, 2700, 2800, 2900, and 3000 psi.
- the contacting is within the range from about 800 to about 1500 psi, for example between and optionally including any two of the following values: 800, 900, 1000, 1 100, 1200, 1300, 1400, and 1500 psi.
- the reaction can be run in a batch or continuous mode, in liquid phase, gas phase, or biphasic conditions.
- the process can be carried out is standard reactors as are known in the art.
- the reaction can be carried out in a trickle bed reactor, wherein the liquid hourly space velocity is between 0.05 and 10 h "1 (ml_ liquid feed/mL
- the reaction can be carried out in a trickle bed reactor, wherein the ratio of the gas volumetric flowrate to the liquid volumetric flowrate as measured at ambient conditions (gas to oil ratio) is between 100 and 5,000, for example from 1 ,000 to about 4,000.
- the amount of catalyst used will depend on the specific equipment configuration and reaction conditions.
- the ratio of catalyst weight to 1 ,2,6-hexanetriol weight ranges from about 0.05 to 2. In some embodiments, this ratio is between and optionally includes any two of the following values: 0.05, 0.1 , 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1 .0, 1 .1 , 1 .2, 1 .3, 1 .4, 1 .5, 1 .6, 1 .7, 1 .8, 1 .9, and 2.0.
- the 1 ,2,6-hexanetriol feed in some embodiments is from about 2 wt% to about 50 wt% in water or another suitable solvent. It is anticipated that the reaction could be run at higher concentrations of 1 ,2,6-hexanetriol in solvent or even with neat 1 ,2,6-hexanetriol.
- Suitable solvents include water, a C1 -C20 alcohol, a C 2 -C 2 o ether, a C 2 -C 2 o ester, or mixtures thereof. Examples of suitable alcohols which are commercially available include methanol, ethanol, propanol, butanol, and hexanol.
- Suitable ethers which are commercially available include dibutylether, dihexylether, methyl-f-butyl-ether, tetrahydrofuran, and dioxane.
- suitable esters which are commercially available include ethyl acetate, butyl acetate, methyl butyrate, ethyl butyrate, butyl butyrate and hexyl acetate.
- the catalyst can be separated from the product mixture by methods known in the art, for example by filtration. After separation from the catalyst, the product mixture
- the product mixture comprises 1 ,2-cyclohexanediol. In some embodiments, the product mixture comprises 1 ,6-hexanediol. In some embodiments, the product mixture comprises
- 1 ,2-cyclohexanediol to 1 ,6-hexanediol is in the range of from about 0.1 to about 20.
- the molar ratio of trans-1 ,2-cyclohexanediol to cis-1 ,2-cyclohexanediol is from 1 to 2.5.
- the product mixture further comprises one or more of 2-hydroxymethyltetrahydropyran, 1 ,5-hexanediol, and 1 ,5-pentanediol, which can be useful as chemical intermediates.
- the product mixture further comprises tetrahydropyran-2-methanol.
- the product mixture further comprises 1 ,5-hexanediol.
- the product mixture further comprises 1 ,5-pentanediol.
- the commercial catalysts obtained as shaped materials were crushed and sieved to 0.125-0.160 mm prior to loading into the continuous reactor.
- the commercial catalysts obtained in powder form were press-pelleted, crushed, and sieved to 0.125-0.160 mm prior to loading in the continuous reactor.
- Catalyst samples referred to as "Catalyst A intimately mixed with Catalyst B" were prepared using the following procedure. If either catalyst A or catalyst B was originally a shaped material (tablets, extrudates, spheres, etc.), it was first crushed to powder form ( ⁇ 125 ⁇ ). Four mL of each catalyst were combined and mixed together in a 25 mL glass vial by shaking for a minimum of 30 seconds. The mixture was then screened using a 250 ⁇ sieve. The sieved material was press-pelleted, crushed, and sieved to
- Catalyst B were prepared using the following procedure. If either catalyst A or catalyst B was originally a shaped material (tablets, extrudates, spheres, etc.), it was first crushed and sieved to 0.125-0.160 mm. If either catalyst A or catalyst B was originally in powder form, it was first press-pelleted, crushed, and sieved to 0.125-0.160 mm. Four ml_ of each catalyst were combined and mixed together in a 25 ml_ glass vial by shaking for minimum of 30 seconds.
- Catalyst samples referred to as "supported copper catalysts” were prepared using the following procedure.
- Supports used in this catalyst preparation method include: Sasol Alumina 3%La, Sasol Alumina 10%La, MEL Ce/ZrO 2 MgO, and HY CBV780. If the support was originally a shaped material (tablets, extrudates, spheres, etc.), it was crushed and sieved to 0.125-0.160 mm. If the support was originally in powder form it was press- pelleted, crushed, and sieved to 0.125-0.160 mm. The support was optionally impregnated with La or Ba at ambient conditions, in a porcelain dish mixed in a lab-shaker with the appropriate concentration of La(NO 3 ) 3 x XH 2 O or
- Ba(NOs)2 solution using incipient wetness technique.
- the mixture was dried at 80 °C in a vented oven.
- the dried catalyst was calcined in a muffle furnace at 300 °C for 4h, ramp rate 1 °C /min, in air.
- the support, or the La/Ba impregnated support was subsequently impregnated with Cu at ambient conditions, in a porcelain dish mixed in a lab- shaker with the appropriate concentration of Cu(NOs)2 x 2.5H 2 O solution using incipient wetness technique.
- the mixture was dried at 80 °C in a vented oven.
- the dried catalyst was calcined in a muffle furnace at 300 °C for 4 h at a ramp rate of 1 °C /min in air.
- the calcined Cu impregnated catalyst was sieved to 0.125-0.160 mm.
- the catalyst was reduced using 5% H 2 in N 2 at
- the reactor was packed with approximately 1 ml_ of catalyst. If the catalyst was not pre-reduced, the following procedure was used for in situ reduction: the reactor was heated at a rate of 1 °C /min under forming gas (5% H 2 in N 2 ) to the desired reduction temperature (see examples), where it was held for the desired hold-up time, typically 2-3 hours.
- the pre-reduced or in-situ reduced catalyst was used for running multiple reactions under varying reaction conditions (temperature, pressure, feed concentrations).
- the reactor temperature was adjusted to the target first reaction condition temperature and held overnight under forming gas and either water or aqueous substrate solution. Subsequently the first reaction condition started by changing the gas feed to 100% H 2 and the liquid feed to the desired aqueous substrate concentration.
- the liquid volumetric feed rate was adjusted to correspond to a target liquid hourly space velocity (LHSV), which was measured in units of ml_ liquid feed/mL catalyst/h.
- LHSV liquid hourly space velocity
- GTO gas to oil ratio
- reaction product mixtures were identified by matching their retention times and mass spectra to those of authentic samples.
- the reactor was connected to a high pressure gas manifold and the content was purged with nitrogen gas (1800 psi) 3 times before hydrogen was added.
- the approximate target amount of hydrogen was added and the reactor was heated to 250 °C and final adjustments to the pressure were made by adding more nitrogen (for 1000 psi target pressure) or hydrogen (for 1800 psi target pressure) to reach the target pressure.
- the reactor was allowed to cool to room temperature within 2 h and the reaction solutions were filtered through a standard 5 ⁇ disposable filter, diluted with n-propanol and analyzed by GC and GC/MS. Products were identified by matching retention times and mass spectra using known samples.
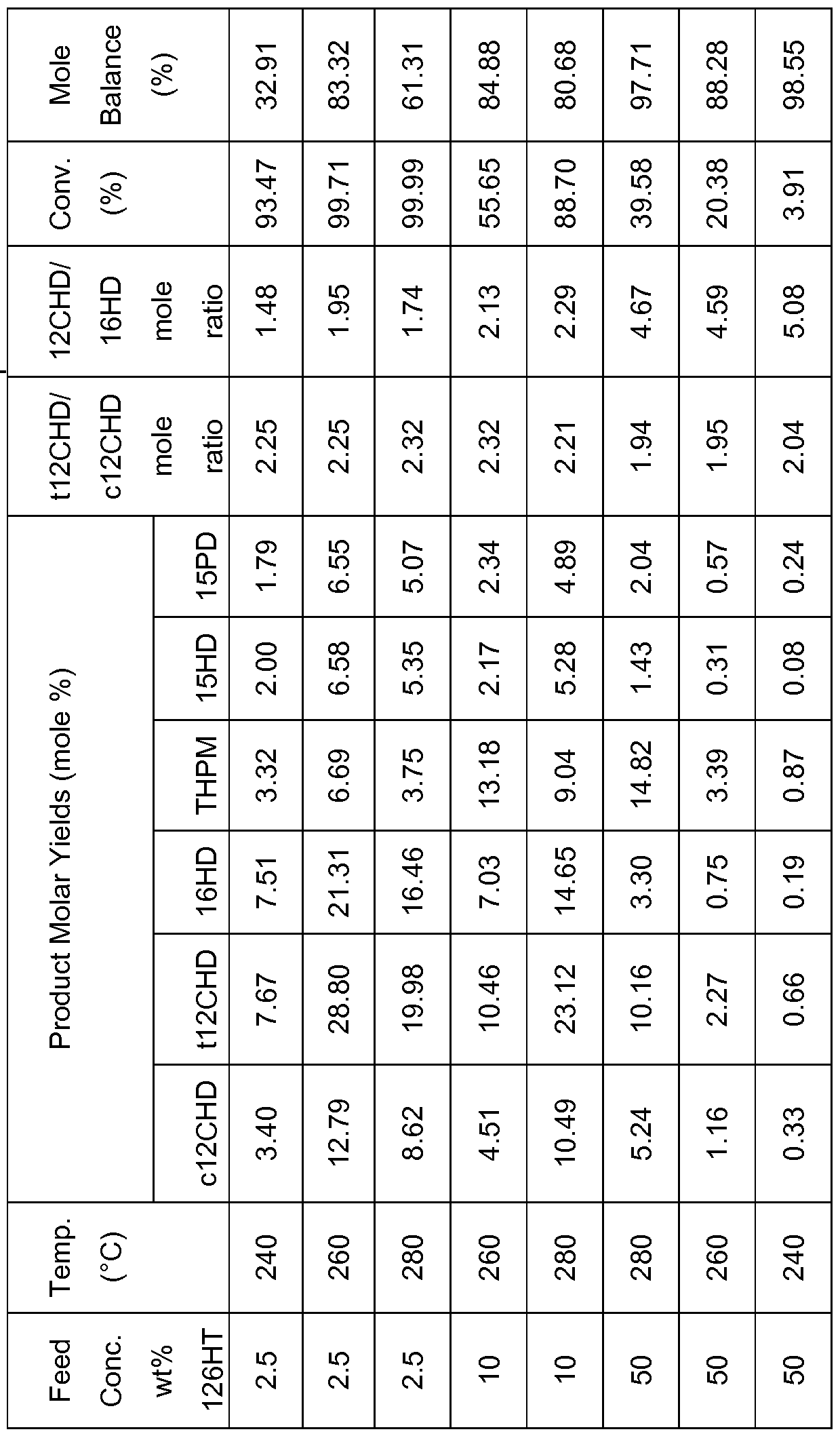
- the continuous reactor was charged with CuO/ZnO (SuedChemie T- 2130) catalyst.
- the catalyst was reduced in situ at 250 °C for 3 h.
- Aqueous solutions of 1 ,2,6-hexanetriol (2.5 wt%, 10 wt% and 50 wt%) were used as the liquid feed.
- the liquid volumetric feed rate corresponded to a liquid hourly space velocity (LHSV) equal to 0.5 ml_ liquid feed/mL catalyst/h.
- Product yields are given in Table 3 for 240-280 °C under 100 bar H 2 pressure.
- the continuous reactor was charged with CuO/ZnO/AI 2 O3
- LHSV liquid hourly space velocity
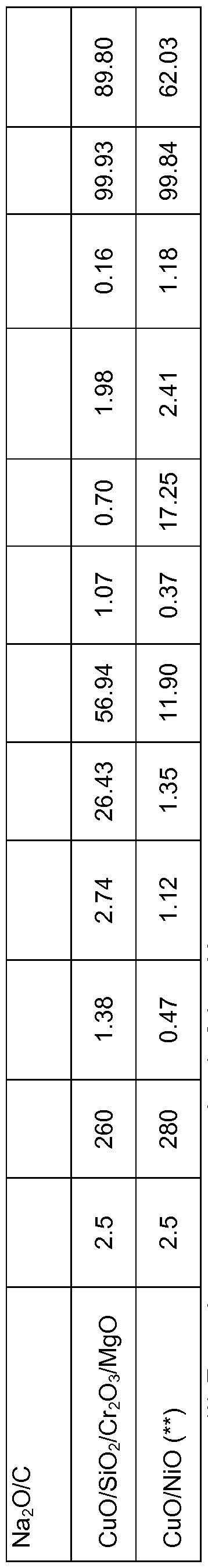
- CuO/AI 2 O 3 (EVONIK CPCAT 9/1597), CuO/ZnO/CeO 2 /AI 2 O 3 /Na 2 O/C (Johnson Matthey PRICAT CZ 30/18 T 6 * 5 mm), CuO/SiO 2 /Cr 2 O 3 /MgO (Johnson Matthey PRICAT CU 60/35 P) and CuO/NiO (Shepherd Chemical LB 3307).
- the catalysts were reduced in situ at 250 °C for 3 h.
- Aqueous solutions of 1 ,2,6-hexanetriol (2.5 wt%, 10 wt% and 50 wt%) were used as the liquid feed.
- the liquid volumetric feed rate corresponded to a liquid hourly space velocity (LHSV) equal to 0.5 mL liquid feed/mL catalyst/h.
- Product yields are given in Table 5 for 240-280 °C under 100 bar H 2 pressure.
- the catalysts were reduced in situ at 300 °C for 2 h.
- a 2.5 wt% aqueous solution of 1 ,2,6-hexanetriol was used as the liquid feed for all the runs.
- the liquid feed volumetric feed rate corresponded to a liquid hourly space velocity (LHSV) equal to 0.5 ml_ liquid feed/mL catalyst/h.
- Product yields at different temperatures are given in Table 6 for 240-280 °C under 100 bar H 2 pressure.
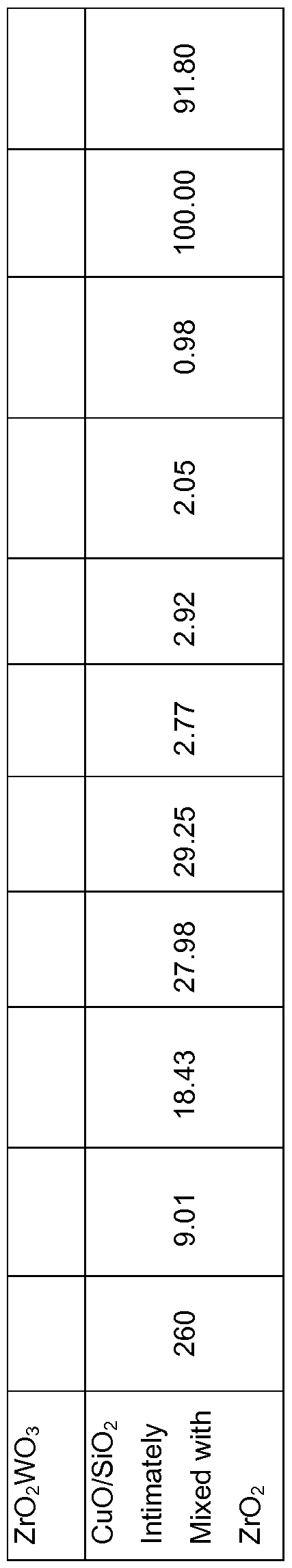
- the following supported copper catalysts were prepared using the Catalyst Preparation Method III: ZrO 2 15%La 7%Cu, Sasol Alumina 10%La 3%Cu, Sasol Alumina 10%La 7%Cu, Sasol Alumina 10%La 15%Cu, MEL Ce/ZrO 2 15%Cu, MgO 3%Cu, MgO 7%Cu, MgO 15%Cu, HY CBV780 6%La 7%Cu and HY CBV780 6%Ba 7%Cu.
- a 2.5 wt% aqueous solution of 1 ,2,6-hexanetriol was used as the liquid feed for all the runs.
- the liquid feed volumetric feed rate corresponded to a liquid hourly space velocity (LHSV) equal to 0.5 mL liquid feed/mL catalyst/h.
- Product yields are given in Table 7 for 260-280 °C under 100 bar H 2 pressure.
Abstract
Description











Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12863411.0A EP2797868A4 (en) | 2011-12-30 | 2012-12-28 | Process for the production of hexanediols |
| JP2014550475A JP2015503569A (en) | 2011-12-30 | 2012-12-28 | Method for producing hexanediol |
| BR112014015999A BR112014015999A8 (en) | 2011-12-30 | 2012-12-28 | process |
| CN201280064374.9A CN104024197A (en) | 2011-12-30 | 2012-12-28 | Process for the production of hexanediols |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161582069P | 2011-12-30 | 2011-12-30 | |
| US61/582,069 | 2011-12-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013101968A1 true WO2013101968A1 (en) | 2013-07-04 |
Family
ID=48695355
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/071891 WO2013101968A1 (en) | 2011-12-30 | 2012-12-28 | Process for the production of hexanediols |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8981130B2 (en) |
| EP (1) | EP2797868A4 (en) |
| JP (1) | JP2015503569A (en) |
| CN (1) | CN104024197A (en) |
| BR (1) | BR112014015999A8 (en) |
| WO (1) | WO2013101968A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8853458B2 (en) | 2012-01-18 | 2014-10-07 | Rennovia, Inc. | Process for production of hexamethylenediamine from carbohydrate-containing materials and intermediates therefor |
| US9586920B2 (en) | 2014-12-02 | 2017-03-07 | Rennovia Inc. | Process for production of hexanetriol from 5-hydroxymethylfurfural |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2797867A4 (en) | 2011-12-30 | 2015-08-05 | Du Pont | Process for preparing 1, 6-hexanediol |
| US8884035B2 (en) | 2011-12-30 | 2014-11-11 | E I Du Pont De Nemours And Company | Production of tetrahydrofuran-2, 5-dimethanol from isosorbide |
| WO2013101970A1 (en) | 2011-12-30 | 2013-07-04 | E. I. Du Pont De Nemours And Company | Production of 5-hydroxymethyl-2-furfural from levoglucosenone |
| US8889912B2 (en) | 2011-12-30 | 2014-11-18 | E I Du Pont De Nemours And Company | Process for preparing 1,6-hexanediol |
| US9018423B2 (en) | 2012-04-27 | 2015-04-28 | E I Du Pont De Nemours And Company | Production of alpha, omega-diols |
| CN109748777B (en) * | 2018-12-26 | 2020-09-11 | 大连理工大学 | Method for preparing 1, 6-hexanediol by catalytic hydrogenolysis of 1,2, 6-hexanetriol |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0441449A (en) * | 1990-06-07 | 1992-02-12 | Daicel Chem Ind Ltd | Production of cyclohexane-1,2-diol |
| JPH0446133A (en) * | 1990-06-12 | 1992-02-17 | Daicel Chem Ind Ltd | Production of cyclohexane-1,2-diol |
| JP2006036653A (en) * | 2004-07-23 | 2006-02-09 | New Japan Chem Co Ltd | Method for producing 1,3-cyclohexanediol |
| KR100645668B1 (en) * | 2002-05-31 | 2006-11-13 | 에스케이 주식회사 | Method for preparing cis-1,3-cyclohexanediol with high yield |
| KR100688765B1 (en) * | 2003-02-12 | 2007-02-28 | 에스케이 주식회사 | Method for preparing cis-cyclohexanediol with a high yield and a high purity from benzendiol |
Family Cites Families (86)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2082025A (en) | 1933-04-13 | 1937-06-01 | Quaker Oats Co | Method for the reduction of furfural and furan derivatives |
| US2201347A (en) | 1936-11-14 | 1940-05-21 | Patchem A G Zur Beteiligung An | Method for the manufacture of hydrogenation products of furfurol |
| US2440929A (en) | 1945-05-25 | 1948-05-04 | Ici Ltd | Production of 1:5-pentanediol |
| US2768213A (en) | 1953-04-13 | 1956-10-23 | Shell Dev | 1, 2, 6-hexanetriol |
| US3070633A (en) | 1958-09-10 | 1962-12-25 | Merck & Co Inc | Process for producing 1, 6-hexanediol |
| US3083236A (en) | 1958-09-10 | 1963-03-26 | Merck & Co Inc | Hydrogenation of 5-hydroxy-methyl furfural |
| US3215742A (en) | 1960-02-02 | 1965-11-02 | Celanese Corp | Process for the preparation of alkylene diamines |
| US3189651A (en) | 1962-01-16 | 1965-06-15 | Merck & Co Inc | Process for preparing hexamethylenediamine compounds from 5-aminomethyl-2-furfurylamine compounds |
| DE1172268B (en) | 1962-02-21 | 1964-06-18 | Basf Ag | Process for the production of diamines |
| US3268588A (en) | 1962-11-16 | 1966-08-23 | Celanese Corp | Process for producing hexamethylenediamine from 1-6-hexanediol |
| US3223714A (en) | 1963-05-22 | 1965-12-14 | Quaker Oats Co | Process of producing furan |
| US3917707A (en) | 1974-07-31 | 1975-11-04 | Du Pont | Suppression of 1,2-diaminocyclohexane formation during production of hexamethylenediamine |
| US3933930A (en) | 1975-03-06 | 1976-01-20 | Celanese Corporation | Hexanediol from cyclohexane |
| US4254059A (en) | 1979-08-01 | 1981-03-03 | Allied Chemical Corporation | Process for hydrogenation of nitriles |
| US4401823A (en) * | 1981-05-18 | 1983-08-30 | Uop Inc. | Hydrogenolysis of polyhydroxylated compounds |
| US4400468A (en) | 1981-10-05 | 1983-08-23 | Hydrocarbon Research Inc. | Process for producing adipic acid from biomass |
| EP0110089B1 (en) | 1982-11-03 | 1988-01-07 | Allied Corporation | Polymer-bound alkyl diarylphosphinite catalyst compositions and processes for making same and using same for selective conversion of acrylonitrile into 1,4-dicyano-1-butene |
| DE3632255A1 (en) | 1986-09-23 | 1988-03-31 | Basf Ag | METHOD FOR PRODUCING FURAN BY DECARBONYLATING FURFURAL |
| DE3818198A1 (en) | 1988-05-28 | 1989-12-21 | Basf Ag | METHOD FOR PRODUCING LESS MORE QUALITY ALCOHOLS |
| DE3843956A1 (en) * | 1988-12-24 | 1990-06-28 | Huels Chemische Werke Ag | METHOD FOR PRODUCING ALIPHATIC AND CYCLOALIPHATIC DIOLS BY CATALYTIC HYDRATION OF DICARBONIC ACID ESTERS |
| JPH0352880A (en) | 1989-07-19 | 1991-03-07 | Japan Tobacco Inc | Production of (s)-gamma-hydroxymethyl alpha,beta-butenolide |
| JPH03109384A (en) | 1989-09-22 | 1991-05-09 | Japan Tobacco Inc | Production of (s)-4-hydroxymethyl-gamma-lactone |
| US5538891A (en) | 1991-09-02 | 1996-07-23 | Boehringer Mannheim Gmbh | Process for enzymatic production of isomerically pure isosorbide-2 and 5-monoesters and their conversion to isosorbide-2 and -5 nitrate |
| JPH0641107A (en) | 1992-07-23 | 1994-02-15 | Japan Tobacco Inc | Production of @(3754/24)3r,4r)-3-hydroxy-4-hydroxymethyl-4-butanolide |
| DE4238493C1 (en) | 1992-11-14 | 1994-04-21 | Degussa | Process for the production of acrolein and its use |
| DE19500783A1 (en) | 1995-01-13 | 1996-07-18 | Bayer Ag | Process for the preparation of aliphatic alpha, omega diols |
| DE19548289A1 (en) | 1995-12-22 | 1997-06-26 | Basf Ag | Process for the simultaneous production of caprolactam and hexamethylenediamine |
| US6008418A (en) | 1996-03-01 | 1999-12-28 | Basf Aktiengesellschaft | Process for preparing 1,6 hexanediol with a level of purity over 99% |
| MY118128A (en) | 1996-03-01 | 2004-09-30 | Basf Ag | The preparation of 1, 6-hexanediol and caprolactone |
| DE19757554A1 (en) | 1997-12-23 | 1999-06-24 | Basf Ag | Production of 1,6-hexane-diol from adipate and/or 6-hydroxy-caproate ester |
| DE19809686A1 (en) | 1998-03-06 | 1999-09-09 | Basf Ag | Process for the hydrogenation of aliphatic alpha, omega-dinitriles |
| DE19818340A1 (en) | 1998-04-23 | 1999-10-28 | Basf Ag | Production of butan-1,4-diol, tetrahydrofuran and gamma-butyrolactone |
| DE19839338A1 (en) | 1998-08-28 | 2000-03-02 | Basf Ag | Improved process for the simultaneous production of 6-aminocapronitrile and hexamethylenediamine |
| US6087296A (en) | 1998-11-05 | 2000-07-11 | E. I. Du Pont De Nemours & Co. | Raney iron catalyst and a process for hydrogenating organic compounds using said catalyst |
| DE19935828A1 (en) | 1999-07-29 | 2001-02-01 | Basf Ag | Production of pentanediols from alkoxydihydropyrans |
| EP1243573B1 (en) | 1999-11-05 | 2014-01-01 | Asahi Kasei Kabushiki Kaisha | Process for the preparation of diol mixtures |
| JP2001226303A (en) * | 2000-02-16 | 2001-08-21 | Asahi Kasei Corp | Oxidizing method for olefin |
| JP4296739B2 (en) | 2001-03-22 | 2009-07-15 | 昭和電工株式会社 | Catalyst for producing both-end diols, method for producing the catalyst, method for producing both-end diols using the catalyst, and both-end diols obtained by the production method |
| EP1243673A1 (en) | 2001-03-24 | 2002-09-25 | Enthone Inc. | Servicing of an electrolyte |
| US6593481B1 (en) | 2001-11-13 | 2003-07-15 | E. I. Du Pont De Nemours And Company | Hydrogenation of 3,4-tetrahydrofurandiol to tetrahydrofuran |
| US7019155B2 (en) | 2001-11-13 | 2006-03-28 | Invista North America S.A.R.L. | Hydrogenation of tetrahydroxybutane to tetrahydrofuran |
| JP2003183200A (en) | 2001-12-14 | 2003-07-03 | Kyowa Yuka Co Ltd | Method for manufacturing 1,6-hexanediol |
| EP1350788A3 (en) | 2002-03-28 | 2003-11-12 | Degussa AG | Process for preparing hexamethylenediamine from butadiene |
| US6818781B2 (en) | 2002-04-17 | 2004-11-16 | E. I. Du Pont De Nemours And Company | Simultaneous reaction and separation process for the manufacture of dianhydro sugar alcohols |
| DE10258316A1 (en) | 2002-12-13 | 2004-06-24 | Basf Ag | High purity 1,6-hexanediol production involves dimerizing an acrylic acid ester then hydrogenating the product with a chromium-free copper catalyst |
| CN105418624A (en) | 2006-03-09 | 2016-03-23 | 阿彻-丹尼尔斯-米德兰公司 | Process for the production of anhydrosugar alcohols |
| US7994347B2 (en) | 2006-06-09 | 2011-08-09 | Battelle Memorial Institute | Hydroxymethylfurfural reduction methods and methods of producing furandimethanol |
| US9045382B2 (en) | 2006-07-25 | 2015-06-02 | Basf Aktiengesellschaft | Process for working up solvent-containing hydrogenation product mixtures |
| DE102007007629A1 (en) | 2007-02-16 | 2008-08-21 | Evonik Degussa Gmbh | Process for the preparation of 5-hydroxymethyl furfural via 5-acyloxymethyl furfural as an intermediate |
| RU2472840C2 (en) | 2007-03-08 | 2013-01-20 | Вайрент, Инк. | Synthesis of liquid fuel and chemical agents from oxygen-containing hydrocarbons |
| CN101279892B (en) * | 2007-04-03 | 2011-04-20 | 微宏动力系统(湖州)有限公司 | Method for preparing 1,2-cyclohexanediol by catalytic oxidation of cyclohexene |
| WO2008129933A1 (en) * | 2007-04-17 | 2008-10-30 | Kao Corporation | Process for producing hydrogenolysis products of polyhydric alcohols |
| GB0713598D0 (en) | 2007-07-13 | 2007-08-22 | Ici Ltd | Cyclic ethers |
| CN101835730B (en) | 2007-10-31 | 2013-06-26 | 三井化学株式会社 | Method for producing propylene glycol |
| JP2011506478A (en) | 2007-12-12 | 2011-03-03 | アーチャー ダニエルズ ミッドランド カンパニー | Conversion of carbohydrates to hydroxymethylfurfural (HMF) and derivatives |
| US9120806B2 (en) | 2008-04-10 | 2015-09-01 | Iowa Corn Promotion Board | Dianhydrosugar production process |
| EP2281795A4 (en) * | 2008-04-30 | 2011-11-16 | Mitsui Chemicals Inc | Method of producing propylene glycol |
| WO2010033789A2 (en) | 2008-09-18 | 2010-03-25 | University Of Massachusetts | Production of hydrogen, liquid fuels, and chemicals from catalytic processing of bio-oils |
| WO2010062689A2 (en) | 2008-10-30 | 2010-06-03 | Archer Daniels Midland Company | Reduction of hmf ethers with metal catalyst |
| EP2401307A4 (en) | 2009-02-24 | 2015-08-05 | Gevo Inc | Methods of preparing renewable butadiene and renewable isoprene |
| WO2010115798A2 (en) | 2009-04-07 | 2010-10-14 | Basf Se | Method for producing 1,6-hexanediol and caprolactone |
| JP5586686B2 (en) | 2009-04-07 | 2014-09-10 | ビーエーエスエフ ソシエタス・ヨーロピア | Method for producing 1,6-hexanediol |
| JP5693561B2 (en) | 2009-04-08 | 2015-04-01 | ビーエーエスエフ ソシエタス・ヨーロピアBasf Se | Process for producing 1,6-hexanediol by hydrogenation of oligo- and polyesters |
| EP2440515B1 (en) | 2009-06-13 | 2018-08-15 | Archer-Daniels-Midland Company | Production of adipic acid and derivatives from carbohydrate-containing materials |
| WO2011014752A2 (en) | 2009-07-31 | 2011-02-03 | Dow Global Technologies Inc. | A process for the conversion of aliphatic cyclic amines to aliphatic diamines |
| CN101628875A (en) | 2009-08-06 | 2010-01-20 | 郸城财鑫糖业有限责任公司 | Method for preparing hexamethylene diamine by starchy material |
| US8263792B2 (en) | 2009-09-24 | 2012-09-11 | Board Of Regents, The University Of Texas System | Biomass refining by selective chemical reactions |
| US8669393B2 (en) | 2010-03-05 | 2014-03-11 | Rennovia, Inc. | Adipic acid compositions |
| CN102190639A (en) | 2010-03-12 | 2011-09-21 | 中国科学院大连化学物理研究所 | Method for preparing 5-hydroxymethyl furfural (5-HMF) by converting carbohydrate compound |
| EP2563880A2 (en) | 2010-04-27 | 2013-03-06 | Phillips 66 Company | Carbohydrates upgrading and hydrotreating to hydrocarbons |
| EP2390247A1 (en) | 2010-05-26 | 2011-11-30 | Netherlands Organisation for Scientific Research (Advanced Chemical Technologies for Sustainability) | Preparation of caprolactone, caprolactam, 2,5-tetrahydrofuran dimethanol, 1,6-hexanediol or 1,2,6-hexanetriol from 5-hydroxymethyl-2-furfuraldehyde |
| US20120059174A1 (en) | 2010-09-08 | 2012-03-08 | Basf Se | Process for preparing epsilon-caprolactone and 1,6-hexanediol |
| CA2820753C (en) | 2010-12-30 | 2020-12-01 | Virent, Inc. | Organo-catalytic biomass deconstruction |
| US8222463B2 (en) | 2011-07-28 | 2012-07-17 | Uop Llc | Process for generation of polyols from saccharide containing feedstock |
| CN103764279A (en) | 2011-08-23 | 2014-04-30 | 宇部兴产株式会社 | Hydrocracking catalyst, method for producing same, and method for producing hydroxy compound using said catalyst |
| US8524925B2 (en) | 2011-10-31 | 2013-09-03 | E I Du Pont De Nemours And Company | Production of furfural from biomass |
| WO2013066776A1 (en) | 2011-10-31 | 2013-05-10 | E. I. Du Pont De Nemours And Company | Processes for producing 5-(hydroxymethyl)furfural |
| US8865940B2 (en) | 2011-12-30 | 2014-10-21 | E I Du Pont De Nemours And Company | Process for preparing 1,6-hexanediol |
| WO2013101970A1 (en) | 2011-12-30 | 2013-07-04 | E. I. Du Pont De Nemours And Company | Production of 5-hydroxymethyl-2-furfural from levoglucosenone |
| US8884035B2 (en) | 2011-12-30 | 2014-11-11 | E I Du Pont De Nemours And Company | Production of tetrahydrofuran-2, 5-dimethanol from isosorbide |
| EP2797867A4 (en) | 2011-12-30 | 2015-08-05 | Du Pont | Process for preparing 1, 6-hexanediol |
| US8889912B2 (en) | 2011-12-30 | 2014-11-18 | E I Du Pont De Nemours And Company | Process for preparing 1,6-hexanediol |
| CN104066710B (en) | 2012-01-18 | 2016-06-29 | 莱诺维亚公司 | From the method that 5 hydroxymethyl furfural produces hexamethylene diamine |
| US9018423B2 (en) | 2012-04-27 | 2015-04-28 | E I Du Pont De Nemours And Company | Production of alpha, omega-diols |
| US8846985B2 (en) | 2012-04-27 | 2014-09-30 | E I Du Pont De Nemours And Company | Production of alpha, omega-diols |
| US8859826B2 (en) | 2012-04-27 | 2014-10-14 | E I Du Pont De Nemours And Company | Production of alpha, omega-diols |
-
2012
- 2012-12-28 CN CN201280064374.9A patent/CN104024197A/en active Pending
- 2012-12-28 JP JP2014550475A patent/JP2015503569A/en not_active Abandoned
- 2012-12-28 US US13/729,390 patent/US8981130B2/en not_active Expired - Fee Related
- 2012-12-28 BR BR112014015999A patent/BR112014015999A8/en not_active Application Discontinuation
- 2012-12-28 EP EP12863411.0A patent/EP2797868A4/en not_active Withdrawn
- 2012-12-28 WO PCT/US2012/071891 patent/WO2013101968A1/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0441449A (en) * | 1990-06-07 | 1992-02-12 | Daicel Chem Ind Ltd | Production of cyclohexane-1,2-diol |
| JPH0446133A (en) * | 1990-06-12 | 1992-02-17 | Daicel Chem Ind Ltd | Production of cyclohexane-1,2-diol |
| KR100645668B1 (en) * | 2002-05-31 | 2006-11-13 | 에스케이 주식회사 | Method for preparing cis-1,3-cyclohexanediol with high yield |
| KR100688765B1 (en) * | 2003-02-12 | 2007-02-28 | 에스케이 주식회사 | Method for preparing cis-cyclohexanediol with a high yield and a high purity from benzendiol |
| JP2006036653A (en) * | 2004-07-23 | 2006-02-09 | New Japan Chem Co Ltd | Method for producing 1,3-cyclohexanediol |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2797868A4 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8853458B2 (en) | 2012-01-18 | 2014-10-07 | Rennovia, Inc. | Process for production of hexamethylenediamine from carbohydrate-containing materials and intermediates therefor |
| US9035109B2 (en) | 2012-01-18 | 2015-05-19 | Rennovia, Inc. | Process for production of hexamethylenediamine from carbohydrate-containing materials and intermediates therefor |
| US9518005B2 (en) | 2012-01-18 | 2016-12-13 | Rennovia Inc. | Process for production of hexamethylenediamine from carbohydrate-containing materials and intermediates therefor |
| US9783473B2 (en) | 2012-01-18 | 2017-10-10 | Rennovia Inc. | Process for production of hexamethylenediamine from carbohydrate-containing materials and intermediates therefor |
| US9586920B2 (en) | 2014-12-02 | 2017-03-07 | Rennovia Inc. | Process for production of hexanetriol from 5-hydroxymethylfurfural |
| US10081612B2 (en) | 2014-12-02 | 2018-09-25 | Archer-Daniels-Midland Company | Process for production of hexanetriol from 5-hydroxymethylfurfural |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2015503569A (en) | 2015-02-02 |
| US20130172579A1 (en) | 2013-07-04 |
| EP2797868A1 (en) | 2014-11-05 |
| EP2797868A4 (en) | 2015-08-12 |
| CN104024197A (en) | 2014-09-03 |
| BR112014015999A8 (en) | 2017-07-04 |
| BR112014015999A2 (en) | 2017-06-13 |
| US8981130B2 (en) | 2015-03-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8981130B2 (en) | Process for the production of hexanediols | |
| US8884035B2 (en) | Production of tetrahydrofuran-2, 5-dimethanol from isosorbide | |
| US8962894B2 (en) | Process for preparing 1, 6-hexanediol | |
| US8846984B2 (en) | Production of α,ω-diols | |
| US9862664B2 (en) | Process for the production of alkenols and use thereof for the production of 1,3-butadiene | |
| US8865940B2 (en) | Process for preparing 1,6-hexanediol | |
| EP2841405A1 (en) | Production of alpha, omega-diols | |
| US8889912B2 (en) | Process for preparing 1,6-hexanediol | |
| KR20110137824A (en) | Method for producing 1,6-hexanediol by hydrogenation of oligo- and polyesters | |
| US20190002381A1 (en) | Hydrogenation of oxygenated molecules from biomass refining | |
| EP2610238A1 (en) | Oxidative catalytic process for the synthesis of lactic acid | |
| WO2018146978A1 (en) | Catalyst for reduction reaction of 3,4-dihydroxytetrahydrofuran, and method for producing 3,4-dihydroxytetrahydrofuran reduced product | |
| US20140121408A1 (en) | Oxidative catalytic process for the synthesis of lactic acid | |
| PL220573B1 (en) | Process for the preparation of a copper-nickel catalyst for hydrogenation of benzene |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12863411 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012863411 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2014550475 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014015999 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014015999 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140627 |