WO2012173219A1 - Novel biaryl ether derivative - Google Patents
Novel biaryl ether derivative Download PDFInfo
- Publication number
- WO2012173219A1 WO2012173219A1 PCT/JP2012/065315 JP2012065315W WO2012173219A1 WO 2012173219 A1 WO2012173219 A1 WO 2012173219A1 JP 2012065315 W JP2012065315 W JP 2012065315W WO 2012173219 A1 WO2012173219 A1 WO 2012173219A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- diabetic
- pharmaceutical composition
- compound
- obesity
- acceptable salt
- Prior art date
Links
- 0 *c(c(C(Nc(cc1)ccc1-c(cn1)cnc1OC1CCC(CC(O)=O)CC1)=O)c1)n[n]1-c1ccccn1 Chemical compound *c(c(C(Nc(cc1)ccc1-c(cn1)cnc1OC1CCC(CC(O)=O)CC1)=O)c1)n[n]1-c1ccccn1 0.000 description 2
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/12—Ophthalmic agents for cataracts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
Definitions
- the present invention relates to a compound having a specific chemical structure having an excellent acylcoenzyme A: diacylglycerol acyltransferase (hereinafter also referred to as DGAT) inhibitory activity and an excellent feeding inhibitory activity, Relates to acceptable salts.
- DGAT diacylglycerol acyltransferase
- triglyceride triacylglycerol or triglyceride, hereinafter also referred to as TG
- TG triglyceride
- TG ingested by the meal is broken down into free fatty acids and monoacylglycerol by the action of bile acids and pancreatic lipase in the lumen of the small intestine.
- Micelles composed of free fatty acid, monoacylglycerol and bile acid are absorbed into small intestinal epithelial cells, and in the endoplasmic reticulum by the action of acylcoenzyme A synthase (hereinafter referred to as ACS), acylcoenzyme A: monoacylglycerol acyltransferase and DGAT.
- ACS acylcoenzyme A synthase
- TG in combination with phospholipids, cholesterol and apolipoprotein, is secreted into the gastrointestinal lymphatic vessels as kilomicrons. Furthermore, TG is secreted into the blood via the lymph main duct and transported to the periphery for use.
- TG is synthesized from glycerol 3-phosphate and free fatty acids by the action of ACS, glycerol 3-phosphate acyltransferase, lysophosphatidic acid acyltransferase, and DGAT (Non-patent Document 2).
- ACS glycerol 3-phosphate acyltransferase
- DGAT Non-patent Document 2
- DGAT is an enzyme that is present in the endoplasmic reticulum in the cell and catalyzes the most important final step reaction in the TG synthesis pathway, that is, the reaction of transferring the acyl group of acylcoenzyme A to the 3-position of 1,2-diacylglycerol.
- Non-Patent Documents 3 to 5 It has been reported that DGAT has two types of isozymes DGAT1 (Non-patent document 6) and DGAT2 (Non-patent document 7).
- DGAT1 is highly expressed in the small intestine and adipose tissue
- DGAT2 is highly expressed in the liver and adipose tissue, respectively
- DGAT1 is mainly used for fat absorption from the small intestine and fat accumulation
- DGAT2 is used for TG synthesis or VLDL in the liver. (Very low density lipoproteins) secretion and fat accumulation in adipose tissue.
- DGAT1 and DGAT2 has not yet been clarified in detail, the relationship between DGAT and obesity, lipid metabolism, sugar metabolism, etc. has been suggested (Non-patent Document 8).
- DGAT is a key enzyme for TG synthesis in gastrointestinal epithelial cells and adipose tissue, and a drug that inhibits DGAT suppresses fat absorption in the gastrointestinal tract and fat accumulation in adipose tissue by suppressing TG synthesis, and obesity , Obesity, hyperlipidemia, hypertriglyceridemia, dyslipidemia, insulin resistance syndrome, diabetes, non-alcoholic steatohepatitis, or obesity-induced hyperlipidemia, hypertriglyceridemia, lipid metabolism It is expected to be useful as a therapeutic or prophylactic agent for abnormal diseases, insulin resistance syndrome, diabetes, nonalcoholic steatohepatitis, hypertension, arteriosclerosis, cerebrovascular disorder, coronary artery disease, etc. 9 to 13).
- An appetite suppressant directly or indirectly regulates the appetite control system, but its mechanism of action is roughly divided into central and peripheral.
- An appetite suppressant acting centrally acts on the hypothalamic nervous system where the feeding center and satiety center exist and the monoamine nervous system in the brain that regulates the nervous system, thereby directly suppressing appetite.
- an appetite suppressant that acts on the periphery acts on a mechanism that senses and transmits the intake of nutrients and the accumulation of surplus energy, and indirectly suppresses appetite.
- Non-patent Document 14 gastrointestinal hormones secreted in close association with the digestion and absorption of food (Non-Patent Document 14) and from fat cells according to the energy accumulation (fat mass)
- Non-patent Document 15 The mechanism by which secreted leptin (Non-patent Document 15) or the like transmits a signal that regulates appetite from the periphery to the center in a hormonal or neurological manner has been clarified.
- These new appetite suppressants associated with peripheral signals are expected to be more effective and less effective for the treatment of obesity.
- Patent Document 1 discloses a compound having 4- (5-carboxamido-2,3'-bipyridin-6'-yloxy) cyclohexanecarboxylic acid.
- this compound is obesity, obesity, hyperlipidemia, hypertriglycerideemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral Neuropathy, including diabetic nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic Hyperlipidemia, hypertriglyceridemia, lipid metabolism resulting from obesity or as an active ingredient of a medicament for the prevention and / or treatment of a disease selected from the group consisting of arteriosclerosis, ischemic heart disease and bulimia Abnormal diseases, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (including diabetic peripheral neuropathy, diabetic nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataracts, Gynecologic diabetes, nonalcoholic steatohepatitis, polycystic ova
- the present invention comprises (1) general formula (I)
- R 1 represents a hydrogen atom, a halogen atom, a C 1 -C 6 alkyl group or a C 3 -C 6 cycloalkyl group
- R 2 independently represents a halogen atom, a C 1 -C 6 alkyl group, a C 1 -C 6 alkoxy group or a hydroxy group
- n represents an integer of 0 to 2.
- a pharmacologically acceptable salt thereof thereof.
- R 1 is a hydrogen atom, a chlorine atom, a methyl group, an ethyl group or a cyclopropyl group, and n is 0, or a pharmacologically acceptable salt thereof. Salt.
- the general formula (I) is the general formula (Ia), R 1 is a hydrogen atom, a chlorine atom, a methyl group, an ethyl group or a cyclopropyl group, n is 1, and R 2 is a methyl group Or a pharmacologically acceptable salt thereof.
- An acyl coenzyme A diacylglycerol acyltransferase inhibitor comprising the compound described in any one of (1) to (9) or a pharmacologically acceptable salt thereof as an active ingredient.
- a pharmaceutical composition comprising as an active ingredient the compound described in any one of (1) to (9) or a pharmacologically acceptable salt thereof.
- the pharmaceutical composition inhibits acylcoenzyme A: diacylglycerol acyltransferase, inhibits synthesis of triglyceride, and suppresses the absorption of triglyceride, thereby treating, improving, reducing and / or preventing symptoms.
- the pharmaceutical composition inhibits acyl coenzyme A: diacylglycerol acyltransferase and inhibits the synthesis of triglyceride, thereby treating and / or treating diseases in which symptoms are treated, ameliorated, reduced and / or prevented.
- the pharmaceutical composition is obesity, obesity, hyperlipidemia, hypertriglyceridosis, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic Nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, false
- the pharmaceutical composition is obesity-induced hyperlipidemia, hypertriglyceridemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic Nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, hypertension (12)
- Obesity obesity, hyperlipidemia, hypertriglyceridemia, lipid metabolism disorder, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic nephropathy, diabetes Retinopathy, including diabetic macroangiopathy), cataract, gestational diabetes, non-alcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, ischemic heart disease or The compound according to any one of (1) to (9) or a pharmacologically acceptable salt thereof for use in the treatment and / or prevention of bulimia.
- the pharmaceutical composition is obesity, obesity, hyperlipidemia, hypertriglyceridemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic Nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, false
- the use according to (28) which is a pharmaceutical composition for the treatment and / or prevention of blood heart disease or bulimia.
- composition is a therapeutic composition for the treatment and / or prevention of hyperlipidemia, hypertriglyceridemia, diabetes, arteriosclerosis or hypertension caused by obesity. .
- (40) Diseases are obesity, obesity, hyperlipidemia, hypertriglycerideemia, lipid metabolism disorders, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic nephropathy , Including diabetic retinopathy, diabetic macrovascular disease), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, ischemic heart.
- the method according to (39) which is a disease or bulimia.
- the method according to (39) which is vascular disorder, coronary artery disease, fatty liver, respiratory disorder, low back pain, knee osteoarthritis, gout or cholelithiasis.
- the “halogen atom” is a fluorine atom, a chlorine atom, a bromine atom or an iodine atom. Preferred is a fluorine atom or a chlorine atom, and more preferred is a chlorine atom.
- the “C 1 -C 6 alkyl group” is a linear or branched alkyl group having 1 to 6 carbon atoms.
- Preferred is a linear or branched alkyl group having 1 to 4 carbon atoms (C 1 -C 4 alkyl group), and more preferred is a methyl group or an ethyl group (C 1 -C 2 alkyl group). And even more preferably a methyl group.
- the “C 1 -C 6 alkoxy group” is a group in which the “C 1 -C 6 alkyl group” is bonded to an oxygen atom, and is a linear or branched alkoxy group having 1 to 6 carbon atoms. It is. For example, a methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, s-butoxy, t-butoxy, pentoxy or hexyloxy group.
- Preferred is a linear or branched alkoxy group having 1 to 4 carbon atoms (C 1 -C 4 alkoxy group), and more preferred is a methoxy group or an ethoxy group (C 1 -C 2 alkoxy group). And even more preferably a methoxy group.
- the “C 3 -C 6 cycloalkyl group” is a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, or a cyclohexyl group, and preferably a cyclopropyl group.
- preferred general formula (I) is general formula (Ia).
- preferred R 1 is a hydrogen atom, a chlorine atom, a methyl group, an ethyl group or a cyclopropyl group.
- preferred R 2 is a methyl group.
- n is preferably 0.
- the compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof has all isomers (diastereoisomers, optical isomers, rotational isomers, etc.).
- the compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof has various isomers because an asymmetric carbon atom exists in the molecule.
- these isomers and mixtures of these isomers are all represented by a single formula, that is, the general formula (I). Therefore, the present invention includes all of these isomers and a mixture of these isomers in an arbitrary ratio.
- an optically active raw material compound is used, or a compound according to the present invention is synthesized using an asymmetric synthesis or asymmetric induction method, or a synthesized compound according to the present invention is synthesized. If desired, it can be obtained by isolation using a conventional optical resolution method or separation method.
- the compound represented by the general formula (Ia) or a pharmacologically acceptable salt thereof is more preferable than the compound represented by the general formula (Ib) or a pharmacologically acceptable salt thereof.
- the compounds of the present invention may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds.
- the atomic isotope include deuterium ( 2 H), tritium ( 3 H), iodine-125 ( 125 I), carbon-14 ( 14 C), and the like.
- the compound can also be radiolabeled with a radioisotope such as, for example, tritium ( 3 H), iodine-125 ( 125 I), or carbon-14 ( 14 C).
- Radiolabeled compounds are useful as therapeutic or prophylactic agents, research reagents such as assay reagents, and diagnostic agents such as in vivo diagnostic imaging agents. All isotope variants of the compounds of the present invention, whether radioactive or not, are intended to be included within the scope of the present invention.
- the pharmacologically acceptable salt refers to a salt that has no significant toxicity and can be used as a medicine.
- the compound represented by the general formula (I) of the present invention can be converted into a salt by reacting with an acid when it has a basic group, or by reacting with a base when it has an acidic group. can do.
- Examples of the salt based on the basic group include hydrohalides such as hydrofluoride, hydrochloride, hydrobromide, and hydroiodide, nitrate, perchlorate, sulfate, Inorganic acid salts such as phosphates; alkyl sulfonates such as methanesulfonate, trifluoromethanesulfonate, and ethanesulfonate; arylsulfonates such as benzenesulfonate and p-toluenesulfonate Organic acids such as acetate, malate, fumarate, succinate, citrate, ascorbate, tartrate, oxalate, maleate; and glycine salt, lysine salt, Examples thereof include amino acid salts such as arginine salt, ornithine salt, glutamate salt and aspartate salt.
- hydrohalides such as hydrofluoride, hydrochloride, hydrobromide, and hydroiodide
- examples of the salt based on the acidic group include alkali metal salts such as sodium salt, potassium salt and lithium salt, alkaline earth metal salts such as calcium salt and magnesium salt, metal salts such as aluminum salt and iron salt.
- Inorganic salts such as ammonium salts, t-octylamine salts, dibenzylamine salts, morpholine salts, glucosamine salts, phenylglycine alkyl ester salts, ethylenediamine salts, N-methylglucamine salts, guanidine salts, diethylamine salts, triethylamine salts , Dicyclohexylamine salt, N, N′-dibenzylethylenediamine salt, chloroprocaine salt, procaine salt, diethanolamine salt, N-benzylphenethylamine salt, piperazine salt, tetramethylammonium salt, tris (hydroxymethyl) aminomethane salt Amine salts such as organic salt
- the compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof is taken in the air or recrystallized to take in water molecules, Such hydrates are also encompassed by the salts of the present invention.
- the compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof may absorb a certain other solvent and become a solvate, and such a solvate is also present. Included in the salts of the invention.
- the compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof has an excellent DGAT inhibitory action and feeding inhibitory action, and is a warm-blooded animal (preferably a mammal, Diseases (including humans): obesity, obesity, hyperlipidemia, hypertriglycerideemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, Diabetic nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis , A disease selected from the group consisting of ischemic heart disease and bulimia, or hyperlipidemia, hypertriglycerideemia, lipid metabolism disorder, insulin resistance syndrome, impaired glucose tolerance, sugar caused by obesity Urinary disease, diabetic complications (including diabetic peripheral neuropathy, diabetic nephropathy, diabetic
- novel compound represented by the general formula (I) provided by the present invention or a pharmacologically acceptable salt thereof has an excellent DGAT inhibitory action, and is a warm-blooded animal (preferably a mammalian animal). And is useful as an active ingredient of a medicament for the prevention and / or treatment of the above-mentioned diseases (including humans). Suitably, it can be used as a medicament for the treatment of the above-mentioned diseases.
- the compound represented by the general formula (I) of the present invention can be produced according to Method A and Method B described below.
- solvent used in the reaction of each step of the following method A and method B is not particularly limited as long as it does not inhibit the reaction and dissolves the starting materials to some extent, and is selected from the following solvent group, for example.
- Solvent groups include hydrocarbons such as pentane, hexane, octane, petroleum ether, ligroin, cyclohexane; formamide, N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidone, N-methyl Amides such as -2-pyrrolidinone and hexamethylphosphoric triamide; ethers such as diethyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, diethylene glycol dimethyl ether and cyclopentyl methyl ether; methanol, ethanol, n-propanol, i -Propanol, n-butanol
- the base used in the reaction of each step of the following method A and B is, for example, alkali metal carbonates such as sodium carbonate, potassium carbonate, lithium carbonate, cesium carbonate; sodium hydrogen carbonate, potassium hydrogen carbonate, lithium hydrogen carbonate Alkali metal bicarbonates such as sodium acetate, potassium acetate, lithium acetate, alkali metal acetates such as cesium acetate; alkali metal hydrides such as lithium hydride, sodium hydride, potassium hydride; Alkali metal hydroxides such as sodium, potassium hydroxide and lithium hydroxide; alkali metal fluorides such as sodium fluoride and potassium fluoride; sodium methoxide, sodium ethoxide, sodium t-butoxide, potassium Alkali metal alcohols such as t-butoxide Cids; alkali metal trialkylsiloxides such as sodium trimethylsiloxide, potassium trimethylsiloxide, lithium trimethylsiloxide; N-methylmorpholine, triethyl
- reaction temperature varies depending on the solvent, starting material, reagent, etc.
- reaction time varies depending on the solvent, starting material, reagent, reaction temperature, and the like.
- each target compound is collected from the reaction mixture according to a conventional method. For example, neutralize the reaction mixture as appropriate, or remove insoluble matter by filtration, add water and an immiscible organic solvent such as ethyl acetate, and separate the organic layer containing the target compound, It can be obtained by washing with water, drying over anhydrous magnesium sulfate, anhydrous sodium sulfate, etc., filtering, and then distilling off the solvent.
- an immiscible organic solvent such as ethyl acetate
- the obtained target compound is eluted with an appropriate eluent by applying a conventional method, for example, recrystallization, reprecipitation, etc., usually using methods commonly used for separation and purification of organic compounds, applying chromatography, and the like. Can be separated and purified.
- a target compound insoluble in a solvent the obtained solid crude product can be purified by washing with a solvent.
- the target compound in each step can be directly used in the next reaction without purification.
- R 1 , R 2 and n have the same meaning as described above.
- X 1 and X 2 each represent a halogen atom (preferably a bromine atom or an iodine atom, and more preferably a bromine atom), and Y 1 and Y 2 are used in the field of synthetic organic chemistry.
- a protecting group for carboxy group preferably a C 1 -C 6 alkyl group or an aralkyl group.
- Y 1 is more preferably a methyl group, and Y 2 is more preferably An ethyl group).
- R 2a is a hydroxy group contained in the group R 2 is other is protected may be hydroxy groups include the same groups as in the definition of group R 2.
- Method A is a method for producing a compound represented by the general formula (I). (Method A)
- Step AI comprises reacting a compound represented by the general formula (II) with a compound represented by the general formula (III) in a solvent in the presence of a Mitsunobu reagent. It is a process of manufacturing the compound represented by these.
- the compound represented by the general formula (II) and the compound represented by the general formula (III) used in this step are known compounds, or a known method using a known compound as a starting material or a method similar thereto. Easily manufactured according to.
- the solvent used in this step is preferably aromatic hydrocarbons or ethers, and more preferably toluene or tetrahydrofuran.
- the Mitsunobu reagent used in this step is preferably an azodicarboxylic acid diester or (cyanomethylene) phosphorane reagent, more preferably diethyl azodicarboxylate (DEAD), diisopropyl azodicarboxylate (DIAD) or ( Cyanomethylene) tributylphosphorane (CMBP).
- DEAD diethyl azodicarboxylate
- DIAD diisopropyl azodicarboxylate
- CMBP Cyanomethylene tributylphosphorane
- the reaction temperature in this step is usually ⁇ 20 ° C. to 180 ° C., preferably 0 ° C. to 120 ° C.
- the reaction time in this step is usually 0.5 hours to 72 hours, preferably 2 hours to 24 hours.
- Step A-II In this step, the compound represented by the general formula (IV) is reacted with the compound represented by the general formula (V) in the presence of a palladium catalyst and a base in a solvent, to thereby obtain a compound represented by the general formula (V).
- This is a process for producing a compound represented by VI).
- the compound represented by the general formula (V) used in this step is a known compound, or can be easily produced according to a known method or a similar method using the known compound as a starting material.
- the solvent used in this step is preferably a mixed solvent of ethers or amides and water, more preferably a mixed solvent of tetrahydrofuran, dioxane or N, N-dimethylacetamide and water.
- the palladium catalyst used in this step is, for example, tetrakis (triphenylphosphine) palladium (0), palladium-activated carbon, palladium acetate (II), palladium trifluoroacetate (II), palladium black, palladium bromide (II ), Palladium (II) chloride, palladium (II) iodide, palladium (II) cyanide, palladium (II) nitrate, palladium (II) oxide, palladium (II) sulfate, dichlorobis (acetonitrile) palladium (II), dichlorobis (Benzonitrile) palladium (II), dichloro (1,5-cyclooctadiene) palladium (II), acetylacetone palladium (II), palladium sulfide (II), [1,1'-bis (diphenylphosphino) ferrocen
- the base used in this step is preferably an alkali metal carbonate, and more preferably potassium carbonate.
- the reaction temperature in this step is usually 20 ° C. to 180 ° C., preferably 60 ° C. to 120 ° C.
- the reaction time in this step is usually 0.5 hours to 72 hours, preferably 2 hours to 24 hours.
- Step A-III the compound represented by the general formula (VI) is converted into the general formula (VII) in a solvent in the presence of a condensing agent, in the presence or absence of a base (preferably in the presence). ) To produce a compound represented by the general formula (VIII).
- the solvent used in this step is preferably an amide, and more preferably N, N-dimethylacetamide.
- the condensing agent used in this step is, for example, azodicarboxylic acid dilower alkyl ester-triphenylphosphine such as azodicarboxylic acid diethyl ester-triphenylphosphine; N, N′-dicyclohexylcarbodiimide (DCC), 1- Carbodiimide derivatives such as ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDCI); 2-halo-1-lower alkylpyridinium halides such as 2-chloro-1-methylpyridinium iodide; diphenylphosphoryl azide ( Diarylphosphoryl azides such as DPPA; phosphoryl chlorides such as diethyl phosphoryl chloride; imidazole derivatives such as N, N′-carbodiimidazole (CDI); benzotriazol-1-yloxy-to (Dimethylamino) phosphonium hexafluoro
- the base used in this step is preferably an organic base, and more preferably triethylamine.
- the reaction temperature in this step is usually -20 ° C to 160 ° C, preferably 0 ° C to 100 ° C.
- the reaction time in this step is usually 0.1 hour to 120 hours, preferably 1 hour to 24 hours.
- Step A-IV This step is based on a known method (for example, “Protective Groups in Organic Synthesis” (the method described in Theodora W. Greene, Peter GMWuts, 1999, published by Wiley-Interscience Publication), etc.). Done.
- Y 1 is a C 1 -C 6 alkyl group.
- This step is represented by the general formula (I) by reacting the compound represented by the general formula (VIII) with a base in a solvent and then removing the hydroxy protecting group in R 2a as required. This is a process for producing a compound.
- the solvent used in this step is preferably an ether or an alcohol, and more preferably tetrahydrofuran, dioxane or methanol.
- the base used in this step is preferably a quaternary ammonium salt, and more preferably tetrabutylammonium hydroxide.
- the reaction temperature in this step is usually 0 ° C. to 150 ° C., preferably 20 ° C. to 100 ° C.
- the reaction time in this reaction is usually 0.5 to 24 hours, preferably 1 to 10 hours.
- Method B is a method for producing a compound represented by the general formula (VII) used in Step A-III of Method A. (Method B)
- Step BI This step is performed by reacting a compound represented by the general formula (IX) with a compound represented by the general formula (X) in a solvent in the presence of a copper catalyst, a base and a ligand. In this step, the compound represented by formula (XI) is produced.
- the compound represented by the general formula (IX) and the compound represented by the general formula (X) used in this step are known compounds, or a known method using a known compound as a starting material or a method similar thereto. Easily manufactured according to.
- the solvent used in this step is preferably an aromatic hydrocarbon, and more preferably toluene.
- the copper catalyst used in this step is 0-valent copper or a complex thereof; 1 such as copper (I) chloride, copper (I) bromide, copper (I) iodide, copper (I) trifluoromethanesulfonate. Or a divalent copper salt such as copper (II) bromide, copper (II) acetate, or copper (II) sulfate, preferably a monovalent copper salt, more preferably Is copper iodide (I).
- 1 such as copper (I) chloride, copper (I) bromide, copper (I) iodide, copper (I) trifluoromethanesulfonate.
- a divalent copper salt such as copper (II) bromide, copper (II) acetate, or copper (II) sulfate, preferably a monovalent copper salt, more preferably Is copper iodide (I).
- the base used in this step is preferably an alkali metal carbonate, and more preferably potassium carbonate.
- the ligand used in this step is, for example, an amine compound, preferably a diamine, and more preferably 1,2-di (methylamino) cyclohexane.
- the reaction temperature in this step is usually 0 ° C to 200 ° C, preferably 80 ° C to 130 ° C.
- the reaction time in this step is usually 0.5 hours to 96 hours, preferably 2 hours to 48 hours.
- Step B-II This step is based on a known method (for example, the method described in “Protective Groups in Organic Synthesis” (Theodora W. Greene, Peter GMWuts, 1999, published by Wiley-Interscience Publication), etc.) Done.
- a known method for example, the method described in “Protective Groups in Organic Synthesis” (Theodora W. Greene, Peter GMWuts, 1999, published by Wiley-Interscience Publication), etc.) Done.
- Y 2 is a C 1 -C 6 alkyl group is shown below.
- This step is a step of producing the compound represented by the general formula (VII) by reacting the compound represented by the general formula (XI) with a base in a solvent.
- the solvent used in this step is preferably ethers, alcohols, water or a mixed solvent thereof, more preferably a mixed solvent of tetrahydrofuran, ethanol and water.
- the base used in this step is preferably an alkali metal hydroxide, and more preferably sodium hydroxide.
- the reaction temperature in this step is usually 0 ° C. to 150 ° C., preferably 20 ° C. to 100 ° C.
- the reaction time in this reaction is usually 0.5 to 24 hours, preferably 1 to 10 hours.
- the protecting group of the “optionally protected hydroxy group” in the definition of R 2a refers to a protecting group that can be cleaved by a chemical method such as hydrogenolysis, hydrolysis, electrolysis, or photolysis, Protective groups commonly used in organic synthetic chemistry are shown (see, for example, TW Greene et al., Protective Groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, Inc. (1999)).
- the “protecting group” of the “hydroxy group that may be protected” in the definition of R 2a is not particularly limited as long as it is a protecting group for a hydroxy group used in the field of synthetic organic chemistry.
- alkylcarbonyl group such as an unsaturated alkylcarbonyl group such as benzoyl, ⁇ -naphthoyl, ⁇ -naphthoyl, propioloyl, methacryloyl, crotonoyl, isocrotonoyl, (E) -2
- Alkoxylated arylcarbonyl groups such as 4-nitrobenzoyl, nitrated arylcarbonyl groups such as 2-nitrobenzoyl, alkoxycarbonylated arylcarbonyl groups such as 2- (methoxycarbonyl) benzoyl, aryls such as 4-phenylbenzoyl
- arylcarbonyl group such as an arylcarbonyl group; an alkoxycarbonyl group such as methoxycarbonyl, ethoxycarbonyl, isopropoxycarbonyl, t-butoxycarbonyl, 2,2,2-trichloroethoxycarbonyl
- An “alkoxycarbonyl group” such as an alkoxycarbonyl group substituted with a halogen or trialkylsilyl group such as 2-trimethylsilylethoxycarbonyl; tetrahydropyran-2-yl, 3-bromotetrahydropyran-2-yl, 4-methoxytetrahydro “Tetrahydro
- the compound of the present invention or a pharmacologically acceptable salt thereof can be administered in various forms.
- the administration form include oral administration by tablets, capsules, granules, emulsions, pills, powders, syrups (solutions), etc., or injections (intravenous, intramuscular, subcutaneous or intraperitoneal administration), Examples include parenteral administration such as instillation and suppository (rectal administration).
- These various preparations are usually used in the pharmaceutical preparation technical field such as excipients, binders, disintegrants, lubricants, flavoring agents, solubilizers, suspension agents, coating agents, etc. as main ingredients in accordance with conventional methods. It can be formulated with the resulting adjuvant.
- excipients such as lactose, sucrose, sodium chloride, glucose, urea, starch, calcium carbonate, kaolin, crystalline cellulose, silicic acid; water, ethanol, propanol, simple syrup, glucose Solution, starch solution, gelatin solution, carboxymethylcellulose, shellac, methylcellulose, potassium phosphate, polyvinylpyrrolidone, etc .; dried starch, sodium alginate, agar powder, laminaran powder, sodium bicarbonate, calcium carbonate, polyoxyethylene sorbitan fatty acid Disintegrators such as esters, sodium lauryl sulfate, monoglyceride stearate, starch, lactose; disintegrators such as sucrose, stearin, cocoa butter, hydrogenated oil; quaternary ammonium salts, sodium lauryl sulfate Moisturizers such as glycerin and starch; Adsorbents such as starch
- the tablet which gave the normal coating for example, a sugar-coated tablet, a gelatin-encapsulated tablet, an enteric-coated tablet, a film-coated tablet, a double tablet, and a multilayer tablet.
- excipients such as glucose, lactose, cocoa butter, starch, hydrogenated vegetable oil, kaolin, talc; binders such as gum arabic powder, tragacanth powder, gelatin, ethanol; laminaran, Disintegrants such as agar can be used.
- a carrier conventionally known in this field can be widely used as a carrier, and examples thereof include polyethylene glycol, cocoa butter, higher alcohol, esters of higher alcohol, gelatin, semi-synthetic glyceride and the like.
- solutions, emulsions or suspensions When used as an injection, it can be used as a solution, emulsion or suspension. These solutions, emulsions or suspensions are preferably sterilized and isotonic with blood.
- the solvent used in the production of these solutions, emulsions or suspensions is not particularly limited as long as it can be used as a medical diluent.
- water, ethanol, propylene glycol, ethoxylated isostearyl alcohol, polyoxylated isoforms are used. Examples include stearyl alcohol and polyoxyethylene sorbitan fatty acid esters.
- a sufficient amount of sodium chloride, glucose or glycerin may be included in the preparation to prepare an isotonic solution, and a normal solubilizing agent, buffer, soothing agent, etc. may be included. You may go out.
- the above-mentioned preparation may contain a coloring agent, a preservative, a fragrance, a flavoring agent, a sweetening agent, and the like as required, and may further contain other medicines.
- the amount of the active ingredient compound contained in the preparation is not particularly limited and is appropriately selected within a wide range, but is usually 0.5 to 70% by weight, preferably 1 to 30% by weight, based on the total composition.
- the amount used varies depending on the symptoms, age, etc. of the patient (warm-blooded animal, particularly human), but in the case of oral administration, the upper limit is 2000 mg (preferably 100 mg) per day, and the lower limit is 0.1 mg ( Preferably 1 mg, more preferably 10 mg) is administered to adults 1 to 6 times per day depending on the symptoms.
- the solvent specified in each example was used at the specified ratio. (Or, the ratio was changed as necessary.)
- the abbreviations used in the examples have the following significance. mg: milligram, g: gram, mL: milliliter, MHz: megahertz.
- 1 H NMR nuclear magnetic resonance
- MS Mass spectrometry
- Example (4b) 3-ethyl-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid
- the compound (75 mg) obtained in Example (4a) was hydrolyzed in the same manner as in Example (1b).
- the title compound 59 mg (90%) was obtained as a colorless solid.
- Example (1e) (cis-4- ⁇ [5- (4- ⁇ [(3-Ethyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino ⁇ phenyl) pyrimidin-2-yl] Oxy ⁇ cyclohexyl) acetic acid
- Example (1e) from the compound (57 mg) obtained in Example (4b) and the compound (90 mg) obtained in Example (1d), 92 mg of the amide compound was obtained. Obtained as an off-white amorphous.
- the amide compound (92 mg) was hydrolyzed to obtain 31 mg (22%, 2 steps) of the title compound as a yellow solid.
- Example (5b) 1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid
- the compound (1.06 g) obtained in Example (5a) was hydrolyzed in the same manner as in Example (1b) to give the title compound 595 mg (64%) was obtained as a colorless solid.
- Example (1e) (cis-4- ⁇ [5- (4- ⁇ [(1-Pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino ⁇ phenyl) pyrimidin-2-yl] oxy ⁇ cyclohexyl) Acetic acid
- 104 mg (78%) of the amide compound was obtained from the compound (50 mg) obtained in Example (5b) and the compound (90 mg) obtained in Example (1d). Obtained as an off-white solid.
- the amide compound (100 mg) was hydrolyzed to obtain 81 mg (84%) of the title compound as a pale yellow solid.
- Example (1e) In the same manner as in Example (1e), 104 mg (78%) of the amide compound was turned off from the compound (50 mg) obtained in Example (5b) and the compound (90 mg) obtained in Example (3a). Obtained as a white solid. In the same manner as in Example (1f), the amide compound (100 mg) was hydrolyzed to obtain 81 mg (84%) of the title compound as a pale yellow solid.
- This biaryl compound (123 mg) was hydrolyzed in the same manner as in Example (1b) to obtain a carboxylic acid compound (3-chloro-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid). .
- Example (7b) (cis-4- ⁇ [5- (4- ⁇ [(3-Chloro-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino ⁇ phenyl) pyrimidin-2-yl] Oxy ⁇ cyclohexyl) acetic acid
- the compound (101 mg) obtained in Example (7a) was hydrolyzed to obtain 82 mg (84%) of the title compound as a pale yellow solid.
- reaction stop solution 70 ⁇ l
- isopropanol / 1-heptane / water 80: 20: 2, v / v / v
- water 30 ⁇ l
- 1-heptane 100 ⁇ l
- a 1-heptane layer 50 ⁇ l was spotted on a TLC plate and developed with a developing solvent consisting of 1-hexane / diethyl ether / acetic acid (85: 15: 1, v / v / v).
- the radioactivity of the triglyceride fraction was quantified with a BAS2000 bioimage analyzer (Fuji Film), and the inhibitory activity of the test compound was calculated by the following formula by comparing with the control. The unreacted (0 minute incubation) radioactivity was used as the background.
- Inhibition rate 100 ⁇ [(radioactivity at the time of addition of test compound) ⁇ (background)] / [(radioactivity of control) ⁇ (background)] ⁇ 100
- the compounds of Examples 1 to 7 showed an inhibition rate of 50% or more at a test compound concentration of 1 ⁇ g / ml.
- the DGAT inhibitory activity test is not limited to the above method.
- microsomes prepared from the small intestine, adipose tissue, or liver of animals such as rats and mice may be used as the DGAT enzyme.
- microsomes prepared from cultured cells (3T3-L1 adipocytes, primary cultured adipocytes, Caco2 cells, HepG2 cells, etc.) or cultured cells highly expressing DGAT can also be used as the DGAT enzyme.
- a flash plate PerkinElmer in which the extraction operation is omitted can be used.
- the compound of the present invention has excellent DGAT1 inhibitory biological activity.
- the DGAT1 enzyme is important for digestion and absorption of neutral fat, and when small intestine DGAT1 is inhibited, the absorption of neutral fat is suppressed.
- the biological activity of the DGAT1 inhibitory action was evaluated using as an index the inhibition of neutral fat absorption after neutral fat loading.
- Male C57BL / 6N mice (7-12 weeks old, body weight 17-25 g, Nippon Charles River) fasted overnight were assigned to Vehicle Group 1, Vehicle Group 2 and each test compound group, respectively vehicle (0.5% Methylcellulose) Alternatively, each test compound (1 to 10 mg / kg) suspended in the vehicle was orally administered (5 mL / kg).
- Lipoprotein lipase inhibitor (Pluronic-F127: Sigma-Aldrich Co., Ltd., 1 g / kg, dissolved in physiological saline at 20% by weight) was intraperitoneally administered (5 mL / kg) Distilled water was orally administered to Vehicle Group 1 and 20% neutral fat-containing emulsion (Intralipid 20%: Terumo Corporation) was orally administered (0.2 mL / mouse) to Vehicle Group 2 and Compound Group.
- Neutral fat absorption inhibitory activity (%) 100-[(Neutral fat concentration of each test compound group)-(Neutral fat concentration of Vehicle group 1)] / [(Neutral fat concentration of Vehicle group 2)-( Vehicle group 1 neutral fat concentration)] ⁇ 100
- the compounds of Examples 1 to 6 showed neutral fat absorption inhibitory activity of 60% or more at a dose of 3 mg / kg or less.
- the compound of the present invention has excellent neutral fat absorption inhibitory activity.
- mice Male C57BL / 6N mice (7-12 weeks old, body weight 17-25 g, Nippon Charles River) are bred individually and fed with a high fat diet (fat content 45 kcal%: Research Diet D12451) for over a week. I got used to it. Allocate the animals evenly to the experimental groups based on the amount of food consumed during the period, fast overnight and then each vehicle (0.5% Methylcellulose) or test compound (1-10 mg / kg) suspended in the vehicle. The group was orally administered (10 mL / kg). A high fat diet was fed 30 minutes after the administration, and the amount of food intake was measured 6 hours after the start of feeding. The feeding inhibitory activity of each test compound was calculated based on the following formula.
- Feeding inhibitory activity (%) [(food consumption of vehicle group) ⁇ (food consumption of each test compound group)] / [(food consumption of vehicle group)] ⁇ 100
- the compound of Example 1 showed an antifeedant activity of 25% or more at a dose of 10 mg / kg or less.
- the compound of the present invention has an excellent antifeedant action.
- the high-fat diet used for the feed is not limited to the above-mentioned high-fat diet, and for example, a rodent feed containing 45 to 60% neutral fat as calories can be used.
- Formulation Example 1 Capsule 50 mg of the compound of Example 1 or 2 Lactose 128mg Corn starch 70mg Magnesium stearate 2mg ------------------ 250mg After mixing the powder of the above formulation and passing through a 60 mesh sieve, this powder is put into a 250 mg gelatin capsule to form a capsule.
- Formulation Example 2 Tablet Example 1 or 2 compound 50 mg Lactose 126mg Corn starch 23mg Magnesium stearate 1mg ------------------ 200mg
- the powder of the above formulation is mixed, granulated and dried using corn starch paste, and then tableted by a tableting machine to make one tablet of 200 mg. This tablet can be sugar-coated if necessary.
- the compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof has an excellent DGAT inhibitory action and antifeeding action and is useful as a medicine.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Ophthalmology & Optometry (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Urology & Nephrology (AREA)
- Emergency Medicine (AREA)
- Heart & Thoracic Surgery (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Cardiology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Child & Adolescent Psychology (AREA)
- Gastroenterology & Hepatology (AREA)
- Reproductive Health (AREA)
- Epidemiology (AREA)
- Vascular Medicine (AREA)
Abstract
The present invention relates to a compound having an excellent DGAT inhibitory activity and an excellent antifeeding activity or a pharmacologically acceptable salt thereof. A compound represented by general formula (I) [wherein R1 represents a hydrogen atom, a halogen atom, a C1-C6 alkyl group or a C3-C6 cycloalkyl group; R2's independently represent a halogen atom, a C1-C6 alkyl group, a C1-C6 alkoxy group or a hydroxy group; and n represents an integer of 0 to 2] or a pharmacologically acceptable salt thereof.
Description
本発明は、優れたアシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼ(Acyl-CoA:diacylglycerol acyltransferase、以下、DGATともいう)阻害作用及び優れた摂食抑制作用を有する特定の化学構造を有する化合物又はその薬理上許容される塩に関する。
The present invention relates to a compound having a specific chemical structure having an excellent acylcoenzyme A: diacylglycerol acyltransferase (hereinafter also referred to as DGAT) inhibitory activity and an excellent feeding inhibitory activity, Relates to acceptable salts.
肥満は、消費エネルギーに比較して摂取エネルギーが過剰な状態が持続することにより、脂肪細胞において中性脂肪(トリアシルグリセロールまたはトリグリセライド、以下、TGともいう)が蓄積し、その結果として体重が標準体重に比較して著しく増加した状態である(非特許文献1)。肥満は、高脂血症、高TG血症、糖尿病、高血圧症、動脈硬化症などの生活習慣病、脳血管障害、冠動脈疾患、呼吸異常、腰痛、変形性膝関節症、痛風、胆石症などをもたらし、肥満のうちこれらの合併症を有するもの、あるいは将来これらの合併症を生じる可能性があるものは、肥満症と定義され、一つの疾患として扱われている。
In obesity, triglyceride (triacylglycerol or triglyceride, hereinafter also referred to as TG) accumulates in adipocytes due to the persistence of excess energy compared to energy consumption, resulting in standard weight gain. It is in a state of significantly increasing compared to body weight (Non-Patent Document 1). Obesity is hyperlipidemia, hyperTGemia, diabetes, hypertension, lifestyle-related diseases such as arteriosclerosis, cerebrovascular disorder, coronary artery disease, respiratory abnormalities, low back pain, knee osteoarthritis, gout, cholelithiasis, etc. Any obesity that has these complications or that may cause these complications in the future is defined as obesity and is treated as a disease.
動物および植物は、脂質を不溶性のTGとして蓄え、必要に応じて、TGを分解してエネルギーを産生する。食事により摂取されたTGは、小腸内腔で胆汁酸および膵リパーゼの作用により、遊離脂肪酸およびモノアシルグリセロールに分解される。遊離脂肪酸、モノアシルグリセロールおよび胆汁酸からなるミセルは、小腸上皮細胞に吸収され、小胞体でアシルコエンザイムA合成酵素(以下、ACSという)、アシルコエンザイムA:モノアシルグリセロールアシルトランスフェラーゼおよびDGATの作用により、新たにTGが合成される。TGは、リン脂質、コレステロールおよびアポリポタンパクと組み合わされて、キロミクロンとして胃腸のリンパ管に分泌される。さらに、TGは、リンパ主管を経て血中に分泌され、末梢に運ばれて利用される。一方、脂肪組織においても、グリセロール3-リン酸および遊離脂肪酸からACS、グリセロール3-リン酸アシルトランスフェラーゼ、リゾホスファチジン酸アシルトランスフェラーゼおよびDGATの作用により、TGが合成される(非特許文献2)。このように過剰に摂取されたTGは、脂肪組織に蓄積され、その結果として肥満が生じる。
Animals and plants store lipid as insoluble TG, and decompose TG as necessary to produce energy. TG ingested by the meal is broken down into free fatty acids and monoacylglycerol by the action of bile acids and pancreatic lipase in the lumen of the small intestine. Micelles composed of free fatty acid, monoacylglycerol and bile acid are absorbed into small intestinal epithelial cells, and in the endoplasmic reticulum by the action of acylcoenzyme A synthase (hereinafter referred to as ACS), acylcoenzyme A: monoacylglycerol acyltransferase and DGAT. TG is newly synthesized. TG, in combination with phospholipids, cholesterol and apolipoprotein, is secreted into the gastrointestinal lymphatic vessels as kilomicrons. Furthermore, TG is secreted into the blood via the lymph main duct and transported to the periphery for use. On the other hand, in adipose tissue, TG is synthesized from glycerol 3-phosphate and free fatty acids by the action of ACS, glycerol 3-phosphate acyltransferase, lysophosphatidic acid acyltransferase, and DGAT (Non-patent Document 2). Thus, TG ingested excessively accumulates in adipose tissue, resulting in obesity.
DGATは、細胞内の小胞体に存在する酵素であり、TG合成経路の最も重要な最終ステップの反応、すなわちアシルコエンザイムAのアシル基を1,2-ジアシルグリセロールの3位へ転移する反応を触媒する酵素である(非特許文献3乃至5)。DGATには、2種類のアイソザイムDGAT1(非特許文献6)およびDGAT2(非特許文献7)が存在することが報告されている。DGAT1は小腸および脂肪組織に、DGAT2は肝臓および脂肪組織にそれぞれ高発現していることから、DGAT1は主として小腸からの脂肪吸収および脂肪組織での脂肪蓄積に、DGAT2は肝臓でのTG合成もしくはVLDL(very low density lipoproteins)分泌、および脂肪組織での脂肪蓄積に関与していると考えられている。DGAT1およびDGAT2の役割の違いはまだ詳細には明らかにされていないが、DGATと肥満、脂質代謝、糖代謝などとの関連性が示唆されている(非特許文献8)。DGATは、消化管上皮細胞および脂肪組織におけるTG合成の鍵酵素であり、DGATを阻害する薬剤は、TG合成を抑制することにより、消化管における脂肪吸収および脂肪組織における脂肪蓄積を抑制し、肥満、肥満症、高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、糖尿病、非アルコール性脂肪肝炎、または、肥満に起因する高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、糖尿病、非アルコール性脂肪肝炎、高血圧症、動脈硬化症、脳血管障害、もしくは、冠動脈疾患などの治療剤もしくは予防剤として有用であると期待される(非特許文献9乃至13)。
DGAT is an enzyme that is present in the endoplasmic reticulum in the cell and catalyzes the most important final step reaction in the TG synthesis pathway, that is, the reaction of transferring the acyl group of acylcoenzyme A to the 3-position of 1,2-diacylglycerol. (Non-Patent Documents 3 to 5). It has been reported that DGAT has two types of isozymes DGAT1 (Non-patent document 6) and DGAT2 (Non-patent document 7). Since DGAT1 is highly expressed in the small intestine and adipose tissue, and DGAT2 is highly expressed in the liver and adipose tissue, respectively, DGAT1 is mainly used for fat absorption from the small intestine and fat accumulation, and DGAT2 is used for TG synthesis or VLDL in the liver. (Very low density lipoproteins) secretion and fat accumulation in adipose tissue. Although the difference in the roles of DGAT1 and DGAT2 has not yet been clarified in detail, the relationship between DGAT and obesity, lipid metabolism, sugar metabolism, etc. has been suggested (Non-patent Document 8). DGAT is a key enzyme for TG synthesis in gastrointestinal epithelial cells and adipose tissue, and a drug that inhibits DGAT suppresses fat absorption in the gastrointestinal tract and fat accumulation in adipose tissue by suppressing TG synthesis, and obesity , Obesity, hyperlipidemia, hypertriglyceridemia, dyslipidemia, insulin resistance syndrome, diabetes, non-alcoholic steatohepatitis, or obesity-induced hyperlipidemia, hypertriglyceridemia, lipid metabolism It is expected to be useful as a therapeutic or prophylactic agent for abnormal diseases, insulin resistance syndrome, diabetes, nonalcoholic steatohepatitis, hypertension, arteriosclerosis, cerebrovascular disorder, coronary artery disease, etc. 9 to 13).
食欲抑制薬は、直接あるいは間接的に食欲制御系を調節するものであるが、その作用メカニズムは中枢性と末梢性に大別される。中枢性に作用する食欲抑制薬は摂食中枢及び満腹中枢の存在する視床下部神経系や同神経系を調節する脳内モノアミン神経系に作用して食欲を直接的に抑制する。一方、末梢性に作用する食欲抑制薬は食事による栄養摂取や余剰エネルギーの蓄積状態を、感知し伝達する機構に作用して間接的に食欲を抑制する。
An appetite suppressant directly or indirectly regulates the appetite control system, but its mechanism of action is roughly divided into central and peripheral. An appetite suppressant acting centrally acts on the hypothalamic nervous system where the feeding center and satiety center exist and the monoamine nervous system in the brain that regulates the nervous system, thereby directly suppressing appetite. On the other hand, an appetite suppressant that acts on the periphery acts on a mechanism that senses and transmits the intake of nutrients and the accumulation of surplus energy, and indirectly suppresses appetite.
近年、食物の消化・吸収と密接に関連して分泌される消化管ホルモン(CCK、GLP-1、PYYなど)(非特許文献14)や、エネルギー蓄積量(脂肪量)に応じて脂肪細胞から分泌されるレプチン(非特許文献15)などが、ホルモン性あるいは神経性に末梢から中枢へ食欲を調節するシグナルを伝えるメカニズムが明らかになってきている。これら末梢性シグナルに関連する新しい食欲抑制薬はより効果的で副作用の少ない肥満症治療薬になることが期待されている。
In recent years, gastrointestinal hormones (CCK, GLP-1, PYY, etc.) secreted in close association with the digestion and absorption of food (Non-Patent Document 14) and from fat cells according to the energy accumulation (fat mass) The mechanism by which secreted leptin (Non-patent Document 15) or the like transmits a signal that regulates appetite from the periphery to the center in a hormonal or neurological manner has been clarified. These new appetite suppressants associated with peripheral signals are expected to be more effective and less effective for the treatment of obesity.
DGAT阻害作用を有する化合物として、特許文献1には、4-(5-カルボキサアミド-2,3’-ビピリジン-6’-イルオキシ)シクロヘキサンカルボン酸を有する化合物が記載されている。
As a compound having a DGAT inhibitory action, Patent Document 1 discloses a compound having 4- (5-carboxamido-2,3'-bipyridin-6'-yloxy) cyclohexanecarboxylic acid.
発明者らは、DGAT阻害作用及び摂食抑制作用を有する化合物について鋭意研究を行った結果、特定の化学構造を有する化合物が、優れたDGAT阻害作用を有しており、特にDGAT1に対して高い阻害作用を有することを見出した。また、本発明者らは、この化合物が、優れた摂食抑制作用を有していることを見出した。更に、本発明者らは、この化合物が、肥満、肥満症、高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、虚血性心疾患及び過食症からなる群から選ばれる疾患の予防及び/又は治療のための医薬の有効成分として、又は肥満に起因する高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、高血圧症、脳血管障害、冠動脈疾患、脂肪肝、呼吸異常、腰痛、変形性膝関節症、痛風、及び胆石症からなる群から選ばれる疾患の治療及び/又は予防のための医薬の有効成分として有用であることを見出した。
As a result of intensive studies on compounds having a DGAT inhibitory action and an antifeedant action, the inventors have found that a compound having a specific chemical structure has an excellent DGAT inhibitory action, particularly high for DGAT1. It was found to have an inhibitory effect. The present inventors have also found that this compound has an excellent antifeeding action. Furthermore, the present inventors have found that this compound is obesity, obesity, hyperlipidemia, hypertriglycerideemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral Neuropathy, including diabetic nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic Hyperlipidemia, hypertriglyceridemia, lipid metabolism resulting from obesity or as an active ingredient of a medicament for the prevention and / or treatment of a disease selected from the group consisting of arteriosclerosis, ischemic heart disease and bulimia Abnormal diseases, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (including diabetic peripheral neuropathy, diabetic nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataracts, Gynecologic diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, hypertension, cerebrovascular disorder, coronary artery disease, fatty liver, respiratory abnormalities, low back pain, deformity It was found useful as an active ingredient of a medicament for the treatment and / or prevention of a disease selected from the group consisting of knee arthropathy, gout, and cholelithiasis.
本発明は、(1)一般式(I)
The present invention comprises (1) general formula (I)

[式中、
R1は、水素原子、ハロゲン原子、C1-C6アルキル基又はC3-C6シクロアルキル基を示し、
R2は、独立して、ハロゲン原子、C1-C6アルキル基、C1-C6アルコキシ基又はヒドロキシ基を示し、
nは、0乃至2の整数を示す。]で表される化合物又はその薬理上許容される塩に関する。 [Where:
R 1 represents a hydrogen atom, a halogen atom, a C 1 -C 6 alkyl group or a C 3 -C 6 cycloalkyl group,
R 2 independently represents a halogen atom, a C 1 -C 6 alkyl group, a C 1 -C 6 alkoxy group or a hydroxy group,
n represents an integer of 0 to 2. Or a pharmacologically acceptable salt thereof.
R1は、水素原子、ハロゲン原子、C1-C6アルキル基又はC3-C6シクロアルキル基を示し、
R2は、独立して、ハロゲン原子、C1-C6アルキル基、C1-C6アルコキシ基又はヒドロキシ基を示し、
nは、0乃至2の整数を示す。]で表される化合物又はその薬理上許容される塩に関する。 [Where:
R 1 represents a hydrogen atom, a halogen atom, a C 1 -C 6 alkyl group or a C 3 -C 6 cycloalkyl group,
R 2 independently represents a halogen atom, a C 1 -C 6 alkyl group, a C 1 -C 6 alkoxy group or a hydroxy group,
n represents an integer of 0 to 2. Or a pharmacologically acceptable salt thereof.
本発明において、好適には、
(2) (1)において、
一般式(I)が、一般式(Ia)である化合物又はその薬理上許容される塩。 In the present invention, preferably,
(2) In (1),
The compound or its pharmacologically acceptable salt whose general formula (I) is general formula (Ia).
(2) (1)において、
一般式(I)が、一般式(Ia)である化合物又はその薬理上許容される塩。 In the present invention, preferably,
(2) In (1),
The compound or its pharmacologically acceptable salt whose general formula (I) is general formula (Ia).

(3) (1)又は(2)において、
R1が、水素原子、塩素原子、メチル基、エチル基又はシクロプロピル基である化合物又はその薬理上許容される塩。 (3) In (1) or (2),
A compound or a pharmacologically acceptable salt thereof, wherein R 1 is a hydrogen atom, a chlorine atom, a methyl group, an ethyl group or a cyclopropyl group.
R1が、水素原子、塩素原子、メチル基、エチル基又はシクロプロピル基である化合物又はその薬理上許容される塩。 (3) In (1) or (2),
A compound or a pharmacologically acceptable salt thereof, wherein R 1 is a hydrogen atom, a chlorine atom, a methyl group, an ethyl group or a cyclopropyl group.
(4) (1)乃至(3)から選択されるいずれか一項において、
nが、0である化合物又はその薬理上許容される塩。 (4) In any one item selected from (1) to (3),
A compound in which n is 0 or a pharmacologically acceptable salt thereof.
nが、0である化合物又はその薬理上許容される塩。 (4) In any one item selected from (1) to (3),
A compound in which n is 0 or a pharmacologically acceptable salt thereof.
(5) (1)乃至(3)から選択されるいずれか一項において、
nが、1であり、R2が、メチル基である化合物又はその薬理上許容される塩。 (5) In any one item selected from (1) to (3),
A compound or a pharmacologically acceptable salt thereof, wherein n is 1, and R 2 is a methyl group.
nが、1であり、R2が、メチル基である化合物又はその薬理上許容される塩。 (5) In any one item selected from (1) to (3),
A compound or a pharmacologically acceptable salt thereof, wherein n is 1, and R 2 is a methyl group.
(6) (1)において、
一般式(I)が、一般式(Ia)であり、R1が、水素原子、塩素原子、メチル基、エチル基又はシクロプロピル基であり、nが、0である化合物又はその薬理上許容される塩。 (6) In (1),
A compound in which general formula (I) is general formula (Ia), R 1 is a hydrogen atom, a chlorine atom, a methyl group, an ethyl group or a cyclopropyl group, and n is 0, or a pharmacologically acceptable salt thereof. Salt.
一般式(I)が、一般式(Ia)であり、R1が、水素原子、塩素原子、メチル基、エチル基又はシクロプロピル基であり、nが、0である化合物又はその薬理上許容される塩。 (6) In (1),
A compound in which general formula (I) is general formula (Ia), R 1 is a hydrogen atom, a chlorine atom, a methyl group, an ethyl group or a cyclopropyl group, and n is 0, or a pharmacologically acceptable salt thereof. Salt.
(7) (1)において、
一般式(I)が、一般式(Ia)であり、R1が、水素原子、塩素原子、メチル基、エチル基又はシクロプロピル基であり、nが、1であり、R2が、メチル基である化合物又はその薬理上許容される塩。 (7) In (1),
The general formula (I) is the general formula (Ia), R 1 is a hydrogen atom, a chlorine atom, a methyl group, an ethyl group or a cyclopropyl group, n is 1, and R 2 is a methyl group Or a pharmacologically acceptable salt thereof.
一般式(I)が、一般式(Ia)であり、R1が、水素原子、塩素原子、メチル基、エチル基又はシクロプロピル基であり、nが、1であり、R2が、メチル基である化合物又はその薬理上許容される塩。 (7) In (1),
The general formula (I) is the general formula (Ia), R 1 is a hydrogen atom, a chlorine atom, a methyl group, an ethyl group or a cyclopropyl group, n is 1, and R 2 is a methyl group Or a pharmacologically acceptable salt thereof.
(8) (cis-4-{[5-(4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(4-{[(3-シクロプロピル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(2-メチル-4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(4-{[(3-エチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(4-{[(1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(2-メチル-4-{[(1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、もしくは、
(cis-4-{[5-(4-{[(3-クロロ-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸
である化合物又はその薬理上許容される塩。 (8) (cis-4-{[5- (4-{[(3-Methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid,
(cis-4-{[5- (4-{[(3-Cyclopropyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} (Cyclohexyl) acetic acid,
(cis-4-{[5- (2-Methyl-4-{[(3-methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl ] Oxy} cyclohexyl) acetic acid,
(cis-4-{[5- (4-{[(3-Ethyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid,
(cis-4-{[5- (4-{[(1-Pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl) acetic acid,
(cis-4-{[5- (2-Methyl-4-{[(1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid, or
(cis-4-{[5- (4-{[(3-Chloro-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl ) A compound which is acetic acid or a pharmacologically acceptable salt thereof.
(cis-4-{[5-(4-{[(3-シクロプロピル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(2-メチル-4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(4-{[(3-エチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(4-{[(1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(2-メチル-4-{[(1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、もしくは、
(cis-4-{[5-(4-{[(3-クロロ-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸
である化合物又はその薬理上許容される塩。 (8) (cis-4-{[5- (4-{[(3-Methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid,
(cis-4-{[5- (4-{[(3-Cyclopropyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} (Cyclohexyl) acetic acid,
(cis-4-{[5- (2-Methyl-4-{[(3-methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl ] Oxy} cyclohexyl) acetic acid,
(cis-4-{[5- (4-{[(3-Ethyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid,
(cis-4-{[5- (4-{[(1-Pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl) acetic acid,
(cis-4-{[5- (2-Methyl-4-{[(1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid, or
(cis-4-{[5- (4-{[(3-Chloro-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl ) A compound which is acetic acid or a pharmacologically acceptable salt thereof.
(9) (8)に記載してある化合物又はその薬理上許容される塩のうちの化合物。
(9) A compound described in (8) or a pharmacologically acceptable salt thereof.
(10) (1)乃至(9)から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩を有効成分として含有するアシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼ阻害剤。
(10) An acyl coenzyme A: diacylglycerol acyltransferase inhibitor comprising the compound described in any one of (1) to (9) or a pharmacologically acceptable salt thereof as an active ingredient.
(11) (1)乃至(9)から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩を有効成分として含有する摂食抑制剤及び/又は食欲抑制剤。
(11) An antifeedant and / or an appetite suppressant containing the compound described in any one of (1) to (9) or a pharmacologically acceptable salt thereof as an active ingredient.
(12) (1)乃至(9)から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩を有効成分として含有する医薬組成物。
(12) A pharmaceutical composition comprising as an active ingredient the compound described in any one of (1) to (9) or a pharmacologically acceptable salt thereof.
(13) 医薬組成物が、アシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼ阻害作用を有する(12)に記載の医薬組成物。
(13) The pharmaceutical composition according to (12), wherein the pharmaceutical composition has an inhibitory action on acylcoenzyme A: diacylglycerol acyltransferase.
(14) 医薬組成物が、摂食抑制作用及び/又は食欲抑制作用を有する(12)に記載の医薬組成物。
(14) The pharmaceutical composition according to (12), wherein the pharmaceutical composition has an antifeedant action and / or an appetite suppressive action.
(15) 医薬組成物が、アシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼ阻害作用により、治療及び/又は予防される疾病の治療及び/又は予防のための(12)に記載の医薬組成物。
(15) The pharmaceutical composition according to (12), for treating and / or preventing a disease wherein the pharmaceutical composition is treated and / or prevented by acylcoenzyme A: diacylglycerol acyltransferase inhibitory action.
(16) 医薬組成物が、アシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼ活性の亢進に起因する疾病の治療及び/又は予防のための(12)に記載の医薬組成物。
(16) The pharmaceutical composition according to (12), wherein the pharmaceutical composition is used for treatment and / or prevention of a disease caused by an increase in acylcoenzyme A: diacylglycerol acyltransferase activity.
(17) 医薬組成物が、アシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼを阻害させ、トリグリセライドの合成を阻害し、トリグリセライドの吸収が抑制されることにより、症状の治療、改善、軽減及び/又は予防がなされる疾病の治療及び/又は予防のための(12)に記載の医薬組成物。
(17) The pharmaceutical composition inhibits acylcoenzyme A: diacylglycerol acyltransferase, inhibits synthesis of triglyceride, and suppresses the absorption of triglyceride, thereby treating, improving, reducing and / or preventing symptoms. The pharmaceutical composition according to (12) for the treatment and / or prevention of certain diseases.
(18) 医薬組成物が、アシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼを阻害させ、トリグリセライドの合成が阻害されることにより、症状の治療、改善、軽減及び/又は予防がなされる疾病の治療及び/又は予防のための(12)に記載の医薬組成物。
(18) The pharmaceutical composition inhibits acyl coenzyme A: diacylglycerol acyltransferase and inhibits the synthesis of triglyceride, thereby treating and / or treating diseases in which symptoms are treated, ameliorated, reduced and / or prevented. The pharmaceutical composition according to (12) for prevention.
(19) 医薬組成物が、肥満、肥満症、高脂血症、高トリグリセライド症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、虚血性心疾患又は過食症の治療及び/又は予防のための(12)に記載の医薬組成物。
(19) The pharmaceutical composition is obesity, obesity, hyperlipidemia, hypertriglyceridosis, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic Nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, false The pharmaceutical composition according to (12) for the treatment and / or prevention of blood heart disease or bulimia.
(20) 医薬組成物が、肥満又は肥満症の治療及び/又は予防のための(12)に記載の医薬組成物。
(20) The pharmaceutical composition according to (12), wherein the pharmaceutical composition is for the treatment and / or prevention of obesity or obesity.
(21) 医薬組成物が、糖尿病の治療及び/又は予防のための(12)に記載の医薬組成物。
(21) The pharmaceutical composition according to (12), wherein the pharmaceutical composition is for the treatment and / or prevention of diabetes.
(22) 医薬組成物が、肥満に起因する高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、高血圧症、脳血管障害、冠動脈疾患、脂肪肝、呼吸異常、腰痛、変形性膝関節症、痛風又は胆石症の治療及び/又は予防のための(12)に記載の医薬組成物。
(22) When the pharmaceutical composition is obesity-induced hyperlipidemia, hypertriglyceridemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic Nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, hypertension (12) The pharmaceutical composition for treatment and / or prevention of symptom, cerebrovascular disorder, coronary artery disease, fatty liver, respiratory abnormality, low back pain, knee osteoarthritis, gout or cholelithiasis.
(23) 医薬組成物が、肥満に起因する高脂血症、高トリグリセライド血症、糖尿病、動脈硬化症又は高血圧症の治療及び/又は予防のための(12)に記載の医薬組成物。
(23) The pharmaceutical composition according to (12), wherein the pharmaceutical composition is for the treatment and / or prevention of hyperlipidemia, hypertriglyceridemia, diabetes, arteriosclerosis or hypertension caused by obesity.
(24) 医薬組成物が、小腸からの脂肪吸収を抑制するための(12)に記載の医薬組成物。
(24) The pharmaceutical composition according to (12), wherein the pharmaceutical composition suppresses fat absorption from the small intestine.
(25) 肥満、肥満症、高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、虚血性心疾患又は過食症の治療及び/又は予防で使用するための、(1)乃至(9)から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩。
(25) Obesity, obesity, hyperlipidemia, hypertriglyceridemia, lipid metabolism disorder, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic nephropathy, diabetes Retinopathy, including diabetic macroangiopathy), cataract, gestational diabetes, non-alcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, ischemic heart disease or The compound according to any one of (1) to (9) or a pharmacologically acceptable salt thereof for use in the treatment and / or prevention of bulimia.
(26) 肥満又は肥満症の治療及び/又は予防で使用するための、(1)乃至(9)から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩。
(26) The compound according to any one of (1) to (9) or a pharmacologically acceptable salt thereof for use in the treatment and / or prevention of obesity or obesity.
(27) 糖尿病の治療及び/又は予防で使用するための、(1)乃至(9)から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩。
(27) The compound according to any one of (1) to (9) or a pharmacologically acceptable salt thereof for use in the treatment and / or prevention of diabetes.
(28) 医薬組成物を製造するための、(1)乃至(9)から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩の使用。
(28) Use of the compound described in any one of (1) to (9) or a pharmacologically acceptable salt thereof for producing a pharmaceutical composition.
(29) 医薬組成物がアシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼを阻害するための医薬組成物である(28)に記載の使用。
(29) The use according to (28), wherein the pharmaceutical composition is a pharmaceutical composition for inhibiting acylcoenzyme A: diacylglycerol acyltransferase.
(30) 医薬組成物が摂食及び/又は食欲を抑制するための医薬組成物である(28)に記載の使用。
(30) Use according to (28), wherein the pharmaceutical composition is a pharmaceutical composition for suppressing eating and / or appetite.
(31) 医薬組成物が肥満、肥満症、高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、虚血性心疾患又は過食症の治療及び/又は予防のための医薬組成物である(28)に記載の使用。
(31) The pharmaceutical composition is obesity, obesity, hyperlipidemia, hypertriglyceridemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic Nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, false The use according to (28), which is a pharmaceutical composition for the treatment and / or prevention of blood heart disease or bulimia.
(32) 医薬組成物が肥満又は肥満症の治療及び/又は予防のための医薬組成物である(28)に記載の使用。
(32) The use according to (28), wherein the pharmaceutical composition is a pharmaceutical composition for the treatment and / or prevention of obesity or obesity.
(33) 医薬組成物が糖尿病の治療及び/又は予防のための医薬組成物である(28)に記載の使用。
(33) Use according to (28), wherein the pharmaceutical composition is a pharmaceutical composition for the treatment and / or prevention of diabetes.
(34) 医薬組成物が肥満に起因する高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、高血圧症、脳血管障害、冠動脈疾患、脂肪肝、呼吸異常、腰痛、変形性膝関節症、痛風又は胆石症の治療及び/又は予防のための医薬組成物である(28)に記載の使用。
(34) Hyperlipidemia due to obesity, hypertriglyceridemia, lipid metabolism disorder, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic kidney) , Including diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, hypertension The use according to (28), which is a pharmaceutical composition for the treatment and / or prevention of cerebrovascular disorder, coronary artery disease, fatty liver, respiratory disorder, low back pain, knee osteoarthritis, gout or cholelithiasis.
(35) 医薬組成物が肥満に起因する高脂血症、高トリグリセライド血症、糖尿病、動脈硬化症又は高血圧症の治療及び/又は予防のための医薬組成物である(28)に記載の使用。
(35) The use according to (28), wherein the pharmaceutical composition is a therapeutic composition for the treatment and / or prevention of hyperlipidemia, hypertriglyceridemia, diabetes, arteriosclerosis or hypertension caused by obesity. .
(36) 医薬組成物が小腸からの脂肪吸収を抑制するための医薬組成物である(28)に記載の使用。
(36) The use according to (28), wherein the pharmaceutical composition is a pharmaceutical composition for suppressing fat absorption from the small intestine.
(37) (1)乃至(9)から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩の薬理的な有効量を温血動物に投与するアシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼ阻害方法。
(37) Acyl coenzyme A: diacylglycerol for administering to a warm-blooded animal a pharmacologically effective amount of the compound described in any one of (1) to (9) or a pharmacologically acceptable salt thereof Acyltransferase inhibition method.
(38) (1)乃至(9)から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩の薬理的な有効量を温血動物に投与する摂食抑制及び/又は食欲抑制方法。
(38) Feeding suppression and / or administration of a pharmacologically effective amount of the compound described in any one of (1) to (9) or a pharmacologically acceptable salt thereof to a warm-blooded animal Appetite suppression method.
(39) (1)乃至(9)から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩の薬理的な有効量を温血動物に投与する疾病の治療及び/又は予防方法。
(39) Treatment of a disease in which a pharmacologically effective amount of the compound described in any one of (1) to (9) or a pharmacologically acceptable salt thereof is administered to a warm-blooded animal and / or Prevention method.
(40) 疾病が肥満、肥満症、高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、虚血性心疾患又は過食症である(39)に記載の方法。
(40) Diseases are obesity, obesity, hyperlipidemia, hypertriglycerideemia, lipid metabolism disorders, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic nephropathy , Including diabetic retinopathy, diabetic macrovascular disease), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, ischemic heart The method according to (39), which is a disease or bulimia.
(41) 疾病が肥満又は肥満症である(39)に記載の方法。
(41) The method according to (39), wherein the disease is obesity or obesity.
(42) 疾病が糖尿病である(39)に記載の方法。
(42) The method according to (39), wherein the disease is diabetes.
(43) 疾病が肥満に起因する高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、高血圧症、脳血管障害、冠動脈疾患、脂肪肝、呼吸異常、腰痛、変形性膝関節症、痛風又は胆石症である(39)に記載の方法。
(43) Hyperlipidemia due to obesity, hypertriglycerideemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic nephropathy, Diabetic retinopathy, including diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, hypertension, brain (39) The method according to (39), which is vascular disorder, coronary artery disease, fatty liver, respiratory disorder, low back pain, knee osteoarthritis, gout or cholelithiasis.
(44) 疾病が肥満に起因する高脂血症、高トリグリセライド血症、糖尿病、動脈硬化症又は高血圧症である(39)に記載の方法。
(44) The method according to (39), wherein the disease is hyperlipidemia, hypertriglyceridemia, diabetes, arteriosclerosis or hypertension caused by obesity.
(45) (1)乃至(9)から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩の薬理的な有効量を温血動物に投与する小腸からの脂肪吸収を抑制する方法。
(45) Absorption of fat from the small intestine in which a pharmacologically effective amount of the compound described in any one of (1) to (9) or a pharmacologically acceptable salt thereof is administered to a warm-blooded animal. How to suppress.
(46) 温血動物がヒトである(37)乃至(45)から選択されるいずれか一項に記載の方法
である。 (46) The method according to any one of (37) to (45), wherein the warm-blooded animal is a human.
である。 (46) The method according to any one of (37) to (45), wherein the warm-blooded animal is a human.
本発明において、「ハロゲン原子」は、フッ素原子、塩素原子、臭素原子又は沃素原子である。好適には、フッ素原子又は塩素原子であり、より好適には、塩素原子である。
In the present invention, the “halogen atom” is a fluorine atom, a chlorine atom, a bromine atom or an iodine atom. Preferred is a fluorine atom or a chlorine atom, and more preferred is a chlorine atom.
本発明において、「C1-C6アルキル基」は、炭素数1乃至6個の直鎖又は分枝鎖アルキル基である。例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、s-ブチル、t-ブチル、ペンチル、イソペンチル、2-メチルブチル、ネオペンチル、ヘキシル、イソヘキシル又は4-メチルペンチル基である。好適には、炭素数1乃至4個の直鎖又は分枝鎖アルキル基(C1-C4アルキル基)であり、より好適には、メチル基又はエチル基(C1-C2アルキル基)であり、更により好適には、メチル基である。
In the present invention, the “C 1 -C 6 alkyl group” is a linear or branched alkyl group having 1 to 6 carbon atoms. For example, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, s-butyl, t-butyl, pentyl, isopentyl, 2-methylbutyl, neopentyl, hexyl, isohexyl or 4-methylpentyl group. Preferred is a linear or branched alkyl group having 1 to 4 carbon atoms (C 1 -C 4 alkyl group), and more preferred is a methyl group or an ethyl group (C 1 -C 2 alkyl group). And even more preferably a methyl group.
本発明において、「C1-C6アルコキシ基」は、前記「C1-C6アルキル基」が酸素原子に結合した基であり、炭素数1乃至6個の直鎖又は分枝鎖アルコキシ基である。例えば、メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、イソブトキシ、s-ブトキシ、t-ブトキシ、ペントキシ又はヘキシルオキシ基である。好適には、炭素数1乃至4個の直鎖又は分枝鎖アルコキシ基(C1-C4アルコキシ基)であり、より好適には、メトキシ基又はエトキシ基(C1-C2アルコキシ基)であり、更により好適には、メトキシ基である。
In the present invention, the “C 1 -C 6 alkoxy group” is a group in which the “C 1 -C 6 alkyl group” is bonded to an oxygen atom, and is a linear or branched alkoxy group having 1 to 6 carbon atoms. It is. For example, a methoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, s-butoxy, t-butoxy, pentoxy or hexyloxy group. Preferred is a linear or branched alkoxy group having 1 to 4 carbon atoms (C 1 -C 4 alkoxy group), and more preferred is a methoxy group or an ethoxy group (C 1 -C 2 alkoxy group). And even more preferably a methoxy group.
本発明において、「C3-C6シクロアルキル基」は、シクロプロピル基、シクロブチル基、シクロペンチル基又はシクロヘキシル基であり、好適には、シクロプロピル基である。
In the present invention, the “C 3 -C 6 cycloalkyl group” is a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, or a cyclohexyl group, and preferably a cyclopropyl group.
本発明において、好適な一般式(I)は、一般式(Ia)である。
In the present invention, preferred general formula (I) is general formula (Ia).
本発明において、好適なR1は、水素原子、塩素原子、メチル基、エチル基又はシクロプロピル基である。
In the present invention, preferred R 1 is a hydrogen atom, a chlorine atom, a methyl group, an ethyl group or a cyclopropyl group.
本発明において、好適なR2は、メチル基である。
In the present invention, preferred R 2 is a methyl group.
本発明において、好適なnは、0である。
In the present invention, n is preferably 0.
本発明の一般式(I)で表される化合物又はその薬理上許容される塩は、全ての異性体(ジアステレオ異性体、光学異性体、回転異性体等)を有する。
The compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof has all isomers (diastereoisomers, optical isomers, rotational isomers, etc.).
本発明の一般式(I)で表される化合物又はその薬理上許容される塩は、その分子内に不斉炭素原子が存在するので、種々の異性体を有する。本発明の化合物においては、これらの異性体およびこれらの異性体の混合物がすべて単一の式、即ち一般式(I)で示されている。従って、本発明はこれらの異性体およびこれらの異性体の任意の割合の混合物をもすべて含むものである。
The compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof has various isomers because an asymmetric carbon atom exists in the molecule. In the compounds of the present invention, these isomers and mixtures of these isomers are all represented by a single formula, that is, the general formula (I). Therefore, the present invention includes all of these isomers and a mixture of these isomers in an arbitrary ratio.
上記のような立体異性体は、光学活性な原料化合物を用いるか、又は不斉合成若しくは不斉誘導の手法を用いて本発明に係る化合物を合成するか、或いは合成した本発明に係る化合物を所望により通常の光学分割法又は分離法を用いて単離することにより得ることができる。
For the stereoisomer as described above, an optically active raw material compound is used, or a compound according to the present invention is synthesized using an asymmetric synthesis or asymmetric induction method, or a synthesized compound according to the present invention is synthesized. If desired, it can be obtained by isolation using a conventional optical resolution method or separation method.
一般式(Ia)で表される化合物又はその薬理上許容される塩は、一般式(Ib)で表される化合物又はその薬理上許容される塩より好適である。
The compound represented by the general formula (Ia) or a pharmacologically acceptable salt thereof is more preferable than the compound represented by the general formula (Ib) or a pharmacologically acceptable salt thereof.

本発明の化合物は、このような化合物を構成する原子の1以上に、原子同位体の非天然割合も含有し得る。原子同位体としては、例えば、重水素(2H)、トリチウム(3H)、ヨウ素-125(125I)又は炭素-14(14C)などが挙げられる。また、前記化合物は、例えば、トリチウム(3H)、ヨウ素-125(125I)又は炭素-14(14C)などの放射性同位体で放射性標識され得る。放射性標識された化合物は、治療又は予防剤、研究試薬、例えば、アッセイ試薬、及び診断剤、例えば、インビボ画像診断剤として有用である。本発明の化合物の全ての同位体変異種は、放射性であると否とを問わず、本発明の範囲に包含されるものとする。
The compounds of the present invention may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds. Examples of the atomic isotope include deuterium ( 2 H), tritium ( 3 H), iodine-125 ( 125 I), carbon-14 ( 14 C), and the like. The compound can also be radiolabeled with a radioisotope such as, for example, tritium ( 3 H), iodine-125 ( 125 I), or carbon-14 ( 14 C). Radiolabeled compounds are useful as therapeutic or prophylactic agents, research reagents such as assay reagents, and diagnostic agents such as in vivo diagnostic imaging agents. All isotope variants of the compounds of the present invention, whether radioactive or not, are intended to be included within the scope of the present invention.
「その薬理上許容される塩」とは、著しい毒性を有さず、医薬として使用され得る塩をいう。本発明の一般式(I)で表される化合物は、塩基性の基を有する場合には酸と反応させることにより、又、酸性の基を有する場合には塩基と反応させることにより、塩にすることができる。
“The pharmacologically acceptable salt” refers to a salt that has no significant toxicity and can be used as a medicine. The compound represented by the general formula (I) of the present invention can be converted into a salt by reacting with an acid when it has a basic group, or by reacting with a base when it has an acidic group. can do.
塩基性基に基づく塩としては、例えば、フッ化水素酸塩、塩酸塩、臭化水素酸塩、ヨウ化水素酸塩のようなハロゲン化水素酸塩、硝酸塩、過塩素酸塩、硫酸塩、燐酸塩等の無機酸塩;メタンスルホン酸塩、トリフルオロメタンスルホン酸塩、エタンスルホン酸塩のようなアルキルスルホン酸塩、ベンゼンスルホン酸塩、p-トルエンスルホン酸塩のようなアリ-ルスルホン酸塩、酢酸塩、りんご酸塩、フマ-ル酸塩、コハク酸塩、クエン酸塩、アスコルビン酸塩、酒石酸塩、シュウ酸塩、マレイン酸塩等の有機酸塩;及び、グリシン塩、リジン塩、アルギニン塩、オルニチン塩、グルタミン酸塩、アスパラギン酸塩のようなアミノ酸塩を挙げることができる。
Examples of the salt based on the basic group include hydrohalides such as hydrofluoride, hydrochloride, hydrobromide, and hydroiodide, nitrate, perchlorate, sulfate, Inorganic acid salts such as phosphates; alkyl sulfonates such as methanesulfonate, trifluoromethanesulfonate, and ethanesulfonate; arylsulfonates such as benzenesulfonate and p-toluenesulfonate Organic acids such as acetate, malate, fumarate, succinate, citrate, ascorbate, tartrate, oxalate, maleate; and glycine salt, lysine salt, Examples thereof include amino acid salts such as arginine salt, ornithine salt, glutamate salt and aspartate salt.
一方、酸性基に基づく塩としては、例えば、ナトリウム塩、カリウム塩、リチウム塩のようなアルカリ金属塩、カルシウム塩、マグネシウム塩のようなアルカリ土類金属塩、アルミニウム塩、鉄塩等の金属塩;アンモニウム塩のような無機塩、t-オクチルアミン塩、ジベンジルアミン塩、モルホリン塩、グルコサミン塩、フェニルグリシンアルキルエステル塩、エチレンジアミン塩、N-メチルグルカミン塩、グアニジン塩、ジエチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、N,N’-ジベンジルエチレンジアミン塩、クロロプロカイン塩、プロカイン塩、ジエタノールアミン塩、N-ベンジルフェネチルアミン塩、ピペラジン塩、テトラメチルアンモニウム塩、トリス(ヒドロキシメチル)アミノメタン塩のような有機塩等のアミン塩;及び、グリシン塩、リジン塩、アルギニン塩、オルニチン塩、グルタミン酸塩、アスパラギン酸塩のようなアミノ酸塩を挙げることができる。
On the other hand, examples of the salt based on the acidic group include alkali metal salts such as sodium salt, potassium salt and lithium salt, alkaline earth metal salts such as calcium salt and magnesium salt, metal salts such as aluminum salt and iron salt. Inorganic salts such as ammonium salts, t-octylamine salts, dibenzylamine salts, morpholine salts, glucosamine salts, phenylglycine alkyl ester salts, ethylenediamine salts, N-methylglucamine salts, guanidine salts, diethylamine salts, triethylamine salts , Dicyclohexylamine salt, N, N′-dibenzylethylenediamine salt, chloroprocaine salt, procaine salt, diethanolamine salt, N-benzylphenethylamine salt, piperazine salt, tetramethylammonium salt, tris (hydroxymethyl) aminomethane salt Amine salts such as organic salts; and include glycine salts, lysine salts, arginine salts, ornithine salts, glutamic acid salts, amino acid salts such as aspartate.
本発明の一般式(I)で表される化合物又はその薬理上許容される塩は、大気中に放置したり、又は、再結晶したりすることにより、水分子を取り込んで、水和物となる場合があり、そのような水和物も本発明の塩に包含される。
The compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof is taken in the air or recrystallized to take in water molecules, Such hydrates are also encompassed by the salts of the present invention.
本発明の一般式(I)で表される化合物又はその薬理上許容される塩は、他のある種の溶媒を吸収し、溶媒和物となる場合があり、そのような溶媒和物も本発明の塩に包含される。
The compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof may absorb a certain other solvent and become a solvate, and such a solvate is also present. Included in the salts of the invention.
本発明の一般式(I)で表される化合物又はその薬理上許容される塩は、優れたDGAT阻害作用及び摂食抑制作用を有しており、温血動物(好ましくは哺乳類動物であり、ヒトを含む)における下記の疾患:肥満、肥満症、高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、虚血性心疾患及び過食症からなる群から選ばれる疾患、又は肥満に起因する高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、高血圧症、脳血管障害、冠動脈疾患、脂肪肝、呼吸異常、腰痛、変形性膝関節症、痛風、及び胆石症からなる群から選ばれる疾患の予防及び/又は治療のための医薬として有用である。また、本発明により提供される一般式(I)で表される新規な化合物またはその薬理上許容される塩は、優れたDGAT阻害作用を有しており、温血動物(好ましくは哺乳類動物であり、ヒトを含む)における上記の疾患の予防及び/又は治療のための医薬の有効成分として有用である。好適には、上記の疾患の治療のための医薬として用いることができる。
The compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof has an excellent DGAT inhibitory action and feeding inhibitory action, and is a warm-blooded animal (preferably a mammal, Diseases (including humans): obesity, obesity, hyperlipidemia, hypertriglycerideemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, Diabetic nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis , A disease selected from the group consisting of ischemic heart disease and bulimia, or hyperlipidemia, hypertriglycerideemia, lipid metabolism disorder, insulin resistance syndrome, impaired glucose tolerance, sugar caused by obesity Urinary disease, diabetic complications (including diabetic peripheral neuropathy, diabetic nephropathy, diabetic retinopathy, diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis Disease selected from the group consisting of infectious disease, atherosclerosis, diabetic arteriosclerosis, hypertension, cerebrovascular disorder, coronary artery disease, fatty liver, respiratory abnormalities, low back pain, knee osteoarthritis, gout, and cholelithiasis It is useful as a medicament for the prevention and / or treatment. Further, the novel compound represented by the general formula (I) provided by the present invention or a pharmacologically acceptable salt thereof has an excellent DGAT inhibitory action, and is a warm-blooded animal (preferably a mammalian animal). And is useful as an active ingredient of a medicament for the prevention and / or treatment of the above-mentioned diseases (including humans). Suitably, it can be used as a medicament for the treatment of the above-mentioned diseases.
本発明の一般式(I)で表される化合物は、以下に記載するA法及びB法に従って製造することができる。
The compound represented by the general formula (I) of the present invention can be produced according to Method A and Method B described below.
下記A法及びB法の各工程の反応において使用される溶媒は、反応を阻害せず、出発原料をある程度溶解するものであれば特に限定はなく、例えば、下記溶媒群より選択される。溶媒群は、ペンタン、ヘキサン、オクタン、石油エーテル、リグロイン、シクロヘキサンのような炭化水素類;ホルムアミド、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチル-2-ピロリドン、N-メチル-2-ピロリジノン、ヘキサメチルリン酸トリアミドのようなアミド類;ジエチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、ジオキサン、ジメトキシエタン、ジエチレングリコールジメチルエーテル、シクロペンチルメチルエーテルのようなエーテル類;メタノール、エタノール、n-プロパノール、i-プロパノール、n-ブタノール、2-ブタノール、2-メチル-1-プロパノール、t-ブタノール、イソアミルアルコール、ジエチレングリコール、グリセリン、オクタノール、シクロヘキサノール、メチルセロソルブのようなアルコール類;ジメチルスルホキシドのようなスルホキシド類;スルホランのようなスルホン類;アセトニトリル、プロピオニトリル、ブチロニトリル、イソブチロニトリルのようなニトリル類;蟻酸エチル、酢酸エチル、酢酸プロピル、酢酸ブチル、炭酸ジエチルのようなエステル類;アセトン、メチルエチルケトン、4-メチル-2-ペンタノン、メチルイソブチルケトン、イソホロン、シクロヘキサノンのようなケトン類;ニトロエタン、ニトロベンゼンのようなニトロ化合物類;ジクロロメタン、1,2-ジクロロエタン、クロロベンゼン、ジクロロベンゼン、クロロホルム、四塩化炭素のようなハロゲン化炭化水素類;ベンゼン、トルエン、キシレンのような芳香族炭化水素類;N-メチルモルホリン、トリエチルアミン、トリプロピルアミン、トリブチルアミン、ジイソプロピルエチルアミン、N-メチルピペリジン、ピリジン、2,6-ルチジン、4-ピロリジノピリジン、ピコリン、4-(N,N-ジメチルアミノ)ピリジン、2,6-ジ(t-ブチル)-4-メチルピリジン、キノリン、N,N-ジメチルアニリン、N,N-ジエチルアニリン、1,5-ジアザビシクロ[4.3.0]ノナ-5-エン(DBN)、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン(DBU)、ピペリジンのようなアミン類;水;及び、これらの混合溶媒からなる。
The solvent used in the reaction of each step of the following method A and method B is not particularly limited as long as it does not inhibit the reaction and dissolves the starting materials to some extent, and is selected from the following solvent group, for example. Solvent groups include hydrocarbons such as pentane, hexane, octane, petroleum ether, ligroin, cyclohexane; formamide, N, N-dimethylformamide, N, N-dimethylacetamide, N-methyl-2-pyrrolidone, N-methyl Amides such as -2-pyrrolidinone and hexamethylphosphoric triamide; ethers such as diethyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, diethylene glycol dimethyl ether and cyclopentyl methyl ether; methanol, ethanol, n-propanol, i -Propanol, n-butanol, 2-butanol, 2-methyl-1-propanol, t-butanol, isoamyl alcohol, diethylene glycol, glycerin, octanol, cyclohexanol, Alcohols such as methyl cellosolve; sulfoxides such as dimethyl sulfoxide; sulfones such as sulfolane; nitriles such as acetonitrile, propionitrile, butyronitrile, isobutyronitrile; ethyl formate, ethyl acetate, propyl acetate, Esters such as butyl acetate and diethyl carbonate; ketones such as acetone, methyl ethyl ketone, 4-methyl-2-pentanone, methyl isobutyl ketone, isophorone and cyclohexanone; nitro compounds such as nitroethane and nitrobenzene; dichloromethane, 1, Halogenated hydrocarbons such as 2-dichloroethane, chlorobenzene, dichlorobenzene, chloroform, and carbon tetrachloride; aromatic hydrocarbons such as benzene, toluene, and xylene; N-methylmorpholine, tri Tylamine, tripropylamine, tributylamine, diisopropylethylamine, N-methylpiperidine, pyridine, 2,6-lutidine, 4-pyrrolidinopyridine, picoline, 4- (N, N-dimethylamino) pyridine, 2,6-di (T-butyl) -4-methylpyridine, quinoline, N, N-dimethylaniline, N, N-diethylaniline, 1,5-diazabicyclo [4.3.0] non-5-ene (DBN), 1, 4-diazabicyclo [2.2.2] octane (DABCO), 1,8-diazabicyclo [5.4.0] undec-7-ene (DBU), amines such as piperidine; water; and mixtures thereof It consists of a solvent.
下記A法及びB法の各工程の反応において使用される塩基は、例えば、炭酸ナトリウム、炭酸カリウム、炭酸リチウム、炭酸セシウムのようなアルカリ金属炭酸塩類;炭酸水素ナトリウム、炭酸水素カリウム、炭酸水素リチウムのようなアルカリ金属炭酸水素塩類;酢酸ナトリウム、酢酸カリウム、酢酸リチウム、酢酸セシウムのようなアルカリ金属酢酸塩類;水素化リチウム、水素化ナトリウム、水素化カリウムのようなアルカリ金属水素化物類;水酸化ナトリウム、水酸化カリウム、水酸化リチウムのようなアルカリ金属水酸化物類;弗化ナトリウム、弗化カリウムのようなアルカリ金属弗化物類;ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム-t-ブトキシド、カリウム-t-ブトキシドのようなアルカリ金属アルコキシド類;ナトリウムトリメチルシロキシド、カリウムトリメチルシロキシド、リチウムトリメチルシロキシドのようなアルカリ金属トリアルキルシロキシド類;N-メチルモルホリン、トリエチルアミン、トリプロピルアミン、トリブチルアミン、ジイソプロピルエチルアミン、ジシクロヘキシルアミン、N-メチルピペリジン、ピリジン、2,6-ルチジン、コリジン、4-ピロリジノピリジン、ピコリン、4-(N,N-ジメチルアミノ)ピリジン、2,6-ジ(t-ブチル)-4-メチルピリジン、キノリン、N,N-ジメチルアニリン、N,N-ジエチルアニリン、1,5-ジアザビシクロ[4.3.0]ノナ-5-エン(DBN)、1,4-ジアザビシクロ[2.2.2]オクタン(DABCO)、1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン(DBU)のような有機塩基類;n-ブチルリチウム、リチウムジイソプロピルアミド、リチウム ビス(トリメチルシリル)アミドのような有機金属塩基類;又は、プロリンのようなアミノ酸である。
The base used in the reaction of each step of the following method A and B is, for example, alkali metal carbonates such as sodium carbonate, potassium carbonate, lithium carbonate, cesium carbonate; sodium hydrogen carbonate, potassium hydrogen carbonate, lithium hydrogen carbonate Alkali metal bicarbonates such as sodium acetate, potassium acetate, lithium acetate, alkali metal acetates such as cesium acetate; alkali metal hydrides such as lithium hydride, sodium hydride, potassium hydride; Alkali metal hydroxides such as sodium, potassium hydroxide and lithium hydroxide; alkali metal fluorides such as sodium fluoride and potassium fluoride; sodium methoxide, sodium ethoxide, sodium t-butoxide, potassium Alkali metal alcohols such as t-butoxide Cids; alkali metal trialkylsiloxides such as sodium trimethylsiloxide, potassium trimethylsiloxide, lithium trimethylsiloxide; N-methylmorpholine, triethylamine, tripropylamine, tributylamine, diisopropylethylamine, dicyclohexylamine, N- Methylpiperidine, pyridine, 2,6-lutidine, collidine, 4-pyrrolidinopyridine, picoline, 4- (N, N-dimethylamino) pyridine, 2,6-di (t-butyl) -4-methylpyridine, quinoline N, N-dimethylaniline, N, N-diethylaniline, 1,5-diazabicyclo [4.3.0] non-5-ene (DBN), 1,4-diazabicyclo [2.2.2] octane ( DABCO), 1,8-diazabicyclo [5 4.0] organic bases such as undec-7-ene (DBU); organometallic bases such as n-butyllithium, lithium diisopropylamide, lithium bis (trimethylsilyl) amide; or amino acids such as proline is there.
下記A法及びB法の各工程の反応において、反応温度は、溶媒、出発原料、試薬等により異なり、反応時間は、溶媒、出発原料、試薬、反応温度等により異なる。
In the reaction of each step of Method A and Method B below, the reaction temperature varies depending on the solvent, starting material, reagent, etc., and the reaction time varies depending on the solvent, starting material, reagent, reaction temperature, and the like.
下記A法及びB法の各工程の反応において、反応終了後、各目的化合物は常法に従って、反応混合物から採取される。例えば、反応混合物を適宜中和し、又、不溶物が存在する場合には濾過により除去した後、水と酢酸エチルのような混和しない有機溶媒を加え、目的化合物を含む有機層を分離し、水等で洗浄後、無水硫酸マグネシウム、無水硫酸ナトリウム等で乾燥、ろ過後、溶剤を留去することによって得られる。得られた目的化合物は必要ならば、常法、例えば再結晶、再沈殿等の通常、有機化合物の分離精製に慣用されている方法を適宜組合せ、クロマトグラフィーを応用し、適切な溶離剤で溶出することによって分離、精製することができる。溶媒に不溶の目的化合物では、得られた固体の粗生成物を溶媒で洗浄して、精製することができる。また、各工程の目的化合物は精製することなくそのまま次の反応に使用することもできる。
In the reaction of each step of the following method A and method B, after completion of the reaction, each target compound is collected from the reaction mixture according to a conventional method. For example, neutralize the reaction mixture as appropriate, or remove insoluble matter by filtration, add water and an immiscible organic solvent such as ethyl acetate, and separate the organic layer containing the target compound, It can be obtained by washing with water, drying over anhydrous magnesium sulfate, anhydrous sodium sulfate, etc., filtering, and then distilling off the solvent. If necessary, the obtained target compound is eluted with an appropriate eluent by applying a conventional method, for example, recrystallization, reprecipitation, etc., usually using methods commonly used for separation and purification of organic compounds, applying chromatography, and the like. Can be separated and purified. For a target compound insoluble in a solvent, the obtained solid crude product can be purified by washing with a solvent. In addition, the target compound in each step can be directly used in the next reaction without purification.
下記A法及びB法の各工程の反応において、R1、R2及びnは、前述したものと同意義を示す。X1及びX2は、ハロゲン原子(好適には、臭素原子又はヨウ素原子であり、より好適には、臭素原子である。)を示し、Y1及びY2は、有機合成化学の分野で使用されるカルボキシ基の保護基(好適には、C1-C6アルキル基又はアラルキル基である。Y1においては、より好適には、メチル基であり、Y2においては、より好適には、エチル基である。)を示す。R2aは、R2の基に含まれるヒドロキシ基が、保護されてもよいヒドロキシ基である他、R2の基の定義における基と同様の基を示す。
In the reaction of each step of the following method A and method B, R 1 , R 2 and n have the same meaning as described above. X 1 and X 2 each represent a halogen atom (preferably a bromine atom or an iodine atom, and more preferably a bromine atom), and Y 1 and Y 2 are used in the field of synthetic organic chemistry. A protecting group for carboxy group (preferably a C 1 -C 6 alkyl group or an aralkyl group. Y 1 is more preferably a methyl group, and Y 2 is more preferably An ethyl group). R 2a is a hydroxy group contained in the group R 2 is other is protected may be hydroxy groups include the same groups as in the definition of group R 2.
A法は、一般式(I)で表される化合物を製造する方法である。
(A法) Method A is a method for producing a compound represented by the general formula (I).
(Method A)
(A法) Method A is a method for producing a compound represented by the general formula (I).
(Method A)

A-I工程
本工程は、溶媒中、光延試薬の存在下、一般式(II)で表される化合物を、一般式(III)で表される化合物と反応させることにより、一般式(IV)で表される化合物を製造する工程である。 Step AI This step comprises reacting a compound represented by the general formula (II) with a compound represented by the general formula (III) in a solvent in the presence of a Mitsunobu reagent. It is a process of manufacturing the compound represented by these.
本工程は、溶媒中、光延試薬の存在下、一般式(II)で表される化合物を、一般式(III)で表される化合物と反応させることにより、一般式(IV)で表される化合物を製造する工程である。 Step AI This step comprises reacting a compound represented by the general formula (II) with a compound represented by the general formula (III) in a solvent in the presence of a Mitsunobu reagent. It is a process of manufacturing the compound represented by these.
本工程において使用される一般式(II)で表される化合物及び一般式(III)で表される化合物は、公知化合物であるか、或いは公知化合物を出発原料に公知の方法又はそれに類似した方法に従って容易に製造される。
The compound represented by the general formula (II) and the compound represented by the general formula (III) used in this step are known compounds, or a known method using a known compound as a starting material or a method similar thereto. Easily manufactured according to.
本工程において使用される溶媒は、好適には、芳香族炭化水素類又はエーテル類であり、より好適には、トルエン又はテトラヒドロフランである。
The solvent used in this step is preferably aromatic hydrocarbons or ethers, and more preferably toluene or tetrahydrofuran.
本工程において使用される光延試薬は、好適には、アゾジカルボン酸ジエステル又は(シアノメチレン)ホスホラン試薬であり、より好適には、アゾジカルボン酸ジエチル(DEAD)、アゾジカルボン酸ジイソプロピル(DIAD)又は(シアノメチレン)トリブチルホスホラン(CMBP)である。
The Mitsunobu reagent used in this step is preferably an azodicarboxylic acid diester or (cyanomethylene) phosphorane reagent, more preferably diethyl azodicarboxylate (DEAD), diisopropyl azodicarboxylate (DIAD) or ( Cyanomethylene) tributylphosphorane (CMBP).
本工程における反応温度は、通常、-20℃乃至180℃であり、好適には、0℃乃至120℃である。
The reaction temperature in this step is usually −20 ° C. to 180 ° C., preferably 0 ° C. to 120 ° C.
本工程における反応時間は、通常、0.5時間乃至72時間であり、好適には、2時間乃至24時間である。
The reaction time in this step is usually 0.5 hours to 72 hours, preferably 2 hours to 24 hours.
A-II工程
本工程は、溶媒中、パラジウム触媒及び塩基の存在下、一般式(IV)で表される化合物を、一般式(V)で表される化合物と反応させることにより、一般式(VI)で表される化合物を製造する工程である。 Step A-II In this step, the compound represented by the general formula (IV) is reacted with the compound represented by the general formula (V) in the presence of a palladium catalyst and a base in a solvent, to thereby obtain a compound represented by the general formula (V). This is a process for producing a compound represented by VI).
本工程は、溶媒中、パラジウム触媒及び塩基の存在下、一般式(IV)で表される化合物を、一般式(V)で表される化合物と反応させることにより、一般式(VI)で表される化合物を製造する工程である。 Step A-II In this step, the compound represented by the general formula (IV) is reacted with the compound represented by the general formula (V) in the presence of a palladium catalyst and a base in a solvent, to thereby obtain a compound represented by the general formula (V). This is a process for producing a compound represented by VI).
本工程において使用される一般式(V)で表される化合物は、公知化合物であるか、或いは公知化合物を出発原料に公知の方法又はそれに類似した方法に従って容易に製造される。
The compound represented by the general formula (V) used in this step is a known compound, or can be easily produced according to a known method or a similar method using the known compound as a starting material.
本工程において使用される溶媒は、好適には、エーテル類又はアミド類と水の混合溶媒であり、より好適には、テトラヒドロフラン、ジオキサン又はN,N-ジメチルアセトアミドと水の混合溶媒である。
The solvent used in this step is preferably a mixed solvent of ethers or amides and water, more preferably a mixed solvent of tetrahydrofuran, dioxane or N, N-dimethylacetamide and water.
本工程において使用されるパラジウム触媒は、例えば、テトラキス(トリフェニルホスフィン)パラジウム(0)、パラジウム-活性炭素、酢酸パラジウム(II)、トリフルオロ酢酸パラジウム(II)、パラジウム黒、臭化パラジウム(II)、塩化パラジウム(II)、沃化パラジウム(II)、シアン化パラジウム(II)、硝酸パラジウム(II)、酸化パラジウム(II)、硫酸パラジウム(II)、ジクロロビス(アセトニトリル)パラジウム(II)、ジクロロビス(ベンゾニトリル)パラジウム(II)、ジクロロ(1,5-シクロオクタジエン)パラジウム(II)、アセチルアセトンパラジウム(II)、硫化パラジウム(II)、[1,1′-ビス(ジフェニルホスフィノ)フェロセン]パラジウム(II)ジクロリド、トリス(ジベンジリデンアセトン)ジパラジウム(0)、テトラキス(アセトニトリル)パラジウム(II)テトラフルオロボレート又は塩化アリールパラジウムダイマーのような2価のパラジウム触媒又は0価のパラジウム触媒であり、好適には、0価のパラジウム触媒であり、より好適には、テトラキス(トリフェニルホスフィン)パラジウム(0)である。
The palladium catalyst used in this step is, for example, tetrakis (triphenylphosphine) palladium (0), palladium-activated carbon, palladium acetate (II), palladium trifluoroacetate (II), palladium black, palladium bromide (II ), Palladium (II) chloride, palladium (II) iodide, palladium (II) cyanide, palladium (II) nitrate, palladium (II) oxide, palladium (II) sulfate, dichlorobis (acetonitrile) palladium (II), dichlorobis (Benzonitrile) palladium (II), dichloro (1,5-cyclooctadiene) palladium (II), acetylacetone palladium (II), palladium sulfide (II), [1,1'-bis (diphenylphosphino) ferrocene] Palladium (II) dichroic A divalent palladium catalyst such as Lido, tris (dibenzylideneacetone) dipalladium (0), tetrakis (acetonitrile) palladium (II) tetrafluoroborate or arylpalladium chloride dimer, preferably a zerovalent palladium catalyst, , A zero-valent palladium catalyst, and more preferably tetrakis (triphenylphosphine) palladium (0).
本工程において使用される塩基は、好適には、アルカリ金属炭酸塩類であり、より好適には、炭酸カリウムである。
The base used in this step is preferably an alkali metal carbonate, and more preferably potassium carbonate.
本工程における反応温度は、通常、20℃乃至180℃であり、好適には、60℃乃至120℃である。
The reaction temperature in this step is usually 20 ° C. to 180 ° C., preferably 60 ° C. to 120 ° C.
本工程における反応時間は、通常、0.5時間乃至72時間であり、好適には、2時間乃至24時間である。
The reaction time in this step is usually 0.5 hours to 72 hours, preferably 2 hours to 24 hours.
A-III工程
本工程は、溶媒中、縮合剤の存在下、塩基の存在下又は非存在下(好適には、存在下)、一般式(VI)で表される化合物を、一般式(VII)で表される化合物と反応させることにより、一般式(VIII)で表される化合物を製造する工程である。 Step A-III In this step, the compound represented by the general formula (VI) is converted into the general formula (VII) in a solvent in the presence of a condensing agent, in the presence or absence of a base (preferably in the presence). ) To produce a compound represented by the general formula (VIII).
本工程は、溶媒中、縮合剤の存在下、塩基の存在下又は非存在下(好適には、存在下)、一般式(VI)で表される化合物を、一般式(VII)で表される化合物と反応させることにより、一般式(VIII)で表される化合物を製造する工程である。 Step A-III In this step, the compound represented by the general formula (VI) is converted into the general formula (VII) in a solvent in the presence of a condensing agent, in the presence or absence of a base (preferably in the presence). ) To produce a compound represented by the general formula (VIII).
本工程において使用される溶媒は、好適には、アミド類であり、より好適には、N,N-ジメチルアセトアミドである。
The solvent used in this step is preferably an amide, and more preferably N, N-dimethylacetamide.
本工程において使用される縮合剤は、例えば、アゾジカルボン酸ジエチルエステル-トリフェニルホスフィンのようなアゾジカルボン酸ジ低級アルキルエステル-トリフェニルホスフィン類;N,N’-ジシクロヘキシルカルボジイミド(DCC)、1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド(EDCI)のようなカルボジイミド誘導体;2-クロル-1-メチルピリジニウムヨージドのような2-ハロ-1-低級アルキルピリジニウムハライド類;ジフェニルホスホリルアジド(DPPA)のようなジアリールホスホリルアジド類;ジエチルホスホリルクロリドのようなホスホリルクロリド類;N,N’-カルボジイミダゾール(CDI)のようなイミダゾール誘導体;ベンゾトリアゾール-1-イルオキシ-トリス(ジメチルアミノ)ホスホニウム ヘキサフルオロホスフェート(BOP)、O-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロホスフェート(HATU)、(1H-ベンゾトリアゾール-1-イルオキシ)トリピロリジノホスホニウムヘキサフルオロホスフェート(PyBOP)のようなベンゾトリアゾール誘導体であり、好適には、ベンゾトリアゾール誘導体であり、より好適には、BOPである。
The condensing agent used in this step is, for example, azodicarboxylic acid dilower alkyl ester-triphenylphosphine such as azodicarboxylic acid diethyl ester-triphenylphosphine; N, N′-dicyclohexylcarbodiimide (DCC), 1- Carbodiimide derivatives such as ethyl-3- (3-dimethylaminopropyl) carbodiimide (EDCI); 2-halo-1-lower alkylpyridinium halides such as 2-chloro-1-methylpyridinium iodide; diphenylphosphoryl azide ( Diarylphosphoryl azides such as DPPA; phosphoryl chlorides such as diethyl phosphoryl chloride; imidazole derivatives such as N, N′-carbodiimidazole (CDI); benzotriazol-1-yloxy-to (Dimethylamino) phosphonium hexafluorophosphate (BOP), O- (7-azabenzotriazol-1-yl) -N, N, N ′, N′-tetramethyluronium hexafluorophosphate (HATU), (1H -Benzotriazol-1-yloxy) tripyrrolidinophosphonium hexafluorophosphate (PyBOP), a benzotriazole derivative, preferably a benzotriazole derivative, more preferably BOP.
本工程において使用される塩基は、好適には、有機塩基類であり、より好適には、トリエチルアミンである。
The base used in this step is preferably an organic base, and more preferably triethylamine.
本工程における反応温度は、通常、-20℃乃至160℃で行われ、好適には、0℃乃至100℃である。
The reaction temperature in this step is usually -20 ° C to 160 ° C, preferably 0 ° C to 100 ° C.
本工程における反応時間は、通常、0.1時間乃至120時間であり、好適には、1時間乃至24時間である。
The reaction time in this step is usually 0.1 hour to 120 hours, preferably 1 hour to 24 hours.
A-IV工程
本工程は、既知の方法(例えば、”Protective Groups in Organic Synthesis” (Theodora W. Greene、Peter G. M.Wuts著、 1999年、Wiley-Interscience Publication発行)等に記載の方法)に準じて行われる。例えば、Y1がC1-C6アルキル基の場合を下記に示す。 Step A-IV This step is based on a known method (for example, “Protective Groups in Organic Synthesis” (the method described in Theodora W. Greene, Peter GMWuts, 1999, published by Wiley-Interscience Publication), etc.). Done. For example, the case where Y 1 is a C 1 -C 6 alkyl group is shown below.
本工程は、既知の方法(例えば、”Protective Groups in Organic Synthesis” (Theodora W. Greene、Peter G. M.Wuts著、 1999年、Wiley-Interscience Publication発行)等に記載の方法)に準じて行われる。例えば、Y1がC1-C6アルキル基の場合を下記に示す。 Step A-IV This step is based on a known method (for example, “Protective Groups in Organic Synthesis” (the method described in Theodora W. Greene, Peter GMWuts, 1999, published by Wiley-Interscience Publication), etc.). Done. For example, the case where Y 1 is a C 1 -C 6 alkyl group is shown below.
本工程は、溶媒中、一般式(VIII)で表される化合物を、塩基と反応させた後、所望によりR2aにおけるヒドロキシ基の保護基を除去することにより、一般式(I)で表される化合物を製造する工程である。
This step is represented by the general formula (I) by reacting the compound represented by the general formula (VIII) with a base in a solvent and then removing the hydroxy protecting group in R 2a as required. This is a process for producing a compound.
本工程において使用される溶媒は、好適には、エーテル類又はアルコール類であり、より好適には、テトラヒドロフラン、ジオキサン又はメタノールである。
The solvent used in this step is preferably an ether or an alcohol, and more preferably tetrahydrofuran, dioxane or methanol.
本工程において使用される塩基は、好適には、四級アンモニウム塩類であり、より好適には、水酸化テトラブチルアンモニウムである。
The base used in this step is preferably a quaternary ammonium salt, and more preferably tetrabutylammonium hydroxide.
本工程における反応温度は、通常、0℃乃至150℃であり、好適には20℃乃至100℃である。
The reaction temperature in this step is usually 0 ° C. to 150 ° C., preferably 20 ° C. to 100 ° C.
本反応における反応時間は、通常、0.5時間乃至24時間であり、好適には1時間乃至10時間である。
The reaction time in this reaction is usually 0.5 to 24 hours, preferably 1 to 10 hours.
B法は、前記A法のA-III工程で用いる一般式(VII)で表される化合物を製造する方法である。
(B法) Method B is a method for producing a compound represented by the general formula (VII) used in Step A-III of Method A.
(Method B)
(B法) Method B is a method for producing a compound represented by the general formula (VII) used in Step A-III of Method A.
(Method B)

B-I工程
本工程は、溶媒中、銅触媒、塩基及びリガンドの存在下、一般式(IX)で表される化合物を、一般式(X)で表される化合物と反応させることにより、一般式(XI)で表される化合物を製造する工程である。 Step BI This step is performed by reacting a compound represented by the general formula (IX) with a compound represented by the general formula (X) in a solvent in the presence of a copper catalyst, a base and a ligand. In this step, the compound represented by formula (XI) is produced.
本工程は、溶媒中、銅触媒、塩基及びリガンドの存在下、一般式(IX)で表される化合物を、一般式(X)で表される化合物と反応させることにより、一般式(XI)で表される化合物を製造する工程である。 Step BI This step is performed by reacting a compound represented by the general formula (IX) with a compound represented by the general formula (X) in a solvent in the presence of a copper catalyst, a base and a ligand. In this step, the compound represented by formula (XI) is produced.
本工程において使用される一般式(IX)で表される化合物及び一般式(X)で表される化合物は、公知化合物であるか、或いは公知化合物を出発原料に公知の方法又はそれに類似した方法に従って容易に製造される。
The compound represented by the general formula (IX) and the compound represented by the general formula (X) used in this step are known compounds, or a known method using a known compound as a starting material or a method similar thereto. Easily manufactured according to.
本工程において使用される溶媒は、好適には、芳香族炭化水素類であり、より好適には、トルエンである。
The solvent used in this step is preferably an aromatic hydrocarbon, and more preferably toluene.
本工程において使用される銅触媒は、0価の銅やその錯体;塩化銅(I)、臭化銅(I)、沃化銅(I)、トリフルオロメタンスルホン酸銅(I)のような1価の銅塩;又は、臭化銅(II)、酢酸銅(II)、硫酸銅(II)のような2価の銅塩であり、好適には、1価の銅塩であり、より好適には、沃化銅(I)である。
The copper catalyst used in this step is 0-valent copper or a complex thereof; 1 such as copper (I) chloride, copper (I) bromide, copper (I) iodide, copper (I) trifluoromethanesulfonate. Or a divalent copper salt such as copper (II) bromide, copper (II) acetate, or copper (II) sulfate, preferably a monovalent copper salt, more preferably Is copper iodide (I).
本工程において使用される塩基は、好適には、アルカリ金属炭酸塩類であり、より好適には、炭酸カリウムである。
The base used in this step is preferably an alkali metal carbonate, and more preferably potassium carbonate.
本工程において使用されるリガンドは、例えば、アミン化合物であり、好適には、ジアミン類であり、より好適には、1,2-ジ(メチルアミノ)シクロヘキサンである。
The ligand used in this step is, for example, an amine compound, preferably a diamine, and more preferably 1,2-di (methylamino) cyclohexane.
本工程における反応温度は、通常、0℃乃至200℃であり、好適には、80℃乃至130℃である。
The reaction temperature in this step is usually 0 ° C to 200 ° C, preferably 80 ° C to 130 ° C.
本工程における反応時間は、通常、0.5時間乃至96時間であり、好適には、2時間乃至48時間である。
The reaction time in this step is usually 0.5 hours to 96 hours, preferably 2 hours to 48 hours.
B-II工程
本工程は、既知の方法(例えば、”Protective Groups in Organic Synthesis” (Theodora W. Greene、Peter G. M.Wuts著、 1999年、Wiley-Interscience Publication発行)等に記載の方法)に準じて行われる。例えば、Y2がC1-C6アルキル基の場合を下記に示す。 Step B-II This step is based on a known method (for example, the method described in “Protective Groups in Organic Synthesis” (Theodora W. Greene, Peter GMWuts, 1999, published by Wiley-Interscience Publication), etc.) Done. For example, the case where Y 2 is a C 1 -C 6 alkyl group is shown below.
本工程は、既知の方法(例えば、”Protective Groups in Organic Synthesis” (Theodora W. Greene、Peter G. M.Wuts著、 1999年、Wiley-Interscience Publication発行)等に記載の方法)に準じて行われる。例えば、Y2がC1-C6アルキル基の場合を下記に示す。 Step B-II This step is based on a known method (for example, the method described in “Protective Groups in Organic Synthesis” (Theodora W. Greene, Peter GMWuts, 1999, published by Wiley-Interscience Publication), etc.) Done. For example, the case where Y 2 is a C 1 -C 6 alkyl group is shown below.
本工程は、溶媒中、一般式(XI)で表される化合物を、塩基と反応させることにより、一般式(VII)で表される化合物を製造する工程である。
This step is a step of producing the compound represented by the general formula (VII) by reacting the compound represented by the general formula (XI) with a base in a solvent.
本工程において使用される溶媒は、好適には、エーテル類、アルコール類、水又はこれらの混合溶媒であり、より好適には、テトラヒドロフラン、エタノール及び水の混合溶媒である。
The solvent used in this step is preferably ethers, alcohols, water or a mixed solvent thereof, more preferably a mixed solvent of tetrahydrofuran, ethanol and water.
本工程において使用される塩基は、好適には、アルカリ金属水酸化物類であり、より好適には、水酸化ナトリウムである。
The base used in this step is preferably an alkali metal hydroxide, and more preferably sodium hydroxide.
本工程における反応温度は、通常、0℃乃至150℃であり、好適には20℃乃至100℃である。
The reaction temperature in this step is usually 0 ° C. to 150 ° C., preferably 20 ° C. to 100 ° C.
本反応における反応時間は、通常、0.5時間乃至24時間であり、好適には1時間乃至10時間である。
The reaction time in this reaction is usually 0.5 to 24 hours, preferably 1 to 10 hours.
上記において、R2aの定義における「保護されてもよいヒドロキシ基」の保護基とは、加水素分解、加水分解、電気分解、光分解のような化学的方法により開裂し得る保護基をいい、有機合成化学で一般的に用いられる保護基を示す(例えば、T. W. Greeneら,Protective Groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, Inc. (1999年)参照)。
In the above, the protecting group of the “optionally protected hydroxy group” in the definition of R 2a refers to a protecting group that can be cleaved by a chemical method such as hydrogenolysis, hydrolysis, electrolysis, or photolysis, Protective groups commonly used in organic synthetic chemistry are shown (see, for example, TW Greene et al., Protective Groups in Organic Synthesis, 3rd Edition, John Wiley & Sons, Inc. (1999)).
上記において、R2aの定義における「保護されてもよいヒドロキシ基」の「保護基」は、有機合成化学の分野で使用されるヒドロキシ基の保護基であれば特に限定はされないが、例えば、ホルミル基、アセチル、プロピオニル、イソブチリル、ピバロイル、バレリルのようなアルキルカルボニル基、クロロアセチル、ジクロロアセチル、トリクロロアセチル、トリフルオロアセチルのようなハロゲン化アルキルカルボニル基、メトキシアセチルのようなアルコキシアルキルカルボニル基、アクリロイル、プロピオロイル、メタクリロイル、クロトノイル、イソクロトノイル、(E)-2-メチル-2-ブテノイルのような不飽和アルキルカルボニル基等の「アルキルカルボニル基」;ベンゾイル、α-ナフトイル、β-ナフトイルのようなアリールカルボニル基、2-ブロモベンゾイル、4-クロロベンゾイルのようなハロゲン化アリールカルボニル基、2,4,6-トリメチルベンゾイル、4-トルオイルのようなアルキル化アリ-ルカルボニル基、4-アニソイルのようなアルコキシ化アリールカルボニル基、4-ニトロベンゾイル、2-ニトロベンゾイルのようなニトロ化アリールカルボニル基、2-(メトキシカルボニル)ベンゾイルのようなアルコキシカルボニル化アリールカルボニル基、4-フェニルベンゾイルのようなアリール化アリールカルボニル基等の「アリールカルボニル基」;メトキシカルボニル、エトキシカルボニル、イソプロポキシカルボニル、t-ブトキシカルボニルのようなアルコキシカルボニル基、2,2,2-トリクロロエトキシカルボニル、2-トリメチルシリルエトキシカルボニルのようなハロゲン又はトリアルキルシリル基で置換されたアルコキシカルボニル基等の「アルコキシカルボニル基」;テトラヒドロピラン-2-イル、3-ブロモテトラヒドロピラン-2-イル、4-メトキシテトラヒドロピラン-4-イル、テトラヒドロチオピラン-2-イル、4-メトキシテトラヒドロチオピラン-4-イルのような「テトラヒドロピラニル又はテトラヒドロチオピラニル基」;テトラヒドロフラン-2-イル、テトラヒドロチオフラン-2-イルのような「テトラヒドロフラニル又はテトラヒドロチオフラニル基」;トリメチルシリル、トリエチルシリル、イソプロピルジメチルシリル、t-ブチルジメチルシリル、メチルジイソプロピルシリル、メチルジ-t-ブチルシリル、トリイソプロピルシリルのようなトリアルキルシリル基、ジフェニルメチルシリル、ジフェニルブチルシリル、ジフェニルイソプロピルシリル、フェニルジイソプロピルシリルのようなアルキルジアリールシリル又はジアルキルアリールシリル基等の「シリル基」;メトキシメチル、1,1-ジメチル-1-メトキシメチル、エトキシメチル、プロポキシメチル、イソプロポキシメチル、ブトキシメチル、t-ブトキシメチルのようなアルコキシメチル基、2-メトキシエトキシメチルのようなアルコキシアルコキシメチル基、2,2,2-トリクロロエトキシメチル、ビス(2-クロロエトキシ)メチルのようなハロゲン化アルコキシメチル等の「アルコキシメチル基」;1-エトキシエチル、1-(イソプロポキシ)エチルのようなアルコキシエチル基、2,2,2-トリクロロエチルのようなハロゲン化エチル基等の「置換エチル基」;ベンジル、α-ナフチルメチル、β-ナフチルメチル、ジフェニルメチル、トリフェニルメチル、α-ナフチルジフェニルメチル、9-アンスリルメチルのような1乃至3個のアリール基で置換されたアルキル基、4-メチルベンジル、2,4,6-トリメチルベンジル、3,4,5-トリメチルベンジル、4-メトキシベンジル、4-メトキシフェニルジフェニルメチル、2-ニトロベンジル、4-ニトロベンジル、4-クロロベンジル、4-ブロモベンジル、4-シアノベンジルのような(アルキル、アルコキシ、ニトロ、ハロゲン、シアノ基でアリール環が置換された1乃至3個のアリール基)で置換されたアルキル基等の「アラルキル基」;ビニルオキシカルボニル、アリルオキシカルボニルのような「アルケニルオキシカルボニル基」;ベンジルオキシカルボニル、4-メトキシベンジルオキシカルボニル、3,4-ジメトキシベンジルオキシカルボニル、2-ニトロベンジルオキシカルボニル、4-ニトロベンジルオキシカルボニルのような、1又は2個のアルコキシ又はニトロ基でアリール環が置換されていてもよい「アラルキルオキシカルボニル基」であり、好適には、アルキルカルボニル基、シリル基又はアラルキル基である。
In the above, the “protecting group” of the “hydroxy group that may be protected” in the definition of R 2a is not particularly limited as long as it is a protecting group for a hydroxy group used in the field of synthetic organic chemistry. Groups, alkylcarbonyl groups such as acetyl, propionyl, isobutyryl, pivaloyl, valeryl, halogenated alkylcarbonyl groups such as chloroacetyl, dichloroacetyl, trichloroacetyl, trifluoroacetyl, alkoxyalkylcarbonyl groups such as methoxyacetyl, acryloyl An “alkylcarbonyl group” such as an unsaturated alkylcarbonyl group such as benzoyl, α-naphthoyl, β-naphthoyl, propioloyl, methacryloyl, crotonoyl, isocrotonoyl, (E) -2-methyl-2-butenoyl; Reel carbonyl group, halogenated arylcarbonyl group such as 2-bromobenzoyl, 4-chlorobenzoyl, alkylated arylcarbonyl group such as 2,4,6-trimethylbenzoyl, 4-toluoyl, 4-anisoyl, etc. Alkoxylated arylcarbonyl groups such as 4-nitrobenzoyl, nitrated arylcarbonyl groups such as 2-nitrobenzoyl, alkoxycarbonylated arylcarbonyl groups such as 2- (methoxycarbonyl) benzoyl, aryls such as 4-phenylbenzoyl An “arylcarbonyl group” such as an arylcarbonyl group; an alkoxycarbonyl group such as methoxycarbonyl, ethoxycarbonyl, isopropoxycarbonyl, t-butoxycarbonyl, 2,2,2-trichloroethoxycarbonyl, An “alkoxycarbonyl group” such as an alkoxycarbonyl group substituted with a halogen or trialkylsilyl group such as 2-trimethylsilylethoxycarbonyl; tetrahydropyran-2-yl, 3-bromotetrahydropyran-2-yl, 4-methoxytetrahydro “Tetrahydropyranyl or tetrahydrothiopyranyl groups” such as pyran-4-yl, tetrahydrothiopyran-2-yl, 4-methoxytetrahydrothiopyran-4-yl; tetrahydrofuran-2-yl, tetrahydrothiofuran-2 A “tetrahydrofuranyl or tetrahydrothiofuranyl group” such as -yl; trimethylsilyl, triethylsilyl, isopropyldimethylsilyl, t-butyldimethylsilyl, methyldiisopropylsilyl, methyldi-t-butylsilyl A “silyl group” such as a trialkylsilyl group such as triisopropylsilyl, an alkyldiarylsilyl or dialkylarylsilyl group such as diphenylmethylsilyl, diphenylbutylsilyl, diphenylisopropylsilyl, phenyldiisopropylsilyl; 1-dimethyl-1-methoxymethyl, ethoxymethyl, propoxymethyl, isopropoxymethyl, butoxymethyl, alkoxymethyl groups such as t-butoxymethyl, alkoxyalkoxymethyl groups such as 2-methoxyethoxymethyl, 2,2, “Alkoxymethyl groups” such as halogenated alkoxymethyl such as 2-trichloroethoxymethyl and bis (2-chloroethoxy) methyl; such as 1-ethoxyethyl and 1- (isopropoxy) ethyl “Substituted ethyl groups” such as alkoxyethyl groups and ethyl halide groups such as 2,2,2-trichloroethyl; benzyl, α-naphthylmethyl, β-naphthylmethyl, diphenylmethyl, triphenylmethyl, α-naphthyldiphenylmethyl Alkyl groups substituted with 1 to 3 aryl groups such as 9-anthrylmethyl, 4-methylbenzyl, 2,4,6-trimethylbenzyl, 3,4,5-trimethylbenzyl, 4-methoxybenzyl , 4-methoxyphenyldiphenylmethyl, 2-nitrobenzyl, 4-nitrobenzyl, 4-chlorobenzyl, 4-bromobenzyl, 4-cyanobenzyl (alkyl, alkoxy, nitro, halogen, cyano groups with an aryl ring Substituted alkyl groups substituted with 1 to 3 aryl groups) Kill group ";" alkenyloxycarbonyl group "such as vinyloxycarbonyl and allyloxycarbonyl; benzyloxycarbonyl, 4-methoxybenzyloxycarbonyl, 3,4-dimethoxybenzyloxycarbonyl, 2-nitrobenzyloxycarbonyl, 4- An “aralkyloxycarbonyl group” in which the aryl ring may be substituted with one or two alkoxy or nitro groups, such as nitrobenzyloxycarbonyl, preferably an alkylcarbonyl group, a silyl group or an aralkyl group. is there.
保護・脱保護が必要な工程は、既知の方法(例えば、”Protective Groups in Organic Synthesis” (Theodora W. Greene、Peter G. M.Wuts著、 1999年、Wiley-Interscience Publication発行)等に記載の方法)に準じて行われる。
Processes that require protection / deprotection are described in known methods (for example, “Protective Groups in Organic Synthesis” (written by Theodora W. Greene, Peter G. M.Wuts, 1999, published by Wiley-Interscience Publication)). Method).
本発明の化合物又はその薬理上許容される塩は、種々の形態で投与することができる。その投与形態としては、例えば、錠剤、カプセル剤、顆粒剤、乳剤、丸剤、散剤、シロップ剤(液剤)等による経口投与、または注射剤(静脈内、筋肉内、皮下または腹腔内投与)、点滴剤、坐剤(直腸投与)等による非経口投与を挙げることができる。これらの各種製剤は、常法に従って主薬に賦形剤、結合剤、崩壊剤、滑沢剤、矯味矯臭剤、溶解補助剤、懸濁剤、コーティング剤等の医薬の製剤技術分野において通常使用し得る補助剤を用いて製剤化することができる。
The compound of the present invention or a pharmacologically acceptable salt thereof can be administered in various forms. Examples of the administration form include oral administration by tablets, capsules, granules, emulsions, pills, powders, syrups (solutions), etc., or injections (intravenous, intramuscular, subcutaneous or intraperitoneal administration), Examples include parenteral administration such as instillation and suppository (rectal administration). These various preparations are usually used in the pharmaceutical preparation technical field such as excipients, binders, disintegrants, lubricants, flavoring agents, solubilizers, suspension agents, coating agents, etc. as main ingredients in accordance with conventional methods. It can be formulated with the resulting adjuvant.
錠剤として使用する場合、担体として、例えば、乳糖、白糖、塩化ナトリウム、グルコース、尿素、デンプン、炭酸カルシウム、カオリン、結晶セルロース、ケイ酸等の賦形剤;水、エタノール、プロパノール、単シロップ、グルコース液、デンプン液、ゼラチン溶液、カルボキシメチルセルロース、セラック、メチルセルロース、リン酸カリウム、ポリビニルピロリドン等の結合剤;乾燥デンプン、アルギン酸ナトリウム、寒天末、ラミナラン末、炭酸水素ナトリウム、炭酸カルシウム、ポリオキシエチレンソルビタン脂肪酸エステル、ラウリル硫酸ナトリウム、ステアリン酸モノグリセリド、デンプン、乳糖等の崩壊剤;白糖、ステアリン、カカオバター、水素添加油等の崩壊抑制剤;第4級アンモニウム塩類、ラウリル硫酸ナトリウム等の吸収促進剤;グリセリン、デンプン等の保湿剤;デンプン、乳糖、カオリン、ベントナイト、コロイド状ケイ酸等の吸着剤;精製タルク、ステアリン酸塩、硼酸末、ポリエチレングリコール等の潤沢剤等を使用することができる。また、必要に応じ通常の剤皮を施した錠剤、例えば糖衣錠、ゼラチン被包錠、腸溶被錠、フィルムコーティング錠あるいは二重錠、多層錠とすることができる。
When used as a tablet, as a carrier, for example, excipients such as lactose, sucrose, sodium chloride, glucose, urea, starch, calcium carbonate, kaolin, crystalline cellulose, silicic acid; water, ethanol, propanol, simple syrup, glucose Solution, starch solution, gelatin solution, carboxymethylcellulose, shellac, methylcellulose, potassium phosphate, polyvinylpyrrolidone, etc .; dried starch, sodium alginate, agar powder, laminaran powder, sodium bicarbonate, calcium carbonate, polyoxyethylene sorbitan fatty acid Disintegrators such as esters, sodium lauryl sulfate, monoglyceride stearate, starch, lactose; disintegrators such as sucrose, stearin, cocoa butter, hydrogenated oil; quaternary ammonium salts, sodium lauryl sulfate Moisturizers such as glycerin and starch; Adsorbents such as starch, lactose, kaolin, bentonite and colloidal silicic acid; Use of lubricants such as purified talc, stearate, boric acid powder and polyethylene glycol can do. Moreover, it can be set as the tablet which gave the normal coating as needed, for example, a sugar-coated tablet, a gelatin-encapsulated tablet, an enteric-coated tablet, a film-coated tablet, a double tablet, and a multilayer tablet.
丸剤として使用する場合、担体として、例えば、グルコース、乳糖、カカオバター、デンプン、硬化植物油、カオリン、タルク等の賦形剤;アラビアゴム末、トラガント末、ゼラチン、エタノール等の結合剤;ラミナラン、寒天等の崩壊剤等を使用することができる。
When used as pills, as carriers, for example, excipients such as glucose, lactose, cocoa butter, starch, hydrogenated vegetable oil, kaolin, talc; binders such as gum arabic powder, tragacanth powder, gelatin, ethanol; laminaran, Disintegrants such as agar can be used.
坐剤として使用する場合、担体としてこの分野で従来公知のものを広く使用でき、例えばポリエチレングリコール、カカオバター、高級アルコール、高級アルコールのエステル類、ゼラチン、半合成グリセリド等を挙げることができる。
When used as a suppository, a carrier conventionally known in this field can be widely used as a carrier, and examples thereof include polyethylene glycol, cocoa butter, higher alcohol, esters of higher alcohol, gelatin, semi-synthetic glyceride and the like.
注射剤として使用する場合、液剤、乳剤または懸濁剤として使用することができる。これらの液剤、乳剤または懸濁剤は、殺菌され、血液と等張であることが好ましい。これら液剤、乳剤または懸濁剤の製造に用いる溶媒は、医療用の希釈剤として使用できるものであれば特に限定はなく、例えば、水、エタノール、プロピレングリコール、エトキシ化イソステアリルアルコール、ポリオキシ化イソステアリルアルコール、ポリオキシエチレンソルビタン脂肪酸エステル類等を挙げることができる。なお、この場合、等張性の溶液を調製するのに充分な量の食塩、グルコースまたはグリセリンを製剤中に含んでいてもよく、また通常の溶解補助剤、緩衝剤、無痛化剤等を含んでいてもよい。
When used as an injection, it can be used as a solution, emulsion or suspension. These solutions, emulsions or suspensions are preferably sterilized and isotonic with blood. The solvent used in the production of these solutions, emulsions or suspensions is not particularly limited as long as it can be used as a medical diluent. For example, water, ethanol, propylene glycol, ethoxylated isostearyl alcohol, polyoxylated isoforms are used. Examples include stearyl alcohol and polyoxyethylene sorbitan fatty acid esters. In this case, a sufficient amount of sodium chloride, glucose or glycerin may be included in the preparation to prepare an isotonic solution, and a normal solubilizing agent, buffer, soothing agent, etc. may be included. You may go out.
また、上記の製剤には、必要に応じて、着色剤、保存剤、香料、風味剤、甘味剤等を含めることもでき、更に、他の医薬品を含めることもできる。
In addition, the above-mentioned preparation may contain a coloring agent, a preservative, a fragrance, a flavoring agent, a sweetening agent, and the like as required, and may further contain other medicines.
上記製剤に含まれる有効成分化合物の量は、特に限定されず広範囲に適宜選択されるが、通常、全組成物中0.5乃至70重量%、好ましくは1乃至30重量%含む。
The amount of the active ingredient compound contained in the preparation is not particularly limited and is appropriately selected within a wide range, but is usually 0.5 to 70% by weight, preferably 1 to 30% by weight, based on the total composition.
その使用量は患者(温血動物、特に人間)の症状、年齢等により異なるが、経口投与の場合には、1日あたり、上限として2000mg(好ましくは100mg)であり、下限として0.1mg(好ましくは1mg、さらに好ましくは10mg)を成人に対して、1日当り1乃至6回症状に応じて投与することが望ましい。
The amount used varies depending on the symptoms, age, etc. of the patient (warm-blooded animal, particularly human), but in the case of oral administration, the upper limit is 2000 mg (preferably 100 mg) per day, and the lower limit is 0.1 mg ( Preferably 1 mg, more preferably 10 mg) is administered to adults 1 to 6 times per day depending on the symptoms.
以下、実施例および試験例を挙げて、本発明をさらに詳細に説明するが、本発明の範囲はこれらに限定されるものではない。
Hereinafter, the present invention will be described in more detail with reference to Examples and Test Examples, but the scope of the present invention is not limited thereto.
実施例のカラムクロマトグラフィーにおける溶出はTLC(Thin Layer Chromatography,薄層クロマトグラフィー)による観察下に行われた。TLC観察においては、TLCプレートとしてメルク(Merck)社製のシリカゲル60F254を、展開溶媒としてはカラムクロマトグラフィーで溶出溶媒として用いられた溶媒を、検出法としてUV検出器を採用した。カラム用シリカゲルは同じくメルク社製のシリカゲルSK-85(230~400メッシュ)、もしくは富士シリシア化学 Chromatorex NH(200 - 350メッシュ)を用いた。通常のカラムクロマトグラフィーの他に、昭光サイエンティフィック社の自動クロマトグラフィー装置(Purif-α2 もしくは Purif-espoir2)を適宜使用した。溶出溶媒は各実施例で指定した溶媒を指定された比率で用いた。(もしくは適宜必要に応じて比率を変化させた。)尚、実施例で用いる略号は、次のような意義を有する。
mg : ミリグラム,g : グラム,mL: ミリリットル,MHz : メガヘルツ。 Elution in the column chromatography of the examples was performed under observation by TLC (Thin Layer Chromatography). In TLC observation, silica gel 60F 254 manufactured by Merck was used as a TLC plate, a solvent used as an elution solvent in column chromatography was used as a developing solvent, and a UV detector was used as a detection method. As silica gel for the column, silica gel SK-85 (230-400 mesh) manufactured by Merck Co., Ltd. or Fuji Silysia Chemical Chromatorex NH (200-350 mesh) was used. In addition to ordinary column chromatography, an automatic chromatography apparatus (Purif-α2 or Purif-espoir2) manufactured by Shoko Scientific was used as appropriate. As the elution solvent, the solvent specified in each example was used at the specified ratio. (Or, the ratio was changed as necessary.) The abbreviations used in the examples have the following significance.
mg: milligram, g: gram, mL: milliliter, MHz: megahertz.
mg : ミリグラム,g : グラム,mL: ミリリットル,MHz : メガヘルツ。 Elution in the column chromatography of the examples was performed under observation by TLC (Thin Layer Chromatography). In TLC observation, silica gel 60F 254 manufactured by Merck was used as a TLC plate, a solvent used as an elution solvent in column chromatography was used as a developing solvent, and a UV detector was used as a detection method. As silica gel for the column, silica gel SK-85 (230-400 mesh) manufactured by Merck Co., Ltd. or Fuji Silysia Chemical Chromatorex NH (200-350 mesh) was used. In addition to ordinary column chromatography, an automatic chromatography apparatus (Purif-α2 or Purif-espoir2) manufactured by Shoko Scientific was used as appropriate. As the elution solvent, the solvent specified in each example was used at the specified ratio. (Or, the ratio was changed as necessary.) The abbreviations used in the examples have the following significance.
mg: milligram, g: gram, mL: milliliter, MHz: megahertz.
以下の実施例において、核磁気共鳴(以下、1H NMR)スペクトルは、テトラメチルシランを標準物質として、ケミカルシフト値をδ値(ppm)にて記載した。分裂パターンは一重線をs、二重線をd、三重線をt、四重線をq、多重線をm、ブロードをbrで示した。
In the following examples, nuclear magnetic resonance (hereinafter, 1 H NMR) spectra are described with chemical shift values expressed as δ values (ppm) using tetramethylsilane as a standard substance. The splitting pattern is indicated by s for single lines, d for double lines, t for triple lines, q for quadruple lines, m for multiple lines, and br for broad lines.
質量分析(以下、MS)は、ESI(Electron Spray Ionization)法で行った。
Mass spectrometry (hereinafter referred to as MS) was performed by ESI (Electron Spray Ionization) method.
(実施例1)
(cis-4-{[5-(4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 Example 1
(cis-4-{[5- (4-{[(3-Methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid
(cis-4-{[5-(4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 Example 1
(cis-4-{[5- (4-{[(3-Methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid

(1a) 3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-カルボン酸エチルエステル
3-メチル-1H-ピラゾール-4-カルボン酸エチルエステル(7.86 g)のトルエン(100 mL)溶液に2-ブロモピリジン(5.9 mL)、炭酸カリウム(14.8 g)、ヨウ化銅(I)(486 mg)、そしてN,N'-ジメチルシクロヘキサン-1,2-ジアミン(725 mg)を室温で加えた。反応混合物を15時間加熱還流し、室温に冷却し、セライトでろ過し、そして濃縮した。残渣物をクロマトグラフィー(ジクロロメタン/酢酸エチル 100:0→80:20)で精製し、標記化合物 4.36 g(37%)を白色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=8.97(1H, s), 8.44(1H, d, J=5.8Hz), 7.97(1H, d, J=8.2Hz), 7.84(1H,ddd, J=7.5,7.5,2.0Hz), 7.23(1H, dd, J=8.4,4.9Hz), 4.32(2H, q, J=7.0Hz), 2.57(3H, s), 1.37(3H, t, J=7.2Hz)。 (1a) 3-methyl-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid ethyl ester solution of 3-methyl-1H-pyrazole-4-carboxylic acid ethyl ester (7.86 g) in toluene (100 mL) 2-bromopyridine (5.9 mL), potassium carbonate (14.8 g), copper (I) iodide (486 mg), and N, N'-dimethylcyclohexane-1,2-diamine (725 mg) at room temperature It was. The reaction mixture was heated to reflux for 15 hours, cooled to room temperature, filtered through celite, and concentrated. The residue was purified by chromatography (dichloromethane / ethyl acetate 100: 0 → 80: 20) to give 4.36 g (37%) of the title compound as a white solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 8.97 (1H, s), 8.44 (1H, d, J = 5.8Hz), 7.97 (1H, d, J = 8.2Hz), 7.84 (1H, ddd, J = 7.5,7.5,2.0Hz), 7.23 (1H, dd, J = 8.4,4.9Hz), 4.32 (2H, q, J = 7.0Hz), 2.57 (3H, s), 1.37 (3H, t , J = 7.2Hz).
3-メチル-1H-ピラゾール-4-カルボン酸エチルエステル(7.86 g)のトルエン(100 mL)溶液に2-ブロモピリジン(5.9 mL)、炭酸カリウム(14.8 g)、ヨウ化銅(I)(486 mg)、そしてN,N'-ジメチルシクロヘキサン-1,2-ジアミン(725 mg)を室温で加えた。反応混合物を15時間加熱還流し、室温に冷却し、セライトでろ過し、そして濃縮した。残渣物をクロマトグラフィー(ジクロロメタン/酢酸エチル 100:0→80:20)で精製し、標記化合物 4.36 g(37%)を白色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=8.97(1H, s), 8.44(1H, d, J=5.8Hz), 7.97(1H, d, J=8.2Hz), 7.84(1H,ddd, J=7.5,7.5,2.0Hz), 7.23(1H, dd, J=8.4,4.9Hz), 4.32(2H, q, J=7.0Hz), 2.57(3H, s), 1.37(3H, t, J=7.2Hz)。 (1a) 3-methyl-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid ethyl ester solution of 3-methyl-1H-pyrazole-4-carboxylic acid ethyl ester (7.86 g) in toluene (100 mL) 2-bromopyridine (5.9 mL), potassium carbonate (14.8 g), copper (I) iodide (486 mg), and N, N'-dimethylcyclohexane-1,2-diamine (725 mg) at room temperature It was. The reaction mixture was heated to reflux for 15 hours, cooled to room temperature, filtered through celite, and concentrated. The residue was purified by chromatography (dichloromethane / ethyl acetate 100: 0 → 80: 20) to give 4.36 g (37%) of the title compound as a white solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 8.97 (1H, s), 8.44 (1H, d, J = 5.8Hz), 7.97 (1H, d, J = 8.2Hz), 7.84 (1H, ddd, J = 7.5,7.5,2.0Hz), 7.23 (1H, dd, J = 8.4,4.9Hz), 4.32 (2H, q, J = 7.0Hz), 2.57 (3H, s), 1.37 (3H, t , J = 7.2Hz).
(1b) 3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-カルボン酸
実施例(1a)で得た化合物 4.36 g のテトラヒドロフラン/エタノール(1:1, 80 mL)溶液に1N水酸化ナトリウム水溶液を室温で加えた。反応混合物を24時間撹拌し、水で希釈し、そして1N塩酸(40 mL)で中和した。析出した固体をろ取し減圧下乾燥し、標記化合物 3.78 g(99%)を白色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=9.07(1H, s), 8.47-8.45(1H, m), 7.99(1H, d, J=8.2Hz), 7.85(1H, ddd, J=7.2,7.2,1.8Hz), 7.26-7.24(1H, m), 2.59(3H, s), 1.8-1.4(1H, brs)。 (1b) 3-methyl-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid 1N water was added to a solution of the compound obtained in Example (1a) 4.36 g in tetrahydrofuran / ethanol (1: 1, 80 mL). Aqueous sodium oxide was added at room temperature. The reaction mixture was stirred for 24 hours, diluted with water and neutralized with 1N hydrochloric acid (40 mL). The precipitated solid was collected by filtration and dried under reduced pressure to obtain 3.78 g (99%) of the title compound as a white solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 9.07 (1H, s), 8.47-8.45 (1H, m), 7.99 (1H, d, J = 8.2 Hz), 7.85 (1H, ddd, J = 7.2,7.2,1.8Hz), 7.26-7.24 (1H, m), 2.59 (3H, s), 1.8-1.4 (1H, brs).
実施例(1a)で得た化合物 4.36 g のテトラヒドロフラン/エタノール(1:1, 80 mL)溶液に1N水酸化ナトリウム水溶液を室温で加えた。反応混合物を24時間撹拌し、水で希釈し、そして1N塩酸(40 mL)で中和した。析出した固体をろ取し減圧下乾燥し、標記化合物 3.78 g(99%)を白色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=9.07(1H, s), 8.47-8.45(1H, m), 7.99(1H, d, J=8.2Hz), 7.85(1H, ddd, J=7.2,7.2,1.8Hz), 7.26-7.24(1H, m), 2.59(3H, s), 1.8-1.4(1H, brs)。 (1b) 3-methyl-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid 1N water was added to a solution of the compound obtained in Example (1a) 4.36 g in tetrahydrofuran / ethanol (1: 1, 80 mL). Aqueous sodium oxide was added at room temperature. The reaction mixture was stirred for 24 hours, diluted with water and neutralized with 1N hydrochloric acid (40 mL). The precipitated solid was collected by filtration and dried under reduced pressure to obtain 3.78 g (99%) of the title compound as a white solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 9.07 (1H, s), 8.47-8.45 (1H, m), 7.99 (1H, d, J = 8.2 Hz), 7.85 (1H, ddd, J = 7.2,7.2,1.8Hz), 7.26-7.24 (1H, m), 2.59 (3H, s), 1.8-1.4 (1H, brs).
(1c) {cis-4-[(5-ブロモピリミジン-2-イル)オキシ]シクロヘキシル}酢酸メチルエステル
(trans-4-ヒドロキシシクロヘキシル) 酢酸メチルエステル (WO2009119534)(2.58 g)と5-ブロモピリミジン-2-オール(1.74 g)のトルエン(30 mL)溶液にシアノメチレントリブチルホスホラン(CMBP)(3.93 mL)を室温で加えた。反応混合物を120℃で9時間加熱し、室温に冷却し、飽和塩化アンモニウム水溶液で希釈し、そして酢酸エチルで抽出した。有機層を水で洗浄し、濃縮した。残渣物をクロマトグラフィーで精製し(自動クロマトグラフィー装置、ヘキサン/酢酸エチル 100:0→80:20)、標記化合物 1.67 g(51%)を薄黄色オイルとして得た。
1H NMR (500MHz, CDCl3):δ(ppm) = 8.51 (2H, s), 5.23-5.18 (1H, m), 3.67 (3H, s), 2.28 (2H, d, J = 6.8 Hz), 2.09-1.89 (3H, m), 1.69-1.43 (6H, m)。 (1c) {cis-4-[(5-Bromopyrimidin-2-yl) oxy] cyclohexyl} acetic acid methyl ester (trans-4-hydroxycyclohexyl) acetic acid methyl ester (WO2009119534) (2.58 g) and 5-bromopyrimidine- Cyanomethylenetributylphosphorane (CMBP) (3.93 mL) was added to a toluene (30 mL) solution of 2-ol (1.74 g) at room temperature. The reaction mixture was heated at 120 ° C. for 9 hours, cooled to room temperature, diluted with saturated aqueous ammonium chloride and extracted with ethyl acetate. The organic layer was washed with water and concentrated. The residue was purified by chromatography (automatic chromatography apparatus, hexane / ethyl acetate 100: 0 → 80: 20) to give 1.67 g (51%) of the title compound as a pale yellow oil.
1 H NMR (500MHz, CDCl 3 ): δ (ppm) = 8.51 (2H, s), 5.23-5.18 (1H, m), 3.67 (3H, s), 2.28 (2H, d, J = 6.8 Hz), 2.09-1.89 (3H, m), 1.69-1.43 (6H, m).
(trans-4-ヒドロキシシクロヘキシル) 酢酸メチルエステル (WO2009119534)(2.58 g)と5-ブロモピリミジン-2-オール(1.74 g)のトルエン(30 mL)溶液にシアノメチレントリブチルホスホラン(CMBP)(3.93 mL)を室温で加えた。反応混合物を120℃で9時間加熱し、室温に冷却し、飽和塩化アンモニウム水溶液で希釈し、そして酢酸エチルで抽出した。有機層を水で洗浄し、濃縮した。残渣物をクロマトグラフィーで精製し(自動クロマトグラフィー装置、ヘキサン/酢酸エチル 100:0→80:20)、標記化合物 1.67 g(51%)を薄黄色オイルとして得た。
1H NMR (500MHz, CDCl3):δ(ppm) = 8.51 (2H, s), 5.23-5.18 (1H, m), 3.67 (3H, s), 2.28 (2H, d, J = 6.8 Hz), 2.09-1.89 (3H, m), 1.69-1.43 (6H, m)。 (1c) {cis-4-[(5-Bromopyrimidin-2-yl) oxy] cyclohexyl} acetic acid methyl ester (trans-4-hydroxycyclohexyl) acetic acid methyl ester (WO2009119534) (2.58 g) and 5-bromopyrimidine- Cyanomethylenetributylphosphorane (CMBP) (3.93 mL) was added to a toluene (30 mL) solution of 2-ol (1.74 g) at room temperature. The reaction mixture was heated at 120 ° C. for 9 hours, cooled to room temperature, diluted with saturated aqueous ammonium chloride and extracted with ethyl acetate. The organic layer was washed with water and concentrated. The residue was purified by chromatography (automatic chromatography apparatus, hexane / ethyl acetate 100: 0 → 80: 20) to give 1.67 g (51%) of the title compound as a pale yellow oil.
1 H NMR (500MHz, CDCl 3 ): δ (ppm) = 8.51 (2H, s), 5.23-5.18 (1H, m), 3.67 (3H, s), 2.28 (2H, d, J = 6.8 Hz), 2.09-1.89 (3H, m), 1.69-1.43 (6H, m).
(1d) (cis-4-{[5-(4-アミノフェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸メチルエステル
実施例(1c)で得た化合物(1.67 g)、4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)アニリン(1.11 g)、テトラキス(トリフェニルホスフィン)パラジウム(0)(295 mg)、そして炭酸カリウム(1.41 g)の1,4-ジオキサン/水(7:3, 30 mL)溶液を80 ℃で2時間加熱した。反応混合物を酢酸エチルで希釈し、水で洗浄し、そして濃縮した。残渣物をクロマトグラフィー(自動クロマトグラフィー装置、ジクロロメタン/酢酸エチル 100:0→85:15)で精製し、標記化合物 1.59 g(92%)を黄色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=8.64(2H, s), 7.32(2H, d, J=8.6Hz), 6.79(2H, d, J=8.2Hz), 5.29(1H, brs), 3.80(2H, brs), 3.68(3H, s), 2.29(2H, d, J=7.0Hz), 2.13-2.09(2H, m), 1.97-1.91(1H, m), 1.72-1.55(6H, m)。 (1d) (cis-4-{[5- (4-Aminophenyl) pyrimidin-2-yl] oxy} cyclohexyl) acetic acid methyl ester Compound (1.67 g) obtained in Example (1c), 4- (4, 4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) aniline (1.11 g), tetrakis (triphenylphosphine) palladium (0) (295 mg), and potassium carbonate (1.41 g) A 1,4-dioxane / water (7: 3, 30 mL) solution was heated at 80 ° C. for 2 hours. The reaction mixture was diluted with ethyl acetate, washed with water and concentrated. The residue was purified by chromatography (automatic chromatography apparatus, dichloromethane / ethyl acetate 100: 0 → 85: 15) to obtain 1.59 g (92%) of the title compound as a yellow solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 8.64 (2H, s), 7.32 (2H, d, J = 8.6Hz), 6.79 (2H, d, J = 8.2Hz), 5.29 (1H, brs), 3.80 (2H, brs), 3.68 (3H, s), 2.29 (2H, d, J = 7.0Hz), 2.13-2.09 (2H, m), 1.97-1.91 (1H, m), 1.72-1.55 (6H, m).
実施例(1c)で得た化合物(1.67 g)、4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)アニリン(1.11 g)、テトラキス(トリフェニルホスフィン)パラジウム(0)(295 mg)、そして炭酸カリウム(1.41 g)の1,4-ジオキサン/水(7:3, 30 mL)溶液を80 ℃で2時間加熱した。反応混合物を酢酸エチルで希釈し、水で洗浄し、そして濃縮した。残渣物をクロマトグラフィー(自動クロマトグラフィー装置、ジクロロメタン/酢酸エチル 100:0→85:15)で精製し、標記化合物 1.59 g(92%)を黄色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=8.64(2H, s), 7.32(2H, d, J=8.6Hz), 6.79(2H, d, J=8.2Hz), 5.29(1H, brs), 3.80(2H, brs), 3.68(3H, s), 2.29(2H, d, J=7.0Hz), 2.13-2.09(2H, m), 1.97-1.91(1H, m), 1.72-1.55(6H, m)。 (1d) (cis-4-{[5- (4-Aminophenyl) pyrimidin-2-yl] oxy} cyclohexyl) acetic acid methyl ester Compound (1.67 g) obtained in Example (1c), 4- (4, 4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) aniline (1.11 g), tetrakis (triphenylphosphine) palladium (0) (295 mg), and potassium carbonate (1.41 g) A 1,4-dioxane / water (7: 3, 30 mL) solution was heated at 80 ° C. for 2 hours. The reaction mixture was diluted with ethyl acetate, washed with water and concentrated. The residue was purified by chromatography (automatic chromatography apparatus, dichloromethane / ethyl acetate 100: 0 → 85: 15) to obtain 1.59 g (92%) of the title compound as a yellow solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 8.64 (2H, s), 7.32 (2H, d, J = 8.6Hz), 6.79 (2H, d, J = 8.2Hz), 5.29 (1H, brs), 3.80 (2H, brs), 3.68 (3H, s), 2.29 (2H, d, J = 7.0Hz), 2.13-2.09 (2H, m), 1.97-1.91 (1H, m), 1.72-1.55 (6H, m).
(1e) (cis-4-{[5-(4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸メチルエステル
実施例(1b)で得た化合物(3.78 g)、実施例(1d)で得た化合物(6.35 g)、トリエチルアミン(3.1 mL)及びBOP試薬(9.05 g)のジメチルアセトアミド溶液(100 mL)を室温で3日間撹拌した。反応混合物を飽和炭酸水素ナトリウム水溶液で希釈し、更に酢酸エチルと水で希釈した。生成した混合物を撹拌した。析出した固体をろ取し、水と酢酸エチルで洗浄し、減圧下乾燥した。標記化合物 6.93 g(71%)をオフホワイト色固体として得た。
1H NMR(400MHz,DMSO-d6):δ(ppm)=10.2(1H, s), 9.50(1H, s), 8.91(2H, s), 8.55(1H, dd, J=4.9 and 1.8Hz), 8.07-8.03(1H, m), 7.96(1H, d, J=8.2Hz), 7.90(2H, d, J=9.0Hz), 7.72(2H, d, J=9.0Hz), 7.44(1H, dd, J=7.3, 4.9Hz), 5.24-5.20(1H, m), 3.61(3H, s), 2.53(3H, s), 2.30(2H, d, J=7.0Hz), 1.99-1.92(2H, m), 1.91-1.81(1H, m), 1.72-1.64(2H, m), 1.60-1.54(2H, m), 1.44-1.33(2H, m)。 (1e) (cis-4-{[5- (4-{[(3-Methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid methyl ester Compound (3.78 g) obtained in Example (1b), Compound (6.35 g) obtained in Example (1d), Triethylamine (3.1 mL) and BOP reagent (9.05 g) in dimethylacetamide The solution (100 mL) was stirred at room temperature for 3 days. The reaction mixture was diluted with saturated aqueous sodium hydrogen carbonate solution, and further diluted with ethyl acetate and water. The resulting mixture was stirred. The precipitated solid was collected by filtration, washed with water and ethyl acetate, and dried under reduced pressure. This gave 6.93 g (71%) of the title compound as an off-white solid.
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 10.2 (1H, s), 9.50 (1H, s), 8.91 (2H, s), 8.55 (1H, dd, J = 4.9 and 1.8 Hz ), 8.07-8.03 (1H, m), 7.96 (1H, d, J = 8.2Hz), 7.90 (2H, d, J = 9.0Hz), 7.72 (2H, d, J = 9.0Hz), 7.44 (1H , dd, J = 7.3, 4.9Hz), 5.24-5.20 (1H, m), 3.61 (3H, s), 2.53 (3H, s), 2.30 (2H, d, J = 7.0Hz), 1.99-1.92 ( 2H, m), 1.91-1.81 (1H, m), 1.72-1.64 (2H, m), 1.60-1.54 (2H, m), 1.44-1.33 (2H, m).
実施例(1b)で得た化合物(3.78 g)、実施例(1d)で得た化合物(6.35 g)、トリエチルアミン(3.1 mL)及びBOP試薬(9.05 g)のジメチルアセトアミド溶液(100 mL)を室温で3日間撹拌した。反応混合物を飽和炭酸水素ナトリウム水溶液で希釈し、更に酢酸エチルと水で希釈した。生成した混合物を撹拌した。析出した固体をろ取し、水と酢酸エチルで洗浄し、減圧下乾燥した。標記化合物 6.93 g(71%)をオフホワイト色固体として得た。
1H NMR(400MHz,DMSO-d6):δ(ppm)=10.2(1H, s), 9.50(1H, s), 8.91(2H, s), 8.55(1H, dd, J=4.9 and 1.8Hz), 8.07-8.03(1H, m), 7.96(1H, d, J=8.2Hz), 7.90(2H, d, J=9.0Hz), 7.72(2H, d, J=9.0Hz), 7.44(1H, dd, J=7.3, 4.9Hz), 5.24-5.20(1H, m), 3.61(3H, s), 2.53(3H, s), 2.30(2H, d, J=7.0Hz), 1.99-1.92(2H, m), 1.91-1.81(1H, m), 1.72-1.64(2H, m), 1.60-1.54(2H, m), 1.44-1.33(2H, m)。 (1e) (cis-4-{[5- (4-{[(3-Methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid methyl ester Compound (3.78 g) obtained in Example (1b), Compound (6.35 g) obtained in Example (1d), Triethylamine (3.1 mL) and BOP reagent (9.05 g) in dimethylacetamide The solution (100 mL) was stirred at room temperature for 3 days. The reaction mixture was diluted with saturated aqueous sodium hydrogen carbonate solution, and further diluted with ethyl acetate and water. The resulting mixture was stirred. The precipitated solid was collected by filtration, washed with water and ethyl acetate, and dried under reduced pressure. This gave 6.93 g (71%) of the title compound as an off-white solid.
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 10.2 (1H, s), 9.50 (1H, s), 8.91 (2H, s), 8.55 (1H, dd, J = 4.9 and 1.8 Hz ), 8.07-8.03 (1H, m), 7.96 (1H, d, J = 8.2Hz), 7.90 (2H, d, J = 9.0Hz), 7.72 (2H, d, J = 9.0Hz), 7.44 (1H , dd, J = 7.3, 4.9Hz), 5.24-5.20 (1H, m), 3.61 (3H, s), 2.53 (3H, s), 2.30 (2H, d, J = 7.0Hz), 1.99-1.92 ( 2H, m), 1.91-1.81 (1H, m), 1.72-1.64 (2H, m), 1.60-1.54 (2H, m), 1.44-1.33 (2H, m).
(1f) (cis-4-{[5-(4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸
実施例(1e)で得た化合物(6.93 g)とテトラブチルアンモニウム水溶液(1 mol/L, 27 mL)のジオキサン溶液(70 mL)を室温で2.5時間撹拌した。反応混合物を濃縮し、水で希釈し、1 N 塩酸(150 mL)で酸性にした。得られた懸濁液にジイソプロピルエーテルを加え、3時間激しく撹拌した。析出した固体をろ取し、減圧下乾燥した。標記化合物 6.63 g (98%)を白色固体として得た。
1H NMR(400MHz,DMSO-d6):δ(ppm)= 10.2(1H, s), 9.50(1H, s), 8.91(2H, s), 8.55(1H, d, J=5.9Hz), 8.05(1H, dd, J=9.8 and 7.9Hz), 7.96(1H, d, J=8.2Hz), 7.90(2H, d, J=9.0Hz), 7.72(2H, d, J=8.6Hz), 7.44(1H, dd, J=7.2 and 4.9Hz), 5.24-5.19(1H, m), 2.53(3H, s), 2.18(2H, d, J=7.1Hz), 1.99-1.92(2H, m), 1.89-1.76(1H, m), 1.71-1.55(4H, m), 1.43-1.35(2H, m).
MS(ESI) m/z:513 (M + H)+。 (1f) (cis-4-{[5- (4-{[(3-Methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid A dioxane solution (70 mL) of the compound (6.93 g) obtained in Example (1e) and a tetrabutylammonium aqueous solution (1 mol / L, 27 mL) was stirred at room temperature for 2.5 hours. The reaction mixture was concentrated, diluted with water and acidified with 1 N hydrochloric acid (150 mL). Diisopropyl ether was added to the resulting suspension and stirred vigorously for 3 hours. The precipitated solid was collected by filtration and dried under reduced pressure. This gave 6.63 g (98%) of the title compound as a white solid.
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 10.2 (1H, s), 9.50 (1H, s), 8.91 (2H, s), 8.55 (1H, d, J = 5.9 Hz), 8.05 (1H, dd, J = 9.8 and 7.9Hz), 7.96 (1H, d, J = 8.2Hz), 7.90 (2H, d, J = 9.0Hz), 7.72 (2H, d, J = 8.6Hz), 7.44 (1H, dd, J = 7.2 and 4.9Hz), 5.24-5.19 (1H, m), 2.53 (3H, s), 2.18 (2H, d, J = 7.1Hz), 1.99-1.92 (2H, m) , 1.89-1.76 (1H, m), 1.71-1.55 (4H, m), 1.43-1.35 (2H, m).
MS (ESI) m / z: 513 (M + H) <+> .
実施例(1e)で得た化合物(6.93 g)とテトラブチルアンモニウム水溶液(1 mol/L, 27 mL)のジオキサン溶液(70 mL)を室温で2.5時間撹拌した。反応混合物を濃縮し、水で希釈し、1 N 塩酸(150 mL)で酸性にした。得られた懸濁液にジイソプロピルエーテルを加え、3時間激しく撹拌した。析出した固体をろ取し、減圧下乾燥した。標記化合物 6.63 g (98%)を白色固体として得た。
1H NMR(400MHz,DMSO-d6):δ(ppm)= 10.2(1H, s), 9.50(1H, s), 8.91(2H, s), 8.55(1H, d, J=5.9Hz), 8.05(1H, dd, J=9.8 and 7.9Hz), 7.96(1H, d, J=8.2Hz), 7.90(2H, d, J=9.0Hz), 7.72(2H, d, J=8.6Hz), 7.44(1H, dd, J=7.2 and 4.9Hz), 5.24-5.19(1H, m), 2.53(3H, s), 2.18(2H, d, J=7.1Hz), 1.99-1.92(2H, m), 1.89-1.76(1H, m), 1.71-1.55(4H, m), 1.43-1.35(2H, m).
MS(ESI) m/z:513 (M + H)+。 (1f) (cis-4-{[5- (4-{[(3-Methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid A dioxane solution (70 mL) of the compound (6.93 g) obtained in Example (1e) and a tetrabutylammonium aqueous solution (1 mol / L, 27 mL) was stirred at room temperature for 2.5 hours. The reaction mixture was concentrated, diluted with water and acidified with 1 N hydrochloric acid (150 mL). Diisopropyl ether was added to the resulting suspension and stirred vigorously for 3 hours. The precipitated solid was collected by filtration and dried under reduced pressure. This gave 6.63 g (98%) of the title compound as a white solid.
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 10.2 (1H, s), 9.50 (1H, s), 8.91 (2H, s), 8.55 (1H, d, J = 5.9 Hz), 8.05 (1H, dd, J = 9.8 and 7.9Hz), 7.96 (1H, d, J = 8.2Hz), 7.90 (2H, d, J = 9.0Hz), 7.72 (2H, d, J = 8.6Hz), 7.44 (1H, dd, J = 7.2 and 4.9Hz), 5.24-5.19 (1H, m), 2.53 (3H, s), 2.18 (2H, d, J = 7.1Hz), 1.99-1.92 (2H, m) , 1.89-1.76 (1H, m), 1.71-1.55 (4H, m), 1.43-1.35 (2H, m).
MS (ESI) m / z: 513 (M + H) <+> .
(実施例2)
(cis-4-{[5-(4-{[(3-シクロプロピル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 (Example 2)
(cis-4-{[5- (4-{[(3-Cyclopropyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} (Cyclohexyl) acetic acid
(cis-4-{[5-(4-{[(3-シクロプロピル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 (Example 2)
(cis-4-{[5- (4-{[(3-Cyclopropyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} (Cyclohexyl) acetic acid

(2a) 3-シクロプロピル-1-ピリジン-2-イル-1H-ピラゾール-4-カルボン酸
実施例(1a)と同様の方法で、5-シクロプロピル-1H-ピラゾール-4-カルボン酸メチルエステル(500 mg)と2-ブロモピリジン(0.29 mL)から、薄オレンジ色オイルを501 mg得た。この薄オレンジ色オイル(500 mg)を実施例(1b)と同様の方法で加水分解し、標記化合物164 mg(24%、2工程)を無色固体として得た。 (2a) 3-Cyclopropyl-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid In the same manner as in Example (1a), 5-cyclopropyl-1H-pyrazole-4-carboxylic acid methyl ester (500 mg) and 2-bromopyridine (0.29 mL) gave 501 mg of a light orange oil. This pale orange oil (500 mg) was hydrolyzed in the same manner as in Example (1b) to obtain 164 mg (24%, 2 steps) of the title compound as a colorless solid.
実施例(1a)と同様の方法で、5-シクロプロピル-1H-ピラゾール-4-カルボン酸メチルエステル(500 mg)と2-ブロモピリジン(0.29 mL)から、薄オレンジ色オイルを501 mg得た。この薄オレンジ色オイル(500 mg)を実施例(1b)と同様の方法で加水分解し、標記化合物164 mg(24%、2工程)を無色固体として得た。 (2a) 3-Cyclopropyl-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid In the same manner as in Example (1a), 5-cyclopropyl-1H-pyrazole-4-carboxylic acid methyl ester (500 mg) and 2-bromopyridine (0.29 mL) gave 501 mg of a light orange oil. This pale orange oil (500 mg) was hydrolyzed in the same manner as in Example (1b) to obtain 164 mg (24%, 2 steps) of the title compound as a colorless solid.
(2b) (cis-4-{[5-(4-{[(3-シクロプロピル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸
実施例(1e)と同様の方法で、実施例(2a)で得た化合物と実施例(1d)で得た化合物からアミド体 135 mg(93%)を薄黄色アモルファスとして得た。このアミド体(130 mg)を実施例(1f)と同様の方法で加水分解し、標記化合物 90 mg (72%)を無色固体として得た。
1H NMR(500MHz,DMSO-d6):δ(ppm)=12.0(1H, s), 10.2(1H, s), 9.43(1H, s), 8.91(2H, s), 8.52(1H, d, J=4.3Hz), 8.02(1H, t, J=7.8Hz), 7.91-7.86(3H, m), 7.72(2H, d, J=8.3Hz), 7.41(1H, t, J=6.1Hz), 5.22(1H, brs), 2.81-2.76(1H, m), 2.19(2H, d, J=7.4Hz), 1.99-1.94(2H, m), 1.86-1.80(1H, m), 1.71-1.65(2H, m), 1.60-1.57(2H, m), 1.42-1.34(2H, m), 1.01-0.98(4H, m)。 (2b) (cis-4-{[5- (4-{[(3-Cyclopropyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl ] Oxy} cyclohexyl) acetic acid In the same manner as in Example (1e), from the compound obtained in Example (2a) and the compound obtained in Example (1d), 135 mg (93%) of the amide compound was converted to a pale yellow amorphous product. Obtained. This amide compound (130 mg) was hydrolyzed in the same manner as in Example (1f) to obtain 90 mg (72%) of the title compound as a colorless solid.
1H NMR (500 MHz, DMSO-d 6 ): δ (ppm) = 12.0 (1H, s), 10.2 (1H, s), 9.43 (1H, s), 8.91 (2H, s), 8.52 (1H, d, J = 4.3Hz), 8.02 (1H, t, J = 7.8Hz), 7.91-7.86 (3H, m), 7.72 (2H, d, J = 8.3Hz), 7.41 (1H, t, J = 6.1Hz) , 5.22 (1H, brs), 2.81-2.76 (1H, m), 2.19 (2H, d, J = 7.4Hz), 1.99-1.94 (2H, m), 1.86-1.80 (1H, m), 1.71-1.65 (2H, m), 1.60-1.57 (2H, m), 1.42-1.34 (2H, m), 1.01-0.98 (4H, m).
実施例(1e)と同様の方法で、実施例(2a)で得た化合物と実施例(1d)で得た化合物からアミド体 135 mg(93%)を薄黄色アモルファスとして得た。このアミド体(130 mg)を実施例(1f)と同様の方法で加水分解し、標記化合物 90 mg (72%)を無色固体として得た。
1H NMR(500MHz,DMSO-d6):δ(ppm)=12.0(1H, s), 10.2(1H, s), 9.43(1H, s), 8.91(2H, s), 8.52(1H, d, J=4.3Hz), 8.02(1H, t, J=7.8Hz), 7.91-7.86(3H, m), 7.72(2H, d, J=8.3Hz), 7.41(1H, t, J=6.1Hz), 5.22(1H, brs), 2.81-2.76(1H, m), 2.19(2H, d, J=7.4Hz), 1.99-1.94(2H, m), 1.86-1.80(1H, m), 1.71-1.65(2H, m), 1.60-1.57(2H, m), 1.42-1.34(2H, m), 1.01-0.98(4H, m)。 (2b) (cis-4-{[5- (4-{[(3-Cyclopropyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl ] Oxy} cyclohexyl) acetic acid In the same manner as in Example (1e), from the compound obtained in Example (2a) and the compound obtained in Example (1d), 135 mg (93%) of the amide compound was converted to a pale yellow amorphous product. Obtained. This amide compound (130 mg) was hydrolyzed in the same manner as in Example (1f) to obtain 90 mg (72%) of the title compound as a colorless solid.
1H NMR (500 MHz, DMSO-d 6 ): δ (ppm) = 12.0 (1H, s), 10.2 (1H, s), 9.43 (1H, s), 8.91 (2H, s), 8.52 (1H, d, J = 4.3Hz), 8.02 (1H, t, J = 7.8Hz), 7.91-7.86 (3H, m), 7.72 (2H, d, J = 8.3Hz), 7.41 (1H, t, J = 6.1Hz) , 5.22 (1H, brs), 2.81-2.76 (1H, m), 2.19 (2H, d, J = 7.4Hz), 1.99-1.94 (2H, m), 1.86-1.80 (1H, m), 1.71-1.65 (2H, m), 1.60-1.57 (2H, m), 1.42-1.34 (2H, m), 1.01-0.98 (4H, m).
(実施例3)
(cis-4-{[5-(2-メチル-4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 (Example 3)
(cis-4-{[5- (2-Methyl-4-{[(3-methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl ] Oxy} cyclohexyl) acetic acid
(cis-4-{[5-(2-メチル-4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 (Example 3)
(cis-4-{[5- (2-Methyl-4-{[(3-methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl ] Oxy} cyclohexyl) acetic acid

(3a) (cis-4-{[5-(4-アミノ-2-メチルフェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸メチルエステル
実施例(1d)と同様の方法で、実施例(1c)で得た化合物(658 mg)、3-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)アニリン(466 mg)から、標記化合物 501 mg(71%)を褐色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=8.44(2H, s), 6.99(1H, d, J=8.2Hz), 6.65-6.60(2H, m), 5.30-5.28(1H, m), 3.68(3H, s), 2.29(2H, d, J=7.4Hz), 2.22(3H, s), 2.14-2.10(2H, m), 1.98-1.92(1H, m), 1.73-1.53(6H, m)。 (3a) (cis-4-{[5- (4-Amino-2-methylphenyl) pyrimidin-2-yl] oxy} cyclohexyl) acetic acid methyl ester In the same manner as in Example (1d), Example (1c ) And 3-methyl-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) aniline (466 mg) to give the title compound 501 mg (71%) was obtained as a brown solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 8.44 (2H, s), 6.99 (1H, d, J = 8.2Hz), 6.65-6.60 (2H, m), 5.30-5.28 (1H, m ), 3.68 (3H, s), 2.29 (2H, d, J = 7.4Hz), 2.22 (3H, s), 2.14-2.10 (2H, m), 1.98-1.92 (1H, m), 1.73-1.53 ( 6H, m).
実施例(1d)と同様の方法で、実施例(1c)で得た化合物(658 mg)、3-メチル-4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)アニリン(466 mg)から、標記化合物 501 mg(71%)を褐色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=8.44(2H, s), 6.99(1H, d, J=8.2Hz), 6.65-6.60(2H, m), 5.30-5.28(1H, m), 3.68(3H, s), 2.29(2H, d, J=7.4Hz), 2.22(3H, s), 2.14-2.10(2H, m), 1.98-1.92(1H, m), 1.73-1.53(6H, m)。 (3a) (cis-4-{[5- (4-Amino-2-methylphenyl) pyrimidin-2-yl] oxy} cyclohexyl) acetic acid methyl ester In the same manner as in Example (1d), Example (1c ) And 3-methyl-4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) aniline (466 mg) to give the title compound 501 mg (71%) was obtained as a brown solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 8.44 (2H, s), 6.99 (1H, d, J = 8.2Hz), 6.65-6.60 (2H, m), 5.30-5.28 (1H, m ), 3.68 (3H, s), 2.29 (2H, d, J = 7.4Hz), 2.22 (3H, s), 2.14-2.10 (2H, m), 1.98-1.92 (1H, m), 1.73-1.53 ( 6H, m).
(3b) (cis-4-{[5-(2-メチル-4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸
実施例(1e)と同様の方法で、実施例(3a)で得た化合物(178 mg)と実施例(1b)で得た化合物(102 mg)から、アミド体 213 mg(79%)を淡褐色固体として得た。 (3b) (cis-4-{[5- (2-Methyl-4-{[(3-methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidine- 2-yl] oxy} cyclohexyl) acetic acid In the same manner as in Example (1e), from the compound (178 mg) obtained in Example (3a) and the compound (102 mg) obtained in Example (1b), amide 213 mg (79%) of body was obtained as a light brown solid.
実施例(1e)と同様の方法で、実施例(3a)で得た化合物(178 mg)と実施例(1b)で得た化合物(102 mg)から、アミド体 213 mg(79%)を淡褐色固体として得た。 (3b) (cis-4-{[5- (2-Methyl-4-{[(3-methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidine- 2-yl] oxy} cyclohexyl) acetic acid In the same manner as in Example (1e), from the compound (178 mg) obtained in Example (3a) and the compound (102 mg) obtained in Example (1b), amide 213 mg (79%) of body was obtained as a light brown solid.
実施例(1f)と同様の方法で、アミド体(212 mg)から、標記化合物 181 mg (88%)を白色固体として得た。
1H NMR(400MHz,DMSO-d6):δ(ppm)=12.1(1H, brs), 10.1(1H, s), 9.51(1H, s), 8.62(2H, s), 8.56-8.54(1H, m), 8.07-8.03(1H, m), 7.96(1H, d, J=8.2Hz), 7.77(1H, d, J=1.9Hz), 7.70(1H, dd, J=8.2 and 2.3Hz), 7.45-7.42(1H, m), 7.26(1H, d, J=8.2Hz), 5.23-5.22(1H, m), 2.53(3H, s), 2.28(3H, s), 2.19(2H, d, J=7.0Hz), 1.99-1.94(2H, m), 1.87-1.82(1H, m), 1.72-1.65(2H, m), 1.62-1.58(2H, m), 1.44-1.34(2H, m).
MS(ESI) m/z:527 (M + H)+。 In the same manner as in Example (1f), 181 mg (88%) of the title compound was obtained as a white solid from the amide compound (212 mg).
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 12.1 (1H, brs), 10.1 (1H, s), 9.51 (1H, s), 8.62 (2H, s), 8.56-8.54 (1H , m), 8.07-8.03 (1H, m), 7.96 (1H, d, J = 8.2Hz), 7.77 (1H, d, J = 1.9Hz), 7.70 (1H, dd, J = 8.2 and 2.3Hz) , 7.45-7.42 (1H, m), 7.26 (1H, d, J = 8.2Hz), 5.23-5.22 (1H, m), 2.53 (3H, s), 2.28 (3H, s), 2.19 (2H, d , J = 7.0Hz), 1.99-1.94 (2H, m), 1.87-1.82 (1H, m), 1.72-1.65 (2H, m), 1.62-1.58 (2H, m), 1.44-1.34 (2H, m ).
MS (ESI) m / z: 527 (M + H) <+> .
1H NMR(400MHz,DMSO-d6):δ(ppm)=12.1(1H, brs), 10.1(1H, s), 9.51(1H, s), 8.62(2H, s), 8.56-8.54(1H, m), 8.07-8.03(1H, m), 7.96(1H, d, J=8.2Hz), 7.77(1H, d, J=1.9Hz), 7.70(1H, dd, J=8.2 and 2.3Hz), 7.45-7.42(1H, m), 7.26(1H, d, J=8.2Hz), 5.23-5.22(1H, m), 2.53(3H, s), 2.28(3H, s), 2.19(2H, d, J=7.0Hz), 1.99-1.94(2H, m), 1.87-1.82(1H, m), 1.72-1.65(2H, m), 1.62-1.58(2H, m), 1.44-1.34(2H, m).
MS(ESI) m/z:527 (M + H)+。 In the same manner as in Example (1f), 181 mg (88%) of the title compound was obtained as a white solid from the amide compound (212 mg).
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 12.1 (1H, brs), 10.1 (1H, s), 9.51 (1H, s), 8.62 (2H, s), 8.56-8.54 (1H , m), 8.07-8.03 (1H, m), 7.96 (1H, d, J = 8.2Hz), 7.77 (1H, d, J = 1.9Hz), 7.70 (1H, dd, J = 8.2 and 2.3Hz) , 7.45-7.42 (1H, m), 7.26 (1H, d, J = 8.2Hz), 5.23-5.22 (1H, m), 2.53 (3H, s), 2.28 (3H, s), 2.19 (2H, d , J = 7.0Hz), 1.99-1.94 (2H, m), 1.87-1.82 (1H, m), 1.72-1.65 (2H, m), 1.62-1.58 (2H, m), 1.44-1.34 (2H, m ).
MS (ESI) m / z: 527 (M + H) <+> .
(実施例4)
(cis-4-{[5-(4-{[(3-エチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 Example 4
(cis-4-{[5- (4-{[(3-Ethyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid
(cis-4-{[5-(4-{[(3-エチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 Example 4
(cis-4-{[5- (4-{[(3-Ethyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid

(4a) 3-エチル-1-ピリジン-2-イル-1H-ピラゾール-4-カルボン酸メチルエステル
実施例(1a)と同様の方法で、メチル 5-エチル-1H-ピラゾール-4-カルボン酸(WO 2009110520)(256 mg)と2-ブロモピリジン(0.147 mL)から標記化合物76 mg(21%)を無色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=8.98(1H, s), 8.43(1H, d, J=5.1Hz), 7.99(1H, d, J=8.2Hz), 7.84(1H, dd, J=8.8 and 6.9Hz), 7.23(1H, dd, J=7.4 and 4.7Hz), 3.86(3H, s), 3.00(2H, q, J=7.7Hz), 1.33(3H, t, J=7.4Hz)。 (4a) 3-Ethyl-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid methyl ester In the same manner as in Example (1a), methyl 5-ethyl-1H-pyrazole-4-carboxylic acid ( WO 2009110520) (256 mg) and 2-bromopyridine (0.147 mL) gave 76 mg (21%) of the title compound as a colorless solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 8.98 (1H, s), 8.43 (1H, d, J = 5.1 Hz), 7.99 (1H, d, J = 8.2 Hz), 7.84 (1H, dd, J = 8.8 and 6.9Hz), 7.23 (1H, dd, J = 7.4 and 4.7Hz), 3.86 (3H, s), 3.00 (2H, q, J = 7.7Hz), 1.33 (3H, t, J = 7.4Hz).
実施例(1a)と同様の方法で、メチル 5-エチル-1H-ピラゾール-4-カルボン酸(WO 2009110520)(256 mg)と2-ブロモピリジン(0.147 mL)から標記化合物76 mg(21%)を無色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=8.98(1H, s), 8.43(1H, d, J=5.1Hz), 7.99(1H, d, J=8.2Hz), 7.84(1H, dd, J=8.8 and 6.9Hz), 7.23(1H, dd, J=7.4 and 4.7Hz), 3.86(3H, s), 3.00(2H, q, J=7.7Hz), 1.33(3H, t, J=7.4Hz)。 (4a) 3-Ethyl-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid methyl ester In the same manner as in Example (1a), methyl 5-ethyl-1H-pyrazole-4-carboxylic acid ( WO 2009110520) (256 mg) and 2-bromopyridine (0.147 mL) gave 76 mg (21%) of the title compound as a colorless solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 8.98 (1H, s), 8.43 (1H, d, J = 5.1 Hz), 7.99 (1H, d, J = 8.2 Hz), 7.84 (1H, dd, J = 8.8 and 6.9Hz), 7.23 (1H, dd, J = 7.4 and 4.7Hz), 3.86 (3H, s), 3.00 (2H, q, J = 7.7Hz), 1.33 (3H, t, J = 7.4Hz).
(4b) 3-エチル-1-ピリジン-2-イル-1H-ピラゾール-4-カルボン酸
実施例(4a)で得た化合物(75 mg)を実施例(1b)と同様の方法で加水分解し、標記化合物59 mg(90%)を無色固体として得た。
1H NMR(500MHz,CDCl3):δ(ppm)=9.06(1H, s), 8.45(1H, d, J=4.3Hz), 8.00(1H, d, J=8.3Hz), 7.85(1H, dd, J=7.8 and 7.8Hz), 7.27-7.24 (1H, m), 3.02(2H, q, J=7.3Hz), 1.85-1.40(1H, brs), 1.36(3H, t, J=8.3Hz)。 (4b) 3-ethyl-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid The compound (75 mg) obtained in Example (4a) was hydrolyzed in the same manner as in Example (1b). The title compound 59 mg (90%) was obtained as a colorless solid.
1 H NMR (500 MHz, CDCl 3 ): δ (ppm) = 9.06 (1H, s), 8.45 (1H, d, J = 4.3 Hz), 8.00 (1H, d, J = 8.3 Hz), 7.85 (1H, dd, J = 7.8 and 7.8Hz), 7.27-7.24 (1H, m), 3.02 (2H, q, J = 7.3Hz), 1.85-1.40 (1H, brs), 1.36 (3H, t, J = 8.3Hz ).
実施例(4a)で得た化合物(75 mg)を実施例(1b)と同様の方法で加水分解し、標記化合物59 mg(90%)を無色固体として得た。
1H NMR(500MHz,CDCl3):δ(ppm)=9.06(1H, s), 8.45(1H, d, J=4.3Hz), 8.00(1H, d, J=8.3Hz), 7.85(1H, dd, J=7.8 and 7.8Hz), 7.27-7.24 (1H, m), 3.02(2H, q, J=7.3Hz), 1.85-1.40(1H, brs), 1.36(3H, t, J=8.3Hz)。 (4b) 3-ethyl-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid The compound (75 mg) obtained in Example (4a) was hydrolyzed in the same manner as in Example (1b). The title compound 59 mg (90%) was obtained as a colorless solid.
1 H NMR (500 MHz, CDCl 3 ): δ (ppm) = 9.06 (1H, s), 8.45 (1H, d, J = 4.3 Hz), 8.00 (1H, d, J = 8.3 Hz), 7.85 (1H, dd, J = 7.8 and 7.8Hz), 7.27-7.24 (1H, m), 3.02 (2H, q, J = 7.3Hz), 1.85-1.40 (1H, brs), 1.36 (3H, t, J = 8.3Hz ).
(4c) (cis-4-{[5-(4-{[(3-エチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸
実施例(1e)と同様の方法で、実施例(4b)で得た化合物(57 mg)と実施例(1d)で得た化合物(90 mg)から、アミド体 92 mgをオフホワイト色アモルファスとして得た。実施例(1f)と同様の方法で、アミド体(92 mg)を加水分解し、標記化合物 31 mg (22%、2工程)を黄色固体として得た。
1H NMR(400MHz,DMSO-d6):δ(ppm)=12.2-12.0(1H, brs), 10.2(1H, s), 9.49(1H, s), 8.91(2H, s), 8.56-8.54(1H, m), 8.07-8.03(1H, m), 7.97(1H, d, J=8.2Hz), 7.90(2H, d, J=8.6Hz), 7.72(2H, d, J=9.0Hz), 7.45-7.42(1H, m), 5.24-5.21(1H, m), 2.99(2H, q, J=7.5Hz), 2.19(2H, d, J=7.0Hz), 1.98-1.93(2H, m), 1.86-1.80(1H, m), 1.72-1.64(2H, m), 1.62-1.56(2H, m), 1.43-1.34(2H, m), 1.28(3H, t, J=7.5Hz)。 (4c) (cis-4-{[5- (4-{[(3-Ethyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid In the same manner as in Example (1e), from the compound (57 mg) obtained in Example (4b) and the compound (90 mg) obtained in Example (1d), 92 mg of the amide compound was obtained. Obtained as an off-white amorphous. In the same manner as in Example (1f), the amide compound (92 mg) was hydrolyzed to obtain 31 mg (22%, 2 steps) of the title compound as a yellow solid.
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 12.2-12.0 (1H, brs), 10.2 (1H, s), 9.49 (1H, s), 8.91 (2H, s), 8.56-8.54 (1H, m), 8.07-8.03 (1H, m), 7.97 (1H, d, J = 8.2Hz), 7.90 (2H, d, J = 8.6Hz), 7.72 (2H, d, J = 9.0Hz) , 7.45-7.42 (1H, m), 5.24-5.21 (1H, m), 2.99 (2H, q, J = 7.5Hz), 2.19 (2H, d, J = 7.0Hz), 1.98-1.93 (2H, m ), 1.86-1.80 (1H, m), 1.72-1.64 (2H, m), 1.62-1.56 (2H, m), 1.43-1.34 (2H, m), 1.28 (3H, t, J = 7.5Hz).
実施例(1e)と同様の方法で、実施例(4b)で得た化合物(57 mg)と実施例(1d)で得た化合物(90 mg)から、アミド体 92 mgをオフホワイト色アモルファスとして得た。実施例(1f)と同様の方法で、アミド体(92 mg)を加水分解し、標記化合物 31 mg (22%、2工程)を黄色固体として得た。
1H NMR(400MHz,DMSO-d6):δ(ppm)=12.2-12.0(1H, brs), 10.2(1H, s), 9.49(1H, s), 8.91(2H, s), 8.56-8.54(1H, m), 8.07-8.03(1H, m), 7.97(1H, d, J=8.2Hz), 7.90(2H, d, J=8.6Hz), 7.72(2H, d, J=9.0Hz), 7.45-7.42(1H, m), 5.24-5.21(1H, m), 2.99(2H, q, J=7.5Hz), 2.19(2H, d, J=7.0Hz), 1.98-1.93(2H, m), 1.86-1.80(1H, m), 1.72-1.64(2H, m), 1.62-1.56(2H, m), 1.43-1.34(2H, m), 1.28(3H, t, J=7.5Hz)。 (4c) (cis-4-{[5- (4-{[(3-Ethyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid In the same manner as in Example (1e), from the compound (57 mg) obtained in Example (4b) and the compound (90 mg) obtained in Example (1d), 92 mg of the amide compound was obtained. Obtained as an off-white amorphous. In the same manner as in Example (1f), the amide compound (92 mg) was hydrolyzed to obtain 31 mg (22%, 2 steps) of the title compound as a yellow solid.
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 12.2-12.0 (1H, brs), 10.2 (1H, s), 9.49 (1H, s), 8.91 (2H, s), 8.56-8.54 (1H, m), 8.07-8.03 (1H, m), 7.97 (1H, d, J = 8.2Hz), 7.90 (2H, d, J = 8.6Hz), 7.72 (2H, d, J = 9.0Hz) , 7.45-7.42 (1H, m), 5.24-5.21 (1H, m), 2.99 (2H, q, J = 7.5Hz), 2.19 (2H, d, J = 7.0Hz), 1.98-1.93 (2H, m ), 1.86-1.80 (1H, m), 1.72-1.64 (2H, m), 1.62-1.56 (2H, m), 1.43-1.34 (2H, m), 1.28 (3H, t, J = 7.5Hz).
(実施例5)
(cis-4-{[5-(4-{[(1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 (Example 5)
(cis-4-{[5- (4-{[(1-Pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl) acetic acid
(cis-4-{[5-(4-{[(1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 (Example 5)
(cis-4-{[5- (4-{[(1-Pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl) acetic acid

(5a) 1-ピリジン-2-イル-1H-ピラゾール-4-カルボン酸エチルエステル
実施例(1a)と同様の方法で、1H-ピラゾール-4-カルボン酸エチルエステル(1.4 g)と2-ブロモピリジン(0.955 mL)から標記化合物1.56 g(72%)を無色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=9.05(1H, s), 8.45(1H, d, J=5.0Hz), 8.11(1H, s), 8.01(1H, d, J=8.2Hz), 7.82(1H, dd, J=8.2 and 7.5Hz), 7.25(1H, dd, J=7.4 and 5.1Hz), 4.35(2H, q, J=7Hz), 1.47(3H, t, J=7Hz)。 (5a) 1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid ethyl ester In the same manner as in Example (1a), 1H-pyrazole-4-carboxylic acid ethyl ester (1.4 g) and 2-bromo The title compound (1.56 g, 72%) was obtained as a colorless solid from pyridine (0.955 mL).
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 9.05 (1H, s), 8.45 (1H, d, J = 5.0 Hz), 8.11 (1H, s), 8.01 (1H, d, J = 8.2 Hz), 7.82 (1H, dd, J = 8.2 and 7.5Hz), 7.25 (1H, dd, J = 7.4 and 5.1Hz), 4.35 (2H, q, J = 7Hz), 1.47 (3H, t, J = 7Hz).
実施例(1a)と同様の方法で、1H-ピラゾール-4-カルボン酸エチルエステル(1.4 g)と2-ブロモピリジン(0.955 mL)から標記化合物1.56 g(72%)を無色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=9.05(1H, s), 8.45(1H, d, J=5.0Hz), 8.11(1H, s), 8.01(1H, d, J=8.2Hz), 7.82(1H, dd, J=8.2 and 7.5Hz), 7.25(1H, dd, J=7.4 and 5.1Hz), 4.35(2H, q, J=7Hz), 1.47(3H, t, J=7Hz)。 (5a) 1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid ethyl ester In the same manner as in Example (1a), 1H-pyrazole-4-carboxylic acid ethyl ester (1.4 g) and 2-bromo The title compound (1.56 g, 72%) was obtained as a colorless solid from pyridine (0.955 mL).
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 9.05 (1H, s), 8.45 (1H, d, J = 5.0 Hz), 8.11 (1H, s), 8.01 (1H, d, J = 8.2 Hz), 7.82 (1H, dd, J = 8.2 and 7.5Hz), 7.25 (1H, dd, J = 7.4 and 5.1Hz), 4.35 (2H, q, J = 7Hz), 1.47 (3H, t, J = 7Hz).
(5b) 1-ピリジン-2-イル-1H-ピラゾール-4-カルボン酸
実施例(5a)で得た化合物(1.06 g)を実施例(1b)と同様の方法で加水分解し、標記化合物595 mg(64%)を無色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=9.15(1H, s), 8.48(1H, d, J=5.0Hz), 8.18(1H, s), 8.04(1H, d, J=8.2Hz), 7.89(1H, dd, J=8.2 and 7.5Hz), 7.30(1H, dd, J=7.4 and 5.1Hz), 2.02-1.40(1H, brs)。 (5b) 1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid The compound (1.06 g) obtained in Example (5a) was hydrolyzed in the same manner as in Example (1b) to give the title compound 595 mg (64%) was obtained as a colorless solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 9.15 (1H, s), 8.48 (1H, d, J = 5.0 Hz), 8.18 (1H, s), 8.04 (1H, d, J = 8.2 Hz), 7.89 (1H, dd, J = 8.2 and 7.5 Hz), 7.30 (1H, dd, J = 7.4 and 5.1 Hz), 2.02-1.40 (1H, brs).
実施例(5a)で得た化合物(1.06 g)を実施例(1b)と同様の方法で加水分解し、標記化合物595 mg(64%)を無色固体として得た。
1H NMR(400MHz,CDCl3):δ(ppm)=9.15(1H, s), 8.48(1H, d, J=5.0Hz), 8.18(1H, s), 8.04(1H, d, J=8.2Hz), 7.89(1H, dd, J=8.2 and 7.5Hz), 7.30(1H, dd, J=7.4 and 5.1Hz), 2.02-1.40(1H, brs)。 (5b) 1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid The compound (1.06 g) obtained in Example (5a) was hydrolyzed in the same manner as in Example (1b) to give the title compound 595 mg (64%) was obtained as a colorless solid.
1 H NMR (400 MHz, CDCl 3 ): δ (ppm) = 9.15 (1H, s), 8.48 (1H, d, J = 5.0 Hz), 8.18 (1H, s), 8.04 (1H, d, J = 8.2 Hz), 7.89 (1H, dd, J = 8.2 and 7.5 Hz), 7.30 (1H, dd, J = 7.4 and 5.1 Hz), 2.02-1.40 (1H, brs).
(5c) (cis-4-{[5-(4-{[(1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸
実施例(1e)と同様の方法で、実施例(5b)で得た化合物(50 mg)と実施例(1d)で得た化合物(90 mg)から、アミド体 104 mg(78%)をオフホワイト色固体として得た。実施例(1f)と同様の方法で、アミド体(100 mg)を加水分解し、標記化合物 81 mg (84%)を淡黄色固体として得た。
1H NMR(400MHz,DMSO-d6):δ(ppm)=12.7-11.8(1H, brs), 10.2(1H, s), 9.46(1H, d, J=0.8Hz), 8.91(2H, s), 8.59-8.57(1H, m), 8.33(1H, d, J=0.7Hz), 8.11-8.06(1H, m), 8.02(1H, d, J=8.3Hz), 7.90(2H, d, J=9.0Hz), 7.74(2H, d, J=8.6Hz), 7.49-7.46(1H, m), 5.24-5.20(1H, m), 2.19(2H, d, J=7.1Hz), 1.99-1.92(2H, m), 1.87-1.80(1H, m), 1.72-1.55(4H, m), 1.43-1.33(2H, m)。 (5c) (cis-4-{[5- (4-{[(1-Pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl) Acetic acid In the same manner as in Example (1e), 104 mg (78%) of the amide compound was obtained from the compound (50 mg) obtained in Example (5b) and the compound (90 mg) obtained in Example (1d). Obtained as an off-white solid. In the same manner as in Example (1f), the amide compound (100 mg) was hydrolyzed to obtain 81 mg (84%) of the title compound as a pale yellow solid.
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 12.7-11.8 (1H, brs), 10.2 (1H, s), 9.46 (1H, d, J = 0.8 Hz), 8.91 (2H, s ), 8.59-8.57 (1H, m), 8.33 (1H, d, J = 0.7Hz), 8.11-8.06 (1H, m), 8.02 (1H, d, J = 8.3Hz), 7.90 (2H, d, J = 9.0Hz), 7.74 (2H, d, J = 8.6Hz), 7.49-7.46 (1H, m), 5.24-5.20 (1H, m), 2.19 (2H, d, J = 7.1Hz), 1.99- 1.92 (2H, m), 1.87-1.80 (1H, m), 1.72-1.55 (4H, m), 1.43-1.33 (2H, m).
実施例(1e)と同様の方法で、実施例(5b)で得た化合物(50 mg)と実施例(1d)で得た化合物(90 mg)から、アミド体 104 mg(78%)をオフホワイト色固体として得た。実施例(1f)と同様の方法で、アミド体(100 mg)を加水分解し、標記化合物 81 mg (84%)を淡黄色固体として得た。
1H NMR(400MHz,DMSO-d6):δ(ppm)=12.7-11.8(1H, brs), 10.2(1H, s), 9.46(1H, d, J=0.8Hz), 8.91(2H, s), 8.59-8.57(1H, m), 8.33(1H, d, J=0.7Hz), 8.11-8.06(1H, m), 8.02(1H, d, J=8.3Hz), 7.90(2H, d, J=9.0Hz), 7.74(2H, d, J=8.6Hz), 7.49-7.46(1H, m), 5.24-5.20(1H, m), 2.19(2H, d, J=7.1Hz), 1.99-1.92(2H, m), 1.87-1.80(1H, m), 1.72-1.55(4H, m), 1.43-1.33(2H, m)。 (5c) (cis-4-{[5- (4-{[(1-Pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl) Acetic acid In the same manner as in Example (1e), 104 mg (78%) of the amide compound was obtained from the compound (50 mg) obtained in Example (5b) and the compound (90 mg) obtained in Example (1d). Obtained as an off-white solid. In the same manner as in Example (1f), the amide compound (100 mg) was hydrolyzed to obtain 81 mg (84%) of the title compound as a pale yellow solid.
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 12.7-11.8 (1H, brs), 10.2 (1H, s), 9.46 (1H, d, J = 0.8 Hz), 8.91 (2H, s ), 8.59-8.57 (1H, m), 8.33 (1H, d, J = 0.7Hz), 8.11-8.06 (1H, m), 8.02 (1H, d, J = 8.3Hz), 7.90 (2H, d, J = 9.0Hz), 7.74 (2H, d, J = 8.6Hz), 7.49-7.46 (1H, m), 5.24-5.20 (1H, m), 2.19 (2H, d, J = 7.1Hz), 1.99- 1.92 (2H, m), 1.87-1.80 (1H, m), 1.72-1.55 (4H, m), 1.43-1.33 (2H, m).
(実施例6)
(cis-4-{[5-(2-メチル-4-{[(1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 (Example 6)
(cis-4-{[5- (2-Methyl-4-{[(1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid
(cis-4-{[5-(2-メチル-4-{[(1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 (Example 6)
(cis-4-{[5- (2-Methyl-4-{[(1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid

実施例(1e)と同様の方法で、実施例(5b)で得た化合物(50 mg)と実施例(3a)で得た化合物(90 mg)から、アミド体 104 mg(78%)をオフホワイト色固体として得た。実施例(1f)と同様の方法で、アミド体(100 mg)を加水分解し、標記化合物 81 mg (84%)を淡黄色固体として得た。
1H NMR(400MHz,DMSO-d6):δ(ppm)=12.2-12.0(1H, brs), 10.2(1H, s), 9.45(1H, s), 8.62(2H, s), 8.58-8.57(1H, m), 8.32(1H, s), 8.08(1H, dt, J=11.1 and 3.9Hz), 8.01(1H, d, J=8.2Hz), 7.75-7.73(1H, m), 7.49-7.46(1H, m), 7.29-7.27(1H, m), 5.24-5.19(1H, m), 2.29(3H, s), 2.19(2H, d, J=7.1Hz), 1.99-1.95(2H, m), 1.87-1.81(1H, m), 1.72-1.58(4H, m), 1.44-1.35(2H, m)。 In the same manner as in Example (1e), 104 mg (78%) of the amide compound was turned off from the compound (50 mg) obtained in Example (5b) and the compound (90 mg) obtained in Example (3a). Obtained as a white solid. In the same manner as in Example (1f), the amide compound (100 mg) was hydrolyzed to obtain 81 mg (84%) of the title compound as a pale yellow solid.
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 12.2-12.0 (1H, brs), 10.2 (1H, s), 9.45 (1H, s), 8.62 (2H, s), 8.58-8.57 (1H, m), 8.32 (1H, s), 8.08 (1H, dt, J = 11.1 and 3.9Hz), 8.01 (1H, d, J = 8.2Hz), 7.75-7.73 (1H, m), 7.49- 7.46 (1H, m), 7.29-7.27 (1H, m), 5.24-5.19 (1H, m), 2.29 (3H, s), 2.19 (2H, d, J = 7.1Hz), 1.99-1.95 (2H, m), 1.87-1.81 (1H, m), 1.72-1.58 (4H, m), 1.44-1.35 (2H, m).
1H NMR(400MHz,DMSO-d6):δ(ppm)=12.2-12.0(1H, brs), 10.2(1H, s), 9.45(1H, s), 8.62(2H, s), 8.58-8.57(1H, m), 8.32(1H, s), 8.08(1H, dt, J=11.1 and 3.9Hz), 8.01(1H, d, J=8.2Hz), 7.75-7.73(1H, m), 7.49-7.46(1H, m), 7.29-7.27(1H, m), 5.24-5.19(1H, m), 2.29(3H, s), 2.19(2H, d, J=7.1Hz), 1.99-1.95(2H, m), 1.87-1.81(1H, m), 1.72-1.58(4H, m), 1.44-1.35(2H, m)。 In the same manner as in Example (1e), 104 mg (78%) of the amide compound was turned off from the compound (50 mg) obtained in Example (5b) and the compound (90 mg) obtained in Example (3a). Obtained as a white solid. In the same manner as in Example (1f), the amide compound (100 mg) was hydrolyzed to obtain 81 mg (84%) of the title compound as a pale yellow solid.
1 H NMR (400 MHz, DMSO-d 6 ): δ (ppm) = 12.2-12.0 (1H, brs), 10.2 (1H, s), 9.45 (1H, s), 8.62 (2H, s), 8.58-8.57 (1H, m), 8.32 (1H, s), 8.08 (1H, dt, J = 11.1 and 3.9Hz), 8.01 (1H, d, J = 8.2Hz), 7.75-7.73 (1H, m), 7.49- 7.46 (1H, m), 7.29-7.27 (1H, m), 5.24-5.19 (1H, m), 2.29 (3H, s), 2.19 (2H, d, J = 7.1Hz), 1.99-1.95 (2H, m), 1.87-1.81 (1H, m), 1.72-1.58 (4H, m), 1.44-1.35 (2H, m).
(実施例7)
(cis-4-{[5-(4-{[(3-クロロ-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 (Example 7)
(cis-4-{[5- (4-{[(3-Chloro-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid
(cis-4-{[5-(4-{[(3-クロロ-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸 (Example 7)
(cis-4-{[5- (4-{[(3-Chloro-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid
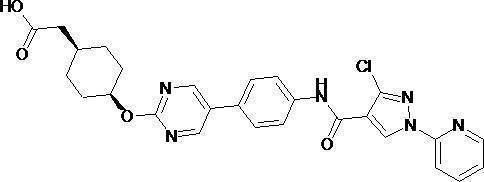
(7a) (cis-4-{[5-(4-{[(3-クロロ-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸メチルエステル
3-クロロ-1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸エチルエステル(WO2010079239)(173 mg)のトリフルオロ酢酸(10 mL)溶液を6時間加熱還流した。反応混合物を濃縮し、ジクロロメタンと飽和炭酸水素ナトリウム溶液で希釈し、ジクロロメタンで抽出した。有機層を水と飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、そして濃縮した。 (7a) (cis-4-{[5- (4-{[(3-Chloro-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid methyl ester 3-chloro-1- (4-methoxybenzyl) -1H-pyrazole-4-carboxylic acid ethyl ester (WO2010079239) (173 mg) in trifluoroacetic acid (10 mL) heated for 6 hours Refluxed. The reaction mixture was concentrated, diluted with dichloromethane and saturated sodium bicarbonate solution and extracted with dichloromethane. The organic layer was washed with water and saturated brine, dried over sodium sulfate, and concentrated.
3-クロロ-1-(4-メトキシベンジル)-1H-ピラゾール-4-カルボン酸エチルエステル(WO2010079239)(173 mg)のトリフルオロ酢酸(10 mL)溶液を6時間加熱還流した。反応混合物を濃縮し、ジクロロメタンと飽和炭酸水素ナトリウム溶液で希釈し、ジクロロメタンで抽出した。有機層を水と飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、そして濃縮した。 (7a) (cis-4-{[5- (4-{[(3-Chloro-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid methyl ester 3-chloro-1- (4-methoxybenzyl) -1H-pyrazole-4-carboxylic acid ethyl ester (WO2010079239) (173 mg) in trifluoroacetic acid (10 mL) heated for 6 hours Refluxed. The reaction mixture was concentrated, diluted with dichloromethane and saturated sodium bicarbonate solution and extracted with dichloromethane. The organic layer was washed with water and saturated brine, dried over sodium sulfate, and concentrated.
この残渣物(3-クロロ-1H-ピラゾール-4-カルボン酸エチルエステル 、102 mg)、2-ブロモピリジン(0.063 mL)、ヨウ化銅(I)(8.9 mg)、N,N'-ジメチルシクロヘキサン-1,2-ジアミン(13 mg)、炭酸カリウム(242 mg)、そしてトルエン(10 mL)の懸濁液を8.5時間加熱還流した。反応混合物を室温に冷却後、セライトでろ過した。ろ液をクロマトグラフィー(ヘキサン/酢酸エチル 5%→10%)で精製し、123 mgの粗生成物(3-クロロ-1-ピリジン-2-イル-1H-ピラゾール-4-カルボン酸エチルエステル)を得た。
This residue (3-chloro-1H-pyrazole-4-carboxylic acid ethyl ester, 102 mg), 2-bromopyridine (0.063 mL), copper (I) iodide (8.9 mg), N, N'-dimethylcyclohexane A suspension of -1,2-diamine (13 mg), potassium carbonate (242 mg), and toluene (10 mL) was heated to reflux for 8.5 hours. The reaction mixture was cooled to room temperature and filtered through celite. The filtrate was purified by chromatography (hexane / ethyl acetate 5% → 10%) to give 123 mg of crude product (3-chloro-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid ethyl ester) Got.
このビアリール体(123 mg)を実施例(1b)と同様の方法で加水分解し、カルボン酸体(3-クロロ-1-ピリジン-2-イル-1H-ピラゾール-4-カルボン酸)を得た。
This biaryl compound (123 mg) was hydrolyzed in the same manner as in Example (1b) to obtain a carboxylic acid compound (3-chloro-1-pyridin-2-yl-1H-pyrazole-4-carboxylic acid). .
実施例(1e)と同様の方法で、このカルボン酸体(109 mg)と実施例(1d)で得た化合物(178 mg)から、アミド体 101 mg(31%、4工程)を淡黄色固体として得た。
1H NMR (500MHz, CDCl3): δ(ppm) = 9.21 (1H, s), 8.70 (2H, s), 8.48 (1H, d, J = 4.9 Hz), 8.40 (1H, brs), 7.96 (1H, d, J = 8.3 Hz), 7.89 (1H, t, J = 8.3 Hz), 7.78 (2H, d, J = 8.8 Hz), 7.54 (2H, d, J = 8.3 Hz), 7.31 (1H, t, J = 5.9 Hz), 5.33-5.29 (1H, m), 3.68 (3H, s), 2.30 (2H, d, J = 6.8 Hz), 2.14-2.09 (2H, m), 1.98-1.92 (1H, m), 1.73-1.53 (6H, m)。 In the same manner as in Example (1e), from this carboxylic acid form (109 mg) and the compound (178 mg) obtained in Example (1d), 101 mg (31%, 4 steps) of amide form was obtained as a pale yellow solid. Got as.
1 H NMR (500MHz, CDCl3): δ (ppm) = 9.21 (1H, s), 8.70 (2H, s), 8.48 (1H, d, J = 4.9 Hz), 8.40 (1H, brs), 7.96 (1H , d, J = 8.3 Hz), 7.89 (1H, t, J = 8.3 Hz), 7.78 (2H, d, J = 8.8 Hz), 7.54 (2H, d, J = 8.3 Hz), 7.31 (1H, t , J = 5.9 Hz), 5.33-5.29 (1H, m), 3.68 (3H, s), 2.30 (2H, d, J = 6.8 Hz), 2.14-2.09 (2H, m), 1.98-1.92 (1H, m), 1.73-1.53 (6H, m).
1H NMR (500MHz, CDCl3): δ(ppm) = 9.21 (1H, s), 8.70 (2H, s), 8.48 (1H, d, J = 4.9 Hz), 8.40 (1H, brs), 7.96 (1H, d, J = 8.3 Hz), 7.89 (1H, t, J = 8.3 Hz), 7.78 (2H, d, J = 8.8 Hz), 7.54 (2H, d, J = 8.3 Hz), 7.31 (1H, t, J = 5.9 Hz), 5.33-5.29 (1H, m), 3.68 (3H, s), 2.30 (2H, d, J = 6.8 Hz), 2.14-2.09 (2H, m), 1.98-1.92 (1H, m), 1.73-1.53 (6H, m)。 In the same manner as in Example (1e), from this carboxylic acid form (109 mg) and the compound (178 mg) obtained in Example (1d), 101 mg (31%, 4 steps) of amide form was obtained as a pale yellow solid. Got as.
1 H NMR (500MHz, CDCl3): δ (ppm) = 9.21 (1H, s), 8.70 (2H, s), 8.48 (1H, d, J = 4.9 Hz), 8.40 (1H, brs), 7.96 (1H , d, J = 8.3 Hz), 7.89 (1H, t, J = 8.3 Hz), 7.78 (2H, d, J = 8.8 Hz), 7.54 (2H, d, J = 8.3 Hz), 7.31 (1H, t , J = 5.9 Hz), 5.33-5.29 (1H, m), 3.68 (3H, s), 2.30 (2H, d, J = 6.8 Hz), 2.14-2.09 (2H, m), 1.98-1.92 (1H, m), 1.73-1.53 (6H, m).
(7b) (cis-4-{[5-(4-{[(3-クロロ-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸
実施例(1f)と同様の方法で、実施例(7a)で得た化合物(101 mg)を加水分解し、標記化合物 82 mg (84%)を淡黄色固体として得た。
1H-NMR (500 MHz, DMSO-d6) : δ (ppm) = 12.04 (1H, brs), 10.31(1H, s), 9.54 (1H, s), 8.91(2H, s), 8.59 (1H, d, J = 3.9 Hz), 8.10 (1H, t, J = 7.3 Hz), 7.95 (1H, d, J = 7.4 Hz), 7.87 (2H, d, J = 8.8 Hz), 7.74 (2H, d, J = 8.8 Hz), 7.52-7.50 (1H, m), 5.24-5.20 (1H, m), 2.20 (2H, d, J = 6.8 Hz), 1.99-1.93 (3H, m), 1.71-1.56 (4H, m), 1.42-1.35 (2H, m)。 (7b) (cis-4-{[5- (4-{[(3-Chloro-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid In the same manner as in Example (1f), the compound (101 mg) obtained in Example (7a) was hydrolyzed to obtain 82 mg (84%) of the title compound as a pale yellow solid.
1 H-NMR (500 MHz, DMSO-d 6 ): δ (ppm) = 12.04 (1H, brs), 10.31 (1H, s), 9.54 (1H, s), 8.91 (2H, s), 8.59 (1H , d, J = 3.9 Hz), 8.10 (1H, t, J = 7.3 Hz), 7.95 (1H, d, J = 7.4 Hz), 7.87 (2H, d, J = 8.8 Hz), 7.74 (2H, d , J = 8.8 Hz), 7.52-7.50 (1H, m), 5.24-5.20 (1H, m), 2.20 (2H, d, J = 6.8 Hz), 1.99-1.93 (3H, m), 1.71-1.56 ( 4H, m), 1.42-1.35 (2H, m).
実施例(1f)と同様の方法で、実施例(7a)で得た化合物(101 mg)を加水分解し、標記化合物 82 mg (84%)を淡黄色固体として得た。
1H-NMR (500 MHz, DMSO-d6) : δ (ppm) = 12.04 (1H, brs), 10.31(1H, s), 9.54 (1H, s), 8.91(2H, s), 8.59 (1H, d, J = 3.9 Hz), 8.10 (1H, t, J = 7.3 Hz), 7.95 (1H, d, J = 7.4 Hz), 7.87 (2H, d, J = 8.8 Hz), 7.74 (2H, d, J = 8.8 Hz), 7.52-7.50 (1H, m), 5.24-5.20 (1H, m), 2.20 (2H, d, J = 6.8 Hz), 1.99-1.93 (3H, m), 1.71-1.56 (4H, m), 1.42-1.35 (2H, m)。 (7b) (cis-4-{[5- (4-{[(3-Chloro-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] Oxy} cyclohexyl) acetic acid In the same manner as in Example (1f), the compound (101 mg) obtained in Example (7a) was hydrolyzed to obtain 82 mg (84%) of the title compound as a pale yellow solid.
1 H-NMR (500 MHz, DMSO-d 6 ): δ (ppm) = 12.04 (1H, brs), 10.31 (1H, s), 9.54 (1H, s), 8.91 (2H, s), 8.59 (1H , d, J = 3.9 Hz), 8.10 (1H, t, J = 7.3 Hz), 7.95 (1H, d, J = 7.4 Hz), 7.87 (2H, d, J = 8.8 Hz), 7.74 (2H, d , J = 8.8 Hz), 7.52-7.50 (1H, m), 5.24-5.20 (1H, m), 2.20 (2H, d, J = 6.8 Hz), 1.99-1.93 (3H, m), 1.71-1.56 ( 4H, m), 1.42-1.35 (2H, m).
(試験例1)
(1)DGAT1酵素の調製
US2007/0249620号公報に記載されている方法に従って、DGAT1酵素を調整、保存した。
(2)DGAT1阻害活性試験
以下の組成の反応液[175 mM Tris-HCl (pH 8.0)、8 mM MgCl2、1 mg/ml BSA、0.3 mM 1,2-dioleoyl-sn-glycerol(10倍濃度のEtOH溶液を10%添加)、10μM [14C]-oleoyl-CoA (約50 mCi/mmol)、0.5% triton X-100、試験例1(1)で得られるDGAT1酵素(10μg)、試験化合物またはビークル(DMSO/MeOH, 7:3溶液、5%添加)、総容量50μl]を室温(23℃)で30分間インキュベーションした。反応液にイソプロパノール/1-ヘプタン/水(80:20:2, v/v/v)からなる反応停止液(70μl)を加えて撹拌し、次いで、水(30μl)および1-ヘプタン(100μl)を加えて撹拌した。1-ヘプタン層(50μl)をTLCプレートにスポットして、1-ヘキサン/ジエチルエーテル/酢酸(85:15:1, v/v/v)からなる展開溶媒にて展開した。BAS2000バイオイメージアナライザ(富士フィルム)によりトリグリセライド画分の放射活性を定量して、コントロールと比較することにより試験化合物の阻害活性を以下の式により算出した。なお、未反応(0分間インキュベーション)の放射活性をバックグラウンドとした。 (Test Example 1)
(1) Preparation of DGAT1 enzyme The DGAT1 enzyme was prepared and stored in accordance with the method described in US2007 / 0249620.
(2) DGAT1 inhibitory activity test Reaction solution [175 mM Tris-HCl (pH 8.0), 8 mM MgCl 2 , 1 mg / ml BSA, 0.3 mM 1,2-dioleoyl-sn-glycerol (10-fold concentration) 10% EtOH solution), 10 μM [ 14 C] -oleoyl-CoA (about 50 mCi / mmol), 0.5% triton X-100, DGAT1 enzyme (10 μg) obtained in Test Example 1 (1), test compound Or a vehicle (DMSO / MeOH, 7: 3 solution, 5% added), total volume 50 μl] was incubated at room temperature (23 ° C.) for 30 minutes. To the reaction mixture was added a reaction stop solution (70 μl) consisting of isopropanol / 1-heptane / water (80: 20: 2, v / v / v) and stirred, then water (30 μl) and 1-heptane (100 μl) Was added and stirred. A 1-heptane layer (50 μl) was spotted on a TLC plate and developed with a developing solvent consisting of 1-hexane / diethyl ether / acetic acid (85: 15: 1, v / v / v). The radioactivity of the triglyceride fraction was quantified with a BAS2000 bioimage analyzer (Fuji Film), and the inhibitory activity of the test compound was calculated by the following formula by comparing with the control. The unreacted (0 minute incubation) radioactivity was used as the background.
(1)DGAT1酵素の調製
US2007/0249620号公報に記載されている方法に従って、DGAT1酵素を調整、保存した。
(2)DGAT1阻害活性試験
以下の組成の反応液[175 mM Tris-HCl (pH 8.0)、8 mM MgCl2、1 mg/ml BSA、0.3 mM 1,2-dioleoyl-sn-glycerol(10倍濃度のEtOH溶液を10%添加)、10μM [14C]-oleoyl-CoA (約50 mCi/mmol)、0.5% triton X-100、試験例1(1)で得られるDGAT1酵素(10μg)、試験化合物またはビークル(DMSO/MeOH, 7:3溶液、5%添加)、総容量50μl]を室温(23℃)で30分間インキュベーションした。反応液にイソプロパノール/1-ヘプタン/水(80:20:2, v/v/v)からなる反応停止液(70μl)を加えて撹拌し、次いで、水(30μl)および1-ヘプタン(100μl)を加えて撹拌した。1-ヘプタン層(50μl)をTLCプレートにスポットして、1-ヘキサン/ジエチルエーテル/酢酸(85:15:1, v/v/v)からなる展開溶媒にて展開した。BAS2000バイオイメージアナライザ(富士フィルム)によりトリグリセライド画分の放射活性を定量して、コントロールと比較することにより試験化合物の阻害活性を以下の式により算出した。なお、未反応(0分間インキュベーション)の放射活性をバックグラウンドとした。 (Test Example 1)
(1) Preparation of DGAT1 enzyme The DGAT1 enzyme was prepared and stored in accordance with the method described in US2007 / 0249620.
(2) DGAT1 inhibitory activity test Reaction solution [175 mM Tris-HCl (pH 8.0), 8 mM MgCl 2 , 1 mg / ml BSA, 0.3 mM 1,2-dioleoyl-sn-glycerol (10-fold concentration) 10% EtOH solution), 10 μM [ 14 C] -oleoyl-CoA (about 50 mCi / mmol), 0.5% triton X-100, DGAT1 enzyme (10 μg) obtained in Test Example 1 (1), test compound Or a vehicle (DMSO / MeOH, 7: 3 solution, 5% added), total volume 50 μl] was incubated at room temperature (23 ° C.) for 30 minutes. To the reaction mixture was added a reaction stop solution (70 μl) consisting of isopropanol / 1-heptane / water (80: 20: 2, v / v / v) and stirred, then water (30 μl) and 1-heptane (100 μl) Was added and stirred. A 1-heptane layer (50 μl) was spotted on a TLC plate and developed with a developing solvent consisting of 1-hexane / diethyl ether / acetic acid (85: 15: 1, v / v / v). The radioactivity of the triglyceride fraction was quantified with a BAS2000 bioimage analyzer (Fuji Film), and the inhibitory activity of the test compound was calculated by the following formula by comparing with the control. The unreacted (0 minute incubation) radioactivity was used as the background.
阻害率 = 100-[(試験化合物添加時の放射活性)-(バックグラウンド)]/[(コントロールの放射活性)-(バックグラウンド)]×100
実施例1乃至7の化合物は、試験化合物濃度1 μg/mlにおいて50%以上の阻害率を示した。 Inhibition rate = 100 − [(radioactivity at the time of addition of test compound) − (background)] / [(radioactivity of control) − (background)] × 100
The compounds of Examples 1 to 7 showed an inhibition rate of 50% or more at a test compound concentration of 1 μg / ml.
実施例1乃至7の化合物は、試験化合物濃度1 μg/mlにおいて50%以上の阻害率を示した。 Inhibition rate = 100 − [(radioactivity at the time of addition of test compound) − (background)] / [(radioactivity of control) − (background)] × 100
The compounds of Examples 1 to 7 showed an inhibition rate of 50% or more at a test compound concentration of 1 μg / ml.
なお、DGAT阻害活性試験は上記の方法に限定されず、例えば、ラット、マウス等の動物の小腸、脂肪組織または肝臓から調製したミクロソームをDGAT酵素として使用してもよい。また、培養細胞(3T3-L1脂肪細胞、初代培養脂肪細胞、Caco2細胞、HepG2細胞等)またはDGATを高発現させた培養細胞から調製したミクロソームをDGAT酵素として使用することもできる。さらに、多数の試験化合物を短時間で効率よく評価するためには、抽出操作を省略したフラッシュプレート(PerkinElmer)を使用することができる。
The DGAT inhibitory activity test is not limited to the above method. For example, microsomes prepared from the small intestine, adipose tissue, or liver of animals such as rats and mice may be used as the DGAT enzyme. Alternatively, microsomes prepared from cultured cells (3T3-L1 adipocytes, primary cultured adipocytes, Caco2 cells, HepG2 cells, etc.) or cultured cells highly expressing DGAT can also be used as the DGAT enzyme. Furthermore, in order to efficiently evaluate a large number of test compounds in a short time, a flash plate (PerkinElmer) in which the extraction operation is omitted can be used.
上記の結果から、本発明の化合物は、優れたDGAT1阻害生物活性を有する。
From the above results, the compound of the present invention has excellent DGAT1 inhibitory biological activity.
(試験例2)
DGAT1酵素は中性脂肪の消化吸収に重要であり、小腸DGAT1が阻害されると中性脂肪の吸収が抑制される。中性脂肪負荷後の中性脂肪吸収抑制を指標として、DGAT1阻害作用の生物活性を評価した。1晩絶食させた雄性C57BL/6Nマウス(7-12週齡、体重17-25 g、日本チャールズリバー)をVehicle群1、Vehicle群2および各試験化合物群に割り付け、それぞれvehicle(0.5% Methylcellulose)またはvehicleに懸濁させた各試験化合物(1乃至10 mg/kg)を経口投与(5 mL/kg)した。一定時間後にリポプロテインリパーゼ阻害剤(Pluronic-F127:シグマアルドリッチ(株)、1 g/kg、重量比20%で生理食塩水に溶解)を腹腔内投与(5 mL/kg)し、直後に、Vehicle群1には蒸留水を、Vehicle群2および化合物群には20%中性脂肪含有エマルジョン (イントラリピッド20%:テルモ(株))を経口投与(0.2 mL/マウス)した。投与後1乃至4時間の一定時間後に、尾静脈または右心室より採血を行い、速やかに血漿を分離回収した後、血漿中の中性脂肪濃度を市販のキット (トリグリセライド E テスト ワコー:和光純薬工業(株))を用いて測定した。本法ではリポプロテインリパーゼ阻害剤の投与により血中に流入した中性脂肪の分解が抑制され、中性脂肪は血中に蓄積するが、その由来は消化管にて吸収された外因性のものと肝臓より放出された内因性のものに二分される。各試験化合物の中性脂肪吸収抑制活性は下記計算式に基づき、内因性中性脂肪の影響を除いて算出した。なお、各試験化合物が内因性中性脂肪濃度に影響しないことは別途確認されている。 (Test Example 2)
The DGAT1 enzyme is important for digestion and absorption of neutral fat, and when small intestine DGAT1 is inhibited, the absorption of neutral fat is suppressed. The biological activity of the DGAT1 inhibitory action was evaluated using as an index the inhibition of neutral fat absorption after neutral fat loading. Male C57BL / 6N mice (7-12 weeks old, body weight 17-25 g, Nippon Charles River) fasted overnight were assigned to Vehicle Group 1, Vehicle Group 2 and each test compound group, respectively vehicle (0.5% Methylcellulose) Alternatively, each test compound (1 to 10 mg / kg) suspended in the vehicle was orally administered (5 mL / kg). Lipoprotein lipase inhibitor (Pluronic-F127: Sigma-Aldrich Co., Ltd., 1 g / kg, dissolved in physiological saline at 20% by weight) was intraperitoneally administered (5 mL / kg) Distilled water was orally administered to Vehicle Group 1 and 20% neutral fat-containing emulsion (Intralipid 20%: Terumo Corporation) was orally administered (0.2 mL / mouse) to Vehicle Group 2 and Compound Group. After 1 to 4 hours after administration, blood is collected from the tail vein or right ventricle, and plasma is promptly separated and collected, and then the neutral fat concentration in the plasma is measured using a commercially available kit (Triglyceride E Test Wako: Wako Pure Chemical Industries, Ltd.) (Industry Co., Ltd.). In this method, the administration of lipoprotein lipase inhibitor suppresses the degradation of neutral fat flowing into the blood, and neutral fat accumulates in the blood, but its origin is exogenous absorbed in the digestive tract And it is divided into endogenous ones released from the liver. The neutral fat absorption inhibitory activity of each test compound was calculated based on the following formula, excluding the influence of endogenous neutral fat. It has been separately confirmed that each test compound does not affect the endogenous triglyceride concentration.
DGAT1酵素は中性脂肪の消化吸収に重要であり、小腸DGAT1が阻害されると中性脂肪の吸収が抑制される。中性脂肪負荷後の中性脂肪吸収抑制を指標として、DGAT1阻害作用の生物活性を評価した。1晩絶食させた雄性C57BL/6Nマウス(7-12週齡、体重17-25 g、日本チャールズリバー)をVehicle群1、Vehicle群2および各試験化合物群に割り付け、それぞれvehicle(0.5% Methylcellulose)またはvehicleに懸濁させた各試験化合物(1乃至10 mg/kg)を経口投与(5 mL/kg)した。一定時間後にリポプロテインリパーゼ阻害剤(Pluronic-F127:シグマアルドリッチ(株)、1 g/kg、重量比20%で生理食塩水に溶解)を腹腔内投与(5 mL/kg)し、直後に、Vehicle群1には蒸留水を、Vehicle群2および化合物群には20%中性脂肪含有エマルジョン (イントラリピッド20%:テルモ(株))を経口投与(0.2 mL/マウス)した。投与後1乃至4時間の一定時間後に、尾静脈または右心室より採血を行い、速やかに血漿を分離回収した後、血漿中の中性脂肪濃度を市販のキット (トリグリセライド E テスト ワコー:和光純薬工業(株))を用いて測定した。本法ではリポプロテインリパーゼ阻害剤の投与により血中に流入した中性脂肪の分解が抑制され、中性脂肪は血中に蓄積するが、その由来は消化管にて吸収された外因性のものと肝臓より放出された内因性のものに二分される。各試験化合物の中性脂肪吸収抑制活性は下記計算式に基づき、内因性中性脂肪の影響を除いて算出した。なお、各試験化合物が内因性中性脂肪濃度に影響しないことは別途確認されている。 (Test Example 2)
The DGAT1 enzyme is important for digestion and absorption of neutral fat, and when small intestine DGAT1 is inhibited, the absorption of neutral fat is suppressed. The biological activity of the DGAT1 inhibitory action was evaluated using as an index the inhibition of neutral fat absorption after neutral fat loading. Male C57BL / 6N mice (7-12 weeks old, body weight 17-25 g, Nippon Charles River) fasted overnight were assigned to Vehicle Group 1, Vehicle Group 2 and each test compound group, respectively vehicle (0.5% Methylcellulose) Alternatively, each test compound (1 to 10 mg / kg) suspended in the vehicle was orally administered (5 mL / kg). Lipoprotein lipase inhibitor (Pluronic-F127: Sigma-Aldrich Co., Ltd., 1 g / kg, dissolved in physiological saline at 20% by weight) was intraperitoneally administered (5 mL / kg) Distilled water was orally administered to Vehicle Group 1 and 20% neutral fat-containing emulsion (Intralipid 20%: Terumo Corporation) was orally administered (0.2 mL / mouse) to Vehicle Group 2 and Compound Group. After 1 to 4 hours after administration, blood is collected from the tail vein or right ventricle, and plasma is promptly separated and collected, and then the neutral fat concentration in the plasma is measured using a commercially available kit (Triglyceride E Test Wako: Wako Pure Chemical Industries, Ltd.) (Industry Co., Ltd.). In this method, the administration of lipoprotein lipase inhibitor suppresses the degradation of neutral fat flowing into the blood, and neutral fat accumulates in the blood, but its origin is exogenous absorbed in the digestive tract And it is divided into endogenous ones released from the liver. The neutral fat absorption inhibitory activity of each test compound was calculated based on the following formula, excluding the influence of endogenous neutral fat. It has been separately confirmed that each test compound does not affect the endogenous triglyceride concentration.
中性脂肪吸収抑制活性(%)=100-[(各試験化合物群の中性脂肪濃度)-(Vehicle群1の中性脂肪濃度)]/[(Vehicle群2の中性脂肪濃度)-(Vehicle群1の中性脂肪濃度)]×100
実施例1乃至6の化合物は、3 mg/kg以下の用量で60%以上の中性脂肪吸収抑制活性を示した。 Neutral fat absorption inhibitory activity (%) = 100-[(Neutral fat concentration of each test compound group)-(Neutral fat concentration of Vehicle group 1)] / [(Neutral fat concentration of Vehicle group 2)-( Vehicle group 1 neutral fat concentration)] × 100
The compounds of Examples 1 to 6 showed neutral fat absorption inhibitory activity of 60% or more at a dose of 3 mg / kg or less.
実施例1乃至6の化合物は、3 mg/kg以下の用量で60%以上の中性脂肪吸収抑制活性を示した。 Neutral fat absorption inhibitory activity (%) = 100-[(Neutral fat concentration of each test compound group)-(Neutral fat concentration of Vehicle group 1)] / [(Neutral fat concentration of Vehicle group 2)-( Vehicle group 1 neutral fat concentration)] × 100
The compounds of Examples 1 to 6 showed neutral fat absorption inhibitory activity of 60% or more at a dose of 3 mg / kg or less.
上記の結果から、本発明の化合物は、優れた中性脂肪吸収抑制活性を有する。
From the above results, the compound of the present invention has excellent neutral fat absorption inhibitory activity.
(試験例3)
雄性C57BL/6Nマウス (7-12週齢、体重17-25 g、日本チャールズリバー)を個体別に飼育し、高脂肪食(脂肪含有率 45 kcal%:リサーチダイエット社D12451)を1週間以上給餌して馴化させた。期間中の摂餌量に基づいて実験群に動物を均等に割り付け、一晩絶食させた後、vehicle (0.5% Methylcellulose)またはvehicleに懸濁させた試験化合物(1乃至10 mg/kg)を各群に経口投与(10 mL/kg)した。投与30分後に高脂肪食を給餌し、給餌開始後6時間での摂餌量を測定した。各試験化合物の摂食抑制活性は下記計算式に基づき算出した。 (Test Example 3)
Male C57BL / 6N mice (7-12 weeks old, body weight 17-25 g, Nippon Charles River) are bred individually and fed with a high fat diet (fat content 45 kcal%: Research Diet D12451) for over a week. I got used to it. Allocate the animals evenly to the experimental groups based on the amount of food consumed during the period, fast overnight and then each vehicle (0.5% Methylcellulose) or test compound (1-10 mg / kg) suspended in the vehicle. The group was orally administered (10 mL / kg). A high fat diet was fed 30 minutes after the administration, and the amount of food intake was measured 6 hours after the start of feeding. The feeding inhibitory activity of each test compound was calculated based on the following formula.
雄性C57BL/6Nマウス (7-12週齢、体重17-25 g、日本チャールズリバー)を個体別に飼育し、高脂肪食(脂肪含有率 45 kcal%:リサーチダイエット社D12451)を1週間以上給餌して馴化させた。期間中の摂餌量に基づいて実験群に動物を均等に割り付け、一晩絶食させた後、vehicle (0.5% Methylcellulose)またはvehicleに懸濁させた試験化合物(1乃至10 mg/kg)を各群に経口投与(10 mL/kg)した。投与30分後に高脂肪食を給餌し、給餌開始後6時間での摂餌量を測定した。各試験化合物の摂食抑制活性は下記計算式に基づき算出した。 (Test Example 3)
Male C57BL / 6N mice (7-12 weeks old, body weight 17-25 g, Nippon Charles River) are bred individually and fed with a high fat diet (fat content 45 kcal%: Research Diet D12451) for over a week. I got used to it. Allocate the animals evenly to the experimental groups based on the amount of food consumed during the period, fast overnight and then each vehicle (0.5% Methylcellulose) or test compound (1-10 mg / kg) suspended in the vehicle. The group was orally administered (10 mL / kg). A high fat diet was fed 30 minutes after the administration, and the amount of food intake was measured 6 hours after the start of feeding. The feeding inhibitory activity of each test compound was calculated based on the following formula.
摂食抑制活性(%)=[(Vehicle群の摂餌量)-(各試験化合物群の摂餌量)]/[(Vehicle群の摂餌量)]×100
実施例1の化合物は、10 mg/kg以下の用量で25%以上の摂食抑制活性を示した。 Feeding inhibitory activity (%) = [(food consumption of vehicle group) − (food consumption of each test compound group)] / [(food consumption of vehicle group)] × 100
The compound of Example 1 showed an antifeedant activity of 25% or more at a dose of 10 mg / kg or less.
実施例1の化合物は、10 mg/kg以下の用量で25%以上の摂食抑制活性を示した。 Feeding inhibitory activity (%) = [(food consumption of vehicle group) − (food consumption of each test compound group)] / [(food consumption of vehicle group)] × 100
The compound of Example 1 showed an antifeedant activity of 25% or more at a dose of 10 mg / kg or less.
上記の結果から、本発明の化合物は、優れた摂食抑制作用を有する。
From the above results, the compound of the present invention has an excellent antifeedant action.
なお、餌に使用する高脂肪食は上記の高脂肪食に限定されず、例えば、カロリーとして45乃至60%の中性脂肪を含有するげっ歯類用飼料を使用することができる。
Note that the high-fat diet used for the feed is not limited to the above-mentioned high-fat diet, and for example, a rodent feed containing 45 to 60% neutral fat as calories can be used.
製剤例1:カプセル剤
実施例1又は2の化合物 50mg
乳糖 128mg
トウモロコシデンプン 70mg
ステアリン酸マグネシウム 2mg
------------------
250mg
上記処方の粉末を混合し、60メッシュのふるいを通した後、この粉末を250mgのゼラチンカプセルに入れ、カプセル剤とする。 Formulation Example 1: Capsule 50 mg of the compound of Example 1 or 2
Lactose 128mg
Corn starch 70mg
Magnesium stearate 2mg
------------------
250mg
After mixing the powder of the above formulation and passing through a 60 mesh sieve, this powder is put into a 250 mg gelatin capsule to form a capsule.
実施例1又は2の化合物 50mg
乳糖 128mg
トウモロコシデンプン 70mg
ステアリン酸マグネシウム 2mg
------------------
250mg
上記処方の粉末を混合し、60メッシュのふるいを通した後、この粉末を250mgのゼラチンカプセルに入れ、カプセル剤とする。 Formulation Example 1: Capsule 50 mg of the compound of Example 1 or 2
Lactose 128mg
Corn starch 70mg
Magnesium stearate 2mg
------------------
250mg
After mixing the powder of the above formulation and passing through a 60 mesh sieve, this powder is put into a 250 mg gelatin capsule to form a capsule.
製剤例2:錠剤
実施例1又は2の化合物 50mg
乳糖 126mg
トウモロコシデンプン 23mg
ステアリン酸マグネシウム 1mg
------------------
200mg
上記処方の粉末を混合し、トウモロコシデンプン糊を用いて造粒、乾燥した後、打錠機により打錠して、1錠200mgの錠剤とする。この錠剤は必要に応じて糖衣を施すことができる。 Formulation Example 2: Tablet Example 1 or 2 compound 50 mg
Lactose 126mg
Corn starch 23mg
Magnesium stearate 1mg
------------------
200mg
The powder of the above formulation is mixed, granulated and dried using corn starch paste, and then tableted by a tableting machine to make one tablet of 200 mg. This tablet can be sugar-coated if necessary.
実施例1又は2の化合物 50mg
乳糖 126mg
トウモロコシデンプン 23mg
ステアリン酸マグネシウム 1mg
------------------
200mg
上記処方の粉末を混合し、トウモロコシデンプン糊を用いて造粒、乾燥した後、打錠機により打錠して、1錠200mgの錠剤とする。この錠剤は必要に応じて糖衣を施すことができる。 Formulation Example 2: Tablet Example 1 or 2 compound 50 mg
Lactose 126mg
Corn starch 23mg
Magnesium stearate 1mg
------------------
200mg
The powder of the above formulation is mixed, granulated and dried using corn starch paste, and then tableted by a tableting machine to make one tablet of 200 mg. This tablet can be sugar-coated if necessary.
本発明の一般式(I)で表される化合物又はその薬理上許容される塩は、優れたDGAT阻害作用及び摂食抑制作用を有し、医薬として有用である。
The compound represented by the general formula (I) of the present invention or a pharmacologically acceptable salt thereof has an excellent DGAT inhibitory action and antifeeding action and is useful as a medicine.
Claims (20)
- 一般式(I)
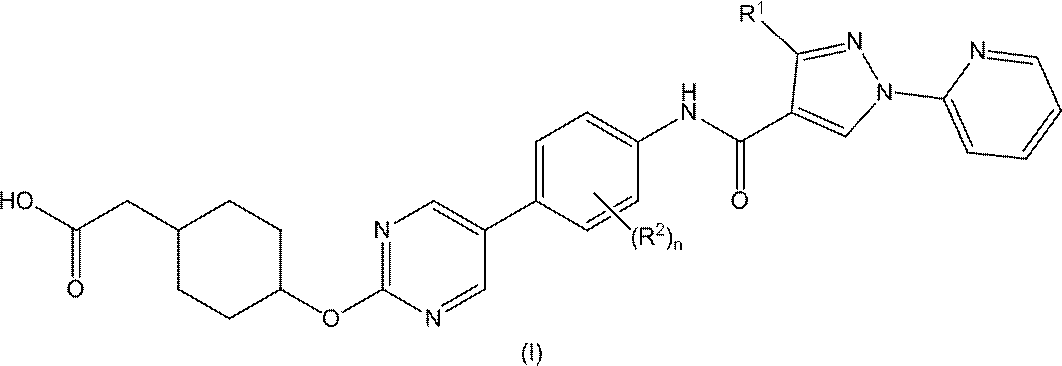
[式中、
R1は、水素原子、ハロゲン原子、C1-C6アルキル基又はC3-C6シクロアルキル基を示し、
R2は、独立して、ハロゲン原子、C1-C6アルキル基、C1-C6アルコキシ基又はヒドロキシ基を示し、
nは、0乃至2の整数を示す。]で表される化合物又はその薬理上許容される塩。 Formula (I)
[Where:
R 1 represents a hydrogen atom, a halogen atom, a C 1 -C 6 alkyl group or a C 3 -C 6 cycloalkyl group,
R 2 independently represents a halogen atom, a C 1 -C 6 alkyl group, a C 1 -C 6 alkoxy group or a hydroxy group,
n represents an integer of 0 to 2. Or a pharmacologically acceptable salt thereof. - 請求項1において、一般式(I)が、一般式(Ia)である化合物又はその薬理上許容される塩。

- 請求項1又は2において、nが、0である化合物又はその薬理上許容される塩。 3. The compound or a pharmacologically acceptable salt thereof according to claim 1 or 2, wherein n is 0.
- (cis-4-{[5-(4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(4-{[(3-シクロプロピル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(2-メチル-4-{[(3-メチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(4-{[(3-エチル-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(4-{[(1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、
(cis-4-{[5-(2-メチル-4-{[(1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸、もしくは、
(cis-4-{[5-(4-{[(3-クロロ-1-ピリジン-2-イル-1H-ピラゾール-4-イル)カルボニル]アミノ}フェニル)ピリミジン-2-イル]オキシ}シクロヘキシル)酢酸
又はその薬理上許容される塩。 (cis-4-{[5- (4-{[(3-Methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid,
(cis-4-{[5- (4-{[(3-Cyclopropyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} (Cyclohexyl) acetic acid,
(cis-4-{[5- (2-Methyl-4-{[(3-methyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl ] Oxy} cyclohexyl) acetic acid,
(cis-4-{[5- (4-{[(3-Ethyl-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid,
(cis-4-{[5- (4-{[(1-Pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl) acetic acid,
(cis-4-{[5- (2-Methyl-4-{[(1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl Acetic acid, or
(cis-4-{[5- (4-{[(3-Chloro-1-pyridin-2-yl-1H-pyrazol-4-yl) carbonyl] amino} phenyl) pyrimidin-2-yl] oxy} cyclohexyl ) Acetic acid or a pharmacologically acceptable salt thereof. - 請求項1乃至4から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩を有効成分として含有するアシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼ阻害剤。 An acyl coenzyme A: diacylglycerol acyltransferase inhibitor containing the compound according to any one of claims 1 to 4 or a pharmacologically acceptable salt thereof as an active ingredient.
- 請求項1乃至4から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩を有効成分として含有する摂食抑制剤及び/又は食欲抑制剤。 An antifeedant and / or an appetite suppressant containing the compound according to any one of claims 1 to 4 or a pharmacologically acceptable salt thereof as an active ingredient.
- 請求項1乃至4から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩を有効成分として含有する医薬組成物。 A pharmaceutical composition comprising the compound according to any one of claims 1 to 4 or a pharmacologically acceptable salt thereof as an active ingredient.
- 医薬組成物が、アシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼ阻害作用を有する請求項7に記載の医薬組成物。 The pharmaceutical composition according to claim 7, which has an acylcoenzyme A: diacylglycerol acyltransferase inhibitory action.
- 医薬組成物が、摂食抑制作用及び/又は食欲抑制作用を有する請求項7に記載の医薬組成物。 The pharmaceutical composition according to claim 7, wherein the pharmaceutical composition has an antifeedant action and / or an appetite suppressive action.
- 医薬組成物が、アシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼを阻害させ、トリグリセライドの合成を阻害し、トリグリセライドの吸収が抑制されることにより、症状の治療、改善、軽減及び/又は予防がなされる疾病の治療及び/又は予防のための請求項7に記載の医薬組成物。 The pharmaceutical composition inhibits acyl coenzyme A: diacylglycerol acyltransferase, inhibits the synthesis of triglyceride, and suppresses the absorption of triglyceride, thereby preventing the treatment, amelioration, reduction and / or prevention of symptoms. 8. A pharmaceutical composition according to claim 7 for treatment and / or prevention.
- 医薬組成物が、アシルコエンザイムA:ジアシルグリセロールアシルトランスフェラーゼを阻害させ、トリグリセライドの合成が阻害されることにより、症状の治療、改善、軽減及び/又は予防がなされる疾病の治療及び/又は予防のための請求項7に記載の医薬組成物。 For the treatment and / or prevention of a disease in which the pharmaceutical composition inhibits acylcoenzyme A: diacylglycerol acyltransferase and the synthesis of triglyceride is inhibited, thereby treating, ameliorating, reducing and / or preventing symptoms. The pharmaceutical composition according to claim 7.
- 医薬組成物が、肥満、肥満症、高脂血症、高トリグリセライド症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、虚血性心疾患又は過食症の治療及び/又は予防のための請求項7に記載の医薬組成物。 The pharmaceutical composition is obesity, obesity, hyperlipidemia, hypertriglyceride disease, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic nephropathy, Diabetic retinopathy, including diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, ischemic heart disease Or the pharmaceutical composition of Claim 7 for the treatment and / or prevention of bulimia.
- 医薬組成物が、肥満又は肥満症の治療及び/又は予防のための請求項7に記載の医薬組成物。 The pharmaceutical composition according to claim 7, wherein the pharmaceutical composition is for the treatment and / or prevention of obesity or obesity.
- 医薬組成物が、糖尿病の治療及び/又は予防のための請求項7に記載の医薬組成物。 The pharmaceutical composition according to claim 7, which is used for the treatment and / or prevention of diabetes.
- 肥満、肥満症、高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、虚血性心疾患又は過食症の治療及び/又は予防で使用するための、請求項1乃至4から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩。 Obesity, obesity, hyperlipidemia, hypertriglycerideemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic nephropathy, diabetic retinopathy Cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, ischemic heart disease or bulimia The compound or a pharmacologically acceptable salt thereof according to any one of claims 1 to 4 for use in therapy and / or prevention.
- 医薬組成物を製造するための、請求項1乃至4から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩の使用。 Use of the compound according to any one of claims 1 to 4 or a pharmacologically acceptable salt thereof for producing a pharmaceutical composition.
- 医薬組成物が肥満、肥満症、高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、虚血性心疾患又は過食症の治療及び/又は予防のための医薬組成物である請求項16に記載の使用。 The pharmaceutical composition is obesity, obesity, hyperlipidemia, hypertriglyceridemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic nephropathy, Diabetic retinopathy, including diabetic macroangiopathy), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, ischemic heart disease The use according to claim 16, which is a pharmaceutical composition for the treatment and / or prevention of bulimia.
- 請求項1乃至4から選択されるいずれか一項に記載された化合物又はその薬理上許容される塩の薬理的な有効量を温血動物に投与する疾病の治療及び/又は予防方法。 A method for treating and / or preventing a disease, wherein a pharmacologically effective amount of the compound or pharmacologically acceptable salt thereof according to any one of claims 1 to 4 is administered to a warm-blooded animal.
- 疾病が肥満、肥満症、高脂血症、高トリグリセライド血症、脂質代謝異常疾患、インスリン抵抗性症候群、耐糖能異常、糖尿病、糖尿病合併症(糖尿病性末梢神経障害、糖尿病性腎症、糖尿病性網膜症、糖尿病性大血管症を含む)、白内障、妊娠糖尿病、非アルコール性脂肪肝炎、多嚢胞卵巣症候群、動脈硬化症、アテローム性動脈硬化症、糖尿病性動脈硬化症、虚血性心疾患又は過食症である請求項18に記載の方法。 Diseases are obesity, obesity, hyperlipidemia, hypertriglyceridemia, dyslipidemia, insulin resistance syndrome, impaired glucose tolerance, diabetes, diabetic complications (diabetic peripheral neuropathy, diabetic nephropathy, diabetic Retinopathy, including diabetic macrovascular disease), cataract, gestational diabetes, nonalcoholic steatohepatitis, polycystic ovary syndrome, arteriosclerosis, atherosclerosis, diabetic arteriosclerosis, ischemic heart disease or binge eating The method according to claim 18, which is a symptom.
- 温血動物がヒトである請求項18又は19に記載の方法。 The method according to claim 18 or 19, wherein the warm-blooded animal is a human.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011-134874 | 2011-06-17 | ||
| JP2011134874 | 2011-06-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012173219A1 true WO2012173219A1 (en) | 2012-12-20 |
Family
ID=47357198
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/065315 WO2012173219A1 (en) | 2011-06-17 | 2012-06-15 | Novel biaryl ether derivative |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW201305140A (en) |
| WO (1) | WO2012173219A1 (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105503730B (en) * | 2015-12-25 | 2018-06-22 | 山东大学 | Pyrazole derivatives and preparation method and application |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090093497A1 (en) * | 2007-05-22 | 2009-04-09 | David Robert Bolin | Diacylglycerol Acyltransferase Inhibitors |
| JP2010132590A (en) * | 2008-12-03 | 2010-06-17 | Astellas Pharma Inc | Oxadiazole compound |
| WO2010077861A1 (en) * | 2008-12-17 | 2010-07-08 | Via Pharmaceuticals, Inc | Inhibitors of diacylglycerol aclytransferase |
| WO2011078102A1 (en) * | 2009-12-22 | 2011-06-30 | 第一三共株式会社 | Novel phenoxypyrimidine derivative |
| WO2012063896A1 (en) * | 2010-11-11 | 2012-05-18 | 第一三共株式会社 | Novel pyrazole amide derivative |
-
2012
- 2012-06-15 TW TW101121436A patent/TW201305140A/en unknown
- 2012-06-15 WO PCT/JP2012/065315 patent/WO2012173219A1/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090093497A1 (en) * | 2007-05-22 | 2009-04-09 | David Robert Bolin | Diacylglycerol Acyltransferase Inhibitors |
| JP2010132590A (en) * | 2008-12-03 | 2010-06-17 | Astellas Pharma Inc | Oxadiazole compound |
| WO2010077861A1 (en) * | 2008-12-17 | 2010-07-08 | Via Pharmaceuticals, Inc | Inhibitors of diacylglycerol aclytransferase |
| WO2011078102A1 (en) * | 2009-12-22 | 2011-06-30 | 第一三共株式会社 | Novel phenoxypyrimidine derivative |
| WO2012063896A1 (en) * | 2010-11-11 | 2012-05-18 | 第一三共株式会社 | Novel pyrazole amide derivative |
Also Published As
| Publication number | Publication date |
|---|---|
| TW201305140A (en) | 2013-02-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2061762B1 (en) | Pyrazine compounds, their use and methods of preparation | |
| JP4054368B2 (en) | Substituted methylaryl or heteroarylamide compounds | |
| WO2016173493A1 (en) | Sulfonylaminocarbonyl derivative, pharmaceutical composition and uses thereof | |
| JP2007210929A (en) | Medicine containing urea compound | |
| JP4531127B2 (en) | Novel tetrahydroisoquinoline derivatives | |
| WO2012063896A1 (en) | Novel pyrazole amide derivative | |
| US20120202847A1 (en) | Novel Tetrahydroisoquinoline Compounds | |
| WO2011078102A1 (en) | Novel phenoxypyrimidine derivative | |
| CA3097752A1 (en) | Imidazopyridines useful as mitochondrial uncouplers | |
| CN102558167A (en) | Thiazolidine derivant with GK and PPAR double excitation activity | |
| WO2012173219A1 (en) | Novel biaryl ether derivative | |
| KR20190140005A (en) | Method of Using Trisubstituted Benzotriazole Derivatives as Dihydroorotate Oxygenase Inhibitors | |
| WO2013039140A1 (en) | Fused heterocyclic derivative | |
| CA2842450A1 (en) | Insulin secretion promoting agents | |
| JP2011088889A (en) | Medicine containing new tetrahydroisoquinoline derivative | |
| WO2012165398A1 (en) | Cycloalkyloxybiaryl compound | |
| CN102786468A (en) | Nicotinic acid derivative having GK (glucokinase) and PPAR (peroxidase proliferator-activated receptor) dual agonist activities | |
| JPH04330060A (en) | Di(nitroxyalkyl)amide of pyridine-2,4-and 2,5-dicarboxylic acid, and preparation and use thereof | |
| JP2006083158A (en) | Substituted urea compound | |
| CN117203211A (en) | Imidazo thiazole derivative, preparation method and application thereof | |
| KR20230066341A (en) | Quinoline Compounds as Selective and/or Dual Modulators of Bile Acid Receptors and Leukotriene Cysteine Receptors | |
| US20230090255A1 (en) | Magl inhibitor, preparation method therefor and use thereof | |
| WO2021098872A1 (en) | Allopregnenolone phosphonamide derivative, preparation method therefor and pharmaceutical use thereof | |
| CN102718727B (en) | The aryl urea derivative of GK and PPAR double excitation activity | |
| TW202030181A (en) | Biaryl derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12800934 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12800934 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |