WO2011133920A1 - Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use - Google Patents
Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use Download PDFInfo
- Publication number
- WO2011133920A1 WO2011133920A1 PCT/US2011/033650 US2011033650W WO2011133920A1 WO 2011133920 A1 WO2011133920 A1 WO 2011133920A1 US 2011033650 W US2011033650 W US 2011033650W WO 2011133920 A1 WO2011133920 A1 WO 2011133920A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- membered
- compound
- cycloalkyl
- optionally substituted
- Prior art date
Links
- 0 CC(C)(*)C(*)(*)N(*)c1c(*)c(*)c(**)c(*)n1 Chemical compound CC(C)(*)C(*)(*)N(*)c1c(*)c(*)c(**)c(*)n1 0.000 description 3
- ZCMPBROLWBJECM-LLVKDONJSA-N C[C@H](CN(C(c1ccccc11)=O)C1=O)c(cc1)ccc1F Chemical compound C[C@H](CN(C(c1ccccc11)=O)C1=O)c(cc1)ccc1F ZCMPBROLWBJECM-LLVKDONJSA-N 0.000 description 2
- DHGRRSSFDBJARB-UHFFFAOYSA-N CC(C)(CN)c(cc1)ccc1F Chemical compound CC(C)(CN)c(cc1)ccc1F DHGRRSSFDBJARB-UHFFFAOYSA-N 0.000 description 1
- VKVNLGSSNKOHNF-UHFFFAOYSA-N CC(C)(c(nccc1)c1F)Nc1ncc(-c(cc(cc2)C(N)=O)c2F)nn1 Chemical compound CC(C)(c(nccc1)c1F)Nc1ncc(-c(cc(cc2)C(N)=O)c2F)nn1 VKVNLGSSNKOHNF-UHFFFAOYSA-N 0.000 description 1
- FZPCDCLZAMCEST-UHFFFAOYSA-N CC(C)(c(nccc1)c1F)Nc1ncc(-c2cc(C#N)ccc2F)nn1 Chemical compound CC(C)(c(nccc1)c1F)Nc1ncc(-c2cc(C#N)ccc2F)nn1 FZPCDCLZAMCEST-UHFFFAOYSA-N 0.000 description 1
- BCKYDHYSRQBQCE-CCAUIXRRSA-N CC1([C@](C2)(C[C@@H]2F)C#N)NC=CC=C1F Chemical compound CC1([C@](C2)(C[C@@H]2F)C#N)NC=CC=C1F BCKYDHYSRQBQCE-CCAUIXRRSA-N 0.000 description 1
- ATNIQYFSDQIEJR-UHFFFAOYSA-N CCOC(CNC(c1cc(Cl)ccc1)=O)=O Chemical compound CCOC(CNC(c1cc(Cl)ccc1)=O)=O ATNIQYFSDQIEJR-UHFFFAOYSA-N 0.000 description 1
- YRBRJLWZJHRXBG-SSDOTTSWSA-N C[C@H](CN)c(cc1)ccc1F Chemical compound C[C@H](CN)c(cc1)ccc1F YRBRJLWZJHRXBG-SSDOTTSWSA-N 0.000 description 1
- DLIGEPKOWMTQQI-SSDOTTSWSA-N C[C@H](CO)c(cc1)ccc1F Chemical compound C[C@H](CO)c(cc1)ccc1F DLIGEPKOWMTQQI-SSDOTTSWSA-N 0.000 description 1
- MZLXAQCGNKZLQA-UHFFFAOYSA-N N#CC(C1)(CC1=O)c(nccc1)c1F Chemical compound N#CC(C1)(CC1=O)c(nccc1)c1F MZLXAQCGNKZLQA-UHFFFAOYSA-N 0.000 description 1
- NQTUTMXQBMACGI-UHFFFAOYSA-N N#CC(C1)(CC1O)c(nccc1)c1F Chemical compound N#CC(C1)(CC1O)c(nccc1)c1F NQTUTMXQBMACGI-UHFFFAOYSA-N 0.000 description 1
- JHQBLYITVCBGTO-UHFFFAOYSA-N N#CCc(cc1)ccc1F Chemical compound N#CCc(cc1)ccc1F JHQBLYITVCBGTO-UHFFFAOYSA-N 0.000 description 1
- WHIHIKVIWVIIER-UHFFFAOYSA-N O=C(c1cc(Cl)ccc1)Cl Chemical compound O=C(c1cc(Cl)ccc1)Cl WHIHIKVIWVIIER-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/74—Amino or imino radicals substituted by hydrocarbon or substituted hydrocarbon radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
- C07D213/82—Amides; Imides in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/38—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/48—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D253/00—Heterocyclic compounds containing six-membered rings having three nitrogen atoms as the only ring hetero atoms, not provided for by group C07D251/00
- C07D253/02—Heterocyclic compounds containing six-membered rings having three nitrogen atoms as the only ring hetero atoms, not provided for by group C07D251/00 not condensed with other rings
- C07D253/06—1,2,4-Triazines
- C07D253/065—1,2,4-Triazines having three double bonds between ring members or between ring members and non-ring members
- C07D253/07—1,2,4-Triazines having three double bonds between ring members or between ring members and non-ring members with hetero atoms, or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the cytoskeleton of skeletal and cardiac muscle cells is unique compared to that of all other cells. It consists of a nearly crystalline array of closely packed cytoskeletal proteins called the sarcomere.
- the sarcomere is elegantly organized as an interdigitating array of thin and thick filaments.
- the thick filaments are composed of myosin, the motor protein responsible for transducing the chemical energy of ATP hydrolysis into force and directed movement.
- the thin filaments are composed of actin monomers arranged in a helical array.
- ADP-Pi to ADP signals a change in structural conformation of the catalytic domain that in turn alters the orientation of the light-chain binding lever arm domain that extends from the globular head; this movement is termed the powerstroke.
- This change in orientation of the myosin head in relationship to actin causes the thick filament of which it is a part to move with respect to the thin actin filament to which it is bound.
- conformation/orientation completes the catalytic cycle, responsible for intracellular movement and muscle contraction.
- Tropomyosin and troponin mediate the calcium effect on the interaction on actin and myosin.
- the troponin complex is comprised of three polypeptide chains: troponin C, which binds calcium ions; troponin I, which binds to actin; and troponin T, which binds to tropomyosin.
- the skeletal troponin- tropomyosin complex regulates the myosin binding sites extending over several actin units at once.
- Troponin a complex of the three polypeptides described above, is an accessory protein that is closely associated with actin filaments in vertebrate muscle.
- the troponin complex acts in conjunction with the muscle form of tropomyosin to mediate the Ca 2+ dependency of myosin ATPase activity and thereby regulate muscle contraction.
- the troponin polypeptides T, I, and C are named for their tropomyosin binding, inhibitory, and calcium binding activities, respectively.
- Troponin T binds to tropomyosin and is believed to be responsible for positioning the troponin complex on the muscle thin filament.
- Troponin I binds to actin, and the complex formed by troponins I and T, and tropomyosin inhibits the interaction of actin and myosin.
- Skeletal troponin C is capable of binding up to four calcium molecules. Studies suggest that when the level of calcium in the muscle is raised, troponin C exposes a binding site for troponin I, recruiting it away from actin. This causes the tropomyosin molecule to shift its position as well, thereby exposing the myosin binding sites on actin and stimulating myosin ATPase activity.
- Human skeletal muscle is composed of different types of contractile fibers, classified by their myosin type and termed either slow or fast fibers. Table 1 summarizes the different proteins that make up these types of muscle. Table 1
- Fast skeletal muscle fibers tend to exert greater force but fatigue faster than slow skeletal muscle fibers and are functionally useful for acute, large scale movements such as rising from a chair or correcting falls.
- Muscle contraction and force generation is controlled through nervous stimulation by innervating motor neurons.
- Each motor neuron may innervate many (approximately 100-380) muscle fibers as a contractile whole, termed a motor unit.
- motor neurons send stimuli as nerve impulses (action potentials) from the brain stem or spinal cord to each fiber within the motor unit.
- the contact region between nerve and muscle fibers is a specialized synapse called the neuromuscular junction (NMJ).
- NMJ neuromuscular junction
- ACh neurotransmitter acetylcholine
- ACh triggers a second action potential in the muscle that spreads rapidly along the fiber and into invaginations in the membrane, termed t-tubules.
- T-tubules are physically connected to Ca2+ stores within the sarcoplasmic reticulum (SR) of muscle via the
- DHPR dihydropyridine receptor
- Muscle function can become compromised in disease by many mechanisms. Examples include the frailty associated with old age (termed sarcopenia) and cachexia syndromes associated with diseases such as cancer, heart failure, chronic obstructive pulmonary disease (COPD), and chronic kidney disease/dialysis. Severe muscular dysfunction can arise from neuromuscular diseases (such as Amyotrophic Lateral Sclerosis (ALS), spinal muscular atrophy (SMA) and myasthenia gravis) or muscular myopathies (such as muscular dystrophies). Additionally, muscle function may become compromised due to rehabilitation-related deficits, such as those associated with recovery from surgery (e.g. post-surgical muscle weakness), prolonged bed rest, or stroke rehabilitation. Additional examples of diseases or conditions where muscle function becomes compromised include peripheral vascular disease (e.g., claudication), chronic fatigue syndrome, metabolic syndrome, and obesity.
- peripheral vascular disease e.g., claudication
- chronic fatigue syndrome e.g., metabolic syndrome, and obesity.
- R 2 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , X, Z 1 , Z 2 , Z 3 , Z 4 and m are as defined herein.
- composition comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof.
- Also provided are methods for treating a disease or condition responsive to modulation of the contractility of the skeletal sarcomere for example, modulation of the troponin complex of the fast skeletal muscle sarcomere through one or more of fast skeletal myosin, actin, tropomyosin, troponin C, troponin I, and troponin T, and fragments and isoforms thereof.
- references to a compound of Formula I includes all subgroups of Formula I defined herein, including all substructures, subgenera, preferences, embodiments, examples and particular compounds defined and/or described herein.
- references to a compound of Formula I and subgroups thereof include polymorphs, solvates, and/or co-crystals thereof. In some embodiments, references to a compound of Formula I and subgroups thereof include isomers, tautomers and/or oxides thereof. In some embodiments, references to a compound of Formula I and subgroups thereof include solvates thereof. Similarly, the term “salts" includes solvates of salts of compounds.
- C-i-6 alkyl When a range of values is given (e.g., C-i-6 alkyl), each value within the range as well as all intervening ranges are included.
- C-i -6 alkyl includes C-i , C2, C3, C 4 , C5, CQ, C-i-6, C2-6, C3-6, C 4- 6, Cs-6, C-i-5, C2-5, C3-5, C 4- 5,
- a moiety When a moiety is defined as being optionally substituted, it may be substituted as itself or as part of another moiety.
- R x is defined as "C-i-6 alkyl or OC-i -6 alkyl, wherein C-i -6 alkyl is optionally subsituted with halogen"
- both the C-i-6 alkyl group alone and the C-i-6 alkyl that makes up part of the OC-i-6 alkyl group may be substituted with halogen.
- Alkenyl groups include, but are not limited to, ethenyl, propenyl (e.g., prop-1 -en-1 -yl, prop-1 -en-2-yl, prop- 2-en-1 -yl (allyl), prop-2-en-2-yl), and butenyl (e.g., but-1 -en-1 -yl, but-1 -en-2-yl, 2-methyl-prop-1 -en-1 -yl, but-2-en-1 -yl, but-2-en-1 -yl, but-2-en-2-yl, buta-1 ,3- dien-1 -yl, buta-1 ,3-dien-2-yl).
- “Lower alkenyl” refers to alkenyl groups having 2 to 6 carbons.
- Cycloalkyl indicates a non-aromatic, fully saturated carbocyclic ring having the indicated number of carbon atoms, for example, 3 to 10, or 3 to 8, or 3 to 6 ring carbon atoms. Cycloalkyl groups may be monocyclic or polycyclic (e.g., bicyclic, tricyclic). Examples of cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl and cyclohexyl, as well as bridged and caged ring groups (e.g., norbornane, bicyclo[2.2.2]octane).
- Cycloalkenyl indicates a non-aromatic carbocyclic ring, containing the indicated number of carbon atoms (e.g., 3 to 10, or 3 to 8, or 3 to 6 ring carbon atoms) and at least one carbon-carbon double bond derived by the removal of one molecule of hydrogen from adjacent carbon atoms of the corresponding cycloalkyl.
- Cycloalkenyl groups may be monocyclic or polycyclic (e.g., bicyclic, tricyclic). Examples of cycloalkenyl groups include cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclopentadienyl, and
- cyclohexenyl as well as bridged and caged ring groups (e.g.,
- one ring of a polycyclic cycloalkenyl group may be aromatic, provided the polycyclic alkenyl group is bound to the parent structure via a non-aromatic carbon atom.
- inden-1 -yl (wherein the moiety is bound to the parent structure via a non-aromatic carbon atom) is considered a cycloalkenyl group
- inden-4-yl (wherein the moiety is bound to the parent structure via an aromatic carbon atom) is not considered a cycloalkenyl group.
- polycyclic cycloalkenyl groups consisting of a cycloalkenyl group fused to an aromatic ring are described below.
- aryl does not encompass or overlap with "heteroaryl", as defined herein, regardless of the point of attachment (e.g., both quinolin-5-yl and quinolin-2-yl are heteroaryl groups).
- aryl is phenyl or naphthyl.
- aryl is phenyl. Additional examples of aryl groups comprising an aromatic carbon ring fused to a non-aromatic ring are described below.
- Heteroaryl indicates an aromatic ring containing the indicated number of atoms (e.g., 5 to 12, or 5 to 10 membered heteroaryl) made up of one or more heteroatoms (e.g., 1 , 2, 3 or 4 heteroatoms) selected from N, O and S and with the remaining ring atoms being carbon. Heteroaryl groups do not contain adjacent S and O atoms. In some embodiments, the total number of S and O atoms in the heteroaryl group is not more than 2. In some
- heteroaryl ring when sulfur is present in a heteroaryl ring, it may, where the nature of the adjacent atoms and groups permits, exist in an oxidized state (i.e., S + -O " or SO2).
- Heteroaryl groups may be monocyclic or polycyclic (e.g., bicyclic, tricyclic).
- a heteroaryl group is monocyclic.
- examples include pyrrole, pyrazole, imidazole, triazole (e.g., 1 ,2,3-triazole, 1 ,2,4- triazole, 1 ,2,4-triazole), tetrazole, furan, isoxazole, oxazole, oxadiazole (e.g., 1 ,2,3-oxadiazole, 1 ,2,4-oxadiazole, 1 ,3,4-oxadiazole), thiophene, isothiazole, thiazole, thiadiazole (e.g., 1 ,2,3-thiadiazole, 1 ,2,4-thiadiazole, 1 ,3,4- thiadiazole), pyridine, pyridazine, pyrimidine, pyrazine, triazine (e.g., 1 ,2,4- triazine, 1 ,3,5-tria
- both rings of a polycyclic heteroaryl group are aromatic.
- examples include indole, isoindole, indazole, benzoimidazole, benzotriazole, benzofuran, benzoxazole, benzoisoxazole, benzoxadiazole, benzothiophene, benzothiazole, benzoisothiazole, benzothiadiazole, 1 H- pyrrolo[2,3-b]pyridine, 1 H-pyrazolo[3,4-b]pyridine, 3H-imidazo[4,5-b]pyridine, 3H-[1 ,2,3]triazolo[4,5-b]pyridine, 1 H-pyrrolo[3,2-b]pyridine, 1 H-pyrazolo[4,3- bjpyridine, 1 H-imidazo[4,5-b]pyridine, 1 H-[1 ,2,3]triazolo[4,5-b]pyridine, 1 H- pyrrolo[2,3-c]pyridine
- quinazoline quinoxaline, phthalazine, naphthyridine (e.g., 1 ,8-naphthyridine, 1 ,7-naphthyridine, 1 ,6-naphthyridine, 1 ,5-naphthyridine, 2,7-naphthyridine, 2,6-naphthyridine), imidazo[1 ,2-a]pyridine, 1 H-pyrazolo[3,4-d]thiazole, 1 H- pyrazolo[4,3-d]thiazole and imidazo[2,1 -b]thiazole.
- naphthyridine e.g., 1 ,8-naphthyridine, 1 ,7-naphthyridine, 1 ,6-naphthyridine, 1 ,5-naphthyridine, 2,7-naphthyridine, 2,6-
- polycyclic heteroaryl groups may include a non- aromatic ring (e.g., cycloalkyl, cycloalkenyl, heterocycloalkyl,
- heterocycloalkenyl fused to a heteroaryl ring
- polycyclic heteroaryl group is bound to the parent structure via an atom in the aromatic ring.
- a 4,5,6,7-tetrahydrobenzo[d]thiazol-2-yl group (wherein the moiety is bound to the parent structure via an aromatic carbon atom) is considered a heteroaryl group
- 4,5,6,7-tetrahydrobenzo[d]thiazol-5-yl is not considered a heteroaryl group.
- polycyclic heteroaryl groups consisting of a heteroaryl ring fused to a non-aromatic ring are described below.
- Heterocycloalkyl indicates a non-aromatic, fully saturated ring having the indicated number of atoms (e.g., 3 to 10, or 3 to 7, membered
- heterocycloalkyl made up of one or more heteroatoms (e.g., 1 , 2, 3 or 4 heteroatoms) selected from N, O and S and with the remaining ring atoms being carbon.
- Heterocycloalkyl groups may be monocyclic or polycyclic (e.g., bicyclic, tricyclic). Examples of heterocycloalkyl groups include oxiranyl, aziridinyl, azetidinyl, pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl and thiomorpholinyl.
- heterocycloalkyl ring When nitrogen is present in a heterocycloalkyl ring, it may, where the nature of the adjacent atoms and groups permits, exist in an oxidized state (i.e., N + -O " ). Examples include piperidinyl N-oxide and morpholinyl-N-oxide. Additionally, when sulfur is present in a heterocycloalkyl ring, it may, where the nature of the adjacent atoms and groups permits, exist in an oxidized state (i.e., S + -O " or -SO2-). Examples include thiomorpholine S-oxide and thiomorpholine S,S-dioxide.
- one ring of a polycyclic heterocycloalkyl group may be aromatic (e.g., aryl or heteroaryl), provided the polycyclic heterocycloalkyl group is bound to the parent structure via a non-aromatic carbon or nitrogen atom.
- a 1 ,2,3,4-tetrahydroquinolin-1 -yl group (wherein the moiety is bound to the parent structure via a non-aromatic nitrogen atom) is considered a heterocycloalkyl group
- 1 ,2,3,4-tetrahydroquinolin-8-yl group is not considered a heterocycloalkyl group.
- heterocycloalkyl groups consisting of a heterocycloalkyl group fused to an aromatic ring are described below.
- Heterocycloalkenyl indicates a non-aromatic ring having the indicated number of atoms (e.g., 3 to 10, or 3 to 7, membered heterocycloalkyl) made up of one or more heteroatoms (e.g., 1 , 2, 3 or 4 heteroatoms) selected from N, O and S and with the remaining ring atoms being carbon, and at least one double bond derived by the removal of one molecule of hydrogen from adjacent carbon atoms, adjacent nitrogen atoms, or adjacent carbon and nitrogen atoms of the corresponding heterocycloalkyl.
- Heterocycloalkenyl groups may be monocyclic or polycyclic (e.g., bicyclic, tricyclic).
- heterocycloalkenyl ring When nitrogen is present in a heterocycloalkenyl ring, it may, where the nature of the adjacent atoms and groups permits, exist in an oxidized state (i.e., N + -O " ). Additionally, when sulfur is present in a heterocycloalkenyl ring, it may, where the nature of the adjacent atoms and groups permits, exist in an oxidized state (i.e., S + -O " or -SO 2 -).
- heterocycloalkenyl groups include dihydrofuranyl (e.g., 2,3-dihydrofuranyl, 2,5-dihydrofuranyl), dihydrothiophenyl (e.g., 2,3-dihydrothiophenyl, 2,5-dihydrothiophenyl), dihydropyrrolyl (e.g., 2,3- dihydro-1 H-pyrrolyl, 2,5-dihydro-1 H-pyrrolyl), dihydroimidazolyl (e.g., 2,3- dihydro-1 H-imidazolyl, 4,5-dihydro-1 H-imidazolyl), pyranyl, dihydropyranyl (e.g., 3,4-dihydro-2H-pyranyl, 3,6-dihydro-2H-pyranyl), tetrahydropyridinyl (e.g., 1 ,2,3,4-tetrahydropyridinyl, 1 ,
- dihydropyridine e.g., 1 ,2-dihydropyridine, 1 ,4-dihydropyridine.
- one ring of a polycyclic heterocycloalkenyl group may be aromatic (e.g., aryl or heteroaryl), provided the polycyclic heterocycloalkenyl group is bound to the parent structure via a non-aromatic carbon or nitrogen atom.
- a 1 ,2-dihydroquinolin-1 -yl group (wherein the moiety is bound to the parent structure via a non-aromatic nitrogen atom) is considered a
- heterocycloalkenyl group while 1 ,2-dihydroquinolin-8-yl group (wherein the moiety is bound to the parent structure via an aromatic carbon atom) is not considered a heterocydoalkenyl group.
- heterocydoalkenyl groups consisting of a heterocydoalkenyl group fused to an aromatic ring are described below.
- polycyclic rings consisting of an aromatic ring (e.g., aryl or heteroaryl) fused to a non-aromatic ring (e.g., cycloalkyl, cycloalkenyl, heterocydoalkyi, heterocydoalkenyl)
- a non-aromatic ring e.g., cycloalkyl, cycloalkenyl, heterocydoalkyi, heterocydoalkenyl
- indenyl 2, 3-dihydro-1 H-indenyl, 1 ,2,3,4-tetrahydronaphthalenyl, benzo[1 ,3]dioxolyl, tetrahydroquinolinyl, 2,3-dihydrobenzo[1 ,4]dioxinyl, indolinyl, isoindolinyl, 2, 3-dihydro-1 H-indazolyl, 2,3-dihydro-1 H-
- each ring is considered an aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocydoalkyi or heterocydoalkenyl group is determined by the atom through which the moiety is bound to the parent structure.
- "Halogen” or “halo” refers to fluorine, chlorine, bromine or iodine.
- “Isomers” are different compounds that have the same molecular formula. “Stereoisomers” are isomers that differ only in the way the atoms are arranged in space. "Enantiomers” are stereoisomers that are non- superimposable mirror images of each other. A 1 :1 mixture of a pair of enantiomers is a “racemic” mixture. The symbol “( ⁇ )” may be used to designate a racemic mixture where appropriate. "Diastereoisomers” are stereoisomers that have at least two asymmetric atoms, but which are not mirror-images of each other. A “meso compound” or “meso isomer” is a non- optically active member of a set of stereoisomers.
- Meso isomers contain two or more stereocenters but are not chiral (i.e., a plane of symmetry exists within the molecule).
- the absolute stereochemistry is specified according to the Cahn-lngold-Prelog R-S system. When a compound is a pure enantiomer the stereochemistry at each chiral carbon can be specified by either R or S.
- Resolved compounds whose absolute configuration is unknown can be designated (+) or (-) depending on the direction (dextro- or levorotatory) which they rotate plane polarized light at the wavelength of the sodium D line.
- compounds disclosed and/or described herein include all such possible enantiomers, diastereomers, meso isomers and other stereoisomeric forms, including racemic mixtures, optically pure forms and intermediate mixtures. Enantiomers, diastereomers, meso isomers and other stereoisomeric forms can be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques. Unless specified otherwise, when the compounds disclosed and/or described herein contain olefinic double bonds or other centers of geometric asymmetry, it is intended that the compounds include both E and Z isomers.
- stereochemistry depicted in the structures of cyclic meso compounds is not absolute; rather the stereochemistry is intended to indicate the positioning of the substituents relative to one another, e.g., cis or trans.
- substituents e.g., cis or trans.
- Tautomers are structurally distinct isomers that interconvert by tautomerization. Tautomerization is a form of isomerization and includes prototropic or proton-shift tautomerization, which is considered a subset of acid-base chemistry. Prototropic tautomerization or proton-shift
- tautomerization involves the migration of a proton accompanied by changes in bond order, often the interchange of a single bond with an adjacent double bond. Where tautomerization is possible (e.g. in solution), a chemical equilibrium of tautomers can be reached.
- An example of tautomerization is keto-enol tautomerization.
- keto-enol tautomerization is the interconverision of pentane-2,4-dione and 4-hydroxypent-3-en-2-one tautomers.
- Another example of tautomerization is phenol-keto
- phenol-keto tautomerization is the interconversion of pyridin-4-ol and pyridin-4(1 H)-one tautomers.
- compounds described herein contain moieties capable of tautomerization, and unless specified otherwise, it is intended that the compounds include all possible tautomers.
- Protecting group has the meaning conventionally associated with it in organic synthesis, i.e., a group that selectively blocks one or more reactive sites in a multifunctional compound such that a chemical reaction can be carried out selectively on another unprotected reactive site, and such that the group can readily be removed after the selective reaction is complete.
- a variety of protecting groups are disclosed, for example, in T.H. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Third Edition, John Wiley & Sons, New York (1999).
- a "hydroxy protected form” contains at least one hydroxy group protected with a hydroxy protecting group.
- amines and other reactive groups may similarly be
- pharmaceutically acceptable salt refers to salts that retain the biological effectiveness and properties of the compounds described herein and are not biologically or otherwise undesirable. Examples of
- compositions described herein are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto.
- Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids.
- Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, and phosphoric acid.
- Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, pyruvic acid, lactic acid, oxalic acid, malic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 2- hydroxyethylsulfonic acid, p-toluenesulfonic acid, stearic acid and salicylic acid.
- Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
- Inorganic bases from which salts can be derived include, for example, sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, and aluminum.
- Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines; substituted amines including naturally occurring substituted amines; cyclic amines; and basic ion exchange resins. Examples of organic bases include isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, and ethanolamine. In some embodiments, the
- pharmaceutically acceptable base addition salt is chosen from ammonium, potassium, sodium, calcium, and magnesium salts.
- the free base can be obtained by basifying a solution of the acid salt.
- an addition salt particularly a pharmaceutically acceptable addition salt
- a suitable organic solvent may be used to dissolve the free base in a suitable organic solvent and treating the solution with an acid, in accordance with conventional procedures for preparing acid addition salts from base compounds (see, e.g., Berge et al., Pharmaceutical Salts, J.
- a “solvate” is formed by the interaction of a solvent and a compound.
- suitable solvents include, for example, water and alcohols (e.g., ethanol).
- Solvates include hydrates having any ratio of compound to water, such as monohydrates, dihydrates and hemi-hydrates.
- a “chelate” is formed by the coordination of a compound to a metal ion at two (or more) points.
- the term “compound” is intended to include chelates of compounds.
- salts includes chelates of salts and “solvates” includes chelates of solvates.
- non-covalent complex is formed by the interaction of a compound and another molecule wherein a covalent bond is not formed between the compound and the molecule.
- complexation can occur through van der Waals interactions, hydrogen bonding, and electrostatic interactions (also called ionic bonding).
- Such non-covalent complexes are included in the term "compound”.
- prodrug refers to a substance administered in an inactive or less active form that is then transformed (e.g., by metabolic processing of the prodrug in the body) into an active compound.
- the rationale behind administering a prodrug is to optimize absorption, distribution, metabolism, and/or excretion of the drug.
- Prodrugs may be obtained by making a derivative of an active compound (e.g., a compound of Formula I or another compound disclosed and/or described herein) that will undergo a
- the transformation of the prodrug to the active compound may proceed spontaneously (e.g., by way of a hydrolysis reaction) or it can be catalyzed or induced by another agent (e.g., an enzyme, light, acid or base, and/or temperature).
- the agent may be endogenous to the conditions of use (e.g., an enzyme present in the cells to which the prodrug is administered, or the acidic conditions of the stomach) or the agent may be supplied
- Prodrugs can be obtained by converting one or more functional groups in the active compound into another functional group, which is then converted back to the original functional group when administered to the body.
- a hydroxyl functional group can be converted to a sulfonate, phosphate, ester or carbonate group, which in turn can be hydrolyzed in vivo back to the hydroxyl group.
- an amino functional group can be converted, for example, into an amide, carbamate, imine, urea, phosphenyl, phosphoryl or sulfenyl functional group, which can be hydrolyzed in vivo back to the amino group.
- a carboxyl functional group can be converted, for example, into an ester (including silyl esters and thioesters), amide or hydrazide functional group, which can be hydrolyzed in vivo back to the carboxyl group.
- prodrugs include, but are not limited to, phosphate, acetate, formate and benzoate derivatives of functional groups (such as alcohol or amine groups) present in the compounds of Formula I and other compounds disclosed and/or described herein.
- the compounds disclosed and/or described herein can be enriched isotopic forms, e.g., enriched in the content of 2 H, 3 H, 11 C, 13 C and/or 14 C.
- the compound contains at least one deuterium atom.
- deuterated forms can be made, for example, by the procedure described in U.S. Patent Nos. 5,846,514 and 6,334,997.
- deuterated compounds may improve the efficacy and increase the duration of action of compounds disclosed and/or described herein.
- Deuterium substituted compounds can be synthesized using various methods, such as those described in: Dean, D., Recent Advances in the Synthesis and Applications of Radiolabeled
- pharmaceutically acceptable carrier or “pharmaceutically acceptable excipient” includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like.
- pharmaceutically acceptable carrier or “pharmaceutically acceptable excipient” includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like.
- the use of such media and agents for pharmaceutically active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, its use in
- compositions is contemplated.
- Supplementary active ingredients can also be incorporated into the pharmaceutical compositions.
- an active agent is used to indicate a compound that has biological activity.
- an “active agent” is a compound having therapeutic utility.
- the compound enhances at least one aspect of skeletal muscle function or activity, such as power output, skeletal muscle force, skeletal muscle endurance, oxygen consumption, efficiency, and/or calcium sensitivity.
- an active agent is a compound of Formula I, or a pharmaceutically acceptable salt thereof.
- patient and “subject” refer to an animal, such as a mammal bird or fish. In some embodiments, the patient or subject is a mammal.
- Mammals include, for example, mice, rats, dogs, cats, pigs, sheep, horses, cows and humans.
- the patient or subject is a human, for example a human that has been or will be the object of treatment, observation or experiment.
- the compounds, compositions and methods described herein can be useful in both human therapy and veterinary applications.
- skeletal muscle includes skeletal muscle tissue as well as components thereof, such as skeletal muscle fibers, the myofibrils comprising the skeletal muscle fibers, the skeletal sarcomere which
- skeletal muscle includes fast skeletal muscle tissue as well as components thereof, such as fast skeletal muscle fibers, the myofibrils comprising the fast skeletal muscle fibers, the fast skeletal sarcomere which comprises the myofibrils, and the various components of the fast skeletal sarcomere described herein, including fast skeletal myosin, actin,
- Skeletal muscle does not include cardiac muscle or a combination of sarcomeric components that occurs in such combination in its entirety in cardiac muscle.
- the term “therapeutic” refers to the ability to modulate the contractility of fast skeletal muscle.
- modulation refers to a change in function or efficiency of one or more components of the fast skeletal muscle sarcomere, including myosin, actin, tropomyosin, troponin C, troponin I, and troponin T from fast skeletal muscle, including fragments and isoforms thereof, as a direct or indirect response to the presence of a compound described herein, relative to the activity of the fast skeletal sarcomere in the absence of the compound.
- the change may be an increase in activity
- modulation is a potentiation of function or efficiency of one or more
- components of the fast skeletal muscle sarcomere including myosin, actin, tropomyosin, troponin C, troponin I, and troponin T from fast skeletal muscle, including fragments and isoforms thereof. Modulation may be mediated by any mechanism and at any physiological level, for example, through
- efficiency means the ratio of mechanical work output to the total metabolic cost.
- therapeutically effective amount refers to that amount of a compound disclosed and/or described herein that is sufficient to affect treatment, as defined herein, when administered to a patient in need of such treatment.
- a therapeutically effective amount of a compound may be an amount sufficient to treat a disease responsive to modulation of fast skeletal muscle.
- the therapeutically effective amount will vary depending upon, for example, the subject and disease condition being treated, the weight and age of the subject, the severity of the disease condition, the particular compound, the dosing regimen to be followed, timing of administration, the manner of administration, all of which can readily be determined by one of ordinary skill in the art.
- the therapeutically effective amount may be ascertained experimentally, for example by assaying blood concentration of the chemical entity, or theoretically, by calculating
- Treatment includes one or more of: preventing a disease or disorder (i.e., causing the clinical symptoms of the disease or disorder not to develop); inhibiting a disease or disorder; slowing or arresting the development of clinical symptoms of a disease or disorder; and/or relieving a disease or disorder (i.e., causing relief from or regression of clinical symptoms).
- compounds described and/or disclosed herein may prevent an existing disease or disorder from worsening, assist in the management of the disease or disorder, or reduce or eliminate the disease or disorder.
- the compounds disclosed and/or described herein may prevent a disease or disorder from developing or lessen the extent of a disease or disorder that may develop.
- power output of a muscle means work/cycle time and may be scaled up from PoLo/cycle time units based on the properties of the muscle. Power output may be modulated by changing, for example, activating parameters during cyclical length changes, including timing of activation (phase of activation) and the period of activation (duty cycle.)
- ATPase refers to an enzyme that hydrolyzes ATP.
- ATPases include proteins comprising molecular motors such as the myosins.
- selective binding refers to preferential binding to a target protein in one type of muscle or muscle fiber as opposed to other types.
- a compound selectively binds to fast skeletal troponin C if the compound preferentially binds troponin C in the troponin complex of a fast skeletal muscle fiber or sarcomere in comparison with troponin C in the troponin complex of a slow muscle fiber or sarcomere or with troponin C in the troponin complex of a cardiac sarcomere.
- Z 1 , Z 2 , Z 3 and Z 4 are each independently selected from N and CR 1 , provided that at least one of Z 1 , Z 2 , Z 3 and Z 4 is N and at least one of Z 1 , Z' Z 3 and Z 4 is CR 1 ;
- R 1 at each occurrence, is independently selected from hydrogen, halogen, CN, C 1-6 alkyl, C 1-6 haloalkyl, C(O)OR a , C(O)NR b R c , OR a , NR b R c , 10 aryl and 5-10 membered heteroaryl;
- R 2 is selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, 5-10 membered heteroaryl and NR b R c , wherein each of the C 3-8 cycloalkyl, C 3-8 cycloalkenyl, 3-8 membered heterocycloalkyi, 3-8 membered
- heterocycloalkenyl, C-6-10 aryl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH 2 ) n OR a , (CH 2 ) n OC(O)R a , (CH 2 ) n OC(O)OR a , (CH 2 ) n OC(O)NR b R c , (CH 2 ) n NR d C(O)R a , (CH 2 ) n NR d C(O)OR a , (CH 2 ) n NR d C(O)NR b R c , (CH 2 ) n NR d C(O)C(O)NR b R c , (CH 2 ) n NR d C(S)R a , (CH 2 ) n NR d C(S)R a , (CH 2
- R 4 is selected from hydrogen, C-i -6 alkyl, C-i -6 haloalkyl, C(O)R a , C(O)OR a , C(O)NR b R c and SO 2 R a ;
- R 5 and R 6 are each independently selected from hydrogen, halogen, C-i-6 alkyl and C-i -6 haloalkyl;
- R 5 and R 6 together with the carbon atom to which they are bound form a group selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyi and 3-8 membered heterocycloalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , NR b R c , C(O)R a , C(O)OR a , C(O)NR b R c , S(O)R a , SO 2 R a , SO 2 NR b R c , Ci -6 alkyl and Ci -6 haloalkyl;
- R 7 is selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyi, 3-8 membered heterocycloalkenyl, C 6- io aryl and 5-10 membered heteroaryl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , OC(O)NR b R c , N R b R c , NR d C(O)R a , NR d C(O)OR a , NR d C(O)NR b R c , NR d C(O)C(O)NR b R c , N R d C(S)R a , NR d C(S)OR a , N R d C(S)NR b R c , N R
- heterocycloalkenyl, C 6- io aryl, C 7- n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R f substituents;
- R 8 and R 9 are each independently selected from hydrogen, halogen and C-i-6 alkyl;
- X is selected from a bond, -(CH 2 ) P -, -(CH 2 ) p C(O)(CH 2 ) q -, - (CH 2 )pO(CH 2 ) q -, -(CH 2 )pS(CH 2 ) q -, -(CH 2 ) p NR d (CH 2 ) q -, -(CH 2 ) p C(O)O(CH 2 ) q -, -(CH 2 )pOC(O)(CH 2 ) q -, -(CH 2 )pNR d C(O)(CH 2 ) q -, -(CH 2 ) p C(O)NR d (CH 2 ) q -, -(CH 2 ) p C(O)NR d (CH 2 ) q -, -(CH 2 )p C(O)NR d (CH 2 ) q -, -
- R a is independently selected from hydrogen, C-i -6 alkyl, C-i-6 haloalkyl, C 2- 6 alkenyl, C 2- 6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C 7- i i aralkyl and 5-10 membered heteroaryl, wherein each of the C-i -6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 3- 8 cycloalkyl, C 3- 8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C 7- n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R f substituent
- R b and R c are each independently selected from hydrogen, C-i-6 alkyl, C-i-6 haloalkyl, C 2- 6 alkenyl, C 2- 6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
- R d at each occurrence, is independently selected from hydrogen and Ci-6 alkyl
- R e at each occurrence, is independently selected from hydrogen, CN, OH, C-i-6 alkoxy, C-i -6 alkyl and C-i -6 haloalkyl;
- R f is independently selected from halogen, CN, OR h , OC(O)R h , OC(O)OR h , OC JNR ⁇ , NR'R j , NR d C(O)R h , NR d C(O)OR h , NR d C(O)NR'R j , NR d C(O)C(O)NR i R j , NR d C(S)R h , NR d C(S)OR h , NR d C(S)NR'R j , NR d C(NR e )NR'R j , NR d S(O)R h , NR d SO 2 R h , NR d SO 2 NR'R j , C(O)R h , C(O)OR h , ⁇ ( ⁇ ) ⁇ , C(S)R h , C(O)OR h ,
- heterocycloalkenyl C 6- io aryl, C 7- n aralkyl and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
- heterocycloalkenyl, C 6- io aryl, C 7- n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R k substituents;
- R f substituents bound to a single carbon atom, together with the carbon atom to which they are both bound, form a group selected from carbonyl, C 3- 8 cycloalkyl and 3-8 membered heterocycloalkyl;
- R g at each occurrence, is independently selected from C-i -6 alkyl, C-i -6 haloalkyl, phenyl, naphthyl, and C 7- n aralkyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, OH, C-i-6 alkoxy, C-i-6 alkyl and C-i -6 haloalkyl;
- R h is independently selected from hydrogen, C-i -6 alkyl, C-i-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C 7- ii aralkyl and 5-10 membered heteroaryl, wherein each of the C-i -6 alkyl, C 2- 6 alkenyl, C 2- 6 alkynyl, C 3- 8 cycloalkyl, C 3- 8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C 7- n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R k substituents
- R 1 and R j are each independently selected from hydrogen, d-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
- heterocycloalkenyl C 6- io aryl, C 7- n aralkyl, 5-10 membered heteroaryl, C(O)R g , and C(O)OR g , wherein each of the C -6 alkyl, C -6 haloalkyl, C 2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C-6-10 aryl, C7-11 aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, OH, C-i -6 alkoxy, C-i -6 alkyl and C-i-6 haloalkyl;
- R k is independently selected from halogen, CN, oxo, OH, C1-6 alkoxy, NH 2 , NH(C 1-6 alkyl), N(C 1-6 alkyl) 2 , NHC(O)C 1-6 alkyl, NHC(0)C 7 -ii aralkyl, NHC(O)OC 1-6 alkyl, NHC(O)OC 7-11 aralkyl, C(O)C 1-6 alkyl, C(0)C 7 -n aralkyl, C(O)OCi -6 alkyl, C(0)OC 7 -n aralkyl, Ci -6 alkyl, Ci -6 haloalkyl, C2-6 alkenyl, and C2-6 alkynyl, wherein each C-i-6 alkyl, C2-6 alkenyl, C 2- 6 alkynyl, and C 7- n aralkyl substituent is optionally substituted with 1 , 2 or 3 substituents
- n 0, 1 or 2;
- n at each occurrence, independently is 0, 1 or 2; p is 0, 1 or 2; and
- q 0, 1 or 2.
- Z 1 and Z 2 are each N, one of Z 3 and Z 4 is N, and the other of Z 3 and Z 4 is CR 1 .
- Z 1 , Z 2 and Z 3 are each N and Z 4 is CR 1 , i.e., a compound of Formula II, or a pharmaceutically acceptable salt thereof:
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , X and m are as defined herein.
- Z 1 , Z 2 and Z 4 are each N and Z 3 is CR 1 , i.e., a compound of Formula III, or a pharmaceutically acceptable salt thereof:
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , X and m are as defined herein.
- one of Z 1 and Z 2 is N, the other of Z 1 and Z 2 is CR 1 , and Z 3 and Z 4 are each CR 1 .
- Z 1 , Z 3 and Z 4 are each CR 1 and Z 2 is N, i.e., a compound of Formula IV, or a pharmaceutically acceptable salt thereof:
- Z 2 , Z 3 and Z 4 are each CR 1 and Z 1 is N, i.e., a compound of Formula V, or a pharmaceutically acceptable salt thereof:
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , X and m are as defined herein.
- m is 0, i.e., a compound of Formula ll(a), lll(a), IV(a) or V(a), or a pharmaceutically acceptable salt thereof:
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 and X are as defined herein.
- m is 1 , i.e., a compound of Formula ll(b), lll(b), IV(b) or V(b), or a pharmaceutically acceptable salt thereof:
- R 1 , R 2 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 and X are as defined herein.
- one of R 5 and R 6 is hydrogen and the other is C-i-6 alkyl.
- R 5 and R 6 are each independently C-i-6 alkyl.
- R 5 and R 6 are each methyl.
- the compounds are of Formula ll(c) or ll(d), or a pharmaceutically acceptable salt thereof:
- R 1 , R 2 , R 4 , R 7 , R 8 , R 9 and X are as defined herein.
- the compounds are of Formula lll(c) or lll(d), or a pharmaceutically acceptable salt thereof:
- R 1 , R 2 , R 4 , R 7 , R 8 , R 9 and X are as defined herein.
- the compounds are of Formula IV(c) or IV(d), or a pharmaceutically acceptable salt thereof:
- R 1 , R 2 , R 4 , R 7 , R 8 , R 9 and X are as defined herein.
- the compounds are of Formula V(c) or V(d), or a pharmaceutically acceptable salt thereof:
- R 1 , R 2 , R 4 , R 7 , R 8 , R 9 and X are as defined herein.
- R 5 and R 6 together with the carbon atom to which they are bound form C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl or 3-8 membered heterocycloalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , NR b R c , C(O)R a , C(O)OR a , C(O)NR b R c , S(O)R a , SO 2 R a , SO 2 NR b R c , Ci -6 alkyl and Ci -6 halo
- C 3-6 cycloalkyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , NR b R c , C(O)R a , C(O)OR a , C(O)NR b R c , S(O)R a , SO 2 R a ,
- cyclobutyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , NR b R c , C(O)R a , C(O)OR a , C(O)NR b R c , S(O)R a , SO 2 R a , SO 2 NR b R c , C 1-6 alkyl and Ci-6 haloalkyl.
- R 5 and R 6 together with the carbon to which they are bound, form cyclobutyl substituted with one substituent selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , NR b R c , C(O)R a , C(O)OR a , C(O)NR b R c , S(O)R a , SO 2 R a , SO 2 NR b R c , C 1-6 alkyl and C 1-6 haloalkyl, wherein the substituent and R 7 are in a cis configuration with respect to one another on the cyclobutyl ring.
- the compounds are of Formula ll(e) or ll(f), or a pharmaceutically acceptable salt thereof:
- R m and R n are each independently selected from hydrogen and halogen, and R 1 , R 2 , R 4 , R 7 , R 8 , R 9 and X are as defined herein.
- the compounds are of Formula lll(e) or lll(f), or a pharmaceutically acceptable salt thereof:
- R m and R n are each independently selected from hydrogen and halogen, and R 1 , R 2 , R 4 , R 7 , R 8 , R 9 and X are as defined herein.
- the compounds are of Formula IV(e) or IV(f), or a pharmaceutically acceptable salt thereof:
- R m and R n are each independently selected from hydrogen and halogen, and R 1 , R 2 , R 4 , R 7 , R 8 , R 9 and X are as defined herein.
- the compounds are of Formula V(e) or V(f), or a pharmaceutically acceptable salt thereof:
- R m and R n are each independently selected from hydrogen and halogen, and R 1 , R 2 , R 4 , R 7 , R 8 , R 9 and X are as defined herein.
- R m and R n are each hydrogen.
- compounds of Formula ll(e), ll(f), lll(e), lll(f), IV(e), IV(f), V(e) or V(f), R m and R n are each halogen.
- compounds of Formula ll(e), ll(f), lll(e), lll(f), IV(e), IV(f), V(e) or V(f), R m and R n are each fluorine.
- the halogen and R 7 are in a trans configuration with respect to one another on the cyclobutyl ring. In some embodiments of such compounds, the halogen and R 7 are in a cis configuration with respect to one another on the cyclobutyl ring.
- one of R m and R n is hydrogen and the other is fluorine.
- the fluorine and R 7 are in a trans configuration with respect to one another on the cyclobutyl ring. In some embodiments of such compounds, the fluorine and R 7 are in a cis configuration with respect to one another on the cyclobutyl ring.
- R 5 and R 6 are each independently C-i-6 alkyl, or R 5 and R 6 together with the carbon atom to which they are bound form C 3- 8 cycloalkyl, C 3- 8 cycloalkenyl, 3-8 membered heterocycloalkyl or 3-8 membered heterocycloalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , NR b R c , C(O)R a , C(O)OR a , C(O)NR b R c , S(O)R a , SO 2 R a , SO 2 NR b R
- R 5 and R 6 are each methyl, or R 5 and R 6 together with the carbon atom to which they are bound form C 3-8 cycloalkyl, C 3-8 cycloalkenyl, 3-8 membered heterocycloalkyl or 3-8 membered heterocycloalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , NR b R c , C(O)R a , C(O)OR a , C(O)NR b R c , S(O)R a , SO 2 R a , SO 2 NR b R c , C
- R 5 and R 6 are each independently C-i-6 alkyl, or R 5 and R 6 , together with the carbon to which they are bound, form a group selected from cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , NR b R c , C(O)R a , C(O)OR a , C(O)NR b R c , S(O)R a , SO 2 R a , SO 2 NR b R c , C 1-6 alkyl and
- R 5 and R 6 are each methyl, or R 5 and R 6 , together with the carbon to which they are bound, form cyclobutyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , NR b R c , C(O)R a , C(O)OR a , C(O)NR b R c , S(O)R a , SO 2 R a , SO 2 NR b R c , Ci -6 alkyl and Ci -6 haloalkyl.
- R 7 is selected from C 3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl and 5-10 membered heteroaryl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , OC(O)
- R 7 is phenyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , OC(O)NR b R c , NR b R c , N R d C(O)R a , N R d C(O)OR a , NR d C(O)N R b R c , N
- R 7 is selected from phenyl, 2- fluorophenyl, 3-fluorophenyl, 2, 4-difluorophenyl, 3,4-difluorophenyl, 3,5- difluorophenyl, 4-fluorophenyl, 2-chlorophenyl, 3-chlorophenyl, 4- chlorophenyl, 2, 4-dichlorophenyl,
- R 7 is 5-10 membered heteroaryl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , OC(O)NR b R c , NR b R c , NR d C(O)R
- NR d C(S)OR a NR d C(S)NR b R c , NR d C(NR e )NR b R c , NR d S(O)R a , NR d SO 2 R a , NR d SO 2 NR b R c , C(O)R a , C(O)OR a , C(O)NR b R c , C(S)R a , C(S)OR a , C(S)NR b R c , C(NR e )NR b R c , SR a , S(O)R a , SO 2 R a , SO 2 NR b R c , Ci -6 alkyl, Ci -6 haloalkyl, C 2-6 alkenyl, C 2-6 alkynyl, C 3- 8 cycloalkyl, C 3- 8 cycloalkenyl, 3-8 membered
- R 7 is pyridyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, OR a , OC(O)R a , OC(O)OR a , OC(O)NR b R c , NR b R c , NR d C(O)R a
- R 7 is selected from 2-pyridyl, 3- pyridyl and 4-pyridyl, each optionally substituted with 1 , 2, 3, 4 or 5
- R 7 is selected from pyrid-2-yl, 3-fluoro-pyrid-2-yl, 4-fluoro-pyrid-2- yl, 5-fluoro-pyrid-2-yl, 6-fluoro-pyrid-2-yl, 3-chloro-pyrid-2-yl, 4-chloro-pyrid-2- yl, 5-chloro-pyrid-2-yl, 6-chloro-pyrid-2-yl, 3-cyano-pyrid-2-yl, 4-cyano-pyrid-2- yl, 5-cyano-pyrid-2-yl, 6-cyano-pyrid-2-yl, 3-methyl-pyrid-2-yl, 4-methyl-pyrid- 2-yl, 5-methyl-pyrid-2-yl, 6-methyl-pyrid-2-yl, 3-difluoromethyl-pyrid
- R 7 is selected from pyrid-3-yl, 2-fluoro-pyrid-3-yl, 4-fluoro-pyrid-3- yl, 5-fluoro-pyrid-3-yl, 6-fluoro-pyrid-3-yl, 2-chloro-pyrid-3-yl, 4-chloro-pyrid-3- yl, 5-chloro-pyrid-3-yl, 6-chloro-pyrid-3-yl, 2-cyano-pyrid-3-yl, 4-cyano-pyrid-3- yl, 5-cyano-pyrid-3-yl, 6-cyano-pyrid-3-yl, 2-methyl-pyrid-3-yl, 4-methyl-pyrid-
- X is selected from a bond, - (CH 2 )p-, -(CH 2 ) P C(O)(CH 2 ) q -, -(CH 2 ) P O(CH 2 ) q -, -(CH 2 ) p S(CH 2 ) q -,
- X is selected from -CH 2 O- and -OCH 2 -.
- X is selected from -CH 2 NR d - and -NR d CH 2 -.
- X is sleeted from -NR d C(O)- and -C(O)NR d -.
- X is sleeted from - CH 2 NR d C(O)- and -C(O)NR d CH 2 -.
- R 2 is selected from C 3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C-6-10 aryl, 5-10 membered heteroaryl and NR b R c , wherein each of the C 3- 8 cycloalkyl, C 3- 8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C-6-10 aryl, 5-10 membered heteroaryl and NR b R c , wherein each of the C 3- 8 cycloalkyl, C 3- 8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalken
- (CH 2 )nC 6- io aryl and (CH 2 ) n 5-10 membered heteroaryl wherein each of the C-i. 6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, (CH 2 ) n C 3- 8 cycloalkyl, (CH 2 ) n 3-8 membered heterocycloalkyl, (CH 2 ) n C6-io aryl and (CH 2 ) n 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R f substituents.
- R 2 is phenyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH 2 ) n OR a , (CH 2 ) n OC(O)R a , (CH 2 ) n OC(O)OR a , (CH 2 ) n OC(O)NR b R c ,
- R 2 is phenyl substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH 2 ) n OR a ,
- substitutent is bonded at the meta position.
- R 2 is phenyl substituted with a substituent selected from (CH 2 ) n C(O)OR a and (CH 2 ) n C(O)NR b R c ; and optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, CN, oxo, (CH 2 ) n OR a , (CH 2 ) n OC(O)R a , (CH 2 ) n OC(O)OR a ,
- (CH 2 )nC 6- io aryl and (CH 2 ) n 5-10 membered heteroaryl wherein each of the C-i. 6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, (CH 2 ) n C 3- 8 cycloalkyl, (CH 2 ) n 3-8 membered heterocycloalkyl, (CH 2 ) n C6-io aryl and (CH 2 ) n 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R f substituents.
- R 2 is phenyl substituted with a substituent selected from C(O)OH, C(O)NH 2 , C(O)OCi -6 alkyl, C(O)NHCi -6 alkyl and C(O)N(Ci -6 alkyl) 2 ; and optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, C-i -6 alkyl and C-i -6 haloalkyl.
- R 2 is phenyl substituted at the meta position with a substituent selected from (CH 2 ) n C(O)OR a and
- heterocycloalkyl (CH 2 ) n C6-io aryl and (CH 2 ) n 5-10 membered heteroaryl, wherein each of the C-i -6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, (CH 2 ) n C 3- 8 cycloalkyl, (CH 2 ) n 3-8 membered heterocycloalkyl, (CH 2 ) n C 6- io aryl and
- (CH 2 ) n 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R f substituents.
- R 2 is phenyl substituted at the meta position with a substituent selected from (CH 2 ) n C(O)OR a and
- R 2 is phenyl substituted at the meta position with a substituent selected from C(O)OH, C(O)NH 2 , C(O)OCi -6 alkyl, C(O)NHCi -6 alkyl and C(O)N(Ci -6 alkyl) 2 ; and optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, hydroxyl, CN
- R 2 is phenyl substituted with (CH 2 ) n NR d C(O)R a , wherein R a is C -6 alkyl or 3-8 membered heterocycloalkyl, each optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, CN, oxo, (CH 2 ) n OR a , (CH 2 ) n
- (CH 2 ) n phenyl, (CH 2 ) n naphthyl and (CH 2 ) n 5-10 membered heteroaryl wherein each of the C-i-6 alkyl, C 2- 6 alkenyl, C 2- 6 alkynyl, (CH 2 ) n C3-8 cycloalkyl, (CH 2 ) n 3- 8 membered heterocycloalkyl, (CH 2 ) n phenyl, (CH 2 ) n naphthyl and (CH 2 ) n 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R f substituents.
- R 2 is phenyl substituted with (CH 2 ) n NR d C(O)R a , wherein R a is selected from C -6 alkyl, C -6 alkyl-OH and Ci-6 alkyl-NH 2 , each optionally substituted with 1 , 2 or 3 additional
- (CH 2 ) n phenyl, (CH 2 ) n naphthyl and (CH 2 ) n 5-10 membered heteroaryl wherein each of the C-i -6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, (CH 2 ) n C 3- 8 cycloalkyl, (CH 2 ) n 3- 8 membered heterocycloalkyl, (CH 2 ) n phenyl, (CH 2 ) n naphthyl and (CH 2 ) n 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R f substituents.
- R 2 is selected from 3- benzamide, N-methyl-3-benzamide, N,N-dimethyl-3-benzamide, 4-fluoro-3- benzamide, N-methyl-4-fluoro-3-benzamide, N,N-dimethyl-4-fluoro-3- benzamide, 3-benzoic acid, methyl-3-benzoate, 4-fluoro-3-benzoic acid, and
- R 2 is 5-10 membered heteroaryl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH 2 ) n OR a , (CH 2 ) n OC(O)R a , (CH 2 ) n OC(O)OR a , (CH 2 ) n OC(O)
- R 2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl, triazyl, furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imi
- heterocycloalkyl (CH 2 ) n C6-io aryl and (CH 2 ) n 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C 2- 6 alkenyl, C 2- 6 alkynyl, (CH 2 ) n C3-8 cycloalkyl, (CH 2 ) n 3-8 membered heterocycloalkyl, (CH 2 ) n C 6- io aryl and
- (CH 2 ) n 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R f substituents.
- R 2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl, triazyl, furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imi
- R 2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl and triazyl, each optionally substituted with (CH 2 ) n C(O)NR b R c .
- R 2 is selected from furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with (CH 2 ) n C(O)NR b R c .
- R 2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl and triazyl, each optionally substituted with (CH 2 ) n C(O)NH 2 .
- R 2 is selected from furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with (CH 2 ) n C(O)NH 2 .
- R 2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl, triazyl, furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with (CH
- heterocycloalkyl (CH 2 ) n phenyl, (CH 2 ) n naphthyl and (CH 2 ) n 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R f substituents.
- R 2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl and triazyl, each optionally substituted with (CH 2 ) n NR d C(O)R a , wherein R a is selected from Ci -6 alkyl, Ci -6 alkyl-OH and C-i-6 alkyl-NH 2 ,
- R 2 is selected from furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with (CH 2 ) n NR
- R 2 is selected from indolyl, indazolyl, benzimidazolyl, benzoxazolyl and benzoisoxazolyl, each optionally substituted with 1 , 2, 3 or 4 substituents selected from halogen, CN, oxo, (CH 2 ) n OR a , (CH 2 ) n OC(O)R a
- (CH 2 ) n naphthyl and (CH 2 ) n 5-10 membered heteroaryl wherein each of the C-i- 6 alkyl, C 2- 6 alkenyl, C 2- 6 alkynyl, (CH 2 ) n C3-8 cycloalkyl, (CH 2 ) n 3-8 membered heterocycloalkyl, (CH 2 ) n phenyl, (CH 2 ) n naphthyl and (CH 2 ) n 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 R f substituents.
- R 2 is selected from 1 H-indazol- 6-yl, 1 H-indazol-5-yl, 1 H-indazol-4-yl, 3-amino(1 H-indazol-5-yl), 3-amino(1 H- indazol-6-yl), 3-amino(1 H-indazol-7-yl), 1 -methyl(1
- R 2 is selected from 3-6 membered heterocycloalkyl and 3-6 membered heterocycloalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH 2 ) n OR a , (CH 2 ) n OC(O)R a , (CH 2 ) n
- R 2 is selected from aziridinyl, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl and morpholinyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH 2 ) n OR a , (CH 2 ) n OC(O)R a
- R 2 is NR b R c , wherein R b and R c are as defined herein.
- R 2 is NR b R c , wherein one of R b and R c is hydrogen and the other is C-i -6 alkyl optionally substituted with 1 , 2, 3, 4 or 5 R f substituents.
- X is -C(O)- and R 2 is NR b R c , wherein R b and R c are as defined herein.
- X is -C(O)- and R 2 is NR b R c , wherein one of R b and R c is hydrogen and the other is C-i -6 alkyl optionally substituted with 1 , 2, 3, 4 or 5 R f substituents.
- X is -(CH 2 ) P - and R 2 is NR b R c , wherein R b and R c are as defined herein.
- X is -(CH 2 ) P - and R 2 is NR b R c , wherein one of R b and R c is hydrogen and the other is C-i-6 alkyl optionally substituted with 1 , 2, 3, 4 or 5 R f substituents.
- R 1 at each occurrence, is independently selected from hydrogen, halogen, CN, C-i-6 alkyl, C-i-6 haloalkyl, C(O)OR a , C(O)NR b R c , OR a , NR b R c , C 6- io aryl and 5-10 membered heteroaryl.
- R 1 at each occurrence, is independently selected from hydrogen, halogen, CN, C-i -6 alkyl, C-i -6 haloalkyl, hydroxyl, C-i -6 alkoxy, NH 2 , NHC-i -6 alkyl, and N(Ci -6 alkyl) 2 .
- R 1 is independently selected from hydrogen, halogen, CN, CF 3 and methyl.
- R 4 is selected from hydrogen, C 1 -6 alkyl, C 1-6 haloalkyl, C(O)R a , C(O)OR a , C(O)NR b R c and SO 2 R a .
- R 4 is hydrogen and each R 1 is hydrogen.
- R 8 and R 9 at each occurrence, are each independently selected from hydrogen, halogen and C-i- 6 alkyl.
- the compound is selected from the compounds in Table 2, or a pharmaceutically acceptable salt thereof.
- the compounds and compositions described and/or disclosed herein modulate the contractility of the skeletal sarcomere. Specifically, the compounds modulate the troponin complex of the fast skeletal muscle sarcomere through one or more of fast skeletal myosin, actin, tropomyosin, troponin C, troponin I, and troponin T, and fragments and isoforms thereof. As used in this context, "modulate” means either increasing or decreasing activity.
- the compounds described and/or disclosed herein potentiate (i.e., increase activity) of one or more of fast skeletal myosin, actin, tropomyosin, troponin C, troponin I, and troponin T, and fragments and isoforms thereof. In other instances, the compounds described and/or disclosed herein inhibit (i.e., decrease activity) of one or more of fast skeletal myosin, actin, tropomyosin, troponin C, troponin I, and troponin T, and fragments and isoforms thereof.
- activators of the fast skeletal troponin complex have been shown to amplify the response of fast skeletal muscle to nerve stimulation, resulting in an increase in muscle force development at sub-maximal muscle activation (see, e.g., Russell et al., "The Fast Skeletal Troponin Activator, CK-2017357, Increases Skeletal Muscle Force in vitro and in situ", 2009 Experimental Biology Conference, New La, LA, April 2009).
- Activators of the fast skeletal troponin complex have been shown to increase the sensitivity of skinned skeletal muscle fibers to calcium, and in living muscle to the frequency of
- a compound or composition described and/or disclosed herein that selectively binds the troponin complex of fast skeletal muscle fiber or sarcomere.
- the compound disclosed and/or described herein activates fast skeletal muscle fibers or sarcomeres.
- administration of a compound disclosed and/or described herein results in an increase in fast skeletal muscle power output.
- administration of a compound disclosed and/or described herein results in increased sensitivity of fast skeletal muscle fibers or sarcomeres to calcium ion, as compared to fast skeletal muscle fibers or sarcomeres untreated with the compound.
- a compound disclosed and/or described herein results in increased sensitivity of fast skeletal muscle fibers or sarcomeres to calcium ion, as compared to fast skeletal muscle fibers or sarcomeres untreated with the compound.
- administration of a compound disclosed and/or described herein results in a lower concentration of calcium ions causing fast skeletal muscle myosin to bind to actin. In some embodiments, administration of a compound disclosed and/or described herein results in the fast skeletal muscle fiber generating force to a greater extent at submaximal levels of muscle activation.
- Also provided is a method for sensitizing a fast skeletal muscle fiber to produce force in response to a lower concentration of calcium ion comprising contacting the fast skeletal muscle fiber with a compound or composition described and/or disclosed herein that selectively binds to troponin complexes in the fast skeletal muscle sarcomere.
- contacting the fast skeletal muscle fiber with the compound results in activation of the fast skeletal muscle fiber at a lower calcium ion concentration than in an untreated fast skeletal muscle fiber.
- contacting the fast skeletal muscle fiber with the compound results in the production of increased force at a lower calcium ion concentration in comparison with an untreated fast skeletal muscle fiber.
- the compound binds to form ligand-troponin-calcium ion complexes that activate the fast skeletal muscle fibers.
- formation of the complexes and/or activation of the fast skeletal muscle fibers results in enhanced force and/or increased time to fatigue as compared to untreated fast skeletal muscle fibers contacted with a similar calcium ion concentration.
- the compounds and pharmaceutical compositions described and/or disclosed herein are capable of modulating the contractility of the fast skeletal sarcomere in vivo, and can have application in both human and animal disease. Modulation would be desirable in a number of conditions or diseases, including, but not limited to, 1 ) neuromuscular disorders, such as Amyotrophic Lateral Sclerosis (ALS), Spinal Muscular Atrophy (SMA), peripheral neuropathies and myasthenia gravis; 2) disorders of voluntary muscle, including muscular dystrophies, myopathies and conditions of muscle wasting, such as sarcopenia and cachexia syndromes (e.g., cachexia syndromes caused by diseases such as cancer, heart failure, chronic obstructive pulmonary disease (COPD), and chronic kidney disease/dialysis), and rehabilitation-related deficits, such as those associated with recovery from surgery (e.g.
- VBD Peripheral Vascular Disease
- PAD Peripheral Arterial Disease
- Neuromuscular diseases include, for example: 1 ) diseases of the motor unit, including but not limited to Amyotrophic Lateral Sclerosis (ALS) including bulbar and primary lateral sclerosis (PLS) variants; spinal muscular atrophy types 1 -4; Kennedy syndrome; post-polio syndrome; motor neuropathies including, for example, critical illness polyneuropathy; multifocal motor neuropathy with conduction block; Charcot-Marie-Tooth disease and other hereditary motor and sensory neuropathies; and Guillain- Barre Syndrome, 2) disorders of the neuromuscular junction, including myasthenia gravis, Lambert-Eaton myasthenic syndrome, and prolonged neuromuscular blockade due to drugs or toxins; and 3) peripheral s of the motor unit, including but not limited to Amyotrophic Lateral Sclerosis (ALS) including bulbar and primary lateral sclerosis (PLS) variants; spinal muscular atrophy types 1 -4; Kennedy syndrome; post-polio syndrome; motor neuropathies including, for example, critical illness polyneuropathy; multifocal
- neuropathies such as acute inflammatory demyelinating
- polyradiculoneuropathy diabetic neuropathy, chronic inflammatory demyelinating polyradiculoneuropathy, traumatic peripheral nerve lesions, neuropathy of leprosy,vasculitic neuropathy, dermatomyositis/polymyositis and neuropathy of Friedreich Ataxia.
- the compounds and compositions described and/or disclosed herein may be used to treat disorders of voluntary muscle.
- Disorders of voluntary muscle include 1 ) muscular dystrophies (including, for example, Duchenne, Becker, Limb-Girdle, Facioscapulohumeral, limb girdle, Emery-Dreyfus, oculopharyngeal and congenital muscular dystrophies); and 2) myopathies, such as nemaline myopathy, central core disease, congenital myopathies, mitochondrial myopathies, acute myopathy; inflammatory myopathies (such as dermatomyositis/polymyositis and inclusion body myositis), endocrine myopathies (such as those associated with hyper- or hypothyroidism), Cushing's or Addison's syndrome or disease and pituitary gland disorders, metabolic myopathies (such as glycogen storage diseases, e.g., McArdle's disease, Pompe disese, etc), drug-induced myopathy (
- ALS Amyotrophic Lateral Sclerosis
- ALS is a disease that generally arises later in life (Age 50+) and has a rapid progression from initial limb weakness to paralysis and death. Common life expectancy after diagnosis is 3-5 years.
- the cause of disease for most ALS patients is unknown (termed the spontaneous form) while a small proportion of patients have an inherited form (familial) of disease.
- the condition causes progressive death of motor neurons through causes that are not clear.
- SMA Spinal Muscular Atrophy
- Myasthenia gravis is a chronic autoimmune neuromuscular disease wherein the body produces antibodies that block, alter, or destroy proteins involved in signaling at the neuromuscular junction, thus preventing muscle contraction from occurring. These proteins include nicotinic acetylcholine receptor (AChR) or, less frequently, a muscle-specific tyrosine kinase (MuSK) involved in AChR clustering (see, e.g., Drachman, N. Eng. J. of Med., 330:1797-1810, 1994). The disease is characterized by varying degrees of weakness of the skeletal (voluntary) muscles of the body.
- AChR nicotinic acetylcholine receptor
- MoSK muscle-specific tyrosine kinase
- myasthenia gravis The hallmark of myasthenia gravis is muscle weakness that increases during periods of activity and improves after periods of rest.
- myasthenia gravis may affect any voluntary muscle, certain muscles, such as those that control eye and eyelid movement, facial expression, chewing, talking, and swallowing are often, but not always, involved in the disorder.
- the muscles that control breathing and neck and limb movements may also be affected.
- the first noticeable symptom is weakness of the eye muscles.
- difficulty in swallowing and slurred speech may be the first signs.
- the degree of muscle weakness involved in myasthenia gravis varies greatly among patients, ranging from a localized form, limited to eye muscles (ocular myasthenia), to a severe or generalized form in which many muscles - sometimes including those that control breathing - are affected.
- Symptoms which vary in type and severity, may include a drooping of one or both eyelids (ptosis), blurred or double vision (diplopia) due to weakness of the muscles that control eye movements, unstable or waddling gait, weakness in arms, hands, fingers, legs, and neck, a change in facial expression, difficulty in swallowing and shortness of breath, and impaired speech (dysarthria).
- ptosis eyelids
- di vision blurred or double vision
- Generalized weakness develops in approximately 85% of patients.
- sarcopenia e.g., sarcopenia associated with aging or disease (e.g. HIV infection).
- Sarcopenia is characterized by a loss of skeletal muscle mass, quality, and strength.
- Clinically a decline in skeletal muscle tissue mass (muscle atrophy) contributes to frailty in older individuals.
- muscle atrophy contributes to frailty in older individuals.
- muscle mass declines by one-third between the ages of 50 and 80.
- extended hospitalization can result in further disuse atrophy leading to a potential loss of the ability for independent living and to a cascade of physical decline.
- Muscle weakness is also a major factor prediposing the elderly to falls and the resulting morbidity and mortality.
- Cachexia is a state often associated with cancer or other serious diseases or conditions, (e.g, chronic obstructive pulmonary disease, heart failure, chronic kidney disease, kidney dialysis), that is characterized by progressive weight loss, muscle atrophy and fatigue, due to the deletion of adipose tissue and skeletal muscle.
- diseases or conditions e.g, chronic obstructive pulmonary disease, heart failure, chronic kidney disease, kidney dialysis
- Muscular dystrophy can be characterized by progressive muscle weakness, destruction and regeneration of the muscle fibers, and eventual replacement of the muscle fibers by fibrous and fatty connective tissue.
- the compounds and compositions described and/or disclosed herein may be used to treat post-surgical muscle weakness, which is a reduction in the strength of one or more muscles following surgical procedure. Weakness may be generalized (i.e. total body weakness) or localized to a specific area, side of the body, limb, or muscle.
- the compounds and compositions described and/or disclosed herein may be used to treat post-traumatic muscle weakness, which is a reduction in the strength of one or more muscles following a traumatic episode (e.g. bodily injury). Weakness may be generalized (i.e. total body weakness) or localized to a specific area, side of the body, limb, or muscle.
- Peripheral vascular disease is a disease or disorder of the circulatory system outside of the brain and heart.
- Peripheral artery disease also known as peripheral artery occlusive disease (PAOD)
- PAOD peripheral artery occlusive disease
- PVD and/or PAD can result from, for example, atherosclerosis, inflammatory processes leading to stenosis, embolus/ thrombus formation, or damage to blood vessels due to disease (e.g., diabetes), infection or injury.
- PVD and/or PAD can cause either acute or chronic ischemia, typically of the legs.
- the symptoms of PVD and/or PAD include pain, weakness, numbness, or cramping in muscles due to decreased blood flow (claudication), muscle pain, ache, cramp, numbness or fatigue that occurs during exercise and is relieved by a short period of rest (intermittent claudication), pain while resting (rest pain) and biological tissue loss (gangrene).
- the symptoms of PVD and/or PAD often occur in calf muscles, but symptoms may also be observed in other muscles such as the thigh or hip.
- Risk factors for PVD and/or PAD include age, obesity, sedentary lifestyle, smoking, diabetes, high blood pressure, and high cholesterol (i.e., high LDL, and/or high triglycerides and/or low HDL). People who have coronary heart disease or a history of heart attack or stroke generally also have an increased frequency of having PVD and/or PAD.
- Activators of the fast skeletal troponin complex have been shown to reduce muscle fatigue and/or to increase the overall time to fatigue in in vitro and in situ models of vascular insufficiency (see, e.g., Russell et al., "The Fast Skeletal Troponin Activator, CK-2017357, Increases Skeletal Muscle Force and Reduces Muscle Fatigue in vitro and in situ", 5th Cachexia Conference, Barcelona, Spain, December 2009; Hinken et al., "The Fast Skeletal Troponin Activator, CK-2017357, Reduces Muscle Fatigue in an in situ Model of Vascular Insufficiency", Society for Vascular Medicine's 2010 Annual Meeting: 21 st Annual Scientific Sessions, Cleveland, OH, April 2010).
- frailty e.g., frailty associated with aging.
- Frailty is characterized by one or more of unintentional weight loss, muscle weakness, slow walking speed, exhaustion, and low physical activity.
- the compounds and compositions described and/or disclosed herein may be used to treat muscle weakness and/or fatigue due to wasting syndrome, which is a condition characterized by involuntary weight loss associated with chronic fever and diarrhea. In some instances, patients with wasting syndrome lose 10% of baseline body weight within one month.
- the compounds and compositions described and/or disclosed herein may be used to treat muscular diseases and conditions caused by structural and/or functional abnormalities of skeletal muscle tissue, including muscular dystrophies, congenital muscular dystrophies, congenital myopathies, distal myopathies, other myopathies (e.g., myofibrillar, inclusion body), myotonic syndromes, ion channel muscle diseases, malignant hyperthermias, metabolic myopathies, congenital myasthenic syndromes, sarcopenia, muscle atrophy and cachexia.
- muscular dystrophies congenital muscular dystrophies, congenital myopathies, distal myopathies, other myopathies (e.g., myofibrillar, inclusion body), myotonic syndromes, ion channel muscle diseases, malignant hyperthermias, metabolic myopathies, congenital myasthenic syndromes, sarcopenia, muscle atrophy and cachexia.
- the compounds and compositions described and/or disclosed herein also may be used to treat diseases and conditions caused by muscle dysfunction originating from neuronal dysfunction or transmission, including amyotrophic lateral sclerosis, spinal muscular atrophies, hereditary ataxias, hereditary motor and sensory neuropathies, hereditary paraplegias, stroke, multiple sclerosis, brain injuries with motor deficits, spinal cord injuries, Alzheimer's disease, Parkinson's disease with motor deficits, myasthenia gravis and Lambert-Eaton syndrome.
- diseases and conditions caused by muscle dysfunction originating from neuronal dysfunction or transmission including amyotrophic lateral sclerosis, spinal muscular atrophies, hereditary ataxias, hereditary motor and sensory neuropathies, hereditary paraplegias, stroke, multiple sclerosis, brain injuries with motor deficits, spinal cord injuries, Alzheimer's disease, Parkinson's disease with motor deficits, myasthenia gravis and Lambert-Eaton syndrome.
- the compounds and compositions described and/or disclosed herein also may be used to treat diseases and conditions caused by CNS, spinal cord or muscle dysfunction originating from endocrine and/or metabolic dysregulation, including claudication secondary to peripheral artery disease, hypothyroidism, hyper- or hypo-parathyroid ism, diabetes, adrenal dysfunction, pituitary dysfunction and acid/base imbalances.
- compositions described and/or disclosed herein may be administered alone or in combination with other therapies and/or therapeutic agents useful in the treatment of the aforementioned disorders.
- the compounds and compositions described and/or disclosed herein may be combined with one or more other therapies to treat ALS.
- suitable therapies include riluzole, baclofen, diazepam, trihexyphenidyl and amitriptyline.
- the compounds and compositions described and/or disclosed herein are combined with riluzole to treat a subject suffering from ALS.
- Suitable therapies include administration of anticholinesterase agents (e.g., neostigmine, pyridostigmine), which help improve neuromuscular transmission and increase muscle strength; administration of anticholinesterase agents (e.g., neostigmine, pyridostigmine), which help improve neuromuscular transmission and increase muscle strength; administration of anticholinesterase agents (e.g., neostigmine, pyridostigmine), which help improve neuromuscular transmission and increase muscle strength; administration of
- immunosuppressive drugs e.g., prednisone, cyclosporine, azathioprine, mycophenolate mofetil
- thymectomy i.e., the surgical removal of the thymus gland, which often is abnormal in myasthenia gravis patients
- plasmapheresis i.e., the surgical removal of the thymus gland, which often is abnormal in myasthenia gravis patients
- intravenous immune globulin e.g., prednisone, cyclosporine, azathioprine, mycophenolate mofetil
- PVD vascular endothelial artery disease
- PAD a progressive neurode deficiency artery disease characterized by a wide range of diseases and conditions in which PVD and PAD are associated.
- Treatment of PVD and PAD is generally directed to increasing arterial blood flow, such as by smoking cessation, controlling blood pressure, controlling diabetes, and exercising.
- Treatment can also include medication, such as medicines to help improve walking distance (e.g., cilostazol, pentoxifylline), antiplatelet agents (e.g., aspirin, ticlopidine, clopidogrel), anticoagulents (e.g., heparin, low molecular weight heparin, warfarin, enoxaparin) throbmolytics, antihypertensive agents (e.g., diuretics, ACE inhibitors, calcium channel blockers, beta blockers, angiotensin II receptor antagonists), and cholesterol-lowering agents (e.g., statins).
- angioplasty, stenting, or surgery e.g., bypass surgery or surgery to remove an atherosclerotic plaque
- surgery e.g., bypass surgery or surgery to remove an atherosclerotic plaque
- Suitable therapeutic agents include, for example, anti-obesity agents, anti-sarcopenia agents, anti-wasting syndrome agents, anti-frailty agents, anti-cachexia agents, anti-muscle spasm agents, agents against post-surgical and post-traumatic muscle weakness, and anti-neuromuscular disease agents.
- Suitable additional therapeutic agents include, for example: orlistat, sibramine, diethylpropion, phentermine, benzaphetamine, phendimetrazine, estrogen, estradiol, levonorgestrel, norethindrone acetate, estradiol valerate, ethinyl estradiol, norgestimate, conjugated estrogens, esterified estrogens, medroxyprogesterone acetate, testosterone, insulin-derived growth factor, human growth hormone, riluzole, cannabidiol, prednisone, albuterol, nonsteroidal anti-inflammatory drugs, and botulinum toxin.
- Suitable therapeutic agents include TRH, diethylstilbesterol, theophylline, enkephalins, E series prostaglandins, compounds disclosed in U.S. Patent No. 3,239,345 (e.g., zeranol), compounds disclosed in U.S.
- Patent No. 4,036,979 e.g., sulbenox
- peptides disclosed in U.S. Patent No. 4,41 1 ,890 growth hormone secretagogues such as GHRP-6, GHRP-1 (disclosed in U.S. Patent No.
- pyridostigmine parathyroid hormone
- PTH(1 -34) parathyroid hormone
- bisphosphonates such as MK-217 (alendronate).
- Still other suitable therapeutic agents include estrogen, testosterone, selective estrogen receptor modulators, such as tamoxifen or raloxifene, other androgen receptor modulators, such as those disclosed in Edwards, J. P. et. al., Bio. Med. Chem. Let., 9, 1003-1008 (1999) and Hamann, L. G. et. al., J. Med. Chem., 42, 210-212 (1999), and progesterone receptor agonists ("PRA”), such as levonorgestrel, medroxyprogesterone acetate (MPA).
- PRA progesterone receptor agonists
- Suitable therapeutic agents include anabolic agents, such as selective androgen receptor modulators (SARMs); antagonists of the activin receptor pathway, such as anti-myostatin antibodies or soluble activin receptor decoys, including ACE-031 (Acceleron Pharmaceuticals, a soluble activin receptor type MB antagonist), MYO-027/PFE-3446879 (Wyeth/Pfizer, an antibody myostatin inhibitor), AMG-745 (Amgen, a peptibody myostatin inhibitor), and an ActRIIB decoy receptor (see Zhou et al., Cell, 142, 531-543, August 20, 2010); and anabolic steroids.
- SARMs selective androgen receptor modulators
- Still other suitable therapeutic agents include aP2 inhibitors, such as those disclosed in U.S. Patent No. 6,548,529, PPAR gamma antagonists, PPAR delta agonists, beta 3 adrenergic agonists, such as AJ9677
- sibutramine topiramate (Johnson & Johnson) or axokine (Regeneron)
- a thyroid receptor beta drug such as a thyroid receptor ligand as disclosed in WO 97/21993, WO 99/00353, and GB98/284425
- anorectic agents such as dexamphetamine, phentermine, phenylpropanolamine or mazindol.
- HIV and AIDS therapies such as indinavir sulfate, saquinavir, saquinavir mesylate, ritonavir,
- lamivudine didanosine, stavudine, and megestrol acetate.
- Still other suitable therapeutic agents include antiresorptive agents, hormone replacement therapies, vitamin D analogues, elemental calcium and calcium supplements, cathepsin K inhibitors, MMP inhibitors, vitronectin receptor antagonists, Src SH.sub.2 antagonists, vacuolar H + -ATPase inhibitors, ipriflavone, fluoride, Tibo lone, pro stanoids, 17-beta hydroxysteroid dehydrogenase inhibitors and Src kinase inhibitors.
- a daily dose ranges from about 0.05 to 100 mg/kg of body weight; in some embodiments, from about 0.10 to 10.0 mg/kg of body weight, and in some embodiments, from about 0.15 to 1 .0 mg/kg of body weight.
- the dosage range would be about from 3.5 to 7000 mg per day; in some embodiments, about from 7.0 to 700.0 mg per day, and in some embodiments, about from 10.0 to 100.0 mg per day.
- an exemplary dosage range for oral administration is from about 70 mg to about 700 mg per day
- an exemplary intravenous administration dosage is from about 70 mg to about 700 mg per day, each depending upon the compound
- Administration of the compounds and compositions disclosed and/or described herein can be via any accepted mode of administration for therapeutic agents including, but not limited to, oral, sublingual,
- the compound or composition is administered orally or intravenously.
- the compound or composition disclosed and/or described herein is administered orally.
- compositions include solid, semi-solid, liquid and aerosol dosage forms, such as tablet, capsule, powder, liquid, suspension, suppository, and aerosol forms.
- the compounds disclosed and/or described herein can also be administered in sustained or controlled release dosage forms (e.g., controlled/sustained release pill, depot injection, osmotic pump, or transdermal (including electrotransport) patch forms) for prolonged timed, and/or pulsed administration at a predetermined rate.
- sustained or controlled release dosage forms e.g., controlled/sustained release pill, depot injection, osmotic pump, or transdermal (including electrotransport) patch forms
- the compositions are provided in unit dosage forms suitable for single administration of a precise dose.
- mannitol e.g., mannitol, lactose, starch, magnesium stearate, sodium saccharine, talcum, cellulose, sodium
- the pharmaceutical composition can also contain minor amounts of nontoxic auxiliary substances such as wetting agents, emulsifying agents, solubilizing agents, pH buffering agents and the like (e.g., sodium acetate, sodium citrate, cyclodextrine derivatives, sorbitan monolaurate,
- the pharmaceutical composition will contain about 0.005% to 95%, or about 0.5% to 50%, by weight of a compound disclosed and/or described herein.
- Actual methods of preparing such dosage forms are known, or will be apparent, to those skilled in this art; for example, see Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pennsylvania.
- the compositions will take the form of a pill or tablet and thus the composition may contain, along with a compounds disclosed and/or described herein, one or more of a diluent (e.g., lactose, sucrose, dicalcium phosphate), a lubricant (e.g., magnesium stearate), and/or a binder (e.g., starch, gum acacia, polyvinylpyrrolidine, gelatin, cellulose, cellulose derivatives).
- a diluent e.g., lactose, sucrose, dicalcium phosphate
- a lubricant e.g., magnesium stearate
- a binder e.g., starch, gum acacia, polyvinylpyrrolidine, gelatin, cellulose, cellulose derivatives.
- Other solid dosage forms include a powder, marume, solution or suspension (e.g., in propylene carbonate, vegetable oils or triglycerides)
- Liquid pharmaceutically administrable compositions can, for example, be prepared by dissolving, dispersing or suspending etc. a compound disclosed and/or described herein and optional pharmaceutical additives in a carrier (e.g., water, saline, aqueous dextrose, glycerol, glycols, ethanol or the like) to form a solution or suspension.
- a carrier e.g., water, saline, aqueous dextrose, glycerol, glycols, ethanol or the like
- injectables can be prepared in conventional forms, either as liquid solutions or suspensions, as emulsions, or in solid forms suitable for dissolution or suspension in liquid prior to injection.
- the percentage of the compound contained in such parenteral compositions depends, for example, on the physical nature of the compound, the activity of the compound and the needs of the subject.
- composition will comprise from about 0.2 to 2% of a compound disclosed and/or described herein in solution.
- compositions of the compounds disclosed and/or described herein may also be administered to the respiratory tract as an aerosol or solution for a nebulizer, or as a microfine powder for insufflation, alone or in combination with an inert carrier such as lactose.
- the particles of the pharmaceutical composition may have diameters of less than 50 microns, or in some embodiments, less than 10 microns.
- compositions can include a compound disclosed and/or described herein and one or more additional medicinal agents, pharmaceutical agents, adjuvants, and the like.
- additional medicinal agents include those described herein.
- 1 -(3-Fluoropyridin-2-yl)cyclobutanecarboxamide To a 250 mL round bottom flask containing DMSO (60 mL), 1 -(3-fluoropyridin-2- yl)cyclobutanecarbonitrile (2.96 g, 16.8 mmol, 1 .0 equiv) was added and the mixture was stirred until homogenous. Potassium carbonate (7.0 g, 50.4 mmol, 3.0 equiv) was then added and the reaction mixture was cooled to 0 °C, followed by the addition of 35% hydrogen peroxide (6.5 mL). The reaction was stirred at 0 °C for 30 min and then warmed to room temperature.
- reaction was diluted with water (50 mL) and ethyl acetate (100 mL). After transferring to a separatory funnel and shaking, the organic layer was separated from the aqueous layer and then washed with brine (3 x 50 mL). The organic layer was then dried over Na 2 SO , filtered, and concentrated to give a crude solid that was purified by silica gel chromatography (10% EtOAC/hexanes) to afford 1 .92 g (59%) of 1 -(3-fluoropyridin-2- yl)cyclobutanecarboxamide as a white solid.
- 1 -(3- fluoropyridin-2-yl)cyclobutanecarboxannide (1 .92 g, 9.88 mmol, 1 .0 equiv) was dissolved in methanol (20 ml_) and potassium hydroxide (1 .1 1 g, 19.8 mmol, 2.0 equiv) was added. The mixture was sonicated until homogeneous, followed by the addition of iodosobenzene diacetate (4.77 g, 14.8 mmol, 1 .5 equiv).
- N-(2-(4-Fluorophenyl)-2-methylpropyl)-6-(2-methylprop-1 - enyl)pyridin-2-amine was added to a 5 mL microwave reaction vessel. To a 5 mL microwave reaction vessel was added 6- bromo-N-(2-(4-fluorophenyl)-2-methylpropyl)pyridin-2-amine (220 mg, 0.7 mmol, 1 .0 equiv), 2-methylprop-1 -enylboronic acid (100 mg, 1 mmol, 1 .5 equiv), CI 2 Pd(dppf) (50 mg, 70 ⁇ , 0.1 equiv), potassium carbonate (300 mg, 2.1 mmol, 3.0 equiv), dioxane (4 mL), and water (1 mL).
- the reaction was heated in a microwave reactor at 135 °C for 35 min and then diluted with saturated sodium bicarbonate (20 mL) and extracted with ethyl acetate (50 mL). The organic layer was then dried over Na 2 SO , filtered, and
- the reaction was heated in a microwave reactor at 140 °C for 20 min and then diluted with saturated sodium bicarbonate (20 ml_) and extracted with ethyl acetate (50 ml_). The organic layer was then dried over Na2SO 4 , filtered, and
- 5-Bromo-N-(1 -phenylcyclobutyl)pyridin-2-amine To a 20 dram vial was added 5-bromo-2-fluoropyridine (250 mg, 5.0 mmol, 1 .0 equiv), 1 - phenylcyclobutanamine (1 .0 g, 7 mmol, 1 .4 equiv), and NMP (1 mL). The reaction was heated to 120 °C, stirred for 72 h, and then diluted with water (20 mL) and extracted with ethyl acetate (50 mL).
- 5-Phenyl-N-(1 -phenylcyclobutyl)pyridin-2-amine To a 3 mL microwave reaction vessel was added 5-bromo-N-(1 -phenylcyclobutyl)pyridin- 2-amine (102 mg, 340 ⁇ , 1 .0 equiv), phenylboronic acid (59 mg, 505 ⁇ , 1 .5 equiv), Cl2Pd(dppf) (25 mg, 34 ⁇ , 0.1 equiv), potassium carbonate (1 16 mg, 842 ⁇ , 2.5 equiv), dioxane (1 mL) and water (500 ⁇ ).
- N-(2-(4-Fluorophenyl)-2-methylpropyl)-5-vinylpyridin-2-amine was added to a 5 mL microwave reaction vessel. To a 5 mL microwave reaction vessel was added 5-bromo-N-(2-(4-fluorophenyl)- 2-methylpropyl)pyrimidin-2-amine (255 mg, 0.8 mmol, 1 .0 equiv), 2,4,6- trivinyl-1 ,3,5,2,4,6-trioxatriborinane (285 mg, 1 .2 mmol, 1 .5 equiv),
- Osmium tetraoxide (15 mg, 0.06 mmol, 0.1 equiv) and morpholine N-oxide (103 mg, 0.9 mmol, 1 .5 equiv) were added, and the reaction was stirred for 1 h. Additional THF (3 mL) was added, and the reaction was stirred for 3 h. The reaction mixture was then diluted with water (10 mL) and extracted with ethyl acetate (30 mL).
- ferf-Butyl 2-(4-fluorophenyl)-2-methylpropyl(5-(2- hydroxyethyl)pyridin-2-yl)carbamate To a 20 dram vial was added tert- butyl 2-(4-fluorophenyl)-2-methylpropyl(5-vinylpyridin-2-yl)carbamate (2.4 g, 6.4 mmol, 1 .0 equiv) and THF (20 mL). Borane (7.7 mL, 7.7 mmol, 4.0 equiv) was added slowly, and the reaction was heated to 35 °C and stirred for 45 min.
- the resultant solid was dissolved in DMF (4 mL), followed by the addition of sodium azide (139 mg, 2.1 mmol, 2.0 equiv). The reaction was stirred for 24 h, then diluted with water (10 mL) and extracted with ethyl acetate (20 mL). The organic layer was dried over Na 2 SO 4 , filtered, and concentrated. The crude solid was dissolved in methanol, followed by the addition of 10% palladium on carbon (80 mg), potassium carbonate (100 mg).
- 6-(2-(4-Fluorophenyl)-2-methylpropylamino)nicotinonitrile To a solution of 6-chloronicotinonitrile (200 mg, 1 .46 mmol, 1 .0 equiv) and 2-(4- fluorophenyl)-2-methylpropan-1 -amine (290 mg, 1 .75 mmol, 1 .2 equiv) in isopropanol (3 mL) in a microwave vial equipped with a stir bar was added potassium carbonate (720 mg, 2.19 mmol, 1 .5 equiv). The vial was fitted with a microwave vial cap and heated to 120 °C for 20 min.
- 6-(2-(4-Fluorophenyl)-2-methylpropylamino)nicotinamide To a cooled (0 °C) suspension of 6-(2-(4-fluorophenyl)-2- methylpropylamino)nicotinonitrile (75 mg, 0.28 mmol, 1 .0 equiv) and K2CO3 (50 mg, 0.36 mmol, 1 .3 equiv) in DMSO (1 mL) was added aq. H 2 O 2 (250 uL, 30% by wt., 2.6 mmol, 9 equiv) by syringe. The reaction was allowed to warm to RT and stirred for 30 minutes.
- reaction mixture was then quenched by the sequential slow addition water (0.6 mL), 3N NaOH (0.6 mL), and water (1 .7 mL).
- the resultant precipitate was filtered and the filtrate was then dried (Na 2 SO 4 ) and concentrated to give 1 .0 g (99%) of 5-(aminomethyl)-N-(2-(4-fluorophenyl)-2- methylpropyl)pyridin-2-amine.
- Methyl (6-(2-(4-fluorophenyl)-2-methylpropylamino)pyridin-3- yl)methylcarbamate.
- 5-(aminomethyl)-N-(2-(4- fluorophenyl)-2-methylpropyl)pyridin-2-amine 100 mg, 0.37 mmol, 1 .0 equiv
- DIPEA 57 mg, 0.44 mmol, 1 .2 equiv
- CH 2 CI 2 (1 .2 mL).
- the mixture was cooled to 0 °C, and methyl carbamate (35 mg, 0.4 mmol, 1 .0 equiv) was then added.
- Example 14 Preparation of 5-(3-(2-(4-fluorophenyl)-2- methylpropylamino)-1, -6-yl)-1 H-indazol-3-amine 3-Amino-1 ,2,4-triazin-2-oxide.
- 1 ,2,4-triazin-3-annine 2.8 g, 30 mmol
- CH 3 CN 25 ml_
- mCPBA 14 g, 81 mmol
- N-(2-(4-Fluorophenyl)-2-methylpropyl)-1 ,2,4-triazin-3-amine-2- oxide A solution of 3-bromo-1 ,2,4-triazin-2-oxide (2.0 g, 1 1 mmol), (2-(4- fluorophenyl)-2-methylpropan-1 -amine (3.0 g, 17 mmol), and
- 6-(5-Bromo-2-fluorophenyl)-1 ,2,4-triazin-3-amine 6-Bromo-1 ,2,4- triazin-3-annine (9.1 g, 40.8 mmol), 5-bromo-2-fluorophenylboronic acid (98.9 g, 40.8 mmol), (dppf)PdCl 2 (3.0 g, 4.0 mmol), nitrogen-sparged dioxane (81 .6 mL), and aq. 2 N K 2 CO 3 (17 mL) were combined and heated in a round bottom flask at 90 °C for 4 h.
- 6-(5-Bromo-2- fluorophenyl)-1 ,2,4-triazin-3-amine (4.5 g, 16.7 mmol) was dissolved in 25 mL of bromoform and heated to 80 °C. Isoamyl nitrite (9.8 g, 83.5 mmol) was then added and stirred at 85 °C for 1 h. The reaction mixture was then
- Example 16 Preparation of (1 -(6-Methoxypyridin-2-yl)cyclobutyl) methanamine 1 -(6-Fluoropyridin-2-yl)cyclobutanecarbonitrile.
- 1,3-(6-Methoxypyridin-2-yl)cyclobutyl) methanamine 1 -(6-Fluoropyridin-2-yl)cyclobutanecarbonitrile.
- NaHMDS sodium hexamethyldisilazide
- reaction was diluted with water (50 mL) and ethyl acetate (100 mL). After transferring to a separatory funnel and shaking, the organic layer was separated from the aqueous layer and then washed with brine (3 x 50 mL). The organic layer was then dried over Na 2 SO , filtered, and concentrated to give a crude solid that was purified by silica gel chromatography (10% EtOAC/hexanes) to afford 1 .92 g of 1 -(3- fluoropyridin-2- l c clobutanecarboxamide as a white solid.
- 1 -(3- fluoropyridin-2-yl)cyclobutanecarboxamide (1 .92 g, 9.88 mmol) was dissolved in methanol (20 mL) and potassium hydroxide (1 .1 1 g, 19.8 mmol, 2.0 equiv) was added. The mixture was sonicated until homogeneous, followed by the addition of iodosobenzene diacetate (4.77 g, 14.8 mmol). The reaction was stirred for 20 min and then diluted with water (100 mL) and ethyl acetate (125 mL).
- Fluoro-3-(3-(((irans)-3-fluoro-1 -(3-fluoropyridin-2-yl)cyclobutyl)methylamino)- 1 ,2,4-triazin-6-yl)benzoic acid (0.5 g, 1 .2 mmol) was diluted in 18 mL DMF and treated with HBTU (1 .8 g, 4.8 mmol), HOBT (0.65 g, 4.8 mmol), methyl amine hydrochloride (0.32 g, 4.8 mmol), and DIEA (8.4 mL, 48 mmol).
- Example 21 Preparation of 4-fluoro-3-(3-(((frans)-3-fluoro-1 -(3- fluoropyridin-2-yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)-2- hydroxybenzamide
- Lithium iodide (1 g), pyridine (20 mL), and 4-fluoro-3-(3-(((frans)-3-fluoro-1 - (3-fluoropyridin-2-yl)cyclobutyl)nnethylannino)-1 ,2,4-triazin-6-yl)-2- methoxybenzamide (210 mg) were added to a round bottom flask. The reaction was heated to 125 °C and stirred for 2 h.
- reaction was stirred at 75 °C for 3 h, followed by cooling to rt, quenching with brine (200 mL). The reaction was extracted four times with CH 2 CI 2 . The combined organic layers were dried over Na 2 SO , concentrated, and purified using silica gel chromatography to afford the 1 .1 g of the title compound as an off-white solid.
- 6-(5-Bromothiazol-2-yl)-1 ,2,4-triazin-3-amine To a vial was added 6-(thiazol-2-yl)-1 ,2,4-triazin-3-amine (600 mg, 3.4 mmol), methanol (1 .2 mL), water (0.6 mL). The mixture was cooled to 0 °C and bromine (816 mg, 5.1 mmol) was added. The reaction was stirred for 45 min and then quenched with saturated sodium bicarbonate solution (20 mL) and extracted three times with ethyl acetate (50 mL).
- 2-(3-Bromo-1,2,4-triazin-6-yl)thiazole-5-carbonitrile 2-(3-Amino- 1 ,2,4-triazin-6-yl)thiazole-5-carbonitrile (30 mg, 0.15 mmol) was dissolved in 1 mL of bromoform and heated to 90 °C. Isoamyl nitrite (88 mg, 0.75 mmol) was added and stirred at 90 °C for 2 h. The reaction mixture was then con ntrated and used directly in the next reaction.
- reaction was then directly purified using silica gel chromatography to afford 2.5 g of 5-bromo-3-fluoro-N-(((frans)-3-fluoro-1 -(3-fluoropyridin-2- l)cyclobutyl)methyl)pyridin-2-amine as a white solid.
- Myofibrils were prepared from rabbit psoas muscle purchased from Pel-Freez Biologicals (Arkansas) within 2 days of ordering, stored on ice. Minced muscle was homogenized in 10 volumes of ice-cold "standard" buffer (50mM Tris, pH 7.4, 0.1 M potassium acetate, 5 mM KCI, 2 mM DTT, 0.2 mM PMSF, 10 ⁇ leupeptin, 5 ⁇ pepstatin, and 0.5 mM sodium azide) containing 5 mM EDTA and 0.5% Triton X-100 using an Omni- Macro homogenizer.
- standard buffer 50mM Tris, pH 7.4, 0.1 M potassium acetate, 5 mM KCI, 2 mM DTT, 0.2 mM PMSF, 10 ⁇ leupeptin, 5 ⁇ pepstatin, and 0.5 mM sodium azide
- Myofibrils were recovered by low speed centrifugation (3000 rpm for 10 minutes) and washed 2 times in the Triton X-100 containing buffer to ensure removal of cellular membrane. Following the Triton washes, myofibrils were washed 3 times in "standard" buffer containing 2 mM magnesium acetate. A final wash in assay buffer (12 mM PIPES, pH 6.8, 60 mM KCI, 1 mM DTT) was performed and brought to 10% sucrose for flash freezing in liquid nitrogen and storage at -80°C.
- Fast fiber activators were identified by measuring the enzymatic activity of muscle myofibril preparations
- Drying Place the filtered residue spread on a cheesecloth in a large glass tray and leave in a hood overnight. When the residue is dry, put in a wide mouth plastic bottle and store at 20 °C.
- Extract buffer see below. Homogenize the meat in a blender, 4 times 15 sec on blend with 15 sees in between. Do this with 1 volume (weight/volume) of buffer taken from the 5 volumes already prepared. Add the homogenate back to the extract buffer and stir until well mixed (5 minutes).
- Step 3 Repeat Step 3 four more times. At the end, do not resuspend in extraction buffer but proceed to Step 5.
- the pellets should be yellow white.
- Step 6 a total of three times.
- EXTRACT BUFFER 50 mM KCI, 5 mM Tris pH 8.0 Prepare as 50 times concentrate. For 2L: 250 mM Tris pH 8.0. Tris Base (121 .14 g/mol, 60.6 g), pH to 8.0 with cone. HCI, then add 2.5 M KCI (74.55 g/mol, 372 g).
- Buffer A 2 mM tris/HCI, 0.2 mM CaCI 2 , 0.5 mM (36 ⁇ /L) 2-mercaptoethanol,
- Solution A 0.3 M KCI, 0.15 M potassium phosphate, 0.02 M EDTA, 0.005 M MgCI 2 , 0.001 M ATP, pH 6.5.
- Solution B 1 M KCI, 0.025 M EDTA, 0.06 M potassium phosphate, pH 6.5.
- Solution C 0.6 M KCI, 0.025 M potassium phosphate, pH 6.5.
- Solution D 0.6 M KCI, 0.05 M potassium phosphate, pH 6.5.
- Solution E 0.15 M potassium phosphate, 0.01 M EDTA, pH 7.5.
- Solution F 0.04 M KCI, 0.01 M potassium phosphate, 0.001 M DTT, pH 6.5.
- Solution G 3 M KCI, 0.01 M potassium phosphate, pH 6.5.
- the myosin is then cut with chymotrypsin or papain in the presence of EDTA to generate the S1 fragment which is soluble at the low salt conditions optimal for ATPase activity (Margossian, supra).
- Myosin is prepared by precipitation from salt extracts of rabbit psoas muscle, and a soluble S1 fraction is prepared by digestion with chymotrypsin (Margossian and Lowey, 1982).
- Actin is purified by first preparing an ether powder of cardiac muscle (Zot HG and Potter J D. (1981 ) Preparative Biochemistry 1 1 :381 -395) as described above. Subsequently, actin is cycled between the filamentous and soluble state through rounds of centrifugation and dialysis (Spudich J A and Watt S. (1971 ) J. Biol. Chem. 246:4866-4871 ).
- Tropomyosin is extracted from the ether powder and separated from the other proteins based on pH dependent precipitations followed by successive ammonium sulfate cuts at 53% and 65% (Smillie LB. (1981 ) Methods Enzymol 85 Pt B:234-41 ).
- the troponins are isolated as an intact complex of TnC, TnT, and Tnl.
- Ether powder is extracted in a high salt buffer. Successive ammonium sulfate cuts of 30% and 45% are done; the precipitate is solubilized by dialysis into a low salt buffer and then further purified on a DEAE Toyopearl column with a 25-350 mM KCI gradient. There is no measurable ATPase in any of the components except for myosin which naturally had a very low basal ATPase in the absence of actin.
- the actin, tropomyosin, and troponin complex Prior to screening, the actin, tropomyosin, and troponin complex are mixed together in the desired ratio (e.g., 7:1 :1 ) to achieve maximal calcium regulation of the actin filament.
- the screen is conducted at a concentration that gives 25% activation. This calcium concentration is in the physiological range during muscle contraction.
- a pyruvate kinase/lactate dehydrogenase/NADH coupled enzyme system (PK/LDH) is added to the actin.
- the myosin is kept separately, and added to the regulated thin filaments to initiate the reaction. Oxidation of NADH is monitored in real time, so that kinetic curves are obtained.
- Compounds are dissolved in DMSO and spotted onto the bottoms of 384 well plates at 10 to 40 g/ml final concentration.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Provided are compounds of Formula I: Formula I or a pharmaceutically acceptable salt thereof, wherein R2, R4, R5, R6, R7, R8, R9, X, Z1, Z2, Z3, Z4 and m are as defined herein. Also provided is a pharmaceutically acceptable composition comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof. Also provided are methods of using a compound of Formula I, or a pharmaceutically acceptable salt thereof.
Description
Certain Amino-Pyridines and Amino-Triazines, Compositions Thereof, and
Methods for Their Use
This application claims the benefit of U.S. Provisional Application Nos. 61/327,538, filed April 23, 2010, 61/327,597, filed April 23, 2010, 61/412,299, filed November 10, 2010, and 61/412,302, filed November 10, 2010, each of which is incorporated by reference in its entirety for all purposes.
The cytoskeleton of skeletal and cardiac muscle cells is unique compared to that of all other cells. It consists of a nearly crystalline array of closely packed cytoskeletal proteins called the sarcomere. The sarcomere is elegantly organized as an interdigitating array of thin and thick filaments. The thick filaments are composed of myosin, the motor protein responsible for transducing the chemical energy of ATP hydrolysis into force and directed movement. The thin filaments are composed of actin monomers arranged in a helical array. There are four regulatory proteins bound to the actin filaments, which allows the contraction to be modulated by calcium ions. An influx of intracellular calcium initiates muscle contraction; thick and thin filaments slide past each other driven by repetitive interactions of the myosin motor domains with the thin actin filaments.
Of the thirteen distinct classes of myosin in human cells, the myosin-ll class is responsible for contraction of skeletal, cardiac, and smooth muscle. This class of myosin is significantly different in amino acid composition and in overall structure from myosin in the other twelve distinct classes. Myosin-ll forms homo-dimers resulting in two globular head domains linked together by a long alpha-helical coiled-coiled tail to form the core of the sarcomere's thick filament. The globular heads have a catalytic domain where the actin binding and ATPase functions of myosin take place. Once bound to an actin filament, the release of phosphate (cf. ADP-Pi to ADP) signals a change in structural conformation of the catalytic domain that in turn alters the orientation of the light-chain binding lever arm domain that extends from the globular head; this movement is termed the powerstroke. This change in orientation of the myosin head in relationship to actin causes the thick filament of which it is a part to move with respect to the thin actin filament to which it is bound. Un-
binding of the globular head from the actin filament (Ca regulated) coupled with return of the catalytic domain and light chain to their starting
conformation/orientation completes the catalytic cycle, responsible for intracellular movement and muscle contraction.
Tropomyosin and troponin mediate the calcium effect on the interaction on actin and myosin. The troponin complex is comprised of three polypeptide chains: troponin C, which binds calcium ions; troponin I, which binds to actin; and troponin T, which binds to tropomyosin. The skeletal troponin- tropomyosin complex regulates the myosin binding sites extending over several actin units at once.
Troponin, a complex of the three polypeptides described above, is an accessory protein that is closely associated with actin filaments in vertebrate muscle. The troponin complex acts in conjunction with the muscle form of tropomyosin to mediate the Ca2+ dependency of myosin ATPase activity and thereby regulate muscle contraction. The troponin polypeptides T, I, and C, are named for their tropomyosin binding, inhibitory, and calcium binding activities, respectively. Troponin T binds to tropomyosin and is believed to be responsible for positioning the troponin complex on the muscle thin filament. Troponin I binds to actin, and the complex formed by troponins I and T, and tropomyosin inhibits the interaction of actin and myosin. Skeletal troponin C is capable of binding up to four calcium molecules. Studies suggest that when the level of calcium in the muscle is raised, troponin C exposes a binding site for troponin I, recruiting it away from actin. This causes the tropomyosin molecule to shift its position as well, thereby exposing the myosin binding sites on actin and stimulating myosin ATPase activity.
Human skeletal muscle is composed of different types of contractile fibers, classified by their myosin type and termed either slow or fast fibers. Table 1 summarizes the different proteins that make up these types of muscle.
Table 1

*MHC Mb is not expressed in human muscle but is present in rodents and other
mammals.
In healthy humans most skeletal muscles are composed of both fast and slow fibers, although the proportions of each vary with muscle type. Slow skeletal fibers, often called type I fibers, have more structural similarity with cardiac muscle and tend to be used more for fine and postural control. They usually have a greater oxidative capacity and are more resistant to fatigue with continued use. Fast skeletal muscle fibers, often called type II fibers, are classified into fast oxidative (I la) and fast glycolytic (type llx/d) fibers. While these muscle fibers have different myosin types, they share many
components including the troponin and tropomyosin regulatory proteins. Fast skeletal muscle fibers tend to exert greater force but fatigue faster than slow skeletal muscle fibers and are functionally useful for acute, large scale movements such as rising from a chair or correcting falls.
Muscle contraction and force generation is controlled through nervous stimulation by innervating motor neurons. Each motor neuron may innervate many (approximately 100-380) muscle fibers as a contractile whole, termed a motor unit. When a muscle is required to contract, motor neurons send
stimuli as nerve impulses (action potentials) from the brain stem or spinal cord to each fiber within the motor unit. The contact region between nerve and muscle fibers is a specialized synapse called the neuromuscular junction (NMJ). Here, membrane depolarizing action potentials in the nerve are translated into an impulse in the muscle fiber through release of the
neurotransmitter acetylcholine (ACh). ACh triggers a second action potential in the muscle that spreads rapidly along the fiber and into invaginations in the membrane, termed t-tubules. T-tubules are physically connected to Ca2+ stores within the sarcoplasmic reticulum (SR) of muscle via the
dihydropyridine receptor (DHPR). Stimulation of the DHPR activates a second Ca2+ channel in the SR, the ryanodine receptor, to trigger the release of Ca2+ from stores in the SR to the muscle cytoplasm where it can interact with the troponin complex to initiate muscle contraction. If muscle stimulation stops, calcium is rapidly taken back up into the SR through the ATP
dependent Ca2+ pump, SERCA.
Muscle function can become compromised in disease by many mechanisms. Examples include the frailty associated with old age (termed sarcopenia) and cachexia syndromes associated with diseases such as cancer, heart failure, chronic obstructive pulmonary disease (COPD), and chronic kidney disease/dialysis. Severe muscular dysfunction can arise from neuromuscular diseases (such as Amyotrophic Lateral Sclerosis (ALS), spinal muscular atrophy (SMA) and myasthenia gravis) or muscular myopathies (such as muscular dystrophies). Additionally, muscle function may become compromised due to rehabilitation-related deficits, such as those associated with recovery from surgery (e.g. post-surgical muscle weakness), prolonged bed rest, or stroke rehabilitation. Additional examples of diseases or conditions where muscle function becomes compromised include peripheral vascular disease (e.g., claudication), chronic fatigue syndrome, metabolic syndrome, and obesity.
Accordingly, there is a need for the development of new compounds that modulate skeletal muscle contractility. There remains a need for agents that exploit new mechanisms of action and which may have better outcomes in terms of relief of symptoms, safety, and patient mortality, both short-term and long-term and an improved therapeutic index.
Provided is a compound of Formula I:

Formula I
or a pharmaceutically acceptable salt thereof, wherein R2, R4, R5, R6, R7, R8, R9, X, Z1, Z2, Z3, Z4 and m are as defined herein.
Also provided is a pharmaceutically acceptable composition comprising a compound of Formula I, or a pharmaceutically acceptable salt thereof.
Also provided are methods for treating a disease or condition responsive to modulation of the contractility of the skeletal sarcomere, for example, modulation of the troponin complex of the fast skeletal muscle sarcomere through one or more of fast skeletal myosin, actin, tropomyosin, troponin C, troponin I, and troponin T, and fragments and isoforms thereof.
As used in the present specification, the following words and phrases are generally intended to have the meanings as set forth below, except to the extent that the context in which they are used indicates otherwise.
Throughout this application, unless the context indicates otherwise, references to a compound of Formula I includes all subgroups of Formula I defined herein, including all substructures, subgenera, preferences, embodiments, examples and particular compounds defined and/or described herein.
References to a compound of Formula I and subgroups thereof include ionic forms, polymorphs, pseudopolymorphs, amorphous forms, solvates, co- crystals, chelates, isomers, tautomers, oxides (e.g., N-oxides, S-oxides), esters, prodrugs, isotopes and/or protected forms thereof. "Crystalline form," "polymorph," and "novel form" may be used interchangeably herein, and are meant to include all crystalline and amorphous forms of the compound, including, for example, polymorphs, pseudopolymorphs, solvates (including hydrates), co-crystals, unsolvated polymorphs (including anhydrates), conformational polymorphs, and amorphous forms, as well as mixtures
thereof, unless a particular crystalline or amorphous form is referred to. In some embodiments, references to a compound of Formula I and subgroups thereof include polymorphs, solvates, co-crystals, isomers, tautomers and/or oxides thereof. In some embodiments, references to a compound of Formula I and subgroups thereof include polymorphs, solvates, and/or co-crystals thereof. In some embodiments, references to a compound of Formula I and subgroups thereof include isomers, tautomers and/or oxides thereof. In some embodiments, references to a compound of Formula I and subgroups thereof include solvates thereof. Similarly, the term "salts" includes solvates of salts of compounds.
By "optional" or "optionally" is meant that the subsequently described event or circumstance may or may not occur, and that the description includes instances where the event or circumstance occurs and instances in which it does not. For example, "optionally substituted alkyl" encompasses both "alkyl" and "substituted alkyl" as defined herein. It will be understood by those skilled in the art, with respect to any group containing one or more
substituents, that such groups are not intended to introduce any substitution or substitution patterns that are sterically impractical, synthetically non- feasible, and/or inherently unstable.
When a range of values is given (e.g., C-i-6 alkyl), each value within the range as well as all intervening ranges are included. For example, "C-i-6 alkyl" includes C-i , C2, C3, C4, C5, CQ, C-i-6, C2-6, C3-6, C4-6, Cs-6, C-i-5, C2-5, C3-5, C4-5,
Ci- , C2-4, C3-4, C-i-3, C2-3, and Ci-2 alkyl.
When a moiety is defined as being optionally substituted, it may be substituted as itself or as part of another moiety. For example, if Rx is defined as "C-i-6 alkyl or OC-i-6 alkyl, wherein C-i-6 alkyl is optionally subsituted with halogen", then both the C-i-6 alkyl group alone and the C-i-6 alkyl that makes up part of the OC-i-6 alkyl group may be substituted with halogen.
"Alkyl" encompasses straight and branched carbon chains having the indicated number of carbon atoms, for example, from 1 to 20 carbon atoms, or 1 to 8 carbon atoms, or 1 to 6 carbon atoms. For example, C-i-6 alkyl encompasses both straight and branched chain alkyl of from 1 to 6 carbon atoms. When an alkyl residue having a specific number of carbons is named, all branched and straight chain versions having that number of carbons are
intended to be encompassed; thus, for example, "propyl" includes n-propyl and isopropyl; and "butyl" includes n-butyl, sec-butyl, isobutyl and t-butyl. Examples of alkyl groups include, but are not limited to, methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, pentyl, 2-pentyl, 3-pentyl, isopentyl, neopentyl, hexyl, 2-hexyl, 3-hexyl, and 3-methylpentyl. "Lower alkyl" refers to alkyl groups having 1 to 6 carbons.
"Haloalkyi" includes straight and branched carbon chains having the indicated number of carbon atoms (e.g., 1 to 6 carbon atoms) substituted with at least one halogen atom. In instances wherein the haloalkyi group contains more than one halogen atom, the halogens may be the same (e.g.,
dichloromethyl) or different (e.g., chlorofluoromethyl). Examples of haloalkyi groups include, but are not limited to, chloromethyl, dichloromethyl,
trichloromethyl, fluoromethyl, difluoromethyl, trifluoromethyl,
chlorofluoromethyl, 2-fluoroethyl, 2,2-difluoroethyl, 2,2,2-trifluoroethyl,
1 ,2-difluoroethyl, 2-chloroethyl, 2,2-dichloroethyl, 2,2,2-trichloroethyl,
1 ,2-dichloroethyl, pentachloroethyl, and pentafluoroethyl.
"Alkenyl" refers to an unsaturated branched or straight-chain alkyl group having the indicated number of carbon atoms (e.g., 2 to 8, or 2 to 6 carbon atoms) and at least one carbon-carbon double bond derived by the removal of one molecule of hydrogen from adjacent carbon atoms of the corresponding alkyl. The group may be in either the cis or trans configuration (Z or E configuration) about the double bond(s). Alkenyl groups include, but are not limited to, ethenyl, propenyl (e.g., prop-1 -en-1 -yl, prop-1 -en-2-yl, prop- 2-en-1 -yl (allyl), prop-2-en-2-yl), and butenyl (e.g., but-1 -en-1 -yl, but-1 -en-2-yl, 2-methyl-prop-1 -en-1 -yl, but-2-en-1 -yl, but-2-en-1 -yl, but-2-en-2-yl, buta-1 ,3- dien-1 -yl, buta-1 ,3-dien-2-yl). "Lower alkenyl" refers to alkenyl groups having 2 to 6 carbons.
"Alkynyl" refers to an unsaturated branched or straight-chain alkyl group having the indicated number of carbon atoms (e.g., 2 to 8 or 2 to 6 carbon atoms) and at least one carbon-carbon triple bond derived by the removal of two molecules of hydrogen from adjacent carbon atoms of the corresponding alkyl. Alkynyl groups include, but are not limited to, ethynyl, propynyl (e.g., prop-1 -yn-1 -yl, prop-2-yn-1 -yl) and butynyl (e.g., but-1 -yn-1 -yl, but-1 -yn-3-yl, but-3-yn-1 -yl). "Lower alkynyl" refers to alkynyl groups having 2
to 6 carbons.
"Cycloalkyl" indicates a non-aromatic, fully saturated carbocyclic ring having the indicated number of carbon atoms, for example, 3 to 10, or 3 to 8, or 3 to 6 ring carbon atoms. Cycloalkyl groups may be monocyclic or polycyclic (e.g., bicyclic, tricyclic). Examples of cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl and cyclohexyl, as well as bridged and caged ring groups (e.g., norbornane, bicyclo[2.2.2]octane). In addition, one ring of a polycyclic cycloalkyl group may be aromatic, provided the polycyclic cycloalkyl group is bound to the parent structure via a non- aromatic carbon. For example, a 1 ,2,3,4-tetrahydronaphthalen-1 -yl group (wherein the moiety is bound to the parent structure via a non-aromatic carbon atom) is a cycloalkyl group, while 1 ,2,3,4-tetrahydronaphthalen-5-yl (wherein the moiety is bound to the parent structure via an aromatic carbon atom) is not considered a cycloalkyl group. Examples of polycyclic cycloalkyl groups consisting of a cycloalkyl group fused to an aromatic ring are described below.
"Cycloalkenyl" indicates a non-aromatic carbocyclic ring, containing the indicated number of carbon atoms (e.g., 3 to 10, or 3 to 8, or 3 to 6 ring carbon atoms) and at least one carbon-carbon double bond derived by the removal of one molecule of hydrogen from adjacent carbon atoms of the corresponding cycloalkyl. Cycloalkenyl groups may be monocyclic or polycyclic (e.g., bicyclic, tricyclic). Examples of cycloalkenyl groups include cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclopentadienyl, and
cyclohexenyl, as well as bridged and caged ring groups (e.g.,
bicyclo[2.2.2]octene). In addition, one ring of a polycyclic cycloalkenyl group may be aromatic, provided the polycyclic alkenyl group is bound to the parent structure via a non-aromatic carbon atom. For example, inden-1 -yl (wherein the moiety is bound to the parent structure via a non-aromatic carbon atom) is considered a cycloalkenyl group, while inden-4-yl (wherein the moiety is bound to the parent structure via an aromatic carbon atom) is not considered a cycloalkenyl group. Examples of polycyclic cycloalkenyl groups consisting of a cycloalkenyl group fused to an aromatic ring are described below.
"Aryl" indicates an aromatic carbon ring having the indicated number of carbon atoms, for example, 6 to 12 or 6 to 10 carbon atoms. Aryl groups may
be monocyclic or polycyclic (e.g., bicyclic, tricyclic). In some instances, both rings of a polycyclic aryl group are aromatic (e.g., naphthyl). In other instances, polycyclic aryl groups may include a non-aromatic ring (e.g., cycloalkyl, cycloalkenyl, heterocycloalkyl, heterocycloalkenyl) fused to an aromatic ring, provided the polycyclic aryl group is bound to the parent structure via an atom in the aromatic ring. Thus, a 1 ,2,3,4- tetrahydronaphthalen-5-yl group (wherein the moiety is bound to the parent structure via an aromatic carbon atom) is considered an aryl group, while 1 ,2,3,4-tetrahydronaphthalen-1 -yl (wherein the moiety is bound to the parent structure via a non-aromatic carbon atom) is not considered an aryl group. Similarly, a 1 ,2,3,4-tetrahydroquinolin-8-yl group (wherein the moiety is bound to the parent structure via an aromatic carbon atom) is considered an aryl group, while 1 ,2,3,4-tetrahydroquinolin-1 -yl group (wherein the moiety is bound to the parent structure via a non-aromatic nitrogen atom) is not considered an aryl group. However, the term "aryl" does not encompass or overlap with "heteroaryl", as defined herein, regardless of the point of attachment (e.g., both quinolin-5-yl and quinolin-2-yl are heteroaryl groups). In some instances, aryl is phenyl or naphthyl. In certain instances, aryl is phenyl. Additional examples of aryl groups comprising an aromatic carbon ring fused to a non-aromatic ring are described below.
"Aralkyl" refers to a residue having the indicated number of carbon atoms (e.g., 7 to 12 or 7 to 10 carbon atoms) in which an aryl moiety is attached to the parent structure via an alkyl residue. The alkyl residue may be straight-chain or branched. Examples include benzyl, phenethyl and 1 -phenylethyl.
"Heteroaryl" indicates an aromatic ring containing the indicated number of atoms (e.g., 5 to 12, or 5 to 10 membered heteroaryl) made up of one or more heteroatoms (e.g., 1 , 2, 3 or 4 heteroatoms) selected from N, O and S and with the remaining ring atoms being carbon. Heteroaryl groups do not contain adjacent S and O atoms. In some embodiments, the total number of S and O atoms in the heteroaryl group is not more than 2. In some
embodiments, the total number of S and O atoms in the heteroaryl group is not more than 1 . Unless otherwise indicated, heteroaryl groups may be bound to the parent structure by a carbon or nitrogen atom, as valency
permits. For example, "pyridyl" includes 2-pyridyl, 3-pyridyl and 4-pyridyl groups, and "pyrrolyl" includes 1 -pyrrolyl, 2-pyrrolyl and 3-pyrrolyl groups. When nitrogen is present in a heteroaryl ring, it may, where the nature of the adjacent atoms and groups permits, exist in an oxidized state (i.e., N+-O"). Additionally, when sulfur is present in a heteroaryl ring, it may, where the nature of the adjacent atoms and groups permits, exist in an oxidized state (i.e., S+-O" or SO2). Heteroaryl groups may be monocyclic or polycyclic (e.g., bicyclic, tricyclic).
In some instances, a heteroaryl group is monocyclic. Examples include pyrrole, pyrazole, imidazole, triazole (e.g., 1 ,2,3-triazole, 1 ,2,4- triazole, 1 ,2,4-triazole), tetrazole, furan, isoxazole, oxazole, oxadiazole (e.g., 1 ,2,3-oxadiazole, 1 ,2,4-oxadiazole, 1 ,3,4-oxadiazole), thiophene, isothiazole, thiazole, thiadiazole (e.g., 1 ,2,3-thiadiazole, 1 ,2,4-thiadiazole, 1 ,3,4- thiadiazole), pyridine, pyridazine, pyrimidine, pyrazine, triazine (e.g., 1 ,2,4- triazine, 1 ,3,5-triazine) and tetrazine.
In some instances, both rings of a polycyclic heteroaryl group are aromatic. Examples include indole, isoindole, indazole, benzoimidazole, benzotriazole, benzofuran, benzoxazole, benzoisoxazole, benzoxadiazole, benzothiophene, benzothiazole, benzoisothiazole, benzothiadiazole, 1 H- pyrrolo[2,3-b]pyridine, 1 H-pyrazolo[3,4-b]pyridine, 3H-imidazo[4,5-b]pyridine, 3H-[1 ,2,3]triazolo[4,5-b]pyridine, 1 H-pyrrolo[3,2-b]pyridine, 1 H-pyrazolo[4,3- bjpyridine, 1 H-imidazo[4,5-b]pyridine, 1 H-[1 ,2,3]triazolo[4,5-b]pyridine, 1 H- pyrrolo[2,3-c]pyridine, 1 H-pyrazolo[3,4-c]pyridine, 3H-imidazo[4,5-c]pyridine, 3H-[1 ,2,3]triazolo[4,5-c]pyridine, 1 H-pyrrolo[3,2-c]pyridine, 1 H-pyrazolo[4,3- cjpyridine, 1 H-imidazo[4,5-c]pyridine, 1 H-[1 ,2,3]triazolo[4,5-c]pyridine, furo[2,3-b]pyridine, oxazolo[5,4-b]pyridine, isoxazolo[5,4-b]pyridine,
[1 ,2,3]oxadiazolo[5,4-b]pyridine, furo[3,2-b]pyridine, oxazolo[4,5-b]pyridine, isoxazolo[4,5-b]pyridine, [1 ,2,3]oxadiazolo[4,5-b]pyridine, furo[2,3-c] pyridine, oxazolo[5,4-c]pyridine, isoxazolo[5,4-c]pyridine, [1 ,2,3]oxadiazolo[5,4- cjpyridine, furo[3,2-c]pyridine, oxazolo[4,5-c]pyridine, isoxazolo[4,5-c]pyridine, [1 ,2,3]oxadiazolo[4,5-c]pyridine, thieno[2,3-b]pyridine, thiazolo[5,4-b]pyridine, isothiazolo[5,4-b]pyridine, [1 ,2,3]thiadiazolo[5,4-b]pyridine, thieno[3,2- bjpyridine, thiazolo[4,5-b]pyridine, isothiazolo[4,5-b]pyridine,
[1 ,2,3]thiadiazolo[4,5-b]pyridine, thieno[2,3-c]pyridine, thiazolo[5,4-c]pyridine,
isothiazolo[5,4-c] pyridine, [1 ,2,3]thiadiazolo[5,4-c]pyridine, thieno[3,2- c]pyridine, thiazolo[4,5-c]pyridine, isothiazolo[4,5-c]pyridine,
[1 ,2,3]thiadiazolo[4,5-c]pyridine, quinoline, isoquinoline, cinnoline,
quinazoline, quinoxaline, phthalazine, naphthyridine (e.g., 1 ,8-naphthyridine, 1 ,7-naphthyridine, 1 ,6-naphthyridine, 1 ,5-naphthyridine, 2,7-naphthyridine, 2,6-naphthyridine), imidazo[1 ,2-a]pyridine, 1 H-pyrazolo[3,4-d]thiazole, 1 H- pyrazolo[4,3-d]thiazole and imidazo[2,1 -b]thiazole.
In other instances, polycyclic heteroaryl groups may include a non- aromatic ring (e.g., cycloalkyl, cycloalkenyl, heterocycloalkyl,
heterocycloalkenyl) fused to a heteroaryl ring, provided the polycyclic heteroaryl group is bound to the parent structure via an atom in the aromatic ring. For example, a 4,5,6,7-tetrahydrobenzo[d]thiazol-2-yl group (wherein the moiety is bound to the parent structure via an aromatic carbon atom) is considered a heteroaryl group, while 4,5,6,7-tetrahydrobenzo[d]thiazol-5-yl (wherein the moiety is bound to the parent structure via a non-aromatic carbon atom) is not considered a heteroaryl group. Examples of polycyclic heteroaryl groups consisting of a heteroaryl ring fused to a non-aromatic ring are described below.
"Heterocycloalkyl" indicates a non-aromatic, fully saturated ring having the indicated number of atoms (e.g., 3 to 10, or 3 to 7, membered
heterocycloalkyl) made up of one or more heteroatoms (e.g., 1 , 2, 3 or 4 heteroatoms) selected from N, O and S and with the remaining ring atoms being carbon. Heterocycloalkyl groups may be monocyclic or polycyclic (e.g., bicyclic, tricyclic). Examples of heterocycloalkyl groups include oxiranyl, aziridinyl, azetidinyl, pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl and thiomorpholinyl. When nitrogen is present in a heterocycloalkyl ring, it may, where the nature of the adjacent atoms and groups permits, exist in an oxidized state (i.e., N+-O"). Examples include piperidinyl N-oxide and morpholinyl-N-oxide. Additionally, when sulfur is present in a heterocycloalkyl ring, it may, where the nature of the adjacent atoms and groups permits, exist in an oxidized state (i.e., S+-O" or -SO2-). Examples include thiomorpholine S-oxide and thiomorpholine S,S-dioxide. In addition, one ring of a polycyclic heterocycloalkyl group may be aromatic (e.g., aryl or heteroaryl), provided the polycyclic heterocycloalkyl group is
bound to the parent structure via a non-aromatic carbon or nitrogen atom. For example, a 1 ,2,3,4-tetrahydroquinolin-1 -yl group (wherein the moiety is bound to the parent structure via a non-aromatic nitrogen atom) is considered a heterocycloalkyl group, while 1 ,2,3,4-tetrahydroquinolin-8-yl group (wherein the moiety is bound to the parent structure via an aromatic carbon atom) is not considered a heterocycloalkyl group. Examples of polycyclic
heterocycloalkyl groups consisting of a heterocycloalkyl group fused to an aromatic ring are described below.
"Heterocycloalkenyl" indicates a non-aromatic ring having the indicated number of atoms (e.g., 3 to 10, or 3 to 7, membered heterocycloalkyl) made up of one or more heteroatoms (e.g., 1 , 2, 3 or 4 heteroatoms) selected from N, O and S and with the remaining ring atoms being carbon, and at least one double bond derived by the removal of one molecule of hydrogen from adjacent carbon atoms, adjacent nitrogen atoms, or adjacent carbon and nitrogen atoms of the corresponding heterocycloalkyl. Heterocycloalkenyl groups may be monocyclic or polycyclic (e.g., bicyclic, tricyclic). When nitrogen is present in a heterocycloalkenyl ring, it may, where the nature of the adjacent atoms and groups permits, exist in an oxidized state (i.e., N+-O"). Additionally, when sulfur is present in a heterocycloalkenyl ring, it may, where the nature of the adjacent atoms and groups permits, exist in an oxidized state (i.e., S+-O" or -SO2-). Examples of heterocycloalkenyl groups include dihydrofuranyl (e.g., 2,3-dihydrofuranyl, 2,5-dihydrofuranyl), dihydrothiophenyl (e.g., 2,3-dihydrothiophenyl, 2,5-dihydrothiophenyl), dihydropyrrolyl (e.g., 2,3- dihydro-1 H-pyrrolyl, 2,5-dihydro-1 H-pyrrolyl), dihydroimidazolyl (e.g., 2,3- dihydro-1 H-imidazolyl, 4,5-dihydro-1 H-imidazolyl), pyranyl, dihydropyranyl (e.g., 3,4-dihydro-2H-pyranyl, 3,6-dihydro-2H-pyranyl), tetrahydropyridinyl (e.g., 1 ,2,3,4-tetrahydropyridinyl, 1 ,2,3,6-tetrahydropyridinyl) and
dihydropyridine (e.g., 1 ,2-dihydropyridine, 1 ,4-dihydropyridine). In addition, one ring of a polycyclic heterocycloalkenyl group may be aromatic (e.g., aryl or heteroaryl), provided the polycyclic heterocycloalkenyl group is bound to the parent structure via a non-aromatic carbon or nitrogen atom. For example, a 1 ,2-dihydroquinolin-1 -yl group (wherein the moiety is bound to the parent structure via a non-aromatic nitrogen atom) is considered a
heterocycloalkenyl group, while 1 ,2-dihydroquinolin-8-yl group (wherein the
moiety is bound to the parent structure via an aromatic carbon atom) is not considered a heterocydoalkenyl group. Examples of polycyclic
heterocydoalkenyl groups consisting of a heterocydoalkenyl group fused to an aromatic ring are described below.
Examples of polycyclic rings consisting of an aromatic ring (e.g., aryl or heteroaryl) fused to a non-aromatic ring (e.g., cycloalkyl, cycloalkenyl, heterocydoalkyi, heterocydoalkenyl) include indenyl, 2, 3-dihydro-1 H-indenyl, 1 ,2,3,4-tetrahydronaphthalenyl, benzo[1 ,3]dioxolyl, tetrahydroquinolinyl, 2,3-dihydrobenzo[1 ,4]dioxinyl, indolinyl, isoindolinyl, 2, 3-dihydro-1 H-indazolyl, 2,3-dihydro-1 H-benzo[d]imidazolyl, 2,3-dihydrobenzofuranyl,
1 ,3-dihydroisobenzofuranyl, 1 ,3-dihydrobenzo[c]isoxazolyl,
2,3-dihydrobenzo[d]isoxazolyl, 2,3-dihydrobenzo[d]oxazolyl,
2,3-dihydrobenzo[b]thiophenyl, 1 ,3-dihydrobenzo[c]thiophenyl,
1 ,3-dihydrobenzo[c]isothiazolyl, 2,3-dihydrobenzo[d]isothiazolyl,
2,3-dihydrobenzo[d]thiazolyl, 5,6-dihydro-4H-cyclopenta[d]thiazolyl,
4,5,6,7-tetrahydrobenzo[d]thiazolyl, 5,6-dihydro-4H-pyrrolo[3,4-d]thiazolyl , 4,5,6,7-tetrahydrothiazolo[5,4-c]pyridinyl, indolin-2-one, indolin-3-one, isoindolin-1 -one, 1 ,2-dihydroindazol-3-one, 1 H-benzo[d]imidazol-2(3H)-one, benzofuran-2(3H)-one, benzofuran-3(2H)-one, isobenzofuran-1 (3H)-one, benzo[c]isoxazol-3(1 H)-one, benzo[d]isoxazol-3(2H)-one, benzo[d]oxazol- 2(3H)-one, benzo[b]thiophen-2(3H)-one, benzo[b]thiophen-3(2H)-one, benzo[c]thiophen-1 (3H)-one, benzo[c]isothiazol-3(1 H)-one,
benzo[d]isothiazol-3(2H)-one, benzo[d]thiazol-2(3H)-one, 4,5- dihydropyrrolo[3,4-d]thiazol-6-one, 1 ,2-dihydropyrazolo[3,4-d]thiazol-3-one, quinolin-4(3H)-one, quinazolin-4(3H)-one, quinazoline-2,4(1 H,3H)-dione, quinoxalin-2(1 H)-one, quinoxaline-2,3(1 H,4H)-dione, cinnolin-4(3H)-one, pyridin-2(1 H)-one, pyrimidin-2(1 H)-one, pyrimidin-4(3H)-one, pyridazin-3(2H)- one, 1 H-pyrrolo[3,2-b]pyridin-2(3H)-one, 1 H-pyrrolo[3,2-c]pyridin-2(3H)-one, 1 H-pyrrolo[2,3-c]pyridin-2(3H)-one, 1 H-pyrrolo[2,3-b]pyridin-2(3H)-one, 1 ,2- dihydropyrazolo[3,4-d]thiazol-3-one and 4,5-dihydropyrrolo[3,4-d]thiazol-6- one. As discussed herein, whether each ring is considered an aryl, heteroaryl, cycloalkyl, cycloalkenyl, heterocydoalkyi or heterocydoalkenyl group is determined by the atom through which the moiety is bound to the parent structure.
"Halogen" or "halo" refers to fluorine, chlorine, bromine or iodine.
"Isomers" are different compounds that have the same molecular formula. "Stereoisomers" are isomers that differ only in the way the atoms are arranged in space. "Enantiomers" are stereoisomers that are non- superimposable mirror images of each other. A 1 :1 mixture of a pair of enantiomers is a "racemic" mixture. The symbol "(±)" may be used to designate a racemic mixture where appropriate. "Diastereoisomers" are stereoisomers that have at least two asymmetric atoms, but which are not mirror-images of each other. A "meso compound" or "meso isomer" is a non- optically active member of a set of stereoisomers. Meso isomers contain two or more stereocenters but are not chiral (i.e., a plane of symmetry exists within the molecule). The absolute stereochemistry is specified according to the Cahn-lngold-Prelog R-S system. When a compound is a pure enantiomer the stereochemistry at each chiral carbon can be specified by either R or S. Resolved compounds whose absolute configuration is unknown can be designated (+) or (-) depending on the direction (dextro- or levorotatory) which they rotate plane polarized light at the wavelength of the sodium D line.
Certain of the compounds disclosed and/or described herein contain one or more asymmetric centers and can thus give rise to enantiomers,
diastereomers, meso isomers and other stereoisomeric forms. Unless otherwise indicated, compounds disclosed and/or described herein include all such possible enantiomers, diastereomers, meso isomers and other stereoisomeric forms, including racemic mixtures, optically pure forms and intermediate mixtures. Enantiomers, diastereomers, meso isomers and other stereoisomeric forms can be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques. Unless specified otherwise, when the compounds disclosed and/or described herein contain olefinic double bonds or other centers of geometric asymmetry, it is intended that the compounds include both E and Z isomers.
The stereochemistry depicted in the structures of cyclic meso compounds is not absolute; rather the stereochemistry is intended to indicate the positioning of the substituents relative to one another, e.g., cis or trans. For example,

is intended to designate a compound wherein the fluorine and pyridyl substituents on the cyclobutyl ring are in a cis configuration to one another, while

is intended to designate a compound wherein the fluorine and pyridyl substituents on the cyclobutyl ring are in a trans configuration to one another.
When a compound can exist as one or more meso isomers, all possible meso isomers are intended to be included. For example, the compound 5-bromo-3-fluoro-N-((3-fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methyl)pyridin-2-amine is intended to include both cis and trans meso isomers

and, and mixtures thereof. Unless otherwise indicated, compounds disclosed and/or described herein include all possible meso isomers and mixtures thereof.
"Tautomers" are structurally distinct isomers that interconvert by tautomerization. Tautomerization is a form of isomerization and includes prototropic or proton-shift tautomerization, which is considered a subset of acid-base chemistry. Prototropic tautomerization or proton-shift
tautomerization involves the migration of a proton accompanied by changes in bond order, often the interchange of a single bond with an adjacent double
bond. Where tautomerization is possible (e.g. in solution), a chemical equilibrium of tautomers can be reached. An example of tautomerization is keto-enol tautomerization. A specific example of keto-enol tautomerization is the interconverision of pentane-2,4-dione and 4-hydroxypent-3-en-2-one tautomers. Another example of tautomerization is phenol-keto
tautomerization. A specific example of phenol-keto tautomerization is the interconversion of pyridin-4-ol and pyridin-4(1 H)-one tautomers. When the compounds described herein contain moieties capable of tautomerization, and unless specified otherwise, it is intended that the compounds include all possible tautomers.
"Protecting group" has the meaning conventionally associated with it in organic synthesis, i.e., a group that selectively blocks one or more reactive sites in a multifunctional compound such that a chemical reaction can be carried out selectively on another unprotected reactive site, and such that the group can readily be removed after the selective reaction is complete. A variety of protecting groups are disclosed, for example, in T.H. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Third Edition, John Wiley & Sons, New York (1999). For example, a "hydroxy protected form" contains at least one hydroxy group protected with a hydroxy protecting group. Likewise, amines and other reactive groups may similarly be
protected.
The term "pharmaceutically acceptable salt" refers to salts that retain the biological effectiveness and properties of the compounds described herein and are not biologically or otherwise undesirable. Examples of
pharmaceutically acceptable salts can be found in Berge et al.,
Pharmaceutical Salts, J. Pharmaceutical Sciences, January 1977, 66(1 ), 1 -19. In many cases, the compounds described herein are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto. Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids. Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, and phosphoric acid. Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, pyruvic acid, lactic acid, oxalic acid, malic acid, maleic acid, malonic
acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, 2- hydroxyethylsulfonic acid, p-toluenesulfonic acid, stearic acid and salicylic acid. Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases. Inorganic bases from which salts can be derived include, for example, sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, and aluminum. Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines; substituted amines including naturally occurring substituted amines; cyclic amines; and basic ion exchange resins. Examples of organic bases include isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, and ethanolamine. In some embodiments, the
pharmaceutically acceptable base addition salt is chosen from ammonium, potassium, sodium, calcium, and magnesium salts.
If the compound described herein is obtained as an acid addition salt, the free base can be obtained by basifying a solution of the acid salt.
Conversely, if the compound is a free base, an addition salt, particularly a pharmaceutically acceptable addition salt, may be produced by dissolving the free base in a suitable organic solvent and treating the solution with an acid, in accordance with conventional procedures for preparing acid addition salts from base compounds (see, e.g., Berge et al., Pharmaceutical Salts, J.
Pharmaceutical Sciences, January 1977, 66(1 ), 1 -19). Those skilled in the art will recognize various synthetic methodologies that may be used to prepare pharmaceutically acceptable addition salts.
A "solvate" is formed by the interaction of a solvent and a compound. Suitable solvents include, for example, water and alcohols (e.g., ethanol). Solvates include hydrates having any ratio of compound to water, such as monohydrates, dihydrates and hemi-hydrates.
A "chelate" is formed by the coordination of a compound to a metal ion at two (or more) points. The term "compound" is intended to include chelates of compounds. Similarly, "salts" includes chelates of salts and "solvates" includes chelates of solvates.
A "non-covalent complex" is formed by the interaction of a compound and another molecule wherein a covalent bond is not formed between the
compound and the molecule. For example, complexation can occur through van der Waals interactions, hydrogen bonding, and electrostatic interactions (also called ionic bonding). Such non-covalent complexes are included in the term "compound".
The term "prodrug" refers to a substance administered in an inactive or less active form that is then transformed (e.g., by metabolic processing of the prodrug in the body) into an active compound. The rationale behind administering a prodrug is to optimize absorption, distribution, metabolism, and/or excretion of the drug. Prodrugs may be obtained by making a derivative of an active compound (e.g., a compound of Formula I or another compound disclosed and/or described herein) that will undergo a
transformation under the conditions of use (e.g., within the body) to form the active compound. The transformation of the prodrug to the active compound may proceed spontaneously (e.g., by way of a hydrolysis reaction) or it can be catalyzed or induced by another agent (e.g., an enzyme, light, acid or base, and/or temperature). The agent may be endogenous to the conditions of use (e.g., an enzyme present in the cells to which the prodrug is administered, or the acidic conditions of the stomach) or the agent may be supplied
exogenously. Prodrugs can be obtained by converting one or more functional groups in the active compound into another functional group, which is then converted back to the original functional group when administered to the body. For example, a hydroxyl functional group can be converted to a sulfonate, phosphate, ester or carbonate group, which in turn can be hydrolyzed in vivo back to the hydroxyl group. Similarly, an amino functional group can be converted, for example, into an amide, carbamate, imine, urea, phosphenyl, phosphoryl or sulfenyl functional group, which can be hydrolyzed in vivo back to the amino group. A carboxyl functional group can be converted, for example, into an ester (including silyl esters and thioesters), amide or hydrazide functional group, which can be hydrolyzed in vivo back to the carboxyl group. Examples of prodrugs include, but are not limited to, phosphate, acetate, formate and benzoate derivatives of functional groups (such as alcohol or amine groups) present in the compounds of Formula I and other compounds disclosed and/or described herein.
The compounds disclosed and/or described herein can be enriched
isotopic forms, e.g., enriched in the content of 2H, 3H, 11C, 13C and/or 14C. In one embodiment, the compound contains at least one deuterium atom. Such deuterated forms can be made, for example, by the procedure described in U.S. Patent Nos. 5,846,514 and 6,334,997. Such deuterated compounds may improve the efficacy and increase the duration of action of compounds disclosed and/or described herein. Deuterium substituted compounds can be synthesized using various methods, such as those described in: Dean, D., Recent Advances in the Synthesis and Applications of Radiolabeled
Compounds for Drug Discovery and Development, Curr. Pharm. Des., 2000; 6(10); Kabalka, G. et al., The Synthesis of Radiolabeled Compounds via Organometallic Intermediates, Tetrahedron, 1989, 45(21 ), 6601 -21 ; and Evans, E., Synthesis of radiolabeled compounds, J. Radioanal. Chem., 1981 , 64(1 -2), 9-32.
The term "pharmaceutically acceptable carrier" or "pharmaceutically acceptable excipient" includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like. The use of such media and agents for pharmaceutically active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, its use in
pharmaceutical compositions is contemplated. Supplementary active ingredients can also be incorporated into the pharmaceutical compositions.
The term "active agent" is used to indicate a compound that has biological activity. In some embodiments, an "active agent" is a compound having therapeutic utility. In some embodiments, the compound enhances at least one aspect of skeletal muscle function or activity, such as power output, skeletal muscle force, skeletal muscle endurance, oxygen consumption, efficiency, and/or calcium sensitivity. In some embodiments, an active agent is a compound of Formula I, or a pharmaceutically acceptable salt thereof.
The terms "patient" and "subject" refer to an animal, such as a mammal bird or fish. In some embodiments, the patient or subject is a mammal.
Mammals include, for example, mice, rats, dogs, cats, pigs, sheep, horses, cows and humans. In some embodiments, the patient or subject is a human, for example a human that has been or will be the object of treatment, observation or experiment. The compounds, compositions and methods
described herein can be useful in both human therapy and veterinary applications.
As used herein, "skeletal muscle" includes skeletal muscle tissue as well as components thereof, such as skeletal muscle fibers, the myofibrils comprising the skeletal muscle fibers, the skeletal sarcomere which
comprises the myofibrils, and the various components of the skeletal sarcomere described herein, including skeletal myosin, actin, tropomyosin, troponin C, troponin I, troponin T and fragments and isoforms thereof. In some embodiments, "skeletal muscle" includes fast skeletal muscle tissue as well as components thereof, such as fast skeletal muscle fibers, the myofibrils comprising the fast skeletal muscle fibers, the fast skeletal sarcomere which comprises the myofibrils, and the various components of the fast skeletal sarcomere described herein, including fast skeletal myosin, actin,
tropomyosin, troponin C, troponin I, troponin T and fragments and isoforms thereof. Skeletal muscle does not include cardiac muscle or a combination of sarcomeric components that occurs in such combination in its entirety in cardiac muscle.
As used herein, the term "therapeutic" refers to the ability to modulate the contractility of fast skeletal muscle. As used herein, "modulation" (and related terms, such as "modulate", "modulated", "modulating") refers to a change in function or efficiency of one or more components of the fast skeletal muscle sarcomere, including myosin, actin, tropomyosin, troponin C, troponin I, and troponin T from fast skeletal muscle, including fragments and isoforms thereof, as a direct or indirect response to the presence of a compound described herein, relative to the activity of the fast skeletal sarcomere in the absence of the compound. The change may be an increase in activity
(potentiation) or a decrease in activity (inhibition), and may be due to the direct interaction of the compound with the sarcomere, or due to the
interaction of the compound with one or more other factors that in turn affect the sarcomere or one or more of its components. In some embodiments, modulation is a potentiation of function or efficiency of one or more
components of the fast skeletal muscle sarcomere, including myosin, actin, tropomyosin, troponin C, troponin I, and troponin T from fast skeletal muscle, including fragments and isoforms thereof. Modulation may be mediated by
any mechanism and at any physiological level, for example, through
sensitization of the fast skeletal sarcomere to contraction at lower Ca2+ concentrations. As used herein, "efficiency" or "muscle efficiency" means the ratio of mechanical work output to the total metabolic cost.
The term "therapeutically effective amount" or "effective amount" refers to that amount of a compound disclosed and/or described herein that is sufficient to affect treatment, as defined herein, when administered to a patient in need of such treatment. A therapeutically effective amount of a compound may be an amount sufficient to treat a disease responsive to modulation of fast skeletal muscle. The therapeutically effective amount will vary depending upon, for example, the subject and disease condition being treated, the weight and age of the subject, the severity of the disease condition, the particular compound, the dosing regimen to be followed, timing of administration, the manner of administration, all of which can readily be determined by one of ordinary skill in the art. The therapeutically effective amount may be ascertained experimentally, for example by assaying blood concentration of the chemical entity, or theoretically, by calculating
bioavailability.
"Treatment" (and related terms, such as "treat", "treated", "treating") includes one or more of: preventing a disease or disorder (i.e., causing the clinical symptoms of the disease or disorder not to develop); inhibiting a disease or disorder; slowing or arresting the development of clinical symptoms of a disease or disorder; and/or relieving a disease or disorder (i.e., causing relief from or regression of clinical symptoms). The term
encompasses situations where the disease or disorder is already being experienced by a patient, as well as situations where the disease or disorder is not currently being experienced but is expected to arise. The term covers both complete and partial reduction or prevention of the condition or disorder, and complete or partial reduction of clinical symptoms of a disease or disorder. Thus, compounds described and/or disclosed herein may prevent an existing disease or disorder from worsening, assist in the management of the disease or disorder, or reduce or eliminate the disease or disorder. When used in a prophylactic manner, the compounds disclosed and/or described herein may prevent a disease or disorder from developing or lessen the extent
of a disease or disorder that may develop.
As used herein, "power output" of a muscle means work/cycle time and may be scaled up from PoLo/cycle time units based on the properties of the muscle. Power output may be modulated by changing, for example, activating parameters during cyclical length changes, including timing of activation (phase of activation) and the period of activation (duty cycle.)
"ATPase" refers to an enzyme that hydrolyzes ATP. ATPases include proteins comprising molecular motors such as the myosins.
As used herein, "selective binding" or "selectively binding" refers to preferential binding to a target protein in one type of muscle or muscle fiber as opposed to other types. For example, a compound selectively binds to fast skeletal troponin C if the compound preferentially binds troponin C in the troponin complex of a fast skeletal muscle fiber or sarcomere in comparison with troponin C in the troponin complex of a slow muscle fiber or sarcomere or with troponin C in the troponin complex of a cardiac sarcomere.
Provided is a compound of Formula I:

Formula I
or a pharmaceutically acceptable salt thereof, wherein:
Z1, Z2, Z3 and Z4 are each independently selected from N and CR1, provided that at least one of Z1, Z2, Z3 and Z4 is N and at least one of Z1, Z' Z3 and Z4 is CR1;
R1, at each occurrence, is independently selected from hydrogen, halogen, CN, C1-6 alkyl, C1-6 haloalkyl, C(O)ORa, C(O)NRbRc, ORa, NRbRc, 10 aryl and 5-10 membered heteroaryl;
R2 is selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, 5-10
membered heteroaryl and NRbRc, wherein each of the C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyi, 3-8 membered
heterocycloalkenyl, C-6-10 aryl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)N RbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra,
(CH2)nSO2NRbRc, C1-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyi, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyi, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents;
R4 is selected from hydrogen, C-i-6 alkyl, C-i-6 haloalkyl, C(O)Ra, C(O)ORa, C(O)NRbRc and SO2Ra;
R5 and R6 are each independently selected from hydrogen, halogen, C-i-6 alkyl and C-i-6 haloalkyl;
or alternatively, R5 and R6 together with the carbon atom to which they are bound form a group selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyi and 3-8 membered heterocycloalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl and Ci-6 haloalkyl;
R7 is selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyi, 3-8 membered heterocycloalkenyl, C6-io aryl and 5-10 membered heteroaryl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, OC(O)NRbRc, N RbRc, NRdC(O)Ra, NRdC(O)ORa, NRdC(O)NRbRc,
NRdC(O)C(O)NRbRc, N RdC(S)Ra, NRdC(S)ORa, N RdC(S)NRbRc,
NRdC(NRe)NRbRc, N RdS(O)Ra, NRdSO2Ra, NRdSO2NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, C(S)Ra, C(S)ORa, C(S)NRbRc, C(NRe)NRbRc, SRa, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl, and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents;
R8 and R9, at each occurrence, are each independently selected from hydrogen, halogen and C-i-6 alkyl;
X is selected from a bond, -(CH2)P-, -(CH2)pC(O)(CH2)q-, - (CH2)pO(CH2)q-, -(CH2)pS(CH2)q-, -(CH2)pNRd(CH2)q-, -(CH2)pC(O)O(CH2)q-, -(CH2)pOC(O)(CH2)q-, -(CH2)pNRdC(O)(CH2)q-, -(CH2)pC(O)NRd(CH2)q-, -(CH2)pNRdC(O)NRd(CH2)q-, -(CH2)pNRdSO2(CH2)q-, and
-(CH2)pSO2NRd(CH2)q-;
Ra, at each occurrence, is independently selected from hydrogen, C-i-6 alkyl, C-i-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-i i aralkyl and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents;
Rb and Rc, at each occurrence, are each independently selected from hydrogen, C-i-6 alkyl, C-i-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
heterocycloalkenyl, C6-io aryl, C7-n aralkyl, 5-10 membered heteroaryl, C(O)Rg, C(O)ORg, C^NR^ and SO2Rg, wherein each of the C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl
and 5-10 mennbered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents;
Rd, at each occurrence, is independently selected from hydrogen and Ci-6 alkyl;
Re, at each occurrence, is independently selected from hydrogen, CN, OH, C-i-6 alkoxy, C-i-6 alkyl and C-i-6 haloalkyl;
Rf, at each occurrence, is independently selected from halogen, CN, ORh, OC(O)Rh, OC(O)ORh, OC JNR^, NR'Rj, NRdC(O)Rh, NRdC(O)ORh, NRdC(O)NR'Rj, NRdC(O)C(O)NRiRj, NRdC(S)Rh, NRdC(S)ORh, NRdC(S)NR'Rj, NRdC(NRe)NR'Rj, NRdS(O)Rh, NRdSO2Rh, NRdSO2NR'Rj, C(O)Rh, C(O)ORh, Ο(Ο)Ν^, C(S)Rh, C(S)ORh, C^NR^, C(NRe)NR'Rj, SRh, S(O)Rh, SO2Rh, SO2NR'Rj, Ci-6 alkyl, C-i-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rk substituents;
or two Rf substituents bound to a single carbon atom, together with the carbon atom to which they are both bound, form a group selected from carbonyl, C3-8 cycloalkyl and 3-8 membered heterocycloalkyl;
Rg, at each occurrence, is independently selected from C-i-6 alkyl, C-i-6 haloalkyl, phenyl, naphthyl, and C7-n aralkyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, OH, C-i-6 alkoxy, C-i-6 alkyl and C-i-6 haloalkyl;
Rh, at each occurrence, is independently selected from hydrogen, C-i-6 alkyl, C-i-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-ii aralkyl and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4
or 5 Rk substituents;
R1 and Rj, at each occurrence, are each independently selected from hydrogen, d-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
heterocycloalkenyl, C6-io aryl, C7-n aralkyl, 5-10 membered heteroaryl, C(O)Rg, and C(O)ORg, wherein each of the C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C-6-10 aryl, C7-11 aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, OH, C-i-6 alkoxy, C-i-6 alkyl and C-i-6 haloalkyl;
Rk, at each occurrence, is independently selected from halogen, CN, oxo, OH, C1-6 alkoxy, NH2, NH(C1-6 alkyl), N(C1-6 alkyl)2, NHC(O)C1-6 alkyl, NHC(0)C7-ii aralkyl, NHC(O)OC1-6 alkyl, NHC(O)OC7-11 aralkyl, C(O)C1-6 alkyl, C(0)C7-n aralkyl, C(O)OCi-6 alkyl, C(0)OC7-n aralkyl, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, and C2-6 alkynyl, wherein each C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, and C7-n aralkyl substituent is optionally substituted with 1 , 2 or 3 substituents selected from OH, d-6 alkoxy, NH2, NH(d-6 alkyl), N(d-6 alkyl)2, NHC(O)Ci-6 alkyl, NHC(O)C7-n aralkyl, NHC(O)OCi-6 alkyl, and NHC(O)OC7- 11 aralkyl;
m is 0, 1 or 2;
n, at each occurrence, independently is 0, 1 or 2; p is 0, 1 or 2; and
q is 0, 1 or 2.
In some embodiments of compounds of Formula I, Z1 and Z2 are each N, one of Z3 and Z4 is N, and the other of Z3 and Z4 is CR1.
In some embodiments of compounds of Formula I, Z1 , Z2 and Z3 are each N and Z4 is CR1 , i.e., a compound of Formula II, or a pharmaceutically acceptable salt thereof:

Formula II
wherein R1, R2, R4, R5, R6, R7, R8, R9, X and m are as defined herein.
In some embodiments of compounds of Formula I, Z1, Z2 and Z4 are each N and Z3 is CR1, i.e., a compound of Formula III, or a pharmaceutically acceptable salt thereof:

Formula III
wherein R1, R2, R4, R5, R6, R7, R8, R9, X and m are as defined herein.
In some embodiments of compounds of Formula I, one of Z1 and Z2 is N, the other of Z1 and Z2 is CR1, and Z3 and Z4 are each CR1.
In some embodiments of compounds of Formula I, Z1, Z3 and Z4 are each CR1 and Z2 is N, i.e., a compound of Formula IV, or a pharmaceutically acceptable salt thereof:
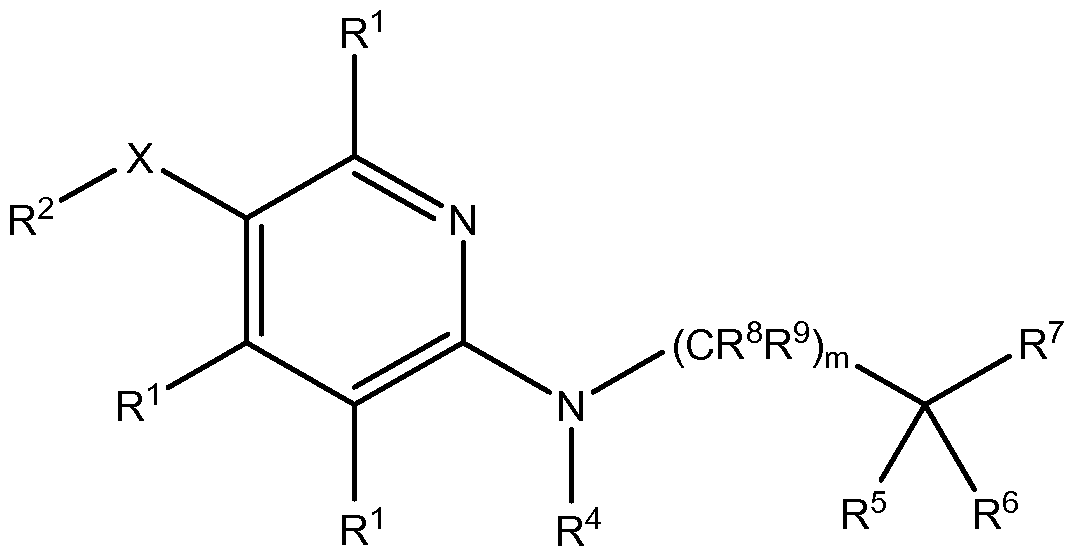
Formula IV
wherein R1, R2, R4, R5, R6, R7, R8, R9, X and m are as defined herein.
In some embodiments of compounds of Formula I, Z2, Z3 and Z4 are each CR1 and Z1 is N, i.e., a compound of Formula V, or a pharmaceutically acceptable salt thereof:

Formula V
wherein R1, R2, R4, R5, R6, R7, R8, R9, X and m are as defined herein.
In some embodiments of compounds of Formula I, II, III, IV or V, m is 0, i.e., a compound of Formula ll(a), lll(a), IV(a) or V(a), or a pharmaceutically acceptable salt thereof:

Formula lll(a)

R1 R4
Formula V(a)
wherein R1, R2, R4, R5, R6, R7 and X are as defined herein.
In some embodiments of compounds of Formula I, II, III, IV or V, m is 1 , i.e., a compound of Formula ll(b), lll(b), IV(b) or V(b), or a pharmaceutically acceptable salt thereof:

Formula lll(b)

Formula V(b)
wherein R1, R2, R4, R5, R6, R7, R8, R9 and X are as defined herein.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), one of R5 and R6 is hydrogen and the other is C-i-6 alkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6 are each independently C-i-6 alkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6 are each methyl.
In some embodiments, the compounds are of Formula ll(c) or ll(d), or a pharmaceutically acceptable salt thereof:

Formula 11(c)

Formula 11(d)
wherein R1, R2, R4, R7, R8, R9 and X are as defined herein.
In some embodiments, the compounds are of Formula lll(c) or lll(d), or a pharmaceutically acceptable salt thereof:

Formula lll(c)

Formula lll(d)
wherein R1 , R2, R4, R7, R8, R9 and X are as defined herein.
In some embodiments, the compounds are of Formula IV(c) or IV(d), or
a pharmaceutically acceptable salt thereof:

Formula IV(c)

Formula IV(d)
wherein R1, R2, R4, R7, R8, R9 and X are as defined herein.
In some embodiments, the compounds are of Formula V(c) or V(d), or a pharmaceutically acceptable salt thereof:

Formula V(c)

Formula V(d)
wherein R1, R2, R4, R7, R8, R9 and X are as defined herein.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6 together with the carbon atom to which they are bound form C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl or 3-8 membered heterocycloalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl and Ci-6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6, together with the carbon to which they are bound, form C3-6 cycloalkyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra,
SO2NRbRc, C -6 alkyl and C -6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6, together with the carbon to which they are bound, form cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl and Ci-6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6, together with the carbon to which they are bound, form cyclobutyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa,
NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, C1-6 alkyl and Ci-6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, 111(a), 111(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6, together with the carbon to which they are bound, form cyclobutyl substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl and C-i-6 haloalkyl, wherein the substituent and R7 are in a trans configuration with respect to one another on the cyclobutyl ring.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6, together with the carbon to which they are bound, form cyclobutyl substituted with one substituent selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, C1-6 alkyl and C1-6 haloalkyl, wherein the substituent and R7 are in a cis configuration with respect to one another on the cyclobutyl ring.
In some embodiments, the compounds are of Formula ll(e) or ll(f), or a pharmaceutically acceptable salt thereof:

Formula ll(e)

Formula 11(f)
wherein Rm and Rn are each independently selected from hydrogen and halogen, and R1, R2, R4, R7, R8, R9 and X are as defined herein.
In some embodiments, the compounds are of Formula lll(e) or lll(f), or a pharmaceutically acceptable salt thereof:

Formula lll(f)
wherein Rm and Rn are each independently selected from hydrogen and halogen, and R1, R2, R4, R7, R8, R9 and X are as defined herein.
In some embodiments, the compounds are of Formula IV(e) or IV(f), or
a pharmaceutically acceptable salt thereof:

Formula IV(e)

Formula IV(f)
wherein Rm and Rn are each independently selected from hydrogen and halogen, and R1, R2, R4, R7, R8, R9 and X are as defined herein.
In some embodiments, the compounds are of Formula V(e) or V(f), or a pharmaceutically acceptable salt thereof:

Formula V(e)

Formula V(f)
wherein Rm and Rn are each independently selected from hydrogen and halogen, and R1, R2, R4, R7, R8, R9 and X are as defined herein.
In some embodiments of compounds of Formula ll(e), ll(f), lll(e), lll(f), IV(e), IV(f), V(e) or V(f), Rm and Rn are each hydrogen.
In some embodiments compounds of Formula ll(e), ll(f), lll(e), lll(f), IV(e), IV(f), V(e) or V(f), Rm and Rn are each halogen.
In some embodiments compounds of Formula ll(e), ll(f), lll(e), lll(f), IV(e), IV(f), V(e) or V(f), Rm and Rn are each fluorine.
In some embodiments compounds of Formula ll(e), ll(f), lll(e), lll(f), IV(e), IV(f), V(e) or V(f), one of Rm and Rn is hydrogen and the other is halogen. In some embodiments of such compounds, the halogen and R7 are in a trans configuration with respect to one another on the cyclobutyl ring. In some embodiments of such compounds, the halogen and R7 are in a cis configuration with respect to one another on the cyclobutyl ring.
In some embodiments compounds of Formula ll(e), ll(f), lll(e), lll(f), IV(e), IV(f), V(e) or V(f), one of Rm and Rn is hydrogen and the other is fluorine. In some embodiments of such compounds, the fluorine and R7 are in a trans configuration with respect to one another on the cyclobutyl ring. In some embodiments of such compounds, the fluorine and R7 are in a cis configuration with respect to one another on the cyclobutyl ring.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6, together with the carbon atom to which they are bound, form 3-6 membered heterocycloalkyi optionally
substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl and Ci-6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6, together with the carbon atom to which they are bound, form aziridine, azetidine, pyrrolidine, oxirane, oxetane or tetrahydrofuran, each of which is optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra,
OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra,
SO2NRbRc, C -6 alkyl and C -6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6 are each independently C-i-6 alkyl, or R5 and R6 together with the carbon atom to which they are bound form C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl or 3-8 membered heterocycloalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, C1-6 alkyl and C-i-6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6 are each methyl, or R5 and R6 together with the carbon atom to which they are bound form C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl or 3-8 membered heterocycloalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, C1-6 alkyl and C1-6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6 are each independently C-i-6 alkyl, or R5 and R6, together with the carbon to which they are bound, form a group selected from cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa,
C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, C1-6 alkyl and C1-6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), III, lll(a), lll(b), IV, IV(a), IV(b) V, V(a) or V(b), R5 and R6 are each methyl, or R5 and R6, together with the carbon to which they are bound, form cyclobutyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl and Ci-6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R7 is selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl and 5-10 membered heteroaryl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, OC(O)NRbRc, NRbRc, N RdC(O)Ra, N RdC(O)ORa, NRdC(O)N RbRc, N RdC(O)C(O)NRbRc, N RdC(S)Ra, N RdC(S)ORa,
NRdC(S)NRbRc, NRdC(NRe)NRbRc, N RdS(O)Ra, NRdSO2Ra, NRdSO2NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, C(S)Ra, C(S)ORa, C(S)NRbRc, C(NRe)NRbRc, SRa, S(O)Ra, SO2Ra, SO2NRbRc, C1-6 alkyl, C1 -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7.11 aralkyl, and 5-10 membered heteroaryl, wherein each of the Ci-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7.11 aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R7 is phenyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, OC(O)NRbRc, NRbRc, N RdC(O)Ra, N RdC(O)ORa, NRdC(O)N RbRc, N RdC(O)C(O)NRbRc, N RdC(S)Ra, N RdC(S)ORa,
NRdC(S)NRbRc, NRdC(NRe)NRbRc, N RdS(O)Ra, NRdSO2Ra, NRdSO2NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, C(S)Ra, C(S)ORa, C(S)NRbRc, C(NRe)NRbRc, SRa, S(O)Ra, SO2Ra, SO2NRbRc, Ci.6 alkyl, Ci.6 haloalkyl, C2.6 alkenyl, C2.6
alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl, and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R7 is selected from phenyl, 2- fluorophenyl, 3-fluorophenyl, 2, 4-difluorophenyl, 3,4-difluorophenyl, 3,5- difluorophenyl, 4-fluorophenyl, 2-chlorophenyl, 3-chlorophenyl, 4- chlorophenyl, 2, 4-dichlorophenyl, 3,4-dichlorophenyl, 3,5-dichlorophenyl, 2- methylphenyl, 3-methylphenyl, 2, 4-dimethylphenyl, 3,4-dimethylphenyl, 3,5- dimethylphenyl, 2-(hydroxymethyl)phenyl, 3-(hydroxymethyl)phenyl, 4- (hydroxymethyl)phenyl, 2-(aminomethyl)phenyl, 3-(aminomethyl)phenyl, 4- (aminomethyl)phenyl, 2-phenol, 3-phenol, 4-phenol, 2-methoxyphenyl, 3- methoxyphenyl, 4-methoxyphenyl, 2-difluoromethoxyphenyl, 3- difluoromethoxyphenyl, 4-difluoromethoxyphenyl, 2-trifluoromethoxyphenyl, 3- trifluoromethoxyphenyl, 4-trifluoromethoxyphenyl, 2-cyanophenyl, 3- cyanophenyl, 4-cyanophenyl, 2-benzamine, 3-benzamide, 4-benzamide, N- mehtyl-2-benzamine, N-methyl-3-benzamide, N-methyl-4-benzamide, N,N- dimethyl-2-benzamine, N,N-dimethyl-3-benzamide, and N,N-dimethyl-4- benzamide.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R7 is 5-10 membered heteroaryl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, OC(O)NRbRc, NRbRc, NRdC(O)Ra, NRdC(O)ORa, NRdC(O)NRbRc, NRdC(O)C(O)NRbRc, NRdC(S)Ra,
NRdC(S)ORa, NRdC(S)NRbRc, NRdC(NRe)NRbRc, NRdS(O)Ra, NRdSO2Ra, NRdSO2NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, C(S)Ra, C(S)ORa, C(S)NRbRc, C(NRe)NRbRc, SRa, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered
heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl, and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R7 is pyridyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, OC(O)NRbRc, NRbRc, NRdC(O)Ra, NRdC(O)ORa, NRdC(O)NRbRc, NRdC(O)C(O)NRbRc, NRdC(S)Ra, NRdC(S)ORa,
NRdC(S)NRbRc, NRdC(NRe)NRbRc, NRdS(O)Ra, NRdSO2Ra, NRdSO2NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, C(S)Ra, C(S)ORa, C(S)NRbRc, C(NRe)NRbRc, SRa, S(O)Ra, SO2Ra, SO2NRbRc, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl, and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R7 is selected from 2-pyridyl, 3- pyridyl and 4-pyridyl, each optionally substituted with 1 , 2, 3, 4 or 5
substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, OC(O)NRbRc, NRbRc, NRdC(O)Ra, NRdC(O)ORa, NRdC(O)NRbRc,
NRdC(O)C(O)NRbRc, NRdC(S)Ra, NRdC(S)ORa, NRdC(S)NRbRc,
NRdC(NRe)NRbRc, NRdS(O)Ra, NRdSO2Ra, NRdSO2NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, C(S)Ra, C(S)ORa, C(S)NRbRc, C(NRe)NRbRc, SRa, S(O)Ra, SO2Ra, SO2NRbRc, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl, and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8
cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, III, IV(a), IV(b), V(a) or V(b), R7 is selected from pyrid-2-yl, 3-fluoro-pyrid-2-yl, 4-fluoro-pyrid-2- yl, 5-fluoro-pyrid-2-yl, 6-fluoro-pyrid-2-yl, 3-chloro-pyrid-2-yl, 4-chloro-pyrid-2- yl, 5-chloro-pyrid-2-yl, 6-chloro-pyrid-2-yl, 3-cyano-pyrid-2-yl, 4-cyano-pyrid-2- yl, 5-cyano-pyrid-2-yl, 6-cyano-pyrid-2-yl, 3-methyl-pyrid-2-yl, 4-methyl-pyrid- 2-yl, 5-methyl-pyrid-2-yl, 6-methyl-pyrid-2-yl, 3-difluoromethyl-pyrid-2-yl, 4-difluoromethyl-pyrid-2-yl, 5-difluoromethyl-pyrid-2-yl, 6-difluoromethyl-pyrid-
2- yl, 3-trifluoromethyl-pyrid-2-yl, 4-trifluoromethyl-pyrid-2-yl, 5-trifluoromethyl- pyrid-2-yl, 6-trifluoromethyl-pyrid-2-yl, 3-hydroxymethyl-pyrid-2-yl,
4- hydroxymethyl-pyrid-2-yl, 5-hydroxymethyl-pyrid-2-yl, 6-hydroxymethyl- pyrid-2-yl, 3-aminomethyl-pyrid-2-yl, 4-aminomethyl-pyrid-2-yl,
5- aminomethyl-pyrid-2-yl, 6-aminomethyl-pyrid-2-yl, 3-hydroxy-pyrid-2-yl, 4-hydroxy-pyrid-2-yl, 5-hydroxy-pyrid-2-yl, 6-hydroxy-pyrid-2-yl, 3-methoxy- pyrid-2-yl, 4-methoxy-pyrid-2-yl, 5-methoxy-pyrid-2-yl, 6-methoxy-pyrid-2-yl,
3- difluoromethoxy-pyrid-2-yl, 4-difluoromethoxy-pyrid-2-yl, 5-difluoromethoxy- pyrid-2-yl, 6-difluoromethoxy-pyrid-2-yl, 3-trifluoromethoxy-pyrid-2-yl,
4- trifluoromethoxy-pyrid-2-yl, 5-trifluoromethoxy-pyrid-2-yl, 6-trifluoromethoxy- pyrid-2-yl, 3-methylthio-pyrid-2-yl, 4-methylthio-pyrid-2-yl, 5-methylthio-pyrid-
2- yl, 6-methylthio-pyrid-2-yl, 3-carboxamide-pyrid-2-yl, 4-carboxamide-pyrid-2- yl, 5- carboxamide-pyrid-2-yl, 6- carboxamide-pyrid-2-yl and 3-fluoro-6- methyl-pyrid-2-yl.
In some embodiments of compounds of Formula I, II, III, IV(a), IV(b), V(a) or V(b), R7 is selected from pyrid-3-yl, 2-fluoro-pyrid-3-yl, 4-fluoro-pyrid-3- yl, 5-fluoro-pyrid-3-yl, 6-fluoro-pyrid-3-yl, 2-chloro-pyrid-3-yl, 4-chloro-pyrid-3- yl, 5-chloro-pyrid-3-yl, 6-chloro-pyrid-3-yl, 2-cyano-pyrid-3-yl, 4-cyano-pyrid-3- yl, 5-cyano-pyrid-3-yl, 6-cyano-pyrid-3-yl, 2-methyl-pyrid-3-yl, 4-methyl-pyrid-
3- yl, 5-methyl-pyrid-3-yl, 6-methyl-pyrid-3-yl, 2-difluoromethyl-pyrid-3-yl,
4- difluoromethyl-pyrid-3-yl, 5-difluoromethyl-pyrid-3-yl, 6-difluoromethyl-pyrid- 3-yl, 2-trifluoromethyl-pyrid-3-yl, 4-trifluoromethyl-pyrid-3-yl, 5-trifluoromethyl- pyrid-3-yl, 6-trifluoromethyl-pyrid-3-yl, 2-hydroxymethyl-pyrid-3-yl,
4- hydroxymethyl-pyrid-3-yl, 5-hydroxymethyl-pyrid-3-yl, 6-hydroxymethyl- pyrid-3-yl, 2-aminomethyl-pyrid-3-yl, 4-aminomethyl-pyrid-3-yl,
5- aminomethyl-pyrid-3-yl, 6-aminomethyl-pyrid-3-yl, 2-hydroxy-pyrid-3-yl, 4-hydroxy-pyrid-3-yl, 5-hydroxy-pyrid-3-yl, 6-hydroxy-pyrid-3-yl, 2-methoxy- pyrid-3-yl, 4-methoxy-pyrid-3-yl, 5-methoxy-pyrid-3-yl, 6-methoxy-pyrid-3-yl,
2- difluoromethoxy-pyrid-3-yl, 4-difluoromethoxy-pyrid-3-yl, 5-difluoromethoxy- pyrid-3-yl , 6-d if luoromethoxy-pyrid-3-yl , 2-trif luoromethoxy-pyrid-3-yl ,
4-trifluoromethoxy-pyrid-3-yl, 5-trifluoromethoxy-pynd-3-yl, 6-trifluoronnethoxy- pyrid-3-yl, 2-methylthio-pyrid-3-yl, 4-methylthio-pyrid-3-yl, 5-methylthio-pyrid-
3- yl, 6-methylthio-pyrid-3-yl, 2-carboxamide-pyrid-3-yl, 4-carboxamide-pyrid-3- yl, 5- carboxamide-pyrid-3-yl and 6- carboxamide-pyrid-3-yl.
In some embodiments of compounds of Formula I, II, 11(a), 11(b), 11(c), 11(d), 11(e), 11(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is selected from a bond, - (CH2)p-, -(CH2)PC(O)(CH2)q-, -(CH2)PO(CH2)q-, -(CH2)pS(CH2)q-,
-(CH2)pNRd(CH2)q-, -(CH2)pC(O)O(CH2)q-, -(CH2)pOC(O)(CH2)q-,
-(CH2)pNRdC(O)(CH2)q-, -(CH2)pC(O)NRd(CH2)q-, -(CH2)pNRdC(O)NRd(CH2)q-, -(CH2)pNRdSO2(CH2)q-, and -(CH2)pSO2NRd(CH2)q-.
In some embodiments of compounds of Formula I, II, 11(a), 11(b), 11(c), 11(d), 11(e), 11(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is a bond.
In some embodiments of compounds of Formula I, II, 11(a), 11(b), 11(c), 11(d), 11(e), 11(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is -O-.
In some embodiments of compounds of Formula I, II, 11(a), 11(b), 11(c), 11(d), 11(e), 11(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is selected from -CH2O- and -OCH2-.
In some embodiments of compounds of Formula I, II, 11(a), 11(b), 11(c), 11(d), 11(e), 11(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is -NRd-.
In some embodiments of compounds of Formula I, II, 11(a), 11(b), 11(c),
11(d), 11(e), 11(f), 111(a), 111(b), l l l(c), 111(d), l l l(e), 111(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is selected from -CH2NRd- and -NRdCH2-.
In some embodiments of compounds of Formula I, II, 11(a), 11(b), 11(c), 11(d), 11(e), 11(f), l l l(a), l l l(b), l l l(c), 111(d), l l l(e), 111(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is sleeted from -NRdC(O)- and -C(O)NRd-.
In some embodiments of compounds of Formula I, II, 11(a), 11(b), 11(c), 11(d), 11(e), 11(f), l l l(a), l l l(b), l l l(c), 111(d), l l l(e), 111(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is sleeted from - CH2NRdC(O)- and -C(O)NRdCH2-.
In some embodiments of compounds of Formula I, II, 11(a), 11(b), 11(c), 11(d), 11(e), 11(f), l l l(a), l l l(b), l l l(c), 111(d), l l l(e), 111(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C-6-10 aryl, 5-10 membered heteroaryl and NRbRc, wherein each of the C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa,
(CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra,
(CH2)nNRdC(S)ORa, (CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc,
(CH2)nNRdS(O)Ra, (CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa,
(CH2)nC(S)NRbRc, (CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra,
(CH2)nSO2Ra, (CH2)nSO2NRbRc, Ci-6 alkyl, d-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl,
(CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i. 6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, 11(a), 11(b), 11(c), 11(d), 11(e), 11(f), 111(a), 111(b), lll(c), 111(d), lll(e), 111(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is phenyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc,
(CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra,
(CH2)nSO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is phenyl substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH2)nORa,
(CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc,
(CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra,
(CH2)nSO2NRbRc, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl groups is optionally
substituted with 1 , 2, 3, 4 or 5 Rf substituents; wherein at least one
substitutent is bonded at the meta position.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), 11(d), 11(e), 11(f), 111(a), lll(b), lll(c), 111(d), lll(e), 111(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is phenyl substituted with a substituent selected from (CH2)nC(O)ORa and (CH2)nC(O)NRbRc; and optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa,
(CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra,
(CH2)nNRdC(S)ORa, (CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc,
(CH2)nNRdS(O)Ra, (CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa,
(CH2)nC(S)NRbRc, (CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra,
(CH2)nSO2Ra, (CH2)nSO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl,
(CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i. 6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is phenyl substituted with a substituent selected from C(O)OH, C(O)NH2, C(O)OCi-6 alkyl, C(O)NHCi-6 alkyl and C(O)N(Ci-6 alkyl)2; and optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, C-i-6 alkyl and C-i-6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is phenyl substituted at the meta position with a substituent selected from (CH2)nC(O)ORa and
(CH2)nC(O)NRbRc; and optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra,
(CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa, (CH2)nNRdC(S)NRbRc,
(CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra, (CH2)nNRdSO2Ra,
(CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc, (CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra, (CH2)nSO2NRbRc, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8
membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and
(CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, ll(a), l l(b), l l(c), l l(d), l l(e), ll(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l ll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is phenyl substituted at the meta position with a substituent selected from (CH2)nC(O)ORa and
(CH2)nC(O)NRbRc, and optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, hydroxyl, CN, C-i-6 alkyl and C-i-6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), l l(b), l l(c), l l(d), l l(e), ll(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l ll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is phenyl substituted at the meta position with a substituent selected from C(O)OH, C(O)NH2, C(O)OCi-6 alkyl, C(O)NHCi-6 alkyl and C(O)N(Ci-6 alkyl)2; and optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, hydroxyl, CN, C-i-6 alkyl and C-i-6 haloalkyl.
In some embodiments of compounds of Formula I, II, ll(a), l l(b), l l(c), l l(d), l l(e), ll(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l ll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is phenyl substituted with (CH2)nNRdC(O)Ra, wherein Ra is C -6 alkyl or 3-8 membered heterocycloalkyl, each optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa,
(CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra,
(CH2)nNRdC(S)ORa, (CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc,
(CH2)nNRdS(O)Ra, (CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa,
(CH2)nC(S)NRbRc, (CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra,
(CH2)nSO2Ra, (CH2)nSO2NRbRc, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl,
(CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl; and optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, CN, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa,
(CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa, (CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc,
(CH2)nNRdS(O)Ra, (CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa,
(CH2)nC(S)NRbRc, (CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra,
(CH2)nSO2Ra, (CH2)nSO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl,
(CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3- 8 membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is phenyl substituted with (CH2)nNRdC(O)Ra, wherein Ra is selected from C -6 alkyl, C -6 alkyl-OH and Ci-6 alkyl-NH2, each optionally substituted with 1 , 2 or 3 additional
substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdSO2Ra,
(CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra, (CH2)nSO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8
membered heterocycloalkyl, (CH2)nphenyl, and (CH2)n5-10 membered heteroaryl; and optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, CN, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra,
(CH2)nNRdC(S)ORa, (CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc,
(CH2)nNRdS(O)Ra, (CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa,
(CH2)nC(S)NRbRc, (CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra,
(CH2)nSO2Ra, (CH2)nSO2NRbRc, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl,
(CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3- 8 membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from 3- benzamide, N-methyl-3-benzamide, N,N-dimethyl-3-benzamide, 4-fluoro-3- benzamide, N-methyl-4-fluoro-3-benzamide, N,N-dimethyl-4-fluoro-3- benzamide, 3-benzoic acid, methyl-3-benzoate, 4-fluoro-3-benzoic acid, and methyl-4-fluoro-3-benzoate.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is 5-10 membered heteroaryl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra, (CH2)nSO2NRbRc, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl, triazyl, furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with 1 , 2, 3 or 4 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra,
(CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa, (CH2)nNRdC(S)NRbRc,
(CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra, (CH2)nNRdSO2Ra,
(CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc, (CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra, (CH2)nSO2NRbRc, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8
membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and
(CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl, triazyl, furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with a substituent selected from
(CH2)nC(O)ORa and (CH2)nC(O)NRbRc; and optionally substituted with 1, 2 or 3 additional substituents selected from halogen, CN, oxo, (CH2)nORa,
(CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc,
(CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra,
(CH2)nSO2NRbRc, C-6 alkyl, C-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl and triazyl, each optionally substituted with (CH2)nC(O)NRbRc.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with (CH2)nC(O)NRbRc.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl and triazyl, each optionally substituted with (CH2)nC(O)NH2.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c),
11(d), 11(e), 11(f), 111(a), 111(b), l l l(c), 111(d), l l l(e), 111(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with (CH2)nC(O)NH2.
In some embodiments of compounds of Formula I, II, 11(a), 11(b), 11(c), 11(d), 11(e), 11(f), l l l(a), l l l(b), l l l(c), 111(d), l l l(e), 111(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl, triazyl, furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with (CH2)nNRdC(O)Ra, wherein Ra is C-i-6 alkyl or 3-8 membered heterocycloalkyl, each optionally substituted with 1 , 2 or 3 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc,
(CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra,
(CH2)nSO2NRbRc, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl,
(CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl, wherein each of the C1-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8
membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), ll(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl and triazyl, each optionally substituted with (CH2)nNRdC(O)Ra, wherein Ra is selected from Ci-6 alkyl, Ci-6 alkyl-OH and C-i-6 alkyl-NH2, each optionally substituted with 1 , 2 or 3 substituents selected
from halogen, CN, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa,
(CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc,
(CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra, (CH2)nSO2NRbRc, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl,
(CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with (CH2)nNRdC(O)Ra, wherein Ra is selected from C -6 alkyl, C -6 alkyl-OH and C-i-6 alkyl-NH2, each optionally substituted with 1 , 2 or 3 substituents selected from halogen, CN, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa,
(CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc,
(CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra, (CH2)nSO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl,
(CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from indolyl, indazolyl, benzimidazolyl, benzoxazolyl and benzoisoxazolyl, each optionally substituted with 1 , 2, 3 or 4 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc,
(CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra,
(CH2)nSO2NRbRc, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl,
(CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i- 6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from 1 H-indazol- 6-yl, 1 H-indazol-5-yl, 1 H-indazol-4-yl, 3-amino(1 H-indazol-5-yl), 3-amino(1 H- indazol-6-yl), 3-amino(1 H-indazol-7-yl), 1 -methyl(1 H-indazol-6-yl), 3- methyl(1 H-indazol-6-yl), 3-amino-1 -methyl(1 H-indazol-5-yl), 3-cyano(1 H- indazol-5-yl), 3-carboxamide(1 H-indazol-5-yl), 3-carboxamidine(1 H-indazol-5- yl), 3-vinyl(1 H-indazol-5-yl), 3-ethyl(1 H-indazol-5-yl), 3-acetamide(1 H-indazol- 5-yl), 3-methylsulfonylamine(1 H-indazol-5-yl), 3-methoxycarboxamide(1 H- indazol-5-yl), 3-methylamino(1 H-indazol-5-yl), 3-dimethylamino(1 H-indazol-5- yl), 3-ethylamino(1 H-indazol-5-yl), 3-(2-aminoethyl)amino(1 H-indazol-5-yl), 3- (2-hydroxyethyl)amino(1 H-indazol-5-yl), 3-[(methylethyl)amino](1 H-indazol-5- yl), 6-benzimidazol-5-yl, 6-(2-methylbenzimidazol-5-yl), 2-aminobenzimidazol- 5-yl, 2-hydroxybenzimidazol-5-yl, 2-acetamidebenzimidazol-5-yl, 3- aminobenzo[3,4-d]isoxazol-5-yl, 3-aminobenzo[d]isoxazol-6-yl, 3- aminobenzo[d]isoxazol-7-yl, 2-methylbenzoxazol-5-yl and 2- methylbenzoxazol-6-yl.
In some embodiments of compounds of Formula I, II, l l(a), l l(b), l l(c), l l(d), l l(e), l l(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l l l(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from 3-6 membered heterocycloalkyl and 3-6 membered heterocycloalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa,
(CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra,
(CH2)nSO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, ll(a), l l(b), l l(c), l l(d), l l(e), ll(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l ll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is selected from aziridinyl, azetidinyl, pyrrolidinyl, piperidinyl, piperazinyl and morpholinyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc,
(CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra,
(CH2)nSO2NRbRc, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, ll(a), l l(b), l l(c), l l(d), l l(e), ll(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l ll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is NRbRc, wherein Rb and Rc are as defined herein.
In some embodiments of compounds of Formula I, II, ll(a), l l(b), l l(c), l l(d), l l(e), ll(f), l l l(a), l l l(b), l l l(c), l l l(d), l l l(e), l ll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R2 is NRbRc, wherein one of Rb
and Rc is hydrogen and the other is C-i-6 alkyl optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is -C(O)- and R2 is NRbRc, wherein Rb and Rc are as defined herein.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is -C(O)- and R2 is NRbRc, wherein one of Rb and Rc is hydrogen and the other is C-i-6 alkyl optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is -(CH2)P- and R2 is NRbRc, wherein Rb and Rc are as defined herein.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), X is -(CH2)P- and R2 is NRbRc, wherein one of Rb and Rc is hydrogen and the other is C-i-6 alkyl optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R1, at each occurrence, is independently selected from hydrogen, halogen, CN, C-i-6 alkyl, C-i-6 haloalkyl, C(O)ORa, C(O)NRbRc, ORa, NRbRc, C6-io aryl and 5-10 membered heteroaryl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R1, at each occurrence, is independently selected from hydrogen, halogen, CN, C-i-6 alkyl, C-i-6 haloalkyl, hydroxyl, C-i-6 alkoxy, NH2, NHC-i-6 alkyl, and N(Ci-6 alkyl)2.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d),
IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R1, at each occurrence, is independently selected from hydrogen, halogen, CN, CF3 and methyl.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R1, at each occurrence, is hydrogen.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R4 is selected from hydrogen, C1-6 alkyl, C1-6 haloalkyl, C(O)Ra, C(O)ORa, C(O)NRbRc and SO2Ra.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R4 is hydrogen.
In some embodiments of compounds of Formula I, II, ll(a), ll(b), ll(c), ll(d), ll(e), ll(f), lll(a), lll(b), lll(c), lll(d), lll(e), lll(f), IV(a), IV(b), IV(c), IV(d), IV(e), IV(f), V(a), V(b), V(c), V(d), V(e) or V(f), R4 is hydrogen and each R1 is hydrogen.
In some embodiments of compounds of Formula I, II, ll(b), ll(d), ll(f), lll(b), lll(d), lll(f), IV(b), IV(d), IV(f), V(b), V(d) or V(f), R8 and R9, at each occurrence, are each independently selected from hydrogen, halogen and C-i- 6 alkyl.
In some embodiments of compounds of Formula I, II, ll(b), ll(d), ll(f), lll(b), lll(d), lll(f), IV(b), IV(d), IV(f), V(b), V(d) or V(f), R8 and R9, at each occurrence, are each hydrogen.
In some embodiments, the compound is selected from the compounds in Table 2, or a pharmaceutically acceptable salt thereof.
The compounds and compositions described and/or disclosed herein modulate the contractility of the skeletal sarcomere. Specifically, the compounds modulate the troponin complex of the fast skeletal muscle sarcomere through one or more of fast skeletal myosin, actin, tropomyosin, troponin C, troponin I, and troponin T, and fragments and isoforms thereof.
As used in this context, "modulate" means either increasing or decreasing activity. In some instances, the compounds described and/or disclosed herein potentiate (i.e., increase activity) of one or more of fast skeletal myosin, actin, tropomyosin, troponin C, troponin I, and troponin T, and fragments and isoforms thereof. In other instances, the compounds described and/or disclosed herein inhibit (i.e., decrease activity) of one or more of fast skeletal myosin, actin, tropomyosin, troponin C, troponin I, and troponin T, and fragments and isoforms thereof.
In both preclinical and clinical settings, activators of the fast skeletal troponin complex have been shown to amplify the response of fast skeletal muscle to nerve stimulation, resulting in an increase in muscle force development at sub-maximal muscle activation (see, e.g., Russell et al., "The Fast Skeletal Troponin Activator, CK-2017357, Increases Skeletal Muscle Force in vitro and in situ", 2009 Experimental Biology Conference, New Orleans, LA, April 2009). Activators of the fast skeletal troponin complex have been shown to increase the sensitivity of skinned skeletal muscle fibers to calcium, and in living muscle to the frequency of
stimulation, each of which results in an increase in muscle force
development at sub-maximal muscle activation. Such activators have also been shown to reduce muscle fatigue and/or to increase the overall time to fatigue in normal and low oxygenated conditions (see, e.g., Russell et al., "The Fast Skeletal Troponin Activator, CK-2017357, Increases Skeletal Muscle Force and Reduces Muscle Fatigue in vitro and in situ", 5th Cachexia Conference, Barcelona, Spain, December 2009; Hinken et al., "The Fast Skeletal Troponin Activator, CK-2017357, Reduces Muscle Fatigue in an in situ Model of Vascular Insufficiency", Society for Vascular Medicine's 2010 Annual Meeting: 21 st Annual Scientific Sessions, Cleveland, OH, April 2010). The increase in muscle force in response to nerve input has been
demonstrated in healthy human volunteers as well (see, e.g., Hansen et al., "CK-2017357, a Novel Activator of Fast Skeletal Muscle, Increases Isometric Force Evoked by Electrical Stimulation of the Anterior Tibialis Muscle in Healthy Male Subjects", Society for Neuroscience 40th Annual Meeting:
Neuroscience 2010, November 2010). Work in additional preclinical models of muscle function suggests that activators of the fast skeletal troponin
complex also cause an increase in muscle power and/or endurance. These pharmacological properties suggest this mechanism of action could have application in conditions, for example, where neuromuscular function is impaired.
Provided are methods for enhancing fast skeletal muscle efficiency in a patient in need thereof, comprising administering to said patient an effective amount of a compound or composition described and/or disclosed herein that selectively binds the troponin complex of fast skeletal muscle fiber or sarcomere. In some embodiments, the compound disclosed and/or described herein activates fast skeletal muscle fibers or sarcomeres. In some embodiments, administration of a compound disclosed and/or described herein results in an increase in fast skeletal muscle power output. In some embodiments, administration of a compound disclosed and/or described herein results in increased sensitivity of fast skeletal muscle fibers or sarcomeres to calcium ion, as compared to fast skeletal muscle fibers or sarcomeres untreated with the compound. In some embodiments,
administration of a compound disclosed and/or described herein results in a lower concentration of calcium ions causing fast skeletal muscle myosin to bind to actin. In some embodiments, administration of a compound disclosed and/or described herein results in the fast skeletal muscle fiber generating force to a greater extent at submaximal levels of muscle activation.
Also provided is a method for sensitizing a fast skeletal muscle fiber to produce force in response to a lower concentration of calcium ion, comprising contacting the fast skeletal muscle fiber with a compound or composition described and/or disclosed herein that selectively binds to troponin complexes in the fast skeletal muscle sarcomere. In some embodiments, contacting the fast skeletal muscle fiber with the compound results in activation of the fast skeletal muscle fiber at a lower calcium ion concentration than in an untreated fast skeletal muscle fiber. In some embodiments, contacting the fast skeletal muscle fiber with the compound results in the production of increased force at a lower calcium ion concentration in comparison with an untreated fast skeletal muscle fiber.
Also provided is a method for increasing time to fast skeletal muscle fatigue in a patient in need thereof, comprising contacting fast skeletal muscle
fibers with a compound or composition described and/or disclosed herein that selectively binds to the troponin complexes of the fast skeletal muscle fibers. In some embodiments, the compound binds to form ligand-troponin-calcium ion complexes that activate the fast skeletal muscle fibers. In some
embodiments, formation of the complexes and/or activation of the fast skeletal muscle fibers results in enhanced force and/or increased time to fatigue as compared to untreated fast skeletal muscle fibers contacted with a similar calcium ion concentration.
The compounds and pharmaceutical compositions described and/or disclosed herein are capable of modulating the contractility of the fast skeletal sarcomere in vivo, and can have application in both human and animal disease. Modulation would be desirable in a number of conditions or diseases, including, but not limited to, 1 ) neuromuscular disorders, such as Amyotrophic Lateral Sclerosis (ALS), Spinal Muscular Atrophy (SMA), peripheral neuropathies and myasthenia gravis; 2) disorders of voluntary muscle, including muscular dystrophies, myopathies and conditions of muscle wasting, such as sarcopenia and cachexia syndromes (e.g., cachexia syndromes caused by diseases such as cancer, heart failure, chronic obstructive pulmonary disease (COPD), and chronic kidney disease/dialysis), and rehabilitation-related deficits, such as those associated with recovery from surgery (e.g. post-surgical muscle weakness) prolonged bed rest or stroke rehabilitation; 3) central nervous system (CNS) disorders in which muscle weakness, atrophy and fatigue are prominent symptoms, such as multiple sclerosis, Parkinson's disease, stroke and spinal cord injury; and 4) muscle symptoms stemming from systemic disorders, including Peripheral Vascular Disease (PVD) or Peripheral Arterial Disease (PAD) (e.g.,
claudication), metabolic syndrome, chronic fatigue syndrome, obesity and frailty due to aging.
The compounds and compositions described and/or disclosed herein may be used to treat neuromuscular diseases, i.e., diseases that affect any part of the nerve-muscle unit. Neuromuscular diseases include, for example: 1 ) diseases of the motor unit, including but not limited to Amyotrophic Lateral Sclerosis (ALS) including bulbar and primary lateral sclerosis (PLS) variants; spinal muscular atrophy types 1 -4; Kennedy syndrome; post-polio syndrome;
motor neuropathies including, for example, critical illness polyneuropathy; multifocal motor neuropathy with conduction block; Charcot-Marie-Tooth disease and other hereditary motor and sensory neuropathies; and Guillain- Barre Syndrome, 2) disorders of the neuromuscular junction, including myasthenia gravis, Lambert-Eaton myasthenic syndrome, and prolonged neuromuscular blockade due to drugs or toxins; and 3) peripheral
neuropathies, such as acute inflammatory demyelinating
polyradiculoneuropathy, diabetic neuropathy, chronic inflammatory demyelinating polyradiculoneuropathy, traumatic peripheral nerve lesions, neuropathy of leprosy,vasculitic neuropathy, dermatomyositis/polymyositis and neuropathy of Friedreich Ataxia.
The compounds and compositions described and/or disclosed herein may be used to treat disorders of voluntary muscle. Disorders of voluntary muscle include 1 ) muscular dystrophies (including, for example, Duchenne, Becker, Limb-Girdle, Facioscapulohumeral, limb girdle, Emery-Dreyfus, oculopharyngeal and congenital muscular dystrophies); and 2) myopathies, such as nemaline myopathy, central core disease, congenital myopathies, mitochondrial myopathies, acute myopathy; inflammatory myopathies (such as dermatomyositis/polymyositis and inclusion body myositis), endocrine myopathies (such as those associated with hyper- or hypothyroidism), Cushing's or Addison's syndrome or disease and pituitary gland disorders, metabolic myopathies (such as glycogen storage diseases, e.g., McArdle's disease, Pompe disese, etc), drug-induced myopathy (statins, ant-retroviral drugs, steroid myopathy) restrictive lung disease, sarcoidosis, Schwartz- Jampel Syndrome, focal muscular atrophies, and distal myopathies.
The compounds and compositions described and/or disclosed herein may be used to treat or Amyotrophic Lateral Sclerosis (ALS). ALS is a disease that generally arises later in life (Age 50+) and has a rapid progression from initial limb weakness to paralysis and death. Common life expectancy after diagnosis is 3-5 years. The cause of disease for most ALS patients is unknown (termed the spontaneous form) while a small proportion of patients have an inherited form (familial) of disease. The condition causes progressive death of motor neurons through causes that are not clear.
Surviving motor units attempt to compensate for dying ones by innervating
more fibers (termed sprouting) but this can only partially correct muscle function, as muscles are subsequently more prone to problems of
coordination and fatigue. Eventually, surviving motor neurons die, resulting in complete paralysis of the affected muscle. The disease is commonly fatal through the eventual loss of innervation to the diaphragm, resulting in respiratory failure. Current treatment options for ALS are limited.
The compounds and compositions described and/or disclosed herein may be used to treat Spinal Muscular Atrophy (SMA). SMA is a genetic disorder that arises through the mutation of a protein, SMN1 , that appears to be required for the survival and health of motor neurons. The disease is most common in children as the majority of patients only survive until 1 1 -12 years of age. There is currently no available treatment for SMA.
The compounds and compositions described and/or disclosed herein may be used to treat myasthenia gravis. Myasthenia gravis is a chronic autoimmune neuromuscular disease wherein the body produces antibodies that block, alter, or destroy proteins involved in signaling at the neuromuscular junction, thus preventing muscle contraction from occurring. These proteins include nicotinic acetylcholine receptor (AChR) or, less frequently, a muscle- specific tyrosine kinase (MuSK) involved in AChR clustering (see, e.g., Drachman, N. Eng. J. of Med., 330:1797-1810, 1994). The disease is characterized by varying degrees of weakness of the skeletal (voluntary) muscles of the body. The hallmark of myasthenia gravis is muscle weakness that increases during periods of activity and improves after periods of rest. Although myasthenia gravis may affect any voluntary muscle, certain muscles, such as those that control eye and eyelid movement, facial expression, chewing, talking, and swallowing are often, but not always, involved in the disorder. The muscles that control breathing and neck and limb movements may also be affected. In most cases, the first noticeable symptom is weakness of the eye muscles. In others, difficulty in swallowing and slurred speech may be the first signs. The degree of muscle weakness involved in myasthenia gravis varies greatly among patients, ranging from a localized form, limited to eye muscles (ocular myasthenia), to a severe or generalized form in which many muscles - sometimes including those that control breathing - are affected. Symptoms, which vary in type and severity,
may include a drooping of one or both eyelids (ptosis), blurred or double vision (diplopia) due to weakness of the muscles that control eye movements, unstable or waddling gait, weakness in arms, hands, fingers, legs, and neck, a change in facial expression, difficulty in swallowing and shortness of breath, and impaired speech (dysarthria). Generalized weakness develops in approximately 85% of patients.
The compounds and compositions described and/or disclosed herein may be used to treat sarcopenia, e.g., sarcopenia associated with aging or disease (e.g. HIV infection). Sarcopenia is characterized by a loss of skeletal muscle mass, quality, and strength. Clinically, a decline in skeletal muscle tissue mass (muscle atrophy) contributes to frailty in older individuals. In human males, muscle mass declines by one-third between the ages of 50 and 80. In older adults, extended hospitalization can result in further disuse atrophy leading to a potential loss of the ability for independent living and to a cascade of physical decline. Moreover, the physical aging process profoundly affects body composition, including significant reductions in lean body mass and increases in central adiposity. The changes in overall adiposity and fat distribution appear to be important factors in many common age-related diseases such as hypertension, glucose intolerance and diabetes,
dyslipidemia, and atherosclerotic cardiovascular disease. In addition, it is possible that the age-associated decrement in muscle mass, and
subsequently in strength and endurance, may be a critical determinant for functional loss, dependence and disability. Muscle weakness is also a major factor prediposing the elderly to falls and the resulting morbidity and mortality.
The compounds and compositions described and/or disclosed herein may be used to treat cachexia. Cachexia is a state often associated with cancer or other serious diseases or conditions, (e.g, chronic obstructive pulmonary disease, heart failure, chronic kidney disease, kidney dialysis), that is characterized by progressive weight loss, muscle atrophy and fatigue, due to the deletion of adipose tissue and skeletal muscle.
The compounds and compositions described and/or disclosed herein may be used to treat muscular dystrophies. Muscular dystrophy can be characterized by progressive muscle weakness, destruction and regeneration of the muscle fibers, and eventual replacement of the muscle fibers by fibrous
and fatty connective tissue.
The compounds and compositions described and/or disclosed herein may be used to treat post-surgical muscle weakness, which is a reduction in the strength of one or more muscles following surgical procedure. Weakness may be generalized (i.e. total body weakness) or localized to a specific area, side of the body, limb, or muscle.
The compounds and compositions described and/or disclosed herein may be used to treat post-traumatic muscle weakness, which is a reduction in the strength of one or more muscles following a traumatic episode (e.g. bodily injury). Weakness may be generalized (i.e. total body weakness) or localized to a specific area, side of the body, limb, or muscle.
The compounds and compositions described and/or disclosed herein may be used to treat muscle weakness and fatigue produced by peripheral vascular disease (PVD) or peripheral artery disease (PAD). Peripheral vascular disease is a disease or disorder of the circulatory system outside of the brain and heart. Peripheral artery disease (PAD), also known as peripheral artery occlusive disease (PAOD), is a form of PVD in which there is partial or total blockage of an artery, usually one leading to a leg or arm. PVD and/or PAD can result from, for example, atherosclerosis, inflammatory processes leading to stenosis, embolus/ thrombus formation, or damage to blood vessels due to disease (e.g., diabetes), infection or injury. PVD and/or PAD can cause either acute or chronic ischemia, typically of the legs. The symptoms of PVD and/or PAD include pain, weakness, numbness, or cramping in muscles due to decreased blood flow (claudication), muscle pain, ache, cramp, numbness or fatigue that occurs during exercise and is relieved by a short period of rest (intermittent claudication), pain while resting (rest pain) and biological tissue loss (gangrene). The symptoms of PVD and/or PAD often occur in calf muscles, but symptoms may also be observed in other muscles such as the thigh or hip. Risk factors for PVD and/or PAD include age, obesity, sedentary lifestyle, smoking, diabetes, high blood pressure, and high cholesterol (i.e., high LDL, and/or high triglycerides and/or low HDL). People who have coronary heart disease or a history of heart attack or stroke generally also have an increased frequency of having PVD and/or PAD.
Activators of the fast skeletal troponin complex have been shown to reduce
muscle fatigue and/or to increase the overall time to fatigue in in vitro and in situ models of vascular insufficiency (see, e.g., Russell et al., "The Fast Skeletal Troponin Activator, CK-2017357, Increases Skeletal Muscle Force and Reduces Muscle Fatigue in vitro and in situ", 5th Cachexia Conference, Barcelona, Spain, December 2009; Hinken et al., "The Fast Skeletal Troponin Activator, CK-2017357, Reduces Muscle Fatigue in an in situ Model of Vascular Insufficiency", Society for Vascular Medicine's 2010 Annual Meeting: 21 st Annual Scientific Sessions, Cleveland, OH, April 2010).
The compounds and compositions described and/or disclosed herein may be used to treat symptoms of frailty, e.g., frailty associated with aging. Frailty is characterized by one or more of unintentional weight loss, muscle weakness, slow walking speed, exhaustion, and low physical activity.
The compounds and compositions described and/or disclosed herein may be used to treat muscle weakness and/or fatigue due to wasting syndrome, which is a condition characterized by involuntary weight loss associated with chronic fever and diarrhea. In some instances, patients with wasting syndrome lose 10% of baseline body weight within one month.
The compounds and compositions described and/or disclosed herein may be used to treat muscular diseases and conditions caused by structural and/or functional abnormalities of skeletal muscle tissue, including muscular dystrophies, congenital muscular dystrophies, congenital myopathies, distal myopathies, other myopathies (e.g., myofibrillar, inclusion body), myotonic syndromes, ion channel muscle diseases, malignant hyperthermias, metabolic myopathies, congenital myasthenic syndromes, sarcopenia, muscle atrophy and cachexia.
The compounds and compositions described and/or disclosed herein also may be used to treat diseases and conditions caused by muscle dysfunction originating from neuronal dysfunction or transmission, including amyotrophic lateral sclerosis, spinal muscular atrophies, hereditary ataxias, hereditary motor and sensory neuropathies, hereditary paraplegias, stroke, multiple sclerosis, brain injuries with motor deficits, spinal cord injuries, Alzheimer's disease, Parkinson's disease with motor deficits, myasthenia gravis and Lambert-Eaton syndrome.
The compounds and compositions described and/or disclosed herein
also may be used to treat diseases and conditions caused by CNS, spinal cord or muscle dysfunction originating from endocrine and/or metabolic dysregulation, including claudication secondary to peripheral artery disease, hypothyroidism, hyper- or hypo-parathyroid ism, diabetes, adrenal dysfunction, pituitary dysfunction and acid/base imbalances.
The compounds and compositions described and/or disclosed herein may be administered alone or in combination with other therapies and/or therapeutic agents useful in the treatment of the aforementioned disorders.
The compounds and compositions described and/or disclosed herein may be combined with one or more other therapies to treat ALS. Examples of suitable therapies include riluzole, baclofen, diazepam, trihexyphenidyl and amitriptyline. In some embodiments, the compounds and compositions described and/or disclosed herein are combined with riluzole to treat a subject suffering from ALS.
The compounds and compositions described and/or disclosed herein may be combined with one or more other therapies to treat myasthenia gravis. Examples of suitable therapies include administration of anticholinesterase agents (e.g., neostigmine, pyridostigmine), which help improve neuromuscular transmission and increase muscle strength; administration of
immunosuppressive drugs (e.g., prednisone, cyclosporine, azathioprine, mycophenolate mofetil) which improve muscle strength by suppressing the production of abnormal antibodies; thymectomy (i.e., the surgical removal of the thymus gland, which often is abnormal in myasthenia gravis patients); plasmapheresis; and intravenous immune globulin.
The compounds and compositions described and/or disclosed herein may be combined with one or more other therapies to treat PVD or PAD (e.g., claudication). Treatment of PVD and PAD is generally directed to increasing arterial blood flow, such as by smoking cessation, controlling blood pressure, controlling diabetes, and exercising. Treatment can also include medication, such as medicines to help improve walking distance (e.g., cilostazol, pentoxifylline), antiplatelet agents (e.g., aspirin, ticlopidine, clopidogrel), anticoagulents (e.g., heparin, low molecular weight heparin, warfarin, enoxaparin) throbmolytics, antihypertensive agents (e.g., diuretics, ACE inhibitors, calcium channel blockers, beta blockers, angiotensin II receptor
antagonists), and cholesterol-lowering agents (e.g., statins). In some patients, angioplasty, stenting, or surgery (e.g., bypass surgery or surgery to remove an atherosclerotic plaque) may be necessary.
Suitable therapeutic agents include, for example, anti-obesity agents, anti-sarcopenia agents, anti-wasting syndrome agents, anti-frailty agents, anti-cachexia agents, anti-muscle spasm agents, agents against post-surgical and post-traumatic muscle weakness, and anti-neuromuscular disease agents.
Suitable additional therapeutic agents include, for example: orlistat, sibramine, diethylpropion, phentermine, benzaphetamine, phendimetrazine, estrogen, estradiol, levonorgestrel, norethindrone acetate, estradiol valerate, ethinyl estradiol, norgestimate, conjugated estrogens, esterified estrogens, medroxyprogesterone acetate, testosterone, insulin-derived growth factor, human growth hormone, riluzole, cannabidiol, prednisone, albuterol, nonsteroidal anti-inflammatory drugs, and botulinum toxin.
Other suitable therapeutic agents include TRH, diethylstilbesterol, theophylline, enkephalins, E series prostaglandins, compounds disclosed in U.S. Patent No. 3,239,345 (e.g., zeranol), compounds disclosed in U.S.
Patent No. 4,036,979 (e.g., sulbenox), peptides disclosed in U.S. Patent No. 4,41 1 ,890, growth hormone secretagogues such as GHRP-6, GHRP-1 (disclosed in U.S. Patent No. 4,41 1 ,890 and publications WO 89/071 10 and WO 89/071 1 1 ), GHRP-2 (disclosed in WO 93/04081 ), NN703 (Novo Nordisk), LY44471 1 (Lilly), MK-677 (Merck), CP424391 (Pfizer) and B-HT920, growth hormone releasing factor and its analogs, growth hormone and its analogs and somatomedins including IGF-1 and IGF-2, alpha-adrenergic agonists, such as clonidine or serotonin 5-HTD agonists, such as sumatriptan, agents which inhibit somatostatin or its release, such as physostigmine,
pyridostigmine, parathyroid hormone, PTH(1 -34), and bisphosphonates, such as MK-217 (alendronate).
Still other suitable therapeutic agents include estrogen, testosterone, selective estrogen receptor modulators, such as tamoxifen or raloxifene, other androgen receptor modulators, such as those disclosed in Edwards, J. P. et. al., Bio. Med. Chem. Let., 9, 1003-1008 (1999) and Hamann, L. G. et. al., J. Med. Chem., 42, 210-212 (1999), and progesterone receptor agonists
("PRA"), such as levonorgestrel, medroxyprogesterone acetate (MPA).
Other suitable therapeutic agents include anabolic agents, such as selective androgen receptor modulators (SARMs); antagonists of the activin receptor pathway, such as anti-myostatin antibodies or soluble activin receptor decoys, including ACE-031 (Acceleron Pharmaceuticals, a soluble activin receptor type MB antagonist), MYO-027/PFE-3446879 (Wyeth/Pfizer, an antibody myostatin inhibitor), AMG-745 (Amgen, a peptibody myostatin inhibitor), and an ActRIIB decoy receptor (see Zhou et al., Cell, 142, 531-543, August 20, 2010); and anabolic steroids.
Still other suitable therapeutic agents include aP2 inhibitors, such as those disclosed in U.S. Patent No. 6,548,529, PPAR gamma antagonists, PPAR delta agonists, beta 3 adrenergic agonists, such as AJ9677
(Takeda/Dainippon), L750355 (Merck), or CP331648 (Pfizer), other beta 3 agonists as disclosed in U.S. Patent Nos. 5,541 ,204, 5,770,615, 5,491 ,134, 5,776,983 and 5,488,064, a lipase inhibitor, such as orlistat or ATL-962 (Alizyme), a serotonin (and dopamine) reuptake inhibitor, such as
sibutramine, topiramate (Johnson & Johnson) or axokine (Regeneron), a thyroid receptor beta drug, such as a thyroid receptor ligand as disclosed in WO 97/21993, WO 99/00353, and GB98/284425, and anorectic agents, such as dexamphetamine, phentermine, phenylpropanolamine or mazindol.
Still other suitable therapeutic agents include HIV and AIDS therapies, such as indinavir sulfate, saquinavir, saquinavir mesylate, ritonavir,
lamivudine, zidovudine, lamivudine/zidovudine combinations, zalcitabine, didanosine, stavudine, and megestrol acetate.
Still other suitable therapeutic agents include antiresorptive agents, hormone replacement therapies, vitamin D analogues, elemental calcium and calcium supplements, cathepsin K inhibitors, MMP inhibitors, vitronectin receptor antagonists, Src SH.sub.2 antagonists, vacuolar H+-ATPase inhibitors, ipriflavone, fluoride, Tibo lone, pro stanoids, 17-beta hydroxysteroid dehydrogenase inhibitors and Src kinase inhibitors.
The above therapeutic agents, when employed in combination with the compounds and compositions disclosed and/or described herein, may be used, for example, in those amounts indicated in the Physicians' Desk
Reference (PDR) or as otherwise determined by one of ordinary skill in the
art.
The compounds and compositions disclosed and/or described herein are administered at a therapeutically effective dosage, e.g., a dosage sufficient to provide treatment for the disease state. While human dosage levels have yet to be optimized for the chemical entities described herein, generally, a daily dose ranges from about 0.05 to 100 mg/kg of body weight; in some embodiments, from about 0.10 to 10.0 mg/kg of body weight, and in some embodiments, from about 0.15 to 1 .0 mg/kg of body weight. Thus, for administration to a 70 kg person, in some embodiments, the dosage range would be about from 3.5 to 7000 mg per day; in some embodiments, about from 7.0 to 700.0 mg per day, and in some embodiments, about from 10.0 to 100.0 mg per day. The amount of the chemical entity administered will be dependent, for example, on the subject and disease state being treated, the severity of the affliction, the manner and schedule of administration and the judgment of the prescribing physician. For example, an exemplary dosage range for oral administration is from about 70 mg to about 700 mg per day, and an exemplary intravenous administration dosage is from about 70 mg to about 700 mg per day, each depending upon the compound
pharmacokinetics.
Administration of the compounds and compositions disclosed and/or described herein can be via any accepted mode of administration for therapeutic agents including, but not limited to, oral, sublingual,
subcutaneous, parenteral, intravenous, intranasal, topical, transdermal, intraperitoneal, intramuscular, intrapulmonary, vaginal, rectal, or intraocular administration. In some embodiments, the compound or composition is administered orally or intravenously. In some embodiments, the compound or composition disclosed and/or described herein is administered orally.
Pharmaceutically acceptable compositions include solid, semi-solid, liquid and aerosol dosage forms, such as tablet, capsule, powder, liquid, suspension, suppository, and aerosol forms. The compounds disclosed and/or described herein can also be administered in sustained or controlled release dosage forms (e.g., controlled/sustained release pill, depot injection, osmotic pump, or transdermal (including electrotransport) patch forms) for prolonged timed, and/or pulsed administration at a predetermined rate. In
some embodiments, the compositions are provided in unit dosage forms suitable for single administration of a precise dose.
The compounds disclosed and/or described herein can be
administered either alone or in combination with one or more conventional pharmaceutical carriers or excipients (e.g., mannitol, lactose, starch, magnesium stearate, sodium saccharine, talcum, cellulose, sodium
crosscarmellose, glucose, gelatin, sucrose, magnesium carbonate). If desired, the pharmaceutical composition can also contain minor amounts of nontoxic auxiliary substances such as wetting agents, emulsifying agents, solubilizing agents, pH buffering agents and the like (e.g., sodium acetate, sodium citrate, cyclodextrine derivatives, sorbitan monolaurate,
triethanolamine acetate, triethanolamine oleate). Generally, depending on the intended mode of administration, the pharmaceutical composition will contain about 0.005% to 95%, or about 0.5% to 50%, by weight of a compound disclosed and/or described herein. Actual methods of preparing such dosage forms are known, or will be apparent, to those skilled in this art; for example, see Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, Pennsylvania.
In some embodiments, the compositions will take the form of a pill or tablet and thus the composition may contain, along with a compounds disclosed and/or described herein, one or more of a diluent (e.g., lactose, sucrose, dicalcium phosphate), a lubricant (e.g., magnesium stearate), and/or a binder (e.g., starch, gum acacia, polyvinylpyrrolidine, gelatin, cellulose, cellulose derivatives). Other solid dosage forms include a powder, marume, solution or suspension (e.g., in propylene carbonate, vegetable oils or triglycerides) encapsulated in a gelatin capsule.
Liquid pharmaceutically administrable compositions can, for example, be prepared by dissolving, dispersing or suspending etc. a compound disclosed and/or described herein and optional pharmaceutical additives in a carrier (e.g., water, saline, aqueous dextrose, glycerol, glycols, ethanol or the like) to form a solution or suspension. Injectables can be prepared in conventional forms, either as liquid solutions or suspensions, as emulsions, or in solid forms suitable for dissolution or suspension in liquid prior to injection. The percentage of the compound contained in such parenteral compositions
depends, for example, on the physical nature of the compound, the activity of the compound and the needs of the subject. However, percentages of active ingredient of 0.01 % to 10% in solution are employable, and may be higher if the composition is a solid which will be subsequently diluted to another concentration. In some embodiments, the composition will comprise from about 0.2 to 2% of a compound disclosed and/or described herein in solution.
Pharmaceutical compositions of the compounds disclosed and/or described herein may also be administered to the respiratory tract as an aerosol or solution for a nebulizer, or as a microfine powder for insufflation, alone or in combination with an inert carrier such as lactose. In such a case, the particles of the pharmaceutical composition may have diameters of less than 50 microns, or in some embodiments, less than 10 microns.
In addition, pharmaceutical compositions can include a compound disclosed and/or described herein and one or more additional medicinal agents, pharmaceutical agents, adjuvants, and the like. Suitable medicinal and pharmaceutical agents include those described herein.
The following examples serve to more fully describe the invention described herein. It is understood that these examples in no way serve to limit the true scope of this invention, but rather are presented for illustrative purposes.
Example 1 : -2-(4-Fluorophenyl)propan-1 -amine

(S)-4-Benzyl-3-(2-(4-fluorophenyl)acetyl)oxazolidin-2-one. To a cooled (-78 °C) solution of (S)-4-benzyloxazolidin-2-one (10 g, 58 mmol, 1 .0 equiv) in 100 ml_ THF was added dropwise n-BuLi (40 ml_, 1 .6 M in hexanes, 64 mmol, 1 .1 equiv). After stirring for 30 minutes, 4-fluorophenylacetyl chloride (10 g, 0.58 mmol, 1 .0 equiv) was added dropwise. After stirring for an additional 30 minutes, the reaction mixture was allowed to warm to room temperature. The reaction was quenched with saturated aq. NH CI, extracted with dichloromethane, and washed with brine. The organic layer was then
dried over sodium sulfate, filtered, and concentrated in vacuo. Purification by silica gel (10 - 20% EtOAc/hexanes) provided the title compound as a thick oil (14.7 g,

(S)-4-Benzyl-3-((S)-2-(4-fluorophenyl)propanoyl)oxazolidin-2-one.
To a room-temperature solution of (S)-4-Benzyl-3-(2-(4- fluorophenyl)acetyl)oxazolidin-2-one (5.1 g, 16.3 mmol, 1 .0 equiv) in dry THF (100 mL) was added iodomethane (1 .0 mL, 16.2 mmol, 1 .0 equiv) by syringe. The resulting mixture was cooled to -78 °C, and NaHMDS (8.15 mL, 2M in THF, 16.3 mmol, 1 .0 equiv) was added dropwise by syringe. After stirring for 15 minutes at -78 °C, the reaction mixture was allowed to warm to room temperature. The reaction was quenched with saturated, aq. NH4CI, and diluted with EtOAc. The organic layer was washed with brine, dried over Na2SO , filtered and concentrated in vacuo. Purification by silica gel chromatography (7 - 20% EtOAc/hexanes) provided the title compound (2.6 g, 49%).

(S)-2-(4-Fluorophenyl)propan-1 -ol. To a room-temperature solution of (S)-4-benzyl-3-((S)-2-(4-fluorophenyl) propanoyl)oxazolidin-2-one (1 .8 g, 5.5 mmol, 1 .0 equiv) in THF (18 mL) was added a solution of NaBH4 (1 .0 g, 26.4 mmol, 4.8 equiv) in water (6.0 mL). The reaction mixture was stirred for 3 h at room temperature and then quenched by the careful addition of aq. 1 M HCI. The reaction mixture was diluted with water and ethyl acetate. The layers were separated and the organic layer was subsequently washed with brine, dried over Na2SO , filtered and concentrated in vacuo. Purification by silica gel chromatography (10 - 75% EtOAc/Hexanes) provided the title compound (0.824 g, 97%).

(S)-2-(2-(4-Fluorophenyl)propyl)isoindoline-1 ,3-dione. To a solution of (S)-2-(4-fluorophenyl)propan-1 -ol (0.82 g, 5.35 mmol, 1 .0 equiv), phthalimide (0.82 g, 5.6 mmol, 1 .05 equiv), and triphenyl phosphine (2.1 g, 8.03 mmol, 1 .5 equiv) in dry THF (18 mL) was added dropwise
diethylazodicarboxylate (3.6 mL, 15% in toluene, 8.0 mmol, 1 .5 equiv). The reaction mixture was stirred over 72 h and then concentrated in vacuo.
Purification by silica gel chromatography (15 - 25% EtOAc/Hexanes) provided the title compound (0.9 g, 59%).

(S)-2-(4-Fluorophenyl)propan-1 -amine. To a room-temperature solution of (S)-2-(2-(4-fluorophenyl)propyl)isoindoline-1 ,3-dione (900 mg, 3.2 mmol, 1 .0 equiv) in toluene (14 mL) was added hydrazine hydrate (1 .4 mL, 45 mmol, 14 equiv) by syringe. The resulting mixture was heated to 80 °C for 30 minutes and then cooled to room temperature. The resulting solution was decanted from the solid in the reaction mixture, and the solid was washed with additional toluene. The combined organic layers were combined and concentrated in vacuo to provide the title compound (491 mg, 99%), which was used without further purification.
Example 2: 2- 4-Fluorophenyl)-2-methylpropan-1 -amine

To a solution of 4-fluorophenylacetonitrile (50 g, 370 mmol, 1 .0 equiv) and iodomethane (70 mL, 1 .1 mol, 3 equiv) in THF (370 mL) was added KOf- Bu (124 g, 1 .1 mol, 3 equiv) as a solid in portions such that the reaction mixture did not exceed 50 °C. The reaction mixture was stirred overnight and
then quenched by the addition of brine. The mixture was diluted with EtOAc and washed twice with brine. The organic layer was dried over Na2SO , filtered, and concentrated in vacuo to provide 2-(4-fluorophenyl)-2- methylpropanenitrile as a yellow oil (57 g, 94%), which was used without further purification in the next step. To a solution of the nitrile in dry THF (800 mL) was added a solution of lithium aluminum hydride (210 mL, 2 M in ether, 420 mmol, 1 .2 equiv). After the mixture was heated at reflux overnight, the reaction was allowed to cool to room temperature, and a Fieser and Fieser work-up (300 uL water/mmol, 1 .0 mL 3N NaOH/mmol, 300 uL water/mmol) was performed. Filtration of the resulting solids provided the title compound as an orange oil (57 g, 92%).
Example : (1 -(4-Fluorophenyl)cyclobutyl)methanamine
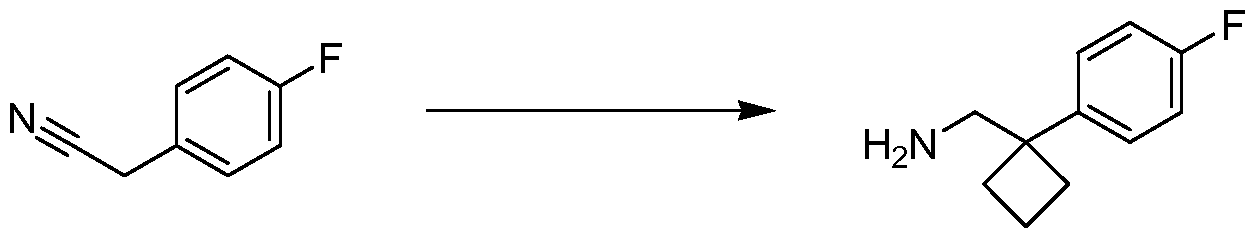
A solution of 4-fluorophenylacetonitrile (6.7 g, 75 mmol, 1 .5 equiv), 1 ,3- dibromopropane (10 mL, 50 mmol, 1 equiv), KOH (27 g, 150 mmol, 3.0 equiv), and tetrabutylammonium bromide (100 mg) in toluene (135 mL) was heated to 100 °C for 3 hours. The organic layer was separated and
concentrated to dryness. Silica gel chromatography using a gradient of 0 - 30% EtOAc/hexanes resulted in partially purified product which was further purified by Kugelrohr distillation at 200 °C to provide 3.76 g (22 mmol) of the intermediate nitrile product as an oil. The residue was dissolved in dry THF (22 mL) and treated with a solution of lithium aluminum hydride (27 mL, 2 M in ether, 55 mmol, 2.5 equiv). The mixture was stirred at 0 °C for 2 hours followed by a Fieser and Fieser work-up (38 uL water/mmol, 1 18 uL 3N NaOH/mmol, 38 uL water/mmol). The organic layer was concentrated to dryness to provide the desired product (3.6 g, 40% overall) as a yellow oil.
Example 4: (1 -(6-Methoxypyridin-2-yl)cyclobutyl)methanamine

2-(3-Fluoropyridin-2-yl)acetonitrile. To a 0 °C solution of 2-chloro-3- fluoropyhdine (3.0 g, 23 mmol, 1 .0 equiv) and acetonitrile (1 .3 ml_, 25 mmol, 1 .1 equiv) in toluene (50 ml_) was added sodium hexamethyldisilazide
(NaHMDS) (2.0 M in THF, 13 ml_, 25 mmol, 1 .1 equiv). The resulting mixture was stirred for 2 hours at 0 °C and then partitioned between EtOAc and water. The aqueous layer was extracted with EtOAc and the combined organic phases were washed with saturated NaCI, dried over Na2SO4 and
concentrated in vacuo to provide the crude desired product as an oil which was used without further purification.

1 -(6-Fluoropyridin-2-yl)cyclobutanecarbonitrile. Following the same procedure as above for 2-(3-fluoropyridin-2-yl)acetonitrile with 2,6- difluoropyridine (5.0 g, 43 mmol, 1 .0 equiv), cyclobutylcarbonitrile (3.5 g, 43 mmol, 1 .0 equiv) and NaHMDS (2.0 M in THF, 24 ml_, 47 mmol, 1 .1 equiv) in toluene (100 ml_) gave the desired product (4.9 g, 64%) as a colorless oil following puri eluent.

1 -(6-Methoxypyridin-2-yl)cyclobutanecarbonitrile. To stirred 6.0 ml_ of anhydrous methanol at 0 °C under nitrogen was added sodium metal (~1 g) and the mixture stirred for 30 minutes. To this was added 1 -(6-fluoropyridin-2- yl)cyclobutanecarbonitrile (1 .6 g, 9.1 mmol, 1 .0 equiv) and the resulting mixture heated to 75 °C for 45 minutes. The solution was cooled to room temperature and partitioned between water and EtOAc. The layers were separated, the aqueous phase was extracted with EtOAc, and the combined
organic phases were washed with saturated NaCI, dried over Na2SO and concentrated in vacuo to give the desired product (1 .7 g, 97%) as a colorless oil.

(1 -(6-Methoxypyridin-2-yl)cyclobutyl)methanamine. To a stirred solution of 1 -(6-methoxypyhdin-2-yl)cyclobutanecarbonithle (1 .7 g, 8.8 mmol, 1 .0 equiv) in THF (20 mL) was added lithium aluminum hydride solution (1 .0 M in THF, 1 1 mL, 1 1 mmol, 1 .1 equiv). The mixture was refluxed for 1 .5 hours and allowed to cool to room temperature. Water (0.43 mL) was added slowly followed by 0.43 mL of 3 M NaOH and then three additions of 0.43 mL of water (Fieser and Fieser workup). The resulting mixture was filtered through diatomaceous earth and rinsed with THF. The combined organics were dried over Na2SO4 and concentrated to dryness to give the desired product (1 .6 g, 97%) as a viscous oil.
Example -(3-Fluoropyridin-2-yl)cyclobutanami

1 -(3-Fluoropyridin-2-yl)cyclobutanecarboxamide. To a 250 mL round bottom flask containing DMSO (60 mL), 1 -(3-fluoropyridin-2- yl)cyclobutanecarbonitrile (2.96 g, 16.8 mmol, 1 .0 equiv) was added and the mixture was stirred until homogenous. Potassium carbonate (7.0 g, 50.4 mmol, 3.0 equiv) was then added and the reaction mixture was cooled to 0 °C, followed by the addition of 35% hydrogen peroxide (6.5 mL). The reaction was stirred at 0 °C for 30 min and then warmed to room temperature. At this time, the reaction was diluted with water (50 mL) and ethyl acetate (100 mL). After transferring to a separatory funnel and shaking, the organic layer was separated from the aqueous layer and then washed with brine (3 x 50 mL). The organic layer was then dried over Na2SO , filtered, and concentrated to give a crude solid that was purified by silica gel chromatography (10%
EtOAC/hexanes) to afford 1 .92 g (59%) of 1 -(3-fluoropyridin-2- yl)cyclobutanecarboxamide as a white solid.

Methyl 1 -(3-fluoropyridin-2-yl)cyclobutylcarbamate. 1 -(3- fluoropyridin-2-yl)cyclobutanecarboxannide (1 .92 g, 9.88 mmol, 1 .0 equiv) was dissolved in methanol (20 ml_) and potassium hydroxide (1 .1 1 g, 19.8 mmol, 2.0 equiv) was added. The mixture was sonicated until homogeneous, followed by the addition of iodosobenzene diacetate (4.77 g, 14.8 mmol, 1 .5 equiv). The reaction was stirred for 20 min and then diluted with water (100 ml_) and ethyl acetate (125 ml_). After transferring to a separatory funnel and shaking, the organic layer was separated from the aqueous layer, and the aqueous layer was extracted with EtOAc (50 ml_). The combined organic layers were then dried over Na2SO4, filtered, and concentrated to give a crude oil that was purified by silica gel chromatography (40% EtOAC/hexanes) to afford 1 .47 g (67%) of methyl 1 -(3-fluoropyridin-2-yl)cyclobutylcarbamate as a white soli

1 -(3-Fluoropyridin-2-yl)cyclobutanamine. To a 20 ml_ microwave reaction vial was added methyl 1 -(3-fluoropyridin-2-yl)cyclobutylcarbamate (1 .47 g, 6.56 mmol, 1 .0 equiv), ethanol (12 ml_) and 3N aqueous sodium hydroxide (7 ml_). The reaction mixture was heated in the microwave reactor at 150 °C for 30 min. The ethanol was evaporated under reduced pressure and the mixture was extracted with ethyl acetate (30 ml_). The aqueous layer was then extracted with ethyl acetate (2 x 30 ml_). The organic layers were combined, dried over Na2SO , filtered, and concentrated to give 1 -(3- fluoropyridin-2-yl)cyclobutanamine (1 .01 g, 93%) as a crude yellow oil that was used without further purification.
Example 6: N-(2-(4-Fluorophenyl)-2-methylpropyl)-6-(2-methylprop-1 - enyl)pyridin-2 -amine

6-Bromo-N-(2-(4-fluorophenyl)-2-methylpropyl)pyridin-2-amine. To a 5 mL microwave reaction vial was added 2,6-dibromopyridine (1 .25 g, 5.0 mmol, 1 .0 equiv), 2-(4-fluorophenyl)-2-methylpropan-1 -amine (1 .06 g, 6 mmol, 1 .2 equiv), DIPEA (1 .9 mL, 1 1 .0 mmol, 2.2 equiv), and acetonitrile (5 mL). The vial was sealed and heated in an oil bath at 137 °C for 48 h,
concentrated, and purified by silica gel column chromatography to give 220 mg (14%) of 6-bromo-N-(2-(4-fluorophenyl)-2-methylpropyl)pyridin-2-amine as a white lid (m/z [M+H] = 323).

N-(2-(4-Fluorophenyl)-2-methylpropyl)-6-(2-methylprop-1 - enyl)pyridin-2-amine. To a 5 mL microwave reaction vessel was added 6- bromo-N-(2-(4-fluorophenyl)-2-methylpropyl)pyridin-2-amine (220 mg, 0.7 mmol, 1 .0 equiv), 2-methylprop-1 -enylboronic acid (100 mg, 1 mmol, 1 .5 equiv), CI2Pd(dppf) (50 mg, 70 μιτιοΙ, 0.1 equiv), potassium carbonate (300 mg, 2.1 mmol, 3.0 equiv), dioxane (4 mL), and water (1 mL). The reaction was heated in a microwave reactor at 135 °C for 35 min and then diluted with saturated sodium bicarbonate (20 mL) and extracted with ethyl acetate (50 mL). The organic layer was then dried over Na2SO , filtered, and
concentrated to give a crude solid that was purified by reverse phase column chromatography to afford 14 mg of N-(2-(4-fluorophenyl)-2-methylpropyl)-6-(2- methylprop-1 -enyl)pyridin-2-amine (m/z [M+H] = 299.4).
Example 7: -Phenyl-N-(1 -phenylcyclobutyl)pyridin-2-amine

4-Bromo-N-(1 -phenylcyclobutyl)pyridin-2-amine. To a 20 dram vial was added 4-bromo-2-chloropyridine (250 mg, 5.0 mmol, 1 .0 equiv), 1 - phenylcyclobutanamine (1 .0 g, 7 mmol, 1 .4 equiv), and NMP (1 ml_). The reaction was heated to 120 °C, stirred for 72 h, and then diluted with water (20 ml_) and ethyl acetate (50 ml_). After transferring to a separatory funnel and shaking, the organic layer was separated from the aqueous layer. The organic layer was then dried over Na2SO , filtered, and concentrated to give a crude solid that was purified by silica gel column chromatography, affording 139 mg (9%) of 4-bromo-N-(1 -phenylcyclobutyl)pyridin-2-amine as an orange solid.

4-Phenyl-N-(1 -phenylcyclobutyl)pyridin-2-amine. To a 3 ml_ microwave reaction vessel was added 4-bromo-N-(1 -phenylcyclobutyl)pyridin- 2-amine (50 mg, 165 μιτιοΙ, 1 .0 equiv), phenylboronic acid (30 mg, 248 μιτιοΙ, 1 .5 equiv), Cl2Pd(dppf) (12 mg, 16 μιτιοΙ, 0.1 equiv), potassium carbonate (57 mg, 413 μιτιοΙ, 2.5 equiv), dioxane (750 μΙ_), and water (250 μΙ_). The reaction was heated in a microwave reactor at 140 °C for 20 min and then diluted with saturated sodium bicarbonate (20 ml_) and extracted with ethyl acetate (50 ml_). The organic layer was then dried over Na2SO4, filtered, and
concentrated to give a crude solid that was purified by reverse phase column chromatography to afford 9 mg of 4-phenyl-N-(1 -phenylcyclobutyl)pyridin-2- amine (m/z [M+H] = 301 .2).
Example -Phenyl-N-(1 -phenylcyclobutyl)pyridin-2 -amine

5-Bromo-N-(1 -phenylcyclobutyl)pyridin-2-amine. To a 20 dram vial was added 5-bromo-2-fluoropyridine (250 mg, 5.0 mmol, 1 .0 equiv), 1 - phenylcyclobutanamine (1 .0 g, 7 mmol, 1 .4 equiv), and NMP (1 mL). The reaction was heated to 120 °C, stirred for 72 h, and then diluted with water (20 mL) and extracted with ethyl acetate (50 mL). The organic layer was then dried over Na2SO , filtered, and concentrated to give a crude solid that was purified by silica gel column chromatography to give 102 mg (26%) of 5- bromo-N-(1 -phenylcyclobutyl)pyridin-2-amine as a pale yellow solid.

5-Phenyl-N-(1 -phenylcyclobutyl)pyridin-2-amine. To a 3 mL microwave reaction vessel was added 5-bromo-N-(1 -phenylcyclobutyl)pyridin- 2-amine (102 mg, 340 μιτιοΙ, 1 .0 equiv), phenylboronic acid (59 mg, 505 μιτιοΙ, 1 .5 equiv), Cl2Pd(dppf) (25 mg, 34 μιτιοΙ, 0.1 equiv), potassium carbonate (1 16 mg, 842 μιτιοΙ, 2.5 equiv), dioxane (1 mL) and water (500 μί). The reaction was heated in a microwave reactor at 140 °C for 20 min and then diluted with saturated sodium bicarbonate (20 mL) and extracted with ethyl acetate (50 mL). The organic layer was then dried over Na2SO4, filtered, and concentrated to give a crude solid that was purified by silica gel column chromatography (10% EtOAc/hexanes), affording 27 mg of 5-phenyl-N-(1 - phenylcyclobutyl)pyridin-2-amine as a white solid (m/z [M+H] = 301 .2).
Example 9: 1 -(6-(2-(4-Fluorophenyl)-2-methylpropylamino)pyridin-3- yl)ethane-1 ,2-diol

N-(2-(4-Fluorophenyl)-2-methylpropyl)-5-vinylpyridin-2-amine. To
a 5 mL microwave reaction vessel was added 5-bromo-N-(2-(4-fluorophenyl)- 2-methylpropyl)pyrimidin-2-amine (255 mg, 0.8 mmol, 1 .0 equiv), 2,4,6- trivinyl-1 ,3,5,2,4,6-trioxatriborinane (285 mg, 1 .2 mmol, 1 .5 equiv),
Cl2Pd(dppf) (58 mg, 79 μιτιοΙ, 0.1 equiv), potassium carbonate (1 .2 mL of a 2N aqueous solution, 2.4 mmol, 3.0 equiv), and dioxane (4 mL). The reaction was heated in a microwave reactor at 1 15 °C for 20 min. The aqueous layer was removed from the reaction, and the organic layer was directly purified by reverse phase column chromatography to give 158 mg (73%) of N-(2-(4- fluor henyl)-2-methylpropyl)-5-vinylpyridin-2-amine.

1 -(6-(2-(4-Fluorophenyl)-2-methylpropylamino)pyridin-3-yl)ethane- 1 ,2-diol. N-(2-(4-fluorophenyl)-2-methylpropyl)-5-vinylpyridin-2-amine (158 mg, 0.6 mmol, 1 .0 equiv) was added to a foil-covered 20 dram vial and then dissolved in a 50% THF/water mixture (6 mL). Osmium tetraoxide (15 mg, 0.06 mmol, 0.1 equiv) and morpholine N-oxide (103 mg, 0.9 mmol, 1 .5 equiv) were added, and the reaction was stirred for 1 h. Additional THF (3 mL) was added, and the reaction was stirred for 3 h. The reaction mixture was then diluted with water (10 mL) and extracted with ethyl acetate (30 mL). The organic layer was dried over Na2SO4, filtered, and concentrated to give a crude solid that was purified by reverse phase column chromatography to give 129 mg (86%) of 1 -(6-(2-(4-fluorophenyl)-2-methylpropylamino)pyridin-3- yl)ethane-1 ,2-diol (m/z [M+H] = 305).
Example 10: N-(2-(6-(2-(4-Fluorophenyl)-2-methylpropylamino)pyridin-3- yl)ethyl)-methanesulfonamide


ferf-Butyl-5-(2-aminoethyl)pyridin-2-yl(2-(4-fluorophenyl)-2- methylpropyl)-carbamate. Methanesulfonyl chloride (87 μί, 1 .1 mmol, 1 .1 equiv) dissolved in 2 mL ethyl acetate was added to a 20 dram vial. To this stirring mixture was added a mixture of te/t-butyl-2-(4-fluorophenyl)-2- methylpropyl(5-(2-hydroxyethyl)pyridin-2-yl)-carbamate (414 mg, 1 .1 mmol, 1 .0 equiv), TMEDA (167 μί, 1 .1 mmol, 1 .1 equiv), and ethyl acetate (3 mL) in a dropwise manner. The reaction was stirred for 1 h. The reaction mixture was then filtered and washed with saturated sodium bicarbonate and brine. The organic layer was dried over Na2SO , filtered, and concentrated. The resultant solid was dissolved in DMF (4 mL), followed by the addition of sodium azide (139 mg, 2.1 mmol, 2.0 equiv). The reaction was stirred for 24 h, then diluted with water (10 mL) and extracted with ethyl acetate (20 mL). The organic layer was dried over Na2SO4, filtered, and concentrated. The crude solid was dissolved in methanol, followed by the addition of 10% palladium on carbon (80 mg), potassium carbonate (100 mg). The reaction was stirred under 45 psi of hydrogen for 24 h, filtered, and concentrated to give terf-butyl 5-(2-aminoethyl)pyridin-2-yl(2-(4-fluorophenyl)-2-
methylpropyl)carbamate.

N-(2-(6-(2-(4-Fluorophenyl)-2-methylpropylamino)pyridin-3- yl)ethyl)-methanesulfonamide. To a 20 dram vial was added terf-butyl 5-(2- aminoethyl)pyridin-2-yl(2-(4-fluorophenyl)-2-methylpropyl)carbamate (140 mg, 0.34 mmol, 1 .0 equiv), TEA (87 μΙ_, 0.67 mmol, 2.0 equiv, THF (3 mL), and methanesulfonyl chloride (29 μΙ_, 0.37 mmol, 1 .1 equiv). The reaction was stirred for 2h and then concentrated, followed by the addition of MeOH (1 mL) and 4N HCI/dioxane (3 mL). The reaction was stirred for 1 h, concentrated, and then purified by preparative TLC (5% MeOH/CH2CI2) to give 5 mg of N-(2- (6-(2-(4-fluorophenyl)-2-methylpropylamino)pyridin-3-yl)ethyl)- methanesulfonamide (m/z [M+H] = 366). -(2-(4-Fluorophenyl)-2-methylpropylamino)nicotinamide

6-(2-(4-Fluorophenyl)-2-methylpropylamino)nicotinonitrile. To a solution of 6-chloronicotinonitrile (200 mg, 1 .46 mmol, 1 .0 equiv) and 2-(4- fluorophenyl)-2-methylpropan-1 -amine (290 mg, 1 .75 mmol, 1 .2 equiv) in isopropanol (3 mL) in a microwave vial equipped with a stir bar was added potassium carbonate (720 mg, 2.19 mmol, 1 .5 equiv). The vial was fitted with a microwave vial cap and heated to 120 °C for 20 min. The reaction mixture was filtered to remove the solid potassium carbonate and concentrated in vacuo. After the residue was redissolved in EtOAc and water, the organic layer was washed with brine, dried over sodium sulfate, and concentrated in vacuo. Purification by silica gel chromatography (1 % - 3% MeOH/DCM) provided the title compound as a beige solid (104 g, 27%), (m/z [M+H] = 270.1 ).

6-(2-(4-Fluorophenyl)-2-methylpropylamino)nicotinamide. To a cooled (0 °C) suspension of 6-(2-(4-fluorophenyl)-2- methylpropylamino)nicotinonitrile (75 mg, 0.28 mmol, 1 .0 equiv) and K2CO3 (50 mg, 0.36 mmol, 1 .3 equiv) in DMSO (1 mL) was added aq. H2O2 (250 uL, 30% by wt., 2.6 mmol, 9 equiv) by syringe. The reaction was allowed to warm to RT and stirred for 30 minutes. The resulting mixture was diluted with EtOAc and washed three times with satd. aq. LiCI and once with brine. The resulting solution was dried over sodium sulfate, filtered, and concentrated in vacuo. Purification by silica gel chromatography (2% - 10% MeOH/DCM) provided the title compound as an off-white foam (54 mg, 77%), (m/z [M+H] = 288.1 ).
Example 12: Methyl (6-(2-(4-fluorophenyl)-2-methylpropylamino)pyridin- 3-yl)methylcarbamate

5-(Aminomethyl)-N-(2-(4-fluorophenyl)-2-methylpropyl)pyridin-2- amine. To a 100 mL round bottom flask was added 6-(2-(4-fluorophenyl)-2- methylpropylamino)nicotinonitrile (1 .0 g, 3.7 mmol, 1 .0 equiv) and THF (15 mL), followed by the addition of 1 M lithium aluminum hydride/THF (14.8 mL, 14.8 mmol, 4.0 equiv). The reaction was heated to reflux for 3 h and allowed to cool to rt. The reaction mixture was then quenched by the sequential slow addition water (0.6 mL), 3N NaOH (0.6 mL), and water (1 .7 mL). The resultant precipitate was filtered and the filtrate was then dried (Na2SO4) and concentrated to give 1 .0 g (99%) of 5-(aminomethyl)-N-(2-(4-fluorophenyl)-2- methylpropyl)pyridin-2-amine.

Methyl (6-(2-(4-fluorophenyl)-2-methylpropylamino)pyridin-3- yl)methylcarbamate. To a 20 dram vial was added 5-(aminomethyl)-N-(2-(4- fluorophenyl)-2-methylpropyl)pyridin-2-amine (100 mg, 0.37 mmol, 1 .0 equiv), DIPEA (57 mg, 0.44 mmol, 1 .2 equiv) and CH2CI2 (1 .2 mL). The mixture was cooled to 0 °C, and methyl carbamate (35 mg, 0.4 mmol, 1 .0 equiv) was then added. The reaction was allowed to warm to rt and stirred for 2 h. The reaction mixture was then diluted with water (2 mL) and extracted with ethyl acetate (15 mL). The organic layer was dried over Na2SO , filtered, and concentrated to give a crude solid that was purified by silica gel column chromatography, affording 20 mg of methyl (6-(2-(4-fluorophenyl)-2- methylpropylamino)pyridin-3-yl)methylcarbamate (m/z [M+H] = 332).
Example 13: Preparation of 3-(6-(2-(4-fluorophenyl)-2- methylpropylamino)-1 ,2,4-triazin-3-yl)benzamide

Ethyl 2-(3-chlorobenzamido)acetate. To a stirred solution of 3- chlorobenzoyl chloride (6.0 mL, 58 mmol) in dichloromethane (150 mL) were added ethyl glycinate hydrochloride (10 g, 72 mmol) and triethylamine (20 mL, 142 mmol). After stirring for 1 h, the mixture was diluted with saturated NaHCO3 and the aqueous layer extracted twice with dichloromethane. The combined organic layers were dried over Na2SO4 and evaporated to dryness in vacuo to give the desired product (9.8 g) as a white solid which was taken on without purification.
 Ethyl 2-(3-chlorophenylthioamido)acetate. To a stirred solution of ethyl 2- (3-chlorobenzamido)acetate (9.8 g, 40 mmol) in toluene (150 mL) was added phosphorus pentasulfide (18 g, 42 mmol), and the mixture was heated to 120 °C for 1 h. The mixture was filtered, evaporated to dryness, and purified using silica gel chromatography (0 - 100 % EtOAc/hexanes) as eluent gave 7.0 g of crude product which was carried forward without further purification
Ethyl 2-(3-chlorophenylthioamido)acetate. To a stirred solution of ethyl 2- (3-chlorobenzamido)acetate (9.8 g, 40 mmol) in toluene (150 mL) was added phosphorus pentasulfide (18 g, 42 mmol), and the mixture was heated to 120 °C for 1 h. The mixture was filtered, evaporated to dryness, and purified using silica gel chromatography (0 - 100 % EtOAc/hexanes) as eluent gave 7.0 g of crude product which was carried forward without further purification

3-(3-Chlorophenyl)-4,5-dihydro-1,2,4-triazin-6(1 H)-one. To a solution of ethyl 2-(3-bromophenylthioamido)acetate (-40 mmol) in ethanol (100 mL) was added hydrazine hydrate (5 mL). The mixture was heated to 90 °C for 2 h and allowed to cool to room temperature. Concentration in vacuo gave a yellow solid that was purified over silica gel using 10% MeOH/DCM as eluent to give the desired product (7.0 g) as an off-white solid.

3-(3-Chlorophenyl)-N-(2-(4-fluorophenyl)-2-methylpropyl)-1 ,2,4- triazin-6-amine. To a solution of 3-(3-chlorophenyl)-4,5-dihydro-1 ,2,4-triazin- 6(1 H)-one (6.0 g, 29 mmol) dissolved in dioxane (75 mL) was added MnO2 (10 g, 120 mmol). The mixture was stirred at 90 °C for 18 h. After filtering through diatomaceous earth and rinsing with hot dioxane, the combined organics were evaporated to dryness in vacuo. The crude residue (1/3 was used) was dissolved in POCI3 (10 mL, 108 mmol) and heated to 70 °C for 2.5 h. The mixture was allowed to cool to rt and evaporated to dryness in vacuo. The residue was dissolved in EtOAc/ether (1 :1 ) and washed with 1 M NaOH. The organic phase was evaporated to dryness and dissolved in NMP (5.0 mL) in a microwave vial. Potassium carbonate (1 .0 g) was added followed by (2- (4-fluorophenyl)-2-methylpropan-1 -amine (2.0 g, 12 mmol), and the mixture was heated in a microwave reactor to 165 °C for 20 min. The solution was diluted with EtOAc and washed with saturated NaCI (4x), dried over Na2SO4,
and evaporated to dryness. Purification using silica gel chromatography (0 - 35 .

3-(6-(2-(4-Fluorophenyl)-2-methylpropylamino)-1 ,2,4-triazin-3- yl)benzonitrile. To a microwave vial containing NiBr2 (280 mg, 1 .3 mmol) and NaCN (120 mg, 2.5 mmol) was added a solution of 3-(3-chlorophenyl)-N- (2-(4-fluorophenyl)-2-methylpropyl)-1 ,2,4-triazin-6-amine (450 mg, 1 .3 mmol) in NMP (4.0 mL). The vial was purged with nitrogen and sealed. The mixture was heated to 200 °C in a microwave for 25 min and allowed to cool to room temperature. The solution was diluted with EtOAc/ether (1 :1 ), washed with water (3x), dried over Na2SO4 and evaporated to dryness. Purification using silica gel chromatography (0 - 65% EtOAc/hexanes) gave the desired product (220 mg) as an off white foam.

3-(6-(2-(4-fluorophenyl)-2-methylpropylamino)-1 ,2,4-triazin-3- yl)benzamide. To a solution of 3-(6-(2-(4-fluorophenyl)-2- methylpropylamino)-1 ,2,4-triazin-3-yl)benzonitrile (220 mg, 0.60 mmol) in DMSO (3.0 mL) was added K2CO3 (300 mg, 2.2 mmol) and 30% hydrogen peroxide (0.60 mL, 7.5 mmol). The mixture was stirred 24 h and partitioned between EtOAc and water. The aqueous layer was extracted with EtOAc (2x) and the combined organics were dried over Na2SO4 and evaporated to dryness in vacuo. Purification using silica gel chromatography (0 - 100% EtOAc/hexanes) gave the desired product (17 mg), m/z = 366.1 [M+H].
Example 14: Preparation of 5-(3-(2-(4-fluorophenyl)-2- methylpropylamino)-1, -6-yl)-1 H-indazol-3-amine
 3-Amino-1 ,2,4-triazin-2-oxide. To a solution of 1 ,2,4-triazin-3-annine (2.8 g, 30 mmol) in CH3CN (25 ml_) was added portionwise mCPBA (14 g, 81 mmol). The mixture was refluxed for 2 h and then evaporated to dryness. The residue was mixed with ether and filtered to give the desired product as a yellow solid that was used in the next step without purification.
3-Amino-1 ,2,4-triazin-2-oxide. To a solution of 1 ,2,4-triazin-3-annine (2.8 g, 30 mmol) in CH3CN (25 ml_) was added portionwise mCPBA (14 g, 81 mmol). The mixture was refluxed for 2 h and then evaporated to dryness. The residue was mixed with ether and filtered to give the desired product as a yellow solid that was used in the next step without purification.
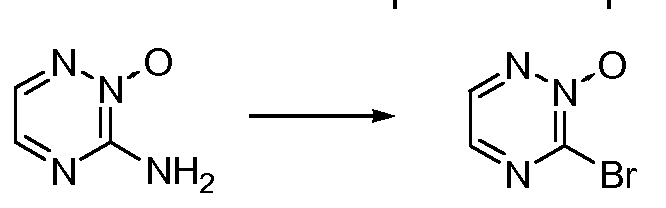
3-Bromo-1 ,2,4-triazin-2 -oxide. To a solution of 48% (wt/wt) HBr (100 ml_) was added 1 ,2,4-triazin-3-amine-2-oxide (30 mmol), and the mixture was stirred at rt. A solution of NaNO2 (30 g, 430 mmol) in water (40 ml_) was added slowly, and the mixture was stirred for 1 h. The reaction was quenched by the addition of an excess of saturated NaHCO3 and extracted with dichloromethane. The combined organic layers were evaporated to dryness. Purification using silica gel chromatography (EtOAc/hexanes) gave 1 .3 g of the intermediate bromide product.

N-(2-(4-Fluorophenyl)-2-methylpropyl)-1 ,2,4-triazin-3-amine-2- oxide. A solution of 3-bromo-1 ,2,4-triazin-2-oxide (2.0 g, 1 1 mmol), (2-(4- fluorophenyl)-2-methylpropan-1 -amine (3.0 g, 17 mmol), and
diisopropylethylamine (3.0 ml_, 17 mmol) in CH3CN (20 ml_) was stirred at rt for 24 h. The solvents were removed and the residue dissolved in EtOAc. The solution was washed with saturated NaHCO3, dried over Na2SO , and evaporated to dryness. The residue was mixed with ether and then filtered to give the desired product (1 .2 g) as a yellow solid.
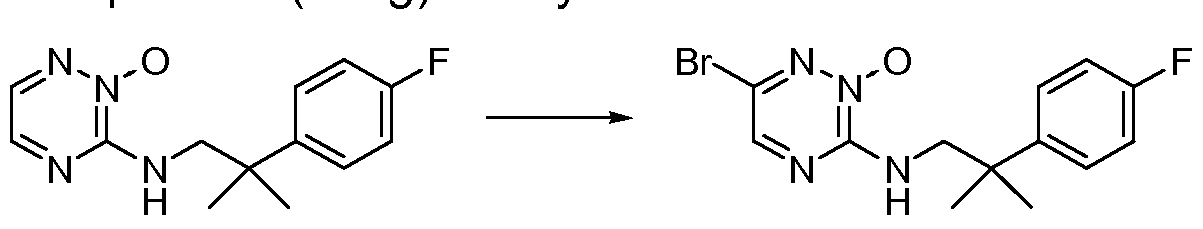
6-Bromo-N-(2-(4-fluorophenyl)-2-methylpropyl)-1 ,2,4-triazin-3- amine-2-oxide. To a solution of N-(2-(4-fluorophenyl)-2-methylpropyl)-1 ,2,4- triazin-3-amine-2-oxide (1 .2 g, 7.4 mmol) in a 1 :1 mixture of dichloromethane and CH3CN (40 ml_) was added Br2 (1 .0 ml_, 19 mmol). The mixture was stirred at room temperature for 72 h. The solvents were evaporated and the
residue was dissolved in ether. The solution was filtered through a plug of silica gel and the solvents evaporated to give the desired product (1 .5 g, 58%) as an orange sticky solid.
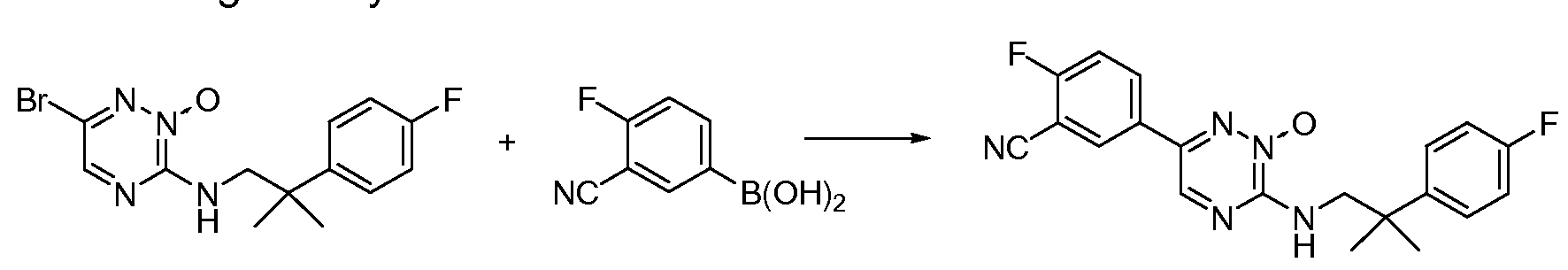
2-Fluoro-5-(3-(2-(4-fluorophenyl)-2-methylpropylamino)-1,2,4- triazin-2-oxide-6-yl)benzonitrile. To a solution of 6-bromo-N-(2-(4- fluorophenyl)-2-methylpropyl)-1 ,2,4-triazin-3-annine-2-oxide (0.48 g, 1 .4 mmol, 1 .0 equiv), 3-cyano-4-fluorophenylboronic acid (0.35 g, 2.1 mmol), and K2CO3 (0.5 g, 3.6 mmol) in DMF (2 ml_) was added Pd(dppf)CI2 (0.10 g, 0.13 mmol). The mixture was stirred at 75 °C for 4 h and then quenched by addition of excess saturated NaHCO3. The mixture was extracted with EtOAc and the organic layer was concentrated. Purification using silica gel chromatography ( - 40% EtOAc/hexanes) gave the desired product (0.38 g) as a yellow solid.

5-(3-(2-(4-fluorophenyl)-2-methylpropylamino)-1 ,2,4-triazin-6-yl)- 1 H-indazol-3-amine. A stirred solution of 2-fluoro-5-(3-(2-(4-fluorophenyl)-2- methylpropylamino)-1 ,2,4-triazin-2-oxide-6-yl)benzonitrile (0.18 g, 0.47 mmol) and hydrazine (0.1 ml_, 2.0 mmol) in n-butanol (0.5 ml_) was sealed and heated to 1 19 °C for 1 h. The solvents were removed and the residue dissolved in benzylamine with a minimal amount of CH3CN. The mixture was sealed and stirred at 177 °C overnight. The mixture was loaded directly onto a reverse phase HPLC for purification using a gradient of CH3CN/water to give the desired product (28 mg), m/z = 378.1 [M+H].
Example 15: Preparation of 4-fluoro-3-(3-(2-(3-fluoropyridin-2-yl)propan- 2-ylamino)-1 ,2,4-triazin-6-yl)benzamide

6-(5-Bromo-2-fluorophenyl)-1 ,2,4-triazin-3-amine. 6-Bromo-1 ,2,4- triazin-3-annine (9.1 g, 40.8 mmol), 5-bromo-2-fluorophenylboronic acid (98.9 g, 40.8 mmol), (dppf)PdCl2 (3.0 g, 4.0 mmol), nitrogen-sparged dioxane (81 .6 mL), and aq. 2 N K2CO3 (17 mL) were combined and heated in a round bottom flask at 90 °C for 4 h. The hot mixture was filtered through a pad of celite and diluted with EtOAc and washed with water. Concentration followed by triturati n with DCM afforded a tan solid (4.5 g), m/z = 269.0 [M+H].

3-Bromo-6-(5-bromo-2-fluorophenyl)-1,2,4-triazine. 6-(5-Bromo-2- fluorophenyl)-1 ,2,4-triazin-3-amine (4.5 g, 16.7 mmol) was dissolved in 25 mL of bromoform and heated to 80 °C. Isoamyl nitrite (9.8 g, 83.5 mmol) was then added and stirred at 85 °C for 1 h. The reaction mixture was then
concentrated and silica gel chromatography afforded a yellow solid (4.4 g), m/z = 331 .1 [M+H].

6-(5-Bromo-2-fluorophenyl)-N-(2-(2-fluorophenyl)propan-2-yl)- 1 ,2,4-triazin-3-amine. 3-Bromo-6-(5-bromo-2-fluorophenyl)-1 ,2,4-triazine (2.8 g, 8.4 mmol), 2-(2-fluorophenyl)propan-2-amine (2.0 g, 12.6 mmol), K2CO3 (2.3 g, 16.8 mmol) and CH3CN (20 mL) was heated to 90 °C. The reaction mixture was diluted with EtOAc and washed with satd. aq. NaHCO3 and brine. The organic layer was dried over sodium sulfate, filtered, concentrated, and purified using silica gel chromatography to afford a yellow
soli = 406.1 [M+H].

4-Fluoro-3-(3-(2-(2-fluorophenyl)propan-2-ylamino)-1 ,2,4-triazin-6- yl)benzonitrile. 6-(5-Bromo-2-fluorophenyl)-N-(2-(2-fluorophenyl)propan-2- yl)-1 ,2,4-triazin-3-annine (2.2 g, 5.6 mmol), zinc cyanide (0.72 g, 6.2 mmol), Pd(PPh3) (2.9 g, 2.8 mmol), and DMF (20 mL) were combined and heated in a round bottom flask at 100 °C for 2 h. The reaction mixture was diluted with EtOAc and washed with satd. aq. NaHCO3 and brine. The organic layer was dried over sodium sulfate, filtered, concentrated and purified using silica gel chromatography to provide a yellow solid (1 .7 g), m/z = 352.1 [M+H].

4-Fluoro-3-(3-(2-(3-fluoropyridin-2-yl)propan-2-ylamino)-1 ,2,4- triazin-6-yl)benzamide. 4-Fluoro-3-(3-(2-(2-fluorophenyl)propan-2-ylamino)- 1 ,2,4-triazin-6-yl)benzonitrile (4.0 g, 1 1 .3 mmol), K2CO3 (6.2 g, 21 .2 mmol), and DMSO (1 13 mL) were combined in a round bottom flask and cooled to 0 °C. H2O2 (38 mL of 35% solution) was then added dropwise and the reaction was warmed to rt and stirred for 1 h. The reaction mixture was diluted with EtOAc and washed with satd. aq. NaHCO3 and brine. The organic layer was dried over sodium sulfate, filtered, concentrated, and purified using silica gel chromatography to afford a white solid (3.3 g), m/z = 371 .1 [M+H].
Example 16: Preparation of (1 -(6-Methoxypyridin-2-yl)cyclobutyl) methanamine
 1 -(6-Fluoropyridin-2-yl)cyclobutanecarbonitrile. To a 0 °C solution of 2,6-difluoropyridine (5.0 g, 43 mmol), cyclobutanecarbonitrile (3.5 g, 43 mmol), and toluene (100 mL) was added sodium hexamethyldisilazide (NaHMDS, 2.0 M in THF, 24 mL, 47 mmol). The resulting mixture was warmed to rt and stirred for 2 h. The mixture was then diluted with EtOAc (200 mL) and water (100 mL). The aqueous layer was extracted with EtOAc and the combined organic phases were washed with brine, dried over Na2SO4, concentrated, and purified using silica gel chromatography to provide the desired product (4.9 g) as a colorless oil.
1 -(6-Fluoropyridin-2-yl)cyclobutanecarbonitrile. To a 0 °C solution of 2,6-difluoropyridine (5.0 g, 43 mmol), cyclobutanecarbonitrile (3.5 g, 43 mmol), and toluene (100 mL) was added sodium hexamethyldisilazide (NaHMDS, 2.0 M in THF, 24 mL, 47 mmol). The resulting mixture was warmed to rt and stirred for 2 h. The mixture was then diluted with EtOAc (200 mL) and water (100 mL). The aqueous layer was extracted with EtOAc and the combined organic phases were washed with brine, dried over Na2SO4, concentrated, and purified using silica gel chromatography to provide the desired product (4.9 g) as a colorless oil.

1 -(6-Methoxypyridin-2-yl)cyclobutanecarbonitrile. To anhydrous methanol (6 mL) at 0 °C under nitrogen was added sodium metal ( ca 1 g) and the mixture stirred for 30 min. 1 -(6-fluoropyridin-2- yl)cyclobutanecarbonitrile (1 .6 g, 9.1 mmol) was then added, and the resulting mixture heated to 75 °C for 45 min. The solution was cooled to room temperature and partitioned between water and EtOAc. The layers were separated, the aqueous phase was extracted with EtOAc, and the combined organic phases were washed with brine, dried over Na2SO4, and concentrated in vacuo to give the desired product (1 .7 g) as an oil.

(1-(6-Methoxypyridin-2-yl)cyclobutyl)methanamine. To a stirred solution of 1 -(6-methoxypyridin-2-yl)cyclobutanecarbonitrile (1 .7 g, 8.8 mmol) in THF (20 mL) was added lithium aluminum hydride solution (1 .0 M in THF, 1 1 mL, 1 1 mmol). The mixture was refluxed for 1 .5 h and allowed to cool to room temperature. Water (0.43 mL) was slowly added, followed by 0.43 mL of 3 M NaOH, and then three additions of 0.43 mL of water. The resulting mixture was filtered through diatomaceous earth and rinsed with THF. The combined organics were dried over Na2SO and concentrated to dryness to give the desired product (1 .6 g) as a viscous oil.
Example 17: Preparation of 1-(3-Fluoropyridin-2-yl)cyclobutanamine Example

1 -(3-Fluoropyridin-2-yl)cyclobutanecarboxamide. To a 250 mL round bottom flask was added DMSO (60 mL) and 1 -(3-fluoropyridin-2- yl)cyclobutanecarbonitrile (2.96 g, 16.8 mmol), and the mixture was stirred until homogenous. Potassium carbonate (7.0 g, 50.4 mmol) was then added and the reaction mixture was cooled to 0 °C, followed by the addition of 35% hydrogen peroxide (6.5 mL). The reaction was stirred at 0 °C for 30 min and then warmed to rt. At this time, the reaction was diluted with water (50 mL) and ethyl acetate (100 mL). After transferring to a separatory funnel and shaking, the organic layer was separated from the aqueous layer and then washed with brine (3 x 50 mL). The organic layer was then dried over Na2SO , filtered, and concentrated to give a crude solid that was purified by silica gel chromatography (10% EtOAC/hexanes) to afford 1 .92 g of 1 -(3- fluoropyridin-2- l c clobutanecarboxamide as a white solid.

Methyl 1 -(3-fluoropyridin-2-yl)cyclobutylcarbamate. 1 -(3- fluoropyridin-2-yl)cyclobutanecarboxamide (1 .92 g, 9.88 mmol) was dissolved in methanol (20 mL) and potassium hydroxide (1 .1 1 g, 19.8 mmol, 2.0 equiv) was added. The mixture was sonicated until homogeneous, followed by the addition of iodosobenzene diacetate (4.77 g, 14.8 mmol). The reaction was stirred for 20 min and then diluted with water (100 mL) and ethyl acetate (125 mL). After transferring to a separatory funnel and shaking, the organic layer was separated from the aqueous layer, and the aqueous layer was extracted with EtOAc (50 mL). The combined organic layers were then dried over Na2SO , filtered, and concentrated to give a crude oil that was purified by silica gel chromatography (40% EtOAC/hexanes) to afford 1 .47 g of methyl 1 - (3-fluoropyridin-2-yl)cyclobutylcarbamate as a white solid.

1 -(3-Fluoropyridin-2-yl)cyclobutanamine. To a 20 mL microwave reaction vial was added methyl 1 -(3-fluoropyridin-2-yl)cyclobutylcarbamate (1 .47 g, 6.56 mmol), ethanol (12 mL) and 3N aqueous sodium hydroxide (7 mL). The reaction mixture was heated in the microwave reactor at 150 °C for 30 min. The ethanol was evaporated under reduced pressure and the mixture was extracted with ethyl acetate (30 mL). The aqueous layer was then extracted with ethyl acetate (2 x 30 mL). The organic layers were combined, dried over Na2SO , filtered, and concentrated to give 1 -(3- fluoropyridin-2-yl)cyclobutanamine (1 .01 g) as a crude yellow oil that was used in the next reaction step without further purification.
Example 18: Preparation of frans-3-Fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methanamine

1 -(3-Fluoropyridin-2-yl)-3-methylenecyclobutanecarbonitrile. To a solution of 3-methylenecyclobutanecarbonitrile (150 g, 1 .61 mol, 1 equiv) and 2-chloro-3-fluoropyridine (212 g, 1 .61 mmol, 1 equiv) in toluene (1 L) was added NaHMDS (2 M in THF, 885 mL, 1 .1 equiv) dropwise at 0-10 °C. Upon completion of addition, the reaction mixture was warmed to rt, stirred overnight, and quenched with NH4CI(Sat.) solution. The organic layer was washed with water (2x500 mL) and brine (500 mL), dried over Na2SO4, filtered, and concentrated to give the crude title compound (272 g) which was used in next step with t further purification, m/z = 189.1 [M+H].


1 -(3-Fluoropyridin-2-yl)-3-hydroxycyclobutanecarbonitrile. To a solution of 1 -(3-fluoropyridin-2-yl)-3-oxocyclobutanecarbonitrile (231 g, 1 .22 mol) in a mixture of DCM (2 L) and MeOH (200 mL) was added NaBH4 portionwise at -78 °C. The reaction mixture was stirred at -78 °C for 1 h and quenched with a mixture of methanol and water (1/1 ). The organic layer was washed with water (500 mL x 3), dried over Na2SO , and concentrated. The residue was purified on silica gel (50% EtOAc/hexanes) to provide the title compound as an am r oil (185.8 g) m/z = 193.2 [M+H].

fraA7s-3-Fluoro-1-(3-fluoropyridin-2-yl)cyclobutanecarbonitrile To a solution of 1 -(3-fluoropyridin-2-yl)-3-hydroxycyclobutanecarbonitrile (185 g, 0.96 mol) in DCM (1 L) was added DAST portionwise at 0-10 °C. Upon the completion of addition, the reaction was refluxed for 6 h. The reaction was cooled to rt and poured onto sat. NaHCO3 solution. The mixture was separated and the organic layer was washed with water, dried over Na2SO , and concentrated. The residue was purified on silica gel (100% DCM) to provide the title compound as a brown oil (1 16 g, 62%) in a 8:1 trans:cis
mixture. The above brown oil (107 g) was dissolved in toluene (1 10 mL) and hexanes (330 mL) at 70 °C. The solution was cooled to 0 °C and stirred at 0 °C overnight. The precipitate was filtered and washed with hexanes to provide the trans isomer as a white solid (87.3 g), m/z = 195.1 [M+H].

fraA7s-3-Fluoro-1-(3-fluoropyridin-2-yl)cyclobutyl)methanamine. A mixture of frans-3-fluoro-1 -(3-fluoropyhdin-2-yl)cyclobutanecarbonitrile (71 g, 0.37 mol) and Raney nickel (~7 g) in 7N ammonia in methanol (700 mL) was charged with hydrogen (60 psi) for 2 days. The reaction was filtered through a celite pad and washed with methanol. The filtrate was concentrated under high vacuum to provide the title compound as a light green oil (70 g), m/z = 199.2 [M+H].
Example 19: Preparation of 4-fluoro-3-(3-(((frans)-3-fluoro-1 -(3- fluoropyridin-2-yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)-N- methylbenzamide

4-Fluoro-3-(3-(((frans)-3-fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)benzoic acid. 4-Fluoro-3-(3- (((frans)-3-fluoro-1 -(3-fluoropyhdin-2-yl)cyclobutyl)methylamino)-1 ,2,4-triazin- 6-yl)benzonitrile (1 .3 g, 74.5 mmol) was diluted in 20 mL of concentrated HCI and heated in a microwave reactor at 1 10 °C for 30 min. The mixture was then concentrated and carried on to next step without further purification, (m/z [M-H] =

Fluoro-3-(3-(((irans)-3-fluoro-1 -(3-fluoropyridin-2-yl)cyclobutyl)methylamino)- 1 ,2,4-triazin-6-yl)benzoic acid (0.5 g, 1 .2 mmol) was diluted in 18 mL DMF and treated with HBTU (1 .8 g, 4.8 mmol), HOBT (0.65 g, 4.8 mmol), methyl amine hydrochloride (0.32 g, 4.8 mmol), and DIEA (8.4 mL, 48 mmol). The mixture was stirred at 24 °C for 30 min, diluted with EtOAc, washed with water and brine, concentrated, and purified using reverse phase chromatography to afford a tan solid (0.25 g, (m/z [M-H] = 429.3).
Example 20: Preparation of 1-(3-chloropyridin-2-yl)-3,3- difluorocyclobutanecarbonitrile

To a 100 mL round bottom flask was added 2,3-dichloropyridine (2.9 g, 20 mmol), 3,3-difluorocyclobutanecarbonitrile (2.1 g, 18 mmol), and toluene (50 mL). The mixture was cooled to °C and sodium hexamethyldisilazide (NaHMDS, 2.0 M in THF, 1 1 mL, 22 mmol) was added. The reaction mixture was warmed to rt and stirred for 2 h. The mixture was then diluted with EtOAc (20 mL) and water (20 mL). The aqueous layer was extracted with EtOAc and the combined organic phases were washed with brine, dried over Na2SO4, and concentrated to provide the desired product (3.4 g) as a colorless oil.
Example 21 : Preparation of 4-fluoro-3-(3-(((frans)-3-fluoro-1 -(3- fluoropyridin-2-yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)-2- hydroxybenzamide
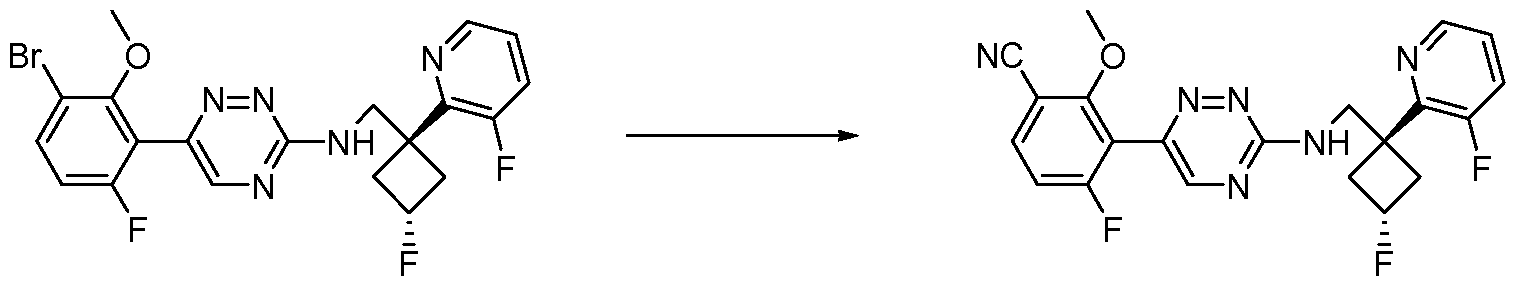
(3-Bromo-6-fluoro-2-methoxyphenyl)-N-(((frans)-3-fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methyl)-1 ,2,4-triazin-3-annine (448 mg, 0.93 mmol), zinc cyanide (131 mg, 1 .1 mmol), Pd(PPh3)4 (296 mg, 0.3 mmol), and DMF (5 mL) were combined and heated to 100 °C for 2 h. The reaction mixture was cooled and filtered through Celite. The reaction mixture was then diluted with EtOAc and washed with satd. aq. NaHCO3 and brine. The organic layer was dried over sodium sulfate, filtered, concentrated, and purified using silica gel
chromatography to afford 4-fluoro-3-(3-(((frans)-3-fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)-2-methoxybenzonitrile (229 mg).

4-Fluoro-3-(3-(((fraA7s)-3-fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)-2-methoxybenzamide. 4-
Fluoro-3-(3-(((frans)-3-fluoro-1 -(3-fluoropyhdin-2-yl)cyclobutyl)methylamino)- 1 ,2,4-triazin-6-yl)-2-methoxybenzonitrile (229 mg, 0.54 mmol), K2CO3 (200 mg, 1 .4 mmol), and DMSO (5 mL) were combined in a round bottom flask and cooled to 0 °C. H2O2 (1 mL of 35% solution) was then added dropwise and the reaction was warmed to rt and stirred for 1 h. The reaction mixture was diluted with EtOAc and washed with satd. aq. NaHCO3 and brine. The organic layer was dried over sodium sulfate, filtered, concentrated, and purified using silica gel chromatography to afford 210 mg of 4-fluoro-3-(3- (((frans)-3-fluoro-1 -(3-fluoropyhdin-2-yl)cyclobutyl)methylamino)-1 ,2,4-triazin- 6- -2-methoxybenzamide.

4-Fluoro-3-(3-(((frans)-3-fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)-2-hydroxybenzamide.
Lithium iodide (1 g), pyridine (20 mL), and 4-fluoro-3-(3-(((frans)-3-fluoro-1 -
(3-fluoropyridin-2-yl)cyclobutyl)nnethylannino)-1 ,2,4-triazin-6-yl)-2- methoxybenzamide (210 mg) were added to a round bottom flask. The reaction was heated to 125 °C and stirred for 2 h. The reaction was concentrated, mixed with methanol, filtered, and purified using reverse phase chromatography to afford 24 mg of 4-fluoro-3-(3-(((frans)-3-fluoro-1 -(3- fluoropyridin-2-yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)-2- hydroxybenzamide, m/z = 430.1 [M+H].
Example 22: Preparation of N-((2-(3-(((fraA7s)-3-fluoro-1 -(3-fluoropyridin-
2-yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)thiazol-5- yl)methyl)acetamide

6-(5-((tert-Butyldimethylsilyloxy)methyl)thiazol-2-yl)-1 ,2,4-triazin-3- amine. Diisopropylamine (13.7 ml_, 98 mmol) and THF (100 ml_) were added to a round bottom flask. The mixture was cooled to -78 °C and n-BuLi (2.5 M, 40 ml_, 100 mmol) was added. The reaction was stirred for 30 min, followed by the addition of (5-((tert-butyldimethylsilyloxy)methyl)thiazol-2-yl) (17.1 g, 75 mmol). The reaction was stirred to 30 min followed by the addition of zinc bromide (18 g, 69 mmol). The reaction was stirred for 1 h at -78 °C and then warmed to rt.
In a separate flask was added 6-bromo-1 ,2,4-triazin-3-amine (10.0 g, 57.5 mmol), (PPh3)4Pd (12.1 g, 1 1 .5 mmol), and THF (200 ml_). The mixture was heated to 80 °C, followed by the dropwise addition (1 h) of (5-((tert- butyldimethylsilyloxy)methyl)thiazol-2-yl)zinc(ll) bromide prepared in the step above. The reaction was then cooled to rt, filtered through a silica pad, concentrated, and then purified using silica gel chromatography to afford 6.7 g the title compound as a light yellow solid.

5-((tert-Butyldimethylsilyloxy)methyl)-2-(3-chloro-1 ,2,4-triazin-6- yl)thiazole. To a vial was added 6-(5-((tert- butyldimethylsilyloxy)methyl)thiazol-2-yl)-1 ,2,4-triazin-3-amine (2.5 g, 10.7
mmol), tetrabutylammonium chloride (6.3 g, 27.7 mmol) and dichloroethane (25 mL). The mixture was heated to 75 °C, followed by the addition of f-butyl nitrite (3.3 mL, 27.7 mmol). The reaction was stirred at 75 °C for 3 h, followed by cooling to rt, quenching with brine (200 mL). The reaction was extracted four times with CH2CI2. The combined organic layers were dried over Na2SO , concentrated, and purified using silica gel chromatography to afford the 1 .1 g of the title compound as an off-white solid.

F
(2-(3-(((frans)-3-Fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)thiazol-5-yl)methanol. To a round bottom flask was added 5-((tert-butyldimethylsilyloxy)methyl)-2-(3- chloro-1 ,2,4-triazin-6-yl)thiazole (1 .1 g, 3.2 mmol), frans-3-fluoro-1 -(3- fluoropyridin-2-yl)cyclobutyl)methanamine (870 mg, 4.3 mmol), potassium carbonate (1 .3 g, 9.4 mmol), and CH3CN (1 1 mL). The reaction mixture was heated to 80 °C and stirred for 2 h. The reaction was then diluted with EtOAc, filtered, and concentrated. The crude residue was suspended in methanol and concentrated HCI (1 mL) was added. The reaction was stirred for 30 min and then concentrated. The crude oil was dissolved in ethyl acetate (50 mL) and washed twice with saturated sodium carbonate (50 mL). The organic layers were dried over Na2SO , concentrated, and purified using silica gel chromatography to afford 470 mg of the title compound as an off- white solid.

6-(5-(Aminomethyl)thiazol-2-yl)-N-(((frans)-3-fluoro-1 -(3- fluoropyridin-2-yl)cyclobutyl)methyl)-1 ,2,4-triazin-3-amine. To a vial containing (2-(3-(((frans)-3-fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)thiazol-5-yl)methanol (457 mg, 1 .17 mmol) was added triphenylphosphine (460 mg, 1 .75 mmol), phthalimide
(257 mg, 1 .75 mmol), DIPEA (1 ml_), and THF (10 ml_).
Diisopropylazodicarboxylate (350 mg, 1 .75 mmol) was then added, and the reaction was stirred for 30 min. The reaction was quenched with saturated sodium bicarbonate solution (10 ml_) and extracted three times with ethyl acetate (50 ml_). The organic layers were dried over Na2SO , concentrated, and purified using silica gel chromatography to afford 670 mg of 2-((2-(3- (((frans)-3-fluoro-1 -(3-fluoropyhdin-2-yl)cyclobutyl)methylamino)-1 ,2,4-triazin- 6-yl)thiazol-5-yl)methyl)isoindoline-1 ,3-dione as an off-white solid. This solid was dissolved in methanol (10 ml_) and hydrazine (1 ml_). The reaction was stirred for 12 h, concentrated, and then purified using reverse phase chromatography to afford 250 mg of 6-(5-(aminomethyl)thiazol-2-yl)-N- (((frans)-3-fluoro-1 -(3-fluoropyhdin-2-yl)cyclobutyl)methyl)-1 ,2,4-thazin-3- amine.

N-((2-(3-(((frans)-3-fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)thiazol-5- yl)methyl)acetamide. To a stirring solution of 6-(5-(aminomethyl)thiazol-2- yl)-N-(((frans)-3-fluoro-1 -(3-fluoropyridin-2-yl)cyclobutyl)methyl)-1 ,2,4-triazin- 3-amine (250 mg, 643 μιτιοΙ) in CH2CI2 (10 ml_) was added acetic anhydride (324 mg, 3.2 μιτιοΙ). The reaction was stirred for 30 min, followed by quenched with saturated sodium bicarbonate solution (10 ml_) and extracted three times with CH2CI2 (50 ml_). The organic layers were dried over Na2SO4, concentrated, and purified using reverse phase chromatography to afford 224 mg of N-((2-(3-(((frans)-3-fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methylamino)-1 ,2,4-triazin-6-yl)thiazol-5-yl)methyl)acetamide as a white solid, m/z = 432.1 [M+H].
Example 23: Preparation of 2-(3-(2-(3-(difluoromethoxy)pyridin-2- yl)propan-2- lamino)-1 ,2,4-triazin-6-yl)thiazole-5-carboxamide

6-(5-Bromothiazol-2-yl)-1 ,2,4-triazin-3-amine. To a vial was added 6-(thiazol-2-yl)-1 ,2,4-triazin-3-amine (600 mg, 3.4 mmol), methanol (1 .2 mL), water (0.6 mL). The mixture was cooled to 0 °C and bromine (816 mg, 5.1 mmol) was added. The reaction was stirred for 45 min and then quenched with saturated sodium bicarbonate solution (20 mL) and extracted three times with ethyl acetate (50 mL). The organic layers were dried over Na2SO4, concentrated, and purified using silica gel chromatography to afford 320 mg of 6-(5-br mothiazol-2-yl)-1 ,2,4-triazin-3-amine as an off-white solid.

2-(3-Amino-1 ,2,4-triazin-6-yl)thiazole-5-carbonitrile. 6-(5- Bromothiazol-2-yl)-1 ,2,4-triazin-3-amine (300 mg, 1 .15 mmol), zinc cyanide (340 mg, 2.30 mmol), Pd(PPh3) (133 mg, 0.13 mmol), and DMF (5 mL) were combined and heated to 160 °C for 12 h. The reaction mixture was cooled, diluted with CH2CI2 (30 mL), filtered through silica gel, eluted with EtOAc, and concentrated. The crude product was then purified using silica gel
chromatography to afford 30 mg of 2-(3-amino-1 ,2,4-triazin-6-yl)thiazole-5- carbonitril .

2-(3-Bromo-1,2,4-triazin-6-yl)thiazole-5-carbonitrile. 2-(3-Amino- 1 ,2,4-triazin-6-yl)thiazole-5-carbonitrile (30 mg, 0.15 mmol) was dissolved in 1 mL of bromoform and heated to 90 °C. Isoamyl nitrite (88 mg, 0.75 mmol) was added and stirred at 90 °C for 2 h. The reaction mixture was then con ntrated and used directly in the next reaction.
 2-(3-(2-(3-(Difluoromethoxy)pyridin-2-yl)propan-2-ylamino)-1,2,4- triazin-6-yl)thiazole-5-carbonitrile. To a scintillation vial was added 2-(3- bronno-1 ,2,4-triazin-6-yl)thiazole-5-carbonithle (directly from above reaction), 2-(3-(difluoromethoxy)pyridin-2-yl)propan-2-amine (100 mg, 500 μιτιοΙ), and CH3CN (2 mL). The reaction was heated to 90 °C and stirred for 12 h. The reaction was then concentrated and purified using silica gel chromatography to afford 15 mg of 2-(3-(2-(3-(difluoromethoxy)pyridin-2-yl)propan-2-ylamino)- 1 2,4-triazin-6-yl)thiazole-5-carbonitrile.
2-(3-(2-(3-(Difluoromethoxy)pyridin-2-yl)propan-2-ylamino)-1,2,4- triazin-6-yl)thiazole-5-carbonitrile. To a scintillation vial was added 2-(3- bronno-1 ,2,4-triazin-6-yl)thiazole-5-carbonithle (directly from above reaction), 2-(3-(difluoromethoxy)pyridin-2-yl)propan-2-amine (100 mg, 500 μιτιοΙ), and CH3CN (2 mL). The reaction was heated to 90 °C and stirred for 12 h. The reaction was then concentrated and purified using silica gel chromatography to afford 15 mg of 2-(3-(2-(3-(difluoromethoxy)pyridin-2-yl)propan-2-ylamino)- 1 2,4-triazin-6-yl)thiazole-5-carbonitrile.

2-(3-(2-(3-(Difluoromethoxy)pyridin-2-yl)propan-2-ylamino)-1,2,4- triazin-6-yl)thiazole-5-carboxamide. 2-(3-(2-(3-(difluoromethoxy)pyridin-2- yl)propan-2-ylamino)-1 ,2,4-triazin-6-yl)thiazole-5-carbonitrile (15 mg, 38 μιτιοΙ), K2CO3 (50 mg, 0.35 mmol), and DMSO (1 mL) were combined in a vial and cooled to 0 °C. H2O2 (0.1 mL of 35% solution) was then added and the reaction was warmed to rt and stirred for 1 h. Acetic acid (0.1 mL) was added, and the reaction was filtered and then purified using reverse phase chromatography to afford 23 mg of 2-(3-(2-(3-(difluoromethoxy)pyridin-2- yl)propan-2-ylamino)-1 ,2,4-triazin-6-yl)thiazole-5-carboxamide as a white solid, m/z = 408.3 [M+H].
Example 24: Preparation of 3-(5-Fluoro-6-(((frans)-3-fluoro-1 -(3- fluoropyridin-2-yl)cyclobutyl)methylamino)pyridin-3-yl)benzamide

5-Bromo-3-fluoro-N-(((fraA7s)-3-fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methyl)pyridin-2-amine. To a microwave tube was added 5- bromo-2-chloro-3-fluoropyridine (1 .5 g, 7.4 mmol), ((frans)-3-fluoro-1 -(3- fluoropyridin-2-yl)cyclobutyl)methanamine (2.2 g, 1 1 .1 mmol), and DIPEA (4.9
mL, 29.7 mmol). The tube was sealed, heated at 1 10 °C, and stirred for 12 h. The reaction was then directly purified using silica gel chromatography to afford 2.5 g of 5-bromo-3-fluoro-N-(((frans)-3-fluoro-1 -(3-fluoropyridin-2- l)cyclobutyl)methyl)pyridin-2-amine as a white solid.

3-(5-Fluoro-6-(((frans)-3-fluoro-1 -(3-fluoropyridin-2- yl)cyclobutyl)methylamino)pyridin-3-yl)benzamide. 5-Bromo-3-fluoro-N- (((frans)-3-fluoro-1 -(3-fluoropyridin-2-yl)cyclobutyl)methyl)pyridin-2-amine (1 .0 g, 2.6 mmol), 3-carbamoylphenylboronic acid (0.54 g, 3.2 mmol), potassium carbonate (1 .5 g, 10.8 mmol), and methanol (25 mL) was added to a 100 mL round bottom flask and then degassed with nitrogen for 5 min. Silica- supported 1 ,1 '-Bis(diphenylphosphino)ferrocene (1 .0 g, 0.28 mmol/g) was added, and the reaction was refluxed in methanol for 2 h. The reaction mixture was then filtered, concentrated, and purified using silica gel chromatography to afford 2.0 g of the title compound as a solid, m/z = 413.3 [M+H].
Example 25: Preparation and assay of fast skeletal myofibrils
Preparation of fast skeletal myofibrils. Rabbit skeletal myofibrils were prepared based upon the method of Herrmann et al. (Biochem.
32(28):7255-7263(1993). Myofibrils were prepared from rabbit psoas muscle purchased from Pel-Freez Biologicals (Arkansas) within 2 days of ordering, stored on ice. Minced muscle was homogenized in 10 volumes of ice-cold "standard" buffer (50mM Tris, pH 7.4, 0.1 M potassium acetate, 5 mM KCI, 2 mM DTT, 0.2 mM PMSF, 10 μΜ leupeptin, 5 μΜ pepstatin, and 0.5 mM sodium azide) containing 5 mM EDTA and 0.5% Triton X-100 using an Omni- Macro homogenizer. Myofibrils were recovered by low speed centrifugation (3000 rpm for 10 minutes) and washed 2 times in the Triton X-100 containing buffer to ensure removal of cellular membrane. Following the Triton washes, myofibrils were washed 3 times in "standard" buffer containing 2 mM
magnesium acetate. A final wash in assay buffer (12 mM PIPES, pH 6.8, 60 mM KCI, 1 mM DTT) was performed and brought to 10% sucrose for flash freezing in liquid nitrogen and storage at -80°C.
Activation of Fast Skeletal Myofibrils. Fast fiber activators were identified by measuring the enzymatic activity of muscle myofibril preparations
TM
using the proprietary PUMA (see, e.g., U.S. Patent Nos. 6,410,254,
6,743,599, 7,202,051 , and 7,378,254) assay system. Myofibril preparations consisted of rabbit skeletal muscle (approximately 90% fast fibers) that had been mechanically homogenized and washed with a detergent (triton X-100) to remove cellular membranes. This preparation retained all of the
sarcomeric components in a native conformation and the enzymatic activity was still regulated by calcium. Compounds were tested using a myofibril suspension and a level of calcium sufficient to increase enzymatic activity of the myofibrils to 25% of their maximal rate (termed pCa25). Enzymatic activity was tracked via a pyruvate kinase and lactate dehydrogenase-coupled enzyme system. This assay regenerates myosin-produced ADP into ATP by oxidizing NADH, producing an absorbance change at 340 nm. The buffering system was 12 mM Pipes, 2 mM MgCI2, 1 mM DTT at pH 6.8 (PM12 buffer). Data was reported as AC1 .4, which is the concentration at which the compound increased the enzymatic activity by 40%. The results are summarized in Table 2 below.
Example 26: Preparation and Assay of Sarcomeric Proteins from
Skeletal Muscle
Powder Preparation
1 . Volumes are given per about 1000 g of the minced muscle.
2. Pre-cut and boil cheesecloth for 10 min in water. Drain and dry.
3. Mince chicken breast in a prechilled meat grinder.
4. Extract with stirring in 2 L of 0.1 M KCI, 0.15 M K-phosphate, pH 6.5 for 10 min at 4 °C. Spin 5000 rpm, 10 min, 4 °C in J LA. Collect the pellet.
5. Extract pellets with stirring with 2 L of 0.05 M NaHCO3 for 5 min. Spin 5000 rpm, 10min, 4 °C in JLA. Collect the pellet. Repeat the extraction once more.
6. Extract the filtered residue with 2 L of 1 mM EDTA, pH 7.0 for 10 min with stirring.
7. Extract with 2 L of H2O for 5 min with stirring. Spin 10000 rpm, 15min, 4 °C in JLA. Carefully collect the pellet, part of which will be loose and gelatinous.
8. Extract 5 times with acetone (2 L of acetone for 10 min each with stirring). Squeeze through cheesecloth gently. All acetone extractions are performed at room temperature. Acetone should be prechilled to 4 °C.
9. Drying: Place the filtered residue spread on a cheesecloth in a large glass tray and leave in a hood overnight. When the residue is dry, put in a wide mouth plastic bottle and store at 20 °C.
Alternate Powder Preparation
(See Zot & Potter (1981 ) Prep. Biochem. 1 1 (4) pp. 381 -395)
1 . Dissect left ventricles of the cardiac muscle. Remove as much of the pericardial tissue and fat as possible. Grind in a prechilled meat grinder.
Weigh.
2. Prepare 5 volumes of Extract buffer (see below). Homogenize the meat in a blender, 4 times 15 sec on blend with 15 sees in between. Do this with 1 volume (weight/volume) of buffer taken from the 5 volumes already prepared. Add the homogenate back to the extract buffer and stir until well mixed (5 minutes).
3. Filter through one layer of cheesecloth in large polypropylene strainer. Resuspend back into 5 volumes of extract buffer as above.
4. Repeat Step 3 four more times. At the end, do not resuspend in extraction buffer but proceed to Step 5. The pellets should be yellow white.
5. Resuspend in 3 volumes (according to original weight) of 95% cold ethanol. Stir for 5 min and squeeze through cheesecloth as above, repeat two more times.
6. Weigh squeezed residue and then resuspend in 3 volumes (new weight/volume) of cold diethyl ether.
7. Repeat Step 6 a total of three times.
8. Leave overnight in a single layer on a cheesecloth in a glass tray.
9. When dry, collect the powder, weigh and store in a wide-mouth jar at 4 °C.
EXTRACT BUFFER: 50 mM KCI, 5 mM Tris pH 8.0 Prepare as 50 times concentrate. For 2L: 250 mM Tris pH 8.0. Tris Base (121 .14 g/mol, 60.6 g), pH to 8.0 with cone. HCI, then add 2.5 M KCI (74.55 g/mol, 372 g).
Actin Preparation
1 . Extract powder (as described above) with 20 ml buffer A (see below, add BME and ATP just prior to use in each of the following steps) per gram of powder (200 ml per 10 g). Use a large 4 L beaker for 150 g of powder. Mix vigorously to dissolve powder. Stir at 4 °C. for 30 min.
2. Separate extract from the hydrated powder by squeezing through several layers of cheesecloth. Cheesecloth should be pre-sterilized by microwaving damp for 1 -2 min.
3. Re-extract the residue with the same volume of buffer A and combine extracts.
4. Spin in JLA10 rotor(s) for 1 hr at 10K rpm (4 °C). Collect supernatant through 2 layers of cheesecloth.
5. Add ATP to 0.2 mM and MgCI2 to 50 mM. Stir on stir plate at 4 °C for 60 minutes to allow actin to polymerize/form para-crystals.
6. Slowly add solid KCI to 0.6 M (45 g/l). Stir at 4 °C for 30 min.
7. Spin in JLA10 rotor(s) at 10K rpm for 1 hr.
8. Depolymerization: Quickly rinse surface of pellets with buffer A and dispose of wash. Soften the pellets by pre-incubation on ice with small amount of buffer A in each tube (use less than half of final resuspension volume total in all tubes). Resuspend by hand first with cell scraper and combine pellets. Wash tubes with extra buffer using a 25 ml pipette and motorized pipettor, aggressively removing actin from sides of tubes.
Homogenize in large dounce in cold buffer A on ice. Use 3 ml per gram of powder originally extracted.
9. Dialyze against buffer A with 4 changes over 48 hour period.
10. Collect dialyzed actin and spin in the 45Ti rotor at 40K rpm for 1 .5 hr (4 °C).
1 1 . Collect supernatant (G-Actin). Save a sample for gel analysis and determination of protein concentration.
12. To polymerize G-actin for storage, add KCI to 50 mM (from 3 M stock), MgCI2 to 1 mM, and NaN3 to 0.02% (from 10% stock). Store at 4 °C. Do not
freeze.
Buffer A: 2 mM tris/HCI, 0.2 mM CaCI2, 0.5 mM (36 μΙ/L) 2-mercaptoethanol,
0.2 mM Na2 ATP (added fresh), and 0.005% Na-azide; pH 8.0.
Purification of Skeletal Muscle Myosin
(See Margossian, S.S. and Lowey, S. (1982) Methods Enzymol. 85, 55-123; and Goldmann, W.H. and Geeves, M.A. (1991 ) Anal. Biochem. 192, 55-58) Solution A: 0.3 M KCI, 0.15 M potassium phosphate, 0.02 M EDTA, 0.005 M MgCI2, 0.001 M ATP, pH 6.5.
Solution B: 1 M KCI, 0.025 M EDTA, 0.06 M potassium phosphate, pH 6.5.
Solution C: 0.6 M KCI, 0.025 M potassium phosphate, pH 6.5.
Solution D: 0.6 M KCI, 0.05 M potassium phosphate, pH 6.5.
Solution E: 0.15 M potassium phosphate, 0.01 M EDTA, pH 7.5.
Solution F: 0.04 M KCI, 0.01 M potassium phosphate, 0.001 M DTT, pH 6.5.
Solution G: 3 M KCI, 0.01 M potassium phosphate, pH 6.5.
All procedures are carried out at 4 °C.
1 . Obtain approx. 1000 g skeletal muscle, such as rabbit skeletal muscle.
2. Grind twice; extract with 2 L solution A for 15 min while stirring; add 4 L cold H2O, filter through gauze; dilute with cold H2O to ionic strength of 0.04, (about 10-fold); let settle for 3 h; collect precipitate at 7,000 rpm in GSA rotor for 15 min.
3. Disperse pellet in 220 ml solution B; dialyze overnight against 6 L solution C; slowly add -400 ml equal volume cold distilled H2O; stir for 30 min; centrifuge at 10,000 rpm for 10 min in GSA rotor.
4. Centrifuge supernatant at 19,000 rpm for 1 h.
5. Dilute supernatant to ionic strength of 0.04 (~8-fold); let myosin settle overnight; collect about 5-6 L fluffy myosin precipitate by centrifuging at 10,000 rpm for 10 min in GSA rotor.
6. Resuspend pellet in minimal volume of solution G; dialyze overnight against 2 L solution D; centrifuge at 19,000 rpm for 2 h, in cellulose nitrate tubes; puncture tubes and separate myosin from fat and insoluble pellet.
7. Dilute supernatant to 5-10 mg/ml and dialyze against solution E extensively, load onto DEAE-sephadex column.
8. Pre-equilibrate with solution E; apply 500-600 g myosin at 30 ml/h; wash with 350 ml solution E; elute with linear gradient of 0-0.5 M KCI in
solution E (2 x 1 liter); collect 10 ml fractions; pool myosin fractions (>0.1 M KCI); concentrate by overnight dialysis against solution F; centrifuge at 25,000 rpm for 30 min; store as above.
9. The myosin is then cut with chymotrypsin or papain in the presence of EDTA to generate the S1 fragment which is soluble at the low salt conditions optimal for ATPase activity (Margossian, supra).
Preparation and Assay
Myosin is prepared by precipitation from salt extracts of rabbit psoas muscle, and a soluble S1 fraction is prepared by digestion with chymotrypsin (Margossian and Lowey, 1982).
Actin is purified by first preparing an ether powder of cardiac muscle (Zot HG and Potter J D. (1981 ) Preparative Biochemistry 1 1 :381 -395) as described above. Subsequently, actin is cycled between the filamentous and soluble state through rounds of centrifugation and dialysis (Spudich J A and Watt S. (1971 ) J. Biol. Chem. 246:4866-4871 ).
Tropomyosin is extracted from the ether powder and separated from the other proteins based on pH dependent precipitations followed by successive ammonium sulfate cuts at 53% and 65% (Smillie LB. (1981 ) Methods Enzymol 85 Pt B:234-41 ). The troponins are isolated as an intact complex of TnC, TnT, and Tnl. Ether powder is extracted in a high salt buffer. Successive ammonium sulfate cuts of 30% and 45% are done; the precipitate is solubilized by dialysis into a low salt buffer and then further purified on a DEAE Toyopearl column with a 25-350 mM KCI gradient. There is no measurable ATPase in any of the components except for myosin which naturally had a very low basal ATPase in the absence of actin.
Prior to screening, the actin, tropomyosin, and troponin complex are mixed together in the desired ratio (e.g., 7:1 :1 ) to achieve maximal calcium regulation of the actin filament. The screen is conducted at a concentration that gives 25% activation. This calcium concentration is in the physiological range during muscle contraction.
To measure the generation of ADP during the reaction, a pyruvate kinase/lactate dehydrogenase/NADH coupled enzyme system (PK/LDH) is added to the actin. The myosin is kept separately, and added to the regulated thin filaments to initiate the reaction. Oxidation of NADH is monitored in real
time, so that kinetic curves are obtained. Compounds are dissolved in DMSO and spotted onto the bottoms of 384 well plates at 10 to 40 g/ml final concentration.
Using procedures similar to those described herein, utilizing reagents and intermediates commercially available (e.g., Sigma-Aldrich, ) or readily synthesized by one of skill in the art, the compounds in Table 2 were synthesized, characterized and tested. AC1 .4 values were determined according to the procedure described in Example 25, and the reported median AC1 .4 values are as follows: A = < 1 uM; B = 1 -10 uM; C = 10-20 uM; D = >20 uM.
Table 2

4-(6-{[2-(4-fluorophenyl)-2- methylpropyl]amino}-3-pyridyl)benzamide
3-(6-{[2-(4-fluorophenyl)-2- methylpropyl]amino}-3-pyridyl)benzamide
6-{[2-(4-f I uoroph enyl )-2- methylpropyl]amino}pyridine-3-carbonitrile
3-(6-{[2-(4-fluorophenyl)ethyl]amino}-3- pyridyl)benzamide
6-{[2-(4-f I uoroph enyl )-2- methylpropyl]amino}pyridine-3- carboxamide







2-fluoro-5-(3-{[1-(3-fluoro(2-pyridyl))- isopropyl]amino}(1 ,2,4-triazin-6- P (* 372.1 D yl))benzoic acid
2-fluoro-5-(3-{[1-(3-fluoro(2-pyridyl))- isopropyl]amino}(1 ,2,4-triazin-6- 371.2 D yl))benzamide
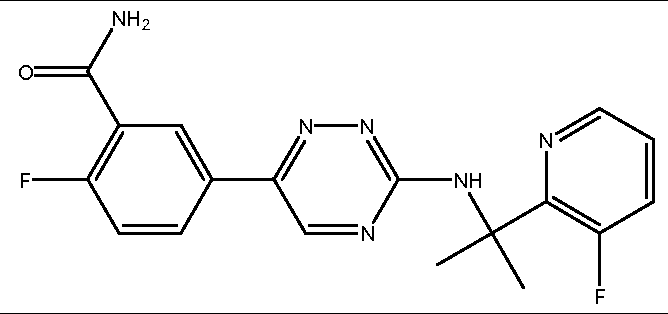
N-((2S)-2,3-dihydroxypropyl)[4-fluoro-3- (3-{[1-(3-fluoro(2-pyridyl))-
445.3 C isopropyl]amino}(1 ,2,4-triazin-6- yl))phenyl]carboxamide
N-[4-fluoro-3-(3-{[1-(3-fluoro(2-pyridyl))- isopropyl]amino}(1 ,2,4-triazin-6- 401.1 B yl ) ) ph e n yl] m ethoxyca rboxa m id e
amino[4-fluoro-3-(3-{[1-(3-fluoro(2- pyridyl))-isopropyl]amino}(1 ,2,4-triazin-6- 422.1 B yl))phenyl]sulfonamide
3-[3-({1-[6-(difluoromethyl)(2-pyridyl)]- isopropyl}am ino)( 1 ,2,4-triazin-6-yl )]-4- 403.1 B fluorobenzamide
3-[3-({1-[6-(difluoromethyl)(2-pyridyl)]- isopropyl}am ino)( 1 ,2,4-triazin-6-yl )]-4- 385.1 B fluorobenzenecarbonitrile

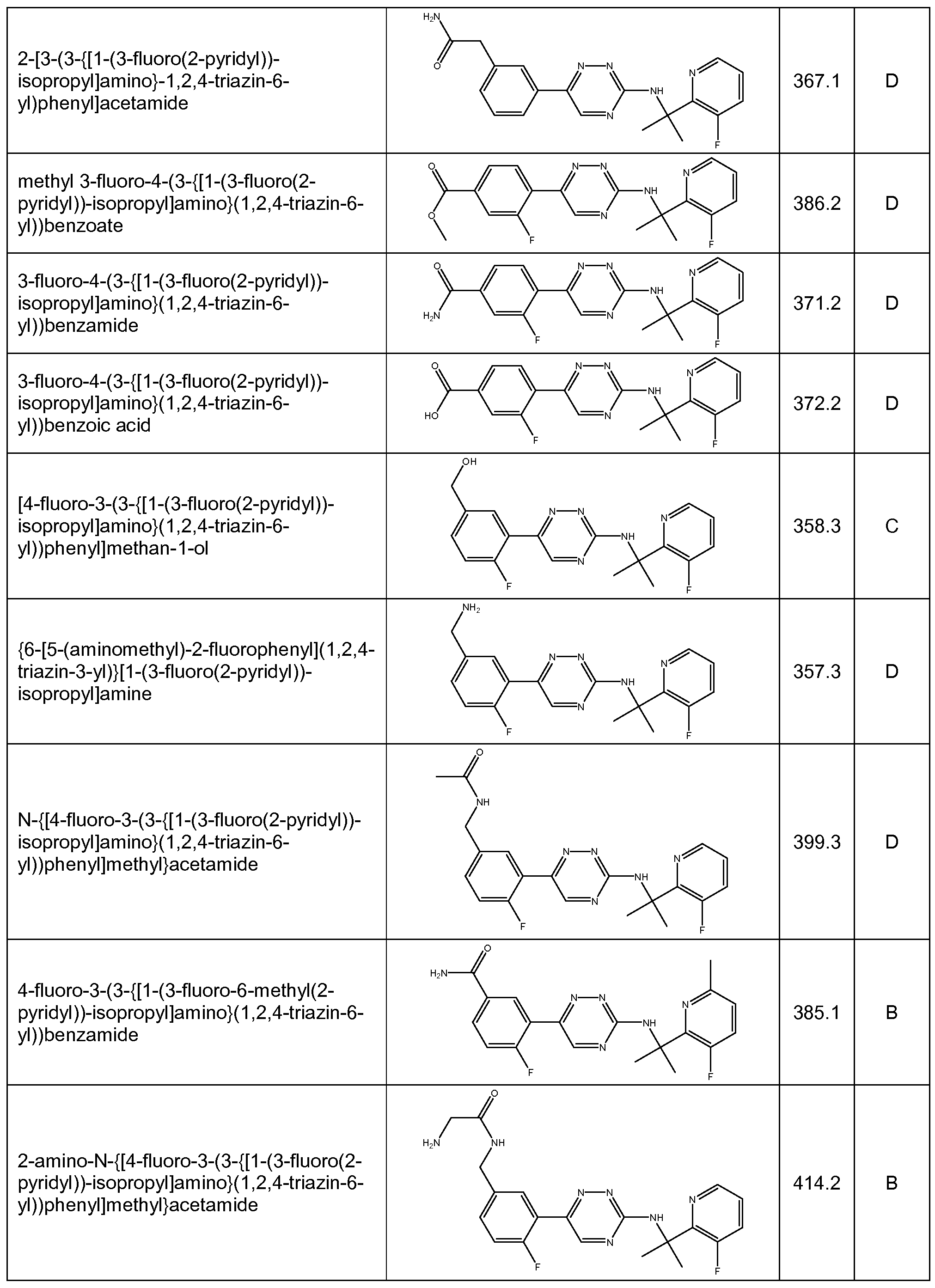






While the present invention has been described with reference to the specific embodiments described herein, it should be understood by those skilled in the art that various changes may be made and equivalents may be substituted without departing from the true spirit and scope of the invention. In addition, modifications may be made to adapt a particular situation, material, composition of matter and/or process to the objective, spirit and scope of the present invention. All such modifications are intended to be within the scope of the claims appended hereto.
Claims
1 . A compound of Formula I:

Formula I or a pharmaceutically acceptable salt thereof, wherein:
Z1, Z2, Z3 and Z4 are each independently selected from N and CR1, provided that at least one of Z1, Z2, Z3 and Z4 is N and at least one of Z1, Z2, Z3 and Z4 is CR1;
R1, at each occurrence, is independently selected from hydrogen, halogen, CN, Ci-6 alkyl, Ci-6 haloalkyl, C(O)ORa, C(O)NRbRc, ORa, NRbRc, C6- 10 aryl and 5-10 membered heteroaryl;
R2 is selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, 5-10
membered heteroaryl and NRbRc, wherein each of the C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
heterocycloalkenyl, C6-io aryl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra,
(CH2)nSO2NRbRc, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 mennbered heterocycloalkyi, (CH2)nC6-io aryl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents;
R4 is selected from hydrogen, d-6 alkyl, Ci-6 haloalkyl, C(O)Ra,
C(O)ORa, C(O)NRbRc and SO2Ra;
R5 and R6 are each independently selected from hydrogen, halogen, C-i-6 alkyl and Ci-6 haloalkyl;
or alternatively, R5 and R6 together with the carbon atom to which they are bound form a group selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyi and 3-8 membered heterocycloalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl and d-6 haloalkyl;
R7 is selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyi, 3-8 membered heterocycloalkenyl, s-io aryl and 5-10 membered heteroaryl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, OC(O)NRbRc, NRbRc, NRdC(O)Ra, NRdC(O)ORa, NRdC(O)NRbRc,
NRdC(O)C(O)NRbRc, NRdC(S)Ra, NRdC(S)ORa, NRdC(S)NRbRc,
NRdC(NRe)NRbRc, NRdS(O)Ra, NRdSO2Ra, NRdSO2NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, C(S)Ra, C(S)ORa, C(S)NRbRc, C(NRe)NRbRc, SRa, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyi, 3-8 membered heterocycloalkenyl, s-io aryl, C7.11 aralkyl, and 5-10 membered heteroaryl, wherein each of the Ci-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyi, 3-8 membered
heterocycloalkenyl, s-io aryl, C7.11 aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents;
R8 and R9, at each occurrence, are each independently selected from hydrogen, halogen and Ci_6 alkyl;
X is selected from a bond, -(CH2)P-, -(CH2)pC(O)(CH2)q-, - (CH2)pO(CH2)q-, -(CH2)pS(CH2)q-, -(CH2)pNRd(CH2)q-, -(CH2)pC(O)O(CH2)q-, -(CH2)pOC(O)(CH2)q-, -(CH2)pNRdC(O)(CH2)q-, -(CH2)pC(O)NRd(CH2)q-, -(CH2)pNRdC(O)NRd(CH2)q-, -(CH2)pNRdSO2(CH2)q-, and
-(CH2)pSO2NRd(CH2)q-;
Ra, at each occurrence, is independently selected from hydrogen, d-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7.11 aralkyl and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents;
Rb and Rc, at each occurrence, are each independently selected from hydrogen, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
heterocycloalkenyl, C6-io aryl, C7-n aralkyl, 5-10 membered heteroaryl, C(O)Rg, C(O)ORg, C^NR^ and SO2Rg, wherein each of the Ci-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents;
Rd, at each occurrence, is independently selected from hydrogen and C1-6 alkyl;
Re, at each occurrence, is independently selected from hydrogen, CN, OH, Ci-6 alkoxy, Ci-6 alkyl and Ci-6 haloalkyl;
Rf, at each occurrence, is independently selected from halogen, CN, ORh, OC(O)Rh, OC(O)ORh, OC(0)NRiRi, N R'Rj, N RdC(O)Rh, N RdC(O)ORh, NRdC(O)N R'Rj, NRdC(O)C(O)NRiRj, NRdC(S)Rh, N RdC(S)ORh, NRdC(S)NR'Rj, NRdC(NRe)NR'Rj, N RdS(O)Rh, N RdSO2Rh, N RdSO2NRiRj, C(O)Rh, C(O)ORh, Ο(Ο)Ν^, C(S)Rh, C(S)ORh, C^NR^, C(NRe)NR'Rj, SRh, S(O)Rh, SO2Rh, SO2NR'Rj, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
heterocycloalkenyl, C-6-10 aryl, C7-11 aralkyl and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
heterocycloalkenyl, Ce-ιο aryl, C7-11 aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rk substituents;
or two Rf substituents bound to a single carbon atom, together with the carbon atom to which they are both bound, form a group selected from carbonyl, C3-8 cycloalkyl and 3-8 membered heterocycloalkyl;
Rg, at each occurrence, is independently selected from d-6 alkyl, Ci-6 haloalkyl, phenyl, naphthyl, and C7-11 aralkyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, OH, d-6 alkoxy, d-6 alkyl and d-6 haloalkyl;
Rh, at each occurrence, is independently selected from hydrogen, d-6 alkyl, d-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7.11 aralkyl and 5-10 membered heteroaryl, wherein each of the d-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, -8 cycloalkyl, -8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rk substituents;
R' and Rj, at each occurrence, are each independently selected from hydrogen, d-6 alkyl, d-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered
heterocycloalkenyl, C-6-10 aryl, C7.11 aralkyl, 5-10 membered heteroaryl, C(O)Rg, and C(O)ORg, wherein each of the Ci_6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, OH, Ci-6 alkoxy, Ci-6 alkyl and Ci-6 haloalkyl;
Rk, at each occurrence, is independently selected from halogen, CN, oxo, OH , d-6 alkoxy, NH2, NH(Ci-6 alkyl), N(Ci-6 alkyl)2, NHC(O)d-6 alkyl, NHC(O)C7-i i aralkyl, N HC(O)OCi-6 alkyl, N HC(O)OC7-n aralkyl, C(O)d-6 alkyl, C(0)C7-n aralkyl, C(O)OC -6 alkyl, C(0)OC7-n aralkyl, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, and C2-6 alkynyl, wherein each C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, and C7.11 aralkyl substituent is optionally substituted with 1 , 2 or 3 substituents selected from OH, d-6 alkoxy, NH2, NH(Ci-6 alkyl), N(Ci-6 alkyl)2, NHC(O)Ci-6 alkyl, NHC(O)C7-n aralkyl, NHC(O)OCi-6 alkyl, and NHC(O)OC7- 11 aralkyl; m is 0, 1 or 2;
n, at each occurrence, independently is 0, 1 or 2;
p is 0, 1 or 2; and
q is 0, 1 or 2.
2. The compound of claim 1 , or a pharmaceutically acceptable salt thereof, wherein m is 0.
3. The compound of claim 1 , or a pharmaceutically acceptable salt thereof, wherein m is 1 .
4. The compound of claim 3, wherein R8 and R9 are each hydrogen.
5. The compound of any one of claims 1 to 4, or a pharmaceutically acceptable salt thereof, wherein R5 and R6 are each Ci-6 alkyl.
6. The compound of claim 5, or a pharmaceutically acceptable salt thereof, wherein R5 and R6 are each methyl.
7. The compound of any one of claims 1 to 4, or a pharmaceutically acceptable salt thereof, wherein R5 and R6 together with the carbon atom to which they are bound form a group selected from C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl and 3-8 membered
heterocydoalkenyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl and Ci-6 haloalkyl.
8. The compound of claim 7, or a pharmaceutically acceptable salt thereof, wherein R5 and R6 together with the carbon atom to which they are bound form C3-8 cycloalkyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, C1-6 alkyl and C-i-6 haloalkyl.
9. The compound of claim 8, or a pharmaceutically acceptable salt thereof, wherein R5 and R6 together with the carbon atom to which they are bound form a group selected from cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl, each optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, C1-6 alkyl and C1-6 haloalkyl.
10. The compound of claim 9, or a pharmaceutically acceptable salt thereof, wherein R5 and R6 together with the carbon atom to which they are bound form cyclobutyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl and Ci-6 haloalkyl.
1 1 . The compound of claim 10, or a pharmaceutically acceptable salt thereof, wherein R5 and R6 together with the carbon atom to which they are bound form cyclobutyl optionally substituted with one or two halogens.
12. The compound of claim 1 1 , or a pharmaceutically acceptable salt thereof, wherein R5 and R6 together with the carbon atom to which they are bound form a group selected from cyclobutyl, 3-fluorocyclobutyl and 3,3- difluorocyclobutyl.
13. The compound of any one of claims 1 to 12, wherein Z1 , Z2 and Z3 are each N and Z4 is CR1.
14. The compound of any one of claims 1 to 12, wherein Z1, Z2 and Z4 are each N and Z3 is CR1.
15. The compound of any one of claims 1 to 12, wherein Z1 , Z3 and Z4 are each CR1 and Z2 is N.
16. The compound of any one of claims 1 to 12, wherein Z2, Z3 and Z4 are each CR1 and Z1 is N.
17. The compound of claim 1 , wherein the compound is of Formula ll(e) or ll(f), or a pharmaceutically acceptable salt thereof:

Formula ll(f)
wherein Rm and Rn are each independently selected from hydrogen, halogen and C-i-6 alkyl.
18. The compound of claim 1 , wherein the compound is of Formula lll(e) or lll(f), or a pharmaceutically acceptable salt thereof:

Formula lll(e) 
Formula 111(f)
wherein Rm and Rn are each independently selected from hydrogen, halogen and C-i-6 alkyl.
19. The compound of claim 1 , wherein the compound is of Formula IV(e) or IV(f), or a pharmaceutically acceptable salt thereof:
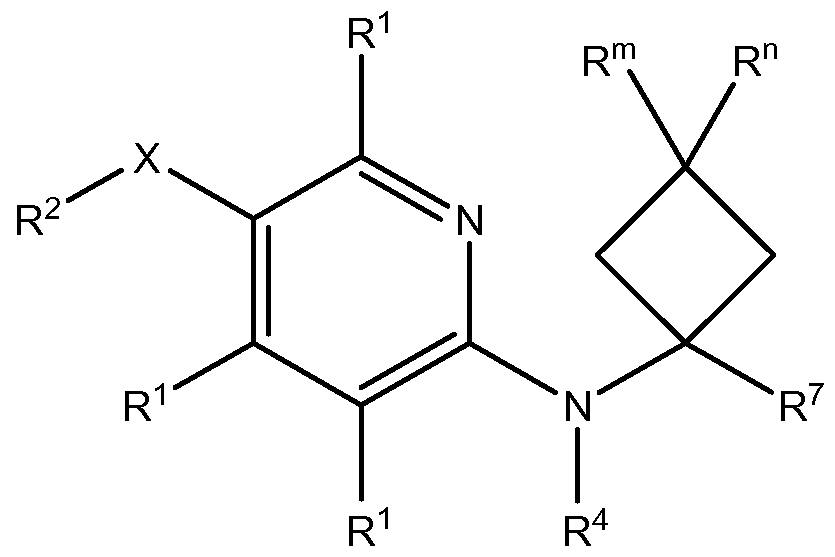
Formula IV(e)

Formula IV(f)
wherein Rm and Rn are each independently selected from hydrogen, halogen and C-i-6 alkyl.
20. The compound of claim 1 , wherein the compound is of Formula V(e) or V(f), or a pharmaceutically acceptable salt thereof:

Formula V(e)

Formula V(f)
wherein Rm and Rn are each independently selected from hydrogen, halogen and C-i-6 alkyl.
21 . The compound of any one of claims 17-20, wherein one of Rm and Rn is hydrogen and the other is halogen.
22. The compound of claim 21 , wherein the halogen and R7 are in a trans configuration with respect to one another on the cyclobutyl ring.
23. The compound of claim 21 , wherein the halogen and R7 are in a cis configuration with respect to one another on the cyclobutyl ring.
24. The compound of claim 22 or 23, wherein one of Rm and Rn is hydrogen and the other is fluorine.
25. The compound of any one of claims 1 to 24, or a pharmaceutically acceptable salt thereof, wherein R7 is phenyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, OC(O)NRbRc, N RbRc, NRdC(O)Ra, NRdC(O)ORa, NRdC(O)NRbRc, NRdC(O)C(O)NRbRc, N RdC(S)Ra, NRdC(S)ORa, N RdC(S)NRbRc,
NRdC(NRe)NRbRc, N RdS(O)Ra, NRdSO2Ra, NRdSO2NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, C(S)Ra, C(S)ORa, C(S)NRbRc, C(NRe)NRbRc, SRa, S(O)Ra, SO2Ra, SO2NRbRc, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 nnennbered heterocycloalkyi, 3-8 nnennbered heterocycloalkenyl, C6-io aryl, C7-n aralkyl, and 5-10 nnennbered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 nnennbered heterocycloalkyi, 3-8 nnennbered
heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 nnennbered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
26. The compound of any one of claims 1 to 24, or a pharmaceutically acceptable salt thereof, wherein R7 is 5-10 membered heteroaryl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, OC(O)NRbRc, NRbRc, N RdC(O)Ra, N RdC(O)ORa, NRdC(O)N RbRc, N RdC(O)C(O)NRbRc, N RdC(S)Ra, N RdC(S)ORa,
NRdC(S)NRbRc, NRdC(NRe)NRbRc, N RdS(O)Ra, NRdSO2Ra, NRdSO2NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, C(S)Ra, C(S)ORa, C(S)NRbRc, C(NRe)NRbRc, SRa, S(O)Ra, SO2Ra, SO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyi, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl, and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyi, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
27. The compound of claim 26, or a pharmaceutically acceptable salt thereof, wherein R7 is pyridyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, OC(O)NRbRc, N RbRc, NRdC(O)Ra, NRdC(O)ORa, NRdC(O)NRbRc,
NRdC(O)C(O)NRbRc, N RdC(S)Ra, NRdC(S)ORa, N RdC(S)NRbRc,
NRdC(NRe)NRbRc, N RdS(O)Ra, NRdSO2Ra, NRdSO2NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, C(S)Ra, C(S)ORa, C(S)NRbRc, C(NRe)NRbRc, SRa, S(O)Ra, SO2Ra, SO2NRbRc, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, C3-6 cycloalkenyl, 3-6 membered heterocycloalkyl, 3-6 membered heterocycloalkenyl, phenyl, naphthyl, C7-n aralkyl, and 5-10 membered heteroaryl, wherein each of the d-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
28. The compound of claim 27, or a pharmaceutically acceptable salt thereof, wherein R7 is 2-pyridyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, ORa, OC(O)Ra, OC(O)ORa, OC(O)NRbRc, NRbRc, NRdC(O)Ra, NRdC(O)ORa, NRdC(O)NRbRc,
NRdC(O)C(O)NRbRc, NRdC(S)Ra, NRdC(S)ORa, NRdC(S)NRbRc,
NRdC(NRe)NRbRc, NRdS(O)Ra, NRdSO2Ra, NRdSO2NRbRc, C(O)Ra, C(O)ORa, C(O)NRbRc, C(S)Ra, C(S)ORa, C(S)NRbRc, C(NRe)NRbRc, SRa, S(O)Ra, SO2Ra, SO2NRbRc, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, C3-6 cycloalkyl, C3-6 cycloalkenyl, 3-6 membered heterocycloalkyl, 3-6 membered heterocycloalkenyl, phenyl, naphthyl, C7-n aralkyl, and 5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, C3-8 cycloalkyl, C3-8 cycloalkenyl, 3-8 membered heterocycloalkyl, 3-8 membered heterocycloalkenyl, C6-io aryl, C7-n aralkyl and 5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
29. The compound of any one of claims 1 to 28, or a pharmaceutically acceptable salt thereof, wherein X is a bond.
30. The compound of any one of claims 1 to 29, or a pharmaceutically acceptable salt thereof, wherein R2 is phenyl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa, (CH2)nNRdC(S)NRbRc,
(CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra, (CH2)nNRdSO2Ra,
(CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc, (CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra, (CH2)nSO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
31 . The compound of any one of claims 1 to 29, or a pharmaceutically acceptable salt thereof, wherein R2 is 5-10 membered heteroaryl optionally substituted with 1 , 2, 3, 4 or 5 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc,
(CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra,
(CH2)nSO2NRbRc, C1-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl,
(CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl, wherein each of the C-|. 6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
32. The compound of claim 31 , or a pharmaceutically acceptable salt thereof, wherein R2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl, triazyl, furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with 1 , 2, 3 or 4 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra, (CH2)nSO2NRbRc, C -6 alkyl, C -6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl,
(CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i- 6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
33. The compound of claim 32, or a pharmaceutically acceptable salt thereof, wherein R2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl, triazyl, furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with a substituent selected from (CH2)nC(O)ORa and (CH2)nC(O)NRbRc; and optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa, (CH2)nNRdC(S)NRbRc,
(CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra, (CH2)nNRdSO2Ra,
(CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc, (CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra, (CH2)nSO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8
membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
34. The compound of claim 33, or a pharmaceutically acceptable salt thereof, wherein R2 is selected from furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with (CH2)nC(O)NRbRc.
35. The compound of claim 32, or a pharmaceutically acceptable salt thereof, wherein R2 is selected from pyridyl, pyrimidyl, pyrazyl, pyridazyl, triazyl, furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with (CH2)nNRdC(O)Ra, wherein Ra is C -6 alkyl or 3-8 membered heterocycloalkyl, each optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, CN, oxo, (CH2)nORa,
(CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc,
(CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa,
(CH2)nNRdC(S)NRbRc, (CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra,
(CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc,
(CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra,
(CH2)nSO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3- 8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl,
(CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl, wherein each of the Ci-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8
membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
36. The compound of claim 35, or a pharmaceutically acceptable salt thereof, wherein R2 is is selected from furanyl, pyrrolyl, thiophenyl, thiazolyl, isothiazolyl, thiadiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, imidazolyl, triazolyl and tetrazolyl, each optionally substituted with (CH2)nNRdC(O)Ra, wherein Ra is selected from Ci-6 alkyl, Ci-6 alkyl-OH and Ci-6 alkyl-NH2, each optionally substituted with 1 , 2 or 3 additional substituents selected from halogen, CN, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc,
(CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdSO2Ra, (CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra, (CH2)nSO2NRbRc, Ci-6 alkyl, Ci-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl.
37. The compound of claim 31 , or a pharmaceutically acceptable salt thereof, wherein R2 is selected from indolyl, indazolyl, benzimidazolyl, benzoxazolyl and benzoisoxazolyl, each optionally substituted with 1 , 2, 3 or 4 substituents selected from halogen, CN, oxo, (CH2)nORa, (CH2)nOC(O)Ra, (CH2)nOC(O)ORa, (CH2)nOC(O)NRbRc, (CH2)nNRbRc, (CH2)nNRdC(O)Ra, (CH2)nNRdC(O)ORa, (CH2)nNRdC(O)NRbRc, (CH2)nNRdC(O)C(O)NRbRc, (CH2)nNRdC(S)Ra, (CH2)nNRdC(S)ORa, (CH2)nNRdC(S)NRbRc,
(CH2)nNRdC(NRe)NRbRc, (CH2)nNRdS(O)Ra, (CH2)nNRdSO2Ra,
(CH2)nNRdSO2NRbRc, (CH2)nC(O)Ra, (CH2)nC(O)ORa, (CH2)nC(O)NRbRc, (CH2)nC(S)Ra, (CH2)nC(S)ORa, (CH2)nC(S)NRbRc, (CH2)nC(NRe)NRbRc, (CH2)nSRa, (CH2)nS(O)Ra, (CH2)nSO2Ra, (CH2)nSO2NRbRc, C1-6 alkyl, C1-6 haloalkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8
membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl, wherein each of the C-i-6 alkyl, C2-6 alkenyl, C2-6 alkynyl, (CH2)nC3-8 cycloalkyl, (CH2)n3-8 membered heterocycloalkyl, (CH2)nphenyl, (CH2)nnaphthyl and (CH2)n5-10 membered heteroaryl groups is optionally substituted with 1 , 2, 3, 4 or 5 Rf substituents.
38. The compound of any one of claims 1 to 37, or a pharmaceutically acceptable salt thereof, wherein R1, at each occurrence, is selected from hydrogen, halogen, CN, CF3 and methyl.
39. The compound of claim 38, or a pharmaceutically acceptable salt thereof, wherein R1, at each occurrence, is hydrogen.
40. The compound of any one of claims 1 to 39, or a pharmaceutically acceptable salt thereof, wherein R4 is hydrogen.
41 . A compound selected from the compounds in Table 2, or a
pharmaceutically acceptable salt thereof.
42. A pharmaceutical composition comprising a compound of any one of claims 1 to 41 , or a pharmaceutically acceptable salt thereof.
43. The pharmaceutical composition of claim 42, wherein the
pharmaceutical composition is formulated for oral, sublingual, subcutaneous, parenteral, intravenous, intranasal, topical, transdermal, intraperitoneal, intramuscular, intrapulmonary, vaginal, rectal, or intraocular administration.
44. The pharmaceutical composition of claim 43, wherein the
pharmaceutical composition is formulated for oral administration.
45. The use of a compound of any one of claims 1 to 41 , or a
pharmaceutically acceptable salt thereof, for the preparation of a medicament for the treatment of a disease or condition selected from neuromuscular disorders, conditions of muscle wasting, muscular myopathies, rehabilitation- related deficits, peripheral vascular disease, peripheral arterial disease, frailty, muscle atrophy and fatigue, metabolic syndrome, chronic fatigue syndrome, and obesity.
46. The use of a compound of any one of claims 1 to 41 , or a
pharmaceutically acceptable salt thereof, for the preparation of a medicament for the treatment of a disease selected from Amyotrophic Lateral Sclerosis (ALS), Spinal Muscular Atrophy (SMA) and myasthenia gravis.
47. The use of a compound of any one of claims 1 to 41 , or a
pharmaceutically acceptable salt thereof, for the preparation of a medicament for the treatment of a disease selected from peripheral vascular disease and peripheral arterial disease.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/642,227 US9133123B2 (en) | 2010-04-23 | 2011-04-22 | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
| EP11772807.1A EP2560488B1 (en) | 2010-04-23 | 2011-04-22 | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
| US14/823,605 US9994528B2 (en) | 2010-04-23 | 2015-08-11 | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US32759710P | 2010-04-23 | 2010-04-23 | |
| US32753810P | 2010-04-23 | 2010-04-23 | |
| US61/327,538 | 2010-04-23 | ||
| US61/327,597 | 2010-04-23 | ||
| US41229910P | 2010-11-10 | 2010-11-10 | |
| US41230210P | 2010-11-10 | 2010-11-10 | |
| US61/412,302 | 2010-11-10 | ||
| US61/412,299 | 2010-11-10 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/642,227 A-371-Of-International US9133123B2 (en) | 2010-04-23 | 2011-04-22 | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
| US14/823,605 Continuation US9994528B2 (en) | 2010-04-23 | 2015-08-11 | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2011133920A1 true WO2011133920A1 (en) | 2011-10-27 |
Family
ID=44834538
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2011/033650 WO2011133920A1 (en) | 2010-04-23 | 2011-04-22 | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
Country Status (3)
| Country | Link |
|---|---|
| US (2) | US9133123B2 (en) |
| EP (1) | EP2560488B1 (en) |
| WO (1) | WO2011133920A1 (en) |
Cited By (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8962632B2 (en) | 2010-04-23 | 2015-02-24 | Cytokinetics, Inc. | Certain amino-pyrimidines, compositions thereof, and methods for their use |
| US8969346B2 (en) | 2010-04-23 | 2015-03-03 | Cytokinetics, Inc. | Amino-pyridazine skeletal muscle modulators |
| US9156831B2 (en) | 2013-01-23 | 2015-10-13 | Astrazeneca Ab | Chemical compounds |
| WO2016202341A1 (en) * | 2015-06-15 | 2016-12-22 | Nmd Pharma Aps | Compounds for use in treating neuromuscular disorders |
| US20170044152A1 (en) * | 2012-10-12 | 2017-02-16 | Calcimedica, Inc. | Compounds that modulate intracellular calcium |
| WO2017139526A1 (en) | 2016-02-12 | 2017-08-17 | Astellas Pharma Inc. | Tetrahydroisoquinoline derivatives |
| US9783522B2 (en) | 2014-04-24 | 2017-10-10 | Mitsubishi Tanabe Pharma Corporation | 2-amino-pyridine and 2-amino-pyrimidine derivatives and medicinal use thereof |
| US9951069B1 (en) | 2017-01-11 | 2018-04-24 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US9994528B2 (en) | 2010-04-23 | 2018-06-12 | Cytokinetics, Inc. | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
| US10308615B2 (en) | 2015-05-29 | 2019-06-04 | Pfizer Inc. | Heterocyclic compounds as inhibitors of Vanin-1 enzyme |
| US10336738B2 (en) | 2010-08-27 | 2019-07-02 | Calcimedica, Inc. | Compounds that modulate intracellular calcium |
| US10357493B2 (en) | 2017-03-10 | 2019-07-23 | Selenity Therapeutics (Bermuda), Ltd. | Metalloenzyme inhibitor compounds |
| US10385028B2 (en) | 2017-12-14 | 2019-08-20 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US10421756B2 (en) | 2015-07-06 | 2019-09-24 | Rodin Therapeutics, Inc. | Heterobicyclic N-aminophenyl-amides as inhibitors of histone deacetylase |
| US10722495B2 (en) | 2017-09-08 | 2020-07-28 | Incyte Corporation | Cyanoindazole compounds and uses thereof |
| US10745388B2 (en) | 2018-02-20 | 2020-08-18 | Incyte Corporation | Indazole compounds and uses thereof |
| US10752635B2 (en) | 2018-02-20 | 2020-08-25 | Incyte Corporation | Indazole compounds and uses thereof |
| US10800761B2 (en) | 2018-02-20 | 2020-10-13 | Incyte Corporation | Carboxamide compounds and uses thereof |
| US10899755B2 (en) | 2018-08-08 | 2021-01-26 | Incyte Corporation | Benzothiazole compounds and uses thereof |
| US10906888B2 (en) | 2016-07-14 | 2021-02-02 | Pfizer Inc. | Pyrimidine carboxamides as inhibitors of Vanin-1 enzyme |
| US10919902B2 (en) | 2015-07-06 | 2021-02-16 | Alkermes, Inc. | Hetero-halo inhibitors of histone deacetylase |
| US10934288B2 (en) | 2016-09-09 | 2021-03-02 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US10980807B2 (en) | 2016-02-09 | 2021-04-20 | Inventisbio Llc | Inhibitor of indoleamine-2,3-dioxygenase (IDO) |
| US11014929B2 (en) | 2016-09-09 | 2021-05-25 | Incyte Corporation | Pyrazolopyrimidine compounds and uses thereof |
| US11066394B2 (en) | 2019-08-06 | 2021-07-20 | Incyte Corporation | Solid forms of an HPK1 inhibitor |
| US11111247B2 (en) | 2018-09-25 | 2021-09-07 | Incyte Corporation | Pyrazolopyrimidine compounds and uses thereof |
| US11142516B2 (en) | 2017-12-26 | 2021-10-12 | Cytokinetics, Inc. | Process for the preparation of an amino-pyrimidine and intermediates thereof |
| US11147788B2 (en) | 2017-12-14 | 2021-10-19 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11225475B2 (en) | 2017-08-07 | 2022-01-18 | Alkermes, Inc. | Substituted pyridines as inhibitors of histone deacetylase |
| US11229205B2 (en) | 2017-09-22 | 2022-01-25 | Syngenta Participations Ag | Herbicidally active pyridyl-/pyrimidyl-pyrazine derivatives |
| US11242343B2 (en) | 2016-09-09 | 2022-02-08 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US11299473B2 (en) | 2018-04-13 | 2022-04-12 | Incyte Corporation | Benzimidazole and indole compounds and uses thereof |
| US11406624B2 (en) | 2017-02-15 | 2022-08-09 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US11427558B1 (en) | 2019-07-11 | 2022-08-30 | ESCAPE Bio, Inc. | Indazoles and azaindazoles as LRRK2 inhibitors |
| WO2022230912A1 (en) | 2021-04-28 | 2022-11-03 | アステラス製薬株式会社 | Substituted triazine compound |
| US11591284B2 (en) | 2017-12-14 | 2023-02-28 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11730714B2 (en) | 2017-12-14 | 2023-08-22 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11780812B2 (en) | 2016-12-30 | 2023-10-10 | Orsobio, Inc. | Oxopyridine derivatives useful as aminocarboxymuconate semialdehyde decarboxylase (ACMSD) inhibitors |
| WO2024064745A1 (en) | 2022-09-21 | 2024-03-28 | Cytokinetics, Incorporated | Synthesis of reldesemtiv |
| WO2024193699A1 (en) * | 2023-03-23 | 2024-09-26 | 成都赜灵生物医药科技有限公司 | Triazine compound and use thereof |
| KR102722949B1 (en) | 2015-06-15 | 2024-10-28 | 엔엠디 파마 에이/에스 | Compounds for use in the treatment of neuromuscular disorders |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011146313A1 (en) | 2010-05-19 | 2011-11-24 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| GEP20166432B (en) | 2011-09-27 | 2016-02-10 | Novartis Ag | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| KR102063098B1 (en) | 2011-10-03 | 2020-01-08 | 더 유니버시티 오브 노쓰 캐롤라이나 엣 채플 힐 | Pyrrolopyrimidine compounds for the treatment of cancer |
| UY34632A (en) | 2012-02-24 | 2013-05-31 | Novartis Ag | OXAZOLIDIN- 2- ONA COMPOUNDS AND USES OF THE SAME |
| US9567326B2 (en) | 2012-05-22 | 2017-02-14 | The University Of North Carolina At Chapel Hill | Pyrimidine compounds for the treatment of cancer |
| US9562047B2 (en) | 2012-10-17 | 2017-02-07 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| US9296733B2 (en) | 2012-11-12 | 2016-03-29 | Novartis Ag | Oxazolidin-2-one-pyrimidine derivative and use thereof for the treatment of conditions, diseases and disorders dependent upon PI3 kinases |
| EP2925752A4 (en) | 2012-11-27 | 2016-06-01 | Univ North Carolina | Pyrimidine compounds for the treatment of cancer |
| CA2903979A1 (en) | 2013-03-14 | 2014-09-18 | Novartis Ag | 3-pyrimidin-4-yl-oxazolidin-2-ones as inhibitors of mutant idh |
| AU2015244171A1 (en) | 2014-04-11 | 2016-11-03 | The University Of North Carolina At Chapel Hill | MerTK-specific pyrimidine compounds |
| HRP20220183T1 (en) | 2014-04-29 | 2022-04-29 | Cytokinetics, Inc. | Methods of reducing decline in vital capacity |
| US10709708B2 (en) | 2016-03-17 | 2020-07-14 | The University Of North Carolina At Chapel Hill | Method of treating cancer with a combination of MER tyrosine kinase inhibitor and an epidermal growth factor receptor (EGFR) inhibitor |
| GB2579747B8 (en) | 2017-08-04 | 2022-10-05 | Skyhawk Therapeutics Inc | Methods and compositions for modulating splicing |
| WO2020163405A1 (en) | 2019-02-05 | 2020-08-13 | Skyhawk Therapeutics, Inc. | Methods and compositions for modulating splicing |
| JP2022519323A (en) | 2019-02-06 | 2022-03-22 | スカイホーク・セラピューティクス・インコーポレーテッド | Methods and compositions for regulating splicing |
| JP2023548342A (en) | 2020-11-06 | 2023-11-16 | サイトキネティックス, インコーポレイテッド | Bicyclic 1,4-diazepanones and their therapeutic uses |
| KR20240133737A (en) * | 2022-01-07 | 2024-09-04 | 트랜스테라 사이언시즈 (난징), 인크. | NLRP3 inflammasome inhibitors and their applications |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4157392A (en) * | 1977-05-17 | 1979-06-05 | Diamond Shamrock Corporation | Pharmacologically active substituted 1,2,4-triazines |
| US6780869B1 (en) * | 1999-11-26 | 2004-08-24 | Smithkline Beecham Corporation | Pyrimidine derivatives |
| US7652009B2 (en) * | 2004-11-30 | 2010-01-26 | Amgem Inc. | Substituted heterocycles and methods of use |
Family Cites Families (250)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CH361573A (en) | 1957-07-12 | 1962-04-30 | Cilag Chemie Aktiengesellschaf | Process for the preparation of new 5-aminomethyl-pyrimidines |
| US3239345A (en) | 1965-02-15 | 1966-03-08 | Estrogenic compounds and animal growth promoters | |
| US4411890A (en) | 1981-04-14 | 1983-10-25 | Beckman Instruments, Inc. | Synthetic peptides having pituitary growth hormone releasing activity |
| GB1345880A (en) | 1971-06-18 | 1974-02-06 | Cepbepe | Pyridazine derivatives |
| US4036979A (en) | 1974-01-25 | 1977-07-19 | American Cyanamid Company | Compositions containing 4,5,6,7-tetrahydrobenz[b]thien-4-yl-ureas or derivatives and methods of enhancing growth rate |
| HU175471B (en) | 1977-06-13 | 1980-08-28 | Gyogyszerkutato Intezet | Process for producing new 3-bracketul-pyrazolyl-bracket closed-pyridazine derivatives |
| DE2730467A1 (en) | 1977-07-06 | 1979-01-18 | Basf Ag | BENZYLPYRIMIDINE, METHOD FOR THE PRODUCTION THEREOF, AND MEDICINAL PRODUCTS CONTAINING THE SAME |
| FR2510997A1 (en) | 1981-08-10 | 1983-02-11 | Sanofi Sa | NOVEL DERIVATIVES OF METHYL-4-PHENYL-6-PYRIDAZINE, PROCESS FOR THEIR PREPARATION AND ACTIVE MEDICINES ON CENTRAL NERVOUS SYSTEM CONTAINING THE SAME |
| FR2540115B1 (en) | 1983-01-28 | 1985-06-07 | Sanofi Sa | PYRIDAZINE DERIVATIVE HAVING PSYCHOTROPIC ACTION, METHOD OF PREPARATION THEREOF AND MEDICAMENTS CONTAINING SAME |
| CA1218655A (en) | 1983-01-28 | 1987-03-03 | Kathleen Biziere | Process for the preparation of pyridazine derivatives having a psychotropic action |
| US4868183A (en) | 1986-07-21 | 1989-09-19 | Otsuka Pharmaceutical Factory, Inc. | N-pyrazinyl substituted P-aminophenols |
| JPS63165376A (en) | 1986-12-27 | 1988-07-08 | Nippon Soda Co Ltd | Oxa(thia)diazole derivative and production thereof and acaricidal agent |
| DK175099B1 (en) | 1987-06-25 | 2004-06-01 | Dow Agrosciences Llc | Process for the preparation of urea derivatives |
| WO1989007110A1 (en) | 1988-01-28 | 1989-08-10 | Eastman Kodak Company | Polypeptide compounds having growth hormone releasing activity |
| ATE113607T1 (en) | 1988-01-28 | 1994-11-15 | Polygen Holding Corp | POLYPEPTIDE COMPOUNDS WITH GROWTH HORMONE RELEASING ACTIVITY. |
| JPH01261381A (en) | 1988-04-12 | 1989-10-18 | Nippon Soda Co Ltd | Oxa(thia)diazole derivative, its production and miticide |
| FR2636628B1 (en) | 1988-08-25 | 1990-12-28 | Sanofi Sa | THIADIAZOLE-1,3,4 DERIVATIVES, PROCESS FOR OBTAINING SAME AND PHARMACEUTICAL COMPOSITIONS CONTAINING SAME |
| JPH02164863A (en) | 1988-12-15 | 1990-06-25 | Otsuka Pharmaceut Co Ltd | Production of p-aminophenol derivative |
| FR2663326B2 (en) | 1989-11-17 | 1992-10-16 | Sanofi Sa | PYRIDAZINE DERIVATIVES, PREPARATION METHOD AND PHARMACEUTICAL COMPOSITIONS CONTAINING THE SAME. |
| US5461053A (en) | 1989-02-07 | 1995-10-24 | Sanofi | Pyridazine derivatives |
| US5656631A (en) | 1989-02-07 | 1997-08-12 | Sanofi | Pyridazine derivatives |
| GR900100380A (en) | 1989-05-20 | 1991-10-10 | Fisons Plc | Process for the preparation of anti-inflammatory aminophenol derivatives |
| IL96891A0 (en) | 1990-01-17 | 1992-03-29 | Merck Sharp & Dohme | Indole-substituted five-membered heteroaromatic compounds,their preparation and pharmaceutical compositions containing them |
| GB9008123D0 (en) | 1990-04-10 | 1990-06-06 | Lilly Industries Ltd | Pharmaceutical compounds |
| US5208248A (en) | 1991-01-11 | 1993-05-04 | Merck Sharpe & Dohme, Ltd. | Indazole-substituted five-membered heteroaromatic compounds |
| US5317103A (en) | 1991-01-15 | 1994-05-31 | Merck Sharp & Dohme Limited | Indole-substituted five-membered heteroaromatic compounds as 5-HT1 agonists |
| JP2651755B2 (en) | 1991-03-01 | 1997-09-10 | 富士写真フイルム株式会社 | Silver halide color photographic materials |
| IL101291A0 (en) | 1991-03-22 | 1992-11-15 | Nippon Soda Co | 2-pyridine derivatives,their preparation and their use as fungicides |
| US5114958A (en) | 1991-05-09 | 1992-05-19 | Warner-Lambert Company | 1,2,4-oxadiazole and 1,2,4-thiadiazole derivatives of fenamates as antiinflammatory agents |
| FR2676444B1 (en) | 1991-05-16 | 1995-03-10 | Sanofi Elf | NOVEL AMINO-3 PYRIDAZINE DERIVATIVES ACTIVE IN THE CENTRAL NERVOUS SYSTEM, PREPARATION METHOD AND PHARMACEUTICAL COMPOSITIONS CONTAINING THE SAME. |
| US5492919A (en) | 1991-08-03 | 1996-02-20 | Smithkline Beecham P.L.C. | 5-HT4 receptor antagonists |
| US5663146A (en) | 1991-08-22 | 1997-09-02 | Administrators Of The Tulane Educational Fund | Polypeptide analogues having growth hormone releasing activity |
| JPH05117255A (en) | 1991-10-25 | 1993-05-14 | Nippon Soda Co Ltd | Oxadiazole and thiadiazole derivative and their production |
| BR9305466A (en) | 1992-03-26 | 1994-12-20 | Dowelanco | N-heterocyclic nitroanilines used as fungicides |
| US5654322A (en) | 1992-08-11 | 1997-08-05 | Wakunaga Seiyaku Kabushiki Kaisha | Biphenylmethane derivatives and pharmaceuticals containing the same |
| CA2165344A1 (en) | 1993-06-15 | 1994-12-22 | Peter C. Canning | Immune stimulants in bacterial infections |
| ZW8594A1 (en) | 1993-08-11 | 1994-10-12 | Bayer Ag | Substituted azadioxacycbalkenes |
| US5776983A (en) | 1993-12-21 | 1998-07-07 | Bristol-Myers Squibb Company | Catecholamine surrogates useful as β3 agonists |
| DE4424788A1 (en) | 1993-12-22 | 1995-06-29 | Bayer Ag | Arylacetic acid derivatives |
| US6008257A (en) | 1994-01-28 | 1999-12-28 | Bayer Aktiengesellschaft | Hydroxamic-acid derivatives, method of preparing them and their use as fungicides |
| GB9405347D0 (en) | 1994-03-18 | 1994-05-04 | Agrevo Uk Ltd | Fungicides |
| US6334997B1 (en) | 1994-03-25 | 2002-01-01 | Isotechnika, Inc. | Method of using deuterated calcium channel blockers |
| CN1087725C (en) | 1994-03-25 | 2002-07-17 | 同位素技术有限公司 | Enhancement of the effect of drugs by deuteration |
| US5488064A (en) | 1994-05-02 | 1996-01-30 | Bristol-Myers Squibb Company | Benzo 1,3 dioxole derivatives |
| US5922751A (en) | 1994-06-24 | 1999-07-13 | Euro-Celtique, S.A. | Aryl pyrazole compound for inhibiting phosphodiesterase IV and methods of using same |
| MX9701764A (en) | 1994-09-09 | 1997-06-28 | Bayer Ag | Imidic acid derivatives and their use as pesticides. |
| US5491134A (en) | 1994-09-16 | 1996-02-13 | Bristol-Myers Squibb Company | Sulfonic, phosphonic or phosphiniic acid β3 agonist derivatives |
| US5541204A (en) | 1994-12-02 | 1996-07-30 | Bristol-Myers Squibb Company | Aryloxypropanolamine β 3 adrenergic agonists |
| WO1996036633A1 (en) | 1995-05-17 | 1996-11-21 | E.I. Du Pont De Nemours And Company | Fungicidal cyclic amides |
| TW502026B (en) | 1995-06-20 | 2002-09-11 | Zeneca Ltd | Aromatic compounds useful as antagonists of e-type prostaglandins, processes for the preparation thereof, pharmaceutical compositions comprising the compounds, and intermediates |
| TW434240B (en) | 1995-06-20 | 2001-05-16 | Zeneca Ltd | Aromatic compounds, preparation thereof and pharmaceutical composition comprising same |
| CZ394097A3 (en) | 1995-06-20 | 1998-09-16 | E. I. Du Pont De Nemours And Company | Anthropodal and fungicidal amides |
| DE69635254T2 (en) | 1995-07-07 | 2006-07-13 | Astrazeneca Ab | Ortho-substituted aromatic compounds containing three (het) aryl rings, their preparation and their use as prostaglandin E2 (PGE2) antagonists |
| DE19525969A1 (en) | 1995-07-17 | 1997-01-23 | Bayer Ag | Ether derivatives |
| US20070173465A9 (en) | 1995-10-11 | 2007-07-26 | Monahan Sean D | Expression of zeta negative and zeta positive nucleic acids using a dystrophin gene |
| CN1056370C (en) | 1995-10-17 | 2000-09-13 | 化学工业部沈阳化工研究院 | 4-aryloxy (arylthio or arylamino) pyrimidine derivative with herbicide active, and method for prepn. of same |
| US6236946B1 (en) | 1995-12-13 | 2001-05-22 | Thomas S. Scanlan | Nuclear receptor ligands and ligand binding domains |
| US6114537A (en) | 1996-02-26 | 2000-09-05 | Apotex Inc. | Process for scavenging thiols |
| US5874452A (en) | 1996-04-03 | 1999-02-23 | Merck & Co., Inc. | Biheteroaryl inhibitors of farnesyl-protein transferase |
| US5859035A (en) | 1996-04-03 | 1999-01-12 | Merck & Co., Inc. | Arylheteroaryl inhibitors of farnesyl-protein transferase |
| EP0904076A1 (en) | 1996-04-03 | 1999-03-31 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| AU715606B2 (en) | 1996-04-03 | 2000-02-03 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| US5883105A (en) | 1996-04-03 | 1999-03-16 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| EP0891356A1 (en) | 1996-04-03 | 1999-01-20 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| US5854265A (en) | 1996-04-03 | 1998-12-29 | Merck & Co., Inc. | Biheteroaryl inhibitors of farnesyl-protein transferase |
| AU716381B2 (en) | 1996-04-03 | 2000-02-24 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| US5770615A (en) | 1996-04-04 | 1998-06-23 | Bristol-Myers Squibb Company | Catecholamine surrogates useful as β3 agonists |
| TW414795B (en) | 1996-07-01 | 2000-12-11 | Yamanouchi Pharma Co Ltd | A thiophene derivative and the pharmaceutical composition |
| JP2000516583A (en) | 1996-08-01 | 2000-12-12 | イー・アイ・デユポン・ドウ・ヌムール・アンド・カンパニー | Cyclic amides that are arthropodic and fungicidal and fungicidal |
| WO1998023155A1 (en) | 1996-11-26 | 1998-06-04 | E.I. Du Pont De Nemours And Company | Arthropodicidal and fungicidal cyclic amides |
| US6187797B1 (en) | 1996-12-23 | 2001-02-13 | Dupont Pharmaceuticals Company | Phenyl-isoxazoles as factor XA Inhibitors |
| US5939439A (en) | 1996-12-30 | 1999-08-17 | Merck & Co., Inc. | Inhibitors of farnesyl-protein transferase |
| HRP980093A2 (en) | 1997-02-28 | 1998-12-31 | Lilly Co Eli | Heterocyclic compounds, pharmaceutical compositions comprising same, and methods for inhibiting "beta"-amyloid peptide release and/or its synthesis by use of such compounds |
| DE19725450A1 (en) | 1997-06-16 | 1998-12-17 | Hoechst Schering Agrevo Gmbh | 4-Haloalkyl-3-heterocyclylpyridines and 4-haloalkyl-5-heterocyclylpyrimidines, processes for their preparation, compositions containing them and their use as pesticides |
| US6699853B2 (en) | 1997-06-16 | 2004-03-02 | Hoechst Schering Agrevo Gmbh | 4-haloalkyl-3-heterocyclylpyridines, 4-haloalkyl-5-heterocyclyl-pyrimidines and 4-trifluoromethyl-3-oxadiazolylpyridines, processes for their preparation, compositions comprising them, and their use as pesticides |
| GB9713739D0 (en) | 1997-06-27 | 1997-09-03 | Karobio Ab | Thyroid receptor ligands |
| GB9716446D0 (en) | 1997-08-05 | 1997-10-08 | Agrevo Uk Ltd | Fungicides |
| AU3289299A (en) | 1998-02-19 | 1999-09-06 | Tularik Inc. | Antiviral agents |
| US6506782B1 (en) | 1998-02-27 | 2003-01-14 | Athena Neurosciences, Inc. | Heterocyclic compounds, pharmaceutical compositions comprising same, and methods for inhibiting β-amyloid peptide release and/or its synthesis by use of such compounds |
| DE19824175A1 (en) | 1998-05-29 | 1999-12-02 | Novartis Ag | Amino azole compounds |
| SK1892001A3 (en) | 1998-08-18 | 2001-11-06 | Ucb Sa | Muscarinic agonists and antagonists, pharmaceutical composition them comprising and use thereof |
| WO2000024725A1 (en) | 1998-10-26 | 2000-05-04 | Vertex Pharmaceuticals Incorporated | Pentacyclic compounds useful as inhibitors of hepatitis c virus ns3 helicase |
| WO2000025768A1 (en) | 1998-10-29 | 2000-05-11 | Trega Biosciences, Inc. | Oxadiazole, thiadiazole and triazole derivatives and combinatorial libraries thereof |
| US20040053900A1 (en) | 1998-12-23 | 2004-03-18 | Pharmacia Corporation | Method of using a COX-2 inhibitor and an aromatase inhibitor as a combination therapy |
| GB9828442D0 (en) | 1998-12-24 | 1999-02-17 | Karobio Ab | Novel thyroid receptor ligands and method II |
| SE1035115T5 (en) * | 1999-02-24 | 2015-08-04 | Hoffmann La Roche | 4-phenylpyridine derivatives and their use as NK-1 receptor antagonists |
| JP2000281579A (en) | 1999-03-29 | 2000-10-10 | Sumitomo Pharmaceut Co Ltd | Proteoglycan production-accelerating agent containing oxadiazolyl-1,4-dihydropyridine derivative |
| US6548529B1 (en) | 1999-04-05 | 2003-04-15 | Bristol-Myers Squibb Company | Heterocyclic containing biphenyl aP2 inhibitors and method |
| US6410254B1 (en) | 1999-05-18 | 2002-06-25 | Cytokinetics | Compositions and assays utilizing ADP or phosphate for detecting protein modulators |
| US6743599B1 (en) | 1999-05-18 | 2004-06-01 | Cytokinetics, Inc. | Compositions and assays utilizing ADP or phosphate for detecting protein modulators |
| US7202051B1 (en) | 1999-05-18 | 2007-04-10 | Cytokinetics, Inc. | Compositions and assays utilizing ADP or phosphate for detecting protein modulators |
| AU763976B2 (en) | 1999-07-23 | 2003-08-07 | Shionogi & Co., Ltd. | Th2 differentiation inhibitors |
| CA2381639A1 (en) | 1999-08-12 | 2001-02-22 | Basf Aktiengesellschaft | Substituted benzoxazoles |
| EP1078632A1 (en) | 1999-08-16 | 2001-02-28 | Sanofi-Synthelabo | Use of monoamine oxydase inhibitors for the manufacture of drugs intended for the treatment of obesity |
| JP4144978B2 (en) | 1999-09-09 | 2008-09-03 | 富士フイルム株式会社 | Synthesis method of 1,2,4-thiadiazole derivatives |
| US6844367B1 (en) | 1999-09-17 | 2005-01-18 | Millennium Pharmaceuticals, Inc. | Benzamides and related inhibitors of factor Xa |
| US6632815B2 (en) | 1999-09-17 | 2003-10-14 | Millennium Pharmaceuticals, Inc. | Inhibitors of factor Xa |
| WO2001021160A2 (en) | 1999-09-23 | 2001-03-29 | Axxima Pharmaceuticals Aktiengesellschaft | Carboxymide and aniline derivatives as selective inhibitors of pathogens |
| KR20020058046A (en) | 1999-11-29 | 2002-07-12 | 한스 루돌프 하우스, 헨리테 브룬너, 베아트리체 귄터 | Pesticidal N-Heteroaryl Alpha-Alkoximino-Carboxamides |
| NZ518931A (en) | 1999-12-02 | 2004-02-27 | Novartis Ag | Aminoheterocyclyamides as Pesticides and Antiparasitic Agents |
| US6602872B1 (en) | 1999-12-13 | 2003-08-05 | Merck & Co., Inc. | Substituted pyridazines having cytokine inhibitory activity |
| AU2010601A (en) | 1999-12-16 | 2001-07-03 | Novartis Ag | Organic compounds |
| TWI284639B (en) | 2000-01-24 | 2007-08-01 | Shionogi & Co | A compound having thrombopoietin receptor agonistic effect |
| US20010049373A1 (en) | 2000-01-28 | 2001-12-06 | Chalquest Richard R. | Materials and methods for killing nematodes and nematode eggs |
| DE10006453A1 (en) | 2000-02-14 | 2001-08-16 | Bayer Ag | New piperidylcarboxylic acid derivatives, are integrin antagonists useful for treating inflammatory, autoimmune or immunological diseases, e.g. atherosclerosis, asthma, diabetes, rheumatoid arthritis or transplant rejection |
| US7078536B2 (en) | 2001-03-14 | 2006-07-18 | Genesoft Pharmaceuticals, Inc. | Charged compounds comprising a nucleic acid binding moiety and uses therefor |
| CN1431896A (en) | 2000-04-04 | 2003-07-23 | 盐野义制药株式会社 | Oily compsns. contg. highly fat-soluble drugs |
| AU2001244610A1 (en) | 2000-04-05 | 2001-10-23 | Shionogi And Co., Ltd. | Oil-in-water microemulsions containing tricyclic compounds or preconcentrates thereof |
| JP2004501913A (en) | 2000-06-23 | 2004-01-22 | ブリストル−マイヤーズ スクイブ ファーマ カンパニー | Heteroaryl-phenyl substituted factor Xa inhibitors |
| CN1439008A (en) | 2000-06-23 | 2003-08-27 | 布里斯托尔-迈尔斯斯奎布药品公司 | 1-(heteroaryl-henyl)-condensed pyrazol derivatives as factor XA inhibitors |
| PE20020384A1 (en) | 2000-07-21 | 2002-05-28 | Schering Corp | PEPTIDES AS INHIBITORS OF THE HEPATITIS C VIRUS SERINE NS3 / NS4a PROTEASE |
| EP1178036A1 (en) | 2000-08-04 | 2002-02-06 | Aventis Cropscience S.A. | Fungicidal phenylimidate derivatives |
| FR2812633A1 (en) | 2000-08-04 | 2002-02-08 | Aventis Cropscience Sa | PHENYL (THIO) UREA AND PHENYL (THIO) CARBAMATE FUNGICIDES DERIVATIVES |
| EP1178035B1 (en) | 2000-08-04 | 2008-07-30 | Bayer CropScience S.A. | Fungicidal phenylimine derivatives |
| EP1180512A1 (en) | 2000-08-04 | 2002-02-20 | Aventis Cropscience S.A. | Fungicidal phenylimine derivatives |
| WO2002026712A2 (en) | 2000-09-29 | 2002-04-04 | Millennium Pharmaceuticals, Inc. | Quaternary amines and related inhibitors of factor xa |
| US6667326B1 (en) | 2000-11-16 | 2003-12-23 | Novartis Animal Health Us, Inc. | Pesticidal aminoheterocyclamide compounds |
| AUPR213700A0 (en) | 2000-12-18 | 2001-01-25 | Biota Scientific Management Pty Ltd | Antiviral agents |
| JP2002212169A (en) | 2001-01-12 | 2002-07-31 | Sumitomo Pharmaceut Co Ltd | Five membered heteroaromatic ring compound |
| BR0206670A (en) | 2001-01-26 | 2004-02-25 | Shionogi & Co | Halogenated compounds exhibiting thrombopoietin receptor agonism |
| JP4145654B2 (en) | 2001-01-26 | 2008-09-03 | 塩野義製薬株式会社 | Cyclic compound having thrombopoietin receptor agonist activity |
| KR20030081520A (en) | 2001-03-14 | 2003-10-17 | 오노 야꾸힝 고교 가부시키가이샤 | Remedies for depression containing ep1 antagonist as the active ingredient |
| US6960595B2 (en) | 2001-03-23 | 2005-11-01 | Bristol-Myers Squibb Pharma Company | 5-6 to 5-7 Heterobicycles as factor Xa inhibitors |
| JP2002305083A (en) | 2001-04-04 | 2002-10-18 | Mitsubishi Chemicals Corp | Organic electroluminescent element |
| EP1406632A4 (en) | 2001-06-08 | 2009-11-04 | Cytovia Inc | Substituted 3-aryl-5-aryl- 1,2,4 -oxadiazoles and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| TWI331526B (en) | 2001-09-21 | 2010-10-11 | Bristol Myers Squibb Pharma Co | Lactam-containing compounds and derivatives thereof as factor xa inhibitors |
| US6825221B2 (en) | 2001-10-18 | 2004-11-30 | Allergan, Inc. | Arylsulfanyl and heteroarylsulfanyl derivatives for treating pain |
| US6921762B2 (en) | 2001-11-16 | 2005-07-26 | Amgen Inc. | Substituted indolizine-like compounds and methods of use |
| EP1447096A1 (en) | 2001-11-19 | 2004-08-18 | Ono Pharmaceutical Co., Ltd. | Remedies for urinary frequency |
| US20050113283A1 (en) | 2002-01-18 | 2005-05-26 | David Solow-Cordero | Methods of treating conditions associated with an EDG-4 receptor |
| US6995144B2 (en) | 2002-03-14 | 2006-02-07 | Eisai Co., Ltd. | Nitrogen containing heterocyclic compounds and medicines containing the same |
| US20040110757A1 (en) | 2002-03-21 | 2004-06-10 | Thomas Arrhenius | Flt-1 ligands and their uses in the treatment of diseases regulatable by angiogenesis |
| AU2003222648A1 (en) | 2002-05-13 | 2003-12-02 | Eli Lilly And Company | Multicyclic compounds for use as melanin concentrating hormone antagonists in the treatment of obesity and diabetes |
| US7119111B2 (en) | 2002-05-29 | 2006-10-10 | Amgen, Inc. | 2-oxo-1,3,4-trihydroquinazolinyl derivatives and methods of use |
| US7232616B2 (en) | 2002-06-13 | 2007-06-19 | Tsinghua University | Organic electroluminescent materials and devices made from such materials |
| CA2489458A1 (en) | 2002-06-14 | 2003-12-24 | Altana Pharma Ag | Substituted diaminopyrimidines |
| US7449456B2 (en) | 2002-06-28 | 2008-11-11 | Astellas Pharma, Inc. | Diaminopyrimidinecarboxamide derivative |
| CA2494323C (en) | 2002-08-07 | 2011-01-25 | Neuraxon Inc. | Amino benzothiazole compounds with nos inhibitory activity |
| RU2352568C9 (en) | 2002-08-09 | 2009-06-27 | Астразенека Аб | [1,2,4]oxadiazoles (versions), methods of obtaining them, pharmaceutical composition and method of inhibiting activation of metabotropic glutamate receptors - 5 |
| MXPA05002571A (en) | 2002-09-04 | 2005-09-08 | Schering Corp | Pyrazolopyrimidines as cyclin-dependent kinase inhibitors. |
| WO2004031177A1 (en) | 2002-09-30 | 2004-04-15 | Banyu Pharmaceutical Co., Ltd. | 2-aminobenzimidazole derivative |
| DE60316013T2 (en) | 2002-11-04 | 2008-05-29 | Vertex Pharmaceuticals Inc., Cambridge | HETEROARYL PYRIMIDINE DERIVATIVES AS JAK INHIBITORS |
| EP1575919A1 (en) | 2002-11-11 | 2005-09-21 | Bayer HealthCare AG | Phenyl or heteroaryl amino alkane derivatives as ip receptor antagonist |
| EP1592421A1 (en) | 2002-12-20 | 2005-11-09 | Pharmacia Corporation | Heteroarylalkanoic acids as integrin receptor antagonists |
| US20040147561A1 (en) | 2002-12-27 | 2004-07-29 | Wenge Zhong | Pyrid-2-one derivatives and methods of use |
| US7202257B2 (en) | 2003-12-24 | 2007-04-10 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| TW200418835A (en) | 2003-01-24 | 2004-10-01 | Tanabe Seiyaku Co | A pyrazolopyrimidine compound and a process for preparing the same |
| CN100345853C (en) | 2003-01-24 | 2007-10-31 | 田边制药株式会社 | Pyrazolopyrimidine compound and method for producing the same |
| AR042956A1 (en) | 2003-01-31 | 2005-07-13 | Vertex Pharma | GIRASA INHIBITORS AND USES OF THE SAME |
| US7157455B2 (en) | 2003-02-10 | 2007-01-02 | Hoffmann-La Roche Inc. | 4-Aminopyrimidine-5-one derivatives |
| US7223788B2 (en) | 2003-02-14 | 2007-05-29 | Sanofi-Aventis Deutschland Gmbh | Substituted N-aryl heterocycles, process for their preparation and their use as medicaments |
| JP2006523184A (en) | 2003-02-22 | 2006-10-12 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフトング | Cyanopyridone derivatives as liquid crystals |
| US20050187266A1 (en) | 2003-04-15 | 2005-08-25 | Pfizer Inc | Alpha substituted carboxylic acids |
| WO2005000298A2 (en) | 2003-06-03 | 2005-01-06 | Novartis Ag | 5-membered heterocycle-based p-38 inhibitors |
| EP1628661A2 (en) | 2003-06-05 | 2006-03-01 | Vertex Pharmaceuticals Incorporated | Modulators of vr1 receptor |
| SI1354876T1 (en) | 2003-06-13 | 2005-08-31 | Servier Lab | |
| WO2005000309A2 (en) | 2003-06-27 | 2005-01-06 | Ionix Pharmaceuticals Limited | Chemical compounds |
| AU2004267094A1 (en) | 2003-08-20 | 2005-03-03 | Vertex Pharmaceuticals Incorporated | (4 -amino -1,2, 5-oxadiazol-4-yl) -hetxiroaromatic compounds useful as protein kinase inhibitors |
| US7378409B2 (en) | 2003-08-21 | 2008-05-27 | Bristol-Myers Squibb Company | Substituted cycloalkylamine derivatives as modulators of chemokine receptor activity |
| ES2651730T3 (en) | 2003-09-26 | 2018-01-29 | Exelixis, Inc. | C-Met modulators and methods of use |
| KR101104100B1 (en) | 2003-11-19 | 2012-01-12 | 제이엔씨 석유 화학 주식회사 | Photopolymerizable liquid crystal composition, its polymer or polymer composition, and optical compensation element |
| WO2005051932A1 (en) | 2003-11-28 | 2005-06-09 | Nippon Soda Co., Ltd. | Arylheterocycle derivative and agricultural or horticulrual bactericide and insecticide |
| GB0328295D0 (en) | 2003-12-05 | 2004-01-07 | Muscagen Ltd | Therapeutic compounds |
| US20070191336A1 (en) | 2003-12-24 | 2007-08-16 | Flynn Daniel L | Anti-inflammatory medicaments |
| US7319108B2 (en) | 2004-01-25 | 2008-01-15 | Sanofi-Aventis Deutschland Gmbh | Aryl-substituted heterocycles, process for their preparation and their use as medicaments |
| WO2005077368A2 (en) | 2004-02-03 | 2005-08-25 | Astrazeneca Ab | Treatment of gastro-esophageal reflux disease (gerd) |
| WO2005077373A2 (en) | 2004-02-03 | 2005-08-25 | Astrazeneca Ab | Treatment of gastro-esophageal reflux disease (gerd) |
| WO2005077345A1 (en) | 2004-02-03 | 2005-08-25 | Astrazeneca Ab | Compounds for the treatment of gastro-esophageal reflux disease |
| RU2358969C2 (en) | 2004-02-13 | 2009-06-20 | Баниу Фармасьютикал Ко., Лтд. | Condensed 4-oxopyrimidine derivative |
| US7585881B2 (en) | 2004-02-18 | 2009-09-08 | Astrazeneca Ab | Additional heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists |
| DE102004008141A1 (en) | 2004-02-19 | 2005-09-01 | Abbott Gmbh & Co. Kg | Guanidine compounds and their use as binding partners for 5-HT5 receptors |
| EP1735319B1 (en) | 2004-03-26 | 2017-05-03 | MethylGene Inc. | Inhibitors of histone deacetylase |
| WO2005095382A1 (en) | 2004-03-30 | 2005-10-13 | Kyowa Hakko Kogyo Co., Ltd. | Anti-tumor agent |
| EP1731513A4 (en) | 2004-03-30 | 2007-10-31 | Daiichi Seiyaku Co | Phenoxyacetic acid derivative and medicine containing the same |
| JP2007531755A (en) | 2004-03-31 | 2007-11-08 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Non-imidazole heterocyclic compounds |
| WO2005097751A2 (en) | 2004-03-31 | 2005-10-20 | Janssen Pharmaceutica, N.V. | Non-imidazole heterocyclic compounds as histamine h3-receptor ligands |
| MXPA06011328A (en) | 2004-04-02 | 2006-12-15 | Vertex Pharma | Azaindoles useful as inhibitors of rock and other protein kinases. |
| US7439369B2 (en) | 2004-06-22 | 2008-10-21 | Loa Alamos National Security, Llc | Method and system for hydrogen evolution and storage |
| US7705025B2 (en) | 2004-08-23 | 2010-04-27 | Eli Lilly And Company | Histamine H3 receptor agents, preparation and therapeutic uses |
| CA2577947A1 (en) | 2004-08-31 | 2006-03-09 | Banyu Pharmaceutical Co., Ltd. | Novel substituted imidazole derivative |
| MX2007005247A (en) | 2004-11-02 | 2008-03-13 | Univ Northwestern | Pyridazine compounds, compositions and methods. |
| EP1809290A2 (en) | 2004-11-03 | 2007-07-25 | Vertex Pharmaceuticals Incorporated | Pyrimidine derivatives as ion channel modulators and methods of use |
| JP2008520745A (en) | 2004-11-22 | 2008-06-19 | バーテックス ファーマシューティカルズ インコーポレイテッド | Bicyclic inhibitors of Rho kinase |
| US20070123572A1 (en) | 2005-11-28 | 2007-05-31 | Kalypsys, Inc. | Novel method of preparation of 5-chloro-3-imidazol-1-yl-[1,2,4]thiadiazole and (3-imidazol-1-yl-[1,2,4]thiadiazol-5yl)-dialkyl-amines |
| JP4932735B2 (en) | 2004-12-17 | 2012-05-16 | アムジエン・インコーポレーテツド | Aminopyrimidine compounds and methods of use |
| CA2589830A1 (en) | 2005-01-05 | 2006-07-13 | Rigel Pharmaceuticals, Inc. | Ubiquitin ligase inhibitors |
| AU2006206611A1 (en) | 2005-01-19 | 2006-07-27 | Bristol-Myers Squibb Company | 2-phenoxy-N- (1, 3 , 4-thiadizol-2-yl) pyridin-3-amine derivatives and related compounds as P2Y1 receptor inhibitors for the treatment of thromboembolic disorders |
| US20080175794A1 (en) | 2005-01-25 | 2008-07-24 | Neurogen Corporation | Substituted Pyridazinyl- and Pyrimidinyl-Quinolin-4-Ylamine Analogues |
| WO2006081230A2 (en) * | 2005-01-26 | 2006-08-03 | Schering Corporation | 3-(indazol-5-yl)-(1,2, 4) triazine derivatives and related compounds as protein kinase inhibitors for the treatment of cancer |
| JP4734346B2 (en) | 2005-02-04 | 2011-07-27 | セノミックス インコーポレイテッド | Compounds containing linked heteroaryl moieties, and novel umami flavor modifiers, tastants and taste enhancers for edible compositions |
| BRPI0607455A2 (en) | 2005-02-16 | 2009-09-01 | Astrazeneca Ab | compound, process for preparing same, use of a compound, and pharmaceutical composition |
| JP2006274133A (en) | 2005-03-30 | 2006-10-12 | Fuji Photo Film Co Ltd | Liquid crystal composition, retardation plate, and elliptical polarization plate |
| CA2603748A1 (en) | 2005-04-06 | 2006-10-12 | Exelixis, Inc. | C-met modulators and methods of use |
| EP1890697B1 (en) | 2005-06-07 | 2015-03-04 | Pharmacopeia, LLC | Azinone and diazinone v3 inhibitors for depression and stress disorders |
| BRPI0613564A2 (en) | 2005-07-04 | 2011-01-18 | Novo Nordisk As | compounds, their uses in the preparation of pharmaceutical compositions and pharmaceutical compositions comprising the same |
| WO2007012642A1 (en) | 2005-07-29 | 2007-02-01 | Basf Aktiengesellschaft | 7-amino-6-thiadiazolyl- and -oxadiazolyl- 1, 2, 4-triazolo [1, 5 -a] pyrimidine compounds and use thereof for the prevention of fungal pests |
| PT1910384E (en) | 2005-08-04 | 2013-01-23 | Sirtris Pharmaceuticals Inc | Imidazo [2,1-b]thiazole derivatives as sirtuin modulating compounds |
| US8093401B2 (en) | 2005-08-04 | 2012-01-10 | Sirtris Pharmaceuticals, Inc. | Sirtuin modulating compounds |
| KR20080047410A (en) | 2005-08-23 | 2008-05-28 | 아이알엠 엘엘씨 | Immunosuppressant compounds and compositions |
| JP2007093918A (en) | 2005-09-28 | 2007-04-12 | Fujifilm Corp | Optical low pass filter, and method for manufacturing the same |
| JP2007093919A (en) | 2005-09-28 | 2007-04-12 | Fujifilm Corp | Optical low pass filter, and method for manufacturing the same |
| WO2007037010A1 (en) | 2005-09-29 | 2007-04-05 | Daiichi Pharmaceutical Co., Ltd. | Phenoxyacetic acid derivatives and drugs using the same |
| JP5243960B2 (en) | 2005-10-21 | 2013-07-24 | ダウ アグロサイエンシィズ エルエルシー | Thieno-pyrimidine compounds having bactericidal activity |
| US7531482B2 (en) | 2005-10-21 | 2009-05-12 | Dow Agrosciences Llc | Thieno-pyrimidine compounds having fungicidal activity |
| BRPI0617891A2 (en) | 2005-10-26 | 2011-08-09 | Boehringer Ingelheim Int | (hetero) aryl compounds having mch antagonist activity, physiologically acceptable salts thereof, composition, pharmaceutical composition as well as use and preparation of said compounds |
| EP1945632B1 (en) | 2005-11-08 | 2013-09-18 | Vertex Pharmaceuticals Incorporated | Heterocyclic modulators of atp-binding cassette transporters |
| ES2970354T3 (en) | 2005-12-13 | 2024-05-28 | Incyte Holdings Corp | Pyrrolo[2,3-d]pyrimidine derivatives as Janus kinase inhibitors |
| US7825120B2 (en) | 2005-12-15 | 2010-11-02 | Cytokinetics, Inc. | Certain substituted ((piperazin-1-ylmethyl)benzyl)ureas |
| US20070197505A1 (en) | 2005-12-15 | 2007-08-23 | Morgan Bradley P | Certain chemical entities, compositions and methods |
| EP1959962A2 (en) | 2005-12-16 | 2008-08-27 | Cytokinetics, Inc. | Certain chemical entities, compositions, and methods |
| WO2007075896A2 (en) | 2005-12-22 | 2007-07-05 | Kemia, Inc. | Heterocyclic cytokine inhibitors |
| WO2007076460A2 (en) | 2005-12-23 | 2007-07-05 | Kalypsys, Inc. | Substituted thiazole ureas useful as inhibitors of protein kinases |
| ZA200807715B (en) | 2006-03-09 | 2009-11-25 | Pharmacopeia Inc | 9-Heteroarylpurine MNK2 inhibitors for treating metabolic disorders |
| JP5243696B2 (en) | 2006-03-17 | 2013-07-24 | 田辺三菱製薬株式会社 | Benzene derivatives |
| KR101054325B1 (en) | 2006-03-31 | 2011-08-04 | 얀센 파마슈티카 엔.브이. | Benzoimidazol-2-yl pyrimidine and pyrazine as histamine H4 receptor modulators |
| DK2010528T3 (en) | 2006-04-19 | 2018-01-15 | Novartis Ag | 6-O-Substituted Benzoxazole and Benzothiazole Compounds and Methods for Inhibiting CSF-1R Signaling |
| US8076486B2 (en) | 2006-04-19 | 2011-12-13 | Merck Serono S.A. | Heteroaryl-substituted arylaminopyridine derivatives as MEK inhibitors |
| WO2007127475A2 (en) | 2006-04-28 | 2007-11-08 | Northwestern University | Pyridazines for demyelinating diseases and neuropathic pain |
| WO2007130383A2 (en) | 2006-04-28 | 2007-11-15 | Northwestern University | Compositions and treatments using pyridazine compounds and secretases |
| KR101441146B1 (en) | 2006-04-28 | 2014-09-17 | 시오노기세야쿠 가부시키가이샤 | Amine derivative having npy y5 receptor antagonist activity |
| WO2007146712A2 (en) | 2006-06-09 | 2007-12-21 | Kemia, Inc. | Therapy using cytokine inhibitors |
| US8435774B2 (en) | 2006-06-28 | 2013-05-07 | Qiagen Gmbh | Enhancing reactivation of thermostable reversibly inactivated enzymes |
| MX2008015457A (en) | 2006-07-07 | 2009-01-16 | Boehringer Ingelheim Int | Phenyl substituted heteroaryl-derivatives and use thereof as anti-tumor agents. |
| WO2008013622A2 (en) | 2006-07-27 | 2008-01-31 | E. I. Du Pont De Nemours And Company | Fungicidal azocyclic amides |
| US8227603B2 (en) | 2006-08-01 | 2012-07-24 | Cytokinetics, Inc. | Modulating skeletal muscle |
| JP2010501629A (en) | 2006-08-30 | 2010-01-21 | ビオヴィトルム・アクチボラゲット(プブリクト) | Pyridine compounds for treating GPR119 related disorders |
| WO2008063888A2 (en) | 2006-11-22 | 2008-05-29 | Plexxikon, Inc. | Compounds modulating c-fms and/or c-kit activity and uses therefor |
| EP2102171A1 (en) | 2006-12-11 | 2009-09-23 | Boehringer Ingelheim International GmbH | New pyridazine derivatives with mch antagonistic activity and medicaments comprising these compounds |
| DE102007002717A1 (en) | 2007-01-18 | 2008-07-24 | Merck Patent Gmbh | Heterocyclic indazole derivatives |
| WO2009011285A1 (en) | 2007-07-13 | 2009-01-22 | Taisho Pharmaceutical Co., Ltd. | Heteroarylbenzene compounds |
| GB0720444D0 (en) | 2007-10-18 | 2007-11-28 | Glaxo Group Ltd | Novel compounds |
| US7943619B2 (en) | 2007-12-04 | 2011-05-17 | Hoffmann-La Roche Inc. | Isoxazolo-pyridazine derivatives |
| TW200940537A (en) | 2008-02-26 | 2009-10-01 | Astrazeneca Ab | Heterocyclic urea derivatives and methods of use thereof |
| US20090264433A1 (en) | 2008-04-21 | 2009-10-22 | Institute For Oneworld Health | Compounds, Compositions and Methods Comprising Triazine Derivatives |
| WO2009131947A2 (en) | 2008-04-21 | 2009-10-29 | Institute For Oneworld Health | Compounds, compositions and methods comprising pyridazine derivatives |
| US20090325902A1 (en) | 2008-06-04 | 2009-12-31 | Astrazeneca Ab | Heterocyclic urea derivatives and methods of use thereof |
| EA020545B1 (en) | 2008-06-09 | 2014-12-30 | Авд. Фарма Гмбх Унд Ко. Кг | Carboxylic acid salts of 2-amino-3-carbethoxyamino-6-(4-fluorobenzylamino)pyridine |
| AR072397A1 (en) | 2008-06-30 | 2010-08-25 | Janssen Pharmaceutica Nv | PROCESS FOR THE PREPARATION OF BENZOIMIDAZOL-2-ILPIRIMIDINE DERIVATIVES |
| US20100025641A1 (en) | 2008-08-04 | 2010-02-04 | Fujifilm Corporation | Infrared region selective reflection coat and infrared region selective reflection film |
| AU2009282567B2 (en) | 2008-08-20 | 2014-10-02 | Merck Sharp & Dohme Corp. | Substituted pyridine and pyrimidine derivatives and their use in treating viral infections |
| JPWO2010113834A1 (en) | 2009-03-30 | 2012-10-11 | アステラス製薬株式会社 | Pyrimidine compounds |
| CA2773561A1 (en) | 2009-09-14 | 2011-03-17 | Phusis Therapeutics Inc. | Pharmaceutical compositions and formulations including inhibitors of the pleckstrin homology domain and methods for using same |
| AR081626A1 (en) | 2010-04-23 | 2012-10-10 | Cytokinetics Inc | AMINO-PYRIDAZINIC COMPOUNDS, PHARMACEUTICAL COMPOSITIONS THAT CONTAIN THEM AND USE OF THE SAME TO TREAT CARDIAC AND SKELETIC MUSCULAR DISORDERS |
| EP2560488B1 (en) | 2010-04-23 | 2015-10-28 | Cytokinetics, Inc. | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
| AR081331A1 (en) | 2010-04-23 | 2012-08-08 | Cytokinetics Inc | AMINO- PYRIMIDINES COMPOSITIONS OF THE SAME AND METHODS FOR THE USE OF THE SAME |
| US8759380B2 (en) | 2011-04-22 | 2014-06-24 | Cytokinetics, Inc. | Certain heterocycles, compositions thereof, and methods for their use |
| CN108553467A (en) | 2012-04-02 | 2018-09-21 | 赛特凯恩蒂克公司 | Improve the method for diaphragm function |
| SG11201406359TA (en) | 2012-04-11 | 2014-11-27 | Cytokinetics Inc | Improving resistance to skeletal muscle fatigue |
| HRP20220183T1 (en) | 2014-04-29 | 2022-04-29 | Cytokinetics, Inc. | Methods of reducing decline in vital capacity |
| US9987279B2 (en) | 2014-09-09 | 2018-06-05 | Astellas Pharma Inc. | Pharmaceutical composition for prevention and/or treatment of urinary incontinence |
-
2011
- 2011-04-22 EP EP11772807.1A patent/EP2560488B1/en active Active
- 2011-04-22 US US13/642,227 patent/US9133123B2/en active Active
- 2011-04-22 WO PCT/US2011/033650 patent/WO2011133920A1/en active Application Filing
-
2015
- 2015-08-11 US US14/823,605 patent/US9994528B2/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4157392A (en) * | 1977-05-17 | 1979-06-05 | Diamond Shamrock Corporation | Pharmacologically active substituted 1,2,4-triazines |
| US6780869B1 (en) * | 1999-11-26 | 2004-08-24 | Smithkline Beecham Corporation | Pyrimidine derivatives |
| US7652009B2 (en) * | 2004-11-30 | 2010-01-26 | Amgem Inc. | Substituted heterocycles and methods of use |
Cited By (77)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9730886B2 (en) | 2010-04-23 | 2017-08-15 | Cytokinetics, Inc. | Amino-pyrimidine skeletal muscle modulators |
| US9604965B2 (en) | 2010-04-23 | 2017-03-28 | Cytokinetics, Inc. | Substituted pyridazines as skeletal muscle modulators |
| US9018223B2 (en) | 2010-04-23 | 2015-04-28 | Cytokinetics, Inc. | Certain amino-pyrimidines, compositions thereof, and methods for their use |
| US10076519B2 (en) | 2010-04-23 | 2018-09-18 | Cytokinetics, Inc. | Substituted pyridazines as skeletal muscle modulators |
| US10272030B2 (en) | 2010-04-23 | 2019-04-30 | Cytokinetics, Inc. | Amino-pyrimidine skeletal muscle modulators |
| US9994528B2 (en) | 2010-04-23 | 2018-06-12 | Cytokinetics, Inc. | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
| US8969346B2 (en) | 2010-04-23 | 2015-03-03 | Cytokinetics, Inc. | Amino-pyridazine skeletal muscle modulators |
| US10765624B2 (en) | 2010-04-23 | 2020-09-08 | Cytokinetics, Inc. | Amino-pyrimidine skeletal muscle modulators |
| US11369565B2 (en) | 2010-04-23 | 2022-06-28 | Cytokinetics, Inc. | Amino-pyrimidine skeletal muscle modulators |
| US8962632B2 (en) | 2010-04-23 | 2015-02-24 | Cytokinetics, Inc. | Certain amino-pyrimidines, compositions thereof, and methods for their use |
| US10336738B2 (en) | 2010-08-27 | 2019-07-02 | Calcimedica, Inc. | Compounds that modulate intracellular calcium |
| US20170044152A1 (en) * | 2012-10-12 | 2017-02-16 | Calcimedica, Inc. | Compounds that modulate intracellular calcium |
| US9657008B2 (en) | 2013-01-23 | 2017-05-23 | Astrazeneca Ab | Chemical compounds |
| US9156831B2 (en) | 2013-01-23 | 2015-10-13 | Astrazeneca Ab | Chemical compounds |
| US9783522B2 (en) | 2014-04-24 | 2017-10-10 | Mitsubishi Tanabe Pharma Corporation | 2-amino-pyridine and 2-amino-pyrimidine derivatives and medicinal use thereof |
| US10308615B2 (en) | 2015-05-29 | 2019-06-04 | Pfizer Inc. | Heterocyclic compounds as inhibitors of Vanin-1 enzyme |
| AU2016279486B2 (en) * | 2015-06-15 | 2021-04-01 | Nmd Pharma A/S | Compounds for use in treating neuromuscular disorders |
| CN107820425A (en) * | 2015-06-15 | 2018-03-20 | Nmd制药公司 | For treating the compound of neuromuscular disorder |
| WO2016202341A1 (en) * | 2015-06-15 | 2016-12-22 | Nmd Pharma Aps | Compounds for use in treating neuromuscular disorders |
| KR102722949B1 (en) | 2015-06-15 | 2024-10-28 | 엔엠디 파마 에이/에스 | Compounds for use in the treatment of neuromuscular disorders |
| RU2745065C2 (en) * | 2015-06-15 | 2021-03-18 | ЭнЭмДи ФАРМА A/C | Compounds used for the treatment of neuromuscular disorders |
| US10934244B2 (en) | 2015-06-15 | 2021-03-02 | Nmd Pharma A/S | Compounds for use in treating neuromuscular disorders |
| CN107820425B (en) * | 2015-06-15 | 2021-08-31 | Nmd制药股份公司 | Compounds for the treatment of neuromuscular disorders |
| US10919902B2 (en) | 2015-07-06 | 2021-02-16 | Alkermes, Inc. | Hetero-halo inhibitors of histone deacetylase |
| US10421756B2 (en) | 2015-07-06 | 2019-09-24 | Rodin Therapeutics, Inc. | Heterobicyclic N-aminophenyl-amides as inhibitors of histone deacetylase |
| US11858939B2 (en) | 2015-07-06 | 2024-01-02 | Alkermes, Inc. | Hetero-halo inhibitors of histone deacetylase |
| US11969425B2 (en) | 2016-02-09 | 2024-04-30 | Inventisbio Llc | Inhibitor of indoleamine-2,3-dioxygenase (IDO) |
| US10980807B2 (en) | 2016-02-09 | 2021-04-20 | Inventisbio Llc | Inhibitor of indoleamine-2,3-dioxygenase (IDO) |
| US11479561B2 (en) | 2016-02-12 | 2022-10-25 | Cytokinetics, Incorporated | Tetrahydroisoquinoline derivatives |
| US9914741B2 (en) | 2016-02-12 | 2018-03-13 | Astellas Pharma Inc. | Tetrahydroisoquinoline derivatives |
| US10689393B2 (en) | 2016-02-12 | 2020-06-23 | Astellas Pharma Inc. | Tetrahydroisoquinoline derivatives |
| WO2017139526A1 (en) | 2016-02-12 | 2017-08-17 | Astellas Pharma Inc. | Tetrahydroisoquinoline derivatives |
| US10259821B2 (en) | 2016-02-12 | 2019-04-16 | Astellas Pharma Inc. | Tetrahydroisoquinoline derivatives |
| EP4032877A1 (en) | 2016-02-12 | 2022-07-27 | Cytokinetics, Incorporated | Tetrahydroisoquinoline derivatives |
| US10906888B2 (en) | 2016-07-14 | 2021-02-02 | Pfizer Inc. | Pyrimidine carboxamides as inhibitors of Vanin-1 enzyme |
| US11795166B2 (en) | 2016-09-09 | 2023-10-24 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US11542265B2 (en) | 2016-09-09 | 2023-01-03 | Incyte Corporation | Pyrazolopyrimidine compounds and uses thereof |
| US11242343B2 (en) | 2016-09-09 | 2022-02-08 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US10934288B2 (en) | 2016-09-09 | 2021-03-02 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US11014929B2 (en) | 2016-09-09 | 2021-05-25 | Incyte Corporation | Pyrazolopyrimidine compounds and uses thereof |
| US11891388B2 (en) | 2016-09-09 | 2024-02-06 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US11780812B2 (en) | 2016-12-30 | 2023-10-10 | Orsobio, Inc. | Oxopyridine derivatives useful as aminocarboxymuconate semialdehyde decarboxylase (ACMSD) inhibitors |
| US11225479B2 (en) | 2017-01-11 | 2022-01-18 | Alkermes, Inc. | Bicyclic inhibitors of histone deacetylase |
| US11286256B2 (en) | 2017-01-11 | 2022-03-29 | Alkermes, Inc. | Bicyclic inhibitors of histone deacetylase |
| US10696673B2 (en) | 2017-01-11 | 2020-06-30 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US11987580B2 (en) | 2017-01-11 | 2024-05-21 | Alkermes, Inc. | Bicyclic inhibitors of histone deacetylase |
| US10793567B2 (en) | 2017-01-11 | 2020-10-06 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US9951069B1 (en) | 2017-01-11 | 2018-04-24 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US10519149B2 (en) | 2017-01-11 | 2019-12-31 | Rodin Therapeutics, Inc. | Bicyclic inhibitors of histone deacetylase |
| US11406624B2 (en) | 2017-02-15 | 2022-08-09 | Incyte Corporation | Pyrazolopyridine compounds and uses thereof |
| US11938134B2 (en) | 2017-03-10 | 2024-03-26 | Eikonizo Therapeutics, Inc. | Metalloenzyme inhibitor compounds |
| US10357493B2 (en) | 2017-03-10 | 2019-07-23 | Selenity Therapeutics (Bermuda), Ltd. | Metalloenzyme inhibitor compounds |
| US11225475B2 (en) | 2017-08-07 | 2022-01-18 | Alkermes, Inc. | Substituted pyridines as inhibitors of histone deacetylase |
| US11912702B2 (en) | 2017-08-07 | 2024-02-27 | Alkermes, Inc. | Substituted pyridines as inhibitors of histone deacetylase |
| US10722495B2 (en) | 2017-09-08 | 2020-07-28 | Incyte Corporation | Cyanoindazole compounds and uses thereof |
| US11229205B2 (en) | 2017-09-22 | 2022-01-25 | Syngenta Participations Ag | Herbicidally active pyridyl-/pyrimidyl-pyrazine derivatives |
| US11730714B2 (en) | 2017-12-14 | 2023-08-22 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US10385028B2 (en) | 2017-12-14 | 2019-08-20 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11591284B2 (en) | 2017-12-14 | 2023-02-28 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11147788B2 (en) | 2017-12-14 | 2021-10-19 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11142516B2 (en) | 2017-12-26 | 2021-10-12 | Cytokinetics, Inc. | Process for the preparation of an amino-pyrimidine and intermediates thereof |
| US10800761B2 (en) | 2018-02-20 | 2020-10-13 | Incyte Corporation | Carboxamide compounds and uses thereof |
| US11492354B2 (en) | 2018-02-20 | 2022-11-08 | Incyte Corporation | Indazole compounds and uses thereof |
| US11731958B2 (en) | 2018-02-20 | 2023-08-22 | Incyte Corporation | Carboxamide compounds and uses thereof |
| US10745388B2 (en) | 2018-02-20 | 2020-08-18 | Incyte Corporation | Indazole compounds and uses thereof |
| US10752635B2 (en) | 2018-02-20 | 2020-08-25 | Incyte Corporation | Indazole compounds and uses thereof |
| US11299473B2 (en) | 2018-04-13 | 2022-04-12 | Incyte Corporation | Benzimidazole and indole compounds and uses thereof |
| US11866426B2 (en) | 2018-08-08 | 2024-01-09 | Incyte Corporation | Benzothiazole compounds and uses thereof |
| US10899755B2 (en) | 2018-08-08 | 2021-01-26 | Incyte Corporation | Benzothiazole compounds and uses thereof |
| US11111247B2 (en) | 2018-09-25 | 2021-09-07 | Incyte Corporation | Pyrazolopyrimidine compounds and uses thereof |
| US11427558B1 (en) | 2019-07-11 | 2022-08-30 | ESCAPE Bio, Inc. | Indazoles and azaindazoles as LRRK2 inhibitors |
| US11066394B2 (en) | 2019-08-06 | 2021-07-20 | Incyte Corporation | Solid forms of an HPK1 inhibitor |
| US11787784B2 (en) | 2019-08-06 | 2023-10-17 | Incyte Corporation | Solid forms of an HPK1 inhibitor |
| KR20240004371A (en) | 2021-04-28 | 2024-01-11 | 아스텔라스세이야쿠 가부시키가이샤 | Substituted Triazine Compounds |
| WO2022230912A1 (en) | 2021-04-28 | 2022-11-03 | アステラス製薬株式会社 | Substituted triazine compound |
| WO2024064745A1 (en) | 2022-09-21 | 2024-03-28 | Cytokinetics, Incorporated | Synthesis of reldesemtiv |
| WO2024193699A1 (en) * | 2023-03-23 | 2024-09-26 | 成都赜灵生物医药科技有限公司 | Triazine compound and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US20160200717A1 (en) | 2016-07-14 |
| US9994528B2 (en) | 2018-06-12 |
| EP2560488B1 (en) | 2015-10-28 |
| US9133123B2 (en) | 2015-09-15 |
| EP2560488A1 (en) | 2013-02-27 |
| US20130150368A1 (en) | 2013-06-13 |
| EP2560488A4 (en) | 2013-11-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11369565B2 (en) | Amino-pyrimidine skeletal muscle modulators | |
| AU2018201953B2 (en) | Certain amino-pyridazines, compositions thereof, and methods of their use | |
| US9994528B2 (en) | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 11772807 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2011772807 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 13642227 Country of ref document: US |