WO2010130934A2 - Derives de 2-cycloamino-5-(pyridin-4-yl)imidazo[2,1-b][1,3,4]thiadiazole, leur preparation et leur application en therapeutique - Google Patents
Derives de 2-cycloamino-5-(pyridin-4-yl)imidazo[2,1-b][1,3,4]thiadiazole, leur preparation et leur application en therapeutique Download PDFInfo
- Publication number
- WO2010130934A2 WO2010130934A2 PCT/FR2010/050897 FR2010050897W WO2010130934A2 WO 2010130934 A2 WO2010130934 A2 WO 2010130934A2 FR 2010050897 W FR2010050897 W FR 2010050897W WO 2010130934 A2 WO2010130934 A2 WO 2010130934A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- compound
- general formula
- optionally substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- PJGLMYXULHFDKP-UHFFFAOYSA-N CC(C)(C)OC(N(C)c1nccc(B2OC(C)(C)C(C)(C)O2)c1)=O Chemical compound CC(C)(C)OC(N(C)c1nccc(B2OC(C)(C)C(C)(C)O2)c1)=O PJGLMYXULHFDKP-UHFFFAOYSA-N 0.000 description 1
- KNNPPOJDINBGRM-CYBMUJFWSA-N C[C@H](C1)NCCN1c([s]1)n[n]2c1nc(-c(cc1)ccc1F)c2-c1ccncc1 Chemical compound C[C@H](C1)NCCN1c([s]1)n[n]2c1nc(-c(cc1)ccc1F)c2-c1ccncc1 KNNPPOJDINBGRM-CYBMUJFWSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/06—Peri-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- R 6 represents a phenyl group optionally substituted by one or more substituents selected from halogen atoms and Ci -6 alkyl; Ci -6 alkyloxy;
- A represents a C 1-7 -alkylene group optionally substituted with one or two R 3 groups;
- R ⁇ 2 form with the carbon atom carrying them a cyclic monoamine optionally comprising an oxygen atom, this cyclic monoamine being optionally substituted with one or more groups R f which are identical to or different from each other;
- an eighth group of compounds is constituted by compounds for which:
- - R 5 represents a hydrogen atom or a C 1-3 -alkyl group, -NR 7 R 8 , C 1-3 alkyloxy;
- - R 6 represents a phenyl group optionally substituted with one or more substituents chosen from halogen atoms;
- the cyclic amine formed by -NALB- represents either a group chosen from piperazinyl and hexahydropyrrolo-pyrrolyl groups, optionally substituted with one or more groups chosen from C1 -6 alkyl groups; either a diazaspiro-undecyl group; or a piperidinyl group, optionally substituted with a pyrrolidinyl group;
- an eleventh group of compounds is constituted by the compounds for which:
- R 5 represents a hydrogen atom or a methyl, methylamino or methoxy group
- R 6 represents a phenyl group optionally substituted with one or more substituents chosen from fluorine atoms;
- the cyclic amine formed by -NALB- represents a group chosen from piperazinyl, (3R) -3-methyl-piperazinyl, 4-isopropyl-piperazinyl, (cis) -hexahydropyrrolo [3,4-c] pyrrol- 2 (1 / - /) yl; 2,9-diaza-spiro [5.5] undec-9-yl, 3,9-diaza-spiro [5.5] undec-9-yl; 4-pyrrolidin-1-yl-piperidinyl; in the form of a base or an acid addition salt.
- the subject of the invention is also a process for the preparation of the compounds of the invention of formula (I).
- the 2-cycloamino-5- (pyridin-4-yl) imidazo [2,1-t)] [1,3,4] thiadiazole derivatives of general formula (I) wherein A, L, B, R 5 and R 6 are as defined above may be prepared by metallocatalyzed coupling between a 5-haloimidazo [2,1-b] [1,3,4] derivative.
- Couplings according to the Stille method are for example carried out by heating in the presence of a catalyst such as tetrakis (triphenylphosphine) palladium, copper iodide, in a solvent such as N, N-dimethylacetamide.
- a catalyst such as tetrakis (triphenylphosphine) palladium, copper iodide
- a solvent such as N, N-dimethylacetamide.
- the couplings according to the Suzuki method are for example made by heating in the presence of a catalyst such as 1, 1 '-bis
- the 5-haloimidazo [2,1-b] [1,3,4] thiadiazole derivatives of general formula (II) are obtained by bromination or regioselective iodination of an imidazo [2,1-b] derivative [1] , 3,4] thiadiazole of the general formula (III), wherein A, L, B and R 6 are as defined above.
- This reaction may be carried out using N-bromo or iodosuccinimide or iodine monochloride in a polar solvent such as acetonitrile, tetrahydrofuran, methanol or chloroform.
- imidazo [2,1-b] [1,3,4] thiadiazole derivatives of general formula (III) as defined above can be prepared by condensation between a [1,3,4] thiadiazole derivative.
- This reaction can be carried out by heating the reactants in a polar solvent such as ethanol.
- the imidazo [2,1-t)] [1,3,4] thiadiazole derivatives of the general formula (III) in which A, L, B and R 6 are as defined above can be prepared according to from a 2-haloimidazo [2,1-t)] [1,3,4] thiadiazole derivative of general formula (V), in which R 6 is as defined above and X 2 represents a halogen, more particularly bromine, by treatment with a diamine of general formula (Va) in which A, L and B are as defined above.
- This reaction can be carried out according to the conditions of the Buchwald-Hartwig metallo-catalysis coupling reaction.
- Couplings according to the Buchwald-Hartwig method are for example carried out by heating the reactants in the presence of a catalyst such as palladium acetate, a ligand such as 4,5-bis (diphenylphosphino) -9,9 dimethylxanthene, a base such as cesium carbonate, in a solvent such as dioxane.
- a catalyst such as palladium acetate, a ligand such as 4,5-bis (diphenylphosphino) -9,9 dimethylxanthene, a base such as cesium carbonate, in a solvent such as dioxane.
- Imidazo [2,1-t)] [1,3,4] thiadiazole derivatives of general formula (V) are known (Journal of Heterocyclic Chemistry (2008), 45 (1), 299-302; Chemistry of Heterocyclic Compounds (New York, NY, United States) (2007), 43 (4), 499-504), or may be prepared by analogy according to methods known to those skilled in the art
- the derivatives of general formula (I) for which the amine formed by N, L, A and B comprises a second secondary or tertiary amine can be prepared respectively from the corresponding primary or secondary amine by alkylation or amino-reduction according to usual methods known to those skilled in the art
- this function may optionally be protected during synthesis by a protecting group, for example benzyl or t-butyloxycarbonyl.
- Example 1 (Compound No. 1): 6- (4-Fluorophenyl) -2-piperazin-1-yl-5- (pyridin-4-yl) imidazo [2,1-e] [1, 3,4] thiadiazole
- the brown oil obtained is then chromatographed on a column of 50 g of silica gel, eluting with a mixture of dichloromethane and methanol (95/5), to give 0.9 g of 4- (5-amino- [1 Tert-butyl 3,4-thiadiazol-2-yl) piperazine-1-carboxylate as a white powder after trituration in di-isopropyl ether, filtration and drying.
- the yellowish solid formed is separated by filtration and then purified by column chromatography of 50 g of silica gel, eluting with a mixture of dichloromethane, methanol and ammonia (98/2 / 0.2) to give 2.7 g. 4- [6- (4-Fluorophenyl) imidazo [2,1-b] [1,4,4] thiadiazol-2-yl] piperazine-1-tert-butylcarboxylate in powder form white after trituration in di-isopropyl ether, filtration and drying.
- the residue is purified on a column of 25 g of silica gel, eluting with a mixture of dichloromethane, methanol and ammonia (95/5 / 0.5) to give 0.225 g of 6- (4-fluorophenyl) - 2- (piperazin-1-yl) -5- (pyridin-4-yl) imidazo [2,1-t)] [1,3,4] thiadiazole as a white powder after trituration in diethyl ether; -ethyl, filtration and drying.
- Example 2 (Compound No. 6): 6- (4-fluorophenyl) -2- (4-isopropylpiperazin-1-yl) -5- (pyridin-4-yl) imidazo [2,1-ef] [1, 3,4] thiadiazole
- Example 3 (Compound No. 7): 6- (4-Fluorophenyl) -2- (4-propylpiperazin-1-yl) -5- (2-methylpyridin-4-yl) imidazo [2,1-ef] 1, 3,4] thiadiazole
- Example 4 (Compound No. 8): 4- (6- (4-Fluorophenyl) -2- (4-isopropylpiperazin-1-yl) imidazo [2,1-b] [1,3,4] thiadiazole-5 yl) - ⁇ / -méthylpyridin-2-amine
- the product is then purified by chromatography on a silica column, eluting with dichloromethane and then with a gradient of ethyl acetate.
- the resultant tert-butyl yl ⁇ pyridin-2-yl) methylcarbamate was treated with a solution of 2 ml of dichloromethane and 0.2 ml of trifluoroacetic acid for 26 hours.
- Example 5 (Compound No. 2): 6- (4-Fluorophenyl) -2- (3 (f) -methylpiperazin-1-yl) -5- (pyridin-4-yl) imidazo [2,1-ef] [1, 3,4] thiadiazole
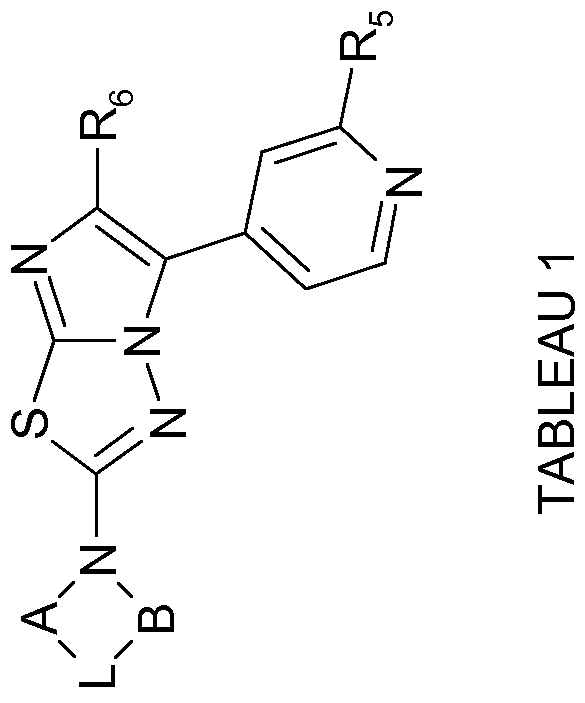
- Table 1 which follows illustrates the chemical structures and the physical properties of some compounds according to the invention.
- HCI represents a compound in hydrochloride form and the ratio in parentheses is the ratio (acid: base)
- HBr represents a compound in hydrobromide form and the ratio in parenthesis is the ratio (acid: base); : base)
- sign "-" means that the compound is in base form
- the column [ ⁇ ] d informs the result of analysis of the rotatory power of the compounds of the table at the wavelength of 589 nM
- the solvent indicated in parentheses corresponds to the solvent used to carry out the measurement of the rotary power in degrees
- the letter "c” indicates the solvent concentration in g / 100 ml.
- NA means that the measurement of the rotatory power is not applicable
- the inhibition in% of the ability of the enzyme to phosphorylate the substrate is determined.
- the most active compounds of the invention have Cl 5 0 (50% concentration inhibiting the enzymatic activity of Casein Kinase 1 Epsilon or Casein Kinase 1 Delta) of between 1 nM and 500 nM.
- the ability of the compounds of the invention to inhibit the phosphorylation of casein by casein kinases 1 epsilon and delta can be evaluated using a FRET fluorescence test ("energy transfer between fluorescent molecules", “Fluorescence Resonance Energy Transfer ”) from the"Z'Lyte TM kinase assay Kit “(reference PV3670; Invitrogen Corporation TM) according to the supplier's instructions.
- the mixture is treated with a specific site protease that specifically cleaves the substrate peptide to form two fluorescent moieties with a large fluorescence emission ratio.
- the observed fluorescence is thus related to the ability of the products of the invention to inhibit the phosphorylation of the substrate peptide by casein kinase 1 epsilon or casein kinase 1 delta.
- the compounds of the invention are dissolved at different concentrations from a 10 mM stock solution in DMSO diluted in a buffer containing 50 mM HEPS, pH 7.5, 1 mMEGTA, 0.01% Brij- 35, 10 mM MgCl for casein kinase 1 epsilon and supplemented with Trizma Base (50 mM), pH 8.0 and NaN3
- TM is made at the final concentration of 2 ⁇ M.
- concentration of ATP is 4 times the K M , which is 2 ⁇ M for casein kinase 1 epsilon and
- Stable transfectants resistant to Zeocin were evaluated for expression of the reporter by adding to the growth medium of the 100 microM luciferin (Promega # E1603 ® ®) and assaying luciferase activity on a TopCount scintillation counter ® (Packard Model # C384V00). Rat-1 cell clones expressing both zeocin resistance and mPeri-directed luciferase activity were synchronized by serum shock with 50% horse serum [HS (Gibco ® # 16050-122) ] and circadian reporter activity was evaluated. Clone P2C4 of Mper1-luc Rat-1 fibroblasts was selected for the test of the compound.
- Mood disorders include depressive disorders, such as unipolar depression, bipolar disorder, mood disorders due to a general medical condition, and mood disorders induced by pharmacological substances. .
- the present invention relates to pharmaceutical compositions comprising, as active principle, a compound according to the invention.
- These pharmaceutical compositions contain an effective dose of at least one compound according to the invention or a pharmaceutically acceptable salt of said compound, as well as at least one pharmaceutically acceptable excipient.
- the dosage appropriate to each patient is determined by the physician according to the mode of administration, the weight and the response of said patient.
- the present invention also relates to a method of treatment of the pathologies indicated above which comprises the administration to a patient of an effective dose of a compound according to the invention, or one of pharmaceutically acceptable salts thereof.
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Addiction (AREA)
- Psychiatry (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
L'invention se rapporte à des dérivés de 2-cycloamino-5-(pyridin-4-yl)imidazo[2,1 - b]{\,3,4]thiadiazole de formule générale (I) dans laquelle; R5 représente un atome d'hydrogène ou un groupe C1-3 alkyle, - NR7R8, ou C1-3 alkyloxy; R6 représente un groupe phényle éventuellement substitué;A représente un groupe C1-7-alkylène éventuellement substitué par un ou deux groupes Ra; B représente un groupe C1-7-alkylène éventuellement substitué par un groupe Rb; L représente soit un atome d'azote éventuellement substitué par un groupe Rc ou Rd, soit un atome de carbone substitué par un groupe Rei et un groupe Rd ou deux groupes Re2; R7 et R8 représentent, indépendamment l'un de l'autre, un atome d'hydrogène ou un groupe C1-3 alkyle; Procédé de préparation et application en thérapeutique.
Description
DERIVES DE 2-CYCLOAMINO-S-(PYRIDI N^-YL)I MIDAZOP1 I - ô][1 ,3,4]THIADIAZOLE, LEUR PREPARATION ET LEUR APPLICATION EN
THERAPEUTIQUE.
La présente invention se rapporte à des dérivés de 2-cycloamino-5-(pyridin-4- yl)imidazo[2,1-ιb][1 ,3,4]thiadiazole, à leur préparation et à leur application en thérapeutique, dans le traitement ou la prévention de maladies impliquant la caséine kinase 1 epsilon et/ou la caséine kinase 1 delta.
La présente invention a pour objet les composés répondant à la formule générale (I)

dans laquelle
- R5 représente un atome d'hydrogène ou un groupe Ci-3 alkyle, -NR7R8, ou Ci-3 alkyloxy ;
- R6 représente un groupe phényle éventuellement substitué par un ou plusieurs substituants choisis parmi les atomes d'halogène et les groupes Ci-6 alkyle ; Ci-6 alkyloxy ;
- A représente un groupe Ci-7-alkylène éventuellement substitué par un ou deux groupes R3 ;
- B représente un groupe Ci-7-alkylène éventuellement substitué par un groupe Rb ; - L représente soit un atome d'azote éventuellement substitué par un groupe Rc ou
Rd, soit un atome de carbone substitué par un groupe Rei et un groupe Rd ou deux groupes Re2 ;
les atomes de carbone de A et de B étant éventuellement substitués par un ou plusieurs groupes Rf identiques ou différents l'un de l'autre ;
R3, Rb et Rc sont définis tels que : deux groupes R3 peuvent former ensemble un groupe Ci-6-alkylène ; R3 et Rb peuvent former ensemble une liaison ou un groupe Ci-6-alkylène; R3 et Rc peuvent former ensemble une liaison ou un groupe Ci-6-alkylène ; Rb et Rc peuvent former ensemble une liaison ou un groupe Ci-6-alkylène ;
Rd représente un groupe choisi parmi l'atome d'hydrogène et les groupes Ci-6- alkyle, C3-7-cycloalkyle, C3-7-cycloalkyl-Ci.6-alkyle, Ci-6-alkylthio-C-i-6-alkyle, Ci- 6-alkyloxy-Ci-6-alkyle, Ci-6-fluoroalkyle, hydroxy-Ci-6-alkyle ;
Rei représente un groupe -NR3R4 ou une monoamine cyclique comportant éventuellement un atome d'oxygène, la monoamine cyclique étant éventuellement substituée par un ou plusieurs substituants choisis parmi l'atome de fluor et les groupes Ci-6-alkyle, Ci-6-alkyloxy, hydroxyle ;
Deux RΘ2 forment avec l'atome de carbone qui les porte une monoamine cyclique comportant éventuellement un atome d'oxygène, cette monoamine cyclique étant éventuellement substituée par un ou plusieurs groupes Rf identiques ou différents l'un de l'autre ;
Rf représente un groupe Ci-6-alkyle, C3-7-cycloalkyle, C3-7-cycloalkyle-Ci-6-alkyle,
Ci-6-alkyloxy-Ci.6-alkyle, hydroxy-Ci-6-alkyle, Ci-6-fluoroalkyle ;
R3 et R4 représentent, indépendamment l'un de l'autre, un atome d'hydrogène ou un groupe Ci-4 alkyle, C3-7-cycloalkyle, C3-7-cycloalkyle-Ci-6-alkyle ;
R7 et R8 représentent, indépendamment l'un de l'autre, un atome d'hydrogène ou un groupe Ci-3 alkyle ;
Les composés de formule (I) peuvent comporter un ou plusieurs atomes de carbone asymétriques. Ils peuvent donc exister sous forme d'énantiomères ou de diastéréoisomères. Ces énantiomères, diastéréoisomères, ainsi que leurs mélanges, y compris les mélanges racémiques, font partie de l'invention.
Les composés de formule (I) peuvent exister à l'état de bases ou de sels d'addition à des acides. De tels sels d'addition font partie de l'invention. Ces sels sont avantageusement préparés avec des acides pharmaceutiquement acceptables,
mais les sels d'autres acides utiles, par exemple, pour la purification ou l'isolement des composés de formule (I) font également partie de l'invention.
Dans le cadre de l'invention, on entend par : - Ct-z où t et z peuvent prendre les valeurs de 1 à 6, une chaîne carbonée pouvant avoir de t à z atomes de carbone, par exemple Ci-6 une chaîne carbonée qui peut avoir de 1 à 6 atomes de carbone ; alkyle, un groupe aliphatique saturé, linéaire ou ramifié; par exemple un groupe Ci-6-alkyle représente une chaîne carbonée de 1 à 6 atomes de carbone, linéaire ou ramifiée, par exemple un méthyle, éthyle, propyle,
/sopropyle, butyle, /sobutyle, terbutyle, pentyle, hexyle ; alkylène, un groupe alkyle divalent saturé, linéaire ou ramifié, par exemple un groupe Ci-6-alkylène représente une chaîne carbonée divalente de 1 à 6 atomes de carbone, linéaire ou ramifiée, par exemple un méthylène, éthylène, 1-méthyléthylène, propylène ; cycloalkyle, un groupe alkyle cyclique, par exemple un groupe C3-7- cycloalkyle représente un groupe carboné cyclique de 3 à 7 atomes de carbone, par exemple un cyclopropyle, cyclobutyle, cyclopentyle, cyclohexyle, cycloheptyle ; - hydroxyle, un groupe -OH ; monoamine cyclique, une chaîne carbonée cyclique saturée comportant 1 atome d'azote ; hydroxyalkyle, un groupe alkyle dont un atome d'hydrogène a été substitué par un groupe hydroxyle ; - alkyloxy, un groupe -O-alkyle ; alkylthio, un groupe -S-alkyle ;
- fluoroalkyle, un groupe alkyle dont un ou plusieurs atomes d'hydrogène ont été substitués par un atome de fluor ;
- fluoroalkyloxy, un groupe alkyloxy dont un ou plusieurs atomes d'hydrogène ont été substitués par un atome de fluor ; un atome d'halogène, un atome de fluor, de chlore, de brome ou d'iode ; aryle, un groupe aromatique mono- ou bicyclique comprenant entre 6 et 10 atomes de carbones. A titre d'exemple de groupe aryle, on peut citer les groupes phényle ou naphtyle.
A titre d'exemples non limitatifs d'aminés ou diamines cycliques formées par N, A, L et B, on peut notamment citer l'aziridine, l'azétidine, la pyrrolidine, la pipéridine,
- A - l'azépine, la morpholine, la thiomorpholine, rhomopipéridine, la décahydro- quinoline, la décahydro-/soquinoline, l'azabicyclo-heptane, l'azabicyclo-octane, l'azabicyclo-nonane, l'aza-oxo-bicyclo-heptane, l'aza-thia-bicyclo-heptane, l'aza- oxo-bicyclo-octane, l'aza-thia-bicyclo-octane ; la pipérazine, l'homopipérazine, le diaza-cyclo-octane, le diaza-cyclo-nonane, le diaza-cyclo-décane, le diaza-cyclo- undécane, l'octahydro-pyrrolo-pyrazine, l'octahydro-pyrrolo-diazepine, l'octahydro- pyrrolo-pyrrole, l'octahydro-pyrrolo-pyridine, le décahydro-naphthyridine, le diaza- bicyclo-heptane, le diaza-bicyclo-octane, le diaza-bicyclo-nonane, le diaza-spiro- heptane, le diaza-spiro-octane, le diaza-spiro-nonane, le diaza-spiro-décane, le diaza-spiro-undécane, l'oxa-diaza-spiro-undécane.
Parmi les composés de formule générale (I) objets de l'invention, un premier groupe de composés est constitué par les composés pour lesquels :
- R5 représente un atome d'hydrogène ou un groupe méthyle, méthyl-amino, méthoxy ; les autres substituants étant tels que définis précédemment.
Parmi les composés de formule générale (I) objets de l'invention, un deuxième groupe de composés est constitué par les composés pour lesquels : - R6 représente un groupe phényle éventuellement substitué par un ou plusieurs substituants choisis parmi les atomes d'halogène ; les autres substituants étant tels que définis précédemment.
Parmi les composés de formule générale (I) objets de l'invention, un troisième groupe de composés est constitué par les composés pour lesquels :
- R6 représente un groupe phényle éventuellement substitué par un ou plusieurs substituants choisis parmi les atomes de fluor ; les autres substituants étant tels que définis précédemment.
Parmi les composés de formule générale (I) objets de l'invention, un quatrième groupe de composés est constitué par les composés pour lesquels :
- l'aminé cyclique formée par -N-A-L-B- représente un groupe choisi parmi les groupes pipérazinyle, hexahydropyrrolo-pyrrolyle, éventuellement substitué par un ou plusieurs groupes choisis parmi les groupes Ci-6-alkyle ; les autres substituants étant tels que définis précédemment.
Parmi les composés de formule générale (I) objets de l'invention, un cinquième groupe de composés est constitué par les composés pour lesquels :
- l'aminé cyclique formée par -N-A-L-B- représente un groupe choisi parmi les groupes pipérazinyle, (3R)-3-méthyl-pipérazinyle, 4-/sopropyl-pipérazinyle, {cis)- hexahydropyrrolo[3,4-c]pyrrol-2(1 /-/)yle ; les autres substituants étant tels que définis précédemment.
Parmi les composés de formule générale (I) objets de l'invention, un sixième groupe de composés est constitué par les composés pour lesquels : - l'aminé cyclique formée par -N-A-L-B- représente un groupe diazaspiro-undécyle ; les autres substituants étant tels que définis précédemment.
Parmi les composés de formule générale (I) objets de l'invention, un septième groupe de composés est constitué par les composés pour lesquels : - l'aminé cyclique formée par -N-A-L-B- représente un groupe choisi parmi les groupes 2,9-diaza-spiro[5.5]undéc-9-yle, 3,9-diaza-spiro[5.5]undéc-9-yle ; les autres substituants étant tels que définis précédemment.
Parmi les composés de formule générale (I) objets de l'invention, un huitième groupe de composés est constitué par les composés pour lesquels :
- l'aminé cyclique formée par -N-A-L-B- représente un groupe pipéridinyle substitué par un groupe pyrrolidinyle ; les autres substituants étant tels que définis précédemment.
Parmi les composés de formule générale (I) objets de l'invention, un neuvième groupe de composés est constitué par les composés pour lesquels :
- l'aminé cyclique formée par -N-A-L-B- représente un groupe 4-pyrrolidin-1-yl- pipéridinyle ; les autres substituants étant tels que définis précédemment.
Parmi les composés de formule générale (I) objets de l'invention, un dixième groupe de composés est constitué par les composés pour lesquels :
- R5 représente un atome d'hydrogène ou un groupe Ci-3-alkyle, -NR7R8, Ci-3 alkyloxy ; - R6 représente un groupe phényle éventuellement substitué par un ou plusieurs substituants choisis parmi les atomes d'halogène ;
- l'aminé cyclique formée par -N-A-L-B- représente soit groupe choisi parmi les groupes pipérazinyle, hexahydropyrrolo-pyrrolyle, éventuellement substitué par un ou plusieurs groupes choisis parmi les groupes Ci-6-alkyle, ; soit un groupe diazaspiro-undécyle ; ou un groupe pipéridinyle, éventuellement substitué par un groupe pyrrolidinyle ;
- R7 et R8 représentent, indépendamment l'un de l'autre, un atome d'hydrogène ou un groupe Ci-3 alkyle ; à l 'état de base ou de sel d'addition à un acide.
Parmi les composés de formule générale (I) objets de l'invention, un onzième groupe de composés est constitué par les composés pour lesquels :
- R5 représente un atome d'hydrogène ou un groupe méthyle, méthyl-amino, méthoxy ;
- R6 représente un groupe phényle éventuellement substitué par un ou plusieurs substituants choisis parmi les atomes de fluor ;
- l'aminé cyclique formée par -N-A-L-B- représente un groupe choisi parmi les groupes pipérazinyle, (3R)-3-méthyl-pipérazinyle, 4-/sopropyl-pipérazinyle, {cis)- hexahydropyrrolo[3,4-c]pyrrol-2(1 /-/)yle ; 2,9-diaza-spiro[5.5]undéc-9-yle, 3,9-diaza- spiro[5.5]undéc-9-yle ; 4-pyrrolidin-1 -yl-pipéridinyle ; à l 'état de base ou de sel d'addition à un acide.
Parmi les composés de formule générale (I) objets de l'invention, on peut notamment citer les composés suivants :
6-(4-Fluoro-phényl)-2-pipérazin-1-yl-5-(pyridin-4-yl)imidazo[2,1-b][1 ,3,4]thiadiazole ; 6-(4-Fluoro-phényl)-2-((3R)-3-méthyl-pipérazin-1-yl)-5-(pyridin-4-yl)imidazo[2,1- fc][1 ,3,4]thiadiazole ;
6-(4-Fluoro-phényl)-2-((3R)-3-méthyl-pipérazin-1-yl)-5-(2-méthyl-pyridin-4-yl)- imidazo[2,1-b][1 ,3,4]thiadiazole ;
{4-[6-(4-Fluoro-phényl)-2-((3R)-3-méthyl-pipérazin-1-yl)-imidazo[2,1- jb][1 ,3,4]thiadiazol-5-yl]-pyridin-2-yl}-méthyl-amine ;
6-(4-Fluoro-phényl)-5-(2-méthoxy-pyridin-4-yl)-2-((/?)-3-méthyl-pipérazin-1-yl)- imidazo[2,1-b][1 ,3,4]thiadiazole ;
6-(4-Fluoro-phényl)-2-(4-isopropyl-pipérazin-1-yl)-5-(pyridin-4-yl)imidazo[2,1- fc][1 ,3,4]thiadiazole ; 6-(4-Fluoro-phényl)-2-(4-/sopropyl-pipérazin-1-yl)-5-(2-méthyl-pyridin-4-yl)- imidazo[2,1-b][1 ,3,4]thiadiazole ;
{4-[6-(4-Fluoro-phényl)-2-(4-/sopropyl-pipérazin-1-yl)-imidazo[2,1-fc][1 ,3,4]thiadiazol-
5-yl]-pyridin-2-yl}-méthyl-amine ;
6-(4-Fluoro-phényl)-2-(4-/sopropyl-pipérazin-1-yl)-5-(2-méthoxy-pyridin-4-yl)- imidazo[2,1-b][1 ,3,4]thiadiazole ; 3-[6-(4-Fluoro-phényl)-5-(pyridin-4-yl)imidazo[2!1-b][1 !3!4]thiadiazol-2-yl]-3,9-diaza- spiro[5.5]undécane et son chlorhydrate ;
9-[6-(4-Fluoro-phényl)-5-(pyridin-4-yl)imidazo[2!1-b][1 !3!4]thiadiazol-2-yl]-2,9-diaza- spiro[5.5]undécane ;
9-[6-(4-Fluoro-phényl)-5-(2-méthyl-pyridin-4-yl)-imidazo[2!1-b][1 !3,4]thiadiazol-2-yl]- 2,9-diaza-spiro[5.5]undécane ;
{4-[2-(2,9-Diaza-spiro[5.5]undéc-9-yl)-6-(4-fluoro-phényl)-imidazo[2,1- fc>][1 ,3,4]thiadiazol-5-yl]-pyridin-2-yl}-méthyl-amine ;
9-[6-(4-Fluoro-phényl)-5-(2-méthoxy-pyridin-4-yl)-imidazo[2, 1 -b][1 ,3,4]thiadiazol-2- yl]-2,9-diaza-spiro[5.5]undécane ; 6-(4-Fluoro-phényl)-2-[(c/s)-hexahydro-pyrrolo[3,4-c]pyrrol-2-yl]-5-(pyridin-4- yl)imidazo[2,1-b][1 ,3,4]thiadiazole ;
6-(4-Fluoro-phényl)-2-[(c/s)-hexahydro-pyrrolo[3,4-c]pyrrol-2-yl]-5-(2-méthyl-pyridin-
4-yl)-imidazo[2,1-b][1 ,3,4]thiadiazole ;
{4-[6-(4-Fluoro-phényl)-2-[(c/s)-hexahydro-pyrrolo[3,4-c]pyrrol-2-yl]-imidazo[2,1- t)][1 ,3,4]thiadiazol-5-yl]-pyridin-2-yl}-méthyl-amine ;
6-(4-Fluoro-phényl)-2-[(c/s)-hexahydro-pyrrolo[3,4-c]pyrrol-2-yl]-5-(2-méthoxy- pyridin-4-yl)-imidazo[2,1-b][1 ,3,4]thiadiazole
6-(4-Fluoro-phényl)-5-(pyridin-4-yl)-2-(4-pyrrolidin-1-yl-pipéridin-1-yl)-imidazo[2,1- b][1 ,3,4]thiadiazole ; 6-(4-Fluoro-phényl)-5-(2-méthyl-pyridin-4-yl)-2-(4-pyrrolidin-1 -yl-pipéridin-1 -yl)- imidazo[2,1-fc][1 ,3,4]thiadiazole ;
{4-[6-(4-Fluoro-phényl)-2-(4-pyrrolidin-1-yl-pipéridin-1-yl)-imidazo[2,1- b][\ ,3,4]thiadiazol-5-yl]-pyridin-2-yl}-méthyl-amine ;
6-(4-Fluoro-phényl)-5-(2-méthoxy-pyridin-4-yl)-2-(4-pyrrolidin-1-yl-pipéridin-1-yl)- imidazo[2,1-ô][1 ,3,4]thiadiazole ;
L'invention a également pour objet un procédé de préparation des composés de l'invention de formule (I).
Conformément à l'invention, on peut préparer les composés de formule générale (I) selon le procédé général décrit dans le schéma 1 ci-après.
SCHEMA 1

(III)

(I)
De manière générale et comme illustré dans le Schéma 1 , les dérivés de 2- cycloamino-5-(pyridin-4-yl)imidazo[2,1-t)][1 ,3,4]thiadiazole de formule générale (I) dans laquelle, A, L, B, R5 et R6 sont tels que définis ci-dessus peuvent être préparés par couplage métallocatalysé entre un dérivé de 5-halogéno-imidazo[2,1- b][1 ,3,4]thiadiazole de formule générale (II), dans laquelle A, L, B et R6 sont tels que définis ci-dessus et X5 représente un halogène tel que le brome ou l'iode, plus
particulièrement l'iode et un dérivé de pyridine de formule générale (lia) dans laquelle R5 est tel que défini ci-dessus et M représente un groupe trialkylstannyle, le plus fréquemment un groupement tributylstannyle ou un groupe dihydroxyboryle ou dialkyloxyboryle, le plus fréquemment un groupe 4,4,5,5-tétraméthyl-1 ,3,3,2- dioxaborolan-2-yle selon les conditions de Stille ou de Suzuki.
Les couplages selon la méthode de Stille sont par exemple réalisés par chauffage en présence d'un catalyseur tel que le tétrakis(triphénylphosphine)palladium, l'iodure de cuivre, dans un solvant tel que le N,N-diméthylacétamide. Les couplages selon la méthode de Suzuki sont par exemple réalisés par chauffage en présence d'un catalyseur tel que le 1 ,1 '-bis
(diphénylphosphino)ferrocènedichloropalladium, d'une base minérale telle que le carbonate de césium, dans un mélange de solvant tels que le dioxane ou le tétrahydrofurane et l'eau.
Les dérivés de 5-halogénoimidazo[2,1-b][1 ,3,4]thiadiazole de formule générale (II) sont obtenu par bromation ou iodation régiosélective d'un dérivé d'imidazo[2,1- b][1 ,3,4]thiadiazole de formule générale (III), dans laquelle dans laquelle A, L, B et R6 sont tels que définis ci-dessus. Cette réaction peut être réalisée au moyen de N- bromo- ou iodosuccinimide ou de monochlorure d'iode, dans un solvant polaire tel que l'acétonitrile, le tétrahydrofurane, le méthanol ou le chloroforme.
Les dérivés d'imidazo[2,1-b][1 ,3,4]thiadiazole de formule générale (III) tels que définis ci-dessus peuvent être préparés par condensation entre un dérivé de [1 ,3,4]thiadiazol-2-ylamine de formule générale (IV) dans laquelle A, L et B sont tels que définis précédemment et un dérivé de 2-halogénoéthanone de formule générale (IVa) dans laquelle R6 est tel que défini précédemment et X représente un groupe partant tel que le brome, le chlore ou l'iode.
Cette réaction peut être effectuée par chauffage des réactifs dans un solvant polaire tel que l'éthanol.
Alternativement, les dérivés d'imidazo[2,1-t)][1 ,3,4]thiadiazole de formule générale (III) dans laquelle A, L, B et R6 sont tels que définis ci-dessus peuvent être préparés à partir d'un dérivé de 2-halogénoimidazo[2,1-t)][1 ,3,4]thiadiazole de formule générale (V), dans laquelle R6 est tel que défini ci-dessus et X2 représente un halogène, plus particulièrement le brome, par traitement au moyen d'une diamine de formule générale (Va) dans laquelle A, L et B sont tels que définis précédemment.
Cette réaction peut être effectuée selon les conditions de la réaction de couplage métallo-catalyse de Buchwald-Hartwig.
Les couplages selon la méthode de Buchwald-Hartwig sont par exemple réalisés par chauffage des réactifs en présence d'un catalyseur tel que l'acétate de palladium, d'un ligand tel que le 4,5-bis(diphénylphosphino)-9,9-diméthylxanthène, d'une base telle que le carbonate de césium, dans un solvant tel que le dioxane.
Les dérivés d'imidazo[2,1-t)][1 ,3,4]thiadiazole de formule générale (V) sont connus (Journal of Heterocyclic Chemistry (2008), 45(1 ), 299-302 ; Chemistry of Heterocyclic Compounds (New York, NY, United States) (2007), 43(4), 499-504), ou peuvent être préparés par analogie selon des méthodes connues de l'Homme du métier
Les dérivés d'imidazo[2,1-t)][1 ,3,4]thiadiazole de formule générale (IV) sont décrits notamment dans les documents WO2006/040569 ; Farmaco, Edizione Scientifica (1986), 41 (10), 737-46 ; Tetrahedron Letters (2008), 49(36), 5241-5243 ; WO1999/47507, ou peuvent être préparés par analogie avec des méthodes connues de l'Homme du métier.
Dans certains cas, les dérivés de formule générale (I) pour lequels l'aminé formée par N, L, A et B comporte une seconde aminé secondaire ou tertiaire peuvent être préparés respectivement à partir de l'aminé primaire ou secondaire correspondante par alkylation ou amino-réduction selon des méthodes usuelles connues de l'Homme du métier
Dans ce qui précède, on entend par groupe partant un groupe pouvant être facilement clivé d'une molécule par rupture d'une liaison hétérolytique, avec départ d'une paire électronique. Ce groupe peut, par exemple, être ainsi remplacé facilement par un autre groupe lors d'une réaction de substitution. De tels groupes partants sont, par exemple, les halogènes ou un groupe hydroxy activé tel qu'un groupe mésyle, tosyle, triflate, acétyle, etc. Des exemples de groupes partants ainsi que des références pour leurs préparations sont données dans « Advances in Organic Chemistry », J. March, 3rd Edition, Wiley Interscience, p. 310-316.
Groupes protecteurs
Pour les composés de formule générale (I), (II), (II), (IV) et (Va) telles que définies ci-dessus et comportant une fonction aminé primaire ou secondaire, cette fonction
peut éventuellement être protégée lors de la synthèse par un groupe protecteur, par exemple un benzyle ou un t-butyloxycarbonyle.
Les exemples suivants décrivent la préparation de certains composés conformes à l'invention. Ces exemples ne sont pas limitatifs et ne font qu'illustrer l'invention. Les numéros des composés exemplifiés renvoient à ceux donnés dans le tableau 1 , ci- après, qui illustrent les structures chimiques et les propriétés physiques respectivement de quelques composés selon l'invention.
Exemple 1 (composé n° 1) : 6-(4-Fluorophényl)-2-pipérazin-1 -yl-5-(pyridin-4- yl)imidazo[2,1 -fe][1 ,3,4]thiadiazole

37566-39-5)

Le mélange de 1 ,00 g (5,55 mmoles) de 5-bromo-[1 ,3,4]thiadiazol-2-ylamine, de 1 ,24 g (6,67 mmoles) de 4-(te/f-butyloxycarbonyl)pipérazine (CAS 57260-71-6) et de 1 ,45 g ( 8,33 mmoles) d'hydrogenocarbonate de potassium dans 10 ml de diméthylformamide est chauffé à 1400C pendant 5 heures. Après refroidissement, le mélange est versé dans 100 ml d'eau et le produit est extrait avec de l'acétate d'éthyle. La solution est ensuite séchée sur sulfate de sodium et le solvant est évaporé sous pression réduite. L'huile brune obtenue est ensuite chromatographiée sur colonne de 50 g de gel de silice en éluant avec un mélange de dichlorométhane et de méthanol (95/5), pour conduire à 0,9 g de 4-(5-amino-[1 ,3,4]thiadiazol-2-yl)- pipérazine-1-carboxylate de te/f-butyle sous forme de poudre blanche après trituration dans l'éther di-/sopropylique, filtration et séchage.
RMN 1H (CDCI3) δ : 4,8 (signal large) ; 3,50 (m, 4H) ; 3,30 (m, 4H) ; 1 ,45 (s, 9H) ppm.
1.2. 4-[6-(4-Fluorophényl)-imidazo[2,1-ά1[1 ,3,41thiadiazol-2-yl1-pipérazine-1- carboxylate de te/f-butyle

Le mélange de 5,59 g (19,6 mmoles) de 4-(5-amino-[1 ,3,4]thiadiazol-2-yl)- pipérazine-1-carboxylate de te/f-butyle et de 5,53 g (25,5 mmoles) de 2-bromo-1-(4- fluorophényl)éthanone (CAS 403-29-2) dans 75 ml d'éthanol est chauffé à reflux pendant 2 heures. Après refroidissement, le solvant est éliminé sous pression réduite et le produit est partagé entre du chloroforme et une solution saturée aqueuse d'hydrogénocarbonate de sodium. La phase organique est séparée, lavée avec de l'eau, séchée et le solvant évaporé sous pression réduite, pour conduire à 9,5 g de bromure de 2-amino-5-(4-te/f-butoxycarbonyl-pipérazin-1-yl)-3-[2-(4- fluorophényl)-2-oxo-éthyl]-[1 ,3,4]thiadiazol-3-ium brut qui sont dissous dans 200 ml d'éthanol. La solution est chauffée à 1200C dans un tube scellé pendant 4 heures puis refroidie. Le solide jaunâtre formé est séparé par filtration puis purifié par chromatographie sur colonne de 50 g de gel de silice en éluant avec un mélange de dichlorométhane, de méthanol et d'ammoniaque (98/2/0,2) pour donner 2,7 g de 4- [6-(4-f luorophényl)-imidazo[2, 1 -b][1 ,3,4]thiadiazol-2-yl]-pipérazine-1 -carboxylate de te/f-butyle sous forme de poudre blanche après trituration dans l'éther di- /sopropylique, filtration et séchage.
RMN 1H (DMSOd6) δ : 8,30 (s, 1 H) ; 7,80 (dd, 2H) ; 7,20 (pseudo t, 1 H) ; 3,5 (m, 8H) ; 1 ,40 (s, 9H) ppm.
1.3. 4-r6-(4-Fluorophényl)-5-iodo-imidazor2,1-άïï1 ,3,4lthiadiazol-2-yll-pipérazine-1- carboxylate de te/f-butyle

A une suspension de 0,245 g (6,07 mmoles) de 4-[6-(4-fluorophényl)-imidazo[2,1- b][\ , 3, 4]thiadiazol-2-yl]-pipérazine-1 -carboxylate de te/f-butyle dans 100 ml d'acétonitrile, on additionne à température ambiante 1 ,50 g (6,50 mmoles) de N- iodosuccinimide (CAS 516-12-1 ). Après 20 minutes de réaction à température ambiante, le mélange est traité avec une solution aqueuse de thiosulfate de sodium
et le produit est extrait avec du dichlorométhane. La phase organique est séparée, séchée sur sulfate de sodium et concentrée sous pression réduite pour donner 3,17 g de 4-[6-(4-fluorophényl)-5-iodo-imidazo[2,1-b][1 ,3,4]thiadiazol-2-yl]-pipérazine-1- carboxylate de te/f-butyle sous forme d'une poudre blanche après trituration dans l'éther di-/sopropylique, filtration et séchage.
RMN 1H (DMSOd6) δ : 7,90 (dd, 2H) ; 7,25 (pseudo t, 1 H) ; 3,45 (m, 8H) ; 1 ,40 (s, 9H) ppm.
1.4. 4-r6-(4-Fluorophényl)-5-(pyridin-4-yl)imidazor2,1-άïï1.3.41thiadiazol-2-yll- pipérazine-1- carboxylate de te/f-butyle

Au mélange de 0,700 g (1 ,32 mmole) de 4-[6-(4-fluorophényl)-5-iodo-imidazo[2,1- b][1 ,3,4]thiadiazol-2-yl]-pipérazine-1-carboxylate de te/f-butyle, de 0,230 g (1 ,59 mmole) d'acide (4-pyridinyl)boronique (CAS 1692-15-5) et de 0,280 g (2,64 mmoles) de bicarbonate de sodium dans 30 ml de Λ/-méthylpyrrolidinone sous argon, on ajoute 0,153 g (0,13 mmole) de tétrakis(triphénylphosphine)palladium(0) (CAS 14221-01-3). La réaction est chauffée à 1500C pendant 5 minutes. Après refroidissement, le solvant est évaporé sous pression réduite et le mélange est partagé entre de l'acétate d'éthyle et de l'eau. La phase organique est ensuite séchée sur sulfate de sodium et le solvant évaporé sous pression réduite. Après recristallisation du solide dans l'alcool /sopropylique, filtration et séchage, on isole 0,52 g 4-[6-(4-fluorophényl)-5-(pyridin-4-yl)imidazo[2,1-b][1 ,3,4]thiadiazol-2-yl]- pipérazine-1-carboxylate de te/f-butyle sous forme de poudre jaune.
RMN 1H (CDCI3) δ : 8,50 (d, 2H) ; 7,50 (m, 4H) ; 7,00 (pseudo t, 2H); 3,55 (m, 4H) ; 3,40 (m, 4H) ; 1 ,30 (s, 9H) ppm.
1.5. 6-(4-Fluorophényl)-2-(pipérazin-1-yl)-5-(pyridin-4-yl)imidazo[2,1- άïï1 ,3,4lthiadiazole

A une solution de 0,650 g (1 ,35 mmole) de 4-[6-(4-fluorophényl)-5-(pyridin-4- yl)imidazo[2,1-b][1 ,3,4]thiadiazol-2-yl]-pipérazine-1-carboxylate de te/f-butyle dans 15 ml de dichlorométhane, on additionne goutte à goutte à 00C, 13,5 ml d'acide trifluoroacétique. Le mélange est versé dans de l'eau et le tout est basifié par addition d'ammoniaque. Le produit est alors extrait avec du dichlorométhane, la phase organique est séchée sur sulfate de sodium et le solvant évaporé sous pression réduite. Le résidu est purifié sur de colonne de 25 g de gel de silice en éluant avec un mélange de dichlorométhane, de méthanol et d'ammoniaque (95/5/0,5) pour donner 0,225 g de 6-(4-fluorophényl)-2-(pipérazin-1-yl)-5-(pyridin-4- yl)imidazo[2,1-t)][1 ,3,4]thiadiazole sous forme d'une poudre blanche après trituration dans l'éther di-éthylique, filtration et séchage.
PF : 233-235°C RMN 1H (CDCI3) δ : 8,60 (d, 2H) ; 7,55 (d, 1 H) ; 7,05 (pseudo t, 2H) ; 3,55 (m, 4H) ; 3,05 (m, 4 H) ppm.
Exemple 2 (composé n° 6) : 6-(4-fluorophényl)-2-(4-/sopropylpipérazin-1 -yl)-5- (pyridin-4-yl)imidazo[2,1 -fe][1 ,3,4]thiadiazole

2.1. 2-Bromo-6-(4-fluorophényl)imidazor2,1-άïï1 ,3,4lthiadiazole (CAS 1094275-22- 5]

A une solution de 14,0 g (78 mmoles) de 5-bromo-1 ,3,4-thiadiazol-2-amine (CAS 37566-39-5) dans 300 ml de butanol, on ajoute par petites portions 22,0 g (101 mmoles) de 2-bromo-1-(4-fluorophényl)-éthanone (CAS 403-29-2). Le mélange est chauffé à reflux pendant une 18 heures. Après refroidissement, le butanol est évaporé et le résidu est trituré dans 100 ml d'acétone puis isolé par filtration après
refroidissement dans un bain de glace. Le solide est repris avec une solution saturée en carbonate de sodium et le produit est extrait avec du chloroforme. La phase organique est séparée, séchée sur sulfate de sodium, filtrée et le solvant est évaporé sous pression réduite pour donner 16,5 g de 2-bromo-6-(4- fluorophényl)imidazo[2,1-b][1 ,3,4]thiadiazole sous forme de poudre après rinçage avec de l'éther diéthylique et séchage. RMN 1H (CDCI3) : 8,01 (1 H, s) ; 7,80 (2H, dd) ; 7,14 (2H, pseudo t) ppm
2.2. 6-(4-Fluorophényl)-2-(4-/sopropylpipérazin-1-yl)imidazo[2,1-ά1[1 ,3,41thiadiazole

A un mélange de 0,298 g (1 ,00 mmole) de 2-bromo-6-(4-fluorophényl)imidazo[2,1- b][1 ,3,4]thiadiazole, de 0,652 g (2,00 mmoles) de carbonate de césium, de 0,011 g (0,05 mmole) d'acétate de palladium (CAS 3375-31-3) et de 0,029 (0,05 mmole) de 4,5-bis(diphénylphosphino)-9,9-diméthylxanthène (CAS 161265-03-8) sous argon, on ajoute 0,21 ml (1 ,5 mmole) de 1-/sopropylpipérazine (CAS 137186-14-2) et 3 ml de 1 ,4-dioxane. Le mélange est chauffé à 100 0C pendant 2 heures dans un tube scellé. Après refroidissement, le solvant est évaporé sous pression réduite et le résidu est purifié par chromatographie sur colonne d'alumine en éluant avec du dichlorométhane, pour donner 0,278 g de 6-(4-fluorophényl)-2-(4-/sopropylpipérazin- 1-yl)imidazo[2,1-fc][1 ,3,4]thiadiazole. PF : 213 0C
RMN 1H (CDCI3) : 7,74 (2H, dd) ; 7,70 (1 H, s) ; 7,05 (2H, t) ; 3,50 (4H, pseudot) ; 2,80 (1 H, m) ; 2,66 (4H, t) ; 1 ,09 (6H, d) ppm
2.3. 6-(4-Fluorophényl)-5-iodo-2-(4-/sopropylpipérazin-1-yl)imidazo[2,1- biπ .3.41thiadiazole

Le mélange de 0,690 g (2,00 mmoles) de 6-(4-fluorophényl)-2-(4-/sopropylpipérazin- 1-yl)imidazo[2,1-b][1 ,3,4]thiadiazole et de 1 ,125 g (5,02 mmoles) de N- iodosuccinimide dans 14 ml d'acétonitrile est agité 16 heures à température ambiante. Le solvant est évaporé sous pression réduite et le résidu est alors purifié par chromatographie sur une colonne d'alumine en éluant avec du dichlorométhane
pour donner 0,548 g de 6-(4-fluorophényl)-5-iodo-2-(4-/sopropylpipérazin-1- yl)imidazo[2,1-b][1 ,3,4]thiadiazole.
RMN 1H (CDCI3) : 7,96 (2H, dd) ; 7,13 (2H, pseudo t) ; 3,54 (4H, t) ; 2,80 (1 H, m) ; 2,67 (4H, t) ; 1 ,09 (6H, d) ppm.
2.4. 6-(4-Fluorophényl)-2-(4-/'sopropylpipérazin-1-yl)-5-(pyridin-4-yl)imidazo[2,1- biπ .3.41thiadiazole

PF : 216,1 0C
RMN 1H (CDCI3) : 8,58 (2H, d) ; 7,55 (4H, m) ; 7,05 (2H, pseudo t) ; 3,53 (4H, t) ; 2,80 (1 H, m) ; 2,67 (4H, t) ; 1 ,08 (6H, d) ppm.
Exemple 3 (composé n° 7) : 6-(4-fluorophényl)-2-(4-/sopropylpipérazin-1 -yl)-5- (2-méthylpyridin-4-yl)imidazo[2,1 -fe][1 ,3,4]thiadiazole

/sopropylpipérazin-1-yl)imidazo[2,1-b][1 ,3,4]thiadiazole, de 0,326 g (1 mmole) de carbonate de césium, de 0,137g (1 mmole) l'acide 2-picolin-4-yl-boronique (CAS 579476-63-4) et 0,041 g (0,05 mmoles) de chlorure de 1 ,1 '- bis(diphénylphosphino)ferrocène palladium (CAS 124268-93-5) dans un mélange de 2,7 ml de tétrahydrofurane et de 0,3 ml d'eau est chauffé à 100 0C pendant 24 heures dans un tube scellé. Après refroidissement, le contenu du tube est repris avec de l'eau et le produit est extrait au dichlorométhane. La phase organique est séchée sur sulfate de sodium, filtrée et le solvant est évaporé sous pression réduite. Le produit est ensuite purifié par chromatographie sur une colonne de silice en éluant avec du dichlorométhane puis avec un gradient d'acétate d'éthyle pour donner 0,055 g de 6-(4-fluorophényl)-2-(4-/sopropylpipérazin-1-yl)-5-(2- méthylpyridin-4-yl)imidazo[2,1-b][1 ,3,4]thiadiazole.
PF : 183,8 0C
RMN 1H (CDCI3) : 8,46 (1 H, d) ; 7,54 (2H, dd) ; 7,40 (1 H, s) ; 7,34 (1 H, d) ; 7,03 (2H, t) ; 3,51 (4H, t) ; 2,79 (1 H, m, CH) ; 2,66 (4H, t) ; 2,53 (3H, s, CH3) ; 1 ,07 (6H, d) ppm.
Exemple 4 (composé n°8): 4-(6-(4-fluorophényl)-2-(4-/sopropylpipérazin-1 - yl)imidazo[2,1 -jb][1 ,3,4]thiadiazol-5-yl)-Λ/-méthylpyridin-2-amine

4.1. (4-Bromopyridin-2-yl)méthylcarbamate de te/f-butyle (CAS 946000-13-1 )

A une suspension de 0,53 g (13 mmoles) d'hydrure de sodium (60% dans l'huile) dans 50 ml de diméthylformamide anhydre à 00C, on additionne par portion 3,00 g (1 1 ,0 mmoles) de (4-bromopyridin-2-yl)carbamate de te/f-butyle (CAS 207799-10- 8). Le mélange est agité entre 0 et 100C pendant 1 heure puis on ajoute goutte à goutte en refroidissant à 0°C, 1 ,79 g (12,6 mmoles) d'iodure de méthyle (CAS74-88-
4) dilué dans 2 à 5 ml de diméthylformamide. La réaction est laissée sous agitation pendant 18 heures à température ambiante puis le mélange est versé sur une solution saturée de chlorure de sodium et le produit est extrait avec de l'acétate d'éthyle. La phase organique est séchée sur sulfate de sodium, filtrée et le solvant est évaporé sous pression réduite pour donner 3,1 g d'huile jaune qui est purifié par chromatographie sur gel de silice en éluant avec un mélange de 20% d'acétate d'éthyle dans le cyclohexane pour donner 2,85 g de (4-bromopyridin-2- yl)méthylcarbamate de te/f-butyle sous forme d'huile incolore.
RMN 1H (DMSO) δ : 8,30 (d, 1 H) ; 8,00 (s, 1 H) ; 7,40 (d, 1 H) ; 3,30 (s, 3H) ; 1 ,50 (s, 9H) ppm.
4.2. Méthyl [4-(4,4,5,5-tétraméthyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2-yl1carbamate de te/f-butyle

A un mélange sous argon de 2,80 g (9,75 mmoles) de (4-bromopyridin-2- yl)méthylcarbamate de te/f-butyle, de 4,95 g (19,5 mmoles) de bis(pinacolato)diboron (CAS 73183-34-3) et de 3,16 g (32,2 mmoles) d'acétate de potassium dans 30 ml de diméthylformamide, on additionne 0,64 g (0,78 mmoles) de complexe de 1 ,1'-bis(diphénylphosphino)ferrocènedichloropalladium (II) et de dichlorométhane (PdCI2(dppf).CH2CI2) (CAS 95464-05-4), puis le mélange est chauffé à 85°C pendant 2 heures. Le mélange est ensuite versé dans de l'eau et le produit est extrait avec de l'acétate d'éthyle. La phase organique est séchée sur sulfate de sodium, filtrée et le solvant est évaporé sous pression réduite pour donner 6,5 g d'huile brune. Celle-ci est reprise dans de l'éther di-/sopropylique et traitée avec du noir animal à reflux du solvant pendant 5 minute pour donner 5,7 g d'huile après filtration sur Bϋchner et évaporation du solvant sous pression réduite. Le résidu est repris et agité pendant 5 minutes avec un mélange de 30 ml d'acétate d'éthyle et de 70 ml de soude 1 N aqueux. La phase aqueuse est séparée puis neutralisée par ajout d'une solution d'acide chlorhydrique aqueux. Le produit est alors extrait avec du dichlorométhane, la phase organique est séchée sur sulfate de sodium, filtrée et le solvant est évaporé sous pression réduite pour donner 2,45 g
de [4-(4,4,5,5-tétraméthyl-1 ,3,2-dioxaborolan-2-yl)pyridin-2-yl]méthylcarbamate de te/f-butyle sous forme d'huile orangée qui cristallise lentement.
PF : 108-1120C RMN 1H (DMSO) δ : 8,30 (d, 1 H) ; 7,85 (s, 1 H) ; 7,25 (d, 1 H) ; 3,30 (s, 3H) ; 1 ,45 (s, 9H) ; 1 ,30 (s, 12 H) ppm.
4.3. 4-(6-(4-fluorophényl)-2-(4-/sopropylpipérazin-1-yl)imidazo[2,1- ά1[1 ,3,41thiadiazol-5-yl)-Λ/-méthylpyridin-2-amine

Un mélange de 0,235 g (0,500 mmole) de 6-(4-fluorophényl)-5-iodo-2-(4- /sopropylpipérazin-1-yl)imidazo[2,1-b][1 ,3,4]thiadiazole, de 0,326 (1 mmole) de carbonate de césium, de 0,334 g de [4-(4,4,5,5-tétraméthyl-1 ,3,2-dioxaborolan-2- yl)pyridin-2-yl]méthylcarbamate de te/f-butyle (1 ,00 mmole) et de 0,041 g (0,05 mmoles) de chlorure de 1 ,1 '-bis(diphénylphosphino)ferrocène palladium (CAS 124268-93-5) dans un mélange de 2,7 ml de tétrahydrofurane et de 0,3 ml d'eau est chauffé à 100 0C pendant 24 heures dans un tube scellé. Après refroidissement, le contenu du tube est repris avec de l'eau et le produit est extrait avec du dichlorométhane. La phase organique est séchée sur sulfate de sodium, filtrée et le solvant est évaporé sous pression réduite. Le produit est ensuite purifié par chromatographie sur une colonne de silice en éluant avec du dichlorométhane puis avec un gradient d'acétate d'éthyle. Le (4-{6-(4-fluorophényl)-2-[4-(propan-2- yl)pipérazin-1-yl]imidazo[2,1-b][1 ,3,4]thiadiazol-5-yl}pyridin-2-yl)méthylcarbamate de te/f-butyle ainsi obtenu est traité avec une solution de 2 ml de dichlorométhane et de 0,2 ml d'acide trifluoroacétique pendant 26 heures. Le mélange est ensuite basifié dans une solution aqueuse saturée de carbonate de sodium et le produit est extrait avec du dichlorométhane. La phase organique est séchée sur sulfate de sodium, filtrée et le solvant est évaporé sous pression réduite et le résidu est alors purifié par chromatographie sur une colonne de gel de silice, en éluant avec un gradient d'acétate d'éthyle dans du dichlorométhane pour donner 0,075 g de 4-(6- (4-fluorophényl)-2-(4-/sopropylpipérazin-1-yl)imidazo[2,1-b][1 ,3,4]thiadiazol-5-yl)-/V- méthylpyridin-2 -aminé.
PF : 205,0 0C
RMN 1H (CDCI3) : 8,08 (1 H, d) ; 7,60 (2H, dd) ; 7,04 (2H, t) ; 6,82 (1 H, d) ; 6,70 (1 H, s) ; 4,72 (1 H, d) ; 3,52 (4H, t) ; 2,87 (3H, d) ; 2,80 (1 H, m) ; 2,66 (4H, t) ; 1 ,08 (6H, d) ppm
Exemple 5 (composé n°2) : 6-(4-Fluorophényl)-2-(3(/?)-méthylpipérazin-1 -yl)-5- (pyridin-4-yl)imidazo[2,1 -fe][1 ,3,4]thiadiazole


A un mélange de 0,298 g (1 ,00 mmole) de 2-bromo-6-(4-fluorophényl)imidazo[2,1- b][1 ,3,4]thiadiazole, de 0,212 g (2,00 mmoles) de carbonate de sodium, de 0,011 g (0.05 mmole) d'acétate de palladium (CAS 3375-31-3) et de 0,029 g (0,05 mmole) de 4,5-bis(diphénylphosphino)-9,9-diméthylxanthène (CAS 161265-03-8) sous argon, on ajoute 0,300 g (1 ,5 mmole) de 2(7?)-méthylpipérazinecarboxylate de te/f- butyle (CAS 170033-47-3)) et 3 ml de 1 ,4-dioxane. Le mélange est chauffé à 100 0C pendant 16 heures dans un tube scellé. Après refroidissement, le solvant est évaporé sous pression réduite et le résidu est purifié par chromatographie sur colonne d'alumine en éluant avec du dichlorométhane pour donner 0,407 g (R)-4-[6- (4-fluorophényl)imidazo[2,1-t)][1 ,3,4]thiadiazol-2-yl]-2-méthylpipérazine-1- carboxylate de te/f-butyle.
RMN 1H (CDCI3) : 7,75 (2H, dd) ; 7,71 (1 H, s) ; 7,10 (2H, t) ; 4,45 (1 H, bs) ; 3,17- 4,05 (6H, m) ; 1 ,52 (9H, s) ; 1 ,29 (3H, d) ppm
5.2. rffl-4-(6-(4-Fluorophényl)-5-iodoimidazor2,1-άïï1 ,3,4lthiadiazol-2-yl)-2- méthylpipérazine-1-carboxylate de te/f-butyle

Le mélange de 4,04 g (9,68 mmoles) de (7?,)-4-[6-(4-fluorophényl)imidazo[2,1- b][1 ,3,4]thiadiazol-2-yl]-2-méthylpipérazine-1-carboxylate de te/f-butyle et de 4,36 g (19,4 mmoles) de Λ/-iodosuccinimide dans 65 ml d'acétonitrile est agité 30 heures à température ambiante. Le solvant est évaporé à sec et le résidu est alors purifié par chromatographie sur une colonne d'alumine en éluant avec du dichlorométhane pour donner 4,9 g de f/?/)-4-(6-(4-fluorophényl)-5-iodoimidazo[2,1-b][1 ,3,4]thiadiazol- 2-yl)-2-méthylpipérazine-1-carboxylate de te/f-butyle.
RMN 1H (CDCI3) : 7,96 (2H, dd) ; 7,15 (2H, t) ; 4,47 (1 H, bs) ; 3,89-4,07 (2H, m) ; 3,22-3,48 (4H, m) ; 1 ,53 (9H, s) ; 1 ,31 (3H, d) ppm
5.3. 6-(4-Fluorophényl)-2-(3(/?)-méthylpipérazin-1-yl)-5-(pyridin-4-yl)imidazo[2,1- biπ .3.41thiadiazole

Un mélange de 0,272 g (0,500 mmole) de (7?j-4-(6-(4-fluorophényl)-5- iodoimidazo[2,1-t)][1 ,3,4]thiadiazol-2-yl)-2-méthylpipérazine-1-carboxylate de te/f- butyle, de 0,326 (1 ,00 mmole) de carbonate de césium, de 0,205 g de 4-(4, 4,5,5- tétraméthyl-1 ,3,2-dioxaborolan-2-yl)pyridine (1 ,00 mmole) et 0,041 g (0,05 mmoles) de chlorure de 1 ,1 '-bis(diphénylphosphino)ferrocène palladium (CAS 124268-93-5) dans un mélange de 2,7 ml de tétrahydrofurane et de 0,3 ml d'eau est chauffé à 100 0C pendant 24 heures dans un tube scellé. Après refroidissement, le contenu du tube est repris avec de l'eau et le produit est extrait au dichlorométhane. La phase organique est séchée sur sulfate de sodium, filtrée et le solvant est évaporé sous pression réduite. Le produit est ensuite purifié par chromatographie sur une colonne de silice en éluant avec du dichlorométhane puis avec un gradient d'acétate d'éthyle. Le (Rj-4-(6-(4-fluorophényl)-5-(pyridin-4-yl)imidazo[2,1-ib][1 ,3,4]thiadiazol- 2-yl)-2-méthylpipérazine-1-carboxylate de te/f-butyle intermédiaire est traité avec une solution de 3 ml de dichlorométhane et de 0,3 ml d'acide trifluoroacétique pendant 4 heures. Le mélange est basifié dans une solution aqueuse saturée de
carbonate de sodium et le produit est extrait avec du dichlorométhane. La phase organique est séchée sur sulfate de sodium, filtrée et le solvant est évaporé sous pression réduite et le résidu est alors purifié par chromatographie sur une colonne de gel de silice, en éluant avec un gradient d'acétate d'éthyle dans le dichlorométhane pour donner 0,097 g de 6-(4-fluorophényl)-2-(3(R)- méthylpipérazin-1-yl)-5-(pyridin-4-yl)imidazo[2,1-t)][1 ,3,4]thiadiazole.
PF : 208,2 0C
RMN 1H (CDCI3) : 8,60 (2H, d) ; 7,57 (4H, m) ; 7,07 (2H, t) ; 3,75 (2H, t) ; 2,77-3,68 (5H, m) ; 1 ,86 (1 H, s) ; 1 ,19 (3H, d) ppm.
Le tableau 1 qui suit illustre les structures chimiques et les propriétés physiques de quelques composés selon l'invention.
Dans ce tableau : - la colonne "PF0C", renseigne les points de fusion des produits en degrés Celsius, "déc." signifie décomposition du produit lors de la mesure du point de fusion,
- dans la colonne "Sel" « HCI » représente un composé sous forme de chlorhydrate et le rapport entre parenthèses est le rapport (acide:base), « HBr » représente un composé sous forme bromhydrate et le rapport entre parenthèse est le rapport (acide : base), le signe « - » signifie que le composé se présente sous forme base,
- la colonne [α]d renseigne le résultat d'analyse du pouvoir rotatoire des composés du tableau à la longueur d'onde de 589 nM, le solvant indiqué entre parenthèses correspond au solvant employé pour réaliser la mesure du pouvoir rotatoire en degrés et la lettre « c » indique la concentration du solvant en g/100 ml. « N.A. » signifie que la mesure du pouvoir rotatoire n'est pas applicable,
- la colonne "(M+H)" renseigne la masse de l'ion moléculaire observée lors de l'analyse des produits par LC-MS (liquid chromatography coupled to Mass Spectroscopy) réalisée sur un appareil Agilent LC-MSD Trap en mode ESI positif ou par MS (Mass Spectroscopy) sur un appareil Autospec M (EBE) en utilisant la technique DCI-NH3,
- « F- » signifie fluoro,
- « CH3- » signifie méthyle,
« CH3OH » signifie méthanol,
- « CH3O- » signifie méthoxy, - « CH3NH- » signifie méthyl-amino,
- « Ph- » signifie phényle ;


K) Ul

K)

Exemples biologiques
La capacité des composés de l'invention à inhiber la phosphorylation de la caséine par les caséine kinase 1 epsilon et delta peut être évaluée selon la procédure décrite dans le document US20050131012.
Dosage sur Plaque-Filtre-d'ATP-33P pour le criblage des inhibiteurs de CK1 epsilon :
On mesure l'effet des composés pour inhiber la phosphorylation de la caséine par l'enzyme caséine kinase 1 epsilon (CK1 epsilon) en utilisant un dosage de la caséine par filtration d'ATP-33P in vitro.
La Caséine Kinase 1 epsilon (0,58 mg/ml) est obtenue par des procédés de fermentation et de purification effectués selon des méthodes bien connues de l'Homme du métier ou peut également être obtenue auprès d'Invitrogen Corporation™ (human CK1 epsilon).
Les composés sont testés à cinq concentrations différentes de manière à générer des Cl50, c'est à dire la concentration à laquelle un composé est capable d'inhiber l'activité enzymatique de 50%, ou bien l'inhibition en % à une concentration de 10 micromolaires.
On prépare des plaques Falcon à fond en « U » en plaçant 5 μL de solutions des composés selon l'invention aux concentrations de 10, 1 , 0,1 , 0,01 ou 0,001 μM dans différents puits. Les solutions des composés selon l'invention à ces différentes concentrations sont préparées par dilution dans un tampon d'essai (Tris 50 mM pH 7,5, MgCI2 10 M, DTT 2 mM et EGTA 1 mM) d'une solution mère dans le DMSO à la concentration de 10 mM. Ensuite, on additionne 5 μL de caséine déphosphorylée à la concentration finale de 0,2 μg/μL, 20 μL de CK1 epsilon à la concentration finale de 3 ng/μL, et 20 μL d'ATP-33P à la concentration finale de 0,02 μCi/μL mélangée avec de l'ATP froide (10 μM final - environ 2x106 CPM par puits). Le volume total final d'essai par puits est égal à 50 μL.
La plaque d'essai Falcon® à fond en « U » citée ci-dessus est agitée au vortex, puis incubée à la température ambiante pendant 2 heures. Après 2 heures, la réaction est arrêtée par addition d'une solution glacée de 65 μL d'ATP froid (2 mM) préparée dans du tampon d'essai.
On transfère ensuite 100 μl_ du mélange réactionnel de la plaque Falcon® à fond en
U dans des plaques de filtration MAPH Millipore®, préalablement imprégnées avec
25 μl_ de TCA glacé à 100 %.
Les plaques de filtration MAPH Millipore sont agitées doucement et on les laisse au repos à la température ambiante pendant au moins 30 minutes pour précipiter les protéines.
Après 30 minutes, les plaques de filtration sont séquentiellement lavées et filtrées avec 2x150 μL de TCA à 20%, 2x150 μL de TCA à 10% et 2x150 μL de TCA à 5%
(6 lavages au total par plaque/900 μL par puits).
On laisse les plaques sécher pendant une nuit à la température ambiante. Ensuite, on ajoute 40 μL de liquide de scintillation Microscint-20 Packard® par puits et les plaques sont fermées de manière étanche. On mesure alors le rayonnement émis par chaque puits pendant 2 minutes dans un compteur à scintillation Topcount NXT
Packard® où les valeurs de CPM /puits sont mesurées.
On détermine l'inhibition en % de la capacité de l'enzyme à phosphoryler le substrat
(caséine) pour chaque concentration de composé testé. Ces données d'inhibition exprimées en % sont utilisées pour calculer la valeur de Cl5o pour chaque composé comparativement aux contrôles.
Les études cinétiques ont déterminé la valeur de KM pour ATP comme étant de 21 μM dans ce système d'essai.
Dans ces conditions, les composés les plus actifs de l'invention présentent des Cl5o (concentration inhibant de 50 % l'activité enzymatique de la Caséine Kinase 1 Epsilon ou Caséine Kinase 1 Delta) comprises entre 1 nM et 500 nM
Le tableau 2 ci-dessous présente les Cl5o d'inhibition de la phosphorylation de la Caséine Kinase 1 Epsilon pour quelques composés selon l'invention.
Tableau 2

Les Caséines Kinases 1 utilisées sont obtenues chez Invitrogen Corporation (human CK1 epsilon PV3500 et human CK1 delta PV3665).
Un peptide substrat, marqué à ses deux extrémités par un groupe fluorophore donneur (la coumarine) et un groupe fluorophore accepteur (la fluorescéine) constituant un système FRET est phosphorylé en présence d'ATP par la caséine kinases 1 epsilon ou delta en présence de concentrations croissantes de composés de l'invention.
Le mélange est traité au moyen d'une protéase site spécifique coupant spéciquement le peptide substrat pour former deux fragments fluorescents présentant un grand ratio d'émission par fluorescence. La fluorescence observée est donc reliée à la capacité des produits de l'invention à inhiber la phosphorylation du peptide substrat par la caséine kinase 1 epsilon ou de la caséine kinase 1 delta.
Les composés de l'invention sont mis en solution à des concentrations différentes à partir d'une solution mère à 10 mM dans le DMSO diluée dans un tampon contenant 50 mM HEPS, pH 7,5, 1 mMEGTA, 0,01% Brij-35, 10 mM MgCI pour la caséine kinase 1 epsilon et supplémenté avec Trizma Base (50 mM), pH 8,0 et NaN3
(0,01% finaux) pour la caséine kinase 1 delta.
La phosphorylation du peptide substrat SER/THR 1 1 obtenu chez Invitrogen
Corporation™ est réalisée à la concentration finale de 2 μM. La concentration en ATP est de 4 fois le KM, celui-ci étant de 2 μM pour la caséine kinase 1 epsilon et de
4 μM pour la caséine kinase 1 delta.
La mesure de la fluorescence émise est mesurée aux longueurs d'onde de 445 et
520 nm (excitation à 400 nm).
Dans ces conditions, les composés les plus actifs de l'invention présentent des Cl5o (concentration inhibant de 50 % l'activité enzymatique de la Caséine Kinase 1
Epsilon ou Caséine Kinase 1 Delta) comprises entre 1 nM et 500 nM.
Protocoles expérimentaux de dosage circadien cellulaire
Des cultures de fibroblastes Mper1-luc Rat-1 (P2C4) ont été réalisées en divisant les cultures tous les 3-4 jours (environ 10-20 % de confluence) sur des flacons de culture de tissus en polystyrène dégazés de 150 cm2 (Falcon® # 35-5001 ) et maintenues en milieu de croissance [EMEM (Cellgro #10-010-CV) ; sérum bovin fœtal à 10 % (FBS; Gibco #16000-044) ; et 50 I.U./ml de pénicilline-streptomycine (Cellgro #30-001 -Cl)] à 37°C et sous CO2 5 %. Des cellules issues de cultures de fibroblastes Rat-1 à 30-50 % de confluence telle que décrite ci-dessus ont été co-transfectées avec des vecteurs contenant le marqueur de sélection pour la résistance à la Zéocine pour une transfection stable et un gène rapporteur de la luciférase dirigé par le promoteur mPer-1. Après 24 à 48 heures, les cultures ont été divisées sur des plaques de 96 puits et maintenues en milieu de croissance additionné de 50 à100 μg/ml de Zéocine (Invitrogen® #45- 0430) pendant 10-14 jours. Les transfectants stables résistant à la Zéocine ont été évalués pour l'expression du rapporteur en ajoutant au milieu de croissance de la luciférine 100 μM (Promega® #E1603®) et en dosant l'activité de la luciférase sur un compteur à scintillation TopCount® (Packard Modèle #C384V00). Les clones de cellule Rat-1 exprimant aussi bien la résistance à la Zéocine que l'activité de la luciférase dirigée par mPeri ont été synchronisés par choc au sérum avec du sérum de cheval à 50 % [HS (Gibco® #16050-122)] et l'activité du rapporteur circadien a été évaluée. Le clone P2C4 de fibroblastes Mper1-luc Rat-1 a été sélectionné pour l'essai du composé.
Des fibroblastes Mper1-luc Rat-1 (P2C4) à 40-50 % de confluence obtenus selon le protocole décrit précédemment ont été étalés sur des plaques de culture de tissu opaques de 96 puits (Perkin Elmer® #6005680). Les cultures sont maintenues en milieu de croissance additionné de 100 μg/ml de Zéocine (Invitrogen #45-0430). jusqu'à ce qu'elles aient atteint 100 % de confluence (48 à 72 h). Les cultures ont ensuite été synchronisées avec 100 μL de milieu de synchronisation [EMEM (Cellgro #10-010-CV) ; 100 LU. /ml de pénicilline-streptomycine (Cellgro #30-001- C1 ) ; HS à 50% (Gibco #16050-122)] pendant 2 heures à 37°C et sous CO2 5%. Après synchronisation, les cultures ont été rincées avec 100 μL d'EMEM (Cellgro #10-010-CV) pendant 10 minutes à température ambiante. Après rinçage, le milieu a été remplacé par 300 μL de milieu indépendant de CO2 [CO2I (Gibco #18045-088) ; L-glutamine 2 mM (Cellgro #25-005-C1 ) ; 100 U.l./ml de pénicilline-streptomycine (Cellgro #30-001 -C1 ) ; luciférine 100 μM (Promega #E 1603)]. Les composés de
l'invention testés pour les effets circadiens ont été ajoutés à du milieu indépendant de CO2 dans du DMSO à 0,3 % (concentration finale). Les cultures ont été fermées immédiatement de manière étanche avec du film TopSeal-A® (Packard #6005185) et transférées pour la mesure de l'activité de luciférase.
Après synchronisation, les plaques d'essai ont été maintenues à 37°C dans une étuve de culture de tissu (Forma Scientific Modèle #3914). L'activité de luciférase in vivo a été estimée en mesurant l'émission relative de lumière sur un compteur à scintillation TopCount (Packard Modèle #C384V00).
L'analyse de périodes a été effectuée en déterminant l'intervalle entre les minimums d'émission relative de lumière sur plusieurs jours, ou par transformation de Fourier. Les deux méthodes ont produit une estimation de période pratiquement identique sur une gamme de périodes circadiennes. La puissance est rapportée en CE Delta (t+1 h), qui est présentée comme la concentration micromolaire efficace qui a induit un prolongement de la période de 1 heure. Les données ont été analysées par ajustement d'une courbe hyperbolique aux données exprimées en changement de période (ordonnée) en fonction de la concentration du composé à tester (abscisse) dans le logiciel XLfit™ et la CE Delta (t+1 h) a été interpolée à partir de cette courbe.
Le tableau 3 ci-dessous présente les CE Delta (t+1 h) pour quelques composés selon l'invention.
Tableau 3

En inhibant les enzymes CK1 epsilon et/ou de CK1 delta, les composés objets de l'invention modulent la rythmicité circadienne, et peuvent être utiles pour le traitement des désordres liés au rythme circadien.
Les composés selon l'invention peuvent notamment être utilisés pour la préparation d'un médicament destiné à prévenir ou à traiter les désordres du sommeil ; les troubles du rythme circadien, tels que notamment ceux dus au décalage horaire, au travail posté.
Parmi les troubles du sommeil, on distingue notamment les troubles primaires du sommeil tels que la dyssomnie, par exemple l'insomnie primaire ; la parasomnie, l'hypersomnie, par exemple la somnolence excessive ; la narcolepsie ; les troubles du sommeil liés à l'apnée du sommeil ; les troubles du sommeil liés au rythme circadien et les dyssomnies non spécifiées par ailleurs ; les troubles du sommeil associés à des troubles médicaux/psychiatriques.
Les composés objets de l'invention provoquent également un déplacement de la phase circadienne et une telle propriété peut être utile dans le cadre d'une monothérapie ou une thérapie combinée potentielle cliniquement efficace pour les troubles de l'humeur.
Parmi les troubles de l'humeur, on distingue notamment les troubles dépressifs, par exemple la dépression unipolaire, les troubles bipolaires, les troubles de l'humeur dus à une affection médicale générale ainsi que les troubles de l'humeur induits par des substances pharmacologiques.
Parmi les troubles bipolaires, on distingue notamment les troubles bipolaires I et troubles bipolaires II, et notamment les troubles affectifs saisonniers.
Les composés objets de l'invention modulant la rythmicité circadienne, peuvent être utiles dans le traitement des troubles anxieux et dépressifs dus en particulier à une altération sur la sécrétion de CRF.
Parmi les troubles dépressifs, on distingue notamment les troubles dépressifs majeurs, troubles dysthymiques, les troubles dépressifs non spécifiés par ailleurs.
Les composés objets de l'invention modulant la rythmicité circadienne, peuvent être utiles pour la préparation d'un médicament destiné à traiter les maladies liées à la dépendance à des substances d'abus telles que la cocaïne, la morphine, la nicotine, l'éthanol, le cannabis.
En inhibant la caséine kinase 1 epsilon et/ou la caséine kinase 1 delta, les composés selon l'invention peuvent être utilisés pour la préparation de médicaments, notamment pour la préparation d'un médicament destiné à prévenir ou à traiter des maladies reliées à l'hyperphosphorylation de la protéine tau, notamment la maladie d'Alzheimer.
Ces médicaments trouvent également leur emploi en thérapeutique, notamment dans le traitement ou la prévention des maladies causées ou exacerbées par la prolifération des cellules et en particulier des cellules tumorales.
Comme inhibiteur de la prolifération des cellules tumorales, ces composés sont utiles dans la prévention et le traitement des tumeurs liquides telles que les leucémies, des tumeurs solides à la fois primaires et métastasiques, des carcinomes et cancers, en particulier : cancer du sein ; cancer du poumon ; cancer de l'intestin grêle, cancer du colon et du rectum ; cancer des voies respiratoires, de l'oropharynx et de l'hypopharynx ; cancer de l'œsophage ; cancer du foie, cancer de l'estomac, cancer des canaux biliaires, cancer de la vésicule biliaire, cancer du pancréas ; cancers des voies urinaires y compris rein, urothélium et vessie; cancers du tractus génital féminin y compris cancer de l'utérus, du col de l'utérus, des ovaires, chloriocarcinome et trophoblastome; cancers du tractus génital masculin y compris cancer de la prostate, des vésicules séminales, des testicules, tumeurs des cellules germinales; cancers des glandes endocrines y compris cancer de la thyroïde, de l'hypophyse, des glandes surrénales ; cancers de la peau y compris hémangiomes, mélanomes, sarcomes, incluant le sarcome de Kaposi ; tumeurs du cerveau, des nerfs, des yeux, des méninges, incluant astrocytomes, gliomes, glioblastomes, rétinoblastomes, neurinomes, neuroblastomes, schwannomes, méningiomes ; tumeurs malignes hématopoïétiques ; leucémies, (Acute Lymphocytic Leukemia (ALL), Acute Myeloid Leukemia (AML), Chronic Myeloid Leukemia (CML), Chronic lymphocytic leukemia (CLL)) chloromes, plasmocytomes, leucémies des cellules T ou B, lymphomes non hodgkiniens ou hodgkiniens, myélomes, hémopathies malignes diverses.
Les composés selon l'invention peuvent également être utilisés pour la préparation de médicaments, notamment pour la préparation d'un médicament destiné à prévenir ou à traiter les maladies inflammatoires, telles que notamment les maladies inflammatoires du système nerveux central comme la sclérose en plaque, encéphalite, myélite et encéphalomyélite et autres maladies inflammatoires comme les pathologies vasculaires, l'athérosclérose, les inflammations des articulations, l'arthrose, l'arthrite rhumatoïde.
Les composés selon l'invention peuvent donc être utilisés pour la préparation de médicaments, en particulier de médicaments inhibiteurs de la caséine kinase 1 epsilon et/ou de la caséine kinase 1 delta.
Ainsi, selon un autre de ses aspects, l'invention a pour objet des médicaments qui comprennent un composé de formule (I), ou un sel d'addition de ce dernier à un acide pharmaceutiquement acceptable du composé de formule (I).
Selon un autre de ses aspects, la présente invention concerne des compositions pharmaceutiques comprenant, en tant que principe actif, un composé selon l'invention. Ces compositions pharmaceutiques contiennent une dose efficace d'au moins un composé selon l'invention ou un sel pharmaceutiquement acceptable dudit composé, ainsi qu'au moins un excipient pharmaceutiquement acceptable.
Lesdits excipients sont choisis selon la forme pharmaceutique et le mode d'administration souhaité, parmi les excipients habituels qui sont connus de l'Homme du métier.
Dans les compositions pharmaceutiques de la présente invention pour l'administration orale, sublinguale, sous-cutanée, intramusculaire, intraveineuse, topique, locale, intra trachéale, intra nasale, transdermique ou rectale, le principe actif de formule (I) ci-dessus, ou son sel, peut être administré sous forme unitaire d'administration, en mélange avec des excipients pharmaceutiques classiques, aux animaux et aux êtres humains pour la prophylaxie ou le traitement des troubles ou des maladies ci-dessus.
Les formes unitaires d'administration appropriées comprennent les formes par voie orale telles que les comprimés, les gélules molles ou dures, les poudres, les granules et les solutions ou suspensions orales, les formes d'administration sublinguale, buccale, intra trachéale, intraoculaire, intra nasale, par inhalation, les formes d'administration topique, transdermique, sous-cutanée, intramusculaire ou intraveineuse, les formes d'administration rectale et les implants. Pour l'application topique, on peut utiliser les composés selon l'invention dans des crèmes, gels, pommades ou lotions.
A titre d'exemple, une forme unitaire d'administration d'un composé selon l'invention sous forme de comprimé peut comprendre les composants suivants :
Composé selon l'invention 50,0 mg
Mannitol 223,75 mg
Croscaramellose sodique 6,0 mg
Amidon de maïs 15,0 mg
Hydroxypropyl-méthylcellulose 2,25 mg Stéarate de magnésium 3,0 mg
Par voie orale, la dose de principe actif administrée par jour peut atteindre 0,1 à 20 mg/kg, en une ou plusieurs prises.
Il peut y avoir des cas particuliers où des dosages plus élevés ou plus faibles sont appropriés ; de tels dosages ne sortent pas du cadre de l'invention. Selon la pratique habituelle, le dosage approprié à chaque patient est déterminé par le médecin selon le mode d'administration, le poids et la réponse dudit patient.
La présente invention, selon un autre de ses aspects, concerne également une méthode de traitement des pathologies ci-dessus indiquées qui comprend l'administration, à un patient, d'une dose efficace d'un composé selon l'invention, ou un de ses sels pharmaceutiquement acceptables.
Claims
1. Composé répondant à la formule générale (I)

- R5 représente un atome d'hydrogène ou un groupe Ci-3 alkyle, -NR7R8, ou Ci-3 alkyloxy ;
- R6 représente un groupe phényle éventuellement substitué par un ou plusieurs substituants choisis parmi les atomes d'halogène et les groupes Ci-6 alkyle ; Ci-6 alkyloxy ;
- A représente un groupe Ci-7-alkylène éventuellement substitué par un ou deux groupes R3 ;
- B représente un groupe Ci-7-alkylène éventuellement substitué par un groupe Rb ;
- L représente soit un atome d'azote éventuellement substitué par un groupe Rc ou Rd, soit un atome de carbone substitué par un groupe Rei et un groupe Rd ou deux groupes Re2 ;
les atomes de carbone de A et de B étant éventuellement substitués par un ou plusieurs groupes Rf identiques ou différents l'un de l'autre ;
R3, Rb et Rc sont définis tels que : deux groupes R3 peuvent former ensemble un groupe Ci-6-alkylène ; R3 et Rb peuvent former ensemble une liaison ou un groupe Ci-6-alkylène; R3 et Rc peuvent former ensemble une liaison ou un groupe Ci-6-alkylène ; Rb et Rc peuvent former ensemble une liaison ou un groupe Ci-6-alkylène ; Rd représente un groupe choisi parmi l'atome d'hydrogène et les groupes Ci-6- alkyle, C3-7-cycloalkyle, C3-7-cycloalkyl-Ci.6-alkyle, Ci-6-alkylthio-C-i-6-alkyle, Ci- 6-alkyloxy-Ci-6-alkyle, Ci-6-fluoroalkyle, hydroxy-Ci-6-alkyle ;
Rei représente un groupe -NR3R4 ou une monoamine cyclique comportant éventuellement un atome d'oxygène, la monoamine cyclique étant éventuellement substituée par un ou plusieurs substituants choisis parmi l'atome de fluor et les groupes Ci-6-alkyle, Ci-6-alkyloxy, hydroxyle ;
Deux RΘ2 forment avec l'atome de carbone qui les porte une monoamine cyclique comportant éventuellement un atome d'oxygène, cette monoamine cyclique étant éventuellement substituée par un ou plusieurs groupes Rf identiques ou différents l'un de l'autre ;
Rf représente un groupe Ci-6-alkyle, C3-7-cycloalkyle, C3-7-cycloalkyle-Ci-6-alkyle, Ci- 6-alkyloxy-Ci-6-alkyle, hydroxy-Ci-6-alkyle, Ci-6-fluoroalkyle ;
R3 et R4 représentent, indépendamment l'un de l'autre, un atome d'hydrogène ou un groupe Ci-4 alkyle, C3-7-cycloalkyle, C3-7-cycloalkyle-Ci-6-alkyle ;
R7 et R8 représentent, indépendamment l'un de l'autre, un atome d'hydrogène ou un groupe Ci-3 alkyle ; à l 'état de base ou de sel d'addition à un acide.
2. Composé selon la revendication 1 , caractérisé en ce que :
- R5 repsésente un atome d'hydrogène ou un groupe Ci-3-alkyle, -NR7R8, Ci-3 alkyloxy ;
- R6 représente un groupe phényle éventuellement substitué par un ou plusieurs substituants choisis parmi les atomes d'halogène ; - l'aminé cyclique formée par -N-A-L-B- représente un groupe choisi parmi les groupes pipérazinyle, hexahydropyrrolo-pyrrolyle, éventuellement substitué par un ou plusieurs groupes choisis parmi les groupes méthyle, /sopropyle ; ou -N-A-L-B- représente un groupe diazaspiro-undécyle ; ou un groupe pipéridinyle, éventuellement substitué par un groupe pyrrolidinyle ; - R7 et R8 représentent, indépendamment l'un de l'autre, un atome d'hydrogène ou un groupe Ci-3 alkyle ; à l 'état de base ou de sel d'addition à un acide.
3. Procédé de préparation d'un composé de formule générale (I) selon la revendication 1 , caractérisé en ce que l'on fait réagir un composé de formule générale (II)

Dans laquelle A, L, B et R6 sont définis selon la revendication 1 et X5 représente un halogène choisi parmi le brome ou l'iode, avec un composé de formule générale (lia)

4. Composé de formule générale (II)
(ll) 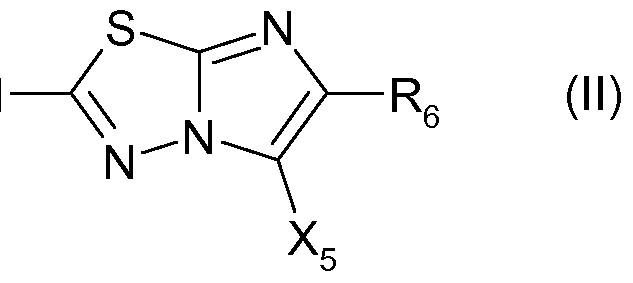 dans laquelle A, L, B et R6 sont définis selon la revendication 1 et X5 représente un halogène choisi parmi le brome ou l'iode.
dans laquelle A, L, B et R6 sont définis selon la revendication 1 et X5 représente un halogène choisi parmi le brome ou l'iode.
5. Médicament, caractérisé en ce qu'il comprend un composé de formule (I) selon l'une quelconque des revendications 1 ou 2, à l'état de base ou de sel d'addition à un acide pharmaceutiquement acceptable.
6. Composition pharmaceutique, caractérisée en ce qu'elle comprend un composé de formule (I) selon l'une quelconque des revendications 1 ou 2, à l'état de base ou de sel d'addition à un acide pharmaceutiquement acceptable, ainsi qu'au moins un excipient pharmaceutiquement acceptable.
7. Utilisation d'un composé de formule générale (I) selon l'une quelconque des revendications 1 ou 2, pour la préparation d'un médicament destiné au traitement ou à la prévention des désordres du sommeil, des troubles du rythme circadien.
8. Utilisation d'un composé de formule générale (I) selon l'une quelconque des revendications 1 ou 2, pour la préparation d'un médicament destiné au traitement ou à la prévention des troubles bipolaires.
9. Utilisation d'un composé de formule générale (I) selon l'une quelconque des revendications 1 ou 2, pour la préparation d'un médicament destiné au traitement ou à la prévention des maladies liées à la dépendance à des substances d'abus.
10. Utilisation d'un composé de formule générale (I) selon l'une quelconque des revendications 1 ou 2, pour la préparation d'un médicament destiné au traitement ou à la prévention des maladies reliées à l'hyperphosphorylation de la protéine tau.
11. Utilisation d'un composé de formule générale (I) selon l'une quelconque des revendications 1 ou 2, pour la préparation d'un médicament destiné au traitement ou à la prévention des maladies causées ou exacerbées par la prolifération des cellules.
12. Utilisation d'un composé de formule générale (I) selon la revendication 11 , caractérisé en ce que les cellules sont des cellules tumorales.
13. Utilisation d'un composé de formule générale (I) selon l'une quelconque des revendications 1 ou 2, pour la préparation d'un médicament destiné au traitement ou à la prévention des maladies inflammatoires.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR0902248A FR2945289A1 (fr) | 2009-05-11 | 2009-05-11 | Derives de 2-cycloamino-5-(pyridin-4-yl)imidazo°2,1-b! °1,3,4!thiadiazole, leur preparation et leur application en therapeutique |
| FR0902248 | 2009-05-11 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2010130934A2 true WO2010130934A2 (fr) | 2010-11-18 |
| WO2010130934A3 WO2010130934A3 (fr) | 2011-01-06 |
Family
ID=41404111
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR2010/050897 Ceased WO2010130934A2 (fr) | 2009-05-11 | 2010-05-10 | Derives de 2-cycloamino-5-(pyridin-4-yl)imidazo[2,1-b][1,3,4]thiadiazole, leur preparation et leur application en therapeutique |
Country Status (3)
| Country | Link |
|---|---|
| AR (1) | AR076665A1 (fr) |
| FR (1) | FR2945289A1 (fr) |
| WO (1) | WO2010130934A2 (fr) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012080729A3 (fr) * | 2010-12-14 | 2012-08-02 | Electrophoretics Limited | Inhibiteurs de caséine kinase 1δ (ck1δ) |
| WO2020005882A1 (fr) * | 2018-06-27 | 2020-01-02 | Ptc Therapeutics, Inc. | Composés hétéroaryles pour le traitement de la maladie de huntington |
| WO2020249717A1 (fr) | 2019-06-13 | 2020-12-17 | Facio Intellectual Property B.V. | Inhibiteurs de la caséine kinase 1 destinés à être utilisés dans le traitement de maladies liées à l'expression de dux4 telles que la dystrophie musculaire et le cancer |
| US11382918B2 (en) | 2017-06-28 | 2022-07-12 | Ptc Therapeutics, Inc. | Methods for treating Huntington's Disease |
| US11395822B2 (en) | 2017-06-28 | 2022-07-26 | Ptc Therapeutics, Inc. | Methods for treating Huntington's disease |
| US11407753B2 (en) | 2017-06-05 | 2022-08-09 | Ptc Therapeutics, Inc. | Compounds for treating Huntington's disease |
| US11638706B2 (en) | 2015-12-10 | 2023-05-02 | Ptc Therapeutics, Inc. | Methods for treating Huntington's disease |
| US11780839B2 (en) | 2018-03-27 | 2023-10-10 | Ptc Therapeutics, Inc. | Compounds for treating Huntington's disease |
| US11858941B2 (en) | 2018-06-27 | 2024-01-02 | Ptc Therapeutics, Inc. | Heterocyclic and heteroaryl compounds for treating Huntington's disease |
| US11939328B2 (en) | 2021-10-14 | 2024-03-26 | Incyte Corporation | Quinoline compounds as inhibitors of KRAS |
| US12139499B2 (en) | 2018-06-27 | 2024-11-12 | Ptc Therapeutics, Inc. | Heteroaryl compounds for treating Huntington's disease |
| US12577226B2 (en) | 2019-05-13 | 2026-03-17 | Ptc Therapeutics, Inc. | Compounds for treating Huntington's disease |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999047507A2 (fr) | 1998-03-19 | 1999-09-23 | Pharmacia & Upjohn Company | 1,3,4-thiadiazoles utiles pour le traitement d'infections a cmv |
| US20050131012A1 (en) | 2003-12-11 | 2005-06-16 | Aventis Pharmaceuticals Inc. | Substituted 1H-pyrrolo[3,2-b, 3,2-c, and 2,3-c]pyridine-2-carboxamides and related analogs as inhibitors of casein kinase lepsilon |
| WO2006040569A1 (fr) | 2004-10-14 | 2006-04-20 | Astex Therapeutics Limited | Composes d'amide thiophenique destines a etre utilises dans le traitement ou la prophylaxie du cancer |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL297170A (fr) * | 1963-04-04 | 1900-01-01 | ||
| US5552422A (en) * | 1995-01-11 | 1996-09-03 | Merck Frosst Canada, Inc. | Aryl substituted 5,5 fused aromatic nitrogen compounds as anti-inflammatory agents |
| PA8595001A1 (es) * | 2003-03-04 | 2004-09-28 | Pfizer Prod Inc | Nuevos compuestos heteroaromaticos condensados que son inhibidores del factor de crecimiento transforante (tgf) |
| WO2009019708A2 (fr) * | 2007-08-09 | 2009-02-12 | Urifer Ltd | Compositions pharmaceutiques et procédés pour le traitement du cancer |
| HRP20151128T1 (hr) * | 2007-09-27 | 2015-11-20 | Fundación Centro Nacional De Investigaciones Oncológicas Carlos Iii | Imidazolotiadiazoli za uporabu kao inhibitori protein kinaze |
-
2009
- 2009-05-11 FR FR0902248A patent/FR2945289A1/fr active Pending
-
2010
- 2010-05-10 AR ARP100101581A patent/AR076665A1/es unknown
- 2010-05-10 WO PCT/FR2010/050897 patent/WO2010130934A2/fr not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999047507A2 (fr) | 1998-03-19 | 1999-09-23 | Pharmacia & Upjohn Company | 1,3,4-thiadiazoles utiles pour le traitement d'infections a cmv |
| US20050131012A1 (en) | 2003-12-11 | 2005-06-16 | Aventis Pharmaceuticals Inc. | Substituted 1H-pyrrolo[3,2-b, 3,2-c, and 2,3-c]pyridine-2-carboxamides and related analogs as inhibitors of casein kinase lepsilon |
| WO2006040569A1 (fr) | 2004-10-14 | 2006-04-20 | Astex Therapeutics Limited | Composes d'amide thiophenique destines a etre utilises dans le traitement ou la prophylaxie du cancer |
Non-Patent Citations (4)
| Title |
|---|
| CHEMISTRY OF HETEROCYCLIC COMPOUNDS, vol. 43, no. 4, 2007, pages 499 - 504 |
| FARMACO, EDIZIONE SCIENTIFICA, vol. 41, no. 10, 1986, pages 737 - 46 |
| JOURNAL OF HETEROCYCLIC CHEMISTRY, vol. 45, no. 1, 2008, pages 299 - 302 |
| TETRAHEDRON LETTERS, vol. 49, no. 36, 2008, pages 5241 - 5243 |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012080729A3 (fr) * | 2010-12-14 | 2012-08-02 | Electrophoretics Limited | Inhibiteurs de caséine kinase 1δ (ck1δ) |
| WO2012080727A3 (fr) * | 2010-12-14 | 2012-08-23 | Electrophoretics Limited | Inhibiteurs de caséine kinase 1 delta (ck1delta) |
| CN103298460A (zh) * | 2010-12-14 | 2013-09-11 | 电泳有限公司 | 酪蛋白激酶1δ(CK1δ)抑制剂 |
| CN103298460B (zh) * | 2010-12-14 | 2016-06-01 | 电泳有限公司 | 酪蛋白激酶1δ(CK1δ)抑制剂 |
| CN105920010A (zh) * | 2010-12-14 | 2016-09-07 | 电泳有限公司 | 酪蛋白激酶1δ(CK1δ)抑制剂 |
| US9763947B2 (en) | 2010-12-14 | 2017-09-19 | Electrophoretics Limited | Casein kinase 1delta (CK1delta) inhibitors |
| US9789111B2 (en) | 2010-12-14 | 2017-10-17 | Electrophoretics Limited | Casein kinase 1δ (CK 1δ) inhibitors |
| US11638706B2 (en) | 2015-12-10 | 2023-05-02 | Ptc Therapeutics, Inc. | Methods for treating Huntington's disease |
| US12384789B2 (en) | 2017-06-05 | 2025-08-12 | Ptc Therapeutics, Inc. | Compounds for treating Huntington's disease |
| US11407753B2 (en) | 2017-06-05 | 2022-08-09 | Ptc Therapeutics, Inc. | Compounds for treating Huntington's disease |
| US11395822B2 (en) | 2017-06-28 | 2022-07-26 | Ptc Therapeutics, Inc. | Methods for treating Huntington's disease |
| US11382918B2 (en) | 2017-06-28 | 2022-07-12 | Ptc Therapeutics, Inc. | Methods for treating Huntington's Disease |
| US11780839B2 (en) | 2018-03-27 | 2023-10-10 | Ptc Therapeutics, Inc. | Compounds for treating Huntington's disease |
| US12103926B2 (en) | 2018-03-27 | 2024-10-01 | Ptc Therapeutics, Inc. | Compounds for treating huntington's disease |
| US11685746B2 (en) | 2018-06-27 | 2023-06-27 | Ptc Therapeutics, Inc. | Heteroaryl compounds for treating Huntington's disease |
| US11858941B2 (en) | 2018-06-27 | 2024-01-02 | Ptc Therapeutics, Inc. | Heterocyclic and heteroaryl compounds for treating Huntington's disease |
| US12139499B2 (en) | 2018-06-27 | 2024-11-12 | Ptc Therapeutics, Inc. | Heteroaryl compounds for treating Huntington's disease |
| WO2020005882A1 (fr) * | 2018-06-27 | 2020-01-02 | Ptc Therapeutics, Inc. | Composés hétéroaryles pour le traitement de la maladie de huntington |
| US12577226B2 (en) | 2019-05-13 | 2026-03-17 | Ptc Therapeutics, Inc. | Compounds for treating Huntington's disease |
| WO2020249717A1 (fr) | 2019-06-13 | 2020-12-17 | Facio Intellectual Property B.V. | Inhibiteurs de la caséine kinase 1 destinés à être utilisés dans le traitement de maladies liées à l'expression de dux4 telles que la dystrophie musculaire et le cancer |
| US11939328B2 (en) | 2021-10-14 | 2024-03-26 | Incyte Corporation | Quinoline compounds as inhibitors of KRAS |
| US12378243B2 (en) | 2021-10-14 | 2025-08-05 | Incyte Corporation | Quinoline compounds as inhibitors of KRAS |
Also Published As
| Publication number | Publication date |
|---|---|
| FR2945289A1 (fr) | 2010-11-12 |
| AR076665A1 (es) | 2011-06-29 |
| WO2010130934A3 (fr) | 2011-01-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2178879B1 (fr) | DERIVES DE 6-CYCLOAMINO-S-(PYRIDAZIN-YL)IMIDAZO[1,2-b]-PYRIDAZINE, LEUR PREPARATION ET LEUR APPLICATION EN THERAPEUTIQUE. | |
| WO2010130934A2 (fr) | Derives de 2-cycloamino-5-(pyridin-4-yl)imidazo[2,1-b][1,3,4]thiadiazole, leur preparation et leur application en therapeutique | |
| EP2170889B1 (fr) | Derives de 6-cycloamino-3-(pyridin-4-yl)imidazo[1,2-b]pyridazine, leur preparation et leur application en therapeutique | |
| KR20120095356A (ko) | Pask의 억제를 위한 복소환 화합물 | |
| US10414769B2 (en) | 5,8-dimethyl-9-phenyl-5,8-dihydro-6H-pyrazolo[3,4-h]quinazolin-2-yl)-(1H-pyrazol-3-yl)-amines as IGF-1R/IR inhibitors | |
| EP2331546B1 (fr) | Derives de 2-alkyl-6-cycloamino-3-(pyridin-4-yl)imidazo[1,2-ib]-pyridazine, leur preparation et leur application en therapeutique | |
| EP3166945B1 (fr) | Nouveaux dérivés triazolopyrimidinone ou triazolopyridinone et leur utilisation | |
| WO2010070238A1 (fr) | DÉRIVÉS DE 6-CYCLOAMINO-2,3-DI-PYRIDINYL-IMIDAZO[1,2-b]-PYRIDAZINE, LEUR PRÉPARATION ET LEUR APPLICATION EN THÉRAPEUTIQUE | |
| EP2398802A1 (fr) | DÉRIVÉS DE 6-CYCLOAMINO-2-THIENYL-3-(PYRIDIN-4-YL)IMIDAZO[1,2-b]-PYRIDAZINE ET 6-CYCLOAMINO-2-FURANYL-3-(PYRIDIN-4-YL)IMIDAZO[1,2-b]-PYRIDAZINE, LEUR PRÉPARATION ET LEUR APPLICATION EN THÉRAPEUTIQUE | |
| EP2370446B1 (fr) | DÉRIVÉS DE 6-CYCLOAMINO-3-(1H-PYRROLO[2,3-b]PYRIDIN-4-YL)IMIDAZO[1,2-b]-PYRIDAZINE, LEUR PRÉPARATION ET LEUR APPLICATION EN THÉRAPEUTIQUE | |
| FR2960876A1 (fr) | Derives de 3,4-dihydropyrrolo[1,2-a]pyrazine-2,8(1h)-dicarboxamide leur preparation et leur application en therapeutique. | |
| EP4543887A2 (fr) | Inhibiteurs à petites molécules de dyrk/clk et leurs utilisations | |
| KR102344185B1 (ko) | 신규한 Pim 키나아제 억제제 및 이의 용도 | |
| FR2846330A1 (fr) | Derives de pyridoindolone substitues en -3 par groupe heterocyclique, leur preparation et leur application en therapeutique | |
| EP4671245A1 (fr) | Composé cyclique hétéroaromatique servant d'inhibiteur de kinase cdk7, sa préparation et son utilisation | |
| MX2011006627A (es) | Derivados de 6-cicloamino-2,3-di-piridinil-imidazo[1,2-b]-piridazi na, su preparacion y su uso en terapeutica. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 10728730 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 10728730 Country of ref document: EP Kind code of ref document: A2 |