Retigabin-Tabletten, bevorzugt mit modifizierter Freisetzung
Die Erfindung betrifft Tabletten, insbesondere Tabletten mit modifizierter Freisetzung, enthaltend (a) Retigabin sowie eine Kombination aus (b) wasserlöslichem Hilfsstoff und (c) nicht-wasserlöslichem Hilfsstoff; sowie ein Verfahren zu deren Herstellung.
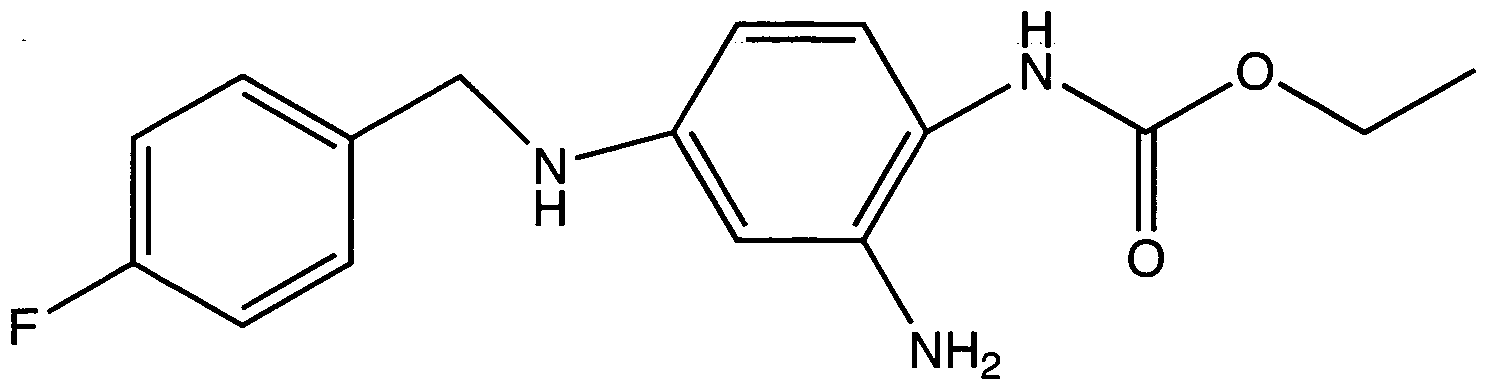
Der IUPAC-Name von Retigabin [INN] ist 2-Amino-4-(4-fluorbenzylamino)- l - ethoxycarbonyl-aminobenzen. Die chemische Struktur von Retigabin wird in nachstehender Formel ( 1 ) dargestellt:
Synthesewege für Retigabin und seine Verwendung als Antiepileptikum wurden in EP O 554 543 beschrieben. Aus WO 01 /22953 A2 ist zudem die Verwendung von Retigabin zur Behandlung des neuropathischen Schmerzes bekannt.
Epilepsie ist eine der häufigsten neurologischen Erkrankungen und betrifft etwa bis zu 1 % der Bevölkerung. Während ein Großteil der Epilepsie-Patienten mit derzeit auf dem Markt verfügbaren Antikonvulsiva behandelt werden kann, sind etwa 30% der Patienten pharmakoresistent. Daher ist es notwendig, neue Antikonvulsiva mit innovativen Wirkmechanismen zu entwickeln. Retigabin, eine antikonvulsive Substanz, erfüllt als Kaliumkanalöffner diese Kriterien. Jedoch sind im Fachgebiet noch keine pharmazeutischen Darreichungsformen bekannt, die eine vorteilhafte, orale Verabreichung von Retigabin in hohen Dosen, insbesondere mit modifizierter Freisetzung, zur Behandlung der Epilepsie erlauben.
WO 02/80898 A2 schlägt vor, Retigabin in Form von Hartgelatinekapseln, enthaltend 50, 100 und 200 mg Wirkstoff, zu formulieren. Hartgelatinekapseln werden von Patienten häufig als unangenehm zum Einnehmen empfunden. Insbesondere ist es problematisch, mit dieser Methode einen hohen Wirkstoffgehalt (z.B. 70 %) in der Kapsel zu verwirklichen. Es hat sich ferner gezeigt, dass Kapseln, die mittels Feuchtgranulierung von Retigabin hergestellt werden, im Hinblick auf ihre pharmakokinetischen Eigenschaften nicht optimal sind und keine modifizierte Freisetzung erlauben.
In WO 01 /66081 A2 werden ferner Retigabin-Retard-Formulierungen vorgeschlagen, die durch Schmelzgranulation hergestellt wurden, wobei eine Zusammensetzung, bestehend ausschließlich aus Retigabin und Saccharosefettsäureester, verwendet wurde. Die Verwendung von größeren Mengen Saccharosefettsäureester ist jedoch aufgrund der Emulgatorwirkung häufig unerwünscht. Auch führen die vorgeschlagenen Formulierungen zu einem häufig unerwünscht langsamen Wirkeintritt.
Aufgabe der vorliegenden Erfindung war es daher, die vorstehend genannten Nachteile zu überwinden.
Eine Aufgabe der Erfindung war es, eine für den Patienten angenehme Darreichungsform bereit zu stellen, die es ermöglicht, auch Wirkstoffmengen von deutlich mehr als 200 mg vorteilhaft zu verabreichen, insbesondere auch mit modifizierter Freisetzung.
Hierbei soll insbesondere sowohl ein schneller Wirkeintritt, als auch eine lang anhaltende Freisetzung (und somit ein konstanter Plasmaspiegel) erreicht werden.
Es soll der Wirkstoff in einer Form bereitgestellt werden, der - gegebenenfalls trotz Mikronisierung - eine gute Fließfähigkeit aufweist und eine gute Verpressung ermöglicht. Die resultierenden Tabletten sollen eine hohe Härte und eine niedrige Friabilität aufweisen.
Insbesondere war es Aufgabe der Erfindung, ein Verfahren zur Herstellung von Retigabin enthaltenden Tabletten bereit zu stellen, die eine vorteilhafte Lackierbar keit zeigen. Bei der Lackierung der erfindungsgemäßen Tabletten soll es zu keinen "Abplatzern" kommen.
Die Erfinder der vorliegenden Anmeldung waren bei der Entwicklung von Retigabin- Formulierungen weiterhin mit der Tatsache konfrontiert, dass kristallines Retigabin in verschiedenen polymorphen Formen existieren kann. Wie in WO 98/31663 beschrieben, sind diese Polymorphe jedoch häufig nicht stabil, sondern neigen dazu, sich in andere polymorphe Formen umzuwandeln. Beispielsweise kann die häufig verwendete Retigabin Form A bei Wärmeinwirkung sich in Form B umwandeln. Die polymorphen Formen A, B und C weisen jedoch ein unterschiedliches Löslichkeitsprofil auf.
Das unterschiedliche Löslichkeitsprofil führt beim Patienten zu einem unerwünscht ungleichmäßigen Anfluten des Wirkstoffs. Aufgabe der vorliegenden Erfindung war es daher, stabile Retigabin-Intermediate bereit zu stellen, die zu einer Darreichungsform
verarbeitet werden können, die eine möglichst gleichmäßige Anflutung beim Patienten ermöglicht. Sowohl interindividuelle als auch intraindividuelle Abweichungen sollen weitgehend vermieden werden.
Im Hinblick auf die Labilität des Wirkstoffs war es daher eine weiteren Aufgabe, Tabletten bereit zu stellen, die eine gute Lagerbeständigkeit aufweisen.
Alle der vorstehend genannten Aufgaben sollen insbesondere für einen hohen Wirkstoffgehalt (drug load) gelöst werden.
Die Aufgaben konnten unerwartet durch die Kombination eines wasserlöslichen und eines nicht-wasserlöslichen Hilfsstoffs gelöst werden.
Gegenstand der Erfindung ist daher eine Tablette enthaltend
(a) Retigabin,
(b) einen wasserlöslichen Hüfsstoff; und
(c) einen nicht-wasserlöslichen Hüfsstoff.
Gegenstand der Erfindung ist ferner ein Verfahren zur Herstellung der erfindungsgemäßen Tabletten, umfassend die Schritte
(I) Bereitstellen von (a) Retigabin, (b) wasserlöslichem Hüfsstoff und (c) nichtwasserlöslichem Hüfsstoff sowie gegebenenfalls (d) Sprengmittel, (II) gegebenenfalls Kompaktierung zu einer Schülpe;
(III) gegebenenfalls Granulierung der Schülpe;
(IV) Kompression zu Tabletten; und
(V) gegebenenfalls Befümung der Tabletten.
Schließlich ist Gegenstand der Erfindung die Verwendung einer Kombination aus wasserlöslichem und nicht-wasserlöslichem Hüfsstoff zur Herstellung einer Retigabin- Tablette mit modifizierter Freisetzung.
Im Rahmen dieser Erfindung umfasst der Begriff "Retigabin" (= Komponenten (a)) 2-Amino-4-(4-fluorbenzylamino)- l -ethoxycarbonyl-aminobenzen gemäß vorstehender Formel ( 1 ). Zudem umfasst der Begriff "Retigabin" alle pharmazeutisch verträglichen Salze, Hydrate und Solvate davon.
Bei den Salzen kann es sich um Säureadditionssalze handeln. Beispiele für geeignete Salze sind Hydrochloride (z.B. Monohydrochlorid, Düiydrochlorid), Carbonate,
Hydrogencarbonate, Acetate, Lactate, Butyrate, Propionate, Sulfate, Methansulfonate,
Citrate, Tartrate, Nitrate, Sulfonate, Oxalate und /oder Succinate. Bevorzugt wird Retigabin im Rahmen dieser Erfindung in Form der freien Base verwendet. Alternativ bevorzugt wird Retigabin im Rahmen dieser Erfindung in Form des Dihydrochlorids verwendet.
Retigabin kann im Rahmen dieser Erfindung sowohl in amorpher als auch in kristalliner Form eingesetzt werden. Ebenfalls kann Retigabin in Form einer festen Lösung eingesetzt werden. Bevorzugt wird kristallines Retigabin verwendet.
Kristallines Retigabin kann gemäß WO 98/31663 in drei verschiedenen polymorphen Formen vorliegen (polymorphe Formen A, B und C). Bevorzugt wird im Rahmen dieser Erfindung im Falle von kristallinem Retigabin die polymorphe Form A verwendet.
Bei dem wasserlöslichen Hilfsstoff (= Komponenten (b)) handelt es sich im Allgemeinen um einen im Europäischen Arzneibuch spezifizierten pharmazeutischen Hilfsstoff, welcher eine Wasserlöslichkeit von mehr als 33 mg/ml, gemessen bei 25 0C, aufweist.
Bevorzugt weist der wasserlösliche Stoff eine Löslichkeit von mehr als 50 mg/ml, noch mehr bevorzugt von mehr als 100 mg/ml, insbesondere von mehr als 250 mg/ml auf, beispielsweise eine Wasserlöslichkeit zwischen 250 mg/ ml und 1 g/ml. Im Rahmen dieser Erfindung wird die Wasserlöslichkeit gemäß Säulenelutionsmethode nach EU-
Richtlinie RL67-548-EWG, Anhang V Kap. A6, bestimmt.
In einer bevorzugten Ausführungsform handelt es sich bei dem wasserlöslichen Hilfsstoff (b) um ein Polymer, insbesondere um ein hydrophiles Polymer. Ferner umfasst der Begriff "wasserlöslicher Hilfsstoff" (b) feste, nicht-polymere Verbindungen, die bevorzugt polare Seitengruppen und die vorstehend genannte Löslichkeit aufweisen. Beispiele hierfür sind Zuckeralkohole.
Bei dem im Rahmen dieser Erfindung verwendeten wasserlösliche Polymer (b) handelt es sich bevorzugt um ein Polymer, das eine Glasübergangstemperatur (Tg) von größer 15 0C, mehr bevorzugt von 40 0C bis 150 0C, insbesondere von 50 0C bis 100 0C aufweist.
Als "Glasübergangstemperatur" (Tg) bezeichnet man die Temperatur, bei der amorphe oder teilkristalline Polymere vom festen Zustand in den flüssigen Zustand übergehen.
Dabei tritt eine deutliche Änderung physikalischer Kenngrößen, z. B. der Härte und der Elastizität, ein. Unterhalb der Tg ist ein Polymer üblicherweise glasartig und hart, oberhalb der Tg geht es in einen gummiartigen bis zähflüssigen Zustand über. Die
Bestimmung der Glasübergangstemperatur erfolgt im Rahmen dieser Erfindung mittels dynamischer Differenzkalorimetrie (DSC). Hierzu kann z. B. ein Gerät von
Mettler Toledo DSC 1 eingesetzt werden. Es wird mit einer Heizrate von 1 -20 °C/min,
bevorzugt 5- 15 °C/min bzw. mit einer Kühlrate von 5-25, bevorzugt 10-20 °C/min, gearbeitet.
Ferner weist das als wasserlösliches Polymer (b) verwendbare Polymer bevorzugt ein gewichtsmittleres oder zahlenmittleres Molekulargewicht von 1.000 bis
100.000 g/mol, mehr bevorzugt von 4.000 bis 70.000 g/mol, insbesondere von 5000 bis 50.000 g/mol auf. Wird das wasserlösliche Polymer (b) in (destilliertem) Wasser in einer Menge von 2 Gew.-% gelöst, so zeigt die resultierende Lösung bevorzugt eine
Viskosität von 0, 1 bis 8 mPa s, mehr bevorzugt von 0,3 bis 6 mPa s, insbesondere von 0,5 bis 4 mPa-s, gemessen bei 25 0C gemäß Ph. Eur. 6. Ausgabe, Kapitel 2.2.10.
Bevorzugt werden, wie vorstehend beschrieben, als wasserlösliche Komponente (b) hydrophile Polymere verwendet. Darunter sind Polymere zu verstehen, die hydrophile Gruppen aufweisen. Beispiele für geeignete hydrophile Gruppen sind Hydroxy, Alkoxy, Acrylat, Methacrylat, Sulfonat, Carboxylat und quartäre Ammoniumgruppen.
Der wasserlösliche Hilfsstoff (b) kann beispielsweise folgende Polymere umfassen: Polysaccharide, wie Hydroxypropylmethylcellulose (HPMC), Carboxymethylcellulose (CMC, insbesondere Natrium- und Calciumsalze), Hydroxyethylcellulose, Ethylhy- droxyethylcellulose, Hydroxypropylcellulose (HPC); Guar-Mehl, Alginsäure und/oder Alginate, Pektin, Tragant; synthetische Polymere wie Polyvinylpyrrolidon (Povidon), Polyvinylacetat (PVAC), Polyvinylalkohol (PVA), Polymere der Acrylsäure und deren Salze, Polyacrylamid, Derivate der Polymethacrylate (Eudragit® E, Eudragit® R, Eudragit® S), Vinylpyrrolidon-Vinylacetat-Copolymere (beispielsweise Kollidon® VA64, BASF), Polyalkylenglykole, wie Polypropylenglykol oder bevorzugt Polyethylenglykol, Co-blockpolymere des Polyethylenglykols, insbesondere Co-blockpolymere aus Polyethylenglykol und Polypropylenglykol (Pluronic®, BASF) sowie Gemische aus den genannten Polymeren. Es könne zudem Dextrine verwendet werden.
Alternativ kann die Komponente (b) auch feste, nicht-polymere Verbindungen, die bevorzugt polare Seitengruppen aufweisen, umfassen. Beispiele hierfür sind Zuckeralkohole oder Disaccharide. Beispiele für geeignete Zuckeralkohole sind Mannitol, Sorbitol, Xylitol, Isomalt, Glucose, Fructose und Gemische daraus. Der Begriff Zuckeralkohole umfasst hier auch Monosaccharide.
Außerdem können auch Fettsäurederivate wie z.B. Natriumlaurylsulfat Verwendung finden, jedoch mit der Maßgabe, dass keine Saccharosefettsäureester verwendet werden. Weiterhin sind Gemische der genannten wasserlöslichen Hilfsstoffe (b) möglich.
Bei dem nicht-wasserlöslichen Hilfsstoff (= Komponenten (c)) handelt es sich im Allgemeinen um einen im Europäischen Arzneibuch spezifizierten pharmazeutischen Hilfsstoff, welcher eine Wasserlöslichkeit von weniger als 33 mg/ml, gemessen bei 25 0C, aufweist. Bevorzugt weist der nicht-wasserlösliche Stoff eine Löslichkeit von 10 mg/ml oder weniger, mehr bevorzugt von 5 mg/ml oder weniger, insbesondere von 0,01 bis 2 mg/ml auf (bestimmt gemäß Säulenelutionsmethode nach EU-Richtlinie RL67-548-EWG, Anhang V Kap. A6).
Bei der Komponente (c) handelt es sich bevorzugt um ein nicht-wasserlösliches Polymer oder um einen nicht-wasserlöslichen pharmazeutischen Hilfsstoff mit polymerähnlichen Eigenschaften. Die Komponente (c) führt bevorzugt zu einer modifizierten Wirkstofffreisetzung. Es hat sich gezeigt, dass das Freisetzungsprofil insbesondere durch Wahl einer Komponente (c) mit geeignetem Molekulargewicht und Quervernetzungsgrad, mit geeigneter Viskosität (bezogen auf eine Lösung der Komponente (c) in Wasser), mit geeignetem Quellverhalten und/oder mit geeigneter Glasübergangs- oder Schmelztemperatur beeinflusst werden kann. Alternativ kann auch eine Komponente (c) verwendet werden, die zu einer sofortigen Freisetzung führt, mit der Maßgabe, dass als Beschichtung eine Retardbeschichtung - wie nachstehend als Komponente (e3) beschrieben - verwendet wird.
Das nicht-wasserlösliche Polymer (c) hat üblicherweise ein gewichtsmittleres Molekulargewicht von 50.000 bis 2.500.000 g/mol, bevorzugt von 250.000 bis 2.000.000 g/mol, mehr bevorzugt von 350.000 bis 1.500.000 g/mol. Das nicht- wasserlösliche Polymer (c) kann linear oder bevorzugt quervernetzt sein. Das nichtwasserlösliche Polymer (c) weist in letzterem Fall bevorzugt einen Quervernetzungsgrad von 0, 1 bis 10 %, insbesondere von 0,5 bis 5 %, auf. (Quervernetzungsgrad = Anzahl an Kohlenstoffatomen, die mehr als an eine Kette anknüpfen / Anzahl an Kohlenstoffatomen in der Polymerkette gesamt).
Wird das nicht-wasserlösliche Polymer (c) in (destilliertem) Wasser in einer Menge von 2 Gew.-% (zumindest teilweise) gelöst, so zeigt die resultierende Lösung bevorzugt eine Viskosität von mehr als 2 mPa s, mehr bevorzugt von 4 mPa s, besonders bevorzugt von mehr als 8 mPa s, insbesondere von mehr als 10 mPa s und beispielsweise bis zu 500 mPa-s, gemessen bei 25 0C gemäß Ph. Eur. 6. Ausgabe, Kapitel 2.2.10.
Bei der Komponente (c) handelt es sich bevorzugt um ein quellbares Polymer oder um einen quellbaren Stoff mit polymerähnlichen Eigenschaften. Das nicht-wasserlösliche
Polymer (c) hat bevorzugt eine Quellungszahl von 1 ,5 bis 4,5, bevorzugt von 2,0 bis 4,0. Die Quellungszahl gibt das Volumen in Millilitern an, das 1 g Stoff einschließlich
des gegebenenfalls anhaftenden Schleims nach Quellen in einer wässrigen Lösung nach 4 Stunden einnimmt. Die Quellungszahl wird nach Ph. Eur. 4. Ausgabe, Kapitel 2.8.4 bestimmt.
Bei der Komponente (c) handelt es sich ferner bevorzugt um ein Polymer oder um einen quellbaren Stoff mit einer Glasübergangstemperatur oder einer Schmelztemperatur von weniger als 200 0C, mehr bevorzugt von 20 0C bis 180 0C, insbesondere von 30 0C bis 170 0C.
Als "Glasübergangstemperatur" (Tg) bezeichnet man die Temperatur, bei der amorphe oder teilkristalline Polymere vom festen Zustand in den flüssigen Zustand übergehen. Dabei tritt eine deutliche Änderung physikalischer Kenngrößen, z. B. der Härte und der Elastizität, ein. Unterhalb der Tg ist ein Polymer üblicherweise glasartig und hart, oberhalb der Tg geht es in einen gummiartigen bis zähflüssigen Zustand über. Die Bestimmung der Glasübergangstemperatur erfolgt im Rahmen dieser Erfindung mittels dynamischer Differenzkalorimetrie (DSC). Hierzu kann z. B. ein Gerät von Mettler Toledo DSC 1 eingesetzt werden. Es wird mit einer Heizrate von 1-20 °C/min, bevorzugt 5- 15 °C/min bzw. mit einer Kühlrate von 5-25, bevorzugt 10-20 °C/min gearbeitet. Die Bestimmung der Schmelztemperatur erfolgt gemäß Ph. Eur., 6. Auflage, Kapitel 2.2.15 (Offene Kapillarmethode).
Beispiele für geeignete nicht-wasserlösliche Polymere (c) sind Polymere auf Acrylatbasis, z.B. Acrylate, Derivate der Methacrylate (Eudragit® NE, Eudragit® RS, Eudragit® RL); Cellulosederivate wie Ethylcellulose (EC), Methylcellulose (MC), Celluloseacetylphathalat, Hydroxypropylmethylcellulosephthalat; synthetische Polymere wie Polyvinylacetat, Polyvinylchlorid, Nylon, Polyamid, Polyethylen, quervernetztes Polyvinylpyrrolidon und Polylactide-co-glycolide. Ebenfalls sind Gemische der genannten Polymere möglich. Die hierbei genannten Polymere besitzen bevorzugt eine oder mehrere der vorstehend genannten funktionellen Eigenschaften (Mw, Quervernetzung, Viskosität in Lösung, Quellungszahl, Schmelz- oder Glasübergangs temperatur ).
Mikrokristalline Cellulose schmilzt bei etwa 250 0C unter Zersetzung. Die Verwendung von mikrokristalliner Cellulose führt üblicherweise zu keiner modifizierten Wirkstofffreisetzung. Daher wird mikrokristalline Cellulose nicht als Komponenten (c) im Rahmen dieser Erfindung verwendet.
Als nicht-wasserlösliche Stoffe (mit polymerähnlichen Eigenschaften) können Wachse und Fette verwendet werden. Geeignete Wachse oder Fette sind bei 25 0C fest. Beispielsweise ist festes Paraffin oder Bienenwachs geeignet. Die Komponente (c) umfasst jedoch keine Saccharosefettsäureester.
In bevorzugten Ausführungsformen der vorliegenden Erfindung werden wasserlöslicher Hilfsstoff (b) und nicht-wasserlöslicher Hilfsstoff (c) in einer Menge verwendet, wobei das Gewichtsverhältnis von Komponente (b) zu Komponente (c) 10 : 1 bis 1 : 10, mehr bevorzugt 5 : 1 bis 1 : 5, noch mehr bevorzugt 4 : 1 bis 1 : 2, insbesondere 3 : 1 bis 1 : 1 , beträgt.
Es ist vorteilhaft, wenn die Komponenten (b) und (c) in partikulärer Form eingesetzt werden, wobei die volumenmittlere Teilchengröße (D50) der Komponenten (b) und (c) weniger als 500 μm, bevorzugt 5 bis 200 μm, beträgt.
In einer möglichen Ausführungsform der vorliegenden Erfindung wird Retigabin in mikronisierter Form verwendet.
Der Begriff "mikronisiertes Retigabin" bezieht sich im Rahmen dieser Erfindung auf partikuläres Retigabin, das im Allgemeinen einen mittleren Teilchendurchmesser von 0, 1 bis 200 μm, bevorzugt von 0,5 bis 100 μm, mehr bevorzugt von 1 bis 50 /an, besonders bevorzugt 1 ,5 bis 25 μm und insbesondere von 2 bis 10 μm, aufweist.
Der Ausdruck "mittlerer Teilchendurchmesser" bezieht sich im Rahmen dieser Erfindung stets auf den D50-Wert des volumenmittleren Teilchendurchmessers, der mittels Laserdiffraktometrie bestimmt wurde. Insbesondere wurde zur Bestimmung ein Mastersizer 2000 von Malvern Instruments verwendet (Nassmessung, mit Ultraschall 60 sek, 2000 rpm, wobei die Auswertung nach dem Fraunhofer Modell erfolgt und bevorzugt ein Dispergiermittel verwendet wird, in dem sich die zu messende Substanz bei 20 0C nicht löst. Der mittlere Teilchendurchmesser, der auch als D50-Wert der integralen Volumenverteilung bezeichnet wird, wird im Rahmen dieser Erfindung als der Teilchendurchmesser definiert, bei dem 50 Volumen-% der Teilchen einen kleineren Durchmesser haben als der Durchmesser, der dem D50-Wert entspricht. Ebenso haben dann 50 Volumen-% der Teilchen einen größeren Durchmesser als der D50-Wert. Die Ausdrücke "mittlere Teilchengröße" und "mittlerer Teilchendurchmesser" werden in Rahmen dieser Anmeldung synonym verwendet.
Die erfindungsgemäße Tablette kann die Komponenten (a), (b) und (c) in üblichen Mengenverhältnissen enthalten. In einer bevorzugten Ausführungsform enthält die erfindungsgemäße Tablette
(a) 25 bis 75 Gew.-%, mehr bevorzugt 35 bis 70 Gew.%, insbesondere mehr als 50 bis 65 Gew.-% Retigabin,
(b) 5 bis 40 Gew.-%, mehr bevorzugt 8 bis 35 Gew.-%, insbesondere 10 bis 30 Gew.-% wasserlöslichen Hilfsstoff und
(C) 20 bis 70 Gew.-%, mehr bevorzugt 22 bis 55 Gew.-%, insbesondere 25 bis 50
Gew. % nicht-wasserlöslichen Hilfsstoff, bezogen auf das Gesamtgewicht der Komponenten (a), (b) und (c).
Die erfindungsgemäße Tablette kann aus den Komponenten (a), (b) und (c) bestehen. Es ist jedoch bevorzugt, dass die erfindungsgemäße Tablette zusätzlich Sprengmittel (= Komponente (d)) enthält. Das Sprengmittel (d) wird bevorzugt in einer Menge von 1 bis 10 Gew.-%, insbesondere 2 bis 8 Gew.-%, bezogen auf das Gesamtgewicht der Komponenten (a) bis (d), verwendet.
Als Sprengmittel werden im Allgemeinen Stoffe bezeichnet, die den Zerfall einer Darreichungsform, insbesondere einer Tablette, nach Einbringen in Wasser beschleunigen. Geeignete Sprengmittel sind z.B. organische Sprengmittel wie Carrageenan, Croscarmellose und Crospovidon. Ebenfalls können alkalische Sprengmittel verwendet werden. Unter alkalischen Sprengmitteln sind Sprengmittel zu verstehen, die beim Lösen in Wasser einen pH-Wert von mehr als 7,0 erzeugen.
Geeignete alkalische Sprengmittel sind Salze von Alkali- und Erdalkalimetallen. Bevorzugt sind hier Natrium, Kalium, Magnesium und Calcium zu nennen. Als Anionen sind Carbonat, Hydrogencarbonat, Phosphat, Hydrogenphosphat und Dihydrogenphosphat bevorzugt. Beispiele sind Natriumhydrogencarbonat, Natrium- hydrogenphosphat, Calciumhydrogencarbonat und dergleichen.
Als Sprengmittel werden bevorzugt Croscarmellose und Crospovidon verwendet.
Die erfindungsgemäße Tablette kann aus den Komponenten (a), (b), (c) und (d) bestehen. Alternativ können jedoch noch weitere pharmazeutische Hilfsstoffe zugesetzt werden. Insbesondere werden noch Mittel zur Verbesserung der Pulverfließfähigkeit und Schmiermittel zugesetzt.
Ein Beispiel für einen Zusatz zur Verbesserung der Pulverfließfähigkeit ist disperses Siliciumdioxid, z.B. bekannt unter dem Handelsnamen Aerosil®. Bevorzugt wird Siliciumdioxid mit einer spezifischen Oberfläche von 50 bis 400 m2/g verwendet, bestimmt nach Gasadsorption gemäß Ph. Eur., 6. Auflage 2.9.26.
Zusätze zur Verbesserung der Pulverfließfähigkeit werden üblicherweise in einer Menge von 0,1 bis 3 Gew.-%, bezogen auf das Gesamtgewicht der Formulierung, verwendet.
Ferner können Schmiermittel verwendet werden. Schmiermittel dienen im Allgemeinen zur Verringerung der Gleitreibung. Insbesondere soll die Gleitreibung vermindert werden, die beim Tablettieren einerseits zwischen den sich in der Matrizenbohrung
auf und ab bewegenden Stempeln und der Matrizenwand sowie andererseits zwischen Tablettensteg und Matrizenwand besteht. Geeignete Schmiermittel stellen z.B. Stearinsäure, Adipinsäure, Natriumstearylfumarat, Zinkstearat und/oder Magnesium- stearat dar.
Schmiermittel werden üblicherweise in einer Menge von 0,1 bis 3 Gew.-%, bezogen auf das Gesamtgewicht der Formulierung, verwendet.
Es liegt in der Natur von pharmazeutischen Hilfsstoffen, dass diese teilweise mehrere Funktionen in einer pharmazeutischen Formulierung wahrnehmen. Im Rahmen dieser Erfindung gilt zur unzweideutigen Abgrenzung daher bevorzugt die Fiktion, dass ein Stoff, der als ein bestimmter Hilfsstoff verwendet wird, nicht zeitgleich auch als weiterer pharmazeutischer Hilfsstoff eingesetzt wird.
Bei der erfindungsgemäßen Tablette, welche die Bestandteile (a), (b), (c) und gegebenenfalls (d) enthält, handelt es sich bevorzugt um eine Tablette mit modifizierter Freisetzung. Unter dem Begriff modifizierte Freisetzung wird im Rahmen dieser Erfindung eine verzögerte Freisetzung [delayed release), eine hinhaltende Freisetzung [prolonged release), eine gleichmäßig hinhaltende Freisetzung {sustalned release) oder eine lang ausgedehnte Freisetzung (extended release) verstanden. Bevorzugt handelt es sich um eine Kinetik, die der sustalned release folgt.
Die erfindungsgemäße Tablette kann befilmt werden. In einer bevorzugten Ausführungsform bilden daher die Bestandteile (a), (b), (c), gegebenenfalls (d) und gegebenenfalls die weiteren, vorstehend beschriebenen Hilfsstoffe einen Tablettenkern, wobei der Tablettenkern bevorzugt mit einem Überzug (= Komponente (e)) beschichtet ist.
Im Allgemeinen sind im Rahmen dieser Erfindung drei unterschiedliche Beschichtungen (e) möglich:
(e 1 ) Beschichtungen ohne Einfluss auf die Wirkstofffreisetzung; (e2) magensaftresistente Beschichtungen; und (e3) Retardbeschichtungen.
Filme ohne Einfluss auf die Wirkstofffreisetzung sind üblicherweise wasserlöslich (bevorzugt weisen sie eine Wasserlöslichkeit von mehr als 250 mg/ml auf). Magensaftresistente Filme besitzen eine pH-abhängige Löslichkeit. Retardfilme sind üblicherweise nicht-wasserlöslich (bevorzugt weisen sie eine Wasserlöslichkeit von weniger als 10 mg /ml auf).
Für die Befϊlmung (e) werden üblicherweise makromolekulare Stoffe verwendet, beispielsweise modifizierte Cellulosen, Polymethacrylate, Polyvinylpyrrolidon, Polyvinylacetatphthalat, Zein und /oder Schellack oder natürliche Gummi, wie z.B. Carrageenan.
Bevorzugte Beispiele für Filmbildner, die keinen Einfluss auf die Wirkstofffreisetzung haben (el) sind Methylcellulose (MC), Hydroxypropylmethylcelluslose (HPMC), Hydroxypropylcellulose (HPC), Hydroxyethylcellulose (HEC), Polvinylpyrrolidon (PVP) und Gemische daraus. Die genannten Polymere sollten üblicherweise ein gewichtsmittleres Molekulargewicht von 10.000 bis 150.000 g/mol aufweisen.
Bevorzugt verwendet wird HPMC, insbesondere HPMC mit einem gewichtsmittleren Molekulargewicht von 10.000 bis 150.000 g/mol und/oder einem durchschnittlichen Substitutionsgrad an -OCH3-Gruppen von 1,2 bis 2,0.
Beispiele für magensaftresistente Beschichtungen (e2) sind Celluloseacetatphthalat (CAP), Hydroxypropylmethylcellulosephthalat und Polyvinylacetatphathalat (PVAP).
Beispiele für Retardbeschichtungen (e3) sind Ethylcellulose (EC, kommerziell z.B. als Surelease® erhältlich) und Polymethacrylate (kommerziell z.B. als Eudragit® RL oder RS und L/S erhältlich).
Die Beschichtung (e) kann frei von Wirkstoff sein. Es ist jedoch auch möglich, dass die Beschichtung (e) Wirkstoff (a) enthält.
In einer bevorzugten Ausführungsform enthält der Überzug (e) Retigabin in einer Menge von 1 bis 45 Gew.-%, mehr bevorzugt von 5 bis 35 Gew.-%, insbesondere von 10 bis 30 Gew.-%, bezogen auf das Gesamtgewicht des in der Tablette enthaltenen Retigabins. In diesem Fall handelt es sich bevorzugt um eine Beschichtung ohne Einfluss auf die Wirkstofffreisetzung (el ).
Die Schichtdicke des Überzugs (e) beträgt im Falle einer Beschichtung ohne Wirkstoff bevorzugt 2 bis 100 μm, insbesondere 20 bis 60 μm. Die Schichtdicke des Überzugs (e) beträgt im Falle einer Beschichtung mit Wirkstoff bevorzugt 10 μm bis 2 mm, insbesondere 50 bis 500 μm.
Im Rahmen dieser Erfindung ist somit eine Ausführungsform bevorzugt, in der 1 bis 45 Gew.-%, mehr bevorzugt von 5 bis 35 Gew.-%, insbesondere von 10 bis 30 Gew.-% der Wirkstoffmenge als Initialdosis mit sofortiger Freisetzung vorliegen und 55 bis 99 Gew.-%, mehr bevorzugt 65 bis 95 Gew.-%, insbesondere 70 bis 90 Gew.-% der Wirkstoffmenge als Matrixformulierung mit verzögerter Freisetzung vorliegen.
Die erfindungsgemäße pharmazeutische Formulierung wird bevorzugt zu Tabletten verpresst. Im Stand der Technik wird hierfür eine Feuchtgranulierung vorgeschlagen (siehe WO 02/080898). Grundsätzlich ist die Feuchtgranulierung auch zur Herstellung der erfindungsgemäßen Tabletten geeignet.
Es hat sich jedoch gezeigt, dass die Eigenschaften der resultierenden Tabletten verbessert werden können, wenn die Feuchtgranulierung vermieden wird.
Die erfindungsgemäßen Intermediate werden daher mittels Direktverpressung zu Tabletten verpresst oder vor dem Verpressen zur Tablette einer Trockengranulierung unterworfen.
Ein weiterer Aspekt der vorliegenden Erfindung betrifft daher ein Trockenverarbeitungs-Verfahren umfassend die Schritte:
(I) Bereitstellen der Komponenten (a), (b), (c) und gegebenenfalls (d);
(II) gegebenenfalls Kompaktierung zu einer Schülpe;
(III) gegebenenfalls Granulierung der Schülpe;
(IV) Kompression zu Tabletten; und
(V) gegebenenfalls Befilmung der Tabletten.
Im Schritt (I) werden das erfindungsgemäße Intermediat und Hilfsstoffe bevorzugt vermischt. Die Vermischung kann in üblichen Mischern erfolgen. Alternativ ist es möglich, dass das Retigabin-Intermediat zunächst nur mit einem Teil der Hilfsstoffe (z.B. 50 % bis 95 %) vor der Kompaktierung (II) vermischt wird, und dass der verbleibende Teil der Hilfsstoffe nach dem Granulierschritt (III) zugegeben wird. Im Falle der Mehrfachkompaktierung sollte das Zumischen der Hilfsstoffe bevorzugt vor dem ersten Kompaktierschritt, zwischen mehreren Kompaktierschritten oder nach dem letzten Granulierschritt erfolgen.
Im Schritt (II) des erfindungsgemäßen Verfahrens wird das Gemisch aus Schritt (I) zu einer Schülpe kompaktiert. Hierbei ist bevorzugt, dass es sich um eine Trockenkompaktierung handelt, d.h. die Kompaktierung erfolgt bevorzugt in Abwesenheit von Lösungsmitteln, insbesondere in Abwesenheit von organischen Lösungsmitteln.
Die Kompaktierungsbedingungen werden üblicherweise so gewählt, dass das erfindungsgemäße Intermediat in Form eines Kompaktats (Schülpe) vorliegt, wobei die Dichte des Intermediats 0,8 bis 1 ,3 g/cm3, bevorzugt 0,9 bis 1,20 g/cm3, insbesondere 1 ,01 bis 1 ,15 g/cm3, beträgt.
Der Ausdruck "Dichte" bezieht sich hierbei bevorzugt auf die "Reindichte" (d.h. nicht auf die Schüttdichte oder Stampfdichte). Die Reindichte kann mit einem Gaspyknometer bestimmt werden. Vorzugsweise handelt es sich bei dem Gaspyknometer um ein Helium-Pyknometer, insbesondere wird das Gerät AccuPyc 1340 Helium Pyknometer des Herstellers Micromeritics, Deutschland, verwendet.
Die Kompaktierung wird bevorzugt in einem Walzengranulator durchgeführt.
Die Walzkraft beträgt üblicherweise 5 bis 70 kN/cm, bevorzugt 10 bis 60 kN/cm, mehr bevorzugt 15 bis 50 kN/cm.
Die Spaltbreite des Walzgranulators beträgt beispielsweise 0,8 bis 5 mm, bevorzugt 1 bis 4 mm, mehr bevorzugt 1 ,5 bis 3 mm, insbesondere 1 ,8 bis 2,8 mm.
In Schritt (III) des Verfahrens wird die Schülpe granuliert. Die Granulierung kann mit im Stand der Technik bekannten Verfahren erfolgen.
In einer bevorzugten Ausführungsform werden die Granulierungsbedingungen so gewählt, dass die resultierenden Teilchen (Granulate) eine volumenmittlere Teilchengröße ((D50)- Wert) von 50 bis 800 μm aufweisen, mehr bevorzugt von 100 bis 750 μm, noch mehr bevorzugt 150 bis 500 μm, insbesondere von 200 bis 450 μm.
In einer bevorzugten Ausführungsform erfolgt die Granulierung in einer Siebmühle. In diesem Fall beträgt die Maschenweite des Siebeinsatzes üblicherweise 0,1 bis 5 mm, bevorzugt 0,5 bis 3 mm, mehr bevorzugt 0,75 bis 2 mm, insbesondere 0,8 bis 1 ,8 mm.
Die aus Schritt (III) resultierenden Granulate können zu pharmazeutischen Darreichungsformen verarbeitet werden. Hierzu wird das Granulat beispielsweise in Sachets oder Kapseln gefüllt. Bevorzugt wird das aus Schritt (III) resultierende Granulat zu Tabletten verpresst (= Schritt IV).
In Schritt (IV) des Verfahrens werden die in Schritt (III) erhaltenen Granulate zu Tabletten verpresst, d.h. es erfolgt eine Kompression zu Tabletten. Die Kompression kann mit im Stand der Technik bekannten Tablettiermaschinen erfolgen.
In Schritt (IV) des Verfahrens können optional den Granulaten aus Schritt (III) pharmazeutische Hilfsstoffe zugegeben werden.
Die Mengen an Hilfsstoffen, die in Schritt (IV) zugesetzt werden, hängen üblicherweise von der Art der herzustellenden Tablette ab und von der Menge an Hilfsstoffen, die bereits in den Schritten (I) oder (II) zugesetzt wurde.
Im Falle der Direktkompression werden lediglich die Schritte (I) und (IV) des vorstehend beschriebenen Verfahrens durchgeführt.
Im optionalen Schritt (V) des erfindungsgemäßen Verfahrens werden die Tabletten aus Schritt (IV) beschichtet. Hierbei können die im Stand der Technik üblichen Verfahren zur Beschichtung, insbesondere zur Befilmung von Tabletten, Anwendung finden. Bezüglich der verwendeten Beschichtungsmaterialien wird auf vorstehende Ausführungen verwiesen.
Im Übrigen sind die vorstehend gemachten Erläuterungen zu bevorzugten Ausführungsformen der erfindungsgemäßen Tablette auch auf das erfindungsgemäße Verfahren übertragbar.
Die Tablettierbedingungen werden in beiden Ausführungsformen des erfindungsgemäßen Verfahrens ferner bevorzugt so gewählt, dass die resultierenden Tabletten ein Verhältnis von Tablettenhöhe zu Gewicht von 0,005 bis 0,3 mm/mg, besonders bevorzugt 0,05 bis 0,2 mm/mg, aufweisen.
Das erfindungsgemäße Verfahren wird bevorzugt so ausgeführt, dass die erfindungsgemäße Tabletten Retigabin in einer Menge von mehr als 200 mg bis 1000 mg, mehr bevorzugt von 250 mg bis 900 mg, insbesondere 300 mg bis 600 mg, enthält. Gegenstand der Erfindung sind somit Tabletten, enthaltend 300 mg, 400 mg, 450 mg, 600 mg oder 900 mg Retigabin.
Ferner weisen die resultierenden Tabletten bevorzugt eine Härte von 50 bis 300 N, besonders bevorzugt von 80 bis 250 N, insbesondere 100 bis 220 N auf. Die Härte wird gemäß Ph.Eur. 6.0, Abschnitt 2.9.8, bestimmt.
Zudem zeigen die resultierenden Tabletten bevorzugt eine Friabilität von kleiner 3 %, besonders bevorzugt von kleiner 2 %, insbesondere kleiner 1 %, auf. Die Friabilität wird gemäß Ph.Eur. 6.0, Abschnitt 2.9.7, bestimmt.
Schließlich weisen die erfindungsgemäßen Tabletten üblicherweise eine Gleichförmigkeit des Gehalts (Content Uniformity) von 95 bis 105 %, bevorzugt von 98 bis 102 %, insbesondere von 99 bis 101 % vom durchschnittlichen Gehalt auf. (Das heißt, sämtliche Tabletten haben einen Wirkstoffgehalt zwischen 95 und 105 % bevorzugt zwischen 98 und 102 %, insbesondere zwischen 99 und 101 % des durchschnittlichen Wirkstoffgehalts.) Die "Content Uniformity" wird gemäß Ph. Eur.6.0, Abschnitt 2.9.6, bestimmt.
Die vorstehenden Angaben zu Härte, Friabilität und Content Untformtty und Freisetzungsprofil beziehen sich hierbei bevorzugt auf die unbefilmte Tablette.
Das Freisetzungsprofil der erfindungsgemäßen Tabletten weist im Falle einer wirkstofffreien Beschichtung (e) gemäß USP-Methode (Paddle) eine gleichmäßige Freisetzung über die Zeit auf. Die Freisetzungskurve zeigt eine gleichmäßig hinhaltende Kinetik. Der Graph weist bevorzugt einen "langsamen" Anstieg auf, d.h. einen Anstieg von weniger als 0,6-0,8% pro Minute. Hierbei ist nach einer Stunde, (im Gegensatz zur schnellen Freisetzung) erst maximal 50% des Wirkstoffes freigesetzt.
Das Freisetzungsprofil der erfindungsgemäßen Tabletten weist im Falle einer wirkstoffhaltigen Beschichtung (e) gemäß USP-Methode (Paddle) eine Kinetik auf, die innerhalb von 15 Minuten eine Initialdosis des Wirkstoffs aufzeigt, d.h. nach 15 Minuten sind mindestens 15 % des Wirkstoffs freigesetzt. Nach den 15 Minuten wird der restliche Wirkstoff "langsam" aus der Formulierung herausdiffundieren, so dass man ab dieser Zeit eine Freisetzungskinetik erhält, die der gleichmäßig hinhaltenden folgt. Nach einer Stunde sind maximal 65 % des Wirkstoffs freigesetzt.
Die erfindungsgemäße Tablette ermöglicht es folglich durch Zusammenspiel von wasserlöslichem Hilfsstoff und nicht-wasserlöslichem Hilfsstoff eine vorteilhafte Formulierung für Retigabin bereit zu stellen, insbesondere eine mit modifizierter Freisetzung. Somit ist Gegenstand der Erfindung die Verwendung einer Kombination aus wasserlöslichem und nicht-wasserlöslichem Hilfsstoff zur Herstellung einer Retigabin-Tablette mit modifizierter Freisetzung. Bei der erfindungsgemäßen Verwendung beträgt der Wirkstoffgehalt der Tablette bevorzugt mehr als 50 Gew.-%. Im Übrigen sind die vorstehend gemachten Erläuterungen zu bevorzugten Ausführungsformen der erfindungsgemäßen Tablette auch auf die erfindungsgemäße Verwendung übertragbar.
Die Erfindung soll anhand der nachfolgenden Beispiele veranschaulicht werden.
BEISPIELE
In allen Beispielen wird Retigabin bevorzugt als Retigabin-Dihydrochlorid eingesetzt, wobei die angegebene Menge sich auf die Menge an Retigabin in Form der freien Base bezieht. Das heißt, die Angabe 300 g Retigabin entspricht etwa 372 g Retigabin - Dihydrochlorid.
Beispiel 1
300 g Retigabin wurde mit 200 g Ethylcellulose und 50 g Polyvinylpyrrolidon versetzt und 15 Minuten im Freifallmischer Turbula® WlOB gemischt. Die erhaltene Mischung wurde über ein Sieb der Größe 500 μm gegeben und mit 2 g Magnesiumstearat versetzt. Die erhaltene Mischung wurde auf einer Rundläuferpresse Fette 102i verpresst. Die Tabletten wurden mit einer Retigabindosierung von 300 mg/Tablette verpresst.
Beispiel 2
200 g Retigabin wurden mit 50 g Hydroxypropylcellulose und 50 g Ethylcellulose 15 Minuten gemischt. (Turbula WlOB). Die Mischung wurde gesiebt und danach mit 3 g Zinkstearat versetzt, gemischt und zu 400 mg Tabletten verpresst.
Beispiel 3
300 g Retigabin wurden mit 75 g PEG (Mw 8000) und mit 75 g Ethylcellulose gemischt. Die Herstellung erfolgt analog Beispiel 1 mit 4 g Magnesiumstearat/Aerosü-Gemisch (5: 1)
Die erhaltene Dosierung betrug 600 mg pro Dosisform.
Beispiel 4
Retigabin 900 g
Pluronic® 200 g
Celluloseacetylphthalat 150 g
AerosÜ® 10 g
Magnesiumstearat 6 g
Retigabin und Pluronic® wurden gemischt und gesiebt.
Das Aerosil und das Magnesiumstearat wurden zugegeben, erneut gemischt und verpresst auf einer Exzenterpresse (Korsch® EKO). Die erhaltene Dosierung betrug 900 mg pro Dosisform.
Beispiel 5a
Retigabin 400 g
Celluloseacetylphthalat 100 g
Sorbitol 200 g
Aerosü® 10 g
Magneslumstearat 3 g
3A des Retlgabins wurde zusammen mit der Hälfte einer Mischung aus Celluloseacetylphthalat und Sorbitol (1 :2) 10 Minuten gemischt und gesiebt. 1A des
Retigabins wurde zusammen mit der anderen Hälfte der Celluloseacetylphthalat
/Sorbitol Mischung 10 Minuten gemischt und Aerosü und Magnesiumstearat wurden zugegeben und für weitere 3 Minuten gemischt. Die beiden hergestellten Mischungen wurden zusammengegeben und 10 Minuten homogenisiert, um dann eine Verpressung durchführen zu können. Die erhaltene Dosierung betrug 400 mg pro Dosisform.
Beispiel 5b
Retigabin 400 g
Celluloseacetylphthalat 150 g
Sorbitol 150 g
Aerosü® 10 g
Magnesiumstearat 3 g
3A des Retigabins wurde zusammen mit der Hälfte einer Mischung aus Celluloseacetylphthalat und Sorbitol (1 :2) 10 Minuten gemischt und gesiebt. 1A des Retigabins wurde zusammen mit der anderen Hälfte der Celluloseacetylphthalat /Sorbitol Mischung 10 Minuten gemischt und Aerosü und Magnesiumstearat wurden zugegeben und für weitere 3 Minuten gemischt. Die beiden hergestellten Mischungen wurden zusammengegeben und 10 Minuten homogenisiert, um dann eine Verpressung durchführen zu können. Die erhaltene Dosierung betrug 400 mg pro Dosisform.
Beispiel 6: Initialdosis in der äußeren Umhüllung
Beispiel 2 wurde so modifiziert, dass nur 3A des Wirkstoffs in den Kern eingearbeitet wurde. 1A des Wirkstoffs wurde mit 30 g eines gängigen Hydroxypropylmethyl- celluloselacks (Opadry® AMB), 10%-ig in Wasser, 3 Minuten in Ultra Turrax® vermengt und auf die Tabletten in Form eines Lacks aufgesprüht.