WO2007141200A1 - Novel n-aryl and n-heteroaryl substituted pyridinone derivatives for use in mch-1 mediated diseases - Google Patents
Novel n-aryl and n-heteroaryl substituted pyridinone derivatives for use in mch-1 mediated diseases Download PDFInfo
- Publication number
- WO2007141200A1 WO2007141200A1 PCT/EP2007/055376 EP2007055376W WO2007141200A1 WO 2007141200 A1 WO2007141200 A1 WO 2007141200A1 EP 2007055376 W EP2007055376 W EP 2007055376W WO 2007141200 A1 WO2007141200 A1 WO 2007141200A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- compound according
- disorders
- compound
- Prior art date
Links
- XTPJCCKHBQLBST-UHFFFAOYSA-N CN(C)C(CC1)CN1c(nc1)ccc1Br Chemical compound CN(C)C(CC1)CN1c(nc1)ccc1Br XTPJCCKHBQLBST-UHFFFAOYSA-N 0.000 description 2
- ZHXUWDPHUQHFOV-UHFFFAOYSA-N Brc(cn1)ccc1Br Chemical compound Brc(cn1)ccc1Br ZHXUWDPHUQHFOV-UHFFFAOYSA-N 0.000 description 1
- PQQMWHUBXVKXBT-UHFFFAOYSA-N CN(C)C(CC1)CN1c(cc1)ccc1N(C=CC(O)=C1)C1=O Chemical compound CN(C)C(CC1)CN1c(cc1)ccc1N(C=CC(O)=C1)C1=O PQQMWHUBXVKXBT-UHFFFAOYSA-N 0.000 description 1
- ROARBGIHGNVCOO-UHFFFAOYSA-N CN(C)C(CC1)CN1c(cc1)ccc1N(C=CC(OCc1ccccc1)=C1)C1=O Chemical compound CN(C)C(CC1)CN1c(cc1)ccc1N(C=CC(OCc1ccccc1)=C1)C1=O ROARBGIHGNVCOO-UHFFFAOYSA-N 0.000 description 1
- PMYHVXRAVRWVHW-UHFFFAOYSA-N CN(C)C(CC1)CN1c(nc1)ccc1N(C=CC(OCc1ccccc1)=C1)C1=O Chemical compound CN(C)C(CC1)CN1c(nc1)ccc1N(C=CC(OCc1ccccc1)=C1)C1=O PMYHVXRAVRWVHW-UHFFFAOYSA-N 0.000 description 1
- AVAWMINJNRAQFS-UHFFFAOYSA-N CN(C)C1CNCC1 Chemical compound CN(C)C1CNCC1 AVAWMINJNRAQFS-UHFFFAOYSA-N 0.000 description 1
- DOVNUEPFPBWTSV-UHFFFAOYSA-N O=C1NC=CC(OCc2ccccc2)=C1 Chemical compound O=C1NC=CC(OCc2ccccc2)=C1 DOVNUEPFPBWTSV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/69—Two or more oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
Definitions
- the present invention concerns N-aryl and N-heteroaryl substituted pyridinone derivatives having antagonistic melanin-concentrating hormone (MCH) activity, in particular MCH-I activity. It further relates to their preparation, compositions comprising them and their use as a medicine.
- MCH melanin-concentrating hormone
- MCH Melanin concentrating hormone
- C ⁇ S central nervous system
- MCH-I and MCH-2 G protein-coupled receptors
- MCH-I receptor was considered a valuable target for the treatment of obesity as MCH promotes feeding behaviour in rodents (Nature (1996), 380, 243-247). Recently however, it was shown that MCH-I antagonism pro- prises anxiolytic and antidepressant profiles in rodents (Nat. Med. (2002) 8, 825-830; Neuropharmacology (2004), 46, 457-467 ; Neuropsychopharmacology (2005), in press). Thus, it is currently generally accepted that MCH receptors, particularly the MCH-I receptor, are a good target for the treatment of affective spectrum disorders (Eur. J. Neuroscience (2000) 12, 1194-1216).
- MCH-I receptor mRNA and protein are distributed in various hypothalamic nuclei including the paraventricular nucleus and several limbic structures all implicated in the regulation of emotion and stress (Eur. J. Neuroscience (2000) 12, 1194-1216). In addition, dense labelling is detected in the nucleus accumbens shell (J. Comp. Neurol. (2001) 435, 26-40). Injection of MCH directly into the paraventricular nucleus has been found to increase plasma adrenocorticotropic hormone (ACTH) and to alter sleep architecture (Verret et al. 2003, BMC Neurosci 4:19).
- ACTH plasma adrenocorticotropic hormone
- MCH corticotro- phin-releasing factor
- CRF corticotro- phin-releasing factor
- MCH-I antagonist SNAP-7941
- antidepressant- and axiolytic-like affects in rodents tests, supporting a role for MCH-I re- ceptor in depression and anxiety.
- MCH-I antagonists A large number of companies is now actively pursuing the development of MCH-I antagonists and a wide range of structural types have been reported in a num- ber of patent publications, mostly in relation to the regulation of food intake and energy expenditure (Expert Opin. Ther. Patents (2005) 15(10)). The majority of reported MCH-antagonists incorporate a basic centre and two (hetero)aromatic parts, joined by linkers.
- WO 2005/085200 (Banyu Pharmaceutical Co., Ltd) discloses pyridinone, pyrimidinone and pyridazinone-derivatives for use as MCH-I antagonists.
- WO 2003/033480, WO 2003/033476 and WO 2005/05042541 disclose different bicyclic heterocycles, such as thieno- pyrimid-4-one-, benzopyrimid-4-one- and ftalimide-derivatives, for use as MCH-I antagonists.
- WO 2003/097047 and WO 2005/040157 (Eli Lilly and Company) and WO 2005/070925 (Aventis Pharma GmbH) report different aromatic 5-membered ring heterocycles, such as oxazole- and oxadiazole-derivatives, for use as MCH-I antagonists.
- WO 2004/011438 and WO 2005/070898 (Aventis Pharma GmbH GMBH) disclose diaryl-substituted cyclic urea derivatives as MCH-I antagonist.
- WO 2003/068230 (Pharmacia Corporation) discloses the preparation of substituted pyridinones as modulators of p38 MAP kinase.
- MCH melanin-concentrating
- a pharmaceutically acceptable acid or base addition salt thereof an TV-oxide form thereof or a quaternary ammonium salt thereof, wherein A is phenyl or pyridinyl ;
- R 1 , R 2 are each, independently from each other, selected from the group of hydrogen, Ci- 3 alkyl, aryl,; arylC 1 . 3 alky 1, Het, HetC 1-3 alkyl,
- R 1 and R 2 are taken together to form an TV-containing heterocycle, selected from the group of pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, and thiomorpholinyl ; wherein each of the afore- mentioned TV-containing heterocycles are optionally substituted by k substitu- ents R 3 ;
- R 3 is selected from the group of hydrogen, halo, cyano, hydroxy, NR a R b , oxo, carboxyl, aminocarbonyl, nitro, thio, formyl, C 1-3 alkyl, aryl, and R a , R b are each, independently from each other, selected from the group of hydrogen, C 1-3 alkyl, aryl, Het, sulphonyl, and ; or R a and R b are taken together to form an TV-containing heterocycle, selected from the group of pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, l-aza-bicyclo[2.2.1]heptyl, and thiomorpholinyl ;
- R 4 , R 5 are each, independently from each other, selected from the group of hydrogen, halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl, C 1-3 alkyl, and k is an integer, equal to zero, 1, 2, 3 or 4 ; 1 is an integer, equal to zero, 1, 2, 3 or 4 ; m is an integer, equal to zero, 1 , 2 or 3 ;
- Y 1 , Y 3 are each, independently from each other, selected from the group of a single bond, O, NR 7 , S, SO, and SO 2 ; wherein R 7 is selected from the group of hydrogen and ; Y 2 is a saturated or unsaturated, straight or branched Ci- 6 -hydrocarbon radical, wherein one or more hydrogen atoms may optionally be replaced by a radical selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, and formyl ;
- B is a 6-membered ring containing zero, 1, 2 or 3 nitrogen atoms, optionally substituted with p substituents R 6 , each independently from each other, selected from the group of hydrogen, halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl, C 1-3 alkyl, and C 1-3 alkyloxy ; and wherein p is an integer, equal to zero, 1 or 2 ; or two substituents R 6 may be combined into a radical -CH 2 CH 2 CH 2 - or -OCH 2 O- ; alkyl is a straight or branched saturated hydrocarbon radical having the indicated number of carbon atoms ; wherein the radical may optionally be substituted on one or more carbon atoms with one or more radicals selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio and formyl ; aryl is naphthyl or pheny
- Het is a heterocyclic radical selected from the group of pyrrolidinyl, imida-zolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, pyrrolyl, pyrrolinyl, imidazolinyl, pyrrazolinyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, azepyl, diazepyl, morpholinyl, thiomorpholinyl, furyl, thienyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, isothiazolyl, dioxolyl, dithianyl, tetrahydrofuryl, tetrahydro- pyranyl, and oxadiazolyl ; where
- compounds according to the invention is meant a compound according to the general Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof or a quaternary ammonium salt thereof, including the above compounds disclosed in WO 2003/068230. Only for establishing the compound scope, it is understood that these compounds are disclaimed by way of a disclaimer.
- the invention also relates to a pharmaceutical composition comprising a pharmaceutically acceptable carrier or diluent and, as active ingredient, a therapeutically effective amount of a compound according to the invention, in particular a compound according to Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof, or a quaternary ammonium salt thereof.
- the invention also relates to the use of a compound according to the invention as a medicament and for the preparation of a medicament for the prevention and/or treatment of a disorder or disease responsive to antagonism of the MCH receptor, in particular to antagonism of the MCH-I receptor.
- the invention relates to the use of a compound according to the in- vention for the preparation of a medicament for the prevention and/or treatment of psychiatric disorders, including but not limited to anxiety, eating disorders, mood disorders, such as bipolar disorders and depression, psychoses, such as schizophrenia, and sleeping disorders. Additionally, the compound can be used for treating obesity, diabetes, sexual disorders and neurological disorders.
- a compound according to the invention, in particular according to Formula (I), may also be suitable as add-on treatment or combination treatment and/or prophylaxis in the above listed diseases, in particular for the prevention and/or treatment of psychiatric disorders, in combination with antidepressants, anxiolytics and/or antipsychotics which are currently available or in development or which will become available in the future, in particular to improve efficacy and/or onset of action.
- antidepressants anxiolytics and/or antipsychotics
- antipsychotics which are currently available or in development or which will become available in the future, in particular to improve efficacy and/or onset of action.
- antidepressants anxiolytics and/or antipsychotics
- compounds are evaluated in combination with antidepressants, anxiolytics and/or antipsychotics for attenuation of stress-induced hyperthermia.
- the invention therefore also relates to the use of the compounds according to the invention in combination with one or more other compounds selected from the group of antidepressants, anxiolytics and antipsychotics, to a pharmaceutical composition comprising the compounds according to the invention and one or more other compounds selected from the group of antidepressants, anxiolytics and antipsychotics, as well as to a process for the preparation of such pharmaceutical compositions.
- the invention also relates to the use of the compounds according to the invention in combination with one or more other compounds selected from the group of lipid- lowering compounds for the prevention and/or treatment of obesity, to a pharmaceutical composition comprising the compounds according to the invention and one or more other compounds selected from the group of lipid-lowering compounds, as well as to a process for the preparation of such pharmaceutical compositions.
- the invention relates to a compound according to the invention, wherein each of R 1 , R 2 , independently from each other, is selected from the group of hydrogen, C 1-3 alkyl, arylC 1 . 3 alky 1, HetC 1-3 alkyl, and NR a R b Ci- 3 alkyl.
- the is substituted with hydroxy or amino .
- the invention relates to a compound according to the invention, wherein R 1 and R 2 are taken together to form an TV-containing heterocycle, selected from the group of pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, and thio- morpholinyl ; and wherein each of the foregoing TV-containing heterocycles is option- ally substituted by k substituents R 3 .
- the invention relates to a compound according to the invention, wherein R 3 is selected from the group of hydroxy, NR a R b , aminocarbonyl, C 1-3 alkyl, aryl, and
- the invention relates to a compound according to the invention, wherein R a , R b are each, independently from each other, selected from the group of hydrogen, C 1-3 alkyl, C 1-3 alkylcarbonyl. C 1-3 alkyloxycarbonyl, and C 1-3 alkyl- sulphonyl; or R a and R b are taken together to form an N-containing heterocycle, selected from the group of pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, l-aza-bicyclo[2.2.1]heptyl, and thiomorpholinyl.
- the invention relates to a compound according to the invention, wherein A is phenyl.
- the invention relates to a compound according to the invention, wherein each of R 4 and R 5 , independently from each other, are selected from the group of hydrogen, halo, C 1-3 alkyl, and
- the invention relates to a compound according to the in- vention, wherein k is 1.
- the invention relates to a compound according to the invention, wherein 1 is zero.
- the invention relates to a compound according to the invention, wherein m is zero. In another embodiment, the invention relates to a compound according to the invention, wherein Y 1 and Y 3 are each, independently from each other, selected from the group of a single bond and O.
- the invention relates to a compound according to the invention, wherein Y 2 is selected from the group of saturated or unsaturated, straight or branched C 1-6 hydrocarbon radicals.
- the invention relates to a compound according to the invention, wherein B is selected from the group of phenyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl and triazinyl.
- B is phenyl, pyridinyl or pyridazinyl. More preferably, B is phenyl.
- the invention relates to a compound according to the invention, wherein the moiety B-Y'-Y ⁇ Y 3 is selected from the radicals (a-1) to (d-5) as listed below, optionally substituted with p substituents R 6 , each independently from each other, selected from the group of hydrogen, fluoro, chloro, hydroxy, methyl, trifluoromethyl, fiuoromethyloxy, difiuoromethyloxy, and methyloxy ; and wherein p is an integer, equal to zero, 1 or 2 ; or two substituents R 6 are combined into a radical -CH 2 CH 2 CH 2 - or -OCH 2 O-.
- the moiety B-Y'-Y 2 ⁇ 3 is selected from the radicals (a-1), (a-2), (a-3) and (b-3).
- alkyl is a straight or branched saturated hydrocarbon radical having the indicated number of carbon atoms, i.e. when is indicated, the alkyl radical may contain from 1 to 3 carbon atom ; and when is indicated, the alkyl radical may contain from 1 to 6 carbon atom.
- Each radical may optionally be substituted on one or more carbon atoms with one or more radicals selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio and formyl.
- alkyl is methyl, ethyl, propyl or isopropyl. Further radicals that are embraced within the scope are e.g.
- aryl is naphthyl or phenyl, each optionally substituted with 1, 2 or 3 substituents, each independently from each other, selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl and
- substituents each independently from each other, selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl
- halo is a substituent selected from the group of fluoro, chloro, bromo and iodo.
- halo is fluoro, chloro or bromo.
- a bond can be any bond, including a covalent bond, a single bond, a double bond, a triple bond, a coordination bond and a hydrogen bond.
- a pharmaceutically acceptable acid addition salt is defined to comprise a therapeutically active non-toxic acid addition salt form that a compound according to Formula (I) is able to form.
- Said salt can be obtained by treating the base form of a compound according to Formula (I) with an appropriate acid, for example an inorganic acid, for example hydrohalic acid, in particular hydrochloric acid, hydrobromic acid, sulphuric acid, nitric acid and phosphoric acid ; an organic acid, for example acetic acid, hydroxyacetic acid, propanoic acid, lactic acid, pyruvic acid, oxalic acid, malonic acid, succinic acid, maleic acid, fumaric acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, cyclamic acid, salicylic acid, p-amino salicylic acid and pamoic acid
- the compound according to Formula (I) containing an acidic proton may also be converted into a therapeutically active non-toxic metal or amine addition salt form
- Ap- intestinal base salt forms comprise, for example, the ammonium salts, the alkaline and earth alkaline metal salts, in particular lithium, sodium, potassium, magnesium and cal- cium salts, salts with organic bases, e.g. the benzathine, TV-methyl-D-glucamine, hy- bramine salts, and salts with amino acids, for example arginine and lysine.
- salt form can be converted into the free form by treatment with an appropriate acid.
- addition salt as used in the framework of this application also comprises a solvate that the compound according to Formula (I), as well as a salt thereof, is able to form.
- solvates are, for example, hydrates and alcoholates.
- the TV-oxide form of the compound according to Formula (I) is meant to comprise a compound of Formula (I) wherein one or several nitrogen atoms are oxidized to so-called TV-oxides, particularly those TV-oxides wherein one or more tertiary nitrogens (e.g. of the piperazinyl or piperidinyl radical) are TV-oxidized.
- TV-oxides can easily be obtained by a skilled person without any inventive skills and they are obvious alternatives for a compound according to Formula (I) since these compounds are metabolites, which are formed by oxidation in the human body upon uptake .
- oxidation is normally the first step involved in drug metabolism (Textbook of Organic Medicinal and Pharmaceutical Chemistry, 1977, pages 70- 75).
- the metabolite form of a compound can also be administered to a human instead of the compound per se, with much the same effects.
- a compound of Formula (I) may be converted to the corresponding TV-oxide form following art-known procedures for converting a trivalent nitrogen into its TV-oxide form.
- Said TV-oxidation reaction may generally be carried out by reacting the compound of Formula (I) with an appropriate organic or inorganic peroxide.
- Appropriate inorganic peroxides comprise, for example, hydrogen peroxide, alkali metal or earth alkaline metal peroxides, e.g. sodium peroxide, potassium peroxide; appropriate or- ganic peroxides may comprise peroxy acids such as, for example, benzenecarboper- oxoic acid or halo substituted benzenecarboperoxoic acid, e.g.
- 3-chlorobenzenecarbo- peroxoic acid peroxoalkanoic acids, e.g. peroxoacetic acid, alkylhydroperoxides, e.g. tert-butyl hydroperoxide.
- Suitable solvents are, for example, water, lower alkanols, e.g. ethanol and the like, hydrocarbons, e.g. toluene, ketones, e.g. 2-butanone, halogenated hydrocarbons, e.g. dichloromethane, and mixtures of such solvents.
- a quaternary ammonium salt of compound according to Formula (I) defines said compound which is able to form by a reaction between a basic nitrogen of a com- pound according to Formula (I) and an appropriate quaternizing agent, such as, for example, an optionally substituted alkylhalide, arylhalide or arylalkylhalide, in particular methyliodide and benzyliodide.
- an appropriate quaternizing agent such as, for example, an optionally substituted alkylhalide, arylhalide or arylalkylhalide, in particular methyliodide and benzyliodide.
- Other reactants with good leaving groups may also be used, such as, for example, alkyl trifluoromethanesulfonates, alkyl methanesulfonates and alkyl p-toluenesulfonates.
- a quaternary ammonium salt has at least one positively charged nitrogen.
- the invention also comprises a derivative compound (usually called "pro-drug") of a pharmacologically-active compound according to the invention, in particular ac- cording to Formula (I), which is degraded in vivo to yield a compound according to the invention.
- Pro-drugs are usually (but not always) of lower potency at the target receptor than the compounds to which they are degraded.
- Pro-drugs are particularly useful when the desired compound has chemical or physical properties that make its administration difficult or inefficient. For example, the desired compound may be only poorly soluble, it may be poorly transported across the mucosal epithelium, or it may have an undesirably short plasma half-life. Further discussion on pro-drugs may be found in Stella, V. J. et al, "Prodrugs", Drug Delivery Systems, 1985, pp. 112-176, and Drugs, 1985, 29, pp. 455-473.
- a pro-drug form of a pharmacologically-active compound according to the inven- tion will generally be a compound according to Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof, or a quaternary ammonium salt thereof, having an acid group which is esterified or amidated. Included in such esterified acid groups are groups of the formula -COOR X , where R x is a C 1-6 alkyl, phenyl, benzyl or one of the following groups :
- Amidated groups include groups of the formula - CONR y R z , wherein R y is H,
- a compound according to the invention having an amino group may be derivatised with a ketone or an aldehyde such as formaldehyde to form a Mannich base. This base will hydrolyze with first order kinetics in aqueous solution.
- stereochemically isomeric form defines all the possible stereochemically isomeric forms that a compound of Formula (I) may possess.
- chemical designation of a compound denotes the mixture of all possible stereochemically isomeric forms, said mixtures containing all di- astereomers and enantiomers of the basic molecular structure. More in particular, stereogenic centers may have the R- or S-configuration; substituents on bivalent cyclic (partially) saturated radicals may have either the cis- or trans-configuration.
- the configuration of the second stereogenic center is indicated using relative descriptors [R*,R* J or [R*,S*J, where R* is always specified as the reference center and [R*,R*] indicates centers with the same chirality and [i?*,5 * *] indicates centers of unlike chirality. For example, if the lowest-numbered chiral center in the molecule has an S configuration and the second center is R, the stereo descriptor would be specified as S-[R*, S*].
- the position of the highest priority sub- stituent on the asymmetric carbon atom in the ring system having the lowest ring num- ber is arbitrarily always in the " ⁇ " position of the mean plane determined by the ring system.
- the position of the highest priority substituent on the other asymmetric carbon atom in the ring system (hydrogen atom in a compound according to Formula (I)) relative to the position of the highest priority substituent on the reference atom is denominated " ⁇ ", if it is on the same side of the mean plane determined by the ring system, or " ⁇ ", if it is on the other side of the mean plane determined by the ring system.
- a compound according to the invention is inherently intended to comprise all isotopic combinations of its chemical elements.
- a chemical element in particular when mentioned in relation to a compound according to Formula (I), comprises all isotopes and isotopic mixtures of this element, either naturally occuring or synthetically produced, either with natural abundance or in an isotopically enriched form.
- hydro- gen when hydro- gen is mentioned, it is understood to refer to 1 H, 2 H, 3 H and mixtures thereof ; when carbon is mentioned, it is understood to refer to 11 C, 12 C, 13 C, 14 C and mixtures thereof ; when nitrogen is mentioned, it is understood to refer to 13 N, 14 N, 15 N and mixtures thereof ; when oxygen is mentioned, it is understood to refer to 14 O, 15 O, 16 O, 17 O, 18 O and mixtures thereof ; and when fluor is mentioned, it is understood to refer to 18 F, 19 F and mixtures thereof.
- a compound according to the invention therefore inherently comprises a compound with one or more isotopes of one or more element, and mixtures thereof, including a radioactive compound, also called radio labelled compound, wherein one or more non-radioactive atoms has been replaced by one of its radioactive isotopes.
- radio labelled compound any compound according to Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof, or a quaternary ammonium salt thereof, which contains at least one radioactive atom.
- a compound can be labelled with positron or with gamma emitting radioactive isotopes.
- the 3 H-atom or the 125 I-atom is the atom of choice to be replaced.
- PET positron emitting
- the most commonly used positron emitting (PET) radioactive isotopes are 11 C, 18 F, 15 O and 13 N, all of which are accelerator produced and have half-lives of 20, 100, 2 and 10 minutes respectively. Since the half-lives of these radioactive isotopes are so short, it is only feasible to use them at institutions which have an accelerator on site for their production, thus limiting their use.
- the most widely used of these are 18 F, 99m Tc, 201 Tl and 123 I.
- the handling of these radioactive isotopes, their production, isolation and incorporation in a molecule are known to the skilled person.
- the radioactive atom is selected from the group of hydrogen, carbon, nitrogen, sulfur, oxygen and halogen.
- the radioactive atom is selected from the group of hydrogen, carbon and halogen.
- the radioactive isotope is selected from the group of 3 H, 11 C, 18 F, 122 I, 123 I, 125 I, 131 I, 75 Br, 76 Br, 77 Br and 82 Br.
- the radioactive isotope is se- lected from the group of ⁇ , 11/ C and 1 1 8T ⁇ .
- a compound according to the invention can generally be prepared by a successive- sion of steps, each of which is known to the skilled person.
- a pyridinone derivative can be prepared according to one or more of the following preparation methods.
- the copper coupling reaction is performed in the presence of a copper salt, such as Cu(OAc) 2 , in an aprotic solvent such as 1 ,2-dichloromethane, in the presence of an amine and amine TV-oxide, such as pyridine and TEMPO, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically overnight at room temperature.
- a copper salt such as Cu(OAc) 2
- an aprotic solvent such as 1 ,2-dichloromethane
- an amine and amine TV-oxide such as pyridine and TEMPO
- the pal- ladium coupling reaction is performed in an aprotic solvent such as toluene, in the presence of a palladium catalyst such as Pd(AcO) 2 , and in the presence of tBuOK, as a base, and a ligand, such as BINAP, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 24 hours at 100 0 C under traditional heating.
- aprotic solvent such as toluene
- a palladium catalyst such as Pd(AcO) 2
- tBuOK as a base
- a ligand such as BINAP
- the copper coupling reaction is performed in the presence of a copper salt, such as CuI, in an aprotic solvent such as 1,4-dioxane, in the presence of an amine, such as ⁇ /, ⁇ -dimethylethylenediamine, in the presence of a base, such as potassium phospate, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 15 minutes at 180 0 C under microwave irradiation.
- a copper salt such as CuI
- an aprotic solvent such as 1,4-dioxane
- an amine such as ⁇ /, ⁇ -dimethylethylenediamine
- a base such as potassium phospate
- the OH-activation can be done via trifiate, in the presence of a sulphonilating agent, such as N-phenyltrifluoromethanesulfonimide, in an aprotic solvent such as THF, in the presence of a base, such as potassium carbonate, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 15 minutes at 120 0 C under microwave irradiation.
- a sulphonilating agent such as N-phenyltrifluoromethanesulfonimide
- THF aprotic solvent
- a base such as potassium carbonate
- the palladium coupling reaction is performed in an aprotic solvent such as toluene or dioxane in the presence of a palladium catalyst such as Pd(AcO) 2 , or Pd(PPh 3 ⁇ and in the presence of a base, such as tBuOK or cessium carbonate, or inorganic aqueous base such as Na 2 CO 3 , and sometimes a ligand, such as BINAP or XANTPHOS, is required, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 24 hours at 100 0 C under traditional heating.
- a palladium catalyst such as Pd(AcO) 2 , or Pd(PPh 3 ⁇
- a base such as tBuOK or cessium carbonate
- inorganic aqueous base such as Na 2 CO 3
- a ligand such as BINAP or XANTPHOS
- Hydrogenation is performed under hydrogen pressure, in a protic or aprotic solvent, such as EtOH, or EtOAc, in the presence of palladium as a catalyst.
- Alkylation is performed in the presence of an organic or inorganic base, such as NaH, or DBU, in an aprotic solvent such as CH 3 CN, or DMF at a convenient temperature to completion of the reaction, typically 130 0 C for 30 minutes under microwave irradiation.
- a Mitsun- obu type reaction can be performed in the presence of a phosphine, such as triphenyl- phosphine, an azodicarboxylate derivative, such as diethylazodicarboxilate in an aprotic solvent, such as THF, at a convenient temperature and time to completion of the reaction, such as 100 0 C for 5 minutes under microwave irradiation.
- a phosphine such as triphenyl- phosphine
- an azodicarboxylate derivative such as diethylazodicarboxilate
- an aprotic solvent such as THF
- Halogenation is per- formed with a suitable halogenating agent, such as POBr 3 , in a suitable solvent, such as DMF, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically overnight at 100 0 C under traditional heating.
- the palladium coupling reaction of boronic acids is performed in an aprotic solvent such as dioxane in the presence of a palladium catalyst such as Pd(PPh 3 )4 and in the presence of an inorganic aqueous base such as Na 2 CO 3 , at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 10 minutes at 150 0 C under microwave heating.
- an aprotic solvent such as dioxane
- a palladium catalyst such as Pd(PPh 3 )4
- an inorganic aqueous base such as Na 2 CO 3
- the copper coupling reaction with boronic acids is performed in the presence of a copper salt, such as Cu(OAc) 2 , in an aprotic solvent such as 1 ,2-dichloromethane, in the presence of an amine and amine TV-oxide, such as pyridine and TEMPO (2,2,6,6-tetramethyl-l-piperidinyloxy), at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically room temperature overnight.
- the reduction in performed with an appropriate reducting agent, such as NaBH 4 , in the presence of a suitable solvent such as MeOH at room temperature for a period of time to ensure the completion of the reaction.
- the palladium coupling with amines is performed in an aprotic solvent such as toluene or dioxane in the presence of a palladium catalyst such as Pd(AcO) 2 , or Pd(PPh 3 ) 4 and in the presence of a base, such as tBuOK or cessium carbonate, or inorganic aqueous base such as Na 2 CO 3 , and sometimes a ligand, such as BINAP or XANTPHOS, is required, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 24 hours at 100 0 C under traditional heating.
- amine substitution is preferably performed using an aprotic solvent, such as DMF, in the presence of the nucleophilic amine, and a base such as DIPEA or DBU, heating at a convenient temperature under traditional heating or microwave irradiation, for a period of time to ensure the completion of the reaction.
- an aprotic solvent such as DMF
- the copper coupling reaction is performed using an aprotic solvent, such as dioxane or DMF, in the presence of CuI, an inorganic base such K3PO4 and MeNH(CH ⁇ ) 2 NHMe as ligand, heating at a convenient temperature under traditional heating or microwave irradiation, for a period of time to ensure the completion of the reaction, typically 25 minutes at 175 0 C under microwave irradiation.
- an aprotic solvent such as dioxane or DMF
- a compound according to the invention in particular compounds according to
- TV-oxide form thereof or a quaternary ammonium salt thereof have surprisingly been shown to have a binding affinity towards the MCH-receptor, in particular towards the MCH-I receptor, in particular as an antagonist.
- a compound according to the invention are suitable for the prevention and/or treatment of diseases where antagonism of the
- MCH-receptor in particular antagonism of the MCH-I receptor is of therapeutic use.
- a compound according to the invention may be suitable for treatment and/or prophylaxis of psychiatric disorders, including but not limited to : anxiety, including but not limited to agoraphobia ; generalized anxiety ; compul- sion ; obsessive-compulsive disorder ; panic disorder ; social phobia ; and stress, such a post-traumatic stress ; attention def ⁇ cit/hyperactivity disorder ; autism ; - dysthymia ; eating disorder, including but not limited to anorexia ; binge eating ; and bulimia nervosa ; - impulse control disorder ; mental retardation, including but not limited to fragile X syndrome ; - mood disorder, including but not limited to agitation ; bipolar disorder, such as bipolar affective disorder, bipolar disorder (I), bipolar disorder (II), hypomania and mania ; depression, such as major depression and suicidal depression ; seasonal mood disorder ; and suicide ; premenstrual syndrome, including but not
- a compound according to the invention may be used for treating sexual disorders, neurological disorders, and most in particular obesity and diabetes.
- the invention therefore relates to a compound according to the general Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof or a quaternary ammonium salt thereof, for use as a medicine.
- the invention also relates to the use of a compound according to the invention for the preparation of a medicament for the prevention and/or treatment of diseases where antagonism of the MCH-receptor, in particular antagonism of the MCH-I receptor is of therapeutic use.
- the invention also relates to the use of a compound according to the invention for the preparation of a medicament for the prevention and/or treatment of anxiety, eating disorders, mood disorders, such as bipolar disorders and depression, psychoses, such as schizophrenia, and sleeping disorders. Additionally, a compound according to the invention may be used for treating sexual disorders and neurological disorders, and in particular obesity and diabetes.
- the compounds according to the invention, in particular according to Formula (I) may be co-administered as add-on treatment and/or prophylaxis in the above listed diseases.
- antidepressants anxiolytics and/or antipsychotics.
- the compounds according to the invention in particular according to Formula (I) may be co-administered in combination with antidepressants, anxiolytics and/or antipsychotics which are currently available or in development or which will become available in the future, in particular to improve efficacy and/or onset of action.
- antidepressants anxiolytics and/or antipsychotics which are currently available or in development or which will become available in the future, in particular to improve efficacy and/or onset of action.
- the compounds of the present invention and the other agents may be present as a combined preparation for simultaneous, separate or sequential use for the prevention and/or treatment of depression and/or anxiety. Such combined preparations may be, for example, in the form of a twin pack.
- the compounds of the present invention and the other agents may be administered as separate pharmaceutical compositions, either simultaneously or sequentially.
- the invention therefore relates to a pharmaceutical composition according to the invention, characterized in that is comprises further one or more other compounds se- lected from the group of antidepressants, anxiolytics and antipsychotics.
- Suitable classes of antidepressant agents include norepinephrine reuptake inhibitors, selective serotonin reuptake inhibitors (SSRTs), monoamine oxidase inhibitors (MAOI's), reversible inhibitors of monoamine oxidase (RIMA's), serotonin and noradrenaline reuptake inhibitors (SNRTs), noradrenergic and specific serotonergic an- tidepressants (NaSSA's), corticotropin releasing factor (CRF) antagonists, ⁇ -adrenoreceptor antagonists and atypical antidepressants.
- SSRTs selective serotonin reuptake inhibitors
- MAOI's monoamine oxidase inhibitors
- RIMA's reversible inhibitors of monoamine oxidase
- SNRTs noradrenergic and specific serotonergic an- tidepressants
- CRF corticotropin releasing factor
- norepinephrine reuptake inhibitors include amitriptyline, clomipramine, doxepin, imipramine, trimipramine, amoxapine, desipramine, maprotiline, nortriptyline, protriptyline, reboxetine and pharmaceutically acceptable salts thereof.
- Suitable examples of selective serotonin reuptake inhibitors include fluoxetine, fluvoxamine, paroxetine, sertraline and pharmaceutically acceptable salts thereof.
- Suitable examples of monoamine oxidase inhibitors include isocarboxazid, phenelzine, tranylcypromine, selegiline and pharmaceutically acceptable salts thereof.
- Suitable examples of reversible inhibitors of monoamine oxidase include mo- clobemide and pharmaceutically acceptable salts thereof.
- Suitable examples of serotonin and noradrenaline reuptake inhibitors include venlafaxine and pharmaceutically acceptable salts thereof.
- Suitable atypical antidepressants include bupropion, lithium, nefazodone, trazodone, viloxazine, sibutramine and pharmaceutically acceptable salts thereof.
- Other suitable antidepressants include adinazolam, alaproclate, amineptine, amitriptyline/chlordiazepoxide combination, atipamezole, azamianserin, apelinaprine, befuraline, bifemelane, binodaline, bipenamol, brofaromine, bupropion, caroxazone, cericlamine, cianopramine, cimoxatone, citalopram, clemeprol, clovoxamine, dazepinil, deanol, demexiptiline, dibenzepin, dothiepin, droxidopa, enefexine, estazolam, etoperi- done, femoxetine, feng
- Suitable classes of anti-anxiety agents include benzodiazepines and 5-HT IA recep- tor agonists or antagonists, especially 5-HT IA partial agonists, corticotropin releasing factor (CRF) antagonists, compounds having muscarinic cholinergic activity and com- pounds acting on ion channels.
- benzodiazepines other suitable classes of anti-anxiety agents are nonbenzodiazepine sedative-hypnotic drugxs such as Zolpidem; mood-stabilizing drugs such as clobazam, gabapentin, lamotrigine, loreclezole, oxcar- bamazepine, stiripentol and vigabatrin; and barbiturates.
- Suitable antipsychotic agents are selected from the group consisting of aceto- phenazine, in particular the maleate salt; alentemol, in particular the hydrobromide salt; alpertine; azaperone; batelapine, in particular the maleate salt; benperidol; benzin- dopyrine, in particular the hydrochloride salt; brofoxine; bromperidol; butaclamol, in particular the hydrochloride salt; butaperazine; carphenazine, in particular the maleate salt; carvotroline, in particular the hydrochloride salt; chlorpromazine; chlorprothixene; cinperene; cintriamide; clomacran, in particular the phosphate salt; clopenthixol; clopi- mozide; clopipazan, in particular the mesylate salt; cloroperone, in particular the hydrochloride salt; clothiapine; clothixamide, in particular the maleate salt; cloza
- the compounds according to the invention, in particular according to Formula (I) may also be used in conjunction with other lipid-lowering agent, thus leading to a so-called combination lipid-lowering therapy for the treatment of obesity.
- the said ad- ditional lipid-lowering agent may be, for instance, a known drug conventionally used for the management of hyperlipidaemia such as e.g. a bile acid sequestrant resin, a fi- bric acid derivative or nicotinic acid as previously mentioned in the background of the invention.
- Suitable additional lipid-lowering agents also include other cholesterol biosynthesis inhibitors and cholesterol absorption inhibitors, especially HMG-CoA reduc- tase inhibitors and HMG-CoA synthase inhibitors, HMG-CoA reductase gene expression inhibitors, CETP inhibitors, ACAT inhibitors, squalene synthetase inhibitors, CB-I antagonists, cholesterol absorption inhibitors such as ezetimibe, and the like.
- HMG-CoA reductase inhibi- tor refers to a compound which inhibits the biotransformation of hydroxymethylglutaryl-coenzyme A to mevalonic acid as catalyzed by the enzyme HMG-CoA reductase.
- HMG-CoA reductase inhibitors are, for example, lovastatin, simvastatin, fiuvastatin, pravastatin, rivastatin, and atorvastatin.
- HMG-CoA synthase inhibitor refers to a compound which inhibits the biosynthesis of hydroxymethylglutaryl-coenzyme A from acetyl-coenzyme A and acetoace- tyl-coenzyme A, catalyzed by the enzyme HMG-CoA synthase
- HMG-CoA reductase gene expression inhibitor may be used as the second compound in the combination therapy aspect of this invention.
- These agents may be HMG-CoA reductase trancription inhibitors that block the transcription of DNA or translation inhibitors that prevent translation of mRNA coding for HMG-CoA reductase into protein.
- Such inhibitors may either affect trancription or translation directly or may be biotransformed into compounds having the above-mentioned attributes by one or more enzymes in the cholesterol biosynthetic cascade or may lead to accumulation of a metabolite having the above-mentioned activities.
- CETP inhibitor refers to a compound which inhibits the cholesteryl ester transfer protein (CETP) mediated transport of various cholesteryl esters and triglycerides from HDL to LDL and VLDL.
- ACAT inhibitor refers to a compound which inhibits the intracellular esterification of dietary cholesterol by the enzyme acyl CoAxholesterol acyltransferase.
- squalene synthetase inhibitor refers to a compound which inhibits the condensation of two molecules of farnesylpyrophosphate to form squalene, catalyzed by the enzyme squalene synthetase.
- compositions also relates to a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier or diluent and, as active ingredient, a therapeutically effective amount of a compound according to the invention, in particular a compound according to Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof or a quaternary ammonium salt thereof.
- a compound according to the invention in particular a compound according to Formula (I)
- Formula (I), the pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof or a quaternary ammonium salt thereof , or any subgroup or combination thereof may be formulated into various pharmaceutical forms for administration purposes.
- compositions there may be cited all compositions usually employed for systemically administering drugs.
- compositions of this invention an effective amount of the particular compound, optionally in addition salt form, as the active ingredient is combined in intimate admixture with a pharmaceutically acceptable carrier, which carrier may take a wide variety of forms depending on the form of preparation desired for administration.
- a pharmaceutically acceptable carrier which carrier may take a wide variety of forms depending on the form of preparation desired for administration.
- These pharmaceutical compositions are desirable in unitary dosage form suitable, in particular, for administration orally, rectally, percutaneously, by parenteral injection or by inhalation.
- any of the usual pharmaceutical media may be employed such as, for example, water, glycols, oils, alcohols and the like in the case of oral liquid preparations such as suspensions, syrups, elixirs, emulsions and solutions; or solid carriers such as starches, sugars, kaolin, diluents, lubricants, binders, disintegrating agents and the like in the case of powders, pills, capsules and tablets. Because of their ease in administration, tablets and capsules represent the most advantageous oral dosage unit forms in which case solid pharmaceutical carriers are obviously employed.
- the carrier will usually comprise sterile water, at least in large part, though other ingredients, for example, to aid solubility, may be included.
- In- jectable solutions for example, may be prepared in which the carrier comprises saline solution, glucose solution or a mixture of saline and glucose solution.
- Injectable suspensions may also be prepared in which case appropriate liquid carriers, suspending agents and the like may be employed.
- solid form preparations that are intended to be converted, shortly before use, to liquid form preparations.
- the carrier optionally comprises a penetration enhancing agent and/or a suitable wetting agent, optionally combined with suitable additives of any nature in minor proportions, which additives do not introduce a significant deleterious effect on the skin.
- Said additives may facilitate the administration to the skin and/or may be helpful for preparing the desired compositions.
- These compositions may be administered in various ways, e.g., as a transdermal patch, as a spot-on, as an ointment.
- Unit dosage form refers to physically discrete units suitable as unitary dosages, each unit containing a predetermined quantity of active ingredient calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier.
- unit dosage forms are tablets (including scored or coated tablets), capsules, pills, powder packets, wafers, suppositories, injectable solutions or suspensions and the like, and segregated multiples thereof. Since the compounds ac- cording to the invention are potent orally administrable dopamine antagonists, pharmaceutical compositions comprising said compounds for administration orally are especially advantageous.
- the invention also relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound according to the invention and one or more other compounds selected from the group of antidepressants, anxiolytics, antipsychotics and lipid- lowering agents as well as to the use of such a composition for the manufacture of a medicament.
- weight ratio of the compounds according to the invention to the one or more other compounds may be in the range of 0.05/0.94 to 0.94/0.05, more in particular 0.10/0.89 to 0.89/0.10, and any number therein between.
- THF tetrahydrofuran
- DMF means 7V,7V-dimethyl- formamide
- EtOAc means ethyl acetate
- DCM means dichloromethane
- DME means 1,2-dimethoxyethane
- DCE means 1 ,2-dichloroethane
- DIPE means diiso- propylether
- DMSO means dimethylsulfoxide
- boc means tert-butoxy carbonyl
- Cu(OAc) 2 means copper acetate
- BINAP is l,l'-[l,l'-binaphthalene]- 2,2'-diylbis[l,l]diphenyl-phosphine
- DBU means l,8-diaza-7-bicyclo[5.4.0]- undecene
- DIPEA means N-ethyl-N-(l-methylethyl)-2-prop
- Microwave assisted reactions were performed in a single-mode reactor: Initia- torTM Sixty EXP microwave reactor (Biotage AB). Description of the instrument can be found in www.biotage.com. And in a multimode reactor: MicroSYNTH Labstation
- intermediate compound 1-2 (0.760 g, 3.3 mmol), potassium carbonate (1.38 g, 10 mmol) in THF, N-phenyltrifluoromethanesulphonimide (1.18 g, 3.3 mmol) was added.
- the reaction heated in a microwave oven for 15 minutes at 120 0 C. Then, the solid was filtered off and washed with DCM. The filtrate solvent was evaporated and the residue was purified by column chromatography over silica gel (eluent: DCM, DCMiEtOAc 9/1 and 4/1). The product fractions were collected and the solvent was evaporated. The residue was treated with DIPE to yield intermediate compound 1-3 (0.900 g, 60 %).
- Tables Ia, 2, 3a, 4 and 5 list the compounds of Formula (I), which were prepared according to one of the above described examples.
- Example “Ex. Nr.” the examples that are described in detail in the experimental section above, have been marked with "*”.
- the analytical data for the compounds, measured according to one or more of the analytical procedures described below, are also present in these tables.
- Tables Ib and 3b list the compounds of Formula (I), which are prepared according to one of the above described examples.
- the HPLC gradient was supplied by a HP 1100 from Agilent Technologies comprising a pump (quaternary or binary) with degasser, an autosampler, a column oven, a diode-array detector (DAD) and a column as specified in the respective methods below.
- Flow from the column was split to a MS spectrometer.
- the MS detector was configured with an electrospray ionization source. Nitrogen was used as the nebulizer gas.
- the source temperature was maintained at 140 °C. Data acquisition was performed with MassLynx-Openlynx software.
- LCMS general procedure B The HPLC gradient was supplied by an Alliance HT 2790 (Waters) system comprising a quaternary pump with degasser, an autosampler, a column oven (set at 40 0 C), a diode-array detector (DAD) and a column as specified in the respective methods below. Flow from the column was split to a MS spectrometer. The MS detector was configured with an electrospray ionization source. Mass spectra were acquired by scanning from 100 to 1000 in 1 second using a dwell time of 0.1 second. The capillary needle voltage was 3 kV and the source temperature was maintained at 140 0 C. Nitrogen was used as the nebulizer gas. Data acquisition was performed with a Waters-Micromass MassLynx-Openlynx data system.
- LCMS method 1 In addition to general procedure A: Reversed phase HPLC was carried out on an ACE-C 18 column (3.0 ⁇ m, 4.6 x 30 mm) from Advanced Chromatography Technologies, with a flow rate of 1.5 ml/min, at 4O 0 C.
- the gradient conditions used are: 80 % A (0.5 g/1 ammonium acetate solution), 10 % B (acetonitrile), 10 % C (methanol) to 50 % B and 50 % C in 6.5 minutes, to 100 % B at 7 minutes and equilibrated to initial conditions at 7.5 minutes until 9.0 minutes. Injection volume 5 ⁇ l.
- High-resolution mass spectra (Time of Flight, TOF) were acquired only in positive ionization mode by scanning from 100 to 750 in 0.5 seconds using a dwell time of 0.1 seconds.
- the capillary needle voltage was 2.5 kV for positive ionization mode and the cone voltage was 20 V.
- Leucine-Enkephaline was the standard substance used for the lock mass calibration.
- Reversed phase HPLC was carried out on an ACE-C 18 column (3.0 ⁇ m, 4.6 x 30 mm) from Advanced Chromatography Technologies, with a flow rate of 1.5 ml/min, at 4O 0 C.
- the gradient conditions used are: 80 % A (0.5 g/1 ammonium acetate solution), 10 % B (acetonitrile), 10 % C (methanol) to 50 % B and 50 % C in 6.5 minutes, to 100 % B at 7 minutes and equilibrated to initial conditions at 7.5 minutes until 9.0 minutes. Injection volume 5 ⁇ l.
- Reversed phase HPLC was carried out on a Chromolith (4.6 x 25 mm) with a flow rate of 3 ml/min.
- Three mobile phases (mobile phase A: 95 % 25 mM ammoniumacetate + 5 % acetonitrile; mobile phase B: acetoni- trile; mobile phase C: methanol) were employed to run a gradient condition from 96 % A, 2 % B and 2 % C, to 2 % A, 49 % B and 49 % C in 0.9 minutes, to 100 % B in 0.3 minutes and hold for 0.2 minutes.
- An injection volume of 2 ⁇ l was used.
- Cone voltage was 10 V for positive ionization mode and 20 V for negative ionization mode.
- melting points were determined in open capillary tubes on a Mettler FP62 apparatus. Melting points were measured with a temperature gradient of 3 or 10 °C/minute. Maximum temperature was 300 0 C. The melting point was read from a digital display.
- MCH-I receptors The interaction of the compounds of Formula (I) with MCH-I receptors was assessed in in vitro transient calcium (Ca 2+ ) mobilization assays in the fiuorimetric imaging plate reader (FLIPR) format (Sullivan et al. 1999, Methods MoI Biol 114:125-133).
- FLIPR fiuorimetric imaging plate reader
- MCH natural agonist
- the interaction of the test compounds with the receptor is assessed in competition experiments.
- concentrations of the test compound are added to the incubation mixture containing the receptor-expressing cells and a submaximal concentra- tion of MCH.
- the test compound in proportion to its antagonist potency and its concentration inhibits MCH-induced Ca 2+ mobilization.
- Example Cl Binding Experiment for MCH-I Cell culture and membrane preparation.
- Chinese Hamster ovary cells (CHO) stably expressing the human MCH-I receptor are grown in a 1:1 mixture of Dulbecco's Modified Eagles Medium (DMEM) and HAM's F 12 medium including GlutamaxTM (Invitrogen), supplemented with 10 % heat-inactivated fetal bovine serum and 400 ⁇ g/ml geneticin.
- DMEM Dulbecco's Modified Eagles Medium
- GlutamaxTM Invitrogen
- MCH-I receptor-expressing CHO cells are seeded in 20 ⁇ l (5,000 cells per well) into 384-well black wall, clear bottom microtiter plates (Costar).
- 20 ⁇ l per well calcium assay kit containing 10 mM probenicide (Molecular Devices) is added. Cells are loaded for 90 min at 37 °C and 5 % CO 2 in a cell culture incubator. After loading, 20 ⁇ l of serial dilutions of the test compound are added and cells are further incubated for 20 min at room temperature in the dark. After 20 min, 20 ⁇ l of a submaximal MCH concentration is added and changes in intracellular calcium are recorded directly in a FLIPR III apparatus (Molecular devices).
- All compounds according to Formula (I) produced an inhibition of more than 50 % (pICso) at a test concentration ranging between 10 "6 M and 10 "9 M in a concentration-dependent manner.
- Example C.2 Determination of brain concentrations
- the dosing formulations were prepared by individually dissolving the compounds in a 10 % hydroxypropyl- ⁇ -cyclodextrin (HP- ⁇ -CD) solution at a final concentration of 1.25 mg/ml for the IV-formulations and they were made isotonic with manni- tol.
- the formulations were stored at room temperature, protected from light and were analysed quantitatively with LC-MS/MS on the day of preparation. The stability of these formulations was checked on the day of dosing. Dose administration was done intravenously in a tail vein at 2 ml/kg to obtain a dose of 2.5 mg/kg.
- Sample time points were 1 and 4 h after intravenous dose administration. Animals were sacrificed by decapitation and blood was collected by exsanguination in 10 ml B-D Sterile EDTA K3 Vacutainer tubes. From each individual animal the whole brain was dissected and weighed. Individual brain samples were homogenized in demineralized water (1/9 w/v or + 3 ml if tissue weight ⁇ 0.33 g). Homogenisation was carried out under dimmed light conditions. The brain samples were analysed using a qualified research LC-MS/MS method. The lower limit of quantification (LLOQ) was in the range of 20 - 50 ng/g.
- LLOQ lower limit of quantification
- Table 7 Comparison of brain levels.
- Active ingredient (a.i.) as used throughout these examples relates to a final compound of formula (i), the pharmaceutically acceptable acid or base addition salts thereof, the stereochemically isomeric forms thereof, the TV-oxide form thereof, a quaternary ammonium salt thereof and prodrugs thereof.
- Example D.I oral drops
- Example D.2 oral solution
- Example D.3 film-coated tablets Preparation of . tablet . . core A mixture of 100 grams of the a.L, 570 grams lactose and 200 grams starch is mixed well and thereafter humidified with a solution of 5 grams sodium dodecyl sulfate and 10 grams polyvinylpyrrolidone in about 200 ml of water. The wet powder mixture is sieved, dried and sieved again. Then there is added 100 grams micro crystalline cellulose and 15 grams hydrogenated vegetable oil. The whole is mixed well and com- pressed into tablets, giving 10,000 tablets, each containing 10 mg of the active ingredient.
- Example D .4 injectable solution
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Hematology (AREA)
- Psychiatry (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Pain & Pain Management (AREA)
- Child & Adolescent Psychology (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pyridine Compounds (AREA)
Abstract
The present invention concerns N-aryl and N-heteroaryl substituted pyridinone derivatives having antagonistic melanin-concentrating hormone (MCH) activity, in particular MCH-1 activity according to the general Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an N-oxide form thereof or a quaternary ammonium salt thereof, wherein the variables are defined in Claim 1. It further relates to their preparation, compositions comprising them and their use as a medicine. The compounds according to the invention are useful for the prevention and/or treatment of psychiatric disorders, including but not limited to anxiety, eating disorders, mood disorders, such as bipolar disorders and depression, psychoses, such as schizophrenia, and sleeping disorders; obesity; diabetes; sexual disorders and neurological disorders.
Description
NOVEL N-ARYL AND N-HETEROARYL SUBSTITUTED PYRIDINONE DERIVATIVES FOR USE IN MCH-I MEDIATED DISEASES
Field of the Invention The present invention concerns N-aryl and N-heteroaryl substituted pyridinone derivatives having antagonistic melanin-concentrating hormone (MCH) activity, in particular MCH-I activity. It further relates to their preparation, compositions comprising them and their use as a medicine.
Background of the Invention
Melanin concentrating hormone (MCH) is a cyclic 19-amino acid polypeptide, which is mainly produced by hypothalamic neurons projecting widely throughout the central nervous system (CΝS) (J. Comp. Neurol. (1992) 319, 218-245). MCH mediates its effects through two G protein-coupled receptors (GPCRs) termed MCH-I and MCH-2 (reviewed in Doggrell, 2003). While in rodents only the MCH-I receptor is expressed, human and primates express both MCH-I and MCH-2 receptors (Genomics (2002), 79, 785-792). Originally, the MCH-I receptor was considered a valuable target for the treatment of obesity as MCH promotes feeding behaviour in rodents (Nature (1996), 380, 243-247). Recently however, it was shown that MCH-I antagonism pro- duces anxiolytic and antidepressant profiles in rodents (Nat. Med. (2002) 8, 825-830; Neuropharmacology (2004), 46, 457-467 ; Neuropsychopharmacology (2005), in press). Thus, it is currently generally accepted that MCH receptors, particularly the MCH-I receptor, are a good target for the treatment of affective spectrum disorders (Eur. J. Neuroscience (2000) 12, 1194-1216). MCH-I receptor mRNA and protein are distributed in various hypothalamic nuclei including the paraventricular nucleus and several limbic structures all implicated in the regulation of emotion and stress (Eur. J. Neuroscience (2000) 12, 1194-1216). In addition, dense labelling is detected in the nucleus accumbens shell (J. Comp. Neurol. (2001) 435, 26-40). Injection of MCH directly into the paraventricular nucleus has been found to increase plasma adrenocorticotropic hormone (ACTH) and to alter sleep architecture (Verret et al. 2003, BMC Neurosci 4:19). MCH also induces corticotro-
phin-releasing factor (CRF) release from hypothalamic explants, an effect that is sensitive to blockade by an MCH-I receptor antagonist (J. Neuroendrocrinol. (2003) 15, 268-2729). Thus it seems likely that stimulation of MCH-I receptor causes activation of the hypothalamus-pituitary-adrenal (HPA) axis through increases in CRF release. Injection of MCH into the nucleus accumbens shell, in which MCH-I receptor is abundant, increased immobility in a forced swim test in rats, suggesting increased depressive behavior (Soc. Neurosci. Abstr. (2004) 763.9). Moreover, Borowsky et al. (Nat. Med. (2002) 8, 825-830) reported the MCH-I antagonist, SNAP-7941, exhibited antidepressant- and axiolytic-like affects in rodents tests, supporting a role for MCH-I re- ceptor in depression and anxiety.
Background prior art
A large number of companies is now actively pursuing the development of MCH-I antagonists and a wide range of structural types have been reported in a num- ber of patent publications, mostly in relation to the regulation of food intake and energy expenditure (Expert Opin. Ther. Patents (2005) 15(10)). The majority of reported MCH-antagonists incorporate a basic centre and two (hetero)aromatic parts, joined by linkers. WO 2005/085200 (Banyu Pharmaceutical Co., Ltd) discloses pyridinone, pyrimidinone and pyridazinone-derivatives for use as MCH-I antagonists. WO 2003/033480, WO 2003/033476 and WO 2005/05042541 (Glaxo Group Limited), WO 2004/024702 (Boehringer Ingelheim Pharma GMBH & Co. KG) and WO 2005/103039 (Neurocrine Biosciences Inc.) disclose different bicyclic heterocycles, such as thieno- pyrimid-4-one-, benzopyrimid-4-one- and ftalimide-derivatives, for use as MCH-I antagonists. WO 2003/097047 and WO 2005/040157 (Eli Lilly and Company) and WO 2005/070925 (Aventis Pharma Deutschland GmbH) report different aromatic 5-membered ring heterocycles, such as oxazole- and oxadiazole-derivatives, for use as MCH-I antagonists. WO 2004/011438 and WO 2005/070898 (Aventis Pharma Deutschland GMBH) disclose diaryl-substituted cyclic urea derivatives as MCH-I antagonist. WO 2003/068230 (Pharmacia Corporation) discloses the preparation of substituted pyridinones as modulators of p38 MAP kinase. Although none of the compounds are disclosed to have MCH-I antagonist activity, some of the exemplified compounds
are excluded from the compound scope only of this application by way of a disclaimer.
Description of the Invention
It is an object of the present invention to provide a compound with a binding affinity towards melanin-concentrating (MCH) receptors, in particular towards MCH-I receptors, in particular as an antagonist.
This object was achieved by a novel TV- aryl and TV-heteroaryl substituted pyridi- none derivative according to the general Formula (I)

a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof or a quaternary ammonium salt thereof, wherein A is phenyl or pyridinyl ;
R1, R2 are each, independently from each other, selected from the group of hydrogen, Ci-3alkyl, aryl,; arylC 1.3 alky 1, Het, HetC1-3alkyl,


NRaRbCi-3alkyl ; or R1 and R2 are taken together to form an TV-containing heterocycle, selected from the group of pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, and thiomorpholinyl ; wherein each of the afore- mentioned TV-containing heterocycles are optionally substituted by k substitu- ents R3 ;
R3 is selected from the group of hydrogen, halo, cyano, hydroxy, NRaRb, oxo, carboxyl, aminocarbonyl, nitro, thio, formyl, C1-3alkyl, aryl,
 and
and
 Ra, Rb are each, independently from each other, selected from the group of hydrogen,
C1-3alkyl, aryl, Het,
Ra, Rb are each, independently from each other, selected from the group of hydrogen,
C1-3alkyl, aryl, Het,
 sulphonyl,
sulphonyl,
 and
and
 ; or Ra and Rb are taken together to form an TV-containing heterocycle, selected from the group of pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, l-aza-bicyclo[2.2.1]heptyl, and thiomorpholinyl ;
; or Ra and Rb are taken together to form an TV-containing heterocycle, selected from the group of pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, l-aza-bicyclo[2.2.1]heptyl, and thiomorpholinyl ;
R4, R5 are each, independently from each other, selected from the group of hydrogen, halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl, C1-3alkyl, and
 k is an integer, equal to zero, 1, 2, 3 or 4 ; 1 is an integer, equal to zero, 1, 2, 3 or 4 ; m is an integer, equal to zero, 1 , 2 or 3 ;
k is an integer, equal to zero, 1, 2, 3 or 4 ; 1 is an integer, equal to zero, 1, 2, 3 or 4 ; m is an integer, equal to zero, 1 , 2 or 3 ;
Y1, Y3 are each, independently from each other, selected from the group of a single bond, O, NR7, S, SO, and SO2 ; wherein R7 is selected from the group of hydrogen and
 ; Y2 is a saturated or unsaturated, straight or branched Ci-6-hydrocarbon radical, wherein one or more hydrogen atoms may optionally be replaced by a radical selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, and formyl ;
; Y2 is a saturated or unsaturated, straight or branched Ci-6-hydrocarbon radical, wherein one or more hydrogen atoms may optionally be replaced by a radical selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, and formyl ;
B is a 6-membered ring containing zero, 1, 2 or 3 nitrogen atoms, optionally substituted with p substituents R6, each independently from each other, selected from the group of hydrogen, halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl, C1-3alkyl, and C1-3alkyloxy ; and wherein p is an integer, equal to zero, 1 or 2 ; or two substituents R6 may be combined into a radical -CH2CH2CH2- or -OCH2O- ; alkyl is a straight or branched saturated hydrocarbon radical having the indicated number of carbon atoms ; wherein the radical may optionally be substituted on one or more carbon atoms with one or more radicals selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio and formyl ; aryl is naphthyl or phenyl, each optionally substituted with 1 , 2 or 3 substituents, each independently from each other, selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl, C1-3 alkyl, and C1-3alkyl- oxy ;
Het is a heterocyclic radical selected from the group of pyrrolidinyl,
imida-zolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, pyrrolyl, pyrrolinyl, imidazolinyl, pyrrazolinyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, azepyl, diazepyl, morpholinyl, thiomorpholinyl, furyl, thienyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, isothiazolyl, dioxolyl, dithianyl, tetrahydrofuryl, tetrahydro- pyranyl, and oxadiazolyl ; wherein each of the aforementioned heterocyclic radicals is optionally substituted with 1, 2 or 3 substituents, each independently from each other, selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl, C1-3alkyl, and
 ; and halo is fluoro, chloro, bromo or iodo.
; and halo is fluoro, chloro, bromo or iodo.
For establishing the scope of protection, the following compounds, as disclosed in WO 2003/068230, are excluded from the compound scope only :
4-[(2,4-difiuorobenzyl)oxy]- 1 -[2,6-difluoro-4-[(2-hydroxyethyl)(methyl)amino]- phenyl]-6-methylpyridin 2(7H)-one ; - 4-[(2,4-difiuorobenzyl)oxy]- 1 -[2,6-difluoro-4-(4-methylpiperazin- 1 -yl)phenyl]-6-m ethylpyridin-2(7H)-one ;
- 4-[(2,4-difiuorobenzyl)oxy]- 1 -[2,6-difluoro-4-(morpholin-4-yl)phenyl]-6-methylpy ridin-2(7H)-one ;
4-[(2,4-difiuorobenzyl)oxy]- 1 -[4-(dimethylamino)-2,6-difluorophenyl]-6-methyl- pyridin-2(7H)-one ;
3-bromo-4-[(2,4-difluorobenzyl)oxy]- 1 -[2,6-difluoro-4-[(2-hydroxyethyl)(methyl)- amino]phenyl]-6-methylpyridin-2(7H)-one ;
3-bromo-4-[(2,4-difluorobenzyl)oxy]- 1 -[2,6-difluoro-4-(4-methylpiperazin- 1 -yl)ph enyl]-6-methylpyridin-2(7H)-one ; - 3-bromo-4-[(2,4-difluorobenzyl)oxy]-l-[4-(dimethylamino)-2,6-difluorophenyl]-6- methylpyridin-2(7H)-one ;
3-bromo-4-[(2,4-difluorobenzyl)oxy]-l-[2,6-difluoro-4-(morpholin-4-yl)phenyl]-6- methylpyridin-2(7H)-one ; and
3-chloro-4-[(2,4-difluorobenzyl)oxy]- 1 -[2,6-difluoro-4-(4-methylpiperazin- 1 -yl)- phenyl]-6-methylpyridin-2(7H)-one.
In the framework of this application, with "compounds according to the invention" is meant a compound according to the general Formula (I), a pharmaceutically
acceptable acid or base addition salt thereof, an TV-oxide form thereof or a quaternary ammonium salt thereof, including the above compounds disclosed in WO 2003/068230. Only for establishing the compound scope, it is understood that these compounds are disclaimed by way of a disclaimer. The invention also relates to a pharmaceutical composition comprising a pharmaceutically acceptable carrier or diluent and, as active ingredient, a therapeutically effective amount of a compound according to the invention, in particular a compound according to Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof, or a quaternary ammonium salt thereof. The invention also relates to the use of a compound according to the invention as a medicament and for the preparation of a medicament for the prevention and/or treatment of a disorder or disease responsive to antagonism of the MCH receptor, in particular to antagonism of the MCH-I receptor.
In particular, the invention relates to the use of a compound according to the in- vention for the preparation of a medicament for the prevention and/or treatment of psychiatric disorders, including but not limited to anxiety, eating disorders, mood disorders, such as bipolar disorders and depression, psychoses, such as schizophrenia, and sleeping disorders. Additionally, the compound can be used for treating obesity, diabetes, sexual disorders and neurological disorders. A compound according to the invention, in particular according to Formula (I), may also be suitable as add-on treatment or combination treatment and/or prophylaxis in the above listed diseases, in particular for the prevention and/or treatment of psychiatric disorders, in combination with antidepressants, anxiolytics and/or antipsychotics which are currently available or in development or which will become available in the future, in particular to improve efficacy and/or onset of action. This is evaluated in rodent models in which antidepressants, anxiolytics and/or antipsychotics are shown to be active. For example, compounds are evaluated in combination with antidepressants, anxiolytics and/or antipsychotics for attenuation of stress-induced hyperthermia.
The invention therefore also relates to the use of the compounds according to the invention in combination with one or more other compounds selected from the group of antidepressants, anxiolytics and antipsychotics, to a pharmaceutical composition comprising the compounds according to the invention and one or more other compounds
selected from the group of antidepressants, anxiolytics and antipsychotics, as well as to a process for the preparation of such pharmaceutical compositions.
The invention also relates to the use of the compounds according to the invention in combination with one or more other compounds selected from the group of lipid- lowering compounds for the prevention and/or treatment of obesity, to a pharmaceutical composition comprising the compounds according to the invention and one or more other compounds selected from the group of lipid-lowering compounds, as well as to a process for the preparation of such pharmaceutical compositions.
Detailed description of the invention
In an embodiment, the invention relates to a compound according to the invention, wherein each of R1, R2, independently from each other, is selected from the group of hydrogen, C1-3alkyl, arylC 1.3 alky 1, HetC1-3alkyl,

 and NRaRbCi-3alkyl. Preferably, the
and NRaRbCi-3alkyl. Preferably, the
 is substituted with hydroxy or amino .
is substituted with hydroxy or amino .
In another embodiment, the invention relates to a compound according to the invention, wherein R1 and R2 are taken together to form an TV-containing heterocycle, selected from the group of pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, and thio- morpholinyl ; and wherein each of the foregoing TV-containing heterocycles is option- ally substituted by k substituents R3.
In another embodiment, the invention relates to a compound according to the invention, wherein R3 is selected from the group of hydroxy, NRaRb, aminocarbonyl, C1-3alkyl, aryl,
 and
and

In another embodiment, the invention relates to a compound according to the invention, wherein Ra, Rb are each, independently from each other, selected from the group of hydrogen, C1-3alkyl, C1-3alkylcarbonyl. C1-3alkyloxycarbonyl, and C1-3alkyl- sulphonyl; or Ra and Rb are taken together to form an N-containing heterocycle, selected from the group of pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, l-aza-bicyclo[2.2.1]heptyl, and thiomorpholinyl. In another embodiment, the invention relates to a compound according to the invention, wherein A is phenyl.
In another embodiment, the invention relates to a compound according to the invention, wherein each of R4 and R5, independently from each other, are selected from the group of hydrogen, halo, C1-3alkyl, and

In another embodiment, the invention relates to a compound according to the in- vention, wherein k is 1.
In another embodiment, the invention relates to a compound according to the invention, wherein 1 is zero.
In another embodiment, the invention relates to a compound according to the invention, wherein m is zero. In another embodiment, the invention relates to a compound according to the invention, wherein Y1 and Y3 are each, independently from each other, selected from the group of a single bond and O.
In another embodiment, the invention relates to a compound according to the invention, wherein Y2 is selected from the group of saturated or unsaturated, straight or branched C1-6 hydrocarbon radicals.
In another embodiment, the invention relates to a compound according to the invention, wherein B is selected from the group of phenyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl and triazinyl. Preferably, B is phenyl, pyridinyl or pyridazinyl. More preferably, B is phenyl. In another embodiment, the invention relates to a compound according to the invention, wherein the moiety B-Y'-Y^Y3 is selected from the radicals (a-1) to (d-5) as listed below, optionally substituted with p substituents R6, each independently from each other, selected from the group of hydrogen, fluoro, chloro, hydroxy, methyl, trifluoromethyl, fiuoromethyloxy, difiuoromethyloxy, and methyloxy ; and wherein p is an integer, equal to zero, 1 or 2 ; or two substituents R6 are combined into a radical -CH2CH2CH2- or -OCH2O-.

(a-1) (b-1) (C-I) (d-1)

(a-2) (b-2) (c-2) (d-2)

(a-3) (b-3) (c-3) (d-3)

(a-4) (b-4) (c-4) (d-4)

(a-5) (b-5) (c-5) (d-5)
Most preferably, the moiety B-Y'-Y2^3 is selected from the radicals (a-1), (a-2), (a-3) and (b-3).
In the framework of this application, alkyl is a straight or branched saturated hydrocarbon radical having the indicated number of carbon atoms, i.e. when
 is indicated, the alkyl radical may contain from 1 to 3 carbon atom ; and when
is indicated, the alkyl radical may contain from 1 to 3 carbon atom ; and when
 is indicated, the alkyl radical may contain from 1 to 6 carbon atom. Each radical may optionally be substituted on one or more carbon atoms with one or more radicals selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio and formyl. Preferably, alkyl is methyl, ethyl, propyl or isopropyl. Further radicals that are embraced within the scope are e.g. hydroxymethyl, 1-hydroxyethyl, 1-hydroxypropyl, fluoromethyl, difluoromethyl and trifluoromethyl.
In the framework of this application, aryl is naphthyl or phenyl, each optionally substituted with 1, 2 or 3 substituents, each independently from each other, selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl and
is indicated, the alkyl radical may contain from 1 to 6 carbon atom. Each radical may optionally be substituted on one or more carbon atoms with one or more radicals selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio and formyl. Preferably, alkyl is methyl, ethyl, propyl or isopropyl. Further radicals that are embraced within the scope are e.g. hydroxymethyl, 1-hydroxyethyl, 1-hydroxypropyl, fluoromethyl, difluoromethyl and trifluoromethyl.
In the framework of this application, aryl is naphthyl or phenyl, each optionally substituted with 1, 2 or 3 substituents, each independently from each other, selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl and
 In the framework of this application, halo is a substituent selected from the group of fluoro, chloro, bromo and iodo. Preferably, halo is fluoro, chloro or bromo.
In the framework of this application, halo is a substituent selected from the group of fluoro, chloro, bromo and iodo. Preferably, halo is fluoro, chloro or bromo.
In the framework of this application, unless otherwise indicated, a bond can be any bond, including a covalent bond, a single bond, a double bond, a triple bond, a coordination bond and a hydrogen bond. In the framework of this application, an unsaturated hydrocarbon radical indicates a hydrocarbon radical which contains one or more double and/or triple bonds, hence comprising partly or completely unsaturated hydrocarbon radicals, such as, for instance -CH=CH-, -CH2CH=CH-, -CH2CH=CHCH2-, -C≡CH-, -C≡CHCH2-,-C≡C-CH=CH-, and the like. A pharmaceutically acceptable acid addition salt is defined to comprise a therapeutically active non-toxic acid addition salt form that a compound according to Formula (I) is able to form. Said salt can be obtained by treating the base form of a compound according to Formula (I) with an appropriate acid, for example an inorganic acid, for example hydrohalic acid, in particular hydrochloric acid, hydrobromic acid, sulphuric acid, nitric acid and phosphoric acid ; an organic acid, for example acetic acid, hydroxyacetic acid, propanoic acid, lactic acid, pyruvic acid, oxalic acid, malonic acid, succinic acid, maleic acid, fumaric acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, cyclamic acid, salicylic acid, p-amino salicylic acid and pamoic acid. Conversely said acid addition salt form may be converted into the free base form by treatment with an appropriate base .
The compound according to Formula (I) containing an acidic proton may also be converted into a therapeutically active non-toxic metal or amine addition salt form
(base addition salt) by treatment with an appropriate organic and inorganic base. Ap- propriate base salt forms comprise, for example, the ammonium salts, the alkaline and earth alkaline metal salts, in particular lithium, sodium, potassium, magnesium and cal-
cium salts, salts with organic bases, e.g. the benzathine, TV-methyl-D-glucamine, hy- bramine salts, and salts with amino acids, for example arginine and lysine.
Conversely, said salt form can be converted into the free form by treatment with an appropriate acid. The term addition salt as used in the framework of this application also comprises a solvate that the compound according to Formula (I), as well as a salt thereof, is able to form. Such solvates are, for example, hydrates and alcoholates.
The TV-oxide form of the compound according to Formula (I) is meant to comprise a compound of Formula (I) wherein one or several nitrogen atoms are oxidized to so-called TV-oxides, particularly those TV-oxides wherein one or more tertiary nitrogens (e.g. of the piperazinyl or piperidinyl radical) are TV-oxidized. Such TV-oxides can easily be obtained by a skilled person without any inventive skills and they are obvious alternatives for a compound according to Formula (I) since these compounds are metabolites, which are formed by oxidation in the human body upon uptake . As is generally known, oxidation is normally the first step involved in drug metabolism (Textbook of Organic Medicinal and Pharmaceutical Chemistry, 1977, pages 70- 75). As is also generally known, the metabolite form of a compound can also be administered to a human instead of the compound per se, with much the same effects.
A compound of Formula (I) may be converted to the corresponding TV-oxide form following art-known procedures for converting a trivalent nitrogen into its TV-oxide form. Said TV-oxidation reaction may generally be carried out by reacting the compound of Formula (I) with an appropriate organic or inorganic peroxide. Appropriate inorganic peroxides comprise, for example, hydrogen peroxide, alkali metal or earth alkaline metal peroxides, e.g. sodium peroxide, potassium peroxide; appropriate or- ganic peroxides may comprise peroxy acids such as, for example, benzenecarboper- oxoic acid or halo substituted benzenecarboperoxoic acid, e.g. 3-chlorobenzenecarbo- peroxoic acid, peroxoalkanoic acids, e.g. peroxoacetic acid, alkylhydroperoxides, e.g. tert-butyl hydroperoxide. Suitable solvents are, for example, water, lower alkanols, e.g. ethanol and the like, hydrocarbons, e.g. toluene, ketones, e.g. 2-butanone, halogenated hydrocarbons, e.g. dichloromethane, and mixtures of such solvents.
A quaternary ammonium salt of compound according to Formula (I) defines said compound which is able to form by a reaction between a basic nitrogen of a com-
pound according to Formula (I) and an appropriate quaternizing agent, such as, for example, an optionally substituted alkylhalide, arylhalide or arylalkylhalide, in particular methyliodide and benzyliodide. Other reactants with good leaving groups may also be used, such as, for example, alkyl trifluoromethanesulfonates, alkyl methanesulfonates and alkyl p-toluenesulfonates. A quaternary ammonium salt has at least one positively charged nitrogen. Pharmaceutically acceptable counterions include chloro, bromo, iodo, trifiuoroacetate and acetate ions.
The invention also comprises a derivative compound (usually called "pro-drug") of a pharmacologically-active compound according to the invention, in particular ac- cording to Formula (I), which is degraded in vivo to yield a compound according to the invention. Pro-drugs are usually (but not always) of lower potency at the target receptor than the compounds to which they are degraded. Pro-drugs are particularly useful when the desired compound has chemical or physical properties that make its administration difficult or inefficient. For example, the desired compound may be only poorly soluble, it may be poorly transported across the mucosal epithelium, or it may have an undesirably short plasma half-life. Further discussion on pro-drugs may be found in Stella, V. J. et al, "Prodrugs", Drug Delivery Systems, 1985, pp. 112-176, and Drugs, 1985, 29, pp. 455-473.
A pro-drug form of a pharmacologically-active compound according to the inven- tion will generally be a compound according to Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof, or a quaternary ammonium salt thereof, having an acid group which is esterified or amidated. Included in such esterified acid groups are groups of the formula -COORX, where Rx is a C1-6alkyl, phenyl, benzyl or one of the following groups :
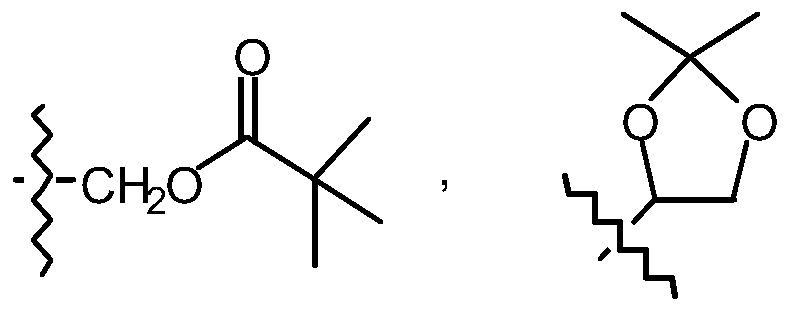
Amidated groups include groups of the formula - CONRyRz, wherein Ry is H,
C1-6alkyl, phenyl or benzyl and Rz is -OH, H, C1-6alkyl, phenyl or benzyl. A compound according to the invention having an amino group may be derivatised with a ketone or
an aldehyde such as formaldehyde to form a Mannich base. This base will hydrolyze with first order kinetics in aqueous solution.
In the framework of this application, a compound according to the invention is inherently intended to comprise all stereochemically isomeric forms thereof. The term "stereochemically isomeric form" as used herein defines all the possible stereochemically isomeric forms that a compound of Formula (I) may possess. Unless otherwise mentioned or indicated, the chemical designation of a compound denotes the mixture of all possible stereochemically isomeric forms, said mixtures containing all di- astereomers and enantiomers of the basic molecular structure. More in particular, stereogenic centers may have the R- or S-configuration; substituents on bivalent cyclic (partially) saturated radicals may have either the cis- or trans-configuration. Compounds encompassing double bonds can have an E or Z-stereochemistry at said double bond. Hence, all stereochemically isomeric forms of a compound of Formula (I) are intended to be embraced within the scope of this invention. Following CAS nomenclature conventions, when two stereogenic centers of known absolute configuration are present in a molecule, an R or S descriptor is assigned (based on Cahn-Ingold-Prelog sequence rule) to the lowest-numbered chiral center, the reference center. The configuration of the second stereogenic center is indicated using relative descriptors [R*,R* J or [R*,S*J, where R* is always specified as the reference center and [R*,R*] indicates centers with the same chirality and [i?*,5**] indicates centers of unlike chirality. For example, if the lowest-numbered chiral center in the molecule has an S configuration and the second center is R, the stereo descriptor would be specified as S-[R*, S*]. If "α" and "β" are used : the position of the highest priority sub- stituent on the asymmetric carbon atom in the ring system having the lowest ring num- ber, is arbitrarily always in the "α" position of the mean plane determined by the ring system. The position of the highest priority substituent on the other asymmetric carbon atom in the ring system (hydrogen atom in a compound according to Formula (I)) relative to the position of the highest priority substituent on the reference atom is denominated "α", if it is on the same side of the mean plane determined by the ring system, or "β", if it is on the other side of the mean plane determined by the ring system.
In the framework of this application, a compound according to the invention is inherently intended to comprise all isotopic combinations of its chemical elements. In
the framework of this application, a chemical element, in particular when mentioned in relation to a compound according to Formula (I), comprises all isotopes and isotopic mixtures of this element, either naturally occuring or synthetically produced, either with natural abundance or in an isotopically enriched form. In particular, when hydro- gen is mentioned, it is understood to refer to 1H, 2H, 3H and mixtures thereof ; when carbon is mentioned, it is understood to refer to 11C, 12C, 13C, 14C and mixtures thereof ; when nitrogen is mentioned, it is understood to refer to 13N, 14N, 15N and mixtures thereof ; when oxygen is mentioned, it is understood to refer to 14O, 15O, 16O, 17O, 18O and mixtures thereof ; and when fluor is mentioned, it is understood to refer to 18F, 19F and mixtures thereof.
A compound according to the invention therefore inherently comprises a compound with one or more isotopes of one or more element, and mixtures thereof, including a radioactive compound, also called radio labelled compound, wherein one or more non-radioactive atoms has been replaced by one of its radioactive isotopes. By the term "radio labelled compound" is meant any compound according to Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof, or a quaternary ammonium salt thereof, which contains at least one radioactive atom. For example, a compound can be labelled with positron or with gamma emitting radioactive isotopes. For radio ligand-binding techniques (membrane receptor assay), the 3H-atom or the 125I-atom is the atom of choice to be replaced. For imaging, the most commonly used positron emitting (PET) radioactive isotopes are 11C, 18F, 15O and 13N, all of which are accelerator produced and have half-lives of 20, 100, 2 and 10 minutes respectively. Since the half-lives of these radioactive isotopes are so short, it is only feasible to use them at institutions which have an accelerator on site for their production, thus limiting their use. The most widely used of these are 18F, 99mTc, 201Tl and 123I. The handling of these radioactive isotopes, their production, isolation and incorporation in a molecule are known to the skilled person.
In particular, the radioactive atom is selected from the group of hydrogen, carbon, nitrogen, sulfur, oxygen and halogen. Preferably, the radioactive atom is selected from the group of hydrogen, carbon and halogen.
In particular, the radioactive isotope is selected from the group of 3H, 11C, 18F, 122I, 123I, 125I, 131I, 75Br, 76Br, 77Br and 82Br. Preferably, the radioactive isotope is se-
lected from the group of Η, 11/ C and 118Tτ .
Preparation
A compound according to the invention can generally be prepared by a succes- sion of steps, each of which is known to the skilled person. In particular, a pyridinone derivative can be prepared according to one or more of the following preparation methods.
Scheme 1

Palladium coupling

The copper coupling reaction is performed in the presence of a copper salt, such as Cu(OAc)2, in an aprotic solvent such as 1 ,2-dichloromethane, in the presence of an amine and amine TV-oxide, such as pyridine and TEMPO, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically overnight at room temperature. The pal- ladium coupling reaction is performed in an aprotic solvent such as toluene, in the presence of a palladium catalyst such as Pd(AcO)2, and in the presence of tBuOK, as a base, and a ligand, such as BINAP, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 24 hours at 100 0C under traditional heating.
Ortho-substituted phenyl derivatives can be prepared according to Scheme 2.
Scheme 2

X= Br, I \ Y= OH, Br, I \ \ If Y= Br, I \
If Y= OH X Palladium \ OH activation / coupling
\
/ \ R2 Palladium coupling
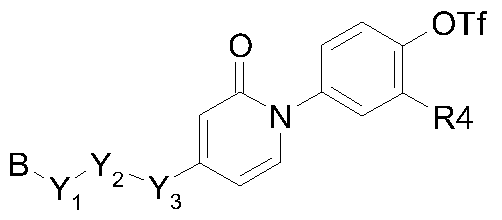

The copper coupling reaction is performed in the presence of a copper salt, such as CuI, in an aprotic solvent such as 1,4-dioxane, in the presence of an amine, such as Λ/,Λ^-dimethylethylenediamine, in the presence of a base, such as potassium phospate, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 15 minutes at 180 0C under microwave irradiation. The OH-activation can be done via trifiate, in the presence of a sulphonilating agent, such as N-phenyltrifluoromethanesulfonimide,, in an aprotic solvent such as THF, in the presence of a base, such as potassium carbonate, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 15 minutes at 120 0C under microwave irradiation. The palladium coupling reaction is performed in an aprotic solvent such as toluene or dioxane in the presence of a palladium catalyst such as Pd(AcO)2, or Pd(PPh3^ and in the presence of a base, such as tBuOK or cessium carbonate, or inorganic aqueous base such as Na2CO3, and sometimes a ligand, such as BINAP or XANTPHOS, is required, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 24 hours at 100 0C under traditional heating.
Compounds according to the invention with substituents other than benzyloxy are prepared according to Scheme 3 and Scheme 4.
Scheme 3

Scheme 4

Hydrogenation is performed under hydrogen pressure, in a protic or aprotic solvent, such as EtOH, or EtOAc, in the presence of palladium as a catalyst. Alkylation is performed in the presence of an organic or inorganic base, such as NaH, or DBU, in an aprotic solvent such as CH3CN, or DMF at a convenient temperature to completion of the reaction, typically 130 0C for 30 minutes under microwave irradiation. A Mitsun- obu type reaction can be performed in the presence of a phosphine, such as triphenyl- phosphine, an azodicarboxylate derivative, such as diethylazodicarboxilate in an aprotic solvent, such as THF, at a convenient temperature and time to completion of the reaction, such as 100 0C for 5 minutes under microwave irradiation. Halogenation is per- formed with a suitable halogenating agent, such as POBr3, in a suitable solvent, such as DMF, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically overnight at 100 0C under traditional heating. The palladium coupling reaction of boronic acids is performed in an aprotic solvent such as dioxane in the presence of a palladium catalyst such as Pd(PPh3)4 and in the presence of an inorganic aqueous base such as Na2CO3, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 10 minutes at 150 0C under microwave heating. The copper coupling reaction with boronic acids is performed in the presence of a copper salt, such as Cu(OAc)2, in an aprotic solvent such as 1 ,2-dichloromethane, in the presence of an amine and amine TV-oxide, such as pyridine and TEMPO (2,2,6,6-tetramethyl-l-piperidinyloxy), at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically room temperature overnight. The reduction in performed with an appropriate reducting agent, such as NaBH4, in the presence of a suitable solvent such as MeOH at room temperature for a period of time to ensure the completion of the reaction. The palladium coupling with amines is performed in an aprotic solvent such as toluene or dioxane in the presence of a palladium catalyst such as Pd(AcO)2 , or Pd(PPh3)4 and in the presence of a base, such as tBuOK or cessium carbonate, or inorganic aqueous base such as Na2CO3, and sometimes a ligand, such as BINAP or XANTPHOS, is required, at a convenient temperature, either by conventional heating or under microwave irradiation, for a period of time to ensure the completion of the reaction, typically 24 hours at 100 0C under traditional heating.
Scheme 5
Br
.->N
Br'
Amine substitution
Copper Coupling
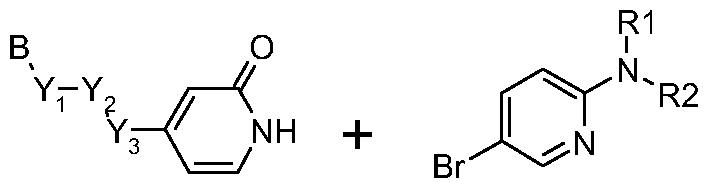

Compounds with 'A' being a pyridinyl can be prepared according to the above scheme 5. The amine substitution is preferably performed using an aprotic solvent, such as DMF, in the presence of the nucleophilic amine, and a base such as DIPEA or DBU, heating at a convenient temperature under traditional heating or microwave irradiation, for a period of time to ensure the completion of the reaction. The copper coupling reaction is performed using an aprotic solvent, such as dioxane or DMF, in the presence of CuI, an inorganic base such K3PO4 and MeNH(CH^)2NHMe as ligand, heating at a convenient temperature under traditional heating or microwave irradiation, for a period of time to ensure the completion of the reaction, typically 25 minutes at 175 0C under microwave irradiation.
Pharmacology
A compound according to the invention, in particular compounds according to
Formula (I), the pharmaceutically acceptable acid or base addition salts thereof, an
TV-oxide form thereof or a quaternary ammonium salt thereof, have surprisingly been shown to have a binding affinity towards the MCH-receptor, in particular towards the MCH-I receptor, in particular as an antagonist.
In view of their above mentioned potency, a compound according to the invention are suitable for the prevention and/or treatment of diseases where antagonism of the
MCH-receptor, in particular antagonism of the MCH-I receptor is of therapeutic use.
In particular, a compound according to the invention may be suitable for treatment and/or prophylaxis of psychiatric disorders, including but not limited to : anxiety, including but not limited to agoraphobia ; generalized anxiety ; compul-
sion ; obsessive-compulsive disorder ; panic disorder ; social phobia ; and stress, such a post-traumatic stress ; attention defϊcit/hyperactivity disorder ; autism ; - dysthymia ; eating disorder, including but not limited to anorexia ; binge eating ; and bulimia nervosa ; - impulse control disorder ; mental retardation, including but not limited to fragile X syndrome ; - mood disorder, including but not limited to agitation ; bipolar disorder, such as bipolar affective disorder, bipolar disorder (I), bipolar disorder (II), hypomania and mania ; depression, such as major depression and suicidal depression ; seasonal mood disorder ; and suicide ; premenstrual syndrome, including but not limited to dysphoria ; - psychosis, including but not limited to aggressiveness ; drug-induced psychosis ; schizoaffective disorder ; schizophrenia, such as delusion, catatonia, catatonic schizophrenia, disorganized schizophrenia, paranoid schizophrenia, residual schizophrenia and schizophreniform disorder ; and dyssomnia, such as secondary dys- somnia ; - sleep disorder, including but not limited to circadian rhythm disorder ; hypersomnia ; insomnia ; narcolepsy and sleep apnea ; stuttering ; and violence.
Additionally, a compound according to the invention may be used for treating sexual disorders, neurological disorders, and most in particular obesity and diabetes.
The invention therefore relates to a compound according to the general Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof or a quaternary ammonium salt thereof, for use as a medicine.
The invention also relates to the use of a compound according to the invention for the preparation of a medicament for the prevention and/or treatment of diseases where antagonism of the MCH-receptor, in particular antagonism of the MCH-I receptor is of therapeutic use.
The invention also relates to the use of a compound according to the invention for the preparation of a medicament for the prevention and/or treatment of anxiety, eating disorders, mood disorders, such as bipolar disorders and depression, psychoses, such as schizophrenia, and sleeping disorders. Additionally, a compound according to the invention may be used for treating sexual disorders and neurological disorders, and in particular obesity and diabetes.
Combination treatments
The compounds according to the invention, in particular according to Formula (I) may be co-administered as add-on treatment and/or prophylaxis in the above listed diseases.
With antidepressants, anxiolytics and/or antipsychotics.
In particular the compounds according to the invention, in particular according to Formula (I) may be co-administered in combination with antidepressants, anxiolytics and/or antipsychotics which are currently available or in development or which will become available in the future, in particular to improve efficacy and/or onset of action. It will be appreciated that the compounds of the present invention and the other agents may be present as a combined preparation for simultaneous, separate or sequential use for the prevention and/or treatment of depression and/or anxiety. Such combined preparations may be, for example, in the form of a twin pack. It will also be appreciated that the compounds of the present invention and the other agents may be administered as separate pharmaceutical compositions, either simultaneously or sequentially.
The invention therefore relates to a pharmaceutical composition according to the invention, characterized in that is comprises further one or more other compounds se- lected from the group of antidepressants, anxiolytics and antipsychotics.
Suitable classes of antidepressant agents include norepinephrine reuptake inhibitors, selective serotonin reuptake inhibitors (SSRTs), monoamine oxidase inhibitors (MAOI's), reversible inhibitors of monoamine oxidase (RIMA's), serotonin and noradrenaline reuptake inhibitors (SNRTs), noradrenergic and specific serotonergic an- tidepressants (NaSSA's), corticotropin releasing factor (CRF) antagonists, α-adrenoreceptor antagonists and atypical antidepressants.
Suitable examples of norepinephrine reuptake inhibitors include amitriptyline, clomipramine, doxepin, imipramine, trimipramine, amoxapine, desipramine, maprotiline, nortriptyline, protriptyline, reboxetine and pharmaceutically acceptable salts thereof. Suitable examples of selective serotonin reuptake inhibitors include fluoxetine, fluvoxamine, paroxetine, sertraline and pharmaceutically acceptable salts thereof.
Suitable examples of monoamine oxidase inhibitors include isocarboxazid, phenelzine, tranylcypromine, selegiline and pharmaceutically acceptable salts thereof.
Suitable examples of reversible inhibitors of monoamine oxidase include mo- clobemide and pharmaceutically acceptable salts thereof.
Suitable examples of serotonin and noradrenaline reuptake inhibitors include venlafaxine and pharmaceutically acceptable salts thereof.
Suitable atypical antidepressants include bupropion, lithium, nefazodone, trazodone, viloxazine, sibutramine and pharmaceutically acceptable salts thereof. Other suitable antidepressants include adinazolam, alaproclate, amineptine, amitriptyline/chlordiazepoxide combination, atipamezole, azamianserin, bazinaprine, befuraline, bifemelane, binodaline, bipenamol, brofaromine, bupropion, caroxazone, cericlamine, cianopramine, cimoxatone, citalopram, clemeprol, clovoxamine, dazepinil, deanol, demexiptiline, dibenzepin, dothiepin, droxidopa, enefexine, estazolam, etoperi- done, femoxetine, fengabine, fezolamine, fiuotracen, idazoxan, indalpine, indeloxazine, iprindole, levoprotiline, litoxetine, lofepramine, medifoxamine, metapramine, metralin- dole, mianserin, milnacipran, minaprine, mirtazapine, monirelin, nebracetam, nefopam, nialamide, nomifensine, norfluoxetine, orotirelin, oxaflozane, pinazepam, pirlindone, pizotyline, ritanserin, rolipram, sercloremine, setiptiline, sibutramine, sulbutiamine, sulpiride, teniloxazine, thozalinone, thymoliberin, tianeptine, tiflucarbine, tofenacin, tofisopam, toloxatone, tomoxetine, veralipride, viqualine, zimelidine and zometapine and pharmaceutically acceptable salts thereof, and St. John's wort herb, or Hypericum perforatum, or extracts thereof.
Suitable classes of anti-anxiety agents include benzodiazepines and 5-HTIA recep- tor agonists or antagonists, especially 5-HTIA partial agonists, corticotropin releasing factor (CRF) antagonists, compounds having muscarinic cholinergic activity and com-
pounds acting on ion channels. In addition to benzodiazepines, other suitable classes of anti-anxiety agents are nonbenzodiazepine sedative-hypnotic drugxs such as Zolpidem; mood-stabilizing drugs such as clobazam, gabapentin, lamotrigine, loreclezole, oxcar- bamazepine, stiripentol and vigabatrin; and barbiturates. Suitable antipsychotic agents are selected from the group consisting of aceto- phenazine, in particular the maleate salt; alentemol, in particular the hydrobromide salt; alpertine; azaperone; batelapine, in particular the maleate salt; benperidol; benzin- dopyrine, in particular the hydrochloride salt; brofoxine; bromperidol; butaclamol, in particular the hydrochloride salt; butaperazine; carphenazine, in particular the maleate salt; carvotroline, in particular the hydrochloride salt; chlorpromazine; chlorprothixene; cinperene; cintriamide; clomacran, in particular the phosphate salt; clopenthixol; clopi- mozide; clopipazan, in particular the mesylate salt; cloroperone, in particular the hydrochloride salt; clothiapine; clothixamide, in particular the maleate salt; clozapine; cyclophenazine, in particular the hydrochloride salt; droperidol; etazolate, in particular the hydrochloride salt; fenimide; fiucindole; fiumezapine; fluphenazine, in particular the decanoate, enanthate and/or hydrochloride salts; fluspiperone; fluspirilene; flutro- line; gevotroline, in particular the hydrochloride salt; halopemide; haloperidol; iloperi- done; imidoline, in particular the hydrochloride salt; lenperone; loxapine; mazapertine, in particular the succinate salt; mesoridazine; metiapine; milenperone; milipertine; molindone, in particular the hydrochloride salt; naranol, in particular the hydrochloride salt; neflumozide, in particular the hydrochloride salt; ocaperidone; olanzapine; oxiperomide; paliperidone; penfluridol; pentiapine, in particular the maleate salt; perphenazine; pimozide; pinoxepin, in particular the hydrochloride salt; pipamperone; piperacetazine; pipotiazine, in particular the palmitate salt; piquindone, in particular the hydrochloride salt; prochlorperazine, in particular the edisylate salt; prochlorperazine, in particular the maleate salt; promazine, in particular the hydrochloride salt; quetiap- ine; remoxipride; risperidone; rimcazol, in particular the hydrochloride salt; seperidol, in particular the hydrochloride salt; sertindole; setoperone; spiperone; sulpiride; thioridazine; thiothixene; thorazine; tioperidone, in particular the hydrochloride salt; tiospi- rone, in particular the hydrochloride salt; trifluoperazine, in particular the hydrochloride salt; trifluperidol; triflupromazine; ziprasidone, in particular the hydrochloride salt; and mixtures thereof.
Lipid-lowering compounds
The compounds according to the invention, in particular according to Formula (I) may also be used in conjunction with other lipid-lowering agent, thus leading to a so-called combination lipid-lowering therapy for the treatment of obesity. The said ad- ditional lipid-lowering agent may be, for instance, a known drug conventionally used for the management of hyperlipidaemia such as e.g. a bile acid sequestrant resin, a fi- bric acid derivative or nicotinic acid as previously mentioned in the background of the invention. Suitable additional lipid-lowering agents also include other cholesterol biosynthesis inhibitors and cholesterol absorption inhibitors, especially HMG-CoA reduc- tase inhibitors and HMG-CoA synthase inhibitors, HMG-CoA reductase gene expression inhibitors, CETP inhibitors, ACAT inhibitors, squalene synthetase inhibitors, CB-I antagonists, cholesterol absorption inhibitors such as ezetimibe, and the like.
Any HMG-CoA reductase inhibitor may be used as the second compound in the combination therapy aspect of this invention. The term "HMG-CoA reductase inhibi- tor" as used herein, unless otherwise stated, refers to a compound which inhibits the biotransformation of hydroxymethylglutaryl-coenzyme A to mevalonic acid as catalyzed by the enzyme HMG-CoA reductase. Such "HMG-CoA reductase inhibitors" are, for example, lovastatin, simvastatin, fiuvastatin, pravastatin, rivastatin, and atorvastatin.
Any HMG-CoA synthase inhibitor may be used as the second compound in the combination therapy aspect of this invention. The term "HMG-CoA synthase inhibitor" as used herein, unless otherwise stated, refers to a compound which inhibits the biosynthesis of hydroxymethylglutaryl-coenzyme A from acetyl-coenzyme A and acetoace- tyl-coenzyme A, catalyzed by the enzyme HMG-CoA synthase
Any HMG-CoA reductase gene expression inhibitor may be used as the second compound in the combination therapy aspect of this invention. These agents may be HMG-CoA reductase trancription inhibitors that block the transcription of DNA or translation inhibitors that prevent translation of mRNA coding for HMG-CoA reductase into protein. Such inhibitors may either affect trancription or translation directly or may be biotransformed into compounds having the above-mentioned attributes by one or more enzymes in the cholesterol biosynthetic cascade or may lead to accumulation of a metabolite having the above-mentioned activities.
Any CETP inhibitor may be used as the second compound in the combination therapy aspect of this invention. The term "CETP inhibitor" as used herein, unless oth-
erwise stated, refers to a compound which inhibits the cholesteryl ester transfer protein (CETP) mediated transport of various cholesteryl esters and triglycerides from HDL to LDL and VLDL.
Any ACAT inhibitor may be used as the second compound in the combination therapy aspect of this invention. The term "ACAT inhibitor" as used herein, unless otherwise stated, refers to a compound which inhibits the intracellular esterification of dietary cholesterol by the enzyme acyl CoAxholesterol acyltransferase.
Any squalene synthetase inhibitor may be used as the second compound in the combination therapy aspect of this invention. The term "squalene synthetase inhibitor" as used herein, unless otherwise stated, refers to a compound which inhibits the condensation of two molecules of farnesylpyrophosphate to form squalene, catalyzed by the enzyme squalene synthetase.
Pharmaceutical compositions The invention also relates to a pharmaceutical composition comprising a pharmaceutically acceptable carrier or diluent and, as active ingredient, a therapeutically effective amount of a compound according to the invention, in particular a compound according to Formula (I), a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof or a quaternary ammonium salt thereof. A compound according to the invention, in particular a compound according to
Formula (I), the pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof or a quaternary ammonium salt thereof , or any subgroup or combination thereof may be formulated into various pharmaceutical forms for administration purposes. As appropriate compositions there may be cited all compositions usually employed for systemically administering drugs.
To prepare the pharmaceutical compositions of this invention, an effective amount of the particular compound, optionally in addition salt form, as the active ingredient is combined in intimate admixture with a pharmaceutically acceptable carrier, which carrier may take a wide variety of forms depending on the form of preparation desired for administration. These pharmaceutical compositions are desirable in unitary dosage form suitable, in particular, for administration orally, rectally, percutaneously, by parenteral injection or by inhalation. For example, in preparing the compositions in
oral dosage form, any of the usual pharmaceutical media may be employed such as, for example, water, glycols, oils, alcohols and the like in the case of oral liquid preparations such as suspensions, syrups, elixirs, emulsions and solutions; or solid carriers such as starches, sugars, kaolin, diluents, lubricants, binders, disintegrating agents and the like in the case of powders, pills, capsules and tablets. Because of their ease in administration, tablets and capsules represent the most advantageous oral dosage unit forms in which case solid pharmaceutical carriers are obviously employed. For parenteral compositions, the carrier will usually comprise sterile water, at least in large part, though other ingredients, for example, to aid solubility, may be included. In- jectable solutions, for example, may be prepared in which the carrier comprises saline solution, glucose solution or a mixture of saline and glucose solution. Injectable suspensions may also be prepared in which case appropriate liquid carriers, suspending agents and the like may be employed. Also included are solid form preparations that are intended to be converted, shortly before use, to liquid form preparations. In the compositions suitable for percutaneous administration, the carrier optionally comprises a penetration enhancing agent and/or a suitable wetting agent, optionally combined with suitable additives of any nature in minor proportions, which additives do not introduce a significant deleterious effect on the skin. Said additives may facilitate the administration to the skin and/or may be helpful for preparing the desired compositions. These compositions may be administered in various ways, e.g., as a transdermal patch, as a spot-on, as an ointment.
It is especially advantageous to formulate the aforementioned pharmaceutical compositions in unit dosage form for ease of administration and uniformity of dosage. Unit dosage form as used herein refers to physically discrete units suitable as unitary dosages, each unit containing a predetermined quantity of active ingredient calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier. Examples of such unit dosage forms are tablets (including scored or coated tablets), capsules, pills, powder packets, wafers, suppositories, injectable solutions or suspensions and the like, and segregated multiples thereof. Since the compounds ac- cording to the invention are potent orally administrable dopamine antagonists, pharmaceutical compositions comprising said compounds for administration orally are especially advantageous.
As already mentioned, the invention also relates to a pharmaceutical composition comprising a compound according to the invention and one or more other compounds selected from the group of antidepressants, anxiolytics, antipsychotics and lipid- lowering agents as well as to the use of such a composition for the manufacture of a medicament.
Depending on the mode of administration, the pharmaceutical composition will include from about 0.05 weight % (weight % = percent by weight) to about 99 weight %, more particular, from about 0.10 weight % to about 99 weight %, of the compounds of the invention, all percentages by weight being based on the total weight of the com- position. When the pharmaceutical composition comprises the compounds according to the invention and one or more other compounds selected from the group of antidepressants, anxiolytics, antipsychotics and lipid-lowering agents, both the compounds according to the invention and the one or more other compounds may be present in a concentration from about 0.05 weight % (weight % = percent by weight) to about 99 weight %, more particular, from about 0.10 weight % to about 99 weight %, all percentages by weight being based on the total weight of the composition. For example, the weight ratio of the compounds according to the invention to the one or more other compounds may be in the range of 0.05/0.94 to 0.94/0.05, more in particular 0.10/0.89 to 0.89/0.10, and any number therein between.
Experimental part
The following examples are intended to illustrate but not to limit the scope of the present invention.
Hereinafter, "THF" means tetrahydrofuran; "DMF" means 7V,7V-dimethyl- formamide; "EtOAc" means ethyl acetate; "DCM" means dichloromethane; "DME" means 1,2-dimethoxyethane; "DCE" means 1 ,2-dichloroethane; "DIPE" means diiso- propylether; "DMSO" means dimethylsulfoxide; "boc" means tert-butoxy carbonyl; "Cu(OAc)2" means copper acetate; "BINAP" is l,l'-[l,l'-binaphthalene]- 2,2'-diylbis[l,l]diphenyl-phosphine; "DBU" means l,8-diaza-7-bicyclo[5.4.0]- undecene; "DIPEA" means N-ethyl-N-(l-methylethyl)-2-propanamine; "XANTPHOS" means 4,5-Bis(diphenylphosphino)-9,9-dimethylxanthene ; "TEMPO" means 2,2,6,6-tetramethyl-l-piperidinyloxy ; and "CELITE" is a diatomaceous earth.
Microwave assisted reactions were performed in a single-mode reactor: Initia- tor™ Sixty EXP microwave reactor (Biotage AB). Description of the instrument can be found in www.biotage.com. And in a multimode reactor: MicroSYNTH Labstation
(Milestone, Inc.). Description of the instrument can be found in www.milestonesci.com.
A. Preparation of the intermediate compounds Al. Preparation of intermediate compound 1-1

To a solution of 4-benzyloxy-2(lH)-pyridone (2 g, 10.0 mmol) in DCM (20 ml), was added 4-bromophenylboronic acid (4 g, 24.0 mmol), Cu(OAc)2 (0.360 g, 2.0 mmol), pyridine (1.51 ml, 20.0 mmol), TEMPO (1.72 g, 11 mmol) and molecular sieves (4 A)
(2 g). The reaction mixture was stirred overnight at room temperature. The solid was filtered off. The filtrate was treated with an aqueous solution of NH4OH. The organic layer was separated, dried (Na2SO4) and the solvent evaporated. The resulting residue was purified by column chromatography (DCM 100 to DCM/EtOAc 4/1). The desired fractions were collected and the solvent was evaporated. The residue was treated with DIPE to yield intermediate compound 1-1 (3.16 g, 89 %).
A2. Preparation of intermediate 1-2

1-2
A mixture of 4-benzyloxy-2(7H)-pyridone (0.5 g, 2.5 mmol),
4-bromo-2-methoxyphenol (0670 g, 3.3 mmol), CuI (0.480 g, 2.5 mmol), 7V,7V'-dimethylethylenediamine (0.530 ml, 5.0 mmol) and K3PO4 (1.06 g, 5.0 mmol) in dioxane/DMF (5/1) was heated for 15 minutes at 180 0C in a microwave oven. Then DCM was added. The solid was filtered off through a CELITE pad and to the filtrate, a 32 % NH3 solution was added. The organic layer was separated, washed with brine, dried (Na2SO4), filtered and the solvent was evaporated. The residue was purified by column chromatography over silica gel (eluent: DCM/MeOH 98/2 and 96/4). The product fractions were collected and the solvent was evaporated. The residue was treated with DIPE to yield intermediate compound 1-2 (0.760 g, 31%).
A3. Preparation of intermediate compound 1-3

A4. Preparation of intermediate compound 1-4

A solution of final compound 1-11 (0.798 g, 2.0 mmol) in ethanol was hydrogenated using Pd/C as a catalyst at room temperature for 4 hours. Then, the catalyst was filtered off through a CELITE pad and the filtrate solvent was evaporated to yield intermediate compound 1-4 (0.570 g, 93 %).
A5. Preparation of intermediate 1-5

To a mixture of intermediate compound 1-4 (0.570 g, 1.9 mmol) in DMF, POBr3 (0.708 g, 2.47 mmol) was added dropwise at 0 0C. The reaction was stirred for 5 hours at
100 0C. Then, additional POBr3 (0.272 g, 0.95 mmol) was added. The reaction was
heated at 100 0C overnight. Then the reaction was poured into ice water. The solvent was evaporated and the residue was purified by column chromatography over silica gel (eluents: DCM/MeOH saturated with ammonia 9/1). The product fractions were collected and the solvent was evaporated to yield intermediate compound 1-5 (0.220 g, 33 %).
A6. Preparation of intermediate 1-6

DBU
1-6
A mixture of 2,5-dibromopyridine (0.4 g, 0.0016 mol), 3-(dimethylamino)pyrrolidine (0.19 g, 0.0016 mol) and DBU (0.263 ml, 0.0017 mol) in DMF (2 ml) was stirred in microwave at 250 0C for 30 minutes. The solvent was evaporated and the residue was purified by short column chromatography over silica gel (eluent: DCM/MeOH 98/2), yielding 0.59 g of intermediate compound 1-6 (100 %).
A7. Preparation of intermediate 1-7
DIPEA
1-7
A mixture of 2,5-dibromopyridine (0.40 g, 0.0016 mol), (3S)-(-)-3-(methylamino) pyrrolidine (0.16 g, 0.0016 mol) in DIPEA (2 ml) and DMF (0.200 ml) was heated at 150 0C for 5 minutes, due to the reaction was not finished the reaction mixture was stirred in a microwave at 175 0C for 15 minutes more. Then, DCM was added and the reaction mixture was treated with NaHCO3 (aqueous saturated solution). The organic layer was separated, dried (Na2SO4) and the solvent evaporated. The residue was puri-
fled by flash column chromatography over silica gel (eluent: DCM/MeOH) 98/2 and 95/5; and DCM/(MeOH/NH3) 95/5), yielding 0.151 g of intermediate compound 1-7
(37 %).
A8. Preparation of intermediate 1-8
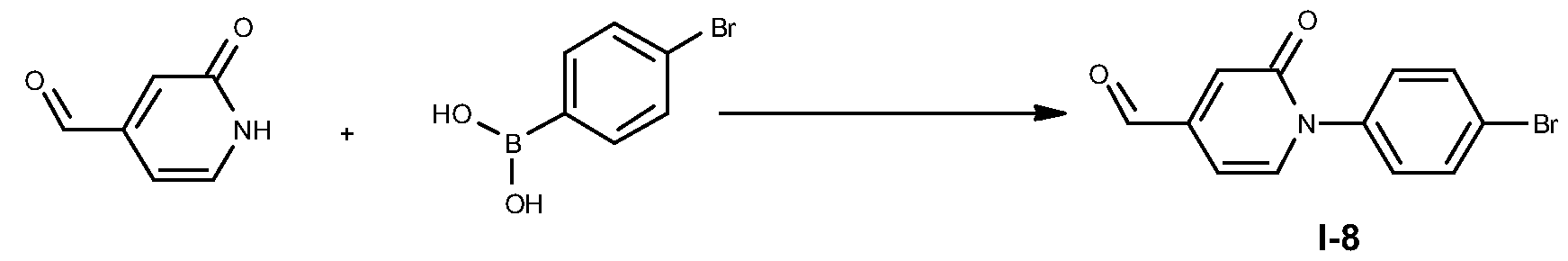
To a solution of 2-oxo-l,2-dihydro-pyridine-4-carbaldehyde (0.123 g, 1.0 mmol) in DCM (2 ml), was added 4-bromophenylboronic acid (0.40 g, 2.0 mmol), Cu(OAc)2 (0.018 g, 0.1 mmol), pyridine (10.162 ml, 2.0 mmol), TEMPO (0.172 g, 1.1 mmol) and molecular sieves (4 A) (0.2 g). The reaction mixture was stirred overnight at room temperature. The solid was filtered off. The filtrate was treated with an aqueous solution of NH4OH. The organic layer was separated, dried (Na2SO4) and the solvent evaporated. The resulting residue was purified by column chromatography in DCM 100 to DCM/EtOAc 4/1. Selected fractions were collected and the solvent evaporated to yield intermediate compound 1-8 (0.180 g, 65 %).
A9. Preparation of intermediate 1-9

To a solution of intermediate 1-8 (0.180 g, 0.65mmol) in MeOH (5.0ml), NaBH4 (0.030 g, 0.78 mmol) was added portionwise at 0 0C. The reaction was stirred for 30min at room temperature. Then a saturated solution OfNH4Cl was added, the mixture was extracted with DCM, the organic layer was separated, dried (Na2SO4) and the solvent evaporated. The residue was precipitated with DIPE to yield intermediate compound 1-9 (0.17O g, 93 %).
AlO. Preparation of intermediate I- 10

1-9 1-10
To a solution of intermediate compound 1-9 (0.170 g, 0.61 mmol) in THF, triphenyl- phosphine (0.241 g, 0.92 mmol), phenol (0.087 g, 0.92 mmol) and diisopropyl azodi- carboxilate (0.181 ml, 0.92 mmol) were added. The reaction was heated at 100 0C for 10 minutes in a microwave oven. Then, the solvent was evaporated and the residue was purified by column chromatography (eluents: DCM and DCM/EtOAc 9/1). The product fractions were collected and the solvent was evaporated. The residue was treated with DIPE to yield intermediate compound I- 10 (0.151 g, 70 %).
Al l. Preparation of intermediate I- 11

1-11 A mixture of 4-benzyloxy-2(lH)-pyridone (0.100 g, 0.50 mmol), 2-bromo-5-iodotoluene (0223 g, 0.75 mmol), CuI (0.048 g, 0.25 mmol), N,N'-dimethylethylenediamine (0.053 ml, 0.5 mmol) and K3PO4 (0.212 g, 1.0 mmol) in dioxane/DMF (4/1) was heated for 15 minutes at 180 0C in a microwave oven. Then DCM was added. The solid was filtered off through a CELITE pad and to the filtrate, a 32 % NH3 solution was added. The organic layer was separated, washed with brine, dried (Na2SO4), filtered and the solvent was evaporated. The residue was purified by column chromatography over silica gel (eluent: DCM). The product fractions were collected and the solvent was evaporated. The residue was treated with DIPE to yield intermediate compound 1-11 (0.130 g, 70 %).
B. Preparation of the final compounds Bl. Preparation of final compound 1-10

To a solution of intermediate compound 1-1 (75 mg, 0.21 mmol) in toluene (previously de-oxygenated) (2 ml) was added 3-[(tert-butoxycarbonyl)(methyl)amino]pyrrolidine (37 mg, 0.32 mmol), Pd(AcO)2 (2.5 mg, 0.011 mmol), BINAP (10 mg, 0.016 mmol) and tBuOK (31 mg, 0.32 mmol). The reaction was heated at 100 0C under nitrogen for 24 hours. The solid was filtered off and the filtrate was concentrated under vacuum. The resulting crude was filtered through silica gel using DCM/ Acetone 2:1. The solvent was evaporated till dryness and the residue was dissolved in DCM (2 ml) and then trifluoroacetic acid (1 ml) was added. The reaction mixture was stirred for 24 hours at room temperature. Then, a solution of aqueous Na2CO3 was added. The organic layer was extracted and dried (Na2SO4), and the solvent evaporated in vacuo. The residue was purified by column chromatography (DCM/MeOH 96/4 to DCM/MeOH(NH3) 96/4) to yield 19 mg of final compound 1-10 (24 %).
B2. Preparation of final compound 1-11

To a solution of intermediate compound 1-1 in toluene (previously deoxygenated) (2 ml) was added 3-dimethylaminopyrrolidine (37 mg, 0.32 mmol), Pd(AcO)2 (2.5 mg, 0.011 mmol), BINAP (10 mg, 0.016 mmol) and ffiuOK (31 mg, 0.32 mmol). The reaction was heated at 100 0C under nitrogen for 24 hours. The solid was filtered off and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (DCM/MeOH 96/4 and DCM/MeOH(NH3) 96/4) to yield 56 mg of final compound 1-11 (68 %).
B3. Preparation of final compound 1-12

To a solution of intermediate compound 1-1 (75 mg, 0.21 mmol) in toluene (previously deoxygenated) (2 ml) was added 3-(Λ/-acetyl-Λ/-methylamino)pyrrolidine (46 mg, 0.32 mmol), Pd(AcO)2 (2.5 mg, 0.011 mmol), BINAP (10 mg, 0.016 mmol) and ffiuOK (31 mg, 0.32 mmol). The reaction was heated at 100 0C under nitrogen for 24 hours. The solid was filtered off and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (DCM/EtOAc 4/1 and DCM/ Acetone 2/1) to yield 52 mg of final compound 1-12 (59 %).
B4. Preparation of final compound 2-2

To a solution of intermediate compound 1-3 (0.200 g, 0.44 mmol) in trifluoro toluene (previously deoxygenated) were added 3-(dimethylamino)pyrrolidine (0.101 g, 0.88 mmol), Pd(AcO)2 (0.01Og mg, 0.044 eq), XANTPHOS (0.051 g, 0.088 mmol) and Cs2CO3 (0.215 g, 0.66 mmol). The reaction was heated at 100 0C under nitrogen for 24 hours. The solid was filtered off through a CELITE pad and the filtrate was concentrated under vacuum. The residue was purified by column chromatography (eluent: DCM/MeOH 98/2, 95/5 and DCM/MeOH saturated with ammonia 96/4). The product fractions were collected and the solvent was evaporated. The residue was treated with DIPE to yield final compound 2-2 (0.036 g, 20 %).
B5. Preparation of final compound 3-1

1-4 3-1
To a solution of intermediate compound 1-4 (0.100 g, 0.33 mmol) in THF, triphenyl- phosphine (0.160 g, 0.59 mmol), 4-fluorobenzyl alcohol (0.090 ml, 0.83 mmol) and diisopropyl azodicarboxilate (0.144 g, 0.63 mmol) were added. The reaction was heated in a microwave oven for 20 minutes at 120 0C. Then, the solvent was evaporated and the residue was purified by column chromatography (eluent: DCM and DCM/MeOH saturated with ammonia 9/1). The product fractions were collected and the solvent was evaporated to yield final compound 3-1 (0.077 g, 56 %).
B6. Preparation of final compound 3-3

To a solution of intermediate compound 1-5 (0.110 g, 0.30 mmol) in dioxane (1.0 ml) (previously de-oxygenated), trαfts-2-phenylvinylboronic acid (0.045 g, 0.30 mmol), tetrakis(triphenylphosphine)palladium(0) (17 mg, 0.015 mmol) and a saturated solution OfNa2CO3 (0.5 ml) were added. The reaction was heated for 10 minutes at 150 0C under microwave irradiation. Then, water and EtOAc were added. Next, the organic layer was separated, dried (Na2SO4), filtered and the solvent was evaporated. The residue was purified by column chromatography (DCM/MeOH saturated with ammonia 9/1 and 4/1). The product fractions were collected and the solvent was evaporated to yield final compound 3-3 (0.090 g, 78 %).
B7. Preparation of final compound 4-1

A mixture of intermediate compound 1-6 (0.2 g, 0.00099 mol), 4-benzyloxy-2(7H)-pyridone (0.32 g, 0.0011 mol), CuI (0.18 g, 0.00099 mol), N,N'-dimethylethylenediamine (0.211 ml, 0.0019 mol, and K3PO4 (0.4 g, 0.0019 mol) in dioxane/DMF 9:1 (12 ml) was stirred in microwave at 175 0C for 20 minutes. Then DCM was added. The solid was filtered through celite and the filtrate was washed with aqueous solution NH4Cl. The organic layer was separated, dried (Na2SO4) and the sol-
vent evaporated. The residue was purified by short column chromatography over silica gel (eluent: DCM/MeOH from 98/2 till 90/10) to give a product, which was further purified by HPLC (Column: Sunfϊre Prep C 18, 5μm particle size. Eluent: Formic acid (0.1 %) and CH3CN). Finally the residue was dissolved in DCM and filtered through Isolute SPE columns "Si-Carbonate". Yield: 0.080 g of pure compound 4-1 (0.080 g; 21 %).
B8. Preparation of final compound 4-2

4-2 A mixture of 4-benzyloxy-2(lH)-pyridone (0.119 g, 0.00059 mol), intermediate compound 1-7 (0.151 g, 0.00059 mol), CuI (0.112 g, 0.00059 mol), ΛζiV-dimethylethylenediamine (0.126 ml, 0.00118 mol), and K3PO4 (0.250 g, 0.00118 mol) in dioxane:DMF 9:1 (2.5 ml) was stirred at 185 0C for 15 minutes. Then, CH2Cl2 was added. The solid was filtered off through celite and the filtrate was treated with NH3 (30 %). The organic layer was separated, dried (Na2SO4) and the solvent evaporated. The residue was purified by flash column chromatography over silica gel (eluent: DCM(MeOH) 95/5 and DCM/(MeOH/NH3) 95/5). Afterwards the residue was treated with ethyl ether, yielding 0.127 g of final compound 4-2 (56 %; S-enantiomer).
B9. Preparation of final compound 3-8
tBuOK


3-8
To a solution of intermediate compound I- 10 in toluene (previously deoxygenated) (2 ml) was added 3-(dimethylamino)pyrrolidine (96 mg, 0.84 mmol), Pd(AcO)2 (4.7 mg, 0.021 mmol), BINAP (20 mg, 0.032 mmol) and tBuOK (101 mg, 1.05 mmol). The reaction was heated at 100 0C under nitrogen for 24 hours. The solid was filtered off and the filtrate was concentrated under vacuum. The residue was purified by column chromatography in DCM/MeOH 97/3 and DCM/MeOH(NH3) 97/3. The product fractions were collected and the solvent was evaporated. The residue was treated with ethyl ether to yield 55 mg of final compound 3-8 (34 %).
BlO. Preparation of final compound 2-1

To a solution of intermediate compound I- 11 in toluene (previously deoxygenated) (2 ml) was added 3-(dimethylamino)pyrrolidine (80 mg, 0.70 mmol), Pd(AcO)2 (4.0 mg, 0.018 mmol), BINAP (16 mg, 0.026 mmol) and tBuOK (85 mg, 0.88 mmol). The reaction was heated at 100 0C under nitrogen for 24 hours. The solid was filtered off and the filtrate was concentrated under vacuum. The residue was purified by column chromatography in DCM/MeOH 98/2 and 95/5. The product fractions were collected and the solvent was evaporated. The residue was treated with DIPE to yield 48 mg of final compound 2-1 (34 %).
Tables Ia, 2, 3a, 4 and 5 list the compounds of Formula (I), which were prepared according to one of the above described examples. In the column "Ex. Nr." the examples that are described in detail in the experimental section above, have been marked with "*". The analytical data for the compounds, measured according to one or more of the analytical procedures described below, are also present in these tables.
Tables Ib and 3b list the compounds of Formula (I), which are prepared according to one of the above described examples.
LCMS Procedures LCMS general procedure A
The HPLC gradient was supplied by a HP 1100 from Agilent Technologies comprising a pump (quaternary or binary) with degasser, an autosampler, a column oven, a diode-array detector (DAD) and a column as specified in the respective methods below. Flow from the column was split to a MS spectrometer. The MS detector was configured with an electrospray ionization source. Nitrogen was used as the nebulizer gas. The source temperature was maintained at 140 °C. Data acquisition was performed with MassLynx-Openlynx software.
LCMS general procedure B The HPLC gradient was supplied by an Alliance HT 2790 (Waters) system comprising a quaternary pump with degasser, an autosampler, a column oven (set at 40 0C), a diode-array detector (DAD) and a column as specified in the respective methods below. Flow from the column was split to a MS spectrometer. The MS detector was configured with an electrospray ionization source. Mass spectra were acquired by scanning from 100 to 1000 in 1 second using a dwell time of 0.1 second. The capillary needle voltage was 3 kV and the source temperature was maintained at 140 0C. Nitrogen was used as the nebulizer gas. Data acquisition was performed with a Waters-Micromass MassLynx-Openlynx data system.
LCMS method 1
In addition to general procedure A: Reversed phase HPLC was carried out on an ACE-C 18 column (3.0 μm, 4.6 x 30 mm) from Advanced Chromatography Technologies, with a flow rate of 1.5 ml/min, at 4O0C. The gradient conditions used are: 80 % A (0.5 g/1 ammonium acetate solution), 10 % B (acetonitrile), 10 % C (methanol) to 50 % B and 50 % C in 6.5 minutes, to 100 % B at 7 minutes and equilibrated to initial conditions at 7.5 minutes until 9.0 minutes. Injection volume 5 μl. High-resolution mass spectra (Time of Flight, TOF) were acquired only in positive ionization mode by scanning from 100 to 750 in 0.5 seconds using a dwell time of 0.1 seconds. The capillary needle voltage was 2.5 kV for positive ionization mode and the cone voltage was 20 V. Leucine-Enkephaline was the standard substance used for the lock mass calibration.
LCMS method 2
In addition to general procedure A: Same as LCMS method 1 using 10 μl of injection volume.
LCMS method 3
In addition to general procedure A: Reversed phase HPLC was carried out on an ACE-C 18 column (3.0 μm, 4.6 x 30 mm) from Advanced Chromatography Technologies, with a flow rate of 1.5 ml/min, at 4O0C. The gradient conditions used are: 80 % A (0.5 g/1 ammonium acetate solution), 10 % B (acetonitrile), 10 % C (methanol) to 50 % B and 50 % C in 6.5 minutes, to 100 % B at 7 minutes and equilibrated to initial conditions at 7.5 minutes until 9.0 minutes. Injection volume 5 μl. Low-resolution mass spectra (Waters ZQ™ detector; quadrupole) were acquired by scanning from 100 to 1000 in 1.0 second using a dwell time of 0.3 seconds. The capillary needle voltage was 3 kV. The cone voltage was 20 V and 50 V for positive ionization mode and 20 V for negative ionization mode.
LCMS method 4
In addition to general procedure B: Reversed phase HPLC was carried out on a Chromolith (4.6 x 25 mm) with a flow rate of 3 ml/min. Three mobile phases (mobile phase A: 95 % 25 mM ammoniumacetate + 5 % acetonitrile; mobile phase B: acetoni- trile; mobile phase C: methanol) were employed to run a gradient condition from 96 % A, 2 % B and 2 % C, to 2 % A, 49 % B and 49 % C in 0.9 minutes, to 100 % B in 0.3 minutes and hold for 0.2 minutes. An injection volume of 2 μl was used. Cone voltage was 10 V for positive ionization mode and 20 V for negative ionization mode.
Melting points (mp)
For a number of compounds, melting points were determined in open capillary tubes on a Mettler FP62 apparatus. Melting points were measured with a temperature gradient of 3 or 10 °C/minute. Maximum temperature was 300 0C. The melting point was read from a digital display.
Table Ia
1
1
1
1
1
1
1
1
1

1
1
3
1
1
1
 . Nr. Ex. Nr. Physico-chemical data
. Nr. Ex. Nr. Physico-chemical data
.CF3COOH -26 Bl LCMS: Rt 2.90, (MH)+ 362, method 1
-18 Bl
-19 B2 or LCMS: Rt 3.74, (MH)+ 376, method 1
-20 B2 LCMS: Rt 3.95, (MH)+ 404, method 1
-21 B2 LCMS: Rt 4.71, (MH)+ 402, method 1
O -22 B2 LCMS: Rt 3.75, (MH)+ 404, method 1
-23 B2 LCMS: Rt 5.18, (MH)+ 462, method 1
-24 B2 LCMS: Rt 5.30, (MH)+ 438, method 2
-25 B2 LCMS: Rt 4.03, (MH)+ 363, method 1
 Table Ib
Table Ib

Table 2

Table 3 a


Table 3b


Table 5
1

The interaction of the compounds of Formula (I) with MCH-I receptors was assessed in in vitro transient calcium (Ca2+) mobilization assays in the fiuorimetric imaging plate reader (FLIPR) format (Sullivan et al. 1999, Methods MoI Biol 114:125-133). In gen- eral, the natural agonist (MCH) is incubated with cells expressing the MCH-I receptor, which elicits a concentration-dependent transient mobilization of Ca2+ from internal stores. The interaction of the test compounds with the receptor is assessed in competition experiments. Various concentrations of the test compound are added to the incubation mixture containing the receptor-expressing cells and a submaximal concentra- tion of MCH. The test compound in proportion to its antagonist potency and its concentration inhibits MCH-induced Ca2+ mobilization.
Example Cl : Binding Experiment for MCH-I Cell culture and membrane preparation. Chinese Hamster ovary cells (CHO) stably expressing the human MCH-I receptor are grown in a 1:1 mixture of Dulbecco's Modified Eagles Medium (DMEM) and HAM's F 12 medium including Glutamax™ (Invitrogen), supplemented with 10 % heat-inactivated fetal bovine serum and 400 μg/ml geneticin.
Ca2+ mobilization experiment for the MCH-I receptor Twenty- four hours before the experiment, MCH-I receptor-expressing CHO cells are seeded in 20 μl (5,000 cells per well) into 384-well black wall, clear bottom microtiter plates (Costar). On the day of the experiment, 20 μl per well calcium assay kit containing 10 mM probenicide (Molecular Devices) is added. Cells are loaded for 90 min at 37 °C and 5 % CO2 in a cell culture incubator. After loading, 20 μl of serial dilutions of the test compound are added and cells are further incubated for 20 min at room temperature in the dark. After 20 min, 20 μl of a submaximal MCH concentration is added and changes in intracellular calcium are recorded directly in a FLIPR III apparatus (Molecular devices).
Data analysis and results
Data from assays in the presence of compound were calculated as a percentage of total
Ca +-responses measured in the absence of test compound. Inhibition curves, plotting percent of total Ca +-responses versus the log value of the concentration of the test compound, were automatically generated, and sigmoidal inhibition curves were fitted using non-linear regression. The pICso values of test compounds were derived from individual curves.
All compounds according to Formula (I) produced an inhibition of more than 50 % (pICso) at a test concentration ranging between 10"6 M and 10"9 M in a concentration-dependent manner.
For a selected number of compounds, covering most of the various embodiments of Formula (I), the results of the in vitro studies are given in Table 6.
Table 6. Pharmacological data for compounds according to the invention.



Example C.2 : Determination of brain concentrations
For the brain distribution study, three male SD rats (214 ± 10 g) were used for each time point. The dosing formulations were prepared by individually dissolving the compounds in a 10 % hydroxypropyl-β-cyclodextrin (HP-β-CD) solution at a final concentration of 1.25 mg/ml for the IV-formulations and they were made isotonic with manni- tol. The formulations were stored at room temperature, protected from light and were analysed quantitatively with LC-MS/MS on the day of preparation. The stability of these formulations was checked on the day of dosing. Dose administration was done intravenously in a tail vein at 2 ml/kg to obtain a dose of 2.5 mg/kg. Sample time points were 1 and 4 h after intravenous dose administration. Animals were sacrificed by decapitation and blood was collected by exsanguination in 10 ml B-D Sterile EDTA K3 Vacutainer tubes. From each individual animal the whole brain was dissected and weighed. Individual brain samples were homogenized in demineralized water (1/9 w/v or + 3 ml if tissue weight < 0.33 g). Homogenisation was carried out under dimmed light conditions. The brain samples were analysed using a qualified research LC-MS/MS method. The lower limit of quantification (LLOQ) was in the range of 20 - 50 ng/g. A limited pharmacokinetic analysis was performed using WinNonlin™ Pro- fessional (Version 4.0.1). A non-compartmental analysis using the lin/log trapezoidal rule with lin/log interpolation was used for all data. It was shown that the compound according to the invention showed a better brain concentration, and hence a better brain penetration, than the compounds from the prior art.
Table 7 : Comparison of brain levels.

D. Composition examples
"Active ingredient" (a.i.) as used throughout these examples relates to a final compound of formula (i), the pharmaceutically acceptable acid or base addition salts thereof, the stereochemically isomeric forms thereof, the TV-oxide form thereof, a quaternary ammonium salt thereof and prodrugs thereof.
Example D.I : oral drops
500 Grams of the a.i. is dissolved in 0.5 1 of 2-hydroxypropanoic acid and 1.5 1 of the polyethylene glycol at 60-80 °c. After cooling to 30-40 0C there are added 35 1 of polyethylene glycol and the mixture is stirred well. Then there is added a solution of
1750 grams of sodium saccharin in 2.5 1 of purified water and while stirring there are added 2.5 1 of cocoa flavor and polyethylene glycol q.s. to a volume of 50 1, providing an oral drop solution comprising 10 mg/ml of a.i. The resulting solution is filled into suitable containers.
Example D.2 : oral solution
9 Grams of methyl 4-hydroxybenzoate and 1 gram of propyl 4-hydroxybenzoate are dissolved in 4 1 of boiling purified water. In 3 1 of this solution are dissolved first 10 grams of 2,3-dihydroxybutanedioic acid and thereafter 20 grams of the a.i. The latter
solution is combined with the remaining part of the former solution and 12 1 1,2,3-propanetriol and 3 1 of sorbitol 70 % solution are added thereto. 40 Grams of sodium saccharin are dissolved in 0.5 1 of water and 2 ml of raspberry and 2 ml of gooseberry essence are added. The latter solution is combined with the former, water is added q.s. to a volume of 20 1 providing an oral solution comprising 5 mg of the active ingredient per teaspoonful (5 ml). The resulting solution is filled in suitable containers.
Example D.3 : film-coated tablets Preparation of .tablet ..core A mixture of 100 grams of the a.L, 570 grams lactose and 200 grams starch is mixed well and thereafter humidified with a solution of 5 grams sodium dodecyl sulfate and 10 grams polyvinylpyrrolidone in about 200 ml of water. The wet powder mixture is sieved, dried and sieved again. Then there is added 100 grams micro crystalline cellulose and 15 grams hydrogenated vegetable oil. The whole is mixed well and com- pressed into tablets, giving 10,000 tablets, each containing 10 mg of the active ingredient.
Coating
To a solution of 10 grams methyl cellulose in 75 ml of denaturated ethanol there is added a solution of 5 grams of ethyl cellulose in 150 ml of dichloromethane. Then there are added 75 ml of dichloromethane and 2.5 ml 1,2,3-propanetriol. 10 grams of polyethylene glycol is molten and dissolved in 75 ml of dichloromethane. The latter solution is added to the former and then there are added 2.5 grams of magnesium octa- decanoate, 5 grams of polyvinylpyrrolidone and 30 ml of concentrated color suspension and the whole is homogenated. The tablet cores are coated with the thus obtained mix- ture in a coating apparatus.
Example D .4 : injectable solution
1.8 grams methyl 4-hydroxybenzoate and 0.2 grams propyl 4-hydroxybenzoate are dissolved in about 0.5 1 of boiling water for injection. After cooling to about 50 0C there are added while stirring 4 grams lactic acid, 0.05 grams propylene glycol and 4 grams of the a.L. The solution is cooled to room temperature and supplemented with water for injection q.s. ad 1 1, giving a solution comprising 4 mg/ml of a.i.. The solution is steril-
ized by filtration and filled in sterile containers.
Claims
1. Compound according to general Formula (I)

a pharmaceutically acceptable acid or base addition salt thereof, an TV-oxide form thereof or a quaternary ammonium salt thereof, wherein A is phenyl or pyridinyl ;
R1, R2 are each, independently from each other, selected from the group of hydrogen, C1-3alkyl, aryl,; arylCi-3alkyl, Het, HetC1-3alkyl, C1-3alkyl- carbonyl, C1-3alkyloxycarbonyl, C1-3alkyloxyC1-3alkyl, C1-3alkyl- carbonylCi-3alkyl, and NRaRbCi-3 alky 1 ; or R1 and R2 are taken together to form an TV-containing heterocycle, selected from the group of pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, and thiomorpholinyl ; wherein each of the aforementioned TV-containing heterocycles are optionally substituted by k substituents R3 ;
R3 is selected from the group of hydrogen, halo, cyano, hydroxy, NRaRb, oxo, carboxyl, aminocarbonyl, nitro, thio, formyl, C1-3alkyl, aryl, Ci-3alkyloxy, and Ci-3alkyloxycarbonyl ; Ra, Rb are each, independently from each other, selected from the group of hydrogen, C1-3alkyl, aryl, Het, C1-3alkylcarbonyl, C1-3alkyloxycarbonyl, C1-3alkylsulphonyl, C1-3alkylcarbonylC1-3alkyl, arylCi-3alkyl and HetCi-3alkyl ; or Ra and Rb are taken together to form an TV-containing heterocycle, se- lected from the group of pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, l-aza-bicyclo[2.2.1]heptyl, and thiomorpholinyl ;
R4, R5 are each, independently from each other, selected from the group of hydrogen, halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl,  ; k is an integer, equal to zero, 1, 2, 3 or 4 ;
; k is an integer, equal to zero, 1, 2, 3 or 4 ;
1 is an integer, equal to zero, 1, 2, 3 or 4 ; m is an integer, equal to zero, 1 , 2 or 3 ;
Y1, Y3 are each, independently from each other, selected from the group of a single bond, O, NR7, S, SO, and SO2 ; wherein R7 is selected from the group of hydro gen and C i .3 alky 1 ;
Y is a saturated or unsaturated, straight or branched Ci-6-hydrocarbon radical, wherein one or more hydrogen atoms may optionally be replaced by a radical selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, and formyl ; B is a 6-membered ring containing zero, 1, 2 or 3 nitrogen atoms, optionally substituted with p substituents R6, each independently from each other, selected from the group of hydrogen, halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl, C1-3alkyl, and  ; and wherein p is an integer, equal to zero, 1 or 2 ; or two substituents R6 may be combined into a radical -CH2CH2CH2- or -OCH2O- ; alkyl is a straight or branched saturated hydrocarbon radical having the indicated number of carbon atoms ; wherein the radical may optionally be substituted on one or more carbon atoms with one or more radicals selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, ni- tro, thio and formyl ; aryl is naphthyl or phenyl, each optionally substituted with 1 , 2 or 3 substituents, each independently from each other, selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl, C1-3 alkyl, and
; and wherein p is an integer, equal to zero, 1 or 2 ; or two substituents R6 may be combined into a radical -CH2CH2CH2- or -OCH2O- ; alkyl is a straight or branched saturated hydrocarbon radical having the indicated number of carbon atoms ; wherein the radical may optionally be substituted on one or more carbon atoms with one or more radicals selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, ni- tro, thio and formyl ; aryl is naphthyl or phenyl, each optionally substituted with 1 , 2 or 3 substituents, each independently from each other, selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl, C1-3 alkyl, and  Het is a heterocyclic radical selected from the group of pyrrolidinyl, imida-zolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, pyrrolyl, pyr- rolinyl, imidazolinyl, pyrrazolinyl, pyrrolyl, imidazolyl, pyrazolyl, tria- zolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, azepyl, di- azepyl, morpholinyl, thiomorpholinyl, furyl, thienyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, isothiazolyl, dioxolyl, dithianyl, tetrahydrofuryl, tetrahydropyranyl, and oxadiazolyl ; wherein each of the aforementioned heterocyclic radicals is optionally substituted with 1, 2 or 3 substituents, each independently from each other, selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl, C1-3alkyl, and Ci-3alkyloxy ; and halo is fluoro, chloro, bromo or iodo ; with the proviso that the following compounds are excluded : 4-[(2,4-difiuorobenzyl)oxy]- 1 -[2,6-difluoro-4-[(2-hydroxyethyl)(methyl)amino] phenyl] -6-methylpyridin 2(7H)-one ;
Het is a heterocyclic radical selected from the group of pyrrolidinyl, imida-zolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, pyrrolyl, pyr- rolinyl, imidazolinyl, pyrrazolinyl, pyrrolyl, imidazolyl, pyrazolyl, tria- zolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, azepyl, di- azepyl, morpholinyl, thiomorpholinyl, furyl, thienyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, isothiazolyl, dioxolyl, dithianyl, tetrahydrofuryl, tetrahydropyranyl, and oxadiazolyl ; wherein each of the aforementioned heterocyclic radicals is optionally substituted with 1, 2 or 3 substituents, each independently from each other, selected from the group of halo, cyano, hydroxy, amino, oxo, carboxyl, nitro, thio, formyl, C1-3alkyl, and Ci-3alkyloxy ; and halo is fluoro, chloro, bromo or iodo ; with the proviso that the following compounds are excluded : 4-[(2,4-difiuorobenzyl)oxy]- 1 -[2,6-difluoro-4-[(2-hydroxyethyl)(methyl)amino] phenyl] -6-methylpyridin 2(7H)-one ;
4-[(2,4-difiuorobenzyl)oxy]- 1 -[2,6-difluoro-4-(4-methylpiperazin- 1 -yl)phenyl]-
6-methylpyridin-2(7H)-one ;
4-[(2,4-difluorobenzyl)oxy]-l-[2,6-difluoro-4-(morpholin-4-yl)phenyl]-6-methy lpyridin-2(7H)-one ;
4-[(2,4-difluorobenzyl)oxy]-l-[4-(dimethylamino)-2,6-difluorophenyl]-6-methy lpyridin-2(7H)-one ;
3-bromo-4-[(2,4-difluorobenzyl)oxy]-l-[2,6-difluoro-4-[(2-hydroxyethyl)
(methyl)amino]phenyl]-6-methylpyridin-2(7H)-one ; 3-bromo-4-[(2,4-difiuorobenzyl)oxy]- 1 -[2,6-difluoro-4-(4-methylpiperazin- 1 -yl
)phenyl]-6-methylpyridin-2(7H)-one ;
3-bromo-4-[(2,4-difluorobenzyl)oxy]-l-[4-(dimethylamino)-2,6-difluorophenyl]
-6-methylpyridin-2(7H)-one ;
3-bromo-4-[(2,4-difluorobenzyl)oxy]-l-[2,6-difluoro-4-(morpholin-4-yl)- phenyl] -6-methylpyridin-2(7H)-one ; and
3-chloro-4-[(2,4-difluorobenzyl)oxy]- 1 -[2,6-difluoro-4-(4-methylpiperazin- 1 -yl
)phenyl]-6-methylpyridin-2(7H)-one.
2. Compound according to claim 1, characterized in that each of R1, R2, independ- ently from each other, is selected from the group of hydrogen, C1-3alkyl,  HetC1-3alkyl,
HetC1-3alkyl,  and NRaRbCi-3alkyl.
and NRaRbCi-3alkyl.
3. Compound according to any one of claims 1 to 2, characterized in that R1 and R are taken together to form an TV-containing heterocycle, selected from the group of pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, and thiomor- pholinyl ; and wherein each of the foregoing TV-containing heterocycles is op- tionally substituted by k substituents R3.
4. Compound according to any one of claims 1 to 3, characterized in that R3 is selected from the group of hydroxy, NRaRb, aminocarbonyl, C1-3alkyl, aryl, 
5. Compound according to any one of claims 1 to 4, characterized in that Ra, Rb are each, independently from each other, selected from the group of hydrogen,
C1-3alkyl,  and
and  or Ra and Rb are taken together to form an N-containing heterocycle, selected from the group of pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, l-aza-bicyclo[2.2.1]heptyl, and thiomorpholinyl.
or Ra and Rb are taken together to form an N-containing heterocycle, selected from the group of pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidinyl, piperazinyl, morpholinyl, l-aza-bicyclo[2.2.1]heptyl, and thiomorpholinyl.
6. Compound according to any one of claims 1 to 5, characterized in that A is phenyl.
7. Compound according to any one of claims 1 to 6, characterized in that each of R4 and R5, independently from each other, are selected from the group of hydrogen, halo, C1-3alkyl, and 
8. Compound according to any one of claims 1 to 7, characterized in that k is 1.
9. Compound according to any one of claims 1 to 8, characterized in that 1 is zero.
10. Compound according to any one of claims 1 to 9, characterized in that m is zero.
11. Compound according to any one of claims 1 to 10, characterized in that Y1 and Y3 are each, independently from each other, selected from the group of a single bond and O.
12. Compound according to any one of claims 1 to 11, characterized in that Y2 is selected from the group of saturated or unsaturated, straight or branched C1-6 hydrocarbon radicals.
13. Compound according to any one of claims 1 to 12, characterized in that B is selected from the group of phenyl, pyridinyl, pyridazinyl, pyrimidinyl, pyraz- inyl and triazinyl.
14. Compound according to any one of claims 1 to 13, characterized in that the moiety B-Y'-Y^Y3 is selected from the radicals (a-1) to (d-5) as listed below, optionally substituted with p substituents R6, each independently from each other, selected from the group of hydrogen, fiuoro, chloro, hydroxy, methyl, trifluoromethyl, fiuoromethyloxy, difluoromethyloxy, and methyloxy ; and wherein p is an integer, equal to zero, 1 or 2 ; or two substituents R6 are combined into a radical -CH2CH2CH2- or -OCH2O-.

(a-1) (b-1) (C-I) (d-1)

(a-2) (b-2) (c-2) (d-2)

(a-3) (b-3) (c-3) (d-3)

(a-4) (b-4) (c-4) (d-4)
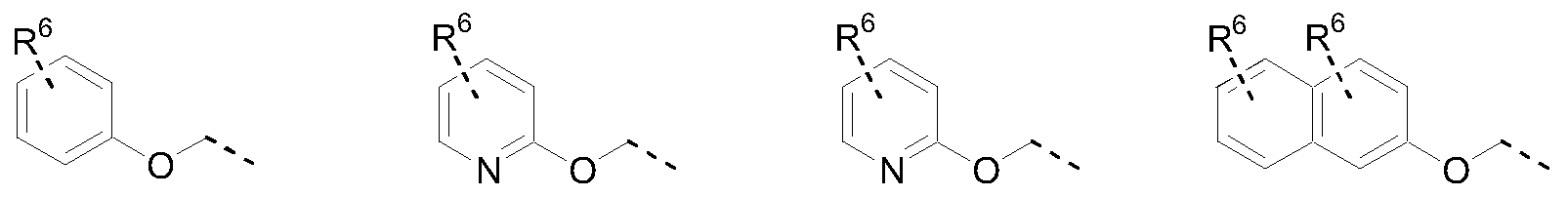
(a-5) (b-5) (c-5) (d-5)
15. Compound according to any one of claims 1 to 14 for use as a medicine.
16. Pharmaceutical composition comprising a pharmaceutically acceptable carrier or diluent and, as active ingredient, a therapeutically effective amount of a compound according to any one of claims 1 to 15.
17. Pharmaceutical composition according to claim 16, characterized in that it is in a form suitable to be orally administered.
18. Pharmaceutical composition according to any one of claims 16 to 17, without the proviso, characterized in that is comprises further one or more other compounds selected from the group of antidepressants, anxiolytics and antipsychotics.
19. Pharmaceutical composition according to any one of claims 16 to 17, without the proviso, characterized in that is comprises further one or more other compounds selected from the group of lipid- lowering compounds.
20. Process for the preparation of a pharmaceutical composition as claimed in any one of claims 16 to 17, characterized in that a pharmaceutically acceptable car- rier is intimately mixed with a therapeutically effective amount of a compound as claimed in any one of claims 1 to 15.
21. Process for the preparation of a pharmaceutical composition as claimed in claim 18, characterized in that a pharmaceutically acceptable carrier is intimately mixed with a therapeutically effective amount of a compound as claimed in any one of claims 1 to 15, without the proviso, and one or more other compounds selected from the group of antidepressants, anxiolytics and antipsychotics.
22. Process for the preparation of a pharmaceutical composition as claimed in claim 19, characterized in that a pharmaceutically acceptable carrier is intimately mixed with a therapeutically effective amount of a compound as claimed in any one of claims 1 to 15, without the proviso, and one or more other compounds selected from the group of lipid- lowering compounds.
23. Use of a compound according to any one of claims 1 to 15, without the proviso, for the preparation of a medicament for the prevention and/or treatment of dis- eases where antagonism of the MCH-receptor, in particular antagonism of the
MCH-I receptor is of therapeutic use.
24. Use of a compound according to any one of claims 1 to 15, without the proviso, for the preparation of a medicament for the prevention and/or treatment of psychiatric disorders, including but not limited to anxiety, eating disorders, mood disorders, such as bipolar disorders and depression, psychoses, such as schizo- phrenia, and sleeping disorders ; obesity ; diabetes ; sexual disorders and neurological disorders.
25. Use of a compound according to any one of claims 1 to 15, without the proviso, in combination with one or more other compounds selected from the group of antidepressants, anxiolytics and antipsychotics for the preparation of a medica- ment for the prevention and/or treatment of anxiety, eating disorders, mood disorders, such as bipolar disorders and depression, psychoses, such as schizophrenia, and sleeping disorders.
26. Use of a compound according to any one of claims 1 to 15, without the proviso, in combination with one or more other compounds selected from the group of lipid- lowering compounds for the preparation of a medicament for the prevention and/or treatment of obesity.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06114917.5 | 2006-06-02 | ||
| EP06114917 | 2006-06-02 | ||
| EP06123263 | 2006-10-31 | ||
| EP06123263.3 | 2006-10-31 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007141200A1 true WO2007141200A1 (en) | 2007-12-13 |
| WO2007141200A8 WO2007141200A8 (en) | 2008-03-27 |
Family
ID=38266244
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/055376 WO2007141200A1 (en) | 2006-06-02 | 2007-06-01 | Novel n-aryl and n-heteroaryl substituted pyridinone derivatives for use in mch-1 mediated diseases |
Country Status (5)
| Country | Link |
|---|---|
| AR (1) | AR061163A1 (en) |
| PE (1) | PE20080150A1 (en) |
| TW (1) | TW200811146A (en) |
| UY (1) | UY30378A1 (en) |
| WO (1) | WO2007141200A1 (en) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008041090A1 (en) * | 2006-10-06 | 2008-04-10 | Pfizer Limited | Malanin concentrating hormone receptor-1 antagonist pyridinones |
| WO2009089482A1 (en) | 2008-01-11 | 2009-07-16 | Albany Molecular Research, Inc. | (1-azinone) -substituted pyridoindoles as mch antagonists |
| WO2010104818A1 (en) * | 2009-03-09 | 2010-09-16 | Bristol-Myers Squibb Company | Aza pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
| WO2010141545A1 (en) * | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| WO2010141539A1 (en) * | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| WO2011003005A1 (en) * | 2009-07-01 | 2011-01-06 | Albany Molecular Research, Inc. | Azinone-substituted azepino[b]indole and pyrido-pyrrolo-azepine mch-1 antagonists, methods of making, and use thereof |
| WO2011127643A1 (en) * | 2010-04-12 | 2011-10-20 | Merck Sharp & Dohme Corp. | Pyridone derivatives |
| US20120316200A1 (en) * | 2010-04-12 | 2012-12-13 | Merck Sharp & Dohme Corp. | Pyridone derivatives |
| WO2013149362A1 (en) * | 2012-04-06 | 2013-10-10 | Glaxosmithkline Llc | 1-(dihydronaphthalenyl)pyridones as melanin-concentrating hormone receptor 1 antagonists |
| US8618299B2 (en) | 2009-07-01 | 2013-12-31 | Albany Molecular Research, Inc. | Azinone-substituted azapolycycle MCH-1 antagonists, methods of making, and use thereof |
| US8629158B2 (en) | 2009-07-01 | 2014-01-14 | Albany Molecular Research, Inc. | Azabicycloalkane-indole and azabicycloalkane-pyrrolo-pyridine MCH-1 antagonists, methods of making, and use thereof |
| US8697700B2 (en) | 2010-12-21 | 2014-04-15 | Albany Molecular Research, Inc. | Piperazinone-substituted tetrahydro-carboline MCH-1 antagonists, methods of making, and uses thereof |
| US8993765B2 (en) | 2010-12-21 | 2015-03-31 | Albany Molecular Research, Inc. | Tetrahydro-azacarboline MCH-1 antagonists, methods of making, and uses thereof |
| US9073925B2 (en) | 2009-07-01 | 2015-07-07 | Albany Molecular Research, Inc. | Azinone-substituted azabicycloalkane-indole and azabicycloalkane-pyrrolo-pyridine MCH-1 antagonists, methods of making, and use thereof |
| US9115089B2 (en) | 2013-06-07 | 2015-08-25 | Confluence Life Sciences, Inc. | Methyl/fluoro-pyridinyl-methoxy substituted pyridinone-pyridinyl compounds and fluoro-pyrimidinyl-methoxy substituted pyridinone-pyridinyl compounds |
| US9359300B2 (en) | 2010-12-06 | 2016-06-07 | Confluence Life Sciences, Inc. | Methyl/difluorophenyl-methoxy substituted pyridinone-pyridinyl compounds, methyl-pyridinyl-methoxy substituted pyridinone-pyridinyl compounds, and methyl-pyrimidinyl-methoxy substituted pyridinone-pyridinyl compounds |
| US9365547B2 (en) | 2010-12-06 | 2016-06-14 | Confluence Life Sciences Inc. | Substituted pyridinone-pyridinyl compounds |
| US9434695B2 (en) | 2012-07-18 | 2016-09-06 | Sunshine Lake Pharma Co., Ltd | Nitrogenous heterocyclic derivatives and their application in drugs |
| US9902712B2 (en) | 2013-12-19 | 2018-02-27 | Sunshine Lake Pharma Co., Ltd. | Nitrogenous heterocyclic derivatives and their application in drugs |
| JP2019524811A (en) * | 2016-08-08 | 2019-09-05 | ジーエヌアイ−イーピーエス ファーマシューティカルズ インコーポレイテッド | Method for producing hydronidone |
| JP2019525946A (en) * | 2016-08-08 | 2019-09-12 | ジーエヌアイ−イーピーエス ファーマシューティカルズ インコーポレイテッド | Method for producing hydronidone |
| EP4151210A3 (en) * | 2020-01-10 | 2023-06-14 | Harmony Biosciences, LLC | Pyridine-carboline derivatives as mchr1 antagonists for use in therapy |
| US11844801B2 (en) | 2020-03-27 | 2023-12-19 | Aclaris Therapeutics, Inc. | Oral compositions of MK2 pathway inhibitor for treatment of immune conditions |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003068230A1 (en) * | 2002-02-14 | 2003-08-21 | Pharmacia Corporation | Substituted pyridinones as modulators of p38 map kinase |
| WO2005085200A1 (en) * | 2004-03-05 | 2005-09-15 | Banyu Pharmaceutical Co., Ltd. | Pyridone derivative |
| WO2007024004A1 (en) * | 2005-08-24 | 2007-03-01 | Banyu Pharmaceutical Co., Ltd. | Phenylpyridone derivative |
-
2007
- 2007-05-31 PE PE2007000677A patent/PE20080150A1/en not_active Application Discontinuation
- 2007-05-31 UY UY30378A patent/UY30378A1/en unknown
- 2007-06-01 TW TW096119627A patent/TW200811146A/en unknown
- 2007-06-01 WO PCT/EP2007/055376 patent/WO2007141200A1/en active Application Filing
- 2007-06-01 AR ARP070102381A patent/AR061163A1/en not_active Application Discontinuation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003068230A1 (en) * | 2002-02-14 | 2003-08-21 | Pharmacia Corporation | Substituted pyridinones as modulators of p38 map kinase |
| WO2005085200A1 (en) * | 2004-03-05 | 2005-09-15 | Banyu Pharmaceutical Co., Ltd. | Pyridone derivative |
| WO2007024004A1 (en) * | 2005-08-24 | 2007-03-01 | Banyu Pharmaceutical Co., Ltd. | Phenylpyridone derivative |
Non-Patent Citations (3)
| Title |
|---|
| DATABASE CAPLUS [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; XP002444260, retrieved from CAPLUS accession no. 139:197371 * |
| DATABASE WPI DERWENT PUBLICATIONS LTD., LONDON, GB; XP002406966, retrieved from WPI accession no. 2005-725210 * |
| DYKE H J ET AL: "Recent developments in the discovery of MCH-1R antagonists for the treatment of obesity - an update", EXPERT OPINION ON THERAPEUTIC PATENTS, ASHLEY PUBLICATIONS, GB, vol. 15, no. 10, October 2005 (2005-10-01), pages 1303 - 1313, XP002382192, ISSN: 1354-3776 * |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008041090A1 (en) * | 2006-10-06 | 2008-04-10 | Pfizer Limited | Malanin concentrating hormone receptor-1 antagonist pyridinones |
| US9296743B2 (en) | 2008-01-11 | 2016-03-29 | Albany Molecular Research, Inc. | (1-azinone)-substituted pyridoindoles |
| WO2009089482A1 (en) | 2008-01-11 | 2009-07-16 | Albany Molecular Research, Inc. | (1-azinone) -substituted pyridoindoles as mch antagonists |
| AU2009204048B2 (en) * | 2008-01-11 | 2013-08-01 | Albany Molecular Research, Inc. | (1-azinone) -substituted pyridoindoles as MCH antagonists |
| US8716308B2 (en) | 2008-01-11 | 2014-05-06 | Albany Molecular Research, Inc. | (1-azinone)-substituted pyridoindoles |
| JP2014129417A (en) * | 2008-01-11 | 2014-07-10 | Albany Molecular Research Inc | (1-azinone)-substituted pyridoindoles as mch antagonists |
| US9650378B2 (en) | 2008-01-11 | 2017-05-16 | Albany Molecular Research, Inc. | (1-azinone)-substituted pyridoindoles |
| JP2011509937A (en) * | 2008-01-11 | 2011-03-31 | アルバニー モレキュラー リサーチ, インコーポレイテッド | (1-Azinone) substituted pyridoindoles as MCH antagonists |
| KR101614723B1 (en) | 2008-01-11 | 2016-04-22 | 알바니 몰레큘라 리써치, 인크. | (1-azinone) -substituted pyridoindoles as mch antagonists |
| EP2476680A1 (en) * | 2008-01-11 | 2012-07-18 | Albany Molecular Research, Inc. | (1-Azinone)-Substituted Pyridoindoles |
| US8278316B2 (en) | 2009-03-09 | 2012-10-02 | Bristol-Myers Squibb Company | Aza pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
| WO2010104818A1 (en) * | 2009-03-09 | 2010-09-16 | Bristol-Myers Squibb Company | Aza pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
| WO2010141539A1 (en) * | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| WO2010141545A1 (en) * | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| US8637501B2 (en) | 2009-07-01 | 2014-01-28 | Albany Molecular Research, Inc. | Azinone-substituted azepino[b]indole and pyrido-pyrrolo-azepine MCH-1 antagonists, methods of making, and use thereof |
| US9073925B2 (en) | 2009-07-01 | 2015-07-07 | Albany Molecular Research, Inc. | Azinone-substituted azabicycloalkane-indole and azabicycloalkane-pyrrolo-pyridine MCH-1 antagonists, methods of making, and use thereof |
| US8618299B2 (en) | 2009-07-01 | 2013-12-31 | Albany Molecular Research, Inc. | Azinone-substituted azapolycycle MCH-1 antagonists, methods of making, and use thereof |
| US8629158B2 (en) | 2009-07-01 | 2014-01-14 | Albany Molecular Research, Inc. | Azabicycloalkane-indole and azabicycloalkane-pyrrolo-pyridine MCH-1 antagonists, methods of making, and use thereof |
| WO2011003005A1 (en) * | 2009-07-01 | 2011-01-06 | Albany Molecular Research, Inc. | Azinone-substituted azepino[b]indole and pyrido-pyrrolo-azepine mch-1 antagonists, methods of making, and use thereof |
| JP2012532144A (en) * | 2009-07-01 | 2012-12-13 | アルバニー モレキュラー リサーチ, インコーポレイテッド | Azinone-substituted azepino [B] indoles and pyrido-pyrrolo-azepine MCH-1 antagonists and methods for making and using the same |
| EP2558094A1 (en) * | 2010-04-12 | 2013-02-20 | Merck Sharp & Dohme Corp. | Pyridone derivatives |
| EP2558094A4 (en) * | 2010-04-12 | 2013-09-25 | Merck Sharp & Dohme | Pyridone derivatives |
| US20120316200A1 (en) * | 2010-04-12 | 2012-12-13 | Merck Sharp & Dohme Corp. | Pyridone derivatives |
| WO2011127643A1 (en) * | 2010-04-12 | 2011-10-20 | Merck Sharp & Dohme Corp. | Pyridone derivatives |
| US9359300B2 (en) | 2010-12-06 | 2016-06-07 | Confluence Life Sciences, Inc. | Methyl/difluorophenyl-methoxy substituted pyridinone-pyridinyl compounds, methyl-pyridinyl-methoxy substituted pyridinone-pyridinyl compounds, and methyl-pyrimidinyl-methoxy substituted pyridinone-pyridinyl compounds |
| US9365546B2 (en) | 2010-12-06 | 2016-06-14 | Confluence Life Sciences Inc. | Substituted pyridinone-pyridinyl compounds |
| US9365547B2 (en) | 2010-12-06 | 2016-06-14 | Confluence Life Sciences Inc. | Substituted pyridinone-pyridinyl compounds |
| US8697700B2 (en) | 2010-12-21 | 2014-04-15 | Albany Molecular Research, Inc. | Piperazinone-substituted tetrahydro-carboline MCH-1 antagonists, methods of making, and uses thereof |
| US8993765B2 (en) | 2010-12-21 | 2015-03-31 | Albany Molecular Research, Inc. | Tetrahydro-azacarboline MCH-1 antagonists, methods of making, and uses thereof |
| WO2013149362A1 (en) * | 2012-04-06 | 2013-10-10 | Glaxosmithkline Llc | 1-(dihydronaphthalenyl)pyridones as melanin-concentrating hormone receptor 1 antagonists |
| US9434695B2 (en) | 2012-07-18 | 2016-09-06 | Sunshine Lake Pharma Co., Ltd | Nitrogenous heterocyclic derivatives and their application in drugs |
| US9115089B2 (en) | 2013-06-07 | 2015-08-25 | Confluence Life Sciences, Inc. | Methyl/fluoro-pyridinyl-methoxy substituted pyridinone-pyridinyl compounds and fluoro-pyrimidinyl-methoxy substituted pyridinone-pyridinyl compounds |
| US9636333B2 (en) | 2013-06-07 | 2017-05-02 | Confluence Life Sciences, Inc. | Methyl/fluoro-pyridinyl-methoxy substituted pyridinone-pyridinyl compounds and fluoro-pyrimidinyl-methoxy substituted pyridinone-pyridinyl compounds |
| US9902712B2 (en) | 2013-12-19 | 2018-02-27 | Sunshine Lake Pharma Co., Ltd. | Nitrogenous heterocyclic derivatives and their application in drugs |
| JP2019524811A (en) * | 2016-08-08 | 2019-09-05 | ジーエヌアイ−イーピーエス ファーマシューティカルズ インコーポレイテッド | Method for producing hydronidone |
| JP2019525946A (en) * | 2016-08-08 | 2019-09-12 | ジーエヌアイ−イーピーエス ファーマシューティカルズ インコーポレイテッド | Method for producing hydronidone |
| EP4151210A3 (en) * | 2020-01-10 | 2023-06-14 | Harmony Biosciences, LLC | Pyridine-carboline derivatives as mchr1 antagonists for use in therapy |
| US11844801B2 (en) | 2020-03-27 | 2023-12-19 | Aclaris Therapeutics, Inc. | Oral compositions of MK2 pathway inhibitor for treatment of immune conditions |
Also Published As
| Publication number | Publication date |
|---|---|
| TW200811146A (en) | 2008-03-01 |
| PE20080150A1 (en) | 2008-04-11 |
| AR061163A1 (en) | 2008-08-06 |
| WO2007141200A8 (en) | 2008-03-27 |
| UY30378A1 (en) | 2008-01-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007141200A1 (en) | Novel n-aryl and n-heteroaryl substituted pyridinone derivatives for use in mch-1 mediated diseases | |
| EP2102208B1 (en) | Novel substituted diaza spiro pyridinone derivatives for use in mch-1 mediated diseases | |
| EP1966164A1 (en) | Novel substituted pyrazinone derivatives for use in mch-1 mediated diseases | |
| US20100029620A1 (en) | Substituted triazoline, tetrazolone and imidazolone derivatives for use as a medicine | |
| EP2091942B1 (en) | Substituted pyrazinone derivatives for use as a medicine | |
| US20090281099A1 (en) | Substituted pyrazinone derivatives for use as a medicine | |
| EP2013202B1 (en) | Substituted pyrazinone derivatives for use as a medicine | |
| EP1966191B1 (en) | Substituted pyrazinone derivatives as alpha2c-adrenoreceptor antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07729777 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07729777 Country of ref document: EP Kind code of ref document: A1 |