WO2008041090A1 - Malanin concentrating hormone receptor-1 antagonist pyridinones - Google Patents
Malanin concentrating hormone receptor-1 antagonist pyridinones Download PDFInfo
- Publication number
- WO2008041090A1 WO2008041090A1 PCT/IB2007/002886 IB2007002886W WO2008041090A1 WO 2008041090 A1 WO2008041090 A1 WO 2008041090A1 IB 2007002886 W IB2007002886 W IB 2007002886W WO 2008041090 A1 WO2008041090 A1 WO 2008041090A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- disorders
- disorder
- pharmaceutically acceptable
- prodrug
- solvate
- Prior art date
Links
- 0 *c(cc1)ncc1N(C=CC(*[Al])=C1)C1=O Chemical compound *c(cc1)ncc1N(C=CC(*[Al])=C1)C1=O 0.000 description 4
- WIEFWQRJCCJSMT-COXUULNHSA-N CC(C)(C)OC(N[C@H](CC1)CN1/C(/N)=C/C=C(\N)/Br)=O Chemical compound CC(C)(C)OC(N[C@H](CC1)CN1/C(/N)=C/C=C(\N)/Br)=O WIEFWQRJCCJSMT-COXUULNHSA-N 0.000 description 1
- UDQFPZBVHKFRCI-UHFFFAOYSA-N CC(N(CC1)C(C2)C1CN2c1ncc(C)cc1)=O Chemical compound CC(N(CC1)C(C2)C1CN2c1ncc(C)cc1)=O UDQFPZBVHKFRCI-UHFFFAOYSA-N 0.000 description 1
- VQFWXSJDALQZJX-UHFFFAOYSA-N CC(N(CC1)CCN1c(cc1)ncc1N(C=CC(OCc1ccccc1)=C1)C1=O)=O Chemical compound CC(N(CC1)CCN1c(cc1)ncc1N(C=CC(OCc1ccccc1)=C1)C1=O)=O VQFWXSJDALQZJX-UHFFFAOYSA-N 0.000 description 1
- LLIXBRKMUANCFK-UHFFFAOYSA-N CCC(NC(CC1)CN1c(cc1)ncc1I)=O Chemical compound CCC(NC(CC1)CN1c(cc1)ncc1I)=O LLIXBRKMUANCFK-UHFFFAOYSA-N 0.000 description 1
- CYBIVJIMFQQIEJ-UHFFFAOYSA-N O=C(C=C(C=C1)OCc2ccccc2)N1c(cc1)cnc1N1CCCC1 Chemical compound O=C(C=C(C=C1)OCc2ccccc2)N1c(cc1)cnc1N1CCCC1 CYBIVJIMFQQIEJ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/74—Amino or imino radicals substituted by hydrocarbon or substituted hydrocarbon radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/34—Tobacco-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present invention relates to, inter alia, certain substituted pyridinone compounds, their salts, solvates and prodrugs, and their use in treating a variety of conditions More particularly the compounds of interest are antagonists at the melanin concentrating hormone type 1 receptor (MCH-1 or MCHR1 , and the like are terms used herein and which are interchangeable) As such the substances described herein are of use in the treatment of diseases or conditions mediated by MCHR1 , such as obesity, and where antagonistic activity at this receptor would have a beneficial effect
- the hormone MCH as found in humans, is a nonadecapeptide and is found throughout the central nervous system, as well as other tissues, including the gut, gonads, adipose tissue, pancreas, skin, and immune system
- MCH is involved in many functions, including feeding, reproduction, stress, and other behavior patterns (See, e g , Griffon, B & Baker, B I , "Cell and Molecular Cell Biology of Melanin-Concentrating Hormone," lnt Rev Cytol , 213 233-277 (2002), Kawano, H , et al , "Melanin-concentrating hormone neuron system the wide web that controls the feeding," Anatom Sci International, 77 149-160 (2002) (disclosing effect of MCH on appetite, arousal and anxiety, food-searching behavior, olfaction, regulation of energy balance, swallowing and mastication), Borowsky, B , et al , "Antidepressant, anx
- MCH receptor 1 antagonists eating disorders (e g , binge eating disorder, anorexia, and bulimia), weight loss or control (e g , reduction in calorie or food intake, and/or appetite suppression), obesity, depression, atypical depression, bipolar disorders, psychoses, schizophrenia, behavioral addictions, suppression of reward-related behaviors (e g , conditioned place avoidance, such as suppression of cocaine- and morphine-induced conditioned place preference), substance abuse, addictive disorders, impulsivity, alcoholism (e g , aJcohol abuse, addiction and/or dependence including treatment for abstinence, craving reduction and relapse prevention of alcohol intake), tobacco abuse (e g , smoking addiction, cessation and/or dependence including treatment for craving reduction and relapse prevention of tobacco smoking), dementia (including memory loss, Alzheimer's disease, dementia of aging, vascular dementia, mild cognitive impairment, age-related cognitive decline, and mild neurocognitive disorder), sexual dysfunction
- eating disorders e g , binge eating disorder, anorexia, and
- Another object of the invention is that the compounds of the invention have low inhibitory activity at the HERG potassium channel
- Prolongation of the cardiac action potential duration (QT prolongation) has been identified as being due to action at the HERG potassium channel (Expert Opinion of Pharmacotherapy, 2, pp947-973, 2000)
- QT prolongation is known to have a potential liability to produce fatal cardiac arrhythmias of Torsades de Pointes (TdP)
- the invention aims to provide compounds which are therapeutically effective MCHR1 antagonists with good cardiac safety
- MCH receptor 1 antagonists include premenstrual syndrome or late luteal phase syndrome, migraines, panic disorder, anxiety, posttraumatic syndrome, social phobia, cognitive impairment in non-demented individuals, non-amnestic mild cognitive impairment, post operative cognitive decline, disorders associated with impulsive behaviours (such as, disruptive behaviour disorders (e g , anxiety/depression, executive function improvement, tic disorders, conduct disorder and/or oppositional defiant disorder), adult personality disorders (e g , borderline personality disorder and antisocial personality disorder), diseases associated with impulsive behaviours (e g , substance abuse, paraphilias and self-mutilation), and impulse control disorders (e g , intermittent explosive disorder, kleptomania, pyromania, pathological gambling, and trichotillomania)), obsessive compulsive disorder, chronic fatigue syndrome, premature ejaculation, sexual dysfunction in females, disorders of sleep (e g , sleep apnea), autism, mut
- Obesity is a major public health concern because of its increasing prevalence and associated health risks Obesity and overweight are generally defined by body mass index (BMI), which is correlated with total body fat and estimates the relative risk of disease BMI is calculated by weight in kilograms divided by height in meters squared (kg/m 2 )
- BMI body mass index
- Overweight is typically defined as a BMI of 25-29 9 kg/m 2
- obesity is typically defined as a BMI of 30 kg/m 2 or more See, e g , National Heart, Lung, and Blood Institute, Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults, The Evidence Report, Washington, DC U S Department of Health and Human Services, NIH publication no 98-4083 (1998)
- the increase in obesity is of concern because of the excessive health risks associated with obesity, including coronary heart disease, strokes, hypertension, type 2 diabetes mellitus, dyshpidemia, sleep apnea, osteoarthritis, gall bladder disease, depression, and certain
- Adrenergic agents e g , diethylpropion, benzphetamine, phendimetrazme, mazindol, and phentermine
- Adrenergic agents act by modulating central norepinephrine and dopamine receptors through the promotion of catecholamine release Older adrenergic weight-loss drugs (e g , amphetamine, methamphetamine, and phenmetrazme), which strongly engage
- CB1 cannabinoid receptor antagonists/inverse agonists have been suggested as potential appetite suppressants See, e g , Arnone, M , et al , "Selective Inhibition of Sucrose and Ethanol Intake by SR141716, an Antagonist of Central Cannabinoid (CB1 ) Receptors," Psvchopharmacol. 132, 104-106 (1997), Colombo, G , et al , "Appetite Suppression and Weight Loss after the Cannabinoid Antagonist SR141716.” L ⁇ fe Sc ⁇ .
- the present invention encompasses a method for promoting weight loss (including prevention or inhibition of weight gam), or treatment of obesity and related eating disorders which comprises the step of administering to an animal (preferably, human) in need thereof a therapeutically effective amount of a MCHR1 antagonist as described herein
- a MCHR1 antagonist as described herein
- "eating disorders” refer to illnesses in which the patient suffers disturbances in their eating behaviors and related thoughts and emotions
- Representative examples of obesity-related eating disorders include overeating, bulimia, binge-eating disorder, compulsive dieting, nocturnal sleep-related eating disorder, pica, Prader-Willi Syndrome, and night-eating syndrome
- Bulimia also referred to as Bulimia Nervosa
- Individuals with binge eating disorder (BED) binge eat but do not regularly use compensatory weight control behaviors such as vomiting, fasting, over-exercise, or abuse of laxatives
- BED binge eating disorder
- the person with BED is often genetically predisposed to weigh more than the "average" person, let alone the unrealistic cultural ideal Due to culturally-reinforced body dissatisfaction, the person diets, making her or himself hungry, and then binges in response to that hunger
- the person may also eat for emotional reasons to comfort themselves, avoid uncomfortable situations, and numb feelings
- Symptoms of night-eating syndrome include little or no appetite for breakfast, eating more food after dinner than during the meal, eating more than half of daily food intake after the dinner hour, the pattern persists for at least two months, feeling tense, anxious, upset, or guilty while eating, difficulty falling asleep or staying asleep, unlike bingeing (which is done in relatively short episodes) continual eating throughout evening hours, and eating produces guilt and shame, not enjoyment
- a person suffering from nocturnal sleep-related eating disorder is somewhere between wakefulness and sleep, and may binge or consume strange combinations of food or non-food items When awake, the person has little or no memory of the episodes Pica is a craving for non-food items, most commonly dirt, clay, chalk, paint chips, cornstarch, baking soda, coffee grounds, cigarette ashes, rust, plastic, etc Pica is usually found in pregnant women, people whose diets are deficient in minerals contained in the consumed substances, people who have psychiatric disturbances, or people whose family or ethnic customs including eating certain non-food substances
- Prader-Willi syndrome is an uncommon inherited disorder characterized by mental retardation, decreased muscle tone, short stature, emotional lability and an insatiable appetite which can lead to life- threatening obesity
- terapéuticaally effective amount means an amount of a drug substance that ( ⁇ ) treats or prevents the particular disease, condition, or disorder, ( ⁇ ) attenuates, ameliorates, or eliminates one or more symptoms of the particular disease, condition, or disorder, or (in) prevents or delays the onset of one or more symptoms of the particular disease, condition, or disorder described herein
- treating embrace both preventative, i e , prophylactic, and palliative treatment
- animal refers to humans (male or female, adults, adolescents and/or children), companion animals (e g , dogs, cats and horses), food-source animals, zoo animals, marine animals, birds and other similar animal species
- Preferred animals include humans, companion animals, and food-source animals, more preferably, humans
- WO2003068230 Pharmacia
- WO2003076405 Bayer
- WO2003097047 Eh Lilly
- WO2004052848 EIi Lilly
- WO2004072025 Aventis
- X is CH 2 CH 2 , CH 2 O or OCH 2
- a and B are each independently CH or N, with the proviso that 1 or both of A and B is N,
- Ar is phenyl optionally substituted by 1 or 2 substituents independently selected from F and Cl,
- R 6 and R 7 are each independently H, C 1 -C 5 alkyl or C 3 -C 5 cycloalkyl,
- R 8 is H, C 1 -C 5 alkyl or C 3 -C 5 cycloalkyl
- R 9 is C 1 -C 5 alkyl or C 3 -C 5 cycloalkyl, each of which is optionally substituted with one or more fluorine atoms,
- Alkyl may be either straight chain or branched
- Suitable 5-membered aromatic heterocycles include oxazole, isoxazole, imidazole, pyrazole, thiazole, isothiazole and oxadiazole
- Suitable 6-membered aromatic heterocycles include pyridine, py ⁇ dazine, py ⁇ midine, pyrazine and t ⁇ azine
- the pharmaceutically acceptable salts of the compounds of the formula (I) include the acid addition and the base salts thereof
- a pharmaceutically acceptable salt of a compound of the formula (I) may be readily prepared by mixing together solutions of a compound of the formula (I) and the desired acid or base, as appropriate The salt may precipitate from solution and be collected by filtration or may be recovered by evaporation of the solvent Suitable acid addition salts are formed from acids which form non-toxic salts Examples include the acetate, adipate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, cyclamate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chlo ⁇ de, hydrobromide/bromide, hydroiodide/iodide, isethionate,
- solvates refers to a complex of a compound or salt of the present invention with one or more solvent molecules
- Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of formula I with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H Bundgaard (Elsevier, 1985)
- prodrugs in accordance with the invention include
- Certain compounds of formula (I) may have asymmetric centers and therefore exist in different enantiomeric forms All optical isomers and stereoisomers of the compounds of formula (I), and mixtures thereof, are considered to be within the scope of the invention
- Diastereome ⁇ c mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods known to those skilled in the art, for example, by chromatography or fractional crystallization
- Enantiomers can be separated by converting the enantiomenc mixtures into a diastereom ⁇ c mixture by reaction with an appropnate optically active compound (e g , alcohol), separating the diastereomers and converting (e g , hydrolyzing) the individual diastereomers to the corresponding pure enantiomers
- the invention includes the use of a racemate, one or more enantiomeric forms, one or more diastereome ⁇ c forms, or mixtures thereof
- the compounds of formula (I) may also exist as t
- a “pharmacological composition” refers to a mixture of one or more of the compounds described herein, or physiologically acceptable salts and solvates thereof, with other chemical components, such as physiologically acceptable carriers and/or excipients The purpose of a pharmacological composition is to facilitate administration of a compound to an organism
- a “physiologically acceptable carrier” refers to a carrier or diluent that does not cause significant or otherwise unacceptable irritation to an organism and does not unacceptably abrogate the biological activity and properties of the administered compound
- Suitable pharmaceutical carriers include inert diluents or fillers, water and various organic solvents
- the pharmaceutical compositions may, if desired, contain additional ingredients such as flavorings, binders, excipients and the like
- va ⁇ ous excipients such as citric acid
- vanous disintegrants such as starch, alginic acid and certain complex silicates
- binding agents such as sucrose, gelatin and acacia
- lubricating agents such as magnesium stearate, sodium lauryl sulfate and talc are often useful for tableting purposes
- Solid compositions of a similar type may also be employed in soft and hard filled gelatin capsules
- Preferred materials, therefor, include lactose or milk sugar and high molecular weight polyethylene glycols
- the active compound therein may be combined with various sweetening or flavonng agents, colo ⁇ ng matters or dyes and, if desired, emuls
- a maintenance dose is administered if necessary.
- the dosage or the frequency of administration, or both can be reduced, as a function of the symptoms, to a level at which the improved proliferative disorder or condition is retained
- treatment can cease Patients can, however, require intermittent treatment on a long-term basis upon any recurrence of the disease symptoms
- an effective dosage is in the range of about 0 001 to about 100 mg per kg body weight per day, preferably about 1 to about 35 mg/kg/day, in single or divided doses For a 70 kg human, this would amount to about 0 05 to about 7 g/day, preferably about 0 2 to about 2 5 g/day
- dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, provided that such larger doses are first divided into several small doses for administration throughout the day
- Suitable pharmaceutical agents that may be used in combination with the compounds of the present invention include anti-obesity agents such as apohpoprotein-B secretion/microsomal triglyceride transfer protein (apo-B/MTP) inhibitors, 11 ⁇ -hydroxy steroid dehydrogenase-1 (11 ⁇ -HSD type 1) inhibitors, peptide YY ⁇ 36 or analogs thereof, cannabinoid antagonists (e g , CB-1 antagonists, such as ⁇ monabant), MCR-4 agonists, cholecystokinin-A (CCK-A) agonists, monoamine reuptake inhibitors (such as sibutramine), sympathomimetic agents, neurotensin inhibitors, p 3 adrenergic receptor agonists, dop
- anti-obesity agents such as apohpoprotein-B secretion/microsomal triglyceride transfer protein (apo-B/MTP) inhibitors, 11 ⁇ -hydroxy steroid
- agents suitable pharmaceutical agents include agents designed to treat tobacco abuse (e g , nicotine receptor partial agonists, bupropion hypochlo ⁇ de (also known under the tradename ZybanTM) and nicotine replacement therapies), agents to treat erectile dysfunction (e g , dopaminergic agents, such as apomorphine), ADD/ADHD agents (e g , RitalinTM, StratteraTM, ConcertaTM and AdderallTM), and agents to treat alcoholism, such as opioid antagonists (e g , naltrexone (also known under the tradename ReViaTM) and nalmefene), disulfiram (also known under the tradename AntabuseTM), and acamprosate (also known under the tradename CampralTM))
- agents for reducing alcohol withdrawal symptoms may also be co-administered, such as benzodiazepines, beta-blockers, clonidine, carbamazepine, pregabalm, and
- antihypertensive agents include antihypertensive agents, anti-inflammatory agents (e g , COX-2 inhibitors), antidepressants (e g , fluoxetine hydrochloride (ProzacTM)), cognitive improvement agents (e g , donepezil hydrochloride (AirceptTM) and other acetylcholinesterase inhibitors), neuroprotective agents (e g , memantine), antipsychotic medications (e g , ziprasidone (GeodonTM), risperidone (RisperdalTM), and olanzapine (ZyprexaTM)), insulin and insulin analogs (e g , LysPro insulin), GLP-1 (7-37) (insulinotropin) and GLP-1 (7-36)-NH 2 , sulfonylureas and analogs thereof chlorpropamide, glibenclamide, tolbutamide, tolazamide, acetohexamide, Glypizide ® , glimepi
- anti-diabetes agents suitable for combination therapy with the present invention include, by way of example only meglitinides, sulfonylureas, and thiazolidinediones
- meglitinides include, but are not limited to Prandin Tablets (Novo Nordisk
- sulfonylureas include, but are not limited to Amaryl tablets (Hoechst Marion Roussel),D ⁇ abeta tablets (Hoechst Marion Roussel), Diabinese Tablets (Pfizer), Glucotrol Tablets (Pfizer), Glucotrol XL Extended Release Tablets (Pfizer), Glynase PresTab Tablets (Pharmacia & Upjohn), Micronase Tablets (Parke-Davis)
- thiazolidinediones include, but are not limited to Rezuhn Tablets (Parke-Davis) When depression is of concern (e g , treatment of, prevention of, maintenance against, etc
- anxiolytic agents suitable for combination therapy with the present invention include, by way of example only Tranxene T-TAB tablets (Abbott), Tranxene-SD Tablets (Abbott), Tranxene SD Half Strength Tablets (Abbott), Valium Tablets (Roche Products), Xanax Tablets (Pharmacia & Upjohn), Atarax Tablets & Syrup (Pfizer), Effexor XR Capsules (Wyeth), Paxil Oral Suspension (GlaxoSmithKline), Paxil Tablets (GlaxoSmithKline), Sinequan Capsules (Pfizer), Sinequan Oral Concentrate (Pfizer), Vista ⁇ l Capsules (Pfizer), Vista ⁇ l Intramuscular Solution (Pfizer), Vista ⁇ l Oral Suspension (Pfizer), Zoloft Oral Concentrate (Pfizer), Zoloft Tablets (Pfizer)
- centrally-acting compounds when either administered alone or in combination therapies such as those mentioned herein, may cause nausea and/or emesis and so it could be advantageous to administer compounds or combinations of the present invention alongside a suitable anti-emetic agent, for example a 5-HT 3 antagonist or a neurok ⁇ n ⁇ n-1 (NK-1) antagonist
- suitable anti-emetic agent for example a 5-HT 3 antagonist or a neurok ⁇ n ⁇ n-1 (NK-1) antagonist
- Suitable 5-HT 3 antagonists include, but are not limited to, granisetron, ondansetron, tropisetron, ramosetron, palonsetron, indisetron, dolasetron, alosetron and azasetron
- Suitable NK-1 antagonists include, but are not limited to, aprepitant, casopitant, ezlopitant, cilapitant, netupitant, vestipitant, vofopitant and 2-(R)-(1-(R)-3,5-b ⁇ s(tr ⁇ fluoromethyl)phenyl)ethoxy-4-(5- (d ⁇ methylam ⁇ no)methyl-1,2,3-t ⁇ azol-4-yl)methyl-3-(S)-(4-fluorophenyl)mo ⁇ hol ⁇ ne See for example International Patent Application publication number WO2006/049933
- the dosage of the additional pharmaceutical agent (e g , anti-obesity agent) will also be generally dependent upon a number of factors including the health of the subject being treated, the extent of treatment desired, the nature and kind of concurrent therapy, if any, and the frequency of treatment and the nature of the effect desired
- the dosage range of an anti-obesity agent is in the range of from about 0 001 mg to about 100 mg per kilogram body weight of the individual per day, preferably from about 0 1 mg to about 10 mg per kilogram body weight of the individual per day
- some variability in the general dosage range may also be required depending upon the age and weight of the subject being treated, the intended route of administration, the particular anti-obesity agent being administered and the like
- the determination of dosage ranges and optimal dosages for a particular patient is also well within the ability of one of ordinary skill in the art having the benefit of the instant disclosure
- the compounds described herein and, in embodiments where combinational therapy is employed, other agents do not have to be administered in the same pharmaceutical composition, and may, because of different
- compositions/Formulations, Dosaqinq. and Modes of Administration Pharmaceutical (including pharmaceuticals for veterina ⁇ al use) compositions according to the invention may, alternatively or in addition to a compound of Formula (I) 1 comprise as an active ingredient or pharmaceutically acceptable salts of such compounds Such compounds and salts are sometimes referred to herein collectively as “active agents” or “agents " Administration of the compounds of the present invention (hereinafter the “active compound(s)”) can be effected by any method that enables delivery of the compounds to the site of action
- Administration methods include oral routes (e g with a solid or liquid formulation), intraduodenal routes, parenteral injection (including intravenous, subcutaneous, intramuscular, intravascular or infusion), topical, pulmonary, intranasal, and rectal administration
- the therapeutic or pharmaceutical compositions of the invention can be administered locally to the area in need of treatment This may be achieved by, for example, but not limited to, local infusion dunng surgery, topical application, e g , cream, ointment, injection, catheter, or implant, said implant made, e g , out of a porous, non-porous, or gelatinous material, including membranes, such as sialastic membranes, or fibers
- the administration can also be by direct injection at the site (or former site) of a tumor or neoplastic or pre-neoplastic tissue
- the therapeutic or pharmaceutical composition can be delivered in a vesicle, e g , a liposome (see, for example, Langer, 1990, Science
- compositions used in the methods of the instant invention can contain the active ingredient in a form suitable for oral use, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, or syrups or elixirs
- Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations
- Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients, which are suitable for the manufacture of tablets
- excipients may be, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate, granulating and disintegrating agents, such as microcrystalline cellulose, sodium crosscarmellose
- the drug substance For companion animals or food-source animals, it is often more convenient to incorporate the drug substance into the animal's food source This may be accomplished by addition of the drug substance to the food source as a dry powder or as a liquid solution or suspension
- Aqueous suspensions can contain the active material in admixture with excipients suitable for the manufacture of aqueous suspensions
- excipients can act as suspending agents and include, e g , sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethyl-cellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia
- dispersing or wetting agents may be a naturally- occurring phosphatide, for example lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethylene-oxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example poly
- Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in mineral oil such as liquid paraffin
- the oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol Sweetening agents such as those set forth above, and flavo ⁇ ng agents may be added to provide a palatable oral preparation
- a thickening agent for example beeswax, hard paraffin or cetyl alcohol Sweetening agents such as those set forth above, and flavo ⁇ ng agents may be added to provide a palatable oral preparation
- an anti-oxidant e g , butylated hydroxyanisol, alpha-tocopherol, or ascorbic acid
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above Additional excipients, for example sweetening, flavoring and coloring agents, may also be present These compositions may be preserved by the addition of ant ⁇ ox ⁇ dant(s)
- the pharmaceutical compositions used in the methods of the instant invention may also be in the form of o ⁇ l- ⁇ n-water emulsions
- the oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these Suitable emulsifying agents may be naturally-occurring phosphatides, for example soy bean lecithin, and esters or partial esters derived from fatty acids and hexitol anhydrides, for example sorbitan monooleate, and condensation products of the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monooleate
- the emulsions may also contain sweetening, flavoring agents, preservatives and antioxidants Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose Such formulations may also contain a demulcent, a preservative, flavoring and coloring
- Pulmonary administration by inhalation may be accomplished by means of producing liquid or powdered aerosols, for example, by using any of various devices known in the art (see e g Newman, S P , 1984, in Aerosols and the Lung, Clarke and Pavia (Eds ), Butterworths, London, England, pp 197-224, PCT Publication No WO 92/16192 dated Oct 1 , 1992, PCT Publication No WO 91/08760 dated Jun 27, 1991 , NTIS Patent Application 7-504-047 filed Apr 3, 1990 by Roosdorp and Crystal) including but not limited to nebulizers, metered dose inhalers, and powder inhalers
- Various delivery devices are commercially available and can be employed, e g Ultravent nebulizer (Mallinckrodt, Inc, St Louis, Mo ), Acorn Il nebulizer (Marquest Medical Products, Englewood, Colo ), Ventolin metered dose inhalers (Glaxo Inc , Research Triangle
- compositions may be in the form of a sterile injectable aqueous solutions
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution
- the sterile injectable preparation may also be a sterile injectable o ⁇ l- ⁇ n-water microemulsion where the active ingredient is dissolved in the oily phase
- the active ingredient may be first dissolved in a mixture of soybean oil and lecithin The oil solution then introduced into a water and glycerol mixture and processed to form a microemulsion
- the injectable solutions or microemulsions may be introduced into a patient's blood-stream by local bolus injection Alternatively, it may be advantageous to administer the solution or microemulsion in such a way as to maintain a constant circulating concentration of the instant compound
- a continuous intravenous delivery device may be utilized An example of such a device is the Deltec CADD-PLUSTM model 5400 intravenous pump
- the pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleagenous suspension for intramuscular and subcutaneous administration This suspension may be formulated according to the known art using those suitable dispersing or wetting agents
- the compounds of Formula I may also be administered in the form of suppositories for rectal administration of the drug
- suppositories for rectal administration of the drug
- These compositions can be prepared by mixing the inhibitors with a suitable non-imtating excipient, which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug
- suitable non-imtating excipient include cocoa butter, glycerinated gelatin, Hated vegetable oils, mixtures of polyethylene glycols of various molecular weights and fatty acid esters of polyethylene glycol
- creams, ointments, jellies, solutions or suspensions, etc , containing a macrolide can be used as used herein, topical application can include mouth washes and gargles
- the compounds used in the methods and compositions described herein can be administered in intranasal form via topical use of suitable intranasal vehicles and delivery devices, or via transdermal routes, using those forms of transdermal
- X-Ar is (CH 2 ) 2 -Ar CH 2 OAr or OCH 2 -Ar More preferably X-Ar is OCH 2 -Ar
- A is N and B is CH or N More preferably A is N and B is CH
- Ar is phenyl, fluorophenyl or chlorophenyl
- Ar is phenyl, 4-chlorophenyl or 4-fluorophenyl Most preferably Ar is phenyl
- R 1 is pipendine, pyrrolidine, piperazine, octahydro-pyrrolo[3,4-c]pyrrole, octahydro- pyrrolo[3,4-b]pyrrole, or octahydro-pyrrolo[3,4-c]pyr ⁇ d ⁇ ne, attached to the "ABCDEC" ring via a N atom, which ring system is optionally substituted by one or more substituents independently selected from OH,
- R 1 is pipendine, pyrrolidine, piperazine, octahydro-pyrrolo[3,4-c]pyrrole, octahydro- pyrrolo[3,4-b]pyrrole, or octahydro-pyrrolo[3,4-c]py ⁇ d ⁇ ne, attached to the "ABCDEC" ring via a N atom, which ring system is optionally substituted by one or more substituents independently selected from
- R 1 is selected from one of the following groups
- Another preferred aspect of the invention is a group of compounds wherein A, B, X, R 1 and Ar are selected from the values in the compounds of the Examples below, or a pharmaceutically acceptable salt, solvate or prodrug thereof
- Another preferred aspect of the invention is a group of compounds of formula
- Ar and R 1 are as defined above, preferably wherein Ar is phenyl, or a pharmaceutically acceptable salt, solvate or prodrug thereof
- Another preferred aspect of the invention is a group of compounds of formula 1
- R 1 is selected from
- the compounds of the invention are selected from one of the Examples below, or a pharmaceutically acceptable salt, solvate or prodrug thereof.
- the compounds of the invention are selected from the compounds of Examples 17a,
- the compounds of the invention are selected from the compounds of Examples 17a,
- Compounds of Formula I can be prepared by a coupling reaction of a fragment Il with a fragment III, where X 1 is a suitable leaving group, for example iodide, bromide or t ⁇ flate (preferably iodide)
- a suitable solvent such as toluene, DMSO or DMF (preferably DMF), at a temperature between about 50 and about 150 0 C (preferably about 80 0 C) in the presence of a suitable copper catalyst (preferably CuI) and bidentate ligand (preferably trans-1 ,2- diaminocyclohexane)
- a suitable copper catalyst preferably CuI
- bidentate ligand preferably trans-1 ,2- diaminocyclohexane
- X 1 is a suitable group, for example a boron derivative (preferably B(OH) 2 )
- a coupling reaction can be carried out in a suitable solvent, for example dichlorom ethane, tetrahydrofuran or acetonitrile (preferably dichloromethane) mediated by a suitable metal salt, for example a copper(ll) derivative (preferably Cu(OAc) 2 ), in the presence of a suitable base, for example pyridine, triethylamine or DBU (preferably pyr
- Compounds of Formula III can be suitably prepared by a displacement reaction from compound Vl, where X 1 and X 2 are suitable halogen substituents (preferably bromo), using a suitable amine "R 1 H", a suitable base, for example t ⁇ ethylamine, sodium carbonate or potassium carbonate (preferably potassium carbonate), in a suitable solvent, for example acetonit ⁇ le, n-butanol or dimethyl sulfoxide (preferably n-butanol) at a temperature between about 25 and about 150 0 C (preferably about 11O 0 C)
- Compounds of Formula VII can be prepared by a coupling reaction of a fragment Il with a fragment Vl, where X 1 can act a suitable leaving group for such a coupling reaction, for example triflate, iodo or bromo (preferably iodo), and X 2 is a also a suitable leaving group for subsequent displacement by the amine R 1 H, for example chloro or fluoro (preferably fluoro)
- the reactants Il and Vl are mixed in a suitable solvent such as DMSO or N,N-d ⁇ methylformam ⁇ de (preferably N,N-d ⁇ methylformam ⁇ de), at a temperature between about 25 and about 150 0 C (preferably about 80 0 C) in the presence of a suitable copper catalyst (preferably CuI) and ligand (preferably trans- 1 ,2-d ⁇ am ⁇ nocyclohexane)
- a suitable copper catalyst preferably CuI
- ligand preferably trans- 1 ,2-d ⁇ am ⁇ nocyclohexane
- X 1 is a suitable group, for example a boron derivative (preferably B(OH) 2
- a coupling reaction can be earned out in a suitable solvent, for example dichloromethane, tetrahydrofuran or acetonitrile (preferably dichloromethane) mediated by a suitable metal salt (preferably Cu(OAc) 2 ) in the presence of a suitable base, for example pyridine or t ⁇ ethy
- a suitable solvent for example ethanol methanol or acetonitrile (preferably ethanol)
- a suitable hydrogen source for example dihydrotoluene, H 2 gas or ammonium formate (preferably dihydrotoluene) and a suitable metal catalyst (preferably palladium hydroxide on carbon)
- a suitable hydrogen source for example dihydrotoluene, H 2 gas or ammonium formate (preferably dihydrotoluene) and a suitable metal catalyst (preferably palladium hydroxide on carbon)
- a suitable metal catalyst preferably palladium hydroxide on carbon
- a parenteral pharmaceutical composition suitable for administration by injection 100 mg of a water-soluble salt of a compound of Formula I is dissolved in DMSO and then mixed with 10 mL of 0 9% sterile saline The mixture is incorporated into a dosage unit form suitable for administration by injection
- a pharmaceutical composition for oral delivery 100 mg of a compound of Formula I is mixed with 750 mg of lactose The mixture is incorporated into an oral dosage unit for, such as a hard gelatin capsule, which is suitable for oral administration
- Membrane homogenates of HEK-293S (Cell line #15-08) cells expressing the HERG product supplied by (PGRD) Sandwich Laboratories were prepared as follows Cell pellets were thawed at room temperature and kept on ice Buffer (5OmM Tris HCI, 1mM MgCI2, 1OmM KCI, pH 7 4, at 4°C) was added to each cell pellet (10 ml of buffer per 10 g of packed cell pellet) and the mixture homogenised using an Omni LabTek homogeniser (20,000rpm for 30 seconds) The homogenate was cent ⁇ fuged at 48,00Og for 20 minutes between 3 and 5 0 C in a Sorvall Evolution RC centrifuge and the supernatant discarded The pellet was resuspended, homogenised (20,000rpm for 10 seconds), and centrifuged as before The resultant supernatant was discarded and the final pellet resuspended (100 ml of the above buffer per 10 g of packed cell pellet), homogenised (20,000r
- the Cy3B ligand was stored in 100% DMSO and diluted to 6nM in assay buffer (5OmM Tris HCI, 1mM MgCI2, 1OmM KCI, 0 05% Pluronic F127, pH 74 at 4°C) on the day of the experiment
- Test samples and controls were diluted in 6% DMSO, 0 05% Pluronic F127 Cell membranes were removed from the - 8O 0 C freezer and placed on ice after defrosting When required the defrosted membranes were homogenised using a polytronic device for no more than 10 seconds, they were then diluted in the above assay buffer to produce a working solution of 0 3mg/ml
- the assay was compiled by adding 1OmL of test compound or control solution, 1OmL of the Cy3B ligand and 1OmL of cell membranes to a black 384-well plate (Matrix, Cat No 4318) The plates were mixed and then incubated for a minimum of 2 hours prior
- the following screen is used to evaluate the efficacy of test compounds for inhibiting food intake in Sprague-Dawley rats after an overnight fast
- mice Male Sprague-Dawley rats are obtained from Charles River Laborato ⁇ es, lnc (Wilmington, MA) The rats are individually housed and fed powdered chow They are maintained on a 12 hour light/dark cycle and received food and water ad libitum The animals are acclimated to the vivarium for a period of one week before testing is conducted Testing is completed during the light portion of the cycle To conduct the food intake efficacy screen, rats are transferred to individual test cages without food the afternoon prior to testing, and the rats are fasted overnight After the overnight fast, rats are dosed the following morning with vehicle or test compounds A known antagonist is dosed (3 mg/kg) as a positive control, and a control group receives vehicle alone (no compound) The test compounds are dosed at ranges between 0 1 and 100 mg/kg depending upon the compound The standard vehicle is 0 5% (w/v) methylcellulose in water and the standard route of administration is oral However, different vehicles and routes of administration are used to accommodate various compounds when
- Oxygen Consumption Whole body oxygen consumption is measured using an indirect calorimeter (Oxymax from Columbus Instruments, Columbus, OH) in male Sprague Dawley rats (if another rat strain or female rats is used, it will be specified) Rats (300-38Og body weight) are placed in the calorimeter chambers and the chambers are placed in activity monitors These studies are done during the light cycle Prior to the measurement of oxygen consumption, the rats are fed standard chow ad libitum During the measurement of oxygen consumption, food is not available Basal pre-dose oxygen consumption and ambulatory activity are measured every 10 minutes for 2 5 to 3 hours At the end of the basal pre-dosing period, the chambers are opened and the animals are administered a single dose of compound (the usual dose range is 0 001 to 10 mg/kg) by oral gavage (or other route of administration as specified, i e s c , i p , i v ) Drugs are prepared in methylcellulose, water or other specified vehicle
- the Oxymax calorimeter software calculates the oxygen consumption (ml/kg/h) based on the flow rate of air through the chambers and difference in oxygen content at inlet and output ports
- the activity monitors have 15 infrared light beams spaced one inch apart on each axis, ambulatory activity is recorded when two consecutive beams are broken and the results are recorded as counts
- Resting oxygen consumption, during pre- and post-dosing, is calculated by averaging the 10-m ⁇ n O 2 consumption values, excluding periods of high ambulatory activity (ambulatory activity count > 100) and excluding the first 5 values of the pre-dose period and the first value from the post-dose period
- Change in oxyge'n consumption is reported as percent and is calculated by dividing the post-dosing resting oxygen consumption by the pre-dose oxygen consumption *100
- CHO cell lines stably expressing the human MCHR1 (Euroscreen, Brussels, Belgium) were maintained in DMEM containing Glutamax (Invitrogen, Carlsbad, CA), 10% FBS, 400ug/ml Geneticin
- Cyclic AMP assays CHO cells stably expressing the human MCHR 1 receptors were collected and resuspended at a density of 4 0 x 10 ⁇ cells/ml in F12 (Hams) media and seeded (20,000 cells per well) into 384-well, solid white Greiner assay plates
- 11-po ⁇ nt 3-fold dose-response curves were constructed (top final assay concentration 3OuM) in PBS with 001% pluronic and 0 3% DMSO and added to the cells along with 25uM forskolin and 0 2nM MCH (final assay concentration) Cells were incubated at 37°C/5% CO 2 for 90 minutes and cAMP accumulation subsequently determined by DiscoveRx Hithunter c-AMP Il Assay kit (GE Healthcare, Buckinghamshire, UK) in accordance with manufactures instructions Luminescence was measured on a LEADseaker imaging system (GE Healthcare, Buckinghamshire, UK) IC50 values were calculated
- Method B 4 7 minute LC-MS gradient and instrument conditions A 3 75 % triflouroacetic acid in water B 1 88 % triflouroacetic acid in acetonitrile Column Ymc ODS-AQ 2 x 50mm with 5 micron particle size Gradient 0 mm 10% B 1 0 5 mins 10% B, 4 mm 100% B, 4 3 mins 10%B Flow 0 8ml/m ⁇ n UV 210nm DAD Temperature 5OC
- Method C 4 7 minute LC-MS gradient and instrument conditions A 0 5 % ammonium hydroxide in water B acetonitrile Column Welch XB-C18 2 1 x 50mm with 5 micron particle size Gradient O mm 5% B, 0 5 mins 5% B, 3 4 mm 100% B 1 4 2 mins 100%B, 4 2 mm 5% B Flow 0 8ml/m ⁇ n UV 210nm DAD 5 Temperature 5OC
- Examples 34-124 were prepared according to the methods described above for examples 1 , 3 and 11, or routine variation thereof, starting from the appropriate 2-fluoropyr ⁇ d ⁇ ne 1 and the appropriate amine 2 10 1 2-Fluoropyr ⁇ d ⁇ nes are described for example in preparations 2, 13 and 15
- Examples 125-143 were prepared according to the methods described above for examples 8, 13 and 17, starting from the appropriate Boc protected compound 1
- Examples 144-154 were prepared according to the method described above for Example 10, starting from the appropriate 4-hydroxypy ⁇ d ⁇ none 1 and the appropriate benzyl bromide 2
- Examples 155-157 were prepared according to the methods described above for example 16, starting from the appropriate pyridone 1 and the bromide from preparation 25
- Examples 158-195 were prepared according to the methods described above for example 20 starting from the appropriate amine 1 and the appropriate acid chloride or chloroformate 2
- Examples 196-203 were prepared according to the methods described above for examples 21 & 23, starting from the appropriate Pyridone 1 and the appropriate iodide 2 1
- the py ⁇ dones are either commercially available or known in the literature
- Examples 204-206 were prepared according to the method described below
- Examples 207-212 were prepared according to the methods described above for example 26 starting from the appropriate amine 1 and ketone or aldehyde 2 1
- the amines are described in Examples 8, 125, 130 and 131. 2
- the ketones and aldehydes are commercial available
- Examples 213-240 were prepared using the methods indicated in the table below starting from the fluoropyridine of preparation 2
- Example 119 100mg, 2 16mmol in dry DMF (2ml) was treated with sodium hydride (60% dispersion in oil, 17 3mg, 0432mmol) at r t and stirred for 2 hours Methyl iodide (26 9ul, 0432mmol) was added and stirring continued for 2 hours The reaction was carefully diluted with methanol (2ml) and passed down a SCX column, washed with methanol and the product eluted with 2M NH3 in methanol
Abstract
The present invention provides for MCHR1 antagonist compounds of formula (I), and the pharmaceutically acceptable salts, solvates and prodrugs thereof, wherein the substituents are as defined herein, and the pharmaceutically acceptable salts, solvates and prodrugs thereof, which are useful in treating diseases or conditions wherein antagonism of the MCHR1 receptor is beneficial.
Description
MELANIN CONCENTRATING HORMONE RECEPTOR-1 ANTAGONIST PYRIDINONES
The present invention relates to, inter alia, certain substituted pyridinone compounds, their salts, solvates and prodrugs, and their use in treating a variety of conditions More particularly the compounds of interest are antagonists at the melanin concentrating hormone type 1 receptor (MCH-1 or MCHR1 , and the like are terms used herein and which are interchangeable) As such the substances described herein are of use in the treatment of diseases or conditions mediated by MCHR1 , such as obesity, and where antagonistic activity at this receptor would have a beneficial effect
The hormone MCH, as found in humans, is a nonadecapeptide and is found throughout the central nervous system, as well as other tissues, including the gut, gonads, adipose tissue, pancreas, skin, and immune system Recent reviews provide evidence that MCH is involved in many functions, including feeding, reproduction, stress, and other behavior patterns (See, e g , Griffon, B & Baker, B I , "Cell and Molecular Cell Biology of Melanin-Concentrating Hormone," lnt Rev Cytol , 213 233-277 (2002), Kawano, H , et al , "Melanin-concentrating hormone neuron system the wide web that controls the feeding," Anatom Sci International, 77 149-160 (2002) (disclosing effect of MCH on appetite, arousal and anxiety, food-searching behavior, olfaction, regulation of energy balance, swallowing and mastication), Borowsky, B , et al , "Antidepressant, anxiolytic and anorectic effects of a melanin- concentrating hormone-1 receptor antagonist," Nature Med 8 825-830 (2002)) Antagonists of the MCH-1 receptor are being studied as a treatment for obesity and other eating disorder (See, e g , Crowley, V E F , et al, "Obesity Therapy Altering the Energy Intake-and-Expenditure Balance Sheet," Nature Reviews Drug Discovery, 1 26-286 (2002), Hillebrand, J J G , et al , "Neuropeptides, food intake and body weight regulation a hypothalamic focus," Peptides, 23 2283-2306 (2002)), HJ Dyke and NC Ray, Expert Opin Ther Patents (2005) 15(10) 1303-1313)
Preliminary investigations have indicated that the following diseases, conditions, and/or disorders are modulated by MCH receptor 1 antagonists eating disorders (e g , binge eating disorder, anorexia, and bulimia), weight loss or control (e g , reduction in calorie or food intake, and/or appetite suppression), obesity, depression, atypical depression, bipolar disorders, psychoses, schizophrenia, behavioral addictions, suppression of reward-related behaviors (e g , conditioned place avoidance, such as suppression of cocaine- and morphine-induced conditioned place preference), substance abuse, addictive disorders, impulsivity, alcoholism (e g , aJcohol abuse, addiction and/or dependence including treatment for abstinence, craving reduction and relapse prevention of alcohol intake), tobacco abuse (e g , smoking addiction, cessation and/or dependence including treatment for craving reduction and relapse prevention of tobacco smoking), dementia (including memory loss, Alzheimer's disease, dementia of aging, vascular dementia, mild cognitive impairment, age-related cognitive decline, and mild neurocognitive disorder), sexual dysfunction in males (e g , erectile difficulty), seizure disorders, epilepsy, inflammation, gastrointestinal disorders (e g , dysfunction of gastrointestinal motility or intestinal propulsion), attention deficit disorder (ADD including attention deficit hyperactivity disorder (ADHD)), Parkinson's disease, and type Il diabetes
Accordingly, it is an object of the invention to provide compounds of the present invention useful in treating diseases, conditions, or disorders that are modulated by MCH receptor 1 antagonists Consequently, the compounds of the present invention (including the compositions and processes used therein) may be used in the manufacture of a medicament for the therapeutic applications described herein
Another object of the invention is that the compounds of the invention have low inhibitory activity at the HERG potassium channel Prolongation of the cardiac action potential duration (QT prolongation) has been identified as being due to action at the HERG potassium channel (Expert Opinion of Pharmacotherapy, 2, pp947-973, 2000) QT prolongation is known to have a potential liability to produce fatal cardiac arrhythmias of Torsades de Pointes (TdP) In providing compounds which exhibit low inhibitory activity at the HERG potassium channel with comparable or improved pharmacokinetics, the invention aims to provide compounds which are therapeutically effective MCHR1 antagonists with good cardiac safety
Other diseases, conditions and/or disorders for which MCH receptor 1 antagonists may be effective include premenstrual syndrome or late luteal phase syndrome, migraines, panic disorder, anxiety, posttraumatic syndrome, social phobia, cognitive impairment in non-demented individuals, non-amnestic mild cognitive impairment, post operative cognitive decline, disorders associated with impulsive behaviours (such as, disruptive behaviour disorders (e g , anxiety/depression, executive function improvement, tic disorders, conduct disorder and/or oppositional defiant disorder), adult personality disorders (e g , borderline personality disorder and antisocial personality disorder), diseases associated with impulsive behaviours (e g , substance abuse, paraphilias and self-mutilation), and impulse control disorders (e g , intermittent explosive disorder, kleptomania, pyromania, pathological gambling, and trichotillomania)), obsessive compulsive disorder, chronic fatigue syndrome, premature ejaculation, sexual dysfunction in females, disorders of sleep (e g , sleep apnea), autism, mutism, neurodegenerative movement disorders, spinal cord injury, damage of the central nervous system (e g , trauma), stroke, neurodegenerative diseases or toxic or infective CNS diseases (e g , encephalitis or meningitis), cardiovascular disorders (e g , thrombosis)
Obesity is a major public health concern because of its increasing prevalence and associated health risks Obesity and overweight are generally defined by body mass index (BMI), which is correlated with total body fat and estimates the relative risk of disease BMI is calculated by weight in kilograms divided by height in meters squared (kg/m2) Overweight is typically defined as a BMI of 25-29 9 kg/m2, and obesity is typically defined as a BMI of 30 kg/m2 or more See, e g , National Heart, Lung, and Blood Institute, Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults, The Evidence Report, Washington, DC U S Department of Health and Human Services, NIH publication no 98-4083 (1998) The increase in obesity is of concern because of the excessive health risks associated with obesity, including coronary heart disease, strokes, hypertension, type 2 diabetes mellitus, dyshpidemia, sleep apnea, osteoarthritis, gall bladder disease, depression, and certain forms of cancer (e g , endometrial,
breast, prostate, and colon) The negative health consequences of obesity make it the second leading cause of preventable death in the United States and impart a significant economic and psychosocial effect on society See, McGinnis M, Foege WH , "Actual Causes of Death in the United States," JAMA, 270, 2207-12 (1993) Obesity is now recognized as a chronic disease that requires treatment to reduce its associated health risks Although weight loss is an important treatment outcome, one of the main goals of obesity management is to improve cardiovascular and metabolic values to reduce obesity-related morbidity and mortality It has been shown that 5-10% loss of body weight can substantially improve metabolic values, such as blood glucose, blood pressure, and lipid concentrations Hence, it is believed that a 5-10% intentional reduction in body weight may reduce morbidity and mortality
Currently available prescription drugs for managing obesity generally reduce weight by inducing satiety or decreasing dietary fat absorption Satiety is achieved by increasing synaptic levels of norepinephrine, serotonin, or both For example, stimulation of serotonin receptor subtypes 1 B, 1 D, and 2C and 1- and 2-adrenergιc receptors decreases food intake by regulating satiety See, Bray GA, "The New Era of Drug Treatment Pharmacologic Treatment of Obesity Symposium Overview," Obes Res , 3(suppl 4), 415s-7s (1995) Adrenergic agents (e g , diethylpropion, benzphetamine, phendimetrazme, mazindol, and phentermine) act by modulating central norepinephrine and dopamine receptors through the promotion of catecholamine release Older adrenergic weight-loss drugs (e g , amphetamine, methamphetamine, and phenmetrazme), which strongly engage in dopamine pathways, are no longer recommended because of the nsk of their abuse Fenfluramine and dexfenfluramine, both serotonergic agents used to regulate appetite, are no longer available for use
More recently, CB1 cannabinoid receptor antagonists/inverse agonists have been suggested as potential appetite suppressants See, e g , Arnone, M , et al , "Selective Inhibition of Sucrose and Ethanol Intake by SR141716, an Antagonist of Central Cannabinoid (CB1 ) Receptors," Psvchopharmacol. 132, 104-106 (1997), Colombo, G , et al , "Appetite Suppression and Weight Loss after the Cannabinoid Antagonist SR141716." Lιfe Scι . 63, PL113-PL117 (1998), Simiand, J , et al , "SR141716, a CB1 Cannabinoid Receptor Antagonist, Selectively Reduces Sweet Food Intake in Marmose," Behav Pharmacol , 9, 179- 181 (1998), and Chaperon, F , et al , "Involvement of Central Cannabinoid (CB1) Receptors in the Establishment of Place Conditioning in Rats," Psvchopharmacoloqy. 135, 324-332 (1998) For a review of cannabinoid CB1 and CB2 receptor modulators, see Pertwee, R G , "Cannabinoid Receptor Ligands Clinical and Neuropharmacological Considerations, Relevant to Future Drug Discovery and Development." Exp Qpin Invest Drugs, 9(7), 1553-1571 (2000)
Although investigations are on-going, there still exists a need for a more effective and safe therapeutic treatment for reducing or preventing weight-gain
The present invention encompasses a method for promoting weight loss (including prevention or inhibition of weight gam), or treatment of obesity and related eating disorders which comprises the step of administering to an animal (preferably, human) in need thereof a therapeutically effective amount of a MCHR1 antagonist as described herein
As used herein, "eating disorders" refer to illnesses in which the patient suffers disturbances in their eating behaviors and related thoughts and emotions Representative examples of obesity-related eating disorders include overeating, bulimia, binge-eating disorder, compulsive dieting, nocturnal sleep-related eating disorder, pica, Prader-Willi Syndrome, and night-eating syndrome
Bulimia (also referred to as Bulimia Nervosa) is characterized by self-perpetuating and self-defeating cycles of binge-eating and purging A person binges by rapidly consuming a large amount of food (or what s/he perceives to be a large amount) in a discrete period of time and in an automatic and helpless manner Individuals with binge eating disorder (BED) binge eat but do not regularly use compensatory weight control behaviors such as vomiting, fasting, over-exercise, or abuse of laxatives The person with BED is often genetically predisposed to weigh more than the "average" person, let alone the unrealistic cultural ideal Due to culturally-reinforced body dissatisfaction, the person diets, making her or himself hungry, and then binges in response to that hunger The person may also eat for emotional reasons to comfort themselves, avoid uncomfortable situations, and numb feelings
Symptoms of night-eating syndrome include little or no appetite for breakfast, eating more food after dinner than during the meal, eating more than half of daily food intake after the dinner hour, the pattern persists for at least two months, feeling tense, anxious, upset, or guilty while eating, difficulty falling asleep or staying asleep, unlike bingeing (which is done in relatively short episodes) continual eating throughout evening hours, and eating produces guilt and shame, not enjoyment
Unlike night-eating syndrome, a person suffering from nocturnal sleep-related eating disorder is somewhere between wakefulness and sleep, and may binge or consume strange combinations of food or non-food items When awake, the person has little or no memory of the episodes Pica is a craving for non-food items, most commonly dirt, clay, chalk, paint chips, cornstarch, baking soda, coffee grounds, cigarette ashes, rust, plastic, etc Pica is usually found in pregnant women, people whose diets are deficient in minerals contained in the consumed substances, people who have psychiatric disturbances, or people whose family or ethnic customs including eating certain non-food substances Prader-Willi syndrome (PWS) is an uncommon inherited disorder characterized by mental retardation, decreased muscle tone, short stature, emotional lability and an insatiable appetite which can lead to life- threatening obesity
The phrase "therapeutically effective amount" means an amount of a drug substance that (ι) treats or prevents the particular disease, condition, or disorder, (ιι) attenuates, ameliorates, or eliminates one or more symptoms of the particular disease, condition, or disorder, or (in) prevents or delays the onset of one or more symptoms of the particular disease, condition, or disorder described herein
The terms "treating", "treat", or "treatment" embrace both preventative, i e , prophylactic, and palliative treatment
The term "animal" refers to humans (male or female, adults, adolescents and/or children), companion animals (e g , dogs, cats and horses), food-source animals, zoo animals, marine animals, birds and other similar animal species Preferred animals include humans, companion animals, and food-source animals, more preferably, humans
Various pyπdinones and pyπmidinones and MCH antagonists are reported in the art, notably WO2003068230 (Pharmacia) - compounds disclosed for the treatment of conditions caused by unregulated p38 MAP kinase or TNF activity such as inflammation or arthritis etc , WO2003076405 (Bayer) - compounds for the treatment of COPD etc , WO2003097047 (Eh Lilly) - MCH antagonists for treatment of conditions such as obesity, WO2004052848 (EIi Lilly) - MCH antagonists for treatment of conditions such as obesity, WO2004072025 (Aventis) - MCH antagonists for treating conditions such as obesity, WO2005018557 (Pharmacia) - compounds disclosed for the treatment of conditions caused by unregulated p38 MAP kinase or TNF activity such as inflammation or arthritis etc , WO2005042541 (Glaxo) - thιenopyπmιd-4-one derivatives useful as MCHR1 antagonists for treating conditions such as obesity, WO2005070925 (Aventis) - MCH antagonists for the treating conditions such as obesity, WO2005103039 (Neurocπne Bioscience) - MCHR1 antagonists for treating conditions such as obesity, EP481448 (Squibb & Sons) - Compounds disclosed as antihypertensive agents, US5470975 (Squibb & Sons) - Compounds disclosed as antihypertensive agents, WO0121577 (Takeda) - MCH antagonists for the treatment of obesity etc , WO2001082925 (Takeda) - MCH antagonists for the treatment of obesity etc , US20020183324 (Bristol-Myers Squibb) - Compounds disclosed as factor Xa inhibitors for use as anticoagulants, WO2003026652 (Bristol-Myers Squibb) - Compounds disclosed as factor Xa inhibitors for use as anticoagulants, WO2003059884 (X-Ceptor) - Compounds disclosed as liver X receptor modulators for a wide range of diseases, US20040006062 (Bristol-Myers Squibb) - Compounds disclosed as factor Xa inhibitors for use as anticoagulants, US20040132718 (Bristol-Myers Squibb) - Compounds disclosed as factor Xa inhibitors for use as anticoagulants, US20040220174 (Bristol-Myers Squibb) - Compounds disclosed as factor Xa inhibitors for use as anticoagulants, WO2005032472 (Bristol-Myers Squibb) - Compounds disclosed as factor Xa inhibitors for use as anticoagulants, US20050267097 (Bristol-Myers Squibb) - Compounds disclosed as factor Xa inhibitors for use as anticoagulants, WO20060565708 (Institutes for Pharmaceutical Discovery) - protein tyrosine phosphatases for the treatment of diabetes
The present invention provides for compounds of formula (I) below

(I) wherein
X is CH2CH2, CH2O or OCH2,
A and B are each independently CH or N, with the proviso that 1 or both of A and B is N,
Ar is phenyl optionally substituted by 1 or 2 substituents independently selected from F and Cl,
R1 is a saturated 4- to 9-membered heterocyclic ring system containing 1 or 2 ring N atoms, which ring system may incorporate spiro-, fused or bridged rings, which is attached to the "ABCCHCHCπng via a N atom, which ring system is optionally substituted by one or more substituents independently selected from =0, R9, OH, C(O)C1-C5 alkyl, C(O)C3-C5 cycloalkyl, C(O)OC1-C5 alkyl, NR6R7, NR8C(O)R9, NR8C(O)OR9, 0(C1-C5 alkyl) or 0(C3-C5 cycloalkyl),
R6 and R7 are each independently H, C1-C5 alkyl or C3-C5 cycloalkyl,
or Rβ and R7 can be taken together with the N atom to which they are attached to form a 4- to 7- membered saturated ring, optionally substituted by =0,
R8 is H, C1-C5 alkyl or C3-C5 cycloalkyl,
R9 is C1-C5 alkyl or C3-C5 cycloalkyl, each of which is optionally substituted with one or more fluorine atoms,
and the pharmaceutically acceptable salts, solvates and prodrugs thereof
"Alkyl" may be either straight chain or branched
"Me" is methyl, and "Et" is ethyl
Suitable 5-membered aromatic heterocycles include oxazole, isoxazole, imidazole, pyrazole, thiazole, isothiazole and oxadiazole Suitable 6-membered aromatic heterocycles include pyridine, pyπdazine, pyπmidine, pyrazine and tπazine
The pharmaceutically acceptable salts of the compounds of the formula (I) include the acid addition and the base salts thereof A pharmaceutically acceptable salt of a compound of the formula (I) may be readily prepared by mixing together solutions of a compound of the formula (I) and the desired acid or base, as appropriate The salt may precipitate from solution and be collected by filtration or may be recovered by evaporation of the solvent Suitable acid addition salts are formed from acids which form non-toxic salts Examples include the acetate, adipate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, cyclamate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloπde, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate,
palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, pyroglutamate, saccharate, stearate, succinate, tannate, tartrate, tosylate, trifluoroacetate and xinofoate salts Suitable base salts are formed from bases which form non-toxic salts Examples include the aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts Hemisalts of acids and bases may also be formed, for example, hemisulphate and hemicalcium salts For a review on suitable salts, see Handbook of Pharmaceutical Salts Properties, Selection, and Use by Stahl and Wermuth (Wiley- VCH, 2002)
The compounds and salts of the present invention may inherently form solvates with pharmaceutically acceptable solvents (including water) and it is intended that the invention embrace both solvated and unsolvated forms The term "solvate" refers to a complex of a compound or salt of the present invention with one or more solvent molecules
As indicated, so-called 'prodrugs' of the compounds of formula I are also within the scope of the invention Thus certain derivatives of compounds of formula I which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of formula I having the desired activity, for example, by hydrolytic cleavage Such derivatives are referred to as 'prodrugs' Further information on the use of prodrugs may be found in Pro-drugs as
Novel Delivery Systems. VoI 14, ACS Symposium Series (T Higuchi and W Stella) and Bioreversible Carriers in Drug Design. Pergamon Press, 1987 (Ed E B Roche, American Pharmaceutical
Association)
Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of formula I with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H Bundgaard (Elsevier, 1985)
Some examples of prodrugs in accordance with the invention include
(a) where the compound of formula I contains an alcohol functionality (-OH), an ether thereof, for example, a compound wherein the hydrogen of the alcohol functionality of the compound of formula I is replaced by (C,-Cβ)alkanoyloxymethyl, and
(b) where the compound of formula I contains a pnmary or secondary amino functionality (-NH2 or - NHR where R ≠ H), an amide thereof, for example, a compound wherein, as the case may be, one or both hydrogens of the ammo functionality of the compound of formula I is/are replaced by
(C,-C10)alkanoyl
Further examples of replacement groups in accordance with the foregoing examples and examples of other prodrug types may be found in the aforementioned references
Moreover, certain compounds of formula I may themselves act as prodrugs of other compounds of formula I
Certain compounds of formula (I) may have asymmetric centers and therefore exist in different enantiomeric forms All optical isomers and stereoisomers of the compounds of formula (I), and mixtures thereof, are considered to be within the scope of the invention Diastereomeπc mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods known to those skilled in the art, for example, by chromatography or fractional crystallization Enantiomers can be separated by converting the enantiomenc mixtures into a diastereomπc mixture by reaction with an appropnate optically active compound (e g , alcohol), separating the diastereomers and converting (e g , hydrolyzing) the individual diastereomers to the corresponding pure enantiomers With respect to the compounds of formula (I), the invention includes the use of a racemate, one or more enantiomeric forms, one or more diastereomeπc forms, or mixtures thereof The compounds of formula (I) may also exist as tautomers This invention relates to the use of all such tautomers and mixtures thereof The compounds described herein, including the pharmaceutically acceptable salts of such compounds, also include isotopically-labelled compounds, which are identical to those recited in Formula (I), but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature Examples of isotopes that can be incorporated into compounds of the invention include isotopes of H, carbon, nitrogen, oxygen, phosphorous, fluorine and chlorine, such as 2H, 3H, 13C, 14C, 15N, 180, 170, 31P, 32P, 35S, '8F, and 36CI, respectively Compounds of the present invention, prodrugs thereof, and pharmaceutically acceptable salts of said compounds or of said prodrugs which contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of this invention Certain isotopically-labelled compounds of the present invention, for example those into which radioactive isotopes such as 3H and 14C are incorporated, are useful in drug and/or substrate tissue distribution assays Tntiated, i e , 3H, and carbon-14, i e , 14C, isotopes are particularly preferred for their ease of preparation and detectability Further, substitution with heavier isotopes such as deuterium, i e , 2H, can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements and, hence, may be preferred in some circumstances lsotopically labelled compounds of Formula (I) of this invention and prodrugs thereof can generally be prepared by carrying out the procedures disclosed in the Schemes and/or in the Examples and Preparations below, by substituting a readily available lsotopically labelled reagent for a non-isotopically labelled reagent In the case of agents that are solids, it is understood by those skilled in the art that the inventive compounds and salts may exist in different crystal or polymorphic forms, all of which are intended to be within the scope of the present disclosure and specified formulas
A "pharmacological composition" refers to a mixture of one or more of the compounds described herein, or physiologically acceptable salts and solvates thereof, with other chemical components, such as physiologically acceptable carriers and/or excipients The purpose of a pharmacological composition is to facilitate administration of a compound to an organism
A "physiologically acceptable carrier" refers to a carrier or diluent that does not cause significant or otherwise unacceptable irritation to an organism and does not unacceptably abrogate the biological activity and properties of the administered compound
Suitable pharmaceutical carriers include inert diluents or fillers, water and various organic solvents The pharmaceutical compositions may, if desired, contain additional ingredients such as flavorings, binders, excipients and the like Thus for oral administration, tablets containing vaπous excipients, such as citric acid may be employed together with vanous disintegrants such as starch, alginic acid and certain complex silicates and with binding agents such as sucrose, gelatin and acacia Additionally, lubricating agents such as magnesium stearate, sodium lauryl sulfate and talc are often useful for tableting purposes Solid compositions of a similar type may also be employed in soft and hard filled gelatin capsules Preferred materials, therefor, include lactose or milk sugar and high molecular weight polyethylene glycols When aqueous suspensions or elixirs are desired for oral administration the active compound therein may be combined with various sweetening or flavonng agents, coloπng matters or dyes and, if desired, emulsifying agents or suspending agents, together with diluents such as water, ethanol, propylene glycol, glycerin, or combinations thereof
Once improvement of the patient's conditions has occurred, a maintenance dose is administered if necessary Subsequently, the dosage or the frequency of administration, or both, can be reduced, as a function of the symptoms, to a level at which the improved proliferative disorder or condition is retained When the symptoms have been alleviated to the desired level, treatment can cease Patients can, however, require intermittent treatment on a long-term basis upon any recurrence of the disease symptoms
The amount and frequency of administration of the compounds used in the methods described herein and, if applicable, other agents will be regulated according to the judgment of the attending clinician (physician) considering such factors as age, condition and size of the patient as well as severity of the disease being treated
The amount of the active compound administered (e g , for treatment, prophylactic, and/or maintenance) will be dependent on the subject being treated, the severity of the disorder or condition, the rate of administration, the disposition of the compound and the discretion of the prescribing physician However, an effective dosage is in the range of about 0 001 to about 100 mg per kg body weight per day, preferably about 1 to about 35 mg/kg/day, in single or divided doses For a 70 kg human, this would amount to about 0 05 to about 7 g/day, preferably about 0 2 to about 2 5 g/day In some instances, dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, provided that such larger doses are first divided into several small doses for administration throughout the day
The compounds of this invention may also be used in conjunction with other pharmaceutical agents for the treatment of the diseases, conditions and/or disorders described herein Therefore, methods of treatment that include administering compounds of the present invention in combination with other pharmaceutical agents are also provided Suitable pharmaceutical agents that may be used in combination with the compounds of the present invention include anti-obesity agents such as
apohpoprotein-B secretion/microsomal triglyceride transfer protein (apo-B/MTP) inhibitors, 11β-hydroxy steroid dehydrogenase-1 (11 β-HSD type 1) inhibitors, peptide YY^36 or analogs thereof, cannabinoid antagonists (e g , CB-1 antagonists, such as πmonabant), MCR-4 agonists, cholecystokinin-A (CCK-A) agonists, monoamine reuptake inhibitors (such as sibutramine), sympathomimetic agents, neurotensin inhibitors, p3 adrenergic receptor agonists, dopamine agonists (such as bromocriptine), melanocyte- stimυlating hormone receptor analogs, 5HT2c agonists (e g APD356 (Arena)), leptin (the OB protein), leptin analogs, leptin receptor agonists, galanin antagonists, lipase inhibitors (such as tetrahydrolipstatin, i e orhstat), anorectic agents (such as a bombesin agonist), Neuropeptιde-Y receptor antagonists (e g , NPY Y5 receptor antagonists, such as the spiro compounds described in US Patent Nos 6,566,367, 6,649,624, 6,638,942, 6,605,720, 6,495,559, 6,462,053, 6,388,077, 6,335,345, and 6,326,375, US Publication Nos 2002/0151456 and 2003/036652, and PCT Publication Nos WO 03/010175 WO 03/082190 and WO 02/048152), thyromimetic agents, dehydroepiandrosterone or an analog thereof, glucocorticoid receptor agonists or antagonists, orexin receptor antagonists, glucagon-like peptιde-1 receptor agonists, ciliary neurotrophic factors (such as Axokine™ available from Regeneron Pharmaceuticals, lnc , Tarrytown, NY and Procter & Gamble Company, Cincinnati, OH), human agouti- related proteins (AGRP), ghrelin receptor antagonists, histamine 3 receptor antagonists or inverse agonists, neuromedin U receptor agonists, stearoyl Co-A desaturase 1 (SCD1 ) hgands, 5HT6 antagonists, diacylglycerol acyl transferase 1 (DGAT1) inhibitors, diacylglycerol acyl transferase 2 (DGAT2) inhibitors, DPPIV inhibitors, acetyl CoA carboxylase (ACC) inhibitors, sodium dependent glucose transporter 2 (SGLT2) inhibitors, PPAR modulators (alpha, beta, gamma), PDE-10 inhibitors (see e g International Patent Application publication WO 2005/120514), opioid receptor antagonists (mu, kappa and delta), zonisamide, topiramate, radaxafine, and the like Other anti-obesity agents, including the preferred agents set forth hereinbelow, are well known, or will be readily apparent in light of the instant disclosure, to one of ordinary skill in the art Especially preferred are anti-obesity agents selected from the group consisting of orhstat, sibutramine, bromocriptine, ephedπne, leptin, pseudoephedπne, rimonabant, peptide YY^36 Or an analog thereof, and 2-oxo-N-(5-phenylpyrazιnyl)spιro-[ιsobenzofuran-1(3H),4'-pιperιdιne]-1'-carboxamιde Preferably, compounds of the present invention and combination therapies are administered in conjunction with exercise and a sensible diet Representative anti-obesity agents for use in the combinations, pharmaceutical compositions, and methods of the invention can be prepared using methods known to one of ordinary skill in the art, for example, sibutramine can be prepared as described in U S Pat No 4,929,629, bromocriptine can be prepared as descnbed in U S Pat Nos 3,752,814 and 3,752,888, orlistat can be prepared as described in U S Pat Nos 5,274,143, 5,420,305, 5,540,917, and 5,643,874, rimonabant can be prepared as described in U S Pat No 5,624,941, PYY^36 (including analogs) can be prepared as described in US Publication No 2002/0141985 and WO 03/027637, and the NPY Y5 receptor antagonist 2-oxo-N-(5- phenylpyrazιnyl)spιro[ιsobenzofuran-1(3H),4'-pιperιdιne]-1'-carboxamιde can be prepared as described in US Publication No 2002/0151456 Other useful NPY Y5 receptor antagonists include those described in PCT Publication No 03/082190, such as 3-oxo-N-(5-phenyl-2-pyrazιnyl)-spιro[ιsobenzofuran-1 (3H), 4'- pιperιdιne]-1'-carboxamιde, 3-oxo-N-(7-trιfluoromethylpyπdo[3,2-b]pyrιdιn-2-yl)-spιro-[ιsobenzofuran- 1(3H), 4'-pιperιdιne]-1'-carboxamιde, N- [5-(3-fluorophenyl)-2-pyrιmιdιnyl]-3-oxospιro-[ιsobenzofuran-
1(3H)1 [4'-pιperιdιne]-1'-carboxamιde, frans-3'-oxo-N-(5-phenyl-2-pyrιmιdιnyl)] spιro[cyclohexane- 1 ,1'(3'H)-ιsobenzofuran]-4-carboxamιde, frans-S'-oxo-N- li-tS-quinolylH-imidazolyllspirotcyclohexane- 1 ,1'(3'H)-ιsobenzofuran]-4-carboxamιde, frans-3-oxo-N-(5-phenyl-2-pyraztnyl)spιro[4-azaιso- benzofuran-1(3H),1'-cyclohexane]-4'-carboxamide, /rans-N-[5-(3-fluorophenyl)-2-pyrιmιdιnyl]-3- oxospιro[5-azaιsobenzofuran-1(3H), I'-cyclohexaneH'-carboxamide, frans-N-[5-(2-fluorophenyl)-2- pyπmιdιnyl]-3-oxospιro[5-azaιsobenzofuran-1(3H), V-cyclohexaneH'-carboxamide, frans-N-[1-(3,5- difluorophenylM-imidazolylJ-S-oxospiro^-azaisobenzofuran-lfSHJ.I'-cyclohexaneH'-carboxamide, frans-3-oxo-N-(1-phenyl-4-pyrazolyl)spιro[4-azaιsobenzofuran-1(3H),1 '-cyclohexane]-4'-carboxamιde, trans-U-l 1 -(2-fluorophenyl)-3-pyrazolyl]-3-oxospιro[6-azaιsobenzofuran- 1 (3H ), 1 '-cyclohexane]-4'- carboxamide, frans-3-oxo-N-(l-phenyl-3-pyrazolyl)spiro[6-azaisobenzofuran-1(3H),1 '-cyclohexane]-4'- carboxamide, frans-3-oxo-N-(2-phenyl-1 ,2,3-tπazol-4-yl)spιro[6-azaιsobenzofuran-1(3H),1 '- cyclohexane]-4'-carboxamιde, and pharmaceutically acceptable salts and esters thereof All of the above recited U S patents and publications are incorporated herein by reference
Other suitable pharmaceutical agents that may be administered in combination with the compounds of the present invention include agents designed to treat tobacco abuse (e g , nicotine receptor partial agonists, bupropion hypochloπde (also known under the tradename Zyban™) and nicotine replacement therapies), agents to treat erectile dysfunction (e g , dopaminergic agents, such as apomorphine), ADD/ADHD agents (e g , Ritalin™, Strattera™, Concerta™ and Adderall™), and agents to treat alcoholism, such as opioid antagonists (e g , naltrexone (also known under the tradename ReVia™) and nalmefene), disulfiram (also known under the tradename Antabuse™), and acamprosate (also known under the tradename Campral™)) In addition, agents for reducing alcohol withdrawal symptoms may also be co-administered, such as benzodiazepines, beta-blockers, clonidine, carbamazepine, pregabalm, and gabapentin (Neurontin™) Treatment for alcoholism is preferably administered in combination with behavioral therapy including such components as motivational enhancement therapy, cognitive behavioral therapy, and referral to self-help groups, including Alcohol Anonymous (AA)
Other pharmaceutical agents that may be useful include antihypertensive agents, anti-inflammatory agents (e g , COX-2 inhibitors), antidepressants (e g , fluoxetine hydrochloride (Prozac™)), cognitive improvement agents (e g , donepezil hydrochloride (Aircept™) and other acetylcholinesterase inhibitors), neuroprotective agents (e g , memantine), antipsychotic medications (e g , ziprasidone (Geodon™), risperidone (Risperdal™), and olanzapine (Zyprexa™)), insulin and insulin analogs (e g , LysPro insulin), GLP-1 (7-37) (insulinotropin) and GLP-1 (7-36)-NH2, sulfonylureas and analogs thereof chlorpropamide, glibenclamide, tolbutamide, tolazamide, acetohexamide, Glypizide®, glimepiπde, repaglmide, meglitmide, biguanides metformin, phenformin, buformin, α-2-antagonιsts and imidazolines midaglizole, isaghdole, deriglidole, idazoxan, efaroxan, fluparoxan, other insulin secretagogues linogliπde, A-4166, ghtazones ciglitazone, Actos® (pioglitazone), englitazone, troghtazone, darghtazone, Avandia® (BRL49653), fatty acid oxidation inhibitors clomoxir, etomoxir, α-glucosιdase inhibitors acarbose, miglitol, emiglitate, voghbose, MDL-25,637, camiglibose, MDL-73,945, U-agonists BRL 35135, BRL 37344, RO 16-8714, ICI D7114, CL 316,243, phosphodiesterase inhibitors L-386,398, lipid-loweπng agents benfluorex fenfluramine, vanadate and vanadium complexes (e g , Naglivan®) and peroxovanadium complexes,
amylin antagonists, glucagon antagonists, gluconeogenesis inhibitors, somatostatin analogs, antilipolytic agents nicotinic acid, acipimox, WAG 994, pramlintide (Symlin™), AC 2993, nateghnide, aldose reductase inhibitors (e g , zopolrestat), glycogen phosphorylase inhibitors, sorbitol dehydrogenase inhibitors, sodium-hydrogen exchanger type 1 (NHE-1) inhibitors and/or cholesterol biosynthesis inhibitors or cholesterol absorption inhibitors, especially a HMG-CoA reductase inhibitor (e g , atorvastatin or the hemicalcium salt thereof), or a HMG-CoA synthase inhibitor, or a HMG-CoA reductase or synthase gene expression inhibitor, a CETP inhibitor, a bile acid sequesterant, a fibrate, an ACAT inhibitor, a squalene synthetase inhibitor, an anti-oxidant or niacin The compounds of the present invention may also be administered in combination with a naturally occurring compound that acts to lower plasma cholesterol levels Such naturally occurring compounds are commonly called nutraceuticals and include, for example, garlic extract, Hoodia plant extracts, and niacin Examples of anti-diabetes agents suitable for combination therapy with the present invention include, by way of example only Biguanides, Glucophage Tablets (Bristol-Myers-Squibb), Glucosadase Inhibitors, for example, Precose Tablets (Bayer), Intermediate Acting Insulins, for example, Humulin L (Lilly), Humulin N (Lilly), Humilin N NPH(LiIIy), lletin Il (Lilly), lletin Il NPH (pork) (Lilly), Novolin L Human Insulin (Novo Nordisk), Novolin N Human Insulin (Novo Nordisk), Novolin N PenFill (Novo Nordisk), Novolin N prefilled Syringe Disposable Insulin Delivery System (Novo Nordisk), Purified Pork Lente Insulin (Novo Nordisk), Purified Pork NPH lsophane Insulin (Novo Nordisk) Examples of intermediate and rapid acting insulins suitable for combination therapy with the present invention include, by way of example only Humulin 50/50 (Lilly), Humulin 70/30 (Lilly), Humulin 70/30 cartridge (Lilly), Novolin 70/30 PenFill (Novo Nordisk), Novolin 70/30 Prefilled Disposable Insulin Delivery system (Novo Nordisk) Examples of rapid acting insulins suitable for combination therapy with the present invention include, by way of example only Humalog Injection (Lilly), Humalm R Regular (Lilly), lletin Il (Lilly), Regular (pork), Novolin R Human Insulin (Novo Nordisk), Purified Pork Regular Insulin (Novo Nordisk), Velosulin BR Human Insulin (Novo Nordisk)
Further examples of other anti-diabetes agents suitable for combination therapy with the present invention include, by way of example only meglitinides, sulfonylureas, and thiazolidinediones Examples of meglitinides include, but are not limited to Prandin Tablets (Novo Nordisk Examples of sulfonylureas include, but are not limited to Amaryl tablets (Hoechst Marion Roussel),Dιabeta tablets (Hoechst Marion Roussel), Diabinese Tablets (Pfizer), Glucotrol Tablets (Pfizer), Glucotrol XL Extended Release Tablets (Pfizer), Glynase PresTab Tablets (Pharmacia & Upjohn), Micronase Tablets (Parke-Davis) Examples of thiazolidinediones include, but are not limited to Rezuhn Tablets (Parke-Davis) When depression is of concern (e g , treatment of, prevention of, maintenance against, etc ), the patient would be expected to benefit from administration of a compound of Formula I in conjunction with, by way of example only, Effexor Tablets (Wyeth), Effexor XR Capsules (Wyeth), Remeron Tablets (Organon), Remeron SolTab Tablets (Organon), Serzone Tablets (Bristol-Myers Squibb), Wellbutrin Tablets (GlaxoSmithKlme), Wellbutrin SR Sustained-Release Tablets (GlaxoSmithKhne), Nardil Tablets (Parke- Davis), Parnate Tablets (GlaxoSmithKlme), Selective Serotonin Reuptake Inhibitors (SSRI) Celexa Oral Solution (Forest), Celexa Tablets (Forest), Lexapro Tablets (Forest), Paxil CR Controlled-Release Tablets (GlaxoSmithKlme), Paxil Oral Suspension (GlaxoSmithKlme), Paxil Tablets (GlaxoSmithKlme), Prozac Pulvates and Liquid (Dista), Zoloft Oral Concentrate (Pfizer), Zoloft Tablets (Pfizer), Norpramin
Tablets (Aventis), Sinequan Capsules (Pfizer), Sinequan Oral Concentrate (Pfizer), Surmontil Capsules (Odyssey), Vivactil Tablets (Merck and Odyssey)
Examples of anxiolytic agents suitable for combination therapy with the present invention include, by way of example only Tranxene T-TAB tablets (Abbott), Tranxene-SD Tablets (Abbott), Tranxene SD Half Strength Tablets (Abbott), Valium Tablets (Roche Products), Xanax Tablets (Pharmacia & Upjohn), Atarax Tablets & Syrup (Pfizer), Effexor XR Capsules (Wyeth), Paxil Oral Suspension (GlaxoSmithKline), Paxil Tablets (GlaxoSmithKline), Sinequan Capsules (Pfizer), Sinequan Oral Concentrate (Pfizer), Vistaπl Capsules (Pfizer), Vistaπl Intramuscular Solution (Pfizer), Vistaπl Oral Suspension (Pfizer), Zoloft Oral Concentrate (Pfizer), Zoloft Tablets (Pfizer)
In some situations, centrally-acting compounds, when either administered alone or in combination therapies such as those mentioned herein, may cause nausea and/or emesis and so it could be advantageous to administer compounds or combinations of the present invention alongside a suitable anti-emetic agent, for example a 5-HT3 antagonist or a neurokιnιn-1 (NK-1) antagonist Suitable 5-HT3 antagonists include, but are not limited to, granisetron, ondansetron, tropisetron, ramosetron, palonsetron, indisetron, dolasetron, alosetron and azasetron
Suitable NK-1 antagonists include, but are not limited to, aprepitant, casopitant, ezlopitant, cilapitant, netupitant, vestipitant, vofopitant and 2-(R)-(1-(R)-3,5-bιs(trιfluoromethyl)phenyl)ethoxy-4-(5- (dιmethylamιno)methyl-1,2,3-tπazol-4-yl)methyl-3-(S)-(4-fluorophenyl)moφholιne See for example International Patent Application publication number WO2006/049933
The dosage of the additional pharmaceutical agent (e g , anti-obesity agent) will also be generally dependent upon a number of factors including the health of the subject being treated, the extent of treatment desired, the nature and kind of concurrent therapy, if any, and the frequency of treatment and the nature of the effect desired In general, the dosage range of an anti-obesity agent is in the range of from about 0 001 mg to about 100 mg per kilogram body weight of the individual per day, preferably from about 0 1 mg to about 10 mg per kilogram body weight of the individual per day However, some variability in the general dosage range may also be required depending upon the age and weight of the subject being treated, the intended route of administration, the particular anti-obesity agent being administered and the like The determination of dosage ranges and optimal dosages for a particular patient is also well within the ability of one of ordinary skill in the art having the benefit of the instant disclosure In general, the compounds described herein and, in embodiments where combinational therapy is employed, other agents do not have to be administered in the same pharmaceutical composition, and may, because of different physical and chemical characteristics, have to be administered by different routes The determination of the mode of administration and the advisability of administration, where possible, in the same pharmaceutical composition, is well within the knowledge of the skilled clinician The initial administration can be made according to established protocols known in the art, and then, based upon the observed effects, the dosage, modes of administration and times of administration can be modified by the skilled clinician The particular choice of compounds used will depend upon the
diagnosis of the attending physicians and their judgment of the condition of the patient and the appropriate treatment protocol The compounds may be administered concurrently (e g , simultaneously, essentially simultaneously or within the same treatment protocol) or sequentially, depending upon the nature of the proliferative disease, the condition of the patient, and the actual choice of compounds used By sequentially and within the same treatment protocol is meant that the active agents may be administered in any order, as deemed appropriate by the skilled clinician, including for example, a first administration of active agent A, followed by an administration of active agent B1 which could be repeated on a daily or other timeframe Alternatively, acitve agent A could be administered for a period (e g a few days/weeks), and the treatment switched to administration of active agent B for a further period Active agents A and B could be single agents or a combination of agents, where at least one of A and B comprises a compound of formula I1 or a salt, solvate or prodrug thereof according to the invention
Pharmaceutical Compositions/Formulations, Dosaqinq. and Modes of Administration Pharmaceutical (including pharmaceuticals for veterinaπal use) compositions according to the invention may, alternatively or in addition to a compound of Formula (I)1 comprise as an active ingredient or pharmaceutically acceptable salts of such compounds Such compounds and salts are sometimes referred to herein collectively as "active agents" or "agents " Administration of the compounds of the present invention (hereinafter the "active compound(s)") can be effected by any method that enables delivery of the compounds to the site of action
Methods of preparing various pharmaceutical compositions with a specific amount of active compound are known, or will be apparent, to those skilled in this art In addition, those of ordinary skill in the art are familiar with formulation and administration techniques Such topics are discussed, e g , in Goodman and Gilman's The Pharmacological Basis of Therapeutics, current edition, Pergamon Press, and Remington's Pharmaceutical Sciences (current edition ) Mack Publishing Co , Easton, Pa These techniques can be employed in appropriate aspects and embodiments of the methods and compositions described herein The following examples are provided for illustrative purposes only and are not meant to serve as limitations of the present disclosure The compounds utilized in the methods of the instant invention may be administered either alone or in combination with pharmaceutically acceptable carriers, excipients or diluents, in a pharmaceutical composition, according to standard pharmaceutical practice
Administration methods include oral routes (e g with a solid or liquid formulation), intraduodenal routes, parenteral injection (including intravenous, subcutaneous, intramuscular, intravascular or infusion), topical, pulmonary, intranasal, and rectal administration For example, the therapeutic or pharmaceutical compositions of the invention can be administered locally to the area in need of treatment This may be achieved by, for example, but not limited to, local infusion dunng surgery, topical application, e g , cream, ointment, injection, catheter, or implant, said implant made, e g , out of a porous, non-porous, or gelatinous material, including membranes, such as sialastic membranes, or fibers The administration can also be by direct injection at the site (or former site) of a tumor or neoplastic or pre-neoplastic tissue
Still further, the therapeutic or pharmaceutical composition can be delivered in a vesicle, e g , a liposome (see, for example, Langer, 1990, Science, 249 1527-1533, Treat et al , 1989, Liposomes in the Therapy of Infectious Disease and Cancer, Lopez-Bernstein and Fidler (eds ), Liss, N Y , pp 353-365) The pharmaceutical compositions used in the methods of the present invention can be delivered in a controlled release system In one embodiment, a pump may be used (see, Sefton, 1987, CRC Crit Ref Biomed Eng 14 201 , Buchwald et al , 1980, Surgery, 88 507, Saudek er a/ , 1989, N Engl J Med , 321 574) Additionally, a controlled release system can be placed in proximity of the therapeutic target (see, Goodson, 1984, Medical Applications of Controlled Release, VoI 2, pp 115-138) The pharmaceutical composition may be in unit dosage forms suitable for single administration of precise dosages The pharmaceutical composition will include a conventional pharmaceutical earner or excipient and a compound according to the invention as an active ingredient In addition, it may include other medicinal or pharmaceutical agents, carriers, adjuvants, etc
The pharmaceutical compositions used in the methods of the instant invention can contain the active ingredient in a form suitable for oral use, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, or syrups or elixirs Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients, which are suitable for the manufacture of tablets These excipients may be, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate, granulating and disintegrating agents, such as microcrystalline cellulose, sodium crosscarmellose, corn starch, or alginic acid, binding agents, for example starch, gelatin, polyvinylpyrrolidone or acacia, and lubricating agents, for example, magnesium stearate, stearic acid or talc The tablets may be uncoated or they may be coated by known techniques to mask the taste of the drug or delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer peπod For example, a water soluble taste masking material such as hydroxypropylmethyl-cellulose or hydroxypropylcellulose, or a time delay material such as ethyl cellulose, or cellulose acetate butyrate may be employed Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water soluble carrier such as polyethyleneglycol or an oil medium, for example peanut oil, liquid paraffin, or olive oil
For companion animals or food-source animals, it is often more convenient to incorporate the drug substance into the animal's food source This may be accomplished by addition of the drug substance to the food source as a dry powder or as a liquid solution or suspension
Aqueous suspensions can contain the active material in admixture with excipients suitable for the manufacture of aqueous suspensions Such excipients can act as suspending agents and include, e g , sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethyl-cellulose, sodium alginate,
polyvinylpyrrolidone, gum tragacanth and gum acacia, dispersing or wetting agents may be a naturally- occurring phosphatide, for example lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethylene-oxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan monooleate The aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose, saccharin or aspartame
Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in mineral oil such as liquid paraffin The oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol Sweetening agents such as those set forth above, and flavoπng agents may be added to provide a palatable oral preparation These compositions may be preserved by the addition of an anti-oxidant, e g , butylated hydroxyanisol, alpha-tocopherol, or ascorbic acid
Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above Additional excipients, for example sweetening, flavoring and coloring agents, may also be present These compositions may be preserved by the addition of antιoxιdant(s)
The pharmaceutical compositions used in the methods of the instant invention may also be in the form of oιl-ιn-water emulsions The oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these Suitable emulsifying agents may be naturally-occurring phosphatides, for example soy bean lecithin, and esters or partial esters derived from fatty acids and hexitol anhydrides, for example sorbitan monooleate, and condensation products of the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monooleate The emulsions may also contain sweetening, flavoring agents, preservatives and antioxidants Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose Such formulations may also contain a demulcent, a preservative, flavoring and coloring agents and antioxidant
Pulmonary administration by inhalation may be accomplished by means of producing liquid or powdered aerosols, for example, by using any of various devices known in the art (see e g Newman, S P , 1984, in Aerosols and the Lung, Clarke and Pavia (Eds ), Butterworths, London, England, pp 197-224, PCT Publication No WO 92/16192 dated Oct 1 , 1992, PCT Publication No WO 91/08760 dated Jun 27, 1991 , NTIS Patent Application 7-504-047 filed Apr 3, 1990 by Roosdorp and Crystal) including but not limited to nebulizers, metered dose inhalers, and powder inhalers Various delivery devices are commercially available and can be employed, e g Ultravent nebulizer (Mallinckrodt, Inc, St Louis, Mo ), Acorn Il nebulizer (Marquest Medical Products, Englewood, Colo ), Ventolin metered dose inhalers (Glaxo Inc , Research Triangle Park, N C ), Spinhaler powder inhaler (Fisons Corp , Bedford, Mass ) or
Turbohaler (Astra) Such devices typically entail the use of formulations suitable for dispensing from such a device, in which a propellant material may be present Ultrasonic nebulizers may also be used A nebulizer may be used to produce aerosol particles, or any of various physiologically inert gases may be used as an aerosolizing agent Other components such as physiologically acceptable surfactants (e g glycerides), excipients (e g lactose), carriers (e g water, alcohol), and diluents may also be included As will be understood by those skilled in the art of delivering pharmaceuticals by the pulmonary route, a major cnteria for the selection of a particular device for producing an aerosol is the size of the resultant aerosol particles Smaller particles are needed if the drug particles are mainly or only intended to be delivered to the peripheral lung, / e the alveoli (e g 0 1-3 μm), while larger drug particles are needed (e g 3-10 μm) if delivery is only or mainly to the central pulmonary system such as the upper bronchi Impact of particle sizes on the site of deposition within the respiratory tract is generally known to those skilled in the art
The pharmaceutical compositions may be in the form of a sterile injectable aqueous solutions Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution
The sterile injectable preparation may also be a sterile injectable oιl-ιn-water microemulsion where the active ingredient is dissolved in the oily phase For example, the active ingredient may be first dissolved in a mixture of soybean oil and lecithin The oil solution then introduced into a water and glycerol mixture and processed to form a microemulsion The injectable solutions or microemulsions may be introduced into a patient's blood-stream by local bolus injection Alternatively, it may be advantageous to administer the solution or microemulsion in such a way as to maintain a constant circulating concentration of the instant compound In order to maintain such a constant concentration, a continuous intravenous delivery device may be utilized An example of such a device is the Deltec CADD-PLUS™ model 5400 intravenous pump The pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleagenous suspension for intramuscular and subcutaneous administration This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents, which have been mentioned above The sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1 ,3-butanedιol Exemplary parenteral administration forms also include solutions or suspensions of active compounds in sterile aqueous solutions, for example, aqueous propylene glycol or dextrose solutions All such dosage forms can be suitable buffered, if desired In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium For this purpose any bland fixed oil may be employed including synthetic mono- or diglycendes In addition, fatty acids such as oleic acid find use in the preparation of injectables
The compounds of Formula I may also be administered in the form of suppositories for rectal administration of the drug These compositions can be prepared by mixing the inhibitors with a suitable non-imtating excipient, which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug Such materials include cocoa butter, glycerinated gelatin, Hated vegetable oils, mixtures of polyethylene glycols of various molecular weights and fatty acid esters of polyethylene glycol
For topical use, creams, ointments, jellies, solutions or suspensions, etc , containing a macrolide can be used As used herein, topical application can include mouth washes and gargles The compounds used in the methods and compositions described herein can be administered in intranasal form via topical use of suitable intranasal vehicles and delivery devices, or via transdermal routes, using those forms of transdermal skin patches well known to those of ordinary skill in the art To be administered in the form of a transdermal delivery system, the dosage administration will, of course, be continuous rather than intermittent throughout the dosage regimen
Preferably X-Ar is (CH2)2-Ar CH2OAr or OCH2-Ar More preferably X-Ar is OCH2-Ar
Preferably A is N and B is CH or N More preferably A is N and B is CH
Preferably Ar is phenyl, fluorophenyl or chlorophenyl
More preferably Ar is phenyl, 4-chlorophenyl or 4-fluorophenyl Most preferably Ar is phenyl
Preferably R1 is piperidine, pyrrolidine, piperazine, octahydro-pyrrolo[3,4-c]pyrrole, octahydro-pyrrolo[3,4- bjpyrrole, or octahydro-pyrrolo[3,4-c]pyπdιne, attached to the "ABCCHCHC" ring via a N atom, which ring system is optionally substituted by one or more substituents independently selected from =0,
C1-C5 alkyl, C3-C5 cycloalkyl, OH, C(O)C1-C5 alkyl, C(O)C3-C5 cycloalkyl, C(O)OC1-C5 alkyl, NR6R7,
NR8C(O)R9, NR8C(O)OR9, 0(C1-C5 alkyl) or 0(C3-C5 cycloalkyl) More preferably R1 is pipendine, pyrrolidine, piperazine, octahydro-pyrrolo[3,4-c]pyrrole, octahydro- pyrrolo[3,4-b]pyrrole, or octahydro-pyrrolo[3,4-c]pyrιdιne, attached to the "ABCDEC" ring via a N atom, which ring system is optionally substituted by one or more substituents independently selected from OH,
OMe, OEt, Me, Et, NH2, NHMe, NMe2, NMeC(O)Me and C(O)Me Yet more preferably R1 is pipendine, pyrrolidine, piperazine, octahydro-pyrrolo[3,4-c]pyrrole, octahydro- pyrrolo[3,4-b]pyrrole, or octahydro-pyrrolo[3,4-c]pyπdιne, attached to the "ABCDEC" ring via a N atom, which ring system is optionally substituted by one or more substituents independently selected from
NHMe, NMe2, OH, NH2, Me and Et Most preferably R1 is selected from one of the following groups

Another preferred aspect of the invention is a group of compounds wherein A, B, X, R1 and Ar are selected from the values in the compounds of the Examples below, or a pharmaceutically acceptable salt, solvate or prodrug thereof
Another preferred aspect of the invention is a group of compounds of formula

Another preferred aspect of the invention is a group of compounds of formula
1


5 or a pharmaceutically acceptable salt, solvate or prodrug thereof.
Preferably the compounds of the invention are selected from one of the Examples below, or a pharmaceutically acceptable salt, solvate or prodrug thereof.
More preferably the compounds of the invention are selected from the compounds of Examples 17a,
10 17b, 18a, 18b, 19 or 20, or a pharmaceutically acceptable salt, solvate or prodrug thereof
Most preferably the compounds of the invention are selected from the compounds of Examples 17a,
18a, 19 or 20, or a pharmaceutically acceptable salt, solvate or prodrug thereof.
Compounds of the invention may be prepared in a variety of ways, including those mentioned in the 15 Examples and Preparations. The routes below illustrate methods of synthesising compounds of formula (I), wherein, unless otherwise described, the susbtituents have the values mentioned above with regard to formula (I).
The skilled person will appreciate that the compounds of the invention could be made by methods other than those specifically described herein, by the methods described herein and/or adaptation thereof, for example by adaptation of methods known in the art, including the art mentioned earlier herein, such as WO2003068230, WO2005018557 and WO 2005103039 Suitable guides to synthesis, functional group interconversions, use of protetcting groups, etc , are for example Comprehensive Organic Transformations" by RC Larock, VCH Publishers lnc (1989), Advanced Organic Chemistry" by J March, Wiley lnterscience (1985), "Designing Organic Synthesis" by S Warren, Wiley lnterscience (1978), Organic Synthesis - The Disconnection Approach" by S Warren, Wiley lnetrscience (1982), "Guidebook to Organic Synthesis" by RK Mackie and DM Smith, Longman (1982), "Protective Groups in Organic Synthesis" by TW Greene and PGM Wuts, John Wiley and Sons, lnc (1999), and "Protecting Groups" by PJ, Kocienski, Georg Thieme Verlag (1994), and any updated versions of said standard works
Compounds of Formula I (Scheme 1) can be prepared by a coupling reaction of a fragment Il with a fragment III, where X1 is a suitable leaving group, for example iodide, bromide or tπflate (preferably iodide)
Scheme 1

Compounds of Formula Il and III are mixed in a suitable solvent such as toluene, DMSO or DMF (preferably DMF), at a temperature between about 50 and about 150 0C (preferably about 80 0C) in the presence of a suitable copper catalyst (preferably CuI) and bidentate ligand (preferably trans-1 ,2- diaminocyclohexane) This reaction is exemplified in Example 16 Alternatively, where X1 is a suitable group, for example a boron derivative (preferably B(OH)2), a coupling reaction can be carried out in a suitable solvent, for example dichlorom ethane, tetrahydrofuran or acetonitrile (preferably dichloromethane) mediated by a suitable metal salt, for example a copper(ll) derivative (preferably Cu(OAc)2), in the presence of a suitable base, for example pyridine, triethylamine or DBU (preferably pyridine) and a suitable drying agent (e g 4A molecular sieves) at a temperature between about 25 and about 50 0C (preferably about 25 0C)
Compounds of formula (II) and (III) are available commercially, or can be made by methods disclosed herein, for example in relation to the Examples and Preparations below, or suitable adaptation thereof by methods known in the art
Compounds of Formula III (Scheme 2) can be suitably prepared by a displacement reaction from compound Vl, where X1 and X2 are suitable halogen substituents (preferably bromo), using a suitable
amine "R1H", a suitable base, for example tπethylamine, sodium carbonate or potassium carbonate (preferably potassium carbonate), in a suitable solvent, for example acetonitπle, n-butanol or dimethyl sulfoxide (preferably n-butanol) at a temperature between about 25 and about 150 0C (preferably about 11O 0C)
Scheme 2

Vl "I
Some of the amines R1H are available commercially (e g see Table below), others are available from the literature or suitable adaptation thereof using methods known to the skilled person This reaction is exemplified in Preparation 6





The reactants Il and Vl are mixed in a suitable solvent such as DMSO or N,N-dιmethylformamιde (preferably N,N-dιmethylformamιde), at a temperature between about 25 and about 150 0C (preferably about 80 0C) in the presence of a suitable copper catalyst (preferably CuI) and ligand (preferably trans- 1 ,2-dιamιnocyclohexane) This reaction is exemplified in Preparation 2 Method B Alternatively, where X1 is a suitable group, for example a boron derivative (preferably B(OH)2), a coupling reaction can be earned out in a suitable solvent, for example dichloromethane, tetrahydrofuran or acetonitrile (preferably dichloromethane) mediated by a suitable metal salt (preferably Cu(OAc)2) in the presence of a suitable base, for example pyridine or tπethylamine (preferably pyridine) and a suitable drying agent (preferably 4A molecular sieves) at a temperature between about 25 and about 100 0C (preferably about 25 0C) Compounds of Formula I can be prepared from fragments of Formula VII by treating with a suitable amine R1H using a suitable solvent, for example acetonitrile, dimethyl sulfoxide or N N- dimethylformamide (preferably dimethyl sulfoxide), using a suitable base, for example potassium carbonate, cesium carbonate or triethylamine (preferably potassium carbonate) at a temperature between about 25 and about 150 0C (preferably about 110 0C) The reaction is exemplified in Examples 1-7
Compounds of Formula VIII (Scheme 4) can be transformed to versatile intermediates of Formulae IX and X, where R100 is a suitable substituent, for example phenyl, 4-bromophenyl or trifluromethyl
Scheme 4

Ar^)P Xl

Treatment of compounds of Formula VIII in a suitable solvent, for example ethanol methanol or acetonitrile (preferably ethanol), with a suitable hydrogen source, for example dihydrotoluene, H2 gas or ammonium formate (preferably dihydrotoluene) and a suitable metal catalyst (preferably palladium hydroxide on carbon), at a temperature between about 25 and about 100 0C, (preferably about 60 0C) gives compounds of Formula IX The reaction is exemplified in Preparation 4 Compounds of Formula IX can be converted to compounds of Formula X using a suitable source of R100-sulfonyl, e g trifluromethane sulfonic anhydride, in a suitable solvent, for example dichloromethane, ethyl acetate or tetrahydrofuran (preferably dichloromethane), using a suitable base, for example triethylamine or pyridine, at a temperature between about -40 0C and about 25 0C (preferably about 25 0C) Compounds of Formula IX can be converted to compounds of Formula XIII using a range of conditions For example, treatment of fragments IX with a suitable alkylating agent ArCH2X3 (Xl), where X3 is a suitable leaving group, for example a halide, mesylate, tosylate or tπflate (preferably bromide), in a suitable solvent, for example acetonitrile or N,N-dιmethylformamιde (preferably acetonitrile) with a suitable base, for example cesium carbonate or potassium carbonate (preferably cesium carbonate), at a temperature between about 25 and about 150 0C (preferably about 80 0C) This reaction is exemplified in Example 10 Alternatively fragments of Formula IX can be alkylated via a Mitsonobu reaction using a suitable alcohol of Formula XII using suitable reagents, for example di-isopropyldiazodicarboxylate or diethyldiazodicarboxylate (preferably dι-ιsopropyldιazodιcarboxylate) and tπphenylphosphine or
tributylphosphine (preferably tπphenylphosphine), in a suitable solvent, for example tetrahydrofuran or acetonitrile (preferably tetrahydrofuran) at a temperature between about 0 0C and about 100 0C (preferably about 25 0C) Compounds of Formula X can also be transformed to compounds of Formula XV using a variety of conditions For example, treatment of compounds of Formula X with a suitable organozinc reagent of Formula XIV in a suitable solvent, for example tetrahydrofuran or 2-methyl tetrahydrofuran (preferably tetrahydrofuran), optionally with a suitable co-solvent, for example N-methyl pyrrohdinone, with a suitable metal catalyst, (preferably bιs(trι-t-butylphosphιne)palladιum) at a temperature between about 25 0C and about 100 0C, (preferably about 50 0C) gives compounds of Formula XV
Some compounds of formula (I) may be converted into other compounds of formula (I) by known functional group interconversions
All of the above reactions and the preparations of novel starting materials used in the preceding methods are conventional and appropriate reagents and reaction conditions for their performance or preparation as well as procedures for isolating the desired products will be well-known to those skilled in the art with reference to literature precedents and the Examples and Preparations herein described Furthermore, it will be clear to the skilled person that the compounds of formula (I) may be synthesised either directly from, or by analogy to, the schemes, literature precedents and the Examples and Preparations herein described, as well as the common general knowledge
EXAMPLES OF PHARMACEUTICAL COMPOSITIONS
Example 1 Parenteral Composition
To prepare a parenteral pharmaceutical composition suitable for administration by injection, 100 mg of a water-soluble salt of a compound of Formula I is dissolved in DMSO and then mixed with 10 mL of 0 9% sterile saline The mixture is incorporated into a dosage unit form suitable for administration by injection
Example 2 Oral Composition
To prepare a pharmaceutical composition for oral delivery, 100 mg of a compound of Formula I is mixed with 750 mg of lactose The mixture is incorporated into an oral dosage unit for, such as a hard gelatin capsule, which is suitable for oral administration
BIOLOGICAL TESTING
hERG Binding Assay
Membrane homogenates of HEK-293S (Cell line #15-08) cells expressing the HERG product supplied by (PGRD) Sandwich Laboratories were prepared as follows Cell pellets were thawed at room temperature and kept on ice Buffer (5OmM Tris HCI, 1mM MgCI2, 1OmM KCI, pH 7 4, at 4°C) was added to each cell pellet (10 ml of buffer per 10 g of packed cell pellet) and the mixture homogenised using an Omni
LabTek homogeniser (20,000rpm for 30 seconds) The homogenate was centπfuged at 48,00Og for 20 minutes between 3 and 50C in a Sorvall Evolution RC centrifuge and the supernatant discarded The pellet was resuspended, homogenised (20,000rpm for 10 seconds), and centrifuged as before The resultant supernatant was discarded and the final pellet resuspended (100 ml of the above buffer per 10 g of packed cell pellet), homogenised (20,000rpm for 10 seconds), dispensed in to tubes in 1,2 and 5 ml ahquots and stored between -750C and -850C until use Protein concentration was determined using a Coomassie Blue kit as per manufacturer's instructions (Sigma 610A & 610-11)
The Cy3B ligand was stored in 100% DMSO and diluted to 6nM in assay buffer (5OmM Tris HCI, 1mM MgCI2, 1OmM KCI, 0 05% Pluronic F127, pH 74 at 4°C) on the day of the experiment Test samples and controls were diluted in 6% DMSO, 0 05% Pluronic F127 Cell membranes were removed from the - 8O0C freezer and placed on ice after defrosting When required the defrosted membranes were homogenised using a polytronic device for no more than 10 seconds, they were then diluted in the above assay buffer to produce a working solution of 0 3mg/ml The assay was compiled by adding 1OmL of test compound or control solution, 1OmL of the Cy3B ligand and 1OmL of cell membranes to a black 384-well plate (Matrix, Cat No 4318) The plates were mixed and then incubated for a minimum of 2 hours prior to reading on a Tecan Ultra (Excitation 530nm, Emission 590nm) All IC50 and K1 data were generated using Pfizer proprietary software
The practice of the instant invention for treating obesity or related eating disorders (including promoting weight loss or reducing weight gain) can be evidenced by activity in at least one of the protocols described hereinbelow
Food Intake
The following screen is used to evaluate the efficacy of test compounds for inhibiting food intake in Sprague-Dawley rats after an overnight fast
Male Sprague-Dawley rats are obtained from Charles River Laboratoπes, lnc (Wilmington, MA) The rats are individually housed and fed powdered chow They are maintained on a 12 hour light/dark cycle and received food and water ad libitum The animals are acclimated to the vivarium for a period of one week before testing is conducted Testing is completed during the light portion of the cycle To conduct the food intake efficacy screen, rats are transferred to individual test cages without food the afternoon prior to testing, and the rats are fasted overnight After the overnight fast, rats are dosed the following morning with vehicle or test compounds A known antagonist is dosed (3 mg/kg) as a positive control, and a control group receives vehicle alone (no compound) The test compounds are dosed at ranges between 0 1 and 100 mg/kg depending upon the compound The standard vehicle is 0 5% (w/v) methylcellulose in water and the standard route of administration is oral However, different vehicles and routes of administration are used to accommodate various compounds when required Food is provided to the rats 30 minutes after dosing and the Oxymax automated food intake system (Columbus Instruments, Columbus, Ohio) is started Individual rat food intake is recorded continuously at 10-mιnute intervals for a period of two hours When required, food intake is recorded manually using an electronic scale, food is weighed every 30 minutes after food is provided up to four hours after food is provided
Compound efficacy is determined by comparing the food intake pattern of compound-treated rats to vehicle and the standard positive control
Oxygen Consumption Whole body oxygen consumption is measured using an indirect calorimeter (Oxymax from Columbus Instruments, Columbus, OH) in male Sprague Dawley rats (if another rat strain or female rats is used, it will be specified) Rats (300-38Og body weight) are placed in the calorimeter chambers and the chambers are placed in activity monitors These studies are done during the light cycle Prior to the measurement of oxygen consumption, the rats are fed standard chow ad libitum During the measurement of oxygen consumption, food is not available Basal pre-dose oxygen consumption and ambulatory activity are measured every 10 minutes for 2 5 to 3 hours At the end of the basal pre-dosing period, the chambers are opened and the animals are administered a single dose of compound (the usual dose range is 0 001 to 10 mg/kg) by oral gavage (or other route of administration as specified, i e s c , i p , i v ) Drugs are prepared in methylcellulose, water or other specified vehicle (examples include PEG400, 30% beta-cyclo dextran and propylene glycol) Oxygen consumption and ambulatory activity are measured every 10 minutes for an additional 1-6 hours post-dosing
The Oxymax calorimeter software calculates the oxygen consumption (ml/kg/h) based on the flow rate of air through the chambers and difference in oxygen content at inlet and output ports The activity monitors have 15 infrared light beams spaced one inch apart on each axis, ambulatory activity is recorded when two consecutive beams are broken and the results are recorded as counts
Resting oxygen consumption, during pre- and post-dosing, is calculated by averaging the 10-mιn O2 consumption values, excluding periods of high ambulatory activity (ambulatory activity count > 100) and excluding the first 5 values of the pre-dose period and the first value from the post-dose period Change in oxyge'n consumption is reported as percent and is calculated by dividing the post-dosing resting oxygen consumption by the pre-dose oxygen consumption *100 Experiments will typically be done with n = 4-6 rats and results reported are mean +/- SEM
An increase in oxygen consumption of >10% is considered a positive result Historically, vehicle-treated rats have no change in oxygen consumption from pre-dose basal
MCHR stable cell lines
CHO cell lines stably expressing the human MCHR1 (Euroscreen, Brussels, Belgium) were maintained in DMEM containing Glutamax (Invitrogen, Carlsbad, CA), 10% FBS, 400ug/ml Geneticin
Cell Membrane Preparation CHO frozen pellets (from two 10-layer factories) containing human MCHR1 receptors where thawed on ice and homogenized in buffer A containing 50 mM Tris (pH 7 4),
0 32 M Sucrose, 1mM EDTA, 1mM EGTA, 1mM sodium bicarbonate, 10μg/mg Benzamadme, 10μg/ml
Bacitracin, 5μg/ml Aprotinin, 5μg/ml Leupeptin The homogenate was centrifuged at 4°C at 6000 x g for
10 mm The supernatant was removed and placed on ice The pellet was re-suspended in buffer A and re-centrifuged at 4 0C at 6000 x g for an additional 10 mm Supernatants were pooled and spun at 48,00Ox g for 30 minutes at 4 0C The final pellet was resuspended in 5 mis Buffer A The final
membrane protein was determined with a BCA kit (Pierce, Rockford, IL) Membranes were stored at -70 0C at a protein concentration of approximately 5 mg/ml
Cyclic AMP assays CHO cells stably expressing the human MCHR 1 receptors were collected and resuspended at a density of 4 0 x 10β cells/ml in F12 (Hams) media and seeded (20,000 cells per well) into 384-well, solid white Greiner assay plates For antagonists studies, 11-poιnt 3-fold dose-response curves were constructed (top final assay concentration 3OuM) in PBS with 001% pluronic and 0 3% DMSO and added to the cells along with 25uM forskolin and 0 2nM MCH (final assay concentration) Cells were incubated at 37°C/5% CO2 for 90 minutes and cAMP accumulation subsequently determined by DiscoveRx Hithunter c-AMP Il Assay kit (GE Healthcare, Buckinghamshire, UK) in accordance with manufactures instructions Luminescence was measured on a LEADseaker imaging system (GE Healthcare, Buckinghamshire, UK) IC50 values were calculated from dose-response data fit using an in- house computer graphics and statistics program The functional Ki was calculated using the Cheng- Prusoff equation Ki = IC50/1+[MCH]/EC50(MCH) where [MCH] = 0 2nM and EC50(MCH) = 0 1nM
Biological Data
Example Number MCHR-1 functional Ki (nM)
1 47.7
2 89.9
3 10.1
4 1950
5 111
6 163
7 87.1
8 8.67
10 42.8
11 97.2
12 9.98
13 4.88
14 nd
15 3.62
16 1337b 38.17a 15.38a 8.868b 18.3
19 6.78
20 15.2
21 nd1a nd1b nd2a nd2b nd3a 15.73b 31.4
24 120
25 80.95a 71.15b 69.6
26 6.64
27 71.4
28 nd
29 23.7
30 2470
32 18.3
33 19.4
34 54.3
35 232
36 75.5
37 42.78a 49.98b 7.16
39 42
40 34.11a 19.91b 19.22a 24.42b 127
43 364
44 113
45 270
46 212
47 57.8
48 705
49 15.9
50 271
51 258
52 5.92
3a 47.63b 10
54 5.91
55 3.66
56 3.69
57 3.66
58 28.8
59 68.3
60 2.94
61 18.72a 16.82b 11.4
63 12.1
64 86.7
65 40.2
66 85.5
67 188
68 119
69 124
70 102
71 626
72 74.6
73 517
74 7.37
75 24
76 3840
77 141
78 155
79 416
80 133
81 1170 82a 28.5 82b 36.7
83 851
84 19.3
85 15.7
86 7.75
87 191
88 1140 89a 3.45 89b 5.06
90 96.7
91 396
92 711
93 1090
94 116
95 147
96 195
97 49.5
98 8.23
99 5.31 100a 2.67 100b 9.82
101 25.8
102 34.8
103 25.8
104 36.6
105 6.34
106 65.8
107 5.43
108 16.8
109 22.7
110 41.6
111 161
112 184
113 50.4
114 47.9
115 100
116 26.9
117 1690
118 11.5
119 nd
120 nd
121 37.1
122 110 123a 12.7 123b nd
124 nd
125 4.46
126 9.92
127 417
128 659
129 6.9
130 9.31
131 9.15
132 7.95
133 3.23
134 2.9
135 4.86
136 4.48
137 234
138 26.8 139a 37.8 139b 172
140 94.7
141 100
142 105
143 5.68
144 69.9
145 98.5
146 60.1
147 26.2
148 125
149 3.46
150 3.47
151 6.04
152 2.31
153 2.02
154 3.99
155 3800
156 230
157 3160
158 49.8
159 201
160 26.8
D
161 33.3
162 30.9
163 83.8
164 66.8
165 29.2
166 1210
167 749
168 539
169 19.4
170 29.1
171 38.1
172 11
173 13.1
174 26.2
175 58.5
176 29.9
177 19.3
178 19.2
179 18.9
180 22.2
181 10.9
182 42.9
183 45.9
184 42
185 42.6
186 29.8
187 24.1
188 218
189 72
AO
190 117
191a 292
191b 24.7
192 144
193 196
194 46.6
195 31.3
196 605
197 7.7
198 10.6
199 110
200 57 201a 348 201b 112
202 46.3
203a 431
203b 6.48
204 330
205 498
206 250
207 6.51
208 2.72
209 10.6
210 4.56
211 19.4
212 14.3
213 168
214 106
215 161
216 78
217 11
218 14.9
219 8.45
220 513
221 126
222 680
223 197
224 16.6
225 8.03
226 2480
227 20.8
228 6.18
229 2480
230 69.8
231 752
232 238
233 17.6
234 3.47
235 10.9
236 34.7
237 101
238 76.1
239 41.2
240 77.5
COMPOUND EXAMPLES AND PREPARATIONS
The invention is illustrated by the following non-limiting examples in which the following abbreviations and definitions are used
APCI atmospheric pressure chemical ionisation
Ac acetyl
Arbocel® filter agent
BINAP 2,2'-bιs(dιphenylphosphιno)-1',1-bιnaphthyl br broad
Boc terf-butoxycarbonyl
Bu butyl
CDCI3 chloroform -d1
CD3OD methanol-d4 δ chemical shift d doublet dd double doublet
DCM dichloromethane
DIEA N.N-dnsopropylethylamine
DMF N,N-dιmethylformamιde
DMSO dimethylsulfoxide eq (molar) equivalents
ESI electrospray ionisation
Et ethyl h hours
HATU O-(7-azabenzotπazole-1-yl)-N,N,N',N'-tetramethyluronιum hexafluorophosphate
HBTU O-(1 H-Benzotπazol-1-yl)-N,N,N',N'-tetramethyluronιum hexafluorophospate
HPLC high performance liquid chromatography
HRMS high resolution mass spectrum
IPA isopropylalcohol
LRMS low resolution mass spectrometry m multiplet
Me methyl
MS mass spectrum mm minutes
MTBE methyl tert butyl ether
NMP N-methylpyrrolidone
NMR nuclear magnetic resonance
Ph phenyl
Pr n-propyl
Psi pounds per square inch pTSA p-Toluenesulfonic acid q quartet
RM reaction mixture r t room temperature
S singlet sat saturated
SM starting material soln solution t triplet
TBDMS Tert butyldimethylsilyl td triplet of doublets
Tf tnfluoromethanesulfonyl
TFA trifluoroacetic acid
THF tetrahydrofuran
TIPS triisopropylsilyl
TLC/t I c thin layer chromatography
1H Nuclear magnetic resonance (NMR) spectra were in all cases consistent with the proposed structures Characteristic chemical shifts (δ) are given in parts-per-million δH downfield from tetramethylsilane using conventional abbreviations for designation of major peaks e g s, singlet, d, doublet, t, triplet, q, quartet, m, multiplet, br, broad
Where compounds were analysed by LCMS several methods were used shown below -
6 minute LC-MS gradient and instrument conditions
Acid run
A 0 1 % formic acid in water
B 0 1 % formic acid in acetonitrile
Column C18 phase Phenomenex Gemini 50 x 4 6mm with 5 micron particle size
Gradient 95-5% A over 3mιn, 1 mm hold, 1ml/mιn UV 210nm - 450nm DAD Temperature 50C
Basic run
A 0 1% ammonium hydroxide in water B 0 1% ammonium hydroxide in acetonitπle Column C18 phase Fortis 50 x 4 6mm with 5 micron particle size Gradient 95-5% A over 3mιn, 1 mm hold, 1ml/mιn UV 210nm - 450nm DAD Temperature 5OC
2 minute LC-MS gradient and instrument conditions
Acid run A 0 1 % formic acid in water
B 0 1 % formic acid in acetonitπle
Column C18 phase Fortis Pace 20 x 2 1mm with 3 micron particle size
Gradient 70-2% A over 1 8mιn, 0 2 mm hold, 1 8ml/mιn
UV 210nm - 450nm DAD Temperature 75C
Method A 4 7 minute LC-MS gradient and instrument conditions
A 3 75 % triflouroacetic acid in water
B 1 88 % triflouroacetic acid in acetonitπle
Column Ymc ODS-AQ 2 x 50mm with 5 micron particle size Gradient 0 mm 1% B, 0 6 mins 5% B, 4 mm 100% B1 4 3 mins 1%B
Flow 0 8ml/mιn
UV 210nm DAD
Temperature 50C
Method B 4 7 minute LC-MS gradient and instrument conditions A 3 75 % triflouroacetic acid in water B 1 88 % triflouroacetic acid in acetonitrile Column Ymc ODS-AQ 2 x 50mm with 5 micron particle size Gradient 0 mm 10% B1 0 5 mins 10% B, 4 mm 100% B, 4 3 mins 10%B Flow 0 8ml/mιn UV 210nm DAD Temperature 5OC
Method C 4 7 minute LC-MS gradient and instrument conditions A 0 5 % ammonium hydroxide in water B acetonitrile
Column Welch XB-C18 2 1 x 50mm with 5 micron particle size Gradient O mm 5% B, 0 5 mins 5% B, 3 4 mm 100% B1 4 2 mins 100%B, 4 2 mm 5% B Flow 0 8ml/mιn UV 210nm DAD 5 Temperature 5OC
Where compounds are purified by HPLC, there are two methods used, shown below
Method D Method E
Column Sunfire C18 4 6 x 50 mm id column Xterra 4 6 x 50 mm id column
Temperature Ambient Ambient
Mobile Phase A 0 05% formic acid in water 0 05% ammonia in water
Mobile Phase B 0 05% formic acid in acetonitrile 0 05% ammonia in acetonitrile
Gradient - Initial 5% B 5% B
Time 0 mins 5% B 5% B
Time 3 mins 98% B 98% B
Time 4 mins 98% B 98% B
Time 4 1 mins 5% B 5% B
Time 5 mins 5% B 5% B
Flow rate 1 5 ml / mm 1 5 ml / mm
Injection volume 5 ul 5 ul
Example 1 10 6'-(4-Acetylpiperazin-1-yl)-4-(benzyloxy)-2H-1,3'-bipyridin-2-one

4-(Benzyloxy)-6'-fluoro-2H-1 ,3'-bιpyrιdιn-2-one from Preparation 2 (100 mg, 0 337 mmol), potassium carbonate (140 mg, 1 01 mmol) and N-acetyl piperazine (48 mg, 0 371 mmol) were suspended in N1N- dimethylformamide (1 00 ml) and warmed to 1000C, for 16 hours The reaction mixture was then cooled
15 to room temperature and then the solvents were evaporated in vacuo The residue was then triturated with dichloromethane (20 ml) and the solid was removed by filtration The filtrate was then evaporated to give a brown residue (78 mg) Purification by column chromatography eluting with dichloromethane methanol aqueous ammonia (90 10 1) gave the title compound (30 mg, 22%) as an off-white solid 1H NMR (400 MHz, CD3OD) δ ppm 2 15 (d, 2H), 2 17 (S1 3H), 3 58-3 70 (m, 6H)1 5 18 (s, 2H), 6 09 (s, 1 H),
20 6 24 (dd, 1 H), 6 94 (d, 1 H), 7 32-7 45 (m, 5H), 7 50 (d, 1H)1 7 58 (dd, 1H), 8 10 (s, 1 H) LRMS m/z (APCI) 405 [MH+]
Example 2 4-(Benzyloxy )-6'-piperazin-1 -yl-2H-1 ,3'-bipyridin-2-one
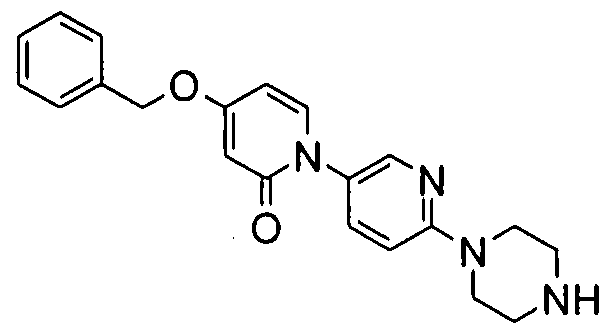
4-(Benzyloxy)-6'-fluoro-2H-1 ,3'-bιpyrιdιn-2-one from Preparation 2 ( 100 mg, 0 337 mmol), potassium carbonate (140 mg, 1 01 mmol) and piperazine (32 mg, 0 371 mmol) were suspended in N1N- dimethylformamide (1 00 ml) and warmed to 100 0C, for 16 hours The reaction mixture was then cooled to room temperature and then the solvents were evaporated in vacuo The residue was then trituated with dichloromethane (20 ml) and the solid was removed by filtration The filtrate was concentrated to give a brown residue (78 mg) Purification by column chromatography eluting with dichloromethane methanol aqueous ammonia (90 10 1) gave an off-white solid This solid was trituated with diethyl ether (5 ml) and then the solid was filtered off and air dried to give the title compound (9 mg, 7%) as a white solid 1H NMR (400 MHz, CDCI3) δ ppm 2 98 (t, 4H), 3 58 (t, 4H), 5 02 (s, 2H), 6 01-6 03 (m, 2H), 6 65 ((J1 1 H), 7 18-7 20 (m, 1 H), 7 31-7 43 (m, 5H), 7 54 (dd, 1H), 8 09 (s, 1 H) LRMS m/z (APCI) 363 [MH+]
Example 3
4-(Benzyloxy)-6l-[(3aR,6aS)-5-methylhexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl]-2H-1,3l-bipyridin-2- one

4-(Benzyloxy)-6'-fluoro-2H-1 ,3'-bιpyrιdιn-2-one from Preparation 2 (100 mg, 0 337 mmol), potassium carbonate (140 mg, 1 01 mmol) and 2-methyl-octahydro-pyrrolo[3,4-c]pyrrole (74 mg, 0 371 mmol, ChemStep) were suspended in dimethyl sulfoxide (1 00 ml) and warmed to 100 0C for 16 hours Warming was ceased and the reaction mixture was diluted with ethyl acetate (30 ml) The reaction mixture was then washed with water (2 x 20 ml) The organic fraction was dried over anhydrous magnesium sulfate, filtered and then evaporated in vacuo to give a yellow oil The oil was purified by column chromatography eluting with dichloromethane methanol aqueous ammonia (90 10 1) to afford the title compound (50 mg, 37%) as a white solid 1H NMR (400 MHz, CDCI3) δ ppm 2 34 (s, 3H), 2 42 (dd, 2H), 2 77 (t, 2H), 2 98-3 03 (m, 2H), 3 42 (dd, 2H), 3 64 (t, 2H), 5 01 (s, 2H), 6 01-6 02 (m, 2H), 6 42 (d, 1H)1 7 18 (d, 1 H), 7 32-7 42 (m, 5H), 7 46 (dd, 1H)1 8 02 (s, 1 H) LRMS m/z (APCI) 403 [MH+]
Example 4
tert-Butyl (3aR,6aS)-5-[4-(benzyloxy)-2-oxo-2H-1,3'-bipyridin-6'-yl]hexahydropyrrolo[3,4-c]pyrrole- 2(1H)-carboxylate

4-(Benzyloxy)-6'-fluoro-2H-1 ,3'-bιpyrιdιn-2-one from Preparation 2 (150 mg, 0 506 mmol), potassium carbonate (210 mg, 1 52 mmol) and hexahydro-pyrrolo[3,4-c]pyrrole-2-carboxyhc acid tert-butyl ester (107 mg, 0 506 mmol, Chembasics) were suspended in dimethyl sulfoxide (1 00 ml) and warmed to 100 0C for 16 hours Heating was ceased and the reaction was cooled The reaction mixture was then diluted with water (20 ml) An off-white solid precipitated and was filtered and dried under air, then placed in the vacuum oven at 55 0C for 1 hour The precipitate was purified by column chromatography eluting with dichloromethane methanol aqueous ammonia 95 5 0 5 to afford the title compound (111 mg, 45%) as a white solid 1H NMR (400MHz, CDCI3) δ ppm 1 43 (s, 9H), 2 99-3 04 (m, 2H), 3 22-3 78 (m, 8H), 5 02 (s, 2H), 6 01-6 04 (m, 2H), 6 41 (d, 1H), 7 19 (d, 1H), 7 34-7 42 (m, 5H), 7 50-7 55 (m, 1H), 8 03 (s, 1 H) LCMS m/z (APCI) 489 [MH+]
Example 5
4-(Benzyloxy)-6'-[4-(dimethylamino)piperidin-1-yl]-2H-1,3'-bipyridin-2-one
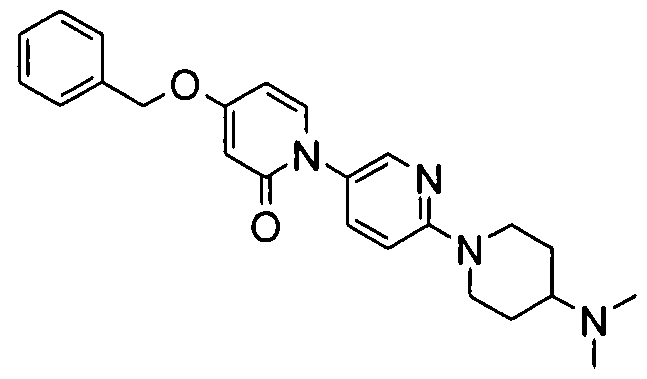
A mixture of 4-(benzyloxy)-6'-chloro-2H-1 ,3'-bιpyrιdιn-2-one from Preparation 3 (50 mg, 0 16 mmol), dιmethyl-pιperιdιne-4-yl-amιne (62 mg, 0 48 mmol) and cesium fluoride (25 mg, 0 16 mmol) in DMSO (0 5 ml) was heated in microwave oven at 150 0C for 45 minutes After cooling to room temperature EtOAc (10 ml) and water (10 ml) were added The organic component was washed with water (2 x 5 ml), brine (5 ml), dried (MgSO4), filtered and concentrated The residue was purified by column chromatography (DCM MeOH 0 880 NH3, 95 5 0 5 then 90 10 1) The product-containing fractions were evaporated and the resulting solid was triturated (Et2O), filtered, washed (pentane) and dried to give the desired product as a white solid (32 mg, 49%) 1H NMR (400 MHz, CDCI3,) δ ppm 1 51 (m, 2H), 1 93 (m, 2H), 2 34 (s, 6H), 2 43 (m, 1 H), 2 88 (m, 2H), 4 38 (m, 2H), 5 02 (s, 2H), 6 03 (m, 2H), 6 70 (d, 1 H), 7 17 (d, 1H), 7 32-7 43 (m, 5H), 7 51 (dd, 1 H), 8 06 (d, 1 H) LRMS m/z (APCI & ES) 405 [MH+]
Example 6 ^(BenzyloxyJ-β'^-methylpiperazin-i-yO^H-I.S'-bipyridin-Σ-one

A mixture of the 4-(benzyloxy)-6'-chloro-2H-1,3'-bιpyrιdιn-2-one from Preparation 3 (53 mg, 0 16 mmol), 1-methyl piperazine (53 μl, 048 mmol) and cesium fluoride (25 mg, 0 16 mmol) in DMSO (0 5 ml) was heated in a microwave oven at 150 0C for 45 minutes After cooling to room temperature ethyl acetate (10 ml) and water (10 ml) were added The organic component was separated, washed water (2 x 5 ml) and then brine (5 ml), dried (MgSO4), filtered and concentrated The residue was purified by column chromatography (DCM MeOH 0 880 NH3, 95 5 0 5 then 90 10 1) The fractions containing product were concentrated and the resulting solid was triturated (Et2O), filtered, washed (pentane) and dried to give the desired product as a white solid (25 mg, 42%) 1H NMR (400 MHz, CDCI3) δ ppm 2 35 (s, 3H), 2 53 (m, 4H), 3 62 (m, 4H), 5 02 (s, 2H), 6 03 (m, 2H), 6 70 (d, 1H), 7 17 (d, 1 H), 7 32-7 44 (m, 5H), 7 53 (dd, 1H), 8 10 (d, 1 H) LRMS m/z (APCI & ES) 377 [MH*]
Example 7
4-(Benzyloxy)-6'-(4-ethylpiperazin-1 -yl)-2H-1 ,3'-bipyridin-2-one

4-(Benzyloxy)-6'-fluoro-2H-1 ,3'-bιpyrιdιn-2-one from Preparation 2 (100 mg, 0 337 mmol), potassium carbonate (140 mg, 1 01 mmol) and N-ethylpiperazine (0 047 mL, 0 371 mmol) were suspended in N1N- dimethylformamide (0 50 ml) and warmed to 100 0C with stirring under N2 for 16 hours The reaction mixture was then cooled to room temperature and then the solvents were removed in vacuo The residue was then trituated with dichloromethane (20 ml) and the insoluble solid was filtered off The filtrate was then evaporated in vacuo to give a brown residue This was purified by column chromatography eluting with dichloromethane methanol aqueous ammonia (90 10 1) to give an off-white solid (100 mg) The solid was triturated with diethyl ether (20 ml) then filtered to give the title compound (70 mg, 53%) as a white solid 1H NMR (400 MHz1 CD3OD) δ ppm 1 17 (t, 3H), 2 50 (q, 2H), 2 60 (t, 4H), 3 62 (t, 4H), 5 18 (S, 2H), 6 05 (S, 1H), 6 23 (dd, 1 H), 6 90 (d, 1 H), 7 32-7 44 (m, 5H), 7 50 (d, 1 H), 7 55 (dd, 1H)1 8 03 (s, 1H) LRMS m/z (APCI) 391 [MH+]
Example 8
4-(Benzyloxy)-6'-[(3aR,6aS)-hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl]-2H-1,3'-bipyridin-2-one dihydrochloride
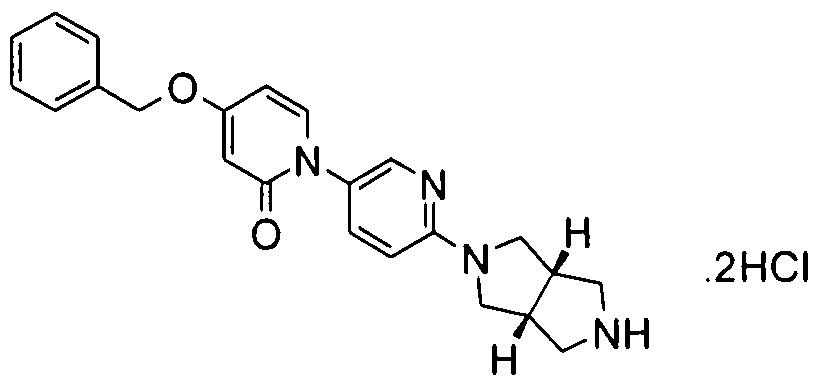
A solution of 4M hydrogen chloride in dioxane (1 09 ml, 4 38 mmol) was added to a solution of ferf-butyl (3aR,6aS)-5-[4-(benzyloxy)-2-oxo-2H-1 ,3'-bιpyπdιn-6'-yl]hexahydropyrrolo[3,4-c]pyrrole-2(1H)- carboxylate from Example 4 (107 mg, 0 22 mmol) in dichloromethane (2 ml) at room temperature Immediately after addition a white precipitate appeared The reaction mixture was stirred for 2 hours then the solvents were evaporated in vacuo The resultant white solid was azeotroped with dichloromethane (2 x 20 ml) then dried in vacuo to afford the title compound (102 mg, 100%) as a white solid 1H NMR (400 MHz, CD3OD) δ ppm 3 37-3 49 (m, 4H), 3 62-3 70 (m, 2H), 3 79 (dd, 2H), 3 98 (dd, 2H), 5 19 (s, 2H), 6 10 (d, 1 H), 6 35 (dd, 1 H), 7 22 (d, 1 H), 7 33-7 44 (m, 5H), 7 59 (d, 1 H), 8 09 (dd, 1 H), 8 18 (s, 1 H) LRMS m/z (APCI) 389 [MH+]
Example 9 4-[(4-Chlorobenzyl)oxy]-6'-t3-(methylamino)pyrrolidin-1-yl]-2H-1 ,3'-bipyridin-2-one

(3R)-1-{4-[(4-Chlorobenzyl)oxy]-2-oxo-2H-1 ,3'-bιpyrιdιn-6'-yl}pyrrolιdιn-3-yl methanesulfonate from Preparation 5 (20 mg, 0 04 mmol) and 2M methylamine in THF (0 53ml, 1 1mmol) were heated at 100 0C in a reacti-vial for 10 days After cooling to room temperature the reaction mixture was concentrated and purified by preparative HPLC, which gave the desired product as a white solid (3 mg, 18%) 1H NMR (CD3OD, 400MHz) δ ppm 1 93-2 02 (m, 1H), 2 27-2 35 (m, 1 H), 2 43 (s, 3H), 3 32-3 38 (m, 1 H), 3 41- 3 58 (m, 2H), 360-3 66 (m, 1H), 3 71-3 78 (m, 1 H), 5 14 (s, 2H), 6 07 (d, 1 H), 6 22 (dd, 1 H), 6 58 (d, 1H), 7 38-7 55 (m, 6H), 7 98 (d, 1 H) LRMS m/z (APCI) 411 [MH+]
Example 10
4-[(4-Chlorobenzyl)oxy]-6'-[(3R)-3-hydroxypyrrolidin-1-yπ-2H-1,3'-bipyridin-2-one

To a stirred solution of 4-hydroxy-6'-[(3R)-3-hydroxypyrrolιdιn-1-yl]-2H-1 ,3'-bιpyrιdιn-2-one from Preparation 4 (66 mg, 0 32 mmol) in acetonitπle (1 ml) was added 4-chlorobenzyl bromide (66 mg, 0 32 mmol) and cesium carbonate (105 mg, 0 32 mmol) The reaction mixture was heated at reflux for 6 5 hours After cooling to room temperature, the reaction mixture was concentrated and the residue was partitioned between dichloromethane (5 ml) and water (5 ml) The organic component was separated using a phase separation cartridge and the resulting crude product mixture was purified by column chromatography (eluting with a gradient from EtOAc to 95 5 0 5 EtOAc MeOH NH3) which gave the desired product as a white solid (51 mg, 44%) 1H NMR (400MHz, CD3OD) δ ppm 2 01-2 09 (m, 1H), 10 2 12-2 21 (m, 1 H), 3 41-3 48 (m, 1 H), 3 58-3 63 (m, 3H), 4 50-4 52 (m, 1H), 5 12 (s, 2H) , 6 05 (d, 1 H), 6 22 (dd, 1 H), 6 59 (d, 1 H), 7 39-7 51 (m, 6H), 8 00 (d, 1 H) LRMS m/z (APCI) 398 [MH*]
Example 11 4-(Benzyloxy)-6l-[(3R)-3-hydroxypyrrolidin-1-yl]-2H-1,3l-bipyridin-2-one
15

To a stirred solution of 4-(benzyloxy)-6'-ftuoro-2H-1 ,3'-bιpyrιdιn-2-one from Preparation 2 (1 50 g, 5 06 mmol) in DMSO (5 ml) was added (R)-pyrrolιdιn-3-ol (1 32 g, 15 2 mmol, Aldnch) The reaction mixture was heated at 150 0C for 30 minutes After cooling to room temperature the reaction mixture was diluted with water (20 ml) and a slight excess of saturated aqueous sodium bicarbonate solution was added
20 The solid was collected by filtration, washed with water (3 x 10 ml) and air-dπed The solid was heated at reflux in acetonitrile (40 ml), cooled to room temperature and the solid was collected by filtration After washing with acetonitrile (10 ml) and air drying, the desired product was isolated as an off-white solid (1 66 g, 90%) 1H NMR (400MHz, CD3OD) δ ppm 2 02-2 09 (m, 1 H), 2 11-2 21 (m, 1 H), 343-3 49 (m, 1H), 3 55-3 65 (m, 3H), 4 50-4 54 (m, 1 H), 5 15 (s, 2H), 6 07 (d, 1 H), 6 24 (dd, 1 H), 6 58 (d, 1 H), 7 31-
25 7 53 (m, 7H), 7 98 (d, 1 H) LRMS m/z (APCI) 364 [MH*]
Example 12 tert-Butyl {(3S)-1-[4-(benzyloxy)-2-oxo-2H-1,3'-bipyridin-6'-yl]pyrrolidin-3-yl}methylcarbamate

A mixture of 4-(benzyloxy)-6'-fluoro-2H-1 ,3'-bιpyrιdιn-2-one from Preparation 2 (500 mg, 1 69 mmol), (S)- methyl-pyrrolιdιnyl-3-yl-carbamιc acid ferf-butyl ester (372 mg, 1 86 mmol, ECA International) and potassium carbonate (700 mg, 5 06 mmol) in dimethylformamide (15 ml) was heated to 110 0C After 14 hours the reaction mixture was concentrated and the crude product mixture was purified by column chromatography eluting with ethyl acetate, followed by 9 1 ethyl acetate methanol which gave the desired product as a white powder (693 mg, 86%) 1H NMR (400MHz, CD3OD) δ ppm 1 45 (s, 9H), 2 21 (m, 2H), 2 81 (s, 3H), 3 43 (m, 2H), 3 84 (m, 2H), 4 82 (b, 1 H), 5 15 (s, 2H), 6 08 (s, 1 H), 6 25 (d, 1H), 6 05 (d, 1 H), 7 31-7 45 (m, 7H), 8 02 (s, 1 H) LRMS 477 [MH*]
Example 13
4-(Benzyloxy)-6'-[(3S)-3-(methylamino)pyrrolidin-1-yl]-2H-1,3'-bipyridin-2-one dihydrochloride

To a stirred solution of fert-butyl {(3S)-1-[4-(benzyloxy)-2-oxo-2H-1 ,3'-bιpyrιdιn-6'-yl]pyrrolιdιn-3- yl}methylcarbamate from example 12 (600 mg, 1 26 mmol) in dioxane (15 ml) was added 4M HCI in dioxane (20 ml, 80 mmol) After stirring for 15 hours at room temperature, the mixture was concentrated and air dried to give a light brown solid The crude product was recrystallised from propan-2-ol/ethanol and dried to give the desired product as a light brown solid (350 mg, 50%) 1H NMR (400 MHz, CD3OD) δ ppm 2 45(m, 1 H), 2 65(m, 1 H), 2 85(s, 3H), 3 85(m, 1 H), 3 94(m, 2H), 4 14(m, 2H), 5 18(s, 2H), 6 08(s, 1H), 6 31(d, 1 H), 7 21(d, 1 H), 7 31-7 45(m, 5H), 7 58(d, 1 H), 8 08(d,1H), 8 19(s, 1 H) LRMS m/z (APCI)
377 [MH+]
Example 14 tert-Butyl ^aRJ-i-IA^benzyloxy^-oxo-ΣH-i.a'-bipyridin-β'-yllpyrrolidin-S-ylJmethylcarbamate

A mixture of 4-(benzyloxy)-6'-fluoro-2H-1,3'-bιpyrιdιn-2-one from Preparation 2 (500 mg, 1 69 mmol), (R)- methyl-pyrrolιdιnyl-3-yl-carbamιc acid tert-butyl ester (372 mg, 1 86 mmol, see Example 34b in WO2003/106462) and potassium carbonate (700 mg, 5 06 mmol) in N,N-dιmethylformamιde (15 ml) was 5 heated to 110 0C After 14 hours the reaction mixture was concentrated and the crude product mixture was purified by column chromatography eluting with ethyl acetate, followed by 9 1 ethyl acetate methanol which gave the desired product as a white powder (693 mg, 86%) 1H NMR (400 MHz, CD3OD) δ ppm 1 45(s, 9H), 2 21(m, 2H), 2 81(s, 3H), 3 43(m, 2H), 3 84(m, 2H), 4 82(b, 1H), 5 15(s, 2H), 6 08((s, 1H), 6 25(d, 1 H), 6 05(d, 1 H), 7 31-7 45(m, 7H), 8 02(s, 1 H) LRMS m/z 477 [MH+] 10
Example 15
4-(benzyloxy)-6'-[(3R)-3-(methylamino)pyrrolidin-1-yl]-2H-1,3'-bipyridin-2-one dihydrochloride

To a stirred solution of fert-butyl {(3R)-1-[4-(benzyloxy)-2-oxo-2H-1 ,3'-bιpyrιdιn-6'-yl]pyrrolιdιn-3-
15 yl}methylcarbamate from Example 14 (650 mg, 1 36 mmol) in dioxane (15 ml) was added 4M HCI in dioxane (27 ml, 109 mmol) After stirring for 15 hours at room temperature, the mixture was concentrated and air dried to give a light brown solid The crude product was recrystallised from propan-
2-ol/ethanol and dried to give the desired product as a light brown solid (430 mg, 70%) 1H NMR
(400MHz, CD3OD) δ ppm 2 45(m, 1 H), 2 65(m, 1 H), 2 85(s, 3H), 3 85(m, 1H), 3 94(m, 2H), 4 14(m, 2H),
20 5 18(s, 2H), 6 08(s, 1 H), 6 31(d, 1 H), 7 21(d, 1 H), 7 31-7 45(m, 5H), 7 58(d, 1 H), 8 08(d,1 H), 8 19(s, 1 H)
LRMS m/z (APCI) 377 [MH+]
Example 16 4-(benzyloxy)-6'-t(3S)-3-hydroxypyrrolidin-1-yl]-2H-1,3'-bipyridin-2-one
 H
H
A stirred solution of 4-benzyloxy-1H-pyrιd-2-one (25 mg, 0 13 mmol), (3S)-1-(5-Bromopyrιdιn-2- yl)pyrrolιdιn-3-ol from Preparation 6 (30 mg, 0 13 mmol), trans-1 ,2-dιamιnocyclohexane (2 μl, 0 02 mmol), potassium carbonate (17 mg, 0 13 mmol) and copper(l) iodide (28 mg, 0 13 mmol) in N-methyl pyrrolidinone (1 ml) was heated at 150 0C for 15 hours in a reacti-vial After cooling to room temperature, the mixture was diluted with DCM (10 ml), filtered through celite and the filtrate was washed with water (5 ml) The aqueous phase was extracted with DCM (2 x 5 ml) and the combined organic layers were passed through a phase separation cartridge The crude product mixture was purified by preparative HPLC (C18 phenomenex® luna 10 μ C18(2) 100 A, mobile phase, gradient using H2O with 0 1% formic acid and MeCN with 0 1% formic acid) to give the desired product as a brown solid (5 mg, 17%) 1H NMR (400MHz, CD3OD) δ ppm 2 01-2 09 (m, 1H), 2 12-2 21 (m, 2H), 3 41-3 48 (m, 1 H), 3 58-3 63 (m, 3H), 4 50-4 52 (m, 1 H), 6 05 (d, 1H), 6 15 (dd, 1 H), 6 62 (d, 1H), 7 32-7 51 (m, 6H), 7 53 (dd, 1 H), 8 00 (d, 1 H) LRMS m/z (APCI) 364 [MH+]
Example 17a
6'-(3-Amιno-3-methylpyrrolιdιn-1 -yl)-4-(benzyloxy)-2H-1 ,3'-bιpyrιdιn-2-one (enantiomer 1 )

A solution of protected amine of Preparation 8a (29 mg, 0 06 mmol) in dichloromethane (5 ml) was cooled with an ice bath and treated with trifluoroacetic acid (2 ml) The cold bath was removed and the solution was stirred at room temperature for 4 hours The solution was concentrated in vacuo, the residue was dissolved in methanol and loaded onto a SCX-2 cartridge Non basic products were eluted with methanol and the desired product was eluted with 2N NH3/MeOH and concentrated in vacuo to give the title compound as a colourless solid 21 mg (91%) 1H NMR (400 MHz, CD3OD) δ ppm 1 35 (s, 3H) 1 99 (t, 2H) 3 40 (s, 2H) 3 53 - 3 59 (m, 1H) 3 63 - 3 69 (m, 1 H) 5 14 (s, 2H) 6 07 (d, 1H) 6 24 (dd, 1 H) 6 55 (d, 1 H) 7 34 - 7 50 (m, 7H) 7 97 (d, 1H) LRMS m/z (APCI) 377 [MH+]
Example 17b
6'-(3-Amιno-3-methylpyrro!ιdιn-1 -yl)-4-(benzyloxy)-2H-1 ,3'-bιpyrιdιn-2-one (enantiomer 2)
Prepared using the same method as 17a from Preparation 8b to give the title compound as a colourless solid 1H NMR (400MHz, CD3OD) δ ppm 1 35 (s, 3H) 1 99 (t, 2H) 3 40 (s, 2H) 3 53 - 3 59 (m, 1H) 3 63 - 3 69 (m, 1 H) 5 14 (s, 2H) 6 07 (d, 1H) 6 24 (dd, 1H) 6 55 (d, 1 H) 7 34 - 7 50 (m, 7H) 7 97 (d, 1 H) LRMS m/z (APCI) 377 [MH*]
Example 18a
4-(Benzyloxy)-6'-[3-methyl-3-(methylamιno)pyrrolιdιn-1-yl]-2H-1,3'-bιpyπdιn-2-one (enantiomer 1)

A solution of protected amine of Preparation 9a (50 mg, 0 1 mmol) in dichloromethane (3 ml) was cooled 10 with an ice bath and treated with tπfluoroacetic acid (2 ml) The cold bath was removed and the solution was stirred at room temperature for 4 hours The solution was concentrated in vacuo and the residue was dissolved in methanol and loaded onto a SCX-2 cartridge Non basic products were eluted with methanol and the desired product was eluted with 2N NH3/MeOH and concentrated in vacuo to give the title compound as a colourless solid 29mg (72%) 1H NMR (400MHz, CD3OD) δ ppm 1 30 (s, 3H) 1 95 - 15 2 00 (m, 1H) 2 06 - 2 13 (m, 1H) 2 37 (s, 3H) 336 - 3 55 (m, 3H) 3 60 - 366 (m, 1H) 5 14 (s, 2H) 6 07 (d, 1H) 6 24 (dd, 1 H) 6 56 (d, 1H) 7 34 - 7 51 (m, 7H) 7 98 (d, 1 H) LRMS m/z (APCI) 391 [MH+]
Example 18b
4-(Benzyloxy)-6'-[3-methyl-3-(methylamιno)pyrrolιdιn-1-yl]-2H-1 ,3'-bιpyπdιn-2-one (enantiomer 2) 20
Prepared using the same method as 18a from preparation 9b to give the title compound as a colourless solid 1H NMR (400MHz, CD3OD) δ ppm 1 30 (s, 3H) 1 95 - 2 00 (m, 1H) 2 06 - 2 13 (m, 1H) 2 37 (s, 3H) 3 36 - 3 55 (m, 3H) 3 60 - 3 66 (m, 1 H) 5 14 (s, 2H) 6 07 (d, 1 H) 6 24 (dd, 1H) 6 56 (d, 1 H) 7 34 - 7 51 (m, 7H) 7 98 (d, 1H) LRMS m/z (APCI) 391 [MH+]
25
Example 19
4-(Benzyloxy)-6'-[(3R)-3-(ethylamιno)pyrrolιdιn-1-yl]-2H-1 ,3'-bιpyrιdιn-2-one

Example 20
Λ/-{(3S)-1-[4-(Benzyloxy)-2-oxo-2H-1,3'-bιpyπdιn-6'-yl]pyrrolιdιn-3-yl}-N-methylacetamιde

Example 21. 21a. 21b (= Preparation 8. 8a. 8b) tert-Butyl {1-[4-(benzyloxy)-2-oxo-2H-1 ,3'-bιpyπdιn-6'-yl]-3-methylpyrrolιdιn-3-yl}carbamate

A solution of 4-benzyloxy-2(1 H)-pyπdone (186 mg, 0 92 mmol), the iodide of preparation 7 (373 mg, 0 92 mmol), copper iodide (176 mg, 0 92 mmol), potassium carbonate (256 mg, 1 85 mmol) and rac-trans- N,N'-dιmethyl cyclohexane-1 ,2-dιamιne (146ml, 0 92 mmol) in 1 ,4-dιoxan (5ml) was degassed via a nitrogen/vacuum cycle The resulting mixture was the heated to 11O0C for 16 hours The reaction was cooled to room temperature and concentrated in vacuo The residue was treated with 0 880NH3/H2o (10ml, 1 1 ratio) and stirred for 15mιn The resulting solid was extracted with ethyl acetate (2x20ml) The combined organics were washed with brine (20ml), dned (Na2SO4), filtered and concentrated in vacuo to give a brown oil Purification by column chromatography (eluting with 100% DCM → 95 5 0 5) gave the title compound (Prep8/Ex.21 as racemate) as a pale yellow solid, 307 mg (69%) 1H NMR (400MHz, CD3OD) δ ppm 1 43 (s, 9H), 1 48 (s, 3H), 1 96-2 04 (m, 1H), 2 33-2 41 (m, 1 H), 3 42-3 58 (bm, 3H), 3 81-3 84 (m, 1 H), 5 15 (s, 2H), 6 08 (d, 1 H), 6 25 (dd, 1 H), 6 55 (d, 1H), 7 32-7 51 (bm, 7H), 7 97 (d, 1H) LRMS m/z (APCI) 477 [MH*]
Chiral prep HPLC (Chiralcel OJ-H, 250x21 2mm id, 100% MeOH, 20ml/mιn rt) enabled separation of the individual enantiomers
Peak 1 Retention time 9 72 minutes Prep. 8a / Ex. 21a Peak 2 Retention time 15 14 minutes Prep. 8b / Ex.21b
Example 22a: 22b (=Preparation 9a: 9b) /erf-Butyl {1 -[4-(benzyloxy)-2-oxo-2H-1 ,3'-bιpyrιdιn-6'-yl]-3-methylpyrrolιdιn-3-yl}methyl carbamate - single enantiomer

A solution of the pyrrolidinone of preparation 8a (57 mg, 0 12 mmol) in DMF (2 ml) was cooled in an ice bath and treated with sodium hydride (7 2 mg of a 60% dispersion in mineral oil, 0 12 mmol) under a nitrogen atmosphere The mixture was allowed to warm gradually to room temperature over 2hours before iodomethane (9 μl, 0 14 mmol) was added Once added, the solution was stirred at room
temperature overnight The reaction mixture was quenched with a few drops of water, diluted with methanol and loaded onto a SCX-2 cartridge, rinsing with methanol The desired product was eluted with 2N NH3/MeOH and concentrated in vacuo to give the title compound (Prep. 9a/Ex.22a) as a colourless oil, 50mg (85%) 1H NMR (400MHz, CD3OD) δ ppm 1 34 (s, 3H) 1 50 (s, 9H) 2 23 - 2 37 (m, 2H) 2 93 (s, 5 3H) 3 39 - 345 (m, 1 H) 3 56 (d, 1H) 3 62 (t, 1H) 4 00 (d, 1H) 5 14 (s, 2H) 6 07 (d, 1H) 6 24 (dd, 1H) 6 54 (d, 1 H) 7 32 - 7 52 (m, 7H) 8 00 (d, 1 H) LRMS m/z (APCI) 491 [MH*]
The opposite enantiomer Prep. 9b (Example 22b) was prepared via the same method with preparation 8b as starting material to give the title compound as a colourless oil 1H NMR (400MHz, CD3OD) δ ppm 10 1 34 (s, 3H) 1 50 (S, 9H) 2 23 - 2 37 (m, 2H) 2 93 (s, 3H) 3 39 - 3 45 (m, 1 H) 3 56 (d, 1H) 3 62 (t, 1H) 4 00 (d, 1H) 5 14 (s, 2H) 6 07 (d, 1 H) 6 24 (dd, 1 H) 6 54 (d, 1 H) 7 32 - 7 52 (m, 7H) 8 00 (d, 1 H) LRMS m/z (APCI) 491 [MH*]
Example 23a; 23b (^Preparation 11a: 11b)
15 fert-Butyl {(3R)-1 -[4-(benzyloxy)-2-oxo-2H-1 ,3'-bιpyrιdιn-6'-yl]pyrrolιdιn-3-yl}ethylcarbamate

A solution of 4-benzyloxy-2(1H)-pyrιdone (274 mg, 1 36 mmol), the iodide of preparation 10 (567 mg, 1 36 mmol), copper iodide (53 mg, 027 mmol), potassium carbonate (376 mg, 2 72 mmol) and rac- trans-N,N'-dιmethyl cyclohexane-1 ,2-dιamιne (86ml, 0 54 mmol) in 1 ,4-dιoxan (10ml) was degassed via
20 a nitrogen/vacuum cycle The resulting mixture was then heated to 1000C under a nitrogen atmosphere for 16 hours The reaction was cooled to room temperature and concentrated in vacuo The residue was treated with 0 880NH3/H2O (10ml, 1 1 v/v ratio) and stirred for 15mιn The resulting solid was filtered off and dried in vacuo giving the title compound (Prep 11 a/Ex 23a) as a brown solid 550mg (82%) 1H NMR (400MHz, CDCI3) δ ppm 1 13 (t, 3H), 1 43 (s, 9H) 2 09-2 25 (m, 2H), 3 12-3 30 (m, 2H), 3 31 (t, 1H),
25 3 40 (q, 1H), 3 65 (q, 2H), 4 60-4 77 (m, 1 H), 5 01 (s, 2H), 6 00-6 01 (m, 2H), 6 40 (d, 1 H), 7 18 (d, 1 H), 7 30-7 41 (m, 5H), 7 48 (dd, 1H), 8 03 (d, 1 H) LRMS m/z (ESI) 491 [MH+]
The opposite enantiomer Prep. 11b (Example 23b) was prepared via the same method with preparation 10b as starting material to give the title compound as a brown solid 1 H NMR (400MHz, CDCI3) δ ppm 1 13 (t, 3H), 1 43 (s, 9H) 2 09-2 25 (m, 2H), 3 12-3 30 (m, 2H), 3 31 (t, 1H), 3 40 (q, 1 H), 3 65 (q, 2H), 30 4 60-4 77 (m, 1H), 5 01 (s, 2H), 6 00-6 01 (m, 2H), 6 40 (d, 1 H), 7 18 (d, 1 H), 7 30-7 41 (m, 5H), 7 48 (dd, 1H), 8 03 (d, 1 H) LRMS m/z (ESI) 491 [MH+]
Example 25. 25a. 25b
1-[4-(Benzyloxy)-2-oxo-2H-1 ,3'-bιpyrιdιn-6'-yl]-N,N-dιmethylpyrrolιdιne-3-carboxamιde

Prepared according to Preparation 28 from 1-[4-(Benzyloxy)-2-oxo-2H-1 ,3'-bιpyπdιn-6'-yl]pyrrolιdιne-3- carboxylic acid from Preparation 31 and dimethylamine-hydrochloπde to to give the title compound (Ex 25 as a racemate) as a colourless solid 1H NMR (400 MHz1 CDCI3) δ ppm 2 19-2 26 (m, 1 H), 2 33-2 44 (m, 1 H), 2 98 (s, 3H), 3 10 (s, 3H), 3 36-3 43 (m, 1 H), 3 46 (q, 1 H), 3 65-3 73 (m, 2H), 3 81 (t, 1 H), 5 01 (S, 2H), 6 01-6 02 (m, 2H), 642 (d, 1 H), 7 19 (d, 1 H), 7 33-7 43 (m, 5H), 7 48 (del, 1 H), 8 03 (d, 1H) LRMS m/z (ESI) 419 [MH+]
Chiral prep HPLC (Chiralpak OJ-H, 250x21 2mm id, 100% MeOH, 18ml/mιn r t ) enabled separation of the individual enantiomers
Peak 1 Retention time 17 41 minutes, Ex. 25a Rotation [α]D25 = -15 16o (3 10 mg/mL MeOH)
Peak 2 Retention time 2894 minutes, Ex. 25b Rotation [α]D 25 = 1962° (3 15 mg/mL MeOH)
Example 26
4-(Benzyloxy)-6'-[(3aR,6aS)-5-ethylhexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl]-2H-1 ,3'-bιpyπdιn-2-one

Example 27
4-(Benzyloxy)-6'-[(3'S)-(2-oxo-1 ,3'-bιpyrrolιdιn-1 '-yl]-2H-1 ,3'bιpyrιdιn-2-one

N-{(3S)-1-[4-(Benzyloxy)-2-oxo-2H-1 ,3'-bipyndin-6'-yl]pyrrolidin-3-yl}-4-chlorobutanamide from
10 Preparation 54 (120 mg, 0 26mmol) was added to a solution of sodium hydride (60% dispersion in mineral oil, 25 mg, 0 65mmol) in NMP (5ml)and the RM was stirred at r t for 16 hours MS showed evidence for product and no SM TLC (EtOAc MeOH NH3 90 10 1) showed both product and SM remaining A further 0 5 eq of NaH (60% disp in oil, 10 mg) was added and the solution was warmed to
4O0C for 4 hours then cooled to r t overnight The reaction mixture was quenched with sat NH4CI (aq)
15 (20 ml) then diluted with EtOAc (30 ml) Layers were separated and the organic layer was washed with brine (30 ml) before drying (MgSO4), filtered and evaporated to give a yellow oil The oil was purified by cartridge chromatography (20 g cartridge) eluting with EtOAc MeOH NH3 98 2 0 2 (100 mL) to 96 4 0 4
(100 mL) to 96 5 0 5 (200 mL) to 93 7 0 7 (300 mL) which gave the title compound as a colorless oil which crystallised on standing (44 0 mg, 39%) 1H NMR (400 MHz, CDCI3) δ ppm 2 00-2 07 (m, 2H),
20 2 09-2 18 (m, 1H), 2 25-2 35 (m, 1H)1 2 42 (t, 2H), 3 40 (t, 2H), 345-3 56 (m, 2H), 3 63-3 73 (m, 2H),
4 89-4 95 (m, 1 H), 5 03 (S, 2H), 6 02-6 03 (m, 2H), 6 42 (d, 1H), 7 19-7 21 (d, 1 H), 7 32-7 42 (m, 5H),
7 52 (dd, 1 H), 8 03 (d, 1H) LRMS m/z (ESI) 431 [MH+]
Example 28
25
(3S)-1-{4-[(4-chlorobenzyl)oxy]-2-oxo-2H-1 ,3'-bιpyrιdιn-6'-yl}pyrrolιdιn-3-yl acetate

Example 29
4-[(4-Chlorobenzyl)oxy]-6'-[(3S)-3-hydroxypyrrolιdιn-1-yl]-2H-1 ,3'-bιpyrιdιn-2-one

(3S)-1-{4-[(4-chlorobenzyl)oxy]-2-oxo-2H-1 ,3'-bιpyrιdιn-6'-yl}pyrrolιdιn-3-yl acetate from Example 28 (7 mg, 0 0159 mmol) and K2CO3 (11 mg, 0 796 mmol) were stirred in methanol at r t overnight TLC, MS and LCMS indicated a bit of starting material not consumed 5 Eq of K2CO3 were added and the mixture was stirred at r t for 2 h TLC and MS indicated completion of the reaction The mixture was diluted with DCM (2ml) The organics were washed with water (1ml) and dried over a separating phase cartridge to give the title compound as a white solid (5 mg, 79%) 1H NMR (400 MHz, CD3OD) δ ppm 2 06 (m, 1H)1 2 14 (m, 1H), 346 (d, 1H), 3 53-3 64 (m, 3H), 4 53 (m, 1H), 5 15 (s, 2H), 6 07 (d, 1 H), 6 23 (dd, 1 H), 6 58 (d, 1 H), 7 31-7 55 (m, 7H), 7 98 (d, 1H) LRMS m/z (APCI & ESI) 398 [MH*]
Example 30
4-(Benzyloxy)-6'-(3-oxopyrrolιdιn-1-yl)-2H-1 ,3'-bιpyrιdιn-2-one
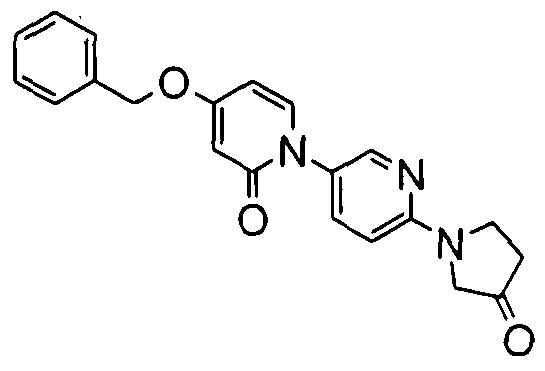
Example 32 ( = Preparation 53) tert-Butyl methyl{(3S)-1-[2-oxo-4-(2-phenylethyl)-2H-1 ,3'-bιpyπdιn-6'-yl]pyrrolιdιn-3-yl}- carbamate

To a stirred solution of 6'-{(3S)-3-[(tert-Butoxycarbonyl)(methyl)amιno]pyrrolιdιn-1-yl}-2-oxo-2H-1 3'- bιpyrιdιnyl-4-yl tπfluoromethanesulfonate from Preparation 52 (50 mg, 0 096 mmol) in THF (784 uL) was added NMP (139 uL), phenethylzinc bromide (289 uL, 0 145 mmol) and bιs(trι-t- butylphosphιne)palladιum(O) (1 01 mg, 0 002 mmol) and the reaction mixture was stirred at 5O0C overnight The reaction mixture was cooled to r t , diluted with EtOAc (5 mL), washed with water (3 x 5ml) The organics were dried (Na2SO4) and evaporated in vacuo The residue was purified by column chromatography using ISCO (4g column) and eluant EtOAc/MeOH/NH3 from EtOAc only to 95/5/0 5 to give the title compound as a colourless solid (12 mg, 27%) 1H-NMR (400MHz, CD3OD) δ ppm 1 42 (s, 9H), 2 17-2 21 (m, 2H), 2 81-2 86 (m, 5H), 2 90-2 95 (m, 2H), 3 39-3 42 (m, 2H), 3 62-369 (m, 2H)1 4 78-4 82 (m, 1 H), 6 37-6 39 (m, 2H), 6 59 (d, 1H), 7 11-7 26 (m, 5H), 7 42 (d, 1H), 7 51 (dd, 1H), 7 98 (d, 1 H) LRMS m/z (APCI & ESI) 475 [MH*]
Example 33 ( = Preparation 56)
4-(Benzyloxy)-5'-[(3R)-3-(methylamιno)pyrrolιdιn-1-yl]-2H-1 ,2'-bιpyrιdιn-2-one

Prepared according to Example 17a from ferf-Butyl {(3R)-1-[4-(benzyloxy)-2-oxo-2H-1 ,2'-bιpyπdιn-5'- yl]pyrrolιdιn-3-yl}methylcarbamate from Preparation 55 to give the title compound as a brownish solid 1H NMR (400 MHz, d6-DMSO) δ ppm 1 90-1 95 (m, 1 H), 2 16-2 21 (m, 1 H), 2 41 (s, 3H), 3 18-3 52 (m, 5H), 5 5 13 (s, 2H)1 5 94 (d, 1H), 6 10 (dd, 1H), 7 04 (dd, 1 H), 7 36-7 46 (m, 6H), 7 68 (d, 1 H), 7 80 (d, 1 H) LRMS m/z (FIA) 377 [MH+]
Examples 34-124 were prepared according to the methods described above for examples 1 , 3 and 11, or routine variation thereof, starting from the appropriate 2-fluoropyrιdιne1 and the appropriate amine2 10 1 2-Fluoropyrιdιnes are described for example in preparations 2, 13 and 15
2 The required amines are either commercially available, described in the literature or in preparations 17, 18, 20 and 21, or represent a routine modification thereof "De-Bocylatιon" is generally carried out where necessary according to the procedure of Example 13

15








 3 3 3 3
3 3 3 3
3
3
3
3
 3
3
3 3
3
3
 3, by 13
3, by 13
3
11
11 by
by



Examples 125-143 were prepared according to the methods described above for examples 8, 13 and 17, starting from the appropriate Boc protected compound1
1 The appropriate Boc protected starting materials are listed in table below





Examples 144-154 were prepared according to the method described above for Example 10, starting from the appropriate 4-hydroxypyπdιnone1 and the appropriate benzyl bromide2
1 4-Hydroxy-6'-[(3R)-3-hydroxypyrrolιdιn-1-yl]-2H-1 ,3'-bιpyrιdιn-2-one from Preparation 4 and tert-Butyl [(3S)-1-(4-hydroxy-2-oxo-2H-1,3'-bιpyrιdιn-6'-yl)pyrrolιdιn-3-yl]methylcarbamate from Preparation 24
2 Benzyl bromides were commercially available



Examples 155-157 were prepared according to the methods described above for example 16, starting from the appropriate pyridone1 and the bromide from preparation 25
1 The pyndones are either commercially available or known in the literature


Examples 158-195 were prepared according to the methods described above for example 20 starting from the appropriate amine1 and the appropriate acid chloride or chloroformate2
1 The starting amines are listed in the table below
2 Acid chlonde or chloroformate are commercially available









Examples 196-203
Examples 196-203 were prepared according to the methods described above for examples 21 & 23, starting from the appropriate Pyridone1 and the appropriate iodide2 1 The pyπdones are either commercially available or known in the literature
2 The iodides are either known in the literature or described in Preparations 32, 33, 34, 35, 36, 39 or 42



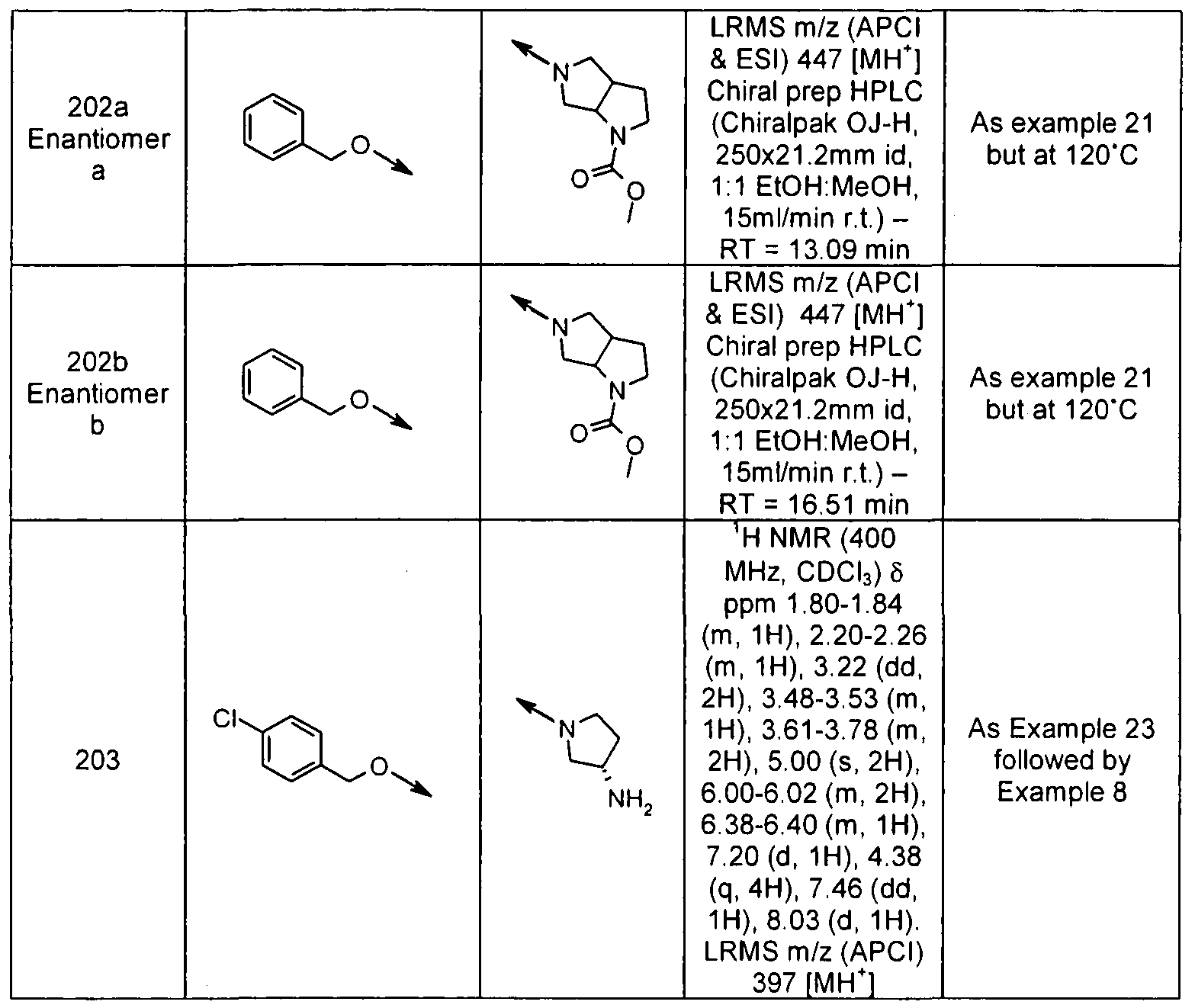
Examples 204-206 were prepared according to the method described below
6'-[(3R)-3-{[tert-Butyl(dιmethyl)sιlyl]oxy}pyrrolιdιn-1-yl]-2-oxo-2H-1 l3l-bιpyπdιn-4-yl 4- bromobenzenesulfonate from Preparation 47 (197mg, 0 324mmol), the appropriate benzyl alcohol (0 982mmol) and potassium hydroxide (55mg) in DMSO (3ml) were heated at 130"C under nitrogen for 1 hour then allowed to stand at r t overnight The reactions were diluted with methanol (3ml) and passed down a SCX column, washed with methanol and the product eluted with 2M NH3 in methanol, evapourated to dryness The residue was chromatographed on Biotage 12 x 150mm silica column eluting with DCM/ MeOH/ NH3 98/2/0 to 93/7/1 Solvent removed in vacuo to give the title compound as solids



Examples 207-212 were prepared according to the methods described above for example 26 starting from the appropriate amine1 and ketone or aldehyde2 1 The amines are described in Examples 8, 125, 130 and 131. 2 The ketones and aldehydes are commercial available



Examples 213-240 were prepared using the methods indicated in the table below starting from the fluoropyridine of preparation 2




Example 241 (= Preparation 23) tert-Butyl (1-{4-[(4-fluorobenzyl)oxy]-2-oxo-2H-1 ,3'-bιpyπdm-6'-yl}-3-methylazetιdιn-3-yl) methylcarbamate

Prepared according to Preparation 22 from fert-butyl (1-{4-[(4-fluorobenzyl)oxy]-2-oxo-2H-1 ,3'-bιpyrιdιn- 6'-yl}-3-methylazetιdιn-3-yl)carbamate from Example 120 to give the title compound as a solid LC-MS RT = 1 52 mm m/z (APCI & ESI) 439 [MH+] (2 mm run)
10 Example 242 ^Preparation 22) fert-Butyl {1-[4-(benzyloxy)-2-oxo-2H-1 ,3'-bιpyπdιn-6'-yl]-3-methylazetιdιn-3-yl}methyl carbamate

A solution of tert-butyl {1-[4-(benzyloxy)-2-oxo-2H-1 ,3'-bιpyπdιn-6'-yl]-3-methylazetιdιn-3-ylcarbarnate
15 from Example 119 (100mg, 2 16mmol) in dry DMF (2ml) was treated with sodium hydride (60% dispersion in oil, 17 3mg, 0432mmol) at r t and stirred for 2 hours Methyl iodide (26 9ul, 0432mmol) was added and stirring continued for 2 hours The reaction was carefully diluted with methanol (2ml) and passed down a SCX column, washed with methanol and the product eluted with 2M NH3 in methanol
The solution was evaporated to dryness to give the title compound as a solid (91 mg, 88%) LC-MS RT =
20 1 50 mm m/z (APCI & ESI) 421 [MH+] (2 mm run)
Example 243 (=Preparation 55) tert-Butyl {(3R)-1-[4-(benzyloxy)-2-oxo-2H-1 ,2'-bιpyrιdιn-5'-yl]pyrrolιdιn-3-yl}methylcarbamate

A suspension of 4-(Benzyloxy)-5'-ιodo-2H-1 ,2'-pyrιdιn-2-one from Preparation 2a (100mg, 0 24mmol), tert-butyl methyl[(3R)-pyrrolιdιn-3-yl]-carbamate (60mg, 0 29mmol, see Example 34b in
WO2003/106462), Pd(OAc)2 (2 5mg, 0 009mmol), BINAP (6 1mg, 0 009mmol) and NaO1Bu (33mg, O 35mmol) in toluene (5ml) was purged with nitrogen for 30mιns Reaction mixture was heated under reflux condition for 12h After cooling to room temperature, water was added and reaction mixture was extracted with ethyl acetate (3 x 20ml) Organic layer was washed with water (30ml), brine (30ml) and dried (Na2SO4) Concentration and column purification (silica, 30% EtOAc/hexane) provided the title compound as a light yellow solid (100mg, 84%) 1H NMR (400 MHz, CDCI3) δ ppm 1 47 (s, 9H), 2 13-2 22 (m, 2H), 2 80 (s, 3H)1 3 23-3 33 (m, 2H), 3 46-3 52 (m, 2H), 5 02 (s, 2H), 6 02 (d, 1H), 6 06 (dd, 1 H), 6 94 (dd, 1H), 7 34-7 40 (m, 5H), 7 59-7 65 (m, 2H), 7 80 (d, 1H) LRMS m/z (FIA) 477 [MH*]
Preparation 1 6'-Amino-4-(benzyloxy)-2H-1,3'-bipyridin-2-one

A stirred solution of 4-benzyloxy-1 H-pyrιd-2-one (5 0 g, 24 8 mmol, Aldnch), 5-ιodo-pyrιdιn-2-ylamιne (5 7 g, 26 1 mmol, Aldnch), trans-N,N'-dιmethylcyclohexane-1 ,2-dιamιne (0 71 g, 4 97 mmol), potassium carbonate (10 3 g, 74 5 mmol) and copper(l) iodide (0 95 g, 4 97 mmol) in toluene (100 ml) was purged with nitrogen for 30 minutes The mixture was then heated at reflux for 3 hours The toluene was then evaporated and the residue was stirred with water (500 ml) for 30 minutes The solids were collected by filtration and washed two more times with water (2 x 500 ml), collecting the solids by filtration after each wash The solids were slurried in toluene/methanol (300 ml of a 3 7 mixture) and then concentrated, which was then repeated a further two times to give the desired product as a dark grey powder (7 06 g, 97%) which was used without further purification 1H NMR (400MHz, d6-DMSO) δ ppm 5 13 (s, 2H), 6 00
(d, 1 H), 6 14 (dd, 1H), 7 30-7 43 (m, 6H) 7 65 (d, 1H), 8 07 (m, 1H), 8 28 (d, 1H) LRMS m/z (APCI/ES) 294 [MH+]
Preparation 2. 2a 5 4-(Benzyloxy)-6'-fluoro-2H-1,3'-bipyridin-2-one (Prep2)

Method A
Pyridine (5 ml) was added dropwise to pyπdine-HF solution (70% HF, 20 ml) CAUTION EXOTHERMIC 6'-Amιno-4-(benzyloxy)-2H-1 ,3'-bιpyπdιn-2-on from Preparation 1 (3 5 g, 12 0 mmol) was added in one
10 portion and the solution cooled to -78 0C under nitrogen Sodium nitrite (1 0 g, 14 4 mmol) was added in one portion and the solution was allowed to warm to room temperature with stirring under nitrogen Evolution of nitrogen began at -20 0C The mixture was poured into 10% aqueous potassium carbonate solution (200 ml) and the mixture was extracted with DCM (30 x 50 ml) The combined extracts were dried (MgSO4), filtered and concentrated to give the desired product (Prep2) as a pink/brown amorphous
15 powder (3 4 g, 96%) 1H NMR (400 MHz, CD3OD) δ ppm 5 18 (s, 2H), 6 16 (s,1H), 6 35 (d, 1H), 7 21 (d, 1H), 7 35-7 45 (m, 5H), 7 61 (d,1H), 8 04 (m,1H), 8 27 (s, 1H) LRMS m/z (APCI/ES) 297 [MH+]
Method B
A stirred solution of 4-benzyloxy-1 H-pyπd-2-one (1 8 g, 9 0 mmol, Aldπch), 2-fluoro-5-ιodo-pyrιdιne (2 0
20 g, 9 0mmol, Aldπch), trans-N,N'-dιmethylcyclohexane-1,2-dιamιne (0 26 g, 1 79 mmol), potassium carbonate (3 70g, 26 9mmol) and copper(l) iodide (0 17 g, 0 90 mmol) in DMF (100 ml) was purged with nitrogen for 30 minutes then heated at 110 0C for 12 hours The mixture was cooled to ambient temperature and filtered The filtrate was evaporated and the residue was vigorously stirred with water
(500 ml) for 3 hours The solids were filtered off and dned by azeotroping with methaπol/toluene (3 1 , 3 x
25 400ml) on a rotary evaporator The crude residue was then purified by column chromatography eluting with a gradient from DCM to 9 1 DCM MeOH to give the desired product (Prep2) as a white powder (1 18 g, 44%)
4-(Benzyloxy)-5'-iodo-2H-1,2'-pyridin-2-one (Prep2a)
30
 4-(Benzyloxy)-5'-ιodo-2H-1,2'-bιpyrιdιn-2-one (Prep2a) was obtained as a by product from Method B as a colourless solid (3 6g) 1H NMR (400 MHz, d6-DMSO) δ ppm 5 14 (s, 2H)1 5 98 (d, 1H), 6 15 (dd, 1 H), 7 35-7 46 (m, 5H), 7 61 (d, 1 H), 7 82 (d, 1 H), 8 29 (dd, 1 H), 8 79 (d, 1 H) LRMS m/z (APCI & ES) 405 [MH+]
4-(Benzyloxy)-5'-ιodo-2H-1,2'-bιpyrιdιn-2-one (Prep2a) was obtained as a by product from Method B as a colourless solid (3 6g) 1H NMR (400 MHz, d6-DMSO) δ ppm 5 14 (s, 2H)1 5 98 (d, 1H), 6 15 (dd, 1 H), 7 35-7 46 (m, 5H), 7 61 (d, 1 H), 7 82 (d, 1 H), 8 29 (dd, 1 H), 8 79 (d, 1 H) LRMS m/z (APCI & ES) 405 [MH+]
Preparation 3 4-(Benzyloxy)-6'-chloro-2H-1,3'-bipyridin-2-one

Preparation 4
4-Hydroxy-6'-[(3R)-3-hydroxypyrrolidin-1-yl]-2H-1,3'-bipyridin-2-one

To a stirred solution of 4-(benzyloxy)-6'-[(3R)-3-hydroxypyrrolιdιn-1-yl]-2H-1 ,3'-bιpyrιdιn-2-one from Example 11 (1 65 g, 4 54 mmol) in ethanol (15 ml) was added 1-methyl-1 ,4-cyclohexadιene (2 20 g, 23 20 mmol) and 20% palladium hydroxide on carbon (190 mg) After heating at 70 0C for 3 hours the mixture was further diluted with ethanol (approximately 100 ml) until all of the reaction mixture other than the catalyst was in solution The hot mixture was then filtered through arbocel under nitrogen, the catalyst was washed with ethanol (2 x 25 ml) and the filtrates were combined The solution was concentrated and the residue was slurried with ethanol (10 ml) The solid was collected by filtration, washed with ethanol (10 ml) then diethyl ether (10 ml) and air-dπed to give the desired product as an off- white solid (1 10g, 89%) 1H NMR (400MHz, CD3OD) δ ppm 2 00-2 09 (m, 1H), 2 10-2 21 (m, 1 H), 3 43-
3 49 (m, 1 H), 3 55-3 65 (m, 3H), 4 51-4 55 (m, 1 H), 5 85 (d, 1H), 6 12 (dd, 1 H), 6 58 (d, 1 H), 7 42 (d, 1H), 4 46 (dd, 1H), 7 98 (d, 1 H) LRMS m/z (APCI) 275 [MH+]
Preparation 5 (3R)-1-{4-[(4-Chlorobenzyl)oxy]-2-oxo-2H-1,3'-bipyridin-6'-yl}pyrrolidin-3-yl methanesulfonate

To a stirred solution of 4-[(4-chlorobenzyl)oxy]-6'-[(3R)-3-hydroxypyrrolιdιn-1-yl]-2H-1 ,3'-bιpyπdιn-2-one from Example 10 (40 mg, 0 10 mmol) in dichloromethane (1 ml) was added tπethylamine (28 μl, 0 20 mmol), followed by methanesulfonyl chloride (9 μl, 0 11 mmol) After stirring at room temperature for 15 hours, second portions of triethylamine (28 μl, 0 20 mmol) and methanesulfonyl chloride (9 μl, 0 11 mmol) were added After 5 hours the reaction mixture was quenched by addition of water (2 ml) and extracted with dichloromethane (2 ml) using a phase separation cartridge The organic component was concentrated to give a clear oil (50 mg) 1H NMR analysis suggested the presence of some unreacted starting material The crude product mixture was dissolved in dichloromethane (1 ml), then triethylamine (28 μl, 0 20 mmol) followed by methanesulfonyl chloride (9 μl, 0 11 mmol) were added After 2 hours the mixture was diluted with water, the organic phase was separated and the aqueous layer was extracted with dichloromethane (2 ml) The organic components were combined, dried using a phase separation cartridge and concentrated to give a clear oil (48 mg, 100%) which solidified on standing 1H NMR (CDCI3, 400MHz) δ ppm 2 37-2 46 (m, 1 H), 2 57-2 65 (m, 1H), 3 09 (s, 3H), 4 85-5 32 (m, 4H), 5 01 (s, 2H), 5 52 (br s, 1 H), 6 01 (d, 1 H), 6 12 (dd, 1H), 6 86 (d, 1 H)1 7 26-7 42 (m, 5H), 8 03 (dd, 1H), 8 25 (s 1H) LRMS m/z (APCI) 476 [MH+]
Preparation 6 (3S)-1-(5-Bromopyridin-2-yl)pyrrolidin-3-ol

To a stirred solution of 2,5-dιbromopyrιdιne (1 0 g, 4 2 mmol) in terf-butanol (5 ml) was added (S)-3- hydroxypyrrolidine (0 74 g, 8 44 mmol, Aldrich) and sodium carbonate (1 34 g, 12 70 mmol) The mixture was heated at 140 0C for 3 hours in a reacti-vial After cooling to room temperature the mixture was diluted with water (20 ml) and extracted with ethyl acetate (20 ml) The aqueous component was separated and extracted with ethyl acetate (20 ml) The combined organic components were dried
(Na2SO4), filtered and concentrated to give a brown oil The crude product mixture was purified by column chromatography (eluting with 100% DCM → 90 10 1 DCM MeOH NH3) to give the desired product as a white solid (1 0 g, 97%) 1H NMR (400MHz, CDCI3) δ ppm 2 01-2 20 (m, 3H), 3 41-3 60 (m, 4H), 4 58-4 61 (m, 1 H), 6 23 (d, 1H), 7 45 (dd, 1H), 8 11 (d, 1 H) LRMS m/z (APCI) 377 [MH*]
Preparation 7 tert-Butyl [1-(5-iodopyridin-2-yl)-3-methylpyrrolidin-3-yl]carbamate

A solution of 2-fluoro-5-ιodo pyridine (590 mg, 2 65 mmol) in DMSO (3 ml) was treated with potassium carbonate (731 mg, 5 3 mmol) and tert-butyl (3-methylpyrrolιdιn-3-yl)carbamate (Chem Pharm Bull 1996, 44(7) 1376, 583 mg, 2 91 mmol) The reaction was heated to 100°C for 16 hours and then cooled to room temperature Water (20 ml) was added and the product was extracted with ethyl acetate (2x20ml) The combined organics were washed with brine (3x20ml), dried (Na2SO4) and concentrated in vacuo to a brown oil Purification on an SCX column (non-basic impurities with methanol, basic products eluted with 2M ammonia in methanol) gave the title compound as a pale brown oil (790 mg, 74%) 1H NMR (400MHz, CD3OD) δ ppm 1 42 (s, 9H), 1 45 (s, 3H), 1 93-2 01 (m, 1H), 2 30-2 38 (m, 1H), 3 33-3 36 (m, 1H), 3 43-3 51 (m, 2H), 3 71-3 74 (d, 1 H), 6 33-6 35 (d, 1 H), 7 67-7 70 (dd, 1H), 8 14 (d, 1H) LRMS m/z (APCI) 404 [MH+]
Preparation 10a: 10b fert-Butyl ethyl[(3f?)-1-(5-ιodopyπdιn-2-yl)pyrrolιdιn-3-yl]carbamate

Potassium carbonate (1 45 g, 10 5 mmol) was added to a mixture of 2-fluoro-5-ιodopyπdιne (780 mg, 3 50 mmol) and fert-butyl ethyl[(3R)-pyrrolιdιn-3-yl]carbamate (900 mg, 4 20 mmol) in DMSO (25 ml) at room temperature The reagents were then heated at 100°C for 5 hours and then cooled to room temperature The reaction was diluted with water (20 ml) and extracted with EtOAc (50 ml) The organic phase was separated and the aqueous phase re-extracted with EtOAc (2 x 30 ml) All organic fractions were combined then dried (MgSO4), filtered and evaporated to give a yellow oil Purification by chromatography (eluting with heptane → heptane EtOAc (70 30)) gave the title compound (Prep. 10a) as a colourless solid 567mg (39%) 1H NMR (400MHz, CDCI3) δ ppm 1 11 (t, 3H), 1 42 (s, 9H), 2 07-2 21 (m, 2H), 3 10-3 38 (m, 4H), 3 59-3 66 (m, 2H), 4 60-4 71 (bm, 1H), 6 19 (d, 1 H), 7 60 (dd, 1 H), 8 25 (s, 1 H) LRMS m/z (APCI) 418 [MH*]
The opposite enantiomer Prep. 10b was prepared via the same method with tert-butyl ethyl[(3S)- pyrrolιdιn-3-yl]carbamate to give the title compound as a colourless solid 1 H NMR (400MHz, CDCI3) δ ppm 1 11 (t, 3H), 1 42 (s, 9H), 2 07-2 21 (m, 2H), 3 10-3 38 (m, 4H), 3 59-3 66 (m, 2H), 4 60-4 71 (bm, 1H), 6 19 (d, 1 H), 7 60 (dd, 1 H), 8 25 (s, 1 H) LRMS m/z (APCI) 418 [MH+]
Preparation 12
6'-Amιno-4-[(4-fluorobenzyl)oxy]-2H-1 ,3'-bιpyrιdιn-2-one

Prepared according to Preparation 1 from 4-[(4-fluorobenzyl)oxy]pyπdιn-2(1 H)-one (WO2005018557) to give the title compound as a dark brown solid 1HNMR (400 MHz DMSO) δ ppm 5 18(s, 2H), 5 95 (s, 1H), 6 05(d, 1 H), 6 12-6 22 (br m, 1 H), 7 18-7 25 (m, 3H), 7 31-7 38 (m, 1 H), 7 41-7 52 (m, 3H) LC-MS RT = 1 89 m/z (APCI & ESI) 312 [MH+] (6 mm acidic run)
Preparation 13 6'-Fluoro-4-[(4-fluorobenzyl)oxy]-2H-1 ,3'-bιpyrιdιn-2-one

Prepared according to Preparation 2 Method A from 6'-Amιno-4-[(4-fluorobenzyl)oxy]-2H-1 ,3'-bιpyπdιn-2- one from Preparation 12 to give the title compound as a pale brown solid 1HNMR (400 MHz dβ-DMSO) δ ppm 5 12(s, 2H), 6 01 (s, 1 H), 6 14 (d, 1 H), 7 23-7 35 (t, 2H), 7 31(d, 1 H), 7 49-7 52 (m, 2H)1 7 64 (d, 1H) 8 04-8 08 (m, 1 H), 8 28 (s, 1 H) LC-MS RT = 1 74 m/z (APCI & ESI) 315 [MH+] (6 mm acidic run)
Preparation 14
6'-Amιno-4-[(4-chlorobenzyl)oxy]-2H-1 ,3'-bιpyπdιn-2-one

4-[(4-chlorobenzyl)oxy]- 6'-fluoro-2H-1 ,3'bιpyπdιn-2-one

5 Prepared according to Preparation 2 Method A from 6'-Amιno-4-[(4-chlorobenzyl)oxy]-2H-1,3'-bιpyrιdιn- 2-one from Preparation 14 to give the title compound as a pale brown solid 1HNMR (400 MHz dβ-DMSO) δ ppm 5 18(s, 2H), 6 01 (s, 1 H), 6 21 (d, 1 H), 7 35 (d, 1H), 7 45-7 51 (m, 4H), 7 69 (d, 1H), 8 00-8 10 (m, 1 H), 8 31 (s, 1 H)
10 Preparation 16
1-Benzyl-3-ιsopropylpyrrolιdιn-3-ol

Cerium (III) chloride (4 4g, 186mmol) was suspended in anhydrous THF ( 62ml) and stirred at r t overnight The reaction was then cooled to 0*C-5'C and isopropylmagnesium chloride (2 OM in THF,
15 9 34ml, 18 6mmol) was added and the reaction was stirred at O'C for 1 V- hours 1-Benzyl-3-pyrrolιdone (2 17g/2ml, 12 4mmol, Aldrich) was added and the reaction was stirred over night allowing to warm to r t The reaction was poured into sat NH4CI(ac,) (300ml) and extracted with ethyl acetate (2 x 200ml) The combined extracts were dried (MgSO4) and concentrated to give the title compound as an oil (2 705g, 99%) 1H NMR (400 MHz, CDCI3) δ ppm 0 91 (d, 3H), 0 95 (d, 3H) 1 65-1 72 (m, 1H)1 1 74-1 81 (m,
20 1H), 1 87-1 94 (m, 1H), 2 28 (d, 1 H), 2 30-2 34 (m, 1H), 2 70 (d, 1 H), 2 96-3 05 (m, 1H), 3 65 (s, 2H), 7 21-7 34 (m, 5H) LRMS m/z (ESI) 220 [MH+]
Preparation 17
3-lsopropylpyrrolιdιn-3-ol hydrochloride
HCI

The hydrochloride salt of 1-Benzyl-3-ιsopropylpyrrolιdιn-3-ol from Preparation 16 (944mg, 3 7mmol) and
10% palladium on carbon (900mg) were placed in a flask under N2 Absolute ethanol (20ml) was carefully added followed by acetic acid (0 63ml, 11 1mmol) and 1 ,4-cyclohexadιene (1 75ml, 18 5mmol)
The mixture was heated to 50'C, once the reaction was complete by LCMS the reaction was cooled and filtered through celite, the filtrate was concentrated to give the title compound LRMS m/z (ESI) 130 [MH+]
Preparation 18
(3S)-N-lsopropyl-N-methylpyrro!ιdιn-3-amιne hydrochloride
HCI
NQ N-
-\
(S)-(-)-N-Boc-3-amιnopyrrohdιne (500mg, 2 68mmol, Lancaster) and acetone (10ml) were added to 4 1 methanol toluene (500ml), the solution was then evaporated to dryness over a period of 5 hours at 35'C The residue was taken up in DCM (100ml), sodium triacetoxyborohydπde (1 14g) was added and the mixture stirred for 10 minutes Acetic acid (20ml) was added and the mixture stirred for 1 hour at r t MS gave [MH+] 228 as single component, corresponding to mono isopropyl secondary amine derivative Formaldehyde solution (4ml) was added and the mixture stirred overnight Aqueous potassium carbonate 10% (150ml) was added and the mixture stirred for 1 hour The DCM layer was separated, dried (MgSO4), then evaporated to give as a colourless viscous oil which partially solidified on standing to a waxy solid The compound was then added to 4N HCI/dioxane (100ml) and stirred overnight The dioxane/HCI was evaporated and the residue dried overnight in vacuo at 50'C to give the title compound as a solid (460mg, 70%) 1H NMR (400MHz , CD3OD) δ ppm 1 31 (s, 3H), 1 45 (s, 3H), 2 38-2 51 (m, 1H), 2 59-2 70 (m, 1H), 2 84 (s, 3H), 321-349 (m, 1H), 3 60-3 91 (m, 4H), 4 21-4 38 (m, 1H)
Preparation 19 tert-Buty) 3-azetιd in- 1 -ylpyrrolidine- 1 -carboxylate
o*
0^1O-O
1-N-Boc-3-pyrrolιdιnone (400mg, 2 16mmol, Aldπch), azetidine hydrochloride (337mg, 2 59mmol, Aldπch) and sodium triacetoxyborohydride (915mg, 4 32mmol) were added to DCM (4ml), acetic acid
(40ml) was then added dropwise over a period of 5 minutes with stirring The mixture was then stirred overnight at r t 10% aqueous potassium carbonate solution (50ml) was then added and the mixture stirred for 30 minutes The DCM layer was separated, dried (MgSO4), then evaporated to give the title compound as an oil (451mg, 92%) 1H NMR (400MHz , CD3OD) δ ppm 1 42(s, 9H), 1 62-1 75 (br m, 1H), 1 80-1 93 (br m, 1H), 2 09-2 19 (m, 2H), 2 58-2 62 (m, 1H), 3 25-3 40 (m, 8H) LRMS.m/z (APCI &
ESI) 227 [MH+]
Preparation 20
3-Azetιdιn-i-ylpyrrolιdιne hydrochloride
.HCI

Prepared according to Example 8 from terf-Butyl 3-azetιdιn-i-ylpyrrolιdιne-i-carboxylate from Preparation 19 to give the title compound as colourless plates 1H NMR (400MHz , CD3OD) δ ppm 1HNMR (400 MHz CD3OD) δ ppm 2 20-2 40 (m, 2H), 2 40-2 60 (m, 2H), 3 20-3 37 (m, 2H), 3 40-3 52 (m, 2H), 3 60-3 80 (m, 2H), 4 10-4 40 (m 3H)
Preparation 21 terf-Butyl (3-methylazetιdιn-3-yl)carbamate
A solution of fert-butyl [1-(dιphenylmethyl)-3-methylazetιdιn-3-yl]carbamate (1 397g, 3 963mmol) in a mixture of ethanol/ethyl acetate (25ml/5ml) was hydrogenated over pearlmans catalyst (300mg) at 50psι and 6O-C overnight The catalyst removed by filtration and the filtrate reduced in vacuo to give a dark oil The oil was taken up in methanol and passed down a SCX column, washed with methanol and the product washed off the column with 2M NH3 in methanol The solvent was evaporated to dryness to give a reddish oil which slowly solidified (730mg, 99%) 1H-NMR(400MHz, CDCI3) δ ppm 1 4 (s, 9H), 1 47 (s, 3H), 3 3 (d, 2H)1 3 85 (br s, 2H), 4 87
Preparation 24 tert-Butyl [(3S)-1-(4-hydroxy-2-oxo-2H-1 ,3'-bipyridin-6'-yl)pyrrolidin-3-yl]methylcarbamate

Prepared according to Preparation 4 from terf-Butyl {(3S)-1-[4-(benzyloxy)-2-oxo-2H-1 ,3'-bιpyπdιn-6'- yl]pyrrolιdιn-3-yl} methyl carbamate from Example 12 to give the title compound as a white foam 1H NMR (400 MHz, CD3OD) δ ppm 1 50 (s, 9 H), 2 19 - 2 29 (m, 2H), 2 85 (s, 3H), 3 42 - 3 52 (m, 2H), 3 65 - 3 75 (m, 2H), 4 83 - 4 91 (m, 1 H), 5 88 (d, 1 H), 6 13 (dd, 1H), 6 62 (d, 1 H), 7 46 (d, 1 H), 7 53 (dd, 1 H), 8 01 (d,1 H) LRMS m/z (APCI & ESI) 387 [MH*]
Preparation 25 terf-Butyl [(3R)-1-(6-bromopyrιdazιn-3-yl)pyrrolιdιn-3-yl]carbamate

3,6-Dιbromopyrιdazιne (800mg, 3 36mmol, Maybπdge), (3R)-(+)-3-(Boc-amιno)pyrrolιdιne (689mg, 3 70mmol, Lancaster), cesium fluoride (102mg, 0673mmol) and potassium carbonate (1 39g, 10 1mmol) were added to DMSO (10ml) and the stirred mixture heated at 100'C for 2 hours, effervescence was initially observed between 60-70'C The mixture was poured into brine (50ml) and the mixture stirred for 30 minutes The solids were filtered off rinsed with water and sucked dry overnight on a sinter to give the title compound as a grey-brown solid (0 99g, 86%) 1H NMR(400MHz, CD3OD) δ ppm 1 41 (s, 9H), 1 93-2 06 (m, 1 H), 2 21-2 30 (m, 1H), 3 31-3 38 (m, 1 H), 3 42-3 61 (m, 2H), 3 69-3 75 (m, 1 H), 4 20-4 29 (m, 1 H), 6 85 (d, 1 H), 7 48 (m, 1 H)
Preparation 26 fert-Butyl [(3S)-1-(6-bromopyrιdazιn-3-yl)pyrrolιdιn-3-yl]carbamate

Prepared according to Preparation 25 from tert-butyl methyl[(3S)-pyrrolιdιn-3-yl]-carbamate (ECA International) to give the title compound as a buff solid 1H NMR (400 MHz, CDCI3) δ ppm 1 44 (s, 9H), 2 10-2 29 (m, 2H), 2 82 (s, 3H), 3 41 (dd, 1H), 3 46 (dd, 1H), 3 67-3 78 (m, 2H), 4 84-4 95 (br m, 1H), 6 55 (d, 1 H), 7 29 (d, 1 H) LRMS m/z (APCI/ESI) 359 [MH+]
Preparation 27 (3R)-1-(6-Bromopyπdazιn-3-yl)pyrrolιdιn-3-ol

Prepared according to Preparation 25 from (R)-(+)-3-hydroxypyrrolιdιne (Aldπch) to give the title compound as a pale yellow solid 1H NMR (400 MHz, CDCI3) δ ppm 1 60-1 80 (br s, water +OH), 2 10- 2 22 (m, 2H), 3 58-3 70 (m, 4H), 463-4 70 (m, 1 H), 6 58 (d, 1H), 7 29 (d, 1 H) LRMS m/z (APCI) 246 [MH+]
Preparation 29
Methyl 1 -(5-ιodopyrιdιn-2-yl)pyrrolιdιπe-3-carboxylate

Prepared according to Preparation 7 from methyl pyrrolιdιne-3-carboxylate (Bioorganic Med Chem Letts 14(19), 4861-4866, 2004) to give the title compound as an off-white solid 1H NMR (400 MHz, CDCI3) δ ppm 2 25 (q, 2H), 3 17-3 22 (m, 1H)1 3 39-3 43 (m, 1H), 3 51-3 59 (m, 1 H), 3 60-3 66 (m 2H), 3 68 (s, 3H), 6 20 (d, 1 H), 7 60 (dd, 1 H), 8 22 (s, 1H) LRMS m/z (APCI & ESI) 333 [MH+]
Preparation 30 Methyl 1-[4-(benzyloxy)-2-oxo-2H-1 ,3'-bιpyπdιn-6'-yl]pyrrolιdιne-3-carboxylate

Prepared according to Preparation 2 Method B from methyl 1-(5-ιodopyπdιn-2-yl)pyrrolιdιne-3- carboxylate from Preparation 29 to give the title compound as an off white solid 1H NMR (400 MHz, CDCI3) δ ppm 2 24-2 32 (m, 2H), 3 18-3 23 (m, 1 H), 3 44-3 51 (m, 1 H), 3 59-3 64 (m, 1H), 3 65-3 79 (m, 5H), 5 01 (S, 2H), 6 00-6 02 (m, 2H), 6 40 (d, 1H), 7 18 (d, 1 H), 7 31-7 41 (m, 5H), 7 46 (dd, 1 H)1 8 02 (s, 1H) LRMS m/z (APCI & ESI) 406 [MH+]
Preparation 31
1-[4-(Benzyloxy)-2-oxo-2H-1 ,3'-bιpyrιdιn-6'-yl]pyrrolιdιne-3-carboxylιc acid

Sodium hydroxide (1M aq soln) (5 92 ml, 5 92 mmol) was added to a solution of methyl 1-[4-(benzyloxy)- 2-oxo-2H-1 ,3'-bιpyπdιn-6'-yl]pyrrolιdιne-3-carboxylate from Preparation 30 (1 20 g, 2 96 mmol) in MeOH (6 00 ml) and THF (4 00 ml) at r t under N2 Stirring was continued for 3 hours and then the solvents were evaporated in vacuo, and the residue was partitioned between CH2CI2 (10 ml) and water (10 mL) The aqueous layer was then acidified to pH2 by addition of 2M HCI (aq) (2 mL) Solvents were again
removed in vacuo and the residue was azeotroped with toluene ( 2 x 10 mL) to give the title compound as an off white solid (1 37 g, 108%) 1H NMR (400 MHz, CD3OD) δ ppm 2 37-2 50 (m, 2H), 3 39-3 45 (m, 1 H), 3 67-3 76 (m, 2H), 3 82-3 91 (m, 2H), 5 13 (s, 2H), 6 03 (d, 1 H), 6 25 (dd, 1 H), 7 18 (d, 1 H), 7 30- 7 43 (d, 5H), 7 52 (d, 1 H), 7 99 (dd, 1 H), 8 06 (d, 1 H) LRMS m/z (APCI) 390 [MH+]
Preparation 32 tert-Butyl [(3S)-1 -(5-ιodopyπdιn-2-yl)pyrrolιdιn-3-yl]carbamate

Prepared according to Preparation 7 from (S)-(-)-3-(Boc-amιno)pyrrolιdιne (Lancaster) to give the title compound as a tan solid 1H NMR (300MHz, CDCI3) δ ppm 1 44 (s, 9H), 1 8-2 0 (m, 1 H), 2 2-2 29 (m, 1H), 3 3 (d, 1H), 349-3 55 (m,2H), 3 65-3 70 (m, 1H), 4 33 (br s, 1H), 4 68 (br s, 1H), 6 20 (d, 1 H), 7 62 (d, 1 H), 8 27 (s, 1 H) LRMS m/z (ESI) 390 [MH+]
Preparation 33 (3S)-1-(5-lodopyrιdιn-2-yl)pyrrolιdιn-3-amιne hydrochloride

Prepared according to Example 13 from /ert-Butyl [(3S)-1-(5-ιodopyrιdιn-2-yl)pyrrolιdιn-3-yl]carbamate from Preparation 32 to give the title compound as a white solid 1H NMR (300MHz, CD3OD) δ ppm 2 25- 2 4 (m, 1 H), 2 5-2 63 (m, 1H), 3 69-3 85 (m, 3H), 3 91-4 02 (m, 1 H), 4 16 (br s, 1 H), 6 98 (d, 1 H), 8 18 (s, 1 H), 8 20 (S, 1 H) LRMS m/z (ESI) 290 [MH+]
Preparation 34
Methyl [(3S)-1 -(5-ιodopyrιdιn-2-yl)pyrrolιdιn-3-yl]carbamate

10 Preparation 35
Λ/-[(3R)-1-(5-lodopyπdιn-2-yl)pyrrolιdιn-3-yl]propanamιde

Prepared according to Preparation 7 from N-[(3R)-pyrrolιdιn-3-yl]propanamιde (EP391169 A2) to give the title compound as a cream solid 1H NMR (400 MHz, CDCI3) δ ppm 1 14 (t, 3H), 1 91-1 99 (m, 1H), 15 2 16 (q, 2H), 2 22-2 31 (m, 1 H), 3 25 (dd, 1H), 3 43-3 54 (m, 2H), 3 65 (dd, 1 H), 4 57-4 62 (m, 1 H), 5 55 (br s, 1H), 6 20 (d, 1H), 7 61 (dd, 1H), 825 (s, 1H) LRMS m/z (APCI & ESI) 346 [MH+]
Preparation 36 tert-Butyl [(3R)-1-(5-ιodopyπdιn-2-yl)pyrrolιdιn-3-yl]methylcarbamate

Prepared according to Preparation 7 from (R)-(+)-3-(Boc-amιno)pyrrolιdιne (Lancaster) to give the title compound as an off white solid 1H NMR (300MHz, CDCI3) δ ppm 1 44 (s, 9H), 2 01-2 21 (m, 2H), 2 80 (S, 3H), 3 25-34 (m, 2H), 3 56-3 6 (m, 2H), 4 86 (br s, 1 H), 6 19 (d, 1 H), 7 62 (d, 1 H), 8 27 (s, 1H) LRMS m/z (ESI) 404 [MH+] 25
Preparation 37 tert-Butyl (3aRS,6aRS)-1-acetylhexahydropyrrolo[3,4-b]pyrrole-5(1 H)-carboxylate

A solution of fert-butyl (3aRS,6aRS)-hexahydropyrrolo[3,4-b]pyrrole-5(1H)-carboxylate (WO2001081347,
2 0Og, 9 42mmol) in dichloromethane (20ml) was cooled to 0°C and treated with triethylamine (1 58ml,
11 3mmol), followed by acetyl chloride (0 703ml, 9 89mmol) The cold bath was removed and the solution stirred at r t under nitrogen overnight The reaction mixture was washed with sat NaHCO3 solution (2 x 20ml) then brine (20ml) The organics were dried (Na2SO4) and concentrated in vacuo to give the title compound as a pale amber oil (2 17g, 90%) 1H-NMR(400MHz, CD3OD) δ ppm 1 44 (s, 9H),
1 80-1 89 (m, 1H), 2 06 (s, 3H), 2 07-2 14 (m, 1H), 2 91-2 98 (m, 1 H), 3 22 -3 27 (m, 1H), 343-348 (m,
1 H), 3 49-3 66 (br m, 4H), 3 32-3 39 (m, 1H) LC-MS RT = 2 45 mm m/z (APCI & ESI) 155 [MH+-BoC] (6 mm acidic run)
Preparation 38
(3aRS,6aRS)-1-Acetyloctahydropyrrolo[3,4-b]pyrrole

Preparation 39
(3aRS, 6aRS)-1-Acetyl-5-(5-ιodopyπdιn-2-yl)-octahydropyrrolo[3,4-b]pyrrole

Prepared according to Preparation 7 from (3aRS,6aRS)-1-acetyloctahydropyrrolo[3,4-b]pyrrole from Preparation 38 to give the title compound as a golden oil which was used without further purification 1H- NMR(400MHz, CD3OD) δ ppm 1 89-1 97 (m, 1 H), 2 07 (s, 3H), 2 13-2 23 (m, 1 H), 3 05-3 14 (m, 1H), 3 34-3 39 (m, 1 H), 5 54-3 69 (m, 5H) 4 47-4 55 (m, 1 H), 6 37 (d, 1 H), 7 70 (dd, 1H), 8 17 (d, 1 H) LC-MS RT = 2 15 mm m/z (APCI & ESI) 358 [MH+] (6 mm acidic run)
Preparation 40
5-fert-Butyl 1 -methyl (3aRS, 6aRS)-hexahydropyrrolo[3,4-b]pyrrole-1,5-dιcarboxylate

Prepared according to Preparation 37 using methyl chloroformate to give the title compound as a pale golden oil 1H-NMR(400MHz, CD3OD) 1 44 (s, 9H)1 1 73-1 82 (m, 1 H), 2 00-2 07 (m, 1H), 2 92-3 01 (m, 1H), 3 18-3 26 (m, 1 H), 3 41-3 59 (br m, 5H), 3 69 (s, 3H), 4 21-4 28 (m, 1 H) LC-MS RT = 2 82 mm m/z (APCI) 171[MH+-BoC] (6 mm acidic run)
Preparation 41
Methyl (3aRS,6aRS)-hexahydropyrrolo[3,4-b]pyrrole-1(2H)-carboxylate

Prepared according to Example 17a from 5-fert-butyl 1 -methyl (3aRS,6aRS)-hexahydropyrrolo[3,4- b]pyrrole-1 ,5-dιcarboxylate from Preparation 40 to give the title compound as a pale yellow oil 1H- NMR(400MHz, CD3OD) δ ppm 1 69-1 77 (m, 1H), 1 98-2 07 (m, 1H), 265-2 71 (dd, 1H), 2 81-2 89 (br m, 1 H), 2 91-2 99 (m, 3H), 3 38-3 46 (m, 1 H), 3 49-3 55 (m, 1 H), 3 70 (br s, 3H), 4 17-4 22 (m, 1H) LRMS m/z (APCI & ESI) 171 [MH+]
Preparation 42
Methyl (3aRS, 6aRS)-5-(5-ιodopyrιdιn-2-yl)hexahydropyrrolo[3,4-b]pyrrole-1 (2H)-carboxylate

Preparation 43 4-Hydroxy-6'-pyrrolιdιn-1-yl-2H-1 ,3'-bιpyπdιn-2-one

Prepared according to Preparation 4 from 4-(benzyloxy)-6'-pyrrolιdιn-1-yl-2H-1,3'-bιpyπdιn-2-one example 34 to give the title compound as a solid which was used without further purification LC-MS RT = 0 59 mm m/z (APCI & ESI) 258 [MH']
Preparation 44
2-Oxo-6'-pyrrolιdιn-1-yl-2H-1 ,3'-bιpyrιdιn-4-yl 4-bromobenzenesulfonate

4-Hydroxy-6'-pyrrolιdιn-1-yl-2H-1 ,3'-bιpyπdιn-2-one from Preparation 43 (1 0g, 3 887mmol), A- bromobenzenesulfonyl chloride (1 15g, 4 δOmmol) and potassium carbonate (1 07g, 7 77mmol) in dimethylacetamide (10ml) under N2 was stirred together overnight at ambient temperature (very thick reaction mixture observed in the morning) EtOAc (30ml) and water (20ml) was added and the mixture was stirred - incomplete solution (other than catalyst) Addition of DCM (50 ml) gave solution The mixture was filtered through Arbocel, washing with DCM (50ml), then water (50ml) The layers were separated and the aqueous extracted with EtOAC (50ml), bulked organic solution was washed water (3 x
5OmI)1 brine (50ml), dried (MgSO4) and evaporated The residue recrystallized from MeCN (needed ~
100ml) to give the title compound as a cream solid (1 39g, 75%) 1H-NMR(400MHz, CDCI3) δ ppm 2 02
(m, 4H), 3 47 (m, 4H), 6 18-6 21 (m, 2H), 6 40 (d, 1H), 7 32 (d, 1H), 7 44 (dd, 1H), 7 75 (m 2H), 7 82 (m,
2H), 8 03 (d, 1 H) LRMS m/z (ESI) 476/478 [MH+]
Preparation 45
4-(Benzyloxy)-6'-[(3R)-3-{[rert-butyl(dιmethyl)sιlyl]oxy}pyrrolιdιn-1-yl]-2H-1 ,3'-bιpyrιdιn-2-one

4-(Benzyloxy)-6'-[(3R)-3hydroxypyrrohdιn-1-yl]-2H-1 ,3'-bιpyrιdιn-2-one from Example 11 (4 00g, 11 01mmol), imidazole (1 5g, 22mmol) and DMAP (53 8mg, 0 44mmol) were added to acetonitrile (100ml) TBDMS-tπflate (3 2g, 12 1mmol) was then added dropwise and the mixture was then heated at 50'C for 1 hour Mixture cooled back to r t and acetonitrile evaporated, residue stirred with water (200ml) for 1 hour, mixture then extracted with EtOAc (3 x 200ml) EtOAc solution then washed successively with 10% K2CO3 (aq) (3 x 200ml), water (3 x 200ml) then brine (200ml) The EtOAc solution was then dried (MgSO4) and evaporated to give the title compound as a white amorphous powder (4 15g, 78%) 1H-NMR(400MHz, d6-DMSO) δ ppm 0 05 (d, 6H), 0 78 (s, 9H), 1 78-1 84 (m,1H), 1 97-2 06 (m, 1 H), 3 18-3 21 (m, 1H), 3 34-3 44 (m, 2H), 3 49-3 53 (m, 1 H), 4 54-4 59 (m, 1H), 5 04 (s, 2H), 5 86 (s, 1H)1 6 18 (d, 1 H), 6 49 (d, 1 H), 7 35-7 58 (m, 7H), 7 99 (s, 1 H) LRMS m/z (ESI) 478 [MH+]
Preparation 46
6'-[(3R)-3-{[fert-Butyl(dιmethyl)sιlyl]oxy}pyrrolιdιn-1-yl]-4-hydroxy-2H-1 ,3l-bιpyrιdιn-2-one

4-(Benzyloxy)-6'-[(3R)-3-{[terf-butyl(dιmethyl)sιlyl]oxy}pyrrolιdιn-1-yl]-2H-1 ,3'-bιpyrιdιn-2-one from Preparation 45 (4 Og1 8 376mmol) in ethanol (100ml) was hydrogenation over 10% palladium on carbon (1 0g) at 50psι and r t for 12 hours The mixture filtered through celite, and the ethanol evaporated to give the title compound as a light brown amorphous solid (3 06g, 94%) 1H-NMR(400MHz, d6-DMSO) δ ppm 0 05 (s, 6H)1 0 82 (s, 9H), 1 80-1 94 (m, 1H), 2 05-2 18 (m, 1H), 3 25-3 32 (m, 1H), 3 40-3 51(m, 2H), 3 56-3 62 (m, 1H), 4 52-4 59 (m, 1H), 5 60 (s, 1H), 5 91 (d, 1 H)1 6 43 (d, 1H), 7 43 (d, 2H), 7 98 (s, 1 H), 10 69 (br s, 1 H) LRMS m/z (ESI) 388 [MH+]
Preparation 47 6'-[(3R)-3-{[tert-Butyl(dιmethyl)sιlyl]oxy}pyrrolιdιn-1-yl]-2-oxo-2H-1 ,3'-bιpyπdιn-4-yl 4-bromo benzenesulfonate

6'-[(3R)-3-{[fe/t-Butyl{dιmethyl)sιlyl]oxy}pyrrolιdιn-1-yI]-4-hydroxy-2H-1,3'-bιpyπdιn-2-one from Preparation 46 (6 185g, 15 96mmol) was suspended in DCM (100ml), tπethylamine (4 45ml, 31 9mmol) was added in one portion and the mixture stirred for 20 minutes at r t to give a homogenous solution The mixture was cooled to 0"C and 4-bromobenzenesulfonyl chloride (4 28Og, 16 8mmol) was added in one portion, reaction exothermed to 5'C The mixture was stirred overnight at r t LC-MS indicated reaction complete 10% aqueous potassium carbonate (200ml) was added and the mixture stirred for 30 minutes The DCM layer was separated washed with water (3 x100ml) then brine (100ml), dried (MgSO4) then evaporated to give the title compound as a white amorphous powder (7 6Og, 78%) Η-NMR(400MHz, dβ-DMSO) δ ppm 0 08 (S, 6H), 0 81 (s, 9H), 1 80-1 92 (m, 1H), 2 05-2 15 (m, 1H), 3 21-3 30 (m, 1H), 3 40-3 49 (m, 2H), 3 57-3 63 (m, 1 H), 4 57-4 61 (m, 1H), 6 18 (d, 2H), 6 52 (d, 1 H), 7 48 (d, 1 H), 7 75 (d, 1 H), 7 98 (s, 4H), 8 0 (S, 1H) LRMS m/z (ESI) 606/608 [MH*]
Preparation 48 4-(Methylthιo)pyπdιn-2( 1 H)-one

4-(Methylthιo)pyπdιne 1 -oxide (Jpn Kokai Tokkyo Koho (1990), 5 pp CODEN JKXXAF JP 02124872, 100mg, 0 708mmol) was added to acetic anhydride (72 3mg, 0 708mmol) and refluxed for 6 hours, the acetic anhydride was then evaporated and the residue refluxed with methanol (5ml) for 3 hours, the mixture was then evaporated and the residue triturated with MTBE (3 x 1ml) to give the title compound as an amorphous dark brown solid (99mg, 99%) 1H-NMR(400MHz, CD3OD) δ ppm 2 41 (s, 3H), 6 25 (d, 2H), 7 21 (s, 1 H)
Preparation 49 6'-[(3R)-3-Hydroxypyrrolιdιn-1-yl]-4-(methylthιo)-2H-1,3'-bιpyrιndιn-2-one
 H
H
Prepared according to Preparation 1 using 4-(Methylthιo)pyrιdιn-2(1 H)-one from Preparation 48 and (3R)-1-(5-lodopyπdιn-2-yl)pyrrolιdιn-3-ol (WO2004039780) to give the title compound as a dark brown amorphous solid which was used without further purification
Preparation 50
6'-[(3R)-3-{[fert-Butyl(dιmethyl)sιlyl]oxy}pyrrolιdιn-1-yl]-4-(methylthιo)-2H-1 ,3'-bιpyrιdιn-2-one

Prepared according to Preparation 45 using 6'-[(3R)-3-Hydroxypyrrohdιn-1-yl]-4-(methylthιo)-2H-1 ,3 - bιpyrιndιn-2-one from Preparation 49, triethylamine as base to give the title compound as a grey solid
1H-NMR(400MHz, d6-DMSO) δ ppm 009 (s, 15H)1190-205 (m, 1H)1209-220 (m, 1H), 167 (s, 3H),
259 Xd, 1H), 274-280 (nm, 2H), 282 (dd, 1H), 379-387 (m, 1H), 551 (s, 1H), 555 (d, 1H), 577 (d,
1 H), 6 60 (d, 1H), 6 72 (d, 1 H), 7 20 (s, 1 H) LC-MS RT = 3 34 mm m/z (APCI & ESI) 418 [MH*] (6 mm acidic run)
Preparation 51
6'-[(3R)-3-{[tert-Butyl(dιmethyl)sιlyl]oxy}pyrrolιdιn-1-yl]-4-(methanesulfonyl)-2H-1 ,3'-bιpyπdιn-2-one

An aqueous mixture of 30% H2O2 (12 2ml, 120mmol) and a 02M solution of NaHCO3 (408ml) previously prepared at O-C was added to a vigorously stirred solution of 6'-[(3R)-3-{[tert-
Butyl(dιmethyl)sιlylJoxy}pyrrolιdιn-1-yl]-4-(methylthιo)-2H-1,3'-bιpyπdιn-2-one from Preparation 50 (10 0g,
23 944mmol) in acetonitrile (552ml) in a single addition The mixture was vigorously stirred with a
mechanical overhead stirrer for 8 hours Reaction complete by LC-MS Reaction mixture was poured in brine (1000ml) the mixture was then extracted with EtOAc (2 x 500ml) The extracts were then evaporated to give a glassy green amorphous solid which was purified on an ISCO 33Og column eluting from 100% DCM 100% EtOAC over 10 CV to give the title compound as a solid (9 0Og, 83%) 1H- NMR(400MHz, CD3OD) δ ppm 0 12 (d, 6H), 0 90 (s, 9H), 1 93-2 02 (m, 1 H), 2 18-2 20 (m, 1 H), 3 19 (s, 3H), 3 39 (d, 1 H), 3 57-3 60 (m, 2H), 3 60-3 67(m, 1 H), 4 78-4 83 (m, 1 H), 6 59 (d, 1 H), 6 79 (d, 1 H), 7 19 (s, 1 H), 7 58 (d, 1 H), 7 83 (d, 1H), 8 04 (s, 1H)
Preparation 52 6'-{(3S)-3-[(fert-Butoxycarbonyl)(methyl)amιno]pyrrolιdιn-1-yl}-2-oxo-2H-1 ,3'-bιpyrιdιnyl-4-yl tπfluoromethanesulfonate
O=S=O
-

To a cold (-5O0C) solution of tert-Butyl [(3S)-1-(4-hydroxy-2-oxo-2H-1 ,3'-bιpyπdιn-6'-yl)pyrrolιdιn-3- yl]methylcarbamate from Preparation 24 (100 mg, 0 259 mmol) in DCM (2 mL) was added triethylamine (57 7 uL, 0 414 mmol) followed by addition of trifluoromethanesulphonic anhydride (65 3 uL, 0 388 mmol) The resulting mixture was stirred at -300C under nitrogen for 2 h The reaction mixture was then poured into ice/water mixture (5 mL) and the products were extracted with DCM (2 x 5mL) The combined organic extracts were washed with water (2 x 5mL), dried over a separating phase cartridge and evaporated to give the title compound as a yellow oil (115 mg, 85%) Η-NMR(400MHz, CDCI3) δ ppm 1 48 (s, 9H), 2 06 - 2 35 (m, 2H), 2 84 (s, 3H), 3 37 - 3 57 (m, 2H), 3 68 - 3 79 (m, 2H), 4 88 (br s, 1H), 628 (dd, 1H), 649 (d, 1H), 6 57 (d, 1H), 744 (d, 1H), 7 58 (dd, 1H), 8 11 (d, 1H) LRMS m/z (APCI & ESI) 519 [MH+]
Preparation 54 N-{(3S)-1-[4-(Benzyloxy)-2-oxo-2H-1 ,3'-bιpyπdιn-6'-yl]pyrrolιdιn-3-yl}-4-chlorobutanamιde

4-Chlorobutyryl chloπde (0 081 ml, 0 73 mmol) was added to a mixture of 6'-[(3S)-3-amιnopyrrolιdιn-1- yl]-4-(benzyloxy)-2H-1 ,3'-bιpyπdιn-2-one Example 55 (239 mg, 0 66 mmol) and tπethylamine (0 092 ml, 066 mmol) in tetrahydrofuran (5 ml), and the opaque reaction mixture was stirred at r t for 2 hours MS showed product mass ion with no SM remaining Solvents were evaporated then the residue was dissolved in DCM (20 mL) and washed with water (30 mL) then brine (20 mL) before drying the organics (MgSO,) filtering and evaporating in vacuo to give the title compound as a yellow powder which was used without further purification (447 mg) LRMS m/z (ESI) 467 [MH+]
It is to be understood that the foregoing description is exemplary and explanatory in nature, and is intended to illustrate the invention and its preferred embodiments Through routine experimentation, the artisan will recognize apparent modifications and variations that may be made without departing from the spirit of the invention Thus, the invention is intended to be defined not by the above description, but by the following claims and their equivalents
Claims
1 A compound of formula (I)

(I) wherein
X is CH2CH2, CH2O or OCH2,
A and B are each independently CH or N, with the proviso that 1 or both of A and B is N,
Ar is phenyl optionally substituted by 1 or 2 substituents independently selected from F and Cl,
R1 is a saturated 4- to 9-membered heterocyclic ring system containing 1 or 2 ring N atoms, which ring system may incorporate spiro-, fused or bridged rings, which is attached to the "ABCCHCHCπng via a N atom, which ring system is optionally substituted by one or more substituents independently selected from =0,
R9 OH, C(O)C1-C5 alkyl, C(O)C3-C5 cycloalkyl, C(O)OC-C5 alkyl, NR6R7, NR8C(O)R9, NR8C(O)OR9, 0(C1-C5 alkyl) or 0(C3-C5 cycloalkyl),
R6 and R7 are each independently H, C, -C5 alkyl or C3-C5 cycloalkyl,
or R6 and R7 can be taken together with the N atom to which they are attached to form a 4- to 7- membered saturated ring, optionally substituted by =0,
R8 is H, C1-C5 alkyl or C3-C5 cycloalkyl,
R9 is C1-C5 alkyl or C3-C5 cycloalkyl, each of which is optionally substituted with one or more fluorine atoms, or a pharmaceutically acceptable salt, solvate or prodrug thereof
2 A compound according to claim 1 wherein X-Ar is OCH2-Ar, or a pharmaceutically acceptable salt, solvate or prodrug thereof
3 A compound according to claim 1 or 2 wherein A is N and B is CH or N, or a pharmaceutically acceptable salt, solvate or prodrug thereof
4 A compound according to claim 1 , 2 or 3 wherein Ar is phenyl, fluorophenyl or chlorophenyl, or a pharmaceutically acceptable salt, solvate or prodrug thereof
5 A compound according to claim 1 , 2, 3 or 4 wherein R1 is piperidine, pyrrolidine, piperazine, octahydro- pyrrolo[3,4-c]pyrrole, octahydro-pyrrolo[3,4-b]pyrrole, or octahydro-pyrrolo[3,4-c]pyπdιne, attached to the "ABCCHCHC" ring via a N atom, which ring system is optionally substituted by one or more substituents independently selected from =0, C1-C5 alkyl, C3-C5 cycloalkyl, OH, C(O)C1-C5 alkyl, C(O)C3-C5 cycloalkyl, C(O)OC1-C5 alkyl, NR6R7, NR8C(O)R9, NR8C(O)OR9, 0(C1-C5 alkyl) or 0(C3-C5 cycloalkyl), or a pharmaceutically acceptable salt, solvate or prodrug thereof
6 A compound according to claim 1 , 2, 3, 4 or 5 wherein A is N and B is CH, or a pharmaceutically acceptable salt, solvate or prodrug thereof
7 A compound according to claim 1 , 2, 3, 4, 5 or 6 wherein Ar is phenyl, 4-chlorophenyl or 4- fluorophenyl, or a pharmaceutically acceptable salt, solvate or prodrug thereof
8 A compound according to claim 1, 2, 3, 4, 5, 6 or 7 whereinR1 is piperidine, pyrrolidine, piperazine, octahydro-pyrrolo[3,4-c]pyrrole, octahydro-pyrrolo[3,4-b]pyrrole, or octahydro-pyrrolo[3,4-c]pyπdιne attached to the "ABCDEC" ring via a N atom, which ring system is optionally substituted by one or more substituents independently selected from OH, OMe, OEt, Me, Et, NH2, NHMe, NMe2, NMeC(O)Me and C(O)Me, or a pharmaceutically acceptable salt, solvate or prodrug thereof
9 A compound according to claim 1 , 2, 3, 4, 5, 6, 7 or 8 wherein Ar is phenyl, or a pharmaceutically acceptable salt, solvate or prodrug thereof
10 A compound according to claim 1 , 2, 3, 4, 5, 6, 7, 8 or 9 wherein R1 is piperidine, pyrrolidine, piperazine, octahydro-pyrrolo[3,4-c]pyrrole, octahydro-pyrrolo[3,4-b]pyrrole, or octahydro-pyrrolo[3,4- φyπdine, attached to the "ABCDEC" ring via a N atom, which ring system is optionally substituted by one or more substituents independently selected from
NHMe, NMe2, OH, NH2, Me and Et, or a pharmaceutically acceptable salt, solvate or prodrug thereof
11 A compound according to claim 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 wherein R1 is piperidine, pyrrolidine, piperazine, octahydro-pyrrolo[3,4-c]pyrrole, octahydro-pyrrolo[3,4-b]pyrrole, or octahydro-pyrrolo[3,4- cjpyπdine, attached to the "ABCDEC" ring via a N atom, which ring system is optionally substituted by one or more substituents independently selected from NHMe, NMe2, OH, NH2, Me and Et, or a pharmaceutically acceptable salt, solvate or prodrug thereof
12 A compound according to claim 1 , 2, 3, 4,, 5, 6, 7, 8, 9, 10 or 11 wherein R' is selected from one of the following groups

or a pharmaceutically acceptable salt, solvate or prodrug thereof
13 A compound according to claim 1 wherein A, B, X, R1 and Ar are selected from the values in the compounds of the Examples below, or a pharmaceutically acceptable salt, solvate or prodrug thereof
14 A compound according to claim 1 of formula
 wherein R1 is selected from
wherein R1 is selected from

5 or a pharmaceutically acceptable salt, solvate or prodrug thereof
15 A compound selected from one of the Examples herein, or a pharmaceutically acceptable salt, solvate or prodrug thereof
10 16 A compound according to claim 15 selected from the compounds of Examples 17a, 17b, 18a, 18b, 19 or 20 herein, or a pharmaceutically acceptable salt, solvate or prodrug thereof
17 A compound according to claim 16 selected from the compounds of Examples 17a, 18a, 19 or 20 herein, or a pharmaceutically acceptable salt, solvate or prodrug thereof
15
18 A pharmaceutical composition comprising a compound, pharmaceutically acceptable salt, solvate or prodrug thereof according to any one of claims 1 to 17, and a pharmaceutically acceptable diluent or carrier
19 A compound, pharmaceutically acceptable salt, solvate or prodrug thereof according to any one of claims 1 to 17 for use as a medicament
20 A compound, pharmaceutically acceptable salt, solvate or prodrug thereof according to any one of claims 1 to 17, for use as a medicament in the treatment or prevention of a disease or condition mediated by the MCH-1 receptor
21 A compound, pharmaceutically acceptable salt, solvate or prodrug thereof according to any one of claims 1 to 17, for use as a medicament in the treatment or prevention of a disease or condition selected from eating disorders (e g , binge eating disorder, anorexia, and bulimia), weight loss or control (e g , reduction in calorie or food intake, and/or appetite suppression), obesity, depression, atypical depression, bipolar disorders, psychoses, schizophrenia, behavioral addictions, suppression of reward- related behaviors (e g , conditioned place avoidance, such as suppression of cocaine- and morphine- induced conditioned place preference), substance abuse, addictive disorders, impulsivity, alcoholism (e g , alcohol abuse, addiction and/or dependence including treatment for abstinence, craving reduction and relapse prevention of alcohol intake), tobacco abuse (e g , smoking addiction, cessation and/or dependence including treatment for craving reduction and relapse prevention of tobacco smoking), dementia (including memory loss, Alzheimer's disease, dementia of aging, vascular dementia, mild cognitive impairment, age-related cognitive decline, and mild neurocognitive disorder), sexual dysfunction in males (e g , erectile difficulty), seizure disorders, epilepsy, inflammation, gastrointestinal disorders (e g , dysfunction of gastrointestinal motility or intestinal propulsion), attention deficit disorder (ADD including attention deficit hyperactivity disorder (ADHD)), Parkinson's disease, type Il diabetes, premenstrual syndrome or late luteal phase syndrome, migraines, panic disorder, anxiety, post-traumatic syndrome, social phobia, cognitive impairment in non-demented individuals, non-amnestic mild cognitive impairment, post operative cognitive decline, disorders associated with impulsive behaviours (such as, disruptive behaviour disorders (e g , anxiety/depression, executive function improvement, tic disorders, conduct disorder and/or oppositional defiant disorder), adult personality disorders (e g , borderline personality disorder and antisocial personality disorder), diseases associated with impulsive behaviours (e g , substance abuse, paraphilias and self-mutilation), and impulse control disorders (e g , intermittent explosive disorder, kleptomania, pyromania, pathological gambling, and trichotillomania)), obsessive compulsive disorder, chronic fatigue syndrome, premature ejaculation, sexual dysfunction in females, disorders of sleep (e g , sleep apnea), autism, mutism, neurodegenerative movement disorders, spinal cord injury, damage of the central nervous system (e g , trauma), stroke, neurodegenerative diseases or toxic or infective CNS diseases (e g , encephalitis or meningitis), cardiovascular disorders (e g , thrombosis)
22 The use of a compound, pharmaceutically acceptable salt, solvate or prodrug thereof according to any one of claims 1 to 17, in the preparation of a medicament for the treatment or prevention of disease or condition selected from eating disorders (e g , binge eating disorder, anorexia, and bulimia), weight loss or control (e g , reduction in calorie or food intake, and/or appetite suppression), obesity,
depression, atypical depression, bipolar disorders, psychoses, schizophrenia, behavioral addictions, suppression of reward-related behaviors (e g , conditioned place avoidance, such as suppression of cocaine- and morphine-induced conditioned place preference), substance abuse, addictive disorders, impulsivity, alcoholism (e g , alcohol abuse, addiction and/or dependence including treatment for abstinence, craving reduction and relapse prevention of alcohol intake), tobacco abuse (e g , smoking addiction, cessation and/or dependence including treatment for craving reduction and relapse prevention of tobacco smoking), dementia (including memory loss, Alzheimer's disease, dementia of aging, vascular dementia, mild cognitive impairment, age-related cognitive decline, and mild neurocognitive disorder), sexual dysfunction in males (e g , erectile difficulty), seizure disorders, epilepsy, inflammation, gastrointestinal disorders (e g , dysfunction of gastrointestinal motility or intestinal propulsion), attention deficit disorder (ADD including attention deficit hyperactivity disorder (ADHD)), Parkinson's disease, type Il diabetes, premenstrual syndrome or late luteal phase syndrome, migraines, panic disorder, anxiety, post-traumatic syndrome, social phobia, cognitive impairment in non-demented individuals, non- amnestic mild cognitive impairment, post operative cognitive decline, disorders associated with impulsive behaviours (such as, disruptive behaviour disorders (e g , anxiety/depression, executive function improvement, tic disorders, conduct disorder and/or oppositional defiant disorder), adult personality disorders (e g , borderline personality disorder and antisocial personality disorder), diseases associated with impulsive behaviours (e g , substance abuse, paraphilias and self-mutilation), and impulse control disorders (e g , intermittent explosive disorder, kleptomania, pyromania, pathological gambling, and trichotillomania)), obsessive compulsive disorder, chronic fatigue syndrome, premature ejaculation, sexual dysfunction in females, disorders of sleep (e g , sleep apnea), autism, mutism, neurodegenerative movement disorders, spinal cord injury, damage of the central nervous system (e g , trauma), stroke, neurodegenerative diseases or toxic or infective CNS diseases (e g , encephalitis or meningitis), cardiovascular disorders (e g , thrombosis)
23 A method for treating or preventing a disease or condition selected from eating disorders (e g , binge eating disorder, anorexia, and bulimia), weight loss or control (e g , reduction in calorie or food intake, and/or appetite suppression), obesity, depression, atypical depression, bipolar disorders, psychoses, schizophrenia, behavioral addictions, suppression of reward-related behaviors (e g , conditioned place avoidance, such as suppression of cocaine- and morphine-induced conditioned place preference), substance abuse, addictive disorders, impulsivity, alcoholism (e g , alcohol abuse, addiction and/or dependence including treatment for abstinence, craving reduction and relapse prevention of alcohol intake), tobacco abuse (e g , smoking addiction, cessation and/or dependence including treatment for craving reduction and relapse prevention of tobacco smoking), dementia (including memory loss, Alzheimer's disease, dementia of aging, vascular dementia, mild cognitive impairment, age-related cognitive decline, and mild neurocognitive disorder), sexual dysfunction in males (e g , erectile difficulty), seizure disorders, epilepsy, inflammation, gastrointestinal disorders (e g , dysfunction of gastrointestinal motility or intestinal propulsion), attention deficit disorder (ADD including attention deficit hyperactivity disorder (ADHD)), Parkinson's disease, type Il diabetes, premenstrual syndrome or late luteal phase syndrome, migraines, panic disorder, anxiety, post-traumatic syndrome, social phobia, cognitive impairment in non-demented individuals, non-amnestic mild cognitive impairment, post operative
cognitive decline, disorders associated with impulsive behaviours (such as, disruptive behaviour disorders (e g , anxiety/depression, executive function improvement, tic disorders, conduct disorder and/or oppositional defiant disorder), adult personality disorders (e g , borderline personality disorder and antisocial personality disorder), diseases associated with impulsive behaviours (e g , substance abuse, paraphilias and self-mutilation), and impulse control disorders (e g , intermittent explosive disorder, kleptomania, pyromania, pathological gambling, and trichotillomania)), obsessive compulsive disorder, chronic fatigue syndrome, premature ejaculation, sexual dysfunction in females, disorders of sleep (e g , sleep apnea), autism, mutism, neurodegenerative movement disorders, spinal cord injury, damage of the central nervous system (e g , trauma), stroke, neurodegenerative diseases or toxic or infective CNS diseases (e g , encephalitis or meningitis), cardiovascular disorders (e g , thrombosis) which comprises the step of administering to an animal (preferably, human) in need thereof a therapeutically effective amount of a MCHR1 antagonist according to any one claims 1 to 17
24 A method for promoting weight loss (including prevention or inhibition of weight gain), or treatment of obesity and related eating disorders which comprises the step of administering to an animal (preferably human) in need thereof a therapeutically effective amount of a MCHR 1 antagonist according to any one claims 1 to 17
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US82843206P | 2006-10-06 | 2006-10-06 | |
| US60/828,432 | 2006-10-06 | ||
| US93974307P | 2007-05-23 | 2007-05-23 | |
| US60/939,743 | 2007-05-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008041090A1 true WO2008041090A1 (en) | 2008-04-10 |
Family
ID=38895954
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2007/002886 WO2008041090A1 (en) | 2006-10-06 | 2007-09-24 | Malanin concentrating hormone receptor-1 antagonist pyridinones |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20080085884A1 (en) |
| AR (1) | AR063119A1 (en) |
| TW (1) | TW200825061A (en) |
| UY (1) | UY30625A1 (en) |
| WO (1) | WO2008041090A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7728031B2 (en) | 2006-02-24 | 2010-06-01 | Abbott Laboratories | Octahydro-pyrrolo[3,4-b]pyrrole derivatives |
| WO2010104830A1 (en) * | 2009-03-09 | 2010-09-16 | Bristol-Myers Squibb Company | Pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
| US8026240B2 (en) | 2007-09-11 | 2011-09-27 | Abbott Laboratories | Octahydro-pyrrolo[3,4-b]pyrrole N-oxides |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2406233B1 (en) | 2009-03-09 | 2013-11-13 | Bristol-Myers Squibb Company | Aza pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
| WO2010141538A1 (en) * | 2009-06-03 | 2010-12-09 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| EP2437599A4 (en) * | 2009-06-03 | 2012-10-31 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| EP2437600A4 (en) * | 2009-06-03 | 2012-11-07 | Glaxosmithkline Llc | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| JP2012528872A (en) * | 2009-06-03 | 2012-11-15 | グラクソスミスクライン・リミテッド・ライアビリティ・カンパニー | Bis-pyridylpyridones as melanin-concentrating hormone receptor 1 antagonists |
| ES2873029T3 (en) * | 2010-12-06 | 2021-11-03 | Aclaris Therapeutics Inc | Substituted pyridinone-pyridinyl compounds |
| CN103570630B (en) | 2012-07-18 | 2016-04-20 | 广东东阳光药业有限公司 | Nitrogen heterocyclic derivative and the application in medicine thereof |
| US9273033B2 (en) | 2012-11-20 | 2016-03-01 | Merck Sharp & Dohme Corp. | Substituted pyridone derivatives as PDE10 inhibitors |
| CN106220613A (en) * | 2016-07-11 | 2016-12-14 | 孙剑 | The anti-frightened fault additive of a kind of anesthetis |
| US20180008575A1 (en) * | 2016-07-11 | 2018-01-11 | Neurovance, Inc. | Methods of treating binge eating disorder |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002003370A (en) * | 1999-09-20 | 2002-01-09 | Takeda Chem Ind Ltd | Melanin coagulating hormone antagonistic agent |
| WO2003068230A1 (en) * | 2002-02-14 | 2003-08-21 | Pharmacia Corporation | Substituted pyridinones as modulators of p38 map kinase |
| WO2005085200A1 (en) * | 2004-03-05 | 2005-09-15 | Banyu Pharmaceutical Co., Ltd. | Pyridone derivative |
| WO2007024004A1 (en) * | 2005-08-24 | 2007-03-01 | Banyu Pharmaceutical Co., Ltd. | Phenylpyridone derivative |
| WO2007141200A1 (en) * | 2006-06-02 | 2007-12-13 | Janssen Pharmaceutica N.V. | Novel n-aryl and n-heteroaryl substituted pyridinone derivatives for use in mch-1 mediated diseases |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5470975A (en) * | 1990-10-16 | 1995-11-28 | E.R. Squibb & Sons, Inc. | Dihydropyrimidine derivatives |
| WO2001082925A1 (en) * | 2000-04-28 | 2001-11-08 | Takeda Chemical Industries, Ltd. | Melanin concentrating hormone antagonists |
| US7223788B2 (en) * | 2003-02-14 | 2007-05-29 | Sanofi-Aventis Deutschland Gmbh | Substituted N-aryl heterocycles, process for their preparation and their use as medicaments |
-
2007
- 2007-09-24 WO PCT/IB2007/002886 patent/WO2008041090A1/en active Application Filing
- 2007-09-27 US US11/862,284 patent/US20080085884A1/en not_active Abandoned
- 2007-10-03 AR ARP070104387A patent/AR063119A1/en unknown
- 2007-10-04 UY UY30625A patent/UY30625A1/en not_active Application Discontinuation
- 2007-10-05 TW TW096137528A patent/TW200825061A/en unknown
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2002003370A (en) * | 1999-09-20 | 2002-01-09 | Takeda Chem Ind Ltd | Melanin coagulating hormone antagonistic agent |
| WO2003068230A1 (en) * | 2002-02-14 | 2003-08-21 | Pharmacia Corporation | Substituted pyridinones as modulators of p38 map kinase |
| WO2005085200A1 (en) * | 2004-03-05 | 2005-09-15 | Banyu Pharmaceutical Co., Ltd. | Pyridone derivative |
| EP1741703A1 (en) * | 2004-03-05 | 2007-01-10 | Banyu Pharmaceutical Co., Ltd. | Pyridone derivative |
| WO2007024004A1 (en) * | 2005-08-24 | 2007-03-01 | Banyu Pharmaceutical Co., Ltd. | Phenylpyridone derivative |
| WO2007141200A1 (en) * | 2006-06-02 | 2007-12-13 | Janssen Pharmaceutica N.V. | Novel n-aryl and n-heteroaryl substituted pyridinone derivatives for use in mch-1 mediated diseases |
Non-Patent Citations (2)
| Title |
|---|
| DATABASE WPI Week 2003, Derwent World Patents Index; AN 656582, XP002406967, 3 * |
| DYKE H J ET AL: "Recent developments in the discovery of MCH-1R antagonists for the treatment of obesity - an update", EXPERT OPINION ON THERAPEUTIC PATENTS, ASHLEY PUBLICATIONS, GB, vol. 15, no. 10, October 2005 (2005-10-01), pages 1303 - 1313, XP002382192, ISSN: 1354-3776 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7728031B2 (en) | 2006-02-24 | 2010-06-01 | Abbott Laboratories | Octahydro-pyrrolo[3,4-b]pyrrole derivatives |
| US8399468B2 (en) | 2006-02-24 | 2013-03-19 | Abbott Laboratories | Octahydro-pyrrolo[3,4-B]pyrrole derivatives |
| US8026240B2 (en) | 2007-09-11 | 2011-09-27 | Abbott Laboratories | Octahydro-pyrrolo[3,4-b]pyrrole N-oxides |
| WO2010104830A1 (en) * | 2009-03-09 | 2010-09-16 | Bristol-Myers Squibb Company | Pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
| US8563583B2 (en) | 2009-03-09 | 2013-10-22 | Bristol-Myers Squibb Company | Pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
Also Published As
| Publication number | Publication date |
|---|---|
| AR063119A1 (en) | 2008-12-30 |
| US20080085884A1 (en) | 2008-04-10 |
| UY30625A1 (en) | 2008-05-31 |
| TW200825061A (en) | 2008-06-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008041090A1 (en) | Malanin concentrating hormone receptor-1 antagonist pyridinones | |
| US9556187B2 (en) | Substituted pyrrolo[3′,2′:5,6]pyrido[4,3-d]pyrimidines and JAK inhibitors comprising the same | |
| CN112409334A (en) | Novel heterocyclic derivatives useful as SHP2 inhibitors | |
| JP4284181B2 (en) | Adenosine A2a receptor antagonist | |
| AU2018237123B2 (en) | Bruton's tyrosine kinase inhibitors | |
| BG106678A (en) | PYRAZOLO[4,3-d]PYRIMIDINE DERIVATIVES | |
| JP2001505567A (en) | Novel substituted pyrazole derivatives for the treatment of cardiovascular diseases | |
| CA2585557C (en) | Pyrazolo[4,3-d] pyrimidine derivatives useful as pde-5 inhibitors | |
| EP1742950B1 (en) | Pyrazolo[4,3-d] pyrimidines | |
| AU2008231543B2 (en) | Pyrimido [4, 5-D] azepine derivatives as 5-HT2C agonists | |
| TW201444821A (en) | Substituted piperidine compounds and their use as orexin receptor modulators | |
| JP2020523332A (en) | Piperidinone formyl peptide 2 receptor agonist | |
| CN111936144A (en) | JAK inhibitors | |
| JP2017100951A (en) | Oxazolidinone and oxazinanone derivative | |
| EP4117784A1 (en) | Gpr52 modulator compounds | |
| JP2002513382A (en) | Azolotriazines and azolopyrimidines | |
| BG103685A (en) | Quinoxalindions | |
| AU2021330379A1 (en) | GPR52 modulator compounds | |
| WO2023105387A1 (en) | Melanocortin 4 receptor antagonists and uses thereof | |
| JP2007512298A (en) | Remedy | |
| NZ620037B2 (en) | Tricyclic heterocyclic compounds and jak inhibitors | |
| MXPA00010388A (en) | Pyrazolopyrimidinone cgmp pde5 inhibitors for the treatment of sexual dysfunction | |
| NZ709835B2 (en) | Tricyclic heterocyclic compounds and jak inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07805004 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07805004 Country of ref document: EP Kind code of ref document: A1 |