Beschreibung
Verfahren zur Vorhersage des individuellen Krankheitsverlaufs bei Sepsis.
Die vorliegende Erfindung betrifft die Verwendung von in vitro aus einer Patientenprobe erhaltenen Genexpressionsprofilen für die Erstellung von Kriterien zur Vorhersage eines individuellen Krankheitsverlaufs einer Sepsis, ein Verfahren zur in vitro Messung derartiger Genexpressionsprofile gemäß Anspruch 10 sowie die Verwendung der Genexpressionsprofile und/oder von den hierfür verwendeten Sonden zum Ausschalten und/oder zur Aktivitätsveränderung von Zielgenen und/oder zur Bestimmung der Genaktivität zum Screening von Wirkstoffen gegen Sepsis und/oder zur Beurteilung der Wirkung gegen Sepsis und/oder der Wirkstoffqualität und/oder der Wirkstoffintegrität in zellulären und zellfreien Sepsis- Modellsystemen und in Sepsis-Tiermodellen.
Weiterhin betrifft die vorliegende Erfindung neue Möglichkeiten der Vorhersage der Überlebenswahrscheinlichkeit und der Entwicklung tödlicher Komplikationen von Sepsispatienten, die sich aus experimentell abgesicherten Erkenntnissen im Zusammenhang mit dem Auftreten von Änderungen der Genaktivitäten (Transkription) bei Patienten mit Sepsis ableiten lassen.
Trotz Fortschritten im pathophysiologischen Verständnis und der supportiven Behandlung von Intensivpatienten sind generalisierte inflammatorische Zustände wie SIRS und Sepsis, definiert entsprechend der ACCP/SCCM Konsensuskonferenz aus dem Jahre 1992 [1], bei Patienten auf Intensivstationen sehr häufig auftretende und erheblich zur Sterblichkeit beitragende Erkrankungen [2-3]. Die Sterblichkeit beträgt ca. 20 % bei SIRS, ca. 40 % bei Sepsis und steigt bei Entwicklung von multiplen
Organdysfunktionen bis auf 70-80 % an [4-6]. Der Morbiditäts- und Letalitätsbeitrag von SIRS und Sepsis ist von fachübergreifender klinischmedizinischer Bedeutung, denn dadurch werden in zunehmendem Maße die Behandlungserfolge der fortgeschrittensten Therapieverfahren zahlreicher medizinischer Fachgebiete (z.B. Traumatologie, Neurochirurgie, Herz- /Lungenchirurgie, Viszeralchirurgie, Transplantationsmedizin, Hämatologie/ Onkologie, etc.) gefährdet, denen ohne Ausnahme eine Erhöhung des Krankheitsrisikos für SIRS und Sepsis immanent ist. Dies drückt sich auch im kontinuierlichen Anstieg der Häufigkeit der Sepsis aus: zwischen 1979 und 1987 um 139% von 73,6 auf 176 Krankheitsfälle je 100.000 Krankenhauspatienten) [7]. Die Senkung der Morbidität und Letalität einer Vielzahl von schwer erkrankten Patienten ist daher an einen gleichzeitigen Fortschritt in der Vorbeugung, Behandlung und insbesondere der Erkennung und Verlaufsbeobachtung der Sepsis und schweren Sepsis gebunden.
Auf molekularer Ebene wird als Sepsis ein Krankheitsbild bezeichnet, welches durch pathogene Mikroorganismen verursacht wird. Auf dem Boden der Erschöpfung Infektionsort-naher, molekularer Kontroll- und Regulationsmöglichkeiten entwickelt sich eine generalisierte, den ganzen Organismus umfassende Entzündungsreaktion, die für die vom Arzt nachgewiesenen klinischen Symptome/Diagnosekriterien/SIRS-Kriterien nach [1] verantwortlich ist. Dieser generalisierte, inflammatorische Zustand (als Sepsis nach [1] definiert) geht mit Zeichen der Aktivierung verschiedener Zellsysteme (endotheliale Zellen, aber auch aller leukozytären Zellsysteme und vor allem des Monozyten/ Makrophagensystems) einher. Schließlich schädigen molekulare Mechanismen, die eigentlich den Wirt gegen invasive Mikroorganismen schützen sollen, dessen eigene Organe/Gewebe und tragen so entscheidend zur Entwicklung der vom Kliniker gefürchteten Organdysfunktionen bei [8-11].
Der Sepsisbegriff hat im Laufe der Zeit einen erheblichen Bedeutungswandel erfahren. Eine Infektion bzw. der dringliche Verdacht auf eine Infektion sind auch heute noch wesentlicher Bestandteil aktueller Sepsisdefinitionen. Besondere Berücksichtigung findet jedoch dabei die Beschreibung Infektionsort-ferner Organfehlfunktionen im Rahmen der inflammatorischen Wirtsreaktion. Im internationalen Schrifttum haben sich zwischenzeitlich die Kriterien der Konsensuskonferenz des „American College of Chest Physicians/Society of Critical Gare Medicine Consensus Conference (ACCP/SCCM)" aus dem Jahr 1992 am breitesten zur Definition des Sepsis- Begriffs durchgesetzt [1]. Entsprechend dieser Kriterien [1] werden die klinisch definierten Schweregrade „systemic inflammatory response syndrom" (SIRS), „Sepsis", „severe Sepsis" und „septic shock" unterschieden. Als SIRS wird dabei die systemische Antwort des inflammatorischen Systems auf einen infektiösen oder nichtinfektiösen Reiz definiert. Dazu müssen mindestens zwei der folgenden klinischen Kriterien erfüllt sein: Fieber >38°C oder Hypothermie <36°C, eine Leukozytose >12G/I oder eine Leukopenie <4G/I bzw. eine Linksverschiebung im Differentialblutbild, eine Herzfrequenz von über 90/min, eine Tachypnoe >20 Atemzüge/min oder ein PaC02 (Partialdruck des Kohlendioxid im arteriellen Blut) <4,3 kPa. Als Sepsis werden solche klinischen Zustände definiert, bei denen die SIRS-Kriterien erfüllt sind und ursächlich eine Infektion nachgewiesen wird oder zumindest sehr wahrscheinlich ist. Eine schwere Sepsis ist vom zusätzlichen Auftreten von Organfehlfunktionen gekennzeichnet. Häufige Organfehlfunktionen sind Änderungen der Bewusstseinslage, eine Oligurie, eine Laktazidose oder eine Sepsisinduzierte Hypotension mit einem systolischen Blutdruck von weniger als 90 mmHg bzw. ein Druckabfall um mehr als 40 mmHg vom Ausgangswert. Wenn eine solche Hypotension nicht durch die Verabreichung von Kristalloiden und/oder Kolloiden zu beheben ist und es zusätzlich zu einer Katecholaminpflichtigkeit des Patienten kommt, so spricht man von einem
septischen Schock. Dieser wird bei etwa 20 % aller Sepsispatienten nachgewiesen.
Sepsis ist das klinische Ergebnis von komplexen und stark heterogenen molekularen Vorgängen, die gekennzeichnet sind durch eine Einbeziehung von vielen Komponenten und deren Wechselwirkungen auf jeder organisatorischen Ebene des menschlichen Körpers: Gene, Zellen, Gewebe, Organe. Die Komplexität der zugrunde liegenden biologischen und immunologischen Prozesse haben viele Arten von Forschungsstudien hervorgerufen, die einen weiten Bereich klinischer Aspekte umfassen. Eines der hieraus zu erkennenden Ergebnisse war, dass die Bewertung neuer Sepsis-Therapien durch relativ unspezifische, klinisch-basierte Einschlusskriterien, welche die molekularen Mechanismen in nicht ausreichender Weise wiedergeben, erschwert wird [12]. Gleichfalls bestehen auf Grund der mangelnden Spezifität der heutigen Sepsis- und SIRS- Diagnose beim Kliniker große Unsicherheiten, ab welchem Zeitpunkt ein Patient einer spezialisierten Therapie, beispielsweise mit Antibiotika, die ihrerseits beträchtliche Nebenwirkungen haben können, zugeführt werden soll [12]. So zeigte eine von der European Society of Intensive Care Medicine (ESICM) durchgeführte Umfrage, dass 71 % der befragten Ärzte Unsicherheit bei der Diagnosestellung einer Sepsis, trotz langjähriger klinischer Erfahrungen, hatten [22].
Bahnbrechende Entdeckungen in Molekularbiologie und Immunologie während der letzten zwei Jahrzehnte ließen ein vertieftes, mehr an den grundlegenden Mechanismen orientiertes Verständnis der Sepsis entstehen. Das dadurch entstandene Wissen um relevante Targets bildete wiederum die Basis für die Entwicklung gezielter und adjuvanter Therapiekonzepte, welche hauptsächlich auf der Neutralisierung wesentlicher Sepsismediatoren beruhen [13-16]. Eine Ursache für das Scheitern fast aller immunmodulatorischer Therapieansätze in klinischen Studien - trotz
Effektivität im Tierexperiment - wird in der nur schlechten Korrelation zwischen den klinischen, eher symptomatisch orientierten Diagnosekriterien und den grundlegenden Mechanismen einer generalisierten Immunantwort gesehen [12, 17-18].
Rückblickend erstaunt dies nicht, da bereits gesunde Menschen bei alltäglichen Verrichtungen Veränderungen der Herz- bzw. Atemfrequenz aufweisen können, welche per Definition bereits die Diagnose eines SIRS zuließen. Bei Berücksichtigung unserer heutigen biomedizinischen Möglichkeiten muss es als Anachronismus erscheinen, dass jährlich 751.000 Patienten in den USA anhand o.g. ACCP/SCCM Kriterien diagnostiziert, klassifiziert und behandelt werden. Von namhaften Autoren wird deshalb schon lange kritisiert, dass zu Lasten einer verbesserten Sepsisdiagnose in der vergangenen Dekade zuviel Energie und finanzielle Ressourcen für die Suche nach einem „magic bullet" der Sepsistherapie aufgewendet wurden [19]. Auch fordern kürzlich publizierte Expertenmeinungen, dass zu einem besseren pathophysiologischen Verständnis der Sepsis eine Modifizierung der Konsensuskriterien nach [1] erforderlich ist [20-21]. Außerdem besteht unter vielen Medizinern Einigung darüber, dass die Konsensuskriterien nach [1] keiner spezifischen Definition von Sepsis entsprechen. So zeigte eine von der European Society of Intensive Care Medicine (ESICM) durchgeführte Umfrage, dass 71 % der befragten Ärzte Unsicherheit bei der Diagnosestellung einer Sepsis, trotz langjähriger klinischer Erfahrungen, hatten [22].
Aufgrund der oben genannten Probleme mit der Anwendung der Konsensuskriterien nach [1] werden unter Intensivmedizinern Vorschläge für eine sensitivere und spezifische Definitionen der verschiedenen Schweregrade der Sepsis diskutiert [2,23]. Neu ist dabei vor allem, dass molekulare Veränderungen direkt in die Beurteilung der Schwere einer Sepsis, aber auch den Einschluss in innovative Behandlungsverfahren der
Sepsis (wie z.B. die Therapie mit aktiviertem rekombinanten Protein C) einbezogen werden sollen. Dieser Konsensusprozess [23], der gegenwärtig von fünf internationalen Fachgesellschaften getragen wird, ist zum gegenwärtigen Zeitpunkt noch längst nicht abgeschlossen. Ziel ist die Etablierung eines Systems zur Schweregradbeurteilung der Sepsis, das es ermöglicht, Patienten anhand ihrer individuellen Patientenreaktion auf der Basis ihrer prädisponierenden Bedingungen, der Art und des Ausmaßes der Infektion, der Art und der Schwere der Wirtsantwort sowie des Grads der begleitenden Organdysfunktionen zu klassifizieren. Das beschriebene System wird mit PIRO, abkürzt nach den englischen Begriffen für „Predisposition", „Insult Infection", „Response" und „Organ dysfunction", bezeichnet. Davon kann dann die individuelle Wahrscheinlichkeit des Überlebens sowie des potentiellen Ansprechens auf die Therapie abgeleitet werden [23]. Gleichfalls sollen nichtinfektiöse Zustände, die gegenwärtig nach [1] unter dem Begriff SIRS subsummiert werden, entsprechend der individuellen Schwere des SIRS genauer klassifiziert werden. Auch hierfür werden Biomarker gesucht, die die Schwere des SIRS auch auf molekularer Ebene widerspiegeln und eine klare Abgrenzung von infektiösen Zuständen (gegenwärtig als Sepsis nach [1] klassifiziert) ermöglichen. Ähnliche Stadieneinteilungen werden bereits heute von anderen medizinischen Fachdisziplinen mit Erfolg angewendet, beispielsweise zur Klassifizierung der verschiedenen Krankheitsstadien im Bereich der Onkologie verwendet (TNM System, [24]).
Verglichen mit den Konsensuskriterien nach [1] sollen in der Zukunft zusätzliche molekulare Parameter in die Diagnosestellung einbezogen werden [23], um so eine verbesserte Korrelation der molekularen inflammatorischen/ immunologischen Wirtsantwort mit dem Schweregrad der Sepsis zu ermöglichen, aber auch Aussagen zur individuellen Prognose abzuleiten. Nach solchen molekularen Biomarkern wird derzeit von verschiedenen wissenschaftlichen und kommerziellen Gruppen intensiv
gesucht, da bisherige Parameter wie z.B. die Bestimmung des C-reaktiven Proteins oder des Procalcitonins nicht allen klinischen Anforderungen gerecht werden und insbesondere nur schlecht in der Lage sind, zwischen Überlebenden und NichtÜberlebenden einer Sepsis zu unterscheiden [25]. Auch aufgrund der unzureichenden Spezifität und Sensivität der Konsensuskriterien nach [1] und des mangelhaften oder verspäteten Nachweises der Ursache der Infektion besteht daher ein dringender Bedarf für neue diagnostische Verfahren, welche die Fähigkeit des Fachmanns verbessern sollen, den Krankheitsverlauf bei Sepsis frühzeitig vorherzusagen, im klinischem Verlauf vergleichbar zu gestalten und bezüglich der individuellen Prognose und dem Ansprechen auf spezifische Behandlungen Aussagen abzuleiten.
Technologische Fortschritte, insbesondere die Entwicklung der Microarray- Technologie, versetzen den Fachmann nun in die Lage, 10000 oder mehr Gene und deren Genprodukte gleichzeitig zu vergleichen. Die Anwendung solcher Microarray-Technologien kann nun Hinweise auf den Status von Gesundheit, Regulationsmechanismen, biochemischer Wechselwirkungen und Signalübertragungsnetzwerken geben. Das Verbessern des Verständnisses darüber, wie ein Organismus auf Infektionen reagiert, sollte die Entwicklung von verstärkten Erkennungs-, Diagnose- und Behandlungsmodalitäten für Sepsis- Erkrankungen erleichtern.
Microarrays stammen vom „Southern blotting" [26] ab, was die erste Herangehensweise darstellt, DNA-Moleküle in einer räumlich ansprechbaren Art und Weise auf einer festen Matrix zu immobilisieren. Die ersten Microarrays bestanden aus DNA-Fragmenten, oft mit unbekannter Sequenz, und wurden auf eine poröse Membran (normalerweise Nylon) punktweise aufgebracht. Routinegemäß wurden cDNA, genomische DNA oder Plasmid- Bibliotheken verwendet, und das hybridisierte Material wurde mit einer radioaktiven Gruppe markiert [27-29].
Kürzlich hat es die Verwendung von Glas als Substrat und Fluoreszenz zur Detektion zusammen mit der Entwicklung neuer Technologien für die Synthese und für das Aufbringen der Nukleinsäuren in sehr hohen Dichten erlaubt, die Nukleinsäurearrays zu miniaturisierten bei gleichzeitiger Erhöhung des experimentellen Durchsatzes und des Informationsgehaltes [30-32].
Weiterhin ist aus WO 03/002763 bekannt, dass die Messung der Genexpression mittels Microarrays grundsätzlich für die Diagnose von Sepsis und sepsisähnlichen Zuständen verwendet werden können.
Eine Begründung für die Anwendbarkeit der Microarray-Technologie wurde zunächst durch klinische Untersuchungen auf dem Gebiet der Krebsforschung geliefert. Hier haben Expressionsprofile ihre Nützlichkeit bei der Identifizierung von Aktivitäten einzelner Gene oder Gengruppen gezeigt, die mit bestimmten klinischen Phänotypen korrelieren [33]. Durch die Analyse vieler Proben, die von Individuen mit oder ohne akute Leukämie oder diffusen B-Zell Lymphomen stammten, wurden Genexpressionsmarker (RNA) gefunden und anschließend für die klinisch relevante Klassifizierung dieser Krebsarten angewandt [33,34]. Golub et al. haben herausgefunden, daß verlässliche Vorhersagen nicht aufgrund von irgendeinem einzelnen Gen gemacht werden können, aber daß Vorhersagen, die auf der Veränderung der Transkritiption von 53 Genen (ausgewählt aus über 6000 Genen, die auf den Arrays vertreten waren) basieren, sehr genau sind [33].
Alisadeh et al. [34] untersuchten große B-Zell Lymphome (DLBCL). Die Autoren erarbeiteten Expressionsprofile mit einem „Lymphochip", einem Microarray, der 18 000 Klone komplementärer DNA trug und entwickelt worden war, um Gene zu überwachen, die in normale und abnormale Lymphozytenentwicklung involviert sind. Unter Anwendung von Cluster-
Analysen waren sie in der Lage, DILBCL in zwei Kategorien einzuteilen, welche starke Unterschiede bezüglich der Überlebenschancen der Patienten aufzeigten. Die Genexpressionsprofile dieser Untergruppen entsprachen zwei bedeutsamen Stadien der B-Zelldifferenzierung.
Die Anwendung von Genexpressionsprofilen, die mittels Microarrays erarbeitet wurden, für die Vorhersage des Krankheitsverlaufs bei Leukämie wurde von Yeoh E. et al. beschrieben [35]. Aus verschiedenen anderen Studien an Krebspatienten sind ebenfalls weitere Beispiele für die Nutzung von Genexpressionsprofilen für die Vorhersage der Überlebenswahrscheinlichkeit bekannt [36-38]. Diese Untersuchungen belegen, dass mittels Messung der Genexpressionsprofile die Wahrscheinlichkeit des Rückfalls der Patienten vorhergesagt werden kann. Dies wäre von erheblicher klinisch-medizinischer Bedeutung, da solchen Patienten einerseits ein höheres Maß an Aufmerksamkeit z.B. durch häufigere Vorstellungstermine im Rahmen der Tumornachsorge entgegengebracht werden könnte. Im Ergebnis könnte ein Rückfall früher diagnostiziert und gezielt behandelt werden. Andererseits könnten Patienten, die ein Genexpressionsprofil aufweisen, das nicht auf eine erhöhtes Rückfallrisiko schließen lässt, weniger häufig einbestellt werden. Auch könnten solche Genexpressionsprofile in Entscheidungen über ein mehr oder weniger aggressives Vorgehen zur Therapie des Tumorleidens einbezogen werden. Als Konsequenz der Ergebnisse wurden von den Autoren vorgeschlagen, Genexpressionsprofile regelmäßig bei Tumorpatienten zu erheben, um so frühzeitig deren individuelles Risiko auf therapiebezogene Komplikationen zu identifizieren [35].
Die Verwendung von Genexpressionsprofilen zur Vorhersage des individuellen Krankheitsverlaufs bei Sepsis und insbesondere zur Vorhersage des individuellen Risikos zur Entwicklung tödlicher Komplikationen wurde noch nicht beschrieben.
Die grundsätzliche Verwendbarkeit von Genexpressionsprofilen, welche beispielsweise mittels der Microarray-Technik erhalten werden können, zur Diagnose von SIRS, generalisierten inflammatorischen Entzündungen, Sepsis und schwerer Sepsis ist in den nicht vorveröffentlichten deutschen Patentanmeldungen DE 103 40 395.7, DE 103 36 511.7, DE 103 150 31.5 sowie 10 2004 009 952.9, auf die hiermit vollinhaltlich Bezug genommen wird, der Anmelderin der vorliegenden Erfindung beschrieben.
Ausgangspunkt für die in der vorliegenden Patentanmeldung offenbarten Erfindung ist die Erkenntnis, daß sich die Genaktivitäten von überlebenden Sepsispatienten von Genaktivitäten verstorbener Sepsispatienten unterscheiden. Diese Unterschiede der Genaktivitäten lassen es somit zu, Vorhersagen für die Wahrscheinlichkeit des Überlebens und der zukünftigen Entwicklung tödlicher KompOlikationen bei Sepsispatienten anhand der Genexpression zu treffen. Diese Unterscheidung ist mit den bisher zur Diagnose verwendeten Proteinmarkem (z.B. Procalcitonin (PCT) oder C- Reaktives Protein (CRP)) klinischen Parametern nicht möglich, aber für die Einleitung einer spezialisierten intensivmedizinischen Therapie und damit für das Verbessern der individuellen Prognose für das Überleben sehr bedeutungsvoll.
Der vorliegenden Erfindung liegt somit die Aufgabe zugrunde, eine Vorhersage des zukünftigen Krankheitsverlaufs bei Sepsis unter Verwendung von Genexpressionsprofilen zu ermöglichen.
Diese Aufgabe wird durch die Merkmale der Ansprüche 1 , 10 und 23 gelöst.
Insbesondere betrifft die vorliegende Erfindung die Verwendung von in vitro aus einer Patientenprobe erhaltenen Genexpressionsprofilen für die
Erstellung von Kriterien für die Vorhersage eines individuellen Krankheitsverlaufs bei Sepsis.
Eine bevorzugte Ausführungsform der Erfindung betrifft die Verwendung der erhaltenen Genexpressionsprofilen für die Bestimmung der Überlebenswahrscheinlichkeit bei Sepsis.
Ferner dient die vorliegende Erfindung der therapiebegleitenden Verlaufsbeurteilung von Sepsis sowie der Klassifizierung von Sepsispatienten.
Die vorliegende Erfindung ist ferner nützlich als Ein- oder Ausschlußkriterium von Patienten mit Sepsis in klinische Studien der Phasen 2-4.
Eine bevorzugte Ausführungsform der Erfindung liegt in der Erstellung von Genaktivitätsdaten für die elektronische Weiterverarbeitung sowie zur Herstellung von Software für die Beschreibung der individuellen Prognose eines Sepsispatienten, für Diagnosezwecke und/oder
Patientendatenmangement-sytstemen.
Die vorliegende Erfindung kann auch zur Herstellung von Expertensystemen und/oder zur Modellierung von zelluläreren Signalübertragungswegen verwendet werden.
Zur Erstellung des Genexpressionsprofiles gemäß der vorliegenden Erfindung wird eine Mehrzahl von spezifischen Genen und/oder Genfragmenten verwendet, welche ausgewählt werden aus der Gruppe bestehend aus SEQ-ID No. 1 bis SEQ-ID No. 247 sowie Genfragmenten davon mit wenigstens 5-2000, bevorzugt 20-200, mehr bevorzugt 20-80 Nukleotiden.
Diese Sequenzen mit der Sequenz ID: 1 bis zur Sequenz ID: 247 sind durch den Umfang der vorliegenden Erfindung mit umfaßt und sind dem angefügten 56 -seifigen, 247 Sequenzen umfassenden, Sequenzprotokoll, das somit Bestandteil der Beschreibung der vorliegenden Erfindung ist, im Einzelnen offenbart und ist somit ebenfalls Bestandteil der Offenbarung der Erfindung. Dieses Sequenzprotokoll beinhaltet zudem eine Zuordnung der einzelnen Sequenzen mit der Sequenz ID: 1 bis zur Sequenz ID: 247 zu deren GenBank Accession Nr. (Internet-Zugang über http://www.ncbi.nlm.nih.gov/).
Darüber hinaus betrifft die vorliegende Erfindung die Verwendung von in vitro aus einer Patientenprobe erhaltenen Genexpressionsprofilen und/oder von den hierfür verwendeten Sonden, welche ausgewählt werden aus der Gruppe bestehend aus SEQ-ID No. 1 bis SEQ-ID No. 247 sowie Genfragmenten davon mit wenigstens 5-2000, bevorzugt 20-200, mehr bevorzugt 20-80 Nukleotiden, zum Ausschalten und/oder zur Aktivitätsveränderung von Zielgenen und/oder zur Bestimmung der Genaktivität zum Screening von Wirkstoffen gegen Sepsis und/oder zur Beurteilung der Wirkung gegen Sepsis und/oder der Wirkstoffqualität und/oder der Wirkstoffintegrität in zellulären und zellfreien Sepsis- Modellsystemen und in Sepsis-Tiermodellen.
Hierzu können ebenfalls hybridisierungsfähige synthetische Analoga der aufgelisteten Sonden verwendet werden.
Ferner kann man bei Sepsispatienten die Genaktivitäten in einer biologischen Flüssigkeit bestimmen und aus deren „Wert" Schlüsse hinsichtlich des Krankheitsverlaufs, der Überlebenswahrscheinlichkeit, des Therapieverlaufs oder der Ein- oder Ausschlussmöglichkeit der Sepsispatienten für klinische Studien ziehen.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß ein spezifisches Gen und/oder Genfragment ausgewählt wird aus der Gruppe bestehend aus SEQ-ID No. 1 bis SEQ-ID No. 247 sowie Genfragmenten davon mit wenigstens 5-2000, bevorzugt 20-200, mehr bevorzugt 20-80 Nukleotiden.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, dass wenigstens 2 bis 100 unterschiedliche cDNAs verwendet werden.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß wenigstens 200 unterschiedliche cDNAs verwendet werden.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß wenigstens 200 bis 500 unterschiedliche cDNAs verwendet werden.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß wenigstens 500 bis 1000 unterschiedliche cDNAs verwendet werden.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß wenigstens 1000 bis 2000 unterschiedliche cDNAs verwendet werden.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß die in Anspruch 11 aufgelisteten Gene oder Genfragmente und/oder von deren RNA abgeleiteten Sequenzen ersetzt werden durch synthetische Analoga, Aptamere sowie Peptidonukleinsäuren.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß die synthetischen Analoga der Gene 5-100, insbesondere ca. 70 Basenpaare umfassen.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß die Genaktivitäten mittels Hybridisierungsverfahren bestimmt wird.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß die Genaktivität mittels Microarrays bestimmt wird.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß die Genaktivität durch Hybridisierungs-unabhängige Verfahren, insbesondere enzymatische und/oder chemische Hydrolyse und/oder Amplifikationsverfahren, vorzugsweise PCR, anschließende Quantifizierung der Nukleinsäuren und/oder von Derivaten und/oder Fragmenten derselben, bestimmt wird.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß die Probe ausgewählt wird aus: Körperflüssigkeiten, insbesondere Blut, Liquor, Urin, Ascitesflüssigkeit, Seminalflüssigkeit, Speichel, Punktat; Zellinhalt oder eine Mischung davon.
Eine weitere Ausführungsform der Erfindung ist dadurch gekennzeichnet, daß Zellproben gegebenenfalls einer lytischen Behandlung unterzogen werden, um deren Zellinhalte freizusetzen.
Es ist dem Fachmann klar, daß die in den Ansprüchen dargelegten einzelnen Merkmale der Erfindung ohne Einschränkung beliebig miteinander kombinierbar sind.
Als Markergene im Sinne der Erfindung werden alle abgeleiteten DNA- Sequenzen, Partialsequenzen und synthetischen Analoga (beispielsweise Peptido-Nukleinsäuren, PNA) verstanden. Die auf Bestimmung der Genexpression auf RNA-Ebene bezogene Beschreibung der Erfindung stellt keine Einschränkung sondern nur eine beispielhafte Anwendung dar.
Die auf Blut bezogene Beschreibung der Erfindung stellt nur eine beispielhafte Anwendung der Erfindung dar. Als biologische Flüssigkeiten im Sinne der Erfindung werden alle Körperflüssigkeiten des Menschen verstanden.
Weitere Vorteile und Merkmale der vorliegenden Erfindung ergeben sich aufgrund der Beschreibung eines Ausführungsbeispiels. Ausführungsbeispiel
Untersuchungen zur differentiellen Genexpression bei Sepsis zur Unterscheidung von nichtÜberlebenden und überlebenden Patienten.
Für die Messung der differentiellen Genexpression bei Sepsis zur Unterscheidung von nichtÜberlebenden und überlebenden Patienten wurden Untersuchungen von Vollblutproben von 28 Patienten, welche auf operativen Intensivstationen behandelt wurden, durchgeführt.
Es wurden Vollblutproben von 12 überlebenden (9 männliche und 3 weibliche Patienten) und 16 verstorbenen Patienten (13 männliche und 3 weibliche Patienten) während deren gesamten Aufenthalts auf der Intensivstation abgenommen (Patientenproben). Jeder dieser Patienten entwickelte im Rahmen seiner intensivmedizinischen Betreuung eine Sepsis unterschiedlich schwerer Ausprägung. Genexpressionsprofile wurden von denjenigen Patientenproben ausgewertet, die am Behandlungstag mit dem schwersten Sepsisgrad nach [1] während der ersten septischen Komplikation (manche Intensivpatienten erleiden mehr als eine Sepsis im Behandlungsverlauf) entnommen wurden. Ausgewählte Charakteristika der Patienten mit Sepsis sind in Tabelle 1 dargestellt. Dabei werden Angaben zum Alter, Geschlecht, der Ursache der Sepsis (siehe Diagnose) sowie klinischer Schwere am Aufnahmetag auf Intensivstation, gemessen anhand des im klinischen Schrifttum gut belegten APACHE-Il-Scores sowie am Behandlungstag von dessen Proben Genexpressionsprofile erhoben wurden, gemessen anhand des im klinischen Schrifttum gut belegten SOFA-Scores (beide Scores jeweils in Punkten),
gemacht. Gleichfalls sind die Plasmaproteinspiegel von Procalcitonin (PCT), einem neuartigen Sepsismarker, am Behandlungstag von dessen Proben Genexpressionsprofile erhoben wurden und der individuelle Überlebensstatus angegeben.
Als Kontrollproben dienten die totale RNA aus Zelllinien SIG-M5. Alle Patientenproben wurden mit der Kontrollprobe jeweils auf einem Microarray ko-hybridisiert.
Tabelle 1 : Daten der Patientengruppe (Fortführung)
Experimentelle Beschreibung:
Nach Abnahme des Vollblutes wurde die totale RNA der Proben unter Anwendung des PAXGene Blood RNA Kit gemäß den Vorgaben des Herstellers (Qiagen) isoliert.
Zellkultivierung
Für die Zellkultivierung (Kontrollproben) wurden 19 Kryozellkulturen (SIGM5)
(eingefroren in flüssigem Stickstoff) genutzt. Die Zellen wurden jeweils mit 2 ml Iscove's Medium (Biochrom AG) beimpft ergänzt mit 20% fetalen Kälber Serum (FCS). Die Zellkuturen wurden anschliessend für 24 Stunden bei 37° C unter 5% C02 in 12-well Platten inkubiert. Danach wurde der Inhalt von 18 Wells in 2 Teile mit jeweils dem gleichen Volumen geteilt, sodass schliesslich 3 Platten des gleichen Formats (insgesamt 36 Wells) zur Verfügung standen. Die Kultivierung wurde anschliessend für 24 Stunden unter den gleichen Bedingungen fortgeführt. Im Anschluss daran wurden die resultierenden Kulturen von 11 Wells jeder Platte vereint und zentrifugiert (1000 x g, 5 min, Raumtemperatur). Der Überstand wurde verworfen und das Zellpellet in 40 ml des o.g. Mediums geloest. Diese 40 ml geloeste Zellen wurden in zwei 250 ml Kolben zu gleichen Teilen aufgeteilt und nach 48 Stunden Inkubation und Zugabe von 5 ml des o.g. Mediums wiederum inkubiert. Von den restlichen 2 ml der zwei verbleibendenden Platten wurden 80 μl in leere Wells der gleichen Platten gegeben, welche bereits vorher mit 1 ml des o.g. Mediums präpariert waren. Nach 48 Stunden Inkubation wurde nur eine der 12 Well-Platten wie folgt prozessiert: Aus jedem Well wurden 500 μl entnommen und vereint. Die daraus resultierenden 6 ml wurden in einen 250 ml Kolben gegeben, welcher ca. 10 ml frisches Medium enthielt. Dieses Gemisch wurde mit 1000 x g 5 Minuten bei Raumtemperatur zentrifugiert und in 10 ml des o.g. Mediums geloest. Die anschliessende Zellzählung ergab folgendes Ergebnis: 1 ,5 x 107 Zellen pro ml, 10 ml Gesamtvolumen, Gesamtzahl der Zellen: 1 ,5 x 108. Da die Zellzahl noch nicht ausreichend war, wurden 2,5 ml des o.g. Zellsuspension in 30 ml des o.g. Mediums in einen 250 ml (75 cm2) Kolben gegeben (insgesamt 4 Kolben). Nach 72 Sunden Inkubationszeit wurden jeweils 20 ml frischen Mediums in die Kolben gegeben. Nach folgender 24-stündiger Inkubation
erfolgte die Zellzählung wie oben beschrieben, die eine Gesamtzellzahl von 3,8 x 108 Zellen ergab. Um die gewünschte Zellzahl von 2 x 106 Zellen zu errreichen wurden die Zellen in 47,5 ml des o.g. Mediums in 4 Kolben resuspendiert. Nach einer Inkubationszeit von 24 Stunden wurden die Zeil zentrifugiert und zweimal mit Phosphatpuffer ohne Ca2+ und Mg2+ (Biochrom AG) gewaschen.
Die Isolation der totalen RNA erfolgt mittels des NucleoSpin RNA L Kits (Machery&Nagel) entsprechend den Angaben des Herstellers. Die oben beschriebene Prozedur wurde wiederholt bis die erforderliche Zellzahl erreicht wurde. Dies war erforderlich, um die erforderliche Menge von 6 mg totale RNA zu erreichen, was etwa einer Effϊziens von 600 μg RNA pro 108 Zellen entspricht.
Reverse Transkription / Markierung / Hybridisierung
Anschliessend wurde aus der totalen RNA der Patienten- und Kontrollproben mittels Reverser Transkription die komplementäre cDNA unter Substitution der dTTP-Fraktion durch synthetisiertes Aminoallyl-deoxyuridintriphosphat (AA- dUTP) hergestellt. Durch RNA-Hydrolyse wurde der RNA/cDNA-Komplex in Einzelstrang-cDNA umgewandelt. Im Anschluss daran wurden die cDNA- Proben mit den Fluoreszenzfarbstoffen Cy3 und Cy5 (Amersham) durch chemische Bindung an AA-dUTP markiert.
Für die Hybridisierung der Proben wurde ein Microarray der Firma SIRS-Lab verwendet. Auf dem verwendeten Microarray sind 5308 verschiedene Polynukleotide mit jeweils einer Länge zwischen 55-70 Basenpaaren immobilisiert. Jedes Polynukleotid repräsentiert ein humanes Gen. Die Spots wurden inklusive einer Vielzahl verschiedener Kontrollspots innerhalb von 28 Subarrays immobilisiert, wobei jedes Subarray in einem Raster von 15x15 Spots angeordnet war. Die Hybridisierung wurde unter Nutzung der Hybridisierungsstation HS 400 (Tecan) nach Angaben des Herstellers durchgeführt. Der Hybridisierungsloesung setzte sich zusammen aus 3,5 x SSC (1xSSC beinhaltet 150 mM NaCI und 15 mM Natriumzitrat), 0,3% SDS, 25% Formamid, jeweils 0,8 μg/μl cot-1 DNA, Hefe tRNA und polyA und den jeweiligen cDNA-Proben. Die Arrays wurden 10,5 Stunden bei 42°C hybridisiert.
Die anschliessende Waschprozedur folgt folgendem Programm: Zugabe von Waschpuffer I (2xSSC, 0,003%SDS) in die Hybridisierungskammer, 1 ,5 Minuten waschen bei Raumtemperatur, Zugabe von Waschpuffer II (1xSSC) in Hybridisierungskammer, Waschen mit Waschpuffer II für 1 ,5 Minuten bei Raumtemperatur, Zugabe von Waschpuffer III (0,2xSSC) in Hybridisierungskammer, Waschen mit Waschpuffer III für 1 ,5 Minuten bei Raumtemperatur. Danach wurde die Oberfläche der Microarrays unter Stickstoff bei einem Druck von 2,5 bar für 2,5 Minuten bei 30°C getrocknet.
Die Hybridisierungssignale der prozessierten Microarrys wurden anschliessend unter Nutzung des Scanners GenePix 4000B (Axon) ausgelesen und die Expressionsverhältnisse der differentiert exprimierten Gene mittels der Software GenePix Pro 4.0 (Axon) bestimmt.
Auswertung:
Für die Auswertung wurde die mittlere Intensität eines Spots als der Medianwert der zugehörigen der Spotpixel bestimmt.
Korrektur systematischer Fehler:
Von dem Mediän der Spotpixel wurde der Mediän der Pixel des lokalen Hintergrunds abgezogen. Für alle weiteren Berechnungen wurden die Signale mittels arcus sinus hyperbolicus transformiert. Die Normalisierung erfolgte nach dem Ansatz von Huber et al. [39]. Dabei wurde der additive und der multiplikative Bias innerhalb eines Microarrays aus 70% der vorhandenen Genproben geschätzt. Für die Analyse wurden die transformierten relativen Verhältnisse der Signale der Patientenproben gegen die Kontrolle berechnet.
Das bedeutet für das j-te Gen (j=1 ,...,5308) des n-ten Patienten (n=1 28) ergab die Berechnung den Wert Gj,n = arcsinh(Scy5(j,n)) - arcsinh(Scy30',n)), wobei [SCy3(j,n), SCy5(j,n)] das zugehörige Signalpaar bezeichnet. War für einen Patienten ein Spot nicht auswertbar (keine nachweisbare Signalintensität in beiden Kanälen), so wurde der zugehörige Wert als nicht vorhanden (.missing value') gekennzeichnet.
Statistischer Vergleich:
Für den Vergleich wurde der zweiseitige Zwei-Stichproben Student-Test pro Gen verwendet. Beide Stichproben enthielten die Werte der Patientengruppen überlebende Patienten bzw. nichtÜberlebenden Patienten. Für die Auswahl der differenziert exprimierten Gene wurde der zugehörige p-Wert und die Anzahl der nicht vorhandenen Werte bewertet.
Ergebnisse:
Für die Gruppe der ausgewählten Gene war der zugehörige p-Wert kleiner als 0.05, wobei in die Auswertung mindestens 5 auswertbare Signale pro Patientengruppe eingingen.
Die Höhe des Expressionsverhältnisses jedes Gens stellte das Kriterium für eine Sortierung der untersuchten Gene dar. Von Interesse waren die Gene, die zwischen den überlebenden und verstorbenen Patienten am meisten überexprimiert bzw. unterexprimiert wurden.
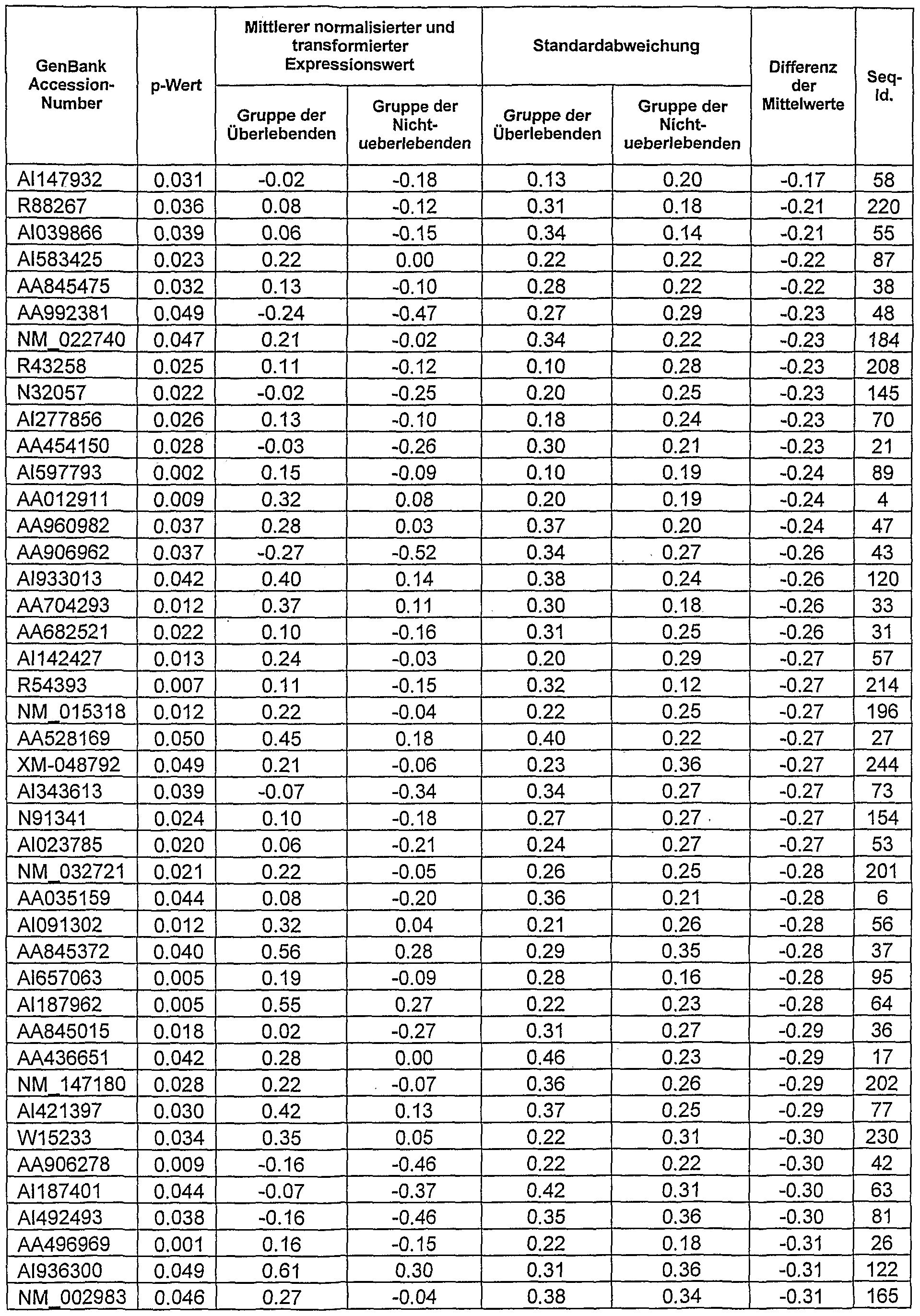
Aus Tabelle 2 ist ersichtlich, dass 65 Gene den Patientenprobe gefunden wurden, die in den nicht überlebenden Patienten gegenüber den überlebenden Patienten signifikant überexprimiert waren. Weiterhin ist aus Tabelle 3 ersichtlich, dass 182 Gene der nicht überlebenden Patienten gegenüber den überlebenden Patienten signifikant unterexprimiert waren. Aus den Ergebnissen wird deutlich, dass die in Tabelle 2 und Tabelle 3 aufgeführten Genaktivitäten zwischen überlebenden und nichtÜberlebenden Patienten bei Sepsis (klassifiziert nach [1]) unterscheiden. Somit stellen die aufgeführten Genaktivitäten Marker für eine Vorhersage der Überlebenswahrscheinlichkeit und der Entwicklung tödlicher Komplikationen von Sepsispatienten dar.
Tabelle 2: Signifikant gesteigerte Genaktivitäten in Proben von überlebenden Patienten mit Sepsis nach [1], dargestellt als deren relatives Verhältnis zu den korrespondierenden Genaktivitäten von nicht überlebenden Patienten mit Se sis nach 1
Tabelle 2 - Fortsetzung
Tabelle 3: Signifikant reduzierte Genaktivitäten in Proben von überlebenden Patienten mit Sepsis nach [1], dargestellt als deren relatives Verhältnis zu den korrespondierenden Genaktivitäten von nicht überlebenden Patienten mit Sepsis nach [1]
Diese in Tabelle 2 und 3 charakteristischen Veränderungen sind für die erfindungsgemäße Verwendung gemäß Anspruch 1 ausnutzbar.
Die in den Tabellen 2 und 3 aufgeführten GenBank Accession Nummern (Internet-Zugang über http://www.ncbi.nim.nih.gov/) der einzelnen Sequenzen sind in dem dieser Anmeldung angefügten 56-seitigen Sequenzprotokol im Einzelnen jeweils einer Sequenz ID (Sequenz ID: 1 bis zur Sequenz ID: 247) zugeordnet.
Referenzen
1. Bone RC, Balk RA, Cerra FB, Dellinger EP, Fein AM, Knaus WA, Schein RM, Sibbald WJ, the ACCP/SCCM Consensus Conference Committee (1992) Definitions for Sepsis and organ failure and guidelines for the use of innovative therapies in Sepsis. Chest 101 ,1656-1662; und Crit Care Med 1992; 20: 864-874.
2. Marshall JC, Vincent JL, Fink MP, Cook DJ, Rubenfeld G, Foster D, Fisher CJ Jr, Faist E, Reinhart K (2003) Measures, markers, and mediators: toward a staging system for clinical Sepsis. A report of the Fifth Toronto Sepsis Roundtable, Toronto, Ontario, Canada, October 25-26, 2000. Crit Care Med. 31 :1560-7.
3. Alberti C, Brun-Buisson C, Goodman SV, Guidici D, Granton J, Moreno R, Smithies M, Thomas O, Artigas A, Le Gall JR; European Sepsis Group (2003) Influence of systemic inflammatory response syndrome and Sepsis on outcome of critically ill infected patients. Am J Respir Crit Care Med. 168:77-84.
4. Brun-Buisson C, Doyon F, Carlet J, Dellamonica P, Gouin F, Lepoutre A, Mercier JC, Offenstadt G, Regnier B: Incidence, risk factors, and outcome of severe Sepsis and septic shock in adults. A multicenter prospective study in intensive care units. French ICU Group for Severe Sepsis. JAMA 1995; 274: 968-974
Le-Gall JR, Lemeshow S, Leleu G, Klar J, Huillard J, Rue M, Teres D, Artigas A: Customized probability modeis for early severe Sepsis in adult intensive care patients. Intensive Care Unit Scoring Group. JAMA 1995; 273: 644-650
Brun-Buisson C, Roudot-Thoraval F, Girou E, Grenier-Sennelier C, Durand- Zaleski I. (2003) The costs of septic syndromes in the intensive care unit
and influence of hospital-acquired Sepsis. Intensive Care Med. [Epub ahead of print]
7. Increase in National Hospital Discharge Survey rates for septicemia-United States, 1979-1987. MMWR Morb Mortal Wkly Rep 1990 ; 39: 31-34
8. Bone, R. C. Sepsis, the sepsis syndrome, multi-organ failure: a plea for comparable definitions. Ann Intern Med 1991 ; 114: 332-333
9. Matot, I., C. L. Sprung, et al. Definition of sepsis. Intensive Care Med 2001; 27 (suppl): S3-S9.
10. Friedland, J. S., J. C. Porter, et al. Plasma proinflammatory cytokine concentrations, Acute Physiology and Chronic Health Evaluation (APACHE) III scores and survival in patients in an intensive care unit. Crit Care Med 1996; 24: 1775-1781.
11. Beutler, B., A. Poltorak, et al. Sepsis and evolution of the innate immune response. Crit Care Med 2001; 29: S2-S6.
12. Vincent JL, Angus D, Annane D, et al. (2001) Clinical expert round table discussion (session 5) at the Margaux Conference on Critical lllness: outcomes of clinical trials in Sepsis: lessons learned. Crit Care Med 29:S136-137.
13. Abraham, E., Laterre P. F., et al. Lenercept (p55 tumor necrosis factor receptor fusion protein) in severe sepsis and early septic shock: a randomized, double-blind, placebo-controlled, multicenter phase III trial with 1 ,342 patients. Crit Care Med 2001 ; 29: 503-510
14. Abraham, E., Reinhart K., et al. Assessment of the safety of recombinant tissue factor pathway inhibitor in patients with severe sepsis: a multicenter, randomized, placebo-controlled, single-blind, dose escalation study. Crit Care Med 2001; 29: 2081-2089
15. Pittet, D., Harbarth S., et al. Impact of immunomodulating therapy on morbidity in patients with severe sepsis. Am J Respir Crit Care Med 1999; 160: 852-857
16. Abraham, E., Marshall J. C, et al. Sepsis and mediator-directed therapy: rethinking the target populations. Mediator-directed therapy in sepsis: rethinking the target populations. Toronto, Canada, 31 October-1 November 1998. Mol Med Today 1999; 5: 56-58.40-43
17. Abraham, E., Raffin T. A. Sepsis therapy trials. Continued disappointment or reason for hope? JAMA 1994; 271 : 1876-1878.
Iδ.Zeni F., Freeman B., et al. Anti-inflammatory therapies to treat sepsis and septic shock: a reassessment. Crit Care Med 1997; 25: 1095-1100
19. Bone, R. C. The pathogenesis of sepsis. Ann Intern Med 1991 ; 115: 457- 469
20. Marshall JC (2000) SIRS and MODS: What is there relevance to the science and practise of intensive care?, Shock 14:586-589
21. Vincent J-L (1997) Dear SIRS, l'm sorry to say that I don't like you. Crit Car Med 25:372-374
22. Ramsay G, Gerlach H, Levy MM et al (2003) An international sepsis survey: As tudy of doctor's knowledge and perception about sepsis. Crit Care Med 31
23. Levy MM, Fink MP, Marshall JC et al. (2003) 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Cri Car Med Vol 31 , No 4
24. http://www.krebsinformation.de/tnm-system.html (Stand 1. März 2004)
25. Rußwurm S. (2002) Procaicitonin als Marker bakterieller Infektionen und Sepsis: Einfluss sepsisrelevanter Bedingungen auf die Expression von Procaicitonin, Habilitationsschrift eingereicht bei der Medizinischen Fakultät der Friedrich-Schiller-Universität Jena
26. Southern EM (1974) An improved method for transferring nucleotides from electrophoresis Strips to thin layers of ion-exchange cellulose. Anal Biochem 62:317-318
27. Gillespie D, Spiegelman S (1965) A quantitative assay for DNA-RNA hybrids with DNA immobilized on a membrane. J Mol Biol 12:829-842
28. Lennon GG, Lehrach H (1991) Hybridization analyses of arrayed cDNA libraries. Trends Genet 7: 314-317
29. Kafatos FC, Jones CW, Efstratiadis A (1979) Determination of nucleic acid sequence homologies and relative concentrations by a dot hybridization procedure. Nucl Acid Res 7:1541-1552
30. Fodor SP, Read JL, Pirrung MC, Stryer L, Lu AT, Solas D (1991) Light- directed, spatially addressable parallel chemical synthesis. Science 251 :767-773
31. Pease AC, Solas D, Sullivan EJ, Cronin MT, Holmes CP, Fodor SP (1994) Light-generated oligonucleotide arrays for rapid DNA sequence analysis. Proc Natl Acad Sei USA 91 :5022-5026
32. Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative monitoring of gene expression pattems with a complementary DNA microarray. Science 270:467-470
33. Golub TR, Slonim DK, Tamayo P, et al. (1999) Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 286:531-537
34.Alizadeh AA, Eisen MB, Davis RE, et al. (2000) Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403:503-511
35.Yeoh E, Ross ME, Shurtleff SA, et al. (2002) Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 1(2): 109-10.
36. Kari L, et al., (2003) Classification and prediction of survival in patients with the leukemic phase of cutaneous T cell lymphoma. Journal of Experimental Medicine, 197[11]: 1477-88
37. van't Veer LJ, et al., (2002) Gene expression profiling predicts clinical outcome of breast cancer. Nature 415:530-6,
38. Beer DG, et al., (2002) Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nature Medicine 8:816-24
39. Huber W, Heydebreck A, Sueltmann H, et al. (2003) Parameter estimation for the calibration and variance stabilization of microarray data. Stat. Appl. in Gen. and Mol. Bio V