WO2004087727A1 - 縮合複素環誘導体、それを含有する医薬組成物およびその医薬用途 - Google Patents
縮合複素環誘導体、それを含有する医薬組成物およびその医薬用途 Download PDFInfo
- Publication number
- WO2004087727A1 WO2004087727A1 PCT/JP2004/004009 JP2004004009W WO2004087727A1 WO 2004087727 A1 WO2004087727 A1 WO 2004087727A1 JP 2004004009 W JP2004004009 W JP 2004004009W WO 2004087727 A1 WO2004087727 A1 WO 2004087727A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- inhibitor
- alkyl
- amino
- solvent
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/20—Carbocyclic rings
- C07H15/203—Monocyclic carbocyclic rings other than cyclohexane rings; Bicyclic carbocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/7036—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin having at least one amino group directly attached to the carbocyclic ring, e.g. streptomycin, gentamycin, amikacin, validamycin, fortimicins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/10—Antioedematous agents; Diuretics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/02—Heterocyclic radicals containing only nitrogen as ring hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
Definitions
- Fused heterocyclic derivative pharmaceutical composition containing the same, and pharmaceutical use thereof
- the present invention relates to a fused heterocyclic derivative or a pharmacologically acceptable salt thereof, or a prodrug thereof, a pharmaceutical composition containing the same, and a pharmaceutical use thereof, which is useful as a pharmaceutical.
- the present invention is useful, for example, as a drug for preventing or treating urinary illness, impaired glucose tolerance, hyperglycemia such as diabetic complications or obesity.
- Technology-Diabetes is one of lifestyle-related diseases due to changes in diet and lack of exercise. Therefore, '' diabetic patients are on diet or ii therapy, but when sufficient control and continuous implementation are difficult, pharmacotherapy is used. Large-scale clinical studies have confirmed that long-term strict glycemic control is necessary to prevent the onset and progress of chronic complications by treating diabetes
- antidiabetic drugs such as biguanides, sulfonylurea drugs, insulin sensitizers and ⁇ -darcosidase inhibitors are being developed. It is used.
- biguanides biguanides
- sulfonylurea drugs insulin sensitizers
- ⁇ -darcosidase inhibitors lactic acidosis for biguanides and low levels for sulfonylprea drugs
- Side effects such as edema may be observed in blood sugar and insulin sensitivity enhancers, and there is concern that they may promote obesity.
- Darcosidase inhibitors are used to improve postprandial hyperglycemia, one of which is acarbose, which is suitable for people with impaired glucose tolerance. It has been reported that this has the effect of preventing or delaying the onset of diabetes (for example, see Reference 4 below).
- .alpha.-dalcosidase inhibitors do not affect the rise of blood glucose due to ingestion of the monosaccharide, dal-3-ose (see, for example, Reference 5 below), and therefore, recent changes in dietary sugar-protein composition. Accordingly, officials are required to have a wide range of carbohydrate absorption inhibitory effects.
- urinary glucose excretion enhancers excrete excessive blood glucose from urine, the accumulation of sugar in the body is reduced, so that the effect of preventing or reducing obesity and the effect of diuresis can be expected. Furthermore, it is considered to be useful for various related diseases caused by the development of diabetes and obesity due to hyperglycemia.
- SGLT1 sodium-dependent glucose transporter 1
- glucose and galactose absorption is poor in patients with dysfunction due to congenital abnormalities of human SGLT1 (for example, see references 8 to 10 below), and SGLT1 is involved in glucose and galactose absorption. (See, for example, references 11 and 12 below).
- SGRN1 mRNA and protein increased, It has been confirmed that the absorption of water and the like has been enhanced (for example, see the following References 13 and 14). Also, diabetic patients generally have enhanced digestion and absorption of carbohydrates.
- SGLT1 mRNA and protein are highly expressed in the human small intestine (see, for example, the following literature). 15). Therefore, by inhibiting human SGLT1, it is possible to inhibit the absorption of carbohydrates such as glucose in the small intestine and to suppress an increase in blood glucose level. It is considered possible to correct postprandial hyperglycemia.
- the fused heterocyclic derivative according to the present invention is a completely novel compound, and the fused heterocyclic derivative has SGLT1 inhibitory activity and / or SGLT2 inhibitory activity and inhibits absorption of glucose and galactose in the small intestine. It has never been reported to be useful as a drug that inhibits the reabsorption of excessive glucose in the kidney.
- Literature 9 Michihiro Kasahara, et al., “Latest Medicine”, January 1996, Vol. 51, No. 1, p.
- Literature 10 Tsuchiya Yufusa, 1 other, “Japanese clinical practice”, August 1999, Vol. 55, No. 8, pp. 213 1-213
- Literature 1 Yoshikatsu Kanai, “Kidney and dialysis", December 1998, Vol. 45, extra edition, p. 232-237-Reference 12: E.Turk, foreign name, "Nafure", 1991.3 Mon, Volume 350, pp. 354-356.
- the present inventors have conducted intensive studies to find compounds that exhibit human SGLT activity inhibitory activity. As a result, certain fused heterocyclic derivatives represented by the following general formula (I) were converted to human SGLT 1 and Alternatively, the present inventors have found that it is an excellent drug that expresses SGLT2 inhibitory activity and has a blood sugar level-suppressing action or a blood sugar-lowering action, and has accomplished the present invention.
- the present invention provides a novel compound exhibiting a human SGLT activity inhibitory action, a pharmaceutical composition containing the same, and a pharmaceutical use thereof.
- R 2 is a hydrogen atom, 7, a logene atom or an alkyl group
- R 3 and R 4 are each independently a hydrogen atom, a hydroxyl group, a halogen atom,
- U is — 0_, one S— or a single bond (however, when U is — ⁇ or — S—, V and W are not simultaneously a single bond);
- V is an alkylene group which may have a hydroxyl group, a C 2 _ 6 alkenylene group or a single bond;
- Z is a hydrogen atom, C 2 _ 7 alkoxycarbonyl group, -.
- Ariru (C 2 - 7 alkoxy force Ruponiru) group, a formyl group, one R A, - COR B, One S_ ⁇ 2 R B, one CON (R c) R D, one CSN (R c) R D, one S ⁇ 2 NHR A or one C ( NR E ) N (R F ) R G ;
- R 5 , R A , R c, and R D are each independently a hydrogen atom, an alkyl group optionally having 1 to 5 arbitrary groups selected from the following substituent groups, or The following substituents (xx ix) to (x X ii) which may have 1 to 3 arbitrary groups selected from the following substituent group ⁇ ;
- Z and R 5 combine with an adjacent nitrogen atom to form an alicyclic amino group which may have 1 to 3 arbitrary groups selected from the following substituent groups; or RG and RD form, together with the adjacent nitrogen atom, an alicyclic amino group optionally having 1 to 3 optional groups selected from the following substituent group ⁇ ;
- R B is, C 2 _ 7 alkoxycarbonyl group, an alkylsulfonyl ⁇ amino group, C 6 - 10 ⁇ reel sulfonyl ⁇ amino group, Substituent Group; have 1-5 pieces of any group selected from 8 May be — a 6-alkyl group or the following substituents (xxxiii) to (xxxvi) which may have 1 to 3 arbitrary groups selected from the following substituent group;
- R E , R F and R G are each independently a hydrogen atom, a cyano group, a carbamoyl group, a C 2 _ 7 acyl group, a C 2 _ 7 alkoxycarbonyl group, and C 6 -i. 1 to 5 arbitrary groups selected from aryl (C 2 _ 7 alkoxyl carbonyl) groups, nitro groups, ( ⁇ -6 alkylsulfonyl groups, sulfamoyl groups, carbamimidyl groups, or the following substituent groups / 3)
- An alkyl group which may have;
- R E and R F combine to form an ethylene group
- R F and R G combine with an adjacent nitrogen atom to form an alicyclic amino group which may have an arbitrary group selected from the following substituent group ⁇ ; —, —S—, or one NH— which may be substituted with a C- 6 alkyl group or a halo (C- 6 alkyl) group;
- Q is, -Ci- 6 alkylene - one C 2 - 6 alkenylene one, - Ji 6 alkylene one hundred and one, - 6 alkylene - S-, -O- CI- 6 alkylene mono-, - S- 6 - one alkylene - O-C ⁇ e ⁇ , or -C ⁇ - 6 ⁇ alkylene one S_C - e alkylene -; and
- Ring A is C. An aryl group or a heteroaryl group
- Harokenin atom - a hydroxyl group, an amino group, (6 alkyl group, - 6 alkoxy group, Bruno V. port '((: alkyl) group, a halo (- s alkoxy) group, hydroxy' (C ⁇ s alkyl) groups, C 2 - 7 alkoxycarbonyl (C ⁇ e alkyl) group, a hydroxy (C 6 alkoxy) group, Amino (CI- 6 alkyl) group, Amino (C: - 6 alkoxy) group, a mono- or di (( ⁇ _ 6 alkyl) amino group, mono- or di [hydroxy (( ⁇ _ 6 alkyl)] amino group, Ci-s alkylsulfonyl group, ( ⁇ _ 6 alkyl sulfonyl Ruamino group, ( ⁇ _ 6 alkylsulphonyl ⁇ amino (C ⁇ s alkyl) group, carboxyl group, C 2 _ 7
- Halogen atom a hydroxyl group, an amino group, _ 6 alkoxy group, _ 6 alkylthio groups, halo ( ⁇ e alkoxy) group, a halo (C - 5 alkylthio) group, a hydroxy (C 6 alkoxy) group, hydroxy (( ⁇ - 6 alkylthio ) group, Amino ((: s ⁇ Alkoxy) group, Amino (C - 6 alkylthio) group, a mono- or di (C ⁇ - 6 alkyl) amino group, mono- or di [hydroxy (( ⁇ -6 alkyl)] amino group, Ureido group, Surufuamido group, mono- or di (C ⁇ 6 alkyl) ureido group, a mono or di [hydrin proxy (( ⁇ alkyl)] ureido group, a mono- or di (( ⁇ - 6 alkyl) Sulf amide group, a mono or di [hydroxy
- R H and R 1 are independently each a hydrogen atom, or an arbitrary may have three 1 groups ( ⁇ _ 6 alkyl group selected from the following substituent group ⁇ ; Alternatively, both bond together with an adjacent nitrogen atom to form an alicyclic amino group which may have 1 to 3 arbitrary groups selected from the following substituent group ⁇ ; [Substituent group A)
- Halogen atom a hydroxyl group, an amino group, an alkoxy group, halo an alkoxy) group, hydroxy (C ⁇ - 6 alkoxy) group, Amino (C ⁇ - 6 alkoxy) group, a mono- or di-alkyl) amino group, mono- or di [hydroxy (C ⁇ e alkyl)] amino group, perido group, sulfamide group, mono or di (( ⁇ alkyl) ureido group, mono or di [hydroxyalkyl)] ureido group, mono or di ((to 6 alkyl) Sulfamide group, mono- or di- Kill)] Surufuamido groups, C 2 - 7 Ashiruamino group, Amino (C 2 _ 7 Ashiruamino) group, 6 alkylsulfonyl group, C ⁇ - 6 alkylsulfonyl ⁇ amino group, a force Luba Moyle alkylsulfon
- Halogen atom a hydroxyl group, an amino group, an alkyl group, an alkoxy group, ha - Mouth - 6 alkyl) group, a halo ((6 alkoxy) group, hydroxy (C 6 ⁇ alkyl) group, C 2 _ 7 alkoxycarbonyl Cal Poni Le alkyl) group , hydroxy (C ⁇ -6 alkoxy) group, amino ((: E alkyl) group, amino (C ⁇ 6 alkoxy) group, a mono- or - di-alkyl) amino group, mono- or di [hydroxy (C -e alkyl)]
- R 2 is a hydrogen atom; Y is —O—, —S— or —NH—; Q is an ethylene group; Pharmaceutically acceptable salts, or prodrugs thereof;
- a pharmaceutical composition comprising, as an active ingredient, the fused heterocyclic derivative according to any one of [1] to [5] or a pharmaceutically acceptable salt thereof, or a prodrug thereof;
- a human SGLT activity inhibitor comprising the condensed heterocyclic derivative according to any one of the above [1] to [5], or a pharmaceutically acceptable salt thereof, or a prodrug thereof as an active ingredient;
- Diseases caused by glycemia are diabetes, impaired glucose tolerance, diabetic complications, obesity, hyperinsulinemia, hyperlipidemia, hypercholesterolemia, hypertridaricelidemia, dyslipidemia A disease selected from the group consisting of atherosclerosis, hypertension, congestive heart failure, edema, hyperuricemia and gout,
- a method for treating hyperglycemia comprising administering an effective amount of the fused heterocyclic derivative according to any one of the above [1] to [5], a pharmaceutically acceptable salt thereof, or a prodrug thereof.
- a method of preventing or treating the resulting disease comprising administering an effective amount of the fused heterocyclic derivative according to any one of the above [1] to [5], a pharmaceutically acceptable salt thereof, or a prodrug thereof.
- Diseases caused by hyperglycemia include diabetes, impaired glucose tolerance, diabetic complications, obesity, hyperinsulinemia, hyperlipidemia, hypercholesterolemia, hypertridariceridemia, lipid metabolism disorders
- the method according to [16], wherein the disease is selected from the group consisting of atherosclerosis, hypertension, congestive heart failure, edema, hyperuricemia and gout;
- Diseases caused by hyperglycemia include diabetes, impaired glucose tolerance, diabetic complications, obesity, hyperinsulinemia, hyperlipidemia, hypercholesterolemia, hypertridariceridemia, and lipid metabolism disorders
- the disease is selected from the group consisting of atherosclerosis, hypertension, congestive heart failure, edema, hyperuricemia and gout;
- Glucose tolerance The condensed heterocyclic derivative or the pharmacologically acceptable derivative thereof according to any one of the above [1:] to [5], for producing a pharmaceutical composition for preventing transition of normal subjects to diabetes.
- the human SGLT activity inhibitor according to any of [7] to [12], which is a combination of at least one drug selected from the group of drugs according to [23];
- 6 alkyl group means methyl group, ethyl group, propyl group, isopropyl group, butyl group, isobutyl group, sec-butyl group, tert-butyl group, pentyl group, isopentyl group, neopentyl group, er refers to a linear or branched alkyl group having 1 to 6 carbon atoms, such as a pentyl group, a hexyl group, etc.
- a 6 alkylene group or a monoalkylene is a methylene group, an ethylene group, a trimethylene group, a tetramethylene group.
- Alkylene groups refer to linear or branched alkylene groups having 1 to 4 carbon atoms such as methylene, ethylene, trimethylene, tetramethylene, propylene, and 1,1-dimethylethylene.
- the hydroxy (C ⁇ 6 alkyl) group means the Ci-e alkyl group substituted by a hydroxyl group.
- C ⁇ - 6 Alkoxy group a methoxy group, an ethoxy group, a propoxy group, isoproterenol 1 to 1 carbon atoms such as oxy, butoxy, isobutoxy, sec-butoxy, tert-butoxy, pentyloxy, isopentyloxy, neopentyloxy, terter_pentyloxy, hexyloxy, etc. 6 means a linear or branched alkoxy group.
- the hydroxy (C ⁇ e alkoxy) group refers to the above alkoxy group substituted with a hydroxyl group.
- the Karupokishi (C _ 6 alkoxy) group (meaning a ⁇ _ 6 alkoxy group.
- Amino alkoxy substituted above Karupokishi group
- the group substituted with Amino groups above Refers to an alkoxy group.
- C 6 alkylthio groups include methylthio, ethylthio, propylthio, isopropyletyl, butylthio, isobutylthio, sec-butylthio, fer-butylthio, pentylthio, isopentylthio, neopentylthio
- Bok 6 alkylthio) group the C E is substituted with a hydroxyl group - refers to 6 alkylthio group.
- the carboxy (( ⁇ _ 6 alkylthio) group refers to the above-mentioned 6 alkylthio group substituted with a carboxy group.
- the amino-6alkylthio) group refers to the above-mentioned C i- 6 alkylthio group substituted with an amino group.
- C 2 - 6 The alkenyl group, a vinyl group, Ariru group, 1 one propenyl group, isopropenyl group, 1-heptenyl, 2-butenyl, 2-Mechiruariru carbon number 2 to 6 straight such groups A chain or branched alkenyl group.
- C 2 - 6 alkenylene group or - C 2 _ 6 alkenylene - A, a vinylene group refers to a straight-chained or branched alkenylene group having 2 to 6 carbon atoms, such as propenylene group.
- the C 2 _ 4 alkenylene group refers to a straight-chained or branched Aruke two alkylene group having 2 to 4 carbon atoms such as Purobe two alkylene groups.
- the Karupokishi (C 2 _ 6 alkenyl) groups, the C 2 substituted with Karupokishi group - refers to 6 alkenyl group.
- a C 2 _ 6 alkenyloxy group is defined as a vinyloxy group, an aryloxy group, a 1-propenyloxy group, an isopropenyloxy group, a 1-butenyloxy group, a 2-butenyloxy group, a 2-carbonyl group such as a 2-methylaryloxy group.
- Linear or branched alke of ⁇ 6 Refers to a niloxy group.
- a C 2 _ 6 alkenylthio group refers to a group having 2 to 2 carbon atoms such as a vinylthio group, an arylthio group, a 1-probenylthio group, an isoprobenylthio group, a 1-butenylthio group, a 2-butenylthio group, and a 2-methylarylthio group.
- the C 2 _ 6 alkynyl group refers to a linear or branched alkynyl group having 2 to 6 carbon atoms, such as an ethynyl group and a 2-propyl group.
- di or dialkyl amino group refers to an amino group mono-substituted with the above-mentioned alkyl group or an amino group di-substituted with the above-mentioned C ⁇ -ealkyl group of the same or different kind.
- the mono- or di [hydroxy ( "3 ⁇ 6 alkyl)] Amino groups are di-substituted by the above hydroxy (_ 6 alkyl) Amino monosubstituted with a group group or any of the above hydroxy (( ⁇ -6 alkyl) group
- Mono or di (C ⁇ e alkyl) ureido group refers to a perido group mono-substituted with the above alkyl group or a perido group disubstituted with any of the above alkyl groups.
- a di [hydroxy (0 ⁇ -6 alkyl)] ureido group which is di-substituted by the above hydroxy (C ⁇ e alkyl) monosubstituted ureido group or any of the above b Dorokishi (C 6 alkyl) with a group group
- Mono- or di-alkyl) sulfamide group means a sulfamide group mono-substituted with the above-mentioned alkyl group or a sulfamide group di-substituted with any of the above-mentioned alkyl groups. Refers to a mid group.
- a mono- or di- [hydroxy (( ⁇ -6 alkyl)] sulfamide group is a sulfamide group monosubstituted with the above-mentioned hydroxyalkyl group or a sulfamide group di-substituted with any of the above-mentioned hydroxy (C ⁇ ealkyl) groups.
- the C 2 _ 7 Ashiru group, Asechiru group, a propionyl group refers to a straight-chained or branched Ashiru group with carbon number 2-7 such as Puchiriru group, I Sobuchiriru group, valeryl group, Kisanoiru group pivaloyl group to, .
- the C 2 _ 7 Ashiruamino group refers to an Amino group substituted by the above C 2 _ 7 Ashiru group.
- a C- 6 alkylsulfinyl group is a group having 1 to 1 carbon atoms such as a methylsulfinyl group and an ethylsulfinyl group. Refers to 6 linear or branched alkylsulfinyl groups.
- the C 6 alkyl sulfonyl group means a linear or branched alkylsulfonyl group having 1 to 6 carbon atoms such as a methanesulfonyl group and an ethanesulfonyl group.
- C DOO 6 The Arukirusu Ruhoniruamino group, the A C i _ 6 alkylsulfonyl group refers to an amino group substituted with P force Rubamoiru (C 6 alkylsulfonyl ⁇ amino) group, a force.
- the alkylsulfonylamino (C ⁇ 6 alkyl) group is The above alkyl group substituted by an alkylsulfonylamino group.
- the halogen atom means a fluorine atom, a chlorine atom, a bromine atom or an iodine atom.
- the halo (C ⁇ ealkyle) group refers to the above-mentioned alkyl group substituted by 1 to 3 of any of the above halogen atoms.
- a halo (( ⁇ -6 alkoxy) group refers to the above alkoxy group substituted with 1 to 3 of any of the above halogen atoms, and a halo (C e alkylthio) group represents 1 to 3 of any of the above halogen atoms.
- C 3 _ 7 alkoxycarbonyl group means a methoxycarbonyl group, an ethoxycarbonyl group, a propoxycarbonyl group, an isopropoxycarbonyl group, a butoxycarbonyl group, an isobutyloxycarbonyl group, sec butoxycarbonyl group, ⁇ e ⁇ -butoxycarbonyl group, pentyloxycarbonyl group, isopentyloxycarbonyl group, neopentyloxycarbonyl group, ter_pentyloxycarbonyl group group refers to a straight-chained or branched alkoxy force Ruponiru group having a carbon number of 2-7 such as Kishiruokishi force Ruponiru groups to C 2 -.
- the Le Koki deer Lupo sulfonyl (d 6 alkyl) group refers to substituted the alkyl group in the above C 2 _ 7 Arukokishikaru Poniru group.
- a C 2 _ 7 alkoxy force Lupo two Le (C ⁇ 6 alkoxy) group refers to a 6 alkoxy C 2 _ 7 alkoxycarbonyl Cal Poni Le (( ⁇ - - above substituted by the above C 2 _ 7 alkoxycarbonyl group. 6 the alkyl thio) group, substituted by the above C 2 _ 7 alkoxycarbonyl Cal Poni Le group refers to the C i 6 ⁇ alkylthio group.
- the C 2 7 alkoxycarbonyl Cal Poni Le (C 2 _ 6 alkenyl) groups have the C 2 6 alkenyl group substituted by the above C 2 _ 7 alkoxycarbonyl Cal Poni Le group La.
- C 3 _ 7 cycloalkyl or C 3- 7 cycloalkyl - refers to a cyclopropyl group, refers to a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, or heptyl group cycloheteroalkyl.
- C 3 - 7 and cycloalkyl alkoxy) group the C 3 - refers to 7 the C i _ 6 alkoxy group substituted with a cycloalkyl group.
- the C 3 _ 7 cycloalkyl (C i _ 6 alkylthio) group the C substituted by the above C 3 one 7 consequent opening alkyl group! — 6 alkylthio group.
- heterocycloalkyl group or heterocycloalkyl means morpholine, thiomorpholine, tetrahydrofuran, tetrahydropyran, aziridine, azetidin, pyrrolidine, imidazolidin, oxazoline, piperidine, piperazine, pyrazolidine, pyrroline, imidazoline, etc.
- the heterocyclyl alkyl (C e alkyl) group refers to the above 6 alkyl group substituted with the above heterocycloalkyl group.
- the term “heterocycloalkylalkoxy” group refers to the above alkoxy group substituted with the above heterocycloalkyl group.
- the heterocycloalkyl (C ⁇ ealkylthio) group is the above-mentioned C! Substituted with the above heterocycloalkyl group.
- _ 6 refers to an alkylthio group.
- C 6 - 1 () Ariru group or C 6 - The 1 Q Ariru refers to phenyl group, an aromatic cyclic hydrocarbon group having a carbon number of 6 or 1 0, such as naphthyl.
- Ariru - The ( ⁇ 6 ⁇ alkyl) group means a substituted the alkyl group in the above C 6 _ 1 Q 7 aryl group.
- C 6 - The - (6 alkoxy C) group, the C 6 - 1 0 Ariru refers to 1 0 Ariru substituted the alkoxy group group. .
- Aryl (C ⁇ 'alkyl) The O) group, the C 6 - refers to one () Ariru substituted the alkylthio group with a group.
- C 6 _ 1 Q ⁇ Li Lumpur refers to 7 alkoxy force Ruponiru group - 1 0 above substituted with Ariru group C 2.
- Heteroaryl groups or heteroaryls are thiazole, oxazole, isothiazole, isoxazole, pyridine, pyrimidine, pyrazine, pyridazine, pyrrole, thiophene, furan, imidazole, pyrazol, oxaziazol, thiodizole, 5- or 6-membered aromatic hetero ring containing 1-4 hetero atoms selected from oxygen, sulfur and nitrogen atoms derived from tolazole, furazane, etc.
- Heteroariru - The (C ⁇ 6 alkyl) group refers to substituted the alkyl group in heteroaryl groups described above.
- the Heteroa reel (C ⁇ 6 alkoxy) group means the above C ⁇ 6 alkoxy group substituted with Teroari Ichiru groups to above.
- the heteroaryl (C 6 alkylthio) — group refers to the above C, — 6 alkylthio group substituted with the heteroaryl group.
- the alicyclic amino group includes a morpholino group, a thiomorpholino group, a monoaziridinyl group, a azetidinyl group, a 1-pyrrolidinyl group, a piperidino group, a 1-imidazolidinyl group, a 1-piperazinyl group, a pyrazolidinyl group, and the like.
- a 5- or 6-membered aliphatic cyclic amino group which may have one heteroatom selected from oxygen, sulfur and nitrogen atoms in addition to the nitrogen atom at the site .
- Aromatic cyclic amino group means 1 to 3 nitrogen atoms in addition to the nitrogen atom at the binding site, such as 1-imidazolyl group, 1-pyrrolyl group, pyrazolyl group, 1-tetrazolyl group, etc. Refers to a 5-membered aromatic aromatic amino group which may be contained in The aromatic cyclic amino (C alkyl) group refers to the above alkyl group substituted with the aromatic cyclic amino group.
- the aromatic cyclic amino (C ⁇ e alkoxy) group, the ⁇ substituted by the above aromatic cyclic ⁇ amino group - refers to 6 alkoxy groups.
- Aromatic cyclic amino - The ( ⁇ 6 alkylthio) group means the above C r _ 6 alkyl thio group substituted by the above aromatic cyclic amino group.
- a hydroxyl-protecting group is generally a methyl group, a benzyl group, a methoxymethyl group, an acetyl group, a bivaloyl group, a benzoyl group, an erf-butyldimethylsilyl group, a ter ⁇ "-butyldiphenylsilyl group, a aryl group, etc.
- a protecting group for a hydroxyl group used in an organic synthesis reaction is a protecting group for an amino group, such as a benzyloxy carbonyl group, fe / ⁇ f-butoxycarbonyl group, a benzyl group, an acetyl group, or a trifluoroacetyl group.
- a prodrug is a protective group for a carboxy group used in the reaction.
- the compound represented by the above general formula (I) of the present invention can be produced according to the following method or a method analogous thereto, or a method described in other documents or a method analogous thereto.
- G 1 is the above-mentioned G in which any hydroxyl group is protected;
- G 2 is the above-mentioned G in which any hydroxyl group may be protected; and
- R 6 is a methyl group or an ethyl group.
- X 1 is a leaving group such as a halogen atom, etc.
- RR 3 , R 4 , G and ring A have the same meaning as described above, provided that a hydroxyl group, an amino group and a hydroxyl group or When a xy group is present, one having an appropriate protecting group may be used.
- the phenol derivative represented by the general formula (II) is subjected to O-benzylation using benzyl chloride or benzylpromide in an inert solvent in the presence of a base such as potassium carbonate or cesium carbonate.
- the compound represented by (III) can be produced.
- the solvent to be used include N, N-'dimethylformamide, acetone and a mixed solvent thereof.
- the reaction temperature is usually from 0 ° C to reflux temperature, and the reaction time is based on the starting materials used and The time is usually 1 hour to 2 days, depending on the solvent and reaction temperature.
- the ketone derivative represented by the general formula (III) and the arylaldehyde derivative represented by the general formula (IV) are combined with potassium hydroxide, sodium hydroxide, potassium er—butoxide, sodium er in an inert solvent.
- the compound represented by the above general formula (V) can be produced by performing an aldole reaction in the presence of a base such as butoxide, sodium methoxide, and sodium ethoxide.
- a base such as butoxide, sodium methoxide, and sodium ethoxide.
- the solvent used include methanol, ethanol, 2-propanol, 3-butanol, tetrahydrofuran, water, a mixed solvent thereof, and the like.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is preferably The time is usually 1 hour to 2 days, depending on the starting material, solvent and reaction temperature.
- the phenol derivative represented by the general formula (V) can be converted to a phenol derivative represented by the haloacetate represented by the general formula (VI) such as methyl bromoacetate, ethyl bromoacetate, methyl methyl acetate, and ethyl ethyl acetate.
- the compound represented by the general formula (VII) can be produced by ⁇ -alkylation in an active solvent in the presence of a base such as potassium carbonate or cesium carbonate.
- a base such as potassium carbonate or cesium carbonate.
- the solvent used for example,
- reaction temperature is usually from room temperature to reflux temperature
- reaction time is the starting material used. It usually varies from 1 hour to 5 days, depending on the quality, solvent and reaction temperature.
- the compound represented by the general formula (VII) is catalytically reduced in an inert solvent using a palladium-based catalyst such as palladium carbon powder, and the double bond is reduced and debenzylated to obtain the compound represented by the general formula (VII).
- the compound represented by the formula (VIII) can be produced.
- the solvent used in the catalytic reduction include methanol, ethanol, 2-propanol, tetrahydrofuran, ethyl acetate, acetic acid, and a mixed solvent thereof.
- the reaction temperature is usually from room temperature to reflux temperature.
- the reaction time varies depending on the starting materials used, the solvent and the reaction temperature, but is usually 1 hour to 2 days.
- the compound represented by the general formula (VIII) is converted into a ring in an inert solvent in the presence of a base such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, or sodium tert-butoxide.
- a base such as sodium methoxide, sodium ethoxide, potassium tert-butoxide, or sodium tert-butoxide.
- 1) appropriately adding water to the reaction mixture and treating it with sodium hydroxide or a hydroxide power and 2) treating the obtained compound in the presence of copper powder in quinoline.
- the benzofuran derivative represented by the general formula (XII) can be produced.
- the solvent used for the cyclization reaction for example, methanol, ethanol, 2-propanol, butanol, a mixed solvent thereof and the like can be illustrated.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is The time is usually 1 hour to 2 days, depending on the starting material used, the solvent and the reaction temperature
- the compound represented by the above general formula (IX) can be produced by subjecting the compound to a -alkylation in an inert solvent in the presence of a base such as potassium carbonate or cesium carbonate.
- a base such as potassium carbonate or cesium carbonate.
- reaction temperature is usually from room temperature to reflux temperature
- reaction time is usually from 1 hour to 5 days, varying based on the used starting materials, solvent and reaction temperature.
- the ketone derivative represented by the general formula (IX) and the arylaldehyde derivative represented by the general formula (IV) are reacted in an inert solvent in the presence of a base such as potassium hydroxide or sodium hydroxide.
- the compound represented by the general formula (X) can be produced by performing an aldol reaction and simultaneously performing hydrolysis.
- the solvent to be used include methanol, ethanol, 2-propanol, n-butanol, tetrahydrofuran, water, a mixed solvent thereof and the like.
- the reaction temperature is usually from room temperature to reflux temperature,
- the reaction time varies depending on the raw materials used, the solvent, the reaction temperature, and the like, but is usually 1 hour to 2 days.
- the double bond of the compound represented by the general formula (X) is catalytically reduced using a palladium-based catalyst such as palladium carbon powder in an inert solvent to give the compound represented by the general formula (XI).
- a palladium-based catalyst such as palladium carbon powder in an inert solvent
- the solvent used include methanol, ethanol, 2-propanol, tetrahydrofuran, ethyl acetate, acetic acid, water, and a mixed solvent thereof.
- the reaction temperature is usually from room temperature to reflux temperature.
- the time is usually 1 hour to 2 days, depending on the starting material used, the solvent and the reaction temperature.
- a double bond of the compound represented by the general formula (X) is converted to a reagent such as triethylsilane in an inert solvent in the presence of a rhodium-based catalyst such as tris (triphenylphosphine) rhodium (I) chloride.
- a rhodium-based catalyst such as tris (triphenylphosphine) rhodium (I) chloride.
- the compound represented by the general formula (XI) can also be produced by hydrogenation using the compound.
- the solvent used include benzene, toluene, and a mixed solvent thereof.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is the starting material used, the solvent, the reaction temperature, etc. It usually varies from 1 hour to 2 days.
- the compound represented by the above general formula (XI) was treated with sodium acetate and
- the benzofuran derivative represented by the general formula (XII) can be obtained by cyclizing in the presence of acetic acid and acetic anhydride, and, if necessary, deprotecting the acetylated hydroxyl group by alkali hydrolysis during the ring reaction.
- the solvent used in the cyclization reaction include acetic acid and the like.
- the reaction temperature is usually from 50 ° C to reflux temperature, and the reaction time varies depending on the used starting materials, solvent, reaction temperature and the like. , Usually one hour to three days.
- Examples of the solvent used for the alkali hydrolysis reaction include water, methanol, ethanol, and a mixed solvent thereof.
- the basic substance examples include sodium hydroxide, sodium methoxide, and sodium methoxide. Sid and the like can be used.
- the treatment temperature is usually from o ° C to reflux temperature, and the treatment time is usually from 30 minutes to 1 day, depending on the raw materials used, the solvent and the treatment temperature.
- the compound represented by the general formula (XI I) is converted to 2,3,4,6-tetra-O-acetyl-1-1 O-trichloroacetimidoyl-Q! —D-dalcoviranose, 3,4,6-tetra-O-acetyl-1-O-trichloroacetimidoyl / 3 / 3-D-darcopyranose, 1,2,3,4,6-penter 0-acetyl-3-D-darcopyranose, 2 , 3,4,6-tetra-O-acetyl-D-darcopyranosylbutamide, 2,3,4,6-tetra-O-acetyl-O-trichloroacetimidoyl-a-D-galactopyranose , 2,3,4,6-tetra-acetyl-l- (9-trichloroacetimidoyl-J3-D-galactopyranose, 1,2,3,4,6-pentene O-acety
- Examples of the solvent used include methylene chloride, toluene, acetonitrile, nitromethane, ethyl acetate, ethyl ether, chloroform, and a mixture thereof, and the reaction temperature is usually -30 ° C to reflux.
- the reaction time is usually 10 minutes to 1 day, depending on the starting materials used, solvent and reaction temperature.
- the compound represented by the general formula (Ia) of the present invention can be produced by subjecting the glycoside represented by the general formula (XIII) to alkaline hydrolysis to remove the protecting group.
- the solvent used include water, methanol, ethanol, tetrahydrofuran, a mixed solvent thereof and the like.
- the basic substance include sodium hydroxide, sodium methoxide, sodium methoxide and the like.
- the processing temperature is usually from 0 ° C to the reflux temperature, and the processing time is usually from 30 minutes to 1 day, depending on the used starting materials, solvent and processing temperature.
- the compound represented by the general formula (II) is converted to 2,3,4,6-tetra-O-acetyl-a-D-darcopyranosylbutamide, 2,3,4,6-tetra-C > -A cetyl- a- D-galactopyranosyl bromide, 2,3,4,6-tetra-O-pivaloyl-D-darcopyranosyl bumid, 2,3,4'6-tetra —Li-pivaloylu ⁇ - D-galactopyranosyl bromide, 2,3,4,6-tetra- (9-benzoyl- ⁇ -D-darcopyranosyl buclamide, 2,3,4,6-tetra-O— Benzyl mono-D-galactopyranosyl bromide, etc., in a hydrated inert solvent, in a hydrated inert solvent, benzyltri (-butyl) ammonium chloride, benzyltri
- the inert solvent used examples include methylene chloride, chloroform, toluene, benzotrifluoride, and a mixture thereof. It can gel, the reaction temperature is usually o ° c to the reflux temperature, the reaction time starting material, solvent, varying based on a used and reaction temperature, is usually for 30 minutes to 1 day.
- the phenol derivative represented by the general formula (XIV) is converted to a haloacetate represented by the general formula (VI) such as methyl bromoacetate, ethyl bromoacetate, methyl methyl acetate, and ethyl ethyl acetate.
- the compound represented by the general formula (XV) can be produced by performing O-alkylation in an inert solvent in the presence of a base such as potassium carbonate or cesium carbonate.
- a base such as potassium carbonate or cesium carbonate.
- the solvent used for example,
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time varies depending on the raw materials used, the solvent, the reaction temperature, and the like. 1 hour to 5 days.
- the ketone derivative represented by the general formula (XV) and the arylaldehyde derivative represented by the general formula (IV) are reacted in an inert solvent in the presence of a base such as potassium hydroxide or sodium hydroxide.
- the compound represented by the general formula (XVI) can be produced by performing an aldol reaction and simultaneously performing hydrolysis.
- the solvent used include methanol, ethanol, 2-propanol, n-butanol, tetrahydrofuran, water, and a mixed solvent thereof.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is usually from 1 hour to 2 days, varying depending on the used starting materials, solvent and reaction temperature.
- the compound represented by the general formula (XVI) By subjecting the double bond of the compound represented by the general formula (XVI) to catalytic reduction using a palladium-based catalyst such as palladium carbon powder in an inert solvent, the compound represented by the general formula (XVI) ) Can be produced.
- the solvent used include methanol, ethanol, 2-propanol, tetrahydrofuran, ethyl acetate, acetic acid, water, and a mixed solvent thereof.
- the reaction temperature is usually from room temperature to reflux temperature.
- the time varies depending on the starting materials, solvent, reaction temperature, etc., but is usually 1 hour to 2 days.
- a double bond of the compound represented by the general formula (XVI) is reacted with a reagent such as triethylsilane in an inert solvent in the presence of a rhodium-based catalyst such as tris (triphenylphosphine) rhodium (I) chloride.
- a reagent such as triethylsilane in an inert solvent
- a rhodium-based catalyst such as tris (triphenylphosphine) rhodium (I) chloride.
- the compound represented by the above general formula (XVII) can also be produced by hydrogenation using Examples of the solvent used include benzene, toluene, and a mixed solvent thereof.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is the starting material, solvent, and reaction temperature. It usually varies from 1 hour to 2 days.
- Cyclizing the compound represented by the general formula (XVII) in an inert solvent in the presence of sodium acetate and acetic anhydride to produce the benzofuran derivative represented by the general formula (XIII) can be.
- the solvent used in the reaction include acetic acid.
- the reaction temperature is usually 50 ° C. to reflux temperature, and the reaction time varies depending on the used starting materials, solvent and reaction temperature. , Usually one hour to three days.
- R 1 is a hydroxyl group
- R 2 is a hydrogen atom
- Y is — Y
- Q is an ethylene group
- R 3 , R 4 , R 6 , G, G ⁇ X 1 and ring A have the same meanings as described above.
- a compound having a protecting group may be used as appropriate.
- the phenol derivative represented by the general formula (XVI II) is converted to potassium carbonate or cesium carbonate in an inert solvent using benzyl chloride or benzylbutamide.
- the compound represented by the general formula (XIX) can be produced by O-benzylation in the presence of such a base.
- the solvent to be used include N, -dimethylformamide, acetone, a mixed solvent thereof and the like. It usually varies from 1 hour to 2 days, depending on the reaction temperature. .
- the ketone derivative represented by the general formula (XIX) and the arylaldehyde derivative represented by the general formula (IV) are combined with potassium hydroxide, sodium hydroxide, potassium er—butoxide, sodium er in an inert solvent.
- the compound represented by the general formula (XX) can be produced by carrying out an aldole reaction in the presence of a base such as butoxide, sodium methoxide and sodium ethoxide.
- a base such as butoxide, sodium methoxide and sodium ethoxide.
- the solvent used include methanol, ethanol, 2-propanol, methanol, tetrahydrofuran, water, and a mixed solvent thereof.
- the reaction temperature is usually from room temperature to reflux temperature.
- the time varies depending on the starting materials, solvent, reaction temperature, etc., but is usually 1 hour to 2 days.
- the phenol derivative represented by the general formula (XX) is converted to a haloacetate represented by the general formula (VI) such as methyl bromoacetate, ethyl acetate bromo, methyl ethyl chloroacetate, and ethyl chloroacetate,
- a haloacetate represented by the general formula (VI) such as methyl bromoacetate, ethyl acetate bromo, methyl ethyl chloroacetate, and ethyl chloroacetate
- reaction temperature is usually from room temperature to reflux temperature, and the reaction time is based on the starting material and It usually varies from 1 hour to 5 days, depending on the solvent and reaction temperature.
- the compound represented by the general formula (XXI) is catalytically reduced in an inert solvent using a palladium-based catalyst such as palladium-carbon powder to reduce and remove the double bond.
- the compound represented by the general formula (XXII) can be produced by carrying out the reaction.
- the solvent used in the catalytic reduction include methanol, ethanol, 2-propanol, tetrahydrofuran, ethyl acetate, acetic acid, a mixed solvent thereof, and the like
- the reaction temperature is usually from room temperature to reflux temperature.
- the reaction time varies depending on the starting materials used, the solvent used, the reaction temperature and the like, but is usually 1 hour to 2 days.
- the phenol derivative represented by the general formula (XXII) is subjected to ⁇ -benzylation using benzyl chloride or benzylpromide in an inert solvent in the presence of a base such as potassium carbonate or cesium carbonate.
- a base such as potassium carbonate or cesium carbonate.
- the compound represented by the formula (XXIII) can be produced. Examples of the solvent used include, for example,
- reaction temperature is usually from o ° c to the reflux temperature, and the reaction time varies depending on the raw materials used, the solvent, the reaction temperature, etc. , Usually one hour to three days.
- the compound represented by the general formula (XXIII) is cyclized in an inert solvent in the presence of a base such as sodium methoxide, sodium ethoxide, potassium er-butoxide, and sodium terf-butoxide. Thereafter, if necessary, 1) by appropriately adding water to the reaction mixture and treating it with sodium hydroxide or hydroxydehydric acid, and 2) treating the obtained compound in the presence of copper powder in quinoline.
- a base such as sodium methoxide, sodium ethoxide, potassium er-butoxide, and sodium terf-butoxide.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time varies depending on the starting materials used, the solvent, the reaction temperature, and the like. Usually 1 hour to 2 days.
- the compound represented by the general formula (XXIV) is converted to 2,3,4,6-tetra-(-1-acetyl-O-trichloroacetimidoyl-CK-D-glucopyranose, 2,3,4,6-tetra-O-acetyl-l-O-trichloroacetimidyl-i3-D-darcopyranose, 1,2,3,4,6_ pen-(-acetyl — / 3— D-Darcopyranose, 2,3,4,6-tetra-(-acetyl- ⁇ -D-darcopyranosylbutamide, 2,3,4,6-tetra- ⁇ -acetyl-1— —-trichloroacetimidoyl ⁇ -D-galactopyranose, 2,3,4,6-tetra-(-acetyl-11-C> -trichloroacetimidoyl-1-i3-D-galactopyranose, 1,2,3,4,6-pen Yu
- reaction time varies depending on the starting materials used, the solvent, the reaction temperature and the like, but is usually from 10 minutes to 1 day.
- the compound represented by the general formula (XXVI) is catalytically reduced in an inert solvent using a palladium-based catalyst such as palladium carbon powder and debenzylated to give the compound represented by the general formula (XXVI).
- Solvents used in the catalytic reduction include, for example, methanol, ethanol, 2-propanol, tetrahydrofuran, ethyl acetate, acetic acid, and mixed solvents thereof.
- the reaction temperature is usually from room temperature to reflux temperature.
- the reaction time varies depending on the starting materials used, the solvent used, the reaction temperature, and the like, but is usually 1 hour to 2 days.
- the compound represented by the general formula (lb) of the present invention can be produced by removing the protective group by subjecting the glycoside represented by the general formula (XXVI) to alkaline hydrolysis.
- the solvent used include water, methanol, ethanol, tetrahydrofuran, and a mixed solvent thereof.
- the basic substance include sodium hydroxide, sodium methoxide, and sodium methoxide. can do.
- the treatment temperature is usually from 0 ° C to the reflux temperature, and the treatment time is usually from 30 minutes to 1 day, depending on the used starting material, solvent and treatment temperature.
- R 2 is a hydrogen atom'; Y is ⁇ - 6 alkyl or halo (C ⁇ 6 alkyl) may be substituted with a group
- a compound wherein Q is an ethylene group can be produced according to the method of the following steps 26 to 34.
- T is a hydrogen atom, an alkyl group or a halo (Ci 6 alkyl) group
- X is a bromine, chlorine or iodine atom; R 1 , R 3 , R 4 , G, G 1 and ring A have the same meaning as described above. However, when a hydroxyl group, an amino group, and Z or a carboxy group are present in each compound, those having an appropriate protecting group may be used. )
- the phenol derivative represented by the general formula (XXVI I) is converted to benzyl chloride
- the compound represented by the general formula (XXV III) can be produced by O-benzylation using benzylbutamide in an inert solvent in the presence of a base such as potassium carbonate or cesium carbonate. it can.
- a base such as potassium carbonate or cesium carbonate. it can.
- the solvent used for example,
- reaction temperature is usually from 0 to reflux temperature, and the reaction time varies depending on the raw material used, the solvent, the reaction temperature, etc. Usually 1 hour to 2 days.
- the compound represented by the general formula (XXVIII) is subjected to a Vi 1 smeier reaction in an inert solvent using phosphorus oxychloride and TV, T-dimethylformamide to introduce a formyl group.
- the compound represented by the general formula (XX IX) can be produced.
- the solvent used include N, iV-dimethylformamide, acetonitrile, methylene chloride, 1,2-dichloroethane, a mixed solvent thereof, and the like, and the reaction temperature is usually 120 ° C to reflux temperature.
- the reaction time varies depending on the starting materials used, the solvent and the reaction temperature, but is usually 30 minutes to 1 day.
- the compound represented by the general formula (XX IX) and the phosphonium salt represented by the general formula (XX X) are combined with an inert solvent such as sodium hydride, sodium hydroxide, potassium fer—butoxide, -butyllithium, iei
- an inert solvent such as sodium hydride, sodium hydroxide, potassium fer—butoxide, -butyllithium, iei
- the olefin compound represented by the general formula (XXI) can be produced.
- the solvent used include tetrahydrofuran, N, TV-dimethylformamide, dimethylsulfoxide, a mixed solvent thereof and the like.
- the reaction temperature is usually from 120 ° C to reflux temperature, and the reaction time is The time is usually 30 minutes to 1 day, depending on the starting material used, the solvent and the reaction temperature.
- the compound represented by the general formula (XXXI) is catalytically reduced in an inert solvent using a palladium-based catalyst such as palladium-carbon powder to reduce and remove the double bond.
- the compound represented by the general formula (XXXV) can be produced by performing the zirination.
- the solvent used in the catalytic reduction include: methanol, ethanol, 2-propanol, tetrahydrofuran, ethyl acetate, acetic acid, and a mixed solvent thereof.
- the reaction temperature is usually from room temperature to reflux temperature.
- the reaction time varies depending on the starting materials used, the solvent, the reaction temperature, and the like, but is usually 1 hour to 2 days.
- the compound represented by the general formula (XXIX) is subjected to a Grignard reaction in an inert solvent using a Grignard reagent represented by the general formula (XXXII), whereby The compound represented by the general formula (XXXIII) can be produced.
- the solvent used include tetrahydrofuran, ethyl ether, a mixed solvent thereof, and the like.
- the reaction temperature is usually from o ° C to reflux temperature, and the reaction time is the starting material, solvent, reaction temperature, etc. It usually varies from 30 minutes to 1 day.
- Step 31-A reducing agent such as porane tetrahydrofuran complex or porane dimethyl sulfide complex in an inert solvent in the presence of an additive such as 4-dimethylaminopyridine in an inert solvent
- the compound represented by the above general formula (XXX IV) can be produced by reduction using Examples of the solvent used include tetrahydrofuran, getyl ether, a mixed solvent thereof, and the like.
- the reaction temperature is usually from o ° C to reflux temperature, and the reaction time is the starting material, solvent, and reaction temperature. It usually varies from 30 minutes to 5 days, depending on the temperature.
- the compound represented by the general formula (XXXIII) is reduced with a reagent such as triethylsilane in an inert solvent in the presence of an acid such as trifluoroacetic acid, boron trifluoride and getyl ether complex.
- a reagent such as triethylsilane in an inert solvent in the presence of an acid such as trifluoroacetic acid, boron trifluoride and getyl ether complex.
- the compound represented by the general formula (XXX IV) can also be produced.
- the solvent used include methylene chloride, 1,2-dichloroethane, and a mixed solvent thereof.
- the reaction temperature is usually from -20 ° C to reflux temperature, and the reaction time is usually from 30 minutes to 5 days, varying depending on the starting materials used, the solvent and the reaction temperature.
- reaction temperature is usually from room temperature to reflux temperature.
- reaction time is based on the starting materials and solvent used. It usually varies from 1 hour to 2 days, depending on the reaction temperature.
- the glycoside represented by the general formula (XX XVI) can be produced by glycosylation in the presence of an agent.
- Solvents used include, for example, methylene chloride, toluene, acetonitrile, nitromethane, ethyl acetate, diethyl ether, chloroform, mixed solvents thereof, and the like.
- the reaction temperature ranges from 30 ° C to the reflux temperature, and the reaction time varies depending on the material used, the solvent, the reaction temperature, etc., but is usually from 10 minutes to 1 day.
- the compound represented by the general formula (Ic) of the present invention can be produced by subjecting the glycoside represented by the general formula (XXVI) to alkaline hydrolysis to remove the protecting group.
- the solvent used include water, methanol, ethanol, tetrahydrofuran, and a mixed solvent thereof.
- the basic substance include sodium hydroxide, sodium methoxide, and sodium ethoxide. Etc. can be used.
- the treatment temperature is usually from 0 ° C to reflux temperature, and the treatment time is usually from 30 minutes to 1 day, depending on the used starting materials, solvent and treatment temperature.
- R 2 is hydrogen atom; Y is - a S-; Q gar (: Bok 6 alkylene one, -C 2 - 6 alkenylene one , —
- R 4 , R 6 , G, G 1 , X 1 and ring A have the same meaning as described above. However, when a hydroxyl group, an amino group, a Z or a carboxy group is present in each compound, a compound having an appropriate protecting group may be used. )
- the compound represented by the general formula (XXXV II) was converted to N, N, N,, N, After treatment with a lithiating reagent such as butyllithium, sec-butyllithium, t6r-butyllithium in an inert solvent in the presence of additives such as tramethylethylenediamine and hexamethylphosphoric triamide
- a lithiating reagent such as butyllithium, sec-butyllithium, t6r-butyllithium in an inert solvent in the presence of additives such as tramethylethylenediamine and hexamethylphosphoric triamide
- the amide derivative represented by the general formula (XXXV III) in an inert solvent
- the compound represented by the general formula (XXX IX) can be produced.
- the solvent to be used include cyclohexane, / 2-hexane, tetrahydrofuran, getyl ether, and a mixed solvent thereof.
- the thiophenol derivative represented by the general formula (XXX IX) is converted to a haloacetate represented by the general formula (VI) such as methyl bromoacetate, ethyl bromoacetate, methyl acetate, and ethyl ethyl acetate.
- a base such as triethylamine or N
- V-diisopropylethylamine in an inert solvent
- reaction temperature is usually from o ° C to reflux temperature
- reaction time is the starting material, solvent, and reaction temperature. It usually varies from 1 hour to 2 days, depending on the temperature.
- the compound represented by the general formula (XL) is cyclized in an inert solvent in the presence of a base such as sodium methoxide, sodium ethoxide, potassium er-butoxide, and sodium erf-butoxide to form the compound.
- a benzothiophene derivative represented by the general formula (XLI) can be produced.
- the solvent used for the cyclization reaction for example, methanol, ethanol, 2-propanol, -butanol, a mixed solvent thereof and the like can be illustrated.
- the reaction temperature is usually from room temperature to reflux temperature,
- the reaction time varies depending on the starting materials used, the solvent, the reaction temperature, etc., but is usually 1 hour to 2 days.
- the compound represented by the general formula (XLII) can be produced by subjecting the compound represented by the general formula (XLII) to alkaline hydrolysis.
- the solvent used include methanol, ethanol, 2-propanol, tetrahydrofuran, water, and a mixed solvent thereof.
- the basic substance include sodium hydroxide and potassium hydroxide. can do.
- the treatment temperature is usually from room temperature to reflux temperature, and the treatment time is usually from 1 hour to 1 day, depending on the raw materials used, the solvent and the treatment temperature.
- the compound represented by the general formula (XLIII) is produced by decarboxylating the compound represented by the general formula (XLII) in an inert solvent in the presence of a catalyst such as copper powder.
- a catalyst such as copper powder.
- the solvent used include quinoline.
- the reaction temperature is usually from 100 to reflux temperature, and the reaction time varies depending on the used starting materials, solvent and reaction temperature, but is usually from 30 minutes to 1 day. Between.
- the compound represented by the general formula (XLIV) is demethylated by using a reagent such as boron tribromide or boron trichloride in an inert solvent in an inert solvent.
- a reagent such as boron tribromide or boron trichloride in an inert solvent in an inert solvent.
- Compounds can be prepared.
- the solvent used include methylene chloride, 1,2-dichloroethane, benzene, toluene, and a mixed solvent thereof.
- the reaction temperature is usually from 178 ° C to reflux temperature, and the reaction time is It usually varies from 30 minutes to 1 day, depending on the starting material used, solvent and reaction temperature.
- the solvent used include methylene chloride, toluene, acetonitrile, nitromethane, ethyl acetate, ethyl ether, chloroform, and a mixed solvent thereof.
- the reaction temperature is usually from 130 ° C. to the reflux temperature, and the reaction time is usually from 10 minutes to 1 day, varying based on the used starting materials, solvent and reaction temperature.
- the glycoside represented by the general formula (XLV) is subjected to alkaline hydrolysis to form a protecting group.
- the compound of the present invention represented by the general formula (Id) can be produced.
- the solvent used include water, methanol, ethanol, tetrahydrofuran, a mixed solvent thereof, and the like.
- the basic substance include sodium hydroxide, sodium methoxide, sodium methoxide, and the like.
- the treatment temperature is usually from 0 ° C to the reflux temperature, and the treatment time is usually from 30 minutes to 1 day, depending on the raw materials used, the solvent and the treatment temperature.
- R 2 is hydrogen atom; Q is located at an ethylene group; R 3 Gar U- V 1 - N (R 5 ) - R A or - U- V 1 - ⁇ ⁇ ( V 1 in the formula which may have a hydroxyl group ( ⁇ _ 6 is an alkylene group or c 2 _ 6 alkenylene group;!! R 5, R a and U Has the same meaning as described above) can be produced according to the method of the following steps 43 to 50.
- L 1 is a mesyloxy group or a tosyloxy group
- M is a silyl protecting group for a hydroxyl group
- RR 4 , R 5 , R A , G, GU, V 1 , Y and ring A have the same meanings as described above.
- a hydroxyl group, an amino group and a Z or hydroxyl group are present in each compound, those having an appropriate protecting group may be used.
- the compound represented by the general formula (XLV I) is inactivated by using a silylating reagent such as ter-butyldiphenylsilyl chloride, er-butyldimethylsilyl chloride, triisopropylsilyl chloride, and triethylsilyl chloride.
- a silylating reagent such as ter-butyldiphenylsilyl chloride, er-butyldimethylsilyl chloride, triisopropylsilyl chloride, and triethylsilyl chloride.
- the compound represented by the general formula (XLVII) can be produced by O-protection in a solvent in the presence of a base such as imidazole, triethylamine or ⁇ , V-diisopropylethylamine.
- the solvent to be used include N, V-dimethylformamide, methylene chloride, a mixed solvent thereof, and the like.
- the solvent used examples include methylene chloride, toluene, acetonitrile, nitromethane, ethyl acetate, ethyl ether, chloroform, and a mixed solvent thereof. It is the reflux temperature, and the reaction time varies depending on the starting materials used, the solvent and the reaction temperature, but is usually from 10 minutes to 1 day.
- the compound represented by the general formula (XLVI-I) is desilylated by using a reagent such as tetra (-butyl) ammonium fluoride in an inert solvent to obtain the compound represented by the general formula (XLVI-I).
- a reagent such as tetra (-butyl) ammonium fluoride in an inert solvent to obtain the compound represented by the general formula (XLVI-I).
- XLIX can be produced.
- the solvent to be used include tetrahydrofuran.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time varies depending on the used starting materials, solvent, reaction temperature, etc., but is usually 1 hour. ⁇ 2 days.
- the compound represented by the general formula (XLIX) is eliminated using an acid chloride such as mesyl chloride or tosyl chloride in an inert solvent in the presence of a base such as triethylamine or N, V-diisopropylethylamine.
- a base such as triethylamine or N, V-diisopropylethylamine.
- the compound represented by the general formula (L) can be produced.
- the solvent used in the introduction reaction for example, methylene chloride salt, ethyl acetate, tetrahydrofuran, pyridine, a mixed solvent thereof and the like can be illustrated.
- the reaction temperature is usually from 0 ° C. to room temperature, and the reaction time is usually from 30 minutes to 1 day, varying based on a used starting material, solvent and reaction temperature.
- a compound represented by the general formula (L ⁇ ) is produced by subjecting the compound represented by the general formula (L) to azidation in an inert solvent using an azide reagent such as sodium azide.
- an azide reagent such as sodium azide.
- the solvent used for the azidation reaction include methylene chloride, ethyl acetate, N, iV-dimethylformamide, dimethyl sulfoxide, V-methylpyrrolidone, N, iV-dimethylimidazolidinone, and a mixed solvent thereof. be able to.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is usually from 30 minutes to 1 day, varying based on a used starting material, solvent and reaction temperature.
- the compound represented by the general formula (LIIa) can be produced by catalytically reducing the compound represented by the general formula (LI) using a palladium-based catalyst such as palladium carbon powder in an inert solvent. It can.
- a palladium-based catalyst such as palladium carbon powder in an inert solvent.
- the solvent used for the catalytic reduction reaction include tetrahydrofuran, methanol, ethanol, ethyl acetate, and a mixed solvent thereof.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is usually from 30 minutes to 1 day, varying based on a used starting material, solvent and reaction temperature.
- the compound represented by the general formula (Ie) of the present invention can be produced by subjecting the compound represented by the general formula (LIIa) to alkaline hydrolysis to remove the protecting group.
- the solvent used for the hydrolysis reaction include methanol, ethanol, tetrahydrofuran, acetonitrile, water, and a mixed solvent thereof.
- the base include, for example, sodium hydroxide, sodium methoxide , Sodium ethoxide, methylamine, dimethylamine and the like.
- the reaction temperature is usually from 0 ° C to reflux temperature, and the reaction time is usually from 30 minutes to 1 day, varying based on a used starting material, solvent and reaction temperature.
- Process 5 0 The compound represented by the general formula (L) is treated in an inert solvent with triethylamine, N, diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4.0] pendane-7-cene, and hydrogenated.
- the compound represented by the above general formula (If) of the present invention can be produced by condensing with a salt thereof and, if necessary, subjecting it to alkaline hydrolysis in the same manner as in Step 49.
- Examples of the solvent used for the condensation reaction include acetonitrile, N, -dimethylformamide, dimethylsulfoxide, TV-methylpyrrolidone, methanol, ethanol, tetrahydrofuran, water, a mixed solvent thereof and the like.
- the reaction temperature is usually from room temperature to reflux temperature, and the reaction time is usually from 1 hour to 3 days, varying depending on a used starting material, solvent and reaction temperature.
- R 2 is a hydrogen atom;
- Q is an ethylene group;
- R B, R c, R D, R E , R F , U and V have the same meanings as described above
- L 2 in the formula is a leaving group such as a pyrazolyl group, a methylthio group or a benzotriazolyl group
- RR 4 , R B , R c , R D , R E , R F , G, GU, V, Y, Z 1 and ring A have the same meaning as described above, provided that, when a hydroxyl group, an amino group and / or a carboxy group are present in each compound, those having an appropriate protecting group may be used. I do not care.
- the compound represented by the general formula (LIX) can be produced from the compound represented by the general formula (LII).
- Compounds represented by the above general formula (LII) can be prepared by mixing triethylamine, N, iV-diisopropylethylamine in an inert solvent such as methylene chloride, ethyl acetate, tetrahydrofuran, pyridine, acetonitrile, or a mixed solvent thereof. , Pyridine, 1,8-diazabicyclo [5.4.0] pendant 7-cene and the like in the presence of an acid chloride represented by the above general formula (LIV) or (LV), usually at a reflux temperature of 0. The reaction is usually performed for 30 minutes to 1 day.
- an inert solvent such as methylene chloride, ethyl acetate, tetrahydrofuran, pyridine, acetonitrile, or a mixed solvent thereof.
- Pyridine, 1,8-diazabicyclo [5.4.0] pendant 7-cene and the like in the presence of an acid chloride represented by the above general
- the compound represented by the above general formula (LII) is treated with triethylamine, V, diisopropylethylamine in an inert solvent such as methylene chloride, ethyl acetate, tetrahydrofuran, pyridine, acetonitrile, toluene, and a mixed solvent thereof.
- an isocyanate compound represented by the above general formula (LVI) is usually used at 0 ° C. The reaction is usually performed at a reflux temperature for 30 minutes to 1 day.
- a compound represented by the general formula (LII) is mixed with 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, In the presence of a condensing agent such as hexylcarposimide, and in the presence or absence of a base such as triethylamine, N, TV-diisopropylethylamine or the like, 1-hydroxybenzotriazole is appropriately added as necessary.
- the reaction is carried out with the sulfonic acid compound represented by the general formula (LVII) at a temperature of usually 0 ° C. to reflux temperature for usually 1 hour to 3 days.
- the compound represented by the above general formula (LII) was treated with N- (benzyloxycarbonyl) 1-1 //-pyrazole-1 in an inert solvent such as tetrahydrofuran, methanol, ethanol, toluene, or a mixed solvent thereof.
- the reaction is carried out with a guanidinating reagent represented by the above general formula (LVIII) such as one-pot lipoxamidine, usually at room temperature to reflux temperature for usually 1 hour to 5 days.
- LVIII guanidinating reagent represented by the above general formula (LVIII)
- the compound represented by the general formula (Ig) of the present invention can be produced by subjecting the compound represented by the general formula (LIX) to alkaline hydrolysis.
- Examples of the solvent used for the hydrolysis reaction include methanol, ethanol, tetrahydrofuran, water, a mixed solvent thereof, and the like.
- Examples of the base include sodium hydroxide, sodium methoxide, and sodium ethoxide. , Methylamine, dimethylamine and the like.
- the reaction temperature is usually from 0 ° C to reflux temperature, and the reaction time is usually from 30 minutes to 1 day, varying depending on a used starting material, solvent and reaction temperature.
- a compound represented by the above general formula (LII) is mixed with a base such as triethylamine, N, -diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4.0] nde-7-cene or the like.
- a base such as triethylamine, N, -diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4.0] nde-7-cene or the like.
- an active ester compound represented by the above general formula (LXI) can be produced.
- the solvent used for the condensation reaction include methylene chloride, tetrahydrofuran, ethyl acetate, acetonitrile, pyridine, and a mixed solvent thereof.
- the reaction temperature is usually from o ° C to reflux temperature, and the reaction time is usually from 30 minutes to 1 day, varying based on a used starting material, solvent and reaction temperature.
- Step 54 ′ The compound represented by the above general formula (LXI) is mixed with an inert solvent such as triethylamine, N, -diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4.0]
- an inert solvent such as triethylamine, N, -diisopropylethylamine, pyridine, 1,8-diazabicyclo [5.4.0]
- Examples of the solvent used for the condensation reaction include methylene chloride, methanol, ethanol, tetrahydrofuran, ethyl acetate, acetonitrile, pyridine, ⁇ , V-dimethylformamide, and a mixed solvent thereof.
- Its reaction temperature The reaction temperature is usually from room temperature to reflux temperature, and the reaction time is usually from 30 minutes to 2 days, depending on the used starting materials, solvent and reaction temperature.
- the compound represented by the general formula (Ih) of the present invention can be produced by subjecting the compound represented by the general formula (LXIII) to alkaline hydrolysis.
- the solvent used for the hydrolysis reaction include, for example, tanol, ethanol, tetrahydrofuran, water, and a mixed solvent thereof.
- the base include, for example, sodium hydroxide, sodium methoxide, sodium ethoxide, Methylamine, dimethylamine and the like can be mentioned.
- the reaction temperature is usually from 0 to reflux temperature, and the reaction time is usually from 30 minutes to 1 day, varying based on a used starting material, solvent and reaction temperature.
- R 2 is a hydrogen atom
- Q is an ethylene group
- R 5 A compound of the formula R A (wherein R 5 , R A , U and V have the same meanings as described above) can be produced according to the following steps 56 to 58.
- Step 56 In an inert solvent, a compound represented by the general formula (LX IV) is mixed with a condensing agent such as 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, dicyclohexyl carbodiimide, and triethylamine, N, -diisopropylamine.
- a condensing agent such as 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, dicyclohexyl carbodiimide, and triethylamine, N, -diisopropylamine.
- the general formula (LXV) By adding 1-hydroxybenzotriazole as needed in the presence or absence of a base such as thiamine and condensing with the amine derivative represented by the general formula (LIII), the general formula The compound represented by (LXV) can be produced.
- the solvent used for the condensation reaction include N, N-dimethylformamide, tetrahydrofuran, methylene chloride, and a mixed solvent thereof.
- the reaction temperature is usually from 0 ° C to reflux temperature, and the reaction time is usually from 1 hour to 3 days, varying depending on a used starting material, solvent and reaction temperature.
- the glycoside represented by the above general formula (LXVI) can be produced by glycosylation in the presence of the compound.
- Examples include acetonitrile, nitromethane, ethyl acetate, ethyl ether, chloroform, mixed solvents thereof, and the like.
- the reaction temperature is usually _30 ° C to reflux temperature, and the reaction time is the starting material and solvent used. Although it varies depending on the reaction temperature and the like, it is usually 10 minutes to 1 day.
- the compound represented by the general formula (Ii) of the present invention can be produced by removing the protective group by subjecting the glycoside represented by the general formula (LXVI) to alkaline hydrolysis.
- the solvent used include water, methanol, ethanol, tetrahydrofuran, a mixed solvent thereof, and the like.
- the basic substance include sodium hydroxide, sodium methoxide, and sodium ethoxide.
- the treatment temperature is usually from o ° C to the reflux temperature, and the treatment time is usually from 30 minutes to 6 hours, depending on the used starting materials, solvent and treatment temperature.
- L 3 is a chlorine atom, a bromine atom, an iodine atom or a trifluoromethanesulfonyloxy group
- RR 4 , R 5 , R A , G, GV 2 , Y and ring A have the same meanings as described above.
- a compound having an appropriate protecting group may be used.
- a compound represented by the general formula (LXVII) is combined with an olefin derivative represented by the general formula (LXVIII), in an inert solvent, palladium carbon powder, palladium acetate, tetrakis (triphenylphosphine) palladium, dibenzylide
- a palladium-based catalyst such as acetone palladium or bis (triphenylphosphine) palladium dichloride
- a phosphine ligand such as tris (2-methylphenyl) phosphine or triphenylphosphine
- Triethylamine, sodium er—butoxide, potassium erf The compound represented by the general formula (LXIX) can be produced by performing the Heck reaction in the presence of a base such as butoxide and cesium fluoride.
- Examples of the solvent used for the reaction include acetonitrile, toluene, tetrahydrofuran, triethylamine, a mixed solvent thereof and the like.
- the reaction temperature is usually from 0 ° C to reflux temperature, and the reaction time is usually from 1 hour to 2 days, varying depending on a used starting material, solvent and reaction temperature.
- the compound represented by the general formula (L XVII) is combined with the olefin derivative represented by the general formula (LXX) in an inert solvent, palladium carbon powder, palladium acetate, tetrakis (triphenylphosphine) palladium, Using a palladium-based catalyst such as benzylideneacetone palladium or bis (triphenylphosphine) palladium dichloride in the presence or absence of a phosphine ligand such as tris (2-methylphenyl) phosphine and triphenylphosphine; and By carrying out the Heck reaction in the presence of a base such as triethylamine, sodium er-butoxide, power, dimethyl-butoxide, or cesium fluoride, the olefin derivative represented by the general formula (LXXI) can be obtained.
- a palladium-based catalyst such as benzylideneacetone palladium or bis (triphen
- the solvent used for the reaction include acetonitrile, toluene, tetrahydrofuran, triethylamine, and a mixed solvent thereof.
- the reaction temperature is usually from 0 ° C to reflux temperature, and the reaction time is usually from 1 hour to 2 days, varying depending on a used starting material, solvent and reaction temperature.
- a compound represented by the general formula (LXXI) is mixed with a condensing agent such as 11-ethyl-3- (3-dimethylaminopropyl) carbodiimide hydrochloride, dicyclohexyl carbodiimide, and triethylamine; By adding 1-hydroxybenzotriazole as needed in the presence or absence of a base such as V-diisopropylethylamine and condensing it with the amine derivative represented by the general formula (LIII).
- the compound represented by the general formula (LXIX) can be produced.
- the solvent used for the condensation reaction for example, 7V, N- Examples thereof include dimethylformamide, tetrahydrofuran, methylene chloride, and a mixed solvent thereof.
- the reaction temperature is usually from 0 ° C to reflux temperature, and the reaction time is usually from 1 hour to 3 days, varying depending on a used starting material, solvent and reaction temperature.
- the compound represented by the general formula (I j) of the present invention can be produced by subjecting the compound represented by the general formula (LIX) to alkaline hydrolysis to remove the protecting group.
- the solvent used for the hydrolysis reaction include methanol, ethanol, tetrahydrofuran, water, and a mixed solvent thereof.
- the base include, for example, sodium hydroxide, sodium methoxide, sodium Ethoxide and the like can be mentioned.
- the reaction temperature is usually 0 ° C
- the reaction time is usually 30 minutes to 1 day, depending on the starting materials used, the solvent and the reaction temperature.
- the compound represented by the general formula (I k) of the present invention is obtained by catalytically reducing the compound represented by the general formula (I j) using a palladium-based catalyst such as palladium carbon powder in an inert solvent.
- a palladium-based catalyst such as palladium carbon powder in an inert solvent.
- the solvent used for the catalytic reduction reaction include methanol, ethanol, tetrahydrofuran, ethyl acetate, a mixed solvent thereof and the like.
- the reaction temperature is usually from 0 ° C to reflux temperature
- the reaction time is usually from 1 hour to 2 days, varying depending on a used starting material, solvent and reaction temperature.
- the compound represented by the general formula (LXXII) can be produced by catalytically reducing the compound represented by the general formula (LXIX) using a palladium-based catalyst such as palladium carbon powder in an inert solvent.
- a palladium-based catalyst such as palladium carbon powder in an inert solvent.
- the solvent used for the catalytic reduction reaction include methanol, ethanol, tetrahydrofuran, ethyl acetate, a mixed solvent thereof and the like.
- the reaction temperature is usually from the reflux temperature to the reflux temperature, and the reaction time depends on the starting materials used, the solvent, the reaction temperature, etc. It usually varies from 1 hour to 2 days.
- the compound represented by the general formula (Ik) of the present invention can be produced by subjecting the compound represented by the general formula (LXXII) to alkaline hydrolysis to remove a protecting group.
- the solvent used for the hydrolysis reaction include methanol, ethanol, tetrahydrofuran, water, a mixed solvent thereof, and the like.
- the base include, for example, sodium hydroxide, sodium methoxide, and sodium ethoxide. And the like.
- the reaction temperature is usually from 0 to reflux temperature, and the reaction time is usually from 30 minutes to 1 day, varying based on a used starting material, solvent and reaction temperature.
- a compound having a hydroxyl group, an amino group and a Z or carboxy group can be subjected to a reaction after optionally introducing a protecting group according to a conventional method as needed.
- the protecting group can be appropriately removed in a later step according to a conventional method.
- the compound represented by the general formula (I) of the present invention obtained in the above-mentioned production method may be a fractionation recrystallization method which is a conventional separation means, a purification method using chromatography, a solvent extraction method, a solid phase extraction method, etc. Can be isolated and purified.
- the fused heterocyclic derivative represented by the general formula (I) of the present invention can be converted into a pharmacologically acceptable salt thereof by a conventional method.
- salts include acid addition salts with mineral acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, methanesulfonic acid, benzenesulfonic acid, and J5— Acid addition salts with organic acids such as toluenesulfonic acid, propionic acid, citric acid, succinic acid, tartaric acid, fumaric acid, butyric acid, oxalic acid, malonic acid, maleic acid, lactic acid, malic acid, carbonic acid, glutamic acid, and aspartic acid , Sodium salts, salts with inorganic bases such as potassium salts, N-methyl-D-dalcamine, N, ⁇ '-dibenzylethylenediamine, 2-amino
- the compound represented by the general formula (I) of the present invention includes a drug such as water or ethanol. Solvates with pharmaceutically acceptable solvents are also included.
- compounds having an unsaturated bond include cis (Z) isomers, which are two geometric isomers, Although trans () -form compounds exist, any of these compounds may be used in the present invention.
- compounds having an asymmetric carbon atom except for a darcopyranosyloxy moiety or a galactopyranosyloxy moiety include: There are two kinds of optical isomers, a compound of? Configuration and a compound of S configuration. In the present invention, any of the optical isomers may be used, and a mixture of those optical isomers may be used. It does not matter.
- the prodrug of the compound represented by the general formula (I) of the present invention can be obtained by a conventional method using a corresponding prodrug-forming reagent such as an octalogenated compound in the compound represented by the general formula (I).
- a group constituting a prodrug into one or more arbitrary groups selected from a hydroxyl group and an amino group as appropriate according to a conventional method, and then isolating and purifying according to a conventional method as needed.
- a group forming a prodrug used in a hydroxy group or an amino group for example, c 2 - 7 Ashiru groups, alkoxy (C 2 _ 7 Ashiru) group, C 2 7 alkoxycarbonyl Cal Poni Le
- the (C 2 7 alkoxy force Ruponiru) group means the c 2 _ 7 alkoxy force Ruponiru group substituted with an alkoxy group.
- the group constituting the prodrug include a darcopyranosyl group and a galactopyranosyl group, for example, a 4- or 6-position hydroxyl group of the dalcopyranosyloxy group or the galactopyranosyloxy group. It is more preferably introduced into the hydroxyl group at the 4- or 6-position of the darcopyranosyloxy group.
- the fused heterocyclic derivative represented by the general formula (I) of the present invention has, for example, a potent human SGLT1 or SGLT2 activity inhibitory activity in the following test for confirming the activity of human SGLT1 or SGLT2. Indicated. Therefore, the fused heterocyclic derivative represented by the general formula (I) of the present invention exhibits excellent SGLT1 activity inhibitory activity in the small intestine or excellent SGLT2 activity inhibitory activity in the kidney, The increase in the blood glucose level can be significantly suppressed, or the blood glucose level can be significantly reduced.
- the fused heterocyclic derivative represented by the general formula (I) of the present invention a pharmacologically acceptable salt thereof and a prodrug thereof are used as a postprandial hyperglycemic inhibitor, diabetes mellitus in a person with impaired glucose tolerance.
- SGLT 1 activity in the small intestine and SGLT 2 activity in the kidney such as diabetes, impaired glucose tolerance, diabetic complications (eg, retinopathy, neuropathy, nephropathy, ulcers, large vessels Disease), obesity, hyperinsulinemia, hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, dyslipidemia, atherosclerosis, hypertension, congestive heart failure, edema, hyperuricemia, It is extremely useful as an agent for preventing or treating diseases caused by hyperglycemia such as gout.
- the compound of the present invention can be used in appropriate combination with at least one of the following drugs.
- drugs that can be used in combination with the compound of the present invention include insulin sensitivity enhancers, glucose absorption inhibitors, biguanides, insulin secretagogues, SGLT2 activity inhibitors, insulin or insulin analogs, and glucagon receptor anions.
- gonist insulin receptor kinase stimulant, triptidyl peptidase II inhibitor, dipeptidyl peptidase IV inhibitor, protein tick synphosphatase-1B inhibitor, glycogen phosphorylase inhibitor, glucose-1 6-phosphatase inhibitor, fructose-bisphosphatase inhibitor, pyruvate dehydrogenase inhibitor, hepatic gluconeogenesis inhibitor, D-chiroinositol, glycogen synthase kinase-3 Inhibitor, glucagon-like beptide 1, glucagon-like beptide 1 ⁇ , glucagon-like peptide - 1 Agonisuto, amylin, an amylin analogue, ⁇ Mirinagonisuto, Aldo Ichisu reductase inhibitors, advanced glycation end products (ad V anc edg 1 ycatione ndp roduc ts) production inhibitor, protein kina
- the present invention provides that Any of the administration forms of staggered administration by the same or different administration routes as a preparation of the compound of the present invention,
- the medicament comprising a combination of drugs includes a dosage form as a single preparation as described above and a dosage form as a combination of separate preparations.
- the compound of the present invention When the compound of the present invention is used in combination with one or more of the above-mentioned drugs as appropriate, it is possible to obtain more advantageous effects than additive effects in preventing or treating the above-mentioned diseases. Or, similarly, the use amount can be reduced as compared with the case of using alone, or the side effect of the concomitant drug can be avoided or reduced.
- Specific compounds of the drugs used in combination and suitable diseases to be treated are exemplified below, but the content of the present invention is not limited to these, and the specific compounds are free of charge.
- Insulin sensitivity enhancers include troglitazone, pioglitazone hydrochloride, rosiglitazone maleate, dalglitazone sodium, GI-262570, isaglitazone (isaglitazone), LG-100641, NC-2100, T-174, DRF-2189, CLX -0921, CS-011, GW-1929, peroxisome proliferator-activated receptor argonist such as ciglitazone, englitazone sodium stream, NIP-221, peroxisome proliferator-activated drug such as GW-9578, BM-170744 Receptor ⁇ agonist, GW—409544, KRP—297, ⁇ —622, CLX—0940, LR-90, SB—2191999, DRF—4158, DRF—MDX8, etc.
- peroxisome proliferator-activated receptor argonist such as ciglitazone, englitazone sodium stream, NIP-22
- retinoide X receptor agonists such as bexa rote ne, and reglixan, ⁇ NO-5816, MBX-102, CRE—1625, FK—614, CLX—0901, CRE—1633, NN—2344, BM—13125, BM—501050, HQ L—975, CLX—0900, M BX—668, MBX—675, Other insulin sensitizers such as S-15261, GW-544, AZ-242, LY-510929, AR-HO 49020 GW-501516.
- Insulin sensitivity enhancers are Treatment of diabetes, impaired glucose tolerance, diabetic complications, obesity, hyperinsulinemia, hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, dyslipidemia, atherosclerosis.
- it promotes the uptake of blood glucose into tissues and lowers blood glucose levels, and is therefore effective in treating diabetes, impaired glucose tolerance, and hyperinsulinemia. It is more preferable for the position.
- sugar absorption inhibitors examples include calcarose, pogliose, miglitol, CKD—711, emiglitate, MDL—25, 63,7, force miglyose, and MHD-73, 945, etc. , AZM-127, etc.—amylase inhibitors, International Publication No. WO 02/09893 pamphlet, International Publication No. WO02 / 004/014932 Pamphlet, International Compounds such as SGL T.1 activity inhibitor and the like described in WO200004 / 018949Panfret, International Publication WO2004 / 0199589Panfret, etc.
- Glucose absorption inhibitors are particularly suitable for the treatment of diabetes, impaired glucose tolerance, diabetic complications, obesity, and hyperinsulinemia, and inhibit the digestion of carbohydrates contained in food in the gastrointestinal tract. It is more preferable for treating glucose intolerance because it delays or inhibits the absorption of glucose and the like into glucose.
- biguanides examples include phenformin, buformin hydrochloride, metformin hydrochloride and the like.
- Biguanide drugs are particularly suitable for the treatment of diabetes, impaired glucose tolerance, diabetic complications, and hyperinsulinemia.They also suppress gluconeogenesis in the liver, promote anaerobic glycolysis in tissues, and improve insulin resistance in peripheral exclusion. Since it lowers blood sugar levels due to its beneficial effects, it is more preferable for the treatment of diabetes, impaired glucose tolerance, and hyperinsulinemia.
- Insulin secretagogues include tolptamide, chlorpropamide, tolazamide, acetohexamide, daliclopyramide, glyburide (dalibenclamide), daliclazide, 1-butyl-3-methanilylprea, carbamide, glipol nulide, glipizide, gliquidone, Darisoxepide, Gribthiazol, Daribazole, Dalihexamide, Glymidine Sodium, Daripinamide, 'Fenbu Tamide, tolcyclamide, glimepiride, nateglinide, mitiglinide calcium hydrate, repaglinide and the like, as well as glucokinase activators such as RO-28-1675.
- Insulin secretagogues are particularly suitable for the treatment of diabetes, impaired glucose tolerance, and diabetic complications.In addition, they act on kidney / 3 cells to increase insulin secretion and lower blood glucose levels, thus reducing diabetes. It is more preferable for the treatment of impaired glucose tolerance.
- Examples of SGLT2 activity inhibitors include T-1095, JP-A-10_237089, JP-A-2001-288178, WO 01/16147 pamphlet, WO WO 1X27128 pamphlet, international Publication WOO 1Z68660 pamphlet, International Publication WO 01 Z 74 834 pamphlet, International publication WOO 1Z74835 pamphlet, International publication WOO 2/28872 pamphlet, International publication WO 02 Z36602 pamphlet, International Compounds described in WOO 2Z44192 pamphlet, WO02 / 53573 pamphlet in International Publication No. WO03 / 000712 pamphlet, WO3Z020737 in WO3Z0737, and the like can be mentioned.
- SGLT2 activity inhibitors are particularly suitable for the treatment of diabetes, impaired glucose tolerance, diabetic complications, obesity, and hyperinsulinemia, and reduce blood glucose levels by inhibiting reabsorption of glucose in the renal tubules. It is more preferable for treating diabetes, impaired glucose tolerance, obesity, and hyperinsulinemia.
- Insulin or insulin analogs include human insulin, animal-derived insulin, and human or animal-derived insulin analogs. These drugs are particularly preferable for the treatment of diabetes, impaired glucose tolerance and diabetic complications, and more preferably for the treatment of diabetes and impaired glucose tolerance.
- Glucagon receptor antagonists include BAY-27-9955, NNC-192-1687, and the like, and insulin receptor kinase stimulants include TER-17411, L-783281, KRX-613, and the like.
- insulin receptor kinase stimulants include TER-17411, L-783281, KRX-613, and the like.
- triptidyl peptidase II inhibitors include UCL-1397 and the like
- dipeptidyl peptidase IV inhibitors include NVP-DPP 728A, TSL-225, P-32 / 98, and the like.
- Protein tyrosine phosphatase-1B inhibitors include PTP-112, ⁇ C-86839, PNU-1 77496, and the like; glycogen phosphorylase inhibitors include NN-4201, CP-368296; and fructose-bisphosphatase inhibitors include R-132917.
- Examples of pyruvate dehydrogenase inhibitors include AZD-7545 and the like, examples of hepatic gluconeogenesis inhibitors include FR-225659 and the like, and examples of glucagon-like peptide-1 analogs include , Exendin-4, CJC-1131 and the like.
- Glucagon-like peptide-1 agonists include AZM-134, LY-315902, amylin, amylin analog or amylinago As a second strike And pramlintide acetate. These drugs, darcos-6-phosphatase inhibitor, D-caylinositol, glycogen synthase kinase-3 inhibitor, and glucagon-like peptide-1 are particularly diabetic, impaired glucose tolerance, diabetic complications, It is preferable for the treatment of insulinemia, and more preferable for the treatment of diabetes and impaired glucose tolerance.
- Aldose reductase inhibitors include ascorbyl gamolate, tolres sunset, epalrestat, ADN_138, BAL-ARI8, ZD-5522, ADN-311, GP-1447, IDD-598, fidalestat, solvin, Ponaresu evening (pona 1 restat), risarestat (risarestat), zenarestat (z en arestat), minarestat (minalrestat), methsol vinyl, AL-1567, imirestat, M-16209, TAT, AD-5467, Zopolrestat, AS-3201, NZ-314, SG-210, JTT-811, and Lindlestat (1 ind ⁇ 1 restat).
- Aldose reductase inhibitors reduce intracellular sorbitol, which is excessively accumulated due to enhancement of the polyol metabolic pathway in sustained hyperglycemic conditions observed in diabetic complication tissues, by inhibiting aldose reductase. Therefore, it is particularly preferable for treating diabetic complications.
- Examples of the advanced glycation end product inhibitor include pyridoxamine, ⁇ P B-919, ALT-946, ALT-711, pimagedin hydrochloride and the like.
- An advanced glycation endogenous product inhibitor is particularly preferable for the treatment of diabetic complications because it inhibits the end glycation endogenous production that is promoted by sustained hyperglycemia in a diabetic state, thereby reducing cytotoxicity.
- protein kinase C inhibitors examples include LY-333531, midosulin and the like. Protein kinase C inhibitors are particularly preferable for treating diabetic complications because they suppress the increase in protein kinase C activity observed due to sustained hyperglycemia in diabetic conditions.
- Rp-aminobutyric acid receptor antagonists include topiramate, etc.
- sodium channel antagonists include mexiletine hydrochloride, oxforce rubazepine, etc.
- transcription factor NF- ⁇ inhibitors include dexliposide.
- lipid peroxidase inhibitors include tilirazad mesylate
- acetylated-hyperlinked-acid dipeptidase inhibitors include GPI-5
- carnitine derivatives include carnitine, levasecarnin hydrochloride, repocarnitine chloride, repocarnitine, ST-261 and the like.
- insulin-like growth factor-I platelet-derived growth factor
- platelet-derived growth factor analog epithelial growth factor
- nerve growth factor peridine
- 5-hydroxy-1-methylhydantoin EGB-761
- bimoclomol Sulodexide and Y-128 are particularly preferred for the treatment of diabetic complications.
- antidiarrheal or laxative examples include polypyrrophyll calcium, albumin tannate, bismuth subnitrate and the like. These drugs are particularly preferable for the treatment of diarrhea and constipation associated with diabetes and the like.
- a reductase inhibitors include seribas dutin sodium, pravath ditin sodium, oral bath dutin (lovastatin), simbas dutin, flupastatin sodium, and atorbas du sodium Tin calcium hydrate, SC—45355, SQ—33600, CP—83011, BB—476, L—669262, S-2468, DMP-565, U
- + Hydroxymethyldaryl rilcoenzyme A reductase inhibitors are particularly suitable for the treatment of hyperlipidemia, hypercholesterolemia, hypertension, glyceridemia, dyslipidemia, and atherosclerosis. It is more preferable for the treatment of hyperlipidemia, hypercholesterolemia, and atherosclerosis because it lowers blood cholesterol by inhibiting methyldalyl rilcoenzyme A reductase.
- fibrate compounds include bezafibrate, beclobrate, vini fibrate, ciprofibrate, clinofibrate, clofibrate, clofibrate aluminum, clofibric acid, ethoibrate, fuenovibrate, gemfibrate, nikorifibrate. , Ronifibrato, Simfibrato, Theofibrato, Hachi! ! ! — :! ? And the like.
- Fibrate compounds are particularly preferred for treating hyperinsulinemia, hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, dyslipidemia, atherosclerosis, and lipoprotein lipase in the liver. It is more preferable for the treatment of hyperlipidemia, hypertriglyceridemia, and atherosclerosis, because it lowers blood triglycerides by activating lipoproteins and enhancing fatty acid oxidation.
- BRL As a second strike, BRL—28410, SR-58611A, ICI—198157, ZD—2079, BMS—194444, BRL-37344, CP—331679, CP—114271, L -7 50 355, BMS—187413, SR-59062 A, BMS-2102 85, LY—377604, SWR-0342 SA, AZ—40140, SB—226552, D—7114, BRL-35135, FR—149175, BRL—26830A, CL-316243, AJ—9677, GW-427
- j3 3 -a-drenergic receptor agonists are especially preferred for the treatment of obesity, hyperinsulinemia, hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, dyslipidemia, and in fat.
- Asilcoenzyme A As cholesterol acetyltransferase inhibitors, NTE-122, MCC-147, PD-132301-2, DUP- ⁇ 29, U-73482, U-76807, RP-70676, P-06139, CP-113113, RP-73163, FR-129169, FY-038, EAB-309, KY-455, LS-3115, FR-145237, T-2591, J1-2104127, R-755 , FCE 28654, YI C-C8 — 434, Avasimibe (av as imi be), CI-1 976, RP-644 77, F—1394, Eldasimimi (eldac imi be), CS—505, CL—283546, YM—17E , Recimibide (lecimi bide;),
- a cholesterol acyltransferase inhibitor is particularly preferable for treating hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, and abnormal lipid metabolism. Since the blood cholesterol is lowered by inhibiting the group transferase, it is more preferable for the treatment of hyperlipidemia and hypercholesterolemia.
- Thyroid hormone receptor agonists include liothyronine sodium, repothyroxine sodium, KB-2611, etc., and cholesterol absorption inhibitors include ezetimibe, SCH-48461, etc., and lipase inhibition.
- examples of the drug include Orris evening, ATL-962, AZM-131, RED-1 03004 and the like.
- Examples of the carnitine palmitoyltransferase inhibitor include etomoxil
- examples of the squalene synthase inhibitor include SDZ- 268-198, BMS-188494, A-87049, RPR-101821, ZD-9720, RPR-107393, ER-27.856, TAK-475 and the like.
- Nicotinic acid derivatives include nicotinic acid, nicotinamide, Nicomol, niceritrol, acipimox, nicorandil, etc., and bile acid adsorbents such as cholestyramine, cholestyrane, colesevelam hydrochloride, GT-102-279, etc.
- One inhibitor includes 264W94, S-8921, SD-5613, and the like.
- As a cholesterol ester transfer protein inhibitor PNU-107368 E, SC-7 95, JTT-705, CP-529414 and the like.
- These drugs are particularly hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, lipid metabolism. Preferred for treatment of abnormalities. .
- Appetite suppressants include monoamine reuptake inhibitors, serotonin reuptake inhibitors, serotonin release stimulants, serotonin agonists (especially 5HT 2C —agonist), noradrenaline reuptake inhibitors, noradrenaline release stimulants, i-ad'renaline receptor Agonisuto, ⁇ 2 - adrenergic receptor Agonisuto, Dopaminago two strike, Kan'napinoido receptor Anne evening agonist, Aamino acid receptor Anne evening agonist, ⁇ 3 - histamine Anne evening agonist, L- histidine, Rebuchin, replica Chin such ⁇ , Lebutin receptor agonist, melanocortin receptor agonist (especially MC3-Ragonist, MC4-Ragonist), ⁇ -melanocyte stimulating hormone, cocaine and amphetamine-regular transcript, mahogany protein, hen Ross evening Chinagonisuto, calcitonin, calcitonin gene-related peptide,
- monoamine reuptake inhibitors include mazindol and the like
- serotonin reuptake inhibitors include dexfenfluramine hydrochloride, fenfluramine hydrochloride, sibudramin hydrochloride, fluvoxamine maleate, sertraline hydrochloride, etc.
- Serotonin agonists include inotributane, (+) norfenfluramine, and the like
- noradrenaline reuptake inhibitors include bupropion, GW-320659, and the like
- noradrenaline release stimulants include: rolipram, ⁇ 1 ⁇ - 992 and the like, / 3 2 - adrenoreceptor Agonisuto, amphetamine, de kiss Toro amphetamine, phentermine, Benzufuetamin, Metaanfue evening Min, phendimetrazine, phenmetrazine, Jechirupuropio Phenylpropanolamine, clobenzolex, and the like; dopamine agonists include ER-230, dobrexin, and promocribtin mesylate; and cannapinoid receptor antagonists include rimonabant and the like.
- Examples of butyric acid receptor antagonists include topiramate and the like.
- Examples of H 3 -histamine antagonists include GT-2394.
- Examples of lebutin, lebutin analogs or lebutin receptor agonists include LY-355101.
- Cholecystokinin agonists include SR-146131, SSR-125180, BP-3.200, A-71623, FPL-15849, GI-248573, GW-7178, GI- 181771 , GW-7854, A-71378, and the like.
- PD-160170 NGD-95-1, BI BP-3226, 122 9-U-91, CGP-71683, BI BO-3304, CP-67190 6-01, J-115814.
- Appetite suppressants are particularly useful for diabetes, impaired glucose tolerance, diabetic complications, obesity, hyperlipidemia, hypercholesterolemia, hypertriglyceridemia, dyslipidemia, atherosclerosis, hypertension, Preferable for the treatment of congestive heart failure, edema, hyperuricemia, and gout.Suppresses appetite by promoting or inhibiting the action of monoamines and bioactive peptides in the brain in the central appetite control system, and reduces appetite. Therefore, it is more preferable for treating obesity.
- Angiotensin converting enzyme inhibitors include captopril, enalapril maleate, alacepril, delapril hydrochloride, ramipril, lisinoburil, imidabril hydrochloride, benazepril hydrochloride, seronapril monohydrate, cilazapril, fosinobulil sodium, perindopril erupmine , Mobertipril calcium, quinapril hydrochloride, spirapril hydrochloride, temocapril hydrochloride, trandethos, zofenopril calcium, moexipril hydrochloride (moeXipri 1), lentil april and the like.
- Angiotensin converting enzyme inhibitors are particularly preferred for the treatment of diabetic complications and hypertension.
- Neutral endopeptidase inhibitors include: omapatrilat, MDL-100240, fasidotril (fasi do tri 1), sampatrilat, 'GW-66051 IX, mixanpril, SA-7060, E-4030 , SLV-306, ecadotril and the like. Neutral endopeptidase inhibitors are particularly preferred for the treatment of diabetic complications and hypertension.
- Angiotensin II receptor antagonists include candesartan cilexetil, candesartan cilexetil hydroclo thiazide, oral sultan potassium, eprosartan mesilate, valsartan, telmisartan, irbesartan, EXP-3174, L-158809, EXP-312, Olmesartan, Tasosartan, KT-3-671, GA-0113, RU-64276, EMD-90423, BR-9701 and the like.
- Angiotensin II receptor Antagonists are particularly preferred for the treatment of diabetic complications and hypertension.
- Endothelin converting enzyme inhibitors include CGS-31447, CGS-35 066, SM-19712 and the like.
- Endothelin receptor antagonists include L-1 7805, TBC-3214, BMS-182874, BQ-610. , TA-0201, SB-215355, PD-1809.88, Sitaxen tuna (sita X sentan) BMS-193884, Darusentan (darus en tan), TBC-3711, Bosentan, Tezosentan sodium (tezos ent an) J-104132, YM-598, S-0139, SB-234551, RPR-118031 A, AT Z_1993, RO-61-1790, ABT-546, Enracentan, BMS-207940 and the like. These drugs are particularly preferable for treating diabetic complications and hypertension, and more preferably for treating hypertension.
- Diuretics include chlorthalidone, metrazone, cyclopentiazide, trichloromethiazide, hydroclothiazide, hydroflumethiazide, ventil hydroclo mouth thiazide, penflutide, meticlothiazide, indapamide, tripamide, mefluside, ezazomide, esazomide, ezazomide Nido, furosemide, bumetanide, methicran, potassium canrenoate, spironolactone, triamterene, aminophylline, cicleclenin hydrochloride, LLU-a, PNU-80873A, isosorbide, D-mannitol, D-sorbitol, fructose, glycerin, Acetozolamide, metazolamide, FR-179544, ⁇ PC-31260, lixipapant (1 ixi vaptan), and conipaptan hydrochloride.
- Diuretics are particularly suitable for the treatment of diabetic complications, hypertension, congestive heart failure and edema, and also increase blood excretion to lower blood pressure and improve edema, resulting in hypertension and congestive heart failure It is more preferable for the treatment of edema.
- Calcium antagonists include: aranidipine, efonidipine hydrochloride, dicardipine hydrochloride, barnidipine hydrochloride, benidipine hydrochloride, manidipine hydrochloride, cilnidipine, dissoldipine, nitrendipine, nifendipine, nilvadipine, felodipine, Amlodipine besylate, branidipine, lercanidipine hydrochloride, isradipine, ergodipine, azelnidipine, lacidipine, batanidipine hydrochloride, remildi.
- vasodilator antihypertensives include indapamide, todralazine hydrochloride, hydralazine hydrochloride, hydralazine, budralazine, etc .
- Antiplatelet agents include ticlopidine hydrochloride, dipyridamole, cilostazol, ethyl icosapentate, salpodalelate hydrochloride, dilazep hydrochloride, trapidil, beraprost sodium, aspirin and the like. Antiplatelet drugs are particularly preferred for the treatment of atherosclerosis and congestive heart failure.
- Uric acid production inhibitors include aloprinol and oxypurinol; uric acid excretion enhancers include benzbromarone and probenecid; urine alkalinizing agents include sodium bicarbonate, potassium citrate, and quencher Sodium acid and the like can be mentioned. These drugs are particularly preferable for treating hyperuricemia and gout.
- insulin sensitizers when used in combination with the compounds of the present invention, in the treatment of diabetes are insulin sensitizers, glucose absorption inhibitors, biguanides, insulin secretagogues, SGLT2 activity inhibitors, insulin or insulin analogs, glucagon receptor antagonists, insulin receptor kinase stimulants, tribe Petityl peptidase II inhibitor, dipeptidyl peptidase IV inhibitor, protein tyrosine phosphatase-IB inhibitor, glycogen phosphorylase inhibitor, glucose 6-phosphatase inhibitor, fruc] S-bisphosphatase inhibitor, pyruvate dehydrogenase inhibitor, hepatic gluconeogenesis inhibitor, D-chiroinositol, glycogen synthase kinase-3 inhibitor, glucagon-like peptide 1-1, dal gongon-like peptide 1-1 analog, glucagon Peptides-1agonist, amylin, amylin analogs, amylin At least is preferably
- SGLT 2 activity inhibitor insulin or insulin analog, glucagon receptor antagonist, insulin receptor kinase stimulant, tripeptidyl peptidase-II inhibitor, dipeptidyl peptidase IV inhibitor, protein tyrosine phosphatase — 1B inhibitor, glycogen phosphorylase inhibitor, glucose 16-phosphatase inhibitor, fructose-bisphosphatase inhibitor, pyruvate dehydrogenase inhibitor, hepatic gluconeogenesis inhibitor, D-chiroinositol, glycogen Synthase kinase-3 inhibitor, glucagon-like peptide-11, glucagon-like peptide-11 analog, glucagon-like peptide-1 agonist, amylin, at least one selected from the group consisting of amylin analog and amylin agonist Better to combine with other drugs Combination with at least one drug selected from the group consisting of insulin sensitizers, glucose absorption inhibitors, biguanides, insulin secretago
- insulin sensitivity enhancers in the treatment of diabetic complications, insulin sensitivity enhancers, glucose absorption inhibitors, biguanides, insulin secretagogues, SGLT2 activity inhibitors, insulin or insulin analogs, and glucagon receptor antagonists Evening gonist, insulin receptor kinase stimulant, 'trip Tidyl peptidase II inhibitor, dipeptidyl peptidase IV inhibitor, protein-tin mouth phosphatase-1B inhibitor, glycogen phosphorylase inhibitor, glucose-6-phosphatase inhibitor, fructose-bisphosphatase inhibitor , Pyruvate dehydrogenase inhibitor, hepatic gluconeogenesis inhibitor, D-chiro-inositol, glycogen synthase kinase-3 inhibitor, glucagon-like peptide 1, glucagon-like peptide-1 analog, glucagon-like peptide-1ago-2 Strike, amylin, amylin analogs, amylin agonists
- insulin sensitivity enhancers In the treatment of obesity, insulin sensitivity enhancers, glucose absorption inhibitors, biguanides, insulin secretagogues, SGLT2 activity inhibitors, insulin or insulin analogs, glucagon receptor antagonists, insulin receptor kinases Stimulant, tripeptidyl peptidase II inhibitor, dipeptidyl peptidase IV inhibitor, protein tyrosine phosphatase-1 ⁇ inhibitor, glycogen phosphorylase inhibitor, glucose-16-phosphatase inhibitor, fructose Bisphosphatase inhibitor, pyruvate dehydrogenase inhibitor, hepatic gluconeogenesis inhibitor, D-potency iono-inositol, glycogen synthase quina Over Ze - 3 inhibitor, glucagon-like base Puchido 1, glucagon-like base Puchido 1 analog • body, glucagon-like base peptide - 1 Agonisuto, amylin, Amirin analogs, Ami Rina
- the pharmaceutical composition of the present invention When the pharmaceutical composition of the present invention is used for actual treatment, various dosage forms are used depending on the usage. Such dosage forms include, for example, powders, granules, fine granules, dry syrups, tablets, capsules, injections, solutions, ointments, suppositories, patches, and the like. It is administered orally.
- the pharmaceutical composition of the present invention includes a sustained-release preparation containing a preparation adhering to the gastrointestinal mucosa (for example, International Publication No. WO99 / 10010 pamphlet, International Publication No. WO99 / No. 26606 pamphlet and Japanese Patent Application Laid-Open No. 2000-25767) are also included.
- compositions can be prepared by appropriate excipients, disintegrants, binders, lubricants, diluents, buffers, isotonic agents, preservatives, It can be produced by appropriately mixing, diluting and dissolving with pharmaceutical additives such as wetting agents, emulsifiers, dispersants, stabilizers, and solubilizing agents, and dispensing according to a conventional method.
- pharmaceutical additives such as wetting agents, emulsifiers, dispersants, stabilizers, and solubilizing agents, and dispensing according to a conventional method.
- they can be produced by formulating each active ingredient simultaneously or separately in the same manner as described above.
- the pharmaceutical composition of the present invention When the pharmaceutical composition of the present invention is used for actual treatment, administration of the compound represented by the above general formula (I), a pharmacologically acceptable salt thereof, or a prodrug thereof as an active ingredient thereof is performed.
- the dose is determined according to the patient's age, gender, body weight, disease and degree of treatment, etc.
- Adults can be administered in a dose of about 0.01 to 30 mg per day, in single or divided doses.
- the dose of the compound of the present invention can be reduced according to the dose of the other drug.
- the title compound was obtained in the same manner as in Reference Example 2, except that o-tolualdehyde was used instead of tolualdehyde. .
- This was dissolved in methanol (5 mL), sodium methoxide (0.043 mL of a 28% methanol solution) was added, and the mixture was stirred at room temperature for 30 minutes.
- reaction mixture was acidified with concentrated hydrochloric acid, and the precipitated crystals were collected by filtration, washed with water, dried under reduced pressure, and dried under reduced pressure to give 2 ', 4, dibenzyloxy 6'-hydroxychalcone (4.85) g) was obtained.
- This was dissolved in iV, iV-dimethylformamide (4 OmL) -acetone (12 mL), and potassium carbonate (2.3 g) and methyl bromoacetate (1.1 mL) were added, followed by stirring at room temperature for 8 hours.
- Reaction mixture The mixture was poured into water and extracted with Jetil ether. The extract was washed with water, dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure.
- Benzyltriphenylphosphonium chloride (0.47 g) was added to a suspension of sodium hydride (60%, 48 mg) in dimethyl sulfoxide (3 mL), and the mixture was stirred at 65 ° C for 1 hour.
- the reaction mixture was ice-cooled, 4-benzyloxy-3-formylindole (0.25 g) was added, and the mixture was stirred at 85 ° C for 3 hours. Reaction mixing After the product was cooled to room temperature, water was added, and the mixture was extracted three times with ethyl acetate.
- the extract was washed successively with water (twice), a saturated aqueous solution of sodium hydrogencarbonate and a saturated saline solution, dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure.
- the extract was washed successively with water (twice) and brine, dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure.
- the residue was dissolved in methylene chloride (8 mL) and 2,3,4,6-tetra-O-acetyl- 1-trichloroacetimidoyl- ⁇ -D-dalcopyranose (0.42 g) and boron trifluoride. Getyl ether complex (0.1 mL) was added, and the mixture was stirred at room temperature for 30 minutes.
- the title compound was obtained in the same manner as in Example 21 except for using 11- (2-hydroxyethyl) pidazine instead of 2-aminoenol.
- the reaction mixture was poured into water and extracted with ethyl acetate.
- the organic layer was washed sequentially with water, a saturated aqueous solution of sodium hydrogen carbonate, water and saturated saline, dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure.
- the residue was dissolved in methylene chloride (5 mL), and 2,3,4,6-tetra-O-acetyl- 1-0-trichloroacetimidoyl- ⁇ -D-dalcoviranose (0.12 g) was added.
- boron trifluoride-getyl ether complex (0.032 mL) was added under ice cooling.
- the extract was washed with water and a saturated saline solution, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure.
- the residue was dissolved in methanol (10 mL), sodium methoxide (28% methanol solution 5 mL) was added, and the mixture was stirred at room temperature for 1 hour.
- methanol 10 mL
- sodium methoxide (28% methanol solution 5 mL) was added, and the mixture was stirred at room temperature for 1 hour.
- 1 mo 1 / L hydrochloric acid (30 mL) and ethyl acetate were added to the residue, and the mixture was stirred for 1 hour.
- the reaction mixture was poured into a saturated aqueous solution of sodium hydrogencarbonate and extracted with ethyl acetate.
- the extract was washed with saturated saline, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure.
- the residue was treated with methylene chloride-methanol, and the precipitated crystals were collected by filtration, washed with methylene chloride, and dried under reduced pressure to obtain the title compound (0.86 g).
- the extract was washed with saturated saline, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure.
- the residue was treated with 3-hexane-ethyl acetate, and the precipitated crystals were collected by filtration and dried under reduced pressure to obtain the title compound (0.51 g).
- Tris (triphenylphosphine) rhodium (I) was added to a suspension of the obtained 4-bromo-2 '-(potoxymethoxy) -6'-hydroxychalcone (3.7 g) in benzene (15 OmL). ) Chloride (1.82 g) and triethylsilane (6.2 mL) were added, and the mixture was stirred at 70 ° C overnight. To the reaction mixture was added 2 m 01 ZL aqueous sodium hydroxide solution and getyl ether, and the aqueous layer was separated. The aqueous layer was washed with getyl ether, made acidic by adding concentrated hydrochloric acid, and extracted with ethyl acetate.
- the reaction mixture was poured into water and extracted with ethyl acetate.
- the extract was washed with water and saturated saline, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure.
- the residue was dissolved in methanol (1 OmL), sodium methoxide (1.5 mL of a 28% methanol solution) was added, and the mixture was stirred at room temperature for 1 hour.
- the residue 1 mo 1 / L hydrochloric acid was added, and the mixture was extracted with ethyl acetate.
- the extract was washed with water and brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure.
- the reaction solution was added to the above resin and stirred at room temperature for 20 hours. Excess solvent was removed, and the resin was washed three times with methylene chloride, three times with N, iV-dimethylformamide, and three times with methylene chloride. The same washing operation was further repeated twice.
- the resulting resin was treated with a 2% solution of 1,8-diazabicyclo [5.4.0] indene-7-cene in V, iV-dimethylformamide at room temperature for 1 hour. After removing the solvent, the mixture was further treated with a 2% solution of 1,8-diazabicyclo [5.4.0] indene-7-cene in iV, iV-dimethylformamide for 30 minutes.
- the obtained resin was suspended in TV, V-dimethylformamide and stirred at room temperature for 30 minutes, after which excess solvent was removed.
- a solution of 6'-hydroxy-2 '-(tetrahydropyran-1-yloxy) acetophenone (2.03 g) in AT, V-dimethylformamide (35 mL) was added to the above resin, and potassium carbonate (2. 08 g) and stirred at room temperature for 20 hours. After removing the solvent, the plate was washed 5 times with a 50% aqueous solution of tetrahydrofuran, 3 times with methanol, 3 times with N, iV-dimethylformamide, and 3 times with methylene chloride. The obtained resin was dried under reduced pressure.
- the resin (0.70 g) obtained in step 1 was suspended in ethanol and allowed to stand at room temperature for 30 minutes, after which excess solvent was removed.
- Tris (triphenylphosphine) mouth Dimethyl (I) chloride 0.084 g) suspension in benzene (5 mL), benzene (2 mL) and triethylsilane (0.48 mL) are added to the above resin, and the mixture is added at 70 ° C. For 3 hours. After removing the solvent, the resultant was washed five times with methylene chloride, five times with N, iV-dimethylformamide, five times with methanol, and three times with N, iV-dimethylformamide. V, N-dimethylformamide was added to the obtained resin, and the mixture was stirred for 5 minutes, and excess solvent was removed.
- Example 40 The title compound was obtained in the same manner as in Example 40, except that .4-monofluorobenzaldehyde was used instead of 2-furaldehyde.
- the reaction mixture was cooled on ice, Water (0.6 mL), a 15% aqueous sodium hydroxide solution (0.6 mL) and water (1.8 mL) were sequentially added, and the mixture was stirred at room temperature for 10 minutes. After filtering off the insoluble matter, the filtrate was concentrated under reduced pressure. Sodium hydride (60%, 0.46 g) was added to a solution of the residue in N, —dimethylformamide (30 mL) under ice cooling, and the mixture was stirred for 10 minutes. Then, benzylbromide (0.99 mL) was added, and the mixture was stirred at room temperature overnight. . Ice water was added to the reaction mixture, and extracted with getyl ether.
- Acetic anhydride (9 mL) was added to a solution of orcinol (30 g) in pyridine (240 mL) at room temperature, and the mixture was stirred for 16 hours. After concentrating the reaction mixture under reduced pressure, the residue is dissolved in ethyl acetate, washed successively with ImolZL hydrochloric acid, water, a saturated aqueous solution of sodium hydrogencarbonate and brine, dried over anhydrous sodium sulfate, and the solvent is distilled off under reduced pressure. This gave orcinol diacetate (43.7 g).
- reaction mixture was cooled to room temperature, it was poured into water and extracted with ethyl acetate. The extract was washed successively with a saturated aqueous solution of sodium hydrogencarbonate (twice), water and saturated saline, dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure.
- Table 1 was prepared in the same manner as in Example 12 or Example 66 using the corresponding starting materials. Was obtained.
- the reaction mixture was poured into lmo 1ZL hydrochloric acid and extracted with ethyl acetate.
- the extract was washed successively with water, a saturated aqueous solution of sodium hydrogencarbonate and brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Molecular Biology (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Obesity (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Hospice & Palliative Care (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Description










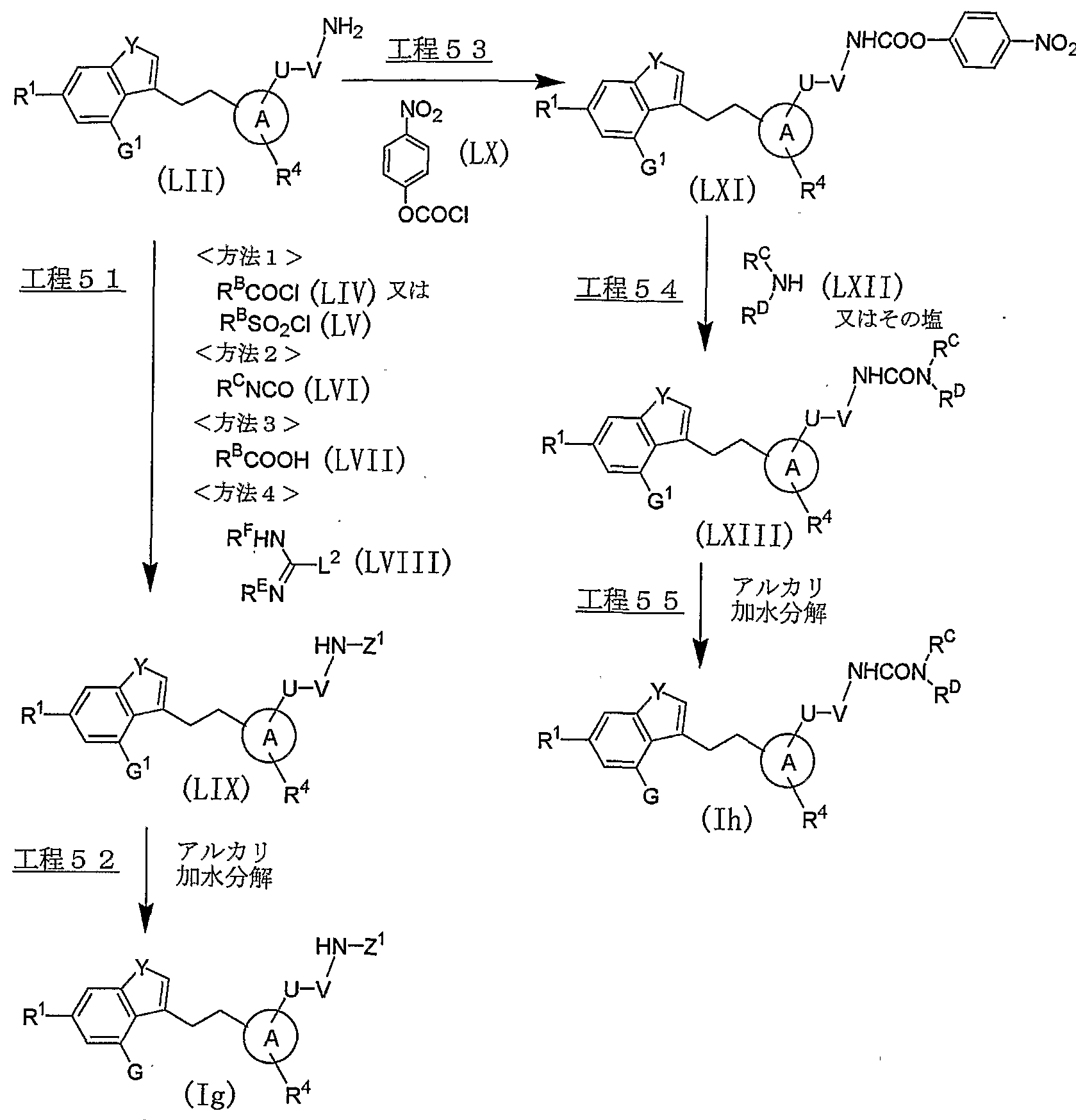









Claims



Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/551,648 US7566699B2 (en) | 2003-03-31 | 2004-03-24 | Fused heterocyclic derivative, medicinal composition containing the same, and medicinal use thereof |
| JP2005504177A JP4753716B2 (ja) | 2003-03-31 | 2004-03-24 | 縮合複素環誘導体、それを含有する医薬組成物およびその医薬用途 |
| ES04722965T ES2383892T3 (es) | 2003-03-31 | 2004-03-24 | Derivado heterocíclico condensado, composición medicinal que lo contiene y el uso medicinal del mismo |
| KR1020057018698A KR101170715B1 (ko) | 2003-03-31 | 2004-03-24 | 축합 복소환 유도체, 그것을 함유하는 의약조성물 및 그의약용도 |
| EP04722965A EP1609798B1 (en) | 2003-03-31 | 2004-03-24 | Fused heterocyclic derivative, medicinal composition containing the same, and medicinal use thereof |
| CA2520436A CA2520436C (en) | 2003-03-31 | 2004-03-24 | Fused heterocyclic derivative, medicinal composition containing the same, and medicinal use thereof |
| AT04722965T ATE557030T1 (de) | 2003-03-31 | 2004-03-24 | Kondensiertes heterocyclisches derivat, dieses enthaltende medizinische zusammensetzung und deren medizinische verwendung |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003-97152 | 2003-03-31 | ||
| JP2003097152A JP2004300102A (ja) | 2003-03-31 | 2003-03-31 | 縮合複素環誘導体、それを含有する医薬組成物およびその医薬用途 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004087727A1 true WO2004087727A1 (ja) | 2004-10-14 |
Family
ID=33127540
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2004/004009 WO2004087727A1 (ja) | 2003-03-31 | 2004-03-24 | 縮合複素環誘導体、それを含有する医薬組成物およびその医薬用途 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US7566699B2 (ja) |
| EP (1) | EP1609798B1 (ja) |
| JP (2) | JP2004300102A (ja) |
| KR (1) | KR101170715B1 (ja) |
| CN (1) | CN100418977C (ja) |
| AT (1) | ATE557030T1 (ja) |
| CA (1) | CA2520436C (ja) |
| ES (1) | ES2383892T3 (ja) |
| TW (1) | TWI377210B (ja) |
| WO (1) | WO2004087727A1 (ja) |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005085267A1 (ja) * | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | 含窒素縮合環誘導体、それを含有する医薬組成物およびその医薬用途 |
| US7084124B2 (en) | 2003-08-01 | 2006-08-01 | Janssen Pharmaceutica, N.V. | Substituted indazole-O-glucosides |
| US7094763B2 (en) | 2003-08-01 | 2006-08-22 | Janssen Pharaceutica, N.V. | Substituted fused heterocyclic C-glycosides |
| US7094764B2 (en) | 2003-08-01 | 2006-08-22 | Janssen Pharmaceutica N.V. | Substituted benzimidazole-, Benztriazole-, and benzimidazolone-O-glucosides |
| US7129220B2 (en) | 2003-08-01 | 2006-10-31 | Janssen Pharmaceutica N.V | Substituted indole-O-glucosides |
| WO2007128761A2 (de) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Verwendungen von dpp iv inhibitoren |
| US7732596B2 (en) | 2004-03-04 | 2010-06-08 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| US7767651B2 (en) | 2005-01-28 | 2010-08-03 | Chugai Seiyaku Kabushiki Kaisha | Spiroketal derivatives and use thereof as diabetic medicine |
| WO2011051864A1 (en) | 2009-11-02 | 2011-05-05 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| US7943582B2 (en) | 2006-12-04 | 2011-05-17 | Mitsubishi Tanabe Pharma Corporation | Crystalline form of 1-(β-D-glucopyransoyl)-4-methyl-3-[5-(4-fluorophenyl)-2- thienylmethyl]benzene hemihydrate |
| US7943788B2 (en) | 2003-08-01 | 2011-05-17 | Mitsubishi Tanabe Pharma Corporation | Glucopyranoside compound |
| WO2011070592A2 (en) | 2009-12-09 | 2011-06-16 | Panacea Biotec Ltd. | Novel sugar derivatives |
| US8772512B2 (en) | 2009-07-10 | 2014-07-08 | Janssen Pharmaceutica Nv | Crystallisation process for 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl] benzene |
| US8785403B2 (en) | 2003-08-01 | 2014-07-22 | Mitsubishi Tanabe Pharma Corporation | Glucopyranoside compound |
| US8853385B2 (en) | 2008-01-17 | 2014-10-07 | Mitsubishi Tanabe Pharma Corporation | Combination therapy comprising SGLT inhibitors and DPP4 inhibitors |
| US9024009B2 (en) | 2007-09-10 | 2015-05-05 | Janssen Pharmaceutica N.V. | Process for the preparation of compounds useful as inhibitors of SGLT |
| US9035044B2 (en) | 2011-05-09 | 2015-05-19 | Janssen Pharmaceutica Nv | L-proline and citric acid co-crystals of (2S, 3R, 4R, 5S,6R)-2-(3-((5-(4-fluorophenyl)thiopen-2-yl)methyl)4-methylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol |
| US9056850B2 (en) | 2008-10-17 | 2015-06-16 | Janssen Pharmaceutica N.V. | Process for the preparation of compounds useful as inhibitors of SGLT |
| US9174971B2 (en) | 2009-10-14 | 2015-11-03 | Janssen Pharmaceutica Nv | Process for the preparation of compounds useful as inhibitors of SGLT2 |
| US9573959B2 (en) | 2013-03-14 | 2017-02-21 | Msd International Gmbh | Methods for preparing SGLT2 inhibitors |
| US10544135B2 (en) | 2011-04-13 | 2020-01-28 | Janssen Pharmaceutica Nv | Process for the preparation of compounds useful as inhibitors of SGLT2 |
| CN110804079A (zh) * | 2019-11-05 | 2020-02-18 | 广东省农业科学院蚕业与农产品加工研究所 | 一种具有dppiv酶抑制活性的呋喃香豆素及其制备方法 |
| US10617668B2 (en) | 2010-05-11 | 2020-04-14 | Janssen Pharmaceutica Nv | Pharmaceutical formulations |
| US11207337B2 (en) | 2015-09-15 | 2021-12-28 | Janssen Pharmaceutica Nv | Co-therapy comprising canagliflozin and phentermine for the treatment of obesity and obesity related disorders |
| US11576894B2 (en) | 2009-07-08 | 2023-02-14 | Janssen Pharmaceutica Nv | Combination therapy for the treatment of diabetes |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI388345B (zh) * | 2005-06-27 | 2013-03-11 | Sankyo Co | 用於高血壓之預防或治療之包含血管緊張素ⅱ受體拮抗劑及鈣通道阻斷劑之固體劑型 |
| WO2007140191A2 (en) | 2006-05-23 | 2007-12-06 | Theracos, Inc. | Glucose transport inhibitors and methods of use |
| BRPI0809607A2 (pt) * | 2007-04-02 | 2014-09-30 | Theracos Inc | Composto, composição farmacêutica, combinação farmacêutica, e, método para tratar uma doença |
| WO2009026537A1 (en) * | 2007-08-23 | 2009-02-26 | Theracos, Inc. | Benzylbenzene derivatives and methods of use |
| GB0718023D0 (en) * | 2007-09-14 | 2007-10-24 | Univ London Pharmacy | Anti-bacterial agents |
| UA101004C2 (en) | 2007-12-13 | 2013-02-25 | Теракос, Инк. | Derivatives of benzylphenylcyclohexane and use thereof |
| BRPI0917196B8 (pt) | 2008-08-22 | 2021-05-25 | Theracos Sub Llc | forma cristalina de um composto sendo complexo de (2s,3r,4r,5s,6r)- 2- (4- cloro- 3- (4- (2- ciclopropoxietóxi) benzil)fenil)- 6- (hidroximetil) tetraidro- 2h- piran- 3,4,5- triol, bis(l-prolina) |
| CN102149717B (zh) | 2008-08-28 | 2014-05-14 | 辉瑞大药厂 | 二氧杂-双环[3.2.1]辛烷-2,3,4-三醇衍生物 |
| JP5637865B2 (ja) * | 2009-01-19 | 2014-12-10 | 国立大学法人富山大学 | 1,5−ag含有組成物 |
| WO2010128152A1 (en) * | 2009-05-07 | 2010-11-11 | Novartis Ag | Fused heterocyclic c-glycosides for the treatment of diabetes |
| WO2011153712A1 (en) | 2010-06-12 | 2011-12-15 | Theracos, Inc. | Crystalline form of benzylbenzene sglt2 inhibitor |
| US9562029B2 (en) | 2011-06-25 | 2017-02-07 | Xuanzhu Pharma Co., Ltd. | C-glycoside derivatives |
| US9193751B2 (en) | 2012-04-10 | 2015-11-24 | Theracos, Inc. | Process for the preparation of benzylbenzene SGLT2 inhibitors |
| US10011627B2 (en) * | 2013-09-09 | 2018-07-03 | Youngene Therapeutics Co., Ltd | C-aryl glucoside derivative, preparation methods thereof, and medical applications thereof |
| CN105611920B (zh) | 2013-10-12 | 2021-07-16 | 泰拉科斯萨普有限责任公司 | 羟基-二苯甲烷衍生物的制备 |
| US9315438B2 (en) | 2014-01-03 | 2016-04-19 | Xuanzhu Pharma Co., Ltd | Optically pure benzyl-4-chlorophenyl-C-glucoside derivative |
| GB201421010D0 (en) * | 2014-11-26 | 2015-01-07 | N Gene Res Lab Inc | Method |
| JP6447557B2 (ja) * | 2016-03-24 | 2019-01-09 | 日亜化学工業株式会社 | 発光装置の製造方法 |
| CN110590722B (zh) * | 2019-10-22 | 2022-11-04 | 温州大学 | 2-三氟甲基苯并呋喃衍生物的合成方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001074834A1 (en) * | 2000-03-30 | 2001-10-11 | Bristol-Myers Squibb Company | O-aryl glucoside sglt2 inhibitors and method |
| WO2002064606A1 (fr) * | 2001-02-14 | 2002-08-22 | Kissei Pharmaceutical Co., Ltd. | Derives glucopyranosyloxybenzylbenzene et leur utilisation medicale |
| JP2003511458A (ja) * | 1999-10-12 | 2003-03-25 | ブリストル−マイヤーズ スクイブ カンパニー | C−アリールグルコシドsglt2抑制剤および方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6515117B2 (en) | 1999-10-12 | 2003-02-04 | Bristol-Myers Squibb Company | C-aryl glucoside SGLT2 inhibitors and method |
| US6600015B2 (en) * | 2000-04-04 | 2003-07-29 | Hoffmann-La Roche Inc. | Selective linear peptides with melanocortin-4 receptor (MC4-R) agonist activity |
| EP1369422B1 (en) * | 2001-02-14 | 2008-07-23 | Japan Science and Technology Agency | Process for preparation of alkenylphosphine oxides or alkenylphosphinic acid esters |
| WO2002080936A1 (en) * | 2001-04-04 | 2002-10-17 | Ortho Mcneil Pharmaceutical, Inc. | Combination therapy comprising glucose reabsorption inhibitors and ppar modulators |
| WO2002088157A1 (fr) * | 2001-04-27 | 2002-11-07 | Ajinomoto Co., Inc. | Derives pyrazolyl-o-glycoside n-substitues et medicament contre le diabete en contenant |
| EP1400529A4 (en) * | 2001-05-30 | 2007-12-19 | Kissei Pharmaceutical | GLUCOPYRANOSYLOXYPYRAZOLE DERIVATIVE, MEDICINAL COMPOSITION CONTAINING THE SAME, AND MEDICINAL AND INTERMEDIARY USE THEREOF |
| WO2003011880A1 (fr) * | 2001-07-31 | 2003-02-13 | Kissei Pharmaceutical Co., Ltd. | Derive de glucopyranosyloxybenzylbenzene, composition medicinale contenant ce derive, usage medicinal de cette composition et produit intermediaire pour produire cette composition |
| EP1680131A4 (en) * | 2003-08-01 | 2009-05-27 | Janssen Pharmaceutica Nv | SUBSTITUTED INDOLE-O-GLUCOSIDES |
-
2003
- 2003-03-31 JP JP2003097152A patent/JP2004300102A/ja active Pending
-
2004
- 2004-03-24 EP EP04722965A patent/EP1609798B1/en not_active Expired - Lifetime
- 2004-03-24 WO PCT/JP2004/004009 patent/WO2004087727A1/ja active Application Filing
- 2004-03-24 KR KR1020057018698A patent/KR101170715B1/ko not_active IP Right Cessation
- 2004-03-24 ES ES04722965T patent/ES2383892T3/es not_active Expired - Lifetime
- 2004-03-24 CA CA2520436A patent/CA2520436C/en not_active Expired - Fee Related
- 2004-03-24 JP JP2005504177A patent/JP4753716B2/ja not_active Expired - Fee Related
- 2004-03-24 US US10/551,648 patent/US7566699B2/en not_active Expired - Fee Related
- 2004-03-24 CN CNB2004800122959A patent/CN100418977C/zh not_active Expired - Fee Related
- 2004-03-24 AT AT04722965T patent/ATE557030T1/de active
- 2004-03-31 TW TW093108839A patent/TWI377210B/zh not_active IP Right Cessation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003511458A (ja) * | 1999-10-12 | 2003-03-25 | ブリストル−マイヤーズ スクイブ カンパニー | C−アリールグルコシドsglt2抑制剤および方法 |
| WO2001074834A1 (en) * | 2000-03-30 | 2001-10-11 | Bristol-Myers Squibb Company | O-aryl glucoside sglt2 inhibitors and method |
| WO2002064606A1 (fr) * | 2001-02-14 | 2002-08-22 | Kissei Pharmaceutical Co., Ltd. | Derives glucopyranosyloxybenzylbenzene et leur utilisation medicale |
Cited By (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7820630B2 (en) | 2003-08-01 | 2010-10-26 | Janssen Pharmaceutica Nv | Substituted indole-O-glucosides |
| US7943788B2 (en) | 2003-08-01 | 2011-05-17 | Mitsubishi Tanabe Pharma Corporation | Glucopyranoside compound |
| US7094763B2 (en) | 2003-08-01 | 2006-08-22 | Janssen Pharaceutica, N.V. | Substituted fused heterocyclic C-glycosides |
| US7094764B2 (en) | 2003-08-01 | 2006-08-22 | Janssen Pharmaceutica N.V. | Substituted benzimidazole-, Benztriazole-, and benzimidazolone-O-glucosides |
| US7129220B2 (en) | 2003-08-01 | 2006-10-31 | Janssen Pharmaceutica N.V | Substituted indole-O-glucosides |
| US7816331B2 (en) | 2003-08-01 | 2010-10-19 | Janssen Pharmaceutica Nv | Substituted indazole-O-glucosides |
| US7816330B2 (en) | 2003-08-01 | 2010-10-19 | Janssen Pharmaceutica Nv | Substituted benzimidazole-, benztriazole-, and benzimidazolone-O-glucosides |
| US7482330B2 (en) | 2003-08-01 | 2009-01-27 | Janssen Pharmaceutica N.V. | Substituted fused heterocyclic C-glycosides |
| US7511022B2 (en) | 2003-08-01 | 2009-03-31 | Janssen Pharmaceutica N.V. | Substituted indole-O-glucosides |
| US7511020B2 (en) | 2003-08-01 | 2009-03-31 | Janssen Pharmaceutica N.V. | Substituted benzimidazole-, benztriazole-, and benzimidazolone-O-glucosides |
| US7511021B2 (en) | 2003-08-01 | 2009-03-31 | Janssen Pharmaceutica N.V. | Substituted indazole-O-glucosides |
| US8785403B2 (en) | 2003-08-01 | 2014-07-22 | Mitsubishi Tanabe Pharma Corporation | Glucopyranoside compound |
| US8222219B2 (en) | 2003-08-01 | 2012-07-17 | Mitsubishi Tanabe Pharma Corporation | Glucopyranoside compound |
| US7816328B2 (en) | 2003-08-01 | 2010-10-19 | Janssen Pharmaceutica Nv | Substituted fused heterocyclic C-glycosides |
| US8202984B2 (en) | 2003-08-01 | 2012-06-19 | Mitsubishi Tanabe Pharma Corporation | Glucopyranoside compound |
| US7084124B2 (en) | 2003-08-01 | 2006-08-01 | Janssen Pharmaceutica, N.V. | Substituted indazole-O-glucosides |
| US7998975B2 (en) | 2004-03-04 | 2011-08-16 | Kissei Pharmaceutical Co., Ltd. | Nitrogenous fused-ring derivatives, medicinal compositions containing the derivatives, and use thereof as drugs |
| US7732596B2 (en) | 2004-03-04 | 2010-06-08 | Kissei Pharmaceutical Co., Ltd. | Fused heterocycle derivative, medicinal composition containing the same, and medicinal use thereof |
| US7375113B2 (en) | 2004-03-04 | 2008-05-20 | Kissei Pharmaceutical Co., Ltd. | Nitrogenous fused-ring derivatives, medicinal compositions containing the derivatives, and use thereof as drugs |
| WO2005085267A1 (ja) * | 2004-03-04 | 2005-09-15 | Kissei Pharmaceutical Co., Ltd. | 含窒素縮合環誘導体、それを含有する医薬組成物およびその医薬用途 |
| US7767651B2 (en) | 2005-01-28 | 2010-08-03 | Chugai Seiyaku Kabushiki Kaisha | Spiroketal derivatives and use thereof as diabetic medicine |
| EP2351568A2 (de) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Verwendungen von dpp iv Inhibitoren |
| WO2007128761A2 (de) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Verwendungen von dpp iv inhibitoren |
| US7943582B2 (en) | 2006-12-04 | 2011-05-17 | Mitsubishi Tanabe Pharma Corporation | Crystalline form of 1-(β-D-glucopyransoyl)-4-methyl-3-[5-(4-fluorophenyl)-2- thienylmethyl]benzene hemihydrate |
| US8513202B2 (en) | 2006-12-04 | 2013-08-20 | Mitsubishi Tanabe Pharma Corporation | Crystalline form of 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl]benzene hemihydrate |
| US9024009B2 (en) | 2007-09-10 | 2015-05-05 | Janssen Pharmaceutica N.V. | Process for the preparation of compounds useful as inhibitors of SGLT |
| US8853385B2 (en) | 2008-01-17 | 2014-10-07 | Mitsubishi Tanabe Pharma Corporation | Combination therapy comprising SGLT inhibitors and DPP4 inhibitors |
| US9056850B2 (en) | 2008-10-17 | 2015-06-16 | Janssen Pharmaceutica N.V. | Process for the preparation of compounds useful as inhibitors of SGLT |
| US11576894B2 (en) | 2009-07-08 | 2023-02-14 | Janssen Pharmaceutica Nv | Combination therapy for the treatment of diabetes |
| US8772512B2 (en) | 2009-07-10 | 2014-07-08 | Janssen Pharmaceutica Nv | Crystallisation process for 1-(β-D-glucopyranosyl)-4-methyl-3-[5-(4-fluorophenyl)-2-thienylmethyl] benzene |
| US9174971B2 (en) | 2009-10-14 | 2015-11-03 | Janssen Pharmaceutica Nv | Process for the preparation of compounds useful as inhibitors of SGLT2 |
| WO2011051864A1 (en) | 2009-11-02 | 2011-05-05 | Pfizer Inc. | Dioxa-bicyclo[3.2.1]octane-2,3,4-triol derivatives |
| WO2011070592A2 (en) | 2009-12-09 | 2011-06-16 | Panacea Biotec Ltd. | Novel sugar derivatives |
| US10617668B2 (en) | 2010-05-11 | 2020-04-14 | Janssen Pharmaceutica Nv | Pharmaceutical formulations |
| US10544135B2 (en) | 2011-04-13 | 2020-01-28 | Janssen Pharmaceutica Nv | Process for the preparation of compounds useful as inhibitors of SGLT2 |
| US9035044B2 (en) | 2011-05-09 | 2015-05-19 | Janssen Pharmaceutica Nv | L-proline and citric acid co-crystals of (2S, 3R, 4R, 5S,6R)-2-(3-((5-(4-fluorophenyl)thiopen-2-yl)methyl)4-methylphenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol |
| US9573959B2 (en) | 2013-03-14 | 2017-02-21 | Msd International Gmbh | Methods for preparing SGLT2 inhibitors |
| EP3466431A1 (en) | 2013-03-14 | 2019-04-10 | MSD International GmbH | Crystalline forms and methods for preparing sglt2 inhibitors |
| US11207337B2 (en) | 2015-09-15 | 2021-12-28 | Janssen Pharmaceutica Nv | Co-therapy comprising canagliflozin and phentermine for the treatment of obesity and obesity related disorders |
| CN110804079A (zh) * | 2019-11-05 | 2020-02-18 | 广东省农业科学院蚕业与农产品加工研究所 | 一种具有dppiv酶抑制活性的呋喃香豆素及其制备方法 |
| CN110804079B (zh) * | 2019-11-05 | 2021-02-12 | 广东省农业科学院蚕业与农产品加工研究所 | 一种具有dppiv酶抑制活性的呋喃香豆素及其制备方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1609798A4 (en) | 2009-06-17 |
| KR20050119179A (ko) | 2005-12-20 |
| KR101170715B1 (ko) | 2012-08-03 |
| ATE557030T1 (de) | 2012-05-15 |
| CA2520436C (en) | 2012-01-24 |
| CN1784415A (zh) | 2006-06-07 |
| JPWO2004087727A1 (ja) | 2006-06-29 |
| JP4753716B2 (ja) | 2011-08-24 |
| CN100418977C (zh) | 2008-09-17 |
| TW200510442A (en) | 2005-03-16 |
| EP1609798A1 (en) | 2005-12-28 |
| ES2383892T3 (es) | 2012-06-27 |
| EP1609798B1 (en) | 2012-05-09 |
| CA2520436A1 (en) | 2004-10-14 |
| US20060247179A1 (en) | 2006-11-02 |
| TWI377210B (en) | 2012-11-21 |
| US7566699B2 (en) | 2009-07-28 |
| JP2004300102A (ja) | 2004-10-28 |
| EP1609798A9 (en) | 2012-01-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2004087727A1 (ja) | 縮合複素環誘導体、それを含有する医薬組成物およびその医薬用途 | |
| JP4902348B2 (ja) | 含窒素縮合環誘導体、それを含有する医薬組成物およびその医薬用途 | |
| TWI298071B (ja) | ||
| JP4540475B2 (ja) | ピラゾール誘導体、それを含有する医薬組成物、その医薬用途及びその製造中間体 | |
| JP4950657B2 (ja) | 縮合複素環誘導体、それを含有する医薬組成物およびその医薬用途 | |
| JP4708187B2 (ja) | ピラゾール誘導体、それを含有する医薬組成物及びその製造中間体 | |
| JP4651934B2 (ja) | ベンジルフェノール誘導体、それを含有する医薬組成物およびその医薬用途 | |
| JP5066512B2 (ja) | ピラゾール誘導体の製造方法およびその製造用中間体 | |
| WO2005085265A1 (ja) | 縮合ヘテロ環誘導体、それを含有する医薬組成物およびその医薬用途 | |
| JPWO2003011880A1 (ja) | グルコピラノシルオキシベンジルベンゼン誘導体、それを含有する医薬組成物、その医薬用途及びその製造中間体 | |
| WO2004019958A1 (ja) | ピラゾール誘導体、それを含有する医薬組成物及びその医薬用途 | |
| WO2002053573A1 (fr) | Derives de glucopyranosiloxypyrazole et leur utilisation dans des medicaments | |
| WO2005095429A1 (ja) | フェノール誘導体、それを含有する医薬組成物及びその医薬用途 | |
| JPWO2002068439A1 (ja) | グルコピラノシルオキシピラゾール誘導体及びその医薬用途 | |
| WO2004058790A1 (ja) | 含窒素複素環誘導体、それを含有する医薬組成物およびその医薬用途 | |
| WO2006054629A1 (ja) | 1-置換-3-(β-D-グリコピラノシル)含窒素ヘテロ環化合物、及びそれを含有する医薬 | |
| JPWO2005095372A1 (ja) | ナフタレン誘導体、それを含有する医薬組成物及びその医薬用途 | |
| WO2005095373A1 (ja) | ナフタレン誘導体、それを含有する医薬組成物およびその医薬用途 | |
| MXPA06009899A (en) | Nitrogenous fused-ring derivatives, medicinal compositions containing the derivatives, and use thereof as drugs |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005504177 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2520436 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006247179 Country of ref document: US Ref document number: 10551648 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020057018698 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4473/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004722965 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20048122959 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057018698 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004722965 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10551648 Country of ref document: US |