Process for the manufacture of a crosslinked. transparent, hydrophilic and photochromic polymeric material, and optical and ophthalmic article obtained.
The invention generally relates to a process for the manufacture of crosslinked, transparent, hydrophilic and photochromic polymeric materials and to the use of the latter in the manufacture of optical and ophthalmic articles having photochromic properties and in particular contact lenses.
It is known to manufacture photochromic contact lenses by incorporation of a photochromic compound, such as a spirooxazine compound, in the mixture of polymerizable monomers resulting, after polymerization in a mould, in the final contact lens.
Thus, Patent Application WO 96/04590 discloses the manufacture of photochromic contact lenses made of methyl methacrylate (MMA) and N- vinylpyrrolidone (NVP) copolymer, which consists in introducing a spirooxazine compound into a mixture of MMA and NVP monomers, in the presence of alkyl methacrylate as crosslinking agent and of azobisisobutyronitrile (AIBN) as thermal initiator, and in subsequently thermally polymerizing the mixture.
This technique, commonly known as "cast in place", is also used in Patent EP-A-277,639, which discloses the incorporation, in a base monomeric mixture (in particular, based on hydroxyethyl methacrylate (HEMA), of a spirooxazine compound comprising an organic functional group which can be polymerized by addition or by ring opening. The mixture is subsequently polymerized. The photochromic compound, thus fixed within the material constituting the lens, is assumed not to be eluted in the lacrymal medium. This "cast in place" process for the photochromic compounds exhibits numerous disadvantages.
Photochromic compounds are compounds which are sensitive to the action of the free radicals formed during the polymerization of mixtures of monomers by activation of the initiator. Under the effect of these free radicals, photochromic compounds, in particular spirooxazine compounds, are liable to decompose, generating coloured by-products. The result of this is. on the one hand, an overall decrease in the effectiveness of the photochromic compound, a portion of which has been destroyed, and, on the other hand, a permanent colouring brought about in the lens by these coloured by-products, which is not desired. In addition, a significant disadvantage is that the by-products can exhibit a toxic nature, while, because of their low molar masses ; they can diffuse through the
lens towards the eye of the wearer.
Thus, while this "cast in place" technique could be used with some success in the manufacture of ophthalmic glasses intended for spectacles, this does not apply in the production of contact lenses and, to the knowledge of the inventors, no photochromic hydrophilic contact lens has been marketed to date.
The possibly toxic nature of the by-products formed during the polymerization renders this "cast in place" technique virtually unusable in the case of contact lenses.
The fact that, in the "cast in place" process for the photochromic compound of Patent EP-A-277,639, the latter is fixed to the polymeric network constituting the contact lens is invalid with regard to the decomposition by-products.
Furthermore, the technique of Patent EP-A-277,639 limits the possible choice of the photochromic compounds which can be used.
The "cast in place" technique lacks flexibility at the industrial level insofar as it is impossible to render photochromic already existing lenses, which necessitates disposing of large stocks of photochromic contact lenses.
Finally, the "cast in place" technique does not make it possible to incorporate the photochromic compound in predetermined chosen regions of the lens. In particular, hydrophilic contact lenses exhibit a diameter greater than that of the iris and it may be aesthetically advantageous to render photochromic just the central part of the contact lens covering the pupillary region of the eye of the wearer.
The object of the invention is therefore to provide a process for the production of crosslinked, transparent, hydrophilic and photochromic polymeric materials which solves the above problems, materials which make it possible to prepare photochromic contact lenses which are preferably resistant to the sterilization treatments, in particular thermal sterilization treatments, conventionally used. According to the invention, the process for producing a crosslinked, transparent, hydrophilic and photochromic polymeric material comprises :
(a) dissolving a photochromic agent in a solvent or mixture of solvents capable of at least partially dissolving the photochromic agent, in order to obtain a photochromic impregnating solution ; (b) impregnating a crosslinked, transparent and hydrophilic polymeric material with the photochromic impregnating solution, in order to obtain a material
impregnated with photochromic solution ;
(c) rinsing the impregnated material with an aqueous solution, in order to replace, with the aqueous solution, the solvent impregnated in the material ; and
(d) recovering the crosslinked, transparent, hydrophilic and photochromic polymeric material.
The process of the invention applies to any type of crosslinked, transparent and hydrophilic polymeric material suitable for the manufacture of contact lenses.
In the context of the present invention, the term "hydrophilic material" is understood to mean any material having a degree of hydrophilicity of greater than or equal to 10 % and preferably of greater than or equal to 35 %.
Particularly preferred materials are those having a degree of hydrophilicity of 50 % or more.
As is conventional, the term "degree of hydrophilicity", or water content, is understood to mean the maximum percentage, by weight, of water which a polymeric material can fix.
The hydrophilic polymeric materials are generally obtained by polymerization, preferably in the presence of a crosslinking agent, of at least one of the following monomers : hydroxyalkyl (meth)acrylates, alkoxy derivatives of hydroxyalkyl (meth)acrylates, aminoalkyl (meth)acrylates, monovinyl ethers, monovinyl polyethers, hydroxylated vinyl ethers, N-vinyllactams, amido derivatives of
(meth)acrylic compounds, ionic monomers, zwitterionic monomers, oligomers of the above mentioned monomers and their mixtures.
The recommended hydroxyalkyl (meth)acrylates are those in which the alkyl group generally comprises from 1 to 4 carbon atoms.
Specific examples of hydroxyalkyl (meth)acrylate are 2 -hydroxyethyl methacrylate (HEMA), hydroxypropyl acrylate, hydroxypropyl (meth)acrylate and 2,3-dihydroxypropyl methacrylate (glyceryl methacrylate).
The recommended alkoxy derivatives of hydroxyalkyl (mefh)acrylates are the mono-, di- or triethoxylated compounds having an alkyl group generally comprising from 1 to 4 carbon atoms.
Mention may be made, among the monomers of the N-vinyllactam type, of N-vinyl-2-pyrrolidone (NVP), N-vinyl-2-piperidone and N-vinylcaprolactam.
Mention may be made, among the amido derivatives of (meth)acrylic compounds which are of use, of (meth)acrylamide, N-methyl(meth)acrylamide, N- isopropyl(meth)acrylamide. N-diacetone-(meth)acrylamide. N.N-
dimethyl(meth)acrylamide, N,N-dimethylamino-methyl(meth)acrylamide, N,N- dimethylaminoethyl(meth)acrylamide and N- methylaminoisopropyl(meth)acrylamide. Mention may be made, among the aminoalkyl(meth)acrylates, of aminoethyl(meth)acrylate, dimethyl-aminoethyl methacrylate, methylaminoethyl methacrylate and diethylaminoethyl methacrylate.
Mention may be made, among the hydrophilic ionic monomers, of (meth)acrylic acid, as well as of cationic monomers, such as quaternary ammonium derivatives of (meth)acrylic acid.
Mention may be made, among the zwitterionic monomers, of those disclosed in Patent Application WO 92/07885. The latter monomers generally make it possible to reduce the affinity of the hydrogel with respect to the proteins in the lacrymal medium.
Examples of conventional crosslinking agents are ethylene glycol di(meth)acrylate, polyethylene glycol di(meth)acrylate, such as diethylene glycol di(meth)acrylate or triethylene glycol di(meth)acrylate, long-chain di(meth)acrylates, such as hexamethylene di(meth)acrylate, vinyl (meth)acrylate, allyl (meth)acrylate, divinylbenzene, diallyl phthalate and trimethylolpropane trimethacrylate.
These crosslinking agents are generally present in a concentration of 0.1 to 2 % by weight in the starting monomeric mixture resulting in the hydrophilic polymers which are used in the context of the invention.
The preferred hydrophilic polymeric materials are those obtained from vinyllactam (including N-vinylpyrrolidone) and/or from N,N-dimethylacrylamide.
A preferred class of polymeric materials is composed of multiphase polymeric materials, in particular two-phase polymeric materials. Multiphase polymeric materials are materials in which the major part of the polymeric network is formed of distinctive microdomains (with a size generally of 0.005 to 0.25 μm) of two or more different materials.
Such networks are generally obtained by copolymerization of a mixture of monomers exhibiting reactive functional groups of different natures, for example by copolymerization of a (meth)acrylic or (meth)acrylamide monomer with an allylic or vinyl monomer.
The multiphase polymeric material is preferably formed by polymerization of a monomeric mixture comprising at least one hydrophobic monomer and one hydrophilic monomer.
The preferred hydrophobic monomers are CΪ-C^ alkyl (meth)acrylates, in
particular methyl (meth)acrylate, and their fluorinated or silicone derivatives, ethoxyethyl methacrylate, cyclohexyl methacrylate, dimethyloxobutyl methacrylate, vinyl derivatives, such as styrene or divinylbenzene, hydrocarbon-comprising dienes, vinyl chloride and acrylonitrile. Mention may be made, among the hydrophilic monomers, of vinyl monomers, such as N-vinylpyrrolidone (NVP), hydroxyalkyl methacrylates, such as hydroxyethyl methacrylate (HEMA), and N,N-dimethylacrylamide.
Generally, hydrophilic monomers which do not comprise protic groups will preferably be chosen. Examples of two-phase hydrophilic polymeric materials which are preferred are methyl methacrylate (MMA)/N-vinylpyrrolidone (NVP) copolymers and methyl methacrylate (MMA)/N,N-dimethylacrylamide copolymers.
The preferred two-phase polymeric material is an MMA/NVP copolymer in a ratio by weight of approximately 30/70, which is the material constituting the Lunelle® lenses from the Company Essilor, or else the material constituting the
Rythmic® lenses from the company Essilor, which is an MMA/NVP copolymer in a ratio by weight of approximately 28/72.
This two-phase polymeric material is a hydrogel composed of MMA (hydrophobic) microdomains of approximately 0.02 μm, which are crosslinked by triethylene glycol dimethacrylate (TEGDMA), within a swollen NVP gel, crosslinked by triallyl isocyanurate (TAIC) bridges. Without wishing to be bound by any one theory, it is believed that, during the implementation of the process of the invention with two-phase polymeric materials as described above, the photochromic agent, overall hydrophobic in nature, is for the most part concentrated in the hydrophobic regions of the two-phase gel, in this case the PMMA regions, which will explain the stability and the effectiveness of the photochromic agent (in particular the good kinetics), even in an aqueous medium.
Besides, the inventors have found that specific hydrophilic materials, when treated according to the process of the invention, gave photochromic materials having very high transparency and very good photochromic performances.
These materials are obtained by polymerising a base composition comprising one or more hydrophilic monomer(s) and at least one (meth)acrylate monomer bearing an alicyclic hydrocarbon group, the alicyclic hydrocarbon group preferably bearing Cι-C
8 alkyl groups. More preferably, the at least one (meth)acrylate monomer bearing an alicyclic hydrocarbon group has the formula :
Each R2, one independently from each other, is a divalent alkylene radical chosen from the group consisting of -CH2-, -CHOH- and -CHR4- where R4 is a Ci-Cg alkyl group R3 is H or CH3 and wherein at least one of R2 radical is -CHR4- and n is 4, 5, 6 or 7.
Specific examples of such monomers are 4-t-butyl, 2- hydroxycyclohexylmethacrylate and 3 ,3 ,5 -trimethylcyclohexyl-methacrylate.
The hydrophilic monomer may be chosen between those mentioned previously.
Hydroxyalkyl(meth)acrylates, and particularly hydroxyethylmethacrylate, are preferred hydrophilic monomers.
It is most preferred that the hydrophilic monomer comprises (meth)acrylic acid and especially methacrylic acid. Preferably, the methacrylic acid is present in an amount of 0.2 to 10 % by weight of the base composition.
In a preferred embodiment, the base composition comprises 0.2 to 20 % by weight of the at least one (me h)acrylate monomer bearing an alicyclic hydrocarbon group. The invention is consequently also directed on new hydrophilic materials especially suited for being treated with the process of the invention.
The new hydrophilic materials are obtained by polymerising a base composition comprising (meth)acrylic acid - at least one radically polymerizable hydrophilic monomer different from
(meth)acrylic acid
at least one (meth)acrylate monomer bearing an alicyclic hydrocarbon group.
The base composition preferably comprises conventional crosslinking agents such as those mentioned previously. The hydrophilic polymers used in the context of the invention can comprise conventional additives, in particular UV absorbers, in proportions such that the photochromic effect is not affected.
In the process of the invention, the photochromic agent can be any photochromic compound or mixture of photochromic compounds conventionally employed in ophthalmic optics.
The preferred photochromic compounds are spirroxazine and chromene compounds.
Photochromic spirooxazine compounds are compounds which are well known in the art and are disclosed, inter alia, in Patents US-A-5, 139,707 and 5,1 14,621 (spiro(indoline-quinazolinoxazine) and spiro(indoline- benzothiazolooxazine)), EP-A-245,020 (spiro[indoline-[2,3']-benzoxazine]), JP-A- 03251587 (spiro[indoline-[2,3']-benzoxazine] substituted at the 6' position) and WO-96/04590 (spiro[indoline-[2,3']-benzoxazine] having a cyano or phenylsulphonyl group at the 6' position). Chromenes are also well known photochromic compounds. These compounds are disclosed, inter alia, in Patents US-A-5,066,818, WO 92/09593, EP- A-401,958, EP-A-562,915 and WO 93/17071.
The preferred chromenes are naphthopyrans. More preferably, the naphthopyrans carry a free hydroxyl group. Particularly preferred naphthopyrans are those comprising a structure selected from : a 2H-naphtho[l,2-b]pyran, a 3H-naphtho[2,l-b]pyran, an indeno[2, 1 -f]naphtho[ 1 ,2-b]pyran and having at least one substituent R represented by one of the following formulas : (V) -DEZ (IV) -DZ (III') -EZ (IV) -z wherein
D is -C- or -CH,-
O with the proviso that in formula (IF), D is -CH2- when Z is OH. E is represented by the following formula
-[(OC2H4)x(OC3H6)y(OC4H8)z]- wherein x, y, z are each a number between 0 and 50 with the sum x+y+z being between 1 and 50.
Z is OH or is a residue of an organic polyol having at least 2 hydroxyl groups.
The group, -(OC2H4)x-, represents poly(ethylene oxide) ; -(OC3H6)y-, represents poly(propylene oxide) ; and, -(OC H8)z-, represents poly(butylene oxide). When used in combination, the poly(ethylene oxide), poly(propylene oxide) and poly(butylene oxide) groups of R may be in a random or block order within the R moiety. The letters x, y and z are each a number between 0 and 50 and the sum of x, y and z is between 1 and 50. The sum of x, y and z may be any number that falls within the range or 1 to 50, e.g., 1, 2, 3... 50. The sum may also range from any lower number to any higher number within the range of 1 to 50, e.g., 6 to 50, 31 to 50. The numbers for x, y, and z are average values and can be partial numbers, e.g., 9.5.
Preferred 2H-naphto[l,2-b]pyran structures have the following formula :
5 4
wherein
B and B' are aryl groups, preferably phenyl, substituted or not.
Preferred 3H-naphfho[2,l-b]pyran structures have the following formula
wherein B and B' have the same meaning as above.
Preferred indeno[2,l-f]naphtho[l,2-b]pyran structures have the following formula
wherein B and B' have the same meanings as above.
The preferred naphthopyran corresponds to the formula of an indeno[2,l- f]naphtho[l,2-b]pyran and, in this case, the free hydroxyl group is attached to the indeno group at the 13 position. Finally, the preferred naphthopyrans comprise two phenyl groups at the 3 position of the pyran group. Each of naphthopyrans comprising one of the 3 naphthopyran structures I",
II", III" as defined above comprises one or several substituants R.
The one or several substituent(s) R may be on the naphthopyran structure or on the B and/or B' groups.
If one or several substituent(s) R are on the B and/or B' groups, the B and/or B' groups are preferably mono-R-substituted phenyls.
The number of R groups (including the mono-R-substituted phenyls) may be 2, 3, 4, 5 or a number equal to the total number of substituents possible on the naphthopyran.
When there is more than one R group or mono-R-substituted phenyl on the naphthopyran, the R groups may be the same or different, e.g., there may be two different groups selected from formulas F to IV.
Preferably there is only one substituent R on each naphthopyran. Most preferred are the naphthopyrans wherein R represents -EZ or -Z, and wherein y=z=0 in formula E.
Preferably the organic polyol used to form the residue -Z has the formula G(OH)k wherein G is the backbone of the polyol and k is at least 2. Preferably, k is an integer from 2 to 5 and most preferably 4. Examples of organic polyols that may be used to form the residue -Z include polyols having at least 3 hydroxy groups such as (a) low molecular weight polyols, i.e., polyols having an average molecular weight less than 500, e.g, aliphatic triols, such as C2-Cι0 aliphatic triols, polyhydric alcohols and alkoxylated low molecular weight polyols ; (b) polyester polyols ; (c) polyether polyols ; (d) amide-containing polyols ; (e) epoxy polyols ; (f) polyhydric polyvinyl alcohols ; (g) urethane polyols ; (h) polyacrylic polyols ; (i) polycarbonate polyols ; and (j) mixtures of such polyols.
Preferably, the organic polyol is selected from low molecular weight polyols and extended polyols. Examples of low molecular weight organic polyols that can be used in the preparation of the hydroxylated photochromic compounds preferably used in the present invention include : tetramethylolmethane, i.e., pentaerythritol, dipentaerythritol, tripentaerythritol ; trimethylolethane ; trimethylolpropane ; ditrimethylolpropane ; 1,2,3-propanetriol, i.e., glycerol ; 2-(hydroxymethyl)-2- methyl- 1, 3 -propanediol ; 2-(hydroxymethyl)-2-ethyl-l,3-propanediol. Extended polyols are reaction products having therminal hydroxyl groups of the polyol and a suitable reactant. e.g., an alkylene oxide, or a lactone. Examples of such extended polyols include ε-caprolactone extended trimethylol methane and ethoxylated or propoxylated trimethylolpropane or pentaerythritol having a number average molecular weisht less than 500.
The residue -Z is formed by the reaction of one of the hydroxyl groups on the polyol with a precursor of group D, such as carboxylic acid or a methylene halide, a precursor of group E, such as polyalkyleneglycol or a hydroxyl group as a substituent of the naphthopyran structure, especially in naphthopyran structures of formula I", II" or III'.
The sheme of synthesis is given hereafter in a specific case wherein the naphthopyran is an indeno[2,l-fJnaphtho[l,2-b]pyran and the organic polyol is an ethoxylated pentaerythritol containing 4 randomly distributed ethoxy equivalents per mole.
(III"')
In reaction scheme, the compound represented by formula (I'") is reduced with lithium aluminum hydride (LAH) to produce the compound represented by formula (IF").
Procedures for preparing the compound of formula (F") are disclosed in US patent 5,645.767. An ethoxylated pentaerythritol containing 4 randomly distributed
ethoxy equivalents per mole is reacted with the compound of formula (IF") using an acid (FT) to form several ethoxylated isomers including the polyhydroxylated indeno-fused naphthopyran of formula (III'").
The preferred chromenes are the photochromic compound (I) and (IA) described hereinbelow.
The photochromic compound comprising a nucleus of chromene type exhibit better stability and better spectrokinetic performances within a hydrogel than the spirooxazines.
The solvents for dissolving the photochromic agent can be any solvent or mixture of solvents in which the photochromic agent is at least partially soluble. The solvent is preferably a dipolar aprotic solvent. The preferred solvents are N- methylpyrrolidone (NMP), dimethyl sulphoxide (DMSO), diethylene glycol and tetrahydrofuran (THF). The particularly preferred solvents are NMP and DMSO. The most preferred solvent is a mixture of DMSO/water. The concentration of photochromic agent in the impregnating solution is generally from 10"4 to 10 %, preferably from 10"4 to 1 % by weight, more preferably from 0.05 to 0.25 % by weight.
The impregnating of the material is generally carried out by immersion, at room temperature, of the polymeric material in the photochromic solution with stirring. The duration of immersion varies according to the nature of the polymeric material and of the photochromic agents solution and is generally of the order of one minute to 2 hours, preferably 5 minutes to one hour.
The impregnating is carried out into the body, that is to say to the core, of the hydrogel and, consequently, the hydrogel, and in particular the final contact lens as well, comprises the photochromic compound incorporated throughout its body.
After impregnating, the polymeric material is treated with an aqueous solution, for example physiological saline, in order to replace the impregnated solvent with the aqueous solution.
The treatment may be performed in two steps : 1st step : treatment with deionized water to replace the impregnated solvent with the deionized water.
2nd step : treatment with saline solution to replace the deionized water with the saline solution.
The whole treatment with aqueous solutions may take 30 minutes to 4 hours, preferably around one hour.
It is performed at a temperature varying from 15 to 80°C, but preferably at ambient temperature (about 20°C).
The following examples illustrate the present invention. In the examples, except when otherwise indicated, all the percentages and parts are expressed by weight.
Examples 1 to 9
Commercial lenses in the hydrated state were immersed in 2 ml of different photochromic solutions of a photochromic compound (I) and subjected to the treatment for 5 minutes while stirring with a plate.
On conclusion of the steeping, the lenses are rinsed with physiological saline and then immersed in this saline until they have recovered their initial diameter (approximately ten minutes).
The lenses are then exposed to UV radiation for 2 minutes. A UV- visible absorption spectrum is run on the lenses before and after exposure to UV radiation.
The lenses are sterilized at 121 °C for 20 minutes.
A UV-visible absorption spectrum is again run before and after exposure to UV radiation. The results observed are combined in TABLE I below.
Example Lens Hydrophilicity Photochromic Appearance of the lens after Preservation of the
No. % solution UV exposure photochromism after sterilisation
HEMA 37 DMSO/ (I) (0.05 %) Homogeneous colouring Yes, but highly attenuated photochromic effect
HEMA 37 NMP/ (I) (0.05 %) Homogeneous colouring Yes, but highly
« H attenuated photo¬
H chromic effect
H M 3 Lunelle© 70 NMP/ (I) (0.05 %) Homogeneous blue colouring Yes a 4 Lunelle® 70 DMSO/ (I) (0.05 %) Homogeneous blue colouring Yes
W M H 5 Menicon® 72 DMSO/ (I) (0.1 %) Purple colouring Yes
Soft 72
6 Menicon® 72 NMP/ (I) (0.109 %) Homogeneous purple colouring Yes
Soft 72
7 Rythmic© 73 DMSO/ (I) (0.05 %) Homogeneous blue colouring Yes
8 Rythmic® 73 NMP/ (I) (0.05 %) Homogeneous blue colouring Yes
9 Gentle Touch® 65 NMP/ (I) (0.102 %) Navy blue colouring Yes
TABLE I
HEMA (Essilor) : h Lunelle® (Essilor) :
Menicon Soft® 72 methacrylate/N-vinyl Rythmic® (Essilor) :
Gentle Touch® (P copolymer.
Photochromic compo h
The synthesis of this photochromic compound is disclosed in Patent
US 5,645,767.
Examples 10 to 27
This compound is disclosed in Patent Application WO 93/17071.
The synthesis of this photochromic compound is disclosed in Patent0,853.
The synthesis of this photochromic compound is disclosed in a general way in Patent US 5,645,767. Spirooxazines
The compound h is disclosed in European Patent EP 277,639. The compounds c, e, f and g are commercially available from the company James Robinson.
The results are given in TABLE II.
Example Lens Solvent Photochromic Appearance of the lens after Preservation of
No. Agent UV exposure the photochromism after sterilisation
10 Lunelle® DMSO a (0.10 %) Vivid orange red colouring Yes
1 1 Lunelle® Diethylene a (0.105 %) Orange colouring Yes glycol
12 Lunelle® THF a (0.106 %) Orange colouring Yes
CΛ a 13 Menicon Soft ®72 DMSO a (0.101 %) Homogeneous orange colouring Yes w CΛ 14 Menicon Soft® 72 NMP a (0.104 %) Homogeneous orange colouring Yes
H »— < 15 Lunelle® NMP b (0.05 %) Homogeneous orange red colouring Yes
H c! 16 Lunelle® DMSO b (0.101 %) Blood red colouring Yes H M 17 Menicon Soft®72 DMSO b (0.101 %) Homogeneous orange colouring Yes
CΛ
K 18 Menicon Soft® 72 NMP b (0.106 %) Homogeneous orange colouring Yes W M 19 Lunelle® NMP c (0.102 %) Homogeneous red colouring Yes H 20 Lunelle® DMSO d (0.104 %) Homogeneous bottle green colouring Yes
21 Lunelle® NMP d (0.106 %) Bottle green colouring Yes
M 22 Menicon Soft® 72 DMSO d (0.104 %) Homogeneous blue-green colouring Yes
23 Menicon Soft® 72 NMP d(0.106 %) Homogeneous blue-green colouring Yes
24 Lunelle® NMP e (0.103 %) Royal blue colouring Yes
25 Lunelle® NMP f (0.1 %) Turquoise blue colouring Yes
26 Lunelle® NMP g (0.101 %) Turquoise blue colouring Yes
27 Lunelle® NMP h (0.101 %) Intense blue-green colouring Yes
TABLE II
Comparative tests (comparison of the kinetics of colouring in different materials)
Three contact lenses are rendered photochromic by the process of the invention (the photochromic compound incorporated is the compound (I)) using a 0.1 % solution of compound (I).
Their kinetic performances are compared with those of contact lenses obtained by the cast in place process. Unless indicated, the performances are measured at 35 °C, in all the examples of this application.
For this, each of the contact lenses is subjected to UV radiation of 6.87 W/m2 and radiation in the visible region of 50.47 klux, for a period of 10 minutes.
The photochromic compound reaches its maximum colouring in this period of time and the graph of T (transmission) against f (time) and D
(optical density) against f (time) is recorded during the period of colouring, the transmission values being measured for a wavelength λ corresponding to the absorption maximum of the photochromic compound (λmax).
The half-colouring time T
J 2 colo, that is to say the time necessary to pass from DO (initial optical density in the unexcited state) to
where Dχ
0 represents the optical density of the contact lens after irradiating for 10 minutes at the wavelength λ
max, is subsequently measured.
Contact lenses Tχ/2 colo According to the invention (1/2 Colouring time)
- Gentle Touch® 15 seconds MMA/DMAA
- Lunelle® 21 seconds
MMA/NVP
- Menicon® 72 48 seconds MMA/DMAA/NVP
Lenses according to the prior art
Lenses are manufactured by the cast in place process.
Three HEMA-based formulations are tested :
HEMA 1 HEMA 2 HEMA 3
HEMA 100 100 100
Ethylene glycol 0.2 % 0.2 % 0.2 % dimethacrylate
AIBN 0.5 % 0.1 % 0.1 %
Photochromic compoud (I) 0.5 % 1 % 0.5 %
The polymerization of the lenses is carried out in bulk and thermally.
The temperature cycle varies from 40 °C to 120 °C (rise increasing according to a cycle of 50-60 hours).
The 1/2 colouring times are measured on the lenses obtained :
T1 2 colo HEMA 1 65 seconds
HEMA 2 90 seconds
HEMA 3 80 seconds
Other comparative tests are carried out.
An MMA (30)/NVP (70)/allyl methacrylate/AIBN/photochromic compound (I) mixture is polymerised.
None of the tests carried out resulted in a lens exhibiting acceptable photochromic characteristics (irremediably coloured lenses, loss of the photochromic effect).
It is therefore seen that the process of the invention results, via mild conditions, in lenses with photochromic properties which are improved with respect to the prior art with regard to the photochromic performances and the stability.
In the case of ophthalmic lenses, the photochromic agent is incorporated at least in the central optical region of the lens. The central optical region is that which confers the corrective properties of the contact lens, if it is a corrective contact lens.
Examples 28 to 3 1
Several commercial contact lenses are rendered photochromic by using the process of the invention.
Each lens is soaked at room temperature during 20 minutes in a solution of dimethylsulfoxide/water (respective weight ratio 70/30) containing 0.0125 % of compound I.
Then the lenses are rinsed with a saline solution.
The obtained lenses are transparent and photochromic.
The spectrokinetic performances of each of the obtained lenses are measured and reported in table 4.
Table 4
Ex. Treated lens T1 2 colo T1 4 colo ΔT colo
(seconds) (seconds) (%)
Ex 28 Rythmic® 16.9 7.1 42.8
Ex 29 Rythmic® UV 20.1 8.3 38.6
(contact lens as example
28 but including an UV absorber)
Ex 30 Permaflex® natural 16.3 6.9 35.6
Ex 31 Precision UV 25.6 9.9 27.7
Rythmic® Copolymer of N-vinylpyrrolidone/methyl methacrylate/allylmethacrylate Permaflex® natural Copolymer of N-vinylpyrrolidone/methyl methacrylate Precision® UV Copolymer of N-vinylpyrrolidone/methyl methacrylate
T
1 4 colo is the time necessary to pass from D
0 (initial optical density in the unexcited state) to
wherein Tin is the transmission of the contact lens after 10 minutes irradiation ( (IUTV 6.87 W/m
2 - visible 50.47 klux) and T
0 is the transmission of the contact lens in the unexcited state.
Examples 32 to 34
Several commercial contact lenses are rendered photochromic by using the process of the invention.
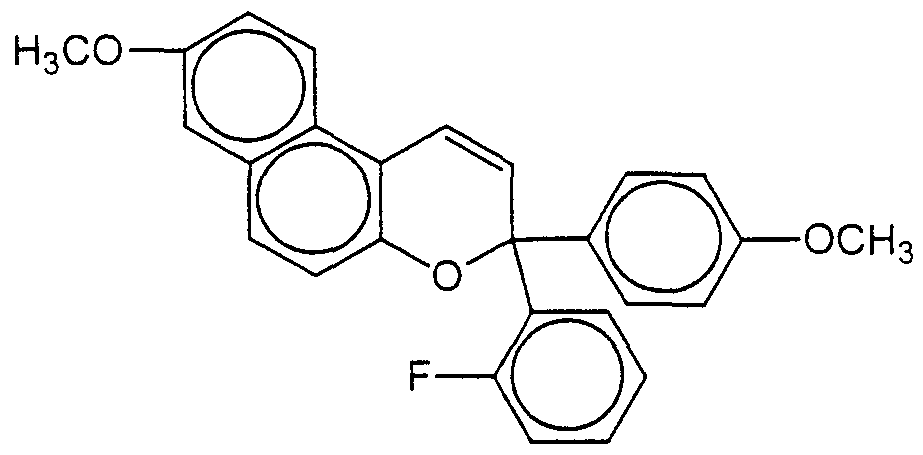
Each contact lens is treated during 5 minutes at ambient temperature with a 0.1 % solution of the compound LA having the formula :
(The method of synthesis of (IA) is described hereafter.) IA being dissolved in N-methylpyrrolidone. Then the contact lens in rinsed with a saline solution. The obtained final contact lens is transparent and photochromic.
Transparency and photochromic performances are maintained even after a sterilization treatment in heated water.
The photochromic performances are measured and reported in table 5.
Table 5
Ex. Treated lens T1 2 colo Tι 4 colo ΔT colo
Ex 32 Review® 38 4.9 9.9 21.8
Ex 33 Rythmic® 4.9 9.9 42.2
Ex 34 Soflens® 66 4.8 9.6 42.5
Review® 38 hydroxyethylmethacrylate polymer (HEMA) Soflens® 66 HEMA/NVP/4-t-butyl, 2 hydroxycyclohexylmethacrylate
Preparation of photochromic compound IA
STEP 1 Potassium t-butoxide (75 grams, 0.67 mole) was added to a reaction flask containing 200 milliliters (mL) of toluene. The reaction flask was equipped with an overhead stirrer, dropping funnel, and a condenser with nitrogen inlet. The contents of the reaction flask was heated to reflux temperature and a mixture of 4,4'-dimethylbenzophenone (105 grams, 0.5 mole), dimethyl succinate (90 grams, 0.62 mole), and toluene (200 grams) was added over a period of one-half hour. The resulting pasty mixture was refluxed an additional two hours, cooled, and about 400 mL of water was added and mixed well. The aqueous layer was separated, acidified with dilute hydrochloric acid, and extracted with 200 mL of toluene. The solvents, toluene and residual t-butanol, were removed on the rotary evaporator to produce a near quantitative yield of crude half-ester, 4,4-di(4- methylphenyl)-3-methoxycarbonyl-3-butenoic acids. This material was not purified further but was used directly in the next step.
STEP 2
The crude half-ester from Step 1 was added to a reaction flask containing 200 mL of toluene. Acetic anhydride (100 grams) and anhydrous sodium acetate (15 grams) were added and the mixture was refluxed for 17 hours. The mixture was cooled and the solvent, toluene, was removed on a rotary evaporator. The resulting residue was dissolved in 200 mL of
methylene chloride and stirred. Water (200 mL) was added followed by the slow addition of solid sodium carbonate until carbon dioxide evolution ceased. The methylene chloride layer was separated and washed with water. The solvent, methylene chloride, was removed on a rotary evaporator to yield about 100 grams of crystalline solid. The recovered product, l-(4- methylphenyl)-2-methoxycarbonyl-4-acetoxy-6-methyl naphtalene, had a melting point of 144-146°C.
STEP 3 The product from Step 2 (about 100 grams) was added to a reaction flask containing 350 mL of a 10 weight percent aqueous sodium hydroxide solution and 50 mL of methanol. The mixture was refluxed for one hour, cooled, then slowly poured into a beaker containing approximately one liter of cold (approx. 4°C) dilute hydrochloric acid. About 100 grams of the resulting crystalline product, l-(4-methylphenyl)-4-hydroxy-6-methyl-2- naphthoic acid, having a melting point of 210-213°C, was collected by vacuum filtration.
STEP 4 The product from Step 3 (about 100 grams) was added to a reaction flask containing xvlene (250 grams) and 250 grams of a 85 weight percent phosphoric acid solution. The stirred mixture was refluxed in a one liter flask equipped with a Dean-Stark trap for 20 hours. During this time a solid product formed. The mixture was cooled and 200 mL of water was added. The solid was broken up with a spatula, filtered, and washed successively with water, 5 weight percent aqueous sodium bicarbonate, and water. Ninety grams of the product, 3,9-dimethyl-5-hydroxy-7H-benzo[C]- fluoren-7-one, were recovered by vacuum filtration.
STEP 5 The product from Step 4 (10 grams) was added to a reaction flask containing l,l-di(4-methoxyphenyl)-2-propyn-l-ol (10 grams) and 100 mL of toluene. The resulting mixture was stirred and heated to 50°C. three drops of dodecylbenzene sulfonic acid were added, and the reaction mixture was kept at 50°C for five hours. After the reaction mixture cooled to room temperature, it was filtered and the collected filtrate was washed three times
with 5 weight percent aqueous sodium hydroxide. The solvent, toluene, was removed on a rotary evaporator and the desired product crystallized on the addition of acetone to the residue. The solid was vacuum filtered, washed with fresh acetone, and dried to yield 16 grams of a product having a melting point of 227-229°C. An NMR showed the product to have a structure consistent with 3,3-di(4-methoxyphenyl)-6,l 1 -dimethyl- 13 -oxo- indeno[2, 1 -f]naphtho[ 1 ,2-b]pyran.
STEP 6 The product of Step 5 (10 grams) was added to a reaction flask containing 50 mL of anhydrous tetrahydrofuran. The mixture was cooled in an ice bath and protected from moisture with a nitrogen pad while an excess of methyl Grignard reagent was added to the reaction with stirring After stirring an additional ten minutes, 200 mL of 5 weight percent aqueous hydrochloric acid was added and the organic layer was separated and washed with water. The solvent, tetrahydrofuran, was removed on a rotary evaporator. The addition of approximately ten milliliters of a 2:1 mixture of hexane:ethyl acetate to the residue caused the crystallization of a non- photochromic material. This material was separated by filtration. The filtrate was column chromatographed on silica using a 3:1 mixture of hexane: ethyl acetate as elutant. The desired product, which crystallized from a methanol mixture, was filtered and dried to yield 8 grams of a product having a melting point of 233-235°C. An NMR spectrum showed the product to have a structure consistent with 3,3-di(4-methoxyphenyl)- 6,1 l,13-trimethyl-13-hydroxy-indeno[2,l-f]naphtho[l,2-b]pyran.
STEP 7
The product from Step 6 (6.0 grams) was added to a reaction flask containing 150 mL of pentaerythritol ethoxylate (3/4 EO/OH) (from Aldrich), 100 mL of tetrahydrofuran, and 2 mL of 37 % hydrochloric acid.
The reaction was heated to 60°C and maintained at that temperature for
8 hours with stirring. The reaction mixture was added to 300 mL of water and 100 mL of ethyl acetate was added. The organic layer was separated, washed with water, filtered, and the solvent, ethyl acetate, was removed on a rotary evaporator. The resulting residue was chromatographed on silica
using acetonitrile (95 %) and methanol (5 %) as the eluant. The recovered oil was dried to yield 2 grams of product. An NMR spectrum showed the product to have a structure consistent with 3,3-di(4-methoxyphenyl)- 6, 11 , 13 -trimethyl- 13 -(2,2-di(2-hydroxyethoxy)methyl-3-hydroxy- propoxy)ethoxy)-indeno[2, 1 -f]naphtho[ 1 ,2-b]pyran.
Examples 35 to 36 :
Two commercial contact lenses are treated using the same procedure as in examples 32 to 34 except that the treatment solution is compound IA in DSMO/water (respective ratio by weight 70/30).
ΔT is measured
Treated lens ΔT (%)
Review® 38 37.8
Rythmic®UV 56.2
(same as Rythmic but including an UV absorber)
ΔT is higher than in examples 32 and 33 indicating that higher amounts of photochromic compounds have been incorporated in the contact lenses.
Exemple 37 A mixture of
HEMA 93.037
Methacrylic acid 2.128
3,3 ,5-trimethylcyclohexylmethacrylate 4.836
EGDMA (ethyleneglycoldimethacrylate) 0.580
AIBN (Azobisisobutyronitrile) 0.967
The mixture is stirred and a contact lens mold is filled with this mixture.
A polymerization is performed by heating the filled mold at 105°C during 60 minutes and then at 130°C during 60 minutes.
After demolding, the lens is hydrated in a NaCl solution. The hydrated lens is soaked at ambient temperature in a 2 ml DMSO/Deionized water solution (70/30 (weight ratio)) of compound IA (concentration of IA : 0.025 % by weight) for 60 minutes with stirring and then rinsed in 25 ml of saline solution at room temperature for 30 minutes.
Then a sterilization is made at 121 °C.
A transparent, clear and photochromic contact lens is finally obtained.