WO1994003634A1 - Sequences d'oligonucleotides pour la detection specifique de mollicutes par amplification de genes conserves - Google Patents
Sequences d'oligonucleotides pour la detection specifique de mollicutes par amplification de genes conserves Download PDFInfo
- Publication number
- WO1994003634A1 WO1994003634A1 PCT/FR1993/000784 FR9300784W WO9403634A1 WO 1994003634 A1 WO1994003634 A1 WO 1994003634A1 FR 9300784 W FR9300784 W FR 9300784W WO 9403634 A1 WO9403634 A1 WO 9403634A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- primers
- primer
- nucleotides
- dna
- mollicute
- Prior art date
Links
- 241001430197 Mollicutes Species 0.000 title claims abstract description 194
- 230000003321 amplification Effects 0.000 title claims description 61
- 238000003199 nucleic acid amplification method Methods 0.000 title claims description 61
- 108090000623 proteins and genes Proteins 0.000 title claims description 41
- 238000011895 specific detection Methods 0.000 title abstract description 13
- 108091034117 Oligonucleotide Proteins 0.000 title description 11
- 239000000523 sample Substances 0.000 claims abstract description 74
- 238000001514 detection method Methods 0.000 claims abstract description 47
- 239000012472 biological sample Substances 0.000 claims abstract description 23
- 239000002773 nucleotide Substances 0.000 claims description 148
- 125000003729 nucleotide group Chemical group 0.000 claims description 148
- 108020004414 DNA Proteins 0.000 claims description 133
- 239000012634 fragment Substances 0.000 claims description 71
- 241000202892 Mycoplasma pirum Species 0.000 claims description 68
- 108091036078 conserved sequence Proteins 0.000 claims description 64
- 238000009396 hybridization Methods 0.000 claims description 34
- 241000202934 Mycoplasma pneumoniae Species 0.000 claims description 31
- 238000006243 chemical reaction Methods 0.000 claims description 31
- 241000894007 species Species 0.000 claims description 31
- 241000204048 Mycoplasma hominis Species 0.000 claims description 30
- 108020004465 16S ribosomal RNA Proteins 0.000 claims description 26
- 238000000034 method Methods 0.000 claims description 25
- 241000202921 Ureaplasma urealyticum Species 0.000 claims description 18
- 241000202952 Mycoplasma fermentans Species 0.000 claims description 16
- 108091028043 Nucleic acid sequence Proteins 0.000 claims description 15
- 241000203022 Acholeplasma laidlawii Species 0.000 claims description 14
- 241000204031 Mycoplasma Species 0.000 claims description 13
- 102000039446 nucleic acids Human genes 0.000 claims description 13
- 108020004707 nucleic acids Proteins 0.000 claims description 13
- 150000007523 nucleic acids Chemical class 0.000 claims description 13
- 108091032973 (ribonucleotides)n+m Proteins 0.000 claims description 12
- 241000204028 Mycoplasma arginini Species 0.000 claims description 12
- 210000004027 cell Anatomy 0.000 claims description 10
- 239000002299 complementary DNA Substances 0.000 claims description 10
- 238000003786 synthesis reaction Methods 0.000 claims description 7
- 230000015572 biosynthetic process Effects 0.000 claims description 6
- 230000000295 complement effect Effects 0.000 claims description 6
- 102000042567 non-coding RNA Human genes 0.000 claims description 6
- 102100034343 Integrase Human genes 0.000 claims description 5
- 241001465754 Metazoa Species 0.000 claims description 5
- 108010092799 RNA-directed DNA polymerase Proteins 0.000 claims description 5
- 230000007717 exclusion Effects 0.000 claims description 4
- 208000015181 infectious disease Diseases 0.000 claims description 4
- 230000000977 initiatory effect Effects 0.000 claims description 4
- 102000053602 DNA Human genes 0.000 claims description 3
- 241000202917 Spiroplasma Species 0.000 claims description 3
- 239000003242 anti bacterial agent Substances 0.000 claims description 3
- 238000000926 separation method Methods 0.000 claims description 3
- 108020004635 Complementary DNA Proteins 0.000 claims description 2
- 108020004682 Single-Stranded DNA Proteins 0.000 claims description 2
- 230000003115 biocidal effect Effects 0.000 claims description 2
- 238000010804 cDNA synthesis Methods 0.000 claims description 2
- 238000012545 processing Methods 0.000 claims description 2
- 241000204018 Anaeroplasma Species 0.000 claims 1
- 238000000338 in vitro Methods 0.000 claims 1
- 238000012360 testing method Methods 0.000 description 31
- 239000000047 product Substances 0.000 description 24
- 239000000203 mixture Substances 0.000 description 22
- 230000035945 sensitivity Effects 0.000 description 21
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 20
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 19
- 239000000243 solution Substances 0.000 description 18
- 241001135743 Mycoplasma penetrans Species 0.000 description 17
- 239000012528 membrane Substances 0.000 description 15
- 241000894006 Bacteria Species 0.000 description 14
- 241000588724 Escherichia coli Species 0.000 description 14
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 14
- 241000202938 Mycoplasma hyorhinis Species 0.000 description 14
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 14
- 239000002751 oligonucleotide probe Substances 0.000 description 14
- 241000282553 Macaca Species 0.000 description 13
- 241000186588 Erysipelatoclostridium ramosum Species 0.000 description 12
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 12
- 241000193462 [Clostridium] innocuum Species 0.000 description 12
- 235000014469 Bacillus subtilis Nutrition 0.000 description 11
- 241000282412 Homo Species 0.000 description 11
- 238000010790 dilution Methods 0.000 description 11
- 239000012895 dilution Substances 0.000 description 11
- 241000202936 Mycoplasma mycoides Species 0.000 description 10
- 241000202915 Spiroplasma citri Species 0.000 description 10
- 239000002585 base Substances 0.000 description 10
- 238000001962 electrophoresis Methods 0.000 description 9
- 238000012546 transfer Methods 0.000 description 9
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 8
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 8
- 239000008188 pellet Substances 0.000 description 8
- 239000006228 supernatant Substances 0.000 description 8
- 241000202967 Mycoplasma iowae Species 0.000 description 7
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 7
- 239000011543 agarose gel Substances 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 238000000605 extraction Methods 0.000 description 7
- 238000010186 staining Methods 0.000 description 7
- 241000204025 Mycoplasma capricolum Species 0.000 description 6
- 239000004677 Nylon Substances 0.000 description 6
- 238000004113 cell culture Methods 0.000 description 6
- 230000009089 cytolysis Effects 0.000 description 6
- 239000000499 gel Substances 0.000 description 6
- 229920001778 nylon Polymers 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- 239000011550 stock solution Substances 0.000 description 6
- 210000001519 tissue Anatomy 0.000 description 6
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 238000003745 diagnosis Methods 0.000 description 5
- 239000012153 distilled water Substances 0.000 description 5
- 239000013642 negative control Substances 0.000 description 5
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 4
- 238000007400 DNA extraction Methods 0.000 description 4
- 108010067770 Endopeptidase K Proteins 0.000 description 4
- 229920001917 Ficoll Polymers 0.000 description 4
- 241000192125 Firmicutes Species 0.000 description 4
- 229920002527 Glycogen Polymers 0.000 description 4
- 241000202946 Mycoplasma pulmonis Species 0.000 description 4
- 108010021757 Polynucleotide 5'-Hydroxyl-Kinase Proteins 0.000 description 4
- 102000008422 Polynucleotide 5'-hydroxyl-kinase Human genes 0.000 description 4
- 108020001027 Ribosomal DNA Proteins 0.000 description 4
- 241000713311 Simian immunodeficiency virus Species 0.000 description 4
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 4
- 229940096919 glycogen Drugs 0.000 description 4
- 210000004698 lymphocyte Anatomy 0.000 description 4
- 239000003550 marker Substances 0.000 description 4
- 244000005700 microbiome Species 0.000 description 4
- 239000002480 mineral oil Substances 0.000 description 4
- 235000010446 mineral oil Nutrition 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 238000005406 washing Methods 0.000 description 4
- 241000972773 Aulopiformes Species 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 241000202957 Mycoplasma agalactiae Species 0.000 description 3
- 241000202954 Mycoplasma californicum Species 0.000 description 3
- 241000204022 Mycoplasma gallisepticum Species 0.000 description 3
- 241000202889 Mycoplasma salivarium Species 0.000 description 3
- 241000204003 Mycoplasmatales Species 0.000 description 3
- 108020002230 Pancreatic Ribonuclease Proteins 0.000 description 3
- 102000005891 Pancreatic ribonuclease Human genes 0.000 description 3
- 229920005654 Sephadex Polymers 0.000 description 3
- 239000012507 Sephadex™ Substances 0.000 description 3
- 108010006785 Taq Polymerase Proteins 0.000 description 3
- 229920004890 Triton X-100 Polymers 0.000 description 3
- 108010046334 Urease Proteins 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 238000005119 centrifugation Methods 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- 239000003184 complementary RNA Substances 0.000 description 3
- 238000011109 contamination Methods 0.000 description 3
- 210000003527 eukaryotic cell Anatomy 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 235000019515 salmon Nutrition 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 244000063299 Bacillus subtilis Species 0.000 description 2
- 108020004394 Complementary RNA Proteins 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 241000202956 Mycoplasma arthritidis Species 0.000 description 2
- 241000204051 Mycoplasma genitalium Species 0.000 description 2
- 241000202898 Ureaplasma Species 0.000 description 2
- 238000000376 autoradiography Methods 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- SUYVUBYJARFZHO-RRKCRQDMSA-N dATP Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-RRKCRQDMSA-N 0.000 description 2
- SUYVUBYJARFZHO-UHFFFAOYSA-N dATP Natural products C1=NC=2C(N)=NC=NC=2N1C1CC(O)C(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-UHFFFAOYSA-N 0.000 description 2
- RGWHQCVHVJXOKC-SHYZEUOFSA-J dCTP(4-) Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)C1 RGWHQCVHVJXOKC-SHYZEUOFSA-J 0.000 description 2
- HAAZLUGHYHWQIW-KVQBGUIXSA-N dGTP Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 HAAZLUGHYHWQIW-KVQBGUIXSA-N 0.000 description 2
- NHVNXKFIZYSCEB-XLPZGREQSA-N dTTP Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)C1 NHVNXKFIZYSCEB-XLPZGREQSA-N 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 239000003599 detergent Substances 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 238000012869 ethanol precipitation Methods 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 239000012139 lysis buffer Substances 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 210000005087 mononuclear cell Anatomy 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- -1 pH 7.5 Chemical compound 0.000 description 2
- 244000052769 pathogen Species 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 229920002401 polyacrylamide Polymers 0.000 description 2
- 238000006116 polymerization reaction Methods 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 210000003705 ribosome Anatomy 0.000 description 2
- 238000004062 sedimentation Methods 0.000 description 2
- 238000011896 sensitive detection Methods 0.000 description 2
- 239000001509 sodium citrate Substances 0.000 description 2
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 241001430294 unidentified retrovirus Species 0.000 description 2
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- 108020005075 5S Ribosomal RNA Proteins 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 101150029129 AR gene Proteins 0.000 description 1
- 241000203024 Acholeplasma Species 0.000 description 1
- 241000024188 Andala Species 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 241000276408 Bacillus subtilis subsp. subtilis str. 168 Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 108010054814 DNA Gyrase Proteins 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 241001646716 Escherichia coli K-12 Species 0.000 description 1
- QTANTQQOYSUMLC-UHFFFAOYSA-O Ethidium cation Chemical compound C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 QTANTQQOYSUMLC-UHFFFAOYSA-O 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 241000754031 Mycoplasma agalactiae PG2 Species 0.000 description 1
- 241000202955 Mycoplasma bovigenitalium Species 0.000 description 1
- 241000614656 Mycoplasma fermentans PG18 Species 0.000 description 1
- 241001531204 Mycoplasma gallisepticum ATCC 19610 Species 0.000 description 1
- 241000999862 Mycoplasma genitalium G37 Species 0.000 description 1
- 241000754030 Mycoplasma hominis ATCC 23114 Species 0.000 description 1
- 241000204045 Mycoplasma hyopneumoniae Species 0.000 description 1
- 241001397205 Mycoplasma iowae 695 Species 0.000 description 1
- 241000202966 Mycoplasma lipophilum Species 0.000 description 1
- 241000202964 Mycoplasma mobile Species 0.000 description 1
- 241000202896 Mycoplasma neurolyticum Species 0.000 description 1
- 241000462791 Mycoplasma pneumoniae FH Species 0.000 description 1
- 241001539527 Mycoplasma salivarium ATCC 23064 Species 0.000 description 1
- 241000202887 Mycoplasma sualvi Species 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 239000012807 PCR reagent Substances 0.000 description 1
- 102000002508 Peptide Elongation Factors Human genes 0.000 description 1
- 108010068204 Peptide Elongation Factors Proteins 0.000 description 1
- 102000005877 Peptide Initiation Factors Human genes 0.000 description 1
- 108010044843 Peptide Initiation Factors Proteins 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 235000014676 Phragmites communis Nutrition 0.000 description 1
- 241000908471 Pirum Species 0.000 description 1
- 108010066717 Q beta Replicase Proteins 0.000 description 1
- 101000897961 Rattus norvegicus Endothelial cell-specific molecule 1 Proteins 0.000 description 1
- 108010000605 Ribosomal Proteins Proteins 0.000 description 1
- 102000002278 Ribosomal Proteins Human genes 0.000 description 1
- 238000002105 Southern blotting Methods 0.000 description 1
- 101710183280 Topoisomerase Proteins 0.000 description 1
- 108020004566 Transfer RNA Proteins 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 238000011166 aliquoting Methods 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000003637 basic solution Substances 0.000 description 1
- RDHPKYGYEGBMSE-UHFFFAOYSA-N bromoethane Chemical compound CCBr RDHPKYGYEGBMSE-UHFFFAOYSA-N 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- TWFZGCMQGLPBSX-UHFFFAOYSA-N carbendazim Chemical compound C1=CC=C2NC(NC(=O)OC)=NC2=C1 TWFZGCMQGLPBSX-UHFFFAOYSA-N 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 230000037029 cross reaction Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000004925 denaturation Methods 0.000 description 1
- 230000036425 denaturation Effects 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 238000007834 ligase chain reaction Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 108091027963 non-coding RNA Proteins 0.000 description 1
- 210000003463 organelle Anatomy 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 210000004976 peripheral blood cell Anatomy 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 238000013081 phylogenetic analysis Methods 0.000 description 1
- 238000002264 polyacrylamide gel electrophoresis Methods 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000037452 priming Effects 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108700022487 rRNA Genes Proteins 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000007894 restriction fragment length polymorphism technique Methods 0.000 description 1
- 108020004418 ribosomal RNA Proteins 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- ZFXYFBGIUFBOJW-UHFFFAOYSA-N theophylline Chemical compound O=C1N(C)C(=O)N(C)C2=C1NC=N2 ZFXYFBGIUFBOJW-UHFFFAOYSA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 101150099542 tuf gene Proteins 0.000 description 1
- 238000009281 ultraviolet germicidal irradiation Methods 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6888—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms
- C12Q1/689—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for detection or identification of organisms for bacteria
Definitions
- Oligonucleotide sequences for specific detection of mollicutes by amplification of conserved genes Oligonucleotide sequences for specific detection of mollicutes by amplification of conserved genes
- the subject of the invention is means and in particular oligonucleotide sequences, for the specific detection of determined mollicutes, this detection calling for an amplification reaction of gene fragments conserved in this class of microorganisms, when a mollicute determined is present in a biological sample.
- Mollicutes represent a class of micro ⁇ organisms including in particular microorganisms belonging to the genera Myco lasma, Ureaplasma, Acholeplasma, Spiroplasma.
- groups of mollicutes have also been distinguished.
- mollicutes can constitute pathogens of their various hosts, in particular plants, humans, animals, or cell cultures, in particular laboratory lines.
- Various studies aimed at detecting mollicutes in biological samples have been the subject of publications.
- European patent application EP 0250662 proposes probes for the detection of mycoplasmas, these probes being derived from the ribosomal AR gene (rRNA) of mycoplasmas, in particular 16S rRNA. This request proposes so-called specific probes for mycoplasmas, as they differ from the E. coli 16S rRNA gene or other prokaryotic organisms.
- rRNA ribosomal AR gene
- A. Blanchard et al FEMS Microbiology Letters, 1991 n ° 81, p37-42, FEM 04480 used the alignment of the 16S rRNA sequences in order to define an appropriate pair of oligonucleotides, for a PCR reaction (Polymerase Chain Reaction). This pair of primers would allow the amplification, at the level of a biological sample, of several species belonging to the genus ycoplasma. According to Blanchard et al, the two primers described do not amplify the DNA of bacteria or eukaryotic cells and they are obtained from the alignment of conserved regions among the species of the genus Mycoplasma.
- the mycoplasmas detected must be identified after the amplification reaction, using a species-specific probe or by sequencing the amplified DNA.
- a probe of M. pirum whose natural host remains undetermined according to Blanchard et al, is described.
- the inventors posed the problem of specific and sensitive detection of mollicutes and in particular of mycoplasmas in biological samples, using the techniques of amplification of fragments of genes conserved within mollicutes or prokaryotes in general.
- M. pirum Mycoplasma pirum
- ribosomal RNA gene for example 16S ribosomal RNA, 5S ribosomal RNA, 5.8S, 23S, transfer RNA, the tuf gene, the genes involved in the cellular function of the translation (initiation factor, elongation factor, for example the tuff gene, termination gene, ribosomal proteins), or enzyme genes such as topoisomerases such as DNA gyrases.
- 16S ribosomal RNA of mollicutes and in particular of mycoplasmas is strongly conserved between the different genera and species.
- This homology between the nucleotide sequences of the genes transcribed into 16S rRNA of mollicutes can be between 80% and 100% of the sequence and it is stronger within the same group of mycoplasmas belonging to different species.
- the homology between M. pirum and M. pneumoniae, M. qallisepticum, U urealyticum is approximately 92%, 94% and 86% respectively.
- the subject of the invention is therefore nucleotide sequences which can be used as primers for the amplification of a specific nucleic acid of a desired mollicute.
- the invention also relates to nucleotide sequences which can be used as probes for the detection of amplification products.
- the means proposed in the invention are also very sensitive, allowing the detection of determined mollicutes without prior culture of the sample.
- the sensitivity of the means proposed in the invention decreases reproducibly to 10 organisms present in the sample.
- the invention defines a first group of primers.
- the subject of the invention is, according to a first embodiment, a pair of primers for the amplification of a DNA sequence of a determined mollicute, from a biological sample, characterized in that it comprises :
- a) a first primer and a second primer each having the following properties:
- each primer comprises n nucleotides, n having a value equal to or greater than 10, preferably n being between 18 and 25,
- each primer has an overall homology equal to or greater than 50% with a nucleotide sequence known as "corresponding sequence”, of a determined conserved gene of a mollicute known as a “reference mollicute", said "corresponding sequence” belonging to a region variable of the conserved gene, this homology being distributed within the primer in such a way that:
- the last 2 nucleotides of the primer, located at the 3 • end, are identical to the nucleotides of 1 • corresponding sequence of the conserved gene
- ol 1 homology of the nucleotides of the 5 ′ part of the primer corresponding to approximately one third of the n nucleotides of the primer is at least 55%, preferably at least 60% with the fragment corresponding to the corresponding sequence of the conserved gene of the reference mollicute and;
- the first primer being further such that:
- o the homology of the 3 ′ part of the primer corresponding to approximately one third of the n nucleotides of the primer is, after exclusion of the last 2 nucleotides of the 3 ′ end, at least 60% with the corresponding fragment of the corresponding sequence of the conserved gene of the reference mollicute and,
- o at least one of the 2 nucleotides of its 3 ′ end is different from the corresponding nucleotide of the aligned nucleotide sequence of the conserved gene present in another mollicute capable of being detected in humans;
- the second primer being further such that:
- the homology of the 3 ′ part of the primer corresponding to approximately one third of the n nucleotides of the primer is at least 50% with the corresponding fragment of the conserved gene of the reference mollicute.
- the primer called "first primer” is characterized with regard to the property identified under b), in that at least the last nucleotide of its 3 • end, preferably the the last two nucleotides from its 3 'end is (are) different from the corresponding nucleotide of the aligned nucleotide sequence of the conserved gene present in another mollicute (another species) capable of being detected in humans.
- the first and second primers are further characterized in that at least the last nucleotide of the 3 'end of 1 • one of the first or second primers is different from the corresponding nucleotide of the aligned nucleotide sequence of the conserved gene present in another mollicute (another species) capable of being detected in humans, and two additional nucleotides distributed among the last three nucleotides of the 3 'end of one of the primers or the two primers, are also different from the corresponding nucleotide of the aligned nucleotide sequence of the conserved gene present in another mollicute (another species) capable of being detected in humans.
- the primers called "second primers” have a lower second level homology than the first primer with regard to their 3 'part.
- the condition relating to the difference with respect to a mollicute different from the reference mollicute, of at least one of the two nucleotides forming the 3 'end of the primer may not be satisfied, as soon as a sufficient homology, that is to say greater than 50% is established in the remaining part of the sequence and in particular for the 3 ′ terminal half of the primer.
- the said first and / or second primer is (are) further characterized in that the homology of the nucleotides of the central part of the primer corresponding to approximately one third of the n nucleotides of the primer is at least 15% with the corresponding fragment of the corresponding sequence of the conserved gene of the reference mollicute.
- the first primer and the second primer can be, as the case may be, the primer hybridizing with the 5 ′ end of the fragment to be amplified, or the primer hybridizing with the 3 ′ end of the fragment to be amplified.
- this central part is deleted in whole or in part or has a weaker homology.
- At least one primer of the couple defined above is chosen for its ability to be present at a very low frequency, less than 1, within the human genome.
- the frequency of appearance of this primer in the human genome can be verified using the technique described by Griffais et al (Nucl. Acids. Res. 1991, vol. 19 no. 14 p 3887-3891) or in Meth in Enzymology 1990, vol 183 p 237- Claverie et al.
- Griffais et al Nucl. Acids. Res. 1991, vol. 19 no. 14 p 3887-3891
- Meth in Enzymology 1990, vol 183 p 237- Claverie et al One can also use a computer research program available on the market for example the FIND program of the company GCG Inc, (W Biotechnology Center, 1710 Univ. Avenue, Madison Wisconsin 53705 - USA)
- the primers whose definition has been given previously can also be used for the amplification of RNA sequences corresponding to the DNA of a mollicute, provided that primers capable of hybridizing with said RNA are used.
- the “matching sequence” is the nucleotide sequence of the conserved gene chosen to define primers, which can be aligned with the primer or its complementary reverse sequence.
- the "corresponding fragment” is a fragment included in the "corresponding sequence” which can be aligned with the fragment forming the 5 'part, the central part or the 3' part of the primer, or its complementary reverse sequence.
- the term homology referred to above refers to the identity of the nucleotides of the primer, with respect to the aligned nucleotides of the sequence of the selected conserved gene of the reference mollicute.
- a homology equal to or greater than 50% with the nucleotide sequence, said corresponding sequence means that at least half of the nucleotides of the primer are identical to the corresponding nucleotides when the primer is aligned with the corresponding sequence of the gene. preserved from which the amplification is carried out in order to detect a determined mollicute (reference mollicute).
- variable region of the DNA of the conserved gene from which the primers for a mollicute are derived is called a region, the rate (percentage) of conservation between different corresponding sequences obtained in mollicutes of distinct genera or species, is less than 80 %, preferably less than 50%.
- variable regions were defined in Gutell et al, 1985 Prog. Nucleic Acid Res Mol Biol 32: 155-216; Woese et al 1983, Microbol Rev 47: 621-669; Neefs et al 1991.
- primer derived from a gene means that this primer is defined, selected from the data available on the chosen gene and in particular on its sequence.
- a step for the selection of a primer corresponding to the preceding characteristics consists, after having chosen a conserved mollicute gene, in establishing a compilation of the different gene sequences for several determined mollicutes and, if appropriate, for other organisms having related genes , and to select from the variable regions of the sequences thus aligned, the nucleotide sequences corresponding to the characteristics defined above.
- reference mollicute or "determined mollicute” corresponds to a particular species of mollicutes, taken inside a genus, for example the genus Mycoplasma or the genus Spiroplasma or even the genus Ureaplasma.
- phylogenetic studies and in particular those reported by Weisburg et al have made it possible to determine the existence of certain groups such as the group of M. pneumoniae, the group of M. hominis, the group of spiroplasms, or the group of acholeplasms to which Acholeplasma laidlawii belongs.
- primers as defined above are used, in the form of pairs of primers.
- the invention also relates to another group of primers (group 2), capable of forming part of a pair of primers, the two primers of which correspond to the definition given of the abovementioned first primer of the first group above.
- group 2 another group of primers
- the reference mollicute is a mycoplasma capable of being detected in humans.
- this mycoplasma is M. pirum.
- the primer is defined from a reference mollicute chosen from M. fermentans, M. hominis, U. urealyticum, M. pneumoniae or A. laidlawii.
- a reference mollicute chosen from M. fermentans, M. hominis, U. urealyticum, M. pneumoniae or A. laidlawii.
- the sequences of the previously mentioned mollicutes were described in the catalog of bacteria and bacteriophages distributed by 1 ⁇ TCC (American Type Culture Collection) in the I8 th Edition in 1992. The access numbers to these strains are given in this catalog and the publications describing them are also given for reference.
- primers described above can be modified by extension or deletion of certain nucleotides or replacement.
- a primer of the invention can be extended at its 5 ′ end and / or at its 3 * end, in particular by the nucleotides adjacent to the corresponding sequence of the conserved gene from which it is defined.
- this gene is the mollicute gene transcribed into 16S ribosomal RNA.
- a first primer preferred according to the invention is the sequence MYCPIRP corresponding to the formula:
- Another preferred primer according to the invention is the primer MYCPIRN corresponding to the formula:
- the primer MYCPIRP is used as primer 5 ', that is to say at the N-terminal end of the fragment to be amplified and the primer MYCPIRN is used as primer 3 •, that is to say at 1 C-terminal end of the fragment to be amplified.
- DNA size of the RNA gene 16S ribosome of M. pirum from which these primers are defined is approximately 173 nucleotides.
- FIG. 1 represents the position of the sequences of the 16S rRNA gene of different mollicutes and makes it possible in particular to locate the sequences of this gene corresponding to the primers MYCPIRP and MYCPIRN of M. pirum between positions 52 and 82 for the first, 207 and 239 for the second. These positions are given with reference to the sequence of the 16S ribosomal RNA gene from E. coli (Brosius et al, 1978, PNAS, USA, vol 75, p4801-4805).
- Figure 1 represents a compilation with alignment of the sequences for the different mollicutes.
- the invention relates to the primers defined from the gene corresponding to the 16S RNA of mollicutes different from M. pirum, these primers being derived from the nucleotide fragments of FIG. 1 for each of the mollicutes, located between positions 52 and 82 d on the one hand, 207 and 239 on the other.
- primers have the advantageous property of being specific for the detection of specific bacterial strains or defined phylogenetic groups.
- the invention also relates to a pair of primers characterized in that it comprises a primer corresponding to the definition given above and having in particular the characteristics identified by a) (1 and 2), b) or c), and a primer capable of hybridizing under strong stringency conditions with a DNA or cDNA fragment obtained from a conserved gene of the specified mollicute called "reference mollicute".
- strong stringency is meant the hybridization of the primers with the DNA of the sample, when this hybridization is carried out at 55 ° C. for 1 minute in the "Taq" buffer marketed by Amersham.
- the pair of primers is chosen such that each of the primers is distant from the other by approximately 150 to 300 nucleotides.
- a particular pair of primers includes MYCPIRP and MYCPIRN.
- the specific primers according to the invention can be used in pairs for the detection of specific strains or families.
- the following pairs of primers are advantageously used for the detection of the strains identified below:
- PENETP 5 'CATGCAAGTCGGACGAAGCA 3' PENETN: 5 ” AGCATTTCCTCTTCTTACAA 3 'for the detection of M. penetrans
- TIRE 5 'CCTGCAAGGGTTCGTTATTT 3' TIRE: 5 ' ATGGTACAGTCAAACTCTAG 3 ' for the detection of M. pneumoniae
- HOI- ⁇ IP 5'-ATACATGCATGTCGAGCGAG-3 'HOMIN: 5'-CATCTTTTAGTGGCGCCTTAC-3' for the detection of M. hominis
- UREUREP 5'-CGTAAAGTTGCTTATGGACGTCGTT-3 • UREUREN: 5'-TTGTGTCTCCTAATCTAACGCTATC-3 'for the detection of U. urealyticum
- MYCARGP 5'-ATGCATGTCGAGCGAGGTTC-3 'MYCARGN: 5'-TTAGCTGCGTCAGTGAACTC-3' for the detection of M. arqinini
- MYCORAP 5 • -CGTTGTGAAAGGAGCGTTTC-3 'MYCORAN: 5'-GAAGCATTTCCTCAACTAAC-3' for the detection of oral M.
- MYCHYOP 5'-TAAATTCTAAAAAAGATCGACTAAC-3 'MYCHYON: 5'-TTCAGTAATTGCTTCTAAATTAACG-3' for the detection of M. hyorhinis
- the primers UREUREP and UREUREN allow the amplification and the detection of a fragment of the urease gene.
- the primers MYCHYOP and MYCHYON make it possible to amplify and detect a fragment of the gene for the 115 kDa protein.
- the invention also relates to nucleotide sequences, which can be used in particular as probes when they are labeled, these sequences being chosen from the conserved or variable regions of the mollicutes which it is sought to detect. These probes are also determined relative to the primers used in the amplification reactions; they are internal fragments with respect to these primers.
- Two probes of interest in the context of the invention are probes suitable for the detection of an amplification product of the 16S ribosomal RNA gene of M. pirum.
- a nucleotide sequence capable of being used as a probe in the context of the invention is characterized by the following properties: it comprises at least 10 nucleotides, preferably from 25 to 30 nucleotides, it specifically hybridizes under the following conditions with the DNA sequence amplified from the conserved gene of a reference mollicute, when the amplification is carried out using 2 primers chosen according to the invention for a reference mollicute.
- 5X Denhardt solution a basic solution 5 times concentrated.
- Such sequences may be derived from or derived from the variable regions or from the constant regions of the reference mollicute.
- a sequence thus constituted When a sequence thus constituted is derived, that is to say defined from a variable region of the reference mollicute, it hybridizes in a specific manner, with the nucleotide chain amplified from the two primers.
- the term “specific hybridization” is understood to mean a hybridization which takes place with a nucleotide sequence of the particular mollicute gene from which the sequence is defined and which does not hybridize with any fragment of the corresponding gene of a gene from another mollicute or d 'a bacterium or a eukaryotic cell in particular which does not hybridize with human genoi ⁇ DNA likely to be present in a biological sample.
- nucleotide sequences derived from the constant regions of a reference mollicute can exhibit homologies with corresponding sequences of other mollicutes or possibly other organisms.
- these sequences can be used as probes in a specific reaction for the detection of a specific mollicute, insofar as the fragment which it is desired to amplify from the primers, capable of hybridizing with this sequence derived from a constant region, will have a known size. When the size of the amplified fragment corresponds to the expected size, the presence of the desired mollicute is confirmed.
- MYCPIRS1 5'CAAATGTACTATCGCATGAGAAACATTT3 'OR MYCPIRS2 5'GGGTGAGTAACACGTATCCAATCTACC3'.
- PENETS 5 'CATGAGAAAATGTTTAAAGTCTGTTTG 3' TIRES: 5 'GAAGAATGACTTTAGCAGGTAATGG 3' HOMINIS: 5 'CGCATGGAACCGCATGGTTCCGTTG 3' UREURES: 5'-CCATCAGGTACTGCTATTCGTTTTGAACC- 3 'MYCARGS: 5'-TTTTGAACCTAGCGGCGAATGGGTGAGT -3 "MYCORAS: 5'-GTCCGCTAAGAGATGAGGGTGCGGAACA -3' MYCHYOS: 5'-AAATTTAATTAGTGAGCAAG-3 'ALAIDLS: 5'-TGACCGAGCAACGCCGCGTGAACGACGA -3'
- sequences described above can be modified by addition, deletion and / or replacement of nucleotides.
- these probes can be extended at their 5 'end and / or at their 3' end by the addition of one or more nucleotides, in particular nucleotides adjacent to the probe sequence, in the corresponding sequence of the gene. of the reference mollicute.
- These probes can, if necessary, be extended at their 5 ′ end and / or at their 3 ′ end by adjacent nucleotides in the corresponding fragment of the corresponding region of the reference mollicute gene used.
- the distance separating two selected primers can be known, either by reference to the data available on the sequence of the mollicute in question, or when the sequence is not known, by reference to molecular weight markers. Consequently, an amplified fragment which would have a size different from the size expected for the amplification of a fragment of a reference mollicute, would correspond to DNA or to RNA of another organism.
- the invention also relates to a nucleotide sequence characterized in that it is reverse and complementary DNA to a previously defined sequence or a nucleotide sequence characterized in that it is a reverse and complementary RNA of a sequence defined above.
- Such a reverse and complementary RNA is capable of being used as antisense RNA, if necessary to block the transcription of the gene for the reference mollicute used for the definition of the primers and of the above sequences or to block the RNA transcribed from of this gene.
- the subject of the invention is also a DNA fragment as obtained by amplification of a DNA from a conserved gene of a reference mollicute or of a corresponding cDNA, the amplification being carried out by means of a pair of primers defined above, the amplified DNA fragment specifically hybridizing with a probe constituted by a specific sequence of a variable region or of a constant region of the gene preserved reference mollicute, internal relative to the positions of the 2 primers.
- the invention also relates to a complementary and reverse RNA with respect to the DNA described above.
- kits for the detection of a specific mollicute said
- reference mollicute in a biological sample, characterized in that it comprises: at least one pair of primers derived from a gene conserved from a reference mollicute, capable of hybridizing under stringent conditions at the 5 'ends and 3 ′ of a DNA fragment of the reference mollicute, these 2 primers being such that: a) the first primer and the second primer each have the following properties:
- each primer comprises n nucleotides, n having a value equal to or greater than 10, preferably n being between 18 and 25,
- each primer has an overall homology equal to or greater than 50% with a nucleotide sequence known as "corresponding sequence”, of a determined conserved gene of a mollicute known as a “reference mollicute", said "corresponding sequence” belonging to a region variable of the conserved gene, this homology being distributed within the primer in such a way that:
- the last 2 nucleotides of the primer, located at the 3 • end, are identical to the nucleotides of the corresponding sequence of the conserved gene
- o the homology of the nucleotides of the 5 ′ part of the primer corresponding to approximately one third of the n primer nucleotides is at least 55% preferably at least 60% with the corresponding fragment of the corresponding sequence of the conserved gene of the reference mollicute and;
- the first primer being further such that:
- o the homology of the 3 ′ part of the primer corresponding to approximately one third of the n nucleotides of the primer is, after exclusion of the last 2 nucleotides of the 3 ′ end, at least 60% with the corresponding fragment of The corresponding sequence of the conserved gene of the reference mollicute and,
- o at least one of the 2 nucleotides of its 3 ′ end is different from the corresponding nucleotide of the aligned nucleotide sequence of the conserved gene present in another mollicute capable of being detected in humans;
- the second primer being further such that:
- the homology of the 3 ′ part of the primer corresponding to approximately one third of the n nucleotides of the primer is at least 50% with the corresponding fragment of the conserved gene of the reference mollicute.
- the reagents necessary for the polymerization of the DNA fragment of the reference mollicute from the nucleotide primers in particular sufficient polymerization enzymes to carry out the amplification; - At least one probe corresponding to a nucleotide sequence of the conserved gene of the reference mollicute, internal with respect to the primers.
- the primer called "first primer” is characterized with regard to the property identified under b), in that at least the last nucleotide of its 3 • end, preferably the last two nucleotides at its 3 'end is (are) different from the corresponding nucleotide of the aligned nucleotide sequence of the conserved gene present in another mollicute (another species) capable of being detected in humans.
- the first and second primers of the kit are further characterized in that at least the last nucleotide of the 3 'end of one of the first or second primers is different from the corresponding nucleotide of the aligned nucleotide sequence of the conserved gene present in another mollicute (another species) capable of being detected in humans, and two additional nucleotides distributed among the last three nucleotides of the 3 'end of one primers, or both primers, are also different from the corresponding nucleotide of the aligned nucleotide sequence of the conserved gene present in another mollicute (another species) capable of being detected in humans.
- the two primers of the pair meet the definition given for the aforementioned first primer and therefore, they verify the properties identified by a) 1 and 2 and b).
- this kit may contain the primers corresponding to the definition of the variants given in the preceding pages.
- the primers can be marked with a chemical, physical or enzymatic marker and / or fixed to a solid support, in particular a particulate or membrane support, for example magnetic beads.
- the probes used for producing the kit of the invention can also be labeled at their 5 • end and / or at their 3 ′ end with a substance which can be detected, or fixed to a solid support.
- radioactive isotopes examples include radioactive isotopes, suitable chemical enzymes or markers, fluorochromes, haptens, antibodies, basic analogs or even physical markers.
- the attachment to a support can be made on a solid particulate or membrane support, for example on magnetic beads.
- a particular kit of the invention is characterized in that the primers that • it contains are MYCPIRP and MYCPIRN, the probe being MYCPIRS1 or MYCPIRS2 or both.
- kits according to the invention are defined using the following primers and specific probes, for detecting the strains indicated below:
- PENETP primer 5'-CATGCAAGTCGGACGAAGCA-3 '
- MYCARGS oligonucleotide probe 5 ⁇ -TTTTGAACCTAGCGGCGAATGGGTGAGT-3 '
- MYCORAS oligonucleotide probe 5'-GTCCGCTAAGAGATGAGGGTGCGGAACA -3 '
- - MYCFERS oligonucleotide probe 5'-GAAGCTTTCTTCGCTGGAGGAGCGGGGT-3 '
- the kit also comprises: an internal control of the amplification reaction, consisting of a DNA fragment capable of being easily detected, for example a fragment containing a gene for resistance to an antibiotic, this fragment being different from the DNA of the reference mollicute capable of being amplified from the sample, this fragment being additionally provided at each of its ends with an amplification primer described above;
- an internal control of the amplification reaction consisting of a DNA fragment capable of being easily detected, for example a fragment containing a gene for resistance to an antibiotic, this fragment being different from the DNA of the reference mollicute capable of being amplified from the sample, this fragment being additionally provided at each of its ends with an amplification primer described above;
- Means for detecting the amplification of internal control for example a probe capable of specifically hybridizing with the DNA contained in the internal control.
- a kit according to the invention can comprise several pairs of specific primers of different mollicutes and, in particular, a pair specific primers for each of the following mollicutes: M. pirum, M. pneumoniae, M. fer entans, U urealyticum and A. laidlavii.
- the kit also includes a specific probe for each of these mollicutes.
- the subject of the invention is also a kit corresponding to one of the definitions above, characterized in that it further comprises a reverse transcriptase for obtaining cDNA from the 16S ribosomal RNA present in the biological sample tested.
- a method for the detection of an infection by a mollicute or several determined mollicutes in particular of the species M. pirum or several determined mollicutes, on a biological sample characterized in that it comprises the steps of: contact of the nucleic acid of the biological sample tested, if necessary under conditions allowing accessibility in the form of single-stranded DNA, with at least one pair of nucleotide primers described above, said primers being able to hybridize with the the nucleic acid of the specific mollicutes sought if they are present, and initiating the synthesis of the elongation product of said primers, each strand of DNA of mollicutes serving as template when it is paired with the primers; b) separation of the strands of DNA synthesized from their template; c) repetition of the synthesis of elongation product, from each DNA strand present at the end of step b) and capable of hybridizing with the primers, until an amplification of l DNA sought, sufficient to be detected, d)
- the amplification techniques include, for example, the PCR (Polymerase Reaction Chain) technique described in the European patent applications of CETUS (n ° 0200363, 0201184, and 0229701), or even the "Q ⁇ replicase” technique described in Boitechnology (vol. 6, October 1988).
- Other techniques are “LCR” (ligase chain reaction), “3SR” (self sustained sequence replication), “Ampliprobe” (amplification from RNA). These techniques are described in J of Virological methods, 1991, 35, 117-126.
- the contacting of the biological sample tested is preceded by a step of processing the sample so as to extract the nucleic acid therefrom.
- this method is characterized in that prior to contacting with the primers, o treats the nucleic acid of the sample with a reverse transcriptase, to obtain the synthesis of cDNA from the RNA possibly present in the sample testé.Il poss the carrying out the method according 1'invention equal ⁇ nt by using primers together with the reverse transcriptase.
- Different samples can be used for the implementation of the invention and advantageously a culture of the sample will not be carried out. prior to the mollicute detection operation. Indeed, the sensitivity of the means defined for this detection allows the realization of the invention on tissues or other samples without prior culture. Thus, it will be possible to use, as samples, human or animal tissues, or alternatively cell lines used for example in the laboratory, in order to detect an infection or contamination by a mollicute or several determined mollicutes.
- the invention allows the amplification, from primers, of the genomic DNA of mollicutes contained in the samples tested, after extraction of this DNA or on the contrary directly without prior extraction, by "in situ" PCR.
- the invention relates in particular to the detection in a patient infected with an HIV retrovirus, of an infection with one or more mollicutes and in particular determined mycoplasmas.
- the primers and other sequences of the invention can be obtained by any suitable technique, in particular by chemical synthesis.
- Figure 1 Alignment of 16S ribosomal DNA sequences from M. pirum and representative species of mollicutes and bacteria.
- the numbers indicate the nucleotide positions relative to the sequence of E. coli 16S rDNA (Brosius et al. 1978).
- the asterisks indicate the nucleotides conserved.
- the sequences (direct or complementary depending on the case) of the oligonucleotides MYCPIRP, MYCPIRN, MYCPIRS1 and MYCPIRS2) are underlined.
- Figure 2 hybridizations with the MYCPIRS1 (A) or MYCPIRS2 (B) probe of the PCR products, after electrophoresis and transfer to the membrane. Track 1: M.
- PCR was carried out from 100 fg of DNA for M. pirum, and from 10 ng of DNA for the other mollicutes.
- Figure 3 hybridization with the MYCPIRS1 probe of the PCR products, after electrophoresis and transfer to the membrane.
- Track l M. hominis
- track 2 M. oral
- track 3 M. arginini
- track 4 M. fermentans
- track 5 M. hyorhinis
- track 6 A. laidlawii
- track 7 C. innocuum
- track 8 M. pirum 70-159
- track 9 M. pirum BER
- track 10 C. ramosum
- track 11 B. subtilis
- track 12 E.coli
- track 13 M. pirum NOU
- track 14 M VUC pirum.
- the PCR was carried out from 100 fg of DNA for M. pirum, and from 10 ng of DNA for the other bacteria.
- Figure 4 polyacrylamide gel electrophoresis of PCR products from the genomic DNA of M. pirum.
- Tracks 2 to 8 dilutions of the DNA of M. pirum by 10 in
- Tracks 10 to 15 and 17 dilutions of the DNA of M. pirum from 10 to 10 from 1 ng to 1 fg, in the presence of DNA from PBMCs.
- Lane 18 DNA control of PBMCs alone (1 ⁇ g).
- Figure 5 Hybridizations with the MYCPIRS1 probe of the PCR products carried out from the genomic DNA of M. pirum. Tracks 1 to 7: 10-fold dilutions of M. pirum's DNA
- Lane 8 M. pirum's DNA is replaced by water.
- Track 1 to 5 different subclones of the line.
- Lane 6 M. pirum (10 pg).
- Tracks 2 to 8 and 10 to 16 PCR on dilution of M. penetrans from 10 to 10 from lfg (1 genome) to lng (10 6 genomes), in the absence (lane 2 to 8) or in the presence (lane 10 to 16) of 1 ⁇ g of PMBCs DNA with the primers PENETP and PENETN. Lane 7: negative control (sterile distilled water). Lane 9: PCR carried out on 1 ⁇ g of DNA from PBMCs. The PCR products were subjected to electrophoresis, transferred to a nylon membrane and hybridized with the internal probe to the amplified fragment PENETS.
- Lanes 1 and 24 PCR using 100 fg of DNA (100 genomes) from M. penetrans.
- Tracks 3 to 22 PCR from 10 ng (10 7 genomes) of the following mycoplasmas: M. gallisepticum, M. pirum, M. pneumoniae, M. génitalium, M. iowae, M. matris, U. urealyticu, M. hyorhinis, M. arginini, M. oral, M. hominis, M. fermentans, M. mycoides, M. capricolum, S. citri, A. laidlawii, C. innocuum, C. ramosum, B. subtilis and E. coli.
- Tracks 2 and 23 negative controls (sterile distilled water).
- Tracks 1 to 6 and 8 to 13 PCR on dilution of M. pneumoniae from 10 to 10 from 100 pg (10 5 genomes) to 1 fg (1 genome), in the absence (tracks 1 to 6) or in the presence ( lanes 8 to 13) of 1 ⁇ g of DNA from PBMCs with the primers PNEUP and PNEUN. Lane 7: negative control (sterile distilled water). Lane 14: PCR carried out on 1 ⁇ g of DNA from PBMCs. The PCR products were subjected to electrophoresis, transferred to a nylon membrane and hybridized with the internal probe to the amplified fragment PNEUS.
- Tracks 2 to 8 'and 10 to 17 PCR on dilutions of M. hominis from 10 to 10 from 10 ng (10 7 genomes) to 1 fg (1 genome), in the absence (tracks 2 to 8') or in the presence (lanes 10 to 17) of 1 ⁇ g of DNA from PBMCs with the primers HOMIP and HOMIN. Lane 1: negative control (sterile distilled water). Lane 9: PCR carried out on 1 ⁇ g of DNA from PBMCs. The PCR products were subjected to electrophoresis, transferred to a nylon membrane and hybridized with the internal probe to the amplified fragment HOMIS. EXAMPLES
- M. mycoides PG1 (ATCC 10114), M. hominis PG21 (ATCC 23114), M. arqinini G230 (ATCC23838), M. oral CH19299 (ATCC 23714), M. hyorhinis BS7 (ATCC 17981), M. pirum 70-159 (ATCC 25960), M. pneumoniae FH (ATCC 15531), M. genitalium G37 (ATCC 33530) M. fermentans PG18 (ATCC 19989), M. penetrans GTU-54-6A1 (ATCC 55252), Ureaplasma urealyticum 960 (NCTC 10177 ), M. arthritidis MY5121TR, M.
- agalactiae PG2 (ATCC 10123), M. pulmonis My 5031, M. salivarium PG20 (ATCC 23064), M. ripened RIII4 (ATCC 33757) and Acholesplasma laidlawii PG8 (ATCC 23206) standard known as Escherichia coli K12 and Bacillus subtilis 168.
- M. pirum strains BER, NOU, and VUC
- strain AOU clinical strain of M. fermentans
- M. matured RIII4 (ATCC 33757), Spiroplasma citri R8A2 (ATCC 27556), M. capricolum Cal. Kid (ATCC 27343), Clostridium innocuum B-3 (ATCC 14501) and C. ramosum 113-1 (ATCC 25582), M. gallisepticum PG31 (ATCC 19610) and M. iowae 695 (ATCC 33552) were used.
- PBMC peripheral blood cells
- the tubes are centrifuged for a few seconds at 1800 rpm to drop the drops, then the samples containing 2 x (10 ml on 5 ml of Ficoll) are centrifuged for 30 minutes at 1800 rpm at 4 ° C.
- the plasma, the red cells and the lymphocytes are thus separated.
- the band corresponding to the lymphocytes (4 2 ml Safelock R tubes) is recovered and also 2 ml samples of plasma (in a 2 ml Safelock R tube). These samples are centrifuged for 30 minutes at 20,000 g at 4 ° C.
- the supernatant is removed and the centrifugation pellet is washed with PBS (phosphate buffered saline) with removal of the supernatant and sedimentation of the cells from the pellet at 20,000 g after each washing. Centrifugation at 20,000 g allows the sedimentation of mycoplasmas which may have adhered to eukaryotic cells. The centrifugation pellets are stored at -20 ° C.
- PBS phosphate buffered saline
- lysis buffer 10 mM Tris-HCl, pH 7.5, EDTA ImM, 1% SDS.
- the cells are resuspended gently with the flexible disposable pipettes.
- the base remains compact. Take care not to splash liquid which could contaminate the other tubes.
- Phenol extraction, chlorophor extraction and precipitation in 2 volumes of absolute ethanol in the presence of 0.1 M NaCl are carried out.
- Washing is carried out with 70% ethanol. Gently vortex and unhook the pellet. Centrifuge at 15,000 g. Remove the supernatant. If too much ethanol remains at the bottom of the tube, dry the open tubes for a few tens of minutes under the laminar flow hood.
- 200 ⁇ l of DNA at 1 ⁇ g / 10 ⁇ l and 200 ⁇ l of DNA at 10 ng / 10 ⁇ l are prepared (two quantities of DNA 1 ⁇ g and 10 ng).
- the sterile distilled water which was used to make the dilutions is stored in a tube and will constitute the "water” control of the PCR reaction (tube n ° 1 of the PCR reaction) (see “PCR reaction”).
- the reaction is carried out in a volume of 50 ⁇ l, consisting of 40 ⁇ l of "mix” to which the Taq DNA polymerase (Amersha International pic) has been added, and of 10 ⁇ l of DNA.
- the reaction volume contains: the DNA to be amplified; the 4 dNTPs at 200 ⁇ M each; the 2 primers at 0.4 ⁇ M each (20 pmol); Taq buffer;
- a stock of 10 mM dNTPs is prepared from the 100 mM stock solution.
- the stock solution at 100 mM of each of the dNTPs is aliquoted in 100 ⁇ l tubes.
- the stock of 10 mM dNTPs is prepared by adding 900 ⁇ l of water to the 100 ⁇ l of stock solution. It is aliquoted into 250 ⁇ l tubes.
- 33 is the quantity in ⁇ g of DNA for an OD of 1 330 is the average mass of a nucleotide
- the concentration thus obtained is 20 ⁇ M.
- a dozen tubes containing volume v are prepared from the stock solution and stored at -20 ° C. It will remain to add the volume 1000-v of water to obtain the stock solution at 20 ⁇ M.
- the stock solution at 20 ⁇ M is itself aliquoted by 250 ⁇ l.
- dNTPs 10 mM 200 ⁇ l of each, i.e. 800 ⁇ l primers 20 ⁇ M: 200 ⁇ l of each, ie 400 ⁇ l 10X Taq: 1000 ⁇ l
- the mix is aliquoted in 1 ml tubes.
- tube n ° 1 "water” control corresponding to the dilution of the DNA.
- tube n ° 2 witness "lysis solution”.
- tubes n ° 3 to 2n + 2 DNA samples from the series.
- tubes n ° 2n + 3 witness "lysis solution”.
- tubes n ° 2n + 4 "water” indicator corresponding to the handling present.
- each cycle consists of a denaturation step at 94 ° C for 1 minute, a priming hybridization step (primers) at 55 ° C for 1 minute and an elongation step from the primer and the matrix to 72 "C for 2 minutes and 30 seconds.
- the DNA (17 ⁇ l of the PCR reaction) was analyzed by electrophoresis on a 1.5% agarose gel and transferred to a nylon membrane (Hybond N + Amersham International pic), according to the alkaline transfer technique ( Reed et al, 1985, Nucleic Acids Res. 3,2: 7207-7221). After the transfer, the filters were air dried for 1 hour and the DNA was fixed by UV irradiation for 3 minutes. Prehybridization was carried out in a solution containing 5 x SSC, 5 x Denhardt solution, 0.1% SDS and 200 ⁇ g or (10 mg / ml DNA) of denatured salmon sperm, at 42 ° C for 2 hours.
- Hybridization was carried out overnight at the same temperature in the same mixture, but by adding one of the two oligonucleotide probes MYCPIRS1 and MYCPIRS2, labeled in 5 ′ with ( 7 "32 P) ATP and the kinase polynucleotide T4 (United States Biochemical Corp.) and purified by chromatography on a sephadex column (Pharmacia LKB Biotechnology). After hybridization, the membranes were washed twice for 15 minutes at room temperature in 2 x SSC, 1% SDS, 2 times for 10 minutes at 55 "C in 0.2 SSC, 0.1% SDS, then quickly rinsed in 2 x SSC at room temperature. The hybridization signals were visualized by autoradiography on photographic film.
- the primers MYCPIRP and MYCPRIN were developed from the 16S rDNA (ribosomal DNA) sequence of M. pirum) (Weisburg et al, 1989, J. Bacteriol. 171: 6455-6467).
- 16S rDNA ribosomal DNA sequence of M. pirum
- the conserved sequences can be used to detect a group of organisms (Lane et al, 1985, Proc. Natl. Acad. Sci. USA.
- variable regions can be used to specifically detect a given organism.
- the rDNA sequences of different mollicutes were analyzed (Weisburg et al, 1989) the ribosomal DNA sequence of gram-positive bacteria phylogenetically related to mollicutes (C. innocuum, C. ramosum and B.
- FIG. 1 shows the alignment of part of the rDNAs of the 16S subunits (corresponding to nucleotides 49 to 233, based on the numbering of the rDNAs of the 16S subunit of E. coli (Brosius and al, 1978, Proc. Natl. Acad. Sci. USA 7_5: 4801-4805)), of all mollicutes belonging to the same phylogenetic group as M. pirum and designated by Weisburg as the "M. pneumoniae group” (Weisburg and al, 1989 cited above) (i.e. M. pneumoniae, M. pirum, M. matris, M. qallisepticum, M.
- the objective being the specific detection of M. pirum in animal or human tissues, which means the detection of a rare event in an environment of animal or human DNA, the primers have have been defined to achieve the best sensitivity in such an environment. This is especially necessary when it comes to detecting M. pirum in the blood, a tissue known to contain inhibitors of the PCR reaction such as hemoglobin.
- the best sensitivity is obtained when human DNA is not amplified with the primers used, that is to say when no additional band (due to non-specific amplification of human DNA) appears on the gel. after coloring with ethydium bromide. Consequently, primers were sought whose sequence corresponds to nucleotide sequences present with a low frequency in the human genome. Such primers were determined from octamers constituted by a nucleotide chosen initially and the 7 nucleotides preceding it in the sequence of M. pirum.
- MYCPIRP Two primers MYCPIRP and MYCPIRN corresponding respectively to the following sequences: MYCPIRP:
- CATCCTATAGCGGTCCAAAC3 were chosen in the regions of the ribosomal DNA of the 16S subunit containing the variable regions V6 for the first primer and Vil' for the last primer (Neefs et al, 1991, Nucleic ACid Res. 19: 1987-1999).
- the first nucleotide of MYCPIRP corresponds to position 52 of the rRNA of the 16S subunit of E. coli; the first nucleotide of MYCPIRN corresponds to position 207 of this same RNA (Brosius et ai, 1983).
- the 3 'end of these primers is specific for M. pirum.
- MYCPIRS1 and MYCPIRS2 located inside the amplified fragment, were chosen for perform Southern analyzes.
- MYCPIRS1 (5 » CAAATGTACTATCGCATGAGAAACATTT3 ', position 182) corresponds to the variable regions VIO and VIO' and is specific for M. pirum.
- MYCPIRS2 (5'GGGTGAGTAACACG TATCCAATCTACC3 *, position 110) corresponds to the conserved regions 7 and 8 and is not a specific sequence for M. pirum (FIG. 1).
- genomic DNA 10 ng, i.e. approximately 10 million genomes
- M. pirum M. pneumoniae, M. génitaliu, M. matured, M. iovae, M. qallisepticum and U. urealyticum
- MYCPIRP and MYCPIRN the primers MYCPIRP and MYCPIRN and the internal probes MYCPIRS1 and MYCPIRS2.
- No amplification signal of the expected size that is to say approximately 173 nucleotides was detected either on the agarose gels stained with ethydium bromide, or after Southern and hybridization with either other probe (figure 2).
- the signal obtained was comparable in intensity with the signal corresponding to the amplification of 100 fg (ie 10 3 genomes) of DNA from M. pirum.
- This signal can be explained by the fact that one of the two primers is homologous to the rDNA of the 16S subunit of M. qallisepticum.
- 10 ng of DNA from mollicutes belonging to other phylogenetic groups were also tested, again without obtaining an amplification signal.
- No amplification signal was obtained with 10 ng of DNA from two gram-positive bacteria with a low percentage of G + C bases, phylogenetically very close to the mollicutes, such as for example C. innocuum and C. ramosum, nor d another grain-positive bacteria B. subtilis.
- DNA from E. coli and mononuclear cells were also tested, leading to negative results ( Figures 2 and 3).
- the means described above make it possible to amplify and detect the DNA of M. pirum in the PBMCs of infected macaques.
- Detection of pathogens by PCR is a powerful diagnostic tool, provided that appropriate technical means are used to avoid obtaining a false positive.
- the detection of mycoplasmas used in these techniques is particularly indicated as their culture is tedious.
- the invention makes it possible for the first time to use 16S rDNA sequences of mollicutes for the specific detection by PCR of a given species of mollicutes, thus showing that the ribosomal sequences constitute a powerful tool for the detection of microorganisms.
- a couple of primers and two internal probes have been developed to detect specifically and with high sensitivity M. pirum by PCR in human tissues. M. pirum has also been detected in the blood of macaques experimentally infected with this mycoplasma. Detection of M.
- Example 2 Primers for the diagnosis of M. penetrans, M. pneumoniae and M. hominis.
- M. penetrans and M. pneumoniae which belong to the same phylogenetic group
- 16S ribosomal DNAs were aligned (Weisburg et al., 1989): - Phylogenetic group of M. pneumoniae: M. penetrans, M. pneumoniae, M. pirum, M. qallisepticum, M. matris, M. iowae and Ureaplasma urealyticum.
- M. hominis M. hominis and M. hyorhinis.
- M. hominis M. hominis, M. oral, M. salivarium, M. arthritidis, M. arginini, M. lipophilum, M. bovigenitalium, M. californicum, M. fermentans, M. agalactiae, M. pulmonis, M. sualvi, M. mobile, M. neurolyticum, M. hyopneumoniae and M. hyorhinis.
- M. pneumoniae M. pneumoniae, M. pirum and U. urealyticum.
- an amplification signal of the expected size (407 base pairs) was obtained after electrophoresis of the amplification products and staining with ethydium bromide, as well as after transfer and hybridization with the internal PENETS probe, radioactively labeled with 32 P.
- the sensitivity descended reproducibly to 10 fg of DNA (ie 10 genomes) of M. penetrans.
- the same sensitivity was obtained in the presence of 1 ⁇ g of human genomic DNA extracted from PBMCs (mononuclear cells of peripheral blood) (Fig 7). Assessment of the specificity of the test:
- No amplification signal (neither after staining with ethydium bromide nor after hybridization with PENETS) was obtained with excess DNA (10 ng, ie 10 7 genomes) of the following mycoplasmas: M. pirum, M. pneumoniae, M. genitalium, M. gallisepticum, M. iowae, M. matris, U. urealyticum, M. hyorhinis, M. arginini, M. oral, M. hominis, M. fermentans, M. mycoides, M. capricolum, S. citri, A. laidlawii, C. innocuum, C. ramosum, B. subtilis and E. coli (Fig 8).
- no amplification was obtained (neither after staining with ethydium bromide, nor after hybridization), from 1 ⁇ g of human PBMCs (FIG. 7).
- An amplification signal of 286 base pairs is obtained in a reproducible manner up to 10 fg (ie 10 genomes) of DNA from M. pneumoniae, after transfer and hybridization with PNEUS and this even in the presence of 1 ⁇ g of DNA human genomics extracted from PBMCs (F: ' i 9).
- An amplification signal of 170 bp of the rDNA16S fragment is obtained up to 10 fg of DNA (ie 10 genomes), after transfer and hybridization with HOMIS. This is verified even when the PCR is carried out in the presence of 1 ⁇ g of human genomic DNA extracted from PBMCs (FIG. 10).
- No amplification signal (after staining with ethyl bromide or after hybridization with the HOMIS probe) is obtained with excess DNA (10 ng, ie 10 7 genomes) of the following mycoplasmas: M. penetrans, M. pirum, M. pneumoniae, M. qenitalium, M. gallisepticum, M. iowae, M. matris, U. urealyticum, M. hyorhinis, M. arginini, M. oral, M. fermentans, M. mycoides, M. capricolum, S. citri, A. laidlawii, C. innocuum, C. ramosum, B. subtilis and E. coli (not shown).
- no amplification is obtained (neither after staining with ethydium bromide, nor after hybridization), from 1 ⁇ g of human PBMCs (FIG. 10).
- Example 3 Detection by PCR of Mycoplasma hominis and Ureaplasma urealyticum.
- This test was carried out using: a pair of primers specific for M. hominis (MYCHOMP and MYCHOMN), determined from the 16S rRNA sequence.
- MYCHOMS an oligonucleotide probe, MYCHOMS, for the detection of the amplification products obtained with MYCHOMP and MYCHOMN (Table 1).
- Amplified gene Fragment of the urease gene The components of this test can be combined in the form of a kit.
- Negative primer 400 nM dATP, dTTP, dGTP, dCTP 200 ⁇ M each
- the mixture was covered with 50 ⁇ l of mineral oil.
- the gel was then transferred onto a nylon membrane (Genescreen +, Du Pont, NEN) according to the alkaline transfer protocol recommended by the suppliers.
- the membrane was pre-hybridized for 3 h at 37 ° C in the following solution:
- the labeled probe was added to the prehybridization solution and the hybridization carried out for 16 h.
- the mixture was left at 37 ° C for 30 min, then at 65 ° C for 10 min.
- the labeled oligonucleotide was purified by chromatography on a Sephadex column (Pharmacia LKB), then added to the pre-hybridization solution.
- the mollicutes DNA was extracted according to conventional techniques. For example, lysis using a detergent (triton X100 or SDS) and proteinase K, phenol, chloroform, RNase A, phenol, chloroform, ethanol precipitation in the presence of 0.1M NaCl and glycogen which acted as a trainer, washing with 70% ethanol, drying and resuspension in sterile water. For example, 10 ⁇ l of the suspension obtained was used in the PCR reaction.
- a detergent triton X100 or SDS
- the M. hominis strains tested were as follows:
- Test sensitivity Reproducible results have been obtained up to 10 fg of DNA, or approximately 10 organisms (or 1 CFU), even in the presence of 1 ⁇ g of human genomic DNA.
- Example 4 PCR detection kit for the five main contaminating mycoplasmas of cell cultures.
- M. arginini MYCARGP and MYCARGN
- M. oral MYCORAP and MYCORAN
- M. hyorhinis MYCHYOP and MYCHYON
- M. fermentans MYCFERP and MYCFERN
- Acholeplas a laidlawii ALAIDLN
- oligonucleotide probes for the detection of the amplification products obtained from MYCARGP and MYCARGN, MYCORAP and MYCORAN, MYCHYOP and MYCHYON, MYCFERP and MYCFERN, ALADP (See Tables 3 to 7).
- the components of these tests can be grouped together in a kit.
- Table 3 Specific primers of M. arginini,
- Table 5 Specific primers of M. hyorhinis.
- Table 7 Specific primers of A. laidlawii.
- Negative primer 400 nM dATP, dTTP, dGTP, dCTP 200 ⁇ M each
- the mixture was covered with 50 ⁇ l of mineral oil.
- reaction mixture ie 17 ⁇ l
- a third of the reaction mixture ie 17 ⁇ l
- the gel was then transferred onto a nylon membrane (Genescreen +, Du Pont, NEN) according to the alkaline transfer protocol recommended by the suppliers.
- the membrane was pre-hybridized for 3 h at 37 ° C in the following solution:
- the labeled probe was added to the prethybridization solution and the hybridization carried out for 16 h.
- the mixture was left at 37 "C for 30 min, then at 65" C for 10 min.
- the labeled oligonucleotide was purified by chromatography on a Sephadex column (Pharmacia LKB), then added to the pre-hybridization solution.
- the membrane was then washed at 55 ° C: (4 X 5 min in 2XSSC, 1% SDS 4 X 5 min in 0.2XSSC, 0.1% SDS) then dried at room temperature and autoradiography.
- the DNA was extracted according to conventional techniques (for example, lysis using a detergent (triton X100 or SDS) and proteinase K, phenol, chloroform, RNase A, phenol, chloroform, precipitation with ethanol in the presence of 0.1M NaCl and glycogen which acts as a trainer, washing with 70% ethanol drying and resuspension in a few tens of ⁇ l of sterile water. For example, 10 ⁇ l of the suspension obtained in the reaction was used of PCR.
- a detergent triton X100 or SDS
- the results were reproducible up to 10 fg of DNA, or approximately 10 organisms (or 1 CFU), even in the presence of 1 ⁇ g of human genomic DNA.
- Another subject of the invention is the primers contained in the pairs of primers defined in the above pages.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Analytical Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description


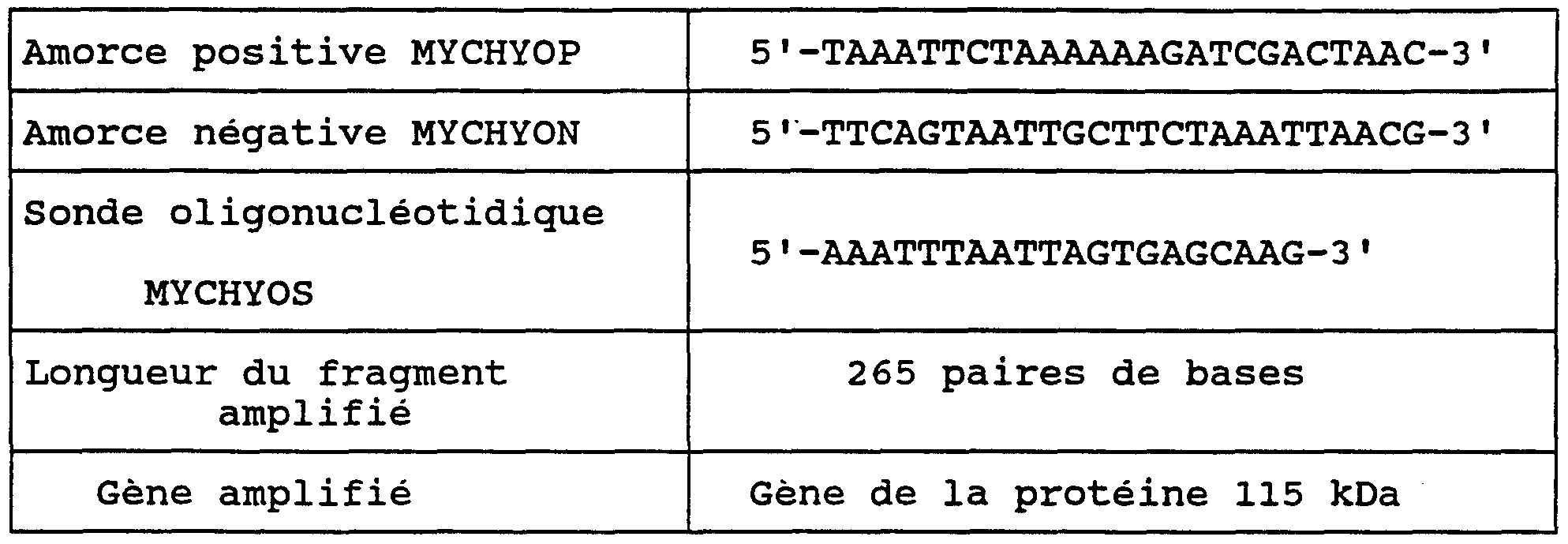


Claims
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP93917845A EP0654094A1 (fr) | 1992-07-30 | 1993-07-30 | Sequences d'oligonucleotides pour la detection specifique de mollicutes par amplification de genes conserves |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FR92/09486 | 1992-07-30 | ||
| FR9209486A FR2694768B1 (fr) | 1992-07-30 | 1992-07-30 | Séquences d'oligonucléotides pour la détection spécifique de mollicutes par amplication de gènes conservés. |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994003634A1 true WO1994003634A1 (fr) | 1994-02-17 |
Family
ID=9432479
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR1993/000784 WO1994003634A1 (fr) | 1992-07-30 | 1993-07-30 | Sequences d'oligonucleotides pour la detection specifique de mollicutes par amplification de genes conserves |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP0654094A1 (fr) |
| FR (1) | FR2694768B1 (fr) |
| WO (1) | WO1994003634A1 (fr) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996006949A2 (fr) * | 1994-08-29 | 1996-03-07 | Gen-Probe Incorporated | Sondes de detection par hybridation d'acides nucleiques ciblant les acides nucleiques de mycoplasma pneumoniae |
| US5965157A (en) * | 1993-04-02 | 1999-10-12 | Anticancer Inc. | Method to provide for production of hair coloring pigments in hair follicles |
| US6733776B1 (en) | 1992-04-02 | 2004-05-11 | Anticancer, Inc. | Method for promoting hair growth |
| WO2007136303A1 (fr) | 2006-05-23 | 2007-11-29 | Closed Company "Molecular-Medicine Technologies" | Procédé d'identification d'une infection uro-génitale, oligonucléotide, combinaison d'oligonucléotides, biopuce et ensemble sur cette base |
| WO2007083147A3 (fr) * | 2006-01-23 | 2007-12-21 | Stirus Global Solutions Ltd | Recherche à haut débit de micro-organismes dans un échantillon biologique |
| US7556825B2 (en) | 1993-04-02 | 2009-07-07 | Anticancer, Inc. | Method for promoting hair growth |
| US10557157B2 (en) | 2014-12-19 | 2020-02-11 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US10597689B2 (en) | 2014-04-30 | 2020-03-24 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US10717995B2 (en) | 2012-11-09 | 2020-07-21 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US10731192B2 (en) | 2012-11-09 | 2020-08-04 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1524320A1 (fr) * | 2003-10-15 | 2005-04-20 | Deutsches Krebsforschungszentrum Stiftung des öffentlichen Rechts | Détection et quantification de contamination par des mycoplasmes basée sur la technogie employant des réseaux d'ADN |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0250662A1 (fr) | 1986-06-25 | 1988-01-07 | The Regents Of The University Of California | Détection de mycoplasma par hybridation d'ADN |
| WO1988003957A1 (fr) | 1986-11-24 | 1988-06-02 | Gen-Probe Incorporated | Sondes d'acide nucleique pour la detection et/ou la quantification d'organismes non viraux |
| EP0305145A2 (fr) | 1987-08-24 | 1989-03-01 | Ortho Diagnostic Systems Inc. | Méthodes et sondes pour détecter des acides nucléiques |
| WO1990006374A1 (fr) * | 1988-12-09 | 1990-06-14 | Amrad Corporation Limited | Examen par adn amplifies |
| EP0457685A1 (fr) * | 1990-05-18 | 1991-11-21 | Institut Pasteur | Nouveaux mycoplasmes - moyens pour détecter et caractériser des mycoplasmes in vitro |
| EP0487218A1 (fr) | 1990-10-31 | 1992-05-27 | Tosoh Corporation | Procédé pour la détection ou détermination quantitative d'acides nucléiques cibles |
-
1992
- 1992-07-30 FR FR9209486A patent/FR2694768B1/fr not_active Expired - Fee Related
-
1993
- 1993-07-30 WO PCT/FR1993/000784 patent/WO1994003634A1/fr not_active Application Discontinuation
- 1993-07-30 EP EP93917845A patent/EP0654094A1/fr not_active Ceased
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0250662A1 (fr) | 1986-06-25 | 1988-01-07 | The Regents Of The University Of California | Détection de mycoplasma par hybridation d'ADN |
| WO1988003957A1 (fr) | 1986-11-24 | 1988-06-02 | Gen-Probe Incorporated | Sondes d'acide nucleique pour la detection et/ou la quantification d'organismes non viraux |
| EP0305145A2 (fr) | 1987-08-24 | 1989-03-01 | Ortho Diagnostic Systems Inc. | Méthodes et sondes pour détecter des acides nucléiques |
| WO1990006374A1 (fr) * | 1988-12-09 | 1990-06-14 | Amrad Corporation Limited | Examen par adn amplifies |
| EP0457685A1 (fr) * | 1990-05-18 | 1991-11-21 | Institut Pasteur | Nouveaux mycoplasmes - moyens pour détecter et caractériser des mycoplasmes in vitro |
| EP0487218A1 (fr) | 1990-10-31 | 1992-05-27 | Tosoh Corporation | Procédé pour la détection ou détermination quantitative d'acides nucléiques cibles |
Non-Patent Citations (8)
| Title |
|---|
| A. BLANCHARD ET AL., FEMS MICROBIOLOGY LETTERS, vol. 81, 1991, pages 37 - 42 |
| CLAVERIE, METH IN ENZYMOLOGY, vol. 183, 1990, pages 237 |
| DENG S ET AL.: "AMPLIFICATION OF 16S RRNA GENES FROM CULTURABLE AND NONCULTURABLE", J MICROBIOL METHODS 14 (1). 1991. 53-62. CODEN: JMIMDQ ISSN: 0167-7012 * |
| DENG S. ET AL., JOURNAL OF MICROBIOLOGICAL METHODS, vol. 14, September 1991 (1991-09-01), pages 53 - 61 |
| GÔBEL U.B., JOURNAL OF GENERAL MICROBIOLOGY, vol. 133, 1987, pages 1969 - 1974 |
| GRIFFAIS ET AL., NUCL. ACIDS. RES., vol. 19, no. 14, 1991, pages 3887 - 3891 |
| S. DENG ET AL.: "PCR Methods and Applications", 1992, COLD SPRING HARBOR LABORATORY PRESS, pages: 202 - 204 |
| U. B. GÖBEL ET AL.: "Oligonucleotide probes complementary to variable regions of ribosomal RNA discriminate between Mycoplasma species", J. GEN. MICROBIOL., vol. 133, 1987, pages 1969 - 1974 * |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6733776B1 (en) | 1992-04-02 | 2004-05-11 | Anticancer, Inc. | Method for promoting hair growth |
| US5965157A (en) * | 1993-04-02 | 1999-10-12 | Anticancer Inc. | Method to provide for production of hair coloring pigments in hair follicles |
| US7556825B2 (en) | 1993-04-02 | 2009-07-07 | Anticancer, Inc. | Method for promoting hair growth |
| WO1996006949A2 (fr) * | 1994-08-29 | 1996-03-07 | Gen-Probe Incorporated | Sondes de detection par hybridation d'acides nucleiques ciblant les acides nucleiques de mycoplasma pneumoniae |
| WO1996006949A3 (fr) * | 1994-08-29 | 1996-05-09 | Gen Probe Inc | Sondes de detection par hybridation d'acides nucleiques ciblant les acides nucleiques de mycoplasma pneumoniae |
| EP0713920A3 (fr) * | 1994-08-29 | 1996-07-31 | Gen Probe Inc | Sondes d'hybridation pour acide nucléique de mycoplasma pneumoniae |
| US5969122A (en) * | 1994-08-29 | 1999-10-19 | Gen-Probe Incorporated | Nucleic acid hybridization assay probes, helper probes and amplification oligonucleotides targeted to mycoplasma pneumoniae nucleic acid |
| EP0713920A2 (fr) * | 1994-08-29 | 1996-05-29 | Gen-Probe Incorporated | Sondes d'hybridation pour acide nucléique de mycoplasma pneumoniae |
| US5656427A (en) * | 1994-08-29 | 1997-08-12 | Gen-Probe Incorporated | Nucleic acid hybridization assay probes, helper probes and amplification oligonucleotides targeted to Mycoplasma pneumoniae nucleic acid |
| WO2007083147A3 (fr) * | 2006-01-23 | 2007-12-21 | Stirus Global Solutions Ltd | Recherche à haut débit de micro-organismes dans un échantillon biologique |
| WO2007136303A1 (fr) | 2006-05-23 | 2007-11-29 | Closed Company "Molecular-Medicine Technologies" | Procédé d'identification d'une infection uro-génitale, oligonucléotide, combinaison d'oligonucléotides, biopuce et ensemble sur cette base |
| US11434508B2 (en) | 2012-11-09 | 2022-09-06 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US11427844B2 (en) | 2012-11-09 | 2022-08-30 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US10717995B2 (en) | 2012-11-09 | 2020-07-21 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US10724057B2 (en) | 2012-11-09 | 2020-07-28 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US10731192B2 (en) | 2012-11-09 | 2020-08-04 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US11434507B2 (en) | 2012-11-09 | 2022-09-06 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US10597689B2 (en) | 2014-04-30 | 2020-03-24 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US10947573B2 (en) | 2014-04-30 | 2021-03-16 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US10907183B2 (en) | 2014-04-30 | 2021-02-02 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US11512334B2 (en) | 2014-04-30 | 2022-11-29 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US11773420B2 (en) | 2014-04-30 | 2023-10-03 | Versalis S.P.A. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US11091784B2 (en) | 2014-12-16 | 2021-08-17 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
| US10557157B2 (en) | 2014-12-19 | 2020-02-11 | Dsm Ip Assets B.V. | Process for enzymatic hydrolysis of lignocellulosic material and fermentation of sugars |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0654094A1 (fr) | 1995-05-24 |
| FR2694768B1 (fr) | 1994-12-23 |
| FR2694768A1 (fr) | 1994-02-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5849901A (en) | DNA fragments of mycobacteria, amplification primers hybridization probes, reagents and method for the detection of mycobacteria | |
| EP0490951B1 (fr) | Fragments d'acides nucleiques derives d'un genome de mycobacterie appropriee, leurs applications dans le diagnostic des infections a mycobacteries et plasmides contenant lesdits fragments | |
| JPH08510386A (ja) | ポリメラーゼ鎖反応によるサルモネラ菌の同定 | |
| WO1994003634A1 (fr) | Sequences d'oligonucleotides pour la detection specifique de mollicutes par amplification de genes conserves | |
| EP0719861B1 (fr) | Séquences nucléics issues du génome de Salmonella Typhi, et leurs utilisations notamment pour le diagnostic in vitro de la présence de bactéries du genre salmonella dans les produits alimentaires | |
| EP0461045B1 (fr) | Détection spécifique du mycobacterium tuberculosis | |
| EP1694858B1 (fr) | Procede de detection de mycobacteries pathogenes dans les specimens cliniques | |
| Fujihara et al. | Occurrence ofCandidatus Mycoplasma turicensis' infection in domestic cats in Japan | |
| JPH11508137A (ja) | 真菌細胞dnaの抽出、増幅および逐次ハイブリッド形成、並びに臨床物質中における真菌細胞の検出法 | |
| López et al. | Simultaneous identification of five marine fish pathogens belonging to the genera Tenacibaculum, Vibrio, Photobacterium and Pseudomonas by reverse line blot hybridization | |
| WO2003097815A2 (fr) | Identification moleculaire de l'espece aspergillus | |
| EP0745691A2 (fr) | Fragment nucléotidique de l'ARN ribosomique 16s de corynébactéries, sondes et amorces dérivées, réactif et procédé de détection | |
| FR2659086A1 (fr) | Fragments nucleotidiques caracteristiques de fragments d'adn de mycobacteries. leur utilisation comme amorce pour la detection d'une infection due a des mycobacteries. | |
| JP5621992B2 (ja) | カンジダ種を検出および同定するための方法 | |
| EP1254253B1 (fr) | Oligonucleotides monocatenaires, sondes, amorces et procede de detection des spirochetes | |
| Moraes et al. | Bacteroides fragilis isolates compared by AP-PCR | |
| FR2764293A1 (fr) | Fragment nucleotidique de l'arn 23s de bacteries du genre chlamydia, utilisations comme sonde, amorce, et dans un reactif et un procede de detection | |
| Avila‐Campos et al. | Specific primer for AP‐PCR identification of Actinobacillus actinomycetemcomitans | |
| CA2306445A1 (fr) | Moyens de documentation de repertoires en immunorecepteurs nkr et/ou en immunorecepteurs contreparties activatrices ou non inhibitrices d'immunorecepteurs nkr | |
| WO1996000299A1 (fr) | Fragments d'acides nucleiques, derives du genome de mycobacterium xenopi et leurs applications |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): JP US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1993917845 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993917845 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref country code: US Ref document number: 1995 367142 Date of ref document: 19950703 Kind code of ref document: A Format of ref document f/p: F |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1993917845 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1993917845 Country of ref document: EP |