WO1993018001A1 - Preparation of intermediates in the synthesis of quinoline antibiotics - Google Patents
Preparation of intermediates in the synthesis of quinoline antibiotics Download PDFInfo
- Publication number
- WO1993018001A1 WO1993018001A1 PCT/US1993/000008 US9300008W WO9318001A1 WO 1993018001 A1 WO1993018001 A1 WO 1993018001A1 US 9300008 W US9300008 W US 9300008W WO 9318001 A1 WO9318001 A1 WO 9318001A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- process according
- benzyl
- alkyl
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/52—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring condensed with a ring other than six-membered
Definitions
- This invention relates to novel processes for the preparation of intermediates in the synthesis of the quinoline antibiotic 7-(1 ⁇ ,5 ⁇ ,6 ⁇ )-(6-amino-3-azabicyclo[3.1.0]hex-3- yl)-1 -(2,4-difiuorophenyl)-6-fluoro-1 ,4-dihydro-4-oxo-1 ,8-naphthyridine-3-carboxylic acid and related antibiotic compounds.
- the quinoline antibiotic 7-(1 ⁇ ,5 ⁇ .6 ⁇ )-(6-amino-3- azabicyclo[3.1.0]hex-3-yl)-1-(2,4-difluorophenyl)-6-fluoro-1 ,4-dihydro-4-oxo-1 ,8- naphthyridine-3-carboxylic acid has the chemical formula
- novel methods of this invention may be used to prepare compounds of the formula
- R is (C,-C ⁇ ) aJkyl, (C 3 -C ⁇ )cycloalkyl or benzyl, wherein the phenyl moiety of said benzyl group may be substituted, optionally, with one or more substituents independently selected from halo (e.g., chloro, fiuoro, bromo or iodo), nitro, (C.-C ⁇ ) alkyl, (C,-C ⁇ ) alkoxy, amino and trifluoromethyl, comprising reacting a compound of the formula
- the compound of formula III formed in the above process is a compound wherein R is (C 1 -C ⁇ )alkyl or benzyl. In a more preferred embodiment, R is benzyl.
- halo refers to chloro, fiuoro, bromo or iodo.
- This invention also relates to the process described above, further comprising reacting the compound of formula III so formed with a reducing agent to form a compound of the formula
- This invention also relates to compounds having the formula
- reaction of a compound having formula II with a halonitromethane preferably chloronitromethane (CICH 2 N0 2 ) or bromonitromethane (BrCH 2 NO 2 ), in the presence of a base yields the corresponding compound of the formula III.
- a halonitromethane preferably chloronitromethane (CICH 2 N0 2 ) or bromonitromethane (BrCH 2 NO 2 )
- This reaction is generally conducted in an inert, polar, aprotic solvent such as dimethytformamide (DMF), dimethylsutfoxide (DMSO) ordimethylacetamide (DMAC), an inert ethereal solvent such as ethyl ether, glyme or tetrahydrofuran (THF), or another inert solvent such as benzene, toluene or a chlorinated benzene or toluene. Toluene is preferred. Suitable reaction temperatures range from about -78 °C to about 80° C, with about 0°C being preferred. It is preferable to add the base last.
- aprotic solvent such as dimethytformamide (DMF), dimethylsutfoxide (DMSO) ordimethylacetamide (DMAC), an inert ethereal solvent such as ethyl ether, glyme or tetrahydrofuran (THF), or another inert
- bases include carbonate bases such as potassium or sodium carbonate, phosphorine amide bases such as 2-tert-butylimino-2-diethylamino-1 ,3- dimethylperhydro-1,3,2-diaza-phosphorine, and amine bases such as triethylamine, quanidine, diisopropylethylamine.tetramethyl quanidine, 1 ,8-diazobicyclo-[5.4.0]undec- 7-ene (DBU) and 1 ,5-diazobicyclo-[4.3.0]non-5-ene (DBN). It is advantageous to use an amine base and, most preferably, to use DBU.
- reducing agents include borane/dimethyisulfide, borane/THF, sodium borohydride and aborontrifluoride «etherate mixture.
- the preferred reducing agent is borane THF.
- the reduction is typically carried out at temperatures ranging from about 45°C to about 90°C, in an inert ethereal solvent such as glyme, diglyme, diethylether, diisopropyl ether or THF. It is preferably carried out at about 66°C in THF.
- the resulting compound of the formula IV may be converted into the corresponding amine of formula V by treating H with a metal and an inorganic acid.
- a metal is zinc.
- Suitable inorganic acids include hydrochloric acid, sutfuric acid. Hydrochloric acid is preferred.
- This reaction is generally conducted in a lower alcohol solvent such as ethanol, methanol, 1-propanoi or 2-propanol, preferably ethanol, at a temperature from about 0°C to about 80 °C, preferably at about 25 °C.
- the corresponding compound of formula VI, wherein X is a nitrogen protecting group is then formed by adding a suitable nitrogen protecting group to the unsubstituted- amino nitrogen of the compound of formula V.
- suitable nitrogen protecting group include (C 2 -C ⁇ ) alkoxycarbonyl, optionally substituted benzyloxycarbonyl, aryioxycarbonyl, silyl, trityl, vinyloxycarbonyl, O-nitrophenylsulfonyl, diphenylphosphinyl, p-toluenesulfonyl, and benzyl.
- di-t-butyldicarbonate or 2-t-butoxycarbonyloxyimino-2- phenylacetonitrile is advantageous to use.
- the addition of the nitrogen protecting group is usually earned out in a chlorinated hydrocarbon solvent such as methylene chloride or 1 ,2-dichloroethane, or an ethereal solvent such as glyme, diglyme or THF, in the presence or absence of a catalytic amount of an amine base such as triethylamine, diisopropylethylamine or pyridine, preferably triethylamine, at a temperature from about 0°C to about 50°C, preferably at about 25 °C.
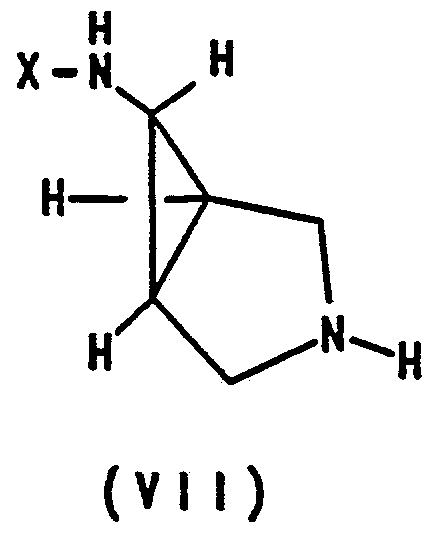
- the hydrogenolytic removal of the R group from the compound of formula VI formed in the foregoing step yields the desired compound of formula VII.
- This is generally accomplished by reacting the compound of formula VI, wherein R is benzyl, with hydrogen gas at a pressure from about 0 psi to about 2000 psi, preferably about 50 psi, in the presence of a noble catalyst such as palladium, platinum or rhodium. Palladium on carbon or palladium hydroxide on carbon is preferred.
- the temperature may range from about 20°C to about 80°C, and is preferably about 25 °C.
- the solvent is usually a lower alcohol and is preferably methanol.
- R is (C,-C ⁇ ) alkyl or (C 3 -C ⁇ ) cycloalkyl
- the R group may be removed by reaction with o ⁇ chloroethylchloroformate (ACE-Cl).
- ACE-Cl o ⁇ chloroethylchloroformate
- the antibacterial compound having formula I and the related azabicyclo quinoline carboxylic acid antibiotics that can be synthesized using the methods and compounds of this invention are useful in the treatment of animals, including humans, having bacterial infections. They are useful in treating bacterial infections of broad spectrum, particularly in treating granvpositive bacterial strains.
- Unrted States Patent Application 07/551,212 and World Patent Application WO 91/02526 set forth in detail the appropriate dosage ranges and methods of administration of such antibiotic compounds. These references also set forth a method by which the antibacterial activity of such compounds may be determined.
- the following examples illustrate the methods and compounds of the present invention, ft will be understood, however, that the invention is not limited to the specific details of these examples.
- Example 4 (2.0 g, 6.94 mmol) in 50 ml of methanol was added palladium hydroxide on carbon (Pd(OH)- C) (50% wet) (1.0 g, 50% by weight). The mixture was hydrogenated at 50 PSI for 6 hours and was then filtered through Celite* and the solvent was evaporated to provide 1.36 g of the product in 99% yield.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description








Claims







Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HU9402530A HU215837B (en) | 1992-03-02 | 1993-01-07 | Preparation of intermediates in the synthesis of quinoline antibiotics and intermediates of this preparation |
| EP93902893A EP0629189B1 (en) | 1992-03-02 | 1993-01-07 | Preparation of intermediates in the synthesis of quinoline antibiotics |
| DE69312913T DE69312913T2 (en) | 1992-03-02 | 1993-01-07 | METHOD FOR PRODUCING INTERMEDIATE PRODUCTS FOR THE SYNTHESIS OF CHINOLINE ANTIBIOTICS |
| JP5515648A JP2564247B2 (en) | 1992-03-02 | 1993-01-07 | Preparation of intermediates in the synthesis of quinoline antibiotics |
| AU34300/93A AU667872B2 (en) | 1992-03-02 | 1993-01-07 | Preparation of intermediates in the synthesis of quinoline antibiotics |
| KR1019940703051A KR950700251A (en) | 1992-03-02 | 1993-01-07 | PREPARATION OF INTERMEDIATES IN THE SYNTHESIS OF QUINOLINE ANTIBIOTICS |
| KR1019940703051A KR0135626B1 (en) | 1992-03-02 | 1993-01-07 | Preparation of intermediates in the synthesis of quinoline |
| FI944013A FI106022B (en) | 1992-03-02 | 1994-09-01 | Process for the preparation of (1a, 5a, 6a) -3- (N-substituted) -6 - [(N-protected) amino] -3-azabicyclo [3.1.0] hexane intermediates in the synthesis of quinoline antibiotics and intermediates in the process |
| NO943243A NO300681B1 (en) | 1992-03-02 | 1994-09-01 | Process for the preparation of intermediates for the synthesis of quinoline antibiotics |
| GR970402413T GR3024789T3 (en) | 1992-03-02 | 1997-09-17 | Preparation of intermediates in the synthesis of quinoline antibiotics. |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US844,367 | 1992-03-02 | ||
| US07/844,367 US5256791A (en) | 1992-03-02 | 1992-03-02 | Preparation of intermediates in the synthesis of quinoline antibiotics |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1993018001A1 true WO1993018001A1 (en) | 1993-09-16 |
Family
ID=25292537
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1993/000008 WO1993018001A1 (en) | 1992-03-02 | 1993-01-07 | Preparation of intermediates in the synthesis of quinoline antibiotics |
Country Status (22)
| Country | Link |
|---|---|
| US (2) | US5256791A (en) |
| EP (1) | EP0629189B1 (en) |
| JP (1) | JP2564247B2 (en) |
| KR (2) | KR950700251A (en) |
| CN (1) | CN1037100C (en) |
| AT (1) | ATE156480T1 (en) |
| AU (1) | AU667872B2 (en) |
| CA (1) | CA2131160C (en) |
| DE (1) | DE69312913T2 (en) |
| DK (1) | DK0629189T3 (en) |
| ES (1) | ES2105217T3 (en) |
| FI (1) | FI106022B (en) |
| GR (1) | GR3024789T3 (en) |
| HU (1) | HU215837B (en) |
| IL (1) | IL104818A (en) |
| MX (1) | MX9301138A (en) |
| MY (1) | MY107723A (en) |
| NO (1) | NO300681B1 (en) |
| NZ (1) | NZ246768A (en) |
| TW (1) | TW211002B (en) |
| WO (1) | WO1993018001A1 (en) |
| ZA (1) | ZA931428B (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997019921A1 (en) * | 1995-11-30 | 1997-06-05 | Pfizer Limited | Process for preparing dioxoazabicyclohexanes |
| EP0818446A1 (en) * | 1996-07-09 | 1998-01-14 | Pfizer Limited | Process for preparing 2,4-dioxo-3-azabicyclo(3.1.0)hexanes |
| WO1999006368A1 (en) * | 1997-08-02 | 1999-02-11 | Bayer Aktiengesellschaft | Special 3-azabicyclo[3.1.0]hexanes, method for producing and modifying the same, and their use |
| EP0930297A1 (en) * | 1998-01-16 | 1999-07-21 | Pfizer Products Inc. | A process for preparing naphthyridones and intermediates |
| US7019142B2 (en) | 1998-01-16 | 2006-03-28 | Pfizer Inc. | Process for preparing naphthyridones and intermediates |
| WO2008010061A2 (en) * | 2006-07-17 | 2008-01-24 | Glenmark Pharmaceuticals S.A. | 3-azabicyclo [3.1.0] hexane vanilloid receptor ligands, pharmaceutical compositions containing them, and processes for their preparation |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5475116A (en) * | 1994-04-29 | 1995-12-12 | Pfizer Inc. | Aza bicyclo[3,1,0]hexane intermediates useful in the synthesis of quinolones |
| US5929240A (en) * | 1994-12-12 | 1999-07-27 | Pfizer Inc. | Process and intermediates for preparing naphthyridonecarboxylic acid salts |
| JPH0912547A (en) * | 1995-06-23 | 1997-01-14 | Chisso Corp | Production of intermediate for new quinolone-based compound |
| JPH1087617A (en) * | 1996-07-09 | 1998-04-07 | Pfizer Inc | Production of intermediate useful for synthesizing quinoline-based antibiotic |
| US6057455A (en) * | 1996-07-09 | 2000-05-02 | Pfizer, Inc. | Preparation of intermediates useful in the synthesis of quinoline antibiotics |
| HN1998000106A (en) | 1997-08-01 | 1999-01-08 | Pfizer Prod Inc | PARENTERAL COMPOSITIONS OF ALATROFLAXACINO |
| US6184380B1 (en) * | 1999-01-25 | 2001-02-06 | Pfizer Inc. | Process for preparing naphthyridones and intermediates |
| MX2021014441A (en) | 2019-05-31 | 2022-01-06 | Ikena Oncology Inc | Tead inhibitors and uses thereof. |
| CA3142351A1 (en) | 2019-05-31 | 2020-12-03 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| WO2023151188A1 (en) * | 2022-02-08 | 2023-08-17 | 上海皓元医药股份有限公司 | Green synthesis method of antiviral drug intermediate |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0007128A1 (en) * | 1978-07-06 | 1980-01-23 | Shell Internationale Researchmaatschappij B.V. | Derivatives of 3-azabicyclo(3.1.0)hexane and processes for their preparation |
| EP0010799A1 (en) * | 1978-10-27 | 1980-05-14 | Shell Internationale Researchmaatschappij B.V. | A process for the preparation of 3-azabicyclo(3.1.0)hexane derivatives and modifications thereof |
| EP0413455A2 (en) * | 1989-08-16 | 1991-02-20 | Pfizer Inc. | Azabicyclo quinolone carboxylic acids |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0676400B2 (en) * | 1987-08-25 | 1994-09-28 | 大日本製薬株式会社 | Novel pyridonecarboxylic acid derivative, its ester and its salt |
| US5196548A (en) * | 1989-05-11 | 1993-03-23 | Pfizer Inc. | Preparation of diazabicyclic Intermediates |
| US5200527A (en) * | 1991-04-08 | 1993-04-06 | Lonza Ltd. | Process for the production of 2-azabicyclo [2.2.1] hept-5-en-3-one |
-
1992
- 1992-03-02 US US07/844,367 patent/US5256791A/en not_active Expired - Fee Related
-
1993
- 1993-01-07 HU HU9402530A patent/HU215837B/en not_active IP Right Cessation
- 1993-01-07 KR KR1019940703051A patent/KR950700251A/en not_active IP Right Cessation
- 1993-01-07 DK DK93902893.2T patent/DK0629189T3/en active
- 1993-01-07 CA CA002131160A patent/CA2131160C/en not_active Expired - Fee Related
- 1993-01-07 DE DE69312913T patent/DE69312913T2/en not_active Expired - Fee Related
- 1993-01-07 JP JP5515648A patent/JP2564247B2/en not_active Expired - Lifetime
- 1993-01-07 NZ NZ246768A patent/NZ246768A/en unknown
- 1993-01-07 KR KR1019940703051A patent/KR0135626B1/en active
- 1993-01-07 ES ES93902893T patent/ES2105217T3/en not_active Expired - Lifetime
- 1993-01-07 AU AU34300/93A patent/AU667872B2/en not_active Ceased
- 1993-01-07 EP EP93902893A patent/EP0629189B1/en not_active Expired - Lifetime
- 1993-01-07 AT AT93902893T patent/ATE156480T1/en active
- 1993-01-07 WO PCT/US1993/000008 patent/WO1993018001A1/en active IP Right Grant
- 1993-01-11 TW TW082100123A patent/TW211002B/en active
- 1993-02-22 IL IL104818A patent/IL104818A/en not_active IP Right Cessation
- 1993-03-01 CN CN93102103A patent/CN1037100C/en not_active Expired - Fee Related
- 1993-03-01 ZA ZA931428A patent/ZA931428B/en unknown
- 1993-03-01 MX MX9301138A patent/MX9301138A/en not_active IP Right Cessation
- 1993-03-01 MY MYPI93000368A patent/MY107723A/en unknown
- 1993-08-04 US US08/101,879 patent/US5298629A/en not_active Expired - Fee Related
-
1994
- 1994-09-01 NO NO943243A patent/NO300681B1/en not_active IP Right Cessation
- 1994-09-01 FI FI944013A patent/FI106022B/en active
-
1997
- 1997-09-17 GR GR970402413T patent/GR3024789T3/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0007128A1 (en) * | 1978-07-06 | 1980-01-23 | Shell Internationale Researchmaatschappij B.V. | Derivatives of 3-azabicyclo(3.1.0)hexane and processes for their preparation |
| EP0010799A1 (en) * | 1978-10-27 | 1980-05-14 | Shell Internationale Researchmaatschappij B.V. | A process for the preparation of 3-azabicyclo(3.1.0)hexane derivatives and modifications thereof |
| EP0413455A2 (en) * | 1989-08-16 | 1991-02-20 | Pfizer Inc. | Azabicyclo quinolone carboxylic acids |
Non-Patent Citations (5)
| Title |
|---|
| JOURNAL OF ORGANIC CHEMISTRY. vol. 44, no. 8, 13 April 1979, EASTON US pages 1195 - 1199 * |
| JOURNAL OF THE AMERICAN CHEMICAL SOCIETY. vol. 114, 1992, GASTON, PA US pages 344 - 345 'Spectroscopic detection of two neutral ÄCHNO2Ü isomers: Nitrocarbene and Nitrosoformaldehyde' * |
| ORGANIC REACTIONS vol. 13, 1963, pages 55 - 90 W. E. PARHAM, E. E. SCHWEIZER 'Halocyclopropanes from halocarbenes' * |
| TETRAHEDRON LETTERS. vol. 29, no. 9, 1988, OXFORD GB pages 987 - 990 * |
| TETRAHEDRON, (INCL. TETRAHEDRON REPORTS) vol. 46, no. 24, 1990, OXFORD GB pages 8117 - 8130 * |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997019921A1 (en) * | 1995-11-30 | 1997-06-05 | Pfizer Limited | Process for preparing dioxoazabicyclohexanes |
| AP746A (en) * | 1995-11-30 | 1999-04-29 | Pfizer Res & Development Company N V /S A | Process for preparing dioxoazabicyclohexanes. |
| EP0818446A1 (en) * | 1996-07-09 | 1998-01-14 | Pfizer Limited | Process for preparing 2,4-dioxo-3-azabicyclo(3.1.0)hexanes |
| US5847158A (en) * | 1996-07-09 | 1998-12-08 | Pfizer Inc. | Process for preparing 2,4-dioxo-3-azabicyclo 3.1.0!Hexanes |
| WO1999006368A1 (en) * | 1997-08-02 | 1999-02-11 | Bayer Aktiengesellschaft | Special 3-azabicyclo[3.1.0]hexanes, method for producing and modifying the same, and their use |
| EP0930297A1 (en) * | 1998-01-16 | 1999-07-21 | Pfizer Products Inc. | A process for preparing naphthyridones and intermediates |
| US7019142B2 (en) | 1998-01-16 | 2006-03-28 | Pfizer Inc. | Process for preparing naphthyridones and intermediates |
| WO2008010061A2 (en) * | 2006-07-17 | 2008-01-24 | Glenmark Pharmaceuticals S.A. | 3-azabicyclo [3.1.0] hexane vanilloid receptor ligands, pharmaceutical compositions containing them, and processes for their preparation |
| WO2008010061A3 (en) * | 2006-07-17 | 2008-04-17 | Glenmark Pharmaceuticals Sa | 3-azabicyclo [3.1.0] hexane vanilloid receptor ligands, pharmaceutical compositions containing them, and processes for their preparation |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0629189B1 (en) | Preparation of intermediates in the synthesis of quinoline antibiotics | |
| EP0001500A1 (en) | 1-Carbocyclic aryl-2-mono or -bis(alkoxycarbonyl) guanidino ethanes, and methods for their preparation and the preparation therefrom of 4,5-dihydro-2-alkoxycarbonylamino-5-carbocyclic aryl imidazoles | |
| KR910007887B1 (en) | Process for the preparation of 1,4 - diazabicyclo (3,2,2) nonane | |
| CN103180308B (en) | Novel method of preparing benzoimidazole derivatives | |
| US5475116A (en) | Aza bicyclo[3,1,0]hexane intermediates useful in the synthesis of quinolones | |
| FR2649100A1 (en) | NOVEL AZETIDINES, THEIR PREPARATION AND THEIR APPLICATION AS INTERMEDIATES FOR THE PREPARATION OF COMPOUNDS WITH ANTIMICROBIAL ACTIVITY | |
| MX2007008214A (en) | Preparation of ketone amides. | |
| US6423845B1 (en) | Process and intermediates to a tetrahydro-[1,8]- Naphthyridine | |
| JP4173599B2 (en) | Process for producing 6-hydroxy-2-oxo-1,2,3,4-tetrahydroquinoline | |
| EP1179532B1 (en) | Process for the production of indole derivatives and intermediates therefor | |
| JP3836777B2 (en) | Production method of fluorine-containing compounds | |
| US5929240A (en) | Process and intermediates for preparing naphthyridonecarboxylic acid salts | |
| AU703541B2 (en) | An intermediate for preparing naphthyridonecarboxylic acid salts | |
| US5026845A (en) | 1,4-diazabicyclo[3.2.2.]nonane and intermediates for the preparation thereof | |
| US20020019532A1 (en) | Process for the synthesis of (2S)-phenyl-3-piperidone | |
| EP4061799A1 (en) | Process of preparing arachidonoylethanolamine analogues | |
| JP2739505B2 (en) | Method for producing optically active 3,4-dehydropyrrolidine compound | |
| EP2125733A1 (en) | Process for preparing ccr-5 receptor antagonists utilizing 4-substituted 1-cyclopropane-sulfonyl-piperidinyl compounds | |
| JPH01319496A (en) | Uracil derivative | |
| JP2002053536A (en) | Spiroaminopyrrolidine derivative and method for producing the same | |
| KR20180093307A (en) | Preparing Method of 4,5-diamino substituted pyrimidine derivatives and Novel Compound for Preparing thereof | |
| JP2002179665A (en) | Method for producing 2(5h)-furanone derivative | |
| JPH06220052A (en) | Production of imide derivative | |
| JPS6157581A (en) | 1,4-dihydro-2-piperazinopyridopyrimidine derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA FI HU JP KR NO NZ US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 246768 Country of ref document: NZ Ref document number: 1993902893 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2131160 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 944013 Country of ref document: FI |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993902893 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1993902893 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 944013 Country of ref document: FI |