USRE44987E1 - Bromo-phenyl substituted thiazolyl dihydropyrimidines - Google Patents
Bromo-phenyl substituted thiazolyl dihydropyrimidines Download PDFInfo
- Publication number
- USRE44987E1 USRE44987E1 US13/869,981 US201313869981A USRE44987E US RE44987 E1 USRE44987 E1 US RE44987E1 US 201313869981 A US201313869981 A US 201313869981A US RE44987 E USRE44987 E US RE44987E
- Authority
- US
- United States
- Prior art keywords
- hepatitis
- salt
- formula
- infection
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 [1*]C1=C(C2N=C([6*])NC(CN3CCOCC3)=C2C([3*])=O)C=CC([2*])=C1 Chemical compound [1*]C1=C(C2N=C([6*])NC(CN3CCOCC3)=C2C([3*])=O)C=CC([2*])=C1 0.000 description 16
- GFWWQGUAHZKPML-UHFFFAOYSA-N CC1=NC=C(F)C=C1F Chemical compound CC1=NC=C(F)C=C1F GFWWQGUAHZKPML-UHFFFAOYSA-N 0.000 description 3
- VZWOXDYRBDIHMA-UHFFFAOYSA-N CC1=NC=CS1 Chemical compound CC1=NC=CS1 VZWOXDYRBDIHMA-UHFFFAOYSA-N 0.000 description 3
- FGCZYICKZZNEEU-VHEBQXMUSA-N O=C(N/C(C(=O)N1CCCCC1)=C(/Cl)C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound O=C(N/C(C(=O)N1CCCCC1)=C(/Cl)C1=CC=CC=C1)C1=CC=CC=C1 FGCZYICKZZNEEU-VHEBQXMUSA-N 0.000 description 3
- SQGRDKSRFFUBBU-UHFFFAOYSA-N [H]N1C(C2=NC=CS2)=NC(C2=C(Br)C=C(F)C=C2)C(C(=O)OCC)=C1CN1CCOCC1 Chemical compound [H]N1C(C2=NC=CS2)=NC(C2=C(Br)C=C(F)C=C2)C(C(=O)OCC)=C1CN1CCOCC1 SQGRDKSRFFUBBU-UHFFFAOYSA-N 0.000 description 2
- ZIGBVTIJGLEPKF-UHFFFAOYSA-N [H]N1C(C2=NC=CS2)=NC(C2=C(Br)C=C(F)C=C2)C(C(=O)OC)=C1CN1CCOCC1 Chemical compound [H]N1C(C2=NC=CS2)=NC(C2=C(Br)C=C(F)C=C2)C(C(=O)OC)=C1CN1CCOCC1 ZIGBVTIJGLEPKF-UHFFFAOYSA-N 0.000 description 1
- SLUQDVUBZBWZMD-UHFFFAOYSA-N [H]N1C(C2=NC=CS2)=NC(C2=C(Br)C=C(F)C=C2)C(C(=O)OCC)=C1C Chemical compound [H]N1C(C2=NC=CS2)=NC(C2=C(Br)C=C(F)C=C2)C(C(=O)OCC)=C1C SLUQDVUBZBWZMD-UHFFFAOYSA-N 0.000 description 1
- ACVCAKOXDIMMMW-UHFFFAOYSA-N [H]N1C(C2=NC=CS2)=NC(C2=C(Br)C=C(F)C=C2)C(C(=O)OCC)=C1CBr Chemical compound [H]N1C(C2=NC=CS2)=NC(C2=C(Br)C=C(F)C=C2)C(C(=O)OCC)=C1CBr ACVCAKOXDIMMMW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the invention relates to a new bromo-phenyl substituted thiazolyl dihydropyrimidine, its preparation method and use as a medicament especially for treating and preventing hepatitis B infections.
- the invention also relates to a composition comprising the dihydropyrimidine, other antiviral agent and, when appropriate, an immunomodulator and a medicament comprising the composition especially for treating and preventing HBV infections such as hepatitis B infections.
- the hepatitis B virus belongs to the family of hepadna viruses. It can cause acute and/or persistent or progressive chronic diseases. Many other clinical manifestations in the pathological state are also caused by the hepatitis B virus—in particular chronic inflammation of the liver, cirrhosis of the liver and hepatocellular carcinoma. In addition, coinfection with the heptatitis delta virus may have adverse effects on the progress of the disease.
- the interferon and lamivudine are conventional medicaments approved to be used for treating chronic hepatitis.
- the interferon has just moderate activity but has an adverse side reaction.
- lamivudine has good activity, its resistance develops rapidly during the treatment and relapse effects often appear after the treatment is stopped.
- the IC 50 value of lamivudine (3-TC) is 300 nM (Science, 299 (2003), 893-896).
- U.S. Pat. No. 7,074,784 discloses 6-amidoalkyldihydropyrimidine and its use as a medicament especially for treating and preventing hepatitis B infection.
- R 1 is o-chlorine
- R 2 is p-chlorine
- R 6 is 3,5-difluoro-pyridin-2-yl
- X is —CH 2 —
- Z is morpholinyl.
- the compound can inhibit the growth of hepatitis B virus during cell culturing.
- the IC 50 value is 2 nM (tested by themselves).
- U.S. Pat. No. 7,074,784 B2 also discloses an example, wherein a difluoro residue is substituted for thiazol-2-yl (described in Example 45 of the patent).
- the derivative has a similar IC 50 value (2 nM) (see Table 1).
- This invention relates to a compound having formula (I) and its isomer (Ia),
- R 1 is o-bromine
- R 2 is p-fluorine
- R 3 is C 1 -C 4 alkyl
- R 6 is thiazol-2-yl
- X is methylene
- Z is morpholinyl.
- R 1 of the compound of the invention having formula (I) and (Ia) is o-bromine
- R 2 is p-fluorine
- R 3 is methyl or ethyl
- R 6 is thiazol-2-yl
- X is methylene
- Z is morpholinyl.
- This invention also relates to an enantiomer of the compound disclosed herein and a mixture thereof.
- the racemate can be separated by a known method, and fundamentally it is a homogeneous component in a stereoisomer mixture.
- the compounds of the invention comprise an isomer having formula (I) and (Ia) and a mixture thereof.
- the compound of the invention can also be in a form of a salt, preferably a physiologically acceptable salt.
- the physiologically acceptable salt can be an inorganic acid salt or organic acid salt.
- it is an inorganic acid salt such as chloride, bromide, phosphate or sulfate, etc., or a carboxylate or a sulfonate, such as acetate, maleate, fumarate, malate, citrate, tartarate, lactate, benzoate or methanesulfonate, ethanesulfonate, benzenesulfonate, toluenesulfonate or naphthalenedisulfonate, etc.
- the physiologically acceptable salt can also be a metal salt or an ammonium salt of the compound of the invention.
- it is a sodium salt, potassium salt, magnesium salt or calcium salt, and an ammonium salt produced by ammonia or organic amine such as ethylamine, diethylamine or triethylamine, diethanolamine or triethanolamine, dicyclohexylamine, dimethylaminoethyl alcohol, arginine, lysine, ethylenediamine or 2-phenylethylamine, etc.
- the compound (I) of the invention can be prepared by the following methods:
- R 1 , R 2 , R 3 , X and Z are as defined herein, and then the benzylidene compound reacts with an amidine having formula (V) or a salt thereof (such as hydrochloride or acetate) with or without the addition of an alkali or an acid, and, when appropriate, in the presence of an inert organic solvent:
- R 6 is as defined herein;
- R 1 , R 2 , R 3 and R 6 are as defined herein and Y is a nucleophilic substituent, such as chloro, bromo, iodo, methylsulfonyl or toluenesulfonyl; or
- R 3 , X and Z are as defined herein.
- Compound of formula (VI) can be prepared by, for example, reacting a compound having formula (VIII)
- R 1 , R 2 , R 3 and R 6 are as defined herein, with a brominating agent such as N-bromosuccinimide, preferably in an inert organic solution, to produce a compound having formula (IX):
- R 3 is as defined herein.
- ⁇ -keto carboxylate (III) is well-known, or can be prepared by known methods published in the literature [for example, D. Baumann, “Um GmbH von Diketen mit mecanicen, Phenolen und Mercaptanen”, in “Methoden der organischen Chemie” (Houben-Weyl), vol. VII/4, 230 ff (1968); Y. Oikawa, K. Sugano und O. Yonemitsu, J. Org. Chem. 43, 2087 (1978)].
- the compound (V) is known and can be prepared according to the descriptions of WO-A-99/54326 and WO-A-99/54329.
- Morpholine (VII) is commercially available.
- the inert organic solvent is preferably an alcohol such as methanol, ethanol and isopropyl alcohol, an ether such as dioxane, diethyl ether, tetrahydrofuran, ethylene glycol monomethyl ether, ethylene glycol dimethyl ether, a carboxylic acid such as acetic acid, dimethylformamide, dimethyl sulfoxide, acetonitrile, pyridine or hexamethyl phosphoric triamide.
- an alcohol such as methanol, ethanol and isopropyl alcohol
- an ether such as dioxane, diethyl ether, tetrahydrofuran, ethylene glycol monomethyl ether, ethylene glycol dimethyl ether, a carboxylic acid such as acetic acid, dimethylformamide, dimethyl sulfoxide, acetonitrile, pyridine or hexamethyl phosphoric triamide.
- the reaction temperature can be varied within quite a wide range. Usually the temperature is between 20° C. and 150° C. Preferably, the temperature is the boiling temperature of the selected solvent.
- the reaction can be carried out under the atmospheric pressure or under a high pressure. It is usually carried out under the atmospheric pressure.
- the reaction can be carried out with or without an acid or alkali. It is preferable to carry out the reaction in the presence of a weak acid such as acetic acid, formic acid or the like.
- a weak acid such as acetic acid, formic acid or the like.
- An embodiment of the invention relates to a composition
- a composition comprising A) at least one of the above dihydropyrimidines and B) at least one of other antiviral agents different from A).
- a certain embodiment of the invention relates to a composition
- a composition comprising A) the above dihydropyrimidine, B) an HBV polymerase inhibitor and, when appropriate, C) an immunomodulator.
- the immunomodulator C) is selected from, for example, all the interferons such as ⁇ -interferon, ⁇ -interferon and ⁇ -interferon, especially ⁇ -2a-interferon and ⁇ -2b-interferon, an interleukin such as interleukin-2, a polypeptide such as thymosin- ⁇ -1 and a thymoctonan, an imidazoquinoline derivative such as levamisole, an immunoglobulin and a therapeutic vaccine.
- interferons such as ⁇ -interferon, ⁇ -interferon and ⁇ -interferon, especially ⁇ -2a-interferon and ⁇ -2b-interferon, an interleukin such as interleukin-2, a polypeptide such as thymosin- ⁇ -1 and a thymoctonan, an imidazoquinoline derivative such as levamisole, an immunoglobulin and a therapeutic vaccine.
- this invention also relates to a composition for treating and preventing HBV infections and its use for treating diseases induced by HBV.
- the use of the combinations of the invention provides valuable advantages for the treatment of HBV-induced diseases compared with monotherapy with the individual compounds, namely principally a synergistic antiviral activity, but also good tolerability of the combinations of the invention in Tox-50 (the range of toxicity at which 50% of the cells survive).
- HBV polymerase inhibitors B for the purposes of the invention are those which, in the endogenous polymerase assay which was published by Ph. A. Furman et al. in Antimicrobial Agents and Chemotherapy, Vol. 36 (No. 12), 2688 (1992) and which is described hereinafter, lead to an inhibition of the formation of an HBV DNA double strand, so as to result in a maximum of 50% of the activity of the zero value.
- HBV polymerase inhibitors B for use in the invention are the substances disclosed in the endogenous polymerase experiment published in “Antimicrobial Agents and Chemotherapy” Vol. 36 (No. 12), 2688 (1992) by Ph. A. Furman, and the substances described below for inhibiting the formation of double-stranded HBV DNA thereby resulting in the maximum 50% activity value to be zero.
- HBV virions from culture supernatants incorporate nucleoside 5′-triphosphates into the plus strand of the HBV DNA in vitro.
- agarose gel electrophoresis By using agarose gel electrophoresis, the incorporation of [ ⁇ - 32 P]-deoxynucleoside 5′-triphosphate into the viral 3.2 kb DNA product is observed in the presence and absence of a substance potentially having HBV polymerase-inhibiting properties.
- HBV virions are obtained from the cell culture supernatant of HepG2.2.15 cells by precipitation with polyethyleneglycol and are concentrated. One part by volume of clarified cell culture supernatant is mixed with 1 ⁇ 4 by volume of an aqueous solution containing 50% by weight polyethylene glycol 8000 and 0.6 M sodium chloride.
- the virions are sedimented by centrifugation at 2500 ⁇ g/15 minutes.
- the sediments are resuspended in 2 ml of buffer containing 0.05 M tris-HCl (pH 7.5) and dialyzed against the same buffer containing 100 mM potassium chloride.
- the samples can be frozen at ⁇ 80° C.
- Each reaction mixture (100 ⁇ l) contains at least 105 HBV virions; 50 mM tris-HCl (pH 7.5); 300 mM potassium chloride; 50 mM magnesium chloride; 0.1% Nonident® P-40 (nonionic detergent from Boehringer Mannheim); 10 ⁇ M dATP, 10 ⁇ M dGTP, 10 ⁇ M dTTP; 10 ⁇ Ci [ 32 P]dCTP (3000 Ci/mmol; final concentration 33 nM) and 1 ⁇ M of the potential polymerase inhibitor in its triphosphorylated form.
- the samples are incubated at 37° C. for one hour and then the reaction is stopped by adding 50 mM EDTA.
- a 10% weight/volume SDS solution (containing 10 g of SDS per 90 ml of water) is added to a final concentration of 1% by volume (based on the total volume), and proteinase K is added to a final concentration of 1 mg/ml. After incubation at 37° C. for one hour, samples are extracted with the same volume of phenol/chloroform/isoamyl alcohol (ratio 25:24:1 by volume), and the DNA is precipitated from the aqueous phase with ethanol.
- Adefovir dipivoxil 9- ⁇ 2-[[bis[(pivaloyloxy)-methoxy]-phosphinyl]-methoxy]-ethyl ⁇ -a-denine, cf.
- Abacavir ( ⁇ )-(1S-cis)-4-[2-amino-6-(cyclopropylamino)-9H-purin-9-yl]-2-cyclopentene-1-methanol, cf.
- FTC (2R-cis)-4-amino-5-fluoro-1-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]-pyrimidin-2(1H)-one, cf.
- ⁇ -L-FDDC 5-(6-amino-2-fluoro-9H-purin-9-yl)-tetrahydro-2-furanmethanol, cf.
- L-FMAU 1-(2-deoxy-2-fluoro- ⁇ -L-arabinofuranosyl)-5-methyl-pyrimidine-2,4(1H,3H)-dione, cf. WO 99/05157, WO 99/05158 and U.S. Pat. No. 5,753,789.
- a further preferred embodiment of the invention relates to a composition
- a composition comprising A) the above dihydropyrimidines having formula (I) and (Ia); and B) lamivudine.
- HBV antiviral agents B comprise, for example, phenylpropenamides of the following formula:
- R 1 and R 2 are, each independently, C 1-4 alkyl or, together with the nitrogen atom on which they are located, form a ring having 5 to 6 ring atoms which comprise carbon and/or oxygen;
- R 3 to R 12 are each independently hydrogen, halogen, C 1-4 alkyl, optionally substituted C 1-4 alkoxy, nitro, cyano or trifluoromethyl;
- R 13 is hydrogen, C 1-4 alkyl, C 1-7 acyl or aralkyl and X is halogen or optionally substituted C 1-4 alkyl.
- AT-61 is the compound
- Preferred immunomodulators C) comprise, for example, all interferons such as ⁇ -, ⁇ - and ⁇ -interferons, in particular also ⁇ -2a- and ⁇ -2b-interferons, interleukins such as interleukin-2, polypeptides such asthymosin- ⁇ -1 and thymoctonan, imidazoquinoline derivatives such as Levamisole®, immunoglobulins and therapeutic vaccines.
- interferons such as ⁇ -, ⁇ - and ⁇ -interferons, in particular also ⁇ -2a- and ⁇ -2b-interferons, interleukins such as interleukin-2, polypeptides such asthymosin- ⁇ -1 and thymoctonan, imidazoquinoline derivatives such as Levamisole®, immunoglobulins and therapeutic vaccines.
- a further preferred embodiment of the invention relates to combinations of A) above dihydropyrimidines (I) and (Ia), B) lamivudine and, where appropriate, C) an interferon.
- the antiviral action of the compounds of the invention on hepatitis B virus is investigated by methods based on those described by M. A. Sells et al., Proc. Natl. Acad. Sci., 84, 1005-1009 (1987) and B. E. Korba et al., Antiviral Research 19, 55-70 (1992).
- the antiviral tests are carried out in 96-well microtiter plates.
- the first vertical row of the plate receives only growth medium and HepG2.2.15 cells. It serves as virus control.
- test compounds 50 mM
- DMSO dimethyl methoxysulfoxide
- test concentration 100 ⁇ M (1st test concentration) in each case into the second vertical test row of the microtiter plate and subsequently diluted in twofold steps 210 times in growth medium plus 2% by weight of fetal calf serum (volume 25 ⁇ l)
- Each well of the microtiter plate then contains 225 ⁇ l of HepG2.2.15 cell suspension (5 ⁇ 104 cells/ml) in growth medium plus 2% by weight of fetal calf serum.
- the test mixture is incubated at 37° C. and 5% CO2 (v/v) for 4 days.
- the supernatant is then aspirated off and discarded, and the wells receive 225 ⁇ l of freshly prepared growth medium.
- the compounds according to the invention are each added anew as 10-fold concentrated solution in a volume of 25 ⁇ l. The mixtures are incubated for a further 4 days
- the HepG2.2.15 cells are examined under the light microscope or by means of biochemical detection methods (for example Alamar Blue stain or Trypan Blue stain) for cytotoxic changes
- the supernatant and/or cells are then harvested and sucked by means of a vacuum onto 96-well dot-blot chambers covered with a nylon membrane (in accordance with the manufacturer's information).
- Substance-induced cytotoxic or cytostatic changes in the HepG2.2.15 cells are detected, for example, under the light microscope as changes in cell morphology. Such substance-induced changes in the HepG2.2.15 cells compare with untreated cells are visible, for example, as cytolysis, vacuolation or altered cell morphology.
- a 50% cytotoxicity (Tox.-50) means that 50% of the cells show a morphology comparable to the corresponding cell control.
- the tolerability of some of the compounds according to the invention is additionally tested on other host cells such as, for example, HeLa cells, primary human peripheral blood cells or transformed cell lines such as H-9 cells.
- the intra- or extracellular supernatants of the HepG2.2.15 cells are denatured (1.5 M NaCl/0.5 N NaOH), neutralized (3 M NaCl/0.5M Tris HCl, pH 7.5) and washed (2 ⁇ SSC).
- the DNA is then baked onto the membrane by incubating the filters at 120° C. for 2-4 hours.
- Detection of the viral DNA from the treated HepG2.2.15 cells on the nylon filters is usually carried out with nonradioactive, digoxigenin-labeled hepatitis B-specific DNA probes, each of which is labeled with digoxigenin, purified and employed for the hybridization in accordance with the manufacturer's information.
- the prehybridization and hybridization take place in 5 ⁇ SSC, 1 ⁇ blocking reagent, 0.1% by weight N-lauroylsarcosine, 0.02% by weight SDS and 100 ⁇ g of herring sperm DNA.
- the prehybridization takes place at 60° C. for 30 minutes, and the specific hybridization takes place with 20 to 40 ng/ml of the digoxigenized, denatured HBV-specific DNA (14 hours, 60° C.). The filters are then washed.
- the filters were washed and prehybridized in a blocking reagent (in accordance with the manufacturer's information). Hybridization was then carried out with an anti-DIG antibody coupled to alkaline phosphatase for 30 minutes. After a washing step, the substrate of alkaline phosphatase, CSPD, was added, incubated with the filters for 5 minutes, then packed in plastic film and incubated at 37° C. for a further 15 minutes. The chemiluminescence of the hepatitis B-specific DNA signals was visualized by exposing the filters to an X-ray film (incubation depending on signal strength: 10 minutes to 2 hours).
- the half-maximum inhibitory concentration (IC 50 , 50% inhibitory concentration) was determined as the concentration at which the intra- or extracellular hepatitis B-specific band was reduced by the compound according to the invention by 50% compared with an untreated sample.
- the compound of the invention exhibits an effective antiviral effect with an IC 50 less than 1 nM. Therefore, the compound of the invention is suitable for use in treating the diseases induced by viruses, especially acute and chronic persistent HBV infections.
- Chronic viral diseases induced by HBV can worsen the morbidity and the chronic hepatitis B virus infection can cause liver cirrhosis and/or hepatocellular carcinoma in many cases.
- Areas of indication which may be mentioned for the compounds of the invention are, for example: the treatment of acute and chronic viral infections which may lead to infectious hepatitis, for example infections with heptatitis B viruses.
- the compounds of the invention are particularly suitable for the treatment of chronic hepatitis B infections and the treatment of acute and chronic hepatitis B viral infections.
- the present invention includes pharmaceutical preparations which, besides nontoxic, inert pharmaceutically suitable carriers, comprise one or more compounds (I) or (Ia) or a combination of the invention or which consist of one or more active ingredients (I) or (Ia) or of a combination of the invention.
- the active ingredients (I) and (Ia) are intended to be present in the pharmaceutical preparations mentioned above in a concentration of about 0.1 to 99.5% by weight, preferably of about 0.5 to 95% by weight, of the complete mixture.
- compositions mentioned above may also comprise other active pharmaceutical ingredients apart from the compounds (I) and (Ia).
- the ratio of the amounts of the components A, B and, where appropriate, C in the compositions of the invention may vary within wide limits; it is preferably 5 to 500 mg of A/10 to 1000 mg of B, in particular 10 to 200 mg of A/20 to 400 mg of B.
- Component C which is also to be used where appropriate, may be used in amounts of, preferably, 1 to 10 million, in particular 2 to 7 million, I.U. (international units), about three times a week over a period of up to one year.
- the compounds or compositions of the invention are intended to be present in the pharmaceutical preparations mentioned above in general in a concentration of about 0.1 to 99.5, preferably about 0.5 to 95, % by weight of the complete mixture.
- the pharmaceutical preparations mentioned above can be produced in a conventional way by known methods, for example by mixing the active ingredient(s) with the carrier(s).
- a single dose contains the active ingredient(s) preferably in amounts of about 1 to about 80, in particular 1 to 30 mg/kg of body weight.
- the dosages mentioned may be necessary to deviate from the dosages mentioned, in particular depending on the species and body weight of the subject to be treated, the nature and severity of the disorder, the type of preparation and mode of administration of the medicament, and the time or interval within which administration takes place.
- the invention therefore relates further to the compounds and compositions defined above for controlling diseases.
- the invention further relates to medicaments comprising at least one of the compounds or compositions defined above and, where appropriate, one or more other active pharmaceutical ingredient(s).
- the invention further relates to the use of the compounds and compositions defined above for producing a medicament for the treatment and prophylaxis of the diseases described above, preferably of viral diseases, in particular of hepatitis B.
- the percentage data in the following examples relate in each case to weight unless indicated otherwise.
- the ratios of solvents in solvent mixtures are in each case based on volume.
- the anti-HBV active compounds in the two examples are enantiomers having a relatively long retention time.
- the treatment of the hepatitis B virus-producing HepG2.2.15 cells with the compounds of the invention can lead to a reduction in intra- and/or extracellular viral DNA.
- the compounds disclosed herein exhibit an effective antiviral effect with the IC 50 less than 1 nM. Therefore, the compounds can be used for the treatment of a disease induced by viruses, especially acute and chronic persistent HBV infections according to the methods of the invention or any method known to a person skilled in the art.
Abstract
Description
| TABLE 1 |
| Example 2 of U.S. Pat. No. 7,074,784 B2 |
| Example | R1 | R2 | R3 | R6 | IC50 (nM) |
| 12 | Cl | Cl | CH3 |
 |
2 (self-tested) |
| 9 | Br | F | CH3 |
 |
7 |
| 5 | Cl | F | CH3 |
 |
2-4 |
| 45 | Cl | Cl | CH3 |
|
2 |

| TABLE 2 |
| Some Examples of this Invention |
| Example | R1 | R2 | R3 | R6 | IC50 (nM) | ||
| 6 | Br | F | CH3 |
|
0.3 | ||
| 5 | Br | F | CH2CH3 |
|
0.2 | ||

- [A] firstly a benzaldehyde having formula (II) reacts with a β-ketoester having formula (III) with or without the addition of an alkali or an acid, and, when appropriate, in the presence of an inert organic solvent to produce a benzylidene compound having formula (IV):

wherein R1, R2, R3, X and Z are as defined herein, and then the benzylidene compound reacts with an amidine having formula (V) or a salt thereof (such as hydrochloride or acetate) with or without the addition of an alkali or an acid, and, when appropriate, in the presence of an inert organic solvent:

wherein R6 is as defined herein; or
- [B] the β-ketoester having formula (III) reacts with the benzaldehyde having formula (II) and the amidine having formula (V) or a salt thereof (such as hydrochloride or acetate) with or without the addition of an alkali or an acid, and, when appropriate, in the presence of an inert organic solvent in one step; or
- [C] if X in formula (I) is methylene, a compound having formula (VI) reacts with morpholine having formula (VII) with or without the addition of an alkali, and, when appropriate, in the presence of an inert organic solvent,
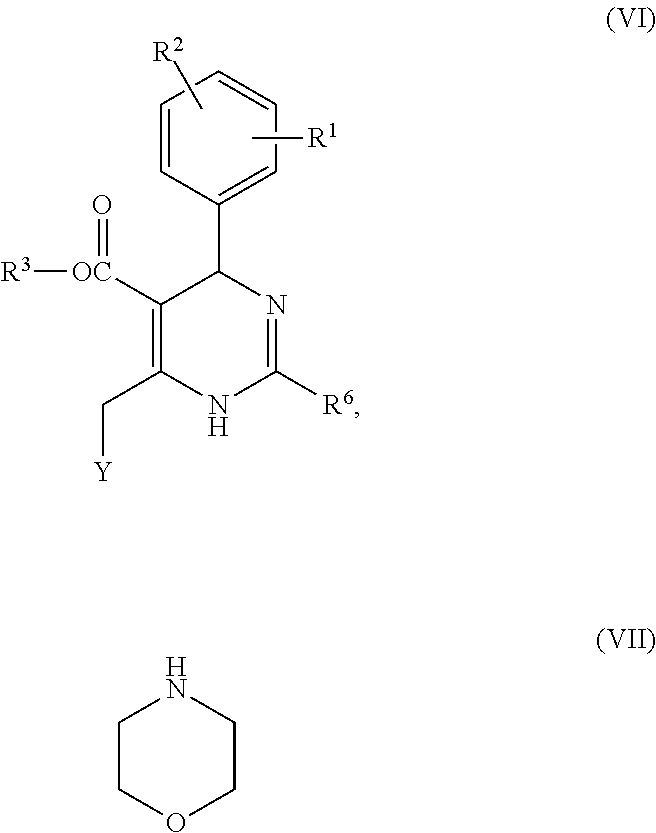
wherein R1, R2, R3 and R6 are as defined herein and Y is a nucleophilic substituent, such as chloro, bromo, iodo, methylsulfonyl or toluenesulfonyl; or
- [D] the benzaldehyde having formula (II) reacts with a compound having formula (X) and the amidine having formula (V) with or without the addition of an alkali and, when appropriate, in an inert organic solvent,

wherein R3, X and Z are as defined herein.

wherein R1, R2, R3 and R6 are as defined herein, with a brominating agent such as N-bromosuccinimide, preferably in an inert organic solution, to produce a compound having formula (IX):

and reacting the compound having a nucleophilic substituent, directly or after the compound being further converted according to a conventional method as described in a literature, with the morpholine having formula (VII).

wherein R3 is as defined herein.

wherein R1 and R2 are, each independently, C1-4 alkyl or, together with the nitrogen atom on which they are located, form a ring having 5 to 6 ring atoms which comprise carbon and/or oxygen; R3 to R12 are each independently hydrogen, halogen, C1-4 alkyl, optionally substituted C1-4 alkoxy, nitro, cyano or trifluoromethyl; and R13 is hydrogen, C1-4 alkyl, C1-7 acyl or aralkyl and X is halogen or optionally substituted C1-4 alkyl.





| Example No. | IC50 (nM) | ||
| 5 | 0.2 | ||
| (−)-5 | 0.1 | ||
| 6 | 0.3 | ||
| (−)-6 | 0.2 | ||
Claims (18)








Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US13/869,981 USRE44987E1 (en) | 2007-06-18 | 2013-04-25 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN200710119019 | 2007-06-18 | ||
| CN200710119019 | 2007-06-18 | ||
| PCT/CN2008/001187 WO2008154817A1 (en) | 2007-06-18 | 2008-06-18 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
| US13/550,601 US8343969B2 (en) | 2007-06-18 | 2012-07-17 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
| US13/869,981 USRE44987E1 (en) | 2007-06-18 | 2013-04-25 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/550,601 Reissue US8343969B2 (en) | 2007-06-18 | 2012-07-17 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| USRE44987E1 true USRE44987E1 (en) | 2014-07-01 |
Family
ID=40155886
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/869,947 Active 2028-12-24 USRE45004E1 (en) | 2007-06-18 | 2008-06-18 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
| US12/664,392 Ceased US8236797B2 (en) | 2007-06-18 | 2008-06-18 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
| US13/550,601 Ceased US8343969B2 (en) | 2007-06-18 | 2012-07-17 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
| US13/869,981 Active USRE44987E1 (en) | 2007-06-18 | 2013-04-25 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
Family Applications Before (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/869,947 Active 2028-12-24 USRE45004E1 (en) | 2007-06-18 | 2008-06-18 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
| US12/664,392 Ceased US8236797B2 (en) | 2007-06-18 | 2008-06-18 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
| US13/550,601 Ceased US8343969B2 (en) | 2007-06-18 | 2012-07-17 | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
Country Status (15)
| Country | Link |
|---|---|
| US (4) | USRE45004E1 (en) |
| EP (2) | EP2159224B1 (en) |
| JP (2) | JP5361879B2 (en) |
| KR (1) | KR101173892B1 (en) |
| CN (2) | CN101328171A (en) |
| AU (1) | AU2008265397C1 (en) |
| BR (1) | BRPI0813237B8 (en) |
| CA (1) | CA2691056C (en) |
| DK (2) | DK2159224T3 (en) |
| ES (2) | ES2442907T3 (en) |
| HK (1) | HK1174035A1 (en) |
| PL (2) | PL2514750T3 (en) |
| PT (2) | PT2159224E (en) |
| RU (1) | RU2443703C2 (en) |
| WO (1) | WO2008154817A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9340538B2 (en) | 2012-08-24 | 2016-05-17 | Sunshine Lake Pharma Co., Ltd. | Dihydropyrimidine compounds and their application in pharmaceuticals |
| US9403814B2 (en) | 2012-09-27 | 2016-08-02 | Sunshine Lake Pharma Co., Ltd. | Crystalline forms of dihydropyrimidine derivatives |
| US9498479B2 (en) | 2013-11-19 | 2016-11-22 | Sunshine Lake Pharma Co., Ltd. | Dihydropyrimidine compounds and their application in pharmaceuticals |
| US9573941B2 (en) | 2013-11-27 | 2017-02-21 | Sunshine Lake Pharma Co., Ltd. | Processes for preparing dihydropyrimidine derivatives and intermediates thereof |
| US9771358B2 (en) | 2014-03-28 | 2017-09-26 | Sunshine Lake Pharma Co., Ltd. | Dihydropyrimidine compounds and their application in pharmaceuticals |
| US10098889B2 (en) | 2015-02-07 | 2018-10-16 | Sunshine Lake Pharma Co., Ltd. | Complexes and salts of dihydropyrimidine derivatives and their application in pharmaceuticals |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101328171A (en) * | 2007-06-18 | 2008-12-24 | 张中能 | Bromophenyl-substituted thiazole dihydropyrimidine |
| CN101744823B (en) * | 2008-12-17 | 2013-06-19 | 广东东阳光药业有限公司 | Solid dispersion of dihydropyrimidine compounds and preparation thereof for medical purpose |
| WO2010069147A1 (en) * | 2008-12-17 | 2010-06-24 | 张中能 | Dihydropyrimidine derivatives, compositions thereof and their use |
| CN101575318B (en) | 2009-06-25 | 2012-02-08 | 中国人民解放军军事医学科学院毒物药物研究所 | Novel dihydropyridine compound and application thereof on preparing drugs for curing and/or preventing virus diseases |
| US9233933B2 (en) | 2012-01-06 | 2016-01-12 | Janssen Sciences Ireland Uc | 4,4-disubstituted-1,4-dihydropyrimidines and the use thereof as medicaments for the treatment of hepatitis B |
| US20130267517A1 (en) | 2012-03-31 | 2013-10-10 | Hoffmann-La Roche Inc. | Novel 4-methyl-dihydropyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| CN103664897B (en) * | 2012-09-01 | 2018-04-03 | 广东东阳光药业有限公司 | Dihydropyrimidines and its application in medicine |
| CN103664925B (en) * | 2012-09-07 | 2018-01-23 | 广东东阳光药业有限公司 | The Dihydropyrimidines of heteroaryl substitution and its application in medicine |
| KR20150054795A (en) * | 2012-09-10 | 2015-05-20 | 에프. 호프만-라 로슈 아게 | 6-amino acid heteroaryldihydropyrimidines for the treatment and prophylaxis of hepatitis b virus infection |
| CN103664899B (en) * | 2012-09-11 | 2017-06-16 | 广东东阳光药业有限公司 | The Dihydropyrimidines of heteroaryl substitution and its application in medicine |
| KR20150133792A (en) * | 2013-03-20 | 2015-11-30 | 인디애나 유니버시티 리서치 앤드 테크놀로지 코포레이션 | Fluorescent-hap: a diagnostic stain for hbv cores in cells |
| EP3139954A4 (en) * | 2014-05-09 | 2018-02-28 | Indiana University Research and Technology Corporation | Methods and compositions for treating hepatitis b virus infections |
| WO2017076791A1 (en) | 2015-11-03 | 2017-05-11 | F. Hoffmann-La Roche Ag | Combination therapy of an hbv capsid assembly inhibitor and an interferon |
| JP2019526562A (en) | 2016-08-24 | 2019-09-19 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | Combination therapy of HBV capsid assembly inhibitor and nucleoside (Thi) analogue |
| SI3587420T1 (en) | 2017-02-23 | 2021-09-30 | Fujian Cosunter Pharmaceutical Co., Ltd. | Tri-cycle compound and applications thereof |
| WO2018181883A1 (en) | 2017-03-31 | 2018-10-04 | 富士フイルム株式会社 | 4-pyridone compound or salt thereof, and pharmaceutical composition and formulation including same |
| CN110809574A (en) | 2017-06-27 | 2020-02-18 | 詹森药业有限公司 | Heteroaryl dihydropyrimidine derivatives and methods for treating hepatitis b infection |
| CN107501257B (en) * | 2017-08-17 | 2020-05-29 | 山东大学 | Dihydropyrimidine-triazole derivative and preparation method and application thereof |
| AU2019272481B2 (en) | 2018-05-25 | 2024-03-21 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | 2,3-dihydro-1H-pyrrolizine-7-formamide derivative and application thereof |
| US11053235B2 (en) | 2018-08-09 | 2021-07-06 | Janssen Sciences Ireland Unlimited Company | Substituted 1,4-dihydropyrimidines for the treatment of HBV infection or HBV-induced diseases |
| EP3854797B1 (en) | 2018-08-23 | 2023-03-15 | Fujian Akeylink Biotechnology Co., Ltd. | Crystal form of tri-cycle compound and application thereof |
| WO2020255016A1 (en) | 2019-06-18 | 2020-12-24 | Janssen Sciences Ireland Unlimited Company | Combination of hepatitis b virus (hbv) vaccines and dihydropyrimidine derivatives as capsid assembly modulators |
| US20230026869A1 (en) | 2019-11-22 | 2023-01-26 | Chia Tai Tianqing Pharmaceutical Group Co., Ltd. | Crystal form of nucleoprotein inhibitor and use thereof |
| WO2022166923A1 (en) | 2021-02-05 | 2022-08-11 | 和博医药有限公司 | Phenyldihydropyrimidine compound and use thereof |
| CN117136187A (en) * | 2021-06-24 | 2023-11-28 | 上海齐鲁制药研究中心有限公司 | Novel anti-hepatitis B compound |
Citations (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4822798A (en) | 1982-09-18 | 1989-04-18 | Bayer Aktiengesellschaft | Circulation-active 4-phenyl-6-substituted dihydropyrimidines |
| WO2000058302A1 (en) | 1999-03-25 | 2000-10-05 | Bayer Aktiengesellschaft | Dihydropyrimidines and their use in the treatment of hepatitis b |
| WO2001045712A1 (en) | 1999-12-22 | 2001-06-28 | Bayer Aktiengesellschaft | Combinations of medicaments for treating viral diseases |
| WO2001068642A1 (en) | 2000-03-16 | 2001-09-20 | Bayer Aktiengesellschaft | Dihydropyrimidines and the use thereof as medicaments for the treatment of hepatitis b |
| WO2001068639A1 (en) | 2000-03-17 | 2001-09-20 | Bayer Aktiengesellschaft | Dihydropyrimidine-5-carboxylic acid esters and use thereof as medicaments against viral diseases |
| WO2001068647A1 (en) | 2000-03-15 | 2001-09-20 | Bayer Aktiengesellschaft | Medicaments against viral diseases |
| WO2001068641A1 (en) | 2000-03-17 | 2001-09-20 | Bayer Aktiengesellschaft | 6-aminoalkyl-dihydropyrimidines and the use thereof as medicaments against viral diseases |
| US6436943B1 (en) | 1998-04-18 | 2002-08-20 | Bayer Aktiengesellschaft | Use of dihydropyrimidines as medicaments, and novel substances |
| US6503913B1 (en) | 1998-04-18 | 2003-01-07 | Bayer Aktiengesellschaft | 2-heterocyclically substituted dihydropyrimidines |
| US6696451B1 (en) | 1998-04-18 | 2004-02-24 | Bayer Aktiengesellschaft | Dihydropyrimidines |
| WO2005008302A1 (en) | 2003-07-22 | 2005-01-27 | National University Corporation Tokyo University Of Agriculture And Technology | Reflection type polarizer, laminate optical member and liquid crystal display unit |
| US7074784B2 (en) | 2000-03-16 | 2006-07-11 | Siegfried Goldmann | Medicaments against viral diseases |
| WO2008009209A1 (en) | 2006-07-10 | 2008-01-24 | Beijing Molecule Science And Technology Co., Ltd | Dihydropyrimidine compounds and their uses in preparation of medicaments for treating and preventing antiviral diseases |
| WO2008154818A1 (en) | 2007-06-18 | 2008-12-24 | Zhang, Zhongneng | Fluorophenyl-substituted thiazolyl dihydropyrimidines |
| WO2008154819A1 (en) | 2007-06-18 | 2008-12-24 | Zhang, Zhongneng | Carbethoxy-substituted thiazolyl dihydropyrimidines |
| WO2008154820A1 (en) | 2007-06-18 | 2008-12-24 | Zhang, Zhongneng | Carbethoxy-substituted thiazolyl dihydropyrimidines |
| US20100004268A1 (en) | 2006-07-10 | 2010-01-07 | Song Li | Optically Pure Dihydropyrimidine Compounds and Their Uses for the Preparation of a Medicament for Treatment and Prevention of Viral Diseases |
| US20100010013A1 (en) | 2007-01-16 | 2010-01-14 | Beijing Molecule Science And Technology Co., Ltd. | Dihydropyrimidine compounds and their uses in manufacture of a medicament for treatment and prevention of viral diseases |
| US8236797B2 (en) | 2007-06-18 | 2012-08-07 | Sunshine Lake Pharma Co., Ltd. | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
Family Cites Families (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6458772B1 (en) | 1909-10-07 | 2002-10-01 | Medivir Ab | Prodrugs |
| CS263952B1 (en) | 1985-04-25 | 1989-05-12 | Holy Antonin | Remedy with antiviral effect |
| CS263951B1 (en) | 1985-04-25 | 1989-05-12 | Antonin Holy | 9-(phosponylmethoxyalkyl)adenines and method of their preparation |
| US5047407A (en) | 1989-02-08 | 1991-09-10 | Iaf Biochem International, Inc. | 2-substituted-5-substituted-1,3-oxathiolanes with antiviral properties |
| GB8815265D0 (en) | 1988-06-27 | 1988-08-03 | Wellcome Found | Therapeutic nucleosides |
| MY104575A (en) | 1989-12-22 | 1994-04-30 | The Wellcome Foundation Ltd | Therapeutic nucleosides. |
| US5827727A (en) | 1990-02-01 | 1998-10-27 | Emory University | Method of resolution of 1,3-oxathiolane nucleoside enantiomers |
| US5700937A (en) | 1990-02-01 | 1997-12-23 | Emory University | Method for the synthesis, compositions and use of 2'-deoxy-5-fluoro-3'-thiacytidine and related compounds |
| US5204466A (en) | 1990-02-01 | 1993-04-20 | Emory University | Method and compositions for the synthesis of bch-189 and related compounds |
| US5276151A (en) | 1990-02-01 | 1994-01-04 | Emory University | Method of synthesis of 1,3-dioxolane nucleosides |
| US5914331A (en) | 1990-02-01 | 1999-06-22 | Emory University | Antiviral activity and resolution of 2-hydroxymethyl-5-(5-fluorocytosin-1-yl)-1,3-oxathiolane |
| EP0481214B1 (en) | 1990-09-14 | 1998-06-24 | Institute Of Organic Chemistry And Biochemistry Of The Academy Of Sciences Of The Czech Republic | Prodrugs of phosphonates |
| US5206244A (en) | 1990-10-18 | 1993-04-27 | E. R. Squibb & Sons, Inc. | Hydroxymethyl (methylenecyclopentyl) purines and pyrimidines |
| US5340816A (en) | 1990-10-18 | 1994-08-23 | E. R. Squibb & Sons, Inc. | Hydroxymethyl(methylenecyclopentyl) purines and pyrimidines |
| IL100965A (en) | 1991-02-22 | 1999-12-31 | Univ Emory | 2-Hydroxymethyl-5-(5-fluorocytosin-l-yl)-1,3-oxathiolane its resolution and pharmaceutical compositions containing it |
| WO1992018517A1 (en) | 1991-04-17 | 1992-10-29 | Yale University | Method of treating or preventing hepatitis b virus |
| US5627160A (en) | 1993-05-25 | 1997-05-06 | Yale University | L-2',3'-dideoxy nucleoside analogs as anti-hepatitis B (HBV) and anti-HIV agents |
| TW374087B (en) | 1993-05-25 | 1999-11-11 | Univ Yale | L-2',3'-dideoxy nucleotide analogs as anti-hepatitis B(HBV) and anti-HIV agents |
| US5753789A (en) | 1996-07-26 | 1998-05-19 | Yale University | Oligonucleotides containing L-nucleosides |
| AU4090697A (en) | 1996-09-03 | 1998-03-26 | Bristol-Myers Squibb Company | Improved process for preparing the antiviral agent {1s-(1alpha, 3alpha, 4beta)}-2-amino-1,9-dihydro-9-{4-hydroxy-3-(hydroxymethyl)-2 -methylenecyclopentyl}-6h-purin-6-one |
| AU5923998A (en) | 1997-01-31 | 1998-08-25 | Avid Therapeutics Inc. | 2-benzoylamino-3-phenylpropenamide derivatives and methods of using the same |
| TW434252B (en) | 1997-07-23 | 2001-05-16 | Univ Georgia Res Found | Process for the preparation of 2'-fluoro-5-methyl-β-L-arabino-furanosyluridine |
| US6636943B1 (en) * | 1999-07-30 | 2003-10-21 | Hewlett-Packard Development Company, L.P. | Method for detecting continuity modules in a direct Rambus DRAM subsystem |
| SE0001836D0 (en) | 2000-05-18 | 2000-05-18 | Inovacor Ab | Computer based system |
-
2008
- 2008-06-18 CN CNA2008101257258A patent/CN101328171A/en not_active Withdrawn
- 2008-06-18 DK DK08757463.8T patent/DK2159224T3/en active
- 2008-06-18 CN CN2008800208411A patent/CN102066369B/en active Active
- 2008-06-18 AU AU2008265397A patent/AU2008265397C1/en active Active
- 2008-06-18 EP EP08757463A patent/EP2159224B1/en active Active
- 2008-06-18 PL PL12176861T patent/PL2514750T3/en unknown
- 2008-06-18 WO PCT/CN2008/001187 patent/WO2008154817A1/en active Application Filing
- 2008-06-18 DK DK12176861.8T patent/DK2514750T5/en active
- 2008-06-18 ES ES12176861.8T patent/ES2442907T3/en active Active
- 2008-06-18 US US13/869,947 patent/USRE45004E1/en active Active
- 2008-06-18 EP EP12176861.8A patent/EP2514750B1/en active Active
- 2008-06-18 PT PT08757463T patent/PT2159224E/en unknown
- 2008-06-18 RU RU2010101212/04A patent/RU2443703C2/en active
- 2008-06-18 PL PL08757463T patent/PL2159224T3/en unknown
- 2008-06-18 KR KR1020107000850A patent/KR101173892B1/en active IP Right Review Request
- 2008-06-18 BR BRPI0813237A patent/BRPI0813237B8/en active IP Right Grant
- 2008-06-18 JP JP2010512492A patent/JP5361879B2/en active Active
- 2008-06-18 US US12/664,392 patent/US8236797B2/en not_active Ceased
- 2008-06-18 PT PT121768618T patent/PT2514750E/en unknown
- 2008-06-18 CA CA2691056A patent/CA2691056C/en active Active
- 2008-06-18 ES ES08757463T patent/ES2391597T3/en active Active
-
2012
- 2012-07-17 US US13/550,601 patent/US8343969B2/en not_active Ceased
-
2013
- 2013-01-31 HK HK13101360.5A patent/HK1174035A1/en unknown
- 2013-04-25 US US13/869,981 patent/USRE44987E1/en active Active
- 2013-06-03 JP JP2013116979A patent/JP5970421B2/en active Active
Patent Citations (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4822798A (en) | 1982-09-18 | 1989-04-18 | Bayer Aktiengesellschaft | Circulation-active 4-phenyl-6-substituted dihydropyrimidines |
| US6436943B1 (en) | 1998-04-18 | 2002-08-20 | Bayer Aktiengesellschaft | Use of dihydropyrimidines as medicaments, and novel substances |
| US6503913B1 (en) | 1998-04-18 | 2003-01-07 | Bayer Aktiengesellschaft | 2-heterocyclically substituted dihydropyrimidines |
| US6696451B1 (en) | 1998-04-18 | 2004-02-24 | Bayer Aktiengesellschaft | Dihydropyrimidines |
| WO2000058302A1 (en) | 1999-03-25 | 2000-10-05 | Bayer Aktiengesellschaft | Dihydropyrimidines and their use in the treatment of hepatitis b |
| WO2001045712A1 (en) | 1999-12-22 | 2001-06-28 | Bayer Aktiengesellschaft | Combinations of medicaments for treating viral diseases |
| WO2001068647A1 (en) | 2000-03-15 | 2001-09-20 | Bayer Aktiengesellschaft | Medicaments against viral diseases |
| US7074784B2 (en) | 2000-03-16 | 2006-07-11 | Siegfried Goldmann | Medicaments against viral diseases |
| WO2001068642A1 (en) | 2000-03-16 | 2001-09-20 | Bayer Aktiengesellschaft | Dihydropyrimidines and the use thereof as medicaments for the treatment of hepatitis b |
| WO2001068639A1 (en) | 2000-03-17 | 2001-09-20 | Bayer Aktiengesellschaft | Dihydropyrimidine-5-carboxylic acid esters and use thereof as medicaments against viral diseases |
| WO2001068641A1 (en) | 2000-03-17 | 2001-09-20 | Bayer Aktiengesellschaft | 6-aminoalkyl-dihydropyrimidines and the use thereof as medicaments against viral diseases |
| WO2005008302A1 (en) | 2003-07-22 | 2005-01-27 | National University Corporation Tokyo University Of Agriculture And Technology | Reflection type polarizer, laminate optical member and liquid crystal display unit |
| WO2008009209A1 (en) | 2006-07-10 | 2008-01-24 | Beijing Molecule Science And Technology Co., Ltd | Dihydropyrimidine compounds and their uses in preparation of medicaments for treating and preventing antiviral diseases |
| US20100004268A1 (en) | 2006-07-10 | 2010-01-07 | Song Li | Optically Pure Dihydropyrimidine Compounds and Their Uses for the Preparation of a Medicament for Treatment and Prevention of Viral Diseases |
| US20100010013A1 (en) | 2007-01-16 | 2010-01-14 | Beijing Molecule Science And Technology Co., Ltd. | Dihydropyrimidine compounds and their uses in manufacture of a medicament for treatment and prevention of viral diseases |
| WO2008154818A1 (en) | 2007-06-18 | 2008-12-24 | Zhang, Zhongneng | Fluorophenyl-substituted thiazolyl dihydropyrimidines |
| WO2008154819A1 (en) | 2007-06-18 | 2008-12-24 | Zhang, Zhongneng | Carbethoxy-substituted thiazolyl dihydropyrimidines |
| WO2008154820A1 (en) | 2007-06-18 | 2008-12-24 | Zhang, Zhongneng | Carbethoxy-substituted thiazolyl dihydropyrimidines |
| US8236797B2 (en) | 2007-06-18 | 2012-08-07 | Sunshine Lake Pharma Co., Ltd. | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
| US8343969B2 (en) | 2007-06-18 | 2013-01-01 | Sunshine Lake Pharma Co., Ltd. | Bromo-phenyl substituted thiazolyl dihydropyrimidines |
Non-Patent Citations (1)
| Title |
|---|
| Huff, Joel R., "HIV Protease: A Novel Chemotherapeutic Target for AIDS," Journal of Medicinal Chemistry, Aug. 1991, pp. 2305-2314, vol. 34, No. 8. |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9340538B2 (en) | 2012-08-24 | 2016-05-17 | Sunshine Lake Pharma Co., Ltd. | Dihydropyrimidine compounds and their application in pharmaceuticals |
| US9403814B2 (en) | 2012-09-27 | 2016-08-02 | Sunshine Lake Pharma Co., Ltd. | Crystalline forms of dihydropyrimidine derivatives |
| US9498479B2 (en) | 2013-11-19 | 2016-11-22 | Sunshine Lake Pharma Co., Ltd. | Dihydropyrimidine compounds and their application in pharmaceuticals |
| US9573941B2 (en) | 2013-11-27 | 2017-02-21 | Sunshine Lake Pharma Co., Ltd. | Processes for preparing dihydropyrimidine derivatives and intermediates thereof |
| US9617252B2 (en) | 2013-11-27 | 2017-04-11 | Sunshine Lake Pharma Co., Ltd. | Processes for preparing dihydropyrimidine derivatives and intermediates thereof |
| US9643962B2 (en) | 2013-11-27 | 2017-05-09 | Sunshine Lake Pharma Co., Ltd. | Processes for preparing dihydropyrimidine derivatives and intermediates thereof |
| US9771358B2 (en) | 2014-03-28 | 2017-09-26 | Sunshine Lake Pharma Co., Ltd. | Dihydropyrimidine compounds and their application in pharmaceuticals |
| US10098889B2 (en) | 2015-02-07 | 2018-10-16 | Sunshine Lake Pharma Co., Ltd. | Complexes and salts of dihydropyrimidine derivatives and their application in pharmaceuticals |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| USRE44987E1 (en) | Bromo-phenyl substituted thiazolyl dihydropyrimidines | |
| US7074784B2 (en) | Medicaments against viral diseases | |
| US6503913B1 (en) | 2-heterocyclically substituted dihydropyrimidines | |
| WO2008154819A1 (en) | Carbethoxy-substituted thiazolyl dihydropyrimidines | |
| US6436943B1 (en) | Use of dihydropyrimidines as medicaments, and novel substances | |
| WO2008154820A1 (en) | Carbethoxy-substituted thiazolyl dihydropyrimidines | |
| WO2008154818A1 (en) | Fluorophenyl-substituted thiazolyl dihydropyrimidines | |
| WO2001068647A1 (en) | Medicaments against viral diseases | |
| DE10013126A1 (en) | New 6-aminoalkyl-dihydropyrimidine-5-carboxylate ester derivatives, useful as antiviral agents having strong activity against hepatitis B virus and low cytotoxicity | |
| DE10012824A1 (en) | New 6-hydroxyhydrocarbyl or 6-thiohydrocarbyl-dihydropyrimidine-5-carboxylic acid derivatives, useful for the treatment of viral infections, especially hepatitis B infections | |
| WO2001068639A1 (en) | Dihydropyrimidine-5-carboxylic acid esters and use thereof as medicaments against viral diseases |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AS | Assignment |
Owner name: ZHANG, ZHONGNENG, CHINA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:GOLDMANN, SIEGFRIED;REEL/FRAME:031133/0818 Effective date: 20100130 Owner name: ZHANG, ZHONGNENG, CHINA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:LI, JING;REEL/FRAME:031133/0800 Effective date: 20100130 Owner name: SUNSHINE LAKE PHARMA CO., LTD., CHINA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:ZHANG, ZHONGNENG;REEL/FRAME:031133/0845 Effective date: 20130826 Owner name: ZHANG, ZHONGNENG, CHINA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:LIU, YI SONG;REEL/FRAME:031133/0685 Effective date: 20100205 |
|
| FPAY | Fee payment |
Year of fee payment: 4 |
|
| AS | Assignment |
Owner name: NORTH & SOUTH BROTHER PHARMACY INVESTMENT COMPANY Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:SUNSHINE LAKE PHARMA CO., LTD.;REEL/FRAME:050781/0929 Effective date: 20190906 |
|
| AS | Assignment |
Owner name: SUNSHINE LAKE PHARMA CO., LTD., CHINA Free format text: ASSIGNMENT OF ASSIGNORS INTEREST;ASSIGNOR:NORTH & SOUTH BROTHER PHARMACY INVESTMENT COMPANY LIMITED;REEL/FRAME:052921/0778 Effective date: 20200525 |
|
| MAFP | Maintenance fee payment |
Free format text: PAYMENT OF MAINTENANCE FEE, 8TH YEAR, LARGE ENTITY (ORIGINAL EVENT CODE: M1552); ENTITY STATUS OF PATENT OWNER: LARGE ENTITY Year of fee payment: 8 |