RU2762111C1 - Соединения для лечения рака - Google Patents
Соединения для лечения рака Download PDFInfo
- Publication number
- RU2762111C1 RU2762111C1 RU2017102002A RU2017102002A RU2762111C1 RU 2762111 C1 RU2762111 C1 RU 2762111C1 RU 2017102002 A RU2017102002 A RU 2017102002A RU 2017102002 A RU2017102002 A RU 2017102002A RU 2762111 C1 RU2762111 C1 RU 2762111C1
- Authority
- RU
- Russia
- Prior art keywords
- another embodiment
- cancer
- compound
- alkyl
- formula
- Prior art date
Links
Images











































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/64—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with substituted hydrocarbon radicals attached to ring carbon atoms, e.g. histidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/04—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D233/20—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D233/22—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/56—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms
- C07D233/60—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, attached to ring carbon atoms with hydrocarbon radicals, substituted by oxygen or sulfur atoms, attached to ring nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/04—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D263/06—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by oxygen atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/08—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D263/10—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D263/14—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms with radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/32—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/24—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/28—Radicals substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/42—Amino or imino radicals substituted by hydrocarbon or substituted hydrocarbon radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/56—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Изобретение относится к новым соединениям, обладающим противораковым действием, и их применению для лечения рака и лекарственно-устойчивых опухолей, таких как меланома, метастатическая меланома, лекарственно-устойчивая меланома, рак простаты и лекарственно-устойчивый рак простаты. 5 н. и 4 з.п. ф-лы, 25 пр., 24 табл., 38 ил.
Description
Область техники, к которой относится изобретение
Настоящее изобретение относится к новым соединениям, обладающим противораковым действием, способам получения этих соединений и их применению для лечения рака, лечения лекарственно-устойчивых опухолей, лекарственно-устойчивого рака, метастатического рака, метастатической меланомы, лекарственно-устойчивой меланомы, рака простаты и лекарственно-устойчивого рака простаты.
Уровень техники
Рак является второй наиболее распространенной причиной смерти в Соединенных Штатах, уступая лишь сердечным заболеваниям. В Соединенных Штатах на долю рака приходится 1 из 4 смертей. 5-летний коэффициент относительной выживаемости всех пациентов, имеющих рак, диагностированных в 1996-2003 годах, составляет 66%, по сравнению с 50% в 1975-1977 годах (Cancer Facts & Figures American Cancer Society: Атланта, штат Джорджия (2008)). Это улучшение выживаемости отражает совершенствование диагностирования на ранних стадиях и улучшение лечения. Открытие высокоэффективных противораковых средств с низкой токсичностью является основной целью онкологических исследований.
Микротрубочки являются волокнами цитоскелета, состоящими из гетеродимеров αβ-тубулина и участвующими в широком ряде клеточных функций, включая сохранение формы, транспорт везикул, подвижность и деление клеток. Тубулин является основным структурным компонентом микротрубочек и хорошо подтвержденной мишенью для различных весьма успешных противораковых лекарств. Соединения, способные влиять на равновесие микротрубочек-тубулина в клетках, являются эффективными для лечения раковых заболеваний. Противораковые лекарства, такие как таксол и винбластин, которые способны влиять на равновесие микротрубочек-тубулина в клетках, широко используются в химиотерапии рака. Существует три основных класса антимитотических средств. Средства, стабилизирующие микротрубочки, которые связываются с полностью сформировавшимися микротрубочками и препятствуют деполимеризации субъединиц тубулина, представлены таксанами и эпотилонами. Другие два класса средств являются средствами, дестабилизирующими микротрубочки, которые связываются с димерами тубулина и ингибируют их полимеризацию в микротрубочки. Алкалоиды барвинка, такие как винбластин, связываются с сайтом барвинка и представляют один из этих классов. Колхицин и средства, связывающие сайт колхицина, взаимодействуют с другим сайтом тубулина и определяют третий класс антимитотических средств.
Как таксаны, так и алкалоиды барвинка широко используются для лечения раковых заболеваний у людей, тогда как в настоящее время для химиотерапии рака до сих пор не одобрено ни одного средства, связывающего сайт колхицина. Однако колхицин-связывающие средства, такие как комбретастатин А-4 (CA-4) и ABT-751 (Фигура 19), в настоящее время проходят клинические исследования в качестве новых потенциальных химиотерапевтических средств (Luo, Y.; Hradil, V. P.; Frost, D. J.; Rosenberg, S. H.; Gordon, G. B.; Morgan, S. J.; Gagne, G. D.; Cox, B. F.; Tahir, S. K.; Fox, G. B., ABT-751, "A novel tubulin-binding agent, decreases tumor perfusion and disrupts tumor vasculature". Anticancer Drugs 2009, 20(6), 483-92.; Mauer, A. M.; Cohen, E. E.; Ma, P. C.; Kozloff, M. F.; Schwartzberg, L.; Coates, A. I.; Qian, J.; Hagey, A. E.; Gordon, G. B., "A phase II study of ABT-751 in patients with advanced non-small cell lung cancer". J Thorac Oncol 2008, 3(6), 631-6.; Rustin, G. J.; Shreeves, G.; Nathan, P. D.; Gaya, A.; Ganesan, T. S.; Wang, D.; Boxall, J.; Poupard, L.; Chaplin, D. J.; Stratford, M. R.; Balkissoon, J.; Zweifel, M., "A Phase Ib trial of CA4P (combretastatin A-4 phosphate), carboplatin, and paclitaxel in patients with advanced cancer". Br J Cancer 2010, 102(9), 1355-60.).
К сожалению, противораковые лекарства, взаимодействующие с микротрубочками, при их клиническом применении, имеют две основные проблемы: резистентность и нейротоксичность. Общий механизм множественной лекарственной устойчивости (МЛУ), а именно отток лекарства, опосредованный белком АТФ-связывающего кассетного (ABC) транспортера, ограничивает их эффективность (Green, H.; Rosenberg, P.; Soderkvist, P.; Horvath, G.; Peterson, C., "beta-Tubulin mutations in ovarian cancer using single strand conformation analysis-risk of false positive results from paraffin embedded tissues". Cancer Letters 2006, 236(1), 148-54.; Wang, Y.; Cabral, F., "Paclitaxel resistance in cells with reduced beta –tubulin". Biochimica et Biophysica Acta, Molecular Cell Research 2005, 1744(2), 245-255.; Leslie, E. M.; Deeley, R. G.; Cole, S. P. C., "Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense". Toxicology and Applied Pharmacology 2005, 204(3), 216-237.).
Р-гликопротеины (P-gp, кодируемые геном MDR1) являются важными членами суперсемейства ABC. P-gp препятствует внутриклеточному накоплению многих противораковых лекарств за счет увеличения их оттока из раковых клеток, внося также вклад в печеночные, почечные или кишечные выводящие пути. Попытки совместного введения модуляторов или ингибиторов P-gp для увеличения клеточной доступности за счет блокирования действия P-gp имели лишь ограниченный успех (Gottesman, M. M.; Pastan, I., "The multidrug transporter, a double-edged sword". J Biol Chem 1988, 263(25), 12163-6.; Fisher, G. A.; Sikic, B. I., "Clinical studies with modulators of multidrug resistance". Hematology/Oncology Clinics of North America 1995, 9(2), 363-82).
Другой основной проблемой с таксанами, как и со многими биологически активными природными продуктами, является их липофильность и недостаточная растворимость в водных системах. Это приводит к применению в клинических композициях эмульгаторов, таких как Cremophor EL и Tween 80. Описан ряд биологических эффектов, связанных с этими носителями лекарственных композиций, включая острые реакции гиперчувствительности и периферические невропатии (Hennenfent, K. L.; Govindan, R., "Novel formulations of taxanes: a review. Old wine in a new bottle?" Ann Oncol 2006, 17(5), 735-49.; ten Tije, A. J.; Verweij, J.; Loos, W. J.; Sparreboom, A., "Pharmacological effects of formulation vehicles : implications for cancer chemotherapy". Clin Pharmacokinet 2003, 42(7), 665-85.).
По сравнению с соединениями, связывающими паклитаксел-связывающий сайт или связывающий сайт алкалоида барвинка, колхицин-связывающие средства обычно обладают относительно простыми структурами. За счет этого обеспечивается лучшая возможность для пероральной биодоступности за счет структурной оптимизации для улучшения растворимости и фармакокинетических (РК) параметров. Кроме того, многие из этих лекарств преодолевают P-gp-опосредованную МЛУ. Следовательно, эти новые соединения, нацеленные на колхицин-связывающий сайт, являются многообещающими в качестве терапевтических средств, в частности, поскольку они обладают улучшенной растворимостью в воде и преодолевают P-gp-опосредованную МЛУ.
Рак простаты является одним из наиболее часто диагностируемых кожных раковых заболеваний среди мужчин в США, и является второй наиболее распространенной причиной смерти от рака, составляя, по прогнозам, свыше 180000 новых случаев и почти 29000 смертей в этом году. Пациенты с распространенным раком простаты проходят антиандрогенную терапию (ADT), как правило, при помощи агонистов лютеинизирующего релизинг-гормона (LHRH) или билатеральной орхиэктомии. Антиандрогенная терапия не только снижает тестостерон, но понижает также уровни эстрогена, поскольку эстроген получается ароматизацией тестостерона, уровни которого истощаются под действием ADT. Дефицит эстрогена, вызванный антиандрогенной терапией, обусловливает существенные побочные эффекты, которые включают приливы крови, гинекомастию и масталгию, рарефикацию костей, снижение качества и прочности костей, остеопороз и опасные для жизни переломы, неблагоприятные жировые изменения и более частые сердечнососудистые заболевания, и инфаркт миокарда, а также депрессию и другие изменения настроения.
Лейпролид ацетат (Lupron®) является синтетическим нонапептидным аналогом природного гонадотропин-высвобождающего гормона (GnRH или LHRH). Лейпролид ацетат является суперагонистом LHRH, который в результате подавляет секрецию LH гипофизом. Лейпролид ацетат действует как потенциальный ингибитор секреции гонадотропина, что приводит к подавлению овариального и тестикулярного стероидогенеза. У людей введение лейпролид ацетата приводит к первоначальному увеличению циркулирующих уровней лютеинизирующего гормона (LH) и фолликулостимулирующего гормона (FSH), приводя к временному повышению уровней гонадных стероидов (тестостерона и дигидротестостерона у мужчин, и эстрона и эстрадиола у предклимактерических женщин). Однако постоянное введение лейпролид ацетата приводит к снижению уровней LH и FSH. У мужчин тестостерон снижается до уровней кастрата (ниже 50 нг/дл). У предклимактерических женщин эстрогены снижаются до постклимактерических уровней. Как известно, тестостерон является стимулом для раковых клеток простаты. Следовательно, подавление секреции тестостерона или ингибирование действия тестостерона является необходимым компонентом терапии рака простаты. Лейпролид ацетат может использоваться для подавления LH, которым является уменьшение и понижение тестостерона в сыворотке до уровней кастрата для лечения рака простаты.
Злокачественная меланома является наиболее опасной формой рака кожи, насчитывающей около 75% смертей от рака кожи. Заболеваемость меланомой неуклонно растет среди западных жителей. За последние 20 лет количество случаев удвоилось. Каждый год во всем мире диагностируется около 160000 новых случаев, и наиболее часто встречается у мужчин и жителей Кавказа. Согласно отчету ВОЗ, ежегодно во всем мире случается около 48000 смертей, связанных с меланомой.
В настоящее время не существует эффективного способа лечения метастатической меланомы. Она является высоко резистентной к существующей химиотерапии, лучевой терапии и иммунотерапии. Метастатическая меланома имеет очень неблагоприятный прогноз, со средним коэффициентом выживаемости 6 месяцев и 5-летним коэффициентом выживаемости менее 5%. За последние 30 лет дакарбазин (DTIC) является единственным одобренным FDA лекарством для лечения метастатической меланомы. Однако он обеспечивает лишь менее 5% полной ремиссии у пациентов. В последние годы были предприняты значительные усилия в борьбе с метастатической меланомой. Ни комбинации DTIC с другими химиотерапевтическими лекарствами (например, цисплатином, винбластином и кармустином), ни добавление к DTIC интерферон-α2b не продемонстрировали преимущества для выживаемости по сравнению с лечением только DTIC. Совсем недавно клинические испытания с антителами и вакцинами для лечения метастатической меланомы также не смогли показать удовлетворительную эффективность.
Клетки меланомы имеют низкие уровни спонтанного апоптоза in vivo по сравнению с другими типами опухолевых клеток, и они являются относительно резистентными к апоптозу, индуцированному лекарствами in vitro. Естественной ролью меланоцитов является защита внутренних органов от УФ света, потенциального ДНК-разрушающего агента. Поэтому не удивительно, что клетки меланомы могут иметь специальные системы восстановления поврежденной ДНК и обладать улучшенными свойствами выживания. Более того, в недавних исследованиях показано, что при прогрессировании меланомы она приобретает комплекс генетических изменений, которые приводят к гиперактивации насосов оттока, ферментов детоксикации и многофакторному изменению путей выживания и апоптоза. Было предположено, что все это опосредует фенотип меланомы, обладающей множественной лекарственной устойчивостью (МЛУ). При быстро растущей заболеваемости этим заболеванием и высокой устойчивости к существующим терапевтическим средствам, разработка более эффективных лекарств для лечения распространенной меланомы и других типов рака, которые могут эффективно преодолевать МЛУ, обеспечит существенные преимущества для онкологических пациентов.
Раскрытие изобретения
В одном варианте настоящее изобретение относится к соединению, представленному структурой формулы XI:

где
X является связью, NH или S;
Q является O, NH или S; и
A является замещенными или незамещенными одно-, конденсированными или многокольцевыми, арильными или (гетеро)циклическими кольцевыми системами; замещенными или незамещенными, насыщенными или ненасыщенными N-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными S-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными O-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными циклическими углеводородами; или замещенными или незамещенными, или насыщенными или ненасыщенными смешанными гетероциклами;
где указанное кольцо А необязательно замещено 1-5 заместителями, которые независимо являются O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN,
-CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом,
-OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2; и
i является целым числом от 0 до 5;
где если Q является S, то X не является связью.
В одном варианте настоящее изобретение относится к соединению, представленному структурой формулы VIII:

(VIII)
R4, R5 и R6 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
Q является S, O или NH;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 3.
В одном варианте настоящее изобретение относится к соединению, представленному структурой формулы XI(b):

где R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4.
В одном варианте настоящее изобретение относится к соединению, представленному структурой формулы XI(c):

где R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4.
В одном варианте настоящее изобретение относится к соединению, представленному структурой формулы XI(e):

где R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4.
В другом варианте настоящее изобретение относится к следующим соединениям: (2-(фениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанону (5a), (2-(п-толиламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанону (5b), (2-(п-фторфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанону (5c), (2-(4-хлорфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанону (5d), (2-(фениламино)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанону (5e), 2-(1H-индол-3-ил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанону (17ya); и (2-(1H-индол-5-иламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанону (55).
В другом варианте соединение настоящего изобретения является его изомером, фармацевтически приемлемой солью, фармацевтическим продуктом, таутомером, гидратом, N-оксидом или их комбинациями.
В одном варианте настоящее изобретение относится к фармацевтической композиции, включающей соединение настоящего изобретения и фармацевтически приемлемый носитель.
В одном варианте настоящее изобретение относится к способу (а) лечения, подавления, снижения степени, уменьшения риска или ингибирования рака; (б) лечения лекарственно-устойчивой опухоли или опухолей; и (в) разрушения раковых клеток, включающему введение соединения настоящего изобретения. В другом варианте рак выбран из группы, состоящей из рака простаты, рака молочной железы, рака яичников, рака кожи, меланомы, рака легких, рака ободочной кишки, лейкоза, рака почек, рака ЦНС и их комбинаций.
Краткое описание чертежей
Рассмотрен предмет обсуждения, поскольку настоящее изобретение отдельно подчеркнуто и четко заявлено в заключительной части настоящего изобретения. Однако настоящее изобретение относительно организации и способа действий, вместе с его объектами, особенностями и преимуществами, может быть более понятно со ссылкой на следующее подробное описание при чтении с сопровождающими чертежами, в которых:
На Фигуре 1 изображен синтез разнообразных моделей кольца В: оксазол. Реагенты и условия: (a) MeOH, CH3COCl, 83%; (b) этиловый эфир бензимидокислоты, CH2Cl2, Et3N, 96%; (c) LiOH, MeOH, H2O, 65%; (d) EDCI, HOBt, NMM, CH3OCH3NH•HCl, 61%; (e) 3,4,5-триметоксифенилмагния бромид, ТГФ, 48%-71%; (f) CBrCl3, DBU, CH2Cl2, 56%.
На Фигуре 2 изображен синтез разнообразных моделей кольца В. Реагенты и условия: (a) EDCI, HOBt, NMM, CH3OCH3NH•HCl, CH2Cl2, 51-95%; (b) 3,4,5-триметоксифенилмагния бромид, ТГФ, 48-78%; (c) LAH, -78 oC, ТГФ, 85%; (d) реагент Десс-Мартина, CH2Cl2, 81%; (e) EDCI, HOBt, NMM, 3,4,5-триметоксибензойная кислота, CH2Cl2, 58%.
На Фигуре 3 изображена схема синтеза соединений настоящего изобретения. Реагенты и условия: (a) MeOH/pH=6,4 фосфатный буфер, комнатная температура; (b) EDCI, HOBt, NMM, HNCH3OCH3; (c) CBrCl3, DBU, CH2Cl2; (d) 3,4,5-триметоксифенилмагния бромид, ТГФ; (e) изопропил трифенилфосфония йодид, n-BuLi, ТГФ; (f) LAH, ТГФ; (g) для 2e-цис и 2e-транс, NH2OH•HCl, C2H5OH, H2O, NaOH; для 2g и 2h, NH2OMe•HCl, пиридин; (h) TsCl, NaH, основной Al2O3; (i) NH2NH2•xH2O, CH2Cl2, t-BuOH; (j) диэтил цианометилфосфонат, n-BuLi, ТГФ; (k) бис-триметилсилилкарбодиимид, TiCl4, CH2Cl2; (l) EDCI, HOBt, Et3N, 3,4,5-триметоксианилин, CH2Cl2.
На Фигуре 4 изображена схема синтеза соединений настоящего изобретения. Реагенты и условия: (a) бром, EtOH; (b) бензотиоамид, EtOH, дефлегмация; (c) EDCI, HOBt, NMM, HNCH3OCH3, CH2Cl2; (d) CBrCl3, DBU, CH2Cl2; (e) LAH, ТГФ; (f) 5-(бромметил)-1,2,3-триметоксибензол, Ph3P, ТГФ; (g) n-BuLi, ТГФ; (h) (1) HCl, H2O; (2) NaNO2, H2O, 0 oC; (i) этилкалия ксантат; (j) KOH/EtOH; (k) H2O, HCl; (l) 5-йод-1,2,3-триметоксибензол, CuI, t-BuONa; (m) 2 экв. или 1 экв. m-CPBA, CH2Cl2; (n) 3,4,5-триметоксианилин, NEt3, ДМФ.
На Фигуре 5 изображена схема синтеза соединений настоящего изобретения. Реагенты и условия: (a) L-цистеин, EtOH, 65 oC; (b) EDCI, HOBt, NMM, HNCH3OCH3, CH2Cl2; (c) TBDMSCl, имидазол, ТГФ; (d) 3,4,5-триметоксифенилбромид, BuLi, ТГФ; (e) TBAF, ТГФ; (f) SOCl2, Et2O; (g) NH3, MeOH; (h) POCl3; (i) PhSO2Cl, Bu4NHSO4, толуол, 50% NaOH; (j) 1 н. NaOH, EtOH, дефлегмация; (k) Boc2O, 1 н. NaOH, 1,4-диоксан; (l) CBrCl3, DBU, CH2Cl2; (m) 4 н. HCl в 1,4-диоксане; (n) NaH, ДМФ, MeI; (o) HCHO, NaBH3CN, Et3N.
На Фигуре 6 изображена схема синтеза соединений настоящего изобретения. Реагенты и условия: (a) EtOH, 65 oC; (b) NaOH, C2H5OH, дефлегмация; (c) EDCI, HOBt, NMM, HNCH3OCH3, CH2Cl2; (d) 3,4,5-триметоксифенилбромид, BuLi, ТГФ; (e) 2 н. HCl в 1,4-диоксане.
На Фигуре 7 изображена схема синтеза для получения арил-бензоил-имидазольных (ABI) соединений настоящего изобретения. Реагенты и условия: (a) t-BuOH, I2, этилендиамин, K2CO3, дефлегмация; (b) PhI (OAc)2, K2CO3, ДМСО; (c) DBU, CBrCl3, ДМФ; (d) NaH, PhSO2Cl, ТГФ, 0°C – комнатная температура; (e) t-BuLi, замещенный бензоилхлорид, ТГФ, -78°C; (f) Bu4NF, ТГФ, комнатная температура.
На Фигуре 8 изображена схема синтеза для получения арил-бензоил-имидазольных (ABI) соединений настоящего изобретения. Реагенты и условия: (a) NH4OH, оксалальдегид, этанол, комнатная температура; (b) NaH, PhSO2Cl, ТГФ, 0°C – комнатная температура; (c) t-BuLi, замещенный бензоилхлорид, ТГФ, -78°C; (d) Bu4NF, ТГФ, комнатная температура; (e) BBr3, CH2Cl2; (f) c-HCl, AcOH, дефлегмация.
На Фигуре 9 изображена схема синтеза для получения арил-бензоил-имидазольных (ABI) соединений настоящего изобретения. Реагенты и условия: (a) NaH, замещенный бензоилхлорид, ТГФ.
На Фигуре 10 изображена схема синтеза соединений 12dc, 12fc, 12daa, 12dab, 12cba. (a) AlCl3, ТГФ, дефлегмация; (b) NaH, CH3I для 12dab и 12cba, и BnBr для 12daa, ТГФ, дефлегмация.
На Фигуре 11 изображена схема синтеза соединений 11gaa, 12la. (a) NH4OH, этанол, глиоксаль, комнатная температура; (b) NaH, замещенный PhSO2Cl, ТГФ, 0°C – комнатная температура; (c) t-BuLi (1,7 M в пентане), замещенный бензоилхлорид, ТГФ, -78°C; (d) Bu4NF, комнатная температура.
На Фигуре 12 изображена схема синтеза соединений 15xaa и 12xa. (a) 1. KOH, этанол; 2. PhSO2Cl, ацетон; (b) NH4OH, глиоксаль, этанол, комнатная температура; (c) NaH, PhSO2Cl, ТГФ, 0°C – комнатная температура; (d) t-BuLi (1,7 M в пентане), бензоилхлорид, ТГФ, -78°C; (e) NaOH, этанол, H2O, дефлегмация.
На Фигуре 13 изображена схема синтеза 17ya. (a) 1. KOH, этанол; 2. PhSO2Cl, ацетон, комнатная температура; (b) NH4OH, глиоксаль, этанол, комнатная температура; (c) NaH, PhSO2Cl, ТГФ, 0°C – комнатная температура; (d) t-BuLi (1,7 M в пентане), бензоилхлорид, ТГФ, -78°C; (e) NaOH, этанол, H2O, дефлегмация.
На Фигуре 14 изображена схема синтеза 12fa. (a) NH4OH, оксалальдегид, этанол, комнатная температура; (b) NaH, PhSO2Cl, ТГФ, 0°C – комнатная температура; (c) t-BuLi, 3,4,5-триметоксибензоилхлорид, ТГФ, -78°C; (d) Bu4NF, ТГФ, комнатная температура.
На Фигуре 15 изображена схема синтеза соединения 55.
На Фигуре 16 изображена схема синтеза соединений на основе изохинолина и хинолина. На Фигуре 16А изображена схема синтеза производных изохинолина. Реагенты и условия: a) арилбороновая кислота (1 экв.), Pd(PPh3)4 (0,01 экв.), K2CO3, H2O, ДМФ, 5 часов; b) арилбороновая кислота (2,4 экв.), Pd(PPh3)4 (0,04 экв.), K2CO3, H2O, ДМФ, 16 часов; c) арилбороновая кислота (1,2 экв.), Pd(PPh3)4 (0,04 экв.), K2CO3, H2O, ДМФ, 16 часов. На Фигуре 16B изображена схема синтеза соединений 41 и 44. Реагенты и условия: a) п-фторбензолсульфонилхлорид, пиридин, пиридин, 80oC, 3 часа; b) 5-индолбороновая кислота (1,2 экв.), Pd(PPh3)4 (0,02 экв.), K2CO3, H2O, ДМФ, 16 часов. На Фигуре 16C изображена схема синтеза производного изохинолина 6d. На Фигуре 16D изображена схема синтеза производного изохинолина 6c. На Фигуре 16E изображена схема синтеза производного изохинолина 6b.
На Фигуре 17 изображена кривая стандартной растворимости ABI соединения 12ga (растворенного в ацетонитриле). Ось х является количеством соединения, а ось y является площадью пика m/z.
На Фигуре 18 изображена измеренная растворимость в воде антитубулиновых соединений 1h, 1c, 66a, 2r-HCl, 5a и 5c.
На Фигуре 19 изображены структуры тубулиновых ингибиторов колхицин-связывающего сайта.
На Фигуре 20 изображена способность антитубулиновых соединений 1h, 1c, 2j, 66a и 5a ингибировать полимеризацию тубулина in vitro (Фигура 20a), а также связывание 5c (Фигура 20b) и 5Hc с колхициновым сайтом (Фигура 20c).
На Фигуре 21 изображены доза-зависимые кривые 2-арил-4-бензоил-имидазольных соединений (ABI) по сравнению с другими противораковыми лекарствами и соединениями на клеточную линию меланомы, обладающей множественной лекарственной устойчивостью (МЛУ клетки), а также соответствующую восприимчивую исходную клеточную линию (клетки нормальной меланомы). Большое расстояние между двумя кривыми для паклитаксела, винбластина и колхицина указывает, что они являются субстратами для Р-гликопротеина (P-gp). Перекрывание двух кривых каждого ABI соединения указывает, что эти ABI соединения не являются субстратами для P-gp и они преодолевают множественную лекарственную устойчивость.
На Фигуре 22 является действие ABI соединений на полимеризацию тубулина in vitro. Тубулин (0,4 мг/анализ) обрабатывали 10 мкМ ABI соединений (контроль с носителем, 5% ДМСО). Поглощение при 340 нм контролировали при 37°C каждую минуту в течение 15 минут, и было показано, что ABI соединения 12da, 12db и 12cb ингибируют полимеризацию тубулина in vitro.
На Фигуре 23 изображен анализ образования колонии меланомы B16-F1 в мягком агаре, которые показал, что ABI соединения ингибируют образование колонии в зависимости от концентрации. На Фигуре 23A представлены иллюстративные изображения контрольного образца и каждого из исследованных соединений (12cb, 12da и 12fb) при 100 нМ. Диаметр каждой лунки составил 35 мм. На Фигуре 23B изображено количественное представление анализов результата для каждого исследованного соединения (12cb, 12da и 12fb). Значение Р рассчитали по сравнению с контрольным образцом, используя t-критерий Стьюдента при помощи программы GraphPad Prism. Колонки, средние значения трех воспроизведений; столбики, стандартное отклонение.
На Фигуре 24 изображено исследование ABI соединений in vivo. На Фигуре 24А изображена in vivo активность 12cb против опухолей меланомы B16-F1 у мышей C57/BL. На Фигуре 24B изображена in vivo активность 12fb против меланомы B16-F1 у мышей C57BL/6 и «голых» мышей SHO. Результаты показали, что 12fb ингибирует рост опухоли меланомы доза-зависимым образом. Мыши C57BL/6 с аллотрансплантатом меланомы B16-F1 (n=5 на группу). Каждой мыши ввели 0,5x106 клеток подкожной инъекцией в бок. Когда размер опухоли достиг ~100 мм3 начали ежедневное лечение внутрибрюшинными инъекциями по 30 мкл. На Фигуре 24C изображена in vivo активность 12fb против ксенотрансплантата меланомы человека A375. «Голые» мыши SHO с ксенотрансплантатом меланомы человека A375 (n = 5 на группу). Каждой мыши ввели 2,5x106 клеток подкожной инъекцией в бок. Когда размер опухоли достиг ~150 мм3 начали ежедневное лечение внутрибрюшинными инъекциями по 30 мкл. Контроль, только раствор носителя; точки, средние значения; столбики, стандартное отклонение. DTIC, (5-(3,3,-диметил-1-триазенил)-имидазол-4-карбоксамид, дакарбазин.
На Фигуре 25 изображен анализ конкурентного связывания колхицина. На Фигуре 25A изображен сцинтилляционный анализ сближения конкурентного связывания [3H]-колхицина, который покзаал, что 12cb конкурентно связывается с тубулиновым колхицин-связывающим сайтом. На Фигуре 25B изображены иллюстративные диаграммы анализов клеточного цикла с использованием проточной цитометрии, которые показали, что ABI соединения (показаны примеры для 12da и 12fb) останавливают клетки A375 на фазе G2/M через 24 часа инкубирования. Действие и сила были такими же, как у колхицина. На Фигуре 25C представлены количественные графические изображения анализов клеточного цикла. Все исследованные соединения (показаны примеры для 12cb, 12da и 12fb) останавливают клетки А375 на фазе G2/M доза-зависимым образом. ABI 12da продемонстрировало более высокую эффективность, чем колхицин. На Фигуре 25D изображены анализы клеточных циклов с использованием проточной цитометрии клеток А375 после инкубирования с 12cb, 12da и 12fb при различных концентрациях в течение 24 часов. Колхицин останавливает большинство клеток на фазе G2/M, начиная с 50 нМ. 12cb, 12da и 12fb также останавливают большинство клеток на фазе G2/M, начиная с 200, 50 и 200 нМ, соответственно.
На Фигуре 26 показано действие 17ya и 55 на полимеризацию тубулина. Соединения 17ya и 55 связываются с колхицин-связывающим сайтом тубулина и ингибируют полимеризацию тубулина. Фигура 26A, связывание конкурентного большинства. Тубулин (1 мг/мл) и колхицин (1,2 мкМ) инкубировали с различными концентрациями подофиллтоксина, винбластина, соединений 17ya и 55. N = 3; среднее ± стандартное отклонение. Подофиллтоксин и винбластин использовались в качестве положительного и отрицательного контроля, соответственно. Фигура 26В, влияние на полимеризацию тубулина. Тубулин (0,4 мг) обрабатывали исследуемыми соединениями (5 мкМ). В качестве положительного контроля использовали колхицин. Фигуры 26C и 26D, способность 17ya и 55 усиливать образование цитоплазматическoго комплекса ДНК-гистон (апоптоз) через 24 часа в клетках PC-3 (C) и PC-3/TxR (D) (N =3); среднее ± стандартное отклонение. В качестве положительного контроля использовали доцетаксел.
На Фигуре 27 изображена противораковая эффективность in vivo. Фигура 27А, «голые» мыши с опухолями PC-3 обрабатывались доцетакселем (внутривенно, 10 или 20 мг/кг) на 1 и 9 день. (N = 5-6). Столбики, стандартная погрешность. Фигура 27В, «голые» мыши с опухолями PC-3/TxR обрабатывались доцетакселем (внутривенно, 10 или 20 мг/кг) на 1 и 9 день, обработки соединением 17ya (перорально, 6,7 мг/кг) один раз в день, пять дней в неделю. (N = 4-5). Столбики, стандартная погрешность. Фигура 27С, «голые» мыши с опухолями PC-3/TxR обрабатывались соединением 17ya (перорально, 3,3 мг/кг) дважды в день, в течение четырех дней на первой неделе, а затем получали дозу один раз в день, пять дней в неделю, в течение недель 2-4 (N = 7), с обработками соединением 55 (перорально, 10 или 30 мг/кг) дважды в день, пять дней в неделю, в течение четырех недель (N = 7). Столбики, стандартная погрешность. Фигура 27D, «голые» мыши с опухолями PC-3/TxR обрабатывались соединением 17ya (перорально, 10 мг/кг) три раза в неделю, в течение четырех недель (N = 5). Столбики, стандартная погрешность.
На Фигуре 28 показано, что соединения 1h, 2k и 2l ингибируют полимеризацию тубулина за счет связывания с колхицин-связывающим сайтом тубулина. (Фигура 28A) Структуры 1h (-H), 2k (-F) и 2l (–OH). (Фигура 28B) Влияние соединений на полимеризацию тубулина. Тубулин (0,4 мг) обрабатывался соединениями 1h, 2k и 2l (10 мкМ). Поглощение при 340 нм контролировали каждую минуту в течение 15 минут. (Фигура 28C) Способность 1h конкурировать за тубулиновые связывающие сайты колхицина, винбластина и паклитаксела с использованием масс-спектрометрического анализа конкурентного связывания (n = 3); столбики, стандартное отклонение.
На Фигуре 29 показано, что соединения 1h, 2k и 2l останавливают клетки на фазе G2/M и индуцируют апоптоз. (Фигура 29A) Иллюстративные диаграммы анализов клеточного цикла после обработки соединениями клеток PC-3 и A375 в течение 24 часов. (Фигура 29B) Изменения соотношения G2/M, вызванные 1h, 2k и 2l в клетках PC-3 и A375 через 24 часа обработки. (Фигура 29C) Способность 1h, 2k и 2l усиливать образование цитоплазматического комплекса ДНК-гистон через 24 часа (n = 3); столбики, стандартное отклонение. В качестве положительных контролей использовались колхицин и винбластин.
На Фигуре 30 изображены фармакокинетические исследования 1h, 2k и 2l, введенных мышам и крысам внутрибрюшинно. (Фигура 30A) Кривая концентрации-времени соединений SMART у мышей ICR (n = 3); столбики, стандартное отклонение. Соединения SMART вводились по 15 мг/кг внутривенно, инъекцией в хвостовую вену. (Фигура 30B) Кривая концентрации-времени 1h и 2k у крыс SD (n = 4); столбики, стандартное отклонение. Крысам породы Sprague-Dawley внутривенно вводили 2,5 мг/кг композиции ДМСО/ПЭГ300 (1/4).
На Фигуре 31 представлена in vivo противораковая эффективность (при внутрибрюшинном введении) и нейротоксичность соединений SMART у мышей. (Фигура 31A) Эффективность соединений SMART для опухоли простаты PC-3, ксенотрансплантированной «голым» мышам (n = 6-8). (Фигура 31B) Эффективность винбластина для опухоли простаты PC-3, ксенотрансплантированной «голым» мышам (n = 8). Он служил положительным контролем. (Фигура 31C) Эффективность in vivo 1h и 2k у «голых» мышей с ксенотрансплантатами меланомы A375 (n = 10). «Голым» мышам инокулировали 2,5 x 106 клеток PC-3 или A375 и ежедневно обрабатывали внутрибрюшинно (соединениями SMART) и q2d (винбластином) после образования опухоли (150-200 мм3). Каждая точка представляет средний объем опухоли для животных в каждой группе. (Фигура 31D) Нейротоксичность in vivo (тест вращающегося стержня) соединения 1h у мышей ICR (n = 7 или 8). 1h (5 и 15 мг/кг), винбластин (0,5 мг/кг) и носитель вводили внутрибрюшинно, ежедневно, а винбластин использовали в качестве положительного контроля. Дозирование прекратили на 31 день. *, p < 0,05. Столбики, стандартная погрешность.
На Фигуре 32 изображено молекулярное моделирование ABI соединений, направленных на тубулин в колхицин-связывающем сайте. На Фигурах 32А и 32В изображено молекулярное моделирование соединений 12cb и 11cb, соответственно.
На Фигуре 33 представлены микроскопические изображения микротрубочек с иммунофлуоресцентной меткой в клетках меланомы WM-164, которые показывают, что модальность микротрубочек резко изменилась после обработки соединением в течение 18 часов. Это дает визуальное подтверждение того, что ABI соединения направлены на тубулин и нарушают функциональное образование микротрубочек.
На Фигуре 34 изображена эффективность и переносимость 6b и 6c в моделях ксенотрансплантата после внутрибрюшинной инъекции. Фигура 34А. Ксенотрансплантаты PC-3 обрабатывались носителем (один раз в день), 6b (40 мг/кг, один раз в день) или 6c (40 мг/кг, один раз в день) в течение 3 недель. Дозировка носителя состояла из 20% Captex200 в Tween80. Объемы опухолей (мм3) нанесли на график в зависимости от времени, и они представляют собой средние значение ± стандартное отклонение для восьми животных. Объемы опухолей показаны на левом графике, а вес тела показан на правом графике. Фигура 34B. Размер печени (г) каждой «голой» мыши измерили через 3 недели лечения. Фигура 34С. Количество белых кровяных клеток подсчитали в цельной крови, собранной у животных через 3 недели лечения.
Фигура 35 - Соединение 17ya демонстрирует потенциальное ингибирование роста эндотелиальных клеток. Ингибирование клеточного роста доксорубицином (Фигура 35А) и соединением 17ya (Фигура 35В) исследовали в нескольких клеточных линиях анализом SRB. Определение HUVEC-активных и HUVEC-неактивных представляет эндотелиальные клеточные культуры, дополненные фактором роста и лишенные фактора роста, соответственно.
Фигура 36 - Разрушение предварительно образованного капилляра под действием 17ya. Клетки HUVEC, загруженные на Matrigel, оставили образовывать трубки на 16 часов, а предварительно образованные трубки обработали исследуемым соединением. Количество трубок (A, B и C) и узлов (D, E и F) подчитывали до 25 часов после обработки лекарством. Графики А и D являются условиями в присутствии CA4, графики В и Е являются условиями в присутствии доксорубицина, а графики С и F являются условиями в присутствии 17ya.
Фигура 37 - Ингибирование эндотелиального капиллярного образования и разрушение предварительно образованных капилляров. Ингибирование образования капилляров ( ) и разрушение предварительного образованных капилляров ( ) сравнили исследованием in vitro с использованием клеток HUVEC через 15 часов после обработки CA4 (A и D), DOX (B и E), и 17ya (C и F). Стрелка показывает значение IC50 каждого соединения при ингибировании роста клеток HUVEC.
Фигура 38 - 17ya и 55 увеличивают проницаемость монослоев эндотелиальных клеток. Конфлюентные монослои HUVEC обрабатывали исследуемым соединением. Просачивание FITC-конъюгированного декстрана через монослой оценивали по измерениям относительной флуоресценции при возбуждении л = 485 нм и излучении л = 530 нм в ресивере для определения изменений проницаемости монослоя после воздействия.
Следует понимать, что для простоты и ясности иллюстрации, элементы, показанные на фигурах, не обязательно изображены в масштабе. Например, для ясности размеры некоторых элементов могут быть преувеличены по сравнению с другими элементами. Кроме того, если это считается целесообразным, цифры ссылок на фигурах могут повторяться для указания соответствующих или аналогичных элементов.
Осуществление изобретения
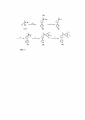
В одном варианте настоящее изобретение относится к соединению формулы (I)

(I)
где
каждый A и C независимо являются замещенными или незамещенными одно-, конденсированными или многокольцевыми, арильными или (гетеро)циклическими кольцевыми системами; замещенными или незамещенными, насыщенными или ненасыщенными N-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными S-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными O-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными циклическими углеводородами; или замещенными или незамещенными, или насыщенными или ненасыщенными смешанными гетероциклами;
B является



















R10 и R11 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
X является связью, NH, C1 - C5 углеводородом, O или S;
Y является связью, -C=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN,
-C=N-CN, -CH=CH-, -C=C(CH3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), –(C=O)-O,
-O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
где указанные кольца А и С необязательно замещены 1-5 заместителями, которые независимо являются O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN,
-CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом,
-OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 0 до 5;
l является целым числом от 1 до 2;
где
если В является бензольным кольцом, тиофеновым кольцом, фурановым кольцом или индольным кольцом, то Х не является связью или CH2, и А не является индолом;
если В является индолом, то Х не является О; и
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, если B формулы I является тиазольным кольцом, то X не является связью.
В одном варианте, A в соединении Формулы I является индолилом. В другом варианте A является 2-индолилом. В другом варианте A является фенилом. В другом варианте A является пиридилом. В другом варианте A является нафтилом. В другом варианте A является изохинолином. В другом варианте, C в соединении Формулы I является индолилом. В другом варианте C является 2-индолилом. В другом варианте C является 5-индолилом. В другом варианте, B в соединении Формулы I является тиазолом. В другом варианте, B в соединении Формулы I является тиазолом; Y является CO, и X является связью. Не ограничивающие примеры соединения формулы I выбраны из: (2-(1H-индол-2-ил)тиазол-4-ил)(1H-индол-2-ил)метанона (8) и (2-(1H-индол-2-ил)тиазол-4-ил)(1H-индол-5-ил)метанона (21).
В одном варианте настоящее изобретение относится к соединению формулы (Ia)

(Ia)
где
A является замещенными или незамещенными одно-, конденсированными или многокольцевыми, арильными или (гетеро)циклическими кольцевыми системами; замещенными или незамещенными, насыщенными или ненасыщенными N-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными S-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными O-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными циклическими углеводородами; или замещенными или незамещенными, насыщенными или ненасыщенными смешанными гетероциклами;
B является


R1, R2 и R3 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
R10 и R11 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
X является связью, NH, C1 - C5 углеводородом, O или S;
Y является связью, -C=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN,
-C=N-CN, -CH=CH-, -C=C(CH3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), –(C=O)-O,
-O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
где указанное кольцо А необязательно замещено 1-5 заместителями, которые независимо являются O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN,
-CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом,
-OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 0 до 5;
l является целым числом от 1 до 2;
m является целым числом от 1 до 3;
где
если В является бензольным кольцом, тиофеновым кольцом, фурановым кольцом или индольным кольцом, то Х не является связью или CH2, и А не является индолом;
если В является индолом, то Х не является О;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, если B формулы Ia является тиазольным кольцом, то X не является связью.
В одном варианте настоящее изобретение относится к соединению формулы (II)

II
где
B является


R1, R2, R3, R4, R5 и R6 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом,
-(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
R10 и R11 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
X является связью, NH, C1 - C5 углеводородом, O или S;
Y является связью, -C=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN,
-C=N-CN, -CH=CH-, C=C(CH3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), –(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
i является целым числом от 0 до 5;
l является целым числом от 1 до 2;
n является целым числом от 1 до 3; и
m является целым числом от 1 до 3;
где
если В является индолом, то Х не является О;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, если B формулы II является тиазольным кольцом, то X не является связью.
В одном варианте настоящее изобретение относится к соединению формулы (III)

(III)
где
B является


R4, R5 и R6 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2; и
R10 и R11 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
X является связью, NH, C1 - C5 углеводородом, O или S;
Y является связью, -C=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN,
-C=N-CN, -CH=CH-, C=C(CH3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
i является целым числом от 0 до 5;
l является целым числом от 1 до 2; и
n является целым числом от 1 до 3;
где
если В является индолом, то Х не является О;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, если B формулы III является тиазольным кольцом, то X не является связью.
В одном варианте настоящее изобретение относится к соединению формулы (IV)

(IV)
где кольцо А является индолилом;
B является


R1 и R2 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
R10 и R11 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
X является связью, NH, C1 - C5 углеводородом, O или S;
Y является связью, C=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN,
-C=N-CN, -CH=CH-, C=C(CH3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
где указанный А необязательно замещен O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2; и
i является целым числом от 0 до 5;
l является целым числом от 1 до 2; и
m является целым числом от 1 до 4;
где
если В является бензольным кольцом, тиофеновым кольцом, фурановым кольцом или индольным кольцом, то Х не является связью или CH2;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, если B формулы IV является тиазольным кольцом, то X не является связью.
В другом варианте, индолил кольца А формулы IV присоединен к одному из его 1-7 положений к Х или непосредственно к В, если Х является связью (то есть отсутствует).
В одном варианте настоящее изобретение относится к соединению формулы IV(a)

IV(a)
B является


R1, R2, R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2; и
R10 и R11 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
X является связью, NH, C1 - C5 углеводородом, O или S;
Y является связью или C=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN,
-C=N-CN, -CH=CH-, C=C(CH3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
i является целым числом от 0 до 5;
l является целым числом от 1 до 2;
n является целым числом от 1 до 2; и
m является целым числом от 1 до 4;
где
если В является бензольным кольцом, тиофеновым кольцом, фурановым кольцом или индольным кольцом, то Х не является связью или CH2;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, если B формулы IVa является тиазольным кольцом, то X не является связью.
В одном варианте настоящее изобретение относится к соединению формулы (V)

(V)
B является


R4, R5 и R6 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
R10 и R11 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 1 до 5;
l является целым числом от 1 до 2; и
n является целым числом от 1 до 3;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В другом варианте, B формулы V не является тиазолом . В другом варианте, B формулы V не является оксазолом. В другом варианте, B формулы V не является оксазолином. В другом варианте, B формулы V не является имидазолом. В другом варианте, B формулы V не является тиазолом, оксазолом, оксазолином или имидазолом.
В одном варианте настоящее изобретение относится к следующим соединениям:
Формула V  |
Соединение | B | R4, R5 и R6 | |
| 1a |
 |
H | ||
| 1b |
 |
H | ||
| 1c |
 |
H | ||
| 1d |
|
H | ||
| 1e |
|
 |
H | |
| 1f |
 |
 |
H | |
| 1g |
 |
 |
H | |
| 1h |
 |
H | ||
| 1i |
 |
H | ||
| 1k |
 |
 |
H | |
| 1l |
|
 |
H | |
| 35a |
 |
 |
H | |
| 36a |
 |
 |
H |
В одном варианте настоящее изобретение относится к соединению формулы (VI)

(VI)
где
R4, R5 и R6 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2; и
Y является связью или C=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN,
-C=N-CN, -CH=CH-, C=C(CH3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
n является целым числом от 1 до 3; и
i является целым числом от 1 до 5;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте настоящее изобретение относится к следующим соединениям:
Формула VI  |
Соединение | Y | R4, R5 и R6 |
| 1h | -C=O | H | |
| 2a | -C=C(CH3)2 | H | |
| 2b | -CH-OH | H | |
| 2c | -C=CH-CN (цис и транс) | H | |
| 2d | -C=N-NH2 (цис и транс) |
H | |
| 2e | -C=N-OH (цис и транс) |
H | |
| 2f | -C=N-OMe (цис и транс) | H | |
| 2g | -(C=O)-NH- | H | |
| 2h | -NH-(C=O)- | H | |
| 2i | Отсутствует | H | |
| 2j | -C=N-CN (цис и транс) | H | |
| 2k | C=O | R4=R6=H R5=п-F |
|
| 2l | C=O | R4=R6=H R5=п-OH |
|
| 2m | C=O | R4=R6=H R5=п-CH3 |
|
| 2n | C=O | R4=R6=H R5=п-CH2-CN |
|
| 2o | C=O | R4=R6=H R5=п-N(CH3)2 |
|
| 2p | C=O | R4=м-F; R5=п-F; R6=м-F; n=1 |
|
| 2q | C=O | R4=R6=H R5= п-CH2-(C=O)NH2 |
|
| 2r | C=O | R4=R6=H R5=п-CH2NH2 |
|
| 2s | C=O | R4=R6=H R5=п-CH2NH-CH3 |
|
| 2t | C=O | R4=м-OMe; R5=п-OMe; R6=м-OMe; n=1 |
|
| 2u | C=O | R4=R6=H R5=п-CH2NMe2 |
В одном варианте настоящее изобретение относится к соединению 3a:

В одном варианте настоящее изобретение относится к соединению 3b:

В одном варианте настоящее изобретение относится к соединению формулы (VII)

(VII)
где
Y является связью или C=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN,
-C=N-CN, -CH=CH-, C=C(CH3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте настоящее изобретение относится к следующим соединениям:
| Формула VII | Соединение | Y |
|
|
4a | S |
| 4b | SO2 | |
| 4c | SO | |
| 4d | -( SO2)-NH- |
В одном варианте настоящее изобретение относится к соединению формулы (VIII)
(VIII)
где
R4, R5 и R6 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
Q является S, O или NH;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 3;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте настоящее изобретение относится к следующим соединениям:
| Формула VIII | Соединение | R4 | R5 | R6 | Q |
 |
5a | H n=1 |
H | H | S |
| 5b | H n=1 |
П-CH3 | H | S | |
| 5c | H n=1 |
П-F | H | S | |
| 5d | H n=1 |
П-Cl | H | S | |
| 5e | H n=1 |
H | H | N |
В одном варианте настоящее изобретение относится к соединению формулы (IX)

(IX)
где
R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -(O)NH2 или NO2;
A’ является галогеном; замещенными или незамещенными одно-, конденсированными или многокольцевыми, арильными или (гетеро)циклическими кольцевыми системами; замещенными или незамещенными, насыщенными или ненасыщенными N-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными S-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными O-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными циклическими углеводородами; или замещенными или незамещенными, насыщенными или ненасыщенными смешанными гетероциклами; где указанное кольцо A’ необязательно замещено 1-5 заместителями, которые независимо являются O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 1 до 5; и
n является целым числом от 1 до 3;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте соединение Формулы IX является структурами следующих соединений:
| Формула IX | Соединение | A’ | R4, R5 |
 |
6a |
 |
H |
| 6b |
|
H | |
| 6c |
|
H | |
| 6d | Cl | H |
В другом варианте A' в формуле IX является галогеном. В одном варианте A’ в формуле IX является фенилом. В другом варианте A' в формуле IX является замещенным фенилом. В другом варианте замещением A' является галоген. В другом варианте замещением является 4-F. В другом варианте замещением является 3,4,5-(OCH3)3. В другом варианте, A’ в формуле IX является замещенным или незамещенным 5-индолилом. В другом варианте, A’ в формуле IX является замещенным или незамещенным 2-индолилом. В другом варианте, A' в формуле IX является замещенным или незамещенным 3-индолилом. В другом варианте, соединения формулы IX представлены на Фигуре 16A.
В одном варианте настоящее изобретение относится к соединению формулы (IXa)

(IXa)
где
R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -(O)NH2 или NO2;
A’ является галогеном; замещенными или незамещенными одно-, конденсированными или многокольцевыми, арильными или (гетеро)циклическими кольцевыми системами; замещенными или незамещенными, насыщенными или ненасыщенными N-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными S-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными O-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными циклическими углеводородами; или замещенными или незамещенными, насыщенными или ненасыщенными смешанными гетероциклами; где указанное кольцо A’ необязательно замещено 1-5 заместителями, которые независимо являются O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 1 до 5; и
n является целым числом от 1 до 3;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В другом варианте A' в формуле IXa является галогеном. В одном варианте A’ в формуле IXa является фенилом. В другом варианте A' в формуле IXa является замещенным фенилом. В другом варианте замещением A' является галоген. В другом варианте замещением является 4-F. В другом варианте замещением является 3,4,5-(OCH3)3. В другом варианте, A’ в формуле IXa является замещенным или незамещенным 5-индолилом. В другом варианте, A’ в формуле IXa является замещенным или незамещенным 2-индолилом. В другом варианте, A' в формуле IXa является замещенным или незамещенным 3-индолилом.
В другом варианте, соединение формулы IXa является 1-хлор-7-(4-фторфенил)изохинолином. В другом варианте, соединение формулы IXa является 7-(4-фторфенил)-1-(1Н-индол-5-ил)изохинолином. В другом варианте, соединение формулы IXa является 7-(4-фторфенил)-1-(3,4,5-триметоксифенил)изохинолином. В другом варианте, соединение формулы IXa является 1,7-бис(4-фторфенил)изохинолином (40). В другом варианте, соединение формулы IXa является 1,7-бис(3,4,5-триметоксифенил)изохинолином. В другом варианте, соединение формулы IXa является 1-(4-фторфенил)-7-(3,4,5-триметоксифенил)изохинолином. В другом варианте, соединение формулы IXa является 1-(1Н-индол-5-ил)-7-(3,4,5-триметоксифенил)изохинолином. В другом варианте, соединение формулы IXa является 1-хлор-7-(3,4,5-триметоксифенил)изохинолином.
В одном варианте настоящее изобретение относится к соединению, представленному структурой формулы XI:
где
X является связью, NH или S;
Q является O, NH или S; и
A является замещенными или незамещенными одно-, конденсированными или многокольцевыми, арильными или (гетеро)циклическими кольцевыми системами; замещенными или незамещенными, насыщенными или ненасыщенными N-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными S-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными O-гетероциклами; замещенными или незамещенными, насыщенными или ненасыщенными циклическими углеводородами; или замещенными или незамещенными, насыщенными или ненасыщенными смешанными гетероциклами; где указанное кольцо A необязательно замещено 1-5 1-5 заместителями, которые независимо являются O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом,
-(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2; и
i является целым числом от 0 до 5;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, если Q в формуле XI является S, то X не является связью.
В одном варианте, A соединения Формулы XI является Ph. В другом варианте, A соединения Формулы XI является замещенным Ph. В другом варианте замещением является 4-F. В другом варианте замещением является 4-Me. В другом варианте, Q соединения формулы XI является S. В другом варианте, X соединения формулы XI является NH. Не ограничивающие примеры соединений формулы XI выбраны из: (2-(фениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (5a), (2-(п-толиламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (5b), (2-(п-фторфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (5c), (2-(4-хлорфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (5d), (2-(фениламино)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (5e), (2-(фениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлоридной соли (5Ha), (2-(п-толиламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлоридной соли (5Hb), (2-(п-фторфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлоридной соли (5Hc), (2-(4-хлорфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлоридной соли (5Hd), (2-(фениламино)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлоридной соли (5He).
В одном варианте настоящее изобретение относится к соединению, представленному структурой формулы XI(a):

где R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте настоящее изобретение относится к соединению, представленному структурой формулы XI(b):

где R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте настоящее изобретение относится к соединению, представленному структурой формулы XI(c):
где R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте настоящее изобретение относится к соединению, представленному структурой формулы XI(d):

где R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте настоящее изобретение относится к соединению, представленному структурой формулы XI(e):
где R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В другом варианте соединение формулы XI является структурой соединения 55:

В другом варианте соединение формулы XI является структурой соединения 17ya:

В одном варианте настоящего изобретения является соединение, представленное следующими структурами:
| Соединение | Структура |
| 8 |
 |
| 9 |
 |
| 10 |
 |
| 11 |
 |
| 12 |
 |
| 13 |
 |
| 14 |
 |
| 16 |
 |
| 17 |
 |
| 18 |
 |
| 19 |
 |
| 20 |
 |
| 21 |
 |
| 22 |
 |
| 23 |
 |
| 24 |
 |
| 25 |
 |
| 26 |
 |
| 27 |
 |
| 28 |
 |
| 29 |
 |
| 30 |
 |
| 32 |
 |
| 33 |
 |
| 34 |
 |
| 35 |
 |
| 40 |
 |
| 41 |
 |
| 42 |
 |
| 43 |
  |
| 44 |
 |
| 45 |
 |
| 46 |
 |
| 47 |
 |
| 48 |
 |
| 49 |
 |
| 50 |
 |
| 51 |
 |
| 52 |
 |
| 53 |
 |
| 54 |
 |
Вполне понятно, что в структурах, представленных в настоящем изобретении, где атомы азота имеют менее 3 связей, присутствуют атомы H для завершения валентности азота.
В одном варианте, группы A, A' и/или C формулы I, I(a), IV, IX, IX(a) и XI независимо являются замещенным и незамещенным фуранилом, индолилом, пиридинилом, фенилом, бифенилом, трифенилом, дифенилметаном, адамантан-илом, флуорен-илом и другими гетероциклическими аналогами, такими как, например, пирролил, пиразолил, имидазолил, пиридинил, пиримидинил, пиразинил, пиридазинил, триазинил, тетразинил, пирролизинил, индолил, изохинолинил, хинолинил, изохинолинил, бензимидазолил, индазолил, хинолизинил, циннолинил, хиналолинил, фталазинил, нафтиридинил, хиноксалинил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, диоксанил, фуранил, пирилий, бензофуранил, бензодиоксолил, тиранил, тиетанил, тетрагидротиофен-ил, дитиоланил, тетрагидротиопиранил, тиофен-ил, тиепинил, тианафтенил, оксатиоланил, морфолинил, тиоксанил, тиазолил, изотиазолил, тиадиазолил, оксазолил, изоксазолил, оксадиазиолил).
В одном варианте, группы A, A' и/или C являются замещенным или незамещенным фенилом. В другом варианте, группы A, A' и/или C являются фенилом, замещенным Cl, F или метилом. В одном варианте, группы A, A' и/или C являются замещенным или незамещенным изохинолинилом. В одном варианте, группы A, A' и/или C включают замещенные и незамещенные индолиловые группы; наиболее предпочтительно, замещенный и незамещенный 3-индолил и 5-индолил.
В одном варианте, группы A, A' и/или C в формулах I, I(a), IV, IX, IX(a) и XI могут быть замещенными или незамещенными. Поэтому хотя иллюстративные группы, перечисленные в предыдущем параграфе, являются незамещенными, специалисту в данной области понятно, что эти группы могут быть замещены одним или более, двумя или более, тремя или более, и даже до пяти заместителями (отличными от водорода).
В одном варианте, наиболее предпочтительные группы A, A’ и/или C замещены 3,4,5-триметоксифенилом. В другом варианте группы A, A’ и/или C замещены алкокси-группой. В другом варианте группы A, A’ и/или C замещены метокси-группой. В другом варианте группы A, A’ и/или C замещены алкилом. В другом варианте группы A, A’ и/или C замещены метилом. В другом варианте группы A, A’ и/или C замещены галогеном. В другом варианте группы A, A’ и/или C замещены F. В другом варианте группы A, A’ и/или C замещены Cl. В другом варианте кольца A, A’ и/или C замещены Br.
Заместители групп A, A’ и/или C в формулах I, I(a), IV, IX, IX(a) и XI независимо выбраны из группы, состоящей из водорода (например, отсутствие замещения в конкретном положении), гидроксила, алифатического C1 - C10 углеводорода прямого или разветвленного строения, алкокси, галоалкокси, арилокси, нитро, циано, алкил-CN, галогена (например, F, Cl, Br, I), галоалкила, дигалоалкила, тригалоалкила, COOH, C(O)Ph, C(O)-алкила, C(O)O-алкила, C(O)H, C(O)NH2, -OC(O)CF3, OCH2Ph, амино, аминоалкила, алкиламино, мезиламино, диалкиламино, ариламино, амидо, NHC(O)-алкила, мочевины, алкил-мочевины, алкиламидо (например, ацетамид), галоалкиламидо, ариламидо, арила и C5 - C7 циклоалкила, арилалкила и их комбинаций. Одиночные заместители могут присутствовать в орто-, мета- или пара-положениях. Если присутствует два или более заместителей, то один из них, предпочтительно, хотя и не обязательно, находится в пара-положении.
В одном варианте группа B в формулах I, I(a), II, III, IV, IVa и V выбрана из замещенного или незамещенного тиазола, тиазолидина, оксазола, оксазолина, оксазолидина, бензола, пиримидина, имидазола, пиридина, фурана, тиофена, изоксазола, пиперидина, пиразола, индола и изохинолина, где указанное кольцо В связано через любое из двух положений кольца с X и Y, или непосредственно с кольцами А и/или С.
В одном варианте группа B в формулах I, I(a), II, III, IV, IVa и V является незамещенной. В другом варианте группа B в формулах I, I(a), II, III, IV, IVa и V является:
















В другом варианте группа B в формулах I, I(a), II, III, IV, IVa и V является замещенной. В другом варианте группа B в формулах I, I(a), II, III, IV, IVa и V является:


где R10 и R11 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, галоалкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2.
В другом варианте группа B является (тиазол). В другом варианте группа B является (тиазол). В другом варианте группа B является  (тиазолидин). В другом варианте группа B является (оксазол). В другом варианте группа B является (оксазолин). В другом варианте группа B является
(тиазолидин). В другом варианте группа B является (оксазол). В другом варианте группа B является (оксазолин). В другом варианте группа B является  (оксазолидин). В другом варианте группа B является (бензол). В другом варианте группа B является
(оксазолидин). В другом варианте группа B является (бензол). В другом варианте группа B является  (бензол). В другом варианте группа B является (пиримидин). В другом варианте группа B является (имидазол). В другом варианте группа B является (пиридин). В другом варианте группа B является
(бензол). В другом варианте группа B является (пиримидин). В другом варианте группа B является (имидазол). В другом варианте группа B является (пиридин). В другом варианте группа B является  (фуран). В другом варианте группа B является (тиофен). В другом варианте группа B является (изоксазол). В другом варианте группа B является (пиперидин). В другом варианте группа B является (пиперидин). В другом варианте группа B является (пиразол). В другом варианте группа B является (индол). В другом варианте группа B является (изохинолин).
(фуран). В другом варианте группа B является (тиофен). В другом варианте группа B является (изоксазол). В другом варианте группа B является (пиперидин). В другом варианте группа B является (пиперидин). В другом варианте группа B является (пиразол). В другом варианте группа B является (индол). В другом варианте группа B является (изохинолин).
В одном варианте группа B в формулах I, I(a), II, III, IV, IVa и V замещена R10 и R11. В другом варианте оба R10 и R11 являются водородами. В другом варианте R10 и R11 независимо являются O-алкилом. В другом варианте R10 и R11 независимо являются O-галоалкилом. В другом варианте R10 и R11 независимо являются F. В другом варианте R10 и R11 независимо являются Cl. В другом варианте R10 и R11 независимо являются Br. В другом варианте R10 и R11 независимо являются I. В другом варианте R10 и R11 независимо являются галоалкилом. В другом варианте R10 и R11 независимо являются CF3. В другом варианте R10 и R11 независимо являются CN. В другом варианте R10 и R11 независимо являются -CH2CN. В другом варианте R10 и R11 независимо являются NH2. В другом варианте R10 и R11 независимо являются гидроксилом. В другом варианте R10 и R11 независимо являются -(CH2)iNHCH3. В другом варианте R10 и R11 независимо являются
-(CH2)iNH2. В другом варианте R10 и R11 независимо являются -(CH2)iN(CH3)2. В другом варианте R10 и R11 независимо являются -OC(O)CF3. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным алкилом. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным галоалкилом. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным алкиламино. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным аминоалкилом. В другом варианте R10 и R11 независимо являются -OCH2Ph. В другом варианте R10 и R11 независимо являются -NHCO-алкилом. В другом варианте R10 и R11 независимо являются COOH. В другом варианте R10 и R11 независимо являются -C(O)Ph. В другом варианте R10 и R11 независимо являются C(O)O-алкилом. В другом варианте R10 и R11 независимо являются C(O)H. В другом варианте R10 и R11 независимо являются
-C(O)NH2. В другом варианте R10 и R11 независимо являются NO2.
В другом варианте группа B в формулах I, I(a), II, III, IV, IVa и V является (тиазол), где R10 и R11 независимо являются H, и l равен 1. В другом варианте, R10 и R11 независимо являются O-алкилом. В другом варианте R10 и R11 независимо являются O-галоалкилом. В другом варианте R10 и R11 независимо являются F. В другом варианте R10 и R11 независимо являются Cl. В другом варианте R10 и R11 независимо являются Br. В другом варианте R10 и R11 независимо являются I. В другом варианте R10 и R11 независимо являются галоалкилом. В другом варианте R10 и R11 независимо являются CF3. В другом варианте R10 и R11 независимо являются CN. В другом варианте R10 и R11 независимо являются -CH2CN. В другом варианте R10 и R11 независимо являются NH2. В другом варианте R10 и R11 независимо являются гидроксилом. В другом варианте R10 и R11 независимо являются -(CH2)iNHCH3. В другом варианте R10 и R11 независимо являются
-(CH2)iNH2. В другом варианте R10 и R11 независимо являются -(CH2)iN(CH3)2. В другом варианте R10 и R11 независимо являются -OC(O)CF3. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным алкилом. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным галоалкилом. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным алкиламино. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным аминоалкилом. В другом варианте R10 и R11 независимо являются -OCH2Ph. В другом варианте R10 и R11 независимо являются -NHCO-алкилом. В другом варианте R10 и R11 независимо являются COOH. В другом варианте R10 и R11 независимо являются -C(O)Ph. В другом варианте R10 и R11 независимо являются C(O)O-алкилом. В другом варианте R10 и R11 независимо являются C(O)H. В другом варианте R10 и R11 независимо являются
-C(O)NH2. В другом варианте R10 и R11 независимо являются NO2.
В другом варианте группа B в формулах I, I(a), II, III, IV, IVa и V является (имидазол), где R10 и R11 независимо являются H, и l равен 1. В другом варианте, R10 и R11 независимо являются O-алкилом. В другом варианте R10 и R11 независимо являются O-галоалкилом. В другом варианте R10 и R11 независимо являются F. В другом варианте R10 и R11 независимо являются Cl. В другом варианте R10 и R11 независимо являются Br. В другом варианте R10 и R11 независимо являются I. В другом варианте R10 и R11 независимо являются галоалкилом. В другом варианте R10 и R11 независимо являются CF3. В другом варианте R10 и R11 независимо являются CN. В другом варианте R10 и R11 независимо являются -CH2CN. В другом варианте R10 и R11 независимо являются NH2. В другом варианте R10 и R11 независимо являются гидроксилом. В другом варианте R10 и R11 независимо являются -(CH2)iNHCH3. В другом варианте R10 и R11 независимо являются -(CH2)iNH2. В другом варианте R10 и R11 независимо являются
-(CH2)iN(CH3)2. В другом варианте R10 и R11 независимо являются -OC(O)CF3. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным алкилом. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным галоалкилом. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным алкиламино. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным аминоалкилом. В другом варианте R10 и R11 независимо являются -OCH2Ph. В другом варианте R10 и R11 независимо являются -NHCO-алкилом. В другом варианте R10 и R11 независимо являются COOH. В другом варианте R10 и R11 независимо являются -C(O)Ph. В другом варианте R10 и R11 независимо являются C(O)O-алкилом. В другом варианте R10 и R11 независимо являются C(O)H. В другом варианте R10 и R11 независимо являются -C(O)NH2. В другом варианте R10 и R11 независимо являются NO2.
В другом варианте группа B в формулах I, I(a), II, III, IV, IVa и V является (изохинолин), где R10 и R11 независимо являются H, и l равен 1. В другом варианте, R10 и R11 независимо являются O-алкилом. В другом варианте R10 и R11 независимо являются O-галоалкилом. В другом варианте R10 и R11 независимо являются F. В другом варианте R10 и R11 независимо являются Cl. В другом варианте R10 и R11 независимо являются Br. В другом варианте R10 и R11 независимо являются I. В другом варианте R10 и R11 независимо являются галоалкилом. В другом варианте R10 и R11 независимо являются CF3. В другом варианте R10 и R11 независимо являются CN. В другом варианте R10 и R11 независимо являются -CH2CN. В другом варианте R10 и R11 независимо являются NH2. В другом варианте R10 и R11 независимо являются гидроксилом. В другом варианте R10 и R11 независимо являются -(CH2)iNHCH3. В другом варианте R10 и R11 независимо являются -(CH2)iNH2. В другом варианте R10 и R11 независимо являются
-(CH2)iN(CH3)2. В другом варианте R10 и R11 независимо являются -OC(O)CF3. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным алкилом. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным галоалкилом. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным алкиламино. В другом варианте R10 и R11 независимо являются C1-C5 линейным или разветвленным аминоалкилом. В другом варианте R10 и R11 независимо являются -OCH2Ph. В другом варианте R10 и R11 независимо являются -NHCO-алкилом. В другом варианте R10 и R11 независимо являются COOH. В другом варианте R10 и R11 независимо являются -C(O)Ph. В другом варианте R10 и R11 независимо являются C(O)O-алкилом. В другом варианте R10 и R11 независимо являются C(O)H. В другом варианте R10 и R11 независимо являются -C(O)NH2. В другом варианте R10 и R11 независимо являются NO2.
В одном варианте мостик X в формулах I, Ia, II, III, IV, IVa и XI является связью. В другом варианте, мостик X является NH. В другом варианте, мостик X является C1 - C5 углеводородом. В другом варианте, мостик X является CH2. В другом варианте, мостик X является –CH2-CH2-. В другом варианте, мостик X является O. В другом варианте, мостик X является S.
В одном варианте, мостик Y в формулах I, Ia, II, III, IV, IVa, VI и VII является C=O. В другом варианте, мостик Y является C=S. В другом варианте мостик Y является C=N(NH2)-. В другом варианте, мостик Y является -C=NOH. В другом варианте, мостик Y является –CH-OH. В другом варианте, мостик Y является -C=CH-(CN). В другом варианте, мостик Y является -C=N(CN). В другом варианте, мостик Y является -C=C(CH3)2. В другом варианте, мостик Y является -C=N-OMe. В другом варианте, мостик Y является
-(C=O)NH-. В другом варианте, мостик Y является -NH(C=O)-. В другом варианте, мостик Y является –(C=O)-O. В другом варианте, мостик Y является –O-(C=O). В другом варианте, мостик Y является –(CH2)1-5-(C=O). В другом варианте, мостик Y является
-(C=O)-(CH2)1-5. В другом варианте, мостик Y является S. В другом варианте, мостик Y является SO. В другом варианте, мостик Y является SO2. В другом варианте, мостик Y является -CH=CH-. В другом варианте, мостик Y является –(SO2)-NH-. В другом варианте, мостик Y является –NH-(SO2)-.
В одном варианте, R1, R2, R3, R4, R5 и R6 в формулах Ia, II, III, IV, IV(a), V, VI, VIII, IX, IX(a), XI(a), XI(b), XI(c), XI(d) и XI(e) независимо являются водородом. В другом варианте R1, R2, R3, R4, R5 и R6 независимо являются O-алкилом. В другом варианте R1, R2, R3, R4, R5 и R6 независимо являются O-галоалкилом. В другом варианте R1, R2, R3, R4, R5 и R6 независимо являются F. В другом варианте R1, R2, R3, R4, R5 и R6 независимо являются Cl. В другом варианте R1, R2, R3, R4, R5 и R6 независимо являются Br. В другом варианте R1, R2, R3, R4, R5 и R6 независимо являются I. В другом варианте R1, R2, R3, R4, R5 и R6 независимо являются галоалкилом. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются CF3. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются CN. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются -CH2CN. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются NH2. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются гидроксилом. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются -(CH2)iNHCH3. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются
-(CH2)iNH2. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются -(CH2)iN(CH3)2. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются -OC(O)CF3. В другом варианте R1, R2, R3, R4, R5 и R6 независимо являются C1-C5 линейным или разветвленным алкилом. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются галоалкилом. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются алкиламино. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются аминоалкилом. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются -OCH2Ph. В другом варианте R1, R2, R3, R4, R5 и R6 независимо являются -NHCO-алкилом. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются COOH. В другом варианте R1, R2, R3, R4, R5 и R6 независимо являются -C(O)Ph. В другом варианте R1, R2, R3, R4, R5 и R6 независимо являются C(O)O-алкилом. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются C(O)H. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются -C(O)NH2. В другом варианте, R1, R2, R3, R4, R5 и R6 независимо являются NO2.
В одном варианте настоящее изобретение относится к соединению формулы XII:

(XII)
где
P и Q независимо являются H или

W является C=O, C=S, SO2 или S=O;
где, по меньшей мере, один из Q и P не является водородом;
R1 и R4 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, -(CH2)iNHCH3, -(CH2)iNH2,
-(CH2)iN(CH3)2; C(O)O-алкилом или C(O)H; где, по меньшей мере, один из R1 и R4 не является водородом;
R2 и R5 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
m является целым числом от 1 до 4;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте настоящее изобретение относится к соединению формулы XIII:

(XIII)
где
Z является O или S;
R1 и R4 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, галоалкилом, аминоалкилом,
-(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2; COOH, C(O)O-алкилом или C(O)H; где, по меньшей мере, один из R1 и R4 не является водородом;
R2 и R5 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2; OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
m является целым числом от 1 до 4;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте настоящее изобретение относится к соединению формулы XIV:

(XIV)
где R1 и R4 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H; где, по меньшей мере, один из R1 и R4 не является водородом;
R2 и R5 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
m является целым числом от 1 до 4;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, R1 соединения формулы XII, XIII и XIV является OCH3. В другом варианте, R1 соединения формулы XII, XIII и XIV является 4-F. В другом варианте, R1 соединения формулы XII, XIII и XIV является OCH3, и m равен 3. В другом варианте, R4 соединения формулы XII, XIII и XIV является 4-F. В другом варианте, R4 соединения формулы XII, XIII и XIV является OCH3. В другом варианте, R4 соединения формулы XIV является CH3. В другом варианте, R4 соединения формулы XII, XIII и XIV является 4-Cl. В другом варианте, R4 соединения формулы XII, XIII и XIV является 4-N(Me)2. В другом варианте, R4 соединения формулы XII, XIII и XIV является OBn. В другом варианте, R4 соединения формулы XII, XIII и XIV является 4-Br. В другом варианте, R4 соединения формулы XII, XIII и XIV является 4-CF3. Не ограничивающие примеры соединений формулы XIV выбраны из: (2-фенил-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12aa), (4-фторфенил)(2-фенил-1H-имидазол-4-ил)метанона (12af), (2-(4-фторфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ba), (2-(4-метоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ca), (4-фторфенил)(2-(4-метоксифенил)-1H-имидазол-4-ил)метанона (12cb), (2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12da), (4-фторфенил)(2-(п-толил)-1H-имидазол-4-ил)метанона (12db), (4-гидрокси-3,5-диметоксифенил)(2-(п-толил)-1H-имидазол-4-ил)метанона (12dc), (2-(4-хлорфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12fa), (2-(4-хлорфенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12fb), (2-(4-хлорфенил)-1H-имидазол-4-ил)(4-гидрокси-3,5-диметоксифенил)метанона (12fc), (2-(4-(диметиламино)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ga); (2-(4-(диметиламино)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12gb), (2-(3,4-диметоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ha), (2-(4-(бензилокси)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12jb), (2-(4-бромфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12la), (2-(4-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12pa).
В одном варианте настоящее изобретение относится к соединению формулы XIVa:

(XIVa)
где R1 и R4 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H; где, по меньшей мере, один из R1 и R4 не является водородом;
R2 и R5 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
R9 является H, линейным или разветвленным, замещенным или незамещенным алкилом, замещенным или незамещенным арилом, CH2Ph, замещенным бензилом, галоалкилом, аминоалкилом, OCH2Ph, замещенным или незамещенным SO2-арилом, замещенным или незамещенным -(C=O)-арилом или OH;
где замещения независимо выбраны из группы, состоящей из водорода (например, отсутствие замещения в конкретном положении), гидроксила, алифатического C1 - C10 углеводорода прямого или разветвленного строения, алкокси, галоалкокси, арилокси, нитро, циано, алкил-CN, галогена (например, F, Cl, Br, I), галоалкила, дигалоалкила, тригалоалкила, COOH, C(O)Ph, C(O)-алкила, C(O)O-алкила, C(O)H, C(O)NH2, -OC(O)CF3, OCH2Ph, амино, аминоалкила, алкиламино, мезиламино, диалкиламино, ариламино, амидо, NHC(O)-алкила, мочевины, алкил-мочевины, алкиламидо (например, ацетамид), галоалкиламидо, ариламидо, арила и C5 - C7 циклоалкила, арилалкила и их комбинаций;
m является целым числом от 1 до 4;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, R9 соединения формулы XIVa является CH3. В другом варианте, R9 соединения формулы XIVa является CH2Ph. В другом варианте, R9 соединения формулы XIVa является (SO2)Ph. В другом варианте, R9 соединения формулы XIVa является (SO2)-Ph-OCH3. В другом варианте, R9 соединения формулы XIVa является H. В другом варианте, R4 соединения формулы XIVa является H. В другом варианте, R4 соединения формулы XIVa является CH3. В другом варианте, R4 соединения формулы XIVa является OCH3. В другом варианте, R4 соединения формулы XIVa является OH. В другом варианте, R4 соединения формулы XIVa является 4-Cl. В другом варианте, R4 соединения формулы XIVa является 4-N(Me)2. В другом варианте, R4 соединения формулы XIVa является OBn. В другом варианте, R1 соединения формулы XIVa является OCH3; m равен 3, и R2 является H. В другом варианте, R1 соединения формулы XIVa является F; m равен 1, и R2 является H. Не ограничивающие примеры соединений формулы XIVa выбраны из: (4-фторфенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11af), (4-фторфенил)(2-(4-метоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11cb), (4-фторфенил)(1-(фенилсульфонил)-2-(п-толил)-1H-имидазол-4-ил)метанона (11db), (2-(4-хлорфенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11fb), (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ga), (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11gb), (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ha), (2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11jb), (2-(4-(диметиламино)фенил)-1-((4-метоксифенил)сульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12gba), (1-бензил-2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12daa), (1-метил-2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12dab), (4-фторфенил)(2-(4-метоксифенил)-1-метил-1H-имидазол-4-ил)метанона (12cba).
В одном варианте настоящее изобретение относится к соединению формулы XV:

(XV)
где R4 и R5 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, R4 соединения формулы XV является H. В другом варианте, R4 соединения формулы XV является F. В другом варианте, R4 соединения формулы XV является Cl. В другом варианте, R4 соединения формулы XV является Br. В другом варианте, R4 соединения формулы XV является I. В другом варианте, R4 соединения формулы XV является N(Me)2. В другом варианте, R4 соединения формулы XV является OBn. В другом варианте, R4 соединения формулы XV является OCH3. В другом варианте, R4 соединения формулы XV является CH3. В другом варианте, R4 соединения формулы XV является CF3. Не ограничивающие примеры соединений формулы XV выбраны из: (2-фенил-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12aa), (2-(4-фторфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ba), (2-(4-метоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ca), (2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12da), (3,4,5-триметоксифенил)(2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)метанона (12ea), (2-(4-хлорфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12fa), (2-(4-(диметиламино)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ga), (2-(3,4-диметоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ha), (2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ia), (2-(4-(бензилокси)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ja), (2-(4-гидроксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ka), (2-(4-бромфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12la), (2-(4-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12pa).
В одном варианте настоящее изобретение относится к соединению формулы XVI:

(XVI)
где R4 и R5 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
R3 является I, Br, Cl или F;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, R3 соединения формулы XVI является галогеном. В другом варианте, R3 является F. В другом варианте, R3 является Cl. В другом варианте, R3 является Br. В другом варианте, R3 является I. В другом варианте, R4 является H. В другом варианте, R4 является OCH3. В другом варианте, R4 является OCH3; n равен 3, и R5 является H. В другом варианте, R4 является CH3. В другом варианте, R4 является F. В другом варианте, R4 является Cl. В другом варианте, R4 является Br. В другом варианте, R4 является I. В другом варианте, R4 является N(Me)2. В другом варианте, R4 является OBn. В другом варианте, R3 является F; R5 является водородом; n равен 1, и R4 является 4-Cl. В другом варианте, R3 является F; R5 является водородом; n равен 1, и R4 является 4-OCH3. В другом варианте, R3 является F; R5 является водородом; n равен 1, и R4 является 4-CH3. В другом варианте, R3 является F; R5 является водородом; n равен 1, и R4 является 4-N(Me)2. В другом варианте, R3 является F; R5 является водородом; n равен 1, и R4 является 4-OBn. Не ограничивающие примеры соединений формулы XVI выбраны из: (4-фторфенил)(2-фенил-1H-имидазол-4-ил)метанона (12af), (4-фторфенил)(2-(4-метоксифенил)-1H-имидазол-4-ил)метанона (12cb), (4-фторфенил)(2-(п-толил)-1H-имидазол-4-ил)метанона (12db), 4-фторфенил)(2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)метанона (12eb), (2-(4-хлорфенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12fb), (2-(4-(диметиламино)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12gb), (2-(4-(бензилокси)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12jb).
В одном варианте настоящее изобретение относится к соединению формулы XVII:

(XVII)
где R4 является H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
где R1 и R2 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
и
m является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, R4 соединения формулы XVII является галогеном. В другом варианте, R4 является F. В другом варианте, R4 является Cl. В другом варианте, R4 является Br. В другом варианте, R4 является I. В другом варианте, R4 является OCH3. В другом варианте R4 является CH3. В другом варианте, R4 является N(Me)2. В другом варианте, R4 является CF3. В другом варианте R4 является ОН. В другом варианте R4 является OBn. В другом варианте, R1 соединения формулы XVII является галогеном. В другом варианте, R1 соединения формулы XVII является F. В другом варианте, R1 соединения формулы XVII является Cl. В другом варианте, R1 соединения формулы XVII является Br. В другом варианте, R1 соединения формулы XVII является I. В другом варианте, R1 соединения формулы XVII является OCH3. В другом варианте, R1 соединения формулы XVII является OCH3, m равен 3, и R2 является H. В другом варианте, R1 соединения формулы XVII является F, m равен 1, и R2 является H. В другом варианте, R4 является F; R2 является водородом; n равен 3, и R1 является OCH3. В другом варианте, R4 является OCH3; R2 является водородом; n равен 3, и R1 является OCH3. В другом варианте, R4 является CH3; R2 является водородом; n равен 3, и R1 является OCH3. В другом варианте, R4 является Cl; R2 является водородом; n равен 3, и R1 является OCH3. В другом варианте, R4 является N(Me)2; R2 является водородом; n равен 3, и R1 является OCH3. В одном варианте, R4 соединения формулы XVII является галогеном; R1 является водородом, и R2 является галогеном. В одном варианте, R4 соединения формулы XVII является галогеном, R1 является галогеном, и R2 является H. В одном варианте, R4 соединения формулы XVII является алкокси, R1 является галогеном, и R2 является H. В одном варианте, R4 соединения формулы XVII является метокси, R1 является галогеном, и R2 является H. Не ограничивающие примеры соединений формулы XVII выбраны из: (2-(4-фторфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ba), (2-(4-метоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ca), (4-фторфенил)(2-(4-метоксифенил)-1H-имидазол-4-ил)метанона (12cb), (2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12da), (4-фторфенил)(2-(п-толил)-1H-имидазол-4-ил)метанона (12db), (4-гидрокси-3,5-диметоксифенил)(2-(п-толил)-1H-имидазол-4-ил)метанона (12dc), (2-(4-хлорфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12fa), (2-(4-хлорфенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12fb), (2-(4-хлорфенил)-1H-имидазол-4-ил)(3,4,5-тригидроксифенил)метанона (13fa), (2-(4-(диметиламино)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ga), (2-(4-(диметиламино)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12gb), (2-(4-(бензилокси)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12jb), (2-(4-гидроксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ka), (2-(4-бромфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12la), (2-(4-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12pa).
В другом варианте соединение формулы XVII является структурой формулы 12fb:

В другом варианте соединение формулы XVII представлено структурой формулы 12cb:

В одном варианте настоящее изобретение относится к соединенияю формулы XVIII:

(XVIII)
где
W является C=O, C=S, SO2 или S=O;
R4 и R7 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
R5 и R8 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
n является целым числом от 1 до 4;
i является целым числом от 0 до 5; и
q является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, W соединения формулы XVIII является C=O. В другом варианте, W соединения формулы XVIII является SO2. В другом варианте, R4 соединения формулы XVIII является H. В другом варианте, R4 соединения формулы XVIII является NO2. В другом варианте, R4 соединения формулы XVIII является OBn. В другом варианте, R7 соединения формулы XVIII является H. В другом варианте, R7 соединения формулы XVIII является OCH3. В другом варианте, R7 соединения формулы XVIII является OCH3, и q равен 3. Не ограничивающие примеры соединений формулы XVII выбраны из: (4-метоксифенил)(2-фенил-1H-имидазол-1-ил)метанона (12aba), (2-фенил-1H-имидазол-1-ил)(3,4,5-триметоксифенил)метанона (12aaa), 2-фенил-1-(фенилсульфонил)-1H-имидазола (10a), 2-(4-нитрофенил)-1-(фенилсульфонил)-1H-имидазола (10x), 2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1H-имидазола (10j).
В одном варианте настоящее изобретение относится к соединению формулы XIX:

(XIX)
где
W является C=O, C=S, SO2, S=O;
R1, R4 и R7 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
R2, R5 и R8 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
m является целым числом от 1 до 4;
n является целым числом от 1 до 4;
i является целым числом от 0 до 5; и
q равен 1-4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, R1, R4 и R7 в формуле XIX независимо являются H. В другом варианте, R1, R4 и R7 в формуле XIX независимо являются O-алкилом. В другом варианте, R1, R4 и R7 в формуле XIX независимо являются галогеном. В другом варианте, R1, R4 и R7 в формуле XIX независимо являются CN. В другом варианте, R1, R4 и R7 в формуле XIX независимо являются OH. В другом варианте, R1, R4 и R7 в формуле XIX независимо являются алкилом. В другом варианте, R1, R4 и R7 в формуле XIX независимо являются OCH2Ph. В одном варианте, R2, R5 и R8 в формуле XIX независимо являются H. В другом варианте, R2, R5 и R8 в формуле XIX независимо являются O-алкилом. В другом варианте, R2, R5 и R8 в формуле XIX независимо являются галогеном. В другом варианте, R2, R5 и R8 в формуле XIX независимо являются CN. В другом варианте, R2, R5 и R8 в формуле XIX независимо являются OH. В другом варианте, R2, R5 и R8 в формуле XIX независимо являются алкилом. В другом варианте, R2, R5 и R8 в формуле XIX независимо являются OCH2Ph. В другом варианте, R5, R2 и R8 в формуле XIX являются H, R4 является 4-N(Me)2, R1 является OCH3, m равен 3, и R7 является OCH3. В другом варианте, R5, R2, R7 и R8 в формуле XIX являются H, R4 является 4-Br, R1 является OCH3, и m равен 3. В другом варианте W является SO2. В другом варианте W является C=O. В другом варианте W является C=S. В другом варианте W является S=O. Не ограничивающие примеры соединений формулы XIX выбраны из: (2-(4-(диметиламино)фенил)-1-((4-метоксифенил)сульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11gaa); (2-(4-бромфенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11la), (4-фторфенил)(2-(4-метоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11cb), (2-(4-хлорфенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11fb), (4-фторфенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11af), (4-фторфенил)(1-(фенилсульфонил)-2-(п-толил)-1H-имидазол-4-ил)метанона (11db), (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ga), (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11gb), (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ha), (2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11jb), (2-(4-(диметиламино)фенил)-1-((4-метоксифенил)сульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12gba).
В другом варианте соединение формулы XIX представлено структурой формулы 11cb:

В другом варианте соединение формулы XIX представлено структурой формулы 11fb:

В одном варианте настоящее изобретение относится к соединению формулы XX:

(XX)
где
R4 является H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом,
-(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H; и
i является целым числом от 0 до 5;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, R4 соединения формулы XX является H. В другом варианте, R4 соединения формулы XX является галогеном. В другом варианте, R4 является F. В другом варианте, R4 является Cl. В другом варианте, R4 является Br. В другом варианте, R4 является I. В другом варианте, R4 является алкилом. В другом варианте R4 является метилом. Не ограничивающие примеры соединений формулы XX выбраны из: (2-фенил-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12aa), (2-(4-фторфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ba), (2-(4-метоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ca), (2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12da), (2-(4-хлорфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12fa), (2-(4-(диметиламино)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ga), (2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ia), (2-(4-(бензилокси)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ja), (2-(4-гидроксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ka), (2-(4-бромфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12la), (2-(4-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12pa).
В другом варианте соединение формулы XX представлено структурой формулы 12da:

В другом варианте соединение формулы XX представлено структурой формулы 12fa:

В одном варианте настоящее изобретение относится к соединению формулы XXI:

(XXI)
где
A является индолилом;
Q является NH, O или S;
R1 и R2 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H; и
где указанный А необязательно замещен замещенным или незамещенным O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом,
-(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -OC(O)CF3, замещенным или незамещенным
-SO2-арилом, замещенным или незамещенным C1-C5 линейным или разветвленным алкилом, замещенным или незамещенным галоалкилом, замещенным или незамещенным алкиламино, замещенным или незамещенным аминоалкилом, -OCH2Ph, замещенным или незамещенным -NHCO-алкилом, COOH, замещенным или незамещенным -C(O)Ph, замещенным или незамещенным C(O)O-алкилом, C(O)H, -C(O)NH2, NO2 или их комбинацией;
i является целым числом от 0 до 5; и
m является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, R1 соединения формулы XXI является OCH3; m равен 3, и R2 является водородом. В другом варианте, R1 является F; m равен 1, и R2 является водородом. В одном варианте, Q в формуле XXI является O. В другом варианте Q в формуле XXI является NH. В другом варианте Q в формуле XXI является S.
В одном варианте, кольцо A соединения формулы XXI является замещенным 5-индолилом. В другом варианте замещением является -(C=O)-арил. В другом варианте, арилом является 3,4,5-(OCH3)3-Ph.
В другом варианте, кольцо A соединения формулы XXI является 3-индолилом. В другом варианте, кольцо A соединения формулы XXI является 5-индолилом. В другом варианте, кольцо A соединения формулы XXI является 2-индолилом. Не ограничивающие примеры соединений формулы XXI выбраны из: (5-(4-(3,4,5-триметоксибензоил)-1H-имидазол-2-ил)-1H-индол-2-ил)(3,4,5-триметоксифенил)метанона (15xaa); (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-2-(3,4,5-триметоксибензоил)-1H-индол-5-ил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (16xaa); 2-(1H-индол-3-ил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17ya); (2-(1H-индол-2-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (62a); и (2-(1H-индол-5-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (66a).
В одном варианте настоящее изобретение относится к соединению формулы XXIa:

(XXIa)
где
W является C=O, C=S, SO2, S=O;
A является индолилом;
R1 и R2 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
R7 и R8 независимо являются H, O-алкилом, I, Br, Cl, F, алкилом, галоалкилом, аминоалкилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкилом, COOH, C(O)O-алкилом или C(O)H;
где указанный А необязательно замещен замещенным или незамещенным O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом,
-(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -OC(O)CF3, замещенным или незамещенным
-SO2-арилом, замещенным или незамещенным C1-C5 линейным или разветвленным алкилом, замещенным или незамещенным галоалкилом, замещенным или незамещенным алкиламино, замещенным или незамещенным аминоалкилом, -OCH2Ph, замещенным или незамещенным -NHCO-алкилом, COOH, замещенным или незамещенным -C(O)Ph, замещенным или незамещенным C(O)O-алкилом, C(O)H, -C(O)NH2, NO2 или их комбинацией;
i является целым числом от 0 до 5; и
m является целым числом от 1 до 4;
q является целым числом от 1 до 4;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, R1 соединения формулы XXIa является OCH3; m равен 3, и R2 является водородом. В другом варианте, R1 является F; m равен 1, и R2 является водородом. В другом варианте, кольцо A соединения формулы XXIa является замещенным 5-индолилом. В другом варианте, кольцо A соединения формулы XXIa является 3-индолилом. Не ограничивающие примеры соединений формулы XXIa выбраны из: (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-2-(3,4,5-триметоксибензоил)-1H-индол-5-ил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (16xaa); (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-1H-индол-3-ил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17yaa).
В одном варианте настоящее изобретение относится к соединению формулы XXII:

(XXII)
где
A является индолилом;
где указанный А необязательно замещен замещенным или незамещенным O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CF3, CN, -CH2CN, NH2, гидроксилом,
-(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -OC(O)CF3, замещенным или незамещенным
-SO2-арилом, замещенным или незамещенным C1-C5 линейным или разветвленным алкилом, замещенным или незамещенным галоалкилом, замещенным или незамещенным алкиламино, замещенным или незамещенным аминоалкилом, -OCH2Ph, замещенным или незамещенным -NHCO-алкилом, COOH, замещенным или незамещенным -C(O)Ph, замещенным или незамещенным C(O)O-алкилом, C(O)H, -C(O)NH2, NO2 или их комбинацией;
i является целым числом от 0 до 5;
или его фармацевтически приемлемой соли, гидрату, полиморфу, метаболиту, таутомеру или изомеру.
В одном варианте, кольцо A соединения формулы XXII является замещенным 5-индолилом. В другом варианте замещением является –(C=O)-арил. В другом варианте, арилом является 3,4,5-(OCH3)3-Ph.
В другом варианте, кольцо A соединения формулы XXII является 3-индолилом. Не ограничивающие примеры соединений формулы XXII выбраны из: (5-(4-(3,4,5-триметоксибензоил)-1H-имидазол-2-ил)-1H-индол-2-ил)(3,4,5-триметоксифенил)метанона (15xaa); (2-(1H-индол-3-ил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17ya).
В другом варианте соединение формулы XXI или XXII представлено структурой формулы 17ya:

В одном варианте, Q соединения формулы XII является H, и P является . В другом варианте, P соединения формулы XII является H, и Q является. В другом варианте, P соединения формулы XII является , и Q является SO2-Ph. В одном варианте Q соединения формулы XII является H, и P является
. В другом варианте, P соединения формулы XII является H, и Q является. В другом варианте, P соединения формулы XII является , и Q является SO2-Ph. В одном варианте Q соединения формулы XII является H, и P является  , где W является C=O. В другом варианте, W соединения формулы XII, XVIII, XIX или XXIa является C=O. В другом варианте, W соединения формулы XII, XVIII, XIX или XXIa является SO2. В другом варианте, W соединения формулы XII, XVIII, XIX или XXIa является C=S. В другом варианте, W соединения формулы XII, XVIII, XIX или XXIa является S=O.
, где W является C=O. В другом варианте, W соединения формулы XII, XVIII, XIX или XXIa является C=O. В другом варианте, W соединения формулы XII, XVIII, XIX или XXIa является SO2. В другом варианте, W соединения формулы XII, XVIII, XIX или XXIa является C=S. В другом варианте, W соединения формулы XII, XVIII, XIX или XXIa является S=O.
В одном варианте, Z соединения формулы XIII является кислородом. В другом варианте, Z соединения формулы XIII является серой.
В одном варианте, R5 соединения формулы XII-XVI, XVIII или XIX является водородом, n равен 1, и R4 находится в пара-положении.
В одном варианте, R4 соединения формулы XII-XX является алкилом. В другом варианте, R4 соединения формулы XII-XX является H. В другом варианте, R4 соединения формулы XII-XX является метилом (CH3). В другом варианте, R4 соединения формулы XII-XX является O-алкилом. В другом варианте, R4 соединения формулы XII-XX является OCH3. В другом варианте, R4 соединения формулы XII-XX является I. В другом варианте, R4 соединения формулы XII-XX является Br. In В другом варианте, R4 соединения формулы XII-XX является F. В другом варианте, R4 соединения формулы XII-XX является Cl. В другом варианте, R4 соединения формулы XII-XX является N(Me)2. В другом варианте, R4 соединения формулы XII-XX является OBn. В другом варианте, R4 соединения формулы XII-XX является OH. В другом варианте, R4 соединения формулы XII-XX является CF3.
В одном варианте, R2 соединения формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa является водородом; R1 является OCH3, и m равен 3. В другом варианте, R2 соединения формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa является водородом; m равен 1, и R1 находится в пара-положении. В другом варианте, R2 соединения формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa является водородом; m равен 1, и R1 является I. В другом варианте, R2 соединения формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa является водородом; m равен 1, и R1 является Br. В другом варианте, R2 соединения формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa является водородом; m равен 1, и R1 является F. В другом варианте, R2 соединения формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa является водородом; m равен 1, и R1 является Cl. В другом варианте, R1 соединения формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa является I. В другом варианте, R1 соединения формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa является Br. В другом варианте, R1 соединения формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa является Cl. В другом варианте, R1 соединения формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa является F.
В одном варианте Q соединения формулы XII является H, и P является , где W является C=O. Не ограничивающие примеры соединений формулы XII-XVII и XX-XXII выбраны из (2-фенил-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12aa); (4-метоксифенил)(2-фенил-1H-имидазол-4-ил)метанона (12ab); (3-метоксифенил)(2-фенил-1H-имидазол-4-ил)метанона (12ac); (3,5-диметоксифенил)(2-фенил-1H-имидазол-4-ил)метанона (12ad); (3,4-диметоксифенил)(2-фенил-1H-имидазол-4-ил)метанона (12ae); (4-фторфенил)(2-фенил-1H-имидазол-4-ил)метанона (12af); (3-фторфенил)(2-фенил-1H-имидазол-4-ил)метанона (12ag); (2-фенил-1H-имидазол-4-ил)(п-толил)метанона (12ah); (2-фенил-1H-имидазол-4-ил)(м-толил)метанона (12ai); (2-(4-фторфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ba); (2-(4-метоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ca); (4-фторфенил)(2-(4-метоксифенил)-1H-имидазол-4-ил)метанона (12cb); (2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12da); (4-фторфенил)(2-(п-толил)-1H-имидазол-4-ил)метанона (12db); (4-фторфенил)(2-(п-толил)-1H-имидазол-4-ил)метанона гидрохлорида (12db-HCl); (4-гидрокси-3,5-диметоксифенил)(2-(п-толил)-1H-имидазол-4-ил)метанона (12dc); (3,4,5-триметоксифенил)(2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)метанона (12ea); (4-фторфенил)(2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)метанона (12eb); (2-(4-хлорфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12fa); (2-(4-хлорфенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12fb); (2-(4-хлорфенил)-1H-имидазол-4-ил)(4-гидрокси-3,5-диметоксифенил)метанона (12fc); (2-(4-(диметиламино)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ga); (2-(4-(диметиламино)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12gb); (2-(3,4-диметоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ha); (2-(3,4-диметоксифенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12hb); (2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ia); (4-фторфенил)(2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)метанона (12ib); (2-(4-(бензилокси)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ja); (2-(4-(бензилокси)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12jb); (2-(4-гидроксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12ka); (2-(4-(гидроксифенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (12kb); (2-(4-бромфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12la); (2-(4-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12pa); (3,4,5-тригидроксифенил)(2-(3,4,5-тригидроксифенил)-1H-имидазол-4-ил)метанона (13ea); (2-(4-хлорфенил)-1H-имидазол-4-ил)(3,4,5-тригидроксифенил)метанона (13fa); и 2-(3,4-дигидроксифенил)-1H-имидазол-4-ил)(3,4,5-тригидроксифенил)метанона (13ha).
В одном варианте, P соединения формулы XII является  , и Q является SO2-Ph. Не ограничивающие примеры соединения формулы XII, где P соединения формулы XII является
, и Q является SO2-Ph. Не ограничивающие примеры соединения формулы XII, где P соединения формулы XII является  , и Q является SO2-Ph, выбраны из (4-метоксифенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11ab); (3-метоксифенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11ac); (2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)(п-толил)метанона (11ah); (4-фторфенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11af); (3-фторфенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11ag); (4-фторфенил)(2-(4-метоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11cb); (1-(фенилсульфонил)-2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11da); (4-фторфенил)(1-(фенилсульфонил)-2-(п-толил)-1H-имидазол-4-ил)метанона (11db); (1-(фенилсульфонил)-2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ea); (4-фторфенил)(1-(фенилсульфонил)-2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)метанона (11eb); (2-(4-хлорфенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11fb); (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ga); (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11gb); (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ha); (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11hb); (1-(фенилсульфонил)-2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ia); (1-(фенилсульфонил)-2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11ib); и (2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11jb); (2-(4-бромфенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11la); (1-(фенилсульфонил)-2-(4-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11pa).
, и Q является SO2-Ph, выбраны из (4-метоксифенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11ab); (3-метоксифенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11ac); (2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)(п-толил)метанона (11ah); (4-фторфенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11af); (3-фторфенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11ag); (4-фторфенил)(2-(4-метоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)метанона (11cb); (1-(фенилсульфонил)-2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11da); (4-фторфенил)(1-(фенилсульфонил)-2-(п-толил)-1H-имидазол-4-ил)метанона (11db); (1-(фенилсульфонил)-2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ea); (4-фторфенил)(1-(фенилсульфонил)-2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)метанона (11eb); (2-(4-хлорфенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11fb); (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ga); (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11gb); (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ha); (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11hb); (1-(фенилсульфонил)-2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11ia); (1-(фенилсульфонил)-2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11ib); и (2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанона (11jb); (2-(4-бромфенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11la); (1-(фенилсульфонил)-2-(4-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11pa).
В одном варианте, R4 и R5 соединений формулы XIII-XVI являются водородами. Не ограничивающие примеры соединений формулы XIII-XVI, где R4 и R5 являются водородами, выбраны из (2-фенил-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12aa); (4-метоксифенил)(2-фенил-1H-имидазол-4-ил)метанона (12ab); (3-метоксифенил)(2-фенил-1H-имидазол-4-ил)метанона (12ac); (3,5-диметоксифенил)(2-фенил-1H-имидазол-4-ил)метанона (12ad); (3,4-диметоксифенил)(2-фенил-1H-имидазол-4-ил)метанона (12ae); (4-фторфенил)(2-фенил-1H-имидазол-4-ил)метанона (12af); (3-фторфенил)(2-фенил-1H-имидазол-4-ил)метанона (12ag); (2-фенил-1H-имидазол-4-ил)(п-толил)метанона (12ah); и (2-фенил-1H-имидазол-4-ил)(м-толил)метанона (12ai).
В одном варианте, P соединения формулы XII является H, и Q является. В другом варианте W является C=O. В другом варианте , W соединения формулы XVIII является C=O. Не ограничивающие примеры соединения формулы XVIII, где W является C=O, выбраны из (4-метоксифенил)(2-фенил-1H-имидазол-1-ил)метанона (12aba) и (2-фенил-1H-имидазол-1-ил)(3,4,5-триметоксифенил)метанона (12aaa).
В другом варианте, W соединения формулы XVIII является SO2. Не ограничивающие примеры соединения формулы XVIII, где W является SO2, выбраны из 2-фенил-1-(фенилсульфонил)-1H-имидазола (10a); 2-(4-нитрофенил)-1-(фенилсульфонил)-1H-имидазола (10x) и 2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1H-имидазола (10j).
При использовании в настоящем документе, «одно-, конденсированные или многокольцевые, арильные или (гетеро)циклические кольцевые системы» могут быть любым таким кольцом, включая, но не ограничиваясь этим, фенил, бифенил, трифенил, нафтил, циклоалкил, циклоалкенил, циклодиенил, флуорен, адамантан и так далее.
«Насыщенные или ненасыщенные N-гетероциклы» могут быть любым таким N-содержащим гетероциклом, включая, но не ограничиваясь этим, аза- и диаза-циклоалкилы, такие как азиридинил, азетидинил, диазатидинил, пирролидинил, пиперидинил, пиперазинил и азоканил, пирролил, паризолил, имидазолил, пиридинил, пиримидинил, пиразинил, пиридазинил, триазинил, тетразинил, пирролизинил, индолил, хинолинил, изохинолинил, бензимидазолил, индазолил, хинолизинил, циннолинил, хинололинил, фталазинил, нафтиридинил, хиноксалинил и так далее.
«Насыщенные или ненасыщенные O-гетероциклы» могут быть любым таким O-содержащим гетероциклом, включая, но не ограничиваясь этим, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, диоксанил, фуранил, пирилий, бензофуранил, бензодиоксолил и так далее.
«Насыщенные или ненасыщенные S-гетероциклы» могут быть любым таким S-содержащим гетероциклом, включая, но не ограничиваясь этим, тиранил, тиетанил, тетрагидротиофен-ил, дитиоланил, тетрагидротиопиранил, тиофен-ил, тиепинил, тианафтенил и так далее.
«Насыщенные или ненасыщенные смешанные гетероциклы» могут быть любым гетероциклом, содержащим два или более S-, N- или O-гетероатомов, включая, но не ограничиваясь этим, оксатиоланил, морфолинил, тиоксанил, тиазолил, изотиазолил, тиадиазолил, оксазолил, изоксазолил, оксадиазолил и так далее.
При использовании в настоящем документе, «алифатический углеводород прямого или разветвленного строения» относится как к алкиленовым группам, которые содержат от одного углерода до определенного верхнего предела, так и к алкенильным группам и алкинильным группам, которые содержат от двух углеродов до верхнего предела, независимо от того, находятся ли эти углероды в одной цепи или в разветвленной цепи. Если отдельно не указано, то углеводород может включать до около 30 углеродов или до около 20 углеводородов, или до около 10 углеводородов. Алкенильные и алкинильные группы могут быть мононенасыщенными или полиненасыщенными. В другом варианте алкил включает C1-C6 углероды. В другом варианте алкил включает C1-C8 углероды. В другом варианте алкил включает C1-C10 углероды. В другом варианте алкилом являются C1-C12 углероды. В другом варианте алкилом являются C1-C5 углероды.
При использовании в настоящем документе, термин «алкил» может быть любой алкильной группой прямого или разветвленного строения, содержащей до около 30 углеродов, если не указано иное. В другом варианте алкил включает C1-C6 углероды. В другом варианте алкил включает C1-C8 углероды. В другом варианте алкил включает C1-C10 углероды. В другом варианте алкилом являются C1-C12 углероды. В другом варианте алкилом являются C1-C20 углероды. В другом варианте циклическая алкильная группа имеет 3-8 атомов углерода. В другом варианте разветвленный алкил является алкилом, замещенным алкильными боковыми цепями из 1-5 атомов углерода.
Алкильная группа может быть единственным заместителем, или она может быть составляющей более крупного заместителя, так как в алкокси, галоалкиле, арилалкиле, алкиламино, диалкиламино, алкиламидо, алкилмочевине и так далее. Предпочтительными алкиловыми группами являются метил, этил и пропил, и, следовательно, галометил, дигалометил, тригалометил, галоэтил, дигалоэтил, тригалоэтил, галопропил, дигалопропил, тригалопропил, метокси, этокси, пропокси, арилметил, арилэтил, арилпропил, метиламино, этиламино, пропиламино, диметиламино, диэтиламино, метиламидо, ацетамидо, пропиламидо, галометиламидо, галоэтиламидо, галопропиламидо, метил-мочевина, этил-мочевина, пропил-мочевина и так далее.
При использовании в настоящем документе, термин «арил» относится к любому ароматическому кольцу, которое напрямую связано с другой группой. Арильная группа может быть единственным заместителем, или арильная группа может быть составляющей более крупного заместителя, так как в арилалкиле, ариламино, ариламидо и так далее. Иллюстративные арильные группы включают, без ограничений, фенил, толил, ксилил, фуранил, нафтил, пиридинил, пиримидинил, пиридазинил, пиразинил, триазинил, тиазолил, оксазолил, изооксазолил, пиразолил, имидазолил, тиофен-ил, пирролил, фенилметил, фенилэтил, фениламино, фениламидо и так далее.
При использовании в настоящем документе, термин «аминоалкил» относится к аминогруппе, замещенной алкильной группой, как оговорено выше. Аминоалкил относится к моноалкиламину, диалкиламину или триалкиламину. Не ограничивающими примерами аминоалкильных групп являются -N(Me)2, -NHMe, -NH3.
«Галоалкильная» группа относится, в другом варианте, к алкильной группе, как оговорено выше, которая замещена одним или несколькими атомами галогена, например, F, Cl, Br или I. Не ограничивающими примерами галоалкильных групп являются CF3, CF2CF3, CH2CF3.
В одном варианте настоящего изобретения представлено соединение настоящего изобретения или его изомер, метаболит, фармацевтически приемлемая соль, фармацевтический продукт, таутомер, гидрат, N-оксид, полиморф или кристалл, или их комбинации. В одном варианте настоящего изобретения представлен изомер соединения настоящего изобретения. В другом варианте настоящего изобретения представлен метаболит соединения настоящего изобретения. В другом варианте настоящего изобретения представлена фармацевтически приемлемая соль соединения настоящего изобретения. В другом варианте настоящего изобретения представлен фармацевтический продукт соединения настоящего изобретения. В другом варианте настоящего изобретения представлен таутомер соединения настоящего изобретения. В другом варианте настоящего изобретения представлен гидрат соединения настоящего изобретения. В другом варианте настоящего изобретения представлен N-оксид соединения настоящего изобретения. В другом варианте настоящего изобретения представлен полиморф соединения настоящего изобретения. В другом варианте настоящего изобретения представлен кристалл соединения настоящего изобретения. В другом варианте настоящего изобретения представлена композиция, включающая соединение настоящего изобретения, как описано в настоящем документе, или, в другом варианте, комбинация изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа или кристалла соединения настоящего изобретения.
В одном варианте термин «изомер» включает, но не ограничиваясь этим, оптические изомеры и аналоги, структурные изомеры и аналоги, конформационные изомеры и аналоги, и тому подобное.
В одном варианте соединения настоящего изобретения являются чистыми (E)-изомерами. В другом варианте соединения настоящего изобретения являются чистыми (Z)-изомерами. В другом варианте соединения настоящего изобретения являются смесью (E) и (Z) изомеров. В одном варианте соединения настоящего изобретения являются чистыми (R)-изомерами. В другом варианте соединения настоящего изобретения являются чистыми (S)-изомерами. В другом варианте соединения настоящего изобретения являются смесью (R) и (S) изомеров.
Соединения настоящего изобретения могут также существовать в форме рацемической смеси, содержащей, в основном, эквивалентные количества стереоизомеров. В другом варианте соединения настоящего изобретения могут быть получены или выделены другим образом, с использованием известных способов получения стереоизомера, в основном, свободного от его соответствующего стереоизомера (то есть, в основном, чистые). Под «в основном, чистым» подразумевается, что чистота стереоизомера составляет, по меньшей мере, около 95%, более предпочтительно, по меньшей мере, около 98%, наиболее предпочтительно, по меньшей мере, около 99%.
Соединения настоящего изобретения могут также быть в форме гидрата, что означает, что это соединение дополнительно включает стехиометрическое или нестехиометрическое количество воды, связанной нековалентными внутримолекулярными силами.
Соединения настоящего изобретения могут существовать в форме одного или нескольких из возможных таутомеров и, в зависимости от конкретных условий, существует возможность разделить некоторые или все таутомеры на индивидуальные и отдельные вещества. Следует понимать, что все возможные таутомеры, включая все дополнительные енольные и кето-таутомеры и/или изомеры, входят в настоящее изобретение. Например, включены, но не ограничиваясь ими, следующие таутомеры.

Настоящее изобретение включает «фармацевтически приемлемые соли» соединений настоящего изобретения, которые могут быть получены по реации соединения настоящего изобретения с кислотой или основанием. Некоторые соединения, в частности, те, которые имеют кислотную или основную группы, также могут быть в форме соли, предпочтительно, фармацевтически приемлемой соли. Термин «фармацевтически приемлемая соль» относится к таким солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислоты, которые не являются биологически или другим образом нежелательными. Эти соли образуются с неорганическими кислотами, такими как хлороводородная кислота, бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобными, и органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, N-ацетилцистеин и тому подобными. Другие соли известны специалистам в данной области, и могут быть легко подобраны для применения в соответствии с настоящим изобретением.
Соответствующие фармацевтически приемлемые соли аминов соединений настоящего изобретения могут быть получены из неорганической кислоты или из органической кислоты. В одном варианте примерами неорганических солей аминов являются бисульфаты, бораты, бромиды, хлориды, гемисульфаты, гидроброматы, гидрохлораты, 2-гидроксиэтилсульфонаты (гидроксиэтансульфонаты), йодаты, йодиды, изотионаты, нитраты, персульфаты, фосфаты, сульфаты, сульфаматы, сульфанилаты, сульфоновые кислоты (алкилсульфонаты, арилсульфонаты, галоген-замещенные алкилсульфонаты, галоген-замещенные арилсульфонаты), сульфонаты и тиоцианаты.
В одном варианте примеры органических солей аминов могут быть выбраны из алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоциклических и сульфоновых классов органических кислот, примерами которых являются ацетаты, аргинины, аспартаты, аскорбаты, адипинаты, антранилаты, альгенаты, алканкарбоксилаты, замещенные алканкарбоксилаты, альгинаты, бензолсульфонаты, бензоаты, бисульфаты, бутираты, бикарбонаты, битартраты, цитраты, камфораты, камфорсульфонаты, циклогексилсульфаматы, циклопентанпропионаты, эдетаты кальция, камзилаты, карбонаты, клавуланаты, циннаматы, дикарбоксилаты, диглюконаты, додецилсульфонаты, дигидрохлориды, деканоаты, энантгуаты, этансульфонаты, эдетаты, эдизилаты, эстолаты, эзилаты, фумараты, формиаты, фториды, галактуронаты, глюконаты, глутаматы, гликоляты, глюкораты, глюкогептаноаты, глицерофосфаты, глюцептаты, гликолиларсанилаты, глутараты, глутаматы, гептаноаты, гексаноаты, гидроксималеаты, гидроксикарбоновые кислоты, гексилрезорцинаты, гидроксибензоаты, гидроксинафтоаты, гидрофториды, лактаты, лактобионаты, лаураты, малаты, малеаты, метиленбис(бета-оксинафтоаты), малонаты, манделаты, мезилаты, метансульфонаты, метилбромиды, метилнитраты, метилсульфонаты, монокалия малеаты, мукаты, монокарбоксилаты, нафталинсульфонаты, 2-нафталинсульфонаты, никотинаты, напзилаты, N-метилглюкамины, оксалаты, октаноаты, олеаты, памоаты, фенилацетаты, пикраты, фенилбензоаты, пивалаты, пропионаты, фталаты, фенилаццетаты, пектинаты, фенилпропионаты, пальмитаты, пантотенаты, полигалактураты, пируваты, хинаты, салицилаты, сукцинаты, стеараты, сульфанилаты, субацетаты, тартраты, теофиллинацетаты, п-толуолсульфонаты (тозилаты), трифторацетаты, терефталаты, таннаты, теоклаты, тригалоацетаты, триэтиодиды, трикарбоксилаты, ундеканоаты и валераты.
В одном варианте примеры неорганических солей карбоновых кислот или гидроксилов могут быть выбраны из аммония, щелочных металлов, включающих литий, натрий, калий, цезий; щелочноземельных металлов, включающих кальций, магний, алюминий; цинка, бария, холинов, четвертичных аммониевых солей.
В другом варианте примеры органических солей карбоновых кислот или гидроксилов могут быть выбраны из аргинина, органических аминов, включающих алифатические органические амины, алициклические органические амины, ароматические органические амины, бензатины, трет-бутиламины, бенетамины (N-бензилфенетиламин), дициклогексиламины, диметиламины, диэтаноламины, этаноламины, этилендиамины, гидрабамины, имидазолы, лизины, метиламины, мегламины, N-метил-D-глюкамины, N,N’-дибензилэтилендиамины, никотинамиды, органические амины, орнитины, пиридины, пиколины, пиперазины, прокаин, трис(гидроксиметил)метиламины, триэтиламины, триэтаноламины, триметиламины, трометамины и карбамиды.
В одном варианте соли могут быть образованы общепринятыми способами, такими как взаимодействие продукта в форме свободного основания или свободной кислоты с одним или несколькими эквивалентами соответствующей кислоты или основания в растворителе или среде, в которой эта соль нерастворима, или в растворителе, таком как вода, который удаляется in vacuo или сушкой с замораживанием, или заменой ионов существующей соли на другой ион или соответствующую ионообменную смолу.
В некоторых вариантах настоящего изобретения представлен способ получения соединений настоящего изобретения. В одном варианте арил-имидазол получается взаимодействием соответствующим образом замещенного бензальдегида с этилендиамином с образованием имидазолинового кольца с последующим окислением имидазолина окислительным агентом до соответствующего имидазола. В другом варианте окислительным агентом является диацетоксийодбензол, бромтрихлорметан и 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), система углерод-O2 или система палладий-углерод. В другом варианте, арил-имидазол получается взаимодействием соответствующим образом замещенного бензальдегида с этилендиамином в присутствии йода и карбоната калия для образования имидазолиного кольца, с последующим окислением имидазолинового кольца, катализируемым диацетоксийодбензолом, бромтрихлорметаном и 1,8-диазабицикло[5.4.0]ундец-7-еном (DBU), системой углерод-O2 или системой палладий-углерод, до соответствующего имидазола. В другом варианте, арил-имидазол получается взаимодействием соответствующим образом замещенного бензальдегида с этилендиамином в присутствии йода и карбоната калия для образования имидазолинового кольца, с последующим окислением имидазолинового кольца, катализируемым диацетоксийодбензолом, до соответствующего имидазола. В другом варианте, арил-имидазол получается взаимодействием соответствующим образом замещенного бензальдегида с этилендиамином в присутствии йода и карбоната калия для образования имидазолиного кольца, с последующим окислением имидазолинового кольца, катализируемым бромтрихлорметаном и 1,8-диазабицикло[5.4.0]ундец-7-еном (DBU), до соответствующего имидазола. В одном варианте, арил-имидазол получается взаимодействием соответствующего бензальдегида в этаноле с оксалальдегидом и гидроксидом аммония для образования имидазольной кольцевой системы.
В одном варианте, арил-бензоил-имидазольное соединение настоящего изобретения получается защитой арил-имидазола с последующим связыванием с соответствующим образом замещенным бензоилхлоридом, с последующим удалением защитной группы. В другом варианте, защитной группой является фенилсульфониловая группа, фталимид, ди-трет-бутил дикарбонат (Boc), флуоренилметилоксикарбонил (Fmoc), бензилоксикарбонил (Cbz) или монометокситритил (MMT). В другом варианте, защита арил-имидазола осуществляется фенилсульфонилом с образованием N-сульфонил-защищенного арил-имидазола. В другом варианте, защищенное арил-имидазольное соединение получается взаимодействием арил-имидазола с фенилсульфонил хлоридом и гидридом натрия в ТГФ. В другом варианте, защищенный арил-имидазол получается в соответствии с Фигурами 7 и 8.
В одном варианте, защищенный арил-имидазол связывается с соответствующим образом замещенным бензоилхлоридом с образованием защищенного арил-бензоил-имидазола. В другом варианте, арил-имидазол связывается с соответствующим образом замещенным бензоилхлоридом в присутствии трет-бутиллития с образованием арил-фенилсульфонила (2-арил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанона. В другом варианте, (2-арил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанон получается в соответствии с Фигурами 7 и 8, этапы е и с, соответственно.
В одном варианте, арил-бензоил-имидазол получается удалением защитной группы арил-бензоил-имидазола. В другом варианте, удаление защитной группы зависит от используемой защитной группы, и она может быть удалена при условиях, известных в данной области. В другом варианте, фенилсульфониловая защитная группа удаляется фторидом тетрабутиламмония в ТГФ. В другом варианте, фенилсульфонил удаляется в соответствии с Фигурами 7 и 8.
В одном варианте, соединения формулы I, Ia, II, III, V и XI получаются в соответствии с Фигурой 1. В другом варианте, соединения формулы I, Ia, II, III, V, VI, VII и XI получаются в соответствии с Фигурой 2. В другом варианте, соединения формулы I, Ia, II, III, V и VI получаются в соответствии с Фигурой 3. В другом варианте, соединения формулы I, Ia, II, III, V и VI получаются в соответствии с Фигурой 4. В другом варианте, соединения формулы I, Ia, II, III, IV, IVa, V, VI и XI получаются в соответствии с Фигурой 5. В другом варианте, соединения формулы I, Ia, II, III, VIII и XI получаются в соответствии с Фигурой 6.
В одном варианте, соединения формулы XII и XVIII получаются в соответствии с Фигурой 9. В другом варианте, соединения формулы XII, XIII, XIV, XIVa, XV, XVI, XVII, XIX и XX получаются в соответствии с Фигурой 10. В другом варианте, соединения формулы XIVa и XIX получаются в соответствии с Фигурой 11. В другом варианте, соединения формулы I, Ia, IV, IVa, XI, XXI, XXIa и XXII получаются в соответствии с Фигурой 12. В другом варианте, соединения формулы I, Ia, IV, IVa, XI, XIb, XXI, XXIa и XXII получаются в соответствии с Фигурой 13. В другом варианте, соединения формулы I, Ia, II, III, V, XI, XII, XIII, XIV, XV, XVII, XIX и XX получаются в соответствии с Фигурой 14. В другом варианте, соединения формулы I, Ia, II, IV, IVa, XI и XIc, получаются в соответствии с Фигурой 15.
В одном варианте, соединения формулы IX и IXa получаются в соответствии с Фигурой 16.
Фармацевтические композиции
Другой аспект настоящего изобретения относится к фармацевтической композиции, включающей фармацевтически приемлемый носитель и соединение в соответствии с аспектами настоящего изобретения. Фармацевтическая композиция может содержать одно или несколько из определенных выше соединений настоящего изобретения. Как правило, фармацевтическая композиция настоящего изобретения включает соединение настоящего изобретения или его фармацевтически приемлемую соль, а также фармацевтически приемлемый носитель. Термин «фармацевтически приемлемый носитель» относится к любым соответствующим адъювантам, носителям, формообразующим средствам или стабилизаторам, и они могут быть в твердой или жидкой форме, такой как таблетки, капсулы, порошки, растворы, суспензии или эмульсии.
Как правило, композиция содержит от около 0,01 до 99 процентов, предпочтительно, от около 20 до 75 процентов активного соединения(-ий), вместе с адъювантами, носителями и/или формообразующими средствами. Хотя индивидуальные потребности могут варьироваться, определение оптимальных диапазонов эффективного количества каждого компонента является известным в данной области. Типичные дозы включают от около 0,01 до около 100 мг/кг веса тела. Предпочтительные дозы включают от около 0,1 до около 100 мг/кг веса тела. Наиболее предпочтительные дозы включают от около 1 до около 100 мг/кг веса тела. Схема лечения для введения соединений настоящего изобретения также может быть легко определена специалистом в данной области. То есть частота введения и размер дозы могут быть установлены стандартной оптимизацией, предпочтительно, при минимизации побочных эффектов.
Твердые единичные лекарственные формы могут быть обычного типа. Твердая форма может быть капсулой и тому подобными, такими как в обычном желатиновом типе, содержащем соединения настоящего изобретения и носитель, например, смазывающие вещества и инертные наполнители, такие как лактоза, сахароза или кукурузный крахмал. В другом варианте, эти соединения таблетируются с обычными основами для таблеток, такими как лактоза, сахароза или кукурузный крахмал, в комбинации со связующими веществами, такими как гуммиарабик, кукурузный крахмал или желатин, средствами для улучшения распадаемости таблеток, такими как кукурузный крахмал, картофельный крахмал или альгиновая кислота, и смазывающим веществом, таким как стеариновая кислота или стеарат магния.
Таблетки, капсулы и тому подобные могут также содержать связующее вещество, такое как трагакантовая камедь, гуммиарабик, кукурузный крахмал или желатин; формообразующие средства, такие как дикальция фосфат; средство для улучшения распадаемости таблеток, такое как кукурузный крахмал, картофельный крахмал, альгиновая кислота; смазывающее вещество, такое как стеарат магния; и подсластитель, такой как сахароза, лактоза или сахарин. Если единичная лекарственная форма является капсулой, она может содержать, помимо материалов перечисленных выше типов, жидкий носитель, такой как жирное масло.
Могут присутствовать различные другие материалы, в качестве покрытий или для модификации физической формы дозированной единицы. Например, таблетки могут быть покрыты шеллаком, сахаром или ими обоими. Сироп может содержать, помимо активного компонента, сахарозу в качестве подсластителя, метил- и пропилпарабены в качестве консервантов, краситель и ароматизатор, такой как вишневый или апельсиновый ароматизатор.
Для перорального терапевтического введения эти активные соединения могут быть внедрены в формообразующие средства и использоваться в форме таблеток, капсул, эликсиров, суспензий, сиропов и тому подобного. Такие композиции и препараты должны содержать, по меньшей мере, 0,1% активного соединения. Процентное содержание соединения в этих композициях может, конечно, варьироваться, и для удобства составляет от около 2% до около 60% веса единицы. Количество активного соединения в таких терапевтически полезных композициях является таким, чтобы получалась соответствующая доза. Предпочтительные композиции в соответствии с настоящим изобретением приготавливаются так, чтобы пероральная дозированная единица содержала от около 1 мг до 800 мг активного соединения.
Активные соединения настоящего изобретения могут вводиться перорально, например, с инертным разбавителем, или с усвояемым съедобным носителем, или они могут быть инкапсулированы в твердые или мягкие капсулы, или они могут быть спрессованы в таблетки, или они могут быть введены непосредственно в пищу рациона.
Фармацевтические формы, пригодные для инъекционного применения, включают стерильные водные растворы или дисперсии и стерильные порошки для приготовления стерильных инъекционных растворов или дисперсий для немедленного приема. Во всех случаях форма должна быть стерильной и должна быть жидкой настолько, чтобы обеспечивать простое введение через шприц. Она должна быть устойчивой в условиях производства и хранения и должна быть предохранена против заражения микроорганизмами, такими как бактерии и грибки. Носителем может быть растворитель или дисперсионная среда, содержащая, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их соответствующие смеси и растительные масла.
Соединения или фармацевтические композиции настоящего изобретения также могут вводиться в инъецируемых дозах в виде растворов или суспензий этих материалов в физиологически приемлемом разбавителе с фармацевтическим адъювантом, носителем или формообразующим средством. Такие адъюванты, носители /или формообразующие средства включают, но не ограничиваясь этим, стерильные жидкости, такие как вода и масла, с добавлением или без добавления поверхностно-активного вещества или других фармацевтически и физиологически приемлемых компонентов. Иллюстративными маслами являются масла нефтяного, животного, растительного или синтетического происхождения, например, арахисовое масло, соевое масло или минеральное масло. В общем, вода, солевой раствор, водный раствор декстрозы и родственные растворы сахаров, а также гликоли, такие как пропиленгликоль или полиэтиленгликоль, являются предпочтительными жидкими носителями, особенно для растворов для инъекций.
Эти активные соединения также могут вводиться парентерально. Растворы или суспензии этих активных соединений могут быть получены в воде, надлежащим образом смешанной с поверхносто-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии могут быть также получены в глицерине, жидких полиэтиленгликолях и их смесях в маслах. Иллюстративными маслами являются масла нефтяного, животного, растительного или синтетического происхождения, например, арахисовое масло, соевое масло или минеральное масло. В общем, вода, солевой раствор, водный раствор декстрозы и растворы родственных сахаров, а также гликоли, такие как пропиленгликоль или полиэтиленгликоль, являются предпочтительными жидкими носителями, особенно для растворов для инъекций. В обычных условиях хранения и использования эти композиции содержат консервант для предотвращения роста микроорганизмов.
Для применения в виде аэрозолей, соединения настоящего изобретения в растворе или суспензии могут быть упакованы в аэрозольную тару под давлением, вместе с соответствующими газами-вытеснителями, например, углеводородными газами-вытеснителями, такими как пропан, бутан или изобутан, с обычными адъювантами. Материалы настоящего изобретения также могут вводиться в форме без давления, такой как в распылителе или ингаляторе.
В одном варианте соединения настоящего изобретения вводятся в комбинации с противораковым средством. В одном варианте противораковым средством является моноклональное антитело. В некоторых вариантах моноклональные антитела используются для диагностики, мониторинга или лечения рака. В одном варианте моноклональные антитела действуют против специфических антигенов на раковых клетках. В одном варианте моноклональное антитело действует как антагонист рецептора раковой клетки. В одном варианте моноклональные антитела усиливают иммунный ответ пациента. В одном варианте моноклональные антитела действуют против клеточных факторов роста, блокируя за счет этого рост раковой клетки. В одном варианте противораковые моноклональные антитела конъюгированы или связаны с противораковыми лекарствами, радиоизотопами, другими модификаторами биологического ответа, другими токсинами или их комбинацией. В одном варианте противораковые моноклональные антитела конъюгированы или связаны с соединением настоящего изобретения, как описано выше в настоящем документе.
Другой аспект настоящего изобретения относится к способу лечения рака, который включает выбор пациента, нуждающегося в лечении рака, и введение этому пациенту фармацевтической композиции, включающей соединение по первому аспекту настоящего изобретения и фармацевтически приемлемый носитель при условиях, эффективных для лечения рака.
При введении соединений настоящего изобретения, они могут вводиться системно или, альтернативно, они могут вводиться непосредственно в определенный участок, где присутствуют раковые клетки или предраковые клетки. Следовательно, введение может выполняться любым способом, эффективным для доставки соединений или фармацевтических композиций к раковым клеткам или предраковым клеткам. Иллюстративные режимы введения включают, без ограничения, введение соединений или композиций перорально, локально, трансдермально, парентерально, подкожно, внутривенно, внутримышечно, внутрибрюшинно, внутриносовым закапыванием, внутриполостным или внутрипузырным закапыванием, внутриглазным введением, внутриартериально, внутрь повреждения или нанесением на слизистые оболочки, такие как слизистые оболочки носа, горла и бронхиол.
Биологическая активность
В одном варианте настоящего изобретения представлены соединения и композиции, включая любые варианты, описанные в настоящем документе, для применения в любых способах настоящего изобретения. В одном варианте применение соединения настоящего изобретения или композиции, включающей это соединений, имеет полезность при ингибировании, подавлении, усилении или стимуляции заданной реакции у пациента, что является понятным специалисту в данной области. В другом варианте композиции могут дополнительно включать дополнительные активные компоненты, активность которых является полезной для конкретного применения, для которого вводится соединение настоящего изобретения.
В одном варианте настоящее изобретение относится к способу лечения, подавления, снижения степени, уменьшения риска развития или ингибирования рака, включающему введение соединения настоящего изобретения пациенту, страдающему раком, в условиях, эффективных для лечения рака.
Устойчивость к лекарственным средствам является основной причиной неудач химиотерапии рака. Одним важнейшим действующим фактором во множественной лекарственной устойчивости является сверхэкспрессия Р-гликопротеина (P-gp). Этот белок является клинически важным транспортером белка, приндлежащим к АТФ-связывающему кассетному семейству транспортеров клеточных мембран. Он может выкачивать вещества, включая противораковые лекарства, из клеток опухоли по АТФ-зависимому механизму.
В одном варианте настоящего изобретения представлены способы: а) лечения, подавления, снижения степени, уменьшения риска или ингибирования лекарственно-устойчивых опухолей; б) лечения, подавления, снижения степени, уменьшения риска или ингибирования метастатического рака; в) лечения, подавления, снижения степени, уменьшения риска или ингибирования лекарственно-устойчивого рака; г) лечения, подавления, снижения степени, уменьшения риска или ингибирования лекарственно-устойчивого рака, где раком является меланома; д) способ лечения, подавления, снижения степени, уменьшения риска или ингибирования лекарственно-устойчивого рака, где раком является рак простаты; е) способ лечения, подавления, снижения степени, уменьшения риска или ингибирования метастатической меланомы; ж) способ лечения, подавления, снижения степени, уменьшения риска или ингибирования рака простаты; з) лечения, подавления, снижения степени, уменьшения риска или ингибирования рака у пациента, где пациент проходил предварительное лечение химиотерапией, лучевой терапией или биологической терапией; включающие этап введения указанному пациенту соединения настоящего изобретения и/или изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа или кристалла указанного соединения, или их комбинации.
Соединения настоящего изобретения являются пригодными при лечении, снижении степени, уменьшении риска или ингибировании рака, метастатического рака, лекарственно-устойчивых опухолей, лекарственно-устойчивого рака и различных форм рака. В предпочтительном варианте раком является рак простаты, рак молочной железы, рак яичников, рак кожи (например, меланома), рак легких, рак ободочной кишки, лейкоз, лимфома, рак головы и шеи, рак поджелудочной железы, рак пищевода, рак почек или рак ЦНС (например, глиома, глиобластома). Лечение этих различных видов рака подтверждается представленными в настоящем документе Примерами. Более того, на основании их предполагаемого способа действия как тубулиновых ингибиторов, предполагается, что другие формы рака точно так же могут лечиться или предупреждаться введением пациенту соединений или композиций настоящего изобретения. Предпочтительные соединения настоящего изобретения являются селективно разрушительными для раковых клеток, предпочтительно вызывая разрушение раковых клеток, а не нормальных клеток. Важно, что вред для нормальных клеток минимизирован, поскольку раковые клетки восприимчивы к разрушению при гораздо более низких концентрациях соединений настоящего изобретения.
В некоторых вариантах настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, полиморфа, кристалла, N-оксида, гидрата или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска или ингибирования рака у пациента. В другом варианте раком является рак коры надпочечников, рак анального канала, рак мочевого пузыря, опухоль мозга, опухоль ствола головного мозга, рак молочной железы, глиома, церебральная астроцитома, эпендимома, медуллобластома, супратенториальная примитивная нейроэктодермальная опухоль, опухоль шишковидного тела, глиома гипоталамуса, рак молочной железы, карциноидная опухоль, карцинома, рак шейки матки, рак ободочной кишки, рак центральной нервной системы (ЦНС), рак эндометрия, рак пищевода, рак внепеченочных желчных протоков, семейство опухолей Юинга (Pnet), экстракраниальная опухоль зародышевых клеток, рак глаз, внутриглазная меланома, рак желчного пузыря, рак желудка, опухоль половых клеток, внегонадная, гестационная трофобластическая опухоль, рак головы и шеи, гипофарингеальный рак, карцинома островковых клеток, рак гортани, лейкоз, острый лимфобластный лейкоз, рак ротовой полости, рак печени, рак легких, немелкоклеточный рак легких, мелкоклеточная лимфома, лимфома, связанная со СПИДом, лимфома центральной нервной системы (первичная), лимфома Т-клеток кожи, болезнь Ходжкина, болезнь не-Ходжкина, злокачественная мезотелиома, меланома, карцинома из клеток Меркеля, метастатическая сквамозная карцинома, множественная миелома, опухоли плазматических клеток, грибовидный микоз, миелодиспластический синдром, миелопролиферативные заболевания, рак носоглотки, нейробластома, рак ротоглотки, остеосаркома, эпителиальный рак яичников, опухоль половых клеток яичников, пограничная опухоль яичников, рак поджелудочной железы, рак экзокринной поджелудочной железы, карцинома островковых клеток, рак придаточных пазух носа и полости носа, рак паращитовидных желез, рак полового члена, рак феохромоцитомы, рак гипофиза, новообразования плазматических клеток, рак предстательной железы, рабдомиосаркома, рак прямой кишки, рак почек, рак клеток почек, рак слюнных желез, синдром Сезари, рак кожи, лимфома Т-клеток кожи, рак кожи, саркома Капоши, рак кожи, меланома, рак тонкого кишечника, саркома мягких тканей, саркома мягких тканей, рак яичек, тимома, злокачественный рак щитовидной железы, рак уретры, рак матки, саркома, необычный рак детского возраста, рак влагалища, рак вульвы, опухоль Вильмса, или любая их комбинация. В другом варианте пациент предварительно проходил лечение химиотерапией, лучевой терапией или биологической терапией.
В некоторых вариантах настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, полиморфа, кристалла, N-оксида, гидрата или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска или ингибирования метастатического рака у пациента. В другом варианте раком является рак коры надпочечников, рак анального канала, рак мочевого пузыря, опухоль мозга, опухоль ствола головного мозга, рак молочной железы, глиома, церебральная астроцитома, эпендимома, медуллобластома, супратенториальная примитивная нейроэктодермальная опухоль, опухоль шишковидного тела, глиома гипоталамуса, рак молочной железы, карциноидная опухоль, карцинома, рак шейки матки, рак ободочной кишки, рак центральной нервной системы (ЦНС), рак эндометрия, рак пищевода, рак внепеченочных желчных протоков, семейство опухолей Юинга (Pnet), экстракраниальная опухоль зародышевых клеток, рак глаз, внутриглазная меланома, рак желчного пузыря, рак желудка, опухоль половых клеток, внегонадная, гестационная трофобластическая опухоль, рак головы и шеи, гипофарингеальный рак, карцинома островковых клеток, рак гортани, лейкоз, острый лимфобластный лейкоз, рак ротовой полости, рак печени, рак легких, немелкоклеточный рак легких, мелкоклеточная лимфома, лимфома, связанная со СПИДом, лимфома центральной нервной системы (первичная), лимфома Т-клеток кожи, болезнь Ходжкина, болезнь не-Ходжкина, злокачественная мезотелиома, меланома, карцинома из клеток Меркеля, метастатическая сквамозная карцинома, множественная миелома, опухоли плазматических клеток, грибовидный микоз, миелодиспластический синдром, миелопролиферативные заболевания, рак носоглотки, нейробластома, рак ротоглотки, остеосаркома, эпителиальный рак яичников, опухоль половых клеток яичников, пограничная опухоль яичников, рак поджелудочной железы, рак экзокринной поджелудочной железы, карцинома островковых клеток, рак придаточных пазух носа и полости носа, рак паращитовидных желез, рак полового члена, рак феохромоцитомы, рак гипофиза, новообразования плазматических клеток, рак предстательной железы, рабдомиосаркома, рак прямой кишки, рак почек, рак клеток почек, рак слюнных желез, синдром Сезари, рак кожи, лимфома Т-клеток кожи, рак кожи, саркома Капоши, рак кожи, меланома, рак тонкого кишечника, саркома мягких тканей, саркома мягких тканей, рак яичек, тимома, злокачественный рак щитовидной железы, рак уретры, рак матки, саркома, необычный рак детского возраста, рак влагалища, рак вульвы, опухоль Вильмса, или любая их комбинация.
В некоторых вариантах настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, полиморфа, кристалла, N-оксида, гидрата или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска или ингибирования лекарственно-устойчивого рака или резистентного рака у пациента. В другом варианте раком является рак коры надпочечников, рак анального канала, рак мочевого пузыря, опухоль мозга, опухоль ствола головного мозга, рак молочной железы, глиома, церебральная астроцитома, эпендимома, медуллобластома, супратенториальная примитивная нейроэктодермальная опухоль, опухоль шишковидного тела, глиома гипоталамуса, рак молочной железы, карциноидная опухоль, карцинома, рак шейки матки, рак ободочной кишки, рак центральной нервной системы (ЦНС), рак эндометрия, рак пищевода, рак внепеченочных желчных протоков, семейство опухолей Юинга (Pnet), экстракраниальная опухоль зародышевых клеток, рак глаз, внутриглазная меланома, рак желчного пузыря, рак желудка, опухоль половых клеток, внегонадная, гестационная трофобластическая опухоль, рак головы и шеи, гипофарингеальный рак, карцинома островковых клеток, рак гортани, лейкоз, острый лимфобластный лейкоз, рак ротовой полости, рак печени, рак легких, немелкоклеточный рак легких, мелкоклеточная лимфома, лимфома, связанная со СПИДом, лимфома центральной нервной системы (первичная), лимфома Т-клеток кожи, болезнь Ходжкина, болезнь не-Ходжкина, злокачественная мезотелиома, меланома, карцинома из клеток Меркеля, метастатическая сквамозная карцинома, множественная миелома, опухоли плазматических клеток, грибовидный микоз, миелодиспластический синдром, миелопролиферативные заболевания, рак носоглотки, нейробластома, рак ротоглотки, остеосаркома, эпителиальный рак яичников, опухоль половых клеток яичников, пограничная опухоль яичников, рак поджелудочной железы, рак экзокринной поджелудочной железы, карцинома островковых клеток, рак придаточных пазух носа и полости носа, рак паращитовидных желез, рак полового члена, рак феохромоцитомы, рак гипофиза, новообразования плазматических клеток, рак предстательной железы, рабдомиосаркома, рак прямой кишки, рак почек, рак клеток почек, рак слюнных желез, синдром Сезари, рак кожи, лимфома Т-клеток кожи, рак кожи, саркома Капоши, рак кожи, меланома, рак тонкого кишечника, саркома мягких тканей, саркома мягких тканей, рак яичек, тимома, злокачественный рак щитовидной железы, рак уретры, рак матки, саркома, необычный рак детского возраста, рак влагалища, рак вульвы, опухоль Вильмса, или любая их комбинация.
В одном варианте «метастатический рак» относится к раку, распространившемуся (метастазированному) из его исходного очага в другую область тела. Практически все виды рака способны распространяться. Метастазы развиваются в зависимости от сложного взаимодействия многих факторов опухолевых клеток, включая тип рака, степень зрелости (дифференцировка) клеток опухоли, расположение и продолжительность наличия рака, а также от других не до конца понятных факторов. Метастазы распространяются тремя путями - локальным увеличением из опухоли в окружающие ткани, через кровоток в удаленные участки или через лимфатическую систему к соседним или удаленным лимфатическим узлам. Каждый тип рака может иметь типичный путь распространения. Опухоль получает название по первичному очагу (например, рак молочной железы, который распространяется в мозг, называется метастатическим раком молочной железы в мозг).
В одном варианте «лекарственно-устойчивый рак» относится к раковым клеткам, которые приобретают резистентность к химиотерапии. Раковые клетки могут приобретать резистентность к химиотерапии по различным механизмам, включая мутацию или сверхэкспрессию мишени лекарства, инактивацию лекарства или удаление лекарства из клетки. Опухоли, которые рецидивируют после первоначальной реакции на химиотерапию, могут быть устойчивыми ко многим лекарствам (они обладают множественной лекарственной устойчивостью). С общепринятой точки зрения лекарственной устойчивости, одна или несколько клеток в популяции опухоли приобретают генетические изменения, которые придают лекарственную устойчивость. Соответственно, причинами лекарственной усточивости, inter alia, являются: а) некоторые клетки, которые не были уничтожены химиотерапией, мутируют (изменяются) и становятся устойчивыми к лекарству. Поскольку они размножаются, они могут быть более устойчивыми клетками, чем клетки, восприимчивые к химиотерапии; б) генная амплификация. Раковая клетка может создавать сотни копий определенного гена. Этот ген запускает сверхвыработку белка, который делает противораковое лекарство неэффективным; в) раковые клетки могут выкачивать лекарство из клетки так быстро, как это происходит при использовании молекулы, называемой р-гликопротеином; г) раковые клетки могут останавливать поглощение лекарств из-за того, что белок, который переносит лекарство через стенки клетки, перестает работать; д) раковые клетки могут научиться воспроизводить ДНК-разрывы, обусловленные некоторыми противораковыми лекарствами; е) раковые клетки могут разработать механизм, инактивирующий лекарство. Одним важнейшим действующим фактором во множественной лекарственной устойчивости является сверхэкспрессия Р-гликопротеина (P-gp). Этот белок является клинически важным транспортером белка, приндлежащим к АТФ-связывающему кассетному семейству транспортеров клеточных мембран. Он может выкачивать вещества, включая противораковые лекарства, из клеток опухоли по АТФ-зависимому механизму. Следовательно, резистентность к противораковым средствам, используемым в химиотерапии, является основной причиной неудачи лечения злокачественных расстройств, вызывающей резистентность опухолей. Устойчивость к лекарственным средствам является основной причиной неудач химиотерапии рака.
В одном варианте «резистентный рак» относится к лекарственно-устойчивому раку, как описано выше в настоящем документе. В другом варианте «резистентный рак» относится к раковым клеткам, которые приобрели устойчивость к любому лечению, такому как химиотерапия, лучевая терапия или биологическая терапия.
В одном варианте настоящее изобретение относится к лечению, подавлению, снижению степени, уменьшению риска или ингибированию рака у пациента, где пациент предварительно проходил лечение химиотерапией, лучевой терапией или биологической терапией.
В одном варианте «химиотерапия» относится к химическому лечению рака, такому как лечение лекарствами, которые напрямую уничтожают раковые клетки. Такие лекарства упоминаются как «противораковые» лекарства или «противоопухолевые средства». В современной терапии используется более 100 лекарств для лечения рака. Для лечения определенного рака. Химиотерапия используется для контроля роста опухоли, если лечение невозможно; для уменьшения опухолей перед хирургической или лучевой терапией; для облегчения симптомов (таких как боль); и для разрушения микроскопических раковых клеток, которые могут присутствовать после удаления известной опухоли хирургией (так называемые дополнительные лечебные мероприятия). Дополнительные лечебные мероприятия проводятся для предупреждения возможного повторного возникновения рака.
В одном варианте «лучевая терапия» относится к рентгеновским и аналогичным (таким как электроны) лучам высокой энергии для лечения заболевания. Многие люди, страдающие раком, проходят лечение лучевой терапией как часть их лечения. Она может проводиться как внешней лучевой терапией из источника, находящегося за пределами организма, с использованием рентгеновских лучей, или из источника, находящегося внутри организма, как в случае внутренней лучевой терапии. Лучевая терапия действует путем разрушения раковых клеток в зоне воздействия. Хотя нормальные клетки также могут быть повреждены лучевой терапией, они обычно могут самовосстанавливаться. Обработка лучевой терапией может лечить некоторые виды рака и может также снижать вероятность рецидива рака после хирургической операции. Ее можно использовать для уменьшения симптомов рака.
В одном варианте «биологическая терапия» относится к веществам, которые природным образом встречаются в организме для разрушения раковых клеток. Существует несколько типов лечения, включая: {0>monoclonal antibodies, cancer growth inhibitors, vaccines and gene therapy.<}0{>моноклональные антитела, ингибиторы роста рака, вакцины и генную терапию. Биологическая терапия известна также как иммунотерапия.
В одном варианте настоящего изобретения представлен способ лечения пациента, страдающего от рака простаты, метастатического рака простаты, резистентного рака простаты или лекарственно-устойчивого рака простаты, включающий этап введения указанному пациенту соединения настоящего изобретения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, или композиции, включающей их, в количестве, эффективном для лечения рака простаты у этого пациента. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В одном варианте настоящего изобретения представлен способ подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования рака простаты, метастатического рака простаты, резистентного рака простаты или лекарственно-устойчивого рака простаты у пациента, включающий введение пациенту соединения настоящего изобретения и/или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, или композиции, включающей их. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В одном варианте настоящего изобретения представлен способ лечения пациента, страдающего от рака молочной железы, метастатического рака молочной железы, резистентного рака молочной железыили лекарственно-устойчивого рака молочной железы, включающий этап введения указанному пациенту соединения настоящего изобретения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, или композиции, включающей их. В другом варианте пациентом является самец или самка. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В одном варианте настоящего изобретения представлен способ подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования рака молочной железы, метастатического рака молочной железы, резистентного рака молочной железы или лекарственно-устойчивого рака молочной железы у пациента, включающий этап введения указанному пациенту соединения настоящего изобретения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, или композиции, включающей их. В другом варианте пациентом является самец или самка. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования рака яичников, метастатического рака яичников, резистентного рака яичников или лекарственно-устойчивого рака яичников у пациента. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В одном варианте настоящего изобретения представлен способ лечения, подавления, снижения степени, уменьшения риска или ингибирования меланомы, метастатической меланомы, резистентной меланомы или лекарственно-устойчивой меланомы у пациента, включающий введение пациенту соединения настоящего изобретения и/или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования рака легких, метастатического рака легких, резистентного рака легких или лекарственно-устойчивого рака легких. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования немелкоклеточного рака легких, метастатического мелкоклеточного рака легких, резистентного мелкоклеточного рака легких или лекарственно-устойчивого мелкоклеточного рака легких. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования рака ободочной кишки, метастатического рака ободочной кишки, резистентного рака ободочной кишки или лекарственно-устойчивого рака ободочной кишки. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования лейкоза, метастатического лейкоза, резистентного лейкоза или лекарственно-устойчивого лейкоза. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования лимфомы, метастатической лимфомы, лимфомы или лекарственно-устойчивой лимфомы. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования рака головы и шеи, метастатического рака головы и шеи, резистентного рака головы и шеи или лекарственно-устойчивого рака головы и шеи. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования рака поджелудочной железы, метастатического рака поджелудочной железы, резистентного рака поджелудочной железы или лекарственно-устойчивого рака поджелудочной железы. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования рака пищевода, метастатического рака пищевода, резистентного рака пищевода или лекарственно-устойчивого рака пищевода. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования рака почек, метастатического рака почек, резистентного рака почек или лекарственно-устойчивого рака почек. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристалла или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска, отсрочки прогрессирования или ингибирования рака ЦНС, метастатического рака ЦНС, резистентного рака ЦНС или лекарственно-устойчивого рака ЦНС. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В некоторых вариантах настоящего изобретения представлено применение соединения, описанного в настоящем документе, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, полиморфа, кристалла, N-оксида, гидрата или любой их комбинации, для лечения, подавления, снижения степени, уменьшения риска или ингибирования лекарственно-устойчивой раковой опухоли или опухолей у пациента. В другом варианте раком является рак коры надпочечников, рак анального канала, рак мочевого пузыря, опухоль мозга, опухоль ствола головного мозга, рак молочной железы, глиома, церебральная астроцитома, эпендимома, медуллобластома, супратенториальная примитивная нейроэктодермальная опухоль, опухоль шишковидного тела, глиома гипоталамуса, рак молочной железы, карциноидная опухоль, карцинома, рак шейки матки, рак ободочной кишки, рак центральной нервной системы (ЦНС), рак эндометрия, рак пищевода, рак внепеченочных желчных протоков, семейство опухолей Юинга (Pnet), экстракраниальная опухоль зародышевых клеток, рак глаз, внутриглазная меланома, рак желчного пузыря, рак желудка, опухоль половых клеток, внегонадная, гестационная трофобластическая опухоль, рак головы и шеи, гипофарингеальный рак, карцинома островковых клеток, рак гортани, лейкоз, острый лимфобластный лейкоз, рак ротовой полости, рак печени, рак легких, немелкоклеточный рак легких, мелкоклеточная лимфома, лимфома, связанная со СПИДом, лимфома центральной нервной системы (первичная), лимфома Т-клеток кожи, болезнь Ходжкина, болезнь не-Ходжкина, злокачественная мезотелиома, меланома, карцинома из клеток Меркеля, метастатическая сквамозная карцинома, множественная миелома, опухоли плазматических клеток, грибовидный микоз, миелодиспластический синдром, миелопролиферативные заболевания, рак носоглотки, нейробластома, рак ротоглотки, остеосаркома, эпителиальный рак яичников, опухоль половых клеток яичников, пограничная опухоль яичников, рак поджелудочной железы, рак экзокринной поджелудочной железы, карцинома островковых клеток, рак придаточных пазух носа и полости носа, рак паращитовидных желез, рак полового члена, рак феохромоцитомы, рак гипофиза, новообразования плазматических клеток, рак предстательной железы, рабдомиосаркома, рак прямой кишки, рак почек, рак клеток почек, рак слюнных желез, синдром Сезари, рак кожи, лимфома Т-клеток кожи, рак кожи, саркома Капоши, рак кожи, меланома, рак тонкого кишечника, саркома мягких тканей, саркома мягких тканей, рак яичек, тимома, злокачественный рак щитовидной железы, рак уретры, рак матки, саркома, необычный рак детского возраста, рак влагалища, рак вульвы, опухоль Вильмса, или любая их комбинация. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте опухолью является опухоль рака простаты. В другом варианте опухолью является опухоль рака яичников. В другом варианте опухолью является опухоль меланомы. В другом варианте опухолью является опухоль меланомы, обладающая множественной лекарственной устойчивостью (МЛУ).
В одном варианте настоящее изобретение относится к способу разрушения раковых клеток, включающему: предоставление соединения настоящего изобретения и соприкосновение раковой клетки с этим соединением в условиях, эффективных для разрушения затронутой раковой клетки. В соответствии с различными вариантами разрушения раковых клеток, клетки, подлежащие разрушению, могут быть расположены как in vivo, так и ex vivo (то есть в культуре). В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
В другом варианте рак выбран из группы, состоящей из рака простаты, рака молочной железы, рака яичников, рака кожи, меланомы, рака легких, рака ободочной кишки, лейкоза, рака почек, рака ЦНС и их комбинаций.
В следующем аспекте настоящее изобретение относится к способу лечения или предупреждения ракового состояния, который включает: обеспечение соединения настоящего изобретения, а затем введение эффективного количества этого соединения пациенту таким способом, который является эффективным для лечения или предупреждения ракового состояния.
В соответствии с одним вариантом, пациент, подлежащий лечению, характеризуется наличием предракового состояния, а введение соединения является эффективным для предупреждения развития предракового состояния в раковое состояние. Это может осуществляться за счет разрушения предраковых клеток до или параллельно с их дальнейшим развитием в раковое состояние.
В соответствии с другим вариантом, пациент, подлежащий лечению, характеризуется наличием ракового состояния, а введение соединения является эффективным для вызывания регрессии ракового состояния или для ингибирования развития ракового состояния, то есть, полной остановки его развития или снижения скорости его развития. Это, предпочтительно, происходит за счет разрушения раковых клеток, независимо от их местоположения в организме пациента. То есть независимо от того, где находятся раковые клетки - в первичном очаге опухоли, или раковые клетки метастазировали и создали вторичные опухоли в организме пациента.
При использовании в настоящем документе, субъект или пациент относится к любому млекопитающему пациенту, включая, без ограничений, людей и других приматов, собак, котов, лошадей, коров, овец, свиней, крыс, мышей и других грызунов. В одном варианте пациентом является самец. В другом варианте пациентом является самка. В некоторых вариантах способы, описанные в настоящем документе, могут быть полезными как для лечения самцов, так и для самок.
При введении соединений настоящего изобретения, они могут вводиться системно или, альтернативно, они могут вводиться непосредственно в определенный участок, где присутствуют раковые клетки или предраковые клетки. Следовательно, введение может выполняться любым способом, эффективным для доставки соединений или фармацевтических композиций к раковым клеткам или предраковым клеткам. Иллюстративные схемы введения включают, без ограничения, введение соединений или композиций перорально, локально, трансдермально, парентерально, подкожно, внутривенно, внутримышечно, внутрибрюшинно, внутриносовым закапыванием, внутриполостным или внутрипузырным закапыванием, внутриглазным введением, внутриартериально, внутрь повреждения или нанесением на слизистые оболочки, такие как слизистые оболочки носа, горла и бронхиол.
Соединения настоящего изобретения являются полезными при лечении или предупреждении различных форм рака, в частности, рака простаты, рака молочной железы, рака яичников, рака кожи (например, меланомы), рака легких, рака ободочной кишки, лейкоза, рака почек и рака ЦНС (например, глиомы, глиобластомы). Лечение этих различных видов рака подтверждается представленными в настоящем документе Примерами. Более того, на основании их предполагаемого способа действия как тубулиновых ингибиторов, предполагается, что другие формы рака точно так же могут лечиться или предупреждаться введением пациенту соединений или композиций настоящего изобретения. Предпочтительные соединения настоящего изобретения являются селективно разрушительными для раковых клеток, предпочтительно вызывая разрушение раковых клеток, а не нормальных клеток. Важно, что вред для нормальных клеток минимизирован, поскольку раковые клетки восприимчивы к разрушению при гораздо более низких концентрациях соединений настоящего изобретения.
Соединения настоящего изобретения являются пригодными при лечении, снижении степени, уменьшении риска или ингибировании рака, метастатического рака, резистентного рака или лекарственно-устойчивого рака. В другом варианте раком является рак простаты, рак молочной железы, рак яичников, рак кожи (например, меланома), рак легких, рак ободочной кишки, лейкоз, лимфома, рак головы и шеи, рак поджелудочной железы, рак пищевода, рак почек или рак ЦНС. Лечение этих различных видов рака подтверждается представленными в настоящем документе Примерами. Более того, на основании их предполагаемого способа действия как тубулиновых ингибиторов, предполагается, что другие формы рака точно так же могут лечиться или предупреждаться введением пациенту соединений или композиций настоящего изобретения. Предпочтительные соединения настоящего изобретения являются селективно разрушительными для раковых клеток, предпочтительно вызывая разрушение раковых клеток, а не нормальных клеток. Важно, что вред для нормальных клеток минимизирован, поскольку раковые клетки восприимчивы к разрушению при гораздо более низких концентрациях соединений настоящего изобретения. В другом варианте соединением является соединение 12db. В другом варианте соединением является соединение 11cb. В другом варианте соединением является соединение 11fb. В другом варианте соединением является соединение 12da. В другом варианте соединением является соединение 12fa. В другом варианте соединением является соединение 12fb. В другом варианте соединением является соединение 12cb. В другом варианте соединением является соединение 55.В другом варианте соединением является соединение 6b. В другом варианте соединением является соединение 17ya.
При использовании в настоящем документе, субъект или пациент относится к любому млекопитающему пациенту, включая, без ограничений, людей и других приматов, собак, котов, лошадей, коров, овец, свиней, крыс, мышей и других грызунов. В некоторых вариантах способы, описанные в настоящем документе, могут быть полезными как для лечения самцов, так и для самок.
В одном варианте соединение вводится в комбинации с противораковым средством путем введения соединений, описанных в настоящем документе, отдельно или в комбинации с другими средствами.
Если соединения или фармацевтические композиции настоящего изобретения вводятся для лечения, подавления, снижения степени, уменьшения риска или ингибирования ракового состояния, то фармацевтическая композиция может также содержать, или может вводиться в сочетании с другими терапевтическими средствами или схемами лечения, известными в настоящее время или разработанными в будущем для лечения различных типов рака. Примеры других терапевтических средств или схем лечения включают, без ограничений, лучевую терапию, иммунотерапию, химиотерапию, хирургическое вмешательство и их комбинации.
Следующие примеры представлены в целях более полной иллюстрации предпочтительных вариантов настоящего изобретения. Однако их никоим образом не следует понимать как ограничение широких границ настоящего изобретения.
ПРИМЕРЫ
Представленные ниже Примеры предназначены только для иллюстративных целей и никоим образом не предназначены для ограничения рамок настоящего изобретения.
Материалы и способы:
Общая информация. Все реагенты были закуплены у компаний Sigma-Aldrich Chemical Co., Fisher Scientific (Питтсбург, штат Пенсильвания), AK Scientific (Маунтин-Вью, штат Калифорния), Oakwood Products (Вест-Колумбия, штат Южная Каролина) и так далее, и использовались без дополнительной очистки. Реакции, чувствительные к действию влаги, выполняли под атмосферой аргона. ABT-751 получили в соответствии со способами, описанными Yoshino et al.26 Обычную тонкослойную хроматографию (ТСХ) выполнили на пластинах Uniplates с алюминиевой подложкой (Analtech, Ньюарк, штат Делавер). Точки плавлении измерили на приборе для измерения температур плавления Fisher-Johns (без поправок). Спектры ЯМР записали на спектрометре Bruker AX 300 (Биллерика, штат Массачусетс) или спектрометре Varian Inova-500 (Вернон-Хиллз, штат Иллинойс). Химические сдвиги записали в миллионных долях (ppm) относительно ТМС в CDCl3. Масс-спектральные данные получили на электрораспылительном приборе с ионной ловушкой Bruker ESQUIRE в режимах с положительными и отрицательными ионами. Элементные анализы были выполнены компанией Atlantic Microlab Inc.
Анализ клеточной культуры и цитотоксичности для рака простаты и меланомы. Все клеточные линии были закуплены у компании ATCC (American Type Culture Collection, Манассас, штат Вирджиния, США), а клеточные культуры были приобретены у компании Cellgro Mediatech (Херндон, штат Вирджиния, США). Авторы изобретения исследовали антипролиферативную активность изобретенных антитубулиновых соединений в четырех клеточных линиях рака простаты человека LNCaP, DU 145, PC-3 и PPC-1) и двух клеточных линиях меланомы человека (A375 и WM-164). Клеточную линию рака яичников человека OVCAR-8 и ее резистентную клеточную линию, которая сверхэкспрессирует P-gp (NCI/ADR-RES), использовали в качестве моделей МЛУ. Обе клеточные линии рака яичников приобрели у компании National Cancer Institutes (NCI). Все клеточные линии протестировали и установили подлинность в ATCC или NCI. Все клеточные линии рака простаты и рака яичников выращивали в RPMI 1640, с дополнением 10% бычьей плодной сыворотки (FBS). Клетки меланомы выращивали в DMEM, с дополнением 5% FBS, 1% смеси антибиотика/фунгицида (Sigma-Aldrich, Inc., Сент-Луис, штат Миссури, США) и бычьего инсулина (5 мкг/мл; Sigma-Aldrich). Цитотоксичный потенциал антитубулиновых соединений оценили с использованием анализа с сульфородамином В (SRB) через 96 часов обработки.
Растворимость в воде. Растворимость лекарств определили по фильтровальной пластине растворимости Multiscreen (Millipore Corporate, Биллерика, штат Массачусетс), подключенной к ЖХ-МС/МС. Вкратце, 198 мкл фосфатно-солевого буферного раствора (PBS) (pH 7,4) поместили в 96-луночный планшет, дозировали по 2 мкл 10 мМ раствора исследуемых соединений (в ДМСО) и перемешивали при осторожном встряхивании (200-300 об./мин.) в течение 1,5 часов при комнатной температуре (N = 3). Планшет центрифугировали при 800g в течение 5 минут, а фильтрат использовали для определения его концентрации и растворимости исследуемого соединения по ЖХ-МС/МС, как описано ниже.
Фармакокинетическое исследование. Самок крыс Sprague-Dawley (n = 3 или 4; 254 ± 4 г) приобрели у компании Harlan Inc. (Индианаполис, штат Индиана). Торакальные катетеры яремной вены для крыс приобрели у компании Braintree Scientific Inc. (Брэйнтри, штат Массачусетс). По прибытию в виварий, животные прошли акклиматизацию в течение 3 дней в помещении с контролируемой температурой (20–22°C) с циклом освещения/темноты 12 часов, перед началом какой-либо обработки. Соединение 1h вводили внутривенно (i.v.) в катетеры яремной вены в дозировке 2,5 мг/кг (в ДМСО/ПЭГ300, 2/8), тогда как 5Ha и 5Hc дозировали по 5 мг/кг (в ДМСО/ПЭГ300, 1/9). Инъекцией ввели равный объем гепаринизированного солевого раствора для замещения крови и взяли образцы крови (250 мкл) через катетеры яремной вены через 10, 20, 30 минут и 1, 2, 4, 8, 12, 24 часа. Соединения 1h, 5Ha и 5Hc вводили (p.o.) через ротовой зонд в дозировке 10 мг/кг (в Tween80/ДМСО/H2O, 2/1/7). Все образцы крови (250 мкл) после перорального введения собрали через катетеры яремной вены через 30, 60, 90 минут, 120 минут, 150 минут, 180 минут, 210 минут, 240 минут и 8, 12, 24 часа. Перед сбором крови приготовили гепаринизированные шприцы и пробирки. Образцы плазмы приготовили центрифугированием образцов крови при 8000g в течение 5 минут. Все образцы плазмы сразу хранили при -80°C до анализов.
Аналиты экстрагировали из 100 мкл плазмы при помощи 200 мкл ацетонитрила, содержащего 200 нМ внутреннего стандарта ((3,5-диметоксифенил)(2-фенил-1Н-имидазол-4-ил)метанона). Образцы тщательно перемешали, центрифугировали, а органический экстракт перенесли в автоматический пробоотборник для анализа ЖХ-МС/МС. Для получения наиболее чувствительных сигналов использовали режим многократного мониторинга реакции (MRM), сканирование при m/z 356 → 188 (соединение 1h), m/z 371 → 203 (соединение 5Ha), m/z 389 → 221 (соединение 5Hc) и m/z 309 → 171 (внутренний стандарт). Фармакокинетические параметры определили при помощи анализа без использования камерной модели (WinNonlin, Pharsight Corporation, Маунтин-Вью, штат Калифорния).
Аналитический способ. Раствор образца (10 мкл) ввели инъекцией в систему ВЭЖХ серии Agilent (Agilent 1100 Series Agilent 1100 Chemstation, Agilent Technology Co, Ltd). Все аналиты разделили на колонке С18 с узким отверстием (Alltech Alltima HP, 2,1Ч100 мм, 3 мкм, Fisher, Фейр Лоан, штат Нью-Джерси). Использовали два градиентных режима. Градиентный режим использовали для достижения разделения аналитов с использованием смесей подвижной фазы А [ACN/H2O (5%/95%, об./об.), содержащей 0,1% муравьиной кислоты] и подвижной фазы B [ACN/H2O (95%/5%, об./об.), содержащей 0,1% муравьиной кислоты] при скорости потока 300 мкл/мин. Подвижную фазу А использовали при концентрации 15% от 0 до 1 минуты с последующим линейно запрограммированным градиентом до 100% подвижной фазы В за 6 минут, 100% подфижной фазы В выдерживали в течение 0,5 минут перед быстрым сбросом до 15% подвижной фазы А. Подвижную фазу А выдерживали еще 12 минут перед окончанием анализа.
Анализ полимеризации тубулина in vitro. Бычий тубулин мозга (0,4 мг, чистота >97%) (Cytoskeleton, Денвер, штат Колорадо) смешали с 10 мкМ исследуемых соединений и инкубировали в 100 мкл общего тубулинового буфера (80 мМ PIPES, 2,0 мМ MgCl2, 0,5 мМ EGTA и 1 мМ ГТФ) при pH 6,9. Поглощение при длине волны 340 нм контролировали каждую 1 минуту в течение 20 минут при помощи микропланшет-ридера SYNERGY 4 (Bio-Tek Instruments, Винооски, штат Вермонт). Спектрофотометр был установлен на 37°C для полимеризации тубулина.
Использовали трехквадрупольный масс-спектрометр, API Qtrap 4000TM (Applied Biosystems/MDS SCIEX, Конкорд, Онтарио, Канада), работающий с источником TurboIonSpray. Напряжение иглы-распылителя установили на 5 кВ для положительного режима. «Газовую завесу» установили на 10; газ 1 и газ 2 установили на 50. Активированную столкновениями диссоциацию (CAD) газа на середину, а температуру источника нагревания пробы на 500°C. Получение данных и количественную обработку выполнили при помощи программы AnalystTM версии 1.4.1 (Applied Biosystems).
Чистоту конечных соединений исследовали при помощи обращенно-фазовой ВЭЖХ на системе ВЭЖХ Waters 2695, оснащенной фотодиодным матричным детектором. Два метода обращенно-фазовой ВЭЖХ выполнили с использованием колонки Supelco AscentisTM 5 мкМ C-18 (250 х 4,6 мм) при комнатной температуре и скорости потока 0,7 мл/мин. ВЭЖХ1: Градиент: растворитель А (вода) и растворитель В (метанол): 0-20 минут 40-100% В (линейный градиент), 20-27 минут 100% В. ВЭЖХ2: Градиент: растворитель А (вода) и растворитель В (метанол): 0-15 минут 40-100% В (линейный градиент), 15-25 минут 100% В. УФ-обнаружение при 254 нм.
Соединения настоящего изобретения были получены в соответствии с Фигурами 1-17.
ПРИМЕР 1
СИНТЕЗ СОЕДИНЕНИЙ С ВАРИАНТАМИ КОЛЬЦА В
Соединения с вариантами кольца В были синтезированы в соответствии с Фигурами 1 и 2.
Оксазольное кольцо В:
Синтез (2-фенил-оксазол-4-ил)-(3,4,5-триметокси-фенил)метанона (36а) (Фигура 1):
(2R)-2-Фенил-4,5-дигидро-оксазол-4-карбоновой кислоты метиловый эфир (32a). Ацетилхлорид (6,8 мл) по каплям добавили к ледяному метанолу (30 мл). После добавления L-серина (0,48 ммоль) реакционную смесь нагрели до комнатной температуры и перемешивали в течение ночи. В результате выпаривания растворителя получили белую твердую HCl соль (2R)-3-гидрокси-2-метил-пропионовой кислоты метилового эфира, которую использовали без очистки на следующем этапе. Триэтиламин (11 мл, 72,3 ммоль) медленно добавили к раствору этилбензимидат гидрохлорида (11,6 г, 62,8 ммоль) в CH2Cl2 (150 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут и одной порцией добавили HCl соль (2R)-3-гидрокси-2-метил-пропионовой кислоты метилового эфира (13,5 г, 79,6 ммоль). Полученную смесь перемешивали в течение 48 часов и концентрировали под пониженным давлением. Соединение 32a выделили из флэш-колонки в виде желтого маслянистого вещества (12,3 г, 95,9%). 1H ЯМР (CDCl3) δ 7.99 -7.38 (m, 5 H), 4.97 (dd, 1 H, J = 7.8 Hz, J = 10.5 Hz), 4.70 (t, 1 H, J = 8.7 Hz), 4.62 (dd, 1 H, J = 8.7 Hz, J = 10.5 Hz), 3.82 (s, 3 H); МС (ESI) m/z 206.1 (M + H)+.
(2R)-2-Фенил-4,5-дигидро-оксазол-4-карбоновая кислота (33a). К ледяному раствору 32a в MeOH/H2O добавили LiOH (2,5 экв.) при перемешивании. Смесь оставили нагреваться до комнатной температуры на 1 час, концентрировали in vacuo, а твердое белое вещество растворили в H2O и подкислили 1 н. HCl до рН 2,0, и экстрагировали при помощи MgSO4, отфильтровали и концентрировали in vacuo для получения кислоты 33a в виде твердого белого вещества (95,8%). 1H ЯМР (CDCl3) δ 7.98 (d, 2 H), 7.57-7.42 (m, 3 H), 5.04 (dd, 1 H, J = 7.8 Hz, J = 10.8 Hz), 4.80 (t, 1 H, J = 8.7 Hz), 4.70 (dd, 1 H, J = 9.0 Hz, J= 10.8 Hz); МС (ESI) m/z 191.9 (M + H)+, 189.7 (M - H)-, 145.8 (M - COOH)-.
(2R)-2-Фенил-4,5-дигидро-оксазол-4-карбоновой кислоты метокси-метил-амид (34a). К смеси 33a (5 ммоль), EDCI (6 ммоль), HOBt (5 ммоль) и Et3N (5 ммоль) в CH2Cl2 (50 мл) добавили HNCH3OCH3 (5 ммоль) и продолжали перемешивание при комнатной температуре в течение 6-8 часов. Реакционную смесь разбавили CH2Cl2 (100 мл) и последовательно промыли водой, насыщенным раствором NaHCO3, насыщенным солевым раствором и высушили над MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта 34a, который очистили колоночной хроматографией в виде твердого белого вещества (61,0%). 1H ЯМР (CDCl3) δ 7.98-7.36 (m, 5 H), 7.57-7.42 (m, 3 H), 5.35 (br, t, 1 H), 4.81 (br, t, 1 H), 4.52 (dd, 1 H, J = 8.7 Hz, J = 10.2 Hz), 3.90 (s, 3 H), 3.27 (s, 3 H); МС (ESI) m/z 257.0 (M + H)+.
(2R)-(2-Фенил-4,5-дигидро-оксазол-4-ил)-(3,4,5-триметокси-фенил)-метанон (35a). К раствору n-BuLi (1,6 M, 0,713 мл) в 8 мл ТГФ добавили раствор 3,4,5-триметоксибромбензола (1,09 ммоль) в 3 мл ТГФ при -78oC. Смесь оставили перемешиваться на 2 часа и добавили раствор амида Вайнреба 34a (1,14 ммоль) в 3 мл ТГФ. Температуру оставили увеличиваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 35a в виде твердого белого вещества (47,9%). 1H ЯМР (CDCl3) δ 7.97 -7.94 (m, 2 H), 7.62 (s, 2 H), 7.54-7.37 (m, 3 H), 5.61 (q, 1 H, J = 7.5 Hz, 9.9 Hz), 5.12 (t, 1 H, J = 7.5 Hz), 4.57 (q, 1 H, J = 7.8 Hz, 9.9 Hz), 3.96 (s, 6 H), 3.95 (s, 3 H); МС (ESI) m/z 364.1(M + Na)+, 340.1 (M - H)-.
(2-Фенил-оксазол-4-ил)-(3,4,5-триметокси-фенил)метанон (36а). Смесь 35a (1,48 ммоль), CBrCl3 (2,59 ммоль) и DBU (2,97 ммоль) в CH2Cl2 (20 мл) перемешивали в течение ночи. Реакционную смесь абсорбировали на силикагель и очистили колоночной хроматографией для получения чистого 36a, что и было задано (61,6%). 1H ЯМР (CDCl3) δ 8.37 (s, 1 H), 8.14-8.12 (m, 2 H), 7.74 (s, 2 H), 7.52-7.49 (m, 3 H), 3.97 (s, 9 H); МС (ESI) m/z 362.1(M + Na)+.
Бензольные, пиримидиновые, пиридиновые, фурановые, тиофеновые, тиазоловые, пиразоловые и пиперидиновые варианты кольца В (Фигура 2): Варианты кольца B (1a-1d, 1k) были получены из их соответствующих кислот (37a-37d, 37k). Соединение 1f с тиофеном в В-кольцевом положении не может быть выделено из смеси 1f и побочного продукта связывания с реактивом Гриньяра 3,4,5,3',4',5'-гексаметоксибифенила, с использованием флэш-колонки. Поэтому для получения 1f использовали альтернативный способ: Амид Вайнреба 38f преобразовали в его соответствующий альдегид, который затем реагировал с 3,4,5-триметоксифенилмагния бромидом с образованием спирта 40f, который может быть легко отделен от 3,4,5,3',4',5'-гексаметоксибифенила с использованием колоночной флэш-хроматографии. Окисление дихроматом пиридиния (PDC) или ДМС не приводит к получению 1f из вторичного спирта 40f с хорошим выходом. Однако при использовании реактива периодинана Десс-Мартина в качестве окислителя, успешно образуется заданное кетоновое соединение 1f. 1e и 1i были получены из спиртов 40e и 40i с использованием аналогичного способа. Соединение 1g было получено по реакции связывания из пиперидина 41g и 3,4,5-триметоксибензойной кислоты.
Бензольное кольцо В:
Синтез бифенил-3-ил(3,4,5-триметоксифенил)метанона (1а) (Фигура 2)

N-Метокси-N-метилбифенил-3-карбоксамид (38а). К смеси 37a (5 ммоль), EDCI (6 ммоль), HOBt (5 ммоль) и NMM (11 ммоль) в CH2Cl2 (50 мл) добавили соль HNCH3OCH3HCl (5 ммоль) и продолжали перемешивание при комнатной температуре в течение 2 часов. Реакционную смесь разбавили CH2Cl2 (100 мл) и последовательно промыли водой, насыщенным раствором NaHCO3, насыщенным солевым раствором и высушили над MgSO4. Растворитель удалили под пониженным давлением для получения бесцветного маслянистого вещества, которое использовали на следующем этапе (58,4%). МС (ESI) m/z 264.0 (M + Na)+.
Бифенил-3-ил(3,4,5-триметоксифенил)метанон (1а). К раствору 38a (Фигура 2) (0,174 г, 0,72 ммоль) в 5 мл ТГФ добавили раствор 3,4,5-триметоксифенилмагния бромида в ТГФ (0,5 н., 1,08 ммоль) при 0oC. Смесь оставили перемешиваться на 30 минут и погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 1a в виде твердого белого вещества (43,8%). 1H ЯМР (CDCl3) δ 8.02 (t, 1 H), 7.84-7.74 (m, 2 H), 7.64-7.38 (m, 6 H), 7.11 (s, 2 H), 3.95 (s, 3 H), 3.88 (s, 6 H); МС (ESI) m/z 371.1(M + Na)+.
Пиримидиновое кольцо В:
Синтез (6-фенилпиримидин-4-ил)(3,4,5-триметоксифенил)метанона (1b) (Фигура 2)

N-Метокси-N-метил-6-фенилпиримидин-4-карбоксамид (38b). К смеси 37b (5 ммоль), EDCI (6 ммоль), HOBt (5 ммоль) и NMM (11 ммоль) в CH2Cl2 (50 мл) добавили соль HNCH3OCH3HCl (5 ммоль) и продолжали перемешивание при комнатной температуре в течение ночи. Реакционную смесь разбавили CH2Cl2 (100 мл) и последовательно промыли водой, насыщенным раствором NaHCO3, насыщенным солевым раствором и высушили над MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 38b в виде твердого желтого вещества (62,3%). 1H ЯМР (CDCl3) δ 9.28 (s, 1 H), 8.14-8.06 (m, 2 H), 7.96 (br, s, 1 H), 7.54-7.50 (m, 3 H), 5.35 (br, t, 1 H), 4.81 (br, t, 1 H), 4.52 (dd, 1 H, J = 8.7 Hz, J = 10.2 Hz), 3.79 (s, 3 H), 3.42 (s, 3 H); МС (ESI) m/z 266.0 (M + Na)+.
(6-Фенилпиримидин-4-ил)(3,4,5-триметоксифенил)метанон (1b). К раствору 38b (0,243 г, 1 ммоль) в 5 мл ТГФ добавили раствор 3,4,5-триметоксифенилмагния бромида в ТГФ (0,5 н., 5,6 мл, 1,4 ммоль) при 0oC. Смесь оставили перемешиваться на 30 минут и погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 1b (52,3%). 1H ЯМР (CDCl3) δ 9.40 (d, 1 H, J = 1.5 Hz), 8.29 (d, 1 H, J = 1.5 Hz), 8.22-8.18, 7.57-7.54 (m, 5 H), 7.46 (s, 2 H), 3.96 (s, 3 H), 3.91 (s, 6 H); МС (ESI) m/z 351.1(M + H)+.
Пиридиновое кольцо В:
Синтез (6-фенилпиридин-2-ил)(3,4,5-триметоксифенил)метанона (1с) (Фигура 2)

N-Метокси-N-метил-6-фенилпиколинамид (38с). К смеси 37c (1,77 ммоль), EDCI (2,12 ммоль), HOBt (1,86 ммоль) и NMM (3,54 ммоль) в CH2Cl2 (20 мл) добавили соль HNCH3OCH3HCl (1,86 ммоль) и продолжали перемешивание при комнатной температуре в течение ночи. Реакционную смесь разбавили CH2Cl2 (40 мл) и последовательно промыли водой, насыщенным раствором NaHCO3, насыщенным солевым раствором и высушили над MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 38c в виде бесцветного маслянистого вещества (51,2%). 1H ЯМР (CDCl3) δ 8.02 (d, 1 H, J = 7.0 Hz), 7.86-7.81 (m, 2 H), 7.55 (br, 1 H), 7.48 (t, 2 H), 7.44-7.41 (m, 1 H), 3.82 (s, 3 H), 3.44 (s, br, 3 H); МС (ESI) m/z 265.0 (M + Na)+.
(6-Фенилпиридин-2-ил)(3,4,5-триметоксифенил)метанон (1с). К раствору 38c (0,210 г, 0,86 ммоль) в 5 мл ТГФ добавили раствор 3,4,5-триметоксифенилмагния бромида в ТГФ (0,5 н., 3,5 мл, 1,73 ммоль) при 0 oC. Смесь оставили перемешиваться на 30 минут и погасили водой, экстрагировали этилацетатом и высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого 1c в виде белых игольчатых кристаллов (78%). 1H ЯМР (CDCl3 ) δ 8.10 (d, br, 2 H), 8.02-8.00 (m, 1 H), 7.97-7.96 (m, 2 H), 7.66 (s, 2 H), 7.49-7.43 (m, 3 H), 3.97 (s, 3 H), 3.89 (s, 6 H); МС (ESI) m/z 372.6 (M + Na)+.
Фурановое кольцо В:
Синтез (5-фенилфуран-2-ил)(3,4,5-триметоксифенил)метанона (1d) (Фигура 2)

N-Метокси-N-метил-5-фенилфуран-2-карбоксамид (38d). К смеси 37d (10 ммоль), EDCI (12 ммоль), HOBt (11 ммоль) и NMM (21 ммоль) в CH2Cl2 (200 мл) добавили соль HNCH3OCH3HCl (10,5 ммоль) и продолжали перемешивание при комнатной температуре в течение ночи. Реакционную смесь разбавили CH2Cl2 (200 мл) и последовательно промыли водой, насыщенным раствором NaHCO3, насыщенным солевым раствором и высушили над MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 38d. (95,2%). 1H ЯМР (CDCl3 ) δ 7.82 (d, 1 H, J = 7.0 Hz), 7.46-7.43 (t, 2 H), 7.37-7.34 (m, 1 H), 7.25 (d, 1 H, J = 4.0 Hz), 6.78 (d, 1 H, J = 4.0 Hz), 3.86 (s, 3 H), 3.41 (s, 3 H); МС (ESI) m/z 254.1 (M + Na)+.
(5-Фенилфуран-2-ил)(3,4,5-триметоксифенил)метанон (1d). К раствору 38d (0,231 г, 1 ммоль) в 5 мл ТГФ добавили раствор 3,4,5-триметоксифенилмагния бромида в ТГФ (0,5 н., 4,0 мл, 2 ммоль) при 0oC. Смесь оставили перемешиваться на 30 минут и погасили водой, экстрагировали этилацетатом и высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 1d в виде белых кристаллов (35,5%). 1H ЯМР (CDCl3) δ 7.85-7.82 (m, 1 H), 7.48-7.36 (m, 4 H), 7.35 (s, 2 H), 7.25 (d, 1 H, J = 4.0 Hz), 6.86 (d, 1 H, J = 4.2 Hz), 3.96 (s, 3 H), 3.95 (s, 6 H); МС (ESI) m/z 339.1 (M + H)+.
Тиазольное кольцо В:
Синтез of (2-фенилтиазол-5-ил)(3,4,5-триметоксифенил)метанона (1e) (Фигура 2)  (1e)
(1e)
(2-Фенилтиазол-5-ил)(3,4,5-триметоксифенил)метанол (40e). К раствору 2-фенилтиазол-5-карбальдегида 38e (0,567 г, 3 ммоль) в 15 мл ТГФ добавили раствор 3,4,5-триметоксифенилмагния бромида в ТГФ (0,5 н., 6,5 мл, 3,25 ммоль) при 0oC. Смесь оставили перемешиваться на 30 минут и погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 40e (72,9%). 1H ЯМР (CDCl3 ) δ 7.90 (m, 2 H), 7.64 (s, 1 H), 7.41 (m, 3 H), 6.69 (s, br, 2 H), 6.04 (s, 1 H), 3.86 (s, 6 H), 3.85 (s, 3 H), 1.57 (d, 1 H, J = 5.5 Hz); МС (ESI) m/z 358.1 (M + Na)+.
(2-Фенилтиазол-5-ил)(3,4,5-триметоксифенил)метанон (1e). К раствору 40e (0,357 г, 1 ммоль) в 40 мл безводного CH2Cl2 добавили реагент Десс-Мартина (0,848 г, 2 ммоль). Смесь оставили перемешиваться на 30 минут и погасили насыщенным раствором Na2S2O3, экстрагировали этилацетатом и высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 1e (80,1%). 1H ЯМР (CDCl3 ) δ 8.33 (s, 1 H), 8.04 (m, 2 H), 7.51 (m, 3 H), 7.18 (s, 2 H), 3.96 (s, 3 H), 3.93 (s, 6 H); МС (ESI) m/z 378.1 (M + H)+.
Тиофеновое кольцо В:
Синтез (5-фенилтиофен-3-ил)(3,4,5-триметоксифенил)метанона (1f) (Фигура 2)

N-Метокси-N-метил-5-фенилтиофен-3-карбоксамид (38f). К смеси 37f (2,5 ммоль), EDCI (2,9 ммоль), HOBt (2,6 ммоль) и NMM (5,3 ммоль) в CH2Cl2 (30 мл) добавили соль HNCH3OCH3HCl (2,6 ммоль) и продолжали перемешивание при комнатной температуре в течение ночи. Реакционную смесь разбавили CH2Cl2 (20 мл) и последовательно промыли водой, насыщенным раствором NaHCO3, насыщенным солевым раствором и высушили над MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 38f. (90,8%). 1H ЯМР (CDCl3 ) δ 8.28 (d, 1 H, J = 1.5 Hz), 7.69 (d, 1 H, J = 1.5 Hz), 7.64 (d, 2 H, J = 7.0 Hz), 7.44 (t, 2 H, J = 7.0 Hz), 7.35-7.32 (m, 1 H), 6.78 (d, 1 H, J = 4.0 Hz), 3.86 (s, 3 H), 3.41 (s, 3 H); МС (ESI) m/z 270.0 (M + Na)+.
(5-Фенилтиофен-3-ил)(3,4,5-триметоксифенил)метанол (40f). При -78oC, к раствору 38f (2,5 ммоль) в 5 мл ТГФ под защитой аргона добавили раствор LiAlH4 в ТГФ (1 н., 1,42 мл) и продолжали перемешивание в течение 1 часа при -20oC. Реакционную смесь поместили на ледяную баню и погасили 20% раствором H2SO4, экстрагировали этилацетатом и высушили над MgSO4. Растворитель удалили под пониженным давлением и очистили колоночной хроматографией для получения 5-фенилтиофен-3-карбальдегида (не показан) (84,8%). 1H ЯМР (CDCl3 ) δ 9.98 (s, 1 H), 8.04 (d, 1 H, J = 1.5 Hz), 7.86 (br, 1 H), 7.61-7.58 (br, 2 H), 7.47-7.33 (m, 3 H), 7.35-7.32 (m, 1 H), 6.78 (d, 1 H, J = 4.0 Hz); МС (ESI) m/z 210.9 (M + Na)+. К раствору 5-фенилтиофен-3-карбальдегида (0,195 г, 1,04 ммоль) в 5 мл ТГФ добавили раствор 3,4,5-триметоксифенилмагния бромида в ТГФ (0,5 н., 2,3 мл, 1,14 ммоль) при 0oC. Смесь оставили перемешиваться на 30 минут и погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 40f. (70,5%).1H ЯМР (CDCl3) δ 7.55-7.52 (m, 2 H), 7.40-7.35 (m, 3 H), 7.30 (br, 1 H), 7.20 (br, 1 H), 6.72 (s, 2 H), 6.01 (d, 1 H, J = 3.9 Hz), 3.86 (s, 6 H), 3.85 (s, 3 H), 2.42 (d, 1 H, J = 3.9 Hz); МС (ESI) m/z 339.1 (M - OH)-.
(5-Фенилтиофен-3-ил)(3,4,5-триметоксифенил)метанон (1f). К раствору 40f (0,260 г, 0,73 ммоль) в 20 мл безводного CH2Cl2 добавили реагент Десс-Мартина (0,465 г, 1,36 ммоль). Смесь оставили перемешиваться на 30 минут и погасили насыщенным раствором Na2S2O3, экстрагирвали этилацетатом и высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 1f в виде светло-желтых кристаллов (60,9%). 1H ЯМР (CDCl3 ) δ 7.97 (d, 1 H, J = 1.5 Hz), 7.82 (d, 1 H, J = 1.5 Hz), 7.59-7.57 (m, 2 H), 7.45-7.34 (m, 3 H), 7.19 (s, 2 H), 3.95 (s, 3 H), 3.93 (s, 6 H); МС (ESI) m/z 355.1 (M + H)+.
Пиперидиновое кольцо В:
Синтез (4-фенилпиперидин-1-ил)(3,4,5-триметоксифенил)метанона (1g) (Фигура 2)

(4-Фенилпиперидин-1-ил)(3,4,5-триметоксифенил)метанон (1g). К смеси 4-фенилпиперидина 41g (5 ммоль), EDCI (6 ммоль), HOBt (5,5 ммоль) и NMM (6 ммоль) в CH2Cl2 (50 мл) добавили 3,4,5-триметоксибензойную кислоту (5,3 ммоль) и продолжали перемешивание при комнатной температуре в течение ночи. Реакционную смесь разбавили CH2Cl2 (100 мл) и последовательно промыли водой, насыщенным раствором NaHCO3, насыщенным солевым раствором и высушили над MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 1g. (57,9%). 1H ЯМР (CDCl3) δ 7.35-7.21 (m, 5 H), 6.66 (s, 2 H), 4.84 (br, 1 H), 3.95 (br, 1 H), 3.88 (s, 6 H), 3.86 (s, 3 H), 3.20-2.87 (br, 2 H), 2.85-2.74 (tt, 1 H, J = 3.6 Hz, J = 15.6 Hz) 1.92 (br, 2 H), 1.70 (br, 2 H); МС (ESI) m/z 378.1 (M + Na)+.
Изоксазольное кольцо В:
Синтез (5-фенилизоксазол-3-ил)(3,4,5-триметоксифенил)метанона (1i) Фигура 2)

(5-Фенилизоксазол-3-ил)(3,4,5-триметоксифенил)метанол (40i). К раствору 5-фенилизоксазол-3-карбальдегида 38i (0,365 г, 2,1 ммоль) в 15 мл ТГФ добавили раствор 3,4,5-триметоксифенилмагния бромида в ТГФ (0,5 н., 5,5 мл, 2,74 ммоль) при 0oC. Смесь оставили перемешиваться на 30 минут и погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 40i в виде твердого белого вещества. (48,8%).1H ЯМР (CDCl3) δ 7.78-7.77 (m, 2 H), 7.48-7.46 (m, 3 H), 6.74 (s, 2 H), 6.45 (s, 1 H), 5.98 (d, 1 H, J = 3.5 Hz) 3.89 (s, 6 H), 3.86 (s, 3 H), 2.77 (d, 1 H, J = 3.5 Hz); МС (ESI) m/z 364.1 (M + Na)+.
(5-Фенилизоксазол-3-ил)(3,4,5-триметоксифенил)метанон (1i). К раствору 40i (0,110 г, 0,73 ммоль) в 8 мл безводного CH2Cl2 добавили реагент Десс-Мартина (0,274 г, 0,645 ммоль). Смесь оставили перемешиваться на 30 минут и погасили насыщенным раствором Na2S2O3, экстрагирвали этилацетатом и высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 1i (70,1%). 1H ЯМР (CDCl3 ) δ 7.87-7.85 (m, 2 H), 7.72 (s, 2 H), 7.53-7.49 (m, 3 H), 7.05 (s, 1 H), 7.82 (d, 1 H, J = 1.5 Hz), 3.97 (s, 3 H), 3.96 (s, 6 H); МС (ESI) m/z 362.1 (M + H)+.
Пиразольное кольцо В:
Синтез (3-фенил-1Н-пиразол-5-ил)(3,4,5-триметоксифенил)метанона (1k) (Фигура 2)
(3-Фенил-1H-пиразол-5-ил)(3,4,5-триметоксифенил)метанон (1k) получили с использованием такого же способа, который использовался для получения соединения 1c, из 3-фенил-1H-пиразол-5-карбоновой кислоты. 1H ЯМР (500MHz, CDCl3) δ 10.97 (br, 1 H), 7.77 (s, br, 2 H), 7.48- 7.38 (m, 5 H), 7.14 (s, br, 1 H), 3.96 (s, 3 H), 3.94 (s, 6 H); МС (ESI) m/z 361.1(M + Na)+, 337.0 (M - H)-.
ПРИМЕР 2
СИНТЕЗ СОЕДИНЕНИЙ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ, ИМЕЮЩИХ РАЗЛИЧНЫЕ ЛИНКЕРЫ Y
Соединения настоящего изобретения имеют различные линкеры Y. Такие соединения, с различными линкерами Y, были синтезированы в соответствии с Фигурами 3 и 4.
Соединение 1h синтезировали из 2-фенил-4,5-дихлор-тиазол-4-карбоновой кислоты 42a за три этапа, описанных ранее (Lu, Y.; Wang, Z.; Li, C. M.; Chen, J.; Dalton, J. T.; Li, W.; Miller, D. D., Synthesis, in vitro structure-activity relationship, and in vivo studies of 2-arylthiazolidine-4-carboxylic acid amides as anticancer agents. Bioorg Med Chem 2010, 18, (2), 477-95, который включен в настоящий документ путем ссылки в полном объеме). 1h преобразовали в оксимовые изомеры 2e-цис,транс и 2f-цис,транс по реакции с гидроксиламинами, NH2OH или NH2OCH3. Интерпретацию выполнили на основании химических и спектральных данных, как описано infra. В результате усовершенствованной перегруппировки Бекмана легко получили перегруппированные амиды 2g и 2h из двух геометрических стереоизомеров 2e-цис и 2e-транс по их реакции с тозилхлоридом с последующим пропусканием через колонку с основным оксидом алюминия. Гидразидные производные 2d-цис и 2d-транс получили смешиванием 1h с гидразингидратом в этаноле и дефлегмацией в течение 24 часов. Акрилонитрилы 2c-транс,цис получили по реакции Виттига 1h с диэтилцианометилфосфонатом. Цианоимин 2j получили с использованием способа, описанного Cuccia (Cuccia, S. J.; Fleming, L. B.; France, D. J., A novel and efficient synthesis of 4-phenyl-2-chloropyrimidines from acetophenone cyanoimines. Synthetic Communications 2002, 32, (19), 3011-3018., включенного в настоящий документ путем ссылки в полном объеме). Карбонильную группу в соединении 1h также восстановили до вторичного спирта 2b или преобразовали в алкен (2a), как показано на Фигуре 3.
Попытки удалить карбонильную группу между кольцами В и С в 1h привели к образованию соединения 2i, как показано на Фигуре 4. Внедрив цис- и транс- двойные связи в карбонильное положение, получили соединения (3a и 3b), которые синтезировали по реакции Виттига с 2-фенилтиазол-4-карбальдегидом. Сульфидное соединение 4a, сульфон 4b и сульфоксид 4c получили с использованием 3-аминобифенила в качестве исходного соединения, через первоначальную реакцию Зандмейера с образованием карбонодитиоата 52a, с последующей реакцией связывания, катализируемой CuI, и оксилением m-CPBA. Сульфонамид-связанное соединение 4d получили по реакции 3-бифенилсульфонил хлорида с 3,4,5-триметоксианилином в присутствии NEt3 в ДМФ.
Синтез (2-фенил-тиазол-4-ил)-(3,4,5-триметокси-фенил)-метанона (1h)
[Фигура 3]

(2-Фенил-тиазол-4-ил)-(3,4,5-триметокси-фенил)метанон (1h). Смесь 2-фенил-4,5-дигидротиазол-4-карбоновой кислоты (5 ммоль), EDCI (6 ммоль) и HOBt (5 ммоль) в CH2Cl2 (50 мл) перемешивали в течение 10 минут. К этому раствору добавили NMM (5 ммоль) и HNCH3OCH3 (5 ммоль) и продолжали перемешивание при комнатной температуре в течение 6-8 часов. Реакционную смесь разбавили CH2Cl2 (100 мл) и последовательно промыли водой, насыщенным раствором NaHCO3, насыщенным солевым раствором и высушили над MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения 2-фенил-4,5-дигидротиазол-4-карбоновой кислоты метоксиметиламида. Раствор 2-фенил-4,5-дигидротиазол-4-карбоновой кислоты метоксиметиламида (1 экв.) в CH2Cl2 охладили до 0oC и добавили дистиллированный DBU (2 экв.). Затем через шприц, по каплям ввели бромтрихлорметан (1,7 экв.) за 10 минут. Реакционную смесь оставили нагреваться до комнатной температуры и перемешивали в течение ночи. После промывания насыщенным водным раствором NH4Cl (2 Ч 50 мл), водную фазу экстрагировали EtOAc (3Ч 50 мл). Объединенные органические слои высушили на MgSO4, отфильтровали и концентрировали in vacuo. Остаток по необходимости очистили флэш-хроматографией, получив 2-фенил-тиазол-4-карбоновой кислоты метоксиметиламид (73,6%). 1H ЯМР (300MHz, CDCl3) δ 8.01 (s, 1 H), 7.99-7.96 (m, 2 H), 7.47-7.44 (m, 3 H), 3.88 (s, 3 H), 3.49 (s, 3 H). МС (ESI) m/z 271.0 (M + Na)+. К раствору 3,4,5-триметоксифенилмагния бромида (0,5 н., 3 мл) в 2 мл ТГФ добавили раствор 2-фенил-тиазол-4-карбоновой кислоты метоксиметиламида (1 ммоль) в 3 мл ТГФ при 0 oC. Смесь перемешивали в течение 30 минут до исчезновения амидов, по данным ТСХ пластин. Реакционную смесь погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 1h. Выход: 27,3 %. 1H ЯМР (300 MHz, CDCl3) δ 8.29 (s, 1 H), 8.03 (q, 2 H), 7.80 (s, 2 H), 7.49-7.47 (m, 3 H), 3.96 (s, 6 H), 3.97 (s, 3 H). МС (ESI) m/z 378.1 (M + Na)+.
Синтез 4-(2-метил-1-(3,4,5-триметоксифенил)проп-1-енил)-2-фенилтиазола (2а) [Фигура 3]

4-(2-Метил-1-(3,4,5-триметоксифенил)проп-1-енил)-2-фенилтиазол (2а) [Фигура 3]. При -78oC, к раствору 223 мг изопропил трифенилфосфония йодида (0,52 ммоль) в 5 мл ТГФ по каплям добавили 0,4 мл 1,6 н. n-BuLi в гексане под защитой Ar2. И перемешивали смесь при 0oC в течение 40 минут. По каплям добавили раствор 140 мг (0,39 ммоль) 1h в 5 мл ТГФ при 0oC и перемешивали смесь в течение 1 часа при комнатной температуре. Реакционную смесь обработали насыщенным раствором NH4Cl. После стандартного выделения продукта, колоночной хроматографии (силикагель, петролейный эфир/этилацетат) получили соединение 2a (86 мг, 57,3%). 1H ЯМР (300 MHz, CDCl3) δ 7.98-7.97 (m, 2 H), 7.45-7.40 (m, 3 H), 6.77 (s, 1 H), 6.48 (s, 2 H), 3.86 (s, 3 H), 3.82 (s, 6 H), 2.15 (s, 3 H), 1.81 (s, 3 H). МС (ESI) m/z 404.1 (M + Na)+.
Синтез (2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанола (2b) [Фигура 3]

2-Фенил-4,5-дигидротиазол-4-карбоновая кислота (42а). Бензонитрил (40 ммоль) смешали с L-цистеином (45 ммоль) в 100 мл смеси 1:1 MeOH/фосфатно-солевого буферного раствора с рН 6,4. Реакционную смесь перемешивали при 40oC в течение 3 дней. Осадок удалили фильтрацией, а MeOH удалили, используя ротационное испарение. Оставшемуся раствору добавили 1 М HCl, чтобы довести рН до 2, при 0oC. Полученный осадок отфильтровали для получения твердого белого вещества 2-фенил-4,5-дигидротиазол-4-карбоновой кислоты 42a, которую использовали непосредственно на следующем этапе без очистки.
2-Фенилтиазол-4-карбальдегид (42b). При -78oC, к раствору 2-фенил-тиазол-4-карбоновой кислоты метоксиметиламида (1 экв.) в ТГФ добавили LiAlH4 (1 экв., 1 н. в ТГФ) и перемешивали в течение 1 часа при -20oC. Реакционную смесь поместили на ледяную баню и погасили 20% раствором H2SO4, экстрагировали этилацетатом и высушили над MgSO4. Растворитель удалили под пониженным давлением и очистили колоночной хроматографией для получения 42b (45,8%). 1H ЯМР (300 MHz, CDCl3) δ 10.1 (s, 1 H), 8.17 (s, 1 H), 8.02-8.00 (m, 2 H), 7.50-7.48 (m, 3 H). МС (ESI) m/z 244.1 (M + Na + MeOH)+.
(2-Фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанол (2b) [Фигура 3]. При 0oC, к раствору 104 мг 42b (0,55 ммоль, 1 экв.) в 6 мл ТГФ добавили 3,4,5-триметоксифенилмагния бромид (0,5 н. в ТГФ, 2,9 мл). Смесь перемешивали в течение 30 минут до исчезновения альдегида, по данным ТСХ пластин. Реакционную смесь погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения (2b). 1H ЯМР (300 MHz, CDCl3) δ 7.95-7.92 (m, 2 H), 7.44-7.43 (m, 4 H), 6.97 (s, 1 H), 6.76 (s, 2 H), 5.93 (d, 1 H, J = 3.6 Hz), 3.86 (s, 9 H). МС (ESI) m/z 402.1 (M + Na)+.
Синтез (Z)-3-(2-фенилтиазол-4-ил)-3-(3,4,5-триметоксифенил)акрилонитрила (2c-транс) и (E)-3-(2-фенилтиазол-4-ил)-3-(3,4,5-триметоксифенил)акрилонитрила (2c-цис) [Фигура 3]


(2c-цис) (2c-транс)
(Z)-3-(2-фенилтиазол-4-ил)-3-(3,4,5-триметоксифенил)акрилонитрил (2c-транс). К раствору 0,4 мл 2,5 н. n-BuLi в гексане и 10 мл ТГФ по каплям добавили раствор 177 мг (1 ммоль) диэтилцианометилфосфоната в 5 мл ТГФ при 0oC под Ar2. Ледяную баню убрали, а смесь перемешивали смесь при 25oC в течение 40 минут. По каплям добавили раствор 200 мг (0,56 ммоль) 1h в 10 мл ТГФ при 0oC и перемешивали смесь в течение 1 часа при комнатной температуре. Реакционную смесь обработали насыщенным раствором NH4Cl. После стандартного выделения продукта, колоночной хроматографии (силикагель, петролейный эфир/этилацетат) получили соединения 2c-транс (83 мг) и 2c-цис (76 мг). 1H ЯМР (300 MHz, CDCl3) δ 8.01-7.99 (m, 2 H), 7.44-7.40 (m, 3 H), 7.21 (s, 1 H), 6.74 (s, 2 H), 6.67 (s, 1 H), 3.93 (s, 3 H), 3.89 (s, 6 H). МС (ESI) m/z 401.1 (M + Na)+.
(E)-3-(2-фенилтиазол-4-ил)-3-(3,4,5-триметоксифенил)акрилонитрил (2c-цис). 1H ЯМР (300 MHz, CDCl3) δ 8.07-8.05 (m, 2 H), 7.49-7.46 (m, 4 H), 6.66 (s, 2 H), 5.64 (s, 1 H), 3.91 (s, 3 H), 3.86 (s, 6 H). МС (ESI) m/z 401.1 (M + Na)+.
Синтез (Z)-4-(гидразоно(3,4,5-триметоксифенил)метил)-2-фенилтиазола (2d-цис) и (E)-4-(гидразоно(3,4,5-триметоксифенил)метил)-2-фенилтиазола (2d-транс) [Фигура 3]


(2d-цис) (2d-транс)
(Z)-4-(гидразоно(3,4,5-триметоксифенил)метил)-2-фенилтиазол (2d-цис). К смеси 1h (230 мг, 0,65 ммоль) в 3 мл CH2Cl2 в 3 мл этанола добавили гидразингидрат (2 мл). Затем смесь дефлегмировали в течение ночи. После завершения реакции остаток абсорбировали на силикагель и очистили колоночной хроматографией для получения соединений 2d-цис (80 мг) и 2d-транс (56 мг). 1H ЯМР (300 MHz, CDCl3) δ 8.01-7.98 (m, 2 H), 7.49-7.46 (m, 5 H), 7.33 (s, 1 H), 6.82 (s, 2 H), 3.87 (s, 3 H), 3.85 (s, 6 H). МС (ESI) m/z 370.1 (M + H)+.
(E)-4-(гидразоно(3,4,5-триметоксифенил)метил)-2-фенилтиазол (2d-транс). 1H ЯМР (300 MHz, CDCl3) δ 8.04-8.01 (m, 2 H), 7.44-7.40 (m, 3 H), 6.95 (s, 1 H), 6.65 (s, 2 H), 5.62 (s, 2 H), 3.93 (s, 3 H), 3.87 (s, 6 H). МС (ESI) m/z 370.1 (M + H)+.
Синтез (Z)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон оксима (2e-цис) и (E)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон оксима (2e-транс) [Фигура 3]


(2e-цис) (2e-транс)
(Z)-(2-Фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон оксим (2e-цис). К суспензии 1h (210 мг, 0,59 ммоль) в 10 мл этанола добавили водный раствор (2 мл) гидроксиламина гидрохлорида (127 мг, 1,83 ммоль). Затем к реакционной смеси по каплям добавили 2 мл 1 н. NaOH и перемешивали смесь при 55oC в течение 3 часов. После завершения реакции остаток абсорбировали на силикагель и очистили колоночной хроматографией для получения соединений 2e-цис (85 мг) и 2e-транс (50 мг). 1H ЯМР (300 MHz, ДМСО-d6) δ 11.95 (s, 1 H), 8.35 (s, 1 H), 7.91-7.89 (m, 2 H), 7.50-7.44 (br, 3 H), 6.85 (s, 2 H), 3.73 (s, 6 H), 3.70 (s, 3 H). МС (ESI) m/z 393.1 (M + Na)+; 368.9 (M – H)-.
(E)-(2-Фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон оксим (2e-транс). 1H ЯМР (300 MHz, ДМСО-d6) δ 11.49 (s, 1 H), 7.92-7.89 (m, 2 H), 7.64 (s, 1 H), 7.51-7.49 (m, 3 H), 7.34 (s, 1 H), 6.75 (s, 2 H), 3.75 (s, 6 H), 3.72 (s, 3 H). МС (ESI) m/z 393.1 (M + Na)+; 368.9 (M – H)-.
Синтез (Z)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон О-метилоксима (2f-цис) и (E)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон О-метилоксима (2f-транс) [Фигура 3]


(2f-цис) (2f-транс)
(Z)-(2-Фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон О-метилоксим (2f-цис). К суспензии 1h (110 мг, 0,59 ммоль) в 10 мл пиридина добавили О-метилгидроксиламина гидрохлорид (52 мг, 0,63 ммоль) и перемешивали смесь при 60oC в течение ночи. Реакцию погасили 1 н. раствором HCl, экстрагировали этилацетатом и высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистых соединений 2f-цис (41 мг) и 2f-транс (33 мг). 1H ЯМР (500 MHz, CDCl3) δ 8.13 (s, 1 H), 7.96-7.94 (m, 2 H), 7.45-7.44 (m, 3 H), 6.94 (s, 2 H), 4.13 (s, 3 H), 3.91 (s, 6 H), 3.88 (s, 3 H). МС (ESI) m/z 407.2 (M + Na)+.
(E)-(2-Фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон О-метилоксим (2f-транс). 1H ЯМР (500 MHz, CDCl3) δ 8.00-7.98 (m, 2 H), 7.44-7.43 (m, 3 H), 7.28 (s, 1 H), 6.70 (s, 2 H), 4.08 (s, 3 H), 3.91 (s, 6 H), 3.85 (s, 3 H). МС (ESI) m/z 407.0 (M + Na)+.
Синтез 2-фенил-N-(3,4,5-триметоксифенил)тиазол-4-карбоксамида (2g)
[Фигура 3]

2-Фенил-N-(3,4,5-триметоксифенил)тиазол-4-карбоксамид (2g). К раствору 2e-цис (21 мг, 0,06 ммоль) в 5 мл CH2Cl2 добавили п-толуолсульфонилхлорид (23 мг, 0,12 ммоль) и NaH (5 мг, 60% в светлом минеральном масле). Затем реакционную смесь перемешивали в течение 20 минут. После завершения реакции остаток абсорбировали на силикагель и очистили колоночной хроматографией на Al2O3 для получения соединения 2g (15 мг). 1H ЯМР (300 MHz, CDCl3) δ 9.22 (s, 1H), 8.19 (s, 1 H), 8.02-7.99 (m, 2 H), 7.52-7.50 (m, 3 H), 7.07 (s, 2 H), 3.92 (s, 6 H), 3.85 (s, 3 H). МС (ESI) m/z 371.1 (M + H)+.
Синтез 3,4,5-триметокси-N-(2-фенилтиазол-4-ил)бензамида (2h) [Фигура 3]

3,4,5-Триметокси-N-(2-фенилтиазол-4-ил)бензамид (2h). К раствору 2e-транс (26 мг, 0,07 ммоль) в 5 мл CH2Cl2 добавили п-толуолсульфонилхлорид (27 мг, 0,14 ммоль) и NaH (5 мг, 60% в светлом минеральном масле). Затем реакционную смесь перемешивали в течение 20 минут. После завершения реакции остаток абсорбировали на силикагель и очистили колоночной хроматографией на Al2O3 для получения соединения 2h (15 мг). 1H ЯМР (300 MHz, CDCl3) δ 8.88 (s, 1H), 7.94-7.91 (m, 2 H), 7.83 (s, 1 H), 7.48-7.46 (m, 3 H), 7.18 (s, 2 H), 3.97 (s, 6 H), 3.94 (s, 3 H). МС (ESI) m/z 393.1 (M + Na)+.
Синтез N-((2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метилен)цианамида (2j) [Фигура 3]

N-((2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метилен)цианамид (2j). 100 мг 1h (0,28 ммоль, 1 экв.) растворили в 10 мл метиленхлорида. По каплям добавили тетрахлорид титана в метиленхлориде (1,0 н., 0,7 мл, 2,5 экв.) при 0oC и перемешивали в течение 30 минут. Добавили бис-триметилсилилкарбодиимид (2,4 экв.) в 2 мл метиленхлорида и перемешивали в течение ночи реакционную смесь, защищенную от воздуха и влаги. Реакционную смесь обработали смесью льда и воды, а затем экстрагировали метиленхлоридом. Органическую фазу высушили над сульфатом магния, отфильтровали через целит и концентрировали для получения неочищенных цианоиминов ацетофенона, которые очистили флэш-колонкой в виде изомеров с соотношением 3:7. 1H ЯМР (300 MHz, CDCl3) δ 8.72 (br, 0.3 H), 8.63 (s, 0.7 H), 8.09-8.07 (m, 1.4 H), 7.99 (br, 0.6 H), 7.58-7.56 (br, 3 H), 7.26 (s, 1.4 H), 7.18 (s, 0.6 H), 3.84, 3.83 (s, s, 6 H), 3.82 (s, 3 H). МС (ESI) m/z 402.1 (M + Na)+.
Синтез N-((4-гидрокси-3,5-диметоксифенил)(2-фенилтиазол-4-ил)метилен)цианамида (32).

N-((4-гидрокси-3,5-диметоксифенил)(2-фенилтиазол-4-ил)метилен)цианамид (32) получили как побочный продукт синтеза 2j. 1H ЯМР (500MHz, CDCl3 ) δ 8.23 (s, 1 H), 8.02 (m, 2 H), 7.92 (s, 2 H), 7.55 (m, 3 H), 6.02 (s, 1 H), 3.99 (s, 6 H). МС (ESI) m/z 364.1(M + H)+.
Синтез (Z)-2-фенил-4-(3,4,5-триметоксистирил)тиазола (3а) и (E)-2-фенил-4-(3,4,5-триметоксистирил)тиазола (3b) [Фигура 4]


(3a) (3b)
(Z)-2-Фенил-4-(3,4,5-триметоксистирил)тиазол (3а). Трифенилфосфин (3,41 г, 13 ммоль) добавили к раствору 5-(бромметил)-1,2,3-триметоксибензола (2,61 г, 10 ммоль) в сухом ТГФ (30 мл). Смесь дефлегмировали при перемешивании в течение 6 часов. Полученное твердое белое вещество отфильтровали и промыли эфиром/гексаном для получения продукта 3,4,5-триметоксибензилтрифенилфосфония бромида с выходом 96,4%. 1H ЯМР (500 MHz, CDCl3) δ 7.77-7.73, 7.65-7.61 (m, 15 H), 6.44 (d, 2 H, J = 1.5 Hz), 5.37 (d, 2 H, J = 14 Hz), 3.76 (s, 3 H), 3.51 (d, 6 H); МС (ESI) m/z 443.1 (M - Br]+. При -78oC, n-BuLi (0,42 мл, 2,5 н. в гексане) добавили к раствору 3,4,5-триметоксибензилтрифенилфосфония бромида (500 мг, 0,96 ммоль) в 10 мл ТГФ. После перемешивания при комнатной температуре в течение 2 часов добавили альдегид 42b (109 мг, 0,58 ммоль) в 3 мл ТГФ и перемешивали в течение 30 минут. Реакционную смесь обработали насыщенным раствором NH4C1. После стандартного выделения продукта, колоночной хроматографии (силикагель, петролейный эфир/этилацетат) получили соединения 3a (57 мг) и 3b (99 мг). 1H ЯМР (500 MHz, CDCl3) δ 7.90-7.89 (m, 2 H), 7.42-7.40 (m, 3 H), 7.07 (s, 1 H), 6.71 (s, 2 H), 6.66 (s, 1 H), 3.87 (s, 6 H), 3.75 (s, 3 H); МС (ESI) m/z 376.1 (M + Na)+.
(E)-2-Фенил-4-(3,4,5-триметоксистирил)тиазол (3b). 1H ЯМР (500 MHz, CDCl3) δ 8.03-8.01 (m, 2 H), 7.52 (d, 1 H, J = 16 Hz), 7.47-7.44 (m, 3 H), 7.16 (s, 1 H), 7.05 (d, 1 H, J = 16 Hz), 6.79 (s, 2 H), 3.92 (s, 6 H), 3.88 (s, 3 H). МС (ESI) m/z 354.1 (M + H)+.
Синтез бифенил-3-ил(3,4,5-триметоксифенил)сульфана (4а), 3-(3,4,5-триметоксифенилсульфонил)бифенила (4b) и 3-(3,4,5-триметоксифенилсульфинил)бифенила (4с) [Фигура 4]



(4a) (4b) (4c)
S-Бифенил-3-ил О-этил карбонодитиоат (52а). К раствору 1 экв. бифенил-3-амина (1 г, 5,92 ммоль) в воде (7,3 мл) при 0°C добавили концентрированную хлороводородную кислоту (1 мл). Медленно добавили холодный раствор 1,1 экв. нитрита натрия (450 мг, 6,5 ммоль) в воде (3 мл) и перемешивали в течение 15 минут. Холодный раствор диазония медленно добавили к раствору 1,3 экв. этилксантата калия (1,16 г, 1,3 ммоль) в воде (1,3 мл) при 45°C. Реакционную смесь перемешивали еще 30 минут при 45°C, а затем охладили до комнатной температуры. Реакционную смесь экстрагировали диэтиловым эфиром (3 х 50 мл). Объединенные органические экстракты промыли 1 н. раствором NaOH (100 мл), водой (3 мл х 50 мл), насыщенным солевым раствором (50 мл), высушили над MgSO4, отфильтровали и выпарили под пониженным давлением. Полученный неочищенный ксантат 52a использовали непосредственно на следующем этапе без дополнительной очистки. МС (ESI) m/z 275.0 (M + H)+.
Бифенил-3-ил(3,4,5-триметоксифенил)сульфан (4а). К раствору 52a (1,1 г, неочищенное соединение) в этаноле (8 мл) добавили гидроксид калия (2,1 г, 12 мл) и нагревали с дефлегматором в течение ночи. Раствор охладили до комнатной температуры и выпарили этанол под пониженным давлением. Остаток растворили в воде и промыли диэтиловым эфиром (10 мл). Водный слой подкислили 2 н. HCl и экстрагировали диэтиловым эфиром (3 х 50 мл). Органические экстракты промыли водой (50 мл), насыщенным солевым раствором (50 мл), высушили над MgSO4, отфильтровали и выпарили под пониженным давлением для получения 0,85 г (77,3%) неочищенного продукта бифенил-3-тиола (в целом, за 3 этапа). В круглодонную колбу с магнитной мешалкой поместили 0,1 г (1,04 ммоль) трет-бутоксида натрия и 83 мг йодида меди (0,43 ммоль). После герметизации реакционного сосуда через септу влили 0,13 г (0,71 ммоль) 4-метоксибензолтиола и 0,19 г (0,65 ммоль) 5-йод-1,2,3-триметоксибензола в 3,0 мл толуола. Реакционную смесь нагревали в течение ночи при 110oC. Очистку выполнили флэш-хроматографией и получили аморфное твердое вещество (выход 40%). 1H ЯМР (500 MHz, CDCl3) δ 7.54-7.52 (m, 3 H), 7.44-7.41 (m, 3 H), 7.37-7.33 (m, 2 H), 7.23 (s, br, 1 H), 6.69 (s, 2 H), 3.86 (s, 3 H), 3.80 (s, 6 H). МС (ESI) m/z 353.2 (M + H)+.
3-(3,4,5-Триметоксифенилсульфонил)бифенил (4b). К раствору 60 мг (0,17 ммоль) соединения 4a и 5 мл дихлорметана очень медленно добавили 2 экв. m-CPBA за 3 часа. Образование сульфоксида контролировали по тонкослойной хроматографии. Очистку выполнили флэш-хроматографической колонкой и получили аморфный порошок соединения (4b) (выход 73%). 1H ЯМР (500 MHz, CDCl3) δ 8.14 (br, 1 H), 7.89 (d, 1 H), 7.78 (d, 1 H), 7.59-7.56 (m, 3 H), 7.49-7.39 (m, 3 H), 7.19 (s, 2 H), 3.89 (s, 6 H), 3.87 (s, 3 H). МС (ESI) m/z 385.0 (M + Na)+.
3-(3,4,5-Триметоксифенилсульфинил)бифенил (4с). При 0oC, к раствору 500 мг (1,42 ммоль) соединения (4a) и 5 мл дихлорметана очень медленно добавили 1 экв. m-CPBA за 3 часа. Образование сульфоксида контролировали по тонкослойной хроматографии. Очистку выполнили флэш-хроматографической колонкой и получили аморфный порошок соединения (4с) (выход 87%). 1H ЯМР (500 MHz, CDCl3) δ 7.92 (br, 1 H), 7.71 (d, 2 H), 7.62-7.60 (m, 3 H), 7.58-7.40 (m, 4 H), 6.94 (s, 2 H), 3.79 (s, 3 H), 3.74 (s, 6 H). МС (ESI) m/z 369.1 (M + H)+.
Синтез N-(3,4,5-триметоксифенил)бифенил-3-сульфонамида (4d) [Фигура 4]

N-(3,4,5-Триметоксифенил)бифенил-3-сульфонамид (4d). Смесь 65 мг бифенил-3-сульфонилхлорида (0,25 ммоль), 44 мг 3,4,5-триметоксианилина (0,24 ммоль) и 0,3 ммоль триэтиламина в 5 мл ДМФ перемешивали в течение ночи. Реакционную смесь обработали водой и экстрагировали этилацетатом. После стандартного выделения продукта, колоночной хроматографии (силикагель, петролейный эфир/этилацетат) получили 88 мг соединения (4d) (91,7%). 1H ЯМР (500 MHz, CDCl3) δ 7.96 (t, 1 H, J = 1.8 Hz), 7.81-7.74 (m, 2 H), 7.57-7.40 (m, 6 H), 6.33 (s, 2 H), 3.86 (s, 3 H), 3.80 (s, 6 H). МС (ESI) m/z 422.1 (M + Na)+.
2-Фенил-4-(3,4,5-триметоксифенил)тиазол (2i) [Фигура 4]

2-Фенил-4-(3,4,5-триметоксифенил)тиазол (2i). Бром (160 мг, 1 ммоль) по каплям добавили к перемешанному раствору 1-(3,4,5-триметоксифенил)этанона (210 мг, 1 ммоль) в этаноле (30 мл) и перемешивали раствор при 0oC в течение 1 часа, а затем вылили в воду для образования осадка. Его перекристаллизовали из этанола для получения бромацетофенона (70%) и использовали непосредственно на следующем этапе. Смесь бромацетофенона (288 мг, 1 ммоль) и бензотиоамида (137 мг, 1 ммоль) в этаноле дефлегмировали в течение 1 часа. Реакционную смесь концентрировали in vacuo и очистили флэш-колонкой для получения 2i (167 мг, 51,1%). 1H ЯМР (500 MHz, CDCl3) δ 8.05-8.03 (m, 2 H), 7.48-7.44 (m, 3 H), 7.41 (s, 1 H), 7.22 (s, 2 H), 3.97 (s, 6 H), 3.89 (s, 3 H). МС (ESI) m/z 350.1 (M + Na)+.
ПРИМЕР 3
СИНТЕЗ МЕТОКСИ-БЕНЗОИЛ-ТИАЗОЛЬНЫХ СОЕДИНЕНИЙ, ИМЕЮЩИХ РАЗНЫЕ КОЛЬЦА «А» И/ИЛИ ЗАМЕЩЕННОЕ КОЛЬЦО «А»
Соединения настоящего изобретения имеют различные замещенные или незамещенные кольца А, такие как фенил или индолил. Такие соединения были синтезированы в соответствии с Фигурами 5 и 6.
Гидроксил и аминометил внедрили в пара-положение фенилового кольца А, а также заменили фенил на 5-индолиловое и 2-индолиловое кольца. Амиды Вайнреба 57a, 61a, 65a и 67a получили по способу, представленному на Фигуре 5, с использованием арилнитрилов в качестве исходных материалов. 2-Циано-индол 60a получили в соответствии со стандартным способом (Pletnev, A. A.; Tian, Q.; Larock, R. C., Carbopalladation of nitriles: synthesis of 2,3-diarylindenones and polycyclic aromatic ketones by the Pd-catalyzed annulation of alkynes and bicyclic alkenes by 2-iodoarenenitriles. J Org Chem 2002, 67(26), 9276-87; влюченным в настоящий документ путем ссылки в полном объеме). При получении использовали защиту гидроксильной (TBDMSCl), индолиловой (PhSO2Cl) и амино-групп (Boc2O). Снятие защиты TBDMS и окисление из тиазолина (58a) до тиазола (2l) происходило в одну стадию с использованием раствора TBAF/ТГФ. Это тиазолин-тиазоловое окисление происходило самопроизвольно в реакции тиазолинового амида Вайнреба и реактива Гриньяра. Такое же явление наблюдали при получении индоловых соединений 62a и 66a.
Соединение 62a выделили в виде чистого тиазолового соединения после реакции с 3,4,5-триметоксифениллитием без необходимости в дальнейшем окислении. Соединение 66a получили удалением фенилсульфониловых защитных групп в горячем растворе NaOH в этаноле. пара-OH и NH2 в кольце А соединений 2l и 2r получили по таким же реакциям Гриньяра из амидов Вайнреба 58a и 68a. Соединение 2r затем преобразовали в HCl соль (2r-HCl) и HCl соль монометиламина 2s-HCl, используя условия NaH/MeI и диметиламин 2u в условиях HCHO/NaBH3CN.
Замещенное кольцо А:
Синтез (2-(4-гидроксифенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (2l) [Фигура 5]

(R)-2-(4-Гидроксифенил)-N-метокси-N-метил-4,5-дигидротиазол-4-карбоксамид (57a) синтезировали по такому же способу, который использовался для 38d. Количественный выход. 1H ЯМР (500 MHz, CDCl3) δ 7.56 (d, 2 H, J = 8.5 Hz), 6.84 (br, 1 H), 6.73 (d, 2 H, J = 8.5 Hz), 5.64 (t, br, 1 H), 3.87 (s, 3 H), 3.30 (s, 3 H). МС (ESI) m/z 289.0 (M + Na)+, 264.9 (M -H)-.
(R)-(2-(4-(трет-Бутилдиметилсилилокси)фенил)-4,5-дигидротиазол-4-ил)(3,4,5-триметоксифенил)метанон (58a) синтезировали по такому же способу, который использовался для (35a) - смотри Пример 1. Выход 67,0%. 1H ЯМР (300 MHz, CDCl3) δ 7.73 (d, 2 H, J = 8.7 Hz), 7.61 (s, 2 H), 6.83 (d, 2 H, J = 8.7 Hz), 5.95 (dd, 1 H, J = 8.1 Hz, 9.0 Hz), 4.09, (dd, 1 H, J = 7.8 Hz, 11.1 Hz), 3.95 (s, 3 H), 3.94 (s, 6 H), 3.55 (dd, 1 H, J = 9.3 Hz, 11.1 Hz), 0.97 (s, 9 H), 0.19 (s, 6 H). МС (ESI) m/z 510.4 (M + Na)+, 486.0 (M -H)-.
(2-(4-Гидроксифенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (2l). При 0oC, к раствору 58a (0,2 ммоль) в 5 мл CH2Cl2 добавили раствор тетрабутиламмония фторида в ТГФ (1 н., 0,6 ммоль) и перемешивали при комнатной температуре около 14 часов до завершения реакции, по данным ТСХ. Выход 67,0%. 1H ЯМР (500 MHz, ДМСО-d6) δ 10.1 (s, 1 H), 8.51 (s, 1 H), 7.85 (d, 2 H, J = 8.50 Hz), 7.62 (s, 2 H), 6.91 (d, 2 H, J = 8.5 Hz), 3.86 (s, 6 H), 3.79 (s, 3 H). МС (ESI) m/z 394.1 (M + Na)+, 369.9 (M -H)-.
(2-(4-(Аминометил)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлорид (2r или 2r-HCl) [Фигура 5]

(R)-трет-Бутил 4-(4-(метокси(метил)карбамоил)-4,5-дигидротиазол-2-ил)бензил карбамат (67a). 4-(Аминометил)бензонитрил (25,09 г, 0,149 моль) и L-цистеин (18,1 г, 0,149 моль) суспендировали в 500 мл MeOH и раствора буфера с рН 6,4 (1:1) и перемешивали в течение 3 дней при комнатной температуре. К смеси добавили триэтиламин (30 мл), а к этой смеси добавили Boc2O (68 г, 0,31 ммоль) и перемешивали в течение 2 часов. Растворители удалили и отфильтровали для получения твердой белой (R)-2-(4-((трет-бутоксикарбониламино)метил)фенил)-4,5-дигидротиазол-4-карбоновой кислоты (38,4 г, 76,8%). Соединение 67a получили из этой кислоты по такому же способу, как использовался для 38d. Выход: 84,4 %. 1H ЯМР (500 MHz, CDCl3) δ 7.75 - 7.77 (d, 2 H, J = 7.5 Hz), 7.27 - 7.26 (d, 2 H, J = 7.5 Hz), 7.23 (s, 1 H), 5.62 (br, 1 H), 4.87 (br, 1 H), 4.30 (br, 2 H), 3.86 (s, 3 H), 3.78 (t, J = 10.0 Hz, 1 H), 3.48 - 3.4 (m, 1 H), 3.25 (s, 3 H), 1.42 (s, 9 H). МС (ESI) m/z 402.1(M + Na)+, 378.0 (M - H)-.
трет-Бутил 4-(4-(3,4,5-триметоксибензоил)тиазол-2-ил)бензилкарбамат (68а). Смесь 67a (2,5 ммоль), CBrCl3 (3,2 ммоль) и DBU (5,0 ммоль) в CH2Cl2 (20 мл) перемешивали в течение ночи. Реакционную смесь абсорбировали на силикагель и очистили колоночной хроматографией для получения промежуточного тиазолового амида Вайнреба. К раствору (3,4,5-триметоксифенил)магния бромида (0,5 М, 5,5 мл) в ТГФ добавили раствор промежуточного тиазолового амида Вайнреба (1,83 ммоль) в 10 мл ТГФ при 0oC и перемешивали в течение 30 минут. Реакционную смесь погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения в виде твердого светло-желтого вещества (32,3%). 1H ЯМР (300M, CDCl3) δ 8.27 (s, 1 H), 7.98 (d, 2 H, J = 8.1 Hz), 7.78 (s, 2 H), 7.39 (d, 2 H, J = 8.1 Hz), 7.27 - 7.26 (d, 2 H, J = 7.5 Hz), 7.23 (s, 1 H), 4.93 (br, 1 H), 4.37 (br, d, 1 H), 3.96 (s, 3 H), 3.95 (s, 6 H), 1.47 (s, 9 H); МС (ESI) m/z 507.1(M + Na)+.
(2-(4-(Аминометил)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлорид (2r или 2r-HCl). При 0 oC, к раствору 68a (200 мг) в 10 мл CH2Cl2 добавили раствор HCl в 1,4-диоксане (4 н., 2 мл) и перемешивали при комнатной температуре в течение 4 часов. Осадок (2r) отфильтровали и промыли диэтиловым эфиром. Выход: 81,3%. 1H ЯМР (500 MHz, ДМСО-d6) δ 8.68 (s, 1 H), 8.38 (br, 3 H), 8.10 (d, 2 H, J = 8.4 Hz), 7.66 (d, 2 H, J = 8.4 Hz), 7.62 (s, 2 H), 4.11 (s, 2 H), 3.87 (s, 6 H), 3.80 (s, 3 H). МС (ESI) m/z 385.1 (M + H)+.
(2-(4-((Диметиламино)метил)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлорид (2u или 2u-HCl) [Фигура 5]

трет-Бутил метил(4-(4-(3,4,5-триметоксибензоил)тиазол-2-ил)бензил)карбамат (71а). При 0oC, к раствору соединения 68a (100 мг, 0,2 ммоль) в 5 мл ДМФ добавили гидрид натрия (10 мг, 0,2 ммоль), затем к реакционной смеси добавили йодметан (77 мг, 0,4 ммоль) и перемешивали при комнатной температуре в течение ночи. Смесь погасили насыщенным раствором NaHCO3, экстрагирвали этилацетатом и высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 71a. Выход: 61,3%. 1H ЯМР (500 MHz, ДМСО-d6) δ 8.30 (s, 1 H), 8.02 (d, 2 H, J = 8.0 Hz), 7.82 (s, 2 H), 7.36 (br, 2 H), 4.50 (s, 2 H), 4.00 (s, 3 H), 3.98 (s, 6 H), 2.90 (d, br, 3 H), 1.50 (s, 9 H). МС (ESI) m/z 521.2 (M + Na)+, 496.9 (M – H)-.
(2-(4-((Метиламино)метил)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлорид (2s или 2s-HCl). При 0oC, к раствору 71a (60 мг) в 5 мл CH2Cl2 добавили раствор HCl в 1,4-диоксане (4 н., 2 мл) и перемешивали при комнатной температуре в течение ночи. Осадок (2s-HCl) отфильтровали и промыли диэтиловым эфиром. Выход: 81,3%. 1H ЯМР (500 MHz, CDCl3) δ 10.0 (s, 1 H), 8.29 (s, 1 H), 8.05 (d, 2 H, J = 6.0 Hz), 7.74 (s, 2 H), 7.72 (d, 2 H, J = 6.0 Hz), 4.15 (s, 2 H), 3.99 (s, 3 H), 3.96 (s, 6 H), 2.61 (s, 3 H). МС (ESI) m/z 399.1 (M + H)+.
(2-(4-((Диметиламино)метил)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлорид (2u или 2u-HCl). К раствору 2r (53 мг, 0,14 ммоль) в 5 мл CH2Cl2 добавили раствор формальдегида (37% в H2O, 340 мг, 4,2 ммоль) и цианоборгидрид натрия (34 мг, 0,55 ммоль), реакционную смесь абсорбировали на силикагель и очистили свободное основание после флэш-колонки (41 мг, 70,9%). При 0oC, к раствору свободного основания (41 мг) в 5 мл CH2Cl2 добавили раствор HCl в 1,4-диоксане (4 н., 2 мл) и перемешивали при комнатной температуре в течение ночи. Осадок (2u) отфильтровали и промыли диэтиловым эфиром. Выход: 71,3%. 1H ЯМР (500 MHz, CDCl3) δ 13.0 (s, 1 H), 8.34 (s, 1 H), 8.13 (d, 2 H, J = 7.0 Hz), 7.82 (d, 2 H, J = 7.5 Hz), 7.75 (s, 2 H), 4.24 (s, 2 H), 3.99 (s, 3 H), 3.97 (s, 6 H), 2.83 (s, 6 H). МС (ESI) m/z 413.1 (M + H)+.
2-(4-(4-(3,4,5-Триметоксибензоил)тиазол-2-ил)фенил)ацетонитрил (2n)

2-(4-(4-(3,4,5-Триметоксибензоил)тиазол-2-ил)фенил)ацетонитрил (2n) получили по такому же способу, как использовался для получения соединения 1h, из терефталонитрила и цистеина. 1H ЯМР (500MHz, CDCl3 ) δ 8.30 (s, 1 H), 8.04 (d, 2 H), 7.76 (s, 2 H), 7.46 (d, 2 H), 3.97 (s, 3 H), 3.95 (s, 6 H), 3.83 (s, 2 H).
Синтез (2-(4-(диметиламино)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (2o)

2-(4-(Диметиламино)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (2o) получили по такому же способу, как использовался для получения соединения 1h, из 4-(диметиламино)бензонитрила и цистеина. 1H ЯМР (300MHz, CDCl3) δ 8.12 (s, 1 H), 7.88 (d, 2 H), 7.80 (s, 2 H), 6.73 (d, 2 H), 3.96 (s, 3 H), 3.95 (s, 6 H), 3.05 (s, 6 H); МС (ESI) m/z 421.1(M + Na)+.
Индолильное кольцо А:
Синтез (2-(1Н-индол-2-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (62a) [Фигура 5]

1Н-Индол-2-карбонитрил (60а). К охлажденному раствору индол-2-карбоновой кислоты (2,0 г, 12,4 ммоль) в 60 мл безводного Et2O добавили 1,9 мл SOCl2 (26 ммоль). После перемешивания в течение 40 минут при комнатной температуре, эфир удалили под пониженным давлением при температуре, не превышающей 35oC. Полученный ацилхлорид растворили в 40 мл безводного Et2O, а полученный раствор сразу добавили к перемешанному раствору жидкого аммиака в 80 мл Et2O. Реакционную смесь перемешивали при комнатной температуре в течение 24 часов. Затем растворитель выпарили под пониженным давлением, а белый индол-2-карбоксамид кристаллизовали из 50% водного EtOH и высушили на воздухе, после чего его растворили в POCl3 и нагревали с дефлегматором в течение 5 минут. Охлажденный раствор вылили на дробленый лед и добавили водный раствор NH4OH для поддержания щелочного рН. Водную смесь экстрагировали Et2O, экстракты высушили над Na2SO4 и выпарили. Получили коричневый индол-2-карбонитрил 60a (общий выход из индол-2-карбоновой кислоты 63,3%). 1H ЯМР (500 MHz, CDCl3) δ 8.56 (br, s, 1 H), 7.68 (d, 1 H, J = 8.0 Hz), 7.43-7.34 (m, 2 H), 7.24-7.21 (m, 2 H). МС (ESI) m/z 144.0 (M + H)+, 140.8 (M -H)-.
(R)-2-(1Н-индол-2-ил)-N-метокси-N-метил-4,5-дигидротиазол-4-карбоксамид (61a) синтезировали по такому же способу, который использовался для 38d. Выход 67,1%. 1H ЯМР (300 MHz, CDCl3) δ 9.06 (s, br, 1 H), 7.64 (d, 2 H, J = 8.1 Hz), 7.36-7.24 (m, 2 H), 7.12 (dt, 1 H, J = 8.1 Hz, 1.2 Hz), 6.95 (d, 1 H, J = 1.8 Hz), 5.60 (t, br, 1 H, J = 8.7 Hz), 3.86 (s, 3 H), 3.78 (t, 1 H, J = 10.2 Hz), 3.58 (dd, 1 H, J = 9.0 Hz, 10.2 Hz), 3.30 (s, 3 H). МС (ESI) m/z 312.1 (M + Na)+, 287.9 (M -H)-.
(2-(1Н-индол-2-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (62а) синтезировали из 61а, используя такой же способ, как использовался для 35а. Выход 45,8%. 1H ЯМР (500 MHz, ДМСО-d6) δ 9.26 (s, 1 H), 8.11 (s, 1 H), 7.66 (d, 1 H, J = 8.0 Hz), 7.46 (s, 2 H), 7.42 (d, 1 H, J = 8.0 Hz), 7.29 (t, 1 H, J = 7.5 Hz), 7.16 (t, 1 H, J = 7.5 Hz), 7.10 (s, 1 H), 3.97 (s, 3 H), 3.93 (s, 6 H). МС (ESI) m/z 417.1 (M + Na)+, 392.9 (M -H)-.
Синтез (2-(1Н-индол-5-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (66a) [Фигура 5]

(R)-2-(1-(Фенилсульфонил)-1Н-индол-5-ил)-4,5-дигидротиазол-4-карбоновая кислота (64а). (R)-2-(1Н-индол-5-ил)-4,5-дигидротиазол-4-карбоновую кислоту 63а синтезировали, используя такой же способ, как использовался для 42а, из 1Н-индол-5-карбонитрила, и использовали без дополнительной очистки. К энергично перемешанному раствору 63а (1 ммоль) и тетрабутиламмония гидросульфата (0,15 ммоль) в толуоле (10 мл) при 0°C добавили 50% водный раствор гидроксида натрия (10 мл) и сульфонилхлорид (2 ммоль). Полученный раствор перемешивали при комнатной температуре в течение 6 часов. Затем добавили 1 н. HCl, чтобы подкислить смесь до рН=2, и экстрагировали CH2Cl2, органический слой отделили и высушили (MgSO4); затем выпарили до сухости для получения соединения 64а, которое использовали на следующих этапах без дополнительной очистки.
(R)-N-метокси-N-метил-2-(1-(фенилсульфонил)-1Н-индол-5-ил)-4,5-дигидротиазол-4-карбоксамид (65a) получили из 64а по такому же способу, который использовался для 38d. Выход 57,1%. 1H ЯМР (500 MHz, CDCl3) δ 7.92 (m, 2 H), 7.77 (m, 3 H), 7.51 (d, 1 H, J = 3.0 Hz), 7.46 (t, 1 H), 7.35 (t, 1H), 6.61 (d, 1 H), 5.58 (br, t, 1 H) 3.82 (s, 3 H), 3.73 (t, 1 H), 3.43 (m, 1 H), 3.21 (s, 3 H). МС (ESI) m/z 452.1 (M + Na)+.
(2-(1Н-индол-5-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (66a). К раствору n-BuLi (1,6 M, 1,7 мл) в 8 мл ТГФ добавили раствор 3,4,5-триметоксибромбензола (2,47 ммоль) в 3 мл ТГФ при -78oC. Смесь оставили перемешиваться на 2 часа и добавили раствор амида Вайнреба 65a (1,24 ммоль) в 3 мл ТГФ. Температуру оставили увеличиваться до комнатной температуры и перемешивали в течение ночи. Реакционную смесь погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который дефлегмировали в 1 н. растворе NaOH в 5 мл этанола для получения соединения 66a со снятой защитой, и очистили колоночной хроматографией для получения чистого соединения в виде светло-желтого твердого вещества (36,3%). 1H ЯМР (300M, CDCl3) δ 8.36 (br, s, 1 H), 8.31 (s, 1 H), 8.21 (s, 1 H), 7.92, 7.89 (dd, 1 H, J = 1.8, 2.7 Hz), 7.46 (d, 1 H) 7.62 (s, 2 H, J = 8.7 Hz), 7.29 (t, 1 H, J = 2.7 Hz), 6.64 (br, 1 H), 3.97 (s, 6 H), 3.97 (s, 3 H); МС (ESI) m/z 417.1(M + Na)+, 392.9 (M - H)-.
Синтез (2-(1Н-индол-2-ил)тиазол-4-ил)(1Н-индол-2-ил)метанона (8).

(2-(1Н-Индол-2-ил)тиазол-4-ил)(1Н-индол-2-ил)метанон (8) получили по такому же способу, как использовался для соединения 1h, из 2-(1Н-индол-2-ил)-4,5-дигидротиазол-4-карбоновой кислоты и цистеина. 1H ЯМР (500MHz, CDCl3 ) δ 9.39 (s, 1 H), 8.54 (s, 1 H), 8.46 (s, 1 H), 8.06 (s, 1 H), 8.03 (dd, 1 H), 7.66 (d, 1 H), 7.51 (d, 1 H), 7.41 (d, 1 H), 7.33 (t, 1 H), 7.29 (d, 1 H), 7.15 (t, 1 H), 7.09 (d, 1 H), 6.72 (s, 1 H). МС (ESI) m/z 366.1(M + Na)+, 341.9 (M - H)-.
Синтез (2-(1Н-Индол-2-ил)тиазол-4-ил)(1Н-индол-5-ил)метанона (21).

(2-(1Н-Индол-2-ил)тиазол-4-ил)(1Н-индол-5-ил)метанон (21) получили по такому же способу, как использовался для соединения 1h, из 2-(1Н-индол-2-ил)-4,5-дигидротиазол-4-карбоновой кислоты и цистеина. 1H ЯМР (500MHz, CDCl3) δ 9.60 (s, 1 H), 9.26 (s, 1 H), 8.31 (s, 1 H), 8.03 (s, 1 H), 7.83 (dd, 1 H), 7.69 (d, 1 H), 7.53-7.49 (m, 2 H), 7.41 (t, 1 H), 7.33 (t, 1 H), 7.21-7.18 (m, 2 H), 7.13 (s, 1 H). МС (ESI) m/z 366.1(M + Na)+, 341.9 (M - H)-.
ПРИМЕР 4
СИНТЕЗ СОЕДИНЕНИЙ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ, ИМЕЮЩИХ АЗОТНЫЙ ЛИНКЕР (Х=NH)
Для улучшения биодоступности, между фениловым кольцом А и тиазоловым кольцом В внедрили линкер NH. Эти новые серии соединений синтезировали так, как показано на Фигуре 6. По реакции 3-бром-2-оксопропановой кислоты этилового эфира и арилтиомочевины в этаноле при 65oC получили 2-(ариламино)-тиазол-4-карбоновые кислоты 73a-d с высокими выходами. Эти кислоты преобразовали в амиды Вайнреба 74a-d, с последующими реакциями с 3,4,5-триметоксифениллитием, в результате которых получили анилин-связанные свободные основания 5a-d, которые могут быть превращены в соли HCl 5Ha-d.
Синтез (2-(фениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метаноновых производных (5a-d) и их HCl солей [Фигура 6]




(5a) (5b) (5c) (5d)



(5Ha) (5Hb) (5Hc)
Общий способ синтеза 2-(ариламино)тиазол-4-карбоновых кислот (37a-d). N-Арилтиомочевину (0,01 моль) и этил бромпируват (0,011 моль) растворили в 3 мл этанола и выдерживали при дефлегмации в течение 2 часов. Реакционную смесь охладили, кристаллический этил 2-(замещенный фениламино)тиазол-4-карбоксилат собрали фильтрацией и промыли этанолом. В результате дефлегмации смеси этиловых эфиров с раствором NaOH в этаноле получили конечные соединения 73a-d, которые использовали непосредственно на следующих этапах.
N-Метокси-N-метил-2-(ариламино)тиазол-4-карбоксамиды (74а-d) синтезировали, используя такой же способ, как использовался для 38d (смотри Пример 1, Фигуру 2).
N-Метокси-N-метил-2-(фениламино)тиазол-4-карбоксамид (74а). Выход 90,2%. 1H ЯМР (500 MHz, CDCl3) δ 7.39 (s, 2 H), 7.38 (br, 1 H), 7.36-7.33 (m, br, 4 H), 7.09 (t, br, 1 H), 3.77 (s, 3 H), 3.43 (s, 3 H), 2.33 (s, 3 H). МС (ESI) m/z 286.0 (M + Na)+.
N-Метокси-N-метил-2-(п-толиламино)тиазол-4-карбоксамид (74b). Выход 93,3%. 1H ЯМР (500 MHz, CDCl3) δ 7.35 (s, 1 H), 7.31 (br, 1 H), 7.22 (d, 2 H), 7.16 (d, 2 H), 3.76 (s, 3 H), 3.42 (s, 3 H), 2.33 (s, 3 H). МС (ESI) m/z 278.0 (M + H)+.
2-(4-Фторфениламино)-N-метокси-N-метилтиазол-4-карбоксамид (74с). Выход 89,7%. 1H ЯМР (500 MHz, CDCl3) δ 7.36 (s, 1 H), 7.36-7.31 (m, 2 H), 7.07-7.04 (m, 6 H), 3.76 (s, 3 H), 3.42 (s, 3 H). МС (ESI) m/z 282.0 (M + Na)+, 280.8 (M - H)-.
2-(4-Хлорфениламино)-N-метокси-N-метилтиазол-4-карбоксамид (74d). 1H ЯМР (500 MHz, CDCl3) δ 7.66 (s, br, 1 H), 7.41 (s, 1 H), 7.34 (d, 2 H), 7.29 (d, 2 H), 3.76 (s, 3 H), 3.42 (s, 3 H). МС: 295.8 (M-1)-; 320.0 (M+Na)+.
Общий способ синтеза (2-(ариламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанонов (5a-d). При -78oC, к раствору 5-бром-1,2,3-триметоксибензола (1,235 г, 5,0 ммоль) в 30 мл ТГФ добавили n-BuLi в гексане (2,5 н., 2,4 мл, 6 ммоль) под защитой Ar2 и перемешивали в течение 10 минут. Амиды Вайнреба 74a-d (1 ммоль) в 10 мл ТГФ добавили к литиевому реагенту и оставили перемешиваться при комнатной температуре на 2 часа. Реакционную смесь погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения (5a-d).
(2-(Фениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (5a). Выход 33,3%. 1H ЯМР (500 MHz, ДМСО-d6) δ 10.4 (s, 1 H), 7.85 (s, 1 H), 7.68 (d, 2 H, J = 8.0 Hz), 7.31 (t, 2 H, J = 8.0 Hz), 6.98 (t, 1 H, J = 8.0 Hz), 3.83 (s, 6 H), 3.78 (s, 3 H). МС (ESI) m/z 393.1 (M + H)+, 368.9 (M -H)-.
(2-(п-Толиламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (5b). Выход 40,6%. 1H ЯМР (500 MHz, CDCl3) δ 7.48 (s, 1 H), 7.47 (s, 2 H), 7.30 (br, 1 H), 7.27 (d, 2 H, J = 8.5 Hz), 7.17 (d, 2 H, J = 8.5 Hz), 3.93 (s, 3 H). 3.90 (s, 6 H), 2.34 (s, 3 H). МС (ESI) m/z 385.1 (M + H)+, 382.9 (M -H)-.
(2-(п-Фторфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (5с). Выход 39,6%. 1H ЯМР (500 MHz, CDCl3) δ 7.52 (br, 1 H), 7.49 (s, 1 H), 7.45 (s, 2 H), 7.40-7.37 (q, 2 H, J = 4.5 Hz), 7.08-7.04 (t, 2 H, J = 8.0 Hz), 3.93 (s, 3 H), 3.89 (s, 6H). МС (ESI) m/z 389.3 (M + H)+, 386.9 (M -H)-.
2-((4-Хлорфенил)амино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (5d) получили, используя такой же способ, как использовался для 5a, из 1-(4-хлорфенил)тиомочевины и этил бромпирувата. Температура плавления: 165-166oC. 1H ЯМР (500 MHz, CDCl3) δ 7.60 (s, br, 1 H), 7.56 (s, 1 H), 7.47 (s, 2 H), 7.38 (d, 2 H), 7.31 (d, 2 H), 3.94 (s, 3 H), 3.89 (s, 6 H). МС: 402.9 (M-1)-; 427.0 (M+Na)+.
Общий способ синтеза гидрохлоридных солей (5Ha-c). При 0oC, к раствору соединения 5a-с (0,1 ммоль) в 5 мл CH2Cl2 добавили раствор HCl в 1,4-диоксане (4 н., 2 мл) и перемешивали при комнатной температуре в течение ночи. Осадки 5Ha-c собрали и промыли диэтиловым эфиром.
(2-(Фениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлоридная соль (5Ha). Выход 91,6%. 1H ЯМР (500 MHz, ДМСО-d6) δ 12.9 (br, 1 H), 7.49-7.46 (m, 2 H), 7.42-7.40 (m, 2 H),7.37-7.34 (m, br, 2 H), 7.11 (s, 2 H), 3.94 (s, 3 H), 3.92 (s, 6 H). МС (ESI) m/z 389.1 (M + H)+.
(2-(п-Толиламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлоридная соль (5Hb). Выход 39,6%. 1H ЯМР (500 MHz, CDCl3) δ 7.30-7.25 (m, br, 5 H), 7.12 (s, 2 H), 3.94 (s, 3 H), 3.92 (s, 6 H), 2.38 (s, 3 H). МС (ESI) m/z 389.1 (M + H)+.
(2-(п-Фторфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлоридная соль (5Hс). Выход 89,3%. 1H ЯМР (500 MHz, CDCl3) δ 10.55 (s, 1 H), 7.85 (s, 1 H), 7.72-7.69 (q, 2 H, J = 4.5 Hz), 7.50 (s, 2 H), 7.18-7.15 (t, 2 H, J = 8.5 Hz), 4.30 (br, 1 H), 3.82 (s, 6H), 3.78 (s, 3 H). МС (ESI) m/z 389.3 (M + H)+.
Синтез (2-(фениламино)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (5e)

Схема 1: получение соединения 5e.
2,2-Диэтокси-N-(иминометилен)этанамин (а). Раствор аминоацетальдегида диэтилацеталя (5,32 г, 40 ммоль) в эфире (20 мл) добавили к суспензии CNBr (4,22 г, 40 ммоль) в гексане (20 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Твердое вещество удалили фильтрацией и промыли эфиром. Объединенный фильтрат концентрировали. В результате флэш-хроматографии концентрированного остатка получили 2,82 г (45%) N-(2,2-диэтоксиэтил)карбодиимида (а). 1H ЯМР (500 MHz, CDCl3): 4.58 (t, J = 5.5 Hz, 1 H), 3.85 (br s, 1 H), 3.73 (m, 2 H), 3.56 (m, 2 H), 3.16 (J = 5.5 Hz, 2 H), 1.23 (t, J = 7.0 Hz, 3 H), МС: 156.8 (M-H)-; 180.9 (M+Na)+.
1-(2,2-Диэтоксиэтил)-3-фенилгуанидин (b). Анилин (1,66 г, 17,8 ммоль) растворили в этаноле (25 мл) и по каплям добавили N-(2,2-диэтоксиэтил)карбодиимид (а) (2,82 г, 17,8 ммоль). Затем добавили метансульфоновую кислоту (1,71 г, 17,8 ммоль) и нагревали смесь с дефлегматором в течение 24 часов. Реакционную смесь вылили в NaOH (0,5 М) и экстрагировали CH2Cl2. В результате высушивания и концентрирования получили продукт, который подвергли флэш-хроматографии для получения промежуточного гуанидина (b) (3,3 г, 73,8%). 1H ЯМР (500 MHz, ДМСО-d6) δ 7.27-6.90 (m, 5 H), 4.55 (t, 1 H), 3.76-3.70 (m, 2 H), 3.60-3.54 (m, 2 H), 3.35-3.34 (d, 2 H), 1.22 (pent, 6 H). МС: 249.8 (M-H)-; 252.1(M+H)+.
N-Фенил-1Н-имидазол-2-амин (с). Гуанидин (b) растворили в HCl (5 мл, 6 М) при 0°C, а затем перемешивали в течение 2 часов. После того, как исходный материал был израсходован, добавляли NaOH (25%) до образования осадка. Эту смесь перемешивали в течение 30 минут. Затем реакционную смесь вылили в NaOH (0,5 М), экстрагировали CH2Cl2, высушили и концентрировали. В результате флэш-хроматографии получили (с) (0,95 г, 50%). 1H ЯМР (500 MHz, ДМСО-d6) δ 8.58 (s, br, 1 H), 7.34-6.74 (m, 5 H), 6.68 (s, 2 H), 6.62 (br, 2 H), 3.82 (s, 6 H), 3.73 (s, 3 H). МС: 157.6 (M-H)-; 160.0 (M+H)+.
N-Фенил-1-тритил-1Н-имидазол-2-амин (d). Тритилхлорид (2,79 г, 10 ммоль) добавили к ледяному раствору фениламиноимидазола (с) (1,59 г, 10 ммоль) и триэтиламина (1,01 г, 10 ммоль) в метилендихлориде (50 мл). Реакционную смесь оставили нагреваться до комнатной температуры и перемешивали в течение ночи. Смесь разбавили метилендихлоридом, последовательно промыли H2O, насыщенным раствором NaHCO3, насыщенным солевым раствором и высушили при помощи MgSO4. В результате фильтрации и выпаривания растворителя с последующим хроматографическим разделением получили продукт (d). 1H ЯМР (500 MHz, (CDCl3) δ 7.52-7.35 (m, 5 H), 7.28-7.43 (m, 15 H), 6.85 (s, 2 H), 6.41 (s, 1 H), 6.08 (s, 1 H). МС: 1399.8 (M-H)-; 402.8 (M+H)+.
(2-(Фениламино)-1-тритил-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (e). При -78oC, t-BuLi в ТГФ (1,7 М, 0,34 мл, 0,58 ммоль) добавили к раствору тритил-защищенного соединения (d) (116 мг, 0,289 ммоль) в ТГФ. Затем добавили 3,4,5-триметоксибензоил хлорид (66,5 мг, 0,289 ммоль) и перемешивали в течение ночи. Реакционную смесь погасили насыщенным раствором NH4Cl и высушили при помощи MgSO4. В результате фильтрации и выпаривания растворителя с последующей хроматографией получили соединение (e) (75 мг, 43,7%). 1H ЯМР (500 MHz, CDCl3) δ 7.55-7.41 (m, 5 H), 7.32 (s, 1 H), 7.28-7.18 (m, 15 H), 6.94 (s, 2 H), 3.78 (s, 6 H), 3.70 (s, 3 H). МС: 594.2 (M-H)-; 596.3 (M+H)+.
(2-(Фениламино)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (5e). К раствору тритил-защищенного соединения (e) (50 мг, 0,084 ммоль) в этиловом эфире добавили 2 М HCl в эфире (1 мл, 1 ммоль). Реакционную смесь перемешивали в течение ночи и промыли насыщенным раствором NaHCO3 и высушили при помощи MgSO4. В результате фильтрации и выпаривания растворителя с последующей флэш-хроматографией получили соединение 5e со снятой защитой (18 мг, 63%). 1H ЯМР (500 MHz, ДМСО-d6) δ 7.54 (s, br, 1 H), 7.51-7.43 (m, 3 H), 7.33 (d, 2 H), 7.04 (s, 2 H), 6.62 (br, 2 H) 3.82 (s, 6 H), 3.73 (s, 3 H). МС: 352.1 (M-H)-; 354.3 (M+H)+.
ПРИМЕР 5
СИНТЕЗ НЕКОТОРЫХ АРИЛ-БЕНЗОИЛ-ИМИДАЗОЛЬНЫХ СОЕДИНЕНИЙ

Получение 2-арил-4,5-дигидро-1Н-имидазолов 14b, 14c, 14x (Фигура 7).

К раствору соответствующего бензальдегида 8(b, c, x) (60 ммоль) в t-BuOH (300 мл) добавили этилендиамин (66 ммоль) и перемешивали в течение 30 минут при комнатной температуре. К реакционной смеси последовательно добавили карбонат калия (75 ммоль) и йод (180 ммоль), а затем перемешивали при 70°C в течение 3 часов. Добавили сульфит натрия (Na2SO3) и экстрагировали смесь хлороформом. Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (хлороформ: метанол 20:1) для получения твердого белого вещества. Выход: 50-60%.
Получение 2-арил-1Н-имидазолов (9a-j, p, x; Фигуры 7 и 8).

Способ A (необходимый только для 9b, 9x; Фигура 7): К раствору 2-арил-4,5-дигидро-1Н-имидазола 14b, x (35 ммоль) в ДМСО (100 мл) добавили карбонат калия (38,5 ммоль) и диацетоксийодбензол (38,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в темноте. Добавили воду, а затем экстрагировали дихлорметаном. Органический слой высушили над сульфатом магния и концентрировали. Остаток подвергли колоночной флэш-хроматографии (гексан: этилацетат 3:2) для получения твердого белого вещества. Выход: 30%-50%.
Способ В (необходимый только для 9c; Фигура 7): К раствору 2-арил-4,5-дигидро-1Н-имидазола 14c (50 ммоль) в ДМФ (70 мл) добавили DBU (55 ммоль) и CBrCl3 (55 ммоль). Реакционную смесь перемешивали в течение ночи и добавили насыщенный (водный) раствор NaHCO3, а затем экстрагировали дихлорметаном. Органический слой высушили над сульфатом магния и концентрировали. Остаток подвергли колоночной флэш-хроматографии (хлороформ: метанол 50:1) для получения твердого белого вещества. Выход: 7%.
Способ С (необходимый для 9a, 9d-j, 9p; Фигура 8): К раствору соответствующего бензальдегида (8a, 8d-j, 8p) (100 ммоль) в этаноле (350 мл) при 0°C добавили раствор 40% оксалальдегида в воде (12,8 мл, 110 ммоль) и раствор 29% гидроксида аммония в воде (1000 ммоль, 140 мл). После перемешивания в течение 2-3 дней при комнатной температуре, реакционную смесь концентрировали, а остаток подвергли колоночной флэш-хроматографии с дихлорметаном в качестве элюента для получения указанного в заголовке соединения в виде желтого порошка. Выход: 20%- 40%.
Получение 2-арил-1-(фенилсульфонил)-1Н-имидазолов (10a-j, p, x; Фигуры 7 и 8).

К раствору 2-арил-1Н-имидазола 9a-j, p, x (20 ммоль) в безводном ТГФ (200 мл) при 0°C добавили гидрид натрия (60% дисперсия в минеральном масле, 1,2 г, 30 ммоль) и перемешивали в течение 30 минут. Добавили бензолсульфонилхлорид (2,82 мл, 22 ммоль) и перемешивали реакционную смесь в течение ночи. После разбавления 100 мл насыщенного (водного) раствора NaHCO3, реакционную смесь экстрагировали этилацетатом (500 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 2:1) для получения бледного твердого вещества. Выход: 50%-70%.
Получение арил (2-арил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанонов (11aa-ai, ba, ca, cb, da, db, ea, eb, fa, fb, ga, gb, ha, hb, ia, ib, ja, jb, pa; Фигуры 7 и 8).

К раствору 2-арил-1-(фенилсульфонил)-1H-имидазола (6,0 ммоль) 10a-j, p, x в безводном ТГФ (30 мл) при -78°C добавили 1,7 M трет-бутиллитий в пентане (5,3 мл, 9,0 ммоль) и перемешивали в течение 10 минут. Добавили соответвующий замещенный бензоилхлорид (7,2 ммоль) при -78°C и перемешивали в течение ночи. Реакционную смесь разбавили 100 мл насыщенного (водного) раствора NaHCO3 и экстрагировали этилацетатом (200 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 4:1) для получения твердого белого вещества. Выход: 15%-40%.
Общий способ получения арил (2-арил-1Н-имидазол-4-ил)метанонов (12aa-ai, ba, ca, cb, da, db, ea, eb, fa, fb, ga, gb, ha, hb, ia, ib, ja, jb, pa; Фигуры 7 и 8).

К раствору арил (2-арил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанонов (2,0 ммоль) 11aa-ai, ba, ca, cb, da, db, ea, eb, fa, fb, ga, gb, ha, hb, ia, ib, ja, jb, pa в ТГФ (20,0 мл) добавили 1,0 М тетрабутиламмония фторид (4,0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавили 50 мл насыщенного (водного) раствора NaHCO3 и экстрагировали этилацетатом (100 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 3:1) или перекристаллизовали из воды и метанола для получения твердого белого вещества. Выход: 80-95%.
Получение (2-(4-гидроксифенил)-1Н-имидазол-4-ил) (арил)метанонов (12ka, 12kb; Фигура 8).

К раствору (2-(4-(бензилокси)фенил)-1H-имидазол-4-ил)(арил)метанона 12ja или 12jb, (1 ммоль) в AcOH (20 мл) добавили концентрированную HCl (2 мл) и дефлегмировали в течение ночи. После удаления растворителя остаток перекристаллизовали из дихлорметана для получения указанного в заголовке соединения в виде твердого желтого вещества. Выход: 70-85%.
Получение (2-арил-1Н-имидазол-4-ил) (3,4,5-тригидроксифенил)метанонов 13ea, 13fa, 13ha (Фигура 8).

К раствору арил (2-арил-1H-имидазол-4-ил)метанона 12ea, 12fa или 12ha (0,5 ммоль) в CH2Cl2 (6,0 мл) добавили 1,0 M BBr3 (2 ммоль) в CH2Cl2 и перемешивали в течение 1 часа при комнатной температуре. Добавили воду для разрушения избытка BBr3. Осажденное твердое вещество отфильтровали и перекристаллизовали из MeOH для получения твердого желтого вещества. Выход: 60-80%.
Получение арил (2-арил-1Н-имидазол-4-ил)метанон-HCl соли (12db-HCl).

К раствору 12db (0,5 ммоль) в метаноле (20 мл) добавили 2 М раствор хлороводорода (5 ммоль) в этиловом эфире и перемешивали в течение ночи при комнатной температуре. Реакционную смесь концентрировали, а остаток промыли CH2Cl2 для получения указанного в заголовке соединения. Выход: 95%.
Получение арил (2-фенил-1Н-мидазол-1-ил)метанона (12aba, 12aaa; Фигура 9).

К раствору 2-фенил-1Н-имидазола 9a (10 ммоль) в ТГФ (20 мл) добавили NaH (15 ммоль) и замещенный бензоилхлорид (12 ммоль) при 0°C. Реакционную смесь перемешивали в течение ночи и разбавили насыщенным раствором NaHCO3, а затем экстрагировали этилацетатом. Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (хлороформ) для получения твердого белого вещества. Выход: 12-16%.
Получение 1-замещенный-(2-фенил-1H-имидазол-1-ил)-арил-метанона (12dc, 12fc, 12daa, 12 dab, 12 cba, 11gaa, 12la; Фигуры 10-11).

Синтез 12dc, 12fc и 12daa, 12dab и 12cba резюмирован на Фигуре 10. Соединения 12da, 12cb и 12fa были синтезированы в соответствии с синтезом, описанным выше и на Фигурах 7 и 8. Обработка 12da и 12fa хлоридом алюминия дает пара-деметилированные 12dc, 12fc с незатронутым 3,5-диметокси. Соединение 12daa получили бензилированием N-1 положения 12da. Тогда как метилирование N-1 положения 12da и 12cb дает соединения 12dab и 12cba, соответственно.
Синтез 12dc, 12fc, 12daa, 12dab, 12cba: Способ D. (для 12dc и 12fc) [Фигура 10]:

R1=CH3 (12dc)
R1=Cl (12fc)
К раствору 12da и 12fa (200 мг) в ТГФ (20 мл) добавили хлорид алюминия (10 экв.). Реакционную смесь перемешивали в течение ночи. Добавили воду, а затем экстрагировали этилацетатом. Органический слой высушили над сульфатом магния и концентрировали. Остаток подвергли колоночной флэш-хроматографии (гексан: этилацетат 1:1) для получения желтовато-белого твердого вещества. Выход: 60%-80%.
Синтез 12daa, 12dab, 12cba, Способ E: [Фигура 10]:

R1=Me; R2=Bn; R3=3,4,5-(OMe)3 (12daa)
R1=Me; R2=CH3; R3=3,4,5-(OMe)3(12dab)
R1=OMe; R2=CH3; R3=F (12cba)
К раствору 12da и 12cb (100 мг) в ТГФ (10 мл) на ледяной бане добавили гидрид натрия (1,2 экв.), а затем добавили метилйодид (для 12dab, 12cba) или бензилбромид (для 12daa) (2 экв.). Полученную реакционную смесь перемешивали в течение 5 часов в условиях дефлегмации. После разбавления 50 мл насыщенного (водного) раствора NaHCO3, реакционную смесь экстрагировали этилацетатом (100 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 2:1) для получения твердого белого вещества. Выход: 50%-98%.
Синтез 11gaa и 12la (Фигура 11):

R1=N(Me)2; R2=(4-OMe)PhSO2 (11gaa)
R1=Br; R2=H (12la)
Замещенные бензальдегидные соединения 8(l, g) преобразовали в соединения 9(l, g) в присутствии гидроксида аммония и глиоксаля для образования имидазоловой структуры. Имидазоловые кольца соединений 9(l, g) защитили соответствующей фенилсульфониловой группой с последующим связыванием с 3,4,5-триметоксибензоил хлоридом для получения соединения 11(la,gaa). В результате обработки 11la трет-бутиламмония фторидом для удаления защитной группы получили 12la.
Структурная характеристика (1-бензил-2-(п-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12daa) (Фигура 11).

Выход: 92,8%; т.пл. 135-137°C. 1H ЯМР (CDCl3, 500 MHz) δ 7.81 (s, 1 H), 7.80 (d, J = 6.5 Hz, 2 H), 7.58 (d, J = 8.0 Hz, 2 H), 7.41-7.45 (m, 3 H), 7.31-7.33 (m, 2 H), 7.20 (d, J = 7.0 Hz, 2 H), 5.33 (s, 2 H), 3.99 (s, 3 H), 3.98 (s, 6 H), 2.47 (s, 3 H). МС (ESI) рассчитано для C27H26N2O4 442.2, найдено 443.1 [M + Na]+. ВЭЖХ1: tR 4,28 мин., чистота > 99%.
Структурная характеристика (2-(4-(диметиламино)фенил)-1-((4-метоксифенил)сульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанона (12gba).

Выход: 34,1%; т.пл. 147-149°C. 1H ЯМР (CDCl3, 500 MHz) δ 8.07 (q, J = 8.5 Hz, 5.5 Hz, 2 H), 7.78 (d, J = 9.0 Hz, 2 H), 7.41 (d, J = 8.5 Hz, 2 H), 7.39 (s, 1 H), 7.23 (t, J = 8.5 Hz, 2 H), 6.91 (d, J = 9.0 Hz, 2 H), 6.68 (d, J = 9.0 Hz, 2 H), 3.89 (s, 3 H), 3.08 (s, 3 H). МС (ESI) рассчитано для C25H22FN3O4S 479.1, найдено 502.1 [M + Na]+. ВЭЖХ2: tR 18,6 мин., чистота 96.9%.
Синтез (2-(4-бромфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12la) (Фигура 11)

Синтез 9l, 9g: К раствору соответствующего бензальдегида (8l и 8g, 100 ммоль) в этаноле (400 мл) при 0°C добавили раствор 40% оксалальдегида (глиоксаля) в воде (1,1 экв.) и раствор 29% гидроксида аммония в воде (10 экв.). После перемешивания в течение 2-3 дней при комнатной температуре, реакционную смесь концентрировали, а остаток подвергли колоночной флэш-хроматографии с дихлорметаном в качестве элюента для получения указанного в заголовке соединения в виде желтого порошка. Выход: 10%- 30%.
Синтез 10la, 10gb: К раствору имидазолов (9l, 9g) (10 ммоль) в безводном ТГФ (200 мл) при 0°C добавили гидрид натрия (60% дисперсия в минеральном масле, 1,2 экв.) и перемешивали в течение 20 минут. Добавили 4-метоксибензолсульфонил хлорид (для 10gb) или бензолсульфонил хлорид (для остальных) (1,2 экв.) и перемешивали реакционную смесь в течение ночи. После разбавления 200 мл насыщенного (водного) раствора NaHCO3, реакционную смесь экстрагировали этилацетатом (600 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 2:1) для получения бледного твердого вещества. Выход: 40%-95%.
Синтез 11la, 11gaa: К раствору 2-арил-1-(фенилсульфонил)-1H-имидазола (10la, 10gb) (5,0 ммоль) в безводном ТГФ (30 мл) при -78°C добавили 1,7 M трет-бутиллитий в пентане (1,2 экв.) и перемешивали в течение 10 минут. Добавили 3,4,5-триметоксибензоил хлорид (1,2 экв.) при -78°C и перемешивали в течение ночи. Реакционную смесь разбавили 100 мл насыщенного (водного) раствора NaHCO3 и экстрагировали этилацетатом (300 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 3:1) для получения твердого белого вещества. Выход: 5%-45%.
Синтез 12la: К раствору арил (2-арил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанона (11la), 2,0 ммоль) в ТГФ (25,0 мл) добавили 1,0 М тетрабутиламмония фторид (2 экв.) и перемешивали в течение ночи. Реакционную смесь разбавили 60 мл насыщенного (водного) раствора NaHCO3 и экстрагировали этилацетатом (150 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 4:1) или перекристаллизовали из воды и метанола для получения твердого белого вещества. Выход: 80-98%.
Синтез (4-фторфенил)(2-(4-метоксифенил)-1Н-имидазол-4-ил)метанона (12cb) (Фигура 7).

К раствору (4-фторфенил)(2-(4-метоксифенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанона (11cb, 872 мг, 2,0 ммоль) в ТГФ (20,0 мл) добавили 1,0 М тетрабутиламмония фторид (4,0 мл, 4,0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавили 50 мл насыщенного (водного) раствора NaHCO3 и экстрагировали этилацетатом (100 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток перекристаллизовали из воды и метанола для получения твердого белого вещества. Выход: 90%; т.пл. 245 – 247oC.
Синтез (2-(п-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12da) (Фигура 8).

К раствору (1-(фенилсульфонил)-2-(п-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11da, 492 мг, 1,0 ммоль) в ТГФ (15,0 мл) добавили 1,0 М тетрабутиламмония фторид (2,0 мл, 2,0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавили 30 мл насыщенного (водного) раствора NaHCO3 и экстрагировали этилацетатом (80 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток перекристаллизовали из воды и метанола для получения твердого белого вещества. Выход: 88,5%.
Синтез (2-(4-хлорфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12fa) (Фигуры 8 и 14).

2-(4-Хлорфенил)-1Н-имидазол (9f): К раствору 4-хлорбензальдегида (8f) (100 ммоль) в этаноле (350 мл) при 0°C добавили раствор 40% оксалальдегида в воде (12,8 мл, 110 ммоль) и раствор 29% гидроксида аммония в воде (1000 ммоль, 140 мл). После перемешивания в течение 2-3 дней при комнатной температуре, реакционную смесь концентрировали, а остаток подвергли колоночной флэш-хроматографии с дихлорметаном в качестве элюента для получения указанного в заголовке соединения в виде желтого порошка. Выход: 19,8 %. 1H ЯМР (500 MHz, ДМСО-d6) δ 13.60 (br, 1H), 7.94 (d, J = 8.5 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 7.27 (s, 1H), 7.03 (s, 1H). МС (ESI): рассчитано для C9H7ClN2, 178.0, найдено 178.9 [M + H]+.
2-(4-Хлорфенил)-1-(фенилсульфонил)-1Н-имидазол (10f): К раствору 2-(4-хлорфенил)-1Н-имидазола (9f) (20 ммоль) в безводном ТГФ (200 мл) при 0°C добавили гидрид натрия (60% дисперсия в минеральном масле, 1,2 г, 30 ммоль) и перемешивали в течение 30 минут. Добавили бензолсульфонилхлорид (2,82 мл, 22 ммоль) и перемешивали реакционную смесь в течение ночи. После разбавления 100 мл насыщенного (водного) раствора NaHCO3, реакционную смесь экстрагировали этилацетатом (500 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 2:1) для получения бледного твердого вещества. Выход: 54,9%. 1H ЯМР (500 MHz, CDCl3) δ 7.65 (d, J = 2.0 Hz, 1H), 7.58 (t, J = 7.5 Hz, 1H), 7.43 (d, J = 8.5 Hz, 2H), 7.38 (t, J = 8.0 Hz, 2H), 7.34-7.36 (m, 4H), 7.12 (d, J = 1.5 Hz, 1H). МС (ESI): рассчитано для C15H11ClN2O2S, 318.0, найдено 341.0 [M + Na]+.
(2-(4-Хлорфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11fa): К раствору 2-(4-хлорфенил)-1-(фенилсульфонил)-1H-имидазола (10f) (6,0 ммоль) в безводном ТГФ (30 мл) при -78°C добавили 1,7 M трет-бутиллитий в пентане (5,3 мл, 9,0 ммоль) и перемешивали в течение 10 минут. Добавили 3,4,5-триметоксибензоил хлорид (7,2 экв.) при -78°C и перемешивали в течение ночи. Реакционную смесь разбавили 100 мл насыщенного (водного) раствора NaHCO3 и экстрагировали этилацетатом (200 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 4:1) для получения твердого белого вещества. Выход: 36,8%; 1H ЯМР (500 MHz, CDCl3) δ 8.05 (d, J = 7.5 Hz, 2H), 7.77 (t, J = 7.5 Hz, 1H), 7.62 (t, J = 8.0 Hz, 2H), 7.48 (s, 1H), 7.44 (d, J = 9.0 Hz, 2H), 7.39 (d, J = 8.5 Hz, 2H), 7.37 (s, 2H). МС (ESI): рассчитано для C25H21ClN2O6S, 512.1, найдено 513.1 [M + H]+.
(2-(4-Хлорфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12fa): К раствору (2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11fa) (2,0 ммоль) в ТГФ (20,0 мл) добавили 1,0 М тетрабутиламмония фторид (4,0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавили 50 мл насыщенного (водного) раствора NaHCO3 и экстрагировали этилацетатом (100 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 3:1) или перекристаллизовали из воды и метанола для получения твердого белого вещества. Выход: 80-95%. Выход: 36,9%; т.пл. 193 – 195°C. 1H ЯМР (500 MHz, CDCl3) δ 10.75 (br, 1H), 7.96 (d, J = 8.5 Hz, 2H), 7.83 (s, 1H), 7.47 (d, J = 9.0 Hz, 2H), 7.23 (s, 2H), 3.97 (s, 3H), 3.94 (s, 6H), 2.43 (s, 3H). МС (ESI): рассчитано для C19H17ClN2O4, 372.1, найдено 395.1 [M + Na]+, 370.9 [M – H]-. ВЭЖХ градиент: растворитель А (вода) и растворитель В (метанол): 0-15 минут 40-100% В (линейный градиент), 15-25 минут 100% В: tR 16.36 мин., чистота > 99%.
Синтез (2-(4-хлорфенил)-1Н-имидазол-4-ил)(4-фторфенил)метанона (12fb) (Фигура 8).

К раствору (2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанона (11fb, 440 мг, 1,0 ммоль) в ТГФ (12,0 мл) добавили 1,0 М тетрабутиламмония фторид (2,0 мл, 2,0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавили 20 мл насыщенного (водного) раствора NaHCO3 и экстрагировали этилацетатом (60 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток перекристаллизовали из воды и метанола для получения твердого белого вещества. Выход: 83,7%.
Физико-химическая характеристика арил-бензоил-имидазольных
соединений и промежуточных соединений
| Соединение | Физико-химическая характеристика |
| 2-фенил-1H-имидазол (9a) | Выход: 36.8%. 1H ЯМР (500 MHz, ДМСО-d 6 ) δ 12.52 (br, 1 H), 7.95 (d, J = 7.0 Hz, 2 H), 7.44 (t, J = 7.5 Hz, 2 H), 7.34 (t, J = 7.0 Hz, 1H), 7.25-7.27 m, 1 H), 7.04 - 7.07 m, 1 H). МС (ESI): рассчитано для C9H8N2, 144.1, найдено 167.1 [M + Na]+. |
| 2-(4-фторфенил)-1H-имидазол (9b) | Выход: 56.5%. 1H ЯМР (300 MHz, ДМСО-d 6 ) δ 12.46 (br, 1 H), 7.94-7.99 (m, 2 H), 7.24-7.30 (m, 2 H), 7.00- 7.03 (m, 2 H). МС (ESI): рассчитано для C9H7FN2, 162.1, найдено 163 [M + H]+, 160.6 [M – H]-. |
| 2-(4-метоксифенил)-1H-имидазол (9c) | Выход: 22.2%. 1H ЯМР (500 MHz, CDCl3) δ 7.80 (d, J = 10.0 Hz, 2 H), 7.15 (s, 2 H), 3.86 (s, 3 H). МС (ESI): рассчитано для C10H10N2O, 174.1, найдено 175 [M + H]+, 172.8 [M – H]-. |
| 2-(п-толил)-1H-имидазол (9d) | Выход: 36.1%. 1H ЯМР (500 MHz, CDCl3) δ 7.64 (d, J = 7.5 Hz, 2 H), 7.16 (d, J = 7.5 Hz, 2 H), 7.12 (s, 1 H), 7.02 (s, 1 H). МС (ESI): рассчитано для C10H10N2, 158.1, найдено 159.0 [M + H]+, 156.8 [M – H]-. |
| 2-(3,4,5-триметоксифенил)-1H-имидазол (9e) | Выход: 26.0%. 1H ЯМР (500 MHz, CDCl3) δ 7.26 (s, 2 H), 7.08 (d, J = 1.5 Hz, 2 H), 3.86 (s, 3 H), 3.82 (s, 6 H). МС (ESI): рассчитано для C12H14N2O3, 234.1, найдено 234.9 [M + H]+. |
| 2-(4-хлорфенил)-1H-имидазол (9f) | Выход: 19.8%. 1H ЯМР (500 MHz, ДМСО-d 6 ) δ 13.60 (br, 1 H), 7.94 (d, J = 8.5 Hz, 2 H), 7.51 (d, J = 8.0 Hz, 2 H), 7.27 (s, 1 H), 7.03 (s, 1 H). МС (ESI): рассчитано для C9H7ClN2, 178.0, найдено 178.9 [M + H]+. |
| 4-(1H-имидазол-2-ил)-N,N-диметиланилин (9g) | Выход: 16.5%. 1H ЯМР (300 MHz, CDCl3) δ 7.70 (dd, J = 7.0 Hz, 2.0 Hz, 2 H), 7.10 (s, 2 H), 6.75 (dd, J = 9.0 Hz, 2.0 Hz, 2 H), 3.02 (s, 6 H). МС (ESI): рассчитано для C11H13N3, 187.1, найдено 187.9 [M + H]+, 185.8 [M – H]-. |
| 2-(3,4-диметоксифенил)-1H-имидазол (9h) | Выход: 22.0 %. 1H ЯМР (500 MHz, CDCl3) δ 7.52 (d, J = 1.5 Hz, 1 H), 7.27-7.28 (m, 1 H), 7.14 (s, 2 H), 6.88 (d, J = 8.0 Hz, 1 H), 3.91 (s, 3 H), 3.87 (s, 3 H). МС (ESI): рассчитано для C11H12N2O2, 204.1, найдено 205.1 [M + H]+, 202.8 [M – H]-. |
| 2-(2-(трифторметил)фенил)-1H-имидазол (9i) | Выход: 25.5%. 1H ЯМР (500 MHz, ДМСО-d 6 ) δ 12.31 (br, 1 H), 7.84 (d, J = 8.0 Hz, 1 H), 7.76 (t, J = 8.0 Hz, 1 H), 7.65 (t, J = 7.5 Hz, 1 H), 7.16 (br, 2 H). МС (ESI): рассчитано для C10H7F3N2, 212.1, найдено 212.9 [M + H]+, 210.7 [M – H]-. |
| 2-(4-(бензилокси)фенил)-1H-имидазол (9j) | Выход: 12.1%. 1H ЯМР (500 MHz, CDCl3) δ 7.77 (d, J = 8.5 Hz, 2 H), 7.36-7.47 (m, 5 H), 7.10-7.18 (m, 2 H), 7.06 (d, J = 9.0 Hz, 2 H), 5.13 (s, 2 H). МС (ESI): рассчитано для C16H14N2O, 250.1, найдено 251.1 [M + H]+, 248.8 [M – H]-. |
| 2-(4-Бромфенил)-1H-имидазол (9l) | Выход: 19.5%. 1H ЯМР (300 MHz, CDCl3) δ 12.59 (s, 1 H), 7.87 (d, J = 8.1 Hz, 2 H), 7.64 (d, J = 8.1 Hz, 1 H), 7.27 (s, 1 H), 7.04 (s, 1 H). МС (ESI) рассчитано для C9H7BrN2 222.0, найдено 222.8 [M + H]+. |
| 2-(4-(Трифторметил)фенил)-1H-имидазол (9p) | Выход: 26.2%; 1H ЯМР (500 MHz, CDCl3) δ 8.03 (d, J = 8.0 Hz, 2 H), 7.66 (d, J = 8.0 Hz, 2 H), 7.25 (s, 2 H). МС (ESI) рассчитано для C10H7F3N2 212.1, найдено 213.1 [M + H]+ . |
| 2-(4-нитрофенил)-1H-имидазол (9x) | Выход: 53.7%. 1H ЯМР (500 MHz, ДМСО-d 6 ) δ 12.97 (br, 1 H), 8.32 (d, J =9.0 Hz, 2 H), 8.17 (d, J = 9.0 Hz, 2 H), 7.42 (s, 1 H), 7.17 (s, 1H). МС (ESI): рассчитано для C9H7N3O2, 189.1, найдено 189.9 [M + H]+, 187.8 [M – H]-. |
| 2-фенил-1-(фенилсульфонил)-1H-имидазол (10a) | Выход: 50.3%. 1H ЯМР (500 MHz, CDCl3) δ 7.64-7.67 (m, 1 H), 7.56 (t, J = 9.0 Hz, 1 H), 7.32-7.48 (m, 9 H), 7.12-7.16 (m, 1 H). МС (ESI): рассчитано для C15H12N2O2S, 284.1, найдено 307.1 [M + Na]+. |
| 2-(4-фторфенил)-1-(фенилсульфонил)-1H-имидазол (10b) | Выход: 56.9%. 1H ЯМР (500 MHz, CDCl3) δ 7.66 (d, J = 2.0 Hz, 1 H), 7.58 (t, J = 10.0 Hz, 1 H), 7.36-7.42 (m, 6 H), 7.12 (d, J = 2.0 Hz, 1 H), 7.06 (t, J = 10.0 Hz, 2 H). МС (ESI): рассчитано для C15H11FN2O2S, 302.1, найдено 300.8 [M – H]-. |
| 2-(4-метоксифенил)-1-(фенилсульфонил)-1H-имидазол (10c) | Выход: 40.9%. 1H ЯМР (500 MHz, CDCl3) δ 7.62 (d, J = 5.0 Hz, 1 H), 7.56 (tt, J = 15.0 Hz, 5.0 Hz, 1 H), 7.32-7.43 (m, 6 H), 7.10 (d, J = 5.0 Hz, 1 H), 6.88 (dt, J = 16.0 Hz, 6.0 Hz, 2 H), 3.87 (s, 3 H). МС (ESI): рассчитано для C16H14N2O3S, 314.1, найдено 337.1 [M +Na]+, 312.9 [M – H]-. |
| 1-(фенилсульфонил)-2-(п-толил)-1H-имидазол (10d) | Выход: 46.6%. 1H ЯМР (500 MHz, CDCl3) δ 7.63 (d, J = 1.0 Hz, 1 H), 7.55 (t, J = 8.0 Hz, 1 H), 7.42 (d, J = 8.0 Hz, 2 H), 7.35 (t, J = 7.5 Hz, 2 H), 7.27-7.29 (m, 2 H), 7.16 (d, J = 7.5 Hz, 2 H), 7.10 (s, 1 H), 2.41 (s, 3 H). МС (ESI): рассчитано для C16H14N2O2S, 298.1, найдено 321.1 [M + Na]+. |
| 1-(фенилсульфонил)-2-(3,4,5-триметоксифенил)-1H-имидазол (10e) | Выход: 55.7%. 1H ЯМР (500 MHz, CDCl3) δ 7.68 (d, J = 1.5 Hz, 1 H), 7.55 (t, J = 7.0 Hz, 1 H), 7.42 (d, J = 7.5 Hz, 2 H), 7.35 (t, J = 8.5 Hz, 2 H), 7.11 (d, J = 1.5 Hz, 2 H), 6.60 (s, 1 H), 3.90 (s, 3 H), 3.79 (s, 6 H). МС (ESI): рассчитано для C18H18N2O5S, 374.1, найдено 397.1 [M + Na]+. |
| 2-(4-хлорфенил)-1-(фенилсульфонил)-1H-имидазол (10f) | Выход: 54.9%. 1H ЯМР (500 MHz, CDCl3) δ 7.65 (d, J = 2.0 Hz, 1 H), 7.58 (t, J = 7.5 Hz, 1 H), 7.43 (d, J = 8.5 Hz, 2 H), 7.38 (t, J = 8.0 Hz, 2 H), 7.34-7.36 (m, 4 H), 7.12 (d, J = 1.5 Hz, 1 H). МС (ESI): рассчитано для C15H11ClN2O2S, 318.0, найдено 341.0 [M + Na]+. |
| N,N-диметил-4-(1-(фенилсульфонил)-1H-имидазол-2-ил) aniline (10g) | Выход: 48.3%. 1H ЯМР (300 MHz, CDCl3) δ 7.59 (d, J = 2.0 Hz, 1 H), 7.55 (t, J = 8.0 Hz, 1 H), 7.45 (d, J = 7.5 Hz, 2 H), 7.28-7.38 (m, 4 H), 7.07 (d, J = 2.0 Hz, 1 H), 6.68 (d, J = 8.5 Hz, 2 H), 3.04 (s, 3 H). МС (ESI): рассчитано для C17H17N3O2S, 327.10, найдено 350.0 [M + Na]+, 325.9 [M – H]-. |
| 4-(1-((4-Метоксифенил)сульфонил)-1H-имидазол-2-ил)-N,N-диметиланилин (10gb) | Выход: 61.5%. 1H ЯМР (500 MHz, CDCl3) δ 7.58 (d, J = 1.5 Hz, 1 H), 7.36 (t, J = 8.43 Hz, 4 H), 7.03 - 7.09 (m, 1 H), 6.80 (d, J = 9.0 Hz, 2 H), 6.69 (d, J = 8.8 Hz, 2 H), 3.84 (s, 3 H), 3.05 (s, 6 H). МС (ESI): рассчитано для C17H17N3O2S, 327.1, найдено 358.2 [M + Na]+. |
| 2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1H-имидазол (10h) | Выход: 60.3%. 1H ЯМР (500 MHz, CDCl3) δ 7.64 (d, J = 7.0 Hz, 1 H), 7.55 (t, J = 7.5 Hz, 1 H), 7.40 (dd, J = 8.5 Hz, 1.5 Hz, 2 H), 7.35 (t, J = 8.0 Hz, 2H), 7.09 (d, J = 2.0 Hz, 1 H), 7.02 (dd, J = 8.0 Hz, 2.0 Hz, 1 H), 6.89 (d, J = 1.5 Hz, 1 H), 6.86 (d, J = 8.0 Hz, 1 H), 3.95 (s, 3 H), 3.81 (s, 3 H). МС (ESI): рассчитано для C17H16N2O4S, 344.10, найдено 367.0 [M + Na]+. |
| 1-(фенилсульфонил)-2-(2-(трифторметил)фенил)-1H-имидазол (10i) | Выход: 58.6%. 1H ЯМР (500 MHz, CDCl3) δ 7.64-7.67 (m, 2 H), 7.61-7.63 (m, 3 H), 7.40-7.46 (m, 5 H), 7.16 (d, J = 1.5 Hz, 1 H). МС (ESI): рассчитано для C16H11F3N2O2S, 352.10, найдено 353.1 [M + H]+. |
| 2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1H-имидазол (10j) | Выход: 62.0%; т.пл. 102 – 104 oC. 1H ЯМР (500 MHz, CDCl3) δ 7.56 (d, J = 1.0 Hz, 1 H), 7.46 (t, J = 8.0 Hz, 1 H), 7.20-7.40 (m, 11 H), 7.03 (d, J = 1.0 Hz, 1H), 6.89 (t, J = 8.0 Hz, 2 H), 5.08 (s, 2 H). МС (ESI): рассчитано для C22H18N2O3S, 390.10, найдено 413.1 [M + Na]+. ВЭЖХ2: t R 18.22 мин., чистота 95.9%. |
| 2-(4-Бромфенил)-1-(фенилсульфонил)-1H-имидазол (10la) | Выход: 61.2%. 1H ЯМР (500 MHz, CDCl3) δ 7.71 (d, J = 2.0 Hz, 1 H), 7.64 (t, J = 7.0 Hz, 1 H), 7.57 (d, J = 9.0 Hz, 2 H), 7.49 (d, J = 7.0 Hz, 2 H), 7.45 (t, J = 9.0 Hz, 2 H), 7.34 (d, J = 8.5 Hz, 2 H), 7.18 (d, J = 1.5 Hz, 1 H). МС (ESI) рассчитано для C15H11BrN2O2S 362.0, найдено 363.0 [M + H]+. |
| 1-(Фенилсульфонил)-2-(4-(трифторметил)фенил)-1H-имидазол (10p) | Выход: 36.7%; 1H ЯМР (500 MHz, CDCl3) δ 7.75 (d, J = 2.0 Hz, 1 H), 7.69 (d, J = 8.0 Hz, 2 H), 7.65 (t, J = 8.0 Hz, 1 H), 7.60 (d, J = 8.0 Hz, 2 H), 7.48 (d, J = 7.5 Hz, 2 H), 7.43 (t, J = 8.0 Hz, 2 H), 7.22 (d, J = 2.0 Hz, 1 H). МС (ESI) рассчитано для C16H11F3N2O2S 352.1, найдено 553.1 [M + H]+ . |
| 2-(4-нитрофенил)-1-(фенилсульфонил)-1H-имидазол (10x) | Выход: 50%; т.пл. 145 – 147oC. 1H ЯМР (500 MHz, ДМСО-d 6 ) δ 8.28 (d, J =8.5 Hz, 2 H), 8.03 (d, J = 1.5 Hz, 1 H), 7.78 (t, J = 7.5 Hz, 1 H), 7.64-7.68 (m, 4H), 7.60 (t, J = 8.0 Hz, 2 H), 7.30 (d, J = 1.5 Hz, 1 H). МС (ESI): рассчитано для C15H11N3O4S, 329.10, найдено 352.0 [M + Na]+, 327.9 [M – H]-. ВЭЖХ2: t R 14.87 мин., чистота 98.8%. |
| (4-метоксифенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанон (11ab) | Выход: 26.3%; т.пл. 118 – 120oC. 1H ЯМР (500 MHz, ДМСО-d 6 ) δ 8.37 (d, J = 1.0 Hz, 1 H), 8.15-8.18 (m, 2 H), 8.12 (d, J = 9.0 Hz, 2 H), 7.56-7.64 (m, 5 H), 7.46-7.50 (m, 3 H), 7.16 (d, J = 8.0 Hz, 2 H), 3.90 (s, 3 H). МС (ESI): рассчитано для C23H18N2O4S, 418.10, найдено 419.1 [M + H]+. ВЭЖХ2: t R 17.72 мин., чистота 95.7%. |
| (3-метоксифенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанон (11ac) | Выход: 31.2%; т.пл. 136 – 138oC. 1H ЯМР (500 MHz, CDCl3) δ 8.35 (s, 1 H), 7.86 (d, J = 8.0 Hz, 1 H),7.72 (s, 1 H), 7.60 (t, J = 7.5 Hz, 1 H), 7.51 (t, J = 7.5 Hz, 1 H), 7.35-7.42 (m, 9H), 7.14 (dd, J = 8.0 Hz, 2.0 Hz, 1 H), 3.88 (s, 3 H). МС (ESI): рассчитано для C23H18N2O4S, 418.10, найдено 419.1 [M + H]+. ВЭЖХ2: t R 17.72 мин., чистота 95.7%. |
| (2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)(п-толил)метанон (11ah) | Выход: 28.9%; т.пл. 108 – 110oC. 1H ЯМР (500 MHz, CDCl3) δ 8.00 (d, J = 7.5 Hz, 2 H), 7.98 (q, J = 8.0 Hz, 1.5 Hz, 2 H), 7.91 (d, J = 8.0 Hz, 1 H), 7.81 (s, 1 H), 7.44-7.48 (m, 3 H), 7.35-7.40 (m, 2 H), 7.30 (t, J = 8.0 Hz, 2 H), 7.20 (s, 2 H), 2.42 (s, 3 H). МС (ESI): рассчитано для C23H18N2O3S, 402.10, найдено 403.1 [M + H]+. ВЭЖХ2: t R 16.06 мин., чистота 96.2%. |
| (4-фторфенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанон (11af) | Выход: 25.4%; т.пл. 114 – 116oC. 1H ЯМР (500 MHz, CDCl3) δ 8.10 (q, J = 3.5 Hz, 5.5 Hz, 2 H), 7.88 (d, J = 7.5 Hz, 2 H), 7.67 (t, J = 7.5 Hz, 1 H), 7.48 - 7.54 (m, 3 H), 7.38 - 7.41 (m, 5 H), 7.24 (t, J = 8.5 Hz, 2 H). МС (ESI): рассчитано для C22H15FN2O3S, 406.10, найдено 429.1 [M + Na]+. ВЭЖХ2: t R 15.43 мин., чистота 96.1%. |
| (3-фторфенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанон (11ag) | Выход: 18.3%; т.пл. 102 – 104oC. 1H ЯМР (500 MHz, CDCl3) δ 8.14 (d, J = 7.5 Hz, 1 H), 7.76 - 7.87 (m, 3 H), 7.74 (d, J = 9.0 Hz, 1 H), 7.37 - 7.57 (m, 10 H), 7.38 - 7.41 (m, 5 H), 7.24 (t, J = 8.5 Hz, 2 H). МС (ESI): рассчитано для C22H15FN2O3S, 406.10, найдено 429.1 [M + Na]+. ВЭЖХ2: t R 15.75 мин., чистота 96.5%. |
| (4-фторфенил)(2-(4-метоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)метанон (11cb) | Выход: 23.5%; т.пл. 135 – 137oC. 1H ЯМР (500 MHz, CDCl3) δ 8.00 (d, J = 5.5 Hz, 2 H), 7.74 - 7.76 (m, 2 H), 7.54-7.58 (m, 1 H), 7.40 (d, J = 7.0 Hz, 2 H), 7.28-7.30 (m, 3 H), 7.14 - 7.16 (m, 2 H), 6.80-6.82 (m, 2 H), 3.80 (s, 3 H). МС (ESI): рассчитано для C23H17FN2O4S, 436.10, найдено 459.0 [M + Na]+, 434.9 [M – H]-. ВЭЖХ2: t R 16.53 мин., чистота 96.1%. |
| (1-(фенилсульфонил)-2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11da) | Выход: 33.8%; 1H ЯМР (500 MHz, CDCl3) δ 8.00 (d, J = 8.0 Hz, 2 H), 7.70 (t, J = 7.5 Hz, 1 H), 7.55 (t, J = 8.0 Hz, 2 H), 7.44 (s, 2 H), 7.34 (s, 2H), 7.31 (d, J = 8.0 Hz, 2 H), 7.21 (d, J = 8.0 Hz, 2 H), 4.00 (s, 3 H), 3.98 (s, 6 H). МС (ESI): рассчитано для C26H24N2O6S, 492.14, найдено 515.2 [M + Na]+. |
| (4-фторфенил)(1-(фенилсульфонил)-2-(п-толил)-1H-имидазол-4-ил)метанон (11db) | Выход: 18.6%; т.пл. 142 – 144oC. 1H ЯМР (500 MHz, CDCl3) δ 8.07 (q, J = 8.5 Hz, 5.5 Hz, 2 H), 7.88 (d, J = 7.5 Hz, 2 H), 7.64 (t, J = 8.0 Hz, 1 H), 7.49 (d, J = 8.0 Hz, 2 H), 7.38 (s, 1H), 7.30 (d, J = 8.0 Hz, 2 H), 7.18 - 7.24 (m, 4 H), 2.43 (s, 3 H). МС (ESI): рассчитано для C23H17FN2O3S, 420.10, найдено 443.0 [M + Na]+, 418.9 [M – H]-. ВЭЖХ2: t R 17.28 мин., чистота 97.3%. |
| (1-(фенилсульфонил)-2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ea) | Выход: 21.1%; т.пл. 135 – 137oC. 1H ЯМР (500 MHz, CDCl3) δ 7.91 (d, J =8.0 Hz, 2 H), 7.65 (t, J = 7.5 Hz, 1 H), 7.51 (t, J = 8.0 Hz, 2 H), 7.44 (s, 1 H), 7.34 (s, 2 H), 6.60 (s, 2 H), 3.98 (s, 3 H), 3.96 (s, 6 H), 3.91 (s, 3 H), 3.73 (s, 6 H). МС (ESI): рассчитано для C28H28N2O9S, 568.2, найдено 569.2 [M + H]+. ВЭЖХ1: t R 17.86 мин., чистота 98.9%. |
| (4-фторфенил)(1-(фенилсульфонил)-2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)метанон (11eb) | Выход: 18.8%; т.пл. 135 – 137oC. 1H ЯМР (500 MHz, CDCl3) δ 8.11 (q, J =5.5 Hz, 3.0 Hz, 1 H), 8.00 -8.03 (m, 1 H), 7.82 (d, J = 7.5 Hz, 1 H), 7.78 (s, 1 H), 7.64 (t, J =7.0 Hz, 1 H), 7.48 (t, J =8.0 Hz, 1 H), 7.42 (s, 1 H), 7.21 - 7.26 (m, 4 H), 6.62 (s, 1 H), 3.98 (s, 3 H), 3.96 (s, 6 H), 3.93 (s, 3 H). МС (ESI): рассчитано для C25H21FN2O6S, 496.10, найдено 497.1 [M + H]+. ВЭЖХ2: t R 15.26 мин., чистота 98%. |
| (2-(4-хлорфенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанон (11fb) | Выход: 36.8%; т.пл. 153 – 155oC. 1H ЯМР (500 MHz, CDCl3) δ 8.06 (q, J =5.5 Hz, 3.0Hz, 2 H), 7.89 (d, J =7.5 Hz, 2 H), 7.68 (t, J =8.0 Hz, 1 H), 7.52 (t, J = 8.0 Hz, 2 H), 7.34-7.38 (m, 5H), 7.23 (t, J =8.5 Hz, 2 H). МС (ESI): рассчитано для C22H14ClFN2O3S, 440.0, найдено 463.0 [M + Na]+. ВЭЖХ2: t R 17.72 мин., чистота 97.38%. |
| (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ga) | Выход: 32.2%; т.пл. 157 – 159oC. 1H ЯМР (500 MHz, CDCl3) δ 7.89 (d, J =8.0 Hz, 2 H), 7.62 (t, J =7.5 Hz, 1 H), 7.48 (t, J =8.0 Hz, 2 H), 7.43 (s, 1 H), 7.32 (d, J =8.5 Hz, 2 H), 7.30 (s, 2H), 6.62 (d, J =9.0 Hz, 2 H), 3.97 (s, 3 H), 3.95 (s, 6 H), 3.05 (s, 6 H). МС (ESI): рассчитано для C27H27N3O6S, 521.2, найдено 544.1 [M + Na]+, 519.8 [M – H]-. ВЭЖХ2: t R 16.00 мин., чистота 97.9%. |
| (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанон (11gb) | Выход: 38.5%; т.пл. 125 – 127oC. 1H ЯМР (500 MHz, CDCl3) δ 8.04 (q, J =5.5 Hz, 3.5Hz, 2 H), 7.80 (d, J =7.5 Hz, 2 H), 7.61 (t, J =8.0 Hz, 1 H), 7.45 (t, J =8.0 Hz, 2 H), 7.39 (s, 1 H), 7.35 (d, J =9.0 Hz, 2 H), 7.21 (t, J =8.5 Hz, 2 H), 6.62 (d, J =9.0 Hz, 2 H), 3.05 (s, 6 H). МС (ESI): рассчитано для C24H20FN3O3S, 449.10, найдено 472.1 [M + Na]+, 447.9 [M – H]-. ВЭЖХ2: t R 16.85 мин., чистота 96.5%. |
| (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ha) | Выход: 28.6%; т.пл. 136 – 138oC. 1H ЯМР (300 MHz, CDCl3) δ 7.92 (dd, J =8.5 Hz, 1.5 Hz, 2 H), 7.66 (t, J =7.5 Hz, 2 H), 7.51 (t, J =7.5 Hz, 2 H), 7.43 (s, 1 H), 7.33 (s, 2 H), 7.02 (dd, J =8.0 Hz, 2.0 Hz, 1 H), 6.91 (d, J =2.0 Hz, 1 H), 6.86 (d, J =8.5 Hz, 1 H), 3.98 (s, 3 H), 3.96 (s, 9 H), 3.77 (s, 3 H). МС (ESI): рассчитано для C27H26N2O8S, 538.10, найдено 561.1 [M + Na]+, 536.8 [M – H]-. ВЭЖХ2: t R 14.67 мин., чистота 98.2%. |
| (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанон (11hb) | Выход: 31.9%; т.пл. 144 – 145oC. 1H ЯМР (300 MHz, CDCl3) δ 8.09 (q, J =5.5 Hz, 3.5 Hz, 2 H), 7.81 (d, J =8.0 Hz, 2 H), 7.62 (t, J =7.5 Hz, 2 H), 7.48 (t, J =7.5 Hz, 2 H), 7.40 (s, 1 H), 7.21-7.25 (m, 2 H), 7.04 (dd, J =8.0 Hz, 2.0 Hz, 1 H), 6.92 (d, J =2.0 Hz, 1 H), 6.86 (d, J =8.5 Hz, 1 H),3.96 (s, 3 H), 3.79 (s, 6 H). МС (ESI): рассчитано для C24H19FN2O5S, 466.10, найдено 489.1 [M + Na]+, 464.8 [M – H]-. ВЭЖХ2: t R 15.52 мин., чистота 97.4%. |
| (1-(фенилсульфонил)-2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ia) | Выход: 25.0%; т.пл. 155 – 157oC. 1H ЯМР (500 MHz, ДМСО-d 6 ) δ 7.91 (d, J =8.0 Hz, 1 H), 7.84 (q, J =7.5 Hz, 5.0 Hz, 2 H), 7.77-7.80 (m, 2 H), 7.75 (s, 2 H), 7.66 (t, J =8.0 Hz, 2 H), 7.56 (d, J =7.5 Hz, 1 H), 7.18 (s, 2 H), 3.87 (s, 6 H), 3.81 (s, 3 H). МС (ESI): рассчитано для C26H21F3N2O6S, 546.10, найдено 569.0 [M + Na]+. ВЭЖХ2: t R 16.16 мин., чистота 98.9%. |
| (1-(фенилсульфонил)-2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанон (11ib) | Выход: 25.0%; т.пл. 151 – 153 oC. 1H ЯМР (500 MHz, CDCl3) δ 8.03 (q, J =5.5 Hz, 3.0 Hz, 2 H), 7.90 (d, J =8.0 Hz, 2 H), 7.80 (d, J =8.0 Hz, 1 H), 7.69 (q, J =7.0 Hz, 6.5 Hz, 2 H), 7.61 (t, J =8.0 Hz, 1 H), 7.52 (t, J =8.0 Hz, 2 H), 7.34 - 7.36 (m, 2 H), 7.23 (t, J =8.5 Hz, 2 H). МС (ESI): рассчитано для C23H14F4N2O3S, 474.10, найдено 497.0 [M + Na]+. ВЭЖХ2: t R 16.80 мин., чистота 98.2%. |
| (2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанон (11jb) | Выход: 22.3.0%; т.пл. 149 – 151oC. 1H ЯМР (500 MHz, CDCl3) δ 8.09 (q, J =5.5 Hz, 3.5 Hz, 2 H), 7.82 (d, J =7.5 Hz, 2 H), 7.63 (t, 7.5 Hz, 1 H), 7.36-7.50(m, 10 H), 7.25 (t, J =8.5 Hz, 2 H), 6.98 (d, J =8.0 Hz, 2 H), 5.17 (s, 2 H). МС (ESI): рассчитано для C29H21FN2O4S, 512.10, найдено 535.0 [M + Na]+. ВЭЖХ2: t R 18.35 мин., чистота 95.1%. |
| (2-(4-бромфенил)-1-(фенилсульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11la) | Выход: 32.6% 1H ЯМР (500 MHz, CDCl3) δ 8.06 (d, J = 8.0 Hz, 2 H), 7.88 (d, J = 8.5 Hz, 1 H), 7.77 (t, J = 7.0 Hz, 1 H), 7.54-7.63 (m, 4 H), 7.31-7.36 (m, 4 H), 4.04 (s, 3 H), 4.01 (s, 6 H). МС (ESI) рассчитано для C25H21BrN2O6S 556.0, найдено 557.0 [M + H]+. |
| (1-(фенилсульфонил)-2-(4-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11pa) | Выход: 36.7%; 1H ЯМР (500 MHz, CDCl3) δ 8.06 (d, J = 7.5 Hz, 2 H), 7.78 (t, J = 8.0 Hz, 1 H), 7.72 (d, J = 8.0 Hz, 2 H), 7.62 (d, J = 8.0 Hz, 2 H), 7.59 (d, J = 8.0 Hz, 2 H), 7.50 (s, 1 H), 7.37 (s, 2 H), 4.04 (s, 3 H), 4.02 (s, 6 H). МС (ESI) рассчитано для C26H21F3N2O6S 546.1, найдено 547.1 [M + H]+ . |
| (2-(4-(диметиламино)фенил)-1-((4-метоксифенил)сульфонил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11gaa) | Выход: 34.1%; т.пл. 147-149°C. 1H ЯМР (500 MHz, CDCl3) δ 8.07 (q, J = 8.5 Hz, 5.5 Hz, 2 H), 7.78 (d, J = 9.0 Hz, 2 H), 7.41 (d, J = 8.5 Hz, 2 H), 7.39 (s, 1 H), 7.23 (t, J = 8.5 Hz, 2 H), 6.91 (d, J = 9.0 Hz, 2 H), 6.68 (d, J = 9.0 Hz, 2 H), 3.89 (s, 3 H), 3.08 (s, 3 H). МС (ESI) рассчитано для C28H29N3O7S 551.2, найдено 573.1 [M + Na]+. ВЭЖХ2: tR 18.6 мин., чистота 96.9%. |
| (2-фенил-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12aa) | Выход: 10.1%; т.пл. 227-229oC. 1H ЯМР (500 MHz, CDCl3) δ 8.0-8.03 (m, 2 H), 7.83 (s, 1 H), 7.34-7.38 (m, 3 H), 7.21 (s, 2 H), 3.90 (s, 3 H), 3.84 (s, 6 H). МС (ESI): рассчитано для C19H18N2O, 338.1, найдено 337.1 [M – H] -. ВЭЖХ2: t R14.19 мин., чистота 96.3%. |
| (4-метоксифенил)(2-фенил-1H-имидазол-4-ил)метанон (12ab) | Выход: 16.6%; т.пл. 179 – 181oC. 1H ЯМР (500 MHz, CDCl3) δ 11.1 (br, 1 H), 8.07-8.10 (m, 2 H), 8.04 (d, J = 8.5 Hz, 2 H), 7.84 (d, J = 1.0 Hz, 1 H), 7.49-7.51 (m, 3 H), 7.07 (d, J = 9.0 Hz, 2 H), 3.95 (s, 3 H). МС (ESI): рассчитано для C17H14N2O2, 278.10, найдено 279.0 [M + H]+. ВЭЖХ1: t R 15.14 мин., чистота > 99%. |
| (3-метоксифенил)(2-фенил-1H-имидазол-4-ил)метанон (12ac) | Выход: 22.5%; т.пл. 160 – 162oC. 1H ЯМР (500 MHz, CDCl3) δ 11.2 (br, 1 H), 8.10-8.12 (m, 2 H), 7.87 (d, J = 1.0 Hz, 1 H), 7.61 (d, J = 7.5 Hz, 1 H), 7.48 - 7.52 (m, 5 H), 7.21 (dd, J = 2.5 Hz, 8.5Hz, 1 H), 3.91 (s, 3 H). МС (ESI): рассчитано для C17H14N2O2, 278.10, найдено 279.0 [M + H]+. ВЭЖХ2: t R 15.07 мин., чистота > 99%. |
| (3,5-диметоксифенил)(2-фенил-1H-имидазол-4-ил)метанон (12ad) | Выход: 26.2%; т.пл. 168 – 170oC. 1H ЯМР (500 MHz, CDCl3) δ 8.04-8.06 (m, 2 H), 7.88 (s, 1 H), 7.50-7.52 (m, 3 H), 7.15 (d, J = 2.0 Hz, 2 H), 6.75 (t, J = 1.0 Hz, 1 H), 3.89 (s, 6 H). МС (ESI): рассчитано для C18H16N2O3, 308.10, найдено 331.1 [M + Na]+, 306.9 [M – H]-. ВЭЖХ2: t R 15.59 мин., чистота > 99%. |
| (3,4-диметоксифенил)(2-фенил-1H-имидазол-4-ил)метанон (12ae) | Выход: 18.6%; т.пл. 162 – 164oC. 1H ЯМР (500 MHz, CDCl3) δ 10.9 (br, 1 H), 8.05 (dd, J = 1.5 Hz, 8.0 Hz, 2 H), 7.86 (d, J = 1.5 Hz, 1 H), 7.74 (dd, J = 2.0 Hz, 8.5 Hz, 1 H), 7.56 (d, J = 2.0 Hz, 1 H), 7.50-7.52 (m, 3 H), 7.04 (d, J = 8.5 Hz, 1 H), 4.03 (s, 3 H), 3.99 (s, 3 H). МС (ESI): рассчитано для C18H16N2O3, 308.10, найдено 331.1 [M + Na]+, 306.9 [M – H]-. ВЭЖХ2: t R 13.54 мин., чистота > 99%. |
| (4-фторфенил)(2-фенил-1H-имидазол-4-ил)метанон (12af) | Выход: 30.2%; т.пл. 231 – 233oC. 1H ЯМР (500 MHz, CDCl3) δ 10.6 (br, 1 H), 8.02-8.05 (m, 4 H), 7.81 (d, J = 1.0 Hz, 1 H), 7.51-7.54 (m, 3 H), 7.27 (t, J = 8.5 Hz, 2 H). МС (ESI): рассчитано для C16H11FN2O, 266.10, найдено 267.0 [M +H]+, 264.8 [M – H]-. ВЭЖХ1: t R 15.37 мин., чистота 98.9%. |
| (3-фторфенил)(2-фенил-1H-имидазол-4-ил)метанон (12ag) | Выход: 23.4%; т.пл. 212 – 214oC. 1H ЯМР (500 MHz, CDCl3) δ 8.05 (dd, J = 1.5 Hz, 7.5 Hz, 2 H), 7.86 (s, 1 H), 7.84 (d, J = 7.0 Hz, 1 H), 7.74 (d, J = 8.5 Hz, 1 H), 7.52-7.58 (m, 4 H), 7.37 (dt, J =2.0 Hz, 6.0 Hz, 1 H). МС (ESI): рассчитано для C16H11FN2O, 266.10, найдено 267.0 [M +H]+, 264.8 [M – H]-. ВЭЖХ1: t R 15.29 мин., чистота > 99%. |
| (2-фенил-1H-имидазол-4-ил)(п-толил)метанон (12ah) | Выход: 15.6%; т.пл. 225 – 227oC. 1H ЯМР (500 MHz, CDCl3) δ 11.1 (br, 1 H), 8.08 (d, J = 7.5 Hz, 2 H), 7.93 (d, J =9.0 Hz, 2 H), 7.84 (s, 1 H), 7.48-7.52 (m, 3 H), 7.38 (d, J = 10.0 Hz, 2 H), 2.50 (s, 3 H). МС (ESI): рассчитано для C17H14N2O, 262.10, найдено 263.0 [M +H]+, 260.8 [M – H]-. ВЭЖХ2: t R 15.86 мин., чистота 98.7%. |
| (2-фенил-1H-имидазол-4-ил)(м-толил)метанон (12ai) | Выход: 20.5%; т.пл. 168 – 169oC. 1H ЯМР (500 MHz, CDCl3) δ 11.0 (br, 1 H), 8.09-8.11 (m, 2 H), 7.84 (d, J = 1.5 Hz, 1 H), 7.81-7.82 (m, 2 H), 7.47-7.52 (m, 5 H), 2.50 (s, 3 H). МС (ESI): рассчитано для C17H14N2O, 262.10, найдено 285.0 [M +Na]+, 260.8 [M – H]-. ВЭЖХ2: t R 15.89 мин., чистота > 99%. |
| (2-(4-фторфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ba) | Выход: 12.2%. т.пл. 176 – 178oC. 1H ЯМР (500 MHz, CDCl3) δ 10.72 (br, 1 H), 8.02 (q, J = 5.0 Hz, 2 H), 7.84 (s, 1 H), 7.19 (t, J = 10.0 Hz, 2 H), 4.00 (s, 6 H), 3.97 (s, 3 H). МС (ESI): рассчитано для C19H17FN2O4, 356.10, найдено 379.1 [M + Na]+, 354.9 [M – H]-. ВЭЖХ1: t R 17.23 мин., чистота > 99% |
| (2-(4-метоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ca) | Выход: 10.2%; т.пл. 220 – 222oC. 1H ЯМР (300 MHz, CDCl3) δ 10.24 (br, 1 H), 7.93 (d, J = 14.5 Hz, 2 H), 7.81 (s, 1 H), 7.24 (s, 2 H), 7.03 (d, J = 14.5 Hz, 2 H), 3.97 (s, 3 H), 3.95 (s, 6 H), 3.90 (s, 3 H). МС (ESI): рассчитано для C20H20N2O5, 368.10, найдено 391.0 [M + Na]+, 367.0 [M – H]-. ВЭЖХ2: t R 14.46 мин., чистота 98.4%. |
| (4-фторфенил)(2-(4-метоксифенил)-1H-имидазол-4-ил)метанон (12cb) | Выход: 15.2%; т.пл. 245 – 247oC. 1H ЯМР (500 MHz, CDCl3) δ 10.20 (br, 1 H), 7.93-7.96 (m, 2 H), 7.85 (d, J = 5.0 Hz, 2 H), 7.68 (s, 1 H), 7.15-7.17 (m, 2 H), 6.95 (d, J = 6.0 Hz, 2 H), 3.82 (s, 3 H). МС (ESI): рассчитано для C17H13FN2O2, 296.10, найдено 319.1 [M + Na]+, 294.9 [M – H]-. ВЭЖХ2: t R 15.40 мин., чистота 98.8%. |
| (2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12da) | Выход: 48.5%; т.пл. 201 – 203oC. 1H ЯМР (500 MHz, CDCl3) δ 10.40 (br, 1 H), 7.88 (d, J = 8.0 Hz, 2 H), 7.82 (s, 1 H), 7.31 (d, J = 8.0 Hz, 2 H), 7.24 (s, 2 H), 3.96 (s, 3 H), 3.94 (s, 6 H), 2.43 (s, 3 H). МС (ESI): рассчитано для C20H20N2O4, 352.10, найдено 375.2 [M + Na]+. ВЭЖХ2: t R 15.45 мин., чистота 97.4%. |
| (4-фторфенил)(2-(п-толил)-1H-имидазол-4-ил)метанон (12db) | Выход: 56.3%; т.пл. 229 – 231oC. 1H ЯМР (500 MHz, CDCl3) δ 10.50 (br, 1 H), 7.99-8.02 (m, 2 H), 7.88 (d, J = 8.0 Hz, 2 H), 7.60 (d, J = 1.0 Hz, 1 H), 7.30 (d, J = 8.0 Hz, 2 H), 7.23 (t, J = 9.0 Hz, 2 H), 2.43 (s, 3 H). МС (ESI): рассчитано для C17H13FN2O, 280.10, найдено 281.0 [M + H]+, 278.9 [M – H]-. ВЭЖХ2: t R 16.31 мин., чистота > 99%. |
| (4-гидрокси-3,5-диметоксифенил)(2-(п-толил)-1H-имидазол-4-ил)метанон (12dc) | Выход: 56.8%; т.пл. 220-222°C. 1H ЯМР (500 MHz, CDCl3) δ 8.02 (d, J = 8.0 Hz, 2H), 7.91(s, 1H), 7.39 (s, 2H), 7.28 (d, J = 7.5 Hz, 2H), 4.00 (s, 6H), 2.44 (s, 3H). МС (ESI) рассчитано для C19H18 FN2O4 338.1, найдено 339.1 [M + H]+. ВЭЖХ1: tR 3.91 мин., чистота > 99%. |
| (3,4,5-триметоксифенил)(2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)метанон (12ea) | Выход: 86.8%; т.пл. 196 – 198oC. 1H ЯМР (500 MHz, ДМСО-d 6 ) δ 13.3 (br, 0.47 H), 13.50 (br, 0.52 H), 8.19 (s, 0.49 H), 7.90 (s, 1 H), 7.83 (s, 0.5 H), 7.59 (s, 1 H), 7.40 (s, 1 H), 7.18 (s, 1 H), 3.89 (s, 6 H), 3.86 (s, 6 H), 3.77 (s, 3 H), 3.72 (s, 3 H). МС (ESI): рассчитано для C22H24N2O7, 428.2, найдено 451.1[M + Na]+, 426.9 [M – H]-. ВЭЖХ2: t R 14.49 мин., чистота > 99%. |
| (4-фторфенил)(2-(3,4,5-триметоксифенил)-1H-имидазол-4-ил)метанон (12eb) | Выход: 90.2%; т.пл. 153 – 155oC. 1H ЯМР (500 MHz, CDCl3) δ 10.42 (br, 1 H), 8.00 (q, J = 5.5 Hz, 3.0Hz, 2 H), 7.76 (s, 1 H), 7.23 (t, J = 8.5 Hz, 2 H), 7.19 (s, 2 H), 3.94 (s, 3 H), 3.92 (s, 3 H). МС (ESI): рассчитано для C19H17FN2O4, 356.1, найдено 379.0 [M + Na]+, 354.9 [M – H]-. ВЭЖХ2: t R 15.31 мин., чистота > 99%. |
| (2-(4-хлорфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12fa) | Выход: 36.9%; т.пл. 193 – 195oC. 1H ЯМР (500 MHz, CDCl3) δ 10.75 (br, 1 H), 7.96 (d, J = 8.5 Hz, 2 H), 7.83 (s, 1 H), 7.47 (d, J = 9.0 Hz, 2 H), 7.23 (s, 2 H), 3.97 (s, 3 H), 3.94 (s, 6 H), 2.43 (s, 3 H). МС (ESI): рассчитано для C19H17ClN2O4, 372.1, найдено 395.1 [M + Na]+, 370.9 [M – H]-. ВЭЖХ2: t R 16.36 мин., чистота > 99%. |
| (2-(4-хлорфенил)-1H-имидазол-4-ил)(4-фторфенил)метанон (12fb) | Выход: 83.7%; т.пл. 232 – 234oC. 1H ЯМР (500 MHz, CDCl3) δ 10.78 (br, 1 H), 8.00 (q, J = 5.5 Hz, 3.0Hz, 2 H), 7.96 (d, J = 9.0 Hz, 2 H), 7.78 (s, 1 H), 7.47 (d, J = 8.0 Hz, 2 H), 7.24 (t, J = 8.5 Hz, 2 H). МС (ESI): рассчитано для C16H10ClFN2O, 300.1, найдено 323.0 [M + Na]+, 298.8 [M – H]-. ВЭЖХ2: t R 17.08 мин., чистота > 99%. |
| (2-(4-хлорфенил)-1H-имидазол-4-ил)(4-гидрокси-3,5-диметоксифенил)метанон (12fc) | Выход: 80.2%; т.пл. 216-218°C. 1H ЯМР (500 MHz, CD3OD) δ 8.06 (d, J = 8.5 Hz, 2 H), 7.99 (s, 1 H), 7.61 (d, J = 8.0 Hz, 2 H), 7.52 (s, 2 H), 4.01 (s, 6 H). МС (ESI) рассчитано для C18H15ClN2O4 358.1, найдено 359.1 [M + H]+. ВЭЖХ2: tR 4.12 мин., чистота > 99%. |
| (2-(4-(диметиламино)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ga) | Выход: 91.2%; т.пл. 195 – 197oC. 1H ЯМР (500 MHz, CDCl3) δ 10.39 (br, 1 H), 7.87 (d, J = 8.5 Hz, 2 H), 7.80 (s, 1 H), 7.23 (s, 2 H), 6.75(d, J = 9.0 Hz, 2 H), 3.95 (s, 3 H), 3.94 (s, 6 H), 3.05 (s, 6 H). МС (ESI): рассчитано для C21H23N3O4, 381.2, найдено 404.2 [M + Na]+, 380.0 [M – H]-. ВЭЖХ2: t R 15.20 мин., чистота 95.8%. |
| (2-(4-(диметиламино)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанон (12gb) | Выход: 86.7%; т.пл. 278 – 280oC. 1H ЯМР (500 MHz, CDCl3) δ 10.21 (br, 1 H), 7.98 (q, J = 5.0 Hz, 3.5Hz, 2 H), 7.84 (d, J = 8.5 Hz, 2 H), 7.72 (s, 1 H), 7.20 (t, J = 8.5 Hz, 2 H), 6.76 (t, J = 9.0 Hz, 2 H), 3.06 (s, 6 H). МС (ESI): рассчитано для C18H16FN3O, 309.1, найдено 332.1 [M + Na]+, 307.9 [M – H]-. ВЭЖХ2: t R 16.06 мин., чистота 95.6%. |
| (2-(3,4-диметоксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ha) | Выход: 85.0%; т.пл. 100 – 102oC. 1H ЯМР (500 MHz, CDCl3) δ 10.19 (br, 1 H), 7.81 (s, 1 H), 7.58 (d, J = 1.5 Hz, 1 H), 7.48 (d, J = 8.0 Hz, 1 H), 7.25 (s, 2 H), 6.97 (d, J = 8.5 Hz, 1 H), 4.00 (s, 3 H), 3.96 (s, 6 H), 3.95 (s, 6 H). МС (ESI): рассчитано для C21H22N2O6, 398.2, найдено 399.1 [M + H]+, 397.0 [M – H]-. ВЭЖХ2: t R 13.73 мин., чистота > 99%. |
| (2-(3,4-диметоксифенил)-1H-имидазол-4-ил)(4-фторфенил)метанон (12hb) | Выход: 78.3%; т.пл. 174 – 176oC. 1H ЯМР (500 MHz, CDCl3) δ 8.02 (t, J = 9.0 Hz, 2 H), 7.75 (s, 1 H), 7.57 (s, 1 H), 7.48 (d, J = 8.5 Hz, 1 H), 7.23 (t, J = 8.5 Hz, 2 H), 6.95 (d, J = 8.5 Hz, 1 H), 3.99 (s, 3 H), 3.96 (s, 3 H). МС (ESI): рассчитано для C18H15FN2O3, 326.1, найдено 349.0 [M + Na]+, 324.9 [M – H]-. ВЭЖХ2: t R 14.65 мин., чистота > 99%. |
| (2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ia) | Выход: 83.8%; т.пл. 75 – 77oC. 1H ЯМР (500 MHz, CDCl3) δ 10.37 (br, 1 H), 8.00-8.02 (m, 1 H), 7.87 (s, 1 H), 7.82-7.85 (m, 1 H), 7.69-7.74 (m, 1 H), 7.62-7.66 (m, 1 H), 7.25 (s, 2 H), 3.99 (s, 3 H), 3.98 (s, 6 H). МС (ESI): рассчитано для C20H17F3N2O4, 406.1, найдено 429.1 [M + Na]+, 405.0 [M – H]-. ВЭЖХ2: t R 13.98 мин., чистота > 99%. |
| (4-фторфенил)(2-(2-(трифторметил)фенил)-1H-имидазол-4-ил)метанон (12ib) | Выход: 91.1%; т.пл. 152 – 154oC. 1H ЯМР (500 MHz, CDCl3) δ 8.12-8.14 (m, 2 H), 7.97 (d, J = 7.5 Hz, 1 H), 7.82-7.85 (m, 2 H), 7.69 (t, J = 7.5 Hz, 1 H), 7.61 (t, J = 8.0 Hz, 1 H), 7.22 (t, J = 9.0 Hz, 2 H). МС (ESI): рассчитано для C17H10F4N2O, 334.1, найдено 357.1 [M + Na]+, 332.9 [M – H]-. ВЭЖХ2: t R 15.10 мин., чистота > 99%. |
| (2-(4-(бензилокси)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ja) | Выход: 16.5%; т.пл. 191 – 193oC. 1H ЯМР (500 MHz, CDCl3) δ 10.22 (br, 1 H), 7.93 (d, J = 9.0 Hz, 2 H), 7.81 (s, 1 H), 7.37-7.47 (m, 5 H), 7.24 (s, 2 H), 7.11 (d, J = 8.5 Hz, 2 H), 5.16 (s, 2 H), 3.97 (s, 3 H), 3.95 (s, 6 H). МС (ESI): рассчитано для C26H24N2O5, 444.2, найдено 467.1 [M + Na]+, 442.9 [M – H]-. ВЭЖХ2: t R 17.36 мин., чистота 95.5%. |
| (2-(4-(бензилокси)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанон (12jb) | Выход: 84.7%; т.пл. 212 – 214oC. 1H ЯМР (300 MHz, CDCl3) δ 10.28 (br, 1 H), 799-8.04 (m, 2 H), 7.92-7.95 (m, 2 H), 7.76 (d, J = 1.5 Hz, 1 H), 7.38-7.48 (m, 5 H), 7.20-7.25 (m, 2 H), 7.09-7.12 (m, 2 H), 5.16 (s, 2 H). МС (ESI): рассчитано для C23H17FN2O2, 372.1, найдено 395.1 [M + Na]+. ВЭЖХ2: t R 17.97 мин., чистота 97.8%. |
| (2-(4-гидроксифенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ka) | Выход: 72.3%. т.пл. 191-193oC. 1H ЯМР (500 MHz, CD3OD) δ 8.31 (s, 1 H), 7.90 (d, J = 8.5 Hz, 2 H), 7.31 (s, 2 H), 7.05 (s, 2 H), 3.95 (s, 6 H), 3.88 (s, 3 H). МС (ESI): рассчитано для C19H18N2O5, 354.1, найдено 355.1 [M + H]+, 352.9 [M – H]-. ВЭЖХ2: t R 12.25 мин., чистота 98.7%. |
| (2-(4-(гидроксифенил)-1H-имидазол-4-ил)(4-фторфенил)метанон (12kb) | Выход: 89.0%; т.пл. 276 – 278oC. 1H ЯМР (500 MHz, CDCl3) δ 8.31 (s, 1 H), 8.13 (q, J = 5.5 Hz, 3.0 Hz, 2 H), 7.93 (d, J = 8.5 Hz, 2 H), 7.38 (t, J = 8.5 Hz, 2 H), 7.07 (d, J = 8.5 Hz, 2 H). МС (ESI): рассчитано для C16H11FN2O2, 282.1, найдено 283.0 [M + H]+, 280.9 [M – H]-. ВЭЖХ2: t R 13.46 мин., чистота 97.65%. |
| (2-(4-бромфенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12la) | Выход: 25.6%; т.пл. 190-192°C. 1H ЯМР (500 MHz, CDCl3) δ 7.99 (d, J = 8.5 Hz, 2 H), 7.92 (s, 1 H), 7.70 (d, J = 8.5 Hz, 2 H), 7.32 (s, 2 H), 4.03 (s, 3 H), 4.00 (s, 6 H). МС (ESI) рассчитано для C19H17 BrN2O4 416.0, найдено 417.0 [M + H]+. ВЭЖХ2: tR 4.24 мин., чистота 98.8%. |
| (2-(4-(трифторметил)фенил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12pa) | Выход: 85.3%; т.пл. 195 – 196°C. 1H ЯМР (500 MHz, CDCl3) δ 8.22 (d, J = 8.5 Hz, 2 H), 7.96 (s, 1 H), 7.83 (d, J = 8.5 Hz, 2 H), 7.34 (s, 2 H), 4.04 (s, 3 H), 4.00 (s, 6 H). МС (ESI) рассчитано для C20H17F3N2O4 406.1, найдено 407.1 [M + H]+, ВЭЖХ2: tR 18.00 мин., чистота >99%. |
| (2-фенил-1H-имидазол-1-ил)(3,4,5-триметоксифенил)метанон (12aaa) | Выход: 39.8%; т.пл. 113 – 115oC. 1H ЯМР (500 MHz, CDCl3) δ 7.53 (q, J = 5.0 Hz, 3.0 Hz, 2 H), 7.41 (d, J = 1.0 Hz, 1 H), 7.33-7.35 (m, 3 H), 7.23 (d, J = 1.0 Hz, 1 H), 7.03 (s, 2 H), 3.93 (s, 3 H), 3.85 (s, 6 H). МС (ESI): рассчитано для C19H18N2O4, 338.1, найдено 339.1 [M +H]+. ВЭЖХ2: t R 13.8 мин., чистота 95.6%. |
| (4-метоксифенил)(2-фенил-1H-имидазол-1-ил)метанон (12aba) | Выход: 56.3%; т.пл. 68 – 70oC. 1H ЯМР (500 MHz, CDCl3) δ 7.78 (d, J = 9.0 Hz, 2 H), 7.54-7.56 (m, 2 H), 7.32-7.34 (m, 4 H), 7.21 (d, J = 1.0 Hz, 1 H), 6.93 (d, J = 8.5 Hz, 2 H), 3.90 (s, 3 H). МС (ESI): рассчитано для C17H14N2O2, 278.1, найдено 301.0 [M +Na]+, 276.8 [M – H]-. ВЭЖХ2: t R 14.72 мин., чистота 95.7%. |
| (4-фторфенил)(2-(п-толил)-1H-имидазол-4-ил)метанон HCl соль (12db-HCl) | Выход: 95%; т.пл. 115 – 117oC. 1H ЯМР (500 MHz, ДМСО-d 6 ) δ 8.20-8.23 (m, 2 H), 8.18 (s, 1 H), 8.04 (d, J = 6.5 Hz, 2 H), 7.42 (t, J = 8.0 Hz, 2 H), 7.37 (d, J = 7.0 Hz, 2 H), 2.38 (s, 3 H). МС (ESI): рассчитано для C17H14FClN2O, 316.1, найдено 281.0 [M - HCl + H]+. ВЭЖХ2: t R 17.16 мин., чистота >99%. |
| (4-фторфенил)(2-(4-метоксифенил)-1-метил-1H-имидазол-4-ил)метанон (12cba) | Выход: 90.2%; т.пл. 148-150°C. 1H ЯМР (500 MHz, CDCl3) δ 8.45 (q, J = 8.5 Hz, 5.5 Hz, 2 H), 7.79 (s, 1 H), 7.63 (d, J = 8.5 Hz, 2 H), 7.16 (t, J = 8.5 Hz, 2 H), 7.03 (d, J = 9.0 Hz, 2 H), 3.89 (s, 3 H), 3.82 (s, 3 H). МС (ESI) рассчитано для C18H15 FN2O2 310.1, найдено 311.0 [M + H]+. ВЭЖХ2: tR 4.01 мин., чистота 97.6%. |
| (1-бензил-2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12daa) | Выход: 92.8%; т.пл. 135-137°C. 1H ЯМР (500 MHz, CDCl3) δ 7.81 (s, 1 H), 7.80(d, J = 6.5 Hz, 2 H), 7.58 (d, J = 8.0 Hz, 2 H), 7.41-7.45 (m, 3 H), 7.31-7.33 (m, 2 H), 7.20 (d, J = 7.0 Hz, 2 H), 5.33 (s, 2 H), 3.99 (s, 3 H), 3.98 (s, 6 H), 2.47 (s, 3 H). МС (ESI) рассчитано для C27H26N2O4 442.2, найдено 443.1 [M + Na]+. ВЭЖХ1: tR 4.28 мин., чистота > 99%. |
| (1-метил-2-(п-толил)-1H-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12dab) | Выход: 87.4%; т.пл. 110-112°C. 1H ЯМР (500 MHz, CDCl3) δ 7.87 (s, 2 H), 7.86 (d, J = 8.0 Hz, 1 H), 7.65 (d, J = 10 Hz, 2 H), 7.37 (d, J = 10 Hz, 2 H), 4.01 (s, 6 H), 4.00 (s, 3 H), 3.90 (s, 3 H). МС (ESI) рассчитано для C21H22N2O4 366.2, найдено 367.2 [M + H]+. ВЭЖХ1: tR 4.23 мин., чистота > 99%. |
| (2-(4-(диметиламино)фенил)-1-((4-метоксифенил)сульфонил)-1H-имидазол-4-ил)(4-фторфенил)метанон (12gba) | Выход: 34.1%; т.пл. 147-149°C. 1H ЯМР (CDCl3, 500 MHz) δ 8.07 (q, J = 8.5 Hz, 5.5 Hz, 2 H), 7.78 (d, J = 9.0 Hz, 2 H), 7.41 (d, J = 8.5 Hz, 2 H), 7.39 (s, 1 H), 7.23 (t, J = 8.5 Hz, 2 H), 6.91 (d, J = 9.0 Hz, 2 H), 6.68 (d, J = 9.0 Hz, 2 H), 3.89 (s, 3 H), 3.08 (s, 3 H). МС (ESI) рассчитано для C25H22FN3O4S 479.1, найдено 502.1 [M + Na]+. ВЭЖХ2: tR 18.6 мин., чистота 96.9%. |
| (3,4,5-тригидроксифенил)(2-(3,4,5-тригидроксифенил)-1H-имидазол-4-ил)метанон (13ea) | Выход: 66.1%. т.пл. 294 – 296oC. 1H ЯМР (500 MHz, CD3OD) δ 8.07 (s, 1 H), 7.07 (s, 2 H), 7.02 (s, 2 H). МС (ESI): рассчитано для C16H12N2O7, 344.1, найдено 345.0[M + H]+, 342.9 [M – H]-. ВЭЖХ2: t R 3.62 мин., чистота 97.9%. |
| (2-(4-хлорфенил)-1H-имидазол-4-ил)(3,4,5-тригидроксифенил)метанон (13fa) | Выход: 79.3%; т.пл. > 300oC. 1H ЯМР (500 MHz, CD3OD) δ 8.02 (d, J = 8.5 Hz, 2 H), 7.77 (s, 1 H), 7.54 (d, J = 8.5 Hz, 2 H), 7.14 (s, 2 H). МС (ESI): рассчитано для C16H11ClN2O4, 330.0, найдено 331.1 [M + Na]+, 328.9 [M – H]-. ВЭЖХ2: t R 11.9 мин., чистота 95.6%. |
| (2-(3,4-дигидроксифенил)-1H-имидазол-4-ил)(3,4,5-тригидроксифенил)метанон (13ha) | Выход: 62.2 %; т.пл. > 300oC. 1H ЯМР (500 MHz, CD3OD) δ 8.11 (s, 1 H), 7.46 (d, J = 2.0 Hz, 1 H), 7.42 (dd, J = 8.5 Hz, 2.0 Hz, 1 H), 7.10 (s, 2 H), 7.02 (d, J = 8.5 Hz, 1 H). МС (ESI): рассчитано для C16H12N2O6, 328.1, найдено 329.0 [M + H]+, 326.9 [M – H]-. ВЭЖХ2: t R 3.64 мин., чистота 97.9%. |
| 2-(4-нитрофенил)-4,5-дигидро-1H-имидазол (14x) | Выход: 70.3%. 1H ЯМР (500 MHz, CDCl3) δ 8.30 (d, J = 9.0 Hz, 2 H), 7.98 (d, J = 8.5 Hz, 2 H), 3.88-3.95 (m, 4 H). МС (ESI): рассчитано для C9H9N3O2, 191.10, найдено 191.9 [M + H]+, 189.7 [M – H]-. |
| 2-(4-фторфенил)-4,5-дигидро-1H-имидазол (14b) | Выход: 60.2%. 1H ЯМР (500 MHz, CDCl3) δ 7.80 (q, J = 7.0 Hz, 2 H), 7.11 (d, J = 10.0 Hz, 2 H), 3.82 (br, 4 H). МС (ESI): рассчитано для C9H9FN2, 164.10, найдено 165 [M + H]+. |
| 2-(4-метоксифенил)-4,5-дигидро-1H-имидазол (14c) | Выход: 56.9%. 1H ЯМР (500 MHz, CDCl3) δ 7.84 (d, J = 8.5 Hz, 2 H), 6.94 (d, J = 9.0 Hz, 2 H), 3.87 (s, 3 H), 3.85 (br, 4 H). МС (ESI): рассчитано для C10H12N2O, 176.10, найдено 177.0 [M + H]+. |
ПРИМЕР 6
СИНТЕЗ НЕКОТОРЫХ ИНДОЛИЛ-БЕНЗОИЛ-ИМИДАЗОЛЬНЫХ СОЕДИНЕНИЙ
Синтез 15xaa представлен на Фигуре 12. Этот способ был изначально разработан для синтеза 12xa, но неселективность бензоилирования в индол-2 и имидазол-4 положениях привела к образованию соединения 15xaa, которое является близко родственным, но более массивным аналогом 11xaa. Индол-5-карбоксальдегид 8x был защищен фенилсульфониловой группой у индольного NH с образованием промежуточного соединения 8xa. 8xa прореагировал с глиоксалем и гидроксидом аммония с образованием 2-арил-имидазола 9xa. Защита имидазольного NH фенилсульфонилом дала промежуточное соединение 10xaa, которое связали с 3,4,5-триметоксибензоил хлоридом для получения 16xaa. В результате удаления защитной группы из 16xaa получили 15xaa.
Синтез 1-(фенилсульфонил)-1Н-индол-5-карбальдегида (8xa). К раствору индол-3-карбоксальдегида (100 ммоль) в этаноле (500 мл) при комнатной температуре добавили гидроксид калия (110 экв.), смесь перемешивали до полной солюбилизации. Этанол полностью удалили в вакууме и добавили ацетон (250 мл), а затем бензолсульфонил хлорид (110 экв.). Осадок отфильтровали, а фильтрат концентрировали и перекристаллизовали из метанола для получения твердого белого вещества. Выход: 32,6%. 1H ЯМР (500 MHz, CDCl3) δ 10.17 (s, 1 H), 8.25 - 8.39 (m, 2 H), 7.97 - 8.09 (m, 3 H), 7.69 (t, J = 7.33 Hz, 1 H), 7.59 (t, J = 7.5 Hz, 2 H), 7.39 - 7.54 (m, 2 H). МС (ESI) рассчитано для C15H11NO3S 285.1, найдено 286.0 [M + H]+.
Синтез (5-(4-(3,4,5-триметоксибензоил)-1Н-имидазол-2-ил)-1Н-индол-2-ил)(3,4,5-триметоксифенил)метанона (15xaa): К раствору (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-2-(3,4,5-триметоксибензоил)-1Н-индол-5-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (16xaa) (1 ммоль) в этаноле (20 мл) добавили гидроксид натрия (10 экв.) и перемешивали в течение ночи в темноте. Реакционную смесь разбавили 50 мл воды и экстрагировали этилацетатом (250 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 3:1) или перекристаллизовали из воды и метанола для получения твердого белого вещества. Выход: 30-95%.
5-(1Н-Имидазол-2-ил)-1-(фенилсульфонил)-1Н-индол (9xa). Выход: 12,0%. 1H ЯМР (500 MHz, ДМСО-d6) δ 8.33 (d, J = 2.9 Hz, 2 H), 8.13 (d, J = 7.8 Hz, 2 H), 7.98 - 8.04 (m, 1 H), 7.62 - 7.67 (m, 1 H), 7.55 (d, J = 7.82 Hz, 2 H), 7.22 - 7.34 (m, 4 H). МС (ESI) рассчитано для C17H13N3O2S 323.1, найдено 324.0 [M + H]+.
1-(Фенилсульфонил)-5-(1-(фенилсульфонил)-1Н-имидазол-2-ил)-1Н-индол (10xaa). Выход: 23.6%. 1H ЯМР (500 MHz, CDCl3) δ 8.01 (d, J = 8.5 Hz, 1 H), 7.95 (d, J = 7.5 Hz, 2 H), 7.73 (d, J = 1.0 Hz, 1 H), 7.70 (d, J = 4.0 Hz, 1 H), 7.63-7.66 (m, 2 H), 7.52-7.56 (m, 3 H), 7.31-7.34 (m, 3 H), 7.22 (t, J = 8.5 Hz, 2 H), 7.17 (s, 1 H), 6.14 (d, J = 3.5 Hz, 1 H). МС (ESI) рассчитано для C23H17N3O4S2 463.1, найдено 464.0 [M + H]+.
(1-(Фенилсульфонил)-2-(1-(фенилсульфонил)-2-(3,4,5-триметоксибензоил)-1Н-индол-5-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (16xaa). Выход: 15,9%. 1H ЯМР (500 MHz, CDCl3) δ 8.18 - 8.25 (m, 3 H), 8.04 (d, J = 8.1 Hz, 2H), 7.70 - 7.78 (m, 2 H), 7.61 - 7.69 (m, 3 H), 7.55 (t, J = 7.7 Hz, 3 H), 7.50 (s, 1 H), 7.38 (s, 2 H), 7.34 (s, 2 H), 6.94 (s, 1 H), 3.99 - 4.06 (m, 12 H), 3.94 - 3.99 (m, 6 H). МС (ESI) рассчитано для C43H37N3O12S2 851.2, найдено 852.1 [M + H]+.
(5-(4-(3,4,5-триметоксибензоил)-1Н-имидазол-2-ил)-1Н-индол-2-ил)(3,4,5-триметоксифенил)метанон (15xaa). Выход: 45,9%; т.пл. 239-241 °C. 1H ЯМР (500 MHz, CDCl3) δ 10.45 (s, 1 H), 9.44 (s, 1 H), 8.41 (s, 1 H), 8.04 (d, J = 8.5 Hz, 1 H), 7.86 (s, 1 H), 7.61 (d, J = 8.5 Hz, 1 H), 7.29 (s, 2 H), 7.26 (s, 2 H), 3.99 (s, 3 H), 3.95-3.97 (m, 15H). МС (ESI) рассчитано для C31H29N3O8 571.2, найдено 572.2 [M + H]+. ВЭЖХ2: tR 4.09 мин., чистота 96.3%.
ПРИМЕР 7
СИНТЕЗ (2-(1Н-ИНДОЛ-3-ИЛ)-1Н-ИМИДАЗОЛ-4-ИЛ)(3,4,5-ТРИМЕТОКСИФЕНИЛ)МЕТАНОНА (17ya) (ФИГУРА 13)

Синтез 1-(фенилсульфонил)-1Н-индол-3-карбоксальдегида (8ya): К раствору индол 3-карбоксальдегида (8y) (100 ммоль) в этаноле (500 мл) при комнатной температуре добавили гидроксид калия (1,1 экв.). Смесь перемешивали до полной солюбилизации. Этанол полностью удалили в вакууме, а остаток растворили в ацетоне (250 мл), а затем добавили бензолсульфонил хлорид (1,1 экв., 110 ммоль). Реакционную смесь перемешивали в течение получаса. Осадок отфильтровали, а фильтрат концентрировали и перекристаллизовали из метанола для получения твердого белого вещества. Выход: 33%. 1H ЯМР (500 MHz, CDCl3) δ 10.17 (s, 1 H), 8.25-8.39 (m, 2 H), 7.97-8.09 (m, 3 H), 7.69 (t, J = 7.33 Hz, 1 H), 7.59 (t, J = 7.5 Hz, 2 H), 7.39 - 7.54 (m, 2 H). МС (ESI) рассчитано для C15H11NO3S 285.1, найдено 286.0 [M + H]+.
Синтез 3-(1Н-имидазол-2-ил)-1-(фенилсульфонил)-1Н-индола (9ya): К раствору 1-(фенилсульфонил)-1Н-индол-3-карбоксальдегида (8ya) (100 ммоль) в этаноле (400 мл) при 0°C добавили раствор 40% оксалальдегида (глиоксаля) в воде (1,1 экв., 110 ммоль) и раствор 29% гидроксида аммония в воде (10 экв., 1000 ммоль). После перемешивания в течение 2-3 дней при комнатной температуре, реакционную смесь погасили водой и экстрагировали дихлорметаном. Органический слой удалили под вакуумом, а остаток подвергли колоночной флэш-хроматографии с гексаном/этилацетатом (4:1-2:1) в качестве элюента для получения указанного в заголовке соединения в виде желтого порошка. Выход: 12%.1H ЯМР (500 MHz, ДМСО-d6) δ 8.33 (d, J = 2.9 Hz, 2 H), 8.13 (d, J = 7.8 Hz, 2 H), 7.98 - 8.04 (m, 1 H), 7.62 - 7.67 (m, 1 H), 7.55 (d, J = 7.82 Hz, 2 H), 7.22 - 7.34 (m, 4 H). МС (ESI) рассчитано для C17H13N3O2S 323.1, найдено 324.0 [M + H]+.
Синтез 1-(фенилсульфонил)-3-(1-(фенилсульфонил)-1Н-имидазол-2-ил)-1Н-ндола (10ya): К раствору 3-(1Н-имидазол-2-ил)-1-(фенилсульфонил)-1Н-индола (9ya) (20 ммоль) в безводном ТГФ (300 мл) при 0°C добавили гидрид натрия (60% дисперсия в минеральном масле, 1,2 экв., 24 ммоль) и перемешивали в течение 20 минут. Добавили бензолсульфонилхлорид (1,2 экв., 24 ммоль) и перемешивали реакционную смесь в течение ночи. После разбавления 200 мл насыщенного (водного) раствора NaHCO3, реакционную смесь экстрагировали этилацетатом (600 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 5:1) для получения твердого белого вещества. Выход: 40%. 1H ЯМР (CDCl3, 300 MHz) δ 8.02-8.08 (m, 4 H), 7.72 (d, J = 1.5 Hz, 1 H), 7.35-7.60 (m, 8 H), 7.23 (d, J = 1.5 Hz, 1 H), 7.10-7.16 (m, 3 H). МС (ESI) рассчитано для C23H17N3O4S2 463.1, найдено 486.0 [M + Na]+.
Синтез (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17yaa): К раствору 1-(фенилсульфонил)-3-(1-(фенилсульфонил)-1H-имидазол-2-ил)-1Н-индола (10ya) (5,0 ммоль) в безводном ТГФ (100 мл) при -78°C добавили 1,7 M трет-бутиллитий в пентане (1,2 экв., 6,0 ммоль) и перемешивали в течение 10 минут. Добавили 3,4,5-триметоксибензоил хлорид (1,2 экв., 6,0 ммоль) в ТГФ при -78°C и перемешивали в течение ночи. Реакционную смесь погасили 100 мл насыщенного (водного) раствора NaHCO3 и экстрагировали этилацетатом (300 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 3:1) для получения твердого белого вещества. Выход: 30%. 1H ЯМР (500 MHz, CDCl3) δ 8.09 (d, J = 10 Hz, 1 H), 8.04 (d, J = 10 Hz, 2 H), 7.91 (s, 1 H), 7.76 (d, J = 5 Hz, 2 H), 7.65 (t, J = 10 Hz, 1 H), 7.55-7.58 (m, 5 H), 7.40 (s, 2 H), 7.33-7.36 (m, 3 H), 7.25 (t, J = 10 Hz, 1 H),4.05 (s, 3 H), 4.03 (s, 6 H). МС (ESI) рассчитано для C33H27N3O8 657.0, найдено 680.1 [M + Na]+.
Синтез (2-(1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил) метанона (17ya): К раствору (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17yaa) (1 ммоль) в этаноле (40 мл) и воде (4 мл) добавили гидроксид натрия (10 экв., 10 ммоль) и перемешивали в течение ночи в условиях дефлегмации в темноте. Реакционную смесь разбавили 50 мл воды и экстрагировали этилацетатом (200 мл). Органический слой высушили над сульфатом магния и концентрировали. Остаток очистили колоночной флэш-хроматографией (гексан: этилацетат 1:1) для получения желтого твердого вещества. Выход: 60%. 1H ЯМР (500 MHz, CD3OD) δ 8.31 (d, J = 6.5 Hz, 1 H), 7.99 (s, 1 H), 7.90 (s, 1 H), 7.48-7.52 (m, 3 H), 7.24-7.28 (m, 2 H), 4.00 (s, 6 H), 3.93 (s, 3 H). МС (ESI) рассчитано для C21H19N3O4 377.1, найдено 400.1 [M + Na]+. Т.пл. 208-210°C.
ПРИМЕР 8
СИНТЕЗ (2-(1Н-ИНДОЛ-5-ИЛАМИНО)ТИАЗОЛ-4-ИЛ)(3,4,5-ТРИМЕТОКСИФЕНИЛ)МЕТАНОНА (СОЕДИНЕНИЕ 55) (ФИГУРА 15).
Смесь 5-нитро-1Н-индола (11 г, 67,9 ммоль) и Pd/C (5%; 1 г), растворенную в этаноле (50 мл), гидрогенировали в течение 3 часов при 40 psi. Реакционную смесь отфильтровали, а избыток этанола выпарили под пониженным давлением. Твердый продукт перекристаллизовали из гексана для получения чистого соединения 5-аминоиндола (55-1). Выход: 92.5%. 1H ЯМР (500 MHz, CDCl3): δ 7.96 (br, 1 H), 7.20 (d, 1 H), 7.13 (s, 1 H), 6.95 (s, 1 H), 6.67 (dd, 1 H), 6.37 (s, 1 H), 3.50 (s, 2 H). МС (ESI) m/z 133.0 (M + H)+ .
Раствор 5-аминоиндола (8 г, 60,6 ммоль) в ацетоне (150 мл) взаимодействовал с бензоилизотиоцианатом (9,88 г, 60, ммоль) при комнатной температуре в течение около 4 часов. Полученное твердое вещество отфильтровали и обработали 2 н. NaOH в ТГФ (120 мл). Смесь дефлегмировали в течение около 6 часов и оставили нагреваться до комнатной температуры. Растворитель выпарили под вакуумом. Остаток разбавили водой (20 мл) и нейтрализовали до рН 7 при помощи 1 н. HCl. Полученное твердое вещество отфильтровали и высушили под вакуумом для получения 5-индолилтиомочевины (55-2). 5-Индолилтиомочевину (0,01 моль) и этил бромпируват (0,011 моль) растворили в 3 мл этанола и выдерживали при дефлегмации в течение 2 часов. Реакционную смесь охладили, кристаллический этил 2-(1Н-индол-5-иламино)тиазол-4-карбоксилат (55-3) собрали фильтрацией и промыли этанолом. В результате дефлегмации смеси этиловых эфиров с раствором NaOH в этаноле получили 2-(1Н-индол-5-иламино)тиазол-4-карбоновую кислоту (55-4), которую использовали непосредственно на следующих этапах. К смеси неочищенной кислоты (2,5 ммоль), EDCI (2,9 ммоль), HOBt (2,6 ммоль) и NMM (5,3 ммоль) в CH2Cl2 (30 мл) добавили соль HNCH3OCH3HCl (2,6 ммоль) и продолжали перемешивание при комнатной температуре в течение ночи. Реакционную смесь разбавили CH2Cl2 (20 мл) и последовательно промыли водой, насыщенным раствором NaHCO3, насыщенным солевым раствором и высушили над MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 2-(1Н-индол-5-иламино)-N-метокси-N-метилтиазол-4-карбоксамида (55-5) (общий выход за 5 этапов 45,6%). При -78oC, к раствору 5-бром-1,2,3-триметоксибензола (1,235 г, 5,0 ммоль) в 30 мл ТГФ добавили n-BuLi в гексане (2,5 н., 2,4 мл, 6 ммоль) под защитой Ar2 и перемешивали в течение 10 минут. Амид Вайнреба (1 ммоль) в 10 мл ТГФ добавили к литиевому реагенту и оставили перемешиваться при комнатной температуре на 2 часа. Реакционную смесь погасили насыщенным раствором NH4Cl, экстрагировали этиловым эфиром, высушили при помощи MgSO4. Растворитель удалили под пониженным давлением для получения неочищенного продукта, который очистили колоночной хроматографией для получения чистого соединения 55 (выход 51,7%). 1H ЯМР (300 MHz, CDCl3) δ 8.29 (br, 1 H), 7.68 (d, 1 H), 7.46 (s, 2 H), 7.39 (s, 1 H), 7.36 (s, 1 H),7.28 ~ 7.26 (m, 1 H), 7.15~7.12 (m, 1 H), 6.55 (m, 1 H), 3.93 (s, 3 H), 3.89 (s, 6 H). МС (ESI) m/z 432.1 (M + Na)+, 408.0 (M - H)- .
ПРИМЕР 9
СИНТЕЗ ХИНОЛИН- И ИЗОХИНОЛИН-АРИЛЬНЫХ СОЕДИНЕНИЙ
(ФИГУРА 16)
Серии соединений получили по реакции сочетания Сузуки 7-бром-1-хлоризохинолина с различными арилбороновыми кислотами.
Синтез 1-хлор-7-(1Н-индол-5-ил)-изохинолина (6d) (Фигура 16С):

Смесь 7-бром-1-хлоризохинолина (0,50 г, 2,1 ммоль), 5-индолбороновой кислоты (0,40 г, 2,5 ммоль), тетракис(трифенилфосфен)палладия (0,035 г, 0,08 ммоль), карбоната калия (2,1 мл, 2 М, 4,1 ммоль), N,N-диметилформамида (11 мл) перемешивали, продувая аргоном пространство над смесью в течение 30 минут. Затем смесь дефлегмировали в течение 16 часов, а затем оставили остывать до комнатной температуры. Смесь отфильтровали через слой силикагеля, разбавили водой (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой отделили и промыли NaOH (2 х 20 мл, 10% водный раствор), водой (5 х 30 мл, до прекращения видимых рефракционных изменений на поверхности раздела органической и водной фазы) и хлоридом аммония (20 мл, насыщенный). Органический слой затем адсорбировали на силикагель и обработали флэш-хроматографией (этилацетат/гексаны) для получения 0,14 г (25%) твердого желтого вещества. МС (ESI): рассчитано для C17H11ClN2, 278.1, найдено 301.0 [M + Na]+. 1H ЯМР (300 MHz, ДМСО-d6) δ 6.56 - 6.58 (m, 1 H), 7.44 (t, J = 2.77 Hz, 1 H), 7.57 - 7.59 (m, 2 H), 7.93 (m, 1 H), 8.04 (s, 1 H), 8.13 - 8.20 (m, 1 H), 8.27 - 8.31 (m, 2 H), 8.43 (m, 1 H), 11.25 (brs, 1 H).
1,7-Бис-(1Н-индол-5-ил)-изохинолин (6b) (Фигура 16Е:

Смесь 7-бром-1-хлоризохинолина (0,20 г, 2,1 ммоль), 5-индолбороновой кислоты (0,80 г, 5,0 ммоль), тетракис(трифенилфосфен)палладия (0,19 г, 0,16 ммоль), карбоната калия (2,1 мл, 2 М, 4,1 ммоль), N,N-диметилформамида (11 мл) перемешивали, продувая аргоном пространство над смесью в течение 30 минут. Затем смесь дефлегмировали в течение 16 часов, а затем оставили остывать до комнатной температуры. Смесь отфильтровали через слой силикагеля, разбавили водой (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой отделили и промыли NaOH (2 х 20 мл, 10% водный раствор), водой (5 х 30 мл, до прекращения видимых рефракционных изменений на поверхности раздела органической и водной фазы) и хлоридом аммония (20 мл, насыщенный). Органический слой затем адсорбировали на силикагель и обработали флэш-хроматографией (этилацетат/гексаны) для получения 0,29 г (39%) твердого желтого вещества. МС (ESI): рассчитано для C25H17N3, 359.1, найдено 360.2 [M + H]+ 382.1 [M + Na]+, и 358.0 [M - H]-.1H ЯМР (500 MHz, ДМСО-d6) δ 6.46 - 6.50 (m, 1 H) 6.52 - 6.59 (m, 1 H) 7.34 - 7.36 (m, 1 H) 7.36 - 7.41 (m, 2 H) 7.42 - 7.52 (m, 3 H) 7.58 (d, J=8.30 Hz, 1 H) 7.81 (dd, J=5.49, 5.00 Hz, 2 H) 7.92 (s, 1 H) 8.08 - 8.17 (m, 2 H) 8.33 (s, 1 H) 8.54 (d, J=5.61 Hz, 1 H) 11.18 (br. s., 1 H) 11.30 (br. s., 1 H) ppm.
1-(4-Фтор-фенил)-7-(1Н-индол-5-ил)-изохинолин (6С) (Фигура 16D):

Смесь 6d (0,20 г, 0,72 ммоль), 4-фторфенилбороновой кислоты (0,12 г, 0,86 ммоль), тетракис(трифенилфосфен)палладия (0,033 г, 0,03 ммоль), карбоната калия (0,72 мл, 2 М, 1,4 ммоль), N,N-диметилформамида (22 мл) перемешивали, продувая пространство аргоном над смесью в течение 30 минут. Затем смесь дефлегмировали в течение 16 часов, а затем оставили остывать до комнатной температуры. Смесь отфильтровали через слой силикагеля, разбавили водой (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой отделили и промыли NaOH (2 х 20 мл, 10% водный раствор), водой (5 х 30 мл, до прекращения видимых рефракционных изменений на поверхности раздела органической и водной фазы) и хлоридом аммония (20 мл, насыщенный). Органический слой затем адсорбировали на силикагель и обработали флэш-хроматографией (этилацетат/гексаны) для получения 0,038 г (16%) твердого желтого вещества. МС (ESI): рассчитано для C23H15FN2, 338.12, найдено 339.2 [M + H]+ и 361.2 [M + Na]+. 1H ЯМР (300 MHz, ДМСО-d6) δ 6.47 - 6.55 (m, 1 H), 6.80 (d, J = 9.16 Hz, 2 H), 7.38 - 7.45 (m, 2 H), 7.47 - 7.62 (m, 3 H), 7.72 (d, J = 8.85 Hz, 2 H), 7.79 - 7.96 (m, 3 H), 11.18 (br. s., 1 H).
1,7-Бис-(4-фтор-фенил)-изохинолин (40) (Фигура 16А).

МС (ESI): рассчитано для C21H13F2N, 317.10, найдено 318.1 [M + H]+, 340.1 [M + Na]+, и 315.9 [M - H]-. 1H ЯМР (500 MHz, ДМСО-d6) δ 7.31 (br. s., 1 H) 7.31 - 7.37 (m, 2 H) 7.39 (br. s., 1 H) 7.41 (t, J = 8.54 Hz, 2 H) 7.72 - 7.77 (m, 2 H) 7.78 - 7.84 (m, 2 H) 7.89 (br. s., 1 H) 7.90 - 7.99 (m, 1 H) 8.09 - 8.19 (m, 3 H) 8.59 (br. s., 1 H) 8.60 - 8.65 (m, 1 H).
Синтез 7-бром-1-(4-фтор-бензолсульфонил)-1,2,3,4-тетрагидро-хинолина (43) и 1-(4-фтор-бензолсульфонил)-7-(1Н-индол-5-ил)-1,2,3,4-тетрагидрохинолина (41). (Фигура 16В).

7-Бром-1,2,3,4-тетрагидрохинолин (0,60 г, 2,8 ммоль) перемешивали с 4-фторфенилсульфонил хлоридом (1,65 г, 8,49 ммоль) в пиридине (5 мл) при 80oC в течение 3 часов. Смесь охладили, концентрировали, а остаток хроматографировали (EtOAc/гексаны на SiO2) для получения 845 мг коричневого твердого вещества (81%) соединения 43. C15H13BrFNO2S 368.98, найдено 394.0 [M + Na]+ и 367.8 [M - H]-. 1H ЯМР (500 MHz, CDCl3) δ 1.58 - 1.67 (m, 2 H) 2.41 (t, J = 6.71 Hz, 2 H) 3.72 - 3.82 (m, 2 H) 6.89 (d, J = 8.30 Hz, 1 H) 7.08 - 7.17 (m, 2 H) 7.18 - 7.24 (m, 1 H) 7.59 - 7.68 (m, 2 H) 7.92 - 8.01 (m, 1 H).
43 (0,46 г, 1,3 ммоль), 5-индолбороновую кислоту (0,26 г, 1,6 ммоль), тетракис(трифенилфосфен)палладий (0,031 г, 0,03 ммоль), карбонат калия (1,35 мл, 2 М, 2,7 ммоль), N,N-диметилформамид (135 мл) перемешивали, продувая аргоном пространство над смесью в течение 30 минут. Затем смесь дефлегмировали в течение 16 часов, а затем оставили остывать до комнатной температуры. Смесь отфильтровали через слой силикагеля, разбавили водой (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой отделили и промыли NaOH (2 х 20 мл, 10% водный раствор), водой (5 х 30 мл, до прекращения видимых рефракционных изменений на поверхности раздела органической и водной фазы) и хлоридом аммония (20 мл, насыщенный). Органический слой затем адсорбировали на силикагель и обработали флэш-хроматографией (этилацетат/гексаны) для получения 0,38 г (77%) белого кристаллического вещества соединения 41. МС (ESI): рассчитано для C23H19FN2O2S, 406.12, найдено 404.9 [M - H]- и 429.1 [M + Na]+. 1H ЯМР (500 MHz, ДМСО-d6) δ 1.56 - 1.66 (m, 2 H) 2.48 (t, J = 6.59 Hz, 2 H) 3.76 - 3.86 (m, 2 H) 6.46 - 6.56 (m, 1 H) 7.14 (m, J = 7.81 Hz, 1 H) 7.33 - 7.37 (m, 1 H) 7.38 - 7.45 (m, 4 H) 7.49 (m, J = 8.54 Hz, 1 H) 7.66 - 7.74 (m, 2 H) 7.74 - 7.81 (m, 1 H) 7.85 - 7.94 (m, 1 H) 11.17 (br. s., 1 H).
7-Бром-2-(4-фтор-бензолсульфонил)-1,2,3,4-тетрагидро-изохинолин (42) (Фигура 16В).

Выход 23%. C15H13BrFNO2S, 369.0, найдено 392.0 [M + Na]+ и 367.7 [M - H]-. 1H ЯМР (500 MHz, ДМСО-d6) δ 2.75 - 2.82 (m, 2 H) 3.32 (t, J = 6.10 Hz, 2 H) 4.24 (s, 2 H) 7.07 (d, J = 8.30 Hz, 1 H) 7.29 - 7.37 (m, 1 H) 7.37 - 7.43 (m, 1 H) 7.47 (t, J = 8.79 Hz, 2 H) 7.87 - 7.93 (m, 2 H).
2-(4-Фтор-бензолсульфонил)-7-(1Н-индол-5-ил)-1,2,3,4-тетрагидро-изохинолин (44).
Выход 77%. 1H ЯМР (500 MHz, ДМСО-d6) δ 2.84 - 2.91 (m, 2 H) 3.35 (t, J=5.98 Hz, 2 H) 4.30 (s, 2 H) 6.44 - 6.48 (m, 1 H) 7.17 (d, J = 7.81 Hz, 1 H) 7.32 - 7.40 (m, 2 H) 7.41 - 7.51 (m, 3 H) 7.75 - 7.79 (m, 1 H) 7.89 - 7.96 (m, 1 H) 11.13 (br. s., 1 H).
ПРИМЕР 10
РАСТВОРИМОСТЬ АРИЛ-БЕНЗОИЛ-ИМИДАЗОЛЬНЫХ (ABI) СОЕДИНЕНИЙ
В ВОДЕ (Фигура 17)
Определение растворимости в воде. Для определения растворимости в воде, 1 мг каждого соединения суспендировали в 1 мл воды и встряхивали в течение 48 часов при комнатной температуре. Суспензию центрифугировали при 10000 об./мин. в течение 10 минут и отфильтровали на фильтре 0,22 мкм. Концентрации каждого соединения измерили по ЖХ-МС, состоящей из ВЭЖХ прибора HP S1100 (Agilent, Фостер сити, штат Калифорния) и МС-датчика Bruker ESQUIRE с электрораспылительным прибором и ионной ловушкой в режиме с положительными ионами (Bruker, Фримонт, штат Калифорния). Для ВЭЖХ использовали обращенно-фазовую колонку Nova-pak C18 (150 м Ч 3,9 мм, Waters, Милфорд, штат Массачусетс). Подвижная фаза состояла из 20:80 об./об. воды/ацетонитрила. Для определения массы, пик получали при m/z 382 (для имидазольных соединений) и m/z 399 (для тиазольных соединений), соответственно. Концентрацию каждого соединения рассчитали по площади МС пика в соответствии со следующим калибровочным уравнением: y=1,3295x + 114,24 (R2=1,00). Для построения стандартной кривой (Фигура 17), по которой было получено это уравнение, 50, 100 мкл каждого раствора 100 мкг/мл, 10 мкг/мл ABI соединения 12ga, и его соответствующего тиазолового аналога, а также СА-4 (структура представлена на Фигуре 19) в ацетонитриле, ввели в хроматограф ВЭЖХ и контролировали по масс-спектроскопии. Количество (нг) каждой введенной пробы нанесли на график в зависимости от относительной площади пика ее массы для построения стандартной кривой на Фигуре 17.
Время удерживания ВЭЖХ ABI соединения 12ga (1,5 минуты) сравнили с его соответствующим тиазоловым аналогом (2,2 минуты), используя подвижную фазу 80/20 метанол/вода, при скорости потока 1 мл/мин. и обращенно-фазовую колонку, что указывает на то, что имидазоловое соединение является более гидрофильным, чем его соответствуюий тиазоловый аналог. Рассчитанные значения logP для ABI соединения 12ga и его соответствующего тиазолового аналога составили примерно 2,9 и 4,4, соответственно. Растворимость в воде соединения 12ga составила 13 мкг/мл, или примерно в 200 раз выше, чем у его тиазолового аналога (72 нг/мл).
ПРИМЕР 11
БИОЛОГИЧЕСКАЯ ОЦЕНКА СОЕДИНЕНИЙ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ:
Пример 11А: Ингибирование клеточного роста in vitro.
Анализ клеточной культуры и цитотоксичности для рака простаты и меланомы. Все клеточные линии были закуплены у компании ATCC (American Type Culture Collection, Манассас, штат Вирджиния, США), а клеточные культуры были приобретены у компании Cellgro Mediatech (Херндон, штат Вирджиния, США). Авторы изобретения исследовали антипролиферативную активность изобретенных антитубулиновых соединений в четырех клеточных линиях рака простаты человека (LNCaP, DU 145, PC-3 и PPC-1) и двух клеточных линиях меланомы человека (A375 и WM-164). Клеточную линию рака яичников человека OVCAR-8 и ее резистентную клеточную линию, которая сверхэкспрессирует P-gp (NCI/ADR-RES), использовали в качестве моделей МЛУ. Обе клеточные линии рака яичников приобрели у компании National Cancer Institutes (NCI). Все клеточные линии протестировали и установили подлинность при помощи ATCC или NCI. Все клеточные линии рака простаты и рака яичников выращивали в RPMI 1640, с дополнением 10% бычьей плодной сыворотки (FBS).
Клетки меланомы выращивали в DMEM, с дополнением 5% FBS, 1% смеси антибиотика/фунгицида (Sigma-Aldrich, Inc., Сент-Луис, штат Миссури, США) и бычьего инсулина (5 мкг/мл; Sigma-Aldrich). Цитотоксичный потенциал антитубулиновых соединений оценили с использованием анализа с сульфородамином В (SRB) через 96 часов после обработки.
Все описанные соединения были сначала оценены на цитотоксичность в клеточной линии меланомы мышей B16-F1, клеточных линиях меланомы человека (A375 и WM-164) и клеточных линиях рака простаты (DU145, PC-3, LNCaP, PPC-1). Соединение 1h и ABT-751 (E7010, Abbott Laboratories/Eisai Co Ltd), для которого начата II фаза клинических испытаний для лечения пациентов, страдающих различными видами рака, были включены в эти анализы в качестве примеров средств, связывающих колхициновый сайт. Значения IC50 для ингибирования клеточного роста показаны в Таблицах 1, 2 и 3.
Результаты:
Таблица 1. Структурно-функциональная взаимосвязь (SAR) соединений с оптимизированным кольцом В
 |
Кольцо B | IC50 ± стандартная погрешность средней величины (SEM) (нМ) | |||||
| B16-F1 | A375 | DU 145 | PC-3 | LNCaP | PPC-1 | ||
| 1a | 1,3-фенил | 500±200 | 87±15 | 178 | 81 | 234 | 85 |
| 1b | 4,6-пиримидин | >30000 | >30000 | 6900 | 8300 | 7000 | 3700 |
| 1c | 2,6-пиридин | 39±12 | 30±14 | 33±3 | 32±2 | 27±2 | 25±1 |
| 1d | 2,5-фуран | 151±24 | 27±8 | 35 | 21 | 23 | 20 |
| 1e | 2,5-тиазол | 12500±5200 | 13600±3800 | >10000 | >10000 | >10000 | >10000 |
| 1f | 2,4-тиофен | 72±15 | 15±6 | 26 | 12 | 17 | 15 |
| 1g | 1,4-пиперидин | >30000 | >30000 | >20000 | >20000 | >20000 | >20000 |
| 1h | 2,4-тиазол | 55±5 | 28±5 | 71±4 | 21±1 | 28±4 | 43±5 |
| 1i | 3,5-изоксазол | >30000 | >30000 | >10000 | >10000 | >10000 | >10000 |
| 36a | 2,4-оксазол | 600±200 | 300±100 | 292 | 294 | 310 | 324 |
| 35a | 2,4-оксазолин | 6500±800 | 500±100 | 1200±100 | 1200±100 | 1200±100 | 1100±100 |
Таблица 2. Структурно-функциональная взаимосвязь (SAR) соединений с оптимизированным карбонильным линкером

Таблица 3. Антипролиферативная активность модифицированных соединений с улучшенной растворимостью в воде
 |
Фрагмент A | IC50 ± стандартная погрешность средней величины (SEM) (нМ) | |||||
| B16-F1 | A375 | DU 145 | PC-3 | LNCaP | PPC-1 | ||
| 58a | 4-OTBDMSPh | 500±200 | 700±300 | 434±30 | 183±24 | 549 | 246±8 |
| 2l | 4-OHPh | 110 | 100 | 116 | 87 | 103 | 76 |
| 62a | 2-индолил | 43±21 | 19±9 | 32 | 24 | 28 | 28 |
| 66a | 5-индолил | 25±13 | 8±1 | 13 | 7 | 10 | 8 |
| 68a | 4-BocNHCH2Ph | 2900±400 | 7900±500 | 4400 | 3100 | 2600 | 2700 |
| 2r | 4-NH2CH2Ph | 38±11 | 41±13 | 25 | 80 | 13 | 34 |
| 2s | 4-NHMeCH2Ph | >10000 | >10000 | ~10000 | >10000 | 114±80 | ~1000 |
| 2u | 4-NMe2CH2Ph | >10000 | >10000 | >10000 | >10000 | 1025±200 | >10000 |
| 5a | PhNH | 65±12 | 45±8 | 70±4 | 57±3 | 51±1 | 54±1 |
| 5Hb | 4-CH3PhNH | НО* | НО | 35±1 | 38±2 | 35±1 | 36±1 |
| 5c | 4-FPhNH | НО | НО | 63±1 | 43±1 | 41±1 | 37±1 |
| 1h | Ph | 55±5 | 28±5 | 71±4 | 21±1 | 28±4 | 43±5 |
| ABT-751 | 2127±351 | 1111±108 | 839±719 | 786±89 | 658±117 | 701±307 | |
*НО = Не определено
Структурно-функциональная взаимосвязь (SAR) молекул с альтернативным кольцом «В». Первая серия была предназначена для альтернативных вариантов тиазолового кольца «В». Соответственно, исследовали серию гетероциклических колец «В». Как показано в Таблице 1, успешными заменами тиазола были пиридин 1c, фуран 1d и тиофен 1f. Значения IC50 (12 нМ ~ 35 нМ против клеток рака простаты) являются близкими к тиазоловому соединению 1h. Внедрение фенила (1a), оксазолина (35a) и оксазола (36a) сохранило активность в диапазоне сотен наномоль. Однако внедрение пиримидина (1b, IC50: 3,7~8,3 мкМM), перевернутого 2,5-тиазола и 3,5-изоксазола (1e и 1i, IC50: > 10 мкМ) вызвало очевидное снижение силы. Модификация кольца «В» на насыщенное кольцо пиперидина (1g) также полностью уничтожило активность (IC50 >20 мкМ).
Структурно-функциональная взаимосвязь (SAR) альтернативных линкеров. Исследования печеночной метаболической устойчивости in vitro показали, что карбонильный линкер между кольцами «В» и «С» в соединениях SMART вызвал короткие периоды полувыведения (5-17 минут), в основном, за счет карбонильного восстановления. Для блокирования восстановления этого кетона до неактивного гидроксильного линкера соединения 2b, этот карбонильный линкер во второй серии соединений был модифицирован (Таблица 2). Карбонильный линкер был заменен двойными связями (2a, 3a и 3b), амидами (2g, 2h), оксимами (2e-цис,транс и 2f-цис,транс), гидразидом (2d-цис, 2d-транс), акрилонитрилами (2c-транс, 2c-цис), цианоимином (2j), сульфониламидом (4d), простым серным эфиром (4a), сульфонильными и сульфинильными соединениями (4b, 4c). Получили также прямую связь соединения 2i без какого-либо линкера между кольцами «В» и «С». Среди этих модификаций линкера лишь цианоиминовое соединение (2j) продемонстрировало многообещающий потенциал (20 ~ 60 нМ) по сравнению с карбонильным соединением 1h, но исследование метаболизма in vitro показало, что период полувыведения 2j в печеночной микросоме человека составляет менее 5 минут. Это позволяет предположить, что хотя кетоновое восстановление заблокировано, это может вызвать новую подверженность соединения 2j неблагоприятным условиям. Пары изомеров соединений, содержащих двойные связи, оксимов и гидразидов были разделены. Соединение 3a было получено для имитации структуры CA-4, (Фигура 19), которая содержит цис-C=C между двумя арильными кольцами, но, к сожалению, 3a и другие пары изомеров потеряли активность после замены линкера C=O. Одным интересным событием является то, что син-изомер 2e-цис (0,1 ~ 0,3 мкМ) продемонстрировал в 10 раз более сильное действие, чем его анти-изомер 2e-транс (>10 мкМ). Период полувыведения 2e-цис в печеночной микросоме человека увеличен до 35 минут, тогда как периоды полувыведения соединений 2d могут быть увеличены до 55 минут. Однако пониженная активность (~1 мкМ) 2d также снижает их потенциал.
Пример 11В: Растворимость соединений настоящего изобретения в воде.
Растворимость лекарств определили по фильтровальной пластине растворимости Multiscreen (Millipore Corporate, Биллерика, штат Массачусетс), подключенной к ЖХ-МС/МС. Вкратце, 198 мкл фосфатно-солевого буферного раствора (PBS) (pH 7,4) поместили в 96-луночный планшет, дозировали по 2 мкл 10 мМ раствора исследуемых соединений (в ДМСО) и перемешивали при осторожном встряхивании (200-300 об./мин.) в течение 1,5 часов при комнатной температуре (N = 3). Планшет центрифугировали при 800g в течение 5 минут, а фильтрат использовали для определения его концентрации и растворимости исследуемого соединения по ЖХ-МС/МС, как описано ниже.
Внедрение полярной и ионизируемой групп в антитубулиновые агенты. Одним основным ограничением агентов SMART является низкая растворимость в воде. Для изучения поведения SMART in vivo, использовали поверхностно-активные вещества, такие как Tween 80, соответственно, были получены благоприятные результаты. Однако эти поверхностно-активные вещества являются биологически активными и отвечают за многие побочные эффекты. Кроме того, предполагается, что низкая растворимость 1h в воде приводит к низкой пероральной биодоступности (3,3%, Таблица 4). В третьей серии соединений растворимость в воде была успешно увеличена без ухудшения силы действия, за счет внедрения полярных групп, таких как гидроксил и индолилы. Кроме того, ионизируемые группы, такие как амино- и алкиламиногруппы, также были внедрены в пара-положение кольца «А». Как показано на Фигуре 5 и в Таблице 3, внедрение индолиловых групп в кольцо «А», особенно, 5-индолила (66а, 7 ~ 25 нМ) увеличило силу действия по сравнению с 4-ОН соединением 2l (76-116 нМ). Аминометил -CH2NH2 в пара-положении кольца «А» также сохранил силу (2r, 13-80 нМ), однако p-NHMe (2s) или p-NMe2 (2u) уничтожили активность. Как показано на Фигуре 18, аналитическое измерение для оценки растворимости в воде показало, что индолиловое соединение 66а увеличило растворимость в PBS с 1,1 мкг/мл (соединение 1h) до 3,8 мкг/мл. Аминометиловое соединение 2r было преобразовано в HCl соль, что увеличило растворимость более, чем в 35 раз (>35 мкг/мл). Хотя соединение 2r продемонстрировало удовлетворительную растворимость воде, фармакокинетические исследования показали, что это соединение обладает все еще очень слабой биодоступностью (F% = 0,2%). Предполагается, что соединение 2r ионизировалось в пищеводе и, следовательно, не было абсорбировано в систему кровотока.
Пример 11С: Фармакокинетические исследования
Фармакокинетическое исследование. Самок крыс Sprague-Dawley (n = 3 или 4; 254 ± 4 г) приобрели у компании Harlan Inc. (Индианаполис, штат Индиана). Торакальные катетеры яремной вены для крыс приобрели у компании Braintree Scientific Inc. (Брэйнтри, штат Массачусетс). По прибытию в виварий, животные прошли акклиматизацию в течение 3 дней в помещении с контролируемой температурой (20–22°C) с циклом освещения/темноты 12 часов, перед началом какой-либо обработки. Соединение 1h вводили внутривенно (i.v.) в катетеры яремной вены в дозировке 2,5 мг/кг (в ДМСО/ПЭГ300, 2/8), тогда как 5Ha и 5Hc дозировали по 5 мг/кг (в ДМСО/ПЭГ300, 1/9). Инъекцией ввели равный объем гепаринизированного солевого раствора для замещения крови и взяли образцы крови (250 мкл) через катетеры яремной вены через 10, 20, 30 минут и 1, 2, 4, 8, 12, 24 часа. Соединения 1h, 5Ha и 5Hc вводили перорально (p.o.) через ротовой зонд в дозировке 10 мг/кг (в Tween80/ДМСО/H2O, 2/1/7). Все образцы крови (250 мкл) после перорального введения собрали через катетеры яремной вены через 30, 60, 90 минут, 120 минут, 150 минут, 180 минут, 210 минут, 240 минут и 8, 12, 24 часа. Гепаринизированные шприцы и пробирки приготовили до сбора крови. Образцы плазмы приготовили центрифугированием образцов крови при 8000g в течение 5 минут. Все образцы плазмы сразу хранили при -80°C до анализов.
Аналиты экстрагировали из 100 мкл плазмы при помощи 200 мкл ацетонитрила, содержащего 200 нМ внутреннего стандарта ((3,5-диметоксифенил)(2-фенил-1Н-имидазол-4-ил)метанона). Образцы тщательно перемешали, центрифугировали, а органический экстракт перенесли в автоматический пробоотборник для анализа ЖХ-МС/МС. Для получения наиболее чувствительных сигналов использовали режим многократного мониторинга реакции (MRM), сканирование при m/z 356 → 188 (соединение 1h), m/z 371 → 203 (соединение 5Ha), m/z 389 → 221 (соединение 5Hc) и m/z 309 → 171 (внутренний стандарт). Фармакокинетические параметры определили при помощи анализа без использования камерной модели (WinNonlin, Pharsight Corporation, Маунтин-Вью, штат Калифорния).
Результаты:
Таблица 4. Фармакокинетические параметры для соединений, исследованных in vivo.
| 1h | 2r | 5Ha | 5Hc | |||||
| Способ введения | Внутривенно (IV) | Перорально (PO) | Внутривенно (IV) | Перорально (PO) | Внутривенно (IV) | Перорально (PO) | Внутривенно (IV) | Перорально (PO) |
| Na | 4 | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| Доза (мг/кг) | 2,5 | 10 | 2,5 | 4 | 5 | 10 | 5 | 10 |
| CLb (мл/мин./кг) | 7,7 ± 1,0 | - | 22 ± 13 | - | 17 ± 3 | - | 13 ± 2 | - |
| Vssc (л/кг) | 4,9 ± 1,9 | - | 0,33± 0,25 | - | 1,4 ± 0,2 | - | 1,4± 0,2 | - |
| AUCd (мин.*мг/мл) | 279 ± 53 | 37 ± 20 | 139± 77 | 0,4 | 296 ± 46 | 65 ± 20 | 381± 65 | 160 ± 13 |
| Cmax e (нм/мл) | 3816±509 | 212 | 3,2 ± 1,6 | 3794±1580 | 4198±438 | 814±255 | 3349±686 | 1262 ± 362 |
| Ff (%) | 3,3 | 0,2 | 11 | 21 | ||||
a Количество крыс. b Общий клиренс. c Объем распределения после внутривенного дозирования. d Площадь под кривой после внутривенного дозирования, интегрированная концентрация лекарства в зависимости от времени и интегрированная концентрация лекарства в зависимости от времени после перорального дозирования. e Максимальная концентрация в плазме после внутривенного дозирования. f Процентная пероральная биодоступность.
Модификация замещенных метоксибензоил-арил-тиазольных (SMART) молекул для улучшения пероральной биодоступности. Многие признанные тубулин-направленные противораковые лекарства, такие как таксаны и винбластин, требуют внутривенного введения из-за низкой пероральной биодоступности. Пероральная биодоступность является сложным параметром, включающим многие химические и физиологические процессы, такие как растворимость, проницаемость и метаболическая устойчивость. Растворимость этих тубулиновых ингибиторов была улучшена за счет вставки амино-линкера между кольцами «А» и «В», как в 5a-d (Фигура 6). В Таблице 3 показано, что NH-мостиковые соединения (5a-c) имеют такой же потенциал (35~ 65 нМ), как и 1h, при увеличенной растворимости (15 и 19 мкг/мл для 5a и 5c, соответственно (Фигура 18), но они являются более, чем в 20 раз более активными, чем ABT-751 (структура АВТ-751 представлена в Таблице 3 и на Фигуре 19).
Фармакокинетические исследования на крысах были выполнены для изучения того, обладают ли эти новые соединения улучшенной биодоступностью по сравнению с соединением 1h (Таблица 4). Данные ясно показали, что 5Hc (HCl соль 5c) демонстрирует более, чем в 4,3 раза более сильное воздействие (AUC) при пероральном способе введения по сравнению с 1h, что позволяет предположить, что улучшенная растворимость в воде за счет амино-линкера успешно увеличивает пероральную биодоступность. Кроме того, максимальная концентрация (Cmax) 5Ha и 5Hc при пероральном введении составила 814 и 1262 нг/мл, соответственно. Тогда как Cmax для 1h составила лишь 212 нг/мл. В целом, биодоступность 5Ha и 5Hc увеличилась с 3,3% для 1h до 11% и 21%, соответственно (Таблица 4). Соединение 5Hc показало умеренный клиренс, умеренный объем распределения и приемлемую пероральную биодоступность. Эти данные позволяют предположить, что эти новые синтезированные амино-связанные соединения обладают потенциалом и ФК профилем, подлежащим разработке как новый класс перорально биодоступных антитубулиновых средств.
Пример 11D: Ингибирование полимеризации тубулина соединениями настоящего изобретения in vitro.
Анализ полимеризации тубулина in vitro. Бычий тубулин мозга (0,4 мг, чистота >97%) (Cytoskeleton, Денвер, штат Колорадо) смешали с 10 мкМ исследуемых соединений и инкубировали в 100 мкл общего тубулинового буфера (80 мМ PIPES, 2,0 мМ MgCl2, 0,5 мМ EGTA и 1 мМ ГТФ) при pH 6,9. Поглощение при длине волны 340 нм контролировали каждую 1 минуту в течение 20 минут при помощи микропланшет-ридера SYNERGY 4 (Bio-Tek Instruments, Винооски, штат Вермонт). Спектрофотометр был установлен на 37 °C для полимеризации тубулина.
Результаты:
Ингибирование полимеризации тубулина некоторыми потенциальными соединениями 1c, 2j, 66a и 5a исследовали по всем трем стратегиям конструирования (альтернативные кольца В, новые линкеры и солюбилизирующие фрагменты) и сравнили с 1h. Бычий тубулин мозга (чистота > 97%) инкубировали с отдельными соединениями (10 мкМ) для испытания их действия на полимеризацию тубулина (Фигура 20а). Через 20 минут инкубирования полимеризация тубулина была ингибирована на 47% действием 1h, по сравнению с носителем. 1c и 2j ингибировали 64% полимеризации через 20 минут с различными профилями ингибирования. Соединения 5a и 66a обеспечили более сильное ингибирование, 78% и 81%, соответственно. Эти данные позволяют предположить, что эти новые соединения демонстрируют сильное действие против полимеризации тубулина, что хорошо согластуется с их цитотоксичностью.
Ингибирование полимеризации тубулина соединениес 5с за счет связывания с колхицин-связывающим сайтом и по сравненению с соединением 1h представлено на Фигурах 20b и 20c.
Пример 11Е: Новые антитубулиновые соединения преодолевают множественную лекарственную устойчивость, опосредованную Р-гликопротеином.
Система Р-гликопротеина (P-gp) является основным физиологическим механизмом множественной лекарственной устойчивости (МЛУ), который действует как АТФ-зависимый насос откачивания лекарства, активно удаляя разнообразные структурно различные цитотоксичные соединения. Усиленный отток этих соединений снижает их внутриклеточное накопление и, таким образом, снижает их цитотоксичность. Поэтому новые соединения, не чувствительные к лекарственной устойчивости, могут обладать более высокой терапевтической и экономической ценностью. Помимо P-gp, клинически используемые антитубулиновые средства обладают другими механизмами устойчивости, такими как изменение динамики микротрубочек и мутации в в-тубулине, которые, как известно, ограничивают восприимчивость к таксанам. Антитубулиновые соединения настоящего изобретения были испытаны против клеточной линии рака яичников OVCAR-8 (исходной) и клеточной линии NCI/ADR-RES, сверхэкспрессирующей P-gp (Таблицы 5A, 5B).
Результаты:
Таблица 5А. Антипролиферативная активность некоторых соединений против МЛУ клеточных линий, сверхэкспрессирующих P-gp.
| Соединение | IC50 (нМ) | Фактор устойчивости | |
| OVCAR-8 | NCI/ADR-RES | ||
| 1c | 33±3 | 13±0,8 | 0,4 |
| 2j | 34±2 | 14±1 | 0,4 |
| 66a | 10±3 | 4±2 | 0,4 |
| 2r | 26±2 | 11±2 | 0,4 |
| 5a | 46 ±6 | 27 | 0,6 |
| 5b | 28 | 21 | 0,8 |
| 5c | 44±3 | 25±6 | 0,6 |
| 1h | 35±2 | 13±1 | 0,4 |
| паклитаксел* | 4,7±0,1 | 6263±634 | 1333 |
| винбластин | 3,9±0,1 | 582±57 | 149 |
| колхицин | 17±1 | 1113±79 | 65 |
Заметно, что антитубулиновые соединения настоящего изобретения показали равносильное антипролиферативное действие против клеточных линий OVCAR-8 и NCI/ADR-RES, что позволяет предположить, что они не являются субстратами P-gp и что они действуют P-gp-независимым образом. Эта характеристика отличается от характеристик паклитаксела, винбластина и колхицина в клетках NCI/ADR-RES.
Таблица 5В. Антипролиферативное действие некоторых фенил-амино-тиазольных соединений
| R | IC50 ± стандартная погрешность средней величины (SEM) (нМ) | ||||||
| B16-F1 | A375 | DU 145 | PC-3 | LNCaP | PPC-1 | ||
| 5a | H | 65±12 | 45±8 | 70±4 | 57±3 | 51±1 | 54±1 |
| 5Hb | 4-CH3 | НО* | НО | 35±1 | 38±2 | 35±1 | 36±1 |
| 5с | 4-F | НО | НО | 63±1 | 43±1 | 41±1 | 37±1 |
| 5d | 4-Cl | НО | 25±7 | 73±1 | 33±1 | 45±1 | 36±1 |
| 1h | - | 55±5 | 28±5 | 71±4 | 21±1 | 28±4 | 43±5 |
| ABT-751 | - | 2127±351 | 1111±108 | 839±719 | 786±89 | 658±117 | 701±307 |
Фенил-амино-тиазоловые соединения 5a, 5Hb, 5c и 5d продемонстрировали потенциальную активность в ряде клеточных линий рака простаты. Неожиданно, фенил-амино-имидазоловое соединение 5e показало отсутствие активности (IC50 > 1000 нМ в LNCaP, PC-3, DU-145 и PPC-1) в этих клеточных линиях рака простаты. Положительными контролями для этого эксперимента были 55 и 17ya, которые показали значения IC50 между 7,5 нМ и 24,1 нМ в тех же клеточных линиях (Таблица 5С).
Таблица 5С
| Таблица 5C. | IC 50 ± стандартная погрешность средней величины (SEM) (нМ) | |||||
| B16-F1 | A375 | DU 145 | PC-3 | LNCaP | PPC-1 | |
| 5a | 65±12 | 45±8 | 70±4 | 57±3 | 51±1 | 54±1 |
| 5Hb | НО | НО | 35±1 | 38±2 | 35±1 | 36±1 |
| 5c | НО | НО | 63±1 | 43±1 | 41±1 | 37±1 |
| 5d | НО | 25±7 | 73±1 | 33±1 | 45±1 | 36±1 |
| 1h | 55±5 | 28±5 | 71±4 | 21±1 | 28±4 | 43±5 |
| ABT-751 | 2127±351 | 1111±108 | 839±719 | 786±89 | 658±117 | 701±307 |
| D | НО | НО | >1000 | >1000 | >1000 | >1000 |
| E | НО | НО | >1000 | >1000 | >1000 | >1000 |
| 5e | НО | НО | >1000 | >1000 | >1000 | >1000 |
| 55 | НО | НО | 24±6 | 12±1 | 13±1 | 15±1 |
| 17ya | НО | НО | 11±1 | 5±2 | 8±2 | 8±1 |
Была открыта новая серия ингибиторов полимеризации тубулина с приемлемой пероральной биодоступностью и равносильной активностью в клеточных линиях опухолей, обладающих множественной лекарственной устойчивостью. В медицинской химии делаются попытки, начинающиеся с оптимизации SMART соединения 1h. Были исследованы химические модификации различных замещенных арилов в кольце «В» и линкеров между кольцами «В» и «С» на основании биологической оценки против раковых клеток in vitro. Исследования структурно-функциональной взаимосвязи (SAR) показали, что оптимальные кольца «В» включают пиридин (1с), тиофен (1f) и фуран (1d), которые сохраняют превосходный потенциал in vitro. Замена карбонильного линкера цианоимином (2j) между кольцами «В» и «С» увеличивает активность. Были выполнены модификации структуры для увеличения растворимости в воде и биодоступности. Внедрение аминогруппы между кольцами «А» и «В» дало соединения 5а-с, которые показали одинаковое антипролиферативное действие in vitro против исследованных раковых клеток, а также МЛУ (+) и МЛУ (-) клеточных линий, более того, растворимость и биодоступность in vivo была существенно увеличена по сравнению с этими характеристиками соединения 1h. Следовательно, эти новые антитубулиновые соединения представляют новое семейство соединений, которые могут быть очень полезными при лечении рака.
ПРИМЕР 12
АНТИПРОЛИФЕРАТИВНАЯ АКТИВНОСТЬ СОЕДИНЕНИЙ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ.
Антипролиферативная активность аналогов, полученных по способам настоящего изобретения, представлена в Таблицах 6 и 6А.
Таблица 6
IC50 ± стандартная погрешность средней величины (SEM) (нМ)
| ID | LNCaP | PC-3 | DU 145 | PPC-1 | A375 | B16-F1 | WM164 | MES-SA | MES-SA/Dx5 | OVCAR-8 | NCI/ADR-RES |
| Паклитаксел | 1,7 | 4,8 | 5,1 | 2,3 | 12 | 17 | 2,7 | 6,6 | 4,7 | 6263 | |
| Винбластин | 1,1 | 2,1 | 1,8 | 1,1 | 1 | 4,7 | 1,4 | 16 | 3,9 | 582 | |
| Колхицин | 16 | 11 | 10 | 20 | 20 | 29 | 8,4 | 22 | 17 | 1113 | |
| 1k | 101 | 101 | 140 | 84 | 100 | 245 | 220 | ||||
| 2k | 6 | 13 | 12 | 8 | 33 | 43 | 11 | 19 | 34 | 12 | |
| 2m | 19 | 8,7 | 6,9 | 6,2 | 11 | 21 | |||||
| 2n | 101 | 131 | 143 | 99 | 210 | 290 | |||||
| 2o | 65 | 73 | 121 | 73 | 38 | 42 | |||||
| 2p | >10000 | 2385 | 1899 | 1079 | 2200 | 16560 | |||||
| 2q | >10000 | >10000 | >10000 | >10000 | >20000 | >20000 | |||||
| 5c-HCl | 53 | 53 | 70 | 43 | |||||||
| 6d | 703 | 908 | 1637 | 929 |
*НО: не определено
Таблица 6A
IC50 (нМ)
| Структура | ID | LNCaP | PC-3 | DU 145 | PPC-1 | A375 | B16-F1 | WM164 | MES-SA | MES-SA/Dx5 | OVCAR-8 | NCI/ADR-RES |
 |
8 | 346 | 704 | 580 | 230 | 318 | 570 | 404 | ||||
 |
9 | ~10000 | ~10000 | ~10000 | ~10000 | |||||||
 |
10 | 658 | 786 | 839 | 701 | 1111 | 2127 | 661 | ||||
 |
11 | >10000 | >10000 | ~10000 | ~10000 | 3470 | 4900 | 4700 | ||||
 |
12 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |||||
 |
13 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | ||||
 |
14 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |||||
 |
16 | >10000 | >10000 | >10000 | >10000 | 15200 | 6900 | |||||
 |
17 | 2100 | 1900 | 2600 | 1300 | 4300 | 9800 | |||||
 |
18 | ~10000 | ~10000 | ~10000 | ~10000 | |||||||
 |
19 | >20000 | >20000 | >20000 | >20000 | >20000 | >20000 | |||||
 |
20 | 1452 | >10000 | 642 | 633 | 2300 | 3100 | 1300 | ||||
 |
21 | 314 | 403 | 435 | 216 | 383 | 924 | 408 | ||||
 |
22 | >20000 | >20000 | >20000 | >20000 | >20000 | >20000 | |||||
 |
23 | ~10000 | ~10000 | ~10000 | ~10000 | |||||||
 |
24 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | ||||
 |
25 | 48 | 44 | 24 | 13 | 20 | 38 | |||||
 |
26 | 23 | 16 | 16 | 15 | 11 | 14 | |||||
 |
29 | 1788 | >10000 | >10000 | >10000 | >10000 | >10000 | |||||
 |
30 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |||||
 |
32 | 1664 | 2291 | 4601 | 1170 | 2700 | >10000 | 2600 | ||||
 |
33 | >2000 | >2000 | >2000 | >2000 | 9800 | >20000 | |||||
 |
34 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | ||||
 |
35 | 1500 | 40100 | 21900 | 15000 | |||||||
 |
39 | 4300 | 32500 | 16800 | 21400 | |||||||
 |
40 | 13400 | 19600 | 18400 | 6200 | |||||||
 |
41 | 15750 | 18170 | 17040 | >20000 | |||||||
 |
42 | 43590 | 23790 | 24880 | >20000 | |||||||
 |
43 | 12690 | 14720 | 17210 | >20000 | |||||||
 |
17ya | 12 | 10 | 17 | 21 | 17.35 | 32.94 | 12.08 | ||||
 |
17yaa | 233.7 | 148.3 | 592.1 | 208.9 | 481.2 | 538.7 | 467.6 | ||||
 |
15xaa | 1068 | 2628 | 5917 | 4575 | 1800 | 1390 | 1700 | ||||
 |
16xaa | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 |
ПРИМЕР 13
БИОЛОГИЧЕСКАЯ ОЦЕНКА ИЗОХИНОЛИНОВЫХ ПРОИЗВОДНЫХ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Клеточная культура
LNCaP, PC-3, DU-145, PPC-1, MES-SA и MES-SA/DX5 были изначально приобретены у компании ATCC (Роквилл, штат Мэриленд). Все клетки, приобретенные у ATCC, сразу распаковали и заморозили, так что все клеточные линии могли быть восстановлены каждые 2-3 месяца из замороженной емкости с той же партией клеток. Для исследований ксенотрансплантатов in vivo, подлинность PC-3 установили в компании Research Animal Diagnostic Laboratory (Колумбия, штат Миссури) за четыре месяца до начала исследований. Внутривидовую контаминацию исследовали по ПЦР, а идентичность клеточных линий проверили созданием генетического профиля. MES-SA и MES-SA/DX5 выдерживали в среде McCoy 5A, содержащей 2 мМ L-глутамина с добавлением 10% бычьей плодной сыворотки (FBS). Все остальные клетки выдерживали в среде RPMI-1640 с 2 мМ L-глутамина и 10% FBS.
Анализ ингибирования роста
Цитотоксическую или антипролиферативную активность исследуемых соединений изучили в нескольких клеточных линиях с использованием анализа с сульфородамином В (SRB). Культивированные клетки поместили в 96-луночные планшеты и инкубировали со средой, содержащей различные концентрации исследуемых соединений, в течение 96 часов. Клетки окрасили раствором SRB. Оптическую плотность определили при 540 нм на микропланшет-ридере (Dynex Technologies, Шантильи, штат Вирджиния). Построили графики процентного ингибирования клеточного роста в зависимости от концентрации лекарства, и определили концентрацию, которая ингибирует клеточный рост на 50% по сравнению с необработанным контролем (IC50) по нелинейной регрессии по методу наименьших квадратов с использованием программы WinNonlin (Pharsight Corporation, Кэри, штат Северная Каролина).
Анализ клеточного цикла
Распределение клеточного цикла определили по окрашиванию йодидом пропидия (PI). Обработанные клетки промыли PBS и фиксировали 70% ледяным этанолом в течение ночи. Фиксированные клетки затем окрасили при помощи 20 мкг/мл PI в присутствии РНКазы А (300 мкг/мл) при 37 єC в течение 30 минут. Распределение клеточного цикла анализировали при помощи анализа сортировки флуоресцентно-активированных клеток (FACS) в рамках базовых услуг University of Tennessee Health Science Center, штат Теннесси.
Исследования метаболизма in vitro
Для обеих I фаз, инкубационная смесь в 65 мМ фосфатно-калиевого буфера (рН 7,4), состояла из 1 мг/мл печеночных микросомальных белков, 3 мМ НАДФH и 0,5 мкМ исследуемого соединения. Концентрация метанола (используемого для растворения субстрата) составила 1% (об./об.). Общий объем инкубации составил 200 мкл, и реакционные смеси инкубировали при 37°C. Для построения кривых устойчивости исследуемых соединений, различные инкубации остановили через 10, 20, 30, 60 и 90 минут для анализа оставшихся соединений. Все реакции останавливали добавлением 200 мкл ледяного ацетонитрила. Затем образцы центрифугировали при 3000g в течение 5 минут, а надосадочную жидкость анализировали по ЖХ-МС/МС.
Фармакокинетические исследования на мышах
Использовали самцов вышей ICR (5-6 недель, 20-25 г). Для 6a, 6b и 6c вводили дозу 5 мг/кг внутривенным (i.v.), внутрибрюшинным (i.p.) и пероральным (p.o.) путем. Внутривенные дозы вводили через хвостовую вену. Пероральные дозы вводили через зонд. В каждый момент времени три-четыре мыши умертвляли изофлураном (Baxter Healthcare, Дирфилд, штат Иллинойс) и брали образцы крови (до 600 мкл каждый) из нижней полой вены. Образцы плазмы хранили при -20°C до анализов. Белки плазмы осадили добавлением ацетонитрила (150 мкл, содержащего внутренний стандарт) к 100 мкл плазмы мыши. Образцы вращали на вортексе, а затем центрифугировали при 8000g в течение 10 минут. Надосадочную жидкость перенесли в чистую пробирку, чтобы ввести в масс-спектрометр для анализа.
Исследование противоопухолевой эффективности in vivo
Клетки PC-3 (2,5Ч106 клеток/участок) плюс Matrigel (BD biosciences, Сан-Хосе, штат Калифорния) ввели подкожной инъекцией в боковые стороны самцов мышей nu/nu. Размер опухоли измеряли штангенциркулем каждые 2-4 дня и рассчитали как V = р / 6 Ч (длина) Ч (ширина)2. Когда опухоли достигли объема около 100~150 мм3, начали обработку лекарством. Контрольную группу обрабатывали носителем (20% Captex200 в Tween80). Во время обработки размер опухоли и вес тела измеряли каждые 2-4 дня.
Подсчет белых кровяных клеток
Цельную кровь собрали из «голых» мышей в конце исследования эффективности. Для подсчета белых кровяных клеток (WBC) с использованием гемацитометра, 10 мкл образца цельной крови разбавили 190 мкл 2% уксусной кислоты. При надлежащей регулировке освещения лейкоциты появляются на гематоцитометре как темные точки. WBC в каждом образце подсчитали дважды в течение одного часа после разбавления и рассчитали среднее значение.
Результаты
Таблица 7. Противораковая эффективность изохинолиновых соединений в различных раковых клеточных линиях и клетках, обладающих МЛУ, опосредованной P-гликопротеином

ПРИМЕЧАНИЕ: P-gp сверхэкспрессируется в MES-SA/DX5. Фактор устойчивости (RF) рассчитали как отношение значений IC50 для устойчивой клеточной сублинии к значениям IC50 исходной клеточной линии. Все эксперименты выполнили, по меньшей мере, в трех экземплярах
Таблица 8. Соединения 6a, 6b и 6c останавливают клетки PC-3 на фазе G2M.
| EC50 остановки фазы G2M (нМ) | |
| 6a | 53,4 |
| 6b | 91,9 |
| 6c | 23,3 |
Таблица 9. Сводка периодов полувыведения (путь I фазы) 6a, 6b и 6c в печеночных микросомах мышей, крыс, хомяков, кроликов, морских свинок, собак, обезьян и людей
| T ½ (мин.) | ||||
| 6a | 6b | 6c | ||
| Мыши | 3,4 | 10 | 13 | |
| Крысы | 12 | 9 | 14 | |
| Хомяки | 6 | 11 | 20 | |
| Кролики | 17 | 16 | 16 | |
| Морские свинки | 15 | 15 | 8 | |
| Собаки | 13 | 30 | 29 | |
| Обезьяны | 16 | 13 | 9 | |
| Люди | 32 | 40 | 47 | |
Таблица 10. Сводка фармакокинетических свойств соединений 6a, 6b и 6c у мышей
| 6a | 6b | 6c | ||
 |
 |
 |
||
| Молекулярный вес | 410,5 | 359,4 | 338,4 | |
| IV CL (мл*мин-1кг-1) | 5 мг/кг | 51 | 14 | 30 |
| IV Vd (л*кг-1) | 5 мг/кг | 2,3 | 1,1 | 1,8 |
| IP Cmax (нг/мл) | 5 мг/кг | 678,4 | 1500 | 1100 |
| IP AUC (мин*мкг/мл) | 5 мг/кг | 59 | 218 | 55 |
| IP биодоступность | Fip% | 60 | 60 | 33 |
| PO Cmax (нг/мл) | 5 мг/кг | 6,7 | 50 | 50 |
| AUC (мин*мкг/мл) | 5 мг/кг | 5 | 7 | 4 |
| PO биодоступность | Fpo% | 5 | 2,1 | 2,7 |
Эффективность и переносимость 6b и 6c измерили в моделях ксенотрансплантата после внутрибрюшинной инъекции (Фигура 34). Ксенотрансплантаты PC-3 обрабатывались носителем (один раз в день), 6b (40 мг/кг, один раз в день) или 6c (40 мг/кг, один раз в день) в течение 3 недель. Дозировка носителя состояла из 20% Captex200 в Tween80. Объемы опухолей (мм3) нанесли на график в зависимости от времени, и они представляют собой средние значение ± стандартное отклонение для восьми животных. Объемы опухоли и коэффициент выживаемости или вес тела представлены на Фигуре 34А. Размер печени (г) каждой «голой» мыши измерили через 3 недели лечения, и он показан на Фигуре 34В. Количество белых кровяных клеток подсчитали в цельной крови, собранной у животных через 3 недели лечения, и оно показано на Фигуре 34С.
ПРИМЕР 14
АНТИПРОЛИФЕРАТИВНАЯ АКТИВНОСТЬ НЕКОТОРЫХ ABI СОЕДИНЕНИЙ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Анализ цитотоксичности в клеточной культуре
Материалы и способы
Антипролиферативную активность ABI соединений исследовали в трех клеточных линиях меланомы (A375 и WM-164, клеточная линия меланомы человека; B16-F1, клеточная линия меланомы мышей) и в четырех клеточных линиях рака простаты человека (LNCaP, DU 145, PC-3 и PPC-1). Все эти линии были закуплены у компании ATCC (American Type Culture Collection, Манассас, штат Вирджиния), кроме клеточной линии PPC-1. Клетки MDA-MB-435 и MDA-MB-435/LCCMDR1 были любезно предоставлены д-ром Robert Clarke медицинского факультета Джорджтаунского университета, Вашингтон, округ Колумбия. Клетки меланомы выращивали в среде DMEM (Cellgro Mediatech, Inc., Херндон, штат Вирджиния), а клетки рака простаты выращивали в RPMI 1640 (Cellgro Mediatech, Inc., Херндон, штат Вирджиния) с добавлением 10% FBS (Cellgro Mediatech). Культуры выдерживали при 37 °C в увлажненной атмосфере, содержащей 5% CO2. 1000-5000 клеток поместили в каждую лунку 96-луночных планшетов, в зависимости от скорости роста, и воздействовали на них различными концентрациями исследуемых соединений в течение 48 часов (быстро растущие клетки меланомы) или 96 часов (медленно растущие клетки рака простаты), в трех-пяти экземплярах. Количество клеток в конце обработки лекарством измерили анализом с сульфородамином В (SRB). Вкратце, клетки фиксировали 10% трихлоруксусной кислотой и окрасили 0,4% SRB, и измерили поглощение при 540 нм с использованием планшет-ридера (DYNEX Technologies, Шантильи, штат Вирджиния). Процент выживаемости клеток в зависимости от концентрации лекарства нанесли на график и получили значения IC50 (концентрации, которая ингибирует клеточный рост на 50% от необработанного контроля) анализом нелинейной регрессии с использованием GraphPad Prism (GraphPad Software, Сан-Диего, штат Калифорния).
Результаты
Результаты антипролиферативной активности in vitro соединений настоящего изобретения с использованием трех клеточных линий меланомы (одной мышиной клеточной линии меланомы, B16-F1, и двух метастатических клеточных линий меланомы человека, A375 и WM-164), а также четырех клеточных линий рака простаты человека (LNCaP, PC-3, Du 145 и PPC-1) подытожены в Таблицах 11-13.
Таблица 11. Эффект ингибирования роста in vitro соединений без замещения в кольце А.
| Структура | ID | R | IC 50 (нМ) | ||||||
| A375 | B16-F1 | WM164 | LNCaP | PC-3 | Du 145 | PPC-1 | |||
 |
12aa | 3,4,5-(OMe)3 | 160 | 120 | 10 | 152 | 288 | 196 | 133 |
| 12ab | 4-OMe | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 12ac | 3-OMe | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 12ad | 3,5-(OMe)2 | 2800 | 5400 | 2100 | 3611 | 3274 | 2590 | 2129 | |
| 12ae | 3,4-(OMe)2 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 12af | 4-F | 580 | 930 | 630 | 613 | 2197 | 846 | 575 | |
| 12ag | 3-F | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 12ah | 4-Me | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 12ai | 3-Me | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
 |
12aba | 4-OMe | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 |
| 12aaa | 3,4,5-(OMe)3 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
 |
10a | H | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 |
| 10x | 4-NO2 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 10j | 4-OBn | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
Из Таблицы 11 видно, что соединения 12aa-12ai демонстрируют умеренную активность со значениями IC50 в микромолярном диапазоне (в среднем для всех семи клеточных линий). Наиболее сильным соединением в этой серии является 12aa со средним значением IC50 160 нМ. Удаление одной из метокси-групп из 3,4,5-триметокси в кольце С (12ad, 12ae) приводит к существенной потере активности (IC50 > 10 мкМ для 12ae, и среднее значение IC50 3,1 мкМ для 12ad). Соединение с 4-фтором в кольце С (12af) также показало относительно хорошую активность (IC50 = 0,91 мкМ), открытие, которое имеет важное значение, поскольку замена триметокси-фрагмента на 4-фтор-группу может давать хорошую активность и улучшенную метаболическую устойчивость. Положение фтора в кольце С является критичным для активности, поскольку сдвиг с 4-фтор на 3-фтор приводит к полной потере активности (IC50 >10 мкМ для 12ag по сравнению с 0,91 мкМ для 12af). Этот результат позволяет предположить, вблизи 4-положения этого кольца существует потенциальный донор водородной связи.
Как ясно показано в Таблице 11, положения колец А и С являются критичными. Простой сдвиг С-кольцевого фрагмента из положения 4 в положение 1 в имидазоловом кольце (кольцо В) приводит к полной потере активности (IC50 >10 мкМ для 12aba, 12aaa, 10a, 10x, 10j).
Таблица 12. Эффект ингибирования роста in vitro соединений с замещениями в кольце А
| ID | R 1 | R 2 | IC50 ± стандартная погрешность средней величины (SEM) (нМ) | |||||||||
| A375 | B16-F1 | WM164 | LNCaP | PC-3 | Du 145 | PPC-1 | OVCAR-8 | NCI/ADR-RES | ||||
|
|
12ba | 4-F | 3,4,5-(OMe)3 | 205±19 | 320±41 | 73±8 | 98±2 | 169±12 | 132±24 | 81±1 | ||
| 12ca | 4-OMe | 3,4,5-(OMe)3 | 30±5 | 108±12 | 31±4 | 31±1 | 45±1 | 48±0,5 | 34±0,3 | |||
| 12cb | 4-OMe | 4-F | 31±5 | 63±7 | 28±3 | 28±2 | 31±2 | 41±38 | 29±1 | |||
| 12da | 4-Me | 3,4,5-(OMe)3 | 9±2 | 46±5 | 8±2 | 12±1 | 9±0,4 | 15±0,5 | 11±0,1 | |||
| 12db | 4-Me | 4-F | 143±13 | 222±10 | 156±19 | 45±2 | 66±3 | 78±5 | 54±1 | |||
| 12db-HCl | 108±11 | 297±23 | 112±9 | НО | НО | НО | НО | |||||
| 12dc | 4-Me | 3,5-(OMe)2-4-OH | 105 | 387 | 123 | 134 | 127 | 174 | 110 | |||
| 12ea | 3,4,5-(OMe)3 | 3,4,5-(OMe)3 | 4800 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |||
| 12eb | 3,4,5-(OMe)3 | 4-F | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |||
| 12fa | 4-Cl | 3,4,5-(OMe)3 | 43±5 | 168±14 | 26±3 | 24±1 | 35±1 | 36±0,4 | 26±0,2 | 47 | 19 | |
| 12fb | 4-Cl | 4-F | 52±4 | 73±6 | 74±9 | 49±2 | 81±2 | 65±1 | 52±1 | |||
| 13fa | 4-Cl | 3,4,5-(OH)3 | 3900 | 1810 | 2100 | 10000 | 10000 | 10000 | >10000 | |||
| 12ga | 4-N(Me)2 | 3,4,5-(OMe)3 | 82±9 | 361±29 | 80±11 | 58±2 | 92±4 | 95±1 | 67±0,7 | |||
| 12gb | 4-N(Me)2 | 4-F | 56±7 | 129±11 | 62±8 | 57±6 | 81±3 | 72±0,4 | 45±0,3 | |||
| 12ha | 3,4-(OMe)2 | 3,4,5-(OMe)3 | 113±14 | 1400±200 | 191±18 | 121±10 | 203±7 | 168±15 | 117±1 | |||
| 12hb | 3,4-(OMe)2 | 4-F | 10000 | 4210 | 1400 | 2533 | 10000 | 10000 | 2172±48 | |||
| 12ia | 2-CF3 | 3,4,5-(OMe)3 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |||
| 12ib | 2-CF3 | 4-F | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |||
| 13ea | 3,4,5-(OH)3 | 3,4,5-(OH)3 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |||
| 12ja | 4-OBn | 3,4,5-(OMe)3 | 5200 | 10000 | 5500 | 2786 | 10000 | 10000 | 2844 | |||
| 12jb | 4-OBn | 4-F | 93±8 | 117±16 | 90±12 | 44±7 | 79±0,4 | 60±3 | 43±0,2 | |||
| 12ka | 4-OH | 3,4,5-(OMe)3 | 1600 | 2400 | 1800 | НО | НО | НО | НО | |||
| 12kb | 4-OH | 4-F | 10000 | >10000 | >10000 | 10000 | >10000 | >10000 | >10000 | |||
| 12kc | 4-OH | 3-OH, 4,5-(OMe)2 | 10000 | 5600 | 6400 | |||||||
| 12la | 4-Br | 3,4,5-(OMe)3 | 32 | 74 | 36 | 34 | 36 | 49 | 33 | |||
| 12pa | 4-CF3 | 3,4,5-(OMe)3 | 163,1 | 468,7 | 175 | 134 | 127 | 174 | 110 | |||
| 13ha | 3,4-(OH)2 | 3,4,5-(OH)3 | >10000 | >10000 | >10000 | НО | НО | НО | НО | |||
| Колхицин | 20±3 | 29±5 | НО | 16±4 | 11±1 | 10±2 | 20±1 | |||||
НО - не определено
Из Таблицы 12 видно, что соединения с 3,4,5-триметокси и 4-фтор-замещениями в кольце А демонстрируют хорошую активность при различных замещениях в кольце А. Эти соединения демонстрируют превосходную антипролиферативную активность со значениями IC50 лишь 8,0 нМ в клеточной линии WM164 (12da). В целом, соединения, включающие единственный заместитель в пара-положении кольца А, являются более сильными, что можно видеть из активностей 12ca, 12cb, 12da, 12db, 12fa, 12fb, 12ga и 12gb (IC50 = 7,9-110 нМ). Соль 12db-HCl (IC50 = 172 нМ) показала немного меньшую активность по сравнению с соответствующим свободным основанием 12db (IC50 = 109 нМ). Соединение 12fb (IC50 = 63,7 нМ), с единственным галогеновым заместителем в пара-положении колец А и С, продемонстрировало силу действия, и оно не имеет метокси-группы. Соединения с 3,4,5-триметокси-заместителями в кольце А полностью теряют активность (IC50 > 10 мкМ для 12ea, 12eb), что позволяет предположить весьма различные связывающие условия возле кольца А и кольца С. Удаление 5-метокси-заместителя из кольца А существенно улучшает активность (IC50 = 330 нМ и >10 мкМ для 12ha, 12ea, соответственно). Деметилирование 3,4,5-триметокси-группы резко снижает активность с 43 нМ (12fa) до 3,89 мкМ (13fa). Такие же результаты наблюдались для 13ea, 12ka, 12kb и 13ha в результате деметилирования заместителей в кольце А или С. Электронодонорные группы (4-метокси, 4-диметиламино, 4-метил) и электроноакцепторные группы (4-хлор, 2-трифторметил) в кольце А не показали существенных различий в активности. Внедрение трифторметиловой группы в орто-положение кольца А обусловило полную потерю активности (IC50 >10 мкМ для 12ia, 12ib). Наличие бензилокси-группы в пара-положении кольца А (IC50 = 75 нМ для 12jb) привело к 440-кратному увеличению активности по сравнению с пара-гидрокси-соединением 12kb (IC50=33 мкМ). Необходимо отметить, что соединение 12jb, с 4-фтор в кольце С, обладает лучшей активностью, чем его аналог 12ja, который имеет 3,4,5-триметокси-группу в кольце С (IC50 составляет 75 нМ для 12jb и 7,3 мкМ для 12ja).
Таблица 13. Эффект ингибирования роста in vitro соединений с защитой в кольце В.
| Структура | ID | R 1 | R 2 | R 3 | IC 50 ± стандартная погрешность средней величины (SEM) (нМ) | ||||||
| A375 | B16-F1 | WM164 | LNCaP | PC-3 | Du 145 | PPC-1 | |||||
 |
11ab | H | 4-OMe | SO2Ph | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 |
| 11ac | H | 3-OMe | SO2Ph | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 11ah | H | 4-Me | SO2Ph | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 11af | H | 4-F | SO2Ph | 630±72 | 946±86 | 596±61 | 573 | 2233 | 846 | 575 | |
| 11ag | H | 3-F | SO2Ph | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 11cb | 4-OMe | 4-F | SO2Ph | 36±5 | 71±8 | 43±6 | 31±2 | 33±2 | 52±3 | 32±0,7 | |
| 11db | 4-Me | 4-F | SO2Ph | 113±14 | 287±31 | 107±14 | 55±3 | 80±1 | 80±1 | 57±1 | |
| 11ea | 3,4,5-(OMe)3 | 3,4,5-(OMe)3 | SO2Ph | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 11eb | 3,4,5-(OMe)3 | 4-F | SO2Ph | 3840 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 11fb | 4-Cl | 4-F | SO2Ph | 88±9 | 107±12 | 70±6 | 48±1 | 76±2 | 64±1 | 54±1 | |
| 11ga | 4-N(Me)2 | 3,4,5-(OMe)3 | SO2Ph | 162±13 | 1200±90 | 308±32 | 62±2 | 93±6 | 99±2 | 72±0,4 | |
| 11gb | 4-N(Me)2 | 4-F | SO2Ph | 55±7 | 242±26 | 56±4 | 56±6 | 83±3 | 74±0,5 | 48±0,3 | |
| 11ha | 3,4-(OMe)2 | 3,4,5-(OMe)3 | SO2Ph | 192±15 | 970±68 | 139±15 | 114±6 | 197±9 | 144±29 | 117±2 | |
| 11hb | 3,4-(OMe)2 | 4-F | SO2Ph | 960±59 | 2000±400 | 1400±30 | 1915±77 | 10000 | 3312 | 1441±49 | |
| 11ia | 2-CF3 | 3,4,5-(OMe)3 | SO2Ph | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 11ib | 2-CF3 | 4-F | SO2Ph | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 11jb | 4-OBn | 4-F | SO2Ph | 64±7 | 110±15 | 48±5 | 35±1 | 75±0,5 | 58±1 | 38±0,2 | |
| 12dab | 4-Me | 3,4,5-(OMe)3 | Me | 32 | 134 | 40 | 32 | 46 | 36 | 28 | |
| 12cba | 4-OMe | 4-F | Me | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | >10000 | |
| 12daa | 4-Me | 3,4,5-(OMe)3 | CH2Ph | 683,2 | 465,8 | 1501 | 777,9 | ||||
| 12gba | 4-N(Me)2 | 4-F | SO2PhOMe | ~100 | ~100 | ~100 | 73,2 | 44,14 | 129,4 | 63,4 | |
Из Таблицы 13 видно, что соединения с фенилсульфониловой защитной группой, присоединенной к азоту имидазолового кольца (11cb, 11db, 11fb, 11ga, 11gb, 11ha, 11jb) также являются очень активными со значениями IC50 в наномолярном диапазоне (Таблица 13). В целом, активность этих соединений сравнима с их соответствующими незащищенными аналогами, что проиллюстрировано сравнением активностей 11cb (43 нМ), 11db (111 нМ), 11fb (72 нМ), 11ga (285 нМ), 11gb (87 нМ), 11ha (268 нМ) и 11jb (61 нМ) с их соответствующими незащищенными аналогами 12cb (36 нМ), 12db (109 нМ), 12fb (64 нМ), 12ga (131 нМ), 12gb (72 нМ), 12ha (330 нМ) и 12jb (75 нМ). Другие соединения (11ab-11ag, 11ea, 11eb, 11hb, 11ia и 11ib, 1-50 мкМ), в целом, являются гораздо менее активными, и также сравнимыми с их аналогами (12ab-12ag, 12ea, 12eb, 12hb, 12ia и 12ib, 1-50 мкМ).
ПРИМЕР 15
АКТИВНОСТЬ АРИЛ-БЕНЗОИЛ-ИМИДАЗОЛЬНЫХ (ABI) СОЕДИНЕНИЙ В КЛЕТКАХ ЛЕКАРСТВЕННО-УСТОЙЧИВОЙ МЕЛАНОМЫ
Р-гликопротеин (P-gp)-опосредованный отток лекарства представляет основной механизм, по которому раковые клетки предотвращают накопление эффективных противораковых концентраций лекарства внутри клетки. Активность ABI соединений сравнили против клеток меланомы, обладающей множественной лекарственной устойчивостью (МЛУ) (MDA-MB-435/LCCMDR1) и их исходных неустойчивых раковых клеток (MDA-MB-435). Хотя MDA-MB-435 первоначально определяется как клеточная линия рака молочной железы, было окончательно показано, что она происходит из клеточной линии меланомы М14. Соединения 12da, 12fb, 12cb, 11cb и 11fb вместе с другими тубулин-направленными агентами, включая колхицин, паклитаксел и винбластин, были исследованы в клеточной линии МЛУ меланомы и ее исходной клеточной линии меланомы (Таблица 14А). Паклитаксел и винбластин являются клинически используемыми противораковыми средствами, которые, как известно, направлены на клеточный тубулин. Хотя колхицин не является одобренным FDA лекарством для лечения рака, его пролекарство, ZD6126, проходит клинические испытания для лечения солидных опухолей. Бортезомиб является первым терапевтическим ингибитором протеасом, и он был одобрен FDA в 2003 году для применения при лечении множественной миеломы. Известно, что ABT-751 направлен на тубулиновый колхицин-связывающий сайт. Он является многообещающим потенциальным лекарством в клинических испытаниях для лечения детей с рецидивной или рефрактерной нейробластомой. Соединения 12da, 12fb, 12cb, 11cb, 11fb имеют гораздо более хорошие показатели устойчивости (3,0 для 12da, 0,9 для 12fb, 1,3 для 12cb, 0,8 для 11cb, 0,7 для 11fb), чем колхицин (65,8), паклитаксел (69,3) и винбластин (27,5). Хотя колхицин, паклитаксел и винбластин демонстрируют превосходную активность в клеточных линиях неустойчивой меланомы (0,5-10 нМ), эти соединения обладают существенно меньшей силой в клеточной линии МЛУ меланомы (277-658 нМ). Напротив, 12cb, 11cb, 11fb обладают, по существу, эквивалентным действием на клеточные линии МЛУ (15 нМ, 38 нМ, 30 нМ, 30 нМ, 35 нМ для 12da, 12fb, 12cb, 11cb и 11fb, соответственно) и неустойчивой меланомы (5 нМ, 41 нМ, 24 нМ, 38 нМ, 50 нМ для 12da, 12fb, 12cb, 11cb и 11fb, соответственно). Соединение 12da является более активным, чем паклитаксел и колхицин при действии на клетки A375 и WM-164.
Таблица 14А. In vitro эффект ингибирования роста ABI соединений по сравнению с другими противораковыми лекарствами на клеточную линию меланомы, обладающей множественной лекарственной устойчивостью (МЛУ клетки), и соответствующую восприимчивую исходную клеточную линию (клетки нормальной меланомы)
| ID соединения | IC 50 ± стандартная погрешность средней величины (SEM) ( n=3 ) | ||||||
| A375 | B16-F1 | WM-164 |
Связывание тубулина
(мкМ) |
MDA-MB-435 |
MDA-MB-435
/LCC6MDR1 |
Коэффициент устойчивости* | |
| 12da | 9±2 | 46±5 | 8±2 | 0,2±0,1 | 5±1 | 15±2 | 3,0 |
| 12fb | 52±4 | 73±6 | 74±9 | 3,9±2,1 | 41±2 | 38±2 | 0,9 |
| 12cb | 31±5 | 63±7 | 28±3 | 3,4±1,5 | 24±2 | 30±4 | 1,3 |
| 11cb | 36±5 | 71±8 | 43±6 | НО | 38±3 | 30±2 | 0,8 |
| 11fb | 88±9 | 107±12 | 74±8 | НО | 50±6 | 35±3 | 0,7 |
| Паклитаксел | 12±3 | 17±2 | 18±3 | N/A | 4±1 | 277±41 | 69,3 |
| Винбластин | 1,1±0,2 | 4,7±0,7 | 0,6±0,1 | НО | 0,4±0,1 | 11±1 | 27,5 |
| Колхицин | 20±3 | 29±5 | 10±2 | 1,8±0,5 | 10±1 | 658±50 | 65,8 |
| Бортезомиб | 8±1 | 24±2 | 8±1 | НО | НО | НО | НО |
| ABT-751 | 1111±108 | 2127±351 | 661±56 | НО | НО | НО | НО |
*Коэффициенты устойчивости рассчитали делением значений IC50 для клеточной линии, обладающей множественной лекарственной устойчивостью, MDA-MB-435/LCC6MDR1, на значения IC50 для соответствующей восприимчивой исходной клеточной линии MDA-MB-435. Аббревиатуры: N/A, значение отсутствует; НО, не определено
Таблица 14В. Противораковая эффективность и связывающая способность колхицинового сайта ABI в различных раковых и МЛУ клеточных линиях с различными механизмами устойчивости. ABI демонстрирую превосходный потенциал против всех исследованных клеточных линий меланомы, включая высоко метастатическую клеточную линию и клеточную линию с множественной лекарственной устойчивостью. Высокая связывающая способность ABI с колхицин-связывающим сайтом тубулина подтверждает, что они направлены внутрь клеток
| IC50 ± стандартная погрешность средней величины (SEM) (ммоль/л) (n=3) | ||||||||
| 12cb | 12da | 12fb | Паклитаксел | Винбластин | Колхицин | ABT-751 | SN-38 | |
| A375 | 31±5 | 9±2 | 52±4 | 12±3 | 1±0,1 | 20±3 | 685±108 | НО |
| A375MA2 | 44±5 | 8±1 | 55±4 | 8±1 | 1±0,2 | 18±2 | 265±36 | НО |
| B16-F1 | 63±7 | 46±5 | 73±6 | 17±2 | 5±1 | 29±5 | 2127±351 | НО |
| WM-164 | 28±3 | 8±2 | 74±9 | 18±3 | 0,6±0,1 | 10±2 | 661±56 | НО |
| MDR1 | ||||||||
| MDA-MB-435* | 24±2 | 5±1 | 41±2 | 4±1 | 0,4±0,1 | 10±1 | 417±23 | НО |
| MDA-MB-435/LCC6MDR1 | 30±4 (1) |
11±2 (2) |
38±2 (1) |
277±4 (69) |
11±1 (28) |
658±50 (66) |
577±31 (1) |
НО |
| OVCAR-8* | 25±2 | 11 ±1 | 45±2 | 10±0,2 | 2±0,1 | 12±1 | 785±17 | 2±0,2 |
| NCI/ADR-RES | 13±1 (0,5) |
5±0,1 (0,5) |
20±6 (0,4) |
5109±170 (511) |
570±84 (285) |
737±51 (61) |
864±42 (1) |
10±1 (5) |
| MRP | ||||||||
| HEK293 -pcDNA3.1* | 12±2 | 9±1 | 54±0,3 | 9±0,3 | 5±0,1 | 3±0,4 | 645±153 | 3±0,4 |
| HEK293-MRP1 | 16±2 (1) |
8±1 (0,9) |
33±7 (0,6) |
30±3 (3) |
24±1 (5) |
5±0,1 (2) |
717±28 (1) |
9±0,04 (3) |
| HEK293-MRP2 | 14±4 (1) |
8±0,3 (0,9) |
39±12 (0,7) |
37±2 (4) |
28±2 (6) |
3±0,3 (1) |
747±7 (1) |
7±0,1 (2) |
| BCRP | ||||||||
| HEK293-482R2 | 17±1 (1) |
8±1 (0,9) |
23±3 (0,4) |
50±1 (6) |
25±1 (5) |
5±0,1 (2) |
653±72 (1) |
123±28 (41) |
| Связывание тубулина (мкМ)+ | 3±1 | 0,2±0,1 | 4±1 | N/A | НО | 2±1 | 3,1++ | НО |
Примечания: *: исходная клеточная линия для лекарственно-устойчивой клеточной сублинии; MDR1 сверхэкспрессировался в MDA-MB-435/LCC6MDR1 и NCI/ADR-RES; MRP1, MRP2 и BCRP сверхэкспрессировались в HEK293-MRP1, HEK293-MRP2 и HEK293-482R2. Коэффициенты устойчивости (значения в скобках) рассчитали делением значений IC50 для устойчивой клеточной сублинии на IC50 для соответствующей исходной клеточной линии. +: IC50 для связывания тубулина рассчитали сцинтилляционным анализом сближения конкурентного связывания [3H]-колхицина. ++: связывающая способность описана в литературе для ABT-751. Аббревиатуры: N/A, не применимо, поскольку они связываются с тубулином у различных сайтов.
Результаты в Таблице 14А показывают, что клеточная линия MDA-MB-435/LCCMDR1 является очень устойчивой к колхицину, паклитакселу и винбластину. Однако ABI настоящего изобретения показали равное действие на лекарственно-устойчивую клеточную линию и восприимчивую исходную клеточную линию. Этот результат является мощным толчком к мысли, что ABI не являются субстратом для P-gp. Следовательно, они преодолевают множественную устойчивость, встречающуюся в клетках MDA-MB-435/LCCMDR1. Дозазависимые кривые представлены на Фигуре 21 для 12fb, 12da и 12cb. В Таблице 14В дополнительно изучаются механизмы устойчивости для паклитаксела, SN-38, винбластина и колхицина, по сравнению с ABI 12cb, 12da и 12fb. MRP и BCRP дают умеренную устойчивость к паклитакселу (коэффициенты устойчивости 4 и 6, соответственно), винбластину (коэффициенты устойчивости 6 и 5, соответственно), а BCRP дает существенную устойчивость к SN-38 (коэффициент устойчивости 41). Однако ни один из ABI не оказался восприимчивым к MRP- или BCRP-опосредованной устойчивости (коэффициенты устойчивости в диапазоне от 0,4 до 1,0). ABT-751, как и ABI, оказался не восприимчивым к MDR1, MRP или BCRP.
ПРИМЕР 16
АНАЛИЗ ПОЛИМЕРИЗАЦИИ МИКРОТРУБОЧЕК IN VITRO
Материалы и способы
Бычий тубулин мозга (0,4 мг) (Cytoskeleton, Денвер, штат Колорадо) смешали с 10 мкМ исследуемого соединения и инкубировали в 110 мкл общего тубулинового буфера (80 мМ PIPES, 2,0 мМ MgCl2, 0,5 мМ EGTA и 1 мМ ГТФ) при pH 6,9. Поглощение при 340 нм контролировали каждую 1 минуту в течение 15 минут при помощи микропланшет-ридера SYNERGY 4 (Bio-Tek Instruments, Винооски, штат Вермонт). Спектрофотометр был установлен на 37 °C для полимеризации тубулина.
Результаты
Было изучено ингибирование полимеризации тубулина арил-бензоил-имидазоловыми (ABI) соединениями. Бычий тубулин мозга (чистота >97%) инкубировали с тремя сильнодействующими ABI соединениями, 12cb, 12da и 12db в концентрации 10 мкМ, для определения эффекта этих ABI соединений на полимеризацию тубулина (Фигура 22). Полимеризация тубулина полностью ингибировалась соединением 12da, тогда как при инкубировании с соединениями 12cb и 12db наблюдалось ~ 80% ингибирования.
Этот эффект дестабилизации микроторубочек был таким же, как и для колхицина и винбластина, но противоположным паклитакселу. Эти результаты не только подтвердили, что ABI могут напрямую взаимодействовать с тубулином, но также позволяют предположить, что они могут разделять одинаковые связывающие сайты с колхицином (или винбластином).
ПРИМЕР 17
ИНГИБИРОВАНИЕ МЕЛАНОМЫ IN VITRO
Материалы и способы
Клетки меланомы B16-F1 поместили при колониеобразующей плотности (2000 клеток на лунку в шестилуночные планшеты) поверх 0,8% агаровой основы. Клетки выращивали в 0,4% агаре вместе со средой DMEM, дополненной бычьей плодной сывороткой и раствором антибиотика/фунгицида при 37 °C в атмосфере 95% воздуха и 5% CO2. Клетки обработали соединениями 12da, 12cb и 12fb в различных концентрациях (20, 100 и 500 нМ). Соединения добавляли в среду из 1 мМ маточных растворов в ДМСО, а соответствующее разбавление ДМСО использовали в качестве контроля. Клетки выращивали в течение 14 дней. Планшеты сфотографировали, а количество колоний измерили автоматическим счетчиком колоний Artek 880 (Artek Systems Corporation, Фармингдейл, штат Нью-Йорк).
Результаты
Четыре иллюстративные фотографии представлены на Фигуре 23. Через 14 дней инкубирования в контрольных образцах (без обработки) образовалось около 130 обнаруживаемых колоний (диаметр более 100 мкм).
Соединения 12cb и 12da эффективно ингибировали образование колоний меланомы B16-F1 даже при самой низкой исследованной концентрации, 20 нМ (р<0,05 по сравнению с контролем). 12fb продемонстрировало эффективное ингибирование при 100 нМ. Все три исследованные соединения показали полное ингибирование образования колоний при 0,5 мкМ, дополнительно подтверждая эффективность ABI против меланомы.
ПРИМЕР 18
ПРОТИВООПУХОЛЕВАЯ АКТИВНОСТЬ IN VIVO
Материалы и способы
Животные: Самок мышей C57/BL возрастом 4-6 недель приобрели у компании Harlan Laboratories (Harlan Laboratories Inc., Индианаполис, штат Индиана). Условия содержания животных удовлетворяли Ассоциации по оценке и аккредитации и нормативам по уходу за лабораторными животными. Все процедуры проводились в соответствии с рекомендациями институционального комитета по уходу за животными и их использованию.
Оценка эффективности in vivo. Клетки меланомы мышей B16-F1 подготовили в среде DMEM, не содержащей FBS (Cellgro Mediatech) в концентрации 5 x 106 жизнеспособных клеток/мл. Суспензию клеток (100 мкл) ввели подкожной инъекцией в правый дорсальный бок каждой мыши. Когда размер опухоли достиг около 100-150 мм3, примерно через 7 дней после инокуляции клеток, всех мышей с опухолями разделили на контрольную и экспериментальную группу обработки по размеру опухоли (n = 5). Каждая группа имела одинаковый средний размер опухоли. Мышам в контрольной группе (отрицательный контроль) внутрибрюшинно вводили инъекции 50 мкл только раствора носителя или DTIC в дозировке 60 мг/кг (положительный контроль) один раз в день. Размер опухоли изеряли каждые 2 дня при помощи контролируемого штангенциркуля с электронным цифровым отсчётом (Fisher Scientific, Inc., Питтсбург, штат Пенсильвания) и рассчитали по формуле a Ч b2 Ч0,5, где a и b представляют наибольший и наименьший диаметры, соответственно. Объем опухоли выразили в кубических миллиметрах. Данные выразили как среднее значение ± стандартная ошибка для каждой группы и нанесли на график как функцию от времени. Процентное уменьшение опухоли по завершению эксперимента (через 14 дней после начала обработки) рассчитали по формуле 100–100 Ч [(T - T0)/(C - C0)], где Т представляет средний объем опухоли обработанной группы в определенный день, T0 представляет средний объем опухоли той же группы в первый день обработки, С представляет средний объем опухоли контрольной группы в определенный день, и С0 представляет средний объем опухоли той же группы в первый день обработки. Активность животных и средний вес тела в каждой группе контролировали в течение всего экспериментального периода для оценки токсичности соединения. В конце обработки всех мышей умертвили CO2 с последующим смещением шейных позвонков, а опухоли собрали для дальнейших исследований.
Результаты
Для оценки эффективности ABI аналогов in vivo, авторы изобретения исследовали противоопухолевую активность соединения 12cb на ксенотрансплантате меланомы мышей B16-F1 по сравнению с DTIC, золотым стандартом при лечении злокачественной меланомы, который использовался в качестве положительного контроля (Фигура 24А). Двадцать самок мышей C57/BL разделили на четыре группы: контрольная группа носителя, группа обработки DTIC (60 мг/кг), группа обработки 12cb (10 мг/кг) и группа обработки 12cb (30 мг/кг). Каждой мыши вели 0,5 миллионов клеток меланомы B16-F1 подкожной инъекцией. Через семь дней после инокуляции опухоли начали лечение, при котором каждое соединение ежедневно вводили внутрибрюшинно (Фигура 24). Объем опухоли существенно снизился (p<0,05), на 47%, 51% и 73% для 12cb (10 мг/кг), DTIC (60 мг/кг) и 12cb (30 мг/кг), соответственно, через 14 дней лечения. Во всех группах лечения не наблюдалось какого-либо существенного снижения веса в ходе эксперимента.
Было выбрано два уровня дозирования 12fb, 15 и 45 мг/кг. DTIC при 60 мг/кг использовался в качестве положительного контроля. Сначала для исследования выбрали модель аллотрансплантата меланомы B16-F1 на мышах C57BL/6. Через 13 дней лечения (Фигура 24В) соединение 12fb ингибировало рост опухоли меланомы (значение TGI) на 32% при 15 мг/кг и на 82% при 45 мг/кг. Значение р t-критерия Стьюдента для 12fb при 45 мг/кг по сравнению с контролем составило менее 0,001, указывая на существенное различие. Значение р t-критерия для 12fb при 15 мг/кг по сравнению с контролем составило 0,08, позволяя предположить, что эта доза не является эффективной. Сравнивая 12fb при 45 мг/кг с DTIC при 60 мг/кг, который имеет TGI, равный 51%, значение р t-критерия составило 0,001, позволяя предположить, что 12fb обладает существенно более высокой активностью, чем DTIC. Для группы контрольного образца и группы обработки 12fb при 15 мг/кг, средний все тела немного увеличился в течение экспериментального периода.
Для дальнейшего подтвердждения активности ABI in vivo, использовали модель ксенотрансплантата меланомы человека А375 на мышах SHO, и исследовали 12fb при 25 мг/кг. DTIC при 60 мг/кг снова использовали в качестве положительного контроля. Через 31 день обработки (Фигура 24С) 12fb ингибировал рост опухоли меланомы (значение TGI) на 69%, тогда как DTIC ингибировал рост на 52%. Значение р t-критерия для обработки 12fb по сравнению с контрольным образцом составило менее 0,001, позволяя предположить, что 12fb существенно ингибирует рост опухоли меланомы при 25 мг/кг. Значение р t-критерия для обработки 12fb по сравнению с DTIC составило менее 0,05, снова позволяя предположить, что 12fb имеет лучшую активность, чем DTIC. В течение экспериментального периода средний вес тела во всех группах незначительно увеличился. Физическая активность мышей также выглядела нормальной, позволяя предположить, что 25 мг/кг является хорошо переносимой дозой для мышей SHO.
ПРИМЕР 19
СВЯЗЫВАНИЕ С КОЛХИЦИНОМ
Материалы и способы
Каждое исследуемое соединение приготовили в 20 Ч концентрации в буфере G-PEM (Cytoskeleton Inc., Денвер, штат Колорадо), а затем пипетировали 10 мкл исследуемого соединения в 96-луночные планшеты. Десять микролитров меченного литием колхицина (Perkin-Elmer, Уолтем, штат Массачусетс) также добавили в каждую экспериментальную лунку. Затем в каждую лунку последовательно добавили 180 мкл суспензии шариков/тубулина (GE Healthcare Bio-Sciences Corp., Пискатавэй, штат Нью-Джерси). Планшет инкубировали в течение 45 минут при 37°C, а затем считывали на планшет-ридере Topcount NXT (Perkin-Elmer, Уолтем, штат Массачусетс). «Холодный» колхицин без радиоактивной метки включили в качестве положительного контроля, а паклитаксел - в качестве отрицательного контроля, поскольку паклитаксел связывается с другим сайтом тубулина и не конкурирует за связывание колхицинового сайта. Данные обработали с использованием программы GraphPad Prism.
Анализ клеточного цикла
Выполнили анализ проточной цитометрии для исследования распределения фаз клеточного цикла. Клетки А375 выращивали в 10-см тканевых культуральных чашках до слияния около 80%, а затем клетки обрабатывали 0, 10, 50, 200 и 1000 нМ колхицина, 12da, 12fb и 12cb в течение 24 часов в питательной среде. Клеточную ДНК окрасили PBS, содержащим 50 мкг/мл йодида пропидия и 100 мкг/мл РНКазы А. Клеточный цикл определили с использованием цитометра BD LSR-II (BD Biosciences, Сан-Хосе, штат Калифорния) при сравнении 10000 клеток. Данные проанализировали, а графики построили с использованием программы Modfit 2.0 (Verity Software House, Топшам, штат Мэн).
Результаты
Было отмечено три лиганд-связывающих сайта в тубулиновом б/в-гетеродимере: паклитаксел-связывающий сайт, винбластин-связывающий сайт и колхицин-связывающий сайт. Измерили связывающую способность соединения 12cb с использованием 3H-меченного колхицина и сцинтилляционного анализа сближения конкуреного связывания (SPA). Результаты подтвердили прочное связывание 12cb со связывающей способностью 3,4±1,5 мкМ (Фигура 25А). Колхицин в этих условиях связывает тубулин со значением IC50 1,8±0,5. Эти результаты ясно показывают, что ABI соединения эффективно ингибируют полимеризацию тубулина.
Диаграмма связывания (Фигура 25А) ясно показывает, что ABI могут конкурентно связываться с колхицин-связывающим сайтом тубулина. По мере увеличения концентрации трех исследуемых соединений с 0,03 мкМ до 100 мкМ, повышенное количество меченного тритием колхицина конкурентно уходило с тубулина и испускало более слабые импульсы SPA. Отрицательный контроль, паклитаксел, дал лишь прямую линию, поскольку теоретически он не должен связываться с колхицин-связывающим сайтом тубулина. Во-вторых, ABI имеют относительно высокую связывающую способность к тубулиновому колхицин-связывающему сайту. Значения IC50, рассчитанные GraphPad Prism для связывания, показали, что 12da имеет самую высокую связывающую способность. Связывающая способность положительно коррелировала с активностью против меланомы in vitro; чем выше связывающая способность, тем выше активность против меланомы.
ABI продемонстрировали, что они останавливают клетки в анализе клеточного цикла на физе G2/M, что указывает на то, что они направлены на тубулин. Соединения 12da, 12fb и 12cb исследовались вместе с колхицином в качестве положительного контроля на клетках А375 (Фигура 25В). Чтобы показать влияние дозы, использовали четыре различные концентрации - 10, 50, 200 и 1000 нМ - каждого соединения (Фигура 25С и 25D). Для контрольных образцов (без обработки) без воздействия, около 16% клеток А375 сгруппировали на фазе G2/M. Для группы обработки колхицином, по мере увеличения концентрации с 10 нМ до 50 нМ, процентное содержание клеток, сгруппированных на фазе G2/M, увеличилось с 14% до 85%. ABI дали такие же результаты для клеток А375, останавливая их на фазе G2/M доза-зависимым образом. Эффективность различных концентраций для остановки клеток на фазе G2/M положительно коррелирует с активностью in vitro.
ПРИМЕР 20
ФАРМАКОЛОГИЯ СОЕДИНЕНИЙ 17ya, 12fa И 55 IN VITRO И IN VIVO
Материалы и способы
Анализ клеточной культуры и цитотоксичности рака простаты. Все клеточные линии рака простаты (LNCaP, PC-3 и DU145, PPC-1) приобрели у компании ATCC (American Type Culture Collection, Манассас, штат Вирджиния, США). PC-3_TxR человека был устойчивым к паклитакселу и использовался в качестве МЛУ модели по сравнению с PC-3. Клеточную культуру приобрели у компании Cellgro Mediatech (Херндон, штат Вирджиния, США). Все клеточные линии использовали для исследования антипролиферативной активности соединений 17ya, 12fa и 55 анализом с сульфородамином В (SRB). Все клеточные линии выдерживали в среде RPMI 1640 с 2 мМ глутамином и 10% бычьей плодной сывороткой (FBS).
Анализ полимеризации микротрубочек in vitro. Свиной тубулин мозга (0,4 мг) (Cytoskeleton, Денвер, штат Колорадо) смешали с 1 и 5 мкМ исследуемого соединения или носителем (ДМСО) и инкубировали в 100 мкл буфера (80 мМ PIPES, 2,0 мМ MgCl2, 0,5 мМ EGTA, рН 6,9 и 1 мМ ГТФ). Поглощение при длине волны 340 нм контролировали каждую минуту в течение 15 минут (микропланшет-ридер SYNERGY 4, Bio-Tek Instruments, Винооски, штат Вермонт). Спектрофотометр поддерживали при 37°C для полимеризации тубулина.
Метаболические инкубации. Исследования метаболической устойчивости осуществляли инкубацией 0,5 мкМ исследуемых соединений в общем реакционном объеме 1 мл, содержащем 1 мг/мл микросомального белка в реакционном буфере [0,2 М раствор фосфатного буфера (рН 7,4), 1,3 мМ НАДФ+, 3,3 мМ глюкоза-6-фосфата и 0,4 ед./мл глюкоза-6-фосфат дегидрогеназы] при 37 °C на водяной бане-шейкере. Систему регенерации НАДФH (раствор А и В) приобрели у компании BD Biosciences (Бедфорд, штат Массачусетс). Для исследований глюкуронирования, 2 мМ кофактора УДФ-глюкуроновой кислоты (Sigma, Сент-Луис, штат Миссури) в деионизированной воде инкубировали с 8 мМ MgCl2, 25 мкг аламетицина (Sigma, Сент-Луис, штат Миссури) в деионизированной воде и регенерирующих растворах НАДФН (BD Biosciences, Бедфорд, штат Массачусетс), как описано ранее. Общая концентрация ДМСО в реакционном растворе составила около 0,5% (об./об.). Аликвоты (100 мкл) реакционных смесей, используемые для определения метаболической устойчивости, отбирали через 5, 10, 20, 30, 60 и 90 минут. Добавили ацетонитрил (150 мкл), содержащий 200 нМ внутреннего стандарта, чтобы погасить реакцию и осадить белки. Затем образцы центрифугировали при 4000g в течение 30 минут при комнатной температуре, а надосадочную жидкость непосредственно анализировали по ЖХ-МС/МС.
Аналитический способ. Раствор образца (10 мкл) ввели инъекцией в систему ВЭЖХ серии Agilent (Agilent 1100 Series Agilent 1100 Chemstation, Agilent Technology Co, Ltd). Все аналиты разделили на колонке С18 с узким отверстием (Alltech Alltima HP, 2,1Ч100 мм, 3 мкм, Fisher, Фейр Лоан, штат Нью-Джерси). Использовали два градиентных режима. Для исследований метаболической устойчивости, использовали градиентный режим для достижения разделения аналитов с использованием смесей подвижной фазы А [ACN/H2O (5%/95%, об./об.), содержащей 0,1% муравьиной кислоты] и подвижной фазы B [ACN/H2O (95%/5%, об./об.), содержащей 0,1% муравьиной кислоты] при скорости потока 300 мкл/мин. Подвижную фазу А использовали при концентрации 10% от 0 до 1 минуты с последующим линейно запрограммированным градиентом до 100% подвижной фазы В за 4 минут, 100% подфижной фазы В выдерживали в течение 0,5 минут перед быстрым сбросом до 10% подвижной фазы А. Подвижную фазу А выдерживали еще 10 минут перед окончанием анализа.
Использовали трехквадрупольный масс-спектрометр, API Qtrap 4000TM (Applied Biosystems/MDS SCIEX, Конкорд, Онтарио, Канада), работающий с источником TurboIonSpray. Напряжение иглы-распылителя установили на 5 кВ для положительного режима. «Газовую завесу» установили на 10; газ 1 и газ 2 установили на 50. Активированную столкновениями диссоциацию (CAD) газа на середину, а температуру источника нагревания пробы на 500°C. Для получения наиболее чувствительных сигналов использовали режим многократного мониторинга реакции (MRM), сканирование при m/z 378 → 210 (17ya), m/z 373 → 205 (12fa), m/z 410 → 242 (55) и m/z 309 → 171 (внутренний стандарт). Получение данных и количественную обработку выполнили при помощи программы AnalystTM версии 1.4.1 (Applied Biosystems).
Растворимость в воде. Растворимость лекарств определили по фильтровальной пластине растворимости Multiscreen (Millipore Corporate, Биллерика, штат Массачусетс), подключенной к ЖХ-МС/МС. Вкратце, 198 мкл фосфатно-солевого буферного раствора (PBS) (pH 7,4) поместили в 96-луночный планшет, дозировали по 2 мкл 10 мМ раствора исследуемых соединений (в ДМСО) и перемешивали при осторожном встряхивании (200-300 об./мин.) в течение 1,5 часов при комнатной температуре (N = 3). Планшет центрифугировали при 800g в течение 10 минут, а фильтрат использовали для определения его концентрации и растворимости исследуемого соединения по ЖХ-МС/МС, как описано ранее.
Фармакокинетическое исследование. Самцов мышей ICR (n = 3 на группу) возрастом от 6 до 8 недель приобрели у компании Harlan Inc. и использовали для изучения фармакокинетики (ФК) 17ya, 12fa и 55. Все соединения (10 мг/кг) растворили в ДМСО/ПЭГ300 (1/9) и ввели однократной внутривенной (i.v.) инъекцией (50 мкл) в хвостовую вену. Образцы крови собрали через 5, 15 и 30 минут, 1, 1,5, 2, 3, 4, 8, 12 и 24 часа после внутривенного введения. Мышам перорально (p.o.), через ротовой зонд ввели по 20 мг/кг (в Tween80/ДМСО/H2O, 2/2/6) каждого исследуемого соединения для оценки их пероральной биодоступности. Образцы крови собрали через 0,5, 1, 1,5, 2, 3, 4, 8, 12 и 24 часа после перорального введения.
Самок крыс Sprague-Dawley (n = 3; 254 ± 4 г) приобрели у компании Harlan Inc. (Индианаполис, штат Индиана). Торакальные катетеры яремной вены для крыс приобрели у компании Braintree Scientific Inc. (Брэйнтри, штат Массачусетс). По прибытию в виварий, животные прошли акклиматизацию в течение 3 дней в помещении с контролируемой температурой (20–22 °C) с циклом освещения/темноты 12 часов, перед началом какой-либо обработки. Соединения 17ya, 12fa и 55 вводили внутривенно в торакальную яремную вену в дозировке 5 мг/кг (в ДМСО/ПЭГ300, 1/9). Инъекцией ввели равный объем гепаринизированного солевого раствора для замещения крови и взяли образцы крови (250 мкл) через катетер яремной вены через 10, 20, 30 минут и 1, 2, 4, 8, 12, 24 часа. Крысам перорально (p.o.), через ротовой зонд вводили по 10 мг/кг (в Tween80/ДМСО/H2O, 2/2/6) каждого исследуемого соединения для оценки их пероральной биодоступности. Все образцы крови (250 мкл) после перорального введения собрали через катетер яремной вены через 30, 60, 90 минут, 120 минут, 150 минут, 180 минут, 210 минут, 240 минут и 8, 12, 24 часа. Гепаринизированные шприцы и пробирки приготовили до сбора крови. Образцы плазмы приготовили центрифугированием образцов крови при 8000g в течение 5 минут. Все образцы плазмы сразу хранили при
-80°C до анализов.
Аналиты экстрагировали из 100 мкл плазмы с 200 мкл ацетонитрила, содержащего 200 нМ внутреннего стандарта. Образцы тщательно перемешали, центрифугировали, а органический экстракт перенесли в автоматический пробоотборник для анализа ЖХ-МС/МС.
Исследования ксенотрансплантата PC-3_TxR. Клетки PC-3_TxR (10×107 на мл) приготовили в питательной среде RPMI1640, содержащей 10% FBS, и смешали с Matrigel (BD Biosciences, Сан-Хосе, штат Калифорния) в соотношении 1:1. Опухоли инициировали подкожной (s.c.) инъекцией 100 мкл смеси (5×106 клеток на одно животное) в бок 6-8-недельных самцов бестимусных «голых» мышей. Измерили длину и ширину опухолей и рассчитали объем опухоли (мм3) по формуле р/6 × L × W2, где длина (L) и ширина (W) были определены в мм. Когда объемы опухолей достигли 300 мм3, животных с опухолями PC-3_TxR обработали носителем [Tween80/ДМСО/H2O (2/2/6)] или 17ya (10 мг/кг) перорально. Схема применения включала 3 раза в неделю в течение четырех недель.
Результаты
17a и 55 демонстрируют широкую цитотоксичность в клетках, включая клетки с множественной лекарственной устойчивостью. Способность 17ya и 55 ингибировать рост клеточных линий рака была оценена с использованием анализа SRB (Таблица 15). Оба соединения ингибируют рост нескольких клеточных линий рака человека, включая пять клеточных линий рака простаты и одну клеточную линию глиомы, со значениями IC50 в низком наномолярном диапазоне. 17ya в этих клеточных линиях демонстрирует в 1,7-4,3 раза более высокую активность, чем 55. Паклитаксел-резистентную клеточную линию PC-3 (PC-3/TxR), которая сверхэкспрессирует Р-гликопротеин (P-gp) использовали для изучения эффекта лекарственной устойчивости на 17ya и 55 и для сравнения с исходной клеточной линией, PC-3. Значения IC50 доцетаксела составили 1,2 ± 0,1 нМ и 17,7 ± 0,7 нМ в клетках PC-3 и PC-3/TxR, соответственно. 17ya и 55 были равносильными против исходной PC-3 и PC-3/TxR, тогда как паклитаксел и доцетаксел показали относительную устойчивость в 85- и 15 раз, соответственно. Эти данные показывают, что 17ya и 55 преодолевают P-gp-опосредованную лекарственную устойчивость.
Таблица 15. Данные цитотоксичности 17ya и 55.
| Клеточная линия | Тип | Цитотоксичность [значения IC50 ± стандартное отклонение (SD), нМ] | ||
| 17ya | 55 | Паклитаксел | ||
 |
||||
| PC-3 | Простаты | 5,2 ± 0,2 | 16 ± 1,5 | 0,6 ± 0,05 |
| PC-3/TxR | Простаты | 2,1 ± 0,1 (0,4) | 6,7 ± 0,5 (0,4) | 51 ± 2,3 (85) |
| LNCaP | Простаты | 12 ± 0,1 | 27 ± 0,6 | 1,7 ± 0,2 |
| Du-145 | Простаты | 17 ± 0,2 | 38 ± 0,6 | 5,1 ± 0,1 |
| PPC-1 | Простаты | 21 ± 0,1 | 36 ± 0,4 | 2,3 ± 0,8 |
| U87MG | Глиома | 10 ± 1,6 | 22 ± 3,0 | NR |
Значения IC50 (среднее ± стандартное отклонение) определили через 96 часов после обработки (N = 3). В качестве положительного контроля использовали паклитаксел. Значения в скобках показывают фактор устойчивости при сравнении значений IC50 в PC-3 и PC-3/TxR. NR, не сообщается.
17ya и 55 связываются с колхицин-связывающим сайтом тубулина, ингибируют полимеризацию тубулина и вызывают апоптоз клеток (Фигура 26). Для изучения взаимодействия низкомолекулярных ингибиторов с тубулином разработали масс-анализ конкурентного связывания. В этом исследовании для конкурирования с колхицин-тубулиновым связыванием использовали различные концентрации 17ya или 55. Оба соединения эффективно конкурировали с колхицином за связывание тубулина (Фигура 26А); однако кривые их конкурентного связывания существенно отклонялись от нуля при более высоких концентрациях, по сравнению с подофиллтоксином, известным мощным лигандом, связывающим колхициновый сайт. Это позволяет предположить, что оба 17ya и 55 обладают меньшим сродством, чем подофиллтоксин, или что они частично связываются с колхицин-связывающим сайтом. Винбластин, отрицательный контроль, не ингибирует колхицин-тубулиновое связывание, успешно демонстрируя специфичность этого масс-анализа конкурентного связывания.
Свиной тубулин мозга (чистота >97%) инкубировали с 17ya или 55 (5 мкМ) для испытания их действия на полимеризацию тубулина (Фигура 26В). 17ya и 55 ингибируют полимеризацию тубулина на 47% и 40% за 15 минут, соответственно. Колхицин при 5 мкМ использовался в качестве положительного контроля и ингибировал полимеризацию тубулина на 32%. Эти данные позволяют предположить, что оба 17ya и 55 обладают немного более сильным ингибированием полимеризации тубулина, чем колхицин. Следовательно, молекулярным механизмом этих соединений является связывание с колхицин-связывающим сайтом, ингибирование полимеризации тубулина и индуцирование цитотоксичности.
Клетки PC-3 и PC-3/TxR обрабатывали 0,8-600 нмоль/л 17ya, 55 или доцетаксела в течение 24 часов. Уровни комплексов ДНК-гистона использовали для отображения клеточного апоптоза. 17ya и 55 обладают равным действием для индукции клеточного апоптоза в PC-3 (Фигура 26С) и PC-3/TxR (Фигура 26D) через 24 часа. Хотя доцетаксел является более сильным для индуцирования апоптоза клеток PC-3, он оказался слабее в клетках PC-3/TxR из-за сверхэкспрессии P-gp.
17ya и 55 показали благоприятные подобные лекарствам свойства. Были изучены подобные лекарствам свойства, такие как метаболическая устойчивость, проницаемость, растворимость в воде и взамодействия лекарств между собой для 17ya и 55 (Таблица 16А). 17ya показало более высокую метаболическую устойчивость и растворимость в воде, чем 55. Оба химических вещества продемонстрировали более чем достаточные значения проницаемости, позволяя предположить, что их можно применять перорально. Кроме того, оба 17ya и 55 показали значения IC50 в микромолярном диапазоне в анализах ферментного ингибирования на CYP, указывая, что оба соединения способны избежать взаимодействий лекарств между собой за счет основных печеночных ферментов CYP. В целом, оба соединения продемонстрировали благоприятные подобные лекарствам свойства.
Таблица 16А. Подобные лекарствам свойства соединения 17а и 55. Были оценены метаболическая устойчивость, проницаемость, растворимость и потенциальные взаимодействия лекарств между собой. Каждое значение представляет среднее из двукратных экспериментов
| Измерение | Единицы | 17ya | 55 | Положительные контроли (среднее) |
| Метаболическая устойчивость | ||||
| Период полувыведения в печеночных микросомах человека | минуты | >60 | 28 | Верапамил (12) |
| Проницаемость | ||||
| Papp(A→B) в анализе CaCO-2 | 10-6 см/с | 36 | 99 | Пропранолол (19) |
| Растворимость в воде | мкг/мл | >75 | 19 | 1h (1,1) |
| Взаимодействие лекарств между собой | ||||
| Значение IC50 в Cyp3A4 (субстрат: тестостерон) | мкМ | 20 | 5,5 | Кетоконазол (0,02) |
| Значение IC50 в Cyp2D6 (субстрат: декстрометорфан) | мкМ | >50 | 34 | Хининдин (0,1) |
| Значение IC50 в Cyp2С19 (субстрат: (S)-мефенитоин) | мкМ | 6,6 | 5,3 | Тиклопидин (0,37) |
| Значение IC50 в Cyp2С9 (субстрат: диклофенак) | мкМ | 17 | 4,9 | Сульфафеназол (0,5) |
| Значение IC50 в Cyp1А2 (субстрат: фенацетин) | мкМ | 9,2 | 8,1 | Фурафиллин (2,2) |
Таблица 16В. Сводка подобных лекарствам и фармакокинетических свойств 17ya, 12fa, 55 и 1h.

Как показано в Таблице 16В, 17ya имеет период полувыведения 80 минут по реакции I фазы, позволяя предположить, что 17ya является устойчивым на I фазе метаболических процессов. Период полувыведения (90 минут) в присутствии УДФ-глюкуроновой кислоты является таким же, как наблюдался без нее. Эти данные позволяют предположить, что 17ya является устойчивым в печеночных микросомах человека и дают надежду, что у людей может быть достигнут низкий клиренс и продолжительный период полувыведения. С другой стороны, 55 продемонстрировало 30 и 43 минуты периода полувыведения, в присутствии и в отсутствие УДФ-глюкуроновой кислоты, соответственно. Соединение 12fa показало период полувыведения 44 на фазе I. Эти данные позволяют предположить, что все три соединения показали приемлемую устойчивость в печеночных микросомах человека, и 17ya является более устойчивым, чем 12fa и 55. При исследовании их метаболизма было обнаружено, что 12fa и 55 демонстрируют более высокие уровни восстановления кетона (данные не показаны), позволяя предположить, что 12fa и 55 являются более лабильными, чем 17ya.
Соединение 17ya демонстрирует превосходную растворимость в воде, 12fa и 55 показали приемлемую растворимость.
Соединение 17ya содержит имидазоловое кольцо, и это кольцо улучшает растворимость в воде, приводя к растворимости в воде >75 мкг/мл (Таблица 16А). Соединения 12fa и 55 демонстрируют более низкую растворимость в воде, и имеют растворимость 12 и 19 мкг/мл, соответственно. В целом, 17ya демонстрирует превосходную растворимость в воде, а 12fa и 55 показали приемлемую растворимость в воде, которая гораздо лучше, чем у 1h. Более высокая растворимость 12fa переносится на гораздо более высокую пероральную биодоступность по сравнению с 1h (35% против 3,3% у крыс). Точно так же, для 17ya и 55 растворимость в воде коррелирует с гораздо более высокой пероральной биодоступностью, как рассмотрено infra (Таблица 17).
Фармакокинетические исследования 17ya и 55 на мышах, крысах и собаках. Фармакокинетические параметры 17ya и 55, введенных однократной (i.v. или p.o.) дозой мышам ICR, крысам Sprague-Dawley и собакам породы бигль, сведены в Таблице 17. 17ya демонстрирует низкий клиренс у мышей и крыс, позволяя предположить, что 17ya обладает метаболической устойчивостью и минимальным пресистемным метаболизмом у этих видов. Кроме того, 17ya имеет умеренный объем распределения у мышей и крыс, указывая, что оно может правильно распределяться в тканях, включая опухоли. В отличие от мышей и крыс, как ни странно, общий клиренс 17ya у собак был высоким. Два распространенных метаболита в плазме собак, гидроксилированный метаболит и неизвестный метаболит со значением m/z +34 от исходного соединения (данные не показаны), согласовались метаболитами, обнаруженными в печеночных микросомах собак. В итоге для 17ya был получен более высокий клиренс и более низкое пероральное воздействие по сравнению с 55 у собак, но не у мышей и крыс. Кроме того, 17ya продемонстрировало распространенные метаболиты только в печеночных микросомах собак, но не в печеночных микросомах мышей, крыс или людей (данные не показаны). 17ya показало приемлемые значения пероральной биодоступности: 21%, 36% и 50% у крыс, мышей и собак, соответственно. В то же время 55 имеет низкий клиренс у крыс и умеренный клиренс у мышей и собак. Как и 17ya, 55 демонстрирует умеренный объем распределения у этих видов. 55 имеет постоянные значения пероральной биодоступности среди трех видов (24%-36%). Эти свойства показывают, что оба 17ya и 55 являются потенциальными перорально доступными ингибиторами тубулина.
Таблица 17. Фармакокинетические исследования соединений 17ya и 55 на мышах, крысах и собаках.
| 17ya | 55 | |||
| IV | PO | IV | PO | |
| ФК мышей (N=3) | ||||
| Доза, мг/кг | 10 | 20 | 10 | 20 |
| Клиренс, мл/мин./кг | 19 | NR | 40 | NR |
| Vss, л/кг | 2,9 | NR | 1,3 | NR |
| t1/2, мин. | 101 | 339 | 46 | 126 |
| AUC, мин.*мкг/мл | 540 | 384 | 249 | 171 |
| Сmax, нг/мл | 4800 | 1560 | 7739 | 1253 |
| F, % | 36% | 34% | ||
| ФК крыс (N=3) | ||||
| Доза, мг/кг | 5 | 10 | 5 | 10 |
| Клиренс, мл/мин./кг | 9,5 ± 2,3 | NR | 10 ± 1,4 | NR |
| Vss, л/кг | 1,8 ± 0,2 | NR | 1,0 ± 0,1 | NR |
| t1/2, мин. | 139 ± 24 | 206 ± 12 | 73 ± 5,0 | 350 ± 214 |
| AUC, мин.*мкг/мл | 553 ± 143 | 233 ± 134 | 509 ± 73 | 246 ± 163 |
| Сmax, нг/мл | 3672 ± 519 | 999 ± 445 | 4609 ± 55 | 757 ± 520 |
| F, % | 21% | 24% | ||
| ФК собак (N=4) | ||||
| Доза, мг/кг | 2 | 5 | 2 | 5 |
| Клиренс, мл/мин./кг | 109 ± 29 | NR | 15 ± 3,2 | NR |
| Vss, л/кг | 94 ± 95 | NR | 0,9 ± 0,2 | NR |
| t1/2, мин. | 2757 ± 1573 | 1695 ± 439 | 82 ± 15 | 191 ± 9,0 |
| AUC, мин.*мкг/мл | 18,5 ± 4,7 | 23,1 ± 11,3 | 141 ± 30 | 128 ± 154 |
| Сmax, нг/мл | 400 ± 118 | 210 ± 133 | 2552 ± 576 | 862 ± 1010 |
| F, % | 50% | 36% | ||
17ya и 55 ингибируют рост ксенотрансплантатов паклитаксел-резистентного рака простаты (PC-3/TxR). Клетки PC-3 (Фигура 27А) и клетки паклитаксел-резистентного рака простаты (PC-3/TxR) (Фигура 27В) инокулировали «голым» мышам и оставили опухоли достигать объемов около 150-300 мм3. Доцетаксел (10 или 20 мг/кг), который клинически используется для лечения рака простаты, использовался для оценки его эффективности в моделях P-gp-опосредованной лекарственной устойчивости in vivo. Было обнаружено, что опухоль PC-3/TxR является быстрорастущей, и ее объем в конце исследования достиг 1500-2500 мм3. Хотя 10 и 20 мг/кг при внутривенном введении доцетаксела продемонстрировали доза-зависимый ответ в обеих моделях (Фигуры 27А и 27В), эффект ингибирования роста опухоли (TGI) снизился с 84% TGI в опухолях PC-3 до 14% TGI в опухолях PC-3/TxR при внутривенном введении при дозе 10 мг/кг (Таблица 18). Кроме того, более высокая доза (20 мг/кг) доцетаксела вызвала частичную регрессию (>100% TGI) опухолей PC-3, но лишь 56% TGI в опухолях PC-3/TxR. Эффективность доцетаксела в опухолях PC-3/TxR резко снизилась по сравнению с эффективностью в опухолях PC-3, позволяя предположить, что эффективность ухудшилась за счет P-gp-опосредованной лекарственной устойчивости, и эти результаты хорошо согласуются с данными цитотоксичности или апоптоза, полученными авторами изобретения in vitro. В отличие от недостатка эффективности доцетаксела в опухолях PC-3/TxR, перорально введенное 17ya (6,7 мг/кг) продемонстрировало более 100% TGI без влияния на вес тела (Фигура 27В и Таблица 18). Кроме того, 2 из 4 «голых» мышей с опухолями PC-3/TxR не имели опухолей на 19 день (данные не показаны).
Модель ксенотрансплантата PC-3/TxR дополнительно использовали для оценки эффективности 17ya (при других схемах применения) и 55. Максимально переносимая доза (снижение веса тела >20%) для 17ya составила 10 мг/кг при пероральном дозировании один раз в день в течение четырех дней; или 3,3 мг/кг дважды в день (b.i.d.) в течение пяти дней (данные не показаны). Как показано на Фигуре 27С, 3,3 мг/кг 17ya вводили дважды в день в течение первых четырех последовательных дней в течение первой недели, а затем схему изменили на один раз в день между 2 и 4 неделями. Результаты показали, что частичная регрессия была достигнута в течение 4-19 дней, и TGI составил 97%, а одна из семи мышей оказалась без опухоли на 26 день. Более высокая доза (10 мг/кг) при более низкой частоте дозирования (q2d) 17ya (Фигура 27D) вызвала частичную регрессию между 13 и 29 днем. Эти данные позволяют предположить, что режимы с оптимизированными дозами и схемами применения способствуют успешному ингибированию 17ya опухолей PC-3/TxR. 55 вводили «голым» мышам перорально при дозировке 10 или 30 мг/кг дважды в день и пять раз в неделю между 1 и 4 неделями. Как показано на Фигуре 27С, профили ингибирования демонстрируют зависимость от дозы в опухолях PC-3/TxR. Значение TGI составило 59% для группы лечения более низкой дозой (10 мг/кг). Более того, более высокая доза (30 мг/кг) начала показывать частичную регрессию (TGI >100%) с 19 дня и до окончания исследования (26 день). Некоторые мыши в группе с носителем в конечной точке потеряли вес, частично из-за раковой кахексии. Напротив, мыши, обработанные 17ya (3,3 мг/кг) или 55 (30 мг/кг) прибавили в весе (Таблица 18), позволяя предположить, что эти оптимизированные дозы 17ya или 55 могут быть хорошо переносимыми и являются профилактическими для раковой кахексии.
Таблица 18. Противораковая активность соединений 17ya и 55 по сравнению с параллельно оцененным доцетакселем in vivo.
| Схема применения | Конечная точка | Количество в конце/начале | Вес тела (г) | Размер опухоли (мм3) | TGI (%) | |||
| В начале | В конце | В начале | В конце | |||||
| Ксенотрансплантат PC-3 | ||||||||
| Носитель_IV | 1 и 9 день | 19 день | 6/6 | 30 ± 2 | 32 ± 4 | 271 ± 83 | 875 ± 292 | -- |
| Доцетаксел_IV_10 мг/кг | 1 и 9 день | 19 день | 5/5 | 29 ± 2 | 24 ± 2 | 247 ± 49 | 341 ± 101 | 84 |
| Доцетаксел_IV_20 мг/кг | 1 и 9 день | 19 день | 5/5 | 28 ± 3 | 24 ± 3 | 243 ± 68 | 172 ± 62 | >100 |
| Ксенотрансплантат PC-3/TxR | ||||||||
| Носитель_IV | 1 и 9 день | 19 день | 5/5 | 33 ± 1 | 26 ± 5 | 171 ± 57 | 2061 ± 858 | -- |
| Доцетаксел_IV_10 мг/кг | 1 и 9 день | 19 день | 4/4 | 31 ± 2 | 25 ± 2 | 143 ± 20 | 1774 ± 183 | 14 |
| Доцетаксел_IV_20 мг/кг | 1 и 9 день | 19 день | 4/4 | 30 ± 1 | 25 ± 4 | 170 ± 86 | 999 ± 905 | 56 |
| 17ya_PO_6,7 мг/кг | qd x 5/w | 19 день | 4/4 | 33 ± 3 | 34 ± 3 | 172 ± 69 | 126 ± 100 | >100 |
| Носитель_PO | b.i.d. x 5/w | 26 день | 6/7 | 30 ± 2 | 25 ± 2 | 156 ± 30 | 2591 ± 1423 | -- |
| 55_PO_10 мг/кг | b.i.d. x 5/w | 26 день | 7/7 | 29 ± 2 | 26 ± 3 | 143 ± 44 | 1152 ± 433 | 59 |
| 55_PO_30 мг/кг | b.i.d. x 5/w | 26 день | 7/7 | 29 ± 3 | 30 ± 2 | 134 ± 34 | 101 ± 19 | >100 |
| 17ya_PO_3,3 мг/кгa | qd x 5/w | 26 день | 7/7 | 29 ± 2 | 30 ± 2 | 139 ± 44 | 214 ± 172 | 97 |
| Носитель_PO | q2d x 3/w | 29 день | 5/5 | 24 ± 2 | 21 ± 1 | 299 ± 40 | 1521 ± 580 | -- |
| 17ya_PO_10 мг/кг | q2d x 3/w | 29 день | 5/5 | 24 ± 2 | 28 ± 2 | 294 ± 156 | 237 ± 103 | >100 |
Схема применения: qd × 5/w = одно введение в течение пяти последовательных дней в неделю; b.i.d. × 5/w = два введения в течение пяти последовательных дней в неделю; или q2d × 3/w = введение через день или три раза в неделю.
а Схема применения составила два введения в течение четырех последовательных дней в течение первой недели, и схема применения была изменена (из-за токсичности) на одно введение в течение пяти последовательных дней в неделю со второй до четвертой недели.
Проникновение 17ya и 55 в мозг «голых» мышей. Было установлено, что концентрации во всем мозге «голых» мышей через 1 час и 4 часа после перорального введения составляют 20 мг/кг 17ya и 55 (Таблица 19). Определили отношения концентраций в мозге и плазме и сравнили с доцетакселем у «голых» мышей. 55 демонстрирует более высокую проницаемость в мозг, чем 17ya и доцетаксел. 17ya демонстрирует лишь незначительно более высокие отношения концентраций в мозге/плазме, чем доцетаксел через 1 час и через 4 часа. Концентрации 55 в мозге достигли 14-19% от концентраций в плазме через 1 час и 4 часа, соответственно, демонстрируя в 3,2 раза более высокое отношение в мозге/плазме через 1 час и 4 часа по сравнению с доцетакселем. Эти данные позволяют предположить, что 55 обладает потенциально благоприятными свойствами для лечения глиомы, поскольку оно обладает более высокой проницаемостью в мозг и большой эффективностью (22 нМ, Таблица 15) в клетках глиомы.
Таблица 19. Исследования гемато-энцефалического барьера (BBB) соединений 17ya и 55. Концентрации в мозге и плазме были определены у «голых» мышей через 1 час и 4 часа после введения доцетаксела (IP, 10 мг/кг), 17ya (PO, 20 мг/кг) и 55 (PO, 20 мг/кг). Каждое значение представляет среднее ± стандартное отклонение для 3 «голых» мышей
| Доцетаксел | 17ya | 55 | ||||
| Измерение | 1 час | 4 часа | 1 час | 4 часа | 1 час | 4 часа |
| Мозг (нг/мл) | 33 ± 14 | 20 ± 9 | 124 ± 108 | 49 ± 32 | 180 ± 44 | 73 ± 18 |
| Плазма (нг/мл) | 768 ± 92 | 345 ± 94 | 2058 ± 1252 | 570 ± 438 | 1669 ± 867 | 380 ± 32 |
| Мозг/плазма (%) | 4,4 ± 2,0 | 6,0 ± 2,9 | 5,4 ± 1,9 | 8,9 ± 1,7 | 14 ± 7,9 | 19 ± 3,1 |
ПРИМЕР 21
ФАРМАКОКИНЕТИКА СОЕДИНЕНИЙ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
Таблица 20
| ID соединения | Период полувыведения в печеночной микросоме человека (мин.) | Период полувыведения в печеночной микросоме мышей (мин.) | Период полувыведения в печеночной микросоме крыс (мин.) | Период полувыведения в печеночной микросоме собак (мин.) | Период полувыведения в печеночной микросоме обезьян (мин.) |
| 1h | 17 | <5 | 31 | 19 | <5 |
| 2e-цис | 35 | ||||
| 2i | 32 | ||||
| 2k | 10 | 9 | 32 | 16 | <5 |
| 2l | 20 | 11 | 49 | 30 | 8 |
| 6a | 32 | 3,43 | 12 | 13 | 16 |
| 6b | 40 | 10 | 9 | 30 | 13 |
| 6c | 47 | 13 | 14 | 29 | 9 |
| 7d | 24 | 37 | 42 | 29 | 15 |
| 12da | 23 | 8 | 28 | 17 | |
| 12fa | 56 | 23 | 46 | 26 | |
| 12fb | 37 | ||||
| 12dab | 21 | <5 | 12 | 46 |
ПРИМЕР 22
БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ 4-ЗАМЕЩЕННЫХ МЕТОКСИБЕНЗОИЛ-АРИЛ-ТИАЗОЛЬНЫХ (SMART) СОЕДИНЕНИЙ 1h, 2k и 2l: АКТИВНЫЕ ИНГИБИТОРЫ МИКРОТРУБОЧЕК
Материалы и способы
Анализ полимеризации микротрубочек in vitro. Бычий тубулин мозга (0,4 мг) (Cytoskeleton, Денвер, штат Колорадо) смешали с 10 мкМ исследуемого соединения или носителем (ДМСО) и инкубировали в 100 мкл буфера (80 мМ PIPES, 2,0 мМ MgCl2, 0,5 мМ EGTA, рН 6,9 и 1 мМ ГТФ). Поглощение при длине волны 340 нм контролировали каждую минуту в течение 15 минут (микропланшет-ридер SYNERGY 4, Bio-Tek Instruments, Винооски, штат Вермонт). Спектрофотометр поддерживали при 37 °C для полимеризации тубулина.
МС анализ конкурентного связывания. Колхицин, винбластин и паклитаксел (1,2 мкМ для каждого) инкубировали с тубулином (1,2 мг/мл) в инкубационном буфере (80 мМ PIPES, 2,0 мМ MgCl2, 0,5 мМ EGTA, рН 6,9) при 37 °C в течение 1 часа. 1h (0,5-125 мкМ) исследовали на индивидуальное конкурирование с колхицин-, винбластин- и паклитаксел-тубулиновым связыванием. Лиганды в свободной форме отделили от тубулина или микротрубочек с использованием метода ультрафильтрации (микроконцентратор) (Microcon, Бедфорд, штат Массачусетс) с размером молекулярного отсечения 30 кДа. Колхицин, винбластин и паклитаксел определили по методу ЖХМС/МС. Способность 1h ингибировать связывание лигандов выразили как процент от контрольного связывания без какого-либо конкурента. Каждую реакцию выполнили в трех экземплярах.
Анализ клеточной культуры и цитотоксичности рака простаты и меланомы. Все клеточные линии рака простаты и меланомы были закуплены у компании ATCC (American Type Culture Collection, Манассас, штат Вирджиния, США), а клеточные культуры были приобретены у компании Cellgro Mediatech (Херндон, штат Вирджиния, США). Антипролиферативную активность соединений исследовали в четырех клеточных линиях рака простаты человека LNCaP, DU 145, PC-3 и PPC-1) и двух клеточных линиях меланомы человека (A375 и WM-164). Клеточную линию яичников человека OVCAR-8 и ее резистентную клеточную линию, которая сверхэкспрессирует P-gp, NCI/ADR-RES, использовали в качестве моделей МЛУ. Обе клеточные линии яичников приобрели у компании National Cancer Institutes (NCI). Все клеточные линии рака простаты выращивали с 10% бычьей плодной сыворотки (FBS).
Анализ клеточного цикла. Для изучения эффекта соединений на распределение клеточного цикла выполнили проточную цитометрию. Клетки PC-3 и A375 обрабатывали в питательной среде указанными концентрациями соединений 1h, 2k, 2l в течение 24 часов. Клеточную ДНК окрасили 100 мкл/мл йодида пропидия и 100 мкл/мл РНКазы А в PBS, и выполнили проточную цитометрию для определения распределения клеточного цикла клеток.
Обнаружение апоптоза по иммуноферментному твердофазному анализу. Количественную оценку прироста моно- и олигонуклеосом в цитоплазме использовали для определения способности соединений вызывать апоптоз (обнаружение гибели клеток ELISA PLUS, Roche, Германия), следуя инструкциям производителя.
Фармакокинетическое исследование. Самцов мышей ICR (n = 3 или 4 на группу) возрастом от 6 до 8 недель приобрели у компании Harlan Inc. и использовали для изучения фармакокинетики (ФК) соединений. 1h, 2k, 2l (15 мг/кг) растворили в ПЭГ300/ДМСО (1/4) и ввели однократной i.v. инъекцией в хвостовую вену. Образцы крови собрали через 2, 5, 15 и 30 минут, 1, 2, 4, 8, 16 и 24 часа после введения. Самцов крыс Sprague-Dawley (n = 4; 254 ± 4 г) приобрели у компании Harlan Inc. (Индианаполис, штат Индиана). 1h, 2k вводили внутривенно в катетеры яремной вены при 2,5 мг/кг (в ДМСО/ПЭГ300, 1/4). Образцы крови (250 мкл) собрали через 10, 20, 30 минут и 1, 2, 4, 8, 12, 24, 48 часов. Для подготовки образцов использовали метод осаждения белков. Аликвоту (200 мкл) ацетонитрила (ACN) добавили к 100 мкл плазмы, а затем тщательно перемешивали на вортексе в течение 15 секунд. После центрифугирования надосадочную жидкость анализировали по объединенной жидкостной хромато-масс-спектрометрии (ЖХ-МС/МС). Фармакокинетические параметры определили при помощи анализа без использования камерной модели (WinNonlin, Pharsight Corporation, Маунтин-Вью, штат Калифорния).
Исследования ксенотрансплантата опухоли PC-3 и A375. Клетки PC-3 и А375 (5х107 на мл) приготовили в питательной среде без фенолового красного, содержащей 10% FBS, и смешали с Matrigel (BD Biosciences, Сан-Хосе, штат Калифорния) в соотношении 1:1. Опухоли инициировали подкожной (s.c.) инъекцией 100 мкл смеси (2,5х106 клеток на одно животное) в бок 6-8-недельных самцов бестимусных «голых» мышей. Измерили длину и ширину опухолей и рассчитали объем опухоли (мм3) по формуле р/6 Ч L Ч W2, где длина (L) и ширина (W) были определены в мм. Когда объемы опухолей достигли 150 мм3, животных с опухолями PC-3 обрабатывали носителем [Captex200/Tween80 (1/4)], 1h (5 и 15 мг/кг), 2k (5 и 15 мг/кг) и 2l (50 мг/кг) внутрибрюшинно в течение 21 дня. Винбластин (0,5 мг/кг) использовали в качестве положительного контроля и вводили q2d с носителем [ДМСО/ПЭГ300 (1/9)]. С другой стороны, мышей с опухолью А375 обрабатывали в течение 34 дней носителем [Captex200/Tween80 (1/4)], 1h (20 мг/кг) или 2k (15 мг/кг). Дозы выбрали на основании исследований острой токсичности 1h и 2k у мышей ICR) (n = 2/группу), которые показали, что дозы до 30 мг/кг и 15 мг/кг, соответственно, не вызывают более 10% снижения веса тела после 4 последовательных дней внутрибрюшинного дозирования.
Противоопухолевая активность in vivo [ингибирование роста опухоли (% Т/С], отсрочка роста опухоли (значение Т-С) и киллинг клеток опухоли (общий log гибели клеток)]. Данные о влиянии лекарства описаны следующими параметрами: % T/C = [Д объема опухоли в группе обработки] / [Д объема опухоли в контрольной группе] Ч 100%. Значения Т-С (отсрочка роста опухоли) основывались на среднем времени (в днях), необходимом для того, чтобы опхуоли в группе обработки (Т) и контрольной группе (С) достигли определенного размера (в данном исследовании 600 мм3). Эти значения затем использовали для количественной оценки киллинга клеток опухоли по следующему уравнению: log гибели клеток = (T-C) / (3,32 Ч Td). Td является временем удвоения объема опухоли в днях. В этом исследовании авторы изобретения определили время удвоения, необходимое для увеличения опухоли с 300 до 600 мм3.
Испытание методом вращающегося стержня. Мышей ICR обучали три раза в день в течение двух дней, чтобы они могли оставаться на вращающемся стержне >120 секунд при 12 об./мин. Затем мышей рандомизировали по продолжительности времени, в течение которого они могут оставаться на вращающемся стержне, и разделили на 7-8 мышей на группу. 1h в дозе 5 или 15 мг/кг в Captex200/Tween80 (1/4) вводили внутрибрюшинной инъекцией. Винбластин в дозе 0,5 мг/кг/день использовали в качестве положительного контроля в тех же условиях. Испытание методом вращающегося стержня выполняли дважды в неделю. Обработку прекратили на 31 день, а последующее наблюдение выполнили на 1, 2 и 4 неделю после прекращения обработки. Скорость стержня увеличили с 59 об./мин. до 40 об./мин. за период 5 минут. Показатель измерили как продолжительность времени, в течение которого мышь может удерживаться на вращающемся стержне.
Исследования усточивости лекарства in vivo. По окончанию исследований ксенотрансплантата PC-3, солидные опухоли из контрольной и обработанной 1h (15 мг/кг) групп удалили и расщепили 0,1% коллагеназой (тип I) и 50 мг/мл ДНКазой (Worthington Biochemical Corp., Фрихолд, штат Нью-Джерси). Диспергированные клетки поместили на планшет в среде RPMI + 10% FBS и инкубировали при 37єC и 5% CO2 в течение 24 часов для прикрепления. Сравнили антипролиферативное действие 1h для определения того, остаются ли клетки опухоли в ксенотрансплантатах PC-3, сохраняющих восприимчивость к лекарству. Клетки PC-3, закупленные у ATCC, использовали в качестве контроля in vitro. Статистические анализы выполнили с использованием простого t-критерия.
Результаты
На основании исследований структурно-функциональной взаимосвязи, для биологической характеристики выбрали три соединения (Фигура 28А). Тогда как 1h и 2k являются сильнодействующими молекулами со свойствами цитотоксичности в низком наномолярном диапазоне, 2l, которое было специально разработано в качестве потенциального метаболита с улучшенной растворимостью, имеет наименьшее антипролиферативное действие (Таблица 21).
Таблица 21. Эффективность соединений in vitro против клеточных линий рака простаты, меланомы и лекарственно-устойчивых клеточных линий (n = 3, среднее ± стандартная погрешность). Паклитаксел, винбластин и колхицин использовали в качестве положительных контролей, как описано ранее.
| IC50 ± стандартная погрешность средней величины (SEM) (нМ) | |||||||
| Клеточная линия | Тип клеток | SMART-H | SMART-F | SMART-OH | Паклитаксел | Винбластин | Колхицин |
| LNCaP | Простаты | 28 ± 4а | 6 ± 1а | 103 ± 9 | 1,7 ± 0,2 | 1,1 ± 0,1 | 16 ± 4 |
| PC-3 | Простаты | 21 ± 1а | 13 ± 1а | 87 ± 5 | 4,8 ± 0,3 | 2,1 ± 0,2 | 11 ± 1 |
| Du-145 | Простаты | 71 ± 4а | 12 ± 1а | 116 ± 14 | 5,1 ± 0,1 | 1,8 ± 1,1 | 10 ± 2 |
| PPC-1 | Простаты | 43 ± 5а | 8 ± 1а | 76 ± 2 | 2,3 ± 0,8 | 1,1 ± 0,4 | 20 ± 1 |
| B16-F1 | Меланома | 55 ± 5а | 43 ± 21а | 113 ± 6 | 17 ± 2 | 4,7 ± 0,7 | 29 ± 5 |
| А375 | Меланома | 28 ± 5а | 33 ± 14а | 93 ± 11 | 12 ± 3 | 1,1 ± 0,2 | 20 ± 3 |
| OVCAR-8 | Яичников | 35 ± 2 | 34 ± 3 | 110 ± 8 | 4,7 ± 0,1 | 3,9 ± 0,1 | 17 ± 1 |
| NCI/ADR-RES | Яичников | 13 ± 1 | 12 ± 1 | 45 ± 5 | 6263 ± 634 | 582 ± 57 | 1113 ± 79 |
| Фактор устойчивости | 0,4 | 0,4 | 0,4 | 1333 | 149 | 65 | |
SMART-H в Таблице 21 является 1h; SMART-F в Таблице 21 является 2k; и SMART-OH в Таблице 21 является 2l.
SMART ингибируют полимеризацию микротрубочек, связываясь с колхицин-
связывающим сайтом тубулина.
Бычий тубулин мозга (чистота > 97%) инкубировали с отдельными соединениями (10 мкМ) для испытания их действия на полимеризацию тубулина (Фигура 28В). Тогда как 1h и 2k ингибируют полимеризацию тубулина на 90%, 2l ингибирует полимеризацию лишь на 55%. Предыдущие исследования показали ингибирование полимеризации в зависимости от концентрации 1h. Кроме того, при тех же экспериментальных условиях IC50 для 1h (4,23 мкМ) является таким же, как для колхицина (4,91 мкМ). Эти данные позволяют предположить, что эти новые соединения демонстрируют сильное действие против полимеризации тубулина, что хорошо согластуется с их цитотоксичностью (Таблица 21). Способность этих соединений конкурировать за известные связывающие сайты тубулина была определена с использованием нового МС анализа конкурентного связывания, который был разработан в лаборатории авторов изобретения. Для этих исследований конкурентного связывания использовались три тубулиновых лиганда, соответствующих этим трем связывающим сайтам тубулина: колхицин, винбластин и паклитаксел. Было обнаружено, что в диапазоне концентраций 0,1-125 мкМ, 1h специфически конкурирует со связыванием колхицина с тубулином, но не конкурирует со связыванием винбластина и паклитаксела с тубулином (Фигура 28С).
SMART cоединения ингибируют рост клеточных линий рака с множественной лекарственной устойчивостью.
Способность соединений ингибировать рост клеточных линий рака была оценена с использованием анализа SRB. Как показано в Таблице 21, эти соединения ингибируют рост нескольких клеточных линий рака человека, включая четыре клеточные линии рака простаты и две клеточные линии меланомы, со значениями IC50 в низком наномолярном диапазоне. Из этих трех соединений 2l обладает наименьшей эффективностью (IC50 76~116 нМ). 2k демонстрирует наилучшее антипролиферативное действие со значениями IC50 6 и 43 в клеточных линиях рака простаты и меланомы. Кроме того, было оценена также эффективность этих соединений в клеточных линиях OVCAR-8 и NCI/ADR-RES (Таблица 21). Соединения являются равносильными против МЛУ клеток (NCI-ADR-RES) и исходной клеточной линии (OVCAR-8). Паклитаксел, винбластин и колхицин показали значения относительной устойчивости в 1333, 149 и 65 раз, соответственно (Таблица 21). Эти данные показывают, что эти соединения преодолевают P-gp-опосредованную лекарственную устойчивость.
SMART соединения останавливают клетки PC-3 (рака простаты) и А375 (меланомы) на фазе G2/M клеточного цикла и вызывают клеточный апоптоз. Клетки PC-3 и А375 обрабатывали 10, 50, 200 и 1000 нМ соединений в течение 24 часов. Обработка SMART соединениями привела к зависящему от концентрации накоплению обеих клеток PC-3 и А375 на фазе G2/M с сопутствующим снижением процентного содержания клеток на фазе G0/G1) (Фигуры 29А и 29В). Доля клеток на фазе G2/M существенно увеличилась при обработке от 50 до 200 нМ 1h, 2k, 2l. Затем исследовали апоптоз по измерению уровня цитоплазматических комплексов ДНК-гистона в клетках PC-3 и А375 через 24 часа обработки. Увеличение концентрации SMART соединений повысило уровень цитоплазматических комплексов ДНК-гистона в клетках PC-3 и А375 (Фигура 29С). Этот эффект был более выраженным в клетках А375, чем в клетках PC-3, однако апоптоз был очевидным в обоих типах клеток. 1h и 2k вызывают умеренный апоптоз при концентрации 50 нМ, тогда как 2l вызывает апоптоз только при концентрациях, больших или равных 200 нМ.
Фармакокинетический профиль SMART соединений in vivo. Однократную болюсную дозу каждого соединения (15 мг/кг) ввели инъекцией в хвостовую вены мышам ICR для описания их фармакокинетики (Фигура 30А). 1h и 2k демонстрируют одинаковые фармакокинетические свойства, а 2l демонстрирует немного большее значение AUC, чем 1h и 2k, указывая на более низкий клиренс для 2l (Таблица 22). 2l также имеет в 2-3 раза более высокое значение Vss, чем 1h и 2k. Значения клиренса для всех трех соединений были равными или более 90 мл/мин./кг, скорости печеночного кровотока у мышей, позволяя предположить, что помимо удаления из печени в выведении соединений могут участвовать другие пути разложения. Фармакокинетика 1h и 2k (2,5 мг/кг) также была изучена у крыс (Фигура 30В). Интересно, что низкие значения клиренса и скорости печеночной экскреции были получены для обоих соединений, позволяя предположить, что эти соединения демонстрируют видовые различия клиренса. У крыс 1h демонстрирует благоприятные фармакокинетические свойства, которыми являются низкий клиренс (6 мл/мин./кг), умеренный объем распределения (7,6 л/кг), продолжительный период полувыведения (24 часа) и высокое воздействие (AUC, 5,8 ч*мкг/мл) (Таблица 22) при внутривенном введении.
Таблица 22. Фармакокинетические параметры SMART соединений. SMART вводились внутривенно, 15 мг/кг и 2,5 мг/кг у мышей и крыс, соответственно
| Фармакокинетические параметры SMART соединений in vivo | |||||
| Виды | Параметр | Единица | SMART-H | SMART-F | SMART-OH |
| Мыши | AUC | ч*мкг/мл | 1,9 | 2,2 | 2,6 |
| t1/2 | мин. | 140 | 141 | 740 | |
| Vss | л/кг | 4,9 | 6,6 | 16,5 | |
| CL | мл/мин./кг | 130 | 112 | 90 | |
| Крысы | AUC | ч*мкг/мл | 5,8 | 1,6 | NA |
| t1/2 | мин. | 1431 | 2410 | NA | |
| Vss | л/кг | 7,6 | 34 | NA | |
| CL | мл/мин./кг | 6 | 11 | NA | |
NA, нет данных
SMART-H в Таблице 22 является 1h; SMART-F в Таблице 22 является 2k; и SMART-OH в Таблице 22 является 2l.
Соединения SMART ингибируют рост ксенотрансплантатов рака простаты и меланомы без нейротоксичности. Опухоли рака простаты PC-3 и меланомы А375 у мышей оставили достигать объема 150 мм3, а затем мышей с опухолью обработали SMART соединениями. Как показано на Фигуре 31А, объемы опухолей в контрольной группе увеличились до 680 мм3 за 21 день исследования. Объемы опухолей в группе, обработанной 1h, увеличились до 370 мм3 (обработка 5 мг/кг) и 176 мм3 (обработка 15 мг/кг) на 21 день, указывая на сильное противоопухолевое действие этого соединения. Опухоли у животных, обработанных 2k, увеличились до 269 мм3 (обработка 5 мг/кг) и 292 мм3 (обработка 15 мг/кг), тогда как животные в группе, обработанной 2l (50 мг/мг) имели опухоли 331 мм3 на 21 день. Это снижение объема опухоли аннулировалось при отмене SMART соединений (данные не показаны). В Таблице 23 сведены данные эффективности in vivo (% Т/С, значения Т-С и log гибели клеток) SMART соединений.
Таблица 23. In vivo влияние соединений SMART (введенных внутрибрюшинно) на рак простаты (PC-3) и меланому (А375). Подытожены %T/C, значения T-C и log гибели клеток. Время удвоения ксенотрансплантата меланомы составило 4,6 дня. В качестве положительного контроля использовали винбластин. % T/C ≤ 42% считается умеренно активным по критериям Национального института онкологии США. NA, нет данных
| Соединение | Доза (мг/кг) | Модель ксенотрансплантата | % T/C | Среднее время для достижения 600 мм3 | Т-С (дни) | Общий log гибели клеток |
| Носитель | NA | Простаты | 100 | 19 дней | NA | NA |
| Винбластин | 0,5 | Простаты | 29 | NA | NA | NA |
| SMART-H | 5 | Простаты | 29 | NA | NA | NA |
| SMART-H | 15 | Простаты | 4 | NA | NA | NA |
| SMART-F | 5 | Простаты | 21 | NA | NA | NA |
| SMART-F | 15 | Простаты | 24 | NA | NA | NA |
| SMART-OH | 50 | Простаты | 34 | NA | NA | NA |
| Носитель | NA | Меланома | 100 | 18 дней | NA | NA |
| SMART-H | 20 | Меланома | 30 | 28 дней | 10 | 0,7 |
| SMART-F | 15 | Меланома | 28 | 29 дней | 11 | 0,7 |
SMART-H в Таблице 23 является 1h; SMART-F в Таблице 23 является 2k; и SMART-OH в Таблице 23 является 2l.
1h в опухоли вызвало %T/C = 29% и 4% при обработке 5 и 15 мг/кг (все дозы вводились внутрибрюшинно (i.p.)), соответственно, тогда как 2k вызвало %T/C = 21% и 24% при обработке 5 и 15 мг/кг, соответственно. Высокая доза 2l (50 мг/кг) продемонстрировала %T/C 34%. Винбластин, положительный контроль, показал %T/C 29% на 22 день в ксенотрансплантатах PC-3 (Фигура 31В). Измерения веса тела, для контролирования токсичности, показали, что лишь 1 из 8 мышей, обработанных 1h (15 мг/кг) и 2 из 7 мышей, обработанных 2k (15 мг/кг) потеряли более 15% веса тела. Помимо противоопухолевого действия соединений на опухоли простаты PC-3, 1h (20 мг/кг) и 2k (15 мг/кг) демонстрируют существенное снижение опухолей А375. Как показано на Фигуре 31С, объемы опухолей в контрольной группе увеличились до 2183 мм3, тогда как 14 объемов в группах обработки 1h и 2k увеличились до 775 мм3 и 722 мм3, соответственно. Обработка 1h и 2k вызвала %T/C 28% и 29%, соответственно. Для оценки нейротоксичного эффекта 1h in vivo были выполнены испытания методом вращающегося стержня. На основании результатов испытаний эффективности in vivo, для исследования влияния 1h на моторную координацию было выбрано 5 или 15 мг/кг [внутрибрюшинное введение, Captex200/Tween80 (1/4)]. Обработку 0,5 мг/кг винбластина использовали в качестве положительного контроля при тех же условиях. Как показано на Фигуре 31D, винбластин постепенно снижает время (в секундах), которое мышь может удерживаться на вращающемся стержне, и приобретает значимость на 27 и 31 день (p < 0,05) по сравнению с группой, обработанной носителем. Однако в группах обработки 1h не было существенных отличий, что позволяет предположить, что 1h не вызывает нейротоксичности у мышей ICR в дозах, связанных с противоопухолевым действием.
1h не вызывает устойчивости к лекарствам у мышей с опухолью PC-3. Авторы изобретения иссекли опухоли PC-3 из «голых» мышей через 21 день обработки носителем (n = 3) или 15 мг/кг 1h (n = 3). Солидные опухоли расщепили и диспергировали в клетках, как описано в разделе способов. Клеточную линию PC-3, закупленную у ATCC (American Type Culture Collection, Манассас, штат Вирджиния, США) использовали в качестве контроля. Значения IC50 составили 29,1 ± 1,1, 29,1 ± 0,8 и 30,4 ± 0,5 нМ в клетках PC-3 производства ATCC и диссоциированных клетках из носителя и опухолей, обработанных 1h, соответственно. Эти данные показывают, что 1h не вызывает устойчивости к лекарствам в опухолях PC-3 через 21 день непрерывной обработки 1h.
ПРИМЕР 23
МОЛЕКУЛЯРНОЕ МОДЕЛИРОВАНИЕ
Способы
Все исследования молекулярного моделирования выполнили при помощи программы Schrödinger Molecular Modeling Suite 2008 (Schrödinger LLC, Нью-Йорк, штат Нью-Йорк), работающей на платформе Dell Linux. Поскольку размер ABI соединений гораздо ближе к размеру ABT-751, чем к DAMA-колхицину, авторы изобретения выбрали тубулиновый комплекс с ABT-751 (код PDB: 3KHC) в качестве системы моделирования. ABI были построены и получены с использованием модуля Ligprep, и они были присоединены к сайту ABT-751 с использованием модуля Glide программы Schrödinger Suite. Наилучшие комплексы присоединения были подвержены ограниченной динамике молекулы для выделения любых цепей с использованием модуля Macromodel с силовым полем OPLS-2005. Лиганд и окружающие его остатки в пределах 15 Ǻ могли двигаться свободно, тогда как остатки радиусом свыше 15 Ǻ жестко удерживались.
Результаты
Исследовали молекулярное моделирование для связывания ABI соединений в тубулине. В базе данных PDB было несколько кристаллических структур лиганд-тубулинового комплекса, из которых самым последним был комплекс Dorleans et al. В целом, колхицин-связывающий карман допускает различные молекулярные структуры, что может указывать на существенные конформационные изменения при связывании лиганда. Фактически, Dorleans et al. расшифровали кристаллические структуры пустых тубулиновых димеров и лиганд-тубулинового комплекса. Ими было обнаружено, что в отсутствие лиганда, петля 7 (T7, остатки 244-251, Фигура 32) в бета-мономере сворачивается и занимает связывающий карман, тогда как при связывании лиганда она разворачивается. Связанная спираль 7 (H7, остатки 224-243) и спираль 8 (H8, остатки 252-260) смещаются при связывании лиганда. Вероятно, что предел, до которого смещается Т7, зависит от размера конкретного лиганда. Эта подвижность представляет собой важную задачу для понимания точного способа связывания конкретных лигандов без расшифровки фактических кристаллических структур. Тем не менее, тщательный анализ возможных способов связывания может дать некоторые понимание связывания различных лигандов.
Схемы связывания 12cb и 11cb (реберная модель) показаны на Фигуре 32А и 32В. Для сравнения, комплексы кристаллических структур ABT-751 и DAMA-колхицина (каркасные модели) вместе с ABI-12cb/тубулиновым комплексом представлены на Фигуре 32А. Для ясности, на Фигуре 32А показаны только относительные вторичные структуры, образующие связывающий карман в-тубулина. Общие структуры 12cb, ABT-751 и DAMA-колхицина хорошо накладываются на связывающий карман. Было определено несколько потенциальных водородных связей между соединением 12cb и тубулином. Карбонильная группа 12cb находится в достаточной близости для образования двух водородных связей со скелетным NH Leu-252 в H8 и боковой цепью Asp-251 в T7 в-мономера тубулина. пара-Замещение фтором в кольце С расположено близко к боковой цепи Cys241 в T7 и Tyr202 в S6, возможно, образовывая одну или две водородные связи. Протон имидазола расположен очень близко и, вероятно, образует водородную связь с Thr179 в петле Т5 (остатки 173-182) б-мономера тубулина (Фигура 32А). Вместе с гидрофобными взаимодействиями, которые обеспечиваются ароматическими кольцами, вероятное образование этих водородных связей способствует высокой связывающей способности с тубулиновым димером, что приводит к высокой антипролиферативной способности.
Способ связывания 11cb, предположительно, является менее определенным, поскольку два из трех ароматических колец могут занимать связывающий карман в-мономера, тогда как третье кольцо может быть направлено в сторону поверхности раздела б/в-мономеров, подобно тому, как связывается боковая цепь DAMA-колхицина. Моделирование авторов изобретения показало, что защитная группа, вероятно, направлена в сторону поверхности раздела димера тубулина, тогда как кольца А и С в 11cb занимают такой же карман и расположение, как в 12cb (Фигура 32В). Это может объяснить схожую активность этих двух соединений, даже несмотря на то, что 11cb имеет наружную кольцевую систему. Из исследований молекулярного моделирования, представленных на Фигурах 32А и 32В, донор водородной связи, вероятно, является тиоловой группой в Cys-241 в петле 7 в-субъединицы б/в-тубулинового димера.
Был смоделирован способ связывания ABI 12fb (не показан) и сравнен с DAMA-колхицином (смотри Фигуру 19, на которой представлена структура колхицина) в б/в-тубулиновом гетеродимере. Общие структуры 12fb и DAMA-колхицина хорошо перекрываются. п-Фторфениловый фрагмент перекрывается с триметоксифениловым фрагментом, который взаимодействует с петлей Т7 в в-субъединице. Точно так же, п-хлорфениловый фрагмент занимает другую сторону кармана, где присутствует семичленное кольцо DAMA-колхицина, где атом хлора занимает карман, в котором взаимодействует метокси-группа.
ПРИМЕР 24
ВИЗУАЛИЗАЦИЯ МИКРОТРУБОЧЕК
Материалы и способы
Для получения визуально приемлемого подтверждения взаимодействия ABI с тубулином внутри клеток использовали набор перегруппировки Cellomics Cytoskeleton (Thermo Scientific, Рокфорд, штат Иллинойс). Клетки меланомы WM-164 обрабатывали каждым соединением в течение 18 часов в двух экземплярах, используя покрытые коллагеном 96-луночные планшеты (Becton Dickinson Labware, Бедфорд, штат Массачусетс). Затем клетки фиксировали 4% параформальдегидом (Thermo Scientific, Рокфорд, штат Иллинойс) и нарушили проницаемость мембран при помощи пермеабилизирующего буфера, поставляемого в наборе. Затем к клеткам добавили первичные антитела тубулина и вторичные антитела с флуоресцентной меткой. Ядра клеток окрасили при помощи DAPI. Для всех клеток использовали также зеленое окрашивание цельных клеток. Все изображения были получены при помощи инвертированного флуоресцентного микроскопа Olympus IX71 (Olympus Corp., Токио, Япония) наложением отдельных изображений тубулина (красный), ядер (синий) и цельных клеток (зеленый). Для сравнения вместе с ABI включили паклитаксел, колхицин и ABT-751.
Результаты
Исследовали визуальное подтверждение взаимодействия ABI с тубулином внутри клеток. Расположение микротрубочек в клетках меланомы человека WM-164 при обработке различными соединениями представлено на Фигуре 33. Изображения микротрубочек ясно показывают, что все пять исследованных соединений привели к перегруппировке цитоскелета. Существует значительное различие между паклитакселем и четырьмя другими соединениями (колхицином, ABT-751, 12cb и 12da). Обработка паклитакселем привела к конденсации микротрубочек, упорядоченно расположенных вокруг ядер, по сравнению с контрольными образцами, что согласуется с его механизмом действия для стабилизации микротрубочек. Напротив, обработка колхицином, ABT-751, 12cb и 12da дала схожее действие на микротрубочки и привела к некоторой степени фрагментации микротрубочек, что согласуется с их общим механизмом действия для дестабилизации микротрубочек. Эти результаты также подтверждают, что ABI имеют одинаковую клеточную мишень с колхицином и вызывают такой же клеточный эффект.
ПРИМЕР 25
ДЕЙСТВИЕ СОЕДИНЕНИЕ 17ya и 55 НА РАЗРУШЕНИЕ СОСУДОВ
Способ
Клетки. HUVEC (эндотелиальные клетки пупочной вены человека) культивировали и выращивали в среде EGM-2 BulletKit (Lonza, Cat No. CC-3162), содержащей добавки для роста, включающие гидрокортизон, основной фактор роста фибробластов человека с гепарином (hFGF-B), фактор роста эндотелия сосудов (VEGF), R3-инсулин-подобный фактор роста 1 (IGF-1), аскорбиновую кислоту, гепарин, бычью плодную сыворотку, эпидермальный фактор роста человека (hEGF) и GA-1000 (гентамицин и амфтотерицин В) в базальной среде эндотелиальных клеток 2. Для экспериментов использовали клетки между третьим и четвертым пассажем. Клетки рака простаты человека PC-3 и клетки рака молочной железы человека T47D культивировали в среде RPMI-1640 с 5% бычьей плодной сыворотки.
Испытания ингибирования клеточного роста. Цитотоксическую или антипролиферативную активность исследуемых соединений изучили в нескольких клеточных линиях с использованием анализа с сульфородамином В (SRB). Культивированные клетки поместили в 96-луночные планшеты и инкубировали в среде, содержащей различные концентрации исследуемых соединений, в течение 24 или 48 часов. Клетки окрасили раствором сульфородамина В (SRB). Оптическую плотность определили при 540 нм на микропланшет-ридере (Dynex Technologies, Шантильи, штат Вирджиния). Построили графики процентного ингибирования клеточного роста в зависимости от концентрации лекарства, и определили концентрацию, которая ингибирует клеточный рост на 50% по сравнению с контролем, обработанным носителем, (IC50) по нелинейной регрессии по методу наименьших квадратов с использованием программы WinNonlin (Pharsight Corporation, Кэри, штат Северная Каролина).
Анализы формирования и разрушения капилляров. Анализы формирования капилляров выполнили в 96-луночных планшетов, поместив 12000 клеток/лунку HUVEC на слое Matrigel (BD Biosciences). Для оценки анти-капиллярного действия, капилляры оставили формироваться на период 16 часов перед добавлением исследуемого соединения или носителя-контроля. Кроме того, ингибирующее действие исследуемых соединений на формирование капилляров исследовали обработкой клеток HUVEC исследуемыми соединениями до формирования капилляров. Изображения получили сразу после добавления соединения, через 5, 10, 15 и 25 часов после воздействия исследуемых соединений. Формирование капилляров количественно определили подсчетом количества трубочек и узлов, имеющих, по меньшей мере, три грани.
Анализ проницаемости эндотелиального монослоя. Проницаемость эндотелиального клеточного слоя оценили в системе трансвелов. HUVEC поместили на планшеты в концентрации 2 Ч 106 на один вкладыш в 24-луночные планшеты в среде EGM-2 и инкубировали в течение 72 часов для достижения 100% слияния. Исследуемые соединения разбавили средой EGM-2 и добавили в верхнюю ячейку прибора. Через 1, 2, и 4 часа инкубации соединения удалили и на 5 минут добавили 75 мкг/мл FITC-конъюгированного декстрана (молекулярный вес 40000). Измерения флуоресценции нижней ячейки получили после возбуждения при 485 нм, и измерили испускание при 520 нм с использованием микропланшет-ридера BioTek Synergy 4.
Результаты
17ya и 55 демонстрируют высокую антипролиферативную активность против эндотелиальных клеток. 17ya и 55 были оценены на цитотоксическую активность против эндотелиалных клеток с добавлением фактора роста и культур эндотелиальных клеток без фактора роста. Комбретастатин А-4 (СА4) и доксорубицин использовали в качестве положительного и отрицательного контроля, соответственно. Соединение 17ya демонстрирует более высокий потенциал, чем соединение 55 против активно разрастающихся эндотелиальных клеток (Таблица 24 и Фигура 35). Оба 17ya и 55 демонстрируют селективность для эндотелиальных клеток, оказывая более низкие значения IC50 по сравнению со значениями для клеток рака простаты. CA4, 17ya и 55 являются в 8, 5 и 3 раза более активными против эндотелиальных клеток, чем против раковых клеток, соответственно, тогда как доксорубицин не является специфическим для эндотелиальных клеток (Таблица 24 и Фигура 35). Однако между покоящимися и активными эндотелиальными клетками с этими соединениями селективности не наблюдалось (данные не показаны).
Таблица 24. Ингибирование роста эндотелиальных клеток соединениями 17ya и 55. N=3

17ya нарушает образование эндотелиальных капилляров, но не разрушает уже сформированные капилляры. Активность 17ya была исследована на эндотелиальных клетках, участвующих в формировании капиллярных трубок in vitro. Эндотелиальные клетки поместили на матрикс Matrigel и наблюдали формирование и построение капиллярных трубок в присутствии или в отсутствие соединений (CA4, доксорубицина и 17ya).
Во избежание путаницы между формированием трубок на ранней стадии и нарушением построения трубок, клетки HUVEC на матриксе в присутствии лекарственной обработки инкубировали в течение 15 часов. Затем разрушение капилляров определили подсчетом количества трубок и узлов в каждой группе обработки. С другой стороны, для оценки действия исследуемых соединений в уже сформированных капиллярах, клетки HUVEC на матриксе оставили формировать капиллярную трубку на 16 часов, и эти капилляры обработали исследуемыми соединениями.
В результате количество трубок и узлов постепенно снижалось с течением времени за счет недостатка или потребления питательного вещества клетками HUVEC (Фигура 36). Этот тренд наблюдали в каждой группе обработки лекарствами (Фигура 36). Для изучения различия между необработанными и предварительно обработанными капиллярами, сравнивали группы инкубации по 15 часов (Фигура 36).
Эндотелиальные клетки, которые подвергались воздействию различных концентраций 17ya (от 0 до 50 мкМ) поместили на планшеты Matrigel, что привело к ингибированию формирования трубок доза-зависимым образом. 17ya с соответствующим значением IC50 5 нМ в исследованиях ингибирования клеточного роста ингибирует более 50% формирования трубок, по сравнению с носителем-контролем (Фигура 37). 17ya при 10 нМ полностью ингибирует формирование трубок (Фигура 37). Однако в уже сформированных капиллярах в группе, обработанной 10 нМ 17ya, это не привело к разрушению структуры капилляров за 15 часов (Фигура 36). Эти результаты позволяют предположить, что 17ya значительно ингибирует формирование эндотелиальных капилляров, но является недостаточно эффективным для разрушения уже сформированных капилляров. Такие же результаты наблюдались в группе обработки CA4 (Фигура 37). Однако доксорубицин не повлиял на построение капилляров при токсичной концентрации.
17ya и 55 увеличивают проницаемость монослоев эндотелиальных клеток. Антитубулиновые средства могут модифицировать целостность слоев эндотелиальных клеток, выстилающих кровеносные сосуды, действуя на цитоскелет эндотелиальных клеток. Таким образом, эффект разрушения сосудов антитубулинового средства, как известно, увеличивает проницаемость кровеносных сосудов и поэтому может приводить к потере белков и высокой вязкости крови. Это может привести к снижению кровотока, вызывая последующую гибель опухоли из-за гипоксии и лишения питательных веществ.
Было оценено влияние 17ya и 55 на сосудистую проницаемость с использованием исследования in vitro, при помощи системы трансвел с конфлюентными монослоями HUVEC. Изменение проницаемости за счет исследуемых соединений измерили по снижению декстрана (молекулярный вес 40000) через 1, 2 и 4 часа после обработки лекарством. В качестве положительного контроля использовали CA4. CA4, 17ya и 55 привели к повышенной проницаемости, и этот эффект было более выраженным через 1 час инкубирования (данные не показаны). 17ya демонстрирует такую же эффективность, как и CA4 (Фигура 38). Доксорубицин не вызывает каких-либо изменений в проницаемости монослоя эндотелиальных клеток (Фигура 38).
Все детали, описанные в настоящем документе (включая все сопровождающие пункты формулы изобретения, реферат и чертежи) и/или все описанные этапы всех способов или процессов могут комбинироваться с любыми из представленных выше аспектов в любых комбинациях, за исключением тех комбинаций, где, по меньшей мере, некоторые из таких деталей и/или этапов являются взаимоисключающими. Несмотря на то, что были отображены и подробно описаны предпочтительные варианты, специалисту в соответствующей области понятно, что могут быть сделаны различные модификации, дополнения, замены и тому подобное, без отклонения от идеи настоящего изобретения, и поэтому подразумевается, что они входят в рамки настоящего изобретения, как определено в формуле изобретения, представленной далее.
Claims (15)
1. Соединение, представленное структурой формулы XI(e):

где R4 и R5 независимо являются водородом, O-алкилом, O-галоалкилом, F, Cl, Br, I, галоалкилом, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3,
-(CH2)iNH2, -(CH2)iN(CH3)2, -OC(O)CF3, C1-C5 линейным или разветвленным алкилом, алкиламино, аминоалкилом, -OCH2Ph, -NHCO-алкилом, COOH, -C(O)Ph, C(O)O-алкилом, C(O)H, -C(O)NH2 или NO2;
i является целым числом от 0 до 5; и
n является целым числом от 1 до 4;
при условии, что R4 и R5 оба одновременно не являются водородом.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, фармацевтический продукт, таутомер или их комбинации.
3. Фармацевтическая композиция, содержащая соединение по п. 1 в терапевтически эффективном количестве для лечения рака и фармацевтически приемлемый носитель.
4. Применение соединения по п. 1 для изготовления лекарственного средства для лечения, подавления, снижения степени, уменьшения риска, ингибирования рака, где указанный рак выбран из группы, состоящей из рака простаты, рака молочной железы, рака яичников, рака кожи, меланомы, рака легких, рака ободочной кишки, лейкоза, рака почек, рака ЦНС и их комбинаций.
5. Применение по п. 4, где указанное лекарственное средство пригодно для использования в комбинации с другой терапией рака.
6. Применение соединения по п. 1 для изготовления лекарственного средства для лечения лекарственно-устойчивой опухоли или опухолей; где указанная опухоль выбрана из группы, состоящей из раковой опухоли меланомы, опухоли рака простаты и опухоли рака яичников и их комбинаций.
7. Применение по п. 6, где указанная опухоль является раковой опухолью меланомы.
8. Применение по п. 6 или 7, где указанное лекарственное средство пригодно для использования в комбинации с другой терапией рака.
9. Применение соединения по п. 1 для изготовления лекарственного средства для разрушения раковых клеток.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US37667510P | 2010-08-24 | 2010-08-24 | |
| US61/376,675 | 2010-08-24 | ||
| PCT/US2010/062418 WO2011109059A1 (en) | 2010-03-01 | 2010-12-29 | Compounds for treatment of cancer |
| USPCT/US2010/062418 | 2010-12-29 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2013112871A Division RU2609018C2 (ru) | 2010-08-24 | 2011-08-24 | Соединения для лечения рака |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| RU2762111C1 true RU2762111C1 (ru) | 2021-12-15 |
Family
ID=45723794
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2013112871A RU2609018C2 (ru) | 2010-08-24 | 2011-08-24 | Соединения для лечения рака |
| RU2017102002A RU2762111C1 (ru) | 2010-08-24 | 2011-08-24 | Соединения для лечения рака |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2013112871A RU2609018C2 (ru) | 2010-08-24 | 2011-08-24 | Соединения для лечения рака |
Country Status (11)
| Country | Link |
|---|---|
| EP (1) | EP2608671B1 (ru) |
| JP (2) | JP5997156B2 (ru) |
| KR (1) | KR101806255B1 (ru) |
| CN (1) | CN103442566B (ru) |
| AU (1) | AU2011293353B2 (ru) |
| CA (2) | CA3030689C (ru) |
| ES (1) | ES2718637T3 (ru) |
| IL (1) | IL224863B (ru) |
| MX (1) | MX336761B (ru) |
| RU (2) | RU2609018C2 (ru) |
| WO (1) | WO2012027481A1 (ru) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9447049B2 (en) | 2010-03-01 | 2016-09-20 | University Of Tennessee Research Foundation | Compounds for treatment of cancer |
| US11084811B2 (en) | 2010-03-01 | 2021-08-10 | Oncternal Therapeutics, Inc. | Compounds for treatment of cancer |
| AU2011293353B2 (en) * | 2010-08-24 | 2015-02-26 | Oncternal Therapeutics, Inc. | Compounds for treatment of cancer |
| EP3708164A1 (en) * | 2014-05-06 | 2020-09-16 | Oncternal Therapeutics, Inc | Compounds for treatment of cancer |
| WO2016077648A1 (en) * | 2014-11-11 | 2016-05-19 | Neymeyer Calvin E | Method of treating pre-cancerous lesion with glyphosate, and compositions thereof |
| JP2018531924A (ja) | 2015-09-24 | 2018-11-01 | ファイザー・インク | テトラヒドロピラノ[3,4−d][1,3]オキサジン誘導体、およびbace阻害剤としてのその使用 |
| JPWO2019188456A1 (ja) * | 2018-03-26 | 2021-03-18 | 学校法人 川崎学園 | 新規抗腫瘍剤 |
| MX2020012291A (es) | 2018-05-15 | 2021-03-25 | Univ Tennessee Res Found | Compuestos para el tratamiento de cancer de mama triple negativo y cancer de ovario. |
| JP2021523915A (ja) * | 2018-05-15 | 2021-09-09 | ユニバーシティ オブ テネシー リサーチ ファウンデーション | 膵臓癌の治療のための化合物 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999024048A1 (de) * | 1997-11-06 | 1999-05-20 | Pharmaceutical Research Center | Arzneimittel für krebs-therapie |
| RU2008105663A (ru) * | 2005-07-18 | 2009-08-27 | Бипар Сайенсиз, Инк. (Us) | Способ лечения рака (варианты) |
| US20090246291A1 (en) * | 2008-03-27 | 2009-10-01 | Angelika Burger | Method and compositions for treatment of cancer |
| CA2962524A1 (en) * | 2008-06-16 | 2010-07-01 | University Of Tennessee Research Foundation | (2-(1-h-indolyl)imidazol-4-yl)(3,4,5-trimethoxyphenyl)methanone useful for the treatment of cancer |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2433018A1 (en) * | 2000-12-21 | 2002-06-27 | Joel C. Barrish | Thiazolyl inhibitors of tec family tyrosine kinases |
| WO2003016338A1 (en) | 2001-08-15 | 2003-02-27 | Parker Hughes Institute | Crystal structure of the btk kinase domain |
| US6818663B2 (en) * | 2002-05-17 | 2004-11-16 | Hoffmann-La Roches | Diaminothiazoles |
| CN102883607B (zh) * | 2010-03-01 | 2015-07-22 | Gtx公司 | 用于治疗癌的化合物 |
| AU2011293353B2 (en) * | 2010-08-24 | 2015-02-26 | Oncternal Therapeutics, Inc. | Compounds for treatment of cancer |
-
2011
- 2011-08-24 AU AU2011293353A patent/AU2011293353B2/en active Active
- 2011-08-24 KR KR1020137007388A patent/KR101806255B1/ko active IP Right Grant
- 2011-08-24 CA CA3030689A patent/CA3030689C/en active Active
- 2011-08-24 CA CA2809256A patent/CA2809256C/en active Active
- 2011-08-24 EP EP11820603.6A patent/EP2608671B1/en active Active
- 2011-08-24 ES ES11820603T patent/ES2718637T3/es active Active
- 2011-08-24 MX MX2013002155A patent/MX336761B/es unknown
- 2011-08-24 CN CN201180051231.XA patent/CN103442566B/zh active Active
- 2011-08-24 JP JP2013526130A patent/JP5997156B2/ja active Active
- 2011-08-24 RU RU2013112871A patent/RU2609018C2/ru active
- 2011-08-24 RU RU2017102002A patent/RU2762111C1/ru active
- 2011-08-24 WO PCT/US2011/048980 patent/WO2012027481A1/en active Application Filing
-
2013
- 2013-02-21 IL IL224863A patent/IL224863B/en active IP Right Grant
-
2016
- 2016-08-25 JP JP2016164420A patent/JP2017008091A/ja active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999024048A1 (de) * | 1997-11-06 | 1999-05-20 | Pharmaceutical Research Center | Arzneimittel für krebs-therapie |
| RU2008105663A (ru) * | 2005-07-18 | 2009-08-27 | Бипар Сайенсиз, Инк. (Us) | Способ лечения рака (варианты) |
| US20090246291A1 (en) * | 2008-03-27 | 2009-10-01 | Angelika Burger | Method and compositions for treatment of cancer |
| CA2962524A1 (en) * | 2008-06-16 | 2010-07-01 | University Of Tennessee Research Foundation | (2-(1-h-indolyl)imidazol-4-yl)(3,4,5-trimethoxyphenyl)methanone useful for the treatment of cancer |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2013538213A (ja) | 2013-10-10 |
| JP5997156B2 (ja) | 2016-09-28 |
| CA3030689A1 (en) | 2012-03-01 |
| JP2017008091A (ja) | 2017-01-12 |
| KR101806255B1 (ko) | 2017-12-07 |
| WO2012027481A9 (en) | 2013-10-10 |
| AU2011293353B2 (en) | 2015-02-26 |
| AU2011293353A1 (en) | 2013-03-14 |
| CA2809256A1 (en) | 2012-03-01 |
| EP2608671A1 (en) | 2013-07-03 |
| EP2608671B1 (en) | 2018-12-12 |
| EP2608671A4 (en) | 2013-08-07 |
| KR20130119912A (ko) | 2013-11-01 |
| RU2609018C2 (ru) | 2017-01-30 |
| MX2013002155A (es) | 2013-08-01 |
| CA2809256C (en) | 2019-03-05 |
| IL224863B (en) | 2019-02-28 |
| RU2013112871A (ru) | 2014-09-27 |
| CN103442566B (zh) | 2015-12-09 |
| WO2012027481A1 (en) | 2012-03-01 |
| ES2718637T3 (es) | 2019-07-03 |
| CN103442566A (zh) | 2013-12-11 |
| CA3030689C (en) | 2021-03-02 |
| MX336761B (es) | 2016-01-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11465987B2 (en) | Compounds for treatment of cancer | |
| US9334242B2 (en) | Compounds for treatment of cancer | |
| US8822513B2 (en) | Compounds for treatment of cancer | |
| RU2762111C1 (ru) | Соединения для лечения рака | |
| US20220024912A1 (en) | Compounds for treatment of cancer | |
| AU2015227531B2 (en) | Compounds for treatment of cancer | |
| AU2015202828B2 (en) | Compounds for treatment of cancer |