RU2659388C1 - Нуклеотиды, включающие N-[(S)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги и их применение - Google Patents
Нуклеотиды, включающие N-[(S)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги и их применение Download PDFInfo
- Publication number
- RU2659388C1 RU2659388C1 RU2017106611A RU2017106611A RU2659388C1 RU 2659388 C1 RU2659388 C1 RU 2659388C1 RU 2017106611 A RU2017106611 A RU 2017106611A RU 2017106611 A RU2017106611 A RU 2017106611A RU 2659388 C1 RU2659388 C1 RU 2659388C1
- Authority
- RU
- Russia
- Prior art keywords
- propanoate
- cyclobutyl
- phosphorylamino
- phenoxy
- carbon
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/664—Amides of phosphorus acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
- A61K31/7072—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid having two oxo groups directly attached to the pyrimidine ring, e.g. uridine, uridylic acid, thymidine, zidovudine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
- A61K31/708—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid having oxo groups directly attached to the purine ring system, e.g. guanosine, guanylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/30—Phosphinic acids R2P(=O)(OH); Thiophosphinic acids, i.e. R2P(=X)(XH) (X = S, Se)
- C07F9/36—Amides thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids RP(=O)(OH)2; Thiophosphonic acids, i.e. RP(=X)(XH)2 (X = S, Se)
- C07F9/44—Amides thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65586—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system at least one of the hetero rings does not contain nitrogen as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic System
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
- C07F9/65616—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings containing the ring system having three or more than three double bonds between ring members or between ring members and non-ring members, e.g. purine or analogs
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/10—Pyrimidine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/20—Purine radicals with the saccharide radical esterified by phosphoric or polyphosphoric acids
Abstract
Изобретение относится к пролекарству, соответствующему общей формуле 1, его стереоизомеру, фармацевтически приемлемой соли, возможно в кристаллической и поликристаллической форме, которое обладает противовирусной активностью. Пролекарство может быть использовано при лечении вирусных заболеваний опосредованных активностью NS5B HCV полимеразы HBV, ДНК-полимеразы и ВИЧ-1 обратной транскриптазы (RT), в частности для лечения гепатита B и C-инфекции. В общей формуле 1

n имеет значение 1 или 0; Nuc представляет собой


R1 представляет собой водород или метил; R2, R3 представляют собой необязательно одинаковые заместители, выбранные из Н, F, Cl, СН3 при условии, когда сплошная линия и сопровождающая ее пунктирная линия ( ) вместе представляют одинарную углерод-углеродную (С-С) связь, или R2 и R3 представляют собой водород при условии, когда сплошная линия и сопровождающая ее пунктирная линия (
) вместе представляют одинарную углерод-углеродную (С-С) связь, или R2 и R3 представляют собой водород при условии, когда сплошная линия и сопровождающая ее пунктирная линия ( ) вместе представляют собой двойную углерод-углеродную связь (С=С); R4 представляет собой заместитель, выбранный из R4.1-R4.5:
) вместе представляют собой двойную углерод-углеродную связь (С=С); R4 представляет собой заместитель, выбранный из R4.1-R4.5:

R3.6 представляет собой заместитель, выбранный из Н, F, Cl, СН3 или CF3; R3,7 представляет собой водород, С1-С4-алкил или С3-С6-циклоалкил; X представляет собой О, СН2 или С=СН2; Y представляет собой S, СН2 или НО-СН-группу при условии, когда непрерывная линия и сопровождающая ее пунктирная линия ( ) вместе представляют собой одинарную углерод-углеродную (С-С) связь, или Y представляет собой СН-группу при условии, когда сплошная линия и сопровождающая ее пунктирная линия (
) вместе представляют собой одинарную углерод-углеродную (С-С) связь, или Y представляет собой СН-группу при условии, когда сплошная линия и сопровождающая ее пунктирная линия (
Description
Настоящее изобретение относится к химиотерапевтическим средствам для лечения вирусных и раковых заболеваний. Эти соединения являются пролекарствами ингибиторов вируса иммунодефицита человека (ВИЧ), вируса гепатита C (ВГС, HCV) полимеразы, вируса гепатита B (ВГВ, HBV) ДНК-полимеразы и предназначены для лечения вируса иммунодефицита человека, гепатита C, гепатита B, а также ко-инфекций ВИЧ/ВГС, ВИЧ/ ВГВ, ВИЧ/ВГС/ВГВ и ВГС/ВГВ.
Вирус иммунодефицита человека (ВИЧ), полученный из примата лентивирусов, является этиологическим агентом синдрома приобретенного иммунодефицита (СПИД). Болезнь была впервые описана в 1981 году, и ВИЧ-1 был выделен в конце 1983 г. С тех пор СПИД стал всемирной эпидемией, расширяясь по масштабам и величине, такие инфекции, как ВИЧ-инфекции повлияли на различные группы населения и географические регионы. Миллионы людей теперь заражены во всем мире; в случае заражения, люди остаются инфицированными на всю жизнь. В течение десяти лет, если его не лечить, подавляющее большинство ВИЧ-инфицированных индивидуумов заканчивают смертельным исходом в результате развития оппортунистических инфекций в результате ВИЧ-индуцированных недостатков в иммунной системе. СПИД является одной из самых важных проблем общественного здравоохранения во всем мире в начале 21-го века. Развитие высокоактивной антиретровирусной терапии (ВААРТ) при хроническом подавлении репликации и профилактики СПИДа ВИЧ является крупным достижением в области медицины ВИЧ [http://basicmedicalkey.com/aids-and-lentiviruses/].
ВИЧ продолжает оставаться одним из основных глобальных проблем общественного здравоохранения. В 2015 году было 36,7 миллиона человек, живущих с ВИЧ (в том числе 1,8 миллиона детей) - глобальная распространенность ВИЧ на уровне 0,8. Подавляющее большинство из этого числа живут в странах с низким и средним уровнем доходов. В том же году 1,1 миллиона человек умерли от связанных со СПИДом заболеваний. С начала эпидемии, по оценкам, 78 миллионов человек заразились ВИЧ, и 35 миллионов человек умерли от связанных со СПИДом заболеваний. По оценкам, 25,5 миллиона человек, живущих с ВИЧ, живут в странах Африки к югу от Сахары. Подавляющее большинство из них (по оценкам, 19 миллионов) живут на востоке и на юге Африки, это составило 46% новых случаев ВИЧ-инфекции во всем мире в 2015 году. Около 40% всех людей, живущих с ВИЧ, не знают, что у них есть вирус [http://www.avert.org/global-hiv-and-aids-statistics].
Развитие антиретровирусных препаратов существенно изменило восприятие ВИЧ / СПИДа от фатальной до хронической и потенциально управляемой болезни, также, наличие и введение антиретровирусной терапии (APT) привело к значительному снижению смертности и заболеваемости, связанной с ВИЧ и СПИДом. Существует взаимосвязь между APT и качеством жизни людей, живущих с ВИЧ и СПИДом, а также ряд исследований отметили положительную связь между APT и улучшением качества жизни среди людей, живущих с ВИЧ и СПИДом как в развитых, так и в развивающихся странах [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3418767/].
ВИЧ-инфицированные пациенты живут дольше со времени введения высокоактивной антиретровирусной терапии (ВААРТ). При проведении антиретровирусной терапии продолжительность жизни пациента может быть продлена до 70-80 лет. Однако сопутствующая инфекция с ВГВ или/и ВГС приводит к повышенной заболеваемости и смертности от болезней печени. Неконтролируемая ВИЧ-инфекция ускоряет прогрессирование ВГС-индуцированного склероза печени.
Сопутствующая инфекция ВИЧ/ВГВ является обычным явлением. Хроническая инфекция ВГВ встречается у 5-10% ВИЧ-инфицированных лиц, которые подвергаются ВГВ, со скоростью в 10 раз выше, чем население в целом [http://hivinsite.ucsf.edu/InSite?page=kb-05-03-04#SlX]. ВИЧ/ВГС-инфицированные больные имеют в три раза больший риск развития цирроза или декомпенсации заболевания печени, чем пациенты с ВГС - моноинфекцией [https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-arv-guidelines/26/hcv-hiv]. В 2006 году в многонациональной когорте, включающей более 25000 ВИЧ-инфицированных людей в США и Европе, 14% смертей были связаны с заболеваниями печени и, из них 66% произошло у лиц с сопутствующей инфекцией ВИЧ/ВГС [http://hivinsite.ucsf.edu/InSite?page=kb-00&doc=kb-05-03-05].
Моноинфекция ВГС или ВГВ представляет собой одну из основных причин хронических заболеваний печени во всем мире. Тем не менее в эндемичных районах значительное число пациентов, инфицированных обоими вирусами в основном в результате общих путей передачи. Многочисленные исследования показали, что ВГС/ВГВ-инфицированные пациенты несут больший риск заболевания циррозом печени и гепатоцеллюлярной карциномы по сравнению с моноинфицированными пациентами. Поразительно, что примерно у 60% пациентов с неактивной инфекцией ВГВ до начала лечения гепатита C может происходить ВГВ реактивация, в то время как другие пациенты вырабатывают антиген гепатита B в сероконверсии после излечения ВГС [https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4367211/].
Нуклеозиды (и нуклеотиды уже используются в клинической практике в течение почти 50 лет и стали краеугольными камнями при лечении пациентов с вирусными инфекциями или раком. Разработка нескольких новых препаратов за последнее десятилетие показывает, что эта семья все еще обладает большим потенциалом.
Нуклеозиды и их аналоги (Nuc), такие как 2'-деокси-L-уридин (Nuc1), 2'-деокси-D-уридин (Nuc2), телбивудин (Nuc3), зидовудин (Nuc4), трифлуридин (Nuc5), клевудин (Nuc6), PSI-6206 (Nuc7), 2'-(S)-2'-хлор-2'-деокси-2'-фторуридин (Nuc8), ND06954 (Nuc9), ставудин (Nuc10), фестинавир (Nuc11), торцитабин (Nuc12), (-)-β-D-(2R,4R)-диоксолантимин (Nuc13), 2-(6-амино-пурин-9-ил)-этанол (Nuc14), (11)-1-(6-амино-9Н-пурин-9-ил)пропан-2-ол (Nuc15), 2'-С-метилцитидин (Nuc16), PSI-6130 (Nuc17), гемцитабин (Nuc18), 2'-хлор-2'-деокси-2'-фторцитидин (Nuc19), 2',2'-дихлор-2'-деоксицитидин (Nuc20), ламивудин (lamivudine) (Nuc21), эмтрицитабин (emtricitabine) (Nuc22), 2'-деоксиаденозин (Nuc23), 2'-деокси-β-L-аденозин (Nuc24), 2'-деокси-4'-С-этинил-2-фтораденозид (Nuc25), [(2R,4R)-4-(6-циклопропиламино-пурин-9-ил)-[1,3]диоксолан-2-ил]-метанол (Nuc26), амдоксовир (Nuc27), энтекавир (Nuc28), FMCA (Nuc29), диоксолан-G (Nuc30), β-D-2'-деокси-2'-(R)-фтор-2'-β-С-метилгуанозин (Nuc31), абакавир (Nuc32), диданозин (Nuc33) и другие представляют большой интерес в качестве перспективных химиотерапевтических агентов [M.J. Sofia. Nucleosides and Nucleotides for the treatment of viral diseases. In Annual Reports in Medicinal Chemistry 2014, Volume 49, Editor-in-Chief M.C. Desai, p 221-247. L.P. Jordheim et al. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug. Discov. 2013 Jun; 12 (6), 447-464].











Нуклеоз(т)иды сыграли важную роль в лечении вирусных заболеваний. Для пациентов с вирусом иммунодефицита человека (ВИЧ) они оказались основой в ряде комбинированных схем лечения. В настоящее время нуклеоз(т)иды являются предпочтительным вариантом и стандартом лечения пациентов, инфицированных вирусом гепатита B (HBV). Они используются также в качестве ключевого компонента в терапии для лечения вируса гепатита C (HCV) инфекции. Они также играют центральную роль при лечении других вирусных инфекций, вызванных вирусами герпеса (HSV-1 и HSV-2), ветряной оспы, вируса Эпштейна-Барр и цитомегаловируса [Е. De Clercg. Ed. Antiviral Agents 2013, Vol. 67: Academic Press: New York. 2013. L.P. Jordheim et al. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nal. Rev. 2013, 12, 447-464]. Привлекательность нуклеоз(т)идной стратегии при разработке терапевтических средств для лечения заболеваний обусловлена тем, что все вирусы требуют полимеразу либо для ДНК либо для РНК репликации.
Другой фактор, который необходимо учитывать при разработке нуклеоз(т)идного ингибитора, относится к нуклеоз(т)идной метаболической активации. Это аналог трифосфата нуклеотида в качестве функционального субстрата для вирусной полимеразы, которая включается в растущую РНК или ДНК цепи, как правило, приводящая к обрыву цепи и в конечном счете прекращению вирусной репликации. Следовательно, эффективность, с помощью которой нуклеоз(т)ид преобразуется в активный трифосфат, а также концентрация и период полураспада трифосфата внутри клетки являются важными факторами, определяющими эффективность нуклеоз(т)ида в качестве ингибитора вирусной репликации. В общем, первый шаг фосфорилирования является определяющим, необходимым для создания активного трифосфата. В тех случаях, когда сам нуклеозид не является хорошим субстратом для киназ, участвующих в начальной стадии фосфорилирования, поставка монофосфата желательна, но это, как правило, требует использования пролекарственной технологии, чтобы замаскировать неблагоприятные характеристики фосфатной группы и облегчить проницаемость. Следовательно, нуклеотидная пролекарственная стратегия весьма полезна в развитии нуклеотидов для лечения вирусных и онкологических заболеваний.
Ряд нуклеоз(т)идных ингибиторов RT были одобрены для лечения ВИЧ-инфекции [R.F. Shinazi et al. Pharmacology of current and promising nucleosides for the treatment of human immunodeficiency viruses. J. Antiviral Res. 2006, 71, 322-334. E.De Clereq. he nucleoside reverse transcriptase inhibitors, nonnucleoside reverse transcriptase inhibitors, and protease inhibitors in the treatment of HIV infections (AIDS). Adva Pharmacol. 2013, 67, 317-358]. Некоторые из них используют в адъювантной терапии совместно с другими ингибиторами репликации ВИЧ, дающими удобные терапевтические режимы, которые стали стандартом в высокоактивной антиретровирусной терапии (ВААРТ). Эти комбинации включают Комбивир (Combivir®), Тризивир (Trizivir®), Эпзиком (Epzicom®),Трувадас (Truvadas), Атрипл (Atriple), Стрибил (Stribile), и Комплерас (Compleras). Трувадас, Атрипл, Комплерас и Стрибил включают нуклеозид эмтрицитабин и ациклический нуклеотид тенофовир дизопроксилфумарат (TDF), в то время как Комбивир (CombivirTM), Тризивир (Trizivir) и Эпзиком (Epzicom) включают в себя комбинацию из двух или трех лекарств, содержащих нуклеозиды Зидовудин (Zidovudine (AZT)), Ламивудин (Lamivudine, 3ТС, Nuc21), и/или Абакавир (Abacavir (ABC)) [R.F. Shinazi et al. Pharmacology of current and promising nucleosides for the treatment of human immunodeficiency viruses. J. Antiviral Res. 2006, 71, 322-334]. Успех антиретровирусной терапии ВИЧ привел к резкому увеличению продолжительности жизни людей, инфицированных этой когда-то неизлечимой болезнью. Тем не менее, даже с этим успехом продолжается поиск новых агентов, направленных на лечение хронически инфицированного населения, снижение резистенции и побочных эффектов, связанных с долгосрочным использованием лекарственных средств.



Достаточно новым пролекарством противовирусного ациклического нуклеозида тенофовир фосфоната (TFV) является тенофовир алафенамид (TAF, известный как Vemlidy и GS-7340). Он обладает улучшенными свойствами по сравнению с известным дизопроксилфумарат тенофовира (TDF). TAF является противовирусным препаратом для комбинированного лечения пациентов, нуждающихся в этом. TAF был разработан с целью повышения целенаправленного воздействия тенофовира (TFV) в мононуклеарных клетках периферической крови и, следовательно, уменьшения нефротоксичности, связанной с TDF. TAF в 400 раз более эффективен, чем TFV. Использование этого пролекарственного подхода привело к повышению коэффициента экспозиции исходного нуклеозида TFV в P13MCs по отношению к плазме, что приводит к более высокой эффективности [WO 2002008241. US 7390791. A.S. Ray, M.W. Fordyce, M.J.M. Hitchcock. Tenofovir alafenamide: A novel prodrug of tenofovir for the treatment of Human Immunodeficiency Virus. Antiviral Research Volume 125, January 2016, Pages 63-70. http://www.accessdata.fda.gov/drugsatfdadocs/nda/2015/ 207561Origls000PharmR.pdf]. В 2016 году TAF одобрен FDA в качестве препарата для лечения ВГВ [https://www.hepmag.com/article/fda-approves-vemlidy-tenofovir-alafenamide-taf-hepatitis-b]. TAF является мощным пролекарством против вируса гепатита B (HBV). По сравнению с TDF он имеет меньше побочных воздействий на кости и почки [http://www.aidsmap.com/Tenofovir-alafenamide-works-well-against-hepatitis-B-with-less-effect-on-bones-and-kidneys/page/3051008/].

Дальнейшее улучшение свойств пролекарств для лечения ВИЧ и ВГВ было достигнуто за счет тенофовир алафенамид фумарата [WO 2002008241. US 7390791] и тенофовир алафенамид полуфумарата [WO 2013025788, WO 2013116720, US 9296769. http://www.gilead.ca/pdf/ca/genvoya_pm_english.pdf].

Поиски нуклеотидных фосфонатов, которые при лечении ВИЧ будут обеспечивать улучшенный профиль резистентности по сравнению с существующими нуклеоз(т)идами и показывать лучший профиль безопасности по сравнению ДНК-полимеразами, привели к GS-9148. Этот нуклеозидфосфонат показал улучшенный профиль резистенции в широком диапазоне мутаций по сравнению с нуклеоз(т)идами, используется в клинической практике. Для того, чтобы улучшить свойства клеточной проницаемости и поглощение в лимфоидные клетках, был получен GS-9131 в качестве ведущего лекарственного кандидата. В пробирке и в естественных условиях было показано, что по сравнению с TDF GS-9131 слабо накапливается в почках и не проявляет заметной почечной токсичности [M.J. Sofia. Nucleosides and Nucleotides for the treatment of viral diseases. In Annual Reports in Medicinal Chemistry 2014, Volume 49, Editor-in-Chief M.C. Desai, p 224.]

В качестве пролекарств предназначенных для лечения ВИЧ были исследованы также фосфорамидаты 6-замещенных-2-Н-пуриновых диоксоланов, в частности пролекарства Диоксолан-А монофосфата, Диоксолан-А монофосрамидат. Последний оказался наиболее активным и наименее цитотоксическим с ЕС50=0,086 мкМ [L. Bondada et al. Adenosine Dioxolane Nucleoside Phosphoramidates as Antiviral Agents for Human Immunodeficiency and Hepatitis В Viruses. ACS Med. Chem. Lett. 2013, 4, 747-751].

Во всем мире 400 миллионов человек инфицированы ВГВ [http://www.pkids.org/files/pdf/ phr/02-01hbv.pdf]. В настоящее время стандартом лечения ВГВ является долгосрочная нуклеозидная терапия. Нуклеозиды, одобренные для лечения ВГВ-инфекции, включают Ламивудин (3ТС, Nuc21) (Lamivudine), Адефовир (Adefovir), Дипивоксил (Dipivoxil), Энтекавир (Entecavir), Телбивудин (Telbivudine) и TDF. Энтекавир (Entecavir) и TDF являются наиболее широко используемыми лекарствами. Длительное использование Энтекавир приводит к резистентности у значительной популяции пациентов, a TDF связан с нефротоксичностью и потерей костной массы [D. Grimm et al. HBV life cycle and novel drug targets. Hepatol. Int. 2011. 5. 644-653. G. Borgia, I. Gentile. Treating chronic hepatitis B: today and tomorrow. Curr. Med. Chem. 2006. 13. 2839-2855]. Тем не менее, дальнейшее использование нуклеозидной терапии связано с уменьшением фиброза печени, демонстрирующего, что подавление вирусной репликации имеет положительное долгосрочное значение [Т.Т. Chang et al. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology 2010. 52, 886-893. P. Marcellin et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet 2013, 381, 468-475].
Несмотря на успехи нуклеоз(т)идной HBV терапии работа по выявлению новых ингибиторов, которые могут обеспечить дополнительное преимущество по сравнению с существующими агентами, продолжается. Для этой цели были исследованы в качестве препаратов для лечения HBV некоторые из анти-ВИЧ агентов, как уже упоминалось выше [С.А. Geng et al. Small-molecule inhibitors for the treatment of hepatitis В virus documented in patents. Mini Rev. Med. Chem. 2013, 13, 749-776].
В последнее время получен 2'-фтор-6'-метилен-карбоциклический аденозин (FMCA) (ЕС50=0,55 μM), который оказался сильным ингибитором репликации HBV, который был также активен в отношении ламивудин-энтекавир резистентного клона (L180M + M204V + S202G) [R.K. Rawal et al. 2'-Fluoro-6'-methylene-carbocyclic adenosine phosphoramidate (FMCAP) prodrug: In vitro anti-HBV activity against the lamivudine-entecavir resistant triple mutant and its mechanism of action. Bioorg. Med. Chem. Lett. 2013. 23, 503-506]. Кроме того, получение соответствующей 5'-фосфорной кислоты из FMCA привело к соединению, которое было в 10 раз более мощным, чем FMCA, против обоих мутантов (дикого - ЕС50=0,62 μМ и резистентного - ЕС50=0,054 μM) [R.K. Rawal et al. 2'-Fluoro-6'-methylene-carbocyclic adenosine phosphoramidate (FMCAP) prodrug: In vitro anti-HBV activity against the lamivudine-entecavir resistant triple mutant and its mechanism of action. Bioorg. Med. Chem. Lett. 2013, 23, 503-506].
Известно также, что пролекарство клевудин (EIDD-02173) сохранило высокую анти-HBV активность в моделях клеточных культур HBV инфекции. Фосфорамидатный фрагмент успешно доставляет клевудин-5'-монофосфат в печень, при этом воздействие на другие органы значительно уменьшается [G.R. Bluemling et al. Targeted Delivery of Clevudine-5'-Monophosphate to the Liver After Oral Administration of a Clevudine-5'- Phosphoramidate Conjugate to Rats for the Treatment of HBV Infections. Global Antiviral Journal 2015, 11, Suppl. 3: HEP DART 2015: Abstr. 104, Р.97].

Гемцитабин-5'-фосфорамидат (NUC-1031) [M. Slusarczyk et al. Application of ProTide Technology to Gemcitabine: A Successful Approach to Overcome the Key Cancer Resistance Mechanisms Leads to a New Agent (NUC-1031) in Clinical Development. J. Med. Chem. 2014, 57, 1531-1542] показал высокую противораковую активность. В частности, NUC-1031 значительно снижает объем опухоли in vivo в ксенографных моделях рака поджелудочной железы человека. Важно отметить, что активация NUC-1031 гораздо меньше зависит от нуклеозидных транспортеров и дезоксицитидина, чем гемцитабин. Кроме того, NUC-1031 в отличие от гемцитабина устойчив к цитидиндезаминазной деградации.

Гепатит C, вызываемый вирусом гепатита C (ВГС), является одним из наиболее распространенных заболеваний печени и широко распространен по всему миру. На основе ежегодных докладов Всемирной организации здравоохранения (ВОЗ) более 130-150 миллионов человек инфицированы ВГС, и более 700 тысяч лиц умирают от ВГС [WHO. Hepatitis С. WHO fact sheet №164. Updated July 2016, http://www.who.int/mediacentre /factsheets/fs164/en/]. ВГС демонстрирует высокое генетическое разнообразие и характеризуется региональными вариациями генотипов (gT) ВГС. Генотип 1 (gT1) является наиболее распространенным во всем мире (83,4 миллиона человек, что составляет 46,2% всех случаев инфицированных ВГС, приблизительно одна треть из которых находится в Восточной Азии). Следующим наиболее распространенным является генотип 3 (gT3). Этим генотипом инфицированы во всем мире 54,3 миллиона человек (30,1%). Генотипы 2, 4, 6 составляют 22,8% всех случаев инфицированных ВГС человек, а генотип 5 (gT5) составляет оставшиеся <1%. В то время как генотипы 1 и 3 доминируют в большинстве стран независимо от экономического статуса, наибольшие доли генотипов 4 и 5 находятся в странах с низким уровнем дохода [Messina, J. P. at al. Global Distribution and Prevalence of Hepatitis С Virus Genotypes. Hepatology 2015, 61 (1), 77-87].
PSI-7851 и его диастереоизомер PSI-7977.
Недавно был разработан ингибитор NS5B HCV полимеразы Софосбувир (Sofosbuvir, PSI-7851), представляющий собой изопропил (S)-2-{[(2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-4-фтор-3-гидрокси-4-метил-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропионат, и его 5р-диастереоизомер Совалди (Sovaldi®, PSI-7977, GS-7977), а также Rp-диастереоизомер PSI-7976. [Sofia, М. J. et al. Discovery of a β-D-20-Deoxy-20-rfluoro-2 0-β-C-methyluridine Sovaldi Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis С Virus. J. Med. Chem. 2010, 53, 7202-7218. Sofia, M. J. et al. Nucleoside phosphoramidate prodrugs. Патент US 7964580 (2011), Патент US 8334270 (2012). Патент RU 2478104 (2013)].

Sovaldi® в настоящее время широко используется для комбинированного лечения гепатита C, в том числе совместно с ингибиторами NS5A HCV. Sovaldi® стал первым нуклеотидом, одобренным обоими регулирующими органами FDA и ЕС для комбинированного лечения больных гепатитом С, инфицированных различными генотипами (gT) вируса гепатита C. В клинических испытаниях он показал высокую эффективность против шести генотипов HCV (gT1-gT6) [I.M. Jacobson et al. Sofosbuvir for hepatitis С genotype 2 or 3 in patients without treatment options. Engl. J. Med. 2013, 368, 1867-1877. E. Lewirz et al. Sofosbuvir for previously untreated chronic hepatitis С infection. Engl. J. Med. 2013,368, 1878-1887].
PSI-7851 и его диастереоизомеры PSI-7976 и PSI-7977 метаболизируют до трифосфата PSI-7409, который и является ингибитором NS5B HCV полимеразы [Е. Murakami et al. Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 2010, 285 (45), 34337-34347].

Известны и другие аналоги Sovaldi® [Патент US 8334270 (2012). М.J. Sofia et al. Discovery of a β-D-20-Deoxy-20-r-fluoro-20-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis С Virus. J. Med. Chem. 2010, 53, 7202-7218.], в том числе циклогексил (S)-2-{[(2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)-4-фтор-3-гидрокси-4-метил-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропионат формулы 2.1, который, как и PSI-7851 и его фосфорные стереоизомеры PSI-7976 и PSI-7977 (Sovaldi®), также метаболизируют до трифосфата PSI-7409.

Исследованы и другие пролекарства, включающие фосфорамидатный остаток, для эффективного транспорта в печень. В частности, PSI-353661 продемонстрировал высокую ингибирующую активность (ЕС90=0,008 μМ ->1000-кратное увеличение активности по сравнению с аналогом гуанозина - β-D-2'-дезокси-2'-R-фтор-2'-β-С-метилгуанозин) и новый профиль резистентности, похожий на PSI-352938 [W. Clung et al. Discovery of PSI-353661, a Novel Purine Nucleotide. ACS Med. Chem. Lett. 2011. 2. 130-135].
Структурно-родственными пролекарствами являются IDX-184 (EC50=0,4 μM) [X.-J. Zhou. Et al. Safety and Pharmacokinetics of IDX184, a Liver-Targeted Nucleotide Polymerase Inhibitor of Hepatitis С Virus, in Healthy Subjects Antimicrob. Agents Chemother. 2011, 55, 76-81. J. Lalezari, et al. Short-Term Monotherapy with IDX184, a Liver-Targeted Nucleotide Polymerase Inhibitor, in Patients with Chronic Hepatitis С Virus Infection. Antimicrob. Agents Chemother. 2012. 56, 6372-6378.] и INX-08189 (BMS-986094, EC50=0.010 μM) [C. McGuigan et al. Phosphorodiamidates as a Promising New Phosphate Prodrug Motif for Antiviral Drug Discovery: Application to Anti-HCV Agents. J. Med. Chem. 2011, 54, 8632-8645. J.H. Vernachio et al. INX-08189, a phosphoramidate prodrug of 6-O-methyl-2'-C-methyl guanosine, is a potent inhibitor of hepatitis С virus replication with excellent pharmacokinetic and pharmacodynamic properties. Antimicrob. Agents Chemother. 2011. 55, 1843-1851]. Эти пролекарства производит идентичный трифосфат, но показали в клинике сердечно-сосудистую токсичность, связанную с INX-08189 [J.J. Arnold et al. Sensitivity of Mitochondrial Transcription and Resistance of RNA Polymerase II Dependent Nuclear Transcription to Antiviral Ribonucleosides. PLOS Pathog. 2012. 8, DOI: 10.1371/journal.ppat. 1003030]. Сильная сердечно-сосудистая токсичность, которая наблюдается у INX-08189, похоже, сократила интерес к разработке гуанозин нуклеозидов для лечения пациентов с HCV.

Следует отметить, что структура фосфорамидатного фрагмента оказывает существенное влияние на стабильность фосфорамидатных нуклеозидов в различных средах, на их фармакокинетику, биодоступность, распределение в органах тела и на избирательность их действия [М.J. Sofia et al. 2010. P. Wang et al. Phosphoramidate prodrugs of (-)-β-D-(2R,4R)-dioxolane-thymine (DOT) as potent anti-HIV agents. Antiviral Chem. Chemotherapy 2012, 22, 217-238. L. Bondada et al. Adenosine Dioxolane Nucleoside Phosphoramidates as Antiviral Agents for Human Immunodeficiency and Hepatitis В Viruses. ACS Med. Chem. Lett. 2013, 4, 747-751. M. Slusarczyk et al. Application of ProTide Technology to Gemcitabine: A Successful Approach to Overcome the Key Cancer Resistance Mechanisms Leads to a New Agent (NUC-1031) in Clinical Development. J. Med. Chem. 2014, 57, 1531-1542].
Подавляющее большинство наиболее эффективных и наименее цитотоксичных фосфорамидатных пролекарств, представляющих практический интерес, являются изопропиловыми эфирами (TDF, TAF, GS-9131, PSI-7851, PSI-7977, EIDD-02173), что объясняется, по-видимому, наиболее эффективной его адресной доставкой к очагам инфекции и эффективным метаболизмом в лекарство.
Несмотря на прогресс, достигнутый в последние годы в антивирусной терапии, создание новых пролекарств с улучшенными характеристиками и их использование в качестве химиотерапевтических средств для лечения вирусных заболеваний и рака являются весьма актуальными.
Авторы неожиданно обнаружили, что неизвестные ранее нуклеотиды, включающие N-[(S)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги общей формулы 1 и их стереоизомеры, изотопно-обогащенные аналоги, их фармацевтически приемлемые соли, гидраты, сольваты, кристаллические и поликристаллические формы являются эффективными пролекарствами для лечения вирусных и раковых заболеваний.

где:
n имеет значение 1 или 0;
Nuc представляет собой  или
или 
R1 представляет собой водород или метил;
R2, R3 представляют собой необязательно одинаковые заместители, выбранные из Н, F, Cl, CH3, ОН при условии, когда сплошная линия и сопровождающая ее пунктирная линия ( ) вместе представляют одинарную углерод-углеродную (С-С) связь, или R2 и R3 представляют собой водород при условии, когда сплошная линия и сопровождающая ее пунктирная линия () вместе представляют собой двойную углерод-углеродную связь (С=C);
) вместе представляют одинарную углерод-углеродную (С-С) связь, или R2 и R3 представляют собой водород при условии, когда сплошная линия и сопровождающая ее пунктирная линия () вместе представляют собой двойную углерод-углеродную связь (С=C);
R4 представляет собой заместитель, выбранный из R4.1- R4.5:

R3.6 представляет собой заместитель, выбранный из Н, F, Cl, CH3 или CF3;
R3.7 представляет собой водород, C1-C4-алкил или C3-C6-Циклоалкил;
X представляет собой O, CH2 или C=CH2;
Y представляет собой O, S, CH2 или НО-СН-группу при условии, когда непрерывная линия и сопровождающая ее пунктирная линия () вместе представляют собой одинарную углерод-углеродную (С-С) связь, или Y представляет собой СН-группу при условии, когда сплошная линия и сопровождающая ее пунктирная линия () вместе представляют собой двойную углерод-углеродную связь (С=С).
Ниже приведены определения различных терминов, используемых для описания данного изобретения. Эти определения применимы к терминам, как они использованы в данном описании и формуле изобретения, если иным не ограничены в конкретных случаях либо по отдельности, либо как часть большей группы.
Термин «пролекарство» относится к соединениям по изобретению, которые расщепляются химически или метаболически и становятся, путем сольволиза или в физиологических условиях, соединением по настоящему изобретению, которое фармацевтически активно в естественных условиях. Пролекарства часто имеют более высокую растворимость, тканевую совместимость, доставку или замедленное высвобождение у млекопитающих (Bungard, Н., Desing of products, pp. 7-9, 21-24, Elsevier, Amsterdam 1985). Пролекарства включают кислотные производные, хорошо известные специалистам в данной области техники, такие как, например, сложные эфиры, полученные реакцией исходного кислотного соединения с подходящим спиртом, или амиды, полученные реакцией соединения исходной кислоты с подходящим амином. Примеры пролекарств включают, но не ограничиваются ими, ацетат, формиат, бензоат или другие ацилированные производные спиртов или аминов функциональных групп в соединениях по настоящему изобретению.
Термин «циклоалкил» означает моновалентную насыщенную карбоциклическую группу, которая может быть моноциклической или мультициклической. Репрезентативные циклоалкильные группы включают в себя в качестве примеров, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и пр.
Термин «активный компонент» (лекарственное вещество) относится к физиологически активному веществу синтетического или иного (биотехнологического, растительного, животного, бактерицидного и так далее) происхождения, обладающему фармакологической активностью, которое является активным ингредиентом фармацевтической композиции.
Термин «лекарственный препарат» означает вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
Термин «терапевтический коктейль» представляет одновременно администрируемую комбинацию двух и более лекарственных препаратов, обладающих различным механизмом фармакологического действия и направленных на различные биомишени, участвующие в патогенезе заболевания.
Термин «фармацевтическая композиция» обозначает композицию, включающую в себя соединение формулы 1 и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного компонента, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного компонента, одного или в комбинации с другим активным компонентом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
Термин «инертный наполнитель», используемый в данном описании, относится к соединению, которое используют для получения фармацевтической композиции, и, как правило, безопасному, нетоксичному и ни биологически, ни иным образом нежелательному, и включает в себя вспомогательные вещества, которые являются приемлемыми для применения в ветеринарии, а также фармакологически приемлемыми для человеческого использования. Соединения по данному изобретению могут быть введены отдельно, но обычно их будут вводить в смеси с одним или более фармацевтически приемлемыми эксципиентами, разбавителями или носителями, выбранными с учетом предполагаемого пути введения и стандартно фармацевтической практики.
Термин «фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные. (Подробное описание свойств таких солей дано в Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19.) Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Термин «кристаллическая форма» означает структуру вещества, характеризующуюся упаковкой образующих ее молекул в один из видов кристаллической решетки.
Термин «поликристаллическая форма» означает структуру вещества, имеющую поликристаллическое строение, т.е. состоящую из множества мелких монокристаллов, т.е. кристаллитов определенной кристаллической формы.
Термин «терапевтически эффективное количество», используемый здесь, означает количество субстанции, пролекарства или лекарства, необходимое для уменьшения симптомов заболевания у субъекта. Доза субстанции, пролекарства или лекарства будет соответствовать индивидуальным требованиям в каждом конкретном случае. Эта доза может варьироваться в широких пределах в зависимости от многочисленных факторов, таких как тяжесть заболевания, подлежащего лечению, возраста и общего состояния здоровья пациента, других лекарственных средств, с помощью которых пациент проходит лечение, способа и формы введения и опыта лечащего врача. Для перорального введения суточная доза составляет приблизительно от 0,01 до 10 г, включая все значения между ними, в день в монотерапии и/или в комбинированной терапии. Предпочтительная суточная доза составляет примерно от 0,1 до 7 г в день. Как правило, лечение начинают с большой начальной «нагрузочной дозы», чтобы быстро уменьшить или устранить вирус, сопровождающей убывающую дозу до уровня, достаточного для предотвращения всплеска инфекции.
Термин «субъект» означает млекопитающее, которое включает, но не ограничивается ими, крупный рогатый скот, свиней, овец, куриц, индеек, буйволов, лам, страусов, собак, кошек и человека, предпочтительно субъектом является человек. Предполагается, что в способе лечения субъекта может быть любое из пролекарств общей формулы 1, его стереоизомер, изотопно-обогащенный аналог, его фармацевтически приемлемая соль, гидрат, сольват, кристаллическая и полиморфная форма, либо в сочетании их с другим соединением, в том числе с ингибитором NS5A HCV.
Термин «сольват» означает комплекс или агрегат, образуемый одной или более молекулами растворенного вещества, т.е. соединением согласно изобретению или его фармацевтически приемлемой солью и одной или более молекулами растворителя. Такие сольваты являются типичными твердыми кристаллами, имеющими, по существу, фиксированное молярное отношение растворенного вещества и растворителя. Репрезентативные растворители включают в себя, не ограничиваясь перечисленными, воду, этанол, изопропанол, уксусную кислоту и пр. Когда растворителем является вода, образуемый сольват представляет собой гидрат.
Настоящее изобретение относится к новому пролекарству, представляющему собой неизвестный ранее нуклеотид общей формулы 1, включающий N-[(S)-1-циклобутоксикарбонил]-фосфорамидатный фрагмент, его стереоизомеру, их изотопно-обогащенному аналогу, фармацевтически приемлемой соли, гидрату, сольвату, кристаллической или поликристаллической форме:
где: 
n имеет значение 1 или 0;
Nuc представляет собой  или
или 
R1 представляет собой водород или метил;
R2, R3 представляют собой необязательно одинаковые заместители, выбранные из Н, F, О, CH3, ОН при условии, когда сплошная линия и сопровождающая ее пунктирная линия () вместе представляют одинарную углерод-углеродную (С-С) связь, или R2 и R3 представляют собой водород при условии, когда сплошная линия и сопровождающая ее пунктирная линия () вместе представляют собой двойную углерод-углеродную связь (С=С);
R4 представляет собой заместитель, выбранный из R4.1-R4.5:

R3.6 представляет собой заместитель, выбранный из Н, F, Cl, CH3 или CF3;
R3.7 представляет собой водород, C1-C4-алкил или C3-C6-циклоалкил;
X представляет собой O, CH2 или C=CH2;
Y представляет собой O, S, СН2 или НО-СН-группу при условии, когда непрерывная линия и сопровождающая ее пунктирная линия () вместе представляют собой одинарную углерод-углеродную (С-С) связь, или Y представляет собой СН-группу при условии, когда сплошная линия и сопровождающая ее пунктирная линия () вместе представляют собой двойную углерод-углеродную связь (С=С).
Предпочтительными про лекарствам и являются соединения общих формул 1А и 1В и их стереоизомеры, а также изотопно-обогащенные аналоги, фармацевтически приемлемые соли, гидраты, сольваты, кристаллические или поликристаллические формы соединений общих формул 1А и 1В и их стереоизомеров.


где: R1, R2, R3, R4, X и Y имеют вышеуказанные значения.
Более предпочтительными пролекарствами являются:
(S)-циклобутил 2-((S)-(((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-илокси)метил)(фенокси)фосфориламино)-пропаноат (1А.1),
(S)-циклобутил 2-((R)-(((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-илокси)метил)(фенокси)фосфориламино)-пропаноат (1А.2),
(S)-циклобутил 2-{(S)-[(2S',3R,5S)-3-гидрокси-5-(5-метил-3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.1),
(S)-циклобутил 2-{(S)-[(2S,3S,4R,5S)-3-гидрокси-4-фтор-5-(5-метил-3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.2),
(S)-циклобутил 2-{(S)-[(2R,3R,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-3-гидрокси-4,4-дифтор-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.3),
(S)-циклобутил 2-{(S)-[(1R,3S,5S)-3-(2-амино-6-оксо-1,6-дигидро-пурин-9-ил)-5-гидрокси-2-метилен-циклопентилметокси]-фенокси-фосфориламино}-пропаноат (1В.4),
(S)-циклобутил 2-{(S)-[(2R,5S)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-[1,3]оксатиолан-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.5),
(S)-циклобутил 2-{(S)-[(2R,5S)-5-(4-амино-5-фтор-2-оксо-2H-пиримидин-1-ил)-[1,3]оксатиолан-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.6),
(S)-циклобутил 2-{(S)-[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.7),









Неожиданно новые пролекарства формул 1А.1 и 1А.2 оказались более эффективными, чем известные пролекарства TAF, использующиеся в настоящее время в комбинированной терапии ВИЧ инфицированных субъекте. Действительно, пролекарства формул 1А.1 и 1А.2 метаболизируют в TVF и дифосфата TVF в периферических мононуклеарных клетках крови (РВМС) с образованием более высокой концентрации и AUClast метаболитов, чем образуется этих метаболитов в сопоставимых условиях при метаболизме TDF и TAF. Как видно из Таблицы 1, при метаболизме пролекарства формулы 1А.1 Cmax и AUClast дифосфата TVF (лекарства) почти в 2 раза выше, чем значения этих параметров, наблюдаемых при метаболизме его изопропильного аналога TAF. Более высокие значения Cmax и AUClast (Таблица 1) наблюдаются и при метаболизме пролекарства формулы 1А.2 по сравнению со значениями этих параметров, наблюдаемых при метаболизме изопропильного аналога TAF.

Оценка антивирусной активности фумаратов соединений формулы 1А.1, 1А.2 и TAF в ВИЧ-тесте с использованием клеток SupT1, зараженных штаммом NL4.3 ВИЧ, несущим вирус GFP-репортер (NL4.3GFP), показала, что фумарат соединения формулы 1А.1 оказался наиболее эффективным (Таблица 2). Его активность и селективность в 1,4 раза выше активности и селективности TAF.

Неожиданно также новое пролекарство формулы 1В.7, его изотопно-обогащенный аналог, кристаллическая и поликристаллическая формы оказались более эффективными ингибиторами NS5B HCV, чем известные пролекарства - ингибиторы NS5B HCV, в том числе более эффективными, чем Sovaldi® и циклогексиловый эфир формулы 2.1.
Действительно, Sovaldi® по отношению к генотипу 1b (gT1b) HCV имеет ЕС50=0.045-0.170 μМ [http://www.hcvdruginfo.ca/downloads/HCV_Sofosbuvir.pdf] и ЕС90=0.59 μM, а новое пролекарство формулы 1В.7 имеет ЕС50=15.0-27.0 нМ и ЕС90=128.0 нМ (Таблица 3), т.е. новое пролекарство формулы 1В.7 более чем в три раза активнее, чем Sovaldi®.

Время полураспада пролекарства формулы 1В.7 в S9 фракции микросом печени человека составляет T1/2 hS9=0,05 ч, а у Sovaldi® соответственно T1/2 hS9=0.54 ч (Таблица 4), т.е. новое пролекарство формулы 1В.7 в 11 раз быстрее метаболизирует в S9 фракции микросом печени человека, чем Sovaldi®.

Кроме того, концентрация и AUC24 ч трифосфата PSI-7409, образующегося при метаболизме пролекарства формулы 1В.7 в печени крыс, составляют Сmax=3,224.0 нг/г и AUC24 ч=30,487.0 нг ч/г, в то время как при аналогичном метаболизме Sovaldi® имеет_Сmax=1,934.0 нг/г и AUC24 ч=16,796.0 нг ч/г (Таблица 5). Это свидетельствует о том, что новое пролекарство формулы 1В.7 почти в 2 раза более эффективно метаболизирует в печени в нужный трифосфат PSI-7409 (лекарство). Еще большую эффективность новое пролекарство формулы 1В.7 имеет по сравнению с известным циклогексиловым эфиром формулы 2.1 (Таблица 5), поскольку последний имеет: ЕС90=250.0 нМ, T1/2 hS9=1,4 ч, Сmax=557 нг/г и AUC24 ч=6,484.0 нг⋅ч/г.

Полученные результаты (эффект) являются неожиданными, поскольку пролекарство формулы 1 В.7, представляет собой циклобутиловый эфир, который не только значительно эффективнее своего аналога - циклогексилового эфира формулы 2.1, но и активнее другого аналога - циклопропилового эфира формулы 2.2 (ЕС90=73.0 нМ, ЕС90=410.0 нМ представлен в Таблице 3), который был специально получен изобретателями для большей наглядности неожиданного эффекта.

Неожиданность состоит в том, что в ряду циклоалкиловых эфиров наиболее эффективным оказался циклобутиловый эфир формулы А1.1 с промежуточным размером циклоалкила, а менее эффективными оказались эфиры с большим размером циклоалкила (известный циклогексиловый эфир формулы 2.1) и меньшим размером циклоалкила (специально полученный изобретателями циклопропиловый эфир формулы 2.2).
Приведенные выше данные убедительно подтверждают новизну изобретения и его изобретательский уровень (эффективность).
Предметом настоящего изобретения является фармацевтическая композиция, содержащая пролекарство общей формулы 1 или его стереоизомер, или их изотопно-обогащенный аналог, фармацевтически приемлемую соль, гидрат, сольват, кристаллическую или поликристаллическую форму, необязательно в комбинации с фармацевтически приемлемым наполнителем, носителем, добавкой, разбавителем для лечения вирусных инфекций и/или опухолевых заболеваний у млекопитающих.
Пролекарства общей формулы 1 могут быть приготовлены в виде самых разнообразных перорально вводимых лекарственных форм и носителей, пероральное введение может быть в форме таблеток, таблеток с покрытием, твердых и мягких желатиновых капсул, растворов, эмульсий, сиропа или суспензии. Соединения по настоящему изобретению являются эффективными при введении в виде суппозитория. Наиболее удобным способом введения обычно является пероральный с использованием обычной суточной схемы приема лекарственных доз, который можно регулировать в зависимости от тяжести заболевания и реакции пациента на противовирусное и противоопухолевое лекарство.
Пролекарство общей формулы 1, его стереоизомер, их изотопно-обогащенный аналог, их фармацевтически приемлемая соль, гидрат, сольват, кристаллическая или поликристаллическая форма вместе с одним или несколькими обычными эксципиентами, носителями или разбавителями могут быть представлены в форме фармацевтических композиций и их единичных доз. Фармацевтические композиции и стандартные лекарственные формы могут состоять из обычных ингредиентов в обычных пропорциях с или без дополнительных активных соединений и лекарственных форм. Фармацевтическая композиция может содержать любое соответствующее эффективное количество активного ингредиента, соразмерное с назначенной суточной дозой. Фармацевтические композиции могут быть использованы в виде твердых веществ, таких как таблетки или заполненные капсулы, в виде полутвердых порошков, препаратов с замедленным высвобождением или жидкостей, таких как суспензии, эмульсии или заполненные капсулы для перорального применения; или в форме суппозиториев для ректального или вагинального введения. Типичный препарат будет содержать примерно от 5% вес. до 95% вес. активного соединения или соединения. Термин «препарат» или «лекарственная форма» предназначен для включения как твердых, так и жидких композиций активного соединения, и специалисту в данной области техники будет понятно, что активный ингредиент может существовать в виде различных препаратов в зависимости от требуемой дозы и фармакокинетических параметров.
Препараты в твердой форме включают порошки, таблетки, пилюли, капсулы, суппозитории и диспергируемые гранулы. Твердый носитель может представлять собой одно или несколько веществ, которые могут также действовать как разбавители, корригенты, солюбилизаторы, смазывающие вещества, суспендирующие агенты, связующие вещества, консерванты, дезинтегрирующие таблетки агенты или инкапсулирующий материал. В порошках носитель обычно представляет собой тонкоизмельченное твердое вещество, которое представляет собой смесь с тонкоизмельченным активным компонентом. В таблетках активный компонент обычно смешивают с носителем, имеющим необходимую связывающую способность, в подходящих пропорциях и спрессовывают в желаемую форму желаемого размера. Подходящие носители включают, но не ограничиваются ими, карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлозу, натрийкарбоксиметилцеллюлозу, легкоплавкий воск, масло какао и тому подобное. Препараты в твердой форме могут содержать в дополнение к активному компоненту красители, ароматизаторы, стабилизаторы, буферы, искусственные и природные подсластители, диспергаторы, загустители, солюбилизирующие агенты и тому подобное.
Жидкие составы также пригодны для перорального введения. Жидкие лекарственные формы представляют собой эмульсии, сиропы, эликсиры и водные суспензии. Они включают твердые формы препаратов, которые предназначены для превращения в жидкие препараты непосредственно перед использованием. Эмульсии могут быть приготовлены в растворах, например, в водных растворах пропиленгликоля или могут содержать эмульгаторы, такие как лецитин, моноолеат сорбита или гуммиарабик. Водные суспензии могут быть приготовлены диспергированием тонкоизмельченного активного компонента в воде с вязкими материалами, такими как природные или синтетические камеди, смолы, метилцеллюлоза, натрийкарбоксиметилцеллюлоза и другими хорошо известными суспендирующими агентами.
Пролекарство общей формулы 1, его стереоизомер, их изотопно-обогащенный аналог, его фармацевтически приемлемая соль, гидрат, сольват, кристаллическая или поликристаллическая форма могут быть приготовлены для введения в виде суппозиториев. Низкоплавкий воск, такой как смесь глицеридов жирных кислот или масло какао, сначала расплавляют и активный компонент гомогенно диспергируют, например, перемешиванием. Расплавленную гомогенную смесь затем выливают в формы удобного размера, дают остыть и затвердеть.
Пролекарство общей формулы 1, его стереоизомер, их изотопно-обогащенный аналог, его фармацевтически приемлемая соль, гидрат, сольват, кристаллическая или поликристаллическая форма могут быть приготовлены для вагинального введения. Применение суппозиториев, тампонов, кремов, гелей, паст, пен или спреев, содержащих в дополнение к активному ингредиенту такие носители, которые известны в данной области техники, будет уместно.
Предметом настоящего изобретения является использование пролекарства общей формулы 1, его стереоизомера, или их изотопно-обогащенного аналога, его фармацевтически приемлемой соли, гидрата, сольвата, кристаллической или поликристаллической формы в производстве лекарственного средства для лечения вирусных и раковых заболеваний. Предполагается, что пролекарство, представленное общей формулой 1, его стереоизомер, или их изотопно-обогащенный аналог, его фармацевтически приемлемая соль, гидрат, сольват, кристаллическая или поликристаллическая форма, используемые в производстве лекарственного средства для лечения любого противовирусного и противоракового заболевания, описанного здесь, могут представлять собой любое из соединений общей формулы 1, выбранное из:
(S)-циклобутил 2-((S)-(((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-илокси)метил)(фенокси)фосфориламино)-пропаноат (1А.1),
(S)-циклобутил 2-((R)-(((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-илокси)метил)(фенокси)фосфориламино)-пропаноат (1А.2),
(S)-циклобутил 2-{(S)-[(2S,3R,5S)-3-гидрокси-5-(5-метил-3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.1),
(S)-циклобутил 2-{(S)-[(2S,3S,4R,5S)-3-гидрокси-4-фтор-5-(5-метил-3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.2),
(S)-циклобутил 2-{(S)-[(2R,3R,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-3-гидрокси-4,4-дифтор-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.3),
(S)-циклобутил 2-{(S)-[(1R,3S,5S)-3-(2-амино-6-оксо-1,6-дигидро-пурин-9-ил)-5-гидрокси-2-метилен-циклопентилметокси]-фенокси-фосфориламино}-пропаноат (1В.4),
(S)-циклобутил 2-{(S)-[(2R,5S)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-[1,3]оксатиолан-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.5),
(S)-циклобутил 2-{(S)-[(2R,5S)-5-(4-амино-5-фтор-2-оксо-2H-пиримидин-1-ил)-[1,3]оксатиолан-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.6),
(S)-циклобутил 2-{(S)-[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.7),
их стереоизомеров, изотопно-обогащенных аналогов, их фармацевтически приемлемых солей, гидратов, сольватов или кристаллических или поликристаллических форм либо по отдельности, либо в сочетании с другим соединением по настоящему изобретению. Лекарственное средство включает в себя, не ограничиваясь ими, любую из композиций, предусматриваемых по настоящему изобретению.
Предметом настоящего изобретения является способ комбинированного лечения и/или профилактики субъекта, нуждающегося в этом, причем указанный способ включает в себя введение терапевтически эффективного количества пролекарства, представленного общей формулой 1, его стереоизомера, или их изотопно-обогащенного аналога, его фармацевтически приемлемой соли, гидрата, сольвата, кристаллической или поликристаллической формы.
Предметом настоящего изобретения является способ комбинированного лечения и/или профилактики ВИЧ инфицированного субъекта, причем указанный способ включает в себя введение терапевтически эффективного количества пролекарства общей формулы 1А или 1В или их изотопно-обогащенного аналога, фармацевтически приемлемой соли, гидрата, сольвата, кристаллической или поликристаллической формы.
Предметом данного изобретения являются фармацевтическая композиция, содержащая в качестве пролекарства ингибитора NS5B HCV полимеразы циклобутил (S)-2-{(S)-[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.7), или его изотопно-обогащенный аналог, кристаллическую или поликристаллическую форму.
Более предпочтительной является фармацевтическая композиция, которая вместе с новым пролекарством формулы 1В.7, или его стереоизомером, изотопно-обогащенным аналогом, гидратом, сольватом, кристаллической или полиморфной формой дополнительно содержит терапевтически эффективное количество ингибитора NS5A HCV, выбранного из группы, включающей: Даклатасвир (Daclatasvir) (Daklinza, BMS790052) [С. Wang et al. 2012], Гепавивир (Hepavivir) (AV-4025) [A.V. Ivachtchenko et al. 2014. US 9428491 B2], AV-4067 и AV-4084 [Pat. Appl. US 14/845,333.], AV-4056 и AV-4058 [US 9428491 В2], Омбитасвир (Ombitasvir) (ABT-267) [C. Gardelli et al. Phosphoramidate Prodrugs of 20-C-Methylcytidine for Therapy of Hepatitis С Virus Infection. J. Med. Chem. 2014, 57, 2047-2057; WO 2010/144646,], Элбасвир (Elbasvir) (MK-8742) [Coburn C.A. et al. ChemMedChem. 2013, 8, 1930-1940; WO 2012/040923; WO 2012/041014]] или Велпатасвир (Velpatasvir) (VEL, GS-5816) [WO 2015110048 A1; http://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/208341 Origls000PharmR.pdf; http://www.gilead.com/~/media/files/pdfs/medicines/liver-disease/epclusa/epclusa_pi.pdf].









Предметом настоящего изобретения является способ комбинированного лечения и/или профилактики субъекта, нуждающегося в этом, причем указанный способ включает введение субъекту терапевтически эффективного количества нуклеотида общей формулы 1, включающего N-[(S)-1-циклобутоксикарбонил]-фосфорамидатный фрагмент, или его стереоизомера, или их изотопно-обогащенного аналога, его фармацевтически приемлемой соли, гидрата, сольвата, кристаллической или поликристаллической формы или фармацевтической композиции по данному изобретению, которая может включать дополнительно терапевтически эффективное количество одного или нескольких других противовирусных или противораковых агентов, в котором используют параллельное или альтернативное введение агентов. Понятно, что время между последовательным введением агентов может находиться в любом временном диапазоне.
Предметом настоящего изобретения является также способ комбинированного лечения и профилактики ВИЧ инфицированного субъекта, причем указанный способ включает введение субъекту терапевтически эффективного количества пролекарства формулы 1В.1, его стереоизомера, изотопно-обогащенного аналога, его фармацевтически приемлемой соли, гидрата, сольвата, кристаллической или поликристаллической формы или фармацевтической композиции по данному изобретению и терапевтически эффективного количества одного или нескольких противо-ВИЧ препаратов, в котором используют параллельное или альтернативное введение препаратов. Понятно, что время между последовательным введением препаратов может находиться в любом диапазоне.
Предметом настоящего изобретения является также способ лечения инфицированного ВГС субъекта, нуждающегося в этом, причем указанный способ включает введение субъекту терапевтически эффективного количества пролекарства формулы 1В.7, его стереоизомера, изотопно-обогащенного аналога, его фармацевтически приемлемой соли, гидрата, сольвата, кристаллической или поликристаллической формы или фармацевтической композиции по данному изобретению и терапевтически эффективного количества другого противовирусного препарата - ингибитора NS5A ВГС, в котором используют параллельное или альтернативное введение агентов. Понятно, что время между последовательным введением агентов может находиться в любом диапазоне.
Когда пролекарство общей формулы 1, его стереоизомер, изотопно-обогащенный аналог, его фармацевтически приемлемую соль, гидрат, сольват, кристаллическую или поликристаллическую форму вводят в комбинации с другим антивирусным или противораковым агентом, активность может быть увеличена по сравнению с исходным пролекарством. Когда лечение комбинированное, то введение препаратов может быть одновременным или последовательным по отношению к пролекарству, представленному общей формулой 1. Понятие «одновременное введение», используемое здесь, таким образом, включает в себя введение агентов в то же время или в разное время. Прием двух или более агентов в то же время может быть достигнут с помощью одной препаративной формы, содержащей два или более активных ингредиентов, или, по существу, одновременного введения двух или более лекарственных форм с одним активным агентом. Следует понимать, что ссылки здесь на лечение распространяются и на профилактику. Кроме того, термин «лечение» вирусной инфекции, как он использован здесь, включает также лечение или профилактику заболевания или состояния, связанного с опосредованной вирусной инфекцией, или их клинических симптомов.
Настоящее изобретение далее будет описано в связи с определенными вариантами осуществления, которые не предназначены для ограничения его объема. Напротив, настоящее изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут быть включены в объем формулы изобретения. Таким образом, следующие примеры, которые включают в себя конкретные варианты, иллюстрируют, но не ограничивают настоящее изобретение.
Пример 1. Общий протокол синтеза пролекарств общей формулы 1А (Схема 1).
Схема 1

где R1 имеет вышеуказанное значение.
К суспензии 3,63 г (10 ммоль) [(R)-2-(6-амино-пурин-9-ил)-1-метил-этоксиметил]фосфоновой кислоты монофениловый эфир (3) [WO 2013116720] в 14 мл сульфолана и 12 мл дихлорметана прикапывали при перемешивании 3 мл (40 ммоль) тионилхлорида. Затем смесь перемешивали с обратным холодильником при температуре 50-55°C под слабым током аргона в течение 15 ч. После этого к колбе подключали вакуум (мембранный насос) и в течение 2 часов удаляли летучие компоненты при 50-55°C. Массе давали охладиться до 30°C и при перемешивании вливали смесь 10 мл дихлорметана и 40 мл сухого ацетонитрила. Реакционную массу, содержащую хлорид (4), охлаждали до (-60)-(-50)°C и приливали раствор 1,546 г (12 ммоль) циклобутилового эфира L-аланина (5) и 4,172 мл (30 ммоль) триэтиламина в 6 мл ацетонитрила. Смеси давали медленно нагреться до комнатной температуры, затем разбавляли 100 мл дихлорметана и наносили на ~100 мл силикагеля на стеклянном фильтре. Далее проводили выделение продукта методом сухой флэш-хроматографии, элюируя сначала дихлорметаном, 30% раствором ацетона в дихлорметане и затем чистым тетрагидрофураном. Получали 2 г (S)-циклобутил 2-(((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-илокси)метил)(фенокси)фосфориламино)-пропаноата (1А), представляющего собой смесь фосфорных стереоизомеров формул 1А.1 и 1А.2. Разделение стереоизомеров формул 1А.1 и 1А.2 проводили методом ВЭЖХ на оптической колонке Phenomenex Amylose-2 AXIA-Pac 250×21.20 мм в изократической системе AcCN:EtOH:HCOOH=200:20:0.5 со скоростью потока 20 мл/мин с УФ-детектором 254 нм. Полученные стереоизомеры формул 1А.1 и 1А.2 перекристаллизовывали с эквимолярным количеством фумаровой кислоты из 100 мл ацетонитрила. Получали Sp-изомер: (S)-циклобутил 2-((S)-(((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-илокси)метил)(фенокси)фосфориламино)пропаноат фумарат (1А.1), LC-MS (ESI) 489 (М+Н)+. 1Н NMR (DMSO-d6, 300 MHz) δ 8.14 (s, 1Н), 8.10 (s, 1H), 7.30 (m, 2H), 7.19 (s, 2H), 7.14 (m, 1H), 7.06 (m, 2H), 6.63 (s, 2H), 5.64 (t, J=11.1 Hz, 1H), 4.86 (p, J=7.2 Hz, 1H), 4.27 (dd, J1=14.4 Hz, J2=3.0 Hz, 1H), 4.14 (dd, J1=14.4 Hz, J2=6.6 Hz, 1H), 3.85 (m, 4H), 2.23 (m, 2H), 1.94 (m, 2H), 1.72 (m, 1H), 1.59 (m, 1H), 1.13 (d, J=6.9 Hz, 3H), 1.07 (d, J=6.6 Hz, 3H). 31P NMR (DMSO-d6, 121.5 MHz) δ 22.05 и Rp-изомер: (S)-циклобутил 2-((R)-(((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-илокси)метил)(фенокси)фосфориламино)пропаноат фумарат (1А.2), LC-MS (ESI) 489 (М+Н)+. 1Н NMR (DMSO-d6, 300 MHz) δ 8.14 (s, 1Н), 8.12 (s, 1H), 7.34 (m, 2H), 7.21 (s, 2H), 7.15 (m, 1H), 7.11 (m, 2H), 6.63 (s, 2H), 5.53 (dd, J1=12.0 Hz, J2=10.5 Hz, 1H), 4.82 (p, J=7.5 Hz, 1H), 4.29 (dd, J1=14.4 Hz, J2=3.6 Hz, 1H), 4.20 (dd, J1=14.4 Hz, J2=5.7 Hz, 1H), 3.98 (m, 1H), 3.86 (m, 3H), 2.21 (m, 2H), 1.91 (m, 2H), 1.69 (m, 1H), 1.57 (m, 1H), 1.13 (d, J=6.9 Hz, 3H), 1.05 (d, J=6.3 Hz, 3H). 31P NMR (DMSO-d6, 121.5 MHz) δ 22.86.
Пример 2. Протокол синтеза (S)-циклобутил 2-(пентафторфенокси-фенокси-фосфориламино)-пропаноатов (7, 7.1) (Схема 2).
Схема 2

К раствору 14.4 г (80.2 ммоль, 1 экв) циклобутил L-аланина гидрохлорида (5.1) [WO 2014033617 A1] в 214 мл DCM добавляли 16.9 г (80.2 ммоль, 1 экв) фенил дихлорфосфата. Смесь охлаждали до (-75)-(-70)°C и добавляли при (-75)-(-70)°C раствор 16.2 г (160.4 ммоль, 2 экв) триэтиламина в 16 мл дихлорметана. Смесь перемешивали при -70°C 30 мин, затем нагревали до -20°C. К реакционной смеси, содержащей хлорид 6, добавляли при (-20)-(-10)°C раствор 14.6 г (79.4 ммоль, 0.99 экв) пентафторфенола в 105 мл дихлорметана, затем добавляли при (-20)-(-10)°C раствор 8.1 г (80.2 ммоль, 1 экв) триэтиламина в 8 мл дихлорметана и смесь перемешивали при комнатной температуре в течение ночи. Полученную смесь упаривали в вакууме досуха и добавляли 500 мл этилацетата и 500 мл воды. Органический слой отделяли и промывали 200 мл воды, затем промывали 5% водным раствором NaНCO3, насыщенным раствором соли, сушили над Na2SO4 и упаривали в вакууме досуха. К остатку, представляющему собой (S)-циклобутил 2-(пентафторфенокси-фенокси-фосфориламино)-пропаноат формулы 7, прибавляли 200 мл смеси гексан-этилацетата (6:1) и смесь перемешивали при комнатной температуре в течение ночи, затем фильтровали, промывали 50 мл смеси гексан-этилацетат (6:1) и сушили на воздухе. Получали 16.7 г продукта, который перекристаллизовывали из 500 мл смеси гексана и этилацетата (4:1). Получали 13.8 г (S)-циклобутил 2-((R)-(пентафторфенокси-фенокси-фосфориламино)-пропаноата (7.1). 1Н NMR (400 MHz, CDCl3) δ 7.42-7.32 (m, 2Н), 7.28-7.19 (m, 3Н), 5.02 (p, J=7.6Hz, 1Н), 4.23-4.1 (m, 1Н), 4.01-3.88 (m, 1H), 2.42-2.3 (m, 2H), 2.14-1.99 (m, 2H), 1.89-1.77 (m, 1H), 1.72-1.59 (m, 1H), 1.48 (d, J=7.2Hz, 3H).
В аналогичных условиях, но исходя из циклопропил L-аланина гидрохлорида (8), получали (S)-циклопропил 2-(пентафторфенокси-фенокси-фосфориламино)-пропаноата 9 и (S)-циклопропил 2-((R)-(пентафторфенокси-фенокси-фосфориламино)-пропаноат (9.1). 1Н NMR (400 MHz, CDCl3) δ 0.71-0.77 (m, 4Н), 1.45 (d, J=6.8 Hz, 3Н), 3.98 (m, 1H), 4.17 (m, 1H), 7.25 (m, 3H), 7.36 (m, 2H).

Пример 3. Общий протокол синтеза пролекарств общей формулы 1В (Схема 3).
Схема 3
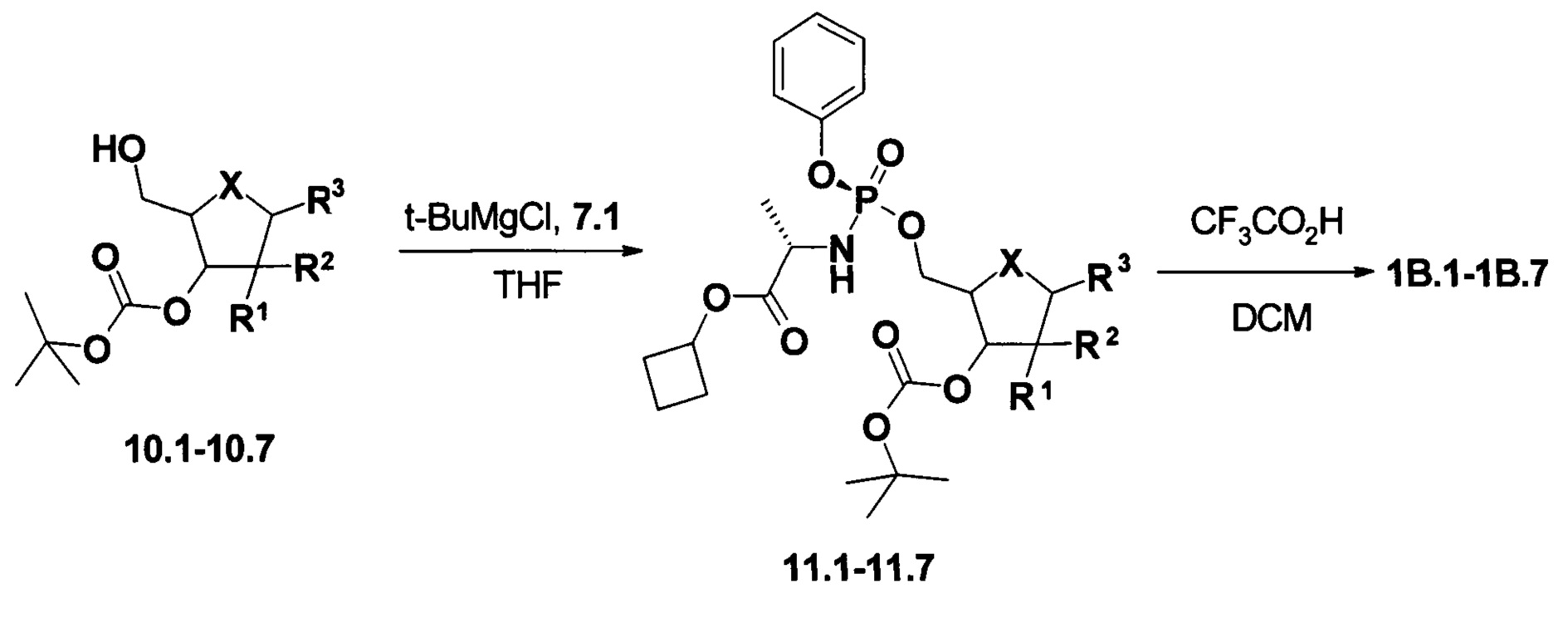
К раствору 5 г (13.9 ммоль, 1 экв) трет-бутил (2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-(гидроксиметил)-4-метил-4-фтортетрагидрофуран-3-илкарбоната (10.7) в 165 мл тетрагидрофурана добавляли 1 М раствор (31.3 мл, 31.3 ммоль, 2.25 экв) трет-бутилмагний хлорида под аргоном и перемешивали полученную смесь 30 мин. при комнатной температуре. К полученной смеси прибавляли раствор 7.8 г (16.7 ммоль, 1.2 экв) (S)-циклобутил 2-((R)-(пентафторфенокси-фенокси-фосфориламино)-пропаноата (7.1) в 30 мл тетрагидрофурана при перемешивании и 0-5°C. Полученную реакционную массу перемешивали 24 ч при комнатной температуре под аргоном, затем постепенно добавляли 10 мл метанола и концентрировали в вакууме. Остаток растворяли в 500 мл этилацетата, промывали 5%-ной лимонной кислотой, раствором NaHCO3, насыщенным раствором соли, сушили над Na2SO4 и выпаривали на роторном испарителе досуха. Получали 12,7 г (S)-циклобутил 2-((R)-(((2R,3R,4R,5R)-3-(трет-бутоксикарбонилокси)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-4-метил-4-фтортетрагидрофуран-2-ил)метокси)(фенокси)фосфориламино)пропаноата (11.7) в виде желтого масла. 1Н NMR (400 MHz, CDCl3) δ 8.77 (s, 1Н), 7.52 (d, J=8Hz, 1H), 7.39-7.29 (m, 2H), 7.25-7.13 (m, 3H), 6.19 (d, J=18.4Hz, 1H), 5.52 (d, J=8Hz, 1H), 5.08-4.93 (m, 2H), 4.63-4.53 (m, 1H), 4.4-4.25 (m, 2H), 4.08-3.91 (m, 2H), 2.42-2.28 (m, 2H), 2.13-1.97 (m, 2H), 1.88-1.75 (m, 1H), 1.71-1.58 (m, 1H), 1.52 (s, 9H), 1.46-1.36 (m, 6H). К раствору 8,9 г (13.8 ммоль, 1 экв) (S)-циклобутил 2-((R)-(((2R,3R,4R,5R)-3-(трет-бутоксикарбонилокси)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-4-метил-4-фтортетрагидрофуран-2-ил)метокси)(фенокси)фосфориламино) пропаноата (11.7) в 120 мл дихлорметана добавляли 120 мл трифторуксусной кислоты при (-10)-(0)°C. Смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь упаривали на роторном испарителе досуха. Остаток растворяли в 500 мл дихлорметана, добавляли 5%-ный водный раствор Na2CO3 до pH ~8. Органический слой отделяли, сушили над Na2SO4, фильтровали и выпаривали в вакууме на роторном испарителе досуха. Продукт очищали с помощью колоночной хроматографии (силикагель, этилацетат:гексан = 1:1-2:11:0). Продукт повторно очищали с помощью колоночной хроматографии (силикагель, дихлорметан:метанол = 19:1-9:1). Получали 5,4 г (S)-циклобутил 2-((S)-(((2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-гидрокси-4-метил-4-фтортетрагидрофуран-2-ил)метокси)(фенокси)фосфориламино)пропаноата (1В.7). LC-MS (ESI) 542 (М+Н)+. 1Н ЯМР (400 МГц, ДМСО-d6) δ 11.51 (brs, 1Н), 7.56 (d, J=8.0 Hz, 1Н), 7.38 (m, 2H), 7.23 (m, 2H), 7.19 (m, 1H), 6.03 (m, 2H), 5.84 (d, J=6.8 Hz, 1H), 5.55 (dd, J1=8.0 Hz, J2=1.2 Hz, 1H), 4.85 (p, J=7.2 Hz, 1H), 4.37 (m, 1H), 4.27 (m, 1H), 4.01 (m, 1H), 3.83 (m, 2H), 2.23 (m, 2H), 1.95 (m, 2H), 1.71 (m, 1H), 1.56 (m, 1H), 1.25 (d, J=22.8 Hz, 3H), 1.23 (d, J=6.8 Hz, 3H).
Перекристаллизация пролекарства формулы 1B.7 из различных растворителей приводит к получению поликристаллических или кристаллической форм. При перекристаллизации из смеси этилацетата с метил-трет-бутиловым эфиром (1:1), этанола, этилацетата и смеси уксусной кислоты с водой получают пролекарство формулы 1В.7 в поликристаллических формах, включающих в основном ромбическую фазу с параметрами элементарной ячейки а=28.1056 (8) А, b=16.8998(4) А, с=5.25380(12) А и моноклинную фазу с параметрами элементарной ячейки а=16.2770(6) А, b=16.9117(8) А, с=5.20429(15) А, β=117.822(2)°.
При перекристаллизации пролекарства формулы 1В.7 из смеси диметилсульфоксида с водой получают вещество в белой кристаллической форме, состоящей из ромбической фазы с параметрами элементарной ячейки а=28.1056(8) А, b=16.8998(4) А, с=5.25380(12) А.
Растворимость кристаллической и поликристаллических форм после перекристаллизации из различных растворителей при pH=2 и pH=7 близка и составляет 0,18-0,25 мг/мл. Исключение составляет поликристаллический образец, полученный при перекристаллизации из диметилсульфоксида, растворимость которого несколько выше и имеет значение 0,63-0,67 мг/мл (Таблица 6).

Аналогично 1В.7 получали (S)-циклобутил 2-{(S)-[(2S,3R,5S)-3-гидрокси-5-(5-метил-3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.1), LC-MS (ESI) 524 (М+Н)+; (S)-циклобутил 2-{(S)-[(2S,3S,4R,5S)-3-гидрокси-4-фтор-5-(5-метил-3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.2), LC-MS (ESI) 542 (М+Н)+; (S)-циклобутил 2-{(S)-[(2R,3R,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-3-гидрокси-4,4-дифтор-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.3), LC-MS (ESI) 545 (М+Н)+ и (S)-циклобутил 2-{(S)-[(1R,3S,5S)-3-(2-амино-6-оксо-1,6-дигидро-пурин-9-ил)-5-гидрокси-2-метилен-циклопентилметокси]-фенокси-фосфориламино}-пропаноат (1В.4), (S)-циклобутил 2-{(S)-[(2R,5S)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-[1,3]оксатиолан-2-илметокси]-фенокси-фосфориламино-пропаноат (1В.5), LC-MS (ESI) 511 (М+Н)+ и (S)-циклобутил 2-{(S)-[(2R,5S)-5-(4-амино-5-фтор-2-оксо-2H-пиримидин-1-ил)-[1,3]оксатиолан-2-илметокси]-фенокси-фосфориламино}-пропаноат (1В.6), LC-MS (ESI) 529 (M+H)+.
Пример 4. Протокол синтеза пролекарства (S)-циклопропил 2-((S)-(((2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-гидрокси-4-метил-4-фтор-тетрагидрофуран-2-ил)метокси)-(фенокси)-фосфориламино)-пропаноата (2.2) (Схема 4).
Схема 4

Циклопропиламин (12: 4,06 мл, 58,8 ммоль) и Boc-L-аланин (22,2 г, 58,8 ммоль) растворяют в 250 мл хлороформа и при охлаждении льдом прибавляют изоамил нитрит (7,9 мл, 58,8 ммоль). Смесь перемешивают 16 ч, не убирая охлаждение, упаривают досуха и хроматографируют на силикагеле (элюент этилацетат: гексан 1:8). Получают 7,68 г (57%) смеси циклопропилового эфира формулы 13 и аллилового эфира формулы 14, соотношение 1:4 (по данным 1Н ЯМР). Полученную смесь эфиров формул 13 и 14 растворяют в 120 мл ацетонитрила и под аргоном добавляют трифенилфосфин (446 мг, 1,7 ммоль) и Pd(PPh3)4 (984 мг, 0,85 ммоль), раствор охлаждают льдом и добавляют раствор пирролидина (2,49 г, 35 ммоль) в 30 мл ацетонитрила. Смесь перемешивают под аргоном при 0-4°C в течение 16 ч, затем упаривают досуха и хроматографируют на силикагеле (элюент этилацетат: гексан 1:8). Получают 1,38 г (S)-циклопропил 2-(трет-бутоксикарбониламино)пропаноат (15). 1Н ЯМР (CDCl3, 400 МГц) δ 5,04 (уш. с, 1Н), 4,26 (м, 1Н), 4,19 (м, 1Н), 1,46 (с, 9Н), 1,37 (д, J=7,2 Гц, 3Н), 0,74 (м, 4Н).
К раствору соединения формулы 15 (3,5 г, 15,3 ммоль) в 10 мл диоксана добавляют 20 мл 3 М раствора HCl в диоксане. Смесь перемешивают 2 ч при комнатной температуре, затем упаривают досуха. Получают (S)-циклопропил 2-амино-пропаноат гидрохлорид (16) с количественным выходом (2,53 г). 1Н ЯМР (ДМСО-d6, 300 МГц) δ 8,65 (уш. с, 3Н), 4,19 (м, 1Н), 3,99 (к, J=6,9 Гц, 1Н), 1,39 (d, J=6,9 Гц, 3Н), 0,73 (м, 4Н).
К охлажденному до -70°C раствору соединения формулы 16 (2,53 г, 15,3 ммоль) и фенилдихлорфосфата (3,23 г, 15,3 ммоль) в 50 мл дихлорметана добавляют по каплям раствор триэтиламина (4,26 мл, 30,6 ммоль) в 10 мл дихлорметана. Затем охлаждение убирают и дают реакционной смеси нагреться до -10°C, после чего прикапывают отдельно приготовленную смесь пентафторфенола (2,82 г, 15,3 ммоль) и триэтиламина (2,13 мл, 15,3 ммоль) в 15 мл дихлорметана. По окончании прибавления реакционную массу перемешивают 12 ч при комнатной температуре, после чего упаривают и остаток обрабатывают 50 мл бензола. Осадок отфильтровывают и промывают 15 мл бензола. Фильтрат промывают насыщенным раствором гидрокарбоната натрия, сушат над сульфатом натрия и упаривают. К остатку прибавляют смесь этилацетат: гексан 1:4 из расчета 8 мл на 1 г вещества и интенсивно перемешивают в течение 16 ч. Осадок отфильтровывают и перекристаллизовывают из смеси этилацетат: гексан 1:4. Получают 1,02 г (14%) (S)-циклопропил 2-((S)-(перфторфенокси)-(фенокси)-фосфориламино)-пропаноата (17). LC-MS (ESI) 452 (М+Н)+. 1Н ЯМР (CDCl3, 300 МГц) δ 7,38 (м, 2Н), 7,26 (м, 3Н), 4,17 (м, 1Н), 3,96 (м, 1H), 1,46 (д, J=7,2 Гц, 3Н), 0,74 (м, 4Н).
К раствору трет-бутил (2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-2-(гидроксиметил)-4-метил-4-фтор-тетрагидрофуран-3-ил карбоната (820 мг, 1,85 ммоль) в 25 мл сухого ТГФ при охлаждении льдом прикапывают 1 М раствор t-BuMgCl в ТГФ (4 мл, 0,4 ммоль). Охлаждение убирают и реакционную массу перемешивают полчаса при комнатной температуре. Затем снова охлаждают льдом и прикапывают раствор соединения формулы 17 (1,02 г, 2,18 ммоль) в ТГФ. Реакционную массу перемешивают 12 ч при комнатной температуре, после чего обрабатывают насыщенным раствором хлорида аммония. Органическую фазу отделяют, водную экстрагируют дихлорметаном. Объединенные экстракты сушат над сульфатом натрия и фильтруют. Получают ~1,16 г (S)-циклопропил 2-((S)-(((2R,3R,4R,5R)-3-(трет-бутоксикарбонилокси)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-4-метил-4-фтор-тетрагидрофуран-2-ил)метокси)-(фенокси)-фосфориламино)-пропаноата (18), который использовали в следующей стадии без дополнительной очистки. LC-MS (ESI) 628 (М+Н)+.
К раствору соединения формулы 18 (~1,16 г, 1,85 ммоль) в 20 мл дихлорметана при охлаждении льдом добавляют 20 мл трифторуксусной кислоты. Реакционную массу перемешивают 3 ч, после чего концентрируют в вакууме. Остаток растворяют в дихлорметане, добавляют воду и нейтрализуют гидрокарбонатом натрия. Органический слой отделяют, сушат над сульфатом натрия и упаривают досуха. Остаток хроматографируют на силикагеле (хлороформ : метанол), а затем прекристаллизовывают из смеси этилацетат: МТБЭ. Получают 505 мг (S)-циклопропил 2-((S)-(((2R,3R,4R,5R)-5-(2,4-диоксо-3,4-дигидропиримидин-1(2H)-ил)-3-гидрокси-4-метил-4-фтор-тетрагидрофуран-2-ил)метокси)-(фенокси)-фосфориламино)-пропаноата (2.2). (52%). LC-MS (ESI) 528 (М+Н)+. 1Н ЯМР (ДМСО-d6, 400 МГц) δ 11,24 (уш. с, 1Н), 7,56 (д, J=8,1 Гц, 1Н), 7,38 (м, 2Н), 7,20 (м, 3Н), 6,05 (м, 2Н), 5.86 (м, 1Н), 5,54 (д, J=8,1 Гц, 1Н), 4,36 (м, 1Н), 4,23 (м, 1Н), 4,02 (м, 2Н), 3,82 (м, 2Н), 1,25 (д, J=22,2 Гц, 3Н), 1,21 (д, J=6,6 Гц, 3Н), 0,66 (м, 2Н), 0,57 (м, 2Н).
Пример 5. Получение фармацевтической композиции в виде таблетки.
Крахмал (1600 мг), молотую лактозу (1600 мг), тальк (400 мг) и 1000 мг пролекарства формул 1А.1, 1В.4, 1В.5, 1В.6 или 1В.7 смешивали друг с другом и прессовали в бар. Полученный брусок измельчали в гранулы и просеивали через сито, чтобы собрать гранулы размером 14-16 меш. Полученные таким образом гранулы были сформированы в таблетки подходящей формы весом 300 или 600 мг каждая.
Пример 6. Получение фармацевтической композиции в виде капсул.
Пролекарство, формул 1А.1, 1В.4, 1В.5, 1В.6 или 1В.7 тщательно смешивали с порошком лактозы в соотношении 2:1. Полученную порошкообразную смесь упаковывали в желатиновые капсулы подходящего размера по 300 или 600 мг в каждой капсуле.
Пример 7. Получение фармацевтической композиции в форме композиций для внутримышечных, внутрибрюшинных или подкожных инъекций.
Смешивали 500 мг пролекарства формул 1А.1, 1В.4, 1В.5, 1В.6 или 1В.7, 300 мг хлорбутанола, 2 мл пропиленгликоля и 100 мл воды для инъекций. Полученный раствор фильтровали, помещали в ампулы по 5 мл и запечатывали.
Пример 8. Определение параметров метаболизма пролекарств формул 1А.1, 1А.2 и прототипа TAF в периферических мононуклеарных клетках крови (РВМС) человека.
Исходные растворы тестируемых соединений формул 1А.1, 1А.2 и TAF были приготовлены в DMSO (Sigma) и хранились на -20°C. РВМС были выделены из крови человека центрифугированием в градиенте Ficoll-Paque Premium (GE Healthcare), до использования хранились в жидком азоте. РВМС сажали в 24-луночные планшеты (Greiner Bio-one) по 1,5 млн. клеток в лунку (4,2 млн/мл) в среде RPMI-1640, содержащей 2 мМ L-глутамина, 0,11 мг/мл пирувата натрия, заменимые и незаменимые аминокислоты, 50 Ед/мл Пенициллина, 50 мкг/мл. Стрептомицина (все реактивы ПанЭко) и 5% HI (Heat Inactivated) фетальной бычьей сыворотки (HyClone). Инкубировали в течение ночи при 37°C и 5% CO2. На следующий день к клеткам добавляли тестируемые и референсные соединения в финальной концентрации 30 мкМ. Инкубировали клетки с соединениями при 37°C и 5% СO2. После 2, 4, 8, 24, 48 и 72 часов инкубации неприкрепившиеся клетки со средой переносили в 1,5 мл пробирки (Eppendorf), центрифугировали 5 минут при 1000 g и удаляли среду. Клетки промывали 1 мл фосфатным буфером (Gibco), лизировали 200 мкл охлажденного до -20°C 70% метанола. Клетки, прикрепившиеся к лункам планшета, промывали 1 мл PBS (Gibco) и лизировали 200 мкл охлажденного до -20°C 70% метанола. Лизаты прикрепившихся и неприкрепившихся клеток из соответствующих лунок объединяли и перемешивали.
Анализ клеточных лизатов на содержание тенофовира (TVF) и дифосфата тенофовира (DP-TVF) проводили методом UPLC-MS/MS с использованием хроматографа 1290 UPLC System (Agilent) и масс-спектрометра с тройным квадруполем QTrap5500 System (АВ Sciex). Разделение аналитов проводили на колонке Thermo Hypercarb (50×3.0 мм, 5 мкм, Thermo Scientific) в подвижной фазе следующего состава: А - 0,5% аммиак в 25 мМ ацетата аммония и В - 0,5% аммиак в 25 мМ ацетата аммония: 2-пропанол: метанол (1:1:3) при скорости потока 0,8 мл/мин. В качестве источника ионов использовали электрораспыление (TurboIonSpray) в режиме регистрации отрицательных ионов. Детектирование аналитов проводили в режиме MRM по переходам для TVF 286>107, 286>79, 286>63 m/z, для DP-TVF 446>348, 446>176, 446>158, 446>79 m/z. Обработку хроматограмм проводили в Analyst 1.5.2 Software (АВ Sciex). Концентрацию TVF и дифосфата TVF в клеточных лизатах оценивали по калибровочным кривым, полученным с помощью стандартных образцов TVF и DP-TVF в 70% метаноле. Полученные результаты представлены в Таблице 1.
Пример 9. Определение анти-ВИЧ активности пролекарств общей формулы 1 и прототипа (TAF).
Определение антивирусной активности тестируемых соединений проводили на линии Т-лимфоцитов, SupT1. Клетки были инфицированы ВИЧ штаммом NL4.3, несущим ген зеленого флуоресцентного белка (NL4.3-GFP). Препарат вируса получали путем трансфекции клеток 293Т провирусной ДНК. Через 48 часов после трансфекции препарат замораживали и хранили до использования. Для повышения эффективности инфекции, суспензию клеток SupT1 осаждали из инфекционной смеси центрифугированием. Тестируемые вещества добавляли к клеткам непосредственно перед добавлением вируса. Через 2 часа инкубации инфекционную смесь заменяли свежей культуральной средой с тестируемыми препаратами. Эффективность инфекции определяли через 45 часов путем подсчета процента флуоресцирующих клеток, по сравнению с неинфицированными клеточными культурами. Цитотоксичность тестируемых соединений определяли параллельно на той же, но не инфицированной клеточной линии SupT1, используя реагент ХТТ. Использовали серийные десятикратные разведения препаратов (начиная от 10 мкМ для определения антивирусной активности, или от 100 мкМ - для цитотоксичности). В качестве негативного контроля использовали 0,1% ДМСО. Были рассчитаны значения EC50, СС50 и СИ (селективный индекс). Качество тестов определяли при помощи следующих контролей: отношение сигнала к фону, интегразный ингибитор ралтегравир (1 мкМ), а также воспроизводимость теста. Контролем для определения цитотоксичности служил препарат эметин (0,03, 0,09 и 0,2 мкМ). Полученные результаты представлены в Таблице 2.
Пример 10. Определение анти-HCV активности и цитотоксичности пролекарства формулы 1В.7 и соединения формулы 2.2 и Sovaldi®.
Для определения противовирусной активности тестируемых композиций, включающих пролекарства, использовалась клеточная линия гепатоцеллюлярной карциномы человека Huh7, стабильно трансфецированная репликоном вируса гепатита C (HCV). По 50 мкл клеточной суспензии в полной культуральной среде (DMEM IX, Cellgro; кат.# 10-013-CV) переносили в 96-луночные плашки с итоговой плотностью 7500 клеток на лунку. Серийное разведение тестируемых пролекарств готовили из свежеприготовленного 200-кратного исходного раствора в ДМСО с 11 концентрационными точками с шагом разведения 3 от 20 нМ в полной среде и использовали в качестве 2-кратных растворов. Не менее чем через 4 часа после посадки клеток добавляли по 50 мкл растворов серийных разведений пролекарств. Конечная концентрация пролекарств составляла от 10 нМ до 0.1 пМ, а ДМСО - 0.5%. Плашку с клетками инкубировали в течение 3 дней при 37°C в увлажненной атмосфере 5% CO2. После инкубации среда была удалена переворачиванием плашки с аккуратным встряхиванием. Клетки фиксировали 100 мкл 1:1 раствора ацетон : метанол в течение 1 минуты, промывали 3 раза фосфатным буфером (PBS) и затем блокировали 150 мкл/лунку 10% бычьей фетальной сывороткой (FBS) в PBS в течение часа при комнатной температуре. Далее клетки промывали 3 раза PBS и инкубировали в течение 2 часов при 37°C с 100 мкл/лунку антителами к неструктурному белку 5В (NS5B) HCV (Affinity BioReagents; кат.# MA 1-080, исходный раствор (1 мг/мл) разводили 1:4000 в 10% FBS-PBS). Клетки промывались 3 раза PBS проявлялись 100 мкл/лунку раствора OPD (на 1 плашку: 1 таблетка OPD, растворенная в 12 мл цитрат/фосфатного буфера с добавлением 5 мкл 30% Н2О2) в течение 30 минут в темноте при комнатной температуре. Реакцию останавливали 100 мкл/лунку 2 N H2SO4, после чего измеряли OD490 с помощью мультифункционального ридера Victor3 V 1420 (Perkin Elmer). Значения ЕС50 тестируемых пролекарств определяли построением кривой активности в программе GraphPad Prizm. Новое пролекарство формулы 1В.7 по отношению к генотипу lb (gTlb) HCV имеет ЕС50=15.0-27.0 нМ и ЕС90=128.0 нМ, Sovaldi® имеет ЕС50=45-170 нМ и ЕС90=590 нМ, циклогексиловый эфир формулы 2.1 имеет ЕС90=250.0 нМ, а циклопропиловый эфир формулы 2.2 имеет ЕС90=73.0 нМ и ЕС90=410.0 нМ (Таблица 3), т.е. новое пролекарство формулы 1В.7 более чем в три раза активнее, чем Sovaldi®, в два раза активнее, чем соединение формулы 2.1, и более чем в три раза активнее, чем соединение 2.2. Полученные результаты представлены в Таблице 3.
Цитотоксичность тестируемых композиций, включающих пролекарства, определялась параллельно на той же клеточной линии Huh7 с помощью набора ATPLite (Perkin-Elmer, Boston, USA) в соответствии с инструкцией производителя. По 50 мкл клеточной суспензии в полной культуральной среде (DMEM IX, Cellgro; кат.# 10-013-CV) переносили в 96-луночные плашки с черными стенками и прозрачным дном с итоговой плотностью 7500 клеток на лунку. Через 18 часов после посадки клеток добавляли по 50 мкл растворов серийных разведений соединений. Плашку с клетками инкубировали в течение 4 дней при 37°C в увлажненной атмосфере 5% CO2. Далее клетки промывали 2 раза 200 мкл/лунку PBS и лизировали добавлением 50 мкл/лунку лизирующего буфера (все реактивы из набора ATPLite). После помешивания в течение 5 минут на шейкере добавляли 50 мкл/лунку субстрата. После дополнительной 5-минутной инкубации плашку хранили 10 минут в темноте и считывали люминесценцию в лунках с помощью мультифункционального ридера Victor3 V 1420 (Perkin Elmer). Значения СС50 тестируемых соединений определяли построением кривой цитотоксичности в программе GraphPad Prizm. В частности, для нового пролекарства формулы 1В.7 найдено значение цитотоксичности СС50>100 мкМ (Таблица 3) и терапевтического окна (терапевтический индекс TI=ЕС50/СС50) TI>6,000.0.
Пример 11. Определение кинетической растворимости веществ.
Принцип метода. Изучаемое вещество растворяют в ДМСО до концентрации 10 мМ, затем вносят в водный растворитель (фосфатный буфер, вода, универсальные буферы с различными pH) до концентрации 200 мкМ. Полученный раствор инкубируют в течение часа при комнатной температуре на шейкере в 96-луночной фильтровальной плашке (Millipore's Multiscreen Solubility Filter Plate), после этого осадок отфильтровывают под вакуумом. Спектр поглощения вещества регистрируют на спектрофотометре в диапазоне 240-400 нм с шагом 10 нм. Для количественных расчетов растворимости используют калибровочную кривую стандартных растворов (0-200 мкМ) с содержанием 40% ацетонитрила. Диапазон определяемых концентраций 3-200 мкМ. Тестирование проводится в дубликатах.
Приготовление калибровочных стандартов. Калибровочные стандарты готовили из 50-кратных сток-растворов в ДМСО с последующим разбавлением их в буфере с 40% содержанием ацетонитрила, который добавлялся чтобы обеспечить полную растворимость тестируемого соединения в калибровочном образце. 6 стандартных образцов с концентрациями 0, 3,125, 12,5, 50, 100 и 200 мкМ были приготовлены в лунках 96-луночного UV-планшета путем добавления 4 мкл соответствующих 50х сток-растворов в ДМСО к 196 мкл буфера с 40% содержанием ацетонитрила. При этом концентрация ДМСО во всех точках оставалась постоянной и была равна 2% (v/v).
Для построения калибровочных кривых сняли оптический спектр UV-планшета в диапазоне длин волн от 250 нм до 400 нм с шагом 10 нм. Из полученных спектров для каждого соединения были выбраны длины волн, отвечающие следующим критериям:
- При минимальной концентрации вещества OD>0.1 (AU)
- При максимальной концентрации вещества OD<2.0
Калибровочная кривая была построена для каждого соединения по зависимости OD при выбранной длине волны от концентрации.
Определение кинетической растворимости соединений. Растворимость определяли в фильтровальном планшете MultiScreen Solubility (Millipore Corp.) следующим образом:
- В лунку фильтровального планшета MultiScreen Solubility добавили 196 мкл буфера (без ацетонитрила) и 4 мкл 10 мМ вещества в ДМСО либо 4 мкл ДМСО (для холостого образца). Планшет инкубировали в течение часа на шейкере (400 об/мин) при комнатной температуре.
- После инкубации растворы отфильтровали через фильтровальный планшет с помощью вакуума (10'' Hg) в полипропиленовый планшет с U-образным дном.
- Из планшета с U-образным дном перенесли 120 мкл/лунку фильтрата в новый UV-планшет, после чего добавили туда 80 мкл/лунку ацетонитрила.
- Измерили оптическую плотность полученных растворов при выбранной ранее длине волны для каждого соединения.
Расчеты. Финальная концентрация вещества в фильтрате рассчитывалась по формуле:
Сфильтрат=(ODλFiltrate-ODλBlank)/Slope×1,67,
где:
ODλFiltrate - оптическая плотность фильтрата при выбранной длине волны,
ODλBlank - OD холостого образца,
Slope - тангенс угла наклона калибровочной прямой,
1.67 - коэффициент разбавления фильтрата ацетонитрилом.
Полученные результаты представлены в Таблице 6.
Пример 12. Порошковые рентгенофазовые исследования образцов пролекарств.
Все дифрактограммы зарегистрированы на дифрактометре Bruker D8 Advance Vario, оснащенном рентгеновской трубкой с медным анодом и Ge(111)-монохроматором (CuKo^) и позиционно-чувствительным детектором LynxEye, в установках на просвет. Интервал съемки составил 3-90° 26 для образца s5 и 5.7-90° 26 для остальных образцов, шаг 0.01° 26. Анализ проводили в программе Bruker Topas5 ['Bruker TOPAS 5 User Manual. - Karlsruhe, Germany: Bruker AXS GmbH, 2015].
Образцы, полученные перекристаллизацией соединения формулы 1В.7 из смеси этилацетата с метил-трет-бутиловым эфиром (1:1), этанола, этилацетата и смеси уксусной кислоты с водой, имеют поликристаллическую форму. Порошковые рентгенофазовые исследования этих образцов имеют одинаковый качественный фазовый состав и незначительно различаются соотношением фаз. Образцы содержат ромбическую фазу с параметрами элементарной ячейки а=28.1056(8) А, b=16.8998(4) А, с=5.25380(12) А. Анализ систематических погасаний позволяет предположить пространственную группу P212121. Объем элементарной ячейки 2495.45(11) А3 соответствует заявленному составу и Z'=1. Образцы содержат также моноклинную фазу с параметрами элементарной ячейки а=16.2770(6) А, b=16.9117(8) А, с=5.20429(15) А, β=117.822(2)°. Систематические погасания позволяют предположить пространственную группу Р21. Объем элементарной ячейки 1266.98(9) А3 соответствует заявленному составу и Z'=1. Оценка соотношения фаз, основанная на сравнении интегральных интенсивностей пиков, предсказывает содержание моноклинной фазы от 30 до 50%.
Образец, полученный перекристаллизацией соединения формулы 1В.7 из смеси диметилсульфоксида с водой, представляет собой белое вещество кристаллической формы. Образец этой формы по данным порошковых рентгенофазовых исследований однофазен и состоит из ромбической фазы с параметрами элементарной ячейки а=28.1056(8) А, b=16.8998(4) А, с=5.25380(12) А. Анализ систематических погасаний позволяет предположить пространственную группу P212121. Объем элементарной ячейки 2495.45(11) А соответствует заявленному составу и Z'=1.
Пример 13. Определение стабильности в биологических средах композиций, включающих пролекарство формулы 1В.7.
Исходные композиции, включающие пролекарство формулы 1В.7 (тестируемое соединение), были приготовлены с концентрацией 10 мМ в ДМСО, из которых затем готовили 100-кратные рабочие растворы с концентрацией 100 мкМ в смеси ацетонитрил: вода с объемным отношением 1:1.
а) Стабильность в S9 фракции. Реакционная смесь была приготовлена в 0,1 М калия фосфатном буфере (pH 7,4 BD Gentest) в общем финальном объеме 250 мкл и содержала 1 мМ NADPH-тетранатриевую соль (AppliChem), 7 мМ глюкозо-6-фосфат натриевую соль (Sigma), 1.5 U/мл глюкозо-6-фосфат дегидрогеназу (Sigma), 3.3 мМ MgCl3 (Sigma), 5 мМ уридин-5-дифосфат-глюкуроновой кислоты тринатриевую соль (УДФГК, Sigma) и 1 мкМ тестируемого соединения (указаны финальные концентрации). Метаболическая реакция была инициирована путем добавления суспензии S9 фракции печени человека (BD Gentest), финальная концентрация белка составляла 1 мг/мл. Реакционная смесь инкубировалась при 37°C на шейкере (Vortemp56) с перемешиванием при 400 об/мин. Через определенные промежутки времени (0, 0.25, 0.5, 1, 2, 4, 6 8, 24 ч), проводился отбор проб в объеме 30 мкл, реакция останавливалась путем добавления к отобранной пробе 180 мкл холодного ацетонитрила, содержащего внутренний стандарт. Осаждение белков проводилось на льду в течение 15 мин. После чего образцы центрифугировались, и в течение 10 мин при 3000 об/мин. 150 мкл супернатанта отбиралось для анализа. Инкубация проводилась в двух повторах, каждая проба измерялась дважды.
б) Стабильность в искусственном желудочном и кишечном соках. Тестируемую композицию, включающую пролекарство формулы 1В.7 в конечной концентрации 1 мкМ инкубировали в искусственном желудочном соке (0,2% NaCl в 0,7% v/v HCl) и искусственном кишечном соке (0,05 М KН2РО4, pH б) 6,75). Инкубацию проводили в шейкере-инкубаторе (Vortemp56) при 37°C и перемешивании при 300 об/мин. Через определенные промежутки времени (0, 0.25, 0.5, 1, 2, 4, 6 8, 24 ч) проводился отбор проб в объеме 30 мкл, реакция останавливалась путем добавления к отобранной пробе 180 мкл холодного ацетонитрила, содержащего внутренний стандарт. После чего образцы центрифугировались, и в течение 10 мин при 3000 об/мин. 150 мкл супернатанта отбиралось для анализа. Инкубация проводилась в двух повторах, каждая проба измерялась дважды.
в) Стабильность в плазме крови. Тестируемое соединение в конечной концентрации 1 мкМ инкубировали в пулированной плазме крови человека (Innovative Research). Инкубацию проводили в шейкере-инкубаторе (Vortemp56) при 37°C и перемешивании при 300 об/мин. Через определенные промежутки времени (0, 0.25, 0.5, 1, 2, 4, 6 8, 24 ч) проводился отбор проб в объеме 30 мкл, реакция останавливалась путем добавления к отобранной пробе 180 мкл холодного ацетонитрила, содержащего внутренний стандарт. После чего образцы центрифугировались, и в течение 10 мин при 3000 об/мин. 150 мкл супернатанта отбиралось для анализа. Инкубация проводилась в двух повторах, каждая проба измерялась дважды.
Анализ проб. Анализ проб проводился методом ВЭЖХ-МС/МС, разработанным для каждого тестируемого пролекарства, с использованием хроматографической системы 1290 Infinity II (Agilent Technologies), сопряженной с тандемным масс-спектрометром QTRAP5500 (АВ Sciex). При разработке условий масс-спектрометрического детектирования растворы тестируемых соединений в смеси ацетонитрил-вода 1:1 с концентрацией 100 нг/мл анализировали путем прямого ввода в масс-спектрометр при помощи шприцевого насоса при ионизации электрораспылением в режиме регистрации положительных ионов. При сканировании в режиме полного ионного тока (MS1) определяли молекулярный ион для каждого соединения, основные ионы-продукты регистрировали в режиме MS2. Далее была проведена оптимизация МС/МС метода в режиме MRM для достижения максимальной чувствительности. При количественной обработке хроматограмм использовали наиболее интенсивный MRM-переход для аналита и внутреннего стандарта. Хроматографическое разделение проводили на колонке YMC Triart С18, 50×2.мм, 1,.9 мкм в градиентном режиме элюирования в подвижной фазе состава 0,1% муравьиная кислота в воде - 0.1% муравьиная кислота в ацетонитриле. В качестве внутреннего стандарта использовали толбутамид (Fluka).
Вычисления. По кинетике убыли тестируемого пролекарства в противовирусной композиции в процессе инкубации в биологической среде рассчитывали время полураспада (Т1/2). Для расчетов использовали нормированные на сигнал внутреннего стандарта значения площадей хроматографических пиков веществ в опытных образцах. По линейной зависимости лог-нормированных площадей хроматографических пиков от времени рассчитывали константу скорости элиминации (k - наклон линейного участка). Далее рассчитывали время полураспада: T1/2=0.693/k. В частности, было найдено (Таблица 4), что пролекарство формулы 1В.7, соединение формулы 2.1 и Sovaldi® имеют сопоставимую стабильность в человеческом желудочный соке (T1/2 SGF=12.7 ч-17 ч), в человеческом кишечном соке (T1/2 S1F>20 ч) и в человеческой плазме (T1/2 HPL>24 ч). В то же время пролекарство формулы 1В.7 более быстро метаболизирует в S9 фракции микросом печени человека и имеет время полураспада T1/2 HS9=0.05 ч, в то время как его прототип Sovaldi® имеет T1/2 HS9=0.57 ч, а циклогексиловый эфир формулы 2.1 имеет Т1/2 HS9=1.4 ч (Таблица 4), т.е. пролекарство формулы 1В.7 в 11 раз быстрее метаболизирует в S9 фракции микросом печени человека, чем Sovaldi® и в 28 раз быстрее, чем соединение формулы 2.1. Полученные результаты представлены в Таблице 4.
Пример 14. Изучение фармакокинетики (РК) композиций, включающих пролекарство формулы 1В.7 и Sovaldi®, в печени крыс.
Приготовления композиций, включающих пролекарство формулы 1В.7 и Sovaldi®, для введения крысам. Исследуемую композицию вводили в дозе 50 мг/кг. Для этого готовили композиции с концентрацией пролекарства формулы 1В.7 или Sovaldi® 5,0 мг/мл в 0,5% р-ре гидроксипропил-метилцеллюлозе (НРМС) с добавкой 5% этанола следующим образом: к навеске пролекарства формулы 1В.7 или Sovaldi® добавляли необходимое количество НРМС и насухую перетирали в ступке, затем постепенно порционно добавляли необходимое количество 5%-го этанола в дистиллированной воде, тщательно перемешивали до получения суспензии, пригодной для внутрижелудочного введения.
Введение композиций, включающих пролекарство формулы 1В.7 и Sovaldi®, животным. Получение образцов плазмы крови и печени. Исследование проводилось на лабораторных крысах линии Sprague Dawley. Крыс разделили на группы соответственно выбранным временным точкам (1, 2, 4, 6, 8, 10, 12, 16 и 24 ч) по 6 животных в группе. Животных взвешивали, рассчитывали объем вводимых композиций, включающих пролекарство формулы 1В.7 или Sovaldi®, для каждого животного из расчета 10 мл/кг. Введение композиций, включающих пролекарство формулы 1 В.7 или Sovaldi®, проводили внутрижелудочно через зонд. Между введениями животным одной временной точки делали промежутки, достаточные для взятия образца печени и крови. Через необходимый промежуток времени после введения крысу эвтаназировали ингаляцией CO2. Немедленно после эвтаназии животное как можно быстрее вскрывали, отрезали верхнюю долю печени и сразу бросали ее в жидкий азот. Замороженный фрагмент печени помещали в промаркированную охлажденную в жидком азоте пробирку. До завершения эксперимента образцы хранили в жидком азоте, затем перемещали в низкотемпературный морозильник на -80°C.
Пробоподготовка. Навеску печени массой около 1 г перетирали в ступке при охлаждении жидким азотом, полученный порошок заливали трехкратным объемом метанола с 70% метанола с ЭДТА и гомогенизировали с помощью гомогенизатора Omni Bead Ruptor 24 на скорости 6.3 м/сек по 45 сек 2 раза с интервалом 10 сек. К 360 мкл полученного гомогената добавляли 40 мкл десятикратного стандартного раствора, содержащего PSI-7409 и Н027-4261 (или метанола в случае экспериментальных проб), и 100 мкл раствора внутреннего стандарта (5-бромуридин трифосфата) с концентрацией 25 т нг/мл. После перемешивания и центрифугирования 400 мкл супернатанта разбавляли 400 мкл раствора 1% муравьиной кислотой в смеси метанол - вода (1:1). Проводили твердофазную экстракцию на картриджах Waters Oasis WAX, элюировали 800 мкл 5% раствора аммиака в метаноле, элюат упаривали и перерастворяли в 200 мкл метанола.
Условия ВЭЖХ-МС/МС анализа. Анализ проводили методом ВЭЖХ-МС/МС с помощью ВЭЖХ системы Agilent 1290 Infinity II, соединенной с масс-спектрометром АВ Sciex QTrap 5500. Разделение проводили на колонке Thermo Hypercarb (50×3 мм, 5 мкм), в качестве подвижной фазы A (ПФА) использовали раствор 25 мМ ацетата аммония с добавкой 0.5% аммиака, в качестве подвижной фазы Б (ПФБ) - раствор 25 мМ ацетата аммония в смеси вода-изопропанол-ацетонитрил (1:1:3) с добавкой 0.5% аммиака. Разделение проводили в градиентном режиме: 0-0.3 мин - 5% ПФБ; 3-3.4 мин - 50% ПФБ; 3.6-4.5 мин - 5% ПФБ, Регистрацию PSI-7409 и Н027-4261 проводили в MRM режиме по ионным переходам 499/159 и 410/150 соответственно.
Фармакокинетический анализ. Фармакокинетический анализ данных «концентрация в печени - время» выполнялся модельно-независимым методом с помощью программы Phoenix™ WinNonlin® 6.3 (Pharsight Corp.) и программы GraphPad Prizm. Рассчитывались следующие фармакокинетические параметры: максимальная концентрация в печени (Cmax) и время ее достижения (Tmax), период полувыведения (Т1/2), площадь под ФК кривой (AUC0-t, AUC0-inf). Полученные данные представлены в Таблице 5. Как видно из Таблицы 5, концентрация и AUC24 ч трифосфата PSI-7409, образующегося при метаболизме пролекарства формулы 1В.7 в печени крыс, составляют Cmax=3,224.0 нг/г и AUC24 ч=30,487.0 нг⋅ч/г, в то время как при аналогичном метаболизме Sovaldi® имеет Cmax=1,934.0 нг/г и AUC24 ч=16,796.0 нг⋅ч/г, а циклогексиловый эфир формулы 2.1 имеет Сmax=557 нг/г и AUC24 ч=6,484.0 нг ч/г (Таблица 5). Это свидетельствует о том, что новое пролекарство формулы 1В.7 почти в 2 раза более эффективно метаболизирует в печени в нужный трифосфат PSI-7409 (лекарство), чем Sovaldi®, и в 4,7 раза более эффективно метаболизирует, чем циклогексиловый эфир формулы 2.1. Полученные результаты представлены в Таблице 5.
Claims (37)
1. Пролекарство общей формулы 1, его стереоизомер, фармацевтически приемлемая соль, кристаллическая и поликристаллическая форма,

где:
n имеет значение 1 или 0;
Nuc представляет собой  или
или  ;
;
R1 представляет собой водород или метил;
R2, R3 представляют собой необязательно одинаковые заместители, выбранные из Н, F, Cl, СН3 при условии, когда сплошная линия и сопровождающая ее пунктирная линия ( ) вместе представляют одинарную углерод-углеродную (С-С) связь, или R2 и R3 представляют собой водород при условии, когда сплошная линия и сопровождающая ее пунктирная линия (
) вместе представляют одинарную углерод-углеродную (С-С) связь, или R2 и R3 представляют собой водород при условии, когда сплошная линия и сопровождающая ее пунктирная линия ( ) вместе представляют собой двойную углерод-углеродную связь (С=С);
) вместе представляют собой двойную углерод-углеродную связь (С=С);
R4 представляет собой заместитель, выбранный из R4.1-R4.5:

R3.6 представляет собой заместитель, выбранный из Н, F, Cl, СН3 или CF3;
R3,7 представляет собой водород, С1-С4-алкил или С3-С6-циклоалкил;
X представляет собой О, СН2 или С=СН2;
Y представляет собой S, СН2 или НО-СН-группу при условии, когда непрерывная линия и сопровождающая ее пунктирная линия ( ) вместе представляют собой одинарную углерод-углеродную (С-С) связь, или Y представляет собой СН-группу при условии, когда сплошная линия и сопровождающая ее пунктирная линия (
) вместе представляют собой одинарную углерод-углеродную (С-С) связь, или Y представляет собой СН-группу при условии, когда сплошная линия и сопровождающая ее пунктирная линия ( ) вместе представляют собой двойную углерод-углеродную связь (С=С).
) вместе представляют собой двойную углерод-углеродную связь (С=С).
2. Пролекарство по п. 1, представляющее собой соединение общей формулы 1А или 1В, их стереоизомер, фармацевтически приемлемую соль, кристаллическую и поликристаллическую формы,


где: непрерывная линия и сопровождающая ее пунктирная линия ( ), R1, R2, R3, R4, X и Y имеют вышеуказанные значения.
), R1, R2, R3, R4, X и Y имеют вышеуказанные значения.
3. Пролекарство по п. 1, представляющее собой соединение, выбранное из
(S)-циклобутил 2-((S)-(((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-илокси)метил)(фенокси)фосфориламино)-пропаноата (1А.1),
(S)-циклобутил 2-((R)-(((R)-1-(6-амино-9H-пурин-9-ил)пропан-2-илокси)метил)(фенокси)фосфориламино)-пропаноата (1А.2),
(S)-циклобутил 2-{(S)-[(2S,3R,5S)-3-гидрокси-5-(5-метил-3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноата (1В.1),
(S)-циклобутил 2-{(S)-[(2S,3S,4R,5S)-3-гидрокси-4-фтор-5-(5-метил-3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноата (1В.2),
(S)-циклобутил 2-{(S)-[(2R,3R,5R)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-3-гидрокси-4,4-дифтор-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноата (1В.3),
(S)-циклобутил 2-{(S)-[(1R,3S,5S)-3-(2-амино-6-оксо-1,6-дигидро-пурин-9-ил)-5-гидрокси-2-метилен-циклопентилметокси]-фенокси-фосфориламино}-пропаноата (1В.4),
(S)-циклобутил 2-{(S)-[(2R,5S)-5-(4-амино-2-оксо-2H-пиримидин-1-ил)-[1,3]оксатиолан-2-илметокси]-фенокси-фосфориламино}-пропаноата (1В.5),
(S)-циклобутил 2-{(S)-[(2R,5S)-5-(4-амино-5-фтор-2-оксо-2H-пиримидин-1-ил)-[1,3]оксатиолан-2-илметокси]-фенокси-фосфориламино}-пропаноата (1В.6),
(S)-циклобутил 2-{(S)-[(2R,3R,4R,5R)-5-(3,4-дигидро-2,4-диоксо-2H-пиримидин-1-ил)-3-гидрокси-4-метил-4-фтор-тетрагидрофуран-2-илметокси]-фенокси-фосфориламино}-пропаноата (1В.7),









их фармацевтически приемлемая соль, кристаллическая или поликристаллическая форма.
4. Фармацевтическая композиция, обладающая противовирусной активностью, которая содержит терапевтически эффективное количество пролекарства по пп. 1-3 и фармацевтически приемлемый носитель.
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2017106611A RU2659388C1 (ru) | 2017-02-28 | 2017-02-28 | Нуклеотиды, включающие N-[(S)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги и их применение |
| EP17791915.6A EP3473636A4 (en) | 2017-02-28 | 2017-04-07 | NUCLEOTIDES CONTAINING A FRAGMENT OF N - [(S) -1-CYCLOBUTOXYCARBONYL] PHOSPHORAMIDATE, ANALOGUES AND USE |
| MA047152A MA47152A (fr) | 2017-02-28 | 2017-04-07 | Nucléotides contenant un fragment de n-[(s)-1-cyclobutoxycarbonyl]phosphoramidate, analogues et utilisation |
| PCT/RU2017/000209 WO2018160088A1 (ru) | 2017-02-28 | 2017-04-07 | Нуклеотиды, включающие n-[(s)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги и их применение |
| US15/746,823 US20200079814A1 (en) | 2017-02-28 | 2017-04-07 | Nucleotides comprising an N-[(S)-1-cyclobutoxycarbonyl]phosphoramidate moiety and analogs and application thereof |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2017106611A RU2659388C1 (ru) | 2017-02-28 | 2017-02-28 | Нуклеотиды, включающие N-[(S)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги и их применение |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| RU2659388C1 true RU2659388C1 (ru) | 2018-07-02 |
Family
ID=62815806
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2017106611A RU2659388C1 (ru) | 2017-02-28 | 2017-02-28 | Нуклеотиды, включающие N-[(S)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги и их применение |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20200079814A1 (ru) |
| EP (1) | EP3473636A4 (ru) |
| MA (1) | MA47152A (ru) |
| RU (1) | RU2659388C1 (ru) |
| WO (1) | WO2018160088A1 (ru) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020246910A1 (ru) | 2019-06-03 | 2020-12-10 | Александл Васильевич ИВАЩЕНКО | АНЕЛИРОВАННЫЕ 9-ГИДРОКСИ- 1,8 - ДИОКСО- 1, 3,4, 8 -ТЕТРАГИДРО-2Н-ПИРИДО [1,2-α] ПИРАЗИН-7 - КАРБОКСАМИДЫ - ИНГИБИТОРЫ ИНТЕГРАЗЫ ВИЧ |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113166190B (zh) * | 2018-11-23 | 2023-05-23 | 正大天晴药业集团股份有限公司 | 低聚核苷酸及前体药物 |
| CN113493481A (zh) * | 2020-04-08 | 2021-10-12 | 北京君科华元医药科技有限公司 | 单磷酸恩替卡韦丙氨酰胺酚酯及其医药用途 |
| MX2023008004A (es) * | 2021-01-25 | 2023-07-13 | Brii Biosciences Inc | Derivado de adenosina y composicion farmaceutica que lo comprende. |
| WO2023250015A1 (en) * | 2022-06-21 | 2023-12-28 | The Children's Medical Center Corporation | Pyrimidine nucleoside treatments |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008121634A2 (en) * | 2007-03-30 | 2008-10-09 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| CN106317116A (zh) * | 2016-08-19 | 2017-01-11 | 张红利 | 磷酰胺核苷类化合物及其药学上可接受的盐与应用、药物组合物 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9821058D0 (en) * | 1998-09-28 | 1998-11-18 | Univ Cardiff | Chemical compound |
| AP1466A (en) | 2000-07-21 | 2005-09-22 | Gilead Sciences Inc | Prodrugs of phosphonate nucleotide analogues and methods for selecting and making same. |
| WO2007002808A1 (en) * | 2005-06-29 | 2007-01-04 | Gilead Sciences, Inc. | Anti-nonmelanoma carcinoma compounds, compositions, and methods of use thereof |
| DK2455376T3 (en) | 2009-06-11 | 2015-03-02 | Abbvie Bahamas Ltd | Heterocyclic compounds as inhibitors of hepatitis C virus (HCV) |
| BR112013007696A2 (pt) | 2010-09-29 | 2019-09-24 | Merck Sharp & Dohme | composto, composição farmacêutica, usos do referido composto e da referida composição |
| WO2012040923A1 (en) | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic indole derivatives and methods of use thereof for the treatment of viral diseases |
| RU2452735C1 (ru) | 2010-11-30 | 2012-06-10 | Александр Васильевич Иващенко | Замещенные азолы, противовирусный активный компонент, фармацевтическая композиция, способ получения и применения |
| JP5651275B2 (ja) | 2011-08-16 | 2015-01-07 | ギリアード サイエンシス インコーポレーテッド | テノホビルアラフェナミドヘミフマレート |
| US20130143835A1 (en) * | 2011-12-05 | 2013-06-06 | Medivir Ab | HCV Polymerase Inhibitors |
| EP2809323A1 (en) | 2012-02-03 | 2014-12-10 | Gilead Sciences, Inc. | Combination therapy comprising tenofovir alafenamide hemifumarate and cobicistat for use in the treatment of viral infections |
| WO2014033617A1 (en) | 2012-08-31 | 2014-03-06 | Novartis Ag | 2'-ethynyl nucleoside derivatives for treatment of viral infections |
| CN104803989B (zh) | 2014-01-23 | 2017-12-22 | 广东东阳光药业有限公司 | 作为丙型肝炎抑制剂的桥环化合物及其在药物中的应用 |
| RU2644156C1 (ru) * | 2017-02-28 | 2018-02-08 | Александр Васильевич Иващенко | Пролекарство ингибитора NS5B HCV полимеразы, способ его получения и применения |
-
2017
- 2017-02-28 RU RU2017106611A patent/RU2659388C1/ru active
- 2017-04-07 EP EP17791915.6A patent/EP3473636A4/en not_active Withdrawn
- 2017-04-07 WO PCT/RU2017/000209 patent/WO2018160088A1/ru unknown
- 2017-04-07 MA MA047152A patent/MA47152A/fr unknown
- 2017-04-07 US US15/746,823 patent/US20200079814A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008121634A2 (en) * | 2007-03-30 | 2008-10-09 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| RU2478104C2 (ru) * | 2007-03-30 | 2013-03-27 | Гилеад Фармассет Ллс | Нуклеозидфосфорамидаты в качестве противовирусных агентов |
| CN106317116A (zh) * | 2016-08-19 | 2017-01-11 | 张红利 | 磷酰胺核苷类化合物及其药学上可接受的盐与应用、药物组合物 |
Non-Patent Citations (3)
| Title |
|---|
| Wang Peiyuan et al. Phosphoramidate prodrugs of (-)-β-D-(2R,4R)-dioxolane-thymine DOT) as potent anti-HIV agents. Antiviral Chemistry & Chemotherapy, 2012, 22(5), 217-238 * |
| Wang Peiyuan et al. Phosphoramidate prodrugs of (-)-β-D-(2R,4R)-dioxolane-thymine DOT) as potent anti-HIV agents. Antiviral Chemistry & Chemotherapy, 2012, 22(5), 217-238 (English). (Zeng Debin et al. Discovery of 2'-α-C-Methyl-2'-β-C-fluorouridine Phosphoramidate Prodrugs as Inhibitors of Hepatitis C Virus, ACS Medicinal Chemistry Letters (English), 2016, 7(12), 1197-1201. Wang Peiyuan et al. Phosphorimidate Progrugs of (-)-β-D-(2R,SR)dioxоlane-thymine (DOT) as potent anti-HIV agents, Antiviral Chemistry Chemotherapy, 2012, 22(5), 217-238. * |
| Zeng Debin et al. Discovery of 2'-α-C-Methyl-2'-β-C-fluorouridine Phosphoramidate Prodrugs as Inhibitors of Hepatitis C Virus, ACS Medicinal Chemistry Letters (English), 2016, 7(12), 1197-1201. * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020246910A1 (ru) | 2019-06-03 | 2020-12-10 | Александл Васильевич ИВАЩЕНКО | АНЕЛИРОВАННЫЕ 9-ГИДРОКСИ- 1,8 - ДИОКСО- 1, 3,4, 8 -ТЕТРАГИДРО-2Н-ПИРИДО [1,2-α] ПИРАЗИН-7 - КАРБОКСАМИДЫ - ИНГИБИТОРЫ ИНТЕГРАЗЫ ВИЧ |
Also Published As
| Publication number | Publication date |
|---|---|
| US20200079814A1 (en) | 2020-03-12 |
| EP3473636A4 (en) | 2019-06-26 |
| MA47152A (fr) | 2019-11-06 |
| EP3473636A1 (en) | 2019-04-24 |
| WO2018160088A1 (ru) | 2018-09-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| RU2659388C1 (ru) | Нуклеотиды, включающие N-[(S)-1-циклобутоксикарбонил]фосфорамидатный фрагмент, их аналоги и их применение | |
| TWI498117B (zh) | 核苷氨基磷酸酯 | |
| TWI598358B (zh) | 核苷磷醯胺 | |
| JP2012514657A (ja) | ウィルス感染を治療するためのグアノシンヌクレオシド化合物のホスホルアミダート誘導体 | |
| CN107427530B (zh) | 用于HCV治疗的β-D-2’-脱氧-2’α-氟-2’-β-C-取代的-2-改性的-N6-取代的嘌呤核苷酸 | |
| MX2013013570A (es) | Profarmacos de monofosfato de purina para el tratamiento de infecciones virales. | |
| BRPI0709127A2 (pt) | 2'-fluoronucleosìdeo fosfonatos como agentes antivirais | |
| JP2010515680A (ja) | Rna依存性rnaウイルス感染症の治療用としてのヌクレオシドアリールホスホロアミデート | |
| TW201002733A (en) | Nucleoside cyclicphosphates | |
| WO2018022221A1 (en) | Phosphoramidate nucleoside prodrug for treating viral diseases and cancer, processes for their preparation and their use | |
| RU2731385C1 (ru) | Макрогетероциклические нуклеозидные производные и их аналоги, получение и применение | |
| CN108350008B (zh) | 一种新型的无环核苷类似物及其药物组合物 | |
| EP2265626B1 (en) | Nucleotide analogues with quaternary carbon stereogenic centers and methods of use | |
| RU2644156C1 (ru) | Пролекарство ингибитора NS5B HCV полимеразы, способ его получения и применения | |
| RU2647576C1 (ru) | Циклобутил (S)-2-[[[(R)-2-(6-аминопурин-9-ил)-1-метил-этокси]метил-фенокси-фосфорил]амино]-пропаноаты, способ их получения и применения | |
| NO324263B1 (no) | Kjemiske forbindelser, anvendelse derav ved behandling av kreft, samt farmasoytiske preparater som omfatter slike forbindelser | |
| RU2650610C1 (ru) | Противовирусная композиция и способ ее применения | |
| EA043026B1 (ru) | Пролекарство ингибитора ns5b hcv полимеразы, способ его получения и применения | |
| BR122013007556A2 (pt) | compostos de fosforamidatos de nucleosídeo e processos para preparação destes compostos |