RU2637312C1 - Способ получения кетона малины - Google Patents
Способ получения кетона малины Download PDFInfo
- Publication number
- RU2637312C1 RU2637312C1 RU2016148283A RU2016148283A RU2637312C1 RU 2637312 C1 RU2637312 C1 RU 2637312C1 RU 2016148283 A RU2016148283 A RU 2016148283A RU 2016148283 A RU2016148283 A RU 2016148283A RU 2637312 C1 RU2637312 C1 RU 2637312C1
- Authority
- RU
- Russia
- Prior art keywords
- raspberry ketone
- tert
- ketone
- raspberry
- compound
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/65—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by splitting-off hydrogen atoms or functional groups; by hydrogenolysis of functional groups
Landscapes
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Изобретение относится к способу получения кетона малины, который используют в парфюмерии в качестве душистого компонента и как отдушку лекарственных средств. Способ заключается в том, что 4-(3',5'-ди-трет-бутил-4'-гидроксифенил)-бутан-2-он и толуол нагревают с раствором AlCl3 в нитрометане при температуре 30-50°C и после разложения реакционной массы водой целевой продукт выделяют кристаллизацией органического слоя. Предлагаемый способ позволяет получить кетон малины, не содержащий трудно отделяемых от него примесей с портящим его запахом при использовании простой технологии. 3 пр.
Description
Обладая запахом и вкусом малины, содержащийся в ее плодах 4-(4'-гидроксифенил) бутан-2-он (1, он же кетон малины, зарубежные его названия: Raspberry ketone, «rheosmin», «фрамбинон». «оксифеналон»), широко используется в парфюмерии [ЕР 12624473 (2002)]в качестве душистого компонента в получении кулинарных изделий и как отдушка лекарственных средств [US 6222062(2001)].
Соединение 1 малотоксично: ЛД50>5 г/кг (крысы, перорально) [Хейфиц Л.А. Душистые вещества и другие продукты в парфюмерной промышленности. С. 157-158]. Оно - действующее начало средств косметики, используемых с целью защиты кожи человека от лейкодермитов, воспалений, эритерм, возникающих на незащищенной коже под действием солнечных лучей и радиационного облучения [WO 2008/47382 (2008)].
Обладая противовоспалительным [Jin Boo Jeong. Hyung Jin Jeong. Food and Chtv. Toxicology v.48 (2010) 2148-2153] и антимикробным действием US 6248765(2001), кетон малины может выступить как противораковое средство [Glu K.U., Inti H.S., Ozgur A., Seen Н., Tutar Y.// Turkich J. Chem v. 39, N1 (2015) P. 179-193] при лечении меланомы. Кетон малины рекомендуется при лечении диабета второго типа [US 6046227(2000) А-1] и как компонент диеты при похудении тучных лиц [Cube R.V., Vernier J.M., Hutchinson J.U., Gardntr M.F. at all.// Life Sci. v. 77 N2 (2005) P. 194-204]. Он используется в качестве промежуточного продукта в синтезе новых душистых веществ. А.И. Кузнецов, Р.Т. Аласади, И.М. Сенон, Т.М. Серова.// Изв. АН сер. хим. 2015. №4. С. 962-964.
Для внедрения в промышленность разработаны отечественные технологии получения синтетического кетона малины высшего качества, обладающего вкусом и запахом малины.
Наиболее привлекательным казался одностадийный способ получения кетона малины взаимодействием фенола и метилвинилкетона в присутствии различных кислотных катализаторов (H2SO4, Н3РО4, AlCl3, FeCl3). Из них наиболее удобным катализатором оказалась серная кислота [Г.В. Шимайская, Л.А. Хейфиц, Г.И. Молдаванская, О.И. Пахомова //Масло-жировая промышленность 1978. №3. С. 34-36]. Содержание кетона малины в реакционной массе достигает 63% при проведении реакции при -5°С в течение 3.5 часов и молярном соотношении метилвинилкетон, фенол и серная кислота 1:4:0,5.

Однако авторы от дальнейшей технологической проработки этого способа отказались из-за высокой токсичности его для работников, находящихся в контакте с высокотоксичными фенолом и метилвинилкетоном; больших потерь кетона малины в процессе его очистки до требований высшего качества (приобретения им малинового запаха и вкуса), а также из-за наличия фенола в сточных водах производства.
Современное состояние развития этого направления представлено в работе [4] Guo Hui, Zhuang Yu Wei, Cao Jian, Guo Bao/Bulletin of the Korean Chemical Society vol. 34. nb.9 (2013) p. 2594-259, в которой описана конденсация метилвинилкетона с фенолом использованием кислотного катализатора конденсации: 1-метил-3-метилимидозониум гидросульфата [BMIM]HSO4. Выход продукта конденсации не превышает 51%.
В работе [Jun-ichi Tateiwa, Hiroki Horiuchi at all. J. Org, Chem. 1994, 59, N20, 5901-5904] изучены побочные соединения (эфиры, продукты орто- замещения ароматического кольца), которые образуются из фенола в процессе синтеза кетона малины. Сам кетон малины предложено получать селективно взаимодействием фенола с 4-гидроксибутан-2-оном в присутствии катализатора Zr+4 или Fe+3 - монтмориллонита (montmorillonite). Выход продукта, очищенного методом колоночной хроматографии, составил 35%. Недостатком метода явилась невысокая степень превращения, которую не удалось улучшить увеличением времени контакта с катализатором, что связано, по-видимому, с образованием реакционной воды, уменьшающей активность катализатора.
Из-за этих недостатков способа исследователи вынужденно переходили к разработке других методов получения кетона малины без использования фенола. Известен способ получения кетона малины взаимодействием фенола с 4-гидроксибутан-2-оном в присутствии большого избытка серной кислоты ФРГ 2145308 (1971) РЖХим 23Р379П. Главный недостаток метода - необходимость тщательной очистки кетона малины от фенола, что приводит к большим потерям продукта.
Известен способ получения кетона малины в две стадии. Конденсация 4-гидроксибензальдегида (2) с ацетоном в присутствии NaOH (мольное соотношение 1:2:2-5 при комнатной температуре в течение суток) приводит к получению 4-(4-гидроксифенил)-2-бутен-3-она (3) с выходом 70-80% [Г.И. Молдаванская, А.И. Платова, О.И. Пахомова, Г.В. Шимайская, Л.А. Хейфиц. Масло-жировая промышленность 1981. №11. С. 27-28]. Как показано авторами этой работы, восстановление ненасыщенного кетона (3) водородом с образованием кетона малины (1) наиболее селективно протекает в присутствии формиатного никеля или никеля на кизельгуре.
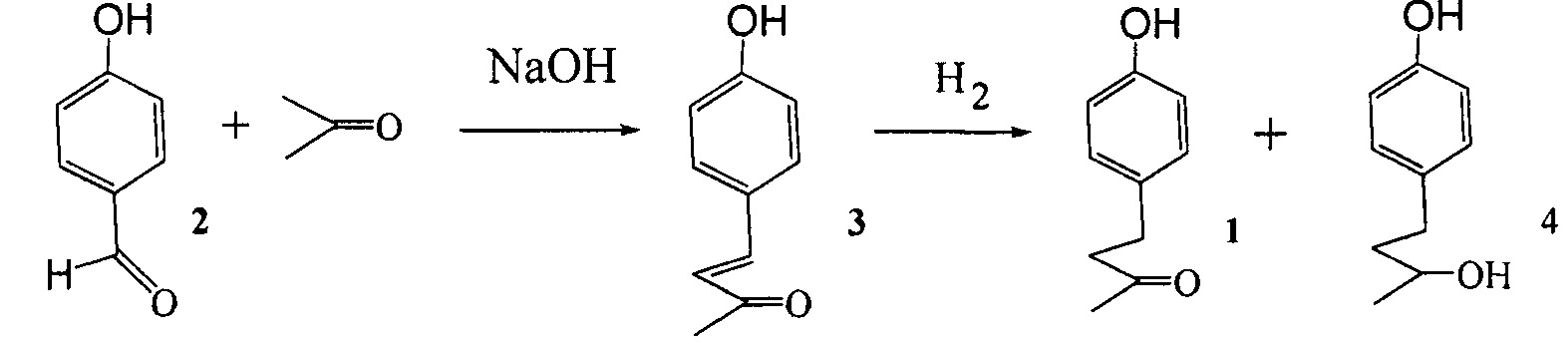
Образование побочного спирта 4 (с восстановлением кетогруппы до спиртовой) в присутствии этих катализаторов заметно не протекает. Выход технического кетона малины по двум стадиям, исходя из гидроксибензальдегида, составляет 40%. Использование других катализаторов гидрирования соединения (3) приводит наряду с кетоном малины (1) к образованию заметных количеств соединения (4).
Получение и использование никелевого катализатора для восстановления ненасыщенного кетона (3) демонстрируется в работе [Bandarenko, М.; Kovalenko, V.: Zeitschrift fur Naturforschung. Section В. Journal of Chemical Sciences; vol. 69; nb. 8; (2014); p.885-888]. Выход чистого кетона малины на этой стадии восстановления составляет 75%. Одним из недостатков способа получения кетона малины, исходя из 4-гидроксибензальдегида, является его малая масштабность получения как побочного продукта при производстве кумарина.
Известен способ получения соединения 1 путем окисления 4-(4'-гидроксифенил)-2-бутанола (4) [Kosjek, Birgit; Stampfer, Wolfgang; Van Deursen, Ruud; Faber, Kurt; Kroutil, Wolfgang; Tetrahedron, vol. 59. N48 (2003). p. 9517-9521]. К этому способу получения кетона малины приходится практически прибегать в случае, когда используют методы восстановления 4-(4'-гидроксифенил)-1-бутен-2-она (3) с образованием побочного спирта 4. При наличии в России высокоселективных никелевых катализаторов восстановления соединения 3, открытых в работе [Г.И. Молдаванская, А.И. Платова, О.И. Пахомова, Г.В. Шимайская, Л.А. Хейфиц. Масло-жировая промышленность 1981. №11. С. 27-28], этот метод получения 1 не является актуальным.
Отмечены высокие потери кетона малины в процессе очистки его от технического образца до высокочистого (приобретения им малинового запаха и вкуса [Г.В. Шимайская, Л.А. Хейфиц. Г.И. Молдаванская, О.И. Пахомова. Масло-жировая промышленность 1978, №3. С. 34].
Известны патенты, цель которых направлена на получение высокочистьгх образцов соединения 1. Цель достигается получением из технического кетона малины 1 его метилового [Nomura Nozawa Sci. Rep. Tohoku Univ., Ser. 1.Phys. Chem. Astron. vol. 7. p. 84.9], трет-бутилового [BASF Aktiengesellschaft. Patent; US 4908481(1990)/ A-1] или кремнийсодержащего эфиров [Carlson E.E., Trader D. Patent US 2014/ 107328 (2014) А-1]. После очистки эфиров проводят их гидролиз с получением кетона малины высокой степени чистоты. Недостатком этих способов получения чистого кетона малины является введение в технологический процесс получения 1 дополнительных химических стадий. Наша цель: получение технических образцов кетона малины, не содержащих трудно отделяемых от него примесей с портящим его запахом. К ним относятся продукты орто- и мета- замещения фенола и их эфиры. Отсутствие этих примесей и самого фенола обеспечивает эффективность очистки кетона малины его кристаллизацией.
Цель достигается применением в качестве исходного соединения синтеза кетона малины доступного и дешевого 2,6-ди-трет-бутилфенола, обеспечивающего любой масштаб производства соединений, полученных на его основе. Наличие в этом соединении в орто- положениях трет-бутильных групп блокирует образование эфиров и побочных продуктов: орто - и мета- замещения ароматического кольца.
Процесс получения кетона малины осуществляется по схеме:
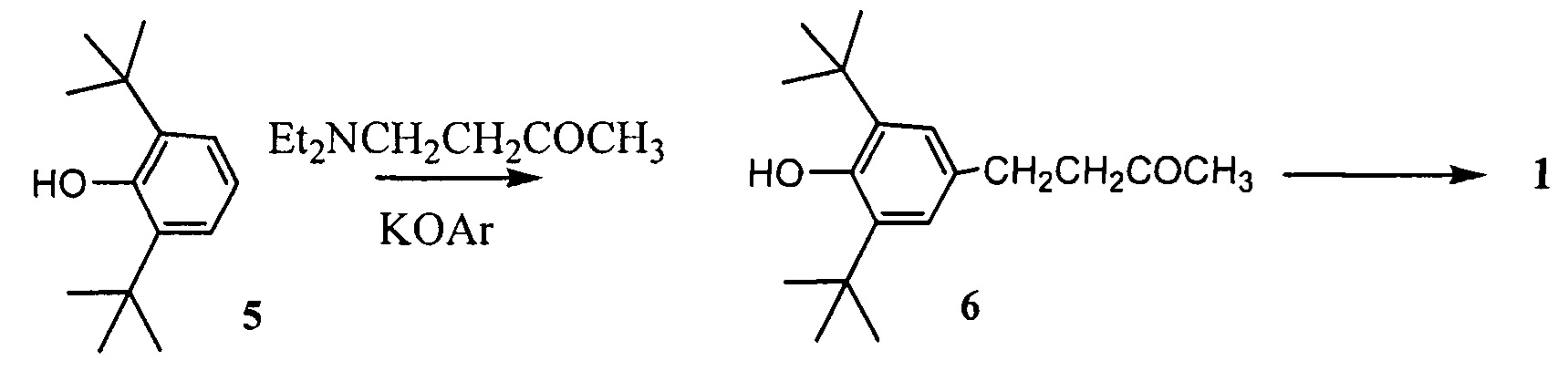
На первой стадии 2,6- ди-трет-бутилфенол (5) нагревают с 4- N,N-диэтиламино-бутан-2-оном в присутствии катализаторов: калиевых или натриевых фенолятов соединений 5 или 6. Процесс получения соединения 6 протекает при нагревании без образования побочных продуктов и сопровождается высвобождением диэтиламина из зоны реакции, что очень важно для сохранения ее реакционной способности. Из реакционной массы перегонкой выделяют последовательно соединение 5, а затем целевой продукт 6, который очищают от следов исходного соединения кристаллизацией. Кубовый остаток, содержащий калиевую соль соединения 6, используют при его повторных наработках в качестве катализатора. В этом случае исключается процесс предварительного синтеза катализатора, а получение соединения 6 реализуется без образования сточных вод.
Де- трет-бутилирование соединения 6 проводят в токе инертного газа толуолом в нитрометане под действием сухого хлорида алюминия при температуре 30-50°С в течение 0,5 - 3-х часов. Методом ГЖХ контролируют отсутствие в реакционной массе фенолов, содержащих трет-бутильные группы. По окончании времени протекания процесса из реакционной массы отгоняют под вакуумом нитрометан, органический слой отделяют и нейтрализуют, промывая теплой водой.
Из органического слоя (кетон малины в растворе толуола и трет-бутилтолуола) продукт выпадает с содержанием 95-98% основного вещества. При необходимости получения чистого вещества 1 проводят повторную кристаллизацию. Выход кетона малины на этой стадии составляет 80%. Параллельно мы применили и рекомендованный ранее метод очистки, который заключается в перегонке технического кетона малины с последующей двойной кристаллизацией. В этом случае получен кетон малины высокой степени чистоты с выходом 50%. Как видим, перегонка и двойная кристаллизация существенно уменьшают выход продукта.
Получение кетона малины на основе очищенного от исходного 2.6-ди-трет-бутилфенола образца соединения 6 существенно облегчает очистку соединения 1 от образующихся трет-бутилфенолов из примеси соединения 5 в продукте 6 в ходе их де- трет-бутилирования.
Способ отличается простой технологией, доступностью сырья и упрощенной методикой очистки продукта от примесей и демонстрируется следующими примерами.
Пример 1
4-(N,N-диэтиламин)бутан-2-он. В стальной вращающийся автоклав помещают 253 мл (3.44 мол) ацетона, 100 г (3.33 мол) параформа, 400 г (3.65 мол) солянокислого диэтиламина, 20 мл 36%-ной соляной кислоты и 100 мл воды, закрывают автоклав и при его вращении содержимое нагревают 1.5 часа при температуре 100°С. Из реакционной массы под вакуумом отгоняют ацетон и 90 г воды, остаток охлаждают до комнатной температуры и осторожно в течение 15 минут переливают в 2-литровую емкость, содержащую 160 г NaOH в виде мелких шариков. В емкость добавляют 0.5 кг NaCl и отделяют органический слой. Неорганический остаток отжимают. Всего получают 365 г 95%-ного 4-(N,N,-диэтиламин-2- бутанона (2.5 мол). Продукт хранят в холодильнике при температуре не выше 4°С.
Пример 2.
4-(2',6'-ди-трет-бутил- гидроксифенил)-бутан-2-он (6). В перегонную колбу с дефлегматором длиной 10 см, капельной воронкой, широким капилляром, доходящим до дна колбы, приемным устройством с холодильником с выходом на вакуумную систему, помещают 70 г (0.34 мол) 2.6-ди-трет-бутилфенола 5, 7 г растертого КОН и 100 мл диметилформамида (ДМФА). Содержимое доводят до кипения и отгоняют 8 г фракции, содержащей преимущественно воду, образующееся в ходе синтеза фенолята 5. Затем при слабом кипении реакционной массы в реактор под вакуумом 70 мм рт.ст. в течение 2.5 часа прикапывают в токе азота 70 г 4-(N,N-диэтиламин)бутан-2-она с одновременной отгонкой в приемник диэтиламина. После чего реакционную массу выдерживают еще 0.5 часа. В нейтральной пробе реакционной массы по данным ГЖХ кроме растворителя содержится 80% продукта и 20% 2.6-ди-трет-бутилфенола.
гидроксифенил)-бутан-2-он (6). В перегонную колбу с дефлегматором длиной 10 см, капельной воронкой, широким капилляром, доходящим до дна колбы, приемным устройством с холодильником с выходом на вакуумную систему, помещают 70 г (0.34 мол) 2.6-ди-трет-бутилфенола 5, 7 г растертого КОН и 100 мл диметилформамида (ДМФА). Содержимое доводят до кипения и отгоняют 8 г фракции, содержащей преимущественно воду, образующееся в ходе синтеза фенолята 5. Затем при слабом кипении реакционной массы в реактор под вакуумом 70 мм рт.ст. в течение 2.5 часа прикапывают в токе азота 70 г 4-(N,N-диэтиламин)бутан-2-она с одновременной отгонкой в приемник диэтиламина. После чего реакционную массу выдерживают еще 0.5 часа. В нейтральной пробе реакционной массы по данным ГЖХ кроме растворителя содержится 80% продукта и 20% 2.6-ди-трет-бутилфенола.
Из реактора под вакуумом 5-7 мм рт.ст. отгоняют последовательно 50 г ДМФА, 15.5 г (0.075 мол) 2.6-ди-трет-бутилфенола и в интервале 170-180°С/5 мм рт.ст. собирают фракцию 50.5 г (0.19 мол) 4-(2',6'-ди-трет-бутил-гидроксифенил)-бутан-2-она. Кристаллизацией ее из спирта получают чистый образец 6 с т. пл. 45-46°С. Точка плавления и спектральные характеристики соответствуют литературным данным образца, полученного нами ранее взаимодействием 2.6-ди-трет-бутилфенола с метилвинилкетоном [Т.Ф. Титова, А.П. Крысин, М.М. Шакиров, В.И. Маматюк. ЖОрХ. Т. 20 №2 (1984) С. 331-338].
При повторной наработке соединения 6 вместо калиевой соли 2.6-ди-трет-бутилфенола, приготовляемой в качестве катализатора, используют полученный ранее кубовый остаток перегонки реакционной массы, состоящей в основном из калиевой соли 4-(2',6'-ди-трет-бутил--гидроксифенил)-бутан-2-она. Этот подход исключает стадию приготовления катализатора, исходя из соединения 5, и увеличивает выход продукта 1. Процесс получения продукта 6 реализуется без образования сточных вод.
Последовательность получения кетона малины демонстрируется на следующем примере.
Пример 3.
4-(4'гидроксифенил)-бутан-2-он, 1. В трехгорлую колбу вместимостью 0.5 л с мешалкой, обратным холодильником капельной воронкой и вводом инертного газа загружают 20 г. AlCl3 и 40 мл нитрометана и при перемешивании получают прозрачный раствор. Из капельной воронки к нему в течение 0.5 часа прикапывают раствор 50 г 4-(2',6'-ди-трет-бутил-гидроксифенил)-бутан-2-она (0.17 мол) в 100 мл толуола, следя за тем, чтобы температура реакционной массы не превышала 50°С. Реакционную массу выдерживают 2 часа при температуре 30-50°С и выливают на 0.5 кг льда. Органический слой отделяют и из него перегонкой выделяют 35 г нитрометана. Из кубового остатка (кетон малины в толуоле и трет-бутилтолуоле) выпадает светлый кристаллический осадок 23.5 г (0.145 мол) 95%-ного 4-(-гидроксифенил)-бутан-2-она, который кристаллизуют из толуола, получая 20 г (0.21 мол) кетона малины с запахом малины с т.пл. 83-85°С.
Claims (1)
- Способ получения кетона малины, отличающийся тем, что 4-(3',5'-ди-трет-бутил-4'-гидроксифенил)-бутан-2-он и толуол нагревают с раствором AlCl3 в нитрометане при температуре 30-50°C и после разложения реакционной массы водой продукт выделяют кристаллизацией органического слоя.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2016148283A RU2637312C1 (ru) | 2016-12-08 | 2016-12-08 | Способ получения кетона малины |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2016148283A RU2637312C1 (ru) | 2016-12-08 | 2016-12-08 | Способ получения кетона малины |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| RU2637312C1 true RU2637312C1 (ru) | 2017-12-04 |
Family
ID=60581441
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2016148283A RU2637312C1 (ru) | 2016-12-08 | 2016-12-08 | Способ получения кетона малины |
Country Status (1)
| Country | Link |
|---|---|
| RU (1) | RU2637312C1 (ru) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113666814A (zh) * | 2021-09-14 | 2021-11-19 | 江西开源香料有限公司 | 一种高纯度覆盆子酮的合成方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4908481A (en) * | 1980-04-22 | 1990-03-13 | Basf Aktiengesellschaft | Preparation of 1-(4-hydroxy-phenyl)-butan-3-one and novel intermediates |
| CN104193607A (zh) * | 2014-09-10 | 2014-12-10 | 曹仪山 | 一种覆盆子酮的合成方法 |
-
2016
- 2016-12-08 RU RU2016148283A patent/RU2637312C1/ru not_active IP Right Cessation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4908481A (en) * | 1980-04-22 | 1990-03-13 | Basf Aktiengesellschaft | Preparation of 1-(4-hydroxy-phenyl)-butan-3-one and novel intermediates |
| CN104193607A (zh) * | 2014-09-10 | 2014-12-10 | 曹仪山 | 一种覆盆子酮的合成方法 |
Non-Patent Citations (3)
| Title |
|---|
| M.Bandarenko et al, Synthesis of Raspberry and Ginger Ketones by Nickel Boride-catalyzed Hydrogenation of 4-Arylbut-3-en-2-ones. Zeitschrift fur Naturforschung. Section B. Journal of Chemical Sciences, 2014, 69(8), 885-888. * |
| M.Bandarenko et al, Synthesis of Raspberry and Ginger Ketones by Nickel Boride-catalyzed Hydrogenation of 4-Arylbut-3-en-2-ones. Zeitschrift fur Naturforschung. Section B. Journal of Chemical Sciences, 2014, 69(8), 885-888. Колтунов К.Ю. и др. Ионное гидрирование α , β -кетонов циклогексаном в присутствии галогенидов алюминия. ЖОрХ, 2001, том 37, N 11, 1610-1617. * |
| Колтунов К.Ю. и др. Ионное гидрирование α , β -кетонов циклогексаном в присутствии галогенидов алюминия. ЖОрХ, 2001, том 37, N 11, 1610-1617. * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113666814A (zh) * | 2021-09-14 | 2021-11-19 | 江西开源香料有限公司 | 一种高纯度覆盆子酮的合成方法 |
| CN113666814B (zh) * | 2021-09-14 | 2024-04-23 | 江西开源香料有限公司 | 一种高纯度覆盆子酮的合成方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU745789B2 (en) | A method for producing para-menthane-3,8-diol | |
| Baran | Method for the cleavage of osmate esters | |
| JP4587549B2 (ja) | 1−ハロゲノ−3−1−メントキシプロパン−2−オール | |
| JP3766591B2 (ja) | シクロヘキセノン長鎖アルコール及びこれを含有する医薬 | |
| AU3433502A (en) | Method for producing 3-1-menthoxypropane-1,2-diol | |
| RU2727202C2 (ru) | Улучшенный синтез гонокиола | |
| RU2637312C1 (ru) | Способ получения кетона малины | |
| ES2754351T3 (es) | Proceso | |
| EP0214426B1 (en) | Intermediates in the synthesis of carboxylic acids | |
| Kumar et al. | Antimicrobial activity of the major isolates of mentha oil and derivatives of menthol | |
| US4517382A (en) | 1-Formyl-tri- and tetramethyl-cyclohex-1-en-3-one oximes | |
| KR101038184B1 (ko) | 화장 활성물질의 제조 방법 | |
| EP2373606B1 (en) | Processes for epimerizing cyclohexenyl ketones with subsequent aldol condensation to produce fragrance compounds | |
| RU2478606C1 (ru) | Способ получения 1-(2-метил-4-феноксифенил)-бутан-1,3-диона | |
| JP5220403B2 (ja) | カテコール基が導入されたジオキサビシクロ[3.3.0]オクタン誘導体の製造方法 | |
| Uchil et al. | Selective reductions of substituted α-(1, 2, 4-triazol-l-yl) chalcones with NaBH 4 and Al-isopropoxide: Synthesis of substituted (±) α-(4-chlorophenyl)-β-(phenylmethylene)-1H-1, 2, 4-triazole-l-ethanols having potential bacteriostatic and agro-based fungicidal activity | |
| JP5080776B2 (ja) | エステル化合物 | |
| JP2010065014A (ja) | 抗アクネ菌化合物及びその製造方法 | |
| Conant et al. | GAMMA-CHLOROPROPYL-PHENYLKETONE | |
| KR20050114238A (ko) | 2-(l-멘톡시)에탄올류의 제조 방법 | |
| SU884562A3 (ru) | Способ получени вторичных амидов дихлоруксусной кислоты | |
| JPS6033371B2 (ja) | トランス−p−メンタン−2,3−ジオ−ルの製造方法 | |
| US8093432B2 (en) | Processes for epimerizing cyclohexenyl ketones with subsequent aldol condensation to produce fragrance compounds | |
| KR100468237B1 (ko) | 멘톨의 제조방법 | |
| Hochstetler et al. | Hydrochlorination of thujopsene |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20181209 |