RU2509084C2 - Получение промежуточных соединений, используемых в синтезе 2'-циано-2'-дезокси-n4-пальмитоил-1-бета-d-арабинофуранозилцитозина - Google Patents
Получение промежуточных соединений, используемых в синтезе 2'-циано-2'-дезокси-n4-пальмитоил-1-бета-d-арабинофуранозилцитозина Download PDFInfo
- Publication number
- RU2509084C2 RU2509084C2 RU2010150153/04A RU2010150153A RU2509084C2 RU 2509084 C2 RU2509084 C2 RU 2509084C2 RU 2010150153/04 A RU2010150153/04 A RU 2010150153/04A RU 2010150153 A RU2010150153 A RU 2010150153A RU 2509084 C2 RU2509084 C2 RU 2509084C2
- Authority
- RU
- Russia
- Prior art keywords
- formula
- compound
- treating
- converting
- methanol
- Prior art date
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 187
- 238000004519 manufacturing process Methods 0.000 title claims description 12
- 230000015572 biosynthetic process Effects 0.000 title description 11
- 238000003786 synthesis reaction Methods 0.000 title description 8
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 claims abstract description 87
- 238000000034 method Methods 0.000 claims abstract description 52
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims abstract description 23
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 claims abstract description 18
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims abstract description 18
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims abstract description 16
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims abstract description 9
- NGEZPLCPKXKLQQ-VOTSOKGWSA-N (e)-4-(3-methoxyphenyl)but-3-en-2-one Chemical compound COC1=CC=CC(\C=C\C(C)=O)=C1 NGEZPLCPKXKLQQ-VOTSOKGWSA-N 0.000 claims abstract description 8
- 239000012453 solvate Substances 0.000 claims abstract description 8
- 229910019093 NaOCl Inorganic materials 0.000 claims abstract description 7
- GDOPTJXRTPNYNR-UHFFFAOYSA-N methyl-cyclopentane Natural products CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 claims abstract description 7
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 claims abstract description 7
- 238000001953 recrystallisation Methods 0.000 claims abstract description 5
- 239000007800 oxidant agent Substances 0.000 claims abstract description 3
- 239000000203 mixture Substances 0.000 claims description 35
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 claims description 32
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 claims description 24
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 claims description 22
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 claims description 21
- SCHZCUMIENIQMY-UHFFFAOYSA-N tris(trimethylsilyl)silicon Chemical compound C[Si](C)(C)[Si]([Si](C)(C)C)[Si](C)(C)C SCHZCUMIENIQMY-UHFFFAOYSA-N 0.000 claims description 11
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 claims description 6
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 claims description 6
- INFPIPCTRVDPJG-UHFFFAOYSA-N o-naphthalen-2-yl chloromethanethioate Chemical compound C1=CC=CC2=CC(OC(=S)Cl)=CC=C21 INFPIPCTRVDPJG-UHFFFAOYSA-N 0.000 claims description 6
- 150000003254 radicals Chemical class 0.000 claims description 6
- MNWBNISUBARLIT-UHFFFAOYSA-N sodium cyanide Chemical compound [Na+].N#[C-] MNWBNISUBARLIT-UHFFFAOYSA-N 0.000 claims description 6
- QYTDEUPAUMOIOP-UHFFFAOYSA-N TEMPO Chemical group CC1(C)CCCC(C)(C)N1[O] QYTDEUPAUMOIOP-UHFFFAOYSA-N 0.000 claims description 3
- 238000006243 chemical reaction Methods 0.000 abstract description 43
- 206010028980 Neoplasm Diseases 0.000 abstract description 6
- 201000011510 cancer Diseases 0.000 abstract description 4
- 239000000126 substance Substances 0.000 abstract description 4
- 239000002718 pyrimidine nucleoside Substances 0.000 abstract description 3
- 230000000694 effects Effects 0.000 abstract description 2
- 230000006872 improvement Effects 0.000 abstract description 2
- 238000004140 cleaning Methods 0.000 abstract 1
- 238000000605 extraction Methods 0.000 abstract 1
- 230000002265 prevention Effects 0.000 abstract 1
- LBGFKUUHOPIEMA-PEARBKPGSA-N sapacitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](C#N)[C@H](O)[C@@H](CO)O1 LBGFKUUHOPIEMA-PEARBKPGSA-N 0.000 description 41
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 35
- 239000000543 intermediate Substances 0.000 description 26
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 17
- 239000000047 product Substances 0.000 description 17
- IIEWJVIFRVWJOD-UHFFFAOYSA-N ethylcyclohexane Chemical compound CCC1CCCCC1 IIEWJVIFRVWJOD-UHFFFAOYSA-N 0.000 description 16
- 239000011541 reaction mixture Substances 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 9
- 239000007787 solid Substances 0.000 description 8
- 239000000243 solution Substances 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- 238000005119 centrifugation Methods 0.000 description 6
- 235000019439 ethyl acetate Nutrition 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 230000003647 oxidation Effects 0.000 description 5
- 238000007254 oxidation reaction Methods 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 4
- 229960002949 fluorouracil Drugs 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 239000000725 suspension Substances 0.000 description 4
- MWFMGBPGAXYFAR-UHFFFAOYSA-N 2-hydroxy-2-methylpropanenitrile Chemical compound CC(C)(O)C#N MWFMGBPGAXYFAR-UHFFFAOYSA-N 0.000 description 3
- 238000006646 Dess-Martin oxidation reaction Methods 0.000 description 3
- 230000000259 anti-tumor effect Effects 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 238000002425 crystallisation Methods 0.000 description 3
- 230000008025 crystallization Effects 0.000 description 3
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 3
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- 238000004821 distillation Methods 0.000 description 3
- 229960005277 gemcitabine Drugs 0.000 description 3
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 150000002576 ketones Chemical class 0.000 description 3
- 239000002244 precipitate Substances 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- 0 C*C(NC(C=CN1[C@]([C@]2C=NC)OC(CO)[C@]2O)=NC1=O)=O Chemical compound C*C(NC(C=CN1[C@]([C@]2C=NC)OC(CO)[C@]2O)=NC1=O)=O 0.000 description 2
- -1 CNDAC nucleoside Chemical class 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 2
- KXKVLQRXCPHEJC-UHFFFAOYSA-N acetic acid trimethyl ester Natural products COC(C)=O KXKVLQRXCPHEJC-UHFFFAOYSA-N 0.000 description 2
- 230000010933 acylation Effects 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- 229910001508 alkali metal halide Inorganic materials 0.000 description 2
- 150000008045 alkali metal halides Chemical class 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 238000009835 boiling Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 208000029742 colonic neoplasm Diseases 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000004093 cyano group Chemical group *C#N 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- RCBVKBFIWMOMHF-UHFFFAOYSA-L hydroxy-(hydroxy(dioxo)chromio)oxy-dioxochromium;pyridine Chemical compound C1=CC=NC=C1.C1=CC=NC=C1.O[Cr](=O)(=O)O[Cr](O)(=O)=O RCBVKBFIWMOMHF-UHFFFAOYSA-L 0.000 description 2
- 239000003999 initiator Substances 0.000 description 2
- 239000002777 nucleoside Substances 0.000 description 2
- KOSYAAIZOGNATQ-UHFFFAOYSA-N o-phenyl chloromethanethioate Chemical compound ClC(=S)OC1=CC=CC=C1 KOSYAAIZOGNATQ-UHFFFAOYSA-N 0.000 description 2
- 239000012044 organic layer Substances 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 239000003208 petroleum Substances 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- OOSZCNKVJAVHJI-UHFFFAOYSA-N 1-[(4-fluorophenyl)methyl]piperazine Chemical compound C1=CC(F)=CC=C1CN1CCNCC1 OOSZCNKVJAVHJI-UHFFFAOYSA-N 0.000 description 1
- CKTSBUTUHBMZGZ-SHYZEUOFSA-N 2'‐deoxycytidine Chemical class O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 CKTSBUTUHBMZGZ-SHYZEUOFSA-N 0.000 description 1
- AVTLBBWTUPQRAY-UHFFFAOYSA-N 2-(2-cyanobutan-2-yldiazenyl)-2-methylbutanenitrile Chemical compound CCC(C)(C#N)N=NC(C)(CC)C#N AVTLBBWTUPQRAY-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- GBAHDBQTXWIRJT-UHFFFAOYSA-N 4-amino-4-hexadecanoyl-1,3-dihydropyrimidin-2-one Chemical compound CCCCCCCCCCCCCCCC(=O)C1(N)NC(=O)NC=C1 GBAHDBQTXWIRJT-UHFFFAOYSA-N 0.000 description 1
- 206010065553 Bone marrow failure Diseases 0.000 description 1
- PMSMULGILVYFGA-AUGDVEQESA-N C=N/C=C1/[C@H](N(C=CC(N)=N2)C2=O)O[C@H](CO)[C@H]1O Chemical compound C=N/C=C1/[C@H](N(C=CC(N)=N2)C2=O)O[C@H](CO)[C@H]1O PMSMULGILVYFGA-AUGDVEQESA-N 0.000 description 1
- ZSEZLADXDFONRC-ZHHKINOHSA-N CC(C)[Si+](C(C)C)(OC[C@H]1O[C@H]2N(C=CC(NC(C)=O)=N3)C3=O)O[Si+](C(C)C)(C(C)C)O[C@H]1[C@H]2O Chemical compound CC(C)[Si+](C(C)C)(OC[C@H]1O[C@H]2N(C=CC(NC(C)=O)=N3)C3=O)O[Si+](C(C)C)(C(C)C)O[C@H]1[C@H]2O ZSEZLADXDFONRC-ZHHKINOHSA-N 0.000 description 1
- JYYTZOGVMGVABJ-RKZKBVABSA-N CC(C)[Si](C(C)C)(O)O[Si+](C(C)C)(C(C)C)OC[C@H](CC1(C#N)OC(Oc2ccc(cccc3)c3c2)=S)O[C@H]1N(C)C(/N=C(\C)/NC(C)=O)=O Chemical compound CC(C)[Si](C(C)C)(O)O[Si+](C(C)C)(C(C)C)OC[C@H](CC1(C#N)OC(Oc2ccc(cccc3)c3c2)=S)O[C@H]1N(C)C(/N=C(\C)/NC(C)=O)=O JYYTZOGVMGVABJ-RKZKBVABSA-N 0.000 description 1
- SUIDQKCEDFKSTL-PIIVXVCUSA-N CC(C)[Si](C(C)C)(OCC1O[C@H]2N(C=CC(NC(C)=O)=N3)C3=O)O[Si+](C(C)C)(C(C)C)O[C@H]1C2(C#N)O Chemical compound CC(C)[Si](C(C)C)(OCC1O[C@H]2N(C=CC(NC(C)=O)=N3)C3=O)O[Si+](C(C)C)(C(C)C)O[C@H]1C2(C#N)O SUIDQKCEDFKSTL-PIIVXVCUSA-N 0.000 description 1
- SPCFAQYAFILSAC-IPKCRJEZSA-N CC(C)[Si](C(C)C)(OCC1O[C@H]2N(C=CC(NC(C)=O)=N3)C3=O)O[Si](C(C)C)(C(C)C)O[C@H]1C2=O Chemical compound CC(C)[Si](C(C)C)(OCC1O[C@H]2N(C=CC(NC(C)=O)=N3)C3=O)O[Si](C(C)C)(C(C)C)O[C@H]1C2=O SPCFAQYAFILSAC-IPKCRJEZSA-N 0.000 description 1
- YIZQXPUKALQEIW-MAMNIGPBSA-N CC(C)[Si](C(C)C)(OC[C@H]([C@@H]12)O[C@H]3N(C=CC(NC(C)=O)=N4)C4=O)O[Si](C(C)C)(C(C)C)[O]1=[O]C23C#N Chemical compound CC(C)[Si](C(C)C)(OC[C@H]([C@@H]12)O[C@H]3N(C=CC(NC(C)=O)=N4)C4=O)O[Si](C(C)C)(C(C)C)[O]1=[O]C23C#N YIZQXPUKALQEIW-MAMNIGPBSA-N 0.000 description 1
- BYGXNMXHFLBDJX-UHFFFAOYSA-N CC(C)[Si](C(C)C)([O](OCC(CC1O)OC1N(C=CC(N)=N1)C1=O)S(C(C)C)C(C)C)[O]#C Chemical compound CC(C)[Si](C(C)C)([O](OCC(CC1O)OC1N(C=CC(N)=N1)C1=O)S(C(C)C)C(C)C)[O]#C BYGXNMXHFLBDJX-UHFFFAOYSA-N 0.000 description 1
- XXTDGTXAUSSCHH-XCWAXFADSA-N CC([C@@H]1[C@H](N(C=CC(N)=N2)C2=O)O[C@H](CO)[C@H]1O)=N Chemical compound CC([C@@H]1[C@H](N(C=CC(N)=N2)C2=O)O[C@H](CO)[C@H]1O)=N XXTDGTXAUSSCHH-XCWAXFADSA-N 0.000 description 1
- 238000003109 Karl Fischer titration Methods 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 101100010166 Mus musculus Dok3 gene Proteins 0.000 description 1
- DCYBPMFXJCWXNB-UXXCCHNYSA-N NC(C=CN1[C@@H]([C@H]2C#N)O[C@@H](CO)[C@H]2O)=NC1=O Chemical compound NC(C=CN1[C@@H]([C@H]2C#N)O[C@@H](CO)[C@H]2O)=NC1=O DCYBPMFXJCWXNB-UXXCCHNYSA-N 0.000 description 1
- 238000006859 Swern oxidation reaction Methods 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- CAHGKCFTENMQGB-UHFFFAOYSA-N cyanic acid propan-2-one Chemical compound C(#N)O.CC(=O)C CAHGKCFTENMQGB-UHFFFAOYSA-N 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 230000001066 destructive effect Effects 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 1
- 231100000371 dose-limiting toxicity Toxicity 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- BCDGQXUMWHRQCB-UHFFFAOYSA-N glycine methyl ketone Natural products CC(=O)CN BCDGQXUMWHRQCB-UHFFFAOYSA-N 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- XEFNBUBDJCJOGM-OTUUDDROSA-N n-[1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-2-oxopyrimidin-4-yl]hexadecanamide Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCC)C=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 XEFNBUBDJCJOGM-OTUUDDROSA-N 0.000 description 1
- 229940127073 nucleoside analogue Drugs 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 238000009520 phase I clinical trial Methods 0.000 description 1
- 238000005191 phase separation Methods 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 229940074545 sodium dihydrogen phosphate dihydrate Drugs 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- DBGVGMSCBYYSLD-UHFFFAOYSA-N tributylstannane Chemical compound CCCC[SnH](CCCC)CCCC DBGVGMSCBYYSLD-UHFFFAOYSA-N 0.000 description 1
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0834—Compounds having one or more O-Si linkage
- C07F7/0838—Compounds with one or more Si-O-Si sequences
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
Настоящее изобретение относится к способу получения соединения формулы 682, которое находится в кристаллической форме, где указанный способ включает: (i) обработку соединения формулы 682-9 пальмитиновым ангидридом в смеси Н2О/диоксан с образованием соединения формулы 682; (ii) обработку продукта, полученного на стадии (i), метанолом с получением соединения формулы 682 в форме сольвата с метанолом (форма К); (iii) выделение полученного на стадии (ii) соединения формулы 682 в форме сольвата с метанолом (форма К); (iv) необязательную очистку продукта стадии (iii) с помощью перекристаллизации. Также заявлен способ получения соединения формулы 682-4, где указанный способ включает (i) превращение соединения формулы 682-1 в соединение формулы 682-2' путем обработки указанного соединения формулы 682-1 1,3-дихлор-1,1,4,4-тетраизопропилдисилоксаном (CIPS) в пиридине; (ii) превращение указанного соединения формулы 682-2' в соединение формулы 682-3 путем обработки указанного соединения формулы 682-2' уксусным ангидридом в EtOH; и (iii) превращение указанного соединения формулы 682-3 в соединение формулы 682-4 путем обработки указанного соединения формулы 682-3 окислителем, предпочтительно свободным радикалом 2,2,6,6-тетраметилпиперидинилокси (TEMPO) и NaOCl. Дальнейшие аспекты изобретения относятся к применению вышеназванных способов для получения 2'-циано-2'-дезокси-N4-пальмитоил-1-β-D-арабинофуранозилцитозина, пиримидинового нуклеозида, который является пригодным для лечения и/или предупреждения рака. 7 н. и 8 з.п. ф-лы, 1 табл., 1 пр., 2 ил.
Description
Настоящее изобретение относится к получению промежуточных соединений, используемых в синтезе 2'-циано-2'-дезокси-N4-пальмитоил-1-β-D-арабинофуранозилцитозина, пиримидинового нуклеозида, который применяют для лечения и/или предупреждения рака. В частности, изобретение обеспечивает улучшенный способ получения 2'-циано-2'-дезокси-N4-пальмитоил-1-β-D-арабинофуранозилцитозина.
УРОВЕНЬ ТЕХНИКИ ИЗОБРЕТЕНИЯ
Терапевтическое применение пиримидиновых нуклеозидов для лечения пролиферативных расстройств подробно описано в уровне техники. В качестве примера, коммерчески доступные противоопухолевые средства пиримидинового ряда включают 5-фторурацил (Duschinsky, R., и др. J. Am. Chem. Soc., 79, 4559 (1957)), тегафур (Hiller, SA., и др., Dokl. Akad. Nauk USSR, 176, 332 (1967)), УФТ (Fujii, S., и др., Gann, 69, 763 (1978)), кармофур (Hoshi, A., и др., Gann, 67, 725 (1976)), доксифлуридин (Cook, A. F., и др., J. Med. Chem., 22, 1330 (1979)), цитарабин (Evance, J. S., и др., Proc. Soc. Exp. Bio. Med., 106. 350 (1961)), анцитабин (Hoshi, A., и др., Gann, 63, 353, (1972)) и эноцитабин (Aoshima, M., и др., Cancer Res., 36, 2726 (1976)).
ЕР 536936 (Sankyo Company Limited) раскрывает различные 2'-циано-2'-дезокси-производные 1-β-D-арабинофуранозилцитозина, которые, как было показано, обладают значительной противоопухолевой активностью. Одним конкретным соединением, раскрытым в ЕР 536936, является 2'-циано-2'-дезокси-N4-пальмитоил-1-β-D-арабинофуранозилцитозин (называемый в дальнейшем «682» или «CYC682»); в настоящее время данное соединение проходит дальнейшее исследование.
CYC682, также известный как 1-(2-С-циано-2-диокси-β-D-арабинофуранозил)-N4-пальмитоилцитозин, (Hanaoka, K., и др., Int. J. Cancer, 1999:82:226-236; Donehower R, и др., Proc Am Soc Clin Oncol, 2000: реферат 764; Burch, PA, и др., Proc Am Soc Clin Oncol, 2001: реферат 364), представляет собой антиметаболит 2'-дезоксицитидина и является новым предназначенным для перорального приема пролекарством нуклеозида CNDAC (1-(2-С-циано-2'-дезокси-β-D-арабинопентафуранозил)цитозин.

В отличие от других метаболитов нуклеозидов, таких как гемцитабин, CYC682 обладает другим механизмом действия, который заключается в том, что он обладает самопроизвольным разрушающим действием по отношению к нитям ДНК, что приводит к высокой противоопухолевой активности на различных линиях клеток, ксенотрансплантате и метастатических раковых моделях.
CYC682 находился в центре внимания ряда исследований из-за его оральной биологической доступности и его улучшенной активности по сравнению с гемцитабином (лидирующий на рынке нуклеозидный аналог) и 5-ФУ(5-FU) (широко применяемое антиметаболитное лекарственное средство) на основании результатов предварительных клинических испытаний на солидных опухолях. Недавно исследователи сообщили, что CYC682 обладает сильной противоопухолевой активностью на модели рака толстой кишки. На этой же модели было обнаружено, что как по увеличению выживаемости, так и по предотвращению распространения метастаз рака толстой кишки в печень, CYC682 превосходит как гемцитабин, так и 5-ФУ (Wu M, и др., Cancer Research, 2003:63:2477-2482). На сегодняшний день, данные фазы I клинических испытаний на пациентах с различными раковыми заболеваниями подтверждают, что CYC682 хорошо переносится людьми, обладая миелосупрессией в качестве лимитирующей дозу токсичности.
Последние исследования были направлены на различные кристаллические формы CYC682 (например, WO 02/064609, Sankyo Company Limited) и оптимизированные композиции, содержащие CYC682, которые обладают улучшенной стабильностью и которые позволяют облегчить обработку (например, WO 07/072061, Cyclacel Limited).
Получение CYC682, описанное в EP 536936 (см. схему 1 далее), включает реакцию цитидина [1] с пальмитиновым ангидридом в ДМФА с образованием N4-пальмитоилцитидина [2], и последующую защиту с использованием 1,3-дихлор-1,1,4,4-тетраизопропилдисилоксаном (CIPS) с образованием промежуточного соединения [3]. Окисление [3] с использованием дихромата пиридиния/уксусного ангидрида в дихлорметане дает промежуточный кетон [4], который затем взаимодействует с цианидом натрия и дигидратом дигидрофосфата натрия в этилацетате, давая циангидрин [5]. Промежуточное соединение [5] затем взаимодействует с N,N-диметиламинопиридином, фенокситиокарбонилхлоридом и триэтиламином, с образованием промежуточного соединения [6], который далее вводят в реакцию с AIBN и трибутилолом в толуоле, получая промежуточное соединение [7]. Снятие защиты в [7] под действием уксусной кислоты и тетрабутиламмонийфторида в ТГФ(THF) дает целевой продукт, CYC682.

Схема 1: Получение CYC682, как описано в EP 536936
Дальнейшие модификации вышеописанного подхода были описаны в JP 07053586 (Sankyo Company Limited). В частности, в JP 07053586 раскрыто, что стадия окисления может быть проведена с использованием свободного радикала 2,2,6,6-тетраметилпиперидинилокси (ТЕМРО), NaOCl и галогенида щелочного металла (см. превращение [3а] в [4а] на схеме 2, приведенное далее). Кроме этого, превращение кетона [4a] в циангидриновое промежуточное соединение [5a] может быть достигнуто за счет обработки [4a] вместо NaCN циангидрином ацетона. Полученный циангидрин [5a] затем может быть обработан 2-нафтилхлортиоформиатом, давая промежуточное соединение [6a].

Схема 2: Альтернативные условия, описанные в JP 07053586
Однако, несмотря на данные модификации, вышеуказанные пути связаны с относительно низкими выходами и/или высоким уровнем вариативности, что подчеркивает необходимость улучшения стратегий синтеза.
Таким образом, задача настоящего изобретения состоит в обеспечении улучшенного способа получения CYC682. Более конкретно, задача изобретения состоит в обеспечении синтетического пути, который приводит к лучшим выходам CYC682 и/или который является пригодным для крупномасштабного получения соединения.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Первый аспект изобретения относится к способу получения соединения формулы 682-4,

где указанный способ включает стадии:
(i) превращение соединения формулы 682-1 в соединение формулы 682-2';
(ii) превращение указанного соединения формулы 682-2' в соединение формулы 682-3; и
(iii) превращение указанного соединения формулы 682-3 в соединение формулы 682-4

Предпочтительно использование обратного порядка двух первых стадий синтеза для введения защитной группы CIPS перед защитой NH2-группы, что приводит к улучшению качества промежуточного соединения 682-4, который дает субстрат для последующей реакции с циангидрином при получении CYC682.
Второй аспект изобретения относится к способу получения соединения формулы 682-9 или 682

где указанный способ включает стадии:
(А) получение промежуточного соединения формулы 682-4, как описано выше;
(В) превращение соединения формулы 682-4 в соединение формулы 682-9; и
(С) необязательное превращение указанного соединения формулы 682-9 в соединение формулы 682.
Третий аспект изобретения относится к способу получения соединения формулы 682-5, где указанный способ включает обработку соединения формулы 682-4 циангидрином ацетона и NEt3 в гептане

Предпочтительно использование циангидрина ацетона и NEt3 в гептане, что приводит к улучшенному выходу и более легкой очистке промежуточного соединения 682-5 по сравнению с ранее известными в области техники условиями реакций.
Четвертый аспект изобретения относится к способу получения соединения формулы 682-9 или 682

где указанный способ включает стадии:
(А'') получение промежуточного соединения формулы 682-5, как описано выше;
(В'') превращение соединения формулы 682-5 в соединение формулы 682-9; и
(С'') необязательное превращение указанного соединения формулы 682-9 в соединение формулы 682.
ПОДРОБНОЕ ОПИСАНИЕ
Как было указано выше, первый аспект изобретения относится к способу получения соединения формулы 682-4, где указанный способ включает стадии:
(i) превращение соединения формулы 682-1 в соединение формулы 682-2';
(ii) превращение указанного соединения формулы 682-2' в соединение формулы 682-3; и
(iii) превращение указанного соединения формулы 682-3 в соединение формулы 682-4.
Предпочтительно введение защитной группы CIPS на первой стадии (i), что дает твердый продукт 682-2', который может быть легче очищен (например, за счет промывания) для удаления нежелательных побочных продуктов и любого избытка реагента для введения защитной группы CIPS. После очистки полученное таким образом твердое промежуточное соединение 682-2' далее ацилируют, с получением промежуточного соединения 682-3, которое затем окисляют, с получением промежуточного соединения 682-4. Возможность очистки 682-2' в твердом виде приводит к лучшему качеству вещества для использования в последующих стадиях способа, давая более высокие выходы и улучшенную воспроизводимость. Более конкретно, вышеуказанный путь приводит к лучшему качеству промежуточного соединения 682-4, который является субстратом для последующей реакции с образованием циангидрина в синтезе CYC682.
В одном предпочтительном варианте осуществления изобретения стадия (i) включает обработку указанного соединения формулы 682-1 1,3-дихлор-1,1,4,4-тетраизопропилдисилоксаном (CIPS) в пиридине. Более подробно данная реакция описана в Org. Process Dev., 4, 172 (2000); патенте США 6531584 B1 (2003); Org. Lett., 8, 55 (2006).
В одном предпочтительном варианте осуществления изобретения стадия (ii) включает обработку указанного соединения формулы 682-2' уксусным ангидридом в EtOH. Альтернативно в качестве растворителя может использоваться ДМФА [см. Angew. Chem. Int. Ed, 43, 3033 (2004)].
Окислители для превращения соединения 682-3 в соединение 682-4 на стадии (iii) известны специалисту в данной области. В качестве примера, превращение может быть осуществлено за счет окисления с использованием периодинана Десс-Мартин [аналоги способов описаны в Helv. Chim. Acta, 85, 224 (2002) и J. Org. Chem., 55, 5186 (1990)], окисления по Сверну [Org. Process Res. Dev., 4, 172 (2000) и J. Med. Chem., 48, 5504 (2005)], окисления под действием дихромата пиридиния или с использованием свободного радикала 2,2,6,6-тетраметилпиперидинилокси (TEMPO) и NaOCl.
В одном конкретном предпочтительном варианте осуществления изобретения стадия (iii) включает окисление указанного соединения формулы 682-3 с использованием свободного радикала 2,2,6,6-тетраметилпиперидинилокси (TEMPO) в присутствии галогенида щелочного металла и NaOCl. Более подробно данная реакция описана в JP 07053586 (Sankyo Company Limited).
Второй аспект изобретения относится к способу получения соединения формулы 682-9 или 682, где указанный способ включает стадии:
(А) получение промежуточного соединения формулы 682-4, как описано выше;
(В) превращение соединения формулы 682-4 в соединение формулы 682-9; и
(С) необязательное превращение указанного соединения формулы 682-9 в соединение формулы 682.
В одном предпочтительном варианте осуществления стадия (В) включает стадии:


(В1) превращение указанного соединения формулы 682-4 в соединение формулы 682-5;
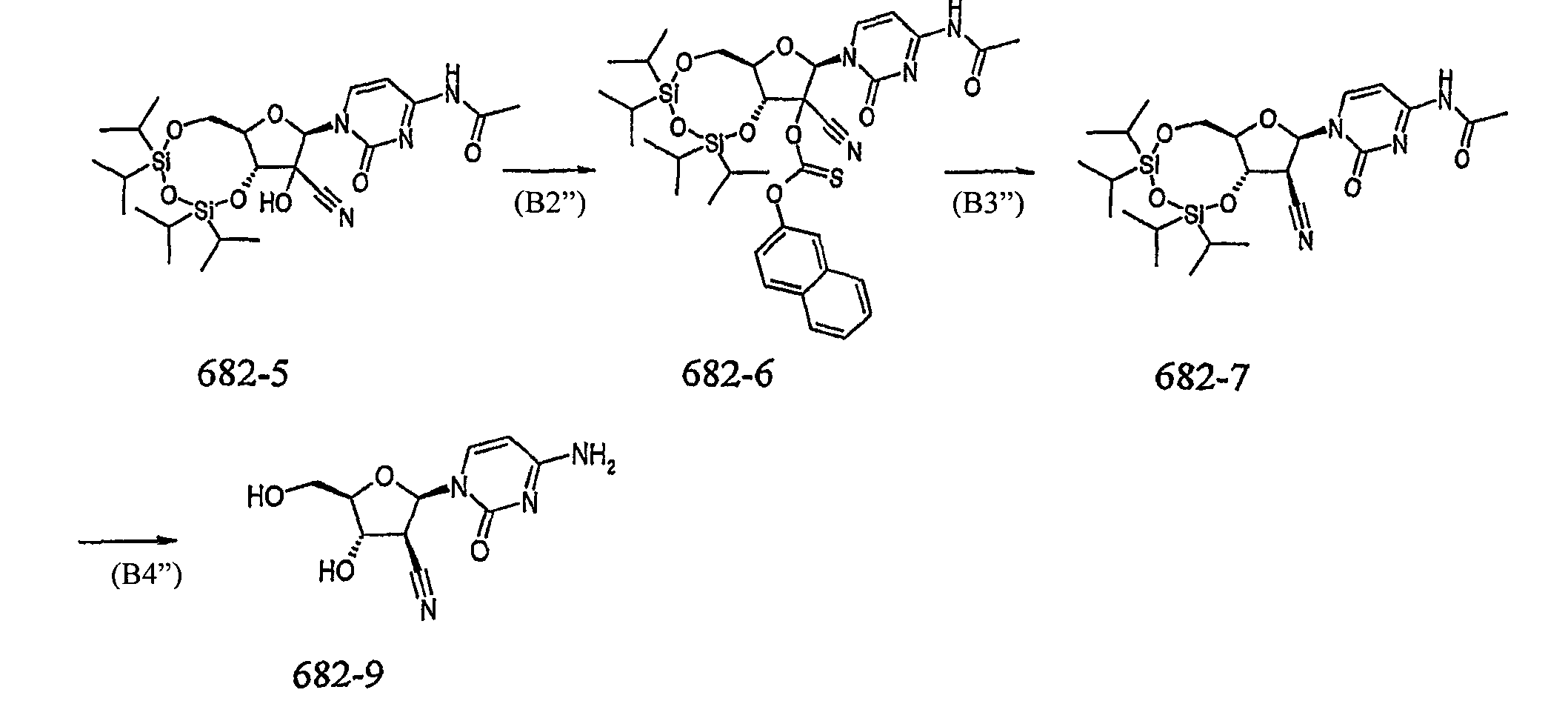
(В2) превращение указанного соединения формулы 682-5 в соединение формулы 682-6;
(В3) превращение указанного соединения формулы 682-6 в соединение формулы 682-7; и
(В4) превращение указанного соединения формулы 682-7 в соединение формулы 682-9.
В одном предпочтительном варианте осуществления стадия (В1) включает обработку указанного соединения формулы 682-4 NaCN/NaHCO3 в H2O/EtOH.
В другом предпочтительном варианте осуществления стадия (В1) включает обработку указанного соединения формулы 682-4 NaCN/NaH2PO4.2H2O в этилацетате. Более подробное описание данной реакции можно найти в EP 536936 (Sankyo Company Limited).
В другом предпочтительном варианте осуществления стадия (В1) включает обработку указанного соединения формулы 682-4 циангидрином ацетона/KH2PO4 в дихлорметане. Более подробное описание данной реакции можно найти в JP 07053586 (Sankyo Company Limited).
В одном конкретном предпочтительном варианте осуществления стадия (В1) включает обработку указанного соединения формулы 682-4 циангидртном ацетона и Net3 в гептане. Более подробное описание данной реакции описано ниже, во втором аспекте настоящего изобретения.
В еще одном альтернативном варианте осуществления стадия (В1) включает обработку указанного соединения формулы 682-4 TMSCN и AlCl3 в дихлорметане. Более подробное описание данной реакции приведено в Tet, 60, 9197 (2004).
Предпочтительно, стадия (В2) включает обработку указанного соединения формулы 682-5 2-нафтилхлортиоформиатом в присутствии NEt3 и диметиламинопиридина. Более подробное описание данной реакции приведено в JP 07053586 (Sankyo Company Limited).
Альтернативно, стадия (В2) включает обработку указанного соединения формулы 682-5 фенокситиокарбонилхлоридом в присутствии NEt3 и диметиламинопиридина. Более подробно данная реакция описана в EP 536936 (Sankyo Company Limited).
Предпочтительно, стадия (В3) включает обработку указанного соединения формулы 682-6 трис(триметилсилил)силаном (TTMSS) и азобисизобутиронитрилом (AIBN) в толуоле. Более подробно использование данных реагентов описано в J. Org. Chem., 53, 3641 (1988) и Tett. Lett., 44, 4027 (2003).
Альтернативно, стадия (В3) включает обработку указанного соединения формулы 682-6 гидридом трибутилолова и азобисизобутиронитрилом (AIBN) в толуоле, как описано в EP 536936 (Sankyo Company Limited).
Удаление CIPS защитной группы в указанном соединении формулы 682-7 на стадии (В4) и последующее выделение свободного основания 682-9 может быть достигнуто с использованием способов, известных специалисту в области техники. Предпочтительно, стадия (В4) включает обработку указанного соединения формулы 682-7 HCl/MeOH, и затем обработку полученного таким образом промежуточного соединения основанием для образования соединения формулы 682-9. Более подробное описание данной реакции можно найти в EP 536936 (Sankyo Company Limited).
Предпочтительно, стадия (С) включает обработку указанного соединения формулы 682-9 пальмитиновым ангидридом в смеси H2O/диоксан. Другие подходящие условия для данного превращения известны специалисту в области техники.
Следующий аспект изобретения относится к способу получения соединения формулы 682-5, где указанный способ включает обработку соединения формулы 682-4 циангидрином ацетона и NEt3 в гептане.

Предпочтительно использование циангидрина ацетона и NEt3 в гептане, что приводит к улучшенному выходу и более легкой очистке промежуточного соединения 682-5 по сравнению с ранее известными в области техники условиями реакций.
Условия предшествующего уровня техники обычно включают использование NaCN или циангидрина ацетона и триэтиламина в 2-фазной реакционной смеси (например, вода/этилацетат), что приводит к достижению равновесия между исходным соединением 682-4 и двумя возможными изомерами циангидрина. С другой стороны, использование циангидрина и NEt3 в гептане способствует образованию только одного из двух возможных циангидриновых продуктов; целевой циангидриновый продукт нерастворим в гептане и осаждается из раствора, в то время как другой изомер и исходный кетон 682-4 остаются в растворе. Такое осаждение сдвигает равновесие в сторону завершения реакции в соответствии с Принципом Ле Шателье, что приводит к улучшенным выходам целевого циангидрина. Более того, образование твердого осадка позволяет легче проводить обработку промежуточного соединения 682-5.
В одном предпочтительном варианте осуществления способ также включает стадию получения указанного соединения формулы 682-4 из соединения формулы 682-3.

Подходящие условия окисления описаны в первом аспекте изобретения. Более предпочтительно, способ включает взаимодействие соединения формулы 682-3 со свободным радикалом 2,2,6,6-тетраметилпиперидинилокси (ТЕМРО) и NaOCl.
В одном предпочтительном варианте осуществления способ также включает стадию получения указанного соединения формулы 682-3 из соединения формулы 682-2.

Более предпочтительно, способ включает взаимодействие указанного соединения формулы 682-2 с 1,3-дихлор-1,1,4,4-тетраизопропилдисилоксаном (CIPS) в пиридине. Подходящие условия данного превращения описаны ранее для первого аспекта изобретения.
В одном предпочтительном варианте осуществления способ также включает стадию получения указанного соединения формулы 682-2 из соединения формулы 682-1.

Более предпочтительно, способ включает взаимодействие указанного соединения формулы 682-1 с Ас2О в EtOH. Подходящие условия данного превращения описаны ранее для первого аспекта изобретения.
В одном предпочтительном варианте осуществления способ также включает стадию получения указанного соединения формулы 682-3 из соединения формулы 682-2'.

Более предпочтительно, способ включает реакцию указанного соединения формулы 682-2' с Ас2О в EtOH.
В одном предпочтительном варианте осуществления способ также включает стадию получения указанного соединения формулы 682-2' из соединения формулы 682-1.

Более предпочтительно, способ включает взаимодействие указанного соединения формулы 682-1 с 1,3-дихлор-1,1,4,4-тетраизопропилдисилоксаном (CIPS) в пиридине.
Следующий аспект изобретения относится к способу получения соединения формулы 682-9 или 682,

где указанный способ включает стадии:
(А'') получение промежуточного соединения формулы 682-5, как описано выше;
(В'') превращение соединения формулы 682-5 в соединение формулы 682-9; и
(С'') необязательное превращение указанного соединения формулы 682-9 в соединение формулы 682.
Предпочтительно, для данного варианта осуществления, стадия (B'') включает стадии:

(В2'') превращение указанного соединения формулы 682-5 в соединение формулы 682-6;
(В3'') превращение указанного соединения формулы 682-6 в соединение формулы 682-7; и
(В4'') превращение указанного соединения формулы 682-7 в соединение формулы 682-9.
Предпочтительно, стадия (В2'') включает обработку указанного соединения формулы 682-5 2-нафтилхлортиоформиатом в присутствии NEt3 и диметиламинопиридина. Подходящие условия данного превращения описаны ранее для первого аспекта изобретения.
Предпочтительно, стадия (В3'') включает обработку указанного соединения формулы 682-6 трис(триметилсилил)силаном (TTMSS) и азобисизобутиронитрилом (AIBN) в толуоле. Подходящие условия данного превращения описаны ранее для первого аспекта изобретения.
Предпочтительно, стадия (В4'') включает обработку указанного соединения формулы 682-7 HCl/MeOH, и затем обработку полученного таким образом промежуточного соединения основанием для образования соединения формулы 682-9. Подходящие условия данного превращения описаны ранее для первого аспекта изобретения.
Предпочтительно, стадия (С'') включает обработку указанного соединения формулы 682-9 пальмитиновым ангидридом в смеси H2O/диоксан.
Далее настоящее изобретение описано с помощью неограничивающих его примеров и со ссылками на следующие фигуры, где
На Фиг. 1 приведен синтез CYC682 по Пути 1, который является модификацией процедуры предшествующего уровня техники.
На Фиг. 2 приведен синтез CYC682 по Пути 1а, в соответствии с предпочтительным вариантом осуществления изобретения.
ПРИМЕРЫ
Стадия 1: 682-1 → 682-2'

Org. Process Dev., 4, 172 (2000); патент США 6531584 B1 (2003); Org. Lett., 8, 55 (2006).
Цитидин (8,0 г, 32,89 ммоль) предварительно сушат за счет отгонки азеотропа с пиридином (2×5 мл), затем суспендируют в пиридине (22 мл), и сосуд продувают аргоном. При комнатной температуре в течение 20 минут по каплям добавляют 1,3-дихлор-1,1,4,4-тетраизопропилдисилоксан (12,0 мл, 35,40 ммоль). Наблюдается небольшое выделение тепла до 32°С. Тяжелый белый осадок оседает на дно колбы. Его разбивают за счет интенсивного перемешивания, и полученную плотную суспензию перемешивают в течение ночи. Смесь выливают в воду (200 мл) и экстрагируют EtOAc (3×200 мл). Объединенные органические экстракты промывают (соляным раствором), сушат (MgSO4), фильтруют и упаривают, получая белое твердое вещество. Его растирают с гептаном, отфильтровывают и промывают гептаном (100 мл), а затем низкокипящим петролейным эфиром (2×50 мл). Получают 13,46 г (84%). На последнем этапе обработки изопропилацетат может быть заменен на гептан.
Стадия 2: 682-2' → 682-3

682-2' (10,0 г, 20,59 ммоль) суспендируют в этаноле (200 мл), и по каплям (без разогрева) добавляют уксусный ангидрид (6,9 мл, 72,06 ммоль). Смесь нагревают (масляная баня 65°С - внутренняя температура 50-53°С) в течение 2 часов. На ТСХ (7% MeOH/ДХМ) виден продукт лишь со следами исходного вещества. Добавляют еще 3 мл уксусного ангидрида (без разогрева) и продолжают нагревать еще 1,5 часа. На ТСХ не наблюдается исходного вещества. Смесь охлаждают до комнатной температуры, и EtOH упаривают на роторном испарителе. Добавляют 5% NaHCO3 (100 мл) (СО2↑), и смесь экстрагируют 1:1 TBDME/гептан (3×100 мл). Объединенные органические слои промывают (солевой раствор), сушат (MgSO4), фильтруют и упаривают, получая белое пенообразное вещество 10,43 г, 96%.
Стадия 3: 682-3 → 682-4

682-3 (8,0 г, 15,15 ммоль) растворяют в ДХМ (120 мл) и охлаждают до 10°С в ледяной бане. Небольшими порциями добавляют Десс-Мартин периодинан (12,58 г, 28,78 ммоль), и капельную воронку промывают ДХМ (20 мл). Полученный мутный раствор перемешивают при охлаждении в течение 10 минут, затем в течение ночи при комнатной температуре. Смесь разбавляют Et2O (450 мл) и промывают водным раствором NaHCO3 (200 мл) в котором растворен Na2S2O3·5H2O (38,5 г). Водную фазу экстрагируют Et2O (200 мл). Объединенные органические слои промывают (насыщенный NaHCO3, затем солевой раствор), сушат (MgSO4), фильтруют и упаривают, получая хрустящую белую пену. ЯМР показал наличие примерно 7,5% оставшегося исходного вещества. Неочищенный продукт повторно растворяют в ДХМ (150 мл) и обрабатывают еще 2,5 г (5,89 ммоль) Десс-Мартин периодинана, как указано выше. Реакционную смесь обрабатывают, как указано выше (с использованием 9 г Na2S2O3·5H2O), получая 7,54 г (95%) целевого продукта в виде белого пенообразного вещества.
Стадия 4: 682-4→682-5

682-4 (700 мг, 1,33 ммоль) частично растворяют в гептане (7 мл), получая мутный раствор. Равномерным потоком добавляют циангидрин ацетона (0,25 мл, 2,66 ммоль), после чего по каплям добавляют триэтиламин (19 мкл, 0,13 ммоль). При комнатной температуре перемешивают смесь, которая постепенно становится все более мутной. Спустя примерно 20 минут реакционная смесь становится вязкой суспензией, похожей на пасту. Спустя 1 час с помощью жидкостной хроматомасс-спектрометрии исходного вещества обнаружено не было. Смесь охлаждают на ледяной бане и фильтруют. Собранное белое твердое вещество промывают холодным гептаном (примерно 15 мл), затем низкокипящим петролейным эфиром (примерно 5 мл). Продукт сушат под вакуумом при 40°С. Получают 673 мг (91%).
Стадия 5: 682-5 → 682-6

Раствор 2-нафтилхлортиоформиата в толуоле (2-NTF) (25% раствор, 1,82 кг/кг 682-5) добавляют к 682-5 и 4-диметиламинопиридину (0,022 кг/кг 682-5) в дихлорметане (10 л/кг 682-5) при 5°С или ниже. К реакционной смеси медленно прибавляют триэтиламин (0,22 кг/кг 682-5) при температуре от 0°С до 10°С, при такой скорости, чтобы температура поддерживалась при 10°С или ниже. Смесь выдерживают при температуре от 0°С до 10°С и контролируют с помощью ВЭЖХ. Реакцию продолжают до тех пор, пока содержание 682-5 не составит 52,0%. После завершения реакции добавляют 1% масс./масс. водного раствора дигидрофосфата (10 кг/кг 682-5) при такой скорости, чтобы температура поддерживалась в диапазоне от 10 до 25°С. Фазы разделяются, и водную фазу экстрагируют дополнительным количеством дихлорметана (4,5 л/кг 682-5). После расслоения фаз, органические фазы однократно промывают апирогенной водой (10 л/кг 682-5), объединяют и переносят для дистилляции с использованием промывочной линией с дихлорметаном. Органические фазы концентрируют при пониженном делении при не более чем 30°С. Загружают метанол (3 л/кг 682-5), и продолжают концентрировать. Загружают дополнительную порцию метанола (10 л/кг 682-5), и продукт гранулируют в течение по меньшей мере 1 часа при 5°С или ниже. Продукт выделяют с помощью центрифугирования с использованием до двух загрузок. Пред сушкой под вакуумом при температуре до 45°С до достижения постоянного веса, каждую загрузку промывают холодным метанолом (1,5 л/кг 682-5) при диапазоне температур от 0 до 5°С.
Стадия 6: 682-6 → 682-7

К промежуточному соединению 682-6 в толуоле (4,5 л/кг 682-6) добавляют радикальный инициатор Vazo67 (2,2'-азобис[2-метилбутиронитрил]) (0,05 кг/кг 682-6) и трис(триметилсилил)силан (TTMSS) (0,41 кг/кг 682-6). Реакционную смесь нагревают до 70°С и перемешивают в течение 1 часа при температуре от 65 до 75°С перед контролированием хода реакции. Контроль реакции осуществляют с помощью ВЭЖХ. Реакцию продолжают до тех пор, пока содержание 682-5 не составит Х2,0%. При необходимости можно добавить дополнительное количество инициатора и TTMSS. После достижения окончания реакции, смесь медленно добавляют к этилциклогексану (20 л/кг 682-6) при температуре от 65 до 75°С. Реакционную смесь охлаждают по меньшей мере в течение 2,5 часов до температуры от 0 до 5°С и выдерживают при данной температуре. Полученное твердое вещество выделяют центрифугированием с использованием до трех загрузок. Каждую загрузку промывают холодным этилциклогексаном (1 л/кг 682-6) при температуре от 0 до 5°С. Продукт сушат под вакуумом при 45°С до достижения постоянного веса.
Стадия 7: 682-7 → 682-8

Для проведения гидролиза, 682-7 растворяют в метаноле (2,34 л/кг 682-7) и соляной кислоте (36%, 0,48 л/кг 682-7) при температуре от 48 до 52°С. Предварительно перед загрузкой в реакционную смесь готовят затравку 682-8 за счет обработки 682-9 (5 г/кг 682-7) соляной кислотой (29 мл/кг 682-7) в метаноле (140 мл/кг 682-7). Реакционную массу нагревают при температуре от 53 до 60°С в течение по меньшей мере 2 часов и контролируют с помощью ВЭЖХ. Реакцию продолжают до тех пор, пока пик при времени удержания около 5,25 не составит 512,0%. По завершению реакции смесь охлаждают до температуры от 10 до 15°С в течение по меньшей мере 100 минут. В течение по меньшей мере 25 минут добавляют этилацетат (10 л/кг 682-7) при температуре от 10 до 15°С, и смесь охлаждают по меньшей мере в течение 30 минут до температуры от 0 до 5°С. Смесь гранулируют в течение по меньшей мере 1 часа при менее чем 5°С. Продукт выделяют центрифугированием с использованием до двух загрузок, и каждую загрузку промывают холодной смесью метанола (0,38 л/кг 682-7) и этилацетата (1,11 л/кг 682-7) при температуре от 0 до 5°С. Продукт сушат под вакуумом при 45°С до достижения постоянного веса.
Стадия 8: 682-8 → 682-9

Хлористоводородную соль 682-8 нейтрализуют добавлением триэтиламина (0,41 кг/кг 682-8) к суспензии 682-8 в смеси метанол (3,9 л/кг 682-8):дихлорметан (10 л/кг 682-8) при температуре от 15 до 30°С. При добавление триэтиламина происходит растворение. Реакционную смесь перемешивают при температуре от 15 до 30°С в течение по меньшей мере 10 минут, и проверяют рН образца после разбавления водой. Ожидается, что его значение будет находиться в диапазоне от 9 до 9,5. Промежуточное соединение 682-9 может подвергаться эпимеризации при высоких значениях рН. Для доведения рН до диапазона от 4,0 до 4,5 и для индуцирования кристаллизации медленно при перемешивании добавляют уксусную кислоту (0,25 кг/кг 682-8) при такой скорости, чтобы поддерживать температуру менее 30°С. При необходимости добавляют дополнительное количество уксусной кислоты. Затем смесь разбавляют дихлорметаном (25 л/кг 682-8) и охлаждают до температуры от 0 до 5°С. Смесь перемешивают при температуре от 0 до 5°С в течение по меньшей мере 1 часа, продукт выделяют центрифугированием с использованием до двух загрузок. Каждую загрузку промывают холодной смесью метанола (0,63 л/кг 682-8) и дихлорметана (4,4 л/кг 682-8). Продукт сушат под вакуумом при 45°С до достижения постоянного веса.
Стадия 9: 682-9 → 682

682 может быть получено согласно способам, описанным в примерах 1-4 ЕР 536936. Промежуточное соединение 682-9 превращают в CYC682 и первоначально выделяют в Форме К, которая является сольватным комплексом с метанолом. Форму К переводят в Форму В, которая является полугидратом, за счет суспендирования в реакции по изменению формы. Форма К или Форма В далее может быть очищена за счет перекристаллизации. Перекристаллизация дает Форму К, которую затем переводят или переводят повторно в Форму В.
(i)682: Форма К
К смеси 682-9 в 1,4-диоксане (20 л/кг 682-9) и апирогенной воды (1,0 л/кг 682-9) добавляют пальмитиновый ангидрид (3,53 кг/кг 682-9), и реакционную смесь нагревают до температуры от 80 до 90°С (целевой диапазон от 80 до 85°С). Контроль реакции осуществляют с помощью ВЫЭЖХ и реакцию продолжают до тех пор, пока содержание 682-9 не достигнет 52,0%. После завершения реакции смесь подвергают горячему фильтрованию, и фильтр промывают 1,4-диоксаном (10 л/кг 682-9) при температуре от 70 до 90°С. Полученный объединенный фильтрат концентрируют до менее чем 30% от его начального объема (7,3 л/кг 682-9) при 60°С или ниже (целевая внутренняя температура от 45 до 55°С или менее). Содержание воды проверяют с помощью титрования по методу Карла Фишера. Если содержание воды <2%, то дополнительно добавляют диоксан, и дистилляцию повторяют. При необходимости, 1,4-диоксан добавляют для разбавления смеси до 30% от ее исходного объема. Добавляют этилциклогексан (48,3 л/кг 682-9) и 1,4-диоксан (3,66 л/кг 682-9) и температуру устанавливают в диапазоне от 43 до 47°С. В течение по меньшей мере 5 минут при температуре от 40 до 45°С добавляют метанол (3,23 л/кг 682-9).
В отдельном реакторе зародыши кристаллов CYC682 (Форма В) (10 г/кг 682-9) добавляют к смеси этилциклогексана (1333 мл/кг 682-9), 1,4-диоксана (177 мл/кг 682-9) и метанола (89 мл/кг 682-9) (15:2:1 об./об./об.). Полученную смесь перемешивают в диапазоне от 20 до 25°С в течение по меньшей мере 1 часа, затем добавляют к исходному реакционному раствору при температуре от 40 до 45°С. После того как происходит кристаллизация Формы К, реакционную смесь перемешивают при температуре от 40 до 45°С в течение по меньшей мере еще 30 минут. Реакционную смесь охлаждают до температуры от 20 до 23°С в течение по меньшей мере 120 минут и выдерживают в диапазоне от 20 до 23°С в течение по меньшей мере 1 часа. Полученное твердое вещество выделяют центрифугированием с использованием до двух загрузок, и каждую загрузку промывают смесью этилциклогексана (7,5 л/кг 682-9), 1,4-диоксана (1,0 л/кг 682-9) и метанола (0,5 л/кг 682-9) при температуре от 0 до 5°С. Продукт сушат под вакуумом при температуре в диапазоне от 35 до 40°С до достижения постоянного веса, получая CYC682 (Форма К).
(ii)682: Форма В
CYC682 (Форма К) суспендируют в метилацетате (8,9 л/кг CYC682), содержащем примерно от 1,5 до 2% апирогенной воды (169,3 мл/кг CYC682). Суспензию перемешивают при температуре от 20 до 25°С (целевая от 22 до 24°С) в течение 1,5 часов и подвергают преобразованию формы. Продукт выделяют с использование нутч-фильтра и промывают смесью метилацетата (2,2 л/кг CYC682) и апирогенной воды (42,3 мл/кг CYC682) при температуре от 20 до 25°С. Продукт сушат под вакуумом при 40°С или ниже до достижения постоянного веса, получая CYC682 (Форма В).
Перекристаллизация CYC682 (Форма К или В)
CYC682 (Форма К или В) суспендируют в смеси 1,4-диоксана (3,33 л/кг CYC682) и этилциклогексана (25 л/кг CYC682), и смесь доводят до температуры в диапазоне от 43 до 47°С. Добавляют метанол (1,66 л/кг CYC682) при температуре от 40 до 50°С в течение по меньшей мере 5 минут до достижения растворения. Для достижения растворения Формы В CYC682 может понадобиться дополнительное нагревание вплоть до 60°С.
В отдельном реакторе зародыши кристаллов CYC682 (от 4 до 15 г/кг CYC682) добавляют к смеси этилциклогексана, 1,4-диоксана и метанола (15:2:1 об./об./об.) как в вышеуказанном разделе (i). Полученную смесь перемешивают в диапазоне от 20 до 25°С в течение по меньшей мере 1 часа, затем добавляют к исходному реакционному раствору при температуре от 40 до 45°С. После того как происходит кристаллизация Формы К, реакционную смесь перемешивают при температуре от 40 до 45°С в течение по меньшей мере еще 30 минут. Реакционную смесь охлаждают до температуры от 20 до 23°С в течение по меньшей мере 120 минут и выдерживают в диапазоне от 20 до 23°С в течение по меньшей мере 1 часа. Полученное твердое вещество выделяют центрифугированием с использованием до двух загрузок, и каждую загрузку промывают смесью этилциклогексана (3,852 л/кг CYC682), 1,4-диоксана (0,514 л/кг CYC682) и метанола (257 мл/кг CYC682) при температуре от 0 до 5°С. Продукт сушат под вакуумом при температуре в диапазоне от 35 до 40°С до достижения постоянного веса, получая CYC682 (Форма К).
Сравнительные исследования
Исследования, проведенные авторами изобретения, показали, что предложенные в настоящем изобретении стадии способа приводят к улучшенным выходам по сравнению методикой, которая ранее использовалась. В качестве примера, в таблице 1 далее приведено сравнение выходов для каждой стадии Пути 1 (см. Фиг. 1; методология предшествующего уровня техники) Пути 1а (см. Фиг. 2; в соответствии с изобретением).
| Таблица 1 | |||||||||||
| Сравнение выходов для Пути 1 и Пути 1а | |||||||||||
| → 2 | → 3 | → 4 | → 5 | → 6 | → 7 | → 8 | → 9 | → К | → B | Общий | |
| Путь 1 | 98 | 38 | 90 | 91 | 85 | 97 | 89 | 89 | 19,9 | ||
| Путь 1а | 86 | 99 | 95 | 92 | 90 | 91 | 85 | 97 | 89 | 89 | 39,8 |
В таблице 1 показано, что обратный порядок первых двух стадий в синтезе, (Путь 1а, а именно введение защитной группы CIPS до стадии ацилирования) и использование циангидрина ацетона/гептана на стадии введения цианогруппы приводит к промежуточному соединению 682-5 с высоким выходом. Для сравнения, проведение стадии ацилирования перед введением защитной группы CIPS (Путь 1) и использование стандартных условий для введения цианогруппы, известных в области техники (например, NaCN, NaHCO3 в H2O/EtOAC) приводит к гораздо меньшему выходу 682-5 (38%). В общем, сравнение двух путей дает 19,9% CYC682 для Пути 1, по сравнению с 39,8% CYC682 для Пути 1а.
Различные модификации и изменения описанных аспектов изобретения будут очевидны специалисту в данной области техники без отклонения от объема и сущности изобретения. Несмотря на то, что изобретение было описано с использованием конкретных предпочтительных вариантов осуществления, следует понимать, что изобретение, как оно заявлено, не должно быть ненадлежащим образом ограничено такими конкретными вариантами осуществления. В действительности, различные модификации описанных способов осуществления изобретения, которые очевидны специалисту в соответствующих областях, охватываются объемом последующей формулы изобретения.
Claims (15)
1. Соединение формулы 682, которое находится в форме сольвата с метанолом

2. Соединение по п.1, которое находится в кристаллической форме.
3. Способ получения соединения формулы 682 в форме сольвата с метанолом по п.1, где указанный способ включает:

(i) обработку соединения формулы 682-9 пальмитиновым ангидридом в смеси Н2О/диоксан с образованием соединения формулы 682;
(ii) обработку продукта, полученного на стадии (i), метанолом с получением соединения формулы 682 в форме сольвата с метанолом (форма К);
(iii) выделение полученного на стадии (ii) соединения формулы 682 в форме сольвата с метанолом (форма К);
(iv) необязательная очистка продукта стадии (iii) с помощью перекристаллизации.
(i) обработку соединения формулы 682-9 пальмитиновым ангидридом в смеси Н2О/диоксан с образованием соединения формулы 682;
(ii) обработку продукта, полученного на стадии (i), метанолом с получением соединения формулы 682 в форме сольвата с метанолом (форма К);
(iii) выделение полученного на стадии (ii) соединения формулы 682 в форме сольвата с метанолом (форма К);
(iv) необязательная очистка продукта стадии (iii) с помощью перекристаллизации.
4. Соединение формулы 682 в форме сольвата с метанолом по п.1, полученное способом по п.3.
5. Способ получения соединения формулы 682-4

где указанный способ включает
(i) превращение соединения формулы 682-1 в соединение формулы 682-2' путем обработки указанного соединения формулы 682-1 1,3-дихлор-1,1,4,4-тетраизопропилдисилоксаном (CIPS) в пиридине;
(ii) превращение указанного соединения формулы 682-2' в соединение формулы 682-3 путем обработки указанного соединения формулы 682-2' уксусным ангидридом в EtOH и
(iii) превращение указанного соединения формулы 682-3 в соединение формулы 682-4 путем обработки указанного соединения формулы 682-3 окислителем, предпочтительно свободным радикалом 2,2,6,6-тетраметилпиперидинилокси (TEMPO) и NaOCl,

где указанный способ включает
(i) превращение соединения формулы 682-1 в соединение формулы 682-2' путем обработки указанного соединения формулы 682-1 1,3-дихлор-1,1,4,4-тетраизопропилдисилоксаном (CIPS) в пиридине;
(ii) превращение указанного соединения формулы 682-2' в соединение формулы 682-3 путем обработки указанного соединения формулы 682-2' уксусным ангидридом в EtOH и
(iii) превращение указанного соединения формулы 682-3 в соединение формулы 682-4 путем обработки указанного соединения формулы 682-3 окислителем, предпочтительно свободным радикалом 2,2,6,6-тетраметилпиперидинилокси (TEMPO) и NaOCl,
6. Способ получения соединения формулы 682-9 или 682

где указанный способ включает
(A) получение промежуточного соединения формулы 682-4 по п.5;
(B) превращение соединения формулы 682-4 в соединение формулы 682-9, включающее стадии:

(B1) превращение указанного соединения формулы 682-4 в соединение формулы 682-5 путем обработки указанного соединения формулы 682-4 NaCN/NaHCO3 в Н2О/ЕtOН или путем обработки указанного соединения формулы 682-4 циангидрином ацетона и NEt3 в гептане;
(B2) превращение указанного соединения формулы 682-5 в соединение формулы 682-6 путем обработки указанного соединения формулы 682-5 2-нафтилхлортиоформиатом в присутствии Net3 и диметиламинопиридина;
(B3) превращение указанного соединения формулы 682-6 в соединение формулы 682-7 путем обработки указанного соединения формулы 682-6 трис(триметилсилил)силаном (TTMSS) и азобисизобутиронитрилом (AIBN) в толуоле; и
(B4) превращение указанного соединения формулы 682-7 в соединение формулы 682-9 путем обработки указанного соединения формулы 682-7 HCl/MeOH и затем обработки полученного таким образом промежуточного соединения основанием для образования соединения формулы 682-9; и
(С) необязательное превращение указанного соединения формулы 682-9 в соединение формулы 682.
где указанный способ включает
(A) получение промежуточного соединения формулы 682-4 по п.5;
(B) превращение соединения формулы 682-4 в соединение формулы 682-9, включающее стадии:
(B1) превращение указанного соединения формулы 682-4 в соединение формулы 682-5 путем обработки указанного соединения формулы 682-4 NaCN/NaHCO3 в Н2О/ЕtOН или путем обработки указанного соединения формулы 682-4 циангидрином ацетона и NEt3 в гептане;
(B2) превращение указанного соединения формулы 682-5 в соединение формулы 682-6 путем обработки указанного соединения формулы 682-5 2-нафтилхлортиоформиатом в присутствии Net3 и диметиламинопиридина;
(B3) превращение указанного соединения формулы 682-6 в соединение формулы 682-7 путем обработки указанного соединения формулы 682-6 трис(триметилсилил)силаном (TTMSS) и азобисизобутиронитрилом (AIBN) в толуоле; и
(B4) превращение указанного соединения формулы 682-7 в соединение формулы 682-9 путем обработки указанного соединения формулы 682-7 HCl/MeOH и затем обработки полученного таким образом промежуточного соединения основанием для образования соединения формулы 682-9; и
(С) необязательное превращение указанного соединения формулы 682-9 в соединение формулы 682.
7. Способ по п.6, где стадия (С) включает обработку указанного соединения формулы 682-9 пальмитиновым ангидридом в смеси Н2О/диоксан.
8. Способ получения соединения формулы 682-5, где указанный способ включает обработку соединения формулы 682-4 циангидрином ацетона и Net3 в гептане:

9. Способ по п.8, который дополнительно включает стадию получения указанного соединения формулы 682-4 из соединения формулы 682-3 путем взаимодействия реакции соединения формулы 682-3 со свободным радикалом 2,2,6,6-тетраметилпиперидинилокси (TEMPO) и NaOCl:

10. Способ по п.9, который дополнительно включает стадию получения указанного соединения формулы 682-3 из соединения формулы 682-2 путем реакции указанного соединения формулы 682-2 с 1,3-дихлор-1,1,4,4-тетраизопропилдисилоксаном (CIPS) в пиридине:

11. Способ по п.10, который дополнительно включает стадию получения указанного соединения формулы 682-2 из соединения формулы 682-1 путем взаимодействия указанного соединения формулы 682-1 с Ас2О в ЕtOН:

12. Способ по п.9, который дополнительно включает стадию получения указанного соединения формулы 682-3 из соединения формулы 682-2' путем взаимодействия указанного соединения формулы 682-2' с Ас2О в ЕtOН:

13. Способ по п.12, который дополнительно включает стадию получения указанного соединения формулы 682-2' из соединения формулы 682-1 путем взаимодействия соединения формулы 682-1 с 1,3-дихлор-1,1,4,4-тетраизопропилдисилоксаном (CIPS) в пиридине:

14. Способ получения соединения формулы 682-9 или 682:

где указанный способ включает
(А") получение промежуточного соединения формулы 682-5 по любому из пп.8-13;
(В") превращение указанного соединения формулы 682-5 в соединение формулы 682-9, включающее стадии
(В2") превращения указанного соединения формулы 682-5 в соединение формулы 682-6 путем обработки указанного соединения формулы 682-5 2-нафтилхлортиоформиатом в присутствии NEt3 и диметиламинопиридина;
(В3") превращения указанного соединения формулы 682-6 в соединение формулы 682-7 путем обработки указанного соединения формулы 682-6 трис(триметилсилил)силаном (TTMSS) и азобисизобутиронитрилом (AIBN) в толуоле; и
(В4") превращения указанного соединения формулы 682-7 в соединение формулы 682-9 путем обработки указанного соединения формулы 682-7 HCl/MeOH и с последующей обработкой полученного таким образом промежуточного соединения основанием для образования соединения формулы 682-9; и
(С") необязательное превращение указанного соединения формулы 682-9 в соединение формулы 682.
где указанный способ включает
(А") получение промежуточного соединения формулы 682-5 по любому из пп.8-13;
(В") превращение указанного соединения формулы 682-5 в соединение формулы 682-9, включающее стадии
(В2") превращения указанного соединения формулы 682-5 в соединение формулы 682-6 путем обработки указанного соединения формулы 682-5 2-нафтилхлортиоформиатом в присутствии NEt3 и диметиламинопиридина;
(В3") превращения указанного соединения формулы 682-6 в соединение формулы 682-7 путем обработки указанного соединения формулы 682-6 трис(триметилсилил)силаном (TTMSS) и азобисизобутиронитрилом (AIBN) в толуоле; и
(В4") превращения указанного соединения формулы 682-7 в соединение формулы 682-9 путем обработки указанного соединения формулы 682-7 HCl/MeOH и с последующей обработкой полученного таким образом промежуточного соединения основанием для образования соединения формулы 682-9; и
(С") необязательное превращение указанного соединения формулы 682-9 в соединение формулы 682.
15. Способ по п.14, где стадия (С") включает обработку указанного соединения формулы 682-9 пальмитиновым ангидридом в смеси Н2О/диоксан.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0808357.8A GB0808357D0 (en) | 2008-05-08 | 2008-05-08 | Process |
| GB0808357.8 | 2008-05-08 | ||
| PCT/GB2009/001134 WO2009136158A1 (en) | 2008-05-08 | 2009-05-08 | Preparation of intermediates useful in the syntheis of 2'-cyano-2'-deoxy-n4-palmitoyl-1-beta-d-arabinofuranosylcytosine |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2010150153A RU2010150153A (ru) | 2012-06-20 |
| RU2509084C2 true RU2509084C2 (ru) | 2014-03-10 |
Family
ID=39570991
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2010150153/04A RU2509084C2 (ru) | 2008-05-08 | 2009-05-08 | Получение промежуточных соединений, используемых в синтезе 2'-циано-2'-дезокси-n4-пальмитоил-1-бета-d-арабинофуранозилцитозина |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US8884001B2 (ru) |
| EP (2) | EP3424510A1 (ru) |
| JP (1) | JP5563558B2 (ru) |
| CN (1) | CN102088982B (ru) |
| AU (1) | AU2009245482B8 (ru) |
| BR (1) | BRPI0912535A2 (ru) |
| CA (1) | CA2723702C (ru) |
| ES (1) | ES2691262T3 (ru) |
| GB (1) | GB0808357D0 (ru) |
| HU (1) | HUE041624T2 (ru) |
| IL (1) | IL209152A (ru) |
| MX (1) | MX2010012156A (ru) |
| PT (1) | PT2291193T (ru) |
| RU (1) | RU2509084C2 (ru) |
| WO (1) | WO2009136158A1 (ru) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2743691T3 (es) | 2011-04-14 | 2020-02-20 | Cyclacel Ltd | Régimen de dosis para sapacitabina y decitabina en combinación para tratar la leucemia mieloide aguda |
| US9872874B2 (en) | 2012-05-15 | 2018-01-23 | Cyclacel Limited | Dosage regimen for sapacitabine and seliciclib |
| CN109651472B (zh) * | 2018-12-29 | 2020-08-18 | 湖南千金湘江药业股份有限公司 | 合成索非布韦关键中间体的方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2085557C1 (ru) * | 1991-09-30 | 1997-07-27 | Санкио Компани Лимитед | Производные нуклеозидов пиримидина или их фармацевтически приемлемые соли и способ их получения |
| RU2116306C1 (ru) * | 1990-06-15 | 1998-07-27 | Санкио Компани Лимитед | Пиримидин-нуклеозиды, способы их получения, фармацевтическая композиция |
| US6335322B1 (en) * | 1993-04-05 | 2002-01-01 | Norsk Hydro Asa | Therapeutic agents |
| RU2256666C2 (ru) * | 2001-02-09 | 2005-07-20 | Санкио Компани, Лимитед | Кристаллические формы производного пиримидинового нуклеозида (варианты), фармацевтическая композиция, способ профилактики и лечения опухолевых заболеваний и применение кристаллической формы |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2287910A1 (fr) * | 1974-10-15 | 1976-05-14 | Asahi Chemical Ind | Derives nucleotidiques et procede de preparation de ceux-ci |
| US6005087A (en) | 1995-06-06 | 1999-12-21 | Isis Pharmaceuticals, Inc. | 2'-modified oligonucleotides |
| JPH0753586A (ja) * | 1993-08-23 | 1995-02-28 | Sankyo Co Ltd | ピリミジンヌクレオシド誘導体の製法 |
| US6908906B2 (en) * | 2001-02-09 | 2005-06-21 | Sankyo Company, Limited | Crystalline forms of pyrimidine nucleoside derivative |
| GB0526419D0 (en) | 2005-12-23 | 2006-02-08 | Cyclacel Ltd | Formulation |
-
2008
- 2008-05-08 GB GBGB0808357.8A patent/GB0808357D0/en not_active Ceased
-
2009
- 2009-05-08 RU RU2010150153/04A patent/RU2509084C2/ru active
- 2009-05-08 AU AU2009245482A patent/AU2009245482B8/en not_active Ceased
- 2009-05-08 WO PCT/GB2009/001134 patent/WO2009136158A1/en active Application Filing
- 2009-05-08 ES ES09742354.5T patent/ES2691262T3/es active Active
- 2009-05-08 BR BRPI0912535A patent/BRPI0912535A2/pt not_active IP Right Cessation
- 2009-05-08 EP EP18173901.2A patent/EP3424510A1/en not_active Withdrawn
- 2009-05-08 HU HUE09742354A patent/HUE041624T2/hu unknown
- 2009-05-08 MX MX2010012156A patent/MX2010012156A/es active IP Right Grant
- 2009-05-08 EP EP09742354.5A patent/EP2291193B1/en not_active Not-in-force
- 2009-05-08 US US12/991,582 patent/US8884001B2/en active Active
- 2009-05-08 CA CA2723702A patent/CA2723702C/en active Active
- 2009-05-08 CN CN2009801269455A patent/CN102088982B/zh not_active Expired - Fee Related
- 2009-05-08 PT PT09742354T patent/PT2291193T/pt unknown
- 2009-05-08 JP JP2011507983A patent/JP5563558B2/ja not_active Expired - Fee Related
-
2010
- 2010-11-04 IL IL209152A patent/IL209152A/en active IP Right Grant
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2116306C1 (ru) * | 1990-06-15 | 1998-07-27 | Санкио Компани Лимитед | Пиримидин-нуклеозиды, способы их получения, фармацевтическая композиция |
| RU2085557C1 (ru) * | 1991-09-30 | 1997-07-27 | Санкио Компани Лимитед | Производные нуклеозидов пиримидина или их фармацевтически приемлемые соли и способ их получения |
| US6335322B1 (en) * | 1993-04-05 | 2002-01-01 | Norsk Hydro Asa | Therapeutic agents |
| RU2256666C2 (ru) * | 2001-02-09 | 2005-07-20 | Санкио Компани, Лимитед | Кристаллические формы производного пиримидинового нуклеозида (варианты), фармацевтическая композиция, способ профилактики и лечения опухолевых заболеваний и применение кристаллической формы |
Also Published As
| Publication number | Publication date |
|---|---|
| BRPI0912535A2 (pt) | 2015-10-13 |
| JP2011519907A (ja) | 2011-07-14 |
| IL209152A (en) | 2015-05-31 |
| EP2291193B1 (en) | 2018-08-08 |
| US20110224421A1 (en) | 2011-09-15 |
| PT2291193T (pt) | 2018-11-08 |
| CA2723702A1 (en) | 2009-11-12 |
| CN102088982A (zh) | 2011-06-08 |
| AU2009245482A1 (en) | 2009-11-12 |
| RU2010150153A (ru) | 2012-06-20 |
| US8884001B2 (en) | 2014-11-11 |
| GB0808357D0 (en) | 2008-06-18 |
| AU2009245482B8 (en) | 2015-06-04 |
| ES2691262T3 (es) | 2018-11-26 |
| AU2009245482B2 (en) | 2015-05-28 |
| JP5563558B2 (ja) | 2014-07-30 |
| CN102088982B (zh) | 2013-08-21 |
| MX2010012156A (es) | 2011-02-15 |
| CA2723702C (en) | 2016-06-21 |
| EP3424510A1 (en) | 2019-01-09 |
| EP2291193A1 (en) | 2011-03-09 |
| HUE041624T2 (hu) | 2019-05-28 |
| IL209152A0 (en) | 2011-01-31 |
| WO2009136158A1 (en) | 2009-11-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7280248B2 (ja) | アミダイト化合物及び該化合物を用いたポリヌクレオチドの製造方法 | |
| US20040186283A1 (en) | Synthesis of 5-azacytidine | |
| CN102325783A (zh) | 嘌呤核苷的合成 | |
| JP2016175933A (ja) | 2’−o−修飾rna | |
| EP3660021B1 (en) | Photoresponsive nucleotide analog capable of photocrosslinking in visible light region | |
| RU2509084C2 (ru) | Получение промежуточных соединений, используемых в синтезе 2'-циано-2'-дезокси-n4-пальмитоил-1-бета-d-арабинофуранозилцитозина | |
| KR20090094800A (ko) | 아졸 뉴클레오시드 및 알엔에이와 디엔에이 바이러스성 폴리머라제 억제제로의 이용 | |
| US20040033967A1 (en) | Alkylated hexitol nucleoside analogues and oligomers thereof | |
| JPH10506621A (ja) | ジヌクレオチド及びオリゴヌクレオチドアナログのための中間体 | |
| US5760210A (en) | Process for the preparation of ribonucleotide reductase inhibitors | |
| EP0640092B1 (en) | A process for the preparation of ribonucleotide reductase inhibitors | |
| WO2009136162A1 (en) | Intermediate and processes involved in the preparation of 2 ' -cyano-2 ' -de0xy-n4-palmit0yl-1-beta-arabin0furan0sylcytosine | |
| EP4349846A1 (en) | Chimeric nucleic acid oligomer including phosphorothioate and boranophosphate, and method for producing same | |
| CN1812995A (zh) | 工业化规模的核苷合成 | |
| GB2459779A (en) | Process for the preparation of 2'-cyano-2'-deoxy-N4-palmitoyl-1-beta-D-arabinofuranosylcytosine | |
| Horton et al. | Synthesis of 3′-C-substituted thymidine derivatives by free-radical techniques: scope and limitations | |
| CN106336443A (zh) | 一类核苷类化合物的合成方法 | |
| RU2237064C1 (ru) | Способ получения фенилхлоргерманов | |
| CN115038790A (zh) | 3’-rna寡核苷酸的合成 | |
| RU2063977C1 (ru) | Способ получения 2',3'-дидезоксинуклеозидов | |
| US5231175A (en) | Process for the preparation of 3'- or 2'-halo-substituted-2',3'-dideoxynucleosides | |
| JPWO2010079813A1 (ja) | イノシン誘導体の製造方法 | |
| WO2008144472A1 (en) | Novel chromophoric silyl protecting groups and their use in the chemical synthesis of oligonucleotides | |
| EP0738273A1 (en) | Monohydrate of (e)-2'-deoxy-2'-(fluoromethylene)cytidine | |
| JPH07157496A (ja) | デオキシヌクレオシドの製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PD4A | Correction of name of patent owner | ||
| PD4A | Correction of name of patent owner | ||
| TC4A | Change in inventorship |
Effective date: 20161227 |