KR101296264B1 - 전이성 유방암의 치료 - Google Patents
전이성 유방암의 치료 Download PDFInfo
- Publication number
- KR101296264B1 KR101296264B1 KR1020087020271A KR20087020271A KR101296264B1 KR 101296264 B1 KR101296264 B1 KR 101296264B1 KR 1020087020271 A KR1020087020271 A KR 1020087020271A KR 20087020271 A KR20087020271 A KR 20087020271A KR 101296264 B1 KR101296264 B1 KR 101296264B1
- Authority
- KR
- South Korea
- Prior art keywords
- epcam
- patients
- weeks
- treatment
- dose
- Prior art date
Links
Images







Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39558—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against tumor tissues, cells, antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Immunology (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Cell Biology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biomedical Technology (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
Abstract
본 발명은 전이성 유방암 치료용 의약품의 제조에 항-EpCAM 항체를 이용하는 것에 관한 것이다. 또한 본 발명은 상기 항-EpCAM 항체를 투여하는 단계를 포함 하는 전이성 유방암의 치료방법에 관한 것이다.
전이성 유방암 치료, 항-EpCAM 항체
Description
본 발명은 항-EpCAM 항체를 이용하여 전이성 유방암 치료용 의약품을 제조하는 것에 관한 것이다. 또한 본 발명은 상기 항-EpCAM 항체를 투여하는 단계를 포함 하는 전이성 유방암의 치료방법에 관한 것이다.
유방암은 여성에 있어 가장 흔한 암이며, 두 번째로 사망률이 높은 암이다. 2001년 유방암의 유병율은 미국에서 100,000 명당 90-100이었으며, 유럽에서는 100,000 명당 50-70이었다. 이 질환의 발병은 세계적으로 점점 증가하는 추세에 있다. 유방암의 위험 인자는 인종, 나이 및 암 억제 유전자 BRCA-1 및 BRCA-2 및 p53에서의 돌연변이 등을 포함한다. 알코올 섭취, 고지방 식이, 운동 부족, 외인성 폐경후 호르몬 및 이온화 방사선 또한 유방암의 발명 위험을 증가시킨다. 에스트로겐 수용체 및 프로게스테론 수용체 음성 유방암(각각 "ER-" 및 "PR-"), 큰 종양 크기, 높은 등급의 세포진단결과, 및 35세 이하인 경우 그 예후가 나쁘다(Goldhirsch et al. (2001). J. Clin. Oncol. 19: 3817-27). 2005년 약 212,000건의 새로운 침습성 유방암 케이스 및 58,000건의 새로운 비침습성 유방암 케이스가 진단될 것으로 추 산되며, 40,000명의 여성이 유방암으로 치사할 것으로 예상된다.
유방암은 일반적으로 몇개의 주요 단계로 분류된다: 초기 단계, 국소 진행 단계, 국소 지속 단계 및 전이 단계. 초기 유방암은 상피내 소엽성 암종(lobular carcinoma i n s itu; "LCIS") 및 관상 암종(ductile carcinoma i n s itu; "DCIS")와 같은 비침습성 유방암을 포함한다. 일반적으로 유방암은 미국 암 위원회(American Joint Committee on Cancer)에 의해 제안된 암전이 분류 시스템(Tumor Node Metastasis; "TNM")에 따라 단계화된다(AJCC Cancer Staging Manual, 6th Edition). TNM 분류 시스템은 유방암을 7개의 분리된 단계로 정의한다: 0, I, IIA, IIB, IIIA, IIIB 및 IV. 단계 0, I 및 단계 II의 서브 타입은 일반적으로 초기 단계의 유방암이다. 단계 II의 일부 서브 타입 및 단계 III은 진전된 유방암이다. 단계 IV는 일반적으로 전이성 유방암을 말한다. 유방암의 TNM 분류에 대한 보다 자세한 정보를 도 1에 도시하였다. 종양 크기는 고형 종양에서의 반응 평가 기준 (Response Evaluation Criteria in Solid Tumors : "RECIST")으로 측정 및 모니터될 수 있다(Therasse et al. (2000). J. Natl. Cancer Inst. 92: 205-16).
초기 유방암의 경우 5년 생존 예후가 일반적으로 60%를 넘지만, 진전된 유방암의 경우 이 수치는 40-60%로 떨어진다. 전이성 유방암의 경우 5년 생존율은 일반적으로 15% 내외에 불과하다. 유방암에 있어서 가장 흔한 원격 전이 부위는 폐, 간, 뼈, 림프절, 피부 및 중추신경계(뇌)를 포함한다. 일단 전이성 유방암으로 진단되면, 환자들은 평균 18 내지 24 개월 정도 살 수 있는 것으로 예측된다. 전이성 유방암의 치료는 거의 불가능하며, 이 질환에 대한 치료 방식은 본질적으로 통증을 완화하는 것에 지나지 않는다.
이상에서 본 바와 같이 유방암의 치료, 특히 전이성 유방암의 치료에 대한 새로운 개발은 매우 중요하다.
현재 전이성 유방암을 포함한 유방암에 대한 치료 옵션은 수술(예컨대, 절제, 자가골수이식 등), 방사선 치료, 화학 요법(예컨대, 독소루빈신과 같은 안트라사이클린, 사이클로포스파미드 및 마이토마이신 C와 같은 알킬화제, 파클리탁셀 및 도세탁셀과 같은 탁산제제, 카페시타빈과 같은 대사저해제, 빈카 알칼로이드 나벨빈과 같은 미세소관 저해제), 내분비 치료(예컨대, 타목시펜과 같은 항에스트로겐 제제, 메드록시프로게스테론 아세테이트 및 메가스트롤 아세테이트와 같은 프로게스틴제제, 아미노글루테타마이드 및 레트로졸과 같은 아로마타제 저해제) 및 생물학적 제제(예컨대, 사이토킨, 모노 클론 항체와 같은 면역 치료제) 등을 포함한다. 가장 일반적인 전이성 유방암 치료 방법은 화학요법(사이클로포스파미드, 독소루비신, 나벨빈, 카페시타빈, 마이토마이신 C를 포함하는 가장 효과적인 약물) 단독 또는 화학요법과 내분비 치료의 조합으로 수행한다.
유방암에 대한 표준적인 치료는 외과적으로 종양을 제거하고, 방사선 치료를 하는 것이며, 이에 선행하거나 후행하여 종양 단계 및 위험 인자에 따라 호르몬 치료 또는 화학요법을 수행하는 것이다. 단계 I 내지 단계 IIIA(하기 및 도 1 참조)의 환자는 화학요법 및 호르몬 치료를 같이 시행할 수 있다. 수술 불가능한 침습성 단계 IIIB 또는 단계 IV 전이성 유방암 상태의 환자에 있어서, 화학요법은 단지 증 상을 경감시키는데 지나지 않는다.
최근 탁산 및 안트라사이클린 제제가 유방암 환자의 생존율을 현저히 개선시켰다. 카페시타빈(Xeloda®, capecitabine, 로슈: 제품 설명 요약)는 안트라사이클린 및/또는 탁산을 포함한 세포독성 화학요법제 처치에 실패한 환자들을 위한 2차 치료 또는 고 품질 치료를 위해 승인되었으며, 이는 특히 낮은 독성 및 경구 포뮬레이션 때문이다(O'Shaughnessy (2002). Oncology 16: 17-22). 그러나 이러한 처치양식의 개선에도 불구하고, 진전된 유방암 환자의 생존율은 여전히 낮으며 화학요법은 오직 그 증상을 경감시키는데 지나지 않는다.
모노클론 항-Her-2/neu 항체 트라스투주마브(Herceptin®, trastuzumab, 로슈:제품 설명 요약, 2002년 3월)는 Her-2/neu를 과발현하는 유방암을 앓고 있는 환자들의 치료를 위해 승인된 첫 번째 생물학적 표적 치료제이다. 파클리탁셀과 조합하여, 전이성 유방암 환자를 위한 1차 치료제로 사용되며, 동일 환자군에 대해 단독으로 2차 또는 고품질 처치로 사용된다(Cardoso et al. (2002). Clin. Breast Cancer 3: 258-9; Tan-Chiu & Piccart (2002). Oncology 63: 57-63). 그러나 오직 소수의 유방암 환자들 만이(약 20%) Her2/neu을 고수준으로 과발현하며, 따라서 이 환자들만이 상기 항체를 사용한 치료에 적합하다.
따라서, 특히 트라스투주마브 적응증이 아닌 유방암 환자들을 위한 새로운 항암제의 개발이 의학적으로 요망된다.
유망한 면역치료제로 중쇄가변영역에 서열: 3, 4, 및 5의 아미노서열을 포함하고, 및/또는 경쇄가변영역에 서열: 6, 7 및 8의 아미노산 서열을 포함하는 인간 항체이다. 이하 이 항체는 "항-EpCAM"으로 지칭되며, 이들의 중쇄 및 경쇄는 각각 서열: 1 및 서열: 2의 아미노산에 의해 더욱 특정된다. 이들 항체는 상피세포부착분자( ep ithelial c ell a dhesion m olecule; "EpCAM", 또는 17-1A 항원, KSA, EGP40, GA733-2, ks 1-4 및 esa로 지칭되기도 한다)에 결합한다. EpCAM은 유방암을 포함한 상이한 기원의 많은 암종에 과발현되는 매우 보존적인 표면 당단백질이다. 대장암, 위암, 폐암, 난소암 또는 전립선암 환자 3722 명에서 채취한 암 샘플을 조직 마이크로어레이상에서 민감성 면역조직화학 염색 분석을 사용하여 EpCAM 발현을 분석하였다. 모든 암 샘플 중 88% 이상에서, 난소암의 94% 이상, 대장암의 94% 이상, 위암의 92% 이상, 전립선암의 90% 이상, 폐암의 71% 이상에서 중등도 내지 강한 EpCAM 발현이 있는 것으로 보고되었다. 이러한 결과는 EpCAM이 상피성 종양 세포에 빈번히 존재하며, 항-EpCAM 제제가 잠재적인 진단 및 치료 표적으로 사용될 수 있음을 확인한다.
원발성 유방암에 대한 두 연구에서, 각각 384 섹션 중 36%에서 (Tandon et al. (1990) Cancer Res. 50: 3317-24) 그리고, 128 샘플 중 59%(Edwards et al. (1986) Cancer Res. 46: 1306-17)에서 강한 EpCAM 발현이 보고되었다. 다른 연구에서(Spizzo et al. (2002) Int. J. Cancer 98: 883-8), 205개의 원발성 유방암 시료 중 73개(36%)에서 강한 EpCAM 발현이 보고되었으며, 저자는 유방암에서의 EpCAM 과발현이 경감된 질병이 없는 상태 및 전체적인 생존과 관련됨을 보고하였다. EpCAM 과발현은 또한 종양 크기 및 호르몬 수용체 음성과 상관성이 있었다; 관상 유방암 및 조직학적 등급 III 서브타입에서 가장 높았다. 다른 연구에서, 약 90%의 유방암 샘플에서 EpCAM이 어느 정도까지 발현되었고, 40% 이상에서 강한 EpCAM 발현을 나타내었다.
인 비트로에서, 항-EpCAM는 항체-의존 세포성 세포 독성(antibody-dependent cellular cytotoxicity; "ADCC") 및 보체-의존성 세포 독성(complement-dependent cytotoxicity; "CDC") 양자를 유발한다. 가장 가능한 작용 기전으로는 항-EpCAM가 EpCAM-양성 종양 세포와 결합함으로써, 환자의 자연 살해 세포들을 종양 부위로 모이게 하는 것이다. 환자의 면역 효과 세포들을 활성화함으로써 EpCAM-양성 세포들은 이후 제거된다.
항-EpCAM을 사용하는 특정 치료 요법이 본 기술분야에 공지되었다(WO 2005/080428). 특히 WO 2005/080428호는 항-EpCAM를 암 환자에 투여하는 치료 요법을 기재하고 있다. 항-EpCAM을 예를 들면 유방암 또는 최소 잔여 질병(minimal residual disease)의 치료에 투여하는 것이 숙고되었다. 맥락상 최소 잔여 질병은 단일 종양 세포의 생존에 의해 유발된 종양의 국소 및 비국소 재발을 의미하는 것으로 이해된다.
본 발명의 목적은 기존의 유방암 치료를 개선하는 것이다.
따라서, 본 발명의 한 태양은 서열번호: 3, 4, 5, 6, 7 및/또는 8의 아마노산 서열을 포함하는 항-EpCAM 항체("항-EpCAM")를 인간 전이성 유방암 치료용 의약품 제조에 사용하는 용도에 관한 것이다.
본 명세서에서 "전이성 유방암"은 최소한 하나의 원발성 유방 종양에서 유래된 변형된, 즉 암성 세포가 그 원발성 종양에서 분리되어 원발성 종양과 떨어진 위치(이하 "원격 부위"라 한다)에서 암으로 계속 성장하는 질병을 말한다. 상기 원격 부위는 예를 들면 원발성 종양이 위치된 동일한 유방(동측 유방; ipsilateral breast) 또는 다른 유방(반대측 유방; contralateral breast) 내일 수 있다. 또한 예를 들어 상기 원격 부위는 하나 이상의 림프절 내일 수 있으며, 이들은 이동성이거나 고정되어 있을 수 있고, 상기 원발성 종양에 대해 동측이거나 반대측일 수 있으며, 쇄골위이거나 겨드랑이 등에 있는 것일 수 있다. TNM 암 분류 기준(도 1 참조)에 따라, 본 발명에서의 "전이성 유방암"은 M=1 (즉 단계 IV 유방암; 도 1 참조) 단계의 모든 암들, 즉 예를 들어 폐, 간, 뼈, 림프절, 피부, 뇌와 같은 원격 부위 및/또는 동측 유방 및/또는 반대측 유방 내의 원격 부위에 전이가 어떤 정도로든 존재하는 모든 암들을 포함한다.
본 명세서에서 "유방암"은 원발성 종양 또는 복수의 개별 원발성 종양이 유방에 존재하는 것을 의미한다. 일반적으로 이는 유방내의 원발성 종양에서 어떠한 암성 세포도 아직 분리되지 않고, 따라서 "원격 부위"로 전이되지 않은 것을 의미한다. 본 발명에서, 복수의 원발성 암이 동일한 또는 동측 및 반대측 유방내에 존재하는 것은 그 자체로 "전이성 유방암"의 정의에 해당되는 것은 아님이 주목되어야 한다. 이는 하나 또는 양쪽의 유방내 복수의 세포가 복수의 원발성 종양을 일으키기 때문이며, 이들 중 어느 것도 아직 전이성이 되지는 않았기 때문이다. 다른 한편으로, 유방 중 최소한 하나에 하나 이상의 원발성 종양의 존재 또는 비존재와 무관하게 단일 유방 내 복수의 원발성 종양들 중 오직 하나의 종양에서 유래 되는 암성 세포가 분리되어 이 단일 세포가 "원격 부위"에서 발전하여 독립된 종양으로 되는 경우 이는 본 발명의 "전이성 유방암"을 구성한다.
본 발명에서 사용되는 "전이성 유방암"은 "원격 부위"에 존재하는 전이가 반드시 유방의 특정 원발성 종양에서 유래하여야 하는 것을 의미하는 것은 아니다. 즉, 원발성 종양이 유방 조직내 기원되는 전이를 일으키는 한, "원격 부위"의 전이 기원은 중요하지 않으며, 이 질병을 "전이성 유방암"으로 지정할 수 있다. 이를 위해 본 발명에서 "유방 조직"은 유방의 소엽(lobules)과 유선(duct), 즉 가장 빈번히 유방 종양이 유발되는 조직을 포함하는 것으로 이해되어야 한다.
본 발명자는 놀랍게도 항-EpCAM이 유방암 즉 유방내 최소한 하나의 원발성 종양이 있는 유방암의 치료에 적합할 뿐아니라 전이성 유방암의 치료에도 적합하다는 것을 발견하였다. 전이성 유방암에 수반되는 종양 로드(즉 암 세포의 수, 종양의 크기, 또는 체내 암의 양; 소위 "종양 부하량(tumor burden)")는 비-전이성 유방암에서 관찰되는 것에 비해 일반적으로 훨씬 더 크기 때문에 항-EpCAM이 이러한 방식으로 사용될 수 있는 것은 전혀 예기치 못한 것이다. 이는 단일 원발성 유방암은 몸을 통해 복수의 분산된 전이를 매우 그리고 자주 발생시키기 때문이다. 몸 전체를 통해 악성 세포의 표면에 존재하는 EpCAM 분자의 절대수는 따라서 비전이성 유방암보다 전이성 유방암에서 일반적으로 더 크다. 첨부된 실시예에서 제공되는 데이타들은 항-EpCAM 항체를 포함하는 약학적 조성물을 투여함으로써 처치되는 질환의 TTP(진행기간; time to progression)이 상당히 연장됨을 보여준다. 이러한 실질적인 효과는 치료될 악성 세포의 표면상의 EpCAM 발현 수준과 관련되는 것으로 보인다. 환자는 Gastl 등의 (2000) Lancet 356, 1981-2 에 보고된 방법에 따라 상이한 그룹의 EpCAM 발현자 그룹들로 분류될 수 있다. 간략하게 설명하면 Gastl 등은 서로 다른 환자들로부터 분리된 종양 세포의 EpCAM 발현을 면역조직화학 염색으로 분석하였다. 전체 면역 염색 점수는 비율점수와 강도점수의 곱으로 계산될 수 있다. 비율점수는 Gastl 등의 개시에 따라 양성-염색된 종양 세포의 산출된 부분을 기술한 것이다(0, 없음; 1, <10%; <10%; 2, 10%-50%; 3, 50%-80%; 4, >80%). 강도 점수는 Gastl 등의 개시에 따라 산출된 염색 강도를 표시한 것이다(0, 염색되지 않음; 1, 약함; 2, 중등; 3, 강함). 산출되는 전체 점수는 0 내지 12이다. 환자 샘플이 전체 점수 3 내지 4에서 식별되는 최저점을 갖는 바이모달 분포(bimodal distribution)의 EpCAM 발현(저 및 고 EpCAM 발현자)를 나타내기 때문에, 고 EpCAM는 전체점수가 4 이상인 경우로 정의된다. 항-EpCAM 항체의 투여를 포함하는 치료를 받는 환자의 예후는 고 EpCAM 발현자로 동정된 경우, 중등 또는 저 EpCAM 발현자로 동정된 환자보다 더 낙관적이다. 이러한 관찰 선상에서, 매우 높은 EpCAM 발현자로 확인된 환자(고 EpCAM 발현자의 평균보다 더 많은 양의 EpCAM을 악성 세포 표면에 보이는 환자, 즉 전체 점수가 8이상인 환자)는 동일한 치료를 받는 고 EpCAM 발현자로 확인된 환자에 비하여 훨씬 TTP가 연장되었다. 또한 환자에게 투여된 약학적 조성물내 항-EpCAM 항체의 양은 직접적으로 예후와 관련된다. 특히 고 용량의 항-EpCAM 항체 투여는 저용량의 투여에 비하여 동일 그룹의 EpCAM 발현자에 있어 치료될 질환의 TTP를 더 연장시킨다.
본 발명의 바람직한 일실시예에서 항-EpCAM 항체 항-EpCAM은 인간 항체이다.
본 발명의 더욱 바람직한 일 실시예에서 항-EpCAM은 서열번호: 3, 4, 5, 6, 7 및 8의 아미노산서열을 모두 포함한다. 더욱 바람직한 실시예에서, 항-EpCAM는 서열번호: 1 및/또는 2를 포함한다. 특히 바람직한 실시예에서, 항-EpCAM는 서열번호: 1 및 2를 모두 포함한다.
본 발명의 바람직한 실시예에 따르면, 전이성 유방암의 치료는 전이성 유방암의 장기 안정화를 포함한다. "장기 안정화(long-term stabilization)"는 항-EpCAM 항체로 치료하는 동안 질병의 진행이 처음 수준 또는 그 이하로 안정화되는 경우를 의미한다. 이는 질병 진행에 소요되는 시간을 연장하는 것으로도 이해된다. "장기 안정화"는 또한 종양이 축소되는 경우(부분 반응)을 포함한다. "장기 안정화"는 또한 질병 진행이 검출 수준 이하로 감소되는 경우, 즉 환자가 치료에 전적으로 반응하여 질병이 치료되는 경우(완전 반응)를 포함한다. 이러한 경우 치료는 질병의 재발을 방지하기 위해 필요한 경우에는 계속 진행할 수 있고, 또는 의사의 판단에 의해 종결될 수 있다.
바람직한 일 실시예에서, 의약품은 소위 "저용량 투여"에 적합하다. 저용량 투여를 위해서는 각 투여 용량이 체중 1 kg당 1 내지 3 mg의 항-EpCAM 항체 범위여야 한다. 바람직하게, 저 용량 투여는 최소한 체중 1 kg 당 1 내지 3 mg의 로딩 용량으로 투여되고, 이후 복수의 유지 용량이 투여되며, 이때 각 유지 용량은 체중 1 kg 당 1 내지 3 mg이다. 더욱 바람직하게 저 용량 투여의 각 용량은 체중 1 kg 당 1.5 내지 2.5 mg이며, 보다 바람직하게는 체중 1 kg 당 1.75 내지 2.25 mg이다. 가장 바람직하게는 저 용량 투여에서 각 용량은 체중 1 kg 당 2 mg이다. 저용량 투여를 위해 바람직하게 최소한 체중 1 kg 당 2 mg의 로딩 용량으로 투여되고, 이후 복수의 유지 용량이 투여되며, 이때 각 유지 용량은 체중 1 kg 당 2 mg이다. 또는 본 의약품은 소위 "고 용량 투여"에 적합하다. 고 용량 투여를 위해서는 각 투여 용량이 체중 1 kg당 4.5 내지 8 mg의 항-EpCAM 항체 범위여야 한다. 바람직하게, 고 용량 투여는 최소한 체중 1 kg 당 4.5 내지 8 mg의 로딩 용량으로 투여되고, 이후 복수의 유지 용량이 투여되며, 이때 각 유지 용량은 체중 1 kg 당 4.5 내지 8 mg이다. 더욱 바람직하게 고 용량 투여의 각 용량은 체중 1 kg 당 5 내지 7 mg이며, 더욱 바람직하게는 체중 1 kg 당 5.5 내지 6.5 mg이며, 보다 바람직하게는 체중 1 kg 당 5.75 내지 6.25 mg이다. 가장 바람직하게는 고 용량 투여에서 각 용량은 체중 1 kg 당 6 mg이다. 고 용량 투여를 위해 바람직하게 최소한 체중 1 kg 당 6 mg의 로딩 용량으로 투여되고, 이후 복수의 유지 용량이 투여되며, 이때 각 유지 용량은 체중 1 kg 당 6 mg이다. 이와 같은 로딩 및 유지 용량이 전이성 유방암의 치료에 치료적 이점을 나타내며, 몸 전체에 걸친 복수 전이가 근절된다는 것은 전혀 예기치 않은 것이다.
본 발명에 따른 다른 실시예에 따라, 전이성 유방암 환자가 EpCAM를 고수준으로 또는 저수준으로 발현하는지를 확인하는 것은 매우 유용하다. 환자는 Gastl et al. (2000) Lancet 356, 1981-2에 개시된 바와 같이 비-EpCAM 발현자, 중등 EpCAM 발현자, 저 EpCAM 발현자 및 고 EpCAM 발현자로 분류될 수 있다. 일반적으로 전이성 유방암 환자에 투여되는 항-EpCAM 양과 치료될 환자에서 관찰된 EpCAM의 발현수준을 연관시켜, 고 EpCAM 발현자에는 고 용량의 항-EpCAM을 투여하고 저 EpCAM 발현자는 저용량의 항-EpCAM을 투여하는 것이 바람직하다. 특히 EpCAM을 고수준으로 발현하는 환자에게는 더 높은 용량의 항-EpCAM, 즉 6 mg/kg체중을 투여하는 것이 바람직하다.
본 발명의 다른 실시형태에 따르면, 각각의 로딩 용량과 다른 연속적인 로딩 용량 또는 첫 유지 용량 간의 시간 간격은 1주일 이하이며, 각 유지 용량 및 후속하는 유지 용량 간의 시간 간격은 2 주일 이하이다. 바람직하게 로딩 용량은 매주 투여되고, 상기 각 유지 용량은 2주마다 한번 씩 투여된다. "로딩기(loading phase)"에 항-EpCAM의 로딩 용량의 매주 투여는 혈청내 최소 항-EpCAM 농도가(배출 및 배설 형태의 연속적인 클리어런스를 고려하여) 원하는 치료적 효과를 유발할 수 있을 정도로 높게 유지되는 것을 보증한다. 이 치료효과를 나타내는데 요구되는 최소한의 항-EpCAM 농도는 "혈청 최저 농도(serum trough level)"로 알려져 있으며, 이하 혈청 최저 농도로 지칭된다. 이 혈청 농도가 도달한 후에는 후속하는 "유지기"에서의 항-EpCAM의 유지 용량의 2 주 간격의 투여에 의해(다시, 연속적인 클리어런스를 고려하여), 항-EpCAM의 혈청 농도가 계속적으로 치료 효과를 나타내는데 요구되는 농도 이하로 떨어지지 않도록 보증한다. 항-EpCAM에 대한 혈청 최저 농도를 측정하는데 요구되는 약물동력학적 계산법은 본 기술분야에 잘 알려져 있다(WO 2005/080428 참조).
특히 바람직한 실시형태에 따라, 로딩 용량은 각 치료 1주, 2주, 및 3주가 시작될 때 투여되고, 이후 11회의 유지 용량이 투여되며, 각 유지 용량은 각 치료 4주, 6주, 8주, 10주, 12주, 14주, 16주, 18주, 20주, 22주 및 24주가 시작될 때 투여된다. 놀랍게도 상기와 같은 시간 간격으로 3번 로딩 용량을 투여하고 이어 상기와 같은 간격으로 11번의 유지 용량을 투여함으로써 이러한 조합이 특히 전이성 유방암의 치료에 효과적임이 밝혀졌다. 이는 시작에서 끝날 때까지 전체 24주의 치료기간을 의미한다(모든 종류의 치료에 수반되는 통상적인 모든 치료후 확인은 고려하지 않음). 더욱 바람직한 실시형태에서 치료 시작에서 끝날 때까지의 전체 기간은 30주, 40주, 50주, 60주이다. 또한 전술된 계획에 따라 EpCAM 항체 투여기간을 가지고, 후속하여 EpCAM 항체를 투여하지 않는 기간을 두고 다시 EpCAM 항체를 투여하는 기간을 두는 것이 바람직하다. 이러한 투여 순서는 수회 반복될 수 있다.
또는 본 발명의 다른 실시형태로서, 전술한 바와 같은 로딩 용량으로 항-EpCAM을 투여하고, 이어 암 진전을 조절하는데 요구되는 회수의 유지 용량을 투여할 수 있다. 이 실시형태에서, 하나 이상의 모니터되는 전이성 암의 크기가 더이상 증가되지 않는 한 암의 진전이 조절되는 것으로 본다. 가장 바람직한 경우에서 모니터 되는 하나 이상의 종양의 크기는 실제로 줄어든다(부분 반응에서와 같이). 여기에서 종양은 완전히 수축, 즉 없어질 수 있다(완전 반응에서와 같이). 모니터 되는 종양의 크기는 동일하게 유지되나 질병이 진행되는 시간이 연장될 수 있다(안정 질병에서와 같이). 따라서 이 실시형태에 따르면, 부분 또는 안정화 반응이 있는 한 전술한 간격으로 항-EpCAM의 유지 용량을 무기한으로 계속 투여할 수 있고, 완전 반응이 보일 때까지 계속될 수 있다. 종양이 계속 진행되는 경우(즉 모니터되는 종양의 크기나 수가 치료되는 동안 증가함), 항-EpCAM의 치료를 중단하고, 적합한 경우, 다른 형태의 치료로 대체할 수 있다.
본 발명의 다른 실시형태에 따라, 항-EpCAM은 0.9% 소듐 클로라이드를 포함하는 용액의 형태로 투여될 수 있다.
본 발명의 다른 실시형태에 따라, 항-EpCAM은 전이성 유방암 환자에게 정맥 투여될 수 있다.
본 발명의 다른 측면은 항-EpCAM을 전이성 유방암의 치료에 사용하는 것이다.
본 발명의 다른 측면은 인간 전이성 유방암의 치료방법에 관한 것으로, 상기 방법은 서열번호: 3, 4, 5, 6, 7 및/또는 8을 포함하는 항-EpCAM 항체를 인체에 투여하는 것을 포함한다. 이 항체는 중쇄 및 경쇄 아미노산 서열이 각각 서열번호: 1 및 2인 것을 추가적인 특징으로 한다.
본 발명의 바람직한 전이성 유방암 치료 방법은 전술한 바와 같으며 이러한 실시형태는 필요한 변경을 가하여(mutatis mutandi ), 본 발명의 진보적인 방법에 적용된다.
이하 본 발명을 도면과 실시예를 사용하여 더욱 자세히 설명하며, 하기 실시예는 본 발명을 제한하지 않는다.
도 1은 유방암에 대한 TNM 분류/단계 시스템의 개략도이다.
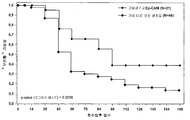
도 2는 각각 항-EpCAM이 저용량 및 고용량으로 투여된 32명 및 35명의 환자에 대하여 질병 진행이 일어나지 않는 가능성을 보여주는 진행기간(TTP)을 시간에 대해 도시한 것이다.
도 3은 각각 항-EpCAM이 저용량 및 고용량으로 투여된 27명 및 40명의 환자에 대하여 질병 진행이 일어나지 않는 가능성을 보여주는 진행기간(TTP)을 시간에 대해 도시한 것이다.
도 4는 고수준 및 저수준 EpCAM 발현 환자 모두에 대해 항-EpCAM이 저용량 및 고용량으로 투여된 경우에 대하여 질병 진행이 일어나지 않는 가능성을 보여주는 진행기간(TTP)을 시간에 대해 도시한 것이다.
도 5는 고수준으로 EpCAM을 발현하는 환자에 항-EpCAM을 고용량으로 투여한 경우에 대하여 기타 다른 환자들과 비교하여, 질병 진행이 일어나지 않는 가능성을 보여주는 진행기간(TTP)을 시간에 대해 도시한 것이다.
도 6은 각각 항-EpCAM이 저용량 및 고용량으로 투여된 54명 및 55명의 환자에 대하여 질병 진행이 일어나지 않는 가능성을 보여주는 진행기간(TTP)을 시간에 대해 도시한 것이다.
도 7은 고수준 및 저수준 EpCAM 발현 환자들에 대해 항-EpCAM이 저용량 및 고용량으로 투여된 경우에 대하여 질병 진행이 일어나지 않는 가능성을 보여주는 진행기간(TTP)을 시간에 대해 도시한 것이다.
도 8은 고수준으로 EpCAM을 발현하는 환자에 항-EpCAM을 고용량으로 투여한 경우에 대하여 저수준으로 EpCAM을 발현하는 환자에 항-EpCAM을 저용량으로 투여한 경우과 비교하여, 질병 진행이 일어나지 않는 가능성을 보여주는 진행기간(TTP)을 시간에 대해 도시한 것이다.
일반론
하기 실시예들은 본 발명의 다양한 측면을 기술하기 위함이고 본 발명의 범위를 제한하지 않는다. 일반적으로 하기 실시예는 "항-EpCAM"으로 명명되는 전적으로 인간 IgG1 항체를 위해 디자인된 임상 연구 프로그램과 이 임상연구 프로그램에서 얻어진 결과를 기술한다. 항-EpCAM의 중쇄 가변영역의 첫번째, 두번째 및 세번째 상보성 결정부위(complementarity determining regions; CDRs)의 아미노산 서열들은 각각 서열번호:3, 4 및 5에 나타내었다. 상기 항-EpCAM의 경쇄 가변영역의 첫번째, 두번째 및 세번째 상보성 결정부위의 아미노산 서열들은 각각 서열번호:6, 7 및 8에 나타내었다. 항-EpCAM의 중쇄 아미노산 서열은 서열번호: 1에 나타내었고, 항-EpCAM의 경쇄 아미노산 서열은 서열번호:2에 나타내었다. 하기 실시예 전체에서, 아래의 용어 및 약어들이 사용된다:
| ADCC | 항체 의존성 세포 매개 세포독성(antibody-dependent cell-mediated cytotoxicity) |
| AE | 부작용(adverse event) |
| ALT | 알라닌 아미노트랜스퍼라지(alanine aminotransferase) |
| ANCOVA | 공분산 분석(analysis of covariance) |
| AP | 알칼리성 포스파타제(alkaline phosphatase) |
| AST | 아스파르트산 아미노트랜스퍼라제(aspartate aminotransferase) |
| AUC | 곡선하곡선(area-under-the-curve) |
| BRCA | 유방암억제유전자(breast cancer tumor suppressor gene) |
| CBA | 사이토메트릭 비드 어레이(cytometric bead array) |
| CDC | 보체 의존성 세포독성(complement-dependent cytotoxicity) |
| CHO | 중국 햄스터 난자(Chinese hamster ovary) |
| Cmin | 최소 약물 농도(minimum drug concentration) |
| CNS | 중추 신경계(central nervous system) |
| CR | 완전 반응(complete Response) |
| CRF | 사례 보고서(case report form) |
| CRP | C-반응성 단백질(C-reactive protein) |
| CT | 컴퓨터단층촬영(computerized tomography) |
| CTCAE | 부작용에 대한 일반적 용어 기준(common terminology criteria for adverse events) |
| ECOG | 이스턴코오퍼레이티브옹콜로지그룹(Eastern Cooperative Oncology Group) |
| ELISA | 효소-결합 면역흡착 어세이(enzyme-linked immunosorbent assay) |
| EpCAM | 상피세포부착분자(epithelial cell adhesion molecule) |
| FAS | 전체분석세트(full analysis set) |
| GCP | 굳 클리니칼 프랙티스(good clinical practice) |
| GGT | 감마-글루타밀트랜스퍼라제(gamma-glutamyltransferase) |
| HAHA | 인간 항-인간 항체(human anti-human antibodies) |
| HBsAg | B형 간염 표면 항원(hepatitis B surface antigen) |
| HCV | C형 간염 바이러스(hepatitis C virus) |
| HIV | 인간 면역 결핍 바이러스(human immunodeficiency virus) |
| ICF | 피험자 동의서(informed consent form) |
| IEC | 독립윤리위원회(independent ethics committee) |
| INR | 국제정규화비율(international normalized ratio) |
| IRB | 임상시험심사위원회(institutional review board) |
| LDH | 락트산데하이드로게나제(lactic dehydrogenase) |
| NK | 자연살해(natural killer) |
| OTR | 전체종양반응(overall tumor response) |
| PK | 약물동력학(pharmacokinetic) |
| PP | 프로토콜분석세트당(per protocol analysis set) |
| PR | 부분반응(partial response) |
| PT | 프로트롬빈시간(prothrombin time) |
| PTT | 부분트롬보플라스틴시간(partial thromboplastin time) |
| PVP | 폴리비닐피롤리돈(polyvinylpyrrolidone) |
| RBC | 적혈구(red blood cell) |
| RECIST | 고형종양에서의 반응평가기준(Response Evaluation Criteria in Solid Tumors) |
| SAE | 심각한 부작용(serious adverse event) |
| SAF | 안전성 분석 세트(safety analysis set) |
| SAP | 통계분석계획(statistical analysis plan) |
| SD | 안정질환(stable disease) |
| SGOT | 혈청글루탐산옥살로아세트산트랜스아미나제(serum glutamic oxaloacetic transaminase) |
| SGPT | 혈청글루탐산피루브산트랜스아미나제(serum glutamic pyruvic transaminase) |
| ULN | 정상상한(upper limits of normal) |
| WBC | 백혈구(white blood cell) |
| WHO | 세계보건기구(World Health Organization) |
실시예
1: 항-
EpCAM
에 대해 디자인된
Phase
II
임상연구프로그램
실시예
1.1: 임상연구 요약
항 -EpCAM에 대해 디자인된 임상 연구를 하기 표에 요약하였다(표 1).
[표 1] 임상 연구 요약
| 연구목적( Study Rationale ) | 항-EpCAM은 신규한 전적으로 인간 IgG1 모노클로날 항체로서, 인간 IgD-양성 B 세포 레퍼토리에서 유래된 것이며, 특이적으로 상피세포부착분자(EpCAM)에 결합한다. EpCAM은 고도로 보존된 표면 당단백질이며, 유방암을 포함하는 다양한 기원의 많은 암종에서 과발현된다. 항-EpCAM는 전임상 연구에서 항체-의존 세포성 세포독성(ADCC)에 의해 유방암 세포를 효과적으로 살해하며 인간에서 안전한 것으로 나타났다. 본 연구는 항-EpCAM이 전이성 유방암을 앓고 있는 환자에서도 항암활성을 나타내는지 여부와 이러한 환자들을 위한 새로운 치료 옵션을 제공할 수 있는지를 조사한다. |
| 연구 디자인( Study Design ) | 랜덤화된, 다기관, 병행 그룹, 공개 임상 phase II 연구. 본 연구는 EpCAM 테스트에서 양성 반응을 나타낸 두개의 서로 다른 용량의 24주일간 치료를 수행하고, 그 효능과 EpCAM 안전성을 평가하기 위해 디자인되었다. 중앙 랜덤화 과정은 스크리닝단계에서 수행된 EpCAM 테스트 결과에 따라 계층화된다. EpCAM 계층 중 하나에 등록에 있어 환자들은 무작위로 저용량 치료 그룹 또는 고용량 치료 그룹으로 할당된다. |
| 수반 의약품( Concomitant Medication ) | 금지: 대상 제품 항- Anti-EpCAM이외의 어떠한 다른 항암 치료 예를 들어 호르몬 치료, 생물학적 치료, 화학요법, 방사선 치료. 만성적이고 체계적인 고-용량 코티코스테로이드 및 다른 면역억제 약물에 의한 치료. 기타 다른 약제. |
|
환자참여지속기간/연구수행기간(
Duration
of
Patient
Participation/
Duration
of
the
Study
)
|
각 환자들에 대해, 연구는 4-주 스크리닝 기간, 24주 치료 기간 및 4주 안전성 팔로업 기간 및 최종 PK/PD 방문(치료 종결후 12주)으로 이루어졌다. 추산 누적 기간은 9개월이며 전체 연구 기간은 22달로 예상되었다. |
| 측정 및 평가( Measurements and Evaluation ) | 각 환자들은 한번의 스트리닝 방문, 치료기간 중 14번, 치료 종결후 12주에 한번의 최종 PK/PD을 포함하여, 최대 18번 방문한다. 환자는 또한 의학적 상태의 변화가 있는 경우, 미리 계획되지 않는 방문을 할 수 있다. |
| 효능( Efficacy ) | 효능 평가는 첫번째 항-EpCAM 투여후 매 6주마다 24주까지 시행하며 그 이후에는 매 8주 마다 시행하며(팔로압 연구 기간동안), 하기를 포함한다: ?흉부 CT 스캔 또는 흉부 X-ray ?복부 CT 스캔 또는 MRI ?뼈 신티그래피 (스크리닝시 뼈 전이가 있는 환자 대상) 반응은 빨라도 4주 후의 팔로압 평가에서 확인되어야 한다. |
| 안전성( Safety ) | 신체 검사 또는 증상 관련 검사로서, 바이탈 사인, 실험실 파라메타에 대한 검사(혈액학적 검사, 임상화학적 검사, 응집 프로파일, 및 뇨 분석)를 18번째 방문을 제외하고 매번 방문시마다 수행한다. ECG는 스크리닝시 수행한다.. ECOG 성능 상태는 스크리닝시 평가한다. 면역원성 분석(HAHA)을 위한 샘플은 스크리닝시 채취한다. |
|
약물동력학(
Pharmacodynamics
)
|
자연 살해 세포(NK)측정을 위한 혈액 샘플은 2, 3 및 15번째 방문시 수집한다. |
| 약물속도학( Pharmacokinetics ) | 항-EpCAM 혈청 최저 농도 및 최고 농도는 2 내지 6번째 방문시, 치료기간 매 6주 내지 8주 및 팔로압 방문 16 내지 18째 방문시 측정된다. |
|
통계학적 방법(
Statistical
Methods
)
|
샘플크기: 플래밍의 표준 단일 단계 과정에 기초하나 정확한 바이노미널 분포를 사용하여(A'Hern)수행한 샘플 크기 계산에 따라, 각 치료단위당 85%의 가능성(즉, 파워=80%)을 제공하기 위해 효능평가 가능한 24명의 환자가 필요하며, 이는 진실한 임상 이점 비율이 25%인 경우, 95% 일측 신뢰 구간(즉 1종오류 (type 1 error)=5%)가 5% 배제함을 입증한다. 약 10% 환자가 효능에 대해 평가가 불가능하다(드롭-아웃)고 가정할 때 적어도 전체 108명(각 치료단위당 27명)이 본 연구에서 랜덤화되어야 한다. 통계분석: 1차 분석은 전체 분석 세트에 기초한다. 제1단계로, 네개의 치료 단위 각각에서 임상 이점 비율(> 24주안정 질환 환자 + CR +PR (RECIST에 따름, 실시예 1.18 참조))을 분리 평가한다. 각 치료 단위의 임상 이점 비율에서 95% 일측 신뢰 구간이 산출된다. 반응률의 95% 일측 신뢰 구간의 하한이 치료단위에서 p0 = 5% 보다 더 큰 경우, 이 치료단위에 대해서는 귀무가설(null hypothesis)이 거부된다. 이 연구에서 컷-오프 포인트는 4이며; 이는 치료단위에서 임상적 이점을 나타내는 4명의 환자가 나타나자마자 각 치료단위에 대한 귀무가설은 거부될 수 있다는 것을 의미한다. 모든 치료 단위가 충분한 활성을 나타내는 경우, 동일 용량 수준의 치료 단위는 풀(pool)이 되며, 그 용량 수준은 저/중등 EpCAM 발현을 나타내는 환자 또는 고 EpCAM 발현을 나타내는 환자의 비동등 임상적 이점 비율을 비교하는데 사용된다. 두 용량 수준의 비교는 두개의 인자, EpCAM 발현 및 용량 수준을 사용한 논리 회귀 모델로 수행하며, 이들에서 두 용량 수준에 대한 적합한 교차비(odds ratio)를 산출한다. 만약 양 용량 수준이 오직 하나의 환자군(저/중등 EpCAM 발현을 나타내는 환자 또는 고 EpCAM 발현을 나타내는 환자 중 어느 군에서라도)에서 충분한 활성을 나타낸다면, 이 두 용량 수준은 비교 설명된다. 모든 제2 종결점(효능 및 안전성)은 분석 설명된다. |
실시예
1.2: 항-
EpCAM
을 사용한 비-임상 연구의 요약
항-EpCAM 및 트라스투주마브의 ADCC 유도 효능을 9개의 유방암세포주를 사용하여 조사하였다. 항-EpCAM은 트라스투주마브와 동등한 수준으로 ADCC 특이적 세포용해를 매개할 수 있었다. 세포 표면상의 EpCAM 분자와 Anti-EpCAM을 사용한 ADCC 에 대한 감수성 사이에는 강한 상관관계가 있었다. 모든 경우에서, 최대 특이적 세포 용해는 항-EpCAM 농도 10 ㎍/mL에서 일어났으며, 이는 환자에서 표적하는 최소 최저 농도이다.
항-EpCAM는 정맥 투여시 토끼에서 매우 양호한 국소 내약성을 나타내었으며, 어떠한 거시적 변화도 일어나지 않았으며, 단지 부차적인 미세한 변화만이 관찰되었다.
실시예
1.3: 용량 선택
전임상 실험을 기초해 볼때, 항-EpCAM의 항암 활성에 혈청 최저 농도 10 ㎍/mL이 효과적인 것으로 예측되었다. 그러나 더 높은 용량이 더욱 효과적일 가능성도 배제할 수 없었다. 따라서 혈청 최저 농도 30 ㎍/mL을 나타낼 수 있도록 산출된 용량을 두번째 용량으로 하여 같이 평가하였다.
본 연구에서 사용된 용량들은 phase I 임상 연구에서 환자들에게 투여된 최고 용량을 초과하지 않는다. 로딩기 및 유지기는 짧은 기간내에 표적하는 최저 혈청 농도를 나타내고, 또한 최대 혈장 농도가 phase I 임상연구에서 평가된 농도를 초과하지 않도록 약물 속도 모델링을 사용하여 산출하였다.
실시예
1.4: 알려진 이점
전 임상 데이타는 전이성 유방암 및 다른 암을 앓고 있는 환자들이 EpCAM을 발현하는 종양 세포들이 항-EpCAM을 통해 제거됨에 따라 진행을 늦출 수 있는(안정 질환) 이점을 가질 수 있음을 제안하였다.
실시예
1.5: 임상 실험 목적
제1목적
● EpCAM 양성의 전이성 유방암을 앓고 있는 환자에서 두 상이한 용량의 항-EpCAM의 임상적 이점을 평가하기 위함이다.
제2목적
● 두 상이한 용량의 항-EpCAM에 대한 다른 반응 파라메터의 평가
● 두 상이한 용량의 항-EpCAM에 대한 안전성 및 내약성 평가
● 두 상이한 용량의 항-EpCAM의 약물 속도론적 평가
● 두 상이한 용량의 항-EpCAM의 약물 동력학적 평가
● 전이성 유방암 환자에서 항-EpCAM의 약물 속도론적 평가
● 항-EpCAM의 약물 동력학적 평가(NK 세포)
실시예
1.6: 조사 계획
연구 종결; 1차 종결 24주에서 임상 이점 비율(SD+PR+CR)
임상 이점 비율은 RECIST에 의해 안정 질환(SD) + 부분 반응(PR) + 완전 반 응(CR)을 보이는 환자 비율로 정의된다(실시예 1.18 참조).
연구 종결; 2차 종결
● 12 주에서의 임상 이점 비율(SD+PR+CR)
● 최선 전체 암 반응 비율(Best Overall Tumor Response; OTR)
● 반응 지속/진행 시간
● 부작용 및 실험실적 이상의 발생
● 항-EpCAM의 혈청 농도
● 말초 자연 살해 세포(NK)의 수
전체 연구 디자인
본 연구는 공개된, 다기관의 랜덤화 임상 phase II 연구로서 저/중등 또는 고 EpCAM 발현을 나타내는 환자들에서 24주의 치료 기간에 걸쳐 두 치료 용량의 항-EpCAM의 활성 및 안전성을 조사하기 위함이다.
전체 112명의 환자들이 본 연구에 참가하였고, 스크리닝 기간 후, 모든 적합 기준을 만족하는 환자들은 각 EpCAM 계층 내에서 무작위로 두 치료군 중 하나에 할당되었다:저용량군 및 고용량군. 항-EpCAM은 로딩기 동안은 각주에 1번씩(제1, 8, 15일), 그 후 매 2주마다, 전체 24주 동안 또는 질병 진행 때까지 60분간 정맥(i.v) 주입으로 투여되었다. 환자들은 EpCAM 발현 수준에 따라 두 그룹으로 계층화되었다: 저/중등도 EpCAM 발현을 나타내는 그룹 및 고 EpCAM 발현을 나타내는 그 룹. 도 2는 이러한 부가적인 계층화의 개요를 나타낸 것이다.
[표 2] 계층화된 치료 그룹 및 항-EpCAM 용량
| 치료군 | EpCAM 발현 | 항- EpCAM 용량 |
| Group I | 원발성 종양상에 EpCAM 중등도 발현 | 매 2주 마다 2 mg/kg 항-EpCAM i.v. |
| Group II | 원발성 종양상에 EpCAM 중등도 발현 | 매 2주 마다 6 mg/kg 항-EpCAM i.v. |
| Group III | 원발성 종양상에 EpCAM 고 발현 | 매 2주 마다 2 mg/kg 항-EpCAM i.v. |
| Group IV | 원발성 종양상에 EpCAM 고 발현 | 매 2주 마다 6 mg/kg 항-EpCAM i.v. |
환자들은 매 6주마다 24주까지 그 후 매 8주마다(팔로압 연구 기간 동안) 임상 평가 및 실험실 테스트로 모니터되었다. 흉부 CT 스캔 또는 흉부 X-ray, 복부 CT 스캔 또는 MRI, 및 스크리닝시 뼈 손상이 검출된 경우 뼈 신티그래피를 포함하여 추가적인 평가를 수행하여 암 반응을 평가하였다.
각 그룹에 대해 효능이 평가되며, 반응 지속/진행 시간을 이 환자 집단에서의 예상치와 비교하였다. RECIST 기준에 의해 측정한 암 반응(실시예 1.18 참조)을 평가하고 통계분석에 사용하였다.
연구의 제1 종결점은 상기 네 그룹 각각에서 24주시의 임상 이점 비율(CR+PR+SD)이다. 12주에서의 임상 이점 비율(SD+PR+CR), 최선 전체 암 반응 비율(OTR), 반응 지속/진행 시간이 제2 종결점으로서 평가된다. 안전성 및 내약성이 24주째에 각 치료 그룹간에 비교된다.
연구중 어느때라도 상기 용량 중 하나가 효능 또는 내약성에서 더욱 선호됨 이 명백한 경우에는 연구 프로토콜은 이에 따라 수정된다.
24주 치료 후, SD, PR 또는 CR이 문서화되고, 허용될 수 없는 독성이 보고되지 않은(그리고 4주 이상 치료 방해가 없는) 환자들은 계속적인 항-EpCAM 치료에 의한 팔로우-온 연구에 참여하였다. 장기 내약성과 같은 파라메터, 임상적 진전 및 모든 생존이 평가된다.
실시예
1.7: 임상연구집단
포함 기준
1. 하기 모든 기준을 만족하는 환자들만이 본 연구에 참여할 수 있다:
2. 스크리닝시 면역조직화학법에 의해 결정된 보존대상 조직 샘플(archived tissue samples)에서 양성 EpCAM 발현을 나타내는 전이성 유방암을 갖는 것으로 조직학적으로 확인.
3, 최소한 한 단위(dimension)에서 측정가능한 최소한 하나의 손상부위(즉 전이)가 존재할 것(RECIST에 따라 (실시예 1.18 참조)).
4. 기대 수명 ≥ 12 개월
5. ECOG 성능 상태 0-1
6. 나이 ≥18 세
7. 피험자 동의 서면을 이해하고 서명할 능력이 있을 것.
배제 기준
하기 기준 중 어느 것 하나에도 해당되는 환자는 본 연구에 참여할 수 없다:
1. 포함 결정시 실험자 평가에 의해 다른 치료가 추천되거나 선호됨.
2. CNS 전이 이력.
3. 실험자 평가에 의해 트라스투주마브(Herceptin®) 치료 징후.
4.치료 시작 전 4주 내 면역치료, 방사선 치료, 화학요법 또는 기타 다른 항암 치료가 있는 경우, 단 하기 제외:
● 1차 방문전 시작된 국소 방사선 치료(표적 손상부위는 방사선 조사 영역내여서는 않되며, 방사선 치료는 배제 기준 6에 정의된 골수 억제를 초래하는 것으로 예측되어서는 안됨)
● 호르몬 치료하에서 환자가 진행성이며, 본 연구 치료 시작 전에 종결된 경우
5. 본 치료 시작 4주 내 다른 연구 의약품.
6. 하기와 같이 기관 또는 골수 기능 이상:
● 헤모글로빈 농도 < 90 g/L 또는 9.0 g/dL
● 류코사이트 < 3 x109/L (3000/mm3)
● 혈소판 수 < 100 x109/L (100,000/mm3)
● AST(SGOT) 또는 ALT(SGPT) > 2 x 정상의 상한 (ULN)
(간 전이가 존재하는 경우 > 5 x ULN)
● 혈청 크레아티닌 > 1.5 x ULN
● 혈청 리파제 > 1.5 x ULN
● 혈청 아밀라제 > 1.5 x ULN
7. 치료 시작 전 5년 이내에 유방암 이외의 악성 종양 이력을 가진 경우로, 피부의 기저 세포 암 또는 경부의 인 시투 암종 예외.
8. 조사자 판단시 연구 수행에 방해가 될 것으로 판단되는 기타 다른 공존 질환 또는 의학적 상태.
9. 전신 코르티코스테로이드제와 같은 면역 억제제를 사용할 필요가 예상되거나, 치료전 4주 이내 면역 억제제의 정기적 사용.
10. 인간 면역결핍 바이러스(HIV) 및/또는 B형 간염 바이러스 (HbsAg 양성) 또는 C형 간염 바이러스 (항-HCV 양성)에 감염된 경우.
11. 임신 또는 수유부, 또는 임신가능성이 있으나, 치료중 또는 치료후 최소한 3개월 동안 효과적인 형태의 피임을 할 의사가 없는 경우.
12. 면역글로불린 또는 본 연구 의약 포뮬레이션의 기타 다른 구성성분에 과민인 것이 알려져 있는 경우.
실시예
1.8: 임상 시험 물질
준비
임상 시험에 사용될 시험 제제 항-EpCAM은 GMP 품질의 용액, 예를 들면 등장성 인산 버퍼내 10 mg/mL 항-EpCAM를 함유하는 용액으로 제공되었으며, +2 내지 +8 ℃에서 보관되었다.
환자에 주입될 최종 용액의 제조를 위해 항-EpCAM의 양은 환자 체중 및 치료 그룹에 기초하여 산출되었다(상기 표 2 참조).
항-EpCAM은 살균 청정 환경(라미나 플루 후드)내에서 500 mL 0.9% 소듐 클로라이드 용액내에 희석되었다. 거품이 생기는 것을 피하면서 백을 부드럽게 반전하여 항-EpCAM 용액을 혼합하여 주입을 위한 최종 용액을 준비하였다. 주입을 위한 농축액 및 최종 용액 모두는 오직 단일 사용을 위한 것이다.
치료 할당 과정
항-EpCAM 치료 개시에 가능한 한 가깝게 랜덤화를 실시하였다. 랜덤화 시점에서 모든 적합성 기준은 충족되어야만 한다. ICR(상호 컴퓨터 반응 시스템; interactive computer response system)에 의해 중앙 랜덤화 과정이 제공되었다. 조사자는 안전 웹사이트상에 각 개별 확인 번호 및 파스워드로 로그인하여 환자 기본 데이타(환자 번호, 스크리닝 데이타, 출생일, EpCAM 테스트 결과)를 제공함으로써 처치 할당에 대한 즉각적 반응을 받았다. 랜덤화 과정은 센터에 의해서 치료 할당을 계층화하지 않으며, EpCAM 발현에 의하여 계층화되어 양 EpCAM 발현 계층에서 고 용량 및 저 용량 치료가 균형있게 분포되도록 하였다. 치료 단위에서 요구된 수의 환자들이 할당되면 더 이상의 환자들은 이 각 단위로 랜덤화 될 수 없다.
상기 용액은 각 환자에게 60분에 걸쳐, 500 mL/h의 유속으로 환자에 정맥 투 여되었다.
실시예
1.9: 처치
처치 계획
질병 진전 또는 처치를 제한하는 독성이 나타나지 않는 한, 24 주의 치료에 걸쳐 전체 14회의 항-EpCAM를 각 환자들에게 주입하였다. 주입용 항-EpCAM 용액은 60분에 걸쳐 정맥 투여되며, 로딩기에는 매주, 유지기 동안에는 격주마다 주입하였다. 2회 방문 전(1일째), 환자들은 각 EpCAM 발현 계층내에서 하기 치료 그룹 중 하나로 랜덤화되었다:
● 그룹 I 및 III (저용량): 매주(제1, 2 및 3주) 2 mg 항-EpCAM/체중 1 kg의 로딩기, 및 이후 격주마다 2 mg 항-EpCAM/체중 1 kg의 유지용량의 11회 투여.
● 그룹 II 및 IV (고용량): 매주(제1, 2 및 3주) 6 mg 항-EpCAM/체중 1 kg의 로딩기, 및 이후 격주마다 6 mg 항-EpCAM/체중 1 kg의 유지용량의 11회 투여.
랜덤화 과정은 중앙화되며 스크리닝 시 수행된 EpCAM 테스트 결과에 따라 계층화된다. 상호 컴퓨터 반응 시스템을 사용하여 EpCAM 결과 및 환자 데이터를 등록하였다. 각 환자에 대해 데이터 처치 할당이 등록된다. 각 센터에서 환자마다 개별적 용량을 산출하고 주입용 최종 용액의 제조가 수행된다.
치료 24 주 후 SD, PR 또는 CR이 문서화되고 허용되지 못하는 독성이 보고되지 않은 환자 (및 4주 이상 처치 중단 없음)들은 계속되는 항-EpCAM 치료에 의한 팔로압 연구에 참여하였다.
부작용에 대한 치료 중단
연구 의약품은 부작용의 심각성 및 인과관계에 따라 중지되거나 중단되거나, 고 용량 그룹의 환자에서 그 용량이 경감될 수 있다.
조사자가 부작용이 연구 의약품에 의해 유발된 것이 아니라는 확증이 있는 경우에는 상기 처치는 계속될 수 있다. 그러나 부작용과 연구 의약품과의 관계가 완전히 배제될 수 없는 경우, 용량은 변경되어야 한다.
환자 중단 기준
연구 약품의 처치는 하기 중 어떤 것이라도 발생되는 경우에는 중단되어야 한다:
● RECIST(실시예 1.18 참조)에 의해 정의된 질병 진전
● 환자 동의 철회
● 환자 또는 조사자가 연구 프로토콜에 순응하지 않음
● 의학적 상태의 진전으로, 조사자 의견에 의해 더 이상의 연구에의 환자 참여가 배제되어야 하는 경우
● 허용되지 않는 의약품의 동시 투여
● 치료 변경이 환자에게 가장 이익이 된다는 조사자의 결정
● 3 급 부작용의 발생과
○ 임상적으로 중요한 부작용으로 조사자에 의해 판단되고 및
○ 다음 투여 전에 2 급 이하로 경감되지 않으며 및
○ 연구 의약품과 관련되었을 최소한의 가능성이 있는 경우
● 4급 부작용의 발생으로 4주 이상 동안 용량 중지가 있는 경우
● 부작용의 발생에 의해 중단이 요망되거나 필요하다는 조사자 및/또는 환자 의견이 있는 경우
환자 중단 전에, 안전성 팔로압을 위해 계획된 모든 시험 또한 수행되어 연구 종말의 평가에 사용된다(실시예 1.11 참조).
동시 투여 의약품
모든 동시투여 의약품은 사례보고서(CRF)에 기록하여야 한다. 하기 의약품 및 치료는 전체 연구 기간동안 허용되지 않는다:
● 조사 의약품 이외의 하기와 같은 모든 항암 치료:
○ 호르몬 치료
○ 생물학적 치료
○ 화학요법
○ 방사선 치료(예외: 1차 방문전 시작된 국소 방사선 치료(표적 손상부위는 방사선 조사 영역내이여서는 않되며, 방사선 치료는 배제 기준 6에 정의된 골수 억제를 초래하는 것으로 예측되어서는 안됨)), LHRH 아날로그 처치된 폐경후 환자(호르몬 수용체+)들은 이들이 연구 시작전에 LHRH 아날로그에서 진전된 경우에 한하여 본 연구 치료를 계속할 수 있다.
● 만성적이고 체계적인 고 용량 코르티코스테로이드 치료 및 기타 다른 면 역억제 치료
● 기타 다른 조사 약품
필요한 경우, 표준 프랙티스에 따라 보조 치료를 의학적으로 필요한 형태로 투여할 수 있으며, 이는 CRF에 기록하여야 한다. 비스포스포네이트 치료는 연구 시작시 뼈 전이를 보이는 환자들에 허용된다.
실시예
1.10: 평가
적합성/효능 평가
피험자 동의: 모든 특정 연구 과정 수행전에 서면의 피험자 동의서를 각 환자들로부터 받아야 한다.
포함/배제: 환자 적합성 평가는 실시예 1.7에 전술한바와 같이 수행된다-임상 연구 집단, 스크리닝을 위해 수행된 모든 실험실 측정의 리뷰를 포함.
의학적 이력/현재 의학적 상태: 과거 이력 및 현재 의학적 상태를 포함하여 일반적이고 질병 특이적 의학적 이력; 환자의 암 경과에 대한 전체 이력, 및 암 단계, 다른 예후 마커를 포함하여 이전 항암 치료에 대한 정보들이 스크리닝시 기록된다.
동시투여 의약품: 모든 동시투여되는 의약품은 연구 기간 내내 기록된다.
B형 및 C형 간염 테스트: B형 간염 표면 항원(HBsAg) 및 C형 간염 바이러스(HCV) 항체 테스트를 스크리닝 기간 중 수행하여 B형 및 C형 간염의 활성 감염을 배제한다.
종양 평가: 종양 반응은 RECIST 기준에 의해 정의된다 (실시예 1.18 참조).
● 뼈 신티그래피 : 뼈 신티그래피를 수행하여 스크리닝시 및 최종 방문시의 뼈 전이의 존재를 평가한다. 스크리닝시 뼈 전이가 존재하거나 임상적으로 지시되는 경우(예를 들면 통증의 발생 및 알칼라인 포스파타제의 상승)에는 추가적인 스캔이 수행된다.
● 흉부 컴퓨터 단층촬영( CT ) 또는 흉부 X- ray : 흉부 CT 또는 흉부 X-ray를 수행하여 스크리닝시 원격 전이의 존재를 서류화하고, 처치기 동안 및 최종 방문시 규칙적으로 반응을 평가한다.
● 복부 CT 스캔 또는 MRI : 복부 CT 스캔 또는 MRI를 수행하여 스크리닝시 원격 전이의 존재를 서류화하고, 처치기 동안 및 최종 방문시 규칙적으로 반응을 평가한다.
안전성 평가
부작용: 처치 기간 동안 및 마지막 항-EpCAM 주입후 4주까지 발생하는 모든 부작용(AE)을 기록한다. 스크리닝 기간 동안, 연구 과정에 관련되는 모든 부작용 또한 기록된다.
심각한 부작용: 모든 심각한 부작용(SAE)은 스크리닝 기간 및 팔로압 기간을 포함하여 전체 연구 기간 동안 기록된다.
신체 검사: 바이탈 사인을 포함하여 모든 신체 시스템의 완전한 신체 검사를 스크리닝시, 처치기 동안 규칙적으로, 그리고 마지막 방문시 수행한다. 신체 검사 에 관한 증상은 연구 내내 적합한 방식으로 수행되며, 모든 임상적으로 관련 있는 발견들은 문서화된다. 주입 일에는 항-EpCAM 주입 전에 신체 검사를 수행한다.
바이탈 사인 모니터링 : 체온(입안 및 고막), 심박수 및 혈압(수축기/이완기)은 하기와 같이 연구 내내 측정된다:
● 주입전
● 제2회 내지 제6회 방문시 주입 동안 매 15분 마다
● 제2회 내지 제6회 방문시 주입이 끝나고 4시간 후까지 매시간
ECOG 점수: 환자 성능 상태의 평가는 이스턴코오퍼레이티브옹콜로지그룹(ECOG) 점수를 사용하여 수행한다(실시예 1.19의 표 6 참조).
심전도( ECG ): 표준 12-리드 ECG는 스크리닝시, 처치기 동안 및 마지막 방문시 수행하였다. 각 ECG를 두 개 출력하여 하나는 현장에서 서류화하는데 사용하고, 나머지 하나는 본부 심장학자가 평가하는데 사용한다.
안전성 실험실적 평가: 안전성 실험실적 평가를 위해 혈액 샘플은 매 방문시마다 오전(주입날은 주입전에)에 채취하여, 하기와 같은 분석을 수행하였다:
● 임상화학적 분석: 아스파르트산 트랜스아미나제(AST), 알라닌 트랜스아미나제 (ALT), γ-글루타밀 트랜스퍼라제 (GGT), 알칼린 포스파타제 (AP), 락테이트 데하이드로게나제(LDH), 전체 빌리루빈, 전체 단백질, 크레아티닌, 요소, 요산, 글루코스, 칼슘, 소듐, 포타슘, 클로라이드, 포스페이트, 아밀라제, 리파제, 알부민, C-반응성 단백질(CRP).
● 혈액학적 분석: 적혈구 카운트 (RBC), 헤모글로빈, 헤마토크리트, 백혈구 카운트 (WBC), 감별 혈액 카운트 및 혈소판 카운트.
● 응고: 프로트롬빈 시간(PT, 국제정규화비율 [INR]), 부분 트롬보플라스틴 시간 (PTT) 및 피브리노겐.
뇨분석 : 뇨내 글루코스, 단백질 및 혈액의 존재를 매 방문시 마다 딥스틱(dipstick)으로 측정. 항-EpCAM 주입하는 날에는 항-EpCAM 주입전에 뇨분석을 시행하였다.
임신 테스트: 모든 임신 가능한 여성에 대해서는 스크리닝시 및 최종 방문시 임신 테스트(β-인간 융모성 고나도트로핀[β-HCG])을 수행하였다.
면역원성: 항-EpCAM 면역원성 평가를 위한 혈액 샘플은 스크리닝시, 6주 및 24주 및 마지막 두번의 팔로업 17 및 18번째 방문시 채취하였다.
약물동력학적 평가
자연 살해 세포: 자연 살해 세포(NK)수는 형광-활성 세포 분류기(FACS)에 의해 측정되었다.
약물속도론적
평가
항-EpCAM 혈청 최저 및 최고 수준은 2 내지 6 번째 방문시에, 처치 기간 중 매 6주 내지 8주 및 마지막 세번의 팔로압 방문인 16 내지 18번째 방문시 측정되었다.
EpCAM
발현 분석
EpCAM 발현은 스크리닝시 환자의 보관 종량 관련물(archived tumor material)을 사용하여 평가되었다. EpCAM 발현은 중앙 실험실에서 면역조직화학적 방법에 의해 측정되었다.
EpCAM 결과가 있는 환자들만(저/중등도 또는 높은) 모든 스크리닝 과정을 진행한다. 음성 EpCAM 테스트 결과를 나타내는 환자들은 적합성 기준에 맞지 않으며 스크리닝 실패로 간주되었다.
연구 동안 생검(예를 들면 새로이 검출된 전이의 경우)이 수행되는 경우, 종양 조직은 EpCAM 발현을 위한 분석을 위해 수집되어야 한다.
실시예
1.11: 방문 계획
모든 연구 방문의 산출은 첫 용량의 항-EpCAM 주입일로 정의되는 기준일(1일)에 기초한다.
스크리닝 기간(-
28일 에서
-1일)
모든 적합성 기준을 만족하는 환자들에만 환자 번호를 할당하였다(실시예 1.8- 치료 할당 과정 참조).
모든 스크리닝 평가는 조사 산물의 첫 투여전 28일 이내에 수행되어야 한다(1일). EpCAM 발현 테스트를 포함하여 모든 결과가 입수된 후 환자가 연구 참여에 적합하다고 선언되어야 한다. 실험실 평가에서 결과가 입수되고 환자가 모든 적합성 기준을 만족한다고 판단되는 경우, 조사자는 중앙화된 랜덤화 과정(ICRS)을 수행한다.
처치 기간(방문 2 내지 15)
처치 기간 중에, 하기 과정 및 평가가 수행되었다:
○ 바이탈 사인
○ 신체 검사
○ 안전성에 관한 실험실적 평가
○ 소변 검사
○ 약물 속도론적 검사
○ NK 세포
○ 면역원성
○ 동시 투여 의약품
○ 부작용 및 심각한 부작용
○ 흉부 X-ray / CT 스캔
○ 복부 CT 스캔 또는 MRI
○ 뼈 신티그래피(뼈 결손이 스크리닝시 검출된 경우)
○ ECG (6번째 및 15번째 방문시에만)
안전성
팔로압
기간 및 최종
PK
/
PD
평가 (방문 16-18)
안전성 팔로압 방문이 치료 종결후 2주 및 8주에 수행되며, 방문 17은 최종 한전성 팔로압 방문이된다.
마지막 연구 방문(방문 18)은 치료후 12주에 이루어지며 최종 PK/PD 평가가 이루어진다.
연구 방문의 종결(방문 17 / 최종)
연구 방문의 종결은 마지막 팔로압 방문 또는 환자가 연구에서 미리 이탈한다면 언제든지 이루어진다.
● 전체 신체 검사
● 바이탈 사인
● 12-리드 ECG(12-lead ECG)
● ECOG
● 안전성에 관한 실험실적 평가
● 소변 검사
● 임신 검사
● 흉부 X-ray / CT 스캔
● 뼈 신티그래피
● 복부 CT 스캔 또는 MRI
● 약물 속도론적 분석
● 면역원성
● 동시 투여 의약품
● 부작용 및 심각한 부작용
효능
팔로업
기간
연구 참여가 끝나고 1년 후 까지 환자는 매 3개월마다 질환의 진전, 다른 암 치료 및 생존에 대해 재조사한다. 24주에 안정 질환, 부분 또는 완전 반응을 나타내었던 환자는 공개 임상 연구에 들어갈 기회를 가지며, 여기에서 질병 진행 및 전체적인 생존에 대한 평가가 수행된다.
실시예
1.12: 샘플 보관
면역원성, NK 세포 계수 및 PK를 위한 혈청 샘플은 -20℃에서 냉동 상태로 보관된다.
실시예
1.13: 안전성 고려
조사자는 부작용(AE) 또는 심각한 부작용(SAE)의 정의를 만족시키는 사건을 검출하고 문서화할 책임이 있다. 이는 그 심각성, 격렬함의 정도 및 조사 제품 및/또는 동시에 이루어지는 치료와의 우발적인 관계의 평가를 포함한다.
실시예
1.14: 부작용, 심각한 부작용
부작용
조사 의약품의 투여중 또는 투여후 발견되거나 진단되는 하기의 사건은 부작용이다:
● 기존에 존재하였던 병 또는 영구적인 병적 상태의 악화
● 기존에 존재하였던 일과성 사건 또는 상태의 빈도수나 강도의 증가
● 연구 참여전 존재하였거나 발병한 적이 있더라도, 조사 의약품의 투여후 검출되거나 진단된 사인 또는 증상
● 현저한 혈액학적 및 기타 다른 실험적 이상 및 시험 약물/조사 의약품 처치의 포기, 용량 감소, 또는 중요한 추가적인 부수 치료를 포함하는 조정(intervention)에 이르게 하는 모든 사건.
심각한 부작용
심각한 부작용(경험) 또는 반응은 어떠한 용량에서건 하기와 같은 상태의 의학적 발생 또는 효과를 일으키는 것을 의미한다:
● 치사 초래(1)
● 생명에 위협을 줌(2)
● 입원이 요망되거나 또는 기존의 입원환자의 경우 입원의 연장
● 지속적이고 중요한 불능 또는 무능을 초래
● 선천적 이상 또는 선천성 결손
심각도의 평가: 부작용의 심각도(또는 강도)는 암 치료 평가 프로그램, 부작용에 대한 일반적 용어기준(CTCAE), 3.0판에 제공되어 있는 등급 점수에 따라 평가한다.
실시예
1.15: 통계 분석
통계적 방법 및 연구 변수의 결정
본 연구의 목적은 항-EpCAM의 유효성 및 안전성을 결정하기 위함이다. 따라서, 두 치료단위(저 및 고 용량의 항-EpCAM)에 대한 랜덤 II상 시험이 수행되고, 여기에서 각 치료 단위는 표준 단일-단계 II 상 연구로 간주된다. 치료 증거가 충분한 활성을 나타내는가의 결정은 각 치료 단위마다 별도로 이루어진다. 샘플 크기 측정 및 1차 통계 분석은 표준 단일-단계 단일-단위 II 상 디자인에 기초한다.
표 3은 제1 및 제2 연구 종결을 결정하기 위해 수행되는 측정 및/또는 데이터 관리 과정을 목록화한 것이다.
[표 3] 제1 및 제2 연구 목적, 유도 변수 및 측정 방법
| 목적 | 종결 | 측정 |
| 1차 | ||
| 두 상이한 양의 항-EpCAM의 EpCAM 양성, 전이성 유방암 환자에의 임상적 이점의 평가 | 24주에서의 임상적 비율 | 24주에 SD, PR 및 CR을 나타내는 환자의 비율 |
| 2차 | ||
| 두 상이한 양의 항-EpCAM에 대한 기타 다른 반응 파라메터의 평가 |
12주에서의 임상적 비율 | 12주 및 48주에 SD, PR 및 CR을 나타내는 환자의 비율 |
| 최선 암 반응 비율(OTR) | 연구 기간에 걸쳐 PR/CR을 성취한 환자 비율 | |
| 반응 지속/ 진행 시간 | 카플란-메이어 방법을 사용하여 산출된 질병 진행에 이르느Me는 평균(메디안) 시간 | |
| 두 상이한 양의 항-EpCAM에 대한 안전성 및 내약성 평가 | 부작용 및 실험실적 이상의 발생 | 부작용을 나타내는 환자의 비비율: 수, 강도 및 실험실적 이상, 임상적 부작용 및 심각한 부작용의 연구 처치와의 관계(조사자에 따라) |
| 두 상이한 양의 항-EpCAM의 약물속도론적 분석 | 항-EpCAM의 혈청 농도 | 지정된 시점에서의 항-EpCAM의 혈청 농도 |
| 두 상이한 양의 항-EpCAM의 약물동력학적 분석 | 말초 자연살해 세포수 | 시간에 따른 NK 세포 계수의 증가/감소% |
통계적 가설: 본 통계적 가설을 위해 하기 가정이 수립되었다:
양호한 지지 간호(best supportive care)에 대한 배경 임상 이점 비율은 5% 이하로 추산된다(p0). 기술되는 환자 집단(양성 EpCAM 발현)에서의 항-EpCAM의 차후 사용은 진임상이점비율(π)가 25% 이상(p1)인 경우 상당한 이점을 갖는다.
각 치료 단위(저/중등도 및 고 EpCAM 계층 내 저 또는 고 용량 그룹)에 대하여, 하기 가설이 적용된다:
H0ij(π≤p0ij): pij ≤ 5% (배경 임상 이점 가능성)
H1ij(π≥p1ij): pij ≥ 25% (항-EpCAM에 대한 관심 임상 이점 가능성)
여기에서 pi는 용량 수준 i로 처리된 저/중등도 EpCAM 발현 계층 (j=1) 및 고 EpCAM 발현 계층 환자(j=2) 내에서 관찰되는 임상 이점 비율이며, 낮은 수준은 i = 1 로 높은 수준은 i = 2로 지시된다.
유의성 수준, 복수 비교 및 중복도: 5%의 1종 오류 및 15%의 2종 오류(85% 파워)는 임상 이점 비율을 결정하는데 적합한 것으로 간주된다. 어떠한 유의성 수준도 복수 비교 또는 중복도로 인하여 조정될 필요가 없다(치료 단위 간에 어떠한 확정적 비교도 수행되지 않음).
샘플 크기 결정: 샘플 크기 추산은 플래밍의 표준 단일 단계 과정에 기초하나 바이노미널 분포에 대한 정규 추정(Fleming (1982). Biometrics 38: 143-51)이 아닌 정확한 바이노미널 분포(A'Hern (2001). Statistics in Medicine 20: 859-66)를 사용하여 이루어진다. 상기 정규 추정이 작은 샘플 크기에 정확하지 않고, 정확한 분포에 기초한 샘플 크기 및 컷-오프 포인트는 컷-오프 포인트가 달성되는 경우 산출된 신뢰 구간이 p0를 포함하지 않는 이점을 갖기 때문에 이러한 접근이 선호된다. 따라서, 본 연구에서는 A'Hern (A'Hern (2001). Statistics in Medicine 20: 859-66)에 의해 제공된 샘플 크기표를 사용하였다.
이러한 계산에 따르면, 본 연구가 24주에 걸쳐 진짜 전체 반응 비율이 25%(p1)인 경우 반응 비율에 대한 95% 일측 신뢰 구간(즉 1종 오류=5%)이 5%(p0)를 배제하는 것을 입증하는 85%의 가능성(즉 파워=85%)을 갖기 위해서는 각 치료단위당 24명의 효능평가 가능한 환자가 필요하다.
약 10% 환자가 효능 평가가 불가능하다(드롭-아웃)고 가정할 때 적어도 전체 108명(저/중등도 EpCAM 계층 54명에서 27명은 저 용량으로 27명은 고용량으로, 고 EpCAM 발현 계층 54명에서 27명은 저 용량으로 27명은 고용량으로)이 본 연구에서 랜덤화되어야 한다.
계획분석:
제1 및 제2 변수가 예비적으로 평가되었다. 환자들에 대해 모든 관련 데이터(CRF 데이터, 실험실 데이터)가 치료단위 및 방문으로 그룹화되어 분석되었다.
각 환자 데이터는 목록으로 제공되었다(치료 단위 및 환자수로 분류). CRF 및 데이타베이스에 포함된 모든 데이터는 목록화되었다.
통계적 특성 및 기타 베이스라인(baseline) 특성: 통계적 및 기타 베이스라인 특성이 전체 및 치료단위별로 연속 변수를 위해 요약 통계학에 의해(환자수, 평 균, 표준편차, 최소, 메디안, 최대)에 의해 요약되고, 카테고리 변수를 위해 절대 및 상대 빈도수에 의해 요약되었다. 베이스라인 특성은 제1 항-EpCAM 투여전에 수행된 모든 검사 결과에 의해 정의되었다.
제1 종결점을 위한 계획 분석: 제 분석은 전체 분석 세트에 기초하며, 각 프로토콜 세트에 기초된 분석은 민감성 분석으로 수행된다. 본 연구의 제1 종결점은 임상 이점 비율(RECIST에 따라(실시예 1.18 참조) 안정 질환+CR+PR을 나타내는 환자)이다.
첫 단계로, 각 치료 단위에서의 임상 이점 비율이 별개로 평가되었다. 각 치료단위의 임상 이점 비율에 대해 95% 일측 신뢰 구간이 산출되었다. 반응 비율의 95% 일측 신뢰 구간의 하한이 치료 단위에서 p0 = 5% 보다 더 크면, 이 치료 단위에 대해서는 귀무가설(null hypothesis)이 거부된다. 이 연구에서 컷-오프 포인트는 4이며; 이는 치료단위에서 임상적 이점을 나타내는 4명의 환자가 나타나자마자 각 치료단위에 대한 귀무가설은 거부될 수 있다는 것을 의미한다.
제1 종결점의 추가적인 분석은 하기를 포함한다:
● 모든 치료 단위가 충분한 활성을 나타내는 경우, 동일 용량 수준의 치료 단위가 풀(pool)이 되며, 그 용량 수준은 저/중등 EpCAM 발현을 나타내는 환자 및 고 EpCAM 발현을 나타내는 환자에 대한 비동등 임상적 이점 비율을 비교하는데 사용된다. 두 용량 수준의 비교는 두개의 인자, EpCAM 발현 및 용량 수준을 사용한 논리 회귀 모델로 수행하며, 이들에서 두 용량 수준에 대한 적합한 교차 비(odds ratio)를 산출한다.
● 만약 양 용량 수준이 오직 하나의 환자군(저/중등 EpCAM 발현을 나타내는 환자 또는 고 EpCAM 발현을 나타내는 환자 중 어느 군에서라도)에서 충분한 활성을 나타낸다면, 이 두 용량 수준은 비교 설명된다.
제2 종결점에 대한 계획 분석: 각 치료 단위에 대해 24주시의 최선 전체 종양 반응 비율을 상기 제1 종결점에서 기술된 것과 동일한 방식으로 평가하였다.
부작용은 치료단위 및 용량 수준에 의해 전체로 요약되며, 제1 시스템 장기 클래스, 고 수준 기간, 선호 기간 및 심각도에 의해 그룹화되었다.
치료 단위 및 용량 수준 사이에 대한 부작용의 전체 발병율의 통계적 비교는 수행되지 않았다.
모든 샘플 일수 전체에 대해 치료 단위 및 용량 수준에 따라 데이터를 요약하였다. 모든 후-베이스라인 샘플 일수들에 대한 베이스라인 값에서의 절대적인 변경을 전체로 치료 단위 및 용량 수준에 의해 요약하였다. 베이스라인에서의 변동표를 전체 뇨분석 데이터에 대해 치료 단위 및 용량 수준에 의해 만들었다.
약물동력학적 파라메터를 원데이타의 요약 통계(평균, 표준편차, 최소, 메디안, 및 최대) 및 모든 연구 일수 전체에 대한 베이스라인에서의 변경을 치료 단위 및 용량 수준에 의해 제시함으로써 분석하였다.
관리상의 데이터
리뷰
항-EpCAM에 의한 후속 연구를 위한 추가적 전략을 결정하기 위하여, 최선 전 체 반응 비율의 관리상 분석을 하기 사항이 확인될 때 수행하였다.
● 70명의 환자에 최소한 한번의 주입을 하고 방문 9/12 주가 지난 후 또는 미리 연구가 중단된 후 및
● 최선 전체 반응 비율에 대한 데이터가 합리적으로 흠이 없다고 간주됨.
이러한 관리상 분석을 위해 소집은 중단되지 않는다.
실시예
1.16: 품질관리 및 품질 보증
연구 장소에 환자들을 등록하기 전에, 독립적 윤리 위원회(IEC) 승인 및 조사자나 연구 스텝의 이력서와 같은 특정 등록 서류들이 입수되어야 한다.
본 연구는 연구 스폰서에서 임명된 자격을 갖추고 적절하게 훈련된 사람에 의해 모니터되었다.
실시예
1.17: 법적 및 윤리적 보증
본 연구는 임상시험 관리기준(GOOD CLINICAL PRACTICE)에 대한 ICH Harmonized Tripartite 가이드라인 및 헬싱키 선언(Declaration of Helsinki, June 1964, as modified by the 48th World Medical Association, Somerset West, Republic of South Africa, October 1996)을 포함하여 관련 법 및 규칙에 따라 수행되었다.
환자 정보 및
피험
동의
환자 동의를 얻는 과정은 모든 적용가능한 등록 요건에 일치하여 수행된다. 환자들을 임상 연구에 포함시키기 전에, 그/그녀의 자유롭고 공표된 피험동의는 반드시 서면으로 입수되어야 한다.
실시예
1.18:
RECIST
기준
하기는 본 연구 전반에 걸쳐 사용되는 RECIST 기준을 요약한 것이다.
적격성
베이스라인에서 측정가능한 질병을 갖는 환자들만이 객관적 종양 반응이 제1 종결점인 프로토콜에 포함되어야 한다.
측정가능한 질병( Measurable disease ) - 하나 이상의 측정가능한 병소의 존재. 만약 측정가능한 질병이 고립 병소에 국한되는 경우, 그 종양성 본질이 세포학/조직학적으로 확인되어야 한다.
측정가능한 병소( Measurable lesions )- 통상 기술 사용시 최장 직경 ≥ 20 mm 또는 나선식 전산화 단층촬영(spiral CT scan) 사용시 최장 직경 ≥ 10 mm 인 최소한 한 디멘젼에서 정확히 측정가능한 병소.
측정가능하지 않은 병소( Non - measurable lesions ): 작은 병소(통상 기술 사용시 최장 직경 ≤ 20 mm 또는 나선식 전산화 단층촬영(spiral CT scan) 사용시 최장 직경 < 10 mm)을 포함하는 기타 다른 병소, 즉 뼈 병소, 연수막 질환(leptomeningeal disease), 복수(ascites), 늑막/심막 유출(pleural/pericardial effusion), 염증성 유방 질환, 림프관염 진피/풀모니스(lymphangitis cutis/pulmonis), 낭종성 병소, 및 이미징 기술에 의해 따르는 미확인 복부 종괴(abdominal masses) 등이다.
모든 측정은 자나 칼리퍼를 사용하여 미터 단위로 측정 및 기록되었다. 모든 베이스라인 평가는 되도록 처치의 시작에 근접하여 수행되며, 처치 시작전 4주를 넘지 않는다.
확인되고 보고된 병소를 특정하기 위해서 베이스라인 및 팔로압 동안에 동일한 평가 방법 및 동일한 기술을 사용한다.
임상적 병소는 오직 표재성(예를 들면 피부 소절 및 촉진가능한 림프절)인 경우에 측정가능한 것으로 간주된다. 피부 병소의 경우 컬러 사진으로 서류를 작성하고, 병소의 크기는 자로 잴 것이 추천된다.
측정 방법
CT 및 MRI는 반응 평가를 위해 표적 병소를 측정하기 위해 선택되는 가장 좋고 재현성 있는 방법이다. 통상의 CT 및 MRI는 인접하는 슬라이스 두께 10 mm 내외의 절편으로 수행하여야 한다. 나선식 CT는 5 mm 인접 재구성 알고리즘을 사용하여 수행된다. 이는 흉부, 복부 및 골반의 종양에도 적용된다. 두부 및 경부 종양 및 말단의 종양은 통상 특별한 프로토콜을 요한다.
흉부 X-ray 상의 병소는 이들이 명확히 확인되고 공기함유 허파에 의해 둘러 싸인 경우 측정가능한 병소로서 받아들여진다. 그러나 CT가 더욱 선호된다.
연구의 제1 종결점이 객관적 반응 평가인 경우, 종양 병소를 측정하기 위해 초음파(US)를 사용해서는 안 된다. 그러나 이는 표재성 접촉성 림프절, 피하 병소 및 갑상선 노듈의 임상적 측정을 위해서는 가능한 대안으로 사용될 수 있다. 초음파는 또한 임상 검사에 의해 통상 평가되는 표재성 병소의 완전한 소멸을 확인하는데 유용하다.
객관적인 종양 평가에 대해 내시경 및 복강경 검사의 사용은 아직 완전하게 그리고 광범위하게 검증되지 않았다. 이러한 특정 맥락에서 이들의 사용은 정교한 장비 및 오직 특정 센터에서만 가능한 고 수준의 전문성을 요구한다. 따라서 객관적 종양 반응을 위한 이들 기술의 사용은 특별한 센터에서의 검증목적으로 제한되어야 한다. 그러나 이러한 기술은 생검이 얻어질 때 완전한 병리적 반응을 확인하는데 유용할 수 있다.
종양 마커 단독으로는 반응을 평가하는데 사용될 수 없다. 마커가 초기에 정상의 상한 이상으로 있었다가 정상화되는 경우, 환자는 모든 병소가 소멸되는 완전한 임상 반응을 나타내는 것으로 간주된다.
세포학적 및 조직학적 기술은 몇몇 드문 경우에 PR 과 CR을 분별하기 위해 사용될 수 있다(예를 들면 처치후 잔여하는 양성 병소와 배세포 종양과 같은 종양 타입에서 잔여하는 음성 병소와의 분별).
"표적" 및 "
비표적
" 병소의 베이스라인
서류화
장기당 최대 5 병소 및 전체 10 병소에 이르는 모든 측정가능한 병소, 모든 대표적인 관련 장기는 표적 병소로서 확인되고 기록되고 베이스라인에서 측정되어야 한다.
표적 병소는 그 크기(최장 직경을 갖는 병소) 및 신속한 반복 측정에의 적합성(이미징 기술 또는 임상적으로)을 기초로 선택되어야 한다.
모든 표적 병소에 대한 최장 직경(LD)의 총합을 계산하여 베이스라인 합계(LD)로 기록한다. 베이스라인 합계(LD)는 객체 종양을 특정화하는데 기준으로 사용된다.
다른 모든 병소(또는 질병 부위)는 비-표적 병소로서 확인되고 베이스라인에서 기록되어야 한다. 이들 병소의 측정은 요구되지 않지만, 각각의 존재 및 비존재는 팔로압에 걸쳐 주목되어야 한다.
반응 기준
표적 병소 평가
* 완전 반응(CR): 모든 표적 병소의 소멸
* 부분 반응(PR): 표적 병소의 LD 합이 베이스라인 합 LD를 기준으로 최소한 30% 감소
* 진행성 질환(PD): 치료 시작후 기록된 최소 합 LD를 기준으로, 표적 병소 의 LD 합이 최소한 20% 증가하거나 하나 이상의 새로운 병소의 출현
* 안정 질환(SD): 치료 시작후 최소 합 LD를 기준으로 하여 볼때, PR에 적합한 정도로 충분히 수축하지도 않고, PD에 적합한 정도로 충분히 증가하지도 않은 상태
비-표적 병소의 평가
* 완전 반응(CR): 모든 비-표적 병소의 소멸 및 종양 마커 수준의 정상화
* 부분 반응/안정 질환(SD): 하나 이상의 비표적 병소의 지속 또는/및 종양 마커 수준이 정상 한계 이상으로 유지
* 진행성 질환(PD): 하나 이상의 새로운 병소의 출현 및/또는 기존의 비-표적 손상(1)의 명백한 진행
(1) 이러한 상황에서 "비표적" 병소의 명백한 진행이 예외적인 경우, 치료 의사의 의견은 설득적이어야 하며 진행 상황은 이후 리뷰 패널(또는 연구 수장)에 의해 확인되어야 한다.
최선 전체 반응의 평가
최선 전체 반응은 치료 시작에서 질병 진행/재발때 까지 기록된 가장 양호한 반응이다(치료가 시작된 이후부터 기록된 가장 최소 측정이 PD를 위해 기준으로 사용된다). 통상 환자의 가장 양호한 반응인가의 판단은 측정 및 확인 기준 양쪽을 모두 만족하는가에 따른다.
| 표적 병소 | 비표적 병소 | 새로운 병소 | 전체반응 |
| CR | CR | 없음 | CR |
| CR | 불완전 반응/SD | 없음 | PR |
| PR | 비-PD | 없음 | PR |
| SD | 비-PD | 없음 | SD |
| PD | 모든 경우 | 있음 또는 없음 | PD |
| Any | PD | 있음 또는 없음 | PD |
| Any | 모든 경우 | 있음 | PD |
그 당시 질병 진행에 대한 명백한 증거 없이 치료 중단이 요구되는 전체적인 건강 상태의 악화를 나타내는 환자는 "증상적 악화(symptomatic deterioration)"을 갖는 것으로 분류된다. 치료 중단 후에도 모든 노력을 기울여 객관적인 진전을 기록화하여야 한다.
어떤 경우에는 정상 조직과 잔여 질병을 구분하기 힘들다. 완전 반응의 평가가 이러한 결정에 의존하는 경우, 잔여 병소를 조사(극세 니들 흡인/생검)에 의하여 완전 반응 상태를 확인하는 것이 추천된다.
확인
객관적 반응 확인의 중요한 목적은 관찰되는 반응 비율을 과대평가하는 것을 피하기 위함이다. 반응의 확인이 가능하지 않은 경우, 반응이 확인되지 않은 연구를 보고할 때는 명확히 하여야 한다.
PR 또는 CR 상태로 할당되기 위해서는 종양 측정의 변화가 반복 평가에 의해 확인되어야 하며, 반복 평가는 반응 기준이 처음 만족된 때부터 최소한 4주 이내에 수행되어야 한다. 연구 프로토콜에 의해 결정된 더 긴 구간도 적합할 수 있다.
SD의 경우, 팔로압 측정은 연구 프로토콜에 정의된 최소 구간(통상, 6-8주 이상)에서 연구 시작 후 최소한 한번은 SD 기준을 만족하여야 한다.
전체 반응의 지속
전체 반응의 지속은 CR 또는 PR에 대한 측정 기준이 만족하는 때(둘 중 어떠한 상황이든 우선 기록된다)부터 재발 또는 PD가 객관적으로 서류화되는 첫날까지 측정되며, 측정이 시작된 후 기록된 가장 최소 측정값을 PD에 대한 기준값으로 한다.
안정 질환의 지속
SD는 치료가 시작될때부터 질환 진행에 대한 기준이 만족될때까지 측정되며, 치료 시작이후로 기록된 가장 최소 측정값을 기준으로 한다. SD 지속의 임상적 관련성은 종양 타입과 등급에 따라 다양하다. 따라서 프로토콜은 SD 측정을 위한 두개의 측정에 요구되는 최소 시간 간격을 특정하는 것이 추천된다. 이 시간 간격은 예기되는 임상 이점이 연구가 수행되는 집단에 미치는 현황을 고려하여야 한다.
반응
리뷰
반응 비율이 제1 종결점인 시도에서는 모든 반응이 연구의 종결시 연구와 독립한 전문가에 의해 리뷰될 것이 강력히 추천된다. 환자 파일 및 방사선 이미지의 리뷰를 동시에 수행하는 것이 가장 좋은 접근 방법이다.
결과 보고
본 연구에 포함된 모든 환자는 주 프로토콜 치료에서 다소 변경이 있거나 환자들이 적합하지 않은 경우에도 치료에 대한 반응을 평가하여야 한다. 각 환자는 하기 기준 중 하나로 할당된다: 1) 완전 반응, 2) 부분 반응, 3) 안정 질병, 4) 진행성 질병, 5) 악성 질병으로 인한 조기 치사, 6) 독성으로 인한 조기 치사, 7) 다른 원인으로 인한 조기 치사, 또는 9) 미상(불충분한 데이터로 인해 평가불가).
적합성 기준을 만족한 모든 환자들은 반응 비율 주분석에 포함되어야 한다. 반응 카테고리 4-9에 속하는 환자들은 치료에 대해 반응 실패로 간주된다(질병 진행). 따라서 정확하지 않은 치료 계획 또는 약물 투여는 반응 비율 분석에서 배제되지 않는다. 카테고리 4-9에 대한 정확한 정의는 프로토콜 특이적이다.
모든 결론은 모든 적합 환자들에 기초하여 내려진다.
이후 하부세트 환자를 기초로 하부 분석이 수행되며, 주 프로토콜에서 벗어나는 것으로 확인된 환자들은 배제된다(즉 다른 이유로 조기 치사, 치료의 조기 중단, 중요한 프로토콜 위반 등). 그러나 이 하부 분석들은 치료 효용성에 관한 결론을 도출하는데는 기초로 사용되지 않으며, 분석에서 환자를 배제시킨 이유는 명확하게 보고되어야 한다. 95% 신뢰 구간이 제공되어야 한다.
실시예
1.19:
ECOG
성능 상태 개관
ECOG 성능 상태 계수는 Oken, M.M. 등. (1982) Am J Clin Oncol 5:649-655에 개시되어있다.
실시예
2: 항-
EpCAM
의 임상 제
II
상 연구에 대한 관리 데이터
리뷰
실시예
2.1: 연구 서론 및 요약
임상 연구는 실시예 1에 전술한 대로 수행되었다. 이 연구의 결과를 실시예 2에 나타내었다.
방법:
랜덤화되며, 공개되며, 다기관의 병행 그룹의 임상 제 II상 연구. 본 연구는 EpCAM 테스트 양성의 두개의 상이한 용량에서 24주에 걸친 치료에 대한 항-EpCAM의 효능과 안전성을 평가하기 위해 디자인되었다. 중앙 랜덤화 과정은 스크리닝시 수행된 EpCAM 테스트 결과에 따라 계층화되었다. EpCAM 계층 중 하나에 등록함에 있어, 환자는 저용량 치료군 또는 고용량 치료군에 랜덤하게 할당되었다.
랜덤화된 치료 환자수 : 112 명(28명의 환자는 현재 진행중)
분석된 환자수 : 73 명의 처치 환자(37명; 항-EpCAM 고용량, 36명: 항-EpCAM 저용량)
전술한 분석을 위해 분석된 데이터:
결과를 위한 모든 데이터는 데이터 클리닝을 완료하지 않고 GCP-요건에 따라 모니터되었다. 12주까지 환자의 모든 방문을 기록하고 6주 및 12주(n = 23) 에 모든 요구되는 종양 평가를 기록한 환자의 사례보고서들을 특히 중요한 프로토콜 변경의 검출 및 안전성 데이터의 결점 유무를 중점적으로 리뷰하였다. 이들 23명의 환자들에 대한 방사선 데이터는 중앙에서 분석 리뷰하였다.
분석 집단의 정의:
● 안전성 분석 세트(SAF): 최소한 한 용량의 할당된 연구 의약품을 투여받은 모든 환자.
● 전체 분석 세트(FAS): EpCAM 양성(저/중등도 또는 고 발현) 종양을 가진 안전성 분석 세트내 환자이며, 임상적 질병 진행 이외의 다른 이유 때문에 조기에 포기한 경우 치료 시작 후 한번 이상 종양 평가를 받은 환자.
안전성 데이터의 분석은 SAF를 기초로 시행되었다. 베이스라인 데이터 및 효능 종결점에 대한 분석은 전체 분석 세트를 기초로 시행되었다.
분석된 종결점(전술한
실시예
1.1 참조):
● 12주의 최선 전체 종양 반응(OTR)비율(RECIST에 따라 완전 반응[CR] 또는 부분 반응[PR]을 나타내는 환자)
● 12주에 임상 이점 비율(CBR)(RECIST에 따라 안정 질환 또는 완전 반응[CR] 또는 부분 반응[PR]을 나타내는 환자)
● 24주에 임상 이점 비율(CBR) (RECIST에 따라 안정 질환 또는 완전 반응[CR] 또는 부분 반응[PR]을 나타내는 환자)
● 랜덤화부터의 시간 또는 치료 시작부터의 시간으로서의 진행 시간(TTP).
분석 서브그룹:
고, 중등도 및 저 EPCAM 발현자들은 Gastl 등. (2000). Lancet 356, 1981-2 에 따라 확인되었다.
하기 서브그룹이 분석되었다:
● 저용량 항-EpCAM 처치된 EpCAM 저/중등도 발현자 그룹("저용량 항-EpCAM/저 EpCAM")
● 고용량 항-EpCAM 처치된 EpCAM 저/중등도 발현자 그룹("고용량 항-EpCAM/저 EpCAM")
● 저용량 항-EpCAM 처치된 EpCAM 고 발현자 그룹("저용량 항-EpCAM/고 EpCAM")
● 고용량 항-EpCAM 처치된 EpCAM 고 발현자 그룹("고용량 항-EpCAM/고EpCAM")
● 저용량 또는 고용량 항-EpCAM 처치된 EpCAM 저/중등도 발현자 그룹("저 EpCAM")
● 저용량 또는 고용량 항-EpCAM 처치된 EpCAM 고 발현자 그룹("고 EpCAM")
● EpCAM 저/중등도 또는 고 발현자에 저용량 항-EpCAM 처치 그룹("저용량 항-EpCAM")
● EpCAM 저/중등도 또는 고 발현자에 고용량 항-EpCAM 처치 그룹("고용량 항-EpCAM").
분석 수행:
전술한 반응(CBR 또는 OTR) 종결점은 하기와 같이 분석되었다:
● 각 서브그룹(상기 참조) 및 모든 환자에 대한 CBR/OTR 비율.
● 하기 비교를 위한 CBR/OTR 비율에 대한 피셔의 정확 검정(Fisher's Exact Test) rate for the following comparisons:
● 각 서브그룹(상기 참조) 대 모든 다른 조합 환자
● "저용량 항-EpCAM/저 EpCAM" 대 "저용량 항-EpCAM/고 EpCAM"
● "저용량 항-EpCAM/저 EpCAM" 대 "고용량 항-EpCAM/저 EpCAM"
● "고용량 항-EpCAM/고 EpCAM" 대 "저용량 항-EpCAM/고 EpCAM"
● "고용량 항-EpCAM/고 EpCAM" 대 "고용량 항-EpCAM/저 EpCAM"
임상적 질병 진행 시간은 처음 항-EpCAM을 주입한 날(민감성 분석의 경우:랜덤화한 날)과 임상 질병 진행일, 즉 진행성 질병이 처음 나타난 날과의 기간으로 정의되었다. 만약 임상 질병 진행이 관찰되지 않은 경우에는 각 시간 간격은 연구 종결일로 하였다. 연구 종결일이 확실치 않은 경우 마지막으로 방문한 날짜를 사용하였다.
진행 시간(TTP) 종결점은 하기와 같이 분석되었다:
● 각 서브그룹 및 모든 환자들에 대한 메디안 TTP(측정가능한 경우)
● 하기 비교를 위한 TTP에 대한 로그 랭크 테스트(Log-Rank test):
○ 각 서브그룹 대 모든 다른 조합 환자
○ "저용량 항-EpCAM/저 EpCAM" 대 "저용량 항-EpCAM/고 EpCAM"
○ "저용량 항-EpCAM/저 EpCAM" 대 "고용량 항-EpCAM/저 EpCAM"
○ "고용량 항-EpCAM/고 EpCAM" 대 "저용량 항-EpCAM/고 EpCAM"
○ "고용량 항-EpCAM/고 EpCAM" 대 "고용량 항-EpCAM/저 EpCAM"
실시예
2.2: 결과 - 연구 환자들
분석된 데이터세트들
분석 집단을 표 4에 나타내었다.
전체 분석 세트(FAS) (n = 67)는 반응 및 진행 시간 분석에 대한 분석 집단을 나타낸 것이다. EpCAM 상태는 두 항-EpCAM 용량군 내에 동등하게 분포되었으며, 각 치료군에서 저 -EpCAM 발현자는 약 38% 이고 고 EpCAM 발현자는 약 57% 이었다.
[표 4] 분석 집단1
| 환자 | 치료군 | |||||
|
전체
(N=73) |
저용량
항-
EpCAM
(N=36) |
고용량
항- EpCAM (N=37) |
||||
| n | % | n | % | n | % | |
| 전체 안전성 분석 세트(SAF) | 73 | 100.0 | 36 | 100.0 | 37 | 100.0 |
| 전체 분석 세트(FAS)2 | 67 | 91.8 | 32 | 88.9 | 35 | 94.6 |
| EpCAM 음성 전체 | 3 | 4.1 | 1 | 2.8 | 2 | 5.4 |
| 안전성 분석 세트(SAF) | 3 | 100.0 | 1 | 100.0 | 2 | 100.0 |
| 전체 분석 세트(FAS)2 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 |
| EpCAM 저/중등도 전체 | 28 | 38.4 | 14 | 38.9 | 14 | 37.8 |
| 안전성 분석 세트(SAF) | 28 | 100.0 | 14 | 100.0 | 14 | 100.0 |
| 전체 분석 세트(FAS)2 | 27 | 96.4 | 13 | 92.9 | 14 | 100.0 |
| EpCAM 고 전체 | 42 | 57.5 | 21 | 58.3 | 21 | 56.8 |
| 안전성 분석 세트(SAF) | 42 | 100.0 | 21 | 100.0 | 21 | 100.0 |
| 전체 분석 세트(FAS)2 | 40 | 95.2 | 19 | 90.5 | 21 | 100.0 |
| 1 관리데이터 리뷰내 포함된 환자수 (%) 2 전체 분석 세트 (FAS): 안전성 분석 세트 중 EpCAM 양성(저/중등도 또는 고 발현) 종양을 가진 환자이며 임상적 질병 진행 이외의 다른 이유 때문에 조기에 포기한 경우 치료 시작 후 한번 이상 종양 평가를 받은 환자. |
||||||
실시예
2.3: 결과-전체 종양 반응(
OTR
) 및 임상 이점 비율(
CBR
)에 대한 효능 분석
연구 프로토콜에 따라 본 연구의 제1 종결점은 24주에서의 임상 이점 비율이다.
"최선 전체 종양 반응(
OTR
)"
EpCAM 고 및 저 발현자 및 전부에 대한 FAS에서 "12주에서의 최선 전체 종양 반응(OTR)", "12주에서의 임상 이점 비율(CBR)", 및 "24주에서의 임상 이점 비율(CBR)"에 대한 결과를 하기 표 5 및 6에 나타내었다.
RECIST 기준에 따라 PR 또는 CR로 나타낸 반응은 중앙 방사선 평가에서의 FAS 내 어떠한 환자에서도 확인될 수 없었다.
"12주에서의 임상 이점 비율(
CBR
)"
전체, FAS의 67명 중 16명(24 %)의 환자가 12주에 질병 안정화(SD)를 나타내었다. 치료군간에는 12주에서의 임상 이점 비율이 저용량군에서는 21.9%, 고용량군에서는 25.7%로서, 이 임상 이점 비율에 있어서는 어떠한 유의성 있는 차이도 없었다. CBR은 고 EpCAM군이 저 EpCAM군에서보다 높았으나, 이 차이는 통계적으로 유의하지 않았다.
임상 이점 비율은 오직 RECIST에 의한 경감이 발견되지 않아 안정 질환을 나타내는 환자들만 포함하였다(위 참조).
[표 5] 12주에서의 임상 이점 비율(CBR) (RESICST에 따라 안정 질환 또는 완 전 반응[CR] 또는 부분 반응[PR]을 나타내는 환자) - 완전 분석 세트
| 저용량 항- EpCAM 1 | 고용량 항- EpCAM 1 | P- 값 2 | |||
| n | % | n | % | ||
| 저 EpCAM ( N 3 = 13 / 14) | 2 | 15.4 | 2 | 14.3 | 1.0 |
| 고 EpCAM ( N 3 = 19 / 21) | 5 | 26.3 | 7 | 33.3 | 0.7365 |
| 전체 ( N 3 = 32 / 35) | 7 | 21.9 | 9 | 25.7 | 0.7797 |
| 1 임상 이점이 RECIST에 따라 평가될 수 있는 환자중 임상 이점을 나타내는 환자수 2 이측 피셔 정확 검증 3 N 저용량군 /고용량군 |
|||||
24주에서의 "임상 이점 비율(
CBR
)"
24주의 임상 이점 비율에서 고용량군(14.3%)는 저용량군(6.3%)에 비해 더 높은 임상 이점 비율을 나타내었다. 고용량군에서 CBR은 저 및 고 EpCAM 서브그룹에서 동일하였다(14.3%). 저용량군에서 CBR은 고 및 저 EpCAM 발현자에 대해 유사하였다(5.3 대 7.7 %).
CBR은 오직 PR 또는 CR이 검출되지 않는 안정 질환을 나타내는 환자들만을 기초로 하였다.
[표 6] 24주에서의 임상 이점 비율(CBR) (RESICST에 따라 안정 질환 또는 완전 반응[CR] 또는 부분 반응[PR]을 나타내는 환자)
|
저용량
항- EpCAM 1 |
고용량
항- EpCAM 1 |
P- 값 2 | |||
| n | % | n | % | ||
| 저 EpCAM ( N 3 = 13 / 14) | 1 | 7.7 | 2 | 14.3 | 1.0 |
| 고 EpCAM ( N 3 = 19 / 21) | 1 | 5.3 | 3 | 14.3 | 0.6039 |
| 전체 ( N 3 = 32 / 35) | 2 | 6.3 | 5 | 14.3 | 0.4266 |
| 1 임상 이점이 RECIST에 따라 평가될 수 있는 환자중 임상 이점을 나타내는 환자수 2 이측 피셔 정확 검증 3 N 저용량군 /고용량군 |
효능 분석: 진행 시간
임상적 질병 진행 시간은 처음 항-EpCAM을 주입한 날(민감성 분석의 경우:랜덤화한 날)과 임상 질병 진행일, 즉 진행성 질병이 처음 나타난 날과의 기간으로 정의되었다. 만약 임상 질병 진행이 관찰되지 않은 경우에는 각 시간 간격은 연구 종결일로 하였다. 연구 종결일이 확실치 않은 경우 마지막으로 방문한 날짜를 사용하였다.
저 및 고 용량군 및 EpCAM 서브그룹에 대한 FAS 내 진행 시간(메디안)을 표 7(치료 시작에서 임상적 질병 진행 까지의 시간) 및 8(랜덤화에서 임상적 질병 진행 까지의 시간)에 나타내었다.
전체 항-EpCAM 고용량군은 항-EpCAM 저용량군에 비교하여 볼 때 진행 시간 메디안(첫 주입에서부터의 시간으로 산출)이 43일에서 78일로 명백히 연장되었다(도 2 참조). 이러한 차이는 "생존" 커브 테스트시 통계적으로 유의하였다(p=0.0348; 로그 랭크 테스트) (도 3 참조). 유사하게 저 대 고 EpCAM 발현 환자 비교시 진행 시간 메디안(첫 주입에서부터의 시간으로 산출)에서 차이점이 관찰되었다(각각 42 에서 80일; p=0.0431; 로그 랭크 테스트). 고 EpCAM 발현환자를 고 용량의 항-EpCAM으로 처리시 가장 높은 진행 시간 메디안(첫 주입에서부터의 시간으로 산출)을 나타내었다(90 일; p=0.0238; 로그 랭크 테스트-기타 다른 모든 환자 들에 비교하여) (도 5 참조).
[표 7] 진행 시간 메디안(주입 시작에서 임상적 질병 진행이 나타나는 시점까지의 시간[일])-전체 분석 세트
| 저용량항 - EpCAM 1 | 고용량항 - EpCAM 1 | P- 값 2 | |
| 저 EpCAM ( N 3 = 13 / 14) | 41일 | 47일 | 0.1451 |
| 고 EpCAM ( N 3 = 19 / 21) | 49일 | 90일 | 0.1262 |
| 전체 ( N 3 = 32 / 35) | 43일 | 78일 | 0.0348 |
| 1 일수로 표시된 진행 시간 메디안 2 치료군 간 차이점에 대한 로그 랭크 테스트 3 N 저용량군 / 고용량군 |
|||
표 7의 데이터를 도 4에 도시하였다.
랜덤화날에서부터 산출된 진행시간도 유사한 결과가 나타났다. 다시 전체 항-EpCAM 고용량군은 항-EpCAM 저용량군에 비교하여볼때 진행 시간 메디안(첫 주입에서부터의 시간으로 산출)이 46일에서 79일로 명백히 연장되었다. 이러한 차이는 "생존" 커브 테스트시 통계적으로 유의하였다(p=0.0441; 로그 랭크 테스트). 이러한 처치간 차이는 고 EpCAM군에 더욱 현저하게 나타났는데, 그 진행시간 메디안은 저용량 및 고용량 EpCAM 그룹에서 각각 63일 및 91일이었으며, 저EpCAM군은 저용량 및 고용량 EpCAM 그룹에서 각각 43일 및 53일이었다.
[표 8] 진행 시간 메디안(랜덤화에서 임상적 질병 진행에까지의 시간[일])-전체 분석 세트
| 저용량 항- EpCAM 1 | 고용량 항- EpCAM 1 | P- 값 2 | |
| 저 EpCAM ( N 3 = 13 / 14) | 43일 | 53일 | 0.2353 |
| 고 EpCAM ( N 3 = 19 / 21) | 63일 | 91일 | 0.1350 |
| 전체 ( N 3 = 32 / 35) | 46일 | 79일 | 0.0441 |
효능 결론
연구 프로토콜에 기초하여
- 최선 전체 반응(OTR)
- 임상 이점 비율(CBR); 및
- 진행 시간(TTP)
에 대해 전체 집단내 고용량 항-EpCAM 군 및 저용량 항-EpCAM 군 및 저- 및 고- EpCAM 서브그룹을 비교 분석하였다.
12주 및 24주에서의 임상 이점 비율(각각 W12 및 W24)은 각 시간에서 안정 질환을 나타내는 모든 환자를 포함하여 수립될 수 있었다.
- W12에 CBR는 저 용량군에서보다 고 용량 항-EpCAM군에서 약간 더 높았으며(25.7% 대 21.9%), 또한 고 EpCAM 서브그룹에서는 양 용량군에서 높은 비율을 나타내었다.
- W24에 CBR은 고 용량 항-EpCAM 군에서 더 높았다(14.3% 대 6.3%(저용량 항-EpCAM군)). EpCAM 서브그룹간에는 어떠한 유의성 있는 차이도 나타나지 않았다.
모든 샘플에서 진행 시간 메디안은 저용량의 항-EpCAM군에 비해 고용량의 항-EpCAM군에서 명백한 연장이 관찰되었으며(43일에서 78일로), 생존 커브 테스트시 이 차이는 통계적으로 유의하였다(p = 0.0348, 로그 랭크 테스트). 진행 시간 메디안(첫 주입에서의 시간으로 산출)은 고 EpCAM 발현 환자를 고용량의 항-EpCAM 로 투여시 가장 높게 관찰되었다(90 일; p = 0.0238; 로그 랭크 테스트 - 기타 다른 환자들과 비교).
총 결론
수득 데이터는 몇몇 환자는 지금도 진행중이며 최소한 7명의 환자에서 장기 질병 안정화(> 24주)를 나타내었다.
도 2-5에서 명백하게 보인바와 같이, 진행 시간 평가는 현저한 통계적으로 유의한 "생존 시간"의 연장을 나타내었으며, 특히 고 용량 항-EpCAM 집단에서 그러하다. 특히 도 5에 보인바와 같이, 고용량 항-EpCAM을 처치받은 고 EpCAM 발현 환자들은 상당히 연장된 진행이 없는 생존을 보였다(90일 대 41-49(기타 다른 군)).
실시예
3: 항-
EpCAM
임상 제
II
상 최종 연구 보고서
실시예
3.1: 연구 서론 및 요약
임상 연구는 실시예 1에 전술한 대로 수행되었다. 이 연구의 결과를 실시예 3에 나타내었다.
방법:
랜덤화되며, 공개되며, 다기관의 병행 그룹의 임상 제 II상 연구. 본 연구는 EpCAM 테스트 양성의 두개의 상이한 용량에서 24주에 걸친 치료에 대한 항-EpCAM의 효능과 안전성을 평가하기 위해 디자인되었다. 중앙 랜덤화 과정은 스크리닝시 수행된 EpCAM 테스트 결과에 따라 계층화되었다. EpCAM 계층 중 하나에 등록함에 있어, 환자는 저용량 치료군 또는 고용량 치료군에 랜덤하게 할당되었다.
랜덤화된 치료 환자수 : 112 명
분석된 환자수 : 112 명의 처치 환자(56명; 항-EpCAM 고용량, 56명: 항-EpCAM 저용량), 이중 109명의 환자가 EpCAM+로 검사되었다.
전술한 분석을 위해 분석된 데이터:
결과를 위한 모든 데이터는 GCP-요건에 따라 모니터되고 크리닝되었으며 데이터베이스는 최종 분석이 수행되기 전에 로킹되었다.
분석 집단의 정의:
● 안전성 분석 세트(SAF): 최소한 한 용량의 할당된 연구 의약품을 투여받은 모든 환자.
● 전체 분석 세트(FAS): EpCAM 양성(저/중등도 또는 고 발현) 종양을 가진 안전성 분석 세트 중 환자이며 임상적 질병 진행 이외의 다른 이유 때문에 조기에 포기한 경우 치료 시작 후 한번 이상 종양 평가를 받은 환자.
안전성 데이터의 분석은 SAF를 기초로 시행되었다. 베이스라인 데이터 및 효능 종결점에 대한 분석은 전체 분석 세트를 기초로 시행되었다.
분석된 종결점(전술한
실시예
1.1 참조):
● 24주에 임상 이점 비율(CBR)(RECIST에 따라 안정 질환[SD] 또는 완전 반응[CR] 또는 부분 반응[PR]을 나타내는 환자)
● 12주의 최선 전체 종양 반응(OTR)비율(RECIST에 따라 완전 반응[CR] 또는 부분 반응[PR]을 나타내는 환자)
● 12주에 임상 이점 비율(CBR) (RECIST에 따라 안정 질환[SD] 또는 완전 반응[CR] 또는 부분 반응[PR]을 나타내는 환자)
● 랜덤화부터의 시간 또는 처치 시작부터의 시간으로서의 진행 시간(TTP).
분석 서브그룹:
고 및 저/중등도 EPCAM 발현자들은 Gastl 등. (2000). Lancet 356, 1981-2에 따라 확인되었다.
하기 서브그룹이 분석되었다:
● 저용량 항-EpCAM 처치된 EpCAM 저/중등도 발현자 그룹("저용량 항-EpCAM/저 EpCAM")
● 고용량 항-EpCAM 처치된 EpCAM 저/중등도 발현자 그룹("고용량 항-EpCAM/저 EpCAM")
● 저용량 항-EpCAM 처치된 EpCAM 고 발현자 그룹("저용량 항-EpCAM/고 EpCAM")
● 고용량 항-EpCAM 처치된 EpCAM 고 발현자 그룹("고용량 항-EpCAM/고EpCAM")
● 저용량 또는 고용량 항-EpCAM 처치된 EpCAM 저/중등도 발현자 그룹("저 EpCAM")
● 저용량 또는 고용량 항-EpCAM 처치된 EpCAM 고 발현자 그룹("고 EpCAM")
● EpCAM 저/중등도 또는 고 발현자에 저용량 항-EpCAM 처치 그룹("저용량 항-EpCAM")
● EpCAM 저/중등도 또는 고 발현자에 고용량 항-EpCAM 처치 그룹("고용량 항-EpCAM").
분석 수행:
정의된 바와 같은 반응(CBR 또는 OTR) 종결점은 하기와 같이 분석되었다:
● 각 서브그룹 및 모든 환자에 대한 CBR/OTR 비율.
임상적 질병 진행 시간은 처음 항-EpCAM을 주입한 날(민감성 분석의 경우:랜덤화한 날)과 임상 질병 진행일, 즉 진행성 질병이 처음 나타난 날과의 기간으로 정의되었다. 만약 임상 질병 진행이 관찰되지 않은 경우에는 각 시간 간격은 연구 종결일로 하였다. 연구 종결일이 확실치 않은 경우 마지막으로 방문한 날짜를 사용하였다.
진행 시간(TTP) 종결점은 하기와 같이 분석되었다:
● 각 서브그룹 및 모든 환자들에 대한 메디안 TTP(측정가능한 경우)
● 하기 비교를 위한 TTP에 대한 로그 랭크 테스트(Log-Rank test):
○ 각 서브그룹 대 기타 모든 다른 환자 조합
○ "저용량 항-EpCAM/저/중등도 EpCAM" 대 "저용량 항-EpCAM/고 EpCAM"
○ "저용량 항-EpCAM/저/중등도 EpCAM" 대 "고용량 항-EpCAM/저/중등도 EpCAM"
○ "고용량 항-EpCAM/고 EpCAM" 대 "저용량 항-EpCAM/고 EpCAM"
○ "고용량 항-EpCAM/고 EpCAM" 대 "고용량 항-EpCAM/저/중등도 EpCAM"
○ "저용량 항-EpCAM/저/중등도 EpCAM" 대 "고용량 항-EpCAM/고 EpCAM"
○ "저용량 항-EpCAM" 대 "고용량 항-EpCAM"
○ "고 EpCAM" 대 "저/중등도 EpCAM"
실시예
3.2: 결과 - 연구 환자들
분석된 데이터세트들
분석 집단을 표 9에 나타내었다.
전체 분석 세트(FAS) (n = 109)는 반응 및 진행 시간 분석에 대한 분석 집단을 나타낸 것이다.
[표 9] 분석 집단1
|
|
저용량
MT201
(N=55) |
고용량
MT201
(N=54) |
||||||
|
저/중등도
EpCAM (N=19) |
고
EpCAM (N=36) |
저/중등도
EpCAM (N=16) |
고
EpCAM (N=38) |
|||||
| n | % | n | % | n | % | n | % | |
| 인종 2 코카시안 | 19 | 100 | 36 | 100 | 16 | 100 | 38 | 100 |
| 나이 [ years ] 1 | n=19 60.3 ± 9.8 (42.0-79.0,61.0) |
n=36 59.2 ± 9.6 (42.0-75.0, 61.5) |
n=16 57.8 ±11.5 (40.0-80.0, 54.5) |
n=38 59.1 ± 11.5 (38.0-81.0, 60.5) |
||||
| 신장 [ cm ] 1 | n=19 160.8 ± 7.9 (149.0-177.0, 161.0) |
n=36 161.8 ± 6.6 (150.0-175.0, 162.5) |
n=16 160.4 ± 6.9 (150.0-170.0, 161.5) |
n=38 162.4 ± 7.5 (145.0-176.0, 162.5) |
||||
| 체중 [ kg ] 1 | n=19 67.1 ± 6.5 (57.0-75.0, 69.0) |
n=36 70.1 ± 13.2 (50.0-108.0, 68.5) |
n=16 68.1 ± 7.9 (53.5-84.0, 66.5) |
n=38 69.0 ± 15.3 (42.7-115.0, 65.0) |
||||
| BMI [ kg /m 2 ] 1 | n=19 26.1 ± 3.1 (20.4-31.6, 26.2) |
n=36 26.8 ± 4.8 (19.3-39.7, 26.7) |
n=16 26.7 ± 4.1 (18.7-33.8, 27.2) |
n=38 26.2 ± 5.9 (17.1-49.1, 25.4) |
||||
| 1 환자수, 평균 ± 표준편차(최소-최대, 메디안) 2 환자수, % |
||||||||
실시예
3.3: 결과-전체 종양 반응(
OTR
) 및 임상 이점 비율(
CBR
)에 대한 효능 분석
연구 프로토콜에 따라 본 연구의 제1 종결점은 24주에서의 임상 이점 비율이다.
"최선 전체 종양 반응(
OTR
)"
EpCAM 고 및 저 발현자 및 전부에 대한 FAS에서 "12주에서의 최선 전체 종양 반응(OTR)", "12주에서의 임상 이점 비율(CBR)", 및 "24주에서의 임상 이점 비율(CBR)"에 대한 결과를 하기 표 10 및 11에 나타내었다.
두 반응(RECIST 기준에 따라 PR 또는 CR로 나타낸)은 지역 방사선 평가에 의해 진단되었으며, FAS 내 어떠한 환자에서도 중앙 방사선 평가에서 확인될 수 없었 다.
"24주에서의 임상 이점 비율(
CBR
)"
그렇게 중요하지 않으나 고용량군(7.9%)은 저용량군(4.5%)에 비교하여 24주에 더 높은 임상 이점 비율(CBR)을 나타내는 경향을 나타내었다. 유사하게 EpCAM 고 발현자에 대한 CBR은 저/중등도 EpCAM 발현자에 비해 더 높은 CBR 비율을 나타내는 경향을 보였다(7.3% 대 3.7%).
CBR은 PR이나 CR이 검출되지 않은 안정 질환을 나타내는 환자들만을 기초로 하였다.
[표 11] 24주에서의 임상 이점 비율(CBR) (RESICST에 따라 안정 질환 또는 완전 반응[CR] 또는 부분 반응[PR]을 나타내는 환자)
|
저용량
MT201
(N=44) |
고용량
MT201
(N=38) |
|||||||
|
저/중등도
EpCAM (N=15) |
고
EpCAM (N=29) |
저/중등도
EpCAM (N=12) |
고
EpCAM (N=26) |
|||||
| n1 | %1 | n1 | %1 | n1 | %1 | n1 | %1 | |
| 임상 이점 2 ( CR , PR 및 SD) | 0 | 0.0 | 2 | 6.9 | 1 | 8.3 | 2 | 7.7 |
| 임상 이점 나타내지 않음2 (PD 및 수득불가) | 15 | 100.0 | 26 | 89.7 | 11 | 91.7 | 24 | 92.3 |
| 평가불가 2 ,3 | 0 | 0.0 | 1 | 3.4 | 0 | 0.0 | 0 | 0.0 |
| CBR 에대한 95% 신뢰 한계 이하 | 0.0% | 1.2% | 0.4% | 1.4% | ||||
| 1 환자수 (%) 2 "최종" 평가에 따름 3 연구내에 존재하나 중앙/IRAV 평가에서 각 반응 평가가 '평가불가'로 분류된 환자 |
||||||||
12주에서의 "임상 이점 비율(
CBR
)"
전체 FAS 내 109명의 환자 중 17명(16 %)이 12주에 질병 안정화(SD)를 나타내었다. 12주에서의 임상 이점 비율(CBR)이 저용량군(14.5%)에 비해 고용량군(16.7%)에서 더 높은 경향을 나타내었다. 유사하게 EpCAM 고발현군의 CBR이 저/중등도 EpCAM 발현자에 비해 더 높은 CBR 비율을 나타내는 경향을 보였다(18.9% 대 8.6%).
[표 10] 12주에서의 임상 이점 비율(CBR) (RESICST에 따라 안정 질환 또는 완전 반응[CR] 또는 부분 반응[PR]을 나타내는 환자) -전체 분석 세트
|
저용량MT201
(N=55) |
고용량
MT201
(N=54) |
|||||||
|
저/중등도
EpCAM (N=19) |
고
EpCAM (N=36) |
저/중등도
EpCAM (N=16) |
고
EpCAM (N=38) |
|||||
| 임상 이점 2 ( CR , PR 및 SD ) | n1 | %1 | n1 | %1 | n1 | %1 | n1 | %1 |
| 1 | 5.3 | 7 | 19.4 | 2 | 12.5 | 7 | 18.4 | |
| 임상 이점 나타내지 않음2 (PD 및 수득불가) | 18 | 94.7 | 28 | 77.8 | 14 | 87.5 | 28 | 73.7 |
| 평가불가 2 ,3 | 0 | 0.0 | 1 | 2.8 | 0 | 0.0 | 3 | 7.9 |
| 1 환자수 (%) 2 "최종" 평가에 따름 3 연구내에 존재하나 중앙/IRAV 평가에서 각 반응 평가가 '평가불가'로 분류된 환자 |
||||||||
효능 분석: 진행 시간
임상적 질병 진행 시간은 처음 항-EpCAM을 주입한 날(민감성 분석의 경우:랜덤화한 날)과 임상 질병 진행일, 즉 진행성 질병이 처음 나타난 날과의 기간으로 정의되었다. 만약 임상 질병 진행이 관찰되지 않은 경우에는 각 시간 간격은 연구 종결일로 하였다. 연구 종결일이 확실치 않은 경우 마지막으로 방문한 날짜를 사용 하였다.
진행시간에 대한 카플란-마이어(KM)커브 분석이 도 6-8에 도시되었다(처치시작에서 임상적 질병 진행까지의 시간)
전체 항-EpCAM 고용량군은 항-EpCAM 저용량군에 비교하여 볼 때 시간에 따른 진행 시간의 상당한 연장을 나타내었다(도 6 참조; 하자드 비율(Hazard ration; HR)=0.666). 이러한 차이는 "생존" 커브 테스트시 통계적으로 유의하였다(p=0.0465; 로그 랭크 테스트). 유사하게 저/중등도 대 고 EpCAM 발현 환자 비교시, 진행 시간에 있어 차이(첫 주입부터의 시간으로 산출)가 관찰되었다(도 7; HR=0.706; p=0.1157; 로그 랭크 테스트). 진행시간의 가장 높은 위험 경감은 고 용량의 항-EpCAM을 처치한 고 EpCAM 발현환자에서 관찰되었다(HR=0.433; p=0.0057; 로그 랭크 테스트-저용량의 항-EpCAM을 처치한 저 EpCAM 발현환자들과 비교) (도 8 참조).
효능 결론
연구 프로토콜에 기초하여
- 최선 전체 반응(OTR)
- 임상 이점 비율(CBR); 및
- 진행 시간(TTP)
에 대해 전체 집단내 고용량 항-EpCAM 군 및 저용량 항-EpCAM 군 및 저- 및 고- EpCAM 서브그룹을 비교 분석하였다.
12주 및 24주에서의 임상 이점 비율(각각 W12 및 W24)은 각 시간 포인트에서 안정 질환을 나타내는 모든 환자를 포함하여 수립될 수 있었다.
- W12에 CBR는 저 용량군보다 고 용량 항-EpCAM군에서 약간 더 높았으며(16.7% 대 14.5%), 또한 고 EpCAM 서브그룹에서는 양 용량군에서 높은 비율을 나타내었다(18.9% 대 8.6%(저/중등도 EpCAM 발현자)).
- W24에 CBR은 고용량 항-EpCAM 군에서 더 높았으며(7.9% 대 4.5%(저용량 항-EpCAM군)), 고 EpCAM 서브그룹에서는 양 용량군에서 더 높은 비율을 나타내었다(7.3% 대 3.7%(저/중등도 EpCAM 발현자)).
모든 샘플에서 진행 시간은 저용량 항-EpCAM군에 비해 고용량의 항-EpCAM군에서 연장이 관찰되었으며(R=0.666), 생존 커브 테스트시 이 차이는 통계적으로 유의하였다(p = 0.0465, 로그 랭크 테스트). 고용량의 항-EpCAM를 처치한 고 EpCAM 발현 환자에 있어 진행 시간이 가장 길게 연장되었다(HR=0.433; p = 0.0057; 로그 랭크 테스트- 저용량의 항-EpCAM을 처치한 저 EpCAM 발현환자들과 비교).
총 결론
수득 데이터는 중앙 방사선 리뷰에 따라 최소한 6명의 환자에서 장기 질병 안정화(> 24주)를 나타내었다.
도 6-8에서 명백하게 알 수 있는 바와 같이, 진행 시간 평가는 현저한 통계적으로 유의한 "생존 시간"의 연장을 나타내었으며, 특히 고 용량 항-EpCAM 집단에서 그러하다. 특히 도 8에 보인바와 같이, 고용량 항-EpCAM을 처치받은 고 EpCAM 발현 환자들은 상당히 연장된 진행이 없는 생존을 보였다.
SEQUENCE LISTING
<110> Micromet AG
<120> Treatment of metastatic breast cancer
<130> IP20081189
<140>
<141>
<150> EP 06 002 680.4
<151> 2006-02-09
<150> US 60/772,421
<151> 2006-02-09
<160> 8
<170> KopatentIn 1.71
<210> 1
<211> 457
<212> PRT
<213> artificial sequence
<220>
<223> Anti-EpCAM Heavy Chain
<400> 1
Glu Val Gln Leu Leu Glu Ser Gly Gly Gly Val Val Gln Pro Gly Arg
1 5 10 15
Ser Leu Arg Leu Ser Cys Ala Ala Ser Gly Phe Thr Phe Ser Ser Tyr
20 25 30
Gly Met His Trp Val Arg Gln Ala Pro Gly Lys Gly Leu Glu Trp Val
35 40 45
Ala Val Ile Ser Tyr Asp Gly Ser Asn Lys Tyr Tyr Ala Asp Ser Val
50 55 60
Lys Gly Arg Phe Thr Ile Ser Arg Asp Asn Ser Lys Asn Thr Leu Tyr
65 70 75 80
Leu Gln Met Asn Ser Leu Arg Ala Glu Asp Thr Ala Val Tyr Tyr Cys
85 90 95
Ala Lys Asp Met Gly Trp Gly Ser Gly Trp Arg Pro Tyr Tyr Tyr Tyr
100 105 110
Gly Met Asp Val Trp Gly Gln Gly Thr Thr Val Thr Val Ser Ser Ala
115 120 125
Ser Thr Lys Gly Pro Ser Val Phe Pro Leu Ala Pro Ser Ser Lys Ser
130 135 140
Thr Ser Gly Gly Thr Ala Ala Leu Gly Cys Leu Val Lys Asp Tyr Phe
145 150 155 160
Pro Glu Pro Val Thr Val Ser Trp Asn Ser Gly Ala Leu Thr Ser Gly
165 170 175
Val His Thr Phe Pro Ala Val Leu Gln Ser Ser Gly Leu Tyr Ser Leu
180 185 190
Ser Ser Val Val Thr Val Pro Ser Ser Ser Leu Gly Thr Gln Thr Tyr
195 200 205
Ile Cys Asn Val Asn His Lys Pro Ser Asn Thr Lys Val Asp Lys Lys
210 215 220
Val Glu Pro Lys Ser Cys Asp Lys Thr His Thr Cys Pro Pro Cys Pro
225 230 235 240
Ala Pro Glu Leu Leu Gly Gly Pro Ser Val Phe Leu Phe Pro Pro Lys
245 250 255
Pro Lys Asp Thr Leu Met Ile Ser Arg Thr Pro Glu Val Thr Cys Val
260 265 270
Val Val Asp Val Ser His Glu Asp Pro Glu Val Lys Phe Asn Trp Tyr
275 280 285
Val Asp Gly Val Glu Val His Asn Ala Lys Thr Lys Pro Arg Glu Glu
290 295 300
Gln Tyr Asn Ser Thr Tyr Arg Val Val Ser Val Leu Thr Val Leu His
305 310 315 320
Gln Asp Trp Leu Asn Gly Lys Glu Tyr Lys Cys Lys Val Ser Asn Lys
325 330 335
Ala Leu Pro Ala Pro Ile Glu Lys Thr Ile Ser Lys Ala Lys Gly Gln
340 345 350
Pro Arg Glu Pro Gln Val Tyr Thr Leu Pro Pro Ser Arg Asp Glu Leu
355 360 365
Thr Lys Asn Gln Val Ser Leu Thr Cys Leu Val Lys Gly Phe Tyr Pro
370 375 380
Ser Asp Ile Ala Val Glu Trp Glu Ser Asn Gly Gln Pro Glu Asn Asn
385 390 395 400
Tyr Lys Thr Thr Pro Pro Val Leu Asp Ser Asp Gly Ser Phe Phe Leu
405 410 415
Tyr Ser Lys Leu Thr Val Asp Lys Ser Arg Trp Gln Gln Gly Asn Val
420 425 430
Phe Ser Cys Ser Val Met His Glu Ala Leu His Asn His Tyr Thr Gln
435 440 445
Lys Ser Leu Ser Leu Ser Pro Gly Lys
450 455
<210> 2
<211> 214
<212> PRT
<213> artificial sequence
<220>
<223> Anti-EpCAM Light Chain
<400> 2
Glu Leu Gln Met Thr Gln Ser Pro Ser Ser Leu Ser Ala Ser Val Gly
1 5 10 15
Asp Arg Val Thr Ile Thr Cys Arg Thr Ser Gln Ser Ile Ser Ser Tyr
20 25 30
Leu Asn Trp Tyr Gln Gln Lys Pro Gly Gln Pro Pro Lys Leu Leu Ile
35 40 45
Tyr Trp Ala Ser Thr Arg Glu Ser Gly Val Pro Asp Arg Phe Ser Gly
50 55 60
Ser Gly Ser Gly Thr Asp Phe Thr Leu Thr Ile Ser Ser Leu Gln Pro
65 70 75 80
Glu Asp Ser Ala Thr Tyr Tyr Cys Gln Gln Ser Tyr Asp Ile Pro Tyr
85 90 95
Thr Phe Gly Gln Gly Thr Lys Leu Glu Ile Lys Arg Thr Val Ala Ala
100 105 110
Pro Ser Val Phe Ile Phe Pro Pro Ser Asp Glu Gln Leu Lys Ser Gly
115 120 125
Thr Ala Ser Val Val Cys Leu Leu Asn Asn Phe Tyr Pro Arg Glu Ala
130 135 140
Lys Val Gln Trp Lys Val Asp Asn Ala Leu Gln Ser Gly Asn Ser Gln
145 150 155 160
Glu Ser Val Thr Glu Gln Asp Ser Lys Asp Ser Thr Tyr Ser Leu Ser
165 170 175
Ser Thr Leu Thr Leu Ser Lys Ala Asp Tyr Glu Lys His Lys Val Tyr
180 185 190
Ala Cys Glu Val Thr His Gln Gly Leu Ser Ser Pro Val Thr Lys Ser
195 200 205
Phe Asn Arg Gly Glu Cys
210
<210> 3
<211> 5
<212> PRT
<213> artificial sequence
<220>
<223> H-CDR1
<400> 3
Ser Tyr Gly Met His
1 5
<210> 4
<211> 17
<212> PRT
<213> artificial sequence
<220>
<223> H-CDR2
<400> 4
Val Ile Ser Tyr Asp Gly Ser Asn Lys Tyr Tyr Ala Asp Ser Val Lys
1 5 10 15
Gly
<210> 5
<211> 18
<212> PRT
<213> artificial sequence
<220>
<223> H-CDR3
<400> 5
Asp Met Gly Trp Gly Ser Gly Trp Arg Pro Tyr Tyr Tyr Tyr Gly Met
1 5 10 15
Asp Val
<210> 6
<211> 11
<212> PRT
<213> artificial sequence
<220>
<223> L-CDR1
<400> 6
Arg Thr Ser Gln Ser Ile Ser Ser Tyr Leu Asn
1 5 10
<210> 7
<211> 7
<212> PRT
<213> artificial sequence
<220>
<223> L-CDR2
<400> 7
Trp Ala Ser Thr Arg Glu Ser
1 5
<210> 8
<211> 9
<212> PRT
<213> artificial sequence
<220>
<223> L-CDR3
<400> 8
Gln Gln Ser Tyr Asp Ile Pro Tyr Thr
1 5
Claims (20)
- 서열번호: 3, 4, 5, 6, 7 및/또는 8을 포함하는 인간 항-EpCAM 항체를 포함하는, 인간 전이성 유방암 치료용 약학 조성물.
- 제1항에 있어서,상기 항-EpCAM 항체는 서열번호: 1 및/또는 2를 포함하는 것을 특징으로 하는 약학 조성물.
- 제1항 또는 제2항에 있어서,상기 치료는 전이성 유방암의 장기 안정화를 포함하는 것을 특징으로 하는 약학 조성물.
- 제1항 또는 제2항에 있어서,상기 전이성 유방암은 암전이분류(Tumor Node Metastasis: TNM) 시스템에 따라 단계 IV로 분류되는 것을 특징으로 하는 약학 조성물.
- 제1항 또는 제2항에 있어서,상기 항-EpCAM 항체는 적어도 체중 1 kg 당 2 mg의 로딩 용량으로 투여되고, 이후 복수의 유지 용량이 투여되며, 이 때 각 유지 용량은 체중 1 kg 당 2 mg인 것을 특징으로 하는 약학 조성물.
- 제1항 또는 제2항에 있어서,상기 항-EpCAM 항체는 적어도 체중 1 kg 당 6 mg의 로딩 용량으로 투여되고, 이후 복수의 유지 용량이 투여되며, 이 때 각 유지 용량은 체중 1 kg 당 6 mg인 것을 특징으로 하는 약학 조성물.
- 제5항에 있어서,상기 각 로딩 용량은 매주 투여되고, 상기 각 유지 용량은 2주마다 한번씩 투여되는 것을 특징으로 하는 약학 조성물.
- 제6항에 있어서,상기 각 로딩 용량은 매주 투여되고, 상기 각 유지 용량은 2주마다 한번씩 투여되는 것을 특징으로 하는 약학 조성물.
- 제7항에 있어서,로딩 용량은 각 치료 1주, 2주 및 3주가 시작될 때 투여되고, 이후 11회의 유지 용량이 투여되며, 이 때 유지 용량은 각 치료 4주, 6주, 8주, 10주, 12주, 14주, 16주, 18주, 20주, 22주 및 24주가 시작될 때 투여되는 것을 특징으로 하는 약학 조성물.
- 제8항에 있어서,로딩 용량은 각 치료 1주, 2주 및 3주가 시작될 때 투여되고, 이후 11회의 유지 용량이 투여되며, 이 때 유지 용량은 각 치료 4주, 6주, 8주, 10주, 12주, 14주, 16주, 18주, 20주, 22주 및 24주가 시작될 때 투여되는 것을 특징으로 하는 약학 조성물.
- 제1항 또는 제2항에 있어서,상기 항체는 0.9% 소듐클로라이드 용액을 포함하는 용액으로 투여되는 것을 특징으로 하는 약학 조성물.
- 제1항 또는 제2항에 있어서,상기 항체는 정맥 투여되는 것을 특징으로 하는 약학 조성물.
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
- 삭제
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US77242106P | 2006-02-09 | 2006-02-09 | |
| EP06002680.4 | 2006-02-09 | ||
| US60/772,421 | 2006-02-09 | ||
| EP06002680 | 2006-02-09 | ||
| PCT/EP2007/001127 WO2007090670A1 (en) | 2006-02-09 | 2007-02-09 | Treatment of metastatic breast cancer |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20080091817A KR20080091817A (ko) | 2008-10-14 |
| KR101296264B1 true KR101296264B1 (ko) | 2013-08-14 |
Family
ID=38110762
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020087020271A KR101296264B1 (ko) | 2006-02-09 | 2007-02-09 | 전이성 유방암의 치료 |
Country Status (9)
| Country | Link |
|---|---|
| US (3) | US7976842B2 (ko) |
| EP (1) | EP1981539B1 (ko) |
| JP (2) | JP2009526770A (ko) |
| KR (1) | KR101296264B1 (ko) |
| AU (1) | AU2007213920B2 (ko) |
| CA (1) | CA2638040A1 (ko) |
| IL (1) | IL192944A0 (ko) |
| RU (1) | RU2434640C2 (ko) |
| WO (1) | WO2007090670A1 (ko) |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10254601A1 (de) | 2002-11-22 | 2004-06-03 | Ganymed Pharmaceuticals Ag | Differentiell in Tumoren exprimierte Genprodukte und deren Verwendung |
| US8921102B2 (en) | 2005-07-29 | 2014-12-30 | Gpb Scientific, Llc | Devices and methods for enrichment and alteration of circulating tumor cells and other particles |
| RU2434640C2 (ru) * | 2006-02-09 | 2011-11-27 | Микромет Аг | Лечение метастатического рака молочной железы |
| US20080050739A1 (en) | 2006-06-14 | 2008-02-28 | Roland Stoughton | Diagnosis of fetal abnormalities using polymorphisms including short tandem repeats |
| US8372584B2 (en) | 2006-06-14 | 2013-02-12 | The General Hospital Corporation | Rare cell analysis using sample splitting and DNA tags |
| US8137912B2 (en) | 2006-06-14 | 2012-03-20 | The General Hospital Corporation | Methods for the diagnosis of fetal abnormalities |
| US20080070792A1 (en) | 2006-06-14 | 2008-03-20 | Roland Stoughton | Use of highly parallel snp genotyping for fetal diagnosis |
| DE102006035617A1 (de) * | 2006-07-31 | 2008-02-21 | Siemens Ag | Automatische Bestimmung von Tumorlast |
| US9623021B2 (en) * | 2007-01-22 | 2017-04-18 | Gtx, Inc. | Nuclear receptor binding agents |
| US9604931B2 (en) | 2007-01-22 | 2017-03-28 | Gtx, Inc. | Nuclear receptor binding agents |
| MX2009007831A (es) * | 2007-01-22 | 2010-01-15 | Gtx Inc | Agentes de union de receptor nuclear. |
| JP5130999B2 (ja) * | 2008-03-31 | 2013-01-30 | 住友ベークライト株式会社 | 抗癌剤の有効性予測方法 |
| EP2334812B1 (en) | 2008-09-20 | 2016-12-21 | The Board of Trustees of The Leland Stanford Junior University | Noninvasive diagnosis of fetal aneuploidy by sequencing |
| WO2011091154A2 (en) * | 2010-01-21 | 2011-07-28 | The Regents Of The University Of California | Use of human erythrocytes for prevention and treatment of cancer dissemination and growth |
| BR112013014076A2 (pt) * | 2010-12-06 | 2016-11-22 | Cure Cancer Worldwide Corp | métodos de direcionamento metabólico de células de câncer usando quimio- e imunotherapia para tratamento de câncer |
| FR2977674B1 (fr) * | 2011-07-06 | 2015-08-14 | Cisbio Bioassays | Methode amelioree de detection et/ou de quantification d'un analyte present a la surface d'une cellule |
| JP2015527869A (ja) | 2011-08-26 | 2015-09-24 | メリマック ファーマシューティカルズ インコーポレーティッド | タンデムFc二重特異性抗体 |
| US20130179184A1 (en) * | 2012-01-06 | 2013-07-11 | Katherine L. Hurst | Individualized Dosing Technique With Multiple Variables |
| WO2013142390A1 (en) * | 2012-03-21 | 2013-09-26 | Gtx, Inc. | Aldo-keto reductase subfamily 1c3 (akr1c3) inhibitors |
| CN105073776B (zh) | 2012-11-13 | 2019-04-12 | 拜恩科技股份公司 | 用于治疗表达紧密连接蛋白的癌症疾病的制剂 |
| EA201591652A1 (ru) | 2013-03-06 | 2016-02-29 | Мерримак Фармасьютикалз, Инк. | ТАНДЕМНЫЕ БИСПЕЦИФИЧЕСКИЕ Fc-АНТИТЕЛА ПРОТИВ c-MET |
| WO2014146672A1 (en) | 2013-03-18 | 2014-09-25 | Ganymed Pharmaceuticals Ag | Therapy involving antibodies against claudin 18.2 for treatment of cancer |
| WO2015065505A1 (en) * | 2013-10-29 | 2015-05-07 | Duke University | Use of cyp27a1 inhibitors, statin, or lxr antagonists alone or in combination with conventional therapy for the treatment of breast cancer |
| US10975164B2 (en) | 2018-02-26 | 2021-04-13 | Emory University | Antibodies useful for detection of human carcinoma antigen |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005080428A2 (en) * | 2004-02-13 | 2005-09-01 | Micromet Ag | Anti-epcam immunoglobulins |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050009097A1 (en) | 2003-03-31 | 2005-01-13 | Better Marc D. | Human engineered antibodies to Ep-CAM |
| RU2434640C2 (ru) * | 2006-02-09 | 2011-11-27 | Микромет Аг | Лечение метастатического рака молочной железы |
| HUE040467T2 (hu) * | 2007-04-03 | 2019-03-28 | Amgen Res Munich Gmbh | Keresztfaj-specifikus kötõdomén |
-
2007
- 2007-02-09 RU RU2008130963/15A patent/RU2434640C2/ru not_active IP Right Cessation
- 2007-02-09 US US12/162,102 patent/US7976842B2/en not_active Expired - Fee Related
- 2007-02-09 CA CA002638040A patent/CA2638040A1/en not_active Abandoned
- 2007-02-09 WO PCT/EP2007/001127 patent/WO2007090670A1/en active Application Filing
- 2007-02-09 KR KR1020087020271A patent/KR101296264B1/ko not_active IP Right Cessation
- 2007-02-09 EP EP07703377.7A patent/EP1981539B1/en active Active
- 2007-02-09 JP JP2008553685A patent/JP2009526770A/ja active Pending
- 2007-02-09 AU AU2007213920A patent/AU2007213920B2/en not_active Ceased
-
2008
- 2008-07-22 IL IL192944A patent/IL192944A0/en unknown
-
2011
- 2011-07-11 US US13/180,093 patent/US8337843B2/en not_active Expired - Fee Related
-
2012
- 2012-12-13 JP JP2012272352A patent/JP2013067645A/ja active Pending
- 2012-12-19 US US13/719,866 patent/US8658172B2/en not_active Expired - Fee Related
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005080428A2 (en) * | 2004-02-13 | 2005-09-01 | Micromet Ag | Anti-epcam immunoglobulins |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2638040A1 (en) | 2007-08-16 |
| EP1981539A1 (en) | 2008-10-22 |
| WO2007090670A1 (en) | 2007-08-16 |
| RU2008130963A (ru) | 2010-03-20 |
| US20130177578A1 (en) | 2013-07-11 |
| US8658172B2 (en) | 2014-02-25 |
| IL192944A0 (en) | 2009-02-11 |
| RU2434640C2 (ru) | 2011-11-27 |
| EP1981539B1 (en) | 2014-07-23 |
| US20120009204A1 (en) | 2012-01-12 |
| JP2013067645A (ja) | 2013-04-18 |
| US8337843B2 (en) | 2012-12-25 |
| US20090304716A1 (en) | 2009-12-10 |
| US7976842B2 (en) | 2011-07-12 |
| AU2007213920B2 (en) | 2013-08-29 |
| AU2007213920A1 (en) | 2007-08-16 |
| JP2009526770A (ja) | 2009-07-23 |
| KR20080091817A (ko) | 2008-10-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101296264B1 (ko) | 전이성 유방암의 치료 | |
| US20230277663A1 (en) | Uses for and article of manufacture including her2 dimerization inhibitor pertuzumab | |
| JP6695003B2 (ja) | 癌のための併用療法 | |
| JP6914336B2 (ja) | 進行したher2発現がんの治療 | |
| US20180134803A1 (en) | Treatment of her2-positive breast cancer | |
| JP2020514281A5 (ko) | ||
| CN101405029A (zh) | 转移性乳腺癌的治疗 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A201 | Request for examination | ||
| E701 | Decision to grant or registration of patent right | ||
| GRNT | Written decision to grant | ||
| LAPS | Lapse due to unpaid annual fee |