JP7437395B2 - 細胞傷害性ベンゾジアゼピン誘導体の調製方法 - Google Patents
細胞傷害性ベンゾジアゼピン誘導体の調製方法 Download PDFInfo
- Publication number
- JP7437395B2 JP7437395B2 JP2021524303A JP2021524303A JP7437395B2 JP 7437395 B2 JP7437395 B2 JP 7437395B2 JP 2021524303 A JP2021524303 A JP 2021524303A JP 2021524303 A JP2021524303 A JP 2021524303A JP 7437395 B2 JP7437395 B2 JP 7437395B2
- Authority
- JP
- Japan
- Prior art keywords
- formula
- compound
- iia
- reaction
- reacting
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000000034 method Methods 0.000 title claims description 142
- 229940053197 benzodiazepine derivative antiepileptics Drugs 0.000 title description 2
- 231100000433 cytotoxic Toxicity 0.000 title description 2
- 230000001472 cytotoxic effect Effects 0.000 title description 2
- 125000003310 benzodiazepinyl group Chemical class N1N=C(C=CC2=C1C=CC=C2)* 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims description 746
- 238000006243 chemical reaction Methods 0.000 claims description 151
- MGNCLNQXLYJVJD-UHFFFAOYSA-N cyanuric chloride Chemical compound ClC1=NC(Cl)=NC(Cl)=N1 MGNCLNQXLYJVJD-UHFFFAOYSA-N 0.000 claims description 129
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 claims description 78
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 claims description 34
- 239000003795 chemical substances by application Substances 0.000 claims description 29
- 150000002466 imines Chemical class 0.000 claims description 28
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 claims description 22
- 239000003638 chemical reducing agent Substances 0.000 claims description 21
- 150000003459 sulfonic acid esters Chemical class 0.000 claims description 21
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 claims description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 17
- 229910000027 potassium carbonate Inorganic materials 0.000 claims description 17
- 239000012190 activator Substances 0.000 claims description 13
- 125000005604 azodicarboxylate group Chemical group 0.000 claims description 13
- XQPRBTXUXXVTKB-UHFFFAOYSA-M caesium iodide Chemical compound [I-].[Cs+] XQPRBTXUXXVTKB-UHFFFAOYSA-M 0.000 claims description 9
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical group CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 claims description 8
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 claims description 7
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical group CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 claims description 7
- QKSQWQOAUQFORH-UHFFFAOYSA-N tert-butyl n-[(2-methylpropan-2-yl)oxycarbonylimino]carbamate Chemical compound CC(C)(C)OC(=O)N=NC(=O)OC(C)(C)C QKSQWQOAUQFORH-UHFFFAOYSA-N 0.000 claims description 7
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical group [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 claims description 6
- 230000008569 process Effects 0.000 claims description 5
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 claims description 3
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 3
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 3
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 claims description 3
- 229910000105 potassium hydride Inorganic materials 0.000 claims description 3
- 229910000029 sodium carbonate Inorganic materials 0.000 claims description 3
- 239000012312 sodium hydride Substances 0.000 claims description 3
- 229910000104 sodium hydride Inorganic materials 0.000 claims description 3
- OQJBFFCUFALWQL-UHFFFAOYSA-N n-(piperidine-1-carbonylimino)piperidine-1-carboxamide Chemical compound C1CCCCN1C(=O)N=NC(=O)N1CCCCC1 OQJBFFCUFALWQL-UHFFFAOYSA-N 0.000 claims description 2
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical group CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 438
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 82
- 239000000243 solution Substances 0.000 description 63
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 51
- 239000002798 polar solvent Substances 0.000 description 43
- 239000002904 solvent Substances 0.000 description 42
- 239000000725 suspension Substances 0.000 description 36
- 239000000203 mixture Substances 0.000 description 28
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 21
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 20
- 239000000047 product Substances 0.000 description 20
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 16
- 239000011541 reaction mixture Substances 0.000 description 16
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 14
- 230000035484 reaction time Effects 0.000 description 14
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical group OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 239000003153 chemical reaction reagent Substances 0.000 description 12
- -1 sodium triacetoxyborohydride Chemical compound 0.000 description 12
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 11
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 10
- 239000012074 organic phase Substances 0.000 description 10
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 10
- ZFSLODLOARCGLH-UHFFFAOYSA-N isocyanuric acid Chemical compound OC1=NC(O)=NC(O)=N1 ZFSLODLOARCGLH-UHFFFAOYSA-N 0.000 description 9
- FEJUGLKDZJDVFY-UHFFFAOYSA-N 9-borabicyclo(3.3.1)nonane Chemical compound C1CCC2CCCC1B2 FEJUGLKDZJDVFY-UHFFFAOYSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 8
- 238000004128 high performance liquid chromatography Methods 0.000 description 8
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 7
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 7
- 229910052757 nitrogen Inorganic materials 0.000 description 7
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 238000010898 silica gel chromatography Methods 0.000 description 6
- UHPMCIAYHJHCON-UHFFFAOYSA-N COCCOCCOCCN(CC(C)(C)SSC)C1=CC(CCl)=CC(CO)=C1 Chemical compound COCCOCCOCCN(CC(C)(C)SSC)C1=CC(CCl)=CC(CO)=C1 UHPMCIAYHJHCON-UHFFFAOYSA-N 0.000 description 5
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 5
- 239000008367 deionised water Substances 0.000 description 5
- 229910021641 deionized water Inorganic materials 0.000 description 5
- IZDROVVXIHRYMH-UHFFFAOYSA-N methanesulfonic anhydride Chemical compound CS(=O)(=O)OS(C)(=O)=O IZDROVVXIHRYMH-UHFFFAOYSA-N 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 4
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 4
- 229940126214 compound 3 Drugs 0.000 description 4
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- UIFGGABIJBWRMG-UHFFFAOYSA-N (4-chlorophenyl)methyl n-[(4-chlorophenyl)methoxycarbonylimino]carbamate Chemical compound C1=CC(Cl)=CC=C1COC(=O)N=NC(=O)OCC1=CC=C(Cl)C=C1 UIFGGABIJBWRMG-UHFFFAOYSA-N 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 229940125898 compound 5 Drugs 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- JBXRLVPILRXPNH-UHFFFAOYSA-N indol-6-one Chemical compound O=C1C=CC2=CC=NC2=C1 JBXRLVPILRXPNH-UHFFFAOYSA-N 0.000 description 3
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- OBYVSQNTHFQXAY-NSHDSACASA-N (12as)-9-hydroxy-8-methoxy-11,12,12a,13-tetrahydroindolo[2,1-c][1,4]benzodiazepin-6-one Chemical compound N1C[C@@H]2CC3=CC=CC=C3N2C(=O)C2=C1C=C(O)C(OC)=C2 OBYVSQNTHFQXAY-NSHDSACASA-N 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- OZMLUMPWPFZWTP-UHFFFAOYSA-N 2-(tributyl-$l^{5}-phosphanylidene)acetonitrile Chemical compound CCCCP(CCCC)(CCCC)=CC#N OZMLUMPWPFZWTP-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- 239000012448 Lithium borohydride Substances 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- JNEIAVFYEBYNTL-UHFFFAOYSA-N [3-(hydroxymethyl)-5-[2-[2-(2-methoxyethoxy)ethoxy]ethyl-[2-methyl-2-(methyldisulfanyl)propyl]amino]phenyl]methanol Chemical compound COCCOCCOCCN(CC(C)(C)SSC)C1=CC(CO)=CC(CO)=C1 JNEIAVFYEBYNTL-UHFFFAOYSA-N 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 229910000085 borane Inorganic materials 0.000 description 2
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 2
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 2
- 239000006227 byproduct Substances 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- VHILMKFSCRWWIJ-UHFFFAOYSA-N dimethyl acetylenedicarboxylate Chemical compound COC(=O)C#CC(=O)OC VHILMKFSCRWWIJ-UHFFFAOYSA-N 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 239000012280 lithium aluminium hydride Substances 0.000 description 2
- 229940098779 methanesulfonic acid Drugs 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 230000000269 nucleophilic effect Effects 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- 239000003880 polar aprotic solvent Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000009257 reactivity Effects 0.000 description 2
- 239000012279 sodium borohydride Substances 0.000 description 2
- 229910000033 sodium borohydride Inorganic materials 0.000 description 2
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- YWWDBCBWQNCYNR-UHFFFAOYSA-N trimethylphosphine Chemical group CP(C)C YWWDBCBWQNCYNR-UHFFFAOYSA-N 0.000 description 2
- COIOYMYWGDAQPM-UHFFFAOYSA-N tris(2-methylphenyl)phosphane Chemical compound CC1=CC=CC=C1P(C=1C(=CC=CC=1)C)C1=CC=CC=C1C COIOYMYWGDAQPM-UHFFFAOYSA-N 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- ROVBYDCETXMZLN-NSHDSACASA-N (12as)-9-hydroxy-8-methoxy-12a,13-dihydroindolo[2,1-c][1,4]benzodiazepin-6-one Chemical compound N1=C[C@@H]2CC3=CC=CC=C3N2C(=O)C2=C1C=C(O)C(OC)=C2 ROVBYDCETXMZLN-NSHDSACASA-N 0.000 description 1
- VLSDXINSOMDCBK-BQYQJAHWSA-N (E)-1,1'-azobis(N,N-dimethylformamide) Chemical compound CN(C)C(=O)\N=N\C(=O)N(C)C VLSDXINSOMDCBK-BQYQJAHWSA-N 0.000 description 1
- USVVENVKYJZFMW-ONEGZZNKSA-N (e)-carboxyiminocarbamic acid Chemical compound OC(=O)\N=N\C(O)=O USVVENVKYJZFMW-ONEGZZNKSA-N 0.000 description 1
- YAXWOADCWUUUNX-UHFFFAOYSA-N 1,2,2,3-tetramethylpiperidine Chemical compound CC1CCCN(C)C1(C)C YAXWOADCWUUUNX-UHFFFAOYSA-N 0.000 description 1
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 1
- LIEOEYTUTSDYKB-BUHFOSPRSA-N 2,2,2-trichloroethyl (ne)-n-(2,2,2-trichloroethoxycarbonylimino)carbamate Chemical compound ClC(Cl)(Cl)COC(=O)\N=N\C(=O)OCC(Cl)(Cl)Cl LIEOEYTUTSDYKB-BUHFOSPRSA-N 0.000 description 1
- RKMGAJGJIURJSJ-UHFFFAOYSA-N 2,2,6,6-Tetramethylpiperidine Substances CC1(C)CCCC(C)(C)N1 RKMGAJGJIURJSJ-UHFFFAOYSA-N 0.000 description 1
- KJFJIEHJCVSAKJ-UHFFFAOYSA-N 2-(trimethyl-$l^{5}-phosphanylidene)acetonitrile Chemical compound CP(C)(C)=CC#N KJFJIEHJCVSAKJ-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- PGHKJMVOHWKSLJ-UHFFFAOYSA-N 2-methoxyethyl n-(2-methoxyethoxycarbonylimino)carbamate Chemical compound COCCOC(=O)N=NC(=O)OCCOC PGHKJMVOHWKSLJ-UHFFFAOYSA-N 0.000 description 1
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 1
- NKMKDPVCESDJCN-UHFFFAOYSA-N 3-[di(propan-2-yl)carbamoylimino]-1,1-di(propan-2-yl)urea Chemical compound CC(C)N(C(C)C)C(=O)N=NC(=O)N(C(C)C)C(C)C NKMKDPVCESDJCN-UHFFFAOYSA-N 0.000 description 1
- DYPPJKHPJMGHKW-QJGAVIKSSA-N 4,4,5,5,6,6,7,7,8,8,9,9,9-tridecafluorononyl (ne)-n-(4,4,5,5,6,6,7,7,8,8,9,9,9-tridecafluorononoxycarbonylimino)carbamate Chemical compound FC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)CCCOC(=O)\N=N\C(=O)OCCCC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F DYPPJKHPJMGHKW-QJGAVIKSSA-N 0.000 description 1
- WDBQJSCPCGTAFG-QHCPKHFHSA-N 4,4-difluoro-N-[(1S)-3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-pyridin-3-ylpropyl]cyclohexane-1-carboxamide Chemical compound FC1(CCC(CC1)C(=O)N[C@@H](CCN1CCC(CC1)N1C(=NN=C1C)C(C)C)C=1C=NC=CC=1)F WDBQJSCPCGTAFG-QHCPKHFHSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-M 4-bromobenzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-M 0.000 description 1
- SBJJKEMOEDKEMT-KBPBESRZSA-N COC(=O)CCCCC(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)Nc1cc(CO)cc(CO)c1 Chemical compound COC(=O)CCCCC(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)Nc1cc(CO)cc(CO)c1 SBJJKEMOEDKEMT-KBPBESRZSA-N 0.000 description 1
- ACGVZAPYSDAYGF-PMERELPUSA-N COCCOCCOCCN(CC(C)(C)SSC)c1cc(CCl)cc(COc2cc3NC[C@@H]4Cc5ccccc5N4C(=O)c3cc2OC)c1 Chemical compound COCCOCCOCCN(CC(C)(C)SSC)c1cc(CCl)cc(COc2cc3NC[C@@H]4Cc5ccccc5N4C(=O)c3cc2OC)c1 ACGVZAPYSDAYGF-PMERELPUSA-N 0.000 description 1
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- WGVWLKXZBUVUAM-UHFFFAOYSA-N Pentanochlor Chemical compound CCCC(C)C(=O)NC1=CC=C(C)C(Cl)=C1 WGVWLKXZBUVUAM-UHFFFAOYSA-N 0.000 description 1
- 241000350481 Pterogyne nitens Species 0.000 description 1
- 101100226004 Rattus norvegicus Erc2 gene Proteins 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- UPXCRVHPPXSSDG-UHFFFAOYSA-N [4-(3,3,4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-heptadecafluorodecyl)phenyl]-diphenylphosphane Chemical compound C1=CC(CCC(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)C(F)(F)F)=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UPXCRVHPPXSSDG-UHFFFAOYSA-N 0.000 description 1
- JEDZLBFUGJTJGQ-UHFFFAOYSA-N [Na].COCCO[AlH]OCCOC Chemical compound [Na].COCCO[AlH]OCCOC JEDZLBFUGJTJGQ-UHFFFAOYSA-N 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical compound [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical group 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229940049706 benzodiazepine Drugs 0.000 description 1
- 150000001557 benzodiazepines Chemical class 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- ZDKRMIJRCHPKLW-UHFFFAOYSA-N benzyl methanesulfonate Chemical compound CS(=O)(=O)OCC1=CC=CC=C1 ZDKRMIJRCHPKLW-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- HHPKBHOIUWPRIE-UHFFFAOYSA-N borane 1,4-oxathiane Chemical compound B.C1CSCCO1 HHPKBHOIUWPRIE-UHFFFAOYSA-N 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000013058 crude material Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- XUPMSLUFFIXCDA-UHFFFAOYSA-N dipyridin-4-yldiazene Chemical compound C1=NC=CC(N=NC=2C=CN=CC=2)=C1 XUPMSLUFFIXCDA-UHFFFAOYSA-N 0.000 description 1
- 125000002228 disulfide group Chemical group 0.000 description 1
- 238000001962 electrophoresis Methods 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 239000012456 homogeneous solution Substances 0.000 description 1
- 150000004678 hydrides Chemical group 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000006317 isomerization reaction Methods 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 231100000682 maximum tolerated dose Toxicity 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- QARBMVPHQWIHKH-KHWXYDKHSA-N methanesulfonyl chloride Chemical group C[35S](Cl)(=O)=O QARBMVPHQWIHKH-KHWXYDKHSA-N 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- XWGSCRBSSWZLBD-UHFFFAOYSA-N n-[tert-butyl(dimethyl)silyl]oxy-4-methylbenzenesulfonamide Chemical compound CC1=CC=C(S(=O)(=O)NO[Si](C)(C)C(C)(C)C)C=C1 XWGSCRBSSWZLBD-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 239000012419 sodium bis(2-methoxyethoxy)aluminum hydride Substances 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- LDSMXGPRCFQXSK-UHFFFAOYSA-N tripyridin-2-ylphosphane Chemical compound N1=CC=CC=C1P(C=1N=CC=CC=1)C1=CC=CC=N1 LDSMXGPRCFQXSK-UHFFFAOYSA-N 0.000 description 1
- HAEUOUZZVSHGKX-UHFFFAOYSA-N tripyridin-3-ylphosphane Chemical compound C1=CN=CC(P(C=2C=NC=CC=2)C=2C=NC=CC=2)=C1 HAEUOUZZVSHGKX-UHFFFAOYSA-N 0.000 description 1
- RIBNXKYQMSBWFD-UHFFFAOYSA-N tripyridin-4-ylphosphane Chemical compound C1=NC=CC(P(C=2C=CN=CC=2)C=2C=CN=CC=2)=C1 RIBNXKYQMSBWFD-UHFFFAOYSA-N 0.000 description 1
- LFNXCUNDYSYVJY-UHFFFAOYSA-N tris(3-methylphenyl)phosphane Chemical compound CC1=CC=CC(P(C=2C=C(C)C=CC=2)C=2C=C(C)C=CC=2)=C1 LFNXCUNDYSYVJY-UHFFFAOYSA-N 0.000 description 1
- WXAZIUYTQHYBFW-UHFFFAOYSA-N tris(4-methylphenyl)phosphane Chemical compound C1=CC(C)=CC=C1P(C=1C=CC(C)=CC=1)C1=CC=C(C)C=C1 WXAZIUYTQHYBFW-UHFFFAOYSA-N 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
Images





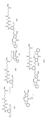







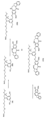








Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/12—Preparation of carboxylic acid amides by reactions not involving the formation of carboxamide groups
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/02—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
- C07C233/04—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with carbon atoms of carboxamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C233/07—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with carbon atoms of carboxamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/22—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton having nitrogen atoms of amino groups bound to the carbon skeleton of the acid part, further acylated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/14—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of sulfides
- C07C319/20—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of sulfides by reactions not involving the formation of sulfide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C319/00—Preparation of thiols, sulfides, hydropolysulfides or polysulfides
- C07C319/22—Preparation of thiols, sulfides, hydropolysulfides or polysulfides of hydropolysulfides or polysulfides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C321/00—Thiols, sulfides, hydropolysulfides or polysulfides
- C07C321/12—Sulfides, hydropolysulfides, or polysulfides having thio groups bound to acyclic carbon atoms
- C07C321/20—Sulfides, hydropolysulfides, or polysulfides having thio groups bound to acyclic carbon atoms of an unsaturated carbon skeleton containing rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C323/00—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups
- C07C323/23—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton
- C07C323/24—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton
- C07C323/25—Thiols, sulfides, hydropolysulfides or polysulfides substituted by halogen, oxygen or nitrogen atoms, or by sulfur atoms not being part of thio groups containing thio groups and nitrogen atoms, not being part of nitro or nitroso groups, bound to the same carbon skeleton having the sulfur atoms of the thio groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Description
本出願は、米国特許法第119条(e)に基づき、2018年11月12日に出願された米国仮特許出願第62/758,814号の出願日の利益を主張するものである。上述の出願の全内容は、参照により本明細書に組み込まれる。



1)式(IA)の化合物:


2)式(IIA)の化合物を式(a)の化合物:


3)式(IIIA)の化合物を式(b)の化合物:


1)式(IA)の化合物:


2)式(IIA)の化合物をスルホン化剤と反応させて、式(VIA)の化合物:

3)式(VIA)の化合物を式(a)の化合物:


4)式(IIIA)の化合物を式(b)の化合物:




1)式(IB)の化合物:


2)式(IIB)の化合物を式(a)の化合物:


3)式(IIIB)の化合物を式(b)の化合物:


1)式(IB)の化合物:


2)式(IIB)の化合物をスルホン化剤と反応させて、式(VIB)の化合物:

3)式(VIB)の化合物を式(a)の化合物:


4)式(IIIB)の化合物を式(b)の化合物:

本明細書で使用される場合、「化合物」という用語は、本発明において構造または式またはその任意の誘導体が開示されている化合物、または参照により組み込まれている構造または式またはその任意の誘導体を含むことが意図される。この用語はまた、立体異性体、幾何異性体、または互変異性体を含む。本出願に記載の本発明のある特定の態様における「立体異性体」、「幾何異性体」、「互変異性体」の特異的な記述は、「化合物」という用語がこれらの他の形態を記述することなく使用される本発明の他の態様におけるこれらの形態の意図的な省略と解釈されてはならない。

本発明は、ベンゾジアゼピン二量体化合物の合成前駆体を作製するための選択的一塩素化のための方法を提供する。驚くべきことに、出発ジオール化合物と比較してある特定のモル当量のシアヌル酸塩化物を使用することにより、より高い収率で一塩素化生成物をもたらすことが発見される。













1)式(IA’)の化合物:


2)式(IIA’)の化合物を式(a)の化合物:


3)式(IIIA’)の化合物を式(b)の化合物:


1)式(IA)の化合物:


2)式(IIA)の化合物を式(a)の化合物:


3)式(IIIA)の化合物を式(b)の化合物:


1)式(IA’)の化合物:


2)式(IIA’)の化合物を式(b)の化合物:


3)式(VA’)の化合物を式(a)の化合物:


1)式(IA)の化合物:


2)式(IIA)の化合物を式(b)の化合物:


3)式(VA)の化合物を式(a)の化合物:


1)式(IA’)の化合物:


2)式(IIA’)の化合物を式(b)の化合物:


3)式(VA’)の化合物をイミン還元剤と反応させて、式(IIIA’)の化合物:

4)式(IIIA’)の化合物を式(b)の化合物:


1)式(IA)の化合物:


2)式(IIA)の化合物を式(b)の化合物:


3)式(VA)の化合物をイミン還元剤と反応させて、式(IIIA)の化合物:

4)式(IIIA)の化合物を式(b)の化合物:


1)式(IA’)の化合物:


2)式(IIA’)の化合物をスルホン化剤と反応させて、式(VIA’)の化合物:

3)式(VIA’)の化合物を式(b)の化合物:


4)式(VA’)の化合物を式(a)の化合物:

式中、X1はスルホン酸エステルである。

1)式(IA)の化合物:


2)式(IIA)の化合物をスルホン化剤と反応させて、式(VIA)の化合物:

3)式(VIA)の化合物を式(b)の化合物:


4)式(VA)の化合物を式(a)の化合物:

式中、X1はスルホン酸エステルである。

1)式(IA’)の化合物:


2)式(IIA’)の化合物をスルホン化剤と反応させて、式(VIA’)の化合物:

3)式(VIA’)の化合物を式(a)の化合物:


4)式(IIIA’)の化合物を式(b)の化合物:

式中、X1はスルホン酸エステルである。

1)式(IA)の化合物:


2)式(IIA)の化合物をスルホン化剤と反応させて、式(VIA)の化合物:

3)式(VIA)の化合物を式(a)の化合物:


4)式(IIIA)の化合物を式(b)の化合物:

式中、X1はスルホン酸エステルである。

1)式(IA’)の化合物:


2)式(IIA’)の化合物をスルホン化剤と反応させて、式(VIA’)の化合物:

3)式(VIA’)の化合物を式(b)の化合物:


4)式(VA’)の化合物をイミン還元剤と反応させて、式(IIIA’)の化合物:

5)式(IIIA’)の化合物を式(b)の化合物:

式中、X1はスルホン酸エステルである。

1)式(IA)の化合物:


2)式(IIA)の化合物をスルホン化剤と反応させて、式(VIA)の化合物:

3)式(VIA)の化合物を式(b)の化合物:


4)式(VA)の化合物をイミン還元剤と反応させて、式(IIIA)の化合物:

5)式(IIIA)の化合物を式(b)の化合物:

式中、X1はスルホン酸エステルである。

1)式(IA’)の化合物:


2)式(IIA’)の化合物を式(b)の化合物:

3)式(VIIA’)の化合物をスルホン化剤と反応させて、式(VIIIA’)の化合物:

4)式(VIIIA’)の化合物を式(a)の化合物:

式中、X1はスルホン酸エステルである。

1)式(IA)の化合物:


2)式(IIA)の化合物を式(b)の化合物:


3)式(VIIA)の化合物をスルホン化剤と反応させて、式(VIIIA)の化合物:

4)式(VIIIA)の化合物を式(a)の化合物:

式中、X1はスルホン酸エステルである。

1)式(IA’)の化合物:


2)式(IIA’)の化合物を式(a)の化合物:


3)式(IXA’)の化合物をスルホン化剤と反応させて、式(XA’)の化合物:

4)式(XA’)の化合物を式(b)の化合物:

式中、X1はスルホン酸エステルである。

1)式(IA)の化合物:


2)式(IIA)の化合物を式(a)の化合物:


3)式(IXA)の化合物をスルホン化剤と反応させて、式(XA)の化合物:

4)式(XA)の化合物を式(b)の化合物:

式中、X1はスルホン酸エステルである。

1)式(IA’)の化合物:


2)式(IIA’)の化合物を式(b)の化合物:


3)式(VIIA’)の化合物をイミン還元剤と反応させて、式(IXA’)の化合物:

4)式(IXA’)の化合物をスルホン化剤と反応させて、式(XA’)の化合物:

5)式(XA’)の化合物を式(b)の化合物:

式中、X1はスルホン酸エステルである。

1)式(IA)の化合物:


2)式(IIA)の化合物を式(b)の化合物:


3)式(VIIA)の化合物をイミン還元剤と反応させて、式(IXA)の化合物:

4)式(IXA)の化合物をスルホン化剤と反応させて、式(XA)の化合物:

5)式(XA)の化合物を式(b)の化合物:

式中、X1はスルホン酸エステルである。







1)式(IB)の化合物:


2)式(IIB)の化合物を式(a)の化合物:


3)式(IIIB)の化合物を式(b)の化合物:


1)式(IB)の化合物:


2)式(IIB)の化合物を式(b)の化合物:


3)式(VB)の化合物を式(a)の化合物:


1)式(IB)の化合物:


2)式(IIB)の化合物を式(b)の化合物:


3)式(VB)の化合物をイミン還元剤と反応させて、式(IIIB)の化合物:

4)式(IIIB)の化合物を式(b)の化合物:

1)式(IB)の化合物:


2)式(IIB)の化合物をスルホン化剤と反応させて、式(VIB)の化合物:

3)式(VIB)の化合物を式(b)の化合物:


4)式(VB)の化合物を式(a)の化合物:

式中、X1はスルホン酸エステルである。
1)式(IB)の化合物:


2)式(IIB)の化合物をスルホン化剤と反応させて、式(VIB)の化合物:

3)式(VIB)の化合物を式(a)の化合物:


4)式(IIIB)の化合物を式(b)の化合物:

式中、X1はスルホン酸エステルである。
1)式(IB)の化合物:


2)式(IIB)の化合物をスルホン化剤と反応させて、式(VIB)の化合物:

3)式(VIB)の化合物を式(b)の化合物:


4)式(VB)の化合物をイミン還元剤と反応させて、
式(IIIB)の化合物:

5)式(IIIB)の化合物を式(b)の化合物:

式中、X1はスルホン酸エステルである。
1)式(IB)の化合物:


2)式(IIB)の化合物を式(b)の化合物と反応させて、式(VIIB)の化合物:

3)式(VIIB)の化合物をスルホン化剤と反応させて、式(VIIIB)の化合物:

4)式(VIIIB)の化合物を式(a)の化合物と反応させて、式(IVB)の化合物を形成するステップとを含み、
式中、X1はスルホン酸エステルである。
1)式(IB)の化合物:


2)式(IIB)の化合物を式(a)の化合物と反応させて、式(IXB)の化合物:

3)式(IXB)の化合物をスルホン化剤と反応させて、式(XB)の化合物:

4)式(XB)の化合物を式(b)の化合物:

式中、X1はスルホン酸エステルである。
1)式(IB)の化合物:


2)式(IIB)の化合物を式(b)の化合物と反応させて、式(VIIB)の化合物:

3)式(VIIB)の化合物をイミン還元剤と反応させて、式(IXB)の化合物:

4)式(IXB)の化合物をスルホン化剤と反応させて、式(XB)の化合物:

5)式(XB)の化合物を式(b)の化合物と反応させて、式(IVB)の化合物を形成するステップとを含み、
式中、X1はスルホン酸エステルである。







撹拌子及び熱電対を備えた乾燥した250mL丸底フラスコ(RBF)に、窒素をゆっくりと充填した。乾燥した50mL丸底フラスコで、(5-((2-(2-(2-メトキシエトキシ)エトキシ)エチル)(2-メチル-2-(メチルジスルファニル)プロピル)アミノ)-1,3-フェニレン)ジメタノール(化合物3)10.12g(22.26mmol)をDMF(40mL、4.0体積)に溶解した。得られた溶液を500mL RBFに移した。化合物3を含有する50mRBFをDMF(30.0mL、3.0体積)ですすぎ、この溶液を500mL RBFに添加した。500mL RBF中の溶液を25±2℃で、600rpmで1時間以上撹拌して均質な懸濁液を得た。別の乾燥した100mL RBFに、2,4,6-トリクロロ-1,3,5-トリアジン(TCT)3.14g(16.70mmol)を添加した。RBFに窒素を充填し、次いで20.0mL(2.0体積)のDMFを添加した。得られた混合物を3±2℃で30±5分間撹拌して溶液を形成し、これを別の漏斗に移し、25±3℃で温度を維持しながら20±3分間にわたって500mL RBFに滴下した。得られた反応混合物を50±5分間撹拌した。反応の完了時、別の漏斗を介して酢酸エチル(100.0mL、10体積)を反応物に添加して反応をクエンチし、続いて、反応温度を30℃未満に維持しながら0.5MのNaOH(50.0mL、5体積)を添加した。得られた混合物を30±5分間撹拌し、次いで分液漏斗に移した。追加の800mL(80体積)の酢酸エチル、続いて100mL(10体積)の脱イオン水を分液漏斗に添加した。有機相を分離し、水(2×200.0mL、2×20体積)で洗浄した。混合した有機相を、ロータリーエバポレーターを使用して濃縮した。粗生成物を、0~55%酢酸エチル/ヘキサンの勾配で溶出するシリカゲルカラムクロマトグラフィーを使用して40分間にわたって精製し、次いで5分間にわたって100%酢酸エチルに増加させて、所望の生成物(3-(クロロメチル)-5-((2-(2-(2-メトキシエトキシ)エトキシ)エチル)(2-メチル-2-(メチルジスルファニル)プロピル)アミノ)フェニル)メタノール(化合物4)(4.9429g、10.72mmol、収率48.1%)を得た。
化合物4(4.0788g、8.81mmol)をジクロロメタン(20.0mL、5体積)に溶解した。得られた透明な黄色溶液を-10℃に冷却し、メタンスルホン酸無水物(4.90g、27.3mmol)を添加した。N-エチル-N-イソプロピルプロパン-2-アミン(9.23mL、52.9mmol)をジクロロメタン(20.0mL、5体積)に溶解し、得られた溶液を、反応温度を-10℃に維持しながら、化合物4及び無水メタンスルホン酸の混合物にゆっくりと添加した。15分後、氷水とメタノール(20.0mL、5体積)の1:1混合物を添加することによって反応をクエンチし、-10℃で、5分間1000rpmで撹拌した。次いで、混合物を予め冷却した(5℃)水(36.0mL、9.0体積)に添加し、-10℃で撹拌し、続いてメタンスルホン酸水溶液(5%溶液、10.0mL、2.5体積)を添加した。得られた溶液を0℃で5分間撹拌し、DCM(16.0mL、40.0体積)を添加し、この混合物を1分間撹拌した。有機層を分離して、反応リアクターに戻し、予め冷却した(5℃)水(36.0mL、9.0体積)及びメタンスルホン酸水溶液(5%溶媒、13.2mL、3.3体積)をリアクターに加えた。混合物を8分間撹拌し、有機層を分離し、氷/水(2x42.7mL、2x10.5体積)で洗浄した。混合した有機層を無水硫酸ナトリウムで乾燥させ、濾過し、溶媒を真空オーブン中で除去して、所望の生成物3-(クロロメチル)-5-((2-(2-(2-メトキシエトキシ)エトキシ)エチル)(2-メチル-2-(メチルジスルファニル)プロピル)アミノ)ベンジルメタンスルホン酸塩(化合物5)(4.8740g、8.81mmol、収率100%)を得た。
(S)-9-ヒドロキシ-8-メトキシ-11,12,12a,13-テトラヒドロ-6H-ベンゾ[5,6][1,4]ジアゼピノ[1,2-a]インドール-6-オン(化合物5)(0.124g、0.418mmol)及びN,N-ジメチルアセトアミド(1.0mL、4体積)を、撹拌子及び熱電対を備えた100mLのRBFに添加し、この溶液を超音波処理した。炭酸カリウム(0.114g、0.828mmol)を溶液に添加した。20mLガラスバイアル中で、化合物5(0.2263g、0.414mmol)をN,N-ジメチルアセトアミド(1.5mL、6体積)に溶解し、得られた溶液を1時間にわたって100mLのRBF反応容器にゆっくりと添加した。反応混合物を、反応温度を20℃±3℃に維持しながら、800rpmで11時間撹拌した。水(3mL、10体積)をゆっくりと添加し、15±5分間撹拌して反応をクエンチした。次いで酢酸エチル(9mL、30体積)を反応混合物に添加し、15±5分間撹拌した。得られた混合物を分液漏斗に移し、有機相を単離した。水層を酢酸エチル(3×12mL、3×40体積)で洗浄した。混合した有機層を半飽和ブライン溶液(1×9mL、1×30体積)で洗浄し、濃縮して粗生成物を得た。粗生成物を、0~70%酢酸エチル/ヘキサンの勾配で溶出するシリカゲルカラムクロマトグラフィーにより35分間にわたって精製して、所望の生成物(S)-9-((3-(クロロメチル)-5-((2-(2-(2-メトキシエトキシ)エトキシ)エチル)(2-メチル-2-(メチルジスルファニル)プロピル)アミノ)ベンジル)オキシ)-8-メトキシ-11,12,12a、13-テトラヒドロ-6H-ベンゾ[5,6][1,4]ジアゼピノ[1,2-a]インドール-6-オン(化合物6)(0.1549g、0.212mmol、収率51.2%)を得た。
撹拌子、熱電対及び加熱マンテルを備えた乾燥した50mL丸底フラスコ(RBF)中で、(S)-9-((3-(クロロメチル)-5-((2-(2-(2-メトキシエトキシ)エトキシ)エチル)(2-メチル-2-(メチルジスルファニル)プロピル)アミノ)ベンジル)オキシ)-8-メトキシ-11,12,12a,13-テトラヒドロ-6H-ベンゾ[5,6][1,4]ジアゼピノ[1,2-a]インドール-6-オン(0.150g、0.195mmol)を充填した。N,N-ジメチルアセトアミド(3.0mL、6体積)を添加し、得られた混合物を20℃±5℃で撹拌して、透明な淡褐色溶液を得た。淡褐色溶液にヨウ化カリウム(0.032g、0.195mmol)、続いて炭酸カリウム(0.054g、0.390mmol)を添加した。20mLガラスバイアル中で、(S)-9-ヒドロキシ-8-メトキシ-12a,13-ジヒドロ-6H-ベンゾ[5,6][1,4]ジアゼピノ[1,2-a]インドール-6-オン(0.071g、0.234mmol)をN,N-ジメチルアセトアミド(2.0mL、4体積)に溶解した。得られた溶液をRBF中の反応混合物にゆっくりと添加すると、反応溶液が黄褐色に変わった。反応混合物を30℃±3℃で加熱しながら24時間撹拌し、次いで20℃±5℃まで冷却した。脱イオン水(6mL、20体積)を反応混合物に添加して反応をクエンチし、生成物を沈殿させた。生成物を濾過し、脱イオン水(2×3mL、10体積)で洗浄し、ジクロロメタン(12mL、40体積)に再溶解した。ジクロロメタン溶液を脱イオン水(6mL、20体積)と混合し、有機相を分離し、半飽和ブライン(2×6mL、2×20体積)及び水(2×6mL、2×20体積)で洗浄した。有機相を無水硫酸ナトリウム上で乾燥させ、濾過し、真空下で約0.45mL(1.5体積)まで濃縮した。粗生成物を、0~100%酢酸エチル/ヘキサンの勾配で溶出するシリカゲルカラムクロマトグラフィーにより50分間にわたって精製し、次いで5分間で100%酢酸エチルに増加して、所望の生成物(S)-8-メトキシ-9-((3-(((S)-8-メトキシ-6-オキソ-11,12,12a,13-テトラヒドロ-6H-ベンゾ[5,6][1,4]ジアゼピノ[1,2-a]インドール-9-イル)オキシ)メチル)-5-((2-(2-(2-メトキシエトキシ)エトキシ)エチル)(2-メチル-2-(メチルジスルファニル)プロピル)アミノ)ベンジル)オキシ)-12a、13-ジヒドロ-6H-ベンゾ[5,6][1,4]ジアゼピノ[1,2-a]インドール-6-オン(0.1033g、0.105mmol、収率53.6%)を得る。
TCEP(3.1g)を数滴の水で湿らせた。飽和NaHCO3(85mL)を、pH6.5に達するまで添加した。0.1Mリン酸緩衝液(34mL)を新たに調製し、pH6.5に調整した後、TCEP溶液に添加した。化合物7(10.1g)をアセトニトリル(588mL)及びMeOH(257mL)に溶解し、TCEP溶液と混合した。2時間後、追加のTCEP(310mg)を添加し(NaHCO3溶液に溶解し、pHを6.5に調整した)、この混合物をさらに2時間撹拌した。DCM(1.3L)及び水(600mL)を添加した。分離後、水相をDCM(250mL)で抽出した。混合した有機相を水(2×250mL)で洗浄し、蒸発乾燥させて、9.6gの還元化合物を得た。
Claims (18)
- 式(IIA)の化合物:
式(IA)の化合物:
- 式(IIIA)の化合物を調製する方法であって、
ii)式(IIA)の化合物を式(a)の化合物と反応させて、式(IIIA)を形成する工程、
- 式(IVA)の化合物を調製する方法であって、
iii)式(IIIA)の化合物を式(b)の化合物と反応させて、式(IVA)を形成する工程、
- 前記式(IIIA)の化合物が、塩基の存在下で前記式(b)の化合物と反応させられる、請求項3記載の方法。
- 前記塩基が、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、水素化ナトリウム、または水素化カリウムである、請求項4記載の方法。
- 前記塩基が、炭酸カリウムである、請求項4記載の方法。
- 前記式(IIIA)の化合物と前記式(b)の化合物との間の前記反応が、ヨウ化カリウムまたはヨウ化セシウムの存在下で行われる、請求項3~6のいずれか一項に記載の方法。
- 前記式(IIA)の化合物と前記式(a)の化合物との間の前記反応が、アルコール活性化剤及びアゾジカルボキシレートの存在下で行われる、請求項2~7の何れか一項に記載の方法。
- 前記アルコール活性化剤が、トリブチルホスフィンまたはトリフェニルホスフィンであり、前記アゾジカルボキシレートが、ジエチルアゾジカルボキシレート(DEAD)、ジイソプロピルアゾジカルボキシレート(DIAD)、1,1’-(アゾジカルボニル)ジピペリジン(ADDP)、及びジ-tert-ブチルアゾジカルボキシレート(DTAD)からなる群から選択される、請求項8に記載の方法。
- 前記アルコール活性化剤が、トリフェニルホスフィンであり、前記アゾジカルボキシレートが、ジイソプロピルアゾジカルボキシレート(DIAD)である、請求項8に記載の方法。
- 前記トリフェニルホスフィン及び前記ジイソプロピルアゾジカルボキシレートが混合されて複合体が形成され、その後に前記式(IIA)の化合物および前記式(a)の化合物と前記複合体が混合される、請求項10に記載の方法。
- 式(IVA)の化合物を調製する方法であって、
iv)式(VA)の化合物を式(a)の化合物と反応させて、式(IVA)を形成する工程、
を含む、前記方法。 - 式(IVA)の化合物を調製する方法であって、
iv)式(IIIA)の化合物を式(b)の化合物と反応させて、式(IVA)を形成する工程、
を含む、前記方法。 - 式(IVA)の化合物を調製する方法であって、
v)前記式(IIIA)の化合物を式(b)の化合物と反応させて、式(IVA)の化合物:
式中、X 1 はスルホン酸エステルである、
を含む、前記方法。 - 0.7~0.8モル当量のシアヌル酸塩化物と反応させる、請求項1~14のいずれか一項に記載の方法。
- 0.85モル当量のシアヌル酸塩化物と反応させる、請求項1~14のいずれか一項に記載の方法。
- 0.75モル当量のシアヌル酸塩化物と反応させる、請求項1~14のいずれか一項に記載の方法。
- 反応がDMF中で行われる、請求項1~17のいずれか一項に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862758814P | 2018-11-12 | 2018-11-12 | |
| US62/758,814 | 2018-11-12 | ||
| PCT/US2019/060677 WO2020102051A1 (en) | 2018-11-12 | 2019-11-11 | Methods of preparing cytotoxic benzodiazepine derivatives |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2024018294A Division JP2024056819A (ja) | 2018-11-12 | 2024-02-09 | 細胞傷害性ベンゾジアゼピン誘導体の調製方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2022512935A JP2022512935A (ja) | 2022-02-07 |
| JPWO2020102051A5 JPWO2020102051A5 (ja) | 2022-11-17 |
| JP7437395B2 true JP7437395B2 (ja) | 2024-02-22 |
Family
ID=68835289
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021524303A Active JP7437395B2 (ja) | 2018-11-12 | 2019-11-11 | 細胞傷害性ベンゾジアゼピン誘導体の調製方法 |
Country Status (11)
| Country | Link |
|---|---|
| US (3) | US10851117B2 (ja) |
| EP (2) | EP4361128A2 (ja) |
| JP (1) | JP7437395B2 (ja) |
| KR (1) | KR20210098470A (ja) |
| AU (1) | AU2019379104A1 (ja) |
| CA (1) | CA3119358A1 (ja) |
| IL (1) | IL283045A (ja) |
| MA (1) | MA54228A (ja) |
| SG (1) | SG11202104360YA (ja) |
| TW (1) | TW202033530A (ja) |
| WO (1) | WO2020102051A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2019379104A1 (en) | 2018-11-12 | 2021-06-03 | Immunogen, Inc. | Methods of preparing cytotoxic benzodiazepine derivatives |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017004025A1 (en) | 2015-06-29 | 2017-01-05 | Immunogen, Inc. | Conjugates of cysteine engineered antibodies |
| WO2017004026A1 (en) | 2015-06-29 | 2017-01-05 | Immunogen, Inc. | Anti-cd 123 antibodies and conjugates and derivatives thereof |
| WO2017015496A1 (en) | 2015-07-21 | 2017-01-26 | Immunogen, Inc. | Methods of preparing cytotoxic benzodiazepine derivatives |
| WO2017091745A1 (en) | 2015-11-25 | 2017-06-01 | Immunogen, Inc. | Pharmaceutical formulations and methods of use thereof |
| WO2018140435A1 (en) | 2017-01-25 | 2018-08-02 | Immunogen, Inc. | Methods of preparing cytotoxic benzodiazepine derivatives |
| WO2019133652A1 (en) | 2017-12-28 | 2019-07-04 | Immunogen, Inc. | Benzodiazepine derivatives |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX346635B (es) | 2011-02-15 | 2017-03-27 | Immunogen Inc | Derivados citotoxicos de la benzodiazepina. |
| AU2019379104A1 (en) | 2018-11-12 | 2021-06-03 | Immunogen, Inc. | Methods of preparing cytotoxic benzodiazepine derivatives |
-
2019
- 2019-11-11 AU AU2019379104A patent/AU2019379104A1/en active Pending
- 2019-11-11 EP EP24163044.1A patent/EP4361128A2/en active Pending
- 2019-11-11 EP EP19817455.9A patent/EP3880681B1/en active Active
- 2019-11-11 WO PCT/US2019/060677 patent/WO2020102051A1/en unknown
- 2019-11-11 CA CA3119358A patent/CA3119358A1/en active Pending
- 2019-11-11 SG SG11202104360YA patent/SG11202104360YA/en unknown
- 2019-11-11 KR KR1020217017972A patent/KR20210098470A/ko unknown
- 2019-11-11 JP JP2021524303A patent/JP7437395B2/ja active Active
- 2019-11-11 MA MA054228A patent/MA54228A/fr unknown
- 2019-11-11 US US16/679,563 patent/US10851117B2/en active Active
- 2019-11-11 TW TW108140769A patent/TW202033530A/zh unknown
-
2020
- 2020-10-01 US US17/060,611 patent/US11396519B2/en active Active
-
2021
- 2021-05-09 IL IL283045A patent/IL283045A/en unknown
-
2022
- 2022-06-15 US US17/841,046 patent/US20230045079A1/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017004025A1 (en) | 2015-06-29 | 2017-01-05 | Immunogen, Inc. | Conjugates of cysteine engineered antibodies |
| WO2017004026A1 (en) | 2015-06-29 | 2017-01-05 | Immunogen, Inc. | Anti-cd 123 antibodies and conjugates and derivatives thereof |
| WO2017015496A1 (en) | 2015-07-21 | 2017-01-26 | Immunogen, Inc. | Methods of preparing cytotoxic benzodiazepine derivatives |
| WO2017091745A1 (en) | 2015-11-25 | 2017-06-01 | Immunogen, Inc. | Pharmaceutical formulations and methods of use thereof |
| WO2018140435A1 (en) | 2017-01-25 | 2018-08-02 | Immunogen, Inc. | Methods of preparing cytotoxic benzodiazepine derivatives |
| WO2019133652A1 (en) | 2017-12-28 | 2019-07-04 | Immunogen, Inc. | Benzodiazepine derivatives |
Also Published As
| Publication number | Publication date |
|---|---|
| US20210122768A1 (en) | 2021-04-29 |
| EP3880681A1 (en) | 2021-09-22 |
| US11396519B2 (en) | 2022-07-26 |
| TW202033530A (zh) | 2020-09-16 |
| US20200172555A1 (en) | 2020-06-04 |
| EP4361128A2 (en) | 2024-05-01 |
| CA3119358A1 (en) | 2020-05-22 |
| EP3880681B1 (en) | 2024-03-13 |
| WO2020102051A8 (en) | 2020-06-18 |
| JP2022512935A (ja) | 2022-02-07 |
| US10851117B2 (en) | 2020-12-01 |
| MA54228A (fr) | 2021-09-22 |
| SG11202104360YA (en) | 2021-05-28 |
| AU2019379104A1 (en) | 2021-06-03 |
| KR20210098470A (ko) | 2021-08-10 |
| WO2020102051A1 (en) | 2020-05-22 |
| IL283045A (en) | 2021-06-30 |
| US20230045079A1 (en) | 2023-02-09 |
| CN112996792A (zh) | 2021-06-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10392407B2 (en) | Methods of preparing cytotoxic benzodiazepine derivatives | |
| JP7400005B2 (ja) | 細胞傷害性ベンゾジアゼピン誘導体の調製方法 | |
| EP3704097B1 (en) | Bicyclic sulfones and sulfoxides and methods of use thereof | |
| JP7437395B2 (ja) | 細胞傷害性ベンゾジアゼピン誘導体の調製方法 | |
| JP2024056819A (ja) | 細胞傷害性ベンゾジアゼピン誘導体の調製方法 | |
| RU2813137C2 (ru) | Способы получения цитотоксических производных бензодиазепина | |
| CN112996792B (zh) | 制备细胞毒性苯并二氮杂卓衍生物的方法 | |
| Hollá | Synthesis of polycyclic lactones and lactams | |
| CN103965198A (zh) | 替卡格雷的中间体及其制备方法以及利用该中间体制备替卡格雷的方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20221109 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20221109 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20230911 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20231211 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20240111 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20240209 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7437395 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |