JP6557140B2 - ペプチドコンジュゲート粒子 - Google Patents
ペプチドコンジュゲート粒子 Download PDFInfo
- Publication number
- JP6557140B2 JP6557140B2 JP2015518611A JP2015518611A JP6557140B2 JP 6557140 B2 JP6557140 B2 JP 6557140B2 JP 2015518611 A JP2015518611 A JP 2015518611A JP 2015518611 A JP2015518611 A JP 2015518611A JP 6557140 B2 JP6557140 B2 JP 6557140B2
- Authority
- JP
- Japan
- Prior art keywords
- particles
- antigen
- plg
- mice
- cells
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000002245 particle Substances 0.000 title claims description 412
- 239000000863 peptide conjugate Substances 0.000 title 1
- 239000000427 antigen Substances 0.000 claims description 216
- 108091007433 antigens Proteins 0.000 claims description 213
- 102000036639 antigens Human genes 0.000 claims description 213
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 claims description 152
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 105
- 201000010099 disease Diseases 0.000 claims description 94
- 239000000203 mixture Substances 0.000 claims description 75
- 108090000623 proteins and genes Proteins 0.000 claims description 57
- 235000018102 proteins Nutrition 0.000 claims description 52
- 102000004169 proteins and genes Human genes 0.000 claims description 52
- 238000000034 method Methods 0.000 claims description 50
- 210000001519 tissue Anatomy 0.000 claims description 35
- 230000001939 inductive effect Effects 0.000 claims description 31
- 230000004044 response Effects 0.000 claims description 30
- 208000023275 Autoimmune disease Diseases 0.000 claims description 28
- 239000013566 allergen Substances 0.000 claims description 27
- 108010058846 Ovalbumin Proteins 0.000 claims description 22
- 229940092253 ovalbumin Drugs 0.000 claims description 22
- 229920001577 copolymer Polymers 0.000 claims description 20
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 20
- -1 fp130-RAPS Proteins 0.000 claims description 14
- 108090001061 Insulin Proteins 0.000 claims description 11
- 102000004877 Insulin Human genes 0.000 claims description 10
- 229940125396 insulin Drugs 0.000 claims description 10
- 206010020751 Hypersensitivity Diseases 0.000 claims description 9
- 108010000123 Myelin-Oligodendrocyte Glycoprotein Proteins 0.000 claims description 9
- 102000002233 Myelin-Oligodendrocyte Glycoprotein Human genes 0.000 claims description 9
- KISWVXRQTGLFGD-UHFFFAOYSA-N 2-[[2-[[6-amino-2-[[2-[[2-[[5-amino-2-[[2-[[1-[2-[[6-amino-2-[(2,5-diamino-5-oxopentanoyl)amino]hexanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-(diaminomethylideneamino)p Chemical compound C1CCN(C(=O)C(CCCN=C(N)N)NC(=O)C(CCCCN)NC(=O)C(N)CCC(N)=O)C1C(=O)NC(CO)C(=O)NC(CCC(N)=O)C(=O)NC(CCCN=C(N)N)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=C(O)C=C1 KISWVXRQTGLFGD-UHFFFAOYSA-N 0.000 claims description 8
- 108010035532 Collagen Proteins 0.000 claims description 8
- 102000008186 Collagen Human genes 0.000 claims description 8
- 229920001436 collagen Polymers 0.000 claims description 8
- 239000003937 drug carrier Substances 0.000 claims description 8
- 102000008214 Glutamate decarboxylase Human genes 0.000 claims description 7
- 108091022930 Glutamate decarboxylase Proteins 0.000 claims description 7
- 208000026935 allergic disease Diseases 0.000 claims description 7
- 230000001363 autoimmune Effects 0.000 claims description 7
- 108091003079 Bovine Serum Albumin Proteins 0.000 claims description 6
- 102000047918 Myelin Basic Human genes 0.000 claims description 6
- 101710107068 Myelin basic protein Proteins 0.000 claims description 6
- 102000007999 Nuclear Proteins Human genes 0.000 claims description 6
- 108010089610 Nuclear Proteins Proteins 0.000 claims description 6
- 108010010974 Proteolipids Proteins 0.000 claims description 6
- 102000016202 Proteolipids Human genes 0.000 claims description 6
- 229940098773 bovine serum albumin Drugs 0.000 claims description 6
- 208000027866 inflammatory disease Diseases 0.000 claims description 6
- 235000013336 milk Nutrition 0.000 claims description 6
- 239000008267 milk Substances 0.000 claims description 6
- 210000004080 milk Anatomy 0.000 claims description 6
- 230000004936 stimulating effect Effects 0.000 claims description 6
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 claims description 5
- 206010052779 Transplant rejections Diseases 0.000 claims description 5
- 230000007815 allergy Effects 0.000 claims description 5
- 102000005962 receptors Human genes 0.000 claims description 5
- 108020003175 receptors Proteins 0.000 claims description 5
- 101710088194 Dehydrogenase Proteins 0.000 claims description 4
- 102000009093 Dihydrolipoyllysine-residue acetyltransferase Human genes 0.000 claims description 4
- 108010073112 Dihydrolipoyllysine-residue acetyltransferase Proteins 0.000 claims description 4
- 102000003886 Glycoproteins Human genes 0.000 claims description 4
- 108090000288 Glycoproteins Proteins 0.000 claims description 4
- 108010033040 Histones Proteins 0.000 claims description 4
- 101000883515 Homo sapiens Chitinase-3-like protein 1 Proteins 0.000 claims description 4
- 108010029485 Protein Isoforms Proteins 0.000 claims description 4
- 102000001708 Protein Isoforms Human genes 0.000 claims description 4
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 claims description 4
- 102000005937 Tropomyosin Human genes 0.000 claims description 4
- 108010030743 Tropomyosin Proteins 0.000 claims description 4
- 239000012645 endogenous antigen Substances 0.000 claims description 4
- 102000005525 fibrillarin Human genes 0.000 claims description 4
- 108020002231 fibrillarin Proteins 0.000 claims description 4
- 210000003780 hair follicle Anatomy 0.000 claims description 4
- 102000054350 human CHI3L1 Human genes 0.000 claims description 4
- 210000001685 thyroid gland Anatomy 0.000 claims description 4
- 102000009366 Alpha-s1 casein Human genes 0.000 claims description 3
- 108050000244 Alpha-s1 casein Proteins 0.000 claims description 3
- 244000144730 Amygdalus persica Species 0.000 claims description 3
- 108010009685 Cholinergic Receptors Proteins 0.000 claims description 3
- 240000009226 Corylus americana Species 0.000 claims description 3
- 235000001543 Corylus americana Nutrition 0.000 claims description 3
- 235000007466 Corylus avellana Nutrition 0.000 claims description 3
- 101800001467 Envelope glycoprotein E2 Proteins 0.000 claims description 3
- 241001330451 Paspalum notatum Species 0.000 claims description 3
- 235000006040 Prunus persica var persica Nutrition 0.000 claims description 3
- 101800001271 Surface protein Proteins 0.000 claims description 3
- 102000034337 acetylcholine receptors Human genes 0.000 claims description 3
- 244000205479 Bertholletia excelsa Species 0.000 claims description 2
- 235000012284 Bertholletia excelsa Nutrition 0.000 claims description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 2
- 230000000521 hyperimmunizing effect Effects 0.000 claims description 2
- 102000006395 Globulins Human genes 0.000 claims 1
- 108010044091 Globulins Proteins 0.000 claims 1
- 241000699670 Mus sp. Species 0.000 description 148
- 210000004027 cell Anatomy 0.000 description 123
- 108090000765 processed proteins & peptides Proteins 0.000 description 112
- 108010064578 myelin proteolipid protein (139-151) Proteins 0.000 description 82
- 241001465754 Metazoa Species 0.000 description 58
- 101000609767 Dromaius novaehollandiae Ovalbumin Proteins 0.000 description 52
- 102000004196 processed proteins & peptides Human genes 0.000 description 47
- 230000028993 immune response Effects 0.000 description 41
- 238000011282 treatment Methods 0.000 description 40
- 239000003814 drug Substances 0.000 description 39
- 230000000890 antigenic effect Effects 0.000 description 38
- 230000006698 induction Effects 0.000 description 36
- 210000001744 T-lymphocyte Anatomy 0.000 description 30
- 108010019759 OVA 323-339 Proteins 0.000 description 29
- 239000002502 liposome Substances 0.000 description 29
- 241000699666 Mus <mouse, genus> Species 0.000 description 26
- 229940079593 drug Drugs 0.000 description 25
- 239000004793 Polystyrene Substances 0.000 description 24
- 230000027455 binding Effects 0.000 description 24
- 239000002105 nanoparticle Substances 0.000 description 24
- 102000004127 Cytokines Human genes 0.000 description 23
- 108090000695 Cytokines Proteins 0.000 description 23
- 230000001900 immune effect Effects 0.000 description 23
- 229920000642 polymer Polymers 0.000 description 23
- 229920002223 polystyrene Polymers 0.000 description 23
- 201000006417 multiple sclerosis Diseases 0.000 description 21
- 238000012384 transportation and delivery Methods 0.000 description 21
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 21
- 239000000126 substance Substances 0.000 description 20
- 239000011324 bead Substances 0.000 description 19
- 150000001875 compounds Chemical class 0.000 description 18
- 239000000463 material Substances 0.000 description 18
- 230000007423 decrease Effects 0.000 description 17
- 230000000694 effects Effects 0.000 description 17
- 102000003814 Interleukin-10 Human genes 0.000 description 16
- 108090000174 Interleukin-10 Proteins 0.000 description 16
- 239000000872 buffer Substances 0.000 description 16
- 210000002540 macrophage Anatomy 0.000 description 16
- 210000000056 organ Anatomy 0.000 description 15
- 210000000278 spinal cord Anatomy 0.000 description 15
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 14
- 238000009472 formulation Methods 0.000 description 14
- 239000002953 phosphate buffered saline Substances 0.000 description 14
- 239000003124 biologic agent Substances 0.000 description 13
- 210000004369 blood Anatomy 0.000 description 13
- 239000008280 blood Substances 0.000 description 13
- 239000013043 chemical agent Substances 0.000 description 13
- 238000011161 development Methods 0.000 description 13
- 230000018109 developmental process Effects 0.000 description 13
- 239000008194 pharmaceutical composition Substances 0.000 description 13
- 229920000747 poly(lactic acid) Polymers 0.000 description 13
- 230000000638 stimulation Effects 0.000 description 13
- 108090000978 Interleukin-4 Proteins 0.000 description 12
- 102000004388 Interleukin-4 Human genes 0.000 description 12
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 12
- 229940037003 alum Drugs 0.000 description 12
- 230000005756 apoptotic signaling Effects 0.000 description 12
- 238000001990 intravenous administration Methods 0.000 description 12
- 229920001223 polyethylene glycol Polymers 0.000 description 12
- 229920001184 polypeptide Polymers 0.000 description 12
- 239000000243 solution Substances 0.000 description 12
- 229940124597 therapeutic agent Drugs 0.000 description 12
- 230000004913 activation Effects 0.000 description 11
- 210000004443 dendritic cell Anatomy 0.000 description 11
- 230000006870 function Effects 0.000 description 11
- 238000002347 injection Methods 0.000 description 11
- 239000007924 injection Substances 0.000 description 11
- 238000010172 mouse model Methods 0.000 description 11
- 239000000546 pharmaceutical excipient Substances 0.000 description 11
- 238000002360 preparation method Methods 0.000 description 11
- 230000001225 therapeutic effect Effects 0.000 description 11
- 230000003614 tolerogenic effect Effects 0.000 description 11
- 238000012546 transfer Methods 0.000 description 11
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 10
- 108090000176 Interleukin-13 Proteins 0.000 description 10
- 108010002616 Interleukin-5 Proteins 0.000 description 10
- 229920000954 Polyglycolide Polymers 0.000 description 10
- 230000008901 benefit Effects 0.000 description 10
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 10
- 239000000969 carrier Substances 0.000 description 10
- 210000003169 central nervous system Anatomy 0.000 description 10
- 208000035475 disorder Diseases 0.000 description 10
- 239000012634 fragment Substances 0.000 description 10
- 108020001507 fusion proteins Proteins 0.000 description 10
- 102000037865 fusion proteins Human genes 0.000 description 10
- 230000003993 interaction Effects 0.000 description 10
- 210000004185 liver Anatomy 0.000 description 10
- 230000001404 mediated effect Effects 0.000 description 10
- 239000004626 polylactic acid Substances 0.000 description 10
- 210000003289 regulatory T cell Anatomy 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- 239000007929 subcutaneous injection Substances 0.000 description 10
- 238000010254 subcutaneous injection Methods 0.000 description 10
- 239000000725 suspension Substances 0.000 description 10
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 9
- 235000001014 amino acid Nutrition 0.000 description 9
- 150000001413 amino acids Chemical class 0.000 description 9
- 238000004458 analytical method Methods 0.000 description 9
- 210000000612 antigen-presenting cell Anatomy 0.000 description 9
- 210000004556 brain Anatomy 0.000 description 9
- 230000000981 bystander Effects 0.000 description 9
- 210000004899 c-terminal region Anatomy 0.000 description 9
- 239000012636 effector Substances 0.000 description 9
- 125000000524 functional group Chemical group 0.000 description 9
- 210000004698 lymphocyte Anatomy 0.000 description 9
- 238000004519 manufacturing process Methods 0.000 description 9
- 239000002096 quantum dot Substances 0.000 description 9
- 230000002829 reductive effect Effects 0.000 description 9
- 210000002966 serum Anatomy 0.000 description 9
- 210000000952 spleen Anatomy 0.000 description 9
- 239000012049 topical pharmaceutical composition Substances 0.000 description 9
- NIXOWILDQLNWCW-UHFFFAOYSA-N Acrylic acid Chemical compound OC(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 8
- 206010002198 Anaphylactic reaction Diseases 0.000 description 8
- 230000036783 anaphylactic response Effects 0.000 description 8
- 208000003455 anaphylaxis Diseases 0.000 description 8
- 230000001580 bacterial effect Effects 0.000 description 8
- 210000001185 bone marrow Anatomy 0.000 description 8
- 150000007942 carboxylates Chemical group 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 8
- 230000021615 conjugation Effects 0.000 description 8
- 239000006071 cream Substances 0.000 description 8
- 210000003979 eosinophil Anatomy 0.000 description 8
- 125000005647 linker group Chemical group 0.000 description 8
- 206010039073 rheumatoid arthritis Diseases 0.000 description 8
- 210000003491 skin Anatomy 0.000 description 8
- 208000027930 type IV hypersensitivity disease Diseases 0.000 description 8
- 230000009385 viral infection Effects 0.000 description 8
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 7
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 7
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 7
- 206010061218 Inflammation Diseases 0.000 description 7
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 7
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 description 7
- 239000002671 adjuvant Substances 0.000 description 7
- 208000037883 airway inflammation Diseases 0.000 description 7
- 238000010171 animal model Methods 0.000 description 7
- 208000006673 asthma Diseases 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 238000000576 coating method Methods 0.000 description 7
- 206010012601 diabetes mellitus Diseases 0.000 description 7
- 238000002296 dynamic light scattering Methods 0.000 description 7
- 239000008103 glucose Substances 0.000 description 7
- 230000002209 hydrophobic effect Effects 0.000 description 7
- 230000002163 immunogen Effects 0.000 description 7
- 238000000338 in vitro Methods 0.000 description 7
- 230000001965 increasing effect Effects 0.000 description 7
- 230000008595 infiltration Effects 0.000 description 7
- 238000001764 infiltration Methods 0.000 description 7
- 230000004054 inflammatory process Effects 0.000 description 7
- 239000008101 lactose Substances 0.000 description 7
- 210000000265 leukocyte Anatomy 0.000 description 7
- 239000003446 ligand Substances 0.000 description 7
- 239000003921 oil Substances 0.000 description 7
- 235000019198 oils Nutrition 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 230000009467 reduction Effects 0.000 description 7
- 230000028327 secretion Effects 0.000 description 7
- 238000002054 transplantation Methods 0.000 description 7
- 208000030767 Autoimmune encephalitis Diseases 0.000 description 6
- 208000035143 Bacterial infection Diseases 0.000 description 6
- 206010011968 Decreased immune responsiveness Diseases 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- 208000036142 Viral infection Diseases 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 208000037884 allergic airway inflammation Diseases 0.000 description 6
- 208000022362 bacterial infectious disease Diseases 0.000 description 6
- 229920002988 biodegradable polymer Polymers 0.000 description 6
- 239000004621 biodegradable polymer Substances 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 230000003247 decreasing effect Effects 0.000 description 6
- 235000019441 ethanol Nutrition 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 238000000684 flow cytometry Methods 0.000 description 6
- 230000004927 fusion Effects 0.000 description 6
- 210000002865 immune cell Anatomy 0.000 description 6
- 230000006058 immune tolerance Effects 0.000 description 6
- 230000003053 immunization Effects 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 230000002757 inflammatory effect Effects 0.000 description 6
- 150000002632 lipids Chemical class 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 210000001616 monocyte Anatomy 0.000 description 6
- 239000008188 pellet Substances 0.000 description 6
- 230000035515 penetration Effects 0.000 description 6
- 230000002093 peripheral effect Effects 0.000 description 6
- 239000006187 pill Substances 0.000 description 6
- 239000003755 preservative agent Substances 0.000 description 6
- 230000008569 process Effects 0.000 description 6
- 230000000069 prophylactic effect Effects 0.000 description 6
- 230000000306 recurrent effect Effects 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 208000015943 Coeliac disease Diseases 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 5
- 108010074328 Interferon-gamma Proteins 0.000 description 5
- 108050003558 Interleukin-17 Proteins 0.000 description 5
- 102000013691 Interleukin-17 Human genes 0.000 description 5
- 108010002350 Interleukin-2 Proteins 0.000 description 5
- 239000005642 Oleic acid Substances 0.000 description 5
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 5
- 229930006000 Sucrose Natural products 0.000 description 5
- 238000010521 absorption reaction Methods 0.000 description 5
- 239000000443 aerosol Substances 0.000 description 5
- 150000001412 amines Chemical group 0.000 description 5
- 238000013459 approach Methods 0.000 description 5
- 230000036760 body temperature Effects 0.000 description 5
- 210000002798 bone marrow cell Anatomy 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 239000002552 dosage form Substances 0.000 description 5
- 239000000839 emulsion Substances 0.000 description 5
- 239000003623 enhancer Substances 0.000 description 5
- 229940088598 enzyme Drugs 0.000 description 5
- 210000003743 erythrocyte Anatomy 0.000 description 5
- 239000000499 gel Substances 0.000 description 5
- 238000002649 immunization Methods 0.000 description 5
- 210000004072 lung Anatomy 0.000 description 5
- 210000001165 lymph node Anatomy 0.000 description 5
- 150000007523 nucleic acids Chemical class 0.000 description 5
- 239000002674 ointment Substances 0.000 description 5
- 229960002969 oleic acid Drugs 0.000 description 5
- 239000004633 polyglycolic acid Substances 0.000 description 5
- 102000040430 polynucleotide Human genes 0.000 description 5
- 108091033319 polynucleotide Proteins 0.000 description 5
- 239000002157 polynucleotide Substances 0.000 description 5
- 239000011148 porous material Substances 0.000 description 5
- 239000002243 precursor Substances 0.000 description 5
- 238000012545 processing Methods 0.000 description 5
- 230000002685 pulmonary effect Effects 0.000 description 5
- 238000011002 quantification Methods 0.000 description 5
- 239000007787 solid Substances 0.000 description 5
- 239000007909 solid dosage form Substances 0.000 description 5
- 210000004988 splenocyte Anatomy 0.000 description 5
- 239000005720 sucrose Substances 0.000 description 5
- 230000001629 suppression Effects 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 4
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 4
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 4
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 4
- 101000669402 Homo sapiens Toll-like receptor 7 Proteins 0.000 description 4
- 102100037850 Interferon gamma Human genes 0.000 description 4
- 102000000588 Interleukin-2 Human genes 0.000 description 4
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 4
- 206010057249 Phagocytosis Diseases 0.000 description 4
- 239000002202 Polyethylene glycol Substances 0.000 description 4
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 4
- 229920002472 Starch Polymers 0.000 description 4
- 102100039390 Toll-like receptor 7 Human genes 0.000 description 4
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 4
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 4
- ATBOMIWRCZXYSZ-XZBBILGWSA-N [1-[2,3-dihydroxypropoxy(hydroxy)phosphoryl]oxy-3-hexadecanoyloxypropan-2-yl] (9e,12e)-octadeca-9,12-dienoate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OCC(O)CO)OC(=O)CCCCCCC\C=C\C\C=C\CCCCC ATBOMIWRCZXYSZ-XZBBILGWSA-N 0.000 description 4
- 230000003213 activating effect Effects 0.000 description 4
- 201000009961 allergic asthma Diseases 0.000 description 4
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 4
- 210000003719 b-lymphocyte Anatomy 0.000 description 4
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 4
- 239000012620 biological material Substances 0.000 description 4
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N biotin Natural products N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 4
- 230000008499 blood brain barrier function Effects 0.000 description 4
- 210000001218 blood-brain barrier Anatomy 0.000 description 4
- 150000001720 carbohydrates Chemical class 0.000 description 4
- 235000014633 carbohydrates Nutrition 0.000 description 4
- 238000012512 characterization method Methods 0.000 description 4
- 235000012000 cholesterol Nutrition 0.000 description 4
- 230000001684 chronic effect Effects 0.000 description 4
- 230000006378 damage Effects 0.000 description 4
- 230000003111 delayed effect Effects 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 238000005538 encapsulation Methods 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- 235000011187 glycerol Nutrition 0.000 description 4
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 210000000987 immune system Anatomy 0.000 description 4
- 230000003136 immunomodifying effect Effects 0.000 description 4
- 238000011503 in vivo imaging Methods 0.000 description 4
- 239000003701 inert diluent Substances 0.000 description 4
- 238000001802 infusion Methods 0.000 description 4
- 210000004153 islets of langerhan Anatomy 0.000 description 4
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- 230000007774 longterm Effects 0.000 description 4
- 239000000314 lubricant Substances 0.000 description 4
- 239000013642 negative control Substances 0.000 description 4
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 4
- 230000008782 phagocytosis Effects 0.000 description 4
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 4
- 229920000917 poly(propylene sulfide) Polymers 0.000 description 4
- 239000013641 positive control Substances 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- 229960004063 propylene glycol Drugs 0.000 description 4
- 230000007480 spreading Effects 0.000 description 4
- 238000003892 spreading Methods 0.000 description 4
- 238000010186 staining Methods 0.000 description 4
- 235000019698 starch Nutrition 0.000 description 4
- 239000004094 surface-active agent Substances 0.000 description 4
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 230000024664 tolerance induction Effects 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 3
- 102100034540 Adenomatous polyposis coli protein Human genes 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- 235000017060 Arachis glabrata Nutrition 0.000 description 3
- 244000105624 Arachis hypogaea Species 0.000 description 3
- 235000010777 Arachis hypogaea Nutrition 0.000 description 3
- 235000018262 Arachis monticola Nutrition 0.000 description 3
- 108091026890 Coding region Proteins 0.000 description 3
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 3
- 208000016192 Demyelinating disease Diseases 0.000 description 3
- 206010012305 Demyelination Diseases 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- 108010011459 Exenatide Proteins 0.000 description 3
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 description 3
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 3
- 101000924577 Homo sapiens Adenomatous polyposis coli protein Proteins 0.000 description 3
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 3
- 102100030703 Interleukin-22 Human genes 0.000 description 3
- 206010052904 Musculoskeletal stiffness Diseases 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- 208000017787 Paraneoplastic neurologic syndrome Diseases 0.000 description 3
- 229920002732 Polyanhydride Polymers 0.000 description 3
- 230000006044 T cell activation Effects 0.000 description 3
- 108091008874 T cell receptors Proteins 0.000 description 3
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 3
- 241000700605 Viruses Species 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 230000002776 aggregation Effects 0.000 description 3
- 238000004220 aggregation Methods 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 125000004103 aminoalkyl group Chemical group 0.000 description 3
- 230000003110 anti-inflammatory effect Effects 0.000 description 3
- 230000005875 antibody response Effects 0.000 description 3
- 239000003963 antioxidant agent Substances 0.000 description 3
- 235000006708 antioxidants Nutrition 0.000 description 3
- 125000003118 aryl group Chemical group 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 229960002685 biotin Drugs 0.000 description 3
- 235000020958 biotin Nutrition 0.000 description 3
- 239000011616 biotin Substances 0.000 description 3
- 239000006172 buffering agent Substances 0.000 description 3
- 238000005119 centrifugation Methods 0.000 description 3
- JUFFVKRROAPVBI-PVOYSMBESA-N chembl1210015 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N[C@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@]3(O[C@@H](C[C@H](O)[C@H](O)CO)[C@H](NC(C)=O)[C@@H](O)C3)C(O)=O)O2)O)[C@@H](CO)O1)NC(C)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 JUFFVKRROAPVBI-PVOYSMBESA-N 0.000 description 3
- 238000007334 copolymerization reaction Methods 0.000 description 3
- 230000000139 costimulatory effect Effects 0.000 description 3
- VBVAVBCYMYWNOU-UHFFFAOYSA-N coumarin 6 Chemical compound C1=CC=C2SC(C3=CC4=CC=C(C=C4OC3=O)N(CC)CC)=NC2=C1 VBVAVBCYMYWNOU-UHFFFAOYSA-N 0.000 description 3
- GVJHHUAWPYXKBD-UHFFFAOYSA-N d-alpha-tocopherol Natural products OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 3
- 230000034994 death Effects 0.000 description 3
- 235000014113 dietary fatty acids Nutrition 0.000 description 3
- 239000003085 diluting agent Substances 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- JSEJGUNDTVASLD-UHFFFAOYSA-N ethene;methanediimine Chemical compound C=C.N=C=N JSEJGUNDTVASLD-UHFFFAOYSA-N 0.000 description 3
- 229960001519 exenatide Drugs 0.000 description 3
- 239000000194 fatty acid Substances 0.000 description 3
- 229930195729 fatty acid Natural products 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 235000013305 food Nutrition 0.000 description 3
- 238000005194 fractionation Methods 0.000 description 3
- 210000001035 gastrointestinal tract Anatomy 0.000 description 3
- 230000009395 genetic defect Effects 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 125000001188 haloalkyl group Chemical group 0.000 description 3
- 230000036039 immunity Effects 0.000 description 3
- 239000007943 implant Substances 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 230000000977 initiatory effect Effects 0.000 description 3
- 238000007912 intraperitoneal administration Methods 0.000 description 3
- 238000002955 isolation Methods 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 230000001926 lymphatic effect Effects 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 239000004530 micro-emulsion Substances 0.000 description 3
- 210000000274 microglia Anatomy 0.000 description 3
- 239000011859 microparticle Substances 0.000 description 3
- 210000005087 mononuclear cell Anatomy 0.000 description 3
- 230000003472 neutralizing effect Effects 0.000 description 3
- 230000001575 pathological effect Effects 0.000 description 3
- 235000020232 peanut Nutrition 0.000 description 3
- 230000000704 physical effect Effects 0.000 description 3
- 229920000728 polyester Polymers 0.000 description 3
- 230000000770 proinflammatory effect Effects 0.000 description 3
- 230000012743 protein tagging Effects 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 150000003384 small molecules Chemical class 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 239000008247 solid mixture Substances 0.000 description 3
- 230000006641 stabilisation Effects 0.000 description 3
- 238000011105 stabilization Methods 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 208000011580 syndromic disease Diseases 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- 125000004001 thioalkyl group Chemical group 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 239000001993 wax Substances 0.000 description 3
- 239000000080 wetting agent Substances 0.000 description 3
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- 201000004384 Alopecia Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 241000894006 Bacteria Species 0.000 description 2
- 208000023328 Basedow disease Diseases 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 206010009900 Colitis ulcerative Diseases 0.000 description 2
- 229930105110 Cyclosporin A Natural products 0.000 description 2
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 2
- 108010036949 Cyclosporine Proteins 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- 206010014025 Ear swelling Diseases 0.000 description 2
- JOYRKODLDBILNP-UHFFFAOYSA-N Ethyl urethane Chemical compound CCOC(N)=O JOYRKODLDBILNP-UHFFFAOYSA-N 0.000 description 2
- 208000009386 Experimental Arthritis Diseases 0.000 description 2
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 2
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 2
- 239000004606 Fillers/Extenders Substances 0.000 description 2
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 208000009329 Graft vs Host Disease Diseases 0.000 description 2
- 208000015023 Graves' disease Diseases 0.000 description 2
- 241000711549 Hepacivirus C Species 0.000 description 2
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 2
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 2
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 2
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 2
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 206010062016 Immunosuppression Diseases 0.000 description 2
- 102100022338 Integrin alpha-M Human genes 0.000 description 2
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 2
- 108090001005 Interleukin-6 Proteins 0.000 description 2
- 102000004889 Interleukin-6 Human genes 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical group OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- 102100039648 Lactadherin Human genes 0.000 description 2
- 101710191666 Lactadherin Proteins 0.000 description 2
- 102000008192 Lactoglobulins Human genes 0.000 description 2
- 108010060630 Lactoglobulins Proteins 0.000 description 2
- 102000003960 Ligases Human genes 0.000 description 2
- 108090000364 Ligases Proteins 0.000 description 2
- LUWJPTVQOMUZLW-UHFFFAOYSA-N Luxol fast blue MBS Chemical compound [Cu++].Cc1ccccc1N\C(N)=N\c1ccccc1C.Cc1ccccc1N\C(N)=N\c1ccccc1C.OS(=O)(=O)c1cccc2c3nc(nc4nc([n-]c5[n-]c(nc6nc(n3)c3ccccc63)c3c(cccc53)S(O)(=O)=O)c3ccccc43)c12 LUWJPTVQOMUZLW-UHFFFAOYSA-N 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 2
- 201000009906 Meningitis Diseases 0.000 description 2
- 238000011789 NOD SCID mouse Methods 0.000 description 2
- 239000004677 Nylon Substances 0.000 description 2
- 206010033799 Paralysis Diseases 0.000 description 2
- 235000019483 Peanut oil Nutrition 0.000 description 2
- 206010034277 Pemphigoid Diseases 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical group [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- 235000021355 Stearic acid Nutrition 0.000 description 2
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 description 2
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 2
- 230000017274 T cell anergy Effects 0.000 description 2
- 230000005867 T cell response Effects 0.000 description 2
- 108010046722 Thrombospondin 1 Proteins 0.000 description 2
- 102100036034 Thrombospondin-1 Human genes 0.000 description 2
- 102000008235 Toll-Like Receptor 9 Human genes 0.000 description 2
- 108010060818 Toll-Like Receptor 9 Proteins 0.000 description 2
- 102000002689 Toll-like receptor Human genes 0.000 description 2
- 108020000411 Toll-like receptor Proteins 0.000 description 2
- 201000006704 Ulcerative Colitis Diseases 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 206010046851 Uveitis Diseases 0.000 description 2
- 206010047115 Vasculitis Diseases 0.000 description 2
- 101710099833 Venom protein Proteins 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000006978 adaptation Effects 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 230000001270 agonistic effect Effects 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- POJWUDADGALRAB-UHFFFAOYSA-N allantoin Chemical compound NC(=O)NC1NC(=O)NC1=O POJWUDADGALRAB-UHFFFAOYSA-N 0.000 description 2
- 235000011399 aloe vera Nutrition 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 230000003474 anti-emetic effect Effects 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 229940125683 antiemetic agent Drugs 0.000 description 2
- 239000002111 antiemetic agent Substances 0.000 description 2
- 230000030741 antigen processing and presentation Effects 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 208000037979 autoimmune inflammatory disease Diseases 0.000 description 2
- 230000006472 autoimmune response Effects 0.000 description 2
- 239000003659 bee venom Substances 0.000 description 2
- 229960002903 benzyl benzoate Drugs 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 235000019437 butane-1,3-diol Nutrition 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 239000008004 cell lysis buffer Substances 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229920002301 cellulose acetate Polymers 0.000 description 2
- 229960000541 cetyl alcohol Drugs 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- 230000001268 conjugating effect Effects 0.000 description 2
- 239000002872 contrast media Substances 0.000 description 2
- 235000005687 corn oil Nutrition 0.000 description 2
- 239000002285 corn oil Substances 0.000 description 2
- 235000012343 cottonseed oil Nutrition 0.000 description 2
- 239000002385 cottonseed oil Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 238000000354 decomposition reaction Methods 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 239000000412 dendrimer Substances 0.000 description 2
- 229920000736 dendritic polymer Polymers 0.000 description 2
- 239000003599 detergent Substances 0.000 description 2
- 239000010432 diamond Substances 0.000 description 2
- 229910003460 diamond Inorganic materials 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- 230000002222 downregulating effect Effects 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 238000012377 drug delivery Methods 0.000 description 2
- 210000005069 ears Anatomy 0.000 description 2
- 230000002500 effect on skin Effects 0.000 description 2
- 210000003162 effector t lymphocyte Anatomy 0.000 description 2
- 229920001971 elastomer Polymers 0.000 description 2
- 239000012039 electrophile Substances 0.000 description 2
- 206010014599 encephalitis Diseases 0.000 description 2
- 201000002491 encephalomyelitis Diseases 0.000 description 2
- 238000009505 enteric coating Methods 0.000 description 2
- 239000002702 enteric coating Substances 0.000 description 2
- 210000002615 epidermis Anatomy 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 210000003414 extremity Anatomy 0.000 description 2
- 210000001508 eye Anatomy 0.000 description 2
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 2
- 230000037406 food intake Effects 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 208000024908 graft versus host disease Diseases 0.000 description 2
- 210000002216 heart Anatomy 0.000 description 2
- 125000001072 heteroaryl group Chemical group 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 239000003906 humectant Substances 0.000 description 2
- 230000002631 hypothermal effect Effects 0.000 description 2
- 230000005934 immune activation Effects 0.000 description 2
- 230000004957 immunoregulator effect Effects 0.000 description 2
- 230000003308 immunostimulating effect Effects 0.000 description 2
- 230000001506 immunosuppresive effect Effects 0.000 description 2
- 239000000411 inducer Substances 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 108010074108 interleukin-21 Proteins 0.000 description 2
- 238000010212 intracellular staining Methods 0.000 description 2
- 239000007928 intraperitoneal injection Substances 0.000 description 2
- PGHMRUGBZOYCAA-ADZNBVRBSA-N ionomycin Chemical compound O1[C@H](C[C@H](O)[C@H](C)[C@H](O)[C@H](C)/C=C/C[C@@H](C)C[C@@H](C)C(/O)=C/C(=O)[C@@H](C)C[C@@H](C)C[C@@H](CCC(O)=O)C)CC[C@@]1(C)[C@@H]1O[C@](C)([C@@H](C)O)CC1 PGHMRUGBZOYCAA-ADZNBVRBSA-N 0.000 description 2
- PGHMRUGBZOYCAA-UHFFFAOYSA-N ionomycin Natural products O1C(CC(O)C(C)C(O)C(C)C=CCC(C)CC(C)C(O)=CC(=O)C(C)CC(C)CC(CCC(O)=O)C)CCC1(C)C1OC(C)(C(C)O)CC1 PGHMRUGBZOYCAA-UHFFFAOYSA-N 0.000 description 2
- 239000000644 isotonic solution Substances 0.000 description 2
- 238000002372 labelling Methods 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- 239000006194 liquid suspension Substances 0.000 description 2
- SNKMVYBWZDHJHE-UHFFFAOYSA-M lithium;dihydrogen phosphate Chemical compound [Li+].OP(O)([O-])=O SNKMVYBWZDHJHE-UHFFFAOYSA-M 0.000 description 2
- HQRPHMAXFVUBJX-UHFFFAOYSA-M lithium;hydrogen carbonate Chemical compound [Li+].OC([O-])=O HQRPHMAXFVUBJX-UHFFFAOYSA-M 0.000 description 2
- 239000006210 lotion Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 210000005015 mediastinal lymph node Anatomy 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- UQDUPQYQJKYHQI-UHFFFAOYSA-N methyl laurate Chemical compound CCCCCCCCCCCC(=O)OC UQDUPQYQJKYHQI-UHFFFAOYSA-N 0.000 description 2
- 239000004005 microsphere Substances 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 2
- 235000019796 monopotassium phosphate Nutrition 0.000 description 2
- 229910000403 monosodium phosphate Inorganic materials 0.000 description 2
- 235000019799 monosodium phosphate Nutrition 0.000 description 2
- 239000002077 nanosphere Substances 0.000 description 2
- 230000007935 neutral effect Effects 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000002736 nonionic surfactant Substances 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000000346 nonvolatile oil Substances 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 239000012038 nucleophile Substances 0.000 description 2
- 229920001778 nylon Polymers 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- 239000003605 opacifier Substances 0.000 description 2
- 230000000242 pagocytic effect Effects 0.000 description 2
- 239000012188 paraffin wax Substances 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000001717 pathogenic effect Effects 0.000 description 2
- 239000000312 peanut oil Substances 0.000 description 2
- 239000003961 penetration enhancing agent Substances 0.000 description 2
- 210000005259 peripheral blood Anatomy 0.000 description 2
- 239000011886 peripheral blood Substances 0.000 description 2
- 210000004976 peripheral blood cell Anatomy 0.000 description 2
- 229940021222 peritoneal dialysis isotonic solution Drugs 0.000 description 2
- 210000001539 phagocyte Anatomy 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 2
- 229920001983 poloxamer Polymers 0.000 description 2
- 208000005987 polymyositis Diseases 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 239000011736 potassium bicarbonate Substances 0.000 description 2
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 2
- 235000015497 potassium bicarbonate Nutrition 0.000 description 2
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 208000037821 progressive disease Diseases 0.000 description 2
- 239000003380 propellant Substances 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- 239000009342 ragweed pollen Substances 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- 108010078070 scavenger receptors Proteins 0.000 description 2
- 102000014452 scavenger receptors Human genes 0.000 description 2
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 2
- 230000003248 secreting effect Effects 0.000 description 2
- 239000008159 sesame oil Substances 0.000 description 2
- 235000011803 sesame oil Nutrition 0.000 description 2
- 235000002639 sodium chloride Nutrition 0.000 description 2
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 230000003381 solubilizing effect Effects 0.000 description 2
- 239000003549 soybean oil Substances 0.000 description 2
- 235000012424 soybean oil Nutrition 0.000 description 2
- 230000002269 spontaneous effect Effects 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000008117 stearic acid Substances 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000005846 sugar alcohols Polymers 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 238000013268 sustained release Methods 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- ABZLKHKQJHEPAX-UHFFFAOYSA-N tetramethylrhodamine Chemical compound C=12C=CC(N(C)C)=CC2=[O+]C2=CC(N(C)C)=CC=C2C=1C1=CC=CC=C1C([O-])=O ABZLKHKQJHEPAX-UHFFFAOYSA-N 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- 230000003827 upregulation Effects 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical group OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- ALSTYHKOOCGGFT-KTKRTIGZSA-N (9Z)-octadecen-1-ol Chemical compound CCCCCCCC\C=C/CCCCCCCCO ALSTYHKOOCGGFT-KTKRTIGZSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- CULQNACJHGHAER-UHFFFAOYSA-N 1-[4-[(2-iodoacetyl)amino]benzoyl]oxy-2,5-dioxopyrrolidine-3-sulfonic acid Chemical compound O=C1C(S(=O)(=O)O)CC(=O)N1OC(=O)C1=CC=C(NC(=O)CI)C=C1 CULQNACJHGHAER-UHFFFAOYSA-N 0.000 description 1
- OSSNTDFYBPYIEC-UHFFFAOYSA-N 1-ethenylimidazole Chemical compound C=CN1C=CN=C1 OSSNTDFYBPYIEC-UHFFFAOYSA-N 0.000 description 1
- NZJXADCEESMBPW-UHFFFAOYSA-N 1-methylsulfinyldecane Chemical compound CCCCCCCCCCS(C)=O NZJXADCEESMBPW-UHFFFAOYSA-N 0.000 description 1
- RZRNAYUHWVFMIP-KTKRTIGZSA-N 1-oleoylglycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-KTKRTIGZSA-N 0.000 description 1
- NHBKXEKEPDILRR-UHFFFAOYSA-N 2,3-bis(butanoylsulfanyl)propyl butanoate Chemical compound CCCC(=O)OCC(SC(=O)CCC)CSC(=O)CCC NHBKXEKEPDILRR-UHFFFAOYSA-N 0.000 description 1
- 150000003923 2,5-pyrrolediones Chemical class 0.000 description 1
- HDPLHDGYGLENEI-UHFFFAOYSA-N 2-[1-(oxiran-2-ylmethoxy)propan-2-yloxymethyl]oxirane Chemical compound C1OC1COC(C)COCC1CO1 HDPLHDGYGLENEI-UHFFFAOYSA-N 0.000 description 1
- ZVTDEEBSWIQAFJ-KHPPLWFESA-N 2-hydroxypropyl (z)-octadec-9-enoate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(C)O ZVTDEEBSWIQAFJ-KHPPLWFESA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- 238000010600 3H thymidine incorporation assay Methods 0.000 description 1
- HIQIXEFWDLTDED-UHFFFAOYSA-N 4-hydroxy-1-piperidin-4-ylpyrrolidin-2-one Chemical compound O=C1CC(O)CN1C1CCNCC1 HIQIXEFWDLTDED-UHFFFAOYSA-N 0.000 description 1
- IJJWOSAXNHWBPR-HUBLWGQQSA-N 5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]-n-(6-hydrazinyl-6-oxohexyl)pentanamide Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)NCCCCCC(=O)NN)SC[C@@H]21 IJJWOSAXNHWBPR-HUBLWGQQSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 208000020053 Abnormal inflammatory response Diseases 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 208000026872 Addison Disease Diseases 0.000 description 1
- POJWUDADGALRAB-PVQJCKRUSA-N Allantoin Natural products NC(=O)N[C@@H]1NC(=O)NC1=O POJWUDADGALRAB-PVQJCKRUSA-N 0.000 description 1
- 206010027654 Allergic conditions Diseases 0.000 description 1
- 241001116389 Aloe Species 0.000 description 1
- 244000144927 Aloe barbadensis Species 0.000 description 1
- 235000002961 Aloe barbadensis Nutrition 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 208000031873 Animal Disease Models Diseases 0.000 description 1
- 206010002556 Ankylosing Spondylitis Diseases 0.000 description 1
- 108090000663 Annexin A1 Proteins 0.000 description 1
- 102100040006 Annexin A1 Human genes 0.000 description 1
- 108090000672 Annexin A5 Proteins 0.000 description 1
- 102100034283 Annexin A5 Human genes 0.000 description 1
- 239000004475 Arginine Chemical group 0.000 description 1
- 206010003267 Arthritis reactive Diseases 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010003645 Atopy Diseases 0.000 description 1
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 description 1
- 206010003827 Autoimmune hepatitis Diseases 0.000 description 1
- 206010055128 Autoimmune neutropenia Diseases 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 206010060976 Bacillus infection Diseases 0.000 description 1
- 108010077805 Bacterial Proteins Proteins 0.000 description 1
- 208000027496 Behcet disease Diseases 0.000 description 1
- 208000008439 Biliary Liver Cirrhosis Diseases 0.000 description 1
- 208000033222 Biliary cirrhosis primary Diseases 0.000 description 1
- 239000004255 Butylated hydroxyanisole Substances 0.000 description 1
- 239000004322 Butylated hydroxytoluene Substances 0.000 description 1
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 1
- 102100032937 CD40 ligand Human genes 0.000 description 1
- 102100032912 CD44 antigen Human genes 0.000 description 1
- 101100289995 Caenorhabditis elegans mac-1 gene Proteins 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 102100032936 Carboxypeptidase M Human genes 0.000 description 1
- 108090000007 Carboxypeptidase M Proteins 0.000 description 1
- 102000005600 Cathepsins Human genes 0.000 description 1
- 108010084457 Cathepsins Proteins 0.000 description 1
- 229940123150 Chelating agent Drugs 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- 206010008609 Cholangitis sclerosing Diseases 0.000 description 1
- 102000010792 Chromogranin A Human genes 0.000 description 1
- 108010038447 Chromogranin A Proteins 0.000 description 1
- 208000030939 Chronic inflammatory demyelinating polyneuropathy Diseases 0.000 description 1
- 102000029816 Collagenase Human genes 0.000 description 1
- 108060005980 Collagenase Proteins 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 208000019707 Cryoglobulinemic vasculitis Diseases 0.000 description 1
- 229920001651 Cyanoacrylate Polymers 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 208000001840 Dandruff Diseases 0.000 description 1
- 102000016911 Deoxyribonucleases Human genes 0.000 description 1
- 108010053770 Deoxyribonucleases Proteins 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 206010061819 Disease recurrence Diseases 0.000 description 1
- 206010013647 Drowning Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 102100025137 Early activation antigen CD69 Human genes 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 1
- 206010014989 Epidermolysis bullosa Diseases 0.000 description 1
- 239000004593 Epoxy Substances 0.000 description 1
- 206010015226 Erythema nodosum Diseases 0.000 description 1
- 101000759376 Escherichia phage Mu Tail sheath protein Proteins 0.000 description 1
- IMROMDMJAWUWLK-UHFFFAOYSA-N Ethenol Chemical compound OC=C IMROMDMJAWUWLK-UHFFFAOYSA-N 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- 102000001690 Factor VIII Human genes 0.000 description 1
- 108010054218 Factor VIII Proteins 0.000 description 1
- 206010054261 Flavivirus infection Diseases 0.000 description 1
- 208000004262 Food Hypersensitivity Diseases 0.000 description 1
- 206010016946 Food allergy Diseases 0.000 description 1
- 206010017577 Gait disturbance Diseases 0.000 description 1
- 208000007465 Giant cell arteritis Diseases 0.000 description 1
- 108010061711 Gliadin Proteins 0.000 description 1
- 206010018364 Glomerulonephritis Diseases 0.000 description 1
- 102400000321 Glucagon Human genes 0.000 description 1
- 108060003199 Glucagon Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Chemical group OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 229930186217 Glycolipid Natural products 0.000 description 1
- 208000024869 Goodpasture syndrome Diseases 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 206010072579 Granulomatosis with polyangiitis Diseases 0.000 description 1
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 208000031220 Hemophilia Diseases 0.000 description 1
- 208000009292 Hemophilia A Diseases 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 208000007514 Herpes zoster Diseases 0.000 description 1
- 208000029433 Herpesviridae infectious disease Diseases 0.000 description 1
- 239000005057 Hexamethylene diisocyanate Substances 0.000 description 1
- 102000006947 Histones Human genes 0.000 description 1
- 101000868215 Homo sapiens CD40 ligand Proteins 0.000 description 1
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 1
- 101000911390 Homo sapiens Coagulation factor VIII Proteins 0.000 description 1
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 description 1
- 101000917826 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor II-a Proteins 0.000 description 1
- 101000917824 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor II-b Proteins 0.000 description 1
- 101000934372 Homo sapiens Macrosialin Proteins 0.000 description 1
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 1
- 208000000038 Hypoparathyroidism Diseases 0.000 description 1
- 206010021067 Hypopituitarism Diseases 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 206010021639 Incontinence Diseases 0.000 description 1
- 102100022339 Integrin alpha-L Human genes 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010065637 Interleukin-23 Proteins 0.000 description 1
- 102000013264 Interleukin-23 Human genes 0.000 description 1
- 208000011200 Kawasaki disease Diseases 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical group SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical group NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical group OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical group C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical group OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 208000034624 Leukocytoclastic Cutaneous Vasculitis Diseases 0.000 description 1
- 208000032514 Leukocytoclastic vasculitis Diseases 0.000 description 1
- 208000012309 Linear IgA disease Diseases 0.000 description 1
- 102100029204 Low affinity immunoglobulin gamma Fc region receptor II-a Human genes 0.000 description 1
- 108010064548 Lymphocyte Function-Associated Antigen-1 Proteins 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 102000043131 MHC class II family Human genes 0.000 description 1
- 108091054438 MHC class II family Proteins 0.000 description 1
- 102100025136 Macrosialin Human genes 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- MWCLLHOVUTZFKS-UHFFFAOYSA-N Methyl cyanoacrylate Chemical compound COC(=O)C(=C)C#N MWCLLHOVUTZFKS-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 1
- 108010058682 Mitochondrial Proteins Proteins 0.000 description 1
- 102000006404 Mitochondrial Proteins Human genes 0.000 description 1
- 208000003250 Mixed connective tissue disease Diseases 0.000 description 1
- 239000004909 Moisturizer Substances 0.000 description 1
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 206010028372 Muscular weakness Diseases 0.000 description 1
- 206010062207 Mycobacterial infection Diseases 0.000 description 1
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 1
- 102000006386 Myelin Proteins Human genes 0.000 description 1
- 108010083674 Myelin Proteins Proteins 0.000 description 1
- 208000009525 Myocarditis Diseases 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 description 1
- 206010062212 Neisseria infection Diseases 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 206010029155 Nephropathy toxic Diseases 0.000 description 1
- 102000019040 Nuclear Antigens Human genes 0.000 description 1
- 108010051791 Nuclear Antigens Proteins 0.000 description 1
- 208000005225 Opsoclonus-Myoclonus Syndrome Diseases 0.000 description 1
- BPQQTUXANYXVAA-UHFFFAOYSA-N Orthosilicate Chemical compound [O-][Si]([O-])([O-])[O-] BPQQTUXANYXVAA-UHFFFAOYSA-N 0.000 description 1
- 206010068786 Overlap syndrome Diseases 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 208000009608 Papillomavirus Infections Diseases 0.000 description 1
- 208000002606 Paramyxoviridae Infections Diseases 0.000 description 1
- 208000008267 Peanut Hypersensitivity Diseases 0.000 description 1
- 208000008223 Pemphigoid Gestationis Diseases 0.000 description 1
- 201000011152 Pemphigus Diseases 0.000 description 1
- 241000721454 Pemphigus Species 0.000 description 1
- 208000031845 Pernicious anaemia Diseases 0.000 description 1
- 108010081690 Pertussis Toxin Proteins 0.000 description 1
- 239000004264 Petrolatum Substances 0.000 description 1
- BELBBZDIHDAJOR-UHFFFAOYSA-N Phenolsulfonephthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2S(=O)(=O)O1 BELBBZDIHDAJOR-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 101800001357 Potential peptide Proteins 0.000 description 1
- 102400000745 Potential peptide Human genes 0.000 description 1
- 208000002500 Primary Ovarian Insufficiency Diseases 0.000 description 1
- 208000012654 Primary biliary cholangitis Diseases 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 108700008625 Reporter Genes Proteins 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- 206010038997 Retroviral infections Diseases 0.000 description 1
- 206010061494 Rhinovirus infection Diseases 0.000 description 1
- 108010000605 Ribosomal Proteins Proteins 0.000 description 1
- 102000002278 Ribosomal Proteins Human genes 0.000 description 1
- 238000011579 SCID mouse model Methods 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 206010039438 Salmonella Infections Diseases 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 206010070834 Sensitisation Diseases 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Chemical group OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 108010071390 Serum Albumin Proteins 0.000 description 1
- 102000007562 Serum Albumin Human genes 0.000 description 1
- 108010029176 Sialic Acid Binding Ig-like Lectin 1 Proteins 0.000 description 1
- 102100032855 Sialoadhesin Human genes 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 241000589970 Spirochaetales Species 0.000 description 1
- 206010041925 Staphylococcal infections Diseases 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 206010061372 Streptococcal infection Diseases 0.000 description 1
- 238000000692 Student's t-test Methods 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 1
- 208000001106 Takayasu Arteritis Diseases 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- 210000000447 Th1 cell Anatomy 0.000 description 1
- 210000004241 Th2 cell Anatomy 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Chemical group CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Chemical group 0.000 description 1
- 206010043561 Thrombocytopenic purpura Diseases 0.000 description 1
- 102000002938 Thrombospondin Human genes 0.000 description 1
- 108060008245 Thrombospondin Proteins 0.000 description 1
- 102100029337 Thyrotropin receptor Human genes 0.000 description 1
- 101710114011 Thyrotropin receptor Proteins 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 1
- 102100031988 Tumor necrosis factor ligand superfamily member 6 Human genes 0.000 description 1
- 108050002568 Tumor necrosis factor ligand superfamily member 6 Proteins 0.000 description 1
- 206010053613 Type IV hypersensitivity reaction Diseases 0.000 description 1
- 206010047400 Vibrio infections Diseases 0.000 description 1
- BZHJMEDXRYGGRV-UHFFFAOYSA-N Vinyl chloride Chemical compound ClC=C BZHJMEDXRYGGRV-UHFFFAOYSA-N 0.000 description 1
- 108010067390 Viral Proteins Proteins 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- 206010047642 Vitiligo Diseases 0.000 description 1
- 206010057293 West Nile viral infection Diseases 0.000 description 1
- 241000710886 West Nile virus Species 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 229940124532 absorption promoter Drugs 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 208000002552 acute disseminated encephalomyelitis Diseases 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 230000033289 adaptive immune response Effects 0.000 description 1
- 238000001042 affinity chromatography Methods 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 229960000458 allantoin Drugs 0.000 description 1
- 230000000172 allergic effect Effects 0.000 description 1
- 230000000961 alloantigen Effects 0.000 description 1
- 230000000735 allogeneic effect Effects 0.000 description 1
- 231100000360 alopecia Toxicity 0.000 description 1
- 208000004631 alopecia areata Diseases 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 238000000540 analysis of variance Methods 0.000 description 1
- 210000003484 anatomy Anatomy 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 238000011558 animal model by disease Methods 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 239000000730 antalgic agent Substances 0.000 description 1
- 230000003092 anti-cytokine Effects 0.000 description 1
- 230000003409 anti-rejection Effects 0.000 description 1
- 229940124691 antibody therapeutics Drugs 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Chemical group OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- XKRFYHLGVUSROY-UHFFFAOYSA-N argon Substances [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 210000001130 astrocyte Anatomy 0.000 description 1
- 208000010216 atopic IgE responsiveness Diseases 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 208000037896 autoimmune cutaneous disease Diseases 0.000 description 1
- 201000004982 autoimmune uveitis Diseases 0.000 description 1
- 210000001099 axilla Anatomy 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 239000003637 basic solution Substances 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Chemical group OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 210000000013 bile duct Anatomy 0.000 description 1
- 230000009141 biological interaction Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 210000002449 bone cell Anatomy 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- 208000000594 bullous pemphigoid Diseases 0.000 description 1
- DQXBYHZEEUGOBF-UHFFFAOYSA-N but-3-enoic acid;ethene Chemical compound C=C.OC(=O)CC=C DQXBYHZEEUGOBF-UHFFFAOYSA-N 0.000 description 1
- 235000019282 butylated hydroxyanisole Nutrition 0.000 description 1
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 1
- 229940043253 butylated hydroxyanisole Drugs 0.000 description 1
- 235000010354 butylated hydroxytoluene Nutrition 0.000 description 1
- 229940095259 butylated hydroxytoluene Drugs 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 230000021523 carboxylation Effects 0.000 description 1
- 238000006473 carboxylation reaction Methods 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 238000001516 cell proliferation assay Methods 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 230000008614 cellular interaction Effects 0.000 description 1
- 230000036755 cellular response Effects 0.000 description 1
- 210000003850 cellular structure Anatomy 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 208000015114 central nervous system disease Diseases 0.000 description 1
- 208000025434 cerebellar degeneration Diseases 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 230000009920 chelation Effects 0.000 description 1
- 238000001311 chemical methods and process Methods 0.000 description 1
- 108091006116 chimeric peptides Proteins 0.000 description 1
- 201000005795 chronic inflammatory demyelinating polyneuritis Diseases 0.000 description 1
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 description 1
- 239000007979 citrate buffer Substances 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 229960002424 collagenase Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 239000012050 conventional carrier Substances 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 238000010219 correlation analysis Methods 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 201000003278 cryoglobulinemia Diseases 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Chemical group SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 230000016396 cytokine production Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 230000000779 depleting effect Effects 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- 210000004207 dermis Anatomy 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 238000001085 differential centrifugation Methods 0.000 description 1
- 229940008099 dimethicone Drugs 0.000 description 1
- 239000004205 dimethyl polysiloxane Substances 0.000 description 1
- 235000013870 dimethyl polysiloxane Nutrition 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000003651 drinking water Substances 0.000 description 1
- 235000020188 drinking water Nutrition 0.000 description 1
- 238000007877 drug screening Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 239000003221 ear drop Substances 0.000 description 1
- 229940047652 ear drops Drugs 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003974 emollient agent Substances 0.000 description 1
- 238000004945 emulsification Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000003344 environmental pollutant Substances 0.000 description 1
- 238000002641 enzyme replacement therapy Methods 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 229940093499 ethyl acetate Drugs 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 239000005038 ethylene vinyl acetate Substances 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 208000012997 experimental autoimmune encephalomyelitis Diseases 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 229960000301 factor viii Drugs 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000834 fixative Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 1
- XUCNUKMRBVNAPB-UHFFFAOYSA-N fluoroethene Chemical compound FC=C XUCNUKMRBVNAPB-UHFFFAOYSA-N 0.000 description 1
- NBVXSUQYWXRMNV-UHFFFAOYSA-N fluoromethane Chemical compound FC NBVXSUQYWXRMNV-UHFFFAOYSA-N 0.000 description 1
- 239000013568 food allergen Substances 0.000 description 1
- 235000020932 food allergy Nutrition 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 238000010353 genetic engineering Methods 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 1
- 229960004666 glucagon Drugs 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 239000004220 glutamic acid Chemical group 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 229960005150 glycerol Drugs 0.000 description 1
- RZRNAYUHWVFMIP-HXUWFJFHSA-N glycerol monolinoleate Natural products CCCCCCCCC=CCCCCCCCC(=O)OC[C@H](O)CO RZRNAYUHWVFMIP-HXUWFJFHSA-N 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 210000004013 groin Anatomy 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 230000003676 hair loss Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000002443 helper t lymphocyte Anatomy 0.000 description 1
- 238000007490 hematoxylin and eosin (H&E) staining Methods 0.000 description 1
- 208000007475 hemolytic anemia Diseases 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- RRAMGCGOFNQTLD-UHFFFAOYSA-N hexamethylene diisocyanate Chemical compound O=C=NCCCCCCN=C=O RRAMGCGOFNQTLD-UHFFFAOYSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 238000010562 histological examination Methods 0.000 description 1
- 102000057593 human F8 Human genes 0.000 description 1
- 229960000900 human factor viii Drugs 0.000 description 1
- 230000008348 humoral response Effects 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- DCPMPXBYPZGNDC-UHFFFAOYSA-N hydron;methanediimine;chloride Chemical compound Cl.N=C=N DCPMPXBYPZGNDC-UHFFFAOYSA-N 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 125000001165 hydrophobic group Chemical group 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 201000006362 hypersensitivity vasculitis Diseases 0.000 description 1
- 208000003532 hypothyroidism Diseases 0.000 description 1
- 230000002989 hypothyroidism Effects 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- 230000002519 immonomodulatory effect Effects 0.000 description 1
- 230000008102 immune modulation Effects 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 230000002998 immunogenetic effect Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 229940124589 immunosuppressive drug Drugs 0.000 description 1
- 238000002650 immunosuppressive therapy Methods 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- 210000002602 induced regulatory T cell Anatomy 0.000 description 1
- 230000006882 induction of apoptosis Effects 0.000 description 1
- 208000000509 infertility Diseases 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 231100000535 infertility Toxicity 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 210000002074 inflammatory monocyte Anatomy 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 108091005434 innate immune receptors Proteins 0.000 description 1
- 230000015788 innate immune response Effects 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 230000004073 interleukin-2 production Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000002510 keratinocyte Anatomy 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000011813 knockout mouse model Methods 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 210000001821 langerhans cell Anatomy 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- 208000027905 limb weakness Diseases 0.000 description 1
- 231100000861 limb weakness Toxicity 0.000 description 1
- 210000005228 liver tissue Anatomy 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 210000003141 lower extremity Anatomy 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 239000002082 metal nanoparticle Substances 0.000 description 1
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 238000001000 micrograph Methods 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 230000001333 moisturizer Effects 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 210000002864 mononuclear phagocyte Anatomy 0.000 description 1
- 210000000865 mononuclear phagocyte system Anatomy 0.000 description 1
- 208000001725 mucocutaneous lymph node syndrome Diseases 0.000 description 1
- 206010065579 multifocal motor neuropathy Diseases 0.000 description 1
- 230000036473 myasthenia Effects 0.000 description 1
- 206010028417 myasthenia gravis Diseases 0.000 description 1
- 208000027531 mycobacterial infectious disease Diseases 0.000 description 1
- 210000005012 myelin Anatomy 0.000 description 1
- 210000000066 myeloid cell Anatomy 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- 239000002159 nanocrystal Substances 0.000 description 1
- 210000001989 nasopharynx Anatomy 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 230000007694 nephrotoxicity Effects 0.000 description 1
- 231100000417 nephrotoxicity Toxicity 0.000 description 1
- 210000004498 neuroglial cell Anatomy 0.000 description 1
- 201000001119 neuropathy Diseases 0.000 description 1
- 230000007823 neuropathy Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 235000014571 nuts Nutrition 0.000 description 1
- 229940060184 oil ingredients Drugs 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 235000021313 oleic acid Nutrition 0.000 description 1
- 229940055577 oleyl alcohol Drugs 0.000 description 1
- XMLQWXUVTXCDDL-UHFFFAOYSA-N oleyl alcohol Natural products CCCCCCC=CCCCCCCCCCCO XMLQWXUVTXCDDL-UHFFFAOYSA-N 0.000 description 1
- 210000004248 oligodendroglia Anatomy 0.000 description 1
- 238000001543 one-way ANOVA Methods 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007524 organic acids Chemical group 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000002905 orthoesters Chemical class 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 239000006174 pH buffer Substances 0.000 description 1
- 238000002638 palliative care Methods 0.000 description 1
- 206010057056 paraneoplastic pemphigus Diseases 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 238000003921 particle size analysis Methods 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 210000001716 patrolling monocyte Anatomy 0.000 description 1
- 201000010853 peanut allergy Diseases 0.000 description 1
- 201000001976 pemphigus vulgaris Diseases 0.000 description 1
- 230000037368 penetrate the skin Effects 0.000 description 1
- 231100000435 percutaneous penetration Toxicity 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 235000019271 petrolatum Nutrition 0.000 description 1
- 229940066842 petrolatum Drugs 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 231100000719 pollutant Toxicity 0.000 description 1
- 229920000233 poly(alkylene oxides) Polymers 0.000 description 1
- 229920000435 poly(dimethylsiloxane) Polymers 0.000 description 1
- 229920001483 poly(ethyl methacrylate) polymer Polymers 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 229940065514 poly(lactide) Drugs 0.000 description 1
- 229920002492 poly(sulfone) Polymers 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229920000447 polyanionic polymer Polymers 0.000 description 1
- 201000006292 polyarteritis nodosa Diseases 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 229920000570 polyether Polymers 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229940068918 polyethylene glycol 400 Drugs 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 150000004804 polysaccharides Chemical class 0.000 description 1
- 229950008882 polysorbate Drugs 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 206010036601 premature menopause Diseases 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 230000037452 priming Effects 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 238000006862 quantum yield reaction Methods 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 208000002574 reactive arthritis Diseases 0.000 description 1
- 230000021419 recognition of apoptotic cell Effects 0.000 description 1
- 238000005215 recombination Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000012858 resilient material Substances 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 239000005060 rubber Substances 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 206010039447 salmonellosis Diseases 0.000 description 1
- 201000000306 sarcoidosis Diseases 0.000 description 1
- 235000003441 saturated fatty acids Nutrition 0.000 description 1
- 150000004671 saturated fatty acids Chemical class 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 210000003786 sclera Anatomy 0.000 description 1
- 208000010157 sclerosing cholangitis Diseases 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000008313 sensitization Effects 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 229940126586 small molecule drug Drugs 0.000 description 1
- 229940001607 sodium bisulfite Drugs 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 238000000935 solvent evaporation Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000003393 splenic effect Effects 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 210000000434 stratum corneum Anatomy 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 229960005404 sulfamethoxazole Drugs 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- JLKIGFTWXXRPMT-UHFFFAOYSA-N sulphamethoxazole Chemical compound O1C(C)=CC(NS(=O)(=O)C=2C=CC(N)=CC=2)=N1 JLKIGFTWXXRPMT-UHFFFAOYSA-N 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 229920003051 synthetic elastomer Polymers 0.000 description 1
- 239000005061 synthetic rubber Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 238000012385 systemic delivery Methods 0.000 description 1
- 206010043207 temporal arteritis Diseases 0.000 description 1
- BFKJFAAPBSQJPD-UHFFFAOYSA-N tetrafluoroethene Chemical compound FC(F)=C(F)F BFKJFAAPBSQJPD-UHFFFAOYSA-N 0.000 description 1
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 230000002992 thymic effect Effects 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 210000002303 tibia Anatomy 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- 235000010384 tocopherol Nutrition 0.000 description 1
- 229930003799 tocopherol Natural products 0.000 description 1
- 229960001295 tocopherol Drugs 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- IEDVJHCEMCRBQM-UHFFFAOYSA-N trimethoprim Chemical compound COC1=C(OC)C(OC)=CC(CC=2C(=NC(N)=NC=2)N)=C1 IEDVJHCEMCRBQM-UHFFFAOYSA-N 0.000 description 1
- 229960001082 trimethoprim Drugs 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 230000005951 type IV hypersensitivity Effects 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Chemical group OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 241000712461 unidentified influenza virus Species 0.000 description 1
- 150000004670 unsaturated fatty acids Chemical class 0.000 description 1
- 235000021122 unsaturated fatty acids Nutrition 0.000 description 1
- 210000000689 upper leg Anatomy 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 229920003169 water-soluble polymer Polymers 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 229960001296 zinc oxide Drugs 0.000 description 1
- 235000014692 zinc oxide Nutrition 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
- PAPBSGBWRJIAAV-UHFFFAOYSA-N ε-Caprolactone Chemical compound O=C1CCCCCO1 PAPBSGBWRJIAAV-UHFFFAOYSA-N 0.000 description 1
Images

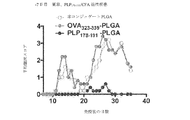

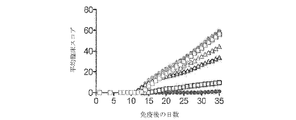

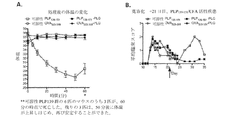





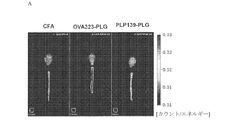


















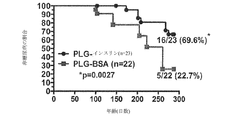













Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0008—Antigens related to auto-immune diseases; Preparations to induce self-tolerance
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/001—Preparations to induce tolerance to non-self, e.g. prior to transplantation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/35—Allergens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/385—Haptens or antigens, bound to carriers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/69—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit
- A61K47/6921—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere
- A61K47/6927—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere the form being a solid microparticle having no hollow or gas-filled cores
- A61K47/6929—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere the form being a solid microparticle having no hollow or gas-filled cores the form being a nanoparticle, e.g. an immuno-nanoparticle
- A61K47/6931—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere the form being a solid microparticle having no hollow or gas-filled cores the form being a nanoparticle, e.g. an immuno-nanoparticle the material constituting the nanoparticle being a polymer
- A61K47/6935—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere the form being a solid microparticle having no hollow or gas-filled cores the form being a nanoparticle, e.g. an immuno-nanoparticle the material constituting the nanoparticle being a polymer the polymer being obtained otherwise than by reactions involving carbon to carbon unsaturated bonds, e.g. polyesters, polyamides or polyglycerol
- A61K47/6937—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the conjugate being characterised by physical or galenical forms, e.g. emulsion, particle, inclusion complex, stent or kit the form being a particulate, a powder, an adsorbate, a bead or a sphere the form being a solid microparticle having no hollow or gas-filled cores the form being a nanoparticle, e.g. an immuno-nanoparticle the material constituting the nanoparticle being a polymer the polymer being obtained otherwise than by reactions involving carbon to carbon unsaturated bonds, e.g. polyesters, polyamides or polyglycerol the polymer being PLGA, PLA or polyglycolic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
- A61K9/1271—Non-conventional liposomes, e.g. PEGylated liposomes, liposomes coated with polymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1641—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poloxamers
- A61K9/1647—Polyesters, e.g. poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
- A61K2039/55511—Organic adjuvants
- A61K2039/55555—Liposomes; Vesicles, e.g. nanoparticles; Spheres, e.g. nanospheres; Polymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/57—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2
- A61K2039/577—Medicinal preparations containing antigens or antibodies characterised by the type of response, e.g. Th1, Th2 tolerising response
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/60—Medicinal preparations containing antigens or antibodies characteristics by the carrier linked to the antigen
- A61K2039/6093—Synthetic polymers, e.g. polyethyleneglycol [PEG], Polymers or copolymers of (D) glutamate and (D) lysine
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Mycology (AREA)
- Microbiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nanotechnology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Dispersion Chemistry (AREA)
- Transplantation (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Rheumatology (AREA)
- Pulmonology (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Inorganic Chemistry (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
Description
本出願は、2012年6月21日出願の米国仮特許出願第61/662,687号の利益を主張するものであり、参照により、その全体が本明細書に組み込まれる。
本発明は、米国国立衛生研究所(National Institutes of Health)から授与されたR01 EB013198の下、政府の支援を受けてなされたものである。米国政府は、本発明において一定の権利を有する。
キメラマウスの生成
6〜8週齢のB6.SJL−PtprcaPep3b/BoyJ(CD45.1)マウスを950ラドの1回線量で照射した。12時間後、C57BL/6−7.2fms−EGFPドナーからの107骨髄細胞でマウスを再構成した。照射後10日間は、飲料水中のスルファメトキサゾール(Sigma Aldrich)およびトリメトプリム(Sigma Aldrich)をマウスに与えた。上述のように、照射の6週間後にマウスをWNVに感染させた。フローサイトメトリーを使用してキメラ化を確認したところ、以前に実証されたように、常にドナー起源の96〜99%であることが分かった。(Getts et al.,J Neurochem.103:1019,2007)。
マウスを麻酔し、50mLの滅菌PBSで灌流した。パラフィンブロックに処理した心臓を除いて(Getts et al.,J.Neurochem 103:10919−1030,2007)、全ての臓器を単離し、Optimum Cutting Temperatureコンパウンド(OCT;Tissue−Tek、日本、東京都)中で瞬間凍結させた。8ミクロンの組織切片を、クリオスタットミクロトーム上で切断し、一晩空気乾燥させた後、必要になるまで−80℃で保存した。凍結切片を解凍し、組織学的検査(標準的なヘマトキシリン およびエオシン染色)または免疫組織化学的検査を行った(Getts et al.,J.Exp Med 205:2319−2337,2008)。MARCO、SIGN−R1およびSIGLEC−1(R&D Systems,MN,USA)、CD68(Abcam,MA,USA)、ならびにKi67(Abcam)に対する抗体を示される通りに使用した。DP−70カメラおよびDPマネージャー2.2.1ソフトウェア(Olympus、日本、東京)を使用して、Olympus BX−51顕微鏡上で画像を取得した。
DP−70カメラおよびDPマネージャー2.2.1ソフトウェア(Olympus)を使用して、Olympus BX−51顕微鏡(Olympus、日本)上で画像を取得した。
以前に記載されているように(Getts et al,J Exp Med.29:2319,2007)、デオキシ−リボヌクレアーゼ(0.005 g/ml;Sigma Aldrich)およびコラゲナーゼ IV(0.05 g/ml;Sigma Aldrich)を含むPBS中、37℃で60分間脳を消化することにより、PBSで灌流したマウスの脳から白血球を得た。10%FCSで消化を停止し、70μmナイロン製細胞濾過器にホモジネートを通過させた(Becton Dickinson、NJ,USA)。340xgで10分間遠心分離した後に得られたペレットを、30% Percoll(Amersham,Norway)中に再懸濁し、80% Percollに重層した。1140xgで25分間室温で遠心分離した後、30%/80% 界面から白血球を回収した。同じプロトコルは、肝臓から白血球を得るためにも用いられ、処理前に組織の重量が計測される。
フローサイトメトリー分析のために、右大腿を切除し、PBSを充填した注射器を使用して骨髄細胞を洗い流した。骨髄前駆体の単離のために、少なくとも4匹のマウスからの大腿および脛骨を用いた。洗い流した後に得られた細胞懸濁液を、70μmの細胞濾過器を通して濾過し、340gで5分間遠心分離した。得られたペレット中の赤血球を、NH4Clベースの赤血球溶解緩衝剤(BD Pharm Lyse(商標);BD Pharmingen)中に溶解した後、340xgで5分間遠心分離した。末梢血の場合、心穿刺により血液を採取し、直ちにクエン酸緩衝液(mMol、Sigma Alrich)中に移した。得られた懸濁液を70% Percoll に重層し、ブレーキをオフにして、室温で20分間1140xgで遠心分離した。界面を回収し、PBS中で細胞を1回洗浄し、340xgで遠心分離した。脾臓の白血球を単離するために、7070μm細胞濾過機に脾臓を通過させ、340gで5分間遠心分離した。得られたペレット中の赤血球を、NH4Clベースの赤血球溶解緩衝剤(BD Pharm Lyse(商標);BD Pharmingen)中に溶解した後、340xgで5分間遠心分離した。
脳、肝臓、血液、および骨髄から(上述のように)採取した細胞を、PBSで洗浄し、抗CD16/CD32 抗体(Biolegend)でブロックした。トリパンブルー排除法を用いて生存細胞を計数したところ、常に>95%の細胞生存率を示していた。
養子移入と称される活性疾患の第2のモデルを調査するために、本発明の実施形態の開発中に実験を行った。動物をペプチドで免疫する代わりに、活性疾患を有するマウスの脾臓からのリンパ球をレシピエントに移植し、その後疾患を発症させた。PLGナノ粒子が、養子性に移植した活性化エフェクター細胞を不活性化する能力を特徴づけるために、本発明の実施形態の開発中に実験を行った。対照ペプチドと結合した粒子または脾細胞で処理したマウスには、4日目に始まって臨床スコアの増加が見られた。PLG−PLP139〜151粒子で処理したマウスは、2日目に、2つの時点を除く全ての時点で40日目までずっと平均臨床スコア0を有し、それらの他の時点での平均臨床スコアは0.25であった。
製造者の指示(Quansys Biosciences、Logan,Utah,USA)に従って多重化プレートELISAを行った。端的に述べると、脳、脾臓、および肝臓組織をPBS中でホモジナイズし、1000xgで回転させて清澄させ、アッセイを行うまで−20℃で保存した。血清試料も使用した。提供された緩衝液中で解凍した試料および標準物質を希釈し、各々30μlを、それぞれが特定の可溶性タンパク質に対する捕捉抗体を含有する16個のスポットを含む各ウェルに播種した。次いで、軌道振盪器上で、プレートを120rpmで1時間インキュベートした。プレートを3回洗浄し、30μlの検出抗体を各ウェルに加え、さらに1時間インキュベートした。3回洗浄した後、ストレプアビジン−HRPを加え、さらに15分間インキュベートした。次いで、プレートを6回洗浄し、基質混合物を加えた。CCDイメージャ(Kodak、Rochester NY,USA)上でプレートを直ちに読み取った。Quansys Q−viewソフトウェア(Quansys Biosciences)を使用してプレートの画像を分析した。
0.1mgのMOGペプチド(MEVGWYRSPFSRVVHLYRNGK(配列番号1);Auspep、Parkville,Victoria,Australia;>95%HPLC精製)を含有するエマルション、および2mg/mLの結核菌(Sigma Aldrich)を含有するフロイント完全アジュバントをマウスに皮下注射した。2日後、マウスに、500μlの百日咳毒素(Sigma Aldrich)を腹腔内投与した。マウスを疾患進行について監視し、以下のスケールで等級分けした:1、尾の弛緩および/または片後肢の脱力;2、複数肢の脱力、歩行障害;3、単一肢の麻痺;4、複数肢の麻痺、失禁;5、瀕死。
グラフを作成し、GraphPad PrismおよびInStatにおいてそれぞれコンピュータ統計的分析を行った(どちらのプログラムもGraphPad software、San Diego,CA,USAから)。データに応じて、対応のない両側スチューデントt検定、またはTukey−Kramerの事後検定を用いた一元配置ANOVA ANOVAを行い、P<0.05を有意と見なした。
負電荷を帯びた免疫修飾性粒子(IMP)の調製
D2O中のポリ(エチレン−無水マレイン酸)(PEMA)の溶液(4mL、1% w/v)に、ジクロロメタン(DCM)中のポリ(ラクチド−co−グリコール酸)(PLG)の溶液(2mL、20% w/v)を滴下で加えた。VC 30 Ultrasonic Processorを使用して、氷上で30秒間16ワットで混合物を超音波処理した。得られたホモジナイズされた粗生成物を、次いで、D2Oの溶液(0.5% w/vのPEMAを含有する200mL)に注ぎ入れた。Bellco Glass,Inc.のBellstir Multi−stir 9 磁気攪拌機を使用して、3.5の速度設定で、ホモジナイズされたスラリを一晩撹拌させた(10Wで10秒間、16Wで10秒間、16Wで30秒間)。
3時間の撹拌後、使い捨てポリスチレン製キュベット中で動的光散乱法を用いて粒径分析を行った。
a.10W、10秒−Z−平均=499.9nm−PdI=0.23、ピーク=634.5nm
b.16W、10秒−Z−平均=528.9nm−PdI=0.227、ピーク=657.5nm
c.16W、30秒−Z−平均=471.6nm−PdI=0.228、ピーク=580.5nm
d.16W、60秒−Z−平均=491.1nm−PdI=0.275、ピーク=600.8nm
新しいD2Oおよび10x炭酸水素ナトリウム緩衝液を一晩4℃で冷却した。40μm細胞濾過器を使用して、各バッチから36mLの粒子懸濁液を濾過し、冷10x炭酸水素ナトリウム緩衝液4mLを含む適切に標示した50mL遠心管内に入れた。各ビーカーから、そのような管を約6本作り出した。全ての管を4℃で15分間7000gで遠心分離し、上清を吸引した。上記手順を用いて懸濁液の調製を繰り返し、できる限り多くの粒子ペレットを1mLの冷D2Oに懸濁した。

抗原を結合させたPLGAビーズの投与は、再発性実験的自己免疫性脳炎を予防する
再発性実験的自己免疫性脳炎(R−EAE)の予防のための寛容を誘導するために、免疫優性プロテオリピドタンパク質PLP139〜151エピトープ(PLG−PLP139〜151)を用いてPLGナノ粒子を調べた。上述のようにR−EAEマウスを作製した。
抗原を結合させたPLG粒子の静脈内注入は、OVA/ミョウバンで事前に感作させた動物においてアナフィラキシー誘導性の体温低下を誘発しない
活性疾患の存在のために、抗原に対するアナフィラキシーが懸念される:これは、即死を引き起こす可能性があり、ポリスチレン結合粒子とともに記載されている。アナフィラキシーは、体温の著しい低下と関連している。OVA−PLGの静脈内投与が、事前に感作させた動物においてアナフィラキシー誘導性の体温低下を誘導するかどうかを調べるために、0日目に、腹腔内注射により10μgのOVA/ミョウバンでマウスを免疫した。14日目に、腹腔内注射により10μgのOVA/ミョウバンでマウスを再び免疫し、次いで、21日目に静脈内投与したOVA−PLGで寛容化した。次いで、28日目に、静脈内投与によりOVA−PLG粒子またはOVAのいずれかでマウスを寛容化した。
PLP−PLG粒子による予防的投与は、長期の抗原特異的寛容を誘導する
疾患誘発の7日前に、濃度を上昇させたPLP139〜151−PLGの静脈内投与によって最適用量を決定し、OVA323〜339−PLGで処理したSJL/Jマウスと比較して臨床疾患の発症について監視した(図6A)。6〜8週齢のメスSJL/Jマウスに、PLP139〜151(四角)またはOVA323〜339(丸)のいずれかを結合させたPLGナノ粒子を静脈内注射した。7日後(図6B)、25日後(図6C)、または50日後(図6D)に、CFA中のPLP139〜151の皮下注射によりEAEを誘導した。パネルBの動物を臨床疾患について100日間追跡した。図6Eは、疾患誘導から8日目に、パネルBに示すマウスのサブセットにおいて遅延型過敏反応(DTH)を行ったことを示す。パネルBのPLP139〜151/CFA予備刺激群から選択された代表の動物(OVA323〜339−PLGおよびPLP139〜151−PLG)の耳を、予備刺激用のPLP139〜151エピトープおよびOVA323〜339対照ペプチドに曝露した。24時間後にDTHの尺度として耳の腫脹を判定し、曝露前の応答を差し引いた。図6Fは、6〜8週齢のメスSJL/Jマウスに、PLP178〜191(三角)、OVA323〜339(丸)、もしくはPLP139〜151(四角)を結合させたPLGナノ粒子、または未結合粒子のみ(縁取りのある丸)を静脈内注射したことを示す。7日後、CFA中のPLP178〜191の皮下注射によりEAEを誘導し、示される時点で疾患を監視した。
抗原結合粒子を用いた再発性実験的自己免疫性脳炎の治療
PLG−PLP139〜151粒子が、疾患を予防するのではなく、むしろ疾患を治療する能力を調査するために、かつ、投与経路が疾患の発症に影響を及ぼしたかどうかを判定するために、本発明の実施形態の開発中に実験を行った。0日目に、PLP139〜151およびアジュバントでマウスを免疫した。マウスは、通常、12〜14日目に最大臨床スコアを有する。このモデルでは、10日目に、静脈内(iv)投与、腹腔内(ip)投与、皮下(sc)投与、または経口的のいずれかにより、PLG−PLP139〜151粒子または対照PLG−OVA323〜339粒子のいずれかでマウスを処理した。図7に示すように、予防的寛容は、PLG−PLP139〜151粒子が静脈内または腹腔内のいずれかで投与された場合に最も効率的である。静脈内投与したPLP139〜151−PLGで処理した動物は、疾患を発症せず、ほとんどの時点で平均臨床スコア0を有していた。これは、70%を超える動物がアナフィラキシーによって死亡したことが観察された、PLP139〜151ポリスチレン粒子を用いて処理した動物とは対称的である。
抗原結合粒子の寛容は、活性な再発性実験的自己免疫性脳炎における抗原特異的Th1およびTh17応答の誘導を阻害する
抗原結合粒子の投与がTヘルパー細胞の誘導を阻害するかどうかを判定するために、−7日目に、MOG35〜55−PLGまたはOVA323〜339−PLG粒子のいずれかをBALB/cマウスに静脈内投与した。0日目に、OVA323〜339−PLG粒子およびフロイント完全アジュバント(CFA)をマウスに皮下投与した。10日目に、MOG35〜55−PLGまたはOVA323〜339−PLG粒子のいずれかで動物を再刺激し、流入領域リンパ節細胞を単離した。10日目に、CPM、ならびにIL−17、GM−CSF、IFN−γ、IL−10、およびIL−4のレベルを測定した。図8に示すように、OVA323〜339−PLG粒子の投与は、処理した動物においてTh1およびTh17応答を阻害した。
PLP−139〜151を結合させたPLGA粒子により寛容を誘導した。
PLP139〜151−PLGまたはOVA323〜339PLGをマウスに送達することにより、さらなる治療的寛容戦略を行った。組織学的分析は、PLP139〜151−PLG粒子の投与が頸髄の炎症および脱髄を阻害することを示した。マウスをPLP−PLGまたはOVA323〜339−PLGで処理し、40日目に組織を回収した。頸髄を単離し、切片化して、R−EAEおよび多発性硬化症の病態の根底にあるCNSにおける免疫応答を調査した。図9は、OVA323〜339−PLGで処理した動物の組織よりも天然組織に類似するPLP139〜151−PLGで処理した動物の脊髄内の免疫細胞浸潤の低下を示す。OVA323〜339−PLGで処理した動物は、CD45、CD4、およびCD11bに陽性染色を示したのに対し、PLP139〜151−PLGで処理した動物は、これらの因子に最小限の染色を示した。
PLP−139〜151を結合させたPLGA粒子によって誘導される寛容は、制御性T細胞の増加/活性化に一部依存する。
−9日目に、制御性T細胞の一般的なマーカーである抗CD25抗体(Tregs)でSJL/Jマウスを処理し、次いで、−7日目に、OVA323〜339PLG粒子および抗CD25抗体、OVA323〜339PLG粒子および対照IgG抗体、PLP139〜151−PLG粒子および抗CD25抗体、またはPLP139〜151−PLG粒子および対照IgG抗体のいずれかで処理した。図13に示すように、PLP139〜151−PLG粒子および抗CD25抗体で処理した動物は、時折、PLP139〜151−PLG粒子および対照IgG抗体で処理した動物よりも高い平均臨床スコアを示した。これは、Treg、または少なくともCD25を発現するT細胞が、寛容の誘導において役割を果たすことを裏付けるものである。
活性および養子性EAEにおいて、PLP139〜151−PLG粒子によって治療的寛容を誘導した。
PLP139〜151−PLG粒子によって誘導された治療的寛容を、活性および養子性EAEにおいて比較した。2.5×106 PLP139〜151で活性化した芽球の養子移入により、6〜8週齢のメスSJL/Jマウスに養子性EAEを誘導した。疾患誘導から2日後(図14A)および14日後(図14C)に、500nmのPLGナノ粒子に結合したPLP139〜151(四角)またはOVA323〜339(丸)ペプチドをマウスに腹腔内注射した。臨床疾患スコアを、抗原を結合させた脾細胞による処理後のスコアと比較した(図14A)。42日目に、PLP139〜151またはOVA323〜339寛容化マウスから、組織学的分析のために脳および脊髄を採取した。パネルAのマウスの切片を、PLPタンパク質およびCD45について染色した(図14B)。パネル(C)のマウスの脊髄切片をLuxol Fast Blueで染色した(図14D)。脱髄および細胞浸潤の領域を矢印で示す。結果は、養子性EAEを有するマウスにおいて、PLP139〜151−PLG粒子によって寛容が誘導されることを示す。
養子移入EAEにおいて、抗PD−1モノクローナル抗体を用いた処理は、PLP139〜151を封入したPLGナノ粒子による寛容誘導を抑制する
養子性EAEを有するマウスにおいて、抗PD−1抗体を用いた処理がPLP139〜151が誘導した寛容に及ぼす影響を調べるために、0日目に、3×106 PLP139〜151で活性化したT細胞芽球を静脈内投与によりマウスに投与した。2日目に、PLG粒子中に封入されたPLP139〜151またはOVA323〜339を、PBSまたは抗PD−1抗体のいずれかとともに静脈内投与により投与した。4日目、6日目、8日目、10日目、および12日目に、全ての動物に250μgの抗PD−1抗体またはPBSを投与した。
養子移入EAEにおいて、作動性抗CD40モノクローナル抗体を用いた処理は、PLP139〜151が封入されたPLGナノ粒子による寛容誘導をIL−12依存性様式で抑制する
養子性EAEを有するマウスにおいて、作動性抗CD40抗体がPLP139〜151が誘導した寛容に及ぼす影響を調べるために、0日目に、3×106 PLP139〜151で活性化したT細胞芽球を静脈内投与によりマウスに投与した。2日目に、PLG粒子内に封入されたPLP139〜151またはOVA323〜339を静脈内投与によりマウスに投与した。3日目に、対照IgG2a抗体、抗CD40抗体、または抗CD40抗体のいずれか、および抗Il−12抗体を動物に投与した。
PLG粒子内に封入されたOVAは、アレルギー性気道炎症および生体内OVA特異的Th2応答を予防的に阻害する。
PLG粒子内に封入されたOVAが気道炎症に及ぼす予防効果を調べるために、−7日目に、マウスをOVA−PLGで静脈内処理した。0日目に、10μg/マウスの用量のOVA/ミョウバンをマウスに腹腔内注射した。7日目に、マウスを再びOVA−PLGで静脈内処理し、14日目に、さらに10μg/マウスのOVA/ミョウバンを腹腔内注射した。28日目〜30日目に、エアロゾル化したOVAでマウスを3回処理した。
PLG粒子内に封入されたOVAは、アレルギー性気道炎症および生体内OVA特異的Th2応答を治療的に阻害する
PLG粒子内に封入されたOVAが気道炎症に及ぼす治療効果を調べるために、0日目および14日目に、10μg/マウスの用量のOVA/ミョウバンでマウスを腹腔内処理した。28日目および42日目に、OVA−PLGをマウスに静脈内投与した。56日目〜58日目に、エアロゾル化したOVAでマウスを3回処理した。
クロモグラニンA p31ペプチド−PLG粒子によって誘導される寛容は、1型糖尿病を阻害する
3週齢マウスから脾臓、腋窩、上腕、鼠径部、および膵臓のリンパ節細胞を単離することにより、BDC2.5マウスにおいて1型糖尿病を誘導した。単離細胞を培養し、2×106細胞/mLを0.5μMのp31ペプチドとともに96時間インキュベートすることにより生体外で活性化した。0時に、静脈内投与により、5×106細胞をNOD.SCIDマウス(6〜8週)に移入した。3日後、SPまたはPLG2に結合したp31またはMOG35〜55ペプチドの静脈内投与によりマウスを寛容化した。
インスリンを結合させたPLG粒子によって誘導される寛容は、NODマウスにおける自然発症性1型糖尿病の発症を阻害する
NODマウスを、6、8、および10週齢で、静脈内投与によりBSA(N=22)またはインスリン(N=23)を結合させたPLG粒子のいずれかで処理した。次いで、>250mg/dLの血糖として定義される糖尿病の発症についてマウスをアッセイした。図25に示すように、インスリンを結合させたPLG粒子の投与は、300日にわたって糖尿病を発症しなかったマウスの割合を有意に増加させた(22.7%と比較して69.6%;p=0.0027)。
生着動態
−7日目に、OVA−PLGまたは対照ペプチドDby−PLG(オスC57BL/6マウスによって発現される主要なH−Y抗原)のいずれかでメスCD45.2マウスを寛容化した。−1日目に、マウスを200ラドで照射し、次いで、0日目に、オスCD45.1マウスの1×106、5×106、または1×107骨髄細胞を移植した。次いで、1日目に、OVA−PLG、Dby−SP、またはDby−PLGのいずれかでレシピエントマウスを寛容化し、キメラ化のFACS分析のために血液を採取した。図26は、レシピエントマウスに観察されたCD45.1ドナー細胞の割合を示す。
クマリン−6 PLGA粒子は、投与の24時間後には検出不可能である。
抗原に結合しているかまたは抗原を含まないかのいずれかであるクマリン−6 PLGA粒子でマウスを処理した。図29に示すように、粒子は、投与後3時間には検出可能であったが、投与後24時間には検出不可能であった。DAPIで対比染色した、注射後3時間(中央の行)および注射後24時間(下の行)の、腹腔内蛍光PLGA/PEMA微粒子を注射したマウス脾臓(左の列)、肝臓(中央の列)、および肺(左の列)切片と比較した非注射ナイーブマウス(上の行)。
ナノ粒子は生体内でマクロファージと会合する
投与後6時間および15時間の肝臓の分析は、PLGA粒子が肝臓内でF4/80+細胞と共局在化していたことを示す(図30)。
コア内に可溶性PLP139〜151を含有する表面官能化ポリ(ラクチド−co−グリコリド)粒子を使用したSJL/JマウスにおけるR−EAEの阻害
0日目のPLP139〜151/CFAによる予備刺激前の−7日目および−1日目に、コア内に可溶性PLP139〜151ペプチドを含む2.5mgの500nm〜700nm表面官能化ポリ(ラクチド−co−グリコリド)粒子をSJL/Jマウスの群に腹腔内注射した。0日目に対照マウスを予備刺激したが、−7日目または−1日目には粒子処理を行わなかった。さらに20日間、R−EAEの臨床徴候についてマウスを観察した。
可溶性卵白アルブミンを含有する表面官能化ポリ(ラクチド−co−グリコリド)粒子によるアレルギー性気道炎症の阻害
マウスにアレルギー性気道炎症(AIA)を誘導した。0日目および+14日目の卵白アルブミン/ミョウバンによる予備刺激前の−7日目および+7日目に、コア内に可溶性卵白アルブミンまたは可溶性ウシ血清アルブミン(対照)を含む2.5mgの500nm〜700nm表面官能化ポリ(ラクチド−co−グリコリド)粒子をBalb/cマウスの群に静脈内注射した。+28−30日目に、エアロゾル化した卵白アルブミンにマウスを曝露した。次いで、マウスを屠殺し、気管支肺胞洗浄液を得た。卵白アルブミン特異的IgEの血清レベルも測定した。
抗原が封入された表面官能化ポリ(ラクチド−co−グリコリド)粒子の合成
本実施例は、自己免疫疾患における免疫誘導およびアレルギーの治療のための、ポリ(ラクチド−co−グリコリド)のシェルに取り囲まれた、高密度のカルボキシレート基で表面官能化され、コア内に可溶性抗原を含有する生分解性ポリ(ラクチド−co−グリコリド)粒子の製剤化および部分的特徴について詳述する。
1.エンドトキシン不含水中の150μLの200mg/mL卵白アルブミンまたはウシ血清アルブミンを、20mLシンチレーションバイアル中のジクロロメタン中の2mLの20% w/vポリ(ラクチド−co−グリコリド)に滴下で加えた。
2.得られた混合物を氷上に置き、プローブ超音波処理器を使用して10ワットで30秒間超音波処理した。
3.10mLの水中1% w/v ポリ(エチレン−alt−無水マレイン酸)を加えた。
4.得られた混合物を氷上に置き、プローブ超音波処理器を使用して16ワットで30秒間超音波処理した。
5.得られた混合物を600mLビーカー中の200mLの0.5% w/v ポリ(エチレン−alt−無水マレイン酸に注ぎ入れ、一晩撹拌して粒子を硬化させた。
6.次いで、硬化した粒子を遠心分離して精製し、炭酸水素緩衝液pH9.6で3回洗浄した。
7.精製した粒子を、水中4% w/vスクロースおよび3% w/v D−マンニトールに再懸濁し、液体窒素中で瞬間凍結し、凍結乾燥させた。
コア内に可溶性PLP139〜151を含有する表面官能化リポソームは、多発性硬化症のマウスR−EAEモデルにおいて免疫学的寛容を誘導する
本発明はまた、高密度の負電荷を帯びた基で表面官能化され、コア内に可溶性抗原を含有する生分解性リポソーム送達ビヒクルが、多発性硬化症のR−EAEマウスモデルにおいて免疫学的寛容を誘導することも発見した。
1)生分解性粒子は、体内に長時間残留せず、完全分解の時間を制御することができる。
2)粒子およびリポソームは、細胞を活性化せずに内部移行を促進するように官能化することができる。この目的のために、我々は、PLGミクロスフェアにホスファチジルセリンを充填した。
3)粒子およびリポソームは、特定の細胞集団のための標的リガンドを組み込むように設計することもできる。
4)粒子内に内部移行する細胞型の活性化を制限し、アネルギーならびに/または制御性T細胞の欠失および活性化を介して寛容の誘発を促進するように、IL−10およびTGF−β等の抗炎症性サイトカインも誘導することができる。
(1)T細胞および抗体媒介性の自己免疫疾患(多発性硬化症、1型糖尿病、関節リウマチ、全身性エリテマトーデス等)−寛容は、特定の自己免疫疾患を駆動する関連自己抗原と複合化させた粒子を用いて誘導される。
(2)食物および肺アレルギー、皮膚アレルギー、ならびに喘息−寛容は、アレルギー反応を誘発する特定の食物(例えば、ピーナツタンパク質等)、注入される物質(ハチ毒タンパク質等)、または吸入される物質(例えば、ブタクサ花粉タンパク質、ペットのフケタンパク質等)と複合化させた粒子を用いて誘導される。
(3)移植拒絶反応−寛容は、レシピエントによる拒絶反応を予防するために、臓器移植の前にドナー臓器またはドナー細胞上の移植抗原に対して誘導される。
(4)酵素補充療法−患者が、特定の欠陥を治療するために投与される組み換え的に生成された酵素に対する中和抗体応答を形成するのを防止するために、遺伝的欠陥を有する患者が産生することができない酵素に対して寛容が誘導される。
Claims (10)
- 対象に投与することによって前記対象において抗原特異的寛容を誘導する方法に使用されるための組成物であって、
負のゼータ電位を有するカルボン酸で表面官能化されたポリ(乳酸−co−グリコール酸)担体粒子を含む組成物であり、
前記担体粒子がそのコア内に抗原を封入し、
前記粒子の前記ゼータ電位が、−75mV〜−40mV、又は−50mV〜−40mVである、
組成物。 - 前記粒子が、約80:20〜約100:0のモル比を有するコポリマーである、請求項1に記載の組成物。
- 前記粒子が、約0.1μm〜約10μm、約0.3μm〜約5μm、約0.5μm〜約3μm、約0.5μm〜約1μm、又は約0.5μmの直径を有する、請求項1に記載の組成物。
- 前記抗原が、自己免疫抗原、対象に移植される組織上に発現される抗原、またはアレルゲンを含む、請求項1に記載の組成物。
- 前記抗原が、ミエリン塩基性タンパク質、アセチルコリン受容体、内在性抗原、ミエリンオリゴデンドロサイト糖タンパク質、膵β細胞抗原、インスリン、グルタミン酸デカルボキシラーゼ(GAD)、11型コラーゲン、ヒト軟骨gp39、fp130−RAPS、プロテオリピドタンパク質、フィブリラリン、低分子核小体タンパク質、甲状腺刺激因子受容体、ヒストン、糖タンパク質gp70、ピルビン酸脱水素酵素ジヒドロリポアミドアセチルトランスフェラーゼ(PCD−E2)、毛包抗原、A−グリアジン、ヒトトロポミオシンアイソフォーム5、バヒアグラス花粉(BaGP)、モモアレルゲンPru p 3、αs−1カゼイン牛乳アレルゲン、Apig1セロリアレルゲン、Bere1ブラジルナッツアレルゲン、B−ラクトグロブリン牛乳アレルゲン、ウシ血清アルブミン、Cor a 1.04ヘーゼルナッツアレルゲン、および卵白アルブミン卵アレルゲンからなる群から選択されるタンパク質を含むか、前記タンパク質の少なくとも一部を含む、請求項4に記載の組成物。
- 前記粒子が、生分解性である、請求項1に記載の組成物。
- 前記粒子が、表面官能化される、請求項1に記載の組成物。
- 薬学的に許容される担体をさらに含む、請求項1〜7のいずれか1項に記載の組成物。
- 前記投与が、疾患または状態を治療または予防するために行われる、請求項1に記載の組成物。
- 前記疾患または状態が、自己免疫疾患、炎症性疾患、アレルギー、移植拒絶反応、または過免疫応答である、請求項9に記載の組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261662687P | 2012-06-21 | 2012-06-21 | |
| US61/662,687 | 2012-06-21 | ||
| PCT/US2013/047079 WO2013192532A2 (en) | 2012-06-21 | 2013-06-21 | Peptide conjugated particles |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019016132A Division JP7210306B2 (ja) | 2012-06-21 | 2019-01-31 | ペプチドコンジュゲート粒子 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2015522569A JP2015522569A (ja) | 2015-08-06 |
| JP2015522569A5 JP2015522569A5 (ja) | 2017-11-09 |
| JP6557140B2 true JP6557140B2 (ja) | 2019-08-07 |
Family
ID=49769721
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015518611A Active JP6557140B2 (ja) | 2012-06-21 | 2013-06-21 | ペプチドコンジュゲート粒子 |
| JP2019016132A Active JP7210306B2 (ja) | 2012-06-21 | 2019-01-31 | ペプチドコンジュゲート粒子 |
| JP2021095735A Active JP7491869B2 (ja) | 2012-06-21 | 2021-06-08 | ペプチドコンジュゲート粒子 |
| JP2023118971A Pending JP2023153869A (ja) | 2012-06-21 | 2023-07-21 | ペプチドコンジュゲート粒子 |
Family Applications After (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019016132A Active JP7210306B2 (ja) | 2012-06-21 | 2019-01-31 | ペプチドコンジュゲート粒子 |
| JP2021095735A Active JP7491869B2 (ja) | 2012-06-21 | 2021-06-08 | ペプチドコンジュゲート粒子 |
| JP2023118971A Pending JP2023153869A (ja) | 2012-06-21 | 2023-07-21 | ペプチドコンジュゲート粒子 |
Country Status (20)
| Country | Link |
|---|---|
| US (4) | US10201596B2 (ja) |
| EP (2) | EP3607973A1 (ja) |
| JP (4) | JP6557140B2 (ja) |
| KR (4) | KR20220166879A (ja) |
| CN (2) | CN110064049B (ja) |
| AU (4) | AU2013278056B2 (ja) |
| BR (1) | BR112014032207B1 (ja) |
| CA (2) | CA2876495C (ja) |
| DK (1) | DK2863942T3 (ja) |
| ES (1) | ES2738481T3 (ja) |
| HK (1) | HK1209039A1 (ja) |
| HR (1) | HRP20191178T1 (ja) |
| IL (3) | IL236191B (ja) |
| MX (1) | MX363889B (ja) |
| PL (1) | PL2863942T3 (ja) |
| PT (1) | PT2863942T (ja) |
| RU (1) | RU2669346C2 (ja) |
| TR (1) | TR201909998T4 (ja) |
| WO (1) | WO2013192532A2 (ja) |
| ZA (1) | ZA201409305B (ja) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2011325966B2 (en) | 2010-11-12 | 2016-09-29 | Oncour Pharma, Inc. | Modified immune-modulating particles |
| US9597385B2 (en) | 2012-04-23 | 2017-03-21 | Allertein Therapeutics, Llc | Nanoparticles for treatment of allergy |
| CN110064049B (zh) | 2012-06-21 | 2024-02-09 | 西北大学 | 肽缀合粒子 |
| EP2928500B1 (en) | 2012-12-04 | 2019-03-06 | Phosphorex Inc. | Microparticles and nanoparticles having negative surface charges |
| IL292823B2 (en) | 2013-03-13 | 2023-11-01 | Cour Pharmaceuticals Dev Company | Secondary vaccine particles for the treatment of inflammation |
| WO2014165679A1 (en) | 2013-04-03 | 2014-10-09 | Allertein Therapeutics, Llc | Novel nanoparticle compositions |
| MX2016001931A (es) * | 2013-08-13 | 2016-09-07 | Univ Northwestern | Particulas conjugadas con peptidos. |
| AU2016379413B2 (en) * | 2015-12-23 | 2024-04-04 | Cour Pharmaceuticals Development Company Inc. | Covalent polymer-antigen conjugated particles |
| IL260296B2 (en) * | 2016-01-04 | 2024-01-01 | Cour Pharmaceuticals Dev Company Inc | Particles thermalize fusion proteins containing associated epitopes |
| GB2550586A (en) | 2016-05-23 | 2017-11-29 | A-Fax Ltd | Racking protection device |
| WO2019217661A1 (en) * | 2018-05-09 | 2019-11-14 | Yale University | Compositions and systems for ex vivo cell modulation and methods of use thereof |
| JP2021523151A (ja) * | 2018-05-11 | 2021-09-02 | ホスホレックス、インコーポレイテッド | 負の表面電荷を有するマイクロ粒子及びナノ粒子 |
| US20220031632A1 (en) * | 2020-05-14 | 2022-02-03 | Op-T Llc | Nanoparticle compositions and uses thereof |
| CA3235813A1 (en) | 2021-10-21 | 2023-04-27 | John J. Puisis | Treatment of primary biliary cholangitis (pbc) with tolerizing nanoparticles |
| WO2024026452A1 (en) | 2022-07-29 | 2024-02-01 | Repertoire Immune Medicines, Inc. | T cell epitopes associated with type 1 diabetes |
Family Cites Families (85)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4565696A (en) * | 1983-08-03 | 1986-01-21 | The Regents Of The University Of California | Production of immunogens by antigen conjugation to liposomes |
| US4751180A (en) | 1985-03-28 | 1988-06-14 | Chiron Corporation | Expression using fused genes providing for protein product |
| US4935233A (en) | 1985-12-02 | 1990-06-19 | G. D. Searle And Company | Covalently linked polypeptide cell modulators |
| WO1993016724A1 (en) | 1992-02-28 | 1993-09-02 | Autoimmune, Inc. | Bystander suppression of autoimmune diseases |
| US6004763A (en) | 1992-09-11 | 1999-12-21 | Institut Pasteur | Antigen-carrying microparticles and their use in the induction of humoral or cellular responses |
| FR2695563B1 (fr) | 1992-09-11 | 1994-12-02 | Pasteur Institut | Microparticules portant des antigènes et leur utilisation pour l'induction de réponses humorales ou cellulaires. |
| JPH06157592A (ja) | 1992-11-24 | 1994-06-03 | Hitachi Chem Co Ltd | ペプチドもしくはその誘導体、それらとタンパク質との結合体、及びこれらを免疫源とする抗エンドセリン−1抗体の製造方法 |
| AU674584B2 (en) | 1993-06-02 | 1997-01-02 | Tvw Telethon Institute For Child Health Research | Cryptic peptides for use in inducing immunologic tolerance |
| US5804201A (en) | 1996-03-11 | 1998-09-08 | The Rockefeller University | Immunomodulatory peptides of vespid antigen 5 |
| EP0991705B1 (en) | 1997-03-31 | 2003-09-10 | The Regents Of The University Of Michigan | Open pore biodegradable matrices |
| US7427602B1 (en) | 1998-05-13 | 2008-09-23 | The Regents Of The University Of Michigan | Sustained DNA delivery from structural matrices |
| EP1212086B8 (en) | 1999-08-18 | 2008-01-02 | Industry-Academic Cooperation Foundation, The Catholic University of Korea | Immunological tolerance-induction agent |
| WO2001094944A2 (en) | 2000-06-02 | 2001-12-13 | Memorial Sloan-Kettering Cancer Center | Artificial antigen presenting cells and methods of use thereof |
| EP1311549A2 (en) | 2000-08-22 | 2003-05-21 | Micromet AG | Composition for the elimination of autoreactive b-cells |
| US7029697B2 (en) | 2001-02-14 | 2006-04-18 | Northwestern University | Controlled surface-associated delivery of genes and oligonucleotides |
| US6890556B1 (en) | 2001-02-14 | 2005-05-10 | Northwestern University | Controlled surface-associated delivery of genes and oligonucleotides |
| NZ532274A (en) * | 2001-10-03 | 2006-02-24 | Chiron Corp | Adjuvanted meningococcus compositions |
| CA2465779A1 (en) | 2001-11-20 | 2003-05-30 | Advanced Inhalation Research, Inc. | Compositions for sustained action product delivery |
| AU2003216851B2 (en) | 2002-03-19 | 2008-04-17 | Powderject Research Limited | Imidazoquinoline adjuvants for vaccines |
| WO2003092654A1 (en) | 2002-05-02 | 2003-11-13 | President And Fellows Of Harvard College | Formulations limiting spread of pulmonary infections |
| WO2004006951A1 (en) | 2002-07-12 | 2004-01-22 | The Johns Hopkins University | Reagents and methods for engaging unique clonotypic lymphocyte receptors |
| US7465463B2 (en) | 2002-09-04 | 2008-12-16 | Polyheal, Ltd. | Compositions comprising microspheres with anti-inflammatory properties for healing of ocular tissues |
| AU2003302226A1 (en) * | 2002-09-24 | 2004-06-30 | University Of Kentucky Research Foundation | Nanoparticle-based vaccine delivery system containing adjuvant |
| WO2005000272A1 (en) | 2003-06-04 | 2005-01-06 | Isis Pharmaceuticals, Inc. | Long-circulating liposomal compositions |
| WO2005015160A2 (en) | 2003-08-07 | 2005-02-17 | The Children's Hospital Of Philadelphia | Functionalized polymeric colloids |
| WO2005019429A2 (en) | 2003-08-22 | 2005-03-03 | Potentia Pharmaceuticals, Inc. | Compositions and methods for enhancing phagocytosis or phagocyte activity |
| US9149440B2 (en) | 2003-09-02 | 2015-10-06 | University Of South Florida | Nanoparticles for drug-delivery |
| CA2545276C (en) | 2003-11-05 | 2016-05-31 | The Government Of The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Carbohydrate antigen-nanoparticle conjugates and uses thereof as antimetastatic agents in treating cancer |
| ES2246695B1 (es) | 2004-04-29 | 2007-05-01 | Instituto Cientifico Y Tecnologico De Navarra, S.A. | Composicion estimuladora de la respuesta inmunitaria que comprende nanoparticulas a base de un copolimero de metil vinil eter y anhidrido maleico. |
| US7846466B2 (en) | 2004-06-10 | 2010-12-07 | Northwestern University | Biodegradable scaffolds and uses thereof |
| US7534448B2 (en) | 2004-07-01 | 2009-05-19 | Yale University | Methods of treatment with drug loaded polymeric materials |
| JP2006248978A (ja) * | 2005-03-10 | 2006-09-21 | Mebiopharm Co Ltd | 新規なリポソーム製剤 |
| AU2006282042B2 (en) | 2005-06-17 | 2011-12-22 | The University Of North Carolina At Chapel Hill | Nanoparticle fabrication methods, systems, and materials |
| ATE458762T1 (de) | 2005-07-01 | 2010-03-15 | Hoffmann La Roche | Carboxylierte latexteilchen |
| WO2007008755A2 (en) | 2005-07-08 | 2007-01-18 | The Board Of Regents, The University Of Texas System | Surface functionalization of polymeric materials |
| GB0514262D0 (en) | 2005-07-12 | 2005-08-17 | Renovo Ltd | Promotion of epithelial regeneration |
| US20070041934A1 (en) | 2005-08-12 | 2007-02-22 | Regents Of The University Of Michigan | Dendrimer based compositions and methods of using the same |
| US20100028450A1 (en) | 2006-01-25 | 2010-02-04 | The Board Of Trustees Of The University Of Illinoi S | Tolerogenic biodegradable artificial antigen presenting system |
| WO2007094003A2 (en) | 2006-02-15 | 2007-08-23 | Ramot At Tel Aviv University Ltd. | Viral display vehicles for treating multiple sclerosis |
| US8431160B2 (en) * | 2006-02-24 | 2013-04-30 | Novartis Ag | Microparticles containing biodegradable polymer and cationic polysaccharide for use in immunogenic compositions |
| JP2009544750A (ja) | 2006-07-28 | 2009-12-17 | ザ ユニヴァーシティ コート オブ ザ ユニヴァーシティ オブ エディンバラ | 治療 |
| AU2008302276B2 (en) * | 2006-09-29 | 2013-10-10 | Takeda Vaccines, Inc. | Method of conferring a protective immune response to norovirus |
| WO2008043157A1 (en) | 2006-10-12 | 2008-04-17 | The University Of Queensland | Compositions and methods for modulating immune responses |
| US20090214474A1 (en) | 2006-11-01 | 2009-08-27 | Barbara Brooke Jennings | Compounds, methods, and treatments for abnormal signaling pathways for prenatal and postnatal development |
| EP2117600B1 (en) | 2006-12-18 | 2012-12-05 | Colorobbia Italia S.p.a. | Magnetic nanoparticles for the application in hyperthermia, preparation thereof and use in constructs having a pharmacological application |
| CN101678090B (zh) | 2007-03-07 | 2012-04-11 | 乌第有限合伙公司 | 用于预防和治疗自身免疫病的组合物和方法 |
| US20080311140A1 (en) | 2007-05-29 | 2008-12-18 | Baylor College Of Medicine | Antigen specific immunosuppression by dendritic cell therapy |
| AU2008314647B2 (en) | 2007-10-12 | 2013-03-21 | Massachusetts Institute Of Technology | Vaccine nanotechnology |
| EP2211841A4 (en) | 2007-10-15 | 2013-03-13 | Cooperative Res Ct For Asthma | PROPHYLAXIS METHOD AND AGENTS FOR USE IN THIS METHOD |
| WO2009052561A1 (en) | 2007-10-22 | 2009-04-30 | The Walter And Eliza Hall Institute Of Medical Research | Compositions and methods for manipulating an immune response |
| EP2057998A1 (en) | 2007-10-31 | 2009-05-13 | Universitätsklinikum Hamburg-Eppendorf | Use of modified cells for the treatment of multiple sclerosis |
| US20090123509A1 (en) | 2007-11-08 | 2009-05-14 | Cory Berkland | Biodegradable Colloidal Gels as Moldable Tissue Engineering Scaffolds |
| US20090238879A1 (en) | 2008-01-24 | 2009-09-24 | Northwestern University | Delivery scaffolds and related methods of use |
| US9669057B2 (en) | 2008-04-25 | 2017-06-06 | Duke University | Regulatory B cells and their uses |
| US20110206773A1 (en) | 2008-05-20 | 2011-08-25 | Yale University | Sustained delivery of drugs from biodegradable polymeric microparticles |
| EP2123261A1 (en) | 2008-05-20 | 2009-11-25 | Stallergenes S.A. | Mucoadhesive particulate formulation for inducing antigen-specific immune tolerance |
| US8323696B2 (en) | 2008-08-29 | 2012-12-04 | Ecole Polytechnique Federale De Lausanne | Nanoparticles for immunotherapy |
| EP2349219A4 (en) * | 2008-09-29 | 2013-05-01 | Univ Mercer | BIOLOGICALLY EFFECTIVE SUBSTANCES CAPSULATING NANO BEADS AND METHOD FOR FORMULATING NANO BEADS |
| CA2771995C (en) | 2008-10-02 | 2016-11-22 | University Of Pittsburgh - Of The Commonwealth System Of Higher Education | Administration of an adborbent polymer for treatment of systemic inflammation |
| CA3042826A1 (en) | 2008-11-30 | 2010-06-03 | Immusant, Inc. | Compositions and methods for treatment of celiac disease |
| EP2376115A4 (en) | 2008-12-11 | 2013-07-03 | Univ Alberta | METHODS AND SYSTEMS FOR INDUCING IMMUNOLOGICAL TOLERANCE TO NON-NONE ANTIGENS |
| SG193843A1 (en) | 2009-01-20 | 2013-10-30 | Univ Northwestern | Compositions and methods for induction of antigen-specific tolerance |
| EP2255831A1 (en) | 2009-05-25 | 2010-12-01 | Institut Pasteur | Liposome based diepitope constructs |
| US8629151B2 (en) | 2009-05-27 | 2014-01-14 | Selecta Biosciences, Inc. | Immunomodulatory agent-polymeric compounds |
| WO2011031441A1 (en) | 2009-08-28 | 2011-03-17 | The Government Of The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Therapy with a chimeric molecule and a pro-apoptotic agent |
| US20110135744A1 (en) | 2009-12-03 | 2011-06-09 | The Regents Of The University Of California | Nanoparticle Based Therapy for Dispersing Mucin |
| US20130209564A1 (en) | 2010-02-22 | 2013-08-15 | Shyam M. Rele | Polysaccharide Particle Vaccines |
| WO2011119262A1 (en) | 2010-03-26 | 2011-09-29 | Cerulean Pharma Inc. | Methods and systems for generating nanoparticles |
| WO2011133617A1 (en) * | 2010-04-23 | 2011-10-27 | The Board Of Trustees Of The University Of Illinois | Nano-hybrid delivery system for sequential utilization of passive and active targeting |
| EP2575869B1 (en) | 2010-06-04 | 2017-07-26 | Flow Pharma Inc. | Peptide particle formulation |
| JP5703372B2 (ja) * | 2010-06-04 | 2015-04-15 | トンジ ユニバーシティTongji University | ピレンおよびピロールのコポリマーならびに該コポリマーの生成方法 |
| CN103068399A (zh) | 2010-06-30 | 2013-04-24 | 卡姆普根有限公司 | 用于治疗多发性硬化、类风湿性关节炎以及其他自身免疫病症的c1orf32 |
| US9522183B2 (en) | 2010-07-31 | 2016-12-20 | The Scripps Research Institute | Compositions and methods for inducing immune tolerance |
| US10131875B2 (en) | 2010-08-04 | 2018-11-20 | Duke University | Regulatory B cells and their uses |
| AU2011325966B2 (en) | 2010-11-12 | 2016-09-29 | Oncour Pharma, Inc. | Modified immune-modulating particles |
| WO2012071014A1 (en) * | 2010-11-24 | 2012-05-31 | Nanyang Technological University | Method for coating particles with calcium phosphate and particles, microparticles and nanoparticles formed thereof |
| US10064406B2 (en) | 2011-01-06 | 2018-09-04 | Cytosorbents Corporation | Polymeric sorbent for removal of impurities from whole blood and blood products |
| EP2667843A2 (en) | 2011-01-24 | 2013-12-04 | Yissum Research Development Company of the Hebrew University of Jerusalem, Ltd. | Nanoparticles based on poly(lactic glycolic) acid for cosmetic applications |
| WO2012149393A2 (en) | 2011-04-29 | 2012-11-01 | Selecta Biosciences, Inc. | Tolerogenic synthetic nanocarriers for antigen-specific deletion of t effector cells |
| CN103702687A (zh) | 2011-07-29 | 2014-04-02 | 西莱克塔生物科技公司 | 产生体液和细胞毒性t淋巴细胞(ctl)免疫应答的合成纳米载体 |
| WO2013036296A1 (en) | 2011-09-06 | 2013-03-14 | Selecta Biosciences, Inc. | Compositions and methods for producing antigen-specific induced tolerogenic dendritic cells with synthetic nanocarriers |
| EP2811980A4 (en) | 2012-01-31 | 2015-12-23 | Cerulean Pharma Inc | POLYMER-AGENT CONJUGATES, PARTICLES, COMPOSITIONS AND METHODS OF USE THEREOF |
| CN110064049B (zh) | 2012-06-21 | 2024-02-09 | 西北大学 | 肽缀合粒子 |
| IL292823B2 (en) | 2013-03-13 | 2023-11-01 | Cour Pharmaceuticals Dev Company | Secondary vaccine particles for the treatment of inflammation |
| MX2016001931A (es) | 2013-08-13 | 2016-09-07 | Univ Northwestern | Particulas conjugadas con peptidos. |
-
2013
- 2013-06-21 CN CN201910226407.9A patent/CN110064049B/zh active Active
- 2013-06-21 ES ES13807832T patent/ES2738481T3/es active Active
- 2013-06-21 EP EP19168035.4A patent/EP3607973A1/en active Pending
- 2013-06-21 KR KR1020227042654A patent/KR20220166879A/ko not_active Application Discontinuation
- 2013-06-21 KR KR1020207021560A patent/KR102283760B1/ko active IP Right Grant
- 2013-06-21 CN CN201380041634.5A patent/CN104684578B/zh active Active
- 2013-06-21 PL PL13807832T patent/PL2863942T3/pl unknown
- 2013-06-21 PT PT13807832T patent/PT2863942T/pt unknown
- 2013-06-21 CA CA2876495A patent/CA2876495C/en active Active
- 2013-06-21 KR KR1020217023650A patent/KR20210096312A/ko active Application Filing
- 2013-06-21 CA CA3211102A patent/CA3211102A1/en active Pending
- 2013-06-21 EP EP13807832.4A patent/EP2863942B1/en active Active
- 2013-06-21 TR TR2019/09998T patent/TR201909998T4/tr unknown
- 2013-06-21 KR KR1020157001460A patent/KR102139267B1/ko active IP Right Grant
- 2013-06-21 DK DK13807832.4T patent/DK2863942T3/da active
- 2013-06-21 US US14/410,011 patent/US10201596B2/en active Active
- 2013-06-21 MX MX2014015843A patent/MX363889B/es active IP Right Grant
- 2013-06-21 JP JP2015518611A patent/JP6557140B2/ja active Active
- 2013-06-21 WO PCT/US2013/047079 patent/WO2013192532A2/en active Application Filing
- 2013-06-21 RU RU2015101758A patent/RU2669346C2/ru active
- 2013-06-21 AU AU2013278056A patent/AU2013278056B2/en active Active
- 2013-06-21 BR BR112014032207-4A patent/BR112014032207B1/pt active IP Right Grant
-
2014
- 2014-12-11 IL IL236191A patent/IL236191B/en active IP Right Grant
- 2014-12-17 ZA ZA2014/09305A patent/ZA201409305B/en unknown
-
2015
- 2015-10-05 HK HK15109722.9A patent/HK1209039A1/xx unknown
-
2018
- 2018-09-28 AU AU2018236881A patent/AU2018236881B2/en active Active
-
2019
- 2019-01-31 JP JP2019016132A patent/JP7210306B2/ja active Active
- 2019-02-11 US US16/272,775 patent/US11413337B2/en active Active
- 2019-05-26 IL IL266880A patent/IL266880B/en unknown
- 2019-07-01 HR HRP20191178TT patent/HRP20191178T1/hr unknown
-
2020
- 2020-04-17 US US16/852,196 patent/US11826407B2/en active Active
- 2020-06-12 AU AU2020203884A patent/AU2020203884A1/en not_active Abandoned
- 2020-09-30 AU AU2020244453A patent/AU2020244453A1/en active Pending
-
2021
- 2021-06-08 JP JP2021095735A patent/JP7491869B2/ja active Active
-
2022
- 2022-02-24 IL IL290884A patent/IL290884A/en unknown
-
2023
- 2023-07-21 JP JP2023118971A patent/JP2023153869A/ja active Pending
- 2023-11-13 US US18/507,837 patent/US20240082371A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7408604B2 (ja) | ペプチドコンジュゲート粒子 | |
| JP7210306B2 (ja) | ペプチドコンジュゲート粒子 | |
| RU2813163C2 (ru) | Частицы, конъюгированные с пептидом |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160617 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20160617 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20170321 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170328 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20170627 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20170928 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20180111 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20180411 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20180608 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20180625 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20181001 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190131 |
|
| C60 | Trial request (containing other claim documents, opposition documents) |
Free format text: JAPANESE INTERMEDIATE CODE: C60 Effective date: 20190131 |
|
| C11 | Written invitation by the commissioner to file amendments |
Free format text: JAPANESE INTERMEDIATE CODE: C11 Effective date: 20190215 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20190416 |
|
| C21 | Notice of transfer of a case for reconsideration by examiners before appeal proceedings |
Free format text: JAPANESE INTERMEDIATE CODE: C21 Effective date: 20190417 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190618 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20190711 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6557140 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |