JP6152391B2 - セラノスティクスにおいて有用なポリマーナノ粒子 - Google Patents
セラノスティクスにおいて有用なポリマーナノ粒子 Download PDFInfo
- Publication number
- JP6152391B2 JP6152391B2 JP2014559046A JP2014559046A JP6152391B2 JP 6152391 B2 JP6152391 B2 JP 6152391B2 JP 2014559046 A JP2014559046 A JP 2014559046A JP 2014559046 A JP2014559046 A JP 2014559046A JP 6152391 B2 JP6152391 B2 JP 6152391B2
- Authority
- JP
- Japan
- Prior art keywords
- polymer
- nanoparticles
- starch
- appendix
- pmaa
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000002105 nanoparticle Substances 0.000 title claims description 451
- 229920000642 polymer Polymers 0.000 title claims description 264
- 229920002472 Starch Polymers 0.000 claims description 155
- 235000019698 starch Nutrition 0.000 claims description 152
- 239000008107 starch Substances 0.000 claims description 152
- 238000000034 method Methods 0.000 claims description 141
- 239000000178 monomer Substances 0.000 claims description 130
- 239000000203 mixture Substances 0.000 claims description 66
- CERQOIWHTDAKMF-UHFFFAOYSA-N Methacrylic acid Chemical compound CC(=C)C(O)=O CERQOIWHTDAKMF-UHFFFAOYSA-N 0.000 claims description 64
- 229920002845 Poly(methacrylic acid) Polymers 0.000 claims description 48
- 238000006116 polymerization reaction Methods 0.000 claims description 40
- 229920000136 polysorbate Polymers 0.000 claims description 29
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 28
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Polymers OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 claims description 27
- 238000010559 graft polymerization reaction Methods 0.000 claims description 27
- 229920001282 polysaccharide Polymers 0.000 claims description 27
- 239000005017 polysaccharide Substances 0.000 claims description 27
- 229950008882 polysorbate Drugs 0.000 claims description 26
- -1 alkylaminoalkyl ester Chemical class 0.000 claims description 22
- 238000004519 manufacturing process Methods 0.000 claims description 17
- JNYAEWCLZODPBN-UHFFFAOYSA-N 2-(1,2-dihydroxyethyl)oxolane-3,4-diol Polymers OCC(O)C1OCC(O)C1O JNYAEWCLZODPBN-UHFFFAOYSA-N 0.000 claims description 16
- SJIXRGNQPBQWMK-UHFFFAOYSA-N 2-(diethylamino)ethyl 2-methylprop-2-enoate Chemical compound CCN(CC)CCOC(=O)C(C)=C SJIXRGNQPBQWMK-UHFFFAOYSA-N 0.000 claims description 16
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 16
- 239000003795 chemical substances by application Substances 0.000 claims description 16
- JNYAEWCLZODPBN-CTQIIAAMSA-N sorbitan Polymers OCC(O)C1OCC(O)[C@@H]1O JNYAEWCLZODPBN-CTQIIAAMSA-N 0.000 claims description 16
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 12
- 239000003124 biologic agent Substances 0.000 claims description 11
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 claims description 10
- 239000003431 cross linking reagent Substances 0.000 claims description 9
- 230000003381 solubilizing effect Effects 0.000 claims description 9
- 239000013543 active substance Substances 0.000 claims description 8
- 239000002904 solvent Substances 0.000 claims description 8
- 125000000524 functional group Chemical group 0.000 claims description 7
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 6
- 229920006037 cross link polymer Polymers 0.000 claims description 6
- 239000007788 liquid Substances 0.000 claims description 5
- NVVDPSZOAIMZLZ-UHFFFAOYSA-N 5-(diethylamino)-2-methylpent-2-enoic acid Chemical group CCN(CC)CCC=C(C)C(O)=O NVVDPSZOAIMZLZ-UHFFFAOYSA-N 0.000 claims description 4
- RWHRFHQRVDUPIK-UHFFFAOYSA-N 50867-57-7 Chemical compound CC(=C)C(O)=O.CC(=C)C(O)=O RWHRFHQRVDUPIK-UHFFFAOYSA-N 0.000 claims description 4
- 150000002148 esters Chemical class 0.000 claims 3
- 150000004676 glycans Chemical class 0.000 claims 3
- 125000005395 methacrylic acid group Chemical group 0.000 claims 1
- 239000002245 particle Substances 0.000 description 190
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 114
- 206010028980 Neoplasm Diseases 0.000 description 94
- 239000003814 drug Substances 0.000 description 88
- 229940079593 drug Drugs 0.000 description 80
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 69
- 229920000053 polysorbate 80 Polymers 0.000 description 69
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 65
- 229940068968 polysorbate 80 Drugs 0.000 description 65
- 210000004027 cell Anatomy 0.000 description 62
- 229960004679 doxorubicin Drugs 0.000 description 57
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 54
- 238000002347 injection Methods 0.000 description 44
- 239000007924 injection Substances 0.000 description 44
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 42
- 239000000243 solution Substances 0.000 description 40
- 238000006243 chemical reaction Methods 0.000 description 39
- 210000004556 brain Anatomy 0.000 description 38
- 238000009826 distribution Methods 0.000 description 37
- 230000015572 biosynthetic process Effects 0.000 description 32
- 230000000694 effects Effects 0.000 description 31
- 241000699670 Mus sp. Species 0.000 description 29
- 210000001519 tissue Anatomy 0.000 description 29
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 26
- 150000003254 radicals Chemical class 0.000 description 25
- 125000002843 carboxylic acid group Chemical group 0.000 description 24
- 150000004804 polysaccharides Chemical class 0.000 description 24
- 239000004094 surface-active agent Substances 0.000 description 23
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 21
- 239000004971 Cross linker Substances 0.000 description 21
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 21
- 230000008859 change Effects 0.000 description 21
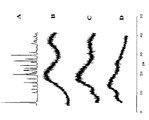
- 238000004448 titration Methods 0.000 description 21
- 238000001727 in vivo Methods 0.000 description 20
- 239000000523 sample Substances 0.000 description 20
- 238000003786 synthesis reaction Methods 0.000 description 20
- 241001465754 Metazoa Species 0.000 description 19
- 239000008280 blood Substances 0.000 description 19
- 210000004369 blood Anatomy 0.000 description 19
- 230000007423 decrease Effects 0.000 description 19
- 210000004185 liver Anatomy 0.000 description 19
- 239000003999 initiator Substances 0.000 description 18
- 238000011534 incubation Methods 0.000 description 17
- 238000011068 loading method Methods 0.000 description 17
- 210000000056 organ Anatomy 0.000 description 17
- 239000000047 product Substances 0.000 description 17
- 238000011282 treatment Methods 0.000 description 17
- QPCDCPDFJACHGM-UHFFFAOYSA-N N,N-bis{2-[bis(carboxymethyl)amino]ethyl}glycine Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(=O)O)CCN(CC(O)=O)CC(O)=O QPCDCPDFJACHGM-UHFFFAOYSA-N 0.000 description 16
- 230000035508 accumulation Effects 0.000 description 16
- 238000009825 accumulation Methods 0.000 description 16
- 239000002872 contrast media Substances 0.000 description 16
- 238000009472 formulation Methods 0.000 description 16
- HZHFFEYYPYZMNU-UHFFFAOYSA-K gadodiamide Chemical compound [Gd+3].CNC(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CCN(CC([O-])=O)CC(=O)NC HZHFFEYYPYZMNU-UHFFFAOYSA-K 0.000 description 16
- 238000006467 substitution reaction Methods 0.000 description 16
- 208000026310 Breast neoplasm Diseases 0.000 description 15
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 15
- 229910052688 Gadolinium Inorganic materials 0.000 description 15
- 238000003917 TEM image Methods 0.000 description 15
- 230000008499 blood brain barrier function Effects 0.000 description 15
- 210000001218 blood-brain barrier Anatomy 0.000 description 15
- 206010006187 Breast cancer Diseases 0.000 description 14
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 14
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 14
- 238000012377 drug delivery Methods 0.000 description 14
- 239000000975 dye Substances 0.000 description 14
- UIWYJDYFSGRHKR-UHFFFAOYSA-N gadolinium atom Chemical compound [Gd] UIWYJDYFSGRHKR-UHFFFAOYSA-N 0.000 description 14
- 239000008103 glucose Substances 0.000 description 14
- 229910052751 metal Inorganic materials 0.000 description 14
- 239000002184 metal Substances 0.000 description 14
- 239000011780 sodium chloride Substances 0.000 description 14
- 208000003174 Brain Neoplasms Diseases 0.000 description 13
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 13
- 239000000725 suspension Substances 0.000 description 13
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 12
- 239000002253 acid Substances 0.000 description 12
- 230000006399 behavior Effects 0.000 description 12
- 238000003384 imaging method Methods 0.000 description 12
- 239000002609 medium Substances 0.000 description 12
- 230000008569 process Effects 0.000 description 12
- 230000035945 sensitivity Effects 0.000 description 12
- 238000001228 spectrum Methods 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 230000008685 targeting Effects 0.000 description 12
- 238000012360 testing method Methods 0.000 description 12
- HPZOOQSXPMEJBV-ODCFVKFUSA-N Tirilazad mesylate Chemical compound CS(O)(=O)=O.O=C([C@@H]1[C@@]2(C)CC=C3[C@@]4(C)C=CC(=O)C=C4CC[C@H]3[C@@H]2C[C@H]1C)CN(CC1)CCN1C(N=1)=CC(N2CCCC2)=NC=1N1CCCC1 HPZOOQSXPMEJBV-ODCFVKFUSA-N 0.000 description 11
- 230000001419 dependent effect Effects 0.000 description 11
- 239000006185 dispersion Substances 0.000 description 11
- 210000003494 hepatocyte Anatomy 0.000 description 11
- 239000004816 latex Substances 0.000 description 11
- 229920000126 latex Polymers 0.000 description 11
- 238000002595 magnetic resonance imaging Methods 0.000 description 11
- 238000005259 measurement Methods 0.000 description 11
- 230000004083 survival effect Effects 0.000 description 11
- 230000007704 transition Effects 0.000 description 11
- 102000011279 Multidrug resistance protein 1 Human genes 0.000 description 10
- 239000007864 aqueous solution Substances 0.000 description 10
- 229910052799 carbon Inorganic materials 0.000 description 10
- 230000003247 decreasing effect Effects 0.000 description 10
- 238000002296 dynamic light scattering Methods 0.000 description 10
- 239000000499 gel Substances 0.000 description 10
- 229920000578 graft copolymer Polymers 0.000 description 10
- 238000000338 in vitro Methods 0.000 description 10
- 229910052757 nitrogen Inorganic materials 0.000 description 10
- 230000001988 toxicity Effects 0.000 description 10
- 231100000419 toxicity Toxicity 0.000 description 10
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 210000000170 cell membrane Anatomy 0.000 description 9
- 229920001519 homopolymer Polymers 0.000 description 9
- 239000000017 hydrogel Substances 0.000 description 9
- 150000002500 ions Chemical class 0.000 description 9
- 230000002829 reductive effect Effects 0.000 description 9
- 239000000126 substance Substances 0.000 description 9
- 229920001897 terpolymer Polymers 0.000 description 9
- 230000036326 tumor accumulation Effects 0.000 description 9
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 8
- 230000003833 cell viability Effects 0.000 description 8
- 239000000306 component Substances 0.000 description 8
- 238000004132 cross linking Methods 0.000 description 8
- 238000009792 diffusion process Methods 0.000 description 8
- 238000012674 dispersion polymerization Methods 0.000 description 8
- 230000029142 excretion Effects 0.000 description 8
- 238000011049 filling Methods 0.000 description 8
- 229910052739 hydrogen Inorganic materials 0.000 description 8
- 210000003734 kidney Anatomy 0.000 description 8
- 239000012528 membrane Substances 0.000 description 8
- 238000005580 one pot reaction Methods 0.000 description 8
- 238000012856 packing Methods 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 8
- 235000019345 sodium thiosulphate Nutrition 0.000 description 8
- 230000006641 stabilisation Effects 0.000 description 8
- 238000011105 stabilization Methods 0.000 description 8
- GZAJOEGTZDUSKS-UHFFFAOYSA-N 5-aminofluorescein Chemical compound C12=CC=C(O)C=C2OC2=CC(O)=CC=C2C21OC(=O)C1=CC(N)=CC=C21 GZAJOEGTZDUSKS-UHFFFAOYSA-N 0.000 description 7
- 229920000856 Amylose Polymers 0.000 description 7
- 238000005481 NMR spectroscopy Methods 0.000 description 7
- 230000002776 aggregation Effects 0.000 description 7
- 230000004700 cellular uptake Effects 0.000 description 7
- 238000012512 characterization method Methods 0.000 description 7
- 150000004665 fatty acids Chemical class 0.000 description 7
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 7
- 238000002073 fluorescence micrograph Methods 0.000 description 7
- 239000001257 hydrogen Substances 0.000 description 7
- 238000002354 inductively-coupled plasma atomic emission spectroscopy Methods 0.000 description 7
- 230000003993 interaction Effects 0.000 description 7
- 210000005229 liver cell Anatomy 0.000 description 7
- 239000000463 material Substances 0.000 description 7
- 238000013421 nuclear magnetic resonance imaging Methods 0.000 description 7
- 108090000765 processed proteins & peptides Proteins 0.000 description 7
- 239000003381 stabilizer Substances 0.000 description 7
- 229940124597 therapeutic agent Drugs 0.000 description 7
- 210000004881 tumor cell Anatomy 0.000 description 7
- 210000005166 vasculature Anatomy 0.000 description 7
- DBCAQXHNJOFNGC-UHFFFAOYSA-N 4-bromo-1,1,1-trifluorobutane Chemical compound FC(F)(F)CCCBr DBCAQXHNJOFNGC-UHFFFAOYSA-N 0.000 description 6
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 6
- 102100029470 Apolipoprotein E Human genes 0.000 description 6
- 101710095339 Apolipoprotein E Proteins 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 102000007330 LDL Lipoproteins Human genes 0.000 description 6
- 108010007622 LDL Lipoproteins Proteins 0.000 description 6
- LXEKPEMOWBOYRF-UHFFFAOYSA-N [2-[(1-azaniumyl-1-imino-2-methylpropan-2-yl)diazenyl]-2-methylpropanimidoyl]azanium;dichloride Chemical compound Cl.Cl.NC(=N)C(C)(C)N=NC(C)(C)C(N)=N LXEKPEMOWBOYRF-UHFFFAOYSA-N 0.000 description 6
- 125000003342 alkenyl group Chemical group 0.000 description 6
- 230000017531 blood circulation Effects 0.000 description 6
- 210000004204 blood vessel Anatomy 0.000 description 6
- 238000000502 dialysis Methods 0.000 description 6
- 235000014113 dietary fatty acids Nutrition 0.000 description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- 238000010494 dissociation reaction Methods 0.000 description 6
- 230000005593 dissociations Effects 0.000 description 6
- STVZJERGLQHEKB-UHFFFAOYSA-N ethylene glycol dimethacrylate Substances CC(=C)C(=O)OCCOC(=O)C(C)=C STVZJERGLQHEKB-UHFFFAOYSA-N 0.000 description 6
- 238000002474 experimental method Methods 0.000 description 6
- 239000000194 fatty acid Substances 0.000 description 6
- 229930195729 fatty acid Natural products 0.000 description 6
- 239000007850 fluorescent dye Substances 0.000 description 6
- 210000002216 heart Anatomy 0.000 description 6
- 238000011835 investigation Methods 0.000 description 6
- 230000005291 magnetic effect Effects 0.000 description 6
- 230000014759 maintenance of location Effects 0.000 description 6
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 6
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 6
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 6
- 238000003918 potentiometric titration Methods 0.000 description 6
- 230000004043 responsiveness Effects 0.000 description 6
- 229960004641 rituximab Drugs 0.000 description 6
- 230000008961 swelling Effects 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 210000003462 vein Anatomy 0.000 description 6
- 230000035899 viability Effects 0.000 description 6
- 238000001157 Fourier transform infrared spectrum Methods 0.000 description 5
- 239000002616 MRI contrast agent Substances 0.000 description 5
- 241001529936 Murinae Species 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 5
- 229930012538 Paclitaxel Natural products 0.000 description 5
- 241000700159 Rattus Species 0.000 description 5
- GLNADSQYFUSGOU-GPTZEZBUSA-J Trypan blue Chemical compound [Na+].[Na+].[Na+].[Na+].C1=C(S([O-])(=O)=O)C=C2C=C(S([O-])(=O)=O)C(/N=N/C3=CC=C(C=C3C)C=3C=C(C(=CC=3)\N=N\C=3C(=CC4=CC(=CC(N)=C4C=3O)S([O-])(=O)=O)S([O-])(=O)=O)C)=C(O)C2=C1N GLNADSQYFUSGOU-GPTZEZBUSA-J 0.000 description 5
- 230000000259 anti-tumor effect Effects 0.000 description 5
- 239000002246 antineoplastic agent Substances 0.000 description 5
- 230000037396 body weight Effects 0.000 description 5
- 239000002738 chelating agent Substances 0.000 description 5
- 238000012937 correction Methods 0.000 description 5
- 231100000135 cytotoxicity Toxicity 0.000 description 5
- 230000003013 cytotoxicity Effects 0.000 description 5
- 239000008367 deionised water Substances 0.000 description 5
- 229910021641 deionized water Inorganic materials 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 238000010586 diagram Methods 0.000 description 5
- ZQPPMHVWECSIRJ-MDZDMXLPSA-N elaidic acid Chemical compound CCCCCCCC\C=C\CCCCCCCC(O)=O ZQPPMHVWECSIRJ-MDZDMXLPSA-N 0.000 description 5
- 230000012202 endocytosis Effects 0.000 description 5
- 238000000799 fluorescence microscopy Methods 0.000 description 5
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 5
- 150000002632 lipids Chemical class 0.000 description 5
- 230000007246 mechanism Effects 0.000 description 5
- 230000001404 mediated effect Effects 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 230000036457 multidrug resistance Effects 0.000 description 5
- 235000021313 oleic acid Nutrition 0.000 description 5
- 125000006353 oxyethylene group Chemical group 0.000 description 5
- 229960001592 paclitaxel Drugs 0.000 description 5
- 230000037361 pathway Effects 0.000 description 5
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 5
- 238000005011 time of flight secondary ion mass spectroscopy Methods 0.000 description 5
- 230000008728 vascular permeability Effects 0.000 description 5
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 4
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 4
- RAZLJUXJEOEYAM-UHFFFAOYSA-N 2-[bis[2-(2,6-dioxomorpholin-4-yl)ethyl]azaniumyl]acetate Chemical compound C1C(=O)OC(=O)CN1CCN(CC(=O)O)CCN1CC(=O)OC(=O)C1 RAZLJUXJEOEYAM-UHFFFAOYSA-N 0.000 description 4
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 4
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 4
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 4
- 229920000945 Amylopectin Polymers 0.000 description 4
- 229940123169 Caspase inhibitor Drugs 0.000 description 4
- 229920002261 Corn starch Polymers 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 102000000853 LDL receptors Human genes 0.000 description 4
- 108010001831 LDL receptors Proteins 0.000 description 4
- 239000005642 Oleic acid Substances 0.000 description 4
- 239000004793 Polystyrene Substances 0.000 description 4
- 229920002125 Sokalan® Polymers 0.000 description 4
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 4
- 238000004220 aggregation Methods 0.000 description 4
- 239000003945 anionic surfactant Substances 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 238000009739 binding Methods 0.000 description 4
- 239000012490 blank solution Substances 0.000 description 4
- 210000004781 brain capillary Anatomy 0.000 description 4
- 201000011510 cancer Diseases 0.000 description 4
- 210000003169 central nervous system Anatomy 0.000 description 4
- 229960003901 dacarbazine Drugs 0.000 description 4
- 230000008021 deposition Effects 0.000 description 4
- 238000003745 diagnosis Methods 0.000 description 4
- 239000012153 distilled water Substances 0.000 description 4
- 230000000977 initiatory effect Effects 0.000 description 4
- 210000000936 intestine Anatomy 0.000 description 4
- 238000010253 intravenous injection Methods 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- 230000000116 mitigating effect Effects 0.000 description 4
- 210000004940 nucleus Anatomy 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- JRKICGRDRMAZLK-UHFFFAOYSA-L persulfate group Chemical group S(=O)(=O)([O-])OOS(=O)(=O)[O-] JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 4
- 239000004584 polyacrylic acid Substances 0.000 description 4
- USHAGKDGDHPEEY-UHFFFAOYSA-L potassium persulfate Chemical compound [K+].[K+].[O-]S(=O)(=O)OOS([O-])(=O)=O USHAGKDGDHPEEY-UHFFFAOYSA-L 0.000 description 4
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 4
- 210000004872 soft tissue Anatomy 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 238000001179 sorption measurement Methods 0.000 description 4
- 210000000952 spleen Anatomy 0.000 description 4
- 238000000954 titration curve Methods 0.000 description 4
- 231100000331 toxic Toxicity 0.000 description 4
- 230000002588 toxic effect Effects 0.000 description 4
- 229960003048 vinblastine Drugs 0.000 description 4
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 4
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 206010015866 Extravasation Diseases 0.000 description 3
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-DYCDLGHISA-M Sodium hydroxide-d Chemical compound [Na+].[2H][O-] HEMHJVSKTPXQMS-DYCDLGHISA-M 0.000 description 3
- GBJVAVGBSGRRKN-JYEBCORGSA-N Z-DEVD-FMK Chemical compound COC(=O)C[C@@H](C(=O)CF)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CCC(=O)OC)NC(=O)[C@H](CC(=O)OC)NC(=O)OCC1=CC=CC=C1 GBJVAVGBSGRRKN-JYEBCORGSA-N 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 238000005054 agglomeration Methods 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- 125000000217 alkyl group Chemical group 0.000 description 3
- 125000003277 amino group Chemical group 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 229940041181 antineoplastic drug Drugs 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 239000007853 buffer solution Substances 0.000 description 3
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 description 3
- 210000003855 cell nucleus Anatomy 0.000 description 3
- 230000001413 cellular effect Effects 0.000 description 3
- 230000000536 complexating effect Effects 0.000 description 3
- 239000008120 corn starch Substances 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 230000009881 electrostatic interaction Effects 0.000 description 3
- 230000007613 environmental effect Effects 0.000 description 3
- 230000005284 excitation Effects 0.000 description 3
- 230000007717 exclusion Effects 0.000 description 3
- 230000036251 extravasation Effects 0.000 description 3
- 239000012467 final product Substances 0.000 description 3
- 238000004108 freeze drying Methods 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 238000011081 inoculation Methods 0.000 description 3
- TWNIBLMWSKIRAT-VFUOTHLCSA-N levoglucosan Chemical group O[C@@H]1[C@@H](O)[C@H](O)[C@H]2CO[C@@H]1O2 TWNIBLMWSKIRAT-VFUOTHLCSA-N 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- 230000001926 lymphatic effect Effects 0.000 description 3
- 229920002521 macromolecule Polymers 0.000 description 3
- 210000004088 microvessel Anatomy 0.000 description 3
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 3
- 239000011022 opal Substances 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 210000003491 skin Anatomy 0.000 description 3
- 238000003860 storage Methods 0.000 description 3
- 239000013589 supplement Substances 0.000 description 3
- 229910052723 transition metal Inorganic materials 0.000 description 3
- 150000003624 transition metals Chemical class 0.000 description 3
- 230000032258 transport Effects 0.000 description 3
- 238000005199 ultracentrifugation Methods 0.000 description 3
- 210000001631 vena cava inferior Anatomy 0.000 description 3
- 229920002554 vinyl polymer Polymers 0.000 description 3
- 238000000733 zeta-potential measurement Methods 0.000 description 3
- YWWVWXASSLXJHU-AATRIKPKSA-N (9E)-tetradecenoic acid Chemical compound CCCC\C=C\CCCCCCCC(O)=O YWWVWXASSLXJHU-AATRIKPKSA-N 0.000 description 2
- HJTAZXHBEBIQQX-UHFFFAOYSA-N 1,5-bis(chloromethyl)naphthalene Chemical compound C1=CC=C2C(CCl)=CC=CC2=C1CCl HJTAZXHBEBIQQX-UHFFFAOYSA-N 0.000 description 2
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- QGJZLNKBHJESQX-UHFFFAOYSA-N 3-Epi-Betulin-Saeure Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C)CCC5(C(O)=O)CCC(C(=C)C)C5C4CCC3C21C QGJZLNKBHJESQX-UHFFFAOYSA-N 0.000 description 2
- CLOUCVRNYSHRCF-UHFFFAOYSA-N 3beta-Hydroxy-20(29)-Lupen-3,27-oic acid Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C(O)=O)CCC5(C)CCC(C(=C)C)C5C4CCC3C21C CLOUCVRNYSHRCF-UHFFFAOYSA-N 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- 208000023275 Autoimmune disease Diseases 0.000 description 2
- DIZWSDNSTNAYHK-XGWVBXMLSA-N Betulinic acid Natural products CC(=C)[C@@H]1C[C@H]([C@H]2CC[C@]3(C)[C@H](CC[C@@H]4[C@@]5(C)CC[C@H](O)C(C)(C)[C@@H]5CC[C@@]34C)[C@@H]12)C(=O)O DIZWSDNSTNAYHK-XGWVBXMLSA-N 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- 206010008874 Chronic Fatigue Syndrome Diseases 0.000 description 2
- 229920000742 Cotton Polymers 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- 206010012289 Dementia Diseases 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- 240000001624 Espostoa lanata Species 0.000 description 2
- 235000009161 Espostoa lanata Nutrition 0.000 description 2
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- 206010019233 Headaches Diseases 0.000 description 2
- 206010019851 Hepatotoxicity Diseases 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 2
- 208000015439 Lysosomal storage disease Diseases 0.000 description 2
- 231100000002 MTT assay Toxicity 0.000 description 2
- 238000000134 MTT assay Methods 0.000 description 2
- 201000009906 Meningitis Diseases 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 2
- BAPJBEWLBFYGME-UHFFFAOYSA-N Methyl acrylate Chemical compound COC(=O)C=C BAPJBEWLBFYGME-UHFFFAOYSA-N 0.000 description 2
- 229930192392 Mitomycin Natural products 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- NIXOWILDQLNWCW-UHFFFAOYSA-N acrylic acid group Chemical group C(C=C)(=O)O NIXOWILDQLNWCW-UHFFFAOYSA-N 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 210000000577 adipose tissue Anatomy 0.000 description 2
- 206010064930 age-related macular degeneration Diseases 0.000 description 2
- DTOSIQBPPRVQHS-PDBXOOCHSA-N alpha-linolenic acid Chemical compound CC\C=C/C\C=C/C\C=C/CCCCCCCC(O)=O DTOSIQBPPRVQHS-PDBXOOCHSA-N 0.000 description 2
- 208000008445 altitude sickness Diseases 0.000 description 2
- JKOQGQFVAUAYPM-UHFFFAOYSA-N amifostine Chemical compound NCCCNCCSP(O)(O)=O JKOQGQFVAUAYPM-UHFFFAOYSA-N 0.000 description 2
- 229960001097 amifostine Drugs 0.000 description 2
- APUPEJJSWDHEBO-UHFFFAOYSA-P ammonium molybdate Chemical compound [NH4+].[NH4+].[O-][Mo]([O-])(=O)=O APUPEJJSWDHEBO-UHFFFAOYSA-P 0.000 description 2
- 235000018660 ammonium molybdate Nutrition 0.000 description 2
- 239000011609 ammonium molybdate Substances 0.000 description 2
- 229940010552 ammonium molybdate Drugs 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 239000012736 aqueous medium Substances 0.000 description 2
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 2
- GOLCXWYRSKYTSP-UHFFFAOYSA-N arsenic trioxide Inorganic materials O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 2
- 238000010560 atom transfer radical polymerization reaction Methods 0.000 description 2
- DVQHYTBCTGYNNN-UHFFFAOYSA-N azane;cyclobutane-1,1-dicarboxylic acid;platinum Chemical compound N.N.[Pt].OC(=O)C1(C(O)=O)CCC1 DVQHYTBCTGYNNN-UHFFFAOYSA-N 0.000 description 2
- 108010071933 benzoylcarbonyl-aspartyl-glutamyl-valyl-aspartyl-fluoromethyl ketone Proteins 0.000 description 2
- QGJZLNKBHJESQX-FZFNOLFKSA-N betulinic acid Chemical compound C1C[C@H](O)C(C)(C)[C@@H]2CC[C@@]3(C)[C@]4(C)CC[C@@]5(C(O)=O)CC[C@@H](C(=C)C)[C@@H]5[C@H]4CC[C@@H]3[C@]21C QGJZLNKBHJESQX-FZFNOLFKSA-N 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 230000029918 bioluminescence Effects 0.000 description 2
- 238000005415 bioluminescence Methods 0.000 description 2
- 229960001561 bleomycin Drugs 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 2
- 229960001467 bortezomib Drugs 0.000 description 2
- 229960003065 bosentan Drugs 0.000 description 2
- GJPICJJJRGTNOD-UHFFFAOYSA-N bosentan Chemical compound COC1=CC=CC=C1OC(C(=NC(=N1)C=2N=CC=CN=2)OCCO)=C1NS(=O)(=O)C1=CC=C(C(C)(C)C)C=C1 GJPICJJJRGTNOD-UHFFFAOYSA-N 0.000 description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 description 2
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 210000000748 cardiovascular system Anatomy 0.000 description 2
- 229960005243 carmustine Drugs 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 2
- 229960000590 celecoxib Drugs 0.000 description 2
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 238000001246 colloidal dispersion Methods 0.000 description 2
- 238000010668 complexation reaction Methods 0.000 description 2
- 238000007334 copolymerization reaction Methods 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- 238000006356 dehydrogenation reaction Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- NIJJYAXOARWZEE-UHFFFAOYSA-N di-n-propyl-acetic acid Natural products CCCC(C(O)=O)CCC NIJJYAXOARWZEE-UHFFFAOYSA-N 0.000 description 2
- PZXJOHSZQAEJFE-UHFFFAOYSA-N dihydrobetulinic acid Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C)CCC5(C(O)=O)CCC(C(C)C)C5C4CCC3C21C PZXJOHSZQAEJFE-UHFFFAOYSA-N 0.000 description 2
- 229960003668 docetaxel Drugs 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 206010014599 encephalitis Diseases 0.000 description 2
- 210000002889 endothelial cell Anatomy 0.000 description 2
- 206010015037 epilepsy Diseases 0.000 description 2
- 238000011067 equilibration Methods 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 229960005167 everolimus Drugs 0.000 description 2
- 238000013213 extrapolation Methods 0.000 description 2
- 210000003608 fece Anatomy 0.000 description 2
- 238000000684 flow cytometry Methods 0.000 description 2
- ZFKJVJIDPQDDFY-UHFFFAOYSA-N fluorescamine Chemical compound C12=CC=CC=C2C(=O)OC1(C1=O)OC=C1C1=CC=CC=C1 ZFKJVJIDPQDDFY-UHFFFAOYSA-N 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 2
- 229960002074 flutamide Drugs 0.000 description 2
- 235000013305 food Nutrition 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 231100000869 headache Toxicity 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 230000007686 hepatotoxicity Effects 0.000 description 2
- 231100000304 hepatotoxicity Toxicity 0.000 description 2
- 125000001183 hydrocarbyl group Chemical group 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 229960003685 imatinib mesylate Drugs 0.000 description 2
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 206010022437 insomnia Diseases 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 238000007917 intracranial administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- 239000004310 lactic acid Substances 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 2
- 229960004942 lenalidomide Drugs 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 208000002780 macular degeneration Diseases 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 229960001428 mercaptopurine Drugs 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- 229960000485 methotrexate Drugs 0.000 description 2
- 238000012703 microemulsion polymerization Methods 0.000 description 2
- 229960004857 mitomycin Drugs 0.000 description 2
- 125000000896 monocarboxylic acid group Chemical group 0.000 description 2
- 210000000865 mononuclear phagocyte system Anatomy 0.000 description 2
- 208000029766 myalgic encephalomeyelitis/chronic fatigue syndrome Diseases 0.000 description 2
- ZIUHHBKFKCYYJD-UHFFFAOYSA-N n,n'-methylenebisacrylamide Chemical compound C=CC(=O)NCNC(=O)C=C ZIUHHBKFKCYYJD-UHFFFAOYSA-N 0.000 description 2
- 201000003631 narcolepsy Diseases 0.000 description 2
- MQYXUWHLBZFQQO-UHFFFAOYSA-N nepehinol Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C)CCC5(C)CCC(C(=C)C)C5C4CCC3C21C MQYXUWHLBZFQQO-UHFFFAOYSA-N 0.000 description 2
- 208000015122 neurodegenerative disease Diseases 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid group Chemical class C(CCCCCCC\C=C/CCCCCCCC)(=O)O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 2
- 229960001756 oxaliplatin Drugs 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 239000006174 pH buffer Substances 0.000 description 2
- SECPZKHBENQXJG-FPLPWBNLSA-N palmitoleic acid Chemical compound CCCCCC\C=C/CCCCCCCC(O)=O SECPZKHBENQXJG-FPLPWBNLSA-N 0.000 description 2
- 230000005298 paramagnetic effect Effects 0.000 description 2
- 229960003330 pentetic acid Drugs 0.000 description 2
- 230000002093 peripheral effect Effects 0.000 description 2
- 229920000771 poly (alkylcyanoacrylate) Polymers 0.000 description 2
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 description 2
- 229920000058 polyacrylate Polymers 0.000 description 2
- 239000011148 porous material Substances 0.000 description 2
- 229920001592 potato starch Polymers 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 2
- 229960004618 prednisone Drugs 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 230000005588 protonation Effects 0.000 description 2
- 238000010926 purge Methods 0.000 description 2
- 239000012264 purified product Substances 0.000 description 2
- 150000003214 pyranose derivatives Chemical group 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000000527 sonication Methods 0.000 description 2
- 210000004003 subcutaneous fat Anatomy 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 230000002459 sustained effect Effects 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- 150000003512 tertiary amines Chemical group 0.000 description 2
- 210000001578 tight junction Anatomy 0.000 description 2
- 230000036962 time dependent Effects 0.000 description 2
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 description 2
- 229960003087 tioguanine Drugs 0.000 description 2
- 231100000041 toxicology testing Toxicity 0.000 description 2
- 230000031998 transcytosis Effects 0.000 description 2
- 238000012546 transfer Methods 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 238000004627 transmission electron microscopy Methods 0.000 description 2
- 229960000575 trastuzumab Drugs 0.000 description 2
- PNYPSKHTTCTAMD-UHFFFAOYSA-K trichlorogadolinium;hexahydrate Chemical compound O.O.O.O.O.O.[Cl-].[Cl-].[Cl-].[Gd+3] PNYPSKHTTCTAMD-UHFFFAOYSA-K 0.000 description 2
- 210000003932 urinary bladder Anatomy 0.000 description 2
- MSRILKIQRXUYCT-UHFFFAOYSA-M valproate semisodium Chemical compound [Na+].CCCC(C(O)=O)CCC.CCCC(C([O-])=O)CCC MSRILKIQRXUYCT-UHFFFAOYSA-M 0.000 description 2
- 229960000604 valproic acid Drugs 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 230000002861 ventricular Effects 0.000 description 2
- 238000012800 visualization Methods 0.000 description 2
- 230000004580 weight loss Effects 0.000 description 2
- 229920001285 xanthan gum Polymers 0.000 description 2
- 239000000230 xanthan gum Substances 0.000 description 2
- 229940082509 xanthan gum Drugs 0.000 description 2
- ORZHVTYKPFFVMG-UHFFFAOYSA-N xylenol orange Chemical compound OC(=O)CN(CC(O)=O)CC1=C(O)C(C)=CC(C2(C3=CC=CC=C3S(=O)(=O)O2)C=2C=C(CN(CC(O)=O)CC(O)=O)C(O)=C(C)C=2)=C1 ORZHVTYKPFFVMG-UHFFFAOYSA-N 0.000 description 2
- 150000007934 α,β-unsaturated carboxylic acids Chemical class 0.000 description 2
- OMDQUFIYNPYJFM-XKDAHURESA-N (2r,3r,4s,5r,6s)-2-(hydroxymethyl)-6-[[(2r,3s,4r,5s,6r)-4,5,6-trihydroxy-3-[(2s,3s,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]methoxy]oxane-3,4,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O[C@H]2[C@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@H](O)[C@H](O)O1 OMDQUFIYNPYJFM-XKDAHURESA-N 0.000 description 1
- DBTMGCOVALSLOR-DEVYUCJPSA-N (2s,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-6-(hydroxymethyl)-4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]oxy-6-(hydroxymethyl)oxane-2,3,5-triol Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](CO)O[C@H](O)[C@@H]2O)O)O[C@H](CO)[C@H]1O DBTMGCOVALSLOR-DEVYUCJPSA-N 0.000 description 1
- OYHQOLUKZRVURQ-NTGFUMLPSA-N (9Z,12Z)-9,10,12,13-tetratritiooctadeca-9,12-dienoic acid Chemical compound C(CCCCCCC\C(=C(/C\C(=C(/CCCCC)\[3H])\[3H])\[3H])\[3H])(=O)O OYHQOLUKZRVURQ-NTGFUMLPSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- YOCIJWAHRAJQFT-UHFFFAOYSA-N 2-bromo-2-methylpropanoyl bromide Chemical compound CC(C)(Br)C(Br)=O YOCIJWAHRAJQFT-UHFFFAOYSA-N 0.000 description 1
- TWJNQYPJQDRXPH-UHFFFAOYSA-N 2-cyanobenzohydrazide Chemical compound NNC(=O)C1=CC=CC=C1C#N TWJNQYPJQDRXPH-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- YWWVWXASSLXJHU-UHFFFAOYSA-N 9E-tetradecenoic acid Natural products CCCCC=CCCCCCCCC(O)=O YWWVWXASSLXJHU-UHFFFAOYSA-N 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- DPUOLQHDNGRHBS-UHFFFAOYSA-N Brassidinsaeure Natural products CCCCCCCCC=CCCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-UHFFFAOYSA-N 0.000 description 1
- 239000007848 Bronsted acid Substances 0.000 description 1
- 229920000018 Callose Polymers 0.000 description 1
- 206010048610 Cardiotoxicity Diseases 0.000 description 1
- 235000000378 Caryota urens Nutrition 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- 229920000887 Chrysolaminarin Polymers 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 240000000163 Cycas revoluta Species 0.000 description 1
- 235000008601 Cycas revoluta Nutrition 0.000 description 1
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 1
- CYCGRDQQIOGCKX-UHFFFAOYSA-N Dehydro-luciferin Natural products OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 1
- MWWSFMDVAYGXBV-RUELKSSGSA-N Doxorubicin hydrochloride Chemical compound Cl.O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-RUELKSSGSA-N 0.000 description 1
- URXZXNYJPAJJOQ-UHFFFAOYSA-N Erucic acid Natural products CCCCCCC=CCCCCCCCCCCCC(O)=O URXZXNYJPAJJOQ-UHFFFAOYSA-N 0.000 description 1
- 206010015548 Euthanasia Diseases 0.000 description 1
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 1
- 229920000855 Fucoidan Polymers 0.000 description 1
- 229920000926 Galactomannan Polymers 0.000 description 1
- 244000068988 Glycine max Species 0.000 description 1
- 235000010469 Glycine max Nutrition 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 244000017020 Ipomoea batatas Species 0.000 description 1
- 235000002678 Ipomoea batatas Nutrition 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 229920001543 Laminarin Polymers 0.000 description 1
- 239000005717 Laminarin Substances 0.000 description 1
- 102000004895 Lipoproteins Human genes 0.000 description 1
- 108090001030 Lipoproteins Proteins 0.000 description 1
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 1
- 235000019759 Maize starch Nutrition 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 229920000057 Mannan Polymers 0.000 description 1
- 235000010804 Maranta arundinacea Nutrition 0.000 description 1
- 235000010103 Metroxylon rumphii Nutrition 0.000 description 1
- 235000021360 Myristic acid Nutrition 0.000 description 1
- TUNFSRHWOTWDNC-UHFFFAOYSA-N Myristic acid Natural products CCCCCCCCCCCCCC(O)=O TUNFSRHWOTWDNC-UHFFFAOYSA-N 0.000 description 1
- 240000002853 Nelumbo nucifera Species 0.000 description 1
- 235000006508 Nelumbo nucifera Nutrition 0.000 description 1
- 235000006510 Nelumbo pentapetala Nutrition 0.000 description 1
- 235000021319 Palmitoleic acid Nutrition 0.000 description 1
- 206010057249 Phagocytosis Diseases 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 240000005893 Pteridium aquilinum Species 0.000 description 1
- 235000009936 Pteridium aquilinum Nutrition 0.000 description 1
- 229920005654 Sephadex Polymers 0.000 description 1
- 239000012507 Sephadex™ Substances 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 244000145580 Thalia geniculata Species 0.000 description 1
- 235000012419 Thalia geniculata Nutrition 0.000 description 1
- UWHZIFQPPBDJPM-FPLPWBNLSA-M Vaccenic acid Natural products CCCCCC\C=C/CCCCCCCCCC([O-])=O UWHZIFQPPBDJPM-FPLPWBNLSA-M 0.000 description 1
- 235000021322 Vaccenic acid Nutrition 0.000 description 1
- 238000005411 Van der Waals force Methods 0.000 description 1
- 230000006682 Warburg effect Effects 0.000 description 1
- 241000021375 Xenogenes Species 0.000 description 1
- UGXQOOQUZRUVSS-ZZXKWVIFSA-N [5-[3,5-dihydroxy-2-(1,3,4-trihydroxy-5-oxopentan-2-yl)oxyoxan-4-yl]oxy-3,4-dihydroxyoxolan-2-yl]methyl (e)-3-(4-hydroxyphenyl)prop-2-enoate Chemical compound OC1C(OC(CO)C(O)C(O)C=O)OCC(O)C1OC1C(O)C(O)C(COC(=O)\C=C\C=2C=CC(O)=CC=2)O1 UGXQOOQUZRUVSS-ZZXKWVIFSA-N 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 125000000278 alkyl amino alkyl group Chemical group 0.000 description 1
- JAZBEHYOTPTENJ-JLNKQSITSA-N all-cis-5,8,11,14,17-icosapentaenoic acid Chemical compound CC\C=C/C\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O JAZBEHYOTPTENJ-JLNKQSITSA-N 0.000 description 1
- WQZGKKKJIJFFOK-DVKNGEFBSA-N alpha-D-glucose Chemical group OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-DVKNGEFBSA-N 0.000 description 1
- 235000020661 alpha-linolenic acid Nutrition 0.000 description 1
- 210000003484 anatomy Anatomy 0.000 description 1
- 238000002583 angiography Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 210000000709 aorta Anatomy 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 229920000617 arabinoxylan Polymers 0.000 description 1
- 229940114079 arachidonic acid Drugs 0.000 description 1
- 235000021342 arachidonic acid Nutrition 0.000 description 1
- XKRFYHLGVUSROY-UHFFFAOYSA-N argon Substances [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 230000003416 augmentation Effects 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 229920001222 biopolymer Polymers 0.000 description 1
- 239000012503 blood component Substances 0.000 description 1
- 230000036770 blood supply Effects 0.000 description 1
- 210000004782 brain capillary endothelium Anatomy 0.000 description 1
- 210000005013 brain tissue Anatomy 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 150000007942 carboxylates Chemical group 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 231100000259 cardiotoxicity Toxicity 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 239000013553 cell monolayer Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 235000013339 cereals Nutrition 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- SECPZKHBENQXJG-UHFFFAOYSA-N cis-palmitoleic acid Natural products CCCCCCC=CCCCCCCCC(O)=O SECPZKHBENQXJG-UHFFFAOYSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 230000008045 co-localization Effects 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000010415 colloidal nanoparticle Substances 0.000 description 1
- 239000000084 colloidal system Substances 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 239000013068 control sample Substances 0.000 description 1
- 239000000599 controlled substance Substances 0.000 description 1
- 229920001577 copolymer Polymers 0.000 description 1
- 210000000877 corpus callosum Anatomy 0.000 description 1
- 230000001054 cortical effect Effects 0.000 description 1
- 239000011557 critical solution Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 210000000172 cytosol Anatomy 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 229940124447 delivery agent Drugs 0.000 description 1
- 239000000412 dendrimer Substances 0.000 description 1
- 229920000736 dendritic polymer Polymers 0.000 description 1
- 230000005595 deprotonation Effects 0.000 description 1
- 238000010537 deprotonation reaction Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- 238000007323 disproportionation reaction Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229960002918 doxorubicin hydrochloride Drugs 0.000 description 1
- 230000035622 drinking Effects 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 229960005135 eicosapentaenoic acid Drugs 0.000 description 1
- 235000020673 eicosapentaenoic acid Nutrition 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000010894 electron beam technology Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 230000008290 endocytic mechanism Effects 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 210000004783 epithelial tight junction Anatomy 0.000 description 1
- DPUOLQHDNGRHBS-KTKRTIGZSA-N erucic acid Chemical compound CCCCCCCC\C=C/CCCCCCCCCCCC(O)=O DPUOLQHDNGRHBS-KTKRTIGZSA-N 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- IDGUHHHQCWSQLU-UHFFFAOYSA-N ethanol;hydrate Chemical compound O.CCO IDGUHHHQCWSQLU-UHFFFAOYSA-N 0.000 description 1
- 238000006266 etherification reaction Methods 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 230000021824 exploration behavior Effects 0.000 description 1
- 210000001723 extracellular space Anatomy 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 239000008394 flocculating agent Substances 0.000 description 1
- 238000012921 fluorescence analysis Methods 0.000 description 1
- 235000012041 food component Nutrition 0.000 description 1
- 239000005417 food ingredient Substances 0.000 description 1
- LGMLJQFQKXPRGA-VPVMAENOSA-K gadopentetate dimeglumine Chemical compound [Gd+3].CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO.OC(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O LGMLJQFQKXPRGA-VPVMAENOSA-K 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000004190 glucose uptake Effects 0.000 description 1
- 125000003827 glycol group Chemical group 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 150000003278 haem Chemical class 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 150000002402 hexoses Chemical class 0.000 description 1
- 231100000086 high toxicity Toxicity 0.000 description 1
- 230000036571 hydration Effects 0.000 description 1
- 238000006703 hydration reaction Methods 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 230000007154 intracellular accumulation Effects 0.000 description 1
- 239000007928 intraperitoneal injection Substances 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 125000003010 ionic group Chemical group 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 229910052747 lanthanoid Inorganic materials 0.000 description 1
- 150000002602 lanthanoids Chemical class 0.000 description 1
- 238000002356 laser light scattering Methods 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 125000005647 linker group Chemical group 0.000 description 1
- OYHQOLUKZRVURQ-AVQMFFATSA-N linoelaidic acid Chemical compound CCCCC\C=C\C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-AVQMFFATSA-N 0.000 description 1
- OYHQOLUKZRVURQ-IXWMQOLASA-N linoleic acid Natural products CCCCC\C=C/C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-IXWMQOLASA-N 0.000 description 1
- 229960004488 linolenic acid Drugs 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 210000001365 lymphatic vessel Anatomy 0.000 description 1
- 210000003712 lysosome Anatomy 0.000 description 1
- 230000001868 lysosomic effect Effects 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 239000005267 main chain polymer Substances 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 239000000693 micelle Substances 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 210000004925 microvascular endothelial cell Anatomy 0.000 description 1
- 230000005012 migration Effects 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 239000007758 minimum essential medium Substances 0.000 description 1
- 230000002107 myocardial effect Effects 0.000 description 1
- 239000002539 nanocarrier Substances 0.000 description 1
- 239000002086 nanomaterial Substances 0.000 description 1
- 210000002569 neuron Anatomy 0.000 description 1
- 230000016273 neuron death Effects 0.000 description 1
- 230000006911 nucleation Effects 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical class CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 230000003534 oscillatory effect Effects 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 210000000963 osteoblast Anatomy 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 230000010627 oxidative phosphorylation Effects 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 230000000149 penetrating effect Effects 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 150000002972 pentoses Chemical class 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 230000008782 phagocytosis Effects 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 1
- 229920000867 polyelectrolyte Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920002643 polyglutamic acid Polymers 0.000 description 1
- 229920002338 polyhydroxyethylmethacrylate Polymers 0.000 description 1
- 229940068965 polysorbates Drugs 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 235000019394 potassium persulphate Nutrition 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000004451 qualitative analysis Methods 0.000 description 1
- 238000004445 quantitative analysis Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 239000012966 redox initiator Substances 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 229940100486 rice starch Drugs 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 230000009919 sequestration Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- UGTZMIPZNRIWHX-UHFFFAOYSA-K sodium trimetaphosphate Chemical compound [Na+].[Na+].[Na+].[O-]P1(=O)OP([O-])(=O)OP([O-])(=O)O1 UGTZMIPZNRIWHX-UHFFFAOYSA-K 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 239000012086 standard solution Substances 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 230000002739 subcortical effect Effects 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- DHCDFWKWKRSZHF-UHFFFAOYSA-N sulfurothioic S-acid Chemical compound OS(O)(=O)=S DHCDFWKWKRSZHF-UHFFFAOYSA-N 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000001360 synchronised effect Effects 0.000 description 1
- 238000001308 synthesis method Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 230000001839 systemic circulation Effects 0.000 description 1
- 230000002123 temporal effect Effects 0.000 description 1
- JGVWCANSWKRBCS-UHFFFAOYSA-N tetramethylrhodamine thiocyanate Chemical compound [Cl-].C=12C=CC(N(C)C)=CC2=[O+]C2=CC(N(C)C)=CC=C2C=1C1=CC=C(SC#N)C=C1C(O)=O JGVWCANSWKRBCS-UHFFFAOYSA-N 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- UWHZIFQPPBDJPM-BQYQJAHWSA-N trans-vaccenic acid Chemical compound CCCCCC\C=C\CCCCCCCCCC(O)=O UWHZIFQPPBDJPM-BQYQJAHWSA-N 0.000 description 1
- 230000007723 transport mechanism Effects 0.000 description 1
- 150000004043 trisaccharides Chemical class 0.000 description 1
- 230000036325 urinary excretion Effects 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 229940100445 wheat starch Drugs 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229920001221 xylan Polymers 0.000 description 1
- 150000004823 xylans Chemical class 0.000 description 1
Images























































Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F251/00—Macromolecular compounds obtained by polymerising monomers on to polysaccharides or derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/197—Carboxylic acids, e.g. valproic acid having an amino group the amino and the carboxyl groups being attached to the same acyclic carbon chain, e.g. gamma-aminobutyric acid [GABA], beta-alanine, epsilon-aminocaproic acid or pantothenic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/36—Polysaccharides; Derivatives thereof, e.g. gums, starch, alginate, dextrin, hyaluronic acid, chitosan, inulin, agar or pectin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/001—Preparation for luminescence or biological staining
- A61K49/0063—Preparation for luminescence or biological staining characterised by a special physical or galenical form, e.g. emulsions, microspheres
- A61K49/0069—Preparation for luminescence or biological staining characterised by a special physical or galenical form, e.g. emulsions, microspheres the agent being in a particular physical galenical form
- A61K49/0089—Particulate, powder, adsorbate, bead, sphere
- A61K49/0091—Microparticle, microcapsule, microbubble, microsphere, microbead, i.e. having a size or diameter higher or equal to 1 micrometer
- A61K49/0093—Nanoparticle, nanocapsule, nanobubble, nanosphere, nanobead, i.e. having a size or diameter smaller than 1 micrometer, e.g. polymeric nanoparticle
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/06—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations
- A61K49/18—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by a special physical form, e.g. emulsions, microcapsules, liposomes
- A61K49/1818—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by a special physical form, e.g. emulsions, microcapsules, liposomes particles, e.g. uncoated or non-functionalised microparticles or nanoparticles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/06—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations
- A61K49/18—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by a special physical form, e.g. emulsions, microcapsules, liposomes
- A61K49/1818—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by a special physical form, e.g. emulsions, microcapsules, liposomes particles, e.g. uncoated or non-functionalised microparticles or nanoparticles
- A61K49/1821—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by a special physical form, e.g. emulsions, microcapsules, liposomes particles, e.g. uncoated or non-functionalised microparticles or nanoparticles coated or functionalised microparticles or nanoparticles
- A61K49/1824—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by a special physical form, e.g. emulsions, microcapsules, liposomes particles, e.g. uncoated or non-functionalised microparticles or nanoparticles coated or functionalised microparticles or nanoparticles coated or functionalised nanoparticles
- A61K49/1878—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by a special physical form, e.g. emulsions, microcapsules, liposomes particles, e.g. uncoated or non-functionalised microparticles or nanoparticles coated or functionalised microparticles or nanoparticles coated or functionalised nanoparticles the nanoparticle having a magnetically inert core and a (super)(para)magnetic coating
- A61K49/1881—Nuclear magnetic resonance [NMR] contrast preparations; Magnetic resonance imaging [MRI] contrast preparations characterised by a special physical form, e.g. emulsions, microcapsules, liposomes particles, e.g. uncoated or non-functionalised microparticles or nanoparticles coated or functionalised microparticles or nanoparticles coated or functionalised nanoparticles the nanoparticle having a magnetically inert core and a (super)(para)magnetic coating wherein the coating consists of chelates, i.e. chelating group complexing a (super)(para)magnetic ion, bound to the surface
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/513—Organic macromolecular compounds; Dendrimers
- A61K9/5161—Polysaccharides, e.g. alginate, chitosan, cellulose derivatives; Cyclodextrin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5192—Processes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
- C08J3/14—Powdering or granulating by precipitation from solutions
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y40/00—Manufacture or treatment of nanostructures
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nanotechnology (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Polymers & Plastics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Radiology & Medical Imaging (AREA)
- Optics & Photonics (AREA)
- Physics & Mathematics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Inorganic Chemistry (AREA)
- Molecular Biology (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Graft Or Block Polymers (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Gastroenterology & Hepatology (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Other Resins Obtained By Reactions Not Involving Carbon-To-Carbon Unsaturated Bonds (AREA)
Description
(a) 溶液中にポリマーを可溶化するステップと、
(b) カルボン酸側基を含む重合可能なモノマーを供給するステップと、
(c) モノマーをグラフト重合して可溶化したポリマー上にポリマー鎖を形成するステップと、
(d) 形成している鎖と反応する官能基を有するエトキシ化分子を供給するステップと、
を含み、
ステップ(c)がエトキシ化分子の存在下で行われて、エトキシ化分子をポリマー鎖に共有結合させる、方法である。
(i) 水溶液中にポリマーを可溶化するステップと、
(ii) カルボン酸側基を含む重合可能なモノマーを供給するステップと、
(iii) モノマーをグラフト重合して可溶化したポリマー上にポリマー鎖を形成するステップと、
(iv) 形成している鎖と反応する官能基を有するポリエトキシ化分子を供給するステップと、
を含み、
重合ステップ(iii)がポリエトキシ化分子の存在下で行われ、ポリエトキシ化分子を形成している鎖に共有結合させ、重合生成物がナノ粒子の外側にポリエトキシ化部分を有するナノ粒子を形成する方法である。
(a) 溶液中でポリマーを可溶化するステップ、
(b) アルキルアミノアルキルエステル側基を含む重合可能なモノマーを供給するステップ、
(c) 架橋剤を供給するステップ、及び
(d) モノマーをグラフト重合して可溶化したポリマー上にポリマー鎖を形成して、ナノ粒子を形成するステップ
を含む方法である。
表1は、ナノ粒子調製レシピ及びポリマー組成を示す。反応収率は、フィード中のMAA、PS80、及びデンプンの全重量に対する精製されたターポリマーの割合と定義された。
化学物質及び試薬
可溶性デンプン(MW2,600-4,500Da)、メタクリル酸(MAA)、N,N'-メチレンビスアクリルアミド(MBA)、チオ硫酸ナトリウム(STS)、過硫酸カリウム(KPS)、ポリソルベート80(PS80)、及びドデシル硫酸ナトリウム(SDS)は、Sigma-Aldrich Canada(カナダ、オンタリオ州オークビル)から購入した。MAA阻害剤は、使用前に真空蒸留によって除去された。全てのその他の化学物質は試薬グレードで、入荷されたままで使用された。
ネズミ乳癌株化細胞EMT6/WTは、Dr. Ian Tannock (オンタリオ州癌協会(Ontario Cancer Institute)、カナダ、オンタリオ州トロント)により最初に提供され、現在我々の研究室で維持されている。細胞の単層が、5%CO2/95%空気の加湿した培養器中37℃の75cm2ポリスチレン組織培養瓶で培養された。癌細胞は、10%ウシ胎仔血清(Cansera Inc.、カナダ、オンタリオ州エトビコーク)を補って、α-最小必須培地中で維持された(オンタリオ州癌協会メディアラボラトリー(Ontario Cancer Institute Media Laboratory)、カナダ、オンタリオ州トロント)。コンフルエンスまで成長した細胞は、0.05%トリプシン-EDTA(Invitrogen Inc.、カナダ、オンタリオ州バーリントン)によりトリプシン処理され、新鮮な成長培地中に希釈され(1/10)、再び蒔かれた。
全ての動物研究は、大学健康ネットワーク(University Health Network)における動物実験委員会(animal care committee)によって認可されており、全ての実験は、動物ケアに関するカナダ評議会(Canadian Council on Animal Care)によって出された全ての指針及び規則に従って実施された。生まれて8週間経った雌のBalb/cマウス(Jackson laboratory、米国メイン州)が使用された。その動物は、研究の期間中、食物及び水への自由な接近を許容された。腫瘍の調査については、100万のネズミのEMT6乳癌細胞が、左脇腹に皮下注射された。腫瘍は成長について観察され、MRI調査は、腫瘍の平均直径が5mmのところで開始された。
[実施例1]
ポリメタクリル酸-グラフト-デンプン(PMMA-g-St)ナノ粒子の合成
PMMA-PS-80-g-Stナノ粒子の合成
フリーラジカル分散重合法が、過硫酸カリウム/チオ硫酸ナトリウム開始(KPS/STS)システムを用いてPMMA-g-Stナノ粒子をワンポットで調製するために、使用された。一連の予備調査が、安定な粒子を得るために必要となる適当な界面活性剤タイプ及び濃度並びにモノマー濃度を特定するために実施された。

を用いて計算された。
を用いて計算された。
FTIRスペクトルが、パーキンエルマースペクトル1000シリーズ分光計(米国マサチューセッツ州)に基づいて記録された。スペクトルは、4cm-1の解像度により取られ、32スキャンの中で平均化された。試料は完全に乾燥したKBrと共に十分に粉砕され、ペレットが真空下での圧縮によって作製された。
1H NMR測定結果は、Varian Mercury 400 MHz(米国カリフォルニア州)を使用して得られた。PMAA-g-St(架橋なし)の試料が、0.01MのNaODに溶解されて、15mg/mlの溶液濃度を得た。スペクトルは、25°のパルス角度、10秒の遅延時間、及び2秒の捕捉時間で得られた。全ての化学シフトは水ピークを基準として100万分の1(ppm)で記録されている。
透過電子顕微鏡法(TEM)が、ナノ粒子の形及び形態を検査するために使用された。PBS(pH=7.4)中のナノ粒子懸濁液がモリブデン酸アンモニウムにより染色され、炭素で被覆されたグリッド上に置かれた。その試料は、ろ紙により吸い取られ、そのまま乾燥された。透過電子顕微鏡写真が、100kVの加速電圧によるHitachi H7000電子顕微鏡(Hitachi Canada, Ltd.、カナダオンタリオ州、ミシサーガ)によって取得された。
1つの例において、粒径は、NICOMP(商標)380ZLS(PSSNICOMP、米国カリフォルニア州、サンタバーバラ)装置を用いる動的光散乱(DLS)によって測定された。粒径は、HeNeレーザー光線により90°の検出角度で、37℃で測定された。精製したラテックスが蒸留水中に分散され、Hielscher UP100Hプローブウルトラソニケーター(Hielscher USA, Inc.、米国ニュージャージー州、リングウッド)を、80%のピーク振幅及び溶液中5mmのプローブ深さで5分間用いて、5mg/mlのストックのラテックス懸濁液が調製された。ストック懸濁液は、さまざまなpH及び0.15Mの一定のイオン強度の水性緩衝溶液により10倍に希釈された。得られた希釈ラテックス懸濁液のpHは、pHメーターにより確認された。各試料についての粒径は、3回測定され、その3つのものの平均が記録された。強度加重平均直径(intensity-weighted mean diameter)が、それはオリジナルデータから直接計算され、体積加重及び数加重平均直径よりもより再現性があるので、流体力学的大きさとして使用された。粒径分布は、多分散指数(PdI)を用いて評価された。一般に0.12より小さいPdI値を有する粒子は、単分散とみなされる。
表面電荷に対する粒子組成及びpHの影響を調査するために、粒子のゼータ電位が電気泳動移動度を用いて測定された。ストックのラテックス懸濁液が、異なるpH及び10mMの一定のイオン強度の緩衝溶液により希釈された。ゼータ電位は、Malvern zeta sizer Nano-ZS(Malvern、英国ウースターシャー)を用いてその後測定された。
を用いてゼータ電位(ξ)と関連付けられる。
電位差滴定が、Fisher Scientific Accumet AB15 pHメーター(Fisher Scientific、カナダ、オンタリオ州トロント)により行われた。試料は、0.050gの精製した粒子を50mLの0.05MのNaCl中に懸濁することによって用意された。滴定は、pH電極(Fisher Scientific)、及び窒素ラインを取り付けた、十分にきれいにした温度制御された(25℃)100mLのビーカー中で行われた。ポリマー懸濁液は、電磁攪拌機を用いて連続的に撹拌された。HCl及びNaOHの0.1Mの体積標準溶液(Fisher Scientific、カナダ、オンタリオ州トロント)が滴定剤として使用された。そのラテックスのpHは、3.0まで下げられ、溶解している二酸化炭素をその系から除去するために滴定の前に20分間窒素がそのラテックス中にバブリングされた。不活性な雰囲気を維持するために滴定中に窒素が試料の上に穏やかに吹付けられた。特に断りのない限り、全てのデータは、正(酸中への塩基)滴定を用いて取得された。懸濁液は、平衡を確保するために各滴定剤添加の間において5分間安定化させた。最初の滴定データは、遊離のH+及びOH-の寄与を考慮することによって補正され、終点をよりはっきりとさせた。その補正は、方程式[26、27]:
に従って実施される。この補正によって、分散液中及びブランクの溶液中のH+及びOH-に対する同じ活量係数を仮定すれば、[V]pHの値は、当量点において一定であるはずである。
最大7.2%までの固形分を有する安定なPMAA-g-Stラテックスが上記の方法を用いて調製された。グラフトは、ワンポット合成工程で同時に起こるグラフト及びナノ粒子形成を可能にする修正された水分散液重合法を用いて実施された。その方法は、油及び有機溶媒の使用を必要としないことが見出された。最初に、モノマー、界面活性剤、及び開始剤が全て水に溶解する。
FTIR及びNMR調査を用いて、グラフトを確認し、ポリマー組成及びグラフトの機構を検討した。デンプン、PMAA及びグラフトしたデンプンのFTIRスペクトルが、図2に示されている。天然デンプンのスペクトルと比較して、大きな変化は、1738cm-1におけるカルボニルC=O吸収周波数の存在である。天然デンプンにおける1166cm-1、1090cm-1、1020cm-1、及び954cm-1のピークは、CO結合伸縮に起因している。1090cm-1及び1020cm-1のピークは、無水グルコース環CO/CC伸縮の特徴を示す。1645cm-1の特性ピークは、デンプン中の結合した水の存在に起因する。水素結合したヒドロキシル基(O-H)によるブロードバンドが3450cm-1に現れており、分子間及び分子内結合した遊離のヒドロキシル基に関連した複雑な振動性伸縮によるものである。2940cm-1のバンドは、C-H伸縮の特徴を示す。天然デンプンにおける3450cm-1の強いOH伸縮バンドは、グラフト反応の後に強度が低下しており、デンプンのOH基を介したMAAとのデンプンの反応を暗示している。また、グラフトポリマーは、520〜920cm-1のピラノース環振動及びさらにPMAAのデンプンへのグラフトを確認するMAAホモポリマーにはない1032cm-1のCO/CC環伸縮の特性ピークも示す。
分析された全てのナノ粒子は、およそ100〜200nmの粒径及びどちらかといえば球状の非常に均一な形態を示した(図6A)。このナノ粒子は、多孔質の綿ボール様の表面形態を有する。多孔質構造を有することは、pH等の環境刺激に応じたより速い相転移とともにより高い薬物充填を促進することに関して有利であり得る。ある程度の粒子の凝集及び融合の存在もあるが、これはTEM試料調製の性質によるものであり得る。TEMグリッド上に近接近して置かれる粒子は、乾燥及び電子線の影響によって部分的に融合し得る。そのような挙動は一般的であり、ポリ(2-ヒドロキシエチルメタクリレート)粒子などの他のポリマーナノ粒子についても頻繁に報告されている[37]。
表3及び図8Aにおける結果は、pHを5から7.4まで増すと共に粒径が増大し、その粒径のpHに依存する変化は、MAA/Stのモル比に関連することを実証している。図8は、粒径がpHを4から7.4まで上げると共に増加し、その増加の規模はMAA含量によって決定されることを示している。例えば、PMAAナノ粒子の平均直径は、pH4における70.5nmからpH7.4における152nmまで2.2倍増加し、一方、PMAA-g-St-4は1.2倍に増加するのみであり、言い換えると、PMAAについては10.1倍の、PMAA-g-St-4については1.5倍の体積比(V7.4/V4)になる。一般に、デンプン含量の増加は、pH感度における減少をもたらす。これは、より低いMAA含量はイオン化されたカルボン酸基のより低い含量に起因するものとされるより小さい静電反発力をもたらし、それゆえ膨れを低下させるという事実のせいにされ得る。異なるpH感度は、異なる量のイオン化可能な及び/又はイオン化されたカルボン酸基を意味する。より低いMAA含量は、より小さい静電反発力及びpH7.4におけるより低い膨れをもたらし、より小さいV7.4/V4値につながる。
ナノ粒子のゼータ電位が、表4及び図8Bにまとめられている。このデータは、全てのナノ粒子が、そのナノ粒子と結合しているカルボン酸基のイオン化によって、2から7までの媒体のpHの増大と共に増加する負の表面電荷を有することを示している。ゼータ電位は、液体のイオンによって作り出される電気二重層における電荷であり、それぞれの粒子の周りに存在する。水溶液中に分散されたナノ粒子は、静電的安定化(表面電荷)によって又は立体安定化(粒子表面における界面活性剤又はその他の分子)によって、或いは両方の組合せによって安定化され得る。一般に、+/-20mVを超えるゼータ電位値が安定なコロイド分散の特徴と考えられる。DLVO理論によれば、凝集は、粒子間の引きつけるファンデルワールス力が、静電気反発力を圧するときに起こる。表4及び図8Bに示されているように、殆どのナノ粒子生成物は、調査されたさまざまなpH値において-20mVに近いかそれを超えるゼータ電位を有し、それ故コロイド的に安定であることが期待される。ナノ粒子中のPMAA含量が減少するとゼータ電位も低下する。pH4において、PMAA-g-St-4ナノ粒子は-2.7mVのゼータ電位を有し、それは、それらがいくらかのコロイド安定性の問題を有するであろうことを意味する。これらのデータは、高いpHにおけるより大きい負電荷と共に、PMAAがナノ粒子の表面電荷に大きく寄与することを示唆する。
図9は、Cs=0.05N NaClでのPMAA-g-St-2ラテックス分散液の電位差滴定の例を示す。特別な定めのない限り、5分の安定化時間が各滴定剤の添加の間で設けられた。これは、平衡を得るために要する緩和時間が対応する低分子量の弱酸と比較して普通は非常に長いので、高分子電解質ラテックス粒子の滴定においては常法である。オリジナルの滴定データは、遊離のH+及びOH-の寄与を考慮に入れて補正され、終点をより明瞭にした。その補正は、方程式2[26、27]:
によるイオン化度(α)と関係している[40、41]。
によって計算される。
図12Aは、粒径及び相転移に対するSDS濃度の影響を示す。PMAA-g-St粒子の直径は、界面活性剤含量が増加すると減少した。SDS界面活性剤の両親媒性構造は、水溶液中のポリマー粒子を安定させる助けとなり、それゆえポリマー鎖がより小さい粒子を形成することを可能にする。粒径に対する界面活性剤量の影響は顕著であった。例えば、SDSが0.17mmolから0.69mmolまで変化すると、粒径は591nmから298nmまで変化した。0.17mmol(0.05g)より下のSDSレベルは粒子の凝集をもたらした(矢印により注釈をつけられている)。PMAA-g-StコポリマーはCOOH基のイオン化のために負に帯電され、SDSはアニオン界面活性剤であるために、粒子表面に吸着されたSDSは粒子の凝集を防ぎながら、粒子が水中に分散することを助ける。pH=4における粒径に対するpH=7.4における粒径の比(D7.4/D4)が、pH応答性を測定するために使用された。その比は、低いSDSレベルにおいて減少したが、しかしながら、これは粒子の凝集及びDLSによる誤解させるような高い読みをもたらす低いpH値での減少された量のSDSによる粒子の貧弱な安定性のためである。
本発明のナノ粒子のさまざまな改変により、特に医療に関連する多数の応用においてそれらの有用性が実証された。ナノ粒子の改変バージョンは、そのナノ粒子の合成中、又はそれらの作製後の両方で得ることが可能である。
本明細書で例示されているように、ポリメタクリル酸-グラフト-デンプン-DTPA(PMAA-g-St-DTPA)を含有するデンプンをベースにしたナノ粒子は、水中の単一のワンポット合成方法で合成され得る。そのポリマーは高い親和性によりガドリニウムに結合することができる。この合成方法は、生理的pHにおいて溶解する適切な分子量のポリマーを得るように調整することができる。
抗癌剤ドキソルビシンの送達のためのSt-g-PMAA-P80ナノ粒子
ナノ粒子のドキソルビシン(カチオン性、Mw=579.98g/mol)を取り込む能力が薬物取り込み調査を用いて評価された。15ミリグラムの凍結乾燥されたナノ粒子が10mlの蒸留した脱イオン水(DDIW)中に懸濁される。薬物は、0.1〜5mg/mlの濃度でその懸濁液に加えられ、24時間そのナノ粒子に吸着させられる。その粒子は、次に30000rpmで30分間の超遠心分離にかけられ、上澄み液中の薬物の量が紫外分光光度計を用いて分析される。その後、粒子中に取り込まれた薬物の量が、充填用溶液中の初期の薬物濃度から最後の薬物濃度を差し引くことによって計算される。薬物充填量及び封入効率は、その時次の方程式を用いて計算される:
カスパーゼ阻害剤ペプチドの送達のためのPMAA-PS 80-g-Stナノ粒子
N-ベンジルオキシカルボニル-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-フルオロメチルケトン(Z-DEVD-FMK)などのカスパーゼ阻害剤ペプチドは神経細胞死を減少させることが実証されているが[95〜97]、血液脳関門(BBB)を横断することはできない
水溶性が乏しい薬物、例えば、カスパーゼ阻害剤ペプチドなどを効果的に充填するために、ポリマーに脂質鎖が導入された。典型的な反応において、ミリスチン酸(10mg、0.043mmol)が、5mlのDMSO中でEDC(34mg、0.175mmol)及びNHS(40mg、0.350mmol)と共に室温で1時間インキュベートされ、5mlのDMSO/H2O混合物中に溶解したPMAA-PS80-g-St(200mg)が加えられた。その混合物は、室温で24時間撹拌され、次いで透析物を12時間毎に変える48時間にわたるDDIW(2L、MWCO=12000kDa)に対する透析によって精製された。固体の脂質ポリマーは凍結乾燥によって得られた。Z-DEVD-FMKがモデルのカスパーゼ阻害剤ペプチドとして使用された。自己集合したナノ粒子を調製するために、10mgの脂質-ポリマーが2.0mlのDDI水中に溶解された。その脂質ポリマー溶液は、次に氷浴に入れられ、Hielscher UP100Hプローブウルトラソニケーター(Hielscher USA, Inc.、ニュージャージー州リングウッド、米国)を用いて超音波処理をしながら、200μL(5×40μL)のペプチド溶液(CH3CN/H2O(2/8、v/v)中1mg/ml)が10秒毎に脂質ポリマー溶液に少しずつ増やしながら加えられた。ペプチドを充填したナノ粒子(NPs)の粒径は、ゼータサイザーナノシステムを使用することによって37nmであると測定された。その充填効率は、91%より高いことが見出された。
MDA-MB435/LCC6ヒト乳癌細胞による粒子の取り込み
MDA-MB435/LCC6細胞によるフルオレスカミンで標識化されたナノ粒子の取り込みが、蛍光顕微鏡検査法及びフローサイトメトリーを用いて調査された。フルオレセインアミン(FA)がNPsに共有結合された。簡潔に言うと、200mgのNPsが20mlのDDIW中に分散され、その後50mgのNHS及びEDCが添加された。45分後、10mgのFAを加えることによって反応が開始された。その混合物は光から保護され、室温で24時間撹拌された。そしてpHが7.4に調整され、粒子は次に3回洗浄され、続いて未反応の染料を除去するために遠心分離にかけられた。
野生型及び多剤耐性ヒト乳癌株化細胞に対するNPs抗腫瘍効果のインビトロ評価
遊離のDox及びDoxを充填したナノ粒子のMDA-MB435/LCC6乳癌株化細胞に対する有効性が、野生型及び耐性細胞の両方において評価された(図19)。細胞は増大する濃度のドキソルビシン又はドキソルビシンを充填したナノ粒子により24時間及び48時間処理され、細胞の生存がMTTアッセイを用いて測定された。遊離のDox及びDox充填ナノ粒子の両方とも野生型株化細胞に対して同様に有効であった(図19A、B)。全ての処理が、野生型細胞に対して細胞毒性を用量依存的形で顕在化させた。24時間処理に対するIC50値は、遊離のDox及び薬物充填ナノ粒子に対してそれぞれ0.34μg/ml及び0.32μg/mlであった。これらの値は、処理の長さを48時間に増すことによって0.07μg/ml(遊離のDox)及び0.06μg/ml(ナノ粒子)に減少した。このデータは、ドキソルビシンのナノ粒子中への充填が薬物活性の損失をもたらさないことを示している。
新規なDTPA含有St-g-PMAA-P80ポリマー及びナノ粒子の合成
デュアルモードナノ粒子の調製
PF-NPs及びSA-NPsの概略構造とそれらの調製手順が図20に示されており以下に示すように調製される。
PMAA-g-St-DTPAは、最初にジエチレントリアミンペンタ酢酸二無水物(DTPAビス無水物)をデンプンに結合させ、その後のMAAのグラフト重合によって合成された。
PMAA-PS80-g-St可溶性ポリマーは、ほんのわずかな修正を加えた上記の方法を用いて合成された。架橋剤(MBA)はなしで、PS80の量は1グラムに増やされた。
St-DTPA-g-PMAA-P80キレートGd+3対遊離のGd+3の細胞生存率についての細胞毒性
インビボでの使用に対するナノ粒子の安全性の初期評価として、それらの毒性が単離したラット肝臓細胞を用いて遊離のガドリニウム溶液と対照して評価された。このモデルは素早い毒性スクリーニングのために使用されており、インビトロ-インビボ毒性外挿を実証している。肝細胞生存率が細胞膜崩壊を測定するトリパンブルー(0.1%w/v)排除試験によって顕微鏡で評価された。肝細胞の生存率が3時間のインキュベーション中30分毎に測定され、その細胞は使用前に少なくとも80%が生存可能であった。800μlの各試料がその肝細胞に加えられた。対照のブランクと、Gd+3を充填したポリマー及びナノ粒子の間で統計的に有意な違いはなかった。遊離のGd+3は、著しい肝細胞毒性を示し、1.5mg/mlのガドリニウム溶液にさらしたとき、240分後にそれぞれ肝細胞の生存が15%未満であった(図24)。ポリマー及びナノ粒子中に充填した同じ用量のGd+3への240分にわたる肝細胞の暴露は、ポリマー及びナノ粒子に対してそれぞれ65%及び68%の生存をもたらした。このデータは、Gd+3をSt-DTPA-PMAA-P80ポリマー/ナノ粒子に充填することが、ガドリニウムの毒性を著しく低下することを示している。
インビボでの使用に対するナノ粒子の安全性の初期評価として、それらの毒性が単離したラット肝臓細胞を用いて遊離のガドリニウム溶液と対照して評価された。このモデルは素早い毒性スクリーニングのために使用されており、インビトロ-インビボ毒性外挿を実証している。肝細胞生存率が細胞膜崩壊を測定するトリパンブルー(0.1%w/v)排除試験によって顕微鏡で評価された。肝細胞の生存率が3時間のインキュベーション中30分毎に測定され、その細胞は使用前に少なくとも80%が生存可能であった。800μlの各試料がその肝細胞に加えられた。対照のブランクと、Gd+3を充填したポリマーの間で統計的に有意な違いはなかった。遊離のGd+3は、著しい肝細胞毒性を示し、1.5mg/mlのガドリニウム溶液にさらしたとき、240分後にそれぞれ肝細胞の生存が15%未満であった(図25)。ポリマー及びナノ粒子中に充填した同じ用量のGd+3への240分にわたる肝細胞の暴露は、ポリマー及びナノ粒子に対してそれぞれ65%及び68%の生存をもたらした。このデータは、Gd+3をPMAA-PS80-g-St-DTPAポリマーに充填することが、ガドリニウムの毒性を著しく低下することを示している。
近赤外色素(HiLyte Fluor750)のSt-g-PMAA-P80ポリマー及びナノ粒子への結合
近赤外色素(HiLyte Fluor750)は、St-g-PMAA-P80にカルボジイミド化学反応(図43A)を用いることで共有結合される。簡潔には、20mgのポリマー/ナノ粒子が、DDIW水中に分散され、続いてEDC(48mg)及びNHS(45mg)が加えられ、90分間撹拌されて、St-g-PMAA-P80上のカルボン酸基を活性化させる。図43Bは、ナノ粒子とのNIR共役の視覚効果を示している。その後、200μlのHilyte Fluor750ヒドラジド(1mg/ml)が添加され、その混合物は暗所で一晩撹拌される。その生成物は、次に水に向けた透析により精製され、続いて凍結乾燥され、その後の使用のために-20℃で保存される。NIR染料で標識化されたSt-g-PMAA-P80は、820nmで蛍光放射を示す(図43C)。
St-g-PMAA-P80の生体内分布及び標的能力
インビボ全身蛍光画像
NIR染料、HiLyte Fluor750、及びドキソルビシンが共充填されたナノ粒子が調製された。上で説明したようにして合成され、凍結乾燥されてデシケーター中で保存されたSt-g-PMAAナノ粒子が使用された。100mgのナノ粒子が、2mlのDDIW中に分散され、30mgのEDC/NHSが加えられた。30分後、0.2mlのHiLyte Fluor750ヒドラジド(1.25mg/ml)が撹拌下で加えられた。その混合物は光から保護され、室温で24時間撹拌された。最後に、その生成物は、pH7.4に中和され、連続する水による洗浄及び遠心分離によって精製された。ナノ粒子は次に上で説明した方法を用いてDoxを充填された。
腫瘍中のナノ粒子に対する薬物動態パラメーターが、図26B中のデータから抽出され表6でまとめられた。腫瘍中のナノ粒子の蓄積、保持、及び排出と関連するこれらのパラメーターは、ナノスケールの薬物送達システムの治療効果を大まかに予測する。一般に、SA-NPsは、PF-NPsについて観察されたものと比較して腫瘍中により高い程度まで蓄積したが、より速い速度で消えた。腫瘍中の蛍光強度のピークは、SA-NPsについては6.15±1.04×108(p/s/cm2/sr)/(μW/cm2)であり、PF-NPsについては0.15±0.25×108(p/s/cm2/sr)/(μW/cm2)であると測定された。SA-NPsについての腫瘍AUCは、PF-NPsよりも3.5倍大きく、腫瘍中におけるPF-NPsよりもSA-NPsのより大規模な蓄積を反映していた。SA-NPsのより大きい腫瘍蓄積は、PF-NPsと比較してそれらのより長い血液循環及び大きさがより小さいことに支えられている。しかしながら、PF-NPsは、277±33hrsの大きいt1/2の値及び0.0025±0.0003hrs-1のより小さいkelに反映される、より遅い腫瘍クリアランスを示し、一方、SA-NPsについてのt1/2及びkelは、92±12hrs及び0.0075±0.0006hrs-1であった。
全体の動物のイメージングが、ナノ粒子の組織分布を測定するために実施された(図27A)。組織分布及び腫瘍蓄積も、集められた血液並びにさまざまな時点で切開された腫瘍及び肝臓、肺、腎臓、脾臓、皮膚、腸及び心臓を含む臓器の生体外のNIR蛍光分析から評価された。そのデータは、組織の平均ベースライン値に対して標準化された蛍光強度の割合として示されている(図27B、C)。SA-NPsは、PF-NPsと比較して血液中でより高い濃度で及びより長い時間にわたって検出された。注射後30分近くで、血液蛍光強度の割合は、SA-NPs及びPF-NPsに対して、それぞれ150.0±12.5及び70.0±6.0であると測定された。注射後4時間でこれらの値は、それぞれ47±8及び4.1±0.5に減少した。これらのデータに基づいて、PF-NPs及びSA-NPsに対する63分及び230分の血液循環半減期が計算された。PF-NPsは、肝臓、脾臓、肺、心臓、及び腸において相当に高レベルの蓄積を示し、その一方で、かなり高レベルのSA-NPsを、腎臓及び腫瘍中で検出することができた。PF-NPsで処理したマウスの腸における高レベルの蛍光は、肝臓に蓄積されている粒子が、糞便により排出されることを示唆している。この排出様式は尿による排出より一般に遅い。このため、肝臓からの蛍光は長期間にわたって非常に強いままである。これらの結果は、生きている動物の全身のイメージングと十分一致している。
光顕レベルでのSA-NPs及びPF-NPsの腫瘍分布が、蛍光顕微鏡検査法を用いて調べられた。媒体のみ(5%ブドウ糖)が注入された腫瘍の組織切片が、FITC(励起:460〜490nm、発光500〜535nm)発光ウインドウに画像化され、自己蛍光の相対的水準が測定された。図28に示されているように、低い水準の自己蛍光が媒体処理組織のいくつかの部位で観察された。このスペクトル放射の性質のより精密な検査は、FITCで標識化されたナノ粒子のよりはっきりとした放射ピークに対比して、幅広い波長分布を示した。そのような自己蛍光は、リポフチン、コラーゲン、及びヘムのような広げられたπ軌道システムを含む組織中に生じる。したがって、FITC発光ウインドウ内の自己蛍光に対するナノ粒子に媒介された蛍光をはっきりと区別するため、組織部位は、FITC及びTRITC(励起540〜565nm、発光575〜620nm)発光ウインドウの両方の面に画像化された。これらの結果の組合せは、図28に示されているように、自己蛍光(黄緑)に対するナノ粒子(緑)に媒介された蛍光の明確な描写を可能にした。図に示されているように、投与後4時間で、SA-NPsは、PF-NPsにおいて見られたものと比較して、マウスの腫瘍組織中でより大きい範囲まで蓄積し、腫瘍の微小血管系から腫瘍を持っている組織中にずっと大きい範囲まで浸出した。対照的に、PF-NPsは、バルクの腫瘍実質内でより限定された分布を示した。PF-NPsは、腫瘍血管のより大きいエレメントに大部分とどめられ、血管周囲の領域に結びつけられた。
インビボのMRI解析
Gd+3-充填PMAA-g-St-PS80(PolyGd)及びドキソルビシンを充填したGd+3-充填PMAA-g-St-PS80(PolyGd-Dox)のインビボのさまざまな臓器中の正のコントラストの向上を生ずる能力が市販されているOmniscan(登録商標)と比較された(図21)。主要な臓器、例えば、肝臓、脾臓、心臓、腸、及び膀胱などが、スライスAに示されており、一方スライスBは、脳、大動脈、及び腎臓についての詳細を提供している。0.05mmol/Kg Gd+3の臨床用量で、Omniscan(登録商標)は、肝臓及び心臓血管系においてごく小さいコントラストの向上を示した。それは脈管構造から急速に浸出させられ、コントラストの向上は注射5分後においてさえも乏しかった。その製剤は、強い膀胱シグナルによって明らかなように、腎臓経路を通って体から急速に除去される。Omniscan(登録商標)用量の半分(0.025mmol/Kg Gd+3)で、PMAA-g-St-P80製剤は、肝臓及び心臓血管系において著しく高いコントラストを生じた。60分後でさえ持続する強い血液プール効果が存在し、この製剤のより長い血液循環半減期に対する証拠を提供している。その造影剤は、腎臓及び次に膀胱において観察される強いコントラストの高まりによって明らかなように、腎臓経路によって排除されるように思われる。その膀胱の信号は時間を超えて急速に増加し続け、腎臓経路を通過するポリマーの排出に対する証拠を提供する。腎臓経路を通って消える製剤を有することは、この排出経路が肝胆道経路よりもずっと速いので臨床的に望ましい。
PMAA-g-St-PS80の脳標的能力
特定のナノ粒子のPS80コーティングは、血液から粒子表面へのApo-Eの高められた吸着を引き起こすことが示唆されている(図30)。その後、ナノ粒子表面のApo-Eの存在は、これらのナノ粒子の脳毛細血管内皮細胞内の内部移行を、これらの細胞によって発現されるLDL受容体によって促進する。LDL受容体によるSt-g-PMAA-P80の微細血管中への取り込みの概略図は、図30に示されている。TOF-SIMSは、表面組成及び表面化学の分析を可能にする感受性の高い表面特定技術である。感度は、ppmのオーダーであり、さらに表面選択性は、サンプリングが試料の表面の1〜2nm以内で行われる、即ち、表面のわずかの原子層のみがサンプリングされるほどのものである。TOF-SIMSは、負のイオンモードにおける255、265、及び283m/zの特性ピークによって明らかにされているようにナノ粒子の表面のPS80の存在をはっきりと示している。これらのピークは一連のオレイン酸及びステアリン酸を示しており、ソルビタン分子の側鎖であり、対照試料(PS80無し)には存在しなかった。St-g-PMAA-P80ナノ粒子表面にポリソルベート80の発現を示しているTof-シムスのデータは図31に示されている。
生体外調査
生体外調査が、PMAA-g-St-PS80の脳標的能力をさらに確認するために行われた。製剤が尾静脈を通して注射され、一定の時点でその動物は犠牲にされ、脳が取り出され、洗浄され、蛍光技術を用いてポリマー含量が測定され、製剤の血中含量も測定された(図22)。血液の蛍光がベースラインに戻ったときの24時間における脳内の蛍光の存在は、製剤の臓器レベルでの脳堆積を示す。さらに、PMAA-g-St-PS80投与後の灌流された脳スライス中の蛍光顕微鏡検査法が、細胞段階における脳中へのポリマー堆積を調査するために使用された(図4)。我々のデータは、PMAA-g-St-PS80ナノ粒子が脳毛細血管から浸出し(内皮の堅固な結合を浸透)、脳の血管周囲の領域内に堆積されることを確認している。
PMAA-g-St-PS80の腫瘍標的の可能性
線状ポリマーの腫瘍標的能力が、ネズミ乳癌腫瘍モデル(同所性ネズミEMT6乳房の腫瘍を持っているBalb/cマウス)を用いて調査された。その腫瘍蓄積は、インビボの蛍光画像及びMRIの両方を用いて調査された(図44)。ポリマーは優れた腫瘍蓄積を示した。腫瘍容積が、処理後のさまざまな時点で算定された。
結合したGdを有するPMAA-g-St-DTPAのpH依存性の緩和能及びMRコントラスト
腫瘍は代謝作用からの乳酸の多量の産生により局所pHの低下を引き起こすことが知られている。ここで我々は、PMAA-g-St-DTPA-Gdナノ粒子が異なるpH値で異なる緩和能を提供することができ、MRイメージングによって、腫瘍組織又は感染病巣中の正常な生理的pH7.4からのpH偏向の検出においてそれらの使用の可能性を示唆することを立証している(表6)。
ポリ(ジエチルアミノエチルメタクリレート)-グラフトデンプン(PDEAEM-g-St)及びPDEAEM-g-St-DTPAナノ粒子の合成及び特性化
フリーラジカル分散重合法が、粒子を調製するために使用された。その重合は、窒素下で、α,α'-アゾジイソブチルアミジン二塩酸塩(V-50)を開始剤として、エチレングリコールジメタクリレート(EGDM)を形成されたナノ粒子のための架橋剤として、そしてポリビニルピロリドン(PVP)を非イオン性安定剤として使用して実施された。その重合は、水-エタノール混合物(9:1)を用いて、70℃で6時間行われた。得られた粒子は、水とエタノールを使用して洗浄及び精製された。PDEAEM-g-Stの線状ポリマーも、架橋剤の使用を除いた同じ合成方法を用いて調製された。
デンプン、PDEAEM及びPDEAEM-g-StのFTIRスペクトルが図37に示されている。天然デンプンのスペクトルと比較すると、大きな変化は、1724cm-1におけるカルボニルC=O吸収周波数及び2962〜2964cm-1における第三級アミン基のストレッチ振動の存在である。天然デンプン中の1166、1090、1020、及び954cm-1におけるピークは、CO結合伸縮に起因している。1090及び1020cm-1におけるピークは、無水のグルコース環CO/CC伸縮の特徴を示している。1645cm-1に発生している特性ピークは、デンプン中に結合した水の存在に起因している。水素結合したヒドロキシル基(O-H)による広帯域は3450cm-1に現れ、原因は、分子間及び分子内結合したヒドロキシル基と関係する複雑な振動伸縮に帰せられる。3450cm-1におけるOH伸縮バンドは、PDEAEMホモポリマーには存在しない。2916〜2920cm-1のバンドは、C-H伸縮の特徴を示している。天然デンプン中の3450cm-1における強いOH伸縮バンドは、グラフト反応の後は強度が減少し、デンプンのOH基を介したデンプンのDEAEMとの反応を暗に示唆した。また、グラフトポリマーは、PDEAEMホモポリマーにはなかった520〜920cm-1におけるピラノース環振動の特性ピークを示し、PDEAEMのデンプンへのグラフトを立証した。
動的光散乱が、異なるpH及び一定のイオン強度の緩衝液中でのさまざまなデンプン:DEAEM比を有するナノ粒子の大きさを測定するために使用された。pH4及び7.4のPBS中のPDEAEM-g-St粒子の強度重量分布を示している典型的な動的レーザー光散乱データが、図39及び40に示されている。その粒子は単分散のものであり、ガウス分布に従った。図41における結果は、粒径がpH及びデンプン含量の関数として変化することを明らかにしている。PDEAEMナノ粒子は、pH4で521nmの平均直径を有した。その大きさはpH7.4でナノ粒子と結合している第三級アミン基の脱プロトン化によって221.8nmに減少した。この平均直径における変化は、pHによる体積における913倍の変化になる。グラフトは場合によっては粒径を2倍を超えて実際に減少することが観察された。DEAEMとデンプンの1:1.4のモル比を有するナノ粒子は、低いpHで250nmの平均直径を有し、7.4のpHでは129nmに縮小した。一般に、デンプン含量の増加は、pH感度における減少をもたらした。しかしながら、デンプンを含む粒子は、生理的pH範囲近くで比較すると、DEAEMナノ粒子と比較してより鋭い相転移を有するようであった。
典型的な重合において、以下のレシピが使用され、Gd3+が上に記載されている方法を用いてPMAA-g-St-DTPA-PS80ナノ粒子に結合された。さまざまなpHの緩衝液中の粒径が、DLSによって測定された。図42中の結果は、pHが低下すると粒径が増大することを示している。粒子容積が増加すると、粒子の水和が増加し、それはMRの下でのより高い緩和能を生むことができ、従って、腫瘍又は感染病巣と健常組織との間のpHの違いを画像化する可能性を有する。レシピ:マルトデキストリン-DTPA(1.4g);水(163.5mL);添加されるNaOH(pHを調整するため(23.5mLでpH8.0);エタノール(5mL);V-50(0.2g);PVP(0.2g);Tween80(0.5g);DEAEM(1.8g);及びEGDM(65μL)。
ドキソルビシンの脳腫瘍への送達
化学療法薬(例えば、ドキソルビシン)及びモノクローナル抗体を含めた多数の治療剤が、BBBを横切ることができず、それため、脳腫瘍の効果的な治療を提供することができない。脳腫瘍への治療剤の送達の1つの例として、乳癌の脳転移が使用された。1万のMDA-MB231-luc 3N2細胞が頭蓋内に注射された。
(付記)
(付記1)
ナノ粒子を製造する方法であって、
(a)溶液中にポリマーを可溶化するステップと、
(b)カルボン酸側基を含む重合可能なモノマーを供給するステップと、
(c)モノマーをグラフト重合して可溶化したポリマー上にポリマー鎖を形成するステップと、
(d)形成している鎖と反応する官能基を有するエトキシ化分子を供給するステップと、を含み、
ステップ(c)がエトキシ化分子の存在下で行われ、エトキシ化分子をポリマー鎖に共有結合させる、方法。
(付記2)
溶液が、ヒドロキシル溶媒を含む請求項1に記載の方法。
(付記3)
ヒドロキシル溶媒が水を含む請求項2に記載の方法。
(付記4)
ポリマーが、ポリマーの構成単位当たり0.05と3の間の置換度を有するポリヒドロキシルポリマーであり、モノマーがアルケニル基を含み、グラフト重合ステップが架橋剤の存在下で行われる付記1、2又は3に記載の方法。
(付記5)
ポリヒドロキシルポリマーが、0.5と3の間の置換度を有する付記4に記載の方法。
(付記6)
ポリヒドロキシルポリマーが、2と3の間の置換度を有する付記4に記載の方法。
(付記7)
ポリヒドロキシルポリマーが、0.5と3の間の置換度を有する多糖を含む付記4に記載の方法。
(付記8)
ポリヒドロキシルポリマーが、グルコースを含む付記7に記載の方法。
(付記9)
多糖が、デンプンを含む付記7に記載の方法。
(付記10)
ステップ(c)のモノマーが、架橋剤の量の1倍と20倍(モル/モル)の間の量で存在する付記4、5又は6に記載の方法。
(付記11)
ステップ(c)のモノマーが、架橋剤の量の3倍と6倍の間の量で存在する付記10に記載の方法。
(付記12)
重合ステップが、フリーラジカル開始剤の存在下で行われるフリーラジカルグラフト重合である付記4から11のいずれかに記載の方法。
(付記13)
開始剤が、遷移金属を実質的に含まない付記4から12のいずれかに記載の方法。
(付記14)
開始剤が、過硫酸塩又はその機能的同等物である付記4から12のいずれかに記載の方法。
(付記15)
エトキシ化分子のエトキシ基が、遊離のヒドロキシル基で終端している付記1から14のいずれかに記載の方法。
(付記16)
エトキシ化分子が、化学反応して界面活性剤分子をポリマー鎖に共有結合させるアルケニル基を含む付記1から15のいずれかに記載の方法。
(付記17)
エトキシ化分子が、R(C9-C31)-C(O)O-基を有するポリエトキシ化ソルビタンであり、ソルビタンが、重合ステップの間にR(C9-C31)-C(O)O-基のC-C共有結合を介して第二のポリマーに結合される付記16に記載の方法。
(付記18)
ソルビタンが、オキシエチレン単位の総数が少なくとも10であるポリソルベートである付記17に記載の方法。
(付記19)
オキシエチレン単位の総数が、10、12、14、16、18、20、22、24、26、28、30又は32である付記15に記載の方法。
(付記20)
前記R(C9-C31)-C(O)O-基が、重合ステップにおいて反応してC-C共有結合を形成する少なくとも1つのC-C不飽和を含む、付記17、18又は19に記載の方法。
(付記21)
ポリエトキシ化分子が、ポリソルベート80である付記20に記載の方法。
(付記22)
ステップ(C)のポリマーの量及びモノマーの量が、ポリマーのモノマー単位に対するポリマー鎖中のモノマー単位のモル比が0.1と10の間であるナノ粒子を製造するように選択される付記1から21のいずれかに記載の方法。
(付記23)
ステップ(C)のポリマーの量及びモノマーの量が、ポリマーのモノマー単位に対するポリマー鎖中のモノマー単位のモル比が0.3と7.0の間であるナノ粒子を製造するように選択される付記22に記載の方法。
(付記24)
ステップ(C)のポリマーの量及びモノマーの量が、ポリマーのモノマー単位に対するポリマー鎖中のモノマー単位のモル比が1と5.0の間であるナノ粒子を製造するように選択される付記22に記載の方法。
(付記25)
ステップ(c)におけるエトキシ化分子の量及びモノマー量が、モノマー単位に対するエトキシ化分子のモル比が0.0005と1の間であるナノ粒子を製造するように選択される付記1から24のいずれかに記載の方法。
(付記26)
ステップ(c)におけるエトキシ化分子の量及びモノマー量が、モノマー単位に対するエトキシ化分子のモル比が0.003と0.01の間であるナノ粒子を製造するように選択される付記25に記載の方法。
(付記27)
重合ステップが、界面活性剤の存在下で行われる付記1から26のいずれかに記載の方法。
(付記28)
界面活性剤が、アニオン界面活性剤である付記27に記載の方法。
(付記29)
生物学的薬剤の送達のためのナノ粒子を製造する方法であって、付記1から28のいずれかに記載の方法を含み、薬剤が、ステップ(a)のポリマーに共有結合している方法。
(付記30)
ポリマーが、ポリヒドロキシル化ポリマーであり、薬剤が、そのヒドロキシル水素原子の置換によってポリマーに共有結合している付記29に記載の方法。
(付記31)
薬剤が、金属を錯体化することができる有機部分を含む付記29に記載の方法。
(付記32)
金属-有機部分錯体を形成するステップをさらに含む付記31に記載の方法。
(付記33)
金属が、核磁気共鳴映像法において信号を提供するように選択される付記31又は32に記載の方法。
(付記34)
金属が、Gd +3 である付記33に記載の方法。
(付記35)
ステップ(c)のモノマーの量が十分に高く、製造されるナノ粒子の水溶液の、pH7.4及び10mMのイオン強度で測定される、ゼータ電位の絶対値が、少なくとも15mVである付記1から34のいずれかに記載の方法。
(付記36)
生物学的薬剤の送達のためのナノ粒子を製造する方法であって、薬剤及び付記1から35のいずれかに記載の方法によって得られたナノ粒子を液体媒体中で分散させて薬剤をナノ粒子中に取り込むステップを含む方法。
(付記37)
生物学的薬剤の送達のためのナノ粒子を製造する方法であって、付記1から35のいずれかに記載の方法によって製造されたナノ粒子に、薬剤をナノ粒子のポリマー鎖の前記カルボン酸基に、共有結合させることを含む方法。
(付記38)
キャリアナノ粒子を製造する方法であって、
(i)水溶液中にポリマーを可溶化するステップと、
(ii)カルボン酸側基を含む重合可能なモノマーを供給するステップと、
(iii)モノマーをグラフト重合して可溶化したポリマー上にポリマー鎖を形成するステップと、
(iv)形成している鎖と反応する官能基を有するポリエトキシ化分子を供給するステップと、
を含み、
重合ステップ(ii)がポリエトキシ化分子の存在下で行われ、ポリエトキシ化分子を形成している鎖に共有結合させ、重合生成物がナノ粒子の外側にポリエトキシ化部分を有するナノ粒子を形成する方法。
(付記39)
第一のポリマー、
第一のポリマーにグラフトした第二のポリマー、及び
第二のポリマーに共有結合したポリエトキシ化部分
を含むナノ粒子。
(付記40)
第二のポリマーが、第二のポリマーの主鎖の2つの炭素当たり約1個のカルボキシル基を有する重合したビニル基を含む付記39に記載のナノ粒子。
(付記41)
第二のポリマーが、ポリアルケニルポリマーである付記39に記載のナノ粒子。
(付記42)
ポリアルケニルポリマーが、ポリアクリル酸である付記41に記載のナノ粒子。
(付記43)
ポリアクリル酸が、ポリ(メタクリル酸)である付記42に記載のナノ粒子。
(付記44)
ポリエトキシ化部分が、R(C9-C31)-C(O)O-基を有するソルビタンであり、ソルビタンが、R-基のC-C共有結合を介して第二のポリマーに結合している付記39から43のいずれかに記載のナノ粒子。
(付記45)
第一のポリマーが、ポリヒドロキシルポリマーを含む付記39から44のいずれかに記載のナノ粒子。
(付記46)
ポリヒドロキシルポリマーが、0.05と3の間の置換度を有する多糖を含む付記45に記載のナノ粒子。
(付記47)
多糖が、デンプンを含む付記46に記載のナノ粒子。
(付記48)
第二のポリマーが、架橋されている付記47に記載のナノ粒子。
(付記49)
付記39から48のいずれかに記載の複数のナノ粒子を含む組成物であって、薬学的に活性な薬剤をさらに含む組成物。
(付記50)
薬剤が、ナノ粒子に吸着されている付記49に記載の組成物。
(付記51)
付記39から48のいずれかに記載の複数のナノ粒子を含む組成物であって、さらにシグナル分子を含む組成物。
(付記52)
シグナル分子が、有機部分によりキレート化された金属であり、部分がナノ粒子に共有結合されている付記51に記載の組成物。
(付記53)
有機部分が、第一のポリマーに共有結合されている付記52に記載の組成物。
(付記54)
シグナル分子が、ナノ粒子に共有結合されており、好ましくはカルボン酸側基に共有結合している付記51に記載の組成物。
(付記55)
シグナル分子が、フルオロフォアである付記51又は54の組成物。
(付記56)
薬学的に活性な薬剤をさらに含む付記51から54のいずれかに記載の組成物。
(付記57)
多糖を含む第一のポリマー、
第一のポリマーにグラフトしたポリ(メタクリル酸)を含む第二の架橋したポリマー、及び
(C9-C31)R-C(O)O-基を含み、第二のポリマーの炭素主鎖とR基との間のC-C結合によって第二のポリマーに共有結合しているポリソルベート
を含むナノ粒子。
(付記58)
(C9-C31)R-C(O)O-基が、Rが直鎖のアルキルである-(C17)R-C(O)O-である付記57に記載のナノ粒子。
(付記59)
ポリソルベートが、基-O(CH 2 CH 2 O) w -C(O)(C17)R、HO(CH 2 CH 2 O) x -、-HO(CH 2 CH 2 O) y -、-HO(CH 2 CH 2 O) z -を含み、但し、w+x+y+z=20である付記58に記載のナノ粒子。
(付記60)
多糖の分子量が、2,600Daと4,500Daの間である付記57、58又は59に記載のナノ粒子。
(付記61)
第一のポリマーがデンプンを含む付記57、58又は59に記載のナノ粒子。
(付記62)
ポリ(メタクリル酸)のメタクリレートモノマー単位に対する多糖のモノマー単位のモル比が、0.2と8.0の間である付記57から61のいずれかに記載のナノ粒子。
(付記63)
ポリソルベート及びポリ(メタクリル酸)のメタクリレートモノマー単位のモル比が、0.002と0.03の間である付記57から62のいずれかに記載のナノ粒子。
(付記64)
ナノ粒子を製造する方法であって、
(a)溶液中でポリマーを可溶化するステップ、
(b)アルキルアミノアルキルエステル側基を含む重合可能なモノマーを供給するステップ、
(c)架橋剤を供給するステップ、及び
(d)モノマーをグラフト重合して可溶化したポリマー上にポリマー鎖を形成して、ナノ粒子を形成するステップ
を含む方法。
(付記65)
ポリマーが、ポリマーの構成単位当たり0.05と3の間の置換度を有するポリヒドロキシルポリマーであり、モノマーが、アルケニル基を含み、グラフト重合ステップが、架橋剤の存在下で行われる付記64に記載の方法。
(付記66)
ポリヒドロキシルポリマーが、ポリマーの構成単位当たり1と3の間の置換度を有する付記65に記載の方法。
(付記67)
ポリヒドロキシルポリマーが、0.5と3の間の置換度を有する多糖を含む付記65に記載の方法。
(付記68)
ポリヒドロキシルポリマーが、グルコースを含む付記67に記載の方法。
(付記69)
多糖が、デンプンを含む付記67に記載の方法。
(付記70)
溶液が、ヒドロキシル溶媒を含む付記64から69のいずれかに記載の方法。
(付記71)
ヒドロキシル溶媒が、水及び/又はアルコールを含む付記70に記載の方法。
(付記72)
ヒドロキシル溶媒が、水及びエタノールを含む付記71に記載の方法。
(付記73)
重合可能なモノマーが、メタクリル酸のアルキルアミノアルキルエステルである付記64から72のいずれかに記載の方法。
(付記74)
モノマーが、ジエチルアミノエチルメタクリル酸である付記73に記載の方法。
(付記75)
重合ステップが、フリーラジカル開始剤の存在下で行われるフリーラジカルグラフト重合である付記64から74のいずれかに記載の方法。
(付記76)
開始剤が、遷移金属を実質的に含まない付記75に記載の方法。
(付記77)
開始剤が、過硫酸塩又はその機能的同等物である付記75又は76に記載の方法。
(付記78)
架橋剤が、エチレングリコールジメタクリレートである付記64から77のいずれかに記載の方法。
(付記79)
ステップ(d)のモノマーが、架橋剤の量の1倍と200倍の間の量(モル/モル)で存在する付記64から78のいずれかに記載の方法。
(付記80)
ステップ(d)のモノマーが、架橋剤の量の3倍と50倍の間の量(モル/モル)で存在する付記79に記載の方法。
(付記81)
ステップ(c)のポリマーの量及びモノマーの量が、ポリマーのモノマー単位に対するポリマー鎖中のモノマー単位のモル比が0.05と20の間であるナノ粒子を製造するように選択される付記64から80のいずれかに記載の方法。
(付記82)
ステップ(c)のポリマーの量及びモノマーの量が、ポリマーのモノマー単位に対するポリマー鎖中のモノマー単位のモル比が2と4の間であるナノ粒子を製造するように選択される付記81に記載の方法。
(付記83)
ポリソルベートが、ステップ(d)において存在する付記64から82のいずれかに記載の方法。
(付記84)
ポリソルベートのR基が、不飽和を含むヒドロカルビル基である付記83に記載の方法。
(付記85)
ポリソルベートが、ポリソルベート80である付記84に記載の方法。
(付記86)
重合可能なモノマーが、ポリソルベートの量の5倍と50倍の間の量(モル/モル)で存在する付記83、84又は85に記載の方法。
(付記87)
重合可能なモノマーが、ポリソルベートの量の20倍と30倍の間の量(モル/モル)で存在する付記86に記載の方法。
(付記88)
非イオン性安定剤がステップ(d)において存在する付記64から87のいずれかに記載の方法。
(付記89)
安定剤が、ポリビニルピロリドンである付記88に記載の方法。
(付記90)
生物学的薬剤の送達のためのナノ粒子を製造する方法であって、付記64から89のいずれかに記載の方法を含み、薬剤がステップ(a)のポリマーに共有結合している方法。
(付記91)
薬剤が、金属を錯体化することができる有機部分を含む付記90に記載の方法。
(付記92)
金属-有機部分錯体を形成するステップをさらに含む付記91に記載の方法。
(付記93)
有機部分が、DTPAである付記92に記載の方法。
(付記94)
金属が、核磁気共鳴映像法において信号を提供するように選択される付記92又は93に記載の方法。
(付記95)
金属が、Gd +3 である付記94に記載の方法。
(付記96)
生物学的薬剤の送達のためのナノ粒子を製造する方法であって、薬剤及び付記64から95のいずれかに記載の方法によって得られたナノ粒子を液体媒体中で分散させて薬剤をナノ粒子中に取り込むステップを含む方法。
(付記97)
多糖を含む第一のポリマー、及び
第一のポリマーにグラフトしたメタクリル酸のアルキルアミノアルキルエステルを含む第二の架橋ポリマーであって、架橋している第二のポリマー
を含むナノ粒子。
(付記98)
第二のポリマーが、重合したジエチルアミノエチルメタクリル酸である付記97に記載のナノ粒子。
(付記99)
第二のポリマーが、第二のポリマーの主鎖の2つの炭素当たり約1個のカルボキシル基を有する重合したビニル基を含む付記97に記載のナノ粒子。
(付記100)
多糖が、デンプンである付記97、98又は99に記載のナノ粒子。
(付記101)
付記97から100のいずれかに記載のナノ粒子の溶液を含む組成物であって、ナノ粒子が、それらの周囲の溶液のpHを約4から約7.4まで変化させたとき、500倍と1500倍の間で増加した体積変化を示す組成物。
(付記102)
ナノ粒子が、それらの周囲の溶液のpHを約4から約7.4まで変化させたとき、700倍と1100倍の間で増加した体積変化を示す付記101に記載の組成物。
(付記103)
付記97から100のいずれかに記載の複数のナノ粒子を含む組成物であって、シグナル分子をさらに含む組成物。
(付記104)
シグナル分子をさらに含む付記101又は102に記載の組成物。
(付記105)
シグナル分子が、有機部分によりキレート化された金属であり、部分がナノ粒子に共有結合されている付記103又は104に記載の組成物。
(付記106)
有機部分が、第一のポリマーに共有結合している付記105に記載の組成物。
(付記107)
金属が、核磁気共鳴映像法において信号を提供するように選択され、多糖のモノマー単位に対するメタクリル酸のアルキルアミノアルキルエステルのモル比が0.5と5の間である付記106に記載の組成物。
(付記108)
多糖のモノマー単位に対するメタクリル酸のアルキルアミノアルキルエステルのモル比が0.5と2の間である付記107に記載の組成物。
(付記109)
付記97から100のいずれかに記載の複数のナノ粒子を含む組成物であって、薬学的に活性な薬剤をさらに含む組成物。
(付記110)
薬剤が、ナノ粒子に吸着されている付記109に記載の組成物。
文献
1. Kerr, .W., Chemistry and industry of starch. Chemistry and industry of starch., 1950(Ed. 2).
2. Ellis, H. and S. Ring, A study of some factors influencing amylose gelation. Carbohydr. Polym., 1985. 5(3): p. 201-213.
3. Cui, S.W., Food carbohydrates: chemistry, physical properties, and applications. 2005, New York: Taylor & Francis.
4. Shalviri, A., Q. Liu, M.J. Abdekhodaie, and X.Y. Wu, Novel modified starch-xanthan gum hydrogels for controlled drug delivery: Synthesis and characterization. Carbohydr. Polym., 2010. 79(4): p. 898-907.
5. Celik, M. and M. Sacak, Synthesis and characterization of starch poly (methyl methacrylate) graft copolymers. J. Appl. Polym. Sci., 2002. 86(1): p. 53-57.
6. Beliakova, M.K., A.A. Aly, and F.A. Abdel Mohdy, Grafting of poly (methacrylic acid) on starch and poly (vinyl alcohol). Starch Starke, 2004. 56(9): p. 407-412.
7. Hebeish, A., M. Beliakova, and A. Bayazeed, Improved synthesis of poly (MAA)-starch graft copolymers. J. Appl. Polym. Sci., 1998. 68(10): p. 1709-1715.
8. Liu, M., R. Cheng, J. Wu, and C. Ma, Graft copolymerization of methyl acrylate onto potato starch initiated by eerie ammonium nitrate. J. Polym. Sci., Part A: Polym. Chem., 1993.
31(13): p. 3181-3186.
9. Mino, G. and S. Kaizerman, A new method for the preparation of graft copolymers. Polymerization initiated by eerie ion redox systems. J. Polym. Sci., 1958. 31(122): p. 242-243.
10. Sangramsingh, N., B. Patra, B. Singh, and C. Patra, Graft copolymerization of methyl methacrylate onto starch using a Ce (IV)-glucose initiator system. J. Appl. Polym. Sci., 2004. 91(2): p. 981-990.
11. Trimnell, D. and E. Stout, Grafting acrylic acid onto starch and poly (vinyl alcohol) by photolysis. J. Appl. Polym. Sci., 1980. 25(10): p. 2431-2434.
12. Zhang, L.M. and D.Q. Chen, Grafting of 2 (Dimethylamino) ethyl Methacrylate onto Potato Starch Using Potassium Permanganate/Sulf uric Acid Initiation System. Starch Starke, 2001. 53(7): p. 311-316.
13. Chan, W.C. and C.Y. Chiang, Flocculation of clay suspensions with water insoluble starch grafting acrylamide/sodium allylsulfonated copolymer powder. Journal of Applied Polymer Science, 1995. 58(10): p. 1721-1726.
14. Elvira, C, J. Mano, J. San Roman, and R. Reis, Starch-based biodegradable hydrogels with potential biomedical applications as drug delivery systems. Biomaterials, 2002. 23(9): p. 1955-1966.
15. Khalil, M., S. Farag, and S. Fattach, Hydrolysis of poly (acrylamide)-starch graft copolymer. Journal of Applied Polymer Science, 1995. 57(3): p. 335-342.
16. Rath, S. and R. Singh, Flocculation characteristics of grafted and ungrafted starch, amylose, and amylopectin. Journal of Applied Polymer Science, 1997. 66(9): p. 1721-1729.
17. Shah, S., B. Patel, C. Patel, and H. Trivedi, Saponification of graft copolymers of sodium salt of partially carboxymethylated amylose and its water absorbency. Starch Starke, 1992. 44(3): p.108-110.
18. Singh, R.P., G. Karmakar, S. Rath, N. Karmakar, S. Pandey, T. Tripathy, J. Panda, K. Kanan, S. Jain, and N. Lan, Biodegradable drag reducing agents and flocculants based on polysaccharides: materials and applications. Polymer Engineering & Science, 2000. 40(1): p. 46-60.
19. Aoki, T., M. Kawashima, H. Katono, K. Sanui, N. Ogata, T. Okano, and Y. Sakurai, Temperature-responsive interpenetrating polymer networks constructed with poly (acrylic acid) and poly (N, N-dimethylacrylamide). Macromolecules, 1994.27(4): p. 947-952.
20. Bearinger, J., D. Castner, and K. Healy, Biomolecular modification of p (AAm-co-EG/AA) IPNs supports osteoblast adhesion and phenotypic expression. J. Biomater. Sci., Polym. Ed., 1998. 9(7): p. 629-652.
21. Vernon, B., S.W. Kim, and Y.H. Bae, Insulin release from islets of Langerhans entrapped in a poly(N-isopropylacrylamide-co-acrylic acid) polymer gel. J Biomater Sci Polym Ed, 1999. 10(2): p. 183-98.
22. Bayazeed, A., M. Elzairy, and A. Hebeish, Synthesis and Application of New Thickerners Part I: Preparation of Poly (Acrylic Acid) Starch Graft Copolymer. Starch Starke, 1989.41(6): p. 233- 236.
23. Hirose, Y., T. Amiya, Y. Hirokawa, and T. Tanaka, Phase transition of submicron gel beads. Macromolecules, 1987.20(6): p. 1342-1344.
24. Saboktakin, M.R., A. Maharramov, and M.A. Ramazanov, pH-sensitive starch hydrogels via free radical graft copolymerization, synthesis and properties. Carbohydr. Polym., 2009.77(3): p. 634-638.
25. Wiersema, P., A. Loeb, and J.T.G. Overbeek, Calculation of the electrophoretic mobility of a spherical colloid particle. J. Colloid Interface Sci., 1966.22(1): p. 78-99.
26. Kawaguchi, S., Y. Nishikawa, T. Kitano, K. Ito, and A. Minakata, Dissociation behavior of poly (itaconic acid) by potentiometric titration and intrinsic viscosity. Macromolecules, 1990. 23(10): p. 2710-2714.
27. Kitano, T., S. Kawaguchi, K. Ito, and A. Minakata, Dissociation behavior of poly (fumaric acid) and poly (maleic acid). 1. Potentiometric titration and intrinsic viscosity. Macromolecules,1987.20(7): p. 1598-1606.
28. Wu, X.Y., Q. Zhang, and R. Arshady, Stimuli sensetive hydrogels: response and release modulation, in Introduction to polymeric biomaterials, R. Arshady, Editor. 2004, Citus Books: London.
29. Zeman, L. and D. Patterson, Pressure effects in polymer solution phase equilibriums. II. Systems showing upper and low critical solution temperatures. J. Phys. Chem. , 1972.76(8): p. 1214-1219.
30. Aggarwal, A., R. Saxena, B. Wang, and G.T. Caneba, Studies of the polymerization of methacrylic acid via free-radical retrograde precipitation polymerization process. J. Appl. Polym. Sci., 1996.62(12): p. 2039-2051.
31. Donbrow, M., E. Azaz, and A. Pillersdorf, Autoxidation of polysorbates. J. Pharm. Sci., 1978. 67(12): p. 1676-1681.
32. Ha, E., W. Wang, and Y.J. Wang, Peroxide formation in polysorbate 80 and protein stability, i. Pharm. Sci., 2002.91(10): p. 2252-2264.
33. Donbrow, M., Nonionic surfactants: physical chemistry. 1987, New York: Marcel Dekker.
34. Nema, S., R. Washkuhn, and R. Brendel, Excipients and their use in injectable products. J. Pharm. Sci. Technol., 1997.51(4): p. 166.
35. Ghosh, P., S.C. Chadha, A.R. Mukherjee, and S.R. Palit, Endgroup studies in persulfate initiated vinyl polymer by dye techniques. Part I. Initiation by persulfate alone. Journal of Polymer Science Part A: General Papers, 1964.2(10): p. 4433-4440.
36. Hohenstein, W. and H. Mark, Polymerization of olefins and diolefins in suspension and emulsion. Part II. Journal of Polymer Science, 1946.1(6): p. 549-580.
37. Tauer, K., A.M .I. Ali, and M. Sedlak, On the preparation of stable poly (2-hydroxyethyl methacrylate) nanoparticles. Colloid Polym. Sci., 2005.283(4): p. 351-358.
38. Maeda, H., J. Wu, T. Sawa, Y. Matsumura, and K. Hori, Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Controlled Release, 2000. 65(1-2): p. 271-284.
39. Wu, X.Y. and P.I. Lee, Preparation and characterization of thermal-and pH-sensitive nanospheres. Pharm. Res., 1993. 10(10): p. 1544-1547.
40. Hoare, T. and R. Pelton, Functional group distributions in carboxylic acid containing poly (N- isopropylacrylamide) microgels. Langmuir, 2004. 20(6): p. 2123-2133.
41. Kawaguchi, S., A. Yekta, and M.A. Winnik, Surface characterization and dissociation properties of carboxylic acid core-shell latex particle by potentiometric and conductometric titration. J Colloid Interface Sci, 1995. 176(2): p. 362-369.
42. Bouma, J., J.H. Beijnen, A. Bult, and W.J.M. Underberg, Anthracycline antitumour agents. Pharm. World Sci., 1986. 8(2): p. 109-133.
43. Adriamycin monograph, in Compendium of pharmaceuticals and specialities, L. Welbanks, Editor. 2000, Canadian pharmacists association: Ottawa, ON. p. 31-32.
44. Alvarez, R.H., Present and future evolution of advanced breast cancer therapy. Breast Cancer Res, 2010. 12(Suppl 2): p. SI.
45. Teraoka, K., M. Hirano, K. Yamaguchi, and A. Yamashina, Progressive cardiac dysfunction in adriamycin-induced cardiomyopathy rats. Eur. J. Heart Failure, 2000. 2(4): p. 373-378.
46. Yeh, E.T.H., A . Tong, D.J. Lenihan, S.W. Yusuf, J. Swafford, C. Champion, J.-B. Durand, H. Gibbs, A.A. Zafarmand, and M.S. Ewer, Cardiovascular Complications of Cancer Therapy. Circulation, 2004. 109(25): p. 3122-3131.
47. Arceci, R.J., Can multidrug resistance mechanisms be modified? Br J Haematol., 2000. 110(2): p. 285-291.
48. Higgins, C.F., Multiple molecular mechanisms for multidrug resistance transporters. Nature, 2007. 446(7137): p. 749-757.
49. Wong, H.L., X.Y. Wu, and R. Bendayan, Multidrug resistance in solid tumors and its reversal., in Pharmaceutical perspectives of cancer therapeutics., Y. LU and R.I. Mahato, Editors. 2007, Springer: New York, NY. p. 121-148.
50. Glavinas, H., P. Krajcsi, J. Cserepes, and B. Sarkadi, The role of ABC transporters in drug resistance, metabolism and toxicity. Curr. Drug Delivery, 2004. 1(1): p. 27-42.
51. Bennis, S., C. Chapey, J. Robert, and P. Couvreur, Enhanced cytotoxicity of doxorubicin encapsulated in polyisohexylcyanoacrylate nanospheres against multidrug-resistant tumour cells in culture. Eur. J. Cancer, 1994. 30(1): p. 89-93.
52. Couvreur, P., L. Grislain, V. Lenaerts, F. Brasseur, P. Guiot, and A. Biernacki, Biodegradable polymeric nanoparticles as drug carrier for antitumor agents, in Polymeric Nanoparticles and Microspheres, P. Guiot and P. Couvreur, Editors. 1986, CRC Press: Boca Raton, FL. p. 27-93.
53. Shuhendler, A.J., R.Y. Cheung, J. Manias, A. Connor, A.M. Rauth, and X.Y. Wu, A novel doxorubicin-mitomycin C co-encapsulated nanoparticle formulation exhibits anti-cancer synergy in multidrug resistant human breast cancer cells. Breast Cancer Res Treat., 2010. 119(2): p. 255-269.
54. Prasad, P., J. Cheng, A. Shuhendler, A.M. Rauth, and X.Y. Wu, A novel nanoparticle formulation overcomes multiple types of membrane efflux pumps in human breast cancer cells. Drug Delivery Transl Res., 2012. 2(2): p. 1-11.
55. Wong, H.L., A.M. Rauth, R. Bendayan, J.L. Manias, M. Ramaswamy, Z. Liu, S.Z. Erhan, and X.Y. Wu, A new polymer-lipid hybrid nanoparticle system increases cytotoxicity of doxorubicin against multidrug-resistant human breast cancer cells. Pharm. Res., 2006. 23(7): p. 1574- 1585.
56. Torchilin, V.P., Passive and active drug targeting: drug delivery to tumors as an example, in Handb Exp Pharmacol., M. schafer-korting, Editor. 2010, Springer: New York, NY. p. 3-53.
57. Maeda, H., The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme egul, 2001.41(1): p. 189-207.
58. Brigger, I., C. Dubernet, and P. Couvreur, Nanoparticles in cancer therapy and diagnosis. Adv Drug Delivery Rev, 2002.54(5): p. 631-651.
59. Panyam, J. and V. Labhasetwar, Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv Drug Delivery Rev, 2003.55(3): p. 329-347.
60. Abbott, N.J. and I.A. Romero, Transporting therapeutics across the blood-brain barrier. Mol. Med. Today, 1996.2(3): p. 106-113.
61. Chauhan, N.B., Trafficking of intracerebroventricularly injected antisense oligonucleotides in the mouse brain. Antisense Nucleic Acid Drug Dev., 2002.12(5): p. 353-357.
62. Neuwelt, E., K. Maravilla, E. Frenkel, S. Rapaport, S. Hill, and P. Barnett, Osmotic blood-brain barrier disruption. Computerized tomographic monitoring of chemotherapeutic agent delivery. J. Clin. Invest., 1979.64(2): p. 684-688.
63. Wohlfart, S., S. Gelperina, and J. Kreuter, Transport of drugs across the blood-brain barrier by nanoparticles. J. Controlled Release, 2011.161(2): p. 264-273.
64. Alyautdin, R.N., V.E. Petrov, K. Langer, A. Berthold, D.A. Kharkevich, and J. Kreuter, Delivery of loperamide across the blood-brain barrier with polysorbate 80-coated polybutylcyanoacrylate nanoparticles. Pharm. Res., 1997.14(3): p. 325-328.
65. Kreuter, J., T. Hekmatara, S. Dreis, T. Vogel, S. Gelperina, and K. Langer, Covalent attachment of apolipoprotein Al and apolipoprotein B-100 to albumin nanoparticles enables drug transport into the brain. J. Controlled Release, 2007.118(1): p. 54-58.
66. Tosi, G., L. Costantino, F. Rivasi, B. Ruozi, E. Leo, A. Vergoni, R. Tacchi, A. Bertolini, M. Vandelli, and F. Forni, Targeting the central nervous system: in vivo experiments with peptide-derivatized nanoparticles loaded with Loperamide and Rhodamine-123. J. Controlled Release, 2007.122(1): p. 1-9.
67. Kurakhmaeva, K.B., I.A. Djindjikhashvili, V.E. Petrov, V.U. Balabanyan, T.A. Voronina, S.S. Trofimov, J. Kreuter, S. Gelperina, D. Begley, and R.N. Alyautdin, Brain targeting of nerve growth factor using poly (butyl cyanoacrylate) nanoparticles. J. Drug Targeting, 2009.17(8): p. 564-574.
68. Zensi, A., D. Begley, C. Pontikis, C. Legros, L. Mihoreanu, S. Wagner, C. Bikhel, H. von Briesen, and J. Kreuter, Albumin nanoparticles targeted with Apo E enter the CNS by transcytosis and are delivered to neurones. J. of Controlled Release, 2009.137(1): p. 78-86.
69. Michaelis, K., M.M. Hoffmann, S. Dreis, E. Herbert, R.N. Alyautdin, M. Michaelis, J. Kreuter, and K. Langer, Covalent linkage of apolipoprotein E to albumin nanoparticles strongly enhances drug transport into the brain. J. Pharmacol. Exp. Ther., 2006.317(3): p. 1246-1253.
70. Lauterbur, P.C., Image formation by induced local interactions: examples employing nuclear magnetic resonance. Nature, 1973.242(5394): p. 190-191.
71. Yan, G.P., L. Robinson, and P. Hogg, Magnetic resonance imaging contrast agents: overview and perspectives. Radiography, 2007.13(suppl 1): p. e5-el9.
72. Lauffer, R.B., Paramagnetic metal complexes as water proton relaxation agents for NMR imaging: theory and design. Chem. Rev., 1987.87(5): p. 901-927.
73. Aime, S., M. Fasano, and E. Terreno, Lanthanide (III) chelates for NMR biomedical applications. Chem. Soc. Rev., 1998.27(1): p. 19-29.
74. Caravan, P., J.J. Ellison, T.J. McMurry, and R.B. Lauffer, Gadolinium (III) chelates as MRI contrast agents: structure, dynamics, and applications. Chem. Rev., 1999. 99(9): p. 2293- 2352.
75. Weinmann, H.J., R.C. Brasch, W.R. Press, and G.E. Wesbey, Characteristics of gadolinium- DTPA complex: a potential NMR contrast agent. Am. J. Roentgenol., 1984. 142(3): p. 619- 624.
76. Haley, T., K. Raymond, N. Komesu, and H. Upham, Toxicological and pharmacological effects of gadolinium and samarium chlorides. Br. J. Pharmacol. Chemother., 1961. 17(3): p. 526- 532.
77. Spencer, A.J., S.A. Wilson, J. Batchelor, A. Reid, J. Pees, and E. Harpur, Gadolinium chloride toxicity in the rat. Toxicol. Pathol., 1997. 25(3): p. 245-255.
78. Graca, J., F. Davison, and J. Feavel, Comparative toxicity of stable rare earth compounds. III. Acute toxicity of intravenous injections of chlorides and chelates in dogs. AMA Arch. Ind. Health, 1964. 8: p. 555-564.
79. Lu, Z.R., A.M. Mohs, Y. Zong, and Y. Feng, Polydisulfide Gd (III) chelates as biodegradable macromolecular magnetic resonance imaging contrast agents. Int. J. Nanomed., 2006. 1(1): p. 31-40.
80. Bogdanov Jr, A., S. Wright, E. Marecos, A. Bogdanova, C. Martin, P. Petherick, and R. Weissleder, A long-circulating co-polymer in "passive targeting" to solid tumors. J. Drug Targeting, 1997. 4(5): p. 321-330.
81. Cai, J., E.M. Shapiro, and A.D. Hamilton, Self-assembling DNA quadruplex conjugated to MRI contrast agents. Bioconjugate Chem., 2009. 20(2): p. 205-208.
82. Doble, D.M.J., M. Botta, J. Wang, S. Aime, A. Barge, and K.N. Raymond, Optimization of the relaxivity of MRI contrast agents: Effect of poly (ethylene glycol) chains on the water- exchange rates of Gd III complexes. J. Am. Chem. Soc, 2001. 123(43): p. 10758-10759.
83. Kobayashi, H., S. Kawamoto, S.K. Jo, H.L. Bryant Jr, M.W. Brechbiel, and A. Robert, Macromolecular MRI contrast agents with small dendrimers: pharmacokinetic differences between sizes and cores. Bioconjugate Chem., 2003. 14(2): p. 388-394.
84. Langereis, S., Q.G. De Lussanet, M.H.P. Van Genderen, W.H. Backes, and E. Meijer, Multivalent contrast agents based on gadolinium-diethylenetriaminepentaacetic acid- terminated poly (propylene imine) dendrimers for magnetic resonance imaging. Macromolecules, 2004. 37(9): p. 3084-3091.
85. Mulder, W.J.M., G.J. Strijkers, A.W. Griffioen, L. van Bloois, G. Molema, G. Storm, G.A. Koning, and K. Nicolay, A liposomal system for contrast-enhanced magnetic resonance imaging of molecular targets. Bioconjugate Chem., 2004. 15(4): p. 799-806.
86. Rebizak, R., M. Schaefer, and E. Dellacherie, Polymeric conjugates of Gd3+- diethylenetriaminepentaacetic acid and dextran. 1. Synthesis, characterization, and paramagnetic properties. Bioconjugate Chem., 1997. 8(4): p. 605-610.
87. Steve, R., M.O. Guler, R.E. Bras, T.J. Meade, and S.I. Stupp, Self-assembled peptide amphiphile nanofibers conjugated to MRI contrast agents. Nano Lett., 2005. 5(1): p. 1-4.
88. Vexler, V.S., O. Clement, H. Schmitt Willich, and R.C. Brasch, Effect of varying the molecular weight of the MR contrast agent Gd-DTPA-polylysine on blood pharmacokinetics and enhancement patterns. J Magn Reson Imaging, 1994. 4(3): p. 381-388.
89. Wen, X., E.F. Jackson, R.E. Price, E.E. Kim, Q. Wu, S. Wallace, C. Charnsangavej, J.G. Gelovani, and C. Li, Synthesis and characterization of poly (L-glutamic acid) gadolinium chelate: a new biodegradable MRI contrast agent. Bioconjugate Chem., 2004. 15(6): p. 1408-1415.
90. Zhang, G., R. Zhang, X. Wen, L Li, and C. Li, Micelles based on biodegradable poly (l-glutamic acid)-b-polylactide with paramagnetic Gd ions chelated to the shell layer as a potential nanoscale MRI- visible delivery system. Biomacromolecules, 2007.9(1): p. 36-42.
91. Maeda, H., J. Wu, T. Sawa, Y. Matsumura, and K. Hori, Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. Journal of Controlled Release, 2000. 65(1-2): p. 271-284.
92. Peters, T., The plasma proteins. 1975, New York: Academic Press.
93. Alhareth, K., C. Vauthier, C. Gueutin, G. Ponchel, and F. Moussa, Doxorubicin loading and in vitro release from poly(alkylcyanoacrylate) nanoparticles produced by redox radical emulsion polymerization. Journal of Applied Polymer Science, 2011.119(2): p. 816-822.
94. Park, J., P.M. Fong, J. Lu, K.S. Russell, C.J. Booth, W.M. Saltzman, and T.M. Fahmy, PEGylated PLGA nanoparticles for the improved delivery of doxorubicin. Nanomedicine: Nanotechnology, Biology and Medicine, 2009.5(4): p. 410-418.
95. Hara, H., R.M. Friedlander, V. Gagliardini, C. Ayata, K. Fink, Z. Huang, M. Shimizu-Sasamata, J. Yuan, and M.A. Moskowitz, Inhibition of interleukin 16 converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proceedings of the National Academy of Sciences, 1997.94(5): p. 2007-2012.
96. Thornberry, N.A. and Y. Lazebnik, Caspases: enemies within. Science, 1998.281(5381): p. 1312-1316.
97. Robertson, G.S., S.J. Crocker, D.W. Nicholson, and J .B. Schulz, Neuroprotection by the inhibition of apoptosis. Brain Pathology, 2000.10(2): p. 283-292.
98. Andersen, E.R., L.T. Holmaas, and V. Olaisen, Process for the production of DTPA-bis anhydride. 2008, EP Patent 1,711,479.
99. Shalviri, A., H.K. Chan, G. Raval, M.J. Abdekhodaie, Q. Liu, H. Heerklotz, and X.Y. Wu, Design of pH-responsive Nanoparticles of Terpolymer of Poly (methacrylic acid), Polysorbate 80 and Starch for Delivery of Doxorubicin. Colloids and Surfaces B: Biointerfaces, 2013.101: p. 405- 413.
100. Barge, A., G. Cravotto, E. Gianolio, and F. Fedeli, How to determine free Gd and free ligand in solution of Gd chelates. A technical note. Contrast Media Mol. Imaging, 2006.1(5): p. 184- 188.
Claims (27)
- ナノ粒子を製造する方法であって、
(a)溶液中にポリマーを可溶化するステップと、
(b)カルボン酸側基又はアルキルアミノアルキルエステル側基を含む重合可能なモノマーを供給するステップと、
(c)モノマーをグラフト重合して可溶化したポリマー上にポリマー鎖を形成するステップと、
(d)形成している鎖と反応する官能基を有するポリエトキシ化ソルビタンをベースとした分子を供給するステップと、を含み、
ステップ(c)がポリエトキシ化ソルビタンをベースとした分子の存在下で行われ、ポリエトキシ化ソルビタンをベースとした分子をポリマー鎖に共有結合させる、方法。 - 溶液が、ヒドロキシル溶媒を含む請求項1に記載の方法。
- 工程(a)のポリマーが、デンプンを含む請求項1に記載の方法。
- ポリエトキシ化ソルビタンをベースとした分子が、R(C9-C31)-C(O)O-基を有するポリエトキシ化ソルビタンエステルであり、ポリエトキシ化ソルビタンエステルが、重合ステップの間にR(C9-C31)-C(O)O-基のC-C共有結合を介して第二のポリマーに結合される請求項1に記載の方法。
- 生物学的薬剤の送達のためのナノ粒子を製造する方法であって、請求項1〜4のいずれか1項に記載の方法を含み、薬剤が、ステップ(a)のポリマーに共有結合している方法。
- 生物学的薬剤の送達のためのナノ粒子を製造する方法であって、薬剤及び請求項1〜4のいずれか1項に記載の方法によって得られたナノ粒子を液体媒体中で分散させて薬剤をナノ粒子中に取り込むステップを含む方法。
- 第一のポリマー、
第一のポリマーにグラフトした第二のポリマー、及び
第二のポリマーに共有結合したポリエトキシ化ソルビタンをベースとした部分
を含むナノ粒子。 - 第二のポリマーが、第二のポリマーの主鎖の2つの炭素当たり約1個のカルボキシル基を有する重合したビニル基を含む請求項7に記載のナノ粒子。
- 第二のポリマーが、ポリ(メタクリル酸)である請求項8に記載のナノ粒子。
- ポリエトキシ化ソルビタンをベースとした部分が、R(C9-C31)-C(O)O-基を有するポリエトキシ化ソルビタンエステルであり、ポリエトキシ化ソルビタンエステルが、R-基のC-C共有結合を介して第二のポリマーに結合している請求項7に記載のナノ粒子。
- 第一のポリマーが、デンプンを含む請求項10に記載のナノ粒子。
- 第二のポリマーが、架橋されている請求項11に記載のナノ粒子。
- 請求項12に記載の複数のナノ粒子を含む組成物であって、薬学的に活性な薬剤をさらに含む組成物。
- 請求項12に記載の複数のナノ粒子を含む組成物であって、さらにシグナル分子、生物学的薬剤、又は薬学的に活性な薬剤を含む組成物。
- 薬学的に活性な薬剤をさらに含む請求項14に記載の組成物。
- 多糖を含む第一のポリマー、
第一のポリマーにグラフトしたポリ(メタクリル酸)を含む第二の架橋したポリマー、及び
第二のポリマーの炭素主鎖とR基との間のC-C結合によって第二のポリマーに共有結合している(C9-C31)R-C(O)O-基を含む、ポリソルベート
を含むナノ粒子。 - ポリソルベートが、基-O(CH2CH2O)w-C(O)(C17)R、HO(CH2CH2O)x-、HO(CH2CH2O)y-、HO(CH2CH2O)z-を含み、但し、w+x+y+z=20である請求項16に記載のナノ粒子。
- 第一のポリマーがデンプンを含む請求項17に記載のナノ粒子。
- ポリ(メタクリル酸)のメタクリレートモノマー単位に対する多糖のモノマー単位のモル比が、0.2と8.0の間である請求項18に記載のナノ粒子。
- ポリソルベート及びポリ(メタクリル酸)のメタクリレートモノマー単位のモル比が、0.002と0.03の間である請求項19に記載のナノ粒子。
- ナノ粒子を製造する方法であって、
(a)溶液中でポリマーを可溶化するステップ、
(b)アルキルアミノアルキルエステル側基を含む重合可能なモノマー又はビニル基及び1個のカルボキシル基を含むモノマーを供給するステップ、
(c)架橋剤を供給するステップ、
(d)モノマーをグラフト重合して可溶化したポリマー上にポリマー鎖を形成して、ナノ粒子を形成するステップ、及び
(e) モノマーのグラフト重合の間にポリエトキシ化ソルビタンをベースとした分子をポリマー鎖に共有結合させるステップ
を含む方法。 - 工程(a)のポリマーが、デンプンを含む請求項21に記載の方法。
- モノマーが、ジエチルアミノエチルメタクリル酸である請求項22に記載の方法。
- 多糖を含む第一のポリマー、
第一のポリマーにグラフトした、メタクリル酸のアルキルアミノアルキルエステル、又はビニル基及び1個のカルボキシル基を含むモノマーを含む第二の架橋ポリマーであって、架橋している第二のポリマー、及び
第二のポリマーに共有結合したポリエトキシ化ソルビタンをベースとした部分
を含むナノ粒子。 - 第二のポリマーが、重合したジエチルアミノエチルメタクリレートである請求項24に記載のナノ粒子。
- 第二のポリマーが、重合したメタクリル酸を含む請求項24に記載のナノ粒子。
- モノマーが、メタクリル酸である請求項22に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261605995P | 2012-03-02 | 2012-03-02 | |
| US61/605,995 | 2012-03-02 | ||
| PCT/CA2013/000203 WO2013127004A1 (en) | 2012-03-02 | 2013-03-04 | Polymeric nanoparticles useful in theranostics |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2015512978A JP2015512978A (ja) | 2015-04-30 |
| JP2015512978A5 JP2015512978A5 (ja) | 2016-04-21 |
| JP6152391B2 true JP6152391B2 (ja) | 2017-06-21 |
Family
ID=49081494
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2014559046A Active JP6152391B2 (ja) | 2012-03-02 | 2013-03-04 | セラノスティクスにおいて有用なポリマーナノ粒子 |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US10233277B2 (ja) |
| EP (1) | EP2820070B1 (ja) |
| JP (1) | JP6152391B2 (ja) |
| CN (1) | CN104470975B (ja) |
| CA (1) | CA2865925C (ja) |
| SG (1) | SG11201405407WA (ja) |
| WO (1) | WO2013127004A1 (ja) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015149188A1 (en) | 2014-04-03 | 2015-10-08 | The Governing Council Of The University Of Toronto | Multifunctional nanoparticle compositions and uses thereof |
| AU2016220103A1 (en) * | 2015-02-18 | 2017-10-05 | Memorial Sloan Kettering Cancer Center | Compositions and methods for nanoparticle-based drug delivery and imaging |
| JP6903055B2 (ja) | 2015-08-03 | 2021-07-14 | イーエヌビー・セラピューティクス・インク | Etbr活性化に関連した癌を治療するための組成物及び方法 |
| CN105732901B (zh) * | 2016-04-22 | 2018-02-02 | 广西大学 | 一种木薯渣磁性微球及其制备方法 |
| CN108003286A (zh) * | 2017-12-15 | 2018-05-08 | 北京思如诺科技有限公司 | 一种用于肿瘤放射性诊疗的共聚物及其制备方法和应用 |
| US10435434B2 (en) | 2018-01-12 | 2019-10-08 | Enb Therapeutics, Inc. | Deuterated compounds, compositions, and methods for treating cancers associated with ETBR activation |
| CN110960508B (zh) * | 2019-11-13 | 2021-11-23 | 湖北大学 | 一种具有抗蛋白吸附和靶向能力的淀粉纳米粒子及其制备方法 |
| US20210361570A1 (en) * | 2020-05-19 | 2021-11-25 | Mcmaster University | In situ gelling polysaccharide-based nanoparticle hydrogel compositions, and methods of use thereof |
| JPWO2022024754A1 (ja) * | 2020-07-27 | 2022-02-03 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57138411A (en) | 1981-02-16 | 1982-08-26 | Nippon Denso Co Ltd | Air intake controller for car air conditioner |
| US4690996A (en) | 1985-08-28 | 1987-09-01 | National Starch And Chemical Corporation | Inverse emulsions |
| GB9411080D0 (en) | 1994-06-02 | 1994-07-20 | Unilever Plc | Treatment |
| AU3243300A (en) * | 1999-02-23 | 2000-09-14 | Isis Pharmaceuticals, Inc. | Multiparticulate formulation |
| CN1388169A (zh) * | 2002-07-26 | 2003-01-01 | 张夫道 | 纳米级废弃塑料-淀粉混聚物生产方法及应用 |
| NO20035745D0 (no) | 2003-12-19 | 2003-12-19 | Amersham Health As | Prosess |
| CN1709935A (zh) * | 2005-06-17 | 2005-12-21 | 天津大学 | 淀粉接枝聚丙烯酰胺及其衍生物反相乳液的制备方法 |
| WO2009049089A1 (en) * | 2007-10-09 | 2009-04-16 | Washington University In St. Louis | Ligand directed toroidal nanoparticles for therapy and diagnostic imaging |
| EP2265174B1 (en) * | 2008-04-04 | 2018-06-06 | The Regents of The University of California | Functionalized magnetic nanoparticles and methods of use thereof |
| EP2310519A2 (de) * | 2008-08-07 | 2011-04-20 | Bioscreen e.K. | Verfahren zur epoxidation ungesättigter fettsäuren und anschliessender kopplung dieser unter verwendung einer lipase |
| WO2010084060A1 (en) | 2009-01-21 | 2010-07-29 | Basf Se | Starch copolymers and nanoparticles thereof for drug delivery systems |
| CN102781965A (zh) * | 2009-10-06 | 2012-11-14 | 安吉奥开米公司 | 用于转运治疗剂的组合物和方法 |
-
2013
- 2013-03-04 WO PCT/CA2013/000203 patent/WO2013127004A1/en active Application Filing
- 2013-03-04 JP JP2014559046A patent/JP6152391B2/ja active Active
- 2013-03-04 CA CA2865925A patent/CA2865925C/en active Active
- 2013-03-04 SG SG11201405407WA patent/SG11201405407WA/en unknown
- 2013-03-04 EP EP13754319.5A patent/EP2820070B1/en active Active
- 2013-03-04 CN CN201380023187.0A patent/CN104470975B/zh active Active
- 2013-03-04 US US14/382,495 patent/US10233277B2/en active Active
-
2018
- 2018-12-21 US US16/229,763 patent/US11155664B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| CN104470975B (zh) | 2018-02-27 |
| US20150132384A1 (en) | 2015-05-14 |
| CA2865925C (en) | 2021-06-01 |
| CA2865925A1 (en) | 2013-09-06 |
| EP2820070A4 (en) | 2016-03-09 |
| SG11201405407WA (en) | 2014-10-30 |
| US20190233567A1 (en) | 2019-08-01 |
| JP2015512978A (ja) | 2015-04-30 |
| CN104470975A (zh) | 2015-03-25 |
| US10233277B2 (en) | 2019-03-19 |
| EP2820070A1 (en) | 2015-01-07 |
| EP2820070B1 (en) | 2023-07-26 |
| US11155664B2 (en) | 2021-10-26 |
| WO2013127004A1 (en) | 2013-09-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11155664B2 (en) | Polymeric nanoparticles useful in theranostics | |
| Liu et al. | Internal stimuli-responsive nanocarriers for drug delivery: Design strategies and applications | |
| Martínez-Jothar et al. | Insights into maleimide-thiol conjugation chemistry: Conditions for efficient surface functionalization of nanoparticles for receptor targeting | |
| US9901616B2 (en) | Apoptosis-targeting nanoparticles | |
| Shen et al. | Dendrimer-based organic/inorganic hybrid nanoparticles in biomedical applications | |
| Zhou et al. | Synthesis and characterization of PEGylated polyethylenimine-entrapped gold nanoparticles for blood pool and tumor CT imaging | |
| Tran et al. | Design of iron oxide nanoparticles decorated oleic acid and bovine serum albumin for drug delivery | |
| Sun et al. | Gadolinium-loaded poly (N-vinylcaprolactam) nanogels: synthesis, characterization, and application for enhanced tumor MR imaging | |
| Hwang et al. | Tumor targetability and antitumor effect of docetaxel-loaded hydrophobically modified glycol chitosan nanoparticles | |
| Díaz-López et al. | The performance of PEGylated nanocapsules of perfluorooctyl bromide as an ultrasound contrast agent | |
| Zhuang et al. | Laponite-polyethylenimine based theranostic nanoplatform for tumor-targeting CT imaging and chemotherapy | |
| Srikar et al. | Polymeric nanoparticles for molecular imaging | |
| US20100168044A1 (en) | Superparamagnetic nanoparticle encapsulated with stimuli responsive polymer for drug delivery | |
| Sun et al. | Macrophages-targeting mannosylated nanoparticles based on inulin for the treatment of inflammatory bowel disease (IBD) | |
| Pei et al. | Design of Janus-like PMMA-PEG-FA grafted fluorescent carbon dots and their nanoassemblies for leakage-free tumor theranostic application | |
| Huang et al. | Design and application of dextran carrier | |
| Gardikis et al. | Dendrimers and the development of new complex nanomaterials for biomedical applications | |
| Shutava et al. | Synergetic effect of polyethylene glycol-grafted chitosan and bovine serum albumin on colloidal stability of polyelectrolyte nanocapsules | |
| Girija | Medical applications of polymer/functionalized nanoparticle systems | |
| Khatik et al. | Integrin αvβ3 receptor overexpressing on tumor-targeted positive MRI-guided chemotherapy | |
| Lim et al. | Preparation of pyrenyl-based multifunctional nanocomposites for biomedical applications | |
| Chen et al. | A dual-targeting nanocarrier based on modified chitosan micelles for tumor imaging and therapy | |
| Narmani et al. | Smart chitosan-PLGA nanocarriers functionalized with surface folic acid ligands against lung cancer cells | |
| Tao et al. | Fe3O4 Nanoparticles Embedded in Pectin–Doxorubicin Composites as pH-Responsive Nanoplatforms for Tumor Diagnosis and Therapy by T 1-Weighted Magnetic Imaging | |
| He et al. | Yeast cell membrane-camouflaged PLGA nanoparticle platform for enhanced cancer therapy |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD01 | Notification of change of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7426 Effective date: 20150320 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20150320 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160302 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20160302 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20170120 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170131 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170426 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20170516 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20170529 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6152391 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |