JP5684787B2 - シチジンベースの抗新生物薬とシチジンデアミナーゼ阻害薬との組合せ、および癌の治療におけるその使用 - Google Patents
シチジンベースの抗新生物薬とシチジンデアミナーゼ阻害薬との組合せ、および癌の治療におけるその使用 Download PDFInfo
- Publication number
- JP5684787B2 JP5684787B2 JP2012504775A JP2012504775A JP5684787B2 JP 5684787 B2 JP5684787 B2 JP 5684787B2 JP 2012504775 A JP2012504775 A JP 2012504775A JP 2012504775 A JP2012504775 A JP 2012504775A JP 5684787 B2 JP5684787 B2 JP 5684787B2
- Authority
- JP
- Japan
- Prior art keywords
- cancer
- compound
- azacytidine
- gemcitabine
- deoxycytidine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 206010028980 Neoplasm Diseases 0.000 title claims description 119
- 201000011510 cancer Diseases 0.000 title claims description 94
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 title claims description 28
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 title claims description 26
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 title claims description 26
- 238000011282 treatment Methods 0.000 title claims description 20
- 239000002246 antineoplastic agent Substances 0.000 title description 10
- 229940123974 Cytidine deaminase inhibitor Drugs 0.000 title description 2
- 229940034982 antineoplastic agent Drugs 0.000 title description 2
- 108010031325 Cytidine deaminase Proteins 0.000 claims description 242
- 102100026846 Cytidine deaminase Human genes 0.000 claims description 239
- 150000001875 compounds Chemical class 0.000 claims description 222
- 239000000758 substrate Substances 0.000 claims description 167
- 229960003603 decitabine Drugs 0.000 claims description 149
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 claims description 145
- 239000000203 mixture Substances 0.000 claims description 135
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 claims description 98
- 229960005277 gemcitabine Drugs 0.000 claims description 98
- 239000008194 pharmaceutical composition Substances 0.000 claims description 69
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 claims description 65
- -1 C 2-6 alkenyl ester Chemical class 0.000 claims description 53
- 150000003839 salts Chemical class 0.000 claims description 40
- 239000003814 drug Substances 0.000 claims description 38
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 claims description 36
- 229960002756 azacitidine Drugs 0.000 claims description 34
- NMUSYJAQQFHJEW-UHFFFAOYSA-N 5-Azacytidine Natural products O=C1N=C(N)N=CN1C1C(O)C(O)C(CO)O1 NMUSYJAQQFHJEW-UHFFFAOYSA-N 0.000 claims description 31
- NGGRGTWYSXYVDK-RRKCRQDMSA-N 4-amino-5-chloro-1-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound C1=C(Cl)C(N)=NC(=O)N1[C@@H]1O[C@H](CO)[C@@H](O)C1 NGGRGTWYSXYVDK-RRKCRQDMSA-N 0.000 claims description 26
- IDYKCXHJJGMAEV-RRKCRQDMSA-N 4-amino-5-fluoro-1-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound C1=C(F)C(N)=NC(=O)N1[C@@H]1O[C@H](CO)[C@@H](O)C1 IDYKCXHJJGMAEV-RRKCRQDMSA-N 0.000 claims description 26
- 125000005907 alkyl ester group Chemical group 0.000 claims description 26
- 239000007787 solid Substances 0.000 claims description 26
- 229950006410 tezacitabine Drugs 0.000 claims description 26
- 229940079593 drug Drugs 0.000 claims description 25
- 208000032839 leukemia Diseases 0.000 claims description 24
- 239000003112 inhibitor Substances 0.000 claims description 22
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 22
- 206010033128 Ovarian cancer Diseases 0.000 claims description 16
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 16
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 15
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 15
- CKTSBUTUHBMZGZ-ULQXZJNLSA-N 4-amino-1-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-tritiopyrimidin-2-one Chemical compound O=C1N=C(N)C([3H])=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 CKTSBUTUHBMZGZ-ULQXZJNLSA-N 0.000 claims description 14
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 14
- 201000002528 pancreatic cancer Diseases 0.000 claims description 14
- OZQDLJNDRVBCST-SHUUEZRQSA-N 5-amino-2-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2,4-triazin-3-one Chemical compound O=C1N=C(N)C=NN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 OZQDLJNDRVBCST-SHUUEZRQSA-N 0.000 claims description 13
- 230000002489 hematologic effect Effects 0.000 claims description 13
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 13
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 13
- 125000003342 alkenyl group Chemical group 0.000 claims description 12
- 229910052739 hydrogen Inorganic materials 0.000 claims description 12
- LJIRBXZDQGQUOO-KVTDHHQDSA-N 6-amino-3-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,4-dihydro-1,3,5-triazin-2-one Chemical compound C1NC(N)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 LJIRBXZDQGQUOO-KVTDHHQDSA-N 0.000 claims description 9
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 9
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 9
- 201000003793 Myelodysplastic syndrome Diseases 0.000 claims description 9
- 206010055113 Breast cancer metastatic Diseases 0.000 claims description 8
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 8
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 8
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 8
- 238000006481 deamination reaction Methods 0.000 claims description 7
- 208000020816 lung neoplasm Diseases 0.000 claims description 7
- 201000002628 peritoneum cancer Diseases 0.000 claims description 7
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 6
- 230000009615 deamination Effects 0.000 claims description 6
- 201000005202 lung cancer Diseases 0.000 claims description 6
- 206010004593 Bile duct cancer Diseases 0.000 claims description 4
- 206010005003 Bladder cancer Diseases 0.000 claims description 4
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 4
- 208000026900 bile duct neoplasm Diseases 0.000 claims description 4
- 208000006990 cholangiocarcinoma Diseases 0.000 claims description 4
- 208000014018 liver neoplasm Diseases 0.000 claims description 4
- 206010044412 transitional cell carcinoma Diseases 0.000 claims description 4
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 4
- 206010009944 Colon cancer Diseases 0.000 claims description 3
- 208000022072 Gallbladder Neoplasms Diseases 0.000 claims description 3
- 241000124008 Mammalia Species 0.000 claims description 3
- 208000000102 Squamous Cell Carcinoma of Head and Neck Diseases 0.000 claims description 3
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 claims description 3
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 3
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 3
- 239000000825 pharmaceutical preparation Substances 0.000 claims description 3
- 229940127557 pharmaceutical product Drugs 0.000 claims description 3
- 238000002560 therapeutic procedure Methods 0.000 claims description 3
- 201000001342 Fallopian tube cancer Diseases 0.000 claims description 2
- 208000013452 Fallopian tube neoplasm Diseases 0.000 claims description 2
- 206010019695 Hepatic neoplasm Diseases 0.000 claims description 2
- 208000029742 colonic neoplasm Diseases 0.000 claims description 2
- 201000010175 gallbladder cancer Diseases 0.000 claims description 2
- 208000024719 uterine cervix neoplasm Diseases 0.000 claims description 2
- GFFXZLZWLOBBLO-ASKVSEFXSA-N tezacitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(=C/F)/[C@H](O)[C@@H](CO)O1 GFFXZLZWLOBBLO-ASKVSEFXSA-N 0.000 claims 11
- RGNOTKMIMZMNRX-XVFCMESISA-N 2-amino-1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-4-one Chemical compound NC1=NC(=O)C=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 RGNOTKMIMZMNRX-XVFCMESISA-N 0.000 claims 2
- 238000000034 method Methods 0.000 description 78
- 239000000243 solution Substances 0.000 description 70
- 239000000651 prodrug Substances 0.000 description 68
- 229940002612 prodrug Drugs 0.000 description 68
- 238000004128 high performance liquid chromatography Methods 0.000 description 35
- 229960000684 cytarabine Drugs 0.000 description 30
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 29
- 241000699670 Mus sp. Species 0.000 description 28
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 27
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 26
- 230000000694 effects Effects 0.000 description 25
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 24
- 239000000872 buffer Substances 0.000 description 24
- 238000002360 preparation method Methods 0.000 description 24
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 23
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 23
- 239000000126 substance Substances 0.000 description 19
- 238000006243 chemical reaction Methods 0.000 description 18
- 201000010099 disease Diseases 0.000 description 18
- 210000004027 cell Anatomy 0.000 description 17
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 16
- 239000000463 material Substances 0.000 description 16
- XPYQFIISZQCINN-QVXDJYSKSA-N 4-amino-1-[(2r,3e,4s,5r)-3-(fluoromethylidene)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one;hydrate Chemical compound O.O=C1N=C(N)C=CN1[C@H]1C(=C/F)/[C@H](O)[C@@H](CO)O1 XPYQFIISZQCINN-QVXDJYSKSA-N 0.000 description 15
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 15
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 15
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 238000009472 formulation Methods 0.000 description 15
- 239000011550 stock solution Substances 0.000 description 15
- 235000019441 ethanol Nutrition 0.000 description 14
- 239000002904 solvent Substances 0.000 description 14
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 13
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 13
- 125000000217 alkyl group Chemical group 0.000 description 13
- 239000011541 reaction mixture Substances 0.000 description 13
- 239000000725 suspension Substances 0.000 description 13
- 239000004480 active ingredient Substances 0.000 description 12
- 239000003795 chemical substances by application Substances 0.000 description 12
- 239000012071 phase Substances 0.000 description 12
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 11
- 208000035475 disorder Diseases 0.000 description 11
- 239000003921 oil Substances 0.000 description 11
- 239000000843 powder Substances 0.000 description 11
- 239000003826 tablet Substances 0.000 description 11
- 241000282414 Homo sapiens Species 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical class C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- 238000004587 chromatography analysis Methods 0.000 description 10
- 239000003937 drug carrier Substances 0.000 description 10
- 239000007924 injection Substances 0.000 description 10
- 238000002347 injection Methods 0.000 description 10
- 235000019198 oils Nutrition 0.000 description 10
- 230000001225 therapeutic effect Effects 0.000 description 10
- 241000699666 Mus <mouse, genus> Species 0.000 description 9
- 238000005481 NMR spectroscopy Methods 0.000 description 9
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 238000010521 absorption reaction Methods 0.000 description 9
- 208000009956 adenocarcinoma Diseases 0.000 description 9
- 230000037396 body weight Effects 0.000 description 9
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Natural products NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 9
- 239000002552 dosage form Substances 0.000 description 9
- 230000002496 gastric effect Effects 0.000 description 9
- 238000001727 in vivo Methods 0.000 description 9
- 208000024891 symptom Diseases 0.000 description 9
- 0 *=C(NCC=CC1)N1[C@@]([C@@]1O)O[C@@](CO)[C@@]1O Chemical compound *=C(NCC=CC1)N1[C@@]([C@@]1O)O[C@@](CO)[C@@]1O 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- 210000004369 blood Anatomy 0.000 description 8
- 239000008280 blood Substances 0.000 description 8
- 239000002775 capsule Substances 0.000 description 8
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 8
- 229920001223 polyethylene glycol Polymers 0.000 description 8
- 239000003981 vehicle Substances 0.000 description 8
- 206010006187 Breast cancer Diseases 0.000 description 7
- 208000026310 Breast neoplasm Diseases 0.000 description 7
- 102000004190 Enzymes Human genes 0.000 description 7
- 108090000790 Enzymes Proteins 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- 206010025323 Lymphomas Diseases 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 206010039491 Sarcoma Diseases 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- 239000000654 additive Substances 0.000 description 7
- 230000000996 additive effect Effects 0.000 description 7
- 238000004458 analytical method Methods 0.000 description 7
- 239000002585 base Substances 0.000 description 7
- 239000012530 fluid Substances 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 239000000047 product Substances 0.000 description 7
- 239000011780 sodium chloride Substances 0.000 description 7
- SNKNSKHSWOOYSB-UHFFFAOYSA-N 1,3,4,7-tetrahydro-1,3-diazepin-2-one Chemical compound O=C1NCC=CCN1 SNKNSKHSWOOYSB-UHFFFAOYSA-N 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- 206010003445 Ascites Diseases 0.000 description 6
- 201000009030 Carcinoma Diseases 0.000 description 6
- 239000002202 Polyethylene glycol Substances 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 6
- 229940041181 antineoplastic drug Drugs 0.000 description 6
- 150000002148 esters Chemical class 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 238000001990 intravenous administration Methods 0.000 description 6
- 230000014759 maintenance of location Effects 0.000 description 6
- 239000002674 ointment Substances 0.000 description 6
- 239000006187 pill Substances 0.000 description 6
- 239000003755 preservative agent Substances 0.000 description 6
- 125000006239 protecting group Chemical group 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- 239000000523 sample Substances 0.000 description 6
- 206010041823 squamous cell carcinoma Diseases 0.000 description 6
- 230000004083 survival effect Effects 0.000 description 6
- 238000002211 ultraviolet spectrum Methods 0.000 description 6
- 239000000080 wetting agent Substances 0.000 description 6
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 5
- 108010010803 Gelatin Proteins 0.000 description 5
- 241000282412 Homo Species 0.000 description 5
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 5
- 229920002472 Starch Polymers 0.000 description 5
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 5
- 229930006000 Sucrose Natural products 0.000 description 5
- 238000002835 absorbance Methods 0.000 description 5
- 239000003963 antioxidant agent Substances 0.000 description 5
- 235000006708 antioxidants Nutrition 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 229910052799 carbon Inorganic materials 0.000 description 5
- 230000034994 death Effects 0.000 description 5
- 231100000517 death Toxicity 0.000 description 5
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 5
- 239000003995 emulsifying agent Substances 0.000 description 5
- 239000008273 gelatin Substances 0.000 description 5
- 229920000159 gelatin Polymers 0.000 description 5
- 235000019322 gelatine Nutrition 0.000 description 5
- 235000011852 gelatine desserts Nutrition 0.000 description 5
- 235000011187 glycerol Nutrition 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- 239000008101 lactose Substances 0.000 description 5
- 239000000314 lubricant Substances 0.000 description 5
- 238000004519 manufacturing process Methods 0.000 description 5
- 239000001301 oxygen Substances 0.000 description 5
- 229910052760 oxygen Inorganic materials 0.000 description 5
- 239000002953 phosphate buffered saline Substances 0.000 description 5
- 229920000642 polymer Polymers 0.000 description 5
- 235000019698 starch Nutrition 0.000 description 5
- 239000005720 sucrose Substances 0.000 description 5
- 239000000829 suppository Substances 0.000 description 5
- 239000000375 suspending agent Substances 0.000 description 5
- 239000003643 water by type Substances 0.000 description 5
- 239000001993 wax Substances 0.000 description 5
- CKTSBUTUHBMZGZ-SHYZEUOFSA-N 2'‐deoxycytidine Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 CKTSBUTUHBMZGZ-SHYZEUOFSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- 241000416162 Astragalus gummifer Species 0.000 description 4
- CKTSBUTUHBMZGZ-UHFFFAOYSA-N Deoxycytidine Natural products O=C1N=C(N)C=CN1C1OC(CO)C(O)C1 CKTSBUTUHBMZGZ-UHFFFAOYSA-N 0.000 description 4
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 4
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 229920001615 Tragacanth Polymers 0.000 description 4
- 235000010443 alginic acid Nutrition 0.000 description 4
- 229920000615 alginic acid Polymers 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 4
- JJWKPURADFRFRB-UHFFFAOYSA-N carbonyl sulfide Chemical compound O=C=S JJWKPURADFRFRB-UHFFFAOYSA-N 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 239000007810 chemical reaction solvent Substances 0.000 description 4
- 238000011260 co-administration Methods 0.000 description 4
- 239000003086 colorant Substances 0.000 description 4
- 229940104302 cytosine Drugs 0.000 description 4
- 230000008034 disappearance Effects 0.000 description 4
- 235000019439 ethyl acetate Nutrition 0.000 description 4
- 239000000796 flavoring agent Substances 0.000 description 4
- 210000004051 gastric juice Anatomy 0.000 description 4
- 239000000499 gel Substances 0.000 description 4
- 239000008103 glucose Substances 0.000 description 4
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 4
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 4
- 239000004615 ingredient Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 239000002777 nucleoside Substances 0.000 description 4
- 239000006072 paste Substances 0.000 description 4
- 230000000144 pharmacologic effect Effects 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- 239000003380 propellant Substances 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- 239000000454 talc Substances 0.000 description 4
- 229910052623 talc Inorganic materials 0.000 description 4
- 235000012222 talc Nutrition 0.000 description 4
- 235000010487 tragacanth Nutrition 0.000 description 4
- 239000000196 tragacanth Substances 0.000 description 4
- 229940116362 tragacanth Drugs 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 230000004614 tumor growth Effects 0.000 description 4
- 238000005303 weighing Methods 0.000 description 4
- YXOIEKXOQGNRAZ-UHFFFAOYSA-N 1,3-diazepin-2-one Chemical compound O=C1N=CC=CC=N1 YXOIEKXOQGNRAZ-UHFFFAOYSA-N 0.000 description 3
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 3
- KGLPWQKSKUVKMJ-UHFFFAOYSA-N 2,3-dihydrophthalazine-1,4-dione Chemical compound C1=CC=C2C(=O)NNC(=O)C2=C1 KGLPWQKSKUVKMJ-UHFFFAOYSA-N 0.000 description 3
- 244000215068 Acacia senegal Species 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 229920001817 Agar Polymers 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- 201000008808 Fibrosarcoma Diseases 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- HESSNRGIEVBPRB-QDDPNBLJSA-N Gemcitabine elaidate Chemical compound FC1(F)[C@H](O)[C@@H](COC(=O)CCCCCCC/C=C/CCCCCCCC)O[C@H]1N1C(=O)N=C(N)C=C1 HESSNRGIEVBPRB-QDDPNBLJSA-N 0.000 description 3
- 208000032612 Glial tumor Diseases 0.000 description 3
- 206010018338 Glioma Diseases 0.000 description 3
- 208000012766 Growth delay Diseases 0.000 description 3
- 229920000084 Gum arabic Polymers 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 208000007766 Kaposi sarcoma Diseases 0.000 description 3
- 208000018142 Leiomyosarcoma Diseases 0.000 description 3
- 206010027406 Mesothelioma Diseases 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 206010043276 Teratoma Diseases 0.000 description 3
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 3
- DRTQHJPVMGBUCF-XVFCMESISA-N Uridine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-XVFCMESISA-N 0.000 description 3
- 235000010489 acacia gum Nutrition 0.000 description 3
- 239000000205 acacia gum Substances 0.000 description 3
- 239000008272 agar Substances 0.000 description 3
- 235000010419 agar Nutrition 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 239000012491 analyte Substances 0.000 description 3
- 235000012216 bentonite Nutrition 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 239000006172 buffering agent Substances 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 230000015556 catabolic process Effects 0.000 description 3
- 235000010980 cellulose Nutrition 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 230000000875 corresponding effect Effects 0.000 description 3
- 239000006071 cream Substances 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 238000006731 degradation reaction Methods 0.000 description 3
- 230000003111 delayed effect Effects 0.000 description 3
- 239000002270 dispersing agent Substances 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 3
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 3
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 3
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 3
- 239000003701 inert diluent Substances 0.000 description 3
- 238000007912 intraperitoneal administration Methods 0.000 description 3
- 235000010445 lecithin Nutrition 0.000 description 3
- 239000000787 lecithin Substances 0.000 description 3
- 229940067606 lecithin Drugs 0.000 description 3
- 210000004185 liver Anatomy 0.000 description 3
- 210000004072 lung Anatomy 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 210000002381 plasma Anatomy 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 3
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 3
- 235000012239 silicon dioxide Nutrition 0.000 description 3
- 235000010356 sorbitol Nutrition 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 229940032147 starch Drugs 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 235000000346 sugar Nutrition 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 239000003765 sweetening agent Substances 0.000 description 3
- 230000002195 synergetic effect Effects 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- 210000003462 vein Anatomy 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 2
- HBBIVXDEBCKFIZ-FNCVBFRFSA-N 3-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-4,7-dihydro-1h-1,3-diazepin-2-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NCC=CC1 HBBIVXDEBCKFIZ-FNCVBFRFSA-N 0.000 description 2
- SNRKAOGJCXPPSV-BWZBUEFSSA-N 3-[(2r,4r,5r)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-4,7-dihydro-1h-1,3-diazepin-2-one Chemical compound FC1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NCC=CC1 SNRKAOGJCXPPSV-BWZBUEFSSA-N 0.000 description 2
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 2
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 2
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 2
- 102100036664 Adenosine deaminase Human genes 0.000 description 2
- 102000055025 Adenosine deaminases Human genes 0.000 description 2
- 239000005995 Aluminium silicate Substances 0.000 description 2
- 201000003076 Angiosarcoma Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 2
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 241000282693 Cercopithecidae Species 0.000 description 2
- 206010008342 Cervix carcinoma Diseases 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 2
- 206010014733 Endometrial cancer Diseases 0.000 description 2
- 206010014759 Endometrial neoplasm Diseases 0.000 description 2
- 241000283073 Equus caballus Species 0.000 description 2
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 2
- 241000282326 Felis catus Species 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 208000001258 Hemangiosarcoma Diseases 0.000 description 2
- 208000017604 Hodgkin disease Diseases 0.000 description 2
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 2
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- 208000000172 Medulloblastoma Diseases 0.000 description 2
- 208000034578 Multiple myelomas Diseases 0.000 description 2
- 206010029260 Neuroblastoma Diseases 0.000 description 2
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 2
- BDJRBEYXGGNYIS-UHFFFAOYSA-N Nonanedioid acid Natural products OC(=O)CCCCCCCC(O)=O BDJRBEYXGGNYIS-UHFFFAOYSA-N 0.000 description 2
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 2
- 235000019483 Peanut oil Nutrition 0.000 description 2
- 241000009328 Perro Species 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 206010035226 Plasma cell myeloma Diseases 0.000 description 2
- 229920002732 Polyanhydride Polymers 0.000 description 2
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- ZTHYODDOHIVTJV-UHFFFAOYSA-N Propyl gallate Chemical compound CCCOC(=O)C1=CC(O)=C(O)C(O)=C1 ZTHYODDOHIVTJV-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 2
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 2
- 239000002250 absorbent Substances 0.000 description 2
- 230000002745 absorbent Effects 0.000 description 2
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 229940072056 alginate Drugs 0.000 description 2
- 239000000783 alginic acid Substances 0.000 description 2
- 229960001126 alginic acid Drugs 0.000 description 2
- 150000004781 alginic acids Chemical class 0.000 description 2
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 235000012211 aluminium silicate Nutrition 0.000 description 2
- 150000001408 amides Chemical class 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 150000005018 aminopurines Chemical class 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 239000000063 antileukemic agent Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- HMFHBZSHGGEWLO-TXICZTDVSA-N beta-D-ribose Chemical group OC[C@H]1O[C@@H](O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-TXICZTDVSA-N 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- 239000012472 biological sample Substances 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 244000309464 bull Species 0.000 description 2
- NRGASKMRNHXWEC-UHFFFAOYSA-N but-2-ene-1,4-diamine;dihydrochloride Chemical compound Cl.Cl.NCC=CCN NRGASKMRNHXWEC-UHFFFAOYSA-N 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- CZBZUDVBLSSABA-UHFFFAOYSA-N butylated hydroxyanisole Chemical compound COC1=CC=C(O)C(C(C)(C)C)=C1.COC1=CC=C(O)C=C1C(C)(C)C CZBZUDVBLSSABA-UHFFFAOYSA-N 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- 201000010881 cervical cancer Diseases 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 235000005687 corn oil Nutrition 0.000 description 2
- 239000002285 corn oil Substances 0.000 description 2
- 235000012343 cottonseed oil Nutrition 0.000 description 2
- 239000002385 cottonseed oil Substances 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 238000004090 dissolution Methods 0.000 description 2
- 238000004821 distillation Methods 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 230000008030 elimination Effects 0.000 description 2
- 238000003379 elimination reaction Methods 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 230000002255 enzymatic effect Effects 0.000 description 2
- 201000004101 esophageal cancer Diseases 0.000 description 2
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 2
- 229940093471 ethyl oleate Drugs 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 230000001605 fetal effect Effects 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000012847 fine chemical Substances 0.000 description 2
- 230000004907 flux Effects 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 150000002334 glycols Chemical class 0.000 description 2
- 229910052736 halogen Inorganic materials 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 208000014829 head and neck neoplasm Diseases 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- 239000003906 humectant Substances 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 150000002430 hydrocarbons Chemical class 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 239000012535 impurity Substances 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 238000007913 intrathecal administration Methods 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- 201000007270 liver cancer Diseases 0.000 description 2
- 239000006210 lotion Substances 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 201000001441 melanoma Diseases 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 239000004530 micro-emulsion Substances 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 150000002772 monosaccharides Chemical class 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 201000008968 osteosarcoma Diseases 0.000 description 2
- 239000000312 peanut oil Substances 0.000 description 2
- 238000003359 percent control normalization Methods 0.000 description 2
- 210000003200 peritoneal cavity Anatomy 0.000 description 2
- 229920005862 polyol Polymers 0.000 description 2
- 150000003077 polyols Chemical class 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 238000004237 preparative chromatography Methods 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000008159 sesame oil Substances 0.000 description 2
- 235000011803 sesame oil Nutrition 0.000 description 2
- 208000007056 sickle cell anemia Diseases 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 238000010898 silica gel chromatography Methods 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 2
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 239000008247 solid mixture Substances 0.000 description 2
- 235000010199 sorbic acid Nutrition 0.000 description 2
- 239000004334 sorbic acid Substances 0.000 description 2
- 229940075582 sorbic acid Drugs 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 229940124597 therapeutic agent Drugs 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 238000000825 ultraviolet detection Methods 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- QIJRTFXNRTXDIP-UHFFFAOYSA-N (1-carboxy-2-sulfanylethyl)azanium;chloride;hydrate Chemical compound O.Cl.SCC(N)C(O)=O QIJRTFXNRTXDIP-UHFFFAOYSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- UCKYOOZPSJFJIZ-FMDGEEDCSA-N (4r)-1-[(2r,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-4-hydroxy-1,3-diazinan-2-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)N[C@H](O)CC1 UCKYOOZPSJFJIZ-FMDGEEDCSA-N 0.000 description 1
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- GVJHHUAWPYXKBD-IEOSBIPESA-N (R)-alpha-Tocopherol Natural products OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- RLFFWWKJSJOYMQ-UHFFFAOYSA-N 1,3-diazepan-2-one Chemical compound O=C1NCCCCN1 RLFFWWKJSJOYMQ-UHFFFAOYSA-N 0.000 description 1
- NQPJDJVGBDHCAD-UHFFFAOYSA-N 1,3-diazinan-2-one Chemical compound OC1=NCCCN1 NQPJDJVGBDHCAD-UHFFFAOYSA-N 0.000 description 1
- MYMSJFSOOQERIO-UHFFFAOYSA-N 1-bromodecane Chemical compound CCCCCCCCCCBr MYMSJFSOOQERIO-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- WNWHHMBRJJOGFJ-UHFFFAOYSA-N 16-methylheptadecan-1-ol Chemical class CC(C)CCCCCCCCCCCCCCCO WNWHHMBRJJOGFJ-UHFFFAOYSA-N 0.000 description 1
- MXHRCPNRJAMMIM-SHYZEUOFSA-N 2'-deoxyuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 MXHRCPNRJAMMIM-SHYZEUOFSA-N 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- JNODDICFTDYODH-UHFFFAOYSA-N 2-hydroxytetrahydrofuran Chemical compound OC1CCCO1 JNODDICFTDYODH-UHFFFAOYSA-N 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical compound BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- SNRKAOGJCXPPSV-UHFFFAOYSA-N 3-[3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-4,7-dihydro-1h-1,3-diazepin-2-one Chemical compound FC1(F)C(O)C(CO)OC1N1C(=O)NCC=CC1 SNRKAOGJCXPPSV-UHFFFAOYSA-N 0.000 description 1
- CSUMTRFXTDNPMI-UHFFFAOYSA-N 3-[4-benzoyl-3,3-difluoro-5-(phenylmethoxymethyl)oxolan-2-yl]-4,7-dihydro-1h-1,3-diazepin-2-one Chemical compound C=1C=CC=CC=1C(=O)C1C(F)(F)C(N2C(NCC=CC2)=O)OC1COCC1=CC=CC=C1 CSUMTRFXTDNPMI-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-YDKYIBAVSA-N 4-amino-1-[(2r,3s,4r,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-YDKYIBAVSA-N 0.000 description 1
- SRRCASSCXPABTR-OIQWARKNSA-N 4-amino-1-[(2s,4r,5r)-3,3-difluoro-2,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound O=C1N=C(N)C=CN1[C@]1(O)C(F)(F)[C@H](O)[C@@H](CO)O1 SRRCASSCXPABTR-OIQWARKNSA-N 0.000 description 1
- 101710169336 5'-deoxyadenosine deaminase Proteins 0.000 description 1
- MFEFTTYGMZOIKO-UHFFFAOYSA-N 5-azacytosine Chemical compound NC1=NC=NC(=O)N1 MFEFTTYGMZOIKO-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 108700040115 Adenosine deaminases Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 206010004146 Basal cell carcinoma Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 206010073106 Bone giant cell tumour malignant Diseases 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- BNRAJXKJCJKQKX-DNRKLUKYSA-N C#[O][C@H]([C@@H](CO)O[C@H]1N(CC=CCN2)C2=O)[C@@]1(F)[F]#C Chemical compound C#[O][C@H]([C@@H](CO)O[C@H]1N(CC=CCN2)C2=O)[C@@]1(F)[F]#C BNRAJXKJCJKQKX-DNRKLUKYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 208000009458 Carcinoma in Situ Diseases 0.000 description 1
- 206010007953 Central nervous system lymphoma Diseases 0.000 description 1
- 208000005243 Chondrosarcoma Diseases 0.000 description 1
- 201000009047 Chordoma Diseases 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- 102000005381 Cytidine Deaminase Human genes 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical class C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 1
- 208000005189 Embolism Diseases 0.000 description 1
- 206010014967 Ependymoma Diseases 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 208000006168 Ewing Sarcoma Diseases 0.000 description 1
- 208000005917 Exostoses Diseases 0.000 description 1
- 206010063599 Exposure to chemical pollution Diseases 0.000 description 1
- 206010016654 Fibrosis Diseases 0.000 description 1
- 206010061968 Gastric neoplasm Diseases 0.000 description 1
- 208000021309 Germ cell tumor Diseases 0.000 description 1
- 208000000527 Germinoma Diseases 0.000 description 1
- 208000007569 Giant Cell Tumors Diseases 0.000 description 1
- 201000010915 Glioblastoma multiforme Diseases 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Polymers OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- 206010061203 Hepatobiliary neoplasm Diseases 0.000 description 1
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- QAQJMLQRFWZOBN-LAUBAEHRSA-N L-ascorbyl-6-palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](O)[C@H]1OC(=O)C(O)=C1O QAQJMLQRFWZOBN-LAUBAEHRSA-N 0.000 description 1
- 239000011786 L-ascorbyl-6-palmitate Substances 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 241000282567 Macaca fascicularis Species 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 1
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 1
- 201000009906 Meningitis Diseases 0.000 description 1
- 102000016397 Methyltransferase Human genes 0.000 description 1
- 108060004795 Methyltransferase Proteins 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 235000005135 Micromeria juliana Nutrition 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 208000014767 Myeloproliferative disease Diseases 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 description 1
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 1
- 208000034176 Neoplasms, Germ Cell and Embryonal Diseases 0.000 description 1
- 241000750004 Nestor meridionalis Species 0.000 description 1
- UTFAIMPZIICABA-FRYXHQTHSA-N O=C(c1ccccc1)OC(C1)[C@@]1([C@H](C1(F)F)OC(c2ccccc2)=O)OC1N(CC=CCN1)C1=O Chemical compound O=C(c1ccccc1)OC(C1)[C@@]1([C@H](C1(F)F)OC(c2ccccc2)=O)OC1N(CC=CCN1)C1=O UTFAIMPZIICABA-FRYXHQTHSA-N 0.000 description 1
- 201000010133 Oligodendroglioma Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 241000282320 Panthera leo Species 0.000 description 1
- 241001504519 Papio ursinus Species 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000004952 Polyamide Substances 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000026149 Primary peritoneal carcinoma Diseases 0.000 description 1
- 238000012356 Product development Methods 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 101710180958 Putative aminoacrylate hydrolase RutD Proteins 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 201000000582 Retinoblastoma Diseases 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 208000026375 Salivary gland disease Diseases 0.000 description 1
- 240000002114 Satureja hortensis Species 0.000 description 1
- 235000007315 Satureja hortensis Nutrition 0.000 description 1
- 201000010208 Seminoma Diseases 0.000 description 1
- 208000000097 Sertoli-Leydig cell tumor Diseases 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- 208000001435 Thromboembolism Diseases 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 102000004142 Trypsin Human genes 0.000 description 1
- 108090000631 Trypsin Proteins 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- 206010046431 Urethral cancer Diseases 0.000 description 1
- 208000028484 Urethral disease Diseases 0.000 description 1
- 206010046458 Urethral neoplasms Diseases 0.000 description 1
- 241000700647 Variola virus Species 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- OIRDTQYFTABQOQ-UHTZMRCNSA-N Vidarabine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O OIRDTQYFTABQOQ-UHTZMRCNSA-N 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 235000009754 Vitis X bourquina Nutrition 0.000 description 1
- 235000012333 Vitis X labruscana Nutrition 0.000 description 1
- 240000006365 Vitis vinifera Species 0.000 description 1
- 235000014787 Vitis vinifera Nutrition 0.000 description 1
- QKYLZBPPSBQAJB-ARIFNINKSA-N [[(2S,3R,5R)-5-(4-amino-2-oxopyrimidin-1-yl)-4,4-difluoro-3-hydroxyoxolan-2-yl]-hydroxymethyl] (E)-octadec-2-enoate Chemical compound FC1([C@@H](O[C@@H]([C@H]1O)C(O)OC(\C=C\CCCCCCCCCCCCCCC)=O)N1C(=O)N=C(N)C=C1)F QKYLZBPPSBQAJB-ARIFNINKSA-N 0.000 description 1
- 230000003187 abdominal effect Effects 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- OIRDTQYFTABQOQ-KQYNXXCUSA-N adenosine Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 210000004100 adrenal gland Anatomy 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 1
- HXBPYFMVGFDZFT-UHFFFAOYSA-N allyl isocyanate Chemical compound C=CCN=C=O HXBPYFMVGFDZFT-UHFFFAOYSA-N 0.000 description 1
- 229940087168 alpha tocopherol Drugs 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 238000012435 analytical chromatography Methods 0.000 description 1
- 210000003484 anatomy Anatomy 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000003474 anti-emetic effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000000118 anti-neoplastic effect Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 229940125683 antiemetic agent Drugs 0.000 description 1
- 239000002111 antiemetic agent Substances 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 235000019789 appetite Nutrition 0.000 description 1
- 230000036528 appetite Effects 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 108010031919 arabinosylcytosine deaminase Proteins 0.000 description 1
- DRTQHJPVMGBUCF-CCXZUQQUSA-N arauridine Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-CCXZUQQUSA-N 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 235000010385 ascorbyl palmitate Nutrition 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- DRTQHJPVMGBUCF-PSQAKQOGSA-N beta-L-uridine Natural products O[C@H]1[C@@H](O)[C@H](CO)O[C@@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-PSQAKQOGSA-N 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 201000008873 bone osteosarcoma Diseases 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 210000003123 bronchiole Anatomy 0.000 description 1
- 230000005587 bubbling Effects 0.000 description 1
- 239000001273 butane Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 150000004652 butanoic acids Chemical class 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 235000012241 calcium silicate Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- RBHJBMIOOPYDBQ-UHFFFAOYSA-N carbon dioxide;propan-2-one Chemical compound O=C=O.CC(C)=O RBHJBMIOOPYDBQ-UHFFFAOYSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 201000007455 central nervous system cancer Diseases 0.000 description 1
- 208000025997 central nervous system neoplasm Diseases 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229940044683 chemotherapy drug Drugs 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 150000005827 chlorofluoro hydrocarbons Chemical class 0.000 description 1
- 201000005217 chondroblastoma Diseases 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- 208000009060 clear cell adenocarcinoma Diseases 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 230000002079 cooperative effect Effects 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 239000001767 crosslinked sodium carboxy methyl cellulose Substances 0.000 description 1
- 239000012043 crude product Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 238000009109 curative therapy Methods 0.000 description 1
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 229960001305 cysteine hydrochloride Drugs 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- MXHRCPNRJAMMIM-UHFFFAOYSA-N desoxyuridine Natural products C1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 MXHRCPNRJAMMIM-UHFFFAOYSA-N 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 150000008050 dialkyl sulfates Chemical class 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 150000001993 dienes Chemical class 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- UGMCXQCYOVCMTB-UHFFFAOYSA-K dihydroxy(stearato)aluminium Chemical compound CCCCCCCCCCCCCCCCCC(=O)O[Al](O)O UGMCXQCYOVCMTB-UHFFFAOYSA-K 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- FSBVERYRVPGNGG-UHFFFAOYSA-N dimagnesium dioxido-bis[[oxido(oxo)silyl]oxy]silane hydrate Chemical compound O.[Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O FSBVERYRVPGNGG-UHFFFAOYSA-N 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 238000002224 dissection Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 229940043264 dodecyl sulfate Drugs 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 238000001952 enzyme assay Methods 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 201000010934 exostosis Diseases 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 239000003885 eye ointment Substances 0.000 description 1
- 235000020937 fasting conditions Nutrition 0.000 description 1
- 239000003925 fat Substances 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000012065 filter cake Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 235000019000 fluorine Nutrition 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 238000003304 gavage Methods 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 229950007898 gemcitabine elaidate Drugs 0.000 description 1
- 229940020967 gemzar Drugs 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 201000003115 germ cell cancer Diseases 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 210000004209 hair Anatomy 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 239000011874 heated mixture Substances 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000019691 hematopoietic and lymphoid cell neoplasm Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 208000006359 hepatoblastoma Diseases 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- YAMHXTCMCPHKLN-UHFFFAOYSA-N imidazolidin-2-one Chemical compound O=C1NCCN1 YAMHXTCMCPHKLN-UHFFFAOYSA-N 0.000 description 1
- 201000004933 in situ carcinoma Diseases 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 239000002198 insoluble material Substances 0.000 description 1
- 206010022498 insulinoma Diseases 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 239000000543 intermediate Substances 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 201000010985 invasive ductal carcinoma Diseases 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 208000003849 large cell carcinoma Diseases 0.000 description 1
- 210000002429 large intestine Anatomy 0.000 description 1
- 239000011981 lindlar catalyst Substances 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 208000037841 lung tumor Diseases 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 230000001926 lymphatic effect Effects 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 201000004593 malignant giant cell tumor Diseases 0.000 description 1
- 208000006178 malignant mesothelioma Diseases 0.000 description 1
- 201000005282 malignant pleural mesothelioma Diseases 0.000 description 1
- 201000000289 malignant teratoma Diseases 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 210000002418 meninge Anatomy 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 208000037843 metastatic solid tumor Diseases 0.000 description 1
- 238000005649 metathesis reaction Methods 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 239000012452 mother liquor Substances 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 229940051866 mouthwash Drugs 0.000 description 1
- 208000010492 mucinous cystadenocarcinoma Diseases 0.000 description 1
- 201000005962 mycosis fungoides Diseases 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical class C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- MRWXACSTFXYYMV-FDDDBJFASA-N nebularine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC=C2N=C1 MRWXACSTFXYYMV-FDDDBJFASA-N 0.000 description 1
- 208000025189 neoplasm of testis Diseases 0.000 description 1
- 210000005170 neoplastic cell Anatomy 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 208000004235 neutropenia Diseases 0.000 description 1
- 150000002814 niacins Chemical class 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 150000003833 nucleoside derivatives Chemical class 0.000 description 1
- 238000011580 nude mouse model Methods 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 235000013348 organic food Nutrition 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 208000029749 ovarian granulosa cell tumor Diseases 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 210000003101 oviduct Anatomy 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 239000006174 pH buffer Substances 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 201000008129 pancreatic ductal adenocarcinoma Diseases 0.000 description 1
- 208000021255 pancreatic insulinoma Diseases 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 235000014594 pastries Nutrition 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000003961 penetration enhancing agent Substances 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 201000002513 peritoneal mesothelioma Diseases 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 235000011007 phosphoric acid Nutrition 0.000 description 1
- 238000006303 photolysis reaction Methods 0.000 description 1
- 230000015843 photosynthesis, light reaction Effects 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 210000004560 pineal gland Anatomy 0.000 description 1
- 210000004224 pleura Anatomy 0.000 description 1
- 239000003880 polar aprotic solvent Substances 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 229920002647 polyamide Polymers 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 239000004417 polycarbonate Substances 0.000 description 1
- 229920000728 polyester Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 239000008057 potassium phosphate buffer Substances 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000012746 preparative thin layer chromatography Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 208000016800 primary central nervous system lymphoma Diseases 0.000 description 1
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 239000000473 propyl gallate Substances 0.000 description 1
- 235000010388 propyl gallate Nutrition 0.000 description 1
- 229940075579 propyl gallate Drugs 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 201000005825 prostate adenocarcinoma Diseases 0.000 description 1
- 208000023958 prostate neoplasm Diseases 0.000 description 1
- 229950008679 protamine sulfate Drugs 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- 239000002212 purine nucleoside Substances 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 239000002516 radical scavenger Substances 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 210000005000 reproductive tract Anatomy 0.000 description 1
- 238000004007 reversed phase HPLC Methods 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 201000000306 sarcoidosis Diseases 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 239000011986 second-generation catalyst Substances 0.000 description 1
- 208000011581 secondary neoplasm Diseases 0.000 description 1
- 230000035807 sensation Effects 0.000 description 1
- 235000019615 sensations Nutrition 0.000 description 1
- 208000004548 serous cystadenocarcinoma Diseases 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 210000003625 skull Anatomy 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- 208000014680 small intestine neoplasm Diseases 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- WBHQBSYUUJJSRZ-UHFFFAOYSA-M sodium bisulfate Chemical compound [Na+].OS([O-])(=O)=O WBHQBSYUUJJSRZ-UHFFFAOYSA-M 0.000 description 1
- 229910000342 sodium bisulfate Inorganic materials 0.000 description 1
- 229940100996 sodium bisulfate Drugs 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229940001482 sodium sulfite Drugs 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 210000004872 soft tissue Anatomy 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 238000000638 solvent extraction Methods 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 238000003797 solvolysis reaction Methods 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 239000004544 spot-on Substances 0.000 description 1
- 238000000528 statistical test Methods 0.000 description 1
- 125000004079 stearyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003206 sterilizing agent Substances 0.000 description 1
- 210000001562 sternum Anatomy 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 210000002536 stromal cell Anatomy 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- 208000001608 teratocarcinoma Diseases 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- 150000003567 thiocyanates Chemical class 0.000 description 1
- AOBORMOPSGHCAX-DGHZZKTQSA-N tocofersolan Chemical compound OCCOC(=O)CCC(=O)OC1=C(C)C(C)=C2O[C@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C AOBORMOPSGHCAX-DGHZZKTQSA-N 0.000 description 1
- 229960000984 tocofersolan Drugs 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 125000005490 tosylate group Chemical group 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000027 toxicology Toxicity 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 239000012588 trypsin Substances 0.000 description 1
- 208000025421 tumor of uterus Diseases 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 238000001946 ultra-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical class CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 1
- 210000003708 urethra Anatomy 0.000 description 1
- 229940045145 uridine Drugs 0.000 description 1
- 208000029584 urinary system neoplasm Diseases 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 206010046766 uterine cancer Diseases 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 210000001215 vagina Anatomy 0.000 description 1
- 239000006216 vaginal suppository Substances 0.000 description 1
- OMOMUFTZPTXCHP-UHFFFAOYSA-N valpromide Chemical compound CCCC(C(N)=O)CCC OMOMUFTZPTXCHP-UHFFFAOYSA-N 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 235000019871 vegetable fat Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 229960003636 vidarabine Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 230000021542 voluntary musculoskeletal movement Effects 0.000 description 1
- 210000003905 vulva Anatomy 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 210000002268 wool Anatomy 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
- 239000011787 zinc oxide Substances 0.000 description 1
- 239000002076 α-tocopherol Substances 0.000 description 1
- 235000004835 α-tocopherol Nutrition 0.000 description 1
Images



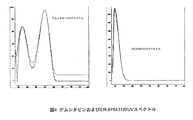








Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description

(ここで、および他において、化合物の名称と化合物の構造との間に矛盾が存在する場合、化学構造が優先される)。
下記の定義を、本明細書を通して使用する。
明細書および請求項中で使用される場合、単数形「a」、「an」および「the」は、文脈で他に明確に示されていない限り、複数形についての言及を包含する。したがって例えば、「1種の(a)化合物」を含む医薬組成物に関する言及は、2種以上の化合物を包含し得る。
心臓:肉腫(例えば、血管肉腫、線維肉腫、横紋筋肉腫、脂肪肉腫など)、横紋筋腫および奇形腫、
肺:気管支癌(例えば、扁平上皮細胞、未分化小細胞、未分化大細胞、腺癌など)、肺胞腺(例えば、細気管支など)癌、肉腫、リンパ腫、非小細胞肺癌および中皮腫、
胃腸:食道(例えば、扁平上皮細胞癌、腺癌、平滑筋肉腫、リンパ腫など)、胃(例えば、癌、リンパ腫、平滑筋肉腫など)、膵臓(例えば、導管性腺癌、インスリノーマ、類癌腫、ビポーマなど)、小腸(例えば、腺癌、リンパ腫、類癌腫、カポジ肉腫など)、大腸(例えば、腺癌など)、
尿生殖路:腎臓(例えば、腺癌、リンパ腫、白血病など)、膀胱および尿道(例えば、扁平上皮細胞癌、移行上皮癌、腺癌など)、前立腺(例えば、腺癌、肉腫など)、精巣(例えば、精上皮腫、奇形腫、胎児性癌、奇形癌、絨毛上皮腫、肉腫、間質細胞癌など)、
肝臓:肝癌(例えば、肝細胞癌腫など)、胆管癌、肝芽腫および血管肉腫、
骨:骨原性肉腫(例えば、骨肉腫など)、線維肉腫、悪性線維性組織球腫、軟骨肉腫、ユーイング肉腫、悪性リンパ腫(例えば、小神経膠腫など)、多発性骨髄腫、悪性巨細胞腫瘍脊索腫(例えば、骨軟骨性外骨症など)、軟骨芽細胞腫および巨細胞腫、
神経系:頭蓋、髄膜(例えば、髄膜肉腫、神経膠腫症など)、脳(例えば、神経膠星状細胞腫、髄芽細胞腫、膠腫、脳室上衣細胞腫、胚細胞腫[松果体腫]、多形性神経膠芽細胞腫、乏突起神経膠腫、網膜芽細胞腫、先天性腫瘍など)、脊髄(例えば、肉腫など)、
乳癌、
婦人科:子宮(例えば、子宮内膜癌など)、子宮頸(例えば、子宮頸癌など)、卵巣(例えば、卵巣癌[漿液性嚢胞腺癌、粘液性嚢胞腺癌、非分類癌腫]、セルトリ−ライディッヒ細胞腫、未分化胚細胞腫、悪性奇形腫など)、外陰(例えば、扁平上皮細胞癌、上皮内癌、腺癌、線維肉腫、黒色腫など)、膣(例えば、明細胞癌、扁平上皮細胞癌、ブドウ状肉腫(胎児性横紋筋肉腫]、ファロピウス管(癌腫)など)、
血液:血液(例えば、骨髄性白血病[急性および慢性]、急性リンパ芽球性白血病、慢性リンパ球性白血病、慢性骨髄球性白血病、骨髄増殖性疾患、多発性骨髄腫、骨髄異形成症候群など)、ホジキン病、非ホジキンリンパ腫、
皮膚:悪性黒色腫、基底細胞癌、扁平上皮細胞癌、カポジ肉腫など、および
副腎:神経芽細胞腫とが挙げられる。
本発明は、CDAの活性を阻害する化合物を提供する。他の実施形態では、これらの化合物は、癌(例えば、骨髄異形成症候群、急性骨髄性白血病、慢性骨髄球性白血病、非小細胞肺癌、膵臓癌、卵巣癌および乳癌)を治療する目的で、他の抗癌医薬品(例えば、非デシタビンCDA基質、非デシタビンCDA基質のプロドラッグまたは非デシタビンCDA基質の前駆体)と組み合わせて投与することができる。
R1およびR2の一方はFであり、他方はHおよびFから選択され、
R3およびR4の一方はHであり、他方はHおよびOHから選択され、
-----は共有結合であるか、または存在せず、-----が共有結合である場合、R4は存在せず、R3は平面上にある]
または薬学的に許容されるその塩、C1〜6アルキルエステルもしくはC2〜6アルケニルエステルに関する。
R3およびR4の一方はHであり、他方はHおよびOHから選択される]
または薬学的に許容されるその塩、C1〜6アルキルエステルもしくはC2〜6アルケニルエステルによって表される。
R1およびR2の一方はFであり、他方はHおよびFから選択され、
R3およびR4の一方はHであり、他方はHおよびOHから選択され、
-----は共有結合であるか、または存在せず、-----が共有結合である場合、R4は存在せず、R3は平面上にある]
または薬学的に許容されるその塩、C1〜6アルキルエステルもしくはC2〜6アルケニルエステルと、薬学的に許容される担体とを含む医薬組成物に関する。
R1およびR2の一方はFであり、他方はHおよびFから選択され、
R3およびR4の一方はHであり、他方はHおよびOHから選択され、
-----は共有結合であるか、または存在せず、-----が共有結合である場合、R4は存在しない]
または薬学的に許容されるその塩、C1〜6アルキルエステルもしくはC2〜6アルケニルエステルとを含む医薬組成物に関する。
A)非デシタビンCDA基質の放出および生物学的利用能、続く、式I〜VIIIのいずれか1つの化合物の放出および生物学的利用能、
B)式I〜VIIIのいずれか1つの化合物の放出および生物学的利用能、続く、非デシタビンCDA基質の放出および生物学的利用能、
C)式I〜VIIIのいずれか1つの化合物の放出および生物学的利用能と同時の(または、実質的に同時の)非デシタビンCDA基質の放出および生物学的利用能、が包含される。
本テキストの範囲内において、本発明の化合物の特に所望される最終生成物の成分ではない容易に除去可能な基は、「保護基」と名付けられている。そのような保護基による官能基の保護、保護基自体およびその開裂反応は例えば、Science of Synthesis:Houben−Weyl Methods of Molecular Transformation.Georg Thieme Verlag、Stuttgart、Germany.2005年、41627頁(URL:http://www.science−of−synthesis.com(電子版、48巻));J.F.W.McOmie、「Protective Groups in Organic Chemistry」、Plenum Press、London and New York 1973年、T.W.Greene and P.G.M.Wuts、「Protective Groups in Organic Synthesis」、第3版、Wiley、New York 1999年、「The Peptides」;3巻(編者:E.Gross and J.Meienhofer)、Academic Press、London and New York 1981年、「Methoden der organischen Chemie(Methods of Organic Chemistry)」、Houben Weyl、第4版、Volume 15/I、Georg Thieme Verlag、Stuttgart 1974年、H.−D.Jakubke and H.Jeschkeit、「Aminosauren,Peptide,Proteine」(Amino acids,Peptides,Proteins)」、Verlag Chemie、Weinheim、Deerfield Beach,and Basel 1982年およびJochen Lehmann、「Chemie der Kohlenhydrate:Monosaccharide und Derivate(Chemistry of Carbohydrates:Monosaccharides and Derivatives)」、Georg Thieme Verlag、Stuttgart 1974年などの標準的な参照文献に記載されている。保護基の特徴は、それらを容易に(即ち、望ましくない副反応を起こすことなく)、例えば、加溶媒分解、還元、光分解によって、または別法では生理学的条件下で(例えば、酵素開裂によって)除去することができることである。
ある種の他の実施形態では、いずれもその全体を引用することにより本明細書の一部をなすものとする米国特許第6,001,994号、米国特許第6,469,058号および米国特許第6,555,518号に記載されている製剤および方法を使用して、本発明の組成物(例えば、非デシタビンCDA基質と組み合わせた式Iの化合物、例えば、ゲムシタビンと組み合わせたER−876437)を、それを必要とする対象に投与することができる。
別段に述べられていない限り、実施例I.B.〜I.C.では、溶媒除去を、Buchi回転蒸発器を使用して実施した。分析クロマトグラフィーは、Hewlett Packardシリーズ1100HPLCを使用して実施し、分取クロマトグラフィーは、Biotage SP4装置またはWaters 4000装置を使用して、Chiralpak IAカラムを用いて、別段に示されていない限り中性条件下で実施した。質量スペクトルは、Waters Acquity UPLC/MSシステムを使用して記録した。残りの実施例については、同様か、または匹敵する装置を使用した。
≪I.A.1.:ER−878899(1,3,4,7−テトラヒドロ−2H−1,3−ジアゼピン−2−オン)の調製≫
ER−878899を、下記のスキームIに概説されている通りに調製した。この調製は、J.Med.Chem.1981年、24巻、662〜666頁;J.Org.Chem.1980年、45巻、485〜489頁およびBull.Soc.Chim.Fr.1973年、198〜292頁に記載されており、これらは全て、その全体を引用することにより本明細書の一部をなすものとする。
I.A.1に従って調製されたER−878899を、下記の通りにスキームIIで使用した。
≪I.B.1.:ER−878899の調製(1,3,4,7−テトラヒドロ−2H−1,3−ジアゼピン−2−オン)≫
Feigenbaum,A.およびLehn,J.M.、Bull.Soc.Chim.Fr.、1973年、198〜202頁およびLiu,P.S.、Marquez,V.E.、Driscoll,J.S.およびFuller,R.W.、J.Med.Chem.、1981年、24巻、662〜666頁に記載された手順に従って、ER−878705(下記に示されている)を調製した。
≪I.C.1.:ER−879381の調製≫
ER−879381を、下記に示されている通りスキームVIIに従って製造した。ER−878899は、実施例I.B.1.に記載の通り調製した。
ER−876437を下記のスキームVIIIに示されている通りに調製した。
≪I.D.1.:ER−878890の調製≫
Cacciamani,T.ら、Arch.Biochem.Biophys.1991年、290巻、285〜92頁;Cohen R.ら、J.Biol.Chem.、1971年、246巻、7566〜8頁;およびVincenzetti S.ら、Protein Expr.Purif.1996年、8巻、247〜53頁に記載されているシチジンデアミナーゼ(CDA)酵素アッセイを使用して、本明細書に記載されている化合物の阻害活性(IC50)を決定した。このアッセイを使用し、CDAによって触媒される脱アミノ化反応が原因である基質(シチジン)の減少を追うことによって、これらの化合物のIC50を決定した。反応の280nmでの吸光度によって、時間の経過に伴う基質(シチジン)の消失をモニタリングした。
ER−876437およびER−876400を両方ともマウスに、10mg/kgで静脈内(IV)に尾静脈を介して、および10mg/kgで経口的に(POまたは経口で)胃管栄養法を介して投与した。全ての用量をリン酸緩衝食塩水(PBS)中で調製し、5mL/kgの体積で投与した。1群当たりマウス5匹をこれらの研究では使用した。血液試料を各マウスの尾静脈から、予め決定された時点で連続して採取した。血漿のために処理する前に、各群中のマウス全てからの血液試料を一緒に貯留した。抜き出した後に、貯留した血液試料を30〜60分以内遠心し、血漿を回収し、アッセイのために凍結させた。調製および抽出の後に、試料をLC/MS/MSによってアッセイした。観察された濃度(ng/mL)を下記の表3に報告する。
この実施例では、室温(約25℃)および37℃で1.45のpHを有する人工胃液中でのER−876400およびER−876437の安定性を記載する。絶食条件下のヒトでは、胃内pHは、1.4から2.1の範囲であると報告されている。引用することにより全体が本明細書の一部をなすものとするKararli、T.T.Comparison of the GI anatomy、physiology,and biochemistry of humans and commonly used laboratory animals.BioPharm & DrugDispos.16巻:351〜380頁、1995年を参照されたい。絶食しているサルの胃内pHは、1〜3の同様の範囲を有すると報告されている。引用することにより全体が本明細書の一部をなすものとするKondo,H.ら、Characteristics of the gastric pH profiles of unfed and fed cynomolgus monkeys as pharmaceutical product development subjects.BioPharm & DrugDispos.24巻:45〜51頁、2003年を参照されたい。
<勾配> 時間(分) 水% MeCN%
0〜2 98 2
2〜2.5 (水98%/MeCN2%)から
(水60%/MeCN40%)への直線勾配
2.5〜3.5 60 40
この研究を使用して、ER−876437がマウスのL1210生存モデルにおける非デシタビンCDA基質(またはそのプロドラッグ)の経口効力を増大させるかを決定することができる。
この研究によって、A2780ヒト卵巣癌異種移植片モデルでの経口ゲムシタビン治療に対するER−876437の増大活性を評価した。ゲムシタビンの30分前にER−876437を投与したが、その際、両方の化合物を経口投与した。動物に月曜から金曜までの毎日、2週間にわたって投与した。
ER−876437およびゲムシタビン−HCl(Gemzar(R)注射用、Eli Lilly)を0.5%メチルセルロース(Sigma)中で処方した。雌のヌードマウス(NU/NU、株コード088、6週齢、Charles River Laboratory)に、マウス1匹当たりA2780癌細胞5×106を皮下移植した。腫瘍が約150mm3になった13日目に、表9に記載されている通りに治療を開始した。
ER−876437単独(群3)は、腫瘍増殖に対して全く効果を有さなかった(図3)。1mg/kgをPOで、qd×5で2週間の計画でのゲムシタビンの経口投与(群2)は、治療の第2週目の後に限られた効力を示したが(図3)、ER−876437単独(群3)は、治療期間全体を通して何ら効力を示さなかった(図3)。腫瘍の2倍化時間を腫瘍増殖遅延(TGD)を定義するために使用すると、ゲムシタビン単独(群2)およびER−876437単独(群3)の両方は、ビヒクル(群1)と比較して僅か2日の遅延を示した(表10)。群1、2および3の間に統計的に重大な差違はなく(Mann−Whitney検定、GraphPad Prism 5、La Jolla、CA)、41日目に、退縮または腫瘍消失を示した生存体はない。
この実施例は、Phenomenex Luna C18(2)HPLCカラム(100Å 4.6×250mm 5μm)を使用した。溶媒送達系は、HPLC四成分ポンプ、低圧混合を使用した。可変ループ、0.1から100μL範囲および温度制御恒温器を有するオートサンプラーを使用した。UV検出器は、二波長検出器、ダイオードアレー検出器、可変波長検出器または同等物を使用することができ、クロマトグラフィーソフトウェア(例えば、HPLCまたは同等物でWaters Empower 2 Build 2154、Agilent ChemStation ソフトウェアバージョンA.09.03以上)を使用して記録することができる。
この実施例では、37℃のトリス−HCl緩衝液中、シチジンデアミナーゼ(CDA)の存在下でのシタラビン(Sigma)の半減期(T1/2)に対するER−876437の効果を記載する。
Claims (21)
- シチジン、デオキシシチジン、5−アザシチジン、ゲムシタビン、ara−C、テザシタビン、5−フルオロ−2’−デオキシシチジン、シトクロル、5,6−ジヒドロ−5−アザシチジン、6−アザシチジン、および1−メチル−Ψ−イソシチジンからなる群から選択される非デシタビンCDA基質と、
式Iの化合物
R1およびR2の一方はFであり、他方はHおよびFから選択され、
R3およびR4の一方はHであり、他方はHおよびOHから選択され、
-----は共有結合であるか、または存在せず、-----が共有結合である場合、R4は存在しない]
または薬学的に許容されるその塩、C1〜6アルキルエステルもしくはC2〜6アルケニルエステルと
を含む組成物。 - シチジン、デオキシシチジン、5−アザシチジン、ゲムシタビン、ara−C、テザシタビン、5−フルオロ−2’−デオキシシチジン、シトクロル、5,6−ジヒドロ−5−アザシチジン、6−アザシチジン、および1−メチル−Ψ−イソシチジンからなる群から選択される非デシタビンCDA基質を含む組成物と、
式Iの化合物
R1およびR2の一方はFであり、他方はHおよびFから選択され、
R3およびR4の一方はHであり、他方はHおよびOHから選択され、
-----は共有結合であるか、または存在せず、-----が共有結合である場合、R4は存在しない]
または薬学的に許容されるその塩、C1〜6アルキルエステルもしくはC2〜6アルケニルエステルを含む組成物と
を含む、癌の治療において同時に、個別に、または連続して使用するための部品のキット。 - 式Iの化合物
R1およびR2の一方はFであり、他方はHおよびFから選択され、
R3およびR4の一方はHであり、他方はHおよびOHから選択され、
-----は共有結合であるか、または存在せず、-----が共有結合である場合、R4は存在しない]
または薬学的に許容されるその塩、C1〜6アルキルエステルもしくはC2〜6アルケニルエステルを用いる、シチジン、デオキシシチジン、5−アザシチジン、ゲムシタビン、ara−C、テザシタビン、5−フルオロ−2’−デオキシシチジン、シトクロル、5,6−ジヒドロ−5−アザシチジン、6−アザシチジン、および1−メチル−Ψ−イソシチジンからなる群から選択される非デシタビンCDA基質で治療される対象において癌を治療するための医薬品。 - 前記癌が、血液学的癌および固形癌からなる群から選択される、請求項3に記載の医薬品。
- 前記癌が、骨髄異形成症候群および白血病からなる群から選択される血液学的癌である、請求項4に記載の医薬品。
- 前記癌が、急性骨髄性白血病および慢性骨髄性白血病からなる群から選択される白血病である、請求項5に記載の医薬品。
- 前記癌が、膵臓癌、卵巣癌、腹膜癌、非小細胞肺癌、転移性乳癌、膀胱癌、移行上皮癌、胆管癌、胆嚢癌、ファロピウス管癌、頭頚部の扁平上皮細胞癌、肝細胞癌、肝臓腫瘍、肺癌、子宮頸腫瘍および結腸癌からなる群から選択される固形癌である、請求項4に記載の医薬品。
- 式Iの化合物
R1およびR2の一方はFであり、他方はHおよびFから選択され、
R3およびR4の一方はHであり、他方はHおよびOHから選択され、
-----は共有結合であるか、または存在せず、-----が共有結合である場合、R4は存在しない]
または薬学的に許容されるその塩、C1〜6アルキルエステルもしくはC2〜6アルケニルエステルを用いる、シチジン、デオキシシチジン、5−アザシチジン、ゲムシタビン、ara−C、テザシタビン、5−フルオロ−2’−デオキシシチジン、シトクロル、5,6−ジヒドロ−5−アザシチジン、6−アザシチジン、および1−メチル−Ψ−イソシチジンからなる群から選択される非デシタビンCDA基質の脱アミノ化防止薬。 - 式Iの化合物
R1およびR2の一方はFであり、他方はHおよびFから選択され、
R3およびR4の一方はHであり、他方はHおよびOHから選択され、
-----は共有結合であるか、または存在せず、-----が共有結合である場合、R4は存在しない]
または薬学的に許容されるその塩、C1〜6アルキルエステルもしくはC2〜6アルケニルエステルを用いる、シチジンデアミナーゼの阻害薬。 - (i)シチジン、デオキシシチジン、5−アザシチジン、ゲムシタビン、ara−C、テザシタビン、5−フルオロ−2’−デオキシシチジン、シトクロル、5,6−ジヒドロ−5−アザシチジン、6−アザシチジン、および1−メチル−Ψ−イソシチジンからなる群から選択される非デシタビンCDA基質と、
(ii)式Iの化合物
R1およびR2の一方はFであり、他方はHおよびFから選択され、
R3およびR4の一方はHであり、他方はHおよびOHから選択され、
-----は共有結合であるか、または存在せず、-----が共有結合である場合、R4は存在しない]
または薬学的に許容されるその塩、C1〜6アルキルエステルもしくはC2〜6アルケニルエステルと、
(iii)薬学的に許容される賦形剤と
を含む医薬組成物。 - 非デシタビンCDA基質を含む前記組成物と、式Iの化合物を含む前記組成物とが同時に投与される、請求項2に記載のキット。
- 非デシタビンCDA基質を含む前記組成物と、式Iの化合物を含む前記組成物とが連続して投与される、請求項2に記載のキット。
- 前記式Iの化合物が、式VIIIの化合物
- 前記式Iの化合物が、式VIIIの化合物
- 前記非デシタビンCDA基質が、5−アザシチジン、ara−C、ゲムシタビン、テザシタビン、5−フルオロ−2’−デオキシシチジン、およびシトクロルからなる群から選択される、請求項1に記載の組成物。
- 式Iの化合物
R1およびR2の一方はFであり、他方はHおよびFから選択され、
R3およびR4の一方はHであり、他方はHおよびOHから選択され、
-----は共有結合であるか、または存在せず、-----が共有結合である場合、R4は存在しない]
または薬学的に許容されるその塩、C1〜6アルキルエステルもしくはC2〜6アルケニルエステルを用いる、
(i)治療を必要とする哺乳動物に式Iの化合物を投与するステップと、
(ii)治療を必要とする哺乳動物に、シチジン、デオキシシチジン、5−アザシチジン、ゲムシタビン、ara−C、テザシタビン、5−フルオロ−2’−デオキシシチジン、シトクロル、5,6−ジヒドロ−5−アザシチジン、6−アザシチジン、および1−メチル−Ψ−イソシチジンからなる群から選択される非デシタビンCDA基質薬剤を投与するステップと
を含む方法において癌を治療するための医薬品。 - 治療において使用するための請求項1に記載の組成物。
- 前記非デシタビンCDA基質が、5−アザシチジン、ara−C、ゲムシタビン、テザシタビン、5−フルオロ−2’−デオキシシチジン、およびシトクロルからなる群から選択される、請求項2に記載のキット。
- 前記非デシタビンCDA基質が、5−アザシチジン、ara−C、ゲムシタビン、テザシタビン、5−フルオロ−2’−デオキシシチジン、およびシトクロルからなる群から選択される、請求項3または請求項16に記載の医薬品。
- 前記非デシタビンCDA基質が、5−アザシチジン、ara−C、ゲムシタビン、テザシタビン、5−フルオロ−2’−デオキシシチジン、およびシトクロルからなる群から選択される、請求項8に記載の脱アミノ化防止薬。
- 前記非デシタビンCDA基質が、5−アザシチジン、ara−C、ゲムシタビン、テザシタビン、5−フルオロ−2’−デオキシシチジン、およびシトクロルからなる群から選択される、請求項10に記載の医薬組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16711709P | 2009-04-06 | 2009-04-06 | |
| US61/167,117 | 2009-04-06 | ||
| PCT/US2010/030081 WO2010118013A1 (en) | 2009-04-06 | 2010-04-06 | Combination of cytidine-based antineoplastic drugs with cytidine deaminase inhibitor and use thereof in the treatment of cancer |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2012522842A JP2012522842A (ja) | 2012-09-27 |
| JP2012522842A5 JP2012522842A5 (ja) | 2013-05-23 |
| JP5684787B2 true JP5684787B2 (ja) | 2015-03-18 |
Family
ID=42372314
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2012504775A Active JP5684787B2 (ja) | 2009-04-06 | 2010-04-06 | シチジンベースの抗新生物薬とシチジンデアミナーゼ阻害薬との組合せ、および癌の治療におけるその使用 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US8329665B2 (ja) |
| EP (1) | EP2416781B1 (ja) |
| JP (1) | JP5684787B2 (ja) |
| AU (2) | AU2010234562B2 (ja) |
| CA (1) | CA2757745C (ja) |
| ES (1) | ES2628609T3 (ja) |
| WO (1) | WO2010118013A1 (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JO2778B1 (en) | 2007-10-16 | 2014-03-15 | ايساي انك | Certain vehicles, installations and methods |
| JP5730854B2 (ja) * | 2009-04-06 | 2015-06-10 | 大塚製薬株式会社 | デシタビンとシチジンデアミナーゼ阻害剤との組合せ、およびがんの治療におけるその使用 |
| EP2417146B1 (en) * | 2009-04-06 | 2016-07-13 | Otsuka Pharmaceutical Co., Ltd. | (2'-deoxy-ribofuranosyl)-1,3,4,7-tetrahydro-(1,3)diazepin-2-one derivatives for treating cancer |
| US8609631B2 (en) | 2009-04-06 | 2013-12-17 | Eisai Inc. | Compositions and methods for treating cancer |
| EP3383406B1 (en) | 2015-12-03 | 2021-10-20 | Epidestiny, Inc. | Compositions containing decitabine, 5-azacytidine and tetrahydrouridine and uses thereof |
| WO2017192863A1 (en) * | 2016-05-04 | 2017-11-09 | L.E.A.F. Holdings Group Llc | Targeted liposomal gemcitabine compositions and methods thereof |
| KR20190097240A (ko) | 2016-12-28 | 2019-08-20 | 트랜스진 에스.에이. | 종양용해성 바이러스 및 치료 분자 |
| WO2023076332A1 (en) * | 2021-11-01 | 2023-05-04 | St. John's University | Injectable liquid pharmaceutical compostion containing gemcitabine and a cytidine deaminase inhibitor |
Family Cites Families (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4017606A (en) | 1973-10-04 | 1977-04-12 | The Upjohn Company | Organic compounds and process |
| US4275057A (en) * | 1980-01-28 | 1981-06-23 | The United States Of America As Represented By The Department Of Health, Education And Welfare | Seven-membered ring compounds as inhibitors of cytidine deaminase |
| US4526988A (en) | 1983-03-10 | 1985-07-02 | Eli Lilly And Company | Difluoro antivirals and intermediate therefor |
| JPS61500224A (ja) | 1983-10-26 | 1986-02-06 | グリ−ル、シエルダン ビ− | 腫瘍性組織を放射線に対して増感する医薬組成物 |
| DE3587500T2 (de) * | 1984-12-04 | 1993-12-16 | Lilly Co Eli | Tumorbehandlung bei Säugetieren. |
| US5223608A (en) | 1987-08-28 | 1993-06-29 | Eli Lilly And Company | Process for and intermediates of 2',2'-difluoronucleosides |
| US4965374A (en) | 1987-08-28 | 1990-10-23 | Eli Lilly And Company | Process for and intermediates of 2',2'-difluoronucleosides |
| US5968914A (en) | 1987-10-28 | 1999-10-19 | Pro-Neuron, Inc. | Treatment of chemotherapeutic agent and antiviral agent toxicity with acylated pyrimidine nucleosides |
| WO1992018517A1 (en) | 1991-04-17 | 1992-10-29 | Yale University | Method of treating or preventing hepatitis b virus |
| FR2682112B1 (fr) | 1991-10-08 | 1993-12-10 | Commissariat A Energie Atomique | Procede de synthese d'acide ribonucleique (arn) utilisant un nouveau reactif de deprotection. |
| US5821357A (en) | 1992-06-22 | 1998-10-13 | Eli Lilly And Company | Stereoselective glycosylation process for preparing 2'-deoxy-2',2'-difluoropurine and triazole nucleosides |
| US5606048A (en) | 1992-06-22 | 1997-02-25 | Eli Lilly And Company | Stereoselective glycosylation process for preparing 2'-Deoxy-2', 2'-difluoronucleosides and 2'-deoxy-2'-fluoronucleosides |
| US5371210A (en) | 1992-06-22 | 1994-12-06 | Eli Lilly And Company | Stereoselective fusion glycosylation process for preparing 2'-deoxy-2',2'-difluoronucleosides and 2'-deoxy-2'-fluoronucleosides |
| US5594124A (en) | 1992-06-22 | 1997-01-14 | Eli Lilly And Company | Stereoselective glycosylation process for preparing 2'-Deoxy-2',2'-difluoropyrimidine nucleosides and 2'-deoxy-2'-fluoropyrimidine nucleosides and intermediates thereof |
| US5426183A (en) | 1992-06-22 | 1995-06-20 | Eli Lilly And Company | Catalytic stereoselective glycosylation process for preparing 2'-deoxy-2',2'-difluoronucleosides and 2'-deoxy-2'-fluoronucleosides |
| DE69403588T2 (de) * | 1993-02-23 | 1998-01-22 | Hope City | 4-ethoxy-5-fluor-2'-deoxyuridine |
| AU6081294A (en) | 1993-05-14 | 1994-12-12 | Pro-Neuron, Inc. | Treatment of chemotherapeutic agent and antiviral agent toxicity with acylated pyrimidine nucleosides |
| GB9311252D0 (en) | 1993-06-01 | 1993-07-21 | Hafslund Nycomed As | Cell growth regualtors |
| US5637688A (en) | 1994-12-13 | 1997-06-10 | Eli Lilly And Company | Process for preparing 1-(2'-deoxy-2'-difluoro-d-ribofuranosyl)-4-aminopyrimidin-2-one hydrochloride |
| US5521294A (en) | 1995-01-18 | 1996-05-28 | Eli Lilly And Company | 2,2-difluoro-3-carbamoyl ribose sulfonate compounds and process for the preparation of beta nucleosides |
| US6001994A (en) | 1995-12-13 | 1999-12-14 | Eli Lilly And Company | Process for making gemcitabine hydrochloride |
| US5760208A (en) | 1996-08-14 | 1998-06-02 | The Board Of Governors For Higher Education, State Of Rhode Island And Providence Plantations | Process to prepare pyrimidine nucleosides |
| ATE339960T1 (de) | 1999-03-01 | 2006-10-15 | Halogenetics Inc | Verwendung von zusammensetzungen enthaltend cldc als strahlungssensibilisatoren in der behandlung von neoplastischen erkrankungen |
| US6933287B1 (en) * | 1999-03-01 | 2005-08-23 | Sheldon B. Greer | Dramatic simplification of a method to treat neoplastic disease by radiation |
| US6462191B1 (en) | 2000-07-13 | 2002-10-08 | Air Products And Chemicals, Inc. | Synthesis of 2-deoxy-2-fluoro-arabinose derivatives |
| US7125983B2 (en) | 2000-11-29 | 2006-10-24 | Mitsui Chemicals, Inc. | L-nucleic acid derivatives and process for the synthesis thereof |
| ATE290882T1 (de) * | 2001-01-16 | 2005-04-15 | Glaxo Group Ltd | Pharmazeutische mischung gegen krebs, die ein 4- chinazolinamin in kombination mit paclitaxel, carboplatin or vinorelbine enthält |
| EP1387850A2 (en) | 2001-05-18 | 2004-02-11 | Rakesh Kumar | Antiviral nucleosides |
| CA2499036A1 (en) | 2002-09-24 | 2004-04-08 | Koronis Pharmaceuticals, Incorporated | 1,3,5-triazines for treatment of viral diseases |
| US8158770B2 (en) | 2004-05-06 | 2012-04-17 | University Of Rochester | Content dependent inhibitors of cytidine deaminases and uses thereof |
| TW200606159A (en) | 2004-07-30 | 2006-02-16 | Pharmaessentia Corp | Stereoselective synthesis of β-nucleosides |
| JP2007522151A (ja) | 2004-12-08 | 2007-08-09 | シコール インコーポレイティド | ジフルオロヌクレオシド及びその調製方法 |
| WO2008085611A2 (en) | 2006-11-27 | 2008-07-17 | University Of Miami | Designer theraphy of pancreatic tumors |
| WO2009021551A1 (en) | 2007-08-13 | 2009-02-19 | Universite De La Mediterranee | A method for assessing the risk of toxicity in a chemotherapy |
| JO2778B1 (en) * | 2007-10-16 | 2014-03-15 | ايساي انك | Certain vehicles, installations and methods |
| WO2010047698A1 (en) | 2008-10-22 | 2010-04-29 | University Of Alabama At Birmingham | Activation-induced cytidine deaminase inhibitor suppression of autoimmune diseases |
| JP5730854B2 (ja) | 2009-04-06 | 2015-06-10 | 大塚製薬株式会社 | デシタビンとシチジンデアミナーゼ阻害剤との組合せ、およびがんの治療におけるその使用 |
| EP2417146B1 (en) | 2009-04-06 | 2016-07-13 | Otsuka Pharmaceutical Co., Ltd. | (2'-deoxy-ribofuranosyl)-1,3,4,7-tetrahydro-(1,3)diazepin-2-one derivatives for treating cancer |
-
2010
- 2010-04-06 AU AU2010234562A patent/AU2010234562B2/en active Active
- 2010-04-06 JP JP2012504775A patent/JP5684787B2/ja active Active
- 2010-04-06 EP EP10713756.4A patent/EP2416781B1/en active Active
- 2010-04-06 WO PCT/US2010/030081 patent/WO2010118013A1/en active Application Filing
- 2010-04-06 US US12/755,116 patent/US8329665B2/en active Active
- 2010-04-06 CA CA2757745A patent/CA2757745C/en active Active
- 2010-04-06 ES ES10713756.4T patent/ES2628609T3/es active Active
-
2016
- 2016-08-12 AU AU2016213880A patent/AU2016213880B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| EP2416781B1 (en) | 2017-03-08 |
| ES2628609T3 (es) | 2017-08-03 |
| US20100279977A1 (en) | 2010-11-04 |
| JP2012522842A (ja) | 2012-09-27 |
| AU2010234562A1 (en) | 2011-11-10 |
| CA2757745A1 (en) | 2010-10-14 |
| CA2757745C (en) | 2018-02-13 |
| EP2416781A1 (en) | 2012-02-15 |
| WO2010118013A1 (en) | 2010-10-14 |
| AU2010234562B2 (en) | 2016-05-12 |
| AU2016213880A1 (en) | 2016-09-01 |
| US8329665B2 (en) | 2012-12-11 |
| AU2016213880B2 (en) | 2018-08-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5684787B2 (ja) | シチジンベースの抗新生物薬とシチジンデアミナーゼ阻害薬との組合せ、および癌の治療におけるその使用 | |
| JP5730854B2 (ja) | デシタビンとシチジンデアミナーゼ阻害剤との組合せ、およびがんの治療におけるその使用 | |
| KR20100091978A (ko) | 시티딘 탈아미노효소 억제제로서 2'-플루오로-2'-데옥시테트라하이드로우리딘 | |
| AU2016213881B2 (en) | (2’-deoxy-ribofuranosyl)-1,3,4,7-tetrahydro-(1,3)diazepin-2-one derivatives for treating cancer | |
| US9040501B2 (en) | Compositions and methods for treating cancer | |
| EP2258378A1 (en) | Anti-tumor agent comprising cytidine derivative and carboplatin |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130404 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20130404 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A132 Effective date: 20140422 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20140711 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20140718 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20140821 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20141107 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20141224 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20150115 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5684787 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |