JP5314600B2 - 造血器癌および増殖性疾患の治療のための固定した薬物比 - Google Patents
造血器癌および増殖性疾患の治療のための固定した薬物比 Download PDFInfo
- Publication number
- JP5314600B2 JP5314600B2 JP2009550160A JP2009550160A JP5314600B2 JP 5314600 B2 JP5314600 B2 JP 5314600B2 JP 2009550160 A JP2009550160 A JP 2009550160A JP 2009550160 A JP2009550160 A JP 2009550160A JP 5314600 B2 JP5314600 B2 JP 5314600B2
- Authority
- JP
- Japan
- Prior art keywords
- cytarabine
- daunorubicin
- dose
- cpx
- day
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 238000011282 treatment Methods 0.000 title abstract description 31
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 title abstract description 25
- 230000002062 proliferating effect Effects 0.000 title abstract description 14
- 201000005787 hematologic cancer Diseases 0.000 title abstract description 6
- 239000003814 drug Substances 0.000 title description 61
- 229940079593 drug Drugs 0.000 title description 58
- 201000010099 disease Diseases 0.000 title description 14
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 title description 6
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 claims abstract description 123
- 229960000684 cytarabine Drugs 0.000 claims abstract description 118
- 238000000034 method Methods 0.000 claims abstract description 39
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 16
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 claims description 79
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 claims description 79
- 229960000975 daunorubicin Drugs 0.000 claims description 78
- 239000002502 liposome Substances 0.000 claims description 36
- 239000000203 mixture Substances 0.000 claims description 28
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 23
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 12
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 claims description 12
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 11
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 11
- 208000032839 leukemia Diseases 0.000 claims description 11
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 claims description 10
- 208000033826 Promyelocytic Acute Leukemia Diseases 0.000 claims description 10
- NRJAVPSFFCBXDT-HUESYALOSA-N 1,2-distearoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCCCCCCCCCCC NRJAVPSFFCBXDT-HUESYALOSA-N 0.000 claims description 6
- 235000012000 cholesterol Nutrition 0.000 claims description 6
- 230000001093 anti-cancer Effects 0.000 claims description 4
- 238000001990 intravenous administration Methods 0.000 claims description 4
- 101001000212 Rattus norvegicus Decorin Proteins 0.000 claims 1
- FVJZSBGHRPJMMA-UHFFFAOYSA-N distearoyl phosphatidylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OCC(O)CO)OC(=O)CCCCCCCCCCCCCCCCC FVJZSBGHRPJMMA-UHFFFAOYSA-N 0.000 claims 1
- 229940045799 anthracyclines and related substance Drugs 0.000 abstract description 43
- 206010028980 Neoplasm Diseases 0.000 abstract description 42
- 230000003042 antagnostic effect Effects 0.000 abstract description 39
- 201000011510 cancer Diseases 0.000 abstract description 32
- 238000001802 infusion Methods 0.000 description 34
- 230000004044 response Effects 0.000 description 23
- 210000004027 cell Anatomy 0.000 description 21
- 239000003981 vehicle Substances 0.000 description 21
- 210000004369 blood Anatomy 0.000 description 20
- 239000008280 blood Substances 0.000 description 20
- 230000001988 toxicity Effects 0.000 description 20
- 231100000419 toxicity Toxicity 0.000 description 20
- 238000002560 therapeutic procedure Methods 0.000 description 19
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 17
- 210000001185 bone marrow Anatomy 0.000 description 16
- 230000000694 effects Effects 0.000 description 16
- 241001465754 Metazoa Species 0.000 description 14
- 241000282472 Canis lupus familiaris Species 0.000 description 13
- 230000006698 induction Effects 0.000 description 13
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 12
- 238000004458 analytical method Methods 0.000 description 11
- 230000003285 pharmacodynamic effect Effects 0.000 description 11
- 208000035475 disorder Diseases 0.000 description 10
- 230000009467 reduction Effects 0.000 description 10
- 230000007423 decrease Effects 0.000 description 9
- 230000002489 hematologic effect Effects 0.000 description 9
- 230000002459 sustained effect Effects 0.000 description 9
- 230000000259 anti-tumor effect Effects 0.000 description 8
- 210000001772 blood platelet Anatomy 0.000 description 8
- 230000001472 cytotoxic effect Effects 0.000 description 8
- 238000009093 first-line therapy Methods 0.000 description 8
- 150000002632 lipids Chemical class 0.000 description 8
- 230000036961 partial effect Effects 0.000 description 8
- 241000700159 Rattus Species 0.000 description 7
- 239000002246 antineoplastic agent Substances 0.000 description 7
- 231100000433 cytotoxic Toxicity 0.000 description 7
- 238000009472 formulation Methods 0.000 description 7
- 239000000890 drug combination Substances 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 238000011084 recovery Methods 0.000 description 6
- 238000011272 standard treatment Methods 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- FVJZSBGHRPJMMA-IOLBBIBUSA-N PG(18:0/18:0) Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@@H](O)CO)OC(=O)CCCCCCCCCCCCCCCCC FVJZSBGHRPJMMA-IOLBBIBUSA-N 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 210000000440 neutrophil Anatomy 0.000 description 5
- 210000005259 peripheral blood Anatomy 0.000 description 5
- 239000011886 peripheral blood Substances 0.000 description 5
- 230000002035 prolonged effect Effects 0.000 description 5
- 206010003694 Atrophy Diseases 0.000 description 4
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 4
- 206010058314 Dysplasia Diseases 0.000 description 4
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 4
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 4
- 206010025323 Lymphomas Diseases 0.000 description 4
- 206010028116 Mucosal inflammation Diseases 0.000 description 4
- 201000010927 Mucositis Diseases 0.000 description 4
- 230000002411 adverse Effects 0.000 description 4
- 230000037444 atrophy Effects 0.000 description 4
- 238000001574 biopsy Methods 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 238000002512 chemotherapy Methods 0.000 description 4
- 229910052802 copper Inorganic materials 0.000 description 4
- 239000010949 copper Substances 0.000 description 4
- 229940127089 cytotoxic agent Drugs 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- 230000003394 haemopoietic effect Effects 0.000 description 4
- 229960000908 idarubicin Drugs 0.000 description 4
- 239000002207 metabolite Substances 0.000 description 4
- 229960001156 mitoxantrone Drugs 0.000 description 4
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 4
- 230000017074 necrotic cell death Effects 0.000 description 4
- 238000010606 normalization Methods 0.000 description 4
- 230000036470 plasma concentration Effects 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 210000000952 spleen Anatomy 0.000 description 4
- 230000004083 survival effect Effects 0.000 description 4
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 4
- 201000004384 Alopecia Diseases 0.000 description 3
- 108020004414 DNA Proteins 0.000 description 3
- 206010011906 Death Diseases 0.000 description 3
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 3
- 241000283984 Rodentia Species 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 102000007537 Type II DNA Topoisomerases Human genes 0.000 description 3
- 108010046308 Type II DNA Topoisomerases Proteins 0.000 description 3
- 230000002159 abnormal effect Effects 0.000 description 3
- 229940041181 antineoplastic drug Drugs 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 210000002798 bone marrow cell Anatomy 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 238000009108 consolidation therapy Methods 0.000 description 3
- 230000034994 death Effects 0.000 description 3
- 231100000517 death Toxicity 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- -1 gemtuzumab Chemical compound 0.000 description 3
- 208000024963 hair loss Diseases 0.000 description 3
- 230000003676 hair loss Effects 0.000 description 3
- 208000019691 hematopoietic and lymphoid cell neoplasm Diseases 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 230000000977 initiatory effect Effects 0.000 description 3
- 230000002045 lasting effect Effects 0.000 description 3
- 210000000265 leukocyte Anatomy 0.000 description 3
- 210000004698 lymphocyte Anatomy 0.000 description 3
- 208000037821 progressive disease Diseases 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 230000000306 recurrent effect Effects 0.000 description 3
- 230000001954 sterilising effect Effects 0.000 description 3
- 238000004659 sterilization and disinfection Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- HJEZFVLKJYFNQW-PRFXOSGESA-N (13S)-13-dihydrodaunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)[C@H](C)O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 HJEZFVLKJYFNQW-PRFXOSGESA-N 0.000 description 2
- 208000019838 Blood disease Diseases 0.000 description 2
- OCUCCJIRFHNWBP-IYEMJOQQSA-L Copper gluconate Chemical compound [Cu+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O OCUCCJIRFHNWBP-IYEMJOQQSA-L 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- PCDQPRRSZKQHHS-CCXZUQQUSA-N Cytarabine Triphosphate Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 PCDQPRRSZKQHHS-CCXZUQQUSA-N 0.000 description 2
- HJEZFVLKJYFNQW-UHFFFAOYSA-N Daunorubicinol Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)O)CC1OC1CC(N)C(O)C(C)O1 HJEZFVLKJYFNQW-UHFFFAOYSA-N 0.000 description 2
- OWCHPBVMSHIYCQ-UHFFFAOYSA-N Dihydro-dauno-mycinon Natural products C1C(O)(C(C)O)CC(O)C2=C1C(O)=C1C(=O)C(C=CC=C3OC)=C3C(=O)C1=C2O OWCHPBVMSHIYCQ-UHFFFAOYSA-N 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 208000010201 Exanthema Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 2
- 201000007224 Myeloproliferative neoplasm Diseases 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- 206010038111 Recurrent cancer Diseases 0.000 description 2
- DVKFVGVMPLXLKC-PUGXJXRHSA-N [(2s,3r,4s,5s,6r)-2-[(2s,3s,4s,5r)-3,4-dihydroxy-2,5-bis(hydroxymethyl)oxolan-2-yl]-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl] dihydrogen phosphate Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@]1(CO)[C@@]1(OP(O)(O)=O)[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 DVKFVGVMPLXLKC-PUGXJXRHSA-N 0.000 description 2
- 230000005856 abnormality Effects 0.000 description 2
- 230000000719 anti-leukaemic effect Effects 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- DRTQHJPVMGBUCF-CCXZUQQUSA-N arauridine Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-CCXZUQQUSA-N 0.000 description 2
- 238000001815 biotherapy Methods 0.000 description 2
- 238000010322 bone marrow transplantation Methods 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 229960000928 clofarabine Drugs 0.000 description 2
- WDDPHFBMKLOVOX-AYQXTPAHSA-N clofarabine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1F WDDPHFBMKLOVOX-AYQXTPAHSA-N 0.000 description 2
- 238000009096 combination chemotherapy Methods 0.000 description 2
- 229940108925 copper gluconate Drugs 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical class O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- 229950000950 daunorubicinol Drugs 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 2
- 229960003957 dexamethasone Drugs 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 238000012377 drug delivery Methods 0.000 description 2
- 238000005538 encapsulation Methods 0.000 description 2
- 210000003979 eosinophil Anatomy 0.000 description 2
- 229960005420 etoposide Drugs 0.000 description 2
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 2
- 201000005884 exanthem Diseases 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 210000004907 gland Anatomy 0.000 description 2
- 239000011521 glass Substances 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 208000014951 hematologic disease Diseases 0.000 description 2
- 208000018706 hematopoietic system disease Diseases 0.000 description 2
- 238000011419 induction treatment Methods 0.000 description 2
- 231100000518 lethal Toxicity 0.000 description 2
- 230000001665 lethal effect Effects 0.000 description 2
- 231100000225 lethality Toxicity 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 231100000682 maximum tolerated dose Toxicity 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 210000004379 membrane Anatomy 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 230000000877 morphologic effect Effects 0.000 description 2
- 201000005962 mycosis fungoides Diseases 0.000 description 2
- 230000002085 persistent effect Effects 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 238000001959 radiotherapy Methods 0.000 description 2
- 206010037844 rash Diseases 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 230000003007 single stranded DNA break Effects 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 230000004797 therapeutic response Effects 0.000 description 2
- 210000001541 thymus gland Anatomy 0.000 description 2
- 229960003087 tioguanine Drugs 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- DRTQHJPVMGBUCF-UHFFFAOYSA-N uracil arabinoside Natural products OC1C(O)C(CO)OC1N1C(=O)NC(=O)C=C1 DRTQHJPVMGBUCF-UHFFFAOYSA-N 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- GVJHHUAWPYXKBD-IEOSBIPESA-N α-tocopherol Chemical compound OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-IEOSBIPESA-N 0.000 description 2
- NAALWFYYHHJEFQ-ZASNTINBSA-N (2s,5r,6r)-6-[[(2r)-2-[[6-[4-[bis(2-hydroxyethyl)sulfamoyl]phenyl]-2-oxo-1h-pyridine-3-carbonyl]amino]-2-(4-hydroxyphenyl)acetyl]amino]-3,3-dimethyl-7-oxo-4-thia-1-azabicyclo[3.2.0]heptane-2-carboxylic acid Chemical compound N([C@@H](C(=O)N[C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C=1C=CC(O)=CC=1)C(=O)C(C(N1)=O)=CC=C1C1=CC=C(S(=O)(=O)N(CCO)CCO)C=C1 NAALWFYYHHJEFQ-ZASNTINBSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- HJTAZXHBEBIQQX-UHFFFAOYSA-N 1,5-bis(chloromethyl)naphthalene Chemical compound C1=CC=C2C(CCl)=CC=CC2=C1CCl HJTAZXHBEBIQQX-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- CYDQOEWLBCCFJZ-UHFFFAOYSA-N 4-(4-fluorophenyl)oxane-4-carboxylic acid Chemical compound C=1C=C(F)C=CC=1C1(C(=O)O)CCOCC1 CYDQOEWLBCCFJZ-UHFFFAOYSA-N 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- UOTZEAIDLOJMDN-UHFFFAOYSA-N 6,8,11-trihydroxy-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione Chemical compound C1C(O)CCC2=C1C(O)=C1C(=O)C(C=CC=C3OC)=C3C(=O)C1=C2O UOTZEAIDLOJMDN-UHFFFAOYSA-N 0.000 description 1
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 1
- VHRSUDSXCMQTMA-PJHHCJLFSA-N 6alpha-methylprednisolone Chemical compound C([C@@]12C)=CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2[C@@H](O)C[C@]2(C)[C@@](O)(C(=O)CO)CC[C@H]21 VHRSUDSXCMQTMA-PJHHCJLFSA-N 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 241000269627 Amphiuma means Species 0.000 description 1
- 206010050247 Anal inflammation Diseases 0.000 description 1
- 101000719121 Arabidopsis thaliana Protein MEI2-like 1 Proteins 0.000 description 1
- 102000015790 Asparaginase Human genes 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 208000008035 Back Pain Diseases 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 206010007882 Cellulitis Diseases 0.000 description 1
- 206010008469 Chest discomfort Diseases 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- 229940124087 DNA topoisomerase II inhibitor Drugs 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 208000000059 Dyspnea Diseases 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- 206010018276 Gingival bleeding Diseases 0.000 description 1
- 108010044091 Globulins Proteins 0.000 description 1
- 102000006395 Globulins Human genes 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 206010019233 Headaches Diseases 0.000 description 1
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 1
- 102000001554 Hemoglobins Human genes 0.000 description 1
- 108010054147 Hemoglobins Proteins 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 102000003964 Histone deacetylase Human genes 0.000 description 1
- 108090000353 Histone deacetylase Proteins 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101001000104 Homo sapiens Myosin-11 Proteins 0.000 description 1
- 101000857677 Homo sapiens Runt-related transcription factor 1 Proteins 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 206010051792 Infusion related reaction Diseases 0.000 description 1
- 102100036532 Interferon alpha-8 Human genes 0.000 description 1
- 239000011786 L-ascorbyl-6-palmitate Substances 0.000 description 1
- QAQJMLQRFWZOBN-LAUBAEHRSA-N L-ascorbyl-6-palmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](O)[C@H]1OC(=O)C(O)=C1O QAQJMLQRFWZOBN-LAUBAEHRSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 208000022435 Light chain deposition disease Diseases 0.000 description 1
- 208000020647 Light chain disease Diseases 0.000 description 1
- 108090001030 Lipoproteins Proteins 0.000 description 1
- 102000004895 Lipoproteins Human genes 0.000 description 1
- 208000008930 Low Back Pain Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 208000034578 Multiple myelomas Diseases 0.000 description 1
- 208000014767 Myeloproliferative disease Diseases 0.000 description 1
- 102100036639 Myosin-11 Human genes 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 206010030201 Oesophageal ulcer Diseases 0.000 description 1
- 206010030216 Oesophagitis Diseases 0.000 description 1
- 208000007222 Physiological Sexual Dysfunction Diseases 0.000 description 1
- 206010035226 Plasma cell myeloma Diseases 0.000 description 1
- 208000033501 Refractory anemia with excess blasts Diseases 0.000 description 1
- 102100023606 Retinoic acid receptor alpha Human genes 0.000 description 1
- 102100025373 Runt-related transcription factor 1 Human genes 0.000 description 1
- 230000018199 S phase Effects 0.000 description 1
- 208000009359 Sezary Syndrome Diseases 0.000 description 1
- 208000021388 Sezary disease Diseases 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 241000220254 Streptomyces coeruleorubidus Species 0.000 description 1
- 208000000389 T-cell leukemia Diseases 0.000 description 1
- 208000028530 T-cell lymphoblastic leukemia/lymphoma Diseases 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- 239000000317 Topoisomerase II Inhibitor Substances 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 102000015098 Tumor Suppressor Protein p53 Human genes 0.000 description 1
- 108010078814 Tumor Suppressor Protein p53 Proteins 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 208000017733 acquired polycythemia vera Diseases 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 229960000548 alemtuzumab Drugs 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 229940087168 alpha tocopherol Drugs 0.000 description 1
- 206010002022 amyloidosis Diseases 0.000 description 1
- 208000022531 anorexia Diseases 0.000 description 1
- 239000003817 anthracycline antibiotic agent Substances 0.000 description 1
- 230000002927 anti-mitotic effect Effects 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 229940034982 antineoplastic agent Drugs 0.000 description 1
- 229940091658 arsenic Drugs 0.000 description 1
- 229910052785 arsenic Inorganic materials 0.000 description 1
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 1
- 229960002594 arsenic trioxide Drugs 0.000 description 1
- GOLCXWYRSKYTSP-UHFFFAOYSA-N arsenic trioxide Inorganic materials O1[As]2O[As]1O2 GOLCXWYRSKYTSP-UHFFFAOYSA-N 0.000 description 1
- 235000010385 ascorbyl palmitate Nutrition 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 238000010256 biochemical assay Methods 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 230000023555 blood coagulation Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 190000008236 carboplatin Chemical compound 0.000 description 1
- 210000004534 cecum Anatomy 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000018486 cell cycle phase Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 201000010902 chronic myelomonocytic leukemia Diseases 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 238000007596 consolidation process Methods 0.000 description 1
- 238000011443 conventional therapy Methods 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- 229940127071 cytotoxic antineoplastic agent Drugs 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 229960003109 daunorubicin hydrochloride Drugs 0.000 description 1
- 206010061428 decreased appetite Diseases 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 230000009429 distress Effects 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 231100000371 dose-limiting toxicity Toxicity 0.000 description 1
- 229940115080 doxil Drugs 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 239000003937 drug carrier Substances 0.000 description 1
- 210000001198 duodenum Anatomy 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- 208000006881 esophagitis Diseases 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 238000011010 flushing procedure Methods 0.000 description 1
- VVIAGPKUTFNRDU-ABLWVSNPSA-N folinic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-ABLWVSNPSA-N 0.000 description 1
- 235000008191 folinic acid Nutrition 0.000 description 1
- 239000011672 folinic acid Substances 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 229960000578 gemtuzumab Drugs 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 231100000869 headache Toxicity 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000005534 hematocrit Methods 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 238000011134 hematopoietic stem cell transplantation Methods 0.000 description 1
- 230000011132 hemopoiesis Effects 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- 231100000171 higher toxicity Toxicity 0.000 description 1
- 238000001794 hormone therapy Methods 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- 210000003405 ileum Anatomy 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000009830 intercalation Methods 0.000 description 1
- 230000002687 intercalation Effects 0.000 description 1
- 108010047126 interferon-alpha 8 Proteins 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 210000004347 intestinal mucosa Anatomy 0.000 description 1
- 210000000936 intestine Anatomy 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 210000001630 jejunum Anatomy 0.000 description 1
- 238000002386 leaching Methods 0.000 description 1
- 229960001691 leucovorin Drugs 0.000 description 1
- 201000002364 leukopenia Diseases 0.000 description 1
- 231100001022 leukopenia Toxicity 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 229950002950 lintuzumab Drugs 0.000 description 1
- 230000003859 lipid peroxidation Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 210000003563 lymphoid tissue Anatomy 0.000 description 1
- 238000007433 macroscopic evaluation Methods 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 238000009115 maintenance therapy Methods 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 201000000638 mature B-cell neoplasm Diseases 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 229960004584 methylprednisolone Drugs 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 208000016586 myelodysplastic syndrome with excess blasts Diseases 0.000 description 1
- 206010028537 myelofibrosis Diseases 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 229960000801 nelarabine Drugs 0.000 description 1
- IXOXBSCIXZEQEQ-UHTZMRCNSA-N nelarabine Chemical compound C1=NC=2C(OC)=NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O IXOXBSCIXZEQEQ-UHTZMRCNSA-N 0.000 description 1
- 208000010915 neoplasm of mature B-cells Diseases 0.000 description 1
- 208000021119 neoplasm of mature T-cells or NK-cells Diseases 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 231100000062 no-observed-adverse-effect level Toxicity 0.000 description 1
- 230000036963 noncompetitive effect Effects 0.000 description 1
- 231100001160 nonlethal Toxicity 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 231100000915 pathological change Toxicity 0.000 description 1
- 230000036285 pathological change Effects 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 208000010626 plasma cell neoplasm Diseases 0.000 description 1
- 208000037244 polycythemia vera Diseases 0.000 description 1
- 230000005195 poor health Effects 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 210000001948 pro-b lymphocyte Anatomy 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 230000014493 regulation of gene expression Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 108091008726 retinoic acid receptors α Proteins 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000007423 screening assay Methods 0.000 description 1
- 208000013220 shortness of breath Diseases 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 235000002639 sodium chloride Nutrition 0.000 description 1
- 239000001540 sodium lactate Substances 0.000 description 1
- 229940005581 sodium lactate Drugs 0.000 description 1
- 235000011088 sodium lactate Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000003393 splenic effect Effects 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000003319 supportive effect Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229960000984 tocofersolan Drugs 0.000 description 1
- 231100000816 toxic dose Toxicity 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 230000036269 ulceration Effects 0.000 description 1
- 230000004222 uncontrolled growth Effects 0.000 description 1
- 238000002562 urinalysis Methods 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 230000008673 vomiting Effects 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 239000002076 α-tocopherol Substances 0.000 description 1
- 235000004835 α-tocopherol Nutrition 0.000 description 1
Images


Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/704—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin attached to a condensed carbocyclic ring system, e.g. sennosides, thiocolchicosides, escin, daunorubicin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T436/00—Chemistry: analytical and immunological testing
- Y10T436/14—Heterocyclic carbon compound [i.e., O, S, N, Se, Te, as only ring hetero atom]
- Y10T436/142222—Hetero-O [e.g., ascorbic acid, etc.]
- Y10T436/143333—Saccharide [e.g., DNA, etc.]
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Dispersion Chemistry (AREA)
- Dermatology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Diabetes (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Saccharide Compounds (AREA)
Description
本出願は2007年2月16日に出願された米国出願番号第60/901,772号、および2007年8月17日に出願された米国出願番号第60/965,196号の利益を主張し、それらのそれぞれはそのまま参照として本明細書に援用される。
[1]対象の癌または血液増殖性疾患を治療するための方法における固定した、非拮抗モル比のシタラビンとアントラサイクリンを含む医薬組成物の使用であって、
該方法は12時間以下の期間にわたり該組成物を静脈内投与することを含み、
固定した比は少なくとも4時間血漿中で維持され、そして
固定した、非拮抗モル比のシタラビンとアントラサイクリンはリポソーム内に被包される、前記使用。
[2]組成物が8時間以下で投与される、[1]に記載の使用。
[3]組成物が3時間以下で投与される、[2]に記載の使用。
[4]前記組成物が90分間以下で投与される、[3]に記載の使用。
[5]シタラビン対アントラサイクリンの前記の固定した非拮抗モル比が約1:1〜25:1の間である、[1]〜[4]のいずれかに記載の使用。
[6]前記の固定した、非拮抗モル比が約5:1である、[5]に記載の使用。
[7]シタラビンが250mg/m 2 以下の用量で投与される、[1]〜[4]のいずれかに記載の使用。
[8]前記リポソームがDSPC、DSPGおよびコレステロールを含む、[1]〜[4]のいずれかに記載の使用。
[9]前記リポソームが7:2:1のモル比でDSPC:DSPG:コレステロールを含む、[8に記載の使用。
[10]アントラサイクリンがダウノルビシンである、[1]〜[4]のいずれかに記載の使用。
[11]前記組成物の用量が32〜134ユニット/m 2 であり、1ユニットが1mgシタラビンおよび0.44mgダウノルビシンである、[10]のいずれかに記載の使用。
[12]前記投与がIVドリップによるものである、[1]〜[4]のいずれかに記載の使用。
[13]癌または血液増殖性疾患が、急性リンパ性白血病(ALL)、急性骨髄性白血病(AML)および急性前骨髄球白血病(APL)から選択される進行した血液癌であり、そして前記血液増殖性疾患が骨髄異形成症候群(MDS)である、[1]〜[4]のいずれかに記載の使用。
[14]前記対象が少なくとも1種の抗癌投薬計画を以前に受けている、および/または以前に寛解を経験している、[1]〜[4]のいずれかに記載の使用。
[15]前記対象が前記抗癌投薬計画後18ヶ月以内に再発を経験している、[14]に記載の使用。
[16]前記対象が前記抗癌投薬計画後6ヶ月以内に再発を経験している、[15]に記載の使用。
[17]完全寛解率の増加の関数として、および/または完全寛解継続期間の延長および/または進行までの時間の延長および/または生存の延長の関数として治療効果を測定することをさらに含む、[1]〜[4]のいずれかに記載の使用。
[18]前記治療効果が、シタラビンとアントラサイクリンが標準治療プロトコルで投与される場合に得られるものより大きい、[17]に記載の使用。
[19]非造血毒性の減少としての改善された安全性結果を測定することをさらに含む、[1]〜[4]のいずれかに記載の使用。
[20]非造血毒性が粘膜炎および/または脱毛である、[19]に記載の使用。
[21]安全性結果が、シタラビンとアントラサイクリンが標準治療プロトコルで投与される場合に得られるものより大きい、[19]に記載の使用。
一側面では、対象における癌または血液増殖性疾患を治療するための方法が本明細書に提供され、該方法は固定した、非拮抗モル比のシタラビンと、ダウノルビシンのようなアントラサイクリンを含む医薬組成物を該対象に投与することを含み、ここでシタラビン:アントラサイクリンの比は少なくとも約4時間、血漿中において非拮抗比で維持される。別の態様では、固定した、非拮抗モル比は少なくとも約8時間、少なくとも約16時間、または少なくとも約24時間維持される。アントラサイクリンはダウノルビシンまたはミトキサントロンでありうる。特定の態様では、アントラサイクリンはダウノルビシンである。典型的には、シタラビンとアントラサイクリンは1以上の送達ベヒクルと安定的に結合している。送達ベヒクル中の被包は調和した様式で疾患部位に2以上の薬剤が送達されることを可能にし、それによって薬剤が非拮抗比で疾患部位に存在することを確実にする。この結果は、薬剤が送達ベヒクル内に一緒に被包されるか、または非拮抗比が疾患部位で維持されるように投与される送達ベヒクル内に別個に被包されるかのいずれでも達成されることになる。(送達システムの薬物動態学(PK)が同程度の場合)調和した送達が達成されるように、PKは送達ベヒクル自体によって制御される。一態様では、送達ベヒクルはリポソームである。
シタラビンとアントラサイクリンの場合、in vitroでの非拮抗モル比は25:1〜約1:1の間であり、ここで5:1のモル比が最適であることが見いだされた。適切なアントラサイクリンはどれも使用することができる。アントラサイクリンは、ダウノルビシン、イダルビシンまたはミトキサントロンでありうる。特定の態様では、アントラサイクリンはダウノルビシンである。
開示された方法はまた、再発した癌を治療することにおいて治療的に有効である。“再発した癌”は以前の治療に反応して、以前に完全に、または部分的に寛解した後に再発した癌を表す。再発は、臨床的、放射線的、もしくは生化学的アッセイによって、または癌マーカーの増大したレベルによって検出されるような腫瘍細胞の再出現または再成長を含む、任意のやり方で定義することができる。先行治療には、化学療法、生物学的またはホルモン療法、放射線療法、および骨髄移植を挙げることができるが、それらに限定されない。
CPX−351によるin vivo研究
CPX−351(シタラビン:ダウノルビシン)リポソーム注入液の毒性は、単回用量および反復(3用量を1日おき、2週間後に繰り返す)用量を与えられたラットおよびイヌにおいて研究されてきた。

第1相治験
物理的、化学的および薬学的情報 CPX−351は抗新生物薬シタラビンおよびダウノルビシンの固定した組み合わせのリポソーム製剤である。2種の薬物は、前臨床研究において非拮抗活性を有することが示された5:1のモル比でリポソームの内側に存在する。リポソーム膜は7:2:1モル比のジステアロイルホスファチジルコリン、ジステアロイルホスファチジルグリセロールおよびコレステロールから構成される。これらのリポソームはおよそ100nmの名目上の直径を有し、スクロース−リン酸−バッファー、pH7.4に懸濁される。滅菌は0.22μmフィルターによる濾過により達成される。
・骨髄の細胞密度減少(>50%減少、芽球の減少を伴う)
および/または
・ことによると、おそらくまたは明確にCPX−351に関連する、非血液学的治療から出現した事象(≧グレード2<DLT)。
結果。この研究は、非ランダム化、オープンラベル、単群、用量エスカレーション第1相治験である。現在までの研究登録期間はおよそ18ヶ月間を要した。すべて以前に治療を受けている33人の対象(男性22人;女性11人)、年齢中央値62歳(24〜81)が10コホートに登録された。初めの10コホートの実態的人口統計および配置は以下の表6に要約される:
・骨髄細胞密度減少(>50%減少、芽球減少を伴う)
および/または
・ことによると、おそらくまたは明確にCPX−351に関連する、非血液学的治療から出現した事象(≧グレード2<DLT)。
CPX−351第1相治験−症例研究
以下は、進行中のCPX−351第1相治験において治療された患者の5例の症例研究である。
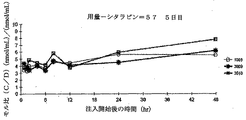
CPX−351第1相治験−薬物動態学
研究の薬物動態学(PK)解析の目標
・白血病患者におけるCPX−351投与後のシタラビンおよびダウノルビシンならびに選択された代謝産物(Ara−Uおよびダウノルビシノール)の単回投与および複数回投与の薬物動態学を決定すること。
・癌患者におけるCPX−351の全体的暴露、用量釣り合い、および蓄積に関する予備情報を集めること。
・可能ならば、CPX−351成分への暴露強度と効果(安全性および有効性)間の相関関係を樹立すること。
・1日目の投与前、注入中45分(または注入の中間点)および90分(または注入の終了時)、その後注入開始に対応して2、4、6、8、12、および24時間において。
・3日目の投与前、注入中45および90分において。
・5日目の投与前、注入中45分(または注入の中間点)および90分(または注入の終了時)において、その後注入開始に対応して2、4、6、8、12、24、48、72、96、および168時間において。
Cmax 最大の観察された濃度
Tmax Cmax出現時期
λz 最終相(5日目の投与後だけ)の自然体数(In)に変換された濃度対時間データの直線回帰から得られた排出速度定数
t1/2 最終半減期、In(2)/λzとして計算
AUC(0〜最後) 直線台形法により得られた、0時から最後の投与後の定量可能な血漿中濃度の時間までの血漿中濃度−時間曲線下面積
AUC(0〜無限) 0時から無限時まで外挿した血漿中濃度−時間曲線下面積
CL (研究5日目だけのシタラビンおよびダウノルビシンに対して)用量/AUC(0〜無限)としてコンピュータにより算出された全身クリアランス
また薬物動態学解析のコンパートメント法を利用して、代謝産物配置動態学を評価し、および/または薬物動態学/薬力学モデリングを実施してもよい。
Claims (5)
- ヒトを対象とする白血病を治療するための方法において使用する、シタラビン対ダウノルビシンが5:1の固定したモル比である、シタラビンとダウノルビシンから実質的になる医薬組成物であって、
該方法は、シタラビンが32〜134mg/m2の単回投与で供給され、投与サイクルを通じて96〜402mg/m2の合計量で供給される投与サイクルにおいて、8時間以下の期間にわたり該組成物を単回投与で静脈内投与することから実質的になり、
固定したモル比は少なくとも4時間血漿中で維持され、そして
固定したモル比のシタラビンとダウノルビシンは、7:2:1のモル比であるDSPC:DSPG:コレステロールを含むリポソーム内に被包され、
該投与サイクルは、第1日目の最初の投与、第3日目の2回目の投与および第5日目の3回目の投与からなる、前記組成物。 - 3時間以下で投与される、請求項1に記載の組成物。
- 90分間以下で投与される、請求項2に記載の組成物。
- 白血病が、急性リンパ性白血病(ALL)、急性骨髄性白血病(AML)または急性前骨髄球白血病(APL)である、請求項1〜3のいずれかに記載の組成物。
- 前記対象が少なくとも1種の抗癌投薬計画を以前に受けている、請求項1〜3のいずれかに記載の組成物。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US90177207P | 2007-02-16 | 2007-02-16 | |
| US60/901,772 | 2007-02-16 | ||
| US96519607P | 2007-08-17 | 2007-08-17 | |
| US60/965,196 | 2007-08-17 | ||
| PCT/US2008/054168 WO2008101214A2 (en) | 2007-02-16 | 2008-02-15 | Fixed drug ratios for treatment of hematopoietic cancers and proliferative disorders |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2010519224A JP2010519224A (ja) | 2010-06-03 |
| JP2010519224A5 JP2010519224A5 (ja) | 2011-04-07 |
| JP5314600B2 true JP5314600B2 (ja) | 2013-10-16 |
Family
ID=39690832
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009550160A Active JP5314600B2 (ja) | 2007-02-16 | 2008-02-15 | 造血器癌および増殖性疾患の治療のための固定した薬物比 |
Country Status (20)
| Country | Link |
|---|---|
| US (2) | US20100303895A1 (ja) |
| EP (3) | EP3300601B1 (ja) |
| JP (1) | JP5314600B2 (ja) |
| KR (2) | KR20100014441A (ja) |
| CN (1) | CN105998046A (ja) |
| AU (1) | AU2008216083B2 (ja) |
| CA (1) | CA2678332C (ja) |
| CY (1) | CY1119631T1 (ja) |
| DK (2) | DK3300601T3 (ja) |
| ES (2) | ES2650167T3 (ja) |
| FR (1) | FR22C1034I2 (ja) |
| HK (1) | HK1253378A1 (ja) |
| HR (1) | HRP20220428T3 (ja) |
| HU (2) | HUE058334T2 (ja) |
| NL (1) | NL301185I2 (ja) |
| NO (1) | NO2120568T3 (ja) |
| PL (2) | PL3300601T3 (ja) |
| PT (2) | PT3300601T (ja) |
| SI (1) | SI3300601T1 (ja) |
| WO (1) | WO2008101214A2 (ja) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100075937A1 (en) * | 2008-09-24 | 2010-03-25 | Hollis-Eden Pharmaceuticals, Inc. | Patient populations and treatment methods |
| CN104114156A (zh) | 2011-10-21 | 2014-10-22 | 切拉托尔制药公司 | 冻干脂质体 |
| US20140154304A1 (en) * | 2012-11-30 | 2014-06-05 | Boehringer Ingelheim International Gmbh | Combination therapy with volasertib |
| WO2015127173A1 (en) * | 2014-02-20 | 2015-08-27 | Agios Pharmaceuticals, Inc | Therapeutically active compounds and their methods of use |
| WO2015127172A1 (en) * | 2014-02-20 | 2015-08-27 | Agios Pharmaceuticals, Inc. | Therapeutically active compounds and their methods of use |
| JP6976941B2 (ja) * | 2015-11-11 | 2021-12-08 | セレーター ファーマシューティカルズ インコーポレイテッド | 白血病に罹患している対象の治療レジメンを選択するためのアッセイ及び方法 |
| US11602513B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11504347B1 (en) | 2016-07-22 | 2022-11-22 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| UY37341A (es) | 2016-07-22 | 2017-11-30 | Flamel Ireland Ltd | Formulaciones de gamma-hidroxibutirato de liberación modificada con farmacocinética mejorada |
| US11602512B1 (en) | 2016-07-22 | 2023-03-14 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11986451B1 (en) | 2016-07-22 | 2024-05-21 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| WO2019023873A1 (zh) * | 2017-07-31 | 2019-02-07 | 江苏竞诺择生物医药科技有限公司 | 一种用于治疗造血系统增殖性疾病的纳米脂质微粒组合物及药用组合物 |
| AU2019350759A1 (en) * | 2018-09-25 | 2021-04-22 | Celator Pharmaceuticals, Inc. | Low-intensity treatment of hematological disorders |
| JP2022514463A (ja) | 2018-12-04 | 2022-02-14 | デル-ヤン ティエン, | 抗がん剤の送達のための立体錯体 |
| CN113473980A (zh) | 2019-03-01 | 2021-10-01 | 弗拉梅尔爱尔兰有限公司 | 在进食状态下具有改善的药代动力学的γ-羟基丁酸酯组合物 |
| US11980636B2 (en) | 2020-11-18 | 2024-05-14 | Jazz Pharmaceuticals Ireland Limited | Treatment of hematological disorders |
| US11779557B1 (en) | 2022-02-07 | 2023-10-10 | Flamel Ireland Limited | Modified release gamma-hydroxybutyrate formulations having improved pharmacokinetics |
| US11583510B1 (en) | 2022-02-07 | 2023-02-21 | Flamel Ireland Limited | Methods of administering gamma hydroxybutyrate formulations after a high-fat meal |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2383259A1 (en) * | 2002-04-23 | 2003-10-23 | Celator Technologies Inc. | Synergistic compositions |
| US7850990B2 (en) * | 2001-10-03 | 2010-12-14 | Celator Pharmaceuticals, Inc. | Compositions for delivery of drug combinations |
| MXPA05004777A (es) * | 2002-11-06 | 2005-07-22 | Celgene Corp | Metodos de uso y composiciones que comprenden farmacos inhibidores selectivos de citocina para el tratamiento y el manejo de padecimientos mieloproliferativos. |
| US20040152632A1 (en) * | 2002-11-06 | 2004-08-05 | Wyeth | Combination therapy for the treatment of acute leukemia and myelodysplastic syndrome |
| EP1575582A4 (en) * | 2002-11-06 | 2009-03-11 | Wyeth Corp | COMBINED THERAPY FOR THE TREATMENT OF ACUTE LEUKEMIA AND MYELODYSPLASIC SYNDROME |
| US8022279B2 (en) * | 2004-04-22 | 2011-09-20 | Celator Pharmaceuticals, Inc. | Liposomal formulations of anthracycline agents and cytidine analogs |
| SE0402025D0 (sv) | 2004-08-13 | 2004-08-13 | Active Biotech Ab | Treatment of hyperproliferative disease with superantigens in combination with another anticancer agent |
| WO2007076117A2 (en) | 2005-12-22 | 2007-07-05 | Celator Pharmaceuticals, Inc. | Liposomal formulations comprising secondary and tertiary amines and methods for preparing thereof |
-
2008
- 2008-02-15 PL PL17200498T patent/PL3300601T3/pl unknown
- 2008-02-15 ES ES08730048.9T patent/ES2650167T3/es active Active
- 2008-02-15 DK DK17200498.8T patent/DK3300601T3/da active
- 2008-02-15 US US12/526,930 patent/US20100303895A1/en not_active Abandoned
- 2008-02-15 WO PCT/US2008/054168 patent/WO2008101214A2/en active Application Filing
- 2008-02-15 ES ES17200498T patent/ES2909903T3/es active Active
- 2008-02-15 DK DK08730048.9T patent/DK2120568T3/en active
- 2008-02-15 JP JP2009550160A patent/JP5314600B2/ja active Active
- 2008-02-15 US US12/032,583 patent/US8092828B2/en active Active
- 2008-02-15 CN CN201610324431.2A patent/CN105998046A/zh active Pending
- 2008-02-15 AU AU2008216083A patent/AU2008216083B2/en active Active
- 2008-02-15 KR KR1020097019393A patent/KR20100014441A/ko active Application Filing
- 2008-02-15 EP EP17200498.8A patent/EP3300601B1/en active Active
- 2008-02-15 PT PT172004988T patent/PT3300601T/pt unknown
- 2008-02-15 SI SI200832194T patent/SI3300601T1/sl unknown
- 2008-02-15 KR KR20157007810A patent/KR20150038752A/ko active Search and Examination
- 2008-02-15 HU HUE17200498A patent/HUE058334T2/hu unknown
- 2008-02-15 NO NO08730048A patent/NO2120568T3/no unknown
- 2008-02-15 EP EP08730048.9A patent/EP2120568B1/en not_active Revoked
- 2008-02-15 PT PT87300489T patent/PT2120568T/pt unknown
- 2008-02-15 PL PL08730048T patent/PL2120568T3/pl unknown
- 2008-02-15 EP EP22150734.6A patent/EP4046640A1/en active Pending
- 2008-02-15 CA CA2678332A patent/CA2678332C/en active Active
- 2008-02-15 HR HRP20220428TT patent/HRP20220428T3/hr unknown
-
2017
- 2017-11-29 CY CY20171101251T patent/CY1119631T1/el unknown
-
2018
- 2018-10-03 HK HK18112675.7A patent/HK1253378A1/zh unknown
-
2022
- 2022-07-05 NL NL301185C patent/NL301185I2/nl unknown
- 2022-07-05 FR FR22C1034C patent/FR22C1034I2/fr active Active
- 2022-07-07 HU HUS2200032C patent/HUS2200032I1/hu unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5314600B2 (ja) | 造血器癌および増殖性疾患の治療のための固定した薬物比 | |
| ES2388064T3 (es) | Formulaciones liposomales de agentes de antraciclina y análogos de citidina | |
| US20090074848A1 (en) | Combination formulations of cytidine analogs and platinum agents | |
| WO2007050784A2 (en) | Fixed ratio drug combination treatments for solid tumors | |
| Eckardt et al. | A phase II trial of DaunoXome, liposome-encapsulated daunorubicin, in patients with metastatic adenocarcinoma of the colon | |
| Sorensen et al. | A dose escalating study of topotecan preceding cisplatin in previously untreated patients with small-cell lung cancer | |
| CN101657098A (zh) | 用于治疗造血性癌症和增殖性病症的固定药物比例 | |
| Tonetti et al. | Use of glutaraldehyde treated autologous human erythrocytes for hepatic targeting of doxorubicin | |
| CN1383382A (zh) | 甲氧基吗啉代阿霉素在治疗肝肿瘤方面的应用 | |
| Reiser et al. | DIZE (dexamethasone, idarubicin, and continuous infusion of ifosfamide and etoposide): an effective and well-tolerated new regimen for patients with relapsed lymphoma | |
| WO2019032437A1 (en) | LIPOSOMAL TAXANES FOR TREATMENT OF CPPC | |
| US20110207680A1 (en) | Administration of Glufosfamide For The Treatment of Cancer | |
| EP0874630A1 (en) | Topoisomerase ii poison and bis-dioxypiperazine derivative combination therapy |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20110214 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110214 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130208 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20130508 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130514 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20130515 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130607 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130705 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 5314600 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |