JP5306238B2 - アデノシン誘導体、その合成方法、並びにこれを有効成分として含有する炎症性疾患の予防及び治療用医薬組成物 - Google Patents
アデノシン誘導体、その合成方法、並びにこれを有効成分として含有する炎症性疾患の予防及び治療用医薬組成物 Download PDFInfo
- Publication number
- JP5306238B2 JP5306238B2 JP2009552570A JP2009552570A JP5306238B2 JP 5306238 B2 JP5306238 B2 JP 5306238B2 JP 2009552570 A JP2009552570 A JP 2009552570A JP 2009552570 A JP2009552570 A JP 2009552570A JP 5306238 B2 JP5306238 B2 JP 5306238B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- diol
- purin
- formula
- tetrahydrothiophene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000003835 adenosine derivatives Chemical class 0.000 title claims abstract description 58
- 208000027866 inflammatory disease Diseases 0.000 title claims abstract description 14
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 9
- 239000004480 active ingredient Substances 0.000 title abstract description 8
- 230000002265 prevention Effects 0.000 title abstract description 7
- 238000001308 synthesis method Methods 0.000 title 1
- 238000000034 method Methods 0.000 claims abstract description 45
- 150000001875 compounds Chemical class 0.000 claims description 177
- -1 1-naphthylmethyl Chemical group 0.000 claims description 67
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 claims description 39
- OIRDTQYFTABQOQ-KQYNXXCUSA-N Adenosine Natural products C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OIRDTQYFTABQOQ-KQYNXXCUSA-N 0.000 claims description 36
- 239000000126 substance Substances 0.000 claims description 30
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 21
- 150000003839 salts Chemical class 0.000 claims description 21
- 239000002126 C01EB10 - Adenosine Substances 0.000 claims description 19
- 229960005305 adenosine Drugs 0.000 claims description 19
- 239000002904 solvent Substances 0.000 claims description 17
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 16
- 206010061218 Inflammation Diseases 0.000 claims description 16
- 238000006243 chemical reaction Methods 0.000 claims description 14
- 230000004054 inflammatory process Effects 0.000 claims description 14
- KDCGOANMDULRCW-UHFFFAOYSA-N Purine Natural products N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 claims description 13
- 239000003054 catalyst Substances 0.000 claims description 13
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 claims description 12
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 claims description 10
- 239000007858 starting material Substances 0.000 claims description 10
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 claims description 10
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 9
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 9
- QAOWNCQODCNURD-UHFFFAOYSA-N sulfuric acid Substances OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 9
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 8
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 claims description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 claims description 6
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 claims description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 6
- 239000003638 chemical reducing agent Substances 0.000 claims description 6
- 229910052801 chlorine Inorganic materials 0.000 claims description 6
- 239000002253 acid Substances 0.000 claims description 5
- 229910052739 hydrogen Inorganic materials 0.000 claims description 5
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 claims description 5
- XUIYCLQDMOOVSQ-IXPVHAAZSA-N (2r,3r,4s)-2-[2-chloro-6-[(2-chlorophenyl)methylamino]purin-9-yl]thiolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC(Cl)=NC(NCC=3C(=CC=CC=3)Cl)=C2N=C1 XUIYCLQDMOOVSQ-IXPVHAAZSA-N 0.000 claims description 4
- IXGMDIBYUXOUSR-CKUKBARFSA-N (2r,3r,4s)-2-[2-chloro-6-[(2-methoxyphenyl)methylamino]purin-9-yl]thiolane-3,4-diol Chemical compound COC1=CC=CC=C1CNC1=NC(Cl)=NC2=C1N=CN2[C@H]1[C@H](O)[C@H](O)CS1 IXGMDIBYUXOUSR-CKUKBARFSA-N 0.000 claims description 4
- JQUBXCDDRXAMLF-IXPVHAAZSA-N (2r,3r,4s)-2-[2-chloro-6-[(3-chlorophenyl)methylamino]purin-9-yl]thiolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC(Cl)=NC(NCC=3C=C(Cl)C=CC=3)=C2N=C1 JQUBXCDDRXAMLF-IXPVHAAZSA-N 0.000 claims description 4
- YIHCHMWFJKTKDU-IXPVHAAZSA-N (2r,3r,4s)-2-[2-chloro-6-[(3-fluorophenyl)methylamino]purin-9-yl]thiolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC(Cl)=NC(NCC=3C=C(F)C=CC=3)=C2N=C1 YIHCHMWFJKTKDU-IXPVHAAZSA-N 0.000 claims description 4
- QMJFSSNSVVHFPD-IXPVHAAZSA-N (2r,3r,4s)-2-[2-chloro-6-[(3-iodophenyl)methylamino]purin-9-yl]thiolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC(Cl)=NC(NCC=3C=C(I)C=CC=3)=C2N=C1 QMJFSSNSVVHFPD-IXPVHAAZSA-N 0.000 claims description 4
- LJLMHEFJMXWPHX-CKUKBARFSA-N (2r,3r,4s)-2-[2-chloro-6-[(5-chloro-2-methoxyphenyl)methylamino]purin-9-yl]thiolane-3,4-diol Chemical compound COC1=CC=C(Cl)C=C1CNC1=NC(Cl)=NC2=C1N=CN2[C@H]1[C@H](O)[C@H](O)CS1 LJLMHEFJMXWPHX-CKUKBARFSA-N 0.000 claims description 4
- HMQUFUXKXUSFFV-IXPVHAAZSA-N (2r,3r,4s)-2-[6-[(3-bromophenyl)methylamino]-2-chloropurin-9-yl]thiolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC(Cl)=NC(NCC=3C=C(Br)C=CC=3)=C2N=C1 HMQUFUXKXUSFFV-IXPVHAAZSA-N 0.000 claims description 4
- YPCLFHBYRDLMJY-AXAPSJFSSA-N (2r,3r,4s)-2-[6-[(3-bromophenyl)methylamino]purin-9-yl]thiolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC=NC(NCC=3C=C(Br)C=CC=3)=C2N=C1 YPCLFHBYRDLMJY-AXAPSJFSSA-N 0.000 claims description 4
- CKXSTCOOOXXOLA-AXAPSJFSSA-N (2r,3r,4s)-2-[6-[(3-chlorophenyl)methylamino]purin-9-yl]thiolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC=NC(NCC=3C=C(Cl)C=CC=3)=C2N=C1 CKXSTCOOOXXOLA-AXAPSJFSSA-N 0.000 claims description 4
- YDNYUMZDXCVVRT-AXAPSJFSSA-N (2r,3r,4s)-2-[6-[(3-fluorophenyl)methylamino]purin-9-yl]thiolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC=NC(NCC=3C=C(F)C=CC=3)=C2N=C1 YDNYUMZDXCVVRT-AXAPSJFSSA-N 0.000 claims description 4
- IDVXTFYPPUNLDO-AXAPSJFSSA-N (2r,3r,4s)-2-[6-[(3-iodophenyl)methylamino]purin-9-yl]thiolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC=NC(NCC=3C=C(I)C=CC=3)=C2N=C1 IDVXTFYPPUNLDO-AXAPSJFSSA-N 0.000 claims description 4
- HEWZVZIVELJPQZ-UHFFFAOYSA-N 2,2-dimethoxypropane Chemical compound COC(C)(C)OC HEWZVZIVELJPQZ-UHFFFAOYSA-N 0.000 claims description 4
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 claims description 4
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 4
- 229910052736 halogen Inorganic materials 0.000 claims description 4
- 150000002367 halogens Chemical class 0.000 claims description 4
- 239000012279 sodium borohydride Substances 0.000 claims description 4
- 229910000033 sodium borohydride Inorganic materials 0.000 claims description 4
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 claims description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 4
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 claims description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 claims description 3
- 125000006282 2-chlorobenzyl group Chemical group [H]C1=C([H])C(Cl)=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 3
- 125000003852 3-chlorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(Cl)=C1[H])C([H])([H])* 0.000 claims description 3
- 125000006482 3-iodobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(I)=C1[H])C([H])([H])* 0.000 claims description 3
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims description 3
- 239000002841 Lewis acid Substances 0.000 claims description 3
- 239000012359 Methanesulfonyl chloride Substances 0.000 claims description 3
- YLEIFZAVNWDOBM-ZTNXSLBXSA-N ac1l9hc7 Chemical compound C([C@H]12)C[C@@H](C([C@@H](O)CC3)(C)C)[C@@]43C[C@@]14CC[C@@]1(C)[C@@]2(C)C[C@@H]2O[C@]3(O)[C@H](O)C(C)(C)O[C@@H]3[C@@H](C)[C@H]12 YLEIFZAVNWDOBM-ZTNXSLBXSA-N 0.000 claims description 3
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 claims description 3
- 229910052921 ammonium sulfate Inorganic materials 0.000 claims description 3
- 235000011130 ammonium sulphate Nutrition 0.000 claims description 3
- 125000005843 halogen group Chemical group 0.000 claims description 3
- 150000007517 lewis acids Chemical class 0.000 claims description 3
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 claims description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 3
- 230000002194 synthesizing effect Effects 0.000 claims description 3
- SBJORAVUWWLBAK-IDHHARJASA-N (2r,3r,4s)-2-[2-chloro-6-(naphthalen-1-ylmethylamino)purin-9-yl]thiolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC(Cl)=NC(NCC=3C4=CC=CC=C4C=CC=3)=C2N=C1 SBJORAVUWWLBAK-IDHHARJASA-N 0.000 claims description 2
- ANLGHWMSNKUOET-UHFFFAOYSA-N 2-[2-chloro-6-(methylamino)purin-9-yl]thiolane-3,4-diol Chemical compound C1=NC=2C(NC)=NC(Cl)=NC=2N1C1SCC(O)C1O ANLGHWMSNKUOET-UHFFFAOYSA-N 0.000 claims description 2
- UWTHIBRSUGIMNO-IXPVHAAZSA-N 3-[[[2-chloro-9-[(2r,3r,4s)-3,4-dihydroxythiolan-2-yl]purin-6-yl]amino]methyl]benzoic acid Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC(Cl)=NC(NCC=3C=C(C=CC=3)C(O)=O)=C2N=C1 UWTHIBRSUGIMNO-IXPVHAAZSA-N 0.000 claims description 2
- 125000006279 3-bromobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(Br)=C1[H])C([H])([H])* 0.000 claims description 2
- 125000006284 3-fluorobenzyl group Chemical group [H]C1=C([H])C(=C([H])C(F)=C1[H])C([H])([H])* 0.000 claims description 2
- 229910052794 bromium Inorganic materials 0.000 claims description 2
- 229910052731 fluorine Inorganic materials 0.000 claims description 2
- 230000002008 hemorrhagic effect Effects 0.000 claims description 2
- ORTFAQDWJHRMNX-UHFFFAOYSA-N hydroxidooxidocarbon(.) Chemical group O[C]=O ORTFAQDWJHRMNX-UHFFFAOYSA-N 0.000 claims description 2
- 229910052740 iodine Inorganic materials 0.000 claims description 2
- 239000012280 lithium aluminium hydride Substances 0.000 claims description 2
- GPSDUZXPYCFOSQ-UHFFFAOYSA-N m-toluic acid Chemical compound CC1=CC=CC(C(O)=O)=C1 GPSDUZXPYCFOSQ-UHFFFAOYSA-N 0.000 claims description 2
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 claims description 2
- 125000004923 naphthylmethyl group Chemical group C1(=CC=CC2=CC=CC=C12)C* 0.000 claims description 2
- ZWLPBLYKEWSWPD-UHFFFAOYSA-N o-toluic acid Chemical compound CC1=CC=CC=C1C(O)=O ZWLPBLYKEWSWPD-UHFFFAOYSA-N 0.000 claims description 2
- 239000003208 petroleum Substances 0.000 claims description 2
- 230000002062 proliferating effect Effects 0.000 claims description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 2
- 235000010265 sodium sulphite Nutrition 0.000 claims description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims 1
- 125000003545 alkoxy group Chemical group 0.000 claims 1
- 125000000217 alkyl group Chemical group 0.000 claims 1
- 230000003412 degenerative effect Effects 0.000 claims 1
- 230000003301 hydrolyzing effect Effects 0.000 claims 1
- 239000012022 methylating agents Substances 0.000 claims 1
- 102000009346 Adenosine receptors Human genes 0.000 abstract description 41
- 108050000203 Adenosine receptors Proteins 0.000 abstract description 41
- 230000027455 binding Effects 0.000 abstract description 28
- 230000015572 biosynthetic process Effects 0.000 abstract description 24
- 238000003786 synthesis reaction Methods 0.000 abstract description 24
- 230000003110 anti-inflammatory effect Effects 0.000 abstract description 17
- 239000000296 purinergic P1 receptor antagonist Substances 0.000 abstract description 13
- 229940121359 adenosine receptor antagonist Drugs 0.000 abstract description 10
- 101150046889 ADORA3 gene Proteins 0.000 abstract 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 84
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 48
- 238000002360 preparation method Methods 0.000 description 43
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 42
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 36
- 239000000203 mixture Substances 0.000 description 34
- 238000004519 manufacturing process Methods 0.000 description 31
- 238000005160 1H NMR spectroscopy Methods 0.000 description 30
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 28
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 24
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 21
- 239000007787 solid Substances 0.000 description 19
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 15
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- 239000006260 foam Substances 0.000 description 15
- 102000005962 receptors Human genes 0.000 description 15
- 108020003175 receptors Proteins 0.000 description 15
- QVSVMNXRLWSNGS-UHFFFAOYSA-N (3-fluorophenyl)methanamine Chemical compound NCC1=CC=CC(F)=C1 QVSVMNXRLWSNGS-UHFFFAOYSA-N 0.000 description 14
- 239000000243 solution Substances 0.000 description 14
- 239000011541 reaction mixture Substances 0.000 description 13
- 238000010898 silica gel chromatography Methods 0.000 description 13
- WHNPRMGBEYZXND-UWBRJAPDSA-N (2r,3s,4s)-2-(2,6-dichloropurin-9-yl)thiolane-3,4-diol Chemical compound O[C@H]1[C@H](O)CS[C@H]1N1C2=NC(Cl)=NC(Cl)=C2N=C1 WHNPRMGBEYZXND-UWBRJAPDSA-N 0.000 description 11
- 235000008504 concentrate Nutrition 0.000 description 11
- 239000012141 concentrate Substances 0.000 description 11
- 239000003446 ligand Substances 0.000 description 10
- 239000006188 syrup Substances 0.000 description 10
- 235000020357 syrup Nutrition 0.000 description 10
- 241001465754 Metazoa Species 0.000 description 9
- 239000005557 antagonist Substances 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 230000005764 inhibitory process Effects 0.000 description 9
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 8
- 239000003814 drug Substances 0.000 description 8
- 125000001424 substituent group Chemical group 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 7
- IPSYPUKKXMNCNQ-PFHKOEEOSA-N (2s,3s,4r,5r)-5-[2-chloro-6-[(3-iodophenyl)methylamino]purin-9-yl]-3,4-dihydroxy-n-methyloxolane-2-carboxamide Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NC)O[C@H]1N1C2=NC(Cl)=NC(NCC=3C=C(I)C=CC=3)=C2N=C1 IPSYPUKKXMNCNQ-PFHKOEEOSA-N 0.000 description 6
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 6
- 239000000556 agonist Substances 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 210000004556 brain Anatomy 0.000 description 6
- 210000004027 cell Anatomy 0.000 description 6
- 238000009472 formulation Methods 0.000 description 6
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 239000012044 organic layer Substances 0.000 description 6
- 229910052760 oxygen Inorganic materials 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- AANJZFFIJQNQEW-MFCLVGODSA-N (2r,3s,4s)-2-(6-chloropurin-9-yl)thiolane-3,4-diol Chemical compound O[C@H]1[C@H](O)CS[C@H]1N1C2=NC=NC(Cl)=C2N=C1 AANJZFFIJQNQEW-MFCLVGODSA-N 0.000 description 5
- 201000006474 Brain Ischemia Diseases 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 230000006870 function Effects 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 229910052717 sulfur Inorganic materials 0.000 description 5
- GSDDNGCNMOUECD-NQMVMOMDSA-N 9-[(3aR,4R,6aS)-2,2-dimethyl-3a,4,6,6a-tetrahydrothieno[3,4-d][1,3]dioxol-4-yl]-2-chloropurine Chemical compound ClC1=NC=C2N=CN(C2=N1)[C@@H]1SC[C@H]2OC(O[C@H]21)(C)C GSDDNGCNMOUECD-NQMVMOMDSA-N 0.000 description 4
- 206010008120 Cerebral ischaemia Diseases 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 description 4
- 241000700159 Rattus Species 0.000 description 4
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 4
- 238000010171 animal model Methods 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- 206010008118 cerebral infarction Diseases 0.000 description 4
- 239000012153 distilled water Substances 0.000 description 4
- 230000002757 inflammatory effect Effects 0.000 description 4
- 239000004615 ingredient Substances 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- SUYJXERPRICYRX-UHFFFAOYSA-N (3-bromophenyl)methanamine Chemical compound NCC1=CC=CC(Br)=C1 SUYJXERPRICYRX-UHFFFAOYSA-N 0.000 description 3
- BJFPYGGTDAYECS-UHFFFAOYSA-N (3-chlorophenyl)methanamine Chemical compound NCC1=CC=CC(Cl)=C1 BJFPYGGTDAYECS-UHFFFAOYSA-N 0.000 description 3
- LQLOGZQVKUNBRX-UHFFFAOYSA-N (3-iodophenyl)methanamine Chemical compound NCC1=CC=CC(I)=C1 LQLOGZQVKUNBRX-UHFFFAOYSA-N 0.000 description 3
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 3
- ZKHQWZAMYRWXGA-KQYNXXCUSA-J ATP(4-) Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-J 0.000 description 3
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 230000009471 action Effects 0.000 description 3
- 239000000924 antiasthmatic agent Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 150000001982 diacylglycerols Chemical class 0.000 description 3
- FDPIMTJIUBPUKL-UHFFFAOYSA-N dimethylacetone Natural products CCC(=O)CC FDPIMTJIUBPUKL-UHFFFAOYSA-N 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 239000003205 fragrance Substances 0.000 description 3
- 210000002216 heart Anatomy 0.000 description 3
- 229960000890 hydrocortisone Drugs 0.000 description 3
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 239000002777 nucleoside Substances 0.000 description 3
- 150000007524 organic acids Chemical class 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 229940044601 receptor agonist Drugs 0.000 description 3
- 239000000018 receptor agonist Substances 0.000 description 3
- 229940124597 therapeutic agent Drugs 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- BMUNWGDQGOJGOE-IXPVHAAZSA-N (2r,3r,4r)-2-[6-[(3-bromophenyl)methylamino]-2-chloropurin-9-yl]oxolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CO[C@H]1N1C2=NC(Cl)=NC(NCC=3C=C(Br)C=CC=3)=C2N=C1 BMUNWGDQGOJGOE-IXPVHAAZSA-N 0.000 description 2
- MUICQSFRABJJOR-WHSBFGAESA-N (2r,3r,4s)-2-[6-[benzyl(naphthalen-1-ylmethyl)amino]-2-chloropurin-9-yl]thiolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CS[C@H]1N1C2=NC(Cl)=NC(N(CC=3C=CC=CC=3)CC=3C4=CC=CC=C4C=CC=3)=C2N=C1 MUICQSFRABJJOR-WHSBFGAESA-N 0.000 description 2
- LOGOEBMHHXYBID-WBKNRDRNSA-N (2s,3s,4r,5r)-5-[6-[(4-amino-3-iodanylphenyl)methylamino]purin-9-yl]-3,4-dihydroxy-n-methyloxolane-2-carboxamide Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NC)O[C@H]1N1C2=NC=NC(NCC=3C=C([125I])C(N)=CC=3)=C2N=C1 LOGOEBMHHXYBID-WBKNRDRNSA-N 0.000 description 2
- MBZCXOZTJSRAOQ-HSUXUTPPSA-N (3ar,4r,6ar)-2,2-dimethyl-3a,4,6,6a-tetrahydrofuro[3,4-d][1,3]dioxol-4-ol Chemical compound C1O[C@@H](O)[C@@H]2OC(C)(C)O[C@@H]21 MBZCXOZTJSRAOQ-HSUXUTPPSA-N 0.000 description 2
- QDMSJAPLEVJBRD-BDBFLJFWSA-N (3ar,4s,6as)-4-(2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyl-3a,4,6,6a-tetrahydrothieno[3,4-d][1,3]dioxole Chemical compound O1C(C)(C)OCC1[C@H]1[C@@H]2OC(C)(C)O[C@@H]2CS1 QDMSJAPLEVJBRD-BDBFLJFWSA-N 0.000 description 2
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 2
- CKMMCZAVXJINCZ-GCIYTXSZSA-N (r)-(2,2-dimethyl-1,3-dioxolan-4-yl)-[(4r,5s)-5-(hydroxymethyl)-2,2-dimethyl-1,3-dioxolan-4-yl]methanol Chemical compound O1C(C)(C)OCC1[C@@H](O)[C@@H]1[C@H](CO)OC(C)(C)O1 CKMMCZAVXJINCZ-GCIYTXSZSA-N 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- YSGGUGVSXFYQDO-IDWMTUBTSA-N 1-[(3ar,4s,6as)-2,2-dimethyl-3a,4,6,6a-tetrahydrothieno[3,4-d][1,3]dioxol-4-yl]ethane-1,2-diol Chemical compound C1S[C@@H](C(O)CO)[C@@H]2OC(C)(C)O[C@@H]21 YSGGUGVSXFYQDO-IDWMTUBTSA-N 0.000 description 2
- RMFWVOLULURGJI-UHFFFAOYSA-N 2,6-dichloro-7h-purine Chemical compound ClC1=NC(Cl)=C2NC=NC2=N1 RMFWVOLULURGJI-UHFFFAOYSA-N 0.000 description 2
- HUJXGQILHAUCCV-MOROJQBDSA-N 3-iodobenzyl-5'-N-methylcarboxamidoadenosine Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NC)O[C@H]1N1C2=NC=NC(NCC=3C=C(I)C=CC=3)=C2N=C1 HUJXGQILHAUCCV-MOROJQBDSA-N 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 235000005979 Citrus limon Nutrition 0.000 description 2
- 244000131522 Citrus pyriformis Species 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- 241000699802 Cricetulus griseus Species 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 102000008338 G protein-coupled adenosine receptor activity proteins Human genes 0.000 description 2
- 108040002766 G protein-coupled adenosine receptor activity proteins Proteins 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- XKMLYUALXHKNFT-UUOKFMHZSA-N Guanosine-5'-triphosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XKMLYUALXHKNFT-UUOKFMHZSA-N 0.000 description 2
- 102000008070 Interferon-gamma Human genes 0.000 description 2
- 108010074328 Interferon-gamma Proteins 0.000 description 2
- 102000013462 Interleukin-12 Human genes 0.000 description 2
- 108010065805 Interleukin-12 Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 208000028389 Nerve injury Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- OATSQCXMYKYFQO-UHFFFAOYSA-N S-methyl thioacetate Chemical compound CSC(C)=O OATSQCXMYKYFQO-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 2
- 102000014384 Type C Phospholipases Human genes 0.000 description 2
- 108010079194 Type C Phospholipases Proteins 0.000 description 2
- MMWCIQZXVOZEGG-HOZKJCLWSA-N [(1S,2R,3S,4S,5R,6S)-2,3,5-trihydroxy-4,6-diphosphonooxycyclohexyl] dihydrogen phosphate Chemical compound O[C@H]1[C@@H](O)[C@H](OP(O)(O)=O)[C@@H](OP(O)(O)=O)[C@H](O)[C@H]1OP(O)(O)=O MMWCIQZXVOZEGG-HOZKJCLWSA-N 0.000 description 2
- BCTJSQNYHKAOSK-BWZBUEFSSA-N [(3ar,4r,6as)-2,2-dimethyl-3a,4,6,6a-tetrahydrothieno[3,4-d][1,3]dioxol-4-yl] acetate Chemical compound O1C(C)(C)O[C@H]2[C@H](OC(=O)C)SC[C@H]21 BCTJSQNYHKAOSK-BWZBUEFSSA-N 0.000 description 2
- GFCSYKYRSODEGC-FXQIFTODSA-N [(3as,4s,6as)-2,2-dimethyl-3a,4,6,6a-tetrahydrofuro[3,4-d][1,3]dioxol-4-yl] acetate Chemical compound O1C(C)(C)O[C@@H]2[C@H](OC(=O)C)OC[C@@H]21 GFCSYKYRSODEGC-FXQIFTODSA-N 0.000 description 2
- MJOQJPYNENPSSS-XQHKEYJVSA-N [(3r,4s,5r,6s)-4,5,6-triacetyloxyoxan-3-yl] acetate Chemical compound CC(=O)O[C@@H]1CO[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O MJOQJPYNENPSSS-XQHKEYJVSA-N 0.000 description 2
- 239000003377 acid catalyst Substances 0.000 description 2
- 102000030621 adenylate cyclase Human genes 0.000 description 2
- 108060000200 adenylate cyclase Proteins 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 230000001088 anti-asthma Effects 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 229940099112 cornstarch Drugs 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 206010015037 epilepsy Diseases 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 150000002391 heterocyclic compounds Chemical class 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 229960001340 histamine Drugs 0.000 description 2
- 210000003630 histaminocyte Anatomy 0.000 description 2
- 210000002865 immune cell Anatomy 0.000 description 2
- 230000002779 inactivation Effects 0.000 description 2
- 239000005550 inflammation mediator Substances 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 229960003130 interferon gamma Drugs 0.000 description 2
- 229940117681 interleukin-12 Drugs 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 229940041476 lactose 100 mg Drugs 0.000 description 2
- 239000011968 lewis acid catalyst Substances 0.000 description 2
- 230000008764 nerve damage Effects 0.000 description 2
- 230000009871 nonspecific binding Effects 0.000 description 2
- 150000003833 nucleoside derivatives Chemical class 0.000 description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 210000001672 ovary Anatomy 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 150000003905 phosphatidylinositols Chemical class 0.000 description 2
- XNGIFLGASWRNHJ-UHFFFAOYSA-N phthalic acid Chemical compound OC(=O)C1=CC=CC=C1C(O)=O XNGIFLGASWRNHJ-UHFFFAOYSA-N 0.000 description 2
- 230000035790 physiological processes and functions Effects 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 239000003223 protective agent Substances 0.000 description 2
- 239000003379 purinergic P1 receptor agonist Substances 0.000 description 2
- 230000001696 purinergic effect Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 229910052979 sodium sulfide Inorganic materials 0.000 description 2
- GRVFOGOEDUUMBP-UHFFFAOYSA-N sodium sulfide (anhydrous) Chemical compound [Na+].[Na+].[S-2] GRVFOGOEDUUMBP-UHFFFAOYSA-N 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 102000003390 tumor necrosis factor Human genes 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- PXJACNDVRNAFHD-UHFFFAOYSA-N (2-methoxyphenyl)methanamine Chemical compound COC1=CC=CC=C1CN PXJACNDVRNAFHD-UHFFFAOYSA-N 0.000 description 1
- QCQOICUQAJLDKV-DFLNUYFWSA-N (2R,3R,4S,5R)-2-[6-amino-6-[(4-amino-3-(125I)iodanylphenyl)methyl]-8H-purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound NC1=C(C=C(CC2(C3=NCN([C@H]4[C@H](O)[C@H](O)[C@@H](CO)O4)C3=NC=N2)N)C=C1)[125I] QCQOICUQAJLDKV-DFLNUYFWSA-N 0.000 description 1
- XJRJYJGMHHTTNM-BOVONTJDSA-N (2S,3R,4S,5R)-2-(6-aminopurin-9-yl)-3,4-dihydroxy-5-(hydroxymethyl)-N-methyloxolane-2-carboxamide Chemical compound CNC(=O)[C@@]1([C@H](O)[C@H](O)[C@@H](CO)O1)N1C=NC=2C(N)=NC=NC12 XJRJYJGMHHTTNM-BOVONTJDSA-N 0.000 description 1
- IYAFDRKTMVINNA-IXPVHAAZSA-N (2r,3r,4r)-2-[2-chloro-6-[(3-iodophenyl)methylamino]purin-9-yl]oxolane-3,4-diol Chemical compound O[C@@H]1[C@H](O)CO[C@H]1N1C2=NC(Cl)=NC(NCC=3C=C(I)C=CC=3)=C2N=C1 IYAFDRKTMVINNA-IXPVHAAZSA-N 0.000 description 1
- ANLGHWMSNKUOET-QKVARQBESA-N (2r,3s,4r)-2-[2-chloro-6-(methylamino)purin-9-yl]thiolane-3,4-diol Chemical compound C1=NC=2C(NC)=NC(Cl)=NC=2N1[C@@H]1SC[C@H](O)[C@@H]1O ANLGHWMSNKUOET-QKVARQBESA-N 0.000 description 1
- LDYMCRRFCMRFKB-MOROJQBDSA-N (2s,3s,4r,5r)-5-[6-[(4-aminophenyl)methylamino]purin-9-yl]-3,4-dihydroxy-n-methyloxolane-2-carboxamide Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NC)O[C@H]1N1C2=NC=NC(NCC=3C=CC(N)=CC=3)=C2N=C1 LDYMCRRFCMRFKB-MOROJQBDSA-N 0.000 description 1
- ODFSPVXJTALZSK-ZYUZMQFOSA-N (3aR,4R,6aR)-4-[(4R)-2,2-dimethyl-1,3-dioxolan-4-yl]-2,2-dimethyl-3a,4,6,6a-tetrahydrofuro[3,4-d][1,3]dioxole Chemical compound O1C(C)(C)OC[C@@H]1[C@@H]1[C@@H]2OC(C)(C)O[C@@H]2CO1 ODFSPVXJTALZSK-ZYUZMQFOSA-N 0.000 description 1
- JWWCLCNPTZHVLF-BHRXDNSCSA-N (3ar,4r,6r,6ar)-6-(2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyl-3a,4,6,6a-tetrahydrofuro[3,4-d][1,3]dioxol-4-ol Chemical compound O1C(C)(C)OCC1[C@@H]1[C@H]2OC(C)(C)O[C@H]2[C@H](O)O1 JWWCLCNPTZHVLF-BHRXDNSCSA-N 0.000 description 1
- WHPSMBYLYRPVGU-RFZPGFLSSA-N (3ar,6ar)-2,2-dimethyl-6,6a-dihydro-3ah-furo[3,4-d][1,3]dioxol-4-one Chemical compound C1OC(=O)[C@@H]2OC(C)(C)O[C@@H]21 WHPSMBYLYRPVGU-RFZPGFLSSA-N 0.000 description 1
- GIGGUFCYUVFLJZ-UHFFFAOYSA-N (5-chloro-2-methoxyphenyl)methanamine Chemical compound COC1=CC=C(Cl)C=C1CN GIGGUFCYUVFLJZ-UHFFFAOYSA-N 0.000 description 1
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- GYSCBCSGKXNZRH-UHFFFAOYSA-N 1-benzothiophene-2-carboxamide Chemical compound C1=CC=C2SC(C(=O)N)=CC2=C1 GYSCBCSGKXNZRH-UHFFFAOYSA-N 0.000 description 1
- LDMOEFOXLIZJOW-UHFFFAOYSA-N 1-dodecanesulfonic acid Chemical compound CCCCCCCCCCCCS(O)(=O)=O LDMOEFOXLIZJOW-UHFFFAOYSA-N 0.000 description 1
- FXEDIXLHKQINFP-UHFFFAOYSA-N 12-O-tetradecanoylphorbol-13-acetate Natural products CCCCCCCCCCCCCC(=O)OC1CC2(O)C(C=C(CO)CC3(O)C2C=C(C)C3=O)C4C(C)(C)C14OC(=O)C FXEDIXLHKQINFP-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- XHNOLUKXFQKXIR-XXEGWPOOSA-N 2-[(2S,3S,4R,5R)-5-[6-amino-6-[(3-iodophenyl)methyl]-8H-purin-9-yl]-3,4-dihydroxyoxolan-2-yl]-2-hydroxy-N-methylacetamide Chemical compound IC=1C=C(CC2(C3=NCN([C@H]4[C@H](O)[C@H](O)[C@@H](C(O)C(NC)=O)O4)C3=NC=N2)N)C=CC1 XHNOLUKXFQKXIR-XXEGWPOOSA-N 0.000 description 1
- 125000002927 2-methoxybenzyl group Chemical group [H]C1=C([H])C([H])=C(C(OC([H])([H])[H])=C1[H])C([H])([H])* 0.000 description 1
- WLJVXDMOQOGPHL-PPJXEINESA-N 2-phenylacetic acid Chemical compound O[14C](=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-PPJXEINESA-N 0.000 description 1
- GSWYUZQBLVUEPH-UHFFFAOYSA-N 3-(azaniumylmethyl)benzoate Chemical compound NCC1=CC=CC(C(O)=O)=C1 GSWYUZQBLVUEPH-UHFFFAOYSA-N 0.000 description 1
- UWTHIBRSUGIMNO-ITDIGPHOSA-N 3-[[[2-chloro-9-[(2r,3s,4r)-3,4-dihydroxythiolan-2-yl]purin-6-yl]amino]methyl]benzoic acid Chemical compound O[C@H]1[C@@H](O)CS[C@H]1N1C2=NC(Cl)=NC(NCC=3C=C(C=CC=3)C(O)=O)=C2N=C1 UWTHIBRSUGIMNO-ITDIGPHOSA-N 0.000 description 1
- 101710169336 5'-deoxyadenosine deaminase Proteins 0.000 description 1
- ZKBQDFAWXLTYKS-UHFFFAOYSA-N 6-Chloro-1H-purine Chemical compound ClC1=NC=NC2=C1NC=N2 ZKBQDFAWXLTYKS-UHFFFAOYSA-N 0.000 description 1
- UHEWCNSNLNSHGO-AZTOOPQRSA-N 9-[(3ar,4r,6as)-2,2-dimethyl-3a,4,6,6a-tetrahydrothieno[3,4-d][1,3]dioxol-4-yl]-6-chloropurine Chemical compound C1=NC2=C(Cl)N=CN=C2N1[C@@H]1SC[C@H]2OC(C)(C)O[C@H]21 UHEWCNSNLNSHGO-AZTOOPQRSA-N 0.000 description 1
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical group O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 1
- 208000030090 Acute Disease Diseases 0.000 description 1
- 102000055025 Adenosine deaminases Human genes 0.000 description 1
- 229940122614 Adenosine receptor agonist Drugs 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- 208000014644 Brain disease Diseases 0.000 description 1
- 206010006482 Bronchospasm Diseases 0.000 description 1
- QWOJMRHUQHTCJG-UHFFFAOYSA-N CC([CH2-])=O Chemical compound CC([CH2-])=O QWOJMRHUQHTCJG-UHFFFAOYSA-N 0.000 description 1
- PAOANWZGLPPROA-RQXXJAGISA-N CGS-21680 Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NCC)O[C@H]1N1C2=NC(NCCC=3C=CC(CCC(O)=O)=CC=3)=NC(N)=C2N=C1 PAOANWZGLPPROA-RQXXJAGISA-N 0.000 description 1
- 208000018152 Cerebral disease Diseases 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-N Decanoic acid Natural products CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 240000001307 Myosotis scorpioides Species 0.000 description 1
- LFZAGIJXANFPFN-UHFFFAOYSA-N N-[3-[4-(3-methyl-5-propan-2-yl-1,2,4-triazol-4-yl)piperidin-1-yl]-1-thiophen-2-ylpropyl]acetamide Chemical compound C(C)(C)C1=NN=C(N1C1CCN(CC1)CCC(C=1SC=CC=1)NC(C)=O)C LFZAGIJXANFPFN-UHFFFAOYSA-N 0.000 description 1
- JADDQZYHOWSFJD-FLNNQWSLSA-N N-ethyl-5'-carboxamidoadenosine Chemical compound O[C@@H]1[C@H](O)[C@@H](C(=O)NCC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 JADDQZYHOWSFJD-FLNNQWSLSA-N 0.000 description 1
- 208000005141 Otitis Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical compound OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 208000028017 Psychotic disease Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 208000003734 Supraventricular Tachycardia Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 239000002593 adenosine A3 receptor agonist Substances 0.000 description 1
- TTWYZDPBDWHJOR-IDIVVRGQSA-L adenosine triphosphate disodium Chemical compound [Na+].[Na+].C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP([O-])([O-])=O)[C@@H](O)[C@H]1O TTWYZDPBDWHJOR-IDIVVRGQSA-L 0.000 description 1
- 150000003838 adenosines Chemical class 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 230000001270 agonistic effect Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- GZCGUPFRVQAUEE-KVTDHHQDSA-N aldehydo-D-mannose Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)C=O GZCGUPFRVQAUEE-KVTDHHQDSA-N 0.000 description 1
- 125000003282 alkyl amino group Chemical group 0.000 description 1
- SRVFFFJZQVENJC-IHRRRGAJSA-N aloxistatin Chemical compound CCOC(=O)[C@H]1O[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)NCCC(C)C SRVFFFJZQVENJC-IHRRRGAJSA-N 0.000 description 1
- OBETXYAYXDNJHR-UHFFFAOYSA-N alpha-ethylcaproic acid Natural products CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 230000008485 antagonism Effects 0.000 description 1
- 239000003416 antiarrhythmic agent Substances 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 229940030600 antihypertensive agent Drugs 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 208000008784 apnea Diseases 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 230000006793 arrhythmia Effects 0.000 description 1
- 125000001769 aryl amino group Chemical group 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000023555 blood coagulation Effects 0.000 description 1
- 210000000621 bronchi Anatomy 0.000 description 1
- 230000007885 bronchoconstriction Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 235000014121 butter Nutrition 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 238000007675 cardiac surgery Methods 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 208000037976 chronic inflammation Diseases 0.000 description 1
- 208000037893 chronic inflammatory disorder Diseases 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 230000019771 cognition Effects 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000000593 degrading effect Effects 0.000 description 1
- 230000030609 dephosphorylation Effects 0.000 description 1
- 238000006209 dephosphorylation reaction Methods 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- IZEKFCXSFNUWAM-UHFFFAOYSA-N dipyridamole Chemical compound C=12N=C(N(CCO)CCO)N=C(N3CCCCC3)C2=NC(N(CCO)CCO)=NC=1N1CCCCC1 IZEKFCXSFNUWAM-UHFFFAOYSA-N 0.000 description 1
- 229960002768 dipyridamole Drugs 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 210000005069 ears Anatomy 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-N ethanesulfonic acid Chemical compound CCS(O)(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-N 0.000 description 1
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 235000021050 feed intake Nutrition 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000009931 harmful effect Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 201000010235 heart cancer Diseases 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 208000024348 heart neoplasm Diseases 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000012442 inert solvent Substances 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 125000006480 iodobenzyl group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229960003511 macrogol Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 238000003328 mesylation reaction Methods 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910052987 metal hydride Inorganic materials 0.000 description 1
- 150000004681 metal hydrides Chemical class 0.000 description 1
- MBABOKRGFJTBAE-UHFFFAOYSA-N methyl methanesulfonate Chemical compound COS(C)(=O)=O MBABOKRGFJTBAE-UHFFFAOYSA-N 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- KPAKEDUKXVSIAP-UHFFFAOYSA-N n-(naphthalen-1-ylmethyl)-1-phenylmethanamine Chemical compound C=1C=CC2=CC=CC=C2C=1CNCC1=CC=CC=C1 KPAKEDUKXVSIAP-UHFFFAOYSA-N 0.000 description 1
- SQMWSBKSHWARHU-SDBHATRESA-N n6-cyclopentyladenosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(NC3CCCC3)=C2N=C1 SQMWSBKSHWARHU-SDBHATRESA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 230000000324 neuroprotective effect Effects 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000003002 pH adjusting agent Substances 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000006201 parenteral dosage form Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 230000004526 pharmaceutical effect Effects 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- PHEDXBVPIONUQT-RGYGYFBISA-N phorbol 13-acetate 12-myristate Chemical compound C([C@]1(O)C(=O)C(C)=C[C@H]1[C@@]1(O)[C@H](C)[C@H]2OC(=O)CCCCCCCCCCCCC)C(CO)=C[C@H]1[C@H]1[C@]2(OC(C)=O)C1(C)C PHEDXBVPIONUQT-RGYGYFBISA-N 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 239000002287 radioligand Substances 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000007142 ring opening reaction Methods 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 238000012453 sprague-dawley rat model Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008227 sterile water for injection Substances 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 125000000446 sulfanediyl group Chemical group *S* 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 150000007970 thio esters Chemical class 0.000 description 1
- 150000007944 thiolates Chemical class 0.000 description 1
- 239000005450 thionucleoside Substances 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 238000002723 toxicity assay Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- PJVWKTKQMONHTI-UHFFFAOYSA-N warfarin Chemical compound OC=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 PJVWKTKQMONHTI-UHFFFAOYSA-N 0.000 description 1
- 229960005080 warfarin Drugs 0.000 description 1
- 230000004584 weight gain Effects 0.000 description 1
- 235000019786 weight gain Nutrition 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Description
Rは、非置換又は独立的に若しくは選択的に1つ以上のC6〜C10のアリール基で置換された直鎖又は分岐鎖のC1〜C5のアルキル、非置換又は独立的に若しくは選択的にハロゲン若しくは一つ以上の直鎖若しくは分岐鎖のC1〜C4のアルコキシ基で置換されたベンジル、又はヒドロキシカルボニルで置換されたベンジルであり、
Yは、H又はハロゲン原子である。
Aは、O又はSであり、
Rは、メチル、エチル、プロピル、ナフチルメチル、ベンジル、独立的に又は選択的にF、Cl、Br、I、C1〜C3アルコキシ及びそれらの組み合わせからなる群から選択される1つの置換基に置換されたベンジル、又はトルイル酸であり、
Yは、H又はClである。
Aは、O又はSであり、
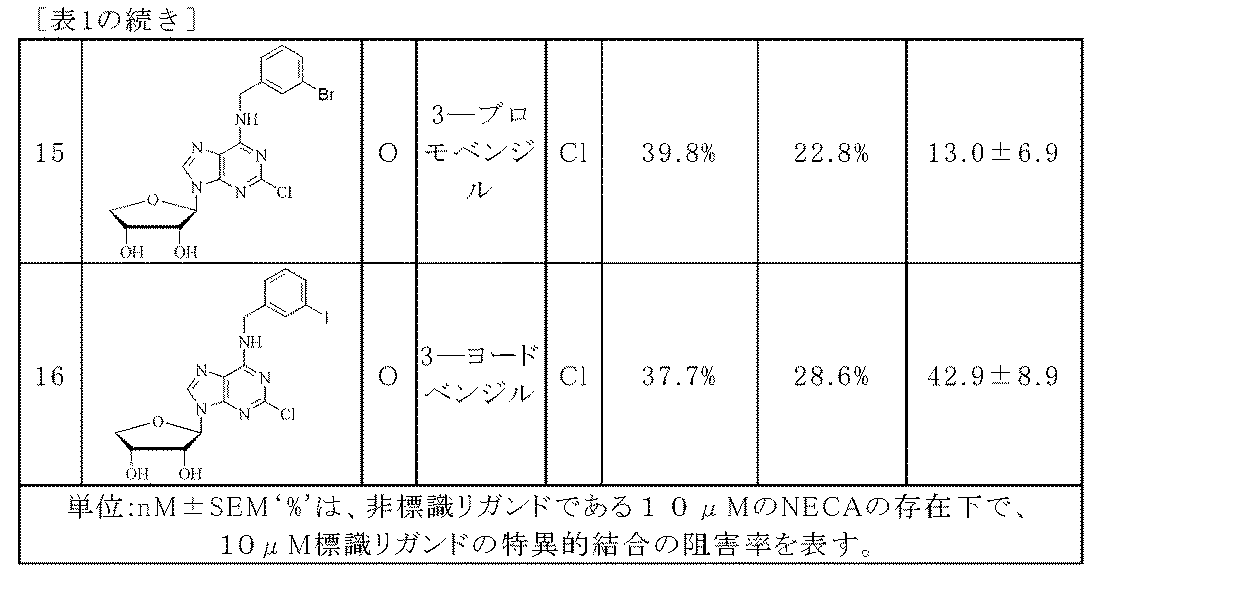
Rは、メチル、エチル、1―ナフチルメチル、ベンジル、2―クロロベンジル、3―フルオロベンジル、3―クロロベンジル、3―ブロモベンジル、3―ヨードベンジル、2―メトキシ―5―クロロベンジル、2―メトキシベンジル又は3―トルイル酸であり、
Yは、H又はClである。
1)(2R,3R,4S)―2―(2―クロロ―6―(3―フルオロベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
2)(2R,3R,4S)―2―(2―クロロ―6―(3―クロロベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
3)(2R,3R,4S)―2―(6―(3―ブロモベンジルアミノ)―2―クロロ―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
4)(2R,3R,4S)―2―(2―クロロ―6―(3―ヨードベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
5)(2R,3R,4S)―2―(2―クロロ―6―(2―クロロベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
6)(2R,3R,4S)―2―(2―クロロ―6―(5―クロロ―2―メトキシベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
7)(2R,3R,4S)―2―(2―クロロ―6―(2―メトキシベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
8)(2R,3R,4S)―2―(2―クロロ―6―(ナフタレン―1―イルメチルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
9)3―((2―クロロ―9―((2R,3R,4S)―3,4―ジヒドロキシテトラヒドロチオフェン―2―イル)―9H―プリン―6―イルアミノ)メチル)安息香酸;
10)2―(2―クロロ―6―メチルアミノ―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
11)(2R,3R,4S)―2―(6―(3―フルオロベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
12)(2R,3R,4S)―2―(6―(3―クロロベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
13)(2R,3R,4S)―2―(6―(3―ブロモベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
14)(2R,3R,4S)―2―(6―(3―ヨードベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
15)(2R,3R,4R)―2―(6―(3―ブロモベンジルアミノ)―2―クロロ―9H―プリン―9―イル)テトラヒドロフラン―3,4―ジオール;及び
16)(2R,3R,4R)―2―(2―クロロ―6―(3―ヨードベンジルアミノ)―9H―プリン―9―イル)テトラヒドロフラン―3,4―ジオール。
1H―NMR(CDCl3)δ 5.34(s,1 H),4.76―4.79(m,1 H),4.58(d,1 H,J=6.0Hz),4.34―4.39(m,1 H),4.15(dd,1 H,J=3.6,7.2Hz),4.00―4.08(m,2 H);
[α]25 D 11.71(c0.11,CH2Cl2);
FAB―MS m/z 261[M+H]+。
[α]25 D―3.88(c0.44,CH2Cl2);
FAB―MS m/z 263[M+H]+。
[α]25 D 38.32(c0.29,CH2Cl2);
FAB―MS m/z 419[M+H]+。
[α]25 D ―96.04(c0.20,CH2Cl2);
FAB―MS m/z 261[M+H]+。
[α]25 D ―96.04(c0.20,CH2Cl2);
FAB―MS m/z 261[M+H]+。
[α]25 D ―258.15 (c0.18,CH2Cl2);
FAB―MS m/z 218[M]+。
1H―NMR(CDCl3)δ 8.17(s,1 H),5.87(s,1 H), 5.32(pseudo t,1 H,J=4.8Hz),5.21(d,1 H,J=5.6Hz),3.79(dd,1 H,J=4.4,12.8Hz),3.26(d,1 H,J=13.2Hz),1.59(s,3 H),1.36(s,3 H);
[α]25 D ―42.04(c0.16,CH2Cl2);
FAB―MS m/z 347[M+H]+。
UV(MeOH)λmax 275.0nm;
1H―NMR (CD3OD)δ 8.87(s,1 H),6.08(d,1 H,J=6.8Hz),4.69(q,1 H,J=3.2Hz),4.48(q,1 H,J=3.6Hz),3.56(dd,1 H,J=4.4,11.2Hz),2.97(dd,1 H,J=3.4,11.2Hz);
[α]25 D ―50.43(c0.12,DMSO);
FAB―MS m/z 307[M+H]+。
UV(MeOH)λmax 275.0nm;
1H―NMR(DMSO―d6)δ 8.91(t,1 H―NH,J=5.8Hz),8.51(s,1 H),7.33―7.39(m,1 H),7.13―7.18(m,2 H),7.06(dt,1 H,J=2.8,11.6Hz),5.82(d,1 H,J=7.2Hz),5.56(d,1 H―OH,J=6.0Hz),5.37(d,1 H―OH,J=4.4Hz),4.65(d,1 H,J=6.0Hz),4.60(m,1 H),4.33―4.35(m,1 H),3.41(dd,1 H,J=4.0,10.8Hz),2.79(dd,1 H,J=2.8,10.8Hz);
[α]25 D ―96.21(c0.12,DMSO);
FAB―MS m/z 396[M+H]+。
UV(MeOH)λmax 274.5nm;
1H―NMR(CD3OD)δ 8.34(s,1 H),7.41(s,1 H), 7.24―7.34(m,3 H),5.94(d,1 H,J=6.4Hz),4.75(brs,2 H),4.61(q,1 H,J=3.2Hz),4.45(q,1 H,J=4.0Hz),3.51(dd,1 H,J=4.8,11.2Hz),2.95(dd,1 H,J=3.6,10.8Hz);
FAB―MS m/z 411 [M]+。
UV(MeOH)λmax 274.0nm;
1H―NMR(DMSO―d6)δ 8.91(brs,1 H―NH)、8.51(s,1 H),7.55(s,1 H),7.43(d,1 H,J=7.6Hz),7.33―7.35(m,1 H),7.26―7.30(m,1 H)、5.82(d,1 H,J=7.2Hz),5.57(d,1 H―OH,J=6.0Hz),5.38(d,1 H―OH,J=4.0Hz),4.60―4.63(m,3 H),4.34(s,1 H),3.41(dd,1 H,J=4.4,11.2Hz),2.80(dd,1 H,J=2.8,10.8Hz);
FAB―MS m/z 456[M+H]+。
UV(MeOH)λmax 274.0nm;
1H―NMR(DMSO―d6)δ 8.90(t,1 H―NH,J=6.4Hz),8.51(s,1 H),7.74(s,1 H),7.60(d,1 H,J=7.6Hz),7.35(d,1 H,J=7.6Hz),7.13(t,1 H,J=8.0Hz),5.82(d,1 H,J=7.6Hz),5.56(d,1 H,J=6.4Hz),5.37(d,1 H,J=4.0Hz),4.60(d,3 H,J=4.4Hz),4.34(brs,1 H),3.38(dd,1 H,J=4.0,10.8Hz),2.80(dd,1 H,J=4.0,10.8Hz);
[α]25 D ―78.91(c0.13,DMSO);
FAB―MS m/z 504[M+H]+。
UV(MeOH)λmax 273.5nm;
1H―NMR(CD3OD)δ 8.35(brs,1 H),7.45―7.47(m,1 H),7.39―7.43(m,1 H),7.25―7.29(m,2 H),5.95(d,1 H,J=6.4Hz),4.60―4.63(m,1 H),4.45(dd,1 H,J=3.6,8.0Hz),3.51(dd,1 H,J=4.8,10.8Hz),2.95(dd,1 H,J=4.0,10.8Hz);
[α]25 D ―96.21(c0.12,DMSO);
FAB―MS m/z 412[M+H]+。
UV(MeOH)λmax 275.5nm;
1H―NMR(DMSO―d6)δ 8.64(t,1 H―NH,J=6.0Hz),8.51(s,1 H),7.21―7.25(m,1 H),7.12(d,1 H,J=7.2Hz),7.00(d,1 H,J=8.0Hz),6.85―6.89(m,1 H),5.82(d,1 H,J=7.6Hz),5.57(d,1 H―OH,J=6.4Hz),5.37(d,1 H―OH,J=4.0Hz),4.61―4.63(m,2 H),4.35(m,1 H),、3.84(s,3 H),3.71(dd,1 H,J=3.6,10.4Hz)、2.80(dd,1 H,J=2.4,10.8Hz);
[α]25 D ―96.10(c0.21,DMSO);
FAB―MS m/z 442[M+H]+。
UV(MeOH)λmax 276.5nm;
1H―NMR(DMSO―d6)δ 8.65(t,1 H―NH,J=6.0Hz),8.51(s,1 H),7.21―7.25(m,1 H),7.12(d,1 H,J=7.2Hz),7.00(d,1 H,J=8.0Hz),6.85―6.89(m,1 H),5.83(d,1 H,J=6.8Hz),5.58(d,1 H―OH,J=6.4Hz),5.39(d,1 H―OH,J=3.6Hz),4.62―4.64(m,2 H),4.35(s,1 H),3.84(s,1 H),3.42(dd,1 H,J=3.6,10.4Hz),2.79―2.82(m,1 H);
[α]25 D ―93.53(c0.17,DMSO);
FAB―MS m/z 407[M+H]+。
UV(MeOH)λmax 281.0nm;
1H―NMR(DMSO―d6)δ 8.96(t,1 H―NH,J=6.0Hz),8.51(s,1 H),8.25(d,1 H,J=8.0Hz),7.95―7.97(m,1 H),7.83―7.85(m,1 H),7.53―7.61(m,2 H),7.43―7.46(m,2 H),5.82(d,1 H,J=7.6Hz),5.56(d,1 H,J=6.4Hz),5.38(d,1 H,J=4.0Hz),5.12(d,1 H,J=6.0Hz)、4.59―4.61(m,1 H),4.34―4.35(m,1 H),3.40―3.44(m,1 H),2.80(dd,1 H,J=2.4,6.8Hz);
FAB―MS m/z 428[M+H]+。
UV(MeOH)λmax 275.5nm;
1H―NMR(DMSO―d6)δ 8.95(t,1 H―NH,J=6.0Hz),8.52(s,1 H),7.89(d,1 H,J=8.4Hz),7.43(d,1 H,J=8.0Hz),5.82(d,1 H,J=7.6Hz),5.57(brs,1 H),5.38(brs,1 H),4.71(d,1 H,J=6.0Hz),4.60(brs,1 H),4.34(brs,1 H),3.41(dd,1 H,J=4.0,10.8Hz),2.80(dd,1 H,J=2.8,10.8Hz);
[α]25 D ―94.55(c0.11,DMSO);
FAB―MS m/z 422[M+H]+。
1H―NMR(CDCl3)δ 2.99(1H,dd,4'―CH,J=4.4,10.8Hz),3.12(3H,brs,NH―CH3),3.44(1H,dd,4'―CH,J=4,10.8Hz),4.41(1H,m,2'―CH,J=5.6Hz),4.47(1H,m,3'―CH),5.89(1H,d,1'―CH,J=5.6Hz),8.40(s,1H,8―CH);
[α]25 D ―34.8(c0.115,DMSO);
FAB―MS m/z 302.3[M+H]+。
13C―NMR(CDCl3)δ 152.05,151.39,151.09,144.34,132.56,111.90,89.60,84.31,70.30,40.76,26.40,24.63;
[α]25 D ―157.64(c0.15,MeOH);
FAB―MS m/z 313[M+H]+。
UV(MeOH)λmax 264.5nm;
1H―NMR(DMSO―d6)δ 9.02(s,1 H),8.82(s,1 H),6.02(d,1 H,J=7.6Hz),5.62(d,1 H―OH,J=6.0Hz),5.43(d,1 H―OH,J=4.0Hz),4.70―4.74(m,1 H),4.36―4.40(m,1 H),3.47(dd,1 H,J=4.0,10.8Hz),3.17(d,1 H,J=5.2Hz),2.84(dd,1 H,J=2.8,11.2Hz);
[α]25 D ―109.15(c0.16,DMSO);
FAB―MS m/z 273[M+H]+。
UV(MeOH)λmax 273.5nm;
1H―NMR(DMSO―d6)δ 8.46(s,1 H),8.22(s,1 H),7.31―7.39(m,1 H),7.12―7.18(m,2 H),7.01―7.05(m,1 H),5.90(d,1 H,J=7.2Hz),5.53(d,1 H―OH,J=6.4Hz),5.35(d,1 H―OH,J=4.0Hz),4.67―4.71(m,2 H),4.35―4.37(m,1 H),3.39―3.43(m,1 H),3.17(d,1 H,J=5.2Hz),2.80(dd,1 H,J=3.2,11.2Hz);
[α]25 D ―141.2(c0.11,DMSO);
FAB―MS m/z 362[M+H]+。
UV(MeOH)λmax 274.5nm;
1H―NMR(DMSO―d6)δ 8.47(s,1 H),8.22(s,1 H),7.39(s,1 H),7.26―7.35(m,3 H),5.91(d,1 H,J=7.2Hz),5.53(d,1 H―OH,J=6.4Hz),5.35(d,1 H―OH,J=4.0Hz),4.67―4.71(m,2 H),4.33―4.37(m,1 H),3.40―3.48(m,2 H),2.80(dd,1 H,J=3.2,10.4Hz);
[α]25 D ―162.5(c0.10,DMSO);
FAB―MS m/z 378[M+H]+。
UV(MeOH)λmax 270.0nm;
1H―NMR(DMSO―d6)δ 8.46(s,1 H),8.22(s,1 H),7.53(s,1 H),7.39―7.42(m,1 H),7.34―7.35(m,1 H),7.24―7.28(m,1 H),5.90(d,1 H,J=7.2Hz),5.53(d,1 H―OH,J=6.4Hz),5.35(d,1 H―OH,J=4.0Hz),4.67―4.71(m,2 H),4.35―4.37(m,1 H),3.41(dd,1 H,J=4.0,10.8Hz),3.06(q,1 H,J=7.2Hz),2.80(dd,1 H,J=2.8,10.8Hz);
[α]25 D ―100.72(c0.14,DMSO);
FAB―MS m/z 422[M+H]+。
UV(MeOH)λmax 271.5nm;
1H―NMR(DMSO―d6)δ 8.46(s,1 H),8.22(s,1 H),7.72(s,1 H),7.56―7.59(m,1 H),7.35―7.36(d,1 H,J=7.6Hz),7.01―7.12(m,1 H),5.90(d,1 H,J=7.2Hz),5.53(d,1 H―OH,J=6.4Hz),5.35(d,1 H―OH,J=4.4Hz),4.67―4.71(m,2 H),4.34―4.38(m,1 H),3.41(dd,1 H,J=4.0,10.8Hz),3.16(d,1 H,J=7.2Hz),2.80(dd,1 H,J=2.8,10.8Hz);
[α]25 D ―97.08(c0.14,DMSO);
FAB―MS m/z 470[M+H]+。
1H―NMR(CDCl3)δ 8.15(s,1 H),6.07(s,1 H),5.41(d,1 H,J=6.0Hz),5.26―5.29(m,1 H),4.25―4.31(m,2 H),1.57(s,3 H),1.41(s,3 H);
[α]25 D ―21.00(c0.10,DMSO);
FAB―MS m/z 331[M+H]+。
UV(MeOH)λmax 276.5nm;
1H―NMR(DMSO―d6)δ 8.98(s,1 H),5.96(d,1 H,J=6.4Hz),5.57(d,1 H―OH,J=6.0Hz),5.32(d,1 H―OH,J=4.0Hz),4.69―4.74(m,1 H),4.41(dd,1 H,J=3.6,9.2Hz),4.29―4.32(m,1 H),3.87(dd,1 H,J=2.0,9.6Hz);
[α]25 D ―68.09(c0.14,DMSO);
FAB―MS m/z 291[M+H]+。
UV(MeOH)λmax 274.5nm;
1H―NMR(DMSO―d6)δ 8.92(t,1 H―NH,J=6.0Hz),8.43(s,1 H),7.55(s,1 H),7.44(d,1 H,J=8.0Hz),7.33―7.35(m,1 H),7.26―7.30(m,1 H),5.81(d,1 H,J=6.4Hz),5.47(d,1 H,J=6.4Hz),5.22(d,1 H,J=4.0Hz),4.66―4.69(m,1 H),4.62(s,2 H),4.32(dd,1 H,J=3.6,9.2Hz),4.25(brs,1 H),3.80(dd,1 H,J=1.6,9.2Hz);
[α]25 D ―62.75(c0.10,DMSO);
FAB―MS m/z 440[M+H]+。
UV(MeOH)λmax 274.0nm;
1H―NMR(DMSO―d6)δ 8.91(t,1 H―NH,J=6.4Hz),8.44(s,1 H),7.75(s,1 H),7.61(d,1 H,J=8.0Hz),7.36(d,1 H,J=7.6Hz),7.13(t,1 H,J=4.0Hz),5.81(d,1 H,J=6.8Hz),5.47(d,1 H―OH,J=6.8Hz),5.23(d,1 H―OH,J=4.0Hz),4.72(dd,1 H,J=6.4,10.8Hz),4.61(d,1 H,J=6.0Hz),4.34(dd,1 H,J=3.6,9.2Hz),3.81(dd,1 H,J=1.2,9.2Hz);
[α]25 D ―68.07(c0.12,DMSO);
FAB―MS m/z 488[M+H]+。
アデノシン誘導体 500mg
コーンスターチ 100mg
ラクトース 100mg
タルク 10mg
上記の成分を混合し、気密袋に充填した。
アデノシン誘導体 100mg
コーンスターチ 100mg
ラクトース 100mg
ステアリン酸マグネシウム 2mg
上記の成分を混合し、通常の方法にしたがって打錠して錠剤を製造した。
アデノシン誘導体 50mg
ラクトース 50mg
ステアリン酸マグネシウム 1mg
上記の成分を混合し、通常の方法にしたがってゼラチンカプセルに充填した。
アデノシン誘導体 10mg
注射用滅菌水 適量
pH調節剤 適量
蒸留水中の活性成分溶液のpHを7.5に調整し、該溶液を滅菌水で希釈して2ml容量とした後アンプルに充填し、滅菌した。
アデノシン誘導体 1g
異性化糖 10g
スクロース 10g
レモン香料 適量
純水 適量
上記成分を純水に溶解させ、レモン香料を適量加えた後、純水を加えて容量を100mlに増大し、褐色のバイアルに充填して滅菌させて液剤を製造した。
Claims (17)
- 下記の化学式1で表示されるアデノシン誘導体またはその薬学的に許容可能な塩。
- 前記Aは、Sであり、
前記Rは、メチル、エチル、プロピル、ナフチルメチル、独立的にまたは選択的にF、Cl、Br、I、C1〜C3のアルコキシ及びこれらの組み合わせからなる群から選択される置換基で置換されたベンジル、またはトルイル酸であり、
前記Yは、HまたはClである、請求項1に記載のアデノシン誘導体またはその薬学的に許容可能な塩。 - 前記Aは、Sであり、
前記Rは、メチル、エチル、1―ナフチルメチル、2―クロロベンジル、3―フルオロベンジル、3―クロロベンジル、3―ブロモベンジル、3―ヨードベンジル、2―メトキシ―5―クロロベンジル、2―メトキシベンジルまたは3―トルイル酸であり、
前記Yは、HまたはClである、請求項1に記載のアデノシン誘導体またはその薬学的に許容可能な塩。 - 前記アデノシン誘導体が、
1)(2R,3R,4S)―2―(2―クロロ―6―(3―フルオロベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
2)(2R,3R,4S)―2―(2―クロロ―6―(3―クロロベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
3)(2R,3R,4S)―2―(6―(3―ブロモベンジルアミノ)―2―クロロ―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
4)(2R,3R,4S)―2―(2―クロロ―6―(3―ヨードベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
5)(2R,3R,4S)―2―(2―クロロ―6―(2―クロロベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
6)(2R,3R,4S)―2―(2―クロロ―6―(5―クロロ―2―メトキシベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
7)(2R,3R,4S)―2―(2―クロロ―6―(2―メトキシベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
8)(2R,3R,4S)―2―(2―クロロ―6―(ナフタレン―1―イルメチルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
9)3―((2―クロロ―9―((2R,3R,4S)―3,4―ジヒドロキシテトラヒドロチオフェン―2―イル)―9H―プリン―6―イルアミノ)メチル)安息香酸;
10)2―(2―クロロ―6―メチルアミノ―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
11)(2R,3R,4S)―2―(6―(3―フルオロベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
12)(2R,3R,4S)―2―(6―(3―クロロベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
13)(2R,3R,4S)―2―(6―(3―ブロモベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;及び
14)(2R,3R,4S)―2―(6―(3―ヨードベンジルアミノ)―9H―プリン―9―イル)テトラヒドロチオフェン―3,4―ジオール;
から成る群から選択される、請求項1又は3に記載のアデノシン誘導体またはその薬学的に許容可能な塩。 - 下記の反応式1に示されるように、
(1)化学式2の出発物質を、触媒としてルイス酸の存在下でシリル化されたプリン化合物と反応させて、β―アノマーを与える工程、
(2)工程(1)で得た化学式3の化合物に塩酸を添加して、化学式4のジオール化合物4を与える工程、及び
(3)工程(2)で得た化学式4のジオール化合物を、触媒として塩基の存在下でアミン化合物と反応させて、化学式1のアデノシン化合物を与える工程、
を包含する、請求項1乃至4のいずれかに記載のアデノシン誘導体またはその薬学的に許容可能な塩の合成方法。
- 工程(1)は、ジクロロエタン、クロロホルム、アセトニトリル及びジクロロメタンからなる群から選択される溶媒中で行われる、請求項5に記載の方法。
- 工程(1)のルイス酸は、トリメチルシリルトリフルオロメタンスルホネートである、請求項5に記載の方法。
- 工程(1)のシリル化されたプリン化合物は、化学式5のプリン化合物を、触媒として硫酸アンモニウムの存在下でヘキサメチルジシラザンと反応させることにより調製される、請求項5に記載の方法。
- 工程(3)の触媒は、トリエチルアミン、ピリジン、N,N―ジメチルアミノピリジン及び1,4―ジオキサンからなる群から選択される、請求項5に記載の方法。
- 化学式2の出発物質は、下記の反応式2に示されるように、
(a1)化学式6のD―マンノースを、触媒として酸の存在下で2,2―ジメトキシプロパンと反応させて、化学式7のジアセトニド化合物を与える工程、
(a2)工程(a1)で得た化学式7の化合物を、還元剤の存在下で開環させて、化学式8のジオール化合物を与える工程、
(a3)工程(a2)で得た化学式8の化合物をメシル化して、化学式9のジメシル化合物にする工程、
(a4)工程(a3)で得た化学式9の化合物を環化させて、化学式10のチオ糖化合物にする工程、
(a5)工程(a4)で得た化学式10の化合物を選択的に加水分解させて、化学式11のジオール化合物にする工程、
(a6)工程(a5)で得た化学式11の化合物を、触媒の存在下で化学式2aのアセテート化合物に変換する工程、
により調製される請求項5に記載の方法。
- 工程(a1)の触媒は、濃硫酸、塩酸ガス及びp―トルエンスルホン酸からなる群から選択される、請求項10に記載の方法。
- 工程(a2)の還元剤は、水素化ホウ素ナトリウム、水素化アルミニウムリチウム及び亜硫酸ナトリウムからなる群から選択される、請求項10に記載の方法。
- 工程(a3)が、メチル化剤として、メタンスルホニルクロライドの存在下で行われる、請求項10に記載の方法。
- 工程(a4)が、エチルエーテル、石油エーテル、ジクロロメタン、テトラヒドロフラン及びN,N―ジメチルホルムアミドからなる群から選択される溶媒中で行われる、請求項10に記載の方法。
- 工程(a5)が、酢酸、硫酸、塩酸及びp―トルエンスルホン酸からなる群から選択される酸の存在下で行われる、請求項10に記載の方法。
- 請求項1の化学式1で表示されるアデノシン誘導体またはその薬学的に許容可能な塩を含む炎症性疾患の予防及び治療用薬学的組成物。
- 前記炎症性疾患は、変質性炎症、滲出性炎症、化膿性炎症、出血性炎症、増殖性炎症及びこれらの組み合わせからなる群から選択される、請求項16に記載の薬学的組成物。
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/KR2007/001131 WO2008108508A1 (en) | 2007-03-07 | 2007-03-07 | Adenosine derivatives, method for the synthesis thereof, and the pharmaceutical compositions for the prevention and treatment of the inflammatory diseases containing the same as an active ingredient |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010520271A JP2010520271A (ja) | 2010-06-10 |
| JP5306238B2 true JP5306238B2 (ja) | 2013-10-02 |
Family
ID=39738369
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009552570A Active JP5306238B2 (ja) | 2007-03-07 | 2007-03-07 | アデノシン誘導体、その合成方法、並びにこれを有効成分として含有する炎症性疾患の予防及び治療用医薬組成物 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9018371B2 (ja) |
| EP (1) | EP2142549B1 (ja) |
| JP (1) | JP5306238B2 (ja) |
| CN (1) | CN101801970B (ja) |
| AU (1) | AU2007348394B2 (ja) |
| CA (1) | CA2680179C (ja) |
| WO (1) | WO2008108508A1 (ja) |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101207755B1 (ko) * | 2010-02-02 | 2012-12-03 | 이화여자대학교 산학협력단 | 염증성 질환의 예방 및 치료용 약제학적 조성물 |
| JP2012196333A (ja) * | 2011-03-22 | 2012-10-18 | Meijo Univ | 高齢者の居眠り運転防止の方法および装置 |
| KR101323413B1 (ko) | 2012-07-26 | 2013-10-29 | 이화여자대학교 산학협력단 | (2r,3r,4s)-2-(2-클로로-6-(3-아이도벤질아미노)-9h-퓨린-9-일)-테트라하이드로싸이오펜-3,4-다이올 및 이의 유도체를 포함하는 만성신장질환 예방 또는 치료용 약학적 조성물 |
| CN108463463A (zh) | 2016-01-14 | 2018-08-28 | 韩德株式会社 | 拮抗a3腺苷受体的化合物、其制备方法、及其医学用途 |
| KR101709307B1 (ko) * | 2016-10-31 | 2017-02-23 | 퓨쳐메디신 주식회사 | 아데노신 유도체를 포함하는 비알콜성 지방간염, 간섬유증 및 간경변증 예방 및 치료용 약학적 조성물 |
| KR101805400B1 (ko) * | 2017-03-21 | 2017-12-07 | 퓨쳐메디신 주식회사 | 아데노신 유도체를 포함하는 녹내장 예방 및 치료용 약학적 조성물 |
| KR101820909B1 (ko) * | 2017-07-07 | 2018-01-23 | 퓨쳐메디신 주식회사 | 아데노신 유도체를 포함하는 만성신장질환 예방 및 치료용 약학적 조성물 |
| IL254535A0 (en) * | 2017-09-17 | 2017-11-30 | Can Fite Biopharma Ltd | Adenosine a3 receptor ligand for use in the management of cytokine release syndrome |
| KR102007640B1 (ko) * | 2017-11-29 | 2019-08-07 | 퓨쳐메디신 주식회사 | 아데노신 유도체를 포함하는 망막 질환 또는 시신경 질환 예방 및 치료용 약학적 조성물 |
| CN107903274A (zh) * | 2017-12-28 | 2018-04-13 | 窦玉玲 | 一种胺类化合物及其在抗肿瘤药物中的应用 |
| CN111943938A (zh) * | 2019-05-17 | 2020-11-17 | 上海再极医药科技有限公司 | 一种a2a腺苷受体拮抗剂的合成方法 |
| EP4186514A1 (en) * | 2021-11-24 | 2023-05-31 | Future Medicine Co., Ltd. | Pharmaceutical composition for preventing or treating cholangitis or liver disease caused by cholangitis comprising adenosine derivative |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5688774A (en) * | 1993-07-13 | 1997-11-18 | The United States Of America As Represented By The Department Of Health And Human Services | A3 adenosine receptor agonists |
| US5688744A (en) * | 1995-11-03 | 1997-11-18 | Rohm And Haas Company | Antimicrobial compounds with quick speed of kill |
| AU2003301589A1 (en) * | 2002-10-25 | 2004-05-13 | Ewha Womans University | Purine nucleosides |
-
2007
- 2007-03-07 US US12/530,086 patent/US9018371B2/en active Active
- 2007-03-07 CN CN200780052031XA patent/CN101801970B/zh active Active
- 2007-03-07 JP JP2009552570A patent/JP5306238B2/ja active Active
- 2007-03-07 AU AU2007348394A patent/AU2007348394B2/en active Active
- 2007-03-07 EP EP07715531.5A patent/EP2142549B1/en active Active
- 2007-03-07 CA CA2680179A patent/CA2680179C/en active Active
- 2007-03-07 WO PCT/KR2007/001131 patent/WO2008108508A1/en active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008108508A1 (en) | 2008-09-12 |
| CN101801970A (zh) | 2010-08-11 |
| CA2680179C (en) | 2016-02-16 |
| EP2142549B1 (en) | 2013-07-10 |
| CN101801970B (zh) | 2013-11-06 |
| US20100137577A1 (en) | 2010-06-03 |
| US9018371B2 (en) | 2015-04-28 |
| AU2007348394B2 (en) | 2013-08-15 |
| JP2010520271A (ja) | 2010-06-10 |
| CA2680179A1 (en) | 2008-09-12 |
| AU2007348394A1 (en) | 2008-09-12 |
| EP2142549A4 (en) | 2011-07-27 |
| EP2142549A1 (en) | 2010-01-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5306238B2 (ja) | アデノシン誘導体、その合成方法、並びにこれを有効成分として含有する炎症性疾患の予防及び治療用医薬組成物 | |
| JP7175525B2 (ja) | アデノシン誘導体を含む非アルコール性脂肪肝炎、肝線維症及び肝硬変症の予防及び治療用薬学的組成物 | |
| US6407076B1 (en) | Adenosine analogues and related method of treatment | |
| KR101396092B1 (ko) | 아데노신 유도체, 이의 제조방법 및 이를 유효성분으로 함유하는 염증성 질환의 예방 및 치료용 약학적 조성물 | |
| US7199127B2 (en) | Purine nucleosides | |
| US12083140B2 (en) | Pharmaceutical composition for preventing and treating glaucoma, containing adenosine derivative | |
| MXPA00004428A (en) | Adensine a1 receptor agonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20100303 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120904 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20121203 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20121218 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130327 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20130507 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130528 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130625 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 5306238 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313115 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |