JP5265915B2 - プロテアソームを阻害するための(−)−エピガロカテキンガラート誘導体 - Google Patents
プロテアソームを阻害するための(−)−エピガロカテキンガラート誘導体 Download PDFInfo
- Publication number
- JP5265915B2 JP5265915B2 JP2007526180A JP2007526180A JP5265915B2 JP 5265915 B2 JP5265915 B2 JP 5265915B2 JP 2007526180 A JP2007526180 A JP 2007526180A JP 2007526180 A JP2007526180 A JP 2007526180A JP 5265915 B2 JP5265915 B2 JP 5265915B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- acetate
- cells
- egcg
- proteasome
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 Cc(cc(C1Oc2c(*)c(*)cc(*)c2CC1O*(C1=CC(OC*)=C(*)C(*)C1)=O)cc1*)c1O Chemical compound Cc(cc(C1Oc2c(*)c(*)cc(*)c2CC1O*(C1=CC(OC*)=C(*)C(*)C1)=O)cc1*)c1O 0.000 description 1
Images































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/58—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4
- C07D311/60—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4 with aryl radicals attached in position 2
- C07D311/62—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4 with aryl radicals attached in position 2 with oxygen atoms directly attached in position 3, e.g. anthocyanidins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pyrane Compounds (AREA)
Description

・R11、R12、R13、R21、R22、R2、R3およびR4は各々独立に、−HおよびC1〜C10アシルオキシル基からなる群から選択され、
・R5は−H、C1〜C10−アルキル、C2〜C10−アルケニル、C2〜C10−アルキニル、C3〜C7−シクロアルキル、フェニル、ベンジルおよびC3〜C7−シクロアルケニルからなる群から選択され、最後に記載した7種の基は各々、任意の組合せの1〜6個のハロゲン原子で置換されていることができ、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個はC1〜C10アシルオキシル基であり、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個は−Hである]。
場合によっては、R11が−Hであり、R12、R13、R21、R22、R2、R3およびR4が各々アセテートまたはベンゾエート基である。
場合によっては、R11およびR13が各々−Hであり、R12、R21、R22、R2、R3およびR4が各々アセテートまたはベンゾエート基である。
本発明の上記化合物の一実施形態では、R5は−Hであり、R11、R12、R13、R21およびR22は各々アセテート基である。この特定の実施形態はまた、以下の3つの変形も提供する:
・R2がアセテート基であり、R3およびR4が各々−Hであるか、
・R3がアセテート基であり、R2およびR4が各々−Hであるか、または
・R2およびR4が各々アセテート基であり、R3が−Hである。
・R11、R12、R13、R21、R22、R2、R3およびR4が各々独立に、−HおよびC1〜C10アシルオキシル基からなる群から選択され、
・R5は−H、C1〜C10−アルキル、C2〜C10−アルケニル、C2〜C10−アルキニル、C3〜C7−シクロアルキル、フェニル、ベンジルおよびC3〜C7−シクロアルケニルからなる群から選択され、最後に記載した7種の基は、任意の組合せの1〜6個のハロゲン原子で置換されていることができ、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個はC1〜C10アシルオキシル基である]。
・R11、R12、R13、R21、R22、R2、R3およびR4が各々独立に、−HおよびC1〜C10アシルオキシル基からなる群から選択され、
・R5は−H、C1〜C10−アルキル、C2〜C10−アルケニル、C2〜C10−アルキニル、C3〜C7−シクロアルキル、フェニル、ベンジルおよびC3〜C7−シクロアルケニルからなる群から選択され、最後に記載した7種の基は、任意の組合せの1〜6個のハロゲン原子で置換されていることができ、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個はC1〜C10アシルオキシル基である]。
・R1は−Hであり、
・R2、R3およびR4は各々独立に、−Hおよび−OHからなる群から選択され、
・R2、R3およびR4のうち少なくとも1個は−Hである]。
・R1は−Hであり、
・R2、R3およびR4は各々独立に、−Hおよび−OHからなる群から選択され、
・R2=R3=R4である場合には、R2は−OHではない]。
・R1は−Hであり、
・R2、R3およびR4は各々独立に、−Hおよび−OHからなる群から選択され、
・R2=R3=R4である場合には、R2は−OHではない]。
本発明の目的、特徴および態様は、以下の説明において開示されるか、または以下の説明から明らかである。当業者には当然のことながら、本議論は、例示的実施形態の説明に過ぎず、本発明のより広い態様を制限しようとするものではなく、より広い態様は例示的構成において具現される。
・R1は−Hであり、
・R2、R3およびR4は各々独立に、−Hおよび−OHからなる群から選択される]。
エステル体の本発明の(−)−EGCGの誘導体の一般式は、次式を有する
・R11、R12、R13、R21、R22、R2、R3およびR4は各々独立に、−HおよびC1〜C10アシルオキシル基からなる群から選択され、
・R5は−H、C1〜C10−アルキル、C2〜C10−アルケニル、C2〜C10−アルキニル、C3〜C7−シクロアルキル、フェニル、ベンジルおよびC3〜C7−シクロアルケニルからなる群から選択され、最後に記載した7種の基は各々、任意の組合せの1〜6個のハロゲン原子で置換されていることができ、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個はC1〜C10アシルオキシル基であり、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個は−Hである]。
C1〜C6アシル:構造−(CO)−Rを有する(式中、Rは、水素または1〜5個の炭素原子を有する直鎖もしくは分岐アルキル基、例えば、メチル、エチル、プロピル、1−メチルエチル、ブチル、1−メチルプロピル、2−メチルプロピル、1,1−ジメチルエチル、ペンチル、2−メチルブチルである)。アルキル基Rは、部分的または完全にハロゲン化されていてもよい。用語「部分的または完全にハロゲン化された」とは、このように特徴づけられた基では、クロロメチル、ジクロロメチル、トリクロロメチル、フルオロメチル、ジフルオロメチル、トリフルオロメチル、クロロフルオロメチル、ジクロロフルオロメチル、クロロジフルオロメチル、1−フルオロエチル、2−フルオロエチル、2,2−ジフルオロエチル、2,2,2−トリフルオロエチル、2−クロロ−2−フルオロエチル、2−クロロ−2,2−ジフルオロエチル、2,2−ジクロロ−2−フルオロエチル、2,2,2−トリクロロエチルおよびペンタフルオロエチルのように、水素原子が、同一または異なるハロゲン原子で部分的または完全に置換されていてもよいことを表すものとする。
試薬
ウシ胎児血清は、Tissue Culture Biologicals(Tulare,C A)から購入した。ペニシリン−ストレプトマイシン−1−グルタクスニン(Glutaxnine)の混合物、RPMIおよびDMEMはInvitrogen(Carlsbad,CA)製である。ジメチルスルホキシド(DMSO)、N−アセチル−L−システイン(NAC)、ヘキスト33342、3−((4,5)−ジメチルチアゾール−2−イル)−2,5−ジフェニルテオリウムブロミド(diphenylteolium bromide)(MTT)、ウシ血清アルブミン(BSA)および(−)−EGCGは、Sigma(St.Louis,MO)から購入した。Suc−Leu−Leu−Val−Tyr−AMC(プロテオソームのキモトリプシン様活性用)は、Biomol(Plymouth Meeting,PA)から入手した。ウサギ由来精製20sプロテアソームは、Boston Biochem(Cambridge,MA)から入手した。Amplex Red H202アッセイキットは、Molecular Probes(Eugene,OR)から購入した。Bax(H280)およびユビキチン(P4D1)に対するモノクローナル抗体、ならびにIKB−a(C15)およびアクチン(C11)に対するポリクローナル抗体、ならびに抗ヤギ、抗ウサギおよび抗マウスIgG−ホースラディッシュペルオキシダーゼは、Santa Cruz Biotechnology(Santa Cruz,CA)から購入した。p27に対するモノクローナル抗体(554069)は、BD Biosciences(San Diego,CA)から購入した。DAPIを含むVectashield Mounting Mediumは、Vector Laboratories Inc.(Burlingame,CA)から購入した。PARP切断部位に対して特異的であり、FITCが結合しているポリクローナル抗体は、Biosource(Camarillo,CA)から入手した。CaspACE FITC−VAD−FMKマーカーはPromega(Madison,WI)から購入した。
1は文献の手順に従って調製したが(Kohri,T.、Nanjo,F.、Suzuki,M.、Seto,R.、Matsumoto,N.、Yamakawa,M.、Hojo,H.、Hara,Y.、Desai,D.、Amin,S.、Conaway,C.C.、Chung,F.L.J Agric Food Chem 2001、49、1042〜8頁)、1の合成をここに示す。1の合成には、出発材料として市販の(−)−EGCGを用いた。(−)−EGCGを無水酢酸およびピリジンで一晩処理することで、目的生成物1が収率82%で得られた(図1)。1の構造は1Hおよび13CNMR、LRMSおよびHRMSによって確認した。
この化合物は文献の手順に従って合成した(Li,L.H.、Chan,T.H.Org.Lett.2001、3、739〜741頁)。
CH2Cl2(10mL)中、3−(ベンジルオキシ)安息香酸(0.12g、0.53mmol)の溶液に、一定量の(COCl)2(0.68mL)を加えた。この混合物を2時間還流した。その後、過剰の(COCl)2と溶媒を蒸留によって除去し、得られた残渣を真空下で一晩乾燥させた。残渣をCH2Cl2(5mL)に再溶解し、0℃でCH2Cl2(10mL)中、5(0.20g、0.26mmol)およびDMAP(0.08g、0.64mmol)の溶液に加えた。次いで、この混合物を室温で一晩撹拌した。飽和NaHCO3を加えた。有機層を分離し、水層を酢酸エチルで抽出した。有機層を合わせ、乾燥させ(Na2SO4)、蒸発させた。残渣を、カラムクロマトグラフィー(ヘキサン:酢酸エチル4:1)によって精製すると、化合物2bが白色固体として得られた(0.22g、88%)。
THF/MeOH(28mL/28mL)およびPd(OH)2(0.19g、炭素上20%)中、2b(0.23g、0.24mmol)の懸濁液を、H2雰囲気下に置いた。得られた混合物を、反応が完了したことをtlcが示すまで室温で撹拌した。次いで、反応混合物を、綿を通して濾過して触媒を除去した。蒸発させた後、残渣をカラムクロマトグラフィー(酢酸エチル:CH2Cl22:1)によって精製すると、生成物2が白色固体として得られた(80mg、79%)。
THF/MeOH(12mL/12mL)およびPd(OH)2(0.08g、炭素上20%)中、2b(0.1g、0.1mmol)の懸濁液を、H2雰囲気下に置いた。得られた混合物を、反応が完了したことをtlcが示すまで室温で撹拌した。次いで、反応混合物を、綿を通して濾過して触媒を除去した。濾液を蒸発させると、脱ベンジル化化合物(2)が得られ、これを精製せずに直ちに次のステップに用いた。得られた脱ベンジル化化合物をピリジン(4mL)および無水酢酸(2mL)に溶解した。得られた混合物を室温で一晩撹拌した。その後、無水酢酸およびピリジンを真空除去した。得られた残渣を20mLのCH2Cl2中に回収し、この溶液を5×H2O5mLおよびブライン5mLで洗浄し、Na2SO4上で乾燥させ、蒸発させた。粗生成物をカラムクロマトグラフィー(ヘキサン:酢酸エチル1:1)によって精製すると、化合物2aが白色粉末として得られた(0.061g、85%)。
標題化合物を、5(0.08g、0.1mmol)および4−(ベンジルオキシ)安息香酸(0.049g、0.22mmol)を用い、2bについて記載した方法と同様に調製すると、3bが白色固体として得られた(0.087g、90%)。
標題化合物を、3b(0.24g、0.25mmol)を用い、2について記載した方法と同様に調製すると、3が白色固体として得られた(79mg、75%)。
標題化合物を、3b(0.15g、0.16mmol)を用い、2aについて記載した方法と同様に調製すると、3aが白色固体として得られた(92.6mg、88%)。
標題化合物を、5(0.3g、0.4mmol)および3,5−ビス(ベンジルオキシ)安息香酸(0.27g、0.81mmol)を用い、2bについて記載した方法と同様に調製すると、4bが白色固体として得られた(0.36g、85%)。
標題化合物を、4b(0.17g、0.16mmol)を用い、2について記載した方法と同様に調製すると、4が白色固体として得られた(50mg、71%)。
標題化合物を、4b(0.15g、0.14mmol)を用い、2aについて記載した方法と同様に調製すると、4aが白色固体として得られた(72mg、70%)。
概要
民間の供給業者から購入した出発材料および試薬を、さらに精製することなく用いた。(2R,3S)トランスおよび(2R,3R)−シス−5,7−ビス(ベンジルオキシ)−2−(4−ベンジルオキシフェニル)クロマン−3−オル、(2R,3S)トランスおよび(2R,3R)−シス−5,7−ビス(ベンジルオキシ)−2−[3,4−ビス(ベンジルオキシ)−フェニル]クロマン−3−オル、(2R,3R)−シス−5,7−ビス(ヒドロキシル)−2−(4−ヒドロキシフェニル)クロマン−3−イル3,4,5−トリヒドロキシベンゾエートの調製には文献の手順を用いた(Sheng Biao Wan、Tak Hang Chan.Tetrahedron,2004、60、8207頁)。
N2雰囲気下、乾燥CH2Cl2(10mL)および1滴のDMF中、4−ベンジルオキシ安息香酸(140mg、0.61mmol)の溶液を、塩化オキサリル(1mL)とともに3時間還流した。過剰の塩化オキサリルおよび溶媒を、蒸留によって除去し、残渣を真空下で3時間乾燥させ、CH2Cl2(2mL)に溶解した。0℃で、CH2Cl2(15mL)中、(2R,3S)−トランス−5,7−ビス(ベンジルオキシ)−2−[3,4−ビス(ベンジルオキシ)フェニル]クロマン−3−オル(195mg、0.3mmol)およびDMAP(75mg、0.62mmol)の溶液に、この溶液を滴下した。混合物を一晩撹拌し、次いで、飽和NaHCO3水溶液を加えた。有機層を分離し、CH2Cl2で水層を抽出した。有機相を合わせ、乾燥させ(MgSO4)、蒸発させた。残渣をシリカゲルでのフラッシュクロマトグラフィー(EtOAc/n−ヘキサン=1/4 v/v)によって精製すると、目的化合物が得られた(220mg、85.0%)。EtOAcおよびn−ヘキサン中で再結晶化させると、白色粉末が得られた:融点148〜150℃、[α]D=+18.3(c=1、CHCl3)、
102の調製手順に続いて、4−ベンジルオキシ安息香酸を用いて、(2R,3R)−シス−5,7−ビス(ベンジルオキシ)−2−[3,4−ビス(ベンジルオキシ)フェニル]クロマン−3−オルをエステル化すると、104が得られ、収率86%であった。融点149〜151℃、[α]D=−3.1(c=1.5、CHCl3)、
102の調製手順に続いて、4−ベンジルオキシ安息香酸を用いて、(2R,3S)−5,7−ビス(ベンジルオキシ)−2−(4−ベンジルオキシフェニル)クロマン−3−オルをエステル化すると、110が得られ、収率86%であった。融点117〜119℃、[α]D=+24.5(c=1.2、CHCl3)、
102の調製手順に続いて、4−ベンジルオキシ安息香酸を用いて、(2R,3R)−5,7−ビス(ベンジルオキシ)−2−(4−ベンジルオキシフェニル)クロマン−3−オルをエステル化すると、111が得られ、収率88%であった。融点129〜131℃、[α]D=−51.8(c=3.9、CHCl3)、
H2雰囲気下、THF/MeOH(1:1 v/v、20mL)の溶媒混合物中、102(200mg、0.23mmol)の溶液に、Pd(OH)2/C(20%、100mg)を加えた。得られた反応混合物をH2下、室温で6時間撹拌し、反応が完了したことはTLCによって示された。反応混合物を濾過して触媒を除去した。濾液を蒸発させ、残渣をシリカゲルでのフラッシュクロマトグラフ(10%MeOH/CH2Cl2、次いで、20%MeOH/CH2Cl2)によって迅速に精製すると、11が得られた(82mg、収率87%):融点220〜222℃(分解)、[α]D=+87.2(c=2.0、EtOH)、
11の調製手順に続いて、104を水素分解すると、12が得られ、収率は85%であった。融点241〜243℃(分解)、[α]D=−145.2(c=0.5、EtOH)、(Lit−144.4、c=1、Me2CO中)、4
11の調製手順に続いて、110を水素分解すると、112が得られ、収率は90%であった。融点253〜255℃(分解)、[α]D=+45.9(c=3.5、EtOH)、
11の調製手順に続いて、111を水素分解すると、10が得られ、収率は89%であった。融点214〜216℃(分解)、[α]D=−116.1(c=2.0、EtOH)、
N2雰囲気下、0℃で、ピリジン(1ml)中、(+)−(2R,3S)−5,7−ジヒドロキシ−2−(3,4−ジヒドロキシフェニル)クロマン−3−イル4−ヒドロキシベンゾエート11(20mg、0.048mmol)の溶液に、無水酢酸(0.2ml)を滴下した。反応混合物を室温で一晩撹拌した。過剰のピリジンを真空下で蒸留した。残渣をシリカゲルでのフラッシュクロマトグラフ(EtOAc/n−ヘキサン、v/vで1/1)によって精製すると、22*が得られた(34mg、収率95%)。融点:融点149〜151℃、[α]D=+42.5(c=1.2、CHCl3)、
22*の調製手順に続いて、12をアセチル化すると、23*が得られ、収率は96%であった。融点91〜93℃、[α]D=−26.5(c=0.5、CHCl3)、
22*の調製手順に続いて、(+)−(2R,3S)−5,7−ジヒドロキシ−2−(3,4−ジヒドロキシフェニル)クロマン−3−イル3,4,5−トリヒドロキシベンゾエートをアセチル化すると(Sheng Biao Wan、Di Chen、Q.Ping Dou and Tak Hang Chan.Bioorganic&Medicinal Chemistry、2004、12、3521頁)、24*が得られ、収率は95%であった。融点140〜142℃、[α]D=+35.3(c=3.0、CHCl3)、
22*の調製手順に続いて、(−)−(2R,3R)−5,7−ジヒドロキシ−2−(3,4−ジヒドロキシフェニル)クロマン−3−イル3,4,5−トリヒドロキシベンゾエートをアセチル化すると(Sheng Biao Wan、Di Chen、Q.Ping Dou and Tak Hang Chan.Bioorganic&Medicinal Chemistry、2004、12、3521頁)、25*が得られ、収率は93%であった。融点105〜107℃、[α]D=−14.5(c=1.2、CHCl3)、
22*の調製手順に続いて、112をアセチル化すると、114が得られ、収率は91%であった。融点167〜169℃、[α]D=+3.2(c=1.0、CHCl3)、
22*の調製手順に続いて、10をアセチル化すると、21*が得られ、収率は89%であった。融点144〜145℃、[α]D=−30.7(c=2.5、CHCl3)、
22*の調製手順に続いて、(+)−(2R,3S)−5,7−ジヒドロキシ−2−(4−ヒドロキシフェニル)クロマン−3−イル3,4,5−トリヒドロキシベンゾエートをアセチル化すると(Sheng Biao Wan、Tak Hang Chan.Tetrahedron、2004、60、8207頁)、16*が得られ、収率は93%であった。融点96〜98℃、[α]D=+17.7(c=1.0、CHCl3)、
22*の調製手順に続いて、(−)−(2R,3R)−5,7−ジヒドロキシ−2−(4−ヒドロキシフェニル)クロマン−3−イル3,4,5−トリヒドロキシベンゾエートをアセチル化すると(Sheng Biao Wan、Tak Hang Chan.Tetrahedron、2004、60、8207頁)、19*が得られ、収率は91%であった。融点152〜154℃、[α]D=−35.5(c=2.5、CHCl3)、
文献の手順(Li,L.、Chan,T.H.Org.Lett.2001、5、739頁)に従って、シンナミルアルコールを、3,5−ジベンジルオキシフェノールと反応させると、(#a)が白色固体として得られた(収率62%)、融点76〜78℃、
出発材料として(#a)およびジヒドロキシル化試薬としてAD−ミックス−αを用いるが、文献の手順(Li,L.、Chan,T.H.Org.Lett.2001、5、739頁)に従って、(+)−#bが白色固体として得られた(収率47%)、融点121〜123℃、[α]D=+4.7(c=0.6、CHCl3)、
出発材料として(+)−#bを用いるが、文献の手順(Li,L.、Chan,T.H.Org.Lett.2001、5、739頁)に従って、(+)−#cが白色固体として得られた(収率47%)、融点60〜62℃、[α]D=+0.9(c=1.0、酢酸エチル)、
出発材料として(+)−#cを用いるが、文献の手順(Li,L.、Chan,T.H.Org.Lett.2001、5、739頁)に従って、化合物(+)−15が得られた(収率90%):融点258〜260℃(分解)、[α]D=+13.9(c=3.5、エタノール)、
0℃で、乾燥DMF(120mL)中、水素化ナトリウム(鉱油中60%分散物、2,4g、100mmol)の撹拌懸濁液に、エタンチオール(10g、216mmol)を滴下した。1時間後、10のバッチに1,3,5−トリベンジルオキシベンゼン(24g、60mmol)を加え、この混合物を150℃で1.5時間加熱した。反応物を冷却した後、水(500mL)を加え、混合物をEtOAcで抽出した。合わせた有機層を乾燥させ(MgSO4)、蒸発させた。残渣をシリカゲルでのフラッシュクロマトグラフィー(ベンゼン)によって精製すると、四塩化炭素から再結晶化させた後に3,5−ジベンジルオキシフェノールが白色固体として得られ(11%)、EtOAcおよびヘキサンから再結晶化させた後に生成物#dが白色固体として得られた(56%)。化合物#dは融点107〜109℃で同定された、
室温、N2雰囲気下、乾燥CH2Cl2(80mL)中、2−ベンジル−3,5−ビス(ベンジルオキシ)フェノール(3.96g、10mmol)および(E)−3,4−ビス(ベンジルオキシ)シンナミルアルコール(3.46g、10mmol)の撹拌混合物に、25%H2SO4/SiO2(1.6g、4mmol)をひとまとめにして加えた。得られた混合物を室温で一晩撹拌した。濾過し、蒸発させた後、残渣をシリカゲルでのカラムクロマトグラフィー(EtOAc/n−ヘキサン=1/7 v/v)によって精製し、EtOAcおよびn−ヘキサンから再結晶化すると、白色固体が得られた(3.4g、収率46.0%):融点93〜95℃、
乾燥DMF(30mL)にプロペン(#e)(3.4g、4.6mmol)を溶解し、この溶液にイミダゾール(1.03g、15.2mmol)およびTBSC1(1.2g、7.8mmol)を逐次加えた。得られた混合物を室温で3日間撹拌し、次いで、飽和Na2CO3溶液を加えて反応をクエンチした。この混合物をEtOAcで抽出した。有機層を合わせ、乾燥させ(MgSO4)、蒸発させた。残渣をシリカゲルでのフラッシュクロマトグラフ(n−ヘキサンおよびEtOAc=9/1 v/v)によって精製すると、[3,5−ビス(ベンジルオキシ)−6−ベンジル−2−[3−[3,4−ビス(ベンジルオキシ)フェニル]アリル]フェノキシ]−t−ブチルジメチルシランが得られた。この物質をさらなる精製を行わずに次のステップに用いた。
1,2−ジクロロエタン(50mL)中、(−)−#f(2.4g、3.1mmol)の懸濁液に、オルトギ酸トリエチル(1mL)、続いてPPTS(450mg、1.8mmol)を加えた。この混合物を、固体が溶解するまで室温で20分間撹拌した。次いで、この混合物を、反応が完了したことがTLCによって示されるまで55℃で5時間加熱した。溶媒を蒸発させた後、残渣をDME(30mL)およびMeOH(30ml)に溶解し、K2CO3(450mg)を加えた。この混合物を室温で一晩撹拌した。溶媒を蒸発させた後、残渣をシリカゲルでのフラッシュクロマトグラフィー(EtOAc/ヘキサン、1/3 v/v)によって精製すると、目的生成物が白色固体として得られた(1.8g、収率77%):融点145〜146℃、[α]D=−20.1(c=1.3、CHCl3)、
N2雰囲気下、CH2Cl2(30mL)中、(−)−#g(900mg、1.2mmol)の撹拌溶液に、デスーマーチン ペルヨージナン(6.3mL、CH2Cl2中15%g/mL、2.2mmol)をひとまとめにして加えた。この混合物を、出発材料がないことがTLCによって示されるまで、室温で約2時間撹拌した。続いて、飽和NaHCO3溶液(15mL)および10%Na2S2O3水溶液(15mL)を加えて反応をクエンチした。有機層を分離し、CH2Cl2で水層を抽出した。合わせた有機相を乾燥させ(MgSO4)、蒸発させた。残渣をシリカゲルでのフラッシュクロマトグラフィー(ベンゼン)によって精製し、次いで、CHCl3およびエーテル中で再結晶化させると、目的化合物が得られた(770mg、86%):融点143〜145℃、[α]D=−17.1(c=1.1、CHCl3)、
N2雰囲気下、乾燥THF(15mL)にケトン(−)−#h(700mg、0.95mmol)を溶解し、この溶液を−78℃に冷却した。次いで、L−セレクトリド(1.5mL、THF中、1M溶液、1.5mmol)を滴下した。得られた溶液を−78℃で8時間撹拌した。反応が完了したことがTLCによって示された時点で、飽和NaHCO3水溶液(10mL)を加えて反応をクエンチした。有機層を分離し、水層をEtOAcで抽出した。合わせた有機相を乾燥させ(MgSO4)、蒸発させた。残渣をシリカゲルでのフラッシュクロマトグラフィー(5%EtOAC/ベンゼン)によって精製し、次いで、エタノールおよびEtOACを用いて再結晶化させると、目的生成物(630mg、90%)が白色固体として得られた:融点129〜131℃、[α]D=+5.3(c=1.2、CHCl3)、
N2雰囲気下、乾燥CH2Cl2(10mL)および1滴のDMF中、3,4,5−トリス(ベンジルオキシ)安息香酸(170mg、0.39mmol)の溶液を塩化オキサリル(1mL)とともに3時間還流した。過剰の塩化オキサリルおよび溶媒を蒸留によって除去し、残渣を真空下で3時間乾燥させ、CH2Cl2(2mL)に溶解した。0℃で、CH2Cl2(15mL)中、(+)−#i(150mg、0.20mmol)およびDMAP(75mg、0.62mmol)の溶液に、この溶液を滴下した。この混合物を室温で一晩撹拌し、次いで、飽和NaHCO3水溶液を加えた。有機層を分離し、水層をCH2Cl2で抽出した。有機相を合わせ、乾燥させ(MgSO4)、蒸発させた。残渣を、シリカゲルでのフラッシュクロマトグラフィー(5%EtOAc/ベンゼン)によって精製すると、目的化合物(215mg、91%)が得られた。CHCl3およびエーテル中で再結晶化させると白色粉末が得られた:融点52〜54℃、[α]D=+37.5(c=1.0、CHCl3)、
出発材料として(−)−#gを用いるが、(−)−#jの調製のために用いた手順に従って、(−)−(2S,3R)−5,7−ビス(ベンジルオキシ)−8−ベンジル−2−[3,4−ビス(ベンジル−オキシ)フェニル]−クロマン−3−イル3,4,5−トリス(ベンジルオキシ)ベンゾエートが白色固体として得られた(収率90%):融点103〜105℃、[α]D=−9.8(c=1.3、CHCl3)、
H2雰囲気下、THF/MeOH(1:1 v/v、20mL)の溶媒混合物中、(+)−#j(200mg、0.17mmol)の溶液に、Pd(OH)2/C(20%、400mg)を加えた。反応混合物を室温、H2下で6時間撹拌し、その時点で反応が完了したことがTLCによって示された。反応混合物を濾過して触媒を除去した。濾液を蒸発させ、残渣を、シリカゲルでのフラッシュクロマトグラフ(10%MeOH/CH2Cl2、次いで、20%MeOH/CH2Cl2)によって迅速に精製すると、(+)−8−ベンジルカテキンガラート((+)−#1)が得られた(82mg、収率90%):融点243〜245℃(分解)、[α]D=+123(c=1.8、アセトン)、
出発材料として(−)−#kを用いるが、(+)−#Iの調製のための手順に従って、(−)−8−ベンジルカテキンガラート((−)−16)が得られた(収率91%):融点239〜241℃(分解)、[α]D=−35(c=2.0、アセトン)、
(−)−EGCGまたは1(0.1mM)を、RPMI1640培養培地とともに37℃でインキュベートした。種々の時点で、15μLの培地を、C−18逆相カラム(CAPCELL PAK C18 UG 120,Shiseido Co.,Ltd.、4.6mm内径×250mm)を備えるHPLCに注入した。流速1mL/分、検出、UV280nm、(−)−EGCG については、時点は0、10、20、40、60、90、120分とし、移動相、20%アセトニトリル水溶液および0.01%TFA、プロドラッグ1については、時点は0、30、60、90、120分とし、移動相、50%アセトニトリル水溶液および0.01%TFA。
2×106個ジャーカットT細胞に溶解バッファー(pH5)(0.25mL)を加えた。これによって、細胞の細胞膜を破壊し、細胞質酵素を放出することができる。培地を酵素にとって最適pH値(pH7)に中和するPBS(0.75mL)を加えた。プロドラッグ1(0.25mM)を反応混合物中に加え、37℃でインキュベートした。上記で概説したように、種々の時点(0、30、60、90、120、150、180、210、240、300および360分)で反応混合物のアリコート(0.06mL)を採取し、濾過し、HPLCに注入し、分析した。
化合物1(35μM)を、ダルベッコの改変イーグル培地(DMEM)(ビタミンC1.67mg/mLを含有する1mL)とともに37℃でインキュベートした。種々の時点で、10μLの溶液を、C−18逆相カラムを備えるHPLCに注入した。流速1mL/分、検出、UV280nm、移動相、0〜8分(20%アセトニトリル水溶液および0.016%TFA)、8〜13分(20%アセトニトリル水溶液および0.016%TFA〜60%アセトニトリル水溶液および0.008%TFAへ変動)。
ヒトジャーカットT細胞およびLNCaP細胞を、10%(v/v)ウシ胎児血清、100U/mlペニシリンおよび100μg/mlストレプトマイシンを補給したRPMIで培養した。非形質転換ナチュラルキラー細胞(YT株)は、10%(v/v)ウシ胎児血清、100U/mlペニシリン、100μg/mlストレプトマイシン、1mM MEMピルビン酸ナトリウムおよび0.1mM MEM非必須アミノ酸溶液を含有するRPMI培地で増殖させた。ヒト乳癌MCF−7細胞、正常(WI−38)およびサルウイルスで形質転換した(VA−13)ヒト線維芽細胞は、10%(v/v)ウシ胎児血清、100U/mlペニシリン、100pg/mlストレプトマイシンを補給したダルベッコの改変イーグル培地で増殖させた。すべての細胞培養は5%CO2雰囲気中、37℃で維持した。
全細胞抽出物を、これまでに記載された通りに調製した(An B,Goldfarb RH、Siman R、Dou QP.Novel dipeptidyl proteasome inhibitors overcome Bcl−2 protective function and selectively accumulate the cyclin−dependent kinase inhibitor p27 and induce apoptosis in transformed,but not normal,human fibroblasts.Cell Death Differ 1998、5、1062〜75頁)。Bax、IKBa、p27、PAWおよびユビキチン化タンパク質発現の分析は、先に報告されたプロトコールに従い、モノクローナルまたはポリクローナル抗体を用いて実施した(An Bら)。
20sプロテアソームのキモトリプシン様活性の測定は、種々の濃度の、先に記載された天然および合成茶ポリフェノール(Nam Sら)とともにか、それらをともなわずに、0.5μgの精製ウサギ20sプロテアソームを40μM蛍光発生ペプチド基質、Suc−Leu−Leu−Val−Tyr−AMCとともにインキュベートすることによって実施した。
VA−13またはWI−38細胞を、24ウェルプレートで70〜80%コンフルエンシーまで増殖させ(2ml/ウェル)、続いて、25pM(−)−EGCG、2または2aで24時間処理した。次いで、40pM Suc−Leu−Leu−Val−Tyr−AMC基質を37℃で2.5時間加え、キモトリプシン様活性を上記の通り測定した。あるいは、細胞を25μMの各化合物で4時間または24時間処理し、回収し、溶解した。次いで、Suc−Leu−Leu−Val−Tyr−AMC(40μM)を、調製した細胞溶解物とともに2.5時間インキュベートし、キモトリプシン様活性を上記の通り測定した。
アポトーシス細胞の免疫染色を、FITCと結合している、切断されたポリ(ADP−リボース)ポリメラーゼ(PARP)を認識するポリクローナル抗体を添加することによって実施し、Axiovert25(Zeiss;Thornwood,NY)顕微鏡で可視化した。細胞は60mmディッシュで約80%コンフルエンシーに増殖させた。次いで、VA−13細胞をVP−16、2または2a(25μM)で24時間処理した。処理後、懸濁液と接着細胞の両方を回収し、PBS pH7.4中で2回洗浄した。以下に列挙したすべてのステップの間で細胞を洗浄し、すべての洗浄はPBSで1分間とした。次いで、氷冷70%エタノールで細胞を固定し、0.1%Triton−X−100で透過処理し、1%ウシ血清アルブミン(BSA)中、室温で30分間ブロッキングした。一次FITC結合p85/PARP抗体とのインキュベーションは、穏やかに撹拌しながら暗所、4℃で30分間とした。次いで、細胞懸濁液を、DAPIを含むVector Shield mouting mediumの存在下、ガラススライドに移した。画像はAxioVision4.1を用いて獲得し、Adobe Photoshop 6.0ソフトウェアを用いて調整した。細胞死は、同一視野中の細胞全数にわたってアポトーシス細胞数を計数することによって定量化した。データは2連の実験の平均±SDである。
トリパンブルーアッセイを用いて、示した天然および合成ポリフェノールで処理したジャーカットT細胞において細胞死を確認した。アポトーシス形態は、先に記載されるように、位相差顕微鏡を用いて評価した(Kazi A、Hill R、Long TE、Kuhn DJ、Twos E、Dou QP.Novel N−thiolated beta−lactam antibiotics selectively induce apoptosis in human tumor and transformed,but not normal or nontransformed,cells.Biochem Pharmacol 2004、67、365〜74頁;Kuhn DJ、Smith DM、Pross S、Whiteside TL、Dou QP.Overexpression of interleukin−2 receptor alpha in a human squamous cell carcinoma of the head and neck cell line is associated with increased proliferation,drug resistance,and transforming ability.J Cell Biochem 2003、89、824〜36頁)。
MTTを用いて、腫瘍細胞の全体的な増殖に対するポリフェノールの効果を調べた。ヒト胸部MCF−7細胞を96ウェルプレートにプレーティングし、70〜80%コンフルエンシーに増殖させ、続いて、24時間類似体を添加した。次いで、ウェルにPBS中、MTT(1mg/ml)を加え、37℃で4時間インキュベートし、代謝的に活性な細胞によってテトラゾリウム塩を完全に切断させた。次いで、MTTを除去し、100μlのDMSOを加え、560nmでマルチラベルプレートリーダー(Victor3;Perkin Elmer)を用いて比色分析を実施した。プロットした吸光度値は、3連の実験から得た平均である。
先に記載されたように(kazi Aら)、細胞の形質転換活性を調べるために、LNCaP細胞(2×104個)を、(−)−EGCGまたは保護された茶類似体(25pM)の存在下、6ウェルプレート上の軟寒天に、またはDMSO(対照)にプレーティングした。
各薬物処理の後、剥離および接着VA−13およびWI−38細胞を、ヘキスト33342で染色し、アポトーシスについて評価した。簡単には、細胞をPBS中で2回洗浄し、4℃で70%エタノールを用いて1時間固定し、PBS中で3回洗浄し、50pMへキストを用い、暗所、室温で30分間染色した。剥離細胞をスライド上にプレーティングし、接着細胞は、10×または40×分解能の蛍光顕微鏡(Zeiss,Germany)を用いてプレート上で可視化した。AxioVision4.1を用いDigital Scientificによって画像を獲得し、Adobe Photoshop6.0を用いて調整した。
細胞を各化合物25μMで4時間または24時間処理し、次いで、回収し、溶解した。細胞溶解物にAc−DEVD−AMC(40μM)を2.5時間加え、カスパーゼ−3活性。
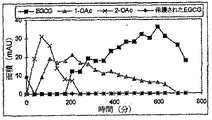
ペルアセタート(−)−EGCG、1の化学安定性および酵素的加水分解
pH値が約8であり体液を模倣する培養培地(RMPI)において、1および(−)−BGCGの安定性が比較されている。0.1mMの(−)−EGCGまたは1を1mL RMPI中、37℃で、示した時間インキュベートした。種々の時点で、HPLCによって残存する試験化合物の量について培地を分析した。図2には分解曲線が示されている。
1が(−)−EGCGのプロドラッグとして機能するなら、細胞の内側で脱アセチル化され、そこでその親化合物に変換するまで生物学的に不活性のままであるはずである。この仮説を調べるために、in vitroおよび無傷のジャーカットT細胞の両方において、1または(−)−EGCG(陽性対照として)のいずれかを用い、プロテアソーム活性を調べた。まず、1および市販の(−)−EGCGをDMSOに溶解し、精製20Sプロテアソームのキモトリプシン様活性に対するその効果を測定した。1は、10μMで精製20Sプロテアソームのキモトリプシン様活性の阻害において完全に不活性であった(図5)。対照的に、(−)−EGCGは、10μMでプロテアソームのキモトリプシン様活性を80〜90%阻害した。したがって、予想したように、細胞系の外側にある1はプロテアソーム阻害剤ではない。
(−)−EGCGおよび1を、5、10および25μMでジャーカットT細胞とともに24時間インキュベートし、続いて、リン酸化Aktに対する特異的抗体を用いてウエスタンブロット解析を行った(図7)。25μMで活性化Aktの73%の減少をもたらすことがデンシトメトリー分析によって示された1での処理と比較して、(−)−EGCGは、25μMで、p−Aktのレベルを32%低減することが分かった(図7)。アクチンはローディングコントロールとして用いた。
(−)−EGCGおよび1の、10μMで24時間処理したジャーカットT細胞において細胞死を誘導する能力も評価されている。(−)−EGCGは細胞死に対して最少の効果しか有さなかった(5%)のに対し、1はその濃度で最大15%の細胞死を誘導できた(図8)。したがって、1の、細胞のプロテアソーム活性を阻害し(図6)、Aktを不活化する(図7)より大きな能力が、その細胞死誘導活性の増大と関連している(図8)。
すべての保護された化合物および保護されていない化合物の最大25μMを、精製20sプロテアソームおよびキモトリプシン活性の蛍光発生基質とともに30分間インキュベートした。次いで、最大半量阻害濃度すなわちIC50を求めた。(−)−EGCGは最も強力であり、IC50は0.2μMであり、続いて、2である(IC50は約9.9μM)と分かった。化合物3および4のIC50値は14〜15μMであると分かった。対照的に、保護された類似体はかなり活性が低かった:25μMで<35%阻害。これは上記の結果と一致する。
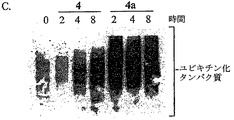
合成茶類似体がin vivoでプロテアソームに対してどのような効果を有していたかを調べるために、ジャーカット細胞を各合成化合物25μMで4時間または24時間処理し、(−)−EGCGを対照として用いた(図9Aおよび9B)。4時間処理した後、ウエスタンブロット解析は、アセタート保護された類似体がより多量のユビキチン化タンパク質を誘導したことを示し(図9A、レーン5、7および9)、このことはプロテアソーム活性が抑制されていることを示す。ペルアセタート保護された(−)−EGCGは天然(−)−EGCGよりもより強力であることを示す上記の先のデータに基づいて、1を対照として用いた(図9A、レーン3対2)。さらに、2種のプロテアソーム標的として知られる、BaxおよびIκBαについてのウエスタンブロットは、これらのタンパク質の消失と、タンパク質のマルチユビキチン化体であると推測されるより高分子量のバンドの出現を示した(図9A、レーン3、5、7および9)。しかし、24時間処理した後、(−)−EGCGおよびその保護されていない類似体は、より多量のユビキチン化タンパク質を示した(図9B、レーン2、4、6および8)。これは、保護された類似体は、より早期の時点ではプロテアソームの強力な阻害剤であるという考えおよび24時間処理した後には、ユビキチン化タンパク質は脱ユビキチン化酵素によって枯渇しているという考えと一致する。p27、別のプロテアソーム標的の蓄積も、保護された類似体2a、3aおよび4aで処理したジャーカット細胞において見られる(図9B、レーン9、5、7)。推定ユビキチン化IκBαバンドは、2a、3aおよび4aで処理した細胞においてもまだ見られる(図9B、レーン9、5、7)。24時間処理では、推定ユビキチン化IκBαバンドは、おそらくは脱ユビキチン化のためにここでは存在しない(図9B)。この実験ではアクチンをローディングコントロールとして用いた。
類似体4および4aの対を用いる速度論実験では、保護されていない類似体4はユビキチン化タンパク質の蓄積を誘導し、最高発現は8時間のポリフェノール処理後であることが分かった(図9C)。反対に、保護された4aは、2時間という早期にユビキチン化タンパク質蓄積の増加を示し、最大8時間まで持続した(図9C)。アセテート保護された類似体が、他の癌細胞系において強力なプロテアソーム阻害剤であるかどうかを調べるために、前立腺癌LNCaP細胞を25μMの(−)−EGCG、1、2aまたは3aで24時間処理し、対照としてDMSOを用いた。実際、ユビキチン結合タンパク質が観察され、2aおよび3aで処理した細胞において最大増加が見られた(図9D)。
プロテアソーム阻害は、多種多様な癌細胞においてアポトーシスを誘導できるが、正常細胞、非形質転換細胞ではそうではないということがわかっている(An Bら)。ジャーカットT細胞を、選択されたポリフェノールおよびその保護された類似体、各25μMで24時間処理し、アポトーシス細胞死を誘導するそれらの能力を調べた。トリパンブルー取り込みアッセイは、2a、3aおよび4aは、それぞれジャーカット細胞の99、57および83%の死を誘導するが、他のものはそうではないことを示した(図10A)。同様に、ウエスタンブロット解析は、2a、3aおよび4aのみが、24時間後にアポトーシス特異的PARP切断を誘導することを示した(図10B)。切断PARP断片(p85:緑色)のみを検出する免疫蛍光染色は、SV40で形質転換したVA−13細胞は、2aによって誘導されるアポトーシスに対して高度に感受性であり、24時間処理した後に73%がアポトーシス細胞であることを示した(図10Cおよび10D)。保護されていない2は、かなり少ないアポトーシスしか誘導しなかった(21%)が、陽性対照として用いた25μMのVP−16は、92%のアポトーシスを誘導した(図10Cおよび10D)。DNAのA−Tリッチ領域の副溝と結合するDAPIを用いる対比染色は、アポトーシス細胞において著しく低下し(図10C)、後期アポトーシスにおけるDNA断片化と一致する。
次いで、処理された乳癌(MCF−7)細胞を5または25μMのペルアセタート保護された類似体で24時間、続いて、MTT類似体で処理し、細胞増殖に対するその効果を調べた。化合物1は、25μMで細胞増殖を40%阻害した(図11A)。保護された化合物2a、3aおよび4aは、それぞれ5μMで50%阻害を引き起こし、25μMで70%阻害を引き起こした(図11A)。
正常細胞ではなく腫瘍細胞においてアポトーシスを誘導する能力は、新規抗癌剤にとって重要な尺度である。保護された化合物が正常細胞にも影響を及ぼすかどうかを調べるために、VA−13およびWI−38細胞の両方を、25μMの(−)−EGCG、2または2aで24時間処理し、プロテアソーム活性、核の形態変化および剥離を調べた。VA−13細胞において、正常WI−38細胞を超えるプロテアソームのキモトリプシン様活性の差示的減少が見られた(図12A)。(−)−EGCGおよび2で処理したVA−13細胞では、プロテアソーム活性の42〜48%減少が観察され、他方、2aはプロテアソーム活性の92%を阻害した。反対に、WI−38細胞ではプロテアソーム活性は、3種のポリフェノール処理すべてで−5%しか低下しなかった。
緑茶ポリフェノール(−)−EGCGは、3つの−OH基を有するB環を含み(図1)、精製20Sプロテアソームに対するIC50値は0.3μMである(表1)。
保護されていないポリフェノール類似体および保護されたポリフェノール類似体が、無傷の細胞においてプロテアソーム活性を阻害できるかどうかを調べるために、白血病ジャーカットT細胞を各化合物25μMで4時間および24時間処理し、続いて、細胞ペレットを回収し、調製した溶解物においてプロテアソームのキモトリプシン様活性を測定した。保護されていない化合物は、無傷のジャーカットT細胞において制限されたプロテアソーム阻害を示すが、10および15は中程度の効力を示した(図14)。これらのデータは、保護されていない化合物のすべてではないがほとんどは、おそらくはその細胞不安定性のために、(−)−EGCG同様、細胞環境において中程度にしか活性でないということを示唆する。
これまでに、プロテアソーム阻害はアポトーシス誘導と関連していることがわかっている。B環/D環保護された類似体が細胞死を誘導できたかどうかを調べるために、ジャーカットT細胞をまず、すべての保護された類似体および保護されていない類似体25μMで24時間処理し、続いて、トリパンブルー排除アッセイを行った(図16)。保護された類似体および保護されていない類似体の両方で4時間処理した後に、細胞形態の変化(縮んだ細胞および特徴的なアポトーシス性ブレブ形成)が観察され、これらの変化は24時間処理した後には、保護された類似体で大幅に増加した(データは示していない)。さらに、ペルアセタート保護された類似体は、4時間インキュベートした後に細胞死の5〜10倍の増加を誘導し、24時間では最大20倍増加した(図16)。対照的に、保護されていないポリフェノールははるかに少ない細胞死しか引き起こさず、24時間処理した後でさえ、3〜5倍の増加にすぎなかった(図16)。
正常細胞においてではなく腫瘍細胞におけるアポトーシス誘導は、抗癌剤の重要な側面である。本発明者らは、ペルアセタート保護された類似体が正常細胞に影響を及ぼすかどうかを調べるために、白血病ジャーカットT細胞および非形質転換不死化ナチュラルキラー(YT)細胞の両方を、外見的に最も強力なペルアセタート保護された類似体19*で処理した。再度、白血病ジャーカットT細胞において、プロテアソーム標的タンパク質、Bax、p27(データは示していない)およびIκB−α(図18)のレベルが上昇した。しかし、非形質転換不死化YT細胞では、これらのタンパク質のレベルは上昇しなかった。さらに、ジャーカットT細胞では、ユビキチン化タンパク質のレベルが用量依存的に蓄積した。対照的に、YT細胞では、ユビキチン化タンパク質のレベルのわずかな、一時的な上昇しか検出されなかった。プロテアソーム活性の選択的阻害と一致して、アポトーシス特異的PARP切断はジャーカットTでのみ見られ、YT細胞では見られなかった。したがって、強力なペルアセタート保護されたポリフェノールは、非形質転換YT細胞において、プロテアソーム阻害と、それに続くアポトーシスを誘導しないことは明らかである。
緑茶由来の天然(−)−EGCGは、そのペルアセタート化合物1に変換した。さらに、ガラート環のヒドロキシル基の欠失を有する、数種の(−)−EGCGの合成類似体を合成した。さらに、ヒドロキシル基をアセテート基に変換してプロドラッグを作出したが、これは細胞の内側でエステラーゼによって切断され、親薬物に変換されることはできなかった。驚くべきことに、保護された類似体は、無傷の腫瘍細胞において、その保護されていない対応物よりもはるかにより強力なプロテアソーム阻害剤であった。白血病(ジャーカット)、充実腫瘍、形質転換細胞株において試験した場合に、保護された類似体が、保護されていないパートナーよりもより強力なアポトーシス誘導因子でもあったことは一貫している。保護された類似体のSAR分析により、その効力の順序は次の通りであると示された:2a=4a>3a>1>(−)−EGCG。
ペルアシルオキシル、特に、ペルアセタート保護された類似体も、その保護されていない、ヒドロキシル化対応物よりもより強力なプロテアソーム阻害剤であるとわかる。プロテアソームのキモトリプシン様サブユニット(β5)のN末端Thrは、(−)−EGCGのエステル結合された炭素に求核攻撃を行い、これによって、β5−サブユニットへの不可逆的なアシル化が開始され、そのプロテアーゼ活性が阻害されるということが示唆された(Nam S、Smith DMおよびDou QP、J Biol Chem 276、13322〜13330頁、2001)。しかし、ペルアセタート部分の付加により、エステル結合炭素の求電子特性が低下し、これによって精製20Sプロテアソームに対する阻害がかなり小さくなる。本研究で調べた保護されたポリフェノールも、精製20Sプロテアソームのキモトリプシン様活性に対して制限された阻害を示したことは驚くべきことではない(表1)。
Claims (9)
- 次式を有する、プロテアソームを阻害するための化合物
・R11、R12、R13、R21、R22、R2、R3およびR4は各々独立に、−HおよびC1〜C10アシルオキシル基からなる群から選択され、
・R5は−H、C1〜C10−アルキル、C2〜C10−アルケニル、C2〜C10−アルキニル、C3〜C7−シクロアルキル、フェニル、ベンジルおよびC3〜C7−シクロアルケニルからなる群から選択され、最後に記載した7種の基は各々、任意の組合せの1〜6個のハロゲン原子で置換されていることができ、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個はC1〜C10アシルオキシル基であり、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個は−Hである]であって、
当該式が、下記の(1)から(5)のいずれか一つを満たす化合物:
(1)R 11 、R 13 、R 2 およびR 4 が各々−Hであり、R 12 、R 21 、R 22 およびR 3 が各々アセテート基である;
(2)R 11 およびR 13 が各々−Hであり、R 12 、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である;
(3)R 11 、R 12 およびR 13 が各々−Hであり、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である;
(4)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 およびR 22 が各々アセテート基であり、R 2 がアセテート基であり、R 3 およびR 4 が各々−Hである;及び
(5)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 およびR 22 が各々アセテート基であり、R 2 およびR 4 が各々アセテート基であり、R 3 が−Hである。 - R11、R13、R2およびR4が各々−Hであり、R12、R21、R22およびR3が各々アセテート基である、請求項1に記載の化合物。
- R11およびR13が各々−Hであり、R12、R21、R22、R2、R3およびR4が各々アセテート基である、請求項1に記載の化合物。
- R11、R12およびR13が各々−Hであり、R21、R22、R2、R3およびR4が各々アセテート基である、請求項1に記載の化合物。
- R2がアセテート基であり、R3およびR4が各々−Hである、請求項1に記載の化合物。
- R2およびR4が各々アセテート基であり、R3が−Hである、請求項1に記載の化合物。
- 次式を有する化合物の有効量を含む、腫瘍細胞増殖を低減するための医薬組成物
・R11、R12、R13、R21、R22、R2、R3およびR4が各々独立に、−HおよびC1〜C10アシルオキシル基からなる群から選択され、
・R5は−H、C1〜C10−アルキル、C2〜C10−アルケニル、C2〜C10−アルキニル、C3〜C7−シクロアルキル、フェニル、ベンジルおよびC3〜C7−シクロアルケニルからなる群から選択され、最後に記載した7種の基は各々、任意の組合せの1〜6個のハロゲン原子で置換されていることができ、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個はC1〜C10アシルオキシル基である]であって、
当該式が、下記の(1)から(9)のいずれか一つを満たす医薬組成物:
(1)R 11 、R 2 およびR 4 が各々−Hであり、R 12 、R 13 、R 21 、R 22 およびR 3 が各々アセテート基である;
(2)R 11 が−Hであり、R 12 、R 13 、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である;
(3)R 11 、R 13 、R 2 およびR 4 が各々−Hであり、R 12 、R 21 、R 22 およびR 3 が各々アセテート基である;
(4)R 11 およびR 13 が各々−Hであり、R 12 、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である;
(5)R 11 、R 12 およびR 13 が各々−Hであり、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である;
(6)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 およびR 22 が各々アセテート基であり、R 2 がアセテート基であり、R 3 およびR 4 が各々−Hである;
(7)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 およびR 22 が各々アセテート基であり、R 2 およびR 4 が各々アセテート基であり、R 3 が−Hである;
(8)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 およびR 22 が各々アセテート基であり、R 3 がアセテート基であり、R 2 およびR 4 が各々−Hである;及び
(9)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である。 - 腫瘍細胞増殖を低減するための医薬の製造における、次式を有する化合物の使用
・R11、R12、R13、R21、R22、R2、R3およびR4が各々独立に、−HおよびC1〜C10アシルオキシル基からなる群から選択され、
・R5は−H、C1〜C10−アルキル、C2〜C10−アルケニル、C2〜C10−アルキニル、C3〜C7−シクロアルキル、フェニル、ベンジルおよびC3〜C7−シクロアルケニルからなる群から選択され、最後に記載した7種の基は各々、任意の組合せの1〜6個のハロゲン原子で置換されていることができ、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個はC1〜C10アシルオキシル基である]であって、
当該式が、下記の(1)から(9)のいずれか一つを満たす使用:
(1)R 11 、R 2 およびR 4 が各々−Hであり、R 12 、R 13 、R 21 、R 22 およびR 3 が各々アセテート基である;
(2)R 11 が−Hであり、R 12 、R 13 、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である;
(3)R 11 、R 13 、R 2 およびR 4 が各々−Hであり、R 12 、R 21 、R 22 およびR 3 が各々アセテート基である;
(4)R 11 およびR 13 が各々−Hであり、R 12 、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である;
(5)R 11 、R 12 およびR 13 が各々−Hであり、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である;
(6)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 およびR 22 が各々アセテート基であり、R 2 がアセテート基であり、R 3 およびR 4 が各々−Hである;
(7)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 およびR 22 が各々アセテート基であり、R 2 およびR 4 が各々アセテート基であり、R 3 が−Hである;
(8)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 およびR 22 が各々アセテート基であり、R 3 がアセテート基であり、R 2 およびR 4 が各々−Hである;及び
(9)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である。 - 次式を有する化合物の有効量を含む、プロテアソームを阻害するための組成物:
・R11、R12、R13、R21、R22、R2、R3およびR4は各々独立に、−HおよびC1〜C10アシルオキシル基からなる群から選択され、
・R5は−H、C1〜C10−アルキル、C2〜C10−アルケニル、C2〜C10−アルキニル、C3〜C7−シクロアルキル、フェニル、ベンジルおよびC3〜C7−シクロアルケニルからなる群から選択され、最後に記載した7種の基は各々、任意の組合せの1〜6個のハロゲン原子で置換されていることができ、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個はC1〜C10アシルオキシル基であり、
・R11、R12、R13、R21、R22、R2、R3およびR4のうち少なくとも1個は−Hである]であって、
当該式が、下記の(1)から(8)のいずれか一つを満たす組成物:
(1)R 11 、R 2 およびR 4 が各々−Hであり、R 12 、R 13 、R 21 、R 22 およびR 3 が各々アセテート基である;
(2)R 11 が−Hであり、R 12 、R 13 、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である;
(3)R 11 、R 13 、R 2 およびR 4 が各々−Hであり、R 12 、R 21 、R 22 およびR 3 が各々アセテート基である;
(4)R 11 およびR 13 が各々−Hであり、R 12 、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である;
(5)R 11 、R 12 およびR 13 が各々−Hであり、R 21 、R 22 、R 2 、R 3 およびR 4 が各々アセテート基である;
(6)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 およびR 22 が各々アセテート基であり、R 2 がアセテート基であり、R 3 およびR 4 が各々−Hである;
(7)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 およびR 22 が各々アセテート基であり、R 2 およびR 4 が各々アセテート基であり、R 3 が−Hである;及び
(8)R 5 が−Hであり、R 11 、R 12 、R 13 、R 21 およびR 22 が各々アセテート基であり、R 3 がアセテート基であり、R 2 およびR 4 が各々−Hである。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/921,332 | 2004-08-19 | ||
| US10/921,332 US7544816B2 (en) | 2004-08-19 | 2004-08-19 | (−)-Epigallocatechin gallate derivatives for inhibiting proteasome |
| US64956905P | 2005-02-04 | 2005-02-04 | |
| US60/649,569 | 2005-02-04 | ||
| PCT/CN2005/001262 WO2006017981A1 (en) | 2004-08-19 | 2005-08-15 | (-)-epigallocatechin gallate derivatives for inhibiting proteasome |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2008509939A JP2008509939A (ja) | 2008-04-03 |
| JP2008509939A5 JP2008509939A5 (ja) | 2008-10-02 |
| JP5265915B2 true JP5265915B2 (ja) | 2013-08-14 |
Family
ID=35907224
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2007526180A Expired - Lifetime JP5265915B2 (ja) | 2004-08-19 | 2005-08-15 | プロテアソームを阻害するための(−)−エピガロカテキンガラート誘導体 |
Country Status (6)
| Country | Link |
|---|---|
| US (4) | US8193377B2 (ja) |
| EP (1) | EP1778663B1 (ja) |
| JP (1) | JP5265915B2 (ja) |
| CN (1) | CN101072764B (ja) |
| ES (1) | ES2660339T3 (ja) |
| WO (1) | WO2006017981A1 (ja) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5265915B2 (ja) | 2004-08-19 | 2013-08-14 | ザ・ホンコン・ポリテクニック・ユニバーシティ | プロテアソームを阻害するための(−)−エピガロカテキンガラート誘導体 |
| EP2088205A1 (en) | 2008-02-11 | 2009-08-12 | Institut National De La Sante Et De La Recherche Medicale (Inserm) | PSMB10: A diagnosis marker and therapeutic target of chronic rejection. |
| US8784873B2 (en) * | 2009-07-22 | 2014-07-22 | Naturewise Biotech & Medicals Corporation | Use of histone deacetylase inhibitors in changing MRJP3 protein in royal jelly |
| US9126997B1 (en) | 2010-09-07 | 2015-09-08 | Northwestern University | Synergistic effect of glucocorticoid receptor agonists in combination with proteosome inhibitors for treating leukemia and myeloma |
| GB2484540B (en) | 2010-10-15 | 2014-01-29 | Microsoft Corp | A loop antenna for mobile handset and other applications |
| CN102078316A (zh) * | 2011-01-24 | 2011-06-01 | 广西医科大学 | 表没食子儿茶素没食子酸酯衍生物在抗肿瘤药物中的应用 |
| CN102153535A (zh) * | 2011-03-04 | 2011-08-17 | 中国海洋大学 | 一种抗肿瘤多药耐药抑制剂苯并吡喃-3-醇酯化衍生物及其制备方法和应用 |
| EP2557079A1 (en) * | 2011-08-09 | 2013-02-13 | Nestec S.A. | Synthesis of catechin and epicatechin conjugates |
| WO2017132775A1 (en) * | 2016-02-05 | 2017-08-10 | Viteava Pharmaceuticals Inc. | Novel compositions and methods for the treatment of leiomyoma |
| CN108129438A (zh) * | 2017-12-25 | 2018-06-08 | 中国海洋大学 | 一种含2-苯色满母核的化合物及其制备方法 |
| KR102057773B1 (ko) | 2018-03-30 | 2019-12-19 | 건국대학교 산학협력단 | 에피갈로카테킨 갈레이트 유도체를 포함하는 유방암 예방 또는 치료용 약학적 조성물 |
| WO2019236765A1 (en) * | 2018-06-05 | 2019-12-12 | Flagship Pioneering Innovations V, Inc. | Acylated catechin polyphenols and methods of their use for the treatment of cancer |
| CN116162078B (zh) * | 2022-12-27 | 2024-10-29 | 佛山病原微生物研究院 | 一种表没食子儿茶素没食子酸酯类化合物及其制备方法和应用 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS57118580A (en) * | 1981-01-16 | 1982-07-23 | Kanebo Ltd | Novel catechin derivative |
| JPS57120584A (en) * | 1981-01-20 | 1982-07-27 | Kanebo Ltd | Production of catechin derivative |
| JPH10254087A (ja) * | 1997-03-14 | 1998-09-25 | Konica Corp | ハロゲン化銀写真感光材料 |
| US6696484B2 (en) * | 1997-10-31 | 2004-02-24 | University Of Chicago Office Of Technology And Intellectual Property | Method and compositions for regulation of 5-alpha reductase activity |
| KR20010031501A (ko) * | 1997-10-31 | 2001-04-16 | 추후제출 | 5-알파 환원효소 활성을 조절하는 방법 및 조성물 |
| US7122573B2 (en) * | 2002-12-06 | 2006-10-17 | Sri International | Analogs of green tea polyphenols as chemotherapeutic and chemopreventive agents |
| US7544816B2 (en) * | 2004-08-19 | 2009-06-09 | The Hong Kong Polytechnic University | (−)-Epigallocatechin gallate derivatives for inhibiting proteasome |
| JP5265915B2 (ja) * | 2004-08-19 | 2013-08-14 | ザ・ホンコン・ポリテクニック・ユニバーシティ | プロテアソームを阻害するための(−)−エピガロカテキンガラート誘導体 |
-
2005
- 2005-08-15 JP JP2007526180A patent/JP5265915B2/ja not_active Expired - Lifetime
- 2005-08-15 ES ES05779886.0T patent/ES2660339T3/es not_active Expired - Lifetime
- 2005-08-15 CN CN2005800354787A patent/CN101072764B/zh not_active Expired - Fee Related
- 2005-08-15 WO PCT/CN2005/001262 patent/WO2006017981A1/en not_active Ceased
- 2005-08-15 EP EP05779886.0A patent/EP1778663B1/en not_active Expired - Lifetime
- 2005-08-15 US US11/660,513 patent/US8193377B2/en active Active
-
2012
- 2012-03-09 US US13/416,657 patent/US8710248B2/en not_active Expired - Lifetime
-
2014
- 2014-03-28 US US14/229,315 patent/US9169230B2/en not_active Expired - Lifetime
-
2015
- 2015-09-15 US US14/854,919 patent/US20160068503A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| US20140213802A1 (en) | 2014-07-31 |
| US9169230B2 (en) | 2015-10-27 |
| CN101072764B (zh) | 2013-07-10 |
| EP1778663A4 (en) | 2008-08-06 |
| CN101072764A (zh) | 2007-11-14 |
| EP1778663A1 (en) | 2007-05-02 |
| US8710248B2 (en) | 2014-04-29 |
| WO2006017981A1 (en) | 2006-02-23 |
| US20120232135A1 (en) | 2012-09-13 |
| US8193377B2 (en) | 2012-06-05 |
| WO2006017981A9 (en) | 2006-05-11 |
| EP1778663B1 (en) | 2017-12-13 |
| US20080176931A1 (en) | 2008-07-24 |
| JP2008509939A (ja) | 2008-04-03 |
| US20160068503A1 (en) | 2016-03-10 |
| ES2660339T3 (es) | 2018-03-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8710248B2 (en) | (−)-epigallocatechin gallate derivatives for inhibiting proteasome | |
| Jeon et al. | Green tea catechins as a BACE1 (β-secretase) inhibitor | |
| AU2009200907A1 (en) | Isolation, purification and synthesis of procyanidin B2 and uses thereof | |
| US7544816B2 (en) | (−)-Epigallocatechin gallate derivatives for inhibiting proteasome | |
| WO2006093547A2 (en) | Novel lipoxygenase inhibitors | |
| WO2012171114A1 (en) | Synthetic epigallocatechin gallate (egcg) analogs | |
| JP2796876B2 (ja) | 置換ピロリジン化合物および医薬組成物 | |
| Tocco et al. | PEG-immobilization of cardol and soluble polymer-supported synthesis of some cardol–coumarin derivatives: Preliminary evaluation of their inhibitory activity on mushroom tyrosinase | |
| SK50062012A3 (sk) | Quercetin derivatives, pharmaceutical compositions comprising them and their use | |
| US9526706B1 (en) | Method of using dihydro-resveratrol for treating acute pancreatitis and associated pulmonary injury | |
| AU2009211300A1 (en) | Phenyl-prenyl derivatives, of marine and synthetic origin, for the treatment of cognitive, neurodegenerative or neuronal diseases or disorders | |
| Macedo et al. | Anti-inflammatory effects of naringenin 8-sulphonate from Parinari excelsa Sabine stem bark and its semi-synthetic derivatives | |
| Chan et al. | (−)-epigallocatechin gallate derivatives for inhibiting proteasome | |
| JP3248183B2 (ja) | 新規な心臓保護剤 | |
| US20090047221A1 (en) | Compounds and compositions useful in the treatment of neoplasia | |
| AU2004257887A1 (en) | Chemical compounds containing tocopherol and at least one additional pharmaceutical active substance | |
| EP3733658B1 (en) | Prenylated benzopyranes as ppar agonists | |
| KR102060412B1 (ko) | 포르툴라카논 화합물과 그 유도체 합성방법 및 이를 포함하는 항염증 약학 조성물 | |
| KR20200033196A (ko) | 약물유도 신장손상 보호활성을 나타내는 신규 세리세틴 전합성법 및 이를 유효성분으로 하는 약학 조성물 | |
| Chen et al. | Design, Synthesis, and Biological Evaluation of Novel Tetramethylpyrazine-nitrone Derivatives as Antioxidants | |
| Yasuda et al. | Preparation and antioxidant/pro-oxidant activities of 3-monosubstituted 5-hydroxyoxindole derivatives | |
| JP2006131594A (ja) | 発癌予防剤 | |
| TWI396533B (zh) | 異戊二烯類黃酮化合物及其用途 | |
| Jaiswal et al. | Synthesis and evaluation of novel heterocyclic chromene derivatives as antimicrobial and antioxidant agents | |
| Lajis et al. | Research Article Depigmenting Effect of Kojic Acid Esters in Hyperpigmented B16F1 Melanoma Cells |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080812 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080812 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20110912 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120125 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120424 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120502 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120514 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20120808 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20121119 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20121122 |
|
| A911 | Transfer to examiner for re-examination before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20130227 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130404 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20130502 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 Ref document number: 5265915 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |