JP5114475B2 - o−キシロールの気相酸化による無水フタル酸の製造 - Google Patents
o−キシロールの気相酸化による無水フタル酸の製造 Download PDFInfo
- Publication number
- JP5114475B2 JP5114475B2 JP2009511484A JP2009511484A JP5114475B2 JP 5114475 B2 JP5114475 B2 JP 5114475B2 JP 2009511484 A JP2009511484 A JP 2009511484A JP 2009511484 A JP2009511484 A JP 2009511484A JP 5114475 B2 JP5114475 B2 JP 5114475B2
- Authority
- JP
- Japan
- Prior art keywords
- reactor
- phthalic anhydride
- catalyst
- reaction product
- xylol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/87—Benzo [c] furans; Hydrogenated benzo [c] furans
- C07D307/89—Benzo [c] furans; Hydrogenated benzo [c] furans with two oxygen atoms directly attached in positions 1 and 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/16—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation
- C07C51/21—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen
- C07C51/255—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of compounds containing six-membered aromatic rings without ring-splitting
- C07C51/265—Preparation of carboxylic acids or their salts, halides or anhydrides by oxidation with molecular oxygen of compounds containing six-membered aromatic rings without ring-splitting having alkyl side chains which are oxidised to carboxyl groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Furan Compounds (AREA)
- Catalysts (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Description
a)その活性材料が銀、バナジウム、および場合によっては1つ以上の促進剤金属を含有する多重金属混合酸化物、殊に銀−酸化バナジウム−ブロンズを含む1つ以上の触媒、
b)酸化バナジウムおよび二酸化チタンを基礎とする1つ以上の触媒、この場合この活性材料のアルカリ金属含量は、0.12質量%以上であり、活性材料の燐含量(Pとして計算した)は、0.20質量%以下であり、および
c)前記定義a)に記載の1つ以上の触媒と前記定義b)に記載の1つ以上の触媒との組合せである。
Aga-cM1 cV2Od*eH2O I
〔式中、aは、0.3〜1.9の値を有し、
M1は、アルカリ金属およびアルカリ土類金属、Bi,Ti、Cu、Zn、Cd、Pb、Cr、Au、Al、Fe、Co、Ni、Mo、Nb、Ce、W、Mn、Ta、Pd、Pt、Ruおよび/またはRhから選択された少なくとも1つの金属を表わし、
cは、0〜0.5の値を有し、但し、(a−c)は、0.1を上廻るものとし、
dは、式I中の酸素とは異なる元素の原子価および頻度によって定められる数を表わし、および
eは、0〜20、特に0〜5の値を有する〕で示される多重金属混合酸化物である。
AgaV2Od*eH2O Ia
[式中、
aは、0.6〜0.9の値であり、
dは、上記の意味を有し、
eは、0〜5の値を有する〕で示される多重金属酸化物である。
触媒の製造
主要反応器触媒Iの製造(選択的触媒)
施糖衣用ドラム中で18時間の攪拌後に蓚酸104.9g、五酸化バナジウム39.4g、酸化アンチモン17.0g、硫酸セシウム2.87g、燐酸水素アンモニウム3.15g、ホルムアミド149.0g、20m2/gのBET表面積を有するアナターゼ変態の二酸化チタン465.9gおよび水721.0gからなる懸濁液236.6gを、160℃で有機結合剤13.0gと一緒に寸法8×6×5mm(外径×高さ×内径)のステアタイトリング1400g上に施こす。第2の工程で、こうして被覆されたリングを、蓚酸56.7g、五酸化バナジウム21.0g、硫酸セシウム2.87g、ホルムアミド198.0g、二酸化チタン501.9gおよび水720.3gからなる、先に同様に18時間攪拌された第2の懸濁液236.2gで有機結合剤12.8gと一緒に被覆する。
施糖衣用ドラム中で18時間の攪拌後に蓚酸106.4g、五酸化バナジウム39.4g、酸化アンチモン17.0g、硫酸セシウム0.63g、燐酸水素アンモニウム3.35g、ホルムアミド149.6g、20m2/gのBET表面積を有するアナターゼ変態の二酸化チタン467.5gおよび水719.1gからなる懸濁液538.0gを、160℃で寸法8×6×5mm(外径×高さ×内径)のステアタイトリング1400g上に施こす。
施糖衣用ドラム中で18時間の攪拌後に蓚酸105.5g、五酸化バナジウム39.4g、酸化アンチモン17.0g、硫酸セシウム0.29g、燐酸水素アンモニウム8.9g、ホルムアミド149.0g、20m2/gのBET表面積を有するアナターゼ変態の二酸化チタン467.0gおよび水720.5gからなる懸濁液540.2gを、160℃で寸法8×6×5mm(外径×高さ×内径)のステアタイトリング1400g上に施こす。
主要反応器として、99本の標準管および2本の熱管を備えた管束型反応器を使用した。標準管は、25mmの内径を有し、熱管は、29mmの内径を有し、ドイツ連邦共和国特許第10110847号明細書に記載された、10cmの間隔で温度測定位置を有する組み込まれた30個の多重要素または試料採取要素を備えたスリーブ(直径10mm)を有する。圧力の調整により、それぞれの管入口に同じ入口圧力が印加されるように準備した。場合によっては、99本の標準管の場合、なお例えば主要反応器触媒Iを添加するかまたは吸着し、2本の熱管の場合には、圧力調整を、球状ステアタイトおよび石英砂の形での不活性材料の添加によって達成した。鉄管は、温度調整のために2つの離隔された塩浴中に存在する塩溶融液によって包囲されていた。下方の塩浴(塩浴B)は、管を下方の管状床によって140cmの高さになるまで包囲し、上方の塩浴(塩浴A)は、管を上方の管状床に到るまで140cmの高さで包囲した。
試験を実施例1〜7に記載の方法に相応して実施したが、しかし、この場合には、後方反応器は、断熱的に運転され、即ち空気は、冷却蛇管に導通されなかった。結果は、第1表中に記載されている。
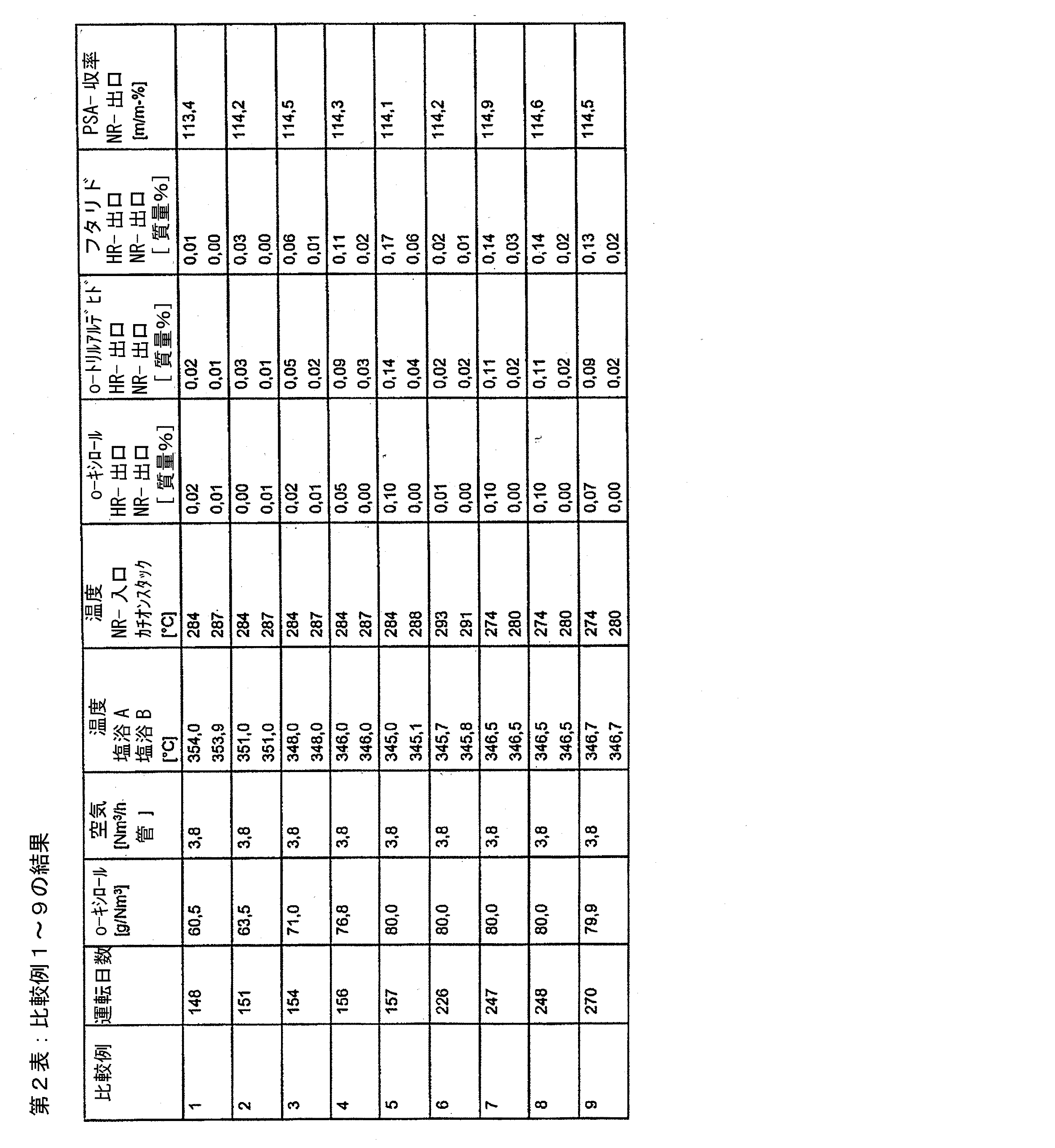
試験を実施例1〜7に記載の方法に相応して実施したが、しかし、この場合には、後方反応器は、断熱的に運転され、主要反応器の2つの塩浴は、本質的同じ温度で運転された。結果は、第2表中に記載されている。
運転日数は、主要反応器触媒の第1の始動時からの運転日数を表わし;
塩浴Aは、反応器入口に向かって置かれた塩浴の塩浴温度を表わし;
塩浴Bは、反応器出口に向かって置かれた塩浴の塩浴温度を表わし;
o−キシロールHR出口、o−トリルアルデヒドHR出口またはフタリドHR出口は、主要反応器出口での粗製生成物ガスの有機成分の質量%でのo−キシロール含量、o−トリルアルデヒド含量またはフタリド含量を表わし;
o−キシロールHR出口、o−トリルアルデヒドHR出口またはフタリドHR出口は、後方反応器出口での粗製生成物ガスの有機成分の質量%でのo−キシロール含量、o−トリルアルデヒド含量またはフタリド含量を表わし;
NR出口でのPSA収量は、後方反応器出口での粗製生成物ガスの分析による100%のo−キシロールに関連する質量%でのPSA収量を表わす。
Claims (12)
- o−キシロールと酸素含有ガスとのガス状混合物を、主要反応器中で、互いに別々の温度調節可能な少なくとも2つの反応帯域に導通させ、その際、最も下流に置かれた反応帯域を、上流と境を接している反応帯域より低い温度で運転し、未反応のo−キシロール、無水フタル酸酸化不足生成物および無水フタル酸を含有するガス状の中間体反応生成物に変換し、および中間体反応生成物を後方反応器中に導入し、主要反応器から流出する中間体反応生成物を後方反応器中への入口前で冷却することにより、無水フタル酸をo−キシロールの接触気相酸化によって製造する方法において、
主要反応器中での反応帯域の温度を、中間体反応生成物中の未反応のo−キシロールの濃度が、中間体反応生成物中の有機成分の質量に対して0.5〜5質量%であり、中間体反応生成物中のo−トリルアルデヒドの濃度は、0.25〜0.6質量%であるように調節することを特徴とする、無水フタル酸をo−キシロールの接触気相酸化によって製造する方法。 - 中間体反応生成物中の無水フタル酸酸化不足生成物の濃度の総和は、少なくとも0.5質量%である、請求項1記載の方法。
- o−トリルアルデヒドおよびフタリドの濃度の総和は、少なくとも0.5質量%である、請求項1または2記載の方法。
- 中間体の反応生成物中のフタリドの濃度は、少なくとも0.25質量%である、請求項1から3までのいずれか1項に記載の方法。
- 反応帯域は、異なる活性の触媒を含む、請求項1から4までのいずれか1項に記載の方法。
- ガス状混合物の流れ方向で最も下流に置かれた反応帯域は、上流と境を接している反応帯域より高い活性を有する触媒を含む、請求項5記載の方法。
- ガス状混合物の流れ方向で最も下流に置かれた反応帯域は、酸化バナジウムおよび二酸化チタンを基礎とする少なくとも1つの触媒を含み、この場合活性材料のアルカリ金属含量は、0.20質量%以下である、請求項5または6記載の方法。
- 上流で、ガス状混合物の流れ方向で最も下流に置かれた反応帯域と境を接している反応帯域は、
a)その活性材料が銀、バナジウム、および場合によっては1つ以上の促進剤金属を含有する多重金属混合酸化物を含む1つ以上の触媒、
b)酸化バナジウムおよび二酸化チタンを基礎とする1つ以上の触媒、この場合この活性材料のアルカリ金属含量は、0.12質量%以上であり、活性材料の燐含量は、0.20質量%以下であるか、または
c)前記定義a)に記載の1つ以上の触媒と前記定義b)に記載の1つ以上の触媒との組合せから選択された触媒を含む、請求項5から7までのいずれか1項に記載の方法。 - 中間体反応生成物中のo−キシロールの濃度に対する測定値が得られ、この測定値から主要反応器中の反応帯域の温度に対する制御の干渉が形成される、請求項1から8までのいずれか1項に記載の方法。
- 主要反応器中に流入するガス状混合物のo−キシロール負荷量は、少なくとも60g/Nm3である、請求項1から9までのいずれか1項に記載の方法。
- 後方反応器中で生じる反応熱の少なくとも一部分を伝熱媒体での間接的な冷却によって導出する、請求項1から10までのいずれか1項に記載の方法。
- 後方反応器を本質的に断熱的に運転する、請求項1から11までのいずれか1項に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06010415.5 | 2006-05-19 | ||
| EP06010415 | 2006-05-19 | ||
| PCT/EP2007/054841 WO2007135104A1 (de) | 2006-05-19 | 2007-05-18 | Herstellung von phthalsäureanhydrid durch gasphasenoxidation von o-xylol |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2009537594A JP2009537594A (ja) | 2009-10-29 |
| JP2009537594A5 JP2009537594A5 (ja) | 2012-10-11 |
| JP5114475B2 true JP5114475B2 (ja) | 2013-01-09 |
Family
ID=38161984
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2009511484A Active JP5114475B2 (ja) | 2006-05-19 | 2007-05-18 | o−キシロールの気相酸化による無水フタル酸の製造 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US8153825B2 (ja) |
| EP (1) | EP2024351B1 (ja) |
| JP (1) | JP5114475B2 (ja) |
| KR (1) | KR101395989B1 (ja) |
| CN (1) | CN101448810B (ja) |
| AT (1) | ATE501132T1 (ja) |
| DE (1) | DE502007006673D1 (ja) |
| ES (1) | ES2361202T3 (ja) |
| MY (1) | MY145398A (ja) |
| SI (1) | SI2024351T1 (ja) |
| WO (1) | WO2007135104A1 (ja) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE502007003524D1 (de) * | 2006-05-19 | 2010-06-02 | Basf Se | Herstellung von phthalsäureanhydrid durch gasphasenoxidation von o-xylol in einem haupt- und nachreaktor |
| US8263789B2 (en) | 2006-12-21 | 2012-09-11 | Basf Se | Catalyst system and method for gas phase oxidation using an upstream layer |
| JP5239995B2 (ja) * | 2008-03-31 | 2013-07-17 | 三菱化学株式会社 | プレート式反応器及び反応生成物の製造方法 |
| WO2009124947A1 (de) * | 2008-04-07 | 2009-10-15 | Basf Se | Verfahren zum anfahren eines gasphasenoxidationsreaktors, der eine katalytisch aktive silber-vanadiumoxid-bronze enthält |
| TWI453190B (zh) | 2008-04-07 | 2014-09-21 | Basf Se | 起動氣相氧化反應器之方法 |
| DE102010012090A1 (de) * | 2010-03-19 | 2011-11-17 | Süd-Chemie AG | Verfahren zur katalytischen Gasphasenoxidation von Kohlenwasserstoffen und Katalysereaktionsvorrichtung |
| CN105339338B (zh) | 2013-06-26 | 2017-11-21 | 巴斯夫欧洲公司 | 起动气相氧化反应器的方法 |
| CN106279084B (zh) * | 2015-05-15 | 2019-01-22 | 中国科学院大连化学物理研究所 | 一种芳香酮氧化裂解制备邻苯二甲酸酐及其芳环取代的衍生物的方法 |
| DE102016002679A1 (de) | 2016-03-04 | 2017-09-21 | Hans-Jürgen Eberle | Vorrichtung und Verfahren zur Durchführung von heterogen katalysierten exothermen Gasphasenreaktionen |
| CN109046412B (zh) * | 2018-08-23 | 2021-05-25 | 常州新日催化剂股份有限公司 | 一种正丁烷氧化制顺酐催化剂及其制备方法 |
Family Cites Families (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2005969A1 (en) * | 1970-02-10 | 1971-08-26 | Badische Anilin & Soda Fabrik AG, 6700 Ludwigshafen | Dicarboxylic acids/and acid anhydridespreparation by isothe - process |
| DE4013051A1 (de) * | 1990-04-24 | 1991-11-07 | Basf Ag | Verfahren zur herstellung von phthalsaeureanhydrid aus o-xylol |
| SE507313C2 (sv) * | 1997-02-25 | 1998-05-11 | Neste Oy | Förfarande för framställning av ftalsyraanhydrid |
| DE19823262A1 (de) | 1998-05-26 | 1999-12-02 | Basf Ag | Verfahren zur Herstellung von Phthalsäureanhydrid |
| DE19823275A1 (de) | 1998-05-26 | 1999-12-02 | Basf Ag | Verfahren zur Herstellung von Phthalsäureanhydrid durch katalytische Gasphasenoxidation von x-Xylol-/-Naphthalin-Gemischen |
| DE19851786A1 (de) | 1998-11-10 | 2000-05-11 | Basf Ag | Silber- und Vanadiumoxid enthaltendes Multimetalloxid und dessen Verwendung |
| JP4557378B2 (ja) * | 1999-06-24 | 2010-10-06 | 株式会社日本触媒 | 無水フタル酸の製造方法 |
| DE10022103A1 (de) | 2000-05-08 | 2001-11-15 | Basf Ag | Silber, Vanadium und ein oder mehrere Elemente der Phosphorgruppe enthaltendes Multimetalloxid und dessen Verwendung |
| DE10040827A1 (de) * | 2000-08-21 | 2002-03-07 | Basf Ag | Verfahren zur Herstellung von Phthalsäureanhydrid |
| DE10110847A1 (de) | 2001-03-07 | 2002-09-12 | Gerhard Olbert | Meßverfahren und -einrichtung zur Überwachung und Steuerung von Reaktionen in Kontaktrohrbündelreaktoren |
| JP4827329B2 (ja) * | 2001-07-04 | 2011-11-30 | 株式会社日本触媒 | 酸無水物の製造方法 |
| DE10144857A1 (de) | 2001-09-12 | 2003-03-27 | Deggendorfer Werft Eisenbau | Reaktoranordnung für die Durchführung katalytischer Gasphasenreaktionen, insbesondere zur Gewinnung von Phthalsäureanhydrid |
| DE10206989A1 (de) | 2002-02-19 | 2003-08-21 | Basf Ag | Verfahren zur Herstellung von Phthalsäureanhydrid |
| DE10323817A1 (de) * | 2003-05-23 | 2004-12-09 | Basf Ag | Verfahren zur Herstellung von Phthalsäureanhydrid |
| DE10334132A1 (de) | 2003-07-25 | 2005-04-07 | Basf Ag | Silber, Vanadium und ein Promotormetall enthaltendes Multimetalloxid und dessen Verwendung |
| DE102004061770A1 (de) | 2004-12-22 | 2006-07-06 | Basf Ag | Verfahren zur Herstellung von Phthalsäureanhydrid |
| WO2007122090A2 (de) | 2006-04-21 | 2007-11-01 | Basf Se | Verfahren zur herstellung von ethylenoxid in einem mikrokanalreaktor |
| EP1852413A1 (de) | 2006-04-27 | 2007-11-07 | Basf Aktiengesellschaft | Verfahren zur Gasphasenoxidation unter Verwendung einer Moderatorlage |
| CN101448571A (zh) | 2006-05-19 | 2009-06-03 | 巴斯夫欧洲公司 | 用于制备羧酸和/或羧酸酐的催化剂体系 |
| DE502007003524D1 (de) * | 2006-05-19 | 2010-06-02 | Basf Se | Herstellung von phthalsäureanhydrid durch gasphasenoxidation von o-xylol in einem haupt- und nachreaktor |
| EP2035138A1 (de) | 2006-06-20 | 2009-03-18 | Basf Se | Katalysatorsystem und verfahren zur herstellung von carbonsäuren und/oder carbonsäureanhydriden |
| EP2056963A1 (de) | 2006-08-23 | 2009-05-13 | Basf Se | Eingebettete katalysatoren basierend auf vpo-vorstufen und siliciumdioxid, deren herstellung und verwendung |
| WO2008022911A1 (de) | 2006-08-23 | 2008-02-28 | Basf Se | KATALYSATOREN AUS VPO-VORSTUFEN MIT DEFINIERTER TEILCHENGRÖßENVERTEILUNG, DEREN HERSTELLUNG UND VERWENDUNG |
| US8263789B2 (en) | 2006-12-21 | 2012-09-11 | Basf Se | Catalyst system and method for gas phase oxidation using an upstream layer |
| EP2114562B1 (de) | 2007-01-19 | 2017-11-01 | Basf Se | Verfahren zur herstellung von katalysatorformkörpern, deren aktivmasse ein multielementoxid ist |
| DE102007010422A1 (de) | 2007-03-01 | 2008-09-04 | Basf Se | Verfahren zur Herstellung eines Katalysators bestehend aus einem Trägerkörper und einer auf der Oberfläche des Trägerkörpers aufgebrachten katalytisch aktiven Masse |
| DE102007028332A1 (de) | 2007-06-15 | 2008-12-18 | Basf Se | Verfahren zum Beschicken eines Reaktors mit einem Katalysatorfestbett, das wenigstens ringförmige Katalysatorformkörper K umfasst |
| DE102007028333A1 (de) | 2007-06-15 | 2008-12-18 | Basf Se | Verfahren zum Einbringen einer wenigstens einer Produktionscharge von ringförmigen Schalenkatalysatoren K entnommenen Teilmenge in ein Reaktionsrohr eines Rohrbündelreaktors |
| DE102007038157A1 (de) | 2007-08-13 | 2009-02-19 | Basf Se | Schüttungs-Tragrost |
| WO2009124947A1 (de) | 2008-04-07 | 2009-10-15 | Basf Se | Verfahren zum anfahren eines gasphasenoxidationsreaktors, der eine katalytisch aktive silber-vanadiumoxid-bronze enthält |
| TWI453190B (zh) | 2008-04-07 | 2014-09-21 | Basf Se | 起動氣相氧化反應器之方法 |
-
2007
- 2007-05-18 AT AT07729285T patent/ATE501132T1/de active
- 2007-05-18 DE DE502007006673T patent/DE502007006673D1/de active Active
- 2007-05-18 EP EP07729285A patent/EP2024351B1/de active Active
- 2007-05-18 KR KR1020087030793A patent/KR101395989B1/ko active IP Right Grant
- 2007-05-18 SI SI200730553T patent/SI2024351T1/sl unknown
- 2007-05-18 ES ES07729285T patent/ES2361202T3/es active Active
- 2007-05-18 CN CN2007800183091A patent/CN101448810B/zh active Active
- 2007-05-18 WO PCT/EP2007/054841 patent/WO2007135104A1/de active Application Filing
- 2007-05-18 JP JP2009511484A patent/JP5114475B2/ja active Active
- 2007-05-18 US US12/301,352 patent/US8153825B2/en active Active
- 2007-05-18 MY MYPI20084679A patent/MY145398A/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| ATE501132T1 (de) | 2011-03-15 |
| WO2007135104A8 (de) | 2008-06-12 |
| US20090198073A1 (en) | 2009-08-06 |
| EP2024351B1 (de) | 2011-03-09 |
| JP2009537594A (ja) | 2009-10-29 |
| DE502007006673D1 (de) | 2011-04-21 |
| KR20090014298A (ko) | 2009-02-09 |
| KR101395989B1 (ko) | 2014-05-16 |
| CN101448810A (zh) | 2009-06-03 |
| ES2361202T3 (es) | 2011-06-14 |
| MY145398A (en) | 2012-02-15 |
| SI2024351T1 (sl) | 2011-05-31 |
| WO2007135104A1 (de) | 2007-11-29 |
| EP2024351A1 (de) | 2009-02-18 |
| CN101448810B (zh) | 2012-07-11 |
| US8153825B2 (en) | 2012-04-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5114475B2 (ja) | o−キシロールの気相酸化による無水フタル酸の製造 | |
| JP5114474B2 (ja) | 主要反応器および後方反応器中でのo−キシロールの気相酸化による無水フタル酸の製造 | |
| US6700000B1 (en) | Method for producing phthalic anhydride | |
| JP4696108B2 (ja) | 銀−酸化バナジウム相およびプロモーター相を有する触媒 | |
| JP5586581B2 (ja) | 気相酸化反応器の始動方法 | |
| US20110028740A1 (en) | Method for starting a gas phase oxidation reactor that contains a catalytically active silver-vanadium oxide bronze | |
| KR101706790B1 (ko) | 촉매 기상 산화반응에 의해 방향족 탄화수소로부터 알데히드, 카르복실산 및 카르복실산 무수물, 특히 프탈산 무수물을 형성하기 위한 촉매 및 상기 유형의 촉매 제조 방법 | |
| JP5973436B2 (ja) | o−キシレン及び/又はナフタレンを無水フタル酸に酸化するための触媒 | |
| JPH08318160A (ja) | 気相酸化反応用担持触媒 | |
| US7462727B2 (en) | Multimetal oxide containing silver, vanadium and a promoter metal and use thereof | |
| US8859459B2 (en) | Multilayer catalyst for preparing phthalic anhydride and process for preparing phthalic anhydride | |
| JP5683105B2 (ja) | 調節体層使用下での気相酸化法 | |
| MXPA03001129A (es) | Metodo para la produccion de anhidrido ftalico. | |
| US10710054B2 (en) | Multi-zoned catalyst system for oxidation of o-xylene and/or naphthalene to phthalic anhydride | |
| US20060235232A1 (en) | Catalysts for gas phase oxidations | |
| US10227319B2 (en) | Catalytic converter arrangement with optimized surface for producing phthalic anhydride | |
| US9656983B2 (en) | Process for starting up a gas phase oxidation reactor | |
| JP5879342B2 (ja) | 無水フタル酸を製造するための多層触媒、及び無水フタル酸の製造方法 | |
| JP2015530228A (ja) | カルボン酸および/またはカルボン酸無水物を製造するための触媒 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20101228 Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20101227 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120224 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120524 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120531 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120625 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120702 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20120719 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20120726 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20120821 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120914 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121015 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151019 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5114475 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |