JP4742483B2 - 変異ERαおよび転写活性化の試験系 - Google Patents
変異ERαおよび転写活性化の試験系 Download PDFInfo
- Publication number
- JP4742483B2 JP4742483B2 JP2001543602A JP2001543602A JP4742483B2 JP 4742483 B2 JP4742483 B2 JP 4742483B2 JP 2001543602 A JP2001543602 A JP 2001543602A JP 2001543602 A JP2001543602 A JP 2001543602A JP 4742483 B2 JP4742483 B2 JP 4742483B2
- Authority
- JP
- Japan
- Prior art keywords
- erα
- mutant
- seq
- gene
- human
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images














































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/72—Receptors; Cell surface antigens; Cell surface determinants for hormones
- C07K14/721—Steroid/thyroid hormone superfamily, e.g. GR, EcR, androgen receptor, oestrogen receptor
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Cell Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Toxicology (AREA)
- Endocrinology (AREA)
- Immunology (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Mushroom Cultivation (AREA)
- Investigating Or Analysing Biological Materials (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
Description
【発明の属する技術分野】
本発明はERαなどのリガンド依存性転写因子およびリガンド依存性転写因子をコードする遺伝子に関する。また本発明は、リガンド依存性転写因子と前記リガンド依存性転写因子用の所定のレポーター遺伝子とを含有する細胞に関する。
【0002】
【従来の技術】
様々な細胞機構はリガンド依存性転写因子によって調節される。リガンド依存性転写因子による調節は通常、当該リガンド依存性転写因子が遺伝子を転写活性化する活性を持っているために達成される。転写活性化では、リガンド依存性転写因子とRNAポリメラーゼII複合体とがある遺伝子上で同時に相互作用して遺伝子発現の速度を上昇させると考えられている。転写活性化は多くの場合、真核細胞中で、ある遺伝子が十分に発現されて様々な細胞機構を調節するかどうかを決定することができる。
【0003】
リガンド依存性転写因子によるこのような転写活性化は、リガンド依存性転写活性化因子がその同系のリガンドおよび同系の応答配列に選択的に結合したときに起こりうる。リガンド依存性転写因子がある遺伝子を転写活性化できるかどうかは、当該遺伝子に同系の応答配列が存在するかどうか、または細胞中にその同系のリガンドが存在するかどうかによって決定されうる。
【0004】
ERαは上記のようなリガンド依存性転写因子の一例である。ERαは、エストロゲンの標的細胞、例えば卵巣細胞、乳腺細胞、子宮細胞、骨細胞などに天然に見出される。ERαの転写活性化活性は、通例、ERαがEREとエストロゲン(E2など)に選択的に結合したときに起こる。ERαによる異常トンラス活性化は様々な疾患の一因になりうると報告されている。正常ERαに対して拮抗性である抗エストロゲンを使用する試みがなされている。上記の疾患に使用されるそのような抗エストロゲンの例には、タモキシフェン、ラロキシフェン、4-ヒドロキシタモキシフェンなどがある。
【0005】
【課題を解決するための手段】
本発明は概して、人工細胞、単離された変異ERα、変異ERαをコードする単離されたポリヌクレオチド、試験ERαによるレポーター遺伝子転写活性化活性を定量的に解析する方法、変異リガンド依存性転写因子をスクリーニングする方法、試験ERαによるレポーター遺伝子転写活性化活性を評価する方法、変異ERαの疾患の処置に有用な化合物をスクリーニングする方法、変異ERαのエストロゲン疾患の処置に有用な医薬、変異ERαの使用、試験ERαをコードするポリヌクレオチドの遺伝子型を診断する方法、試験ERαをコードするポリヌクレオチドの遺伝子型を診断する方法、および試験ERαの表現型を診断する方法を提供する。
【0006】
【発明の実施の形態】
5.1.定義
AR:アンドロゲン受容体タンパク質を意味する。
E2:エストラジオールを意味する。
ERα:エストロゲン受容体αタンパク質を意味する。本明細書では「変異ERα」という語句に続けて文字-数字-文字の組み合わせを記すことによって、特定のERα変異体を表す(例えばK303R、S309F、G390D、M396V、G415V、G494V、K531EおよびS578Pなど)。文字-数字-文字の組み合わせにおいて、数字は変異ERα中の置換アミノ酸の相対位置を示し、数字の前にある文字は表示した相対位置における正常ERα中のアミノ酸を示し、数字の後に続く文字は表示した相対位置における与えられた変異ERα中の置換アミノ酸を示す。変異ERα中に2つの置換アミノ酸がある場合は、「変異ERα」という語句に続けて2組の文字-数字-文字の組み合わせを記す(例えばG390D/S578Pなど)。
ERβ:エストロゲン受容体βタンパク質を意味する。
GR:グルココルチコイド受容体タンパク質を意味する。
MR:鉱質コルチコイド受容体タンパク質を意味する。
PPAR:ペルオキシソーム増殖剤応答性受容体タンパク質を意味する。
PR:プロゲステロン受容体タンパク質を意味する。
PXR:プレグナンX受容体タンパク質を意味する。
TR:甲状腺ホルモン受容体タンパク質を意味する。
VDR:ビタミンD受容体タンパク質を意味する。
DR1:次のヌクレオチド配列を持つ受容体応答配列を意味する:
5'-AGGTCAnAGGTCA-3'
(上記配列中、nはA、C、TまたはGを表す)。
DR3:次のヌクレオチド配列を持つ受容体応答配列を意味する:
5'-AGGTCAnnnAGGTCA-3'
(上記配列中、nはA、C、TまたはGを表す)。
DR4:次のヌクレオチド配列を持つ受容体応答配列を意味する:
5'-AGGTCAnnnnAGGTCA-3'
(上記配列中、nはA、C、TまたはGを表す)。
ERE:エストロゲン応答配列を意味する。
MMTV:マウス乳癌ウイルスを意味する。
【0007】
5.2.細胞
本発明の細胞は、レポーター遺伝子を含む染色体を含む。染色体中のレポーター遺伝子はERE、TATA配列およびレポーター配列を含む。また本細胞は、変異ERαまたは変異ERαをコードする遺伝子も含む。この点で、本細胞は、前記変異ERαが前記レポーター遺伝子を転写活性化する活性を持ちうる生物学的システムになる。E2および部分抗エストロゲンが存在する状態での変異ERαによるレポーター遺伝子転写活性化活性は、E2および前記部分抗エストロゲンが存在する状態での正常ERαによる活性よりも一般に高い。あるいは、変異ERαを刺激する可能性がある単独の薬剤として部分抗エストロゲンが存在する状態での変異ERαによるレポーター遺伝子転写活性化活性は、正常ERαを刺激する可能性がある単独の薬剤として部分抗エストロゲンが存在する状態での正常ERαによる活性よりも一般に高い。
【0008】
通例、ERE、TATA配列およびレポーター配列は、レポーター遺伝子中で、当該レポーター遺伝子の転写活性化が可能になるような構成をとる。例えばレポーター遺伝子は、TATA配列の上流に作動的に配置されたEREと、TATA配列の下流に作動的に配置されたレポーター配列とを持つことができる。所望により、レポーター遺伝子は、当該レポーター遺伝子の発現に有利な通常のヌクレオチド配列をさらに含有してもよい。
【0009】
TATA配列は次のヌクレオチド配列を持ちうる:
5'-TATAA-3'。
【0010】
天然の細胞では、EREは正常ERαの受容体応答配列である。正常ERαがE2に結合し、正常ERα-E2複合体がEREに結合すると、正常ERαは転写活性化活性を発揮する。本細胞では、変異ERαに結合して変異ERαにレポーター遺伝子転写活性化活性を持たせることが、EREの機能である。通例、このようなEREは次のヌクレオチド配列に包含される:
5'-AGGTCAnnnTGACCTT-3'
(上記配列中、nはA、G、CまたはTを表す)。また、レポーター遺伝子におけるEREの縦列反復により、より効率のよいレポーター遺伝子転写活性化活性を得ることができる。レポーター遺伝子にはEREの2〜5回の縦列反復を使用することができる。レポーター遺伝子中に使用することができるEREの一例として、アフリカツメガエル・ビテロゲニン遺伝子由来のERE(Cell,57,1139-1146)がある。レポーター遺伝子用EREは、化学合成によって、またはポリメラーゼ連鎖反応(PCR)増幅法を用いるクローニングによって調製することができる。
【0011】
レポーター遺伝子中のレポーター配列は、EREにとって本来外来であるレポーター配列である。したがってレポーター配列とEREとが天然遺伝子に一緒に見出されることはない。また、かかるレポーター配列がレポータータンパク質をコードする場合、当該レポーター配列は通例、細胞中で多かれ少なかれ活性なレポータータンパク質をコードする。レポータータンパク質の例としては、ルシフェラーゼ、分泌アルカリホスファターゼ、β-ガラクトシダーゼ、クロラムフェニコールアセチルトランスフェラーゼ、成長ホルモンなどがある。
【0012】
ERE、TATA配列およびレポーター配列の結合には、従来の方法を使用することができる。レポーター遺伝子を作製した後、そのレポーター遺伝子を染色体に挿入することができる。レポーター遺伝子を宿主細胞に導入すると、レポーター遺伝子の染色体への挿入が起こりうる。このような宿主細胞へのレポーター遺伝子導入法については後述する。
【0013】
細胞中の変異ERαは通例、E2および部分抗エストロゲンの存在下または変異ERαを刺激する可能性がある単独の薬剤としての部分抗エストロゲンの存在下にレポーター遺伝子を転写活性化する特別な活性を持っている。E2および部分抗エストロゲンの存在下に変異ERαがもたらす転写活性化活性は、通例、E2および前記部分抗エストロゲンの存在下に正常ERαがもたらす活性よりも高い。変異ERαを刺激する可能性がある単独の薬剤として部分抗エストロゲンが存在する状態での変異ERαによるレポーター遺伝子転写活性化活性は、正常ERαを刺激する可能性がある単独の薬剤として当該部分抗エストロゲンが存在する状態での正常ERαによる活性よりも高い。トランス活性は転写速度の増加を伴うので、正常ERαおよび変異ERαによるこのような転写活性化は、レポーター遺伝子の発現レベルを測定することによって観察することができる。変異ERαおよび正常ERαがもたらすレポーター遺伝子の発現レベルを同一の点でゼロになるように調節した場合、変異ERαは正常ERαよりも高い発現レベルをもたらすだろう。
【0014】
また変異ERαは、完全抗エストロゲンの存在下では、そのレポーター遺伝子転写活性化活性が阻害されうることにも注目すべきである。変異ERαがもたらすこのようなレポーター遺伝子転写活性化活性は、完全抗エストロゲンの存在下に正常ERαがもたらすレポーター遺伝子転写活性化活性の阻害に似ている。
【0015】
正常ERαは、ある種(ヒト、サル、マウス、ウサギ、ラットなど)が最も普通に持っているとされるERαを包含する。例えば、ヒト正常ERαは配列番号1に示すアミノ酸配列を持つ。このようなヒト正常ERαはTora L.ら,EMBO,第8巻,第7号,1981-1986(1989)に記載されている。
【0016】
部分抗エストロゲンは通例、正常ERαのAF1領域には拮抗性でなく正常ERαのAF2領域に対して拮抗性である。正常ERαのAF2領域と、正常ERαのAF1領域は、それぞれ正常ERαによる転写活性化に関与する正常ERα中の領域である(Metzger D.ら,J. Biol. Chem.,270:9535-9542(1995))。
【0017】
部分抗エストロゲンのこのような性質は、例えばBerry M.ら,EMBO J.,9:2811-2818(1990)に記載のレポーターアッセイを行うことによって観察することができる。このようなレポーターアッセイでは、例えば初代培養ニワトリ胚線維芽細胞(この細胞は例えばSolomon,J.J.,Tissue Cult. Assoc. Manual.,1:7-11(1975)の説明に従って調製することができる)など、内因性正常ERαのAF1領域が強い転写活性化活性を持つ細胞を使用する。ニワトリ胚線維芽細胞を使用する場合は、同線維芽細胞に改変を施して、当該改変線維芽細胞が正常ERαをコードする遺伝子をその細胞内で発現させ、かつ当該改変線維芽細胞がレポーター遺伝子を持つようにする(以下、AF1評価用線維芽細胞という)。AF1評価用線維芽細胞を十分量の部分抗エストロゲンにばく露すると、当該部分抗エストロゲンが正常ERαのAF1領域に対して拮抗性を示しえないかどうかを決定することができる。このような場合、部分抗エストロゲンはAF1評価用線維芽細胞におけるレポーター遺伝子の発現レベルを増大させる。さらに初代培養ニワトリ胚線維芽細胞に2回目の改変を施して、当該第2改変線維芽細胞がAF1領域を欠失させた切断型正常ERαをコードする遺伝子を発現させ、かつ当該第2改変線維芽細胞がレポーター遺伝子を持つようにする(以下、AF2評価用線維芽細胞という)。AF2評価用線維芽細胞を十分量の部分抗エストロゲンにばく露すると、当該部分抗エストロゲンが正常ERαのAF2領域に対して拮抗性を示すかどうかを決定することができる。このような場合、部分抗エストロゲンはAF2評価用線維芽細胞におけるレポーター遺伝子の発現レベルを増大させることができない。
【0018】
このような部分抗エストロゲンの例にはタモキシフェン、4-ヒドロキシタモキシフェン、ラロキシフェンなどがある。
【0019】
完全抗エストロゲンは通例、正常ERαに対して完全な拮抗性を示す抗エストロゲンである。この点で、完全抗エストロゲンはERαに対して部分的作動性を示すことはできない。AF1評価用線維芽細胞またはAF2評価用線維芽細胞を使ったレポーターアッセイでは、完全抗エストロゲンは、前記細胞中の正常ERαまたは切断型正常ERαによるレポーター遺伝子転写活性化活性を実質上示さない。したがって、完全抗エストロゲンとAF1評価用線維芽細胞またはAF2評価用線維芽細胞のいずれかとを用いるこのようなレポーターアッセイでは、レポーター遺伝子の発現レベルは実質上増加しない。
【0020】
このような完全抗エストロゲンの例にはICI182780(Wakeling AEら,Cancer Res.,512:3867-3873(1991))、ZM189154(Dukes Mら,J. Endocrinol.,141:335-341(1994))などがある。
【0021】
変異ERαは、E2および部分抗エストロゲンの存在下または変異ERαを刺激する可能性がある単独の薬剤としての部分抗エストロゲンの存在下で上述のようなレポーター遺伝子転写活性化活性を付与する1または複数の置換アミノ酸を含む。通例、前記1または複数の置換アミノ酸は、変異ERα中の303から578までの1または複数の相対位置に存在する。例えば変異ERαは303、309、390、396、415、494、531、578などから選択される1または複数の相対位置に置換アミノ酸を含みうる。通例、変異ERα中のこのような相対位置は、配列番号1に示すアミノ酸配列とのホモロジー整列に基づく。
【0022】
一般にホモロジー整列には、与えられたアミノ酸配列のホモロジーに基づくアミノ酸配列の整列が包含される。例えば下記表1に、配列番号1に示すアミノ酸配列(ヒト正常ERα)、マウスERα(Genbankアクセッション番号M38651)、ラットERα(X6)(Genbankアクセッション番号X61098)およびラットERα(Y0)(Genbankアクセッション番号Y00102)のホモロジー整列を記載する。
【0023】
【表1】

表1において「hERa.TXT」は配列番号1に示すアミノ酸配列、「mER.TXT」はマウスERαのアミノ酸配列、「ratER(X6).TXT」はラットERα(X6)のアミノ酸配列、「ratER(Y0)」はラットERα(Y0)のアミノ酸配列を示し、アミノ酸配列はアミノ酸の1文字表記を使って記載されている。この整列は市販のソフトウェアGENETYX-WIN SV/Rバージョン4.0(Software Development Co.)を使って作製した。「*」という記号は相対位置303および578に位置するアミノ酸を示している。
【0025】
ホモロジー整列下での相対位置は配列番号1に示すアミノ酸の絶対位置に対応する。例えば相対位置303はホモロジー整列下に配列番号1に示すアミノ酸配列中のN末端から303番目のアミノ酸と整列する変異ERα中のアミノ酸を包含する。また、相対位置578はホモロジー整列下に配列番号1に示すアミノ酸配列中のN末端から578番目のアミノ酸と整列する変異ERα中のアミノ酸を包含する。表1に関して、相対位置303の例には、配列番号1に示すアミノ酸配列においてアミノ末端から303番目のアミノ酸であるリジン、マウスERαのアミノ酸配列においてアミノ末端から307番目のアミノ酸であるリジン、ラットERα(X6)のアミノ酸配列においてアミノ末端から308番目のアミノ酸であるリジン、およびラットERα(Y0)のアミノ酸配列においてアミノ末端から308番目のアミノ酸であるリジンがある。また、表1に関して相対位置578の例には、配列番号1に示すアミノ酸においてアミノ末端から578番目のアミノ酸であるセリン、マウスERαのアミノ酸配列においてアミノ末端から582番目のアミノ酸であるセリン、ラットERα(X6)のアミノ酸配列においてアミノ末端から583番目のアミノ酸であるセリン、およびラットERα(Y0)のアミノ酸配列においてアミノ末端から583番目のアミノ酸であるセリンがある。
【0026】
本発明に関連して行われるホモロジー整列では、配列番号1に示すアミノ酸配列を、変異ERαと配列番号1に示すアミノ酸配列とのホモロジーに基づいて、変異ERαをコードするアミノ酸配列と整列する。ホモロジー整列で配列番号1のアミノ酸配列に対してアミノ酸配列を整列させると、変異ERαは通例、配列番号1に示すアミノ酸配列と少なくとも80%のホモロジーを持つ。
【0027】
変異ERαは哺乳動物などの動物から得ることができる。前記哺乳動物の例としてはヒト、サル、ウサギ、ラット、マウスなどが挙げられる。ヒト変異ERαの場合、変異ERαは一般に595アミノ酸のアミノ酸長を持つ。
【0028】
相対位置303に置換アミノ酸を持つ場合、変異ERαは、正常ERαの相対位置303に存在するリジンを置換アミノ酸に変化させることによって得ることができる。このような場合、変異ERαは相対位置303に置換アミノ酸アルギニンを持つことができる(変異ERαK303Rなど)。ヒト変異ERαK303Rは配列番号2に示すアミノ酸配列を持つ。
【0029】
相対位置309に置換アミノ酸を持つ場合、変異ERαは、正常ERαの相対位置309に存在するセリンを置換アミノ酸に変化させることによって得ることができる。このような場合、変異ERαは相対位置309に置換アミノ酸フェニルアラニンを持つことができる(変異ERαS309Fなど)。ヒト変異ERαS309Fは配列番号3に示すアミノ酸配列を持つ。
【0030】
相対位置390に置換アミノ酸を持つ場合、変異ERαは、正常ERαの相対位置390に存在するグリシンを置換アミノ酸に変化させることによって得ることができる。このような場合、変異ERαは相対位置390に置換アミノ酸アスパラギン酸を持つことができる(変異ERαG390Dなど)。ヒト変異ERαG390Dは配列番号4に示すアミノ酸配列を持つ。
【0031】
相対位置396に置換アミノ酸を持つ場合、変異ERαは、正常ERαの相対位置396に存在するメチオニンを置換アミノ酸に変化させることによって得ることができる。このような場合、変異ERαは相対位置396に置換アミノ酸バリンを持つことができる(変異ERαM396Vなど)。ヒト変異ERαM396Vは配列番号5に示すアミノ酸配列を持つ。
【0032】
相対位置415に置換アミノ酸を持つ場合、変異ERαは、正常ERαの相対位置415に存在するグリシンを置換アミノ酸に変化させることによって得ることができる。このような場合、変異ERαは相対位置415に置換アミノ酸バリンを持つことができる(変異ERαG415Vなど)。ヒト変異ERαG415Vは配列番号6に示すアミノ酸配列を持つ。
【0033】
相対位置494に置換アミノ酸を持つ場合、変異ERαは、正常ERαの相対位置494に存在するグリシンを置換アミノ酸に変化させることによって得ることができる。このような場合、変異ERαは相対位置494に置換アミノ酸バリンを持つことができる(変異ERαG494Vなど)。ヒト変異ERαG494Vは配列番号7に示すアミノ酸配列を持つ。
【0034】
相対位置531に置換アミノ酸を持つ場合、変異ERαは、正常ERαの相対位置531に存在するリジンを置換アミノ酸に変化させることによって得ることができる。このような場合、変異ERαは相対位置531に置換アミノ酸グルタミン酸を持つことができる(変異ERαK531Eなど)。ヒト変異ERαK531Eは配列番号8に示すアミノ酸配列を持つ。
【0035】
相対位置578に置換アミノ酸を持つ場合、変異ERαは、正常ERαの相対位置578に存在するセリンを置換アミノ酸に変化させることによって得ることができる。このような場合、変異ERαは相対位置578に置換アミノ酸プロリンを持つことができる(変異ERαS578Pなど)。ヒト変異ERαS578Pは配列番号9に示すアミノ酸配列を持つ。
【0036】
相対位置390および578に置換アミノ酸を持つ場合、変異ERαは、正常ERαの相対位置390に存在するグリシンを置換アミノ酸に変化させると共に、相対位置578に存在するセリンをもう1つの置換アミノ酸に変化させることによって得ることができる。このような場合、変異ERαは相対位置390に置換アミノ酸アスパラギン酸を、また相対位置578に置換アミノ酸プロリンを持つことができる(変異ERαG390D/S578Pなど)。ヒト変異ERαG390D/S578Pは配列番号10に示すアミノ酸配列を持つ。
【0037】
変異ERαを得るには、周知の標準遺伝コードに従って変異ERαをコードする遺伝子を、細胞によって発現させることができる。このような変異ERα遺伝子は、通例、変異ERαをコードするポリヌクレオチドおよびプロモーターを含む。変異ERα遺伝子は組織試料から単離することができる。また、正常ERαをコードするポリヌクレオチドを変異導入法により変異ERαをコードするように変異させ、その結果得られる変異ERαをコードするポリヌクレオチドの上流にプロモーターを作動的に連結することによって、変異ERαを作製してもよい。正常ERαポリヌクレオチドに1または複数の変異を導入して、変異ERαポリヌクレオチドを得るには、部位特異的変異導入法などの変異導入法を利用することができる。ヒト正常ERαポリヌクレオチドを変異させる場合は、Tora L.ら,EMBO J.,第8巻,第7号:1981-1986(1989)に記載のヌクレオチド配列を持つヒト正常ERαポリヌクレオチドを利用する。
【0038】
変異ERα遺伝子中のプロモーターは、変異ERαを発現させて細胞中に変異ERαをもたらすことができるように転写を開始する。この点で、通常は、当該細胞内で機能することができるプロモーターが、変異ERαをコードするポリヌクレオチドの上流に作動的に連結される。例えば、細胞が動物宿主細胞または分裂酵母縮重細胞に由来する場合、プロモーターの例としては、ラウス肉腫ウイルス(RSV)プロモーター、サイトメガロウイルス(CMV)プロモーター、シミアンウイルス(SV40)の初期および後期プロモーター、MMTVプロモーターなどを挙げることができる。細胞が出芽酵母宿主細胞由来である場合、プロモーターの例としてADH1プロモーターなどを挙げることができる。
【0039】
変異導入法を使用する場合は、正常ERαをコードするポリヌクレオチドを単離し、次に単離されたERαポリヌクレオチドにオリゴヌクレオチドを使って変異を起こさせることができる。次に、得られた変異ERαポリヌクレオチドを利用して、変異ERα遺伝子を作製することができる。
【0040】
オリゴヌクレオチドは、cDNAライブラリーまたは動物のcDNAから正常ERαをコードするcDNAが特異的に増幅されるように設計し合成する。そのようなオリゴヌクレオチドは、正常ERαをコードする周知のヌクレオチド配列(例えばTora L.ら,EMBO J.,第8巻,第7号:1981-1986(1989)またはGenbankなどのデータベースに見出される正常ERαヌクレオチド配列)に基づいて設計することができる。そのような正常ERαヌクレオチド配列としては、ヒト、サル、ウサギ、ラット、マウスなどに由来する正常ERαヌクレオチド配列を利用することができる。設計したオリゴヌクレオチドは、次にDNAシンセサイザー(モデル394,Applied Biosystems)で合成することができる。次にポリメラーゼ連鎖反応(PCR)増幅を利用して、正常ERαポリヌクレオチドをcDNAライブラリーまたはcDNAから単離することができる。ヒト正常ERα遺伝子の場合は、配列番号11および配列番号12に記載のオリゴヌクレオチドを利用して、Tora L.ら,EMBO J.,第8巻,第7号:1981-1986(1989)に記載のヌクレオチドを配列を持つヒト正常ERαポリヌクレオチドをPCR増幅することができる。
【0041】
cDNAはJ.Sambrook,E.F.Frisch,T.Maniatis「Molecular Cloning(第2版)」(コールドスプリングハーバー研究所,1989)に記載の遺伝子工学的手法に従って動物組織(例えばヒト、サル、ウサギ、ラットまたはマウス組織)から得ることができる。このような手法では、肝臓または子宮などの動物組織中のRNAを当該組織から一括して抽出し、そのRNAを一括して当該動物のcDNAに逆転写する。例えば、まず、グアニジン塩酸塩またはグアニジンチオシアネートなどのタンパク質変性剤を含む緩衝液中で動物組織をホモジナイズする。動物組織をホモジナイズすることによって得たタンパク質を変性させるために、フェノールとクロロホルムを含む混合液(以下フェノール-クロロホルムという)などの試薬をさらに加える。変性したタンパク質を遠心分離によって除去した後、回収した上清画分からRNAを一括して抽出する。RNAは、グアニジン塩酸塩/フェノール法、SDS-フェノール法、グアニジンチオシアネート/CsCl法などの方法によって、一括して抽出することができる。ISOGEN(ニッポンジーン)は、上記のRNA一括抽出法に基づく市販キットの一例である。RNAを一括して抽出した後、オリゴdTプライマーをRNA中のポリA配列にアニールさせて、テンプレートたるRNAを一括して逆転写する。逆転写酵素を利用することで、RNAを一本鎖cDNAに一括して逆転写することができる。大腸菌DNAポリメラーゼIを前記一本鎖cDNAと共に使用することにより、一本鎖cDNAからcDNAを合成することができる。大腸菌DNAポリメラーゼIを使用する際は、大腸菌DNAポリメラーゼIをより効率よく作動させるプライマーが生成するように、大腸菌RNaseHも使用する。cDNAは通常の精製法を使って、例えばフェノール-クロロホルム抽出とエタノール沈殿などによって精製することができる。このような方法に基づく市販キットの例には、cDNA Synthesis System Plus(Amersham Pharmacia Biotech)、TimeSaver cDNA Synthesis Kit(Amersham Pharmacia Biotech)などがある。
【0042】
次に、正常ERαポリヌクレオチドをcDNAから単離する。正常ERαポリヌクレオチドの単離に利用することができる単離法としては、PCR増幅の使用を挙げることができる。PCR増幅では通例、cDNAから正常ERαポリヌクレオチドを増幅する。このPCR増幅におけるPCR混合物は、十分量のcDNA、十分量のフォワードオリゴヌクレオチドとリバースオリゴヌクレオチド、耐熱性DNAポリメラーゼ(LT-Taqポリメラーゼ(宝酒造)など)、dNTP(dATP、dTTP、dGTP、dCTP)およびPCR増幅緩衝液を含みうる。ヒト正常ERαポリヌクレオチドを増幅するPCR混合物には、10ngのcDNAおよび各10pmolのフォワードオリゴヌクレオチドおよびリバースオリゴヌクレオチド(配列番号11および配列番号12)を使用することができる。次に、PCR増幅では、PCR混合物を、アニーリング、伸長および変性のためのインキュベーションサイクルにかける。例えばPCR増幅では、PCRsystem 9700(Applied Biosystems)などのサーマルサイクラーを使って、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを35回繰り返すことができる。cDNAを用いたPCR増幅後に、得られたPCR混合物の全量を低融点アガロースゲル電気泳動(ニッポンジーン、アガロースL)にかける。正常ERαポリヌクレオチドを含むバンドが存在することを確認した後、正常ERαポリヌクレオチドを低融点アガロースゲルから回収する。
【0043】
cDNAライブラリーとしては、QUICK clone cDNAs(CLONETECH製)などの市販の動物由来cDNAライブラリーを利用することができる。cDNAライブラリーは上述のように単離することができる。
【0044】
回収された正常ERαポリヌクレオチドのヌクレオチド配列は、ダイレクトシークエンシング用の正常ERαポリヌクレオチド試料を調製することによって確認することができる。また、DNA蛍光シークエンシング法を使って、正常ERαポリヌクレオチドを配列決定してもよい。この点に関して、正常ERαポリヌクレオチド試料の調製には、Dye Terminator Sequencing Kit FS(Applied Biosystems)などの市販の蛍光シークエンシング用試薬を利用することができる。正常ERαポリヌクレオチドの蛍光シークエンシングは、ABI自動シークエンサー(モデル377、Applied Biosystems)などの自動シークエンサーを使って行うことができる。また、正常ERαポリヌクレオチドは手作業で配列決定してもよい(Biotechniques,7,494(1989))。
【0045】
便宜上、単離された正常ERαポリヌクレオチドを大腸菌などの宿主中で複製する能力を持つベクターに挿入することができる。例えば、与えられた単離正常ERαポリヌクレオチドが突出末端を持つ場合は、単離された正常ERαポリヌクレオチド約1μgの末端をDNA Blunting Kit(宝酒造)で処理することによって平滑化する。次にT4ポリヌクレオチドキナーゼを使って、平滑末端化正常ERαポリヌクレオチドの末端をリン酸化することができる。フェノール処理の後、正常ERαポリヌクレオチドをエタノール沈殿によって精製し、大腸菌中で複製する能力を持つベクターに挿入することができる。正常ERαポリヌクレオチドを含むこの大腸菌ベクターは、大腸菌宿主細胞にクローニングすることができる。
【0046】
次に、正常ERαポリヌクレオチドを含む大腸菌ベクターをクローン化大腸菌細胞から単離することができる。次に、正常ERαポリヌクレオチドを含む単離された大腸菌ベクターをテンプレートとして使用して、最終的に得られる変異ERαポリヌクレオチドが所望の相対位置に置換アミノ酸をコードする変異コドンを含むように、正常ERαポリヌクレオチドに変異を起こさせる(すなわちヌクレオチド置換を導入する)。
【0047】
所望のヌクレオチド置換は、J.Sambrook,E.F. Frisch,T.Maniatis「Molecular Cloning(第2版)」(コールドスプリングハーバー研究所,1989)に記載の部位特異的変異導入法またはMcClary JAら,Biotechniques 1989(3):282-289に記載の部位特異的変異導入法に従って、正常ERαポリヌクレオチドに導入することができる。例えば所望のヌクレオチド置換は、Stratagene製のQuikChange Site-Directed Mutagenesis Kitなどの市販キットを使用することによって、正常ERαポリヌクレオチドに導入することができる。通例、このような部位特異的変異導入法では、所望のヌクレオチド置換を導入するオリゴヌクレオチドを使用する。正常ERαポリヌクレオチドに使用した部位特異的変異導入法を、QuikChange Site-Directed Mutagenesis Kitに関して、以下に詳述する。
【0048】
QuikChange Site-Directed Mutagenesis Kitでは、2つのオリゴヌクレオチドを利用して、正常ERαポリヌクレオチドに所望のヌクレオチド置換をもたらす。このような2つのオリゴヌクレオチドの組み合わせとして、配列番号13に記載のオリゴヌクレオチドと配列番号14に記載のオリゴヌクレオチドとを含む組み合わせ、配列番号15に記載のオリゴヌクレオチドと配列番号16に記載のオリゴヌクレオチドとを含む組み合わせ、配列番号17に記載のオリゴヌクレオチドと配列番号18に記載のオリゴヌクレオチドとを含む組み合わせ、配列番号19に記載のオリゴヌクレオチドと配列番号20に記載のオリゴヌクレオチドとを含む組み合わせ、配列番号21に記載のオリゴヌクレオチドと配列番号22に記載のオリゴヌクレオチドとを含む組み合わせ、配列番号23に記載のオリゴヌクレオチドと配列番号24に記載のオリゴヌクレオチドとを含む組み合わせ、配列番号25に記載のオリゴヌクレオチドと配列番号26に記載のオリゴヌクレオチドとを含む組み合わせ、または配列番号27に記載のオリゴヌクレオチドと配列番号28に記載のオリゴヌクレオチドとを含む組み合わせを、正常ERαポリヌクレオチドに使用することができる。下記表2に、ヌクレオチド置換の位置にコードされるアミノ酸の相対位置と、上述したオリゴヌクレオチドの組み合わせを使用することによって得られる変異コドンを示す。
【0049】
【表2】
【0050】
本発明細胞を作出するには、通常、変異ERα遺伝子とレポーター遺伝子とを宿主細胞に導入する。レポーター遺伝子は、当該レポーター遺伝子が宿主細胞の染色体に挿入されるような形で、宿主細胞に導入される。変異ERα遺伝子は一過性発現が起こるように宿主細胞に導入されるか、または宿主細胞の染色体に挿入される。変異ERα遺伝子を宿主細胞の染色体に挿入する場合、変異ERα遺伝子とレポーター遺伝子とを1つの染色体に導入してもよいし、変異ERα遺伝子をレポーター遺伝子に利用する染色体とは異なる染色体に挿入してもよい。
【0051】
宿主細胞は通例、正常または変異ERαを発現させない。宿主細胞の例としては、CG1945(CLONETECH)などの出芽酵母細胞、HeLa細胞、CV-1細胞、Hepa1細胞、NIH3T3細胞、HepG2細胞、COS1細胞、BF-2細胞、CHH-1細胞および昆虫細胞などの動物細胞を挙げることができる。
【0052】
変異ERα遺伝子およびレポーター遺伝子は、変異ERα遺伝子およびレポーター遺伝子を宿主細胞に導入することができるように、ベクターに挿入することができる。そのようなベクターは通例、当該ベクターが細胞中で複製できるように複製起点を持つ。所望により、ベクターは選択マーカー遺伝子を持ってもよい。
【0053】
出芽酵母細胞を宿主細胞として使用する場合、ベクターの例としては、プラスミドpGBT9、pGAD424、pACT2(CLONETECH)などを挙げることができる。哺乳類細胞を宿主細胞として使用する場合、ベクターの例としては、pRc/RSV、pRc/CMV(Invitrogen)などのプラスミド、ウイルス由来の自律複製起点を含むベクター、例えばウシ乳頭腫ウイルスプラスミドpBPV(Amersham Pharmacia Biotech)、EVウイルスプラスミドpCEP4(Invitrogen)などを挙げることができる。
【0054】
変異ERαをコードするベクター(以下、変異ERαベクターという)を作製する場合、変異ERαポリヌクレオチドをベクターに挿入することで、変異ERαベクターと一緒に変異ERα遺伝子も作製することができるように、当該ベクターはさらにプロモーターを含むことが好ましい。同様に、レポーター遺伝子をコードするベクター(以下、レポーターベクターという)を作製する場合、レポーターベクターと一緒にレポーター遺伝子も作製することができるように、当該ベクターはTATA配列またはEREを含むことが好ましい。
【0055】
変異ERαベクターを動物宿主細胞用の変異ERα遺伝子と一緒に作製する際には、pRc/RSVまたはpRc/CMVを利用することができる。プラスミドpRc/RSVおよびpRc/CMVは、動物宿主細胞由来の細胞内で機能することができるプロモーターと、前記プロモーターの下流に作動的に配置されたクローニング部位とを含んでいる。この点で、変異ERαベクターは、変異ERαポリヌクレオチドをpRc/RSVまたはpRc/CMVのクローニング部位に挿入することにより、変異ERα遺伝子と一緒に作製することができる。pRc/RSVおよびpRc/CMVはSV40の自律複製起点(ori)も含んでいるので、pRc/RSVおよびpRc/CMVは、所望によりori(-)SV40ゲノムで形質転換した動物宿主細胞に変異ERα遺伝子を導入するために使用することができる。ori(-)SV40ゲノムで形質転換したそのような動物宿主細胞としてはCOS細胞が挙げられる。ori(-)SV40ゲノムで形質転換したそのような動物宿主細胞に導入すると、pRc/RSVまたはpRc/CMVから作製した変異ERαベクターは、細胞中でかなり大きいコピー数まで増加することができるので、変異ERα遺伝子を大量に発現させることができる。
【0056】
変異ERαベクターを出芽酵母宿主細胞に導入する場合は、pACT2を使って変異ERαベクターを作製することが好ましい。pACT2はADH1プロモーターを持っているので、変異ERαポリヌクレオチドをADH1プロモーターの下流に挿入することにより、変異ERαベクターと一緒に変異ERα遺伝子を作製することができる。このような場合、変異ERαベクターは変異ER遺伝子を大量に発現させることができる。
【0057】
変異ERα遺伝子の導入には、宿主細胞のタイプに応じて、従来の技術を使用することができる。哺乳類細胞または昆虫細胞を宿主細胞として使用する場合は、例えばリン酸カルシウム法、DEAE-デキストラン法、エレクトロポレーション、リポフェクションなどを使用することができる。酵母細胞を宿主細胞として使用する場合は、リチウム法(例えばYeast Transfomation Kit(CLONETECH)を用いる方法)などを使用することができる。
【0058】
また、変異ERα遺伝子をウイルスDNAとして宿主細胞に導入する場合、変異ERα遺伝子は上記の技術によって宿主細胞に導入することができるばかりでなく、宿主細胞をウイルス型のレポーター遺伝子および変異ERα遺伝子を含む組換えウイルス粒子に感染させることによって、変異ERα遺伝子を宿主細胞に導入することもできる。例えば、動物宿主細胞にはワクシニアウイルスなどのウイルスを利用することができ、昆虫動物細胞を宿主細胞として使用する場合は、バキュロウイルスなどの昆虫ウイルスを利用することができる。
【0059】
変異ERαベクターまたはレポーターベクターが上述のように選択マーカー遺伝子を含む場合は、その選択マーカー遺伝子を使って本発明の細胞をクローニングすることができる。このような場合は、選択マーカー遺伝子を利用することにより、細胞に対して致死的活性を示す選択薬に対する薬剤耐性を付与することができる。この場合は、当該選択薬を添加した培地で細胞を培養することによって、本発明の細胞をクローニングすることができる。選択マーカー遺伝子と選択薬の代表的な組み合わせには、ネオマイシン耐性付与選択マーカー遺伝子とネオマイシンとの組み合わせ、ハイグロマイシン耐性付与選択マーカー遺伝子とハイグロマイシンとの組み合わせ、およびブラストサイジンS耐性付与選択マーカー遺伝子とブラストサイジンSとの組み合わせなどがある。選択マーカー遺伝子が細胞の栄養要求性を補足する栄養素をコードする場合は、栄養素を実質上含まない最少培地を使って細胞を培養することができる。また、エストロゲン結合活性を測定するアッセイも、細胞のクローニングに使用することができる。
【0060】
レポーター遺伝子を宿主細胞に導入する場合、レポーター遺伝子は通常、線状遺伝子として導入される。線状レポーター遺伝子により、宿主細胞染色体へのレポーター遺伝子の挿入が可能になる。レポーターベクターを利用する場合、レポーターベクターは制限酵素消化によって線状化することができる。線状レポーター遺伝子は、リポフェクション法を利用して、宿主細胞に導入することができる。
【0061】
また、変異ERα遺伝子を導入する前にレポーター遺伝子を宿主細胞に導入して、安定形質転換カセット細胞を得ることができることに注意すべきである。安定形質転換カセット細胞はその染色体にレポーター遺伝子を安定に含むので、レポーター遺伝子は遺伝的に子孫世代に受け継がれることができる。安定形質転換カセット細胞を作製するために、レポーター遺伝子を宿主細胞の染色体に導入し、その宿主細胞を数週間培養することができる。選択マーカー遺伝子を利用する場合は、数週間の培養後に、選択マーカー遺伝子を使って、安定形質転換カセット細胞をクローニングすることができる。例えば、選択薬を補足した培地で形質転換宿主細胞を数週間継続的に培養して、安定形質転換カセット細胞をクローニングすることができる。次に変異ERα遺伝子を安定形質転換カセット細胞に導入して、本発明の細胞を作製することができる。
【0062】
また、変異ERα遺伝子は、宿主細胞がレポーター遺伝子と変異ERα遺伝子とで安定に形質転換されるような形で、レポーター遺伝子を持つ宿主細胞に導入することもできる。
【0063】
本発明の細胞は、変異ERαの疾患の処置に有用な化合物をスクリーニングするために利用することができる。そのような変異ERαの疾患として、変異ERαによる異常な転写活性化を伴う疾患(乳癌など)を挙げることができる。そのような化合物をスクリーニングするには、変異ERαに対して拮抗性または作動性であると思われる有効量の試験化合物に細胞をばく露して、レポーター遺伝子の転写活性化レベルを測定する。
【0064】
細胞は通例、1〜数日間にわたって十分量の試験化合物にばく露する。細胞は変異ERαに対する作動条件または拮抗条件下で試験化合物にばく露することができる。作動条件では通例、変異ERαを刺激する可能性がある単独の薬剤としての試験化合物にアッセイ細胞をばく露する。拮抗条件では通例、試験化合物およびE2にアッセイ細胞をばく露する。
【0065】
ばく露後に、レポーター遺伝子の発現レベルを測定することによって、レポーター遺伝子の転写活性化レベルを測定する。このような場合、レポータータンパク質またはレポーターRNA(レポーター配列によってコードされるもの)は細胞内に貯蔵されるか、細胞から分泌されるので、発現レベルはそれらを使って測定することができる。レポーター遺伝子の発現レベルはノーザンブロット解析もしくはウェスタンブロット解析によって、またはレポータータンパク質の活性レベルを測定することによって測定することができる。レポータータンパク質の活性レベルは通例、レポーター遺伝子の発現レベルを示す。
【0066】
例えば、レポーター遺伝子がレポータータンパク質としてルシフェラーゼをコードする場合、レポーター遺伝子の発現レベルは、ルシフェリンとルシフェラーゼとを反応させることによって得られるルミネセンスによって測定することができる。このような場合は、粗細胞抽出物を細胞から調製し、その粗細胞抽出物にルシフェリンを加える。ルシフェリンは細胞抽出物中のルシフェラーゼと室温で反応させることができる。ルシフェリンの添加によって得られるルミネセンスは、通常、レポーター遺伝子の発現レベルの指標として測定される。というのも、粗細胞抽出物は、細胞内で発現され粗細胞抽出物中に存在するルシフェラーゼのレベルに比例する強度のルミネセンスをもたらすからである。得られた粗細胞抽出物中のルミネセンスを測定するには、ルミノメーターを利用することができる。
【0067】
次に、測定された転写活性化レベルをコントロールと比較して、試験化合物の作動または拮抗作用を評価することができる。試験化合物のスクリーニングにおけるそのようなコントロールは、細胞を試験化合物にばく露しなかった場合のレポーター遺伝子の予想転写活性化レベルとすることができる。作動条件下で変異ERαによるレポーター遺伝子の転写活性化レベルがコントロールより高い場合、当該試験化合物は変異ERαに対するアゴニストとして評価される。
【0068】
また、細胞を拮抗条件下でE2および試験化合物にばく露する場合は、試験化合物を変異ERαに対するアンタゴニストとして評価することができる。このような場合、コントロールは、等量のE2の存在下で予想される変異ERαによるレポーター遺伝子の転写活性化レベルとすることができる。変異ERαによるレポーター遺伝子の転写活性化レベルがコントロールより低い場合、当該試験化合物は変異ERαに対して拮抗性であると評価される。
【0069】
次に、変異ERαに対して作動性または拮抗性である上述のような試験化合物を、変異ERαの疾患の処置に有用な化合物として選択することができる。このような場合、細胞を作動条件下でばく露する時は、コントロールより有意に高いレポーター遺伝子の転写活性化レベルを与える試験化合物を通常は選択する。細胞を拮抗条件下でばく露する時は、コントロールよりも有意に低いレポーター遺伝子の転写活性化レベルを与える試験化合物を通常は選択する。
【0070】
また、正常リガンド依存性転写因子の疾患を処置するための化合物をスクリーニングすることもできる。そのような場合は、変異ERα遺伝子の代わりに正常リガンド依存性転写因子をコードする遺伝子を宿主細胞に導入する。そのような正常リガンド依存性転写因子の例には、正常ERβ(Genbankアクセッション番号AB006590)、正常AR(Genbankアクセッション番号M23263)、正常GR(Genbankアクセッション番号M10901)、正常TRα(M24748)、正常PR(Genbankアクセッション番号15716)、正常PXR(Genbankアクセッション番号AF061056)、正常VDR(Genbankアクセッション番号J03258)などの正常親油性ビタミン受容体、正常RAR(Genbankアクセッション番号06538)、正常MR(Genbankアクセッション番号M16801)、正常PPARγ(Genbankアクセッション番号U79012)などがある。このような場合、レポーター遺伝子はEREの代わりに当該正常リガンド依存性転写因子に対応する適切な受容体応答配列を含む。
【0071】
5.3.診断方法
本発明の診断方法では、試験ERαの表現型または試験ERαをコードするポリヌクレオチドの遺伝子型の診断が行われる。遺伝子型診断法では、上記5.2項で述べたように、試験ERαをコードするポリヌクレオチドがその中にレポーター遺伝子転写活性化活性を付与する1または複数の置換アミノ酸を与える変異コドンを含むかどうかを決定することができる。表現型診断法では、上記5.2項で述べたように、試験ERαがその中にレポーター遺伝子転写活性化活性を付与する1または複数の置換アミノ酸を含むかどうかを決定することができる。
【0072】
遺伝子型診断法では通例、試験ERαポリヌクレオチドを調製し、変異コドンを検索し、もし存在すればその変異コドン中の変異を決定する。そのような遺伝子型診断法の例には、PCR増幅およびヌクレオチド配列決定法、一本鎖高次構造多型(SSCP)法、制限酵素切断断片長多型(RFLP)法、ハイブリダイゼーション法などがある。
【0073】
遺伝子型診断法用の試験ERαポリヌクレオチドは、試験ゲノムDNAまたは試験cDNAを調製することによって調製できる。このような場合、試験ERαポリヌクレオチドを含有する試験ゲノムDNAまたは試験cDNAは、試験動物(例えば被験者)から得た試験試料から一括して調製される。そのような試験試料は非外科的方法、外科的方法(例えば細針または生検)などによって得ることができる。そのような試験試料の例には、試験哺乳動物の細胞組織、例えば毛髪、末梢血、口内上皮組織、肝臓、前立腺、卵巣、子宮、乳腺などがあり、そこから試験DNAまたは試験cDNAを抽出することができる。
【0074】
例えば試験ゲノムDNAはTAKARA PCR Technical news No. 2(宝酒造、1991年9月)に記載の方法に従って調製することができる。このような場合、試験哺乳動物から得た毛髪2〜3本の試験試料を滅菌水とエタノールで洗浄し、長さ2〜3mmに切断する。次に毛髪中の試験細胞を十分量(例えば200μl)のBCL緩衝液(10mM トリス-HCl(pH7.5)、5mM MgCl2、0.32Mショ糖、1%トリトンX-100)で溶解する。溶解した試験細胞にプロテイナーゼKおよびSDSをそれぞれ100μl/mlおよび0.5%(w/v)の最終濃度で添加し、混合することによって、試験ゲノムDNAから不要なタンパク質を除去する。反応混合物を70℃でインキュベートした後、試験ゲノムDNAをフェノール-クロロホルム抽出によって精製することができる。
【0075】
また、試験試料が末梢血である場合は、例えばDNA Extraction Kit(Stratagene)で試験試料を処理することにより、試験ゲノムDNAを一括して得ることができる。
【0076】
また、試験試料を生検試料から得る場合は、細胞組織中のRNAを一括して逆転写することにより、試験試料から試験cDNAを調製することができる。RNAは、好ましくは当該細胞組織がまだ新鮮なうちに、TRIZOL試薬(Gibco)を使用することによって、細胞組織から一括して得ることができる。
【0077】
また、試験ゲノムDNAは、村松正實編「ラボマニュアル遺伝子工学」(丸善、1988)に記載の方法に従って調製することもできる。
【0078】
さらに試験cDNAは、上記5.2項で述べたように、J.Sambrook,E.F.Frisch,T.Maniatis「Molecular Cloning(第2版)」(コールドスプリングハーバー研究所,1989)に記載の遺伝子工学的手法に従って調製してもよい。
【0079】
遺伝子型診断法において変異コドンを検索する場合、試験ERαポリヌクレオチド中の検索領域には、変異コドンであると思われるコドンが含まれる。したがって試験ERαポリヌクレオチド中の検索領域は、試験ERα中の相対位置303〜578にあるアミノ酸をコードする試験ERαポリヌクレオチド中のコドンを含みうる。例えばこのような遺伝子型診断法では、303、309、390、396、、415、494、531、578などから選択される相対位置のアミノ酸をコードする試験ERαポリヌクレオチド中のコドンを検索領域に含めることができる。
【0080】
次に、PCR増幅および配列決定法ならびにSSCP法では、調製した試験cDNAまたは試験ゲノムDNAを使って、そこから試験ERαポリヌクレオチド中の検索領域を特異的にPCR増幅することができる。試験ERαポリヌクレオチド中に存在する検索領域を試験cDNAまたは試験ゲノムDNAから特異的にPCR増幅するには、検索オリゴヌクレオチドを利用することができる。
【0081】
このPCR増幅における検索オリゴヌクレオチドは通例、試験ERαポリヌクレオチド中の検索領域を特異的にPCR増幅するように設計される。検索オリゴヌクレオチドは8〜50bp(好ましくは15〜40bp)のサイズと30%〜70%のGC含量を持ちうる。このような検索オリゴヌクレオチドは、DNAシンセサイザーにより、β-シアノエチルホスホアミダイト法、チオホスファイト法などを用いて合成することができる。また、検索オリゴヌクレオチドは標識を持たないか、非放射性の標識を持つか、または32Pなどによる放射性標識を持つことができる。PCR増幅では通例、フォワード検索オリゴヌクレオチドとリバース検索オリゴヌクレオチドとの組み合わせを利用して、試験ERαポリヌクレオチド中の検索領域を特異的にPCR増幅する。ヒト試験ERαポリヌクレオチド用のこのようなフォワードおよびリバース検索オリゴヌクレオチドの例を、検索領域にコードされるアミノ酸の相対位置と共に下記表3に示す。
【0082】
【表3】
試験ERαポリヌクレオチド中の検索領域は、Saikiら,Science,第230巻,1350-1354頁(1985)に記載の方法に従って、試験cDNAまたは試験ゲノムDNAから特異的にPCR増幅することができる。このPCR増幅におけるPCR混合物は、1.5mM〜3.0mMの塩化マグネシウム、耐熱性DNAポリメラーゼ、dNTP(dATP、dTTP、dGTPおよびdCTP)、リバース検索オリゴヌクレオチドの1つと組み合わせたフォワード検索オリゴヌクレオチドの一つ、および試験ゲノムDNAまたは試験cDNAを含みうる。このPCR増幅では、変性インキュベーション、アニーリングインキュベーションおよび伸長インキュベーションを課すインキュベーションサイクルを20〜50回(好ましくは25〜40回)繰り返すことができる。変性インキュベーションでは、90℃〜95℃(好ましくは94℃〜95℃)で1分〜5分間(好ましくは1分〜2分間)PCR混合物をインキュベートすることができる。変性インキュベーションに続くアニーリングインキュベーションでは、30℃〜70℃(好ましくは40℃〜60℃)で3秒〜3分間(好ましくは5秒〜2分間)PCR混合物をインキュベートすることができる。変性インキュベーションに続く伸長インキュベーションでは、70℃〜75℃(好ましくは72℃〜73℃)で約15秒〜5分間(好ましくは30秒〜4分間)PCR混合物をインキュベートすることができる。
【0084】
PCR増幅およびヌクレオチド配列決定法を利用する場合、遺伝子型診断法では、得られたPCR混合物を低融点アガロースゲル電気泳動にかけることができる。増幅された検索領域を含むポリヌクレオチド(以下、検索領域ポリヌクレオチドという)を低融点アガロースゲルから回収し、配列決定することにより、検索領域ポリヌクレオチドのヌクレオチド配列を得る。
【0085】
次に、検索領域ポリヌクレオチドを配列決定し、そのヌクレオチド配列中の変異を決定することにより、変異コドンがあればその変異コドン中の変異を決定することができる。検索領域ポリヌクレオチドを配列決定する際には、ダイレクトシークエンシング法または自動シークエンシング法を利用することができる。ダイレクトシークエンシング法の例には、手作業によるシークエンシング法(Maxam,A.M.およびW.Gilbert,Proc. Natl. Acad. Sci. USA,74,560,1977に記載のMaxam Gilbert法)、Sanger法(Sanger,F.およびA.R.Coulson,J. Mol. Biol.,94,441,1975ならびにSanger,F.,NicklenおよびA.R.,Coulson,Proc. Natl. Acad. Sci. USA.,74,5463,1977に記載)、BioTechniques,7,494(1989)に記載の方法などがある。ABI自動シークエンサー(モデル377、Applied Biosystems)などの自動DNAシークエンサーを使用する場合は、ABI Big Dye Terminator Cycle Sequencing Ready Reaction Kitなどの適当なDNAシークエンシングキットを使って、自動DNAシークエンサー用の検索領域を調製することができる。配列決定が終わったら、次に、検索領域ポリヌクレオチドのヌクレオチド配列を正常ERαをコードするヌクレオチド配列と比較して、検索領域中に変異コドンがあればその中の変異を決定することができる。
【0086】
SSCP法を利用する場合は、得られたPCR混合物を、Hum. Mutation,第2巻,338頁に従って非変性ポリアクリルアミドゲル電気泳動にかける。このような場合、検索領域ポリヌクレオチドが放射標識され、その放射能を使って非変性ポリアクリルアミドゲル中の検索領域ポリヌクレオチドを検出することができるように、上記PCR増幅では放射標識オリゴヌクレオチドを利用することが好ましい。このようなSSCP法では、放射標識された検索領域ポリヌクレオチドを一本鎖ポリヌクレオチドに熱変性し、緩衝液中で非変性ポリアクリルアミドゲル電気泳動にかけて、各一本鎖ポリヌクレオチドを分離することができる。非変性ポリアクリルアミドゲル電気泳動に利用することができる緩衝液の例には、トリス-リン酸(pH7.5〜8.0)、トリス-酢酸(pH7.5〜8.0)、トリス-ホウ酸(pH7.5〜8.3)などがあり、トリス-ホウ酸(pH7.5〜8.3)は好ましい。さらに、EDTAなどの非変性ポリアクリルアミドゲル電気泳動用補助成分を利用してもよい。このような非変性ポリアクリルアミドゲル電気泳動の条件としては、30〜40Wの定電力、4℃〜室温(約20〜25℃)で1時間〜4時間を挙げることができる。
【0087】
非変性ポリアクリルアミドゲル電気泳動後に、その非変性ポリアクリルアミドゲルをろ紙に転写し、X線フィルムと接触させて、放射標識検索領域ポリヌクレオチドからの放射線にX線フィルムを露出する。X線フィルムの露出には適当なカセットを利用することができる。X線フィルムの現像によって得られるオートラジオグラムにより、放射標識検索領域ポリヌクレオチドの移動度を標準物質の移動度と比較することができる。前記標準物質の移動度は、検索領域ポリヌクレオチドが正常ERαポリヌクレオチドの正常コドンだけからなる場合に予想される移動度とすることができる。標準物質の移動度とは異なる放射標識検索領域ポリヌクレオチドの移動度は、通例、当該放射標識検索領域中に1または複数の変異コドンが存在することを示す。
【0088】
次に、熱水または沸騰水を使って、放射標識検索領域ポリヌクレオチドを非変性ポリアクリルアミドゲルから回収することができる。得られた放射標識検索領域は再びPCR増幅した後、配列決定用に調製することができる。変異コドンがある場合は、上記の方法と同様にPCR増幅およびヌクレオチド配列法で、変異コドン中の変異を決定することができる。
【0089】
ハイブリダイゼーション法では、通例、プローブオリゴヌクレオチドを利用して、当該プローブオリゴヌクレオチドが検索領域にハイブリダイズすることができるかどうかを観察する。ハイブリダイゼーション法では、検索領域ポリヌクレオチド、調製した試験cDNA、調製した試験ゲノムDNA、精製した試験ERαポリヌクレオチドなどを利用することによって、検索領域を用意することができる。また、ハイブリダイゼーション法では、検索領域ポリヌクレオチドを制限酵素消化した後、制限酵素消化された検索領域ポリヌクレオチドを利用して、プローブオリゴヌクレオチドがそれにハイブリダイズすることができるかどうかを観察してもよい。
【0090】
プローブオリゴヌクレオチドは15〜40bpのサイズと30%〜70%のGC含量を持つ。このようなプローブオリゴヌクレオチドはDNAシンセサイザーにより、β-シアノエチルホスホアミダイト法、チオホスファイト法などを用いて合成することができる。またプローブオリゴヌクレオチドは通例、非放射性の(例えばビオチンなどによる)標識を持つか、または32Pなどによる放射標識を持つ。
【0091】
検索領域が正常ERαポリヌクレオチドの正常コドンだけから構成される場合、プローブオリゴヌクレオチドは検索領域のヌクレオチド配列から構成することができる。このようなヌクレオチド配列により、プローブオリゴヌクレオチドは、試験ERαポリヌクレオチド中の検索領域が正常ERαポリヌクレオチドの正常コドンだけから構成される場合には、ストリンジェントな条件で試験ERαポリヌクレオチド中の検索領域にハイブリダイズすることができる。ヒト試験ERαポリヌクレオチド用のこのようなプローブオリゴヌクレオチドの例を、検索領域にコードされるアミノ酸の相対位置と共に下記表3に示す。
【0092】
【表4】
通例、ハイブリダイゼーション法はストリンジェントな条件で行われる。前記ストリンジェントな条件として、例えばプレハイブリダイゼーション処理またはハイブリダイゼーション処理を、プレハイブリダイゼーション緩衝液およびハイブリダイゼーション緩衝液中で行い、洗浄緩衝液中で15分間の洗浄を2回行う。ハイブリダイゼーション法では、所望により、0.1×SSC(0.015M NaCl、0.0015Mクエン酸ナトリウム)および0.5%SDSを含む緩衝液中で30分間の洗浄をさらに行ってもよい。プレハイブリダイゼーション緩衝液としては、6×SSC(0.9M NaCl、0.09Mクエン酸ナトリウム)、5×デンハルト液(0.1%(w/v)フィコール400、0.1%(w/v)ポリピロリドンおよび0.1%BSA)、0.5%(w/v)SDSおよび100μg/mlのサケ精子DNAを含む緩衝液を利用することができる。またプレハイブリダイゼーション緩衝液として、サケ精子DNAを100μg/mlの濃度になるように添加したDIG EASY Hyb緩衝液(Boehringer Mannheim)を利用してもよい。さらにプレハイブリダイゼーション緩衝液として、6×SSPE(0.9M NaCl、0.052M NaH2PO4、7.5mM EDTA)、0.5%SDS、5×デンハルト液および0.1mg/mlのサケ精子DNAを含む緩衝液を利用してもよい。ハイブリダイゼーション緩衝液としては、プローブオリゴヌクレオチドを十分量になるように添加したプレハイブリダイゼーション緩衝液を利用することができる。プレハイブリダイゼーション処理およびハイブリダイゼーション処理の温度はプローブオリゴヌクレオチドの長さによって変わりうるが、例えばプローブオリゴヌクレオチドのTm値〜プローブオリゴヌクレオチドのTm値より2〜3℃低い温度とすることができる。洗浄温度もオリゴヌクレオチドの長さによって変わりうるが、例えば室温で行うことができる。このような場合、Tm値は、ハイブリダイゼーション緩衝液中でプローブオリゴヌクレオチド中のヌクレオチド単位と水素結合を形成すべきヌクレオチド単位の量を見積もり、次に、水素結合を形成すべきプローブオリゴヌクレオチド中のAまたはTヌクレオチド単位について2℃を加え、水素結合を形成すべきプローブオリゴヌクレオチド中のGまたはTヌクレオチド単位について4℃を加えることによって得られる温度を加えることによって得ることができる。
【0094】
例えばハイブリダイゼーション法は、ドットブロットハイブリダイゼーション法、ミスマッチ検出法などを伴ってもよい。
【0095】
ドットブロットハイブリダイゼーション法では、通例、試験ERαポリヌクレオチドをメンブレンに固定し、固定された試験ERαポリヌクレオチド中の検索領域にプローブオリゴヌクレオチドがハイブリダイズすることができるかどうかを評価する。試験ERαポリヌクレオチドをメンブレン上に固定する際には、試験ERαポリヌクレオチドとして、検索領域ポリヌクレオチド、調製した試験cDNA、調製した試験ゲノムDNA、精製した試験ERαポリヌクレオチドなどを使用することができる。試験ERポリヌクレオチドは、試験ERαポリヌクレオチドを90〜100℃で3〜5分間インキュベートし、試験ERαポリヌクレオチドをメンブレン上にスポットし、得られたメンブレンを乾燥し、スポットした検索領域をUV光に曝露することによって固定することができる。メンブレンとしては、Hybond N(Amerscham Pharmacia)などのナイロンメンブレンを使用することができる。次に、プローブオリゴヌクレオチドを使って、当該プローブオリゴヌクレオチドが検索領域にハイブリダイズすることができるかどうかを評価することができる。プローブオリゴヌクレオチドは、当該プローブオリゴヌクレオチドと試験ERαポリヌクレオチドとを40〜50℃で10〜20時間インキュベートすることによって利用することができる。次に、得られたメンブレンを洗浄し、ハイブリダイズしたプローブオリゴヌクレオチドが存在するなら、それを検出することができる。
【0096】
プローブオリゴヌクレオチドが32Pで放射標識される場合、ハイブリダイズしたプローブオリゴヌクレオチドは(存在するとすれば)、得られたメンブレンをX線フィルムに曝露することによって検出できる。
【0097】
プローブオリゴヌクレオチドがビオチンを使って非放射性標識される場合、ハイブリダイズしたプローブオリゴヌクレオチドは(存在するとすれば)、スペーサーおよびハイブリダイゼーション検出酵素(ビオチン化アルカリホスファターゼ、ビオチン化ペルオキシダーゼなど)を使って検出することができる。ビオチンで標識されたプローブオリゴヌクレオチドが検索領域にハイブリダイズすることができる場合、ストレプトアビジンなどのスペーサーは、ハイブリダイズしたビオチン標識プローブオリゴヌクレオチドに結合することができ、その結果、ハイブリダイゼーション検出酵素は、スペーサーを介して、ハイブリダイズしたビオチン標識プローブオリゴヌクレオチドに結合することができるようになる。次に、結合したハイブリダイゼーション検出酵素は反応に関与して、当該プローブオリゴヌクレオチドが試験ERαポリヌクレオチド中の検索領域にハイブリダイズしているかどうかを示すことができる。この酵素反応は色の変化またはルミネセンスをもたらすことができる。
【0098】
プローブオリゴヌクレオチドが検索領域にハイブリダイズしない場合は、当該検索領域は1または複数の変異コドンを含むと決定することができる。次に、検索領域を配列決定してもよい。変異コドンが存在する場合、変異コドン中の変異は、上記の方法と同様にして、PCR増幅およびヌクレオチド配列決定法で決定することができる。
【0099】
ミスマッチ検出法はBiswas,I.およびHsieh,P.,J. Biol. Chem.,271(9),5040-5048(1996)ならびに Nippon gene information,1999,No.125(ニッポンジーン、富山)に記載されている。このミスマッチ検出法では、Taq Mut Sなどのミスマッチ検出酵素を利用して、検索領域に対するプローブオリゴヌクレオチドのハイブリダイゼーション中の一定のミスマッチを検索する。ミスマッチ検出酵素により、プローブオリゴヌクレオチドと検索領域とのハイブリダイゼーション中のミスマッチを、高温(例えば75℃以下の温度)で検出することが可能になる。通例、ミスマッチ検出酵素が結合するこのようなハイブリダイゼーション中のミスマッチは、上述のようなゲルシフトアッセイまたはドットブロットハイブリダイゼーション法によって検出することができる。ミスマッチ検出酵素がプローブオリゴヌクレオチドと検索領域とのミスマッチしたハイブリダイゼーションに結合することができる場合は、検索領域は1または複数の変異コドンを含むと決定することができる。検索領域を配列決定してもよい。変異コドンが存在するなら、その変異コドン中の変異を、上記の方法と同様にして、PCR増幅およびヌクレオチド配列決定法で決定することができる。
【0100】
またRFLP法では、制限酵素を反応条件下で試験ERαポリヌクレオチドと混合する。通例、制限酵素は、変異コドンであると疑われる検索領域中のコドンとオーバーラップする制限部位を持つ。前記制限部位での制限消化の成否により、検索領域中の変異コドンの有無を決定することができる。制限消化の結果は例えば低融点アガロースゲル電気泳動などによるゲル電気泳動分析によって評価することができる。次に必要なら検索領域を配列決定することができる。次に、変異コドンが存在するなら、その変異コドン中の変異は上記の方法と同様にPCR増幅およびヌクレオチド配列決定法で決定することができる。
【0101】
表現型診断法では、試験ERαのアミノ酸配列中で、上記5.2項に述べたようなレポーター遺伝子転写活性化活性を付与する1または複数の置換アミノ酸の検索を行うことができる。置換アミノ酸を検索した後、試験ERα中に変異が存在するのであれば、試験ERαのアミノ酸配列を正常ERαのアミノ酸配列と比較することによって、その変異を決定する。試験ERα中の置換アミノ酸を検索するために、試験ERα中の検索領域は、試験ERα中の相対位置303〜578のアミノ酸を含むことができる。例えば、このような表現型診断法では、303、309、390、396、415、494、531、578などから選択される1または複数の相対位置にある試験ERα中のアミノ酸を検索領域に含めることができる。
【0102】
試験ERα中の置換アミノ酸の検索には、試験ERαの検索領域中にエピトープを持つ抗体が役立ちうる。このような抗体の結合の成否により、試験ERαの検索領域に置換アミノ酸が存在するかどうかを決定することができる。次に、試験ERα中の変異は、試験ERαのアミノ酸配列を正常ERαのアミノ酸配列と比較することによって決定することができる。
【0103】
試験ERαは試験試料から細胞抽出技術によって調製することができる。また、表現型診断法用の試験ERαは、組換え試験ERαを精製することによって調製することもできる。
【0104】
5.4.試験ERαを用いるレポーターアッセイ
試験ERαは、レポーター遺伝子を含む染色体と当該試験ERαとを含むアッセイ細胞を利用することにより、上記5.2に記載のレポーター遺伝子転写活性化活性に関してアッセイすることができる。このような場合は、通例、アッセイ細胞をリガンドにばく露し、レポーター遺伝子の転写活性化レベルを測定することで、試験ERαによるレポーター遺伝子転写活性化活性を定量的に解析する。また、試験ERαによるレポーター遺伝子転写活性化活性は、試験ERαによるレポーター遺伝子の転写活性化レベルを標準物質によるレポーター遺伝子の転写活性化レベルと比較することによって評価することもできる。さらにまた、試験ERαによるレポーター遺伝子の転写活性化レベルが標準物質による転写活性化レベルとは異なる試験ERαを選択することによって、試験ERαをスクリーニングすることもできる。
【0105】
アッセイ細胞は、試験ERαをコードする遺伝子とレポーター遺伝子とを宿主細胞に導入することによって作製することができる。レポーター遺伝子は宿主細胞の染色体に挿入される。試験ERα遺伝子は一過性発現が起こるように宿主細胞に導入することができ、また試験ERα遺伝子が宿主細胞の染色体に挿入されるように試験ERα遺伝子を宿主細胞に導入することもできる。試験ERα遺伝子を宿主細胞の染色体に挿入する場合、試験ERα遺伝子は、試験ERαと一緒に当該染色体に挿入してもよいし、当該宿主細胞中の別の染色体に挿入してもよい。また、上記5.2項に記載したように、レポーター遺伝子を宿主細胞に導入して安定形質転換カセット細胞を作製した後、上記5.2項に記載したように、試験ERα遺伝子を前記安定形質転換カセット細胞に導入することもできる。
【0106】
試験ERα遺伝子は、当該試験ERα遺伝子をアッセイ細胞中で発現させて試験ERαを得ることができるように、宿主細胞に導入される。この点で、前記試験ERαは通例、試験ERαをコードするポリヌクレオチドの上流に作動的に連結されたプロモーターを含む。
【0107】
試験ERα遺伝子を宿主細胞に導入するには、上記5.2項に記載したように、宿主細胞のタイプに従って従来の試験ERα遺伝子導入技術を適用することができる。この点に関して、試験ERαを一過性発現用の宿主細胞に導入する場合は、環状の試験ERα遺伝子を導入する。試験ERα遺伝子を宿主細胞の染色体に挿入する場合は、線状の試験ERαを導入する。また、上記5.2項に記載したように、ベクターを利用して宿主細胞に試験ERα遺伝子またはレポーター遺伝子を導入してもよい。
【0108】
また、試験ERα遺伝子を安定形質転換カセット細胞に導入して、アッセイ細胞を得ることもできる。このような場合、試験ERα遺伝子を安定形質転換カセット細胞に導入して安定形質転換バイナリー細胞を得ることもできる。このような安定形質転換バイナリー細胞は、試験ERα遺伝子およびレポーター遺伝子を安定に含む染色体を持っている。
【0109】
アッセイ細胞の作製に利用される宿主細胞は、通例、正常または変異ERαを発現させない。宿主細胞の例には、HeLa細胞、CV-1細胞、Hepa1細胞、NIH3T3細胞、HepG2細胞、COS1細胞、BF-2細胞、CHH-1細胞などがある。
【0110】
レポーターアッセイでは、通例、アッセイ細胞を十分量のリガンドに1〜数日間ばく露する。またリガンドは試験ERαに対する作動条件または拮抗条件下でアッセイ細胞にばく露することができる。作動条件では通例、変異ERαを刺激する可能性がある単独の薬剤としてのリガンドにアッセイ細胞をばく露する。拮抗条件では通例、リガンドおよびE2にアッセイ細胞をばく露する。
【0111】
リガンドとしては、通常、正常ERαに対して純粋にまたは部分的に拮抗性または作動性であるリガンドを利用する。そのようなリガンドの例には、タモキシフェン、4-ヒドロキシタモキシフェンおよびラロキシフェンなどの部分抗エストロゲン、ICI182780(Wakeling AEら,Cancer Res.,512:3867-3873(1991))およびZM189154(Dukes Mら,J. Endocrinol.,141:335-341(1994))などの完全抗エストロゲンがある。
【0112】
ばく露後に、レポーター遺伝子の発現レベルを測定することによって、レポーター遺伝子の転写活性化レベルを測定する。このような場合、レポータータンパク質またはレポーターRNA(レポーター配列によってコードされるもの)は細胞内に貯蔵されるか、細胞から分泌されるので、発現レベルはそれらを使って測定することができる。レポーター遺伝子の発現レベルはノーザンブロット解析もしくはウェスタンブロット解析によって、またはレポータータンパク質の活性レベルを測定することによって測定することができる。タンパク質の活性レベルは通例、レポーター遺伝子の発現レベルを示す。
【0113】
例えば、レポーター遺伝子がレポータータンパク質としてルシフェラーゼをコードする場合、レポーター遺伝子の発現レベルは、ルシフェリンとルシフェラーゼとを反応させることによって得られるルミネセンスによって測定することができる。このような場合は、粗細胞抽出物を細胞から調製し、その粗細胞抽出物にルシフェリンを加える。ルシフェリンは細胞抽出物中のルシフェラーゼと室温で反応させることができる。ルシフェリンの添加によって得られるルミネセンスは、通常、レポーター遺伝子の発現レベルの指標として測定される。というのも、粗細胞抽出物は、細胞内で発現され粗細胞抽出物中に存在するルシフェラーゼのレベルに比例する強度のルミネセンスをもたらすからである。得られた粗細胞抽出物中のルミネセンスを測定するには、ルミノメーターを利用することができる。
【0114】
次に、測定された転写活性化レベルを標準物質によるレポーター遺伝子の転写活性化レベルと比較して、試験ERαによる転写活性化活性を評価することができる。試験ERαによる転写活性化活性を評価する場合、前記標準物質によるレポーター遺伝子の転写活性化レベルは、アッセイ細胞が(試験ERαの代わりに)正常ERαまたは表現型がわかっているERαを発現させる場合に予想されるレポーター遺伝子の転写活性化レベルとすることができる。試験ERαが与える転写活性化レベルの測定値が標準物質によるレポーター遺伝子の転写活性化レベルと異なる場合は、当該試験ERαを変異ERαとして選択することができる。
【0115】
また、変異リガンド依存性転写因子をスクリーニングすることもできる。このような場合は、試験ERα遺伝子の代わりに試験リガンド依存性転写因子をコードする遺伝子を宿主細胞に導入する。このような試験リガンド依存性転写因子の例には、試験ERβ、試験AR、試験GR、試験TR、試験PR、試験PXR、試験親油性ビタミン受容体(例えば試験VDR)および試験RARなどがある。このような場合、レポーター遺伝子は、EREの代わりに、与えられた試験リガンド依存性転写因子にコグネイトである適当な受容体応答配列を含む。
【0116】
【実施例】
6.1.実施例1 ヒト変異ERαをコードするポリヌクレオチド
6.1.1.ヒト正常ERαをコードするプラスミドの生成
ヒト肝臓cDNAライブラリー(CLONETECH、QuickクローンcDNA#7113-1)を利用して、そこからヒト正常ERαをコードするcDNAを特異的にPCR増幅する。このPCR増幅におけるPCR混合物は、ヒト肝臓cDNAライブラリー10ng、配列番号11に記載のオリゴヌクレオチド10pmol、配列番号12に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。配列番号11および配列番号12に記載のオリゴヌクレオチドはDNAシンセサイザー(モデル394、Applied Biosystems)を使って合成する。このPCR増幅では、PCRsystem 9700(Applied Biosystems)を使って、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを35回繰り返す。
【0117】
得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、PCR増幅によって増幅されたcDNAが約1.8kbのサイズを持つことを確認する。増幅されたcDNAを低融点アガロースゲルから回収した後、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って、回収したcDNAの試料を調製する。調製したcDNA試料をABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定することにより、当該cDNAが配列番号1に示すアミノ酸配列を持つヒト正常ERαをコードするヌクレオチド配列を持っていることを明らかにする。
【0118】
次に、もう1つのPCR増幅を同様に行って、上記cDNA中の開始コドン(ATG)のすぐ上流にKozakコンセンサス配列を付加する。このPCR増幅では、100ngの上記cDNAg、配列番号151に記載のオリゴヌクレオチドおよび配列番号12に記載のオリゴヌクレオチドを利用する。得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、このPCR増幅で増幅されたcDNAが約1.8kbのサイズを持つことを確認する。増幅されたcDNAを低融点アガロースゲルから回収した後、増幅されたcDNA 1μgをDNA Blunting Kit(宝酒造)で処理することにより、増幅されたcDNAの末端を平滑化する。次に、前記の処理によって得たcDNAをT4ポリヌクレオチドキナーゼと反応させて、その末端をリン酸化する。リン酸化したcDNAをフェノール処理した後、リン酸化cDNAをエタノール沈殿させて、精製型のリン酸化cDNAを得る。
【0119】
プラスミドpRc/RSV(Invitrogen)を制限酵素HindIIIで制限消化し、次に65℃で1時間、細菌アルカリホスファターゼ(BAP)で処理する。次に、制限消化pRc/RSVをフェノール処理とエタノール沈殿によって精製する。制限消化されたpRc/RSVをDNA Blunting Kit(宝酒造)で処理してその末端を平滑化し、低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。制限消化されたpRc/RSVを低融点アガロースゲルから回収した後、制限消化されたpRc/RSV 100ngおよび上記精製型リン酸化cDNAの全てを、T4 DNAリガーゼによるライゲーション反応に使用する。そのライゲーション反応混合物を使って、大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞は、アンピシリンを50μg/mlの濃度になるように添加したLB(Luria-Bertani)培地(以下、LB-amp培地という;J. Sambrook,E. F. Frisch,T. Maniatis「Molecular Cloning(第2版)」(Cold Springs Harbor Laboratory Publishing,1989))で培養する。次に、アンピシリン耐性を示すクローンを回収する。次に、それらクローンの一部を使って、ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。調製したプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、配列番号1に示すアミノ酸配列を持つヒト正常ERαをコードするヌクレオチドを配列を持っているプラスミドが存在することを確認する。そのようなプラスミドを選択し、pRc/RSV-hERαKozakと名づける。
【0120】
6.1.2.ヒト変異ERαK303R、S309F、M396V、G415V、G494VまたはK531Eをコードするプラスミドの作製
6.1.2.1.変異導入用プラスミドの作製
プラスミドpRc/RSV-hERαKozakを37℃で1時間、制限酵素NotIで制限消化する。その制限消化反応混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、約1.6kbのサイズを持つDNA断片が存在することを確認する。次に、その1.6kb DNA断片を低融点アガロースゲルから回収する。
【0121】
プラスミドpBluescriptII SK(+)(Stratagene)を37℃で1時間、NotIで制限消化した後、65℃で1時間、BAPで処理する。その制限消化反応混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、制限消化されたpBluescriptII SK(+)を低融点アガロースゲルから回収する。次に、上記1.6kb DNA断片100ngと回収されたpBluescriptII SK(+) 100ngとを、T4 DNAリガーゼによるライゲーション反応に使用する。そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-amp培地で培養する。次に、アンピシリン耐性を示すクローンを回収する。次に、それらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を制限酵素NotIおよびHindIIIで制限消化する。その制限消化反応混合物をアガロースゲル電気泳動にかける。次に、プラスミドのプラス鎖がヒト正常ERαをコードするセンス鎖を作動的なM13ミクロファージ複製起点(f1 ori)と共に含んでいるプラスミドが存在することを確認する。これに関連して、f1 oriがプラスミドの一方の鎖を複製する時に、ヒト正常ERαをコードするセンス鎖がそれと共に複製されるような構造を持つプラスミドが存在することを確認する。そのようなプラスミドを選択し、pSK-NNと名づける。
【0122】
6.1.2.2.相対位置303、309、396、415、494および531における部位特異的変異導入
McClary JAら,Biotechniques 1989(3):282-289に記載の方法に従って、ヒト正常ERαをコードするポリヌクレオチドに指定の変異を導入する。そのような方法を本発明との関連で以下に説明する。
【0123】
上記6.1.2.1に記載のプラスミドpSK-NNを利用して大腸菌コンピテントCJ236細胞(宝酒造)を、大腸菌CJ236細胞と一緒に提供されるプロトコールに従って形質転換する。次に、アンピシリン耐性を示すクローンをLB-amp培地で16時間培養する。次に、そのクローンの1コロニーを、M13ヘルパーファージを少なくとも1×1011pfu/ml-培地の濃度になるように加えた10mlの2×YT培地(以下、2×YT-M13という)に懸濁する。クローンを2×YT-M13培地中37℃で2時間培養した後、カナマイシンを50μg/mlの濃度になるように加え、次にそのクローンを22時間培養する。得られた懸濁液を遠心分離し、得られた上清8mlを15mlの試験管に移す。次に、その上清に2mlの2.5M NaCl-40%PEG8000(Sigma)を加え、上清と共に撹拌する。その上清を4℃で1時間冷蔵し、遠心分離(3,000rpm、2,000×g、10分間、4℃)して、そこからファージをペレットとして集める。ファージを400μlの蒸留水に懸濁した後、同体積のフェノールを加え、得られた懸濁液を穏やかに5分間振とうする。得られた上清を遠心分離して、そこから水層を取り出す。次に、2回目のフェノール処理を行うために、水層に同体積のフェノールを加え、激しく振とうする。得られた懸濁液を遠心分離して、そこから水層を取り出す。2回目のフェノール処理によって得た水層に、同体積のクロロホルムを加え、激しく振盪する。得られた懸濁液を遠心分離(15,000rpm、20,000×g、5分、4℃)して、そこから水層を取り出す。クロロホルム処理によって得た水層に、800μlの100%エタノールと、50μlの3M酢酸ナトリウムとを加える。得られた水層を−80℃に20分間冷却した後、その水層を遠心分離する。これによって得られるペレットを70%エタノールですすいだ後、乾燥する。滅菌水中の残渣をペレット化した後、水溶液の吸光度を260nmの波長で測定して、その中に含まれるヒト正常ERαをコードする一本鎖センスDNAの量を計算する。
【0124】
部位特異的変異導入用のオリゴヌクレオチドを合成して、配列番号152、配列番号153、配列番号154、配列番号155、配列番号156および配列番号157に記載のオリゴヌクレオチドを得る。
【0125】
配列番号152に記載のオリゴヌクレオチドを使用すると、相対位置303にあるリジンをコードするAAGコドンが、アルギニンをコードするAGGコドンに変化する。
【0126】
配列番号153に記載のオリゴヌクレオチドを使用すると、相対位置309にあるセリンをコードするTCCコドンが、フェニルアラニンをコードするTTCコドンに変化する。
【0127】
配列番号154に記載のオリゴヌクレオチドを使用すると、相対位置396にあるメチオニンをコードするATGコドンが、バリンをコードするGTGコドンに変化する。
【0128】
配列番号155に記載のオリゴヌクレオチドを使用すると、相対位置415にあるグリシンをコードするGGAコドンが、バリンをコードするGTAコドンに変化する。
【0129】
配列番号156に記載のオリゴヌクレオチドを使用すると、相対位置494にあるグリシンをコードするGGCコドンが、バリンをコードするGTCコドンに変化する。
【0130】
配列番号157に記載のオリゴヌクレオチドを使用すると、相対位置531にあるリジンをコードするAAGコドンが、グルタミン酸をコードするGAGコドンに変化する。
【0131】
各オリゴヌクレオチドを、10pmolのポリヌクレオチドキナーゼ(宝酒造)を使って、ポリヌクレオチドキナーゼと一緒に提供される緩衝液中でリン酸化する。このリン酸化反応では、2mMのATPを各反応混合物に使用し、反応混合物を37℃で30分間インキュベートする。次に、リン酸化されたオリゴヌクレオチド約1pmolを、正常ERαをコードする0.2pmolの一本鎖センスDNA とそれぞれ混合する。次に、10μlのアニーリング反応混合物を調製するために、上記混合物をそれぞれアニーリング緩衝液(20mM トリス-Cl(pH7.4)、2mM MgCl2、50mM NaCl)に加える。そのアニーリング反応混合物を70℃で10分間のインキュベーション、次いで37℃で60分間のインキュベーションに付した後、4℃でインキュベートする。次に、そのアニーリング反応混合物に、それぞれ2単位(0.25μl)のT7 DNAポリメラーゼ(New England Labs)、2単位(0.25μl)のT4 DNAリガーゼ(宝酒造)および1.2μlの合成緩衝液(175mM トリス-Cl(pH7.4)、375mM MgCl2、5mM DTT、4mM dATP、4mM dCTP、4mM dGTP、4mM dTTPおよび7.5mM ATP)を加えることによって、合成反応混合物を調製する。その合成反応混合物を4℃で5分間インキュベートし、室温で5分間インキュベートし、次に37℃で2時間インキュベートして、合成DNAプラスミドを得る。
【0132】
次に2μlの各合成反応混合物を使って大腸菌コンピテントDH5α細胞細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次に、それらクローンの一部を使って上記合成反応で得られたプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定する。
【0133】
上記の配列決定により、配列番号152に記載のオリゴヌクレオチドを利用することによって合成される単離プラスミドは、ヒト変異ERαをコードするヌクレオチド配列中に相対位置303に相当するAGGコドンを持っていてアルギニンをもたらす単離プラスミドを与えることを確認する。このような単離プラスミドを選択し、pSK-NN303と名づける。
【0134】
上記の配列決定により、配列番号153に記載のオリゴヌクレオチドから合成される単離プラスミドは、ヒト変異ERαをコードするヌクレオチド配列中に相対位置309に相当するTTCコドンを持っていてフェニルアラニンをもたらす単離プラスミドを与えることを確認する。このような単離プラスミドを選択し、pSK-NN309と名づける。
【0135】
上記の配列決定により、配列番号154に記載のオリゴヌクレオチドから合成される単離プラスミドは、ヒト変異ERαをコードするヌクレオチド配列中に相対位置396に相当するGTGコドンを持っていてバリンをもたらす単離プラスミドを与えることを確認する。このような単離プラスミドを選択し、pSK-NN396と名づける。
【0136】
上記の配列決定により、配列番号155に記載のオリゴヌクレオチドから合成される単離プラスミドは、ヒト変異ERαをコードするヌクレオチド配列中に相対位置415に相当するGTAコドンを持っていてバリンをもたらす単離プラスミドを与えることを確認する。このような単離プラスミドを選択し、pSK-NN415と名づける。
【0137】
上記の配列決定により、配列番号156に記載のオリゴヌクレオチドから合成される単離プラスミドは、ヒト変異ERαをコードするヌクレオチド配列中に相対位置494に相当するGTCコドンを持っていてバリンをもたらす単離プラスミドを与えることを確認する。このような単離プラスミドを選択し、pSK-NN494と名づける。
【0138】
上記の配列決定により、配列番号157に記載のオリゴヌクレオチドから合成される単離プラスミドは、ヒト変異ERαをコードするヌクレオチド配列中に相対位置531に相当するGAGコドンを持っていてグルタミン酸をもたらす単離プラスミドを与えることを確認する。このような単離プラスミドを選択し、pSK-NN531と名づける。
【0139】
下記表4に、使用する変異導入用オリゴヌクレオチド、それによって生成するプラスミド、およびその結果得られるヒト変異ERαを示す。
【0140】
【表5】
プラスミドpSK-NN303、pSK-NN309、pSK-NN396、pSK-NN415、pSK-NN494およびpSK-NN531をそれぞれ37℃で1時間、制限酵素NotIで制限消化する。次に、各制限消化反応混合物を低融点アガロースゲル電気泳動にかけて、約1.6kbのサイズを持つDNA断片が存在することを確認する。次に、その1.6kb DNA断片を低融点アガロースゲルから回収する。
【0142】
6.1.1項で得たプラスミドpRc/RSV-hERαKozakを37℃で1時間、制限酵素NotIで制限消化し、65℃で1時間、BAPで処理する。次にその制限消化反応混合物を低融点アガロースゲル電気泳動にかけて、約5.5kbのサイズを持つDNA断片が存在することを確認する。次にその5.5kb DNA断片を低融点アガロースゲルから回収する。
【0143】
次に、回収された5.5kb DNA断片100ngをそれぞれ上記1.6kb DNA断片100ngと混合して、T4 DNAリガーゼによるライゲーション反応を行う。そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-amp培地で培養する。次に、アンピシリン耐性を示すクローンを回収する。次に、それらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を制限酵素NotIまたはMluIで制限消化する。次に、その制限消化反応混合物をアガロースゲル電気泳動にかける。各制限消化によって所望のサイズを持つDNA断片を与える単離プラスミドが存在することを確認する。そのような単離プラスミドは、制限酵素NotIによる制限消化では5.5kbと1.6kbのサイズを持つDNA断片を与え、制限酵素MluIによる制限消化では7.1kbのDNA断片を与える。
【0144】
次に、上記各プラスミドを、配列番号158、配列番号159および配列番号160に記載のオリゴヌクレオチドを使ってPCR増幅する。これらのPCR増幅では、PCR混合物は上記プラスミドの1つ、配列番号158に記載のオリゴヌクレオチド、配列番号159に記載のオリゴヌクレオチド、配列番号160に記載のオリゴヌクレオチド、400μM dNTP(100μM dATP、100μM dTTP、100μM dGTPおよび100μM dCTP)、組換えTaq DNAポリメラーゼ(宝酒造)、前記組換えTaq DNAポリメラーゼと一緒に提供されるPCR緩衝液を含む。これらのPCR増幅では、94℃で30秒間のインキュベーション、次いで65℃で1分間のインキュベーションの後、72℃で1分45秒間のインキュベーションを課すインキュベーションサイクルを30回繰り返す。得られた各25μlのPCR混合物のうち10μlを1%アガロースゲル電気泳動(アガロースS、ニッポンジーン)にかけて、得られたプラスミドが約1.2kbのサイズを持つことを確認する。次に、Dye Terminator Sequence Kit FS(Applied Biosystems)を使ってプラスミドを調製する。調製したプラスミドの試料をそれぞれABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定する。
【0145】
上記の配列決定により、pSK-NN303から得られるプラスミドはヒト変異ERαK303R(AAG→AGG;リジン→アルギニン;相対位置303)をコードすることが確認される。このプラスミドをpRc/RSV-hERαK303R Kozakと名づける。
【0146】
上記の配列決定により、pSK-NN309から得られるプラスミドはヒト変異ERαS309F(TCC→TTC;セリン→フェニルアラニン;相対位置309)をコードすることが確認される。このプラスミドをpRc/RSV-hERαS309F Kozakと名づける。
【0147】
上記の配列決定により、pSK-NN396から得られるプラスミドはヒト変異ERαM396V(ATG→GTG;メチオニン→バリン;相対位置396)をコードすることが確認される。このプラスミドをpRc/RSV-hERαM396V Kozakと名づける。
【0148】
上記の配列決定により、pSK-NN415から得られるプラスミドはヒト変異ERαG415V(GGA→GTA;グリシン→バリン;相対位置415)をコードすることが確認される。このプラスミドをpRc/RSV-hERαG415V Kozakと名づける。
【0149】
上記の配列決定により、pSK-NN494から得られるプラスミドはヒト変異ERαG494V(GGC→GTC;グリシン→バリン;相対位置494)をコードすることが確認される。このプラスミドをpRc/RSV-hERαG494V Kozakと名づける。
【0150】
上記の配列決定により、pSK-NN531から得られるプラスミドはヒト変異ERαK531E(AAG→GAG;リジン→グルタミン酸;相対位置531)をコードすることが確認される。このプラスミドをpRc/RSV-hERαK531E Kozakと名づける。
【0151】
下記表5に、プラスミドの生成に使用したプラスミド、およびそのプラスミドから結果として生成するプラスミドを示す。
【0152】
【表6】
6.1.3.ヒト変異ERαG390D、S578PまたはG390D/S578Pをコードするプラスミドの作製
6.1.3.1.ヒト変異ERαG390DおよびS578Pをコードするプラスミドの生成
QuickChange Site-Directed Mutagenesis Kit(Stratagene)を使って、上記6.1.1に記載のプラスミドpRc/RSV-hERαKozakを、変異したプラスミドがヒト変異ERαG390Dまたはヒト変異ERαS578Pをコードするように変異させた。配列番号17および配列番号18に記載のオリゴヌクレオチドを使用すると、相対位置390にあるグリシンをコードするGGTコドンがアスパラギン酸をコードするGAT変異コドンに変化する。配列番号27および配列番号28に記載のオリゴヌクレオチドを使用すると、相対位置578にあるセリンをコードするTCCコドンがプロリンをコードするCCC変異コドンに変化する。QuickChange Site-Directed Mutagenesis Kitと一緒に提供されるマニュアルを使って、プラスミドpRc/RSV-hERαG390D Kozak(GGT→GAT;グリシン→アスパラギン酸;相対位置390)およびpRc/RSV-hERαS578P Kozak(TCC→CCC;セリン→プロリン;相対位置578)を作製する。プラスミドpRc/RSV-hERαG390D KozakとpRc/RSV-hERαS578P Kozakとを配列決定して、ヒト変異ERαをコードするプラスミドが相対位置390または578に所望の変異を含んでいることを確認する。
【0154】
次にQuickChange Site-Directed Mutagensis Kit(Stratagene)を使って、変異したプラスミドがヒト変異ERαG390D/S578Pをコードするように 、pRc/RSV-hERαG390D Kozakを変異させる。配列番号27および配列番号28に記載のオリゴヌクレオチドを使って、プラスミドpRc/RSV-hERαG390D/S578P Kozak(GGT→GAT;グリシン→アスパラギン酸;相対位置390およびTCC→CCC;セリン→プロリン;相対位置578)を作製する。プラスミドpRc/RSV-hERαG390D/S578P Kozakを配列決定して、ヒト変異ERαをコードするプラスミドが所望の変異を相対位置390および578に含んでいることを確認する。
【0155】
6.1.3.2.ヒト変異ERαG390D/S578Pをコードするプラスミドの試験ヒト肝組織試料からの調製
試験ヒト肝組織の凍結試料を使って、ヒト変異ERαG390D/S578Pをコードするポリヌクレオチドを得た。試験ヒト肝組織試料を利用するにあたって、4Mチオシアン酸グアニジウム、0.1M トリス-HCl(pH7.5)および1%β-メルカプトエタノールを含む緩衝液5mlにて、0.1gの試験ヒト肝組織試料をホモジナイザーでホモジナイズした。得られた緩衝液を25mlの5.7M CsCl水溶液に重層し、90,000×gで24時間超遠心分離することにより、RNAペレットを得た。そのRNAペレットを70%エタノールで濯いだ後、RNAペレットを室温で乾燥させた。次にそのRNAペレットを1.2μg/mlの濃度になるように滅菌水10μlに溶解した。次に、RNA溶液中のRNAを一括して逆転写反応におけるテンプレートとして使用することにより、試験cDNAを作製した。試験cDNAを作製するにあたって、逆転写酵素(Superscript II;GibcoBRL)を1μlのRNA溶液、オリゴ-dTオリゴヌクレオチド(Amerscham Pharmacia)、および逆転写酵素と一緒に提供される緩衝液と共に使用した。逆転写反応を37℃で1時間行って、上記試験cDNAを得た。
【0156】
上記6.1.1と同様に、1/50体積の試験cDNAを使ってpRc/RSV-hERαG390D/S578P Kozakを作製した。この場合、上記試験cDNAを使用して、配列番号11および配列番号12に記載のオリゴヌクレオチドにより、ヒト変異ERαG390D/S578PをコードするcDNAを試験cDNAから特異的にPCR増幅した。次に、ヒト変異ERαG390D/S578PをコードするcDNAを、配列番号151および配列番号12に記載のオリゴヌクレオチドを使ってPCR増幅して、当該cDNAの開始コドン(ATG)のすぐ上流にKozakコンセンサス配列を加えた。次に増幅産物をプラスミドpRc/RSVのHindIII部位に挿入して、pRc/RSV-hERαG390D/S578P Kozakを得た。
【0157】
6.2 実施例2 レポーター遺伝子を含有するプラスミドの作製
配列番号161に記載のオリゴヌクレオチドと、それに相補的なヌクレオチド配列を持つオリゴヌクレオチドとを、DNAシンセサイザーで合成した。配列番号161に記載のオリゴヌクレオチドは、アフリカツメガエル・ビテロゲニン遺伝子中の上流領域に由来するEREの一方の鎖をコードするように合成した。もう一つのオリゴヌクレオチドは、配列番号161に記載のオリゴヌクレオチドに相補的なヌクレオチド配列を持つように合成した。これら2つのオリゴヌクレオチドを互いにアニールさせて、EREをコードするDNA(以下、ERE DNAという)を作製した。次にERE DNAをT4 DNAリガーゼで互いに結合させて、EREが5回縦列反復したERE×5 DNAを得た。T4ポリヌクレオチドキナーゼをERE×5 DNAと反応させて、その末端をリン酸化した。
【0158】
次に配列番号162に記載のオリゴヌクレオチドと配列番号163に記載のオリゴヌクレオチドとをDNAシンセサイザーで合成した。配列番号162に記載のオリゴヌクレオチドは、マウスメタロチオネインI遺伝子に由来するTATA配列のヌクレオチド配列およびそのリーダー配列中の一方の鎖をコードするように合成した。配列番号163に記載のオリゴヌクレオチドは、配列番号162に記載のオリゴヌクレオチドに相補的なヌクレオチド配列をコードするように合成した。配列番号162と配列番号163に記載のオリゴヌクレオチドを互いにアニールさせて、TATA配列をコードするDNAを作製した。T4ポリヌクレオチドキナーゼを、TATA配列をコードする前記DNAと反応させて、その末端をリン酸化した。
【0159】
ホタルルシフェラーゼ遺伝子をコードするプラスミドpGL3(Promega)を、制限酵素BglIIおよびHindIIIで制限消化した後、65℃で1時間、BAPで処理した。次にその制限消化反応混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、ホタルルシフェラーゼをコードするヌクレオチド配列を持つDNA断片が存在することを確認した。次に、ホタルルシフェラーゼをコードするヌクレオチド配列を持つDNA断片を低融点アガロースゲルから回収した。次に、回収したDNA断片100ngと、TATA配列をコードするDNA 1μgとをT4 DNAリガーゼによるライゲーション反応に使用して、プラスミドpGL3-TATAを得た。
【0160】
プラスミドpGL3-TATAを制限酵素SmaIで制限消化した後、65℃で1時間、BAPで処理した。次に、その制限消化反応混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、TATA配列とホタルルシフェラーゼとをコードするDNA断片が存在することを確認した。そのようなDNA断片を低融点アガロースゲルから回収した後、回収したDNA断片100ngとERE×5 DNA 1μgとをT4 DNAリガーゼによるライゲーション反応に使用して、プラスミドpGL3-TATA-ERE×5を得た。
【0161】
プラスミドpUCSV-BSD(フナコシ)を制限酵素BamHIで制限消化して、ブラストサイジンSデアミナーゼ遺伝子発現カセットをコードするDNAを調製した。また、プラスミドpGL3-TATA-ERE×5を制限酵素BamHIで制限消化した後、65℃で1時間、BAPで処理した。次にブラストサイジンSデアミナーゼ遺伝子発現カセットをコードするDNA断片を制限消化したpGL3-TATA-ERE×5と混合した。次にその混合物を、T4 DNAリガーゼによるライゲーション反応に使用して、プラスミドを得た。そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞を形質転換した。形質転換細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に単離された各プラスミドの部分試料を制限酵素BamHIで制限消化する。次に、その制限消化反応混合物をアガロースゲル電気泳動にかけて、ブラストサイジンSデアミナーゼ遺伝子発現カセットをコードするDNAがpGL3-TATA-ERE×5のBamHIの制限部位に挿入されている構造を持つプラスミドが存在するかどうかを確認した。そのような構造を持つプラスミドを選択し、pGL3-TATA-ERE×5-BSDと名づけた。
【0162】
6.3.実施例3 安定形質転換カセット細胞の作製
染色体の一つに6.2項で作製したレポーター遺伝子(以下、EREレポーター遺伝子という)を安定に含有する安定形質転換カセット細胞を作製するために、プラスミドpGL3-TATA-ERE×5-BSDを線状化し、HeLa細胞に導入した。
【0163】
プラスミドpGL3-TATA-ERE×5-BSDを線状化するために、pGL3-TATA-ERE×5-BSDを制限酵素SalIで制限消化した。
【0164】
直径約10cmの培養皿(Falcon)を使用し、10%FBSを含むDMEM培地(日水製薬)にて、5%CO2下に37℃で、約5×105個のHeLa細胞を宿主細胞として1日培養した。
【0165】
次に、線状化したpGL3-TATA-ERE×5-BSDをリポフェクトアミン(Life Technologies)を用いるリポフェクション法によって培養HeLa細胞に導入した。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿の上記プラスミドおよび21μl/培養皿のリポフェクトアミンを含めた。
【0166】
リポフェクション処理の後、DMEM培地を、10%FBSを含むDMEM培地と交換し、形質転換したHeLa細胞を約36時間培養した。次に、形質転換HeLa細胞をトリプシン処理によって培養皿から除去、収集し、ブラストサイジンSを16μg/mlの濃度になるように添加した培地を含む容器に移した。ブラストサイジンS含有培地を3、4日毎に新しいバッチのブラストサイジンS含有培地に交換しながら、前記ブラストサイジンS含有培地で形質転換HeLa細胞を1ヶ月培養した。
【0167】
その結果増殖して1〜数mmの直径を持つコロニーを形成することができたクローンを、前もって培地を分注しておいた96ウェルViewPlate(Berthold)のウェルに、丸ごと移した。それらクローンのコロニーをさらに培養した。コロニーが増殖してウェルの底面の50%以上を覆うようになったら(移植の約5日後)、それらのクローンをトリプシン処理によって除去、収集した。次にクローンを2つの継代培養物に分割した。一方の継代培養物は96ウェルViewPlateに移して、それをマスタープレートとした。もう1つの継代培養物は96ウェルViewPlateに移して、これをアッセイプレートとした。マスタープレートとアッセイプレートには、クローンを培養することができるように培地を含めた。マスタープレートは同様の条件で引き続き培養した。
【0168】
アッセイプレート中の継代培養物を2日間培養した後、培地をアッセイプレートのウェルから除去し、ウェル壁に付着したクローンをPBS(-)で2回洗浄した。5倍希釈した溶解緩衝液PGC50(東洋インキ)をアッセイプレートのウェル中の継代培養物に1ウェルあたり20μlずつ加えた。そのアッセイプレートを室温で30分間静置した後、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットした。次に、基質溶液PGL100(東洋インキ)50μlをアッセイプレート中の各溶解クローンに自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで測定した。高いルシフェラーゼ活性を示すクローン10個をそこから選択した。
【0169】
次に、選択した10クローンに相当するマスタープレート中のクローン試料を、直径約10cmの培養皿(Falcon)を用いて、培地中、5%CO2下に、37℃で1〜2週間培養した。
【0170】
次に、リポフェクトアミン(Life Technologies)を用いるリポフェクション法により、プラスミドpRc/RSV-hERαKozakを、上で選択したクローンに導入して、2次クローンを得た。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿の上記プラスミドおよび21μl/培養皿のリポフェクトアミンを含めた。次に、得られた2次クローンに、17β-E2を含有するDMSO溶液を、濃度が10nMになるように添加した。2次クローンを2日間培養した後、各2次クローンについて上記と同様にルシフェラーゼ活性を測定した。最も高いルシフェラーゼ活性の誘導を示す2次クローンを与えるマスタープレート中のクローンを、染色体の一つにEREレポーター遺伝子を安定に含んでいる安定形質転換カセット細胞(以下、安定形質転換EREカセット細胞という)として選択した。
【0171】
6.4.実施例4 安定形質転換バイナリー細胞の作製
EREレポーター遺伝子をヒト変異ERαG390D、S578PもしくはG390D/S578Dまたはヒト正常ERαと共に含有する4つの安定形質転換細胞(以下、安定形質転換EREバイナリー細胞という)を作製した。第1の安定形質転換EREバイナリー細胞は、その染色体中にレポーター遺伝子をコードする線状化pGL3-TATA-ERE×5-BSDと、ヒト正常ERαをコードする線状化pRc/RSV-hERαKozakとを含んだ。第2の安定形質転換EREバイナリー細胞は、その染色体中にEREレポーター遺伝子をコードする線状化pGL3-TATA-ERE×5-BSDと、ヒト変異ERαG390Dをコードする線状化pRc/RSV-hERαG390D Kozakとを含んだ。第3の安定形質転換EREバイナリー細胞は、その染色体中にEREレポーター遺伝子をコードする線状化pGL3-TATA-ERE×5-BSDと、ヒト変異ERαS578Pをコードする線状化pRc/RSV-hERαS578P Kozakとを含んだ。第4の安定形質転換EREバイナリー細胞は、その染色体中にEREレポーター遺伝子をコードする線状化pGL3-TATA-ERE×5-BSDと、ヒト変異ERαG390D/S578Pをコードする線状化pRc/RSV-hERαG390D/S578P Kozakとを含んだ。
【0172】
安定形質転換EREバイナリー細胞を作製するために、プラスミドpGL3-TATA-ERE×5-BSD、pRc/RSV-hERαG390D Kozak、pRc/RSV-hERαS578P KozakおよびpRc/RSV-hERαG390D/S578P Kozakをそれぞれ線状化し、HeLa細胞に導入した。線状化するために、上記プラスミドを制限酵素SalIで制限消化した。
【0173】
直径約10cmの培養皿(Falcon)を使用し、10%FBSを含むDMEM培地(日水製薬)にて、5%CO2下に37℃で、約5×105個のHeLa細胞を宿主細胞として1日培養した。
【0174】
下記表6に示すように、線状pGL3-TATA-ERE×5-BSDをヒト変異ERαまたはヒト正常ERαをコードする線状プラスミドと共にそれぞれHeLa細胞に導入した。線状化したプラスミドは、リポフェクトアミン(Life Technologies)を用いるリポフェクション法によってHeLa細胞に導入した。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法における各処理の条件には、5時間の処理、7μg/培養皿のプラスミド(それぞれ3.5μg)および21μl/培養皿のリポフェクトアミンを含めた。
【0175】
【表7】
リポフェクション処理の後、DMEM培地を、10%FBSを含むDMEM培地と交換し、形質転換HeLa細胞を約36時間培養した。次に、形質転換HeLa細胞をトリプシン処理によって培養皿からそれぞれ除去、収集し、ブラストサイジンSおよびG418を添加した培地を含む容器に移した。各細胞培養物についてブラストサイジンSの濃度は16μg/mlとした。各細胞培養物についてG418 の濃度は800μg/mlとした。培地を3、4日毎に新しいバッチのブラストサイジンSおよびG418 含有培地に交換しながら、前記ブラストサイジンSおよびG418 含有培地で形質転換HeLa細胞を1ヶ月培養した。
【0177】
その結果1〜数mmの直径を持つコロニーまで増殖することができたクローンを、前もって培地を分注しておいた96ウェルViewPlate(Berthold)のウェルに、それぞれ移した。それらのクローンをさらに培養した。クローンが増殖してウェルの底面の50%以上を覆うようになったら(移植の約5日後)、それらのクローンをトリプシン処理によって除去、収集した。次に各クローンを3つの継代培養物に分割した。各クローンについて、1つの継代培養物は96ウェルViewPlateに移して、マスタープレートとした。他の2つの継代培養物はそれぞれ96ウェルViewPlateに移して、アッセイプレートとした。マスタープレートとアッセイプレートには、クローンを培養することができるように培地を含めた。マスタープレートは同様の条件で引き続き培養する。第1アッセイプレート中の各継代培養物に、17β-E2を含有するDMSO溶液を、濃度が10nMになるように添加した。第2アッセイプレート中の継代培養物には等体積のDMSOを添加した。次に第1および第2アッセイプレートを2日間培養した。
【0178】
次に第1および第2アッセイプレートのウェルから培地を除去し、ウェル壁に付着したクローンをPBS(-)で2回洗浄した。5倍希釈した溶解緩衝液PGC50(東洋インキ)を第1および第2アッセイプレートのウェル中のクローンに1ウェルあたり20μlずつ加えた。その第1および第2アッセイプレートを室温で30分間静置した後、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットした。次に、基質溶液PGL100(東洋インキ)50μlをアッセイプレート中の各溶解クローンにそれぞれ自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで測定した。2倍高いルシフェラーゼ活性の誘導(%)を示した第1アッセイプレート中のクローンに相当するマスタープレート中のクローンを、レポーター遺伝子とヒト変異ERαG390D、S578PもしくはG390D/S578P遺伝子またはヒト正常ERα遺伝子とを安定に含有する安定形質転換EREバイナリー細胞として選択した。
【0179】
6.5.実施例5 ヒト変異ERαのレポーターアッセイ
6.5.1.レポーターアッセイ用安定形質転換EREバイナリー細胞の調製
次に、上記6.4で作製した約2×104個の安定形質転換EREバイナリー細胞を96ウェルルミノメータープレート(Corning Coaster)のウェルに移し、活性炭デキストラン処理FBSを濃度が10%(v/v)になるように添加したE-MEM培地(以下、活性炭デキストランFBS/E-MEMという)中で安定形質転換EREバイナリー細胞を終夜培養した。
【0180】
6.5.2.ヒト変異ERαK303R、S309F、M396V、G415V、G494VまたはK531Eをコードするプラスミドの導入
それぞれに6.3項で作製した安定形質転換EREカセット細胞約2×106個を含む7つの継代培養物を、直径約10cmの培養皿(Falcon)を使って、活性炭デキストランFBS/E-MEM培地で1日培養した。
【0181】
一過性発現のために、プラスミドpRc/RSV-hERαKozak(6.1.1項で作製したもの、正常ERαをコードする)および変異ERαをコードするプラスミド(6.1.2.2項で作製したもの、すなわちpRc/RSV-hERαK303R Kozak、pRc/RSV-hERαS309F Kozak、pRc/RSV-hERαM396V Kozak、pRc/RSV-hERαG415V Kozak、pRc/RSV-hERαG494V Kozak、またはpRc/RSV-hERαK531E Kozak、それぞれヒト変異ERαをコードする)を、リポフェクトアミン(Life Technologies)を用いるリポフェクション法により、安定形質転換EREカセット細胞の継代培養物に、それぞれ導入した。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法における各処理の条件には、5時間の処理、7μg/培養皿のプラスミドおよび21μl/培養皿のリポフェクトアミンを含めた。得られた細胞培養物を5%CO2下に37℃で16時間培養した後、その活性炭デキストランFBS/E-MEM培地を新しいバッチの活性炭デキストランFBS/E-MEM培地に交換して、各細胞継代培養物をさらに3時間培養した。次に、その細胞継代培養物をそれぞれ収集し、活性炭デキストランFBS/E-MEM培地に均一に懸濁した。
【0182】
6.5.3.レポーター遺伝子転写活性化活性の測定
大別して4種類のDMSO溶液を使用して、上記6.5.1および6.5.2で調製した継代培養物中の細胞を、様々な濃度の完全抗エストロゲンまたは部分抗エストロゲンにばく露した。第1のDMSO溶液は様々な濃度の部分抗エストロゲン(4-ヒドロキシタモキシフェンまたはラロキシフェン)を含むように調製した。第2のDMSO溶液は様々な濃度の完全抗エストロゲン(ZM189154)を含むように調製した。第3のDMSO溶液は10nMのE2と様々な濃度の部分抗エストロゲン(4-ヒドロキシタモキシフェンまたはラロキシフェン)とを含むように調製した。第4のDMSO溶液は、10nMのE2と様々な濃度の完全抗エストロゲン(ZM189154)を含むように調製した。
【0183】
次に第1、第2、第3または第4DMSO溶液を上記6.5.1および6.5.2で調製した継代培養物に、下記表7、8、9および10に示すように添加した。第1、第2、第3または第4DMSO溶液は、各ウェルにおける第1、第2、第3または第4DMSO溶液の濃度が約0.1%(v/v)になるように、96ウェルViewPlateのウェルに添加した。また、96ウェルViewPlateのウェル中の各継代培養物について、2つのコントロールを調製した。一方のコントロールはDMSO(部分抗エストロゲンまたは完全抗エストロゲンを含まない)にばく露した。他方のコントロールは本質的に100pMのE2からなるDMSO溶液にばく露した。
【0184】
次に、細胞を5%CO2下に37℃で36〜40時間培養した。5倍希釈した溶解緩衝液PGC50(東洋インキ)をウェル中の細胞に1ウェルあたり50μlずつ加えた。その96ウェルViewPlateをときどき穏やかに振とうしながら室温で30分間インキュベートした。次に、溶解した細胞10μlをそれぞれ白色96ウェルサンプルプレート(Berthold)に移し、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットした。次に、基質溶液PGL100(東洋インキ)50μlを白色96ウェルサンプルプレート中の各溶解細胞にそれぞれ自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで直ちに5秒間測定した。
【0185】
6.5.2項で調製した細胞から得られるルシフェラーゼ活性を図1〜32に図示する。
【0186】
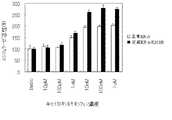
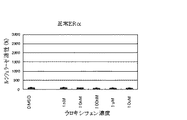
図1および2は、ヒト正常ERαまたはヒト変異ERαK303Rを刺激する可能性がある単独の薬剤として4-ヒドロキシタモキシフェンまたはZM189154が存在する状態でヒト正常ERαまたはヒト変異ERαK303Rがもたらすルシフェラーゼ活性を表す。
【0187】
図3は、E2とZM189154とが存在する状態でヒト正常ERαおよびヒト変異ERαK303Rがもたらすルシフェラーゼ活性を表す。
【0188】
図4および5は、ヒト正常ERαまたはヒト変異ERαS309Fを刺激する可能性がある単独の薬剤として4-ヒドロキシタモキシフェンまたはZM189154が存在する状態でヒト正常ERαおよびヒト変異ERαS309Fがもたらすルシフェラーゼ活性を表す。
【0189】
図6は、E2とZM189154とが存在する状態でヒト正常ERαおよびヒト変異ERαS309Fがもたらすルシフェラーゼ活性を表す。
【0190】
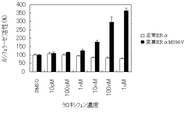
図7および8は、ヒト正常ERαまたはヒト変異ERαM396Vを刺激する可能性がある単独の薬剤として4-ヒドロキシタモキシフェンまたはラロキシフェンが存在する状態でヒト正常ERαおよびヒト変異ERαM396Vがもたらすルシフェラーゼ活性を表す。
【0191】
図9〜11は、E2と4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154とが存在する状態でヒト正常ERαおよびヒト変異ERαM396Vがもたらすルシフェラーゼ活性を表す。
【0192】
図12および13は、ヒト正常ERαまたはヒト変異ERαG415Vを刺激する可能性がある単独の薬剤として4-ヒドロキシタモキシフェンまたはZM189154が存在する状態でヒト正常ERαおよびヒト変異ERαG415Vがもたらすルシフェラーゼ活性を表す。
【0193】
図14および15は、E2と4-ヒドロキシタモキシフェンまたはZM189154とが存在する状態でヒト正常ERαおよびヒト変異ERαG415Vがもたらすルシフェラーゼ活性を表す。
【0194】
図16〜17は、ヒト正常ERαまたはヒト変異ERαG494Vを刺激する可能性がある単独の薬剤として4-ヒドロキシタモキシフェンまたはラロキシフェンが存在する状態でヒト正常ERαおよびヒト変異ERαG494Vがもたらすルシフェラーゼ活性を表す。
【0195】
図18〜20は、E2と4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154とが存在する状態でヒト正常ERαおよびヒト変異ERαG494Vがもたらすルシフェラーゼ活性を表す。
【0196】
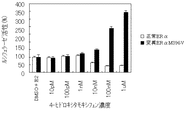
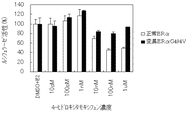
図21〜26は、ヒト正常ERαまたはヒト変異ERαK531Eを刺激する可能性がある単独の薬剤として4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154が存在する状態でヒト正常ERαおよびヒト変異ERαK531Eがもたらすルシフェラーゼ活性を表す。
【0197】
図27〜32は、E2と4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154とが存在する状態でヒト正常ERαおよびヒト変異ERαK531Eがもたらすルシフェラーゼ活性を表す。
【0198】
6.5.1項で調製した細胞から得られるルシフェラーゼ活性を図33〜48に示す。
【0199】
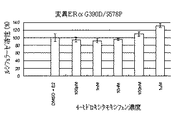
図33〜40は、ヒト正常ERα、ヒト変異ERαG390D、ヒト変異ERαS578Pおよびヒト変異ERαG390D/S578Pを刺激する可能性がある単独の薬剤として4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154が存在する状態でヒト正常ERα、ヒト変異ERαG390D、ヒト変異ERαS578Pおよびヒト変異ERαG390D/S578Pがもたらすルシフェラーゼ活性を表す。
【0200】
図41〜48は、E2と4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154とが存在する状態でヒト正常ERα、ヒト変異ERαG390D、ヒト変異ERαS578Pおよびヒト変異ERαG390D/S578Pがもたらすルシフェラーゼ活性を表す。
【0201】
【表8】
【表9】
【表10】
【表11】
6.6.実施例6 比較用二重一過性レポーターアッセイ
約2×106個のHeLa細胞を、直径約10cmの培養皿(Falcon)を使って、活性炭デキストランFBS/E-MEM培地中、5%CO2下に37℃で、1日培養した。HeLa細胞を培養した後、それらHeLa細胞を2つの継代培養物に分割した。
【0206】
次に、一過性発現のために、3.75μgのpRc/RSV-hERαKozakと3.75μgのpGL3-TATA-ERE×5とを、第1継代培養物中のHeLa細胞に、リポフェクタミンを用いるリポフェクション法で導入した。第2の継代培養物には、一過性発現のために、3.75μgのpRc/RSV-hERαK531E Kozakと3.75μgのpGL3-TATA-ERE×5とを、リポフェクトアミンを用いるリポフェクション法で導入した。次に、第1および第2継代培養物を5%CO2下に37℃で16時間培養した。活性炭デキストランFBS/E-MEM培地を新しいバッチの活性炭デキストランFBS/E-MEM培地と交換した後、第1および第2継代培養物を同様に3時間培養した。次に第1および第2継代培養物中の細胞をそれぞれ収集し、活性炭デキストランFBS/E-MEM培地に均一に懸濁した。
【0207】
第1および第2継代培養物中の細胞をばく露するために、大別して2種類のDMSO溶液を調製した。第1DMSO溶液は様々な濃度の4-ヒドロキシタモキシフェンを含むように調製した。第2DMSO溶液は10nMのE2と様々な濃度の4-ヒドロキシタモキシフェンとを含むように調製した。
【0208】
次に第1および第2DMSO溶液をそれぞれ、96ウェルViewPlate中の第1および第2継代培養物と、各ウェルにおける第1または第2DMSO溶液の濃度が約0.1%(v/v)になるように混合した。
【0209】
次に第1および第2継代培養物を5%CO2下に37℃で36時間培養した。5倍希釈した溶解緩衝液PGC50(ニッポンジーン)を、ウェル中の第1および第2継代培養物に、1ウェルあたり50μlずつ加えた。その96ウェルViewPlateをときどき穏やかに振とうしながら室温で30分間インキュベートした。次に、得られた溶解細胞10μlをそれぞれ白色96ウェルサンプルプレート(Berthold)に移し、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットした。次に、基質溶液PGL100(東洋インキ)50μlを白色96ウェルサンプルプレート中の各溶解細胞にそれぞれ自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで直ちに5秒間測定した。
【0210】
上記二重一過性レポーターアッセイで得られるルシフェラーゼ活性を図49〜52に示す。
【0211】
図49および50は、ヒト正常ERαまたはヒト変異ERαK531Eを刺激する可能性がある単独の薬剤として4-ヒドロキシタモキシフェンが存在する状態でヒト正常ERαおよびヒト変異ERαK531Eがもたらすルシフェラーゼ活性を表す。
【0212】
図51および52は、E2と4-ヒドロキシタモキシフェンとが存在する状態でヒト正常ERαおよびヒト変異ERαK531Eがもたらすルシフェラーゼ活性を表す。
【0213】
6.7.実施例7 検索オリゴヌクレオチド
ヒト試験ERα中の置換アミノ酸をコードする変異コドンを検索するために、ヒト試験ERα遺伝子がヒト正常ERαをコードする場合には検索オリゴヌクレオチドがヒト試験ERα遺伝子中の検索領域にアニールできるように、検索オリゴヌクレオチドを設計する。検索領域は、相対位置303のアミノ酸をコードするコドン、相対位置309のアミノ酸をコードするコドン、相対位置390のアミノ酸をコードするコドン、相対位置396のアミノ酸をコードするコドン、相対位置415のアミノ酸をコードするコドン、相対位置494のアミノ酸をコードするコドン、相対位置531のアミノ酸をコードするコドン、または相対位置578のアミノ酸をコードするコドンを含む。また、上記オリゴヌクレオチドは30〜70%のGC含量と20bpのサイズとを持つように設計する。このように設計したオリゴヌクレオチドに基づいて、上記の本発明オリゴヌクレオチドをDNAシンセサイザー(モデル394、Applied Biosystems)で合成する。
【0214】
6.8.実施例8 PCR増幅およびヌクレオチド配列決定法による遺伝子型診断
試験ヒト肝組織試料を使って、当該試料中の試験ERαポリヌクレオチドの遺伝子型を診断する。試験ヒト肝組織試料を利用するにあたって、4Mチオシアン酸グアニジウム、0.1M トリス-HCl(pH7.5)および1%β-メルカプトエタノールを含む緩衝液5mlにて、0.1gの試験ヒト肝組織試料をホモジナイザーでホモジナイズする。得られた緩衝液を25mlの5.7M CsCl水溶液に重層し、90,000×gで24時間超遠心分離することにより、RNAペレットを得る。そのRNAペレットを70%エタノールで濯いだ後、RNAペレットを室温で風乾する。次にそのRNAペレットを1.2μg/mlの濃度になるように滅菌水10μlに溶解する。次に、RNA溶液中のRNAを逆転写反応におけるテンプレートとして一括して使用することにより、試験cDNAの溶液を作製する。試験cDNAを作製するにあたって、Superscript II(Gibco)を1μlのRNA溶液、オリゴ-dTオリゴヌクレオチド(Amersham-Pharmacia)、およびオリゴ-dTと一緒に提供される緩衝液と共に使用した。逆転写反応は37℃で1時間行った。
【0215】
1/50体積の試験cDNA試料を使用し、下記表11に示す検索オリゴヌクレオチドの組み合わせを使って、PCR増幅を行う。
【0216】
【表12】
これらのPCR増幅では、PCR混合物は試験cDNA、AmpliTaq DNAポリメラーゼ(Perkin Elmer)、100μMのdNTP(dATP、dTTP、dGTP、dCTP)、検索オリゴヌクレオチドの組み合わせの一つ、AmpliTaqポリメラーゼと一緒に提供される緩衝液を含む。このPCR増幅では、95℃で1分間のインキュベーション、次いで55℃で30秒間のインキュベーションの後、72℃で1分間のインキュベーションを課すインキュベーションサイクルを、各PCR増幅について35回繰り返す。得られた検索領域ポリヌクレオチドを1%低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、回収する。回収された検索領域ポリヌクレオチドの全量を用いて、検索領域を配列決定する。検索領域のヌクレオチド配列をヒト正常ERαをコードするヌクレオチド配列と比較する。
【0218】
6.9.実施例9 SSCP法による遺伝子型診断
6.9.1.試験組織試料からの試験ゲノムcDNAの抽出
TAKARA PCR Technical news No.2(宝酒造、1991年9月)に記載の方法に従って試験組織試料から試験ゲノムDNAを調製する。この方法を本発明との関連で以下に説明する。
【0219】
試験対象の毛髪試料2〜3本を滅菌水で洗浄した後、100%エタノールで洗浄する。毛髪試料を風乾した後、毛髪試料を2〜3mmに切断してプラスチック製試験管に移す。そこに200μlのBCL(10mM トリス-HCl(pH7.5)、5mM MgCl2、0.32Mショ糖、1%トリトンX-100)を加える。次に、プロテイナーゼK溶液とSDS溶液をそれぞれ100μg/mlおよび0.5%(w/v)になるように混合する。
【0220】
得られた混合物を70℃で1時間インキュベートした後、その混合物をフェノール-クロロホルム抽出して、そこから水層を回収する。フェノール-クロロホルム抽出では、実質的に等体積のフェノール-クロロホルムを上記混合物に加える。その混合物を激しく振とうし、遠心分離する(15,000rpm、20,000×g、5分、4℃)。そこから、フェノール層を乱さないようにピペットで水層を取り出す。次に、その水層を使って、2回目のフェノール-クロロホルム抽出を同様に行う。
【0221】
実質的に等体積のクロロホルムを2回目のフェノール-クロロホルム抽出で得た水層と混合して、得られたクロロホルム混合物から水層を取り出す。クロロホルムによるこの抽出では、クロロホルム混合物を激しく振とうし、水層をクロロホルム混合物から取り出すことができるように遠心分離する。次に、クロロホルム混合物から取り出した水層に500μlの100%エタノールを加える。その中の試験ゲノムDNAを−80℃で20分間沈殿させた後、遠心分離して試験ゲノムDNAのペレットを得る。得られた試験ゲノムDNAのペレットを乾燥し、試験ゲノムDNAを試験ERαポリヌクレオチドにすることができるように、滅菌水に溶解する。
【0222】
もう1つの選択肢として、末梢血を試験試料として使用し、そこから試験ゲノムDNAを得ることもできる。10mlの血液を試験対象から採取し、DNA Extraction Kit(Stratagene)を使ってその血液から試験ゲノムDNAを抽出する。
【0223】
6.9.2.PCR-SSCP法による試験ゲノムDNAの解析
試験ゲノムDNAを用いるPCR増幅のために、フォワード検索オリゴヌクレオチドとリバース検索オリゴヌクレオチドの組み合わせを選択する。フォワードおよびリバース検索オリゴヌクレオチドは、試験ERαポリヌクレオチド中の検索領域の位置に基づいて選択される。下記表12に、フォワードおよびリバース検索オリゴヌクレオチドの組み合わせを、与えられた相対位置の置換アミノ酸をコードする変異コドンを含有すると疑われる検索領域と共に示す。
【0224】
【表13】
フォワードおよびリバース検索オリゴヌクレオチドの組み合わせをDNAシンセサイザーで合成する。DNA MEGALABEL Kit(宝酒造)を使ってフォワードおよびリバース検索オリゴヌクレオチドのそれぞれを32Pで標識する。次に試験ゲノムDNAをそれぞれPCR増幅に使用して、増幅された検索領域ポリヌクレオチドを得る。これらのPCR増幅において各PCR混合物はAmplitaq DNAポリメラーゼ(Perkin Elmer)、400μMのdNTP(100μM dATP、100μM dTTP、100μM dGTPおよび100μM dCTP)、100pmolの32P標識フォワード検索オリゴヌクレオチド、100pmolの32P標識リバース検索オリゴヌクレオチド、1μgの試験ゲノムDNA、およびAmplitaq DNAポリメラーゼと一緒に提供される緩衝液を含む。各PCR増幅では、94℃で1分間のインキュベーション、次いで55℃で30秒間のインキュベーションの後、72℃で1分間のインキュベーションを課すインキュベーションサイクルを、各PCR増幅について35回繰り返す。
【0226】
PCR増幅後に、増幅された各検索領域ポリヌクレオチドから採取した1/20体積の試料を80%ホルムアミド中、80℃で5分間熱変性させる。次に、熱変性した各検索領域ポリヌクレオチドを、180mM トリス-ホウ酸緩衝液(pH8.0)を用いる5%非変性ポリアクリルアミドゲルでの電気泳動にかける。電気泳動の条件には室温空冷かつ40Wの定電力で60分を含める。電気泳動後に、その5%非変性ポリアクリルアミドゲルをX線フィルムを使って従来の方法でオートラジオグラフィーにかけて、検索領域の放射活性を検出する。
【0227】
変異コドンをコードする産物は5%非変性ポリアクリルアミドゲルでは正常コドンをコードする産物とは異なる移動度を持つので、検索領域ポリヌクレオチドの各移動度を、ヒト正常ERα中の対応する領域をコードする標準ポリヌクレオチドと比較することにより、検索領域中の変異の有無が検出される。
【0228】
6.9.3.変異の決定
検索領域中の変異コドンを検出したら、検索領域ポリヌクレオチドを含む1mm角の切片を5%非変性ポリアクリルアミドゲルから切り出す。1mm角の切片のそれぞれを100μlの滅菌水中90℃で10分間処理して、検索領域ポリヌクレオチドを1mm角の切片から回収する。次に、検索領域ポリヌクレオチドの1/20体積試料をそれぞれ2回目のPCR増幅に使用する。これらのPCR増幅では、上記6.9.2項で使用した検索オリゴヌクレオチドの組み合わせを使用した。これらのPCR増幅における各PCR混合物は、Amplitaq DNAポリメラーゼ(ABI)、400μMのdNTP(100μM dATP、100μM dTTP、100μM dGTPおよび100μM dCTP)、フォワード検索オリゴヌクレオチド、リバース検索オリゴヌクレオチド、試験DNA断片の一つ、およびAmplitaq DNAポリメラーゼと一緒に提供される緩衝液を含む。各PCR増幅では、94℃で1分間のインキュベーション、次いで55℃で30秒間のインキュベーションの後、72℃で1分間のインキュベーションを課すインキュベーションサイクルを、35回繰り返す。
【0229】
反応の完了後に、増幅された検索領域ポリヌクレオチドを、低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。増幅された検索領域ポリヌクレオチドを低融点アガロースゲルから回収した後、回収された検索領域ポリヌクレオチドを、Dye Terminator Cycle Sequence Ready Reaction Kit(Applied Biosystems)を使って調製する。調製した検索領域ポリヌクレオチドの試料をそれぞれABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、検索領域中に変異コドンがあれば、その変異コドン中の変異を決定する。
【0230】
6.10.実施例10 RFLP法による遺伝子型診断
試験ゲノムDNAまたは試験cDNAを用いるPCR増幅のために、フォワード検索オリゴヌクレオチドとリバース検索オリゴヌクレオチドの組み合わせを選択する。フォワードおよびリバース検索オリゴヌクレオチドは、試験ERαポリヌクレオチド中の検索領域の位置に基づいて選択される。下記表13に、フォワードおよびリバース検索オリゴヌクレオチドの組み合わせを、与えられた相対位置の置換アミノ酸をコードする変異コドンを含有すると疑われる検索領域と共に示す。
【0231】
【表14】
PCR増幅に試験ゲノムDNAまたは試験cDNAを使って、約100または160bpのサイズを持つ検索領域ポリヌクレオチドを増幅する。これらのPCR増幅における各PCR混合物は、Amplitaq DNAポリメラーゼ(ABI)、試験ゲノムDNAまたは試験cDNA、dNTP(dATP、dTTP、dGTPおよびdCTP)、フォワード検索オリゴヌクレオチド、リバース検索オリゴヌクレオチド、およびAmplitaq DNAポリメラーゼと一緒に提供される緩衝液を含む。各PCR増幅では、94℃で1分間のインキュベーション、次いで55℃で30秒間のインキュベーションの後、72℃で1分間のインキュベーションを課すインキュベーションサイクルを、35回繰り返す。
【0233】
次に、各検索領域ポリヌクレオチドの試料をそれぞれ制限消化反応用の様々な制限酵素(各制限消化反応につき1制限酵素)と混合し、37℃で1時間インキュベートする。その制限消化反応混合物をアガロースゲル電気泳動にかけて、検索領域ポリヌクレオチドが様々な制限酵素の一つによってうまく制限消化されるかどうかを確認する。下記表14および表15に示す制限酵素による制限消化の成功により、下記表14および表15に示す与えられた相対位置の置換アミノ酸をコードする変異コドンが検索領域中に存在するかどうかが示される。
【0234】
【表15】
表14では、与えられた相対位置のアミノ酸をコードするコドンにおける与えられた制限酵素による制限消化の不成功により、当該コドンは変異コドンであることが示される。このような場合は、ABI自動シークエンサー(モデル377、Applied Biosystems)を使って当該検索領域を配列決定して、検索領域中に変異コドンがあるなら、その変異コドン中の変異を決定する。
【0236】
【表16】
表15の場合は、与えられた相対位置のアミノ酸をコードするコドンにおける与えられた制限酵素による制限消化の成功により、当該コドンは変異コドンであることが示される。このような場合は、検索領域中に変異コドンがあるとすれば、その変異コドン中の変異は上記表15に記載のヌクレオチド配列であると決定される。
【0238】
6.11.実施例11 サザンハイブリダイゼーション法による遺伝子型診断
6.9.1項で得た5μgの試験ゲノムDNAを制限酵素StuIで完全に制限消化する。その制限消化反応混合物を、4%Nusieve 3:1アガロースゲル(FMC BIO)を使って、20Vで16時間の電気泳動にかける。キャピラリーアルカリブロット法(Hybond blotting membrane manual、Amerscham)を使って、4%Nuseive 3:1アガロースゲル中の分離されたDNA断片をナイロンメンブレンに2時間ブロットする。続いて、ブロットしたフィルターを2×SSC緩衝液(0.3M NaCl、0.33M クエン酸Na、pH7.0)で軽く洗浄し、ブロットしたナイロンメンブレンを80℃で90分間乾燥する。
【0239】
ブロットしたナイロンメンブレンを55℃で16時間、プレハイブリダイゼーション緩衝液(6×SSPE(0.9M NaCl、0.052M NaH2PO2、7.5mM EDTA)、0.5%SDS、5×デンハルト液、および0.1mg/mlサケ精子DNA)で処理する。次にプレハイブリダイゼーション緩衝液を等体積のハイブリダイゼーション緩衝液(6×SSPE(0.9M NaCl、0.052M NaH2PO2、7.5mM EDTA)、0.5%SDS、5×デンハルト液、0.1mg/mlサケ精子DNAおよび32P標識プローブオリゴヌクレオチド)と交換する。ハイブリダイゼーション緩衝液中の32P標識プローブオリゴヌクレオチドの放射能濃度は、ハイブリダイゼーション緩衝液150ml毎に少なくとも10×108cpmである。32P標識プローブオリゴヌクレオチドとしては、末端が32Pで標識された配列番号81に記載のオリゴヌクレオチドを利用する。32P標識プローブは、T4ポリヌクレオチドキナーゼと一緒に提供される緩衝液中で、γ-32P-ATP、T4ポリヌクレオチドキナーゼおよび配列番号81に記載のオリゴヌクレオチド1μgを、37℃で1時間インキュベートすることによって作製する。
【0240】
ハイブリダイゼーションの後、ブロットしたナイロンメンブレンを、1×SSC(0.15M NaCl、15mMクエン酸ナトリウム)および0.5%SDSを含む洗浄緩衝液で2回洗浄する。ブロッティングフィルターを2回洗浄する際には、各洗浄後に、ブロットしたナイロンメンブレンを洗浄緩衝液中、62℃で40分間インキュベートする。
【0241】
次にブロットしたメンブレンをX線フィルムで10日間オートラジオグラフィーにかけて、制限酵素StuIが相対位置494の置換アミノ酸をコードする変異コドンと思われる検索領域中のコドンとオーバーラップする制限部位を制限消化することができるかどうかを解析する。制限酵素StuIによる制限消化の成功は、試験ERαポリヌクレオチド中に、相対位置494のアミノ酸をコードするコドンとオーバーラップして、AGGCCTを包含するヌクレオチド配列が存在することを示す。このような場合、試験ERαは正常ERαであると決定される。対応する部位での制限酵素StuIによる制限消化の失敗は、試験ERαポリヌクレオチドの相対位置494に置換アミノ酸をコードする変異コドンが存在することを示す。このような場合は、ABI自動シークエンサー(モデル377、Applied Biosystems)を使って検索領域を配列決定し、検索領域に変異コドンがあれば、その中の変異を決定する。
【0242】
6.12.実施例12 ヒト正常ARをコードするプラスミドの作製
ヒト前立腺cDNAライブラリー(CLONETECH、QUICK clone cDNA#7123-1)を利用して、そこからヒト正常ARをコードするcDNA(Genbankアクセッション番号M23263)をPCR増幅する。このPCR増幅におけるPCR混合物は、ヒト前立腺cDNAライブラリー10ng、配列番号176に記載のオリゴヌクレオチド10pmol、配列番号177に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。配列番号176および配列番号177に記載のオリゴヌクレオチドは、DNAシンセサイザー(モデル394、Applied Biosystems)を使って合成する。このPCR増幅では、PCRsystem 9700(Applied Biosystems)を使って、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを35回繰り返す。
【0243】
得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、ヒト正常ARをコードするcDNAがPCR増幅されることを臭化エチジウム染色で確認する。増幅されたcDNAを低融点アガロースゲルから回収した後、回収したcDNAの試料をDye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。調製したcDNA試料をABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定する。
【0244】
次に、cDNA中の開始コドン(ATG)のすぐ上流にKozakコンセンサス配列を加えるために、新たなPCR増幅を行う。このPCR増幅におけるPCR混合物は、ヒト正常ARをコードするcDNA 100ng、配列番号178に記載のオリゴヌクレオチドと配列番号179に記載のオリゴヌクレオチド、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。このPCR増幅では、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを25回繰り返す。得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。増幅されたcDNAを低融点アガロースゲルから回収した後、1μgの増幅cDNAをDNA Blunting Kit(宝酒造)で処理して、増幅cDNAの末端を平滑化する。次に、それによって得たcDNAをT4ポリヌクレオチドキナーゼと反応させて、その末端をリン酸化する。リン酸化されたcDNAをフェノール処理した後、リン酸化cDNAをエタノール沈殿して、精製型のリン酸化cDNAを得る。
【0245】
プラスミドpRc/RSV(Invitrogen)を制限酵素HindIIIで消化した後、65℃で1時間、BAPで処理する。次に、制限消化pRc/RSVをフェノール処理およびエタノール沈殿によって精製する。次に、制限消化pRc/RSVをDNA Blunting Kit(宝酒造)で処理してその末端を平滑化し、低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。制限消化されたpRc/RSVを低融点アガロースゲルから回収した後、その制限消化pRc/RSV 100ngおよび上記精製型リン酸化cDNAの全てをT4 DNAリガーゼによるライゲーション反応に使用する。そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ヒト正常ARをコードするプラスミドが存在することを確認する。そのようなプラスミドを選択し、pRc/RSV-hAR Kozakと名づける。
【0246】
6.13.実施例13 ヒト正常GRをコードするプラスミドの作製
ヒト肝臓cDNAライブラリー(CLONETECH、QUICK clone cDNA#7113-1)を利用して、そこからヒト正常GRをコードするcDNA(Genbankアクセッション番号M10901)をPCR増幅する。このPCR増幅におけるPCR混合物は、ヒト肝臓cDNAライブラリー10ng、配列番号180に記載のオリゴヌクレオチド10pmol、配列番号181に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。配列番号180および配列番号181に記載のオリゴヌクレオチドは、DNAシンセサイザー(モデル394、Applied Biosystems)を使って合成する。このPCR増幅では、PCRsystem 9700(Applied Biosystems)を使って、95℃で1分間のインキュベーションの後、60℃で3分間のインキュベーションを課すインキュベーションサイクルを35回繰り返す。
【0247】
得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、ヒト正常GRをコードするcDNAがPCR増幅されることを臭化エチジウム染色で確認する。増幅されたcDNAを低融点アガロースゲルから回収した後、回収したcDNAの試料をDye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。調製したcDNA試料をABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定する。
【0248】
次に、cDNA中の開始コドン(ATG)のすぐ上流にKozakコンセンサス配列を加えるために、新たなPCR増幅を行う。このPCR増幅におけるPCR混合物は、ヒト正常GRをコードするcDNA 100ng、配列番号182に記載のオリゴヌクレオチドと配列番号183に記載のオリゴヌクレオチド、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。このPCR増幅では、95℃で1分間のインキュベーションの後、60℃で3分間のインキュベーションを課すインキュベーションサイクルを25回繰り返す。得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。増幅されたcDNAを低融点アガロースゲルから回収した後、1μgの増幅cDNAをDNA Blunting Kit(宝酒造)で処理して、増幅cDNAの末端を平滑化する。次に、それによって得たcDNAをT4ポリヌクレオチドキナーゼと反応させて、その末端をリン酸化する。リン酸化されたcDNAをフェノール処理した後、リン酸化cDNAをエタノール沈殿して、精製型のリン酸化cDNAを得る。
【0249】
プラスミドpRc/RSV(Invitrogen)を制限酵素HindIIIで消化した後、65℃で1時間、BAPで処理する。次に、制限消化pRc/RSVをフェノール処理およびエタノール沈殿によって精製する。次に、制限消化pRc/RSVをDNA Blunting Kit(宝酒造)で処理してその末端を平滑化し、低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。制限消化されたpRc/RSVを低融点アガロースゲルから回収した後、その制限消化pRc/RSV 100ngおよび上記精製型リン酸化cDNAの全てをT4 DNAリガーゼによるライゲーション反応に使用する。その反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ヒト正常GRをコードするプラスミドが存在することを確認する。そのようなプラスミドを選択し、pRc/RSV-hGR Kozakと名づける。
【0250】
6.14.実施例14 ヒト正常PRをコードするプラスミドの作製
ヒト肝臓cDNAライブラリー(CLONETECH、QUICK clone cDNA#7113-1)を利用して、そこからヒト正常PRをコードするcDNA(Genbankアクセッション番号M15716)をPCR増幅する。このPCR増幅におけるPCR混合物は、ヒト肝臓cDNAライブラリー10ng、配列番号184に記載のオリゴヌクレオチド10pmol、配列番号185に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。配列番号184および配列番号185に記載のオリゴヌクレオチドは、DNAシンセサイザー(モデル394、Applied Biosystems)を使って合成する。このPCR増幅では、PCRsystem 9700(Applied Biosystems)を使って、95℃で1分間のインキュベーションの後、55℃で3分間のインキュベーションを課すインキュベーションサイクルを35回繰り返す。
【0251】
得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、ヒト正常PRをコードするcDNAがPCR増幅されることを臭化エチジウム染色で確認する。増幅されたcDNAを低融点アガロースゲルから回収した後、回収したcDNAの試料をDye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。調製したcDNA試料をABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定する。
【0252】
次に、cDNA中の開始コドン(ATG)のすぐ上流にKozakコンセンサス配列を加えるために、新たなPCR増幅を行う。このPCR増幅におけるPCR混合物は、ヒト正常PRをコードするcDNA 100ng、配列番号186に記載のオリゴヌクレオチドと配列番号187に記載のオリゴヌクレオチド、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。このPCR増幅では、95℃で1分間のインキュベーションの後、55℃で3分間のインキュベーションを課すインキュベーションサイクルを25回繰り返す。得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。増幅された試験cDNAを低融点アガロースゲルから回収した後、1μgの増幅試験cDNAをDNA Blunting Kit(宝酒造)で処理して、増幅試験cDNAの末端を平滑化する。次に、それによって得た試験cDNAをT4ポリヌクレオチドキナーゼと反応させて、cDNAの末端をリン酸化する。リン酸化された試験cDNAをフェノール処理した後、リン酸化試験cDNAをエタノール沈殿して、精製型のリン酸化試験cDNAを得る。
【0253】
プラスミドpRc/RSV(Invitrogen)を制限酵素HindIIIで消化した後、65℃で1時間、BAPで処理する。次に、制限消化pRc/RSVをフェノール処理およびエタノール沈殿によって精製する。次に、制限消化pRc/RSVをDNA Blunting Kit(宝酒造)で処理してその末端を平滑化し、低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。制限消化されたpRc/RSVを低融点アガロースゲルから回収した後、その制限消化pRc/RSV 100ngおよび上記精製型リン酸化試験cDNAの全てをT4 DNAリガーゼによるライゲーション反応に使用する。その反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ヒト正常PRをコードするプラスミドが存在することを確認する。そのようなプラスミドを選択し、pRc/RSV-hPR Kozakと名づける。
【0254】
6.15.ヒト正常MRをコードするプラスミドの作製
ヒト肝臓cDNAライブラリー(CLONETECH、QUICK clone cDNA#7113-1)を利用して、そこからヒト正常MRをコードするcDNA(Genbankアクセッション番号M16801)をPCR増幅する。このPCR増幅におけるPCR混合物は、ヒト肝臓cDNAライブラリー10ng、配列番号188に記載のオリゴヌクレオチド10pmol、配列番号189に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。配列番号188および配列番号189に記載のオリゴヌクレオチドは、DNAシンセサイザー(モデル394、Applied Biosystems)を使って合成する。このPCR増幅では、PCRsystem 9700(Applied Biosystems)を使って、95℃で1分間のインキュベーションの後、60℃で3分間のインキュベーションを課すインキュベーションサイクルを35回繰り返す。
【0255】
得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、ヒト正常MRをコードするcDNAがPCR増幅されることを臭化エチジウム染色で確認する。増幅されたcDNAを低融点アガロースゲルから回収した後、回収したcDNAの試料をDye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。調製したcDNA試料をABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定する。
【0256】
次に、cDNA中の開始コドン(ATG)のすぐ上流にKozakコンセンサス配列を加えるために、新たなPCR増幅を行う。このPCR増幅におけるPCR混合物は、ヒト正常MRをコードするcDNA 100ng、配列番号190に記載のオリゴヌクレオチドと配列番号191に記載のオリゴヌクレオチド、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。このPCR増幅では、95℃で1分間のインキュベーションの後、60℃で3分間のインキュベーションを課すインキュベーションサイクルを25回繰り返す。得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。増幅されたcDNAを低融点アガロースゲルから回収した後、1μgの増幅cDNAをDNA Blunting Kit(宝酒造)で処理して、増幅cDNAの末端を平滑化する。次に、それによって得たcDNAをT4ポリヌクレオチドキナーゼと反応させて、その末端をリン酸化する。リン酸化されたcDNAをフェノール処理した後、リン酸化cDNAをエタノール沈殿して、精製型のリン酸化cDNAを得る。
【0257】
プラスミドpRc/RSV(Invitrogen)を制限酵素HindIIIで消化した後、65℃で1時間、BAPで処理する。次に、制限消化pRc/RSVをフェノール処理およびエタノール沈殿によって精製する。次に、制限消化pRc/RSVをDNA Blunting Kit(宝酒造)で処理してその末端を平滑化し、低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。制限消化されたpRc/RSVを低融点アガロースゲルから回収した後、その制限消化pRc/RSV 100ngおよび上記精製型リン酸化cDNAの全てをT4 DNAリガーゼによるライゲーション反応に使用する。その反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ヒト正常MRをコードするプラスミドが存在することを確認する。そのようなプラスミドを選択し、pRc/RSV-hMR Kozakと名づける。
【0258】
6.16.実施例16 MMTVレポーター遺伝子を染色体の一つに安定に含有する安定形質転換細胞の作製
プラスミドpMSG(Pharmacia)を制限酵素HindIIIおよびSmaIで制限消化して、1463bpのサイズを持ちMMTV-LTR領域の部分配列をコードするDNA断片を得る。次にその1463bp DNA断片をDNA Blunting Kit(宝酒造)で処理して、当該1463bp DNA断片の末端を平滑化する。
【0259】
ホタルルシフェラーゼ遺伝子をコードするプラスミドpGL3(Promega)を、制限酵素BglIIおよびHindIIIで制限消化した後、65℃で1時間、BAPで処理する。次にその制限消化反応混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、ホタルルシフェラーゼをコードするヌクレオチド配列を持つDNA断片が存在することを確認した。次に、ホタルルシフェラーゼをコードするヌクレオチド配列を持つDNA断片を低融点アガロースゲルから回収した。次に、ホタルルシフェラーゼをコードするヌクレオチド配列を持つ回収したDNA断片100ngと、上記1463bp DNA断片1μgとを、T4 DNAリガーゼによるライゲーション反応に使用する。次に、そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次に、アンピシリン耐性を示すクローンを回収する。次に、それらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を制限酵素KpnIおよびClaIで制限消化する。その制限消化反応混合物を、アガロースゲル電気泳動にかけて、ホタルルシフェラーゼをコードするヌクレオチド配列を持つDNA断片の上流に作動的に配された上記1463bp DNA断片1コピー(以下MMTVレポーター遺伝子という)を含有するプラスミドが存在することを確認する。そのようなプラスミドを選択し、pGL3-MMTVと名づける。
【0260】
プラスミドpUCSV-BSD(フナコシ)を制限酵素BamHIで制限消化して、ブラストサイジンSデアミナーゼ遺伝子発現カセットをコードするDNAを調製する。また、プラスミドpGL3-MMTVを制限酵素BamHIで制限消化した後、65℃で1時間、BAPで処理する。得られたブラストサイジンSデアミナーゼ遺伝子発現カセットコードDNAと制限消化pGL3-MMTVとを混合して、T4 DNAリガーゼによるライゲーション反応に使用する。そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に単離された各プラスミドの部分試料をDye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ブラストサイジンSデアミナーゼ遺伝子発現カセットをコードするDNAがpGL3-MMTVのBamHIの制限部位に挿入されている構造を持つプラスミドが存在することを確認する。そのような構造を持つプラスミドを選択し、pGL3-MMTV-BSDと名づける。
【0261】
染色体の一つにMMTVレポーター遺伝子を安定に含有する安定形質転換細胞(以下、安定形質転換MMTVカセット細胞という)を作製するために、プラスミドpGL3-MMTV-BSDを線状化し、HeLa細胞に導入した。
【0262】
プラスミドpGL3-MMTV-BSDを線状化するために、pGL3-MMTV-BSDを制限酵素SalIで制限消化する。
【0263】
直径約10cmの培養皿(Falcon)を使用し、10%FBSを含むDMEM培地(日水製薬)にて、5%CO2下に37℃で、約5×105個のHeLa細胞を宿主細胞として1日培養した。
【0264】
次に、線状化したpGL3-MMTV-BSDをリポフェクトアミン(Life Technologies)を用いるリポフェクション法によって培養HeLa細胞に導入する。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿の線状化pGL3-MMTV-BSDおよび21μl/培養皿のリポフェクトアミンを含めた。
【0265】
リポフェクション処理の後、DMEM培地を、10%FBSを含むDMEM培地と交換し、形質転換HeLa細胞を約36時間培養する。次に、形質転換HeLa細胞をトリプシン処理によって培養皿から除去、収集し、ブラストサイジンSを16μg/mlの濃度になるように添加した培地を含む容器に移す。ブラストサイジンS含有培地を3、4日毎に新しいバッチのブラストサイジンS含有培地に交換しながら、前記ブラストサイジンS含有培地で形質転換HeLa細胞を1ヶ月培養する。
【0266】
その結果増殖して1〜数mmの直径を持つコロニーを形成することができるクローンを、前もって培地を分注しておいた96ウェルViewPlate(Berthold)のウェルに、丸ごと移す。それらクローンのコロニーをさらに培養する。クローンが増殖して各ウェルの底面の50%以上を覆うようになったら(移植の約5日後)、それらのクローンをトリプシン処理によって除去、収集する。次にクローンを2つの継代培養物に分割する。一方の継代培養物は96ウェルViewPlateに移して、それをマスタープレートとする。他方の継代培養物は96ウェルViewPlateに移して、これをアッセイプレートとする。マスタープレートとアッセイプレートには、クローンを培養することができるように培地が含まれている。マスタープレートは同様の条件で引き続き培養する。
【0267】
次に、培地をアッセイプレートのウェルから除去し、ウェル壁に付着したクローンをPBS(-)で2回洗浄する。5倍希釈した溶解緩衝液PGC50(東洋インキ)をアッセイプレートのウェル中のクローンに1ウェルあたり20μlずつ加える。そのアッセイプレートを室温で30分間静置した後、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットする。次に、基質溶液PGL100(東洋インキ)50μlをアッセイプレート中の各溶解クローンに自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで測定する。高いルシフェラーゼ活性を示した複数のクローンをそこから選択する。
【0268】
次に、選択したクローンの試料を、直径約10cmの培養皿(Falcon)を用いて、活性炭デキストランFBS/E-MEM培地中、5%CO2下に、37℃で1〜2週間培養する。
【0269】
次に、リポフェクトアミン(Life Technologies)を用いるリポフェクション法により、プラスミドpRc/RSV-hAR Kozakを、上で選択したクローンに導入して、2次クローンを得る。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿の上記プラスミド、および21μl/培養皿のリポフェクトアミンを含めた。次に、得られた2次クローンに、正常ARの天然のコグネイトリガンドであるジヒドロテストステロン(DHT)を含有するDMSO溶液を、DHT濃度が10nMになるように添加する。2次クローンを2日間培養した後、各2次クローンについて上記と同様にルシフェラーゼ活性を測定する。最も高いルシフェラーゼ活性の誘導を示す2次クローンを与えたマスタープレート中のクローンを、安定形質転換MMTVカセット細胞として選択する。
【0270】
ちなみに、安定形質転換MMTVカセット細胞は、AR、GR、PR、MRなどを用いるレポーターアッセイに使用することができる。
【0271】
6.17.実施例17 ヒト試験ARとしてヒト正常ARを用いるレポーターアッセイ
6.17.1.安定形質転換MMTVカセット細胞の調製
6.16項で得た安定形質転換MMTVカセット細胞約2×106個を直径約10cmの培養皿(Falcon)を使って、5%CO2下に、活性炭デキストランFBS/E-MEM培地で1日培養する。
【0272】
一過性発現のために、プラスミドpRc/RSV-hAR Kozakを、リポフェクトアミン(Life Technologies)を用いるリポフェクション法により、安定形質転換MMTVカセット細胞の継代培養物に導入する。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿のpRc/RSV-hAR Kozak、および21μl/培養皿のリポフェクトアミンを含める。得られた細胞継代培養物を5%CO2下に37℃で16時間培養した後、活性炭デキストランFBS/E-MEM培地を新しいバッチの活性炭デキストランFBS/E-MEM培地に交換して、細胞継代培養物をさらに3時間培養する。次に、細胞継代培養物を収集し、活性炭デキストランFBS/E-MEM培地に均一に懸濁して、その継代培養物とする。
【0273】
6.17.2.MMTVレポーター遺伝子転写活性化活性の測定
第1DMSO溶液は様々な濃度のフルタミドを含むように調製する。第1DMSO溶液では、正常ARに対するアゴニストとしてフルタミドを使用する。また、第2DMSO溶液は10nMのDHTと様々な濃度のフルタミドとを含むように調製する。第2DMSO溶液では、正常ARに対するアンタゴニストとしてフルタミドを使用する。
【0274】
次に、96ウェルViewPlateで、第1および第2DMSO溶液をそれぞれ上記6.17.1項で調製した継代培養物と、各ウェル中の第1または第2DMSO溶液の濃度が約0.1%(v/v)になるように混合する。また、標準物質として、6.17.1項で得た細胞継代培養物の試料を96ウェルViewPlateのウェルで、DHTを含有するDMSO溶液と混合する。
【0275】
次に、細胞を5%CO2下に37℃で40時間培養する。5倍希釈した溶解緩衝液PGC50(東洋インキ)をウェル中の継代培養物に1ウェルあたり50μlずつ加える。その96ウェルViewPlateをときどき穏やかに振とうしながら室温で30分間インキュベートする。次に、溶解した細胞10μlをそれぞれ白色96ウェルサンプルプレート(Berthold)に移し、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットする。次に、基質溶液PGL100(東洋インキ)50μlを白色96ウェルサンプルプレート中の各溶解細胞にそれぞれ自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで直ちに5秒間測定する。
【0276】
また、上記レポーターアッセイでは、試験ARとして変異ARを使用することもできる。この場合は、pRc/RSV-hAR Kozakの代わりに変異ARをコードするプラスミドを使用する。変異ARをコードするプラスミドを得るには、上記と同様にして、変異ARをコードするポリヌクレオチドの上流にKozakコンセンサス配列を作動的に付加し、得られたポリヌクレオチドをプラスミドpRc/RSV(Invitrogen)中のHindIIIの制限部位に挿入する。
【0277】
6.18.実施例18 ヒト試験GRとしてヒト正常GRを用いるレポーターアッセイ
6.18.1.安定形質転換MMTVカセット細胞の調製
6.16項で得た安定形質転換MMTVカセット細胞約2×106個を直径約10cmの培養皿(Falcon)を使って、5%CO2下に、活性炭デキストランFBS/E-MEM培地で1日培養する。
【0278】
一過性発現のために、プラスミドpRc/RSV-hGR Kozakを、リポフェクトアミン(Life Technologies)を用いるリポフェクション法により、安定形質転換MMTVカセット細胞の継代培養物に導入する。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿のpRc/RSV-hAR Kozak、および21μl/培養皿のリポフェクトアミンを含める。得られた細胞継代培養物を5%CO2下に37℃で16時間培養した後、活性炭デキストランFBS/E-MEM培地を新しいバッチの活性炭デキストランFBS/E-MEM培地に交換して、細胞継代培養物をさらに3時間培養する。次に、その細胞継代培養物を収集し、活性炭デキストランFBS/E-MEM培地に均一に懸濁する。
【0279】
6.18.2.MMTVレポーター遺伝子転写活性化活性の測定
第1DMSO溶液は様々な濃度のプレグナノロン16αカルボニトリル(PCN)を含むように調製する。第1DMSO溶液では、正常GRに対するアゴニストとしてPCNを使用する。また、第2DMSO溶液は10nMのコルチコステロンと様々な濃度のPCNとを含むように調製する。第2DMSO溶液では、正常GRに対するアンタゴニストとしてPCNを使用する。
【0280】
次に、96ウェルViewPlateで、第1および第2DMSO溶液をそれぞれ上記6.18.1項で調製した細胞継代培養物と、各ウェル中の第1または第2DMSO溶液の濃度が約0.1%(v/v)になるように混合する。また、標準物質として、上記細胞継代培養物の試料を、96ウェルViewPlateのウェルで、コルチコステロンを含有するDMSO溶液と混合する。
【0281】
次に、細胞を5%CO2下に37℃で40時間培養する。5倍希釈した溶解緩衝液PGC50(東洋インキ)をウェル中の継代培養物に1ウェルあたり50μlずつ加える。その96ウェルViewPlateをときどき穏やかに振とうしながら室温で30分間インキュベートする。次に、溶解した細胞10μlをそれぞれ白色96ウェルサンプルプレート(Berthold)に移し、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットする。次に、基質溶液PGL100(東洋インキ)50μlを白色96ウェルサンプルプレート中の各溶解細胞にそれぞれ自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで直ちに5秒間測定する。
【0282】
また、上記レポーターアッセイでは、試験GRとして変異GRを使用することもできる。この場合は、pRc/RSV-hGR Kozakの代わりに変異GRをコードするプラスミドを使用する。変異GRをコードするプラスミドを得るには、上記と同様にして、変異GRをコードするポリヌクレオチドの上流にKozakコンセンサス配列を作動的に付加し、得られたポリヌクレオチドをプラスミドpRc/RSV(Invitrogen)中のH indIIIの制限部位に挿入する。
【0283】
6.19.実施例19 ヒト試験PRとしてヒト正常PRを用いるレポーターアッセイ
6.19.1.安定形質転換MMTVカセット細胞の調製
6.16項で得た安定形質転換MMTVカセット細胞約2×106個を直径約10cmの培養皿(Falcon)を使って、5%CO2下に37℃で、活性炭デキストランFBS/E-MEM培地で1日培養する。
【0284】
一過性発現のために、プラスミドpRc/RSV-hPR Kozakを、リポフェクトアミン(Life Technologies)を用いるリポフェクション法により、安定形質転換MMTVカセット細胞の継代培養物に導入する。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿のpRc/RSV-hAR Kozak、および21μl/培養皿のリポフェクトアミンを含める。得られた細胞継代培養物を5%CO2下に37℃で16時間培養した後、活性炭デキストランFBS/E-MEM培地を新しいバッチの活性炭デキストランFBS/E-MEM培地と交換して、細胞継代培養物をさらに3時間培養する。次に、その細胞継代培養物を収集し、活性炭デキストランFBS/E-MEM培地に均一に懸濁する。
【0285】
6.19.2.MMTVレポーター遺伝子転写活性化活性の測定
第1DMSO溶液は様々な濃度のRU486を含むように調製する。第1DMSO溶液では、正常PRに対するアゴニストとしてRU486を使用する。また、第2DMSO溶液は10nMのプロゲステロンと様々な濃度のRU486とを含むように調製する。第2DMSO溶液では、正常PRに対するアンタゴニストとしてRU486を使用する。
【0286】
96ウェルViewPlateで、第1および第2DMSO溶液をそれぞれ上記6.19.1項で調製した細胞継代培養物と、各ウェル中の第1または第2DMSO溶液の濃度が約0.1%(v/v)になるように混合する。また、標準物質として、上記細胞継代培養物の試料を、プロゲステロンを含有するDMSO溶液と混合する。
【0287】
次に、細胞を5%CO2下に37℃で40時間培養する。5倍希釈した溶解緩衝液PGC50(東洋インキ)をウェル中の細胞に1ウェルあたり50μlずつ加える。その96ウェルViewPlateをときどき穏やかに振とうしながら室温で30分間インキュベートする。次に、溶解した細胞10μlをそれぞれ白色96ウェルサンプルプレート(Berthold)に移し、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットする。次に、基質溶液PGL100(東洋インキ)50μlを白色96ウェルサンプルプレート中の各溶解細胞にそれぞれ自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで直ちに5秒間測定する。
【0288】
また、上記レポーターアッセイでは、試験PRとして変異PRを使用することもできる。この場合は、pRc/RSV-hPR Kozakの代わりに変異PRをコードするプラスミドを使用する。変異PRをコードするプラスミドを得るには、上記と同様にして、変異PRをコードするポリヌクレオチドの上流にKozakコンセンサス配列を作動的に付加し、得られたポリヌクレオチドをプラスミドpRc/RSV(Invitrogen)中のHindIIIの制限部位に挿入する。
【0289】
6.20.実施例20 ヒト正常ERβをコードするプラスミドの作製
ヒト前立腺cDNAライブラリー(CLONETECH、QUICK clone cDNA#7123-1)を利用して、そこからヒト正常ERβをコードするcDNA(Genbankアクセッション番号AB006590)をPCR増幅する。このPCR増幅におけるPCR混合物は、ヒト肝臓cDNAライブラリー10ng、配列番号192に記載のオリゴヌクレオチド10pmol、配列番号193に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。配列番号192および配列番号193に記載のオリゴヌクレオチドは、DNAシンセサイザー(モデル394、Applied Biosystems)を使って合成する。このPCR増幅では、PCRsystem 9700(Applied Biosystems)を使って、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを35回繰り返す。
【0290】
得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、ヒト正常ERβをコードするcDNAがPCR増幅されることを臭化エチジウム染色で確認する。増幅されたcDNAを低融点アガロースゲルから回収した後、回収したcDNAの試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。調製したcDNA試料をABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定する。
【0291】
次に、cDNA中の開始コドン(ATG)のすぐ上流にKozakコンセンサス配列を加えるために、新たなPCR増幅を行う。このPCR増幅におけるPCR混合物は、上記cDNA 100ng、配列番号194に記載のオリゴヌクレオチド10pmol、配列番号195に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。このPCR増幅では、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを25回繰り返す。得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。増幅されたcDNAを低融点アガロースゲルから回収した後、1μgの増幅cDNAをDNA Blunting Kit(宝酒造)で処理して、増幅cDNAの末端を平滑化する。次に、それによって得たcDNAをT4ポリヌクレオチドキナーゼと反応させて、当該cDNAの末端をリン酸化する。リン酸化されたcDNAをフェノール処理した後、リン酸化cDNAをエタノール沈殿して、精製型のリン酸化cDNAを得る。
【0292】
プラスミドpRc/RSV(Invitrogen)を制限酵素HindIIIで消化した後、65℃で1時間、BAPで処理する。次に、制限消化pRc/RSVをフェノール処理およびエタノール沈殿によって精製する。次に、制限消化pRc/RSVをDNA Blunting Kit(宝酒造)で処理してその末端を平滑化し、低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。制限消化されたpRc/RSVを低融点アガロースゲルから回収した後、その制限消化pRc/RSV 100ngおよび上記精製型リン酸化cDNAの全てをT4 DNAリガーゼによるライゲーション反応に使用する。そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ヒト正常ERβをコードするプラスミドが存在することを確認する。そのようなプラスミドを選択し、pRc/RSV-hERβKozakと名づける。
【0293】
6.21.実施例21 ヒト試験ERβとしてヒト正常ERβを用いるレポーターアッセイ
6.21.1.安定形質転換EREカセット細胞の調製
6.3項で得た安定形質転換EREカセット細胞約2×106個を直径約10cmの培養皿(Falcon)を使って、5%CO2下に37℃で、活性炭デキストランFBS/E-MEM培地で1日培養する。
【0294】
一過性発現のために、プラスミドpRc/RSV-hERβKozakを、リポフェクトアミン(Life Technologies)を用いるリポフェクション法により、安定形質転換EREカセット細胞の継代培養物に導入する。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿のpRc/RSV-hERβKozak、および21μl/培養皿のリポフェクトアミンを含める。得られた細胞継代培養物を5%CO2下に37℃で16時間培養した後、活性炭デキストランFBS/E-MEM培地を新しいバッチの活性炭デキストランFBS/E-MEM培地に交換して、細胞継代培養物をさらに3時間培養する。次に、その細胞継代培養物を収集し、活性炭デキストランFBS/E-MEM培地に均一に懸濁する。
【0295】
6.21.2.EREレポーター遺伝子転写活性化活性の測定
第1DMSO溶液は様々な濃度の4-ヒドロキシタモキシフェンを含むように調製する。第1DMSO溶液では、ヒト正常ERβに対するアゴニストとして4-ヒドロキシタモキシフェンを使用する。また、第2DMSO溶液は10nMのE2と様々な濃度の4-ヒドロキシタモキシフェンとを含むように調製する。第2DMSO溶液では、ERβに対するアンタゴニストとして4-ヒドロキシタモキシフェンを使用する。
【0296】
96ウェルViewPlateで、第1および第2DMSO溶液をそれぞれ上記6.21.1項で調製した継代培養物と、各ウェル中の第1または第2DMSO溶液の濃度が約0.1%(v/v)になるように混合する。また、標準物質として、96ウェルViewPlateのウェルで、上記細胞の試料を、E2 を含有するDMSO溶液と混合する。
【0297】
次に、細胞を5%CO2下に37℃で40時間培養する。5倍希釈した溶解緩衝液PGC50(東洋インキ)をウェル中の細胞に1ウェルあたり50μlずつ加える。その96ウェルViewPlateをときどき穏やかに振とうしながら室温で30分間インキュベートする。次に、溶解した細胞10μlをそれぞれ白色96ウェルサンプルプレート(Berthold)に移し、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットする。次に、基質溶液PGL100(東洋インキ)50μlを白色96ウェルサンプルプレート中の各溶解細胞にそれぞれ自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで直ちに5秒間測定する。
【0298】
また、上記レポーターアッセイでは、試験ERβとして変異ERβを使用することもできる。この場合は、pRc/RSV-hERβKozakの代わりに変異ERβをコードするプラスミドを使用する。変異ERβをコードするプラスミドを得るには、上記と同様にして、変異ERβをコードするポリヌクレオチドの上流にKozakコンセンサス配列を作動的に付加し、得られたポリヌクレオチドをプラスミドpRc/RSV(Invitrogen)中のHindIIIの制限部位に挿入する。
【0299】
6.22.実施例22 ヒト正常TRαをコードするプラスミドの作製
ヒト肝臓cDNAライブラリー(CLONETECH、QUICK clone cDNA#7113-1)を利用して、そこからヒト正常TRαをコードするcDNA(Genbankアクセッション番号M24748)をPCR増幅する。このPCR増幅におけるPCR混合物は、ヒト肝臓cDNAライブラリー10ng、配列番号196に記載のオリゴヌクレオチド10pmol、配列番号197に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。配列番号196および配列番号197に記載のオリゴヌクレオチドは、DNAシンセサイザー(モデル394、Applied Biosystems)を使って合成する。このPCR増幅では、PCRsystem 9700(Applied Biosystems)を使って、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを35回繰り返す。
【0300】
得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、ヒト正常TRαをコードするcDNAがPCR増幅されることを臭化エチジウム染色で確認する。増幅されたcDNAを低融点アガロースゲルから回収した後、回収したcDNAの試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。調製したcDNA試料をABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定する。
【0301】
次に、cDNA中の開始コドン(ATG)のすぐ上流にKozakコンセンサス配列を加えるために、新たなPCR増幅を行う。このPCR増幅におけるPCR混合物は、ヒト正常TRαをコードするcDNA 100ng、配列番号198に記載のオリゴヌクレオチド10pmol、配列番号199に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。このPCR増幅では、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを25回繰り返す。得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。増幅されたcDNAを低融点アガロースゲルから回収した後、1μgの増幅cDNAをDNA Blunting Kit(宝酒造)で処理して、増幅cDNAの末端を平滑化する。次に、それによって得たcDNAをT4ポリヌクレオチドキナーゼと反応させて、当該cDNAの末端をリン酸化する。リン酸化されたcDNAをフェノール処理した後、リン酸化cDNAをエタノール沈殿して、精製型のリン酸化cDNAを得る。
【0302】
プラスミドpRc/RSV(Invitrogen)を制限酵素HindIIIで消化した後、65℃で1時間、BAPで処理する。次に、制限消化pRc/RSVをフェノール処理およびエタノール沈殿によって精製する。次に、制限消化pRc/RSVをDNA Blunting Kit(宝酒造)で処理してその末端を平滑化し、低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。制限消化されたpRc/RSVを低融点アガロースゲルから回収した後、その制限消化pRc/RSV 100ngおよび上記精製型リン酸化cDNAの全てをT4 DNAリガーゼによるライゲーション反応に使用する。そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ヒト正常TRαをコードするプラスミドが存在することを確認する。そのようなプラスミドを選択し、pRc/RSV-hTRαKozakと名づける。
【0303】
6.23.実施例23 ヒト正常TRβをコードするプラスミドの作製
ヒト肝臓cDNAライブラリー(CLONETECH、QUICK clone cDNA#7113-1)を利用して、そこからヒト正常TRβをコードするcDNA(Genbankアクセッション番号M26747)をPCR増幅する。このPCR増幅におけるPCR混合物は、ヒト肝臓cDNAライブラリー10ng、配列番号200に記載のオリゴヌクレオチド10pmol、配列番号201に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。配列番号200および配列番号201に記載のオリゴヌクレオチドは、DNAシンセサイザー(モデル394、Applied Biosystems)を使って合成する。このPCR増幅では、PCRsystem 9700(Applied Biosystems)を使って、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを35回繰り返す。
【0304】
得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、ヒト正常TRβをコードするcDNAがPCR増幅されることを臭化エチジウム染色で確認する。増幅されたcDNAを低融点アガロースゲルから回収した後、回収したcDNAの試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。調製したcDNA試料をABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定する。
【0305】
次に、cDNA中の開始コドン(ATG)のすぐ上流にKozakコンセンサス配列を加えるために、新たなPCR増幅を行う。このPCR増幅におけるPCR混合物は、ヒト正常TRαβをコードするcDNA 100ng、配列番号202に記載のオリゴヌクレオチド10pmol、配列番号203に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。このPCR増幅では、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを25回繰り返す。得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。増幅されたcDNAを低融点アガロースゲルから回収した後、1μgの増幅cDNAをDNA Blunting Kit(宝酒造)で処理して、増幅cDNAの末端を平滑化する。次に、それによって得たcDNAをT4ポリヌクレオチドキナーゼと反応させて、当該cDNAの末端をリン酸化する。リン酸化されたcDNAをフェノール処理した後、リン酸化cDNAをエタノール沈殿して、精製型のリン酸化cDNAを得る。
【0306】
プラスミドpRc/RSV(Invitrogen)を制限酵素HindIIIで消化した後、65℃で1時間、BAPで処理する。次に、制限消化pRc/RSVをフェノール処理およびエタノール沈殿によって精製する。次に、制限消化pRc/RSVをDNA Blunting Kit(宝酒造)で処理してその末端を平滑化し、低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。制限消化されたpRc/RSVを低融点アガロースゲルから回収した後、その制限消化pRc/RSV 100ngおよび上記精製型リン酸化cDNAの全てをT4 DNAリガーゼによるライゲーション反応に使用する。そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ヒト正常TRβをコードするプラスミドが存在することを確認する。そのようなプラスミドを選択し、pRc/RSV-hTRβKozakと名づける。
【0307】
6.24.実施例24 DR4レポーター遺伝子を含有するプラスミドの作製
配列番号204に記載のオリゴヌクレオチドと、それに相補的なヌクレオチド配列を持つオリゴヌクレオチドとを、DNAシンセサイザーで合成する。配列番号204に記載のオリゴヌクレオチドは、DR4の一方の鎖をコードするように合成する。もう一つのオリゴヌクレオチドは、第1オリゴヌクレオチドに相補的なヌクレオチド配列を持つように合成する。これら2つのオリゴヌクレオチドを互いにアニールさせて、DR4配列をコードするDNA(以下、DR4 DNAという)を作製する。次に、DR4 DNAをT4ポリヌクレオチドキナーゼと反応させて、DR4 DNAの末端をリン酸化する。次に、DR4 DNAをT4 DNAリガーゼで互いに結合させて、DR4配列が5回縦列反復したDR4×5 DNAを得る。次に、そのライゲーション反応混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、そのゲルからDR4×5 DNAを回収する。
【0308】
6.2項で得たプラスミドpGL3-TATAを制限酵素SmaIで制限消化した後、65℃で1時間、BAPで処理する。次に、その制限消化反応混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。その低融点アガロースゲルからホタルルシフェラーゼをコードするヌクレオチド配列を持つDNA断片を回収した後、回収したDNA断片100ngとDR4×5 DNA 1μgとをライゲーション反応に使用する。次に、得られたライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に単離された各プラスミドの部分試料を制限酵素KpnIおよびXhoIで制限消化する。その制限消化反応混合物をアガロースゲル電気泳動にかけて、DR4×5 DNAがpGL3-TATA中の制限酵素SmaIの制限部位に挿入されている構造を持つプラスミドが存在することを確認する。そのような構造を持つプラスミドを選択し、pGL3-TATA-DR4×5と名づける。
【0309】
次に、プラスミドpGL3-TATA-DR4×5を制限酵素SalIで制限消化する。制限消化されたpGL3-TATA-DR4×5の末端をBlunting Kit(宝酒造)で平滑化した後、その制限消化pGL3-TATA-DR4×5を65℃で1時間、BAPで処理する。また、6.2項で得たブラストサイジンSデアミナーゼ遺伝子をコードするDNA断片(BamHI-BamHI断片)の末端も、Blunting Kitを使って平滑化する。
【0310】
次に、ブラストサイジンSデアミナーゼ遺伝子発現カセットをコードするDNA断片と、制限消化pGL3-TATA-DR4×5とを混合して、T4 DNAリガーゼによるライゲーション反応を行う。そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次に、アンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ブラストサイジンSデアミナーゼ遺伝子発現カセットをコードするDNAがpGL3-TATA-DR4×5中の制限酵素SalIの制限部位に挿入されている構造を持つプラスミドが存在するかどうかを確認する。そのようなプラスミドを選択し、pGL3-TATA-DR4×5-BSDと名づける。
【0311】
6.25.実施例25 DR4レポーター遺伝子を染色体の一つに安定に含有する安定形質転換細胞の作製
DR4レポーター遺伝子を染色体の一つに安定に含有する安定形質転換細胞(以下、安定形質転換DR4カセット細胞という)を作製するために、プラスミドpGL3-TATA-DR4×5-BSDを線状化し、HeLa細胞に導入した。
【0312】
プラスミドpGL3-TATA-DR4×5-BSDを線状化するために、pGL3-TATA-DR4×5-BSDを制限酵素NotIで制限消化する。
【0313】
直径約10cmの培養皿(Falcon)を使用し、10%FBSを含むDMEM培地(日水製薬)にて、5%CO2下に37℃で、約5×105個のHeLa細胞を宿主細胞として1日培養する。
【0314】
次に、線状化pGL3-TATA-DR4×5-BSDを、リポフェクトアミン(Life Technologies)を用いるリポフェクション法によって培養HeLa細胞に導入する。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿の線状化pGL3-TATA-DR4×5-BSD、および21μl/培養皿のリポフェクトアミンを含める。
【0315】
リポフェクション処理の後、DMEM培地を、10%FBSを含むDMEM培地と交換し、形質転換したHeLa細胞を約36時間培養する。次に、形質転換HeLa細胞をトリプシン処理によって培養皿から除去、収集し、ブラストサイジンSを16μg/mlの濃度になるように添加した培地を含む容器に移す。ブラストサイジンS含有培地を3、4日毎に新しいバッチのブラストサイジンS含有培地に交換しながら、前記ブラストサイジンS含有培地で形質転換HeLa細胞を1ヶ月培養する。
【0316】
その結果増殖して1〜数mmの直径を持つコロニーを形成することができるクローンを、前もって培地を分注しておいた96ウェルViewPlate(Berthold)のウェルに、丸ごと移す。それらのクローンをさらに培養する。クローンが増殖してウェルの底面の50%以上を覆うようになったら(移植の約5日後)、それらのクローンをトリプシン処理によって除去、収集する。次に、各クローンを2つの継代培養物に分割する。一方の継代培養物は96ウェルViewPlateに移して、それをマスタープレートとする。他方の継代培養物は96ウェルViewPlateに移して、これをアッセイプレートとする。マスタープレートとアッセイプレートは、クローンを培養することができるように培地を含有する。マスタープレートは同様の条件で引き続き培養する。
【0317】
次に、アッセイプレートのウェル中の培地をウェルから除去し、ウェル壁に付着したクローンをPBS(-)で2回洗浄する。5倍希釈した溶解緩衝液PGC50(東洋インキ)をアッセイプレートのウェル中のクローンに1ウェルあたり20μlずつ加える。そのアッセイプレートを室温で30分間静置した後、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットする。次に、基質溶液PGL100(東洋インキ)50μlをアッセイプレート中の各溶解クローンに自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで測定する。ルシフェラーゼ活性を示した複数のクローンをそこから選択する。
【0318】
次に、選択したクローンの試料を、直径約10cmの培養皿(Falcon)を用いて、活性炭デキストランFBS/E-MEM培地中、5%CO2下に、37℃で1日培養する。
【0319】
次に、リポフェクトアミン(Life Technologies)を用いるリポフェクション法により、プラスミドpRc/RSV-hTRαKozakを、上で選択したクローンに導入して、2次クローンを得る。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿の上記pRc/RSV-hTRαKozak、および21μl/培養皿のリポフェクトアミンを含めた。次に、得られた2次クローンに、ヒト正常TRαの天然のコグネイトリガンドであるトリヨードチロニン(T3)を含有するDMSO溶液を、培地中のT3濃度が10nMになるように加える。それらの2次クローンを2日間培養した後、各2次クローンについて上記と同様にルシフェラーゼ活性を測定する。最も高いルシフェラーゼ活性の誘導を示す2次クローンを与えたマスタープレート中のクローンを、安定形質転換DR4カセット細胞として選択する。
【0320】
ちなみに、安定形質転換DR4カセット細胞は、TRα、TRβ、CAR、LXR、PXRなどを用いるレポーターアッセイに使用することができる。
【0321】
6.26.実施例26 ヒト正常VDRをコードするプラスミドの作製
ヒト肝臓cDNAライブラリー(CLONETECH、QUICK clone cDNA#7113-1)を利用して、そこからヒト正常VDRをコードするcDNA(Genbankアクセッション番号J03258)をPCR増幅する。このPCR増幅におけるPCR混合物は、ヒト肝臓cDNAライブラリー10ng、配列番号205に記載のオリゴヌクレオチド10pmol、配列番号206に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。配列番号205および配列番号206に記載のオリゴヌクレオチドは、DNAシンセサイザー(モデル394、Applied Biosystems)を使って合成する。このPCR増幅では、PCRsystem 9700(Applied Biosystems)を使って、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを35回繰り返す。
【0322】
得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、ヒト正常VDRをコードするcDNAがPCR増幅されることを臭化エチジウム染色で確認する。増幅されたcDNAを低融点アガロースゲルから回収した後、回収したcDNAの試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。調製したcDNA試料をABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定する。
【0323】
次に、正常VDRをコードするcDNA中の開始コドン(ATG)のすぐ上流にKozakコンセンサス配列を加えるために、新たなPCR増幅を行う。このPCR増幅におけるPCR混合物は、正常VDRをコードするcDNA 100ng、配列番号207に記載のオリゴヌクレオチド10pmol、配列番号208に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。このPCR増幅では、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを25回繰り返す。得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。増幅されたcDNAを低融点アガロースゲルから回収した後、1μgの増幅cDNAをDNA Blunting Kit(宝酒造)で処理して、増幅cDNAの末端を平滑化する。次に、それによって得たcDNAをT4ポリヌクレオチドキナーゼと反応させて、当該cDNAの末端をリン酸化する。リン酸化されたcDNAをフェノール処理した後、リン酸化cDNAをエタノール沈殿して、精製型のリン酸化cDNAを得る。
【0324】
プラスミドpRc/RSV(Invitrogen)を制限酵素HindIIIで消化した後、65℃で1時間、BAPで処理する。次に、制限消化pRc/RSVをフェノール処理およびエタノール沈殿によって精製する。次に、制限消化pRc/RSVをDNA Blunting Kit(宝酒造)で処理してその末端を平滑化し、低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。制限消化されたpRc/RSVを低融点アガロースゲルから回収した後、その制限消化pRc/RSV 100ngおよび上記精製型リン酸化cDNAの全てをT4 DNAリガーゼによるライゲーション反応に使用する。そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ヒト正常VDRをコードするプラスミドが存在することを確認する。そのようなプラスミドを選択し、pRc/RSV-hVDR Kozakと名づける。
【0325】
6.27.実施例27 DR3レポーター遺伝子を含有するプラスミドの作製
配列番号209に記載のオリゴヌクレオチドと、それに相補的なヌクレオチド配列を持つオリゴヌクレオチドとを、DNAシンセサイザーで合成する。配列番号209に記載のオリゴヌクレオチドは、DR3の一方の鎖をコードするように合成する。もう一つのオリゴヌクレオチドは、第1オリゴヌクレオチドに相補的なヌクレオチド配列を持つように合成する。これら2つのオリゴヌクレオチドを互いにアニールさせて、DR3配列をコードするDNA(以下、DR3 DNAという)を作製する。次に、DR3 DNAをT4ポリヌクレオチドキナーゼと反応させて、DR3 DNAの末端をリン酸化する。次に、DR3 DNAをT4 DNAリガーゼで互いに結合させて、DR3配列が5回縦列反復したDR3×5 DNAを得る。次に、そのライゲーション反応混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、そのゲルからDR3×5 DNAを回収する。
【0326】
6.2項で得たプラスミドpGL3-TATAを制限酵素SmaIで制限消化した後、65℃で1時間、BAPで処理する。次に、その制限消化反応混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。その低融点アガロースゲルからホタルルシフェラーゼをコードするヌクレオチド配列を持つDNA断片を回収した後、回収したDNA断片100ngとDR3×5 DNA 1μgとをライゲーション反応に使用する。次に、得られたライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に単離された各プラスミドの部分試料を制限酵素KpnIおよびXhoIで制限消化する。その制限消化反応混合物をアガロースゲル電気泳動にかけて、DR3×5 DNAがpGL3-TATA中の制限酵素SmaIの制限部位に挿入されている構造を持つプラスミドが存在することを確認する。そのようなプラスミドを選択し、pGL3-TATA-DR3×5と名づける。
【0327】
次に、プラスミドpGL3-TATA-DR3×5を制限酵素SalIで制限消化する。制限消化されたpGL3-TATA-DR3×5の末端をBlunting Kit(宝酒造)で平滑化した後、その制限消化pGL3-TATA-DR3×5を65℃で1時間、BAPで処理する。また、6.2項で得たブラストサイジンSデアミナーゼ遺伝子発現カセットを含むDNA断片(BamHI-BamHI断片)の末端も、Blunting Kitを使って平滑化する。平滑末端化pGL3-TATA-DR3×5と、ブラストサイジンSデアミナーゼ遺伝子発現カセットを含む平滑末端化DNA断片とを、T4 DNAリガーゼによるライゲーション反応に使用する。次に、得られたライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次に、アンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。ブラストサイジンSデアミナーゼ遺伝子発現カセットを含むDNA断片がpGL3-TATA-DR3×5中の制限酵素SalIの制限部位に挿入されている単離プラスミドを選択し、pGL3-TATA-DR3×5-BSDと名づける。
【0328】
6.28.実施例28 DR3レポーター遺伝子を染色体の一つに安定に含有する安定形質転換カセット細胞の作製
DR3レポーター遺伝子を染色体の一つに安定に含有する安定形質転換細胞(以下、安定形質転換DR3カセット細胞という)を作製するために、プラスミドpGL3-TATA-DR3×5-BSDを線状化し、HeLa細胞に導入した。
【0329】
プラスミドpGL3-TATA-DR3×5-BSDを制限酵素NotIで制限消化することにより、pGL3-TATA-DR3×5-BSDを線状化する。
【0330】
直径約10cmの培養皿(Falcon)を使用し、10%FBSを含むDMEM培地(日水製薬)にて、5%CO2下に37℃で、約5×105個のHeLa細胞を宿主細胞として1日培養する。
【0331】
次に、リポフェクトアミン(Life Technologies)を用いるリポフェクション法によって線状化pGL3-TATA-DR3×5-BSDを培養HeLa細胞に導入する。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿の線状化pGL3-TATA-DR3×5-BSD、および21μl/培養皿のリポフェクトアミンを含める。
【0332】
リポフェクション処理の後、DMEM培地を、10%FBSを含むDMEM培地と交換し、形質転換したHeLa細胞を約36時間培養する。次に、形質転換HeLa細胞をトリプシン処理によって培養皿から除去、収集し、ブラストサイジンSを16μg/mlの濃度になるように添加した培地を含む容器に移す。ブラストサイジンS含有培地を3、4日毎に新しいバッチのブラストサイジンS含有DMEM培地に交換しながら、前記ブラストサイジンS含有培地で形質転換HeLa細胞を1ヶ月培養する。
【0333】
増殖して1〜数mmの直径を持つコロニーを形成することができるクローンを、前もって培地を分注しておいた96ウェルViewPlate(Berthold)のウェルに、丸ごと移す。それらのクローンをさらに培養する。クローンが増殖してウェルの底面の50%以上を覆うようになったら(移植の約5日後)、それらのクローンをトリプシン処理によって除去、収集する。次に、各クローンを2つの継代培養物に分割する。一方の継代培養物は96ウェルViewPlateに移して、それをマスタープレートとする。他方の継代培養物は96ウェルViewPlateに移して、これをアッセイプレートとする。マスタープレートとアッセイプレートは、クローンを培養することができるように培地を含有する。マスタープレートは同様の条件で引き続き培養する。
【0334】
次に、アッセイプレートのウェル中の培地をウェルから除去し、ウェル壁に付着したクローンをPBS(-)で2回洗浄する。5倍希釈した溶解緩衝液PGC50(東洋インキ)をアッセイプレートのウェル中のクローンに1ウェルあたり20μlずつ加える。そのアッセイプレートを室温で30分間静置した後、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットする。次に、基質溶液PGL100(東洋インキ)50μlをアッセイプレート中の各溶解クローンに自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで測定する。ルシフェラーゼ活性を示した複数のクローンをそこから選択する。
【0335】
次に、選択したクローンの試料を、直径約10cmの培養皿(Falcon)を用いて、活性炭デキストランFBS/E-MEM培地中、5%CO2下に、37℃で1〜2週間培養する。
【0336】
次に、リポフェクトアミン(Life Technologies)を用いるリポフェクション法により、プラスミドpRc/RSV-hVDR Kozakを、上で選択したクローンに導入して、2次クローンを得る。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿の上記pRc/RSV-VDR Kozak、および21μl/培養皿のリポフェクトアミンを含めた。次に、得られた2次クローンに、ヒト正常VDRの天然のコグネイトリガンドである1,25-(OH)ビタミンD3を含有するDMSO溶液を、培地中の1,25-(OH)ビタミンD3濃度が10nMになるように加える。それらの2次クローンを2日間培養した後、各2次クローンについて上記と同様にルシフェラーゼ活性を測定する。最も高いルシフェラーゼ活性の誘導を示す2次クローンを与えたマスタープレート中のクローンを、安定形質転換DR3カセット細胞として選択する。
【0337】
6.29.実施例29 正常PPARγをコードするプラスミドの作製
ヒト肝臓cDNAライブラリー(CLONETECH、QUICK clone cDNA#7113-1)を利用して、そこからヒト正常PPARγをコードするcDNA(Genbankアクセッション番号U79012)をPCR増幅する。このPCR増幅におけるPCR混合物は、ヒト肝臓cDNAライブラリー10ng、配列番号210に記載のオリゴヌクレオチド10pmol、配列番号211に記載のオリゴヌクレオチド10pmol、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。配列番号210および配列番号211に記載のオリゴヌクレオチドは、DNAシンセサイザー(モデル394、Applied Biosystems)を使って合成する。このPCR増幅では、PCRsystem 9700(Applied Biosystems)を使って、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを35回繰り返す。
【0338】
得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、ヒト正常PPARγをコードするcDNAがPCR増幅されることを臭化エチジウム染色で確認する。増幅されたcDNAを低融点アガロースゲルから回収した後、回収したcDNAの試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。調製したcDNA試料をABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定する。
【0339】
次に、上記cDNA中の開始コドン(ATG)のすぐ上流にKozakコンセンサス配列を加えるために、新たなPCR増幅を行う。このPCR増幅におけるPCR混合物は、正常PPARγおよびKozakコンセンサス配列をコードするcDNA 100ng、配列番号212に記載のオリゴヌクレオチド、配列番号211に記載のオリゴヌクレオチド、LA-Taqポリメラーゼ(宝酒造)、LA-Taqポリメラーゼと一緒に提供される緩衝液、およびdNTP(dATP、dTTP、dGTP、dCTP)を含む。このPCR増幅では、95℃で1分間のインキュベーションの後、68℃で3分間のインキュベーションを課すインキュベーションサイクルを25回繰り返す。得られたPCR混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。増幅されたcDNAを低融点アガロースゲルから回収した後、1μgの増幅cDNAをDNA Blunting Kit(宝酒造)で処理して、増幅cDNAの末端を平滑化する。次に、それによって得たcDNAをT4ポリヌクレオチドキナーゼと反応させて、当該cDNAの末端をリン酸化する。リン酸化されたcDNAをフェノール処理した後、リン酸化cDNAをエタノール沈殿して、精製型のリン酸化cDNAを得る。
【0340】
プラスミドpRc/RSV(Invitrogen)を制限酵素HindIIIで消化した後、65℃で1時間、BAPで処理する。次に、制限消化pRc/RSVをフェノール処理およびエタノール沈殿によって精製する。次に、制限消化pRc/RSVをDNA Blunting Kit(宝酒造)で処理してその末端を平滑化し、低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。制限消化されたpRc/RSVを低融点アガロースゲルから回収した後、その制限消化pRc/RSV 100ngおよび上記精製型リン酸化cDNAの全てをT4 DNAリガーゼによるライゲーション反応に使用する。そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ヒト正常PPARγをコードするプラスミドが存在することを確認する。そのようなプラスミドを選択し、pRc/RSV-hPPARγKozakと名づける。
【0341】
6.30.実施例30 DR1レポーター遺伝子を含有するプラスミドの作製
配列番号213に記載のオリゴヌクレオチドと、それに相補的なヌクレオチド配列を持つオリゴヌクレオチドとを、DNAシンセサイザーで合成する。配列番号213に記載のオリゴヌクレオチドは、DR1配列の一方の鎖をコードするように合成する。もう一つのオリゴヌクレオチドは、第1オリゴヌクレオチドに相補的なヌクレオチド配列を持つように合成する。これら2つのオリゴヌクレオチドを互いにアニールさせて、DR1配列をコードするDNA(以下、DR1 DNAという)を作製する。T4ポリヌクレオチドキナーゼをDR1 DNAと反応させて、DR1 DNAの末端をリン酸化する。次に、DR1 DNAをT4 DNAリガーゼで互いに結合させて、DR1配列が5回縦列反復したDR1×5 DNAを得る。次に、そのライゲーション反応混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかけて、そのゲルからDR1×5 DNAを回収する。
【0342】
6.2項で得たプラスミドpGL3-TATAを制限酵素SmaIで制限消化した後、65℃で1時間、BAPで処理する。次に、その制限消化反応混合物を低融点アガロースゲル電気泳動(アガロースL、ニッポンジーン)にかける。その低融点アガロースゲルからホタルルシフェラーゼをコードするヌクレオチド配列を持つDNA断片を回収した後、回収したDNA断片100ngとDR1×5 DNA 1μgとをT4 DNAリガーゼによるライゲーション反応に使用する。次に、得られたライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次にアンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に単離された各プラスミドの部分試料を制限酵素KpnIおよびXhoIで制限消化する。その制限消化反応混合物をアガロースゲル電気泳動にかけて、DR1×5 DNAがpGL3-TATA中の制限酵素SmaIの制限部位に挿入されているプラスミドが存在することを確認する。次に、そのプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、DR1配列の5回縦列反復を持つプラスミドが得られたことを確認する。そのようなプラスミドを選択し、pGL3-TATA-DR1×5と名づける。
【0343】
次に、プラスミドpGL3-TATA-DR1×5を制限酵素SalIで制限消化する。制限消化されたpGL3-TATA-DR1×5の末端をBlunting Kit(宝酒造)で平滑化した後、その制限消化pGL3-TATA-DR1×5を65℃で1時間、BAPで処理する。また、6.2項で得たブラストサイジンSデアミナーゼ遺伝子をコードするDNA断片(pUCSV-BSD(フナコシ)由来のBamHI-BamHI断片)の末端も、Blunting Kitを使って平滑化する。
【0344】
次に、ブラストサイジンSデアミナーゼ遺伝子発現カセットをコードするDNA断片と、制限消化したpGL3-TATA-DR1×5とを混合して、T4 DNAリガーゼによるライゲーション反応を行う。次に、そのライゲーション反応混合物を使って大腸菌コンピテントDH5α細胞(東洋紡)を形質転換する。形質転換した大腸菌細胞をLB-ampで培養する。次に、アンピシリン耐性を示すクローンを回収する。次にそれらクローンの一部を使って、上記ライゲーション反応によって生成したプラスミドをそこから単離する。次に、単離された各プラスミドの部分試料を、Dye Terminator Sequence Kit FS(Applied Biosystems)を使って調製する。単離されたプラスミドをABI自動シークエンサー(モデル377、Applied Biosystems)で配列決定して、ブラストサイジンSデアミナーゼ遺伝子発現カセットをコードするDNAがpGL3-TATA-DR1×5中の制限酵素SalIの制限部位に挿入されている構造をプラスミドが持つかどうかを確認する。そのプラスミドを選択し、pGL3-TATA-DR1×5-BSDと名づける。
【0345】
6.31.実施例31 DR1レポーター遺伝子を染色体の一つに安定に含有する安定形質転換カセット細胞の作製
DR1レポーター遺伝子を染色体の一つに安定に含有する安定形質転換カセット細胞(以下、安定形質転換DR1カセット細胞という)を作製するために、プラスミドpGL3-TATA-DR1×5-BSDを線状化し、HeLa細胞に導入した。
【0346】
プラスミドpGL3-TATA-DR1×5-BSDを線状化するために、pGL3-TATA-DR1×5-BSDを制限酵素NotIで制限消化する。
【0347】
直径約10cmの培養皿(Falcon)を使用し、10%FBSを含むDMEM培地(日水製薬)にて、5%CO2下に37℃で、約5×105個のHeLa細胞を宿主細胞として1日培養する。
【0348】
次に、リポフェクトアミン(Life Technologies)を用いるリポフェクション法によって線状化pGL3-TATA-DR1×5-BSDを培養HeLa細胞に導入する。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿の線状化pGL3-TATA-DR1×5-BSD、および21μl/培養皿のリポフェクトアミンを含める。
【0349】
リポフェクション処理の後、DMEM培地を、10%FBSを含むDMEM培地と交換し、形質転換したHeLa細胞を約36時間培養する。次に、形質転換HeLa細胞をトリプシン処理によって培養皿から除去、収集し、ブラストサイジンSを16μg/mlの濃度になるように添加した培地を含む容器に移す。ブラストサイジンS含有培地を3、4日毎に新しいバッチのブラストサイジンS含有培地に交換しながら、前記ブラストサイジンS含有培地で形質転換HeLa細胞を1ヶ月培養する。
【0350】
増殖して1〜数mmの直径を持つコロニーを形成することができるクローンを、前もって培地を分注しておいた96ウェルViewPlate(Berthold)のウェルに、それぞれ丸ごと移す。それらのクローンをさらに培養する。クローンが増殖してウェルの底面の50%以上を覆うようになったら(移植の約5日後)、それらのクローンをトリプシン処理によって除去、収集する。次に、各クローンを2つの継代培養物に分割する。一方の継代培養物は96ウェルViewPlateに移して、それをマスタープレートとする。他方の継代培養物は96ウェルViewPlateに移して、これをアッセイプレートとする。マスタープレートとアッセイプレートは、クローンを培養することができるように培地を含有する。マスタープレートは同様の条件で引き続き培養する。
【0351】
次に、アッセイプレートのウェル中の培地をウェルから除去し、ウェル壁に付着したクローンをPBS(-)で2回洗浄する。5倍希釈した溶解緩衝液PGC50(東洋インキ)をアッセイプレートのウェル中のクローンに1ウェルあたり20μlずつ加える。そのアッセイプレートを室温で30分間静置した後、自動基質注入器を装着したルミノメーターLB96P(Berthold)にセットする。次に、基質溶液PGL100(東洋インキ)50μlをアッセイプレート中の各溶解クローンに自動的に分注して、各ウェル中のルシフェラーゼ活性をルミノメーターLB96Pで測定する。ルシフェラーゼ活性を示した複数のクローンをそこから選択する。
【0352】
次に、選択したクローンの試料を、直径約10cmの培養皿(Falcon)を用いて、活性炭デキストランFBS/E-MEM培地中、5%CO2下に、37℃で1〜2週間培養する。
【0353】
次に、リポフェクトアミン(Life Technologies)を用いるリポフェクション法により、プラスミドpRc/RSV-hPPARγKozakを、上で選択したクローンに導入して、2次クローンを得る。リポフェクトアミンと一緒に提供されるマニュアルに従い、このリポフェクション法の条件には、5時間の処理、7μg/培養皿の上記pRc/RSV-hPPARγKozak、および21μl/培養皿のリポフェクトアミンを含めた。次に、得られた2次クローンに、ヒト正常PPARγの天然のコグネイトリガンドである15dプロスタグランジンJ2を含有するDMSO溶液を、培地中の15dプロスタグランジンJ2濃度が10nMになるように加える。それらの2次クローンを2日間培養した後、各2次クローンについて上記と同様にルシフェラーゼ活性を測定する。最も高いルシフェラーゼ活性の誘導を示す2次クローンを与えたマスタープレート中のクローンを、安定形質転換DR1カセット細胞として選択する。
【0354】
ちなみに、安定形質転換DR1カセット細胞は、PPAR、RAR、レチノインX受容体、HNF-4、TR-2、TR-4などを用いるレポーターアッセイに使用することができる。
[図面の簡単な説明]
図1〜32は、ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。図1、2、4、5、7、8、12、13、16、17および21〜26は、ヒト変異ERαまたはヒト変異ERαを刺激する可能性がある単独の薬剤として様々な濃度の4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154が存在する状態でのルシフェラーゼ活性を表す。図3、6、9〜11、14、15、18〜20、27〜32は、様々な濃度の4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154と共に様々な濃度の100pM E2が存在する状態でのルシフェラーゼ活性を表す。安定形質転換カセット細胞を利用して、ヒト変異ERα遺伝子またはヒト正常ERα遺伝子を一過性に発現させると共に、その染色体からレポーター遺伝子を発現させた。図1、2、4、5、7、8、12、13、16、17および21〜26では、ヒト変異ERαまたはヒト正常ERαを(4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154を含まない)DMSOの存在下に置いたコントロールがもたらすルシフェラーゼ活性を使って、ルシフェラーゼ活性をゼロにする。図3、6、9〜11、14、15、18〜20、27〜32では、100pMのE2を含むDMSO溶液の存在下にヒト変異ERαまたはヒト正常ERαを置いたコントロールがもたらすルシフェラーゼ活性を使って、ルシフェラーゼ活性をゼロにする。ヒト変異ERαおよびヒト正常ERαが与えるルシフェラーゼ活性をゼロにする際には、コントロールによるルシフェラーゼ活性を100%に設定した。図1、2、4、5、7、8、12、13、16、17および21〜26では、コントロールが与えるルシフェラーゼ活性をDMSOとして示している。図3、6、9〜11、14、15、18〜20、27〜32では、コントロールが与えるルシフェラーゼ活性をDMSO+E2として示している。
図33〜48は、ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子、ヒト変異ERα遺伝子およびヒト正常ERαは、細胞の染色体から発現させた。図33〜40は、ヒト変異ERαまたはヒト正常ERαを刺激する可能性がある単独の薬剤として様々な濃度の4-ヒドロキシタモキシフェン、ZM189154またはラロキシフェンが存在する状態でのルシフェラーゼ活性を表す。図41〜48は、様々な濃度の4-ヒドロキシタモキシフェン、ZM189154またはラロキシフェンと共に100pMのE2が存在する状態でのルシフェラーゼ活性を表す。安定形質転換バイナリー細胞を利用して、レポーター遺伝子をヒト変異ERα遺伝子と共に、またはヒト正常ERα遺伝子と共に、染色体から発現させた。図33〜40では、ヒト変異ERαまたはヒト正常ERαを(4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154を含まない)DMSOの存在下に置いたコントロールがもたらすルシフェラーゼ活性をゼロにする。図41〜48では、100pMのE2を含むDMSO溶液の存在下にヒト変異ERαまたはヒト正常ERαを置いたコントロールがもたらすルシフェラーゼ活性をゼロにする。ヒト変異ERαおよびヒト正常ERαが与えるルシフェラーゼ活性をゼロにする際には、コントロールによるルシフェラーゼ活性を100%に設定した。図33〜40では、コントロールが与えるルシフェラーゼ活性をDMSOとして示している。図41〜48では、コントロールが与えるルシフェラーゼ活性をDMSO+E2として示している。
図49〜52は、比較例として、レポーター遺伝子を細胞中で一過性に発現させた場合にヒト変異ERαK531Eまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。図49および50は、ヒト変異ERαを刺激する可能性がある単独の薬剤として様々な濃度の4-ヒドロキシタモキシフェンが存在する状態でのルシフェラーゼ活性を表す。図51および52は、様々な濃度の4-ヒドロキシタモキシフェンと共に100pMのE2が存在する状態でのルシフェラーゼ活性を表す。図49および50では、ヒト正常ERαまたはヒト変異ERαK531Eを(4-ヒドロキシタモキシフェンを含まない)DMSOの存在下に置いたコントロールのルシフェラーゼ活性をゼロにする。図51および52では、100pMのE2を含むDMSO溶液の存在下にヒト変異ERαK531Eを置いたコントロールのルシフェラーゼ活性をゼロにする。ヒト変異ERαおよびヒト正常ERαが与えるルシフェラーゼ活性をゼロにする際には、結果として得られたコントロールによるルシフェラーゼ活性を100%に設定する。図49および50では、コントロールが与えるルシフェラーゼ活性をDMSOとして示している。図51および52では、コントロールが与えるルシフェラーゼ活性をDMSO+E2として示している。
【0355】
[配列表フリーテキスト]
配列番号1
ヒト正常ERα
配列番号2
ヒト変異ERαK303R
配列番号3
ヒト変異ERαS309F
配列番号4
ヒト変異ERαG390D
配列番号5
ヒト変異ERαM396V
配列番号6
ヒト変異ERαG415V
配列番号7
ヒト変異ERαG494V
配列番号8
ヒト変異ERαK531E
配列番号9
ヒト変異ERαS578P
配列番号10
ヒト変異ERαG390D/S578P
配列番号11
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号12
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号13
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号14
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号15
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号16
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号17
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号18
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号19
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号20
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号21
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号22
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号23
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号24
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号25
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号26
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号27
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号28
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号29
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号30
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号31
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号32
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号33
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号34
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号35
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号36
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号37
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号38
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号39
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号40
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号41
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号42
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号43
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号44
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号45
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号46
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号47
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号48
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号49
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号50
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号51
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号52
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号53
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号54
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号55
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号56
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号57
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号58
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号59
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号60
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号61
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号62
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号63
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号64
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号65
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号66
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号67
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号68
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号69
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号70
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号71
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号72
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号73
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号74
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号75
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号76
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号77
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号78
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号79
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号80
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号81
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号82
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号83
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号84
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号85
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号86
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号87
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号88
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号89
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号90
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号91
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号92
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号93
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号94
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号95
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号96
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号97
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号98
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号99
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号100
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号101
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号102
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号103
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号104
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号105
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号106
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号107
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号108
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号109
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号110
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号111
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号112
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号113
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号114
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号115
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号116
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号117
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号118
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号119
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号120
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号121
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号122
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号123
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号124
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号125
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号126
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号127
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号128
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号129
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号130
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号131
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号132
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号133
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号134
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号135
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号136
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号137
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号138
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号139
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号140
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号141
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号142
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号143
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号144
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号145
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号146
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号147
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号148
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号149
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号150
サザンハイブリダイゼーションのために設計されたオリゴヌクレオチドプローブ
配列番号151
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号152
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号153
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号154
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号155
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号156
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号157
変異導入のために設計されたオリゴヌクレオチドプライマー
配列番号158
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号159
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号160
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号161
合成のために設計されたオリゴヌクレオチド
配列番号162
合成のために設計されたオリゴヌクレオチド
配列番号163
合成のために設計されたオリゴヌクレオチド
配列番号164
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号165
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号166
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号167
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号168
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号169
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号170
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号171
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号172
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号173
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号174
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号175
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号176
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号177
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号178
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号179
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号180
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号181
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号181
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号182
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号183
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号184
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号185
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号186
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号187
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号188
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号189
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号190
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号191
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号192
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号193
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号194
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号195
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号196
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号197
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号198
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号199
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号200
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号201
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号202
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号203
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号204
合成のために設計されたオリゴヌクレオチド
配列番号205
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号206
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号207
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号208
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号209
合成のために設計されたオリゴヌクレオチド
配列番号210
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号211
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号212
PCRのために設計されたオリゴヌクレオチドプライマー
配列番号213
合成のために設計されたオリゴヌクレオチド
【0356】
【配列表】
【図面の簡単な説明】
【図1】 ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図2】 ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図3】 ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図4】 ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図5】 ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図6】 ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図7】 ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図8】 ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図9】 ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図10】 ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図11】ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図12】ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図13】ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図14】ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図15】ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図16】ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図17】ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図18】ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図19】ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図20】ヒト変異ERαまたはヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図21】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。
【図22】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図23】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。
【図24】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図25】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。
【図26】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図27】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。
【図28】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図29】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。
【図30】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
【図31】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。
【図32】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子は細胞の染色体から発現させた。変異ERα遺伝子は細胞中で一過性に発現させた。
図1、2、4、5、7、8、12、13、16、17および21〜26は、ヒト変異ERαまたはヒト変異ERαを刺激する可能性がある単独の薬剤として様々な濃度の4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154が存在する状態でのルシフェラーゼ活性を表す。図3、6、9〜11、14、15、18〜20、27〜32は、様々な濃度の4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154と共に様々な濃度の100pM E2が存在する状態でのルシフェラーゼ活性を表す。安定形質転換カセット細胞を利用して、ヒト変異ERα遺伝子またはヒト正常ERα遺伝子を一過性に発現させると共に、その染色体からレポーター遺伝子を発現させた。図1、2、4、5、7、8、12、13、16、17および21〜26では、ヒト変異ERαまたはヒト正常ERαを(4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154を含まない)DMSOの存在下に置いたコントロールがもたらすルシフェラーゼ活性を使って、ルシフェラーゼ活性をゼロにする。図3、6、9〜11、14、15、18〜20、27〜32では、100pMのE2を含むDMSO溶液の存在下にヒト変異ERαまたはヒト正常ERαを置いたコントロールがもたらすルシフェラーゼ活性を使って、ルシフェラーゼ活性をゼロにする。ヒト変異ERαおよびヒト正常ERαが与えるルシフェラーゼ活性をゼロにする際には、コントロールによるルシフェラーゼ活性を100%に設定した。図1、2、4、5、7、8、12、13、16、17および21〜26では、コントロールが与えるルシフェラーゼ活性をDMSOとして示している。図3、6、9〜11、14、15、18〜20、27〜32では、コントロールが与えるルシフェラーゼ活性をDMSO+E2として示している。
【図33】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト正常ERαは、細胞の染色体から発現させた。
【図34】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト変異ERα遺伝子は、細胞の染色体から発現させた。
【図35】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト変異ERα遺伝子は、細胞の染色体から発現させた。
【図36】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト変異ERα遺伝子は、細胞の染色体から発現させた。
【図37】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト正常ERαは、細胞の染色体から発現させた。
【図38】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト変異ERα遺伝子は、細胞の染色体から発現させた。
【図39】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト正常ERαは、細胞の染色体から発現させた。
【図40】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト変異ERα遺伝子は、細胞の染色体から発現させた。
【図41】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト正常ERαは、細胞の染色体から発現させた。
【図42】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト変異ERα遺伝子は、細胞の染色体から発現させた。
【図43】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト変異ERα遺伝子は、細胞の染色体から発現させた。
【図44】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト変異ERα遺伝子は、細胞の染色体から発現させた。
【図45】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト正常ERαは、細胞の染色体から発現させた。
【図46】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト変異ERα遺伝子は、細胞の染色体から発現させた。
【図47】ヒト正常ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト正常ERαは、細胞の染色体から発現させた。
【図48】ヒト変異ERαが与えるルシフェラーゼ活性を表す。レポーター遺伝子およびヒト変異ERα遺伝子は、細胞の染色体から発現させた。
図33〜40は、ヒト変異ERαまたはヒト正常ERαを刺激する可能性がある単独の薬剤として様々な濃度の4-ヒドロキシタモキシフェン、ZM189154またはラロキシフェンが存在する状態でのルシフェラーゼ活性を表す。図41〜48は、様々な濃度の4-ヒドロキシタモキシフェン、ZM189154またはラロキシフェンと共に100pMのE2が存在する状態でのルシフェラーゼ活性を表す。安定形質転換バイナリー細胞を利用して、レポーター遺伝子をヒト変異ERα遺伝子と共に、またはヒト正常ERα遺伝子と共に、染色体から発現させた。図33〜40では、ヒト変異ERαまたはヒト正常ERαを(4-ヒドロキシタモキシフェン、ラロキシフェンまたはZM189154を含まない)DMSOの存在下に置いたコントロールがもたらすルシフェラーゼ活性をゼロにする。図41〜48では、100pMのE2を含むDMSO溶液の存在下にヒト変異ERαまたはヒト正常ERαを置いたコントロールがもたらすルシフェラーゼ活性をゼロにする。ヒト変異ERαおよびヒト正常ERαが与えるルシフェラーゼ活性をゼロにする際には、コントロールによるルシフェラーゼ活性を100%に設定した。図33〜40では、コントロールが与えるルシフェラーゼ活性をDMSOとして示している。図41〜48では、コントロールが与えるルシフェラーゼ活性をDMSO+E2として示している。
【図49】比較例として、レポーター遺伝子を細胞中で一過性に発現させた場合にヒト正常ERαが与えるルシフェラーゼ活性を表す。
【図50】比較例として、レポーター遺伝子を細胞中で一過性に発現させた場合にヒト変異ERαK531Eが与えるルシフェラーゼ活性を表す。
【図51】比較例として、レポーター遺伝子を細胞中で一過性に発現させた場合にヒト正常ERαが与えるルシフェラーゼ活性を表す。
【図52】比較例として、レポーター遺伝子を細胞中で一過性に発現させた場合にヒト変異ERαK531Eが与えるルシフェラーゼ活性を表す。
図49および50は、ヒト変異ERαを刺激する可能性がある単独の薬剤として様々な濃度の4-ヒドロキシタモキシフェンが存在する状態でのルシフェラーゼ活性を表す。図51および52は、様々な濃度の4-ヒドロキシタモキシフェンと共に100pMのE2が存在する状態でのルシフェラーゼ活性を表す。図49および50では、ヒト正常ERαまたはヒト変異ERαK531Eを(4-ヒドロキシタモキシフェンを含まない)DMSOの存在下に置いたコントロールのルシフェラーゼ活性をゼロにする。図51および52では、100pMのE2を含むDMSO溶液の存在下にヒト変異ERαK531Eを置いたコントロールのルシフェラーゼ活性をゼロにする。ヒト変異ERαおよびヒト正常ERαが与えるルシフェラーゼ活性をゼロにする際には、結果として得られたコントロールによるルシフェラーゼ活性を100%に設定する。図49および50では、コントロールが与えるルシフェラーゼ活性をDMSOとして示している。図51および52では、コントロールが与えるルシフェラーゼ活性をDMSO+E2として示している。
Claims (17)
- (i)エストロゲン応答配列(以下、EREという)、TATA配列および前記EREにとって本来外来であるレポーター配列を含むレポーター遺伝子を含む染色体、ならびに
(ii)以下の(ii-a)および(ii-b)から選択される1または複数の因子、
(ii-a)前記レポーター遺伝子を転写活性化する活性を持つ変異エストロゲンレセプターαタンパク質(以下、ERαという)(ここに、部分抗エストロゲンおよびエストラジオール(以下、E2という)の存在下における前記活性は部分抗エストロゲンおよびE2の存在下における正常ERαの活性よりも高いか、または部分抗エストロゲンの存在下における前記活性は部分抗エストロゲンの存在下における正常ERαの活性よりも高いものとする)、および
(ii-b)前記変異ERαをコードする遺伝子、
を含む人工細胞であり、前記変異ERαは以下の(a)〜(e)のいずれかであることを特徴とする人工細胞。
(a)配列番号2で示されるアミノ酸配列からなる変異ERα。
(b)配列番号3で示されるアミノ酸配列からなる変異ERα。
(c)配列番号4で示されるアミノ酸配列からなる変異ERα。
(d)配列番号9で示されるアミノ酸配列からなる変異ERα。
(e)配列番号10で示されるアミノ酸配列からなる変異ERα。
ここで、部分抗エストロゲンとは、正常ERαのエストロゲン受容体活性化機能1(Estrogen Receptor Activation Function1、略称:AF1)領域には拮抗性を示さないが、正常ERαのエストロゲン受容体活性化機能2(Estrogen Receptor Activation Function2、略称:AF2)領域に拮抗性を示すものを意味する。 - 前記部分抗エストロゲンが、タモキシフェン、ラロキシフェン、および4−ヒドロキシタモキシフェンからなる群より選択される一つである請求項1記載の人工細胞。
- (ii-b)の遺伝子が(i)の染色体に含まれる請求項1乃至2記載のいずれかの人工細胞。
- (ii-b)の遺伝子がベクターに含まれる請求項1乃至2記載のいずれかの人工細胞。
- 配列番号2で示されるアミノ酸配列からなる変異エストロゲンレセプターαタンパク質(以下、ERαという)。
- 配列番号3で示されるアミノ酸配列からなる変異エストロゲンレセプターαタンパク質(以下、ERαという)。
- 配列番号4で示されるアミノ酸配列からなる変異エストロゲンレセプターαタンパク質(以下、ERαという)。
- 配列番号9で示されるアミノ酸配列からなる変異エストロゲンレセプターαタンパク質(以下、ERαという)。
- 配列番号10で示されるアミノ酸配列からなる変異エストロゲンレセプターαタンパク質(以下、ERαという)。
- 請求項5記載の変異ERαのアミノ酸配列をコードする塩基配列からなるポリヌクレオチド。
- 請求項6記載の変異ERαのアミノ酸配列をコードする塩基配列からなるポリヌクレオチド。
- 請求項7記載の変異ERαのアミノ酸配列をコードする塩基配列からなるポリヌクレオチド。
- 請求項8記載の変異ERαのアミノ酸配列をコードする塩基配列からなるポリヌクレオチド。
- 請求項9記載の変異ERαのアミノ酸配列をコードする塩基配列からなるポリヌクレオチド。
- 請求項10乃至14記載のいずれかのポリヌクレオチドを含むベクター。
- 請求項15記載のベクターを含むウイルス。
- 変異エストロゲンレセプターαタンパク質(以下、ERαという)に由来する乳癌の処置に有用な化合物をスクリーニングする方法であって、
請求項1乃至4記載のいずれかの人工細胞を試験化合物にばく露すること、
前記人工細胞のレポーター遺伝子の転写活性化量を測定すること、
を含む方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2001543602A JP4742483B2 (ja) | 1999-12-07 | 2000-12-01 | 変異ERαおよび転写活性化の試験系 |
Applications Claiming Priority (26)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP11-348022 | 1999-12-07 | ||
| JP34802299 | 1999-12-07 | ||
| JP1999348022 | 1999-12-07 | ||
| JP1999370667 | 1999-12-27 | ||
| JP37066799 | 1999-12-27 | ||
| JP11-370667 | 1999-12-27 | ||
| JP2000207011 | 2000-07-07 | ||
| JP2000-207011 | 2000-07-07 | ||
| JP2000207011 | 2000-07-07 | ||
| JP2000-220508 | 2000-07-21 | ||
| JP2000220508 | 2000-07-21 | ||
| JP2000220508 | 2000-07-21 | ||
| JP2000234053 | 2000-08-02 | ||
| JP2000234053 | 2000-08-02 | ||
| JP2000-234053 | 2000-08-02 | ||
| JP2000235463 | 2000-08-03 | ||
| JP2000235460 | 2000-08-03 | ||
| JP2000235463 | 2000-08-03 | ||
| JP2000235460 | 2000-08-03 | ||
| JP2000-235463 | 2000-08-03 | ||
| JP2000235461 | 2000-08-03 | ||
| JP2000235461 | 2000-08-03 | ||
| JP2000-235461 | 2000-08-03 | ||
| JP2000-235460 | 2000-08-03 | ||
| PCT/JP2000/008553 WO2001042307A1 (en) | 1999-12-07 | 2000-12-01 | MUTANT ERα AND TEST SYSTEMS FOR TRANSACTIVATION |
| JP2001543602A JP4742483B2 (ja) | 1999-12-07 | 2000-12-01 | 変異ERαおよび転写活性化の試験系 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2003516137A JP2003516137A (ja) | 2003-05-13 |
| JP2003516137A5 JP2003516137A5 (ja) | 2008-02-14 |
| JP4742483B2 true JP4742483B2 (ja) | 2011-08-10 |
Family
ID=27573667
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2001543602A Expired - Fee Related JP4742483B2 (ja) | 1999-12-07 | 2000-12-01 | 変異ERαおよび転写活性化の試験系 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US7582476B2 (ja) |
| EP (1) | EP1237925B1 (ja) |
| JP (1) | JP4742483B2 (ja) |
| AT (1) | ATE366745T1 (ja) |
| AU (1) | AU782142B2 (ja) |
| CA (1) | CA2393045A1 (ja) |
| DE (1) | DE60035522T2 (ja) |
| WO (1) | WO2001042307A1 (ja) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002057283A1 (en) * | 2001-01-19 | 2002-07-25 | Baylor College Of Medecine | Methods and compositions in breast cancer diagnosis and therapeutics |
| EP1533368A4 (en) * | 2002-07-02 | 2006-06-21 | Astellas Pharma Inc | SCREENING METHOD FOR A DRUG TO IMPROVE INSULIN RESISTANCE |
| CA3081071A1 (en) * | 2002-10-16 | 2004-04-29 | Gen-Probe Incorporated | Compositions and methods for detecting west nile virus |
| US7927840B2 (en) | 2006-09-11 | 2011-04-19 | Gen Probe Incorporated | Method for detecting West Nile Virus nucleic acids in the 3′ non-coding region |
| AU2003280793A1 (en) * | 2002-11-15 | 2004-06-15 | Sumitomo Chemical Company, Limited | MUTANT ESTROGEN RECEPTOR-Alpha AND GENE THEREOF |
| JP2007535906A (ja) * | 2003-10-24 | 2007-12-13 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | 直交遺伝子スイッチ |
| FR2863275B1 (fr) * | 2003-12-09 | 2007-08-10 | Biomerieux Sa | Procede pour le diagnostic/pronostic du cancer du sein |
| EP1711606A2 (en) | 2004-01-20 | 2006-10-18 | Isis Pharmaceuticals, Inc. | Modulation of glucocorticoid receptor expression |
| JP2006153656A (ja) * | 2004-11-29 | 2006-06-15 | Olympus Corp | 反応カプセル、反応カプセルを使用した検査方法およびシステム |
| WO2013063313A1 (en) | 2011-10-25 | 2013-05-02 | Isis Pharmaceuticals, Inc. | Antisense modulation of gccr expression |
| WO2014205138A1 (en) | 2013-06-19 | 2014-12-24 | Seragon Pharmaceuticals, Inc. | Estrogen receptor modulator and uses thereof |
| BR112015030595A2 (pt) | 2013-06-19 | 2017-07-25 | Seragon Pharmaceuticals Inc | moduladores de receptor de estrogênio de azetidina e usos dos mesmos |
| SG11201607339VA (en) | 2014-03-13 | 2016-10-28 | Hoffmann La Roche | Methods and compositions for modulating estrogen receptor mutants |
| JP6768711B2 (ja) | 2015-05-26 | 2020-10-14 | エフ・ホフマン−ラ・ロシュ・アクチェンゲゼルシャフト | 複素環式エストロゲン受容体モジュレーター及びその使用 |
| US10179676B1 (en) | 2017-07-26 | 2019-01-15 | William Hagan Taylor | Dispenser with self-closing cap |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1987005049A1 (en) * | 1986-02-20 | 1987-08-27 | California Biotechnology Inc. | Eucaryotic expression of steroid receptor proteins |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2634495B1 (fr) * | 1988-07-01 | 1990-10-26 | Chambon Pierre | Procede de preparation d'une proteine par des levures utilisant un systeme inductible, vecteurs et souches transformees correspondantes |
| AU8951191A (en) * | 1990-10-29 | 1992-05-26 | Dekalb Plant Genetics | Isolation of biological materials using magnetic particles |
-
2000
- 2000-12-01 WO PCT/JP2000/008553 patent/WO2001042307A1/en active IP Right Grant
- 2000-12-01 AU AU18868/01A patent/AU782142B2/en not_active Ceased
- 2000-12-01 DE DE60035522T patent/DE60035522T2/de not_active Expired - Lifetime
- 2000-12-01 US US10/148,835 patent/US7582476B2/en not_active Expired - Fee Related
- 2000-12-01 AT AT00981647T patent/ATE366745T1/de not_active IP Right Cessation
- 2000-12-01 EP EP00981647A patent/EP1237925B1/en not_active Expired - Lifetime
- 2000-12-01 JP JP2001543602A patent/JP4742483B2/ja not_active Expired - Fee Related
- 2000-12-01 CA CA002393045A patent/CA2393045A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1987005049A1 (en) * | 1986-02-20 | 1987-08-27 | California Biotechnology Inc. | Eucaryotic expression of steroid receptor proteins |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2003516137A (ja) | 2003-05-13 |
| ATE366745T1 (de) | 2007-08-15 |
| EP1237925A1 (en) | 2002-09-11 |
| US7582476B2 (en) | 2009-09-01 |
| EP1237925B1 (en) | 2007-07-11 |
| WO2001042307A1 (en) | 2001-06-14 |
| AU1886801A (en) | 2001-06-18 |
| DE60035522T2 (de) | 2008-03-20 |
| US20030207380A1 (en) | 2003-11-06 |
| AU782142B2 (en) | 2005-07-07 |
| DE60035522D1 (de) | 2007-08-23 |
| CA2393045A1 (en) | 2001-06-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4742483B2 (ja) | 変異ERαおよび転写活性化の試験系 | |
| AU616389B2 (en) | Hormone receptor compositions and methods | |
| US6713270B1 (en) | Method for identifying ligand of estrogen receptor beta | |
| US20050079576A1 (en) | Androgen receptor proteins, recombinant DNA molecules coding for such, and use of such compositions | |
| WO1995013373A1 (en) | Ubiquitous nuclear receptor: compositions and methods | |
| US5972643A (en) | Isolated polynucleotide molecules encoding CTCF, a CCCTC-binding factor | |
| AU8724498A (en) | 1-alpha-hydroxylase materials and methods | |
| JP4400183B2 (ja) | 変異型エストロゲンレセプターαおよびその遺伝子 | |
| JP4400182B2 (ja) | 変異型エストロゲンレセプターαおよびその遺伝子 | |
| JP4400181B2 (ja) | 変異型エストロゲンレセプターαおよびその遺伝子 | |
| JP2004254600A (ja) | 変異型アンドロゲンレセプターおよびその活性評価用細胞 | |
| JP2004008141A (ja) | 変異型アンドロゲンレセプターおよびその活性評価用細胞 | |
| WO2004046352A1 (ja) | 変異型エストロゲンレセプターαおよびその遺伝子 | |
| JP2004254601A (ja) | 変異型アンドロゲンレセプターおよびその活性評価用細胞 | |
| JP2004008140A (ja) | 変異型アンドロゲンレセプターおよびその活性評価用細胞 | |
| JP2004254602A (ja) | 変異型アンドロゲンレセプターおよびその活性評価用細胞 | |
| CTCF et al. | Lobanenkov et al. | |
| JP2004008142A (ja) | 変異型アンドロゲンレセプターおよびその活性評価用細胞 | |
| WO1997047172A1 (fr) | Proteine isoforme du recepteur de la vitamine d |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20071116 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20071116 |
|
| RD05 | Notification of revocation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7425 Effective date: 20080128 |
|
| RD05 | Notification of revocation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7425 Effective date: 20080512 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100907 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20101028 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20110412 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20110425 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140520 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140520 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |