JP4303111B2 - 硫黄回収プロセスにおける硫黄成分の回収方法 - Google Patents
硫黄回収プロセスにおける硫黄成分の回収方法 Download PDFInfo
- Publication number
- JP4303111B2 JP4303111B2 JP2003540079A JP2003540079A JP4303111B2 JP 4303111 B2 JP4303111 B2 JP 4303111B2 JP 2003540079 A JP2003540079 A JP 2003540079A JP 2003540079 A JP2003540079 A JP 2003540079A JP 4303111 B2 JP4303111 B2 JP 4303111B2
- Authority
- JP
- Japan
- Prior art keywords
- gas
- content
- sulfur
- cooling
- water
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images
Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B17/00—Sulfur; Compounds thereof
- C01B17/02—Preparation of sulfur; Purification
- C01B17/04—Preparation of sulfur; Purification from gaseous sulfur compounds including gaseous sulfides
- C01B17/05—Preparation of sulfur; Purification from gaseous sulfur compounds including gaseous sulfides by wet processes
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/14—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols by absorption
- B01D53/1456—Removing acid components
- B01D53/1468—Removing hydrogen sulfide
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D53/00—Separation of gases or vapours; Recovering vapours of volatile solvents from gases; Chemical or biological purification of waste gases, e.g. engine exhaust gases, smoke, fumes, flue gases, aerosols
- B01D53/34—Chemical or biological purification of waste gases
- B01D53/46—Removing components of defined structure
- B01D53/48—Sulfur compounds
- B01D53/52—Hydrogen sulfide
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Analytical Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Environmental & Geological Engineering (AREA)
- Health & Medical Sciences (AREA)
- Inorganic Chemistry (AREA)
- Biomedical Technology (AREA)
- Treating Waste Gases (AREA)
- Gas Separation By Absorption (AREA)
- Processing Of Solid Wastes (AREA)
Description
(a)スイートニングされた(sweetened)生成ガスを得るための、H2S吸収剤を使用することによるサワーガス(sour gas)からのH2Sの除去
(b)H2Sを得るための、H2S含有量の多い吸収剤からのH2Sの放出(stripping)
(c)SO2およびH2Sを得るための、H2Sの燃焼
(d)硫黄を生成して回収し、そしてH2SとSO2の含有量が低減された排ガスを形成するための、高温での固体触媒によるH2SのSO2との反応
(e)H2SとSO2の残留量の主要成分を硫黄として回収し、さらに大気に排出しても良い排煙を生成するための、工程(d)からの排ガスの処理
2H2S(気)+SO2(気)→3S(液)+2H2O(気) (1)
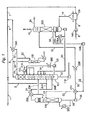
(a)H2S含有量の多いガス中のH2SをSO2と反応させて、硫黄およびH2SとH2Oを含む反応器排ガスを生成する工程、
(b)反応器排ガスを燃焼して、SO2と水蒸気を含む燃焼ガスを生成する工程、
(c)工程(b)からの燃焼ガスを冷却して、水蒸気と硫黄を凝縮し、主として水から成る水性流れを生成する工程、および
(d)工程(c)からの水性流れを反応器に流入させて、工程(a)の反応のための冷却を提供する工程、
を含むプロセスが提供される。
(a)燃焼ガスを冷却して、水蒸気と硫黄を凝縮し、さらに主として水から成る水性流れを生成する工程、および
(b)反応器中に前記水性流れを投入して、H2S含有量の多いガスとSO2間の反応のための冷却を提供する工程、
を含むプロセスが含まれる。
(a)実質上全ての水素がH2Oに変換されて、硫黄の少なくとも90%、好ましくは約98%〜99%がSO2に変換されるが、硫黄の少なくとも0.1%、好ましくは約1%〜2%が硫黄蒸気になるように、炉内で冷却されたH2S含有量の多い排ガスをある量のO2含有量の多いガスで燃焼する工程;
(b)別の接触装置中の冷却された水とまたはSO2吸収装置の下方部中に投入された水と直接接触させて工程(a)からのSO2含有量の多いガスを冷却して、H2Oおよび硫黄の蒸気を凝縮し、懸濁された硫黄を含む水性スラリーを生成し、冷却されたSO2含有量の多いガスを得る工程、
(c)SO2含有量の多いガスをSO2吸収剤と接触させることによってSO2吸収装置内で冷却されたSO2含有量の多いガスからSO2を吸収して、SO2含有量の多い吸収剤および硫黄化合物含有量の低い排煙を得て;さらにSO2含有量の多い吸収剤からSO2を放出させてSO2含有量の多いガスを得る工程;および
(d)反応塔の一箇所以上の位置で注入された水性流れの一つとして、工程(b)からの水性スラリーを使用して、蒸発によって反応熱の一部を吸収する工程、
を含むプロセスが含まれる。
(a)H2S含有量の多いガスからのH2Sを反応塔内でSO2と反応させて、硫黄およびH2Sを含む反応器排ガスを生成する工程、
(b)反応器排ガスを燃焼し、そして好ましくは迂回路を通過するいかなるH2S含有量の多いガスをも燃焼して、水蒸気と他のガスを含むSO2含有量の多いガスを得る工程、
(c)工程(b)からのSO2含有量の多いガスと水蒸気を冷却して、水の流れを得る工程、および
(d)前記水の流れを反応塔に注入して、反応塔内で必要とされる冷却の一部を提供する工程、
を含むプロセスが提供される。
(e)冷却された燃焼ガスからSO2を除去して、硫黄化合物の含有量が低い排煙を得る工程、
が含まれる。
2H2S(気)+SO2(気)→3S(液)+2H2O(気) (1)
Claims (25)
- H2S含有量の多いガスからH2Sを除去して硫黄を生成するためのプロセスであって、
(a)該H2S含有量の多いガスからのH2Sを反応器内でSO2と反応させて、硫黄およびH2SとH2Oを含む反応器排ガスを生成する工程、
(b)該反応器排ガスを燃焼して、SO2、水蒸気および硫黄蒸気を含む燃焼ガスを生成する工程、
(c)工程(b)からの該燃焼ガスを冷却して、水蒸気および硫黄蒸気を凝縮し、硫黄を含む水性流れを生成する工程、および
(d)工程(c)からの該水性流れを該反応器に流入させて、工程(a)の該反応のための冷却を提供する工程、
を含むプロセス。 - 工程(c)からの該水性流れが主として水を含む、請求項1に記載のプロセス。
- 該燃焼ガスの該冷却が直接的な水冷却で遂行される、請求項1に記載のプロセス。
- 該燃焼ガスの該冷却が冷却媒体との間接的な熱交換によって遂行される、請求項1に記載のプロセス。
- (e)該冷却された燃焼ガスからSO2を除去して、硫黄化合物の含有量が低い排煙を得る工程、
を更に含む、請求項1に記載のプロセス。 - 工程(e)がSO2吸収装置内で実施され、そこでは溶剤中への吸収によって該冷却された燃焼ガスからSO2が除去され、SO2含有量の多い溶剤を得る、請求項5に記載のプロセス。
- 工程(a)で使用されるSO2が、少なくとも部分的に、該SO2含有量の多い溶剤からのSO2のストリッピングによって得られる、請求項6に記載のプロセス。
- 工程(c)の該冷却が、該SO2吸収装置の下方部において実施される、請求項7に記載のプロセス。
- 工程(c)の該冷却が直接的な水冷却で遂行される、請求項8に記載のプロセス。
- 工程(a)が溶剤の存在下で行われ、該反応器を出る前の該反応器排ガスが第2の水性流れと接触して溶剤蒸気を回収し、該第2の水性流れ中に含まれる該水の存在下で未反応のSO2が該反応器排ガス中のH2Sと反応して、主として水から成り懸濁化された硫黄を含む第3の水性流れを生成し、さらに該第3の水性流れが該反応器に投入されて、工程(a)の該反応のための冷却を提供する、請求項1に記載のプロセス。
- 工程(c)からの該水性流れ中の該硫黄量が0.1〜10重量%である、請求項1に記載のプロセス。
- 該SO2吸収装置の上方部に投入されるSO2含有量の希薄な溶剤を使用することによってSO2が該燃焼ガスから除去され、SO2含有量の多い溶剤が該SO2吸収装置の中間部から除去される、請求項6に記載のプロセス。
- H2S含有量の多いガスからH2Sを除去するためのプロセスであって、そこでは該H2S含有量の多いガスが有機液体の存在下の反応器内でSO2と反応して硫黄を生成し、さらにH2Sが燃焼されて、SO2、水蒸気および気体状硫黄を含む燃焼ガスを生成し、さらにSO2が該H2S含有量の多いガスとさらに反応するものであり、
(a)該燃焼ガスを冷却して、水蒸気と硫黄蒸気を凝縮し、さらに主として水から成り懸濁化された硫黄を含む水性流れを生成する工程、および
(b)該反応器中に前記水性流れを投入して、該H2S含有量の多いガスと該SO2間の該反応のための冷却を提供する工程、
を含むプロセス。 - 該燃焼ガスの該冷却が、直接的な水冷却によって遂行される、請求項13に記載のプロセス。
- 該燃焼ガスの該冷却が、冷却媒体との間接的な熱交換によって遂行される、請求項13に記載のプロセス。
- (c)該冷却された燃焼ガスからSO2を除去して、100ppm以下のSO2を含有する排煙を得る工程、
をさらに含む、請求項13に記載のプロセス。 - 工程(c)が、上方の部分および下方の部分を有するSO2吸収装置において実施され、そこでは溶剤中への吸収によって該冷却された燃焼ガスからSO2が除去されて、SO2含有量の多い溶剤を得る、請求項16に記載のプロセス。
- 該H2S含有量の多いガスと反応されるSO2が、少なくとも部分的に、該SO2含有量の多い溶剤からのSO2のストリッピングによって得られる、請求項17に記載のプロセス。
- 該燃焼ガスの該冷却が、該SO2吸収装置の該下方の部分において実施される、請求項17に記載のプロセス。
- 該燃焼ガスの該冷却が、直接的な水冷却によって遂行される、請求項19に記載のプロセス。
- 該H2S含有量の多いガスと該SO2の間の該反応が溶剤の存在下で行われて反応器排ガスを生成し、該反応器を出る前の該反応器排ガスが第2の水性流れと接触して溶剤蒸気を回収し、該第2の水性流れ中に含まれる該水の存在下で未反応のSO2が該反応器排ガス中のH2Sと反応して、主として水から成り懸濁化された硫黄を含む第3の水性流れを生成し、さらに該第3の水性流れが該反応器に投入されて、該H2S含有量の多いガスの該SO2との該反応のための冷却を提供する、請求項13に記載のプロセス。
- 該水性流れ中の該硫黄量が0.1〜10重量%である、請求項13に記載のプロセス。
- 該SO2吸収装置の上方の部分に投入されるSO2含有量の希薄な溶剤を使用することによってSO2が該燃焼ガスから除去され、SO2含有量の多い溶剤が該SO2吸収装置の中間部から除去される、請求項17に記載のプロセス。
- H2S含有量の多いガスからH2Sを除去して硫黄を生成するためのプロセスであって、該H2S含有量の多いガスとSO2含有量の多いガスを供給して、それらの反応に触媒として機能する溶剤の存在下の反応塔中で該H2Sが化学量論的に過剰であって、液状の硫黄と水蒸気を形成する工程;但しそこでは、第1の水性流れが該反応塔の一箇所以上の位置で注入されて、水の蒸発で反応熱の一部を吸収し;さらにそこでは該H2S含有量の多い排ガスが該反応塔の上方の部分において第2の水性流れでスクラッビングされて、溶剤蒸気と未反応のSO2を回収し、次いで冷却されて水を凝縮する;該H2S含有量の多い排ガスを燃焼して、該反応塔に供給されるSO2を生成する工程;吸収装置内で該燃焼ガスをSO2吸収剤と接触させることによって該燃焼ガスからSO2を吸収して、SO2含有量の多い吸収剤を得る工程、および該SO2含有量の多い吸収剤からSO2を放出させてSO2含有量の多いガスを得る工程を含み、さらに、
(a)実質上全ての水素がH2Oに変換され、そして該硫黄の少なくとも90%がSO2に変換されるが、硫黄の少なくとも0.1%が硫黄蒸気になるように、炉内で該冷却されたH2S含有量の多い排ガスをある量のO2含有量の多いガスで燃焼する工程、
(b)冷却された水と直接接触させて工程(a)からの該SO2含有量の多いガスを冷却して、固体硫黄を含む水性スラリーを生成する工程、
(c)該SO2含有量の多いガスをSO2吸収剤と接触させることによってSO2吸収装置内で該冷却された該ガスからSO2を吸収して、SO2含有量の多い吸収剤を得る工程、
(d)該SO2含有量の多い吸収剤からSO2を放出させて、SO2含有量の多いガスを得る工程、および
(e)該反応塔の一箇所以上の位置で注入された該第1の水性流れの一部として、工程(b)からの水中に懸濁した固体硫黄の該スラリーを使用して、蒸発によって該反応熱の一部を吸収する工程、
を含むプロセス。 - H2S含有量の多いガスからH2Sを除去して硫黄を生成するためのプロセスであって、
(a)該H2S含有量の多いガスからのH2Sを有機液体の存在下の反応器内でSO2と反応させて、硫黄およびH2SとH2Oを含む反応器排ガスを生成する工程、
(b)該反応器排ガスを燃焼して、SO2および水蒸気を含む燃焼ガスを生成する工程、
(c)工程(b)からの該燃焼ガスを冷却して、水蒸気を凝縮し、水性流れを生成する工程、
(d)該冷却された燃焼ガスからSO2を回収する工程、
(e)工程(d)からのSO2を工程(a)のSO2ガスとして該反応器に流入する工程、および
(f)工程(c)からの該水性流れを該反応器に流入させて、工程(a)の該反応のための冷却を提供する工程、
を含むプロセス。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/999,825 US6645459B2 (en) | 2001-10-30 | 2001-10-30 | Method of recovering sulfurous components in a sulfur-recovery process |
| PCT/US2002/034363 WO2003037789A1 (en) | 2001-10-30 | 2002-10-25 | Method of recovering sulfurous components in a sulfur-recovery process |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2005507844A JP2005507844A (ja) | 2005-03-24 |
| JP2005507844A5 JP2005507844A5 (ja) | 2006-01-05 |
| JP4303111B2 true JP4303111B2 (ja) | 2009-07-29 |
Family
ID=25546703
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003540079A Expired - Fee Related JP4303111B2 (ja) | 2001-10-30 | 2002-10-25 | 硫黄回収プロセスにおける硫黄成分の回収方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US6645459B2 (ja) |
| EP (1) | EP1456123B1 (ja) |
| JP (1) | JP4303111B2 (ja) |
| AT (1) | ATE430114T1 (ja) |
| BR (1) | BR0213753B1 (ja) |
| CA (1) | CA2464726C (ja) |
| DE (1) | DE60232180D1 (ja) |
| ES (1) | ES2322892T3 (ja) |
| MX (1) | MXPA04004100A (ja) |
| WO (1) | WO2003037789A1 (ja) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7038102B2 (en) * | 2003-07-30 | 2006-05-02 | Exxonmobil Chemical Patents Inc. | Liquid contacting of post-quench effluent vapor streams from oxygenate to olefins conversion to capture catalyst fines |
| US7250149B1 (en) | 2004-02-24 | 2007-07-31 | Smith Strom W | Sulfur gas treatment process |
| DE112005001658T5 (de) * | 2004-07-12 | 2007-06-06 | Exxonmobil Upstream Research Co., Houston | Verfahren zum Entfernen schwefel-haltiger Verbindungen |
| FR2897860B1 (fr) * | 2006-02-27 | 2008-05-30 | Total Sa | Procede d'optimisation de la marche des unites claus. |
| AU2009318991A1 (en) * | 2008-11-28 | 2010-06-03 | Shell Internationale Research Maatschappij B.V. | Process for the selective oxidation of hydrogen sulphide |
| US20100303700A1 (en) * | 2009-05-29 | 2010-12-02 | Nagaraju Palla | Process for treating a gas stream or effluent |
| US7811361B2 (en) * | 2009-06-30 | 2010-10-12 | Uop Llc | Process for a gas removal zone |
| US7811544B1 (en) * | 2009-08-26 | 2010-10-12 | Gas Technology Institute | Sulfur recovery process |
| US8241603B1 (en) | 2011-03-22 | 2012-08-14 | Gas Technology Institute | Process and system for removing sulfur from sulfur-containing gaseous streams |
| DE102011002320B3 (de) | 2011-04-28 | 2012-06-21 | Knauf Gips Kg | Verfahren und Vorrichtung zur Erzeugung von Strom aus schwefelwasserstoffhaltigen Abgasen |
| CN105764844A (zh) | 2013-11-08 | 2016-07-13 | 沙特阿拉伯石油公司 | 硫回收单元和工艺 |
| US10793434B2 (en) | 2019-01-25 | 2020-10-06 | Strom W. Smith | System for hydrogen sulfide destruction and sulfur recovery |
| US11548784B1 (en) | 2021-10-26 | 2023-01-10 | Saudi Arabian Oil Company | Treating sulfur dioxide containing stream by acid aqueous absorption |
| US11926799B2 (en) | 2021-12-14 | 2024-03-12 | Saudi Arabian Oil Company | 2-iso-alkyl-2-(4-hydroxyphenyl)propane derivatives used as emulsion breakers for crude oil |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5116186B2 (ja) | 1972-12-15 | 1976-05-22 | ||
| US4053575A (en) * | 1974-08-19 | 1977-10-11 | The United States Of America As Represented By The Secretary Of The Interior | Sulfur recovery from H2 S and SO2 -containing gases |
| US4056606A (en) * | 1975-05-30 | 1977-11-01 | Bayer Aktiengesellschaft | Desulfurization of waste gases containing hydrogen sulfide |
| US4124685A (en) | 1976-06-08 | 1978-11-07 | Tarhan Mehmet O | Method for substantially complete removal of hydrogen sulfide from sulfur bearing industrial gases |
| JPS59204687A (ja) | 1983-05-02 | 1984-11-20 | ストウフア−・ケミカル・カンパニ− | 燃料ガスの脱硫方法 |
| DE3333933A1 (de) | 1983-09-20 | 1985-04-04 | Linde Ag, 6200 Wiesbaden | Verfahren zum reinigen eines gasstromes |
| EP0199815A1 (en) | 1984-11-04 | 1986-11-05 | The Regents Of The University Of California | Process for removal of hydrogen sulfide from gases |
| US5098681A (en) | 1990-08-16 | 1992-03-24 | The Dow Chemical Company | Process for absorption of sulfur compounds from fluids |
| US5397556A (en) | 1992-12-16 | 1995-03-14 | The Regents Of The Unviversity Of California | Process for recovery of sulfur from acid gases |
| US5928620A (en) | 1997-09-10 | 1999-07-27 | The Regents Of The University Of California | Process employing single-stage reactor for recovering sulfur from H2 S- |
-
2001
- 2001-10-30 US US09/999,825 patent/US6645459B2/en not_active Expired - Lifetime
-
2002
- 2002-10-25 WO PCT/US2002/034363 patent/WO2003037789A1/en active Application Filing
- 2002-10-25 JP JP2003540079A patent/JP4303111B2/ja not_active Expired - Fee Related
- 2002-10-25 AT AT02776318T patent/ATE430114T1/de not_active IP Right Cessation
- 2002-10-25 BR BRPI0213753-4A patent/BR0213753B1/pt not_active IP Right Cessation
- 2002-10-25 EP EP02776318A patent/EP1456123B1/en not_active Expired - Lifetime
- 2002-10-25 DE DE60232180T patent/DE60232180D1/de not_active Expired - Lifetime
- 2002-10-25 CA CA2464726A patent/CA2464726C/en not_active Expired - Fee Related
- 2002-10-25 MX MXPA04004100A patent/MXPA04004100A/es active IP Right Grant
- 2002-10-25 ES ES02776318T patent/ES2322892T3/es not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| CA2464726A1 (en) | 2003-05-08 |
| MXPA04004100A (es) | 2004-07-23 |
| EP1456123A1 (en) | 2004-09-15 |
| WO2003037789A1 (en) | 2003-05-08 |
| US6645459B2 (en) | 2003-11-11 |
| ATE430114T1 (de) | 2009-05-15 |
| BR0213753B1 (pt) | 2012-02-07 |
| CA2464726C (en) | 2010-05-11 |
| BR0213753A (pt) | 2005-01-04 |
| ES2322892T3 (es) | 2009-07-01 |
| EP1456123A4 (en) | 2006-03-15 |
| EP1456123B1 (en) | 2009-04-29 |
| JP2005507844A (ja) | 2005-03-24 |
| DE60232180D1 (de) | 2009-06-10 |
| US20030082096A1 (en) | 2003-05-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100810188B1 (ko) | 황화수소 함유 가스 스트림의 처리방법 | |
| CA2302673C (en) | High efficiency process for recovering sulfur from h2s-bearing gas | |
| US10479684B2 (en) | Enhancement of claus tail gas treatment by sulfur dioxide-selective membrane technology and sulfur dioxide-selective absorption technology | |
| JP4303111B2 (ja) | 硫黄回収プロセスにおける硫黄成分の回収方法 | |
| EP1142628A2 (en) | Treatment of gas streams containing hydrogen sulphide | |
| US8361421B2 (en) | Method of treating a syngas stream and an apparatus therefor | |
| KR20120047253A (ko) | 황 회수 플랜트 테일 가스 처리 방법 | |
| EA010565B1 (ru) | Способ удаления содержащих серу соединений из углеводородсодержащих газов (варианты) | |
| CA3059059C (en) | Enhancement of claus tail gas treatment by sulfur dioxide-selective membrane technology | |
| JP2005507844A5 (ja) | ||
| US20030103884A1 (en) | Low-emission method of recovering sulfur from sour industrial gases | |
| CN1206038A (zh) | 煤气的净化方法 | |
| JP4391140B2 (ja) | チオ硫酸アンモニウムの製造方法 | |
| MXPA00002441A (en) | High efficiency process for recovering sulfur from h2 | |
| JPH09278421A (ja) | 天然ガスからの硫黄回収方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20051025 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20051025 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080909 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20081208 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090203 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090223 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090324 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090423 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120501 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120501 Year of fee payment: 3 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130501 Year of fee payment: 4 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20140501 Year of fee payment: 5 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |