JP2017514901A - 6−クロロ置換イミダゾ[1,2−a]ピリジンカルボキサミドおよびその使用 - Google Patents
6−クロロ置換イミダゾ[1,2−a]ピリジンカルボキサミドおよびその使用 Download PDFInfo
- Publication number
- JP2017514901A JP2017514901A JP2017508756A JP2017508756A JP2017514901A JP 2017514901 A JP2017514901 A JP 2017514901A JP 2017508756 A JP2017508756 A JP 2017508756A JP 2017508756 A JP2017508756 A JP 2017508756A JP 2017514901 A JP2017514901 A JP 2017514901A
- Authority
- JP
- Japan
- Prior art keywords
- salts
- oxides
- solvates
- amino
- enantiomer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- BEHYAANJUKYBTH-UHFFFAOYSA-N imidazo[1,2-a]pyridine-2-carboxamide Chemical class C1=CC=CC2=NC(C(=O)N)=CN21 BEHYAANJUKYBTH-UHFFFAOYSA-N 0.000 title 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 44
- 239000003814 drug Substances 0.000 claims abstract description 16
- 201000010099 disease Diseases 0.000 claims abstract description 11
- 238000004519 manufacturing process Methods 0.000 claims abstract description 9
- 150000001875 compounds Chemical class 0.000 claims description 178
- 150000003839 salts Chemical class 0.000 claims description 77
- 238000000034 method Methods 0.000 claims description 72
- 150000001204 N-oxides Chemical class 0.000 claims description 57
- 239000012453 solvate Substances 0.000 claims description 46
- 239000002904 solvent Substances 0.000 claims description 38
- -1 2-Amino-4,4-difluoro-2-methylbutyl Chemical group 0.000 claims description 37
- 206010019280 Heart failures Diseases 0.000 claims description 18
- 239000003112 inhibitor Substances 0.000 claims description 17
- 239000012442 inert solvent Substances 0.000 claims description 15
- 239000002253 acid Substances 0.000 claims description 14
- 206010020772 Hypertension Diseases 0.000 claims description 13
- 239000002585 base Substances 0.000 claims description 13
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 11
- 241001465754 Metazoa Species 0.000 claims description 10
- 230000008085 renal dysfunction Effects 0.000 claims description 10
- 125000006239 protecting group Chemical group 0.000 claims description 9
- 206010002383 Angina Pectoris Diseases 0.000 claims description 8
- 206010003210 Arteriosclerosis Diseases 0.000 claims description 8
- 208000011775 arteriosclerosis disease Diseases 0.000 claims description 8
- 208000028867 ischemia Diseases 0.000 claims description 8
- 208000019553 vascular disease Diseases 0.000 claims description 7
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 claims description 6
- 239000003146 anticoagulant agent Substances 0.000 claims description 6
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 6
- 125000006508 2,6-difluorobenzyl group Chemical group [H]C1=C([H])C(F)=C(C(F)=C1[H])C([H])([H])* 0.000 claims description 5
- CQDLKDLQCOJOLY-UHFFFAOYSA-N NC(CNC(=O)C1=C(N=C2N1C=C(C=C2OCC1=C(C=CC=C1F)F)Cl)C)(CCF)C Chemical compound NC(CNC(=O)C1=C(N=C2N1C=C(C=C2OCC1=C(C=CC=C1F)F)Cl)C)(CCF)C CQDLKDLQCOJOLY-UHFFFAOYSA-N 0.000 claims description 5
- 208000001435 Thromboembolism Diseases 0.000 claims description 5
- 150000001412 amines Chemical class 0.000 claims description 5
- 150000001732 carboxylic acid derivatives Chemical class 0.000 claims description 5
- 239000003054 catalyst Substances 0.000 claims description 4
- 150000001408 amides Chemical class 0.000 claims description 3
- 239000002220 antihypertensive agent Substances 0.000 claims description 3
- 229960004676 antithrombotic agent Drugs 0.000 claims description 3
- 230000008878 coupling Effects 0.000 claims description 3
- 238000010168 coupling process Methods 0.000 claims description 3
- 238000005859 coupling reaction Methods 0.000 claims description 3
- 230000004060 metabolic process Effects 0.000 claims description 3
- 235000005152 nicotinamide Nutrition 0.000 claims description 3
- 239000011570 nicotinamide Substances 0.000 claims description 3
- 231100000252 nontoxic Toxicity 0.000 claims description 3
- 230000003000 nontoxic effect Effects 0.000 claims description 3
- 229940082615 organic nitrates used in cardiac disease Drugs 0.000 claims description 3
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 3
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 2
- 229940030600 antihypertensive agent Drugs 0.000 claims description 2
- 208000001647 Renal Insufficiency Diseases 0.000 claims 1
- 239000003925 fat Substances 0.000 claims 1
- 201000006370 kidney failure Diseases 0.000 claims 1
- 238000002360 preparation method Methods 0.000 abstract description 14
- 238000011282 treatment Methods 0.000 abstract description 11
- 230000002265 prevention Effects 0.000 abstract description 8
- 208000024172 Cardiovascular disease Diseases 0.000 abstract description 5
- ZMBYQTGAXZOMOO-UHFFFAOYSA-N imidazo[1,2-a]pyridine-3-carboxamide Chemical class C1=CC=CN2C(C(=O)N)=CN=C21 ZMBYQTGAXZOMOO-UHFFFAOYSA-N 0.000 abstract description 2
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 144
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 86
- 239000000203 mixture Substances 0.000 description 84
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 75
- 239000012071 phase Substances 0.000 description 74
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 72
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 72
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 63
- 239000000243 solution Substances 0.000 description 59
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 51
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 50
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 39
- 238000006243 chemical reaction Methods 0.000 description 38
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 36
- 208000035475 disorder Diseases 0.000 description 33
- 239000000126 substance Substances 0.000 description 33
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 32
- 238000005160 1H NMR spectroscopy Methods 0.000 description 30
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 30
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical class OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 30
- 239000011541 reaction mixture Substances 0.000 description 28
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 27
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 24
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 24
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 23
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 23
- 238000001514 detection method Methods 0.000 description 23
- 238000012360 testing method Methods 0.000 description 23
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 22
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Chemical class OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 22
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 21
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 21
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 20
- 239000000047 product Substances 0.000 description 20
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 18
- 239000012074 organic phase Substances 0.000 description 18
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 17
- 229910052938 sodium sulfate Inorganic materials 0.000 description 17
- 235000011152 sodium sulphate Nutrition 0.000 description 17
- AFABGHUZZDYHJO-UHFFFAOYSA-N 2-Methylpentane Chemical compound CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 16
- 229920006395 saturated elastomer Polymers 0.000 description 16
- 229910052786 argon Inorganic materials 0.000 description 15
- 239000008346 aqueous phase Substances 0.000 description 14
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 14
- 238000002953 preparative HPLC Methods 0.000 description 14
- 101000610640 Homo sapiens U4/U6 small nuclear ribonucleoprotein Prp3 Proteins 0.000 description 13
- 101001110823 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-A Proteins 0.000 description 13
- 101000712176 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) 60S ribosomal protein L6-B Proteins 0.000 description 13
- 102100040374 U4/U6 small nuclear ribonucleoprotein Prp3 Human genes 0.000 description 13
- ZOOGRGPOEVQQDX-KHLHZJAASA-N cyclic guanosine monophosphate Chemical compound C([C@H]1O2)O[P@](O)(=O)O[C@@H]1[C@H](O)[C@H]2N1C(N=C(NC2=O)N)=C2N=C1 ZOOGRGPOEVQQDX-KHLHZJAASA-N 0.000 description 13
- 230000000694 effects Effects 0.000 description 13
- 238000002474 experimental method Methods 0.000 description 13
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 13
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 13
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 13
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 12
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 12
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- 229910021529 ammonia Inorganic materials 0.000 description 12
- 229910052763 palladium Inorganic materials 0.000 description 12
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical class COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 11
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical class CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 11
- 239000007868 Raney catalyst Substances 0.000 description 11
- NPXOKRUENSOPAO-UHFFFAOYSA-N Raney nickel Chemical compound [Al].[Ni] NPXOKRUENSOPAO-UHFFFAOYSA-N 0.000 description 11
- 229910000564 Raney nickel Inorganic materials 0.000 description 11
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 11
- 102000007637 Soluble Guanylyl Cyclase Human genes 0.000 description 11
- 108010007205 Soluble Guanylyl Cyclase Proteins 0.000 description 11
- 210000004027 cell Anatomy 0.000 description 11
- 235000019253 formic acid Nutrition 0.000 description 11
- 238000000746 purification Methods 0.000 description 11
- 238000010898 silica gel chromatography Methods 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 10
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 10
- 230000003176 fibrotic effect Effects 0.000 description 10
- 230000035699 permeability Effects 0.000 description 10
- 239000002002 slurry Substances 0.000 description 10
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 10
- 235000017557 sodium bicarbonate Nutrition 0.000 description 10
- 239000011780 sodium chloride Substances 0.000 description 10
- 210000001519 tissue Anatomy 0.000 description 10
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 9
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 9
- 239000007821 HATU Substances 0.000 description 9
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 9
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 8
- 241000700159 Rattus Species 0.000 description 8
- 239000005557 antagonist Substances 0.000 description 8
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 8
- 210000004369 blood Anatomy 0.000 description 8
- 239000008280 blood Substances 0.000 description 8
- 208000017169 kidney disease Diseases 0.000 description 8
- 229910052757 nitrogen Inorganic materials 0.000 description 8
- 239000003826 tablet Substances 0.000 description 8
- JCKMSCHJSIGXNH-UHFFFAOYSA-N 6-chloro-8-[(2,6-difluorophenyl)methoxy]-2-methylimidazo[1,2-a]pyridine-3-carboxylic acid Chemical compound C=1C(Cl)=CN2C(C(O)=O)=C(C)N=C2C=1OCC1=C(F)C=CC=C1F JCKMSCHJSIGXNH-UHFFFAOYSA-N 0.000 description 7
- 206010012289 Dementia Diseases 0.000 description 7
- 102000004190 Enzymes Human genes 0.000 description 7
- 108090000790 Enzymes Proteins 0.000 description 7
- 101001047090 Homo sapiens Potassium voltage-gated channel subfamily H member 2 Proteins 0.000 description 7
- 102100022807 Potassium voltage-gated channel subfamily H member 2 Human genes 0.000 description 7
- 208000006011 Stroke Diseases 0.000 description 7
- 230000036772 blood pressure Effects 0.000 description 7
- 229910002091 carbon monoxide Inorganic materials 0.000 description 7
- 230000001276 controlling effect Effects 0.000 description 7
- 239000012043 crude product Substances 0.000 description 7
- 229940088598 enzyme Drugs 0.000 description 7
- 229910000027 potassium carbonate Inorganic materials 0.000 description 7
- 235000011181 potassium carbonates Nutrition 0.000 description 7
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 7
- 238000010992 reflux Methods 0.000 description 7
- 239000003643 water by type Substances 0.000 description 7
- HSINOMROUCMIEA-FGVHQWLLSA-N (2s,4r)-4-[(3r,5s,6r,7r,8s,9s,10s,13r,14s,17r)-6-ethyl-3,7-dihydroxy-10,13-dimethyl-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-17-yl]-2-methylpentanoic acid Chemical compound C([C@@]12C)C[C@@H](O)C[C@H]1[C@@H](CC)[C@@H](O)[C@@H]1[C@@H]2CC[C@]2(C)[C@@H]([C@H](C)C[C@H](C)C(O)=O)CC[C@H]21 HSINOMROUCMIEA-FGVHQWLLSA-N 0.000 description 6
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 6
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 208000027418 Wounds and injury Diseases 0.000 description 6
- 230000001154 acute effect Effects 0.000 description 6
- 239000003613 bile acid Substances 0.000 description 6
- 238000011534 incubation Methods 0.000 description 6
- 239000011591 potassium Substances 0.000 description 6
- 229910052700 potassium Inorganic materials 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 208000011580 syndromic disease Diseases 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 6
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 5
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 5
- 0 Cc1c(C(O*)=O)[n](cc(cc2O)Cl)c2n1 Chemical compound Cc1c(C(O*)=O)[n](cc(cc2O)Cl)c2n1 0.000 description 5
- 108010078321 Guanylate Cyclase Proteins 0.000 description 5
- 102000014469 Guanylate cyclase Human genes 0.000 description 5
- 239000005909 Kieselgur Substances 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- 235000011114 ammonium hydroxide Nutrition 0.000 description 5
- LLRVOKAAOCQGOK-UHFFFAOYSA-N benzyl n-(2-cyano-1-fluoropropan-2-yl)carbamate Chemical compound FCC(C)(C#N)NC(=O)OCC1=CC=CC=C1 LLRVOKAAOCQGOK-UHFFFAOYSA-N 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 230000001684 chronic effect Effects 0.000 description 5
- 230000006378 damage Effects 0.000 description 5
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 238000010931 ester hydrolysis Methods 0.000 description 5
- 238000009472 formulation Methods 0.000 description 5
- 210000002216 heart Anatomy 0.000 description 5
- 150000004677 hydrates Chemical class 0.000 description 5
- 229910052739 hydrogen Inorganic materials 0.000 description 5
- 239000001257 hydrogen Substances 0.000 description 5
- 208000014674 injury Diseases 0.000 description 5
- 239000000543 intermediate Substances 0.000 description 5
- 230000000155 isotopic effect Effects 0.000 description 5
- 238000005259 measurement Methods 0.000 description 5
- 230000002829 reductive effect Effects 0.000 description 5
- 230000009424 thromboembolic effect Effects 0.000 description 5
- 238000000825 ultraviolet detection Methods 0.000 description 5
- PAMIQIKDUOTOBW-UHFFFAOYSA-N 1-methylpiperidine Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- XKMLYUALXHKNFT-UUOKFMHZSA-N Guanosine-5'-triphosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O XKMLYUALXHKNFT-UUOKFMHZSA-N 0.000 description 4
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 4
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical group [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- 208000009525 Myocarditis Diseases 0.000 description 4
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- 229960000583 acetic acid Drugs 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 229940083712 aldosterone antagonist Drugs 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 239000001569 carbon dioxide Substances 0.000 description 4
- 229910002092 carbon dioxide Inorganic materials 0.000 description 4
- 230000008602 contraction Effects 0.000 description 4
- DDRJAANPRJIHGJ-UHFFFAOYSA-N creatinine Chemical compound CN1CC(=O)NC1=N DDRJAANPRJIHGJ-UHFFFAOYSA-N 0.000 description 4
- 239000012065 filter cake Substances 0.000 description 4
- 150000003278 haem Chemical class 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 239000006193 liquid solution Substances 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 230000002503 metabolic effect Effects 0.000 description 4
- 239000002808 molecular sieve Substances 0.000 description 4
- 238000007911 parenteral administration Methods 0.000 description 4
- 230000002093 peripheral effect Effects 0.000 description 4
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 4
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 4
- 230000000638 stimulation Effects 0.000 description 4
- 238000003756 stirring Methods 0.000 description 4
- 239000006228 supernatant Substances 0.000 description 4
- 238000001356 surgical procedure Methods 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- YYGNTYWPHWGJRM-UHFFFAOYSA-N (6E,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene Chemical compound CC(C)=CCCC(C)=CCCC(C)=CCCC=C(C)CCC=C(C)CCC=C(C)C YYGNTYWPHWGJRM-UHFFFAOYSA-N 0.000 description 3
- VRLJFRODHVSTIK-UHFFFAOYSA-N 2-(benzhydrylideneamino)acetonitrile Chemical compound C=1C=CC=CC=1C(=NCC#N)C1=CC=CC=C1 VRLJFRODHVSTIK-UHFFFAOYSA-N 0.000 description 3
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 3
- 239000005541 ACE inhibitor Substances 0.000 description 3
- 206010059245 Angiopathy Diseases 0.000 description 3
- 102000015427 Angiotensins Human genes 0.000 description 3
- 108010064733 Angiotensins Proteins 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- WCXSKRCUQWZGOR-UHFFFAOYSA-N C(#N)C(C)(CC(F)F)NC(OCC1=CC=CC=C1)=O Chemical compound C(#N)C(C)(CC(F)F)NC(OCC1=CC=CC=C1)=O WCXSKRCUQWZGOR-UHFFFAOYSA-N 0.000 description 3
- 229940127291 Calcium channel antagonist Drugs 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 102000004895 Lipoproteins Human genes 0.000 description 3
- 108090001030 Lipoproteins Proteins 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- 102100031545 Microsomal triglyceride transfer protein large subunit Human genes 0.000 description 3
- 206010030113 Oedema Diseases 0.000 description 3
- 102000004861 Phosphoric Diester Hydrolases Human genes 0.000 description 3
- 108090001050 Phosphoric Diester Hydrolases Proteins 0.000 description 3
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- BHEOSNUKNHRBNM-UHFFFAOYSA-N Tetramethylsqualene Natural products CC(=C)C(C)CCC(=C)C(C)CCC(C)=CCCC=C(C)CCC(C)C(=C)CCC(C)C(C)=C BHEOSNUKNHRBNM-UHFFFAOYSA-N 0.000 description 3
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 3
- 230000009102 absorption Effects 0.000 description 3
- 238000010521 absorption reaction Methods 0.000 description 3
- 239000003463 adsorbent Substances 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 3
- 229940127218 antiplatelet drug Drugs 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical class OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- BZZSIZPHNGARLH-UHFFFAOYSA-N benzyl n-(2-cyano-5,5,5-trifluoropentan-2-yl)carbamate Chemical compound FC(F)(F)CCC(C)(C#N)NC(=O)OCC1=CC=CC=C1 BZZSIZPHNGARLH-UHFFFAOYSA-N 0.000 description 3
- 239000002876 beta blocker Substances 0.000 description 3
- 230000005540 biological transmission Effects 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 239000013553 cell monolayer Substances 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 239000003354 cholesterol ester transfer protein inhibitor Substances 0.000 description 3
- 238000004587 chromatography analysis Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical class OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- CYCGRDQQIOGCKX-UHFFFAOYSA-N dehydroluciferin Chemical compound OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 206010012601 diabetes mellitus Diseases 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 238000004090 dissolution Methods 0.000 description 3
- PRAKJMSDJKAYCZ-UHFFFAOYSA-N dodecahydrosqualene Natural products CC(C)CCCC(C)CCCC(C)CCCCC(C)CCCC(C)CCCC(C)C PRAKJMSDJKAYCZ-UHFFFAOYSA-N 0.000 description 3
- 150000002170 ethers Chemical class 0.000 description 3
- 239000010408 film Substances 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 238000002290 gas chromatography-mass spectrometry Methods 0.000 description 3
- 230000002496 gastric effect Effects 0.000 description 3
- 238000003304 gavage Methods 0.000 description 3
- 239000008103 glucose Substances 0.000 description 3
- 230000005802 health problem Effects 0.000 description 3
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 3
- 239000007943 implant Substances 0.000 description 3
- 230000002757 inflammatory effect Effects 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 229910052742 iron Inorganic materials 0.000 description 3
- 229910001629 magnesium chloride Inorganic materials 0.000 description 3
- JLFNLZLINWHATN-UHFFFAOYSA-N pentaethylene glycol Chemical compound OCCOCCOCCOCCOCCO JLFNLZLINWHATN-UHFFFAOYSA-N 0.000 description 3
- WEXRUCMBJFQVBZ-UHFFFAOYSA-N pentobarbital Chemical compound CCCC(C)C1(CC)C(=O)NC(=O)NC1=O WEXRUCMBJFQVBZ-UHFFFAOYSA-N 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 3
- 208000005069 pulmonary fibrosis Diseases 0.000 description 3
- 230000009103 reabsorption Effects 0.000 description 3
- 229940044601 receptor agonist Drugs 0.000 description 3
- 239000000018 receptor agonist Substances 0.000 description 3
- 239000002461 renin inhibitor Substances 0.000 description 3
- 229940086526 renin-inhibitors Drugs 0.000 description 3
- 238000006798 ring closing metathesis reaction Methods 0.000 description 3
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 3
- 238000011699 spontaneously hypertensive rat Methods 0.000 description 3
- 229940031439 squalene Drugs 0.000 description 3
- TUHBEKDERLKLEC-UHFFFAOYSA-N squalene Natural products CC(=CCCC(=CCCC(=CCCC=C(/C)CCC=C(/C)CC=C(C)C)C)C)C TUHBEKDERLKLEC-UHFFFAOYSA-N 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 102000004217 thyroid hormone receptors Human genes 0.000 description 3
- 108090000721 thyroid hormone receptors Proteins 0.000 description 3
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- GQIRIWDEZSKOCN-UHFFFAOYSA-N 1-chloro-n,n,2-trimethylprop-1-en-1-amine Chemical compound CN(C)C(Cl)=C(C)C GQIRIWDEZSKOCN-UHFFFAOYSA-N 0.000 description 2
- OFJRNBWSFXEHSA-UHFFFAOYSA-N 2-(3-amino-1,2-benzoxazol-5-yl)-n-[4-[2-[(dimethylamino)methyl]imidazol-1-yl]-2-fluorophenyl]-5-(trifluoromethyl)pyrazole-3-carboxamide Chemical compound CN(C)CC1=NC=CN1C(C=C1F)=CC=C1NC(=O)C1=CC(C(F)(F)F)=NN1C1=CC=C(ON=C2N)C2=C1 OFJRNBWSFXEHSA-UHFFFAOYSA-N 0.000 description 2
- QOZNHJKSURHSFT-UHFFFAOYSA-N 2-(benzhydrylideneamino)-4,4-difluoro-2-methylbutanenitrile Chemical compound C=1C=CC=CC=1C(=NC(CC(F)F)(C)C#N)C1=CC=CC=C1 QOZNHJKSURHSFT-UHFFFAOYSA-N 0.000 description 2
- ZOFFFVIPDUEFCN-UHFFFAOYSA-N 2-(benzhydrylideneamino)-4-fluoro-2-methylbutanenitrile Chemical compound C=1C=CC=CC=1C(=NC(CCF)(C)C#N)C1=CC=CC=C1 ZOFFFVIPDUEFCN-UHFFFAOYSA-N 0.000 description 2
- QGHJJQCXOHXFCY-UHFFFAOYSA-N 2-(benzhydrylideneamino)-5-fluoro-2-methylpentanenitrile Chemical compound C=1C=CC=CC=1C(=NC(CCCF)(C)C#N)C1=CC=CC=C1 QGHJJQCXOHXFCY-UHFFFAOYSA-N 0.000 description 2
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical class CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 2
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- NCGICGYLBXGBGN-UHFFFAOYSA-N 3-morpholin-4-yl-1-oxa-3-azonia-2-azanidacyclopent-3-en-5-imine;hydrochloride Chemical compound Cl.[N-]1OC(=N)C=[N+]1N1CCOCC1 NCGICGYLBXGBGN-UHFFFAOYSA-N 0.000 description 2
- OCKGFTQIICXDQW-ZEQRLZLVSA-N 5-[(1r)-1-hydroxy-2-[4-[(2r)-2-hydroxy-2-(4-methyl-1-oxo-3h-2-benzofuran-5-yl)ethyl]piperazin-1-yl]ethyl]-4-methyl-3h-2-benzofuran-1-one Chemical compound C1=C2C(=O)OCC2=C(C)C([C@@H](O)CN2CCN(CC2)C[C@H](O)C2=CC=C3C(=O)OCC3=C2C)=C1 OCKGFTQIICXDQW-ZEQRLZLVSA-N 0.000 description 2
- XRSYKYYBXLVPAJ-UHFFFAOYSA-N 5-chloro-2-nitropyridin-3-ol Chemical compound OC1=CC(Cl)=CN=C1[N+]([O-])=O XRSYKYYBXLVPAJ-UHFFFAOYSA-N 0.000 description 2
- AFWNILVXTVCIJQ-UHFFFAOYSA-N 5-chloro-3-[(2,6-difluorophenyl)methoxy]-2-nitropyridine Chemical compound [O-][N+](=O)C1=NC=C(Cl)C=C1OCC1=C(F)C=CC=C1F AFWNILVXTVCIJQ-UHFFFAOYSA-N 0.000 description 2
- VUJSFQIUJVYUNE-UHFFFAOYSA-N 5-chloro-3-[(2,6-difluorophenyl)methoxy]pyridin-2-amine Chemical compound NC1=NC=C(Cl)C=C1OCC1=C(F)C=CC=C1F VUJSFQIUJVYUNE-UHFFFAOYSA-N 0.000 description 2
- 208000004476 Acute Coronary Syndrome Diseases 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- SIYMQNAIHOYHAC-UHFFFAOYSA-N CC(CC(F)F)(CNC(c1c(C)nc(c(OCc(c(F)ccc2)c2F)c2)[n]1cc2Cl)=O)N Chemical compound CC(CC(F)F)(CNC(c1c(C)nc(c(OCc(c(F)ccc2)c2F)c2)[n]1cc2Cl)=O)N SIYMQNAIHOYHAC-UHFFFAOYSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 2
- 206010007556 Cardiac failure acute Diseases 0.000 description 2
- 206010007558 Cardiac failure chronic Diseases 0.000 description 2
- 208000031229 Cardiomyopathies Diseases 0.000 description 2
- 229940122502 Cholesterol absorption inhibitor Drugs 0.000 description 2
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 description 2
- WACWXJHWXRIXAZ-UHFFFAOYSA-N Cl.CC(N)(CC(F)F)C#N Chemical compound Cl.CC(N)(CC(F)F)C#N WACWXJHWXRIXAZ-UHFFFAOYSA-N 0.000 description 2
- GYJXCRTWVSQDQT-UHFFFAOYSA-N Cl.CC(N)(CCCF)C#N Chemical compound Cl.CC(N)(CCCF)C#N GYJXCRTWVSQDQT-UHFFFAOYSA-N 0.000 description 2
- KSHOAHHZZTWGIW-UHFFFAOYSA-N Cl.CC(N)(CCF)C#N Chemical compound Cl.CC(N)(CCF)C#N KSHOAHHZZTWGIW-UHFFFAOYSA-N 0.000 description 2
- 201000003883 Cystic fibrosis Diseases 0.000 description 2
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 2
- 102000003849 Cytochrome P450 Human genes 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- 206010012689 Diabetic retinopathy Diseases 0.000 description 2
- 208000005189 Embolism Diseases 0.000 description 2
- 206010014561 Emphysema Diseases 0.000 description 2
- 206010048554 Endothelial dysfunction Diseases 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- MZXKBYYEROJDAW-UHFFFAOYSA-N FC(F)CC(N=C(c1ccccc1)c1ccccc1)C#N Chemical compound FC(F)CC(N=C(c1ccccc1)c1ccccc1)C#N MZXKBYYEROJDAW-UHFFFAOYSA-N 0.000 description 2
- KTTDGGIHSGGUMK-UHFFFAOYSA-N FCCC(N=C(c1ccccc1)c1ccccc1)C#N Chemical compound FCCC(N=C(c1ccccc1)c1ccccc1)C#N KTTDGGIHSGGUMK-UHFFFAOYSA-N 0.000 description 2
- VASDTJCSAXCJHL-UHFFFAOYSA-N FCCCC(N=C(c1ccccc1)c1ccccc1)C#N Chemical compound FCCCC(N=C(c1ccccc1)c1ccccc1)C#N VASDTJCSAXCJHL-UHFFFAOYSA-N 0.000 description 2
- 206010016654 Fibrosis Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical class OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 208000010412 Glaucoma Diseases 0.000 description 2
- 206010018364 Glomerulonephritis Diseases 0.000 description 2
- 206010020853 Hypertonic bladder Diseases 0.000 description 2
- 102100021711 Ileal sodium/bile acid cotransporter Human genes 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- UQSXHKLRYXJYBZ-UHFFFAOYSA-N Iron oxide Chemical compound [Fe]=O UQSXHKLRYXJYBZ-UHFFFAOYSA-N 0.000 description 2
- 102000007330 LDL Lipoproteins Human genes 0.000 description 2
- 108010007622 LDL Lipoproteins Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 229940086609 Lipase inhibitor Drugs 0.000 description 2
- 208000017170 Lipid metabolism disease Diseases 0.000 description 2
- 206010027525 Microalbuminuria Diseases 0.000 description 2
- 208000034486 Multi-organ failure Diseases 0.000 description 2
- 208000010718 Multiple Organ Failure Diseases 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- 208000009722 Overactive Urinary Bladder Diseases 0.000 description 2
- 102000023984 PPAR alpha Human genes 0.000 description 2
- 108010016731 PPAR gamma Proteins 0.000 description 2
- 229940122054 Peroxisome proliferator-activated receptor delta agonist Drugs 0.000 description 2
- 102100038825 Peroxisome proliferator-activated receptor gamma Human genes 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 2
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 2
- 206010064911 Pulmonary arterial hypertension Diseases 0.000 description 2
- 206010037423 Pulmonary oedema Diseases 0.000 description 2
- 241000700157 Rattus norvegicus Species 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 239000012317 TBTU Substances 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 206010046543 Urinary incontinence Diseases 0.000 description 2
- 206010052428 Wound Diseases 0.000 description 2
- OQQVFCKUDYMWGV-UHFFFAOYSA-N [5-[1-(phenylmethyl)-3-indazolyl]-2-furanyl]methanol Chemical compound O1C(CO)=CC=C1C(C1=CC=CC=C11)=NN1CC1=CC=CC=C1 OQQVFCKUDYMWGV-UHFFFAOYSA-N 0.000 description 2
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 2
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000012190 activator Substances 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 206010069351 acute lung injury Diseases 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 2
- 238000002399 angioplasty Methods 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 229940127219 anticoagulant drug Drugs 0.000 description 2
- 210000000709 aorta Anatomy 0.000 description 2
- YZXBAPSDXZZRGB-DOFZRALJSA-N arachidonic acid Chemical compound CCCCC\C=C/C\C=C/C\C=C/C\C=C/CCCC(O)=O YZXBAPSDXZZRGB-DOFZRALJSA-N 0.000 description 2
- 206010003119 arrhythmia Diseases 0.000 description 2
- 230000006793 arrhythmia Effects 0.000 description 2
- 229960002274 atenolol Drugs 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- BRGSMTUJJHSCLV-UHFFFAOYSA-N benzyl n-(1-amino-3-fluoro-2-methylpropan-2-yl)carbamate Chemical compound NCC(C)(CF)NC(=O)OCC1=CC=CC=C1 BRGSMTUJJHSCLV-UHFFFAOYSA-N 0.000 description 2
- AEDHKYKBURAJOP-UHFFFAOYSA-N benzyl n-(2-cyano-4-fluorobutan-2-yl)carbamate Chemical compound FCCC(C)(C#N)NC(=O)OCC1=CC=CC=C1 AEDHKYKBURAJOP-UHFFFAOYSA-N 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 208000029028 brain injury Diseases 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000000480 calcium channel blocker Substances 0.000 description 2
- 235000011089 carbon dioxide Nutrition 0.000 description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 2
- 208000015114 central nervous system disease Diseases 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 206010008118 cerebral infarction Diseases 0.000 description 2
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 229940125881 cholesteryl ester transfer protein inhibitor Drugs 0.000 description 2
- 230000027288 circadian rhythm Effects 0.000 description 2
- 208000019425 cirrhosis of liver Diseases 0.000 description 2
- 208000010877 cognitive disease Diseases 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N coumarin Chemical compound C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 2
- 229940109239 creatinine Drugs 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 238000007257 deesterification reaction Methods 0.000 description 2
- 230000007850 degeneration Effects 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- 230000035487 diastolic blood pressure Effects 0.000 description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Chemical class CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 239000002934 diuretic Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 230000002526 effect on cardiovascular system Effects 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 230000008694 endothelial dysfunction Effects 0.000 description 2
- 239000002308 endothelin receptor antagonist Substances 0.000 description 2
- 239000000066 endothelium dependent relaxing factor Substances 0.000 description 2
- 229960001208 eplerenone Drugs 0.000 description 2
- JUKPWJGBANNWMW-VWBFHTRKSA-N eplerenone Chemical compound C([C@@H]1[C@]2(C)C[C@H]3O[C@]33[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)C(=O)OC)C[C@@]21CCC(=O)O1 JUKPWJGBANNWMW-VWBFHTRKSA-N 0.000 description 2
- 230000029142 excretion Effects 0.000 description 2
- 208000030533 eye disease Diseases 0.000 description 2
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 2
- 239000010931 gold Substances 0.000 description 2
- 229910052737 gold Inorganic materials 0.000 description 2
- 239000008187 granular material Substances 0.000 description 2
- 208000019622 heart disease Diseases 0.000 description 2
- 210000003709 heart valve Anatomy 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 2
- 238000007327 hydrogenolysis reaction Methods 0.000 description 2
- 238000002513 implantation Methods 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 230000000968 intestinal effect Effects 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical class CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 230000013016 learning Effects 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 230000036244 malformation Effects 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 239000012452 mother liquor Substances 0.000 description 2
- 230000002107 myocardial effect Effects 0.000 description 2
- 208000010125 myocardial infarction Diseases 0.000 description 2
- 201000009925 nephrosclerosis Diseases 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- 208000020629 overactive bladder Diseases 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 108091008725 peroxisome proliferator-activated receptors alpha Proteins 0.000 description 2
- 239000008194 pharmaceutical composition Substances 0.000 description 2
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 230000036470 plasma concentration Effects 0.000 description 2
- 229920000136 polysorbate Polymers 0.000 description 2
- 229950008882 polysorbate Drugs 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000003908 quality control method Methods 0.000 description 2
- 239000003087 receptor blocking agent Substances 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- QEVHRUUCFGRFIF-MDEJGZGSSA-N reserpine Chemical compound O([C@H]1[C@@H]([C@H]([C@H]2C[C@@H]3C4=C(C5=CC=C(OC)C=C5N4)CCN3C[C@H]2C1)C(=O)OC)OC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 QEVHRUUCFGRFIF-MDEJGZGSSA-N 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 201000000306 sarcoidosis Diseases 0.000 description 2
- 231100000241 scar Toxicity 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- 235000015424 sodium Nutrition 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 229960002256 spironolactone Drugs 0.000 description 2
- LXMSZDCAJNLERA-ZHYRCANASA-N spironolactone Chemical compound C([C@@H]1[C@]2(C)CC[C@@H]3[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)SC(=O)C)C[C@@]21CCC(=O)O1 LXMSZDCAJNLERA-ZHYRCANASA-N 0.000 description 2
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 230000008961 swelling Effects 0.000 description 2
- 230000035488 systolic blood pressure Effects 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Chemical class OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- 208000014001 urinary system disease Diseases 0.000 description 2
- 210000002700 urine Anatomy 0.000 description 2
- 206010047302 ventricular tachycardia Diseases 0.000 description 2
- 238000005303 weighing Methods 0.000 description 2
- CEMAWMOMDPGJMB-UHFFFAOYSA-N (+-)-Oxprenolol Chemical compound CC(C)NCC(O)COC1=CC=CC=C1OCC=C CEMAWMOMDPGJMB-UHFFFAOYSA-N 0.000 description 1
- NXWGWUVGUSFQJC-GFCCVEGCSA-N (2r)-1-[(2-methyl-1h-indol-4-yl)oxy]-3-(propan-2-ylamino)propan-2-ol Chemical compound CC(C)NC[C@@H](O)COC1=CC=CC2=C1C=C(C)N2 NXWGWUVGUSFQJC-GFCCVEGCSA-N 0.000 description 1
- DMYZJLOWGSRVKP-RTBURBONSA-N (2r,4r)-1-n-(4-chlorophenyl)-4-hydroxy-2-n-[4-(3-oxomorpholin-4-yl)phenyl]pyrrolidine-1,2-dicarboxamide Chemical compound N1([C@H](C[C@H](C1)O)C(=O)NC=1C=CC(=CC=1)N1C(COCC1)=O)C(=O)NC1=CC=C(Cl)C=C1 DMYZJLOWGSRVKP-RTBURBONSA-N 0.000 description 1
- AMNXBQPRODZJQR-DITALETJSA-N (2s)-2-cyclopentyl-2-[3-[(2,4-dimethylpyrido[2,3-b]indol-9-yl)methyl]phenyl]-n-[(1r)-2-hydroxy-1-phenylethyl]acetamide Chemical compound C1([C@@H](C=2C=CC=C(C=2)CN2C3=CC=CC=C3C3=C(C)C=C(N=C32)C)C(=O)N[C@@H](CO)C=2C=CC=CC=2)CCCC1 AMNXBQPRODZJQR-DITALETJSA-N 0.000 description 1
- LJCBAPRMNYSDOP-LVCYMWGESA-N (2s)-3-(7-carbamimidoylnaphthalen-2-yl)-2-[4-[(3s)-1-ethanimidoylpyrrolidin-3-yl]oxyphenyl]propanoic acid;hydron;chloride;pentahydrate Chemical compound O.O.O.O.O.Cl.C1N(C(=N)C)CC[C@@H]1OC1=CC=C([C@H](CC=2C=C3C=C(C=CC3=CC=2)C(N)=N)C(O)=O)C=C1 LJCBAPRMNYSDOP-LVCYMWGESA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- RWIUTHWKQHRQNP-ZDVGBALWSA-N (9e,12e)-n-(1-phenylethyl)octadeca-9,12-dienamide Chemical compound CCCCC\C=C\C\C=C\CCCCCCCC(=O)NC(C)C1=CC=CC=C1 RWIUTHWKQHRQNP-ZDVGBALWSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical class OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- TWBNMYSKRDRHAT-RCWTXCDDSA-N (S)-timolol hemihydrate Chemical compound O.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1 TWBNMYSKRDRHAT-RCWTXCDDSA-N 0.000 description 1
- KOHIRBRYDXPAMZ-YHBROIRLSA-N (S,R,R,R)-nebivolol Chemical compound C1CC2=CC(F)=CC=C2O[C@H]1[C@H](O)CNC[C@@H](O)[C@H]1OC2=CC=C(F)C=C2CC1 KOHIRBRYDXPAMZ-YHBROIRLSA-N 0.000 description 1
- VMFCTZUYOILUMY-UHFFFAOYSA-N 1,1-difluoro-2-iodoethane Chemical compound FC(F)CI VMFCTZUYOILUMY-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- ZUCFGSAENWHFPO-UHFFFAOYSA-N 1-[4-amino-2,6-di(propan-2-yl)phenyl]-3-[1-butyl-4-[3-(3-hydroxypropoxy)phenyl]-2-oxo-1,8-naphthyridin-3-yl]urea;hydrate;hydrochloride Chemical compound O.Cl.CC(C)C=1C=C(N)C=C(C(C)C)C=1NC(=O)NC=1C(=O)N(CCCC)C2=NC=CC=C2C=1C1=CC=CC(OCCCO)=C1 ZUCFGSAENWHFPO-UHFFFAOYSA-N 0.000 description 1
- PMGZJNCIQHGNLT-UHFFFAOYSA-N 1-[bis(2,2-dimethylpropanoyloxymethoxy)phosphoryl]-4-(3-phenoxyphenyl)butane-1-sulfonic acid Chemical compound CC(C)(C)C(=O)OCOP(=O)(OCOC(=O)C(C)(C)C)C(S(O)(=O)=O)CCCC1=CC=CC(OC=2C=CC=CC=2)=C1 PMGZJNCIQHGNLT-UHFFFAOYSA-N 0.000 description 1
- LVYJIIRJQDEGBR-UHFFFAOYSA-N 1-fluoro-2-iodoethane Chemical compound FCCI LVYJIIRJQDEGBR-UHFFFAOYSA-N 0.000 description 1
- URBUZQPPQLQHBZ-UHFFFAOYSA-N 1-fluoro-3-iodopropane Chemical compound FCCCI URBUZQPPQLQHBZ-UHFFFAOYSA-N 0.000 description 1
- MSWVMWGCNZQPIA-UHFFFAOYSA-N 1-fluoropropan-2-one Chemical compound CC(=O)CF MSWVMWGCNZQPIA-UHFFFAOYSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- DEMLYXMVPJAVFU-UHFFFAOYSA-N 2-(chloromethyl)oxirane;2-methyl-1h-imidazole Chemical compound ClCC1CO1.CC1=NC=CN1 DEMLYXMVPJAVFU-UHFFFAOYSA-N 0.000 description 1
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 1
- HQSRVYUCBOCBLY-XOOFNSLWSA-N 2-[(2r)-butan-2-yl]-4-[4-[4-[4-[[(2s,4r)-2-(4-chlorophenyl)-2-[(4-methyl-1,2,4-triazol-3-yl)sulfanylmethyl]-1,3-dioxolan-4-yl]methoxy]phenyl]piperazin-1-yl]phenyl]-1,2,4-triazol-3-one Chemical compound O=C1N([C@H](C)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@H]3O[C@@](CSC=4N(C=NN=4)C)(OC3)C=3C=CC(Cl)=CC=3)=CC=2)C=C1 HQSRVYUCBOCBLY-XOOFNSLWSA-N 0.000 description 1
- CMLUGNQVANVZHY-POURPWNDSA-N 2-[1-[2-[(3r,5s)-1-(3-acetyloxy-2,2-dimethylpropyl)-7-chloro-5-(2,3-dimethoxyphenyl)-2-oxo-5h-4,1-benzoxazepin-3-yl]acetyl]piperidin-4-yl]acetic acid Chemical compound COC1=CC=CC([C@@H]2C3=CC(Cl)=CC=C3N(CC(C)(C)COC(C)=O)C(=O)[C@@H](CC(=O)N3CCC(CC(O)=O)CC3)O2)=C1OC CMLUGNQVANVZHY-POURPWNDSA-N 0.000 description 1
- FBMYKMYQHCBIGU-UHFFFAOYSA-N 2-[2-hydroxy-3-[[1-(1h-indol-3-yl)-2-methylpropan-2-yl]amino]propoxy]benzonitrile Chemical compound C=1NC2=CC=CC=C2C=1CC(C)(C)NCC(O)COC1=CC=CC=C1C#N FBMYKMYQHCBIGU-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- TXIIZHHIOHVWJD-UHFFFAOYSA-N 2-[7-(2,2-dimethylpropanoylamino)-4,6-dimethyl-1-octyl-2,3-dihydroindol-5-yl]acetic acid Chemical compound CC(C)(C)C(=O)NC1=C(C)C(CC(O)=O)=C(C)C2=C1N(CCCCCCCC)CC2 TXIIZHHIOHVWJD-UHFFFAOYSA-N 0.000 description 1
- QHVBWSIFLCIXBD-UHFFFAOYSA-N 2-[[2-[3-(diaminomethylidene)-6-oxocyclohexa-1,4-dien-1-yl]oxy-3,5-difluoro-6-[3-(1-methyl-4,5-dihydroimidazol-2-yl)phenoxy]pyridin-4-yl]-methylamino]acetic acid Chemical compound N=1C(OC=2C=C(C=CC=2)C=2N(CCN=2)C)=C(F)C(N(CC(O)=O)C)=C(F)C=1OC1=CC(=C(N)N)C=CC1=O QHVBWSIFLCIXBD-UHFFFAOYSA-N 0.000 description 1
- NOIXNOMHHWGUTG-UHFFFAOYSA-N 2-[[4-[4-pyridin-4-yl-1-(2,2,2-trifluoroethyl)pyrazol-3-yl]phenoxy]methyl]quinoline Chemical class C=1C=C(OCC=2N=C3C=CC=CC3=CC=2)C=CC=1C1=NN(CC(F)(F)F)C=C1C1=CC=NC=C1 NOIXNOMHHWGUTG-UHFFFAOYSA-N 0.000 description 1
- AORAEUVRJPUCDZ-UHFFFAOYSA-N 2-amino-3-fluoro-2-methylpropanenitrile Chemical compound FCC(N)(C)C#N AORAEUVRJPUCDZ-UHFFFAOYSA-N 0.000 description 1
- JIVPVXMEBJLZRO-CQSZACIVSA-N 2-chloro-5-[(1r)-1-hydroxy-3-oxo-2h-isoindol-1-yl]benzenesulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC([C@@]2(O)C3=CC=CC=C3C(=O)N2)=C1 JIVPVXMEBJLZRO-CQSZACIVSA-N 0.000 description 1
- JCJODSMQBXMELN-UHFFFAOYSA-N 2-ethyl-5-phenyl-1,2-oxazol-2-ium Chemical compound O1[N+](CC)=CC=C1C1=CC=CC=C1 JCJODSMQBXMELN-UHFFFAOYSA-N 0.000 description 1
- SGUAFYQXFOLMHL-UHFFFAOYSA-N 2-hydroxy-5-{1-hydroxy-2-[(4-phenylbutan-2-yl)amino]ethyl}benzamide Chemical compound C=1C=C(O)C(C(N)=O)=CC=1C(O)CNC(C)CCC1=CC=CC=C1 SGUAFYQXFOLMHL-UHFFFAOYSA-N 0.000 description 1
- BZACBBRLMWHCNM-UHFFFAOYSA-N 2-methylimidazo[1,2-a]pyridine Chemical compound C1=CC=CC2=NC(C)=CN21 BZACBBRLMWHCNM-UHFFFAOYSA-N 0.000 description 1
- YZQLWPMZQVHJED-UHFFFAOYSA-N 2-methylpropanethioic acid S-[2-[[[1-(2-ethylbutyl)cyclohexyl]-oxomethyl]amino]phenyl] ester Chemical compound C=1C=CC=C(SC(=O)C(C)C)C=1NC(=O)C1(CC(CC)CC)CCCCC1 YZQLWPMZQVHJED-UHFFFAOYSA-N 0.000 description 1
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 1
- MPSXGPCFLAGJOM-UHFFFAOYSA-M 2-tert-butyl-5-methyl-1,2-oxazol-2-ium;perchlorate Chemical compound [O-]Cl(=O)(=O)=O.CC1=CC=[N+](C(C)(C)C)O1 MPSXGPCFLAGJOM-UHFFFAOYSA-M 0.000 description 1
- AUYYCJSJGJYCDS-UHFFFAOYSA-N 2/3/6893 Natural products IC1=CC(CC(N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- KLDLRDSRCMJKGM-UHFFFAOYSA-N 3-[chloro-(2-oxo-1,3-oxazolidin-3-yl)phosphoryl]-1,3-oxazolidin-2-one Chemical compound C1COC(=O)N1P(=O)(Cl)N1CCOC1=O KLDLRDSRCMJKGM-UHFFFAOYSA-N 0.000 description 1
- SCJCDNUXDWFVFI-UHFFFAOYSA-N 4,4,4-trifluorobutanal Chemical compound FC(F)(F)CCC=O SCJCDNUXDWFVFI-UHFFFAOYSA-N 0.000 description 1
- WTUCTMYLCMVYEX-UHFFFAOYSA-N 4,4,4-trifluorobutanoic acid Chemical compound OC(=O)CCC(F)(F)F WTUCTMYLCMVYEX-UHFFFAOYSA-N 0.000 description 1
- NOBZETMXGVAWIM-UHFFFAOYSA-N 4-[(2-carbamimidoyl-3,4-dihydro-1h-isoquinolin-7-yl)oxymethyl]-1-pyridin-4-ylpiperidine-4-carboxylic acid;methanesulfonic acid Chemical compound CS(O)(=O)=O.C1=C2CN(C(=N)N)CCC2=CC=C1OCC(CC1)(C(O)=O)CCN1C1=CC=NC=C1 NOBZETMXGVAWIM-UHFFFAOYSA-N 0.000 description 1
- KEWSCDNULKOKTG-UHFFFAOYSA-N 4-cyano-4-ethylsulfanylcarbothioylsulfanylpentanoic acid Chemical compound CCSC(=S)SC(C)(C#N)CCC(O)=O KEWSCDNULKOKTG-UHFFFAOYSA-N 0.000 description 1
- TUIDQYRZDZRHPQ-UHFFFAOYSA-N 5-chloropyridin-3-ol Chemical compound OC1=CN=CC(Cl)=C1 TUIDQYRZDZRHPQ-UHFFFAOYSA-N 0.000 description 1
- 102100031126 6-phosphogluconolactonase Human genes 0.000 description 1
- 108010029731 6-phosphogluconolactonase Proteins 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 208000009304 Acute Kidney Injury Diseases 0.000 description 1
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 201000010053 Alcoholic Cardiomyopathy Diseases 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- 208000000044 Amnesia Diseases 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 206010002199 Anaphylactic shock Diseases 0.000 description 1
- 206010002329 Aneurysm Diseases 0.000 description 1
- 206010002388 Angina unstable Diseases 0.000 description 1
- 208000031295 Animal disease Diseases 0.000 description 1
- 101000988143 Antheraea pernyi Pheromone-binding protein 1 Proteins 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- 206010002921 Aortitis Diseases 0.000 description 1
- QNZCBYKSOIHPEH-UHFFFAOYSA-N Apixaban Chemical compound C1=CC(OC)=CC=C1N1C(C(=O)N(CC2)C=3C=CC(=CC=3)N3C(CCCC3)=O)=C2C(C(N)=O)=N1 QNZCBYKSOIHPEH-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 206010003178 Arterial thrombosis Diseases 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 206010003658 Atrial Fibrillation Diseases 0.000 description 1
- 208000002102 Atrial Premature Complexes Diseases 0.000 description 1
- 206010003662 Atrial flutter Diseases 0.000 description 1
- 206010003671 Atrioventricular Block Diseases 0.000 description 1
- 208000006808 Atrioventricular Nodal Reentry Tachycardia Diseases 0.000 description 1
- 229930003347 Atropine Natural products 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 206010061666 Autonomic neuropathy Diseases 0.000 description 1
- 208000037157 Azotemia Diseases 0.000 description 1
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 description 1
- 239000005528 B01AC05 - Ticlopidine Substances 0.000 description 1
- 239000005711 Benzoic acid Chemical class 0.000 description 1
- 208000018083 Bone metabolism disease Diseases 0.000 description 1
- 201000006474 Brain Ischemia Diseases 0.000 description 1
- 206010048962 Brain oedema Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- DOTHWLHXVOKARQ-UHFFFAOYSA-N C(#N)C(C)(CCCF)NC(OCC1=CC=CC=C1)=O Chemical compound C(#N)C(C)(CCCF)NC(OCC1=CC=CC=C1)=O DOTHWLHXVOKARQ-UHFFFAOYSA-N 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- PDNVUILNJYUBDQ-UHFFFAOYSA-N CC(CCCF)(CNC(c1c(C)nc(c(OCc(c(F)ccc2)c2F)c2)[n]1cc2Cl)=O)N Chemical compound CC(CCCF)(CNC(c1c(C)nc(c(OCc(c(F)ccc2)c2F)c2)[n]1cc2Cl)=O)N PDNVUILNJYUBDQ-UHFFFAOYSA-N 0.000 description 1
- XTMFHGKTWUVYPT-UHFFFAOYSA-N CC(CCCF)(CNC(c1c(C)nc(c(OCc(c(F)ccc2)c2F)c2)[n]1cc2Cl)=O)NC(OCc1ccccc1)=O Chemical compound CC(CCCF)(CNC(c1c(C)nc(c(OCc(c(F)ccc2)c2F)c2)[n]1cc2Cl)=O)NC(OCc1ccccc1)=O XTMFHGKTWUVYPT-UHFFFAOYSA-N 0.000 description 1
- JDEQZXQZZBDTSI-UHFFFAOYSA-N CC(CCF)(C#N)N Chemical compound CC(CCF)(C#N)N JDEQZXQZZBDTSI-UHFFFAOYSA-N 0.000 description 1
- HXBFJIFCOBCCLZ-UHFFFAOYSA-N CC(CNC(c1c(C)nc(c(OCc(c(F)ccc2)c2F)c2)[n]1cc2Cl)=O)(CF)N Chemical compound CC(CNC(c1c(C)nc(c(OCc(c(F)ccc2)c2F)c2)[n]1cc2Cl)=O)(CF)N HXBFJIFCOBCCLZ-UHFFFAOYSA-N 0.000 description 1
- KSFOVUSSGSKXFI-GAQDCDSVSA-N CC1=C/2NC(\C=C3/N=C(/C=C4\N\C(=C/C5=N/C(=C\2)/C(C=C)=C5C)C(C=C)=C4C)C(C)=C3CCC(O)=O)=C1CCC(O)=O Chemical compound CC1=C/2NC(\C=C3/N=C(/C=C4\N\C(=C/C5=N/C(=C\2)/C(C=C)=C5C)C(C=C)=C4C)C(C)=C3CCC(O)=O)=C1CCC(O)=O KSFOVUSSGSKXFI-GAQDCDSVSA-N 0.000 description 1
- 201000002829 CREST Syndrome Diseases 0.000 description 1
- 206010007559 Cardiac failure congestive Diseases 0.000 description 1
- 206010007572 Cardiac hypertrophy Diseases 0.000 description 1
- 208000006029 Cardiomegaly Diseases 0.000 description 1
- 206010007637 Cardiomyopathy alcoholic Diseases 0.000 description 1
- 206010008120 Cerebral ischaemia Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 1
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 1
- 229940127328 Cholesterol Synthesis Inhibitors Drugs 0.000 description 1
- 229940123239 Cholesterol synthesis inhibitor Drugs 0.000 description 1
- 102100037637 Cholesteryl ester transfer protein Human genes 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- 208000026151 Chronic thromboembolic pulmonary hypertension Diseases 0.000 description 1
- 208000032544 Cicatrix Diseases 0.000 description 1
- FTKOTFCLDSFHMT-UHFFFAOYSA-N Cl.Cl.CC(N)(CN)CCF Chemical compound Cl.Cl.CC(N)(CN)CCF FTKOTFCLDSFHMT-UHFFFAOYSA-N 0.000 description 1
- 208000028698 Cognitive impairment Diseases 0.000 description 1
- 229920002905 Colesevelam Polymers 0.000 description 1
- 229920002911 Colestipol Polymers 0.000 description 1
- 208000014526 Conduction disease Diseases 0.000 description 1
- 208000002330 Congenital Heart Defects Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 206010056370 Congestive cardiomyopathy Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 208000011990 Corticobasal Degeneration Diseases 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 229920001651 Cyanoacrylate Polymers 0.000 description 1
- 101710095468 Cyclase Proteins 0.000 description 1
- 208000026292 Cystic Kidney disease Diseases 0.000 description 1
- NBSCHQHZLSJFNQ-GASJEMHNSA-N D-Glucose 6-phosphate Chemical compound OC1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H](O)[C@H]1O NBSCHQHZLSJFNQ-GASJEMHNSA-N 0.000 description 1
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- XUIIKFGFIJCVMT-GFCCVEGCSA-N D-thyroxine Chemical compound IC1=CC(C[C@@H](N)C(O)=O)=CC(I)=C1OC1=CC(I)=C(O)C(I)=C1 XUIIKFGFIJCVMT-GFCCVEGCSA-N 0.000 description 1
- GSNUFIFRDBKVIE-UHFFFAOYSA-N DMF Natural products CC1=CC=C(C)O1 GSNUFIFRDBKVIE-UHFFFAOYSA-N 0.000 description 1
- 206010067889 Dementia with Lewy bodies Diseases 0.000 description 1
- 208000016192 Demyelinating disease Diseases 0.000 description 1
- 206010012305 Demyelination Diseases 0.000 description 1
- 101000783577 Dendroaspis angusticeps Thrombostatin Proteins 0.000 description 1
- 101000783578 Dendroaspis jamesoni kaimosae Dendroaspin Proteins 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical class OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 208000007342 Diabetic Nephropathies Diseases 0.000 description 1
- 206010054044 Diabetic microangiopathy Diseases 0.000 description 1
- 206010056340 Diabetic ulcer Diseases 0.000 description 1
- 208000003037 Diastolic Heart Failure Diseases 0.000 description 1
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 1
- 201000010046 Dilated cardiomyopathy Diseases 0.000 description 1
- 208000032928 Dyslipidaemia Diseases 0.000 description 1
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 102000002045 Endothelin Human genes 0.000 description 1
- 108050009340 Endothelin Proteins 0.000 description 1
- YARKMNAWFIMDKV-UHFFFAOYSA-N Epanolol Chemical compound C=1C=CC=C(C#N)C=1OCC(O)CNCCNC(=O)CC1=CC=C(O)C=C1 YARKMNAWFIMDKV-UHFFFAOYSA-N 0.000 description 1
- 208000010228 Erectile Dysfunction Diseases 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 229940123583 Factor Xa inhibitor Drugs 0.000 description 1
- BBLCJHLNBHNRDE-UHFFFAOYSA-N Fc1cccc(F)c1COC1=CC(Cl)=CNC1 Chemical compound Fc1cccc(F)c1COC1=CC(Cl)=CNC1 BBLCJHLNBHNRDE-UHFFFAOYSA-N 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 206010057671 Female sexual dysfunction Diseases 0.000 description 1
- 108010049003 Fibrinogen Proteins 0.000 description 1
- 102000008946 Fibrinogen Human genes 0.000 description 1
- 108090000331 Firefly luciferases Proteins 0.000 description 1
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 206010017533 Fungal infection Diseases 0.000 description 1
- 206010017711 Gangrene Diseases 0.000 description 1
- VFRROHXSMXFLSN-UHFFFAOYSA-N Glc6P Natural products OP(=O)(O)OCC(O)C(O)C(O)C(O)C=O VFRROHXSMXFLSN-UHFFFAOYSA-N 0.000 description 1
- 208000022461 Glomerular disease Diseases 0.000 description 1
- 206010018366 Glomerulonephritis acute Diseases 0.000 description 1
- 108010018962 Glucosephosphate Dehydrogenase Proteins 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000880514 Homo sapiens Cholesteryl ester transfer protein Proteins 0.000 description 1
- 101001057135 Homo sapiens Melanoma-associated antigen H1 Proteins 0.000 description 1
- RKUNBYITZUJHSG-UHFFFAOYSA-N Hyosciamin-hydrochlorid Natural products CN1C(C2)CCC1CC2OC(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-UHFFFAOYSA-N 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 208000002682 Hyperkalemia Diseases 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- 208000029422 Hypernatremia Diseases 0.000 description 1
- 206010021024 Hypolipidaemia Diseases 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- 101710156096 Ileal sodium/bile acid cotransporter Proteins 0.000 description 1
- 206010021639 Incontinence Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 102100025306 Integrin alpha-IIb Human genes 0.000 description 1
- 101710149643 Integrin alpha-IIb Proteins 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- 206010048858 Ischaemic cardiomyopathy Diseases 0.000 description 1
- 208000032382 Ischaemic stroke Diseases 0.000 description 1
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 1
- UETNIIAIRMUTSM-UHFFFAOYSA-N Jacareubin Natural products CC1(C)OC2=CC3Oc4c(O)c(O)ccc4C(=O)C3C(=C2C=C1)O UETNIIAIRMUTSM-UHFFFAOYSA-N 0.000 description 1
- 208000002260 Keloid Diseases 0.000 description 1
- 206010023330 Keloid scar Diseases 0.000 description 1
- 206010023439 Kidney transplant rejection Diseases 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 206010024119 Left ventricular failure Diseases 0.000 description 1
- 201000002832 Lewy body dementia Diseases 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- 229940127470 Lipase Inhibitors Drugs 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- 208000000185 Localized scleroderma Diseases 0.000 description 1
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 239000004425 Makrolon Substances 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102100027256 Melanoma-associated antigen H1 Human genes 0.000 description 1
- 208000026139 Memory disease Diseases 0.000 description 1
- 208000001145 Metabolic Syndrome Diseases 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- 208000020128 Mitral stenosis Diseases 0.000 description 1
- 206010027727 Mitral valve incompetence Diseases 0.000 description 1
- 238000006751 Mitsunobu reaction Methods 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 206010028594 Myocardial fibrosis Diseases 0.000 description 1
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- 108020001621 Natriuretic Peptide Proteins 0.000 description 1
- 102000004571 Natriuretic peptide Human genes 0.000 description 1
- BMRXXBXPVJOAMM-UHFFFAOYSA-N Nc(ncc(Cl)c1)c1O Chemical compound Nc(ncc(Cl)c1)c1O BMRXXBXPVJOAMM-UHFFFAOYSA-N 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 206010029164 Nephrotic syndrome Diseases 0.000 description 1
- 208000007256 Nevus Diseases 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- SNIOPGDIGTZGOP-UHFFFAOYSA-N Nitroglycerin Chemical compound [O-][N+](=O)OCC(O[N+]([O-])=O)CO[N+]([O-])=O SNIOPGDIGTZGOP-UHFFFAOYSA-N 0.000 description 1
- 239000000006 Nitroglycerin Substances 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- LVICICZQETYOGS-UHFFFAOYSA-N OCc(c(F)ccc1)c1F Chemical compound OCc(c(F)ccc1)c1F LVICICZQETYOGS-UHFFFAOYSA-N 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 208000001132 Osteoporosis Diseases 0.000 description 1
- 108010015181 PPAR delta Proteins 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 206010033799 Paralysis Diseases 0.000 description 1
- 208000007542 Paresis Diseases 0.000 description 1
- 208000018737 Parkinson disease Diseases 0.000 description 1
- 208000000450 Pelvic Pain Diseases 0.000 description 1
- 208000018262 Peripheral vascular disease Diseases 0.000 description 1
- 229940080774 Peroxisome proliferator-activated receptor gamma agonist Drugs 0.000 description 1
- 229940123333 Phosphodiesterase 5 inhibitor Drugs 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 101000622060 Photinus pyralis Luciferin 4-monooxygenase Proteins 0.000 description 1
- 206010063985 Phytosterolaemia Diseases 0.000 description 1
- UJEWTUDSLQGTOA-UHFFFAOYSA-N Piretanide Chemical compound C=1C=CC=CC=1OC=1C(S(=O)(=O)N)=CC(C(O)=O)=CC=1N1CCCC1 UJEWTUDSLQGTOA-UHFFFAOYSA-N 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 208000000418 Premature Cardiac Complexes Diseases 0.000 description 1
- 208000002158 Proliferative Vitreoretinopathy Diseases 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Chemical class OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 206010037596 Pyelonephritis Diseases 0.000 description 1
- 208000003782 Raynaud disease Diseases 0.000 description 1
- 206010067171 Regurgitation Diseases 0.000 description 1
- 206010038423 Renal cyst Diseases 0.000 description 1
- 208000033626 Renal failure acute Diseases 0.000 description 1
- 206010061481 Renal injury Diseases 0.000 description 1
- 206010063897 Renal ischaemia Diseases 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- 206010038934 Retinopathy proliferative Diseases 0.000 description 1
- 208000025747 Rheumatic disease Diseases 0.000 description 1
- 206010039163 Right ventricular failure Diseases 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 108091006614 SLC10A2 Proteins 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- 201000001880 Sexual dysfunction Diseases 0.000 description 1
- 208000002227 Sitosterolemia Diseases 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 208000005392 Spasm Diseases 0.000 description 1
- 208000007718 Stable Angina Diseases 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 206010066218 Stress Urinary Incontinence Diseases 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 206010065342 Supraventricular tachyarrhythmia Diseases 0.000 description 1
- 206010051379 Systemic Inflammatory Response Syndrome Diseases 0.000 description 1
- 208000008253 Systolic Heart Failure Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Chemical class [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 101000712605 Theromyzon tessulatum Theromin Proteins 0.000 description 1
- IUJDSEJGGMCXSG-UHFFFAOYSA-N Thiopental Chemical compound CCCC(C)C1(CC)C(=O)NC(=S)NC1=O IUJDSEJGGMCXSG-UHFFFAOYSA-N 0.000 description 1
- 229940122388 Thrombin inhibitor Drugs 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- AUYYCJSJGJYCDS-LBPRGKRZSA-N Thyrolar Chemical compound IC1=CC(C[C@H](N)C(O)=O)=CC(I)=C1OC1=CC=C(O)C(I)=C1 AUYYCJSJGJYCDS-LBPRGKRZSA-N 0.000 description 1
- 208000009205 Tinnitus Diseases 0.000 description 1
- NGBFQHCMQULJNZ-UHFFFAOYSA-N Torsemide Chemical compound CC(C)NC(=O)NS(=O)(=O)C1=CN=CC=C1NC1=CC=CC(C)=C1 NGBFQHCMQULJNZ-UHFFFAOYSA-N 0.000 description 1
- VXFJYXUZANRPDJ-WTNASJBWSA-N Trandopril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@H]2CCCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 VXFJYXUZANRPDJ-WTNASJBWSA-N 0.000 description 1
- 208000030886 Traumatic Brain injury Diseases 0.000 description 1
- FNYLWPVRPXGIIP-UHFFFAOYSA-N Triamterene Chemical compound NC1=NC2=NC(N)=NC(N)=C2N=C1C1=CC=CC=C1 FNYLWPVRPXGIIP-UHFFFAOYSA-N 0.000 description 1
- XSTXAVWGXDQKEL-UHFFFAOYSA-N Trichloroethylene Chemical group ClC=C(Cl)Cl XSTXAVWGXDQKEL-UHFFFAOYSA-N 0.000 description 1
- 201000001943 Tricuspid Valve Insufficiency Diseases 0.000 description 1
- 206010044640 Tricuspid valve incompetence Diseases 0.000 description 1
- 206010044642 Tricuspid valve stenosis Diseases 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 206010060755 Type V hyperlipidaemia Diseases 0.000 description 1
- 208000007814 Unstable Angina Diseases 0.000 description 1
- 208000000921 Urge Urinary Incontinence Diseases 0.000 description 1
- 208000026723 Urinary tract disease Diseases 0.000 description 1
- 208000012931 Urologic disease Diseases 0.000 description 1
- SECKRCOLJRRGGV-UHFFFAOYSA-N Vardenafil Chemical compound CCCC1=NC(C)=C(C(N=2)=O)N1NC=2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(CC)CC1 SECKRCOLJRRGGV-UHFFFAOYSA-N 0.000 description 1
- 201000004810 Vascular dementia Diseases 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- 206010047249 Venous thrombosis Diseases 0.000 description 1
- 208000008131 Ventricular Flutter Diseases 0.000 description 1
- 208000009729 Ventricular Premature Complexes Diseases 0.000 description 1
- 206010047281 Ventricular arrhythmia Diseases 0.000 description 1
- 206010065341 Ventricular tachyarrhythmia Diseases 0.000 description 1
- 201000008485 Wernicke-Korsakoff syndrome Diseases 0.000 description 1
- 201000008803 Wolff-Parkinson-white syndrome Diseases 0.000 description 1
- 206010048215 Xanthomatosis Diseases 0.000 description 1
- VORIUEAZEKLUSJ-UHFFFAOYSA-M [(6-chlorobenzotriazol-1-yl)oxy-(dimethylamino)methylidene]-dimethylazanium;trifluoroborane;fluoride Chemical compound [F-].FB(F)F.C1=C(Cl)C=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 VORIUEAZEKLUSJ-UHFFFAOYSA-M 0.000 description 1
- 229960000446 abciximab Drugs 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 210000000683 abdominal cavity Anatomy 0.000 description 1
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 1
- 210000003815 abdominal wall Anatomy 0.000 description 1
- 208000004622 abetalipoproteinemia Diseases 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 231100000851 acute glomerulonephritis Toxicity 0.000 description 1
- 201000011040 acute kidney failure Diseases 0.000 description 1
- 201000005180 acute myocarditis Diseases 0.000 description 1
- 208000012998 acute renal failure Diseases 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 229950000221 adaprolol Drugs 0.000 description 1
- IPGLIOFIFLXLKR-AXYNENQYSA-N adaprolol Chemical compound C1=CC(OCC(O)CNC(C)C)=CC=C1CC(=O)OCCC1(C2)C[C@@H](C3)C[C@H]2C[C@@H]3C1 IPGLIOFIFLXLKR-AXYNENQYSA-N 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 239000002170 aldosterone antagonist Substances 0.000 description 1
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 1
- 150000008041 alkali metal carbonates Chemical class 0.000 description 1
- 229910000272 alkali metal oxide Inorganic materials 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 208000006682 alpha 1-Antitrypsin Deficiency Diseases 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Chemical class OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229960002213 alprenolol Drugs 0.000 description 1
- PAZJSJFMUHDSTF-UHFFFAOYSA-N alprenolol Chemical compound CC(C)NCC(O)COC1=CC=CC=C1CC=C PAZJSJFMUHDSTF-UHFFFAOYSA-N 0.000 description 1
- 229960002414 ambrisentan Drugs 0.000 description 1
- OUJTZYPIHDYQMC-LJQANCHMSA-N ambrisentan Chemical compound O([C@@H](C(OC)(C=1C=CC=CC=1)C=1C=CC=CC=1)C(O)=O)C1=NC(C)=CC(C)=N1 OUJTZYPIHDYQMC-LJQANCHMSA-N 0.000 description 1
- 238000010640 amide synthesis reaction Methods 0.000 description 1
- 229960002576 amiloride Drugs 0.000 description 1
- XSDQTOBWRPYKKA-UHFFFAOYSA-N amiloride Chemical compound NC(=N)NC(=O)C1=NC(Cl)=C(N)N=C1N XSDQTOBWRPYKKA-UHFFFAOYSA-N 0.000 description 1
- 229960000528 amlodipine Drugs 0.000 description 1
- ZPBWCRDSRKPIDG-UHFFFAOYSA-N amlodipine benzenesulfonate Chemical compound OS(=O)(=O)C1=CC=CC=C1.CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl ZPBWCRDSRKPIDG-UHFFFAOYSA-N 0.000 description 1
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 1
- MZZLGJHLQGUVPN-HAWMADMCSA-N anacetrapib Chemical compound COC1=CC(F)=C(C(C)C)C=C1C1=CC=C(C(F)(F)F)C=C1CN1C(=O)O[C@H](C=2C=C(C=C(C=2)C(F)(F)F)C(F)(F)F)[C@@H]1C MZZLGJHLQGUVPN-HAWMADMCSA-N 0.000 description 1
- 229950000285 anacetrapib Drugs 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 230000000202 analgesic effect Effects 0.000 description 1
- 208000003455 anaphylaxis Diseases 0.000 description 1
- 208000007502 anemia Diseases 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 239000003698 antivitamin K Substances 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 210000002376 aorta thoracic Anatomy 0.000 description 1
- 206010002906 aortic stenosis Diseases 0.000 description 1
- 229960003886 apixaban Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 229940114079 arachidonic acid Drugs 0.000 description 1
- 235000021342 arachidonic acid Nutrition 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 230000004872 arterial blood pressure Effects 0.000 description 1
- 210000002565 arteriole Anatomy 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- 230000001746 atrial effect Effects 0.000 description 1
- RKUNBYITZUJHSG-SPUOUPEWSA-N atropine Chemical compound O([C@H]1C[C@H]2CC[C@@H](C1)N2C)C(=O)C(CO)C1=CC=CC=C1 RKUNBYITZUJHSG-SPUOUPEWSA-N 0.000 description 1
- 229960000396 atropine Drugs 0.000 description 1
- 230000001363 autoimmune Effects 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical class OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- RROBIDXNTUAHFW-UHFFFAOYSA-N benzotriazol-1-yloxy-tris(dimethylamino)phosphanium Chemical compound C1=CC=C2N(O[P+](N(C)C)(N(C)C)N(C)C)N=NC2=C1 RROBIDXNTUAHFW-UHFFFAOYSA-N 0.000 description 1
- 229960004324 betaxolol Drugs 0.000 description 1
- CHDPSNLJFOQTRK-UHFFFAOYSA-N betaxolol hydrochloride Chemical compound [Cl-].C1=CC(OCC(O)C[NH2+]C(C)C)=CC=C1CCOCC1CC1 CHDPSNLJFOQTRK-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 230000036983 biotransformation Effects 0.000 description 1
- 229960002781 bisoprolol Drugs 0.000 description 1
- VHYCDWMUTMEGQY-UHFFFAOYSA-N bisoprolol Chemical compound CC(C)NCC(O)COC1=CC=C(COCCOC(C)C)C=C1 VHYCDWMUTMEGQY-UHFFFAOYSA-N 0.000 description 1
- OIRCOABEOLEUMC-GEJPAHFPSA-N bivalirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)CNC(=O)CNC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 OIRCOABEOLEUMC-GEJPAHFPSA-N 0.000 description 1
- 229960001500 bivalirudin Drugs 0.000 description 1
- 108010055460 bivalirudin Proteins 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 238000009530 blood pressure measurement Methods 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 229960003065 bosentan Drugs 0.000 description 1
- SXTRWVVIEPWAKM-UHFFFAOYSA-N bosentan hydrate Chemical compound O.COC1=CC=CC=C1OC(C(=NC(=N1)C=2N=CC=CN=2)OCCO)=C1NS(=O)(=O)C1=CC=C(C(C)(C)C)C=C1 SXTRWVVIEPWAKM-UHFFFAOYSA-N 0.000 description 1
- 208000006752 brain edema Diseases 0.000 description 1
- 229950005341 bucindolol Drugs 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- 229960004064 bumetanide Drugs 0.000 description 1
- MAEIEVLCKWDQJH-UHFFFAOYSA-N bumetanide Chemical compound CCCCNC1=CC(C(O)=O)=CC(S(N)(=O)=O)=C1OC1=CC=CC=C1 MAEIEVLCKWDQJH-UHFFFAOYSA-N 0.000 description 1
- 229960000330 bupranolol Drugs 0.000 description 1
- HQIRNZOQPUAHHV-UHFFFAOYSA-N bupranolol Chemical compound CC1=CC=C(Cl)C(OCC(O)CNC(C)(C)C)=C1 HQIRNZOQPUAHHV-UHFFFAOYSA-N 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 229960005069 calcium Drugs 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- LLSDKQJKOVVTOJ-UHFFFAOYSA-L calcium chloride dihydrate Chemical compound O.O.[Cl-].[Cl-].[Ca+2] LLSDKQJKOVVTOJ-UHFFFAOYSA-L 0.000 description 1
- 229940052299 calcium chloride dihydrate Drugs 0.000 description 1
- ZJKZKKPIKDNHDM-UHFFFAOYSA-L calcium;6-(5-carboxylato-5-methylhexoxy)-2,2-dimethylhexanoate Chemical compound [Ca+2].[O-]C(=O)C(C)(C)CCCCOCCCCC(C)(C)C([O-])=O ZJKZKKPIKDNHDM-UHFFFAOYSA-L 0.000 description 1
- SGZAIDDFHDDFJU-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SGZAIDDFHDDFJU-UHFFFAOYSA-N 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- 229960002823 canrenoic acid Drugs 0.000 description 1
- PBKZPPIHUVSDNM-WNHSNXHDSA-N canrenoic acid Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@](CC4)(O)CCC(O)=O)[C@@H]4[C@@H]3C=CC2=C1 PBKZPPIHUVSDNM-WNHSNXHDSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 208000020450 carbohydrate metabolism disease Diseases 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- UBAZGMLMVVQSCD-UHFFFAOYSA-N carbon dioxide;molecular oxygen Chemical compound O=O.O=C=O UBAZGMLMVVQSCD-UHFFFAOYSA-N 0.000 description 1
- 229950005499 carbon tetrachloride Drugs 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 206010007625 cardiogenic shock Diseases 0.000 description 1
- PUXBGTOOZJQSKH-UHFFFAOYSA-N carprofen Chemical compound C1=C(Cl)C=C2C3=CC=C(C(C(O)=O)C)C=C3NC2=C1 PUXBGTOOZJQSKH-UHFFFAOYSA-N 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 229960001222 carteolol Drugs 0.000 description 1
- LWAFSWPYPHEXKX-UHFFFAOYSA-N carteolol Chemical compound N1C(=O)CCC2=C1C=CC=C2OCC(O)CNC(C)(C)C LWAFSWPYPHEXKX-UHFFFAOYSA-N 0.000 description 1
- 229960004195 carvedilol Drugs 0.000 description 1
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 230000003727 cerebral blood flow Effects 0.000 description 1
- 208000026106 cerebrovascular disease Diseases 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229960001701 chloroform Drugs 0.000 description 1
- 229960001523 chlortalidone Drugs 0.000 description 1
- 230000001906 cholesterol absorption Effects 0.000 description 1
- 208000020832 chronic kidney disease Diseases 0.000 description 1
- 208000022831 chronic renal failure syndrome Diseases 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 description 1
- 229960003009 clopidogrel Drugs 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- GMRWGQCZJGVHKL-UHFFFAOYSA-N colestipol Chemical compound ClCC1CO1.NCCNCCNCCNCCN GMRWGQCZJGVHKL-UHFFFAOYSA-N 0.000 description 1
- 229960002604 colestipol Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 208000028831 congenital heart disease Diseases 0.000 description 1
- 208000018631 connective tissue disease Diseases 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 238000007887 coronary angioplasty Methods 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 229960000956 coumarin Drugs 0.000 description 1
- 235000001671 coumarin Nutrition 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- 229960003850 dabigatran Drugs 0.000 description 1
- YBSJFWOBGCMAKL-UHFFFAOYSA-N dabigatran Chemical compound N=1C2=CC(C(=O)N(CCC(O)=O)C=3N=CC=CC=3)=CC=C2N(C)C=1CNC1=CC=C(C(N)=N)C=C1 YBSJFWOBGCMAKL-UHFFFAOYSA-N 0.000 description 1
- 229950004181 dalcetrapib Drugs 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 238000013480 data collection Methods 0.000 description 1
- 229960002887 deanol Drugs 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 229960005227 delapril Drugs 0.000 description 1
- WOUOLAUOZXOLJQ-MBSDFSHPSA-N delapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N(CC(O)=O)C1CC2=CC=CC=C2C1)CC1=CC=CC=C1 WOUOLAUOZXOLJQ-MBSDFSHPSA-N 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 229960001767 dextrothyroxine Drugs 0.000 description 1
- 201000009101 diabetic angiopathy Diseases 0.000 description 1
- 208000033679 diabetic kidney disease Diseases 0.000 description 1
- 230000003205 diastolic effect Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- ZWWWLCMDTZFSOO-UHFFFAOYSA-N diethoxyphosphorylformonitrile Chemical compound CCOP(=O)(C#N)OCC ZWWWLCMDTZFSOO-UHFFFAOYSA-N 0.000 description 1
- FPUQGCOBYOXAED-UHFFFAOYSA-N diethyl 2-[[2-[3-(dimethylcarbamoyl)-4-[[2-[4-(trifluoromethyl)phenyl]benzoyl]amino]phenyl]acetyl]oxymethyl]-2-phenylpropanedioate Chemical compound C=1C=CC=CC=1C(C(=O)OCC)(C(=O)OCC)COC(=O)CC(C=C1C(=O)N(C)C)=CC=C1NC(=O)C1=CC=CC=C1C1=CC=C(C(F)(F)F)C=C1 FPUQGCOBYOXAED-UHFFFAOYSA-N 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 229960002768 dipyridamole Drugs 0.000 description 1
- IZEKFCXSFNUWAM-UHFFFAOYSA-N dipyridamole Chemical compound C=12N=C(N(CCO)CCO)N=C(N3CCCCC3)C2=NC(N(CCO)CCO)=NC=1N1CCCCC1 IZEKFCXSFNUWAM-UHFFFAOYSA-N 0.000 description 1
- 230000006806 disease prevention Effects 0.000 description 1
- XRKMNJXYOFSTBE-UHFFFAOYSA-N disodium;iron(4+);nitroxyl anion;pentacyanide;dihydrate Chemical compound O.O.[Na+].[Na+].[Fe+4].N#[C-].N#[C-].N#[C-].N#[C-].N#[C-].O=[N-] XRKMNJXYOFSTBE-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 230000001882 diuretic effect Effects 0.000 description 1
- 229940030606 diuretics Drugs 0.000 description 1
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 229960000622 edoxaban Drugs 0.000 description 1
- PSMMNJNZVZZNOI-SJILXJHISA-N edoxaban tosylate hydrate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1.N([C@H]1CC[C@@H](C[C@H]1NC(=O)C=1SC=2CN(C)CCC=2N=1)C(=O)N(C)C)C(=O)C(=O)NC1=CC=C(Cl)C=N1 PSMMNJNZVZZNOI-SJILXJHISA-N 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- XFLQIRAKKLNXRQ-UUWRZZSWSA-N elobixibat Chemical compound C12=CC(SC)=C(OCC(=O)N[C@@H](C(=O)NCC(O)=O)C=3C=CC=CC=3)C=C2S(=O)(=O)CC(CCCC)(CCCC)CN1C1=CC=CC=C1 XFLQIRAKKLNXRQ-UUWRZZSWSA-N 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- JJJFUHOGVZWXNQ-UHFFFAOYSA-N enbucrilate Chemical compound CCCCOC(=O)C(=C)C#N JJJFUHOGVZWXNQ-UHFFFAOYSA-N 0.000 description 1
- 206010014665 endocarditis Diseases 0.000 description 1
- ZUBDGKVDJUIMQQ-UBFCDGJISA-N endothelin-1 Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(O)=O)NC(=O)[C@H]1NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@@H](CC=2C=CC(O)=CC=2)NC(=O)[C@H](C(C)C)NC(=O)[C@H]2CSSC[C@@H](C(N[C@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N2)=O)NC(=O)[C@@H](CO)NC(=O)[C@H](N)CSSC1)C1=CNC=N1 ZUBDGKVDJUIMQQ-UBFCDGJISA-N 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 238000006911 enzymatic reaction Methods 0.000 description 1
- 229960002711 epanolol Drugs 0.000 description 1
- 229960003745 esmolol Drugs 0.000 description 1
- AQNDDEOPVVGCPG-UHFFFAOYSA-N esmolol Chemical compound COC(=O)CCC1=CC=C(OCC(O)CNC(C)C)C=C1 AQNDDEOPVVGCPG-UHFFFAOYSA-N 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- RDULEYWUGKOCMR-UHFFFAOYSA-N ethyl 2-chloro-3-oxobutanoate Chemical compound CCOC(=O)C(Cl)C(C)=O RDULEYWUGKOCMR-UHFFFAOYSA-N 0.000 description 1
- YYSVZZWIGYOHLH-UHFFFAOYSA-N ethyl 6-chloro-8-[(2,6-difluorophenyl)methoxy]-2-methylimidazo[1,2-a]pyridine-3-carboxylate Chemical compound C=1C(Cl)=CN2C(C(=O)OCC)=C(C)N=C2C=1OCC1=C(F)C=CC=C1F YYSVZZWIGYOHLH-UHFFFAOYSA-N 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000005713 exacerbation Effects 0.000 description 1
- 210000003722 extracellular fluid Anatomy 0.000 description 1
- OLNTVTPDXPETLC-XPWALMASSA-N ezetimibe Chemical compound N1([C@@H]([C@H](C1=O)CC[C@H](O)C=1C=CC(F)=CC=1)C=1C=CC(O)=CC=1)C1=CC=C(F)C=C1 OLNTVTPDXPETLC-XPWALMASSA-N 0.000 description 1
- 229960000815 ezetimibe Drugs 0.000 description 1
- 235000019197 fats Nutrition 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 210000001105 femoral artery Anatomy 0.000 description 1
- 210000003191 femoral vein Anatomy 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 229940012952 fibrinogen Drugs 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 229950010034 fidexaban Drugs 0.000 description 1
- 238000013100 final test Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 239000006260 foam Substances 0.000 description 1
- 229960001318 fondaparinux Drugs 0.000 description 1
- KANJSNBRCNMZMV-ABRZTLGGSA-N fondaparinux Chemical compound O[C@@H]1[C@@H](NS(O)(=O)=O)[C@@H](OC)O[C@H](COS(O)(=O)=O)[C@H]1O[C@H]1[C@H](OS(O)(=O)=O)[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](OS(O)(=O)=O)[C@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O[C@@H]4[C@@H]([C@@H](O)[C@H](O)[C@@H](COS(O)(=O)=O)O4)NS(O)(=O)=O)[C@H](O3)C(O)=O)O)[C@@H](COS(O)(=O)=O)O2)NS(O)(=O)=O)[C@H](C(O)=O)O1 KANJSNBRCNMZMV-ABRZTLGGSA-N 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 230000002431 foraging effect Effects 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 210000001652 frontal lobe Anatomy 0.000 description 1
- 239000001530 fumaric acid Chemical class 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 208000024386 fungal infectious disease Diseases 0.000 description 1
- 229960003883 furosemide Drugs 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 230000001434 glomerular Effects 0.000 description 1
- 206010061989 glomerulosclerosis Diseases 0.000 description 1
- 125000000291 glutamic acid group Chemical group N[C@@H](CCC(O)=O)C(=O)* 0.000 description 1
- 229960003711 glyceryl trinitrate Drugs 0.000 description 1
- 244000144993 groups of animals Species 0.000 description 1
- RQFCJASXJCIDSX-UUOKFMHZSA-N guanosine 5'-monophosphate Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@H]1O RQFCJASXJCIDSX-UUOKFMHZSA-N 0.000 description 1
- 150000005826 halohydrocarbons Chemical class 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- 210000002837 heart atrium Anatomy 0.000 description 1
- 239000001307 helium Substances 0.000 description 1
- 229910052734 helium Inorganic materials 0.000 description 1
- SWQJXJOGLNCZEY-UHFFFAOYSA-N helium atom Chemical compound [He] SWQJXJOGLNCZEY-UHFFFAOYSA-N 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 239000002628 heparin derivative Substances 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 239000000833 heterodimer Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 229960002003 hydrochlorothiazide Drugs 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 201000005991 hyperphosphatemia Diseases 0.000 description 1
- 230000001631 hypertensive effect Effects 0.000 description 1
- 208000006575 hypertriglyceridemia Diseases 0.000 description 1
- 206010020871 hypertrophic cardiomyopathy Diseases 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 208000026621 hypolipoproteinemia Diseases 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- 208000022368 idiopathic cardiomyopathy Diseases 0.000 description 1
- UTCSSFWDNNEEBH-UHFFFAOYSA-N imidazo[1,2-a]pyridine Chemical compound C1=CC=CC2=NC=CN21 UTCSSFWDNNEEBH-UHFFFAOYSA-N 0.000 description 1
- 150000005234 imidazo[1,2-a]pyridines Chemical class 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 229950005809 implitapide Drugs 0.000 description 1
- 201000001881 impotence Diseases 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 208000017745 inborn carbohydrate metabolic disease Diseases 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- NDDAHWYSQHTHNT-UHFFFAOYSA-N indapamide Chemical compound CC1CC2=CC=CC=C2N1NC(=O)C1=CC=C(Cl)C(S(N)(=O)=O)=C1 NDDAHWYSQHTHNT-UHFFFAOYSA-N 0.000 description 1
- 229960004569 indapamide Drugs 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 239000001023 inorganic pigment Substances 0.000 description 1
- 201000004332 intermediate coronary syndrome Diseases 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 210000002977 intracellular fluid Anatomy 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical group II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 208000002551 irritable bowel syndrome Diseases 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 229960002725 isoflurane Drugs 0.000 description 1
- DXDRHHKMWQZJHT-FPYGCLRLSA-N isoliquiritigenin Chemical compound C1=CC(O)=CC=C1\C=C\C(=O)C1=CC=C(O)C=C1O DXDRHHKMWQZJHT-FPYGCLRLSA-N 0.000 description 1
- JBQATDIMBVLPRB-UHFFFAOYSA-N isoliquiritigenin Natural products OC1=CC(O)=CC=C1C1OC2=CC(O)=CC=C2C(=O)C1 JBQATDIMBVLPRB-UHFFFAOYSA-N 0.000 description 1
- 235000008718 isoliquiritigenin Nutrition 0.000 description 1
- MOYKHGMNXAOIAT-JGWLITMVSA-N isosorbide dinitrate Chemical compound [O-][N+](=O)O[C@H]1CO[C@@H]2[C@H](O[N+](=O)[O-])CO[C@@H]21 MOYKHGMNXAOIAT-JGWLITMVSA-N 0.000 description 1
- 229960000201 isosorbide dinitrate Drugs 0.000 description 1
- YWXYYJSYQOXTPL-SLPGGIOYSA-N isosorbide mononitrate Chemical compound [O-][N+](=O)O[C@@H]1CO[C@@H]2[C@@H](O)CO[C@@H]21 YWXYYJSYQOXTPL-SLPGGIOYSA-N 0.000 description 1
- 229960003827 isosorbide mononitrate Drugs 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical class C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 210000004731 jugular vein Anatomy 0.000 description 1
- 210000001117 keloid Anatomy 0.000 description 1
- 229960001632 labetalol Drugs 0.000 description 1
- 239000004310 lactic acid Chemical class 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 208000030175 lameness Diseases 0.000 description 1
- 229950005241 landiolol Drugs 0.000 description 1
- WMDSZGFJQKSLLH-RBBKRZOGSA-N landiolol Chemical compound O1C(C)(C)OC[C@H]1COC(=O)CCC(C=C1)=CC=C1OC[C@@H](O)CNCCNC(=O)N1CCOCC1 WMDSZGFJQKSLLH-RBBKRZOGSA-N 0.000 description 1
- 230000028252 learning or memory Effects 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- CZRQXSDBMCMPNJ-ZUIPZQNBSA-N lisinopril dihydrate Chemical compound O.O.C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 CZRQXSDBMCMPNJ-ZUIPZQNBSA-N 0.000 description 1
- 210000001853 liver microsome Anatomy 0.000 description 1
- 230000006742 locomotor activity Effects 0.000 description 1
- 229960003566 lomitapide Drugs 0.000 description 1
- QKVKOFVWUHNEBX-UHFFFAOYSA-N lomitapide mesylate Chemical compound CS(O)(=O)=O.C12=CC=CC=C2C2=CC=CC=C2C1(C(=O)NCC(F)(F)F)CCCCN(CC1)CCC1NC(=O)C1=CC=CC=C1C1=CC=C(C(F)(F)F)C=C1 QKVKOFVWUHNEBX-UHFFFAOYSA-N 0.000 description 1
- 239000002171 loop diuretic Substances 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- DLBFLQKQABVKGT-UHFFFAOYSA-L lucifer yellow dye Chemical compound [Li+].[Li+].[O-]S(=O)(=O)C1=CC(C(N(C(=O)NN)C2=O)=O)=C3C2=CC(S([O-])(=O)=O)=CC3=C1N DLBFLQKQABVKGT-UHFFFAOYSA-L 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- WRUGWIBCXHJTDG-UHFFFAOYSA-L magnesium sulfate heptahydrate Chemical compound O.O.O.O.O.O.O.[Mg+2].[O-]S([O-])(=O)=O WRUGWIBCXHJTDG-UHFFFAOYSA-L 0.000 description 1
- 229940061634 magnesium sulfate heptahydrate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical class OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Chemical class 0.000 description 1
- 239000001630 malic acid Chemical class 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- DKWNMCUOEDMMIN-PKOBYXMFSA-N melagatran Chemical compound C1=CC(C(=N)N)=CC=C1CNC(=O)[C@H]1N(C(=O)[C@H](NCC(O)=O)C2CCCCC2)CC1 DKWNMCUOEDMMIN-PKOBYXMFSA-N 0.000 description 1
- 229960002137 melagatran Drugs 0.000 description 1
- 229950008446 melinamide Drugs 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000015654 memory Effects 0.000 description 1
- 230000006984 memory degeneration Effects 0.000 description 1
- 206010027175 memory impairment Diseases 0.000 description 1
- 208000023060 memory loss Diseases 0.000 description 1
- 229960003134 mepindolol Drugs 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- YLGXILFCIXHCMC-JHGZEJCSSA-N methyl cellulose Chemical compound COC1C(OC)C(OC)C(COC)O[C@H]1O[C@H]1C(OC)C(OC)C(OC)OC1COC YLGXILFCIXHCMC-JHGZEJCSSA-N 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 229960002237 metoprolol Drugs 0.000 description 1
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 210000001589 microsome Anatomy 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 208000027061 mild cognitive impairment Diseases 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 239000002394 mineralocorticoid antagonist Substances 0.000 description 1
- 230000000116 mitigating effect Effects 0.000 description 1
- 208000006887 mitral valve stenosis Diseases 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- XLFWDASMENKTKL-UHFFFAOYSA-N molsidomine Chemical compound O1C(N=C([O-])OCC)=C[N+](N2CCOCC2)=N1 XLFWDASMENKTKL-UHFFFAOYSA-N 0.000 description 1
- 229960004027 molsidomine Drugs 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 210000000214 mouth Anatomy 0.000 description 1
- 208000029744 multiple organ dysfunction syndrome Diseases 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 206010028537 myelofibrosis Diseases 0.000 description 1
- 208000031225 myocardial ischemia Diseases 0.000 description 1
- BXGTVNLGPMZLAZ-UHFFFAOYSA-N n'-ethylmethanediimine;hydrochloride Chemical compound Cl.CCN=C=N BXGTVNLGPMZLAZ-UHFFFAOYSA-N 0.000 description 1
- 229960004255 nadolol Drugs 0.000 description 1
- VWPOSFSPZNDTMJ-UCWKZMIHSA-N nadolol Chemical compound C1[C@@H](O)[C@@H](O)CC2=C1C=CC=C2OCC(O)CNC(C)(C)C VWPOSFSPZNDTMJ-UCWKZMIHSA-N 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical class C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- 239000007923 nasal drop Substances 0.000 description 1
- 229940100662 nasal drops Drugs 0.000 description 1
- 239000000692 natriuretic peptide Substances 0.000 description 1
- 229920005615 natural polymer Polymers 0.000 description 1
- 229960000619 nebivolol Drugs 0.000 description 1
- 229940105631 nembutal Drugs 0.000 description 1
- 208000021971 neovascular inflammatory vitreoretinopathy Diseases 0.000 description 1
- 201000008383 nephritis Diseases 0.000 description 1
- 230000007372 neural signaling Effects 0.000 description 1
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 229940100688 oral solution Drugs 0.000 description 1
- 229940100692 oral suspension Drugs 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- AHLBNYSZXLDEJQ-FWEHEUNISA-N orlistat Chemical compound CCCCCCCCCCC[C@H](OC(=O)[C@H](CC(C)C)NC=O)C[C@@H]1OC(=O)[C@H]1CCCCCC AHLBNYSZXLDEJQ-FWEHEUNISA-N 0.000 description 1
- 229960001243 orlistat Drugs 0.000 description 1
- PFGVNLZDWRZPJW-OPAMFIHVSA-N otamixaban Chemical compound C([C@@H](C(=O)OC)[C@@H](C)NC(=O)C=1C=CC(=CC=1)C=1C=C[N+]([O-])=CC=1)C1=CC=CC(C(N)=N)=C1 PFGVNLZDWRZPJW-OPAMFIHVSA-N 0.000 description 1
- 229950009478 otamixaban Drugs 0.000 description 1
- 229960004570 oxprenolol Drugs 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 229950003510 pactimibe Drugs 0.000 description 1
- 230000036407 pain Effects 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 230000001991 pathophysiological effect Effects 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 229960002035 penbutolol Drugs 0.000 description 1
- KQXKVJAGOJTNJS-HNNXBMFYSA-N penbutolol Chemical compound CC(C)(C)NC[C@H](O)COC1=CC=CC=C1C1CCCC1 KQXKVJAGOJTNJS-HNNXBMFYSA-N 0.000 description 1
- 229960001412 pentobarbital Drugs 0.000 description 1
- 230000008447 perception Effects 0.000 description 1
- 208000008494 pericarditis Diseases 0.000 description 1
- 229960002582 perindopril Drugs 0.000 description 1
- IPVQLZZIHOAWMC-QXKUPLGCSA-N perindopril Chemical compound C1CCC[C@H]2C[C@@H](C(O)=O)N(C(=O)[C@H](C)N[C@@H](CCC)C(=O)OCC)[C@H]21 IPVQLZZIHOAWMC-QXKUPLGCSA-N 0.000 description 1
- 208000033808 peripheral neuropathy Diseases 0.000 description 1
- 206010034674 peritonitis Diseases 0.000 description 1
- SONNWYBIRXJNDC-VIFPVBQESA-N phenylephrine Chemical compound CNC[C@H](O)C1=CC=CC(O)=C1 SONNWYBIRXJNDC-VIFPVBQESA-N 0.000 description 1
- 229960001802 phenylephrine Drugs 0.000 description 1
- 150000004031 phenylhydrazines Chemical class 0.000 description 1
- 239000002590 phosphodiesterase V inhibitor Substances 0.000 description 1
- PJNZPQUBCPKICU-UHFFFAOYSA-N phosphoric acid;potassium Chemical compound [K].OP(O)(O)=O PJNZPQUBCPKICU-UHFFFAOYSA-N 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 229960002508 pindolol Drugs 0.000 description 1
- JZQKKSLKJUAGIC-UHFFFAOYSA-N pindolol Chemical compound CC(C)NCC(O)COC1=CC=CC2=C1C=CN2 JZQKKSLKJUAGIC-UHFFFAOYSA-N 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 229960001085 piretanide Drugs 0.000 description 1
- 229960002797 pitavastatin Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 1
- 230000010118 platelet activation Effects 0.000 description 1
- 229920000515 polycarbonate Polymers 0.000 description 1
- 208000014321 polymorphic ventricular tachycardia Diseases 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 239000008057 potassium phosphate buffer Substances 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 239000003286 potassium sparing diuretic agent Substances 0.000 description 1
- 229940097241 potassium-sparing diuretic Drugs 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- IENZQIKPVFGBNW-UHFFFAOYSA-N prazosin Chemical compound N=1C(N)=C2C=C(OC)C(OC)=CC2=NC=1N(CC1)CCN1C(=O)C1=CC=CO1 IENZQIKPVFGBNW-UHFFFAOYSA-N 0.000 description 1
- 229960001289 prazosin Drugs 0.000 description 1
- 201000011461 pre-eclampsia Diseases 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 238000003825 pressing Methods 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000006785 proliferative vitreoretinopathy Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 229950003776 protoporphyrin Drugs 0.000 description 1
- 208000005333 pulmonary edema Diseases 0.000 description 1
- 201000010298 pulmonary valve insufficiency Diseases 0.000 description 1
- 208000009138 pulmonary valve stenosis Diseases 0.000 description 1
- 208000030390 pulmonic stenosis Diseases 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 238000011158 quantitative evaluation Methods 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical class O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- XKMLYUALXHKNFT-UHFFFAOYSA-N rGTP Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O XKMLYUALXHKNFT-UHFFFAOYSA-N 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- 229950010535 razaxaban Drugs 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000002336 repolarization Effects 0.000 description 1
- 208000015658 resistant hypertension Diseases 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- 230000033764 rhythmic process Effects 0.000 description 1
- 229940099315 rimadyl Drugs 0.000 description 1
- 229960001148 rivaroxaban Drugs 0.000 description 1
- KGFYHTZWPPHNLQ-AWEZNQCLSA-N rivaroxaban Chemical compound S1C(Cl)=CC=C1C(=O)NC[C@@H]1OC(=O)N(C=2C=CC(=CC=2)N2C(COCC2)=O)C1 KGFYHTZWPPHNLQ-AWEZNQCLSA-N 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229960000672 rosuvastatin Drugs 0.000 description 1
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 1
- 230000037387 scars Effects 0.000 description 1
- 201000000980 schizophrenia Diseases 0.000 description 1
- 238000002098 selective ion monitoring Methods 0.000 description 1
- 230000036303 septic shock Effects 0.000 description 1
- 231100000872 sexual dysfunction Toxicity 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 208000007056 sickle cell anemia Diseases 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 229960002578 sitaxentan Drugs 0.000 description 1
- PHWXUGHIIBDVKD-UHFFFAOYSA-N sitaxentan Chemical compound CC1=NOC(NS(=O)(=O)C2=C(SC=C2)C(=O)CC=2C(=CC=3OCOC=3C=2)C)=C1Cl PHWXUGHIIBDVKD-UHFFFAOYSA-N 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 208000019116 sleep disease Diseases 0.000 description 1
- 239000000779 smoke Substances 0.000 description 1
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 229940083618 sodium nitroprusside Drugs 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 229960002370 sotalol Drugs 0.000 description 1
- ZBMZVLHSJCTVON-UHFFFAOYSA-N sotalol Chemical compound CC(C)NCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 ZBMZVLHSJCTVON-UHFFFAOYSA-N 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 230000007863 steatosis Effects 0.000 description 1
- 231100000240 steatosis hepatitis Toxicity 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- NCEXYHBECQHGNR-QZQOTICOSA-N sulfasalazine Chemical compound C1=C(O)C(C(=O)O)=CC(\N=N\C=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-QZQOTICOSA-N 0.000 description 1
- 229960001940 sulfasalazine Drugs 0.000 description 1
- NCEXYHBECQHGNR-UHFFFAOYSA-N sulfasalazine Natural products C1=C(O)C(C(=O)O)=CC(N=NC=2C=CC(=CC=2)S(=O)(=O)NC=2N=CC=CC=2)=C1 NCEXYHBECQHGNR-UHFFFAOYSA-N 0.000 description 1
- 150000003460 sulfonic acids Chemical class 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 206010042772 syncope Diseases 0.000 description 1
- 229920001059 synthetic polymer Polymers 0.000 description 1
- 229960000835 tadalafil Drugs 0.000 description 1
- IEHKWSGCTWLXFU-IIBYNOLFSA-N tadalafil Chemical compound C1=C2OCOC2=CC([C@@H]2C3=C([C]4C=CC=CC4=N3)C[C@H]3N2C(=O)CN(C3=O)C)=C1 IEHKWSGCTWLXFU-IIBYNOLFSA-N 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 239000011975 tartaric acid Chemical class 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- 238000004154 testing of material Methods 0.000 description 1
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 1
- 230000000542 thalamic effect Effects 0.000 description 1
- 239000003451 thiazide diuretic agent Substances 0.000 description 1
- 229960003279 thiopental Drugs 0.000 description 1
- 239000003868 thrombin inhibitor Substances 0.000 description 1
- 230000002537 thrombolytic effect Effects 0.000 description 1
- PHWBOXQYWZNQIN-UHFFFAOYSA-N ticlopidine Chemical compound ClC1=CC=CC=C1CN1CC(C=CS2)=C2CC1 PHWBOXQYWZNQIN-UHFFFAOYSA-N 0.000 description 1
- 229960005001 ticlopidine Drugs 0.000 description 1
- 229960004605 timolol Drugs 0.000 description 1
- 231100000886 tinnitus Toxicity 0.000 description 1
- COKMIXFXJJXBQG-NRFANRHFSA-N tirofiban Chemical compound C1=CC(C[C@H](NS(=O)(=O)CCCC)C(O)=O)=CC=C1OCCCCC1CCNCC1 COKMIXFXJJXBQG-NRFANRHFSA-N 0.000 description 1
- 229960003425 tirofiban Drugs 0.000 description 1
- 239000003106 tissue adhesive Substances 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 229960005461 torasemide Drugs 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 230000008733 trauma Effects 0.000 description 1
- 238000011277 treatment modality Methods 0.000 description 1
- 125000005270 trialkylamine group Chemical group 0.000 description 1
- 229960001288 triamterene Drugs 0.000 description 1
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 1
- UBOXGVDOUJQMTN-UHFFFAOYSA-N trichloroethylene Natural products ClCC(Cl)Cl UBOXGVDOUJQMTN-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 210000005239 tubule Anatomy 0.000 description 1
- 208000009852 uremia Diseases 0.000 description 1
- 206010046459 urethral obstruction Diseases 0.000 description 1
- 206010046494 urge incontinence Diseases 0.000 description 1
- 210000001635 urinary tract Anatomy 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 229960004699 valsartan Drugs 0.000 description 1
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 1
- 229960002381 vardenafil Drugs 0.000 description 1
- 230000001196 vasorelaxation Effects 0.000 description 1
- 230000002883 vasorelaxation effect Effects 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 208000003663 ventricular fibrillation Diseases 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- 206010047470 viral myocarditis Diseases 0.000 description 1
- 230000009278 visceral effect Effects 0.000 description 1
- 229940019333 vitamin k antagonists Drugs 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 238000011706 wistar kyoto rat Methods 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 230000031143 xenobiotic glucuronidation Effects 0.000 description 1
- ZXIBCJHYVWYIKI-PZJWPPBQSA-N ximelagatran Chemical compound C1([C@@H](NCC(=O)OCC)C(=O)N2[C@@H](CC2)C(=O)NCC=2C=CC(=CC=2)C(\N)=N\O)CCCCC1 ZXIBCJHYVWYIKI-PZJWPPBQSA-N 0.000 description 1
- 229960001522 ximelagatran Drugs 0.000 description 1
- 229960000537 xipamide Drugs 0.000 description 1
- MTZBBNMLMNBNJL-UHFFFAOYSA-N xipamide Chemical compound CC1=CC=CC(C)=C1NC(=O)C1=CC(S(N)(=O)=O)=C(Cl)C=C1O MTZBBNMLMNBNJL-UHFFFAOYSA-N 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Urology & Nephrology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Vascular Medicine (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
本出願は、新規な6−クロロ置換イミダゾ[1,2−a]ピリジン−3−カルボキサミド、その調製法、疾患を治療および/または予防するためのその単独または組み合わせでのその使用、ならびに疾患を治療および/または予防するため、特に心血管障害を治療および/または予防するための医薬を製造するためのその使用に関する。
Description
本出願は、新規な6−クロロ置換イミダゾ[1,2−a]ピリジン−3−カルボキサミド、その調製法、疾患を治療および/または予防するためのその単独または組み合わせでのその使用、ならびに疾患を治療および/または予防するため、特に心血管障害を治療および/または予防するための医薬を製造するためのその使用に関する。
哺乳動物細胞で最も重要な細胞伝達系の1つが環状グアノシン一リン酸(cGMP)である。内皮から放出され、ホルモンおよび機械的シグナルを伝達する一酸化窒素(NO)と合わせて、これはNO/cGMP系を形成する。グアニル酸シクラーゼは、グアノシン三リン酸(GTP)からのcGMPの生合成を触媒する。現在までに知られているこのファミリーの代表的なものは、構造的特徴またはリガンドの型のいずれかによって、ナトリウム利尿ペプチドによって刺激され得る粒子状グアニル酸シクラーゼ、およびNOによって刺激され得る可溶性グアニル酸シクラーゼの2つのグループに分類することができる。可溶性グアニル酸シクラーゼは、2個のサブユニットからなり、調節中心の一部である、ヘテロ二量体1個当たり1個のヘムをおそらく含む。これは活性化機構にとって非常に重要である。NOはヘムの鉄原子に結合し、したがって酵素の活性を著しく増加させることができる。対照的に、無ヘム製剤はNOによって刺激され得ない。一酸化炭素(CO)もヘムの中心鉄原子に結合することができるが、COによる刺激はNOによる刺激よりもずっと小さい。
cGMPの形成ならびに結果として生じるホスホジエステラーゼ、イオンチャネルおよびプロテインキナーゼの調節によって、グアニル酸シクラーゼは種々の生理学的過程、特に平滑筋細胞の弛緩および増殖、血小板凝集および血小板粘着、ならびに神経シグナル伝達、ならびに上記過程の破壊に基づく障害においても重要な役割を果たす。病態生理学的状態では、NO/cGMP系が抑制され、これが例えば、高血圧、血小板活性化、細胞増殖増加、内皮機能不全、粥状動脈硬化、狭心症、心不全、心筋梗塞、血栓症、脳卒中および性機能障害をもたらし得る。
予想される高い効率および低レベルの副作用のために、生物のcGMPシグナル経路の影響を標的化することによるこのような障害の考えられるNO非依存性治療は有望な手法である。
現在まで、可溶性グアニル酸シクラーゼの治療的刺激に、その効果がNOに基づく有機硝酸塩などの化合物がもっぱら使用されてきた。後者は生物変換によって形成され、ヘムの中心鉄原子での攻撃によって溶性グアニル酸シクラーゼを活性化する。副作用に加えて、耐性の発達がこの治療様式の決定的な欠点の1つである。
近年、直接、すなわち、NOの事前放出なしに可溶性グアニル酸シクラーゼを刺激するいくつかの物質、例えば、3−(5’−ヒドロキシメチル−2’−フリル)−1−ベンジルインダゾール[YC−1;Wuら、Blood 84(1994)、4226;Mulschら、Brit.J.Pharmacol.120(1997)、681]、脂肪酸[Goldbergら、J.Biol.Chem.252(1977)、1279]、ジフェニルヨードニウムヘキサフルオロホスフェート[Pettiboneら、Eur.J.Pharmacol.116(1985)、307]、イソリキリチゲニン[Yuら、Brit.J.Pharmacol.114(1995)、1587]および種々の置換ピラゾール誘導体(国際公開第98/16223号パンフレット)が記載されている。
障害を治療するために使用することができる種々のイミダゾ[1,2−a]ピリジン誘導体は、特に、欧州特許第0266890号明細書、国際公開第89/03833号パンフレット、日本国特許第01258674号明細書[Chem.Abstr.112:178986参照]、国際公開第96/34866号パンフレット、欧州特許第1277754号明細書、国際公開第2006/015737号パンフレット、国際公開第2008/008539号パンフレット、国際公開第2008/082490号パンフレット、国際公開第2008/134553号パンフレット、国際公開第2010/030538号パンフレット、国際公開第2011/113606号パンフレットおよび国際公開第2012/165399号パンフレットに記載されている。
Wuら、Blood 84(1994)、4226
Mulschら、Brit.J.Pharmacol.120(1997)、681
Goldbergら、J.Biol.Chem.252(1977)、1279
Pettiboneら、Eur.J.Pharmacol.116(1985)、307
Yuら、Brit.J.Pharmacol.114(1995)、1587
Chem.Abstr.112:178986
可溶性グアニル酸シクラーゼの刺激物質として作用し、よって、疾患の治療および/または予防に適している新規な物質を提供することが本発明の目的であった。
本発明は、
ent−N−(2−アミノ−3−フルオロ−2−メチルプロピル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
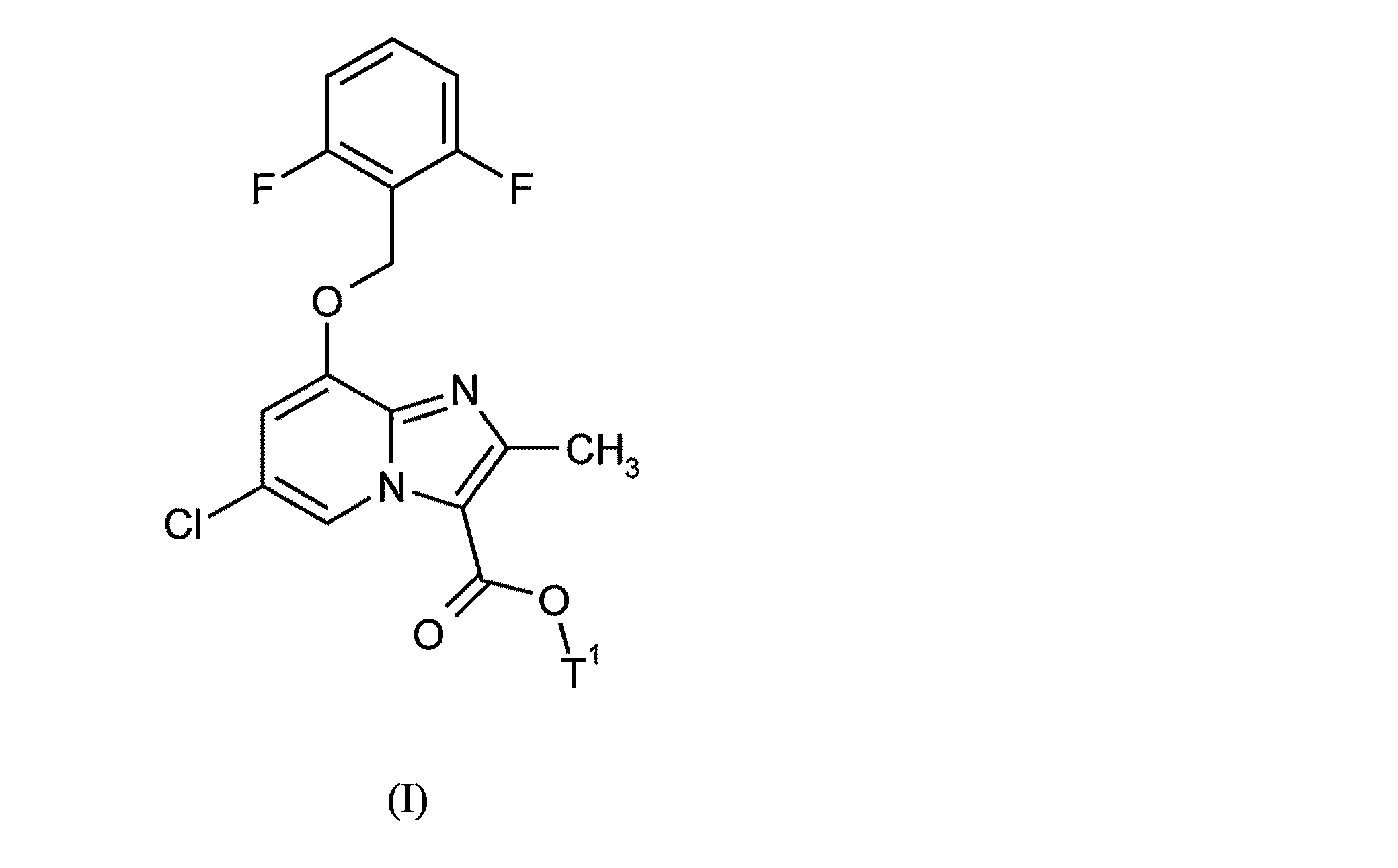
および
ent−N−(2−アミノ−5,5,5−トリフルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
および
ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
および
ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
および
ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
および
ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
およびrac−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エラセミ体)
および
ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
および
ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
ならびにそのN−オキシド、塩、溶媒和物、N−オキシドの塩、ならびにN−オキシドおよび塩の溶媒和物
からなる群から選択される化合物を提供する。
ent−N−(2−アミノ−3−フルオロ−2−メチルプロピル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
ent−N−(2−アミノ−5,5,5−トリフルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
からなる群から選択される化合物を提供する。
本発明の文脈において好ましい塩は、本発明の化合物の生理学的に許容される塩である。それ自体が製薬用途に適していないが、例えば、本発明による化合物を単離または精製するために使用することができる塩も包含される。
本発明の化合物の生理学的に許容される塩には、鉱酸、カルボン酸およびスルホン酸の酸付加塩、例えば、塩酸、臭化水素酸、硫酸、リン酸、メタンスルホン酸、エタンスルホン酸、トルエンスルホン酸、ベンゼンスルホン酸、ナフタレンジスルホン酸、ギ酸、酢酸、トリフルオロ酢酸、プロピオン酸、乳酸、酒石酸、リンゴ酸、クエン酸、フマル酸、マレイン酸および安息香酸の塩が含まれる。
本発明の化合物の生理学的に許容される塩には、従来の塩基の塩、例としておよび好ましくは、アルカリ金属塩(例えば、ナトリウムおよびカリウム塩)、アルカリ土類金属塩(例えば、カルシウムおよびマグネシウム塩)、およびアンモニアまたは1〜16個の炭素原子を有する有機アミンに由来するアンモニウム塩、例としておよび好ましくは、エチルアミン、ジエチルアミン、トリエチルアミン、エチルジイソプロピルアミン、モノエタノールアミン、ジエタノールアミン、トリエタノールアミン、ジシクロヘキシルアミン、ジメチルアミノエタノール、プロカイン、ジベンジルアミン、N−メチルモルホリン、アルギニン、リジン、エチレンジアミンおよびN−メチルピペリジンも含まれる。
本発明の文脈における溶媒和物は、溶媒分子による配位によって固体または液体状態で錯体を形成する本発明の化合物の形態として記載される。水和物は、配位が水によるものである溶媒和物の特別な形態である。水和物が本発明の文脈において好まれる溶媒和物である。
本発明による化合物は、その構造に応じて異なる立体異性型で、すなわち、配置異性体の形態でまたは適当な場合には、配座異性体(アトロプ異性体の場合を含む、エナンチオマーおよび/またはジアステレオマー)として存在し得る。そのため、本発明は、エナンチオマーおよびジアステレオマーならびにこれらのそれぞれの混合物を包含する。立体異性的に均質な成分を、既知の様式でエナンチオマーおよび/またはジアステレオマーのこのような混合物から単離することができ、好ましくはクロマトグラフィー法、特にアキラルまたはキラル相でのHPLCクロマトグラフィーがこの目的のために使用される。
本発明による化合物が互変異性型で生じ得る場合、本発明は全ての互変異性型を包含する。
本発明はまた、本発明による化合物の全ての適当な同位体変種も包含する。本発明による化合物の同位体変種は、ここでは、本発明による化合物中の少なくとも1個の原子が同じ原子番号であるが通常または主に自然に生じる原子質量とは異なる原子質量を有する別の原子と交換された化合物を意味すると理解される。本発明による化合物に組み込まれ得る同位体の例としては、水素、炭素、窒素、酸素、リン、硫黄、フッ素、塩素、臭素およびヨウ素の同位体、例えば、2H(重水素)、3H(トリチウム)、13C、14C、15N、17O、18O、32P、33P、33S、34S、35S、36S、18F、36Cl、82Br、123I、124I、129Iおよび131Iがある。本発明による化合物の特定の同位体変種、特に1種または複数の放射性同位元素が組み込まれたものは、例えば、体内での作用機構または活性化合物分布の調査に有益となり得、比較的容易な調製性(preparability)および検出性のために、特に3Hまたは14Cで標識された化合物がこの目的に適している。さらに、同位体、例えば、重水素の組込みにより、化合物のより大きな代謝安定性の結果としての特定の治療上の利益、例えば、体内での半減期の延長または要求される活性剤用量の減少がもたらされ得るので、本発明による化合物のこのような修飾も、いくつかの場合、本発明の好ましい実施形態を構成し得る。本発明の化合物の同位体変種は、当業者に既知の方法、例えば、以下にさらに記載される方法および実施例に記載の手順によって、それぞれの試薬および/または出発材料の対応する同位体修飾を用いることにより調製することができる。
本発明はさらに、本発明による化合物のプロドラッグも包含する。「プロドラッグ」という用語は、本文脈において、それ自体は生物学的に活性であっても不活性であってもよいが、体内での滞留時間中に(例えば、代謝的にまたは加水分解的に)反応して本発明による化合物を与える化合物を指す。
本発明の文脈において、「治療」または「治療すること」という用語は、疾患、状態、障害、傷害または健康問題の、このような状態および/またはこのような状態の症状の発達、経過または進行の阻害、遅延、検査、軽減、減弱、制限、減少、抑制、忌避または治癒を含む。ここでは、「療法」は「治療」という用語と同義であると理解される。
「防止」、「予防」および「妨害」という用語は本発明の文脈において同義的に使用され、疾患、状態、障害、傷害または健康問題に、あるいはこのような状態および/またはこのような状態の症状の発達または進行に罹患する、を経験する、を患うまたはこれらを有するリスクの回避または減少を指す。
疾患、状態、障害、傷害または健康問題の治療または予防は、部分的であっても完全であってもよい。
本発明の文脈においては、組織名ent−N−(2−アミノ−3−フルオロ−2−メチルプロピル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)および構造式
を有する化合物、ならびにそのN−オキシド、塩、溶媒和物、N−オキシドの塩、ならびにN−オキシドおよび塩の溶媒和物が好まれる。
本発明の文脈においては、組織名ent−N−(2−アミノ−5,5,5−トリフルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)および構造式
を有する化合物、ならびにそのN−オキシド、塩、溶媒和物、N−オキシドの塩、ならびにN−オキシドおよび塩の溶媒和物が好まれる。
本発明の文脈においては、組織名ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)および構造式
を有する化合物、ならびにそのN−オキシド、塩、溶媒和物、N−オキシドの塩、ならびにN−オキシドおよび塩の溶媒和物が好まれる。
本発明の文脈においては、組織名ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)および構造式
を有する化合物、ならびにそのN−オキシド、塩、溶媒和物、N−オキシドの塩、ならびにN−オキシドおよび塩の溶媒和物が好まれる。
本発明の文脈においては、組織名ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)および構造式
を有する化合物、ならびにそのN−オキシド、塩、溶媒和物、N−オキシドの塩、ならびにN−オキシドおよび塩の溶媒和物が好まれる。
本発明の文脈においては、組織名ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)および構造式
を有する化合物、ならびにそのN−オキシド、塩、溶媒和物、N−オキシドの塩、ならびにN−オキシドおよび塩の溶媒和物が好まれる。
本発明の文脈においては、組織名rac−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(ラセミ体)および構造式
を有する化合物、ならびにそのN−オキシド、塩、溶媒和物、N−オキシドの塩、ならびにN−オキシドおよび塩の溶媒和物が好まれる。
本発明の文脈においては、組織名ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)および構造式
を有する化合物、ならびにそのN−オキシド、塩、溶媒和物、N−オキシドの塩、ならびにN−オキシドおよび塩の溶媒和物が好まれる。
本発明の文脈においては、組織名ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)および構造式
を有する化合物、ならびにそのN−オキシド、塩、溶媒和物、N−オキシドの塩、ならびにN−オキシドおよび塩の溶媒和物が好まれる。
本発明はさらに、
[A]式(I)
(式中、
T1は(C1〜C4)−アルキルまたはベンジルを表す)
の化合物を、適当な塩基または酸の存在下、不活性溶媒中で反応させて式(II)
のカルボン酸を得て、
その後、これをアミドカップリング条件下、不活性溶媒中で、
からなる群から選択されるアミンと反応させて、
場合により存在する保護基を、場合により触媒の存在下で、水素化分解的に(hydrogenolytically)除去し、
得られた化合物を場合により適当な(i)溶媒および/または(ii)酸もしくは塩基を用いて、その溶媒和物、塩および/または塩の溶媒和物に変換する
ことを特徴とする、本発明の化合物を調製する方法を提供する。
[A]式(I)
T1は(C1〜C4)−アルキルまたはベンジルを表す)
の化合物を、適当な塩基または酸の存在下、不活性溶媒中で反応させて式(II)
その後、これをアミドカップリング条件下、不活性溶媒中で、
場合により存在する保護基を、場合により触媒の存在下で、水素化分解的に(hydrogenolytically)除去し、
得られた化合物を場合により適当な(i)溶媒および/または(ii)酸もしくは塩基を用いて、その溶媒和物、塩および/または塩の溶媒和物に変換する
ことを特徴とする、本発明の化合物を調製する方法を提供する。
記載される調製法を、以下の合成スキーム(スキーム1)によって例として示すことができる:
スキーム1:
[a):水酸化リチウム、THF/メタノール/H2O、室温;b):HATU、N,N−ジイソプロピルエチルアミン、DMF、室温;c):パラジウム活性炭素(10%)、エタノール、水素、室温]。
スキーム1:
式(III−A)、(III−B)、(III−C)、(III−D)および(III−E)の化合物は商業的に入手可能である、または文献から既知である、または文献の方法と同様に調製することができる。
アミドカップリングに適した不活性溶媒は、例えば、エーテル(ジエチルエーテル、ジオキサン、テトラヒドロフラン、グリコールジメチルエーテルまたはジエチレングリコールジメチルエーテルなど)、炭化水素(ベンゼン、トルエン、キシレン、ヘキサン、シクロヘキサンまたは鉱油留分など)、ハロ炭化水素(ジクロロメタン、トリクロロメタン、テトラクロロメタン、1,2−ジクロロエタン、トリクロロエチレンまたはクロロベンゼンなど)、または他の溶媒(アセトン、酢酸エチル、アセトニトリル、ピリジン、ジメチルスルホキシド、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N,N’−ジメチルプロピレン尿素(DMPU)またはN−メチルピロリドン(NMP)など)である。言及する溶媒の混合物を使用することも同様に可能である。ジクロロメタン、テトラヒドロフラン、ジメチルホルムアミドまたはこれらの溶媒の混合物が好まれる。
アミド形成に縮合剤として使用するのに適しているのは、例えば、場合により1−ヒドロキシベンゾトリアゾール(HOBt)またはN−ヒドロキシスクシンイミド(HOSu)などのさらなる補助剤、および塩基としてのアルカリ金属炭酸塩、例えば、炭酸ナトリウムもしくは炭酸カリウムもしくは重炭酸ナトリウムもしくは重炭酸カリウム、またはトリアルキルアミン、例えば、トリエチルアミン、N−メチルモルホリン、N−メチルピペリジンもしくはN,N−ジイソプロピルエチルアミンなどの有機塩基とも組み合わせた、カルボジイミド(N,N’−ジエチル−、N,N’−ジプロピル−、N,N’−ジイソプロピル−、N,N’−ジシクロヘキシルカルボジイミド(DCC)またはN−(3−ジメチルアミノプロピル)−N’−エチルカルボジイミド塩酸塩(EDC)など)、ホスゲン誘導体(N,N’−カルボニルジイミダゾール(CDI)など)、1,2−オキサゾリウム化合物(2−エチル−5−フェニル−1,2−オキサゾリウム3−硫酸塩または2−tert−ブチル−5−メチルイソオキサゾリウム過塩素酸塩など)、アシルアミノ化合物(2−エトキシ−1−エトキシカルボニル−1,2−ジヒドロキノリンまたはイソブチルクロロホルメートなど)、プロパンホスホン酸無水物(T3P)、1−クロロ−N,N,2−トリメチルプロパ−1−エン−1−アミン、ジエチルシアノホスホネート、ビス(2−オキソ−3−オキサゾリジニル)ホスホリルクロリド、ベンゾトリアゾール−1−イルオキシトリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスフェート、
ベンゾトリアゾール−1−イルオキシトリス(ピロリジノ)ホスホニウムヘキサフルオロホスフェート(PyBOP)、O−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボレート(TBTU)、O−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)、2−(2−オキソ−1−(2H)−ピリジル)−1,1,3,3−テトラメチルウロニウムテトラフルオロボレート(TPTU)、O−(7−アザベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HATU)またはO−(1H−6−クロロベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムテトラフルオロボレート(TCTU)である。N−メチルモルホリンと組み合わせたTBTU、N,N−ジイソプロピルエチルアミンまたは1−クロロ−N,N,2−トリメチルプロパ−1−エン−1−アミンと組み合わせたHATUを用いることが好まれる。
ベンゾトリアゾール−1−イルオキシトリス(ピロリジノ)ホスホニウムヘキサフルオロホスフェート(PyBOP)、O−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボレート(TBTU)、O−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HBTU)、2−(2−オキソ−1−(2H)−ピリジル)−1,1,3,3−テトラメチルウロニウムテトラフルオロボレート(TPTU)、O−(7−アザベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスフェート(HATU)またはO−(1H−6−クロロベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムテトラフルオロボレート(TCTU)である。N−メチルモルホリンと組み合わせたTBTU、N,N−ジイソプロピルエチルアミンまたは1−クロロ−N,N,2−トリメチルプロパ−1−エン−1−アミンと組み合わせたHATUを用いることが好まれる。
縮合は、一般的に−20℃〜+100℃の温度範囲で、好ましくは0℃〜+60℃で行う。変換は、大気圧、高圧または減圧(例えば、0.5〜5bar)で行うことができる。一般に、反応は大気圧で行う。
あるいは、式(II)のカルボン酸を最初に対応する塩化カルボニルに変換することもでき、次いで、これを直接または式(III)のアミンとの別の反応で本発明の化合物に変換することができる。カルボン酸からの塩化カルボニルの形成は、当業者に既知の方法、例えば、適当な塩基の存在下、例えば、ピリジンの存在下、塩化チオニル、塩化スルフリルまたは塩化オキサリルによる処理によって、また場合により適当な不活性溶媒中、ジメチルホルムアミドの添加によって行う。
式(I)の化合物のエステル基T1の加水分解は、不活性溶媒中、エステルを酸または塩基で処理することによって慣用的方法により行い、後者の場合、最初に形成する塩が酸で処理することによって遊離カルボン酸に変換される。tert−ブチルエステルの場合、エステル加水分解を、好ましくは酸を用いて行う。ベンジルエステルの場合、エステル開裂を、好ましくはパラジウム活性炭素またはラネーニッケルを用いて水素化分解によって行う。この反応に適した不活性溶媒は、水またはエステル加水分解のための慣用的な有機溶媒である。これらには、好ましくはアルコール(メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノールまたはtert−ブタノールなど)、またはエーテル(ジエチルエーテル、テトラヒドロフラン、2−メチルテトラヒドロフラン、ジオキサンまたはグリコールジメチルエーテルなど)、または他の溶媒(アセトン、ジクロロメタン、ジメチルホルムアミドまたはジメチルスルホキシドなど)が含まれる。言及する溶媒の混合物を使用することも可能である。塩基性エステル加水分解の場合、水とジオキサン、テトラヒドロフラン、メタノールおよび/またはエタノールの混合物を用いることが好まれる。
エステル加水分解に適した塩基は慣用的無機塩基である。これらには、好ましくはアルカリ金属もしくはアルカリ土類金属水酸化物、例えば、水酸化ナトリウム、水酸化リチウム、水酸化カリウムもしくは水酸化バリウム、またはアルカリ金属もしくはアルカリ土類金属炭酸塩、例えば、炭酸ナトリウム、炭酸カリウムもしくは炭酸カルシウムが含まれる。水酸化ナトリウムまたは水酸化リチウムが特に好まれる。
エステル開裂に適した酸は、一般的に、場合により水を添加した、硫酸、塩化水素/塩酸、臭化水素/臭化水素酸、リン酸、酢酸、トリフルオロ酢酸、トルエンスルホン酸、メタンスルホン酸もしくはトリフルオロメタンスルホン酸またはこれらの混合物である。tert−ブチルエステルの場合には塩化水素またはトリフルオロ酢酸、またメチルエステルの場合には塩酸が好まれる。
エステル加水分解は、一般的に0℃〜+100℃の温度範囲内で、好ましくは+0℃〜+50℃で行う。
これらの変換は、大気圧、高圧または減圧(例えば、0.5〜5bar)で行うことができる。一般に、反応は各場合において大気圧で行う。
使用するアミノ保護基は、好ましくはtert−ブトキシカルボニル(Boc)またはベンジルオキシカルボニル(Z)である。ヒドロキシまたはカルボキシル官能基のために使用する保護基は、好ましくはtert−ブチルまたはベンジルである。これらの保護基は、慣用的方法によって、好ましくは、ジオキサン、ジエチルエーテル、ジクロロメタンまたは酢酸などの不活性溶媒中、塩化水素、臭化水素またはトリフルオロ酢酸などの強酸との反応によって脱離し、追加の不活性溶媒を用いることなく脱離を行うことも場合により可能である。保護基としてのベンジルおよびベンジルオキシカルボニルの場合、これらをパラジウム触媒の存在下、水素化分解によって除去してもよい。言及する保護基の脱離を、場合によりワンポット反応で同時にまたは別々の反応ステップで行うことができる。
ベンジル基の除去を、ここでは、保護基の化学から既知の慣用的方法によって、好ましくは、不活性溶媒、例えば、エタノールまたは酢酸エチル中、パラジウム触媒、例えば、パラジウム活性炭素の存在下での水素化分解によって行う[例えば、T.W.GreeneおよびP.G.M.Wuts、Protective Groups in Organic Synthesis、Wiley、ニューヨーク、1999も参照]。
式(I)の化合物は文献から既知である、または式(IV)
の化合物を、適当な塩基の存在下、不活性溶媒中で式(V)
(式中、
X1は適当な脱離基、特に塩素、臭素、ヨウ素、メシレート、トリフレートまたはトシレートを表す)
の化合物と反応させて、式(VI)
の化合物を得て、
次いで、これを不活性溶媒中で式(VII)
(式中、T1は上に示される意味を有する)
の化合物と反応させることによって調製することができる。
X1は適当な脱離基、特に塩素、臭素、ヨウ素、メシレート、トリフレートまたはトシレートを表す)
の化合物と反応させて、式(VI)
次いで、これを不活性溶媒中で式(VII)
の化合物と反応させることによって調製することができる。
記載される方法は、以下のスキーム(スキーム2)によって代表的な様式で示される:
スキーム2:
[a):i)NaOMe、MeOH、室温;ii)DMSO、室温;b):EtOH、モレキュラーシーブ、還流]。
スキーム2:
示される合成順序は、それぞれの反応ステップが異なる順で行われるように修正することができる。このような修正合成順序の例がスキーム3に示される。
スキーム3:
[a):EtOH、モレキュラーシーブ、還流;b):b)Cs2CO3、DMF、50℃]。
イミダゾ[1,2−a]ピリジン基本骨格を与える閉環(VI)+(VII)→(I)または(IV)+(VII)→(VIII)のための不活性溶媒は慣用的な有機溶媒である。これらには、好ましくはアルコール(メタノール、エタノール、n−プロパノール、イソプロパノール、n−ブタノール、n−ペンタノールまたはtert−ブタノールなど)、またはエーテル(ジエチルエーテル、テトラヒドロフラン、2−メチルテトラヒドロフラン、ジオキサンまたはグリコールジメチルエーテルなど)、または他の溶媒(アセトン、ジクロロメタン、1,2−ジクロロエタン、アセトニトリル、ジメチルホルムアミドまたはジメチルスルホキシドなど)が含まれる。言及する溶媒の混合物を使用することも可能である。エタノールを用いることが好まれる。
閉環は、場合によりマイクロ波中、一般的に+50℃〜+150℃の温度範囲内で、好ましくは+50℃〜+100℃で行う。
閉環(VI)+(VII)→(I)または(IV)+(VII)→(VIII)は、場合により脱水反応添加剤の存在下、例えば、モレキュラーシーブ(孔径4Å)の存在下で、または水分離器を用いて行う。反応(VI)+(VII)→(I)または(IV)+(VII)→(VIII)は、場合により塩基(例えば、重炭酸ナトリウム)を添加した(この場合、この試薬の添加を一度にまたは数回に分けて行うことができる)、過剰の式(VII)の試薬、例えば、1〜20当量の試薬(VII)を用いて行う。
スキーム1〜3に示される2,6−ジフルオロベンジル基の導入の代替として、スキーム4に示されるように、これらの中間体を光延反応の条件下で式(IX)のアルコールと反応させることも可能である。
スキーム4:
(R1は式(III−A)、(III−B)、(III−C)、(III−D)および(III−E)の化合物を表し、
T1は上に示される意味を有する)。
T1は上に示される意味を有する)。
フェノールとアルコールのこのような光延縮合のための典型的な反応条件は、関連文献、例えば、Hughes、D.L.Org.React.1992、42、335;Dembinski、R.Eur.J.Org.Chem.2004、2763に見出され得る。典型的には、反応を、0℃〜使用する溶媒の沸点の間の温度で、不活性溶媒、例えば、THF、ジクロロメタン、トルエンまたはDMF中、活性化剤、例えば、ジエチルアゾジカルボキシレート(DEAD)またはジイソプロピルアゾジカルボキシレート(DIAD)、およびホスフィン試薬、例えば、トリフェニルホスフィンまたはトリブチルホスフィンを用いて行う。
本発明の化合物は、有用な薬理学的特性を有し、ヒトおよび動物の疾患を予防および治療するために使用することができる。本発明の化合物はさらなる治療代替を提供するので、薬学の分野を拡大する。
本発明の化合物は血管弛緩および血小板凝集の阻害を引き起こし、血圧の低下および冠血流量の上昇をもたらす。これらの効果は、可溶性グアニル酸シクラーゼの直接刺激およびcGMPの細胞内増加によって媒介される。さらに、本発明の化合物は、cGMPレベルを上昇させる物質、例えば、EDRF(内皮由来弛緩因子)、NOドナー、プロトポルフィリンIX、アラキドン酸またはフェニルヒドラジン誘導体の作用を強化する。
本発明の化合物は、心血管、肺、血栓塞栓および線維障害の治療および/または予防に適している。
したがって、本発明による化合物を、心血管障害、例えば、高血圧症(高血圧)、抵抗性高血圧、急性および慢性心不全、冠動脈心疾患、安定および不安定狭心症、末梢および心臓血管障害、不整脈、心房性および心室性不整脈ならびに伝導障害、例えば、I〜III度の房室ブロック(ABブロックI〜III)、上室性頻脈性不整脈、心房細動、心房粗動、心室細動、
心室粗動、心室性頻脈性不整脈、多形性心室頻拍、心房性および心室性期外収縮、房室接合部期外収縮、洞不全症候群、失神、房室結節リエントリー性頻拍、ウォルフ・パーキンソン・ホワイト症候群、急性冠動脈症候群(ACS)、自己免疫心疾患(心膜炎、心内膜炎、弁膜炎、大動脈炎、心筋症)、ショック、例えば、心原性ショック、敗血症性ショックおよびアナフィラキシーショック、動脈瘤、ボクサー心筋症(心室性期外収縮(PVC))を治療および/または予防するための、血栓塞栓障害および虚血、例えば、心筋虚血、
心筋梗塞、卒中、心肥大、一過性および虚血性発作、子癇前症、炎症性心血管障害、冠動脈および末梢動脈の攣縮、浮腫形成、例えば、肺浮腫、脳浮腫、腎性浮腫または心不全によって引き起こされる浮腫、末梢循環障害、再灌流損傷、動脈および静脈血栓症、微量アルブミン尿、心筋不全、内皮機能不全を治療および/または予防するための、例えば、血栓溶解療法、経皮的血管形成術(PTA)、経管的冠動脈形成術(PTCA)、心臓移植およびバイパス手術後の再狭窄、ならびに微小および大血管損傷(血管炎)、フィブリノーゲンおよび低密度リポタンパク質(LDL)のレベル上昇ならびにプラスミノーゲン活性化因子阻害因子1(PAI−1)の濃度上昇を予防するための、ならびに勃起不全および女性性機能不全を治療および/または予防するための医薬に使用することができる。
心室粗動、心室性頻脈性不整脈、多形性心室頻拍、心房性および心室性期外収縮、房室接合部期外収縮、洞不全症候群、失神、房室結節リエントリー性頻拍、ウォルフ・パーキンソン・ホワイト症候群、急性冠動脈症候群(ACS)、自己免疫心疾患(心膜炎、心内膜炎、弁膜炎、大動脈炎、心筋症)、ショック、例えば、心原性ショック、敗血症性ショックおよびアナフィラキシーショック、動脈瘤、ボクサー心筋症(心室性期外収縮(PVC))を治療および/または予防するための、血栓塞栓障害および虚血、例えば、心筋虚血、
心筋梗塞、卒中、心肥大、一過性および虚血性発作、子癇前症、炎症性心血管障害、冠動脈および末梢動脈の攣縮、浮腫形成、例えば、肺浮腫、脳浮腫、腎性浮腫または心不全によって引き起こされる浮腫、末梢循環障害、再灌流損傷、動脈および静脈血栓症、微量アルブミン尿、心筋不全、内皮機能不全を治療および/または予防するための、例えば、血栓溶解療法、経皮的血管形成術(PTA)、経管的冠動脈形成術(PTCA)、心臓移植およびバイパス手術後の再狭窄、ならびに微小および大血管損傷(血管炎)、フィブリノーゲンおよび低密度リポタンパク質(LDL)のレベル上昇ならびにプラスミノーゲン活性化因子阻害因子1(PAI−1)の濃度上昇を予防するための、ならびに勃起不全および女性性機能不全を治療および/または予防するための医薬に使用することができる。
本発明の文脈において、「心不全」という用語は、心不全の急性型と慢性型の両方、およびより具体的なまたは関連する型の疾患、例えば、急性非代償性心不全、右心不全、左心不全、全心不全、虚血性心筋症、拡張型心筋症、肥大型心筋症、特発性心筋症、先天性心疾患、心臓弁奇形を伴う心不全、僧帽弁狭窄、僧帽弁閉鎖不全、大動脈弁狭窄、大動脈弁閉鎖不全、三尖弁狭窄、三尖弁閉鎖不全、肺動脈弁狭窄、肺動脈弁閉鎖不全、混合型心臓弁奇形、心筋炎症(心筋炎)、慢性心筋炎、急性心筋炎、ウイルス性心筋炎、糖尿病性心不全、アルコール性心筋症、心臓貯蔵障害、拡張期心不全および収縮期心不全、ならびに既存の慢性心不全の悪化の急性期(心不全憎悪)を包含する。
さらに、本発明の化合物を、動脈硬化、脂質代謝障害、低リポ蛋白血症、脂質異常症、高トリグリセリド血症、高脂血症、高コレステロール血症、無βリポ蛋白血症、シトステロール血症、黄色腫症、タンジール病、脂肪症、肥満ならびに混合型高脂血症およびメタボリックシンドロームを治療および/または予防するために使用することもできる。
本発明の化合物を、一次および二次レイノー現象、微小循環障害、跛行、末梢および自律神経ニューロパチー、糖尿病性細小血管症、糖尿病性網膜症、四肢の糖尿病性潰瘍、壊疽、CREST症候群、エリテマトーデス、爪真菌症、リウマチ性障害を治療および/または予防するためならびに創傷治癒を促進するために使用することもできる。
本発明による化合物は、泌尿器科障害、例えば、良性前立腺症候群(BPS)、良性前立腺肥大(BPH)、良性前立腺腫脹(BPE)、膀胱下尿道閉塞(BOO)、下部尿路症候群(LUTS、ネコ泌尿器科症候群(FUS)を含む)、神経性過活動膀胱(OAB)および(IC)、失禁(UI)、例えば、混合型尿失禁、切迫性尿失禁、ストレス性尿失禁または溢流性尿失禁(MUI、UUI、SUI、OUI)、骨盤痛を含む泌尿生殖器系の障害、男性および女性泌尿生殖器系の器官の良性および悪性障害を治療するのにさらに適している。
本発明の化合物は、腎障害、特に、急性および慢性腎機能不全、ならびに急性および慢性腎不全を治療および/または予防するのにも適している。本発明の文脈において、「腎機能不全」という用語は、腎機能不全の急性症状と慢性症状の両方、ならびに根底にあるまたは関連する腎障害、例えば、腎低灌流、透析時低血圧、閉塞性尿路疾患、糸球体症、糸球体腎炎、急性糸球体腎炎、糸球体硬化、尿細管間質障害、腎症障害、例えば、原発性および先天性腎疾患、腎炎、免疫学的腎障害、例えば、腎移植拒絶反応および免疫複合体誘発性腎障害、
毒性物質によって誘発される腎症、造影剤によって誘発される腎症、糖尿病性および非糖尿病性腎症、腎盂腎炎、腎嚢胞、腎硬化症、高血圧性腎硬化症およびネフローゼ症候群(例えば、異常に低下したクレアチニンおよび/または水分排泄、異常に上昇した尿素、窒素、カリウムおよび/またはクレアチニンの血中濃度、例えば、グルタミルシンテターゼなどの腎臓酵素の変化した活性、変化した尿浸透圧または尿量、上昇した微量アルブミン尿、マクロアルブミン尿、糸球体および細動脈の病変、尿細管拡張、高リン酸塩血症ならびに/あるいは透析の必要性によって診断上特徴付けされ得る)を包含する。本発明はまた、腎機能不全、例えば、肺水腫、心不全、尿毒症、貧血、電解質障害(例えば、高カリウム血症、高ナトリウム血症)ならびに骨および炭水化物代謝の障害を治療および/または予防するための本発明の化合物の使用も包含する。
毒性物質によって誘発される腎症、造影剤によって誘発される腎症、糖尿病性および非糖尿病性腎症、腎盂腎炎、腎嚢胞、腎硬化症、高血圧性腎硬化症およびネフローゼ症候群(例えば、異常に低下したクレアチニンおよび/または水分排泄、異常に上昇した尿素、窒素、カリウムおよび/またはクレアチニンの血中濃度、例えば、グルタミルシンテターゼなどの腎臓酵素の変化した活性、変化した尿浸透圧または尿量、上昇した微量アルブミン尿、マクロアルブミン尿、糸球体および細動脈の病変、尿細管拡張、高リン酸塩血症ならびに/あるいは透析の必要性によって診断上特徴付けされ得る)を包含する。本発明はまた、腎機能不全、例えば、肺水腫、心不全、尿毒症、貧血、電解質障害(例えば、高カリウム血症、高ナトリウム血症)ならびに骨および炭水化物代謝の障害を治療および/または予防するための本発明の化合物の使用も包含する。
さらに、本発明の化合物は、喘息性障害、肺動脈高血圧(PAH)および他の型の肺高血圧(PH)(左心臓疾患、HIV、鎌状赤血球貧血、血栓塞栓症(CTEPH)、サルコイドーシス、COPDまたは肺線維症に関連する肺高血圧を含む)、慢性閉塞性肺疾患(COPD)、急性呼吸窮迫症候群(ARDS)、急性肺傷害(ALI)、α1−抗トリプシン欠乏症(AATD)、肺線維症、肺気腫(例えば、タバコの煙によって誘発される肺気腫)および嚢胞性線維症(CF)を治療および/または予防するのにも適している。
本発明に記載される化合物はまた、NO/cGMP系の障害によって特徴付けられる中枢神経系障害を制御するための有効成分でもある。これらは、特に、例えば、軽度認知障害、加齢性学習および記憶障害、加齢性記憶喪失、血管性認知症、頭蓋脳損傷、脳卒中、脳卒中後に起こる認知症(脳卒中後認知症)、外傷後頭蓋脳損傷、一般的な集中力障害、学習および記憶の問題を有する小児の集中力障害、アルツハイマー病、
レビー小体型認知症、ピック症候群を含む前頭葉変性を伴う認知症、パーキンソン病、進行性核性麻痺、大脳皮質基底核変性症を伴う認知症、筋萎縮性側索硬化症(ALS)、ハンチントン舞踏病、脱髄、多発性硬化症、視床変性、クロイツフェルト−ヤコブ認知症、HIV認知症、認知症を伴う統合失調症、またはコルサコフ精神病などの状態/疾患/症候群に関連して特に生じるものなどの認知障害後の知覚、集中力、学習または記憶を改善するのに適している。これらはまた、中枢神経系障害、例えば、不安、緊張およびうつ病、CNS関連性機能障害および睡眠障害の状態を治療および/または予防する、ならびに食物、嗜好品および習慣性物質の摂取の病理学的障害を制御するのにも適している。
レビー小体型認知症、ピック症候群を含む前頭葉変性を伴う認知症、パーキンソン病、進行性核性麻痺、大脳皮質基底核変性症を伴う認知症、筋萎縮性側索硬化症(ALS)、ハンチントン舞踏病、脱髄、多発性硬化症、視床変性、クロイツフェルト−ヤコブ認知症、HIV認知症、認知症を伴う統合失調症、またはコルサコフ精神病などの状態/疾患/症候群に関連して特に生じるものなどの認知障害後の知覚、集中力、学習または記憶を改善するのに適している。これらはまた、中枢神経系障害、例えば、不安、緊張およびうつ病、CNS関連性機能障害および睡眠障害の状態を治療および/または予防する、ならびに食物、嗜好品および習慣性物質の摂取の病理学的障害を制御するのにも適している。
さらに、本発明の化合物はまた、脳血流を制御するのにも適しており、偏頭痛を制御するのに有効な薬剤となる。これらはまた、脳梗塞(脳卒中)、例えば、卒中、脳虚血および頭蓋−脳外傷の続発症を予防および制御するのにも適している。本発明による化合物を疼痛の状態および耳鳴を制御するために使用することもできる。
さらに、本発明の化合物は、抗炎症作用を有するので、敗血症(SIRS)、多臓器不全(MODS、MOF)、腎臓の炎症性障害、慢性腸炎症(IBD、クローン病、UC)、膵炎、腹膜炎、リウマチ様障害、炎症性皮膚障害および炎症性眼障害を治療および/または予防するための抗炎症剤として使用することができる。
さらに、本発明の化合物を自己免疫疾患を治療および/または予防するために使用することもできる。
本発明の化合物は、内臓、例えば、肺、心臓、腎臓、骨髄および特に、肝臓の線維性障害、ならびに皮膚科学的線維症および線維性眼障害を治療および/または予防するのにも適している。本発明の文脈において、線維性障害という用語は、特に、以下の用語を含む:肝線維症、肝硬変、肺線維症、心内膜心筋線維症、腎症、糸球体腎炎、間質性腎線維症、糖尿病から生じる線維性損傷、骨髄線維症および同様の線維性障害、強皮症、限局性強皮症、ケロイド、肥厚性瘢痕(外科手技後も)、母斑、糖尿病網膜症、増殖性硝子体網膜症および結合組織の障害(例えば、サルコイドーシス)。
本発明の化合物は、例えば、緑内障手術の結果としての術後瘢痕を制御するのにも適している。
本発明の化合物を老化および角質化皮膚のために美容的に使用することもできる。
さらに、本発明による化合物は、肝炎、新生物、骨粗鬆症、緑内障および胃不全麻痺を治療および/または予防するのに適している。
本発明はさらに、障害、特に上記障害を治療および/または予防するための本発明による化合物の使用を提供する。
本発明はさらに、心不全、狭心症、高血圧、肺高血圧、虚血、血管障害、腎機能不全、血栓塞栓性障害、線維性障害および動脈硬化を治療および/または予防するための本発明の化合物の使用を提供する。
本発明はさらに、心不全、狭心症、高血圧、肺高血圧、虚血、血管障害、腎機能不全、血栓塞栓性障害、線維性障害および動脈硬化を治療および/または予防する方法に使用するための本発明の化合物を提供する。
本発明はさらに、障害、特に上記障害を治療および/または予防するための医薬を製造するための本発明の化合物の使用を提供する。
本発明はさらに、心不全、狭心症、高血圧、肺高血圧、虚血、血管障害、腎機能不全、血栓塞栓性障害、線維性障害および動脈硬化を治療および/または予防するための医薬を製造するための本発明の化合物の使用を提供する。
本発明はさらに、有効量の本発明の化合物の少なくとも1種を使用して、障害、特に上記障害を治療および/または予防する方法を提供する。
本発明はさらに、有効量の本発明の化合物の少なくとも1種を使用して心不全、狭心症、高血圧、肺高血圧、虚血、血管障害、腎機能不全、血栓塞栓性障害、線維性障害および動脈硬化を治療および/または予防する方法を提供する。
本発明による化合物は、単独で、または必要に応じて他の活性化合物と組み合わせて使用することができる。本発明はさらに、特に上記障害を治療および/または予防するための、本発明の化合物の少なくとも1種と、1種または複数のさらなる活性化合物とを含む医薬を提供する。組み合わせに適した有効成分の好ましい例には以下が含まれる:
・有機硝酸塩およびNOドナー、例えば、ニトロプルシドナトリウム、ニトログリセリン、一硝酸イソソルビド、二硝酸イソソルビド、モルシドミンまたはSIN−1、およびNO吸入;
・環状グアノシン一リン酸(cGMP)の分解を阻害する化合物、例えば、ホスホジエステラーゼ(PDE)1、2および/または5の阻害剤、特にシルデナフィル、バルデナフィルおよびタダラフィルなどのPDE5阻害剤;
・例としておよび好ましくは、血小板凝集阻害剤、抗凝固剤または線維素溶解促進性物質の群の抗血栓剤;
・例としておよび好ましくは、カルシウム拮抗剤、アンジオテンシンAII拮抗剤、ACE阻害剤、エンドセリン拮抗剤、レニン阻害剤、α受容体遮断薬、β受容体遮断薬、ミネラルコルチコイド受容体拮抗剤および利尿剤の群の降圧活性化合物;および/または
・例えばおよび好ましくは、甲状腺受容体作動薬、コレステロール合成阻害剤、例えば、例としておよび好ましくは、HMG−CoA還元酵素阻害剤またはスクアレン合成阻害剤、ACAT阻害剤、CETP阻害剤、MTP阻害剤、PPARα、PPARγおよび/またはPPARδ作動薬、コレステロール吸収阻害剤、リパーゼ阻害剤、重合胆汁酸吸着剤、胆汁酸再吸収阻害剤およびリポタンパク質拮抗剤の群の脂肪代謝を変化させる活性化合物。
・有機硝酸塩およびNOドナー、例えば、ニトロプルシドナトリウム、ニトログリセリン、一硝酸イソソルビド、二硝酸イソソルビド、モルシドミンまたはSIN−1、およびNO吸入;
・環状グアノシン一リン酸(cGMP)の分解を阻害する化合物、例えば、ホスホジエステラーゼ(PDE)1、2および/または5の阻害剤、特にシルデナフィル、バルデナフィルおよびタダラフィルなどのPDE5阻害剤;
・例としておよび好ましくは、血小板凝集阻害剤、抗凝固剤または線維素溶解促進性物質の群の抗血栓剤;
・例としておよび好ましくは、カルシウム拮抗剤、アンジオテンシンAII拮抗剤、ACE阻害剤、エンドセリン拮抗剤、レニン阻害剤、α受容体遮断薬、β受容体遮断薬、ミネラルコルチコイド受容体拮抗剤および利尿剤の群の降圧活性化合物;および/または
・例えばおよび好ましくは、甲状腺受容体作動薬、コレステロール合成阻害剤、例えば、例としておよび好ましくは、HMG−CoA還元酵素阻害剤またはスクアレン合成阻害剤、ACAT阻害剤、CETP阻害剤、MTP阻害剤、PPARα、PPARγおよび/またはPPARδ作動薬、コレステロール吸収阻害剤、リパーゼ阻害剤、重合胆汁酸吸着剤、胆汁酸再吸収阻害剤およびリポタンパク質拮抗剤の群の脂肪代謝を変化させる活性化合物。
抗血栓剤は、好ましくは、血小板凝集阻害剤、抗凝固剤または線維素溶解促進性物質の群の化合物を意味すると理解される。
本発明の好ましい実施形態では、本発明による化合物を、血小板凝集阻害剤、例としておよび好ましくは、アスピリン、クロピドグレル、チクロピジンまたはジピリダモールと組み合わせて投与する。
本発明の好ましい実施形態では、本発明の化合物を、トロンビン阻害剤、例としておよび好ましくは、キシメラガトラン、ダビガトラン、メラガトラン、ビバリルジンまたはクレキサンと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、GPIIb/IIIa拮抗剤、例としておよび好ましくは、チロフィバンまたはアブシキシマブと組み合わせて投与する。
本発明の好ましい実施形態では、本発明の化合物を、第Xa因子阻害剤、例としておよび好ましくは、リバロキサバン(BAY59−7939)、DU−176b、アピキサバン、オタミキサバン、フィデキサバン、ラザキサバン、フォンダパリヌクス、イドラパリヌクス、PMD−3112、YM−150、KFA−1982、EMD−503982、MCM−17、MLN−1021、DX 9065a、DPC 906、JTV 803、SSR−126512またはSSR−128428と組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、ヘパリンまたは低分子量(LMW)ヘパリン誘導体と組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、ビタミンK拮抗剤、例としておよび好ましくは、クマリンと組み合わせて投与する。
降圧剤は、好ましくは、カルシウム拮抗剤、アンジオテンシンAII拮抗剤、ACE阻害剤、エンドセリン拮抗剤、レニン阻害剤、α受容体遮断薬、β受容体遮断薬、ミネラルコルチコイド受容体拮抗剤および利尿剤の群の化合物を意味すると理解される。
本発明の好ましい実施形態では、本発明による化合物を、カルシウム拮抗剤、例としておよび好ましくは、ニフェジピン、アムロジピン、ベラパミルまたはジルチアゼムと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、α1受容体遮断薬、例としておよび好ましくは、プラゾシンと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、β受容体遮断薬、例としておよび好ましくは、プロプラノロール、アテノロール、チモロール、ピンドロール、アルプレノロール、オクスプレノロール、ペンブトロール、ブプラノロール、メチプラノロール、ナドロール、メピンドロール、カラザロール(carazalol)、ソタロール、メトプロロール、ベタキソロール、セリプロロール、ビソプロロール、カルテオロール、エスモロール、ラベタロール、カルベジロール、アダプロロール、ランジオロール、ネビボロール、エパノロールまたはブシンドロールと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、アンジオテンシンAII拮抗剤、例としておよび好ましくは、ロサルタン、カンデサルタン、バルサルタン、テルミサルタンまたはエンブルサタンと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、ACE阻害剤、例としておよび好ましくは、エナラプリル、カプトプリル、リシノプリル、ラミプリル、デラプリル、フォシノプリル、キノプリル、ペリンドプリルまたはトランドプリルと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、エンドセリン拮抗剤、例としておよび好ましくは、ボセンタン、ダルセンタン、アンブリセンタンまたはシタクスセンタンと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、レニン阻害剤、例としておよび好ましくは、SPP−600またはSPP−800と組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、ミネラルコルチコイド受容体拮抗剤、例としておよび好ましくは、スピロノラクトンまたはエプレレノンと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、ループ利尿剤、例えば、フロセミド、トラセミド、ブメタニドおよびピレタニド、カリウム保持性利尿剤、例えば、アミロライドおよびトリアムテレン、アルドステロン拮抗剤、例えば、スピロノラクトン、カンレノ酸カリウムおよびエプレレノン、ならびにチアジド系利尿剤、例えば、ヒドロクロロチアジド、クロルタリドン、キシパミドおよびインダパミドと組み合わせて投与する。
脂肪代謝調節剤は、好ましくは、CETP阻害剤、甲状腺受容体作動薬、コレステロール合成阻害剤、例えば、HMG−CoA還元酵素阻害剤またはスクアレン合成阻害剤、ACAT阻害剤、MTP阻害剤、PPARα、PPARγおよび/またはPPARδ作動薬、コレステロール吸収阻害剤、重合胆汁酸吸着剤、胆汁酸再吸収阻害剤、リパーゼ阻害剤およびリポタンパク質拮抗剤の群の化合物を意味すると理解される。
本発明の好ましい実施形態では、本発明による化合物を、CETP阻害剤、例としておよび好ましくは、ダルセトラピブ、BAY60−5521、アナセトラピブまたはCETPワクチン(CETi−1)と組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、甲状腺受容体作動薬、例としておよび好ましくは、D−チロキシン、3,5,3’−トリヨードチロニン(T3)、CGS23425またはアキシチロム(CGS26214)と組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、スタチンのクラスのHMG−CoA還元酵素阻害剤、例としておよび好ましくは、ロバスタチン、シンバスタチン、プラバスタチン、フラバスタチン、アトルバスタチン、ロスバスタチンまたはピタバスタチンと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、スクアレン合成阻害剤、例としておよび好ましくは、BMS−188494またはTAK−475と組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、ACAT阻害剤、例としておよび好ましくは、アバシミブ、メリナミド、パクチミブ、エフルチミブまたはSMP−797と組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、MTP阻害剤、例としておよび好ましくは、インプリタピド、BMS−201038、R−103757またはJTT−130と組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、PPARγ作動薬、例としておよび好ましくは、ピオグリタゾンまたはロシグリタゾンと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、PPARδ作動薬、例としておよび好ましくは、GW501516またはBAY68−5042と組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、コレステロール吸収阻害剤、例としておよび好ましくは、エゼチミブ、チクエシドまたはパマクエシドと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、リパーゼ阻害剤、例としておよび好ましくは、オルリスタットと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、重合胆汁酸吸着剤、例としておよび好ましくは、コレスチラミン、コレスチポール、コレソルバム、コレスタゲルまたはコレスチミドと組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、胆汁酸再吸収阻害剤、例としておよび好ましくは、ASBT(=IBAT)阻害剤、例えば、AZD−7806、S−8921、AK−105、BARI−1741、SC−435またはSC−635と組み合わせて投与する。
本発明の好ましい実施形態では、本発明による化合物を、リポタンパク質拮抗剤、例としておよび好ましくは、ゲムカベンカルシウム(CI−1027)またはニコチン酸と組み合わせて投与する。
本発明はさらに、典型的には1種または複数の不活性で、非毒性の、薬学的に適当な賦形剤と共に少なくとも1種の本発明による化合物を含む医薬、および上記目的のためのその使用を提供する。
本発明による化合物は全身的におよび/または局所的に作用することができる。この目的のために、これらを適当な様式で、例えば、経口、非経口、肺、経鼻、舌下、舌、頬側、直腸、真皮、経皮、結膜もしくは耳経路により、またはインプラントもしくはステントとして投与することができる。
本発明による化合物をこれらの投与経路に適した投与形態で投与することができる。
経口投与に適した投与形態は、先行技術により作用し、本発明による化合物を速効性のおよび/または修正された様式で放出し、本発明による化合物を結晶および/または非晶質および/または溶解形態で含むもの、例えば、錠剤(非コーティングあるいは例えば、本発明の化合物の放出を制御する、胃液抵抗性または遅延溶解または不溶性コーティングによるコーティング錠)、口腔で急速に崩壊する錠剤またはフィルム/オブラート、フィルム/凍結乾燥物、カプセル剤(例えば、硬質または軟質ゼラチンカプセル)、糖衣錠、顆粒剤、ペレット剤、散剤、乳剤、懸濁剤、エアゾール剤または液剤である。
非経口投与は、(例えば、静脈内、動脈内、心臓内、髄腔内もしくは腰椎内経路により)吸収ステップを回避して、または(例えば、筋肉内、皮下、皮内、経皮もしくは腹腔内経路により)吸収を含めて達成することができる。非経口投与に適した投与形態には、液剤、懸濁剤、乳剤、凍結乾燥物または滅菌散剤の形態の注射および注入用製剤が含まれる。
他の投与経路については、適当な例に、吸入医薬形態(粉末吸入器、ネブライザーを含む)、点鼻薬、液またはスプレー、舌、舌下または頬側投与のための錠剤、フィルム/ウエハーまたはカプセル剤、坐剤、耳または眼製剤、膣カプセル剤、水性懸濁剤(ローション、振盪混合物)、親油性懸濁剤、軟膏、クリーム、経皮治療システム(例えば、パッチ)、ミルク、ペースト、フォーム、粉剤、インプラントまたはステントがある。
経口または非経口投与、特に、経口投与が好ましい。
本発明による化合物を言及する投与形態に変換することができる。これは、不活性で、非毒性の、薬学的に適当な補助剤と混合することによって、それ自体は既知の様式で達成することができる。これらの賦形剤には、担体(例えば、微結晶セルロース、乳糖、マンニトール)、溶媒(例えば、液体ポリエチレングリコール)、乳化剤および分散剤または湿潤剤(例えば、ドデシル硫酸ナトリウム、ポリオキシソルビタンオレエート)、結合剤(例えば、ポリビニルピロリドン)、合成および天然ポリマー(例えば、アルブミン)、安定剤(例えば、抗酸化剤、例えば、アスコルビン酸)、着色剤(例えば、無機顔料、例えば、酸化鉄)ならびに香味および/または臭気修正剤が含まれる。
一般に、非経口投与の場合、有効な結果を達成するために、約0.001〜1mg/kg、好ましくは約0.01〜0.5mg/kg体重の量を投与することが有利であることが分かった。経口投与の場合、用量は約0.001〜2mg/kg、好ましくは約0.001〜1mg/kg体重である。
それにもかかわらず、具体的には体重、投与経路、活性化合物に対する個体の反応、製剤の性質および投与が行われる時間または間隔の関数として言及する量から逸脱することが必要となり得る場合もある。したがって、上記最小量未満で間に合わせることで十分となり得る場合がある一方で、言及する上限を超過しなければならない場合がある。より多量を投与する場合、これらを1日数回の個々の用量に分割することが賢明となり得る。
以下の実施例は本発明を説明する。本発明はこれらの実施例に制限されない。
特に明言しない限り、以下の試験および実施例中の百分率は、重量百分率であり、部は重量部である。液体/液体溶液の溶媒比、希釈比および濃度データは各場合において体積に基づく。
A.実施例
LC/MSおよびHPLC法:
方法1(LC−MS):
機器:Micromass Quattro Premier with Waters UPLC Acquity;カラム:Thermo Hypersil GOLD 1.9μ 50×1mm;移動相A:水1l+50%ギ酸0.5ml、移動相B:アセトニトリル1l+50%濃度ギ酸0.5ml;勾配:0.0分90%A→0.1分90%A→1.5分10%A→2.2分10%A;オーブン:50℃;流量:0.33ml/分;UV検出:210nm
方法1(LC−MS):
機器:Micromass Quattro Premier with Waters UPLC Acquity;カラム:Thermo Hypersil GOLD 1.9μ 50×1mm;移動相A:水1l+50%ギ酸0.5ml、移動相B:アセトニトリル1l+50%濃度ギ酸0.5ml;勾配:0.0分90%A→0.1分90%A→1.5分10%A→2.2分10%A;オーブン:50℃;流量:0.33ml/分;UV検出:210nm
方法2(LC−MS):
機器:Waters ACQUITY SQD UPLCシステム;カラム:Waters Acquity UPLC HSS T3 1.8μ50×1mm;移動相A:水1l+99%ギ酸0.25ml、移動相B:アセトニトリル1l+99%濃度ギ酸0.25ml;勾配:0.0分90%A→1.2分5%A→2.0分5%A;オーブン:50℃;流量:0.40ml/分;UV検出:210〜400nm。
機器:Waters ACQUITY SQD UPLCシステム;カラム:Waters Acquity UPLC HSS T3 1.8μ50×1mm;移動相A:水1l+99%ギ酸0.25ml、移動相B:アセトニトリル1l+99%濃度ギ酸0.25ml;勾配:0.0分90%A→1.2分5%A→2.0分5%A;オーブン:50℃;流量:0.40ml/分;UV検出:210〜400nm。
方法3(LC−MS):
機器:Micromass Quattro Premier with Waters UPLC Acquity;カラム:Thermo Hypersil GOLD 1.9μ 50×1mm;移動相A:水1l+50%ギ酸0.5ml、移動相B:アセトニトリル1l+50%濃度ギ酸0.5ml;勾配:0.0分97%A→0.5分97%A→3.2分5%A→4.0分5%A;オーブン:50℃;流量:0.3ml/分;UV検出:210nm。
機器:Micromass Quattro Premier with Waters UPLC Acquity;カラム:Thermo Hypersil GOLD 1.9μ 50×1mm;移動相A:水1l+50%ギ酸0.5ml、移動相B:アセトニトリル1l+50%濃度ギ酸0.5ml;勾配:0.0分97%A→0.5分97%A→3.2分5%A→4.0分5%A;オーブン:50℃;流量:0.3ml/分;UV検出:210nm。
方法4(LC−MS):
MS機器:Waters(Micromass)QM;HPLC機器:Agilent 1100シリーズ;カラム:Agilent ZORBAX Extend−C18 3.0×50mm 3.5ミクロン;移動相A:水1l+0.01molの炭酸アンモニウム、移動相B:アセトニトリル1l;勾配:0.0分98%A→0.2分98%A→3.0分5%A→4.5分5%A;オーブン:40℃;流量:1.75ml/分;UV検出:210nm
MS機器:Waters(Micromass)QM;HPLC機器:Agilent 1100シリーズ;カラム:Agilent ZORBAX Extend−C18 3.0×50mm 3.5ミクロン;移動相A:水1l+0.01molの炭酸アンモニウム、移動相B:アセトニトリル1l;勾配:0.0分98%A→0.2分98%A→3.0分5%A→4.5分5%A;オーブン:40℃;流量:1.75ml/分;UV検出:210nm
方法5(LC−MS):
機器:Waters ACQUITY SQD UPLCシステム;カラム:Waters Acquity UPLC HSS T3 1.8μ30×2mm;移動相A:水1l+99%ギ酸0.25ml、移動相B:アセトニトリル1l+99%濃度ギ酸0.25ml;勾配:0.0分90%A→1.2分5%A→2.0分5%A;オーブン:50℃;流量:0.60ml/分;UV検出:208〜400nm。
機器:Waters ACQUITY SQD UPLCシステム;カラム:Waters Acquity UPLC HSS T3 1.8μ30×2mm;移動相A:水1l+99%ギ酸0.25ml、移動相B:アセトニトリル1l+99%濃度ギ酸0.25ml;勾配:0.0分90%A→1.2分5%A→2.0分5%A;オーブン:50℃;流量:0.60ml/分;UV検出:208〜400nm。
方法6(GC−MS):
機器:Thermo Scientific DSQII、Thermo Scientific Trace GC Ultra;カラム:Restek RTX−35MS、15m×200μm×0.33μm;一定ヘリウム流量:1.20ml/分;オーブン:60℃;入口:220℃;勾配:60℃、30℃/分→300℃(3.33分間維持)。
機器:Thermo Scientific DSQII、Thermo Scientific Trace GC Ultra;カラム:Restek RTX−35MS、15m×200μm×0.33μm;一定ヘリウム流量:1.20ml/分;オーブン:60℃;入口:220℃;勾配:60℃、30℃/分→300℃(3.33分間維持)。
特に明言しない限り、以下の試験および実施例中の百分率は、重量百分率であり、部は重量部である。液体/液体溶液の溶媒比、希釈比および濃度データは各場合において体積に基づく。
以下の段落で報告される1H NMRスペクトルのプロトンシグナルの多重度は、各場合で観察されるシグナル型を表しており、いかなる高次シグナル現象も考慮していない。全ての1H NMRスペクトルデータで、化学シフトδをppmで述べる。
さらに、出発材料、中間体および実施例は水和物として存在してもよい。含水量の定量方法はなかった。一定の場合、水和物は1H NMRスペクトルに影響を及ぼし、おそらくは1H NMRの水シグナルをシフトさせるおよび/または有意に広くし得る。
1H NMRスペクトルにおいて、化学系「2−メチルイミダゾ[1,2−a]ピリジン」のメチル基は、一重項(頻繁に、DMSO−d6でおよび2.40〜2.60ppmの範囲で)として現れ、それ自体明確に識別可能である、溶媒シグナルによって重ね合わせられる、または完全に溶媒のシグナルの下にある。1H NMRスペクトルにおいて、このシグナルは存在すると仮定され得る。
本発明の化合物を上記方法による分取HPLC(溶離液は添加剤、例えば、トリフルオロ酢酸、ギ酸またはアンモニアを含む)によって精製する場合、本発明の化合物が十分に塩基性または酸性の官能基を含む場合には、本発明の化合物を、塩型で、例えば、トリフルオロ酢酸塩、ギ酸塩またはアンモニウム塩として得ることができる。このような塩を、当業者に既知の種々の方法によって、対応する遊離塩基または酸に変換することができる。
以下に記載する本発明の合成中間体および実施例の場合、対応する塩基または酸の塩の形態で指定されるいずれの化合物も、一般的にそれぞれの調製および/または精製法によって得られる未知の正確な化学量論的組成の塩である。そのため、より詳細に指定しない限り、「塩酸塩」、「トリフルオロアセテート」、「ナトリウム塩」または「xHCl」、「xCF3COOH」、「xNa+」などの名称および構造式への追加は、このような塩の場合、化学量論的意味で理解されるべきでなく、その中に存在する塩形成成分に関する説明的性質を有するにすぎない。
対応して、合成中間体または実施例またはその塩を、記載される調製および/または精製法によって未知の化学量論的組成の溶媒和物、例えば、水和物の形態で得た場合(これらが定義された型のものである場合)、これを適用する。
出発材料および中間体:
実施例1A
5−クロロ−2−ニトロピリジン−3−オール
氷冷しながら、5−クロロピリジン−3−オール30g(232mmol、1当量)を濃硫酸228mlに溶解し、0℃で、濃硝酸24mlをゆっくり添加した。反応物を室温に加温し、一晩攪拌し、次いで、氷/水混合物中に攪拌し、さらに30分間攪拌した。固体を濾別し、冷水で洗浄し、風乾した。これにより標記化合物33g(理論値の82%)が得られ、これをさらに精製することなく次の反応に使用した。
LC−MS(方法2):Rt=0.60分
MS(ESneg):m/z=172.9/174.9(M−H)−
1H−NMR(400 MHz,DMSO−d6):δ=7.71(d,1 H);8.10(d,1 H);12.14(br.1 H).
実施例1A
5−クロロ−2−ニトロピリジン−3−オール
LC−MS(方法2):Rt=0.60分
MS(ESneg):m/z=172.9/174.9(M−H)−
1H−NMR(400 MHz,DMSO−d6):δ=7.71(d,1 H);8.10(d,1 H);12.14(br.1 H).
実施例2A
5−クロロ−3−[(2,6−ジフルオロベンジル)オキシ]−2−ニトロピリジン
5−クロロ−2−ニトロピリジン−3−オール33g(実施例1A;189mmol、1当量)および炭酸セシウム61.6g(189mmol、1当量)を最初にDMF528mlに装入し、2,6−ジフルオロベンジルブロミド40.4g(189mmol、1当量)を添加し、混合物を室温で一晩攪拌した。反応混合物を水/1N塩酸水溶液中に攪拌した。固体を濾別し、水で洗浄し、風乾した。これにより、標記化合物54.9g(理論値の97%)が得られた。
1H−NMR(400 MHz,DMSO−d6):δ=5.46(s,2 H);7.22(t,2 H);7.58(q,1 H);8.28(d,1 H);8.47(d,1 H).
5−クロロ−3−[(2,6−ジフルオロベンジル)オキシ]−2−ニトロピリジン
1H−NMR(400 MHz,DMSO−d6):δ=5.46(s,2 H);7.22(t,2 H);7.58(q,1 H);8.28(d,1 H);8.47(d,1 H).
実施例3A
5−クロロ−3−[(2,6−ジフルオロベンジル)オキシ]ピリジン−2−アミン
5−クロロ−3−[(2,6−ジフルオロベンジル)オキシ]−2−ニトロピリジン59.7g(実施例2A;199mmol、1当量)を最初にエタノール600mlに装入し、鉄粉末34.4g(616mmol、3.1当量)を添加し、混合物を加熱還流した。濃塩酸152mlをゆっくり滴加し、混合物をさらに30分間還流で沸騰させた。反応混合物を冷却し、氷/水混合物中に攪拌した。得られた混合物を、酢酸ナトリウムを用いてpH5に調整した。固体を濾別し、水で洗浄し、風乾し、次いで、減圧下50℃で乾燥させた。これにより、標記化合物52.7g(理論値の98%)が得られた。
LC−MS(方法2):Rt=0.93分
MS(ESpos):m/z=271.1/273.1(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=5.14(s,2 H);5.82(br.s,2 H);7.20(t,2 H);7.35(d,1 H);7.55(q,1 H);7.56(d,1 H).
5−クロロ−3−[(2,6−ジフルオロベンジル)オキシ]ピリジン−2−アミン
LC−MS(方法2):Rt=0.93分
MS(ESpos):m/z=271.1/273.1(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=5.14(s,2 H);5.82(br.s,2 H);7.20(t,2 H);7.35(d,1 H);7.55(q,1 H);7.56(d,1 H).
実施例4A
エチル6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキシレート
5−クロロ−3−[(2,6−ジフルオロベンジル)オキシ]ピリジン−2−アミン40g(実施例3A;147.8mmol、1当量)を最初にエタノール800mlに装入し、粉末状モレキュラーシーブ3Å30gおよびエチル2−クロロアセトアセテート128g(739mmol、5当量)を添加し、混合物を一晩加熱還流した。反応混合物を濃縮し、残渣を酢酸エチルに溶解し、濾過した。酢酸エチル相を水で洗浄し、乾燥させ、濾過および濃縮した。これにより、標記化合物44g(理論値の78%)が得られた。
LC−MS(方法2):Rt=1.27分
MS(ESpos):m/z=381.2/383.2(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.36(t,3 H);2.54(s,3 H;hidden by DMSO signal);4.37(q,2 H);5.36(s,2 H);7.26(t,2 H);7.38(d,1 H);7.62(q,1 H);8.92(d,1 H).
エチル6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキシレート
LC−MS(方法2):Rt=1.27分
MS(ESpos):m/z=381.2/383.2(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.36(t,3 H);2.54(s,3 H;hidden by DMSO signal);4.37(q,2 H);5.36(s,2 H);7.26(t,2 H);7.38(d,1 H);7.62(q,1 H);8.92(d,1 H).
実施例5A
6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボン酸
エチル6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキシレート44g(実施例4A;115mmol、1当量)をTHF550mlおよびメタノール700mlに溶解し、水酸化リチウム13.8g(水150mlに溶解;577mmol、5当量)を添加し、混合物を室温で一晩攪拌した。1N塩酸水溶液を添加し、混合物を減圧下で濃縮した。得られた固体を濾別し、水で洗浄した。これにより、標記化合物34g(理論値の84%)が得られた。
LC−MS(方法1):Rt=1.03分
MS(ESpos):m/z=353.0/355.0(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=2.54(s,3 H;overlapped by DMSO signal);5.36(s,2 H);7.26(t,2 H);7.34(d,1 H);7.61(q,1 H);8.99(d,1 H);13.36(br.s,1 H).
6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボン酸
LC−MS(方法1):Rt=1.03分
MS(ESpos):m/z=353.0/355.0(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=2.54(s,3 H;overlapped by DMSO signal);5.36(s,2 H);7.26(t,2 H);7.34(d,1 H);7.61(q,1 H);8.99(d,1 H);13.36(br.s,1 H).
実施例6A
rac−2−アミノ−3−フルオロ−2−メチルプロパノニトリル
標記化合物は文献から既知である:
1)McConathy,J.ら、Journal of Medicinal Chemistry 2002、45、2240〜2249。
2)Bergmann,E.D.ら、Journal of the Chemical Society 1963、3462〜3463。
さらなる方法:
フルオロアセトン1.0g(0.94ml;13.15mmol)を最初にメタノール中2Nアンモニア11mlに装入した。室温で、シアン化ナトリウム721mg(14.72mmol)および塩化アンモニウム788mg(14.72mmol)を連続的に添加し、混合物を還流で2時間攪拌した。反応溶液を冷却し、濾過し、塩化メチレンで洗浄した。母液から固体が沈殿し、これを濾別した。塩化メチレンおよびメタノールを標準圧力で母液から留去した。これにより、標的化合物1.32g(理論値の89%、純度約90%)が得られた。生成物をさらに精製することなく次の反応に使用した。
GC−MS(方法6):Rt=1.64分
MS(EIpos):m/z=87(M−CH3)+
rac−2−アミノ−3−フルオロ−2−メチルプロパノニトリル
1)McConathy,J.ら、Journal of Medicinal Chemistry 2002、45、2240〜2249。
2)Bergmann,E.D.ら、Journal of the Chemical Society 1963、3462〜3463。
さらなる方法:
フルオロアセトン1.0g(0.94ml;13.15mmol)を最初にメタノール中2Nアンモニア11mlに装入した。室温で、シアン化ナトリウム721mg(14.72mmol)および塩化アンモニウム788mg(14.72mmol)を連続的に添加し、混合物を還流で2時間攪拌した。反応溶液を冷却し、濾過し、塩化メチレンで洗浄した。母液から固体が沈殿し、これを濾別した。塩化メチレンおよびメタノールを標準圧力で母液から留去した。これにより、標的化合物1.32g(理論値の89%、純度約90%)が得られた。生成物をさらに精製することなく次の反応に使用した。
GC−MS(方法6):Rt=1.64分
MS(EIpos):m/z=87(M−CH3)+
実施例7A
rac−ベンジル(2−シアノ−1−フルオロプロパン−2−イル)カルバメート
THF/水(9/1)29ml中実施例6Aからのrac−2−アミノ−3−フルオロ−2−メチルプロパノニトリル1.34g(11.83mmol、純度約90%)に炭酸カリウム5.07g(36.67mmol)を添加した。0℃で、クロロギ酸ベンジル1.69ml(11.83mmol)をゆっくり滴加し、反応混合物を室温で一晩攪拌した。溶媒をデカントし、水相をTHFで2回抽出し、次いで、THFをデカントした。合わせた有機相を硫酸ナトリウムを用いて乾燥させ、濾過および濃縮した。残渣をシリカゲルクロマトグラフィー(移動相:シクロヘキサン/酢酸エチル勾配)によって分離し、生成物分画をロータリーエバポレーターで濃縮した。これにより、標的化合物1.89g(理論値の66%、純度97%)が得られた。
LC−MS(方法2):Rt=0.89分
MS(ESpos):m/z=237(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.58(d,3H),4.47−4.78(m,2H),5.10(s,2H),7.30−7.43(m,5H),8.34(br.s,1H).
rac−ベンジル(2−シアノ−1−フルオロプロパン−2−イル)カルバメート
LC−MS(方法2):Rt=0.89分
MS(ESpos):m/z=237(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.58(d,3H),4.47−4.78(m,2H),5.10(s,2H),7.30−7.43(m,5H),8.34(br.s,1H).
実施例8A
ent−ベンジル(2−シアノ−1−フルオロプロパン−2−イル)カルバメート(エナンチオマーA)
実施例7Aからのrac−ベンジル(2−シアノ−1−フルオロプロパン−2−イル)カルバメート3.0g(12.69mmol)をキラル相[カラム:Daicel Chiralpak AY−H、5μm、250×20mm、移動相:80%イソヘキサン、20%イソプロパノール、流量:15ml/分;40℃、検出:220nm]で分取法によりエナンチオマーに分離した。
エナンチオマーA:収量:1.18g(>99%ee)
Rt=5.37分[Daicel Chiralcel AY−H、5μm、250×4.6mm;移動相:70%イソヘキサン、30%2−プロパノール;流量1.0ml/分;40℃;検出:220nm]。
ent−ベンジル(2−シアノ−1−フルオロプロパン−2−イル)カルバメート(エナンチオマーA)
エナンチオマーA:収量:1.18g(>99%ee)
Rt=5.37分[Daicel Chiralcel AY−H、5μm、250×4.6mm;移動相:70%イソヘキサン、30%2−プロパノール;流量1.0ml/分;40℃;検出:220nm]。
実施例9A
ent−ベンジル(2−シアノ−1−フルオロプロパン−2−イル)カルバメート(エナンチオマーB)
実施例7Aからのrac−ベンジル(2−シアノ−1−フルオロプロパン−2−イル)カルバメート3.0g(12.69mmol)をキラル相[カラム:Daicel Chiralpak AY−H、5μm、250×20mm、移動相:80%イソヘキサン、20%イソプロパノール、流量:15ml/分;40℃、検出:220nm]で分取法によりエナンチオマーに分離した。
エナンチオマーB:収量:1.18g(>99%ee)
Rt=6.25分[Daicel Chiralcel AY−H、5μm、250×4.6mm;移動相:70%イソヘキサン、30%2−プロパノール;流量1.0ml/分;40℃;検出:220nm]。
ent−ベンジル(2−シアノ−1−フルオロプロパン−2−イル)カルバメート(エナンチオマーB)
エナンチオマーB:収量:1.18g(>99%ee)
Rt=6.25分[Daicel Chiralcel AY−H、5μm、250×4.6mm;移動相:70%イソヘキサン、30%2−プロパノール;流量1.0ml/分;40℃;検出:220nm]。
実施例10A
rac−ベンジル(1−アミノ−3−フルオロ−2−メチルプロパン−2−イル)カルバメート
アルゴン下で、ラネーニッケル(水性スラリー)1.55gを、メタノール中7Nアンモニア14.9ml中実施例7Aからのrac−ベンジル(2−シアノ−1−フルオロプロパン−2−イル)カルバメート1.2g(5.08mmol)に添加し、混合物を約25barの水素圧力および室温で24時間水素付加した。反応混合物を珪藻土を通して濾過し、メタノールで洗浄し、濃縮した。これにより、標的化合物1.2g(理論値の98%)が得られた。
LC−MS(方法2):Rt=0.49分
MS(ESpos):m/z=241(M+H)+
rac−ベンジル(1−アミノ−3−フルオロ−2−メチルプロパン−2−イル)カルバメート
LC−MS(方法2):Rt=0.49分
MS(ESpos):m/z=241(M+H)+
実施例11A
ent−ベンジル(1−アミノ−3−フルオロ−2−メチルプロパン−2−イル)カルバメート(エナンチオマーA)
アルゴン下で、ラネーニッケル(水性スラリー)1.55gを、メタノール中7Nアンモニア14.9ml中実施例8Aからのent−ベンジル(2−シアノ−1−フルオロプロパン−2−イル)カルバメート(エナンチオマーA)1.2g(5.08mmol)に添加し、混合物を約25barの水素圧力および室温で24時間水素付加した。反応混合物を珪藻土を通して濾過し、メタノールで洗浄し、濃縮した。これにより、標的化合物700mg(理論値の57%、純度約85%)が得られた。
LC−MS(方法2):Rt=0.52分
MS(ESpos):m/z=241(M+H)+
ent−ベンジル(1−アミノ−3−フルオロ−2−メチルプロパン−2−イル)カルバメート(エナンチオマーA)
LC−MS(方法2):Rt=0.52分
MS(ESpos):m/z=241(M+H)+
実施例12A
ent−ベンジル(1−アミノ−3−フルオロ−2−メチルプロパン−2−イル)カルバメート(エナンチオマーB)
アルゴン下で、ラネーニッケル(水性スラリー)1.55gを、メタノール中7Nアンモニア14.9ml中実施例9Aからのent−ベンジル(2−シアノ−1−フルオロプロパン−2−イル)カルバメート(エナンチオマーB)1.2g(5.08mmol)に添加し、混合物を約25barの水素圧力および室温で24時間水素付加した。反応混合物を珪藻土を通して濾過し、メタノールで洗浄し、濃縮した。これにより、標的化合物1.2g(理論値の98%、純度約85%)が得られた。
LC−MS(方法2):Rt=0.50分
MS(ESpos):m/z=241(M+H)+
ent−ベンジル(1−アミノ−3−フルオロ−2−メチルプロパン−2−イル)カルバメート(エナンチオマーB)
LC−MS(方法2):Rt=0.50分
MS(ESpos):m/z=241(M+H)+
実施例13A
rac−2−アミノ−5,5,5−トリフルオロ−2−メチルペンタノニトリル
5,5,5−トリフルオロペンタン−2−オン8.0g(57.1mmol)[CAS登録番号:1341078−97−4;商業的に入手可能、またはメチルケトンを当業者に知られている文献方法、例えば、a)Y.BaiらAngewandte Chemie 2012、51、4112〜4116;K.HiroiらSynlett 2001、263〜265;K.Mikamiら1982 Chemistry Letters、1349〜1352による4,4,4−トリフルオロブタナールからの2段階で;またはb)A.A.WubeらBioorganic and Medicinal Chemistry 2011、19、567〜579;G.M.RubottomらJournal of Organic Chemistry 1983、48、1550〜1552;T.ChenらJournal of Organic Chemistry 1996、61、4716〜4719による4,4,4−トリフルオロブタン酸から調製することができる。生成物を蒸留またはクロマトグラフィーによって単離することができる]を最初にメタノール中2Nアンモニア47.8mlに装入し、シアン化ナトリウム3.69g(75.4mmol)および塩化アンモニウム4.03g(75.4mmol)を室温で添加し、混合物を還流下で4時間攪拌した。反応混合物を冷却し、ジエチルエーテルを添加し、存在する固体を濾別した。溶媒を標準圧力下で濾液から留去した。標記化合物8.7g(理論値の92%)が残渣として得られ、これをさらに精製することなく後の段階に使用した。
GC−MS(方法6):Rt=1.90分
MS(ESpos):m/z=151(M−CH3)+
rac−2−アミノ−5,5,5−トリフルオロ−2−メチルペンタノニトリル
GC−MS(方法6):Rt=1.90分
MS(ESpos):m/z=151(M−CH3)+
実施例14A
rac−ベンジル(2−シアノ−5,5,5−トリフルオロペンタン−2−イル)カルバメート
実施例33Aからのrac−2−アミノ−5,5,5−トリフルオロ−2−メチルペンタノニトリル8.7g(52.36mmol)を最初にテトラヒドロフラン/水=9/1 128mlに装入し、炭酸カリウム22.43g(162.3mmol)を添加した。0℃で、クロロギ酸ベンジル8.93g(52.36mmol)をゆっくり滴加した。次いで、混合物を攪拌しながら徐々に室温まで加温させ、室温で一晩攪拌した。上清溶媒をデカントし、残渣を各々100mlのテトラヒドロフランと共に2回攪拌し、次いで、上清溶媒を都度デカントした。合わせた有機相を濃縮し、粗製生成物をシリカゲルクロマトグラフィー(移動相:シクロヘキサン/酢酸エチル勾配9/1〜4/1)によって精製した。標記化合物11.14g(理論値の68%)が得られた。
LC−MS(方法2):Rt=1.01分
MS(ESpos):m/z=301(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.58(s,3H),2.08−2.21(m,2H),2.24−2.52(m,2H),5.09(s,2H),7.29−7.41(m,5H),8.17(br.s,1H).
rac−ベンジル(2−シアノ−5,5,5−トリフルオロペンタン−2−イル)カルバメート
LC−MS(方法2):Rt=1.01分
MS(ESpos):m/z=301(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.58(s,3H),2.08−2.21(m,2H),2.24−2.52(m,2H),5.09(s,2H),7.29−7.41(m,5H),8.17(br.s,1H).
実施例15A
ent−ベンジル(2−シアノ−5,5,5−トリフルオロペンタン−2−イル)カルバメート(エナンチオマーA)
実施例14Aからのrac−ベンジル(2−シアノ−5,5,5−トリフルオロペンタン−2−イル)カルバメート11.14gをキラル相[カラム:Daicel Chiralpak AZ−H、5μm、SFC、250×50mm、移動相:94%二酸化炭素、6%メタノール、流量:200ml/分;温度:38℃、圧力:135bar、検出:210nm]で分取法によってエナンチオマーに分離した。
エナンチオマーA:4.12g(約79%ee)
Rt=1.60分[SFC、Daicel Chiralpak AZ−H、250×4.6mm、5μm、移動相:90%二酸化炭素、10%メタノール、流量:3ml/分;温度:30℃、検出:220nm]。
LC−MS(方法2):Rt=1.01分
MS(ESpos):m/z=301(M+H)+
ent−ベンジル(2−シアノ−5,5,5−トリフルオロペンタン−2−イル)カルバメート(エナンチオマーA)
エナンチオマーA:4.12g(約79%ee)
Rt=1.60分[SFC、Daicel Chiralpak AZ−H、250×4.6mm、5μm、移動相:90%二酸化炭素、10%メタノール、流量:3ml/分;温度:30℃、検出:220nm]。
LC−MS(方法2):Rt=1.01分
MS(ESpos):m/z=301(M+H)+
実施例16A
ent−ベンジル(2−シアノ−5,5,5−トリフルオロペンタン−2−イル)カルバメート(エナンチオマーB)
実施例14Aからのrac−ベンジル(2−シアノ−5,5,5−トリフルオロペンタン−2−イル)カルバメート11.14gをキラル相[カラム:Daicel Chiralpak AZ−H、5μm、SFC、250×50mm、移動相:94%二酸化炭素、6%メタノール、流量:200ml/分;温度:38℃、圧力:135bar、検出:210nm]で分取法によってエナンチオマーに分離した。
エナンチオマーB:4.54g(約70%ee;純度約89%)
Rt=1.91分[SFC、Daicel Chiralpak AZ−H、250×4.6mm、5μm、移動相:90%二酸化炭素、10%メタノール、流量:3ml/分;温度:30℃、検出:220nm]。
LC−MS(方法2):Rt=1.01分
MS(ESpos):m/z=301(M+H)+
ent−ベンジル(2−シアノ−5,5,5−トリフルオロペンタン−2−イル)カルバメート(エナンチオマーB)
エナンチオマーB:4.54g(約70%ee;純度約89%)
Rt=1.91分[SFC、Daicel Chiralpak AZ−H、250×4.6mm、5μm、移動相:90%二酸化炭素、10%メタノール、流量:3ml/分;温度:30℃、検出:220nm]。
LC−MS(方法2):Rt=1.01分
MS(ESpos):m/z=301(M+H)+
実施例17A
ent−ベンジル(1−アミノ−5,5,5−トリフルオロ−2−メチルペンタン−2−イル)カルバメート(エナンチオマーA)
実施例15Aからのent−ベンジル(2−シアノ−5,5,5−トリフルオロペンタン−2−イル)カルバメート(エナンチオマーA)4.12g(13.17mmol)をメタノール中7Nアンモニア溶液39mlに溶解し、ラネーニッケル(50%水性スラリー)4gをアルゴン下で添加した。反応混合物をオートクレーブ中20〜30barで一晩水素付加した。ラネーニッケル(50%水性スラリー)さらに1gを添加し、反応混合物をオートクレーブ中20〜30barで5時間水素付加した。反応混合物を珪藻土を通して濾過し、メタノールですすぎ、濃縮した。標的化合物3.35g(理論値の56%;純度約67%)が得られ、これを精製することなく後の段階に使用した。
LC−MS(方法5):Rt=1.68分
MS(ESpos):m/z=305(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.13(s,3H),1.40(br.s,2H),1.70−1.80(m,1H),1.83−1.95(m,1H),2.08−2.2(m,2H),4.98(s,2H),6.85(br.s,1H),7.28−7.41(m,5H).
ent−ベンジル(1−アミノ−5,5,5−トリフルオロ−2−メチルペンタン−2−イル)カルバメート(エナンチオマーA)
LC−MS(方法5):Rt=1.68分
MS(ESpos):m/z=305(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.13(s,3H),1.40(br.s,2H),1.70−1.80(m,1H),1.83−1.95(m,1H),2.08−2.2(m,2H),4.98(s,2H),6.85(br.s,1H),7.28−7.41(m,5H).
実施例18A
ent−ベンジル(1−アミノ−5,5,5−トリフルオロ−2−メチルペンタン−2−イル)カルバメート(エナンチオマーB)
実施例16Aからのent−ベンジル(2−シアノ−5,5,5−トリフルオロペンタン−2−イル)カルバメート(エナンチオマーB)4.54g(13.45mmol;純度約89%)をメタノール中7Nアンモニア溶液39mlに溶解し、ラネーニッケル(50%水性スラリー)5gをアルゴン下で添加した。反応混合物をオートクレーブ中20〜30barで3時間水素付加した。反応混合物を珪藻土を通して濾過し、メタノールですすぎ、濃縮した。標的化合物4.20g(理論値の97%;純度約95%)が得られ、これをさらに精製することなくその後の段階に使用した。
LC−MS(方法4):Rt=2.19分
MS(ESpos):m/z=305(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.13(s,3H),1.40(br.s,2H),1.69−1.80(m,1H),1.83−1.96(m,1H),2.07−2.22(m,2H),4.98(s,2H),6.85(br.s,1H),7.27−7.40(m,5H).
ent−ベンジル(1−アミノ−5,5,5−トリフルオロ−2−メチルペンタン−2−イル)カルバメート(エナンチオマーB)
LC−MS(方法4):Rt=2.19分
MS(ESpos):m/z=305(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.13(s,3H),1.40(br.s,2H),1.69−1.80(m,1H),1.83−1.96(m,1H),2.07−2.22(m,2H),4.98(s,2H),6.85(br.s,1H),7.27−7.40(m,5H).
実施例19A
rac−2−[(ジフェニルメチレン)アミノ]−4−フルオロブタノニトリル
[(ジフェニルメチレン)アミノ]アセトニトリル16.5g(74.91mmol)を最初に無水THF495mlに装入し、n−ブチルリチウム(ヘキサン中2.5N)35.96ml(89.89mmol)をアルゴン下−78℃で添加し、混合物を−78℃で15分間攪拌した。その後、反応溶液を最高0℃まで加温した。1−ヨード−2−フルオロエタン13.03g(74.91mmol)を反応溶液に滴加し、これを0℃でさらに15分間攪拌した。0℃で、最初に水、次いで、酢酸エチルを反応溶液に添加し、混合物を飽和塩化ナトリウム水溶液で洗浄した。水相を酢酸エチルで2回抽出した。合わせた有機相を硫酸ナトリウム上で乾燥させ、濾過および濃縮した。残渣をシリカゲルクロマトグラフィー(移動相:ジクロロメタン/シクロヘキサン=1/1〜2/1)によって精製した。これにより、標的化合物18.7g(理論値の80%、純度85%)が得られた。
LC−MS(方法3):Rt=2.42分
MS(ESpos):m/z=267(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=2.13−2.41(m,2H),4.40(t,1H),4.43−4.71(m,2H),7.25−7.30(m,2H),7.33−7.63(m,8H).
rac−2−[(ジフェニルメチレン)アミノ]−4−フルオロブタノニトリル
LC−MS(方法3):Rt=2.42分
MS(ESpos):m/z=267(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=2.13−2.41(m,2H),4.40(t,1H),4.43−4.71(m,2H),7.25−7.30(m,2H),7.33−7.63(m,8H).
実施例20A
rac−2−[(ジフェニルメチレン)アミノ]−4,4−ジフルオロブタノニトリル
[(ジフェニルメチレン)アミノ]アセトニトリル18g(81.72mmol)を最初に無水THF500mlに装入し、n−ブチルリチウム(ヘキサン中2.5N)39.22ml(98.06mmol)をアルゴン下−78℃で添加し、混合物を−78℃で15分間攪拌した。その後、反応溶液を最高0℃まで加温した。1,1−ジフルオロ−2−ヨードエタン17.25g(89.89mmol)を反応溶液に滴加し、これを0℃でさらに15分間攪拌した。0℃で、最初に水、次いで、酢酸エチルを反応溶液に添加し、混合物を半飽和塩化ナトリウム水溶液で3回洗浄した。合わせた水相を酢酸エチルで2回抽出した。合わせた有機相を硫酸ナトリウム上で乾燥させ、濾過および濃縮した。残渣をシリカゲルクロマトグラフィー(移動相:ジクロロメタン/シクロヘキサン=1/1)によって精製した。これにより、標的化合物13.57g(理論値の49%、純度84%)が得られた。
LC−MS(方法3):Rt=2.48分
MS(ESpos):m/z=285(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=2.53−2.61(m,2H;partially overlapped with solvent peak),4.50(t,1H),6.08−6.41(m,1H),7.23−7.33(m,2H),7.38−7.47(m,2H),7.49−7.67(m,6H).
rac−2−[(ジフェニルメチレン)アミノ]−4,4−ジフルオロブタノニトリル
LC−MS(方法3):Rt=2.48分
MS(ESpos):m/z=285(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=2.53−2.61(m,2H;partially overlapped with solvent peak),4.50(t,1H),6.08−6.41(m,1H),7.23−7.33(m,2H),7.38−7.47(m,2H),7.49−7.67(m,6H).
実施例21A
rac−2−[(ジフェニルメチレン)アミノ]−5−フルオロペンタノニトリル
[(ジフェニルメチレン)アミノ]アセトニトリル18g(81.72mmol)を最初に無水THF500mlに装入し、n−ブチルリチウム(ヘキサン中2.5N)39.22ml(98.06mmol)をアルゴン下−78℃で添加し、混合物を−78℃でさらに15分間攪拌した。その後、反応溶液を最高0℃まで加温し、1−フルオロ−3−ヨードプロパン16.9g(89.89mmol)を反応溶液に滴加し、これを0℃でさらに15分間攪拌した。0℃で、水、次いで、酢酸エチルを添加し、混合物を飽和塩化ナトリウム水溶液で洗浄した。水相を酢酸エチルで2回抽出した。合わせた有機相を硫酸ナトリウム上で乾燥させ、濾過および濃縮した。残渣をシリカゲルクロマトグラフィー(移動相:トルエン100%、ジクロロメタン/シクロヘキサン=1/1〜2/1により再精製)によって精製した。これにより、標的化合物16.73g(理論値の73%)が得られた。
LC−MS(方法3):Rt=2.50分
MS(ESpos):m/z=281(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.66−1.85(m,2H),1.87−2.00(m,2H),4.26−4.41(m,2H),4.43−4.55(m,1H),7.20−7.33(m,2H),7.38−7.48(m,2H),7.48−7.63(m,6H).
rac−2−[(ジフェニルメチレン)アミノ]−5−フルオロペンタノニトリル
LC−MS(方法3):Rt=2.50分
MS(ESpos):m/z=281(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.66−1.85(m,2H),1.87−2.00(m,2H),4.26−4.41(m,2H),4.43−4.55(m,1H),7.20−7.33(m,2H),7.38−7.48(m,2H),7.48−7.63(m,6H).
実施例22A
rac−2−[(ジフェニルメチレン)アミノ]−4−フルオロ−2−メチルブタノニトリル
実施例19Aからのrac−2−[(ジフェニルメチレン)アミノ]−4−フルオロブタノニトリル19.94g(63.64mmol、純度85%)の無水THF421mlへの初期装入に、n−ブチルリチウム(ヘキサン中2.5N)25.71ml(64.28mmol)をアルゴン下−78℃で添加し、混合物を−78℃でさらに10分間攪拌した。その後、ヨードメタン36.1g(254.57mmol)を−78℃で反応溶液に添加した。反応混合物を4.5時間にわたって徐々に0℃にもっていった。0℃で、最初に水、次いで、酢酸エチルを添加し、混合物を飽和塩化ナトリウム水溶液で2回洗浄した。有機相を硫酸ナトリウム上で乾燥させ、濾過および濃縮した。残渣をシリカゲルクロマトグラフィー(移動相:シクロヘキサン/酢酸エチル=15/1)によって精製した。これにより、標的化合物17.2g(理論値の78%、純度81%)が得られた。
LC−MS(方法3):Rt=2.46分
MS(ESpos):m/z=281(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.65−1.67(s,3H),2.30−2.47(m,2H),4.55−4.84(m,2H),7.27−7.32(m,2H),7.37−7.42(m,2H),7.43−7.52(m,6H).
rac−2−[(ジフェニルメチレン)アミノ]−4−フルオロ−2−メチルブタノニトリル
LC−MS(方法3):Rt=2.46分
MS(ESpos):m/z=281(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.65−1.67(s,3H),2.30−2.47(m,2H),4.55−4.84(m,2H),7.27−7.32(m,2H),7.37−7.42(m,2H),7.43−7.52(m,6H).
実施例23A
rac−2−[(ジフェニルメチレン)アミノ]−4,4−ジフルオロ−2−メチルブタノニトリル
実施例20Aからのrac−2−[(ジフェニルメチレン)アミノ]−4,4−ジフルオロブタノニトリル13.07g(38.62mmol)の無水THF255mlへの初期装入に、n−ブチルリチウム(ヘキサン中2.5N)15.6ml(39.0mmol)をアルゴン下−78℃で添加し、混合物を−78℃でさらに10分間攪拌した。その後、ヨードメタン22.6g(154.46mmol)を−78℃で反応溶液に添加した。反応混合物を3.5時間にわたって徐々に0℃にもっていった。0℃で、最初に水、次いで、酢酸エチルを添加し、混合物を飽和塩化ナトリウム水溶液で2回洗浄した。有機相を硫酸ナトリウム上で乾燥させ、濾過および濃縮した。残渣をシリカゲルクロマトグラフィー(移動相:シクロヘキサン/酢酸エチル=15/1)によって精製した。これにより、標的化合物11.4g(理論値の91%、純度92%)が得られた。
LC−MS(方法3):Rt=2.52分
MS(ESpos):m/z=299(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.67(s,3H),2.55−2.77(m,2H),6.14−6.48(m,1H),7.28−7.34(m,2H),7.36−7.44(m,2H),7.44−7.54(m,6H).
rac−2−[(ジフェニルメチレン)アミノ]−4,4−ジフルオロ−2−メチルブタノニトリル
LC−MS(方法3):Rt=2.52分
MS(ESpos):m/z=299(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.67(s,3H),2.55−2.77(m,2H),6.14−6.48(m,1H),7.28−7.34(m,2H),7.36−7.44(m,2H),7.44−7.54(m,6H).
実施例24A
rac−2−[(ジフェニルメチレン)アミノ]−5−フルオロ−2−メチルペンタノニトリル
実施例21Aからのrac−2−[(ジフェニルメチレン)アミノ]−5−フルオロペンタノニトリル16.73g(59.68mmol)の無水THF394mlへの初期装入に、n−ブチルリチウム(ヘキサン中2.5N)24.11ml(60.27mmol)をアルゴン下−78℃で添加し、混合物を−78℃でさらに10分間攪拌した。その後、ヨードメタン34.93g(238.70mmol)を−78℃で反応溶液に添加した。反応混合物を4.5時間にわたって徐々に0℃にもっていった。0℃で、最初に水、次いで、酢酸エチルを添加し、混合物を飽和塩化ナトリウム水溶液で2回洗浄した。有機相を硫酸ナトリウム上で乾燥させ、濾過および濃縮した。残渣をシリカゲルクロマトグラフィー(移動相:シクロヘキサン/酢酸エチル=15/1)によって精製した。これにより、標的化合物18.94g(理論値の95%、純度88%)が得られた。
LC−MS(方法3):Rt=2.55分
MS(ESpos):m/z=295(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.62(s,3H),1.73−1.90(m,2H),1.94−2.03(m,1H),2.04−2.18(m,1H),4.47(t,1H),4.58(t,1H),7.23−7.33(m,2H),7.35−7.43(m,2H),7.44−7.56(m,6H).
rac−2−[(ジフェニルメチレン)アミノ]−5−フルオロ−2−メチルペンタノニトリル
LC−MS(方法3):Rt=2.55分
MS(ESpos):m/z=295(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.62(s,3H),1.73−1.90(m,2H),1.94−2.03(m,1H),2.04−2.18(m,1H),4.47(t,1H),4.58(t,1H),7.23−7.33(m,2H),7.35−7.43(m,2H),7.44−7.56(m,6H).
実施例25A
rac−2−アミノ−4−フルオロ−2−メチルブタノニトリル塩酸塩
実施例22Aからのrac−2−[(ジフェニルメチレン)アミノ]−4−フルオロ−2−メチルブタノニトリル17.45g(50.45mmol;純度81%)をテトラヒドロフラン235.6mlおよび水9.1mlに溶解し、塩化水素溶液(ジエチルエーテル中0.5N)111ml(55.46mmol)を添加し、混合物を室温で一晩攪拌した。次いで、塩化水素溶液(ジエチルエーテル中2N)25.21ml(50.42mmol)を添加し、混合物を濃縮した。単離した粗生成物をさらに精製することなくさらに直接反応させた。
LC−MS(方法3):Rt=0.22分
MS(ESpos):m/z=117(M−HCl+H)+
rac−2−アミノ−4−フルオロ−2−メチルブタノニトリル塩酸塩
LC−MS(方法3):Rt=0.22分
MS(ESpos):m/z=117(M−HCl+H)+
実施例26A
rac−ベンジル(2−シアノ−4−フルオロブタン−2−イル)カルバメート
実施例25Aからの粗生成物rac−2−アミノ−4−フルオロ−2−メチルブタノニトリル塩酸塩を最初にテトラヒドロフラン/水(1:1)165mlに装入し、炭酸カリウム28.57g(206.71mmol)およびクロロギ酸ベンジル9.46g(55.46mmol)を添加した。反応混合物を室温で一晩攪拌した。クロロギ酸ベンジルさらに1.72g(10.1mmol)を反応物に添加し、混合物を室温でさらに2時間攪拌した。次いで、相を分離し、水相を酢酸エチルで2回抽出した。合わせた有機相を飽和塩化ナトリウム水溶液で1回洗浄し、次いで、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。残渣をシリカゲルクロマトグラフィー(移動相:シクロヘキサン/酢酸エチル勾配20/1〜5/1)によって精製した。これにより、標記化合物5.04g(2ステップにわたって理論値の38%)が得られた。
LC−MS(方法3):Rt=1.95分
MS(ESpos):m/z=251(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.59(s,3H),2.20−2.43(m,2H),4.55(t,1H),4.67(t,1H),5.08(s,2H),7.28−7.45(m,5H),8.12(br.s,1H).
rac−ベンジル(2−シアノ−4−フルオロブタン−2−イル)カルバメート
LC−MS(方法3):Rt=1.95分
MS(ESpos):m/z=251(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.59(s,3H),2.20−2.43(m,2H),4.55(t,1H),4.67(t,1H),5.08(s,2H),7.28−7.45(m,5H),8.12(br.s,1H).
実施例27A
rac−2−アミノ−4,4−ジフルオロ−2−メチルブタノニトリル塩酸塩
実施例23Aからのrac−2−[(ジフェニルメチレン)アミノ]−4,4−ジフルオロ−2−メチルブタノニトリル10.84g(33.43mmol;純度92%)をテトラヒドロフラン156mlおよび水6mlに溶解し、塩化水素溶液(ジエチルエーテル中0.5N)73.5ml(36.77mmol)を添加し、混合物を室温で一晩攪拌した。次いで、塩化水素溶液(ジエチルエーテル中2N)16.71ml(33.43mmol)を反応溶液に添加し、混合物を濃縮した。単離した粗生成物をさらに精製することなくさらに直接反応させた。
LC−MS(方法3):Rt=0.32分
MS(ESpos):m/z=135(M−HCl+H)+
rac−2−アミノ−4,4−ジフルオロ−2−メチルブタノニトリル塩酸塩
LC−MS(方法3):Rt=0.32分
MS(ESpos):m/z=135(M−HCl+H)+
実施例28A
rac−ベンジル(2−シアノ−4,4−ジフルオロブタン−2−イル)カルバメート
実施例27Aからの粗生成物rac−2−アミノ−4,4−ジフルオロ−2−メチルブタノニトリル塩酸塩を最初にテトラヒドロフラン/水(1:1)109mlに装入し、炭酸カリウム18.94g(137.06mmol)およびクロロギ酸ベンジル6.27g(36.77mmol)を添加し、混合物を室温で一晩攪拌した。クロロギ酸ベンジルさらに1.14g(6.69mmol)を反応物に添加し、混合物を室温でさらに2時間攪拌した。次いで、相を分離し、水相を酢酸エチルで2回抽出した。合わせた有機相を飽和塩化ナトリウム水溶液で1回洗浄し、次いで、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。残渣をシリカゲルクロマトグラフィー(移動相:シクロヘキサン/酢酸エチル勾配20/1〜5/1)によって精製した。これにより、標記化合物7.68g(2ステップにわたって理論値の61%、純度71%)が得られた。
LC−MS(方法3):Rt=2.04分
MS(ESpos):m/z=269(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.65(s,3H),2.51−2.65(m,2H),5.10(s,2H),6.08−6.41(m,1H),7.27−7.44(m,5H),8.24(br.s,1H).
rac−ベンジル(2−シアノ−4,4−ジフルオロブタン−2−イル)カルバメート
LC−MS(方法3):Rt=2.04分
MS(ESpos):m/z=269(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.65(s,3H),2.51−2.65(m,2H),5.10(s,2H),6.08−6.41(m,1H),7.27−7.44(m,5H),8.24(br.s,1H).
実施例29A
rac−2−アミノ−5−フルオロ−2−メチルペンタノニトリル塩酸塩
実施例24Aからのrac−2−[(ジフェニルメチレン)アミノ]−5−フルオロ−2−メチルペンタノニトリル18.94g(56.62mmol;純度88%)をテトラヒドロフラン264.6mlおよび水10.2mlに溶解し、塩化水素溶液(ジエチルエーテル中0.5N)124.6ml(62.28mmol)を添加し、混合物を室温で一晩攪拌した。次いで、塩化水素溶液(ジエチルエーテル中2N)28.3ml(56.62mmol)を添加し、混合物を濃縮した。単離した粗生成物をさらに精製することなくさらに直接反応させた。
LC−MS(方法3):Rt=0.25分
MS(ESpos):m/z=131(M−HCl+H)+
rac−2−アミノ−5−フルオロ−2−メチルペンタノニトリル塩酸塩
LC−MS(方法3):Rt=0.25分
MS(ESpos):m/z=131(M−HCl+H)+
実施例30A
rac−ベンジル(2−シアノ−5−フルオロペンタン−2−イル)カルバメート
実施例29Aからの粗生成物rac−2−アミノ−5−フルオロ−2−メチルペンタノニトリル塩酸塩を最初にテトラヒドロフラン/水(1:1)185mlに装入し、炭酸カリウム32.09g(232.18mmol)およびクロロギ酸ベンジル10.63g(62.29mmol)を添加し、混合物を室温で一晩攪拌した。クロロギ酸ベンジルさらに1.93g(11.33mmol)を反応物に添加し、混合物を室温でさらに2時間攪拌した。次いで、相を分離し、水相を酢酸エチルで2回抽出した。合わせた有機相を飽和塩化ナトリウム水溶液で1回洗浄し、次いで、硫酸ナトリウム上で乾燥させ、濾過し、濃縮した。残渣をシリカゲルクロマトグラフィー(移動相:シクロヘキサン/酢酸エチル勾配20/1〜5/1)によって精製した。標的化合物11.77gが得られた(2段階にわたって理論値の72%、純度92%)。
LC−MS(方法3):Rt=2.03分
MS(ESpos):m/z=265(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.55(s,3H),1.66−1.85(m,2H),1.86−2.04(m,2H),4.40(t,1H),4.52(t,1H),5.08(s,2H),7.28−7.44(m,5H),8.05(br.s,1H).
rac−ベンジル(2−シアノ−5−フルオロペンタン−2−イル)カルバメート
LC−MS(方法3):Rt=2.03分
MS(ESpos):m/z=265(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.55(s,3H),1.66−1.85(m,2H),1.86−2.04(m,2H),4.40(t,1H),4.52(t,1H),5.08(s,2H),7.28−7.44(m,5H),8.05(br.s,1H).
実施例31A
ent−ベンジル(2−シアノ−4,4−ジフルオロブタン−2−イル)カルバメート(エナンチオマーA)
実施例28Aからのrac−ベンジル(2−シアノ−4,4−ジフルオロブタン−2−イル)カルバメート7.68g(20.33mmol、純度71%)をキラル相[カラム:Daicel Chiralpak AY−H、5μm、250×20mm、移動相:80%イソヘキサン、20%イソプロパノール、流量:25ml/分;温度:22℃、検出:210nm]で分取法によりエナンチオマーに分離した。
エナンチオマーA:収量:2.64g(>99%ee)
Rt=6.67分[Chiralpak AY−H、5μm、250×4.6mm;移動相:80%イソヘキサン、20%イソプロパノール;流量3ml/分;検出:220nm]。
ent−ベンジル(2−シアノ−4,4−ジフルオロブタン−2−イル)カルバメート(エナンチオマーA)
エナンチオマーA:収量:2.64g(>99%ee)
Rt=6.67分[Chiralpak AY−H、5μm、250×4.6mm;移動相:80%イソヘキサン、20%イソプロパノール;流量3ml/分;検出:220nm]。
実施例32A
ent−ベンジル(2−シアノ−4,4−ジフルオロブタン−2−イル)カルバメート(エナンチオマーB)
実施例28Aからのrac−ベンジル(2−シアノ−4,4−ジフルオロブタン−2−イル)カルバメート7.68g(20.33mmol、純度71%)をキラル相[カラム:Daicel Chiralpak AY−H、5μm、250×20mm、移動相:80%イソヘキサン、20%イソプロパノール、流量:25ml/分;温度:22℃、検出:210nm]で分取法によりエナンチオマーに分離した。
エナンチオマーB:収量:2.76g(93%ee)
Rt=7.66分[Chiralpak AY−H、5μm、250×4.6mm;移動相:80%イソヘキサン、20%イソプロパノール;流量3ml/分;検出:220nm]。
ent−ベンジル(2−シアノ−4,4−ジフルオロブタン−2−イル)カルバメート(エナンチオマーB)
エナンチオマーB:収量:2.76g(93%ee)
Rt=7.66分[Chiralpak AY−H、5μm、250×4.6mm;移動相:80%イソヘキサン、20%イソプロパノール;流量3ml/分;検出:220nm]。
実施例33A
ent−ベンジル(2−シアノ−5−フルオロペンタン−2−イル)カルバメート(エナンチオマーA)
実施例30Aからのrac−ベンジル(2−シアノ−5−フルオロブタン−2−イル)カルバメート11.77g(40.97mmol、純度92%)をキラル相[カラム:SFC Daicel Chiralpak AZ−H、5μm、250×30mm、移動相:90%CO2、10%メタノール、流量:100ml/分;温度:40℃、検出:210nm]で分取法によりエナンチオマーに分離した。
エナンチオマーA:収量:5.7g(>99%ee)
Rt=1.76分[SFC Chiralpak AZ−3、3μm、50×4.6mm;移動相:CO2/メタノール勾配(5%〜60%メタノール);流量:3ml/分;検出:220nm]。
ent−ベンジル(2−シアノ−5−フルオロペンタン−2−イル)カルバメート(エナンチオマーA)
エナンチオマーA:収量:5.7g(>99%ee)
Rt=1.76分[SFC Chiralpak AZ−3、3μm、50×4.6mm;移動相:CO2/メタノール勾配(5%〜60%メタノール);流量:3ml/分;検出:220nm]。
実施例34A
ent−ベンジル(2−シアノ−5−フルオロペンタン−2−イル)カルバメート(エナンチオマーB)
実施例30Aからのrac−ベンジル(2−シアノ−5−フルオロブタン−2−イル)カルバメート11.77g(40.97mmol、純度92%)をキラル相[カラム:SFC Daicel Chiralpak AZ−H、5μm、250×30mm、移動相:90%CO2、10%メタノール、流量:100ml/分;温度:40℃、検出:210nm]で分取法によりエナンチオマーに分離した。
エナンチオマーB:収量:5.0g(>99%ee)
Rt=1.97分[SFC Chiralpak AZ−3、3μm、50×4.6mm;移動相:CO2/メタノール勾配(5%〜60%メタノール);流量:3ml/分;検出:220nm]。
ent−ベンジル(2−シアノ−5−フルオロペンタン−2−イル)カルバメート(エナンチオマーB)
エナンチオマーB:収量:5.0g(>99%ee)
Rt=1.97分[SFC Chiralpak AZ−3、3μm、50×4.6mm;移動相:CO2/メタノール勾配(5%〜60%メタノール);流量:3ml/分;検出:220nm]。
実施例35A
ent−ベンジル(1−アミノ−4,4−ジフルオロ−2−メチルブタン−2−イル)カルバメート(エナンチオマーA)
実施例31Aからのent−ベンジル(2−シアノ−4,4−ジフルオロブタン−2−イル)カルバメート(エナンチオマーA)2.3g(8.57mmol)をメタノール中7Nアンモニア溶液75mlに溶解し、ラネーニッケル(50%水性スラリー)2.66gをアルゴン下で添加した。反応混合物をオートクレーブ中20〜30barで1.5時間水素付加した。反応混合物をCeliteを通して濾過し、メタノールおよびメタノール中2Nアンモニアですすぎ、濃縮した。これにより、標的化合物2.23g(理論値の94%)が得られた。
LC−MS(方法3):Rt=1.48分
MS(ESpos):m/z=273(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.19(s,3H),1.48(br.s,2H),2.08−2.40(m,2H),2.53−2.72(m,2H;partially overlapped with solvent peak),5.00(s,2H),5.90−6.23(m,1H),6.95(br.s,1H),7.25−7.41(m,5H).
ent−ベンジル(1−アミノ−4,4−ジフルオロ−2−メチルブタン−2−イル)カルバメート(エナンチオマーA)
LC−MS(方法3):Rt=1.48分
MS(ESpos):m/z=273(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.19(s,3H),1.48(br.s,2H),2.08−2.40(m,2H),2.53−2.72(m,2H;partially overlapped with solvent peak),5.00(s,2H),5.90−6.23(m,1H),6.95(br.s,1H),7.25−7.41(m,5H).
実施例36A
ent−ベンジル(1−アミノ−4,4−ジフルオロ−2−メチルブタン−2−イル)カルバメート(エナンチオマーB)
実施例32Aからのent−ベンジル(2−シアノ−4,4−ジフルオロブタン−2−イル)カルバメート(エナンチオマーB)2.76g(10.29mmol)をメタノール中7Nアンモニア溶液90mlに溶解し、ラネーニッケル(50%水性スラリー)3.19gをアルゴン下で添加した。反応混合物をオートクレーブ中20〜30barで1.5時間水素付加した。反応混合物をCeliteを通して濾過し、メタノールおよびメタノール中2Nアンモニアですすぎ、濃縮した。これにより、標的化合物2.64g(理論値の88%、純度93%)が得られた。
LC−MS(方法3):Rt=1.49分
MS(ESpos):m/z=273(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.19(s,3H),1.48(br.s,2H),2.08−2.40(m,2H),2.53−2.73(m,2H;partially overlapped with solvent peak),5.00(s,2H),5.90−6.24(m,1H),6.95(br.s,1H),7.25−7.41(m,5H).
ent−ベンジル(1−アミノ−4,4−ジフルオロ−2−メチルブタン−2−イル)カルバメート(エナンチオマーB)
LC−MS(方法3):Rt=1.49分
MS(ESpos):m/z=273(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.19(s,3H),1.48(br.s,2H),2.08−2.40(m,2H),2.53−2.73(m,2H;partially overlapped with solvent peak),5.00(s,2H),5.90−6.24(m,1H),6.95(br.s,1H),7.25−7.41(m,5H).
実施例37A
ent−ベンジル(1−アミノ−5−フルオロ−2−メチルペンタン−2−イル)カルバメート(エナンチオマーA)
実施例33Aからのent−ベンジル(2−シアノ−5−フルオロペンタン−2−イル)カルバメート(エナンチオマーA)5.7g(21.57mmol)をメタノール中7Nアンモニア溶液125mlに溶解し、ラネーニッケル(50%水性スラリー)6.68gをアルゴン下で添加した。反応混合物をオートクレーブ中20〜30barで4.5時間水素付加した。反応混合物をCeliteを通して濾過し、メタノールおよびメタノール中2Nアンモニアですすぎ、濃縮した。これにより、標的化合物5.22g(理論値の77%、純度85%)が得られた。
LC−MS(方法3):Rt=1.51分
MS(ESpos):m/z=269(M+H)+
ent−ベンジル(1−アミノ−5−フルオロ−2−メチルペンタン−2−イル)カルバメート(エナンチオマーA)
LC−MS(方法3):Rt=1.51分
MS(ESpos):m/z=269(M+H)+
実施例38A
ent−ベンジル(1−アミノ−5−フルオロ−2−メチルペンタン−2−イル)カルバメート(エナンチオマーB)
実施例34Aからのent−ベンジル(2−シアノ−5−フルオロペンタン−2−イル)カルバメート(エナンチオマーB)5.0g(18.92mmol)をメタノール中7Nアンモニア溶液110mlに溶解し、ラネーニッケル(50%水性スラリー)5.86gをアルゴン下で添加した。反応混合物をオートクレーブ中20〜30barで4.5時間水素付加した。反応混合物をCeliteを通して濾過し、メタノールおよびメタノール中2Nアンモニアですすぎ、濃縮した。これにより、標的化合物4.6g(理論値の84%、純度93%)が得られた。
LC−MS(方法3):Rt=1.47分
MS(ESpos):m/z=269(M+H)+
ent−ベンジル(1−アミノ−5−フルオロ−2−メチルペンタン−2−イル)カルバメート(エナンチオマーB)
LC−MS(方法3):Rt=1.47分
MS(ESpos):m/z=269(M+H)+
実施例39A
rac−4−フルオロ−2−メチルブタン−1,2−ジアミン二塩酸塩
実施例26Aからのrac−ベンジル(2−シアノ−4−フルオロブタン−2−イル)カルバメート1.00g(4.00mmol)をエタノール/氷酢酸(1/1)114mlに溶解し、パラジウム活性炭素(10%)0.85gを添加した。反応混合物をオートクレーブ中30〜50barで3時間水素付加した。反応混合物を濾過し、濾過ケークをエタノールですすぎ、次いで、濾液をMilliporeフィルタを通してもう一回濾過した。濾液を塩化水素溶液(ジエチルエーテル中2N)10mlと混和し、次いで、濃縮した。標的化合物1.04gが得られ、これをさらに精製することなく後の段階に使用した。
LC−MS(方法3):Rt=0.19分
MS(ESpos):m/z=121(M−2HCl+H)+
rac−4−フルオロ−2−メチルブタン−1,2−ジアミン二塩酸塩
LC−MS(方法3):Rt=0.19分
MS(ESpos):m/z=121(M−2HCl+H)+
実施例40A
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−3−フルオロ−2−メチルプロパン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)
実施例5Aからの6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボン酸150mg(0.43mmol)をDMF2.7mlに溶解し、HATU178mg(0.47mmol)およびN,N−ジイソプロピルエチルアミン0.22ml(1.28mmol)を添加し、混合物を室温で20分間攪拌した。その後、実施例12Aからのent−ベンジル(1−アミノ−3−フルオロ−2−メチルプロパン−2−イル)カルバメート(エナンチオマーB)133mg(0.47mmol、純度85%)を添加した。混合物を室温で一晩攪拌し、次いで、分取HPLC(RP18カラム、移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。これにより、標記化合物216mg(理論値の63%;純度86%)が得られた。
LC−MS(方法2):Rt=1.23分
MS(ESpos):m/z=575(M−TFA+H)+
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−3−フルオロ−2−メチルプロパン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)
LC−MS(方法2):Rt=1.23分
MS(ESpos):m/z=575(M−TFA+H)+
実施例41A
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−5,5,5−トリフルオロ−2−メチルペンタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)
実施例5Aからの6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボン酸150mg(0.43mmol)をDMF2.7mlに溶解し、HATU178mg(0.47mmol)およびN,N−ジイソプロピルエチルアミン0.22ml(1.28mmol)を添加し、混合物を室温で20分間攪拌した。その後、実施例18Aからのent−ベンジル(1−アミノ−5,5,5−トリフルオロ−2−メチルペンタン−2−イル)カルバメート(エナンチオマーB)150mg(0.47mmol、純度95%)を添加した。混合物を室温で一晩攪拌し、次いで、分取HPLC(RP18カラム、移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。標記化合物261mgが得られた(理論値の79%)。
LC−MS(方法2):Rt=1.31分
MS(ESpos):m/z=639(M−TFA+H)+
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−5,5,5−トリフルオロ−2−メチルペンタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)
LC−MS(方法2):Rt=1.31分
MS(ESpos):m/z=639(M−TFA+H)+
実施例42A
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−5−フルオロ−2−メチルペンタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーA)
実施例5Aからの6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボン酸250mg(0.71mmol)をDMF2.36mlに溶解し、HATU350mg(0.92mmol)およびN,N−ジイソプロピルエチルアミン0.62ml(3.54mmol)を添加し、混合物を室温で20分間攪拌した。その後、実施例37Aからのent−ベンジル(1−アミノ−5−フルオロ−2−メチルペンタン−2−イル)カルバメート(エナンチオマーA)291mg(0.92mmol、純度85%)を添加した。30分後、水を添加し、得られた固体を濾別し、水で洗浄した。残渣を分取HPLC(RP18カラム;移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。これにより、標記化合物397mg(理論値の71%;純度91%)が得られた。
LC−MS(方法2):Rt=1.21分
MS(ESpos):m/z=603(M−TFA+H)+
1H−NMR(500 MHz,DMSO−d6):δ[ppm]=1.22(s,3H),1.52−1.75(m,3H),1.80−1.93(m,1H),2.53(s,3H;overlapped with solvent peak),3.49−3.61(m,2H),4.29−4.38(m,1H),4.39−4.51(m,1H),5.00(s,2H),5.37(s,2H),7.07−7.17(m,1H),7.20−7.38(m,8H),7.54−7.64(m,1H),7.90(t,1H),8.76(d,1H).
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−5−フルオロ−2−メチルペンタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーA)
LC−MS(方法2):Rt=1.21分
MS(ESpos):m/z=603(M−TFA+H)+
1H−NMR(500 MHz,DMSO−d6):δ[ppm]=1.22(s,3H),1.52−1.75(m,3H),1.80−1.93(m,1H),2.53(s,3H;overlapped with solvent peak),3.49−3.61(m,2H),4.29−4.38(m,1H),4.39−4.51(m,1H),5.00(s,2H),5.37(s,2H),7.07−7.17(m,1H),7.20−7.38(m,8H),7.54−7.64(m,1H),7.90(t,1H),8.76(d,1H).
実施例43A
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−5−フルオロ−2−メチルペンタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)
実施例5Aからの6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボン酸250mg(0.71mmol)をDMF2.36mlに溶解し、HATU350mg(0.92mmol)およびN,N−ジイソプロピルエチルアミン0.62ml(3.54mmol)を添加し、混合物を室温で20分間攪拌した。その後、実施例38Aからのent−ベンジル(1−アミノ−5−フルオロ−2−メチルペンタン−2−イル)カルバメート(エナンチオマーB)267mg(0.92mmol、純度93%)を添加した。30分後、水を添加し、得られた固体を濾別し、水で洗浄した。残渣を分取HPLC(RP18カラム;移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。これにより、標記化合物328mg(理論値の61%;純度94%)が得られた。
LC−MS(方法2):Rt=1.21分
MS(ESpos):m/z=603(M−TFA+H)+
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−5−フルオロ−2−メチルペンタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)
LC−MS(方法2):Rt=1.21分
MS(ESpos):m/z=603(M−TFA+H)+
実施例44A
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−4,4−ジフルオロ−2−メチルブタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーA)
実施例5Aからの6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボン酸250mg(0.71mmol)をDMF2.36mlに溶解し、HATU350mg(0.92mmol)およびN,N−ジイソプロピルエチルアミン0.62ml(3.54mmol)を添加し、混合物を室温で20分間攪拌した。次いで、実施例35Aからのent−ベンジル(1−アミノ−4,4−ジフルオロ−2−メチルブタン−2−イル)カルバメート(エナンチオマーA)256mg(0.92mmol)を添加した。60分後、水を添加し、得られた固体を濾別し、水で洗浄した。残渣を分取HPLC(RP18カラム;移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。標記化合物439mgが得られた(理論値の86%)。
LC−MS(方法2):Rt=1.22分
MS(ESpos):m/z=607(M−TFA+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.29(s,3H),2.05−2.25(m,2H),2.53(s,3H;overlapped with solvent peak),3.54−3.62(m,2H;overlapped with solvent peak),5.01(s,2H),5.38(s,2H),5.98−6.29(m,1H),7.18−7.39(m,9H),7.54−7.64(m,1H),7.95(t,1H),8.74(d,1H).
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−4,4−ジフルオロ−2−メチルブタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーA)
LC−MS(方法2):Rt=1.22分
MS(ESpos):m/z=607(M−TFA+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.29(s,3H),2.05−2.25(m,2H),2.53(s,3H;overlapped with solvent peak),3.54−3.62(m,2H;overlapped with solvent peak),5.01(s,2H),5.38(s,2H),5.98−6.29(m,1H),7.18−7.39(m,9H),7.54−7.64(m,1H),7.95(t,1H),8.74(d,1H).
実施例45A
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−4,4−ジフルオロ−2−メチルブタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)
実施例5Aからの6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボン酸250mg(0.71mmol)をDMF2.36mlに溶解し、HATU323mg(0.85mmol)およびN,N−ジイソプロピルエチルアミン0.62ml(3.54mmol)を添加し、混合物を室温で20分間攪拌した。次いで、実施例36Aからのent−ベンジル(1−アミノ−4,4−ジフルオロ−2−メチルブタン−2−イル)カルバメート(エナンチオマーB)246mg(0.85mmol、純度93%)を添加した。30分後、水を添加し、得られた固体を濾別し、水で洗浄した。残渣を分取HPLC(RP18カラム;移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。標記化合物431mgが得られた(理論値の82%)。
LC−MS(方法2):Rt=1.21分
MS(ESpos):m/z=607(M−TFA+H)+
1H−NMR(500 MHz,DMSO−d6):δ[ppm]=1.29(s,3H),2.06−2.24(m,2H),2.53(s,3H;overlapped with solvent peak),3.55−3.62(m,2H),5.01(s,2H),5.38(s,2H),6.00−6.29(m,1H),7.19−7.39(m,9H),7.55−7.64(m,1H),7.99(t,1H),8.74(d,1H).
ent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−4,4−ジフルオロ−2−メチルブタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)
LC−MS(方法2):Rt=1.21分
MS(ESpos):m/z=607(M−TFA+H)+
1H−NMR(500 MHz,DMSO−d6):δ[ppm]=1.29(s,3H),2.06−2.24(m,2H),2.53(s,3H;overlapped with solvent peak),3.55−3.62(m,2H),5.01(s,2H),5.38(s,2H),6.00−6.29(m,1H),7.19−7.39(m,9H),7.55−7.64(m,1H),7.99(t,1H),8.74(d,1H).
実施例:
実施例1
ent−N−(2−アミノ−3−フルオロ−2−メチルプロピル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
実施例40Aからのent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−3−フルオロ−2−メチルプロパン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)216mg(0.27mmol、純度86%)をエタノール7mlに溶解し、パラジウム活性炭素(10%)14mgを添加し、混合物を標準圧力で1時間水素化した。反応溶液をMilliporeフィルタを通して濾過し、濾過ケークをエタノールで洗浄し、濾液を濃縮した。残渣を分取HPLC(RP18カラム;移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって2回精製した。生成物分画を合わせ、濃縮した。その後、残渣をジクロロメタンおよび少量のメタノールに溶解し、少量の飽和重炭酸ナトリウム水溶液で2回洗浄した。水相をジクロロメタンで2回抽出した。合わせた有機相を硫酸ナトリウム上で乾燥させ、濾過し、濃縮および凍結乾燥した。これにより、標的化合物53mg(理論値の43%)が得られた。
LC−MS(方法2):Rt=0.70分
MS(ESpos):m/z=441(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.04(s,3H),1.65(br.s,2H),2.55(s,3H;overlapped with solvent peak),3.24−3.39(m,2H;overlapped with solvent peak),4.08−4.14(m,1H),4.19−4.27(m,1H),5.35(s,2H),7.16−7.29(m,3H),7.55−7.65(m,1H),7.72−7.81(m,1H),8.75(d,1H).
実施例1
ent−N−(2−アミノ−3−フルオロ−2−メチルプロピル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
LC−MS(方法2):Rt=0.70分
MS(ESpos):m/z=441(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.04(s,3H),1.65(br.s,2H),2.55(s,3H;overlapped with solvent peak),3.24−3.39(m,2H;overlapped with solvent peak),4.08−4.14(m,1H),4.19−4.27(m,1H),5.35(s,2H),7.16−7.29(m,3H),7.55−7.65(m,1H),7.72−7.81(m,1H),8.75(d,1H).
実施例2
ent−N−(2−アミノ−5,5,5−トリフルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
実施例41Aからのent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−5,5,5−トリフルオロ−2−メチルペンタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)261mg(0.35mmol)をエタノール9mlに溶解し、パラジウム活性炭素(10%)11mgを添加し、混合物を標準圧力で1.5時間水素化した。反応溶液をMilliporeフィルタを通して濾過し、濾過ケークをエタノールで洗浄し、濾液を濃縮した。残渣を分取HPLC(RP18カラム;移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。その後、残渣をジクロロメタンおよび少量のメタノールに溶解し、少量の飽和重炭酸ナトリウム水溶液で2回洗浄した。水相をジクロロメタンで2回抽出した。合わせた有機相を硫酸ナトリウム上で乾燥させ、濾過し、濃縮および凍結乾燥した。これにより、標的化合物105mg(理論値の59%)が得られた。
LC−MS(方法2):Rt=0.80分
MS(ESpos):m/z=505(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.02(s,3H),1.47−1.61(m,4H),2.24−2.47(m,2H),2.53(s,3H;overlapped with solvent peak),3.18−3.28(m,2H),5.35(s,2H),7.17−7.29(m,3H),7.55−7.65(m,1H),7.81−7.92(m,1H),8.70(d,1H).
ent−N−(2−アミノ−5,5,5−トリフルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
LC−MS(方法2):Rt=0.80分
MS(ESpos):m/z=505(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.02(s,3H),1.47−1.61(m,4H),2.24−2.47(m,2H),2.53(s,3H;overlapped with solvent peak),3.18−3.28(m,2H),5.35(s,2H),7.17−7.29(m,3H),7.55−7.65(m,1H),7.81−7.92(m,1H),8.70(d,1H).
実施例3
ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
実施例42Aからのent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−5−フルオロ−2−メチルペンタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーA)397mg(0.50mmol、純度91%)をエタノール12.9mlに溶解し、パラジウム活性炭素(10%)16mgを添加し、混合物を標準圧力で1.5時間水素化した。反応溶液をMilliporeフィルタによって濾過し、濾液を濃縮した。残渣を分取HPLC(RP18カラム;移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。その後、残渣をジクロロメタンおよび少量のメタノールに溶解し、少量の飽和重炭酸ナトリウム水溶液で2回洗浄した。水相をジクロロメタンで2回抽出した。合わせた有機相を硫酸ナトリウム上で乾燥させ、濾過し、濃縮および凍結乾燥した。これにより、標的化合物133mg(理論値の55%)が得られた。
LC−MS(方法2):Rt=0.71分
MS(ESpos):m/z=469(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.02(s,3H),1.30−1.42(m,2H),1.43−1.88(m,4H),2.53(s,3H;overlapped by solvent peak),3.17−3.30(m,2H),4.30−4.39(m,1H),4.42−4.52(m,1H),5.34(s,2H),7.18−7.30(m,3H),7.55−7.64(m,1H),7.78(br.s,1H),8.75(d,1H).
ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
LC−MS(方法2):Rt=0.71分
MS(ESpos):m/z=469(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.02(s,3H),1.30−1.42(m,2H),1.43−1.88(m,4H),2.53(s,3H;overlapped by solvent peak),3.17−3.30(m,2H),4.30−4.39(m,1H),4.42−4.52(m,1H),5.34(s,2H),7.18−7.30(m,3H),7.55−7.64(m,1H),7.78(br.s,1H),8.75(d,1H).
実施例4
ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
実施例43Aからのent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−5−フルオロ−2−メチルペンタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)328mg(0.43mmol、純度94%)をエタノール11.1mlに溶解し、パラジウム活性炭素(10%)14mgを添加し、混合物を標準圧力で3時間水素化した。反応溶液をCeliteを通して濾過し、濾過ケークをエタノールで洗浄し、濾液を濃縮した。残渣を分取HPLC(RP18カラム;移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。その後、残渣をジクロロメタンおよび少量のメタノールに溶解し、少量の飽和重炭酸ナトリウム水溶液で2回洗浄した。水相をジクロロメタンで2回抽出した。合わせた有機相を硫酸ナトリウム上で乾燥させ、濾過し、濃縮および凍結乾燥した。これにより、標的化合物133mg(理論値の63%)が得られた。
LC−MS(方法4):Rt=2.35分
MS(ESpos):m/z=469(M+H)+
1H−NMR(500 MHz,DMSO−d6):δ[ppm]=1.01(s,3H),1.32−1.42(m,2H),1.45−1.83(m,4H),2.53(s,3H;overlapped by solvent peak),3.18−3.29(m,2H),4.32−4.39(m,1H),4.43−4.50(m,1H),5.34(s,2H),7.18−7.28(m,3H),7.56−7.63(m,1H),7.78(br.s,1H),8.75(d,1H).
ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
LC−MS(方法4):Rt=2.35分
MS(ESpos):m/z=469(M+H)+
1H−NMR(500 MHz,DMSO−d6):δ[ppm]=1.01(s,3H),1.32−1.42(m,2H),1.45−1.83(m,4H),2.53(s,3H;overlapped by solvent peak),3.18−3.29(m,2H),4.32−4.39(m,1H),4.43−4.50(m,1H),5.34(s,2H),7.18−7.28(m,3H),7.56−7.63(m,1H),7.78(br.s,1H),8.75(d,1H).
実施例5
ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
実施例44Aからのent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−4,4−ジフルオロ−2−メチルブタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーA)439mg(0.61mmol)をエタノール15.6mlに溶解し、パラジウム活性炭素(10%)19mgを添加し、混合物を標準圧力で75分間水素化した。反応溶液をMilliporeフィルタを通して濾過し、濾液を濃縮した。残渣を分取HPLC(RP18カラム;移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。その後、残渣をジクロロメタンおよび少量のメタノールに溶解し、少量の飽和重炭酸ナトリウム水溶液で2回洗浄した。水相をジクロロメタンで2回抽出した。合わせた有機相を硫酸ナトリウム上で乾燥させ、濾過し、濃縮および凍結乾燥した。これにより、標的化合物152mg(理論値の52%)が得られた。
LC−MS(方法2):Rt=0.75分
MS(ESpos):m/z=473(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.08(s,3H),1.70(br.s,2H),1.83−1.99(m,2H),2.53(s,3H;overlapped by solvent peak),3.20−3.35(m,2H;overlapped with solvent peak),5.34(s,2H),6.08−6.42(m,1H),7.18−7.29(m,3H),7.55−7.64(m,1H),7.82(t,1H),8.73(d,1H).
ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
LC−MS(方法2):Rt=0.75分
MS(ESpos):m/z=473(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ[ppm]=1.08(s,3H),1.70(br.s,2H),1.83−1.99(m,2H),2.53(s,3H;overlapped by solvent peak),3.20−3.35(m,2H;overlapped with solvent peak),5.34(s,2H),6.08−6.42(m,1H),7.18−7.29(m,3H),7.55−7.64(m,1H),7.82(t,1H),8.73(d,1H).
実施例6
ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
実施例45Aからのent−ベンジル{1−[({6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−イル}カルボニル)アミノ]−4,4−ジフルオロ−2−メチルブタン−2−イル}カルバメートトリフルオロアセテート(エナンチオマーB)431mg(0.59mmol、純度97%)をエタノール15.1mlに溶解し、パラジウム活性炭素(10%)19mgを添加し、混合物を標準圧力で75分間水素化した。反応溶液をMilliporeフィルタを通して濾過し、濾液を濃縮した。残渣を分取HPLC(RP18カラム;移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。その後、残渣をジクロロメタンおよび少量のメタノールに溶解し、少量の飽和重炭酸ナトリウム水溶液で2回洗浄した。水相をジクロロメタンで2回抽出した。合わせた有機相を硫酸ナトリウム上で乾燥させ、濾過し、濃縮および凍結乾燥した。これにより、標的化合物149mg(理論値の53%)が得られた。
LC−MS(方法2):Rt=0.74分
MS(ESpos):m/z=473(M+H)+
1H−NMR(500 MHz,DMSO−d6):δ[ppm]=1.08(s,3H),1.70(br.s,2H),1.84−1.98(m,2H),2.53(s,3H;overlapped by solvent peak),3.22−3.32(m,2H;overlapped with solvent peak),5.34(s,2H),6.10−6.38(m,1H),7.18−7.28(m,3H),7.57−7.63(m,1H),7.82(t,1H),8.73(d,1H).
ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
LC−MS(方法2):Rt=0.74分
MS(ESpos):m/z=473(M+H)+
1H−NMR(500 MHz,DMSO−d6):δ[ppm]=1.08(s,3H),1.70(br.s,2H),1.84−1.98(m,2H),2.53(s,3H;overlapped by solvent peak),3.22−3.32(m,2H;overlapped with solvent peak),5.34(s,2H),6.10−6.38(m,1H),7.18−7.28(m,3H),7.57−7.63(m,1H),7.82(t,1H),8.73(d,1H).
実施例7
rac−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(ラセミ体)
実施例5Aからの6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボン酸200mg(0.57mmol)を最初にHATU259mg(0.68mmol)およびN,N−ジイソプロピルエチルアミン0.40ml(2.27mmol)と一緒にDMF2.6mlに装入し、混合物を20分間攪拌した。実施例39Aからのrac−4−フルオロ−2−メチルブタン−1,2−ジアミン塩酸塩311mg(純度約75%と仮定して1.20mmol)のDMF1.3mlおよびN,N−ジイソプロピルエチルアミン0.59ml(3.40mmol)中溶液を最初の反応混合物に添加し、混合物を室温で0.5時間攪拌した。アセトニトリル、水およびTFAを添加し、生成物を分取HPLC(RP18カラム、移動相:0.1%TFAを添加したアセトニトリル/水勾配)によって精製した。生成物分画を合わせ、濃縮した。その後、残渣をジクロロメタンおよび少量のメタノールに溶解し、飽和重炭酸ナトリウム水溶液で2回洗浄した。水相をジクロロメタンで2回抽出した。合わせた有機相を硫酸ナトリウム上で乾燥させ、濾過し、濃縮および凍結乾燥した。これにより、標的化合物100mg(理論値の38%)が得られた。
LC−MS(方法2):Rt=0.69分
MS(ESpos):m/z=455(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.04(s,3H),1.58(br.s,2H),1.66 − 1.84(m,2H),2.56(s,3H),3.19−3.32(m,2H;partially overlapped with solvent peak),4.55−4.63(m,1H),4.66−4.73(m,1H),5.34(s,2H),7.17−7.28(m,3H),7.56−7.64(m,1H),7.72−7.83(m,1H),8.74(d,1H).
rac−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(ラセミ体)
LC−MS(方法2):Rt=0.69分
MS(ESpos):m/z=455(M+H)+
1H−NMR(400 MHz,DMSO−d6):δ=1.04(s,3H),1.58(br.s,2H),1.66 − 1.84(m,2H),2.56(s,3H),3.19−3.32(m,2H;partially overlapped with solvent peak),4.55−4.63(m,1H),4.66−4.73(m,1H),5.34(s,2H),7.17−7.28(m,3H),7.56−7.64(m,1H),7.72−7.83(m,1H),8.74(d,1H).
実施例8
ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
実施例7からのrac−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド135mg(0.30mmol)をキラル相[カラム:Daicel Chiralpak IF、5μm、250×20mm、移動相:100%エタノール+0.2%ジエチルアミン、流量:15ml/分;温度:45℃、検出:220nm]で分取法によりエナンチオマーに分離した。
生成物分画をドライアイス上で回収し、濃縮し(浴温度:30℃)、凍結乾燥した。
エナンチオマーA:収量:50mg(>99%ee)
Rt=6.85分[Chiralpak AZ−H、5μm、250×4.6mm;移動相:100%エタノール+0.2%ジエチルアミン;流量:1ml/分;検出:270nm]。
ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)
生成物分画をドライアイス上で回収し、濃縮し(浴温度:30℃)、凍結乾燥した。
エナンチオマーA:収量:50mg(>99%ee)
Rt=6.85分[Chiralpak AZ−H、5μm、250×4.6mm;移動相:100%エタノール+0.2%ジエチルアミン;流量:1ml/分;検出:270nm]。
実施例9
ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
実施例7からのrac−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド135mgをキラル相[カラム:Daicel Chiralpak IF、5μm、250×20mm、移動相:100%エタノール+0.2%ジエチルアミン、流量:15ml/分;温度:45℃、検出:220nm]で分取法によりエナンチオマーに分離した。
生成物分画をドライアイス上で回収し、濃縮し(浴温度:30℃)、凍結乾燥した。
エナンチオマーB:収量:50mg(96%ee)
Rt=9.80分[Chiralpak AZ−H、5μm、250×4.6mm;移動相:100%エタノール+0.2%ジエチルアミン;流量:1ml/分;検出:270nm]。
ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)
生成物分画をドライアイス上で回収し、濃縮し(浴温度:30℃)、凍結乾燥した。
エナンチオマーB:収量:50mg(96%ee)
Rt=9.80分[Chiralpak AZ−H、5μm、250×4.6mm;移動相:100%エタノール+0.2%ジエチルアミン;流量:1ml/分;検出:270nm]。
B.薬理学的有効性の評価
以下の略語を使用する:
以下の略語を使用する:
本発明の化合物の薬理作用を以下のアッセイで証明することができる:
B−1.PPi検出によるsGC酵素活性の測定
可溶性グアニリルシクラーゼ(sGC)は、刺激されると、GTPをcGMPおよびピロリン酸(PPi)に変換する。PPiは、国際公開第2008/061626号パンフレットに記載されている方法を用いて検出する。アッセイで生じるシグナルは、反応が進行するにつれて増加するので、sGC酵素活性の尺度として働く。PPi基準曲線を用いて、酵素を既知の様式で、例えば、変換速度、刺激性またはミカエリス定数に関して、特徴付けることができる。
B−1.PPi検出によるsGC酵素活性の測定
可溶性グアニリルシクラーゼ(sGC)は、刺激されると、GTPをcGMPおよびピロリン酸(PPi)に変換する。PPiは、国際公開第2008/061626号パンフレットに記載されている方法を用いて検出する。アッセイで生じるシグナルは、反応が進行するにつれて増加するので、sGC酵素活性の尺度として働く。PPi基準曲線を用いて、酵素を既知の様式で、例えば、変換速度、刺激性またはミカエリス定数に関して、特徴付けることができる。
試験の遂行
試験を行うために、酵素溶液29μl(50mM TEA、2mM塩化マグネシウム、0.1%BSA(分画V)、0.005%Brij 35、pH7.5中0〜10nM可溶性グアニリルシクラーゼ(Honickaら、Journal of Molecular Medicine 77(1999)14〜23により調製))を最初にマイクロプレートに装入し、刺激溶液1μl(DMSO中0〜10μM 3−モルホリノシドノンイミン、SIN−1、Merck)を添加した。マイクロプレートを室温で10分間インキュベートした。次いで、検出混合物20μl(50mM TEA、2mM塩化マグネシウム、0.1%BSA(分画V)、0.005%Brij 35、pH7.5中1.2nMホタルルシフェラーゼ(フォティナス・ピラリス(Photinus pyralis)ルシフェラーゼ、Promega)、29μMデヒドロルシフェリン(Bitler&McElroy、Arch.Biochem.Biophys.72(1957)358により調製)、122μMルシフェリン(Promega)、153μM ATP(Sigma)および0.4mM DTT(Sigma))を添加した。基質溶液20μl(50mM TEA、2mM塩化マグネシウム、0.1%BSA(分画V)、0.005%Brij 35、pH7.5中1.25mMグアノシン5’三リン酸(Sigma))を添加することによって、酵素反応を開始し、ルミノメータで連続的に分析した。
試験を行うために、酵素溶液29μl(50mM TEA、2mM塩化マグネシウム、0.1%BSA(分画V)、0.005%Brij 35、pH7.5中0〜10nM可溶性グアニリルシクラーゼ(Honickaら、Journal of Molecular Medicine 77(1999)14〜23により調製))を最初にマイクロプレートに装入し、刺激溶液1μl(DMSO中0〜10μM 3−モルホリノシドノンイミン、SIN−1、Merck)を添加した。マイクロプレートを室温で10分間インキュベートした。次いで、検出混合物20μl(50mM TEA、2mM塩化マグネシウム、0.1%BSA(分画V)、0.005%Brij 35、pH7.5中1.2nMホタルルシフェラーゼ(フォティナス・ピラリス(Photinus pyralis)ルシフェラーゼ、Promega)、29μMデヒドロルシフェリン(Bitler&McElroy、Arch.Biochem.Biophys.72(1957)358により調製)、122μMルシフェリン(Promega)、153μM ATP(Sigma)および0.4mM DTT(Sigma))を添加した。基質溶液20μl(50mM TEA、2mM塩化マグネシウム、0.1%BSA(分画V)、0.005%Brij 35、pH7.5中1.25mMグアノシン5’三リン酸(Sigma))を添加することによって、酵素反応を開始し、ルミノメータで連続的に分析した。
B−2.組換えグアニル酸シクラーゼレポーター細胞系に対する効果
本発明による化合物の細胞活性を、F.Wunderら、Anal.Biochem.339、104〜112(2005)に記載されているように、組換えグアニル酸シクラーゼレポーター細胞系を用いて測定する。
本発明による化合物の細胞活性を、F.Wunderら、Anal.Biochem.339、104〜112(2005)に記載されているように、組換えグアニル酸シクラーゼレポーター細胞系を用いて測定する。
本発明の化合物についての代表的なMEC値(MEC=最小有効濃度)を(いくつかの場合、個々の測定についての平均値として)以下の表に示す:
B−3.インビトロでの血管弛緩効果
ウサギを首への強打によって気絶させ、失血させる。大動脈を取り出し、接着組織を取り除き、幅1.5mmの輪に分割し、これらを以下の組成(それぞれmM):塩化ナトリウム:119;塩化カリウム:4.8;塩化カルシウム二水和物:1;硫酸マグネシウム七水和物:1.4;リン酸二水素カリウム:1.2;重炭酸ナトリウム:25;グルコース:10を有する37℃のカルボゲン注入クレブス−ヘンゼライト液を含む5ml臓器浴に予応力下で個々に入れる。収縮力をStatham UC2セルで測定し、増幅し、A/D変換器(DAS−1802 HC、Keithley Instruments Munich)を用いてデジタル化し、線形レコーダーで並行して記録する。収縮を得るために、フェニレフリンを増加する濃度で累積的に浴に添加する。数回の対照サイクル後、試験する物質を実行毎に増加する投与量で添加し、収縮の大きさをその直前の実行時に得られた収縮の大きさと比較する。これを、対照値の大きさを50%減少させるために要する濃度(IC50値)を計算するために使用する。標準投与体積は5μlであり、浴溶液中のDMSO含量は0.1%に相当する。
ウサギを首への強打によって気絶させ、失血させる。大動脈を取り出し、接着組織を取り除き、幅1.5mmの輪に分割し、これらを以下の組成(それぞれmM):塩化ナトリウム:119;塩化カリウム:4.8;塩化カルシウム二水和物:1;硫酸マグネシウム七水和物:1.4;リン酸二水素カリウム:1.2;重炭酸ナトリウム:25;グルコース:10を有する37℃のカルボゲン注入クレブス−ヘンゼライト液を含む5ml臓器浴に予応力下で個々に入れる。収縮力をStatham UC2セルで測定し、増幅し、A/D変換器(DAS−1802 HC、Keithley Instruments Munich)を用いてデジタル化し、線形レコーダーで並行して記録する。収縮を得るために、フェニレフリンを増加する濃度で累積的に浴に添加する。数回の対照サイクル後、試験する物質を実行毎に増加する投与量で添加し、収縮の大きさをその直前の実行時に得られた収縮の大きさと比較する。これを、対照値の大きさを50%減少させるために要する濃度(IC50値)を計算するために使用する。標準投与体積は5μlであり、浴溶液中のDMSO含量は0.1%に相当する。
B−4.麻酔されたラットにおける血圧測定
体重300〜350gの雄ウィスターラットにチオペンタールで麻酔をかける(100mg/kg腹腔内)。気管切開後、カテーテルを大腿動脈に導入して血圧を測定する。試験する物質を、経管栄養を用いて経口的にまたは大腿静脈を介して静脈内に溶液として投与する(StaschらBr.J.Pharmacol.2002;135:344〜355)。
体重300〜350gの雄ウィスターラットにチオペンタールで麻酔をかける(100mg/kg腹腔内)。気管切開後、カテーテルを大腿動脈に導入して血圧を測定する。試験する物質を、経管栄養を用いて経口的にまたは大腿静脈を介して静脈内に溶液として投与する(StaschらBr.J.Pharmacol.2002;135:344〜355)。
B−5.意識のある自然発症高血圧ラットにおける血圧のラジオテレメトリー測定
DATA SCIENCES INTERNATIONAL DSI、米国から商業的に入手可能なテレメトリシステムを、下記の意識のあるラットでの血圧測定に使用する。
DATA SCIENCES INTERNATIONAL DSI、米国から商業的に入手可能なテレメトリシステムを、下記の意識のあるラットでの血圧測定に使用する。
システムは3つの主要部品からなる:
埋込み型送信機(Physiotel(登録商標)テレメトリ送信機)
データ取得コンピュータ
と多重化装置(DSI Data Exchange Matrix)を介して接続されている受信機(Physiotel(登録商標)受信機)。
埋込み型送信機(Physiotel(登録商標)テレメトリ送信機)
データ取得コンピュータ
と多重化装置(DSI Data Exchange Matrix)を介して接続されている受信機(Physiotel(登録商標)受信機)。
テレメトリシステムによって、その通常の生育環境にいる意識のある動物の血圧、心拍数および体動を連続的に記録することが可能になる。
動物材料
試験は、体重200g超の成体雌、自然発症高血圧ラット(SHR岡本)で行う。岡本 京都大学医学部、1963のSHR/NCrlは、血圧が非常に上昇した雄ウィスター京都ラットと血圧がわずかに上昇した雌の交雑種で、米国国立衛生研究所にF13で渡した。
試験は、体重200g超の成体雌、自然発症高血圧ラット(SHR岡本)で行う。岡本 京都大学医学部、1963のSHR/NCrlは、血圧が非常に上昇した雄ウィスター京都ラットと血圧がわずかに上昇した雌の交雑種で、米国国立衛生研究所にF13で渡した。
送信機埋め込み後、実験動物を別々にMakrolonケージ3型に収容する。これらの動物は標準的な飼料および水を自由に入手できる。
実験室の昼/夜リズムは、午前6:00時と午後7:00時に室内照明によって変化させる。
送信機埋め込み
使用するTA11PA−C40テレメトリ送信機を、最初の実験使用の少なくとも14日前に無菌状態で実験動物に外科的に埋め込む。創傷が治癒し、埋込みが定着した後、このような計装動物を繰り返し使用することができる。
使用するTA11PA−C40テレメトリ送信機を、最初の実験使用の少なくとも14日前に無菌状態で実験動物に外科的に埋め込む。創傷が治癒し、埋込みが定着した後、このような計装動物を繰り返し使用することができる。
埋込みのために、絶食動物にペントバルビタール(Nembutal、Sanofi:50mg/kg腹腔内)で麻酔をかけ、腹部の広域にわたって剪毛および消毒する。白線に沿って腹腔を開いた後、システムの液体充填測定カテーテルを、下行大動脈に頭蓋方向に分岐より上に挿入し、組織接着剤(VetBonD(商標)、3M)で固定する。送信機ハウジングを腹壁筋に腹腔内固定し、創傷を一層ずつ閉じる。
感染症を予防するために抗生物質(Tardomyocel COMP Bayer 1ml/kg皮下)を術後に投与する。
物質および溶液
特に名言しない限り、試験する物質を、それぞれ動物の群(n=6)に経管栄養によって経口投与する。5ml/kg体重の投与量によると、試験物質を適当な溶媒混合物に溶解する、または0.5%チロースに懸濁する。
特に名言しない限り、試験する物質を、それぞれ動物の群(n=6)に経管栄養によって経口投与する。5ml/kg体重の投与量によると、試験物質を適当な溶媒混合物に溶解する、または0.5%チロースに懸濁する。
溶媒で処理した動物群を対照として使用する。
実験概要
本テレメトリ測定装置を24匹の動物に設定する。各実験を実験番号で記録する(V年月日)。
本テレメトリ測定装置を24匹の動物に設定する。各実験を実験番号で記録する(V年月日)。
システムで生きている計装ラットの各々に受信アンテナ(1010 Receiver、DSI)を割り当てる。
埋込み送信機を組み込まれたマグネチックスイッチを用いて外部から起動することができる。実験の準備期間にこれらを送信に切り替える。発信シグナルは、データ取得システム(WINDOWS(登録商標)用のDataquest(商標)A.R.T.、DSI)によってオンラインで検出し、適宜処理することができる。データをこの目的のために作成され、実験番号を有するファイルにそれぞれ保存する。
標準的な手順では、それぞれ10秒間、以下を測定する:
収縮期血圧(SBP)
拡張期血圧(DBP)
平均動脈圧(MAP)
心拍数(HR)
活動(ACT)。
収縮期血圧(SBP)
拡張期血圧(DBP)
平均動脈圧(MAP)
心拍数(HR)
活動(ACT)。
測定値の取得を5分間隔でコンピュータ制御下で繰り返す。絶対値として得たソースデータを、現在測定されている大気圧(Ambient Pressure Reference Monitor;APR−1)を用いた図表で補正し、個々のデータとして保存する。さらなる技術的詳細は、製造企業(DSI)の広範なドキュメントに示される。
特に指示しない限り、試験物質を実験日の午前9:00に投与する。投与後、上記パラメータを24時間にわたって測定する。
評価
実験終了後、取得した個々のデータを分析ソフトウェア(DATAQUEST(商標)A.R.T.(商標)ANALYSIS)を用いてソーティングする。ブランク値はここでは投与2時間前の時間と仮定されるので、選択されたデータセットは実験日の午前7:00〜翌日の午前9:00の期間を包含する。
実験終了後、取得した個々のデータを分析ソフトウェア(DATAQUEST(商標)A.R.T.(商標)ANALYSIS)を用いてソーティングする。ブランク値はここでは投与2時間前の時間と仮定されるので、選択されたデータセットは実験日の午前7:00〜翌日の午前9:00の期間を包含する。
データを、平均値(15分平均)を決定することによって事前定義可能な(predefinable)期間にわたって平滑化し、テキストファイルとして記憶媒体に移す。このように予めソーティングし、圧縮した測定値をエクセルテンプレートに移し、表にする。各実験日について、得られたデータを実験番号を有する専用ファイルに保存する。結果および試験プロトコルをファイルに数字でソーティングした紙の形状で保存する。
文献:
Klaus Witte、Kai Hu、Johanna Swiatek、Claudia Mussig、Georg ErtlおよびBjorn Lemmer:Experimental heart failure in rats:effects on cardiovascular circadian rhythms and on myocardial β−adrenergic signaling.Cardiovasc Res 47(2):203〜405、2000;Kozo Okamoto:Spontaneous hypertension in rats.Int Rev Exp Pathol 7:227〜270、1969;Maarten van den Buuse:Circadian Rhythms of Blood Pressure,Heart Rate,and Locomotor Activity in Spontaneously Hypertensive Rats as Measured With Radio−Telemetry.Physiology&Behavior 55(4):783〜787、1994。
Klaus Witte、Kai Hu、Johanna Swiatek、Claudia Mussig、Georg ErtlおよびBjorn Lemmer:Experimental heart failure in rats:effects on cardiovascular circadian rhythms and on myocardial β−adrenergic signaling.Cardiovasc Res 47(2):203〜405、2000;Kozo Okamoto:Spontaneous hypertension in rats.Int Rev Exp Pathol 7:227〜270、1969;Maarten van den Buuse:Circadian Rhythms of Blood Pressure,Heart Rate,and Locomotor Activity in Spontaneously Hypertensive Rats as Measured With Radio−Telemetry.Physiology&Behavior 55(4):783〜787、1994。
B−6.静脈内投与および経口投与後の薬物動態パラメータの測定
本発明による化合物の薬物動態パラメータを、雄CD−1マウス、雄ウィスターラットおよび雌ビーグルで測定する。マウスおよびラットの場合の静脈内投与は、種特異的血漿/DMSO製剤によって行い、イヌの場合には、水/PEG400/エタノール製剤によって行う。全ての種において、溶解した物質の経口投与は、水/PEG400/エタノール製剤に基づいて、経管栄養を介して行う。ラットからの血液採取は、物質投与前に、シリコーンカテーテルを右外頚静脈に挿入することによって単純化する。手術は、イソフルラン麻酔および鎮痛剤(アトロピン/リマダイル(3/1)0.1ml皮下)の投与により、実験の少なくとも1日前に行う。血液は、物質投与の少なくとも24〜最大で72時間後の終端時点を含む時間窓内で採取する(一般的には10時点超)。血液をヘパリン処理チューブに取り出す。次いで、血漿を遠心分離によって得て、必要に応じて、さらなる処理まで−20℃で保管することができる。
本発明による化合物の薬物動態パラメータを、雄CD−1マウス、雄ウィスターラットおよび雌ビーグルで測定する。マウスおよびラットの場合の静脈内投与は、種特異的血漿/DMSO製剤によって行い、イヌの場合には、水/PEG400/エタノール製剤によって行う。全ての種において、溶解した物質の経口投与は、水/PEG400/エタノール製剤に基づいて、経管栄養を介して行う。ラットからの血液採取は、物質投与前に、シリコーンカテーテルを右外頚静脈に挿入することによって単純化する。手術は、イソフルラン麻酔および鎮痛剤(アトロピン/リマダイル(3/1)0.1ml皮下)の投与により、実験の少なくとも1日前に行う。血液は、物質投与の少なくとも24〜最大で72時間後の終端時点を含む時間窓内で採取する(一般的には10時点超)。血液をヘパリン処理チューブに取り出す。次いで、血漿を遠心分離によって得て、必要に応じて、さらなる処理まで−20℃で保管することができる。
内部標準(化学的に関連のない物質でもあり得る)を本発明の化合物の試料、較正試料および修飾物質に添加し、それに続いて過剰のアセトニトリルを用いてタンパク質を沈殿させる。LC濃度に適合した緩衝液を添加し、その後ボルテックスし、引き続いて1000gで遠心分離する。C18逆相カラムおよび可変的移動相混合物を用いるLC−MS/MSによって上清を分析する。特定の選択したイオンモニタリング実験の抽出イオンクロマトグラムからのピーク高さまたは面積を介して物質を定量化する。
測定した血漿濃度/時間プロットを使用して、検証された薬物動態計算プログラムを用いて、AUC、Cmax、t1/2(終端半減期)、F(生物学的利用能)、MRT(平均滞留時間)およびCL(クリアランス)などの薬物動態パラメータを計算する。
物質定量化を血漿で行うので、薬物動態パラメータを対応して調整することができるようにするために、物質の血液/血漿分布を決定することが必要である。この目的のために、定義された量の物質を揺動ローラーミキサーにおいて20分間当の種のヘパリン処理全血でインキュベートする。1000gでの遠心分離後、血漿濃度を(LC−MS/MS;上記参照によって)測定し、C血液/C血漿の比の値を計算することによって決定する。
B−7.代謝試験
本発明の化合物の代謝プロファイルを決定するため、極めて実質的に完全な肝I相およびII相代謝、ならびに代謝に関与する酵素についての情報を得て、これらを比較するために、本発明による化合物を組換えヒトチトクロムP450(CYP)酵素、種々の動物種(例えば、ラット、イヌ)およびヒト起源からの肝ミクロソームまたは新鮮な初代培養肝細胞と共にインキュベートする。
本発明の化合物の代謝プロファイルを決定するため、極めて実質的に完全な肝I相およびII相代謝、ならびに代謝に関与する酵素についての情報を得て、これらを比較するために、本発明による化合物を組換えヒトチトクロムP450(CYP)酵素、種々の動物種(例えば、ラット、イヌ)およびヒト起源からの肝ミクロソームまたは新鮮な初代培養肝細胞と共にインキュベートする。
本発明の化合物を約0.1〜10μMの濃度でインキュベートした。このために、アセトニトリル中0.01〜1mMの濃度を有する本発明の化合物のストック溶液を調製し、次いで、インキュベーション混合物に1:100希釈でピペットを用いて移す。肝ミクロソームおよび組換え酵素を、1mM NADP+、10mMグルコース−6−リン酸および1ユニットのグルコース−6−リン酸脱水素酵素からなるNADPH産生系を含むおよび含まない50mMリン酸カリウム緩衝液pH7.4中37℃でインキュベートした。初代培養肝細胞を37℃で同様にウィリアムE培地中懸濁でインキュベートした。0〜4時間のインキュベーション時間後、インキュベーション混合物をアセトニトリル(最終濃度約30%)を用いて停止し、タンパク質を約15000×gで遠心分離した。こうして停止した試料を直接分析した、または分析まで−20℃で保存した。
紫外線および質量分析検出を含む高速液体クロマトグラフィー(HPLC−UV−MS/MS)によって分析を行う。このために、インキュベーション試料の上清を、適当なC18逆相カラムおよびアセトニトリルと10mMギ酸アンモニウム水溶液または0.05%ギ酸の可変性移動相混合物を用いてクロマトグラフにかける。質量分析データと合わせたUVクロマトグラムは、代謝産物の同定、構造決定および定量的評価、ならびにインキュベーション混合物中の本発明の化合物の定量的代謝還元に役立つ。
B−8.Caco−2透過性試験
Caco−2細胞系、胃腸障壁での透過性予測のための確立されたインビトロモデルを用いて試験物質の透過性を測定した(Artursson,P.およびKarlsson,J.(1991).Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial(Caco−2)cells.Biochem.Biophys.175(3)、880〜885)。Caco−2細胞(ACC番号169、DSMZ、 Deutsche Sammlung von Mikroorganismen und Zellkulturen、Braunschweig、ドイツ)をインサートを含む24ウェルプレートに蒔き、14〜16日間培養した。透過性試験のために、試験物質をDMSOに溶解し、輸送バッファ(ハンクス緩衝塩類溶液、Gibco/Invitrogen、19.9mMグルコースおよび9.8mM HEPESを含む)を用いて最終試験濃度に希釈した。試験物質の頂端側から基底側への透過性(PappA−B)を測定するために、試験物質を含む溶液を、Caco−2細胞単層の頂端側に適用し、輸送バッファを基底側に適用した。試験物質の基底側から頂端側への透過性(PappB−A)を測定するために、試験物質を含む溶液を、Caco−2細胞単層の基底側に適用し、輸送バッファを頂端側に適用した。質量バランスを確保するために、実験の開始時に、試料をそれぞれのドナー区画から採った。37℃で2時間のインキュベーション時間後、試料を2つの区画から採った。試料をLC−MS/MSを用いて分析し、見かけの透過係数(Papp)を計算した。各細胞単層について、ルシファーイエローの透過性を測定して細胞層完全性を確認した。各試験実行において、アテノロール(低透過性についてのマーカー)およびスルファサラジン(活性排泄についてのマーカー)の透過性も品質管理として測定した。
Caco−2細胞系、胃腸障壁での透過性予測のための確立されたインビトロモデルを用いて試験物質の透過性を測定した(Artursson,P.およびKarlsson,J.(1991).Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial(Caco−2)cells.Biochem.Biophys.175(3)、880〜885)。Caco−2細胞(ACC番号169、DSMZ、 Deutsche Sammlung von Mikroorganismen und Zellkulturen、Braunschweig、ドイツ)をインサートを含む24ウェルプレートに蒔き、14〜16日間培養した。透過性試験のために、試験物質をDMSOに溶解し、輸送バッファ(ハンクス緩衝塩類溶液、Gibco/Invitrogen、19.9mMグルコースおよび9.8mM HEPESを含む)を用いて最終試験濃度に希釈した。試験物質の頂端側から基底側への透過性(PappA−B)を測定するために、試験物質を含む溶液を、Caco−2細胞単層の頂端側に適用し、輸送バッファを基底側に適用した。試験物質の基底側から頂端側への透過性(PappB−A)を測定するために、試験物質を含む溶液を、Caco−2細胞単層の基底側に適用し、輸送バッファを頂端側に適用した。質量バランスを確保するために、実験の開始時に、試料をそれぞれのドナー区画から採った。37℃で2時間のインキュベーション時間後、試料を2つの区画から採った。試料をLC−MS/MSを用いて分析し、見かけの透過係数(Papp)を計算した。各細胞単層について、ルシファーイエローの透過性を測定して細胞層完全性を確認した。各試験実行において、アテノロール(低透過性についてのマーカー)およびスルファサラジン(活性排泄についてのマーカー)の透過性も品質管理として測定した。
B−9.hERGカリウム電流アッセイ
hERG(ヒトether−a−go−go関連遺伝子)カリウム電流は、ヒト心筋活動電位の再分極に有意に寄与する(Scheelら、2011)。医薬によるこの電流の阻害は、稀に、潜在的に致死性の不整脈を引き起こすので、薬物開発中の早期の段階で試験される。
hERG(ヒトether−a−go−go関連遺伝子)カリウム電流は、ヒト心筋活動電位の再分極に有意に寄与する(Scheelら、2011)。医薬によるこの電流の阻害は、稀に、潜在的に致死性の不整脈を引き起こすので、薬物開発中の早期の段階で試験される。
ここで使用される機能的hERGアッセイは、KCNH2(HERG)遺伝子を安定的に発現している組換えHEK293細胞系に基づく(Zhouら、1998)。これらの細胞を、膜電圧を制御し、室温でhERGカリウム電流を測定する自動化システム(Patchliner(商標);Nanion、Munich、ドイツ)で、「細胞全体電位固定」技術(Hamillら、1981)を用いて試験する。PatchControlHT(商標)ソフトウェア(Nanion)は、Patchlinerシステム、データ収集およびデータ分析を制御する。PatchMasterPro(商標)ソフトウェアによって制御される2つのEPC−10クアドロ増幅器(共にHEKA Elektronik、Lambrecht、ドイツ)によって電圧を制御する。中抵抗のNPC−16チップ(約2MΩ;Nanion)は、電位固定実験のための平面基板として働く。
NPC−16チップを細胞内および細胞外液(Himmel、2007参照)ならびに細胞懸濁液で充填する。ギガオームシールを形成し、全細胞モード(いくつかの自動化品質管理ステップを含む)を確立した後、細胞膜を−80mV保持電位で固定する。その後の電位固定プロトコルは、指令電圧を+20mV(1000ms間)、−120mV(500ms間)に変化させ、−80mVの保持電位に戻す;これを12秒毎に繰り返す。初期安定化期(約5〜6分)後、試験物質溶液を、増加する濃度で(例えば、0.1、1および10μmol/l)(暴露約5〜6分/濃度)ピペットによって導入し、引き続いて数回の洗浄ステップを行う。
電位を+20mVから−120mVに変化させることによって生み出される内向き「尾」電流の振幅が、hERGカリウム電流を定量化するのに役立ち、時間の関数として記載される(IgorPro(商標)ソフトウェア)。種々の時間間隔(例えば、試験物質の前の安定化期、試験物質の第1/第2/第3の濃度)の最後での電流振幅が、そこから試験物質の半数阻害濃度IC50が計算される濃度/効果曲線を確立するのに役立つ。
Hamill OP、Marty A、Neher E、Sakmann B、Sigworth FJ.Improved patch−clamp techniques for high−resolution current recording from cells and cell−free membrane patches.Pfluegers Arch 1981;391:85〜100。
Himmel HM.Suitability of commonly used excipients for electrophysiological in−vitro safety pharmacology assessment of effects on hERG potassium current and on rabbit Purkinje fiber action potential.J Pharmacol Toxicol Methods 2007;56:145〜158。
Scheel O、Himmel H、Rascher−Eggstein G、Knott T.Introduction of a modular automated voltage−clamp platform and its correlation with manual human ether−a−go−go related gene voltage−clamp data.Assay Drug Dev Technol 2011;9:600〜607。
Zhou ZF、Gong Q、Ye B、Fan Z、Makielski JC、Robertson GA、January CT.Properties of hERG channels stably expressed in HEK293 cells studied at physiological temperature.Biophys J 1998;74:230〜241。
C.医薬組成物の実施例
本発明の化合物を以下の通り医薬製剤に変換することができる:
錠剤:
組成:
本発明の化合物100mg、乳糖(一水和物)50mg、コーンスターチ(野生)50mg、ポリビニルピロリドン(PVP25)(BASF、Ludwigshafen、ドイツ)10mgおよびステアリン酸マグネシウム2mg。
本発明の化合物を以下の通り医薬製剤に変換することができる:
錠剤:
組成:
本発明の化合物100mg、乳糖(一水和物)50mg、コーンスターチ(野生)50mg、ポリビニルピロリドン(PVP25)(BASF、Ludwigshafen、ドイツ)10mgおよびステアリン酸マグネシウム2mg。
錠剤重量212mg。直径8mm、曲率半径12mm。
製造:
本発明の化合物、乳糖およびスターチの混合物をPVPの水中5%溶液(w/w)を用いて造粒する。顆粒を乾燥させ、次いで、ステアリン酸マグネシウムと5分間混合する。この混合物を従来の打錠機を用いて圧縮する(錠剤のフォーマットについては上記参照)。圧縮に使用するガイド値は押圧力15kNとする。
本発明の化合物、乳糖およびスターチの混合物をPVPの水中5%溶液(w/w)を用いて造粒する。顆粒を乾燥させ、次いで、ステアリン酸マグネシウムと5分間混合する。この混合物を従来の打錠機を用いて圧縮する(錠剤のフォーマットについては上記参照)。圧縮に使用するガイド値は押圧力15kNとする。
経口投与用の懸濁剤:
組成:
本発明の化合物1000mg、エタノール(96%)1000mg、Rhodigel(登録商標)(FMC、ペンシルバニア州、米国製のキサンタンガム)400mgおよび水99g。
組成:
本発明の化合物1000mg、エタノール(96%)1000mg、Rhodigel(登録商標)(FMC、ペンシルバニア州、米国製のキサンタンガム)400mgおよび水99g。
経口懸濁剤10mlは1回量の本発明の化合物100mgに相当する。
製造:
Rhodigelをエタノールに懸濁し、本発明の化合物を懸濁液に添加する。攪拌しながら水を添加する。Rhodigelの膨潤が完了するまで、混合物を約6時間攪拌する。
Rhodigelをエタノールに懸濁し、本発明の化合物を懸濁液に添加する。攪拌しながら水を添加する。Rhodigelの膨潤が完了するまで、混合物を約6時間攪拌する。
経口投与用の液剤:
組成:
本発明の化合物500mg、ポリソルベート2.5gおよびポリエチレングリコール400 97g。経口液剤20gは1回量の本発明の化合物100mgに相当する。
組成:
本発明の化合物500mg、ポリソルベート2.5gおよびポリエチレングリコール400 97g。経口液剤20gは1回量の本発明の化合物100mgに相当する。
製造:
本発明の化合物を、攪拌しながらポリエチレングリコールとポリソルベートの混合物に懸濁する。本発明の化合物の溶解が完了するまで、攪拌操作を継続する。
本発明の化合物を、攪拌しながらポリエチレングリコールとポリソルベートの混合物に懸濁する。本発明の化合物の溶解が完了するまで、攪拌操作を継続する。
静脈内溶液:
本発明の化合物を、生理学的に許容される溶媒(例えば、等張食塩水溶液、グルコース溶液5%および/またはPEG400溶液30%)に飽和溶解度未満の濃度で溶解する。得られた溶液を滅菌濾過に供し、滅菌パイロジェンフリー注射容器に分配する。
本発明の化合物を、生理学的に許容される溶媒(例えば、等張食塩水溶液、グルコース溶液5%および/またはPEG400溶液30%)に飽和溶解度未満の濃度で溶解する。得られた溶液を滅菌濾過に供し、滅菌パイロジェンフリー注射容器に分配する。
Claims (16)
- 組織名ent−N−(2−アミノ−3−フルオロ−2−メチルプロピル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)および構造式
- 組織名ent−N−(2−アミノ−5,5,5−トリフルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)および構造式
- 組織名ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)および構造式
- 組織名ent−N−(2−アミノ−5−フルオロ−2−メチルペンチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)および構造式
- 組織名ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)および構造式
- 組織名ent−N−(2−アミノ−4,4−ジフルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)および構造式
- 組織名rac−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(ラセミ体)および構造式
- 組織名ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーA)および構造式
- 組織名ent−N−(2−アミノ−4−フルオロ−2−メチルブチル)−6−クロロ−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミド(エナンチオマーB)および構造式
- [A]式(I)
T1は(C1〜C4)−アルキルまたはベンジルを表す)
の化合物を、適当な塩基または酸の存在下、不活性溶媒中で反応させて式(II)
その後、これをアミドカップリング条件下、不活性溶媒中で、
場合により存在する保護基を、場合により触媒の存在下で、水素化分解的に除去し、
得られた化合物を場合により適当な(i)溶媒および/または(ii)酸もしくは塩基を用いて、その溶媒和物、塩および/または塩の溶媒和物に変換する
ことを特徴とする、本発明による化合物を調製する方法。 - 疾患を治療および/または予防するための請求項1から9のいずれか一項に記載の化合物。
- 心不全、狭心症、高血圧、肺高血圧、虚血、血管疾患、腎機能不全、血栓塞栓性疾患および動脈硬化を治療および/または予防するための医薬を調製するための請求項1から9のいずれか一項に記載の化合物の使用。
- 不活性で、非毒性の薬学的に適した賦形剤と組み合わせて請求項1から9のいずれか一項に記載の化合物を含む医薬。
- 有機硝酸塩、NOドナー、cGMP−PDE阻害剤、抗血栓剤、降圧剤および脂肪代謝調節剤からなる群から選択されるさらなる活性化合物と組み合わせて請求項1から9のいずれか一項に記載の化合物を含む医薬。
- 心不全、狭心症、高血圧、肺高血圧、虚血、血管疾患、腎不全、血栓塞栓性疾患および動脈硬化を治療および/または予防するための請求項13または14に記載の医薬。
- 有効量の少なくとも1種の請求項1から9のいずれか一項に記載の化合物または請求項13から15のいずれか一項に記載の医薬を用いて、ヒトおよび動物の心不全、狭心症、高血圧、肺高血圧、虚血、血管疾患、腎機能不全、血栓塞栓性疾患および動脈硬化を治療および/または予防する方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14166911 | 2014-05-02 | ||
| EP14166911.9 | 2014-05-02 | ||
| PCT/EP2015/059350 WO2015165970A1 (de) | 2014-05-02 | 2015-04-29 | 6-chlor-substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung als stimulatoren der löslichen guanylatcyclase |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017514901A true JP2017514901A (ja) | 2017-06-08 |
Family
ID=50693467
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017508756A Pending JP2017514901A (ja) | 2014-05-02 | 2015-04-29 | 6−クロロ置換イミダゾ[1,2−a]ピリジンカルボキサミドおよびその使用 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20170101407A1 (ja) |
| EP (1) | EP3137465A1 (ja) |
| JP (1) | JP2017514901A (ja) |
| CN (1) | CN106507673A (ja) |
| CA (1) | CA2947372A1 (ja) |
| WO (1) | WO2015165970A1 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015165933A2 (de) * | 2014-05-02 | 2015-11-05 | Bayer Pharma Aktiengesellschaft | 6-substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung |
| WO2018184976A1 (de) | 2017-04-05 | 2018-10-11 | Bayer Pharma Aktiengesellschaft | Substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung |
| CN108358797A (zh) * | 2018-04-20 | 2018-08-03 | 南京农业大学 | 一种烷基甘氨酸的合成方法 |
| WO2024102699A1 (en) * | 2022-11-07 | 2024-05-16 | ELANCO US, Inc. | Guanylate cyclase (gc) stimulator formulations and uses thereof |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ221996A (en) * | 1986-10-07 | 1989-08-29 | Yamanouchi Pharma Co Ltd | Imidazo-pyridine derivatives and pharmaceutical compositions |
| SE8704248D0 (sv) * | 1987-10-30 | 1987-10-30 | Haessle Ab | Medical use |
| EA201391769A1 (ru) * | 2011-05-30 | 2014-04-30 | Астеллас Фарма Инк. | Имидазопиридиновые соединения |
| US9126998B2 (en) * | 2012-11-05 | 2015-09-08 | Bayer Pharma AG | Amino-substituted imidazo[1,2-a]pyridinecarboxamides and their use |
| US9624214B2 (en) * | 2012-11-05 | 2017-04-18 | Bayer Pharma Aktiengesellschaft | Amino-substituted imidazo[1,2-a]pyridinecarboxamides and their use |
-
2015
- 2015-04-29 EP EP15721635.9A patent/EP3137465A1/de not_active Withdrawn
- 2015-04-29 CN CN201580034595.5A patent/CN106507673A/zh active Pending
- 2015-04-29 US US15/308,200 patent/US20170101407A1/en not_active Abandoned
- 2015-04-29 CA CA2947372A patent/CA2947372A1/en not_active Abandoned
- 2015-04-29 WO PCT/EP2015/059350 patent/WO2015165970A1/de active Application Filing
- 2015-04-29 JP JP2017508756A patent/JP2017514901A/ja active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| CN106507673A (zh) | 2017-03-15 |
| US20170101407A1 (en) | 2017-04-13 |
| CA2947372A1 (en) | 2015-11-05 |
| WO2015165970A1 (de) | 2015-11-05 |
| EP3137465A1 (de) | 2017-03-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2013340726B2 (en) | Amino-substituted imidazo[1,2-a]pyridinecarboxamides and their use | |
| EP3030562B1 (de) | Substituierte pyrazolo[1,5-a]pyridin-3-carboxamide und ihre verwendung | |
| US9771360B2 (en) | Cyano-substituted imidazo[1,2-A]pyridinecarboxamides and their use | |
| EP3107920B1 (de) | 3-(pyrimidin-2-yl)imidazo[1,2-a]pyridine | |
| JP2016539166A (ja) | アリール−およびヘテロアリール置換イミダゾ[1,2−a]ピリジン−3−カルボキサミドおよびその使用 | |
| JP2016527295A (ja) | 置換イミダゾ[1,2−a]ピラジンカルボキサミドおよびその使用 | |
| JP2017508811A (ja) | 置換イミダゾ[1,2−a]ピリジンカルボキサミドおよびその使用 | |
| JP2017514899A (ja) | ヘテロシクリル−およびヘテロアリール−置換イミダゾ[1,2−a]ピリジンおよびその使用 | |
| JP2017514901A (ja) | 6−クロロ置換イミダゾ[1,2−a]ピリジンカルボキサミドおよびその使用 | |
| JP2017514900A (ja) | 6−置換イミダゾ[1,2−a]ピリジンカルボキサミドおよびその使用 | |
| JP2017514898A (ja) | 心血管疾患の治療のためのn−(2−アミノ−5−フルオロ−2−メチルペンチル)−8−[(2,6−ジフルオロベンジル)オキシ]−2−メチルイミダゾ[1,2−a]ピリジン−3−カルボキサミドの、並びにジ−及びトリフルオロ誘導体のエナンチオマー | |
| JP2017529330A (ja) | 置換キノリン−4−カルボキサミドおよびその使用 | |
| US10150773B2 (en) | N-substituted 8-[(2,6-difluorobenzyl)oxy]-2,6- dimethylimidazo[1,2-a]pyrazin-3-carboxamide derivatives as stimulators of soluble guanylate cyclase (SGC) for the treatment of cardiovascular diseases | |
| JP2018505885A (ja) | 置換ピラゾロ[1,5−a]−ピリジン−3−カルボキサミドおよびその使用 | |
| WO2018184976A1 (de) | Substituierte imidazo[1,2-a]pyridincarboxamide und ihre verwendung |