JP2010529068A - Nk3受容体アンタゴニストとしてのプロリンアミド−テトラゾール誘導体 - Google Patents
Nk3受容体アンタゴニストとしてのプロリンアミド−テトラゾール誘導体 Download PDFInfo
- Publication number
- JP2010529068A JP2010529068A JP2010510746A JP2010510746A JP2010529068A JP 2010529068 A JP2010529068 A JP 2010529068A JP 2010510746 A JP2010510746 A JP 2010510746A JP 2010510746 A JP2010510746 A JP 2010510746A JP 2010529068 A JP2010529068 A JP 2010529068A
- Authority
- JP
- Japan
- Prior art keywords
- phenyl
- difluoro
- tetrazol
- carbonyl
- benzonitrile
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 **1C=CC(N2CCN(CC(C3)N(CI)CC3[n]3nnc(C4=C*(*)C=CC=C4)n3)CC2)=CC=C1 Chemical compound **1C=CC(N2CCN(CC(C3)N(CI)CC3[n]3nnc(C4=C*(*)C=CC=C4)n3)CC2)=CC=C1 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
本発明は、式(I)[式中、R1は、シクロアルキルであるか、あるいは非置換であるか又は1個若しくは2個のハロゲン原子により置換されているフェニルであり;R2は、水素又はハロゲンであり;R3は、水素、CN、低級アルコキシ、低級アルキル又はハロゲンであり;nは、0、1又は2である]の化合物、又は薬学的に適切なその酸付加塩に関する。本化合物は、鬱病、疼痛、精神病、パーキンソン病、統合失調症、不安及び注意欠陥多動障害(ADHD)の処置のための有望なNK−3受容体アンタゴニストであることが見出された。
Description
本発明は、式(I):
[式中、
R1は、シクロアルキルであるか、あるいは非置換であるか又は1個若しくは2個のハロゲン原子により置換されているフェニルであり;
R2は、水素又はハロゲンであり;
R3は、水素、CN、低級アルコキシ、低級アルキル又はハロゲンであり;
nは、0、1又は2である]で示される化合物又は薬学的に適切なその酸付加塩に関する。
本発明は、式(I)の化合物の個々のジアステレオ異性体及びエナンチオマー、更にはそのラセミ及び非ラセミ混合物を包含する、全ての立体異性体形を包含する。
本発明の化合物は、鬱病、疼痛、精神病、パーキンソン病、統合失調症、不安及び注意欠陥多動障害(ADHD)の処置のための有望なNK−3受容体アンタゴニストであることが見出された。
3種の主要な哺乳動物のタキキニンである、サブスタンスP(SP)、ニューロキニンA(NKA)及びニューロキニンB(NKB)は、Phe−X−Gly−Leu−Met−NH2の共通のCOOH末端ペンタペプチド配列を共有する神経ペプチドのファミリーに属する。神経伝達物質として、これらのペプチドは、NK−1、NK−2及びNK−3と称される3種の別個のニューロキニン(NK)受容体を介してその生物学的活性を発揮する。SPはNK−1受容体に、NKAはNK−2に、そしてNKBはNK−3受容体に優先的に結合する。
NK−3受容体は、CNSにおける優勢発現を特徴とし、そして中枢モノアミン作動系の調節におけるその関与が証明されている。これらの性質により、NK−3受容体は、不安、鬱病、双極性障害、パーキンソン病、統合失調症及び疼痛のような、中枢神経系疾患の可能性ある標的となっている(Neurosci. Letters, 2000, 283, 185-188; Exp. Opin. Ther. Patents 2000, 10, 939-960; Neuroscience, 1996, 74, 403-414; Neuropeptides, 1998, 32, 481-488)。
統合失調症は、重篤かつ慢性の精神的損傷を特徴とする、主要な神経精神障害の一つである。この深刻な疾患には、世界人口の約1%が罹患している。症状は、成人早期に始まり、次には対人及び社会的機能不全の時期が続く。統合失調症は、幻聴及び幻視、偏執症、妄想(陽性症状)、感情鈍麻、鬱病、快感消失、会話の貧困、記憶及び注意欠陥、並びに社会的離脱(陰性症状)として現れる。
数十年間、科学者や臨床医は、統合失調症の薬理学的処置用の理想的な薬剤の発見を目指して努力してきた。しかし、多様な症状によるこの疾患の複雑さのため、このような努力は阻まれてきた。統合失調症の診断には特異的な重要な特徴がなく、単一の症状が全ての患者に一貫して存在することもない。したがって、単一疾患としての、又は種々の異なる疾患としての統合失調症の診断は、検討はされているが、未だ解決していない。統合失調症用の新しい薬物の開発における主要な問題は、この疾患の原因及び性質に関する知識の欠如である。対応する治療法の開発を理論的に説明するための薬理学的研究に基づいて、幾つかの神経化学的仮説が提唱されている:ドーパミン、セロトニン及びグルタミン酸仮説。しかし統合失調症の複雑さを考慮すると、陽性及び陰性の兆候及び症状に対する有効性には、適切な多重受容体親和性プロフィールが必要となろう。更には、統合失調症に対する理想的薬物は、統合失調症患者の順守が低いため、好ましくは1日1回投与が可能な低用量であろう。
近年、選択的NK1及びNK2受容体アンタゴニストによる臨床試験が文献中に出現し、嘔吐、鬱病、不安、疼痛及び片頭痛(NK1)並びに喘息(NK2及びNK1)の処置に関する結果が示された。最も刺激的なデータは、NK1受容体アンタゴニストによる化学療法誘導性嘔吐、吐き気及び鬱病の処置、並びにNK2受容体アンタゴニストによる喘息の処置において生み出された。対照的に、NK3受容体アンタゴニストに関する臨床データは、2000年までの文献に出現しなかった。Sanofi-SynthelaboのOsanetant(SR 142,801)は、統合失調症の見込みある処置用の、NK3タキキニン受容体について記述された最初に同定された強力かつ選択的な非ペプチドアンタゴニストであったが、これは以下の文献に報告された(Current Opinion in Investigational Drugs, 2001, 2(7), 950-956及びPsychiatric Disorders Study 4, Schizophrenia, June 2003, Decision Recources, Inc., Waltham, Massachusetts)。提案された薬物のSR 142,801は、第II相試験において、行動変化、妄想、幻覚、極端な感情、興奮した運動活動及び思考散乱性言語のような統合失調症の陽性症状に対して活性であるが、陰性症状(これらは、鬱病、快感消失、社会的孤立又は記憶及び注意欠陥である)の処置には不活性であることが示されている。
ニューロキニン−3受容体アンタゴニストは、疼痛又は炎症、更には統合失調症において有用であると記述されている(Exp. Opinion. Ther. Patents (2000), 10(6), 939-960及びCurrent Opinion in Investigational Drugs, 2001, 2(7), 950-956及びPsychiatric Disorders Study 4, Schizophrenia, June 2003, Decision Recources, Inc., Waltham, Massachusetts)。
本発明の目的は、式(I)の新規な化合物、その製造法、本発明の化合物に基づく医薬及びその製造、更には鬱病、疼痛、双極性障害、精神病、パーキンソン病、統合失調症、不安及び注意欠陥多動障害(ADHD)のような病気の制御又は予防における式(I)の化合物の使用である。
本発明の化合物を用いる好ましい適応症は、鬱病、精神病、パーキンソン病、統合失調症、不安及び注意欠陥多動障害(ADHD)である。
本説明において使用される一般用語の下記の定義は、問題の用語が、単独で出現するか、又は組合せて出現するかにかかわらず適用される。
本明細書において使用されるとき、「低級アルキル」という用語は、1〜7個の炭素原子を含有する直鎖又は分岐鎖アルキル基、例えば、メチル、エチル、プロピル、イソプロピル、n−ブチル、i−ブチル、t−ブチルなどを意味する。好ましい低級アルキル基は、1〜4個の炭素原子を持つ基である。
本明細書において使用されるとき、「低級アルコキシ」という用語は、上述のような1〜7個の炭素原子を含有する直鎖又は分岐鎖アルキル基であって、このアルキル基が、酸素原子を介して結合している基を意味する。
「ハロゲン」という用語は、塩素、ヨウ素、フッ素及び臭素を意味する。
「シクロアルキル」という用語は、3〜7個の炭素原子を含有する飽和炭素環、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチルなどを意味する。
「薬学的に許容しうる酸付加塩」という用語は、塩酸、硝酸、硫酸、リン酸、クエン酸、ギ酸、フマル酸、マレイン酸、酢酸、コハク酸、酒石酸、メタンスルホン酸、p−トルエンスルホン酸などのような、無機及び有機酸との塩を包含する。
R1が、非置換であるか又は1個若しくは2個のハロゲン原子により置換されているフェニルである、式(I)の化合物、例えば、下記化合物が好ましい:
2−(4−{(2S,4S)−1−ベンジル−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(2−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル、
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(3−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル、
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(4−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル、
2−(4−{(2S,4S)−1−(2−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、
2−(4−{(2S,4S)−1−(3−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、
2−(4−{(2S,4S)−1−(4−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、又は
2−(4−{(2S,4S)−1−(3,4−ジフルオロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル。
2−(4−{(2S,4S)−1−ベンジル−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(2−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル、
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(3−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル、
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(4−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル、
2−(4−{(2S,4S)−1−(2−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、
2−(4−{(2S,4S)−1−(3−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、
2−(4−{(2S,4S)−1−(4−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、又は
2−(4−{(2S,4S)−1−(3,4−ジフルオロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル。
R1が、シクロアルキルである、式(I)の化合物、例えば、下記化合物もまた好ましい:
2−(4−{(2S,4S)−1−シクロヘキシルメチル−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル。
2−(4−{(2S,4S)−1−シクロヘキシルメチル−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル。
本発明の式(I)の化合物の調製は、逐次又は収束合成経路で実施することができる。本発明の化合物の合成は、下記スキームに示される。本反応及び生じる生成物の精製を実施するのに必要な技能は、当業者には知られている。下記の製造法の説明において使用される置換基及び指数は、特に断りない限り、本明細書に前記の意味を有する。
式(I)の化合物は、後述の方法により、実施例に与えられた方法により、又は類似の方法により製造することができる。個々の反応工程に適切な反応条件は、当業者には知られている。しかし、反応順序は、スキーム1に示されるものに限られるわけではなく、出発物質及びそのそれぞれの反応性に応じて、反応工程の順序は自由に変更することができる。出発物質は、市販されているか、あるいは後述の方法と類似の方法により、説明中若しくは実施例中に引用された参考文献に記載された方法により、又は当該分野において既知の方法により調製することができるかのいずれかである。
本発明の式(I)の化合物及びその薬学的に許容しうる塩は、当該分野において既知の方法により、例えば、後述の製造法により調製することができるが、この製造法は、
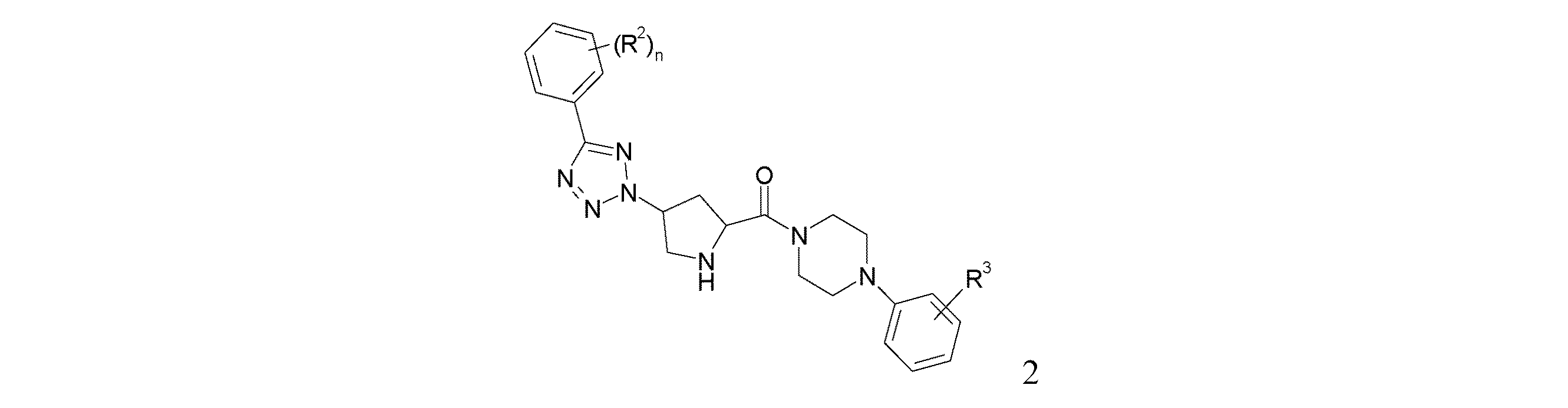
式(2):
式(2):
で示される化合物を、R1C(O)H、酢酸及びNaBH(OAc)3と反応させることにより、式(I):
[式中、置換基は、上記のとおりである]で示される化合物を得ること、そして所望であれば、式(I)の化合物を薬学的に許容しうる塩に変換することを含む。
式(I)の化合物の調製法は、より詳細にはスキーム1及び実施例1〜9に更に記述される。
a) DMF中の対応するフェニル−テトラゾールの溶液に、2−[4−(2−シアノ−フェニル)−ピペラジン−1−カルボニル]−4−(トルエン−4−スルホニルオキシ)−ピロリジン−1−カルボン酸tert−ブチルエステル及び無水炭酸ナトリウムを加える。この混合物は、約60度で一晩撹拌する。この溶液をEAで希釈し、洗浄し、乾燥して濃縮することにより、対応する4−[5−(フェニル)−テトラゾール−2−イル]−ピロリジン−1,2−ジカルボン酸1−tert−ブチルエステル2−メチルエステル(4)を得る。
b) 0度に冷却したMeOH中の4−[5−(フェニル)−テトラゾール−2−イル]−ピロリジン−1,2−ジカルボン酸1−tert−ブチルエステル2−メチルエステルの溶液に、LiOHを加えて、この混合物を一晩撹拌する。MeOHの除去後、残留物はHClで酸性にし、EAで抽出し、乾燥して濃縮することにより、対応する4−[5−(フェニル)−テトラゾール−2−イル]−ピロリジン−1,2−ジカルボン酸1−tert−ブチルエステル(5)を得る。
c) DCM中の4−[5−(フェニル)−テトラゾール−2−イル]−ピロリジン−1,2−ジカルボン酸1−tert−ブチルエステル、HOBt、EDC.HCl及びEt3Nの溶液に、置換フェニル−ピペラジン、例えば、1−(2−シアノフェニル)−ピペラジンを加える。この混合物は一晩撹拌し、次にNa2CO3、ブラインで洗浄し、乾燥して濃縮することにより、2−[4−(2−シアノ−フェニル)−ピペラジン−1−カルボニル]−4−[5−(フェニル)−テトラゾール−2−イル]−ピロリジン−1−カルボン酸tert−ブチルエステル(6)を得る。
d) 2−[4−(2−シアノ−フェニル)−ピペラジン−1−カルボニル]−4−[5−(フェニル)−テトラゾール−2−イル]−ピロリジン−1−カルボン酸tert−ブチルエステルをCF3COOHに加えて、この反応混合物を約5時間撹拌する。CF3COOHの除去後、残留物をDCMに溶解し、洗浄し、乾燥して濃縮することにより、2−(4−{4−[5−(フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(2)を得る。
e) 2−(4−{4−[5−(フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、ベンズアルデヒド及び酢酸(触媒)をDCMに溶解して、この溶液を20分間撹拌する。次にNaBH(OAc)3を加える。生じる混合物は室温まで温めて一晩撹拌する。次にこの溶液を飽和NaHCO3、ブラインで洗浄し、乾燥して濃縮することにより、2−(4−{1−ベンジル−4−[5−(フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(1)を得る。
塩形成は、それ自体既知であり、かつ当業者が精通している方法により、室温で遂行する。無機酸との塩だけでなく、有機酸との塩も考慮される。塩酸塩、臭化水素酸塩、硫酸塩、硝酸塩、クエン酸塩、酢酸塩、マレイン酸塩、コハク酸塩、メタン−スルホン酸塩、p−トルエンスルホン酸塩などは、このような塩の例である。
略語
DCM=ジクロロメタン;
DMF=N,N−ジメチルホルムアミド;
MS=質量分析;
EA=酢酸エチル
EDC=1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド
HOBt=1−ヒドロキシベンゾトリアゾール水和物
DCM=ジクロロメタン;
DMF=N,N−ジメチルホルムアミド;
MS=質量分析;
EA=酢酸エチル
EDC=1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド
HOBt=1−ヒドロキシベンゾトリアゾール水和物
前述のとおり、式(I)の化合物及びその薬学的に利用しうる付加塩は、有用な薬理学的特性を有する。本発明の化合物は、ニューロキニン3(NK−3)受容体のアンタゴニストであることが見い出された。本化合物は、本明細書に後述の試験法により詳しく調べた。
試験の記述:
本化合物は、本明細書に後述の試験法により詳しく調べた。
[3H]SR 142801競合結合アッセイ
hNK3受容体結合実験は、[3H]SR 142801(カタログ番号TRK1035、比活性:74.0 Ci/mmol, Amersham, GE Healthcare UK limited, Buckinghamshire, UK)、及び組換えヒトNK3受容体を一時的に発現するHEK293細胞から単離した膜を用いて実行した。解凍後、膜ホモジネートは、4℃で10分間48,000×gで遠心分離し、ペレットは50mM トリス−HCl、4mM MnCl2、1μMホスホラミドン、0.1% BSA結合緩衝液(pH7.4)に再懸濁して、5μgタンパク質/ウェルの最終アッセイ濃度とした。阻害実験には、膜を、[3H]SR 142801(放射性リガンドのKD値に等しい濃度)及び10倍濃度の阻害化合物(0.0003〜10μM)と共に(500μlの総反応容量として)室温(RT)で75分間インキュベートした。インキュベーションの最後に、膜をユニフィルター(0.3% PEI+0.3% BSA中で1時間プレインキュベートしたGF/Cフィルターを内蔵した96ウェルの白色マイクロプレート、Packard BioScience, Meriden, CT)上でFiltermate 196ハーベスター(Packard BioScience)により濾過して、氷冷50mMトリス−HCl(pH7.4)緩衝液で4回洗浄した。非特異結合は、両方の放射性リガンドについて10μM SB222200の存在下で測定した。フィルター上の放射活性は、45μlのmicroscint 40(Canberra Packard S.A., Zuerich, Switzerland)の添加及び1時間の振盪後、クエンチング補正してPackard Top-countマイクロプレートシンチレーションカウンターで計数を行った(5分間)。阻害曲線は、Excel-fit 4ソフトウェア(Microsoft)を用いて、Hillの式:y=100/(1+(x/IC50)nH)(ここで、nH=傾きである)により当てはめた。IC50値は、阻害曲線から誘導し、そして親和定数(Ki)値は、Cheng-Prussoff式:Ki=IC50/(1+[L]/KD)(ここで、[L]は、放射性リガンドの濃度であり、そしてKDは、飽和等温線から誘導された受容体でのその解離定数である)を用いて算出した。全ての実験は、二重反復で実行し、そして個々のKi値の平均±標準誤差(SEM)を算出した。
本化合物は、本明細書に後述の試験法により詳しく調べた。
[3H]SR 142801競合結合アッセイ
hNK3受容体結合実験は、[3H]SR 142801(カタログ番号TRK1035、比活性:74.0 Ci/mmol, Amersham, GE Healthcare UK limited, Buckinghamshire, UK)、及び組換えヒトNK3受容体を一時的に発現するHEK293細胞から単離した膜を用いて実行した。解凍後、膜ホモジネートは、4℃で10分間48,000×gで遠心分離し、ペレットは50mM トリス−HCl、4mM MnCl2、1μMホスホラミドン、0.1% BSA結合緩衝液(pH7.4)に再懸濁して、5μgタンパク質/ウェルの最終アッセイ濃度とした。阻害実験には、膜を、[3H]SR 142801(放射性リガンドのKD値に等しい濃度)及び10倍濃度の阻害化合物(0.0003〜10μM)と共に(500μlの総反応容量として)室温(RT)で75分間インキュベートした。インキュベーションの最後に、膜をユニフィルター(0.3% PEI+0.3% BSA中で1時間プレインキュベートしたGF/Cフィルターを内蔵した96ウェルの白色マイクロプレート、Packard BioScience, Meriden, CT)上でFiltermate 196ハーベスター(Packard BioScience)により濾過して、氷冷50mMトリス−HCl(pH7.4)緩衝液で4回洗浄した。非特異結合は、両方の放射性リガンドについて10μM SB222200の存在下で測定した。フィルター上の放射活性は、45μlのmicroscint 40(Canberra Packard S.A., Zuerich, Switzerland)の添加及び1時間の振盪後、クエンチング補正してPackard Top-countマイクロプレートシンチレーションカウンターで計数を行った(5分間)。阻害曲線は、Excel-fit 4ソフトウェア(Microsoft)を用いて、Hillの式:y=100/(1+(x/IC50)nH)(ここで、nH=傾きである)により当てはめた。IC50値は、阻害曲線から誘導し、そして親和定数(Ki)値は、Cheng-Prussoff式:Ki=IC50/(1+[L]/KD)(ここで、[L]は、放射性リガンドの濃度であり、そしてKDは、飽和等温線から誘導された受容体でのその解離定数である)を用いて算出した。全ての実験は、二重反復で実行し、そして個々のKi値の平均±標準誤差(SEM)を算出した。
hNK−3受容体親和性を持つ化合物1〜9の結果は、下記表1に示した。
式(I)の化合物及び薬学的に使用されうるその酸付加塩は、医薬として、例えば、医薬製剤の形態で使用することができる。医薬製剤は、例えば、錠剤、コーティング錠、糖衣錠、硬及び軟ゼラチンカプセル剤、液剤、乳剤又は懸濁剤の剤形で、経口投与することができる。しかし投与はまた、例えば坐剤の剤形で直腸内に、又は例えば注射液の剤形で非経口的に行うこともできる。
式(I)の化合物及び薬学的に使用されうるその酸付加塩は、医薬として、例えば、医薬製剤の形態で使用することができる。医薬製剤は、例えば、錠剤、コーティング錠、糖衣錠、硬及び軟ゼラチンカプセル剤、液剤、乳剤又は懸濁剤の剤形で、経口投与することができる。しかし投与はまた、例えば坐剤の剤形で直腸内に、又は例えば注射液の剤形で非経口的に行うこともできる。
軟ゼラチンカプセル剤に適切な賦形剤は、例えば、植物油、ロウ、脂肪、半固体及び液体ポリオール等である。
液剤及びシロップ剤の製造に適切な賦形剤は、例えば、水、ポリオール、ショ糖、転化糖、ブドウ糖等である。
注射液に適切な賦形剤は、例えば、水、アルコール、ポリオール、グリセリン、植物油等である。
坐剤に適切な賦形剤は、例えば、天然又は硬化油、ロウ、脂肪、半液体又は液体ポリオール等である。
更に、医薬製剤は、防腐剤、可溶化剤、安定剤、湿潤剤、乳化剤、甘味料、着色料、香味料、浸透圧を変えるための塩類、緩衝液、マスキング剤又は抗酸化剤を含有することができる。それらは、その他の治療上価値のある物質も更に含有することができる。
投与量は、広い限度内で変更することができ、当然、それぞれの特定の症例ごとに個々の要件に適合されるであろう。一般的に、経口投与の場合、一般式(I)の化合物の、1人当たり約10〜1000mgの1日投与量が適切であるが、必要であれば上記の上限を超えることもできる。
実施例A
通常の方法で、以下の組成の錠剤を製造する:
mg/錠剤
活性物質 5
乳糖 45
トウモロコシデンプン 15
微晶質セルロース 34
ステアリン酸マグネシウム 1
錠剤重量 100
通常の方法で、以下の組成の錠剤を製造する:
mg/錠剤
活性物質 5
乳糖 45
トウモロコシデンプン 15
微晶質セルロース 34
ステアリン酸マグネシウム 1
錠剤重量 100
実施例B
以下の組成のカプセル剤を製造する:
mg/カプセル
活性物質 10
乳糖 155
トウモロコシデンプン 30
タルク 5
カプセル充填重量 200
以下の組成のカプセル剤を製造する:
mg/カプセル
活性物質 10
乳糖 155
トウモロコシデンプン 30
タルク 5
カプセル充填重量 200
活性物質、乳糖及びトウモロコシデンプンを、最初にミキサーで、次に、微粉砕機で混合する。混合物をミキサーに戻し、タルクを加えて、十分に混合する。混合物を機械により硬ゼラチンカプセルに充填する。
実施例C
以下の組成の坐剤を製造する:
mg/坐剤
活性物質 15
坐剤用錬剤 1285
合計 1300
以下の組成の坐剤を製造する:
mg/坐剤
活性物質 15
坐剤用錬剤 1285
合計 1300
坐薬用錬剤をガラス又はスチール容器中で溶解し、十分に混合し、45℃に冷却する。その後、そこに微粉砕した活性物質を加え、それが完全に分散するまで撹拌する。混合物を適切な大きさの坐剤成形型に注ぎ、放置して冷却し、次に坐剤を成形型から取り外し、パラフィン紙又は金属箔で個別に包む。
下記の実施例は、本発明を限定することなく説明する。全ての温度は摂氏で示される。
実施例1
2−(4−{(2S,4S)−1−ベンジル−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
a) (2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−1,2−ジカルボン酸1−tert−ブチルエステル2−メチルエステル
DMF 5ml中の(2,4−ジフルオロ−フェニル)−テトラゾール(0.44g、1.1mmol)の溶液に、(2S,4R)−2−[4−(2−シアノ−フェニル)−ピペラジン−1−カルボニル]−4−(トルエン−4−スルホニルオキシ)−ピロリジン−1−カルボン酸tert−ブチルエステル0.1g(0.55mmol)及び無水炭酸ナトリウム(0.15g、1.4mmol)を加えた。混合物を60度で一晩激しく撹拌した。溶液をEA 30mlで希釈し、1M Na2CO3、5%クエン酸及びブラインで洗浄し、乾燥させ、濃縮して、粗生成物を黄色の油状物(0.14g、0.34mmol)として得た。MS(m/e)=410.3[M+H]+。
2−(4−{(2S,4S)−1−ベンジル−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
a) (2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−1,2−ジカルボン酸1−tert−ブチルエステル2−メチルエステル
DMF 5ml中の(2,4−ジフルオロ−フェニル)−テトラゾール(0.44g、1.1mmol)の溶液に、(2S,4R)−2−[4−(2−シアノ−フェニル)−ピペラジン−1−カルボニル]−4−(トルエン−4−スルホニルオキシ)−ピロリジン−1−カルボン酸tert−ブチルエステル0.1g(0.55mmol)及び無水炭酸ナトリウム(0.15g、1.4mmol)を加えた。混合物を60度で一晩激しく撹拌した。溶液をEA 30mlで希釈し、1M Na2CO3、5%クエン酸及びブラインで洗浄し、乾燥させ、濃縮して、粗生成物を黄色の油状物(0.14g、0.34mmol)として得た。MS(m/e)=410.3[M+H]+。
b) (2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−1,2−ジカルボン酸1−tert−ブチルエステル
0度に冷却したMeOH(25ml)中の4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−1,2−ジカルボン酸1−tert−ブチルエステル2−メチルエステル(0.14g、0.34mmol)の溶液に、LiOH(0.06g、1.36mmol)を加え、混合物を一晩撹拌した。MeOHの除去後、残留物を2M HClで酸性化した。水層をEAで抽出し、有機溶液を乾燥させ、濃縮して、標記生成物を黄色の油状物(0.1g、0.25mmol)として得た。MS(m/e)=396.3[M+H]+。
0度に冷却したMeOH(25ml)中の4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−1,2−ジカルボン酸1−tert−ブチルエステル2−メチルエステル(0.14g、0.34mmol)の溶液に、LiOH(0.06g、1.36mmol)を加え、混合物を一晩撹拌した。MeOHの除去後、残留物を2M HClで酸性化した。水層をEAで抽出し、有機溶液を乾燥させ、濃縮して、標記生成物を黄色の油状物(0.1g、0.25mmol)として得た。MS(m/e)=396.3[M+H]+。
c) (2S,4S)−2−[4−(2−シアノ−フェニル)−ピペラジン−1−カルボニル]−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−1−カルボン酸tert−ブチルエステル
DCM(20ml)中の酸(0.1g、0.25mmol)、HOBt(0.051g、0.38mmol)、EDC.HCl(0.073g、0.38mmol)及びEt3N(0.07ml、0.5mmol)の溶液に、1−(2−シアノフェニル)−ピペラジン(0.056g、0.3mmol)を加えた。混合物を一晩撹拌し、次に飽和Na2CO3、ブラインで洗浄し、乾燥させ、濃縮して、標記生成物を黄色の油状物(0.184g、0.33mmol)として得た。MS(m/e)=565.3[M+H]+。
DCM(20ml)中の酸(0.1g、0.25mmol)、HOBt(0.051g、0.38mmol)、EDC.HCl(0.073g、0.38mmol)及びEt3N(0.07ml、0.5mmol)の溶液に、1−(2−シアノフェニル)−ピペラジン(0.056g、0.3mmol)を加えた。混合物を一晩撹拌し、次に飽和Na2CO3、ブラインで洗浄し、乾燥させ、濃縮して、標記生成物を黄色の油状物(0.184g、0.33mmol)として得た。MS(m/e)=565.3[M+H]+。
d) (2S,4S)−2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
2−[4−(2−シアノ−フェニル)−ピペラジン−1−カルボニル]−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−1−カルボン酸tert−ブチルエステル(0.184g、0.33mmol)を、CF3COOH(0.22g、1.95mmol)に加え、反応混合物を5時間撹拌した。CF3COOHを除去した後、残留物をDCMに溶解し、飽和NaHCO3、ブラインで洗浄し、乾燥させ、濃縮して、標記生成物を黄色の油状物(0.136g、0.29mmol)として得た。MS(m/e)=465.2[M+H]+。
2−[4−(2−シアノ−フェニル)−ピペラジン−1−カルボニル]−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−1−カルボン酸tert−ブチルエステル(0.184g、0.33mmol)を、CF3COOH(0.22g、1.95mmol)に加え、反応混合物を5時間撹拌した。CF3COOHを除去した後、残留物をDCMに溶解し、飽和NaHCO3、ブラインで洗浄し、乾燥させ、濃縮して、標記生成物を黄色の油状物(0.136g、0.29mmol)として得た。MS(m/e)=465.2[M+H]+。
e) (2S,4S)−2−(4−{1−ベンジル−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(0.136g、0.29mmol)、ベンズアルデヒド(0.034g、0.32mmol)及び酢酸(触媒)を、DCM(20ml)に溶解し、溶液を20分間撹拌した。次にNaBH(OAc)3(0.12g、0.58mmol)を注意深く加えた。得られた混合物を室温に温め、一晩撹拌した。次に溶液を飽和NaHCO3、ブラインで洗浄し、乾燥させ、濃縮して、標記生成物を黄色の油状物(0.15g、6mmol)として得た。MS(m/e)=555.4[M+H]+。
2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(0.136g、0.29mmol)、ベンズアルデヒド(0.034g、0.32mmol)及び酢酸(触媒)を、DCM(20ml)に溶解し、溶液を20分間撹拌した。次にNaBH(OAc)3(0.12g、0.58mmol)を注意深く加えた。得られた混合物を室温に温め、一晩撹拌した。次に溶液を飽和NaHCO3、ブラインで洗浄し、乾燥させ、濃縮して、標記生成物を黄色の油状物(0.15g、6mmol)として得た。MS(m/e)=555.4[M+H]+。
実施例2
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(2−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに2−フルオロ−ベンズアルデヒド(17mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(6.1mg、8.2%)に変換した。MS(m/e)=573.2[M+H]+。
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(2−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに2−フルオロ−ベンズアルデヒド(17mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(6.1mg、8.2%)に変換した。MS(m/e)=573.2[M+H]+。
実施例3
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(3−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに3−フルオロ−ベンズアルデヒド(17mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(9.9mg、13.3%)に変換した。MS(m/e)=573.1[M+H]+。
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(3−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに3−フルオロ−ベンズアルデヒド(17mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(9.9mg、13.3%)に変換した。MS(m/e)=573.1[M+H]+。
実施例4
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(4−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに4−フルオロ−ベンズアルデヒド(17mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(8.2mg、11%)に変換した。MS(m/e)=573.1[M+H]+。
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(4−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに4−フルオロ−ベンズアルデヒド(17mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(8.2mg、11%)に変換した。MS(m/e)=573.1[M+H]+。
実施例5
2−(4−{(2S,4S)−1−(2−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに2−クロロ−ベンズアルデヒド(20mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(11mg、14.4%)に変換した。MS(m/e)=589.1(75%);591.1(25%)[M+H]+。
2−(4−{(2S,4S)−1−(2−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに2−クロロ−ベンズアルデヒド(20mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(11mg、14.4%)に変換した。MS(m/e)=589.1(75%);591.1(25%)[M+H]+。
実施例6
2−(4−{(2S,4S)−1−(3−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに3−クロロ−ベンズアルデヒド(20mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(7.7mg、10%)に変換した。MS(m/e)=589.1(75%);591.1(25%)[M+H]+。
2−(4−{(2S,4S)−1−(3−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに3−クロロ−ベンズアルデヒド(20mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(7.7mg、10%)に変換した。MS(m/e)=589.1(75%);591.1(25%)[M+H]+。
実施例7
2−(4−{(2S,4S)−1−(4−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに4−クロロ−ベンズアルデヒド(20mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(8.3mg、11%)に変換した。MS(m/e)=589.1(75%);591.1(25%)[M+H]+。
2−(4−{(2S,4S)−1−(4−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに4−クロロ−ベンズアルデヒド(20mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(8.3mg、11%)に変換した。MS(m/e)=589.1(75%);591.1(25%)[M+H]+。
実施例8
2−(4−{(2S,4S)−1−シクロヘキシルメチル−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりにシクロヘキサンカルバルデヒド(16mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(8.5mg、11.7%)に変換した。MS(m/e)=561.3[M+H]+。
2−(4−{(2S,4S)−1−シクロヘキシルメチル−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりにシクロヘキサンカルバルデヒド(16mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(8.5mg、11.7%)に変換した。MS(m/e)=561.3[M+H]+。
実施例9
2−(4−{(2S,4S)−1−(3,4−ジフルオロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに3,4−ジフルオロ−ベンズアルデヒド(20mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(10.4mg、13.5%)に変換した。MS(m/e)=591.2[M+H]+。
2−(4−{(2S,4S)−1−(3,4−ジフルオロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
実施例1eについて記載されたように、2−(4−{4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル(60mg、0.13mmol)を、ベンズアルデヒドの代わりに3,4−ジフルオロ−ベンズアルデヒド(20mg、0.14mmol)を使用して、明黄色の油状物として標記化合物(10.4mg、13.5%)に変換した。MS(m/e)=591.2[M+H]+。
Claims (11)
- 式(I):
[式中、
R1は、シクロアルキルであるか、あるいは非置換であるか又は1個若しくは2個のハロゲン原子により置換されているフェニルであり;
R2は、水素又はハロゲンであり;
R3は、水素、CN、低級アルコキシ、低級アルキル又はハロゲンであり;
nは、0、1又は2である]で示される化合物又は薬学的に適切なその酸付加塩。 - R1が、非置換であるか又は1個若しくは2個のハロゲン原子により置換されているフェニルである、請求項1記載の式(I)の化合物。
- 化合物が、
2−(4−{(2S,4S)−1−ベンジル−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(2−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル、
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(3−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル、
2−{4−[(2S,4S)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−1−(4−フルオロ−ベンジル)−ピロリジン−2−カルボニル]−ピペラジン−1−イル}−ベンゾニトリル、
2−(4−{(2S,4S)−1−(2−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、
2−(4−{(2S,4S)−1−(3−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、
2−(4−{(2S,4S)−1−(4−クロロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル、又は
2−(4−{(2S,4S)−1−(3,4−ジフルオロ−ベンジル)−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリル
である、請求項2記載の式(I)の化合物。 - R1が、シクロアルキルである請求項1記載の式(I)の化合物。
- 化合物が、2−(4−{(2S,4S)−1−シクロヘキシルメチル−4−[5−(2,4−ジフルオロ−フェニル)−テトラゾール−2−イル]−ピロリジン−2−カルボニル}−ピペラジン−1−イル)−ベンゾニトリルである、請求項4記載の式(I)の化合物。
- 式(I)で示される化合物の製造方法であって、式(2):
で示される化合物を、R1C(O)H、酢酸及びNaBH(OAc)3と反応させることにより、式(I):
[式中、置換基は、請求項1に記載のとおりである]で示される化合物を得ること、そして所望であれば、式(I)の化合物を薬学的に許容しうる塩に変換することを含む方法。 - 請求項6記載の方法によるか又は同等な方法により製造される、請求項1記載の化合物。
- 請求項1〜5のいずれか一項記載の1つ以上の化合物及び薬学的に許容しうる賦形剤を含む医薬。
- 鬱病、疼痛、精神病、パーキンソン病、統合失調症、不安及び注意欠陥多動障害(ADHD)の処置のための、請求項8記載の医薬。
- 鬱病、疼痛、精神病、パーキンソン病、統合失調症、不安及び注意欠陥多動障害(ADHD)の処置用の医薬の製造のための、請求項1〜5のいずれか一項記載の化合物の使用。
- 本明細書に先に記載された発明。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07109783 | 2007-06-07 | ||
| PCT/EP2008/056587 WO2008148688A1 (en) | 2007-06-07 | 2008-05-29 | Prolinamide-tetrazole derivatives as nk3 receptor antagonists |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2010529068A true JP2010529068A (ja) | 2010-08-26 |
Family
ID=39768738
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2010510746A Ceased JP2010529068A (ja) | 2007-06-07 | 2008-05-29 | Nk3受容体アンタゴニストとしてのプロリンアミド−テトラゾール誘導体 |
Country Status (14)
| Country | Link |
|---|---|
| US (1) | US7501422B2 (ja) |
| EP (1) | EP2155729B1 (ja) |
| JP (1) | JP2010529068A (ja) |
| KR (1) | KR101163294B1 (ja) |
| CN (1) | CN101679385A (ja) |
| AT (1) | ATE498622T1 (ja) |
| AU (1) | AU2008258664B2 (ja) |
| BR (1) | BRPI0812253A2 (ja) |
| CA (1) | CA2690292A1 (ja) |
| DE (1) | DE602008005014D1 (ja) |
| ES (1) | ES2359598T3 (ja) |
| IL (1) | IL202282A0 (ja) |
| MX (1) | MX2009013116A (ja) |
| WO (1) | WO2008148688A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023042914A1 (en) * | 2021-09-17 | 2023-03-23 | Otsuka Pharmaceutical Co., Ltd. | Pyrrolidine compounds as histone deacetylase inhibitors |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040034019A1 (en) * | 2002-08-08 | 2004-02-19 | Ronald Tomlinson | Piperazine and piperidine derivatives |
-
2008
- 2008-05-29 CA CA2690292A patent/CA2690292A1/en not_active Abandoned
- 2008-05-29 DE DE602008005014T patent/DE602008005014D1/de active Active
- 2008-05-29 CN CN200880018987A patent/CN101679385A/zh active Pending
- 2008-05-29 MX MX2009013116A patent/MX2009013116A/es active IP Right Grant
- 2008-05-29 KR KR1020107000225A patent/KR101163294B1/ko not_active Expired - Fee Related
- 2008-05-29 AU AU2008258664A patent/AU2008258664B2/en not_active Ceased
- 2008-05-29 JP JP2010510746A patent/JP2010529068A/ja not_active Ceased
- 2008-05-29 ES ES08760179T patent/ES2359598T3/es active Active
- 2008-05-29 BR BRPI0812253-9A2A patent/BRPI0812253A2/pt not_active IP Right Cessation
- 2008-05-29 EP EP08760179A patent/EP2155729B1/en not_active Not-in-force
- 2008-05-29 WO PCT/EP2008/056587 patent/WO2008148688A1/en not_active Ceased
- 2008-05-29 AT AT08760179T patent/ATE498622T1/de active
- 2008-05-30 US US12/129,720 patent/US7501422B2/en not_active Expired - Fee Related
-
2009
- 2009-11-23 IL IL202282A patent/IL202282A0/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040034019A1 (en) * | 2002-08-08 | 2004-02-19 | Ronald Tomlinson | Piperazine and piperidine derivatives |
Non-Patent Citations (1)
| Title |
|---|
| JPN6012043470; J. Med. Chem. vol.45, no.18, 2002, p.3972-3983 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2690292A1 (en) | 2008-12-11 |
| US20080306089A1 (en) | 2008-12-11 |
| KR101163294B1 (ko) | 2012-07-05 |
| EP2155729A1 (en) | 2010-02-24 |
| EP2155729B1 (en) | 2011-02-16 |
| MX2009013116A (es) | 2010-01-15 |
| AU2008258664A8 (en) | 2010-01-14 |
| ES2359598T3 (es) | 2011-05-25 |
| ATE498622T1 (de) | 2011-03-15 |
| BRPI0812253A2 (pt) | 2014-12-23 |
| DE602008005014D1 (de) | 2011-03-31 |
| IL202282A0 (en) | 2010-06-16 |
| WO2008148688A1 (en) | 2008-12-11 |
| CN101679385A (zh) | 2010-03-24 |
| AU2008258664B2 (en) | 2012-07-05 |
| US7501422B2 (en) | 2009-03-10 |
| AU2008258664A1 (en) | 2008-12-11 |
| KR20100020026A (ko) | 2010-02-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8293770B2 (en) | Pyrrolidine derivatives as NK-3 receptor antagonists | |
| JP5410536B2 (ja) | 3−(ベンジルアミノ)−ピロリジン誘導体及びnk−3受容体アンタゴニストとしてのその使用 | |
| RU2625798C2 (ru) | Производные (3-метилпирролидин-3-ил)-метил пиридинилового эфира и их применение в качестве антагонистов рецептора nk-3 | |
| JP2011522855A (ja) | Nk3受容体アンタゴニストとしてのピロリジンエーテル誘導体 | |
| EP2398768B1 (en) | Pyrrolidine derivatives as nk3 receptor antagonists | |
| JP2014501767A (ja) | Nk3アンタゴニストとしてのピロリジン誘導体 | |
| EP2155729B1 (en) | Prolinamide-tetrazole derivatives as nk3 receptor antagonists | |
| JP5362839B2 (ja) | Nk3受容体アンタゴニストとしてのキナゾリン誘導体 | |
| EP2167509A1 (en) | Piperazine and ý1,4¨diazepan derivatives as nk antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120821 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20121116 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121211 |
|
| A045 | Written measure of dismissal of application [lapsed due to lack of payment] |
Free format text: JAPANESE INTERMEDIATE CODE: A045 Effective date: 20130423 |