JP2009522357A - ブプレノルフィンのための出発物質としてのオリパビンの使用 - Google Patents
ブプレノルフィンのための出発物質としてのオリパビンの使用 Download PDFInfo
- Publication number
- JP2009522357A JP2009522357A JP2008549488A JP2008549488A JP2009522357A JP 2009522357 A JP2009522357 A JP 2009522357A JP 2008549488 A JP2008549488 A JP 2008549488A JP 2008549488 A JP2008549488 A JP 2008549488A JP 2009522357 A JP2009522357 A JP 2009522357A
- Authority
- JP
- Japan
- Prior art keywords
- formula
- compound
- buprenorphine
- norbuprenorphine
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D489/00—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula:
- C07D489/09—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula: containing 4aH-8, 9 c-Iminoethano- phenanthro [4, 5-b, c, d] furan ring systems condensed with carbocyclic rings or ring systems
- C07D489/10—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula: containing 4aH-8, 9 c-Iminoethano- phenanthro [4, 5-b, c, d] furan ring systems condensed with carbocyclic rings or ring systems with a bridge between positions 6 and 14
- C07D489/12—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula: containing 4aH-8, 9 c-Iminoethano- phenanthro [4, 5-b, c, d] furan ring systems condensed with carbocyclic rings or ring systems with a bridge between positions 6 and 14 the bridge containing only two carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D489/00—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula:
- C07D489/02—Heterocyclic compounds containing 4aH-8, 9 c- Iminoethano-phenanthro [4, 5-b, c, d] furan ring systems, e.g. derivatives of [4, 5-epoxy]-morphinan of the formula: with oxygen atoms attached in positions 3 and 6, e.g. morphine, morphinone
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
ここに、オリパビンを出発物質として利用する、ノルブプレノルフィンの、そして最終的にはブプレノルフィンの、合成方法が提供される。従来のブプレノルフィンの製造方法は、テバインを出発物質として利用し、O−脱メチル化段階を必要とし、それは、典型的には低収率ないし中収率の変換である。オリパビン分子はO−3メチル基を欠くので、今回の出発物質としてのオリパビンの使用は、O−脱メチル化段階を必要としない。
Description
発明の背景
ブプレノルフィンは、混合アゴニスト/アンタゴニストとして作用し、アヘン中毒および痛覚消失の重要な処置選択肢である。
ブプレノルフィンは、混合アゴニスト/アンタゴニストとして作用し、アヘン中毒および痛覚消失の重要な処置選択肢である。
ブプレノルフィンを製造するために世界中で使用される従来の合成経路は、出発物質としてテバインを利用する。
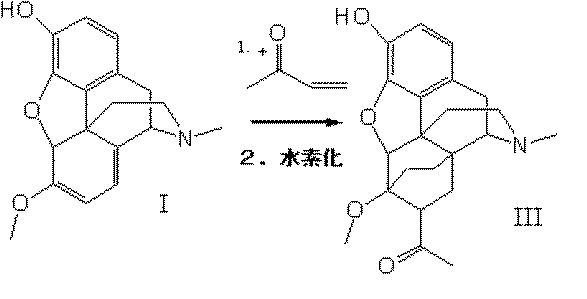
一連の化学反応を通して、テバインは、ブプレノルフィンの直接の前駆物質であるノル−ブプレノルフィンに変換される。最終段階は、シクロプロピルメチル基を窒素に付加し、ノル−ブプレノルフィンからブプレノルフィンを形成させる。
テバインからブプレノルフィンへの従来の一連の反応の概要は、以下の通りである:
1. 4+2反応生成物を形成させる、テバインのメチルビニルケトンとの反応。
2. 炭素−炭素二重結合の水素化。
3. グリニャール反応を介する第3級ブチル基の付加。
4. 2段階の連続反応を介するN−脱メチル化。
5. O−脱メチル化反応およびN−シアノ加水分解。
6. ブプレノルフィンを形成させる、シクロプロピルメチル基の付加。
1. 4+2反応生成物を形成させる、テバインのメチルビニルケトンとの反応。
2. 炭素−炭素二重結合の水素化。
3. グリニャール反応を介する第3級ブチル基の付加。
4. 2段階の連続反応を介するN−脱メチル化。
5. O−脱メチル化反応およびN−シアノ加水分解。
6. ブプレノルフィンを形成させる、シクロプロピルメチル基の付加。
この従来の製造スキームの欠点は、O−脱メチル化段階が、低収率ないし中収率の変換であるとみなされることである。従って、O−脱メチル化段階を含まないノルブプレノルフィン/ブプレノルフィンの製造スキームへの要望がある。
発明の概要
本発明のある態様は、オリパビン(oripavine)を出発物質として利用する、ノルブプレノルフィンの製造方法を提供することである。その方法は:
式Iで示されるオリパビンを、メチルビニルケトンと反応させ、式IIで示される化合物を形成させること;
式IIで示される化合物を水素化し、式IIIで示される化合物を形成させること;
式IIIで示される化合物にt−ブチル基を付加し、式Xで示される化合物を形成させること;および、
式Xで示される化合物の窒素を脱メチル化し、式VIIIのノルブプレノルフィンを形成させること
を含む。
本発明のある態様は、オリパビン(oripavine)を出発物質として利用する、ノルブプレノルフィンの製造方法を提供することである。その方法は:
式Iで示されるオリパビンを、メチルビニルケトンと反応させ、式IIで示される化合物を形成させること;
本発明の他の態様は、オリパビンを出発物質として利用する、ブプレノルフィンの製造方法を提供することである。
詳細な説明
ノル−ブプレノルフィンおよび場合によりブプレノルフィンの合成に好ましい出発物質としてオリパビンを利用する方法が提供される。オリパビンは、天然産生のケシ(Papaver somniferum)のアルカロイドである。従来の技術と今回の出発物質としてのオリパビンの使用との重要な違いは、オリパビン分子はO−3メチル基を欠くので、O−脱メチル化段階(典型的には低収率ないし中収率の変換)が必要ないことである。数段階を含む合成では、総合的な変換が経済的であるために、高収率の反応のみを含むのが有利である。今回のオリパビンをベースとする合成はO−3脱メチル化段階を必要としないので、オリパビンからの総合的な収率は、テバインを出発物質として使用するときに伝統的に達成される収率より、改善された収率をもたらす。ブプレノルフィンをもたらすオリパビンからの変換経路は、便利であり、他の合成経路と比較してより直接的である。
ノル−ブプレノルフィンおよび場合によりブプレノルフィンの合成に好ましい出発物質としてオリパビンを利用する方法が提供される。オリパビンは、天然産生のケシ(Papaver somniferum)のアルカロイドである。従来の技術と今回の出発物質としてのオリパビンの使用との重要な違いは、オリパビン分子はO−3メチル基を欠くので、O−脱メチル化段階(典型的には低収率ないし中収率の変換)が必要ないことである。数段階を含む合成では、総合的な変換が経済的であるために、高収率の反応のみを含むのが有利である。今回のオリパビンをベースとする合成はO−3脱メチル化段階を必要としないので、オリパビンからの総合的な収率は、テバインを出発物質として使用するときに伝統的に達成される収率より、改善された収率をもたらす。ブプレノルフィンをもたらすオリパビンからの変換経路は、便利であり、他の合成経路と比較してより直接的である。
オリパビンをノルブプレノルフィン、および場合によりブプレノルフィンに変換する段階の例示的実施態様は、以下の通りである:
上記で概説した連続反応は、必要な変換を示すために提示する例示的実施態様であるが、変換を用い得る順序について限定されない。代替的な実施態様では、Y保護基の除去が触媒的水素化を介するものであるとき、ディールズ・アルダーの二重結合の水素化を、段階7の一部として達成することもできる。
段階1:
第一段階は、オリパビンのメチルビニルケトンとの反応を含む。この付加反応は、当分野で知られているいかなる常套法により達成してもよい。例示的実施態様は、オリパビンおよびメチルビニルケトンを溶媒に溶解し、反応が実質的に完了するまで還流する、ディールズ・アルダー反応である。例示的な適する溶媒には、イソプロピルアルコール、メタノール、エタノール、トルエンおよびこれらの混合物が含まれる。次いで、反応混合物を濾過し、ディールズ・アルダーの付加物の固体を単離する。典型的な反応は、少なくとも約98%の純度の少なくとも約85%の収率をもたらす。
第一段階は、オリパビンのメチルビニルケトンとの反応を含む。この付加反応は、当分野で知られているいかなる常套法により達成してもよい。例示的実施態様は、オリパビンおよびメチルビニルケトンを溶媒に溶解し、反応が実質的に完了するまで還流する、ディールズ・アルダー反応である。例示的な適する溶媒には、イソプロピルアルコール、メタノール、エタノール、トルエンおよびこれらの混合物が含まれる。次いで、反応混合物を濾過し、ディールズ・アルダーの付加物の固体を単離する。典型的な反応は、少なくとも約98%の純度の少なくとも約85%の収率をもたらす。
段階2:
第2段階は、C−C二重結合の水素化を含む。例示的実施態様では、段階1で形成されたディールズ・アルダーの付加物を、Pd/炭素触媒と共に反応容器に入れ、次いで溶媒に溶解した。現在のところ好ましい溶媒はメタノールであるが、メタノール、エタノール、イソプロピルアルコール、酢酸およびこれらの混合物を含む任意の適する溶媒を使用し得る。水素化は、高い圧力と温度で、窒素下で行う。当分野で周知の通り、反応の実質的な完了を確実にするように、温度および圧力を選択する。この反応に典型的な例示的な温度範囲は約50−90℃であり、約60℃が好ましく、この反応に典型的な例示的な圧力範囲は約20−60psiであり、約35psiが好ましい。反応混合物を濾過して触媒を除去し、得られる濾液を真空下で濃縮し、式IIIで示される生成物を得る。
第2段階は、C−C二重結合の水素化を含む。例示的実施態様では、段階1で形成されたディールズ・アルダーの付加物を、Pd/炭素触媒と共に反応容器に入れ、次いで溶媒に溶解した。現在のところ好ましい溶媒はメタノールであるが、メタノール、エタノール、イソプロピルアルコール、酢酸およびこれらの混合物を含む任意の適する溶媒を使用し得る。水素化は、高い圧力と温度で、窒素下で行う。当分野で周知の通り、反応の実質的な完了を確実にするように、温度および圧力を選択する。この反応に典型的な例示的な温度範囲は約50−90℃であり、約60℃が好ましく、この反応に典型的な例示的な圧力範囲は約20−60psiであり、約35psiが好ましい。反応混合物を濾過して触媒を除去し、得られる濾液を真空下で濃縮し、式IIIで示される生成物を得る。
段階3:
場合による段階3は、式IVで示される化合物を形成させる、保護基Yの付加を開示する。ノル−ブプレノルフィンおよびその後のブプレノルフィンのための出発物質としてオリパビンを使用する好ましい方法は、O−3保護基を利用する。しかしながら、総合的収率は妥協されることがあるが、この反応は、保護基を使用せずに達成できる。さらに、保護基は、他の段階と同時に除去し得、それにより1つの化学的段階を排除し得る。O−3保護基の付加は、3位の保護されないフェノール官能基に関わる望まれない化学反応を最小限にし得る。例示的な適する保護基には、ベンジル、O−t−ブチルおよびシリル基が含まれる。
場合による段階3は、式IVで示される化合物を形成させる、保護基Yの付加を開示する。ノル−ブプレノルフィンおよびその後のブプレノルフィンのための出発物質としてオリパビンを使用する好ましい方法は、O−3保護基を利用する。しかしながら、総合的収率は妥協されることがあるが、この反応は、保護基を使用せずに達成できる。さらに、保護基は、他の段階と同時に除去し得、それにより1つの化学的段階を排除し得る。O−3保護基の付加は、3位の保護されないフェノール官能基に関わる望まれない化学反応を最小限にし得る。例示的な適する保護基には、ベンジル、O−t−ブチルおよびシリル基が含まれる。
ある例示的実施態様では、式IIIで示される還元されたディールズ・アルダーの付加物および粉砕したK2CO3(K2CO3は可溶性ではない)をクロロホルムおよび臭化ベンジルに添加し、加熱し、還流させる。室温に冷却後、反応混合物を濾過し、K2CO3を除去する。次いで、濾液を真空下で濃縮し、トルエン中で共沸乾固(azeo dried)する。
段階4:
第4段階は、段階3で形成された式IVで示される粗製物を、グリニャール反応において利用する。水分の無い条件下で、さらに不活性雰囲気下で、t−BuMgClを添加し、続いて無水トルエンを添加する。この溶液を約100℃のポット温度に達するまで蒸留し、式IVで示される化合物を添加する。反応をクエンチし、反応混合物の温度を低下させる。有機層および水層を分離し、有機層を真空下で濃縮し、油状残渣を得る。次いで、この油状残渣を精製し、約93%までの純度の式Vで示される化合物を得る。
第4段階は、段階3で形成された式IVで示される粗製物を、グリニャール反応において利用する。水分の無い条件下で、さらに不活性雰囲気下で、t−BuMgClを添加し、続いて無水トルエンを添加する。この溶液を約100℃のポット温度に達するまで蒸留し、式IVで示される化合物を添加する。反応をクエンチし、反応混合物の温度を低下させる。有機層および水層を分離し、有機層を真空下で濃縮し、油状残渣を得る。次いで、この油状残渣を精製し、約93%までの純度の式Vで示される化合物を得る。
代替的実施態様では、当分野で周知の通り、t−ブチルリチウム試薬を使用してt−ブチル基を付加する。
N−脱メチル化反応は、当分野で知られている任意の適する方法により達成し得る。段階5および6で示した例示的実施態様では、段階5でメチル基を最初にニトリルに変換し、続いて、段階6でニトリル基を還元する。
段階5:
段階5の例示的実施態様では、式Vで示される第三級アルコール出発物質を溶媒に溶解し、不活性雰囲気でフラッシュし、次いでK2CO3および臭化シアンを添加する。次いで、反応が実質的に完了するまでこの反応混合物を還流し、室温に冷却し、濾過してK2CO3を除去する。次いで、反応混合物を抽出し、有機層を低減させ、真空下で乾燥させる。得られる固体を精製し、乾燥後に約93%までの純粋(clean)物質を得る。
段階5の例示的実施態様では、式Vで示される第三級アルコール出発物質を溶媒に溶解し、不活性雰囲気でフラッシュし、次いでK2CO3および臭化シアンを添加する。次いで、反応が実質的に完了するまでこの反応混合物を還流し、室温に冷却し、濾過してK2CO3を除去する。次いで、反応混合物を抽出し、有機層を低減させ、真空下で乾燥させる。得られる固体を精製し、乾燥後に約93%までの純粋(clean)物質を得る。
段階6:
例示的実施態様では、水酸化カリウムをジエチレングリコールに溶解し、加熱する。式VIIで示されるN−CN化合物を添加し、反応が実質的に完了するまで反応混合物を加熱する。室温に冷却後、蒸留水を添加し、得られる固体を回収し、乾燥させ、収率は約100%までである。
例示的実施態様では、水酸化カリウムをジエチレングリコールに溶解し、加熱する。式VIIで示されるN−CN化合物を添加し、反応が実質的に完了するまで反応混合物を加熱する。室温に冷却後、蒸留水を添加し、得られる固体を回収し、乾燥させ、収率は約100%までである。
N−脱メチル化は、本方法から逸脱せずに、当業者に知られている任意の方法により、達成し得る。
段階7:
第7段階は、場合による保護基の除去を含む。例示的実施態様では、段階3で付加されたY保護基を除去する。この実施態様では、第2級アミン出発物質を、適する溶媒中でPd/炭素により触媒的に除去し得る。適する溶媒には、メタノール、エタノール、酢酸イソプロピルおよびこれらの混合物が含まれる。得られる濾液を真空下で乾燥し、ノルブプレノルフィンを得る。他の実施態様では、Y保護基は、HCl、HOAc、HFまたはF陰イオンなどの酸で除去し得る。
第7段階は、場合による保護基の除去を含む。例示的実施態様では、段階3で付加されたY保護基を除去する。この実施態様では、第2級アミン出発物質を、適する溶媒中でPd/炭素により触媒的に除去し得る。適する溶媒には、メタノール、エタノール、酢酸イソプロピルおよびこれらの混合物が含まれる。得られる濾液を真空下で乾燥し、ノルブプレノルフィンを得る。他の実施態様では、Y保護基は、HCl、HOAc、HFまたはF陰イオンなどの酸で除去し得る。
段階8:
最後に、段階8に例示説明した通り、ノルブプレノルフィンを場合によりブプレノルフィンに変換する。例示的実施態様では、ノルブプレノルフィンはブプレノルフィンに変換される。
最後に、段階8に例示説明した通り、ノルブプレノルフィンを場合によりブプレノルフィンに変換する。例示的実施態様では、ノルブプレノルフィンはブプレノルフィンに変換される。
例示的実施態様では、ノルブプレノルフィン、穏やかな塩基およびシクロプロピルメチルブロミドの混合物を、約80−100℃の油浴中で、反応が実質的に完了するまで加熱する。次いで、反応混合物を、機械的に撹拌しながら、5分間かけて水160mlに添加し、ゴム状物を得る。混合物を撹拌し、濾過し、濾過ケーキを水で洗浄する。HPLCは、面積で約90%の所望の生成物および0.2−0.5%のN−ブテニル置換された不純物を示すであろう。得られる生成物を乾燥し、次いで、アルコール中で沸騰させ、冷却し、濾過し、ブプレノルフィンを得る。
代替法では、還元的アミノ化により、または、アシル化に続くアミドの還元により、ノルブプレノルフィンをブプレノルフィンに変換できる。
代替的実施態様では、水素化段階2を段階1で形成された粗反応混合物で実施し、それにより式IIIで示される化合物を形成させるためのワンポットの反応スキームを提供する。
オリパビン、メチルビニルケトンおよびイソプロピルアルコールを圧力下で加熱する。冷却したら、Pd−C触媒を添加し、反応が実質的に完了するまで、反応混合物を圧力下で加熱する。次いで、生成物を可溶化し、触媒を濾過により除去する。次いで、濾液を真空下で濃縮する。
他の代替的実施態様では、O−3上の保護基を使用しない、オリパビンからのノルブプレノルフィンの、場合によりブプレノルフィンの製造方法を、下記に例示説明する。個々の反応は、上記で詳述した通りである。
その方法は、
a)式Iで示されるオリパビンを、メチルビニルケトンと反応させ、式IIで示される化合物を形成させること;
b)式IIで示される化合物を水素化し、式IIIで示される化合物を形成させること;
c)t−ブチル基を式IIIで示される化合物に添加し、式Xで示される化合物を形成させること;および
d)式Xで示される化合物の窒素を脱メチル化し、式VIIIのノルブプレノルフィンを形成させること、
を含む。
a)式Iで示されるオリパビンを、メチルビニルケトンと反応させ、式IIで示される化合物を形成させること;
実施例
実施例1
オリパビン(150g、505mmol)を、機械的撹拌装置、熱電対および還流冷却器を備えた1Lの3口のジャケット付きフラスコに入れた。反応物をイソプロピルアルコール(IPA)750mLに溶解した(5mL/gに等しい)。90%のテクニカルグレードのメチルビニルケトン(MVK)を、2部に分けて添加した。各部は、68.0mL(747mmol、1.5当量)からなり、2回目の添加は、1回目の約8時間後に行った。1回目のMVKの添加後、反応混合物を約78℃で還流に加熱し、MVKが消費されるにつれて、温度は84℃近くまでゆっくりと高まった。2回目のMVKの添加後、反応を終夜継続させ、ゆっくりと5−10℃に冷却することにより、午前中に停止させた。5−10℃で約2時間撹拌した後、反応混合物を濾過して固体を単離し、冷たいIPAで洗浄した。乾燥後、出発物質のアッセイ結果を用いて補正した後の面積で純度98−99%の付加物を収率85%で回収した。
実施例1
実施例2
a)圧力容器に、オリパビン9g(アッセイ結果77%)、メチルビニルケトン5.6mlおよびイソプロピルアルコール45mlを入れた。容器を密封し、内容物を110℃に加熱し、35psiの圧力をもたらした。4時間後、反応混合物を室温に冷却し、Pd−C(10%)0.6gを添加した。混合物を70℃および40psiで24時間水素化した。メタノールを使用して生成物を可溶化し、hyfloのプラグを通して濾過することにより触媒を除去した。濾液を真空で濃縮し、8.6gを得た。
実施例3
実施例1で形成されたディールズ・アルダーの付加物(13g、35mmol)を、5%チャージ(乾燥ベース)の10%Pd/炭素触媒(1.3g)と共に反応容器に入れた。次いで、付加物を10ml/gのメタノール(130mL)に溶解/懸濁した。容器を40psiのN2で8回、40psiのH2で4回フラッシュした後、圧力を40psiのH2に戻した。温度を60℃に上げ、ここで約8時間かけて、パール−シェイカー(Parr- Shaker)中で水素化を実施した。出発物質が完全に消費されたら、容器をN2でフラッシュして残っているH2を追い出した。次いで、反応混合物を温かい溶液としてセライトのプラグを通して濾過し、触媒を除去した。次いで、濾液を真空下で濃縮し、12.9g(99%)の期待された生成物を得た。
実施例4
実施例4に従い形成された還元されたディールズ・アルダーの付加物(24.0g、65mmol)および粉砕したK2CO3(45.0g、326mmol)を、機械的撹拌装置および還流冷却器を備えた500mLの3口丸底フラスコに入れた。CHCl3200mL、および次いで臭化ベンジル(9.3mL、13.4g、78.3mmol)をフラスコに添加した。次いで、反応混合物を還流に6時間加熱した。反応混合物を室温に放冷した後、それを濾過してK2CO3を除去し、固体を過剰のCHCl3で洗浄した。次いで、濾液を真空で濃縮し、トルエンで共沸乾固した(3x100mL)。次いで、この粗製物をグリニャール反応に直接使用した。
実施例5
使用前に、全てのガラス器具を150℃で終夜オーブン乾燥した。1lの4口丸底フラスコに、機械的撹拌機、目盛り付きの添加用漏斗、蒸留ヘッドおよび熱電対を取り付けた。これらの器具をまだ熱いうちに組み立て、窒素雰囲気下で室温に放冷した。1M t−BuMgCl222ml(222mmol)をフラスコに添加し、続いて無水トルエン250mlを添加した。溶液を100℃のポット温度に達するまで蒸留した。熱い溶液に、無水トルエン50ml中の実施例5に従い形成されたベンジル化ケトン20.4g(44.4mmol)を15分間かけて添加した。反応を100ないし105℃で4時間加熱し、次いで室温に放冷した。氷浴を使用して反応混合物を10℃未満に冷却し、内部温度を30℃未満に保ちながら、4N NH4Cl153mlでクエンチした。濃縮したHCl(22ml)を不均一な混合物に添加し、それによりpHを4に下げる。有機層および水層を分離し、200mlずつのCHCl3で水層および反応容器を2回抽出した。合わせた有機層を水200mlで洗浄し、真空で濃縮し、油状残渣を得た。反応の後処理の後、粗製油状物を酢酸エチル約200mLに溶解し、約30gのシリカゲルを加えた。次いで、得られる混合物を真空で濃縮し、シリカゲルに結合した生成物の固体をもたらした。次いで、この物質を、1%トリエチルアミンを含む2:1ヘプタン:酢酸エチルを使用して、シリカゲルフラッシュカラムクロマトグラフィーにより精製した。溶出物の分画により、純度93%の生成物13.6gを得た。
実施例6
実施例6に従い形成された第3級アルコール出発物質(18.2g、35.2mmol)を、磁気撹拌子を備えた500mLの丸底フラスコに入れた。次いで、出発物質をCHCl3100mLに溶解した。フラスコをN2でフラッシュした後、K2CO3(1.7g、12.3mmol)および臭化シアン(5.6g、52.9mmol)を添加した。反応混合物を還流させ、それを17時間続ける。室温に冷却した後、混合物を濾過してK2CO3を除去した。次いで、濾液を2:1H2O:濃NH4OH90mLと共に45分間激しく撹拌した。次いで、層を分離し、水層をさらに50mLのCHCl3で洗浄した。合わせた有機層を無水MgSO4で乾燥させ、真空で濃縮した。これにより、高真空下でさらに乾燥した後、泡状固体を得る。次いで、泡状固体をトルエン25mLおよびヘプタン70mLに溶解した。反応混合物を水浴中に置き、約90℃とした。さらに60mLのヘプタンを添加した後、溶液をゆっくりと冷却し、約82℃で沈殿が形成された。その浴を氷浴により10℃に下げると、この反応混合物はゆっくりと室温まで冷却した。30分後、固体を減圧濾過により単離し、3:1ヘプタン:トルエン40mLで洗浄した。これにより、乾燥後に93%の純粋物質12.4g(67%)を得た。
実施例7
250mlの丸底フラスコに、水酸化カリウム20.2g(360mmol)およびジエチレングリコール55mlを入れた。反応混合物を110℃に加熱し、加熱した溶液に、実施例6に従い形成されたN−CN化合物9.5g(18mmol)を添加した。27時間加熱した後、反応を室温に冷却し、蒸留水150mlを添加した。15分間の混合の後、固体を回収し、蒸留水100mlで洗浄した。乾燥後、生成物8.9gを得、これは、粗製物収率100%に相当した。
実施例8
実施例7に従い形成された第2級アミン出発物質(4.0g、8mmol)および10%Pd/炭素(0.4g、乾燥ベースで5%w/w)を、パール−シェイカー反応ボトルに入れた。反応物をメタノール30mLに懸濁し、パール−シェイカー器具に取り付けた。次いで、反応混合物を40psiのN2で8回フラッシュし、反応ボトルをH2で40psiまで満たした。反応ボトルを2.5時間60℃で振盪させた。そのとき、LC分析は出発物質が残っていないことを示した。次いで、反応物質をセライトのプラグを通して濾過し、触媒を除去した。次いで、濾液を真空で濃縮し、ノルブプレノルフィンを得た。
実施例9
ノルブプレノルフィン11.5g、重炭酸ナトリウム4.6g、シクロプロピルメチルブロミド4.85g(GCにより、その中に1%臭化ブテニルがある)および乾燥DMF46mlの混合物を、反応が実質的に完了するまで、80−100℃の油浴中で4時間加熱する。反応混合物を、洗浄液としての最小限のDMFと共に、滴下漏斗に移す。機械的に撹拌しながら、反応混合物を5分間かけて水160mlに添加する。(反応に水を添加し、ゴム状物を得る)。混合物を約10分間撹拌し、濾過する。濾過ケーキを水20mlで洗浄する。HPLCは、面積で約90%の所望の生成物および0.2−0.5%のN−ブテニル置換された不純物を示すであろう。得られる生成物を乾燥させ、約12gを得る。得られる生成物を、メタノール30ml中で約30分間沸騰させ、冷却し、濾過し、ブプレノルフィン約7.3gを得る。
ここに、ノルブプレノルフィンおよびブプレノルフィンの新規製造方法を説明した。本発明の方法を特定の反応および実施例を参照して説明したが、明記しない限り、そのような参照により本発明の範囲を限定する意図はない。材料および工程の順序、並びに、方法の組合せにおいて、様々な改変を行い得、それは、本発明から逸脱せずに、様々な工程に適するように改変される。上述の説明は、明確な理解のためにのみ与えられるものであり、改変は当業者に明らかであろうから、そこから不必要な限定を理解するべきではない。
Claims (15)
- ノルブプレノルフィンの製造方法であって、
a)式Iで示されるオリパビンを、メチルビニルケトンと反応させ、式IIで示される化合物を形成させること;
- シクロプロピルメチル基をノルブプレノルフィンに付加し、ブプレノルフィンを形成させることをさらに含む、請求項1に記載の方法。
- a)およびb)が、単一のポットの連続反応に組み合わされている、請求項1に記載の方法。
- ノルブプレノルフィンの製造方法であって、
a)式Iで示されるオリパビンを、メチルビニルケトンと反応させ、式IIで示される化合物を形成させること;
- シクロプロピルメチル基をノルブプレノルフィンに付加し、ブプレノルフィンを形成させることをさらに含む、請求項4に記載の方法。
- 脱メチル化段階が、
a)式Vで示される化合物上の窒素を、式VIで示されるニトリルに変換すること;および、
- 保護基Yが、ベンジル、O−t−ブチルおよびシリル基からなる群から選択される、請求項4に記載の方法。
- a)およびb)が、単一のポットの連続反応に組み合わされている、請求項4に記載の方法。
- ブプレノルフィンの製造方法であって、
a)式Iで示されるオリパビンを、メチルビニルケトンと反応させ、式IIで示される化合物を形成させること;
g)式VIIIで示されるノルブプレノルフィンにシクロプロピルメチル基を付加し、式IXのブプレノルフィンを形成させること、
を含む方法。 - 脱メチル化段階が、
a)式Vで示される化合物上の窒素を、式VIで示される化合物で示されるニトリルに変換すること;および、
- 保護基Yが、ベンジル、Ot−ブチルおよびシリル基からなる群から選択される、請求項9に記載の方法。
- a)およびb)が、単一のポットの連続反応に組み合わされている、請求項9に記載の方法。
- ブプレノルフィンの製造方法であって、
a)式Iで示されるオリパビンを、メチルビニルケトンと反応させ、式IIで示される化合物を形成させること;
を含む、方法。 - 保護基Yが、ベンジル、O−t−ブチルおよびシリル基からなる群から選択される、請求項13に記載の方法。
- a)およびb)が、単一のポットの連続反応に組み合わされている、請求項13に記載の方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US75638006P | 2006-01-05 | 2006-01-05 | |
| PCT/US2006/048479 WO2007081506A1 (en) | 2006-01-05 | 2006-12-18 | The use of oripavine as a starting material for buprenorphine |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2009522357A true JP2009522357A (ja) | 2009-06-11 |
Family
ID=37914270
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008549488A Pending JP2009522357A (ja) | 2006-01-05 | 2006-12-18 | ブプレノルフィンのための出発物質としてのオリパビンの使用 |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US8993764B2 (ja) |
| EP (1) | EP1981891B1 (ja) |
| JP (1) | JP2009522357A (ja) |
| CN (1) | CN101356177A (ja) |
| AT (1) | ATE441649T1 (ja) |
| AU (1) | AU2006335138B2 (ja) |
| CA (1) | CA2636271A1 (ja) |
| DE (1) | DE602006008991D1 (ja) |
| ES (1) | ES2329296T3 (ja) |
| WO (1) | WO2007081506A1 (ja) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080125592A1 (en) * | 2006-10-17 | 2008-05-29 | Penick Corporation | Process for preparing oxymorphone, naltrexone, and buprenorphine |
| CA2674915C (en) | 2006-10-17 | 2015-06-30 | Penick Corporation | Process for preparing oxymorphone |
| WO2009078986A1 (en) * | 2007-12-17 | 2009-06-25 | Mallinckrodt Inc. | Process for the preparation of buprenorphine and derivatives of buprenorphine |
| WO2009122436A2 (en) * | 2008-03-31 | 2009-10-08 | Sun Pharmaceutical Industries Ltd. | An improved process for the preparation of morphinane analogues |
| US8232398B2 (en) | 2008-09-30 | 2012-07-31 | Mallinckrodt Llc | Recycling process for increasing the yield of opiate alkaloid derivatives |
| AU2009300386B2 (en) * | 2008-09-30 | 2015-07-02 | SpecGx LLC | Processes for the alkylation of norbuprenorphine with reduced impurity formation |
| PL2344507T5 (pl) | 2008-09-30 | 2021-10-11 | SpecGx LLC | Sposoby zwiększania wydajności hydrolizy grupy 3-O-metylowej i 17-N-nitrylowej w wytwarzaniu pochodnych alkaloidów opiatowych |
| WO2010039217A1 (en) * | 2008-09-30 | 2010-04-08 | Mallinckrodt Inc. | Processes for the synthesis of opiate alkaloids with reduced impurity formation |
| CA2738761C (en) * | 2008-09-30 | 2019-10-29 | Mallinckrodt Inc. | Processes for the production of buprenorphine with reduced impurity formation |
| JP5824448B2 (ja) | 2009-04-24 | 2015-11-25 | ブロック ユニバーシティ | モルフィナンおよびモルフィノン化合物の調製法 |
| AU2011263417B2 (en) | 2010-06-11 | 2014-03-27 | Rhodes Technologies | Transition metal-catalyzed processes for the preparation of N-allyl compounds and use thereof |
| CA2802294C (en) * | 2010-06-11 | 2016-05-10 | Rhodes Technologies | Process for n-dealkylation of tertiary amines |
| CN102382118A (zh) * | 2011-05-23 | 2012-03-21 | 浙江仙琚制药股份有限公司 | 一种7ɑ-乙酰基-6,14-乙基桥四氢蒂巴因的制备方法 |
| US9315514B2 (en) | 2012-08-27 | 2016-04-19 | Rhodes Technologies | 1,3-dioxanomorphides and 1,3-dioxanocodides |
| EP3023427A1 (en) * | 2014-11-19 | 2016-05-25 | Siegfried AG | Improved method of manufacturing buprenorphine and analogues thereof from oripavine |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3415830A (en) | 1964-01-27 | 1968-12-10 | Shionogi & Co | Process for preparing intermediates of garrya alkaloids |
| US3285914A (en) * | 1964-06-11 | 1966-11-15 | Smith Kline French Lab | 3-n-substituted derivatives of oripavine and thebaine |
| US3318884A (en) | 1965-07-21 | 1967-05-09 | American Cyanamid Co | Substituted 6-amino-6-demethoxythebaine and 6-amino-6-demethoxyoripavine derivatives |
| US4275205A (en) | 1980-05-05 | 1981-06-23 | Miles Laboratories, Inc. | 7,7-Ditosyloxymethyl-4,5α-epoxy-morphinan-6-ols |
| US4347361A (en) | 1980-12-10 | 1982-08-31 | Sisa, Incorporated | 4,5α-Epoxy-3-hydroxy or methoxy-7-(1-hydroxy-alkyl or 1-oxoalkyl)morphinan-6-one compounds |
| US4795813A (en) | 1981-08-17 | 1989-01-03 | The Florida Board Of Regents On Behalf Of The Florida State University | Synthesis of derivatives of codeine and other 3-O-alkylmorphines |
| US5634946A (en) * | 1988-08-24 | 1997-06-03 | Focal, Inc. | Polymeric endoluminal paving process |
| PT95069B (pt) * | 1989-08-24 | 1997-10-31 | Searle & Co | Processo para a preparacao de (+)-isomeros de derivados de endoetano/endoetanoepoximofinano, uteis como agentes anti-tussicos |
| KR100204659B1 (ko) * | 1996-05-28 | 1999-06-15 | 강재헌 | 신규한 부프레노핀계 진통제용 화합물 |
| US6067749A (en) * | 1996-07-11 | 2000-05-30 | Tasmanian Alkaloids Pty. Ltd. | Papaver somniferum strain with high concentration of thebaine and oripavine |
| AUPQ968300A0 (en) | 2000-08-25 | 2000-09-21 | Glaxo Wellcome Australia Ltd | Chemical methods |
| CN1233645C (zh) | 2001-09-14 | 2005-12-28 | 中国人民解放军军事医学科学院毒物药物研究所 | 新的东罂粟碱衍生物及其医药用途 |
| US6818721B2 (en) | 2002-12-02 | 2004-11-16 | Rpo Pty Ltd. | Process for producing polysiloxanes and use of the same |
-
2006
- 2006-12-18 CN CN200680050478.9A patent/CN101356177A/zh active Pending
- 2006-12-18 AT AT06845842T patent/ATE441649T1/de active
- 2006-12-18 WO PCT/US2006/048479 patent/WO2007081506A1/en active Application Filing
- 2006-12-18 DE DE602006008991T patent/DE602006008991D1/de active Active
- 2006-12-18 AU AU2006335138A patent/AU2006335138B2/en not_active Ceased
- 2006-12-18 ES ES06845842T patent/ES2329296T3/es active Active
- 2006-12-18 EP EP06845842A patent/EP1981891B1/en not_active Not-in-force
- 2006-12-18 US US12/159,025 patent/US8993764B2/en active Active
- 2006-12-18 JP JP2008549488A patent/JP2009522357A/ja active Pending
- 2006-12-18 CA CA002636271A patent/CA2636271A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| AU2006335138B2 (en) | 2012-08-16 |
| ATE441649T1 (de) | 2009-09-15 |
| AU2006335138A1 (en) | 2007-07-19 |
| CA2636271A1 (en) | 2007-07-19 |
| US8993764B2 (en) | 2015-03-31 |
| US20080312441A1 (en) | 2008-12-18 |
| ES2329296T3 (es) | 2009-11-24 |
| EP1981891B1 (en) | 2009-09-02 |
| EP1981891A1 (en) | 2008-10-22 |
| WO2007081506A1 (en) | 2007-07-19 |
| DE602006008991D1 (de) | 2009-10-15 |
| CN101356177A (zh) | 2009-01-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2009522357A (ja) | ブプレノルフィンのための出発物質としてのオリパビンの使用 | |
| US7038053B2 (en) | Process for imidazo[4,5-c]pyridin-4-amines | |
| CN101796051B (zh) | 与取代的杂环稠合的γ-咔啉的合成 | |
| CA2738761A1 (en) | Processes for the production of buprenorphine with reduced impurity formation | |
| JP2003513974A (ja) | イミダゾリジノン系αv−インテグリン拮抗薬の製造方法および製造用中間体 | |
| JP3173602B2 (ja) | エンイン誘導体の新規製造中間体及びその製造法 | |
| CA2067200C (en) | Total synthesis of northebaine, normorphine, noroxymorphone enantiomers and derivatives via n-nor intermediates | |
| EP2342206B1 (en) | Processes for the hydrogenation of opiate alkaloid derivatives | |
| TW200831478A (en) | Chromane derivatives, synthesis thereof, and intermediates thereto | |
| KR100399669B1 (ko) | 약제학적으로유용한벤조모르판유도체제조시의중간체인노르벤조모르판,특히(-)-(1r,5s,2'r)-3'-하이드록시-2-(2-메톡시프로필)-5,9,9-트리메틸-6,7-벤조모르판을제조하는방법 | |
| US7423152B2 (en) | Process for the manufacture of intermediates in camptothecin production | |
| JP4874122B2 (ja) | トルテロジンを得るための方法 | |
| US6407241B1 (en) | Process and intermediates for the preparation of imidazolidinone αv integrin antagonists | |
| EP1565421B1 (en) | A method for preparing indan-1,3-dicarboxylic acid | |
| JPH07173153A (ja) | 1−[2−アミノ−5−[1−(トリフェニルメチル)−1h−イミダゾール−4−イル]−1−オキソペンチル]ピペリジン誘導体、その製法およびその合成中間体としての使用法 | |
| MX2008008568A (en) | The use of oripavine as a starting material for buprenorphine | |
| US5380849A (en) | Process for optically pure decahydroisoqiunolines | |
| CN112939848B (zh) | 一种布比卡因及其中间体(s)-2-哌啶甲酸的制备方法 | |
| Riechers et al. | Cyclization of 3‐Aminoacrylates–Total Synthesis of Pumiliotoxin C and Related Stereoisomeric Compounds | |
| CN109575075B (zh) | 一种制备喹啉酮生物碱的中间体 | |
| US4282146A (en) | Preparation of 1,2,4,5-tetrahydro-7-alkoxy-(and 7,8-dialkoxy)-3H,3-benzazepines and 3-substituted derivatives thereof from the corresponding phenethylamines | |
| JP2003171354A (ja) | 光学活性アミン化合物又はその塩の製造方法 | |
| JP3144920B2 (ja) | α−アシルアミノケトン誘導体、その製造方法及びその利用 | |
| JPH11286481A (ja) | ピペリジニリデン誘導体の製造方法 |