JP2007520566A - 自己免疫治療のための抗cd3及び抗原特異的免疫療法 - Google Patents
自己免疫治療のための抗cd3及び抗原特異的免疫療法 Download PDFInfo
- Publication number
- JP2007520566A JP2007520566A JP2006552302A JP2006552302A JP2007520566A JP 2007520566 A JP2007520566 A JP 2007520566A JP 2006552302 A JP2006552302 A JP 2006552302A JP 2006552302 A JP2006552302 A JP 2006552302A JP 2007520566 A JP2007520566 A JP 2007520566A
- Authority
- JP
- Japan
- Prior art keywords
- antigen
- antibody
- cells
- autoantigen
- self
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 239000000427 antigen Substances 0.000 title claims abstract description 282
- 108091007433 antigens Proteins 0.000 title claims abstract description 232
- 102000036639 antigens Human genes 0.000 title claims abstract description 232
- 230000001363 autoimmune Effects 0.000 title abstract description 10
- 238000002560 therapeutic procedure Methods 0.000 title description 11
- 238000009169 immunotherapy Methods 0.000 title description 3
- 238000000034 method Methods 0.000 claims abstract description 121
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims abstract description 53
- 230000008569 process Effects 0.000 claims abstract description 11
- 210000003289 regulatory T cell Anatomy 0.000 claims description 94
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 81
- 206010012601 diabetes mellitus Diseases 0.000 claims description 77
- 108090001061 Insulin Proteins 0.000 claims description 50
- 102000004877 Insulin Human genes 0.000 claims description 45
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 42
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 claims description 39
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 claims description 39
- 229940125396 insulin Drugs 0.000 claims description 39
- 108010076181 Proinsulin Proteins 0.000 claims description 38
- 241000282414 Homo sapiens Species 0.000 claims description 35
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 31
- 239000008103 glucose Substances 0.000 claims description 31
- DEFJQIDDEAULHB-IMJSIDKUSA-N L-alanyl-L-alanine Chemical compound C[C@H](N)C(=O)N[C@@H](C)C(O)=O DEFJQIDDEAULHB-IMJSIDKUSA-N 0.000 claims description 29
- 108010056243 alanylalanine Proteins 0.000 claims description 29
- 108090000623 proteins and genes Proteins 0.000 claims description 23
- 108091022930 Glutamate decarboxylase Proteins 0.000 claims description 22
- 102000004169 proteins and genes Human genes 0.000 claims description 22
- 230000028993 immune response Effects 0.000 claims description 21
- 238000000338 in vitro Methods 0.000 claims description 21
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 claims description 20
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 claims description 20
- 108010002350 Interleukin-2 Proteins 0.000 claims description 20
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 claims description 20
- 108010075254 C-Peptide Proteins 0.000 claims description 19
- 108010044226 Class 8 Receptor-Like Protein Tyrosine Phosphatases Proteins 0.000 claims description 19
- 102000009109 Fc receptors Human genes 0.000 claims description 16
- 108010087819 Fc receptors Proteins 0.000 claims description 16
- 210000004369 blood Anatomy 0.000 claims description 16
- 239000008280 blood Substances 0.000 claims description 16
- 238000004519 manufacturing process Methods 0.000 claims description 16
- 102000008214 Glutamate decarboxylase Human genes 0.000 claims description 15
- 210000000612 antigen-presenting cell Anatomy 0.000 claims description 15
- 210000004153 islets of langerhan Anatomy 0.000 claims description 15
- 101001018097 Homo sapiens L-selectin Proteins 0.000 claims description 14
- 102100033467 L-selectin Human genes 0.000 claims description 14
- 208000024891 symptom Diseases 0.000 claims description 13
- 230000006378 damage Effects 0.000 claims description 12
- 102100038222 60 kDa heat shock protein, mitochondrial Human genes 0.000 claims description 11
- 108010058432 Chaperonin 60 Proteins 0.000 claims description 11
- 102100034091 Receptor-type tyrosine-protein phosphatase-like N Human genes 0.000 claims description 11
- 101710149863 C-C chemokine receptor type 4 Proteins 0.000 claims description 10
- 102100032976 CCR4-NOT transcription complex subunit 6 Human genes 0.000 claims description 10
- 230000007423 decrease Effects 0.000 claims description 10
- 230000000263 nonmitogenic effect Effects 0.000 claims description 10
- -1 CD45TO Proteins 0.000 claims description 9
- 101000801234 Homo sapiens Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 claims description 9
- 102100033728 Tumor necrosis factor receptor superfamily member 18 Human genes 0.000 claims description 9
- 239000013604 expression vector Substances 0.000 claims description 8
- 230000001939 inductive effect Effects 0.000 claims description 8
- 108010021625 Immunoglobulin Fragments Proteins 0.000 claims description 6
- 102000008394 Immunoglobulin Fragments Human genes 0.000 claims description 6
- 230000002401 inhibitory effect Effects 0.000 claims description 6
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 claims description 4
- 102000007079 Peptide Fragments Human genes 0.000 claims description 4
- 108010033276 Peptide Fragments Proteins 0.000 claims description 4
- 108010086646 insulin B (9-23) Proteins 0.000 claims description 4
- 102000002812 Heat-Shock Proteins Human genes 0.000 claims description 3
- 108010004889 Heat-Shock Proteins Proteins 0.000 claims description 3
- 208000013016 Hypoglycemia Diseases 0.000 claims description 3
- 230000002159 abnormal effect Effects 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 230000012010 growth Effects 0.000 claims description 3
- 230000002218 hypoglycaemic effect Effects 0.000 claims description 3
- KISWVXRQTGLFGD-UHFFFAOYSA-N 2-[[2-[[6-amino-2-[[2-[[2-[[5-amino-2-[[2-[[1-[2-[[6-amino-2-[(2,5-diamino-5-oxopentanoyl)amino]hexanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-(diaminomethylideneamino)p Chemical compound C1CCN(C(=O)C(CCCN=C(N)N)NC(=O)C(CCCCN)NC(=O)C(N)CCC(N)=O)C1C(=O)NC(CO)C(=O)NC(CCC(N)=O)C(=O)NC(CCCN=C(N)N)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=C(O)C=C1 KISWVXRQTGLFGD-UHFFFAOYSA-N 0.000 claims description 2
- 208000023328 Basedow disease Diseases 0.000 claims description 2
- 208000019707 Cryoglobulinemic vasculitis Diseases 0.000 claims description 2
- 208000015023 Graves' disease Diseases 0.000 claims description 2
- 208000030836 Hashimoto thyroiditis Diseases 0.000 claims description 2
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 claims description 2
- 101000622137 Homo sapiens P-selectin Proteins 0.000 claims description 2
- 108010036012 Iodide peroxidase Proteins 0.000 claims description 2
- 102000047918 Myelin Basic Human genes 0.000 claims description 2
- 101710107068 Myelin basic protein Proteins 0.000 claims description 2
- 102100023472 P-selectin Human genes 0.000 claims description 2
- 230000024932 T cell mediated immunity Effects 0.000 claims description 2
- 102000009843 Thyroglobulin Human genes 0.000 claims description 2
- 108010034949 Thyroglobulin Proteins 0.000 claims description 2
- 102100027188 Thyroid peroxidase Human genes 0.000 claims description 2
- 108090000253 Thyrotropin Receptors Proteins 0.000 claims description 2
- 102100029337 Thyrotropin receptor Human genes 0.000 claims description 2
- 201000003278 cryoglobulinemia Diseases 0.000 claims description 2
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- 230000028996 humoral immune response Effects 0.000 claims description 2
- 235000003642 hunger Nutrition 0.000 claims description 2
- 201000006417 multiple sclerosis Diseases 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 230000037351 starvation Effects 0.000 claims description 2
- 201000000596 systemic lupus erythematosus Diseases 0.000 claims description 2
- 229960002175 thyroglobulin Drugs 0.000 claims description 2
- 230000004044 response Effects 0.000 abstract description 78
- 238000011260 co-administration Methods 0.000 abstract description 30
- 230000005784 autoimmunity Effects 0.000 abstract description 26
- 230000002195 synergetic effect Effects 0.000 abstract description 18
- 230000001717 pathogenic effect Effects 0.000 abstract description 13
- 238000011084 recovery Methods 0.000 abstract description 13
- 238000002648 combination therapy Methods 0.000 abstract description 7
- 230000007123 defense Effects 0.000 abstract 1
- 230000004936 stimulating effect Effects 0.000 abstract 1
- 210000004027 cell Anatomy 0.000 description 127
- 238000011282 treatment Methods 0.000 description 113
- 241000699670 Mus sp. Species 0.000 description 84
- 238000002474 experimental method Methods 0.000 description 53
- 210000001744 T-lymphocyte Anatomy 0.000 description 46
- 102000003814 Interleukin-10 Human genes 0.000 description 42
- 108090000174 Interleukin-10 Proteins 0.000 description 42
- 230000000694 effects Effects 0.000 description 41
- 108090000695 Cytokines Proteins 0.000 description 26
- 102000004127 Cytokines Human genes 0.000 description 25
- 208000023275 Autoimmune disease Diseases 0.000 description 23
- 210000004698 lymphocyte Anatomy 0.000 description 22
- 230000001105 regulatory effect Effects 0.000 description 22
- 238000012360 testing method Methods 0.000 description 22
- 241000712899 Lymphocytic choriomeningitis mammarenavirus Species 0.000 description 20
- 102000000588 Interleukin-2 Human genes 0.000 description 19
- 235000018102 proteins Nutrition 0.000 description 19
- VOUAQYXWVJDEQY-QENPJCQMSA-N 33017-11-7 Chemical compound OC(=O)CC[C@H](N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)NCC(=O)NCC(=O)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N1[C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)CCC1 VOUAQYXWVJDEQY-QENPJCQMSA-N 0.000 description 18
- 239000012634 fragment Substances 0.000 description 18
- 238000013459 approach Methods 0.000 description 17
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 17
- 201000010099 disease Diseases 0.000 description 16
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 16
- 230000006870 function Effects 0.000 description 16
- 108010047620 Phytohemagglutinins Proteins 0.000 description 15
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 15
- 230000001885 phytohemagglutinin Effects 0.000 description 15
- 108010074328 Interferon-gamma Proteins 0.000 description 14
- 241000699666 Mus <mouse, genus> Species 0.000 description 14
- 238000004458 analytical method Methods 0.000 description 14
- 206010018429 Glucose tolerance impaired Diseases 0.000 description 13
- 102100037850 Interferon gamma Human genes 0.000 description 13
- 239000003814 drug Substances 0.000 description 13
- 230000006698 induction Effects 0.000 description 13
- 230000009885 systemic effect Effects 0.000 description 13
- 229940079593 drug Drugs 0.000 description 12
- 230000001225 therapeutic effect Effects 0.000 description 12
- 230000036039 immunity Effects 0.000 description 11
- 238000012546 transfer Methods 0.000 description 11
- 102000006449 Class 8 Receptor-Like Protein Tyrosine Phosphatases Human genes 0.000 description 10
- 108091008874 T cell receptors Proteins 0.000 description 10
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 10
- 210000000987 immune system Anatomy 0.000 description 10
- 238000001727 in vivo Methods 0.000 description 10
- 208000015181 infectious disease Diseases 0.000 description 10
- 238000010186 staining Methods 0.000 description 10
- 102000004388 Interleukin-4 Human genes 0.000 description 9
- 108090000978 Interleukin-4 Proteins 0.000 description 9
- 230000036783 anaphylactic response Effects 0.000 description 9
- 208000003455 anaphylaxis Diseases 0.000 description 9
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 description 9
- 230000007774 longterm Effects 0.000 description 9
- 238000010172 mouse model Methods 0.000 description 9
- 230000000638 stimulation Effects 0.000 description 9
- 206010002198 Anaphylactic reaction Diseases 0.000 description 8
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 8
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 8
- 241000700605 Viruses Species 0.000 description 8
- 230000004913 activation Effects 0.000 description 8
- 230000006472 autoimmune response Effects 0.000 description 8
- 239000012636 effector Substances 0.000 description 8
- 201000001421 hyperglycemia Diseases 0.000 description 8
- 230000001900 immune effect Effects 0.000 description 8
- 230000001506 immunosuppresive effect Effects 0.000 description 8
- 230000007246 mechanism Effects 0.000 description 8
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 8
- 102100035902 Glutamate decarboxylase 1 Human genes 0.000 description 7
- 238000010171 animal model Methods 0.000 description 7
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 7
- 230000000981 bystander Effects 0.000 description 7
- 230000000977 initiatory effect Effects 0.000 description 7
- 102000004196 processed proteins & peptides Human genes 0.000 description 7
- 230000002829 reductive effect Effects 0.000 description 7
- 241000282412 Homo Species 0.000 description 6
- 108060003951 Immunoglobulin Proteins 0.000 description 6
- 102000007999 Nuclear Proteins Human genes 0.000 description 6
- 108010089610 Nuclear Proteins Proteins 0.000 description 6
- 235000001014 amino acid Nutrition 0.000 description 6
- 150000001413 amino acids Chemical group 0.000 description 6
- 238000000684 flow cytometry Methods 0.000 description 6
- 230000002641 glycemic effect Effects 0.000 description 6
- 102000018358 immunoglobulin Human genes 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 230000003914 insulin secretion Effects 0.000 description 6
- 230000014759 maintenance of location Effects 0.000 description 6
- 206010062016 Immunosuppression Diseases 0.000 description 5
- 230000009471 action Effects 0.000 description 5
- 239000011324 bead Substances 0.000 description 5
- 239000003153 chemical reaction reagent Substances 0.000 description 5
- 230000016396 cytokine production Effects 0.000 description 5
- 230000037213 diet Effects 0.000 description 5
- 235000005911 diet Nutrition 0.000 description 5
- 102000054766 genetic haplotypes Human genes 0.000 description 5
- 230000003053 immunization Effects 0.000 description 5
- 238000002649 immunization Methods 0.000 description 5
- 238000010253 intravenous injection Methods 0.000 description 5
- 108091005601 modified peptides Proteins 0.000 description 5
- 210000000056 organ Anatomy 0.000 description 5
- 230000002062 proliferating effect Effects 0.000 description 5
- 230000011664 signaling Effects 0.000 description 5
- 238000009097 single-agent therapy Methods 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 238000011830 transgenic mouse model Methods 0.000 description 5
- 230000003612 virological effect Effects 0.000 description 5
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 4
- 108010041986 DNA Vaccines Proteins 0.000 description 4
- 229940021995 DNA vaccine Drugs 0.000 description 4
- 238000011510 Elispot assay Methods 0.000 description 4
- 102000003886 Glycoproteins Human genes 0.000 description 4
- 108090000288 Glycoproteins Proteins 0.000 description 4
- 101100223318 Homo sapiens GAD2 gene Proteins 0.000 description 4
- 108010029973 Lymphocytic choriomeningitis virus glycoprotein peptide Proteins 0.000 description 4
- 102000043131 MHC class II family Human genes 0.000 description 4
- 108091054438 MHC class II family Proteins 0.000 description 4
- 241001529936 Murinae Species 0.000 description 4
- 241000699660 Mus musculus Species 0.000 description 4
- 108700019146 Transgenes Proteins 0.000 description 4
- 238000011284 combination treatment Methods 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 230000001419 dependent effect Effects 0.000 description 4
- 238000001514 detection method Methods 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 238000011194 good manufacturing practice Methods 0.000 description 4
- 230000002519 immonomodulatory effect Effects 0.000 description 4
- 230000001976 improved effect Effects 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- PBGKTOXHQIOBKM-FHFVDXKLSA-N insulin (human) Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3C=CC(O)=CC=3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3NC=NC=3)NC(=O)[C@H](CO)NC(=O)CNC1=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)=O)CSSC[C@@H](C(N2)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)[C@@H](C)CC)[C@@H](C)O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C1=CN=CN1 PBGKTOXHQIOBKM-FHFVDXKLSA-N 0.000 description 4
- 230000006362 insulin response pathway Effects 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 238000002955 isolation Methods 0.000 description 4
- 210000001165 lymph node Anatomy 0.000 description 4
- 238000012986 modification Methods 0.000 description 4
- 230000004048 modification Effects 0.000 description 4
- 210000000496 pancreas Anatomy 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- 230000001681 protective effect Effects 0.000 description 4
- 210000004988 splenocyte Anatomy 0.000 description 4
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 3
- 229940122450 Altered peptide ligand Drugs 0.000 description 3
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- 101000738765 Homo sapiens Receptor-type tyrosine-protein phosphatase N2 Proteins 0.000 description 3
- 102100037404 Receptor-type tyrosine-protein phosphatase N2 Human genes 0.000 description 3
- 241000283984 Rodentia Species 0.000 description 3
- 230000005867 T cell response Effects 0.000 description 3
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 3
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 230000033228 biological regulation Effects 0.000 description 3
- 230000002596 correlated effect Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 238000001647 drug administration Methods 0.000 description 3
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 230000008102 immune modulation Effects 0.000 description 3
- 230000006058 immune tolerance Effects 0.000 description 3
- 230000001965 increasing effect Effects 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 230000036961 partial effect Effects 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 238000010254 subcutaneous injection Methods 0.000 description 3
- 239000007929 subcutaneous injection Substances 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 230000002459 sustained effect Effects 0.000 description 3
- 102000006306 Antigen Receptors Human genes 0.000 description 2
- 108010083359 Antigen Receptors Proteins 0.000 description 2
- 108091008875 B cell receptors Proteins 0.000 description 2
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 2
- 229940045513 CTLA4 antagonist Drugs 0.000 description 2
- 208000002691 Choroiditis Diseases 0.000 description 2
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 2
- 108010036949 Cyclosporine Proteins 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- 208000030453 Drug-Related Side Effects and Adverse reaction Diseases 0.000 description 2
- 102100036255 Glucose-6-phosphatase 2 Human genes 0.000 description 2
- 102000001554 Hemoglobins Human genes 0.000 description 2
- 108010054147 Hemoglobins Proteins 0.000 description 2
- 101000976075 Homo sapiens Insulin Proteins 0.000 description 2
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 2
- 101150108784 INSB gene Proteins 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- 108010005991 Pork Regular Insulin Proteins 0.000 description 2
- 208000003971 Posterior uveitis Diseases 0.000 description 2
- 102000002727 Protein Tyrosine Phosphatase Human genes 0.000 description 2
- ZSJLQEPLLKMAKR-UHFFFAOYSA-N Streptozotocin Natural products O=NN(C)C(=O)NC1C(O)OC(CO)C(O)C1O ZSJLQEPLLKMAKR-UHFFFAOYSA-N 0.000 description 2
- 230000006044 T cell activation Effects 0.000 description 2
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 2
- 206010070863 Toxicity to various agents Diseases 0.000 description 2
- 208000036142 Viral infection Diseases 0.000 description 2
- 239000013566 allergen Substances 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 230000003416 augmentation Effects 0.000 description 2
- 230000003190 augmentative effect Effects 0.000 description 2
- 230000001588 bifunctional effect Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 230000020411 cell activation Effects 0.000 description 2
- 238000001516 cell proliferation assay Methods 0.000 description 2
- 239000006285 cell suspension Substances 0.000 description 2
- 229960001265 ciclosporin Drugs 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- VGGRNGOEDNBLPH-YJHCMWSWSA-N diapep277 Chemical compound CC(C)[C@H](N)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)NCC(=O)NCC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1CCC[C@H]1C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@H](C(=O)N[C@@H](C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(O)=O)CCC1 VGGRNGOEDNBLPH-YJHCMWSWSA-N 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- 238000013401 experimental design Methods 0.000 description 2
- 239000013613 expression plasmid Substances 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 230000003832 immune regulation Effects 0.000 description 2
- 239000003018 immunosuppressive agent Substances 0.000 description 2
- 238000000099 in vitro assay Methods 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 238000013383 initial experiment Methods 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 230000000527 lymphocytic effect Effects 0.000 description 2
- 230000002503 metabolic effect Effects 0.000 description 2
- 239000000203 mixture Substances 0.000 description 2
- 239000000178 monomer Substances 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- 230000008506 pathogenesis Effects 0.000 description 2
- 108020000494 protein-tyrosine phosphatase Proteins 0.000 description 2
- 238000000746 purification Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000009711 regulatory function Effects 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 230000007480 spreading Effects 0.000 description 2
- 238000003892 spreading Methods 0.000 description 2
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 2
- 229960001052 streptozocin Drugs 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 238000009121 systemic therapy Methods 0.000 description 2
- 230000024664 tolerance induction Effects 0.000 description 2
- 230000009261 transgenic effect Effects 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 230000009385 viral infection Effects 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical group COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- IJJWOSAXNHWBPR-HUBLWGQQSA-N 5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]-n-(6-hydrazinyl-6-oxohexyl)pentanamide Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)NCCCCCC(=O)NN)SC[C@@H]21 IJJWOSAXNHWBPR-HUBLWGQQSA-N 0.000 description 1
- BZTDTCNHAFUJOG-UHFFFAOYSA-N 6-carboxyfluorescein Chemical compound C12=CC=C(O)C=C2OC2=CC(O)=CC=C2C11OC(=O)C2=CC=C(C(=O)O)C=C21 BZTDTCNHAFUJOG-UHFFFAOYSA-N 0.000 description 1
- 208000033399 Anaphylactic responses Diseases 0.000 description 1
- 108010032595 Antibody Binding Sites Proteins 0.000 description 1
- 102100032912 CD44 antigen Human genes 0.000 description 1
- 241000557626 Corvus corax Species 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 206010011968 Decreased immune responsiveness Diseases 0.000 description 1
- 102100025137 Early activation antigen CD69 Human genes 0.000 description 1
- 108010008177 Fd immunoglobulins Proteins 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 208000034826 Genetic Predisposition to Disease Diseases 0.000 description 1
- 101710172364 Glucose-6-phosphatase 2 Proteins 0.000 description 1
- 102100028970 HLA class I histocompatibility antigen, alpha chain E Human genes 0.000 description 1
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 description 1
- 108010088652 Histocompatibility Antigens Class I Proteins 0.000 description 1
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 1
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 description 1
- 101100223310 Homo sapiens GAD1 gene Proteins 0.000 description 1
- 101000930907 Homo sapiens Glucose-6-phosphatase 2 Proteins 0.000 description 1
- 101000986085 Homo sapiens HLA class I histocompatibility antigen, alpha chain E Proteins 0.000 description 1
- 101000984186 Homo sapiens Leukocyte immunoglobulin-like receptor subfamily B member 4 Proteins 0.000 description 1
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 description 1
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 1
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 1
- 108010001127 Insulin Receptor Proteins 0.000 description 1
- 102100036721 Insulin receptor Human genes 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010002616 Interleukin-5 Proteins 0.000 description 1
- 102000000743 Interleukin-5 Human genes 0.000 description 1
- 208000007976 Ketosis Diseases 0.000 description 1
- 102100025578 Leukocyte immunoglobulin-like receptor subfamily B member 4 Human genes 0.000 description 1
- 102000043129 MHC class I family Human genes 0.000 description 1
- 108091054437 MHC class I family Proteins 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- KHGNFPUMBJSZSM-UHFFFAOYSA-N Perforine Natural products COC1=C2CCC(O)C(CCC(C)(C)O)(OC)C2=NC2=C1C=CO2 KHGNFPUMBJSZSM-UHFFFAOYSA-N 0.000 description 1
- 208000001280 Prediabetic State Diseases 0.000 description 1
- 102100028516 Receptor-type tyrosine-protein phosphatase U Human genes 0.000 description 1
- 101710191614 Receptor-type tyrosine-protein phosphatase-like N Proteins 0.000 description 1
- 241000219061 Rheum Species 0.000 description 1
- 206010072148 Stiff-Person syndrome Diseases 0.000 description 1
- 208000007271 Substance Withdrawal Syndrome Diseases 0.000 description 1
- 102100028644 Tenascin-R Human genes 0.000 description 1
- 206010043376 Tetanus Diseases 0.000 description 1
- 210000004241 Th2 cell Anatomy 0.000 description 1
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 1
- 208000027418 Wounds and injury Diseases 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 208000030961 allergic reaction Diseases 0.000 description 1
- 229940037003 alum Drugs 0.000 description 1
- 230000030741 antigen processing and presentation Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 235000009697 arginine Nutrition 0.000 description 1
- 150000001484 arginines Chemical class 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 238000009395 breeding Methods 0.000 description 1
- 230000001488 breeding effect Effects 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- 229940046731 calcineurin inhibitors Drugs 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 210000004970 cd4 cell Anatomy 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000010307 cell transformation Effects 0.000 description 1
- 230000008614 cellular interaction Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 210000003040 circulating cell Anatomy 0.000 description 1
- 230000007012 clinical effect Effects 0.000 description 1
- 238000011220 combination immunotherapy Methods 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 230000000875 corresponding effect Effects 0.000 description 1
- 230000000139 costimulatory effect Effects 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 210000004748 cultured cell Anatomy 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 206010052015 cytokine release syndrome Diseases 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 230000000779 depleting effect Effects 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000008029 eradication Effects 0.000 description 1
- 230000005713 exacerbation Effects 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 210000004408 hybridoma Anatomy 0.000 description 1
- 238000010166 immunofluorescence Methods 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 230000008975 immunomodulatory function Effects 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 230000001861 immunosuppressant effect Effects 0.000 description 1
- 229940125721 immunosuppressive agent Drugs 0.000 description 1
- 238000005462 in vivo assay Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 230000015788 innate immune response Effects 0.000 description 1
- 239000004026 insulin derivative Substances 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 238000010212 intracellular staining Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 230000004140 ketosis Effects 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 210000005210 lymphoid organ Anatomy 0.000 description 1
- 238000002826 magnetic-activated cell sorting Methods 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 238000001823 molecular biology technique Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 230000003562 morphometric effect Effects 0.000 description 1
- 238000013425 morphometry Methods 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 231100000255 pathogenic effect Toxicity 0.000 description 1
- 230000007918 pathogenicity Effects 0.000 description 1
- 229940023041 peptide vaccine Drugs 0.000 description 1
- 229930192851 perforin Natural products 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 230000008823 permeabilization Effects 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 238000009521 phase II clinical trial Methods 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 201000009104 prediabetes syndrome Diseases 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 230000037452 priming Effects 0.000 description 1
- 238000007639 printing Methods 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 230000009696 proliferative response Effects 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 238000003753 real-time PCR Methods 0.000 description 1
- 230000003252 repetitive effect Effects 0.000 description 1
- 230000004043 responsiveness Effects 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 238000011268 retreatment Methods 0.000 description 1
- 231100000279 safety data Toxicity 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 210000004989 spleen cell Anatomy 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 229960001967 tacrolimus Drugs 0.000 description 1
- 108010020387 tenascin R Proteins 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 238000002255 vaccination Methods 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 230000002747 voluntary effect Effects 0.000 description 1
- 238000001262 western blot Methods 0.000 description 1
- 230000003820 β-cell dysfunction Effects 0.000 description 1
Images










Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
- C07K16/2809—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily against the T-cell receptor (TcR)-CD3 complex
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39541—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against normal tissues, cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
Abstract
本発明は、自己免疫を治療する方法及び寛容を再確立させる方法を提供する。前記方法は、抗CD3抗体及び自己抗原の同時投与を必要とする。前記同時投与は、自己攻撃性免疫プロセスを防御し又は低下させ、及び/又は自己抗原に対する寛容を再確立させる相乗作用の提供に潜在的な力を有する。本方法を支える重要な論理的根拠は、抗CD3抗体と一緒に自己抗原を投与することによって、これら自己抗原に対する応答が変化し、自己免疫の進行を阻止することができるということである。自己抗原で再攻撃し、非病原性応答を刺激することによって、自己免疫プロセスの封鎖を維持することができる。本明細書に提示した前臨床的証拠は、抗CD3と自己抗原の併用は自己免疫糖尿病の回復に相乗作用を示し、したがって、抗CD3と自己抗原との併用療法は、他の自己免疫異常の回復に相乗的な防御を提供しえることを提唱している。
Description
本出願は米国仮特許出願第60/541,959号(2004年2月4日出願)(前記文献は参照により本明細書に含まれる)の優先権を主張する。
本明細書で開示する発明は、国立衛生研究所(National Institute of Health)のグラント1R21 DK069872-01によりアメリカ合衆国政府の援助を受けて達成された。したがって、合衆国政府は本発明において一定の権利を有する。
本明細書に引用した全ての特許、特許出願及び刊行物は参照によりその全体が本明細書に含まれる。これら刊行物のその全内容の開示は、本明細書における本発明の開示及び特許請求の日現在の当業者に公知の技術分野の状態をより十分に明らかにするために、参照により本明細書に含まれる。
本特許文書の内容の一部は著作権保護の対象である文書を含む。本著作権者は、本特許書類又は特許の内容が米国特許商標局のファイル又は記録として存在するとき、前記の何人によるファクシミリ複製についても異議を唱えるものではないが、前記以外に関しては全ての著作権はこれを留保する。
本発明は自己免疫分野に関する。より具体的には、本発明は自己免疫を治療する方法及び寛容を再確立させる方法に関する。
本明細書で開示する発明は、国立衛生研究所(National Institute of Health)のグラント1R21 DK069872-01によりアメリカ合衆国政府の援助を受けて達成された。したがって、合衆国政府は本発明において一定の権利を有する。
本明細書に引用した全ての特許、特許出願及び刊行物は参照によりその全体が本明細書に含まれる。これら刊行物のその全内容の開示は、本明細書における本発明の開示及び特許請求の日現在の当業者に公知の技術分野の状態をより十分に明らかにするために、参照により本明細書に含まれる。
本特許文書の内容の一部は著作権保護の対象である文書を含む。本著作権者は、本特許書類又は特許の内容が米国特許商標局のファイル又は記録として存在するとき、前記の何人によるファクシミリ複製についても異議を唱えるものではないが、前記以外に関しては全ての著作権はこれを留保する。
本発明は自己免疫分野に関する。より具体的には、本発明は自己免疫を治療する方法及び寛容を再確立させる方法に関する。
健常な個体の免疫系はその者の身体の自己抗原に対して寛容である。寛容は、抗原に対する免疫学的な無応答の状態であり、自己免疫は、自己抗原に対して寛容が存在しないときに生じる。自己免疫疾患に対する治療法は、自己免疫の原因が、標的として多数の自己抗原を含む複雑な病因論が関与する、しばしば多因子性であるという事実のために妨げられてきた。
自己免疫の複雑な性質のために、以前の治療法は、免疫系の全般的な抑制を試みてきた。ほとんどの免疫抑制剤は、T細胞の枯渇又は不活化によってT細胞応答を妨げる。例えば、グルココルチコイド及びカルシヌリン阻害物質(例えばシクロスポリンA及びFK-506)は、T細胞増殖因子の産生を妨げながらサイトカイン遺伝子転写を阻止する。他の薬剤、例えばキャンパス1HはT細胞の長期枯渇を生じる。これらのアプローチは短期間においては効果的であるが、それらの作用は抗原特異的ではなく、薬剤の中止後に持続することはできない。したがって、免疫抑制剤を中止した後に免疫応答が生じない、真の免疫寛容はほとんど達成されていない。
対照的に、CD3分子に対するモノクローナル抗体(mAb)は、1型真性糖尿病のマウスモデルで自己免疫に対して寛容を誘発した。抗CD3mAbによる処置はNODマウスで糖尿病を回復させ、移植した同系(syngeneic)の島に対する免疫応答の再発を予防した。前記は、持続的な免疫抑制を必要とすることなく達成され、T細胞数は枯渇せず、更にT細胞数が定量的に正常である時点で持続した。また別のアプローチは、自己抗原を投与することによって特定の免疫学的非応答を誘発することである。しかしながら、抗CD3又は自己抗原の投与は、寛容誘発の期間又は発症後の有効性に限界がありえる。
自己免疫の複雑な性質のために、以前の治療法は、免疫系の全般的な抑制を試みてきた。ほとんどの免疫抑制剤は、T細胞の枯渇又は不活化によってT細胞応答を妨げる。例えば、グルココルチコイド及びカルシヌリン阻害物質(例えばシクロスポリンA及びFK-506)は、T細胞増殖因子の産生を妨げながらサイトカイン遺伝子転写を阻止する。他の薬剤、例えばキャンパス1HはT細胞の長期枯渇を生じる。これらのアプローチは短期間においては効果的であるが、それらの作用は抗原特異的ではなく、薬剤の中止後に持続することはできない。したがって、免疫抑制剤を中止した後に免疫応答が生じない、真の免疫寛容はほとんど達成されていない。
対照的に、CD3分子に対するモノクローナル抗体(mAb)は、1型真性糖尿病のマウスモデルで自己免疫に対して寛容を誘発した。抗CD3mAbによる処置はNODマウスで糖尿病を回復させ、移植した同系(syngeneic)の島に対する免疫応答の再発を予防した。前記は、持続的な免疫抑制を必要とすることなく達成され、T細胞数は枯渇せず、更にT細胞数が定量的に正常である時点で持続した。また別のアプローチは、自己抗原を投与することによって特定の免疫学的非応答を誘発することである。しかしながら、抗CD3又は自己抗原の投与は、寛容誘発の期間又は発症後の有効性に限界がありえる。
発明の要旨
本発明は、自己免疫を治療するために抗CD3抗体(又はCD3リガンド)及び自己抗原の両方の使用を必要とする方法を提供する。抗CD3抗体及び自己抗原の両方の投与(すなわち本明細書で使用される“同時投与”)は、自己免疫疾患の治療に対して、抗CD3抗体又は自己抗原のどちらか単独の投与を超える相乗作用を提供する潜在的能力を有する。前記相乗作用は、自己免疫応答の標的である自己抗原に対する免疫寛容を高めることによって示すことができる。同時投与によって提供されえる相乗作用は予想外及び/又は不確実であるだけでなく、同時投与自体が自己免疫の治療に役立つ可能性もまた予想に反している。なぜならば、(1)抗CD3治療は、自己抗原によってもたらされる寛容の誘発の一切を覆すか帳消しにする全般的な免疫抑制作用を生じる可能性があるか、又は(2)抗CD3又は自己抗原のどちらかの投与は現存の自己攻撃現象の悪化をもたらす可能性があると考えることが理に適っているからである。理論に拘束されないが、本発明は、抗CD3は免疫系を“リセット”し、それによって、抗原特異的治療によるいくつかの治療的介在のための扉を開いて長期的寛容を維持することができる調節の誘発を提供する。
ある特徴では、本発明は、対象者での自己抗原に対する寛容を回復又は誘発する方法を提供する。前記方法は、対象者に(a)抗CD3抗体及び(b)自己抗原を投与することを含み、ここで前記抗CD3抗体及び自己抗原は、対象者で自己抗原に対する寛容を回復又は誘発するために十分な量で投与される。
本発明は、自己免疫を治療するために抗CD3抗体(又はCD3リガンド)及び自己抗原の両方の使用を必要とする方法を提供する。抗CD3抗体及び自己抗原の両方の投与(すなわち本明細書で使用される“同時投与”)は、自己免疫疾患の治療に対して、抗CD3抗体又は自己抗原のどちらか単独の投与を超える相乗作用を提供する潜在的能力を有する。前記相乗作用は、自己免疫応答の標的である自己抗原に対する免疫寛容を高めることによって示すことができる。同時投与によって提供されえる相乗作用は予想外及び/又は不確実であるだけでなく、同時投与自体が自己免疫の治療に役立つ可能性もまた予想に反している。なぜならば、(1)抗CD3治療は、自己抗原によってもたらされる寛容の誘発の一切を覆すか帳消しにする全般的な免疫抑制作用を生じる可能性があるか、又は(2)抗CD3又は自己抗原のどちらかの投与は現存の自己攻撃現象の悪化をもたらす可能性があると考えることが理に適っているからである。理論に拘束されないが、本発明は、抗CD3は免疫系を“リセット”し、それによって、抗原特異的治療によるいくつかの治療的介在のための扉を開いて長期的寛容を維持することができる調節の誘発を提供する。
ある特徴では、本発明は、対象者での自己抗原に対する寛容を回復又は誘発する方法を提供する。前記方法は、対象者に(a)抗CD3抗体及び(b)自己抗原を投与することを含み、ここで前記抗CD3抗体及び自己抗原は、対象者で自己抗原に対する寛容を回復又は誘発するために十分な量で投与される。
ある特徴では、本発明は、対象者で自己抗原に対する免疫応答を低下、阻害又は予防する方法を提供する。前記方法は、対象者に(a)抗CD3抗体及び(b)自己抗原を投与することを含み、ここで前記抗CD3抗体及び自己抗原は、対象者での自己抗原に対する免疫応答を低下、阻害又は予防するために十分な量で投与される。前記免疫応答は液性でも細胞性免疫応答でもよい。
ある特徴では、本発明は、対象者で抗原特異的T調節細胞を産生(又は発生又は誘発)する方法を提供する。前記方法は、対象者に(a)抗CD3抗体及び(b)自己抗原を投与することを含み、ここで前記抗CD3抗体及び自己抗原は、対象者でT調節細胞を産生するために十分な量で前記対象者に投与される。ある特徴では、前記T調節細胞は、前記自己抗原又は他の自己抗原に特異的なT細胞レセプター(TCR)を含むことができる。
ある特徴では、本発明は、自己抗原に対する寛容を回復又は確立(又は誘発)する方法を提供する。前記方法は、(a)対象者に(i)抗CD3抗体及び(ii)自己抗原を投与する工程;(b)T調節細胞(及び別の特徴では、単離されるT調節細胞は、前記自己抗原に特異的なT細胞レセプター(TCR)を含む)を単離する工程;(c)前記T調節細胞を増殖させる条件下でin vitroでインキュベートする(又は単離されたT調節細胞集団の数を増加させる)工程;及び(d)前記対象者に工程(c)のT調節細胞を投与して前記自己抗原に対する寛容を回復させるか又は樹立する工程を含む。工程(c)は、例えばT調節細胞集団をIL-2とインキュベートすることを含むことができる。工程(c)は更に、T調節細胞を抗CD3抗体及び自己抗原とインキュベートすることを含むことができる。更に前記に加えて、又は前記とは別に、工程(c)は更にT調節細胞を抗原提示細胞(APC)及び自己抗原とインキュベートすることを含むことができる。前記APCは、前記対象者から入手してもよいし、前記方法の対象者と同系の他の対象者から入手してもよい。前記T調節細胞は、例えば対象者由来の血液又はリンパサンプルから単離することができる。
ある特徴では、本発明は、対象者で抗原特異的T調節細胞を産生(又は発生又は誘発)する方法を提供する。前記方法は、対象者に(a)抗CD3抗体及び(b)自己抗原を投与することを含み、ここで前記抗CD3抗体及び自己抗原は、対象者でT調節細胞を産生するために十分な量で前記対象者に投与される。ある特徴では、前記T調節細胞は、前記自己抗原又は他の自己抗原に特異的なT細胞レセプター(TCR)を含むことができる。
ある特徴では、本発明は、自己抗原に対する寛容を回復又は確立(又は誘発)する方法を提供する。前記方法は、(a)対象者に(i)抗CD3抗体及び(ii)自己抗原を投与する工程;(b)T調節細胞(及び別の特徴では、単離されるT調節細胞は、前記自己抗原に特異的なT細胞レセプター(TCR)を含む)を単離する工程;(c)前記T調節細胞を増殖させる条件下でin vitroでインキュベートする(又は単離されたT調節細胞集団の数を増加させる)工程;及び(d)前記対象者に工程(c)のT調節細胞を投与して前記自己抗原に対する寛容を回復させるか又は樹立する工程を含む。工程(c)は、例えばT調節細胞集団をIL-2とインキュベートすることを含むことができる。工程(c)は更に、T調節細胞を抗CD3抗体及び自己抗原とインキュベートすることを含むことができる。更に前記に加えて、又は前記とは別に、工程(c)は更にT調節細胞を抗原提示細胞(APC)及び自己抗原とインキュベートすることを含むことができる。前記APCは、前記対象者から入手してもよいし、前記方法の対象者と同系の他の対象者から入手してもよい。前記T調節細胞は、例えば対象者由来の血液又はリンパサンプルから単離することができる。
T調節細胞の単離及び/又は誘発に関する本発明の特徴では、T調節細胞はそれらの細胞表面上に、例えばCD4及びCD25;CD4、CD25及びCD62L;CD25、CD45RO、CD62L及びGITR;CD25、FoxP3、GITR、CTLA4、CD62L及びCD45RO;CD4、CCR4、CD62L及びCD45RO;又はCD25を発現するCD8 T細胞のサブセットを発現することができる。T調節細胞の単離は、例えばフローサイトメトリー又は磁性ビーズ法(前記はそれらの細胞表面発現又は細胞内で産生されたある種のタンパク質、例えばIL-4、IL-10又はTGF-ベータの発現を基にして集団を分離することができる)によって実施することができる。フローサイトメトリーはまた細胞内タンパク質発現を特定することができ、したがって、前記方法は細胞表面上でのタンパク質の細胞外提示に限定されない。
本発明の方法は一般的には自己免疫疾患及び異常の治療を目的とする。例えば、本方法の対象者は、グレーヴズ病、橋本甲状腺炎、低血糖症、多発性硬化症、本態性混合型クリオグロブリン血症、全身性紅斑性狼瘡、I型糖尿病又は前記のいずれかの組合せに罹患していよう。ある特徴では、本方法の対象者は、自己抗原に対して抗原特異性、又はT細胞レセプター(TCR)若しくはB細胞レセプター(BCR)特異性を有するT細胞又はB細胞を必要とする自己免疫応答に異常を有する。
本発明の方法は一般的には自己免疫疾患及び異常の治療を目的とする。例えば、本方法の対象者は、グレーヴズ病、橋本甲状腺炎、低血糖症、多発性硬化症、本態性混合型クリオグロブリン血症、全身性紅斑性狼瘡、I型糖尿病又は前記のいずれかの組合せに罹患していよう。ある特徴では、本方法の対象者は、自己抗原に対して抗原特異性、又はT細胞レセプター(TCR)若しくはB細胞レセプター(BCR)特異性を有するT細胞又はB細胞を必要とする自己免疫応答に異常を有する。
本発明では、自己抗原の投与はタンパク質又はタンパク質のペプチドフラグメントを含む自己抗原を含むことができる。例えば自己抗原は、甲状腺刺激ホルモンレセプター、サイログロブリン、甲状腺ペルオキシダーゼ、ミエリン塩基性タンパク質、グルタミン酸デカルボキシラーゼ(GAD65)、島細胞抗原-2(IA-2)、インシュリン、プロインシュリン又は熱ショックタンパク質60(HSP60)又はその任意の組合せ(そのフラグメント又は変異体を含む)を含むことができる。
ある特徴では、本発明は、I型糖尿病(T1D)の治療方法を提供する。前記方法は、対象者に(a)抗CD3抗体及び(b)自己抗原を投与することを含み、ここで前記抗CD3抗体及び自己抗原は、I型糖尿病の根幹的原因(前記はランゲルハンス島内のインシュリン産生細胞の免疫学的仲介破壊である)の治療に十分な量で投与される。インシュリン産生細胞の破壊は代謝的要求に見合うインシュリン産生の不足を生じ、グルコールレベルを上昇させ、重篤な場合はケトン症もたらす。別の特徴では、抗CD3抗体及び自己抗原は、I型糖尿病又はI型糖尿病に付随する1つ又は2つ以上の症状の治療に十分な量で投与される。I型糖尿病に付随する症状には、インシュリン産生低下、異常な血糖レベル、インシュリン産生細胞の破壊、及び異常なCペプチドレベルが含まれるが、ただしこれらに限定されない。自己抗原は、例えば島細胞抗原を含むことができる。固有の自己抗原には、インシュリン、プロインシュリン、プロインシュリンII、インシュリンB9-23ペプチド、細胞障害性Tリンパ球エピトープをもたないプロインシュリン、インシュリンC13-A5ペプチド、グルタミン酸デカルボキシラーゼ(GAD65)、島細胞抗原512/IA-2、島細胞抗原p69、及び熱ショックタンパク質60(HSP60)が含まれるが、ただしこれらに限定されない。
ある特徴では、本発明は、I型糖尿病(T1D)の治療方法を提供する。前記方法は、対象者に(a)抗CD3抗体及び(b)自己抗原を投与することを含み、ここで前記抗CD3抗体及び自己抗原は、I型糖尿病の根幹的原因(前記はランゲルハンス島内のインシュリン産生細胞の免疫学的仲介破壊である)の治療に十分な量で投与される。インシュリン産生細胞の破壊は代謝的要求に見合うインシュリン産生の不足を生じ、グルコールレベルを上昇させ、重篤な場合はケトン症もたらす。別の特徴では、抗CD3抗体及び自己抗原は、I型糖尿病又はI型糖尿病に付随する1つ又は2つ以上の症状の治療に十分な量で投与される。I型糖尿病に付随する症状には、インシュリン産生低下、異常な血糖レベル、インシュリン産生細胞の破壊、及び異常なCペプチドレベルが含まれるが、ただしこれらに限定されない。自己抗原は、例えば島細胞抗原を含むことができる。固有の自己抗原には、インシュリン、プロインシュリン、プロインシュリンII、インシュリンB9-23ペプチド、細胞障害性Tリンパ球エピトープをもたないプロインシュリン、インシュリンC13-A5ペプチド、グルタミン酸デカルボキシラーゼ(GAD65)、島細胞抗原512/IA-2、島細胞抗原p69、及び熱ショックタンパク質60(HSP60)が含まれるが、ただしこれらに限定されない。
前記方法では、抗CD3抗体及び自己抗原は先ず初めに同じ日に投与することができる。同時投与という用語によれば、抗CD3及び抗原の投与予定が重なるときにそれらは同時投与される。ある特徴では、同時投与予定は、対象者の免疫系によって抗CD3抗体及び抗原が少なくとも1つの時点で本質的に同時に出会うように設定される。したがって、例えば治療の1日目に抗CD3及び抗原は投与され、1日目後、抗CD3及び抗原は別々の日に投与することができる。本発明は、抗CD3抗体及び自己抗原の両者の投与の結果でありえる、相乗的保護作用(すなわち寛容の再樹立/誘発、自己攻撃応答の低下、又は自己免疫の病理発生作用の一般的低下)を提供する潜在的能力を有するので、同時投与は重要である。換言すれば、同時投与は、抗CD3投与が自己抗原投与に対して影響を有し、逆に自己抗原投与が抗CD3投与に影響するシナリオを提供することができる。抗CD3及び自己抗原はしたがって同じときに投与される必要はない。しかしながら、それらは、免疫応答に対するそれらの作用が相乗化されえるように時間的に十分接近して投与されねばならない。
本方法のある特徴では、抗CD3抗体はモノクローナル抗体である。例えば、前記抗体はIgG分子を含むことができる。前記抗体はヒト化されていてもよいし(すなわちげっ歯類とヒトのアミノ酸配列のキメラ)、又は完全にヒト由来でもよい。ある特徴では、前記抗体は少なくとも二価であるべきである(すなわち、同じ特異性をもつ少なくとも2つの抗原結合部位を有する)。抗CD3抗体は抗体の部分配列又はフラグメントを含むことができる。前記抗体フラグメントは、例えば(Fab')2分子を含むことができる。ある特徴では、前記抗体フラグメントはFcレセプターが特異的に結合することができない(すなわち、前記免疫グロブリンのFc-レセプター結合部位は変異しているか、欠失している)。ある特徴では、抗CD3抗体は有糸分裂非誘発性抗体を含むことができる。ある特徴では、抗CD3抗体はOKT3抗体を含むことができる。OKT3抗体は、本来のOKT3抗体の変種又は変異体、例えばヒト(又はヒト化)OKT3γ(Ala-Ala)抗体であってもよい。
本方法のある特徴では、抗CD3抗体はモノクローナル抗体である。例えば、前記抗体はIgG分子を含むことができる。前記抗体はヒト化されていてもよいし(すなわちげっ歯類とヒトのアミノ酸配列のキメラ)、又は完全にヒト由来でもよい。ある特徴では、前記抗体は少なくとも二価であるべきである(すなわち、同じ特異性をもつ少なくとも2つの抗原結合部位を有する)。抗CD3抗体は抗体の部分配列又はフラグメントを含むことができる。前記抗体フラグメントは、例えば(Fab')2分子を含むことができる。ある特徴では、前記抗体フラグメントはFcレセプターが特異的に結合することができない(すなわち、前記免疫グロブリンのFc-レセプター結合部位は変異しているか、欠失している)。ある特徴では、抗CD3抗体は有糸分裂非誘発性抗体を含むことができる。ある特徴では、抗CD3抗体はOKT3抗体を含むことができる。OKT3抗体は、本来のOKT3抗体の変種又は変異体、例えばヒト(又はヒト化)OKT3γ(Ala-Ala)抗体であってもよい。
本発明の方法では、自己抗原の投与は、自己抗原をコードする発現ベクターを、前記発現ベクターが前記自己抗原をin vitroで生成することができるように投与することを含むことができる。
本方法では、抗CD3抗体は、静脈内に投与されるべきである。更にまた、前記自己抗原は経鼻的、経口的、皮下、筋肉内又は静脈内に投与することができる。前記抗CD3抗体及び自己抗原は、医薬的に許容できる担体賦形剤若しくは稀釈剤中で及び/又は前記とともに投与することができる。
したがって、本発明は、本明細書に記載した自己免疫疾患又は自己免疫異常の治療を目的とする抗CD3抗体及び自己抗原の使用を含む。本発明はまた、本明細書に記載した自己免疫疾患又は異常の治療を目的とする医薬の製造における抗CD3抗体及び自己抗原の使用を含む。
本発明はまた、本発明の方法に関連するキットを提供する。例えば、ある特徴では、キットは(a)抗CD3抗体;(b)自己抗原;及び(c)抗CD3抗体及び自己抗原の同時投与のための指示(前記指示は投与スケジュール並びに抗CD3抗体及び自己抗原の投与量を含む)を含むことができる。別の特徴では、キットは(a)抗CD3抗体;(b)島細胞関連抗原;及び(c)抗CD3抗体及び島細胞関連抗原の同時投与のための指示(前記指示は投与スケジュール並びに抗CD3抗体及び島関連抗原の投与量を含む)を含むことができる。
本方法では、抗CD3抗体は、静脈内に投与されるべきである。更にまた、前記自己抗原は経鼻的、経口的、皮下、筋肉内又は静脈内に投与することができる。前記抗CD3抗体及び自己抗原は、医薬的に許容できる担体賦形剤若しくは稀釈剤中で及び/又は前記とともに投与することができる。
したがって、本発明は、本明細書に記載した自己免疫疾患又は自己免疫異常の治療を目的とする抗CD3抗体及び自己抗原の使用を含む。本発明はまた、本明細書に記載した自己免疫疾患又は異常の治療を目的とする医薬の製造における抗CD3抗体及び自己抗原の使用を含む。
本発明はまた、本発明の方法に関連するキットを提供する。例えば、ある特徴では、キットは(a)抗CD3抗体;(b)自己抗原;及び(c)抗CD3抗体及び自己抗原の同時投与のための指示(前記指示は投与スケジュール並びに抗CD3抗体及び自己抗原の投与量を含む)を含むことができる。別の特徴では、キットは(a)抗CD3抗体;(b)島細胞関連抗原;及び(c)抗CD3抗体及び島細胞関連抗原の同時投与のための指示(前記指示は投与スケジュール並びに抗CD3抗体及び島関連抗原の投与量を含む)を含むことができる。
発明の詳細な説明
自己免疫疾患で、特定抗原又は抗CD3抗体の投与によって免疫調節を誘発するこれまでの試みは有効性を示したが、これら作用の持続期間又は疾患表出後のそれらの効き目はこれらのアプローチの臨床応用の制限となった。本発明は、抗原及び抗CD3mAbが同時投与される、自己免疫疾患の新規な治療方法を提供することができる。これらの物質を一緒に投与することによって相乗的な防御効果が提供され、この場合、前記同時投与は自己抗原に対する応答を少なくとも変化させ、自己抗原に対して非病原性応答を誘発し、局所免疫調節を誘発することができる。
自己免疫疾患で、特定抗原又は抗CD3抗体の投与によって免疫調節を誘発するこれまでの試みは有効性を示したが、これら作用の持続期間又は疾患表出後のそれらの効き目はこれらのアプローチの臨床応用の制限となった。本発明は、抗原及び抗CD3mAbが同時投与される、自己免疫疾患の新規な治療方法を提供することができる。これらの物質を一緒に投与することによって相乗的な防御効果が提供され、この場合、前記同時投与は自己抗原に対する応答を少なくとも変化させ、自己抗原に対して非病原性応答を誘発し、局所免疫調節を誘発することができる。
用語
本明細書で用いられる“同時投与”は、抗CD3及び抗原の投与スケジュールが重複するように前記抗CD3及び抗原を投与することを指す。前記抗CD3抗体及び抗原は同じときに投与される必要はない。例えば非限定的な実施態様では、少なくとも1つの時点で本質的に同時にそれらが個体の免疫系によって遭遇させられる投与スケジュールで、それらを投与することができる。したがって、例えば処置の1日目に抗CD3及び抗原の両方を投与することができ、1日目以降に抗CD3及び抗原を別々の日に投与することができる。
本明細書で用いられる“抗体”という用語は、別に指定されなければ、抗体分子及び種々の抗体由来分子を指すために広く用いられる。そのような抗体由来分子は少なくとも1つの可変領域(重鎖又は軽鎖の可変領域のどちらか)を含み、前記には、例えばFabフラグメント、Fab'フラグメント、F(ab')2フラグメント、Fdフラグメント、Fabcフラグメント、Fdフラグメント、Fabcフラグメント、Sc抗体(単鎖抗体)、ジアボディ(diabody)、個々の抗体の軽鎖、個々の抗体の重鎖、抗体鎖と他の分子とのキメラ融合物などが含まれるが、ただしこれらに限定されない。
基本的な抗体の構造ユニットはテトラマーを含むことが知られている。各テトラマーは2つの同一のポリペプチド鎖ペアで構成され、各ペアは1つの“軽鎖”(約25kDa)及び1つの“重鎖”(約50−70kDa)を有する。各鎖のアミノ末端部分は、約100から110またはそれより多いアミノ酸の可変領域(主として抗原認識をもたらす)を含む。各鎖のカルボキシ末端部分は定常領域を規定し、前記は主としてエフェクター機能をもたらす。ヒトの軽鎖はカッパ及びラムダ軽鎖に分類される。重鎖は、ミュー、デルタ、ガンマ、アルファ又はエプシロンに分類され、それぞれ抗体のアイソタイプをIgM、IgD、IgG、IgA及びIgEと規定する。軽鎖及び重鎖内で、可変領域及び定常領域は、約12以上のアミノ酸の“J”領域によって結合され、重鎖はまた約10以上のアミノ酸の“D”領域を含む(全般的には以下を参照されたい:Fundamental Immunology Ch.7(W. Paul, Ed., 2nd ed. Raven Press, NY 1989)、前記文献は参照により本明細書に含まれる)。各軽鎖/重鎖対の可変領域は抗体結合部位を形成する。したがって、完全なIgG抗体は2つの結合部位を有する。二官能性又は二特異性抗体を除いて、2つの結合部位は同じものである。
本明細書で用いられる“同時投与”は、抗CD3及び抗原の投与スケジュールが重複するように前記抗CD3及び抗原を投与することを指す。前記抗CD3抗体及び抗原は同じときに投与される必要はない。例えば非限定的な実施態様では、少なくとも1つの時点で本質的に同時にそれらが個体の免疫系によって遭遇させられる投与スケジュールで、それらを投与することができる。したがって、例えば処置の1日目に抗CD3及び抗原の両方を投与することができ、1日目以降に抗CD3及び抗原を別々の日に投与することができる。
本明細書で用いられる“抗体”という用語は、別に指定されなければ、抗体分子及び種々の抗体由来分子を指すために広く用いられる。そのような抗体由来分子は少なくとも1つの可変領域(重鎖又は軽鎖の可変領域のどちらか)を含み、前記には、例えばFabフラグメント、Fab'フラグメント、F(ab')2フラグメント、Fdフラグメント、Fabcフラグメント、Fdフラグメント、Fabcフラグメント、Sc抗体(単鎖抗体)、ジアボディ(diabody)、個々の抗体の軽鎖、個々の抗体の重鎖、抗体鎖と他の分子とのキメラ融合物などが含まれるが、ただしこれらに限定されない。
基本的な抗体の構造ユニットはテトラマーを含むことが知られている。各テトラマーは2つの同一のポリペプチド鎖ペアで構成され、各ペアは1つの“軽鎖”(約25kDa)及び1つの“重鎖”(約50−70kDa)を有する。各鎖のアミノ末端部分は、約100から110またはそれより多いアミノ酸の可変領域(主として抗原認識をもたらす)を含む。各鎖のカルボキシ末端部分は定常領域を規定し、前記は主としてエフェクター機能をもたらす。ヒトの軽鎖はカッパ及びラムダ軽鎖に分類される。重鎖は、ミュー、デルタ、ガンマ、アルファ又はエプシロンに分類され、それぞれ抗体のアイソタイプをIgM、IgD、IgG、IgA及びIgEと規定する。軽鎖及び重鎖内で、可変領域及び定常領域は、約12以上のアミノ酸の“J”領域によって結合され、重鎖はまた約10以上のアミノ酸の“D”領域を含む(全般的には以下を参照されたい:Fundamental Immunology Ch.7(W. Paul, Ed., 2nd ed. Raven Press, NY 1989)、前記文献は参照により本明細書に含まれる)。各軽鎖/重鎖対の可変領域は抗体結合部位を形成する。したがって、完全なIgG抗体は2つの結合部位を有する。二官能性又は二特異性抗体を除いて、2つの結合部位は同じものである。
鎖は全て、3つの超可変領域(相補性決定領域又はCDRとも称される)が結合した比較的保存されたフレームワーク領域(FR)をもつ同じ一般構造を示す。各対の2つの鎖のCDRは前記フレームワーク領域によって並べられ特異的エピトープと結合することができる。軽鎖及び重鎖の両者は、N-末端からC-末端までの間にドメインFR1、CDR1、FR2、CDR2、FR3、CDR3及びFR4を含む。各ドメインへのアミノ酸の割り当ては、以下の文献の定義に一致する:Kabat Sequences of Proteins of Immunological Interest(National Institutes of Health, Bethesda, Md.,1987 and 1991);Chothia et al. J. Mol. Biol. 1987, 196:901-917;Chothia et al. Nature 1989, 342:878-883。
免疫グロブリン分子に関して本明細書で用いられる“可変領域”という用語は、免疫学分野の当業者が前記用語に付与する一般的な意味を有する。抗体重鎖及び抗体軽鎖の両者は“可変領域”と“定常領域”に分割することができる。可変領域と重鎖領域との間の分割点は、抗体構造について記載した以下の標準的な成書(例えばKabat et al. Sequences of Proteins of Immunological Interest:5th Ed. “U.S. Department of Health and Human Services, U.S. Government Printing Office (1991))を参考にして当業者は容易に決定することができる。
本明細書で用いられる“ヒト化”抗体という用語は、そのCDR(相補性決定領域)が非ヒトの免疫グロブリンに由来し、抗体分子の残りの部分が主としてヒトの免疫グロブリンに由来する分子を指す。
免疫グロブリン分子に関して本明細書で用いられる“可変領域”という用語は、免疫学分野の当業者が前記用語に付与する一般的な意味を有する。抗体重鎖及び抗体軽鎖の両者は“可変領域”と“定常領域”に分割することができる。可変領域と重鎖領域との間の分割点は、抗体構造について記載した以下の標準的な成書(例えばKabat et al. Sequences of Proteins of Immunological Interest:5th Ed. “U.S. Department of Health and Human Services, U.S. Government Printing Office (1991))を参考にして当業者は容易に決定することができる。
本明細書で用いられる“ヒト化”抗体という用語は、そのCDR(相補性決定領域)が非ヒトの免疫グロブリンに由来し、抗体分子の残りの部分が主としてヒトの免疫グロブリンに由来する分子を指す。
“二特異性”又は二官能性抗体は、2つの別個の重/軽鎖対及び2つの別個の結合部位をもつ人工的なハイブリッド抗体である。二特異性抗体は、ハイブリドーマの融合又はFab'フラグメントの連結を含む多様な方法によって作製することができる(例えば以下を参照されたい:Songsivilai et al. Clin. Exp. Immunol. 1990, 79:315-321;Kostelny et al. J. Immunol. 1992, 148:1547-1553)。更にまた、二特異性抗体は、“ジアボディ”(diabody)として(Holliger et al. PNAS USA 1993, 90:6444-6448)、又は“ヤヌシン”として(Janusin)(Traunecker et al. EMBO J 1991, 10:3655-3659;Traunecker et al. Int. J. Cancer Suppl. 1992, 7:51-52)形成することもできる。二特異性抗体は、単一結合部位をもつフラグメント(例えばFab、Fab'及びFv)の形態としては存在しない。本方法では、抗CD3抗体は、少なくとも“二価”であるべきであり、換言すれば前記抗体は、同じ結合特異性を有する少なくとも2つの抗原結合部位を有するべきである。
本明細書で用いられる“島細胞抗原”は、膵のランゲルハンス島(4つの主要細胞タイプ、アルファ、ベータ、デルタ及びガンマ細胞に分けることができる)に由来することができる抗原を指す。島細胞抗原の具体的な例には、島細胞抗原(ICA)512/IA2、島細胞抗原p69(ICA69)、グルタミン酸デカルボキシラーゼ(GAD65)、島特異的グルコース-6-ホスファターゼ触媒性サブユニット関連タンパク質(IGRP)、インシュリン、プロインシュリン及びその誘導体が含まれるが、ただしこれらに限定されない。ICA512/IA2のまた別の名称は、PTPRN2、IA-2、ICA512、R-PTP-N、IA-2/PTP、PTPIA2、島細胞抗原2、島細胞抗原512、島細胞自己抗原3、タンパク質チロシンホスファターゼ様N、タンパク質チロシンホスファターゼレセプターpi、フォグリン、チロシンホスファターゼIA-2ベータ、IAR/レセプター様タンパク質チロシンホスファターゼである。
本明細書で用いられる“島細胞抗原”は、膵のランゲルハンス島(4つの主要細胞タイプ、アルファ、ベータ、デルタ及びガンマ細胞に分けることができる)に由来することができる抗原を指す。島細胞抗原の具体的な例には、島細胞抗原(ICA)512/IA2、島細胞抗原p69(ICA69)、グルタミン酸デカルボキシラーゼ(GAD65)、島特異的グルコース-6-ホスファターゼ触媒性サブユニット関連タンパク質(IGRP)、インシュリン、プロインシュリン及びその誘導体が含まれるが、ただしこれらに限定されない。ICA512/IA2のまた別の名称は、PTPRN2、IA-2、ICA512、R-PTP-N、IA-2/PTP、PTPIA2、島細胞抗原2、島細胞抗原512、島細胞自己抗原3、タンパク質チロシンホスファターゼ様N、タンパク質チロシンホスファターゼレセプターpi、フォグリン、チロシンホスファターゼIA-2ベータ、IAR/レセプター様タンパク質チロシンホスファターゼである。
本発明では、“有糸分裂非誘発性”という用語は、T細胞の分裂を引き起こさない抗CD3抗体のある種の非限定的タイプを指すために用いられる。
本発明では、“自己抗原”という用語は、免疫応答の標的である自己抗原を指す。自己抗原又は自己抗原に対するそのような免疫応答は自己攻撃性応答と呼ばれ(又は自己免疫応答と称され)、病原性応答が含まれる。
本発明では、T細胞レセプターは1つの抗原に特異的であるか、又はMHC(主要組織適合性)分子によって提示される抗原とT細胞レセプターとが特異的に結合するという関係で抗原と特異性を有する。
本発明は、分子生物学分野の当業者が熟知しているポリヌクレオチドを操作する技術を含む一般的な分子生物学的な方法が理解されていることを前提とする。そのような周知の技術の例は以下の文献で見出すことができる:Molecular Cloning: A Laboratory Manual 2nd Edition, Sambrook et al. Cold Spring Harbor, N.Y. (1989)。一般的な分子生物学的技術の例には、in vitro連結、制限エンドヌクレアーゼ消化、PCR、細胞の形質転換、ハイブリダイゼーション、エレクトロポレーション、DNA配列決定などが含まれるが、ただしこれらに限定されない。
本発明はまた、免疫学分野の当業者が熟知している一般的な免疫学的な方法が理解されていることを前提とする。基礎的知識及び方法は以下の文献で見出すことができる:Current Protocols in Immunology, editors(Bierer et al. 4 volumes, John Wiley & Sons, Inc.)。前記文献は以下に関する教示を含む:実験動物の飼育と取り扱い、免疫応答の誘発、リンパ球機能のin vitroアッセイ、リンパ球機能のin vivoアッセイ、免疫蛍光及び細胞分類、サイトカインとその細胞レセプター、ヒトの免疫学的研究、タンパク質、ペプチドの単離と分析、細胞活性化の分子生物学、生化学、補体、先天性免疫、自己免疫及び炎症性疾患の動物モデル(前記はNODマウスモデル、SLEマウスモデル(狼瘡に関する)及び調節T細胞の枯渇による自己免疫疾患の誘発に関する章を含む)、抗原のプロセッシング及び提示、免疫分子及びレセプターの操作、免疫系のリガンド-レセプター相互作用、顕微鏡観察法、並びに、一般的な免疫系遺伝子及びタンパク質(白血球表面分子についてのCD系を含む)の略語と用語。
本発明では、“自己抗原”という用語は、免疫応答の標的である自己抗原を指す。自己抗原又は自己抗原に対するそのような免疫応答は自己攻撃性応答と呼ばれ(又は自己免疫応答と称され)、病原性応答が含まれる。
本発明では、T細胞レセプターは1つの抗原に特異的であるか、又はMHC(主要組織適合性)分子によって提示される抗原とT細胞レセプターとが特異的に結合するという関係で抗原と特異性を有する。
本発明は、分子生物学分野の当業者が熟知しているポリヌクレオチドを操作する技術を含む一般的な分子生物学的な方法が理解されていることを前提とする。そのような周知の技術の例は以下の文献で見出すことができる:Molecular Cloning: A Laboratory Manual 2nd Edition, Sambrook et al. Cold Spring Harbor, N.Y. (1989)。一般的な分子生物学的技術の例には、in vitro連結、制限エンドヌクレアーゼ消化、PCR、細胞の形質転換、ハイブリダイゼーション、エレクトロポレーション、DNA配列決定などが含まれるが、ただしこれらに限定されない。
本発明はまた、免疫学分野の当業者が熟知している一般的な免疫学的な方法が理解されていることを前提とする。基礎的知識及び方法は以下の文献で見出すことができる:Current Protocols in Immunology, editors(Bierer et al. 4 volumes, John Wiley & Sons, Inc.)。前記文献は以下に関する教示を含む:実験動物の飼育と取り扱い、免疫応答の誘発、リンパ球機能のin vitroアッセイ、リンパ球機能のin vivoアッセイ、免疫蛍光及び細胞分類、サイトカインとその細胞レセプター、ヒトの免疫学的研究、タンパク質、ペプチドの単離と分析、細胞活性化の分子生物学、生化学、補体、先天性免疫、自己免疫及び炎症性疾患の動物モデル(前記はNODマウスモデル、SLEマウスモデル(狼瘡に関する)及び調節T細胞の枯渇による自己免疫疾患の誘発に関する章を含む)、抗原のプロセッシング及び提示、免疫分子及びレセプターの操作、免疫系のリガンド-レセプター相互作用、顕微鏡観察法、並びに、一般的な免疫系遺伝子及びタンパク質(白血球表面分子についてのCD系を含む)の略語と用語。
概説
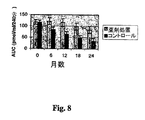
抗CD3抗体の作用に関する研究によって、前記抗体は自己免疫応答を非病原性表現型(おそらく調節T細胞の作用を必要とする)に変化させることができることが示唆される。新規開始T1D患者で抗CD3mAb(hOKT3γ1(Ala-Ala))を用いた最初の実験によれば、前記薬剤は機能的な免疫寛容を誘発することができ、前記寛容は処置後少なくとも1年間持続することが示された。しかしながら、疾患の最初の1年が過ぎた後の患者の追跡実験によれば、抗CD3mAbによる処置の2年後ですら統計的に有意なインシュリン分泌の改善が存在するが、C-ペプチドは最初の年の後では基準応答の71%に減少することが示された。これらの発見は、治療後の最初の年の後で寛容は低下しえること、更に追加の治療的介在が臨床反応の維持に必要とされることを示唆している。
したがって、本方法の重要な論理的根拠は、自己免疫応答の標的として特定された自己抗原(特にT細胞依存自己免疫応答の標的である自己抗原)を抗CD3抗体と一緒に投与することによって、前記自己抗原に対する応答を変化させ、自己免疫の進行を予防することができるということである。自己抗原による再攻撃及び非病原性応答の刺激によって、自己免疫プロセスの阻止を維持することができる。理論に拘束されないが、抗CD3及び自己抗原の同時投与は、これら自己抗原、及び問題となっている具体的な自己免疫疾患の標的である他の自己抗原に対する寛容を再確立させることができると考えられる。本明細書で提供する前臨床の証拠によれば、抗CD3及び自己抗原の併用は、自己免疫糖尿病の回復において相乗作用を有し、したがって同時投与は、他の自己免疫疾患の回復に相乗的防御を提供する潜在的能力を有することが示された。
抗CD3抗体の作用に関する研究によって、前記抗体は自己免疫応答を非病原性表現型(おそらく調節T細胞の作用を必要とする)に変化させることができることが示唆される。新規開始T1D患者で抗CD3mAb(hOKT3γ1(Ala-Ala))を用いた最初の実験によれば、前記薬剤は機能的な免疫寛容を誘発することができ、前記寛容は処置後少なくとも1年間持続することが示された。しかしながら、疾患の最初の1年が過ぎた後の患者の追跡実験によれば、抗CD3mAbによる処置の2年後ですら統計的に有意なインシュリン分泌の改善が存在するが、C-ペプチドは最初の年の後では基準応答の71%に減少することが示された。これらの発見は、治療後の最初の年の後で寛容は低下しえること、更に追加の治療的介在が臨床反応の維持に必要とされることを示唆している。
したがって、本方法の重要な論理的根拠は、自己免疫応答の標的として特定された自己抗原(特にT細胞依存自己免疫応答の標的である自己抗原)を抗CD3抗体と一緒に投与することによって、前記自己抗原に対する応答を変化させ、自己免疫の進行を予防することができるということである。自己抗原による再攻撃及び非病原性応答の刺激によって、自己免疫プロセスの阻止を維持することができる。理論に拘束されないが、抗CD3及び自己抗原の同時投与は、これら自己抗原、及び問題となっている具体的な自己免疫疾患の標的である他の自己抗原に対する寛容を再確立させることができると考えられる。本明細書で提供する前臨床の証拠によれば、抗CD3及び自己抗原の併用は、自己免疫糖尿病の回復において相乗作用を有し、したがって同時投与は、他の自己免疫疾患の回復に相乗的防御を提供する潜在的能力を有することが示された。
その潜在的な相乗効果を凌ぐ、抗CD3及び自己抗原の同時投与のまた別の利点は、自己免疫疾患はしばしば多数の自己抗原に対して自己攻撃性応答を含むという問題と関連がある。前記自己攻撃性リンパ球の全てを、それらの同族抗原の全てを用いて直接的な抗原特異的寛容によって無力化又は抹殺しようとすることは困難であろう。なぜならば前記同族抗原の全てを知ることは不可能であるからである。例えば、1型糖尿病(T1D)は、ランゲルハンス島に入り込み、そこでβ-細胞を破壊する自己攻撃性リンパ球によって引き起こされると考えられる。そのような細胞の活性化はおそらく多因子性であろう(前記多因子には、遺伝的素因、環境的な引き金(例えばウイルス)及びおそらく膵臓(島細胞)の損傷(例えば局所的前炎症性反応によって惹起される膵臓の損傷)が中心的に関与する)。前糖尿病のヒトの個体が島細胞抗体によって特定されるときは、自己攻撃プロセスは通常はかなり進行しているので、2つ以上の島抗原に対する攻撃性応答がこの病期の間に進行しているのであろうと推定することができる。更にまた、非特異的な慢性全身性免疫抑制が任意選択であるとは考えられない。なぜならば、糖尿病はしばしば若年個体が罹患し、生涯にわたる免疫抑制は、インシュリン単独療法と比較したとしても許容不能な副作用を伴う。したがって、特異性が有り全身的服作用が少ない免疫関連の治癒力のある治療的介在が希求される。本発明の方法によれば、抗CD3及びただ1つの自己抗原の同時投与が、1つの自己免疫疾患の標的である複数の自己抗原に対する寛容を再確立するために十分であるので、複数の自己抗原をもつ標的の問題は回避される。長期の非特異的な全身性免疫抑制の問題もまた本方法では回避される。なぜならば、抗CD3及び自己抗原の同時投与は、生涯にわたる連続投与の必要性が無く、長期の寛容を再確立させることができるからである。
理論に拘束されないが、本発明は、抗CD3及び自己抗原の同時投与は、部分的には調節T細胞(及び調節抗原提示細胞(APC))の活性化/増補(expand)を誘発することによって、長期にわたる寛容を相乗的に確立させる潜在的能力をもつことを示す。免疫系では、調節T細胞は、自己免疫疾患を制御する能力を有する。いくつかの細胞、例えばCD25陽性リンパ球(前記に多くの研究室が努力を傾注している)は、全身性非抗原特異的態様で作用するように思われる(Belghith et al. Nat. Med. 2003, 9:1202-8;Chatenoud et al. Immunol. Rev. 2001, 182:149-63;Green et al. Proc. Natl. Acad. Sci. USA 2003, 100:10878-83;Asseman et al. Autoimmun. Rev. 2002, 1:190-7)。これらの細胞は、マウスのいくつかの自己免疫的症状で数が減少することが見出されている。例えば、CD28-/-NODマウスで発生する加速的糖尿病は、調節CD4+CD25+ T細胞が存在しないためであり、これら細胞の輸液によって回復させることができる。
調節T細胞の様々な多数の表現型が記載されてきた。それらは胸腺切除の後で生じ、更に共同刺激ブロッカー又はFcR非結合抗CD3による全身性免疫調節後に誘発することができる(本明細書で更に説明される)。それらのエフェクター機能は完全には判明していない。それらは免疫系の固有の均衡の一部であるように思われ、それらが失われることによって重篤な免疫の失調及び自己免疫が生じる。特定の抗原特異性をもつTh2-様調節細胞が記載された。それらは傍観的サプレッサーとして機能し、抗原特異的免疫の後で生じると考えられる(Homann et al. J.Immunol. 1999, 163:1833-8)。そのエフェクター機能にしたがって、それらはTh3(TGF-β産生細胞)と名付けられた。これらの細胞は特殊化されたエフェクター機能をもつ抗原特異的リンパ球であり、Th2細胞のようには行動しない。したがっていわゆるTh1/Th2の枠組みをこれら細胞に適用することは誤りをもたらすであろう。
調節T細胞の様々な多数の表現型が記載されてきた。それらは胸腺切除の後で生じ、更に共同刺激ブロッカー又はFcR非結合抗CD3による全身性免疫調節後に誘発することができる(本明細書で更に説明される)。それらのエフェクター機能は完全には判明していない。それらは免疫系の固有の均衡の一部であるように思われ、それらが失われることによって重篤な免疫の失調及び自己免疫が生じる。特定の抗原特異性をもつTh2-様調節細胞が記載された。それらは傍観的サプレッサーとして機能し、抗原特異的免疫の後で生じると考えられる(Homann et al. J.Immunol. 1999, 163:1833-8)。そのエフェクター機能にしたがって、それらはTh3(TGF-β産生細胞)と名付けられた。これらの細胞は特殊化されたエフェクター機能をもつ抗原特異的リンパ球であり、Th2細胞のようには行動しない。したがっていわゆるTh1/Th2の枠組みをこれら細胞に適用することは誤りをもたらすであろう。
CD8+調節T細胞の亜集団もまたヒト及びマウスの系で記載された。CD8+CD28low細胞の亜集団は、抗原提示細胞上のILT3分子との相互作用によって移植物寛容を媒介することができる。また別の細胞タイプは、活性化されたCD4+細胞上で発現される非古典的クラスI MHC分子(Qa-1又はHLA-E)の認識によって、CD4+ T細胞を調節するらしい(Colovai et al. Transplant Proc. 2001, 33:104-7;Liu et al. Transplant Proc. 2001, 33:82-3;Chang et al. Nat. Immunol. 2002;H. Jiang et al. Annu. Rev. Immunol. 2000, 18:185-216)。最後に、活性な調節機能をもつAPCが最近報告された(Homann et al. Immunity 2002, 16:403-15;Serreze et al. Curr. Dir. Autoimmun. 2003, 6:212-27;Boudaly et al. Eur. Cytokine Netw. 2002, 13:29-37)。これらは、アネルギーT細胞又は調節細胞との共同刺激、接触又は他の免疫調節のブロック後に生じることができる。それらは、それらの抗原特異的調節エフェクター機能のために重要である。したがって、抗CD3及び自己抗原の同時投与に関するこれら“調節細胞”の分析と追跡は、寛容の誘発及び再確立の根幹にあるメカニズムに関する重要な識見を提供しえる。
上記で述べたように、抗CD3とただ1つの自己抗原の同時投与が、自己免疫疾患の標的である複数の自己抗原に対する寛容の再確立に十分でありえるので、本発明は複数の自己抗原標的に関する問題を回避することができる。理論に拘束されないが、本方法は、攻撃性細胞の抹消及び抗原特異的アネルギーのメカニズムで“傍観者サプレッション”と称されるプロセスによって複数の自己抗原に対する寛容を再確立させることができるかもしれない。
上記で述べたように、抗CD3とただ1つの自己抗原の同時投与が、自己免疫疾患の標的である複数の自己抗原に対する寛容の再確立に十分でありえるので、本発明は複数の自己抗原標的に関する問題を回避することができる。理論に拘束されないが、本方法は、攻撃性細胞の抹消及び抗原特異的アネルギーのメカニズムで“傍観者サプレッション”と称されるプロセスによって複数の自己抗原に対する寛容を再確立させることができるかもしれない。
傍観者サプレッションは抗原拡散の現象と関連がある。抗原拡散は、局所の自己免疫プロセス進行時の必須の成分と考えられる。したがって、患者がいくつかの自己抗体を有するときは、自己攻撃性応答には多くの自己抗原(又は“自己抗原”)が中心的に関与しているであろうと推測することができる。個々の自己免疫疾患について自己抗原の大半は特定できないであろうから、対応するMHC拘束成分及びペプチドの情報を必要とする治療スケジュールで自己攻撃性特異性の各々について寛容をもたらすことは不可能である。本発明の方法による調節細胞の誘発はこの状況でいくつかの利点を有する。例えば、T1Dにおける調節T細胞は、傍観的サプレッサーとしてPDLN及び島で局所的に作用することができることが知られている。このことは、それらが他の自己抗原特異性をもつ攻撃性リンパ球を抑制することができることを意味する。前記は、例えば免疫調節機能をもつサイトカインの分泌により、抗原提示細胞(APC)を調節することによってもたらすことができる。したがって、そのような傍観的サプレッサーT調節細胞は、他のいくつかの自己抗原に対する自己攻撃をそれらの正確な特異性を知ることなく鈍化させることができる。抗CD3は、IL-10のアップレギュレーションも含む、全身性免疫偏向を作り出すので、抗CD3投与時における抗原特異性免疫(すなわち自己抗原の投与)は、T-調節細胞誘発の蓋然性をより高めるであろう。T-調節細胞は続いて傍観的サプレッサーとして機能することができる。
進行中の自己反応性プロセスの全ての成分が標的器官を必ずしも損傷するわけではない。実際、自己攻撃性及び自己反応性の調節応答は、自己免疫疾患の多くの実験モデルで記載されている。これらは比較的もろい平衡で共存し、この平衡は通常は、臨床的に明瞭な自己免疫が生じる前に攻撃性応答側にシフトする。病原性免疫応答の表現型のシフト及び/又は周到に選択された免疫調節による調節性成分の増強は、前記疾患の進行を阻止し、更に回復させるために極めて重要であるはずである。理論に拘束されないが、本発明によって提供されることは、抗原と抗CD3抗体の同時投与が、前記抗原に対する応答を非病原性であるように変化させるか、及び/又は前記併用療法によって誘発される調節T細胞が前記抗原に対する応答を改変して自己免疫を予防し、更に前記同時投与の作用が、寛容の再確立/誘発において、又は有害なT-細胞媒介自己免疫作用の低下において相乗的であるということである。したがって、本発明の非制限的な論理的根拠は、抗CD3抗体は免疫系を“リセット”することができ、いくつかの抗原特異的免疫に長期の寛容を維持することができる調節を誘発させるということである。
抗CD3抗体
本発明以前には、抗CD3及び自己抗原の併用投与が自己抗原に対する寛容の誘発又は再確立に相乗作用をもたらすことができるという証拠はなかった。更に、抗CD3の免疫抑制力が、自己抗原の投与がこれら抗原に対する寛容の確立に対してもちえる一切の前駆的作用を帳消しにすることは極めてありえることであるので、そのような相乗的な成果は予想だにされなかった。
したがって、本発明の方法は、抗CD3抗体及び自己免疫T-依存応答の標的である自己抗原の同時投与を必要とする。本発明が意図する自己抗原の具体的な例は以下に記載されている。このセクションでは、本発明の方法で用いることができる抗CD3抗体の具体的な例が提供される。
一般的には、本発明の方法は、自己抗原と一緒に投与して前記自己抗原に対する寛容を誘発又は再確立することができる任意の抗CD3抗体の使用を意図する。抗CD3抗体のこの普遍的使用に対する1つの制限は、この抗体はモノマー形であってはならないということ、換言すれば、本発明の抗体は少なくとも2つの抗原結合部位を保有しなければならないということである。したがって、Fab抗CD3抗体は、単Fab分子が一緒に連結されないかぎり一般的には本発明では機能しない。本発明の方法は、完全長又はそのマルチマーフラグメントである抗CD3抗体の使用を含むことができる。マルチマー抗体フラグメントには、例えばF(ab')2、単鎖二価抗体を含む二価抗体、ビアボディ抗体、及び二価の単鎖Fv抗体が含まれよう。前記抗体は任意のクラスの抗体、すなわちIgG、IgM、IgE、IgA及びIgDであろう。前記抗体は任意のサブクラスであり、例えばヒトの抗体ではIgG1、IgG2、IgG3、IgG4、IgA1及びIgA2であり、更にマウスの抗体ではIgG1、IgG2a、IgG2bであろう。
本発明以前には、抗CD3及び自己抗原の併用投与が自己抗原に対する寛容の誘発又は再確立に相乗作用をもたらすことができるという証拠はなかった。更に、抗CD3の免疫抑制力が、自己抗原の投与がこれら抗原に対する寛容の確立に対してもちえる一切の前駆的作用を帳消しにすることは極めてありえることであるので、そのような相乗的な成果は予想だにされなかった。
したがって、本発明の方法は、抗CD3抗体及び自己免疫T-依存応答の標的である自己抗原の同時投与を必要とする。本発明が意図する自己抗原の具体的な例は以下に記載されている。このセクションでは、本発明の方法で用いることができる抗CD3抗体の具体的な例が提供される。
一般的には、本発明の方法は、自己抗原と一緒に投与して前記自己抗原に対する寛容を誘発又は再確立することができる任意の抗CD3抗体の使用を意図する。抗CD3抗体のこの普遍的使用に対する1つの制限は、この抗体はモノマー形であってはならないということ、換言すれば、本発明の抗体は少なくとも2つの抗原結合部位を保有しなければならないということである。したがって、Fab抗CD3抗体は、単Fab分子が一緒に連結されないかぎり一般的には本発明では機能しない。本発明の方法は、完全長又はそのマルチマーフラグメントである抗CD3抗体の使用を含むことができる。マルチマー抗体フラグメントには、例えばF(ab')2、単鎖二価抗体を含む二価抗体、ビアボディ抗体、及び二価の単鎖Fv抗体が含まれよう。前記抗体は任意のクラスの抗体、すなわちIgG、IgM、IgE、IgA及びIgDであろう。前記抗体は任意のサブクラスであり、例えばヒトの抗体ではIgG1、IgG2、IgG3、IgG4、IgA1及びIgA2であり、更にマウスの抗体ではIgG1、IgG2a、IgG2bであろう。
抗CD3抗体はポリクローナルでもモノクローナルでもよい。前記抗体はまたキメラ(すなわち2つ以上の種に由来する配列の組合せ、例えばマウス-ヒトキメラ免疫グロブリン)でも、ヒト化されていても又は完全にヒト抗体であってもよい。ヒト抗体は、ネズミ又はラット(又は他の種)の可変及び/又は定常領域を保有する抗体に付随するある種の問題を回避する。そのようなネズミ又はラットに由来するタンパク質の存在は抗体の迅速なクリアランスをもたらすか、又は患者による前記抗体に対する免疫応答の発生をもたらしえる。ネズミ又はラット由来の抗体の利用を回避するために、ヒト化抗体を開発するか、又はヒトの抗体機能をげっ歯類に導入し、それによって前記げっ歯類が完全にヒトの配列を有する抗体を産生させることによって完全にヒトの抗体を作製することができる。例えば、米国特許5,770,429号、同6,150,584号及び同6,677,138号は、標的抗原に対して高い親和性の完全にヒトの抗体を作製するためのトランスジェニックマウス技術に関する(すなわち、HuMab-mouse (商標)又はXenmouse(商標))。
ある実施態様では、抗CD3抗体はFcレセプター(FcR)と結合しない。そのような抗CD3抗体は、本明細書では“FcR非結合抗CD3Ab”と称される。使用することができる具体的なFcR非結合抗CD3AbはOKT3抗体である。本発明はまたOKT3抗体の変異体又は変種(hOKT3γ1(Ala-Ala)及びhOKT3γ3(IgG3)を含む:K. Herold et al. N. Engl. J. Med. 2002, 346:1692-8;D. Xu et al. Cell Immunol. 2000, 200:16-26)の使用を意図する。hOKT3γ1(Ala-Ala)は、ヒトのものと比較して、マウスで類似の機能を示し、変異したFc結合領域を有する。前記は有糸分裂非誘発性であるが、T細胞でシグナリングを誘発する。抗CD3-IgG3はAla-Ala変種と類似している。なぜならば、この抗体はヒトのものと比較してマウスで類似の機能を示し、更に変異したFc-結合領域を有するからである。前記もまた有糸分裂非誘発性であるが、T細胞でシグナリングをまた誘発する。
ある実施態様では、抗CD3抗体はFcレセプター(FcR)と結合しない。そのような抗CD3抗体は、本明細書では“FcR非結合抗CD3Ab”と称される。使用することができる具体的なFcR非結合抗CD3AbはOKT3抗体である。本発明はまたOKT3抗体の変異体又は変種(hOKT3γ1(Ala-Ala)及びhOKT3γ3(IgG3)を含む:K. Herold et al. N. Engl. J. Med. 2002, 346:1692-8;D. Xu et al. Cell Immunol. 2000, 200:16-26)の使用を意図する。hOKT3γ1(Ala-Ala)は、ヒトのものと比較して、マウスで類似の機能を示し、変異したFc結合領域を有する。前記は有糸分裂非誘発性であるが、T細胞でシグナリングを誘発する。抗CD3-IgG3はAla-Ala変種と類似している。なぜならば、この抗体はヒトのものと比較してマウスで類似の機能を示し、更に変異したFc-結合領域を有するからである。前記もまた有糸分裂非誘発性であるが、T細胞でシグナリングをまた誘発する。
更に、抗CD3 Fab'2抗体が意図される。前記は、例えばマウス2C11細胞クローンに由来する抗体で、FcR非結合、有糸分裂非誘発性及びT細胞でシグナリングを誘発する。抗CD3抗体(OKT3抗体及びその変種/変異体を含む)の使用及び作製に関する方法は、米国特許6,113,901号、同6,491,916号及び同5,885,573号に記載されている(前記文献は参照により本明細書に含まれる)。更に、本発明のある実施態様では、抗CD3抗体は免疫消耗性(immune depleting)ではない。
抗CD3抗体は約5μgから約2000μgの量で投与することができる。前記投与は、例えば約1−14日の期間毎日実施することができる。ある実施態様では、前記投与は10日間毎日実施される。別の実施態様では、前記投与は12日間毎日実施される。また別の実施態様では、抗CD3抗体は、1日目に約200-250μg/m2の量で、2日目に約400-500μg/m2の量で、更に3−12日目に約900-1000μg/m2の量で投与される。前記投与は静脈内(i.v.)で実施されるべきである。T1Dの場合は、抗CD3抗体は、例えば高血糖の開始後0−10日目で投与することができる。
抗CD3抗体は約5μgから約2000μgの量で投与することができる。前記投与は、例えば約1−14日の期間毎日実施することができる。ある実施態様では、前記投与は10日間毎日実施される。別の実施態様では、前記投与は12日間毎日実施される。また別の実施態様では、抗CD3抗体は、1日目に約200-250μg/m2の量で、2日目に約400-500μg/m2の量で、更に3−12日目に約900-1000μg/m2の量で投与される。前記投与は静脈内(i.v.)で実施されるべきである。T1Dの場合は、抗CD3抗体は、例えば高血糖の開始後0−10日目で投与することができる。
自己抗原
本発明は、抗CD3抗体及びT依存自己免疫応答の標的である自己抗原を同時投与することによって自己免疫を治療し、或いは寛容を確立又は誘発する方法を目的とする。自己免疫疾患の他に、本発明の方法はまた、アレルゲンに対する寛容を確立するためにも用いることができる。この場合、アレルゲンペプチド又はタンパク質が抗CD3抗体とともに同時投与される。
本発明の方法による治療が意図される、T細胞によって媒介される自己免疫疾患の非限定的な例には、下記の表に提供される疾患が含まれるが、ただしこれらに限定されない(下記の表には自己免疫疾患に関連する自己抗原もまた含まれ、これら自己抗原を抗CD3抗体と同時投与することができる):
本発明は、抗CD3抗体及びT依存自己免疫応答の標的である自己抗原を同時投与することによって自己免疫を治療し、或いは寛容を確立又は誘発する方法を目的とする。自己免疫疾患の他に、本発明の方法はまた、アレルゲンに対する寛容を確立するためにも用いることができる。この場合、アレルゲンペプチド又はタンパク質が抗CD3抗体とともに同時投与される。
本発明の方法による治療が意図される、T細胞によって媒介される自己免疫疾患の非限定的な例には、下記の表に提供される疾患が含まれるが、ただしこれらに限定されない(下記の表には自己免疫疾患に関連する自己抗原もまた含まれ、これら自己抗原を抗CD3抗体と同時投与することができる):
表1:自己免疫疾患及び自己抗原標的

したがって、ある種の実施態様では、本発明は、抗CD3抗体及び自己抗原(例えば上記の表に挙げたようなもの)の同時投与を必要とする、自己免疫を治療し、又は寛容を誘発/確立する方法を提供する。投与される自己抗原は完全なタンパク質又はそのペプチドフラグメントの形態であろう。前記タンパク質又はペプチドフラグメントの配列は野生型でも変異体でもよい。更に、前記タンパク質又はペプチドは、医薬的に許容できる担体中のタンパク質又はペプチドとして対象者に導入してもよいし、また、前記タンパク質又はペプチドは発現ベクターによってコードされてもよい。前記の場合は発現ベクターが導入される(例えば実施例を参照されたい、前記実施例では自己抗原はpCMV-発現ベクターによって対象者で発現される)。発現ベクターによる自己抗原の投与(および投与できる特異的抗原)に関する更なる記述に関しては、米国特許公開US2002/0107210を参照されたい(前記文献は参照により本明細書に含まれる)。
一般的には、抗原は、例えば約100μgから約2000μg/kg体重の量で投与することができる。抗原は、複数日数を含む投与スケジュールで投与することができる。この場合、投与される抗原の総量は、ある実施態様では約100μgから約2000μgである。ある実施態様では、抗原は約50から約200μgの量で毎日、4日間投与される。抗CD3との同時投与に関しては、抗CD3及び抗原は一緒に例えば1日目に投与することができ、1日目以降は投与時間が異なってもよい。更に、抗CD3及び抗原の最初の投与の後、抗CD3及び抗原の両方、抗CD3のみ、又は抗原のみをブースターとして投与することができる。例えば、最初の投与の後、ブースターは、最初の投与後約6ヶ月から約2年の期間に投与することができる。抗原は経鼻的に、皮下に(s.c.)、筋肉内に(i.m.)、又は静脈内に(i.v.)投与することができる。
一般的には、抗原は、例えば約100μgから約2000μg/kg体重の量で投与することができる。抗原は、複数日数を含む投与スケジュールで投与することができる。この場合、投与される抗原の総量は、ある実施態様では約100μgから約2000μgである。ある実施態様では、抗原は約50から約200μgの量で毎日、4日間投与される。抗CD3との同時投与に関しては、抗CD3及び抗原は一緒に例えば1日目に投与することができ、1日目以降は投与時間が異なってもよい。更に、抗CD3及び抗原の最初の投与の後、抗CD3及び抗原の両方、抗CD3のみ、又は抗原のみをブースターとして投与することができる。例えば、最初の投与の後、ブースターは、最初の投与後約6ヶ月から約2年の期間に投与することができる。抗原は経鼻的に、皮下に(s.c.)、筋肉内に(i.m.)、又は静脈内に(i.v.)投与することができる。
上記の表に挙げたように、糖尿病治療(T1D患者での寛容の再確立)のために抗CD3と一緒に同時投与することができる抗原の例には、例えばインシュリン、プロインシュリン、GAD65、ICA512/IA-2及びHSP60が含まれる。前記抗原は先ず初めに、高血糖症の開始後、例えば2ヶ月以内に、1−2ヶ月以内に、6週間以内に、又は10日後に続いて投与することができる。実施例1のマウスについて観察された“治療ウインドウ” とは異なり、T1Dのための自己抗原の投与は、ヒトの患者の特定の血糖値レベル内で実施しなければならないわけではない。
T1Dの治療のある実施例では、10μgの抗原をそれぞれ別々の日に4日間投与できる。前記抗原はミョウバン中で投与することができ、例えば皮下に注射することができる。別の実施態様では、前記抗原は、経鼻的に約1から約2mgの量で、それぞれ別々の日に4日間投与できる。また別の実施態様では、前記抗原は、経鼻的に約1.5 mgの量で、それぞれ別々の日に4日間投与できる。糖尿病の回復に対する治療の効果はグルコースレベルの測定及びインシュリンの判定によって調べることができる。
ある実施態様では、ヒトインシュリン又はヒトインシュリン類似体X38(NovoNordisk)を投与することができる。前記は、例えば10μLのPBS中に約0.05mg/用量の量で、4日間それぞれ別々の日に最初の用量として投与できる。また別の実施態様では、ブタのインシュリン-B鎖を、例えば100μL中に約5mg/kgの量で皮下に投与することができる。吸収がより遅く、アナフィラキシーを誘発しない(アナフィラキシーはインシュリンB9-23ペプチドの単独使用で観察される)改変ペプチドもまた投与することができる。
ある実施態様では、ブタのインシュリン-B鎖APL(変異ペプチドリガンド(altered peptide ligand))を用いることができる。前記は、例えば100μL中に約5mg/kgの量で、処置の0、3、7、10及び15日目に皮下に投与することができる。前記抗原はニューロクリン(Neurocrine, San Diego, CA, USA)から市場で入手することができる(Alleva et al. Diabetes 2002, 51:2126-34)。
ある実施態様では、ヒトのGAD65タンパク質(hGAD65)を投与することができる。前記は、例えば300μLのPBS中に約100μgの量で0、1、7及び12日目に皮下に投与することができる。前記抗原はダイアミド(Diamyd, Stockholm, Sweden)から市場で入手することができる。
ある実施態様では、プロインシュリンIIペプチドを投与することができる。前記は、例えば100μL中に約5mg/kgの量で、処置の0、3、7、10及び15日目に皮下に投与することができる。前記抗原はニューロクリン(Neurocrine, San Diego, CA, USA)から市場で入手することができる。
ある実施態様では、HSP60ペプチド(DiaPep277)を投与することができる(Raz et al. Lancet 2001, 358:1749-53)。
T1Dの治療のある実施例では、10μgの抗原をそれぞれ別々の日に4日間投与できる。前記抗原はミョウバン中で投与することができ、例えば皮下に注射することができる。別の実施態様では、前記抗原は、経鼻的に約1から約2mgの量で、それぞれ別々の日に4日間投与できる。また別の実施態様では、前記抗原は、経鼻的に約1.5 mgの量で、それぞれ別々の日に4日間投与できる。糖尿病の回復に対する治療の効果はグルコースレベルの測定及びインシュリンの判定によって調べることができる。
ある実施態様では、ヒトインシュリン又はヒトインシュリン類似体X38(NovoNordisk)を投与することができる。前記は、例えば10μLのPBS中に約0.05mg/用量の量で、4日間それぞれ別々の日に最初の用量として投与できる。また別の実施態様では、ブタのインシュリン-B鎖を、例えば100μL中に約5mg/kgの量で皮下に投与することができる。吸収がより遅く、アナフィラキシーを誘発しない(アナフィラキシーはインシュリンB9-23ペプチドの単独使用で観察される)改変ペプチドもまた投与することができる。
ある実施態様では、ブタのインシュリン-B鎖APL(変異ペプチドリガンド(altered peptide ligand))を用いることができる。前記は、例えば100μL中に約5mg/kgの量で、処置の0、3、7、10及び15日目に皮下に投与することができる。前記抗原はニューロクリン(Neurocrine, San Diego, CA, USA)から市場で入手することができる(Alleva et al. Diabetes 2002, 51:2126-34)。
ある実施態様では、ヒトのGAD65タンパク質(hGAD65)を投与することができる。前記は、例えば300μLのPBS中に約100μgの量で0、1、7及び12日目に皮下に投与することができる。前記抗原はダイアミド(Diamyd, Stockholm, Sweden)から市場で入手することができる。
ある実施態様では、プロインシュリンIIペプチドを投与することができる。前記は、例えば100μL中に約5mg/kgの量で、処置の0、3、7、10及び15日目に皮下に投与することができる。前記抗原はニューロクリン(Neurocrine, San Diego, CA, USA)から市場で入手することができる。
ある実施態様では、HSP60ペプチド(DiaPep277)を投与することができる(Raz et al. Lancet 2001, 358:1749-53)。
T-調節細胞
T調節細胞は、抗CD3及び抗原の投与によって開始される長期持続寛容を支える主成分である。最初の同時投与の後、更なる抗原の投与は抗原特異的T調節細胞集団の長期間の維持に役立ちえるということは可能である。したがって、本発明はまた、自己抗原に対して抗原特異性を有するT調節細胞の集団を増補(expand)又は生産させるために抗CD3抗体及び自己抗原を投与して、自己免疫を治療又は寛容を誘発/確立する方法を提供する。同時投与を用いてT調節細胞をin vivoで増補又は生産させることができる。本発明はまた、同時投与後にT調節細胞を単離することを意図する。この場合、単離された細胞を更にin vitroで増補/増殖させることができる(単離及び増補の方法については実施例を参照されたい)。in vitroでの増補の他に、T調節細胞は更に抗CD3及び抗原に暴露することができる。in vitroでの増補の後で、前記調節細胞を将来の患者での使用に備えて凍結するか、又は患者に再投与することができる。
ある実施態様では、単離されたT調節細胞は少なくともCD4+及びCD25+である。また別の実施態様では、単離されたT調節細胞は少なくともCD4+、CD25+、GITR+、CTLA-4+及びCD62L+である。また別の実施態様では、単離されたT調節細胞は少なくともCD4+、IL-10+及びTGF-β+である。また別の実施態様では、単離されたT調節細胞は少なくともCD4+、IL-10+、CD45RO+及びCCR4+である。また別の実施態様では、単離されたT調節細胞は少なくともCD4+、IL-10+、CD45RO+、CCR4+及びTGF-β+である。また別の実施態様では、単離されたT調節細胞は少なくともCCR4+、CD62L及びCD45RO+である。また別の実施態様では、単離されたT調節細胞は少なくともCD4+及びIL-4+である。また別の実施態様では、単離されたT調節細胞は少なくともCD8+である。
本発明の範囲から逸脱することなく、上記の方法及び組成物において種々の変更を実施することができるので、上記の記載に含まれているか、添付の図面に示されているか、又は添付の請求の範囲に規定されている全ての事柄は例示であり、制限的な意味をもたないと解されるべきである。
以下の実施例は、本発明の特徴の実施において本発明者らが用いた技術の典型的な例である。これらの技術は本発明の実施のための例であるが、当業者には、本発明の開示を参考にして、多くの改変が本発明の範囲から逸脱することなく実施されえることは理解されよう。したがって、下記に記載する実施例は、本発明の説明のために提供され、本発明の制限のために含まれるものではない。
T調節細胞は、抗CD3及び抗原の投与によって開始される長期持続寛容を支える主成分である。最初の同時投与の後、更なる抗原の投与は抗原特異的T調節細胞集団の長期間の維持に役立ちえるということは可能である。したがって、本発明はまた、自己抗原に対して抗原特異性を有するT調節細胞の集団を増補(expand)又は生産させるために抗CD3抗体及び自己抗原を投与して、自己免疫を治療又は寛容を誘発/確立する方法を提供する。同時投与を用いてT調節細胞をin vivoで増補又は生産させることができる。本発明はまた、同時投与後にT調節細胞を単離することを意図する。この場合、単離された細胞を更にin vitroで増補/増殖させることができる(単離及び増補の方法については実施例を参照されたい)。in vitroでの増補の他に、T調節細胞は更に抗CD3及び抗原に暴露することができる。in vitroでの増補の後で、前記調節細胞を将来の患者での使用に備えて凍結するか、又は患者に再投与することができる。
ある実施態様では、単離されたT調節細胞は少なくともCD4+及びCD25+である。また別の実施態様では、単離されたT調節細胞は少なくともCD4+、CD25+、GITR+、CTLA-4+及びCD62L+である。また別の実施態様では、単離されたT調節細胞は少なくともCD4+、IL-10+及びTGF-β+である。また別の実施態様では、単離されたT調節細胞は少なくともCD4+、IL-10+、CD45RO+及びCCR4+である。また別の実施態様では、単離されたT調節細胞は少なくともCD4+、IL-10+、CD45RO+、CCR4+及びTGF-β+である。また別の実施態様では、単離されたT調節細胞は少なくともCCR4+、CD62L及びCD45RO+である。また別の実施態様では、単離されたT調節細胞は少なくともCD4+及びIL-4+である。また別の実施態様では、単離されたT調節細胞は少なくともCD8+である。
本発明の範囲から逸脱することなく、上記の方法及び組成物において種々の変更を実施することができるので、上記の記載に含まれているか、添付の図面に示されているか、又は添付の請求の範囲に規定されている全ての事柄は例示であり、制限的な意味をもたないと解されるべきである。
以下の実施例は、本発明の特徴の実施において本発明者らが用いた技術の典型的な例である。これらの技術は本発明の実施のための例であるが、当業者には、本発明の開示を参考にして、多くの改変が本発明の範囲から逸脱することなく実施されえることは理解されよう。したがって、下記に記載する実施例は、本発明の説明のために提供され、本発明の制限のために含まれるものではない。
実施例1:マウスの新規開始糖尿病を治療する抗CD3と抗原特異的免疫療法の実験結果
下記で述べる実験結果は、“方法と実験計画”のセクションの実験方法を用いて得られた。
NODモデル:
糖尿病マウスの抗CD3mAb治療の報告された研究によれば、糖尿病の永久的回復における顕著な有効性(概して80%)が示された(Chatenoud et al. Proc. Natl. Acad. Sci. USA 1994, 91:123-7;Chatenoud et al. J. Immunol. 1997, 158:2947-54)。
ヒトでの研究の経験では、1年で71%の患者でインシュリン応答の維持又は改善すらあるが、結局2年目でインシュリン分泌が低下したということであった。したがって、臨床状況と関連性を有するモデルで併用免疫療法の効果を調べるために、併用療法の抗CD3による治療部分を、抗CD3治療のみを用いる以前の実験と比較して改変した。抗CD3単独治療に関しては、新規開始糖尿病後に意図的に至適用量以下(100μgを5回静注)の抗CD3F(ab')2のみで治療したマウスの50%が防御された(図1C;下記図1の図面の簡単な説明を参照されたい)。しかしながら、NODモデルでは、T1Dの回復には抗CD3F(ab')2は治療ウインドウの範囲(血糖値が250から500mg/dLの間)で投与されねばならない。実際のところ、500μg/dLより高い血糖値をもつマウスは、抗CD3治療後にほとんど防御されない。
下記で述べる実験結果は、“方法と実験計画”のセクションの実験方法を用いて得られた。
NODモデル:
糖尿病マウスの抗CD3mAb治療の報告された研究によれば、糖尿病の永久的回復における顕著な有効性(概して80%)が示された(Chatenoud et al. Proc. Natl. Acad. Sci. USA 1994, 91:123-7;Chatenoud et al. J. Immunol. 1997, 158:2947-54)。
ヒトでの研究の経験では、1年で71%の患者でインシュリン応答の維持又は改善すらあるが、結局2年目でインシュリン分泌が低下したということであった。したがって、臨床状況と関連性を有するモデルで併用免疫療法の効果を調べるために、併用療法の抗CD3による治療部分を、抗CD3治療のみを用いる以前の実験と比較して改変した。抗CD3単独治療に関しては、新規開始糖尿病後に意図的に至適用量以下(100μgを5回静注)の抗CD3F(ab')2のみで治療したマウスの50%が防御された(図1C;下記図1の図面の簡単な説明を参照されたい)。しかしながら、NODモデルでは、T1Dの回復には抗CD3F(ab')2は治療ウインドウの範囲(血糖値が250から500mg/dLの間)で投与されねばならない。実際のところ、500μg/dLより高い血糖値をもつマウスは、抗CD3治療後にほとんど防御されない。
抗CD3F(ab')2及び抗原特異的療法(ここでは組換え体ヒトGAD65(rhGAD65)及びCTLエピトープ(43)をもたないマウスプロインシュリンIIペプチド)を併用することによって、防御マウスの数が抗CD3単独治療よりも顕著に増加するように相乗作用が得られた(図2A)。抗CD3F(ab')2及びrhGAD65の同時投与の後、NODマウスの33%が糖尿病を維持し、マウスプロインシュリンIIペプチドを用いることによって、最初の抗CD3注射の2週間後にマウスの100%が防御される。したがって、新規開始糖尿病NODに与えられた抗CD3及び経鼻的プロインシュリンによる治療は、抗CD3単独で観察された50%防御(図1C)と比較して100%の防御をもたらし、更にプロインシュリン単独では全く防御は得られなかった(データは示されていない)。
更にまた、図2Bは、抗CD3mAbが抗原と併用されたとき糖尿病の回復が強化されることを示している。糖尿病であることが判明した(グルコースレベルが>200mg/dLであるときに糖尿病と診断される)雌のNODマウスを、抗CD3mAbのF(ab')2フラグメント(145-2C11、n=9)、プロインシュリンペプチド(Auspen、n=8)、又は抗CD3mAbのF(ab')2フラグメント及びプロインシュリンペプチドの両方(n=8)で処置した。用いたプロインシュリンの用量は0、1、7、12日目に40μg(経鼻)、用いた抗CD3mAbのF(ab')2フラグメントは0−4日目に50μg(静注)であった。血糖レベルは、携帯用グルコース測定計を用いて2−3回/週で7週間の間平日に測定した。グルコースレベルが>200mg/dLであることが判明したマウスを糖尿病ありと判定した。抗CD3mAbのF(ab')2フラグメントとプロインシュリンの併用処置は、F(ab')2抗CD3mAb(p<0.05)又はプロインシュリン(p=0.01)のみで処置されたものと比較して、42日目の糖尿病の割合において顕著な改善を示した。
これらの結果は、抗CD3又は自己抗原単独投与と比較したとき、抗CD3及び自己抗原の同時投与によって提供される自己免疫に対する相乗的防御効果を明瞭に示している。
更にまた、図2Bは、抗CD3mAbが抗原と併用されたとき糖尿病の回復が強化されることを示している。糖尿病であることが判明した(グルコースレベルが>200mg/dLであるときに糖尿病と診断される)雌のNODマウスを、抗CD3mAbのF(ab')2フラグメント(145-2C11、n=9)、プロインシュリンペプチド(Auspen、n=8)、又は抗CD3mAbのF(ab')2フラグメント及びプロインシュリンペプチドの両方(n=8)で処置した。用いたプロインシュリンの用量は0、1、7、12日目に40μg(経鼻)、用いた抗CD3mAbのF(ab')2フラグメントは0−4日目に50μg(静注)であった。血糖レベルは、携帯用グルコース測定計を用いて2−3回/週で7週間の間平日に測定した。グルコースレベルが>200mg/dLであることが判明したマウスを糖尿病ありと判定した。抗CD3mAbのF(ab')2フラグメントとプロインシュリンの併用処置は、F(ab')2抗CD3mAb(p<0.05)又はプロインシュリン(p=0.01)のみで処置されたものと比較して、42日目の糖尿病の割合において顕著な改善を示した。
これらの結果は、抗CD3又は自己抗原単独投与と比較したとき、抗CD3及び自己抗原の同時投与によって提供される自己免疫に対する相乗的防御効果を明瞭に示している。
RIP-LCMVモデル(RIP-GPマウス):
RIP-LCMVトランスジェニックマウスは、ウイルス誘発1型糖尿病のためのマウスモデルとして用いることができる。前記マウスは明瞭に規定された標的自己抗原を主として膵臓のβ-細胞で発現するが、他の器官では全く発現しない(図5参照)。ラットインシュリンプロモーター(RIP)を用いることによって、このアプローチは、自己抗原として膵島でリンパ球性脈絡膜炎ウイルス(LCMV)の糖タンパク質(GP)又は核内タンパク質(NP)のどちらかを発現するトランスジェニック系列を作出する。これらウイルスのトランスジーンの発現はいずれのβ-細胞の機能不全ももたらさず、そのようなトランスジェニックマウスの大半(>95%)はそれらの生涯にわたって健常であり、島の機能不全又は浸潤の徴候を示さない(前記はLCMV-GP株には適用されるが、LCMV-NP株には適用されない)。
これらのマウスはしたがって、規定された1つの自己抗原に対する免疫応答を操作し、更にどの自己反応性応答が臨床的に明瞭な疾患をもたらすかを検査するための理想的なツールを構成する。実際に、LCMVについてもっともドミナント及び前記に次いでドミナントなT細胞(CD4及びCD8)エピトープが、マウスH-2b及びH-2dハプロタイプについてマッピングされた。免疫応答は多くの別個の研究室で正確に定量され、大半の“業界ツール(tools of the trade)”、例えばテトラマー、細胞内サイトカイン産生用FACS及びELISPOTアッセイが確立された。結果として、この実験モデルは、自己反応性免疫応答を追跡し、操作し、特定することができ、始動自己抗原が判明するという点で固有である。それ以来、RIP-LCMVマウスは、特定された自己抗原(トランスジーン)に対する末梢寛容/非応答性は全身的ウイルス感染によって破壊され、β-細胞の攻撃及び最終的なそれらの破壊をもたらすことができるという考え方を実証してきた。自己反応性で攻撃性応答の“成功”のためには、十分に大量の自己反応性リンパ球が全身的に生産される必要がある。
RIP-LCMVトランスジェニックマウスは、ウイルス誘発1型糖尿病のためのマウスモデルとして用いることができる。前記マウスは明瞭に規定された標的自己抗原を主として膵臓のβ-細胞で発現するが、他の器官では全く発現しない(図5参照)。ラットインシュリンプロモーター(RIP)を用いることによって、このアプローチは、自己抗原として膵島でリンパ球性脈絡膜炎ウイルス(LCMV)の糖タンパク質(GP)又は核内タンパク質(NP)のどちらかを発現するトランスジェニック系列を作出する。これらウイルスのトランスジーンの発現はいずれのβ-細胞の機能不全ももたらさず、そのようなトランスジェニックマウスの大半(>95%)はそれらの生涯にわたって健常であり、島の機能不全又は浸潤の徴候を示さない(前記はLCMV-GP株には適用されるが、LCMV-NP株には適用されない)。
これらのマウスはしたがって、規定された1つの自己抗原に対する免疫応答を操作し、更にどの自己反応性応答が臨床的に明瞭な疾患をもたらすかを検査するための理想的なツールを構成する。実際に、LCMVについてもっともドミナント及び前記に次いでドミナントなT細胞(CD4及びCD8)エピトープが、マウスH-2b及びH-2dハプロタイプについてマッピングされた。免疫応答は多くの別個の研究室で正確に定量され、大半の“業界ツール(tools of the trade)”、例えばテトラマー、細胞内サイトカイン産生用FACS及びELISPOTアッセイが確立された。結果として、この実験モデルは、自己反応性免疫応答を追跡し、操作し、特定することができ、始動自己抗原が判明するという点で固有である。それ以来、RIP-LCMVマウスは、特定された自己抗原(トランスジーン)に対する末梢寛容/非応答性は全身的ウイルス感染によって破壊され、β-細胞の攻撃及び最終的なそれらの破壊をもたらすことができるという考え方を実証してきた。自己反応性で攻撃性応答の“成功”のためには、十分に大量の自己反応性リンパ球が全身的に生産される必要がある。
更に、我々のデータは、島の抗原提示細胞(APC)の活性化が(及びおそらくは膵臓の直接的ウイルス感染も同様に)、そのような自己反応性T細胞が病原性を獲得するために通常要求されることを示している。RIP-LCMV-NPマウスの偶発的疾患はつい最近発見され(予備的データを参照されたい)、これまでの研究では精査されなかった。なぜならば、前記疾患は6ヶ月齢後でなければ重度に進行しないからである。今日まで、遺伝的欠損(ノックアウト)マウスの使用によって、パーフォリン、インターフェロン-γはいずれもTNF-αと同様に、これらマウスで糖尿病の進行に必須であると決定された。これらの研究で、全身的作用は局所化された膵/島特異的プロセスから切り離すことはできなかった。RIP-LCMVマウスは、ヒトの糖尿病の多くの局面、例えば臨床症状の開始に至る他のセルフ(島)抗原への自己免疫プロセスの拡散、及び島抗体の産生を反映する。これらの特徴によって、前記モデルはヒトの糖尿病の病理発生の理解のための適切で高度に相関性のあるモデルになる。
LCMV接種マウス:
ウイルス(LCMV)感染は、1型糖尿病をRIP-NP/GPマウスで誘発する。この実験方法の主要な利点は、規定された病理発生及び多数の分析方法での試薬の利用可能性である。コントロール実験には、C57BL6(DbKbハプロタイプ)及びBalbマウス(LdKdハプロタイプ)での実験が含まれる。前記マウスは、NOD及びNOD-NPマウス(DbKdハプロタイプ)とMHCハプロタイプを共有する。ウイルス誘発糖尿病及び免疫調節のためのRIP-NP又はNOD-NP実験:a)糖尿病のより高い発生率及びb)より迅速な糖尿病開始のカイネティクスのために、実験群はより小さくなり、1年当たりより多くの実験を実施できる。各実験群につき15匹のマウス、6−8実験群、1年当たり4−5実験。
図3に示したように(図面の簡単な説明を参照されたい)、有糸分裂非誘発性抗CD3F(ab')2(LCMV感染後15日目から20日目)で処置後、約40%のRIP-LCMV-GPマウスが防御される。hGAD65(真核細胞発現ベクター、pCMVを使用)単独処置は糖尿病からの部分的回復(40%防御マウス)をもたらし、同様な結果が以前に、RIP-LCMV-NPマウスの治療のためにブタインシュリンB発現プラスミド(pCMV/insB)を単独で用いたときにも得られた(Coon et al. J. Clin. Invest. 1999, 104:189-94)。抗CD3及びpCMV/hGAD65処置の併用は、RIP-LCMV-GPモデルで強力な相乗効果を示す(80%のマウスが防御された)。
LCMV接種マウス:
ウイルス(LCMV)感染は、1型糖尿病をRIP-NP/GPマウスで誘発する。この実験方法の主要な利点は、規定された病理発生及び多数の分析方法での試薬の利用可能性である。コントロール実験には、C57BL6(DbKbハプロタイプ)及びBalbマウス(LdKdハプロタイプ)での実験が含まれる。前記マウスは、NOD及びNOD-NPマウス(DbKdハプロタイプ)とMHCハプロタイプを共有する。ウイルス誘発糖尿病及び免疫調節のためのRIP-NP又はNOD-NP実験:a)糖尿病のより高い発生率及びb)より迅速な糖尿病開始のカイネティクスのために、実験群はより小さくなり、1年当たりより多くの実験を実施できる。各実験群につき15匹のマウス、6−8実験群、1年当たり4−5実験。
図3に示したように(図面の簡単な説明を参照されたい)、有糸分裂非誘発性抗CD3F(ab')2(LCMV感染後15日目から20日目)で処置後、約40%のRIP-LCMV-GPマウスが防御される。hGAD65(真核細胞発現ベクター、pCMVを使用)単独処置は糖尿病からの部分的回復(40%防御マウス)をもたらし、同様な結果が以前に、RIP-LCMV-NPマウスの治療のためにブタインシュリンB発現プラスミド(pCMV/insB)を単独で用いたときにも得られた(Coon et al. J. Clin. Invest. 1999, 104:189-94)。抗CD3及びpCMV/hGAD65処置の併用は、RIP-LCMV-GPモデルで強力な相乗効果を示す(80%のマウスが防御された)。
NODマウスで観察されたように(図1及び2)、抗CD3と抗原特異的治療との相乗効果は、最初の抗CD3F(ab')2の注射が治療ウインドウ内(血糖[BG]値が250と500mg/dLの間)で投与されたときにのみ観察された(図3)。興味深いことには、BGが500mmg/dLを超えたとき(治療ウインドウ外)にマウスを処置した場合、それらマウスはそれ以降ずっと糖尿病を維持した。対照的に、BG値が250mg/dLより低い前糖尿病期に抗CD3を注射したとき、前記マウスは決して糖尿病にはならなかった。(これは糖尿病防御と等価であろうが、新規開始試験ではない)。この結論は少なくとも2つの主要な未解決の問題を提起した:(1)そのような治療ウインドウを規定するためのヒトでの最良のマーカー(又はマーカーの組合せ)は何であるか、及び(2)そのようなウインドウは疾患経過中のいつ発生するか。理論に拘束されないが、本発明の場合、新規開始糖尿病患者はこの併用治療の治療的介在のための最良の最初の標的群でありえる。これらの実験を更に拡大するために、RIP-LCMV-NPモデルを用いて、抗CD3とインシュリン(ペプチド又はDNAワクチンとして)間の相乗的潜在能力を調べることができる。
抗CD3全身療法と抗原特異的免疫との併用は、NOD及びRIP-LCMVマウスモデルでの新規開始1型糖尿病(T1D)の治療で強力な相乗効果を示すことができる。
T1Dの治療を目的とする抗原特異的な治療介在の数年後、一般的な結論は、そのような治療は動物モデルでは初期に(前糖尿病期に)与えなければならないということである。これらの治療的介在は予防試験では有効であるかもしれないが、新規開始試験では有効ではない(前記は初期のヒトの治療のためにより安全な時間枠である)。これらの実験は、FcR非結合抗CD3mAbは、抗体と一緒に投与される糖尿病抗原に対する免疫応答を変化させるか否かを試験した。更に、自己免疫及びアナフィラキシー反応の悪化が、MSのヒトで変異ペプチドリガンドの投与後に(ただし糖尿病試験ではそうではない)(Neurocrine, 公開情報及び私的な意見交換;Bielekova et al. Nat. Med. 2000, 6:1167-75;Kppos et al. Nat. Med. 2000, 6:1176-82)、マウスモデルのインシュリンB鎖ペプチド(Liu et al. J. Clin. Invest. 2002, 110:1021-7;Pedotti et al. BMC Immunol. 2003, 4:2)と同様に観察された。理論に拘束されないが、抗CD3mAbによって引き起こされる免疫調節は自己攻撃性応答を回避し、このようにして有効性及び安全性を高めるであろう。したがって、これらの実験では、治療の有用性、最適タイミング及び抗原と併用される抗CD3mAbの安全性を確立するために、有効性と種々の治療パラメーターが試験される。
抗CD3全身療法と抗原特異的免疫との併用は、NOD及びRIP-LCMVマウスモデルでの新規開始1型糖尿病(T1D)の治療で強力な相乗効果を示すことができる。
T1Dの治療を目的とする抗原特異的な治療介在の数年後、一般的な結論は、そのような治療は動物モデルでは初期に(前糖尿病期に)与えなければならないということである。これらの治療的介在は予防試験では有効であるかもしれないが、新規開始試験では有効ではない(前記は初期のヒトの治療のためにより安全な時間枠である)。これらの実験は、FcR非結合抗CD3mAbは、抗体と一緒に投与される糖尿病抗原に対する免疫応答を変化させるか否かを試験した。更に、自己免疫及びアナフィラキシー反応の悪化が、MSのヒトで変異ペプチドリガンドの投与後に(ただし糖尿病試験ではそうではない)(Neurocrine, 公開情報及び私的な意見交換;Bielekova et al. Nat. Med. 2000, 6:1167-75;Kppos et al. Nat. Med. 2000, 6:1176-82)、マウスモデルのインシュリンB鎖ペプチド(Liu et al. J. Clin. Invest. 2002, 110:1021-7;Pedotti et al. BMC Immunol. 2003, 4:2)と同様に観察された。理論に拘束されないが、抗CD3mAbによって引き起こされる免疫調節は自己攻撃性応答を回避し、このようにして有効性及び安全性を高めるであろう。したがって、これらの実験では、治療の有用性、最適タイミング及び抗原と併用される抗CD3mAbの安全性を確立するために、有効性と種々の治療パラメーターが試験される。
方法及び実験プラン:
T1DMの動物モデル:前臨床実験では、例えば1型糖尿病のためには2つの動物モデル、ウイルス誘発自己免疫糖尿病のためのRIP-LCMVモデル及び偶発的疾患のためのNOD(非肥満糖尿病)マウスモデルを用いることができる。目的は、類似性と相違点を見つけ出し、併用療法の難しさに対して良好な印象を得るために、2つの別個のモデル系での治療の有効性を比較することである。これは特に重要である。なぜならば、ヒトの前糖尿病は遺伝的に不均一であり、それらのβ細胞破壊及び自己攻撃応答について別個の根幹的な免疫学的原因を有する可能性があるからである。ウイルス抗原モデルを使用する更に別の理由は、前記モデルは、自己攻撃性リンパ球は容易に追跡することができ(NODでの偶発的自己免疫では困難であろう)、自己免疫反応を開始させる時点は実験的に選択することができる(非糖尿病期、前糖尿病期及び新規糖尿病期の確実な予測及び比較を可能にする)ということである。更にまた、RIP-LCMVマウスは、1型糖尿病の多くの特徴、例えば自己反応性CD4及びCD8リンパ球の中心的関与、APC活性化、臨床症状に先行するインシュリン及びGADに対する自己抗体及び遺伝的因子への依存を示す。
T1DMの動物モデル:前臨床実験では、例えば1型糖尿病のためには2つの動物モデル、ウイルス誘発自己免疫糖尿病のためのRIP-LCMVモデル及び偶発的疾患のためのNOD(非肥満糖尿病)マウスモデルを用いることができる。目的は、類似性と相違点を見つけ出し、併用療法の難しさに対して良好な印象を得るために、2つの別個のモデル系での治療の有効性を比較することである。これは特に重要である。なぜならば、ヒトの前糖尿病は遺伝的に不均一であり、それらのβ細胞破壊及び自己攻撃応答について別個の根幹的な免疫学的原因を有する可能性があるからである。ウイルス抗原モデルを使用する更に別の理由は、前記モデルは、自己攻撃性リンパ球は容易に追跡することができ(NODでの偶発的自己免疫では困難であろう)、自己免疫反応を開始させる時点は実験的に選択することができる(非糖尿病期、前糖尿病期及び新規糖尿病期の確実な予測及び比較を可能にする)ということである。更にまた、RIP-LCMVマウスは、1型糖尿病の多くの特徴、例えば自己反応性CD4及びCD8リンパ球の中心的関与、APC活性化、臨床症状に先行するインシュリン及びGADに対する自己抗体及び遺伝的因子への依存を示す。
マウス及び試薬:RIP-LCMV(6-10週齢、通常LCMV感染(105pfu腹腔内)後11−16日以内にT1Dを発症する)及びNODマウスを、高血糖(血糖>250mg/dL)の開始後0、1、2、3及び4日目にIgG2a抗CD3(Ala-Ala)で処置することができる。F(ab')2抗CD3mAb(前記は非常に類似の特性を有する)を用いる予備実験では、本発明は、100μg/注射(新規開始糖尿病の後に連続して5日間)は、処置されたRIP-LCMV又はNODマウスの約50%を防御することを示した(図1C)。同様な実験を抗CD3mAbのこの形態(IgG2a抗CD3(Ala-Ala))及び他の形態を用いて実施し、前記の発見を再生することができる。理論に拘束されないが、本発明で提供されることは、抗CD3の処置は、防御されなかった残りのマウス(約50%)で抗原特異的療法のための“扉を開け”、それによって防御マウスの数を増加させるであろうということである(これは図3の結果によって支持される)。これは、併用アプローチの研究のために最適な応答率基準ラインである。なぜならば、併用によって惹起されるいずれの小さな相違も明瞭となるからである。更にまた、実験系は、mAb処置後2年の臨床状況と相関性を有し、前記臨床状況では薬剤処置患者の24%がMMTT応答を有していた(これは基準ラインの80%で、前記に対しコントロールでは11%であった)。
マウス(各群に8−10匹)を14の別個の群に分け、前記は、例えば(i)抗CD3単独、(ii)抗原特異的療法単独、又は(iii)抗原特異的療法と併用した抗CD3を受ける。
群1:抗CD3mAb単独(10μg、静注)、高血糖開始後0、1、2、3及び4日目。
群2:抗CD3mAbを抗原(下記のリスト参照)と併用、前記抗原は高血糖の開始後抗CD3mABと一緒に開始される。これらの実験によって、抗CD3mAbを抗原と一緒に投与することの影響が調べられる。抗CD3mAbの投与に続いて、循環T細胞の数が減少し、おそらくは血管区画からの活性化T細胞の退出の反映であり、全T細胞のアポトーシスを反映しているとは思われない。したがって、数は減少するが、残留するT細胞は、抗CD3mAbと同じときに投与された抗原に対する応答の改変に役立ちえる。
群3:抗原との抗CD3mAbの併用は、抗CD3mAbの10日後に開始した。これらのマウスでは、抗CD3mAbによる処置の後T細胞の回復があるだろう。この群では、抗CD3mAb処置の持続効果が試験される。
群4:抗原単独処置(下記リスト参照)が高血糖の開始直後に開始される。
群5:抗原単独処置(下記リスト参照)が高血糖開始の10日後に開始される。
群6:未処置(PBS注射)NOD又はRIP-LCMV。
抗原特異的処置のコントロールとして、pCMV単独及び無関係のペプチド又はタンパク質単独処置を用いることができる。血糖は、前糖尿病期の間および処置の最初の週には週に2回、その後は週毎に評価することができる。
群1:抗CD3mAb単独(10μg、静注)、高血糖開始後0、1、2、3及び4日目。
群2:抗CD3mAbを抗原(下記のリスト参照)と併用、前記抗原は高血糖の開始後抗CD3mABと一緒に開始される。これらの実験によって、抗CD3mAbを抗原と一緒に投与することの影響が調べられる。抗CD3mAbの投与に続いて、循環T細胞の数が減少し、おそらくは血管区画からの活性化T細胞の退出の反映であり、全T細胞のアポトーシスを反映しているとは思われない。したがって、数は減少するが、残留するT細胞は、抗CD3mAbと同じときに投与された抗原に対する応答の改変に役立ちえる。
群3:抗原との抗CD3mAbの併用は、抗CD3mAbの10日後に開始した。これらのマウスでは、抗CD3mAbによる処置の後T細胞の回復があるだろう。この群では、抗CD3mAb処置の持続効果が試験される。
群4:抗原単独処置(下記リスト参照)が高血糖の開始直後に開始される。
群5:抗原単独処置(下記リスト参照)が高血糖開始の10日後に開始される。
群6:未処置(PBS注射)NOD又はRIP-LCMV。
抗原特異的処置のコントロールとして、pCMV単独及び無関係のペプチド又はタンパク質単独処置を用いることができる。血糖は、前糖尿病期の間および処置の最初の週には週に2回、その後は週毎に評価することができる。
抗CD3抗体:3つの別個の供給源由来の抗CD3を、例えば相違点及び類似点を検出するために試験することができる。これは、マウスでの発見をヒトでの状況に移すために重要である。この場合、抗CD3-Ala-Alaが用いられている。
抗CD3-Ala-Ala:この抗体は、ヒトと比較してマウスで類似の機能を示し、変異したFc-結合領域を有する。前記は有糸分裂非誘発性であるが、T細胞でシグナリングを誘発しない。
抗CD3-IgG3:この抗体はヒトと比較してマウスで同様な機能を示し、変異したFc-結合領域を有するので、Ala-Ala型と類似する。前記は有糸分裂非誘発性であるが、T細胞でシグナリングを誘発する。
抗CD3Fab'2(BioSourceから市販されている):この抗体はマウス2C11に由来し、FcR非結合性である。前記は有糸分裂非誘発性であるが、T細胞でシグナリングを誘発する。
抗CD3-Ala-Ala:この抗体は、ヒトと比較してマウスで類似の機能を示し、変異したFc-結合領域を有する。前記は有糸分裂非誘発性であるが、T細胞でシグナリングを誘発しない。
抗CD3-IgG3:この抗体はヒトと比較してマウスで同様な機能を示し、変異したFc-結合領域を有するので、Ala-Ala型と類似する。前記は有糸分裂非誘発性であるが、T細胞でシグナリングを誘発する。
抗CD3Fab'2(BioSourceから市販されている):この抗体はマウス2C11に由来し、FcR非結合性である。前記は有糸分裂非誘発性であるが、T細胞でシグナリングを誘発する。
抗CD3mAbとの相乗性が評価されるべき抗原投与スケジュール:調節細胞を誘発することが示され、更に動物モデルで糖尿病を予防した種々の島自己抗原をテストすることができる。種々の投与ルートも同様にテストすることができる。最終的には、本発明のゴールの1つは、抗CD3と最適な相乗性を示す下記リストの候補物質を特定し、続いて臨床試験のために前記抗原に焦点を合わせることである(実施例3参照)。
鼻内投与ヒトインシュリン及びヒトインシュリン類似体X38(10μLのPBS中に0.05mg/用量)、(0、3、7、12日目)。ノヴォノルディスク(NovoNordisk)から入手。特に興味深いことは、前記のインシュリン類似体X38は1000倍低いインシュリンレセプター結合能を有するということである。
ブタインシュリン-B鎖(100μL中に5mg/kg、皮下注射)、(0、3、7、10、15日目)。ノヴォノルディスク(Novo Nordisk, Bagsvaerd, DK)から入手(Liu et al. J. Clin. Invest. 2002, 110:1021-7)。より吸収が遅く、更にインシュリンB9-23ペプチド単独で認められたアナフィラキシーを誘発しない改変ペプチドが生成された。この改変ペプチドも同様にテストすることができる。
ブタインシュリン-B鎖APL(改変ペプチドリガンド)(100μL中に5mg/kg、皮下注射)、(0、3、7、10、15日目)は、ニューロクライン(Neurocrine, San Diego, CA, USA)から市場で入手される(Alleva et al. Diabetes 2002, 51:2126-34)。
ヒトGAD65タンパク質(hGAD65)(300μLのPBS中に100μg、皮下注射、ディアミド(Diamyd, Stockholm, Sweden)から市場で入手される)、(0、1、7、12日目)。
CTLエピトープを含まないマウスプロインシュリンIIペプチド(プロインシュリンペプチド)単独(10μLのPBS中に40μg、鼻内投与)、(0、3、7、12日目)。オースペップ(Auspep, Melbourne, Australia, 43)から入手される。
ブタインシュリンB鎖又はヒトGADを発現するpCMVベクターによるDNAワクチン:(100mLのPBS中に100mg、筋肉内注射)、0、3、7、12日目に投与。これらは、内毒素非含有条件下で製造される。調節細胞はそのようなDNAワクチンで誘発できる。(Coon et al. J. Clin. Invest. 1999, 104:189-94)。
HSP60ペプチド:HSP60ペプチドは、ヒト及びマウスの糖尿病で重要な抗原であると提唱された。HSP60によるワクチン接種は、マルチドースのストレプトゾトシンでNODマウスに誘発される自己免疫糖尿病を予防することが示された(Elias et al. Proc. Natl. Acad. Sci. USA 1991, 88:3088-91.54;Elias et al. Diabetes 1994, 43:992-8;Birk et al. J. Autoimmun. 1996, 9:159-66)。実際、新規開始糖尿病の患者での臨床試験によって、臨床効果が示された(Raz et al. Lancet 2001, 358:1749-53)。
鼻内投与ヒトインシュリン及びヒトインシュリン類似体X38(10μLのPBS中に0.05mg/用量)、(0、3、7、12日目)。ノヴォノルディスク(NovoNordisk)から入手。特に興味深いことは、前記のインシュリン類似体X38は1000倍低いインシュリンレセプター結合能を有するということである。
ブタインシュリン-B鎖(100μL中に5mg/kg、皮下注射)、(0、3、7、10、15日目)。ノヴォノルディスク(Novo Nordisk, Bagsvaerd, DK)から入手(Liu et al. J. Clin. Invest. 2002, 110:1021-7)。より吸収が遅く、更にインシュリンB9-23ペプチド単独で認められたアナフィラキシーを誘発しない改変ペプチドが生成された。この改変ペプチドも同様にテストすることができる。
ブタインシュリン-B鎖APL(改変ペプチドリガンド)(100μL中に5mg/kg、皮下注射)、(0、3、7、10、15日目)は、ニューロクライン(Neurocrine, San Diego, CA, USA)から市場で入手される(Alleva et al. Diabetes 2002, 51:2126-34)。
ヒトGAD65タンパク質(hGAD65)(300μLのPBS中に100μg、皮下注射、ディアミド(Diamyd, Stockholm, Sweden)から市場で入手される)、(0、1、7、12日目)。
CTLエピトープを含まないマウスプロインシュリンIIペプチド(プロインシュリンペプチド)単独(10μLのPBS中に40μg、鼻内投与)、(0、3、7、12日目)。オースペップ(Auspep, Melbourne, Australia, 43)から入手される。
ブタインシュリンB鎖又はヒトGADを発現するpCMVベクターによるDNAワクチン:(100mLのPBS中に100mg、筋肉内注射)、0、3、7、12日目に投与。これらは、内毒素非含有条件下で製造される。調節細胞はそのようなDNAワクチンで誘発できる。(Coon et al. J. Clin. Invest. 1999, 104:189-94)。
HSP60ペプチド:HSP60ペプチドは、ヒト及びマウスの糖尿病で重要な抗原であると提唱された。HSP60によるワクチン接種は、マルチドースのストレプトゾトシンでNODマウスに誘発される自己免疫糖尿病を予防することが示された(Elias et al. Proc. Natl. Acad. Sci. USA 1991, 88:3088-91.54;Elias et al. Diabetes 1994, 43:992-8;Birk et al. J. Autoimmun. 1996, 9:159-66)。実際、新規開始糖尿病の患者での臨床試験によって、臨床効果が示された(Raz et al. Lancet 2001, 358:1749-53)。
全ての抗原は、以前に証明された(糖尿病期に与えられたときの)それらの動物モデルでの有効性、及びT1DMの患者での抗原特異的臨床試験について考慮中であるという事実を基準に選択された。上記に特記したように、抗原は、高血糖症の開始後又は開始の10日後に投与することができる。糖尿病の回復に対する処置の効果はグルコースレベルの測定及びインシュリン判定によって調べることができる。更に、リアルタイムPCRを種々の群のマウスから単離した島で実施し、投与処置プログラムが島におけるサイトカインの発現を変化させたか否かを決定した。
したがって、NOD及びRIP-LCMVマウスモデルを併用処置で用いることによって、抗CD3及び島特異的抗原投与の強力な相乗作用が予想され、最適な抗原製剤を特定することができる。
結果は、“治療ウインドウ”の概念はT1Dのための治療有効性の達成に重要でありえることを示した(図1C)。NODモデルは、(より遅い疾患経過のために)比較的“広い”治療ウインドウを示したが、RIP-LCMVモデルにおける同様なウインドウは、(LCMV感染後の急速で強力な膵の破壊のために)より狭く、決定がより困難である。1つの可能性は、感染のためにより少量のウイルスを用いるか、又はマウスを出生後6週ではなく8−12週で感染させることによってRIP-LCMVモデルでこのウインドウを広げることである(両方法はT1Dを遅くし、膵臓の攻撃を減らし、必要な場合には、より広い治療ウインドウを作るはずである)。
したがって、上記の実験は、最適な抗原-抗CD3の組合せ及びタイミングを特定し、ベータ細胞の破壊の加速が2つの別個のモデルで生じないことを担保することができる。データから、プロインシュリンはもっとも有望な候補物質であるように思われ、最近、前糖尿病個体におけるTh1様応答に対して正常個体のTH2様応答を示す、プロインシュリンの特異的な応答の追跡がヒトで報告されたので、前記はまた良い選択でもあろう。実際、抗原特異的応答を追跡できることによって、臨床試験のための更に別のガイドラインが提供されるであろう(実施例3参照)。
下記の表は、実施例で用いられる抗CD3及び抗原のいくつかの入手場所のリストを提供する。
したがって、NOD及びRIP-LCMVマウスモデルを併用処置で用いることによって、抗CD3及び島特異的抗原投与の強力な相乗作用が予想され、最適な抗原製剤を特定することができる。
結果は、“治療ウインドウ”の概念はT1Dのための治療有効性の達成に重要でありえることを示した(図1C)。NODモデルは、(より遅い疾患経過のために)比較的“広い”治療ウインドウを示したが、RIP-LCMVモデルにおける同様なウインドウは、(LCMV感染後の急速で強力な膵の破壊のために)より狭く、決定がより困難である。1つの可能性は、感染のためにより少量のウイルスを用いるか、又はマウスを出生後6週ではなく8−12週で感染させることによってRIP-LCMVモデルでこのウインドウを広げることである(両方法はT1Dを遅くし、膵臓の攻撃を減らし、必要な場合には、より広い治療ウインドウを作るはずである)。
したがって、上記の実験は、最適な抗原-抗CD3の組合せ及びタイミングを特定し、ベータ細胞の破壊の加速が2つの別個のモデルで生じないことを担保することができる。データから、プロインシュリンはもっとも有望な候補物質であるように思われ、最近、前糖尿病個体におけるTh1様応答に対して正常個体のTH2様応答を示す、プロインシュリンの特異的な応答の追跡がヒトで報告されたので、前記はまた良い選択でもあろう。実際、抗原特異的応答を追跡できることによって、臨床試験のための更に別のガイドラインが提供されるであろう(実施例3参照)。
下記の表は、実施例で用いられる抗CD3及び抗原のいくつかの入手場所のリストを提供する。
表2:抗体及び抗原試薬の例示的供給源
実施例2:抗CD3及び自己抗原によって誘発されたT調節細胞は自己免疫治療に用いることができる
実施例1のデータは、抗CD3と自己抗原の併用投与は、寛容の誘発及び自己免疫に対する防御で相乗作用を有することを示す。これらの結果を足場として、本発明は、自己抗原及び抗CD3処置に暴露されたT調節細胞を単離及び/又はin vitroで増補し、自己免疫療法のためにそれら細胞を使用することができる方法を意図する。ヒトでの臨床試験の前に前臨床試験を実施し、特定されるいずれの併用療法(すなわち抗CD3のいずれの特定の形態といずれの特定の自己抗原との併用)が調節T細胞(Treg)及び/又は全身性免疫偏向を誘発するか否かを決定することができる。
抗CD3処置又は抗CD3+抗原処置RIP-LCMVドナー由来のリンパ球を用いる予備的な養子細胞移入実験(下記表3参照)では、抗CD3+抗原処置RIP-LCMVマウス由来のCD4+細胞のみが、同系の前糖尿病非免疫抑制ホストを糖尿病の進行から防御することができた。抗CD3のみで処置されたマウス由来の細胞は防御を仲介することができなかった。この実験は、抗原投与後のT調節細胞の誘発は抗CD3及び抗原処置マウスで効果的に生じることを示している。Chatenoud(彼は、抗CD3処置NODドナーマウス由来のリンパ球でT調節活性を検出するために免疫欠損レシピエントを用いた)による以前の観察とは対照的に、免疫コンピテントマウスでのそのような調節はより激しいT調節誘発に左右されえる。
実施例1のデータは、抗CD3と自己抗原の併用投与は、寛容の誘発及び自己免疫に対する防御で相乗作用を有することを示す。これらの結果を足場として、本発明は、自己抗原及び抗CD3処置に暴露されたT調節細胞を単離及び/又はin vitroで増補し、自己免疫療法のためにそれら細胞を使用することができる方法を意図する。ヒトでの臨床試験の前に前臨床試験を実施し、特定されるいずれの併用療法(すなわち抗CD3のいずれの特定の形態といずれの特定の自己抗原との併用)が調節T細胞(Treg)及び/又は全身性免疫偏向を誘発するか否かを決定することができる。
抗CD3処置又は抗CD3+抗原処置RIP-LCMVドナー由来のリンパ球を用いる予備的な養子細胞移入実験(下記表3参照)では、抗CD3+抗原処置RIP-LCMVマウス由来のCD4+細胞のみが、同系の前糖尿病非免疫抑制ホストを糖尿病の進行から防御することができた。抗CD3のみで処置されたマウス由来の細胞は防御を仲介することができなかった。この実験は、抗原投与後のT調節細胞の誘発は抗CD3及び抗原処置マウスで効果的に生じることを示している。Chatenoud(彼は、抗CD3処置NODドナーマウス由来のリンパ球でT調節活性を検出するために免疫欠損レシピエントを用いた)による以前の観察とは対照的に、免疫コンピテントマウスでのそのような調節はより激しいT調節誘発に左右されえる。
抗CD3mAb及び抗原によるT調節細胞の誘発:抗原特異的T調節細胞のin vitro増補及びそれら細胞の1型糖尿病モデルへの再導入によって疾患を予防することができる。以前の公表された研究及び未公表研究の結果は下記の表に要約されている。簡単に記せば、抗CD3とともに、又は抗CD3無しに島抗原特異的免疫を施されたNODマウス又はRIP-LCMVマウスから単離したT調節細胞を、IL-2(及びいくつかの実験ではIL-4)の存在下で、更に表示のようにいくつかの事例ではインシュリンB鎖(RIP-LCMV実験)又はBFDC2.5もしくはGADペプチドを提示するI-Ag7テトラマー担持ビーズの存在下でin vitroで刺激した。in vitroでの増補に続いて、これらの細胞株を同系の前糖尿病RIP-LCMV又はNODレシピエントに再導入し、1型糖尿病の発症をモニターした。
表3:T調節細胞実験の要約
*in vitroでの抗原刺激が無い場合は、抗原刺激有り(ほんの2x105細胞が必要)と比較してより多くの細胞が防御のために移入されねばならないことに留意されたい(刺激無しの場合5x106細胞が必要)。
以下の実験は、どのようにしてT調節細胞をそれらが産生した生成物によって追跡するかを、テトラマー及びELISPOTを用いてそれら細胞を直接特定することによって追跡する方法とともに述べる。
特に、T調節細胞は、例えばそれらが産生するサイトカインによって追跡することができる。予備的実験結果は、T調節細胞は、IL-10及びIL-4、又はIL-10、IL-4、IFN-γおよびTGF-βを含むことができるサイトカイン産生プロフィルを有することを示した。特に、IL-10は、T-調節細胞によって産生される主要なサイトカインであろう。更に、TGF-βもまた非常に重要でありえよう(5)。両方の事例で、in vitro刺激はT調節細胞機能を強化することができ、更にIL-2の存在は必須のようである。
特に、T調節細胞は、例えばそれらが産生するサイトカインによって追跡することができる。予備的実験結果は、T調節細胞は、IL-10及びIL-4、又はIL-10、IL-4、IFN-γおよびTGF-βを含むことができるサイトカイン産生プロフィルを有することを示した。特に、IL-10は、T-調節細胞によって産生される主要なサイトカインであろう。更に、TGF-βもまた非常に重要でありえよう(5)。両方の事例で、in vitro刺激はT調節細胞機能を強化することができ、更にIL-2の存在は必須のようである。
方法
抗CD3(+/-抗原)後の全身性免疫偏向の判定:血清中の全身性サイトカインレベルはELISAによって判定することができる。ポリクローナル態様で生じるサイトカインの産生は、仕分けしたリンパ球集団(膵臓、PDLN、脾臓及びPBMC)をELISPOTアッセイ及び、前記に加えin vitroポリクローナル刺激(抗CD3/CD28及びSEB)に暴露することによってex vivoで直接判定することができる。これらの実験を最初マウスで実施したとき、それら実験は、抗CD3の後で観察された全身的なサイトカインのシフトに関してヒトの臨床的研究に対する類似点を引き出すことを可能にした。
調節T細胞の追跡と分析:防御マウス由来の調節T細胞を追跡し、分析し、レシピエントマウスに移入し、最後に併用療法後の全般的な免疫の偏向及びサイトカインプロフィルを単一療法と比較することができる。以下の技術を用いて調節T細胞を追跡及び分析することができる。ドナー及びレシピエントのT調節細胞の特定のために、本発明では、抗原特異性、サイトカイン産生とともに、フローサイトメトリー及びウェスタンブロット分析によるマーカーとしてCD25+、FoxP3及びGITRの発現を用いることができる。これらの全てが移入前の細胞のソーティングに適しているわけではないが(下記参照)、それにもかかわらず前記は、T調節細胞のプロフィルにおける系統的変化の判定に非常に有用であろう。
抗CD3(+/-抗原)後の全身性免疫偏向の判定:血清中の全身性サイトカインレベルはELISAによって判定することができる。ポリクローナル態様で生じるサイトカインの産生は、仕分けしたリンパ球集団(膵臓、PDLN、脾臓及びPBMC)をELISPOTアッセイ及び、前記に加えin vitroポリクローナル刺激(抗CD3/CD28及びSEB)に暴露することによってex vivoで直接判定することができる。これらの実験を最初マウスで実施したとき、それら実験は、抗CD3の後で観察された全身的なサイトカインのシフトに関してヒトの臨床的研究に対する類似点を引き出すことを可能にした。
調節T細胞の追跡と分析:防御マウス由来の調節T細胞を追跡し、分析し、レシピエントマウスに移入し、最後に併用療法後の全般的な免疫の偏向及びサイトカインプロフィルを単一療法と比較することができる。以下の技術を用いて調節T細胞を追跡及び分析することができる。ドナー及びレシピエントのT調節細胞の特定のために、本発明では、抗原特異性、サイトカイン産生とともに、フローサイトメトリー及びウェスタンブロット分析によるマーカーとしてCD25+、FoxP3及びGITRの発現を用いることができる。これらの全てが移入前の細胞のソーティングに適しているわけではないが(下記参照)、それにもかかわらず前記は、T調節細胞のプロフィルにおける系統的変化の判定に非常に有用であろう。
調節リンパ球の存在を判定するための養子細胞移入:in vivoでの自己免疫抑制活性を有する与えられた器官由来のリンパ球分画を、既に報告されたように(Homann et al. Immunity 2006, 16:403-15;Homann et al. Immunity 1999, 11:463-72)、養子細胞移入及び細胞ソーティングによって評価することができる。簡単に記せば、CD4リンパ球の養子細胞移入は、非免疫抑制、非操作(非照射)前糖尿病同系レシピエントで疾患を予防した。前記の方法は、免疫学的に“エンプティー”の環境ではホメオスタシス作用及び細胞の増補は生じないので、in vivoで調節T細胞の機能及び存在を調べるための現実的、定量的及び信頼に足る方法である。重要なことには、移入直前の表現型決定後のソーティングによって極めて重要な亜集団を特定することができる。
以下の表現型をT調節細胞のソーティングに用いることができる:(a)移入時の細胞表面マーカー:CD25+、CD4+、CD62L(MACSソート)又は(b)T調節細胞が必要とするサイトカイン:IL-2、4、10、IFN-γ(ミルテニー(Miltenyi)ビーズソート)。
更に、in vitro培養は、抗原特異的サブセットの増幅を可能にする。養子移入はRIP-LCMV及びNODモデルを用いて実施することができる。防御マウス(抗CD3又は島抗原の単独又は併用処置)由来の脾細胞及び膵ドレインリンパ節(PDLN)を用いて、仮説的調節細胞を収集することができる。最初に、脾細胞又はPDLN細胞のプールを用いて、非照射同系レシピエントに移入(NODモデルの場合は、6から8週齢マウスの腹腔内又は静脈内注射、RIP-LCMVモデルの場合はLCMV感染後5−6日目)することができる。第二の実験セットでは、潜在的な調節T細胞は、T細胞集団の特別な精製により追跡することができる(CD4、CD25及びCD62細胞表面マーカーの発現にしたがい、例えば磁性ビーズ又はFACS-ヴァンテージ技術による精製)。これらの亜集団を上記のレシピエントに注射して、T調節細胞又は自己免疫の存在を判定することができる。
以下の表現型をT調節細胞のソーティングに用いることができる:(a)移入時の細胞表面マーカー:CD25+、CD4+、CD62L(MACSソート)又は(b)T調節細胞が必要とするサイトカイン:IL-2、4、10、IFN-γ(ミルテニー(Miltenyi)ビーズソート)。
更に、in vitro培養は、抗原特異的サブセットの増幅を可能にする。養子移入はRIP-LCMV及びNODモデルを用いて実施することができる。防御マウス(抗CD3又は島抗原の単独又は併用処置)由来の脾細胞及び膵ドレインリンパ節(PDLN)を用いて、仮説的調節細胞を収集することができる。最初に、脾細胞又はPDLN細胞のプールを用いて、非照射同系レシピエントに移入(NODモデルの場合は、6から8週齢マウスの腹腔内又は静脈内注射、RIP-LCMVモデルの場合はLCMV感染後5−6日目)することができる。第二の実験セットでは、潜在的な調節T細胞は、T細胞集団の特別な精製により追跡することができる(CD4、CD25及びCD62細胞表面マーカーの発現にしたがい、例えば磁性ビーズ又はFACS-ヴァンテージ技術による精製)。これらの亜集団を上記のレシピエントに注射して、T調節細胞又は自己免疫の存在を判定することができる。
類リンパ器官及びPBMC中の抗原特異的攻撃性及び調節性T細胞の追跡:以下のアプローチを用いて、LCMV NP-、GP-及びGAD、プロインシュリン並びにinsB-特異的CD4及びCD8リンパ球をRIP-LCMVマウス及びNODモデルで検出することができる。調節T細胞は、いくつかのT細胞供給源(島及び膵浸潤リンパ球、脾細胞並びにリンパ節細胞)を用いることによって、これら亜集団で追跡することができる。
a)ペプチド:LCMV-NPトランスジーン由来のエピトープはドミナントなDb-拘束NP396-404(アミノ酸FQPQNGQFI(配列番号:1))及びサブドミナントなKb NP314-322((W)PIACRSTI(配列番号:2))である。NP CD8+ T細胞によって認識される他のウイルスエピトープはDb GP33-41(KAVYNFATC(配列番号:3))、Db GP276-286(SGVENPGGYCL(配列番号:4))及びKd GP283-291(GYCLTKWMI(配列番号:5))であり、コントロールとして用いられる。更にまた、Kd INS-B15-23(LYLVCGERG(配列番号:6))、ミモトープKd NRP-A7(KYNKANAFL(配列番号:7))同様I-Ag7-拘束INS-B9-23(SHLVEALYLVCGERG(配列番号:8))、及びインシュリンC13-A5同様GADタンパク質(Diamyd)もまた用いることができる。これらのペプチドはまたELISPOT及び増殖アッセイで利用することができる。測定することができるサイトカインはIFN-γ、IL-10、TNF-α、IL-4及びIL-10である(ELISPOTのセクションを参照されたい)。
in situテトラマー染色:これはもっとも侵襲性が少ないアプローチで、組織切片上でCD8細胞を直接ex vivoで検出することを可能にする。このアプローチは確立され、例は図4に提供されている。フローサイトメトリー又はin situ染色によるβ-細胞抗原特異的CD8+ T細胞の検出のために、以下のフィコエリトリン-(PE)又はアロフィコシアニン-(APC)結合テトラマーを用いることができる:DbNP396-404、KdINS-B15-23及びKdNRP-A7(ミモトープ)。LCMV GP-特異的エピトープ(DbGP33-41及びKdGP283-291)を認識するテトラマーはコントロールとして機能することができる。
a)ペプチド:LCMV-NPトランスジーン由来のエピトープはドミナントなDb-拘束NP396-404(アミノ酸FQPQNGQFI(配列番号:1))及びサブドミナントなKb NP314-322((W)PIACRSTI(配列番号:2))である。NP CD8+ T細胞によって認識される他のウイルスエピトープはDb GP33-41(KAVYNFATC(配列番号:3))、Db GP276-286(SGVENPGGYCL(配列番号:4))及びKd GP283-291(GYCLTKWMI(配列番号:5))であり、コントロールとして用いられる。更にまた、Kd INS-B15-23(LYLVCGERG(配列番号:6))、ミモトープKd NRP-A7(KYNKANAFL(配列番号:7))同様I-Ag7-拘束INS-B9-23(SHLVEALYLVCGERG(配列番号:8))、及びインシュリンC13-A5同様GADタンパク質(Diamyd)もまた用いることができる。これらのペプチドはまたELISPOT及び増殖アッセイで利用することができる。測定することができるサイトカインはIFN-γ、IL-10、TNF-α、IL-4及びIL-10である(ELISPOTのセクションを参照されたい)。
in situテトラマー染色:これはもっとも侵襲性が少ないアプローチで、組織切片上でCD8細胞を直接ex vivoで検出することを可能にする。このアプローチは確立され、例は図4に提供されている。フローサイトメトリー又はin situ染色によるβ-細胞抗原特異的CD8+ T細胞の検出のために、以下のフィコエリトリン-(PE)又はアロフィコシアニン-(APC)結合テトラマーを用いることができる:DbNP396-404、KdINS-B15-23及びKdNRP-A7(ミモトープ)。LCMV GP-特異的エピトープ(DbGP33-41及びKdGP283-291)を認識するテトラマーはコントロールとして機能することができる。
b)テトラマー及びFACS細胞内サイトカイン染色:MHCクラスI又はII両分子のためのテトラマーが利用できる。上記に記載したように、種々の起源(PBL、脾細胞、ドレインリンパ節など)の単一細胞懸濁液を用いて、マルチカラーフローサイトメトリー分析を実施する。in vitroで再刺激した後、CD8、CD4、CD25、CD44、CD62L、CD69など、又は抗原特異的TCRのような選択表面マーカーについて、テトラマー技術(上記に記載)を用いることによって細胞を染色することができる。続いて、固定、透過性付与及びIFN-γ、TNF-α又はIL-4について細胞内染色を実施する。選択した実験について、7つまでの別個の色による染色をFACSヴァンテージで用いることができる。
c)ELISPOTアッセイ:抗原特異的刺激がサイトカイン産生の検出に必要である(IFN-γ、IL-4、IL-10及びIL-5)。しかしながら、このアプローチの強みは、信頼できる結果を得るために極めてわずかの細胞しか必要とされないという事実にある。この技術の実施で良好な成果が得られた(von Herrath et al. J. Immunol. 2002, 168:933-41;Coon et al. J. Clin. Invest. 1999, 104:189-94)。
d)上清中のサイトカインのための増殖及びELISA:この方法は、もっとも多くの細胞及びもっとも強いin vitro刺激を必要とする。分泌されたサイトカインの定量のために、採集された器官の単一細胞懸濁液をペプチドの存在下で培養し、上清を48−72時間後に収集し、IFN-γ、IL-2、IL-4、IL-10及びTGF-βの分析のために-20℃で保存する。
c)ELISPOTアッセイ:抗原特異的刺激がサイトカイン産生の検出に必要である(IFN-γ、IL-4、IL-10及びIL-5)。しかしながら、このアプローチの強みは、信頼できる結果を得るために極めてわずかの細胞しか必要とされないという事実にある。この技術の実施で良好な成果が得られた(von Herrath et al. J. Immunol. 2002, 168:933-41;Coon et al. J. Clin. Invest. 1999, 104:189-94)。
d)上清中のサイトカインのための増殖及びELISA:この方法は、もっとも多くの細胞及びもっとも強いin vitro刺激を必要とする。分泌されたサイトカインの定量のために、採集された器官の単一細胞懸濁液をペプチドの存在下で培養し、上清を48−72時間後に収集し、IFN-γ、IL-2、IL-4、IL-10及びTGF-βの分析のために-20℃で保存する。
上記の技術によって、細胞表面マーカー又はサイトカイン発現に関して、抗CD3及び抗原(例えば単独若しくは併用処置される抗CD3F(ab')2又は島抗原)による種々の処置の間に誘発されたT調節細胞の検出及び性状決定が可能になる。実際、抗CD3又は抗原特異的単独処置と比較して、抗CD3/抗原併用処置によって誘発されたそのようなT調節細胞の表現型に関して利用可能な情報はまだ存在しない。抗原特異的サイトカイン産生細胞のELISPOT(又は利用可能な場合にはMHCクラスIIテトラマーの直接染色)による追跡は、ヒトの臨床試験での同様な追跡の実施の道標として役立ちえる。養子細胞移入はヒトでは可能ではないので、ex vivoで抗原に暴露された、又は暴露されていないときの正確なサイトカインのプロフィルを明らかにすることは必須であろう。理論に拘束されないが、本発明は、抗CD3単独処置は全身的なサイトカインシフトをもたらし、抗原との併用は抗原特異的T調節細胞(サイトカイン例えばIL-10、IL-4及びIL-13を分泌する)の生産をもたらすことを示している。この併用免疫療法を受ける患者の同様なサイトカイン判定を用いて、処置の有効性をモニター/評価/実証することができる。
生じる可能性がある1つの技術的な問題は、T調節細胞はT細胞集団で低率で見出されるということである。しかしながら、感度の高い技術、例えばELISPOT、ELISA及びFACS分析の使用は信頼できる、一定の結果を提供できるはずである。例えば、以前の養子移入実験は、T調節細胞は良好に単離、追跡および分析することができることを示している(Homann et al. Immunity 1999, 11:463-72)。
生じる可能性がある1つの技術的な問題は、T調節細胞はT細胞集団で低率で見出されるということである。しかしながら、感度の高い技術、例えばELISPOT、ELISA及びFACS分析の使用は信頼できる、一定の結果を提供できるはずである。例えば、以前の養子移入実験は、T調節細胞は良好に単離、追跡および分析することができることを示している(Homann et al. Immunity 1999, 11:463-72)。
実施例3:改変抗CD3抗体との同時投与
実施例1のデータは、抗CD3F(ab')2の全身療法と抗原特異的アプローチとの併用は、新規開始T1Dの治療で明瞭な相乗作用を発揮することを示している。いくつかの自己抗原(DNAワクチン、完全タンパク質又はペプチド)が、最大の防御を提供する最良の組合せを決定するために治療的に評価されつつある。したがって、T1Dのためのモデル系(実施例1及び2)を用いて、抗CD3mAb hOKT3γ1(Ala-Ala)で処置されたT1DMの患者で認められる効果に類似する使用を目的とする新規試薬を検査することができる。実施例4で提唱される実験は臨床試験のデザインに関し、前記臨床試験では抗原と抗CD3mABとの組合せによる免疫応答の変化が調べられ、組合せの安全性に関する情報が提供され、更にまた中心的に関与するメカニズムの理解が促進される。
実施例1のデータは、抗CD3F(ab')2の全身療法と抗原特異的アプローチとの併用は、新規開始T1Dの治療で明瞭な相乗作用を発揮することを示している。いくつかの自己抗原(DNAワクチン、完全タンパク質又はペプチド)が、最大の防御を提供する最良の組合せを決定するために治療的に評価されつつある。したがって、T1Dのためのモデル系(実施例1及び2)を用いて、抗CD3mAb hOKT3γ1(Ala-Ala)で処置されたT1DMの患者で認められる効果に類似する使用を目的とする新規試薬を検査することができる。実施例4で提唱される実験は臨床試験のデザインに関し、前記臨床試験では抗原と抗CD3mABとの組合せによる免疫応答の変化が調べられ、組合せの安全性に関する情報が提供され、更にまた中心的に関与するメカニズムの理解が促進される。
実施例4:抗CD3抗体と自己抗原の同時投与による糖尿病治療のための臨床試験計画
以下は、前臨床及び臨床試験によって達成されることを意図した、例示的で具体的な4つのマイルストーンである。
(1)2ヶ所のセンターでの共同前臨床データの再現性:前糖尿病マウスだけでなく新規開始糖尿病マウスも防御されるということは重要である。前糖尿病期に与えたときにはNODマウスを防御する免疫依存的介在処置は多いが、新規開始NODで正常血糖を再確立することができるものはほとんどない。実施例1(図2)では、NODマウス及びLCMVモデルで処置マウスの約50%が開始期の糖尿病を回復させることができるように、抗CD3mAbの投与のための投薬技術が改変された。図1Cのデータは、抗原添加の効果を評価するためのウインドウを提供した。更にまた、前記の効果が維持されない場合(すなわち>3ヶ月)、抗原の反復投与は再発率を低下させるか否かを容易に決定することができる。NOD実験は、臨床試験の進行前に2ヶ所のセンターで別個に確認することができる。
(2)明瞭な抗原候補物質:上記に記載した実験を基にして、糖尿病の回復率を維持するために最適な抗原を選択することができる。2つ又は3つ以上の抗原が、抗CD3との最適な類似する相乗作用を示す場合には、前記抗原はGMP(良好な製造慣行(good manufacturing practice))製剤でもっとも利用性が高い試験候補物質として選択することができる。ここでのデータは、プロインシュリン及び/又はGAD65がこれらの基準を満たすことを示唆している。
以下は、前臨床及び臨床試験によって達成されることを意図した、例示的で具体的な4つのマイルストーンである。
(1)2ヶ所のセンターでの共同前臨床データの再現性:前糖尿病マウスだけでなく新規開始糖尿病マウスも防御されるということは重要である。前糖尿病期に与えたときにはNODマウスを防御する免疫依存的介在処置は多いが、新規開始NODで正常血糖を再確立することができるものはほとんどない。実施例1(図2)では、NODマウス及びLCMVモデルで処置マウスの約50%が開始期の糖尿病を回復させることができるように、抗CD3mAbの投与のための投薬技術が改変された。図1Cのデータは、抗原添加の効果を評価するためのウインドウを提供した。更にまた、前記の効果が維持されない場合(すなわち>3ヶ月)、抗原の反復投与は再発率を低下させるか否かを容易に決定することができる。NOD実験は、臨床試験の進行前に2ヶ所のセンターで別個に確認することができる。
(2)明瞭な抗原候補物質:上記に記載した実験を基にして、糖尿病の回復率を維持するために最適な抗原を選択することができる。2つ又は3つ以上の抗原が、抗CD3との最適な類似する相乗作用を示す場合には、前記抗原はGMP(良好な製造慣行(good manufacturing practice))製剤でもっとも利用性が高い試験候補物質として選択することができる。ここでのデータは、プロインシュリン及び/又はGAD65がこれらの基準を満たすことを示唆している。
(3)臨床試験に用いられる抗原の供給源:臨床的に潜在的に利用できる抗原は下記の表4に挙げられている。新規なペプチドを製造しなければならないという稀な事例では、そのようなペプチドは外部の製造者によってGMP条件の下で製造することができる。このペプチドの使用のためのIND(治験用新薬)申請を登録し、完全な糖尿病の患者で小規模のフェースI投薬/安全性試験を実施することができる。
(4)重篤な副作用又は自己攻撃性リンパ球の検出可能な増加は存在しない:2つのモデル又は場所で、抗原と抗CD3の最適な組合せは疾患を悪化させないということは必須である。更にまた、反復ペプチド注射に続く重篤なアレルギー反応は生じてはいけない。インシュリンペプチド及びGAD65の両者に対する致死的なアナフィラキシー反応の報告はNODマウスで存在しない(E. Liu et. al. J. Clin. Invest. 2002, 110:1021-7.51;Pedotti et al. BMC Immunol. 2003, 4:2)。インシュリンペプチドB9-23の場合、C-末端への2つのアルギニンの結合は吸収プロフィルを遅らせ、前記によってこれまでに観察されたアナフィラキシーは予防されるようである。この改変ペプチドをインシュリンB9-23と平行して試験し、抗CD3mAbと一緒に前記ペプチドを投与したとき、この改変がアナフィラキシーの防止に必要か否かを決定することができる。下記に特記するように、変異B9-23インシュリンペプチドを用いるフェースI試験が既に実施され、アナフィラキシーは観察されなかった。同様に、アナフィラキシーは、ミョウバン-GAD65のフェースI試験でも報告されなかった。にもかかわらず、ここで提唱される前臨床試験には、アナフィラキシーが誘発されるか否か及びこの副作用に対する抗CD3mAbの作用を決定するために、1回分投与量を超える抗原が投与されたマウスが含まれる。
(4)重篤な副作用又は自己攻撃性リンパ球の検出可能な増加は存在しない:2つのモデル又は場所で、抗原と抗CD3の最適な組合せは疾患を悪化させないということは必須である。更にまた、反復ペプチド注射に続く重篤なアレルギー反応は生じてはいけない。インシュリンペプチド及びGAD65の両者に対する致死的なアナフィラキシー反応の報告はNODマウスで存在しない(E. Liu et. al. J. Clin. Invest. 2002, 110:1021-7.51;Pedotti et al. BMC Immunol. 2003, 4:2)。インシュリンペプチドB9-23の場合、C-末端への2つのアルギニンの結合は吸収プロフィルを遅らせ、前記によってこれまでに観察されたアナフィラキシーは予防されるようである。この改変ペプチドをインシュリンB9-23と平行して試験し、抗CD3mAbと一緒に前記ペプチドを投与したとき、この改変がアナフィラキシーの防止に必要か否かを決定することができる。下記に特記するように、変異B9-23インシュリンペプチドを用いるフェースI試験が既に実施され、アナフィラキシーは観察されなかった。同様に、アナフィラキシーは、ミョウバン-GAD65のフェースI試験でも報告されなかった。にもかかわらず、ここで提唱される前臨床試験には、アナフィラキシーが誘発されるか否か及びこの副作用に対する抗CD3mAbの作用を決定するために、1回分投与量を超える抗原が投与されたマウスが含まれる。
同様に、本方法は、抗原に対する改変T細胞応答に対する抗CD3mAbの防御作用を示すが、抗原とT細胞アゴニストとの併用は、前記抗原又は他の細胞に対し望まない反応性応答を増大させるかもしれない。これは、提唱した前臨床実験で調べることができる。平行して、提唱した抗原を患者で用いる別の初期実験を実行することができる。これらの実験から得られたデータを、一切の長期の副作用と同様に上記に特記した作用について特別な注意を払いながら再吟味することができる。スティッフマン症候群はGAD65に対する自己抗体を伴うので、前臨床データ及び初期臨床データは神経学的な事象の一切の形跡について再吟味し、提唱した臨床試験のモニター計画で対象者の追跡期間を配慮することができる。
抗原とFcR非結合抗CD3mAbとのメカニズムの臨床的解釈は、T1DMにおけるインシュリン産生は、FcR非結合抗CD3mAbにより低下が妨げられ(更に改善すらされ)、更に抗原の反復投与によって維持される。前記は、これら2つのアプローチ(上記の実施例1−3で更に発展されている)の併用のメカニズムを理解することによってその根拠を得ている。これらの前臨床実験は、抗原の選択、抗原及び抗CD3mAbの投与タイミング、治療の免疫学的影響の評価に用いることができるマーカー、及びもっとも重要なことには併用の安全性に関する道標を提供する。本方法は臨床試験に用いることができる。
臨床試験は、新規開始1型糖尿病患者でのインシュリン産生低下に対する抗CD3mAbと併用した抗原の影響を調べることである。抗CD3mAbと抗原による処置を徹底的なグルコース管理及び観察と比較することができる。臨床試験の期間は2年である。臨床試験はまた、前記療法及び他の抗原に対するT細胞応答に対する抗原と抗CD3mAbとの併用の影響を決定することである。
抗原とFcR非結合抗CD3mAbとのメカニズムの臨床的解釈は、T1DMにおけるインシュリン産生は、FcR非結合抗CD3mAbにより低下が妨げられ(更に改善すらされ)、更に抗原の反復投与によって維持される。前記は、これら2つのアプローチ(上記の実施例1−3で更に発展されている)の併用のメカニズムを理解することによってその根拠を得ている。これらの前臨床実験は、抗原の選択、抗原及び抗CD3mAbの投与タイミング、治療の免疫学的影響の評価に用いることができるマーカー、及びもっとも重要なことには併用の安全性に関する道標を提供する。本方法は臨床試験に用いることができる。
臨床試験は、新規開始1型糖尿病患者でのインシュリン産生低下に対する抗CD3mAbと併用した抗原の影響を調べることである。抗CD3mAbと抗原による処置を徹底的なグルコース管理及び観察と比較することができる。臨床試験の期間は2年である。臨床試験はまた、前記療法及び他の抗原に対するT細胞応答に対する抗原と抗CD3mAbとの併用の影響を決定することである。
B.背景:
Eisenbarthは、経過中にインシュリン分泌の漸進的直線的減少が存在するT1DMの自然な経過を最初に記載した(Eisenbarth, N. Engl. J. Med. 1986, 314:1360-8)。この過程のいくつかの時点で、臨床症状が出現する。最初の形態測定研究によれば、症状提示時にはβ細胞集団の90%が失われていると示唆されたが、我々のより最近の研究によれば(我々はT1DMの最初の2年のインシュリン分泌速度(ISR)を測定した)、症状提示時の損傷はより少なく、生理学的刺激(混合食耐性試験(MMTT))に対するISRは正常の50%であることを示している(Gepts, Diabetes 1965, 14:619-633;Steele et al. Diabetes 2004, 53:426-433)。にもかかわらず、経過中に、T1MDのほぼ全ての患者(特に小児)が一切の検出可能なインシュリンの生成能力を失う(ただし例外も記載された:Steele et al. 2004, 53:426-433;The DCCT Research Group. J. Clin. Endocrinol Metab. 1987, 65:30-6)。しかしながらインシュリン分泌保持は明らかに重要な臨床的目標である。なぜならば、前記分泌保持は血糖制御の改善と密接に関連し、したがって前記を維持することができるならば長期の合併症のリスクはおそらく減少するからである(Faber et al. Diabetologia 1977, 13:263-8;The Diabetes Control and Complications Trial Research Group. N. Emgl. J. Med. 1993, 329:977-86)。
Eisenbarthは、経過中にインシュリン分泌の漸進的直線的減少が存在するT1DMの自然な経過を最初に記載した(Eisenbarth, N. Engl. J. Med. 1986, 314:1360-8)。この過程のいくつかの時点で、臨床症状が出現する。最初の形態測定研究によれば、症状提示時にはβ細胞集団の90%が失われていると示唆されたが、我々のより最近の研究によれば(我々はT1DMの最初の2年のインシュリン分泌速度(ISR)を測定した)、症状提示時の損傷はより少なく、生理学的刺激(混合食耐性試験(MMTT))に対するISRは正常の50%であることを示している(Gepts, Diabetes 1965, 14:619-633;Steele et al. Diabetes 2004, 53:426-433)。にもかかわらず、経過中に、T1MDのほぼ全ての患者(特に小児)が一切の検出可能なインシュリンの生成能力を失う(ただし例外も記載された:Steele et al. 2004, 53:426-433;The DCCT Research Group. J. Clin. Endocrinol Metab. 1987, 65:30-6)。しかしながらインシュリン分泌保持は明らかに重要な臨床的目標である。なぜならば、前記分泌保持は血糖制御の改善と密接に関連し、したがって前記を維持することができるならば長期の合併症のリスクはおそらく減少するからである(Faber et al. Diabetologia 1977, 13:263-8;The Diabetes Control and Complications Trial Research Group. N. Emgl. J. Med. 1993, 329:977-86)。
下記及び前述の実施例で示されたように、予備的データは、抗CD3mAbに暴露されたT細胞は調節機能を有するという見解を支持する。しかしながら、抗CD3mAB OKT3の使用に対して2つの制限が存在する。前記制限には、ネズミの免疫グロブリンに対するヒトの抗ネズミ応答の発達、及びサイトカイン放出症候群(CD3分子の架橋及びin vivoでのT細胞活性化に続いて生じる)が含まれる(Chatenoud et al. Transplantation 1990, 49:697-702;Chatenoud et al. Transplant Proc. 1990, 22:2605-8;Chatenoud et al. Curr. Top. Microbiol. Immunol. 1991, 174:121-34;Chatenoud et al. Transplant Proc. 1993, 25:47-51)。これらの問題を回避するために、ヒト化抗CD3mAbが開発された。前記抗体はOKT3と同じCDR領域を有するが、前記分子のFc部分に変異を有しFcR結合が低下している(Xu et al. Cell Immunol. 2000, 200:16-26)。この分子、hOKT3γ1(Ala-Ala)は、OKT3と同様な態様でTCRの結合及び調節を惹起するが、同じT細胞活性化をin vitroでもin vivoでも引き起こさない。より最近の研究によれば、実際、前記mAbは活性化シグナルをT細胞にデリバーするが、OKT3による活性化に続いて生じるIFN-γに代わってIL-10の比較的強い産生をもたらすことが示唆される。
新規開始1型糖尿病患者の治療を目的とするhOKT3γ1(Ala-Ala)のフェースI/II臨床試験は1999年に開始した。総計42人の患者をこの任意抽出試験のために募集し、半数を抗CD3mAbの12又は14日単一コースのために任意抽出し、別の半数は観察された(Herold et al. N. Engl. J. Med. 2002, 346:1692-8)。全対象者は6ヶ月毎にMMTTを実施し、疾患の最初の2年間におけるインシュリンの自然低下に対するmAb処置の影響を判定した。1年後、MMTTに対する平均的C-ペプチド(インシュリンと等モルベースで分泌される、インシュリン生成の副産物)応答は基準値の98±9.8%であったが、コントロール群では基準値の54±7.9%であった(p<0.01)。単一処置コースの実験開始から2年後、前記応答は71±12.1%であったが、コントロールでは実験開始時レベルの25±7.7%であった(p<0.01)。したがって、抗CD3mAbによる単一処置コースは2年を越えてもなおインシュリン応答に統計的な改善をもたらしたが、1年後には明瞭に効果の減少があった。ヒトの免疫応答の持続的改変(例えばワクチン)は一般的には反復免疫を必要とするので、前記結果はおそらく驚くべき発見ではない。したがって、これらの発見は、更に追加されるアプローチ(効果的で安全で特異的なもの)が前記疾患の寛容維持に必要であることを示している。残念なことに、寛容の低下はNODマウスでの抗CD3mAbのオリジナル実験では観察されなかった。実際、抗CD3mAb処置後に糖尿病を回復させることができなかったマウスでさえ、疾患の再進行を生じることなく同系の島を受け入れた(L. Chatenoud, E. Thervet, J. Primo, JF. Bach “Anti-CD3 antibody induces long-trem remission of overt autoimmunity in nonobese diabetic mice”, Proc. Natl. Acad. Sci. USA 1994, 91:123-7)。したがって、ここに記載される実験は、部分的には、抗CD3mAbと抗原との間の相互作用を調べることができるモデル系を作り出すという目的で考案された。
抗CD3mAbによって寛容を誘発するメカニズムはエフェクター細胞の枯渇を単純に必要とするわけではないことは明白である。患者を抗CD3mAbで処置した後、循環T細胞数は一過性に減少するが、その後はmAbが連続投与されたとしても増加する。最後の薬剤投与後2週間のうちに、循環リンパ球数は処置前レベルの100%である。しかしながら観察される臨床効果は、薬剤投与後数ヶ月間、場合によって1年以上もたらされ、その時点では循環細胞に識別可能な変化は存在しない。
したがって、現在提唱されている方法は、抗CD3mAbの傘の下での島抗原の投与が、非病原性表現型を特徴とする抗原への応答をもたらすアプローチである(上記実施例1−3を参照されたい)。これらの細胞は、順を追って病原性T細胞を調節すると期待されるかもしれないが、明らかに非病原性は維持することができる。本発明は、抗CD3mAbと一緒に投与されたときには島抗原に対する応答は、最近その性状が明らかにされた、正常コントロールの対象者での応答(前記応答ではIFN-γではなく非病原性サイトカイン(例えばIL-10)が分泌される)に類似すると提唱した。本発明は、前記は、病原性エフェクター細胞の活性化ではなくむしろ免疫調節をもたらすことができると説明する。
したがって、現在提唱されている方法は、抗CD3mAbの傘の下での島抗原の投与が、非病原性表現型を特徴とする抗原への応答をもたらすアプローチである(上記実施例1−3を参照されたい)。これらの細胞は、順を追って病原性T細胞を調節すると期待されるかもしれないが、明らかに非病原性は維持することができる。本発明は、抗CD3mAbと一緒に投与されたときには島抗原に対する応答は、最近その性状が明らかにされた、正常コントロールの対象者での応答(前記応答ではIFN-γではなく非病原性サイトカイン(例えばIL-10)が分泌される)に類似すると提唱した。本発明は、前記は、病原性エフェクター細胞の活性化ではなくむしろ免疫調節をもたらすことができると説明する。
C.予備的データ:
1.T1DMの最初の2年間におけるインシュリン応答に対するhOKT3γ1(Ala-Ala)による処置の影響:新規開始T1MD(診断後6週以内)の患者を、hOKT3γ1(Ala-Ala)の12日若しくは14日コース(n=21)による処置又は観察(n=19)のために任意抽出した(図8参照)。混合食に対するC-ペプチド応答(4時間混合食耐性試験、MMTT)の曲線下の面積を6ヶ月毎に調べ、インシュリン分泌を判定した。平均して、MMTTに対するC-ペプチド応答は、抗CD3mAb処置群の1年では開始応答の97±9.6%であり、一方、コントロール群では開始応答の53±7.6%であった(p=0.01)。2年後、応答は実験開始時のそれぞれ71±12%及び25±7.7%であった(p=0.003)。前記応答は、1年後に薬剤処置患者で下降したが、それらは2年後のコントロール群の場合より顕著に高かった。
2.hOKT3γ1(Ala-Ala)による処置は調節表現型をもつ細胞をin vivoで誘発する:hOKT3γ1(Ala-Ala)で処置した患者の末梢血中でCD4+IL-10+ T細胞の亜集団を特定した。mAb処置の終わりにhOKT3γ1(Ala-Ala)で処置した患者からPBMCを採集し、凍結した。これらの細胞をmAb処置前に採集した細胞も同様に融解し、細胞内サイトカイン、IL-10及びIFN-γについて染色した。マーカーは表面分子CD4及びCD3の周囲に配置された。抗CD3mAb処置後の患者から採集された細胞はin vivoでIL-10を産生していた。図9A(mAb処置前)及び図9B(mAb処置後)は、hOKT3γ1(Ala-Ala)による処置でCD4+IL-10+細胞が誘発されたことを示している。これらの誘発細胞はIFN-γ陰性であり、もっぱらCD45RO+であり、一般的にはCCR4+であった。これら細胞の13%は、表面染色でTGF-βを産生した。IL-10の偶発的なin vivo産生は、これらの細胞は、抗原が提示されるときに存在するならば免疫調節作用を示すことができることを示唆している。
1.T1DMの最初の2年間におけるインシュリン応答に対するhOKT3γ1(Ala-Ala)による処置の影響:新規開始T1MD(診断後6週以内)の患者を、hOKT3γ1(Ala-Ala)の12日若しくは14日コース(n=21)による処置又は観察(n=19)のために任意抽出した(図8参照)。混合食に対するC-ペプチド応答(4時間混合食耐性試験、MMTT)の曲線下の面積を6ヶ月毎に調べ、インシュリン分泌を判定した。平均して、MMTTに対するC-ペプチド応答は、抗CD3mAb処置群の1年では開始応答の97±9.6%であり、一方、コントロール群では開始応答の53±7.6%であった(p=0.01)。2年後、応答は実験開始時のそれぞれ71±12%及び25±7.7%であった(p=0.003)。前記応答は、1年後に薬剤処置患者で下降したが、それらは2年後のコントロール群の場合より顕著に高かった。
2.hOKT3γ1(Ala-Ala)による処置は調節表現型をもつ細胞をin vivoで誘発する:hOKT3γ1(Ala-Ala)で処置した患者の末梢血中でCD4+IL-10+ T細胞の亜集団を特定した。mAb処置の終わりにhOKT3γ1(Ala-Ala)で処置した患者からPBMCを採集し、凍結した。これらの細胞をmAb処置前に採集した細胞も同様に融解し、細胞内サイトカイン、IL-10及びIFN-γについて染色した。マーカーは表面分子CD4及びCD3の周囲に配置された。抗CD3mAb処置後の患者から採集された細胞はin vivoでIL-10を産生していた。図9A(mAb処置前)及び図9B(mAb処置後)は、hOKT3γ1(Ala-Ala)による処置でCD4+IL-10+細胞が誘発されたことを示している。これらの誘発細胞はIFN-γ陰性であり、もっぱらCD45RO+であり、一般的にはCCR4+であった。これら細胞の13%は、表面染色でTGF-βを産生した。IL-10の偶発的なin vivo産生は、これらの細胞は、抗原が提示されるときに存在するならば免疫調節作用を示すことができることを示唆している。
3.抗CD3mAbによる処置は調節T細胞をin vitroで誘発する:機能特性に関する研究のために十分な数の細胞を得るために、抗CD3mAbを利用して調節T細胞をin vitroで増補させた。PBMCをOKT3mAbとともに10日間培養し、CD4+細胞をソーティングによって単離した。続いて前記細胞をIL-10及びIL-2とともに19日間培養した。続いて、これらの細胞を同じ個体由来のPBMCの培養に添加し、PHA(フィトヘマグルチニン)で刺激した。1:5の比での細胞の添加は、図10Aに示したようにCD4+ T細胞の増殖を阻害した。前記調節作用は細胞添加の非特異的作用ではなく、抗CD3刺激を必要とした。なぜならば、IL-10及びIL-2単独で10日間培養した細胞は、PHAで刺激したPBMCを阻害しなかったからである(図10B)。しかしながら、IL-10及びIL-2との培養が調節のために必要であった。なぜならば抗CD3mAbだけで培養した細胞では調節は生じなかったからである。PBMC増殖を阻害するCD4+細胞の大半(94%)がCCR4+であり、46%がCD62L+であり、93%がCD45RO+である。前記細胞の約5%がGITR+である。
同様な結果が、hOKT3γ1(Ala-Ala)と一緒にPBMCを培養することによって得られた。hOKT3γ1(Ala-Ala)を含むか、又はIL-2と一緒にIL-10を含む培養で10日後に、ex vivoでのIL-10産生細胞の我々の発見(4)を基にして、CD4+CCR4+及びCD4+CCR4-の亜集団に細胞を仕分けした。続いてこれらの細胞を更に2週間IL-10及びIL-2中で培養し、その後同じドナー由来のPBMCを加えてPHAで刺激した。図13A及び13Bに示されているように、抗CD3mAbを含む培養に由来するCCR4+細胞はPHA刺激PBMCの増殖を阻害した。
要約すれば、この実施例で提供される予備的データは、抗CD3mAbによる単一処置コースは、T1DMの自然な経過を変えることができることを示している。しかしながら、前記作用の期間はこの時点では明確ではなく、更に追加される処置形態が必要なことは十分にありそうなことである。mAbによる再処置は可能な方法であるが、繰返し投与できる本方法の抗原特異的アプローチが好ましい。手近の試薬を用いた上記の実施例1−3に記載した本発明の方法の予備臨床試験は、目標を達成するためのアプローチを提供する(前記アプローチは、抗CD3mAb及び自己抗原により寛容を誘発し、抗原を繰返し投与することによって寛容を維持することができる)。この試験はまた、抗CD3mAbと抗原の同時投与は、抗原への反復暴露によって刺激することができる抗原特異的調節T細胞を誘発することを示す。このアプローチの臨床的有効性は、必要とされるメカニズムと同様に、この提案の臨床試験で解明することができる。
要約すれば、この実施例で提供される予備的データは、抗CD3mAbによる単一処置コースは、T1DMの自然な経過を変えることができることを示している。しかしながら、前記作用の期間はこの時点では明確ではなく、更に追加される処置形態が必要なことは十分にありそうなことである。mAbによる再処置は可能な方法であるが、繰返し投与できる本方法の抗原特異的アプローチが好ましい。手近の試薬を用いた上記の実施例1−3に記載した本発明の方法の予備臨床試験は、目標を達成するためのアプローチを提供する(前記アプローチは、抗CD3mAb及び自己抗原により寛容を誘発し、抗原を繰返し投与することによって寛容を維持することができる)。この試験はまた、抗CD3mAbと抗原の同時投与は、抗原への反復暴露によって刺激することができる抗原特異的調節T細胞を誘発することを示す。このアプローチの臨床的有効性は、必要とされるメカニズムと同様に、この提案の臨床試験で解明することができる。
D.実験のデザイン及び方法:
新規開始1型糖尿病の患者におけるインシュリン産生低下に対する抗原と抗CD3mAbの同時投与の影響を調べる。この提案の臨床試験は、抗CD3mAbと抗原による処置が徹底的なグルコース管理と観察に対して比較される、標識公開の任意抽出管理臨床試験(open label, randomized controlled trial)である。前記試験の期間は2年間である。
論理的根拠:1型糖尿病のNOD及びLCMVモデルにおける前臨床試験(先行する実施例参照)は、糖尿病予防の相乗作用は抗CD3mAbと抗原を併用することによって誘発できることを示している。ここで強調される本臨床試験の問いは、抗CD3mAb処置とともに糖尿病抗原により免疫することによって新規開始T1DM患者で2年間インシュリン応答を改善することができるか否かである。実施例1−3に記載した実験は、この臨床試験に使用されるべき最適の抗原を確立させ、更に、投薬スケジュール及び追跡することができる分析のための終了点に関する情報とともに、以前に生じた懸念に応える安全性情報を提供した。本提案の臨床試験は、新規開始1型糖尿病患者の4アーム、標識公開試験である。第一の終了点は、抗CD3と抗原との併用効果を攻撃的な糖尿病操作と比較すること、及び試験開始後2年の混合食耐性試験(MMTT)に対するC-ペプチド応答に対する観察である。第二の終了点には、1年後のC-ペプチド応答に対する処置の影響及びインシュリンの使用が含まれる。
新規開始1型糖尿病の患者におけるインシュリン産生低下に対する抗原と抗CD3mAbの同時投与の影響を調べる。この提案の臨床試験は、抗CD3mAbと抗原による処置が徹底的なグルコース管理と観察に対して比較される、標識公開の任意抽出管理臨床試験(open label, randomized controlled trial)である。前記試験の期間は2年間である。
論理的根拠:1型糖尿病のNOD及びLCMVモデルにおける前臨床試験(先行する実施例参照)は、糖尿病予防の相乗作用は抗CD3mAbと抗原を併用することによって誘発できることを示している。ここで強調される本臨床試験の問いは、抗CD3mAb処置とともに糖尿病抗原により免疫することによって新規開始T1DM患者で2年間インシュリン応答を改善することができるか否かである。実施例1−3に記載した実験は、この臨床試験に使用されるべき最適の抗原を確立させ、更に、投薬スケジュール及び追跡することができる分析のための終了点に関する情報とともに、以前に生じた懸念に応える安全性情報を提供した。本提案の臨床試験は、新規開始1型糖尿病患者の4アーム、標識公開試験である。第一の終了点は、抗CD3と抗原との併用効果を攻撃的な糖尿病操作と比較すること、及び試験開始後2年の混合食耐性試験(MMTT)に対するC-ペプチド応答に対する観察である。第二の終了点には、1年後のC-ペプチド応答に対する処置の影響及びインシュリンの使用が含まれる。
方法:
研究の論点及び試験のデザイン:最初の2年間におけるMMTTに対するC-ペプチド応答の低下に対する抗CD3mAbと抗原との影響を徹底的な糖血症の管理及び観察と比較するために、公開標識試験が新規開始1型糖尿病患者で提唱される。これら抗原に対する応答における抗CD3mAb又は抗原単独の作用を調べるために、2つの追加試験群が加えられ、前記試験群ではこれらの応答を調べることができる。これらの試験群は、2つの主要処置の問題群の患者に対するメカニズム研究の比較のために主として加えられる。しかしながら、前記試験は、これら個々の処置が、徹底的なグルコース管理及び観察と比較して、C-ペプチド応答に対して臨床的影響を有するか否かを決定するために力が与えられる。しかしながら主要な臨床的問いは、徹底的なインシュリン管理及び観察と比較される、2年後の抗体と抗原併用の効果である。
臨床で使用される抗原の可能な供給源:上記実施例のLCMV及びNODマウスでの実験は、抗CD3mAbとGAD65又はプロインシュリンとの併用の有効性を示し、更に別の実験はNODモデルでのインシュリンペプチド単独の有効性を示唆した。更にまた、これら提唱された抗原について、抗CD3併用又は抗CD3無しの抗原投与の免疫学的応答に対する影響を追跡するために用いられるin vitroアッセイが利用可能である。熱ショックタンパク質ペプチド(DiaPep277)は、NODマウス、マルチドースのストレプトゾトシン誘発糖尿病、及びヒトの糖尿病での有益な作用を示し、抗CD3mAbとの併用に関して上記実験で調べられている(Elias et al. Diabetes 1994, 43:992-8;Birk et al. J. Autoimmun. 1996, 9:159-66;Raz et al. Lancet 2001, 358:1749-53)。以下の表は提唱される抗原及びそれらの臨床使用のための現在の開発状況を示す。
研究の論点及び試験のデザイン:最初の2年間におけるMMTTに対するC-ペプチド応答の低下に対する抗CD3mAbと抗原との影響を徹底的な糖血症の管理及び観察と比較するために、公開標識試験が新規開始1型糖尿病患者で提唱される。これら抗原に対する応答における抗CD3mAb又は抗原単独の作用を調べるために、2つの追加試験群が加えられ、前記試験群ではこれらの応答を調べることができる。これらの試験群は、2つの主要処置の問題群の患者に対するメカニズム研究の比較のために主として加えられる。しかしながら、前記試験は、これら個々の処置が、徹底的なグルコース管理及び観察と比較して、C-ペプチド応答に対して臨床的影響を有するか否かを決定するために力が与えられる。しかしながら主要な臨床的問いは、徹底的なインシュリン管理及び観察と比較される、2年後の抗体と抗原併用の効果である。
臨床で使用される抗原の可能な供給源:上記実施例のLCMV及びNODマウスでの実験は、抗CD3mAbとGAD65又はプロインシュリンとの併用の有効性を示し、更に別の実験はNODモデルでのインシュリンペプチド単独の有効性を示唆した。更にまた、これら提唱された抗原について、抗CD3併用又は抗CD3無しの抗原投与の免疫学的応答に対する影響を追跡するために用いられるin vitroアッセイが利用可能である。熱ショックタンパク質ペプチド(DiaPep277)は、NODマウス、マルチドースのストレプトゾトシン誘発糖尿病、及びヒトの糖尿病での有益な作用を示し、抗CD3mAbとの併用に関して上記実験で調べられている(Elias et al. Diabetes 1994, 43:992-8;Birk et al. J. Autoimmun. 1996, 9:159-66;Raz et al. Lancet 2001, 358:1749-53)。以下の表は提唱される抗原及びそれらの臨床使用のための現在の開発状況を示す。
表4:臨床試験用抗原の例示的供給源
抗CD3mAbの供給源:抗CD3mAb、hOKT3γ1(Ala-Ala)をGMP条件下で製造される。抗Cd3抗体の他の形態もまた代用することができるが、この場合抗CD3抗体は、本明細書に記載したように前臨床実験で、本実施例の予備的データのセクションに記載したように予備実験で、及び薬剤毒性及び安全性実験で調べることができる。
薬剤毒性及び安全性の問題:安全性データは、hOKT3γ1(Ala-Ala)で処置した32人のT1DM患者及び他の症状のための薬剤で処置された約15人の他の対象者から集められた。T1DMの患者から得られたデータは、前記薬剤に対しては良好な耐性が示されることを示した。フェースI/IIの臨床試験では、新規開始1型糖尿病の患者を、hOKT3γ1(Ala-Ala)の単一コース(n=21)又はコントロール(観察)群(n=20)に割り当てた(注記:コントロール対象者のうちの2人は12ヶ月前に前記臨床試験から外した)。投薬スケジュールは最初の12人の患者の処置後、14日から12日コースに改変した。以下のチャートは、12日から14日の投薬スケジュールを受けた21人の患者で観察された副作用を示す。
薬剤毒性及び安全性の問題:安全性データは、hOKT3γ1(Ala-Ala)で処置した32人のT1DM患者及び他の症状のための薬剤で処置された約15人の他の対象者から集められた。T1DMの患者から得られたデータは、前記薬剤に対しては良好な耐性が示されることを示した。フェースI/IIの臨床試験では、新規開始1型糖尿病の患者を、hOKT3γ1(Ala-Ala)の単一コース(n=21)又はコントロール(観察)群(n=20)に割り当てた(注記:コントロール対象者のうちの2人は12ヶ月前に前記臨床試験から外した)。投薬スケジュールは最初の12人の患者の処置後、14日から12日コースに改変した。以下のチャートは、12日から14日の投薬スケジュールを受けた21人の患者で観察された副作用を示す。
表5:フェースI/II試験でのhOKT3γ1(Ala-Ala)による処置の副作用(WHO基準)
抗CD3mAbのフェースII臨床試験は新規ロットの薬剤を用いて開始された。フェースIの投薬PK試験をこの新規ロットの薬剤を用いて繰返し、提唱される投薬はこれらの試験から得た推奨される投薬を反映している。
患者集団:試験集団は期間が6週間未満のT1DMの患者を含む。このカットオフ期間は任意ではあるが、シクロスポリン臨床試験から得られた発見に基づいている(前記臨床試験では、6週以内で治療を開始した患者のほうが、その時期の後で治療を開始した患者よりも応答率は顕著に高かった)。年齢は8−30歳の範囲である(Stiller et al. Science 1984, 223:1362-7)。以下の採用基準及び排除基準が提唱される。
採用基準には以下が含まれる:1)2ヶ月を超えない、ADA基準によるI型真性糖尿病の診断;2)8−30歳の男性又は女性、最低体重は34kg;3)検出可能な抗GAD、抗ICA512/IA-2、又はインシュリン自己抗体(インシュリン処置前)。患者は、HLA型によっては採用/排除又は任意抽出されない。HLA型をもつ患者の各試験群への均質な分布は、提唱される分析(例えばテトラマー試験が含まれる場合)を可能にする。
患者集団:試験集団は期間が6週間未満のT1DMの患者を含む。このカットオフ期間は任意ではあるが、シクロスポリン臨床試験から得られた発見に基づいている(前記臨床試験では、6週以内で治療を開始した患者のほうが、その時期の後で治療を開始した患者よりも応答率は顕著に高かった)。年齢は8−30歳の範囲である(Stiller et al. Science 1984, 223:1362-7)。以下の採用基準及び排除基準が提唱される。
採用基準には以下が含まれる:1)2ヶ月を超えない、ADA基準によるI型真性糖尿病の診断;2)8−30歳の男性又は女性、最低体重は34kg;3)検出可能な抗GAD、抗ICA512/IA-2、又はインシュリン自己抗体(インシュリン処置前)。患者は、HLA型によっては採用/排除又は任意抽出されない。HLA型をもつ患者の各試験群への均質な分布は、提唱される分析(例えばテトラマー試験が含まれる場合)を可能にする。
サンプルサイズと正当性:現時点では、食事とインシュリン以外にはT1DMのための承認された治療は存在しない。したがって、本試験は抗原と抗CD3mAbの併用を抗CD3mAb単独又は抗原単独と比較するのではなく、観察下での徹底的なグルコース管理と比較する。後者との比較は大きな群サイズ(約62/群)を用いて実施される。基準C-ペプチド応答の80%保持が処置効果の判定基準として選択される。エントリー時のコントロール患者の平均飢餓C-ペプチドは0.21pmol/mLであり、この応答の80%保持でも臨床的には有意である。なぜならば、刺激されたC-ペプチドレベルは>0.2pmol/mLとなり、これは代謝コントロールの改善に付随していたからである(The DCCT Research Group, J Clin Endocrinol Metab 1987, 65:30-6)。このC-ペプチド応答レベルの保持は明瞭に臨床的有意を示す。フェースI/II試験では、薬剤処置対象者の24%(5/21)及びコントロール対象者の11%(2/19)が試験開始後2年でこの基準に該当した。併用処置はこの割合を2倍にするか、又は対象者の48%が2年後に彼らの基準応答の80%を保持しているはずである。この基準の場合、23人/群のサンプルサイズは81%のパワーを提供する(アルファ=0.05,2テール検定)。この提唱サンプルサイズは、1年後にアルファ0.05に対して86%のパワーを提供する。
任意抽出及び処置プラン:同意を得た後で実験室でのスクリーニング試験を実施する。これらの試験の結果が満足すべきものである場合、患者を4群の1つに任意抽出する。全患者は先に記載したように4時間のMMTTを受ける。糖血変動域の平均的増幅を比較することができるように、患者には連続グルコースモニターを装着してもらう。各処置群の全患者に試験期間中ヘモグロビンA1cレベルを7.5%未満に維持するように依頼する。このレベルは、小児内分泌医が重篤で頻繁な低血糖のリスクをもたらさない厳密なグルコース管理の指標として提唱した。この推奨の改変(増加又は減少)はDSMBによって要求されえる。これを実施するために、CDEが全ての患者と約2週間毎に接触する。前記各々が患者の担当医と接触し、試験の目的に合致させるために必要な支援を提供する。前記の努力は、糖血症の管理そのものが1型糖尿病患者のC-ペプチド応答の重要な決定因子であるという証明のために全ての群で要求される。全ての試験対象者が比較可能な糖血管理レベルを維持するように依頼されるので、ヘモグロビンA1cレベルはこの試験の主要な終末点とはならないであろう。しかしながら、所望のグルコース管理レベルに達するために要求されるインシュリンの量は分析されるであろう。
抗CD3mAbと抗原の併用又は抗CD3mAb単独に対して任意抽出された患者はhOKT3γ1(Ala-Ala)の12日コースを受ける。用いられる用量は以下のとおりである:1日目、1日当たり227μg/m2;2日目、1日当たり459μg/m2;3−12日目、1日当たり919μg/m2。薬剤は15分かけて静脈内投与される。患者は12日の全処置期間にわたって入院する必要はないが、最初の3回の輸液のために収容される。併用処置を受ける患者は最初の薬剤投与と一緒に抗原投与を開始する。続いて抗原は約3ヶ月の間隔(予備試験及び前臨床試験の結果に左右される)で再投与される。抗原のみを投与されるように任意抽出された患者は、基準値試験及びMMTTが完了した後で最初の投薬を受けることができる。抗CD3mAb群の患者は抗原を投与されない。
循環リンパ球数、化学的変化及び他の安全性パラメータ(EBV及び/又はCMVウイルス負荷を含む)を2年間のコースの間モニターする。スクリーニングの時点及び2年間の試験期間中、サンプルを下記に記載するT細胞試験のために採取する。許容できる7cc/kgの血液採取容積にしたがって、この試験のための最低体重は約33kgであると概算される。
循環リンパ球数、化学的変化及び他の安全性パラメータ(EBV及び/又はCMVウイルス負荷を含む)を2年間のコースの間モニターする。スクリーニングの時点及び2年間の試験期間中、サンプルを下記に記載するT細胞試験のために採取する。許容できる7cc/kgの血液採取容積にしたがって、この試験のための最低体重は約33kgであると概算される。
終末点の分析:第一の終末点は、2年の時点で基準レベルの80%のMMTTに対するC-ペプチド応答をもつ個体の頻度である。厳密な糖血症管理と観察の群及び抗原と抗CD3mAbの併用群でこの基準に合致する個体の頻度をχ2分析(Chi-squared analysis)によって比較する。
第二の終末点には1年の時点で基準C-ペプチド応答の80%の保持が含まれる。したがって、このデータの分析はこの時点で行われ、厳密な糖血症管理と観察の群及び抗原と抗CD3mAbの併用群でこの基準に合致する個体の頻度が同様にχ2分析によって比較される。同じアプローチを用いて、基準C-ペプチド応答の80%をもつ個体の頻度が抗CD3mAb又は抗原のみを投与された群で1年及び2年の時点でより大きいか否かが判定されるが、ただし前記3つの処置群の間の相違について結論を引き出すのが目的ではない。2つの主要群の個体によって用いられるインシュリンの投与量を、グルコース連続モニターで捕捉された糖血変動域レベルと同様に比較することができる。
調節T細胞が誘発されるか、又は増補されるか否かの検査もまた実施される。
論理的根拠:抗CD3mAbの存在下で、島抗原に対する応答を本発明の方法によって調節することができる。ELISPOT又はMHCクラスIIテトラマーを含む現在利用可能な技術は、抗原と抗CD3mAbの併用が、応答細胞の表現型と同様に抗原反応性前駆細胞の数を変化させることができるか否かの問いに直接答えを出すことを可能にする。更にまた、上記に記載した患者での予備的実験は、mAbは調節T細胞の亜集団を誘発することを示している。したがって、in vivoで誘発されるCD4+のT細胞が機能的な調節特性を有するか否か、及びそれらが投与された抗原に特異的であるか否かが決定される。
第二の終末点には1年の時点で基準C-ペプチド応答の80%の保持が含まれる。したがって、このデータの分析はこの時点で行われ、厳密な糖血症管理と観察の群及び抗原と抗CD3mAbの併用群でこの基準に合致する個体の頻度が同様にχ2分析によって比較される。同じアプローチを用いて、基準C-ペプチド応答の80%をもつ個体の頻度が抗CD3mAb又は抗原のみを投与された群で1年及び2年の時点でより大きいか否かが判定されるが、ただし前記3つの処置群の間の相違について結論を引き出すのが目的ではない。2つの主要群の個体によって用いられるインシュリンの投与量を、グルコース連続モニターで捕捉された糖血変動域レベルと同様に比較することができる。
調節T細胞が誘発されるか、又は増補されるか否かの検査もまた実施される。
論理的根拠:抗CD3mAbの存在下で、島抗原に対する応答を本発明の方法によって調節することができる。ELISPOT又はMHCクラスIIテトラマーを含む現在利用可能な技術は、抗原と抗CD3mAbの併用が、応答細胞の表現型と同様に抗原反応性前駆細胞の数を変化させることができるか否かの問いに直接答えを出すことを可能にする。更にまた、上記に記載した患者での予備的実験は、mAbは調節T細胞の亜集団を誘発することを示している。したがって、in vivoで誘発されるCD4+のT細胞が機能的な調節特性を有するか否か、及びそれらが投与された抗原に特異的であるか否かが決定される。
方法:
抗原反応性T細胞のELISPOT又はテトラマーによる分析:臨床試験の患者から得たサンプルを、Peakmanらが記載した技術を用いるELISPOTによって、又はMHCクラスII GADテトラマーを用いる染色によって分析する(Arif et al. J. Clin. Invest. 2004, 113:451-63)。ELISPOTによって、インシュリンペプチドC13-A5及びIA-2ペプチドと同様にコントロールペプチドに対する応答が分析される。プロインシュリン及びIA-2ペプチドに対するIL-10及びIFN-γ応答を調べることができる。IA-2に対するIL-10応答は、コントロール対象者(57%)と糖尿病対象者(8%)との間でもっとも識別性が高いように思われるが、他のプロインシュリンペプチドに対する応答もまた望ましい。なぜならば、非糖尿病コントロール対象者の7%に対して糖尿病患者の72%が、プロインシュリン又はIA-2ペプチドのどちらかの少なくとも1つに対しIFN-γ応答を有するからである。
テトラマー試験は、先ず初めにin vitroでのペプチド反応性細胞の増補、それに続く前記応答細胞のプレート結合モノマーによる二次刺激、その後のテトラマーの染色を必要とする(Reijonen et al. Ann. NY Acad. Sci. 2003, 1005:82-8;Nepom et al. Arthritis Rheum. 2002, 46:5-12)。サイトカインの測定のために、刺激二次培養から上清を単離する。これらの試験では、凍結されていた細胞のアリコットを同じときに使用し、コントロールテトラマー(例えばHA抗原)を全体的免疫抑制の測定として及び/又は凍結後のサンプルに関する問題として同じときに使用することができる。
両技術はともに、抗原反応性T細胞を列挙し、抗原に応答するサイトカインの相対的産生を示すことを可能にする。抗CD3mAbによる処置は抗原に対するIL-10応答を増加させることができるが、抗原反応性細胞の実際の数に対する影響は不明である。これらの応答は、抗原の反復投与が応答に影響を及ぼすか否かを決定するために、提唱した2年の期間にわたって追跡される。上記に記載したように、応答の表現型は、抗原への持続的暴露により維持されると予想される。重要なことには、これらの発見をまたこの疾患の代謝過程における変化に相関させることもできるであろう。
抗原反応性T細胞のELISPOT又はテトラマーによる分析:臨床試験の患者から得たサンプルを、Peakmanらが記載した技術を用いるELISPOTによって、又はMHCクラスII GADテトラマーを用いる染色によって分析する(Arif et al. J. Clin. Invest. 2004, 113:451-63)。ELISPOTによって、インシュリンペプチドC13-A5及びIA-2ペプチドと同様にコントロールペプチドに対する応答が分析される。プロインシュリン及びIA-2ペプチドに対するIL-10及びIFN-γ応答を調べることができる。IA-2に対するIL-10応答は、コントロール対象者(57%)と糖尿病対象者(8%)との間でもっとも識別性が高いように思われるが、他のプロインシュリンペプチドに対する応答もまた望ましい。なぜならば、非糖尿病コントロール対象者の7%に対して糖尿病患者の72%が、プロインシュリン又はIA-2ペプチドのどちらかの少なくとも1つに対しIFN-γ応答を有するからである。
テトラマー試験は、先ず初めにin vitroでのペプチド反応性細胞の増補、それに続く前記応答細胞のプレート結合モノマーによる二次刺激、その後のテトラマーの染色を必要とする(Reijonen et al. Ann. NY Acad. Sci. 2003, 1005:82-8;Nepom et al. Arthritis Rheum. 2002, 46:5-12)。サイトカインの測定のために、刺激二次培養から上清を単離する。これらの試験では、凍結されていた細胞のアリコットを同じときに使用し、コントロールテトラマー(例えばHA抗原)を全体的免疫抑制の測定として及び/又は凍結後のサンプルに関する問題として同じときに使用することができる。
両技術はともに、抗原反応性T細胞を列挙し、抗原に応答するサイトカインの相対的産生を示すことを可能にする。抗CD3mAbによる処置は抗原に対するIL-10応答を増加させることができるが、抗原反応性細胞の実際の数に対する影響は不明である。これらの応答は、抗原の反復投与が応答に影響を及ぼすか否かを決定するために、提唱した2年の期間にわたって追跡される。上記に記載したように、応答の表現型は、抗原への持続的暴露により維持されると予想される。重要なことには、これらの発見をまたこの疾患の代謝過程における変化に相関させることもできるであろう。
患者の調節T細胞の試験:NODマウスの研究及び以前の研究は、CD4+細胞は免疫調節に必要であるという知見を支持するが、本発明は、調節細胞の正体はなお不明であり、他のサブクラスもまた同様に必要とされるかもしれないという考えを採用する。PBMCは、2年間の追跡期間を通して種々の時点(抗CD3mAb処置の直後及び抗原による免疫後を含む)で患者から単離される。CD4+又はCD8+ T細胞をダイナル(Dynal)ビーズを用いてPBMCから精製する。調節細胞の亜集団をCD25の発現によって選別する。これらの細胞を種々の割合で、単独で又は薬剤処置前に凍結しておいた患者由来のPBMCとともにPHA又は抗原の存在下で添加する。応答細胞をCFSEで標識する。被検抗原には、破傷風菌と同様に、抗CD3mABと一緒に患者に投与される抗原が含まれる。増殖もまたPHAで刺激することができる。このようにして、抑制性応答が存在するのか否か、及び、それは免疫抗原に特異的であるのか否か又はそれは一般的な現象であるのか否かを調べる。これらの試験では、種々の時点で比較することができるように凍結細胞を利用することができる。応答を群の間で比較するだけでなく、処置後のCD4+ T細胞添加の影響を処置前のPBMCから単離した前記細胞と比較する。更に別の培養に、抗IL-10及び/又は抗TGF-βmAb を添加して、阻害作用が観察されるとしたら、前記作用はこれらサイトカインに依存するか否かを決定する。添加した細胞のアリコットをFoxP3の発現についてRNA発現によって調べ、更にGITR、CTLA-4、CD25、CD62L及びCD45RO(これらは調節T細胞の表現型を示す)をフローサイトメトリーによって調べる。
提唱される調節T細胞マーカー(CD25、CD45RO、CD62L及びGITRを含む)を基にして、細胞のサブセットをまた精製する。抗原又はPHA刺激細胞培養へのこれら細胞の添加の影響を調べる。
自己抗原を利用する増殖アッセイでは、抗原に対する応答は、カットオフの3をぎりぎり超えるだけの刺激インデックスを示す、弱い応答の可能性がある。これは、バックグラウンド染色が高い場合、CFSEアッセイ特有の問題かもしれない。したがって、増殖応答を増大させるために、抗CD28mABとともにプレート結合MHCモノマー(前記は一般的にはるかに強い刺激を提供する)に対する応答で細胞の活性化を調べる。また別には、処置前の患者由来の抗原反応性T細胞を増補し、これらの細胞を応答細胞として使用し、更に2年の期間を通して単離されるCD4又はCD8細胞を添加することができる。調節細胞は、抗原反応性T細胞クローンの増補又は前記クローンのプライミングに影響するかもしれない。
提唱される調節T細胞マーカー(CD25、CD45RO、CD62L及びGITRを含む)を基にして、細胞のサブセットをまた精製する。抗原又はPHA刺激細胞培養へのこれら細胞の添加の影響を調べる。
自己抗原を利用する増殖アッセイでは、抗原に対する応答は、カットオフの3をぎりぎり超えるだけの刺激インデックスを示す、弱い応答の可能性がある。これは、バックグラウンド染色が高い場合、CFSEアッセイ特有の問題かもしれない。したがって、増殖応答を増大させるために、抗CD28mABとともにプレート結合MHCモノマー(前記は一般的にはるかに強い刺激を提供する)に対する応答で細胞の活性化を調べる。また別には、処置前の患者由来の抗原反応性T細胞を増補し、これらの細胞を応答細胞として使用し、更に2年の期間を通して単離されるCD4又はCD8細胞を添加することができる。調節細胞は、抗原反応性T細胞クローンの増補又は前記クローンのプライミングに影響するかもしれない。
Claims (38)
- 自己抗原に対する寛容を回復させるか又は誘発する方法であって、
(a)抗CD3抗体及び
(b)前記自己抗原、
を対象者に投与することを含み、前記抗CD3抗体及び自己抗原が、対象者において前記自己抗原に対する寛容を回復させるか又は誘発するのに十分な量で投与されることを特徴とする方法。 - 対象者における自己抗原に対する免疫応答を阻害又は予防する方法であって、対象者に(a)抗CD3抗体及び
(b)前記自己抗原、
を投与することを含み、前記抗CD3抗体及び自己抗原が、前記自己抗原に対する免疫応答を低下、阻害又は予防するのに十分な量で投与されることを特徴とする方法。 - 対象者において抗原特異的T調節細胞を産生させる方法であって、対象者に、
(a)抗CD3抗体及び
(b)自己抗原、
を投与することを含み、前記抗CD3抗体及び自己抗原が、対象者がT調節細胞を産生するのに十分な量で前記対象者に投与されることを特徴とする方法。 - 以下の工程、
(a)対象者に、
(i)抗CD3抗体及び
(ii)自己抗原、
を投与する工程、
(b)T調節細胞を前記対象者から単離する工程、
(c)前記T調節細胞を増殖条件下でin vitroでインキュベートする工程、及び
(d)前記対象者に工程(c)のT調節細胞を投与して前記自己抗原に対する寛容を回復させるか又は樹立する工程、
を含むことを特徴とする自己抗原に対する寛容を回復させるか又は樹立する方法。 - 工程(c)が、前記T調節細胞集団をIL-2とインキュベートすることを含む、請求項4に記載の方法。
- 更に、前記単離T調節細胞を、抗CD3抗体及び自己抗原とインキュベートすることを含む、請求項5に記載の方法。
- 更に、前記T調節細胞を、抗原提示細胞(APC)及び自己抗原とインキュベートすることを含む、請求項5に記載の方法。
- 前記T調節細胞が、その表面にCD4、CD25及びCD62Lを発現する、請求項3又は4に記載の方法。
- 前記T調節細胞が、その表面にCD25、CD45TO、CD62L及びGITRを発現する、請求項3又は4に記載の方法。
- 前記T調節細胞が、その表面にCD25、FoxP3、GITR、CTLA4、CD62L及びCD45ROを発現する、請求項3又は4の方法。
- 前記T調節細胞が、その表面にCD4、CCR4、CD62及びCD45ROを発現する、請求項3又は4に記載の方法。
- 前記免疫応答が、液性免疫応答又は細胞性免疫応答である、請求項2に記載の方法。
- 前記自己抗原が、タンパク質又はそのペプチドフラグメントを含む、請求項1、2、3又は4に記載の方法。
- 前記対象者が、グレーヴズ病、橋本甲状腺炎、低血糖症、多発性硬化症、本態性混合型クリオグロブリン血症、全身性紅斑性狼瘡、I型糖尿病又は前記のいずれかの組合せに罹患している、請求項1、2、3又は4に記載の方法。
- 前記タンパク質が、甲状腺刺激ホルモンレセプター、サイログロブリン、甲状腺ペルオキシダーゼ、ミエリン塩基性タンパク質、グルタミン酸デカルボキシラーゼ(GAD65)、島細胞抗原512/IA-2(ICA512/IA-2)、島細胞抗原p69(ICA69)、インシュリン、プロインシュリン、熱ショックタンパク質60(HSP60)又はその任意の組合せを含む、請求項13に記載の方法。
- I型糖尿病(T1D)を治療する方法であって、対象者に、
(a)抗CD3抗体及び
(b)自己抗原、
を投与することを含み、前記抗CD3抗体及び自己抗原が、I型糖尿病又はI型糖尿病に関連する1つ又は2つ以上の症状を治療するのに十分な量で投与される、前記I型糖尿病の治療方法。 - 前記症状が、インシュリン産生の低下を含む、請求項16に記載の方法。
- 前記症状が、異常な血糖レベルを含む、請求項16に記載の方法。
- 前記症状が、インシュリン産生島細胞の破壊を含む、請求項16に記載の方法。
- 前記症状が、平均的な飢餓Cペプチドレベルを含む、請求項16に記載の方法。
- 前記自己抗原が、インシュリン、プロインシュリン、プロインシュリンII、インシュリンB9-23ペプチド、細胞障害性Tリンパ球エピトープをもたないプロインシュリンペプチド、インシュリンC13-A5ペプチド、グルタミン酸デカルボキシラーゼ(GAD65)、ICA512 /IA-2、ICA69、又は熱ショックタンパク質(HSP)60を含む、請求項16に記載の方法。
- 前記抗CD3抗体及び抗原が、初めに同じ日に投与される、請求項1、2、3、4又は16に記載の方法。
- 前記抗体が、モノクローナル抗体である、請求項1、2、3、4又は16に記載の方法。
- 前記抗体が、IgG分子を含む、請求項23に記載の方法。
- 前記抗体が、ヒト化抗体又は完全なヒトの抗体を含む、請求項23に記載の方法。
- 前記抗体が、少なくとも2つの抗原結合部位を含む、請求項23に記載の方法。
- 前記抗CD3抗体が、抗体フラグメントを含む、請求項23に記載の方法。
- 前記抗体フラグメントが、(Fab')2分子を含む、請求項27に記載の方法。
- 前記抗体フラグメントが、Fcレセプターと結合しない、請求項2に記載7の方法。
- 前記抗CD3抗体が、有糸分裂非誘発性抗体を含む、請求項23に記載の方法。
- 前記抗CD3抗体が、OKT3抗体を含む、請求項1、2、3、4又は16に記載の方法。
- 前記OKT3抗体が、ヒトOKT3γ(Ala-Ala)抗体である、請求項31に記載の方法。
- 前記投与が、前記自己抗原をコードする発現ベクターの投与を含む、請求項1、2、3、4又は16に記載の方法。
- 前記抗CD3抗体が、静脈内に投与される、請求項1、2、3、4又は16に記載の方法。
- 前記自己抗原が、経鼻的、経口的、皮下、筋肉内又は静脈内に投与される、請求項1、2、3、4又は16に記載の方法。
- 前記抗CD3抗体及び自己抗原が、医薬的に許容できる担体賦形剤又は稀釈剤とともに投与される、請求項1、2、3、4又は16に記載の方法。
- (a)抗CD3抗体、
(b)自己抗原、及び
(c)前記抗CD3抗体及び自己抗原の同時投与のための指示、
を含むことを特徴とするキット。 - 前記指示が、投与スケジュール並びに前記抗CD3抗体及び自己抗原の投与量を含む、請求項37に記載のキット。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US54195904P | 2004-02-04 | 2004-02-04 | |
| PCT/US2005/003712 WO2005076965A2 (en) | 2004-02-04 | 2005-02-04 | Anti-cd3 and antigen-specific immunotherapy to treat autoimmunity |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2007520566A true JP2007520566A (ja) | 2007-07-26 |
| JP2007520566A5 JP2007520566A5 (ja) | 2008-03-27 |
Family
ID=34860238
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006552302A Pending JP2007520566A (ja) | 2004-02-04 | 2005-02-04 | 自己免疫治療のための抗cd3及び抗原特異的免疫療法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20070190045A1 (ja) |
| EP (1) | EP1725254A4 (ja) |
| JP (1) | JP2007520566A (ja) |
| AU (1) | AU2005213449A1 (ja) |
| CA (1) | CA2554978A1 (ja) |
| IL (1) | IL177193A0 (ja) |
| WO (1) | WO2005076965A2 (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008528043A (ja) * | 2005-02-04 | 2008-07-31 | ダウ・アグロサイエンシーズ・エルエルシー | 抗t細胞および自己免疫疾患の自己抗原治療 |
| JP2013508392A (ja) * | 2009-10-20 | 2013-03-07 | グラクソ グループ リミテッド | 自己免疫疾患における抗cd3抗体の投薬 |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030108548A1 (en) * | 1993-06-01 | 2003-06-12 | Bluestone Jeffrey A. | Methods and materials for modulation of the immunosuppressive activity and toxicity of monoclonal antibodies |
| US20050004001A1 (en) * | 2002-10-02 | 2005-01-06 | Robert Harris | Formulation of antigen |
| EP1753783B1 (en) * | 2004-06-03 | 2014-08-06 | Novimmune SA | Anti-cd3 antibodies and methods of use thereof |
| SG163615A1 (en) * | 2005-07-11 | 2010-08-30 | Macrogenics Inc | Methods for the treatment of autoimmune disorders using immunosuppressive monoclonal antibodies with reduced toxicity |
| US20100209437A1 (en) * | 2005-09-12 | 2010-08-19 | Greg Elson | Anti-CD3 Antibody Fromulations |
| DK2119450T3 (da) * | 2005-11-29 | 2013-05-06 | Actogenix Nv | Induktion af mucosal tolerance over for pankreatisk ø-beta-celle-autoantigener |
| WO2007084775A2 (en) * | 2006-01-20 | 2007-07-26 | The Trustees Of The University Of Pennsylvania | Compositions and methods for modulation of suppressor t cell activation |
| EP2023955A4 (en) * | 2006-06-06 | 2009-10-28 | Tolerrx Inc | Administration of anti-CD3 antibodies in the treatment of autoimmune diseases |
| SG177907A1 (en) * | 2006-06-14 | 2012-02-28 | Macrogenics Inc | Methods for the treatment of autoimmune disorders using immunosuppressive monoclonal antibodies with reduced toxicity |
| AU2007337082A1 (en) * | 2006-12-21 | 2008-07-03 | Macrogenics Inc. | Methods for the treatment of LADA and other adult-onset autoimmune diabetes using immunosuppressive monoclonal antibodies with reduced toxicity |
| ES2492468T3 (es) | 2007-01-25 | 2014-09-09 | Actogenix N.V. | Tratamiento de enfermedad inmunitaria por administración a través de la mucosa de antígenos usando Lactobacillus genéticamente modificado |
| EP2146741A4 (en) * | 2007-04-24 | 2010-08-04 | Diamyd Therapeutics Ab | DRUGS AND METHODS FOR TREATING AUTOIMMUNE DISEASE AND CANCER |
| WO2011091138A1 (en) * | 2010-01-20 | 2011-07-28 | Bayhill Therapeutics, Inc. | Combination therapy to treat autoimmune diseases |
| US9018006B2 (en) | 2010-07-23 | 2015-04-28 | The University Of Toledo | Stable Tregs and related materials and methods |
| CN106668852B (zh) * | 2012-04-13 | 2020-12-25 | 艾棣维欣(苏州)生物制药有限公司 | 一种治疗和/或预防ⅰ型糖尿病的组合物及其应用 |
| US8735359B2 (en) * | 2012-05-24 | 2014-05-27 | Orban Biotech Llc | Combinations of modalities for the treatment of diabetes |
| EP3760224A3 (en) * | 2014-06-04 | 2021-03-31 | Diamyd Medical AB | Novel combinations for antigen based therapy |
| GB201510056D0 (en) * | 2015-06-10 | 2015-07-22 | King S College London | Multi-peptide composition |
| CN110087530A (zh) | 2016-12-07 | 2019-08-02 | 普罗根尼蒂公司 | 胃肠道检测方法、装置和系统 |
| EP3600416B1 (en) | 2017-03-30 | 2023-06-07 | Biora Therapeutics, Inc. | Treatment of a disease of the gastrointestinal tract with an immune modulatory agent released using an ingestible device |
| US11806386B2 (en) | 2017-08-07 | 2023-11-07 | St. Vincent's Institute Of Medical Research | Type I diabetes therapy |
| WO2019246312A1 (en) | 2018-06-20 | 2019-12-26 | Progenity, Inc. | Treatment of a disease of the gastrointestinal tract with an immunomodulator |
| US20230009902A1 (en) | 2018-06-20 | 2023-01-12 | Progenity, Inc. | Treatment of a disease or condition in a tissue orginating from the endoderm |
| EP3883635A1 (en) | 2018-11-19 | 2021-09-29 | Progenity, Inc. | Methods and devices for treating a disease with biotherapeutics |
| US11434291B2 (en) | 2019-05-14 | 2022-09-06 | Provention Bio, Inc. | Methods and compositions for preventing type 1 diabetes |
| EP3870261B1 (en) | 2019-12-13 | 2024-01-31 | Biora Therapeutics, Inc. | Ingestible device for delivery of therapeutic agent to the gastrointestinal tract |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003028441A1 (en) * | 2001-10-02 | 2003-04-10 | The Board Of Trustees Of The Leland Stanford Junior University | Gene therapy for the prevention of autoimmune disease |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69032484D1 (de) * | 1989-10-27 | 1998-08-20 | Arch Dev Corp | Zusammensetzungen und deren verwendung zur förderung der immunopotentiation |
| US6406696B1 (en) * | 1989-10-27 | 2002-06-18 | Tolerance Therapeutics, Inc. | Methods of stimulating the immune system with anti-CD3 antibodies |
| US6150584A (en) * | 1990-01-12 | 2000-11-21 | Abgenix, Inc. | Human antibodies derived from immunized xenomice |
| US5770429A (en) * | 1990-08-29 | 1998-06-23 | Genpharm International, Inc. | Transgenic non-human animals capable of producing heterologous antibodies |
| US5885573A (en) * | 1993-06-01 | 1999-03-23 | Arch Development Corporation | Methods and materials for modulation of the immunosuppressive activity and toxicity of monoclonal antibodies |
| US6491916B1 (en) * | 1994-06-01 | 2002-12-10 | Tolerance Therapeutics, Inc. | Methods and materials for modulation of the immunosuppresive activity and toxicity of monoclonal antibodies |
| FR2730411B1 (fr) * | 1995-02-14 | 1997-03-28 | Centre Nat Rech Scient | Association medicamenteuse utile pour la transfection et l'expression in vivo d'exogenes |
| US5834597A (en) * | 1996-05-20 | 1998-11-10 | Protein Design Labs, Inc. | Mutated nonactivating IgG2 domains and anti CD3 antibodies incorporating the same |
| US6420140B1 (en) * | 1996-10-11 | 2002-07-16 | Abgenix, Inc. | Production of multimeric protein by cell fusion method |
| US6884785B2 (en) * | 1999-06-17 | 2005-04-26 | The Scripps Research Institute | Compositions and methods for the treatment or prevention of autoimmune diabetes |
| US20040037826A1 (en) * | 2002-06-14 | 2004-02-26 | Michelsen Birgitte Koch | Combined use of a modulator of CD3 and a GLP-1 compound |
-
2005
- 2005-02-04 AU AU2005213449A patent/AU2005213449A1/en not_active Abandoned
- 2005-02-04 EP EP05722771A patent/EP1725254A4/en not_active Withdrawn
- 2005-02-04 JP JP2006552302A patent/JP2007520566A/ja active Pending
- 2005-02-04 CA CA002554978A patent/CA2554978A1/en not_active Abandoned
- 2005-02-04 WO PCT/US2005/003712 patent/WO2005076965A2/en active Application Filing
-
2006
- 2006-08-01 IL IL177193A patent/IL177193A0/en unknown
- 2006-08-03 US US11/498,381 patent/US20070190045A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003028441A1 (en) * | 2001-10-02 | 2003-04-10 | The Board Of Trustees Of The Leland Stanford Junior University | Gene therapy for the prevention of autoimmune disease |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008528043A (ja) * | 2005-02-04 | 2008-07-31 | ダウ・アグロサイエンシーズ・エルエルシー | 抗t細胞および自己免疫疾患の自己抗原治療 |
| JP2013508392A (ja) * | 2009-10-20 | 2013-03-07 | グラクソ グループ リミテッド | 自己免疫疾患における抗cd3抗体の投薬 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20070190045A1 (en) | 2007-08-16 |
| CA2554978A1 (en) | 2005-08-25 |
| EP1725254A4 (en) | 2008-02-13 |
| WO2005076965A3 (en) | 2006-07-06 |
| AU2005213449A1 (en) | 2005-08-25 |
| WO2005076965A2 (en) | 2005-08-25 |
| IL177193A0 (en) | 2006-12-10 |
| EP1725254A2 (en) | 2006-11-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2007520566A (ja) | 自己免疫治療のための抗cd3及び抗原特異的免疫療法 | |
| Bresson et al. | Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs | |
| JP3007977B2 (ja) | 抗原特異的なt細胞寛容の誘導方法 | |
| RU2540018C2 (ru) | Средство для лечения заболевания | |
| US20070190052A1 (en) | Regulatory CD8cells induced with anti-CD3 antibody | |
| EP1421191B1 (en) | Humanised antibodies specific for both CD45RB and CD45RO | |
| McHugh et al. | Control of organ-specific autoimmunity by immunoregulatory CD4+ CD25+ T cells | |
| You et al. | Transforming growth factor‐β and T‐cell‐mediated immunoregulation in the control of autoimmune diabetes | |
| JP2015120724A (ja) | ICOS陽性細胞のinvivo枯渇によるT細胞介在病態の治療方法 | |
| MX2010010028A (es) | Agente para tratar enfermedad. | |
| US20220411510A1 (en) | Anti-CD28 Humanized Antibodies Formulated for Administration to Humans | |
| US7527972B2 (en) | Uses of bispecific antibody coated dendritic cells pulsed with antigens and GM-CSF in immune regulation | |
| Burns et al. | Memory alloreactive B cells and alloantibodies prevent anti-CD154-mediated allograft acceptance | |
| Krishnamurthy et al. | Analysis of antigen specific T cells in diabetes–Lessons from pre-clinical studies and early clinical trials | |
| Chatenoud et al. | Regulatory T cells in the control of autoimmune diabetes: the case of the NOD mouse | |
| US20210236512A1 (en) | Clozapine for the treatment of a immunoglobulin driven b cell disease | |
| WO2021019249A1 (en) | Clozapine for use in treating pathogenic immunoglobulin driven b cell disease | |
| TW202302642A (zh) | 用於治療抗體介導移植物排斥用途之抗cd38抗體 | |
| Berrih-Aknin et al. | Vaccines against myasthenia gravis | |
| WO2009105548A2 (en) | Methods for treating diabetes | |
| Ng et al. | From mice to men: the challenges of developing tolerance-inducing biological drugs for the clinic | |
| Smith | An Investigation of Oral Tolerance and Priming in Vivo | |
| Rosenzweig | Immune Privilege and Tolerance-Therapeutic Antibody Approaches | |
| Vanasek | Cell cycle progression regulates in vivo clonal anergy induction | |
| Graça | Mechanisms of peripheral tolerance in transplantation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080204 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080204 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20110905 |