JP2004161802A - Biodegradable polyester resin composition and method for producing the same - Google Patents
Biodegradable polyester resin composition and method for producing the same Download PDFInfo
- Publication number
- JP2004161802A JP2004161802A JP2002325983A JP2002325983A JP2004161802A JP 2004161802 A JP2004161802 A JP 2004161802A JP 2002325983 A JP2002325983 A JP 2002325983A JP 2002325983 A JP2002325983 A JP 2002325983A JP 2004161802 A JP2004161802 A JP 2004161802A
- Authority
- JP
- Japan
- Prior art keywords
- resin composition
- poly
- phbh
- hydroxybutyrate
- hydroxyhexanoate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Compositions Of Macromolecular Compounds (AREA)
- Biological Depolymerization Polymers (AREA)
Abstract
Description
【0001】
【発明の属する技術分野】
本発明は、生分解性樹脂組成物およびその製造方法に関する。
【0002】
【従来の技術】
プラスチックは軽く、強く、しかも耐久性、成形加工性に優れることから包装材をはじめ、弱電部品、自動車部品、建材、日用雑貨などの多岐の分野で多量に使用されている。しかし、これら多量に使用されているプラスチックスは廃棄物処理の困難さ、燃焼時の有毒ガスの発生などの問題があり、またゴミ埋め立て用地の不足・発生ガスにより大気汚染・酸性雨による樹木の被害などが報告されており、地球規模での環境汚染が進行している現状にある。このため、自然環境下で分解するプラスチックの開発が数多く行なわれており、多くの生分解性を有する樹脂が作り出されてきており、これら生分解性を有する樹脂を利用した成形品を製造する方法も数多く検討がなされている。例えば、微生物発酵法による製造物、天然高分子である澱粉と合成プラスチックとのブレンドによる製造物があげられる。
【0003】
しかしながら、微生物発酵法による製造は、多量製造の困難さおよび一般の合成高分子と比較して樹脂の値段がきわめて高価である点で広く利用されておらず、また、天然高分子である澱粉と合成プラスチックとのブレンド物は、天然高分子の非熱可塑性のために成形性が悪く、成形品製造に利用する試みは広く行なわれていないのが実状である。また、一般の合成技術を利用して生分解性を有する樹脂を製造する方法、例えばカプロラクトン、乳酸あるいはグリコール類の開環重合による製造方法の検討が行なわれてきている。しかしながら、これらの方法により得られる樹脂も成形加工性や、原料もしくは製造価格の問題があり、極く限られた特殊用途のみに用いられているに過ぎない。
【0004】
一方、合成高分子の中でも、脂肪族ポリエステル系樹脂が生分解性を有することは広く知られている。しかしながら、従来の脂肪族ポリエステル系樹脂は、工業上の生産性の点から見ると、その結晶化速度は遅く、成形性の点で充分なものとは言えず改良が要望されていた。合成高分子である脂肪族ポリエステルを用いた例としては、高分子量脂肪族ポリエステルに結晶核剤やアセチレングリコールなどを添加することにより、結晶化速度を改良させることが開示されている(たとえば、特許文献1および2参照)。また、脂肪族ポリエステルの中でも、特に結晶化速度が遅い、ポリ(3−ヒドロキシアルカノエート)類を用いた例としては、3−ヒドロキシブチレート単位97〜85モル%単位および4−ヒドロキシブチレート単位3〜15モル%単位からなるポリエステル共重合体に、結晶核剤として窒化ホウ素を添加することにより、結晶化促進効果が得られることが開示されている(たとえば、特許文献3参照)。また、ポリ(3−ヒドロキシアルカノエート)類であるポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)を用いた例としては、ポリヒドロキシブチレートなど、より高い融解温度を有するポリ(3−ヒドロキシアルカノエート)を添加することで、結晶化速度が速くなることが開示されている(たとえば、特許文献4参照)。しかしながら、いずれの発明も、効果は顕著に得られるものではなく、本発明は、粒径の微細なポリ(3−ヒドロキシアルカノエート)および可塑剤を添加することで、画期的な結晶化促進効果を得るに至った。
【0005】
【特許文献1】
特開平8−120165号公報(段落番号[0005])
【特許文献2】
特開2000−345014号公報(段落番号[0006])
【特許文献3】
特開平6−157878号公報(段落番号[0006])
【特許文献4】
国際公開第02/50156 A2号パンフレット(9頁2〜24行)
【0006】
【発明が解決しようとする課題】
本発明は、ポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)の結晶化速度を増大させることで、フィルム成形、射出成形、押出成形、紡糸などの成形方法利用の際に、融着性防止、離形性向上、成形サイクル時間の短縮、連続的な樹脂ペレット化、など成形性が改善され、また、廃棄処分手段のひとつとしての生分解性、すなわち、微生物などによる分解も可能な、使用後廃棄処分がしやすい環境適合性に優れた生分解性ポリエステル系樹脂組成物およびその製造方法を提供する。
【0007】
【発明が解決するための手段】
すなわち本発明は、(A)融解温度Tm1を有する、ポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)100重量部に対して、(B)平均粒径300μm以下であり融解温度Tm2を有する、一般式(1):
[−CHR−CH2−CO−O−] (1)
(ここで、RはCnH2n+1で表されるアルキル基で、n=1〜15である。)
で表わされるポリ(3−ヒドロキシアルカノエート)0.1〜20重量部を含有し、かつ、Tm2≧Tm1+20℃である生分解性樹脂組成物に関する。
【0008】
さらに、前記生分解性樹脂組成物が、ポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)(A)に由来する融解温度Tm3、およびポリ(3−ヒドロキシアルカノエート)(B)に由来する融解温度Tm4とを有し、かつ、Tm4≧Tm3+20℃であることが好ましい。
【0009】
さらに、前記ポリ(3−ヒドロキシアルカノエート)(B)の平均粒径が、50μm以下であることが好ましい。
【0010】
さらに、前記ポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)(A)の組成比が、3−ヒドロキシブチレート/3−ヒドロキシヘキサノエート=99.9〜80/0.1〜20(モル%)であり、かつ、前記ポリ(3−ヒドロキシアルカノエート)(B)が、ポリ(3−ヒドロキシブチレート)であることが好ましい。
【0011】
さらに、前記ポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)(A)の組成比が、3−ヒドロキシブチレート/3−ヒドロキシヘキサノエート=90〜80/10〜20(モル%)であり、かつ、前記ポリ(3−ヒドロキシアルカノエート)(B)が、ポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)であり、前記(B)の組成比が、3−ヒドロキシブチレート/3−ヒドロキシヘキサノエート=99.99〜90.01/0.01〜9.99(モル%)であることが好ましい。
【0012】
さらに、前記生分解性樹脂組成物が、ポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)(A)100重量部に対して、さらに可塑剤(C)0.1〜50重量部を含有することが好ましい。
【0013】
さらに、前記可塑剤(C)が、エーテル系可塑剤、エステル系可塑剤、フタル酸系可塑剤、リン系可塑剤からなる群から選ばれる少なくとも1種以上であることが好ましい。
【0014】
さらに本発明は、前記ポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)(A)およびポリ(3−ヒドロキシアルカノエート)(B)、またはポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)(A)、ポリ(3−ヒドロキシアルカノエート)(B)および可塑剤(C)を混合する方法が、加熱溶融して混練する方法、可溶溶媒を用いて溶媒中で混練する方法、および前記(A)を生産する微生物の培養中もしくは精製段階で得られるスラリー中に混練する方法からなる群から選ばれる少なくとも1種以上である生分解性樹脂組成物の製造方法に関する。
【0015】
さらに、混合する方法が、ポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)(A)を生産する微生物の培養中もしくは精製段階で得られるスラリー中で混合する方法であることが好ましい。
【0016】
【発明の実施の形態】
本発明は、(A)融解温度Tm1を有する、ポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)100重量部に対して、(B)平均粒径300μm以下であり融解温度Tm2を有する、一般式(1):
[−CHR−CH2−CO−O−] (1)
(ここで、RはCnH2n+1で表されるアルキル基で、n=1〜15である。)
で表わされるポリ(3−ヒドロキシアルカノエート)0.1〜20重量部を含有し、かつ、Tm2≧Tm1+20℃である生分解性樹脂組成物に関する。
【0017】
前記ポリ(3−ヒドロキシブチレート−コ−3−ヒドロキシヘキサノエート)(以下、PHBHと記載する)(A)は、3−ヒドロキシブチレート(以下、3HBと記載する)と3−ヒドロキシヘキサノエート(以下、3HHと記載する)の共重合体である。前記PHBH(A)は、微生物から生産する方法または化学合成法のいずれの方法によって得られてもよく、特に限定されるものではない。なかでも、油脂を原料として微生物を培養することでPHBHを得ることができる点、化学合成法に比べてプロセスが簡単でコストも安価であるという点で、微生物から生産する方法が好ましい。
【0018】
また、微生物から生産されるPHBHは、化学合成法で得られるPHBHに比べて、PHBHの分子量分布が広く、3HBおよび3HHが適度に不均一に重合している点で好ましい。さらに、化学合成法によって得られるPHBHは、未反応のモノマー成分や使用した重合開始剤、乳化重合の場合には乳化剤などが、PHBH中に残存して物性が低下する可能性がある。
【0019】
前記PHBHを生産する微生物としては、細胞内にPHBHを蓄積する微生物であれば特に限定されず、A.lipolytica、A.eutrophus、A.latusなどのアルカリゲネス属(Alcaligenes)、シュウドモナス属(Pseudomonas)バチルス属(Bacillus)、アゾトバクター属(Azotobacter)、ノカルディア属(Nocardia)、アエロモナス属(Aeromonas)などの菌があげられる。なかでも、PHBHを効率よく生産するという点で、特にA.caviaeなどの菌株、さらにはPHA合成酵素群の遺伝子を導入したAlcaligenes eutrophus AC32(FERM P−15786)(J.Bacteriol., 179, 4821−4830頁(1997))などがより好ましく、これらの微生物を適切な条件で培養して菌体内にPHBHを蓄積させた微生物菌体が用いられる。
【0020】
前記PHBH(A)の組成比は、3HBの組成の上限は、99.9モル%が好ましく、99モル%がより好ましく、97モル%がさらに好ましく、さらには90モル%が好ましい。3HBの組成の下限は、80モル%が好ましく、85モル%がより好ましい。一般に、3HHの組成比が高いほどPHBHのポリマー特性はより柔軟となるが、結晶化速度は低下する傾向がある。本発明において、PHBH(A)の3HBの組成が80モル%より少ないと結晶化速度が遅くなる傾向があり、99.9モル%をこえると結晶化度が上昇し、樹脂が脆くなる傾向がある。
【0021】
前記PHBH(A)の重量平均分子量の下限は、5万以上であることが好ましく、10万以上であることがより好ましい。重量平均分子量が5万より低いと加工時の溶融粘度変化が大きく、流動性が高くなり、成形加工性が低下する傾向がある。
【0022】
前記PHBH(A)の融解温度Tm1は、示差走査熱量計(以下、DSCと記す)を用いて、PHBH(A)1〜10mgを10℃/分の昇温速度で、30℃からPHBH(A)が充分に融解する想定融解温度+50〜60℃まで昇温し、ついで10℃/分の降温速度で30℃まで降温した後、再度10℃/分の昇温速度で、PHBH(A)が充分に融解する想定融解温度+50〜60℃まで昇温したときの吸熱曲線のピークトップの温度をいう。前記PHBH(A)は、再度昇温したときの吸熱曲線ピークが、単一または複数のピークを示し、複数の場合、吸熱量の大きいピークトップ温度をTm1とする。
【0023】
前記ポリ(3−ヒドロキシアルカノエート)(以下、PHAと記載する)(B)は、微生物から生産する方法または合成法のいずれの方法によって得られてもよく、特に限定されるものではない。前記PHA(B)は、一般式(1)で表わされ、RはCnH2n+1で表されるアルキル基で、n=1〜15であり、n=1〜3がより好ましい。nが15をこえると結晶核剤としての効果が低下する。
【0024】
前記PHA(B)は、ホモポリマー、または2種以上のポリマーユニットの組み合わせからなる共重合体、ジ−コポリマー、トリ−コポリマー、テトラ−コポリマーなど、またはこれらのホモポリマー、コポリマーなどから選ばれる2種以上のブレンド物があげられる。なかでも、n=1のPHB、n=2のポリ(3−ヒドロキシバリレート)、n=3のポリ(3−ヒドロキシヘキサノエート)、n=5のポリ(3−ヒドロキシオクタノエート)、n=15のポリ(3−ヒドロキシオクタデカノエート)のホモポリマー、または2種以上の組み合わせからなる共重合体、ジ−コポリマー、トリ−コポリマー、またはこれらのブレンド物が、好ましくは使用できる。なかでも、マトリックス樹脂との相溶性・分散性の点で、PHBH、PHB、またはこれらのブレンド物が、好適に使用される。
【0025】
前記PHA(B)が、共重合体であるときの組成比は、3HB/3HH=99.99〜90.01/0.01〜9.99(モル%)が好ましく、99.99〜95/0.01〜5(モル%)がより、好ましい。3HBの組成が95モル%より少ないと結晶化促進の点で効果が弱くなる傾向がある。
【0026】
とくに、PHBH(A)の組成比が、3HB/3HH=99.9〜80/0.1〜20(モル%)であるときには、結晶化促進の点で、前記PHA(B)は、PHBであることが好ましい。
【0027】
また、PHBH(A)の組成比が、3HB/3HH=90〜80/10〜20(モル%)であるときには、均一分散性や相溶性の点では、PHA(B)は、PHBHであるのが好ましく、その組成比は3HB/3HH=99.99〜90.01/0.01〜9.99(モル%)であることが好ましい。一方、結晶化促進の点ではPHA(B)はPHBであってもよい。また、PHA(B)としてPHBとPHBHのブレンドを用いてもかまわない。
【0028】
前記PHA(B)の融解温度Tm2は、前記と同様にDSCを用いて測定される。前記Tm2は、Tm2≧Tm1+20℃である。Tm2が、Tm2≧Tm1+20℃であることは、結晶化速度を著しく増大させる要因の一つである。PHBH(A)を所定の温度で溶融させた場合、PHBH(A)よりも高融解温度を有するPHA(B)の結晶核が融け残るため、それを核点として、結晶が急速に成長するためである。
【0029】
また、前記融解温度Tm1を有するPHBH(A)および前記融解温度Tm2を有するPHA(B)からなる、本発明の生分解性樹脂組成物は、PHBH(A)に由来する融解温度Tm3、およびPHA(B)に由来する融解温度Tm4とを有し、かつ、Tm4≧Tm3+20℃であることが好ましい。
【0030】
Tm3およびTm4は、前記と同様にDSCを用いて測定される。また、PHBH(A)単独のTm1と樹脂組成物中のPHBH(A)に由来するTm3、およびPHA(B)単独のTm2と樹脂組成物中のPHA(B)に由来するTm4とは、組成物中にPHBH(A)またはPHA(B)以外の添加剤が含まれることや、結晶化成長過程が異なることなどから、必ずしも一致しない。
【0031】
前記PHBH(A)100重量部に対して、PHA(B)の含有量は、0.1〜20重量部である。0.1〜10重量部がより好ましい。PHA(B)の含有量が、0.1重量部よりも少ないと、結晶化速度の向上効果が不充分であり、20重量部をこえると、含有量に見合うだけの効果が期待できず、実際的でないばかりか、不経済である。
【0032】
PHA(B)の平均粒径は、マイクロトラック粒度計(日機装製、FRA)など汎用の粒度計を用い、PHA(B)の水懸濁液を所定濃度に調整し、正規分布の全粒子の50%蓄積量に対応する粒径を平均粒径とする。PHA(B)の平均粒径は、300μm以下である。100μm以下がより好ましく、50μm以下がさらに好ましく、10μm以下がさらにさらに好ましく、5μm以下が最も好ましい。PHA(B)の平均粒径が、300μmをこえると、樹脂組成物中でPHA(B)の均一な分散が困難となる。PHA(B)の平均粒径が300μm以下であると、細かい結晶核点を樹脂組成物中に、微分散できる。平均粒径300μm以下のPHA(B)を得る方法として、特に限定されないが、PHAを産出する微生物を培養し、PHAを含有する微生物を物理的破砕処理することによって、容易に粒径の小さい(通常数μm以下)PHAを得ることができる。また、粒径の大きいPHAの場合には、それを機械的に破砕する、あるいは溶媒に再溶解してスプレードライするなどして、所望する300μm以下の粒径とすることもできる。
【0033】
また、前記PHA(B)の結晶核剤以外の、公知の結晶核剤として、ポリエチレンやポリプロピレンなどのポリオレフィン樹脂、ポリエチレンテレフタレートやポリブチレンテレフタレートなどの芳香族ポリエステル樹脂など汎用プラスティック、またはポリ乳酸系樹脂や脂肪族ポリエステル系樹脂など他の生分解性樹脂において結晶核剤として効果を示すものをPHA(B)と併用してもよい。具体的には、カーボンブラック、炭酸カルシウム、合成ケイ酸およびケイ酸塩、亜鉛華、ハイサイトクレー、カオリン、塩基性炭酸マグネシウム、マイカ、タルク、石英粉、ケイ藻土、ドロマイト粉、酸化チタン、酸化亜鉛、酸化アンチモン、硫酸バリウム、硫酸カルシウム、アルミナ、ケイ酸カルシウム、窒化ホウ素、架橋高分子ポリスチレン、ロジン系金属塩などがあげられる。前記結晶核剤は、1種あるいは2種以上用いても構わない。
【0034】
本発明においては、さらに、可塑剤(C)を用いることにより、結晶化速度の向上に効果が得られる。本発明で使用する可塑剤(C)として、ポリ乳酸系樹脂や脂肪族ポリエステル系樹脂など他の生分解性を有する樹脂や、汎用プラスティックにおいて用いられる可塑剤を用いてもよい。可塑剤(C)の具体例としては、エーテル系可塑剤、エステル系可塑剤、フタル酸系可塑剤、リン系可塑剤などが好ましい。なかでも、ポリエステルとの相溶性に優れる点からエーテル系可塑剤、エステル系可塑剤がより好ましい。
【0035】
エーテル系可塑剤としては、例えばポリエチレングリコール、ポリプロピレングリコール、ポリテトラメチレングリコールなどのポリオキシアルキレングリコールなどをあげることができる。また、エステル系可塑剤としては脂肪族ジカルボン酸と脂肪族アルコールとのエステル類などをあげることができる。たとえば、セバシン酸ジメチル、グリセロルトリアセテートなどがあげられる。ここで脂肪族ジカルボン酸として、例えばシュウ酸、コハク酸、セバシン酸、アジピン酸などをあげることができ、脂肪族アルコールとして、例えばメタノール、エタノール、n−プロパノール、イソプロパノール、n−ヘキサノール、n−オクタノール、2−エチルヘキサノール、n−ドデカノール、ステアリルアルコールなどの一価アルコール、エチレングリコール、1,2−プロピレングリコール、1,3−プロピレングリコール、1,3−ブタンジオール、1,5−ペンタンジオール、1,6−ヘキサンジオール、ジエチレングリコール、ネオペンチルグリコール、ポリエチレングリコールなどの2価アルコール、また、グリセリン、トリメチロールプロパン、ペンタエリストールなどの多価アルコールをあげることができる。また、前記ポリエーテルとポリエステルの2種以上の組み合わせからなる共重合体、ジ−コポリマー、トリ−コポリマー、テトラ−コポリマーなど、またはこれらのホモポリマー、コポリマーなどから選ばれる2種以上のブレンド物があげられる。なかでも、結晶化促進の観点から、特に好ましくは、ポリエチレングリコール、セバシン酸ジメチル、グリセロルトリアセテートである。前記可塑剤は、1種あるいは2種以上用いても構わない。
【0036】
可塑剤(C)を用いることにより、樹脂組成物の粘度が低下し、そのことで、結晶核剤の分散性向上に繋がり、さらなる結晶化促進向上の効果が得られる。また、可塑剤(C)は、樹脂組成物のガラス転移温度を低下させ、熱によって分解しやすいという難点があるポリエステル樹脂の加工温度を低くする作用がある。さらに、可塑剤(C)は、樹脂組成物の最大結晶化温度をより低温側にシフトさせ、樹脂組成物をペレット化する場合において、樹脂組成によっては、室温近くで結晶化速度の増大が見込まれ、加熱結晶化養生など他の工程を簡略化できる効果もある。
【0037】
可塑剤(C)の含有量は、PHBH(A)100重量部に対し、0.1〜50重量部が好ましく、0.1〜20重量部がより好ましい。0.1重量部よりも少ないと、結晶化速度の効果は得られない傾向があり、50重量部をこえると、樹脂粘度の著しい低下が見られ、樹脂強度が大きく低下する傾向がある。
【0038】
前記PHBH(A)およびPHA(B)、またはPHBH(A)、PHA(B)および可塑剤(C)を混合する方式は、特に限定されるものではなく、適宜必要に応じて用いればよい。例えば、加熱溶融して混合する方法があげられる。加熱溶融して混合する方法としては、単軸押出機、2軸押出機、ニーダー、ギアポンプ、混練ロール、撹拌機を持つタンクなどの機械的撹拌、流れの案内装置により分流と合流を繰り返す静止混合器の応用などがあげられる。
【0039】
また、PHBH(A)およびPHA(B)、またはPHBH(A)、PHA(B)および可塑剤(C)を混合する方法として、可溶溶媒を用いて溶媒中で混合する方法があげられる。混合は撹拌スターラーなどで行ない、室温で、20時間撹拌溶解させるのが好ましい。つぎに、室温に放置するなどして、溶媒を除去し、本発明の生分解性樹脂組成物を得る。この場合の可溶溶媒とは、主にPHBH(A)、PHA(B)に対する可溶な溶媒であり、例えば、クロロホルムや酢酸エチルなどがあげられる。
【0040】
また、PHBH(A)およびPHA(B)、またはPHBH(A)、PHA(B)および可塑剤(C)を混合する方式として、PHBH(A)を生産する微生物の培養中もしくは精製段階において得られるスラリー中に、PHA(B)および可塑剤(C)を個別あるいは同時に添加してもよく、例えば、PHBH(A)の精製段階の内、メタノール洗浄を行なう工程中に添加する例などがあげられる。また、精製終了後は、遠心分離工程などを経て、液体分と樹脂固形分とを分離し、減圧乾燥後、本発明の生分解性樹脂組成物を得る。
【0041】
これらの混合方法のなかでも、結晶化促進の観点から、特にPHBH(A)を生産する微生物の培養中あるいは精製段階において、PHA(B)および可塑剤(C)を個別あるいは同時に添加、混合する方法が最も好ましい。
【0042】
また、樹脂組成物をペレット化する場合には、冷却側に水槽を用い、結晶化速度をさらに向上させるために、PHBH(A)の最大結晶化温度近辺の温水を用いてもよく、カッティングは、アンダーウオーターカットや、空中ストランドカッティング方式など、いずれを用いることができる。混合温度は、PHBH(A)の融解温度Tm1とPHA(B)の融解温度Tm2の間の温度であることが好ましく、Tm2以上の温度で混合した場合、PHBH(A)およびPHA(B)が熱によって分解する恐れがあるため、極短時間で混練することが好ましい。
【0043】
また、混合手順は、特に限定させるものでないが、PHBH(A)のペレットを作製した後に、PHA(B)および可塑剤(C)を添加混合する方法、PHBH(A)にPHA(B)または可塑剤(C)を先に混合し、マスターバッチを作製した後に、可塑剤(C)またはPHA(B)を添加混合する方法、PHA(B)および可塑剤(C)を先に混合し、マスターバッチを作製した後に、PHBH(A)に混合する方法などがあげられる。
【0044】
また、本発明の生分解性樹脂組成物には、必要に応じて、顔料、染料などの着色剤、無機系または有機系粒子、ガラス繊維、ウイスカー、雲母などの充填剤、酸化防止剤、紫外線吸収剤などの安定剤、滑剤、離型剤、撥水剤、抗菌剤その他の副次的添加剤を含有することができる。
【0045】
本発明の生分解性樹脂組成物は、各種繊維、糸、ロープ、織物、編物、不織布、紙、フィルム、シート、チューブ、板、棒、容器、袋、部品、発泡体などの形状に成形できる。また、2軸延伸フィルムにも加工できる。成形品は、農業、漁業、林業、園芸、医学、衛生品、衣料、非衣料、包装、その他の分野に好適に用いることができる。
【0046】
【実施例】
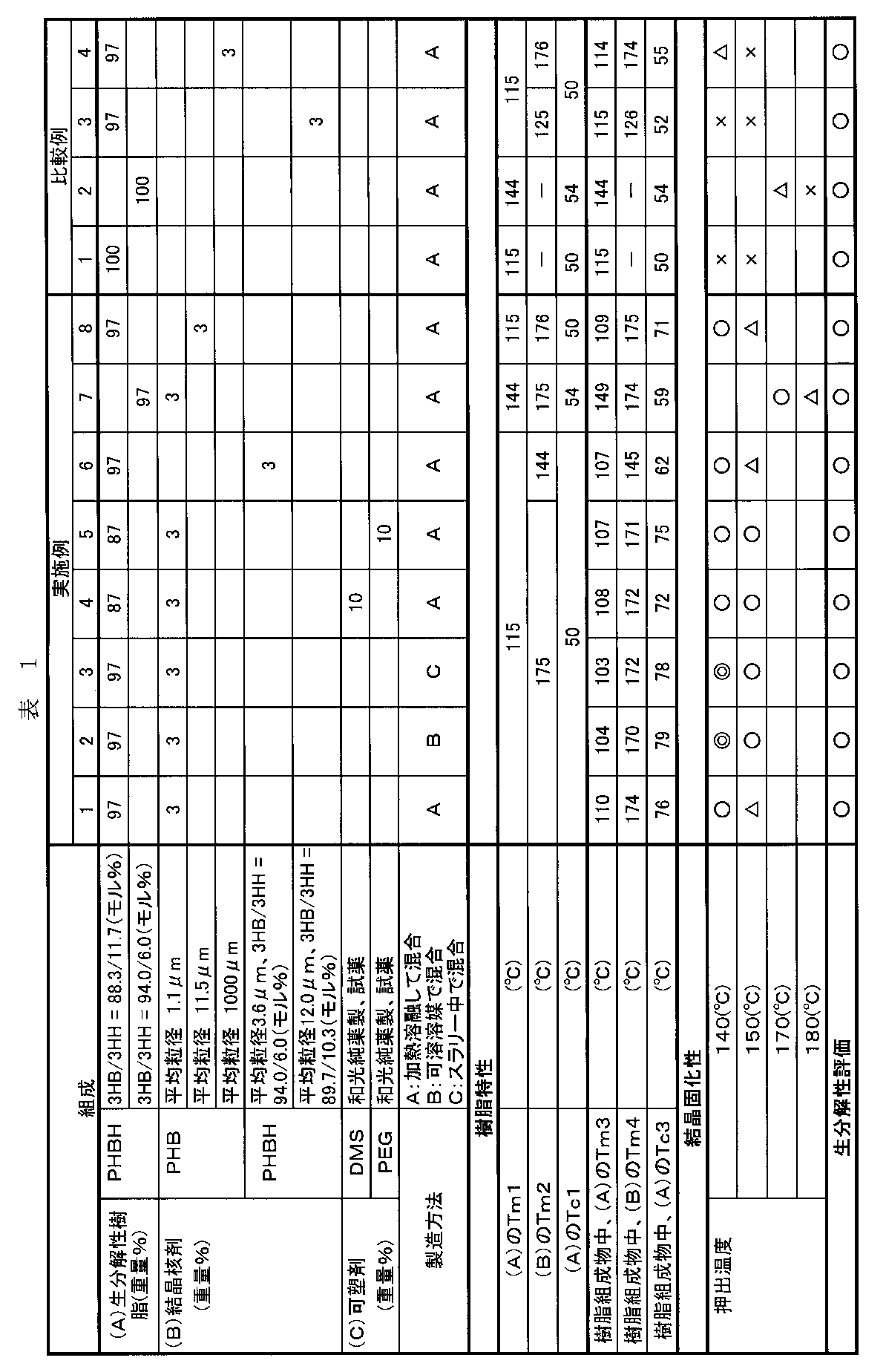
つぎに本発明の生分解性樹脂組成物およびその製造方法について実施例に基づいてさらに詳細に説明するが、本発明はかかる実施例のみに制限されるものではない。なお、特に断らない限り「部」は重量部を、「%」は重量%を表す。実施例で実施した評価方法は以下の通りである。結果はまとめて表1に示した。
【0047】
(1)結晶化温度(Tc)、融解温度(Tm):セイコー電子工業DSC200を用いて、PHBH(A)、またはPHA(B)1〜10mgをそれぞれに、10℃/分の昇温速度で、30℃からPHBH(A)、またはPHA(B)が充分に融解する想定融解温度+50〜60℃まで昇温し、ついで10℃/分の降温速度で30℃まで降温した(このときのPHBH(A)の結晶化に伴う発熱曲線ピークを記録した。PHBH(A)の結晶化温度(Tc1))。つぎに、再度10℃/分の昇温速度で、PHBH(A)、またはPHA(B)が充分に融解する想定融解温度+50〜60℃まで昇温した(このときのPHBH(A)、またはPHA(B)の融解に伴う吸熱曲線ピークを記録した。融解温度(Tm1、Tm2))。なお、PHBH(A)、またはPHA(B)の場合、再度昇温した時の吸熱曲線ピークは、単一または複数のピークを示し、複数の場合、それぞれにおいて、吸熱量の大きいピークトップ温度をTm1、Tm2とした。
【0048】
また、本発明の生分解性樹脂組成物1〜10mgを10℃/分の昇温速度で、30℃から樹脂組成物が充分に融解する想定融解温度+50〜60℃まで昇温し、ついで10℃/分の降温速度で30℃まで降温した(このときの樹脂組成物の結晶化に伴う発熱曲線ピークを記録した。樹脂組成物中のPHBH(A)の結晶化温度(Tc3))。つぎに、再度10℃/分の昇温速度で、樹脂組成物が充分に融解する想定融解温度+50〜60℃まで昇温した(このときの樹脂組成物の融解に伴う吸熱曲線ピークを記録した。融解温度(Tm3およびTm4))。なお、本発明の樹脂組成物の場合、再度昇温した時、PHBH(A)に由来するTm3、およびPHA(B)に由来するTm4の吸熱曲線ピークは、単一または複数のピークを示し、複数の場合、吸熱量の大きいピークトップ温度をそれぞれTm3、Tm4とした。
【0049】
(2)平均粒径:PHA(B)の水懸濁液を所定濃度に調整し、マイクロトラック粒度計(日機装製、FRA)を用いて、正規分布の全粒子の50%蓄積量に対応する粒径を平均粒径とし、3回の平均値を用いた。
【0050】
(3)結晶固化性:キャピログラフ(東洋精機製作所製)を用い、1mmφ×10mmのダイスを使用して、前記(1)で測定したTmを元に、Tm+20℃以上の温度範囲で溶融させ、剪断速度122/秒にて、樹脂組成物を溶融押出し、押出ストランドが押出吐出口から結晶固化するまでの距離および時間で、結晶固化性の評価をした。具体的には、組成比が、3HB/3HH=88.3/11.7(モル%)のPHBH(A)を用いた場合、押出温度を140および150℃にて実施した。また、組成比が、3HB/3HH=94.0/6.0モル%)のPHBH(A)を用いた場合、押出温度を170および180℃にて実施した。
◎:押出ストランドが、押出吐出口から出てきた時点で、結晶固化する
○:押出吐出口から結晶固化するまでの距離が、0〜約20cmである
△:押出吐出口から結晶固化するまでの距離が、約20〜50cmである
×:押出吐出口から結晶固化するまでの距離が、約50cm以上で、かつ、固化時間が、10分間以上である。
【0051】
(4)生分解性評価:樹脂組成物のプレスシートを、長さ115×幅25×厚み2(m)のダンベル状に切り出し、深さ10cmの土中に埋めて6ヶ月後、形状変化を観察し、分解性を以下の基準で評価した。
○:形状が確認できないほど分解
△:かなりの部分分解されているが、形状は何とか確認できる
×:ほとんど形状に変化なく、分解していない
【0052】
実施例1
微生物として、Alcaligenes eutrophusにAeromonas caviae由来のPHA合成酵素遺伝子を導入したAlcaligenes eutrophus AC32(J.Bacteriol.,179,4821(1997))を用いて生産されたPHBH(A)(3HB/3HH=88.3/11.7(モル%))100重量部に対して、PHA(B)として平均粒径1.1μmのPHB粉末(三菱ガス化学製、ビオグリーン)3重量部をドライブレンドした後、ラボプラストミル(東洋精機製作所製)を用いて、50rpm、ヒーター温度130℃の条件下で5分間混練し、餅状の樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。比較例1と比較し、140および150℃における、結晶固化速度が著しく速くなり、かつ、生分解性を有するポリエステル系樹脂組成物を得ることができた。なお、PHBH(A)の重量平均分子量は88万であった。
【0053】
実施例2
実施例1と同じ微生物を用いて生産されたPHBH(A)(3HB/3HH=88.3/11.7(モル%))100重量部に対して、PHA(B)として平均粒径1.1μmのPHB粉末(三菱ガス化学製、ビオグリーン)3重量部をドライブレンドした後、100mlサンプル瓶にクロロホルム溶剤(和光純薬製、試薬)と前記ドライブレンド物とを、クロロホルム溶媒/ブレンド物=90/10重量部の割合で、合計100g仕込み、撹拌スターラーで、室温、20時間撹拌溶解させた後、クロロホルム溶剤を蒸発させ、シート状の樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。比較例1と比較し、140および150℃における、結晶固化速度が著しく速くなり、かつ、生分解性を有するポリエステル系樹脂組成物を得ることができた。なお、PHBH(A)の重量平均分子量は88万であった。
【0054】
実施例3
実施例1と同じ微生物を用いて生産されたPHBH(A)(3HB/3HH=88.3/11.7(モル%))の培養後の精製工程の中で得られる溶媒スラリー中に、前記PHBH(A)100重量部に対して、PHA(B)として平均粒径1.1μmのPHB粉末(三菱ガス化学製、ビオグリーン)3重量部を添加し、撹拌翼で充分に撹拌した後、遠心分離器で溶媒と固形分とを分離して得られた樹脂組成物を、減圧乾燥し、粉末状の樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。比較例1と比較し、140および150℃における、結晶固化速度が著しく速くなり、かつ、生分解性を有するポリエステル系樹脂組成物を得ることができた。なお、PHBH(A)の重量平均分子量は88万であった。
【0055】
実施例4
可塑剤(C)として、セバシン酸ジメチル(以下、DMSと記す)を用いた以外は、実施例1と同様にして樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。比較例1と比較し、140および150℃における、結晶固化速度が著しく速くなり、かつ、生分解性を有するポリエステル系樹脂組成物を得ることができた。なお、PHBH(A)の重量平均分子量は88万であった。
【0056】
実施例5
可塑剤(C)として、ポリエチレングリコール(以下、PEGと記す)を用いた以外は、実施例1と同様にして樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。比較例1と比較し、140および150℃における、結晶固化速度が著しく速くなり、かつ、生分解性を有するポリエステル系樹脂組成物を得ることができた。なお、PHBH(A)の重量平均分子量は88万であった。
【0057】
実施例6
PHA(B)として、 平均粒径3.6μmのPHBH粉末(3HB/3HH=94.0/6.0(モル%))を用いた以外は、実施例1と同様にして樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。比較例1と比較し、140および150℃における、結晶固化速度が著しく速くなり、かつ、生分解性を有するポリエステル系樹脂組成物を得ることができた。なお、PHBH(A)の重量平均分子量は88万であった。
【0058】
実施例7
実施例1と同じ微生物を用いて生産されたPHBH(A)として、3HB/3HH =94.0/6.0(モル%)共重合組成物を用い、ヒーター温度170℃で行なった以外は、実施例1と同様にして樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。比較例2と比較し、170および180℃における、結晶固化速度が著しく速くなり、かつ、生分解性を有するポリエステル系樹脂組成物を得ることができた。なお、PHBH(A)の重量平均分子量は100万であった。
【0059】
実施例8
PHA(B)として、平均粒径11.5μmのPHB粉末(Good Fellow社製)を用いた以外は、実施例1と同様にして樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。比較例1と比較し、140および150℃における、結晶固化速度が著しく速くなり、かつ、生分解性を有するポリエステル系樹脂組成物を得ることができた。
【0060】
比較例1
PHB(B)を用いない以外は、実施例1と同様にして樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。前記実施例1〜6および8と比較し、140および150℃における、押出後のストランドはひどく粘着性を帯び、かつ、結晶固化速度は著しく遅く、押出直後から10分以上の結晶固化時間を要した。なお、PHBH(A)の重量平均分子量は88万であった。
【0061】
比較例2
PHB(B)を用いない以外は、実施例7と同様にして樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。実施例7と比較し、180℃における、押出後のストランドはひどく粘着性を帯び、かつ、結晶固化速度は著しく遅く、押出直後から10分以上の結晶固化時間を要した。
【0062】
比較例3
PHA(B)として、平均粒径12.0μmのPHBH粉末(3HB/3HH=89.7/10.3(モル%))共重合体粉末を用いた以外は、実施例1と同様にして樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。実施例1〜6および8と比較し、140および150℃における、押出後のストランドはひどく粘着性を帯び、かつ、結晶固化速度は著しく遅く、押出直後から10分以上の結晶固化時間を要した。なお、PHBH(A)の重量平均分子量は88万であった。
【0063】
比較例4
PHA(B)として、平均粒径1000μmのPHB粉末を用いた以外は、実施例1と同様にして樹脂組成物を得た。得られた樹脂組成物の樹脂特性、結晶固化性評価、および生分解性評価を表1に示す。実施例1〜6および8と比較し、150℃における、押出後のストランドはひどく粘着性を帯び、かつ、結晶固化速度は著しく遅く、押出直後から10分以上の結晶固化時間を要した。なお、PHBH(A)の重量平均分子量は88万であった。
【0064】
【表1】
【発明の効果】
本発明によれば、(A)融解温度Tm1を有する、PHBH100重量部に対して、(B)平均粒径300μm以下であり融解温度Tm2を有する、一般式(1):
[−CHR−CH2−CO−O−] (1)
(ここで、RはCnH2n+1で表されるアルキル基で、n=1〜15である。)
で表わされるPHA0.1〜20重量部を含有し、かつ、Tm2≧Tm1+20℃である生分解性樹脂組成物であるので、結晶化速度が著しく増大し、生分解性の優れた繊維、発泡品、成型品、フィルムやシートなどへの成形加工が容易である。したがって、本発明で得られる生分解性樹脂組成物は、使い捨ての包装材料や日用雑貨品などに有効に使用できる。[0001]
TECHNICAL FIELD OF THE INVENTION
The present invention relates to a biodegradable resin composition and a method for producing the same.
[0002]
[Prior art]
BACKGROUND ART Plastics are light, strong, and excellent in durability and moldability, so that they are widely used in various fields such as packaging materials, light electric parts, automobile parts, building materials, and daily necessities. However, these plastics, which are used in large quantities, have problems such as difficulties in waste disposal and generation of toxic gas during combustion. Damages have been reported and environmental pollution on a global scale is in progress. For this reason, many plastics that decompose in the natural environment have been developed, and many biodegradable resins have been produced, and a method for producing molded articles using these biodegradable resins has been developed. Many studies have been made. For example, a product produced by a microbial fermentation method and a product produced by blending starch, which is a natural polymer, with a synthetic plastic can be used.
[0003]
However, the production by the microbial fermentation method is not widely used because of the difficulty of mass production and the extremely high price of the resin as compared with general synthetic polymers. Blended products with synthetic plastics have poor moldability due to the non-thermoplastic nature of natural polymers, and attempts to utilize them in the production of molded articles have not been made widely. In addition, a method for producing a biodegradable resin using a general synthesis technique, for example, a production method by ring-opening polymerization of caprolactone, lactic acid or glycols has been studied. However, resins obtained by these methods also have problems in moldability, raw materials or production costs, and are used only for extremely limited special applications.
[0004]
On the other hand, among synthetic polymers, it is widely known that aliphatic polyester resins have biodegradability. However, the conventional aliphatic polyester-based resin has a low crystallization rate from the viewpoint of industrial productivity and cannot be said to be sufficient in terms of moldability, and improvement has been demanded. As an example using an aliphatic polyester which is a synthetic polymer, it is disclosed that the crystallization speed is improved by adding a nucleating agent or acetylene glycol to a high molecular weight aliphatic polyester (for example, see Patent References 1 and 2). Examples of poly (3-hydroxyalkanoates) having a low crystallization rate among aliphatic polyesters include 97-85 mol% of 3-hydroxybutyrate units and 4-hydroxybutyrate units. It is disclosed that a crystallization promoting effect can be obtained by adding boron nitride as a crystal nucleating agent to a polyester copolymer composed of 3 to 15 mol% units (for example, see Patent Document 3). Examples of using poly (3-hydroxybutyrate-co-3-hydroxyhexanoate), which is a poly (3-hydroxyalkanoate), include polyhydroxybutyrate and the like having a higher melting temperature. It is disclosed that the addition of (3-hydroxyalkanoate) increases the crystallization rate (for example, see Patent Document 4). However, no effect is remarkably obtained in any of the inventions, and the present invention provides an epoch-making crystallization promotion by adding poly (3-hydroxyalkanoate) having a fine particle size and a plasticizer. The effect has been obtained.
[0005]
[Patent Document 1]
JP-A-8-120165 (paragraph number [0005])
[Patent Document 2]
JP-A-2000-345014 (paragraph number [0006])
[Patent Document 3]
JP-A-6-157778 (paragraph number [0006])
[Patent Document 4]
WO 02/50156 A2 pamphlet (page 9, lines 2 to 24)
[0006]
[Problems to be solved by the invention]
The present invention provides a method for increasing the crystallization rate of poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) by using a molding method such as film molding, injection molding, extrusion molding, and spinning. Improves moldability such as prevention of fusion, improvement of mold release, shortening of molding cycle time, continuous resin pelletization, and also biodegradability as one of disposal means, that is, degradation by microorganisms etc. Provided is a biodegradable polyester-based resin composition which is easy to dispose after use and has excellent environmental compatibility, and a method for producing the same.
[0007]
Means for Solving the Invention
That is, the present invention relates to (A) 100 parts by weight of poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) having a melting temperature Tm1, and (B) an average particle diameter of 300 μm or less, General formula (1) having Tm2:
[-CHR-CH 2 -CO-O-] (1)
(Where R is C n H 2n + 1 Wherein n = 1 to 15. )
The present invention relates to a biodegradable resin composition containing 0.1 to 20 parts by weight of a poly (3-hydroxyalkanoate) represented by the formula: and Tm2 ≧ Tm1 + 20 ° C.
[0008]
Further, the biodegradable resin composition has a melting temperature Tm3 derived from poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) (A), and poly (3-hydroxyalkanoate) (B). And a melting temperature Tm4 derived from the following formula, and Tm4 ≧ Tm3 + 20 ° C.
[0009]
Further, the average particle size of the poly (3-hydroxyalkanoate) (B) is preferably 50 μm or less.
[0010]
Further, the composition ratio of the poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) (A) is such that 3-hydroxybutyrate / 3-hydroxyhexanoate = 99.9 to 80 / 0.1. -20 (mol%), and the poly (3-hydroxyalkanoate) (B) is preferably poly (3-hydroxybutyrate).
[0011]
Further, the composition ratio of the poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) (A) is such that 3-hydroxybutyrate / 3-hydroxyhexanoate = 90-80 / 10-20 (mol) %), And the poly (3-hydroxyalkanoate) (B) is poly (3-hydroxybutyrate-co-3-hydroxyhexanoate), and the composition ratio of the (B) is It is preferable that 3-hydroxybutyrate / 3-hydroxyhexanoate = 99.99 to 90.01 / 0.01 to 9.99 (mol%).
[0012]
Furthermore, the biodegradable resin composition further contains 0.1 to 50 parts by weight of a plasticizer (C) based on 100 parts by weight of poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) (A). It is preferred to contain a part.
[0013]
Further, the plasticizer (C) is preferably at least one selected from the group consisting of an ether-based plasticizer, an ester-based plasticizer, a phthalate-based plasticizer, and a phosphorus-based plasticizer.
[0014]
Further, the present invention relates to the above-mentioned poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) (A) and poly (3-hydroxyalkanoate) (B), or poly (3-hydroxybutyrate-co- A method of mixing (3-hydroxyhexanoate) (A), poly (3-hydroxyalkanoate) (B) and a plasticizer (C) is a method of kneading by heating and melting, and a method using a soluble solvent in a solvent. And a method for producing a biodegradable resin composition which is at least one selected from the group consisting of kneading during culturing of the microorganism producing (A) or in a slurry obtained in a purification step. About.
[0015]
Furthermore, the method of mixing may be a method of mixing during culturing of a microorganism producing poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) (A) or in a slurry obtained in a purification step. preferable.
[0016]
BEST MODE FOR CARRYING OUT THE INVENTION
The present invention relates to (A) 100 parts by weight of poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) having a melting temperature Tm1, and (B) an average particle size of 300 μm or less and a melting temperature Tm2. Having the general formula (1):
[-CHR-CH 2 -CO-O-] (1)
(Where R is C n H 2n + 1 Wherein n = 1 to 15. )
The present invention relates to a biodegradable resin composition containing 0.1 to 20 parts by weight of a poly (3-hydroxyalkanoate) represented by the formula: and Tm2 ≧ Tm1 + 20 ° C.
[0017]
The poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) (hereinafter referred to as PHBH) (A) comprises 3-hydroxybutyrate (hereinafter referred to as 3HB) and 3-hydroxyhexanoate. Ethate (hereinafter referred to as 3HH). The PHBH (A) may be obtained by any method of producing from a microorganism or a chemical synthesis method, and is not particularly limited. Above all, a method of producing from microorganisms is preferable because PHBH can be obtained by culturing microorganisms using oils and fats as raw materials, and the process is simpler and the cost is lower than in chemical synthesis.
[0018]
PHBH produced from microorganisms is preferable because PHBH has a broader molecular weight distribution than PHBH obtained by a chemical synthesis method, and 3HB and 3HH are polymerized moderately unevenly. Further, in PHBH obtained by a chemical synthesis method, unreacted monomer components, used polymerization initiators, and in the case of emulsion polymerization, emulsifiers and the like may remain in PHBH to deteriorate physical properties.
[0019]
The microorganism that produces PHBH is not particularly limited as long as it is a microorganism that accumulates PHBH in cells. lipolytica, A. eutrophus, A .; Latus and other bacteria of the genus Alcaligenes, the genus Pseudomonas, the genus Bacillus, the genus Azotobacter, the genus Nocardia, and the genus Aeromonas. Above all, in terms of efficiently producing PHBH, A.I. strains such as Caviae, and more preferably Alcaligenes eutrophus AC32 (FERM P-15786) (J. Bacteriol., 179, 4821-2830 (1997)) into which genes of the PHA synthase group have been introduced. A microbial cell obtained by culturing under appropriate conditions and accumulating PHBH in the cell is used.
[0020]
As for the composition ratio of the PHBH (A), the upper limit of the composition of 3HB is preferably 99.9 mol%, more preferably 99 mol%, further preferably 97 mol%, and further preferably 90 mol%. The lower limit of the composition of 3HB is preferably 80 mol%, more preferably 85 mol%. In general, the higher the composition ratio of 3HH, the more flexible the polymer properties of PHBH, but the lower the crystallization rate. In the present invention, if the composition of 3HB of PHBH (A) is less than 80 mol%, the crystallization rate tends to be slow, and if it exceeds 99.9 mol%, the crystallinity increases and the resin tends to become brittle. is there.
[0021]
The lower limit of the weight average molecular weight of the PHBH (A) is preferably 50,000 or more, more preferably 100,000 or more. If the weight average molecular weight is lower than 50,000, the change in the melt viscosity during processing is large, the fluidity tends to be high, and the moldability tends to decrease.
[0022]
The melting temperature Tm1 of the PHBH (A) is determined by using a differential scanning calorimeter (hereinafter, referred to as DSC) to measure 1 to 10 mg of PHBH (A) at a rate of 10 ° C./min from 30 ° C. to PHBH (A). ) Is sufficiently melted, the temperature is raised to +50 to 60 ° C., then the temperature is lowered to 30 ° C. at a rate of 10 ° C./min, and PHBH (A) is again heated at a rate of 10 ° C./min. It refers to the temperature at the peak top of the endothermic curve when the temperature is raised to the assumed melting temperature of sufficient melting + 50 to 60 ° C. In the PHBH (A), the endothermic curve peak when the temperature is raised again indicates a single or a plurality of peaks.
[0023]
The poly (3-hydroxyalkanoate) (hereinafter referred to as PHA) (B) may be obtained by any method of producing from a microorganism or a synthetic method, and is not particularly limited. The PHA (B) is represented by the general formula (1), and R is C n H 2n + 1 Wherein n = 1 to 15, and n = 1 to 3 is more preferable. When n exceeds 15, the effect as a crystal nucleating agent decreases.
[0024]
The PHA (B) is selected from homopolymers, copolymers composed of a combination of two or more polymer units, di-copolymers, tri-copolymers, tetra-copolymers and the like, or homopolymers and copolymers thereof. Blends of more than one species are included. Among them, PHB with n = 1, poly (3-hydroxyvalerate) with n = 2, poly (3-hydroxyhexanoate) with n = 3, poly (3-hydroxyoctanoate) with n = 5, A homopolymer of poly (3-hydroxyoctadecanoate) in which n = 15, or a copolymer, di-copolymer, tri-copolymer or a blend of two or more thereof in combination can be preferably used. Among them, PHBH, PHB, or a blend thereof is preferably used in terms of compatibility and dispersibility with the matrix resin.
[0025]
When the PHA (B) is a copolymer, the composition ratio is preferably 3HB / 3HH = 99.99 to 90.01 / 0.01 to 9.99 (mol%), and 99.99 to 95 / 0.01-5 (mol%) is more preferable. When the composition of 3HB is less than 95 mol%, the effect tends to be weak in promoting crystallization.
[0026]
In particular, when the composition ratio of PHBH (A) is 3HB / 3HH = 99.9 to 80 / 0.1 to 20 (mol%), the PHA (B) is made of PHB in terms of promoting crystallization. Preferably, there is.
[0027]
When the composition ratio of PHBH (A) is 3HB / 3HH = 90 to 80/10 to 20 (mol%), PHA (B) is PHBH in terms of uniform dispersibility and compatibility. It is preferable that the composition ratio is 3HB / 3HH = 99.99 to 90.01 / 0.01 to 9.99 (mol%). On the other hand, PHA (B) may be PHB in terms of promoting crystallization. Further, a blend of PHB and PHBH may be used as PHA (B).
[0028]
The melting temperature Tm2 of the PHA (B) is measured by using DSC as described above. The Tm2 is Tm2 ≧ Tm1 + 20 ° C. The fact that Tm2 satisfies Tm2 ≧ Tm1 + 20 ° C. is one of the factors that significantly increase the crystallization rate. When PHBH (A) is melted at a predetermined temperature, the crystal nucleus of PHA (B) having a higher melting temperature than PHBH (A) remains unmelted, and the crystal grows rapidly using the nucleus as a core point. It is.
[0029]
Further, the biodegradable resin composition of the present invention comprising PHBH (A) having the melting temperature Tm1 and PHA (B) having the melting temperature Tm2 has a melting temperature Tm3 derived from PHBH (A) and a PHA. It is preferable that the melting temperature Tm4 derived from (B) be satisfied, and that Tm4 ≧ Tm3 + 20 ° C.
[0030]
Tm3 and Tm4 are measured using DSC as described above. The Tm1 of PHBH (A) alone and Tm3 derived from PHBH (A) in the resin composition, and the Tm2 of PHA (B) alone and Tm4 derived from PHA (B) in the resin composition have the following composition: They do not always match because the substance contains additives other than PHBH (A) or PHA (B), and the crystallization growth process is different.
[0031]
The content of PHA (B) is 0.1 to 20 parts by weight based on 100 parts by weight of the PHBH (A). 0.1 to 10 parts by weight is more preferable. When the content of PHA (B) is less than 0.1 part by weight, the effect of improving the crystallization rate is insufficient, and when it exceeds 20 parts by weight, the effect corresponding to the content cannot be expected, Not only impractical, but uneconomical.
[0032]
The average particle size of the PHA (B) is determined by adjusting the aqueous suspension of the PHA (B) to a predetermined concentration using a general-purpose particle size analyzer such as a Microtrac particle sizer (manufactured by Nikkiso Co., Ltd., FRA). The particle size corresponding to the 50% accumulation amount is defined as the average particle size. The average particle size of PHA (B) is 300 μm or less. It is more preferably 100 μm or less, further preferably 50 μm or less, still more preferably 10 μm or less, and most preferably 5 μm or less. When the average particle size of PHA (B) exceeds 300 μm, it becomes difficult to uniformly disperse PHA (B) in the resin composition. When the average particle size of PHA (B) is 300 μm or less, fine crystal nuclei can be finely dispersed in the resin composition. The method for obtaining PHA (B) having an average particle diameter of 300 μm or less is not particularly limited, but the PHA-producing microorganism is cultured and the PHA-containing microorganism is subjected to physical crushing treatment to easily reduce the particle size ( PHA can be usually obtained. In the case of PHA having a large particle size, it can be crushed mechanically or redissolved in a solvent and spray-dried to obtain a desired particle size of 300 μm or less.
[0033]
Known nucleating agents other than the nucleating agent of PHA (B) include general-purpose plastics such as polyolefin resins such as polyethylene and polypropylene, aromatic polyester resins such as polyethylene terephthalate and polybutylene terephthalate, and polylactic acid-based resins. Other biodegradable resins, such as resin and aliphatic polyester resin, which exhibit an effect as a crystal nucleating agent may be used in combination with PHA (B). Specifically, carbon black, calcium carbonate, synthetic silicic acid and silicate, zinc white, high cytoclay, kaolin, basic magnesium carbonate, mica, talc, quartz powder, diatomaceous earth, dolomite powder, titanium oxide, Examples include zinc oxide, antimony oxide, barium sulfate, calcium sulfate, alumina, calcium silicate, boron nitride, cross-linked polystyrene, and rosin-based metal salts. The nucleating agent may be used alone or in combination of two or more.
[0034]
In the present invention, the use of the plasticizer (C) can further improve the crystallization rate. As the plasticizer (C) used in the present invention, another biodegradable resin such as a polylactic acid-based resin or an aliphatic polyester-based resin, or a plasticizer used in general-purpose plastics may be used. Specific examples of the plasticizer (C) include an ether plasticizer, an ester plasticizer, a phthalic plasticizer, and a phosphorus plasticizer. Among them, ether-based plasticizers and ester-based plasticizers are more preferable because of their excellent compatibility with polyester.
[0035]
Examples of the ether plasticizer include polyoxyalkylene glycol such as polyethylene glycol, polypropylene glycol, and polytetramethylene glycol. Examples of the ester plasticizer include esters of an aliphatic dicarboxylic acid and an aliphatic alcohol. For example, dimethyl sebacate, glycerol triacetate and the like can be mentioned. Here, examples of the aliphatic dicarboxylic acid include oxalic acid, succinic acid, sebacic acid, and adipic acid. Examples of the aliphatic alcohol include methanol, ethanol, n-propanol, isopropanol, n-hexanol, and n-octanol. , 2-ethylhexanol, n-dodecanol, monohydric alcohols such as stearyl alcohol, ethylene glycol, 1,2-propylene glycol, 1,3-propylene glycol, 1,3-butanediol, 1,5-pentanediol, And dihydric alcohols such as 6,6-hexanediol, diethylene glycol, neopentyl glycol, and polyethylene glycol, and polyhydric alcohols such as glycerin, trimethylolpropane, and pentaeristol. Further, a copolymer, a di-copolymer, a tri-copolymer, a tetra-copolymer, or the like composed of a combination of two or more of the polyether and the polyester, or a blend of two or more selected from homopolymers, copolymers, and the like thereof may be used. can give. Among these, polyethylene glycol, dimethyl sebacate, and glycerol triacetate are particularly preferable from the viewpoint of promoting crystallization. One or more plasticizers may be used.
[0036]
By using the plasticizer (C), the viscosity of the resin composition decreases, which leads to an improvement in the dispersibility of the crystal nucleating agent, and a further effect of promoting and improving crystallization is obtained. Further, the plasticizer (C) has an effect of lowering the glass transition temperature of the resin composition and lowering the processing temperature of the polyester resin, which has a disadvantage that it is easily decomposed by heat. Further, the plasticizer (C) shifts the maximum crystallization temperature of the resin composition to a lower temperature side, and when the resin composition is pelletized, an increase in the crystallization rate near room temperature is expected depending on the resin composition. This also has the effect of simplifying other steps such as heat crystallization curing.
[0037]
The content of the plasticizer (C) is preferably 0.1 to 50 parts by weight, more preferably 0.1 to 20 parts by weight, based on 100 parts by weight of PHBH (A). If the amount is less than 0.1 part by weight, the effect of the crystallization rate tends not to be obtained, and if it exceeds 50 parts by weight, the resin viscosity is remarkably reduced, and the resin strength tends to be greatly reduced.
[0038]
The method of mixing PHBH (A) and PHA (B), or PHBH (A), PHA (B) and plasticizer (C) is not particularly limited, and may be used as needed. For example, there is a method of mixing by heating and melting. As a method for heating and melting and mixing, there are mechanical stirring such as a single-screw extruder, a twin-screw extruder, a kneader, a gear pump, a kneading roll, a tank having a stirrer, and a static mixing in which splitting and joining are repeated by a flow guiding device. Application of the vessel.
[0039]
Further, as a method of mixing PHBH (A) and PHA (B), or PHBH (A), PHA (B) and plasticizer (C), there is a method of mixing in a solvent using a soluble solvent. The mixing is performed with a stirring stirrer or the like, and the mixture is preferably stirred and dissolved at room temperature for 20 hours. Next, the solvent is removed by leaving the mixture at room temperature or the like to obtain the biodegradable resin composition of the present invention. The soluble solvent in this case is a solvent that is mainly soluble in PHBH (A) and PHA (B), and examples thereof include chloroform and ethyl acetate.
[0040]
Further, as a method of mixing PHBH (A) and PHA (B), or PHBH (A), PHA (B) and plasticizer (C), it is obtained during culture of a microorganism producing PHBH (A) or in a purification stage. The PHA (B) and the plasticizer (C) may be added individually or simultaneously to the slurry to be obtained. For example, in the purification step of PHBH (A), an example in which PHA (B) and the plasticizer (C) are added during the step of washing with methanol is given. Can be After the purification, the liquid component and the resin solid component are separated through a centrifugal separation step and the like, and dried under reduced pressure to obtain the biodegradable resin composition of the present invention.
[0041]
Among these mixing methods, from the viewpoint of promoting crystallization, PHA (B) and plasticizer (C) are separately or simultaneously added and mixed during the cultivation of the microorganism producing PHBH (A) or during the purification stage. The method is most preferred.
[0042]
When pelletizing the resin composition, a water tank may be used on the cooling side, and hot water near the maximum crystallization temperature of PHBH (A) may be used to further improve the crystallization speed. , Underwater cut, aerial strand cutting, and the like. The mixing temperature is preferably a temperature between the melting temperature Tm1 of PHBH (A) and the melting temperature Tm2 of PHA (B), and when mixed at a temperature of Tm2 or higher, PHBH (A) and PHA (B) It is preferable to knead the mixture in a very short time because it may be decomposed by heat.
[0043]
In addition, the mixing procedure is not particularly limited, but a method in which PHA (B) and a plasticizer (C) are added and mixed after preparing pellets of PHBH (A), or PHA (B) or PHBH (A) is added to PHBH (A). After mixing the plasticizer (C) first to prepare a master batch, a method of adding and mixing the plasticizer (C) or PHA (B), mixing PHA (B) and the plasticizer (C) first, After a master batch is prepared, a method of mixing the master batch with PHBH (A) can be used.
[0044]
Further, the biodegradable resin composition of the present invention, if necessary, pigments, coloring agents such as dyes, inorganic or organic particles, glass fibers, whiskers, fillers such as mica, antioxidants, ultraviolet rays It may contain stabilizers such as absorbents, lubricants, release agents, water repellents, antibacterial agents and other secondary additives.
[0045]
The biodegradable resin composition of the present invention can be formed into various fibers, yarns, ropes, woven fabrics, knitted fabrics, nonwoven fabrics, papers, films, sheets, tubes, plates, bars, containers, bags, parts, foams, and the like. . It can also be processed into a biaxially stretched film. The molded article can be suitably used in agriculture, fishing, forestry, horticulture, medicine, sanitary goods, clothing, non-clothing, packaging, and other fields.
[0046]
【Example】
Next, the biodegradable resin composition of the present invention and the method for producing the same will be described in more detail with reference to Examples, but the present invention is not limited to only these Examples. Unless otherwise specified, "parts" indicates parts by weight and "%" indicates% by weight. The evaluation methods performed in the examples are as follows. The results are summarized in Table 1.
[0047]
(1) Crystallization temperature (Tc), melting temperature (Tm): Using Seiko Denshi Kogyo DSC200, 1 to 10 mg of PHBH (A) or PHA (B) was added at a rate of 10 ° C./min. The temperature was raised from 30 ° C. to an assumed melting temperature of PHBH (A) or PHA (B) +50 to 60 ° C., and then lowered to 30 ° C. at a rate of 10 ° C./min (PHBH at this time). The exothermic curve peak associated with crystallization of (A) was recorded: crystallization temperature (Tc1) of PHBH (A). Next, at a heating rate of 10 ° C./min, the temperature was increased to the assumed melting temperature of 50 to 60 ° C. at which PHBH (A) or PHA (B) was sufficiently melted (PHBH (A) at this time, or The endothermic curve peak associated with the melting of PHA (B) was recorded: melting temperature (Tm1, Tm2). In the case of PHBH (A) or PHA (B), the endothermic curve peak when the temperature is raised again indicates a single or a plurality of peaks. Tm1 and Tm2 were set.
[0048]
Further, 1 to 10 mg of the biodegradable resin composition of the present invention is heated at a heating rate of 10 ° C./min from 30 ° C. to an assumed melting temperature at which the resin composition sufficiently melts +50 to 60 ° C. The temperature was lowered to 30 ° C. at a temperature lowering rate of 30 ° C./min (the exothermic curve peak accompanying the crystallization of the resin composition was recorded. The crystallization temperature (Tc3) of PHBH (A) in the resin composition). Next, the resin composition was heated again at a heating rate of 10 ° C./min to an assumed melting temperature at which the resin composition sufficiently melts +50 to 60 ° C. (The endothermic curve peak accompanying the melting of the resin composition at this time was recorded. Melting temperature (Tm3 and Tm4)). In the case of the resin composition of the present invention, when the temperature is raised again, the endothermic curve peaks of Tm3 derived from PHBH (A) and Tm4 derived from PHA (B) show a single or multiple peaks, In a plurality of cases, the peak top temperatures having a large endothermic amount were defined as Tm3 and Tm4, respectively.
[0049]
(2) Average particle size: adjust the aqueous suspension of PHA (B) to a predetermined concentration, and use a Microtrac particle sizer (Nikkiso, FRA) to correspond to the 50% accumulation amount of all particles in a normal distribution. The particle diameter was defined as the average particle diameter, and the average value of three times was used.
[0050]
(3) Crystal solidification property: using a Capillograph (manufactured by Toyo Seiki Seisaku-sho, Ltd.), using a 1 mmφ × 10 mm die, based on the Tm measured in the above (1), melting at a temperature range of Tm + 20 ° C. or more, and shearing. The resin composition was melt-extruded at a rate of 122 / sec, and the crystallization solidification was evaluated based on the distance and time until the extruded strand crystallized from the extrusion discharge port. Specifically, when PHBH (A) having a composition ratio of 3HB / 3HH = 88.3 / 11.7 (mol%) was used, the extrusion was performed at 140 and 150 ° C. When PHBH (A) having a composition ratio of 3HB / 3HH (94.0 / 6.0 mol%) was used, the extrusion was performed at 170 and 180 ° C.
◎: Crystallized when the extruded strand comes out of the extrusion discharge port
:: The distance from the extrusion outlet to solidification of the crystal is 0 to about 20 cm.
Δ: The distance from the extrusion outlet to solidification of the crystal is about 20 to 50 cm.
X: The distance from the extrusion discharge port to solidification of the crystal is about 50 cm or more, and the solidification time is 10 minutes or more.
[0051]
(4) Evaluation of biodegradability: A press sheet of the resin composition was cut into a dumbbell shape having a length of 115 × width 25 × thickness of 2 (m), and buried in soil having a depth of 10 cm. Observation was made and the degradability was evaluated according to the following criteria.
○: Decomposed so that the shape cannot be confirmed
△: considerable partial decomposition, but the shape can be confirmed
×: Almost no change in shape and no decomposition
[0052]
Example 1
PHBH (A) (3H88 / H) was produced using Alcaligenes eutrophus AC32 (J. Bacteriol., 179, 4821 (1997)) in which a PHA synthase gene derived from Aeromonas caviae was introduced into Alcaligenes eutrophus. After dry blending 3 parts by weight of PHB powder (Bio Green, manufactured by Mitsubishi Gas Chemical Co., Ltd.) with an average particle diameter of 1.1 μm as PHA (B) with respect to 100 parts by weight of 3 / 11.7 (mol%)), Using a plastmill (manufactured by Toyo Seiki Seisaku-sho, Ltd.), the mixture was kneaded for 5 minutes under the conditions of 50 rpm and a heater temperature of 130 ° C. to obtain a cake-like resin composition. Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. As compared with Comparative Example 1, the crystallization speed at 140 and 150 ° C. was remarkably increased, and a polyester resin composition having biodegradability was obtained. The weight average molecular weight of PHBH (A) was 880,000.
[0053]
Example 2
With respect to 100 parts by weight of PHBH (A) (3HB / 3HH = 88.3 / 11.7 (mol%)) produced using the same microorganism as in Example 1, the average particle size as PHA (B) was 1. After dry blending 3 parts by weight of 1 μm PHB powder (manufactured by Mitsubishi Gas Chemical, Biogreen), a 100 ml sample bottle was mixed with a chloroform solvent (manufactured by Wako Pure Chemical Industries, Ltd.) and the dry blend, and a chloroform solvent / blend = A total of 100 g was charged in a proportion of 90/10 parts by weight, and the mixture was stirred and dissolved with a stirring stirrer at room temperature for 20 hours, and then the chloroform solvent was evaporated to obtain a sheet-shaped resin composition. Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. As compared with Comparative Example 1, the crystallization speed at 140 and 150 ° C. was remarkably increased, and a polyester resin composition having biodegradability was obtained. The weight average molecular weight of PHBH (A) was 880,000.
[0054]
Example 3
The PHBH (A) (3HB / 3HH = 88.3 / 11.7 (mol%)) produced using the same microorganism as in Example 1 was used in the solvent slurry obtained in the purification step after the culturing. To 100 parts by weight of PHBH (A), 3 parts by weight of PHB powder (Biogreen, manufactured by Mitsubishi Gas Chemical Co., Ltd.) having an average particle diameter of 1.1 μm was added as PHA (B), and the mixture was sufficiently stirred with a stirring blade. The resin composition obtained by separating the solvent and the solid content with a centrifugal separator was dried under reduced pressure to obtain a powdery resin composition. Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. As compared with Comparative Example 1, the crystallization speed at 140 and 150 ° C. was remarkably increased, and a polyester resin composition having biodegradability was obtained. The weight average molecular weight of PHBH (A) was 880,000.
[0055]
Example 4
A resin composition was obtained in the same manner as in Example 1, except that dimethyl sebacate (hereinafter, referred to as DMS) was used as the plasticizer (C). Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. As compared with Comparative Example 1, the crystallization speed at 140 and 150 ° C. was remarkably increased, and a polyester resin composition having biodegradability was obtained. The weight average molecular weight of PHBH (A) was 880,000.
[0056]
Example 5
A resin composition was obtained in the same manner as in Example 1, except that polyethylene glycol (hereinafter referred to as PEG) was used as the plasticizer (C). Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. As compared with Comparative Example 1, the crystallization speed at 140 and 150 ° C. was remarkably increased, and a polyester resin composition having biodegradability was obtained. The weight average molecular weight of PHBH (A) was 880,000.
[0057]
Example 6
A resin composition was obtained in the same manner as in Example 1 except that PHBH powder having an average particle size of 3.6 μm (3HB / 3HH = 94.0 / 6.0 (mol%)) was used as PHA (B). Was. Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. As compared with Comparative Example 1, the crystallization speed at 140 and 150 ° C. was remarkably increased, and a polyester resin composition having biodegradability was obtained. The weight average molecular weight of PHBH (A) was 880,000.
[0058]
Example 7
As PHBH (A) produced using the same microorganism as in Example 1, a 3HB / 3HH = 94.0 / 6.0 (mol%) copolymer composition was used, except that the reaction was performed at a heater temperature of 170 ° C. A resin composition was obtained in the same manner as in Example 1. Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. Compared with Comparative Example 2, a polyester resin composition having a significantly higher crystallization solidification rate at 170 and 180 ° C. and having biodegradability was obtained. The weight average molecular weight of PHBH (A) was 1,000,000.
[0059]
Example 8
A resin composition was obtained in the same manner as in Example 1, except that PHB powder (manufactured by Good Fellow) having an average particle size of 11.5 μm was used as PHA (B). Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. As compared with Comparative Example 1, the crystallization speed at 140 and 150 ° C. was remarkably increased, and a polyester resin composition having biodegradability was obtained.
[0060]
Comparative Example 1
A resin composition was obtained in the same manner as in Example 1 except that PHB (B) was not used. Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. Compared with Examples 1 to 6 and 8, the extruded strands at 140 and 150 ° C. were extremely tacky, and had a very low crystallization solidification rate, requiring a crystallization solidification time of at least 10 minutes immediately after extrusion. did. The weight average molecular weight of PHBH (A) was 880,000.
[0061]
Comparative Example 2
A resin composition was obtained in the same manner as in Example 7, except that PHB (B) was not used. Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. Compared with Example 7, the strand after extrusion at 180 ° C. was extremely tacky, and the crystallization solidification rate was extremely slow, requiring a crystallization solidification time of 10 minutes or more immediately after extrusion.
[0062]
Comparative Example 3
A resin was prepared in the same manner as in Example 1 except that PHBH powder (3HB / 3HH = 89.7 / 10.3 (mol%)) copolymer powder having an average particle size of 12.0 μm was used as PHA (B). A composition was obtained. Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. Compared with Examples 1 to 6 and 8, the strand after extrusion at 140 and 150 ° C. was extremely tacky, and the crystallization solidification rate was extremely slow, requiring a crystallization solidification time of 10 minutes or more immediately after extrusion. . The weight average molecular weight of PHBH (A) was 880,000.
[0063]
Comparative Example 4
A resin composition was obtained in the same manner as in Example 1, except that PHB powder having an average particle diameter of 1000 μm was used as PHA (B). Table 1 shows the resin properties, crystal solidification evaluation, and biodegradability evaluation of the obtained resin composition. Compared with Examples 1 to 6 and 8, the strand after extrusion at 150 ° C. was extremely tacky, and the crystallization solidification rate was extremely slow, requiring a crystallization solidification time of 10 minutes or more immediately after extrusion. The weight average molecular weight of PHBH (A) was 880,000.
[0064]
[Table 1]
【The invention's effect】
According to the present invention, (B) 100 parts by weight of PHBH having a melting temperature Tm1 and (B) having an average particle size of 300 μm or less and having a melting temperature Tm2 are represented by the following general formula (1):
[-CHR-CH 2 -CO-O-] (1)
(Where R is C n H 2n + 1 Wherein n = 1 to 15. )
Is a biodegradable resin composition containing 0.1 to 20 parts by weight of PHA represented by the formula: and Tm2 ≧ Tm1 + 20 ° C., so that the crystallization rate is remarkably increased and fibers and foams excellent in biodegradability are obtained. It can be easily formed into molded products, films and sheets. Therefore, the biodegradable resin composition obtained by the present invention can be effectively used for disposable packaging materials, daily necessities and the like.
Claims (9)
[−CHR−CH2−CO−O−] (1)
(ここで、RはCnH2n+1で表されるアルキル基で、n=1〜15である。)
で表わされるポリ(3−ヒドロキシアルカノエート)0.1〜20重量部を含有し、かつ、Tm2≧Tm1+20℃である生分解性樹脂組成物。(A) 100 parts by weight of poly (3-hydroxybutyrate-co-3-hydroxyhexanoate) having a melting temperature Tm1, and (B) an average particle diameter of 300 μm or less and a melting temperature Tm2. Equation (1):
[-CHR-CH 2 -CO-O- ] (1)
(Here, R is an alkyl group represented by C n H 2n + 1 and n = 1 to 15.)
A biodegradable resin composition containing 0.1 to 20 parts by weight of poly (3-hydroxyalkanoate) represented by the formula: and Tm2 ≧ Tm1 + 20 ° C.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002325983A JP2004161802A (en) | 2002-11-08 | 2002-11-08 | Biodegradable polyester resin composition and method for producing the same |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002325983A JP2004161802A (en) | 2002-11-08 | 2002-11-08 | Biodegradable polyester resin composition and method for producing the same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2004161802A true JP2004161802A (en) | 2004-06-10 |
Family
ID=32805041
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2002325983A Pending JP2004161802A (en) | 2002-11-08 | 2002-11-08 | Biodegradable polyester resin composition and method for producing the same |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2004161802A (en) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005162884A (en) * | 2003-12-03 | 2005-06-23 | Kaneka Corp | Film using poly(3-hydroxyalkanoate) composition |
| JP2006045366A (en) * | 2004-08-05 | 2006-02-16 | Kaneka Corp | Poly(3-hydroxybutylate-co-3-hydroxyhexanoate) composition and its molding |
| WO2007049694A1 (en) * | 2005-10-26 | 2007-05-03 | Kaneka Corporation | Expanded polyhydroxyalkanoate resin bead, molded object thereof, and process for producing the expanded resin bead |
| WO2008018567A1 (en) * | 2006-08-10 | 2008-02-14 | Kaneka Corporation | Biodegradable resin composition and molded body thereof |
| JP2009096849A (en) * | 2007-10-15 | 2009-05-07 | Tokyo Institute Of Technology | Biodegradable resin composition |
| US7919549B2 (en) * | 2005-05-09 | 2011-04-05 | Kaneka Corporation | Biodegradable resin composition and molded article produced from the same |
| US7973101B2 (en) * | 2005-05-13 | 2011-07-05 | Kaneka Corporation | Biodegradable resin composition and molded article produced from the same |
| US20120138503A1 (en) * | 2009-10-21 | 2012-06-07 | Innovative Bottles, Llc | Biodegradable pharmacy container and safety cap |
| JP2014227543A (en) * | 2013-05-27 | 2014-12-08 | 独立行政法人理化学研究所 | Polyester resin composition, method for manufacturing the same, and molding formed from the resin composition |
| WO2015146194A1 (en) * | 2014-03-28 | 2015-10-01 | 株式会社カネカ | Polyester resin composition, compact formed from such resin composition, and method for manufacturing such compact |
| EP2881435A4 (en) * | 2012-08-03 | 2016-01-06 | Kaneka Corp | Polyester resin composition and molded body containing this resin composition |
| JP2019166703A (en) * | 2018-03-23 | 2019-10-03 | 株式会社カネカ | Poly(3-hydroxybutyrate)-based resin sheet |
| WO2020195550A1 (en) * | 2019-03-28 | 2020-10-01 | 株式会社カネカ | Polyhydroxy alkanoate resin composition, molded article thereof, and film or sheet |
| CN113573869A (en) * | 2019-03-22 | 2021-10-29 | 株式会社钟化 | Poly (3-hydroxybutyrate) -based resin sheet for thermoforming, molded article thereof, and method for producing same |
| WO2022004637A1 (en) * | 2020-06-29 | 2022-01-06 | 株式会社カネカ | Resin film |
| WO2022009717A1 (en) * | 2020-07-07 | 2022-01-13 | 株式会社カネカ | Resin tube |
| WO2022065182A1 (en) * | 2020-09-28 | 2022-03-31 | 株式会社カネカ | Resin composition for injection molding, and injection-molded object |
| CN115023469A (en) * | 2020-01-29 | 2022-09-06 | 株式会社钟化 | Biodegradable polyester solution and use thereof |
| JP7134524B1 (en) | 2021-08-18 | 2022-09-12 | 川上産業株式会社 | Concrete sealing curing sheet |
| CN115157478A (en) * | 2022-09-02 | 2022-10-11 | 北京蓝晶微生物科技有限公司 | Granulation processing method of degradable material and molded body prepared by granulation processing method |
| CN115698399A (en) * | 2020-06-02 | 2023-02-03 | 三菱瓦斯化学株式会社 | Method for producing polymer molded article |
| WO2023190185A1 (en) * | 2022-03-29 | 2023-10-05 | 株式会社カネカ | Thermoplastic resin composition |
| WO2023190184A1 (en) * | 2022-03-29 | 2023-10-05 | 株式会社カネカ | Thermoplastic resin composition |
-
2002
- 2002-11-08 JP JP2002325983A patent/JP2004161802A/en active Pending
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2005162884A (en) * | 2003-12-03 | 2005-06-23 | Kaneka Corp | Film using poly(3-hydroxyalkanoate) composition |
| JP2006045366A (en) * | 2004-08-05 | 2006-02-16 | Kaneka Corp | Poly(3-hydroxybutylate-co-3-hydroxyhexanoate) composition and its molding |
| US7919549B2 (en) * | 2005-05-09 | 2011-04-05 | Kaneka Corporation | Biodegradable resin composition and molded article produced from the same |
| US7973101B2 (en) * | 2005-05-13 | 2011-07-05 | Kaneka Corporation | Biodegradable resin composition and molded article produced from the same |
| WO2007049694A1 (en) * | 2005-10-26 | 2007-05-03 | Kaneka Corporation | Expanded polyhydroxyalkanoate resin bead, molded object thereof, and process for producing the expanded resin bead |
| WO2008018567A1 (en) * | 2006-08-10 | 2008-02-14 | Kaneka Corporation | Biodegradable resin composition and molded body thereof |
| US8053491B2 (en) | 2006-08-10 | 2011-11-08 | Kaneka Corporation | Biodegradable resin composition and molded article of the same |
| JP5264487B2 (en) * | 2006-08-10 | 2013-08-14 | 株式会社カネカ | Biodegradable resin composition and molded article thereof |
| EP2050790A4 (en) * | 2006-08-10 | 2014-10-01 | Kaneka Corp | Biodegradable resin composition and molded body thereof |
| JP2009096849A (en) * | 2007-10-15 | 2009-05-07 | Tokyo Institute Of Technology | Biodegradable resin composition |
| US20120138503A1 (en) * | 2009-10-21 | 2012-06-07 | Innovative Bottles, Llc | Biodegradable pharmacy container and safety cap |
| US9527619B2 (en) * | 2009-10-21 | 2016-12-27 | Innovative Bottles, Inc. | Biodegradable pharmacy container and safety cap |
| EP2881435A4 (en) * | 2012-08-03 | 2016-01-06 | Kaneka Corp | Polyester resin composition and molded body containing this resin composition |
| JP2014227543A (en) * | 2013-05-27 | 2014-12-08 | 独立行政法人理化学研究所 | Polyester resin composition, method for manufacturing the same, and molding formed from the resin composition |
| EP3124544A4 (en) * | 2014-03-28 | 2017-12-27 | Kaneka Corporation | Polyester resin composition, compact formed from such resin composition, and method for manufacturing such compact |
| JPWO2015146194A1 (en) * | 2014-03-28 | 2017-04-13 | 株式会社カネカ | Polyester resin composition, molded article formed from the resin composition, and method for producing the same |
| WO2015146194A1 (en) * | 2014-03-28 | 2015-10-01 | 株式会社カネカ | Polyester resin composition, compact formed from such resin composition, and method for manufacturing such compact |
| JP2019166703A (en) * | 2018-03-23 | 2019-10-03 | 株式会社カネカ | Poly(3-hydroxybutyrate)-based resin sheet |
| JP7007968B2 (en) | 2018-03-23 | 2022-02-10 | 株式会社カネカ | Poly (3-hydroxybutyrate) resin sheet |
| CN113573869A (en) * | 2019-03-22 | 2021-10-29 | 株式会社钟化 | Poly (3-hydroxybutyrate) -based resin sheet for thermoforming, molded article thereof, and method for producing same |
| CN113631657B (en) * | 2019-03-28 | 2024-03-15 | 株式会社钟化 | Polyhydroxyalkanoate-based resin composition, molded article thereof, and film or sheet |
| CN113631657A (en) * | 2019-03-28 | 2021-11-09 | 株式会社钟化 | Polyhydroxyalkanoate resin composition, molded article thereof, and film or sheet |
| WO2020195550A1 (en) * | 2019-03-28 | 2020-10-01 | 株式会社カネカ | Polyhydroxy alkanoate resin composition, molded article thereof, and film or sheet |
| EP3950838A4 (en) * | 2019-03-28 | 2022-12-28 | Kaneka Corporation | Polyhydroxy alkanoate resin composition, molded article thereof, and film or sheet |
| CN115023469A (en) * | 2020-01-29 | 2022-09-06 | 株式会社钟化 | Biodegradable polyester solution and use thereof |
| CN115698399A (en) * | 2020-06-02 | 2023-02-03 | 三菱瓦斯化学株式会社 | Method for producing polymer molded article |
| WO2022004637A1 (en) * | 2020-06-29 | 2022-01-06 | 株式会社カネカ | Resin film |
| WO2022009717A1 (en) * | 2020-07-07 | 2022-01-13 | 株式会社カネカ | Resin tube |
| WO2022065182A1 (en) * | 2020-09-28 | 2022-03-31 | 株式会社カネカ | Resin composition for injection molding, and injection-molded object |
| JP7134524B1 (en) | 2021-08-18 | 2022-09-12 | 川上産業株式会社 | Concrete sealing curing sheet |
| JP2023027977A (en) * | 2021-08-18 | 2023-03-03 | 川上産業株式会社 | Concrete sealing curing sheet |
| WO2023190185A1 (en) * | 2022-03-29 | 2023-10-05 | 株式会社カネカ | Thermoplastic resin composition |
| WO2023190184A1 (en) * | 2022-03-29 | 2023-10-05 | 株式会社カネカ | Thermoplastic resin composition |
| CN115157478A (en) * | 2022-09-02 | 2022-10-11 | 北京蓝晶微生物科技有限公司 | Granulation processing method of degradable material and molded body prepared by granulation processing method |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP2004161802A (en) | Biodegradable polyester resin composition and method for producing the same | |
| JP5620061B2 (en) | Process for producing polylactic acid block copolymer | |
| JP5183203B2 (en) | Biodegradable resin composition and molded body thereof | |
| JP4078855B2 (en) | Polylactic acid block copolymer, process for producing the same, molded article, and polylactic acid composition | |
| CN102282194B (en) | Polylactic acid resin composition and additive for polylactic acid resin | |
| US6191203B1 (en) | Polymer blends containing polyhydroxyalkanoates and compositions with good retention of elongation | |
| KR101784221B1 (en) | Biodegradable resin composition and biodegradable film prepared from the same | |
| TW200838902A (en) | Method for producing polylactic acid | |
| WO2022142512A1 (en) | Semi-aromatic polyester, and preparation method therefor and application thereof | |
| WO2008018474A1 (en) | Polylactic acid and method for producing the same | |
| JP5353768B2 (en) | Resin composition | |
| JP6401615B2 (en) | Resin composition, resin molded body, and production method thereof | |
| JP5264487B2 (en) | Biodegradable resin composition and molded article thereof | |
| JP2007100104A (en) | Polylactic acid block copolymer, its preparation process, molded article and polylactic acid composition | |
| JPWO2005054366A1 (en) | Poly (3-hydroxyalkanoate) composition and molded article thereof | |
| JP2000017153A (en) | Resin composition and its molded product | |
| JP4326832B2 (en) | Method for producing biodegradable polyester resin composition | |
| KR101690082B1 (en) | Biodegradable resin composition and biodegradable film prepared therefrom | |
| JP2010059354A (en) | Polylactic acid composition | |
| JP2019119840A (en) | Aliphatic polyester resin composition | |
| JP2004331757A (en) | Poly(3-hydroxyalkanoate) composition and method for producing the same | |
| JP2008247956A (en) | Polyester composition | |
| KR20210009844A (en) | Polyester resin blend | |
| JP2006045366A (en) | Poly(3-hydroxybutylate-co-3-hydroxyhexanoate) composition and its molding | |
| JP2008248176A (en) | Method for producing stereocomplex polylactic acid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20050929 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20071130 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080708 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20090224 |