ES2948324A2 - Proceso para preparar (15alfa,16alfa,17beta)-estra-1,3,5(10)-trieno-3,15,16,17-tetrol monohidrato (Estetrol monohidrato) - Google Patents
Proceso para preparar (15alfa,16alfa,17beta)-estra-1,3,5(10)-trieno-3,15,16,17-tetrol monohidrato (Estetrol monohidrato) Download PDFInfo
- Publication number
- ES2948324A2 ES2948324A2 ES202330269A ES202330269A ES2948324A2 ES 2948324 A2 ES2948324 A2 ES 2948324A2 ES 202330269 A ES202330269 A ES 202330269A ES 202330269 A ES202330269 A ES 202330269A ES 2948324 A2 ES2948324 A2 ES 2948324A2
- Authority
- ES
- Spain
- Prior art keywords
- estetrol
- monohydrate
- hours
- reduced pressure
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Steroid Compounds (AREA)
Abstract
Proceso para preparar (15α,16α,17β)-estra-1,3,5(10)-trieno-3,15,16,17-tetrol monohidrato (Estetrol monohidrato). La presente invención se refiere a un proceso para preparar (15α,16α,17β)-estra-1,3,5 (10)-trieno-3,15,16,17-tetrol monohidrato, también conocido como Estetrol monohidrato.
Description
DESCRIPCIÓN
Proceso para preparar (15a,16a,17p)-estra-1,3,5(10)-trieno-3,15,16,17-tetrol monohidrato (Estetrol monohidrato)
CAMPO DE LA INVENCIÓN
La presente invención se refiere al sector de los procesos para la síntesis de ingredientes activos para uso farmacéutico, y en concreto a un proceso para preparar el compuesto a escala industrial (15a,16a,17p)-estra-1,3,5(10)-trieno-3,15,16,17-tetrol, también conocido como Estetrol, en forma de monohidrato.
ESTADO DE LA TÉCNICA
El compuesto Estetrol es un ingrediente activo con actividad farmacológica que hace que resulte útil para la terapia de reemplazo hormonal (Hormone Replacement Therapy, HRT), en la anticoncepción femenina, o en la terapia de disfunciones autoinmunes ligadas a desequilibrios hormonales.
La fórmula estructural del Estetrol se indica a continuación:

Cada una de las posiciones 15, 16 y 17 del esqueleto esteroidal (resaltadas en la fórmula indicada arriba) contienen un hidroxilo que, como se indica en la fórmula estructural, presenta una disposición espacial definida.
El Estetrol es un producto natural aislado de la orina humana y se conoce desde hace años; ha sido descrito en el artículo "Synthesis of epimeric 15-hydroxyestriols, new and potential metabolites of estradiol”, J. Fishman et al., JOC Vol. 33, N.° 8, Agosto 1968, p. 3133-3135 (compuesto la de la figura que se muestra en la página 3133).
En lo que respecta a la obtención de Estetrol, el proceso que se puede obtener a partir de este artículo no presenta aplicabilidad industrial debido al bajo rendimiento del proceso.
Se han publicado recientemente varias solicitudes de patente relacionadas con nuevos procesos de síntesis de Estetrol, pero ninguno de ellos evita la formación del isómero 15p,16p,17p, presentando la fórmula estructural
que se indica a continuación, a partir del cual se debe purificar el Estetrol para utilizarse en preparaciones farmacéuticas.

Por ejemplo, la solicitud WO 2004/041839 A2 (página 6, líneas 5-10) describe un proceso para obtener Estetrol cuya pureza puede alcanzar el 99 %, sin que la suma de las impurezas aisladas sea superior al 1 %. El ejemplo 11 de la pág. 28 describe un Estetrol con pureza HPLC del 99,1 % (HPLC-Ms) que, sin embargo, no aporta información sobre el contenido de las impurezas aisladas; el límite aceptado por las directrices internacionales para sustancias farmacéuticas es del 0,1 % para las desconocidas y del 0,15 % para las identificadas.
El contenido de impurezas en un ingrediente activo (API) es un requisito fundamental e irrevocable para permitir su uso en preparaciones farmacéuticas, y también es una característica esencial para definir un proceso susceptible de aplicación industrial. Cualquier proceso, independientemente del rendimiento, que proporciona un API con un contenido de impurezas que no respete los límites de las directrices internacionales no es un proceso útil desde el punto de vista industrial, ya que el API, el resultado del proceso, no se puede usar.
Solicitudes posteriores relacionadas con la producción de Estetrol son, por ejemplo, WO 2012/164096 A1, WO 2013/050553 A1 y WO 2015/040051 A1.
En el documento WO 2015/040051 A1, la relación Estetrol/isómero 15p,16p,17p es igual a 99:1 en los ejemplos 10 y 15, e igual a 98:2 en los ejemplos 11 y 17. No obstante, en estos ejemplos, no se dan indicaciones para reducir el contenido de isómero 15p,16p,17p a al menos un 0,15 %. Ni siquiera la purificación cromatográfica (ejemplo 15) permite obtener este resultado. En este documento se observa (página 9, líneas 5-15) que los procesos descritos en la técnica anterior descrita (representada, en el caso de este documento, por las solicitudes WO 2012/164096 A1 y WO 2013/050553 A1) proporcionan cantidades aún mayores e inaceptables del isómero 15p,16p,17p.
Por lo tanto, parece evidente que ninguno de los procesos descritos aporta una solución a la limitación de la formación del isómero 15p,16p,17p o un método de purificación de Estetrol a partir de dicho isómero.
SUMARIO DE LA INVENCIÓN
El objeto de la presente invención es proporcionar un proceso para la preparación de Estetrol monohidrato.
BREVE DESCRIPCIÓN DE LOS DIBUJOS
La Figura 1 muestra el cromatograma HPLC de Estetrol monohidrato que se puede obtener con el proceso de la invención.
La Figura 2 muestra el difractograma DRX del Estetrol monohidrato que se puede obtener con el proceso de la invención y de Estetrol anhidro.
La Figura 3 muestra el cromatograma DSC de Estetrol monohidrato que se puede obtener con el proceso de la invención.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
El Estetrol anhidro de pureza adecuada para los objetos de la invención se puede producir con un proceso de síntesis que comprende las etapas A) a D) descritas abajo.
La etapa A) consiste en la oxidación del compuesto (17p)-3-(fenilmetoxi)-estra-1,3,5(10),15-tetraen-17-ol (intermedio 1) para obtener el compuesto (17P)-3-(fenilmetoxi)-estra-1,3,5(10)-trieno-15,16,17-triol (intermedio 2):

donde Bn = bencilo, y en el que la configuración de los átomos de carbono 15 y 16 del esqueleto esteroidal del intermedio 2 no es fija.
El sustrato de partida de esta etapa, el intermedio 1, se puede obtener según lo descrito en la solicitud WO 2004/041839 A2.
Como oxidante en la reacción de la etapa A) es posible usar tetróxido de osmio (OsO4) soportado en un polímero o, preferiblemente, tal cual. Como cooxidante se usa una amina N-óxido orgánica, como trimetilamina N-óxido dihidratada.
Puesto que la oxidación con OsO4 no es estereoselectiva, el intermedio 2 se obtiene como una mezcla de isómeros con la configuración 15a,16a,17p y 15p,16p,17fr el isómero 15a,16a,17p se produce en cantidad preponderante junto con una cantidad minoritaria del isómero 15p,16p,17p.
La reacción se lleva a cabo en un solvente inerte a los derivados del osmio, como tetrahidrofurano (THF), a una temperatura de entre 35 y 60 °C, preferiblemente de entre 45 y 55 °C, y durante un período de al menos 12 horas, preferiblemente al menos 16 horas.
El producto de reacción (intermedio 2) tras el tratamiento final se trata con un producto que secuestra impurezas metálicas en solución para eliminar el contenido de osmio residual. Estos productos, muy conocidos en química, se basan por lo general en un gel de sílice funcionalizado y al que se suele hacer referencia en el sector con el término limpiador, que se empleará en el resto del texto y en las reivindicaciones. El limpiador es preferiblemente QuadraSil® MP.
El tratamiento con el limpiador se puede llevar a cabo y repetirse en cada etapa del proceso; preferiblemente, se lleva a cabo en la etapa A).
La etapa B) consiste en la acetilación del intermedio 2 para proporcionar el compuesto (15a,16a,17p)-3-(fenilmetoxi)-estra-1,3,5(10)-trieno-15,16,17-triol triacetato (intermedio 3) pasando por el intermedio 3’ en el que la configuración de los átomos de carbono 15 y 16 del esqueleto esteroidal no es fija:
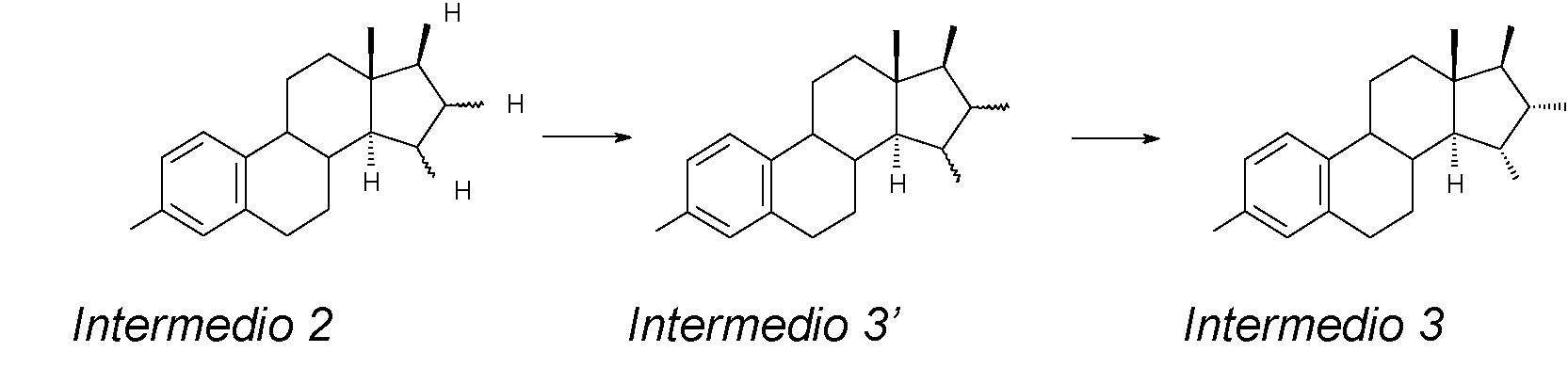
El intermedio 2, el sustrato de partida de la reacción de acetilación, se puede cargar en la reacción como sólido o, preferiblemente, se utiliza directamente la solución obtenida en la etapa A).
El resultado directo de la reacción de acetilación del intermedio 2 es el intermedio 3’, que consiste en una mezcla de los isómeros 15a,16a,17p y 15p,16p,17fr a continuación, dicha mezcla se separa con un proceso de purificación que constituye la segunda parte de la etapa B).
La acetilación exhaustiva de la etapa B) se realiza en un solvente compatible con las condiciones de la propia reacción, como, por ejemplo, acetato de isopropilo, acetato de etilo, tetrahidrofurano, piridina o tolueno. El solvente preferido es piridina.
Para la reacción, se usa anhídrido acético como reactante, en presencia de una base orgánica o inorgánica, de un catalizador y posiblemente de cantidades catalíticas de anhídrido trifluoroacético. Como base orgánica se utiliza preferiblemente piridina, y 4-dimetilaminopiridina como catalizador.
La temperatura de reacción es de entre 5 y 40 °C, preferiblemente de entre
20 y 30 °C; el tiempo de reacción es de al menos 3 horas, preferiblemente de al menos 4 horas.
La purificación del intermedio 3’, con la eliminación del isómero 15p,16p,17p, se obtiene con la secuencia de operaciones descrita a continuación:
B.1) un tratamiento térmico que consiste en reflujar el intermedio 3’ que se quiere purificar en un alcohol alifático lineal o ramificado C1-C6, durante al menos 10 minutos, preferiblemente durante al menos 15 minutos;
B.2) agitar la suspensión del intermedio 3’ que se quiere purificar en un alcohol alifático lineal o ramificado C1-C6, a una temperatura de entre 15 y 35 °C, preferiblemente de entre 20 y 30 °C e incluso más preferiblemente de entre 23 y 27 °C durante un período de entre 2 y 24 horas, preferiblemente durante un período de entre 3 y 18 horas, incluso más preferiblemente durante un período de entre 4 y 16 horas;
B.3) recuperar el intermedio 3 purificado mediante filtración.
El alcohol del tratamiento térmico (operación B.1) y de la suspensión (operación B.2) puede ser el mismo o distinto; preferiblemente, se utiliza el mismo alcohol, que preferiblemente es metanol.
El intermedio 3 que se quiere purificar se puede recuperar mediante filtración tras la operación B.1) y volver a suspenderse en el solvente para obtener la suspensión de la operación B.2), o se puede mantener siempre el mismo solvente operando en el mismo recipiente.
El tratamiento de purificación del intermedio 3 se puede repetir el número de veces que sea necesario para obtener el nivel deseado de pureza según el contenido inicial del isómero 15p,16p,17p. Preferiblemente, el proceso de purificación se repite al menos dos veces.
Los inventores realizaron una serie de pruebas experimentales repitiendo tres veces la secuencia de las operaciones B.1, B.2 y B.3 en muestras del intermedio 3’ que contienen un 5 % del isómero 15p,16p,17fr en la primera de estas pruebas, la operación B.2 de agitación de la suspensión se llevó a cabo tres veces durante 16 h, en una segunda prueba tres veces durante 8 h, y en una tercera prueba tres veces durante 4 h; estas pruebas confirmaron que el proceso que comprende las operaciones B.1 B.2 B.3, llevó en todos los casos a un producto final en el que el contenido del isómero 15p,16p,17p era inferior a un 0,10 % y, en algunos casos, inferior a un 0,05 %.
La etapa C) consiste en dos reacciones consecutivas, una primera desbencilación mediante hidrogenación catalítica del intermedio 3 para formar el intermedio 4, y a continuación la hidrólisis de los acetatos presentes en el
intermedio 4, según el siguiente esquema:

El orden en que se llevan a cabo es el que se ha indicado arriba. En primer lugar se realiza la desbencilación catalítica y, a continuación, la hidrólisis de los acetatos; la inversión del orden de las reacciones hace que sea difícil completar la desbencilación.
El intermedio 4 obtenido a partir de la primera reacción se puede aislar y a continuación reaccionar de nuevo, aunque preferiblemente este intermedio se mantiene disuelto en el solvente de la primera reacción.
Las condiciones de desbencilación e hidrólisis son las que resultan conocidas para los químicos expertos en química orgánica.
La primera reacción, la desbencilación, consiste en una hidrogenación con hidrógeno gaseoso en presencia de un catalizador adecuado. Las condiciones preferidas para esta reacción son:
- uso de paladio en carbón (Pd/C) a un 5 % o preferiblemente a un 10 % en peso como catalizador;
- presión de hidrógeno entre 1 y 6 bar, preferiblemente entre 2 y 4 bar, incluso más preferiblemente entre 2,5 y 3,5 bar;
- un alcohol alifático lineal o ramificado C1-C6, preferiblemente metanol, como el solvente de reacción;
- tiempo de reacción de al menos 16 horas, preferiblemente de al menos 20 horas;
- temperatura de hidrogenación de entre 30 y 60 °C, preferiblemente entre 35 y 55 °C, incluso más preferiblemente entre 40 y 50 °C.
La segunda reacción consiste en la hidrólisis de los acetatos del intermedio 4, utilizando bases. Las condiciones preferidas para esta reacción son:
- uso de carbonato de sodio, carbonato de potasio o carbonato de litio como base; preferiblemente se utiliza carbonato de potasio;
- tiempo de reacción de al menos 2 horas, preferiblemente de al menos 4
horas;
- temperatura de reacción entre 10 y 40 °C, preferiblemente entre 15 y 35 °C, incluso más preferiblemente entre 20 y 30 °C.
La solución que contiene el producto de reacción (Estetrol) se puede tratar con un limpiador a base de gel de sílice funcionalizado para eliminar el contenido residual de paladio. El limpiador es preferiblemente QuadraSil® MP.
Por último, la etapa D) consiste en la purificación del Estetrol obtenido en la etapa C).
Esta etapa se lleva a cabo mediante cristalización caliente-frío, de acuerdo con métodos conocidos por los expertos en química orgánica.
Los solventes utilizados son tetrahidrofurano (THF), metanol y acetonitrilo.
También en esta operación, el Estetrol se puede tratar con un limpiador a base de gel de sílice funcionalizado, preferiblemente QuadraSil® MP, para eliminar el contenido residual de paladio. El solvente en el que se usa el limpiador se selecciona de entre tetrahidrofurano (THF), metanol y acetonitrilo; preferiblemente se utiliza tetrahidrofurano.
Al término de esta operación, se obtiene Estetrol puro en forma «anhidra», esto es, con un contenido de agua residual mínimo, con una relación estequiométrica agua/API muy por debajo de 1.
La invención se dirige a la preparación de Estetrol en forma de monohidrato, que se lleva a cabo con la siguiente secuencia de operaciones:
E.1) disolver Estetrol puro en forma anhidra en un solvente orgánico miscible con agua, como acetona, metanol, etanol, isopropanol, tetrahidrofurano, dimetilformamida o dimetilacetamida hasta su solución completa; el solvente preferido es metanol;
E.2) mezclar la solución del punto E.1) con agua, preferiblemente agua pura; preferiblemente, esta operación se lleva a cabo haciendo gotear el agua en la solución orgánica de Estetrol;
E.3) eliminar el solvente orgánico mediante destilación, preferiblemente a presión reducida;
E.4) mantener la suspensión en agitación, preferiblemente durante al menos 15 minutos a una temperatura comprendida entre 5 y 20 °C;
E.5) filtrar y lavar el sólido; preferiblemente, el sólido filtrado se lava en el filtro con agua;
E.6) secar el sólido durante al menos 5 horas a al menos 40 °C y presión
reducida, preferiblemente durante al menos 6 horas a al menos 45 °C y presión reducida.
La invención se ilustrará ulteriormente con los siguientes ejemplos.
INSTRUMENTOS EXPERIMENTALES, MÉTODOS Y CONDICIONES RMN:
Espectrómetro RMN JEOL 400 YH (400 MHz); software JEOL Delta v5.1.1;
Espectros registrados en DMSO-d6.
MS:
Instrumento: DSQ-trace Thermofisher
Introducción de la muestra - sonda de exposición directa (dep) Ionización química (CI) con metano
Presión de metano: 2,2 psi
Temperatura fuente: 200 °C
HPLC:
Sistema de cromatografía Agilent modelo 1260 Infinity; Detector UV MODELO G1315C DAD VL+
Método HPLC 1:
Condiciones cromatográficas:
Columna: Supelco ascentis express C18 250x4,6 mm, 5^m
- Flujo: 1 ml/min
- Detector: UV 280 nm
- Volumen de inyección: 5 μl
- Temperatura: 25 °C
- Fase móvil A: agua
- Fase móvil B: acetonitrilo

Método HPLC 2:
Condiciones cromatográficas:
- Columna: Supelco discovery C18250x4,6 mm, 5^m - Flujo: 1 ml/min
- Detector: UV 280 nm
- Volumen de inyección: 25 μl
- Temperatura: 22 °C
- Fase móvil A: Solución 4,29 g/l de CH3COONH4 en agua/metanol/acetonitrilo 90/6/4
- Fase móvil B: Solución 38,6 g/l de CH3COONH4 en agua/metanol/acetonitrilo 10/54/36

TLC:
MERCK: TLC Gel de sílice 60 F254 Láminas de aluminio 20 x 20 cm, cód.
1.0554.0001.
Detector TLC:
Fosfomolibdato de cerio: Se disuelven 25 g de ácido fosfomolíbdico y 10 g de sulfato de cerio (IV) en 600 ml de H2O. Se añaden 60 ml de H2SO4 al 98 % y se lleva a 1 l con H2O. La placa se impregna con la solución y posteriormente se calienta hasta que se detectan los productos.
XPRD:
El análisis XPRD se realizó utilizando un difractómetro de polvo Bruker D2 Phaser (2.a edición) operando en geometría Bragg-Brentano, equipado con un muestreador múltiple rotativo y detector lineal de tipo SSD (Lynxeye). La fuente de rayos X es un tubo de rayos X con un ánodo de cobre que funciona a 30 KV y 10 mA. Para el análisis, se utiliza la radiación X que presenta una longitud de onda correspondiente a la Ka media del cobre (A = 1,54184 A). La radiación Kp se filtra a través de un filtro de níquel especial.
Se utlizaron portamuestras de silicio «de fondo cero» con una superficie plana sobre la que se extendió la muestra para formar una capa fina. Durante el análisis, se hace girar el portamuestras a una velocidad de 60 rpm.
La exploración se lleva a cabo en el intervalo 4-40° 20 con incrementos de 0,016° 20 y un tiempo de adquisición de 1,0 s por cada incremento.
Los difractogramas se procesaron utilizando el software Bruker DIFFRAC.EVA.
DSC:
El análisis DSC se realizó en una atmósfera inerte (nitrógeno) utilizando un calorímetro diferencial de barrido Perkin Elmer Diamond DSC. Las muestras se prepararon pesando el polvo en crisoles de aluminio de 40 μl, que posteriormente se sellaron antes del análisis. El análisis se realizó en el intervalo de temperatura 25-250 °C usando una velocidad de calentamiento de 10 °C/min.
NOTAS
El agua utilizada en las descripciones experimentales se debe entender como agua pura a no ser que se indique lo contrario.
Los solventes orgánicos usados en las descripciones experimentales se deben entender como de tipo «técnico» a no ser que se indique lo contrario.
Los reactivos y catalizadores usados en las descripciones experimentales
se deben entender como de calidad comercial a no ser que se indique lo contrario.
El producto QuadraSil® MP está disponible a través de Johnson Matthey.
EJEMPLO 1
Este ejemplo se refiere a la etapa A) anteriormente descrita, desde el intermedio 1 al intermedio 2.

En un matraz con nitrógeno, se cargaron 32,4 del intermedio 1 (89,87 mmol, 1 eq) y 356 ml de tetrahidrofurano. Se añadieron 0,324 g de tetróxido de osmio (1,28 mml, 1 % en peso) y 17,9 g de trimetilamina N-óxido dihidratada (161,26 mmol, 1,8 eq) en este orden a la solución. El sistema se calentó a 50 °C y se mantuvo en agitación durante 16 horas.
La reacción se controló mediante análisis TLC en las siguientes condiciones: Placa TLC: gel de sílice en alúmina; sustrato de partida (intermedio 1) disuelto en diclorometano; mezcla de reacción diluida en diclorometano; eluyente: acetato de etilo (EtOAc); detector: fosfomolibdato de cerio.
Al término de la reacción, la solución se enfrió hasta 25 °C y se hizo gotear una solución de metabisulfito de sodio (18,3 g) en agua (162 ml). El solvente se concentró a presión reducida y se añadieron al residuo 193 ml de acetato de isopropilo y 290 ml de ácido clorhídrico 1M.
Se añadieron 1,6 g de carbón y 1,6 g de dicalite al sistema bifásico y este se mantuvo en agitación a 25 °C durante 15 minutos. Primero se filtró la suspensión sobre una capa de dicalite y posteriormente en un filtro Millipore (0,22 ^m). Las fases se separaron y la fase acuosa se extrajo con 160 ml de acetato de isopropilo. Se añadieron 1,12 g de QuadraSil® MP a la fase orgánica y el sistema se mantuvo en agitación a 25 °C durante 16 horas. La suspensión se filtró en un filtro Millipore (0,22 ^m) lavando con 32 ml de acetato de isopropilo.
La solución obtenida de este modo se utilizó en la reacción posterior.
EJEMPLO 2
Este ejemplo se refiere a la etapa B) anteriormente descrita.

La solución del intermedio 2 obtenida según se ha descrito en el ejemplo anterior se concentró a presión reducida hasta un volumen residual de 50 ml.
Se añadieron 228 ml de piridina y el acetato de isopropilo residual se destiló a presión reducida. Se añadieron 0,877 g de 4-dimetilaminopiridina (7,19 mmol, 0,08 eq) a la solución y a continuación se hizo gotear 29,45 ml de anhídrido acético (312 mmol, 3,47 eq) mientras la temperatura se mantuvo por debajo de 30 °C. La solución se mantuvo en agitación a 25 °C durante 4 horas.
La reacción se controló mediante análisis TLC en las siguientes condiciones: Placa TLC: gel de sílice en alúmina; sustrato de partida (intermedio 2) disuelto en diclorometano; mezcla de reacción apagada en HCl 1M y extraída con EtOAc, la fase orgánica se depositó; eluyente: EtOAc; detector: fosfomolibdato de cerio.
La mezcla de reacción se concentró a presión reducida hasta un volumen residual de 85 ml y se añadieron 250 ml de acetato de isopropilo y 125 ml de agua. Se añadieron 55 ml de ácido clorhídrico al 37 % al sistema bifásico, mientras se mantuvo la temperatura por debajo de 30 °C (pH final de la fase acuosa = 1).
Las fases se separaron y la fase orgánica se lavó dos veces con solución saturada de bicarbonato de sodio (2 x 90 ml) y posteriormente con solución saturada de cloruro de sodio (90 ml).
La fase orgánica se concentró a presión reducida hasta formar un residuo oleoso. Se añadieron 100 ml de metanol y la mezcla se concentró de nuevo a presión reducida hasta formar una pasta. Se añadieron 210 ml de metanol y el sistema se puso en reflujo durante 15 minutos. La suspensión se enfrió hasta 25 °C y se mantuvo en agitación durante 16 horas. El sólido se filtró en un büchner lavando con 35 ml de metanol. El sólido se secó a presión reducida a 45 °C durante 3 horas.
Se obtuvieron 28,4 g de sólido que constituye el intermedio 3’; con un análisis HPLC (método 1) se detectó un contenido de isómero 15p,16p,17p = 1,6 %.
El sólido (28 g) se disolvió con 168 ml de metanol y el sistema se puso en reflujo durante 15 minutos. La suspensión se enfrió hasta 25 °C y se mantuvo en
agitación durante 16 horas. El sólido se filtró en un büchner lavando con 28 ml de metanol, y a continuación se secó a presión reducida a 45 °C durante 3 horas. Se obtuvieron 24 g de producto (HPLC, método 1): isómero 15p,16p,17p = 0,18 %).
El sólido (23,5 g) se disolvió con 140 ml de metanol y el sistema se puso en reflujo durante 15 minutos. La suspensión se enfrió hasta 25 °C y se mantuvo en agitación durante 16 horas. El sólido se filtró en un büchner lavando con 23 ml de metanol y se secó al vacío a 45 °C durante 3 horas.
Se obtuvieron 22,1 g del intermedio 3 (sólido casi blanco).
Pureza HPLC (método 1): 97,5 %, isómero 15p,16p,17p = 0,07 %.
1H-RMN (400 MHz, DMSO-da): 57,39-7,26 (m, 5H); 7,12 (d, 1H, J = 9,2 Hz); 6,72-6,67 (m, 2H); 5,22-5,18 (t, 1H, J = 7,4 Hz); 5,04-4,99 (m, 3H); 4,84 (d, 1H, J = 6,4 Hz); 2,74-2,70 (m, 2H); 2,25-2,20 (m, 2H); 1,99-1,97 (2s, 9H); 1,7-1,2 (m, 7H); 0,85 (s, 3H).
Masa (CI): m/z = 521 [M++1].
EJEMPLO 3
Este ejemplo se refiere a la implementación de la etapa C) anteriormente descrita.

Se cargaron 21,6 g del intermedio 3 obtenido según se ha descrito en el ejemplo anterior y 154 ml de tetrahidrofurano en un matraz.
Se añadieron 2,2 g de QuadraSil® MP a la solución y el sistema se mantuvo en agitación a 25 °C durante 16 horas. La suspensión se filtró en un filtro Millipore (0,22 ^m) lavando con 22 ml de tetrahidrofurano. El solvente se concentró a presión reducida hasta formar una pasta.
El residuo se disolvió con 650 ml de metanol y se cargó en un reactor de hidrogenación. Se añadieron 2,05 g de paladio sobre carbón al 10 % a la suspensión y se llevó a cabo la hidrogenación a 45 °C y 3 bar durante 22 horas.
La reacción se controló mediante análisis TLC en las siguientes condiciones: Placa TLC: gel de sílice en alúmina; sustrato de partida (intermedio 3) disuelto en diclorometano; mezcla de reacción diluida con metanol; eluyente:
heptano/EtOAc 1/1; detector: fosfomolibdato de cerio. Al término de la reacción, el sistema se filtró sobre una capa de dicalite (30 g) mediante lavado con metanol (120 ml).
El solvente se concentró a presión reducida hasta un volumen residual de 430 ml y se añadieron 5,16 g de carbonato de potasio. La mezcla se mantuvo en agitación a 25 °C durante 4 horas. La reacción se controló mediante análisis TLC en las siguientes condiciones: Placa TLC: gel de sílice en alúmina; producto intermedio 4 disuelto en diclorometano; mezcla de reacción apagada en HCl 1M y extraída con EtOAc, la fase orgánica se depositó; eluyente: heptano/EtOAc 1/1; detector: fosfomolibdato de cerio. La suspensión se filtró en un filtro Millipore (0,22 jm ) mediante lavado con metanol (20 ml).
La solución se concentró a presión reducida hasta un volumen residual de 54 ml, se añadieron 162 ml de agua y el metanol residual se eliminó a presión reducida.
La suspensión obtenida se neutralizó con 40 ml de ácido clorhídrico 1M y se enfrió hasta 10 °C agitando durante 30 minutos. El sólido se filtró en un büchner lavando con agua y se secó a presión reducida a 50 °C durante 6 horas.
Se obtuvieron 13 g de Estetrol bruto (sólido blanco).
EJEMPLO 4
Este ejemplo se refiere a la implementación de la etapa D) anteriormente descrita.
El Estetrol bruto, obtenido según lo descrito en el ejemplo anterior, se disolvió en 91 ml de tetrahidrofurano. Se añadieron 0,4 g de QuadraSil® MP a la solución y el sistema se mantuvo en agitación a 25 °C durante 16 horas. La suspensión se filtró en Millipore (0,22 |jm) lavando con 25 ml de tetrahidrofurano. El solvente se eliminó a presión reducida y se añadieron 130 ml de acetonitrilo y 104 ml de metanol. El sistema se mantuvo en agitación a 25 °C hasta su completa disolución.
La solución se concentró a presión reducida hasta un volumen residual de 130 ml y se añadieron 104 ml de acetonitrilo. El sistema se concentró de nuevo a presión reducida hasta un volumen residual de 130 ml y se añadieron 104 ml de acetonitrilo.
El sistema se concentró a presión reducida hasta un volumen residual de 130 ml y se mantuvo en agitación a 25 °C durante 3 horas. La suspensión se enfrió hasta 5 °C y se mantuvo en agitación durante 1 hora. El sólido se filtró en un büchner lavando con acetonitrilo frío, y se secó a presión reducida durante 3 horas a 45 °C.
Se obtuvieron 10,5 g de producto, que se analizó mediante HPLC (método HPLC 2). El producto resultó ser Estetrol de pureza HPLC = 99,91 %, con el isómero 15p,16p,17p no detectable.
Una muestra del producto se sometió a análisis XPRD; el resultado de la prueba es el difractograma que se muestra en la parte superior de la figura 2. La tabla que se muestra a continuación muestra las posiciones (como valores de ángulo 20 ± 0,2°) y las intensidades relativas de los principales picos del difractograma.

Otra muestra que pesaba 8 mg del producto obtenido se sometió a la prueba DSC, mostrando una T de fusión de aproximadamente 244,5 °C.
EJEMPLO 5
Este ejemplo se refiere a la implementación del proceso de la invención.
Se disolvieron 8 g de Estetrol obtenido en el ejemplo 4 en 96 ml de metanol y se hizo gotear 240 ml de agua en la solución preparada de este modo. El sistema se concentró a presión reducida hasta que se eliminó completamente el metanol. La suspensión se mantuvo en agitación a 15 °C durante 30 minutos y el sólido se filtró en un büchner lavando con 56 ml de agua.
El sólido se secó a presión reducida a 45 °C durante 6 horas. Se obtuvieron 8,3 g de Estetrol monohidrato (sólido blanco) y se analizaron mediante HPLC (método 2). Los resultados de la prueba se muestran en la figura 1: el producto ha resultado ser Estetrol monohidrato de pureza HPLC = 100 % (el pico en un tiempo de retención de aproximadamente 18’ no es atribuible al producto, sino a la propia elución cromatográfica).
Una muestra del producto se sometió a análisis XPRD; el resultado de la prueba es el difractograma que se muestra en la parte inferior de la figura 2. La siguiente tabla muestra las posiciones (como valores de ángulo 20 ± 0,2°) y las intensidades relativas de los principales picos del difractograma.

Otra muestra con un peso de 3,4 mg del producto obtenido se sometió a la prueba DSC; el resultado de la prueba se representa en la figura 3, que muestra un primer pico ensanchado con un máximo a aproximadamente 107,4 °C, atribuido a la deshidratación del Estetrol monohidrato, y un segundo pico a aproximadamente 244 °C, esto es, a una temperatura correspondiente fundamentalmente a la temperatura de fusión del Estetrol observada en la prueba del Ejemplo 4.
1H-RMN (400 MHz, DMSO-d6): 59,0 (s, 1H); 7,05 (d, 1H, J = 8,4 Hz); 6,51 6,48 (m, 1H); 6,27 (d, 1H, J = 2,4 Hz); 4,86-4,85 (d, 1H, J = 4,8 Hz); 4,61-4,59 (d, 1H, J = 5,6 Hz); 4,27-4,26 (d, 1H, J = 6 Hz); 3,72-3,66 (m, 2H); 3,26-3,24 (t, 1H, J = 5,6 Hz); 2,72-2,68 (m, 2H); 2,22-2,18 (m, 2H); 2,1-2,05 (m, 1H); 1,76-1,73 (d, 1H, 12 Hz); 1,4-1,03 (m, 5H); 0,66 (s, 3H).
Masa (CI): m/z = 305 [M++1].
Claims (3)
1. Proceso para la transformación de Estetrol en Estetrol monohidrato de acuerdo con la siguiente secuencia de operaciones:
E.1) disolver Estetrol puro en forma anhidra en un solvente orgánico miscible con agua, como acetona, metanol, etanol, isopropanol, tetrahidrofurano, dimetilformamida o dimetilacetamida hasta su solución completa;
E.2) mezclar la solución del punto E.1) con agua;
E.3) eliminar el solvente orgánico mediante destilación;
E.4) mantener la suspensión en agitación, preferiblemente durante al menos 15 minutos a una temperatura comprendida entre 5 y 20 °C;
E.5) filtrar y lavar el sólido;
E.6) secar el sólido durante al menos 5 horas a al menos 40 °C y presión reducida.
2. Proceso según la reivindicación 1, donde el agua utilizada en la etapa E.2) es agua pura.
3. Proceso según la reivindicación 1 o 2, donde la etapa E.3) se lleva a cabo a presión reducida.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES202330269A ES2948324A2 (es) | 2020-09-25 | 2020-09-25 | Proceso para preparar (15alfa,16alfa,17beta)-estra-1,3,5(10)-trieno-3,15,16,17-tetrol monohidrato (Estetrol monohidrato) |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES202330269A ES2948324A2 (es) | 2020-09-25 | 2020-09-25 | Proceso para preparar (15alfa,16alfa,17beta)-estra-1,3,5(10)-trieno-3,15,16,17-tetrol monohidrato (Estetrol monohidrato) |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2948324A2 true ES2948324A2 (es) | 2023-09-08 |
Family
ID=87887930
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES202330269A Pending ES2948324A2 (es) | 2020-09-25 | 2020-09-25 | Proceso para preparar (15alfa,16alfa,17beta)-estra-1,3,5(10)-trieno-3,15,16,17-tetrol monohidrato (Estetrol monohidrato) |
Country Status (1)
| Country | Link |
|---|---|
| ES (1) | ES2948324A2 (es) |
-
2020
- 2020-09-25 ES ES202330269A patent/ES2948324A2/es active Pending
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2915058B2 (es) | Proceso para preparar (15alfa,16alfa,17beta)-estra-1,3,5(10)-trieno-3,15,16,17-tetrol (Estetrol) e intermedios de dicho proceso | |
| US10273263B2 (en) | Pro-drug forming compounds | |
| WO2006120472A2 (en) | Novel beta-steroid compounds | |
| ES2948324A2 (es) | Proceso para preparar (15alfa,16alfa,17beta)-estra-1,3,5(10)-trieno-3,15,16,17-tetrol monohidrato (Estetrol monohidrato) | |
| Tian et al. | Catalytic transfer hydrogenation of 7-ketolithocholic acid to ursodeoxycholic acid with Raney nickel | |
| EP2890705B1 (en) | Process for the preparation of abiraterone or abiraterone acetate | |
| ES2943573A2 (es) | Proceso para la preparación de 21-(acetiloxi)-17-(1-oxopropoxi)-pregn-4-eno-3,20-diona | |
| ES2976651A2 (es) | PROCESO PARA PREPARAR (15alfa,16alfa,17beta)-ESTRA-1,3,5(10)-TRIENO-3,15,16,17-TETROL (ESTETROL) Y ESTETROL MONOHIDRATO | |
| US20230287036A1 (en) | Process for preparing (15alpha,16alpha,17beta)-estra-1,3,5(10)-triene-3,15,16,17-tetrol monohydrate (estetrol monohydrate) | |
| Lemini et al. | A comparative structural study of the steroid epimers: 17β-amino-1, 3, 5 (10)-estratrien-3-ol, 17α-amino-1, 3, 5 (10)-estratrien-3-ol, and some derivatives by 1H NMR, and x-ray diffraction analysis | |
| RU2818561C1 (ru) | СПОСОБ ПОЛУЧЕНИЯ (15α,16α,17β)-ЭСТРА-1,3,5(10)-ТРИЕН-3,15,16,17-ТЕТРОЛА (ЭСТЕТРОЛА) И ИНТЕРМЕДИАТЫ В ЭТОМ СПОСОБЕ | |
| TWI334781B (en) | Stereospecific synthesis of sapogenins | |
| ES2670477A2 (es) | Procedimiento para la preparacion de la 17beta-hidroxi-des-a-androst-9,10-en-5-ona | |
| CN114940695B (zh) | 一种具有抗肿瘤活性的雄诺龙衍生物及其制备方法与应用 | |
| CN116490512A (zh) | 21-(乙酰氧基)-17-(1-丙酰氧基)-孕甾-4-烯-3,20-二酮的制备方法 | |
| TR2023003649A2 (tr) | (15α,16α,17β)-ESTRA-1,3,5(10)-TRİEN-3,15,16,17-TETROL MONOHİDRAT (ESTETROL MONOHİDRAT) HAZIRLANMASINA YÖNELİK PROSES | |
| WO2022207607A1 (en) | PROCESS FOR PREPARING B-[(7α,17β)-17-HYDROXY-7-[9-[(4,4,5,5,5-PENTAFLUOROPENTYL)SULFINYL]NONYL]ESTRA-1,3,5(10)-TRIEN-3-YL]-BORONIC ACID | |
| WO2022238752A1 (en) | Process for the preparation of b-[(7alpha,17beta)-17-hydroxy-7-[9-[(4,4,5,5,5-pentafluoropentyl)sulfinyl]nonyl]estra-1,3,5(10)-trien-3-yl]-boronic acid and intermediates of said process | |
| ES2697706A2 (es) | Proceso Para La Preparación De Galeterona. | |
| TW200831526A (en) | Novel process for diastereoselectively obtaining a chiral primary amine on a steroid |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| BA2A | Patent application published |
Ref document number: 2948324 Country of ref document: ES Kind code of ref document: A2 Effective date: 20230908 |