ES2946673T3 - Agonistas del FPR2 de óxido de biaril dialquil fosfina - Google Patents
Agonistas del FPR2 de óxido de biaril dialquil fosfina Download PDFInfo
- Publication number
- ES2946673T3 ES2946673T3 ES20735802T ES20735802T ES2946673T3 ES 2946673 T3 ES2946673 T3 ES 2946673T3 ES 20735802 T ES20735802 T ES 20735802T ES 20735802 T ES20735802 T ES 20735802T ES 2946673 T3 ES2946673 T3 ES 2946673T3
- Authority
- ES
- Spain
- Prior art keywords
- halo
- pharmaceutically acceptable
- compound
- hydrogen
- acceptable salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229940125634 FPR2 agonist Drugs 0.000 title description 5
- AUONHKJOIZSQGR-UHFFFAOYSA-N oxophosphane Chemical compound P=O AUONHKJOIZSQGR-UHFFFAOYSA-N 0.000 title description 2
- 125000005841 biaryl group Chemical group 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 104
- 238000000034 method Methods 0.000 claims abstract description 47
- 238000011282 treatment Methods 0.000 claims abstract description 38
- 239000000203 mixture Substances 0.000 claims abstract description 19
- 206010019280 Heart failures Diseases 0.000 claims abstract description 18
- 125000005843 halogen group Chemical group 0.000 claims description 47
- 150000003839 salts Chemical class 0.000 claims description 34
- 229910052739 hydrogen Inorganic materials 0.000 claims description 33
- 239000001257 hydrogen Substances 0.000 claims description 33
- 125000000217 alkyl group Chemical group 0.000 claims description 20
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 19
- -1 haloC1-4alkyl Chemical group 0.000 claims description 18
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 17
- 150000002431 hydrogen Chemical group 0.000 claims description 14
- 208000019622 heart disease Diseases 0.000 claims description 10
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 9
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 9
- 125000003545 alkoxy group Chemical group 0.000 claims description 9
- 239000003937 drug carrier Substances 0.000 claims description 9
- 125000001188 haloalkyl group Chemical group 0.000 claims description 8
- 125000001072 heteroaryl group Chemical group 0.000 claims description 8
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 8
- 208000010125 myocardial infarction Diseases 0.000 claims description 8
- 125000004076 pyridyl group Chemical group 0.000 claims description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 6
- 230000000302 ischemic effect Effects 0.000 claims description 6
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 6
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 5
- 125000000229 (C1-C4)alkoxy group Chemical group 0.000 claims description 4
- 206010007556 Cardiac failure acute Diseases 0.000 claims description 4
- 206010007558 Cardiac failure chronic Diseases 0.000 claims description 4
- 206010007559 Cardiac failure congestive Diseases 0.000 claims description 4
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 4
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 4
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 4
- 125000006583 (C1-C3) haloalkyl group Chemical group 0.000 claims description 3
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 3
- 206010002383 Angina Pectoris Diseases 0.000 claims description 3
- 206010002388 Angina unstable Diseases 0.000 claims description 3
- 208000003037 Diastolic Heart Failure Diseases 0.000 claims description 3
- 208000007814 Unstable Angina Diseases 0.000 claims description 3
- 230000001154 acute effect Effects 0.000 claims description 3
- 208000029078 coronary artery disease Diseases 0.000 claims description 3
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 3
- 208000038002 heart failure with reduced ejection fraction Diseases 0.000 claims description 3
- 230000000642 iatrogenic effect Effects 0.000 claims description 3
- 201000004332 intermediate coronary syndrome Diseases 0.000 claims description 3
- 238000002560 therapeutic procedure Methods 0.000 claims description 3
- 208000008253 Systolic Heart Failure Diseases 0.000 claims description 2
- 125000004183 alkoxy alkyl group Chemical group 0.000 claims description 2
- 230000003683 cardiac damage Effects 0.000 claims description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 125000004765 (C1-C4) haloalkyl group Chemical group 0.000 claims 1
- 125000002853 C1-C4 hydroxyalkyl group Chemical group 0.000 claims 1
- 101000983970 Conus catus Alpha-conotoxin CIB Proteins 0.000 claims 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims 1
- 239000000556 agonist Substances 0.000 abstract description 18
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 18
- 201000010099 disease Diseases 0.000 abstract description 12
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 abstract description 9
- 201000001320 Atherosclerosis Diseases 0.000 abstract description 7
- 108090000765 processed proteins & peptides Proteins 0.000 abstract description 4
- 101710176384 Peptide 1 Proteins 0.000 abstract 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 abstract 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 40
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 34
- 238000005160 1H NMR spectroscopy Methods 0.000 description 28
- 239000000543 intermediate Substances 0.000 description 27
- 239000003795 chemical substances by application Substances 0.000 description 25
- 239000011541 reaction mixture Substances 0.000 description 22
- 239000002904 solvent Substances 0.000 description 21
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 19
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 17
- 239000000243 solution Substances 0.000 description 17
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 14
- 239000004480 active ingredient Substances 0.000 description 14
- 239000008194 pharmaceutical composition Substances 0.000 description 14
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 13
- 230000000694 effects Effects 0.000 description 13
- 230000002829 reductive effect Effects 0.000 description 12
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 11
- 239000003814 drug Substances 0.000 description 11
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 10
- 239000002245 particle Substances 0.000 description 10
- 229940124597 therapeutic agent Drugs 0.000 description 10
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- 229910002091 carbon monoxide Inorganic materials 0.000 description 8
- 239000006184 cosolvent Substances 0.000 description 8
- 238000011321 prophylaxis Methods 0.000 description 8
- 239000007787 solid Substances 0.000 description 8
- 229910052786 argon Inorganic materials 0.000 description 7
- 238000003556 assay Methods 0.000 description 7
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 6
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 6
- 125000003118 aryl group Chemical group 0.000 description 6
- 230000007211 cardiovascular event Effects 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- 239000003446 ligand Substances 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 102000014187 peptide receptors Human genes 0.000 description 6
- 108010011903 peptide receptors Proteins 0.000 description 6
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 6
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 239000003472 antidiabetic agent Substances 0.000 description 5
- 210000004027 cell Anatomy 0.000 description 5
- 230000008878 coupling Effects 0.000 description 5
- 238000010168 coupling process Methods 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- 235000019439 ethyl acetate Nutrition 0.000 description 5
- 239000003112 inhibitor Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 102000005962 receptors Human genes 0.000 description 5
- 108020003175 receptors Proteins 0.000 description 5
- 239000005695 Ammonium acetate Substances 0.000 description 4
- 206010061218 Inflammation Diseases 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- 206010028980 Neoplasm Diseases 0.000 description 4
- 208000027418 Wounds and injury Diseases 0.000 description 4
- 235000019257 ammonium acetate Nutrition 0.000 description 4
- 229940043376 ammonium acetate Drugs 0.000 description 4
- 229940125708 antidiabetic agent Drugs 0.000 description 4
- 125000002619 bicyclic group Chemical group 0.000 description 4
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 230000004761 fibrosis Effects 0.000 description 4
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 4
- 230000035876 healing Effects 0.000 description 4
- 230000004054 inflammatory process Effects 0.000 description 4
- 208000014674 injury Diseases 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 239000004033 plastic Substances 0.000 description 4
- 125000006239 protecting group Chemical group 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- 238000013268 sustained release Methods 0.000 description 4
- 239000012730 sustained-release form Substances 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 150000003672 ureas Chemical class 0.000 description 4
- 239000003643 water by type Substances 0.000 description 4
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 3
- YCCMTCQQDULIFE-UHFFFAOYSA-N 3-aminopiperidine-2-one Chemical compound NC1CCCNC1=O YCCMTCQQDULIFE-UHFFFAOYSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 208000032928 Dyslipidaemia Diseases 0.000 description 3
- 206010016654 Fibrosis Diseases 0.000 description 3
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 101000818546 Homo sapiens N-formyl peptide receptor 2 Proteins 0.000 description 3
- 101000818522 Homo sapiens fMet-Leu-Phe receptor Proteins 0.000 description 3
- 208000002193 Pain Diseases 0.000 description 3
- 206010040047 Sepsis Diseases 0.000 description 3
- 208000006011 Stroke Diseases 0.000 description 3
- 235000019270 ammonium chloride Nutrition 0.000 description 3
- 239000003524 antilipemic agent Substances 0.000 description 3
- 208000006673 asthma Diseases 0.000 description 3
- 150000005347 biaryls Chemical group 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 238000013262 cAMP assay Methods 0.000 description 3
- 235000013877 carbamide Nutrition 0.000 description 3
- 238000012512 characterization method Methods 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 206010012601 diabetes mellitus Diseases 0.000 description 3
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 3
- 102000045766 human FPR1 Human genes 0.000 description 3
- 102000057492 human FPR2 Human genes 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 3
- 239000012948 isocyanate Substances 0.000 description 3
- 210000005240 left ventricle Anatomy 0.000 description 3
- 208000031225 myocardial ischemia Diseases 0.000 description 3
- 239000012044 organic layer Substances 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 239000000123 paper Substances 0.000 description 3
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical class OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 230000004044 response Effects 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 241000894007 species Species 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 2
- UUUHXMGGBIUAPW-UHFFFAOYSA-N 1-[1-[2-[[5-amino-2-[[1-[5-(diaminomethylideneamino)-2-[[1-[3-(1h-indol-3-yl)-2-[(5-oxopyrrolidine-2-carbonyl)amino]propanoyl]pyrrolidine-2-carbonyl]amino]pentanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-methylpentanoyl]pyrrolidine-2-carbon Chemical compound C1CCC(C(=O)N2C(CCC2)C(O)=O)N1C(=O)C(C(C)CC)NC(=O)C(CCC(N)=O)NC(=O)C1CCCN1C(=O)C(CCCN=C(N)N)NC(=O)C1CCCN1C(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C1CCC(=O)N1 UUUHXMGGBIUAPW-UHFFFAOYSA-N 0.000 description 2
- SHEGZFCMBWOUCJ-UHFFFAOYSA-N 1-bromo-2-dimethylphosphorylbenzene Chemical compound CP(C)(=O)C1=CC=CC=C1Br SHEGZFCMBWOUCJ-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 208000024827 Alzheimer disease Diseases 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- 102000004145 Annexin A1 Human genes 0.000 description 2
- 108090000663 Annexin A1 Proteins 0.000 description 2
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- 208000011231 Crohn disease Diseases 0.000 description 2
- 201000003883 Cystic fibrosis Diseases 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- 206010052337 Diastolic dysfunction Diseases 0.000 description 2
- 102000016622 Dipeptidyl Peptidase 4 Human genes 0.000 description 2
- OHCQJHSOBUTRHG-KGGHGJDLSA-N FORSKOLIN Chemical compound O=C([C@@]12O)C[C@](C)(C=C)O[C@]1(C)[C@@H](OC(=O)C)[C@@H](O)[C@@H]1[C@]2(C)[C@@H](O)CCC1(C)C OHCQJHSOBUTRHG-KGGHGJDLSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 101000930822 Giardia intestinalis Dipeptidyl-peptidase 4 Proteins 0.000 description 2
- 101001059802 Homo sapiens N-formyl peptide receptor 3 Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 206010020751 Hypersensitivity Diseases 0.000 description 2
- 206010020772 Hypertension Diseases 0.000 description 2
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 2
- 102000036770 Islet Amyloid Polypeptide Human genes 0.000 description 2
- 108010041872 Islet Amyloid Polypeptide Proteins 0.000 description 2
- 208000019693 Lung disease Diseases 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- 102100028130 N-formyl peptide receptor 3 Human genes 0.000 description 2
- 208000036110 Neuroinflammatory disease Diseases 0.000 description 2
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 2
- 208000008589 Obesity Diseases 0.000 description 2
- 102000004270 Peptidyl-Dipeptidase A Human genes 0.000 description 2
- 108090000882 Peptidyl-Dipeptidase A Proteins 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 206010036049 Polycystic ovaries Diseases 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 201000004681 Psoriasis Diseases 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 230000008484 agonism Effects 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 230000009435 amidation Effects 0.000 description 2
- 238000007112 amidation reaction Methods 0.000 description 2
- 150000001412 amines Chemical group 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 2
- 229940125364 angiotensin receptor blocker Drugs 0.000 description 2
- 239000000883 anti-obesity agent Substances 0.000 description 2
- 239000003529 anticholesteremic agent Substances 0.000 description 2
- 229940125710 antiobesity agent Drugs 0.000 description 2
- 239000002830 appetite depressant Substances 0.000 description 2
- 125000004429 atom Chemical group 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 2
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 2
- 230000001275 ca(2+)-mobilization Effects 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 2
- 210000004413 cardiac myocyte Anatomy 0.000 description 2
- 229940107161 cholesterol Drugs 0.000 description 2
- 235000012000 cholesterol Nutrition 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 238000009833 condensation Methods 0.000 description 2
- 230000005494 condensation Effects 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- FMGSKLZLMKYGDP-USOAJAOKSA-N dehydroepiandrosterone Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC=C21 FMGSKLZLMKYGDP-USOAJAOKSA-N 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 235000019253 formic acid Nutrition 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 239000003292 glue Substances 0.000 description 2
- 208000038003 heart failure with preserved ejection fraction Diseases 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 210000000936 intestine Anatomy 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- IXAQOQZEOGMIQS-SSQFXEBMSA-M lipoxin A4(1-) Chemical compound CCCCC[C@H](O)\C=C\C=C/C=C/C=C/[C@@H](O)[C@@H](O)CCCC([O-])=O IXAQOQZEOGMIQS-SSQFXEBMSA-M 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 2
- 210000002540 macrophage Anatomy 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 230000002107 myocardial effect Effects 0.000 description 2
- 210000000440 neutrophil Anatomy 0.000 description 2
- 235000020824 obesity Nutrition 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- MPQXHAGKBWFSNV-UHFFFAOYSA-N oxidophosphanium Chemical class [PH3]=O MPQXHAGKBWFSNV-UHFFFAOYSA-N 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- DHHVAGZRUROJKS-UHFFFAOYSA-N phentermine Chemical compound CC(C)(N)CC1=CC=CC=C1 DHHVAGZRUROJKS-UHFFFAOYSA-N 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 2
- 201000010065 polycystic ovary syndrome Diseases 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 102000004196 processed proteins & peptides Human genes 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000007634 remodeling Methods 0.000 description 2
- 201000002793 renal fibrosis Diseases 0.000 description 2
- 206010039073 rheumatoid arthritis Diseases 0.000 description 2
- 206010039083 rhinitis Diseases 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 230000006641 stabilisation Effects 0.000 description 2
- 238000011105 stabilization Methods 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 2
- 229960004699 valsartan Drugs 0.000 description 2
- 239000003981 vehicle Substances 0.000 description 2
- 238000012800 visualization Methods 0.000 description 2
- DBGIVFWFUFKIQN-VIFPVBQESA-N (+)-Fenfluramine Chemical compound CCN[C@@H](C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-VIFPVBQESA-N 0.000 description 1
- DBGIVFWFUFKIQN-UHFFFAOYSA-N (+-)-Fenfluramine Chemical compound CCNC(C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-UHFFFAOYSA-N 0.000 description 1
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 1
- FXMZVILEJBDPNO-UHFFFAOYSA-N (2-oxopiperidin-3-yl) carbamate Chemical compound NC(=O)OC1CCCNC1=O FXMZVILEJBDPNO-UHFFFAOYSA-N 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 1
- RGXGEFSBDPGCEU-UHFFFAOYSA-N 1,4-dibromo-2,3-difluorobenzene Chemical compound FC1=C(F)C(Br)=CC=C1Br RGXGEFSBDPGCEU-UHFFFAOYSA-N 0.000 description 1
- BOVGTQGAOIONJV-BETUJISGSA-N 1-[(3ar,6as)-3,3a,4,5,6,6a-hexahydro-1h-cyclopenta[c]pyrrol-2-yl]-3-(4-methylphenyl)sulfonylurea Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1C[C@H]2CCC[C@H]2C1 BOVGTQGAOIONJV-BETUJISGSA-N 0.000 description 1
- OIRHKGBNGGSCGS-UHFFFAOYSA-N 1-bromo-2-iodobenzene Chemical compound BrC1=CC=CC=C1I OIRHKGBNGGSCGS-UHFFFAOYSA-N 0.000 description 1
- LLJFMFZYVVLQKT-UHFFFAOYSA-N 1-cyclohexyl-3-[4-[2-(7-methoxy-4,4-dimethyl-1,3-dioxo-2-isoquinolinyl)ethyl]phenyl]sulfonylurea Chemical compound C=1C(OC)=CC=C(C(C2=O)(C)C)C=1C(=O)N2CCC(C=C1)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 LLJFMFZYVVLQKT-UHFFFAOYSA-N 0.000 description 1
- NKOGCJIYHZVBDR-UHFFFAOYSA-N 1-phenylpiperidin-2-one Chemical class O=C1CCCCN1C1=CC=CC=C1 NKOGCJIYHZVBDR-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- ILPUOPPYSQEBNJ-UHFFFAOYSA-N 2-methyl-2-phenoxypropanoic acid Chemical class OC(=O)C(C)(C)OC1=CC=CC=C1 ILPUOPPYSQEBNJ-UHFFFAOYSA-N 0.000 description 1
- BFQSQUAVMNHOEF-UHFFFAOYSA-N 4-bromo-2,6-difluoroaniline Chemical compound NC1=C(F)C=C(Br)C=C1F BFQSQUAVMNHOEF-UHFFFAOYSA-N 0.000 description 1
- SJISCEAZUHNOMD-UHFFFAOYSA-N 4-phenylcyclohexan-1-amine Chemical compound C1CC(N)CCC1C1=CC=CC=C1 SJISCEAZUHNOMD-UHFFFAOYSA-N 0.000 description 1
- FLDSMVTWEZKONL-AWEZNQCLSA-N 5,5-dimethyl-N-[(3S)-5-methyl-4-oxo-2,3-dihydro-1,5-benzoxazepin-3-yl]-1,4,7,8-tetrahydrooxepino[4,5-c]pyrazole-3-carboxamide Chemical compound CC1(CC2=C(NN=C2C(=O)N[C@@H]2C(N(C3=C(OC2)C=CC=C3)C)=O)CCO1)C FLDSMVTWEZKONL-AWEZNQCLSA-N 0.000 description 1
- 239000005541 ACE inhibitor Substances 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- ITZMJCSORYKOSI-AJNGGQMLSA-N APGPR Enterostatin Chemical compound C[C@H](N)C(=O)N1CCC[C@H]1C(=O)NCC(=O)N1[C@H](C(=O)N[C@@H](CCCN=C(N)N)C(O)=O)CCC1 ITZMJCSORYKOSI-AJNGGQMLSA-N 0.000 description 1
- 206010000349 Acanthosis Diseases 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 206010000748 Acute febrile neutrophilic dermatosis Diseases 0.000 description 1
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 1
- 208000007848 Alcoholism Diseases 0.000 description 1
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 229920000856 Amylose Polymers 0.000 description 1
- 229940123413 Angiotensin II antagonist Drugs 0.000 description 1
- 102000008873 Angiotensin II receptor Human genes 0.000 description 1
- 108050000824 Angiotensin II receptor Proteins 0.000 description 1
- 102000015427 Angiotensins Human genes 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 208000009137 Behcet syndrome Diseases 0.000 description 1
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- 102000013585 Bombesin Human genes 0.000 description 1
- 108010051479 Bombesin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 206010006458 Bronchitis chronic Diseases 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000002080 C09CA02 - Eprosartan Substances 0.000 description 1
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- OCTTYPLXPKVODJ-UHFFFAOYSA-N CP(C1=C(C=CC=C1)B1OC(C(O1)(C)C)(C)C)(C)=O Chemical compound CP(C1=C(C=CC=C1)B1OC(C(O1)(C)C)(C)C)(C)=O OCTTYPLXPKVODJ-UHFFFAOYSA-N 0.000 description 1
- 101100101413 Caenorhabditis elegans ubh-4 gene Proteins 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 206010007572 Cardiac hypertrophy Diseases 0.000 description 1
- 208000006029 Cardiomegaly Diseases 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- RKWGIWYCVPQPMF-UHFFFAOYSA-N Chloropropamide Chemical compound CCCNC(=O)NS(=O)(=O)C1=CC=C(Cl)C=C1 RKWGIWYCVPQPMF-UHFFFAOYSA-N 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- 208000014085 Chronic respiratory disease Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 229920002911 Colestipol Polymers 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 206010056370 Congestive cardiomyopathy Diseases 0.000 description 1
- 206010010741 Conjunctivitis Diseases 0.000 description 1
- 102400000739 Corticotropin Human genes 0.000 description 1
- 101800000414 Corticotropin Proteins 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- FMGSKLZLMKYGDP-UHFFFAOYSA-N Dehydroepiandrosterone Natural products C1C(O)CCC2(C)C3CCC(C)(C(CC4)=O)C4C3CC=C21 FMGSKLZLMKYGDP-UHFFFAOYSA-N 0.000 description 1
- SUZLHDUTVMZSEV-UHFFFAOYSA-N Deoxycoleonol Natural products C12C(=O)CC(C)(C=C)OC2(C)C(OC(=O)C)C(O)C2C1(C)C(O)CCC2(C)C SUZLHDUTVMZSEV-UHFFFAOYSA-N 0.000 description 1
- 201000004624 Dermatitis Diseases 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 1
- 201000010046 Dilated cardiomyopathy Diseases 0.000 description 1
- 102000004980 Dopamine D2 Receptors Human genes 0.000 description 1
- 108090001111 Dopamine D2 Receptors Proteins 0.000 description 1
- 239000004131 EU approved raising agent Substances 0.000 description 1
- 208000030814 Eating disease Diseases 0.000 description 1
- 206010014561 Emphysema Diseases 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 108010011459 Exenatide Proteins 0.000 description 1
- HTQBXNHDCUEHJF-XWLPCZSASA-N Exenatide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 HTQBXNHDCUEHJF-XWLPCZSASA-N 0.000 description 1
- 206010073306 Exposure to radiation Diseases 0.000 description 1
- 208000019454 Feeding and Eating disease Diseases 0.000 description 1
- 108010076288 Formyl peptide receptors Proteins 0.000 description 1
- 102000011652 Formyl peptide receptors Human genes 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 108091006027 G proteins Proteins 0.000 description 1
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 1
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 1
- 101710198884 GATA-type zinc finger protein 1 Proteins 0.000 description 1
- 102000030782 GTP binding Human genes 0.000 description 1
- 108091000058 GTP-Binding Proteins 0.000 description 1
- 102400001370 Galanin Human genes 0.000 description 1
- 101800002068 Galanin Proteins 0.000 description 1
- 208000022461 Glomerular disease Diseases 0.000 description 1
- 102400000322 Glucagon-like peptide 1 Human genes 0.000 description 1
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- 201000005569 Gout Diseases 0.000 description 1
- 206010072579 Granulomatosis with polyangiitis Diseases 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 208000013875 Heart injury Diseases 0.000 description 1
- 102000004384 Histamine H3 receptors Human genes 0.000 description 1
- 108090000981 Histamine H3 receptors Proteins 0.000 description 1
- 101000869643 Homo sapiens Relaxin receptor 1 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 206010022489 Insulin Resistance Diseases 0.000 description 1
- 229940122199 Insulin secretagogue Drugs 0.000 description 1
- 229940122355 Insulin sensitizer Drugs 0.000 description 1
- 208000012659 Joint disease Diseases 0.000 description 1
- 208000011200 Kawasaki disease Diseases 0.000 description 1
- 102000000853 LDL receptors Human genes 0.000 description 1
- 108010001831 LDL receptors Proteins 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 206010049694 Left Ventricular Dysfunction Diseases 0.000 description 1
- 229940127470 Lipase Inhibitors Drugs 0.000 description 1
- 208000017170 Lipid metabolism disease Diseases 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- ZPXSCAKFGYXMGA-UHFFFAOYSA-N Mazindol Chemical compound N12CCN=C2C2=CC=CC=C2C1(O)C1=CC=C(Cl)C=C1 ZPXSCAKFGYXMGA-UHFFFAOYSA-N 0.000 description 1
- 239000000637 Melanocyte-Stimulating Hormone Substances 0.000 description 1
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 1
- 101710151321 Melanostatin Proteins 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- IBAQFPQHRJAVAV-ULAWRXDQSA-N Miglitol Chemical compound OCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO IBAQFPQHRJAVAV-ULAWRXDQSA-N 0.000 description 1
- 238000006751 Mitsunobu reaction Methods 0.000 description 1
- 102100035971 Molybdopterin molybdenumtransferase Human genes 0.000 description 1
- 101710119577 Molybdopterin molybdenumtransferase Proteins 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 208000009525 Myocarditis Diseases 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- PVLANTRCJQSBPX-UHFFFAOYSA-N NC1C(N(CCC1)C1=C(C(=C(C=C1)C1=C(C=CC=C1)P(=O)(C)C)F)F)=O Chemical compound NC1C(N(CCC1)C1=C(C(=C(C=C1)C1=C(C=CC=C1)P(=O)(C)C)F)F)=O PVLANTRCJQSBPX-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 206010029216 Nervousness Diseases 0.000 description 1
- 102400000064 Neuropeptide Y Human genes 0.000 description 1
- 241000208125 Nicotiana Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 239000005480 Olmesartan Substances 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 101150014691 PPARA gene Proteins 0.000 description 1
- KFSLWBXXFJQRDL-UHFFFAOYSA-N Peracetic acid Chemical compound CC(=O)OO KFSLWBXXFJQRDL-UHFFFAOYSA-N 0.000 description 1
- 208000005764 Peripheral Arterial Disease Diseases 0.000 description 1
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 208000024777 Prion disease Diseases 0.000 description 1
- 208000004403 Prostatic Hyperplasia Diseases 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 102100032444 Relaxin receptor 1 Human genes 0.000 description 1
- 206010063837 Reperfusion injury Diseases 0.000 description 1
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 description 1
- 208000005074 Retroviridae Infections Diseases 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 229910004298 SiO 2 Inorganic materials 0.000 description 1
- 208000021386 Sjogren Syndrome Diseases 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 208000010265 Sweet syndrome Diseases 0.000 description 1
- 206010071436 Systolic dysfunction Diseases 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 229940123464 Thiazolidinedione Drugs 0.000 description 1
- 208000024799 Thyroid disease Diseases 0.000 description 1
- JLRGJRBPOGGCBT-UHFFFAOYSA-N Tolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(C)C=C1 JLRGJRBPOGGCBT-UHFFFAOYSA-N 0.000 description 1
- NGBFQHCMQULJNZ-UHFFFAOYSA-N Torsemide Chemical compound CC(C)NC(=O)NS(=O)(=O)C1=CN=CC=C1NC1=CC=CC(C)=C1 NGBFQHCMQULJNZ-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 206010072686 Uveitic glaucoma Diseases 0.000 description 1
- 206010047139 Vasoconstriction Diseases 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 229930003779 Vitamin B12 Natural products 0.000 description 1
- FZNCGRZWXLXZSZ-CIQUZCHMSA-N Voglibose Chemical compound OCC(CO)N[C@H]1C[C@](O)(CO)[C@@H](O)[C@H](O)[C@H]1O FZNCGRZWXLXZSZ-CIQUZCHMSA-N 0.000 description 1
- 108010084455 Zeocin Proteins 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 229960001466 acetohexamide Drugs 0.000 description 1
- VGZSUPCWNCWDAN-UHFFFAOYSA-N acetohexamide Chemical compound C1=CC(C(=O)C)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 VGZSUPCWNCWDAN-UHFFFAOYSA-N 0.000 description 1
- WEVYAHXRMPXWCK-FIBGUPNXSA-N acetonitrile-d3 Chemical compound [2H]C([2H])([2H])C#N WEVYAHXRMPXWCK-FIBGUPNXSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 229950010850 acistrate Drugs 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 206010069351 acute lung injury Diseases 0.000 description 1
- 210000005006 adaptive immune system Anatomy 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000000048 adrenergic agonist Substances 0.000 description 1
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 201000007930 alcohol dependence Diseases 0.000 description 1
- 229940083712 aldosterone antagonist Drugs 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 201000009961 allergic asthma Diseases 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- 239000003888 alpha glucosidase inhibitor Substances 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 238000002266 amputation Methods 0.000 description 1
- 206010002022 amyloidosis Diseases 0.000 description 1
- 208000003455 anaphylaxis Diseases 0.000 description 1
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 239000003957 anion exchange resin Substances 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 229940124332 anorexigenic agent Drugs 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000879 anti-atherosclerotic effect Effects 0.000 description 1
- 230000001708 anti-dyslipidemic effect Effects 0.000 description 1
- 230000003510 anti-fibrotic effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000002253 anti-ischaemic effect Effects 0.000 description 1
- 230000003063 anti-neuropathic effect Effects 0.000 description 1
- 230000002946 anti-pancreatic effect Effects 0.000 description 1
- 230000002769 anti-restenotic effect Effects 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940125682 antidementia agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 229940030600 antihypertensive agent Drugs 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 230000003078 antioxidant effect Effects 0.000 description 1
- 229940127218 antiplatelet drug Drugs 0.000 description 1
- 229960004676 antithrombotic agent Drugs 0.000 description 1
- 230000025194 apoptotic cell clearance Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 230000006793 arrhythmia Effects 0.000 description 1
- 206010003119 arrhythmia Diseases 0.000 description 1
- 230000000923 atherogenic effect Effects 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- 230000001746 atrial effect Effects 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 239000002876 beta blocker Substances 0.000 description 1
- 229940097320 beta blocking agent Drugs 0.000 description 1
- 229960000516 bezafibrate Drugs 0.000 description 1
- IIBYAHWJQTYFKB-UHFFFAOYSA-N bezafibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1CCNC(=O)C1=CC=C(Cl)C=C1 IIBYAHWJQTYFKB-UHFFFAOYSA-N 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 229920000080 bile acid sequestrant Polymers 0.000 description 1
- 229940096699 bile acid sequestrants Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 229910052797 bismuth Inorganic materials 0.000 description 1
- JCXGWMGPZLAOME-UHFFFAOYSA-N bismuth atom Chemical compound [Bi] JCXGWMGPZLAOME-UHFFFAOYSA-N 0.000 description 1
- VHYCDWMUTMEGQY-UHFFFAOYSA-N bisoprolol Chemical compound CC(C)NCC(O)COC1=CC=C(COCCOC(C)C)C=C1 VHYCDWMUTMEGQY-UHFFFAOYSA-N 0.000 description 1
- 229960002781 bisoprolol Drugs 0.000 description 1
- DNDCVAGJPBKION-DOPDSADYSA-N bombesin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC=1NC2=CC=CC=C2C=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1NC(=O)CC1)C(C)C)C1=CN=CN1 DNDCVAGJPBKION-DOPDSADYSA-N 0.000 description 1
- 238000006795 borylation reaction Methods 0.000 description 1
- 208000029028 brain injury Diseases 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 150000004768 bromobenzenes Chemical class 0.000 description 1
- 206010006451 bronchitis Diseases 0.000 description 1
- MAEIEVLCKWDQJH-UHFFFAOYSA-N bumetanide Chemical compound CCCCNC1=CC(C(O)=O)=CC(S(N)(=O)=O)=C1OC1=CC=CC=C1 MAEIEVLCKWDQJH-UHFFFAOYSA-N 0.000 description 1
- 229960004064 bumetanide Drugs 0.000 description 1
- 230000003491 cAMP production Effects 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- SGZAIDDFHDDFJU-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SGZAIDDFHDDFJU-UHFFFAOYSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000004432 carbon atom Chemical group C* 0.000 description 1
- 230000009787 cardiac fibrosis Effects 0.000 description 1
- 239000000496 cardiotonic agent Substances 0.000 description 1
- 231100000457 cardiotoxic Toxicity 0.000 description 1
- 230000001451 cardiotoxic effect Effects 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 229960004195 carvedilol Drugs 0.000 description 1
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 230000010094 cellular senescence Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 210000003169 central nervous system Anatomy 0.000 description 1
- 239000002975 chemoattractant Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229960001761 chlorpropamide Drugs 0.000 description 1
- 230000001906 cholesterol absorption Effects 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 208000007451 chronic bronchitis Diseases 0.000 description 1
- 208000037976 chronic inflammation Diseases 0.000 description 1
- 230000006020 chronic inflammation Effects 0.000 description 1
- YZFWTZACSRHJQD-UHFFFAOYSA-N ciglitazone Chemical compound C=1C=C(CC2C(NC(=O)S2)=O)C=CC=1OCC1(C)CCCCC1 YZFWTZACSRHJQD-UHFFFAOYSA-N 0.000 description 1
- 229950009226 ciglitazone Drugs 0.000 description 1
- 230000005796 circulatory shock Effects 0.000 description 1
- 229960001214 clofibrate Drugs 0.000 description 1
- KNHUKKLJHYUCFP-UHFFFAOYSA-N clofibrate Chemical compound CCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 KNHUKKLJHYUCFP-UHFFFAOYSA-N 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- AGVAZMGAQJOSFJ-WZHZPDAFSA-M cobalt(2+);[(2r,3s,4r,5s)-5-(5,6-dimethylbenzimidazol-1-yl)-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl] [(2r)-1-[3-[(1r,2r,3r,4z,7s,9z,12s,13s,14z,17s,18s,19r)-2,13,18-tris(2-amino-2-oxoethyl)-7,12,17-tris(3-amino-3-oxopropyl)-3,5,8,8,13,15,18,19-octamethyl-2 Chemical compound [Co+2].N#[C-].[N-]([C@@H]1[C@H](CC(N)=O)[C@@]2(C)CCC(=O)NC[C@@H](C)OP(O)(=O)O[C@H]3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)\C2=C(C)/C([C@H](C\2(C)C)CCC(N)=O)=N/C/2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O AGVAZMGAQJOSFJ-WZHZPDAFSA-M 0.000 description 1
- 230000019771 cognition Effects 0.000 description 1
- 229960002604 colestipol Drugs 0.000 description 1
- GMRWGQCZJGVHKL-UHFFFAOYSA-N colestipol Chemical compound ClCC1CO1.NCCNCCNCCNCCN GMRWGQCZJGVHKL-UHFFFAOYSA-N 0.000 description 1
- OHCQJHSOBUTRHG-UHFFFAOYSA-N colforsin Natural products OC12C(=O)CC(C)(C=C)OC1(C)C(OC(=O)C)C(O)C1C2(C)C(O)CCC1(C)C OHCQJHSOBUTRHG-UHFFFAOYSA-N 0.000 description 1
- 239000013066 combination product Substances 0.000 description 1
- 229940127555 combination product Drugs 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 210000004351 coronary vessel Anatomy 0.000 description 1
- 229960000258 corticotropin Drugs 0.000 description 1
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 229960004597 dexfenfluramine Drugs 0.000 description 1
- WGLUMOCWFMKWIL-UHFFFAOYSA-N dichloromethane;methanol Chemical compound OC.ClCCl WGLUMOCWFMKWIL-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229960004890 diethylpropion Drugs 0.000 description 1
- XXEPPPIWZFICOJ-UHFFFAOYSA-N diethylpropion Chemical compound CCN(CC)C(C)C(=O)C1=CC=CC=C1 XXEPPPIWZFICOJ-UHFFFAOYSA-N 0.000 description 1
- WQAWEUZTDVWTDB-UHFFFAOYSA-N dimethyl(oxo)phosphanium Chemical compound C[P+](C)=O WQAWEUZTDVWTDB-UHFFFAOYSA-N 0.000 description 1
- 235000014632 disordered eating Nutrition 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- DLNKOYKMWOXYQA-UHFFFAOYSA-N dl-pseudophenylpropanolamine Natural products CC(N)C(O)C1=CC=CC=C1 DLNKOYKMWOXYQA-UHFFFAOYSA-N 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 206010013663 drug dependence Diseases 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 150000002066 eicosanoids Chemical class 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 231100000284 endotoxic Toxicity 0.000 description 1
- 230000002346 endotoxic effect Effects 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 229960001208 eplerenone Drugs 0.000 description 1
- JUKPWJGBANNWMW-VWBFHTRKSA-N eplerenone Chemical compound C([C@@H]1[C@]2(C)C[C@H]3O[C@]33[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)C(=O)OC)C[C@@]21CCC(=O)O1 JUKPWJGBANNWMW-VWBFHTRKSA-N 0.000 description 1
- 229960004563 eprosartan Drugs 0.000 description 1
- OROAFUQRIXKEMV-LDADJPATSA-N eprosartan Chemical compound C=1C=C(C(O)=O)C=CC=1CN1C(CCCC)=NC=C1\C=C(C(O)=O)/CC1=CC=CS1 OROAFUQRIXKEMV-LDADJPATSA-N 0.000 description 1
- SRCZQMGIVIYBBJ-UHFFFAOYSA-N ethoxyethane;ethyl acetate Chemical compound CCOCC.CCOC(C)=O SRCZQMGIVIYBBJ-UHFFFAOYSA-N 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 229960001519 exenatide Drugs 0.000 description 1
- 229960001582 fenfluramine Drugs 0.000 description 1
- 229960002297 fenofibrate Drugs 0.000 description 1
- YMTINGFKWWXKFG-UHFFFAOYSA-N fenofibrate Chemical compound C1=CC(OC(C)(C)C(=O)OC(C)C)=CC=C1C(=O)C1=CC=C(Cl)C=C1 YMTINGFKWWXKFG-UHFFFAOYSA-N 0.000 description 1
- 229940125753 fibrate Drugs 0.000 description 1
- 239000002319 fibrinogen receptor antagonist Substances 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 229960003765 fluvastatin Drugs 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 229960003883 furosemide Drugs 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 230000009395 genetic defect Effects 0.000 description 1
- 208000007565 gingivitis Diseases 0.000 description 1
- 229960004580 glibenclamide Drugs 0.000 description 1
- 229960000346 gliclazide Drugs 0.000 description 1
- 229960004346 glimepiride Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical compound O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- 229960001381 glipizide Drugs 0.000 description 1
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 1
- 229960003468 gliquidone Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000004217 heart function Effects 0.000 description 1
- 210000005003 heart tissue Anatomy 0.000 description 1
- 230000002008 hemorrhagic effect Effects 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 238000004896 high resolution mass spectrometry Methods 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 235000003642 hunger Nutrition 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 230000001227 hypertriglyceridemic effect Effects 0.000 description 1
- 206010020871 hypertrophic cardiomyopathy Diseases 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 201000010659 intrinsic asthma Diseases 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 229960002198 irbesartan Drugs 0.000 description 1
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 1
- 229910052741 iridium Inorganic materials 0.000 description 1
- GKOZUEZYRPOHIO-UHFFFAOYSA-N iridium atom Chemical compound [Ir] GKOZUEZYRPOHIO-UHFFFAOYSA-N 0.000 description 1
- 208000012947 ischemia reperfusion injury Diseases 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- ACRHBAYQBXXRTO-OAQYLSRUSA-N ivabradine Chemical compound C1CC2=CC(OC)=C(OC)C=C2CC(=O)N1CCCN(C)C[C@H]1CC2=C1C=C(OC)C(OC)=C2 ACRHBAYQBXXRTO-OAQYLSRUSA-N 0.000 description 1
- 229960003825 ivabradine Drugs 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- RLAWWYSOJDYHDC-BZSNNMDCSA-N lisinopril Chemical compound C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 RLAWWYSOJDYHDC-BZSNNMDCSA-N 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 239000002171 loop diuretic Substances 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 229960000299 mazindol Drugs 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000006883 memory enhancing effect Effects 0.000 description 1
- 208000030159 metabolic disease Diseases 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- 229960001110 miglitol Drugs 0.000 description 1
- 239000002394 mineralocorticoid antagonist Substances 0.000 description 1
- 238000003032 molecular docking Methods 0.000 description 1
- SLZIZIJTGAYEKK-CIJSCKBQSA-N molport-023-220-247 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)CN)[C@@H](C)O)C1=CNC=N1 SLZIZIJTGAYEKK-CIJSCKBQSA-N 0.000 description 1
- 208000001725 mucocutaneous lymph node syndrome Diseases 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 201000006938 muscular dystrophy Diseases 0.000 description 1
- 210000000066 myeloid cell Anatomy 0.000 description 1
- 210000000107 myocyte Anatomy 0.000 description 1
- DILRJUIACXKSQE-UHFFFAOYSA-N n',n'-dimethylethane-1,2-diamine Chemical compound CN(C)CCN DILRJUIACXKSQE-UHFFFAOYSA-N 0.000 description 1
- 239000006070 nanosuspension Substances 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229960000698 nateglinide Drugs 0.000 description 1
- OELFLUMRDSZNSF-BRWVUGGUSA-N nateglinide Chemical compound C1C[C@@H](C(C)C)CC[C@@H]1C(=O)N[C@@H](C(O)=O)CC1=CC=CC=C1 OELFLUMRDSZNSF-BRWVUGGUSA-N 0.000 description 1
- 230000003959 neuroinflammation Effects 0.000 description 1
- 235000005152 nicotinamide Nutrition 0.000 description 1
- 239000011570 nicotinamide Substances 0.000 description 1
- 229960003966 nicotinamide Drugs 0.000 description 1
- 229960003512 nicotinic acid Drugs 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- ODUCDPQEXGNKDN-UHFFFAOYSA-N nitroxyl Chemical compound O=N ODUCDPQEXGNKDN-UHFFFAOYSA-N 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 239000002664 nootropic agent Substances 0.000 description 1
- URPYMXQQVHTUDU-OFGSCBOVSA-N nucleopeptide y Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 URPYMXQQVHTUDU-OFGSCBOVSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229960005117 olmesartan Drugs 0.000 description 1
- VTRAEEWXHOVJFV-UHFFFAOYSA-N olmesartan Chemical compound CCCC1=NC(C(C)(C)O)=C(C(O)=O)N1CC1=CC=C(C=2C(=CC=CC=2)C=2NN=NN=2)C=C1 VTRAEEWXHOVJFV-UHFFFAOYSA-N 0.000 description 1
- AHLBNYSZXLDEJQ-FWEHEUNISA-N orlistat Chemical compound CCCCCCCCCCC[C@H](OC(=O)[C@H](CC(C)C)NC=O)C[C@@H]1OC(=O)[C@H]1CCCCCC AHLBNYSZXLDEJQ-FWEHEUNISA-N 0.000 description 1
- 229960001243 orlistat Drugs 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 239000001301 oxygen Chemical group 0.000 description 1
- 239000005022 packaging material Substances 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 230000009745 pathological pathway Effects 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 208000008494 pericarditis Diseases 0.000 description 1
- 201000001245 periodontitis Diseases 0.000 description 1
- 210000001539 phagocyte Anatomy 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 229960003562 phentermine Drugs 0.000 description 1
- QARVLSVVCXYDNA-UHFFFAOYSA-N phenyl bromide Natural products BrC1=CC=CC=C1 QARVLSVVCXYDNA-UHFFFAOYSA-N 0.000 description 1
- 229960000395 phenylpropanolamine Drugs 0.000 description 1
- DLNKOYKMWOXYQA-APPZFPTMSA-N phenylpropanolamine Chemical compound C[C@@H](N)[C@H](O)C1=CC=CC=C1 DLNKOYKMWOXYQA-APPZFPTMSA-N 0.000 description 1
- CWCMIVBLVUHDHK-ZSNHEYEWSA-N phleomycin D1 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC[C@@H](N=1)C=1SC=C(N=1)C(=O)NCCCCNC(N)=N)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C CWCMIVBLVUHDHK-ZSNHEYEWSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 208000024335 physical disease Diseases 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 229960003611 pramlintide Drugs 0.000 description 1
- 108010029667 pramlintide Proteins 0.000 description 1
- NRKVKVQDUCJPIZ-MKAGXXMWSA-N pramlintide acetate Chemical compound C([C@@H](C(=O)NCC(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CS)NC(=O)[C@@H](N)CCCCN)[C@@H](C)O)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 NRKVKVQDUCJPIZ-MKAGXXMWSA-N 0.000 description 1
- 229960002847 prasterone Drugs 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 238000009117 preventive therapy Methods 0.000 description 1
- FYPMFJGVHOHGLL-UHFFFAOYSA-N probucol Chemical compound C=1C(C(C)(C)C)=C(O)C(C(C)(C)C)=CC=1SC(C)(C)SC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 FYPMFJGVHOHGLL-UHFFFAOYSA-N 0.000 description 1
- 229960003912 probucol Drugs 0.000 description 1
- 108010070701 procolipase Proteins 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- 108091006082 receptor inhibitors Proteins 0.000 description 1
- DTLOVISJEFBXLX-REAFJZEQSA-N relexan 2 Chemical compound C([C@H]1C(=O)N[C@H](C(=O)NCC(=O)N[C@H]2CSSC[C@@H](C(=O)N[C@H](C(N1)=O)CSSC[C@@H](C(NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](C)C(=O)N[C@H](C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H]([C@@H](C)O)NC2=O)C(O)=O)C(=O)NCC(=O)N[C@@H](CCSC)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CO)C(O)=O)[C@@H](C)CC)[C@@H](C)CC)C(C)C)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(O)=O)C(C)C)[C@@H](C)CC)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1NC(=O)CC1)C(C)C)C1=CN=CN1 DTLOVISJEFBXLX-REAFJZEQSA-N 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 230000010410 reperfusion Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 210000005241 right ventricle Anatomy 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229960000672 rosuvastatin Drugs 0.000 description 1
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 1
- 108010033693 saxagliptin Proteins 0.000 description 1
- QGJUIPDUBHWZPV-SGTAVMJGSA-N saxagliptin Chemical compound C1C(C2)CC(C3)CC2(O)CC13[C@H](N)C(=O)N1[C@H](C#N)C[C@@H]2C[C@@H]21 QGJUIPDUBHWZPV-SGTAVMJGSA-N 0.000 description 1
- 229960004937 saxagliptin Drugs 0.000 description 1
- 230000037390 scarring Effects 0.000 description 1
- 239000008299 semisolid dosage form Substances 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229960002792 serelaxin Drugs 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 229960004425 sibutramine Drugs 0.000 description 1
- UNAANXDKBXWMLN-UHFFFAOYSA-N sibutramine Chemical compound C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UNAANXDKBXWMLN-UHFFFAOYSA-N 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- MFFMDFFZMYYVKS-SECBINFHSA-N sitagliptin Chemical compound C([C@H](CC(=O)N1CC=2N(C(=NN=2)C(F)(F)F)CC1)N)C1=CC(F)=C(F)C=C1F MFFMDFFZMYYVKS-SECBINFHSA-N 0.000 description 1
- 229960004034 sitagliptin Drugs 0.000 description 1
- 201000002859 sleep apnea Diseases 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000001488 sodium phosphate Substances 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 208000020431 spinal cord injury Diseases 0.000 description 1
- 229960002256 spironolactone Drugs 0.000 description 1
- LXMSZDCAJNLERA-ZHYRCANASA-N spironolactone Chemical compound C([C@@H]1[C@]2(C)CC[C@@H]3[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)SC(=O)C)C[C@@]21CCC(=O)O1 LXMSZDCAJNLERA-ZHYRCANASA-N 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000037351 starvation Effects 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 230000035882 stress Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 208000011117 substance-related disease Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 229910052717 sulfur Chemical group 0.000 description 1
- 239000011593 sulfur Chemical group 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 201000000596 systemic lupus erythematosus Diseases 0.000 description 1
- 238000002626 targeted therapy Methods 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 238000011285 therapeutic regimen Methods 0.000 description 1
- 239000003451 thiazide diuretic agent Substances 0.000 description 1
- 150000001467 thiazolidinediones Chemical class 0.000 description 1
- 208000021510 thyroid gland disease Diseases 0.000 description 1
- 229960002277 tolazamide Drugs 0.000 description 1
- OUDSBRTVNLOZBN-UHFFFAOYSA-N tolazamide Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1CCCCCC1 OUDSBRTVNLOZBN-UHFFFAOYSA-N 0.000 description 1
- 229960005371 tolbutamide Drugs 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 229960005461 torasemide Drugs 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 230000000472 traumatic effect Effects 0.000 description 1
- PBIMIGNDTBRRPI-UHFFFAOYSA-N trifluoro borate Chemical compound FOB(OF)OF PBIMIGNDTBRRPI-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- RYFMWSXOAZQYPI-UHFFFAOYSA-K trisodium phosphate Chemical compound [Na+].[Na+].[Na+].[O-]P([O-])([O-])=O RYFMWSXOAZQYPI-UHFFFAOYSA-K 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- GXPHKUHSUJUWKP-UHFFFAOYSA-N troglitazone Chemical compound C1CC=2C(C)=C(O)C(C)=C(C)C=2OC1(C)COC(C=C1)=CC=C1CC1SC(=O)NC1=O GXPHKUHSUJUWKP-UHFFFAOYSA-N 0.000 description 1
- 229960001641 troglitazone Drugs 0.000 description 1
- GXPHKUHSUJUWKP-NTKDMRAZSA-N troglitazone Natural products C([C@@]1(OC=2C(C)=C(C(=C(C)C=2CC1)O)C)C)OC(C=C1)=CC=C1C[C@H]1SC(=O)NC1=O GXPHKUHSUJUWKP-NTKDMRAZSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 229940046728 tumor necrosis factor alpha inhibitor Drugs 0.000 description 1
- 239000002452 tumor necrosis factor alpha inhibitor Substances 0.000 description 1
- 238000000825 ultraviolet detection Methods 0.000 description 1
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 1
- 208000019553 vascular disease Diseases 0.000 description 1
- 230000025033 vasoconstriction Effects 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 239000011715 vitamin B12 Substances 0.000 description 1
- 235000019163 vitamin B12 Nutrition 0.000 description 1
- 229960001729 voglibose Drugs 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/576—Six-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/576—Six-membered rings
- C07F9/59—Hydrogenated pyridine rings
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
La divulgación se refiere a compuestos de Fórmula (I), que son agonistas del receptor del péptido 2 de formilo (FPR2) y/o agonistas del receptor del péptido 1 de formilo (FPR1). La descripción también proporciona composiciones y métodos para usar los compuestos, por ejemplo, para el tratamiento de aterosclerosis, insuficiencia cardíaca, enfermedad pulmonar obstructiva crónica (EPOC) y enfermedades relacionadas. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Agonistas del FPR2 de óxido de biaril dialquil fosfina
Antecedentes de la invención
La presente invención se refiere a compuestos de óxido de biaril dialquil fosfina novedosos, que son agonistas del receptor de péptidos formilados 2 (FPR2, por sus siglas en inglés) y/o agonistas del receptor de péptidos formilados 1 (FPR1, por sus siglas en inglés), y composiciones que contienen los mismos. Los compuestos se pueden usar, por ejemplo, para el tratamiento de la ateroesclerosis, la insuficiencia cardíaca, la enfermedad pulmonar obstructiva crónica (EPOC) y enfermedades relacionadas.
El receptor de péptidos formilados 2 (FPR2) pertenece a un pequeño grupo de siete dominios transmembrana, receptores acoplados a la proteína G que se expresan en múltiples tejidos humanos, incluyendo las células inmunitarias, y se sabe que son importantes en la defensa y la inflamación del hospedador. El FPR2 comparte una homología de secuencia significativa con los FPR1 y FPR3 (Chen K, et al., Journal of Autoimmunity 85, 2017, 64-77). En conjunto, estos receptores se unen a varios agonistas estructuralmente diversos, incluyendo péptidos W-formilados y no formilados que actúan como quimioatrayentes y activan los fagocitos. El péptido endógeno Anexina A1 y sus fragmentos W-terminales son ejemplos de ligandos que se unen a los FPR1 y FPR2 humanos. Los ácidos grasos, tales como la lipoxina A4 eicosanoide, que pertenece a una clase de pequeños mediadores prorresolutivos (SPM, por sus siglas en inglés), también se han sido identificado como agonista para el FPR2 (Ye RD., et al., Pharmacol. Rev., 2009, 61, 119-61).
Los ligandos endógenos prorresolutivos de FPR2, tales como la lipoxina A4 y la Anexina A1, se ha indicado que desencadenan una amplia gama de cadenas citoplasmáticas, tales como el acoplamiento de Gi, la movilización de Ca2+ y la incorporación de p-arrestina. (Cattaneo, F, et al., Int J Mol Sci. Abril de 2013; 14(4): 7193-7230). El FPR2 regula el sistema inmunitario tanto innato adaptativo, incluyendo los neutrófilos, los macrófagos y los linfocitos T y B. En el caso de los neutrófilos, los ligandos del FPR2 modulan el movimiento, la citotoxicidad y la esperanza de vida. En el caso de los macrófagos, el agonismo del FPR2 previene la apoptosis y potencia la eferocitosis. (Chandrasekharan JA, Sharma-Walia N,. J. Inflamm. Res., 2015, 8 , 181-92). El inicio de la resolución de la inflamación mediante el agonismo del FPR2 es responsable de potenciar la cicatrización antifibrótica y el retorno del tejido lesionado a la homeostasis (Romano M., et al., Eur. J. Pharmacol., 2015, 5, 49-63).
La inflamación crónica forma parte de la vía de patogenia de muchas enfermedades humanas y la estimulación de las vías de resolución con agonistas del FPR2 puede tener efectos tanto protectores como reparadores. La lesión por isquemia y reperfusión (I/R) es una característica frecuente de diversas enfermedades asociadas a una alta morbilidad y mortalidad, tales como el infarto de miocardio y el accidente cerebrovascular. La cicatrización no productiva asociada a la muerte de cardiomiocitos y la remodelación patológica resultante de la lesión por isquemia y reperfusión conduce a la formación de cicatrices, la fibrosis y la pérdida progresiva de la función cardíaca. Se propone la modulación del FPR2 para potenciar la cicatrización miocárdica posterior a la lesión y disminuir la remodelación miocárdica adversa (Kain V., et al., J. Mol. Cell. Cardiol., 2015, 84, 24-35). Además, los agonistas prorresolutivos del FPR2, en el sistema nervioso central, pueden ser agentes terapéuticos útiles para el tratamiento de diversas afecciones clínicas de I/R, incluyendo el accidente cerebrovascular en el cerebro (Gavins FN., Trends Pharmacol. Sci., 2010, 31, 266-76) y lesión de la médula espinal inducida por I/R (Liu ZQ., et al., Int. J. Clin. Exp. Med., 2015, 8 , 12826-33).
Además de los efectos beneficiosos de dirigirse al receptor FPR2 con agonistas prorresolutivos novedosos para el tratamiento terapéutico de lesiones inducidas por I/R, la utilidad de estos ligandos también se puede aplicar a otras enfermedades. En el aparato circulatorio, se ha hallado que tanto el receptor de FPR2 como sus agonistas prorresolutivos son responsables de la estabilización y cicatrización de las placas aterógenas (Petri MH., et al., Cardiovasc. Res., 2015, 105, 65-74; y Fredman G., et al., Sci. Trans. Med., 2015, 7(275); 275ra20). Los agonistas del FPR2 también han demostrado ser beneficiosos en modelos preclínicos de enfermedades humanas inflamatorias crónicas, incluyendo: enfermedades infecciosas, psoriasis, dermatitis, síndrome inflamatorio intestinal, enfermedad de Crohn, inflamación ocular, septicemia, dolor, enfermedades metabólicas/diabetes, cáncer, EPOC, asma y enfermedades alérgicas, fibrosis quística, lesión pulmonar aguda y fibrosis, artritis reumatoide y otras enfermedades articulares, enfermedad de Alzheimer, fibrosis renal y trasplante de órganos (Romano M., et al., Eur. J. Pharmacol., 2015, 5, 49-63, Perrett, M., et al., Trends in Pharm. Sci., 2015, 36, 737-755). El documento US2006/074072 A1 divulga compuestos útiles para el tratamiento del infarto de miocardio.
Sumario de la invención
La presente invención proporciona compuestos de óxido de biaril dialquil fosfina novedosos y análogos de los mismos, que resultan útiles como agonistas del FPR2, incluyendo estereoisómeros, tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos.
La presente invención también proporciona composiciones farmacéuticas que comprenden un vehículo farmacéuticamente aceptable y al menos uno de los compuestos de la presente invención o estereoisómeros,
tautómeros, sales farmacéuticamente aceptables o solvatos de los mismos.
Los compuestos de la invención se pueden usar en terapia.
Los compuestos de la invención se pueden usar en el tratamiento y/o la profilaxis de múltiples enfermedades o trastornos asociados al FPR2, tales como las enfermedades inflamatorias, las cardiopatías, las enfermedades crónicas de las vías respiratorias, los cánceres, la septicemia, los síntomas alérgicos, la infección por el retrovirus VIH, los trastornos circulatorios, la neuroinflamación, los trastornos nerviosos, los dolores, las enfermedades priónicas, la amiloidosis y los trastornos inmunitarios. Las cardiopatías se seleccionan del grupo que consiste en angina de pecho, angina inestable, infarto de miocardio, enfermedad coronaria aguda, daño yatrógeno cardíaco e insuficiencia cardíaca, incluyendo, pero sin limitación, insuficiencia cardíaca aguda, insuficiencia cardíaca crónica de origen isquémico y no isquémico, insuficiencia cardíaca sistólica, insuficiencia cardíaca diastólica, insuficiencia cardíaca con fracción de eyección reducida (HFrEF, por sus siglas en inglés) e insuficiencia cardíaca con fracción de eyección conservada (HFpEF, por sus siglas en inglés).
Los compuestos de la invención se pueden usar solos, en combinación con otros compuestos de la presente invención o en combinación con uno o más agentes de otro tipo.
Otras características y ventajas de la invención resultarán evidentes a partir de la siguiente descripción detallada y de las reivindicaciones.
Descripción de la invención
La invención abarca compuestos de la Fórmula (I), que son agonistas del receptor de péptidos formilados 2 (FPR2) y/o agonistas del receptor de péptidos formilados 1 (FPR1), composiciones que contienen los mismos y los compuestos o las composiciones para uso en el tratamiento de la ateroesclerosis, la insuficiencia cardíaca, la enfermedad pulmonar obstructiva crónica (EPOC) y enfermedades relacionadas, por ejemplo.
Un aspecto de la invención es un compuesto de la Fórmula (I):

o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde:
Ar1 es fenilo sustituido con 1-2 R1a y 1-2 R1b o heteroarilo de 6 miembros con 1-3 átomos de nitrógeno y sustituido con 1-2 R1a y 1-2 R1b;
Ar2 es fenilo sustituido con 1-4 R2 o heteroarilo de 6 miembros con 1-3 átomos de nitrógeno y sustituido con 1-4 R2; Ar3 es fenilo sustituido con 0-4 R3 o piridinilo sustituido con 0-4 R3;
R1a es hidrógeno o halo;
R1b es halo, haloalquilo, alcoxi o haloalcoxi;
R2 es hidrógeno, ciano, halo, alquilo, hidroxialquilo, haloalquilo, cicloalquilo, alcoxi o haloalcoxi;
R3 es ciano, halo, alquilo, alcoxi, hidroxialquilo, alcoxialquilo o haloalquilo; y R4 es alquilo o alcoxi.
Otro aspecto de la invención es un compuesto de la Fórmula (II):

o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde:
Ar1 es fenilo sustituido con 1 R1a y 1-2 R1b o heteroarilo de 6 miembros con 1-2 átomos de nitrógeno y sustituido con 1 R1a y 1-2 R1b;
Ar2 es fenilo sustituido con 1-3 R2 o piridinilo sustituido con 1-4 R2;
R1a es hidrógeno o halo;
R1b es halo, haloalquilo C1-4, alcoxi C1.4 o haloalcoxi C1.4;
R2 es hidrógeno, halo, alquilo C1-4, hidroxialquilo C1.4, haloalquilo C1.4, cicloalquilo C3.6, alcoxi C1.4 o haloalcoxi C1-4; y R4 es alquilo C1.3.
Otro aspecto de la invención es un compuesto de la Fórmula (III):

o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde:
Ar1 es fenilo sustituido con 1 R1a y 1-2 R1b, piridinilo sustituido con 1 R1a y 1-2 R1b o pirazinilo sustituido con 1 R1a y 1-2 R1b;
R1a es hidrógeno o halo;
R1b es halo, haloalquilo C1-4, alcoxi C1-4 o haloalcoxi C1-4;
R2 es hidrógeno, halo, alquilo C1.4, hidroxialquilo C1.4, haloalquilo C1-4, cicloalquilo C3-6, alcoxi C1.4 o haloalcoxi C1. 4; y R4 es alquilo C1-2.
Otro aspecto de la invención es un compuesto de la Fórmula (IV):

o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde:
R1a es hidrógeno o F;
R1b es halo, haloalquilo C1-2 o alcoxi C1-2;
R2 es hidrógeno, halo, alquilo C1-3, haloalquilo C1-3 o cicloalquilo C3-6; y
R4 es metilo o etilo.
Otro aspecto de la invención es un compuesto de la Fórmula (IV) o (VI) o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde:
R1a es hidrógeno o F;
R1b es F, Cl o CF3; y
R2 es hidrógeno, F, Cl o ciclopropilo; y
R4 es metilo.
Otro aspecto de la invención es un compuesto de la Fórmula (V):

o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde:
R1a es hidrógeno o F;
R1b es halo, haloalquilo C1-2 o alcoxi C1-2;
R2 es halo; y
R4 es metilo o etilo.
Otro aspecto de la invención es un compuesto de la Fórmula (VI):

o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde: R1a es hidrógeno o F;
R1b es halo, haloalquilo C1-2 o alcoxi C1-2;
R2 es halo, alquilo C1-3, haloalquilo C1-3 o cicloalquilo C3-6; y
R4 es metilo o etilo.
Otro aspecto de la invención es un compuesto de la Fórmula (VII):
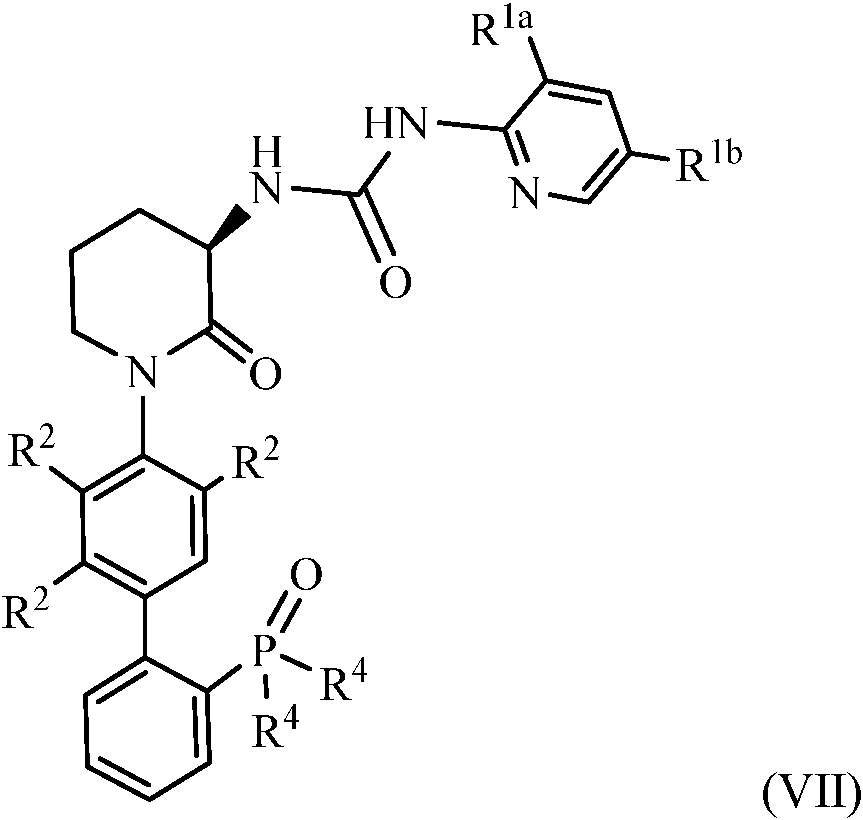
o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde: R1a es hidrógeno o halo;
R1b es halo, haloalquilo C1-2 o alcoxi C1-2;
R2 es hidrógeno, halo, alquilo C1-3 o cicloalquilo C3-6; y
R4 es alquilo C1-2.
Otro aspecto de la invención es un compuesto de la Fórmula (VIII):

o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde:
R1b es halo, haloalquilo C1-2 o alcoxi C1-2;
R2 es hidrógeno, halo, alquilo C1-3 o cicloalquilo C3-6; y
R4 es alquilo C1-2.
Otro aspecto de la invención es un compuesto de la Fórmula (IX):

o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo, en donde:
Ar1 es fenilo sustituido con 1 R1a y 1-2 R1b, piridinilo sustituido con 1 R1a y 1-2 R1b o pirazinilo sustituido con 1 R1a y 1-2 R1b;
R1a es hidrógeno o halo;
R1b es halo, haloalquilo C1-4, alcoxi C1.4 o haloalcoxi C1.4; y
R4 es alquilo C1-2.
Otro aspecto de la invención es un compuesto seleccionado del grupo que consiste en:


o un estereoisómero, un tautómero o una sal farmacéuticamente aceptable del mismo.
En un compuesto de la Fórmula (I), (II), (III), (IV), (V), (VI), (VII), (VIII) o (IX), el alcance de cualquier aparición de un sustituyente variable, incluyendo Ar1, Ar2, Ar3, R1a, R1b, R2, R3 y R4, se puede usar independientemente con el alcance de cualquier otra aparición de un sustituyente variable. Como tal, la invención incluye combinaciones de los diferentes aspectos.
A menos que se especifique otra cosa, estos términos tienen los siguientes significados. El término "alquilo" significa un grupo alquilo lineal o ramificado compuesto por 1 a 6 carbonos. El término "alquenilo" significa un grupo alquilo lineal o ramificado compuesto por 2 a 6 carbonos con al menos un enlace doble. El término "alquinilo" significa un grupo alquilo lineal o ramificado compuesto por 2 a 6 carbonos con al menos un enlace triple. El término "cicloalquilo" significa un sistema de anillos monocíclico compuesto por 3 a 7 carbonos. Los términos con un resto hidrocarburo (por ejemplo, alcoxi) incluyen isómeros lineales y ramificados en la parte de hidrocarburo. El término "halo" incluye fluoro, cloro, bromo y yodo. Los términos "haloalquilo" y "haloalcoxi" incluyen todos los isómeros halogenados, desde monohalo hasta perhalo. El término "arilo" significa un grupo hidrocarburo aromático monocíclico o bicíclico que tiene de 6 a 12 átomos de carbono o un sistema de anillos condensados bicíclico en donde uno o ambos anillos son aromáticos. Los sistemas de anillo condensados bicíclicos consisten en un grupo fenilo condensado con un anillo carbocíclico, aromático o no aromático, de cuatro a siete miembros. Los ejemplos representativos de grupos arilo incluyen, pero sin limitación, fenilo, indanilo, indenilo, naftilo y tetrahidronaftilo. El término "heteroarilo" significa un sistema de anillos aromáticos monocíclico de 5 a 7 miembros o bicíclico de 8 a 11 miembros con 1-5 heteroátomos seleccionados independientemente de nitrógeno, oxígeno y azufre. Cuando no se especifica la ubicación de la fijación de un enlace, la unión se puede fijar en cualquier ubicación adecuada, tal como entienden los expertos en la materia. Las combinaciones de sustituyentes y patrones de enlace son solo aquellos que dan como resultado compuestos estables, tal como entienden los expertos en la materia. Los términos entre paréntesis y paréntesis múltiples están destinados a aclarar las relaciones de enlace a aquellos expertos en la materia. Por ejemplo, un término, tal como ((R)alquilo), significa un sustituyente alquilo que está sustituido adicionalmente con el sustituyente R.
La invención incluye todas las formas de sal farmacéuticamente aceptables de los compuestos. Las sales farmacéuticamente aceptables son aquellas en las que los contraiones no contribuyen significativamente a la actividad fisiológica o a la toxicidad de los compuestos y, como tales, funcionan como equivalentes farmacológicos. Estas sales se pueden elaborar de acuerdo con técnicas orgánicas comunes empleando reactivos disponibles en el mercado. Algunas formas de sales aniónicas incluyen acetato, acistrato, besilato, bromuro, cloruro, citrato, fumarato, glucuronato, bromhidrato, clorhidrato, yodhidrato, yoduro, lactato, maleato, mesilato, nitrato, pamoato, fosfato, succinato, sulfato, tartrato, tosilato y xinofoato. Algunas formas de sales catiónicas incluyen amonio, aluminio, benzatina, bismuto, calcio, colina, dietilamina, dietanolamina, litio, magnesio, meglumina, 4-fenilciclohexilamina, piperazina, potasio, sodio, trometamina y zinc.
Algunos de los compuestos de la invención existen en formas estereoisoméricas que incluyen la siguiente estructura con el carbono indicado. La invención incluye todas las formas estereoisoméricas de los compuestos, incluyendo enantiómeros y diastereómeros. En la técnica, se conocen métodos de elaboración y separación de estereoisómeros. La invención incluye todas las formas tautoméricas de los compuestos. La invención incluye atropisómeros e isómeros rotacionales.
La invención está destinada a incluir todos los isótopos de átomos que aparecen en los compuestos. Los isótopos incluyen aquellos átomos que tienen el mismo número atómico, pero diferentes números másicos. A modo de ejemplo general y sin limitación, los isótopos de hidrógeno incluyen deuterio y tritio. Los isótopos de carbono incluyen 13C y 14C. Los compuestos marcados con isótopos de la invención se pueden preparar generalmente mediante técnicas convencionales conocidas por aquellos expertos en la materia o mediante procesos análogos a aquellos descritos en el presente documento, usando un reactivo marcado con isótopos adecuado en lugar del reactivo no marcado empleado de otro modo. Tales compuestos pueden tener diversos usos posibles, por ejemplo, como patrones y reactivos para determinar la actividad biológica. En el caso de los isótopos estables, tales compuestos pueden tener el potencial de modificar favorablemente las propiedades biológicas, farmacológicas o farmacocinéticas.
MÉTODOS BIOLÓGICOS
Los receptores de péptidos W-formilados (FPR, por sus siglas en inglés) son una familia de receptores quimioatrayentes que facilitan la respuesta de los leucocitos durante la inflamación. Los FPR pertenecen a la superfamilia de receptores acoplados a la proteína G de siete dominios transmembrana y están relacionados con las proteínas G inhibidoras (Gi). Se han identificado tres miembros de la familia (FPR1, FPR2 y FPR3) en seres humanos, y se encuentran predominantemente en células mieloides con una distribución variada y también se ha notificado su presencia en múltiples órganos y tejidos. Después de unirse al agonista, los FPR activan una multitud de vías fisiológicas, tales como la transducción de la señalización celular, la movilización de Ca2+ y la transcripción. La familia interactúa con un conjunto diverso de ligandos que incluyen proteínas, polipéptidos y metabolitos de ácidos grasos que activan respuestas tanto proinflamatorias como prorresolutivas corriente adelante. Se usaron ensayos de monofosfato de adenosina cíclico (cAMP, por sus siglas en inglés) de FPR2 y FPR1 para medir la actividad de los compuestos en la presente patente.
Ensayos de monofosfato de adenosina cíclico (cAMP) de FPR2 y FPR1. Se añadió una mezcla de forskolina (concentración final de 5 μM en el caso del FPR2 o concentración final de 10 μM en el caso del FPR1) e IBMX (concentración final de 200 μM) a placas Proxiplate de 384 pocillos (Perkin-Elmer) en las que previamente se habían aplicado compuestos de ensayo en DMSO (concentración final del 1 %) a concentraciones finales en el intervalo de 0,020 nM a 100 μM. Se cultivaron células de ovario de hámster chino (CHO, por sus siglas en inglés) que sobreexpresaban los receptores FPR1 humanos o FPR2 humanos en medio F-12 (de Ham) complementado con FBS cualificado al 10 %, 250 μg/ml de zeocina y 300 μg/ml de higromicina (Life Technologies). Las reacciones se iniciaron mediante la adición de 2.000 células de FPR2 humano por pocillo o 4.000 células de FPR1 humano por pocillo en PBS de Dulbecco (con calcio y magnesio) (Life Technologies) complementado con BSA al 0,1 % (Perkin-Elmer). Las mezclas de reacción se incubaron durante 30 min a ta. El nivel de cAMP intracelular se determinó usando el kit de reactivos de ensayo HTRF HiRange cAMP (Cisbio) de acuerdo con las instrucciones del fabricante. Se elaboraron soluciones de anti-cAMP conjugado con criptato y cAMP marcado con fluoróforo d2 en un tampón de lisis suministrado por separado. Una vez finalizada la reacción, las células se lisaron con un volumen igual de la solución de d2-cAMP y la solución de anti-cAMP. Después de una incubación de 1 h a ta, se midió la intensidad de fluorescencia resuelta en el tiempo usando el lector Envision (Perkin-Elmer) a una longitud de onda de excitación de 400 nm y a longitudes de onda de emisión duales de 590 nm y 665 nm. Se construyó una curva de calibración con un patrón externo de cAMP a concentraciones que variaban de 1 μM a 0,1 μM mediante la representación gráfica de la relación entre la intensidad de fluorescencia desde una longitud de onda de emisión de 665 nm y la intensidad desde la longitud de onda de emisión de 590 nm frente a las concentraciones de cAMP. A continuación, se determinó la potencia y actividad de un compuesto para inhibir la producción de cAMP mediante el ajuste a una ecuación logística tetraparamétrica a partir de un gráfico del nivel de cAMP frente a las concentraciones de compuestos.
Los Ejemplos que se divulgan a continuación se analizaron en el ensayo de cAMP de FPR2 y FPR1 descritos anteriormente y se halló que tenían actividad agonista de FPR2 y/o FPR1. La siguiente Tabla 1 enumera los valores de CE50 en los ensayos de cAMP de FPR2 y FPR1 medidos para los siguientes Ejemplos.
Tabla 1

COMPOSICIONES FARMACEUTICAS Y METODOS DE USO
Los compuestos de la presente invención se pueden administrar a mamíferos, preferentemente seres humanos, para el tratamiento de diversas afecciones y trastornos asociados al receptor FPR2, tales como la enfermedad de Behcet, el síndrome de Sweet, el lupus eritematoso sistémico (SLE, por sus siglas en inglés), la granulomatosis de Wegener, la infección con virus, la diabetes, las amputaciones, los cánceres, la infección bacteriana, las lesiones externas físicas, los trastornos físicos, incluyendo la exposición a la radiación, la vasoconstricción, las reacciones anafilácticas, las reacciones alérgicas, la rinitis, los choques (endotóxicos, hemorrágicos, traumáticos, isquemia esplácnica y choques circulatorios), la artritis reumatoide, la gota, psoriasis, la hiperplasia benigna de la próstata, la isquemia miocárdica, el infarto de miocardio, la insuficiencia cardíaca, las lesiones cerebrales, las enfermedades pulmonares, la EPOC, la enfermedad de las vías respiratorias obstructiva crónica (COAD, por sus siglas en inglés), la enfermedad del pulmón obstructiva crónica (COLD, por sus siglas en inglés), la lesión pulmonar aguda, el síndrome de dificultad respiratoria aguda, la bronquitis crónica, el enfisema pulmonar, el asma (asma alérgica y asma no alérgica), la fibrosis quística, la fibrosis renal, la nefropatía, las glomerulopatías renales, la colitis ulcerosa, la enfermedad intestinal inflamatoria (EII), la enfermedad de Crohn, la periodontitis, los dolores, la enfermedad de Alzheimer, el SIDA, el glaucoma uveítico, la conjuntivitis, el síndrome de Sjogren, la rinitis, la ateroesclerosis, las enfermedades neuroinflamatorias, incluyendo la esclerosis múltiple, el accidente cerebrovascular, la septicemia, y similares.
A menos que se especifique otra cosa, los siguientes términos tienen los significados indicados. El término "sujeto" se refiere a cualquier especie humana u otra especie de mamífero que se pueda beneficiar potencialmente del tratamiento con un agonista del FPR2 y/o FPR1, tal como lo entienden los expertos en este campo. Algunos sujetos incluyen seres humanos de cualquier edad con factores de riesgo de padecer enfermedad cardiovascular. Los factores de riesgo frecuentes incluyen la edad, el sexo, el peso, los antecedentes familiares, la apnea del sueño, el consumo de alcohol o tabaco, la arritmia por inactividad física o los signos de resistencia a la insulina, tales como la acantosis pigmentaria, la hipertensión, la dislipidemia o el síndrome del ovario poliquístico (PCOS, por sus siglas en inglés). El término "paciente" significa una persona adecuada para la terapia, tal como lo determinan los expertos en el campo. Los
términos "tratar" o "tratamiento abarcan el tratamiento de un paciente o sujeto, tal como lo entienden los expertos en este campo. Los términos "prevenir" o "prevención" abarcan el tratamiento preventivo (es decir, la profilaxis y/o reducción del riesgo) de una enfermedad subclínica en un paciente o sujeto destinado a reducir la probabilidad de aparición de una enfermedad clínica, tal como lo entienden los expertos en este campo. Los pacientes se seleccionan para la terapia preventiva basándose en factores que se sabe que aumentan el riesgo de padecer una enfermedad clínica en comparación con la población general. La expresión "cantidad terapéuticamente eficaz" significa una cantidad de un compuesto que es eficaz, tal como entienden los expertos en este campo.
Otro aspecto de la invención son composiciones farmacéuticas que comprenden una cantidad terapéuticamente eficaz de un compuesto de las Fórmulas (I)-(IX) en combinación con un vehículo farmacéutico.
Otro aspecto de la invención son composiciones farmacéuticas que comprenden una cantidad terapéuticamente eficaz de un compuesto de las Fórmulas (I)-(IX) en combinación con al menos otro agente terapéutico y un vehículo farmacéutico.
La expresión "composición farmacéutica" significa una composición que comprende un compuesto de la invención en combinación con al menos un vehículo farmacéuticamente aceptable adicional. Un "vehículo farmacéuticamente aceptable" se refiere a los medios generalmente aceptados en la técnica para la administración de principios biológicamente activos a los animales, en particular, los mamíferos, incluyendo, por ejemplo, adyuvantes, excipientes o vehículos, tales como diluyentes, agentes conservantes, cargas, agentes reguladores de flujo, agentes disgregantes, agentes humectantes, agentes emulsionantes, agentes de suspensión, agentes edulcorantes, agentes saborizantes, agentes aromatizantes, agentes antibacterianos, agentes antifúngicos, agentes lubricantes y agentes dispensadores, dependiendo de la naturaleza del modo de administración y las formas farmacéuticas.
Los vehículos farmacéuticamente aceptables se formulan de acuerdo con varios factores incluidos dentro del alcance de aquellos expertos habituales en la materia. Estos incluyen, sin limitación: el tipo y la naturaleza del principio activo que se formula; el sujeto al que se va a administrar la composición que contiene el agente; la vía de administración prevista de la composición; y la indicación terapéutica a la que se dirige. Los vehículos farmacéuticamente aceptables incluyen medios líquidos tanto acuosos como no acuosos, así como diversas formas farmacéuticas sólidas y semisólidas. Tales vehículos pueden incluir varios ingredientes y aditivos diferentes, además del principio activo, incluyéndose tales ingredientes adicionales en la formulación por diversas razones, por ejemplo, la estabilización del principio activo, los aglutinantes, etc., bien conocidos por aquellos expertos habituales en la materia. Las descripciones de los vehículos farmacéuticamente aceptables adecuados y de los factores implicados en su selección se encuentran en diversas fuentes fácilmente disponibles, tales como, por ejemplo, Allen, L.V., Jr. et al., Remington: The Science and Practice of Pharmacy (2 volúmenes), 22a Edición, Pharmaceutical Press (2012).
En particular, cuando se proporcionan en forma de unidad de dosificación individual, existe la posibilidad de que se produzca una interacción química entre los principios activos combinados. Por esta razón, cuando el compuesto de la presente invención y un segundo agente terapéutico se combinan en una unidad de dosificación individual, estos se formulan de tal manera que, aunque se combinen los principios activos en una unidad de dosificación individual, se minimice el contacto físico entre los principios activos (es decir, se reduce). Por ejemplo, un principio activo se puede recubrir entéricamente. Mediante el recubrimiento entérico de uno de los principios activos, no solo resulta posible minimizar el contacto entre los principios activos combinados, sino que también resulta posible controlar la liberación de uno de estos componentes en el tubo digestivo de tal manera que uno de estos componentes no se libere en el estómago, sino que, más bien, se libere en los intestinos. También se puede recubrir uno de los principios activos con un material que efectúe una liberación mantenida a lo largo del tubo digestivo y también sirve para minimizar el contacto físico entre los principios activos combinados. Además, el componente de liberación mantenida también se puede recubrir entéricamente de tal manera que la liberación de este componente se produzca únicamente en el intestino. Otra estrategia más podría implicar la formulación de un producto combinado en el que un componente se recubre con un polímero de liberación mantenida y/o entérica y el otro componente también se recubre con un polímero, tal como una hidroxipropil metilcelulosa (HPMC) de bajo grado de viscosidad u otros materiales adecuados, tal como se conoce en la técnica, con el fin de separar adicionalmente los componentes activos. El recubrimiento polimérico sirve para formar una barrera adicional frente a la interacción con el otro componente.
Los compuestos de la presente invención se pueden usar en un método para el tratamiento de cardiopatías que comprende administrar una cantidad terapéuticamente eficaz de un compuesto de las Fórmulas (I)-(IX) a un paciente.
Los compuestos de la presente invención se pueden usar en un método para el tratamiento de cardiopatías en donde la cardiopatía se selecciona del grupo que consiste en angina de pecho, angina inestable, infarto de miocardio, insuficiencia cardíaca, enfermedad coronaria aguda, insuficiencia cardíaca aguda, insuficiencia cardíaca crónica y daño yatrógeno cardíaco.
Se entenderá que el tratamiento o la profilaxis de la insuficiencia cardíaca puede implicar también el tratamiento o la profilaxis de un episodio cardiovascular. El tratamiento o la profilaxis, tal como se hace referencia en el presente documento, se puede referir al tratamiento o la profilaxis de determinados síntomas o afecciones negativos asociados a o que surgen como resultado de un episodio cardiovascular. A modo de ejemplo, el tratamiento o la profilaxis puede
implicar la reducción o prevención de cambios negativos en el acortamiento fraccional, el peso del corazón, el peso del pulmón, el área de sección transversal de miocitos, la fibrosis cardíaca inducida por sobrecarga de presión, la senescencia celular inducida por estrés y/o las propiedades de hipertrofia cardíaca o cualquier combinación de los mismos, que se asocian a o que surgen como resultado de un episodio cardiovascular. El tratamiento se puede administrar como preparación para un episodio cardiovascular o en respuesta al mismo para aliviar los efectos negativos. La prevención puede implicar un tipo de tratamiento proactivo o profiláctico para prevenir el episodio cardiovascular o para reducir la aparición de los efectos negativos de un episodio cardiovascular.
Los compuestos de las Fórmulas (I)-(IX) o una sal farmacéuticamente aceptable de los mismos se pueden usar para la preparación de una composición farmacéutica para el tratamiento o la profilaxis de la insuficiencia cardíaca, por ejemplo, la insuficiencia cardíaca es consecuencia de la hipertensión, una cardiopatía isquémica, una cardiopatía no isquémica, la exposición a un compuesto cardiotóxico, la miocarditis, la enfermedad de Kawasaki, la diabetes de tipo I y de tipo II, la enfermedad tiroidea, la infección vírica, la gingivitis, la drogadicción, el alcoholismo, la pericarditis, la ateroesclerosis, la enfermedad vascular, la miocardiopatía hipertrófica, la miocardiopatía dilatada, el infarto de miocardio, la fibrosis auricular, la disfunción sistólica del ventrículo izquierdo, la disfunción diastólica del ventrículo izquierdo, la cirugía de revascularización coronaria, la cirugía de implantación de marcapasos, la inanición, un trastorno alimentario, las distrofias musculares y un defecto genético. La insuficiencia cardíaca que se vaya a tratar puede ser insuficiencia cardíaca diastólica, insuficiencia cardíaca con fracción de eyección reducida (HFrEF), insuficiencia cardíaca con fracción de eyección conservada (HFpEF), insuficiencia cardíaca aguda e insuficiencia cardíaca crónica de origen isquémico y no isquémico.
Los compuestos de las Fórmulas (I)-(IX) se pueden usar en un método para tratar la disfunción sistólica y/o diastólica, en donde el compuesto se administra en una cantidad terapéuticamente eficaz para aumentar la capacidad de los cardiomiocitos para contraerse y relajarse, aumentando, de ese modo, el llenado y el vaciado de los ventrículos derecho e izquierdo, preferentemente, el ventrículo izquierdo.
Los compuestos de las Fórmulas (I)-(IX) se pueden usar en un método para tratar la insuficiencia cardíaca en donde el compuesto se administra en una cantidad terapéuticamente eficaz para aumentar la fracción de eyección en el ventrículo izquierdo.
Los compuestos de las Fórmulas (I)-(IX) se pueden usar en un método para tratar la insuficiencia cardíaca en donde el compuesto se administra en una cantidad terapéuticamente eficaz para reducir la fibrosis en el tejido cardíaco.
Los compuestos de la presente invención se pueden usar en un método para el tratamiento de cardiopatías, en donde el tratamiento es posterior a un infarto de miocardio.
Los compuestos de la presente invención se pueden usar en un método para el tratamiento de cardiopatías que comprende administrar una cantidad terapéuticamente eficaz de un compuesto de las Fórmulas (I)-(IX) a un paciente junto con otros agentes terapéuticos.
Los compuestos de la presente invención se pueden administrar mediante cualquier medio adecuado, por ejemplo, por vía oral, tal como comprimidos, cápsulas (cada una de las cuales incluye formulaciones de liberación mantenida o de liberación programada), píldoras, polvos, gránulos, elixires, tinturas, suspensiones (incluyendo nanosuspensiones, microsuspensiones, dispersiones liofilizadas), jarabes y emulsiones; por vía sublingual; por vía bucal; por vía parenteral, tal como mediante inyección subcutánea, intravenosa, intramuscular o intraesternal o técnicas de infusión (por ejemplo, como soluciones o suspensiones acuosas o no acuosas inyectables estériles); por vía nasal, incluyendo la administración a las membranas nasales, tal como mediante pulverización por inhalación; por vía tópica, tal como en forma de una crema o pomada; o por vía rectal, tal como en forma de supositorios. Estos se pueden administrar solos o se pueden administrar con un vehículo farmacéutico seleccionado sobre la base de la vía de administración elegida y la práctica farmacéutica convencional.
La pauta posológica de los compuestos de la presente invención, por supuesto, variará dependiendo de factores conocidos, tales como las características farmacodinámicas del agente particular y su modo y vía de administración; la especie, la edad, el sexo, la salud, el estado médico y peso del receptor; la naturaleza y el alcance de los síntomas; la clase de tratamiento concurrente; la frecuencia del tratamiento; la vía de administración, la función renal y hepática del paciente y el efecto deseado.
A modo de orientación general, la dosificación oral diaria de cada principio activo, cuando se usa para los efectos indicados, variará de entre aproximadamente 0,01 y aproximadamente 5.000 mg al día, preferentemente entre aproximadamente 0,1 y aproximadamente 1.000 mg al día y lo más preferentemente entre aproximadamente 0,1 y aproximadamente 250 mg al día. Por vía intravenosa, las dosis más preferidas variarán de aproximadamente 0,01 a aproximadamente 10 mg/kg/minuto durante una infusión a velocidad constante. Los compuestos de la presente invención se pueden administrar en una dosis diaria individual o la dosificación diaria total se puede administrar en dosis divididas de dos, tres o cuatro veces al día.
Las formas farmacéuticas (composiciones farmacéuticas) adecuadas para la administración pueden contener de
aproximadamente 1 miligramo a aproximadamente 2.000 miligramos de principio activo por unidad de dosificación. En estas composiciones farmacéuticas, el principio activo estará habitualmente presente en una cantidad de aproximadamente el 0,1-95% en peso, basándose en el peso total de la composición. Una cápsula típica para administración oral contiene al menos uno de los compuestos de la presente invención (250 mg), lactosa (75 mg) y estearato de magnesio (15 mg). La mezcla se hace pasar a través de un tamiz de malla 60 y se envasa en una cápsula de gelatina n.° 1. Una preparación inyectable típica se produce mediante la colocación en condiciones asépticas de al menos uno de los compuestos de la presente invención (250 mg) en un vial, el liofilizado en condiciones asépticas y el sellado. Para su uso, el contenido del vial se mezcla con 2 ml de solución salina fisiológica, a fin de producir una preparación inyectable.
Los compuestos de la presente invención se pueden emplear en combinación con otros agentes terapéuticos adecuados útiles en el tratamiento de las enfermedades o los trastornos mencionados anteriormente, incluyendo: agentes antiateroescleróticos, agentes antidislipidémicos, agentes antidiabéticos, agentes antihiperglucémicos, agentes antihiperinsulinémicos, agentes antitrombóticos, agentes antirretinopáticos, agentes antineuropáticos, agentes antinefropáticos, agentes antiisquémicos, agentes antihipertensores, agentes antiobesidad, agentes antihiperlipidémicos, agentes hipertrigliceridémicos, agentes antihipercolesterolémicos, agentes antirreestenóticos, agentes antipancreáticos, agentes hipolipemiantes, agentes anorexigénicos, agentes que potencian la memoria, agentes antidemencia, agentes promotores de la cognición, supresores del apetito, agentes para el tratamiento de la insuficiencia cardíaca, agentes para el tratamiento de la enfermedad arterial periférica, agentes para el tratamiento de tumores malignos y agentes antiinflamatorios.
Los compuestos de la invención se pueden usar con al menos uno de los siguientes agentes para la insuficiencia cardíaca seleccionados de diuréticos de asa, inhibidores de la enzima convertidora de la angiotensina (ACE, por sus siglas en inglés), bloqueadores de los receptores de la angiotensina II (ARB, por sus siglas en inglés), inhibidores de los receptores de la angiotensina-neprilisina (ARNI, por sus siglas en inglés), betabloqueantes, antagonistas de los receptores de mineralocorticoides, donadores de nitroxilo, agonistas de RXFP1, agonistas de APJ y agentes cardiotónicos. Estos agentes incluyen, pero sin limitación, furosemida, bumetanida, torsemida, sacubitrial-valsartán, diuréticos de tiazida, captopril, enalapril, lisinopril, carvedilol, metopolol, bisoprolol, serelaxina, espironolactona, eplerenona, ivabradina, candesartán, eprosartán, irbesartán, losartán, olmesartán, telmisartán y valsartán.
Los compuestos de la presente invención se pueden emplear en combinación con al menos uno de los siguientes agentes terapéuticos en el tratamiento de la ateroesclerosis: agentes antihiperlipidémicos, agentes de elevación del HDL en plasma, agentes antihipercolesterolémicos, inhibidores de la biosíntesis del colesterol (tales como inhibidores de la HMG-CoA reductasa), agonistas de LXR, probucol, raloxifeno, ácido nicotínico, niacinamida, inhibidores de la absorción del colesterol, secuestrantes de ácidos biliares (tales como resinas de intercambio aniónico o aminas cuaternarias [por ejemplo, colestiramina o colestipol]), inductores de los receptores de la lipoproteína de baja densidad, clofibrato, fenofibrato, benzofibrato, cipofibrato, gemfibrizol, vitamina Be, vitamina B12, vitaminas antioxidantes, pbloqueantes, agentes antidiabéticos, antagonistas de la angiotensina II, inhibidores de la enzima convertidora de la angiotensina, inhibidores de la agregación plaquetaria, antagonistas de los receptores de fibrinógeno, aspirina o derivados de ácido fíbrico.
Los compuestos de la presente invención se pueden emplear en combinación con al menos uno de los siguientes agentes terapéuticos en el tratamiento: inhibidor de la biosíntesis del colesterol, particularmente un inhibidor de la HMG-CoA reductasa. Los ejemplos de inhibidores de la HMG-CoA reductasa adecuados incluyen, pero sin limitación, lovastatina, simvastatina, pravastatina, fluvastatina, atorvastatina y rosuvastatina.
Los compuestos de la invención se pueden usar en combinación con al menos uno de los siguientes agentes antidiabéticos dependiendo de la terapia diana deseada. Los estudios indican que la modulación de la diabetes y la hiperlipidemia se pueden mejorar adicionalmente mediante la adición de un segundo agente al régimen terapéutico. Los ejemplos de agentes antidiabéticos incluyen, pero sin limitación, sulfonilureas (tales como clorpropamida, tolbutamida, acetohexamida, tolazamida, gliburida, gliclazida, glinasa, glimepirida y glipizida), biguanidas (tales como metformina), tiazolidinadionas (tales como ciglitazona, pioglitazona, troglitazona y rosiglitazona) y sensibilizadores a la insulina relacionados, tales como activadores selectivos y no selectivos de PPARa, PPARp y PPARy; deshidroepiandrosterona (también denominada DHEA o su éster de sulfato conjugado, DHEA-SO4); antiglucocorticoides; inhibidores de TNFα; inhibidores de la dipeptidil peptidasa IV (DPP4) (tales como sitagliptina y saxagliptina), agonistas o análogos de GLP-1 (tales como exenatida), inhibidores de la a-glucosidasa (tales como acarbosa, miglitol y voglibosa), pramlintida (un análogo sintético de la hormona humana amilina), otros secretagogos de insulina (tales como repaglinida, gliquidona y nateglinida), insulina, así como los agentes terapéuticos analizados anteriormente para el tratamiento de la ateroesclerosis.
Los compuestos de la invención se pueden usar en combinación con al menos uno de los siguientes agentes antiobesidad seleccionados de fenilpropanolamina, fentermina, dietilpropión, mazindol, fenfluramina, dexfenfluramina, fentiramina, agentes agonistas del adrenoceptor P3; sibutramina, inhibidores de la lipasa gastrointestinal (tales como orlistato) y leptinas. Otros agentes usados en el tratamiento de la obesidad o los trastornos relacionados con la obesidad incluyen neuropéptido Y, enterostatina, colecitocinina, bombesina, amilina, receptores de la histamina H3, moduladores del receptor de la dopamina D2, hormona estimulante de melanocitos, factor liberador de la corticotropina,
galanina y ácido gamma amino butírico (GABA, por sus siglas en inglés).
Los compuestos de la presente invención también son útiles como compuestos patrón o de referencia, por ejemplo, como patrón o control de calidad, en ensayos o pruebas que implican el FPR2. Tales compuestos se pueden proporcionar en un kit comercial, por ejemplo, para su uso en la investigación farmacéutica que implica la actividad del FPR2. Por ejemplo, un compuesto de la presente invención se podría usar como referencia en un ensayo para comparar su actividad conocida con un compuesto con una actividad desconocida. Esto garantizaría al experimentador que el ensayo se estaba realizando de manera correcta y proporcionaría una base para la comparación, en especial si el compuesto de ensayo era un derivado del compuesto de referencia. Al desarrollar nuevos ensayos o protocolos, se podrían usar los compuestos de acuerdo con la presente invención para analizar su eficacia. Los compuestos de la presente invención también se pueden usar en ensayos diagnósticos que implican el FPR2.
Los compuestos de la presente invención se pueden usar en un artículo manufacturado. Tal como se usa en el presente documento, el artículo manufacturado está destinado a incluir, pero sin limitación, kits y envases. El artículo manufacturado puede comprender: (a) un primer recipiente; (b) una composición farmacéutica localizada dentro del primer recipiente, en donde la composición comprende un primer agente terapéutico, que comprende: un compuesto de la presente invención o una sal farmacéuticamente aceptable del mismo; y, (c) un prospecto de envase que indica que la composición farmacéutica se puede usar para el tratamiento de dislipidemias y las secuelas de las mismas. El prospecto de envase puede indicar que la composición farmacéutica se puede usar en combinación (tal como se ha definido anteriormente) con un segundo agente terapéutico para el tratamiento de dislipidemias y las secuelas de las mismas. El artículo manufacturado puede comprender, además: (d) un segundo recipiente, en donde los componentes (a) y (b) se localizan dentro del segundo recipiente y el componente (c) se localiza dentro o fuera del segundo recipiente. La expresión "localizado dentro del primer y segundo recipientes" significa que el respectivo recipiente contiene el artículo dentro de sus límites. El primer recipiente es un receptáculo usado para contener una composición farmacéutica. Este recipiente puede ser para la fabricación, la conservación, el envío y/o la venta individual/a granel. El primer recipiente está destinado a abarcar un frasco, un bote, un vial, un matraz, una jeringa, un tubo (por ejemplo, para una preparación en crema) o cualquier otro recipiente usado para fabricar, contener, conservar o distribuir un producto farmacéutico. El segundo recipiente es uno que se usa para contener el primer recipiente y, opcionalmente, el prospecto de envase. Los ejemplos del segundo recipiente incluyen, pero sin limitación, cajas (por ejemplo, de cartón o plástico), embalajes, paquetes, bolsas (por ejemplo, bolsas de papel o de plástico), bolsitas y sacos. El prospecto de envase se puede fijar físicamente en el exterior del primer recipiente a través de cinta adhesiva, pegamento, grapas u otro método de fijación o este se puede introducir en el segundo recipiente sin ningún medio físico de fijación al primer recipiente. Como alternativa, el prospecto de envase se localiza en el exterior del segundo recipiente. Cuando se localiza en el exterior del segundo recipiente, resulta preferible que el prospecto de envase se fije físicamente a través de cinta adhesiva, pegamento, grapas u otro método de fijación. Como alternativa, este puede estar adyacente a o en contacto con el exterior del segundo recipiente sin fijarse físicamente. El prospecto de envase es una ficha técnica, una etiqueta, un marcador, etc., que proporciona información referente a la composición farmacéutica localizada dentro del primer recipiente. La información descrita normalmente la determinará el organismo de regulación gubernamental de la zona en la que se vaya a comercializar el artículo manufacturado (por ejemplo, la Administración de Alimentos y Medicamentos de los Estados Unidos). Preferentemente, el prospecto de envase describe específicamente las indicaciones para las que se ha aprobado la composición farmacéutica. El prospecto de envase se puede elaborar de cualquier material sobre el que una persona pueda leer información contenida en el mismo o sobre el mismo. Preferentemente, el prospecto de envase es un material imprimible (por ejemplo, papel, plástico, cartulina, papel de aluminio, papel o plástico con respaldo adhesivo, etc.) sobre el que se ha plasmado (por ejemplo, impreso o aplicado) la información deseada.
MÉTODOS QUÍMICOS
Las abreviaturas, tal como se usan en el presente documento, se definen de la siguiente manera: "1 x" para una vez, "2 x" para dos veces, "3 x" para tres veces, "°C" para grados Celsius, "ac." para acuoso/a, "Col" para columna, "equiv." para equivalente o equivalentes, "g" para gramo o gramos, "mg" para miligramo o miligramos, "l" para litro o litros, "ml" para mililitro o mililitros, " jl" para microlitro o microlitros, "N" para normal, "M" para molar, "nM" para nanomolar, "mol" para mol o moles, "mmol" para milimol o milimoles, "min" para minuto o minutos, "h" para hora u horas, "ta" para temperatura ambiente, "TR" para tiempo de retención, "UN" para durante una noche, "atm" para atmósfera, "kPa (psi)" para kilopascales (libras por pulgada cuadrada), "conc." para concentrado, "ac." para "acuoso/a", "sat." o "satd." para saturado/a, "PM" para peso molecular, "mo" o "jondas" para microondas, "pf' para punto de fusión, "P" para peso, "EM" o "Espec. de masas" para espectrometría de masas, "ESI" para espectroscopía de masas con ionización por electropulverización, "AR" para alta resolución, "EMAR" para espectrometría de masas de alta resolución, "CLEM" para cromatografía líquida-espectrometría de masas, "CLAP" para cromatografía líquida de alta presión, "CLAP FI" para CLAP de fase inversa, "CCF" o "ccf' para cromatografía en capa fina, "RMN" para espectroscopía de resonancia magnética nuclear, "eOn" para espectroscopía de efecto Overhauser nuclear, "1H" para protón, "8" para delta, "s" para singulete, "d" para doblete, "t" para triplete, "c" para cuadruplete, "m" para multiplete, "a" para amplio, "Hz" para hercio y "a", "p", "R", "S", "E" y "Z" son denominaciones estereoquímicas familiares para un experto en la materia.

continuación

Los compuestos divulgados se pueden elaborar mediante diversos métodos conocidos en la materia, incluyendo aquellos de los siguientes esquemas y los de la sección de realizaciones específicas. La numeración de estructuras y la numeración de variables mostradas en los esquemas sintéticos son distintas y no se deben confundir con la numeración de estructuras o variables de las reivindicaciones o del resto de la memoria descriptiva. Las variables de los esquemas solo pretenden ilustrar cómo elaborar algunos de los compuestos de la presente invención.
La divulgación no se limita a los ejemplos ilustrativos anteriores y los ejemplos se deben considerar en todos los aspectos como ilustrativos y no restrictivos y todos los cambios que entran dentro del significado y rango de equivalencia de las reivindicaciones están, por lo tanto, destinados a abarcarse.
Una consideración a tener en cuenta en la planificación de cualquier vía sintética en este campo es la elección del grupo protector usado para la protección de los grupos funcionales reactivos presentes en los compuestos descritos en la presente invención. Un relato autorizado que describe las numerosas alternativas para el experto capacitado es Greene, T.W. et al., Protecting Groups in Organic Synthesis, 4a Edición, Wiley (2007)).
Los compuestos que tienen la Fórmula (I) general se pueden preparar mediante uno o más de los siguientes esquemas sintéticos.
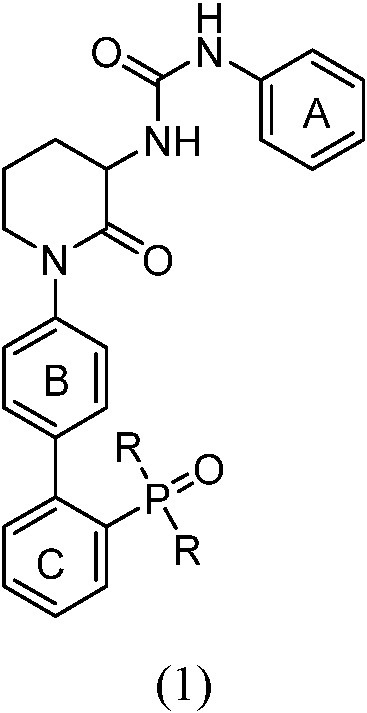
Los compuestos de 1-arilpiperidinona de la presente invención, en donde los Anillos A, B y C son fenilo sustituido, se pueden preparar mediante la vía general que se muestra en el Esquema 1, a partir de una 3-aminopiperidin-2-ona 1a adecuadamente protegida, donde GP es un grupo protector, tal como Boc o Cbz. El acoplamiento catalizado por cobre o paladio de 1a con un bromobenceno sustituido 1b en un disolvente adecuado, tal como butanol o dioxano, en presencia de una base, tal como carbonato de potasio o carbonato de cesio, y un ligando adecuado, tal como N,N-dimetiletilendiamina o xantphos, puede proporcionar 1 -fenilpiperidinonas 1c. Los métodos adicionales para esta transformación incluyen otras variaciones de amidación catalizada por cobre de Buchwald, Ullmann y Goldberg o amidación catalizada por Pd de Buchwald dependiendo de la naturaleza de B, usando métodos conocidos por un experto en la materia para estos tipos de acoplamientos (véase, por ejemplo, Yin & Buchwald Organic Lett. 2000, 2, 1101; Klapers et al. JACS, 2001, 123, 7727; Klapars et al. JACS, 2002, 124, 7421; Yin & Buchwald JACS, 2002, 124, 6043; Kiyomor, Madoux & Buchwald, Tet. Lett., 1999, 40, 2657). La borilación de C-H catalizada por paladio o iridio posterior de 1c de acuerdo con el método de Ishiyama et al. (Angew. Chem. Int. Ed. 2002, 41, 3056) con boronato 1d, seguida de la condensación de las especies de pinacolatoboro resultantes con haluros de arilo o heteroarilo 1e usando procesos catalizados por paladio o cobre, proporciona compuestos de biarilo 1f. La retirada del grupo protector Boc o Cbz de 1f, seguida de la condensación de la amina libre resultante con un isocianato de arilo adecuadamente sustituido, 1g puede proporcionar ureas 1h. Los isocianatos adecuados están disponibles en el mercado o se pueden obtener fácilmente a partir de la anilina correspondiente mediante métodos conocidos por un experto en la materia. Un experto en la materia también reconocerá que los compuestos adicionales de la presente invención, en donde los
Anillos A, B o C son anillos de heteroarilo, tales como piridina, pirimidina, tiazol, etc., también se pueden preparar usando los métodos descritos en el Esquema 1 mediante la sustitución del bromo o yoduro de heteroarilo por 1b y el isocianato de heteroarilo por 1g. Las ureas racémicas 1h se sometieron, además, a la separación enantiomérica usando CLAP quiral o SFC para proporcionar 1i y 1j como enantiómeros individuales.
Esquema 1

Como alternativa, tal como se describe en el Esquema 2, los compuestos de la presente invención se pueden preparar a partir del producto intermedio 1c mediante la realización de un acoplamiento catalizado por paladio con un ácido fenil borónico 2a adecuadamente sustituido o un reactivo de boronato o trifluoroborato adecuado puede proporcionar los compuestos de biarilo 1f. Las transformaciones posteriores para obtener compuestos de urea 1h son similares al Esquema 1.

Adicionalmente, los compuestos de la presente invención se pueden preparar a partir de anilinas 3a sustituidas disponibles en el mercado o se pueden obtener fácilmente mediante métodos conocidos por un experto en la materia mediante la conversión en el producto intermedio 3c usando el acoplamiento de amida con ácido (R)-2-amino-5-(benciloxi)-5-oxopentanoico protegido. La reducción posterior de éster de benciloxi al alcohol 3d correspondiente se puede obtener usando protocolos de la bibliografía, seguida de la reacción de Mitsunobu intramolecular para proporcionar los compuestos 1c. Las transformaciones posteriores para obtener compuestos de urea 1i son similares al Esquema 1.

Otras características de la invención resultarán evidentes en el transcurso de las siguientes descripciones de realizaciones de ejemplo que se ofrecen para ilustrar la invención y que no pretenden limitar la misma.
Los siguientes métodos se usaron en los Ejemplos ilustrados, excepto en los casos en los que se indique otra cosa. La purificación de productos intermedios y productos finales se llevó a cabo a través de cromatografía de fase normal o inversa. La cromatografía de fase normal se llevó a cabo usando cartuchos de SiO2 preenvasados eluyendo o bien con gradientes de hexanos y EtOAc o bien con DCM y MeOH, a menos que se indicara otra cosa. La CLAP preparativa de fase inversa se llevó a cabo usando columnas C18 con detección UV a 220 nm o CLAP preparativa eluyendo con
gradientes de Disolvente A (agua al 90 %, MeOH al 10 %, TFA al 0,1 %) y Disolvente B (agua al 10 %, MeOH al 90 %, TFA al 0,1 %), o con gradientes de Disolvente A (agua al 95 %, ACN al 5 %, TFA al 0,1 %) y Disolvente B (agua al 5 %, ACN al 95 %, TFA al 0,1 %), o con gradientes de Disolvente A (agua al 95 %, ACN al 2 %, HCOOH al 0,1 %) y Disolvente B (ACN al 98 %, agua al 2 %, HCOOH al 0,1 %), o con gradientes de Disolvente A (agua al 95 %, ACN al 5 %, NH4OAc 10 mM) y Disolvente B (ACN al 98 %, agua al 2 %, NH4OAc 10 mM) o con gradientes de Disolvente A (agua al 98 %, ACN al 2 %, NH4OH al 0,1 %) y Disolvente B (ACN al 98 %, agua al 2 %, NH4OH al 0,1 %).
Métodos de CL/EM empleados en la caracterización de los Ejemplos. La CLAP/EM analítica de fase inversa se realizó en un sistema Waters Acquity acoplado con un espectrómetro de masas Waters MICROMASS® ZQ.
Método A: gradiente lineal de B a entre el 0 y el 100 % durante 3 min, con un tiempo de mantenimiento de 0,75 min al 100 % de B;
visualización de UV a 220 nm
columna: Waters BEH C18 de 2,1 x 50 mm
caudal: 1,0 ml/min
Disolvente A: TFA al 0,1 %, agua al 95 %, ACN al 5 %
Disolvente B: TFA al 0,1 %, agua al 5 %, ACN al 95 %
Método B: gradiente lineal de B a entre el 0 y el 100 % durante 3 min, con un tiempo de mantenimiento de 0,75 min al 100 % de B;
visualización de UV a 220 nm
columna: Waters BEH C18 de 2,1 x 50 mm
caudal: 1,0 ml/min
Disolvente A: acetato de amonio 10 mM, agua al 95 %, ACN al 5 %
Disolvente B: acetato de amonio 10 mM, agua al 5 %, ACN al 95 %
CLAP analítica: métodos empleados en la caracterización de los Ejemplos
Los productos se analizaron mediante CLAP analítica de fase inversa: se llevó a cabo en un CLAP analítica Shimadzu: sistema que ejecuta el software Discovery VP. TR = tiempo de retención.
Método C: Ascentis Express C18, de 2,1 x 50 mm, partículas de 2,7 μm; Disolvente A: agua al 95 %, ACN al 5 %, TFA al 0,05 %; Disolvente B: ACN al 95 %, agua al 5 %, TFA al 0,1 %; temperatura: 50 °C; gradiente: B a entre el 0 y 100 % durante 3 minutos, a continuación, con un tiempo de mantenimiento de 1 minuto al 100 % de B; flujo: 1,1 ml/min. Método D: Ascentis Express C18, de 2,1 x 50 mm, partículas de 2,7 μm; Disolvente A: agua al 95 %, ACN al 5 % con acetato de amonio 10 mM; Disolvente B: ACN al 95 %, agua al 5 % con acetato de amonio 10 mM; temperatura: 50 °C; gradiente: B a entre el 0 y 100% durante 3 minutos, a continuación, con un tiempo de mantenimiento de 1 minuto al 100 % de B; flujo: 1,1 ml/min.
Métodos de pureza quiral y SFC
Método I: Chiralpak AD-H, de 250 x 4,6 mm, partículas de 5,0 μm; % de CO2: 60 %, % de codisolvente: 40 % {DEA al 0,2 % en IPA:a Cn (1:1)}, flujo total: 4,0 g/min, contrapresión: 10 MPa (100 bares), temperatura: 25 °C, UV: 218 nm.
Método II: Chiralpak OD-H, de 250 x 4,6 mm, partículas de 5,0 μm; % de CO2: 60 %, % de codisolvente: 40 % {DEA al 0,2% en IPA:ACN (1:1)}, flujo total: 4,0 g/min, contrapresión: 10,4 MPa (104 bares), temperatura: 24,9 °C, UV: 287 nm.
Método III: Chiralpak OJ-H, de 250 x 4,6 mm, partículas de 5,0 μm; % de CO2: 60 %, % de codisolvente: 30 % (DEA al 0,3 % en metanol), flujo total: 4,0 g/min, contrapresión: 10,1 MPa (101 bares), temperatura: 23,6 °C, UV: 272 nm.
Método IV: Chiralpak AS-H, de 250 x 4,6 mm, partículas de 5,0 μm; % de CO2: 60 %, % de codisolvente: 40 % (DEA al 0,3 % en metanol), flujo total: 4,0 g/min, contrapresión: 10,2 MPa (102 bares), temperatura: 25,4 °C, UV: 272 nm.
Método V: Chiralcel OJ-H, de 250 x 4,6 mm, partículas de 5,0 μm; % de CO2: 60 %, % de codisolvente: 40 % (DEA al 0,2 % en metanol), flujo total: 4,0 g/min, contrapresión: 10,2 MPa (102 bares), temperatura: 24,6 °C, UV: 272 nm.
Método VI: Luxcellulose-2, de 250 x 4,6 mm, partículas de 5,0 μm; % de CO2: 60 %, % de codisolvente: 35 % (DEA al 0,2 % en metanol), flujo total: 3,0 g/min, contrapresión: 10,1 MPa (101 bares), temperatura: 23,6 °C, UV: 260 nm.
Método VII: Chiralcel AS-H, de 250 x 4,6 mm, partículas de 5,0 μm; % de CO2: 60 %, % de codisolvente: 40 % (DEA al 0,2 % en metanol), flujo total: 4,0 g/min, contrapresión: 10,1 MPa (101 bares), temperatura: 24,4 °C, UV: 270 nm.
Método VIII: Chiralpak IC, de 250 x 4,6 mm, partículas de 5,0 μm; % de CO2: 60 %, % de codisolvente: 40 % (DEA al 0,2 % en metanol), flujo total: 4,0 g/min, contrapresión: 10,1 MPa (101 bares), temperatura: 24,4 °C, UV: 270 nm.
Método IX: columna: Chiralpak IF (250 x 4,6 mm), de 5 micrómetros, fase móvil: DEA al -0,2 % en etanol, flujo: 1.0 ml/min.
Método X: columna: LUX AMYLOSE 2 (250 x 4,6 mm), de 5 micrómetros, fase móvil: DEA al 0,2 % en n-hexano:etanol: 5:95, flujo: 1,0 ml/min.
Método XI: columna: CHIRALCEL OD-H (250 x 4,6 mm), de 5 micrómetros, fase móvil: DEA al 0,2% en nhexano:etanol: 70:30, flujo: 1,0 ml/min.
Método XII: columna: CHIRALPAK ID (250 x 4,6 mm), de 5 micrómetros, fase móvil: DEA al 0,1 % en metanol, flujo: 1.0 ml/min.
Se empleó la RMN en la caracterización de los Ejemplos. Los espectros de 1H RMN se obtuvieron con espectrómetros de transformada de Fourier Bruker o JEOL® funcionando a las siguientes frecuencias: 1H RMN: 300 MHz (Bruker o JEOL®), o 400 MHz (Bruker o JEOL®) o 500 MHz (Bruker o JEOL®). 13C RMN: 100 MHz (Bruker o JEOL®). Los datos espectrales se indican en el formato: desplazamiento químico (multiplicidad, constantes de acoplamiento y número de hidrógenos). Los desplazamientos químicos se especifican en ppm campo abajo de un patrón interno de tetrametilsilano (unidades 8, tetrametilsilano = 0 ppm) y/o con referencia a picos de disolvente, que en los espectros de 1H RMN aparecen a 2,49 ppm para CD2HSOCD3, 3,30 ppm para CD2HOD, 1,94 para CD3CN y 7,24 ppm para CHCl3 y que en los espectros de 13C RMN aparecen a 39,7 ppm para CD3SOCD3, 49,0 ppm para CD3OD y 77,0 ppm para CDCh. Todos los espectros de 13C RMN se desacoplaron de protones.
Producto intermedio 1: (1-(4-bromo-2,3-difluorofenil)-2-oxopiperidin-3-il)carbamato de ferc-butilo

A una solución agitada de 1,4-dibromo-2,3-difluorobenceno (4,0 g, 15 mmol) en 1,4-dioxano (40 ml) se le añadieron (2-oxopiperidin-3-il)carbamato de ferc-butilo (3,5 g, 16 mmol) y carbonato de cesio (9,6 g, 29 mmol). La mezcla de reacción se purgó con nitrógeno durante 5 min y se cargó con Xantphos (0,85 g, 1,5 mmol) y Pd2(dba)3 (0,67 g, 0,74 mmol). La mezcla de reacción se purgó nuevamente con nitrógeno durante 3 min y se calentó a 110 °C durante 16 h. La mezcla se enfrió y se filtró a través de una almohadilla de Celite y el filtrado se concentró a presión reducida. El producto en bruto se purificó a través de cromatografía en columna (éter de pet-EtOAc) para proporcionar el Producto intermedio 1 (1,9 g, 4,7 mmol, con un rendimiento del 20 %) en forma de un sólido de color amarillento. EM (ESI) m/z: 405,2 (M+H)+. 1H RMN (400 MHz, CDCla) 57,36 (m, 1H), 6,96 (m, 1H), 5,44 (s, 1H), 4,37-4,22 (m, 1H), 3,71-3,58 (m, 2H), 2,69-2,55 (m, 1H), 2,16-2,02 (m, 2H), 1,77 (m, 1H), 1,47 (m, 9H).
Producto intermedio 2: (1-(2,3-difluoro-4-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)-2-oxopiperidin-3-il)carbamato de ferc-butilo

A una solución agitada del Producto intermedio 2 (250 mg, 0,62 mmol) en 1,4-dioxano (10 ml) se le añadieron bis(pinacolato)diboro (240 mg, 0,93 mmol) y acetato de potasio (150 mg, 1,5 mmol). La mezcla de reacción resultante se purgó con argón durante 5 min y se cargó con complejo de dicloruro de 1,1'-bis(difenilfosfino)ferrocenopaladio(II).DCM (35 mg, 0,043 mmol). A continuación, la mezcla de reacción se purgó nuevamente con argón durante 3 min y se calentó hasta 80 °C durante 16 h. La mezcla de reacción se filtró a través de una almohadilla de Celite, se
concentró a presión reducida y se usó para la siguiente etapa sin purificación adicional. EM (ESI) m/z: 453,5 (M+H)+.
Producto intermedio 3: (1-(2'-(dimetilfosforil)-2,3-difluoro-[1,1'-bifenil]-4-il)-2-oxopiperidin-3-il)carbamato de ferc-butilo

A una solución agitada del Producto intermedio 2 (230 mg, 0,50 mmol) en 1,4-dioxano (2 ml) se le añadieron óxido de (2-bromofenil)dimetilfosfina (120 mg, 0,50 mmol) y fosfato de potasio (210 mg, 1,0 mmol). La mezcla de reacción se purgó con nitrógeno durante 5 min y se cargó con complejo de dicloruro de 1,1'-bis(difenilfosfino)ferrocenopaladio(II).DCM (41 mg, 0,050 mmol). La mezcla de reacción se purgó nuevamente con nitrógeno durante 3 min y se calentó a 80 °C durante 16 h. La mezcla de reacción se enfrió y se filtró a través de una almohadilla de Celite y el filtrado se concentró a presión reducida. El producto en bruto se purificó a través de cromatografía ultrarrápida (CHCh-MeOH) para proporcionar el Producto intermedio 3 (140 mg, 0,23 mmol, con un rendimiento del 47 %) en forma de un líquido de color marrón oscuro. EM (ESI) m/z: 496,5 (M+NH4)+. 1H RMN (400 MHz, DMSO-da) 88,08-7,86 (m, 1H), 7,83-7,74 (m, 2H), 7,68-7,50 (m, 3H), 7,29-7,22 (m, 1H), 4,19-4,15 (m, 1H), 3,71-3,68 (m, 1H), 3,64-3,60 (m, 1H), 2,15 1,83 (m, 4H), 1,53-1,46 (s, 9H), 1,42-1,34 (m, 6H).
Producto intermedio 4: 3-amino-1-(2'-(dimetilfosforil)-2,3-difluoro-[1,1'-bifenil]-4-il)piperidin-2-ona

A una solución enfriada con hielo del Producto intermedio 3 (130 mg, 0,27 mmol) en 1,4-dioxano (1,5 ml) en atmósfera de argón a ta se le añadió HCl 4 M (1,0 ml, 4,0 mmol) en 1,4-dioxano y la mezcla se agitó a ta durante 2 h. El disolvente se evaporó a presión reducida para obtener un sólido gomoso. Este se trituró adicionalmente con éter de pet (5 ml x 2) y se secó para proporcionar el Producto intermedio 4 (95 mg, 0,25 mmol, con un rendimiento del 92 %) en forma de un sólido de color marrón. EM (ESI) m/z: 379,2 (M+H)+.
Producto intermedio 5: óxido de (2-bromofenil)dimetilfosfina

A una solución agitada de 1-bromo-2-yodobenceno (10 g, 35 mmol) en DMF (80 ml) se le añadieron óxido de dimetilfosfina (3,3 g, 42 mmol) y K3PO4 (8,3 g, 39 mmol). La mezcla de reacción se purgó con nitrógeno durante 5 min y se cargó con Xantphos (1,2 g, 2,1 mmol) y Pd(OAc)2(0,40 g, 1,8 mmol). La mezcla de reacción se purgó nuevamente con nitrógeno durante 3 min y se calentó a 100 °C durante 6 h. La mezcla de reacción se enfrió, se filtró a través de una almohadilla de Celite y se concentró a presión reducida. El producto en bruto se purificó a través de cromatografía (MeOH-DCM) para producir el Producto intermedio 5 (5,0 g, 21 mmol, con un rendimiento del 60 %) en forma de un sólido de color amarillento. EM (ESI) m/z: 232,9 (M+H)+. 1H RMN (400 MHz, DMSO-d6) 7,98-7,94 (m, 1H), 7,77-7,74 (m, 1H), 7,76-7,48 (m, 2H), 1,83 (d, J = 14 Hz, 6H).
Producto intermedio 6: óxido de dimetil(2-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)fenil)fosfina

A una solución agitada del Producto intermedio 5 (1,0 g, 4,3 mmol) se le añadieron bis(pinacolato)diboro (1,3 g, 5,2 mmol) y acetato de sodio (0,39 g, 4,7 mmol). La mezcla de reacción se purgó con nitrógeno durante 5 min y se cargó con Pd(dba)2 (0,025 g, 0,043 mmol) y X-phos (0,041 g, 0,086 mmol). La mezcla de reacción se purgó nuevamente con nitrógeno durante 3 min y se calentó a 110 °C durante 3 h. La mezcla de reacción se diluyó con EtOAc (20 ml), se filtró a través de una almohadilla de Celite y se evaporó a presión reducida. El producto en bruto se usó en la siguiente etapa sin purificación adicional. EM (ESI) m/z: 281,0 (M+H)+.
Producto intermedio 7: (R)-5-((4-bromo-2,6-difluorofenil)amino)-4-((ferc-butoxicarbonil)amino)-5-oxopentanoato de bencilo

A una solución agitada del Producto intermedio 6 (5,0 g, 15 mmol) en ACN (40 ml) en atmósfera de argón a temperatura ambiente se le añadieron DIEA (7,8 ml, 45 mmol), HATU (6,8 g, 18 mmol) y 4-bromo-2,6-difluoroanilina (3,2 g, 16 mmol). La mezcla de reacción se agitó a 60 °C durante 16 h. A continuación, la mezcla de reacción se concentró a presión reducida, se inactivó con una solución de NH4Cl saturada acuosa (10 ml) y se extrajo con EtOAc (20 ml x 3). Las capas orgánicas combinadas se lavaron con salmuera (20 ml), se secaron sobre Na2SO4, se concentraron a presión reducida y se purificaron a través de cromatografía en columna (35 % de EtOAc-éter de pet) para proporcionar el Producto intermedio 7 (1,4 g, 2,7 mmol, con un rendimiento del 18 %) en forma de un sólido de color blanco. EM (ESI) m/z: 527,1 (M+H)+. 1H RMN (300 MHz, CDCh) 57,36-7,35 (m, 5H), 7,16 (d, J = 5,4 Hz, 2H), 5,35-5,30 (m, 1H), 5,15-5,11 (m, 2H), 2,72-2,48 (m, 2H), 2,31-2,19 (m, 1H), 2,10-1,99 (m, 1H), 1,54 (s, 9H), 1,53-1,50 (m, 1H), 0,90-0,84 (m, 1H).
Producto intermedio 8: (R)-(1-((4-bromo-2,6-difluorofenil)amino)-5-hidroxi-1-oxopentan-2-il)carbamato de ferc-butilo

A una solución agitada del Producto intermedio 7 (1,4 g, 2,7 mmol) en THF (12 ml) con argón a 0 °C se le añadieron MeOH (0,32 ml, 8,0 mmol) y UBH4 (4,0 ml, 8,0 mmol). La mezcla de reacción resultante se calentó gradualmente hasta ta y se agitó durante 30 min más. La mezcla de reacción se inactivó con NH4Cl acuoso (15 ml) gota a gota y se extrajo con EtOAc (20 ml x 3). Las capas orgánicas combinadas se lavaron con agua (15 ml) y salmuera (20 ml), se secaron sobre Na2SO4 y se concentraron a presión reducida. El producto en bruto se purificó a través de cromatografía en columna (70 % de EtOAc-éter de pet) para proporcionar el Producto intermedio 8 (860 mg, 2,0 mmol, con un rendimiento del 77 %) en forma de un sólido de color blanco. EM (ESI) m/z: 423,1 (M+H)+. 1H RMN (400 MHz, CDCla) 5 8,28 (s a, 1H), 7,17-7,12 (m, 2H), 5,36 (s a, 1H), 4,52-4,48 (m, 1H), 3,79 (c, J = 5,5 Hz, 2H), 2,10-1,97 (m, 1H), 1,94
1,68 (m, 3H), 1,52-1,44 (m, 9H).
Producto intermedio 9: (R)-(1-(4-bromo-2,6-d¡fluorofeml)-2-oxop¡pendin-3-¡l)carbamato de ferc-butilo

A una solución enfriada con hielo de azodicarboxilato de di-ferc-butilo (1,0 g, 4,4 mmol) en THF (5 ml) en atmósfera de argón a ta se le añadió fr/-n-butilfosfina (1,1 ml, 4,4 mmol). Después de 5 min, se canuló en el matraz una solución enfriada con hielo del Producto intermedio 8 (1,3 g, 3,0 mmol) en THF (5 ml) y la mezcla de reacción se calentó hasta ta durante 2 h. La mezcla de reacción se inactivó con una solución de NaHCO3 saturada ac. (20 ml) y se extrajo con EtOAc (20 ml x 3). Las capas orgánicas combinadas se lavaron con una solución de NH4Cl ac. (15 ml x 2) y salmuera (20 ml), se secaron sobre Na2SO4 y se concentraron a presión reducida. El producto en bruto se purificó a través de cromatografía en columna (20 % de éter de pet-EtOAc) para proporcionar el Producto intermedio 9 (800 mg, 2,0 mmol, con un rendimiento del 67 %) en forma de un sólido de color blanco. EM (ESI) m/z: 405,1 (M+H)+. 1H RMN (400 MHz, CDCh) 57,20-7,18 (m, 1H), 7,16-7,12 (m, 1H), 5,44 (s a, 1H), 4,36-4,24 (m, 1H), 3,66-3,51 (m, 2H), 2,66-2,56 (m, 1H), 2,12-2,05 (m, 2H), 1,81-1,70 (m, 1H), 1,57-1,45 (s, 9H).
Ejemplo 1: (R)-1-(4-cloro-2-fluorofen¡l)-3-(1-(2'-(d¡met¡lfosfo^l)-2,3-d¡fluoro-[1,1'-b¡fen¡l]-4-¡l)-2-oxop¡pe^d¡n-3-¡l)urea

A una suspensión enfriada con hielo del Producto intermedio 4 (100 mg, 0,26 mmol) en DMF (2 ml) en atmósfera de argón a ta se le añadieron DIEA (0,14 ml, 0,79 mmol) y (4-cloro-2-fluorofenil)carbamato de fenilo (70 mg, 0,26 mmol). La solución resultante se calentó a 45 °C durante 16 h. La mezcla de reacción se concentró a presión reducida y el residuo se purificó mediante CLAP de fase inversa seguida de CLAP quiral para proporcionar el Ejemplo 1 (22 mg, 0,039 mmol, con un rendimiento del 15 %) en forma de un sólido de color blanco. EM (ESI) m/z: 550,2 (M+H)+. 1H RMN (400 MHz, DMSO-d6) 58,67 (d, J = 2,2 Hz, 1H), 8,17 (t, J = 8,9 Hz, 1H), 8,01-7,88 (m, 1H), 7,74-7,57 (m, 2H), 7,50-7,33 (m, 2H), 7,33-7,21 (m, 2H), 7,21-7,05 (m, 2H), 4,48-4,37 (m, 1H), 3,85-3,62 (m, 2H), 2,31 (dd, J = 12,3, 6,5 Hz, 1H), 2,17-1,93 (m, 2H), 1,86 (d, J = 6,5 Hz, 1H), 1,49 (d, J = 13,5 Hz, 6H). TR = 1,539 min, 99,2 % (Método D).
Los siguientes Ejemplos en la Tabla 2 se elaboraron mediante el uso del mismo procedimiento que el mostrado en el

(continuación)

(continuación)

(continuación)
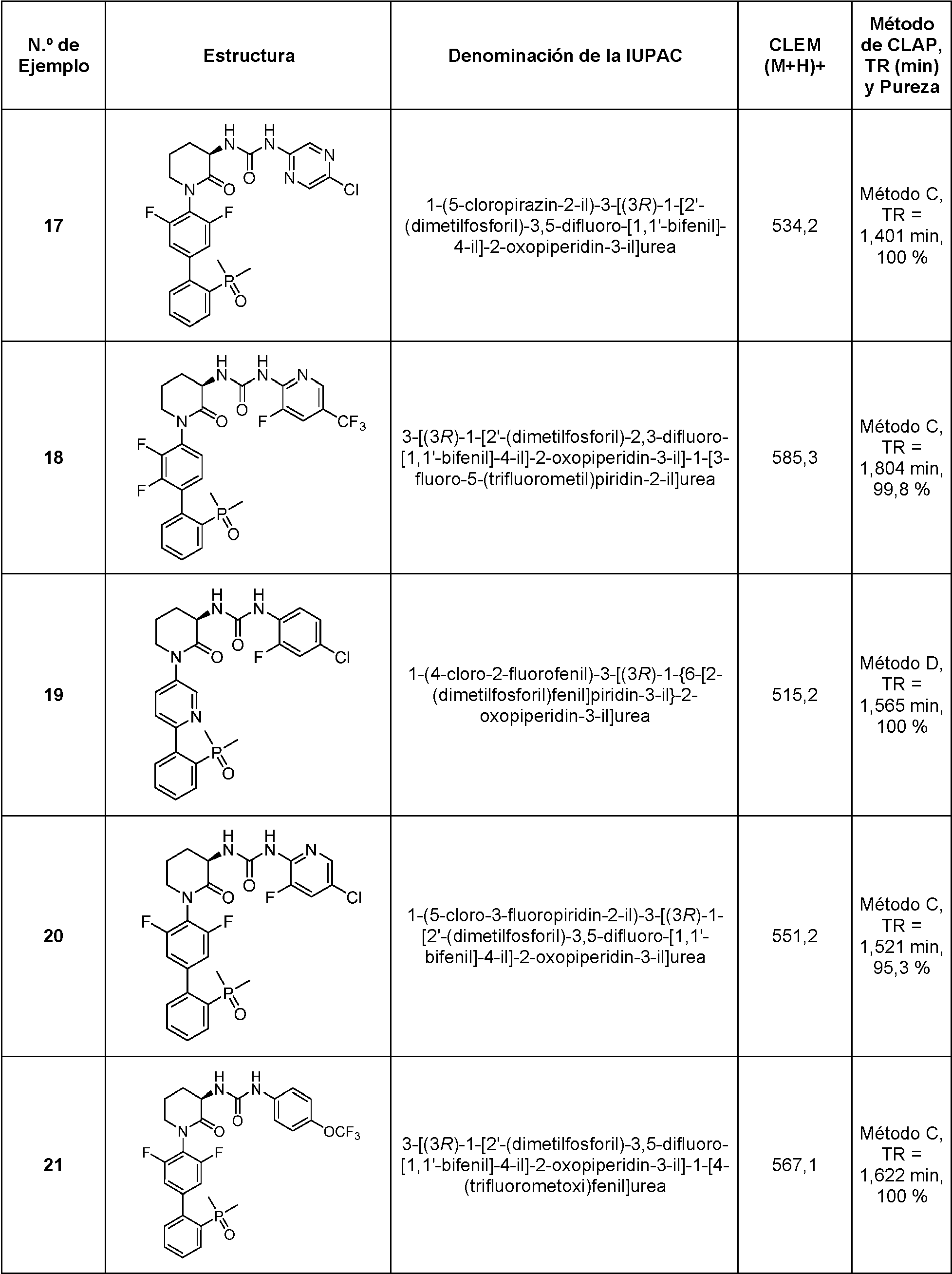
Datos de RMN de los Ejemplos de la Tabla 2:
Ejemplo 2: 1H RMN (400 MHz, DMSO-da) 88,95 (s a, 1H), 8,42 (t, J = 8,3 Hz, 1H), 7,95 (m, 1H), 7,73-7,56 (m, 3H), 7.50 (d, J = 8,8 Hz, 1H), 7,39 (d, J = 3,7 Hz, 1H), 7,34-7,15 (m, 3H), 4,52-4,39 (m, 1H), 3,76-3,68 (m, 2H), 2,39-2,28 (m, 1H), 2,22-1,96 (m, 2H), 1,91-1,85 (m, 1H), 1,49 (d, J = 13,5 Hz, 6H).
Ejemplo 3: 1H RMN (400 MHz, DMSO-d6) 88,64 (s, 1H), 8,17 (t, J = 8,9 Hz, 1H), 7,91 (m, 1H), 7,73-7,44 (m, 2H), 7,44 7,33 (m, 2H), 7,29 (d, J = 9,0 Hz, 2H), 7,23-7,09 (m, 2H), 4,50-4,38 (m, 1H), 3,72-3,59 (m, 2H), 2,35-2,29 (m, 1H), 2,1 2,02 (m, 2H), 1,84 (d, J = 5,6 Hz, 1H), 1,75-1,51 (m, 4H), 1,07-0,79 (m, 6H).
Ejemplo 4: 1H RMN (400 MHz, DMSO-d6) 89,38 (s a, 1H), 8,83 (s, 1H), 8,21-8,09 (m, 1H), 8,02 (m, 1H), 7,94 (m, 1H), 7,61 (t, J = 7,5 Hz, 1H), 7,54 (t, J = 7,5 Hz, 1H), 7,35 (m, 1H), 7,32-7,14 (m, 2H), 7,10 (d, J = 7,1 Hz, 1H), 4,53-4,39 (m, 1H), 3,75-3,56 (m, 2H), 2,42-2,38 (m, 1H), 2,15-1,82 (m, 4H), 1,44-1,14 (m, 6H), 1,04-0,85 (m, 3H), 0,64-0,60 (m, 1H).
Ejemplo 5: 1H RMN (400 MHz, DMSO-d6) 88,69 (s, 1H), 8,17 (m, 1H), 8,01-7,87 (m, 1H), 7,64-7,44 (m, 2H), 7,44-7,31 (m, 2H), 7,28-7,25 (m, 2H), 7,23-6,95 (m, 3H), 4,38-4,34 (m, 1H), 3,66-3,55 (m, 2H), 2,35-2,30 (m, 1H), 2,09-2,03 (m, 2H), 1,92-1,86 (m, 2H), 1,33-1,27 (m, 6H), 0,87-0,84 (m, 3H), 0,64-0,61 (m, 1H).
Ejemplo 6: 1H RMN (400 MHz, DMSO-d6) 88,30 (d, J = 1,5 Hz, 1H), 7,95 (m, 1H), 7,86 (m, 1H), 7,70-7,55 (m, 2H), 7.38 (m, 1H), 7,33-7,19 (m, 2H), 6,90 (d, J = 6,8 Hz, 1H), 6,86 (m, 1H), 6,71 (m, 1H), 4,45-4,33 (m, 1H), 3,82-3,59 (m, 5H), 2,32-2,24 (m, 1H), 2,14-1,97 (m, 2H), 1,88-1,73 (m, 1H), 1,59-1,34 (m, 6H).
Ejemplo 7: 1H RMN (400 MHz, DMSO-d6) 88,63 (d, J = 2,4 Hz, 1H), 8,17 (m, 1H), 7,92 (m, 1H), 7,69-7,52 (m, 2H), 7,49-7,24 (m, 4H), 7,22-7,05 (m, 2H), 4,50-4,40 (m, 1H), 3,77-3,58 (m, 2H), 2,32 (dd, J = 12,0, 5,9 Hz, 1H), 2,17-1,94 (m, 2H), 1,90-1,77 (m, 1H), 1,46 (d, J = 13,5 Hz, 6H).
Ejemplo 8: 1H RMN (400 MHz, DMSO-d6) 88,88 (s, 2H), 8,40 (d, J = 1,2 Hz, 1H), 7,95 (m, 1H), 7,73-7,57 (m, 2H), 7,53 (s, 1H), 7,38 (s, 1H), 7,34-7,17 (m, 2H), 4,47 (m, 1H), 3,79-3,75 (m, 1H), 3,74-3,68 (m, 1H), 2,37-2,33 (m, 1H), 2,18 1,98 (m, 2H), 1,98-1,80 (m, 1H), 1,53-1,46 (m, 6H).
Ejemplo 9: 1H RMN (400 MHz, DMSO-d6) 89,41 (s, 1H), 8,83 (s, 1H), 8,17 (d, J = 2,0 Hz, 1H), 8,03 (m, 1H), 7,98-7,90 (m, 1H), 7,72-7,54 (m, 2H), 7,37 (m, 1H), 7,33-7,18 (m, 2H), 4,56-4,47 (m, 1H), 3,79-3,73 (m, 1H), 3,72-3,66 (m, 1H), 2.38 (m, 1H), 2,16-1,98 (m, 2H), 1,96-1,87 (m, 1H), 1,49 (d, J = 13,5 Hz, 6H).
Ejemplo 10: 1H RMN (400 MHz, DMSO-d6) 88,96 (s, 1H), 8,01-7,89 (m, 1H), 7,73-7,56 (m, 2H), 7,56-7,41 (m, 2H), 7,41-7,34 (m, 1H), 7,33-7,14 (m, 4H), 6,62 (d, J = 7,1 Hz, 1H), 4,45-4,35 (m, 1H), 3,80-3,60 (m, 2H), 2,31 (m, 1H), 2,16-1,94 (m, 2H), 1,88 (m, 1H), 1,48 (d, J = 13,5 Hz, 6H).
Ejemplo 11: 1H RMN (400 MHz, DMSO-d6) 88,92 (d, J = 2,9 Hz, 1H), 8,42 (m, 1H), 7,93 (m, 1H), 7,73-7,55 (m, 3H), 7.51 (d, J = 8,6 Hz, 1H), 7,46-7,35 (m, 3H), 7,33 (d, J = 7,3 Hz, 1H), 4,52-4,43 (m, 1H), 3,75-3,59 (m, 2H), 2,37-2,32 (m, 1H), 2,21-1,94 (m, 2H), 1,92-1,80 (m, 1H), 1,46 (d, J = 13,5 Hz, 6H).
Ejemplo 12: 1H RMN (400 MHz, DMSO-d6) 89,81 (s, 1H), 8,63-8,52 (m, 1H), 8,13 (s, 1H), 8,07 (dd, J = 9,0, 2,4 Hz, 1H), 7,99-7,90 (m, 1H), 7,74 (d, J = 8,8 Hz, 1H), 7,70-7,56 (m, 2H), 7,43-7,34 (m, 1H), 7,34-7,22 (m, 2H), 4,54-4,45 (m, 1H), 3,83-3,74 (m, 1H), 3,74-3,65 (m, 1H), 2,41-2,30 (m, 1H), 2,17-1,97 (m, 2H), 1,95-1,79 (m, 1H), 1,48 (d, J = 13,5 Hz, 6H).
Ejemplo 13: 1H RMN (400 MHz, DMSO-d6) 88,48 (s, 1H), 7,92 (m, 1H), 7,72-7,53 (m, 2H), 7,46-7,36 (m, 3H), 7,35 7,22 (m, 2H), 6,91-6,75 (m, 2H), 6,47 (d, J = 6,8 Hz, 1H), 4,42 (m, 1H), 3,70 (s, 3H), 3,68-3,58 (m, 2H), 2,35-2,27 (m, 1H), 2,18-1,93 (m, 2H), 1,89-1,78 (m, 1H), 1,46 (d, J = 13,5 Hz, 6H).
Ejemplo 14: 1H RMN (400 MHz, DMSO-d6) 88,93-8,82 (m, 1H), 8,16 (d, J = 2,2 Hz, 1H), 8,02 (m, 1H), 7,95-7,86 (m, 1H), 7,69-7,58 (m, 3H), 7,34 (m, 1H), 7,28 (m, 1H), 7,21-7,14 (m, 1H), 4,55-4,48 (m, 1H), 3,81-3,74 (m, 1H), 3,72-3,65 (m, 1H), 2,42-2,34 (m, 1H), 2,18-1,93 (m, 2H), 1,96-1,84 (m, 1H), 1,81-1,66 (m, 3H), 1,65-1,52 (m, 1H), 1,11-0,68 (m, 6H).
Ejemplo 15: 1H RMN (400 MHz, DMSO-d6) 89,42 (s, 1H), 8,81 (d, J = 5,9 Hz, 1H), 8,18 (s, 1H), 8,10-7,98 (m, 1H), 7,90 (m, 1H), 7,74-7,50 (m, 2H), 7,35 (m, 1H), 7,29 (m, 2H), 4,61-4,48 (m, 1H), 3,68 (m, 2H), 2,43-2,37 (m, 1H), 2,18 1,99 (m, 2H), 1,97-1,84 (m, 1H), 1,78-1,54 (m, 4H), 1,03-0,77 (m, 6H).
Ejemplo 16: 1H RMN (400 MHz, DMSO-d6) 88,74 (d, J = 2,4 Hz, 1H), 8,24 (m, 1H), 7,95 (m, 7,6, 1,3 Hz, 1H), 7,70 7,57 (m, 2H), 7,45-7,22 (m, 4H), 7,20-7,09 (m, 2H), 4,47-4,37 (m, 1H), 3,77-3,61 (m, 2H), 2,35-2,30 (m, 1H), 2,12-1,99 (m, 2H), 1,91-1,80 (m, 1H), 1,58-1,41 (m, 6H).
Ejemplo 17: 1H RMN (400 MHz, DMSO-d6) 89,68 (s, 1H), 8,88 (d, J = 1,5 Hz, 1H), 8,40 (d, J = 1,2 Hz, 1H), 7,92 (m, 1H), 7,72-7,54 (m, 2H), 7,50 (d, J = 6,8 Hz, 1H), 7,45-7,26 (m, 3H), 4,55-4,45 (m, 1H), 3,77-3,55 (m, 2H), 2,42-2,34 (m, 1H), 2,21-1,94 (m, 2H), 1,93-1,80 (m, 1H), 1,46 (d, J = 13,5 Hz, 6H).
Ejemplo 18: 1H RMN (400 MHz, DMSO-da) 59,52 (s, 1H), 9,26 (s, 1H), 8,47 (s, 1H), 8,20 (m, 1H), 7,95 (m, 1H), 7,75 7,53 (m, 2H), 7,37 (m, 1H), 7,33-7,19 (m, 2H), 4,60-4,52 (m, 1H), 3,80-3,76 (m, 1H), 3,75-3,66 (m, 1H), 2,41 (m, 1H), 2,07 (d, J = 5,1 Hz, 2H), 1,99-1,88 (m, 1H), 1,49 (d, J = 13,5 Hz, 6 H).
Ejemplo 19: 1H RMN (400 MHz, DMSO-d6) 58,72 (s a, 1H), 8,65 (s, 1H), 8,16 (m, 1H), 8,08 (m, 1H), 7,90 (m, 1H), 7,75-7,55 (m, 3H), 7,55-7,46 (m, 1H), 7,40 (d, J = 12,5 Hz, 1H), 7,14 (d, J = 7,6 Hz, 1H), 7,18 (d, J = 9,0 Hz, 1H), 4,40 (m, 1H), 3,85 (m, 1H), 3,79-3,70 (m, 1H), 2,28 (d, J = 5,9 Hz, 1H), 2,16-1,91 (m, 2H), 1,86 (d, J = 6,4 Hz, 1H), 1,46 (d, J = 13,5 Hz, 6 H).
Ejemplo 20: 1H RMN (400 MHz, DMSO-d6) 59,40-9,15 (s a, 1H), 8,80 (s a, 1H), 8,18 (d, J = 2,0 Hz, 1H), 8,02 (m, 1H), 7,96-7,84 (m, 1H), 7,71-7,51 (m, 2H), 7,48-7,27 (m, 3H), 4,60-4,48 (m, 1H), 3,76-3,57 (m, 2H), 2,39 (m, 1H), 2,17-2,00 (m, 2H), 2,00-1,81 (m, 1H), 1,46 (d, J = 13,5 Hz, 6 H).
Ejemplo 21: 1H RMN (400 MHz, DMSO-d6) 59,76 (s, 1H), 8,58 (s, 1H), 8,17-8,04 (m, 2H), 7,96-7,89 (m, 1H), 7,76 (d, J = 8.8 Hz, 1H), 7,65 (m, 1H), 7,59 (m, 1H), 7,47-7,31 (m, 3H), 4,59-4,49 (m, 1H), 3,68 (m, 2H), 2,37 (m, 1H), 2,20 1,96 (m, 2H), 1,94-1,81 (m, 1H), 1,46 (d, J = 13,5 Hz, 6 H).
Claims (15)
1. Un compuesto de la Fórmula (I):

o una sal farmacéuticamente aceptable del mismo, en la que:
Ar1 es fenilo sustituido con 1-2 R1a y 1-2 R1b o heteroarilo de 6 miembros con 1-3 átomos de nitrógeno y sustituido con 1-2 R1a y 1-2 R1b;
Ar2 es fenilo sustituido con 1-4 R2 o heteroarilo de 6 miembros con 1-3 átomos de nitrógeno y sustituido con 1-4 R2; Ar3 es fenilo sustituido con 0-4 R3 o piridinilo sustituido con 0-4 R3;
R1a es hidrógeno o halo;
R1b es halo, haloalquilo, alcoxi o haloalcoxi;
R2 es hidrógeno, ciano, halo, alquilo, hidroxialquilo, haloalquilo, cicloalquilo, alcoxi o haloalcoxi;
R3 es ciano, halo, alquilo, alcoxi, hidroxialquilo, alcoxialquilo o haloalquilo; y
R4 es alquilo o alcoxi.
2. El compuesto de la reivindicación 1, que tiene la Fórmula (II):

o una sal farmacéuticamente aceptable del mismo, en la que:
Ar1 es fenilo sustituido con 1 R1a y 1-2 R1b o heteroarilo de 6 miembros con 1-2 átomos de nitrógeno y sustituido con 1 R1a y 1-2 R1b;
Ar2 es fenilo sustituido con 1-3 R2 o piridinilo sustituido con 1-3 R2;
R1a es hidrógeno o halo;
R1b es halo, haloalquilo C1-4, alcoxi C1-4 o haloalcoxi C1-4;
R2 es hidrógeno, halo, alquilo C1-4, hidroxialquilo C1-4, haloalquilo C1-4, cicloalquilo C3-6, alcoxi C1-4 o haloalcoxi C1-4; y
R4 es alquilo C1-3.
3. El compuesto de las reivindicaciones 1 o 2, que tiene la Fórmula (III):

o una sal farmacéuticamente aceptable del mismo, en la que:
Ar1 es fenilo sustituido con 1 R1a y 1-2 R1b, piridinilo sustituido con 1 R1a y 1-2 R1b o pirazinilo sustituido con 1 R1a y 1-2 R1b;
R1a es hidrógeno o halo;
R1b es halo, haloalquilo C1.4, alcoxi C1.4 o haloalcoxi C1.4;
R2 es hidrógeno, halo, alquilo C1-4, hidroxialquilo C1.4, haloalquilo C1.4, cicloalquilo C3-6, alcoxi C1-4 o haloalcoxi C1.
4; y
R4 es alquilo C1.2.
4. El compuesto de la reivindicación 3, que tiene la Fórmula (IV):

o una sal farmacéuticamente aceptable del mismo, en la que:
R1a es hidrógeno o F;
R1b es halo, haloalquilo C1-2 o alcoxi C1-2;
R2 es hidrógeno, halo, alquilo C1-3, haloalquilo C1.3 o cicloalquilo C3-6; y
R4 es metilo o etilo.5
5. El compuesto de la reivindicación 4, que tiene la Fórmula (VI):

o una sal farmacéuticamente aceptable del mismo, en la que:
R1a es hidrógeno o F;
R1b es halo, haloalquilo C1-2 o alcoxi C1-2;
R2 es halo, alquilo C1-3, haloalquilo C1-3 o cicloalquilo C3-6; y
R4 es metilo o etilo.
6. El compuesto de las reivindicaciones 4 o 5, o una sal farmacéuticamente aceptable del mismo, en donde:
R1a es hidrógeno o F;
R1b es F, Cl o CF3;
R2 es hidrógeno, F, Cl, isopropilo, CF3 o ciclopropilo; y
R4 es metilo.
7. El compuesto de la reivindicación 3, que tiene la Fórmula (V):

o una sal farmacéuticamente aceptable del mismo, en la que:
R1a es hidrógeno o F;
R1b es halo, haloalquilo C1-2 o alcoxi C1-2;
R2 es halo; y
R4 es metilo o etilo.
8. El compuesto de la reivindicación 3, que tiene la Fórmula (VII):

o una sal farmacéuticamente aceptable del mismo, en la que:
R1a es hidrógeno o halo;
R1b es halo, haloalquilo C1-2 o alcoxi C1-2;
R2 es hidrógeno, halo, alquilo C1-3 o cicloalquilo C3-6; y
R4 es alquilo C1-2.
9. El compuesto de la reivindicación 3, que tiene la Fórmula (VIII):

o una sal farmacéuticamente aceptable del mismo, en la que:
R1b es halo, haloalquilo C1-2 o alcoxi C1-2;
R2 es hidrógeno, halo, alquilo C1-3 o cicloalquilo C3-6; y
R4 es alquilo C1-2.
10. El compuesto de las reivindicaciones 1 o 2, que tiene la Fórmula (IX):

o una sal farmacéuticamente aceptable del mismo, en la que:
Ar1 es fenilo sustituido con 1 R1a y 1-2 R1b, piridinilo sustituido con 1 R1a y 1-2 R1b o pirazinilo sustituido con 1 R1a y 1-2 R1b;
R1a es hidrógeno o halo;
R1b es halo, haloalquilo C1.4, alcoxi C1.4 o haloalcoxi C1.4; y
R4 es alquilo C1-2.
11. Un compuesto de la reivindicación 1 seleccionado del grupo que consiste en:


o una sal farmacéuticamente aceptable del mismo.
12. Una composición que comprende un compuesto de una cualquiera de las reivindicaciones 1-11, o una sal farmacéuticamente aceptable del mismo, y un vehículo, un diluyente o un excipiente farmacéuticamente aceptables.
13. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1-11, o una sal farmacéuticamente aceptable del mismo, o una composición de acuerdo con la reivindicación 12, para su uso en terapia.
14. Un compuesto de cualquiera de las reivindicaciones 1-11, o una sal farmacéuticamente aceptable del mismo, o una composición de acuerdo con la reivindicación 12, para su uso en un método para el tratamiento de una cardiopatía, comprendiendo el método administrar una cantidad terapéuticamente eficaz de un compuesto de una cualquiera de las reivindicaciones 1-11, o una sal farmacéuticamente aceptable del mismo, a un paciente que lo necesita.
15. Un compuesto de cualquiera de las reivindicaciones 1-11, o una sal farmacéuticamente aceptable del mismo, o una composición de acuerdo con la reivindicación 12, para su uso en el tratamiento de una cardiopatía, en donde la cardiopatía se selecciona del grupo que consiste en angina de pecho, angina inestable, infarto de miocardio, insuficiencia cardíaca, enfermedad coronaria aguda y daño yatrógeno cardíaco; preferentemente en donde la insuficiencia cardíaca se selecciona del grupo que consiste en insuficiencia cardíaca congestiva, insuficiencia cardíaca sistólica, insuficiencia cardíaca diastólica, insuficiencia cardíaca con fracción de eyección reducida (HFrEF, por sus siglas en inglés), insuficiencia cardíaca con fracción de eyección conservada (HFpEF, por sus siglas en inglés), insuficiencia cardíaca aguda, insuficiencia cardíaca crónica de origen isquémico y no isquémico.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201962862897P | 2019-06-18 | 2019-06-18 | |
| PCT/US2020/037881 WO2020257161A1 (en) | 2019-06-18 | 2020-06-16 | Biaryl dialkyl phosphine oxide fpr2 agonists |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2946673T3 true ES2946673T3 (es) | 2023-07-24 |
Family
ID=71409568
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES20735802T Active ES2946673T3 (es) | 2019-06-18 | 2020-06-16 | Agonistas del FPR2 de óxido de biaril dialquil fosfina |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20220298186A1 (es) |
| EP (1) | EP3986903B1 (es) |
| KR (1) | KR20220024551A (es) |
| CN (1) | CN114008059B (es) |
| ES (1) | ES2946673T3 (es) |
| WO (1) | WO2020257161A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018227065A1 (en) * | 2017-06-09 | 2018-12-13 | Bristol-Myers Squibb Company | Piperidinone formyl peptide 2 receptor and formyl peptide 1 receptor agonists |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10302500A1 (de) * | 2003-01-23 | 2004-07-29 | Merck Patent Gmbh | Carbonsäureamidderivate |
-
2020
- 2020-06-16 CN CN202080044033.XA patent/CN114008059B/zh active Active
- 2020-06-16 WO PCT/US2020/037881 patent/WO2020257161A1/en unknown
- 2020-06-16 KR KR1020227001291A patent/KR20220024551A/ko unknown
- 2020-06-16 EP EP20735802.9A patent/EP3986903B1/en active Active
- 2020-06-16 ES ES20735802T patent/ES2946673T3/es active Active
- 2020-06-16 US US17/616,748 patent/US20220298186A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| KR20220024551A (ko) | 2022-03-03 |
| WO2020257161A1 (en) | 2020-12-24 |
| EP3986903A1 (en) | 2022-04-27 |
| JP2022537350A (ja) | 2022-08-25 |
| CN114008059B (zh) | 2024-03-12 |
| EP3986903B1 (en) | 2023-05-10 |
| US20220298186A1 (en) | 2022-09-22 |
| CN114008059A (zh) | 2022-02-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2904483T3 (es) | Agonistas de fenilpirrolidinona del receptor 2 de formil péptido | |
| ES2901471T3 (es) | Agonistas del receptor 2 de formilpéptido y del receptor 1 de formilpéptido de piperidinona | |
| KR102623473B1 (ko) | 피페리디논 포르밀 펩티드 2 수용체 및 포르밀 펩티드 1 수용체 효능제 | |
| ES2964299T3 (es) | Derivados de pirrolidinona como agonistas del receptor del péptido formilado 2 | |
| ES2946673T3 (es) | Agonistas del FPR2 de óxido de biaril dialquil fosfina | |
| US20220177487A1 (en) | Fused polycyclic pyridone compounds as influenza virus replication inhibitors | |
| JP7494224B2 (ja) | ビアリールジアルキルホスフィンオキシドfpr2アゴニスト | |
| KR20230038515A (ko) | 옥소피롤리딘 fpr2 효능제 | |
| EA043110B1 (ru) | Фенилпирролидиноновые агонисты формилпептидного рецептора 2 |