ES2934582T3 - Profármacos de CNP con unión de portador en el resto del anillo - Google Patents
Profármacos de CNP con unión de portador en el resto del anillo Download PDFInfo
- Publication number
- ES2934582T3 ES2934582T3 ES17700496T ES17700496T ES2934582T3 ES 2934582 T3 ES2934582 T3 ES 2934582T3 ES 17700496 T ES17700496 T ES 17700496T ES 17700496 T ES17700496 T ES 17700496T ES 2934582 T3 ES2934582 T3 ES 2934582T3
- Authority
- ES
- Spain
- Prior art keywords
- cnp
- moiety
- poly
- drug
- dysplasia
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/56—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule
- A61K47/59—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes
- A61K47/60—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic macromolecular compound, e.g. an oligomeric, polymeric or dendrimeric molecule obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyureas or polyurethanes the organic macromolecular compound being a polyoxyalkylene oligomer, polymer or dendrimer, e.g. PEG, PPG, PEO or polyglycerol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/06—Drugs for disorders of the endocrine system of the anterior pituitary hormones, e.g. TSH, ACTH, FSH, LH, PRL, GH
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Endocrinology (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Physical Education & Sports Medicine (AREA)
- Organic Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Rheumatology (AREA)
- Diabetes (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
La presente invención se refiere a profármacos de CNP en los que el vehículo se une de forma covalente y reversible al resto del anillo de un resto de CNP, a composiciones farmacéuticas que comprenden dichos profármacos de CNP, a sus usos y a métodos para tratar enfermedades que pueden tratarse con los profármacos de CNP. de la presente invención. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Profármacos de CNP con unión de portador en el resto del anillo
La presente invención se refiere a profármacos de péptido natriurético tipo C (“CNP”, por sus siglas en inglés) en los que el portador se une de forma covalente y reversible al resto de anillo de un resto de c Np , a composiciones farmacéuticas que comprenden tales profármacos de CNP, a sus usos en el tratamiento de enfermedades que pueden tratarse con los profármacos de CNP de la presente invención.
Las mutaciones de ganancia de función en FGFR3 conducen a acondroplasia (“ACH”, por sus siglas en inglés), hipocondroplasia (“HCH”, por sus siglas en inglés) y displasia tanatofórica (“TD”, por sus siglas en inglés). Estas afecciones, todas debidas al aumento de la señalización del receptor del factor de crecimiento de fibroblastos 3 (“FGFR3”, por sus siglas en inglés), se caracterizan por un enanismo rizomélico desproporcionado y difieren en gravedad, que varía de leve (HCH) a grave (ACH) y letal (TD). El FGFR3 es un regulador clave del crecimiento óseo endocondral y señales a través de varias vías intracelulares, incluyendo las del transductor de señales y activador de la transcripción (“STAT”, por sus siglas en inglés) y la proteína quinasa activada por mitógeno (“MAPK”, por sus siglas en inglés). La activación constitutiva de FGFR3 afecta la proliferación y la diferenciación terminal de los condrocitos de la placa de crecimiento y la síntesis de la matriz extracelular. La activación de FGFR3 se asocia con una mayor fosforilación de las vías STAT y MAPK. La vía de señalización de MAPK está regulada por el péptido natriurético tipo C (CNP). La unión de CNP a su receptor, el receptor B del péptido natriurético (“NPR-B”, por sus siglas en inglés), inhibe la señalización posterior de FGFR3 y, por lo tanto, desencadena el crecimiento endocondral y el sobrecrecimiento esquelético, como se observa en ratones y humanos que sobreexpresan CNP. La sobreproducción de CNP en el cartílago o la administración continua de CNP mediante infusión intravenosa (iv) normaliza el enanismo de los ratones acondroplásicos, lo que sugiere que la administración de CNP a niveles suprafisiológicos es una estrategia para tratar la ACH.
Sin embargo, dada la corta vida media de CNP-22 (2 minutos después de la administración intravenosa (iv)), el CNP como agente terapéutico es un desafío en una población pediátrica debido a que requeriría una infusión continua. Además, como el CNP se inactiva ampliamente en el tejido subcutáneo, se requiere una infusión iv.
Potter (FEBS Journal 278 (2011) 1808-1817) describe que la eliminación de CNP se produce por dos rutas de degradación: degradación mediada por receptor y degradación por proteasas extracelulares. El CNP se degrada por la acción de la endopeptidasa neutra 24,11 (“NEP”, por sus siglas en inglés) y se elimina de la circulación sistémica por el receptor de depuración del péptido natriurético, NPR-C, que se une y deposita el CNP en los lisosomas, en donde se degrada el CNP.
La reducción de la degradación por una o ambas de estas rutas de eliminación serviría para prolongar la vida media del CNP.
Debido al tamaño limitado de la cavidad de su sitio activo, la NEP reconoce preferiblemente sustratos menores de aproximadamente 3 kDa. En US 8.377.884 B2 se describen variantes de CNP que opcionalmente se conjugan permanentemente con polímero de PEG para aumentar la resistencia a la escisión de NEP. Sin embargo, se encontró que la adición de PEG, incluso tan pequeña como 0,6 kDa, a CNP de tipo silvestre reduce la actividad de CNP, y la adición de más de aproximadamente 2 o 3 kDa de PEG a CNP o variantes del mismo reduce la actividad de CNP en una manera dependiente del tamaño. Por lo tanto, la unión de moléculas de PEG mayores de 2 a 3 kDa para reducir la degradación de NEP se acompaña de una pérdida de actividad, lo que puede reducir el potencial terapéutico de tales moléculas.
Además de afectar negativamente a la actividad del péptido, la conjugación de PEG u otra macromolécula con CNP también puede impedir la distribución eficaz en la placa de crecimiento. Farnum et al. (Anat Rec A Discov Mol Cell Evol Biol. Enero 2006; 288(1): 91-103) demostró que la distribución de moléculas desde la vasculatura sistémica a la placa de crecimiento dependía del tamaño, y que las moléculas pequeñas (hasta 10 kDa) podían distribuirse a la placa de crecimiento, mientras que un tamaño molecular de 40 kDa o mayor impedía la entrada a la placa de crecimiento.
La solicitud internacional WO 2009/156481 A1 se refiere a conjugados de PEG reversibles de BNP cuyo término se definió como que incluye a todos los miembros de la familia de péptidos natriuréticos. Esta solicitud sólo se centra en los efectos cardiovasculares de esta clase de péptidos, que están mediados por el receptor A del péptido natriurético (NPR-A). En WO 2009/156481 A1 no se revelan las propiedades específicas de CNP con respecto a la regulación del crecimiento, la proliferación y la diferenciación de los condrocitos de la placa de crecimiento cartilaginoso, mediada por la activación del receptor B del péptido natriurético (NPR-B).
En The American Journal of Human Genetics 91, 1108-1114 se describió un enfoque diferente para crear una molécula de CNP resistente a NEP y permitir la administración subcutánea. BMN-111 es un péptido natriurético de tipo C (CNP) humano recombinante modificado al que se han añadido 17 aminoácidos para formar un análogo farmacológico de CNP de aminoácido 39. BMN-111 imita la actividad farmacológica del CNP en la placa de crecimiento y tiene una vida media prolongada como resultado de la resistencia a la endopeptidasa neutra (NEP) que permite la administración subcutánea (sc) una vez al día. Como BMN-111 es un péptido que no se produce de forma natural, el riesgo de inducir una respuesta inmunológica aumenta en comparación con el péptido nativo y, tal como lo describe Martz en “sFGFR for achondroplasia”
(SciBx, Biocentury, octubre de 2013), se ha observado una respuesta inmunológica a BMN-111 en estudios con animales, sin que la presencia de anticuerpos afecte a la actividad farmacológica del fármaco. Sin embargo, BMN-111 sólo tiene una vida media de 20 minutos, que cuando se dosifica diariamente se asocia con una exposición de corta duración a niveles eficaces del fármaco.
Para aumentar la exposición a niveles de fármaco eficaces, se puede aumentar la dosis del fármaco que tiene actividad de CNP. Dado que los péptidos natriuréticos son una familia de hormonas que pueden afectar el volumen sanguíneo y la presión arterial, un aumento de la dosis puede estar asociado con efectos adversos cardiovasculares. Los estudios de BMN-111 en animales y humanos han demostrado que a medida que aumenta la dosis, la presión arterial desciende y la frecuencia cardíaca aumenta. Las dosis de BMN-111 de hasta 15 |jg/kg se asociaron con hipotensión leve en voluntarios sanos. Por lo tanto, aumentar la dosis de un fármaco que tiene actividad de CNP para aumentar la exposición al fármaco puede estar asociado con efectos secundarios cardiovasculares inaceptables.
En WO2009/0676639A2 y WO2010/135541A2 se contempla el uso de CNP PEGilado. Sin embargo, los autores consideran que conservar la funcionalidad de dicho CNP PEGilado es una propiedad esencial de sus conjugados. Por lo tanto, los conjugados de CNP PEGilados descritos allí mantienen su capacidad para unirse a NPR-B y, por lo tanto, todavía plantean el problema de la hipotensión.
Wendt et al. (J Pharmacol Exp Ther, April 2005, 353: 132-149) enseña variantes de péptido natriurético tipo C resistentes a endopeptidasa neutra como un nuevo enfoque terapéutico para el tratamiento del enanismo relacionado con el receptor del factor de crecimiento de fribroblastos 3. También describe variantes de CNP PEGiladas en las que el péptido se une al polímero a través de un enlace amida estable.
WO2004/047871A2 describe compuestos natriuréticos modificados, tales como péptidos natriuréticos de tipo cerebral (“BNP”, por sus siglas en inglés), urodilatina, péptido natriurético cerebral canino (“cBNP”, por sus siglas en inglés), péptido natriurético auricular (“ANP”, por sus siglas en inglés), péptido natriurético de dendroaspis (“DNP”, por sus siglas en inglés) o péptido natriurético tipo C (CNP), y conjugados de los mismos y usos de estos en el tratamiento de una enfermedad cardiaca congestiva y afecciones relacionadas con esta afección.
WO2010/033217A1 enseña una nueva versión quimérica de péptido natriurético que se ha creado combinando porciones de péptido natriurético tipo C (CNP) con péptido natriurético de dendroaspis. Por consiguiente, el CD-NP resultante consiste en CNP humano de 22 aminoácidos y C-terminal de 15 aminoácidos de Dendroaspis NP. Dichos conjugados pueden ser útiles en el tratamiento de insuficiencia cardiaca congestiva aguda, crónica y en etapa temprana.
En resumen, existe la necesidad de un tratamiento de CNP más cómodo y seguro con un riesgo reducido de hipotensión.
Por lo tanto, es un objeto de la presente invención superar, al menos parcialmente, las deficiencias descritas anteriormente.
Este objeto se logra con un profármaco de CNP o una sal farmacéuticamente aceptable del mismo que comprende
- un resto de CNP -D que comprende un resto de anillo; y
- un resto portador -Z que está conjugado a través de un resto -L2- a un resto enlazador de profármaco reversible -L1-, cuyo resto enlazador de profármaco reversible -L1- está conjugado de forma covalente y reversible a una cadena lateral de un residuo de aminoácido de dicho resto del anillo de -D; y en donde -L2- es un enlace químico o un espaciador, en donde el resto -L1-L2- es de fórmula (Md-ii):

en donde
la línea punteada no marcada indica la unión a un nitrógeno de una cadena lateral de aminoácido del resto del anillo de -D formando un enlace amida; y la línea punteada marcada con el asterisco indica unión a -Z; en donde -D tiene la secuencia de SEQ ID NO:24 y -L1- está conjugado con la lisina en la posición 26;
en donde -Z es de fórmula (h):

en donde
la línea punteada indica unión a -L2-; y cada -Zc es un resto

en donde
cada c1 es un número entero que oscila independientemente entre 200 y 250.
Sorprendentemente, se descubrió que la unión de un portador -Z a través de un resto enlazador de profármaco reversible al anillo de un resto de CNP reduce significativamente la unión del profármaco al receptor NPR-B en comparación con, por ejemplo, la unión N-terminal o la unión fuera del resto de anillo de CNP. Esto permite la administración de un compuesto de CNP con actividad residual baja o casi nula, es decir, con unión reducida de NPR-B, lo que a su vez reduce el riesgo de hipotensión. Como la unión del polímero -Z al resto de CNP es reversible, la unión de n PR-B se restablece tras la liberación del profármaco de CNP y el CNP liberado puede distribuirse de manera efectiva a la placa de crecimiento.
Esta combinación de inactividad inicial frente a NPR-B y posterior liberación controlada del conjugado de profármaco que convierte la fracción CNP en una forma completamente activa del fármaco con respecto a la unión de NPR-B proporciona varias ventajas, tal como la reducción del riesgo de hipotensión, la opción de administrar dosis más altas y una disminución en la frecuencia de administración, lo que mejora el cumplimiento y la conveniencia para el paciente.
Además, se encontró sorprendentemente que una liberación continua de CNP, tal como desde un sistema de liberación controlada, tales como los profármacos de la presente invención, es más eficaz que una inyección en bolo una vez al día.
Dentro de la presente invención, los términos se utilizan con el siguiente significado.
De la manera que se usa en la presente, el término “CNP” se refiere al polipéptido CNP de SEQ ID NO:24.
El CNP-22 natural (SEQ ID NO:1) tiene la siguiente secuencia: GLSKGCFGLKLDRIGSMSGLGC, en donde las cisteínas en la posición 6 y 22 están conectadas a través de un puente disulfuro, como se ilustra en la Figura 1.
SEQ ID NO:24 tiene la siguiente secuencia: LQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC, en donde las cisteínas en la posición 22 y 38 están conectadas a través de un puente disulfuro.
Para el término “CNP” también se describen las siguientes secuencias de péptidos, en donde sólo la secuencia de SEQ ID NO:24 está de acuerdo con la invención:
SEQ ID NO:2 (CNP-53): DLRVDTKSRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC; SEQ ID NO:3 (G-CNP-53): GDLRVDTKSRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:4 (M-CNP-53): MDLRVDTKSRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:5 (P-CNP-53): PDLRVDTKSRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:6 (CNP-53 M48N): DLRVDTKSRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSNSGLGC;
SEQ ID NO:7 (CNP-53 A15-31): DLRVDTKSRAAWARGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:8 (CNP-52): LRVDTKSRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:9 (CNP-51): RVDTKSRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:10 (CNP-50): VDTKSRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:11 (CNP-49): DTKSRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:12 (CNP-48): TKSRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:13 (CNP-47): KSRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:14 (CNP-46): SRAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:15 (CNP-45): RAAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:16 (CNP-44): AAWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:17 (CNP-44 A14-22): AAWARLLQEHPNAGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:18 (CNP-44 A15-22): AAWARLLQEHPNARGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:19 (CNP-43): AWARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:20 (CNP-42): WARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:21 (CNP-41): ARLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:22 (CNP-40): RLLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:23 (CNP-39): LLQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:24 (CNP-38): LQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:25 (CNP-37): QEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:26 (CNP-37 Q1 pQ, en donde pQ = piroglutamato): pQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC; SEQ ID NO:27 (G-CNP-37): GQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:28 (P-CNP-37): PQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:29 (M-CNP-37): MQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:30 (PG-CNP-37): PGQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:31 (MG-CNP-37): MGQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:32 (CNP-37 M32N): QEHPNARKYKGANKKGLSKGCFGLKLDRIGSNSGLGC;
SEQ ID NO:33 (G-CNP-37 M32N): GQEHPNARKYKGANKKGLSKGCFGLKLDRIGSNSGLGC;
SEQ ID NO:34 (G-CNP-37 K14Q): GQEHPNARKYKGANQKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:35 (G-CNP-37 K14P): GQEHPNARKYKGANPKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:36 (G-CNP-37 K14Q, A15): GQEHPNARKYKGANQGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:37 (G-CNP-37 K14Q, K15Q): GQEHPNARKYKGANQQGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:38 (CNP-36): EHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:39 (CNP-35): HPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:40 (CNP-34): PNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:41 (CNP-33): NARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:42 (CNP-32): ARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:43 (CNP-31): RKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:44 (CNP-30): KYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:45 (CNP-29): YKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:46 (CNP-28): KGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:47 (GHKSEVAHRF-CNP-28): GHKSEVAHRFKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:48 (CNP-27): GANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:49 (CNP-27 K4Q, K5Q): GANQQGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:50 (CNP-27 K4R,K5R): GANRRGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:51 (CNP-27 K4P,K5R): GANPRGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:52 (CNP-27 K4S,K5S): GANSSGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:53 (CNP-27 K4P,K5R): GANGANPRGLSRGCFGLKLDRIGSMSGLGC;
SEQ ID NO:54 (CNP-27 K4R, K5R, K9R): GANRRGLSRGCFGLKLDRIGSMSGLGC;
SEQ ID NO:55 (CNP-27 K4R, K5R, K9R, M22N): GANRRGLSRGCFGLKLDRIGSNSGLGC;
SEQ ID NO:56 (P-CNP-27 K4R, K5R, K9R): PGANRRGLSRGCFGLKLDRIGSMSGLGC;
SEQ ID NO:57 (M-CNP-27 K4R, K5R, K9R): MGANRRGLSRGCFGLKLDRIGSMSGLGC;
SEQ ID NO:58 (fragmento de HSA-CNP-27): GHKSEVAHRFKGANKKGLSKGCFGLKLDRIGSMSGLG;
SEQ ID NO:59 (fragmento de HSA-CNP-27 M22N): GHKSEVAHRFKGANKKGLSKGCFGLKLDRIGSNSGLGC;
SEQ ID NO:60 (fragmento de M-HSA-CNP-27): MGHKSEVAHRFKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:61 (fragmento de P-HSA-CNP-27): PGHKSEVAHRFKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:62 (CNP-26): ANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:63 (CNP-25): NKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:64 (CNP-24): KKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:65 (CNP-23): KGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:66 (R-CNP-22): RGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:67 (ER-CNP-22): ERGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:68 (R-CNP-22 K4R): RGLSRGCFGLKLDRIGSMSGLGC;
SEQ ID NO:69 (ER-CNP-224KR): ERGLSRGCFGLKLDRIGSMSGLGC;
SEQ ID NO:70 (RR-CNP-22): RRGLSRGCFGLKLDRIGSMSGLGC;
SEQ ID NO:71 (fragmento de HRGP-CNP-22): GHHSHEQHPHGANQQGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:72 (fragmento de HRGP-CNP-22): GAHHPHEHDTHGANQQGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:73 (fragmento de HRGP-CNP-22): GHHSHEQHPHGANPRGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:74 (fragmento de IgG1(Fc)-CNP-22): GQPREPQVYTLPPSGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:75 (fragmento de HSA-CNP-22): GQHKDDNPNLPRGANPRGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:76 (fragmento de HSA-CNP-22): GERAFKAWAVARLSQGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:77 (fragmento de inhibidor de osteocrina NPR C-CNP22): FGIPMDRIGRNPRGLSKGCFGLKLDRIGSMSGL-GC;
SEQ ID NO:78 (fragmento de dominio de unión a heparina FGF2-CNP22): GKRTGQYKLGSKTGPGPKGLSKGCFGLKLD-RIGSMSGLGC;
SEQ ID NO:79 (fragmento de IgG1(Fc)-CNP-22 K4R): GQPREPQVYTGANQQGLSRGCFGLKLDRIGSMSGLGC;
SEQ ID NO:80 (fragmento de HSA-CNP-22 K4R): GVPQVSTSTGANQQGLSRGCFGLKLDRIGSMSGLGC;
SEQ ID NO:81 (fragmento de fibronectina-CNP-22 K4R): GQPSSSSQSTGANQQGLSRGCFGLKLDRIGSMSGLGC; SEQ ID NO:82 (fragmento de fibronectina-CNP-22 K4R): GQTHSSGTQSGANQQGLSRGCFGLKLDRIGSMSGLGC; SEQ ID NO:83 (fragmento de fibronectina-CNP-22 K4R): GSTGQWHSESGANQQGLSRGCFGLKLDRIGSMSGLGC; SEQ ID NO:84 (fragmento de dedo de zinc-CNP-22 K4R): GSSSSSSSSSGANQQGLSRGCFGLKLDRIGSMSGLGC; SEQ ID NO:85 (CNP-21): LSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:86 (CNP-20): SKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:87 (CNP-19): KGCFGLKLDRIGSMSGLGC;
SEQ ID NO:88 (CNP-18): GCFGLKLDRIGSMSGLGC;
SEQ ID NO:89 (CNP-17): CFGLKLDRIGSMSGLGC;
SEQ ID NO:90 (fragmento de BNP-CNP-17-fragmento de BNP): SPKMVQGSGCFGLKLDRIGSMSGLGCKVLRRH; SEQ ID NO:91 (CNP-38 L1G): GQEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:92 (Ac-CNP-37; en donde Ac= acetilo): Ac-QEHPNARKYKGANKKGLSKGCFGLKLDRIGSMSGLGC;
SEQ ID NO:93: QEHPNARX1YX2GANX3X4GLSX5GCFGLX6LDRIGSMSGLGC,
en donde X1, X2, X3, X4 , X5 y X6 se seleccionan independientemente entre sí del grupo que consiste en K, R, P, S y Q, siempre que al menos uno de X1, X2 , X3 , X4 , X5 y X6 se seleccione del grupo que consiste en R, P, S y Q; preferiblemente X1, X2, X3 , X4 , X5 y X6 se seleccionan del grupo que consiste en K y R, siempre que al menos uno de X1, X2 , X3 , X4 , X5 y X6 sea R;
SEQ ID NO:94: QEHPNARKYKGANX1X2GLSX3GCFGLX4LDRIGSMSGLGC,
en donde X1, X2 , X3 y X4 se seleccionan independientemente entre sí del grupo que consiste en K, R, P, S y Q, siempre que al menos uno de X1, X2 , X3 y X4 se seleccione del grupo que consiste en R, P, S y Q; preferiblemente X1, X2 , X3 y X4 se seleccionan de K y R, siempre que al menos uno de X 1, X2, X3 y X4 sea R;
SEQ ID NO:95: QEHPNARKYKGANX1X2GLSKGCFGLKLDRIGSMSGLGC,
en donde X1X2 se seleccionan del grupo que consiste en KR, RK, KP, PK, SS, RS, SR, QK, QR, KQ, RQ, RR y QQ.
De la manera que se usa en la presente, el término “variante del polipéptido CNP” se refiere a un polipéptido con la secuencia de s Eq ID NO:24.
De la manera que se usa en la presente, el término “resto de anillo” se refiere al tramo de restos de aminoácidos consecutivos del fármaco o resto de CNP que se localiza entre dos restos de cisteína que forman un puente disulfuro intramolecular. Estas dos cisteínas corresponden a las cisteínas en la posición 22 y la posición 38 en la secuencia de CNP-38 (SEQ ID NO:24). Por consiguiente, los aminoácidos 23 a 37 están ubicados en dicho resto de anillo, si el fármaco o resto de CNP tiene la secuencia de CNP-38.
Independientemente de la longitud del resto de CNP, la secuencia del resto de anillo de CNP de tipo silvestre es FGLKLDRIGSMSGLG (SEQ ID NO:96).
De la manera que se usa en la presente, el término “composición líquida” se refiere a una mezcla que comprende un profármaco de CNP soluble en agua y uno o más disolventes, tal como agua.
De la manera que se usa en la presente, el término “composición seca” significa que una composición farmacéutica se proporciona en forma seca. Los métodos adecuados para el secado son el secado por pulverización y la liofilización, es decir, secado por congelación. Tal composición seca del profármaco de la presente invención tiene un contenido de agua residual de un máximo del 10%, preferiblemente menos del 5% y más preferiblemente menos del 2%, determinado de acuerdo con Karl Fischer. Preferiblemente, la composición farmacéutica de la presente invención se seca mediante liofilización.
De la manera que se usa en la presente, el término “fármaco” se refiere a una sustancia utilizada en el tratamiento, cura, prevención o diagnóstico de una enfermedad, o utilizada para mejorar el bienestar físico o mental. Si un fármaco D-H se conjuga con otro resto, el resto -D del producto resultante que se originó a partir del fármaco se denomina “resto biológicamente activo”.
De la manera que se usa en la presente, el término “profármaco” se refiere a un resto biológicamente activo conectado de forma reversible y covalente a un grupo protector especializado a través de un resto enlazador de profármaco reversible que es un resto enlazador que comprende un enlace reversible con el resto biológicamente activo y en donde el grupo protector especializado altera o elimina propiedades indeseables en la molécula original. Esto también incluye la mejora de las propiedades deseables del fármaco y la supresión de las propiedades indeseables. El grupo protector no tóxico especializado se denomina “portador”. Un profármaco libera el resto biológicamente activo -D unido de forma reversible y covalente en forma de su correspondiente fármaco D-H. En otras palabras, un profármaco es un conjugado que comprende un resto biológicamente activo que se conjuga de forma covalente y reversible con un resto portador a través de un resto enlazador de profármaco reversible, cuya conjugación covalente y reversible del portador con el resto enlazador de profármaco reversible es directa o a través de un espaciador, tal como -L2-. Dicho conjugado libera el resto biológicamente activo anteriormente conjugado en forma de fármaco libre.
Un “enlace biodegradable” o un “enlace reversible” es un enlace que es hidrolíticamente degradable, es decir, escindible, en ausencia de enzimas en condiciones fisiológicas (solución amortiguadora acuosa a pH 7,4, 37 °C) con una vida media que varía de una hora a seis meses, preferiblemente de una hora a cuatro meses, aún más preferiblemente de una hora a tres meses, aún más preferiblemente de una hora a dos meses, aún más preferiblemente de una hora a un mes. Por consiguiente, un “enlace estable” es un enlace que tiene una vida media en condiciones fisiológicas (solución amortiguadora acuosa a pH 7,4, 37 °C) de más de seis meses.
Por lo tanto, un “resto enlazador de profármaco reversible” es un resto que se conjuga covalentemente con un resto biológicamente activo, tal como CNP, a través de un enlace reversible y que también se conjuga covalentemente con un resto portador, tal como -Z, en donde la conjugación covalente con dicho resto portador es directa o a través de un resto espaciador, tal como -L2-. El enlace entre -Z y -L2- es un enlace estable.
De la manera que se usa en la presente, el término “enlazador de profármacos sin trazas” significa un enlazador de profármacos reversible que al escindirse libera el fármaco en su forma libre. De la manera que se usa en la presente, el término “forma libre” de un fármaco significa el fármaco en su forma farmacológicamente activa y no modificada.
De la manera que se usa en la presente, el término “composición farmacéutica” se refiere a una composición que contiene uno o más ingredientes activos, por ejemplo un fármaco o un profármaco, aquí específicamente los profármacos de CNP de la presente invención, y opcionalmente uno o más excipientes, así como cualquier producto que resulte, directa o indirectamente, de la combinación, complejación o agregación de dos o más de los ingredientes de la composición, o de la disociación de uno o más de los ingredientes, o de otros tipos de reacciones o interacciones de uno o más de los ingredientes. Por consiguiente, las composiciones farmacéuticas de la presente invención abarcan cualquier composición preparada mediante el mezclado de uno o más profármacos de CNP de la presente invención y, opcionalmente, un excipiente farmacéuticamente aceptable.
De la manera que se usa en la presente, el término “excipiente” se refiere a un diluyente, adyuvante o vehículo con el que se administra el agente terapéutico, tal como un fármaco o profármaco. Dicho excipiente farmacéutico puede ser líquido estéril, tales como el agua y los aceites, incluyendo los de origen petrolero, animal, vegetal o sintético, tales como el aceite de cacahuate, aceite de soya, aceite mineral o aceite de sésamo. El agua es un excipiente preferido cuando la composición farmacéutica se administra por vía oral. La solución salina y la dextrosa acuosa son excipientes preferidos cuando la composición farmacéutica se administra por vía intravenosa. Las soluciones salinas y las soluciones acuosas de dextrosa y glicerol se emplean preferiblemente como excipientes líquidos para soluciones inyectables. Los excipientes farmacéuticos adecuados incluyen almidón, glucosa, lactosa, sacarosa, manitol, trehalosa, gelatina, malta, arroz, harina, tiza, gel de sílice, estearato de sodio, monoestearato de glicerol, talco, cloruro de sodio, leche desnatada en polvo, glicerol, propilenglicol, agua y etanol. La composición farmacéutica, si se desea, también puede contener cantidades menores de agentes humectantes o emulsionantes, agentes amortiguadores de pH, tales como, por ejemplo, acetato, succinato, tris, carbonato, fosfato, HEPES (ácido 4-(2-hidroxietil)-1-piperazinaetanosulfónico), MES (ácido 2-(N-morfolino)etanosulfónico), o puede contener detergentes, tales como Tween, poloxámeros, poloxaminas, CHAPS, Igepal, o aminoácidos, tales como, por ejemplo, glicina, lisina o histidina. Estas composiciones farmacéuticas pueden adoptar la forma de soluciones, suspensiones, emulsiones, comprimidos, píldoras, cápsulas, polvos o formulaciones de liberación sostenida. La composición farmacéutica puede formularse como un supositorio, con aglutinantes y excipientes tradicionales, tales como los triglicéridos. La formulación oral puede incluir excipientes estándar, tales como grados farmacéuticos de manitol, lactosa, almidón, estearato de magnesio, sacarina de sodio, celulosa y carbonato de magnesio. Dichas composiciones contendrán una cantidad terapéuticamente efectiva del fármaco o del resto biológicamente activo, junto con una cantidad adecuada de excipiente, a fin de proporcionar la forma de administración adecuada al paciente. La formulación debe adaptarse al modo de administración.
De la manera que se usa en la presente, el término “liposoma” se refiere a una vesícula, preferiblemente una vesícula esférica, que tiene al menos una bicapa lipídica. Preferiblemente, los liposomas comprenden fosfolípidos, incluso más preferiblemente fosfatidilcolina. El término “liposoma” se refiere a varias estructuras y tamaños, tales como, por ejemplo, a las vesículas liposomales multilamelares (“MLV”, por sus siglas en inglés) que tienen más de una bicapa lipídica concéntrica con un diámetro promedio de 100 a 1000 nm, vesículas liposómicas unilamelares pequeñas (“SUV”, por sus siglas en inglés) que tienen una bicapa lipídica y un diámetro promedio de 25 a 100 nm, vesículas liposómicas unilamelares grandes (“LUV”, por sus siglas en inglés) que tienen una bicapa lipídica y un diámetro promedio de
aproximadamente 1000 |jm, y vesículas unilamelares gigantes (“GUV”, por sus siglas en inglés) que tienen una bicapa lipídica y un diámetro promedio de 1 a 100 jm . El término “liposoma” también incluye las vesículas elásticas, tales como los transferosomas y los etosomas, por ejemplo.
De la manera que se usa en la presente, el término “acuasoma” se refiere a las nanopartículas esféricas con un diámetro de 60 a 300 nm que comprenden al menos tres capas de estructura autoensamblada, específicamente, un núcleo nanocristalino en fase sólida recubierto por una película oligomérica a la que se adsorben las moléculas del fármaco con o sin modificación del fármaco.
De la manera que se usa en la presente, el término “etosoma” se refiere a las vesículas lipídicas que comprenden fosfolípidos y etanol y/o isopropanol en una concentración relativamente alta y agua, y que tienen un tamaño que oscila entre decenas de nanómetros y micrómetros.
De la manera que se usa en la presente, el término “LeciPlex” se refiere al sistema vesicular a base de fosfolípidos con carga positiva que comprende PC de soya, un agente catiónico y un disolvente biocompatible como PEG 300, PEG 400, éter monoetílico de dietilenglicol, éter de polietilenglicol de alcohol tetrahidrofurfurílico o 2-pirrolidona o N-metil-2-pirrolidona.
De la manera que se usa en la presente, el término “niosoma” se refiere a las vesículas unilamelares o multilamelares que comprenden tensioactivos no iónicos.
De la manera que se usa en la presente, el término “farmacosoma” se refiere a los agregados vesiculares, micelares o hexagonales ultrafinos de lípidos unidos covalentemente a restos biológicamente activos.
De la manera que se usa en la presente, el término “proniosoma” se refiere a formulaciones secas de portadores recubiertos de tensioactivos que, al rehidratarse y agitarse ligeramente, proporcionan niosomas.
De la manera que se usa en la presente, el término “polimerosoma” se refiere a una vesícula esférica artificial que comprende una membrana formada por copolímeros en bloque sintéticos anfifílicos y que, opcionalmente, puede comprender una solución acuosa en su núcleo. Un polimerosoma tiene un diámetro que varía de 50 nm a 5 jm y más. El término también incluye a los sintosomas, que son polimerosomas modificados para incluir canales que permiten el paso de ciertos químicos a través de la membrana hacia el interior o el exterior de la vesícula.
El término “esfingosoma” se refiere a una vesícula concéntrica bicapa en la que un volumen acuoso está totalmente encerrado por una bicapa lipídica membranosa compuesta principalmente por esfingolípidos naturales o sintéticos.
De la manera que se usa en la presente, el término “transferosoma” se refiere a vesículas lipídicas ultraflexibles que comprenden un núcleo acuoso y que están formadas por una mezcla de lípidos polares comunes y lípidos adecuados activados por los bordes que facilitan la formación de bicapas muy curvadas que hacen que el transferosoma sea altamente deformable.
De la manera que se usa en la presente, el término “ufasoma” se refiere a una vesícula que comprende ácidos grasos insaturados.
De la manera que se usa en la presente, el término “reactivo” significa un compuesto químico que comprende al menos un grupo funcional para reaccionar con el grupo funcional de otro compuesto químico o fármaco. Se entiende que un fármaco que comprende un grupo funcional (tal como una amina primaria o secundaria, o un grupo funcional hidroxilo) es también un reactivo.
De la manera que se usa en la presente, el término “resto” significa una parte de una molécula a la que le faltan uno o más átomos en comparación con el reactivo correspondiente. Si, por ejemplo, un reactivo de la fórmula “H-X-H” reacciona con otro reactivo y pasa a formar parte del producto de reacción, el correspondiente resto del producto de reacción tiene la estructura “H-X-” o “-X-”, mientras que cada “-” indica la unión a otro resto. De este modo, un resto biológicamente activo se libera de un profármaco como un fármaco.
Se entiende que si se proporciona la secuencia o estructura química de un grupo de átomos que está unido a dos restos o que interrumpe un resto, dicha secuencia o estructura química puede estar unida a los dos restos en cualquier orientación, a menos que se indique explícitamente lo contrario. Por ejemplo, un resto “-C(O)N(R1)-” puede unirse a dos fracciones o interrumpir un resto como "-C(O)N(R1)-" o como “-N(R1)C(O)-”. De manera similar, un resto

puede unirse a dos restos o puede interrumpir un resto, ya sea como

De la manera que se usa en la presente, el término “grupo funcional” significa un grupo de átomos que puede reaccionar con otros grupos de átomos. Los grupos funcionales incluyen, entre otros, los siguientes grupos: ácido carboxílico (-(C=O)OH), amina primaria o secundaria (-NH2 , -NH-), maleimida, tiol (-SH), ácido sulfónico (-(O=S=O)OH), carbonato, carbamato (-O(C=O)N<), hidroxilo (-OH), aldehído (-(C=O)H), cetona (-(C=O)-), hidrazina (>N-N<), isocianato, isotiocianato, ácido fosfórico (-O(P=O)OHOH), ácido fosfónico (-O(P=O)OHH), haloacetilo, haluro de alquilo, acriloilo, fluoruro de arilo, hidroxilamina, disulfuro, sulfonamidas, ácido sulfúrico, vinilsulfona, vinilcetona, diazoalcano, oxirano y aziridina.
En caso de que los profármacos de la presente invención comprendan uno o más grupos ácidos o básicos, la invención también comprende sus correspondientes sales farmacéutica o toxicológicamente aceptables, en particular sus sales farmacéuticamente utilizables. Por lo tanto, los profármacos de la presente invención que comprenden grupos ácidos pueden utilizarse de acuerdo con la invención, por ejemplo, como sales de metales alcalinos, sales de metales alcalinotérreos o como sales de amonio. Ejemplos más precisos de tales sales incluyen sales de sodio, sales de potasio, sales de calcio, sales de magnesio o sales con amoníaco o aminas orgánicas, tales como, por ejemplo, etilamina, etanolamina, trietanolamina o aminoácidos. Los profármacos de la presente invención que comprenden uno o más grupos básicos, es decir, grupos que pueden protonarse, pueden estar presentes y pueden utilizarse de acuerdo con la invención en forma de sus sales de adición con ácidos inorgánicos u orgánicos. Los ejemplos de ácidos adecuados incluyen cloruro de hidrógeno, bromuro de hidrógeno, ácido fosfórico, ácido sulfúrico, ácido nítrico, ácido metanosulfónico, ácido ptoluenosulfónico, ácidos naftalendisulfónicos, ácido oxálico, ácido acético, ácido tartárico, ácido láctico, ácido salicílico, ácido benzoico, ácido fórmico, ácido propiónico, ácido piválico, ácido dietilacético, ácido malónico, ácido succínico, ácido pimélico, ácido fumárico, ácido maleico, ácido málico, ácido sulfamínico, ácido fenilpropiónico, ácido glucónico, ácido ascórbico, ácido isonicotínico, ácido cítrico, ácido adípico y otros ácidos conocidos por el experto en la técnica. Los expertos en la técnica conocen métodos adicionales para convertir el grupo básico en un catión, como la alquilación de un grupo amina que da como resultado un grupo amonio con carga positiva y un contraión apropiado de la sal. Si los profármacos de la presente invención comprenden simultáneamente grupos ácidos y básicos, la invención también incluye, además de las formas de sal mencionadas, sales internas o betainas (zwitteriones). Las sales respectivas pueden obtenerse por los métodos habituales que conoce el experto en la técnica, como, por ejemplo, poniendo estos profármacos en contacto con un ácido o una base orgánica o inorgánica en un disolvente o dispersante, o mediante intercambio aniónico o catiónico con otras sales. La presente invención también incluye todas las sales de los profármacos de la presente invención que, debido a su baja compatibilidad fisiológica, no son directamente adecuadas para su uso en productos farmacéuticos, pero que pueden utilizarse, por ejemplo, como intermedios para reacciones químicas o para la preparación de sales farmacéuticamente aceptables.
El término “farmacéuticamente aceptable” significa una sustancia que no causa daño cuando se administra a un paciente y, preferiblemente, significa que está aprobada por una agencia reguladora, tal como la EMA (Europa) y/o la FDA (EE. UU.) y/o cualquier otra agencia reguladora nacional para su uso en animales, preferiblemente para su uso en humanos.
De la manera que se usa en la presente, el término “aproximadamente” en combinación con un valor numérico se utiliza para indicar un rango que va desde e incluye el valor numérico más y menos no más del 10% de dicho valor numérico, más preferiblemente no más del 8% de dicho valor numérico, incluso más preferiblemente no más del 5% de dicho valor
numérico, y más preferiblemente no más del 2% de dicho valor numérico. Por ejemplo, la frase “aproximadamente 200” se utiliza para significar un rango que va desde e incluye 200 /- 10%, es decir, que va desde e incluye 180 a 220; preferiblemente 200 /-8%, es decir, que va desde e incluye 184 a 216; aún más preferiblemente que va desde e incluye 200 /-5%, es decir, que va desde e incluye 190 a 210; y más preferiblemente 200 /-2%, es decir, que va desde e incluye 196 a 204. Se entiende que un porcentaje dado como “aproximadamente 20%” no significa “20% /- 10%”, es decir, que va desde e incluye 10 a 30%, sino que “aproximadamente 20%” significa que va desde e incluye 18 y 22%, es decir, más y menos el 10% del valor numérico que es 20.
De la manera que se usa en la presente, el término “polímero” significa una molécula que comprende unidades estructurales de repetición, es decir, los monómeros, conectados por enlaces químicos de forma lineal, circular, ramificada, reticulada o dendrimérica, o una combinación de las mismas, que puede ser de origen sintético o biológico, o una combinación de ambos. Se entiende que un polímero también puede comprender uno o más grupos químicos y/o restos, tales como, por ejemplo, uno o más grupos funcionales.
Preferiblemente, un polímero soluble tiene un peso molecular de al menos 0,5 kDa, por ejemplo, un peso molecular de al menos 1 kDa, un peso molecular de al menos 2 kDa, un peso molecular de al menos 3 kDa, o un peso molecular de al menos 5 kDa. Si el polímero es soluble, es preferible que tenga un peso molecular de como máximo 1000 kDa, como máximo 750 kDa, como máximo 500 kDa, como máximo 300 kDa, como máximo 200 kDa, como máximo 100 kDa. Se entiende que también una proteína es un polímero en el que los aminoácidos son las unidades estructurales que se repiten, aunque las cadenas laterales de cada aminoácido puedan ser diferentes.
De la manera que se usa en la presente, el término “polimérico” significa un reactivo o un resto que comprende uno o más polímeros o restos de polímeros. Un reactivo o resto polimérico puede comprender opcionalmente uno o más restos, que se seleccionan preferiblemente del grupo que consiste en:
• alquilo C1-50, alquenilo C2-50, alquinilo C2-50, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, heterobiciclilo de 8 a 11 miembros, fenilo, naftilo, indenilo, indanilo y tetralinilo; y
• enlaces seleccionados del grupo que comprende

las líneas punteadas indican unión al residuo del resto o reactivo, y
- R y -Ra se seleccionan independientemente entre sí del grupo que consiste en -H, metilo, etilo, propilo, butilo, pentilo y hexilo.
El experto en la técnica entiende que los productos de polimerización obtenidos de una reacción de polimerización no tienen todos el mismo peso molecular, sino que presentan una distribución de peso molecular. Por consiguiente, los rangos de peso molecular, pesos moleculares, rangos de números de monómeros en un polímero y números de monómeros en un polímero, tal como se utilizan en la presente, se refieren al peso molecular promedio en número y al número promedio de monómeros, es decir, a la media aritmética del peso molecular del polímero o del resto polimérico y a la media aritmética del número de monómeros del polímero o del resto polimérico.
Por consiguiente, en un resto polimérico que comprende “x” unidades de monómero, cualquier número entero dado para “x” corresponde al número medio aritmético de monómeros. Cualquier rango de números enteros dado para “x” proporciona el rango de enteros en el que se encuentra la media aritmética del número de monómeros. Un número entero para “x” dado como “aproximadamente x” significa que la media aritmética del número de monómeros se encuentra en un rango de números enteros de x /- 10%, preferiblemente x /- 8%, más preferiblemente x /- 5% y más preferiblemente x /- 2%.
De la manera que se usa en la presente, el término “peso molecular promedio en número” significa la media aritmética ordinaria de los pesos moleculares de los polímeros individuales.
De la manera que se usa en la presente, el término “soluble en agua” con referencia a un portador significa que cuando dicho portador forma parte del profármaco CNP de la presente invención, al menos 1 g del profármaco CNP que comprende dicho portador soluble en agua puede disolverse en un litro de agua a 20 °C para formar una solución homogénea. Por consiguiente, el término “insoluble en agua” con referencia a un portador significa que cuando dicho portador forma parte del profármaco CNP de la presente invención menos de 1 g del profármaco CNP que comprende dicho portador insoluble en agua puede disolverse en un litro de agua a 20 °C para formar una solución homogénea.
De la manera que se usa en la presente, el término “a base de PEG” en relación con un resto o reactivo significa que dicho resto o reactivo comprende PEG. Preferiblemente, un resto o reactivo a base de PEG comprende al menos un 10% (p/p) de PEG, tal como al menos un 20% (p/p) de PEG, tal como al menos un 30% (p/p) de PEG, tal como al menos un 40% (p/p) de PEG, tal como al menos un 50% (p/p), tal como al menos un 60 (p/p) de PEG, tal como al menos un 70% (p/p) de PEG, tal como al menos un 80% (p/p) de PEG, tal como al menos un 90% (p/p) de PEG, tal como al menos un 95% (p/p) de PEG. El porcentaje en peso restante del resto o reactivo a base de PEG son otros restos seleccionados preferiblemente de los siguientes restos y enlaces:
• alquilo C1-50, alquenilo C2-50, alquinilo C2-50, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, heterobiciclilo de 8 a 11 miembros, fenilo, naftilo, indenilo, indanilo y tetralinilo; y
• enlaces seleccionados del grupo que comprende

en donde
las líneas punteadas indican unión al residuo del resto o reactivo, y
-R y -Ra se seleccionan de forma independiente entre el grupo formado por -H, metilo, etilo, propilo, butilo,
pentilo y hexilo.
De la manera que se usa en la presente, el término “a base de PEG que comprende al menos un X% de PEG” en relación con un resto o reactivo significa que dicho resto o reactivo comprende al menos un X% (p/p) de unidades de etilenglicol (-CH2CH2O-), en donde las unidades de etilenglicol pueden estar dispuestas en bloques, alternadas o pueden estar distribuidas aleatoriamente dentro del resto o reactivo y preferiblemente todas las unidades de etilenglicol de dicho resto o reactivo están presentes en un bloque; el porcentaje en peso restante del resto o reactivo a base de PEG son otros restos seleccionados preferiblemente entre los siguientes restos y enlaces:
• alquilo C1-50, alquenilo C2-50, alquinilo C2-50, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, heterobiciclilo de 8 a 11 miembros, fenilo, naftilo, indenilo, indanilo y tetralinilo; y
• enlaces seleccionados del grupo que comprende


las líneas punteadas indican unión al residuo del resto o reactivo, y
- R y -Ra se seleccionan independientemente entre sí del grupo que consiste en -H, metilo, etilo, propilo, butilo, pentilo y hexilo.
El término “a base de ácido hialurónico que comprende al menos un X% de ácido hialurónico” se utiliza en consecuencia.
De la manera que se usa en la presente, el término “sustituido” significa que uno o más átomos de -H de una molécula o resto se sustituyen por un átomo diferente o un grupo de átomos, que se denominan “sustituyentes”.
Preferiblemente, uno o más sustituyentes opcionales adicionales se seleccionan independientemente entre sí del grupo que consiste en halógeno, -CN, -COORx1, -ORx1, -C(O)Rx1, -C(O)N(Rx1Rx1a), -S(O)2N(Rx1Rx1a), -S(O)N(Rx1Rx1a), -S(O)2Rx1, -S(O)Rx1, -N(Rx1)S(O)2N(Rx1aRx1b), -SRx1, -N(Rx1Rx1a), -NO2 , -OC(O)Rx1, -N(Rx1)C(O)R x1a, -N(Rx1)S(O)2R x1a, -N(Rx1)S(O)Rx1a, -N(Rx1)C(O)ORx1a, -N(Rx1)C(O)N(Rx1aRx1b), -OC(O)N(Rx1Rx1a), -T0, alquilo C1-50, alquenilo C2-50 y alquinilo C2-50; en donde -T0, alquilo C1-50, alquenilo C2-50 y alquinilo C2-50 están opcionalmente sustituidos con uno o más -Rx2, que son iguales o diferentes, y en donde alquilo C1-50, alquenilo C2-50 y alquinilo C2-50 están opcionalmente interrumpidos por uno o más grupos seleccionados del grupo que consiste en -T0-, -C(O)O-, -O-, -C(O)-, -C(O)N(Rx3)-, -S(O)2N(Rx3)-, -S(O)N(Rx3)-, -S(O)2-, -S(O)-, -N(Rx3)S(O)2N(Rx3a)-, -S-, -N(Rx3)-, -OC(ORx3)(Rx3a)-, -N(Rx3)C(O)N(Rx3a)-, y -OC(O)N(Rx3)-;
-Rx1, -Rx1a, -Rx1b se seleccionan independientemente entre sí del grupo que consiste en -H, -T0, alquilo C1-50, alquenilo C2-50 y alquinilo C2-50; en donde -T0, alquilo C1-50, alquenilo C2-50 y alquinilo C2-50 están opcionalmente sustituidos con uno o más -Rx2, que son iguales o diferentes, y en donde alquilo C1-50, alquenilo C2-50 y alquinilo C2-50 están opcionalmente interrumpidos por uno o más grupos seleccionados del grupo que consiste en -T0-, -C(O)O-, -O-, -C(O)-, -C(O)N(Rx3)-, -S(O)2N(Rx3)-, -S(O)N(Rx3)-; -S(O)2-, -S(O)-, -N(Rx3)S(O)2N(Rx3a)-, -S-, -N(Rx3)-, -OC(ORx3)(Rx3a)-, -N(Rx3)C(O)N(Rx3a)-,y -OC(O)N(Rx3)-;
cada T0 se selecciona independientemente del grupo que consiste en fenilo, naftilo, indenilo, indanilo, tetralinilo, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, y heterobiciclilo de 8 a 11 miembros; en donde cada T0 está opcionalmente sustituido de forma independiente con uno o más -Rx2, que son iguales o diferentes;
cada -Rx2 se selecciona independientemente del grupo que consiste en halógeno, -CN, oxo (=O), -COORx4, -ORx4, -C(O)Rx4, -C(O)N(Rx4Rx4a), -S(O)2N(Rx4Rx4a), -S(O)N(Rx4Rx4a), -S(O)2Rx4, -S(O)Rx4, -N(Rx4)S(O)2N(Rx4aRx4b), -SRx4, -N(Rx4Rx4a), -NO2 , -OC(O)Rx4, -N(Rx4)C(O)Rx4a, -N(Rx4)S(O)2Rx4a, -N(Rx4)S(O)Rx4a, -N(Rx4)C(O)ORx4a, -N(Rx4)C(O)N(Rx4aRx4b), -OC(O)N(Rx4Rx4a), y alquilo C1-6; en donde el alquilo C1-6 está opcionalmente sustituido con uno o más halógenos, que son iguales o diferentes;
cada -Rx3, -Rx3a, -Rx4, -Rx4a, -Rx4b se selecciona independientemente del grupo que consiste en -H y alquilo C1-6; en donde el alquilo C1-6 está opcionalmente sustituido con uno o más halógenos, que son iguales o diferentes.
Más preferiblemente, uno o más sustituyentes opcionales adicionales se seleccionan independientemente entre sí del grupo que consiste en halógeno, -CN, -COORx1, -ORx1, -C(O)Rx1, -C(O)N(Rx1Rx1a), -S(O)2N(Rx1Rx1a), -S(O)N(Rx1Rx1a), -S(O)2Rx1, -S(O)Rx1, -N(Rx1)S(O)2N(Rx1aRx1b), -SRx1, -N(Rx1Rx1a), -NO2 , -OC(O)Rx1, -N(Rx1)C(O)R x1a, -N(Rx1)S(O)2R x1a, -N(Rx1)S(O)Rx1a, -N(Rx1)C(O)ORx1a, -N(Rx1)C(O)N(Rx1aRx1b), -OC(O)N(Rx1Rx1a), -T0, alquilo C1-10, alquenilo C2-10 y alquinilo C2-10; en donde -T0 , alquilo C1-10, alquenilo C2-10 y alquinilo C2-10 están opcionalmente sustituidos con uno o más -Rx2, que son iguales o diferentes, y en donde alquilo C1-10, alquenilo C2-10 y alquinilo C2-10 están opcionalmente interrumpidos por uno o más grupos seleccionados del grupo que consiste en -T0-, -C(O)O-, -O-, -C(O)-, -C(O)N(Rx3)-, -S(O)2N(Rx3)-, -S(O)N(Rx3)-, -S(O)2-, -S(O)-, -N(Rx3)S(O)2N(Rx3a)-, -S-, -N(Rx3)-, -OC(ORx3)(Rx3a)-, -N(Rx3)C(O)N(Rx3a)-, y -OC(O)N(Rx3)-;
cada -Rx1, -Rx1a, -Rx1b, -Rx3, -Rx3a se selecciona independientemente del grupo que consiste en -H, halógeno, alquilo C1-6, alquenilo C2-6 y alquinilo C2-6;
cada T0 se selecciona independientemente del grupo que consiste en fenilo, naftilo, indenilo, indanilo, tetralinilo, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, y heterobiciclilo de 8 a 11 miembros; en donde cada T0 está opcionalmente sustituido de forma independiente con uno o más -Rx2, que son iguales o diferentes;
cada -Rx2 se selecciona independientemente del grupo que consiste en halógeno, -CN, oxo (=O), -COORx4, -ORx4, -C(O)Rx4, -C(O)N(Rx4Rx4a), -S(O)2N(Rx4Rx4a), -S(O)N(Rx4Rx4a), -S(O)2Rx4, -S(O)Rx4, -N(Rx4)S(O)2N(Rx4aRx4b), -SRx4, -N(Rx4Rx4a), -NO2 , -OC(O)Rx4, -N(Rx4)C(O)Rx4a, -N(Rx4)S(O)2Rx4a, -N(Rx4)S(O)Rx4a, -N(Rx4)C(O)ORx4a, -N(Rx4)C(O)N(Rx4aRx4b), -OC(O)N(Rx4Rx4a), y alquilo C1-6; en donde el alquilo C1-6 está opcionalmente sustituido con uno o más halógenos, que son iguales o diferentes;
cada -Rx4, -Rx4a, -Rx4b se selecciona independientemente del grupo que consiste en -H, halógeno, alquilo C1-6, alquenilo C2-6 y alquinilo C2-6;
Aún más preferiblemente, uno o más sustituyentes opcionales adicionales se seleccionan independientemente entre sí del grupo que consiste en halógeno, -CN, -COORx1, -ORx1, -C(O)Rx1, -C(O)N(Rx1Rx1a), -S(O)2N(Rx1Rx1a), -S(O)N(Rx1Rx1a), -S(O)2Rx1, -S(O)Rx1, -N(Rx1)S(O)2N(Rx1aRx1b), -SRx1, -N(Rx1Rx1a), -NO2 , -OC(O)Rx1, -N(Rx1)C(O)R x1a, -N(Rx1)S(O)2R x1a, -N(Rx1)S(O)Rx1a, -N(Rx1)C(O)ORx1a, -N(Rx1)C(O)N(Rx1aRx1b), -OC(O)N(Rx1Rx1a), -T0, alquilo C1-6, alquenilo C2-6 y alquinilo C2-6; en donde -T0, alquilo C1-6, alquenilo C2-6 y alquinilo C2-6 están opcionalmente sustituidos con uno o más -Rx2, que son iguales o diferentes, y en donde alquilo C1-6, alquenilo C2-6 y alquinilo C2-6 están opcionalmente interrumpidos por uno o más grupos seleccionados del grupo que consiste en -T0-, -C(O)O-, -O-, -C(O)-, -C(O)N(Rx3)-, -S(O)2N(Rx3)-, -S(O)N(Rx3)-, -S(O)2-, -S(O)-, -N(Rx3)S(O)2N(Rx3a)-, -S-, -N(Rx3)-, -OC(ORx3)(Rx3a)-, -N(Rx3)C(O)N(Rx3a)-, y -OC(O)N(Rx3)-;
cada -Rx1, -Rx1a, -Rx1b, -Rx2, -Rx3, -Rx3a se selecciona independientemente del grupo que consiste en -H, halógeno, alquilo C1-6, alquenilo C2-6 y alquinilo C2-6;
cada T0 se selecciona independientemente del grupo que consiste en fenilo, naftilo, indenilo, indanilo, tetralinilo, cicloalquilo C3-10, heterociclilo de 3 a 10 miembros, y heterobiciclilo de 8 a 11 miembros; en donde cada T0 está opcionalmente sustituido de forma independiente con uno o más -Rx2, que son iguales o diferentes.
Preferiblemente, un máximo de 6 átomos de H de una molécula opcionalmente sustituida se sustituyen independientemente con un sustituyente, por ejemplo, 5 átomos de H se sustituyen independientemente con un sustituyente, 4 átomos de H se sustituyen independientemente con un sustituyente, 3 átomos de H se sustituyen independientemente con un sustituyente, 2 átomos de H se sustituyen independientemente con un sustituyente, o 1 átomo de H se sustituye con un sustituyente.
El término “interrumpido” significa que un resto se inserta entre dos átomos de carbono o, si la inserción es en uno de los extremos del resto, entre un carbono o heteroátomo y un átomo de hidrógeno.
De la manera que se usa en la presente, el término “alquilo C1-4” solo o en combinación significa un resto alquilo de cadena lineal o ramificada que tiene de 1 a 4 átomos de carbono. Si está presente al final de una molécula, los ejemplos de alquilo C1-4 de cadena lineal o ramificada son metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo y terc-butilo. Cuando dos restos de una molécula están unidos por el alquilo C1-4, los ejemplos de tales grupos alquilo C1-4 son-CH2-, -CH2-CH2-, -CH(CH3)-, -CH2-CH2-CH2-, -CH(C2H5)-, -C(c H3)2-. Cada hidrógeno de un carbono de alquilo C1-4 puede estar opcionalmente reemplazado por un sustituyente como se definió anteriormente. Opcionalmente, un alquilo C1-4 puede estar interrumpido por uno o más restos como se define a continuación.
De la manera que se usa en la presente, el término “alquilo C1-6” solo o en combinación significa un resto alquilo de cadena lineal o ramificada que tiene de 1 a 6 átomos de carbono. Si están presentes al final de una molécula, los ejemplos de grupos alquilo C1-6 de cadena lineal y ramificados son metilo, etilo, n-propilo, isopropilo, n-butilo, isobutilo, sec-butilo, tercbutilo, n-pentilo, 2-metilbutilo, 2,2-dimetilpropilo, n-hexilo, 2-metilpentilo, 3-metilpentilo, 2,2-dimetilbutilo, 2,3-dimetilbutilo y 3,3-dimetilpropilo. Cuando dos restos de una molécula están unidos por el alquilo C1-6, los ejemplos de tales grupos alquilo C1-6 son -CH2-, -CH2-CH2-, -CH(CH3)-, -CH2-CH2-CH2-, -CH(C2H5)- y -C(CH3) 2-. Cada átomo de hidrógeno de un carbono C1-6 puede ser sustituido opcionalmente por un sustituyente como el definido anteriormente. Opcionalmente, un alquilo C1-6 puede estar interrumpido por uno o más restos como se define a continuación.
Por consiguiente, “alquilo C1-10”, “alquilo C1-20” o “alquilo C1-50” significa una cadena de alquilo que tiene de 1 a 10, de 1 a 20 o de 1 a 50 átomos de carbono, respectivamente, en donde cada átomo de hidrógeno del carbono C1-10, C1-20 o C1-50 puede reemplazarse opcionalmente por un sustituyente como se definió anteriormente. Opcionalmente, un alquilo C1-10 o C1-50 puede estar interrumpido por uno o más restos como se define a continuación.
De la manera que se usa en la presente, el término “alquenilo C2-6” solo o en combinación significa un resto de hidrocarburo de cadena recta o ramificada que comprende al menos un enlace doble carbono-carbono que tiene de 2 a 6 átomos de carbono. Si está presente al final de una molécula, los ejemplos son -CH=CH2 , -CH=CH-CH3, -CH2-CH=CH2 , -CH=CHCH2-CH3 y -CH=CH-CH=CH2. Cuando dos restos de una molécula están unidos por el grupo alquenilo C2-6, entonces un ejemplo de tal alquenilo C2-6 es -CH=CH-. Cada átomo de hidrógeno de un resto de alquenilo C2-6 puede ser sustituido
opcionalmente por un sustituyente como el definido anteriormente. Opcionalmente, un alquenilo C2-6 puede estar interrumpido por uno o más elementos como se define a continuación.
Por consiguiente, el término “alquenilo C2-10”, “alquenilo C2-20” o “alquenilo C2-50”, solo o en combinación, significa un resto de hidrocarburo de cadena recta o ramificada que comprende al menos un enlace doble carbono-carbono que tiene de 2 a 10, 2 a 20 o 2 a 50 átomos de carbono. Cada átomo de hidrógeno de un grupo alquenilo C2-10, alquenilo C2-20 o alquenilo C2-50 puede ser sustituido opcionalmente por un sustituyente como se definió anteriormente. Opcionalmente, un alquenilo C2-10, un alquenilo C2-20 o un alquenilo C2-50 puede estar interrumpido por uno o más elementos como se define a continuación.
De la manera que se usa en la presente, el término “alquinilo C2-6” solo o en combinación significa un resto de hidrocarburo de cadena recta o ramificada que comprende al menos un enlace triple carbono-carbono que tiene de 2 a 6 átomos de carbono. Si está presente al final de una molécula, los ejemplos son -CeCH, -CH2-CECH, CH2-CH2-CECH y CH2-CEC-CH3. Cuando dos partes de una molécula están unidas por el grupo alquilo, un ejemplo es -C=C-. Cada átomo de hidrógeno de un grupo alquinilo C2-6 puede ser sustituido opcionalmente por un sustituyente como el definido anteriormente. Opcionalmente, se pueden producir uno o más enlaces dobles. Opcionalmente, un alquinilo C2-6 puede estar interrumpido por uno o más elementos como se define a continuación.
Por consiguiente, de la manera que se usa en la presente, el término “alquinilo C2-10”, “alquinilo C2-20” y “alquinilo C2-50”, solo o en combinación, significa un resto de hidrocarburo de cadena recta o ramificada que comprende al menos un enlace triple carbono-carbono que tiene de 2 a 10, 2 a 20 o 2 a 50 átomos de carbono, respectivamente. Cada átomo de hidrógeno de un grupo alquinilo C2-10, alquinilo C2-20 o alquinilo C2-50 puede ser sustituido opcionalmente por un sustituyente como el definido anteriormente. Opcionalmente, se pueden producir uno o más enlaces dobles. Opcionalmente, un alquinilo C2-10, un alquinilo C2-20 o un alquinilo C2-50 puede estar interrumpido por una o más moléculas como se define a continuación.
Como se mencionó anteriormente, un alquilo C1-4, alquilo C1-6, alquilo C1-10, alquilo C1-20, alquilo C1-50, alquenilo C2-6, alquenilo C2-10, alquenilo C2-20, alquenilo C2-50, alquinilo C2-6, alquinilo C2-10, alquenilo C2-20 o alquinilo C2-50 pueden estar opcionalmente interrumpidos por uno o más restos que se seleccionan preferiblemente del grupo que consiste en
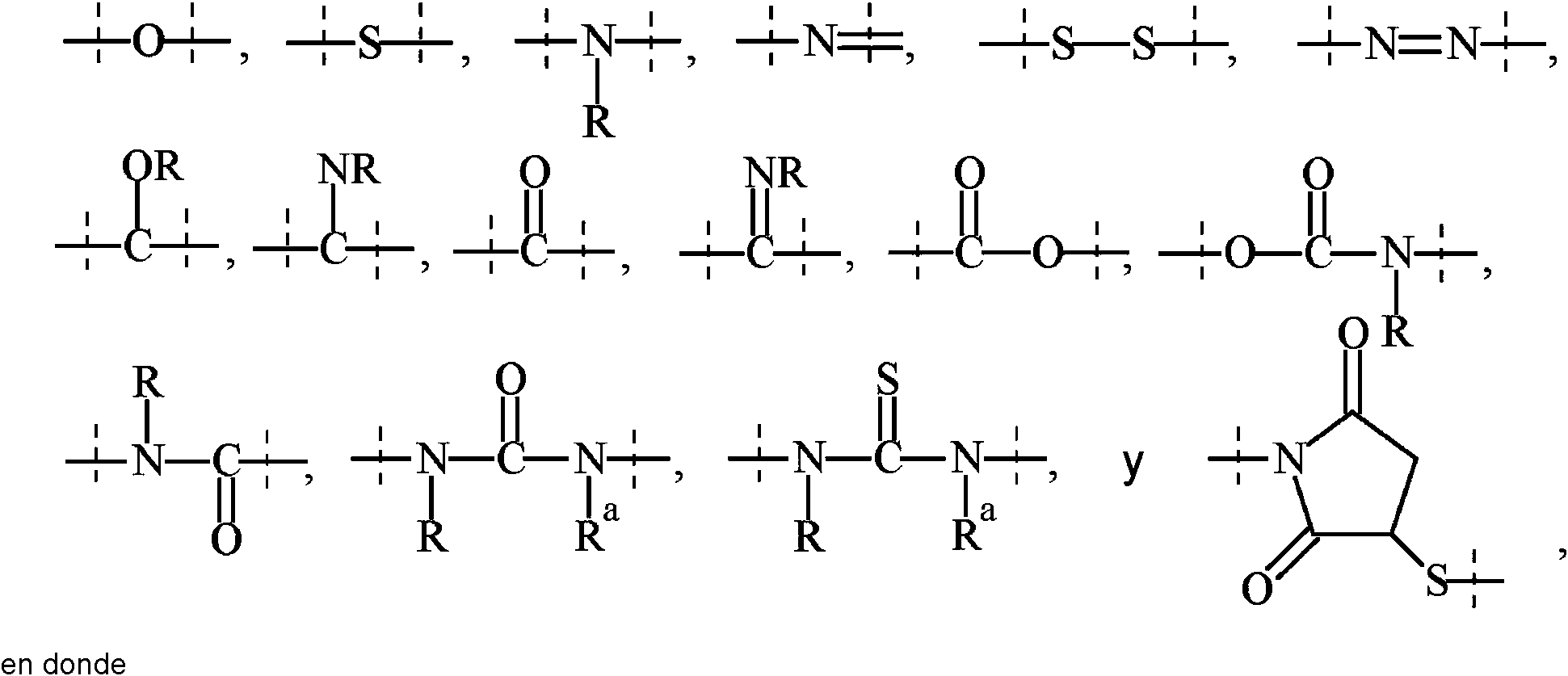
las líneas punteadas indican la unión con el resto dl resto o el reactivo; y
- R y -Ra se seleccionan independientemente entre sí del grupo que consiste en -H, metilo, etilo, propilo, butilo, pentilo y hexilo.
De la manera que se usa en la presente, el término “cicloalquilo C3-10” significa una cadena de alquilo cíclica que tiene de 3 a 10 átomos de carbono, que puede estar saturada o insaturada, por ejemplo, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, ciclohexenilo, cicloheptilo, ciclooctilo, ciclononilo o ciclodecilo. Cada átomo de hidrógeno de un carbono de cicloalquilo C3-10 puede reemplazarse por un sustituyente como se definió anteriormente. El término “cicloalquilo C3-10” también incluye biciclos puenteados como norbornano o norborneno.
El término “carbopoliciclo de 8 a 30 miembros” o “carbopoliciclo de 8 a 30 miembros” significa un resto cíclico de dos o más anillos con 8 a 30 átomos de anillo, en donde dos anillos vecinos comparten al menos un átomo de anillo y que puede contener hasta el número máximo de enlaces dobles (anillo aromático o no aromático saturado, parcialmente saturado o insaturado). Preferiblemente un carbopoliciclo de 8 a 30 miembros significa un resto cíclico de dos, tres, cuatro o cinco anillos, más preferiblemente de dos, tres o cuatro anillos.
De la manera que se usa en la presente, el término “heterociclilo de 3 a 10 miembros” o “heterociclo de 3 a 10 miembros” significa un anillo con 3, 4, 5, 6, 7, 8, 9 o 10 átomos de anillo que puede contener hasta el número máximo de enlaces dobles (anillo aromático o no aromático totalmente saturado, parcialmente saturado o insaturado) en donde al menos un átomo del anillo y hasta 4 átomos del anillo están sustituidos por un heteroátomo seleccionado del grupo que consiste en azufre (incluyendo -S(O)-, -S(O)2-), oxígeno y nitrógeno (incluyendo =N(O)-) y en donde el anillo está unido al resto de la molécula a través de un átomo de carbono o nitrógeno. Los ejemplos de heterociclos de 3 a 10 miembros incluyen, pero no se limitan a, aziridina, oxirano, tiirano, azirina, oxireno, tiireno, azetidina, oxetano, tietano, furano, tiofeno, pirrol, pirrolina, imidazol, imidazolina, pirazol, pirazolina, oxazol, oxazolina, isoxazol, isoxazolina, tiazol, tiazolina, isotiazol, isotiazolina, tiadiazol, tiadiazolina, tetrahidrofurano, tetrahidrotiofeno, pirrolidina, imidazolidina, pirazolidina, oxazolidina, isoxazolidina, tiazolidina, isotiazolidina, tiadiazolidina, sulfolano, pirano, dihidropirano, tetrahidropirano, imidazolidina, piridina, piridazina, pirazina, pirimidina, piperazina, piperidina, morfolina, tetrazol, triazol, triazolidina, tetrazolidina, diazepano, azepina y homopiperazina. Cada átomo de hidrógeno de un heterociclilo de 3 a 10 miembros o de un grupo heterocíclico de 3 a 10 miembros puede ser sustituido por un sustituyente como se define a continuación.
De la manera que se usa en la presente, el término “heterociclo de 8 a 11 miembros” o “heterobiciclo de 8 a 11 miembros” significa un resto heterocíclico de dos anillos con 8 a 11 átomos de anillo, en donde al menos un átomo de anillo es compartido por ambos anillos y que puede contener hasta el número máximo de enlaces dobles (anillo aromático o no aromático totalmente saturado, parcialmente saturado o insaturado) en donde al menos un átomo del anillo y hasta 6 átomos del anillo están sustituidos por un heteroátomo seleccionado del grupo que consiste en azufre (incluyendo -S(O)-, -S(O)2-), oxígeno y nitrógeno (incluyendo =N(O)-), y en donde el anillo está unido al resto de la molécula a través de un átomo de carbono o nitrógeno. Ejemplos de un heterociclo de 8 a 11 miembros son indol, indolina, benzofurano, benzotiofeno, benzoxazol, benzisoxazol, benzotiazol, benzoisotiazol, bencimidazol, bencimidazolina, quinolina, quinazolina, dihidroquinazolina, quinolina, dihidroquinolina, tetrahidroquinolina, decahidroquinolina, isoquinolina, decahidroisoquinolina, tetrahidroisoquinolina, dihidroisoquinolina, benzazepina, purina y pteridina. El término heterociclo de 8 a 11 miembros también incluye las estructuras espiro de dos anillos como el 1,4-dioxa-8-azaspiro[4.5]decano o los heterociclos puenteados como el 8-aza-biciclo[3.2.1]octano. Cada átomo de hidrógeno de un heterobiciclo de 8 a 11 miembros o de un carbono de heterobiciclo de 8 a 11 miembros puede ser sustituido por un sustituyente como se define a continuación.
Del mismo modo, el término “heteropolíclico de 8 a 30 miembros” o “heteropolímero de 8 a 30 miembros” significa un resto heterocíclico de más de dos anillos con 8 a 30 átomos de anillo, preferiblemente de tres, cuatro o cinco anillos, en donde dos anillos vecinos comparten al menos un átomo de anillo y que puede contener hasta el número máximo de enlaces dobles (anillo aromático o no aromático totalmente saturado, parcialmente saturado o insaturado), en donde al menos un átomo del anillo y hasta 10 átomos del anillo están sustituidos por un heteroátomo seleccionado del grupo del azufre (incluyendo -S(O)-, -S(O)2-), oxígeno y nitrógeno (incluyendo =N(O)-), y en donde el anillo está unido al resto de una molécula a través de un átomo de carbono o nitrógeno.
Se entiende que la frase “el par Rx/Ry se une con el átomo al que están unidos para formar un cicloalquilo C3-10 o un heterociclilo de 3 a 10 miembros” en relación con un resto de la estructura

significa que Rx y Ry forman la siguiente estructura:

en donde R es cicloalquilo C3-10 o heterociclilo de 3 a 10 miembros.
También se entiende que la frase “el par Rx/Ry se une con los átomos a los que están unidos para formar un anillo A” en relación con un resto de la estructura

significa que Rx y Ry forman la siguiente estructura:

De la manera que se usa en la presente, el término “alquino terminal” significa un resto

De la manera que se usa en la presente, “halógeno” significa flúor, cloro, bromo o yodo. Generalmente se prefiere que el halógeno sea fluoro o cloro.
En general, el término “comprender” o “que comprende” también abarca “consistir en” o “que consiste en”.
En la presente invención, -L1- se conjuga de forma covalente y reversible con la cadena lateral de la lisina en la posición 26 de dicho resto de anillo de -D.
También se descubrió sorprendentemente que un aumento de las longitudes de la secuencia de PNC es beneficioso con respecto a la estabilidad de NEP: El CNP-22 era más susceptible a la degradación de NEP que CNP-34, que a su vez era más susceptible que CNP-38.
En la presente invención, el resto -L1- se conjuga con el resto de anillo de -D a través de un grupo amina primario. -L1- está conectado al resto de anillo de -D a través de un enlace amida. Se entiende que este enlace puede no ser reversible per se, pero que los grupos vecinos convenientemente elegidos comprendidos en -L1- pueden hacer que tales enlaces sean reversibles.
-D tiene la secuencia de SEQ ID NO:24 y -L1- se conjuga con la lisina en la posición 26. El resto -L1-L2- es de fórmula (IIdii):

en donde
la línea punteada no marcada indica la unión a un nitrógeno de una cadena lateral de aminoácidos del resto de anillo de -D mediante la formación de un enlace amida; la línea punteada marcada con el asterisco indica la unión a -Z, y la línea punteada marcada con el asterisco indica la unión a -Z de la fórmula (h) tal y como se definió anteriormente, en donde -D tiene la secuencia de SEQ ID NO:24 y -L1- se conjuga con la lisina en la posición 26.
En una realización más preferida, el resto -L1-L2- es de fórmula (IId-ii'):

en donde
la línea punteada no marcada indica la unión a -D tal como se define en la fórmula (Md-ii); y
la línea punteada marcada con el asterisco indica la unión a -Z de fórmula (h).
- Z es soluble en agua.
- Z tiene un peso molecular de aproximadamente 40 kDa.
Los solicitantes descubrieron que una unión N-terminal de un resto -L1-L2-Z es significativamente más eficiente con respecto a la estabilidad de NEP que la unión en un sitio interno, y que el sitio de unión menos eficiente con respecto a la estabilidad de NEP es en la parte del anillo de un resto de CNP. Sin embargo, los solicitantes descubrieron sorprendentemente que esta desventaja de la unión al anillo con respecto a la estabilidad de NEP puede compensarse utilizando un resto ramificado -Z que tenga un peso molecular de al menos 10 kDa, tal como al menos 12 kDa, tal como al menos 15 kDa, tal como al menos 18 kDa, tal como al menos 20 kDa, tal como al menos 24 kDa, tal como al menos 25 kDa, tal como al menos 27 kDa, tal como al menos 30 kDa. Preferiblemente, dicho resto ramificado -Z tiene un peso molecular no superior a 500 kDa, preferiblemente no superior a 250 kDa, preferiblemente no superior a 200 Da, preferiblemente no superior a 150 kDa, y más preferiblemente no superior a 100 kDa. Lo más preferible es que dicho resto ramificado -Z tenga un peso molecular de aproximadamente 40 kDa. Por lo tanto, el uso de tal resto ramificado -Z en la parte del anillo del resto de CNP no sólo conduce a un aumento de la estabilidad de NEP, sino que combina el aumento de la estabilidad de NEP con la reducción de la unión de NPR-B asociada con la unión al anillo.
Sorprendentemente, se descubrió que aunque el resto de anillo está implicado en la unión de NPR-C, la unión de un portador de 5 kDa al resto de anillo no tuvo un efecto significativo en la afinidad de NPR-C. Además, se descubrió sorprendentemente que un portador 4x 10 kDa, es decir, un portador ramificado que tiene cuatro brazos de 10 kDa, unido a la fracción de anillo, es más eficiente en la reducción de la afinidad de NPR-C que un portador 2x 20 kDa, es decir, un portador ramificado que tiene dos brazos de 20 kDa, aunque el peso molecular total sea el mismo. Por lo tanto, no es sólo el peso molecular total del portador unido al resto del anillo, sino el patrón de ramificación particular del portador lo que influye en la afinidad de unión de NPR-C.
Este hallazgo también se ve respaldado por la afinidad de NPR-C medida con un portador de 4 brazos de 40 kDa que tiene un patrón de ramificación diferente y que sigue mostrando una alta afinidad de NPR-C.
En resumen, se encontró sorprendentemente que la afinidad de NPR-C puede reducirse eficientemente con un portador multiramificado fijado al resto de anillo que tiene un primer punto de ramificación cerca del resto de CNP, tal como, por ejemplo, a menos de 300 átomos del resto de CNP, preferiblemente a 200 átomos del resto de CNP, incluso más preferiblemente a 100 átomos del resto de CNP, incluso más preferiblemente a menos de 50 átomos del resto de CNP, incluso más preferiblemente a menos de 25 átomos del resto de CNP, y más preferiblemente a menos de 10 átomos del resto de CNP.
Aún más preferiblemente, uno o más puntos de ramificación adicionales están situados a menos de 500 átomos del resto de CNP, aún más preferiblemente a 300 átomos del resto de CNP, aún más preferiblemente a menos de 200 átomos del resto de CNP, aún más preferiblemente a menos de 100 átomos del resto de CNP, aún más preferiblemente a menos de 75 átomos del resto de CNP, aún más preferiblemente a menos de 50 átomos del resto de CNP, aún más preferiblemente a menos de 40 átomos del resto de c Np , y más preferiblemente a menos de 35 átomos del resto de CNP.
Además, se descubrió que este patrón de ramificación es beneficioso para la estabilidad in vivo del resto de CNP, es decir, la protección contra la degradación proteolítica. Se descubrió sorprendentemente que la degradación N-terminal era más fuerte cuando se utilizaba un portador 2x 20 kDa en comparación con un portador 4x 10 kDa. Asimismo, el uso de un portador de 4 brazos de 40 kDa con un patrón de ramificación diferente mostró una degradación N-terminal aún más fuerte.
El resto -Z es un polímero ramificado a base de PEG que comprende al menos un 10% de PEG, tiene un punto de ramificación y dos brazos de polímero a base de PEG, y tiene un peso molecular de aproximadamente 40 kDa. Por
consiguiente, cada uno de los dos brazos de polímero a base de PEG tiene un peso molecular de aproximadamente 20 kDa y el punto de ramificación es -CH<.
-Z es de fórmula (h):

en donde
la línea punteada indica unión a -L2-; y
cada -Zc es un resto

en donde
cada c1 es un número entero que oscila independientemente entre 200 y 250.
preferiblemente, los dos c1 de la fórmula (h) son iguales.
preferiblemente, los dos c1 de la fórmula (h) son aproximadamente 225.
En una realización aún más preferida, el resto -Z tiene la fórmula (h-i):

en donde
la línea punteada indica unión a -L2-; y
cada -Zc es un resto

cada c1 es un número entero que varía independientemente de 200 a 250.
Preferiblemente, los dos c1 de la fórmula (h-i) son iguales.
preferiblemente, los dos c1 de la fórmula (h-i) son aproximadamente 225.
La masa total del profármaco de CNP de la presente invención es de al menos 10 kDa, tal como al menos 12 kDa, tal como al menos 15 kDa, tal como al menos 20 kDa o tal como al menos 30 kDa. Se prefiere que la masa total del profármaco de CNP de la presente invención sea como máximo de 250 kDa, como máximo de 200 kDa, 180 kDa, 150 kDa o 100 kDa.
En una realización preferida, la actividad residual del profármaco de CNP de la presente invención es de menos del 10%, más preferiblemente menos del 1%, incluso más preferiblemente menos del 0,1%, incluso más preferiblemente menos del 0,01%, incluso más preferiblemente menos del 0,001%, y lo más preferiblemente menos del 0,0001%.
De la manera que se usa en la presente, el término “actividad residual” se refiere a la actividad que exhibe el profármaco de CNP de la presente invención con el resto de CNP unido a un portador en relación con la actividad que exhibe el correspondiente CNP libre. En este contexto, el término “actividad” se refiere a la unión de NPR-B. Se entiende que la medición de la actividad residual del profármaco de CNP de la presente invención lleva un tiempo durante el cual se liberará una cierta cantidad de CNP del profármaco de CNP de la presente invención, y que dicho CNP liberado distorsionará los resultados medidos para el profármaco de CNP. Por lo tanto, es una práctica aceptada probar la actividad residual de un profármaco con un conjugado en el que el resto del fármaco, en este caso CNP, está unida de forma no reversible, es decir, estable, a un portador, que se asemeja lo más posible a la estructura del profármaco de CNP cuya
actividad residual se va a medir.
Un ensayo adecuado para medir la actividad de CNP y la actividad residual del profármaco de CNP de la presente invención, preferiblemente en forma de análogo estable, se describe en WO 2010/135541 A1, ejemplo 4, página 143/144. Otro aspecto de la presente invención es una composición farmacéutica que comprende al menos un profármaco de CNP o una sal farmacéuticamente aceptable del mismo de la presente invención y al menos un excipiente, en donde el profármaco de CNP o la sal farmacéuticamente aceptable del mismo comprende:
- un resto de CNP -D que comprende un resto de anillo; y
- un resto portador -Z que se conjuga a través de un resto -L2- a un resto enlazador de profármaco reversible -L1-, cuyo resto enlazador de profármaco reversible -L1- está conjugado de manera covalente y reversible a una cadena lateral de un residuo de aminoácido de dicho anillo resto de -D; y en donde -L2- es un enlace químico o un espaciador, en donde el resto -L1-L2- es de fórmula (IId-ii):
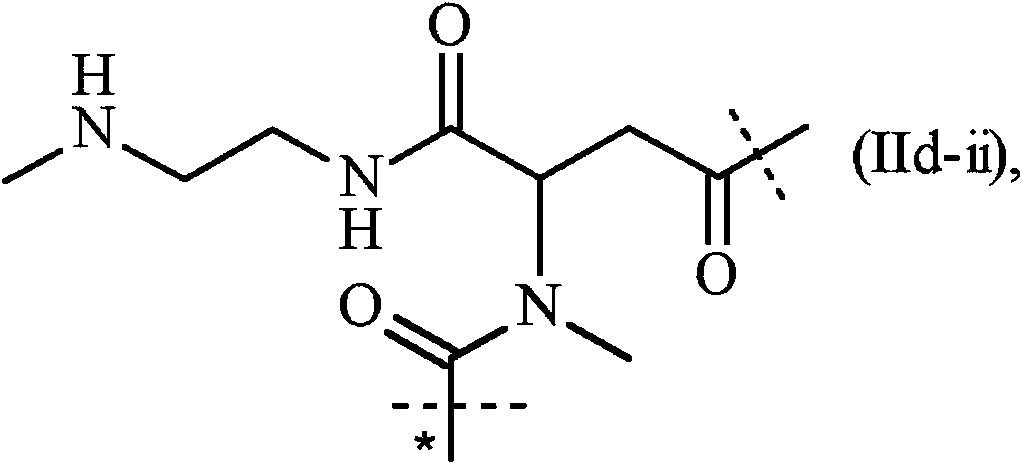
en donde
la línea punteada no marcada indica unión a un nitrógeno de una cadena lateral de aminoácido del resto de anillo de -D mediante la formación de un enlace amida; y la línea punteada marcada con el asterisco indica unión a -Z; en donde -D tiene la secuencia de SEQ ID NO: 24 y -L1- se conjuga con la lisina en la posición 26; en donde -Z es de fórmula (h):

en donde
la línea punteada indica unión a -L2-; y
cada -Zc es un resto

en donde
cada c1 es un número entero que oscila independientemente entre 200 y 250.
Preferiblemente, la composición farmacéutica que comprende al menos un profármaco de CNP o una sal farmacéuticamente aceptable del mismo de la presente invención tiene un pH que va desde e incluye pH 3 a pH 8. Más preferiblemente, la composición farmacéutica tiene un pH que va desde e incluye pH 4 a pH 6. Lo más preferible es que la composición farmacéutica tenga un pH que va desde e incluye pH 4 a pH 5.
En una realización, la composición farmacéutica que comprende al menos un profármaco de CNP o una sal farmacéuticamente aceptable del mismo de la presente invención y al menos un excipiente es una formulación líquida.
En otra realización, la composición farmacéutica que comprende al menos un profármaco de CNP o una sal farmacéuticamente aceptable del mismo de la presente invención y al menos un excipiente es una formulación seca.
Dicha composición farmacéutica líquida o seca comprende al menos un excipiente. Los excipientes utilizados en las formulaciones parenterales pueden clasificarse, por ejemplo, como agentes amortiguadores, modificadores de la isotonicidad, conservantes, estabilizadores, agentes antiadsorción, agentes de protección contra la oxidación, viscosificadores/agentes potenciadores de la viscosidad u otros agentes auxiliares. Sin embargo, en algunos casos, un excipiente puede tener funciones dobles o triples. Preferiblemente, al menos un excipiente comprendido en la composición farmacéutica de la presente invención se selecciona del grupo que consiste en
(i) Agentes amortiguadores: amortiguadores fisiológicamente tolerados para mantener el pH en un rango deseado, tal como fosfato de sodio, bicarbonato, succinato, histidina, citrato y acetato, sulfato, nitrato, cloruro, piruvato; también pueden utilizarse antiácidos, tales como Mg(OH)2 o ZnCO3;
(ii) Modificadores de la isotonicidad: para minimizar el dolor que puede resultar del daño celular debido a las diferencias de presión osmótica en el depósito de la inyección; la glicerina y el cloruro de sodio son ejemplos; las concentraciones efectivas pueden determinarse mediante osmometría utilizando una osmolalidad supuesta de 285-315 mOsmol/kg para el suero;
(iii) Conservantes y/o antimicrobianos: las formulaciones parenterales multidosis requieren la adición de conservantes en una concentración suficiente para minimizar el riesgo de que los pacientes se infecten al ser inyectados, y se han establecido los requisitos regulatorios correspondientes; los conservantes típicos incluyen m-cresol, fenol, metilparabeno, etilparabeno, propilparabeno, butilparabeno, clorobutanol, alcohol bencílico, nitrato fenilmercúrico, timerosol, ácido sórbico, sorbato de potasio, ácido benzoico, clorocresol y cloruro de benzalconio;
(iv) Estabilizadores: La estabilización se consigue mediante el refuerzo de las fuerzas estabilizadoras de la proteína, la desestabilización del estado desnaturalizado o la unión directa de los excipientes a la proteína; los estabilizadores pueden ser aminoácidos tales como alanina, arginina, ácido aspártico, glicina, histidina, lisina, prolina, azúcares tales como glucosa, sacarosa, trehalosa, polioles tales como glicerol, manitol, sorbitol, sales tales como fosfato de potasio, sulfato de sodio, agentes quelantes tales como EDTA, hexafosfato, ligandos tales como iones metálicos divalentes (zinc y calcio), otras sales o moléculas orgánicas, tales como los derivados fenólicos; además, se pueden utilizar oligómeros o polímeros, tales como ciclodextrinas, dextrano, dendrímeros, PEG o PVP, o protamina o HSA;
(v) Agentes antiadsorción: Se utilizan principalmente tensioactivos iónicos o no iónicos u otras proteínas o polímeros solubles para recubrir o adsorber de forma competitiva la superficie interior del recipiente de la formulación; por ejemplo, poloxámero (Pluronic F-68), PEG dodecil éter (Brij 35), polisorbato 20 y 80, dextrano, polietilenglicol, PEG-polihistidina, BSA y HSA, y gelatinas; la concentración y el tipo de excipiente elegidos dependen del efecto que se quiera evitar, pero normalmente se forma una monocapa de tensioactivo en la interfaz justo por encima del valor de CMC;
(vi) Agentes de protección contra la oxidación: antioxidantes, tales como ácido ascórbico, ectoína, metionina, glutatión, monotioglicerol, morina, polietilenimina (PEI), galato de propilo y vitamina E; también pueden utilizarse agentes quelantes, tales como ácido cítrico, EDTA, hexafosfato y ácido tioglicólico;
(vii) Viscosificantes o potenciadores de la viscosidad: retardan la sedimentación de las partículas en el vial y la jeringa, y
se usan para facilitar el mezclado y resuspensión de las partículas, así como para hacer que la suspensión sea más fácil de inyectar (es decir, poca fuerza sobre el émbolo de la jeringa); los viscosificantes o potenciadores de la viscosidad adecuados son, por ejemplo, viscosificantes de carbómero como Carbopol 940, Carbopol Ultrez 10, derivados de celulosa como hidroxipropilmetilcelulosa (hipromelosa, HPMC) o dietilaminoetilcelulosa (DEAE o DEAE-C), silicato de magnesio coloidal (Veegum) o silicato de sodio, gel de hidroxiapatita, gel de fosfato tricálcico, xantanas, carragenanos como la goma Satia UTC 30, poli(hidroxiácidos) alifáticos, tales como poli(ácido D,L- o L-láctico) (“PLA”, por sus siglas en inglés) y poli(ácido glicólico) (“PGA”, por sus siglas en inglés) y sus copolímeros (“PLGA”, por sus siglas en inglés), terpolímeros de D,L-lactida, glicolida y caprolactona, poloxámeros, bloques de poli(oxietileno) hidrofílicos y bloques de poli(oxipropileno) hidrofóbicos para formar un tribloque de poli(oxietileno)-poli(oxipropileno)-poli(oxietileno) (por ejemplo, Pluronic®), copolímero de polieteréster, tal como un copolímero de tereftalato de polietilenglicol/tereftalato de polibutileno, isobutirato de acetato de sacarosa (“SAIB”, por sus siglas en inglés), dextrano o derivados del mismo, combinaciones de dextranos y PEG, polidimetilsiloxano, colágeno, quitosano, alcohol polivinílico (“PVA”, por sus siglas en inglés) y derivados, polialquilimidas, poli(acrilamida-co-dialildimetilamonio (DADMA)), polivinilpirrolidona (PVP), glicosaminoglicanos (GAG), tales como sulfato de dermatán, sulfato de condroitina, sulfato de queratán, heparina, sulfato de heparán, hialuronano, copolímeros de bloque AB o tribloque ABA compuestos por bloques A hidrófobos, tales como poliláctido (PLA) o poli(láctido-co-glicólido) (PLGA), y bloques B hidrófilos, tales como polietilenglicol (PEG) o polivinilpirrolidona; dichos copolímeros de bloque, así como los poloxámeros mencionados anteriormente, pueden exhibir un comportamiento de gelificación térmica inversa (estado fluido a temperatura ambiente para facilitar la administración y estado de gel por encima de la temperatura de transición sol-gel a la temperatura corporal después de la inyección);
(viii) Agente esparcidor o difusor: modifica la permeabilidad del tejido conectivo a través de la hidrólisis de componentes de la matriz extracelular en el espacio intrasticial, tal como ácido hialurónico, un polisacárido que se encuentra en el espacio intercelular del tejido conectivo; un agente esparcidor, tal como hialuronidasa disminuye temporalmente la viscosidad de la matriz extracelular y promueve la difusión de los fármacos inyectados; y
(ix) Otros agentes auxiliares: tales como agentes humectantes, modificadores de la viscosidad, antibióticos, hialuronidasa; ácidos y bases, tales como ácido clorhídrico e hidróxido de sodio, son agentes auxiliares necesarios para el ajuste del pH durante la fabricación.
Otro aspecto de la presente invención es el profármaco de CNP o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica que comprende al menos un profármaco de CNP de la presente invención para su uso como un medicamento.
Preferiblemente, dicho medicamento se usa en el tratamiento de una enfermedad seleccionada del grupo que consiste en acondroplasia, hipocondroplasia, baja estatura, enanismo, osteocondrodisplasias, displasia tanatofórica, osteogénesis imperfecta, acondrogénesis, condrodisplasia punctata, acondroplasia homocigota, displasia campomélica, hipofosfatasia letal congénita, tipo letal perinatal de osteogénesis imperfecta, síndrome de costillas cortas-polidactilia, tipo rizomélico de condrodisplasia punctata, displasia metafisaria tipo Jansen, displasia espondiloepifisaria congénita, atelosteogénesis, displasia diastrófica, fémur corto congénito, displasia mesomélica tipo Langer, displasia mesomélica tipo Nievergelt, síndrome de Robinow, síndrome de Reinhardt, acrodisostosis, disostosis periférica, displasia de Kniest, fibrocondrogénesis, síndrome de Roberts, displasia acromesomélica, micromelia, síndrome de Morquio, síndrome de Kniest, displasia metatrófica, displasia espondiloepimetafisaria, neurofibromatosis, síndrome de Legius, síndrome LEOPARD, síndrome de Noonan, fibromatosis gingival hereditaria, síndrome cardio-facio-cutáneo, síndrome de Costello, deficiencia de SHOX, baja estatura idiopática, deficiencia de la hormona del crecimiento, artrosis, disostosis cleidocraneal, craneosinostosis, braquidactilia, camptodactilia, polidactilia, sindactilia, displasia disegmentaria, encondromatosis, displasia fibrosa, exostosis múltiple hereditaria, raquitismo hipofosfatémico, síndrome de Jaffe-Lichtenstein, síndrome de Marfan, síndrome de McCune-Albright, osteopetrosis y osteopoiquilia.
En otra realización, dicho medicamento se usa en el tratamiento de un trastorno oftálmico, tal como glaucoma y/o presión intraocular elevada.
En otra realización, dicho medicamento se usa en el tratamiento de una enfermedad de cáncer asociada con la sobreactivación de FGFR3, por ejemplo, mieloma múltiple, síndrome mieloproliferativo, leucemia, leucemia de células plasmáticas, linfoma, glioblastoma, cáncer de próstata, cáncer de vejiga o cáncer de mama.
En otra realización, dicho medicamento se utiliza en el tratamiento de un trastorno del músculo liso vascular, preferiblemente seleccionado del grupo que consiste en hipertensión, restenosis, arteriosclerosis, insuficiencia cardíaca aguda descompensada, insuficiencia cardíaca congestiva, edema cardíaco, nefredema, edema hepático, insuficiencia renal aguda e insuficiencia renal crónica.
En otra realización, dicho medicamento se usa en el tratamiento de choque hemorrágico.
Preferiblemente, dicho medicamento se utiliza en el tratamiento de un fenotipo de acondroplasia seleccionado del grupo que consiste en retraso del crecimiento, deformidades del cráneo, defectos de ortodoncia, compresión de la médula cervical, estenosis espinal, hidrocefalia, pérdida de audición debida a la otitis crónica, enfermedades cardiovasculares, enfermedades neurológicas y obesidad.
Más preferiblemente dicho medicamento se utiliza en el tratamiento de acondroplasia.
Otro aspecto de la presente invención es el profármaco de CNP o una sal farmacéuticamente aceptable del mismo, o la composición farmacéutica que comprende al menos un profármaco de CNP de la presente invención para su uso en el tratamiento de una enfermedad que puede tratarse con CNP.
Preferiblemente, dicha enfermedad se selecciona del grupo que consiste en acondroplasia, hipocondroplasia, baja estatura, enanismo, osteocondrodisplasias, displasia tanatofórica, osteogénesis imperfecta, acondrogénesis, condrodisplasia punctata, acondroplasia homocigota, displasia campomélica, hipofosfatasia letal congénita, tipo letal perinatal de osteogénesis imperfecta, síndrome de costillas cortas-polidactilia, tipo rizomélico de condrodisplasia punctata, displasia metafisaria tipo Jansen, displasia espondiloepifisaria congénita, atelosteogénesis, displasia diastrófica, fémur corto congénito, displasia mesomélica tipo Langer, displasia mesomélica tipo Nievergelt, síndrome de Robinow, síndrome de Reinhardt, acrodisostosis, disostosis periférica, displasia de Kniest, fibrocondrogénesis, síndrome de Roberts, displasia acromesomélica, micromelia, síndrome de Morquio, síndrome de Kniest, displasia metatrófica, displasia espondiloepimetafisaria, neurofibromatosis, síndrome de Legius, síndrome LEOPARD, síndrome de Noonan, fibromatosis gingival hereditaria, síndrome cardio-facio-cutáneo, síndrome de Costello, deficiencia de SHOX, baja estatura idiopática, deficiencia de la hormona del crecimiento, artrosis, disostosis cleidocraneal, craneosinostosis, braquidactilia, camptodactilia, polidactilia, sindactilia, displasia disegmentaria, encondromatosis, displasia fibrosa, exostosis múltiple hereditaria, raquitismo hipofosfatémico, síndrome de Jaffe-Lichtenstein, síndrome de Marfan, síndrome de McCune-Albright, osteopetrosis y osteopoiquilia.
En otra realización, la enfermedad es un trastorno oftálmico, tal como glaucoma y/o presión intraocular elevada.
En otra realización, dicha enfermedad está asociada con la sobreactivación de FGFR3 en cáncer, por ejemplo, mieloma múltiple, síndrome mieloproliferativo, leucemia, leucemia de células plasmáticas, linfoma, glioblastoma, cáncer de próstata, cáncer de vejiga o cáncer de mama.
En otra realización, dicha enfermedad es un trastorno del músculo liso vascular, preferiblemente seleccionado del grupo que consiste en hipertensión, restenosis, arteriosclerosis, insuficiencia cardíaca aguda descompensada, insuficiencia cardíaca congestiva, edema cardíaco, nefredema, edema hepático, insuficiencia renal aguda e insuficiencia renal crónica. En otra realización, dicha enfermedad es un choque hemorrágico.
Preferiblemente, dicha enfermedad es un fenotipo de acondroplasia seleccionado del grupo que consiste en retraso del crecimiento, deformidades del cráneo, defectos de ortodoncia, compresión de la médula cervical, estenosis espinal, hidrocefalia, pérdida de audición debida a la otitis crónica, enfermedades cardiovasculares, enfermedades neurológicas y obesidad.
Lo más preferible es que dicha enfermedad sea acondroplasia.
En una realización, el paciente que se somete al método de tratamiento de la presente invención es un paciente mamífero, preferiblemente un paciente humano. En una realización, este paciente humano es un adulto. En una realización preferida, el paciente humano es un paciente pediátrico.
En una realización, la enfermedad a tratar con el profármaco de CNP o una sal farmacéuticamente aceptable del mismo, o la composición farmacéutica que comprende al menos un profármaco de CNP de la presente invención ocurre en un paciente mamífero, preferiblemente en un paciente humano. En una realización, este paciente humano es un adulto. En una realización preferida, el paciente humano es un paciente pediátrico.
En una realización preferida, la presente invención se refiere a un profármaco de CNP o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica de la presente invención, para usar en el tratamiento de la acondroplasia mediante inyección subcutánea.
En otro aspecto, la presente invención se refiere a una composición farmacéutica que comprende al menos un profármaco de CNP de la presente invención o una sal farmacéuticamente aceptable del mismo, en donde la composición farmacéutica comprende al menos un resto biológicamente activo o fármaco adicional.
Al menos un resto biológicamente activo o fármaco adicional puede estar en su forma libre (es decir, en forma de fármaco libre), puede estar en forma de conjugado estable o puede estar en forma de un compuesto de liberación controlada. En una realización, al menos un resto biológicamente activo o fármaco adicional es un fármaco en su forma libre, es decir, la composición farmacéutica de la presente invención comprende al menos un profármaco de CNP y al menos un fármaco adicional.
Preferiblemente, al menos un fármaco adicional se selecciona del grupo que consiste en antihistamínicos; anticuerpos
anti-FGFR3 humanos; formas solubles del receptor del factor de crecimiento de fibroblastos 3 humano; inhibidores de la tirosina quinasa; estatinas; agonistas de CNP; hormona del crecimiento; IGF-1; ANP; BNP; inhibidores de peptidasas y proteasas; e inhibidores de NPR-C.
Un antihistamínico preferido es meclozina.
Un inhibidor de tirosina quinasa preferido es NVP-BGJ398.
Una estatina preferida es rosuvastatina.
Un agonista de CNP preferido para al menos un fármaco adicional es vosoritida.
Los inhibidores preferidos de peptidasas y proteasas son inhibidores de NEP y furina.
Los inhibidores preferidos de NEP son tiorfano y candoxatril.
Los inhibidores preferidos de NPR-C son el fragmento de SEQ ID NO:98 (FGIPMDRIGRNPR) y el anticuerpo B701. Los inhibidores preferidos de las tirosina quinasas son como se describen en las patentes de EE. UU. 6329375 y 6344459. En una realización, al menos un fármaco adicional es un antihistamínico.
En otra realización, al menos un fármaco adicional es un anticuerpo anti-FGFR3 humano.
En otra realización, al menos un fármaco adicional es una forma soluble del receptor del factor de crecimiento de fibroblastos 3 humano (sFGFR3).
En otra realización, al menos un fármaco adicional es un inhibidor de tirosina quinasa.
En otra realización, al menos un fármaco adicional es una estatina.
En otra realización, al menos un fármaco adicional es una hormona del crecimiento.
En otra realización, al menos un fármaco adicional es un agonista de CNP.
En otra realización, al menos un fármaco adicional es IGF-1.
En otra realización, al menos un fármaco adicional es ANP.
En otra realización, al menos un fármaco adicional es BNP.
En otra realización, al menos un fármaco adicional es un inhibidor de peptidasas y proteasas.
En otra realización, al menos un fármaco adicional es un inhibidor de NPR-C.
En otra realización, al menos un resto biológicamente activo o fármaco adicional está en forma de un conjugado estable. En una realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende al menos un resto biológicamente activo conjugado covalentemente a través de un enlace estable a un resto polimérico, preferiblemente a un resto polimérico soluble en agua, ya sea directamente o a través de un resto espaciador.
Preferiblemente, dicho resto polimérico, aún más preferiblemente el resto polimérico soluble en agua, comprende un polímero seleccionado del grupo que consiste en 2-metacriloil-oxietil fosfoil colinas, poli(ácidos acrílicos), poli(acrilatos), poli(acrilamidas), poli(alquiloxi) polímeros, poli(amidas), poli(amidoaminas), poli(aminoácidos), poli(anhídridos), poli(aspartamidas), poli(ácidos butíricos), poli(ácidos glicólicos), tereftalatos de polibutileno, poli(caprolactonas), poli(carbonatos), poli(cianoacrilatos), poli(dimetilacrilamidas), poli(ésteres), poli(etilenos), poli(etilenglicoles), poli(óxidos de etileno), poli(fosfatos de etilo), poli(etiloxazolinas), poli(ácidos glicólicos), poli(hidroxietilacrilatos), poli(hidroxietiloxazolinas), poli(hidroximetacrilatos), poli(hidroxipropilmetacrilamidas), poli(metacrilatos de hidroxipropilo), poli(hidroxipropiloxazolinas), poli(iminocarbonatos), poli(ácidos lácticos), poli(ácidos láctico-co-glicólico), poli(metacrilamidas), poli(metacrilatos), poli(metiloxazolinas), poli(organofosfacenos), poli(ortoésteres), poli(oxazolinas), poli(propilenglicoles), poli(siloxanos), poli(uretanos), poli(alcoholes vinílicos), poli(vinilaminas), poli(vinilmetiléteres), poli(vinilpirrolidonas), siliconas, celulosas, carbometilcelulosas, hidroxipropilmetilcelulosas, quitinas, quitosanos, dextranos, dextrinas, gelatinas, ácidos hialurónicos y derivados, ácidos hialurónicos funcionalizados, mananos, pectinas, ramnogalacturonanos, almidones, almidones de hidroxialquilo, almidones de hidroxietilo y otros polímeros a base de carbohidratos, xilanos y copolímeros de los mismos.
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable se conjuga covalentemente a través de un enlace estable con un resto de unión a albúmina. Preferiblemente, dicho resto que se une a la albúmina es un resto alquilo C8-24 o un derivado de ácido graso. Los derivados de ácidos grasos preferidos son los descritos en WO 2005/027978 A2 y WO 2014/060512 A1.
Preferiblemente, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto biológicamente activo seleccionado del grupo que consiste en antihistamínicos; anticuerpos anti-FGFR3 humanos; formas solubles del receptor del factor de crecimiento de fibroblastos 3 humano (sFGFR3); inhibidores de tirosina quinasa; estatinas; agonistas de CNP; hormona de crecimiento; IGF-1; ANP; BNP; inhibidores de peptidasas y proteasas; e inhibidores de NPR-C.
Un antihistamínico preferido es meclozina.
Un inhibidor de tirosina quinasa preferido es NVP-BGJ398.
Una estatina preferida es rosuvastatina.
Un agonista de CNP preferido para al menos otro resto biológicamente activo es vosoritida.
Los inhibidores preferidos de peptidasas y proteasas son inhibidores de NEP y furina.
Los inhibidores preferidos de NEP son tiorfano y candoxatril.
Los inhibidores preferidos de NPR-C son el fragmento de SEQ ID NO:98 (FGIPMDRIGRNPR) y el anticuerpo B701. Los inhibidores preferidos de las tirosina quinasas son como se describen en las patentes de EE. UU. 6329375 y 6344459. En una realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto de antihistamínico.
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto de anticuerpo anti-FGFR3 humano.
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende formas solubles del resto del receptor del factor de crecimiento de fibroblastos 3 humano (sFGFR3).
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto inhibidor de tirosina quinasa.
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto de estatina.
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto de hormona del crecimiento.
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto agonista de CNP.
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto de IGF-1.
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto de ANP.
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto de BNP.
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto de inhibidor de peptidasas y proteasas.
En otra realización, al menos un resto biológicamente activo adicional en forma de un conjugado estable comprende un resto de inhibidor de NPR-C.
En otra realización, al menos un resto biológicamente activo o fármaco adicional está en forma de un compuesto de liberación controlada.
Preferiblemente, al menos un resto biológicamente activo adicional o fármaco en forma de compuesto de liberación controlada comprende al menos un resto biológicamente activo o fármaco seleccionado del grupo que consiste en antihistamínicos; anticuerpos anti-FGFR3 humanos; formas solubles del receptor del factor de crecimiento de fibroblastos 3 humano; estatinas; agonistas de CNP; hormona del crecimiento; IGF-1; ANP; BNP; inhibidores de peptidasas y proteasas; inhibidores de tirosina quinasas; e inhibidores de NPR-C.
Un antihistamínico preferido es meclozina.
Un inhibidor de tirosina quinasa preferido es NVP-BGJ398.
Una estatina preferida es rosuvastatina.
Un agonista de CNP preferido para al menos un fármaco adicional es vosoritida.
Los inhibidores preferidos de peptidasas y proteasas son inhibidores de NEP y furina.
Los inhibidores preferidos de NEP son tiorfano y candoxatril.
Los inhibidores preferidos de NPR-C son el fragmento de SEQ ID NO:98 (FGIPMDRIGRNPR) y el anticuerpo B701. Los inhibidores preferidos de las tirosina quinasas son como se describen en las patentes de EE. UU. 6329375 y 6344459. En una realización, al menos un resto o fármaco biológicamente activo adicional en forma de un compuesto de liberación controlada comprende un resto o fármaco antihistamínico.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada comprende un resto o fármaco de anticuerpo anti-FGFR3 humano.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada comprende formas solubles del resto o fármaco del receptor del factor de crecimiento de fibroblastos 3 humano (sFGFR3).
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada comprende un resto o fármaco de inhibidor de tirosina quinasa.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada comprende un resto o fármaco de estatina.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada comprende un resto o fármaco de hormona de crecimiento.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada comprende un resto o fármaco de agonista de CNP.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada comprende un resto o fármaco de IGF-1.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada comprende un resto o fármaco de ANP.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada comprende un resto o fármaco de BNP.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada comprende un resto o fármaco de inhibidor de peptidasas y proteasas.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada comprende un inhibidor del resto o fármaco de NPR-C.
En una realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada es insoluble en agua.
Preferiblemente, dicho compuesto de liberación controlada e insoluble en agua se selecciona del grupo formado por cristales, nanopartículas, micropartículas, nanoesferas y microesferas.
En una realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación
controlada insoluble en agua es un cristal que comprende al menos un fármaco o resto biológicamente activo.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua es una nanopartícula que comprende al menos un fármaco o resto biológicamente activo.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua es una micropartícula que comprende al menos un fármaco o resto biológicamente activo.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua es una nanoesfera que comprende al menos un fármaco o resto biológicamente activo.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua es una microesfera que comprende al menos un fármaco o resto biológicamente activo.
En una realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua es una vesícula que comprende al menos un fármaco o resto biológicamente activo. Preferiblemente, dicha vesícula que comprende al menos un fármaco o resto biológicamente activo es una micela, un liposoma o un polimerosoma.
En una realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua es una micela que comprende al menos un fármaco o resto biológicamente activo.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua es un liposoma que comprende al menos un fármaco o resto biológicamente activo. Preferiblemente, dicho liposoma se selecciona del grupo que consiste en acuasomas; vesículas de tensioactivos no iónicos, tales como niosomas y proniosomas; liposomas catiónicos, tal como LeciPlex; transferomas; etosomas; ufasomas; esfingosomas; y farmacosomas.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua es un polimerosoma que comprende al menos un fármaco o resto biológicamente activo.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende al menos un resto biológicamente activo o fármaco incrustado de forma no covalente en un polímero insoluble en agua. Preferiblemente, tal polímero insoluble en agua comprende un polímero seleccionado del grupo que consiste en 2-metacriloil-oxietil fosfoil colinas, poli(ácidos acrílicos), poli(acrilatos), poli(acrilamidas), poli(alquiloxi) polímeros, poli(amidas), poli(amidoaminas), poli(aminoácidos), poli(anhídridos), poli(aspartamidas), poli(ácidos butíricos), poli(ácidos glicólicos), tereftalatos de polibutileno, poli(caprolactonas), poli(carbonatos), poli(cianoacrilatos), poli(dimetilacrilamidas), poli(ésteres), poli(etilenos), poli(etilenglicoles), poli(óxidos de etileno), poli(fosfatos de etilo), poli(etiloxazolinas), poli(ácidos glicólicos), poli(hidroxietilacrilatos), poli(hidroxietiloxazolinas), poli(hidroximetacrilatos), poli(hidroxipropilmetacrilamidas), poli(metacrilatos de hidroxipropilo), poli(hidroxipropiloxazolinas), poli(iminocarbonatos), poli(ácidos lácticos), poli(ácidos láctico-co-glicólico), poli(metacrilamidas), poli(metacrilatos), poli(metiloxazolinas), poli(organofosfacenos), poli(ortoésteres), poli(oxazolinas), poli(propilenglicoles), poli(siloxanos), poli(uretanos), poli(alcoholes vinílicos), poli(vinilaminas), poli(vinilmetiléteres), poli(vinilpirrolidonas), siliconas, celulosas, carbometilcelulosas, hidroxipropilmetilcelulosas, quitinas, quitosanos, dextranos, dextrinas, gelatinas, ácidos hialurónicos y derivados, ácidos hialurónicos funcionalizados, mananos, pectinas, ramnogalacturonanos, almidones, almidones de hidroxialquilo, almidones de hidroxietilo y otros polímeros a base de carbohidratos, xilanos y copolímeros de los mismos.
En una realización preferida, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende al menos un fármaco o resto biológicamente activo incrustado de forma no covalente en ácido poli(láctico-co-glicólico) (PLGA).
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende al menos un resto biológicamente activo conjugado de forma covalente y reversible con un polímero insoluble en agua. Preferiblemente, tal polímero insoluble en agua comprende un polímero seleccionado del grupo que consiste en 2-metacriloil-oxietil fosfoil colinas, poli(ácidos acrílicos), poli(acrilatos), poli(acrilamidas), poli(alquiloxi) polímeros, poli(amidas), poli(amidoaminas), poli(aminoácidos), poli(anhídridos), poli(aspartamidas), poli(ácidos butíricos), poli(ácidos glicólicos), tereftalatos de polibutileno, poli(caprolactonas), poli(carbonatos), poli(cianoacrilatos), poli(dimetilacrilamidas), poli(ésteres), poli(etilenos), poli(etilenglicoles), poli(óxidos de etileno), poli(fosfatos de etilo), poli(etiloxazolinas), poli(ácidos glicólicos), poli(hidroxietilacrilatos), poli(hidroxietiloxazolinas), poli(hidroximetacrilatos), poli(hidroxipropilmetacrilamidas), poli(metacrilatos de hidroxipropilo), poli(hidroxipropiloxazolinas), poli(iminocarbonatos), poli(ácidos lácticos), poli(ácidos láctico-co-glicólico), poli(metacrilamidas), poli(metacrilatos), poli(metiloxazolinas), poli(organofosfacenos), poli(ortoésteres), poli(oxazolinas), poli(propilenglicoles), poli(siloxanos), poli(uretanos), poli(alcoholes vinílicos), poli(vinilaminas), poli(vinilmetiléteres), poli(vinilpirrolidonas), siliconas, celulosas, carbometilcelulosas, hidroxipropilmetilcelulosas, quitinas, quitosanos, dextranos, dextrinas, gelatinas, ácidos hialurónicos y derivados, ácidos hialurónicos funcionalizados, mananos, pectinas,
ramnogalacturonanos, almidones, almidones de hidroxialquilo, almidones de hidroxietilo y otros polímeros a base de carbohidratos, xilanos y copolímeros de los mismos.
Preferiblemente, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende al menos un resto biológicamente activo o fármaco seleccionado del grupo que consiste en antihistamínicos; anticuerpos anti-FGFR3 humanos; formas solubles del receptor del factor de crecimiento de fibroblastos 3 humano; inhibidores de tirosina quinasa; estatinas; agonistas de CNP; hormona de crecimiento; IGF-1; ANP; BNP; inhibidores de peptidasas y proteasas; e inhibidores de NPR-C.
Un antihistamínico preferido es meclozina.
Un inhibidor de tirosina quinasa preferido es NVP-BGJ398.
Una estatina preferida es rosuvastatina.
Un agonista de CNP preferido para al menos un fármaco adicional es vosoritida.
Los inhibidores preferidos de peptidasas y proteasas son inhibidores de NEP y furina.
Los inhibidores preferidos para NEP son tiorfán y candoxatril.
Los inhibidores preferidos de NPR-C son el fragmento de SEQ ID NO:98 (FGIPMDRIGRNPR) y el anticuerpo B701. Los inhibidores preferidos de las tirosina quinasas son como se describen en las patentes de EE. UU. 6329375 y 6344459. En una realización, al menos un resto biológicamente activo o fármaco adicional en forma de una liberación controlada insoluble en agua comprende un resto o fármaco antihistamínico.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende un resto o fármaco de anticuerpo anti-FGFR3 humano.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende formas solubles del resto o fármaco de receptor del factor de crecimiento de fibroblastos 3 humano (sFGFR3).
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende un resto o fármaco de inhibidor de tirosina quinasa.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende un resto o fármaco de estatina.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende un resto o fármaco de hormona del crecimiento.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende un resto o fármaco de agonista de CNP.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende un resto o fármaco de IGF-1.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende un resto o fármaco de ANP.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende un resto o fármaco de BNP.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende un resto o fármaco de inhibidor de peptidasas y proteasas.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada insoluble en agua comprende un resto o fármaco de inhibidor de NPR-C.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada es soluble en agua.
En una realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación
controlada soluble en agua comprende al menos un resto biológicamente activo conjugado covalentemente a través de un enlace reversible a un resto polimérico soluble en agua, ya sea directamente o a través de un resto espaciador.
Preferiblemente, dicho resto polimérico soluble en agua comprende un polímero seleccionado del grupo que consiste en 2-metacriloil-oxietil fosfoil colinas, poli(ácidos acrílicos), poli(acrilatos), poli(acrilamidas), poli(alquiloxi) polímeros, poli(amidas), poli(amidoaminas), poli(aminoácidos), poli(anhídridos), poli(aspartamidas), poli(ácidos butíricos), poli(ácidos glicólicos), tereftalatos de polibutileno, poli(caprolactonas), poli(carbonatos), poli(cianoacrilatos), poli(dimetilacrilamidas), poli(ésteres), poli(etilenos), poli(etilenglicoles), poli(óxidos de etileno), poli(fosfatos de etilo), poli(etiloxazolinas), poli(ácidos glicólicos), poli(hidroxietilacrilatos), poli(hidroxietil-oxazolinas), poli(hidroximetacrilatos), poli(hidroxipropilmetacrilamidas), poli(metacrilatos de hidroxipropilo), poli(hidroxipropiloxazolinas), poli(iminocarbonatos), poli(ácidos lácticos), poli(ácidos láctico-co-glicólico), poli(metacrilamidas), poli(metacrilatos), poli(metiloxazolinas), poli(organofosfacenos), poli(ortoésteres), poli(oxazolinas), poli(propilenglicoles), poli(siloxanos), poli(uretanos), poli(alcoholes vinílicos), poli(vinilaminas), poli(vinilmetiléteres), poli(vinilpirrolidonas), siliconas, celulosas, carbometilcelulosas, hidroxipropilmetilcelulosas, quitinas, quitosanos, dextranos, dextrinas, gelatinas, ácidos hialurónicos y derivados, ácidos hialurónicos funcionalizados, mananos, pectinas, ramnogalacturonanos, almidones, almidones de hidroxialquilo, almidones de hidroxietilo y otros polímeros a base de carbohidratos, xilanos y copolímeros de los mismos.
En otra realización, al menos un resto biológicamente activo adicional en forma de un compuesto de liberación controlada soluble en agua se conjuga covalentemente a través de un enlace estable con un resto de unión a albúmina. Preferiblemente, dicho resto que se une a la albúmina es un resto alquilo C8-24 o un derivado de ácido graso. Los derivados de ácidos grasos preferidos son los descritos en WO 2005/027978 A2 y WO 2014/060512 A1.
Preferiblemente, al menos un resto biológicamente activo adicional en forma un de compuesto de liberación controlada soluble en agua comprende un resto biológicamente activo seleccionada del grupo que consiste en antihistamínicos; anticuerpos anti-FGFR3 humanos; formas solubles del receptor del factor de crecimiento de fibroblastos 3 humano; inhibidores de tirosina quinasa; estatinas; agonistas de CNP; hormona de crecimiento; IGF-1; ANP; BNP; inhibidores de peptidasas y proteasas; e inhibidores de NPR-C.
Un antihistamínico preferido es meclozina.
Un inhibidor de tirosina quinasa preferido es NVP-BGJ398.
Una estatina preferida es rosuvastatina.
Un agonista de CNP preferido para al menos un fármaco adicional es vosoritida.
Los inhibidores preferidos de peptidasas y proteasas son inhibidores de NEP y furina.
Los inhibidores preferidos de NEP son tiorfano y candoxatril.
Los inhibidores preferidos de NPR-C son el fragmento de SEQ ID NO:98 (FGIPMDRIGRNPR) y el anticuerpo B701.
Los inhibidores preferidos de las tirosina quinasas son como se describen en las patentes de EE. UU. 6329375 y 6344459.
En una realización, al menos un resto biológicamente activo o fármaco adicional en forma de una liberación controlada soluble en agua comprende un resto o fármaco antihistamínico.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada soluble en agua comprende un resto o fármaco de anticuerpo anti-FGFR3 humano.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada soluble en agua comprende formas solubles del resto o fármaco de receptor del factor de crecimiento de fibroblastos 3 humano (sFGFR3).
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada soluble en agua comprende un resto o fármaco de inhibidor de tirosina quinasa.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada soluble en agua comprende un resto o fármaco de estatina.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada soluble en agua comprende un resto o fármaco de hormona del crecimiento. Un compuesto de la hormona de crecimiento de liberación controlada soluble en agua preferido es el compuesto 2 del ejemplo 2 de WO2016/079114A1. Por consiguiente, un compuesto preferido de la hormona de crecimiento de liberación controlada soluble en agua tiene la siguiente estructura:

En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada soluble en agua comprende un resto o fármaco de agonista de CNP.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada soluble en agua comprende un resto o fármaco de IGF-1.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada soluble en agua comprende un resto o fármaco de ANP.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada soluble en agua comprende un resto o fármaco de BNP.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada soluble en agua comprende un resto o fármaco de inhibidor de peptidasas y proteasas.
En otra realización, al menos un resto biológicamente activo o fármaco adicional en forma de un compuesto de liberación controlada soluble en agua comprende un resto o fármaco de inhibidor de NPR-C.
Otro aspecto de la presente invención es la composición farmacéutica que comprende al menos un profármaco de CNP y al menos un resto biológicamente activo o fármaco adicional de la presente invención para su uso como un medicamento. Otro aspecto de la presente invención es la composición farmacéutica que comprende al menos un profármaco de CNP y al menos un resto biológicamente activo o fármaco adicional de la presente invención para su uso en el tratamiento de un paciente que padece un trastorno que se beneficia de la estimulación del crecimiento.
Preferiblemente, el paciente es un paciente mamífero, más preferiblemente un paciente humano.
Preferiblemente, los trastornos que se benefician de la estimulación del crecimiento se seleccionan del grupo que comprende acondroplasia, hipocondroplasia, baja estatura, enanismo, osteocondrodisplasias, displasia tanatofórica, osteogénesis imperfecta, acondrogénesis, condrodisplasia punctata, acondroplasia homocigota, displasia campomélica, hipofosfatasia letal congénita, tipo letal perinatal de osteogénesis imperfecta, síndrome de costillas cortas-polidactilia, tipo rizomélico de condrodisplasia punctata, displasia metafisaria tipo Jansen, displasia espondiloepifisaria congénita, atelosteogénesis, displasia diastrófica, fémur corto congénito, displasia mesomélica tipo Langer, displasia mesomélica tipo Nievergelt, síndrome de Robinow, síndrome de Reinhardt, acrodisostosis, disostosis periférica, displasia de Kniest, fibrocondrogénesis, síndrome de Roberts, displasia acromesomélica, micromelia, síndrome de Morquio, síndrome de Kniest, displasia metatrófica, y displasia espondiloepimetafisaria. Lo más preferiblemente, el trastorno que se beneficia de la estimulación del crecimiento es la acondroplasia.
Otro aspecto de la presente invención es un método de tratamiento de un paciente que sufre un trastorno que se beneficia de la estimulación del crecimiento mediante la administración de la composición farmacéutica de la presente invención. Preferiblemente, el paciente es un paciente mamífero, más preferiblemente un paciente humano.
Preferiblemente, los trastornos que se benefician de la estimulación del crecimiento se seleccionan del grupo que comprende acondroplasia, hipocondroplasia, baja estatura, enanismo, osteocondrodisplasias, displasia tanatofórica, osteogénesis imperfecta, acondrogénesis, condrodisplasia punctata, acondroplasia homocigota, displasia campomélica, hipofosfatasia letal congénita, tipo letal perinatal de osteogénesis imperfecta, síndrome de costillas cortas-polidactilia, tipo rizomélico de condrodisplasia punctata, displasia metafisaria tipo Jansen, displasia espondiloepifisaria congénita, atelosteogénesis, displasia diastrófica, fémur corto congénito, displasia mesomélica tipo Langer, displasia mesomélica tipo Nievergelt, síndrome de Robinow, síndrome de Reinhardt, acrodisostosis, disostosis periférica, displasia de Kniest, fibrocondrogénesis, síndrome de Roberts, displasia acromesomélica, micromelia, síndrome de Morquio, síndrome de Kniest, displasia metatrófica, y displasia espondiloepimetafisaria. Lo más preferiblemente, el trastorno que se beneficia de la estimulación del crecimiento es la acondroplasia.
El resto polipeptídico del profármaco de CNP puede prepararse mediante métodos estándar de síntesis de péptidos en fase sólida, por ejemplo, mediante la química de Boc (R. B. Merrifield, J. Am. Chem. Soc., 85(14): 2149-2154 (1963)). Alternativamente, se puede emplear la química Fmoc (fluorenilmetoxicarbonilo).
Se pueden emplear métodos conocidos en la técnica para mejorar la pureza y/o el rendimiento, incluyendo el uso de pseudoprolina u otros bloques de construcción de dipéptidos, el acoplamiento de fragmentos y otros (J.Wade et al., Lett. Pept. Sci., 7(2):107-112 (2000); Y.Fujiwara et al., Chem. Pharm.Bull., 44(7):1326-1331 (1996); P. Cherkupally et al., Eur. J. Org. Chem., 6372-6378 (2013)).
Alternativamente, el resto polipeptídico del profármaco CNP puede producirse mediante procesos de síntesis recombinante.
Figura 1 (no de acuerdo con la invención): Estructura del CNP de acuerdo con SEQ ID NO:1.
Ejemplos
Materiales y métodos
El CNP SEQ ID No:1 se obtuvo de Bachem AG, Bubendorf, Suiza (CNP-22, humano, No. de catálogo: H-1296). El CNP-34 SEQ ID No:40 y CNP-38 SEQ ID No:24 se obtuvieron de CASlO ApS, Kongens Lyngby, Dinamarca.
El CNP-38 protegido por cadena lateral en resina TCP que tiene el extremo N-terminal protegido por Boc y cadena lateral protegida por ivDde de Lys26 (sintetizado mediante estrategia Fmoc) se obtuvo de CASLO ApS, Kongens Lyngby, Dinamarca.
El CNP-34 protegido por cadena lateral en resina CTC Tentagel que tenía el extremo N-terminal protegido por Boc y cadena lateral protegida por ivDde de Lys12, Lys16 o Lys22 (sintetizado mediante estrategia Fmoc) se obtuvo de Peptide Specialty Laboratories GmbH, Heidelberg, Alemania. El CNP-38 protegido por cadena lateral en resina TCP Tentagel que tenía el extremo N-terminal libre (sintetizado por estrategia Fmoc) se obtuvo de Peptide Specialty Laboratories GmbH, Heidelberg, Alemania.
La metoxi PEG amina 5 kDa se obtuvo de Rapp Polymere GmbH, Tuebingen, Alemania. Todos los demás PEG utilizados en este trabajo se adquirieron de NOF Europe N.V., Grobbendonk, Bélgica.
El FmocN-Me-Asp(OtBu)-OH se obtuvo de Bachem AG, Bubendorf, Suiza. El ácido S-tritil-6-mercaptohexanoico se adquirió en Polypeptide, Estrasburgo, Francia. El HATU se obtuvo de Merck Biosciences GmbH, Schwalbach/Ts, Alemania.
El alcohol 2,4-dimetilbencílico se obtuvo de abcr GmbH, Karlsruhe, Alemania.
El Fmoc-N-Me-Asp(OBn)-OH se obtuvo de Peptide International Inc., Louisville, KY, EE. UU.
La endopeptidasa neutra (NEP) se obtuvo de Enzo Life Sciences GmbH, Lorrach, Alemania.
Todos los demás productos químicos y reactivos se adquirieron en Sigma Aldrich GmbH, Taufkirchen, Alemania.
Se utilizaron jeringas equipadas con fritas de polietileno (MultiSynTech GmbH, Witten, Alemania) como recipientes de reacción o para los pasos de lavado de las resinas peptídicas.
Procedimiento general para la eliminación del grupo protector ivDde de los CNP protegidos por cadena lateral en resina La resina se pre-suavizó en DMF durante 30 minutos y el disolvente se desechó. El grupo ivDde se eliminó incubando la resina con DMF/hidrato de hidracina 4/1 (v/v, 2,5 mL/g de resina) durante 8 x 15 min. Para cada paso se utilizó una nueva solución de DMF/hidrato de hidracina. Finalmente, la resina se lavó con DMF (10 x), DCM (10 x) y se secó al vacío.
Purificación mediante RP-HPLC:
Para la RP-HPLC preparativa se utilizó un controlador Waters 600 y un Detector de Absorbancia Dual 2487, equipado con las siguientes columnas: Waters XBridge™ BEH300 Prep C18 5 |jm,150 x 10 mm, tasa de flujo de 6 mL/min, o Waters XBridge™ BEH300 Prep C1810 jm , 150 x 30 mm, tasa de flujo de 40 mL/min. Se utilizaron gradientes lineales del sistema de disolventes A (agua con 0,1% de TFA v/v o 0,01% de HC1 conc. v/v) y del sistema de disolventes B (acetonitrilo con 0,1% de TFA v/v o 0,01% de HC1 conc. v/v).
Las fracciones de HPLC que contenían el producto se agruparon y liofilizaron si no se indica lo contrario.
Cromatografía ultrarrápida
Las purificaciones por cromatografía ultrarrápida se realizaron en un sistema Isolera One de Biotage AB, Suecia, utilizando cartuchos de sílice Biotage KP-Sil y n-heptano y acetato de etilo como eluyentes. Los productos se detectaron a 254 nm.
Métodos analíticos
La cromatografía líquida de ultra-alta resolución (“UPLC”, por sus siglas en inglés)-espectrometría de masas (“MS”, por sus siglas en inglés) se realizó en un sistema Waters Acquity equipado con una columna Waters BEH300 C18 (2,1 x 50 mm, tamaño de partícula de 1,7 jm , flujo: 0,25 mL/min; disolvente A: agua con 0,04% de TFA (v/v), disolvente B: acetonitrilo con 0,05% de TFA (v/v)) acoplado a un espectrómetro de masas LTQ Orbitrap Discovery de Thermo Scientific o acoplado a un instrumento Waters Micromass ZQ.
La cromatografía de exclusión por tamaño (“SEC”, por sus siglas en inglés) se realizó con un sistema AEKTAbasic de Amersham Bioscience equipado con una columna Superdex 2005/150 GL (Amersham Bioscience/GE Healthcare) dotada de un filtro de entrada de 0,45 jm , a menos que se indique lo contrario. Como fase móvil se utilizó fosfato de sodio 20 mM, NaCl 140 mM, pH 7,4.
Debido a la naturaleza reversible de la unión de -L1-a-D, las mediciones de la estabilidad de NEP y de la afinidad del receptor se hicieron utilizando análogos estables de los profármacos de CNP de la presente invención, es decir, se hicieron utilizando estructuras similares a las de los profármacos de CNP de la presente invención que en lugar de una unión reversible de -Z a -D tienen una unión estable.
Esto era necesario, debido a que los profármacos de CNP de la presente invención liberarían CNP en el curso del experimento y dicho CNP liberado habría influido en el resultado.
Cuantificación de las concentraciones plasmáticas totales de CNP-38
Las concentraciones plasmáticas totales de CNP-38 (CNP-38 conjugado y liberado) se determinaron mediante la cuantificación del péptido característico N-terminal (secuencia: LQEHPNAR) y péptido característico C-terminal (secuencia: IGSMSGLGC) tras la digestión tríptica.
El análisis LC-MS se llevó a cabo utilizando un UPLC Agilent 1290 acoplado a un espectrómetro de masas Agilent 6550 iFunnel Q-TOF mediante una sonda ESI. La cromatografía se realizó en una columna analítica Waters Acquity BEH300 C18 (D.I. 50 x 2,1 mm, tamaño de partícula de 1,7 jm ) con prefiltro a una tasa de flujo de 0,25 mL/min (T = 25 °C). Se utilizó agua (grado UPLC) con 0,2% de ácido fórmico (v/v) como fase móvil A y acetonitrilo (grado UPLC) con 0,2% de ácido fórmico como fase móvil B. El sistema de gradiente comprendía un breve paso isocrático a los parámetros iniciales de 0,1% de B durante 3,0 min, seguido de un aumento lineal de 0,1% de B a 16 % de B en 17 min. El análisis de masas se realizó en el modo de monitoreo de iones simples (“SIM”, por sus siglas en inglés), monitoreando los iones m/z 482,75 [M+2H]2+ (N-terminal) y m/z 824,36 [M+H]1+ (C-terminal). Como estándar interno se utilizó el péptido CNP-38 deuterado.
Los estándares de calibración del conjugado CNP-38 en plasma en blanco se prepararon de la siguiente manera: El plasma cinomolecular descongelado con Li-heparina se homogeneizó primero y posteriormente se centrifugó durante 5 minutos. La formulación del conjugado CNP-38 se diluyó hasta una solución de trabajo de 10 jg/mL (eq. del conjugado CNP-38) en DMSO y se introdujo en plasma en blanco a concentraciones entre 9,3 ng/100 jL (eq. del conjugado CNP-38) y 139,5 ng/100 jL (eq. del conjugado CNP-38). Estas soluciones se utilizaron para generar una curva de calibración. Las curvas de calibración se ponderaron 1/x2 para ambos péptidos característicos (N- y C-Terminal). Para el control de calidad, se prepararon tres muestras de control de calidad con contenidos de 116,2 ng/100 jL (QC alto, eq. del conjugado CNP-38), 69,75 ng/100 jL (QC medio, eq. del conjugado CNP-38) y 23,25 ng /100 jL (QC bajo, eq. del conjugado CNP-38).
Para la preparación de la muestra, la precipitación de las proteínas se llevó a cabo mediante la adición de 300 jL de metanol pre-enfriado (0 °C) a 100 jL de la muestra de plasma. Se transfirieron 200 jL del sobrenadante a una nueva placa de pocillos y se evaporaron hasta sequedad (bajo una suave corriente de nitrógeno a 35 °C). Se utilizaron 100 jL de disolvente de reconstitución (solución amortiguadora de digestión Thermo, número de pedido 60109-101, Thermo Fisher Scientific GmbH, Dreieich, Alemania) para disolver el residuo. Se disolvieron 20 jg de tripsina (número de pedido
V5111, Promega GmbH, Mannheim, Alemania) en 20 |jL de ácido acético 10 mM. Se añadieron 2 |jL de la solución de tripsina a cada cavidad.
Tras 4 horas de incubación a 37 °C (baño de agua), se añadieron 5 jiL de una solución de TCEP 0,5 M a cada cavidad y se incubaron de nuevo durante 5 minutos a 96 °C. Después de que las muestras se enfriaran a temperatura ambiente, se añadieron 3 jiL de acetonitrilo. Los eluidos se transfirieron dentro de viales. Se inyectaron 10 ji L en el sistema UPLC-MS. Ejemplo 1 (no según la invención)
Síntesis del reactivo enlazador 1f
El reactivo enlazador 1f se sintetizó de acuerdo con el siguiente esquema:

A una solución de N-metil-N-Boc-etilendiamina (2 g, 11,48 mmol) y NaCNBH3 (819 mg, 12,63 mmol) en MeOH (20 mL) se le añadió 2,4,6-trimetoxibenzaldehído (2,08 g, 10,61 mmol) en porciones. La mezcla se agitó a TA durante 90 min, se acidificó con HCl 3 M (4 mL) y se agitó 15 min más. La mezcla de reacción se añadió a una solución saturada de NaHCO3 (200 mL) y se extrajo 5 veces con CH2O 2. Las fases orgánicas combinadas se secaron sobre Na2SO4 y los disolventes se evaporaron a presión reducida. La N-metil-N-Boc-N'-Tmob-etilendiamina 1a resultante se secó al vacío y se utilizó en el siguiente paso de la reacción sin purificación adicional.
Rendimiento: 3,76 g (11,48 mmol, 89% de pureza, 1a : producto doblemente protegido por Tmob = 8:1)
MS: m/z 355,22 = [M+H]+, (masa monoisotópica calculada =354,21).
A una solución de 1a (2 g, 5,65 mmol) en CH2O 2 (24 mL) COMU (4,84 g, 11,3 mmol), N-Fmoc-N-Me-Asp(OBn)-OH (2,08
g, 4,52 mmol) y se le añadió 2,4,6-colidina (2,65 mL, 20,34 mmol). La mezcla de reacción se agitó durante 3 h a TA, se diluyó con CH2O 2 (250 mL) y se lavó 3 veces con H2SO4 0,1 M (100 mL) y 3 veces con salmuera (100 mL). Las fases acuosas se volvieron a extraer con CH2O 2 (100 mL). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y el residuo se concentró hasta un volumen de 24 mL. 1b se purificó mediante cromatografía ultrarrápida.
Rendimiento: 5,31 g (148%, 6,66 mmol)
MS: m/z 796,38 = [M+H]+, (masa monoisotópica calculada =795,37)
A una solución de 1b (5,31 g, máx. 4,52 mmol) en THF (60 mL) se le añadió DBU (1,8 mL, 3% v/v). La solución se agitó durante 12 min a TA, se diluyó con CH2O 2 (400 mL) y se lavó 3 veces con H2SO40,1 M (150 mL) y 3 veces con salmuera (150 mL). Las fases acuosas se volvieron a extraer con CH2O 2 (100 mL). Las fases orgánicas combinadas se secaron sobre Na2SO4 y se filtraron. 1 c se aisló tras la evaporación del disolvente y se usó en la siguiente reacción sin purificación adicional.
MS: m/z 574,31 = [M+H]+, (masa monoisotópica calculada = 573,30)
1c (5,31 g, 4,52 mmol, crudo) se disolvió en acetonitrilo (26 mL) y se añadieron COMU (3,87 g, 9,04 mmol), ácido 6-tritilmercaptohexanoico (2,12 g, 5,42 mmol) y 2,4,6-colidina (2,35 mL, 18,08 mmol). La mezcla de reacción se agitó durante 4 h a TA, se diluyó con CH2O 2 (400 mL) y se lavó 3 veces con H2SO40,1 M (100 mL) y 3 veces con salmuera (100 mL). Las fases acuosas se volvieron a extraer con CH2O 2 (100 mL). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y 1d se aisló tras la evaporación del disolvente. El producto 1d se purificó mediante cromatografía ultrarrápida.
Rendimiento: 2,63 g (62%, 94% de pureza)
MS: m/z 856,41 = [M+H]+, (masa monoisotópica calculada = 855,41)
A una solución de 1d (2,63 g, 2,78 mmol) en i-PrOH (33 mL) y H2O (11 mL) se añadió LiOH (267 mg, 11,12 mmol) y la mezcla de reacción se agitó durante 70 min a TA. La mezcla se diluyó con CH2O 2 (200 mL) y se lavó 3 veces con H2SO4 0,1 M (50 mL) y 3 veces con salmuera (50 mL). Las fases acuosas se volvieron a extraer con CH2O 2 (100 mL). Las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y 1e se aisló tras la evaporación del disolvente. 1e se purificó mediante cromatografía ultrarrápida.
Rendimiento: 2,1 g (88%)
MS: m/z 878,4 = [M+Na]+, (masa monoisotópica calculada = 855,40)
A una solución de 1e (170 mg, 0,198 mmol) en DCM anhidro (4 mL) se le añadió DCC (123 mg, 0,59 mmol), y una cantidad catalítica de DMAP. Después de 5 min se añadió N-hidroxisuccinimida (114 mg, 0,99 mmol) y la mezcla de reacción se agitó a TA durante 1 h. La mezcla de reacción se filtró, el disolvente se eliminó al vacío y el residuo se absorbió en acetonitrilo al 90% más TFA al 0,1% (3,4 mL). La mezcla en bruto se purificó mediante RP-HPLC. Las fracciones del producto se neutralizaron con solución amortiguadora de fosfato 0,5 M con pH 7,4 y se concentraron. La fase acuosa restante se extrajo con DCM y se aisló 1f al evaporar el disolvente.
Rendimiento: 154 mg (81%)
MS: m/z 953,4 = [M+H]+, (masa monoisotópica calculada = 952,43)
Ejemplo 2 (no según la invención)
Síntesis de tio l 2 de monoenlazador n eK4,eK10-CNP, tio l 2c de monoenlazador NEK4-CNP y tio l 2d de monoenlazador NeK10-CNP

El tiol 2 de monoenlazador NeK4/eK10-CNP (mezcla de regioisómeros con enlazador conjugado en el grupo amino de la cadena lateral de Lys4 o Lys10) se prepara disolviendo CNP-22 (5,2 |jmol) en 0,6 mL de DMSO. Se añaden 0,15 mL de solución amortiguadora de borato 0,375 M, ajustada a pH 8,5 con hidrato de tetrabutilamonio, 60 jL de DIPEA y 1f (6,1 mg, 7,1 jmol) en 0,34 mL de DMSO y se agita la mezcla durante 30 min a TA. La mezcla de reacción se diluye con 2 mL de acetonitrilo/agua 1/1 (v/v) y 200 jL de AcOH, y se aísla el conjugado de monoenlazador NeK4/eK10-CNP protegido de la mezcla de reacción mediante RP-HPLC.
Se pueden utilizar gradientes de RP-HPLC optimizados para aislar el tiol 2a de monoenlazador NeK4-CNP y el tiol 2b de monoenlazador NeK10-CNP.
La eliminación de los grupos protectores se ve afectada por el tratamiento de las fracciones del producto liofilizado con 0,6 mL de 90/10/2/2 (v/v/v/v) HFIP/TFA/TES/agua durante 1 h a TA. El tiol 2 de monoenlazador NeK4/eK10-CNP desprotegido se purifica mediante RP-HPLC. La identidad y la pureza del producto se determinan mediante ESI-LCMS.
El tiol 2c de monoenlazador NeK4-CNP desprotegido y el tiol 2d de monoenlazador NeK10-CNP pueden obtenerse igualmente a partir de 2a y 2b, respectivamente.
Ejemplo 3 (no según la invención)
Síntesis de tio l 3 de monoenlazador NaG1-CNP

El tiol 3 de monoenlazador NaG1-CNP se prepara disolviendo CNP-22 (5,2 jmol) en 0,6 mL de DMSO. Se añaden 0,25 mL de solución amortiguadora de fosfato 0,5 M con pH 7,4 y 1f (6,1 mg, 7,1 jmol) en 0,34 mL de DMSO y se agita la mezcla durante varias horas a TA. La mezcla de reacción se diluye con 2 mL de acetonitrilo/agua 1/1 (v/v) y 200 jL de AcOH, y se aísla el tiol de monoenlazador NaG1-CNP protegido de la mezcla de reacción mediante RP-HPLC.
La eliminación de los grupos protectores se ve afectada por el tratamiento de las fracciones del producto liofilizado con 0,6 mL de 90/10/2/2 (v/v/v/v) HFIP/TFA/TES/agua durante 1 h a TA. El tiol 3 de monoenlazador NaG1-CNP desprotegido se purifica mediante RP-HPLC. La identidad y la pureza del producto se determinan mediante ESI-LCMS.
Ejemplo 4 (no según la invención)
PEGilación de los tioles 2c, 2d y 3 de monoenlazador CNP


Se disuelve 1 |jmol de tiol 2c de monoenlazador CNP en 0,5 mL de acetonitrilo / solución amortiguadora de succinato 0,2 M con pH 3,8 1/1 (v/v), se añade 1,2 jm ol de PEG-maleimida lineal 40 kDa y la mezcla se agita a TA. La reacción se apaga añadiendo 20 jL de AcOH y el conjugado 4 de CNP se purifica mediante RP-HPLC preparativa.
Los conjugados de CNP 5 y 6 se preparan igualmente a partir de 1 jmol de tioles 2d y 3 de monoenlazador CNP.
El contenido de CNP se determina mediante un análisis cuantitativo de aminoácidos tras una hidrólisis total en condiciones ácidas.
Ejemplo 5 (no según la invención)
Cinética de liberación in vitro
Los conjugados 4, 5 y 6 de CNP se disuelven en fosfato de sodio 60 mM, EDTA 3 mM, Tween-20 0,01%, pH 7,4 a una concentración de aproximadamente 2 mg/mL y se filtran de forma estéril. Las mezclas se incuban a 37 °C. Se extraen alícuotas en determinados momentos y se analizan mediante RP-HPLC y ESI-MS. Las señales UV que se correlacionan con el CNP liberado se integran y se trazan en función del tiempo de incubación.
Se aplica un software de ajuste de curvas para estimar el correspondiente tiempo medio de liberación.
Ejemplo 6
Farmacocinética y producción de GMPc en ratas
Se inyectan dosis equimolares de CNP-22, conjugados de 4, 5 o 6 de CNP por vía iv y sc en ratas normales. Los niveles plasmáticos de CNP y cGMP a lo largo del tiempo se determinan según se describe en la literatura (patente de EE. UU.
8.377.884 B2).
Ejemplo 7
Síntesis de ácido 6-mercaptohexanoico protegido con Dmb 7
El compuesto 7 se sintetizó de acuerdo con el siguiente esquema:

A una solución de ácido 6-mercaptohexanoico (7,10 g, 47,90 mmol) en ácido trifluoroacético (20 mL), se le añadió alcohol 2,4-dimetilbenzílico (13,5 g, 95,80 mmol). La mezcla se agitó a TA durante 60 minutos y posteriormente se eliminó el ácido trifluoroacético al vacío. El residuo se disolvió en una mezcla de 95,8 mL de LiOH (3 M) y THF (81 mL) y se agitó a TA durante 60 min. El disolvente se eliminó al vacío y el residuo acuoso se extrajo 3 veces con EtOAc (200 mL). Las fases orgánicas combinadas se secaron sobre MgSO4 y el disolvente se eliminó al vacío. 7 se purificó mediante RP-HPLC. Rendimiento: 2,27 g (8,52 mmol, 18%)
MS: m/z 267,01 = [M+H]+, (masa monoisotópica calculada = 266,13)
Ejemplo 8
Síntesis del reactivo enlazador 8c
El reactivo enlazador 8c se sintetizo de acuerdo con el siguiente esquema:

A una solución de 1b (21,6 g, 27,18 mmol) en isopropanol (401 mL) se le añadió agua (130 mL) y LiOH (3,90 g, 163,06 mmol). La mezcla de reacción se agitó durante 3 h a TA, posteriormente se diluyó con tolueno (300 mL) y se lavó 3 veces con HCl 0,1 M (200 mL). Las fases acuosas combinadas se lavaron 3 veces con tolueno (100 mL). La fase acuosa se alcalinizó con NaOH 4 M (4 mL) a un pH de 8,5 y se extrajo 8 veces con CH2O 2 (200 mL). Las fases de CH2O 2 combinadas se lavaron con salmuera (50 mL), se secaron sobre Na2SO4. 8b se aisló tras la evaporación del disolvente y se usó en la siguiente reacción sin purificación adicional.
Rendimiento: 11,89 g (24,59 mmol, 90%)
MS: m/z 484,16 = [M+H]+, (masa monoisotópica calculada = 483,26)
A una solución de 7 (293 mg, 1,10 mmol) y PyBOP (572 mg, 1,10 mmol) en THF (10 mL) se le añadió DIEA (0,52 mL, 3,00 mmol) bajo una atmósfera de N2. La mezcla de reacción se agitó durante 60 min a TA. Se añadió una solución de 8b (484 mg, 1,00 mmol) en THF (2 mL) y la reacción se agitó durante otros 60 min. La reacción se apagó con una solución de ácido cítrico 2 M (10 mL) y el THF se eliminó al vacío. La fase acuosa resultante se extrajo 2 veces con EtOAc (15 mL) y las capas orgánicas combinadas se lavaron con agua (10 mL) y salmuera (10 mL), y se secaron sobre MgSO4. El disolvente se eliminó al vacío y 8c se purificó mediante HPLC RP.
Ejemplo 9
Síntesis del reactivo enlazador 9
El reactivo enlazador 9 se sintetizó de acuerdo con el siguiente esquema:

A una solución de 8b (2,00 g, 4,14 mmol) y Fmoc-Cl (1,07 g, 4,14 mmol) en dioxano (20 mL) se le añadió una solución de Na2CO31 M (20 mL). La mezcla de reacción se agitó durante 40 min a TA. Se añadió agua (100 mL) y éter dietílico (100 mL) y la fase acuosa se extrajo 2 veces con éter dietílico (100 mL). La fase acuosa se acidificó con HCl conc. hasta pH 1
y se extrajo de nuevo 3 veces con éter dietílico. Las fases orgánicas combinadas se secaron sobre Na2SO4 y el disolvente se eliminó al vacío. 9 se utilizó en el siguiente paso sin purificación adicional.
Rendimiento: 2,63 g (3,73 mmol, 90%)
MS: m/z 728,32 = [M+Na]+, (masa monoisotópica calculada = 705,33)
Ejemplo 10 (no según la invención)
Síntesis del conjugado 10f de Lys26 CNP-38 PEG2x20 kDa reversible
El conjugado 10f se sintetizó de acuerdo con el siguiente esquema:

10a
1


Se desprotegieron 2,00 g (0,21 mmol) de CNP-38 protegido por cadena lateral en resina CTC que tenía el extremo N-terminal protegido por Boc y cadena lateral protegida por ivDde de Lys26 de acuerdo con el procedimiento proporcionado en Materiales y Métodos para obtener 10a. Una solución del reactivo enlazador 8c (336 mg, 0,46 mmol), PyBOP (239 mg, 0,46 mmol) y DIEA (182 |j L,1,04 mmol) en DMF (5 mL) se incubó durante 10 min a TA, y posteriormente se añadió a la resina 10a. La suspensión se agitó durante 2 h a TA. La resina se lavó 10 veces con DMF (10 mL) y 10 veces con DCM (10 mL), y se secó al vacío durante 15 min. La escisión del péptido de la resina y la eliminación de los grupos protectores se logró mediante el tratamiento de la resina con 15 mL de cóctel de escisión pre-enfriado (-18 °C) 68,5/10/5/3,5/1 (v/p/v/v/v/v) TFA/DTT/tiofenol/agua/TIPS. La mezcla se dejó calentar hasta TA y se agitó durante 60 min. La resina se filtró y el producto en bruto 10c se precipitó en éter dietílico pre-enfriado (-18 °C). El precipitado se disolvió en ACN/agua y se purificó mediante RP-HPLC. Las fracciones de HPLC combinadas se usaron directamente en el siguiente paso.
MS: m/z 1124,60 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1124,59).
A las fracciones combinadas de HPLC de 10c (250 mL) se añadieron 40 mL de solución amortiguadora de ácido cítrico 0,5 M (pH = 5,00) y 7 mL de una solución 0,01 M de 2,2'-ditiobis(piridina-N-óxido) en 1/1 (v/v) acetonitrilo/agua. Tras la incubación durante 5 minutos a TA, la reacción se completó. La mezcla se diluyó con 500 mL de agua que contenía 0,1% de TFA (v/v) y se acidificó con AcOH (20 mL) hasta un pH de aproximadamente 2. 10d se purificó mediante RP-HPLC. Las fracciones del producto se combinaron y se liofilizaron.
Rendimiento: 101 mg (17,3 jmol, 9%) CNP-38-enlazador-Dmb * 10 TFA
MS: m/z 1124,10 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1124,09).
La escisión del grupo protector Dmb se consiguió añadiendo 30 mL de cóctel de escisión 100/5/3/2/1 (v/v/w/v/v) TFA/MSA/DTT/agua/tioanisol pre-enfriado (-18 °C) a 10d (101 mg, 17,3 jmol) y agitando durante 3 h a 0 °C. El producto en bruto 10e se precipitó en éter dietílico pre-enfriado (-18 °C). El precipitado se disolvió en agua que contenía 0,1 % de TFA (v/v) y se incubó durante 10 minutos para hidrolizar cualquier éster de TFA. 10e se purificó mediante RP-HPLC.
Rendimiento: 46 mg (8,34 jmol, 48%) CNP-38-enlazador-tiol * 10 TFA
MS: m/z 1094,58 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1094,57).
A una solución de 10e (46 mg, 8,43 jmol) en 1,15 mL de agua que contenía 0,1% de TFA (v/v) se añadió una solución de maleimida PEG 2x20 kDa (Sunbright GL2-400MA, 870 mg, 21,75 jmol) en 4,35 mL de agua que contenía 0,1% de TFA (v/v), seguida de solución amortiguadora de ácido láctico 0,5 M (1,07 mL, pH = 4,20). La mezcla se agitó a TA durante 4 h. El conjugado 10f se purificó mediante RP-HPLC.
Rendimiento: 233 mg (5,21 jmol, 62%) de conjugado 10f * 10 HCl
Ejemplo 11
Síntesis del conjugado 11 i de Lys26 CNP-38 PEG4x10 kDa reversible
El conjugado 11 i se sintetizó de acuerdo con el siguiente esquema:


S Trt STrt
TFA, DTT, tioamsol, fenol, agua, TIPS
11e




A una solución de 9 (353 mg, 0,50 mmol) y PyBOP (260 mg, 0,50 mmol) en DMF (9 mL) se le añadió DIEA (105 |jL, 0,60 mmol). Esta mezcla se vertió sobre la resina de CNP-38 desprotegido de la cadena lateral Lys26 (2,00 g, 0,21 mmol) y la suspensión se agitó durante 2 h a TA para obtener la resina l ia . La resina se lavó 10 veces con DMF (7 mL). La escisión del grupo protector Fmoc en 11a se llevó a cabo con una solución de HOBt (0,68 g, 5,03 mmol) y piperazina (3,00 g, 34,83 mmol) en DMF (47 mL). Por lo tanto, la resina se incubó 5 veces con 10 mL de la mezcla de escisión durante 15 min a TA cada vez. A continuación, la resina se lavó 7 veces con DMF (7 mL).
Se preparó una solución de Fmoc-Lys(Fmoc)-OH (449 mg, 0,76 mmol), COMU (325 mg, 0,76 mmol) y DIEA (165 j L, 0,95 mmol) en DMF (9 mL) y se extrajo sobre la resina. La mezcla se agitó durante 2 h a TA. El procedimiento se repitió dos veces, cada una de ellas durante 1 h, con la mezcla de acoplamiento recién preparada. La resina se lavó 10 veces con DMF (7 mL) y los grupos aminos libres restantes se cubrieron con 8 mL de 1/1/2 (v/v/v) Ac2O/piridina/DMF.
La escisión de los grupos protectores Fmoc en 11c se llevó a cabo con una solución de HOBt (0,68 g, 5,03 mmol), piperazina (3,00 g, 34,83 mmol) en DMF (47 mL). Por lo tanto, la resina se incubó 5 veces con 10 mL de la mezcla de escisión durante 15 min a TA cada vez. La resina se lavó 7 veces con DMF (7 mL).
A una solución de 7 (266 mg, 1,00 mmol) y PyBOP (520 mg, 1,00 mmol) en DMF (9 mL) se le añadió DIEA (209 j L, 1,20 mmol). Esta mezcla se extrajo sobre la resina y se agitó durante 2 h a TA. La resina se lavó 7 veces con DMF (7 mL) para obtener la resina 11e. La escisión del péptido de la resina y la eliminación de los grupos protectores se logró mediante el tratamiento de la resina con 15 mL de cóctel de escisión 68,5/10/10/5/3,5/1 (v/p/v/v/v/v) TFA/DTT/tioanisol/fenol/agua/TIPS pre-enfriado (-18 °C). La mezcla se dejó calentar hasta TA y se agitó durante 3 h. La resina se filtró y el crudo 11f se precipitó en éter dietílico pre-enfriado (- 18 °C) y se purificó por RP-HPLC. Las fracciones de HPLC combinadas se usaron directamente en el siguiente paso.
MS: m/z 1218,66 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1218,65).
A las fracciones del producto de HPLC combinadas de 11f (1 L) se añadieron 160 mL de solución amortiguadora de ácido cítrico 0,5 M (pH = 5,00) y 100 mL de una solución 50 mM de 2,2'-ditiobis(piridina-N-óxido) en 9/1 (v/v) acetonitrilo/agua. La mezcla se agitó durante 4 h a TA y posteriormente se diluyó con 1 L de agua que contenía 0,1% de TFA (v/v). 11g se purificó mediante RP-HPLC. Las fracciones del producto se combinaron y se liofilizaron.
Rendimiento: 64,3 mg (10,7 jmol, 6%) CNP-38-enlazador-DMB * 10 TFA
MS: m/z 1218,15 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1218,14).
La escisión del grupo protector Dmb se consiguió añadiendo 45 mL de cóctel de escisión 100/5/3/2/1 (v/v/w/v/v) TFA/MSA/DTT/agua/tioanisol pre-enfriado (-18 °C) a 11g (61,8 mg, 10,3 jmol), y después agitando durante 4 h a 0 °C. El producto en bruto 11h se precipitó en éter pre-enfriado (-18 °C). El precipitado se disolvió en una solución de 1/1 (v/v) acetonitrilo/agua que contenía 0,1% de TFA (v/v) y se incubó durante 4 h a TA para hidrolizar cualquier éster de TFA. 11h se purificó mediante RP-HPLC.
Rendimiento: 38,4 mg (6,65 jmol, 65%) CNP-38-enlazador-tiol * 10 TFA
MS: m/z 1159,11 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+ = 1159,10).
A una solución de 11h (34,6 mg, 5,99 jmol) en 1 mL de agua que contenía TFA al 0,1% (v/v) se le añadió una solución de PEG 2x10 kDa maleimida (Sunbright GL2-200MA, 1,12 g, 56,03 jmol) en 6,1 mL de agua que contenía TFA al 0,1% (v/v), seguido de solución amortiguadora de ácido láctico 0,5 M (1,46 mL, pH = 4,00). La mezcla se agitó a TA durante 4 h. El conjugado 11i se purificó mediante RP-HPLC.
Rendimiento: 227 mg (4,96 jmol, 83%) de conjugado lli * 10 HCl
Ejemplo 12 (no según la invención)
Síntesis del conjugado 12g de Lys26 CNP-38 PEG4x10 kDa permanente
El conjugado 12g se sintetizó de acuerdo con el siguiente esquema:

12d
12d 

A una solución de Fmoc-Lys(Fmoc)-OH (365 mg, 0,62 mmol) y PyBOP (322 mg, 0,62 mmol) en DMF (4,6 mL) se le añadió
DIEA (0,11 mL, 0,62 mmol). La mezcla se extrajo en la resina 10a (2,0 g, 0,21 mmol). La suspensión se agitó durante 2 h a TA. La resina se lavó 10 veces con DMF (7 mL). La escisión de los grupos protectores Fmoc en 12a se llevó a cabo con una solución de HOBt (1,35 g, 9,99 mmol), piperazina (6,00 g, 69,66 mmol) en DMF (94 mL).
Por lo tanto, la resina se incubó 5 veces con la mezcla de escisión durante 15 minutos a TA cada vez, para proporcionar la resina 12b. A continuación, la resina se lavó 7 veces con DMF (7 mL).
A una solución de 7 (283 mg, 1,06 mmol) y PyBOP (552 mg, 1,06 mmol) en DMF (6,5 mL), se le añadió DIEA (185 |j L, 1,06 mmol) y la mezcla se extrajo en la resina 12b (2,07 g, 0,10 mmol/g, 0,21 mmol). La mezcla se agitó durante 2 h a TA. Posteriormente, la resina se lavó 10 veces cada uno con DMF (7 mL) y CH2O 2 (7 mL), y se secó al vacío.
La escisión del péptido de la resina y la eliminación de los grupos protectores se logró mediante el tratamiento de la resina con 15 mL de cóctel de escisión 68,5/10/10/5/3,5/1 (v/p/v/v/v/v) TFA/DTT/tioanisol/fenol/agua/TIPS pre-enfriado (-18 °C). La mezcla se dejó calentar hasta TA y se agitó durante 2,5 h. La resina se filtró, el producto en bruto 12d se precipitó en éter dietílico pre-enfriado (-18 °C) y se purificó mediante RP-HPLC. Las fracciones de HPLC combinadas se usaron directamente en el siguiente paso.
MS: m/z 1172,37 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1172,37).
A las fracciones del producto de HPLC combinadas de 12d (390 mL) se añadieron 58,5 mL de solución amortiguadora de ácido cítrico 0,5 M (pH = 5,00) y 8,9 mL de una solución de 2,2'-ditiobis(piridina-N-óxido) 10 mM en 1/1 (v/v) acetonitrilo/agua. La mezcla se agitó durante 10 minutos a TA y posteriormente se diluyó con 400 mL de agua que contenía 0,1% de TFA (v/v). 12e se purificó mediante RP-HPLC.
Rendimiento: 100 mg (17,5 jmol, 8% en 6 pasos) CNP-38-enlazador-Dmb * 9 TFA
MS: m/z 1171,87 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1171,86).
La escisión del grupo protector Dmb se consiguió añadiendo 65 mL de cóctel de escisión 100/5/3/2/1 (v/v/w/v/v) TFA/MSA/DTT/agua/tioanisol pre-enfriado (-18 °C) a 12e (100 mg, 17,5 jmol) y agitando durante 3,5 h a 0 °C. El producto en bruto 12f se precipitó en éter dietílico pre-enfriado (-18 °C). El precipitado se disolvió en agua que contenía 0,1% de TFA (v/v) y se incubó durante 2 horas a TA para hidrolizar los ésteres de TFA. 12f se purificó mediante RP-HPLC. Rendimiento: 43,4 mg (7,92 jmol, 45%) CNP-38-enlazador-tiol * 9 TFA
MS: m/z 1112,83 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1112,82).
A una solución de 12f (39,6 mg, 7,22 jmol) en 1 mL de agua que contenía TFA al 0,1% (v/v) se le añadió una solución de PEG 2x10 kDa maleimida (Sunbright GL2-200MA, 1,22 g, 59,94 jmol) en 6,16 mL de agua que contenía TFA al 0,1% (v/v), seguido de solución amortiguadora de ácido láctico 0,5 M (1,41 mL, pH = 4,20). La mezcla se agitó a TA durante 4 h. El conjugado 12g se purificó mediante RP-HPLC.
Rendimiento: 204 mg (4,48 jmol, 57%) de conjugado 12g * 9 HCl
Ejemplo 13 (no según la invención)
Síntesis de PEG5kDa tio l 13c
El PEG5kDa tiol 13c se sintetizó de acuerdo con el siguiente esquema:

A una solución de 13b (58,6 mg, 0,15 mmol), HOBt (22,9 mg, 0,15 mmol) y clorhidrato de EDC (28,8 mg, 0,15 mmol) en DCM (1,00 mL) se le añadió 2,4,6-colidina (121 mg, 1,00 mmol). A continuación, se añadió una solución de metoxi PEG amina 5 kDa 13a (500 mg, 0,10 mmol) en DCM (4,00 mL) y la mezcla se agitó durante 16 h a TA. El disolvente se evaporó, la mezcla se disolvió en ACN/agua y se purificó mediante RP-HPLC. La cantidad de disolvente se redujo al vacío y el residuo acuoso se extrajo con DCM (1 x 100 mL, 2 x 50 mL). Las capas orgánicas combinadas se redujeron al vacío a 20 mL. Se añadieron TFA (1,6 mL) y TES (3,5 mL), y la mezcla se agitó a TA durante 4,5 h. 13c se precipitó en éter dietílico, se almacenó durante la noche a - 20 °C, se filtró y se secó al vacío.
Rendimiento: 372 mg (72 |jmol, 72%)
Ejemplo 14 (no según la invención)
Síntesis del conjugado 14e de CNP-34 PEG 5 kDa permanente N-terminal
El conjugado 14e se sintetizó de acuerdo con el siguiente esquema:

14a

PEG 5kDa O
El CNP-34 protegido por cadena lateral en resina TCP tentagel que tenía el extremo N-terminal libre 14a (0,78 g, 70 jmol)
se pre-hinchó en DMF durante 30 min. Una solución de ácido maleimido hexanoico (85,3 mg, 0,40 mmol), DIC (50,9 mg, 0,40 mmol) y Rendimiento: 372 mg (72 mmol,72%) de Oxyma (57,4 mL, 0,40 mmol) en DMF (6 mL) se extrajo sobre la resina y la mezcla se agitó durante 30 min a TA. A continuación, se repitió el acoplamiento una vez con una solución de acoplamiento recién preparada. La resina se lavó 10 veces cada una con DMF y ChhCh y se secó al vacío proporcionando 14b.
La escisión del péptido de la resina y la eliminación de los grupos protectores se logró mediante el tratamiento de la resina con 6 mL de cóctel de escisión 100/3/2/1 (v/v/v/v) TFA/TES/agua/tioanisol durante 1,5 h a TA. El péptido en bruto se precipitó en éter dietílico pre-enfriado (-18 °C).
MS: m/z 937,77 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 937,74).
El precipitado se disolvió en 15 mL de TFA. Se añadió una solución de difenilsulfóxido (68,06 mg, 0,34 mmol) y anisol (0,18 mL, 1,68 mmol) en 5 mL de TFA. Se añadió triclorometilsilano (0,47 mL, 4,17 mmol) y la mezcla se agitó durante 15 min a TA. Se añadió fluoruro de amonio (0,38 g, 10,3 mmol) y la solución se agitó durante otros 2 min. El material en bruto se precipitó en éter dietílico pre-enfriado (-18 °C) y se purificó mediante RP-HPLC para obtener 14d.
Rendimiento: 8,30 mg (1,78 |jmol, 82% de pureza, 1,4% en 3 pasos) CNP-34-Malhx * 8 TFA
MS: m/z 937,26 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 937,23).
A una solución de 14d (7,34 mg, 1,57 jmol) en 200 mL de 1/1 (v/v) acetonitrilo/agua con 0,1% de TFA (v/v) se le añadió una solución de 13c (20 mg, 3,90 jmol) en 200 jL de agua con 0,1% de TFA (v/v), seguida de 200 jL de solución amortiguadora de acetato 0,5 M (pH = 5,00). La mezcla se incubó a TA durante 30 minutos. El conjugado 14e se purificó mediante RP-HPLC.
Rendimiento: 9,92 mg (1,01 jmol, 57%) conjugado 14e * 8 TFA
Ejemplo 15 (no según la invención)
Síntesis del conjugado 15e de CNP-38 PEG 5kDa permanente N-terminal
El conjugado 15e se sintetizó de acuerdo con el siguiente esquema:


El compuesto 15d se sintetizó como se describió para 14d, excepto que se utilizó como material de partida el CNP-38 protegido por cadena lateral en resina TCP tentagel que tenía el extremo N-terminal libre 15a (1,34 g, 0,12 mmol). Rendimiento: 15,6 mg (2,94 |jmol, 6,6%) CNP-38-Malhx * 9 TFA
MS: m/z 1064,05 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1064,04).
El conjugado 15e se sintetizó como se describió para 14e, excepto que se utilizó 15d (8,34 g, 1,58 mmol) como material de partida.
Rendimiento: 9,47 mg (0,91 jmol, 31%) conjugado 15e * 9 TFA
Ejemplo 16 (no según la invención)
Síntesis del conjugado 16e de Lys12 CNP-34 PEG 5 kDa permanente
El conjugado 16e se sintetizó de acuerdo con el siguiente esquema:


Se desprotegieron 1,00 g ( 0,10 mmol) de CNP-34 protegido por cadena lateral en resina TCP tentagel que tenía el extremo N-terminal protegido por Boc y cadena lateral protegida por ivDde de Lys12 de acuerdo con el procedimiento proporcionado en Materiales y Métodos para obtener 16a.
El compuesto 16d se sintetizó como se describe para el 14d, excepto que se usó la resina 16a (1,00 g, 0,10 mmol) como material de partida.
Rendimiento: 17,0 mg (3,65 jmol, 3,7%) CNP-34-Lys12-Malhx * 8 TFA
MS: m/z 937,25 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 937,23).
El conjugado 16e se sintetizó como se describe para 14e, excepto que se usó 16d (17 mg, 3,65 |jmol) como material de partida.
Rendimiento: 12,2 mg (1,25 jmol, 34%) conjugado 16e * 8 TFA
Ejemplo 17 (no según la invención)
Síntesis del conjugado 17e de Lys16 CNP-34 PEG 5 kDa permanente
El conjugado 17e se sintetizó de acuerdo con el siguiente esquema:

17a

17d

17e
Se desprotegieron 0,78 g (0,07 mmol) de CNP-34 protegido por cadena lateral en resina TCP tentagel que tenía el extremo N-terminal protegido por Boc y cadena lateral protegida por ivDde de Lys16 de acuerdo con el procedimiento proporcionado en Materiales y Métodos para obtener 17a.
El compuesto 17d se sintetizó como se describe para el 14d, excepto que se usó la resina 17a (0,78 g, 0,13 mmol) como material de partida.
Rendimiento: 5,39 mg (1,16 jmol, 1,7%) CNP-34-Lys16-Malhx * 8 TFA
MS: m/z 937,26 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 937,23).
El conjugado 17e se sintetizó como se describe para 14e, excepto que se usó 17d (5,39 mg, 1,16 |jmol) como material de partida.
Rendimiento: 10,7 mg (1,09 jmol, 94%) conjugado 17e * 8 TFA
Ejemplo 18 (no según la invención)
Síntesis del conjugado 18e de Lys22 CNP-34 PEG 5 kDa permanente
El conjugado 18e se sintetizó de acuerdo con el siguiente esquema:

Se desprotegieron 1,07 g (0,11 mmol) de CNP-34 protegido por cadena lateral en resina TCP tentagel que tenia el extremo N-terminal protegido por Boc y cadena lateral protegida por ivDde de Lys22 de acuerdo con el procedimiento proporcionado en Materiales y Métodos para obtener 18a.
El compuesto 18d se sintetizó como se describe para el 14d, excepto que se usó la resina 18a (1,07 g, 0,11 mmol) como material de partida.
Rendimiento: 5,20 mg (1,12 jmol, 1,0% ) CNP-34-Lys22-Malhx * 8 TFA
MS: m/z 937,26 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 937,23).
El conjugado 18e se sintetizó como se describe para 14e, excepto que se usó 18d (5,2 mg, 1,12 |jmol) como material de partida.
Rendimiento: 4,20 mg (0,43 jmol, 38%) conjugado 18e * 8 TFA
Ejemplo 19 (no según la invención)
Síntesis del conjugado 19e de Lys26 CNP-38 PEG 5 kDa permanente
El conjugado 19e se sintetizó de acuerdo con el siguiente esquema:
Se desprotegieron (0,865 g, 0,10 mmol) de CNP-38 protegido por cadena lateral en resina TCP tentagel que tenía el extremo N-terminal protegido por Boc y cadena lateral protegida por ivDde de Lys26 de acuerdo con el procedimiento proporcionado en Materiales y Métodos para obtener 19a.
El compuesto 19d se sintetizó como se describe para el 14d, excepto que se usó la resina 19a (0,865 g, 0,10 mmol) como material de partida.
Rendimiento: 10,3 mg (1,95 jmol, 2,0%) CNP-38-Lys26-Malhx * 9 TFA
MS: m/z 1064,05 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1064,04).
El conjugado 19e se sintetizó como se describe para 14e, excepto que se usó 19d (4,70 mg, 1,10 |jmol) como material de partida.
Rendimiento: 3,20 mg (0,31 jmol, 28%) conjugado 19e * 9 TFA
Ejemplo 20
Cinética de liberación in vitro
Los conjugados 10f y 11 i de CNP se disolvieron en una solución amortiguadora PBS que contenía 3 mM de EDTA y 10 mM de metionina, pH 7,4, a una concentración de aproximadamente 1 mg de conjugado/mL. Las soluciones se filtraron de forma estéril y se incubaron a 37 °C. Se extrajeron alícuotas en determinados puntos de tiempo y se analizaron mediante RP-HPLc y ESI-MS. Las señales UV que se correlacionan con el CNP liberado se integraron y se trazaron en función del tiempo de incubación.
Se aplicó un software de ajuste de curvas para estimar el correspondiente tiempo medio de liberación.
Resultados:
Para el conjugado 10f se obtuvo un tiempo de vida media de liberación de 8,5 d (± 1 d).
Para el conjugado lli se obtuvo un tiempo de vida media de liberación de 9,5 d (± 1,5 d).
Ejemplo 21
Digestión de variantes de CNP por la endopeptidasa neutra in vitro
Para determinar la estabilidad in vitro de diversas variantes de CNP, incluyendo diferentes longitudes de cadena peptídica y PEGilaciones, utilizando diferentes sitios de PEGilación y moléculas de PEG en presencia de Endopeptidasa Neutral (NEP), se estableció un ensayo de digestión de NEP. Este ensayo monitoreo la disminución de la variante de CNP no digerida (normalizada con el estándar interno PFP) a lo largo del tiempo en referencia al punto a tiempo.
De manera más detallada, la NEP humana recombinante (2,5 |jg/mL de concentración final) y el pentafluorofenol estándar (PFP; 40 jg/m L de concentración final) se añadieron a la variante de CNP (100 jg de equivalentes de CNP/mL) en solución amortiguadora de digestión (50 mM Tris-HCl, pH 7,4, 10 mM NaCl). La solución se incubó a 37 °C y 500 rpm durante un máximo de 4 días. Las muestras se tomaron a diferentes intervalos de tiempo. La reacción se detuvo mediante una reducción combinada y desnaturalización por calor añadiendo TCEP (tris(2-carboxietil)fosfina; 25 mM de concentración final) e incubando la mezcla a 95 °C, 500 rpm durante 5 minutos. Los productos de reacción resultantes se asignaron utilizando HPLC-MS. La vida media de cada variante de CNP se calculó a través del cambio de la relación de las áreas de los picos de HPLC-UV de CNP y PFP a lo largo del tiempo. Para compensar las variaciones en la actividad de las proteasas, se realizó una digestión de CNP-38 o CNP-34 en cada medición de lotes como referencia.
La Tabla 1 enumera las vidas medias, con base en el ensayo de escisión in vitro de NEP, de diversas variantes de CNP de diferentes longitudes y que tienen varias moléculas de PEG unidas a diferentes cadenas laterales.

1) Debido a las variaciones en la actividad catalítica de NEP entre experimentos, se formó una media de todas las mediciones de vida media de CNP-34 (4,15 h) y las mediciones de vida media de los conjugados de CNP-34 se normalizaron a esta media usando un coeficiente para calcular el t1/2 ajustado.
2) Debido a las variaciones en la actividad catalítica de NEP entre experimentos, se formó una media de todas las mediciones de vida media de CNP-38 (12,10 h) y las mediciones de vida media de los conjugados de CNP-38 se normalizaron a esta media usando un coeficiente para calcular el t1/2 ajustado.
El orden de rango de resistencia hacia NEP es el siguiente: La variante de CNP más larga (CNP-38) es más estable que la variante de CNP más corta (CNP-34), que a su vez es más estable que la CNP-22 más corta. El orden de los sitios de unión de PEG es el siguiente: N-terminal > junto al anillo > anillo. Por lo tanto, una unión N-terminal de PEG confiere la mayor estabilidad hacia la digestión proteolítica con NEP para los conjugados probados. La estabilidad del CNP-38
PEGilado en Lys26 puede aumentar con el aumento del tamaño del PEG.
Ejemplo 22
Estimulación funcional del GMPc en células NIH-3T3 con variantes de CNP
La actividad funcional de las variantes de CNP se determinó en un ensayo celular con células NIH-3T3 (línea celular de fibroblastos de embrión murino). Estas células expresan endógenamente NPR-B en la superficie celular. La estimulación de NPR-B con CNP conduce a la producción intracelular del segundo mensajero cGMP que se detecta con un ensayo de cGMP disponible en el mercado. Las células NIH-3T3 se cultivaron de forma rutinaria en medio DMEM F-12 con 5% de FBS, 5 mM de glutamina a 37 °C y 5% de CO2. Para cada ensayo, se resuspendieron 50.000 células en solución amortiguadora de estimulación (PBS de Dulbecco con IBMX) y se incubaron con las variantes de CNP en diferentes concentraciones. CNP (las diluciones se realizaron en PBS con 0,2% de BSA). Tras una incubación de 30 minutos a 37 °C y 5% de CO2 , se lisaron las células y se determinaron los niveles de GMPc con un ensayo TR-FRET de GMPc disponible en el mercado (Cisbio, Kit de GMPc, No.de Cat.: 62GM2PEB). Las variantes de CNP con PEG se caracterizaron siempre en comparación con la versión no PEGilada en el mismo lote de experimentos. En la medida de lo posible, la evaluación de la actividad residual se realizó mediante el parámetro CE50 de la curva dosis-respuesta resultante (modelo restringido con pendiente común).
Tabla 2: Actividad NPR-B residual de las variantes de CNP PEGiladas en un ensayo basado en células, determinada frente a la variante de CNP no PEGilada

Comparando los sitios de unión de PEG probados, la unión en Lys26 (anillo-lisina) mostró la mayor reducción de la actividad funcional, mientras que la unión N-terminal mostró valores de actividad funcional residual relativamente altos. El aumento del tamaño de PEG dio como resultado una mejor protección de la molécula de CNP y una menor actividad funcional residual.
Ejemplo 23
Estudio de crecim iento en ratones FVB tras 5 semanas de tratamiento con CNP-38 mediante inyección diaria en bolo subcutáneo o mediante infusión subcutánea continua
Este estudio se realizó para comprobar el efecto de la inyección diaria en bolo subcutáneo frente a la infusión subcutánea continua de CNP-38 en el crecimiento de los animales. A ratones machos FVB de 21 a 22 días de edad (n = 9/grupo) se les administró 50 nmol/kg/d de CNP-38 o un vehículo (acetato 30 mM con pH 4 que contenía 5% de sacarosa y 1% de alcohol bencílico), ya sea mediante una inyección diaria en bolo subcutáneo o mediante una infusión subcutánea continua en la región escapular durante 35 días. La infusión continua se aplicó mediante bombas osmóticas Alzet modelo 1002 durante la semana 1-2, seguida del modelo 1004 durante la semana 3-5. Las concentraciones de CNP-38 en las bombas se ajustaron al peso medio de los animales el día 7 del estudio (modelo de bomba 1002) o el día 25 del estudio (modelo de bomba 1004). El crecimiento se determinó en el día 35 mediante la medición de la longitud total del cuerpo y las mediciones radiográficas del fémur y la tibia derechos.
Resultados de los animales tratados por inyección en bolo subcutánea diaria: En d 35, la longitud corporal total de los animales tratados con CNP-38 fue del 110,2%, la longitud del fémur derecho fue del 105,6%, y la longitud de la tibia derecha fue del 104,0% en comparación con los animales tratados con vehículo.
Resultados de los animales tratados por infusión subcutánea continua: En d 35, la longitud corporal total de los animales tratados con CNP-38 fue del 121,7%, la longitud del fémur derecho fue del 107,5%, y la longitud de la tibia derecha fue del 112,2% en comparación con los animales tratados con vehículo.
Se concluyó que la infusión subcutánea continua o las formulaciones relacionadas de liberación lenta de CNP-38 (por ejemplo, un profármaco de CNP-38 de liberación lenta) son más eficaces que la inyección en bolo subcutánea diaria para provocar el crecimiento del esqueleto apendicular y axial.
Ejemplo 24 (no según la invención)
Estudio farmacocinético del conjugado de Lys26 CNP-38 PEG431O kDa permanente en monos cynomolgus
Este estudio se realizó para demostrar la idoneidad del 12g como compuesto modelo para un profármaco de liberación lenta de CNP-38 en monos cynomolgus. Los monos cynomolgus macho (2-4 años, 3,5-4,1 kg) recibieron una única administración intravenosa (n = 3 animales) o una única administración subcutánea (n = 2 animales) de 12g a una dosis de 0,146 mg de CNP-38 eq/kg. Se recolectaron muestras de sangre hasta 168 h después de la dosis y se generó plasma. Las concentraciones plasmáticas totales de CNP-38 se determinaron mediante la cuantificación del péptido característico N-terminal (secuencia: LQEHPNAR) y péptido característico C-terminal (secuencia: IGSMSGLGC) tras la digestión tríptica descrita en Materiales y Métodos.
Resultados: Las administraciones de las dosis fueron bien toleradas sin signos visibles de malestar durante la administración y después de la misma. No se observaron reacciones en el sitio de la dosis en ningún momento a lo largo del estudio. Tras la inyección intravenosa, el tmáx de CNP-38 se observó a los 15 minutos (punto de tiempo más temprano analizado), seguido de un lento descenso del contenido de CNP-38 con un tiempo de vida media de aproximadamente 24 h. Tras la inyección subcutánea, la concentración de CNP-38 alcanzó un tmáx de 48 h. A las 168 h, la concentración de CNP-38 seguía siendo tan alta como aproximadamente 50% de Cmáx. La biodisponibilidad fue de aproximadamente el 50%. Se obtuvieron curvas FC similares para el péptido característico N- y C-terminal hasta 168 h después de la dosis, lo que indica la presencia de CNP-38 intacto en el conjugado.
La farmacocinética favorable de larga duración durante varios días y la estabilidad de CNP-38 en el conjugado indican la idoneidad del compuesto modelo permanente del conjugado 12g de Lys26 CNP-38 PEG 4x10 kDa como profármaco de CNP-38 de liberación lenta tras la inyección subcutánea. Puede concluirse que conjugados similares que tienen un CNP-38 enlazado transitoriamente a Lys26 (como, por ejemplo, 11 i) son profármacos de CNP-38 adecuados que proporcionan niveles duraderos de CNP-38 bioactivo liberado durante varios días.
Ejemplo 25
Estudio farmacocinético de los conjugados 10f y lli transitorios en monos cynomolgus
Este estudio se realizó para demostrar la idoneidad de 10f y 11 i como profármacos de CNP-38 de liberación lenta en monos cynomolgus. Los monos cynomolgus macho (2-4 años, 3-5 kg) recibieron una única administración subcutánea (n = 3 animales) del compuesto 10f o una única administración subcutánea (n = 3 animales) de lli a una dosis de 0,146 mg de CNP-38 eq/kg. Se recolectaron muestras de sangre hasta 168 h después de la dosis y se generó plasma. Los niveles plasmáticos del contenido total de CNP-38 se analizaron como se describe en el ejemplo 24. Para analizar el contenido plasmático de CNP-38 libre, las muestras de sangre se acidificaron después de la extracción añadiendo un 20% de volumen de solución amortiguadora de citrato de sodio 0,5 M con pH 4 para detener la liberación adicional de CNP-38 del conjugado. Los niveles de CNP-38 libre en el plasma pueden determinarse, por ejemplo, mediante ELISA utilizando un anticuerpo de CNP que se une a la región del anillo de CNP, como se describe en la literatura (patente de EE. UU.
8.377.884 B2), o mediante LCMS/MS.
Resultados: Las administraciones de las dosis fueron bien toleradas sin signos visibles de malestar durante la administración y después de la misma. No se observaron reacciones en el sitio de la dosis en ningún momento a lo largo del estudio. Tras la administración de la dosis, el tmáx total de CNP-38 se observó a las 12 h para el compuesto 10f y a las 24 h para el compuesto 11i. Los niveles plasmáticos totales de CNP-38 estaban por debajo del LC (100 ng/mL, péptido C-terminal) después de 120 h para el compuesto 10f, mientras que el nivel plasmático seguía siendo tan alto como el 30% de Cmáx para el compuesto 11 i después de 168 h (péptido C-terminal). Para el compuesto 11 i se encontró una vida media terminal similar de 3-4 d para el péptido C-terminal y el N-terminal, indicando la presencia de CNP-38 intacto en el conjugado.
Conclusión: La FC favorable de larga duración durante varios días y la estabilidad de CNP-38 en el conjugado lli indican su idoneidad como profármaco de CNP-38 para proporcionar niveles duraderos de CNP-38 bioactivo liberado durante varios días.
Ejemplo 27 (no según la invención)
Síntesis de CNP-3827d etiquetado con fluoresceína y ensayo de afinidad NPR-C
El compuesto 27d se sintetizó de acuerdo con el siguiente esquema:

27a
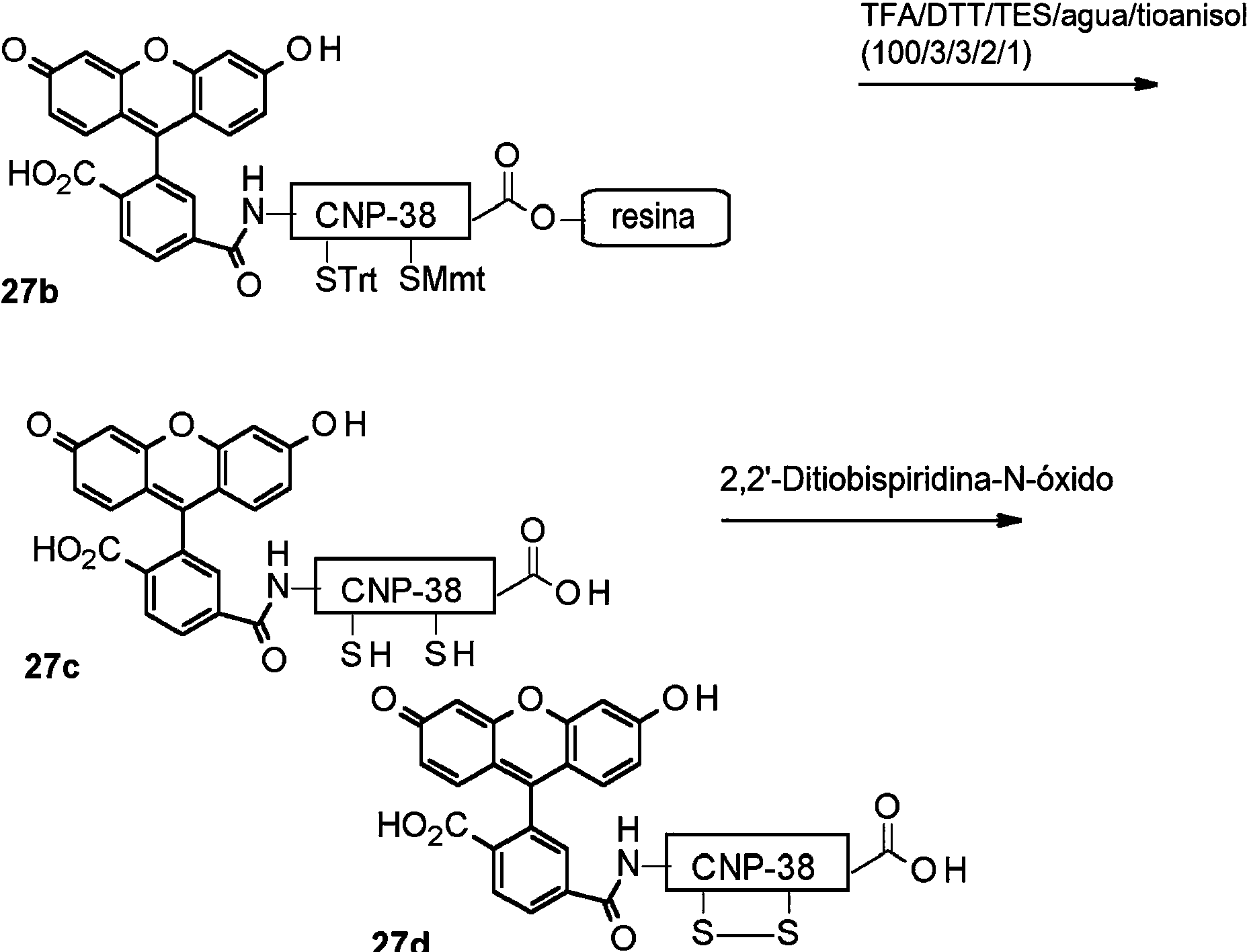
El CNP-38 protegido por cadena lateral en resina TCP tentagel que tenía el extremo N-terminal libre 27a (0,50 g, 35,4 jmol) se pre-hinchó en DMF durante 30 min. Una solución de éster de 5(6)-carboxifluoresceína-N-succinimidilo (41,9 mg, 88,5 | jmol) y DIEA (30,9 |j L,177 jmol) en DMF (1,6 mL) se extrajo en la resina y la mezcla se agitó durante la noche a TA. La resina se lavó 10 veces cada una con DMF y CH2O 2 y se secó al vacío proporcionando 27b.
La escisión del péptido de la resina y la eliminación de los grupos protectores se lograron mediante el tratamiento de la resina con 7 mL de cóctel de escisión 100/3/3/2/1 (v/w/v/v/v) TFA/DTT/TES/agua/ tioanisol durante 1 h a TA. La resina se filtró y el producto en bruto 27c se precipitó en éter dietílico pre-enfriado (-18 °C) y se purificó mediante RP-HPLC ara obtener 27c. Las fracciones de HPLC combinadas se usaron directamente en el siguiente paso.
MS: m/z 1105,80 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1105,81).
A las fracciones del producto de HPLC combinadas de 27c (115 mL), se añadieron 30 mL de solución amortiguadora de ácido cítrico 0,5 M (pH = 5,00) y 8 mL de una solución de 2,2'-ditiobis(piridi na-N-óxido) 10 mM en 1/1 (v/v) acetonitrilo/agua. La mezcla se agitó durante 60 min a TA y posteriormente se diluyó con 350 mL de agua que contenía 0,1% de TFA (v/v).
27d se purificó mediante RP-HPLC.
Rendimiento: 16,1 mg (2,90 jmol, 8,2% en 3 pasos) de CNP-38 etiquetado * 10 TFA
MS: m/z 1105,30 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1105,30).
Para el ensayo de afinidad con NPR-C, se desarrolló una línea celular Hek293 que expresa NPR-C. La región de
codificación de la secuencia NPR-C (BC131540) se clonó en un vector lentiviral bajo el promotor CMV para la expresión constitutiva del receptor. Como marcador de selección eucariota se utilizó un elemento bicistrónico situado en el vector para la resistencia a la puromicina. Tras la transducción, los grupos de células de crecimiento estable se sometieron a qRT-PCR para confirmar la expresión del ARNm del receptor en comparación con las células Hek293 parentales. Se amplió un conjunto de células que expresan NPR-C y se congeló como banco de células maestro para el análisis de muestras de CNP.
Para el ensayo, las células en crecimiento fueron tripsinizadas del fondo del matraz de células, se contaron y sembraron en una placa de 96 pocillos (1,5 x 105/pocillo) y centrifugadas. Los sobrenadantes se descartaron. El estándar y la muestra de CNP se diluyeron en serie en 9 pasos en PBS con 0,2% de BSA, se transfirieron a la microplaca por duplicado y se mezclaron con las células. Tras 30 minutos de incubación a temperatura ambiente, se añadió a cada pocillo CNP 27d etiquetado con fluoresceína con una concentración constante y se incubaron las células durante 45 minutos más a temperatura ambiente. Posteriormente, las células se analizaron mediante citometría de flujo utilizando la intensidad media de fluorescencia del canal FITC (FL1, Beckman Coulter FC500MPL) como lectura.
La curva estándar y la curva de la muestra se generaron en un software de análisis (PLA 2,0) utilizando un ajuste 4PL para el cálculo de la potencia y/o CI50.
Tabla 3: Afinidad de NPR-C residual de las variantes de CNP-38 PEGiladas en un ensayo celular frente a CNP-38

Ejemplo 29
Síntesis del reactivo Asn-enlazador 29b


A una solución de 1b (12,85 g, 16,14 mmol) en isopropanol (238 mL), se le añadió H2O (77,5 mL) y LiOH (2,32 g, 96,87 mmol). La mezcla de reacción se agitó durante 4 h a TA. Después, la mezcla de reacción se diluyó con tolueno (300 mL). Las fases se separaron y la fase orgánica se lavó 3 veces con HCl 0,1 M (200 mL). Las fases se separaron de nuevo. La fase acuosa se extrajo 3 veces con tolueno (100 mL). El producto se encontró en la fase acuosa ácida y el valor del pH de esta fase se ajustó a pH 8,5 mediante la adición de NaOH 4 N. Posteriormente, la fase acuosa se extrajo 3 veces con CH2O 2 (200 mL). La fase orgánica se lavó con salmuera (50 mL), se secó sobre Na2SO4 y se filtró. 29a se aisló tras la evaporación del disolvente y se usó en la siguiente reacción sin purificación adicional.
Rendimiento: 6,46 g (13,36 mmol, 83%)
MS: m/z 484,06 = [M+H]+, (masa monoisotópica calculada =483,26)
A una solución de ácido 6-maleimidohexanoico (1,73 g, 8,19 mmol) en THF (70 mL), se le añadió PyBOP (4,26 g, 8,19 mmol) y DIEA (3,89 mL, 22,33 mmol). A continuación, la mezcla de reacción se agitó durante 2 h a Ta . A continuación, se disolvió 29a (3,60 g, 7,44 mmol) en THF (10 mL) y se añadió a la mezcla de reacción. La reacción se agitó a TA durante la noche. A continuación, se añadió metil-terc-butiléter (300 mL). La fase orgánica se lavó 2 veces con solución de HCl 0,1 M (200 mL). Las fases acuosas combinadas se extrajeron 2 veces con metil-terc-butiléter (200 mL). Las fases orgánicas combinadas se lavaron con salmuera (150 mL), se secaron sobre Na2SO4 y se filtraron. El disolvente se evaporó al vacío. 29b se purificó mediante cromatografía ultrarrápida.
Rendimiento: 3,34 g (4,94 mmol, 66%)
MS: m/z 677,34 = [M+H]+, (masa monoisotópica calculada =676,33)
Ejemplo 30 (no según la invención)
Síntesis de tio l de 4 brazos-PEG 40kDa 30c
El tiol de 4 brazos PEG 30c se sintetizó de acuerdo con el siguiente esquema:

A una solución de ácido 6-tritilmercapto-hexanoico (111,72 mg, 286,02 |jmol), HOBt (43,81 mg, 286,06 jmol) y EDC*HCl (54,84 mg, 286,06 jmol) en CH2CI2 (5 mL) se le añadió 2,4,6 -colidina (251 jL , 1,91 mmol). A continuación, esta solución se añadió a una solución de amino de 4 brazos PEG 40 kDa (NOF, Sunbright PTE-400PA, 1,30 g, 31,78 jmol) en CH2O 2 (10 mL). La mezcla de reacción se agitó durante la noche a TA. A continuación, se evaporó el disolvente (baño de agua a 30 °C). 30b se purificó mediante RP-HPLC.
Rendimiento: 650,5 mg (48%).
La escisión del grupo protector Trt se logró añadiendo el cóctel de escisión (500 mg de DTT/500 mL de TFA/500 jL de agua, 2,5 mL de TES/5,0 mL de HFIP/25,0 mL de CH2O 2) a 30b (500 mg, 11,79 jmol) e incubando durante 30 min a TA.
30c se obtuvo tras la precipitación en éter dietílico pre-enfriado (-18 °C).
Rendimiento: 401,3 mg (82%; 93,3% de pureza)
Ejemplo 31 (no según la invención)
Síntesis del conjugado 31d
El conjugado 31d se sintetizó de acuerdo con el siguiente esquema:

STrt STrt
31a



Se añadió una solución del reactivo enlazador 29b (3,82 g, 5,64 mmol), OxymaPure (802 mg, 5,64 mmol) y DIC (868 |jL, 5,64 mmol) en DMF (42,5 mL) a la resina 10a (18 g, 1,85 mmol). La suspensión se agitó durante 100 minutos a TA para obtener la resina 31a. La resina se lavó 10 veces con DMF (10 mL) y 10 veces con DCM (10 mL), y se secó al vacío durante 15 minutos. La escisión del péptido de la resina y la eliminación de los grupos protectores se logró mediante el tratamiento de la resina con 135 mL de cóctel de escisión 100/3/2/1 (v/v/v/v) TFA/TES/agua/tioanisol. La mezcla se agitó durante 60 minutos a TA. El producto en bruto 31b se precipitó en éter dietílico pre-enfriado (-18 °C).
El precipitado se disolvió en TFA (423 mL). A esta solución, se le añadió una solución de sulfóxido de difenilo (1,87 g, 9,25 mmol) y anisol (5,05 mL, 46,25 mmol) en TFA (40 mL). Después, se añadió triclorometilsilano (13,3 mL, 114,7 mmol) y la mezcla de reacción se agitó durante 15 min a TA. A continuación, se añadió fluoruro de amonio (10,96 g, 296 mmol) y la solución se agitó durante 2 min en un baño de agua a TA. El producto en bruto 31c se precipitó en éter dietílico pre enfriado (-18 °C) y se purificó mediante RP-HPLC.
Rendimiento: 187 mg (34,2 jmol,16%) CNP-38-enlazador * 9 TFA
MS: m/z 1110,33 = [M+4H]4+, (masa monoisotópica calculada para [M+4H]4+= 1110,33).
A una solución de 31c (88,0 mg, 16,1 jmol) en 4,40 mL de MeCN/H2O (1:1) que contenía 0,1% de TFA (v/v) se le añadió una solución de tiol de 4 brazos-PEG 40kDa 30c (107,35 mg, 2,59 jmol) en 1,45 mL de agua que contenía 0,1% de TFA y 1 mM de EDTA, seguido de solución amortiguadora de fosfato 0,5 M que contenía 3 mM de EDTA (1,46 mL, pH 6,0). La mezcla se incubó durante 2 h a TA. El conjugado 31d se purificó mediante RP-HPLC.
Rendimiento: 129 mg (2,09 jmol, 80%) conjugado 16d * 36 TFA
Ejemplo 32
Síntesis alternativa del ácido 6-mercaptohexanoico protegido con Dmb 7
El ácido mercapto hexanoico protegido por Dmb 7 se sintetizó de acuerdo con el siguiente esquema:

A una solución de ácido 6-bromohexanoico (100 g, 0,513 mol) en EtOH (1,0 L) se le añadió tiourea (47 g, 0,615 mol) en una porción a 20 °C. A continuación, la suspensión se calentó hasta 78 °C (se formó una solución transparente) y se agitó durante 12 h. Se añadió gota a gota una solución de NaOH (62 g, 1,54 mol) en H2O (1,0 L) con un embudo de presión constante. Después, se continuó el reflujo durante 2 h adicionales. La mezcla de reacción se vertió en H2O (1 L) y se extrajo con EtOAc (1 L). La fase acuosa se acidificó con HCl concentrado hacia pH = 2 y luego se extrajo 3 veces con EtOAc (500 mL). Las fases orgánicas combinadas se lavaron con salmuera (400 mL). Después, las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y el disolvente se evaporó a presión reducida a 45 °C. El ácido 6-mercaptohexanoico se utilizó en la siguiente reacción sin purificación adicional.
Rendimiento: 62 g (crudo)
1H-NMR (400 MHz, CDCla):
5= 2,50 - 2,55 (q, J = 7,2 Hz, 2H), 2,36 (t, J = 7,6 Hz, 2H), 1,66 - 1,61 (m, 4H), 1,41 - 1,49 (m, 2H), 1,34 (t, J = 7,6 Hz, 1H) ppm.
El ácido 6-mercaptohexanoico (27,0 g, 0,182 mol) se cargó en un matraz de tres cuellos de 1 L con THF anhidro (540 mL). La solución se desgasificó mediante la técnica de congelación-bombeo-descongelación y, a continuación, se enfrió a 0 °C con un baño de hielo externo. Se añadió NaH (18,2, 455,4 mmol, 4,16 mL, 60% de pureza) con cucharas durante 30 min a 0 °C. A continuación, se añadió en una porción el cloruro de 2,6-dimetilbencilo (28,2 g, 0,182 mol). La mezcla de reacción se calentó hasta 20 °C y se agitó durante 12 h. La mezcla de reacción se vertió en H2O (540 mL) y se extrajo 2 veces con MTBE (540 mL). Después, la fase acuosa se acidificó con HCl conc. hasta alcanzar un pH = 2 y se extrajo 3 veces con MTBE (500 mL). Las fases orgánicas combinadas se lavaron con salmuera (500 mL), se secaron sobre Na2SO4 y se filtraron. 7 se aisló tras la evaporación del disolvente a presión reducida a 45 °C como un aceite amarillo.
Rendimiento: 41,5 g (0,16 mol, 85%)
1H-NMR (400 MHz, DMSO-d6):
5 = 11,99 (s, 1 H), 7,05 -7,07 (d, J= 6,8 Hz, 1H), 6,97 (s, 1H), 6,91 - 6,92 (d, J= 6,8 Hz, 1H), 3,66 (s, 2H), 2,38 - 2,39 (m, 2H), 2,29 (s, 3H), 2,23 (s, 3H), 2,16 - 2,19 (m, 2H), 1,40 - 1,55 (m, 4H), 1,22 - 1,38 (m, 2H) ppm.
MS (modo neg.): m/z 265,0 = [M-H]', (masa monoisotópica calculada =265,13).
Ejemplo 33 (no según la invención)
Síntesis del reactivo enlazador 33c
El reactivo enlazador 33c se sintetizó de acuerdo con el siguiente esquema:

Se l levaron a cabo cuatro reacciones en paralelo. A una solución del compuesto 1b (60 g, 75 mmol) en CH2CI2 (300 mL) se le añadió piperidina (58 g, 0,68 mol, 67 mL). La mezcla de reacción se agitó a TA durante 4 h. Las cuatro reacciones que se realizaron en paralelo se combinaron para su elaboración. La mezcla de reacción se diluyó con H2O (500 mL) y se ajustó con una solución de HCl 0,5 N hacia un pH = 3 ~ 4. La fase orgánica se separó y la fase acuosa se extrajo con CH2O 2 (800 mL). Las fases orgánicas combinadas se lavaron con salmuera (400 mL) y solución de NaHCO3 saturada al 5% (400 mL) sucesivamente. A continuación, las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y el disolvente se evaporó al vacío. 33a se purificó mediante cromatografía sobre sílice (malla 100-200) con DCM/MeOH (20/1 a 4/1).
Rendimiento: 150 g (87%)
1H-NMR (400 MHz, DMSO-d6):
5 = 7,34 - 7,38 (m, 4H), 6,25 - 7,29 (m, 2H), 5,08 - 5,19 (m, 2H), 4,60 - 4,68 (m, 1H), 4,32 - 4,40 (m, 2H), 3,73 - 3,79 (m, 9H), 3,10 - 3,27 (m, 3H), 2,65 - 3,05 (m, 8H), 1,36 (s, 9H) ppm.
Se l levaron a cabo dos reacciones en paralelo. A una solución de Fmoc-L-Lys(Fmoc)-OH (79 g, 0,13 mol), 33a (70 g, 0,12 mol), 4-eti l-morfolina (70 g, 0,61 mol, 77 mL) en MeCN (850 mL) se le añadió gota a gota T3P (50% en EtOAc; 140 g, 0,22 mol) a lo largo de un período de 30 min. Tras la adición, la mezcla de reacción se agitó a TA durante 18 h. Las dos reacciones que se realizaron en paralelo se combinaron para su elaboración. La mezcla de reacción se diluyó con H2O/CH2O 2 (1:1,2 L) y posteriormente se ajustó con una solución de HCl 0,5 N hacia un pH = 3 ~ 4. La fase orgánica se separó y la fase acuosa se extrajo con CH2O 2 (1 L). Las fases orgánicas combinadas se lavaron sucesivamente con salmuera (800 mL) y solución de NaHCO3 al 5% (800 mL). A continuación, las fases orgánicas combinadas se secaron
sobre Na2SO4, se filtraron y el disolvente se evaporó al vacío. 33b se purificó mediante cromatografía sobre sílice (malla 100-200) con éter de petróleo/acetato de etilo (5/1 a 1/1).
Rendimiento: 160 g (57%)
1H-NMR (400 MHz, DMSO-d6):
5 = 7,80 - 7,90 (m, 4H), 7,61 - 7,68 (m, 5H), 7,20 - 7,40 (m, 14H), 6,14 - 6,28 (m, 3H), 5,01 - 5,07 (m, 2H), 4,15 - 4,36 (m, 8H), 3,71 - 3,77 (m, 9H), 2,80 - 3,53 (m, 9H), 2,66 - 2,75 (m, 4H), 2,36 - 2,39 (m, 1H), 1,52 - 1,55 (m, 2H), 0,88 - 1,19 (m, 13H)ppm.
Se llevaron a cabo dos reacciones en paralelo. A una solución de 33b (60 g, 52 mmol) en MeOH (1,2 L) se le añadió Pd/C al 10% (18 g) en una botella hidrogenada de 2 L. La mezcla de reacción se desgasificó y se purgó 3 veces con H2 y luego se agitó a 25 °C bajo atmósfera de H2 (3,16 kg/cm2 (45 psi)) durante 2,5 h. Las dos reacciones que se realizaron en paralelo se combinaron para su elaboración. La mezcla de reacción se filtró con diatomita y el filtrado se concentró al vacío para proporcionar el producto en bruto 33c. 33c se purificó mediante cromatografía sobre sílice (malla 100-200) con DCM/MeOH (200/1 a 100/3).
Rendimiento: 70 g (63%)
1H-NMR (400 MHz, DMSO-d6):
5 = 12,15 (s, 1H), 7,87 - 7,89 (m, 4H), 7,50 - 7,70 (m, 5H), 7,31 - 7,40 (m, 9H), 6,20 - 6,23 (m, 2H), 4,13 - 4,36 (m, 10H), 3,70 - 3,77 (m, 9H), 2,62 - 3,10 (m, 12H), 2,30 - 2,34 (m, 1H), 2,14 - 2,18 (m, 1H), 1,50 - 1,58 (m, 2H), 1,25 - 1,34 (m, 13H) ppm.
MS: m/z 1056,4= [M+H]+, (masa monoisotópica calculada =1056,50).
Ejemplo 34 (no según la invención)
Síntesis alternativa de 11c
El compuesto 11c se sintetizó de acuerdo con el siguiente esquema:

Una solución del reactivo enlazador 33c (3,21 g, 3,04 mmol), PyBOP (1,58 g, 3,04 mmol) y DIPEA (848 mL, 4,86 mmol) en DMF (24,0 mL) se incubó durante 5 min a TA, y luego se añadió a la resina 10a (12 g, 1,21 mmol). La suspensión se agitó durante 2,5 h a TA. La resina se lavó 10 veces con DMF (10 mL) y 10 veces con DCM (10 mL), y se secó al vacío durante 60 minutos.
Ejemplo *35
CNP-38 y Conjugado 11i: Evaluación de los efectos cardiovasculares en el ratón consciente (administración subcutánea)
El propósito de este estudio fue evaluar los efectos secundarios hemodinámicos de 11 i a un nivel de dosis equivalente a un nivel de dosis de CNP-38 que provoca efectos secundarios hemodinámicos (disminución de la presión arterial) en el ratón telemetrado.
A los ratones machos Crl:CD1(ICR) (de 8 a 13 semanas de edad y de 27,3 a 35,6 g de peso corporal al inicio de la dosificación) se les implantó quirúrgicamente un transmisor telemétrico TA11PA-C10 (Data Sciences International (DSI)) en la arteria carótida. El cuerpo del transmisor se colocó por vía subcutánea en el flanco lateral del ratón. Los ratones fueron dosificados por vía subcutánea en un diseño cruzado de cuadrado latino con al menos 72 horas entre las ocasiones de dosificación. Los ratones fueron dosificados con 1) vehículo (succinato 10mM, manitol 46,0 g/L, pH 4,00), 2) CNP-38 (800 |jg CNP-38/kg, succinato 10mM, manitol 46,0 g/L, pH 4,00) o 3) 11 i (800 |jg CNP-38 eq/kg, succinato 10mM, manitol 46,0 g/L, pH 4,00). Se incluyeron al menos cuatro ratones en cada nivel de dosis. La presión arterial (sistólica (PAS), diastólica (PAD) y media (PAM)) y la frecuencia cardíaca (FC, derivada de la presión arterial), se registraron utilizando un sistema de captura de datos digital conectado con un sistema de adquisición y análisis de datos DSITM Ponemah. El sistema de captura permitió registrar los parámetros cardiovasculares mientras los ratones estaban en jaulas individuales. El día de cada sesión de prueba se pesaron los animales y se realizó un registro previo a la dosis durante al menos 60 minutos antes de la misma. Cada ratón fue devuelto a la jaula de vivienda y se registraron los parámetros cardiovasculares durante aproximadamente 48 horas después de la dosis. Se informó de la presión arterial y la FC en los siguientes puntos de tiempo: -30, -20, -10, 5, 15 y 30 minutos después de la dosis y 1, 2, 6, 12, 18, 24, 30, 36, 42 y 48 horas después de la dosis. Cada punto de tiempo se presentó como el valor promedio de cinco minutos de registro antes del punto de tiempo. El periodo de seguimiento se seleccionó para cubrir la exposición a los elementos de prueba tanto antes como después de Tmáx.
Resultados: En comparación con los valores antes de la dosis, los animales que recibieron una dosis de vehículo presentaron un aumento de la PAM en los puntos de tiempo de muestreo 5, 15 y 30 minutos después de la dosis. Esto se consideró una respuesta fisiológica normal debida a la manipulación y la dosificación. El mismo aumento fisiológico de la PAM se observó en los animales a los que se les administró la dosis de 11 i en el punto de tiempo de muestreo 5, 15 y 30 minutos después de la dosis. En 3 de los 4 animales dosificados con CNP-38, el aumento fisiológico de la PAM no fue evidente. Por el contrario, 3 de los 4 animales dosificados con CNP-38 mostraron una disminución significativa de la PAM en los puntos de tiempo de muestreo 5, 15 y 30 minutos después de la dosis. Durante los diez puntos de tiempo restantes, no hubo diferencias en la PAM entre los animales dosificados con vehículo, CNP-38 y 11i.
PAM (mmHg) antes de la dosis a 30 min después de la dosis (media ± DE)

Se observaron tendencias similares para la PAS y PAD en todos los niveles de dosis. La FC no se vio afectada por el tratamiento con CNP-38 u 11i.
En conclusión, la administración subcutánea de 11 i no disminuyó la presión arterial como se observó para una dosis equivalente de CNP-38.
Ejemplo 36 (no según la invención)
Perfil farmacocinético de CNP-38 tras la administración subcutánea de una sola dosis a monos cynomolgus
Este estudio se realizó para comprobar la farmacocinética del CNP-38 tras su administración subcutánea (s.c.) en monos cynomolgus. Tres monos machos (2-4 años, 3-5 kg) recibieron una única inyección s.c. a una dosis de 40 jg/kg de CNP-38. Se tomaron muestras de sangre a los 5, 10, 15, 30, 45 min y 1, 2, 4, 8 horas después de la dosis.
Método: Los niveles plasmáticos de CNP se analizaron mediante un radioinmunoanálisis competitivo disponible en el
mercado (RK-012-03, Phoenix Pharmaceuticals, CA). El ensayo se aplicó esencialmente como lo describe el fabricante. El ensayo se basa en la unión competitiva entre CNP etiquetado con 125I (suministrado en el kit) y CNP no etiquetado (procedente de la muestra del estudio o de los calibrantes) con un anticuerpo anti-CNP. Cuando la concentración de CNP en la muestra aumenta, la cantidad de CNP etiquetado con 125I que es capaz de unirse al anticuerpo disminuye. Al medir la cantidad de CNP etiquetado con 125I en función de la concentración de péptido, es posible construir una curva de calibración a partir de la cual se puede determinar la concentración de péptido en la muestra. Se realizaron algunos cambios en el protocolo de ensayo suministrado. Estos cambios incluyeron el uso de calibrantes de CNP internos y muestras de control de calidad para asegurar la consistencia entre los ensayos. Para acortar la duración del ensayo, la incubación inicial de las muestras con los anticuerpos se realizó a temperatura ambiente durante 5 horas (en lugar de 16 24 horas a 4 °C). Debido a los efectos de la matriz en el plasma de los monos, la dilución mínima requerida se fijó en 1:10, lo que produjo un rango de ensayo de 150-1080 pg/mL de CNP.
Resultados: La administración de CNP-38 a monos cynomolgus fue bien tolerada. Tras la inyección s.c., la mediana de Tmáx de CNP-38 se observó a los 10 minutos, con un tiempo medio de vida media de aproximadamente 7 minutos.

Ejemplo 37
Perfil farmacocinético del Conjugado 11 i tras la administración subcutánea de una sola dosis a monos cynomolgus
Este estudio se realizó para investigar la farmacocinética del 11 i tras su administración s.c. en monos cynomolgus. Cuatro animales machos (2-4 años, 3-5 kg) recibieron una única inyección s.c. de 11 i a una dosis de 40 |jg de CNP-38 eq/kg. Se recolectaron muestras de sangre hasta 168 h después de la dosis y se generó plasma (LiHeparin). Las concentraciones totales de CNP-38 se determinaron mediante LC-Ms /MS.
Método: El término “CNP-38 total” se refiere a una combinación de CNP-38 libre y CNP-38 unido en el conjugado de CNP-38. Las concentraciones plasmáticas totales de CNP-38 se determinaron mediante la cuantificación del péptido característico C-terminal (secuencia: IGSMSGLGC) tras la digestión tríptica y la reducción del puente disulfuro.
El análisis LC-MS se llevó a cabo utilizando un UPLC Agilent 1290 acoplado a un espectrómetro de masas Agilent 6460 Triple Quad mediante una sonda ESI. La cromatografía se realizó en una columna analítica Waters Acquity BEH C18 (D.I.
50 x 1,0 mm, tamaño de partícula de 1,7 jm , 130 A) con prefiltro a una tasa de flujo de 0,5 mL/min (T = 45 °C). Se utilizó agua (grado sódico ultrapuro < 500 ppt) con un 0,1% de ácido fórmico (v/v) como fase móvil A y acetonitrilo (grado ULC/MS) con un 0,1% de ácido fórmico como fase móvil B. El sistema de gradiente comprendía un breve paso isocrático a los parámetros iniciales de 0,1% de B durante 0,5 min, seguido de un aumento lineal de 0,1% de B a 30% de B en 1,5 min. El análisis de masas se realizó en el modo de monitoreo de reacción múltiple (MRM), monitoreando las reacciones de la ionización m/z 824,5 [M+H]1+ a 515,2. Como estándar interno se utilizó el conjugado de CNP-38 deuterado.
Los estándares de calibración del conjugado CNP-38 en plasma en blanco se prepararon de la siguiente manera: El plasma de cynomolgus descongelado con Li-heparina primero se homogeneizó y posteriormente se centrifugó durante 5 minutos. La formulación del conjugado CNP-38 se diluyó en ocho soluciones de trabajo de calibración diferentes que contenían entre 0,103 y 51,28 jg/m L (CNP-38 eq.) en 50% de metanol/50% de agua/0,1% de ácido fórmico (v/v/v). Las soluciones de trabajo se añadieron al plasma en blanco a concentraciones entre 10,3 ng/ mL (CNP-38 eq.) y 5128 ng/mL (CNP-38 eq.). Los estándares se utilizaron para la generación de una curva de calibración. Se generó una curva de calibración con base en las relaciones de área de pico del analito y el estándar interno utilizando una regresión lineal ponderada (1/x2) y las concentraciones de la muestra se determinaron mediante un cálculo retrospectivo con respecto a la curva de calibración.
Para la preparación de la muestra, la precipitación de las proteínas se llevó a cabo mediante la adición de 200 jL de acetonitrilo pre-enfriado (0 °C) a 50 jL de la muestra de plasma y 10 jL de solución de estándar interno (2,8 jg/m L de CNP-38 eq. en 50% de metanol/50% de agua/0,1% de ácido fórmico (v/v/v)). Se transfirieron 200 jL del sobrenadante a una nueva placa de pocillos y se evaporaron hasta sequedad (bajo una suave corriente de nitrógeno a 35 °C). Para el disolvente de reconstitución se disolvieron 100 jg de tripsina (número de pedido V5111, Promega GmbH, Mannheim, Alemania) en 100 jL de ácido acético 10 mM. Se añadieron 2,5 mL de solución amortiguadora de Tris y 500 jL de metanol. Se añadieron 50 jL del disolvente de reconstitución resultante a cada cavidad de la placa de pocillos. Tras 3 horas de incubación a 37 °C (Eppendorf ThermoMixer con ThermoTop), se añadieron 5 jL de una solución de TCEP 0,5 M a cada cavidad y se incubaron de nuevo durante 30 minutos a 37 °C. Después de que las muestras se enfriaran a
temperatura ambiente, se añadieron 2 |jL de ácido fórmico al 60% en agua. Se inyectaron 10 |jL en el sistema UHPLC-MS. Resultados: La administración de 11 i a monos cynomolgus fue bien tolerada. Tras la inyección s.c., la mediana de Tmáx de 11 i fue de 36 h, y con un tiempo medio de vida media de 107 h.

Ejemplo 38
Estimulación funcional del GMPc en células NIH-3T3 con CNP liberado
11 i se incubó en condiciones fisiológicas (1 mg de CNP-38 eq/mL), como se describe en el Ejemplo 20. Después de 7 días, se aisló el CNP-38 l iberado mediante RP-HPLC y se analizó su bioactividad como se describe en el Ejemplo 22.

Ejemplo 39
Síntesis alternativa de 11h

Síntesis alternativa del compuesto 11h: 39a se sintetiza mediante síntesis en fase sólida como se describe en Material y Métodos. El grupo protector PG1 para la cadena lateral de lisina del anillo y los grupos protectores PG2 para las cadenas laterales de cisteína son Mmt. La escisión suave de la resina y la formación de disulfuro por tratamiento con yodo proporciona el compuesto 39c. Después del acoplamiento de la molécula enlazadora 39d y la desprotección global, 11h se purifica mediante RP-HPLC.
Abreviaturas:
ACH acondroplasia
ACN acetonitri lo
AcOH ácido acético
AUCtlast Área bajo la curva hasta el último punto de tiempo cuantificable
Bn bencilo
Boc terc-buti loxicarbonilo
BSA albúmina sérica bovina
cGMP monofosfato de guanosina cíclico
Cmáx Concentración máxima
CMV citomegalovirus
CNP péptido natriurético tipo C
COMU hexafluorofosfato de (1-ciano-2-etoxi-2-oxoetilidenaminooxi)dimetilamino-morfolino-carbenio conc. Concentrado
d día
CTC Poliestirol de cloruro de clorotritilo
PAD Presión arterial diastólica
DBU 1,3-diazabiciclo[5.4.0]undeceno
DCC N,N'-diciclohexilcarbodiimida
DCM diclorometano
DIC N,N'-diisopropilcarbodiimida
DIEA N,N-diisopropiletilamina
DIPEA N,N-diisopropiletilamina
DMAP dimetilamino-pi ridina
DMEM medio de Eagle modificado de Dulbecco
Dmb 2,4-dimetilbencilo
DMEM medio de Eagle modificado de Dulbecco
DMF N,N-dimetilformamida
DMSO dimetilsulfóxido
DTT ditiotreitol
CE50 concentración efectiva media
EDC 1-etil-3-(3-dimetilaminopropil)carbodiimida
EDTA ácido etilenodiaminotetracético
ELISA ensayo por inmunoabsorción ligado a enzimas
eq equivalente estequiométrico
ESI-MS ionización por electroaspersión y espectrometría de masas
Et etilo
EtOAc acetato de etilo
EtOH etanol
FBS suero bovino fetal
FGFR3 receptor del factor de crecimiento de fibroblastos 3
FITC isotiocianato de fluoresceína
Fmoc 9-fluorenilmetiloxicarbonilo
h hora
HATU hexafluorofosfato de O-(7-azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio
HCH hipocondroplasia
HFIP hexafluoroisopropanol
HPLC cromatografía líquida de alta resolución
HOBt N-hidroxibenzotriazol
FC Frecuencia cardíaca
IBMX 3-isobutiM-metilxantina
iPrOH 2-propanol
iv intravenoso
ivDde 4,4-dimetil-2,6-dioxociclohex-1-ilideno)-3-metilbutilo
LC cromatografía líquida
LTQ cuadrupolo de trampa lineal
Mal 3-maleimidopropilo
PAM Presión arterial media
Me metilo
MeOH metanol
min minutos
Mmt monometoxitritilo
MS espectro de masas / espectrometría de masas
MSA ácido metanosulfónico
MTBE metil-terc-butiléter
Mtt metiltritilo
PM peso molecular
m/z relación masa-carga
NEP endopeptidasa neutra
NHS N-hidroxisuccinimida
NPR receptor de péptido natriurético
OtBu terc-butiloxi
PBS solución salina amortiguada con fosfato
PEG poli(etilenglicol)
PFP pentafluorofenol
pH potentia Hydrogenii
Pr propilo
PyBOP hexafluorofosfato de benzotriazol-1-il-oxitripirrolidinofosfonio
Q-TOF tiempo de vuelo con cuadrupolo
qRT-PCR reacción en cadena de la polimerasa cuantitativa en tiempo real RP-HPLC cromatografía líquida de alta resolución de fase reversa
rpm rondas por minuto
TA temperatura ambiente
SIM monitoreo de un solo ión
PAS Presión arterial sistólica
SEC cromatografía de exclusión por tamaño
sc subcutáneo
Su succinimidilo
TaP 2.4.6- tri propil-1,3,5,2,4,6-trioxatrifosforinano-2,4,6-trióxido
TCEP tris(2-carboxietil)fosfina
TCP poliestirol de cloruro de tritilo
TD displasia tanatofórica
TES trietilsilano
TFA ácido trifluoroacético
THF tetrahidrofurano
TIPS triisopropilsilano
T máx Tiempo de concentración máxima
TMEDA N,N,N'N'-tetrametiletilendiamina
Tmob 2.4.6- trimetoxibencilo
TR-FRET transferencia de energía de fluorescencia resuelta en el tiempo Trt trifenilmetilo, tritilo
UPLC cromatografía líquida de ultra-alta resolución
UV ultravioleta
vs. versus, frente a
ZQ cuadrupolo simple
Claims (10)
1. Un profármaco de CNP o una sal farmacéuticamente aceptable del mismo, que comprende
- un resto de CNP -D que comprende un resto de anillo; y
- un resto portador -Z que se conjuga a través de un resto -L2- a un resto enlazador de profármaco reversible -L1-, cuyo resto enlazador de profármaco reversible -L1- está conjugado de manera covalente y reversible a una cadena lateral de un residuo de aminoácido de dicho anillo resto de -D; y en donde -L2- es un enlace químico o un espaciador, en donde el resto -L1-L2- es de fórmula (Md-ii):

en donde
la línea punteada no marcada indica la unión a un nitrógeno de una cadena lateral de aminoácido del resto del anillo de -D formando un enlace amida; y la línea punteada marcada con el asterisco indica unión a -Z; en donde -D tiene la secuencia de SEQ ID NO:24 y -L1- está conjugado con la lisina en la posición 26; en donde -Z es de fórmula (h):

en donde
la línea punteada indica unión a -L2-; y
cada -Zc es un resto

en donde
cada c1 es un número entero que oscila independientemente entre 200 y 250.
2. El profármaco de CNP o una sal farmacéuticamente aceptable del mismo de la reivindicación 1, en donde
-L1-L2- es de fórmula (Md-ii '):

en donde
la línea punteada no marcada indica unión a -D como se define en la reivindicación 1 y la línea punteada marcada indica unión a -Z como se define en la reivindicación 1.
3. El profármaco de CNP o una sal farmacéuticamente aceptable del mismo de la reivindicación 1 o 2, en donde - Z es de fórmula (h-i):

en donde
la línea punteada indica unión a -L2-; y
cada -Zc es un resto

en donde
cada c1 es un número entero que varía independientemente de 200 a 250.
4. El profármaco de CNP o una sal farmacéuticamente aceptable del mismo de cualquiera de las reivindicaciones 1 a 3, en donde cada c1 es aproximadamente 225.
5. Una composición farmacéutica que comprende al menos un profármaco de CNP o una sal farmacéuticamente aceptable del mismo de cualquiera de las reivindicaciones 1 a 4 y al menos un excipiente.
6. El profármaco de CNP o una sal farmacéuticamente aceptable del mismo de cualquiera de las reivindicaciones 1 a 4, o la composición farmacéutica de la reivindicación 5 para uso como un medicamento.
7. El profármaco de CNP o una sal farmacéuticamente aceptable del mismo, o la composición farmacéutica para uso de la reivindicación 6, en donde el medicamento se usa en el tratamiento de la acondroplasia mediante inyección subcutánea.
8. El profármaco de CNP o una sal farmacéuticamente aceptable del mismo de cualquiera de las reivindicaciones 1 a 4, o la composición farmacéutica de la reivindicación 5 para uso en el tratamiento de una enfermedad que puede tratarse con CNP.
9. El profármaco de CNP o una sal farmacéuticamente aceptable del mismo, o la composición farmacéutica para uso de la reivindicación 8, en donde la enfermedad que se puede tratar con CNP se selecciona del grupo que consiste en
acondroplasia, hipocondroplasia, baja estatura, enanismo, osteocondrodisplasias, displasia tanatofórica, osteogénesis imperfecta, acondrogénesis, condrodisplasia punctata, acondroplasia homocigota, displasia campomélica, hipofosfatasia letal congénita, tipo letal perinatal de osteogénesis imperfecta, síndrome de costillas cortas-polidactilia, tipo rizomélico de condrodisplasia punctata, displasia metafisaria tipo Jansen, displasia espondiloepifisaria congénita, atelosteogénesis, displasia diastrófica, fémur corto congénito, displasia mesomélica tipo Langer, displasia mesomélica tipo Nievergelt, síndrome de Robinow, síndrome de Reinhardt, acrodisostosis, disostosis periférica, displasia de Kniest, fibrocondrogénesis, síndrome de Roberts, displasia acromesomélica, micromelia, síndrome de Morquio, síndrome de Kniest, displasia metatrófica, displasia espondiloepimetafisaria, neurofibromatosis, síndrome de Legius, síndrome LEOPARD, síndrome de Noonan, fibromatosis gingival hereditaria, síndrome cardio-facio-cutáneo, síndrome de Costello, deficiencia de SHOX, baja estatura idiopática, deficiencia de la hormona del crecimiento, artrosis, disostosis cleidocraneal, craneosinostosis, braquidactilia, camptodactilia, polidactilia, sindactilia, displasia disegmentaria, encondromatosis, displasia fibrosa, exostosis múltiple hereditaria, raquitismo hipofosfatémico, síndrome de Jaffe-Lichtenstein, síndrome de Marfan, síndrome de McCune-Albright, osteopetrosis y osteopoiquilia.
10. El profármaco de CNP o una sal farmacéuticamente aceptable del mismo, o la composición farmacéutica para uso de la reivindicación 9, en donde la enfermedad que se puede tratar con CNP es la acondroplasia.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16150625 | 2016-01-08 | ||
| EP16179288 | 2016-07-13 | ||
| EP16191460 | 2016-09-29 | ||
| PCT/EP2017/050209 WO2017118698A1 (en) | 2016-01-08 | 2017-01-05 | Cnp prodrugs with carrier attachment at the ring moiety |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2934582T3 true ES2934582T3 (es) | 2023-02-23 |
Family
ID=57821955
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES17700496T Active ES2934582T3 (es) | 2016-01-08 | 2017-01-05 | Profármacos de CNP con unión de portador en el resto del anillo |
Country Status (15)
| Country | Link |
|---|---|
| US (2) | US11413351B2 (es) |
| EP (2) | EP3400019B1 (es) |
| JP (2) | JP6968802B2 (es) |
| AU (3) | AU2017205688C1 (es) |
| CA (1) | CA3007979C (es) |
| DK (1) | DK3400019T3 (es) |
| ES (1) | ES2934582T3 (es) |
| HR (1) | HRP20221339T1 (es) |
| HU (1) | HUE060521T2 (es) |
| LT (1) | LT3400019T (es) |
| PL (1) | PL3400019T3 (es) |
| RS (1) | RS63793B1 (es) |
| SI (1) | SI3400019T1 (es) |
| WO (1) | WO2017118698A1 (es) |
| ZA (1) | ZA201804415B (es) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3400019B1 (en) | 2016-01-08 | 2022-09-28 | Ascendis Pharma Growth Disorders A/S | Cnp prodrugs with carrier attachment at the ring moiety |
| US11389511B2 (en) | 2016-01-08 | 2022-07-19 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with reduced side-effects |
| KR20180100624A (ko) | 2016-01-08 | 2018-09-11 | 아센디스 파마 그로우쓰 디스오더스 에이/에스 | 증가된 nep 안정성을 갖는 제어 방출성 cnp 작용제 |
| AU2017205268B2 (en) * | 2016-01-08 | 2022-02-17 | Ascendis Pharma Growth Disorders A/S | CNP prodrugs with large carrier moieties |
| AU2017205693B2 (en) | 2016-01-08 | 2022-03-31 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with low initial NPR-B activity |
| CN115177720A (zh) | 2016-01-08 | 2022-10-14 | 阿森迪斯药物生长障碍股份有限公司 | 具有低npr-c结合的控制释放cnp激动剂 |
| HRP20230385T1 (hr) | 2016-09-29 | 2023-06-23 | Ascendis Pharma Growth Disorders A/S | Kombinirana terapija sa cnp agonistima s kontroliranim oslobađanjem |
| WO2018163131A1 (en) | 2017-03-10 | 2018-09-13 | Quiapeg Pharmaceuticals Ab | Releasable conjugates |
| EP3849660A1 (en) | 2018-09-12 | 2021-07-21 | QuiaPEG Pharmaceuticals AB | Releasable glp-1 conjugates |
| EP3923906A1 (en) | 2019-02-11 | 2021-12-22 | Ascendis Pharma Growth Disorders A/S | Dry pharmaceutical formulations of cnp conjugates |
| TW202124422A (zh) * | 2019-09-16 | 2021-07-01 | 美商拜奧馬林製藥公司 | Cnp變異體及其共軛物 |
| WO2023227505A1 (en) | 2022-05-23 | 2023-11-30 | Ascendis Pharma Growth Disorders A/S | Liquid pharmaceutical formulations of cnp compounds |
| US12077561B2 (en) | 2022-11-02 | 2024-09-03 | Novo Nordisk A/S | CNP compounds |
Family Cites Families (60)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6624142B2 (en) | 1997-12-30 | 2003-09-23 | Enzon, Inc. | Trimethyl lock based tetrapartate prodrugs |
| WO2000018422A1 (fr) | 1998-09-28 | 2000-04-06 | Santen Pharmaceutical Co., Ltd. | Promoteurs de secretion lacrymale ou gouttes ophtalmologiques contenant des peptides natriuretiques comme principes actifs pour traiter la keratoconjonctivite |
| BRPI0203172B8 (pt) | 2001-09-28 | 2021-05-25 | Nakao Kazuwa | composição farmacêutica para acondroplasia |
| CN101724075B (zh) | 2001-10-10 | 2014-04-30 | 诺和诺德公司 | 肽的重构和糖缀合 |
| US7122189B2 (en) | 2002-08-13 | 2006-10-17 | Enzon, Inc. | Releasable polymeric conjugates based on aliphatic biodegradable linkers |
| AU2003297583B2 (en) * | 2002-11-26 | 2010-01-14 | Biocon, Ltd | Modified naturetic compounds, conjugates, and uses thereof |
| US7332164B2 (en) | 2003-03-21 | 2008-02-19 | Enzon Pharmaceuticals, Inc. | Heterobifunctional polymeric bioconjugates |
| WO2004089280A2 (en) | 2003-04-08 | 2004-10-21 | Yeda Research And Development Co. Ltd. | Reversible pegylated drugs |
| US8377917B2 (en) | 2004-03-23 | 2013-02-19 | Complex Biosystems Gmbh | Polymeric prodrug with a self-immolative linker |
| GB2427360A (en) | 2005-06-22 | 2006-12-27 | Complex Biosystems Gmbh | Aliphatic prodrug linker |
| CU23556A1 (es) | 2005-11-30 | 2010-07-20 | Ct Ingenieria Genetica Biotech | Estructura polimérica semejante a dendrímero para la obtención de conjugados de interés farmacéutico |
| US8450269B2 (en) | 2006-02-03 | 2013-05-28 | Prolor Biotech Ltd. | Long-acting growth hormone and methods of producing same |
| JP2010502231A (ja) | 2006-09-08 | 2010-01-28 | メイヨ・ファウンデーション・フォー・メディカル・エデュケーション・アンド・リサーチ | 血管拡張作用を欠く水利尿およびナトリウム利尿ポリペプチド |
| JP2010503707A (ja) | 2006-09-15 | 2010-02-04 | エンゾン ファーマスーティカルズ インコーポレイテッド | オリゴヌクレオチドの送達を目的としたヒンダードエステル系生体分解性リンカー |
| EP2409687B1 (en) | 2006-11-09 | 2014-06-11 | Alcon Research, Ltd. | Water insoluble polymer matrix for drug delivery |
| KR20080098216A (ko) | 2007-05-04 | 2008-11-07 | 한미약품 주식회사 | 캐리어 물질을 이용한 나트륨 배설 펩타이드 약물 결합체 |
| DK2369005T3 (da) | 2007-06-21 | 2013-06-24 | Univ Muenchen Tech | Biologisk aktive proteiner med forøget stabilitet in vivo og/eller in vitro |
| EP2175878A4 (en) | 2007-07-11 | 2014-12-03 | Belrose Pharma Inc | POLYMER DRUG DISPENSING SYSTEM WITH A MULTIPLE SUBSTITUTED AROMATIC PART |
| AR069409A1 (es) | 2007-11-21 | 2010-01-20 | Biomarin Pharm Inc | Variantes de peptidos natriureticos de tipo c |
| RU2010136023A (ru) * | 2008-02-01 | 2012-03-10 | Асцендис Фарма Ас (Dk) | Пролекарство, содержащее саморасщепляемый линкер |
| EP2277890B1 (en) * | 2008-05-23 | 2016-01-06 | Daiichi Sankyo Company, Limited | Peptide capable of extending half-life of peptide of interest in plasma |
| EP2288261A4 (en) | 2008-05-23 | 2013-09-25 | Enzon Pharmaceuticals Inc | POLYMER SYSTEMS CONTAINING INTRA-CELLULAR RELEASE DISULFID LINKERS FOR THE DISPOSAL OF OLIGONUCLEOTIDES |
| WO2009156481A1 (en) | 2008-06-25 | 2009-12-30 | Ascendis Pharma As | Pegylated bnp |
| EP2334335A1 (en) | 2008-09-19 | 2011-06-22 | Nektar Therapeutics | Polymer conjugates of cd-np peptides |
| CN102348715B (zh) | 2009-02-03 | 2017-12-08 | 阿穆尼克斯运营公司 | 延伸重组多肽和包含该延伸重组多肽的组合物 |
| HUE057174T2 (hu) * | 2009-05-20 | 2022-04-28 | Biomarin Pharm Inc | A C-típusú nátriuretikus peptid variánsai |
| US9173953B2 (en) | 2009-07-31 | 2015-11-03 | Ascendis Pharma As | Prodrugs containing an aromatic amine connected by an amido bond to a linker |
| WO2011031441A1 (en) | 2009-08-28 | 2011-03-17 | The Government Of The United States Of America As Represented By The Secretary Of The Department Of Health And Human Services | Therapy with a chimeric molecule and a pro-apoptotic agent |
| WO2011075471A2 (en) | 2009-12-18 | 2011-06-23 | Alcon Research, Ltd. | Novel npr-b agonists and methods of use for the treatment of c-type natriuretic peptide-mediated disorders |
| EP2519228A4 (en) | 2009-12-31 | 2013-06-19 | Enzon Pharmaceuticals Inc | POLYMER CONJUGATES FROM AN AROMATIC AMINE WITH COMPOUNDS WITH A CLEANER UREA CONTAINER |
| EP2525829A1 (en) | 2010-01-22 | 2012-11-28 | Ascendis Pharma A/S | Dipeptide-based prodrug linkers for aromatic amine-containing drugs |
| US9561285B2 (en) | 2010-01-22 | 2017-02-07 | Ascendis Pharma As | Carrier-linked carbamate prodrug linkers |
| WO2011089216A1 (en) | 2010-01-22 | 2011-07-28 | Ascendis Pharma As | Dipeptide-based prodrug linkers for aliphatic amine-containing drugs |
| EP3372617B1 (en) | 2010-04-02 | 2024-07-24 | Amunix Pharmaceuticals, Inc. | Binding fusion proteins, binding fusion protein-drug conjugates, xten-drug conjugates and methods of making and using same |
| WO2011140392A1 (en) | 2010-05-05 | 2011-11-10 | Prolynx Llc | Controlled drug release from solid supports |
| JP5977229B2 (ja) | 2010-05-05 | 2016-08-24 | プロリンクス リミテッド ライアビリティ カンパニー | 巨大分子共役体からの徐放 |
| EP2571510B1 (en) | 2010-05-21 | 2018-08-08 | XL-protein GmbH | Biosynthetic proline/alanine random coil polypeptides and their uses |
| EP3643306A3 (en) | 2011-08-12 | 2020-08-26 | Ascendis Pharma A/S | Carrier-linked prodrugs having reversible carboxylic ester linkages |
| CA2843875C (en) | 2011-08-12 | 2020-01-07 | Ascendis Pharma A/S | High-loading water-soluble carrier-linked prodrugs |
| EP2741782B1 (en) | 2011-08-12 | 2020-05-06 | Ascendis Pharma A/S | Protein carrier-linked prodrugs |
| EP2741778A1 (en) | 2011-08-12 | 2014-06-18 | Ascendis Pharma A/S | Polymeric hyperbranched carrier-linked prodrugs |
| MX349950B (es) | 2011-08-12 | 2017-08-22 | Ascendis Pharma As | Profarmacos de treprostinil enlazados a un portador. |
| WO2013036857A1 (en) | 2011-09-07 | 2013-03-14 | Prolynx Llc | Sulfone linkers |
| EP2853273A4 (en) | 2012-04-25 | 2016-01-13 | Daiichi Sankyo Co Ltd | BONE REPAIR ACCELERATOR |
| US20150087688A1 (en) | 2012-04-25 | 2015-03-26 | Ascendis Pharma A/S | Prodrugs of hydroxyl-comprising drugs |
| US10052366B2 (en) | 2012-05-21 | 2018-08-21 | Alexion Pharmaceuticsl, Inc. | Compositions comprising alkaline phosphatase and/or natriuretic peptide and methods of use thereof |
| JP6536871B2 (ja) | 2013-12-02 | 2019-07-03 | 国立大学法人京都大学 | Fgfr3病の予防および治療剤ならびにそのスクリーニング方法 |
| CA2940835A1 (en) | 2014-02-27 | 2015-09-03 | Daiichi Sankyo Company, Limited | Medicine against growth failure induced by administration of steroid |
| EP3193941B1 (en) | 2014-08-06 | 2024-05-22 | Ascendis Pharma A/S | Prodrugs comprising an aminoalkyl glycine linker |
| WO2016110577A1 (en) | 2015-01-09 | 2016-07-14 | Ascendis Pharma A/S | Cnp prodrugs |
| US11202819B2 (en) | 2015-12-08 | 2021-12-21 | Biomarin Pharmaceutical Inc. | Use of C-type natriuretic peptide variants to treat osteoarthritis |
| CN115177720A (zh) | 2016-01-08 | 2022-10-14 | 阿森迪斯药物生长障碍股份有限公司 | 具有低npr-c结合的控制释放cnp激动剂 |
| AU2017205268B2 (en) | 2016-01-08 | 2022-02-17 | Ascendis Pharma Growth Disorders A/S | CNP prodrugs with large carrier moieties |
| US11389511B2 (en) | 2016-01-08 | 2022-07-19 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with reduced side-effects |
| KR20180100624A (ko) | 2016-01-08 | 2018-09-11 | 아센디스 파마 그로우쓰 디스오더스 에이/에스 | 증가된 nep 안정성을 갖는 제어 방출성 cnp 작용제 |
| EP3400019B1 (en) | 2016-01-08 | 2022-09-28 | Ascendis Pharma Growth Disorders A/S | Cnp prodrugs with carrier attachment at the ring moiety |
| AU2017205693B2 (en) | 2016-01-08 | 2022-03-31 | Ascendis Pharma Growth Disorders A/S | Controlled-release CNP agonists with low initial NPR-B activity |
| HRP20230385T1 (hr) | 2016-09-29 | 2023-06-23 | Ascendis Pharma Growth Disorders A/S | Kombinirana terapija sa cnp agonistima s kontroliranim oslobađanjem |
| CA3109223A1 (en) | 2018-08-16 | 2020-02-20 | Beyondspring Pharmaceuticals, Inc. | Method and composition for stimulating immune response |
| EP3923906A1 (en) | 2019-02-11 | 2021-12-22 | Ascendis Pharma Growth Disorders A/S | Dry pharmaceutical formulations of cnp conjugates |
-
2017
- 2017-01-05 EP EP17700496.7A patent/EP3400019B1/en active Active
- 2017-01-05 RS RS20221110A patent/RS63793B1/sr unknown
- 2017-01-05 HR HRP20221339TT patent/HRP20221339T1/hr unknown
- 2017-01-05 US US16/067,095 patent/US11413351B2/en active Active
- 2017-01-05 AU AU2017205688A patent/AU2017205688C1/en active Active
- 2017-01-05 ES ES17700496T patent/ES2934582T3/es active Active
- 2017-01-05 CA CA3007979A patent/CA3007979C/en active Active
- 2017-01-05 LT LTEPPCT/EP2017/050209T patent/LT3400019T/lt unknown
- 2017-01-05 DK DK17700496.7T patent/DK3400019T3/da active
- 2017-01-05 WO PCT/EP2017/050209 patent/WO2017118698A1/en active Application Filing
- 2017-01-05 JP JP2018535367A patent/JP6968802B2/ja active Active
- 2017-01-05 PL PL17700496.7T patent/PL3400019T3/pl unknown
- 2017-01-05 SI SI201731268T patent/SI3400019T1/sl unknown
- 2017-01-05 HU HUE17700496A patent/HUE060521T2/hu unknown
- 2017-01-05 EP EP22197366.2A patent/EP4162955A1/en active Pending
-
2018
- 2018-07-02 ZA ZA2018/04415A patent/ZA201804415B/en unknown
-
2021
- 2021-10-27 JP JP2021175180A patent/JP2022025104A/ja active Pending
- 2021-11-30 AU AU2021277638A patent/AU2021277638C1/en active Active
-
2022
- 2022-06-16 US US17/842,436 patent/US20220331438A1/en active Pending
-
2023
- 2023-08-02 AU AU2023210599A patent/AU2023210599B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| WO2017118698A1 (en) | 2017-07-13 |
| JP2022025104A (ja) | 2022-02-09 |
| DK3400019T3 (da) | 2022-10-24 |
| LT3400019T (lt) | 2022-12-12 |
| EP3400019B1 (en) | 2022-09-28 |
| AU2021277638B2 (en) | 2023-05-18 |
| JP6968802B2 (ja) | 2021-11-17 |
| HUE060521T2 (hu) | 2023-03-28 |
| PL3400019T3 (pl) | 2023-01-23 |
| ZA201804415B (en) | 2019-03-27 |
| EP3400019A1 (en) | 2018-11-14 |
| US11413351B2 (en) | 2022-08-16 |
| US20220331438A1 (en) | 2022-10-20 |
| CA3007979A1 (en) | 2017-07-13 |
| AU2023210599B2 (en) | 2024-10-03 |
| JP2019510735A (ja) | 2019-04-18 |
| AU2017205688C1 (en) | 2022-03-10 |
| AU2017205688B2 (en) | 2021-11-04 |
| AU2021277638C1 (en) | 2023-11-09 |
| EP4162955A1 (en) | 2023-04-12 |
| AU2021277638A1 (en) | 2021-12-23 |
| AU2017205688A1 (en) | 2018-06-07 |
| US20190008977A1 (en) | 2019-01-10 |
| HRP20221339T1 (hr) | 2023-01-06 |
| RS63793B1 (sr) | 2022-12-30 |
| CA3007979C (en) | 2023-12-19 |
| AU2023210599A1 (en) | 2023-08-24 |
| SI3400019T1 (sl) | 2023-01-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2934582T3 (es) | Profármacos de CNP con unión de portador en el resto del anillo | |
| JP7499807B2 (ja) | 増加したnep安定性を有する徐放cnp作動薬 | |
| US11154593B2 (en) | CNP prodrugs with large carrier moieties | |
| AU2017205693B2 (en) | Controlled-release CNP agonists with low initial NPR-B activity | |
| US20220409700A1 (en) | Controlled-release cnp agonists with reduced side-effects | |
| US20220211817A1 (en) | Controlled-release cnp agonists with low npr-c binding |