ES2918193T3 - Polvo medicamentoso para administración por inhalación y un proceso del mismo - Google Patents
Polvo medicamentoso para administración por inhalación y un proceso del mismo Download PDFInfo
- Publication number
- ES2918193T3 ES2918193T3 ES08775027T ES08775027T ES2918193T3 ES 2918193 T3 ES2918193 T3 ES 2918193T3 ES 08775027 T ES08775027 T ES 08775027T ES 08775027 T ES08775027 T ES 08775027T ES 2918193 T3 ES2918193 T3 ES 2918193T3
- Authority
- ES
- Spain
- Prior art keywords
- drug
- solution
- adjuvant
- particles
- tobramycin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/007—Pulmonary tract; Aromatherapy
- A61K9/0073—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy
- A61K9/0075—Sprays or powders for inhalation; Aerolised or nebulised preparations generated by other means than thermal energy for inhalation via a dry powder inhaler [DPI], e.g. comprising micronized drug mixed with lactose carrier particles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/7036—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin having at least one amino group directly attached to the carbocyclic ring, e.g. streptomycin, gentamycin, amikacin, validamycin, fortimicins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1682—Processes
- A61K9/1688—Processes resulting in pure drug agglomerate optionally containing up to 5% of excipient
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Otolaryngology (AREA)
- Pulmonology (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Se describe un fármaco en polvo y un proceso para su preparación, que comprende partículas de fármacos en cantidades iguales o superiores al 90% p/p, en donde dichas partículas de fármacos están al menos parcialmente recubiertas con una cantidad inferior o igual a 2.0% w/ W de un adyuvante que consiste en una o una mezcla de ácido graso saturado C12-C18 o sus sales. La invención se relaciona particularmente con el polvo de tobramicina. (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Polvo medicamentoso para administración por inhalación y un proceso del mismo
CAMPO DE LA INVENCIÓN
[0001] La presente invención se refiere a un polvo que comprende una sustancia antibiótica de aminoglucósido recubierta al menos parcialmente con un ácido graso o su sal, y a su uso por inhalación para su uso
[0002] en el tratamiento de enfermedades derivadas de infecciones bacterianas.
ESTADO DE LA TÉCNICA
[0003] La administración de los antibióticos absorbidos mal de forma oral, como los aminoglucósidos, a la vía aérea por la vía de aerosoles ha sido analizada en pacientes con fibrosis quística. Gran parte de este trabajo se ha destinado al tratamiento de infecciones pulmonares crónicas con la administración de soluciones mediante nebulización.
[0004] La fibrosis quística (FQ) es una enfermedad hereditaria común y potencialmente mortal que afecta a más de 60.000 personas en todo el mundo. Está asociada con un error en el transporte de iones cloruro a través de las membranas epiteliales de las glándulas exocrinas, lo que provoca una reducción del contenido de agua de sus secreciones (Moskowitz, S.M. y col., 2005, Cystic fibrosis lung disease: genetic influences, microbial interactions, and radiological assessment. Pediatr. Radiol. 35, 739-757). A los cambios morfológicos de dilatación e hipertrofia de las glándulas bronquiales les sigue el taponamiento mucoso. Este moco viscoso en las vías respiratorias permite la colonización bacteriana, con la consiguiente infección del tracto respiratorio, lo que contribuye al daño tisular en curso. Haemophilus influenzae y Staphylococcus aureus son los primeros patógenos que colonizan la vía respiratoria durante la infancia. Conforme progresa la enfermedad pulmonar, a esta le seguirá la colonización por Pseudomonas aeruginosa. Tras un período de colonización intermitente con P.aeruginosa, la colonización se vuelve crónica en la mayoría de pacientes con FQ, y prácticamente imposible de erradicar. Los pacientes con FQ suelen ser tratados con dos antibióticos antipseudomónicos parenterales, uno de los cuales es normalmente un aminoglucósido. Estos antibióticos luchan contra la progresión de la FQ reduciendo el deterioro pulmonar y mejorando la función de los pulmones.
[0005] Los aminoglucósidos son un grupo de antibióticos que incluye al menos ocho fármacos: amikacina, gentamicina, kanamicina, neomicina, netilmicina, paromomicina, estreptomicina y tobramicina. Estos presentan la misma estructura química básica, es decir, anillo de aminociclitol unido a aminoazúcares en su estructura. Su actividad bactericida se atribuye a la unión irreversible a los ribosomas, aunque también se ha considerado su interacción con otras estructuras celulares y procesos metabólicos. Tienen un amplio espectro antimicrobiano. Son activos contra bacilos gramnegativos aerobios y aerobios facultativos, y contra algunas bacterias grampositivas como estafilococos.
[0006] Los aminoglucósidos administrados por vía intravenosa penetran escasamente en el espacio endobronquial, alcanzando concentraciones de esputo máximas medias que son solo del 12 al 20 % de las concentraciones séricas máximas. No obstante, puesto que los componentes del esputo de FQ se unen a los aminoglucósidos, las concentraciones de esputo deben superar de 10 a 25 veces la concentración inhibitoria mínima para ser bactericida (Mendelman PM, Smith AL, Levy J, y col. Aminoglycoside penetration, inactivation, and efficacy in cystic fibrosis sputum. Am Rev Respir Dis 1985; 132:761-765). Por lo tanto, para alcanzar concentraciones farmacéuticas adecuadas en el sitio de infección, es necesario utilizar dosis grandes. Un inconveniente de los aminoglucósidos es su asociación con la nefrotoxicidad y la ototoxicidad, estando ambas asociadas con niveles plasmáticos mínimos elevados y niveles máximos elevados y constantes.
[0007] Estos problemas pueden superarse con el uso de aminoglucósidos en aerosol, que resultan adecuados para inhalación debido a que siguen siendo bioactivos cuando están en aerosol y son escasamente absorbidos a través de las superficies epiteliales. Por lo tanto, pueden conseguirse teóricamente unas concentraciones elevadas en las secreciones bronquiales con un riesgo mínimo para la toxicidad sistémica.
[0008] El objetivo de la terapia con antibióticos inhalados en FQ es llegar a todo el árbol bronquial desde el tracto superior, que puede contener depósitos de Pseudomonas aeruginosa desde donde pueden infectarse las vías respiratorias inferiores, hasta las pequeñas vías respiratorias periféricas donde progresa la enfermedad. Teniendo en cuenta el hecho de que las vías respiratorias comprenden cerca de un 95 % del área de superficie de los pulmones, la actuación de los antibióticos en la terapia de la FQ debería dirigirse principalmente a las pequeñas vías respiratorias periféricas para alcanzar concentraciones de fármaco lo suficientemente elevadas a lo largo del sistema respiratorio. Por otra parte, los aerosoles están sujetos a problemas de sedimentación gravitacional e impactación. La impactación se produce principalmente con partículas más grandes siempre que el transporte sea rápido y cambie de dirección, o sea turbulento. Por consiguiente, la impactación se producirá en las vías respiratorias superiores, es decir, la boca, la faringe y la laringe y en las vías respiratorias grandes del pulmón hasta los 2 pm de diámetro. La sedimentación es un proceso que depende del tiempo, en el que las pequeñas partículas de aerosol se asientan en las vías respiratorias por la influencia de la gravedad. Esto tiene lugar en las vías respiratorias pequeñas y en los alvéolos.
[0009] La medida más apropiada del tamaño de partícula de los aerosoles es el diámetro aerodinámico, ya que este se refiere al comportamiento dinámico de la partícula y depende de los mecanismos principales de depósito de aerosoles en el tracto respiratorio. Este parámetro físico clave predice el sitio de deposición del aerosol dentro de los pulmones para partículas micronizadas inhaladas. El diámetro aerodinámico de las partículas de aerosol es el que define el sitio de deposición de los fármacos en aerosol: las partículas mayores de 10 pm se depositan en el tracto respiratorio superior y
tienen un efecto terapéutico pequeño. Las partículas pequeñas con un diámetro aerodinámico menor de 5 |jm pasan a través de las vías respiratorias superiores y se depositan en las vías respiratorias inferiores por la influencia de la gravedad y el movimiento browniano. En cambio, puede que las partículas menores de 0,5 jm ni siquiera se depositen, ya que estas se asientan muy lentamente y pueden ser arrastradas por el flujo de aire y, finalmente, exhaladas. Los aerosoles suelen ser heterodispersos, y su comportamiento se describe mediante el diámetro aerodinámico mediano de la masa (MMAD). La mitad de la masa del aerosol está contenida en partículas más pequeñas, y la otra mitad de la masa del aerosol está contenida en partículas mayores que el MMAD.
[0010] Existen cinco clases actuales de dispositivos de inhalación que pueden suministrar fármacos al tracto respiratorio: inhaladores de polvo seco, inhaladores de dosis medida, nebulizadores de dosis medida, nebulizadores ultrasónicos y nebulizadores de tipo «jet». En su forma actual, los inhaladores de dosis medida resultan poco prácticos para suministrar la masa de sustancias aminoglucósidas requerida para la actividad. Los nebulizadores de dosis medida son dispositivos que producen aerosoles forzando la salida de una solución a través de un tamiz a alta presión, y no han sido clínicamente analizados para la distribución de antibióticos. Para tratar de manera adecuada la lesión pulmonar en la FQ, donde la implicación comienza en los bronquiolos más pequeños y se extiende hacia los bronquios, el nebulizador debería producir partículas en el intervalo de 1-5 jm . No obstante, existe una gran variación entre distintos nebulizadores en el tamaño medio y el intervalo de tamaños de partículas generado. Newman y col. han evaluado varios nebulizadores de tipo «jet» disponibles en el mercado para su uso con solución de gentamicina (Newman SP, Pellow PGD, Clay MM, Clarke Sw . Evaluation of jet nebulisers for use with gentamicin solution. Thorax 1985; 40: 671-676. 1982; 4: 626-630). Se analizaron la salida de aerosol, el tamaño de gota y el tiempo de nebulización. Se encontró una diferencia de diez veces entre el dispositivo de administración más eficiente y el menos eficiente. La eficiencia aumentó con el caudal de aire y el volumen que se va a nebulizar.
[0011] Hasta la fecha, el único producto antibiótico aminoglucosídico inhalable disponible en el mercado es Tobi®, una solución de tobramicina sin conservantes para nebulización que ha sido aprobada por la Administración de Alimentos y Medicamentos de Estados Unidos (D. E. Geller, W. H. Pitlick, P. A. Nardella, W. G. Tracewell, B. W Ramsey. Pharmacokinetics and bioavailability of aerosolized tobramycin in cystic fibrosis. Chest (2002) 122 (1) págs. 219-26). Los nebulizadores ultrasónicos y los de tipo «jet» se utilizan con frecuencia para administrar solución de tobramicina a los pulmones. La necesidad de una unidad de compresión o aire presurizado para el tipo «jet» y electricidad para el nebulizador ultrasónico inmoviliza al paciente. Se requiere la limpieza y desinfección regular del nebulizador con el fin de prevenir la contaminación con microorganismos y la posterior colonización de la orofaringe del paciente. Un inconveniente aún mayor de los nebulizadores es su baja eficiencia. Para la mayoría de los sistemas nebulizadores, se estima que aproximadamente el 10 % de la masa de fármaco inicialmente nebulizada se deposita en los pulmones, y el 90 % restante permanece en el nebulizador, o impacta en la orofaringe y se traga, o se dispersa en la atmósfera (Touw DJ, Brimicombe RW, Hodson ME, y col. Inhalation of antibiotics in cystic fibrosis. Eur Respir J 1995; 8:1594 -1604).
[0012] La inhalación de polvo seco puede suponer una alternativa a la nebulización de fármacos. En unos pocos estudios, se ha observado el uso de antibióticos micronizados. Goldman y col. han mostrado que el polvo de gentamicina micronizado inhalado produce concentraciones de gentamicina en el líquido de lavado broncoalveolar similares a las producidas por la solución de gentamicina nebulizada (J.M. Goldman, S.M. Bayston, S. O'Connor, R.E. Meigh, Inhaled micronized gentamicin powder: a new delivery system, Thorax 45 (1990) 939-940). La inhalación de una preparación de polvo seco es rápida y cómoda, y puede ser particularmente adecuada para pacientes con fibrosis quística y bronquiectasia que deseen mantener un estilo de vida independiente.
[0013] No obstante, los polvos micronizados muestran poca fluidez y una tendencia extrema a la aglomeración y adhesión. Deben ser procesados en una formulación adecuada de flujo libre para la reproducibilidad precisa de la dosis, especialmente en el intervalo de dosis bajo, utilizado para inhalación.
[0014] Existen tecnologías recientes de ingeniería de partículas, como el proceso PulmoSphere (Inhale Therapeutic Systems; San Carlos, California, EE. UU.), que han dado como resultado polvos fácilmente dispersables con propiedades aerodinámicas mejoradas. Una administración pulmonar de un aerosol de polvo de tobramicina modificado (formulación de tobramicina PulmoSphere, PStob; Inhale Therapuetic Systems) a partir de un IPS (inhalador de polvo seco) simple y portátil ha sido descrita por Newhouse y col. (Newhouse y col., 2003, Newhouse MT, Hirst PH, Duddu SP, Walter YH, Tarara TE, Clark AR, Weers JG: Inhalation of a dry powder tobramycin PulmoSphere formulation in healthy volunteers. Chest 2003;124:360-366). Este estudio ha demostrado que la administración pulmonar de sulfato de tobramicina a partir de un IPS pasivo es posible para la PStob debido a la elevada carga de fármaco (90 % en peso/peso de sulfato de tobramicina), y una deposición pulmonar nueve veces mejorada en comparación con el nebulizador a lo largo de un período de administración de quince minutos. Se puede suministrar una dosis comparable al pulmón con polvo PStob a la del producto nebulizado, pero en aproximadamente una décima parte del tiempo y con un esfuerzo considerablemente menor. Esto tiene el potencial de mejorar la adherencia y la aceptación del paciente. La masa relativamente grande de polvo administrado durante 15 min (150 mg, 52 mg depositados en el pulmón y 65 mg depositados en la orofaringe) se toleró bien.
[0015] Pilcer y col. (Pilcer G., Sebti T., Amighi K: Formulation and characterization of lipid-coated tobramycin particles for dry powder inhalation. Pharm Res (2006) vol. 23 (5) pp. 931-40) demostraron que los 15 minutos de nebulización de 300 mg de Tobi® se sustituirían ventajosamente por la inhalación de cuatro cápsulas de 15 mg o dos cápsulas de 30 mg de tobramicina recubierta con lípidos. Se utilizaron fosfolípidos y partículas de tobramicina recubiertas de colesterol para mejorar la selectividad del fármaco en el pulmón. El recubrimiento lipídico modificó las propiedades de las partículas de
tobramicina micronizadas, permitiendo su deposición en el pulmón. Los lípidos utilizados fueron fosfolípidos y colesterol, dos componentes bien tolerados fisiológicamente utilizados en una concentración del 5 % en tobramicina. El estudio demostró que el uso de composiciones de fosfolípidos, a base de mezclas de colesterol y fosfolípidos, mejoró la administración de tobramicina al tracto pulmonar. Las partículas preparadas eran pequeñas y presentaron unos resultados de FPF (fracción de partícula fina) muy elevados. Se observó una buena fluidez, haciendo que los polvos sean idealmente adecuados para su uso en inhaladores de polvo seco sin vehículo.
[0016] Por consiguiente, los autores seleccionaron dos formulaciones lipídicas en función de su comportamiento aerodinámico y sus valores de dosis de partícula fina (FPD) y los compararon con Tobi® (solución de nebulizador) (Chiron Corporation; Seattle, Washington, EE. UU.) en una evaluación combinada in vivo de centellografía y farmacocinética de tobramicina tras la inhalación de una única dosis oral en nueve pacientes con FQ. Los resultados del estudio han demostrado que las formulaciones de tobramicina para IPS que contienen elevadas concentraciones de fármaco permiten una elevada deposición en el pulmón de los pacientes. La inhalación de una cápsula de 40 mg o dos cápsulas de 20 mg de tobramicina micronizada recubierta con lípido o dos cápsulas de 30 mg de tobramicina micronizada podría ser un sustituto ventajoso a la administración de Tobi®.
[0017] Por consiguiente, para el uso de inhalación en inhaladores de polvo seco, se advierte la necesidad de tener polvos de sustancia farmacológica transformados con técnicas apropiadas en colecciones de partículas respirables. El tamaño, la forma y la densidad permiten que la partícula se deposite en la zona apropiada del pulmón. En concreto, el aspecto relevante es presentar una partícula respirable que contenga la menor cantidad de adyuvante para evitar cargar el pulmón con sustancias no activas y potencialmente inocuas. Por lo tanto, el principal problema para la administración mediante inhalación de algún polvo de fármacos aminoglucósidos es transformar sus características de micromerítica con el fin de adaptarlo para su uso en inhaladores de polvo seco, evitando el uso excesivo de adyuvante. Algunos problemas adicionales, más allá de los implicados en las propiedades aerodinámicas, son la reducción de la cohesión del polvo pequeño y su sensibilidad a la humedad del ambiente, o su comportamiento higroscópico que se opone significativamente a la capacidad del polvo para ser aerosolizado o simplemente dosificado, o también la conservación durante el almacenamiento.
[0018] El aminoglucósido para administración pulmonar presenta problemas en la fabricación de polvos micronizados que deben ser respirables, estables, con una elevada concentración del fármaco y fluidos. Un proceso de micronización utilizado con frecuencia para la realización de polvos para inhalación es el proceso de secado por pulverización, en el que las gotas de solución o dispersión del producto se secan en un flujo de aire caliente.
[0019] Las soluciones propuestas en la literatura a los problemas de los polvos para inhalación de tobramicina fabricados mediante secado por pulverización consistieron en el secado por pulverización de dispersiones de partículas micronizadas de tobramicina o emulsiones de solución acuosa del fármaco en disolventes orgánicos.
[0020] Nunca se ha llevado a cabo el secado por pulverización de una solución acuosa a base de tobramicina.
[0021] Los presentes inventores trataron de secar por pulverización soluciones acuosas de tobramicina sin excipientes, pero obtuvieron polvos demasiado higroscópicos, delicuescentes e inestables en condiciones de almacenamiento normales. En estos casos, la respirabilidad fue inferior al 30 %, pero, lo que es más importante, el polvo no podía ser aerosolizado debido a la elevada cohesión de las partículas y a la baja fluidez.
[0022] El uso de sustancias lipídicas como adyuvante para tratar de reducir la absorción de agua y mejorar la fluidez fue utilizado por Pilcer G., pero el procedimiento descrito requería el uso de polvos de tobramicina previamente micronizados. Puesto que las sustancias lipídicas no son solubles en agua, el proceso para el recubrimiento de las partículas de fármaco requería disolventes orgánicos para la combinación de manera homogénea del adyuvante con el fármaco. Por lo tanto, los autores secaron por pulverización una suspensión de partículas de tobramicina previamente micronizadas en disolventes orgánicos que contenían los lípidos disueltos. Además del problema del uso de disolventes orgánicos para la disolución de lípidos, la tobramicina utilizada en este proceso necesita ser micronizada por adelantado mediante un tratamiento mecánico antes de ser secada por pulverización. Asimismo, los polvos de tobramicina para inhalación contenían un 5 % p/p de fosfolípidos y colesterol, y la gran cantidad de estas sustancias incorpora preocupaciones sobre la estabilidad fisicoquímica.
[0023] El polvo de tobramicina para inhalación también se fabricó utilizando la tecnología PulmoSphere a base de lípidos, produciendo partículas porosas altamente dispersables. En este caso, se han realizado partículas de tobramicina utilizando una solución de sulfato de tobramicina a base de emulsión que contenía un agente espumante volátil (perfluorooctil bromuro) que ayuda en la creación de partículas de tipo esponja con una densidad ultrabaja. El lípido era fosfatidilcolina en una concentración elevada en relación con la tobramicina utilizada como emulsionante, y la sustancia farmacológica era la sal de sulfato.
[0024] En el documento WO 2008053253 se da a conocer el uso de formulaciones de polvo seco que se preparan para conseguir una alta dosis suministrada del agente activo, donde hay presente un agente de control de fuerza en la superficie de las partículas de agente activo.
SUMARIO
[0025] Los problemas anteriormente indicados y los inconvenientes de los sistemas de la técnica anterior se superan mediante un polvo medicamentoso para inhalación que comprende partículas en las que el contenido de fármaco es igual o mayor al 90 % p/p, donde dichas partículas de fármaco están recubiertas al menos parcialmente con una cantidad de un adyuvante menor o igual al 2,0 % p/p con respecto al peso del fármaco, consistiendo dicho adyuvante en uno o una mezcla de ácidos grasos saturados C12-C18 o sus sales, donde dicho polvo medicamentoso se obtiene mediante a) la preparación de una solución límpida de las partículas de fármaco mezclando una solución acuosa de las partículas de fármaco con una solución alcohólica del adyuvante, donde el adyuvante que consiste en uno o una mezcla de ácidos grasos saturados C12-C18 o sus sales está disperso de manera coloidal en la solución; y
b) el secado por pulverización de la solución límpida de la etapa a) utilizando una temperatura de entrada de entre 120-135 °C, una velocidad de alimentación de entre 3,0 y 6,0 ml/min, un caudal de aire de 600 l/h, una boquilla de entre 0,5 y 1 mm, y un aspirador 100 %, donde
la temperatura de la solución límpida en la etapa a) y en la etapa b) es mayor o igual a 28 °C para dispersar de manera coloidal el adyuvante en la solución, y donde dicho polvo medicamentoso es un antibiótico aminoglucósido que comprende partículas de antibiótico que presentan una solubilidad en agua de al menos un 3,0 % p/v.
[0026] En la presente descripción:
- cuando se utiliza el término «fármaco» se pretende hacer referencia a sustancias antibióticas aminoglucósidas que presentan una solubilidad en agua de al menos un 3,0 % p/v;
- cuando se utiliza la expresión «al menos parcialmente recubierto/a(s)» para una partícula de fármaco, se pretende que la partícula de fármaco tenga al menos más de un 20 % de la superficie de la partícula recubierta, preferiblemente más del 50 %; más preferiblemente, el recubrimiento es en forma de manchas.
[0027] En otro aspecto, se describe un proceso para preparar el polvo medicamentoso, que permite la transformación de las propiedades micromeríticas, es decir, las características de densidad, forma y tamaño de las partículas de sustancias antibióticas aminoglucósidas, con el fin de conseguir que el polvo sea respirable, estable, altamente concentrado y fluido, es decir, adecuado para su inhalación como inhaladores de polvo seco.
[0028] El producto dado a conocer en el presente documento comprende lípidos tales como ácidos grasos saturados que tienen una longitud de cadena distinta o sus sales. Estos lípidos no son miscibles con la solución acuosa, y no resulta viable un mezclado profundo capaz de distribuir el lípido sobre la superficie de la partícula de fármaco formada durante el secado por gotas. En el presente documento se describe un proceso que contempla la preparación de una dispersión acuosa para pulverización, en la que el adyuvante lipídico se dispersó de manera coloidal en la solución acuosa de fármaco. El procedimiento inventado fue capaz de proporcionar una solución de fármaco y adyuvante, que siguió siendo límpida sin la separación de los componentes durante el tiempo requerido por el secado por pulverización de la solución.
[0029] Por lo tanto, el proceso para la preparación del polvo medicamentoso comprende las siguientes etapas:
a) la preparación de una solución límpida de partículas de fármaco mezclando una solución acuosa de las partículas de fármaco con una solución alcohólica del adyuvante, donde un adyuvante que consiste en uno o una mezcla de ácidos grasos saturados C12-C18 o sus sales se dispersa de manera coloidal en la solución; y
b) el secado por pulverización de la solución límpida de la etapa a) utilizando una temperatura de entrada de entre 120-135 °C, una velocidad de alimentación de entre 3,0 y 6,0 ml/min, un caudal de aire de 600 l/h, una boquilla de entre 0,5 y 1 mm, y un aspirador 100 %, donde
[0030] la temperatura de la solución límpida en la etapa a) y en la etapa b) es mayor o igual a 28 °C para dispersar de manera coloidal el adyuvante en la solución, y donde dicho polvo medicamentoso es un antibiótico aminoglucósido que comprende partículas de antibiótico que presentan una solubilidad en agua de al menos un 3,0 % p/v.
[0031] En el presente proceso, la solución es límpida, puesto que el adyuvante está disperso de manera coloidal en la solución. Para dispersar de manera coloidal el adyuvante, la temperatura de la solución en la etapa a) y en la etapa b) se seleccionaría en función del tipo de ácido graso saturado C12-C18. Dicha temperatura es mayor o igual a 28 °C, más preferiblemente mayor o igual a 30 °C, aún más preferiblemente de aproximadamente 30 °C.
[0032] Esta solución se obtuvo mediante un mezclado apropiado de la solución acuosa del fármaco con una solución alcohólica del adyuvante. De este modo, durante el secado de la gota producido en el aparato de secado por pulverización, debido a la evaporación del disolvente, el adyuvante precipita en la interfase agua/aire de la gota que forma la partícula de fármaco. El procedimiento adoptado y los ingredientes utilizados determinaron la deposición del ácido graso saturado, preferiblemente como manchas, sobre la superficie de la partícula de fármaco seca.
[0033] El resultado del proceso es la obtención de una partícula de fármaco, destinada al tratamiento de una infección broncopulmonar bacteriana crónica, y que presenta una alta respirabilidad, estabilidad, un elevado contenido de principio activo, sin afectar a la fluidez y disolución del fármaco.
DESCRIPCIÓN DE LAS FIGURAS
[0034] A continuación se describirán ciertas formas de realización en la descripción detallada que se muestra abajo, en referencia a las siguientes figuras, en las cuales:
- la Figura 1 muestra una partícula de tobramicina típica secada por pulverización, que puede obtenerse mediante los presentes Ejemplo 1 y Ejemplo 2, respectivamente; y
- la Figura 2 muestra la presencia del adyuvante en la superficie de la partícula de fármaco obtenida en el presente Ejemplo 3, dependiendo de la cantidad de adyuvante utilizada en la preparación de la solución.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
[0035] La invención se refiere a un polvo medicamentoso según lo definido en la reivindicación 1.
[0036] El polvo de la invención comprende partículas de antibiótico, que es un antibiótico aminoglucósido y presenta una solubilidad en agua de al menos un 3,0 % p/v. Preferiblemente, la sustancia antibiótica aminoglucósida se selecciona del grupo que consiste en amikacina, gentamicina, kanamicina, neomicina, netilmicina, paromomicina, estreptomicina y tobramicina, aún más preferiblemente es tobramicina o amikacina.
[0037] La partícula de fármaco de la invención está recubierta al menos parcialmente con un adyuvante en una cantidad inferior o igual al 2,0 % p/p con respecto al peso del fármaco, que consiste en uno o una mezcla de ácidos grasos saturados C12-C18 o sus sales. El adyuvante se seleccionará preferiblemente del grupo que consiste en ácido láurico, ácido mirístico, ácido palmítico, ácido esteárico, sus mezclas o sus sales. Más preferiblemente, el adyuvante de la invención es ácido esteárico o una sal de este.
[0038] El ácido láurico tiene 12 carbonos y es uno de los tres ácidos grasos saturados más ampliamente distribuidos que se encuentran en la naturaleza (14:0, 16:0 y 18:0). El ácido mirístico tiene 14 carbonos. El ácido palmítico tiene 16 carbonos y es el ácido graso saturado más común en los lípidos de plantas y animales. Por último, el ácido esteárico tiene 18 carbonos y es el ácido graso saturado de mayor peso molecular que se produce abundantemente en grasas y aceites.
[0039] El adyuvante también puede encontrarse en forma de sal de un ácido graso saturado C12-C18. Preferiblemente, la sal está hecha con metales alcalinos o alcalinotérreos, más preferiblemente metales alcalinos y, aún más preferiblemente, es una sal de sodio.
[0040] La partícula de fármaco de la invención está recubierta al menos parcialmente con una cantidad inferior o igual al 2,0 % p/p de un adyuvante con respecto al peso del fármaco. La cantidad de adyuvante está preferiblemente en el intervalo de 0,25 a 2,0 p/p, más preferiblemente en el intervalo de 0,5 a 1,5.
[0041] La presente invención también se refiere a un proceso para la preparación del polvo medicamentoso que comprende las siguientes etapas:
a) preparación de una solución límpida de partículas de fármaco mezclando una solución acuosa de las partículas de fármaco con una solución alcohólica del adyuvante, donde un adyuvante que consiste en uno o una mezcla de ácidos grasos saturados C12-C18 o sus sales se dispersa de manera coloidal en la solución; y
b) secado por pulverización de la solución límpida de la etapa a) utilizando una temperatura de entrada de entre 120 135 °C, una velocidad de alimentación de entre 3,0 y 6,0 ml/min, un caudal de aire de 600 l/h, una boquilla de entre 0,5 y 1 mm, y un aspirador 100 %, donde
la temperatura de la solución límpida en la etapa a) y en la etapa b) es mayor o igual a 28 °C para dispersar de manera coloidal el adyuvante en la solución, y donde
dicho polvo medicamentoso es un antibiótico aminoglucósido que comprende partículas de antibiótico que presentan una solubilidad en agua de al menos un 3,0 % p/v.
[0042] Sin limitarse a ninguna teoría, los inventores han descubierto que, al preparar una solución límpida del fármaco y el adyuvante, podría obtenerse un polvo medicamentoso con una elevada respirabilidad y características físicas y químicas óptimas mediante el secado por pulverización de dicha solución.
[0043] En el presente proceso, la solución es límpida y el adyuvante está disperso de manera coloidal en la solución. Dicha temperatura es mayor o igual a 28 °C, más preferiblemente mayor o igual a 30 °C, aún más preferiblemente de aproximadamente 30 °C. Según una forma de realización preferida, el proceso contempla la preparación de una solución de fármaco y, de manera separada, una solución de adyuvante con el fin de obtener la mezcla límpida de la etapa a).
[0044] Preferiblemente, el fármaco se disuelve en agua, más preferiblemente en una cantidad en el intervalo de 7 a 30 mg/ml y el adyuvante se disuelve en un alcohol, más preferiblemente en una cantidad en el intervalo de 0,15 a 0,70 mg/ml. El alcohol utilizado para la disolución del adyuvante se selecciona preferiblemente del grupo que consiste en etanol, metanol, propanol o isopropanol.
[0045] En la forma de realización preferida, las soluciones de adyuvante y de fármaco se mezclan en una relación en el intervalo de 2:8 a 5:5, más preferiblemente 3:7.
[0046] En la forma de realización más preferida, la solución etanólica de ácido esteárico se mezcla con solución acuosa de tobramicina en una relación de 3:7.
[0047] La solución límpida de la invención en el caso de la tobramicina tiene preferiblemente valores de pH entre 10,0 10,7.
[0048] La solución límpida de la etapa a) contiene una concentración de sólidos que es inferior o igual al 2 % p/v, preferiblemente en el intervalo del 1 % al 2 % p/v.
[0049] En el caso de la forma de realización preferida, donde el fármaco es tobramicina y el adyuvante es estearato de sodio, la concentración de sólidos de aproximadamente un 1 % p/v fue óptima para tener el recubrimiento deseado.
[0050] Según la presente descripción, tras mezclar las dos soluciones pertinentes, se obtiene una solución límpida, donde el adyuvante está disperso de manera coloidal. A continuación, esta solución se pulveriza en seco en la etapa b) para obtener el polvo medicamentoso. La solución obtenida preferiblemente se seca por pulverización en un aparato Buchi 191 utilizando una temperatura de entrada de entre 120-135 °C, preferiblemente a aproximadamente 125 °C, una velocidad de alimentación de entre 3,0 y 6,0, preferiblemente de aproximadamente 3,5 ml/min, un caudal de aire de 600 l/h, una boquilla de entre 0,5 y 1 mm, preferiblemente de 0,7 mm y un aspirador 100 %. Según la invención, la solución que se va a secar por pulverización es límpida y siguió siendo límpida manteniendo su temperatura superior o igual a 28 °C, más preferiblemente superior o igual a 30 °C, aún más preferiblemente de aproximadamente 30 °C.
[0051] De este modo, durante la pulverización de la gota producida en el aparato de secado por pulverización, debido a la evaporación del disolvente, el adyuvante precipita en la interfase agua/aire de la gota formando la partícula de fármaco. El procedimiento adoptado y el disolvente utilizado determinaron la deposición del adyuvante en la superficie de la partícula de fármaco seca.
[0052] Por lo tanto, los inventores descubrieron que el mezclado profundo obtenido era capaz de concentrar el adyuvante al menos parcialmente en la superficie de la partícula de fármaco durante el secado. En el proceso de acuerdo con la invención, se preparó una solución de fármaco y adyuvante, que siguió siendo límpida sin la separación de los componentes durante el tiempo necesario para el secado por pulverización de la solución. Tal y como se ha descrito en la forma de realización más preferida, los inventores descubrieron que las dos soluciones podían mezclarse de manera espontánea en la relación de agua/alcohol 70:30 con un mezclado suave, proporcionando una mezcla límpida a 30 °C en la que el adyuvante está distribuido de manera coloidal. El polvo obtenido útil para inhalación, preferiblemente de tobramicina o amikacina, se caracteriza, por lo tanto, por presentar un alto contenido de sustancia farmacológica. La presente invención solucionó los problemas relacionados con la dificultad para areosolizar polvo medicamentoso en el que las partículas no poseen un comportamiento aerodinámico útil, puesto que son demasiado higroscópicas. El proceso según la invención, que contempla el secado por pulverización de una solución límpida de agua-fármaco en presencia de cantidades muy pequeñas de adyuvante, permitió preparar polvos de fármaco con una elevada respirabilidad, un alto contenido de fármaco, sin ser higroscópicos, fluidos y lo suficientemente resistentes al efecto de la exposición incontrolada al ambiente.
[0053] Debido al proceso de fabricación, los polvos pulmonares de tobramicina objeto de la presente invención están caracterizados por micropartículas con superficie corrugada, como resultará evidente en la Figura 1 anexa. En el caso de la tobramicina, la estructura es completamente distinta a la estructura microcristalina de tobramicina. Estos polvos pulmonares de antibióticos muestran características de empaquetamiento que les permiten introducirse directamente en el depósito de los sistemas de administración IPS. Además, esta invención permite la administración en aerosol de la sustancia en formas secas a una alta concentración, mejorando la estabilidad de almacenamiento y la dosis depositada en los pulmones.
[0054] Según el proceso dado a conocer en el presente documento, la transformación de las propiedades micromeríticas, es decir, la densidad, la forma y el tamaño de partícula, de las sustancias de antibiótico aminoglucósido se obtiene para conseguir que estas sean adecuadas para la inhalación de polvo seco. La característica técnica sorprendente consiste en la preparación de partículas sólidas mediante el secado por pulverización de la solución acuosa del fármaco en la que hay disperso coloidalmente un adyuvante que consiste en uno o una mezcla de ácidos grasos saturados C12-C18 o sus sales. El proceso de mezclado adoptado permite la obtención a una temperatura mayor o igual a 28 °C, más preferiblemente mayor o igual a 30 °C, aún más preferiblemente de aproximadamente 30 °C, de una solución completamente límpida del lípido de ácido graso en agua. Durante el secado, la estructura coloidal de la solución favorece la formación de micropartículas de fármaco cuya superficie está recubierta por el adyuvante, evitando de este modo la necesidad de micronizar por adelantado las partículas del polvo, según lo descrito en la técnica anterior. La cantidad de adyuvante presente en la partícula es muy baja (0,25-2,0 % p/p en tobramicina), definitivamente menor que en la técnica anterior. Para el polvo de la invención, a pesar de la baja cantidad de material adyuvante (preferiblemente de 0,5-1,5 % p/p en tobramicina), la sensibilidad a la humedad se redujo significativamente y se mejoró notablemente la aerosolización del polvo. Por consiguiente, según los inventores, el adyuvante, disperso coloidalmente en la solución acuosa de antibiótico que se depositó en las partículas de antibiótico formadas con el procedimiento descrito proporciona una protección frente al entorno. Además, las características de densidad, forma y tamaño de las partículas secadas por pulverización las hacen favorables para la deposición en el pulmón. Como se describirá en el ejemplo 3, esta deposición se ha demostrado utilizando un microscopio electrónico de barrido asistido por una sonda de campo de emisión de electrones.
[0055] En resumen, el proceso descrito en el presente documento fue capaz de modificar las propiedades del polvo medicamentoso en comparación con la materia prima, manteniendo un elevado contenido de fármaco (>90 %). En concreto, la pequeña cantidad de adyuvante fue capaz de proteger a los polvos de la humedad del ambiente,
proporcionando un comportamiento no higroscópico a los polvos. Además, la partícula de tobramicina obtenida presentaba una forma corrugada por la presencia del adyuvante, una característica que junto con la densidad y el tamaño, proporcionó una alta respirabilidad del producto. La nueva «naturaleza» del polvo medicamentoso permitió mantener estable el producto en el dispositivo para inhalación hasta su uso. Fue más relevante el hecho de que la respirabilidad de los polvos que presentan estas propiedades fue mayor que los valores habituales descritos en la literatura. Esta respirabilidad ha sido evaluada utilizando el impactador de cascada Andersen o el impactador Next Generation, como se describe en la presente Farmacopea de Estados Unidos. Estos aparatos se utilizan para determinar las partículas finas de una nube de aerosol generada por preparaciones para inhalación y permiten medir la masa de fármaco inferior a un tamaño de partícula aerodinámica concreto. La masa de fármaco que tiene un diámetro aerodinámico inferior a 5 pm se considera generalmente como «respirable», aunque el tamaño óptimo para la deposición en el pulmón está en el intervalo de 2-5 micras. El proceso descrito en esta patente contempló la producción de polvos finos en los que más del 90 % de las partículas tienen dimensiones inferiores a 7 micras como volumen-diámetro. Por otra parte, el estudio de respirabilidad realizado por el impactador de cascada Andersen y el impactador Next Generation del polvo de la invención fue, en todos los casos, superior al 50 %.
[0056] Por lo tanto, las características óptimas del polvo medicamentoso de la invención se basan en el descubrimiento de que las micropartículas corrugadas peculiares obtenidas mediante secado por pulverización de una solución límpida de agua-alcohol de un fármaco y una solución de ácido graso están micronizadas, con flujo libre y con una elevada densidad compactada. Las micropartículas son básicamente amorfas y se caracterizan por presentar una forma definida corrugada, como se muestra en la Figura 1 para la tobramicina. Esta forma de partícula hace que los polvos no sean cohesivos, puesto que las micropartículas mantienen su individualidad y no se aglomeran. Además, no muestran sustancialmente pérdidas de actividad si se almacenan en condiciones normales. Estos polvos que no se van a micronizar no son cohesivos, son de flujo bastante libre y fáciles de medir en el IPS. Estas propiedades físicas, junto con el comportamiento aerodinámico favorable debido al tamaño, la forma y la densidad de las partículas, determinan una inesperada y sorprendentemente elevada respirabilidad.
[0057] Por último, el polvo medicamentoso de la invención tiene un contenido de fármaco mayor o igual al 90 %, puesto que la humedad residual era inferior al 8 % p/p y el contenido lipídico menor o igual al 2,0 % p/p. Pueden almacenarse en condiciones normales de humedad y temperatura en un recipiente sellado. El hecho de que el polvo final contenga un porcentaje muy bajo del adyuvante hace que la sustancia que se administra sea bastante pura. La administración descrita en el presente documento puede implicar fármacos en polvo proporcionados como aerosol utilizando un dispositivo capaz de medir y aerosolizar las partículas. Asimismo, el polvo de la presente invención muestra una fracción respirable de su aerosol superior al 40 %, preferiblemente >48 %.
[0058] Los polvos que pueden obtenerse con el procedimiento descrito a continuación poseen las siguientes características, que los hacen muy adecuados para la administración por inhalación mediante un inhalador de polvo seco:
Rendimiento del proceso de secado por pulverización (%): entre 64 % y 79 %
Contenido de agua (%): entre 5,4 y 9,0
Contenido de fármaco (%): entre 90 y 94
Tamaño aerodinámico (MMAD): entre 2,7 pm y 4,6 pm
Respirabilidad (fracción de partícula fina): entre 41 % y 53 %
Empaquetamiento: densidad aparente 0,14-0,26 g/ml; densidad compactada 0,23-0,40 g/ml
Densidad real: entre 1,16 g/ml y 1,60 g/ml
Índice de Carr: (medición de la mejora en el empaquetamiento de un lecho de polvo: valores de aproximadamente 20 30 % indican buen flujo) entre 31,3 % y 37,1 %
[0059] A continuación se describirá la invención haciendo referencia a algunos ejemplos, que se han proporcionado para fines ejemplificadores.
EJEMPLOS
Ejemplo 1
[0060] Este ejemplo describe la fabricación de un polvo de tobramicina adecuado para administración pulmonar. Este ejemplo describe la preparación de la solución que se va a pulverizar, en la que el ácido esteárico, como adyuvante, está disperso de manera coloidal en la solución acuosa de tobramicina para proporcionar una mezcla límpida de los componentes. El resultado del proceso es la obtención de una partícula de fármaco que muestra estabilidad y respirabilidad sin afectar a la disolución del fármaco.
[0061] Los ingredientes y cantidades para preparar 2 litros de solución de fármaco-adyuvante para secado por pulverización fueron los que se indican en la Tabla 1.
Tabla 1

[0062] La relación de porcentajes entre tobramicina y ácido esteárico fue de 99:1. Se prepararon dos soluciones:
- una solución acuosa que contenía el fármaco disuelto y
- una solución etanólica que contenía el adyuvante.
[0063] La relación entre tobramicina-agua y ácido esteárico-solución de etanol fue de 70:30. El ácido esteárico se disolvió en etanol calentando la solución a 30 °C y agitando magnéticamente a 200 rpm en una placa de agitación caliente durante 30 min para conseguir una solución límpida transparente. Se empleó una protección para evitar la evaporación del etanol.
[0064] La tobramicina se disolvió en agua desionizada (conductividad 0,5 - 1,0 j S) como la concentración de 14 mg/ml con agitación magnética. Tras calentar la solución de fármaco a 30 °C, la solución de ácido esteárico/etanol se añadió a la velocidad de 100 ml/min agitando a 200 rpm durante 15 min. La dispersión coloidal aparece incolora, transparente y se mantiene límpida con agitación a una temperatura de aproximadamente 30 °C durante todo el proceso de secado por pulverización. La concentración de sólidos (fármaco adyuvante) en la solución final fue del 1 % p/v.
[0065] La solución coloidal hidroalcohólica resultante, en la que la relación entre agua y alcohol era de 7:3, se secó por pulverización en un secador Mini Spray B-191 (Buchi, Suiza) utilizando una temperatura de entrada de 125 °C, una velocidad de alimentación de material de 3 ml min-1, flujo de atomización de 100 % y flujo de aire en la boquilla de 600 ml min-1. El rendimiento del proceso para preparar polvo de tobramicina-ácido esteárico fue de 64,7 % ± 3,4.
[0066] La Figura 1 adjunta muestra la imagen de microscopía electrónica de barrido de las micropartículas de tobramicina/ácido esteárico. El polvo secado por pulverización de tobramicina-ácido esteárico mostró un valor de diámetro de volumen mediano (dv 0,5) de 3,65 ± 0,19. El análisis morfológico por microscopía electrónica de barrido del polvo indicó que las micropartículas de tobramicina secadas por pulverización presentaban una forma bastante esférica con superficie corrugada. Se investigó el comportamiento aerodinámico utilizando el impactador Next Generation, determinando la distribución granulométrica aerodinámica del polvo. El valor de fracción de partícula fina (FPF) (<5 jm ) fue de 60 % y el diámetro aerodinámico mediano de la masa fue de aproximadamente 3 jm . Este polvo mostró una muy buena respirabilidad, alcanzó las etapas profundas del impactador Next Generation y el porcentaje recuperado en la garganta fue bajo (7 %).
Ejemplo 2
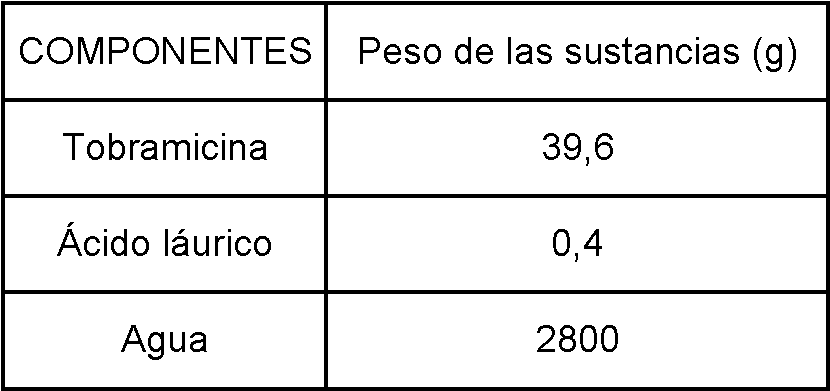
[0067] Este ejemplo describe la preparación de la solución que se va a pulverizar, en la que el ácido láurico se dispersó de manera coloidal en la solución acuosa de tobramicina, proporcionando una mezcla límpida de los componentes. Se produjeron micropartículas de tobramicina-ácido láurico mediante secado por pulverización del fármaco con el adyuvante lipófilo. La relación de porcentajes entre tobramicina y ácido láurico fue de 99:1. La concentración de sólidos (fármaco adyuvante lipófilo) en la solución final fue del 1 % p/v. Se prepararon una solución acuosa de fármaco y una solución etanólica del adyuvante de ácido láurico. La relación de porcentajes entre tobramicina-agua y ácido láurico-solución de etanol fue de 70:30.
[0068] La composición para preparar 4 litros de solución de fármaco-adyuvante para secado por pulverización fue la que se indica en la Tabla 2.
Tabla 2:

[0069] El ácido láurico se disolvió en etanol calentando la solución a 30 °C y agitando a 200 rpm en una placa de agitación caliente durante otros 30 min, consiguiendo una solución límpida transparente. La tobramicina se disolvió en agua desionizada (conductividad 0,5 -1,0 j S) en la concentración de 14 mg/ml. Tras calentar la solución de fármaco a 30 °C, se añadió la solución de ácido láurico/etanol a la velocidad de 100 ml/min agitando a 200 rpm durante 15 min. La solución obtenida a 30 °C era límpida.
[0070] La solución coloidal hidroalcohólica resultante se secó por pulverización en un secador Mini Spray B-191, como en el Ejemplo 1. El rendimiento de la preparación de polvo seco de tobramicina-ácido láurico fue de 68,3 % ± 2,6.
[0071] El polvo secado por pulverización de tobramicina-ácido láurico mostró un valor de diámetro de volumen mediano (dv 0,5) de 3,03 ± 0,12. El análisis morfológico por microscopía electrónica de barrido del polvo indicó que las micropartículas de tobramicina secadas por pulverización presentaban una forma bastante esférica con superficie corrugada, como se muestra en la Figura 1. Se investigó el comportamiento aerodinámico utilizando el impactador Next Generation, obteniendo la distribución granulométrica aerodinámica del polvo. El valor de fracción de partícula fina (<5 jm ) fue de 53% y el diámetro aerodinámico mediano de la masa fue de aproximadamente 3 jm . Este polvo mostró una muy buena respirabilidad, alcanzó las etapas profundas del impactador de cascada Andersen y el porcentaje recuperado en la garganta fue bajo (14 %).
Ejemplo 3
[0072] Este ejemplo describe la producción de un polvo de tobramicina-estearato de sodio (99:1) para inhalación utilizando una sal de ácido graso, es decir, estearato de sodio, añadida como adyuvante para brindar propiedades aerodinámicas y de estabilidad al polvo de tobramicina.
[0073] La composición para preparar 1 litro de solución de fármaco-adyuvante para secado por pulverización fue la que se indica en la Tabla 3:
Tabla 3

[0074] Se produjeron micropartículas de tobramicina-estearato de sodio mediante el secado por pulverización de la solución de fármaco-adyuvante. La relación de porcentajes entre tobramicina y ácido de estearato de sodio fue de 99:1. La concentración de sólidos (fármaco adyuvante) en la solución final fue del 1 % p/v. La solución acuosa de fármaco y la solución etanólica del adyuvante lipófilo a 30 °C se mezclaron en tobramicina-agua y estearato de sodio-solución de etanol en una relación de 70:30.
[0075] El estearato de sodio se disolvió en etanol calentando la solución a 30 °C y agitando a 200 rpm en una placa de agitación caliente durante otros 30 min para conseguir una solución límpida transparente. La tobramicina se disolvió en agua desionizada (conductividad 0,5 - 1,0 j S) a la concentración de 14 mg/ml y se agitó durante otros 10 min. Tras calentar la solución de fármaco a 30 °C, se añadió la solución de estearato de sodio a la velocidad de 100 ml/min agitando a 200 rpm durante 15 min. Se obtuvo una solución límpida.
[0076] La solución coloidal hidroalcohólica resultante se secó por pulverización en un secador Mini Spray B-191, como en el ejemplo 1. El rendimiento del proceso para preparar un polvo seco de tobramicina-estearato de sodio fue de 72,4 % ± 1,9. El contenido de fármaco fue del 90,74 %.
[0077] Como es bien sabido, la tobramicina es un polvo muy higroscópico, los cristales tienden a absorber agua del ambiente para licuarse. El polvo preparado en este ejemplo contenía un adyuvante que ayudó a proteger el polvo frente a la humedad y a reducir la higroscopicidad en comparación con la materia prima de tobramicina.
[0078] El análisis morfológico por microscopía electrónica de barrido del polvo indicó que las micropartículas de tobramicina secadas por pulverización presentaban una forma bastante esférica con superficie corrugada. En análisis de
espectroscopia de dispersión de energía de la distribución de sodio en la superficie de las micropartículas indicó la distribución máxima del adyuvante en la superficie de las micropartículas en el contenido 1 %. El contenido de agua en el polvo de tobramicina/estearato de sodio determinado por el método de Karl Fisher fue de 8,47 ± 0,53. La distribución granulométrica del polvo seco micronizado de tobramicina/estearato de sodio se determinó utilizando difracción láser (Mastersizer X, Malvern, Reino Unido). El tamaño de partícula de la materia prima de tobramicina y del polvo de tobramicina secado por pulverización se describe en la siguiente Tabla 4. Los dv 0,1, dv 0,5, dv 0,9 representan el diámetro de volumen de las partículas a 10, 50 y 90 % en la curva acumulativa de menor tamaño.
Tabla 4

[0079] Se utilizó el impactador de cascada Andersen (ACI) equipado con un puerto de inducción de acero inoxidable de 90 grados conforme a las instrucciones de los fabricantes que incluía el ajuste del caudal a 60 l/min. Durante el impacto, el aerosol se dividió en siete categorías de tamaño según el diámetro aerodinámico de la partícula (USP 30). En este estudio se utilizó un dispositivo Turbospin (Ph&T, MI, IT) para aerosolizar el polvo. La fracción respirable se expresó en término de fracción de partícula fina (FpF), que se consideró que eran aquellas partículas menores de 5 pm. El Mm AD se define como el diámetro de partícula mediano de la formulación que se deposita en el ACI. La siguiente Tabla 5 muestra propiedades de aerosolización de las micropartículas de tobramicina/estearato de sodio. Como se muestra en la Tabla 5, el polvo preparado mostró muy buenas propiedades de aerosolización.
Tabla 5

[0080] La distribución del adyuvante lipófilo, en este caso estearato de sodio, en la superficie de las micropartículas de tobramicina se analizó empleando SEM-EDS (microscopía electrónica de barrido-espectroscopía de dispersión de energía, JSM 6000F JEOL, Japón). Microscopio de emisión de campo de alta resolución (0,6 nm a 30 kV), microanálisis Windowless EDS (de boro).
[0081] El análisis por dispersión de energía de rayos X, también conocido como EDS, EDX o EDAX, es una técnica utilizada para identificar la composición elemental de una muestra o una pequeña zona de interés en la muestra. Durante el EDS, se expone una muestra a un haz de electrones dentro de un microscopio electrónico de barrido (SEM). Estos electrones chocan con los electrones dentro de la muestra, provocando que algunos de ellos sean retirados de sus órbitas. Las posiciones vacantes se rellenan con electrones de mayor energía, que emiten rayos X en el proceso. Al analizar los rayos X emitidos, puede determinarse la composición elemental de la muestra. El EDS es una potente herramienta para el microanálisis de componentes constituyentes elementales.
[0082] Estos estudios se llevaron a cabo para tratar de identificar cualquier diferencia entre la composición de la superficie de las partículas, en nuestro caso, la presencia de estearato de sodio. Resultaría difícil evaluar la topografía y la composición de un material particulado analizando un número relativamente pequeño de muestras; no obstante, estas técnicas pueden permitir algo de información relativa a la composición química de cualquier característica de la superficie de partícula.
[0083] El análisis EDS indicó una distribución de adyuvante en la superficie de las micropartículas de tobramicina. Se observó una relación lineal entre la concentración de estearato de sodio y el recuento de sodio (R2 = 0,97) hasta una concentración del 1 %, como se muestra en la Figura 2.
[0084] El contenido del 1 % se consideró óptimo, puesto que el incremento de adyuvante del 0 al 1 % determina la deposición en la partícula de fármaco de una cantidad de estearato de sodio que aumenta linealmente; un incremento adicional reduce la deposición en la superficie de las micropartículas de tobramicina. Esto indica que se alcanzó la cantidad máxima de cobertura.
Ejemplo 4
[0085] Este ejemplo describe la producción de un polvo de amikacina-estearato de sodio (99,5:0,5) para inhalación utilizando una sal de ácido graso, es decir, estearato de sodio, añadida como adyuvante para brindar propiedades aerodinámicas y de estabilidad al polvo de amikacina.
[0086] La preparación de dispersión coloidal en la que hay una solución etanólica de estearato de sodio y una solución acuosa de amikacina se mezcló utilizando el proceso descrito en el presente documento.
[0087] La composición para preparar 1 litro de solución de fármaco-adyuvante lipófilo para secado por pulverización fue la siguiente:
Tabla 6

[0088] El estearato de sodio se disolvió en etanol a la concentración de 0,17 mg/ml calentando la solución a 30 °C y agitando a 200 rpm en una placa de agitación caliente durante otros 30 min para conseguir una solución límpida transparente. La amikacina se disolvió en agua desionizada (conductividad 0,5 - 1,0 pS). Tras calentar la solución de fármaco a 30 °C, se añadió la solución de estearato de sodio a la velocidad de 100 ml/min agitando a 200 rpm durante 15 min. La solución acuosa de fármaco y la solución etanólica del adyuvante lipófilo se mezclaron en una relación de 70:30. Se obtuvo una mezcla límpida de los componentes. La concentración de sólidos (fármaco adyuvante) en la solución final fue del 1 % p/v.
[0089] La solución coloidal hidroalcohólica resultante se secó por pulverización en un secador Mini Spray B-191 (en las mismas condiciones del ejemplo 1). El rendimiento del proceso para preparar un polvo seco de tobramicina-estearato de sodio fue de 79,4 % ± 2,8.
[0090] El polvo secado por pulverización de amikacina-estearato de sodio mostró un valor de diámetro de volumen mediano (dv 0,5) de 3,28 ± 0,19 menor de 5 pm. El contenido de agua del polvo fue de 5,42 % ± 0,64. Se investigó el comportamiento aerodinámico utilizando el impactador de cascada Andersen, obteniendo la distribución granulométrica aerodinámica del polvo. El valor de fracción de partícula fina (<5 pm) fue de 41,7 % ± 3,6 y el diámetro aerodinámico mediano de la masa fue de aproximadamente 4,6 pm. Este polvo mostró una muy buena respirabilidad, alcanzó las etapas profundas del impactador de cascada Andersen y el porcentaje recuperado en la garganta fue de aproximadamente el 21 %.
Ejemplo 5
[0091] Este ejemplo describe la producción de un polvo de tobramicina-estearato de sodio (99:1) para inhalación utilizando una sal de ácido graso, es decir, estearato de sodio en caso de que la concentración de sólidos (fármaco adyuvante) en la solución final sea del 2 % p/v. Esto permite la reducción del volumen y el tiempo del proceso de secado por pulverización.
[0092] La preparación de dispersión coloidal en la que hay una solución etanólica de estearato de sodio y una solución acuosa de tobramicina se mezcló utilizando el proceso descrito en el presente documento.
[0093] Se produjeron micropartículas de tobramicina-estearato de sodio mediante el secado por pulverización de la solución de fármaco-adyuvante. La relación de porcentajes entre tobramicina y ácido de estearato de sodio fue de 99:1. La solución acuosa de fármaco y la solución etanólica del adyuvante lipófilo se mezclaron a 30 °C. La relación de tobramicina-agua y estearato de sodio-solución de etanol fue de 70:30. La concentración de sólidos (fármaco adyuvante) en la solución final fue del 2 % p/v.
[0094] La composición para preparar 500 ml de solución de fármaco-adyuvante lipófilo para secado por pulverización fue la siguiente:
Tabla 7

[0095] El estearato de sodio se disolvió en etanol a la concentración de 0,6 mg/ml calentando la solución a 30 °C y agitando a 200 rpm en una placa de agitación caliente (Arex Velp Scientifica, MO, IT) durante 30 min adicionales para conseguir una solución límpida transparente. Se empleó una protección para evitar la evaporación del etanol.
[0096] La tobramicina se disolvió en agua desionizada (conductividad 0,5 -1,0 j S) a la concentración de 28,2 mg/ml y se agitó durante otros 10 min. Tras calentar la solución de fármaco a 30 °C, se añadió la solución de estearato de sodio a la velocidad de 100 ml/min agitando a 200 rpm durante 15 min.
[0097] Se obtuvo una mezcla límpida de los componentes.
[0098] La solución coloidal hidroalcohólica resultante se secó por pulverización en un secador Mini Spray B-191 en las mismas condiciones indicadas en el Ejemplo 1.
[0099] El rendimiento del proceso para preparar un polvo seco de tobramicina-estearato de sodio fue de 76,9 % ± 2,1. El polvo secado por pulverización de tobramicina-estearato de sodio mostró un valor de diámetro de volumen mediano (dv 0,5) de 2,59 ± 0,01. El contenido de agua en el polvo de tobramicina/estearato de sodio fue del 9 %. La densidad real (g/ml) y el % del índice de Carr del polvo fueron respectivamente 1,60 g/ml y 35,5 %.
[0100] Se investigó el comportamiento aerodinámico utilizando el impactador Next Generation, obteniendo la distribución granulométrica aerodinámica del polvo. El valor de fracción de partícula fina (<5 jm ) fue de 48,2 % ± 3,9 y el diámetro aerodinámico mediano de la masa fue de aproximadamente 2,7 jm .
[0101] Como se ha mostrado en los ejemplos anteriores, la preparación de partículas sólidas mediante el secado por pulverización de una solución acuosa de la materia prima en la que se dispersa coloidalmente un ácido graso en una cantidad muy baja da lugar a un alto contenido de fármaco, siempre mayor o igual al 90 %. La presente invención ha resuelto los problemas relacionados con la dificultad para areosolizar un polvo antibiótico o quimioterapéutico en el que las partículas no poseen un comportamiento aerodinámico útil. Además, esta invención permite la administración de la sustancia en formas secas, mejorando la estabilidad de almacenamiento y la dosis depositada en los pulmones. Asimismo, puesto que la dosis administrada suele ser muy elevada, la concentración de la sustancia activa permite reducir el número de administraciones y la densidad del aerosol.
[0102] Debido al proceso de fabricación, los polvos pulmonares están caracterizados por la estructura, el tamaño y la forma de las micropartículas constituyentes con superficie corrugada, completamente distintas a la estructura microcristalina. Estos polvos pulmonares muestran características de empaquetamiento que les permiten introducirse directamente en el depósito de los sistemas de administración IPS.
Claims (15)
1. Polvo medicamentoso para inhalación que comprende partículas en las que el contenido de fármaco es igual o mayor al 90 % p/p, donde dichas partículas de fármaco están recubiertas al menos parcialmente con una cantidad de un adyuvante menor o igual al 2,0 % p/p con respecto al peso del fármaco, consistiendo dicho adyuvante en uno o una mezcla de ácidos grasos saturados C12-C18 o sus sales, donde dicho polvo medicamentoso se obtiene mediante a) la preparación de una solución límpida de las partículas de fármaco mezclando una solución acuosa de las partículas de fármaco con una solución alcohólica del adyuvante, donde el adyuvante que consiste en uno o una mezcla de ácidos grasos saturados C12-C18 o sus sales está disperso de manera coloidal en la solución; y
b) el secado por pulverización de la solución límpida de la etapa a) utilizando una temperatura de entrada de entre 120-135 °C, una velocidad de alimentación de entre 3,0 y 6,0 ml/min, un caudal de aire de 600 l/h, una boquilla de entre 0,5 y 1 mm, y un aspirador 100 %, donde
la temperatura de la solución límpida en la etapa a) y en la etapa b) es mayor o igual a 28 °C para dispersar de manera coloidal el adyuvante en la solución, y donde
dicho polvo medicamentoso es un antibiótico aminoglucósido que comprende partículas de antibiótico que presentan una solubilidad en agua de al menos un 3,0 % p/v.
2. Polvo medicamentoso según la reivindicación 1, donde el antibiótico aminoglucósido se selecciona del grupo que consiste en amikacina, gentamicina, kanamicina, neomicina, netilmicina, paromomicina, estreptomicina y tobramicina.
3. Polvo medicamentoso según la reivindicación 2, donde el antibiótico aminoglucósido es tobramicina o amikacina.
4. Polvo medicamentoso según cualquiera de las reivindicaciones 1-3, donde el adyuvante se selecciona del grupo que consiste en ácido láurico, ácido mirístico, ácido palmítico, ácido esteárico, sus mezclas y sus sales.
5. Polvo medicamentoso según la reivindicación 4, donde el adyuvante es ácido esteárico o una sal de este.
6. Polvo medicamentoso según cualquiera de las reivindicaciones 1-5, donde la partícula de fármaco está recubierta al menos parcialmente con un adyuvante en la cantidad de 0,25 a 2,0 p/p con respecto al peso del fármaco.
7. Polvo medicamentoso según la reivindicación 6, donde la partícula de fármaco está recubierta al menos parcialmente con un adyuvante en la cantidad de 0,5 a 1,5 p/p con respecto al peso del fármaco.
8. Polvo medicamentoso según cualquiera de las reivindicaciones 1-7, donde la temperatura de la solución límpida en la etapa a) y en la etapa b) es mayor o igual a 30 °C.
9. Proceso para la preparación del polvo medicamentoso según cualquiera de las reivindicaciones 1-5 que comprende las siguientes etapas:
a) preparación de una solución límpida de partículas de fármaco mezclando una solución acuosa de las partículas de fármaco con una solución alcohólica del adyuvante, donde un adyuvante que consiste en uno o una mezcla de ácidos grasos saturados C12-C18 o sus sales está disperso de manera coloidal en la solución;
b) secado por pulverización de la solución límpida de la etapa a) utilizando una temperatura de entrada de entre 120 135 °C, una velocidad de alimentación de entre 3,0 y 6,0 ml/min, un caudal de aire de 600 l/h, una boquilla de entre 0,5 y 1 mm, y un aspirador 100 %, donde
la temperatura de la solución límpida en la etapa a) y en la etapa b) es mayor o igual a 28 °C para dispersar de manera coloidal el adyuvante en la solución, y donde
dicho polvo medicamentoso es un antibiótico aminoglucósido que comprende partículas de antibiótico que presentan una solubilidad en agua de al menos un 3,0 % p/v.
10. Proceso según la reivindicación 9 donde la solución límpida de partículas de fármaco se obtiene mediante una mezcla de una solución acuosa del fármaco con una solución alcohólica del adyuvante.
11. Proceso según la reivindicación 9 o 10, donde el alcohol utilizado para la disolución del adyuvante se selecciona del grupo que consiste en etanol, metanol, propanol o isopropanol.
12. Proceso según cualquiera de las reivindicaciones 9-11, donde las soluciones del adyuvante y del fármaco se mezclan en una relación en el intervalo de 2:8 a 5:5.
13. Proceso según cualquiera de las reivindicaciones 9-12, donde la temperatura de la solución límpida en la etapa a) y en la etapa b) es mayor o igual a 30 °C.
14. Uso de un polvo medicamentoso según cualquiera de las reivindicaciones 1-8 para la fabricación de un medicamento para el tratamiento de enfermedades derivadas de infecciones bacterianas para su administración mediante inhalación.
15. Uso según la reivindicación 14, donde la enfermedad es fibrosis quística.
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/EP2008/059110 WO2010003465A2 (en) | 2008-07-11 | 2008-07-11 | A drug powder for inhalation administration and a process thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2918193T3 true ES2918193T3 (es) | 2022-07-14 |
Family
ID=40278883
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES08775027T Active ES2918193T3 (es) | 2008-07-11 | 2008-07-11 | Polvo medicamentoso para administración por inhalación y un proceso del mismo |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP2313114B1 (es) |
| ES (1) | ES2918193T3 (es) |
| WO (1) | WO2010003465A2 (es) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG181673A1 (en) | 2009-12-14 | 2012-07-30 | Chiesi Farma Spa | Antibiotic microparticles for inhalation |
| US9572774B2 (en) | 2011-05-19 | 2017-02-21 | Savara Inc. | Dry powder vancomycin compositions and associated methods |
| SG11202103851PA (en) | 2018-10-18 | 2021-05-28 | Deutsches Krebsforsch | Dry pharmaceutical composition for inhalation |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2002335046A1 (en) * | 2001-10-19 | 2003-05-06 | Inhale Therapeutic Systems, Inc. | The use of proton sequestering agents in drug formulations |
| US20040184995A1 (en) * | 2003-03-17 | 2004-09-23 | Yamanouchi Pharmaceutical Co., Ltd. | Novel dry powder inhalation for lung-delivery and manufacturing method thereof |
| GB0525254D0 (en) * | 2005-12-12 | 2006-01-18 | Jagotec Ag | Powder compositions for inhalation |
| EP2083833B1 (en) * | 2006-10-11 | 2018-01-24 | Laboratoires SMB SA | Pharmaceutical anti-infective composition for inhalation. |
| GB0621957D0 (en) * | 2006-11-03 | 2006-12-13 | Vectura Group Plc | Inhaler devices and bespoke pharmaceutical compositions |
-
2008
- 2008-07-11 EP EP08775027.9A patent/EP2313114B1/en active Active
- 2008-07-11 ES ES08775027T patent/ES2918193T3/es active Active
- 2008-07-11 WO PCT/EP2008/059110 patent/WO2010003465A2/en active Application Filing
Also Published As
| Publication number | Publication date |
|---|---|
| WO2010003465A3 (en) | 2010-04-08 |
| EP2313114B1 (en) | 2022-03-23 |
| WO2010003465A2 (en) | 2010-01-14 |
| EP2313114A2 (en) | 2011-04-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9421166B2 (en) | Pulmonary delivery of aminoglycoside | |
| JP5989750B2 (ja) | フルオロキノロンの肺送達 | |
| Zhou et al. | Inhaled formulations and pulmonary drug delivery systems for respiratory infections | |
| Aquino et al. | Dry powder inhalers of gentamicin and leucine: formulation parameters, aerosol performance and in vitro toxicity on CuFi1 cells | |
| ES2909080T3 (es) | Formulación de DPI que contiene sulfoalquil éter ciclodextrina | |
| ES2880271T3 (es) | Polvos pulmonares de ultra baja densidad | |
| ES2959198T3 (es) | Compuestos análogos de pirfenidona y piridona en aerosol | |
| JP2009532489A (ja) | 薬剤微粒子 | |
| BRPI1011721B1 (pt) | Formulações em pó seco inaláveis compreendendo sal de cátion de metal divalente e métodos para tratar doenças pulmonares | |
| US20120128729A1 (en) | Gallium Formulation For The Treatment And Prevention of Infectious Diseases | |
| JP2004535454A (ja) | エーロゾル化用に至適化されたトブラマイシン製剤 | |
| JP2014515356A (ja) | 乾燥粉末バンコマイシン組成物および関連する方法 | |
| KR20120104235A (ko) | 흡입용 항생제 미립자 | |
| WO2012061902A1 (en) | Micronised spray-dried particles comprising polymyxin | |
| WO2006033713A2 (en) | Methods for ciprofloxacin inhalation | |
| KR20170039255A (ko) | 흡입용 건조 분말 제형 | |
| ES2918193T3 (es) | Polvo medicamentoso para administración por inhalación y un proceso del mismo | |
| PT1594500E (pt) | Tratamento para doenças bacterianas dos órgãos respiratórios por aplicação local de fluoroquinolonas | |
| ES2612257T3 (es) | Formulación de suspensión mejorada de dipropionato de beclometasona para administración mediante inhalación | |
| Adhikari et al. | Co-amorphization: a formulation strategy for amorphous high dose dry powder to treat lung infections | |
| KR102259824B1 (ko) | 보센탄을 함유한 약학 제제 | |
| WO2023205389A1 (en) | Dry powder inhalation delivery of pharmaceuticals | |
| AU2002361897B2 (en) | Pulmonary delivery of aminoglycosides | |
| CN116617404A (zh) | 磷脂作为抑晶剂的用途 |