ES2861623T3 - Sistemas, métodos y composiciones de componentes de CRISPR-Cas para manipulación de secuencias - Google Patents
Sistemas, métodos y composiciones de componentes de CRISPR-Cas para manipulación de secuencias Download PDFInfo
- Publication number
- ES2861623T3 ES2861623T3 ES17162030T ES17162030T ES2861623T3 ES 2861623 T3 ES2861623 T3 ES 2861623T3 ES 17162030 T ES17162030 T ES 17162030T ES 17162030 T ES17162030 T ES 17162030T ES 2861623 T3 ES2861623 T3 ES 2861623T3
- Authority
- ES
- Spain
- Prior art keywords
- sequence
- crispr
- cas9
- target
- tracr
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
- C12N15/113—Non-coding nucleic acids modulating the expression of genes, e.g. antisense oligonucleotides; Antisense DNA or RNA; Triplex- forming oligonucleotides; Catalytic nucleic acids, e.g. ribozymes; Nucleic acids used in co-suppression or gene silencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/8509—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells for producing genetically modified animals, e.g. transgenic
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/87—Introduction of foreign genetic material using processes not otherwise provided for, e.g. co-transformation
- C12N15/90—Stable introduction of foreign DNA into chromosome
- C12N15/902—Stable introduction of foreign DNA into chromosome using homologous recombination
- C12N15/907—Stable introduction of foreign DNA into chromosome using homologous recombination in mammalian cells
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/16—Hydrolases (3) acting on ester bonds (3.1)
- C12N9/22—Ribonucleases RNAses, DNAses
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Biomedical Technology (AREA)
- Zoology (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Biophysics (AREA)
- Mycology (AREA)
- Cell Biology (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
Una composición que comprende un sistema de repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas (CRISPR, por sus siglas en inglés), comprendiendo el sistema CRISPR: - una proteína Cas9 que comprende una o más secuencias de localización nuclear (NLS); - una secuencia guía unida a una secuencia de emparejamiento tracr y - una secuencia tracr con 30 o más nucleótidos de longitud; en donde la secuencia guía dirige la unión específica de la secuencia de un complejo CRISPR a una secuencia diana en una célula eucariota.
Description
DESCRIPCIÓN
Sistemas, métodos y composiciones de componentes de CRISPR-Cas para manipulación de secuencias
SOLICITUDES RELACIONADAS
Campo de la invención
La presente descripción se refiere en general a sistemas, métodos y composiciones usados para el control de la expresión de genes que implica fijar como objetivo una secuencia, tal como perturbación del genoma o edición de genes, que pueden usar sistemas vectores relacionados con Repeticiones Palindrómicas Cortas Agrupadas y Regularmente Interespaciadas (CRISPR, por sus siglas en inglés) y componentes de los mismos.
Informe relativo a la investigación patrocinada por el gobierno federal
Esta invención se realizó con apoyo estatal bajo el Premio Pionero de los INS DP1MH100706, otorgado por los Institutos Nacionales de Salud. El gobierno tiene ciertos derechos sobre la invención.
Antecedentes de la invención
Los recientes avances en las técnicas de secuenciación del genoma y métodos de análisis han acelerado significativamente la capacidad para catalogar y cartografiar factores genéticos asociados a un diverso intervalo de funciones biológicas y enfermedades. Se requieren tecnologías precisas para fijar como objetivo genoma para permitir la ingeniería inversa sistemática de variaciones genéticas causales permitiendo la perturbación selectiva de elementos genéticos individuales, así como avanzar las aplicaciones de la biología sintética, biotecnológicas y médicas. Aunque están disponibles técnicas de edición de genoma tales como dedos de cinc diseñadores, efectores del tipo activador de la transcripción (los TALE, por sus siglas en inglés) o meganucleasas de migración para producir perturbaciones del genoma fijado como objetivo, sigue existiendo la necesidad de nuevas tecnologías de ingeniería del genoma que tengan un precio asequible, fáciles de montar, escalables y susceptibles de fijar como objetivo múltiples posiciones dentro del genoma eucariota.
Sumario de la invención
Existe una necesidad apremiante de sistemas y técnicas alternativos y robustos para fijar como objetivo secuencias con una amplia serie de aplicaciones. Esta invención estudia esta necesidad y proporciona ventajas relacionadas. El CRISPR/Cas o el sistema CRISPR-Cas (ambos términos se usan de manera intercambiable por toda esta solicitud) no requiere la generación de proteínas personalizadas para secuencias específicas fijadas como objetivo sino más bien se puede programar una enzima Cas única mediante una molécula de ARN corta para reconocer un objetivo de ADN específico, en otras palabras, la enzima Cas se puede reclutar para un objetivo de ADN específico usando dicha molécula de ARN corta. Añadir el sistema CRISPR-Cas al repertorio de técnicas de secuenciación del genoma y métodos de análisis puede simplificar de manera significativa la metodología y acelerar la capacidad para catalogar y cartografiar factores genéticos asociados a un diverso intervalo de funciones biológicas y enfermedades. Para utilizar el sistema CRISPR-Cas de manera eficaz para editar genoma sin efectos perjudiciales, es crítico comprender aspectos de ingeniería y optimización de estas herramientas de ingeniería del genoma.
En un aspecto, la invención proporciona una composición que comprende un sistema CRISPR, como se expone en la reivindicación 1. También se proporcionan un sistema vector CRISPR como se describe en la reivindicación 12 y usos y métodos como se describe en las otras reivindicaciones independientes. Otras características de la invención se exponen en las reivindicaciones dependientes.
También se describe en la presente memoria un sistema vector que comprende uno o más vectores. En algunas realizaciones, el sistema comprende: (a) un primer elemento regulador ligado de manera operable a una secuencia de emparejado tracr y uno o más sitios de inserción para insertar una o más secuencias guía aguas arriba de la secuencia de emparejado tracr, en la que cuando se expresa, la secuencia guía se dirige a unión específica de secuencias de un complejo CRISPR a una secuencia fijada como objetivo en una célula eucariota, en la que el complejo CRISPR comprende una enzima CRISPR complejada con (1) la secuencia guía que se hibrida a la secuencia fijada como objetivo y (2) la secuencia de emparejado tracr que se hibrida a la secuencia tracr y (b) un segundo elemento regulador ligado de manera operable a una secuencia codificadora de enzimas que codifica dicha enzima CRISPR que comprende una secuencia de localización nuclear; en el que los componentes (a) y (b) están situados en los mismos vectores o diferentes del sistema. En algunas realizaciones, el componente (a) comprende además la secuencia tracr aguas abajo de la secuencia de emparejamiento tracr bajo el control del primer elemento regulador. En algunas realizaciones, el componente (a) comprende además dos o más secuencias guía ligadas de manera operable al primer elemento regulador, en el que cuando se expresan, cada una de las dos o más secuencias guía dirige unión específica de la secuencia de un complejo CRISPR a una
secuencia objetivo diferente en una célula eucariota. En algunas realizaciones, el sistema comprende la secuencia traer bajo el control de un tercer elemento regulador, tal como un activador de polimerasa III. En algunas realizaciones, la secuencia tracr presenta al menos 50%, 60%, 70%, 80%, 90%, 95% o 99% de complementariedad de secuencias a lo largo de la longitud de la secuencia de emparejado tracr cuando se alinean de manera óptima. Determinar el alineamiento óptimo está dentro del alcance de un experto en la materia. Por ejemplo, hay algoritmos y programas de alineamiento disponibles públicamente y comercialmente tales como, pero no limitado a, ClustalW, Smith-Waterman en Matlab, Bowtie, Geneious, Biopython y SeqMan. En algunas realizaciones, el complejo CRISPR comprende una o más secuencias de localización nuclear de suficiente fortaleza para conducir la acumulación de dicho complejo CRISPR en una cantidad detectable en el núcleo de una célula eucariota. Sin desear estar ligados por la teoría, se cree que una secuencia de localización nuclear no es necesariamente para actividad del complejo CRISPR en eucariotas, sino que incluyendo tales secuencias se mejora la actividad del sistema, especialmente en cuanto a moléculas de ácido nucleico fijadas como objetivo en el núcleo. En algunas realizaciones, la enzima CRISPR es una enzima del sistema CRISPR de tipo II. En algunas realizaciones, la enzima CRISPR es una enzima Cas9. En algunas realizaciones, la enzima Cas9 es Cas9 S. pneumoniae, S. pyogenes o S. thermophilus y puede incluir Cas9 mutada derivada de estos organismos. La enzima puede ser un homólogo u ortólogo de Cas9. En algunas realizaciones, la enzima CRISPR es codónoptimizada para expresión en una célula eucariota. En algunas realizaciones, la enzima CRISPR dirige la escisión de una o dos cadenas en la posición de la secuencia objetivo. En algunas realizaciones, la enzima CRISPR carece de actividad de escisión de la cadena de ADN. En algunas realizaciones, el primer elemento regulador es un activador de la polimerasa III. En algunas realizaciones, el segundo elemento regulador es un activador de la polimerasa II. En algunas realizaciones, la secuencia guía tiene al menos 15, 16, 17, 18, 19, 20, 25 nucleótidos o entre 10-30 o entre 15-25 o entre 15-20 nucleótidos de longitud. En general y por toda esta memoria descriptiva, el término "vector" se refiere a una molécula de ácido nucleico capaz de transportar otro ácido nucleico al que se ha unido. Los vectores incluyen, pero no se limitan a, moléculas de ácido nucleico que son monocatenarias, bicatenarias o parcialmente bicatenarias; moléculas de ácido nucleico que comprenden uno o más extremos libres, ningún extremo libre (por ejemplo, circular); moléculas de ácido nucleico que comprenden ADN, ARN o ambos y otras variedades de polinucleótidos conocidos en la técnica. Un tipo de vector es un "plásmido", que se refiere a un bucle de ADN de doble cadena circular en que se pueden insertar segmentos de ADN adicionales, tal como por técnicas de clonación molecular clásicas. Otro tipo de vector es un vector vírico, en el que están presentes secuencias de ADN o ARN derivadas víricamente en el vector para empaquetamiento en un virus (por ejemplo, retrovirus, retrovirus de replicación defectuosa, adenovirus, adenovirus de replicación defectuosa y virus adenoasociados). Los vectores víricos también incluyen polinucleótidos soportados por un virus para transinfección a una célula huésped. Algunos vectores son capaces de replicación autónoma en una célula huésped en que se introducen (por ejemplo, vectores bacterianos con un origen bacteriano de replicación y vectores de mamífero episomales). Otros vectores (por ejemplo, vectores de mamíferos no episomales) se integran en el genoma de una célula huésped en la introducción en la célula huésped y se replican de ese modo junto con el genoma huésped. Por otra parte, algunos vectores son capaces de dirigir la expresión de los genes a los que están unidos de manera operativa. Dichos vectores se refieren en la presente memoria como "vectores de expresión". Los vectores de expresión comunes de utilidad en técnicas de ADN recombinante están con frecuencia en la forma de plásmidos.
Los vectores de expresión recombinantes pueden comprender un ácido nucleico como se describe en la presente memoria en una forma adecuada para expresión del ácido nucleico en una célula huésped, que significa que los vectores de expresión recombinantes incluyen uno o más elementos reguladores, que se pueden seleccionar sobre la base de las células huésped que se tienen que usar para expresión, que está unido de manera operativa a la secuencia de ácidos nucleicos que se tiene que expresar. En un vector de expresión recombinante, "unido de manera operable" significa que la secuencia de nucleótidos de interés está unida al elemento o a los elementos reguladores de una manera que permite la expresión de la secuencia de nucleótidos (por ejemplo, en un sistema de transcripción/traducción in vitro o en una célula huésped cuando el vector se introduce en la célula huésped).
El término "elemento regulador" incluye activadores, potenciadores, sitios de entrada de ribosomas internos (IRES, por sus siglas en inglés) y otros elementos de control de la expresión (por ejemplo, señales de terminación de la transcripción, tales como señales de poliadenilación y secuencias poli-U). Tales elementos reguladores se describen, por ejemplo, en Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990). Los elementos reguladores incluyen aquéllos que dirigen la expresión constitutiva de una secuencia de nucleótidos en muchos tipos de célula huésped y aquéllos que dirigen la expresión de la secuencia de nucleótidos sólo en ciertas células huésped (por ejemplo, secuencias reguladoras específicas del tejido). Un activador específico del tejido puede dirigir la expresión principalmente en un tejido deseado de interés, tal como músculo, neurona, hueso, piel, sangre, órganos específicos (por ejemplo, hígado, páncreas) o tipos de células particulares (por ej., linfocitos). Los elementos reguladores también pueden dirigir la expresión de una manera dependiente temporal, tal como de una manera dependiente del ciclo celular o dependiente de la fase de desarrollo, que puede ser o no también específica del tejido o del tipo de célula. En algunas realizaciones, un vector comprende uno o más activadores de pol III (por ej., 1,2, 3, 4, 5 o más activadores de pol III), uno o más activadores de pol II (por ej., 1,2, 3, 4, 5 o más activadores de pol II), uno o más activadores
de pol I (por ej., 1, 2, 3, 4, 5 o más activadores de pol I) o combinaciones de los mismos. Ejemplos de activadores de pol III incluyen, pero no se limitan a, activadores U6 y H1. Ejemplos de activadores de pol II incluyen, pero no se limitan a, el activador LTR del virus del sarcoma de Rous (RSV, por sus siglas en inglés) retrovírico (opcionalmente con el potenciador de RSV), el activador de citomegalovirus (CMV) (opcionalmente con el estimulador de CMV) [véase, por ej., Boshart et al, Cell, 41: 521-530 (1985)], el activador de SV40, el activador de dihidrofolato reductasa, el activador de p-actina, el activador de fosfoglicerol cinasa (PGK, por sus siglas en inglés) y el activador de EF1a. También están incluidos en el término "elemento regulador" los elementos estimuladores, tales como WPRE; estimuladores de CMV; el segmento R-U5' en LTR de HTLV-1 (Mol. Cell. Biol., Vol. 8 (1), pág. 466-472, 1988); estimulador de SV40 y la secuencia del intrón entre los exones 2 y 3 de p-globina de conejo (Proc. Natl. Acad. Sci. USA., Vol. 78 (3), pág. 1.527-31, 1981). Se apreciará por los expertos en la materia que el diseño del vector de expresión puede depender de factores tales como la elección de la célula huésped que se tiene que transformar, el nivel de expresión deseado, etc. Se puede introducir un vector en células huésped para producir de ese modo transcritos, proteínas o péptidos, incluyendo proteínas o péptidos de fusión, codificados por ácidos nucleicos como se describe en la presente memoria (por ejemplo, transcritos de repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas (CRISPR), proteínas, enzimas, formas mutantes de los mismos, proteínas de fusión de los mismos, etc.).
Vectores ventajosos incluyen lentivirus y virus adenoasociados y también se pueden seleccionar tipos de tales vectores para fijar como objetivo tipos particulares de células.
En un aspecto, se describe en la presente memoria un vector que comprende un elemento regulador unido de manera operable a una secuencia codificadora de enzimas que codifica una enzima CRISPR que comprende una o más secuencias de localización nucleares. En algunas realizaciones, dicho elemento regulador conduce la transcripción de la enzima CRISPR en una célula eucariota de manera que dicha enzima CRISPR se acumula en una cantidad detectable en el núcleo de la célula eucariota. En algunas realizaciones, el elemento regulador es un activador de la polimerasa II. En algunas realizaciones, la enzima CRISPR es una enzima del sistema CRISPR de tipo II. En algunas realizaciones, la enzima CRISPR es una enzima Cas9. En algunas realizaciones, la enzima Cas9 es Cas9 de S. pneumoniae, S. pyogenes o S. thermophilus y puede incluir Cas9 mutada procedente de estos organismos. En algunas realizaciones, la enzima CRISPR es codón-optimizada para expresión en una célula eucariota. En algunas realizaciones, la enzima CRISPR dirige la escisión de una o dos cadenas en la posición de la secuencia objetivo. En algunas realizaciones, la enzima CRISPR carece de actividad de escisión de la cadena de ADN.
En un aspecto, se describe en la presente memoria una enzima CRISPR que comprende una o más secuencias de localización nuclear de suficiente fortaleza para conducir la acumulación de dicha enzima CRISPR en una cantidad detectable en el núcleo de una célula eucariota. En algunas realizaciones, la enzima CRISPR es una enzima del sistema CRISPR de tipo II. En algunas realizaciones, la enzima CRISPR es una enzima Cas9. En algunas realizaciones, la enzima Cas9 es Cas9 de S. pneumoniae, S. pyogenes o S. thermophilus y puede incluir Cas9 mutada procedente de estos organismos. La enzima puede ser un homólogo u ortólogo de Cas9. En algunas realizaciones, la enzima CRISPR carece de capacidad para escindir una o más cadenas de una secuencia objetivo a la que se une.
En un aspecto, se describe en la presente memoria una célula eucariota huésped que comprende: (a) un primer elemento regulador unido de manera operable a una secuencia de emparejamiento tracr y uno o más sitios de inserción para insertar una o más secuencias guía aguas arriba de la secuencia de emparejamiento tracr, en la que cuando se expresa, la secuencia guía dirige la unión específica de la secuencia de un complejo CRISPR a una secuencia objetivo en una célula eucariota, en la que el complejo CRISPR comprende una enzima CRISPR complejada con (1) la secuencia guía que se hibrida a la secuencia objetivo y (2) la secuencia de emparejamiento tracr que se hibrida a la secuencia tracr y/o (b) un segundo elemento regulador unido de manera operable a una secuencia codificadora de enzima que codifica a dicha enzima CRISPR que comprende una secuencia de localización nuclear. En algunas realizaciones, la célula huésped comprende los componentes (a) y (b). En algunas realizaciones, el componente (a), el componente (b) o los componentes (a) y (b) están integrados de manera estable en un genoma de la célula eucariota huésped. En algunas realizaciones, el componente (a) comprende además la secuencia tracr aguas abajo de la secuencia de emparejamiento tracr bajo el control del primer elemento regulador. En algunas realizaciones, el componente (a) comprende además dos o más secuencias guía unidas de manera operable al primer elemento regulador, en el que cuando se expresan, cada una de las dos o más secuencias guía dirige la unión específica de la secuencia de un complejo CRISPR a una secuencia objetivo diferente en una célula eucariota. En algunas realizaciones, la célula huésped eucariota comprende además un tercer elemento regulador, tal como un activador de la polimerasa III, unido de manera operable a dicha secuencia tracr. En algunas realizaciones, la secuencia tracr presenta al menos 50%, 60%, 70%, 80%, 90%, 95% o 99% de complementariedad de secuencias a lo largo de la longitud de la secuencia de emparejamiento tracr cuando se alinean de manera óptima. En algunas realizaciones, la enzima CRISPR comprende una o más secuencias de localización nuclear de suficiente fortaleza para conducir la acumulación de dicha enzima CRISPR en una cantidad detectable en el núcleo de una célula eucariota. En algunas realizaciones,
la enzima CRISPR es una enzima del sistema CRISPR de tipo II. En algunas realizaciones, la enzima CRISPR es una enzima Cas9. En algunas realizaciones, la enzima Cas9 es Cas9 de S. pneumoniae, S. pyogenes o S. thermophilus y puede incluir Cas9 mutada procedente de estos organismos. La enzima puede ser un homólogo u ortólogo de Cas9. En algunas realizaciones, la enzima CRISPR es codón-optimizada para expresión en una célula eucariota. En algunas realizaciones, la enzima CRISPR dirige la escisión de una o dos cadenas en la posición de la secuencia objetivo. En algunas realizaciones, la enzima CRISPR carece de actividad de escisión de la cadena de ADN. En algunas realizaciones, el primer elemento regulador es un activador de la polimerasa III. En algunas realizaciones, el segundo elemento regulador es un activador de la polimerasa II. En algunas realizaciones, la secuencia guía es al menos 15, 16, 17, 18, 19, 20, 25 nucleótidos o entre 10-30 o entre 15-25 o entre 15-20 nucleótidos de longitud. En un aspecto, se describe en la presente memoriaun organismo eucariota no humano; preferiblemente un organismo eucariota multicelular, que comprende una célula eucariota huésped según cualquiera de las realizaciones descritas. En otros aspectos, se describe en la presente memoria un organismo eucariota; preferiblemente un organismo eucariota multicelular, que comprende una célula eucariota huésped según cualquiera de las realizaciones descritas. El organismo en algunas realizaciones de estos aspectos puede ser un animal; por ejemplo, un mamífero. También, el organismo puede ser un artrópodo tal como un insecto. El organismo puede ser también una planta. Además, el organismo puede ser un hongo.
En un aspecto, se describe en la presente memoria un estuche que comprende uno o más de los componentes descritos en la presente memoria. En algunas realizaciones, el estuche comprende un sistema vector e instrucciones para usar el estuche. En algunas realizaciones, el sistema vector comprende: (a) un primer elemento regulador unido de manera operable a una secuencia de emparejamiento tracr y uno o más sitios de inserción para insertar una o más secuencias guía aguas arriba de la secuencia de emparejamiento tracr, en el que cuando se expresan, la secuencia guía dirige la unión específica de la secuencia de un complejo CRISPR a una secuencia fijada como objetivo en una célula eucariota, en la que el complejo CRISPR comprende una enzima CRISPR complejada con (1) la secuencia guía que se hibrida a la secuencia objetivo y (2) la secuencia de emparejamiento tracr que se hibrida a la secuencia tracr y/o (b) un segundo elemento regulador unido de manera operable a una secuencia codificadora de enzima que codifica dicha enzima CRISPR que comprende una secuencia de localización nuclear. En algunas realizaciones, el estuche comprende los componentes (a) y (b) situados en los mismos vectores del sistema o diferentes. En algunas realizaciones, el componente (a) comprende además la secuencia tracr aguas abajo de la secuencia de emparejamiento tracr bajo el control del primer elemento regulador. En algunas realizaciones, el componente (a) comprende además dos o más secuencias guía unidas de manera operable al primer elemento regulador, en el que cuando se expresan, cada una de las dos o más secuencias guía dirige la unión específica de la secuencia de un complejo CRISPR a una secuencia objetivo diferente en una célula eucariota. En algunas realizaciones, el sistema comprende además un tercer elemento regulador, tal como un activador de la polimerasa III, unido de manera operable a dicha secuencia tracr. En algunas realizaciones, la secuencia tracr presenta al menos 50%, 60%, 70%, 80%, 90%, 95% o 99% de complementariedad de secuencias a lo largo de la longitud de la secuencia de emparejamiento tracr cuando se alinea de manera óptima. En algunas realizaciones, la enzima CRISPR comprende una o más secuencias de localización nuclear de suficiente fortaleza para conducir la acumulación de dicha enzima CRISPR en una cantidad detectable en el núcleo de una célula eucariota. En algunas realizaciones, la enzima CRISPR es una enzima del sistema CRISPR de tipo II. En algunas realizaciones, la enzima CRISPR es una enzima Cas9. En algunas realizaciones, la enzima Cas9 es Cas9 de S. pneumoniae, S. pyogenes o S. thermophilus y puede incluir Cas9 mutada procedente de estos organismos. La enzima puede ser un homólogo u ortólogo de Cas9. En algunas realizaciones, la enzima CRISPR es codón-optimizada para expresión en una célula eucariota. En algunas realizaciones, la enzima CRISPR dirige la escisión de una o dos cadenas en la posición de la secuencia objetivo. En algunas realizaciones, la enzima CRISPR carece de actividad de escisión de la cadena de ADN. En algunas realizaciones, el primer elemento regulador es un activador de la polimerasa III. En algunas realizaciones, el segundo elemento regulador es un activador de la polimerasa II. En algunas realizaciones, la secuencia guía es al menos 15, 16, 17, 18, 19, 20, 25 nucleótidos o entre 10-30 o entre 15-25 o entre 15-20 nucleótidos de longitud.
En un aspecto, se describe en la presente memoria un método para modificar un polinucleótido objetivo en una célula eucariota. En algunas realizaciones, el método comprende permitir que se una un complejo CRISPR al polinucleótido objetivo para efectuar la escisión de dicho polinucleótido objetivo modificando de ese modo el polinucleótido objetivo, en el que el complejo CRISPR comprende una enzima CRISPR complejada con una secuencia guía hibridada a una secuencia objetivo dentro de dicho polinucleótido objetivo, en el que dicha secuencia guía está ligada a una secuencia de emparejamiento tracr que a su vez se hibrida a una secuencia tracr. En algunas realizaciones, dicha escisión comprende escindir una o dos cadenas en la posición de la secuencia objetivo mediante dicha enzima CRISPR. En algunas realizaciones, dicha escisión da como resultado la transcripción disminuida de un gen objetivo. En algunas realizaciones, el método comprende además reparar dicho polinucleótido objetivo escindido por recombinación homóloga con un polinucleótido exógeno de plantilla, en el que dicha reparación da como resultado una mutación que comprende una inserción, supresión o sustitución de uno o más nucleótidos de dicho polinucleótido objetivo. En algunas realizaciones, dicha mutación da como resultado uno o más cambios de aminoácido en una proteína expresada a partir de un gen que comprende la secuencia objetivo. En algunas realizaciones, el método comprende además suministrar uno o más vectores a
dicha célula eucariota, en el que uno o más vectores conducen la expresión de uno o más de: la enzima CRISPR, la secuencia guía ligada a la secuencia de emparejamiento tracr y la secuencia tracr. En algunas realizaciones, dichos vectores se suministran a la célula eucariota en un individuo. En algunas realizaciones, dicha modificación tiene lugar en dicha célula eucariota en un cultivo celular. En algunas realizaciones, el método comprende además aislar dicha célula eucariota de un individuo antes de dicha modificación. En algunas realizaciones, el método comprende además devolver dicha célula y/o células eucariotas procedentes de ahí a dicho individuo.
En un aspecto, se describe en la presente memoria un método para modificar la expresión de un polinucleótido en una célula eucariota. En algunas realizaciones, el método comprende permitir que un complejo CRISPR se una al polinucleótido de manera que dicha unión dé como resultado la expresión aumentada o disminuida de dicho polinucleótido; en el que el complejo CRISPR comprende una enzima CRISPR complejada con una secuencia guía hibridada a una secuencia objetivo dentro de dicho polinucleótido, en el que dicha secuencia guía está ligada a una secuencia de emparejamiento tracr que a su vez se hibrida a una secuencia tracr. En algunas realizaciones, el método comprende además suministrar uno o más vectores a dichas células eucariotas, en el que uno o más vectores conducen la expresión de una o más de: la enzima CRISPR, la secuencia guía ligada a la secuencia de emparejamiento tracr y la secuencia tracr.
En un aspecto, se describe en la presente memoria un método para generar una célula eucariota modelo que comprende un gen mutado de la enfermedad. En algunas realizaciones, un gen de la enfermedad es cualquier gen asociado a un aumento del riesgo de tener o desarrollar una enfermedad. En algunas realizaciones, el método comprende (a) introducir uno o más vectores en una célula eucariota, en la que uno o más vectores conducen la expresión de uno o más de: una enzima CRISPR, una secuencia guía ligada a una secuencia de emparejamiento tracr y una secuencia tracr y (b) permitir que un complejo CRISPR se una a un polinucleótido objetivo para efectuar la escisión del polinucleótido objetivo dentro de dicho gen de la enfermedad, en el que el complejo CRISPR comprende la enzima CRISPR complejada con (1) la secuencia guía que se hibrida a la secuencia objetivo dentro del polinucleótido objetivo y (2) la secuencia de emparejamiento tracr que se hibrida a la secuencia tracr, generándose de ese modo una célula eucariota modelo que comprende un gen mutado de la enfermedad. En algunas realizaciones, dicha escisión comprende escindir una o dos cadenas en la posición de la secuencia objetivo mediante dicha enzima CRISPR. En algunas realizaciones, dicha escisión da como resultado la transcripción disminuida de un gen objetivo. En algunas realizaciones, el método comprende además reparar dicho polinucleótido objetivo escindido por recombinación homóloga con un polinucleótido exógeno de plantilla, en el que dicha reparación da como resultado una mutación que comprende una inserción, supresión o sustitución de uno o más nucleótidos de dicho polinucleótido objetivo. En algunas realizaciones, dicha mutación da como resultado uno o más cambios de aminoácido en una expresión de proteína de un gen que comprende la secuencia objetivo.
En un aspecto, se describe en la presente memoria un método para desarrollar un agente biológicamente activo que module un caso de señalización celular asociado a un gen de la enfermedad. En algunas realizaciones, un gen de la enfermedad es cualquier gen asociado a un aumento del riesgo de tener o desarrollar una enfermedad. En algunas realizaciones, el método comprende (a) poner en contacto un compuesto de ensayo con una célula modelo de una cualquiera de las realizaciones descritas y (b) detectar un cambio en una lectura que sea indicativo de una reducción o un aumento del caso de señalización celular asociado a dicha mutación en dicho gen de la enfermedad, desarrollando de ese modo dicho agente biológicamente activo que module dicho caso de señalización celular asociado a dicho gen de la enfermedad.
En un aspecto, se describe en la presente memoria un polinucleótido recombinante que comprende una secuencia guía aguas arriba de una secuencia de emparejamiento tracr, en el que la secuencia guía cuando se expresa dirige la unión específica de la secuencia de un complejo CRISPR a una correspondiente secuencia objetivo presente en una célula eucariota. En algunas realizaciones, la secuencia objetivo es una secuencia vírica presente en una célula eucariota. En algunas realizaciones, la secuencia objetivo es un proto-oncogén o un oncogén.
En un aspecto, se describe en la presente memoria un método para seleccionar una o más células procariotas por introducción de una o más mutaciones en un gen en una o más células procariotas, comprendiendo el método: introducir uno o más vectores en la célula o las células procariotas, en las que uno o más vectores conducen la expresión de uno o más de: una enzima CRISPR, una secuencia guía ligada a una secuencia de emparejamiento tracr, una secuencia tracr y una plantilla de edición; en la que la plantilla de edición comprende una o más mutaciones que anulan la escisión de la enzima CRISPR; permitir la recomendación homóloga de la plantilla de edición con el polinucleótido objetivo en la célula o las células que se tienen que seleccionar; permitir que un complejo CRISPR se una a un polinucleótido objetivo para efectuar la escisión del polinucleótido objetivo dentro de dicho gen, en el que el complejo CRISPR comprende la enzima CRISPR complejada con (1) la secuencia guía que se hibrida a la secuencia objetivo dentro del polinucleótido objetivo y (2) la secuencia de emparejamiento tracr que se hibrida a la secuencia tracr, en la que la unión del complejo CRISPR al polinucleótido objetivo induce la muerte celular, permitiendo de ese modo que se seleccione una o más células procariotas en que se ha introducido una o más mutaciones. En una realización preferida, la enzima CRISPR es Cas9. En otro aspecto que se describe
en la presente memoria, la célula que se tiene que seleccionar puede ser una célula eucariota. Los aspectos de la invención permiten la selección de células específicas sin que se requiera un marcador de selección o un procedimiento de dos etapas que pueda incluir un sistema de contraselección.
Breve descripción de los dibujos
Las nuevas características de la invención se exponen con particularidad en las reivindicaciones adjuntas. Se obtendrá un mejor entendimiento de las características y ventajas de la presente invención por referencia a la siguiente descripción detallada que explica realizaciones ilustrativas, en que se utilizan los principios de la invención, y los dibujos adjuntos de los que:
La Figura 1 muestra un modelo esquemático del sistema CRISPR. La nucleasa Cas9 de Streptococcus pyogenes (amarillo) se fija como objetivo para ADN genómico por un ARN guía sintético (ARNsg) que consiste en una secuencia guía de 20-nt (azul) y un andamio (rojo). Los pares de bases de la secuencia guía con el ADN objetivo (azul), directamente aguas arriba de una unidad adyacente protoespaciadora 5'-NGG (PAM; magenta) requisito y Cas9 media una doble rotura de cadena (DRC) ~3 pb aguas arriba de la PAM (triángulo rojo).
La Figura 2A-F muestra un sistema CRISPR ejemplar, un posible mecanismo de acción, una adaptación de ejemplo para la expresión en células eucariotas y los resultados de los ensayos que evalúan la localización nuclear y la actividad de CRISPR.
La Figura 3 muestra una casete de expresión ejemplar para expresión de elementos del sistema CRISPR en células eucariotas, estructuras previstas de secuencias guía de ejemplo y actividad del sistema CRISPR cuando se mide en células eucariotas y procariotas.
La Figura 4A-D muestra los resultados de una evaluación de especificidad de SpCas9 para un objetivo de ejemplo. La Figura 5A-G muestra un sistema vector ejemplar y los resultados para su uso en dirigir recombinación homóloga en células eucariotas.
La Figura 6 proporciona una tabla de secuencias protoespaciadoras y resume los resultados de eficacia de modificación para objetivos protoespaciadores diseñados basándose en sistemas CRISPR de S. pyogenes y S. thermophilus ejemplares con las correspondientes PAM contra sitios en genomas humanos y de ratón. Se transinfectaron células con Cas9 y pre-ARNcr/ARNtracr o ARN quimérico y se analizaron 72 horas después de transinfección. Se calculan los porcentajes de inserciones o supresiones basándose en los resultados de la prueba Surveyor de estirpes celulares indicadas (N=3 para todos los objetivos protoespaciadores, los errores son E. E. M. , N. D. indica no detectable usando la prueba Surveyor y N. E. indica no ensayado en este estudio).
La Figura 7A-C muestra una comparación de diferentes transcritos de ARNtracr para fijar como objetivo genes mediados por Cas9.
La Figura 8 muestra un esquema de una prueba de nucleasa surveyor para detección de microinserciones y microsupresiones inducidas por doble rotura de cadena.
La Figura 9A-B muestra vectores de expresión bicistrónicos ejemplares para expresión de elementos del sistema CRISPR en células eucariotas.
La Figura 10 muestra un ensayo de interferencia de transformación de plásmidos bacterianos, casetes de expresión y plásmidos usados en el mismo y eficacias de transformación de células usadas en el mismo.
La Figura 11A-C muestra histogramas de distancias entre PAM del sitio 1 SF370 de S. pyogenes (NGG) (Figura 10A) y PAM del sitio 2 de LMD9 de S. thermophilus (NNAGAAW) (Figura 10B) en el genoma humano y las distancias para cada PAM por cromosoma (Chr) (Figura 10C).
La Figura 12A-C muestra un sistema CRISPR ejemplar, una adaptación de ejemplo para expresión en células eucariotas y los resultados de ensayos que evalúan la actividad CRISPR.
La Figura 13A-C muestra manipulaciones ejemplares de un sistema CRISPR para fijar como objetivo sitios genómicos en células de mamífero.
La Figura 14A-B muestra los resultados de un análisis por el método Northern de tratamiento de ARNcr en células de mamífero.
La Figura 15 muestra una selección ejemplar de protoespaciadores en los sitios PVALB humano y Th de ratón.
La Figura 16 muestra protoespaciador de ejemplo y los correspondientes objetivos de secuencia PAM del sistema CRISPR de S. thermophilus en el sitio EMX1 humano.
La Figura 17 muestra una tabla de secuencias para cebadores y sondas usados para pruebas Surveyor, RFLP, secuenciación genómica y método de Northern.
La Figura 18A-C muestra manipulación ejemplar de un sistema CRISPR con ARN quiméricos y los resultados de pruebas SURVEYOR para actividad del sistema en células eucariotas.
La Figura 19A-B muestra una representación gráfica de los resultados de pruebas SURVEYOR para actividad del sistema CRISPR en células eucariotas.
La Figura 20 muestra una visualización ejemplar de algunos sitios objetivo de Cas9 de S. pyogenes en el genoma humano usando el buscador de genomas UCSC.
La Figura 21 muestra estructuras secundarias previstas para ARN quiméricos ejemplares que comprenden una secuencia guía, secuencia de emparejamiento tracr y secuencia tracr.
La Figura 22 muestra vectores de expresión bicistrónicos ejemplares para expresión de elementos del sistema CRISPR en células eucariotas.
La Figura 23 muestra que la actividad de la nucleasa Cas9 frente a objetivos endógenos puede explotarse para edición de genomas. (a) Concepto de edición de genomas usando el sistema CRISPR. La construcción que fija como objetivo CRISPR dirigió la escisión de un sitio cromosómico y se cotransformó con una plantilla de edición que se recombinó con el objetivo para evitar la escisión. Los transformados resistentes a la kanamicina que sobrevivieron al ataque de CRISPR contenían modificaciones introducidas por la plantilla de edición. ARN de CRISPR trans-activante, tracr; gen de resistencia a la kanamicina, aphA-3. (b) Transformación de ADN crR6M en células R682325 sin plantilla de edición, srtA natural R6 o las plantillas de edición de R6370,1. La recombinación de srtA R6 o R63701 evitó la escisión por Cas9. Se calculó la eficacia de transformación como unidades formadoras de colonias (ufc) por pg de ADN de crR6M; se muestran los valores medios con desviaciones estándar de al menos tres experimentos independientes. Se realizó análisis PCR sobre 8 clones en cada transformación. “No.” indica el sitio srtA no editado de la cepa R682325; “Ed.” muestra la plantilla de edición. Los objetivos R682325 y R63701 se distinguen por restricción con EaeI.
La Figura 24 muestra análisis de PAM y secuencias de siembra que eliminan la escisión de Cas9. (a) Se transformaron los productos PCR con secuencias PAM aleatoriadas o secuencias de siembra aleatoriadas en células crR6. Estas células expresaron Cas9 cargado con un ARNcr que fijó como objetivo una región cromosómica de células R682325 (resaltado en rosa) que está ausente a partir del genoma R6. Más de 2x105 transformados resistentes al cloranfenicol, soportando PAM o secuencias de siembra inactivas, se combinaron para multiplicación y secuenciación intensa de la región fijada como objetivo. (b) Proporción relativa de número de lecturas después de la transformación de las construcciones PAM aleatorias en células crR6 (comparado con el número de lecturas en transformados R6). Se muestra la abundancia relativa para cada secuencia PAM de 3 nucleótidos. Se muestran secuencias seriamente infrarrepresentadas (NGG) en rojo; una parcialmente infrarrepresentada en naranja (NAG) (c) Proporción relativa de número de lecturas después de transformación de las construcciones de secuencias de siembra aleatorias en células crR6 (comparado con el número de lecturas en transformados de R6). Se muestra la abundancia relativa de cada nucleótido para cada posición de los primeros 20 nucleótidos de la secuencia protoespaciadora. La abundancia alta indica ausencia de escisión por Cas9, es decir, una mutación que inactiva CRISPR. La línea gris muestra el nivel de la secuencia TN. La línea de puntos representa el nivel por encima del cual una mutación interrumpe significativamente la escisión (Véase la sección “Analysis of deep sequencing data” en el Ejemplo 5)).
La Figura 25 muestra la introducción de mutaciones únicas y múltiples usando el sistema CRISPR en S. pneumoniae, (a) Secuencias de nucleótidos y aminoácidos de bgaA tipo natural y editado (nucleótidos verdes; restos de aminoácidos subrayados). Se muestra el protoespaciador, PAM y sitios de restricción. (b) Eficacia de transformación de células transformadas con construcciones fijadoras de objetivo en presencia de una plantilla o control de edición. (c) Análisis PCR para 8 transformados de cada experimento de edición seguido por digestión con BtgZI (R ^A ) y Tsel (NE^AA). La supresión de bgaA se reveló como un producto PCR más pequeño. (d) Prueba de Miller para medir la actividad de la p-galactosidasa de cepas TN y editadas. (e) Para una doble supresión de una sola etapa, la construcción fijadora de objetivo contenía dos espaciadores (en este caso emparejando srtA y bgaA) y se cotransformó con dos plantillas de edición diferentes (f) Análisis PCR para 8 transformados para detectar supresiones en los sitios srtA y bgaA. Los transformados 6/8 contenían supresiones de los dos genes.
La Figura 26 proporciona mecanismos que subrayan la edición usando el sistema CRISPR. (a) Se introdujo un codón de terminación en el gen de resistencia a eritromicina ermAM para generar cepa JEN53. Se puede restaurar
la secuencia natural fijando como objetivo el codón de terminación con la construcción CRISPR::ermAM(terminación) y usando la secuencia natural ermAM como una plantilla de edición. (b) Secuencias ermAM mutantes y de tipo natural. (c) Fracción de ufc (ermR) resistente a eritromicina calculada a partir de las ufc totales o resistentes a kanamicina (kanR). (d) Fracción de células totales que adquieren tanto la construcción CRISPR como la plantilla de edición. La cotransformación de la construcción que fija como objetivo CRISPR produjo más transformados (ensayo t, p=0,011). En todos los casos los valores muestran la media ± d. e. para tres experimentos independientes.
La Figura 27 ilustra edición de genomas con el sistema CRISPR en E. coli. (a) Un plásmido resistente a kanamicina que soporta la matriz CRISPR (pCRISPR) que fija como objetivo el gen para editar puede transformarse en la cepa de recombinación de HME63 que contiene un plásmido resistente a cloranfenicol que alberga cas9 y tracr (pCas9), junto con un oligonucleótido que especifica la mutación. (b) Se introdujo una mutación de K42T que confería resistencia a la estreptomicina en el gen rpsL (c) Fracción de ufc resistentes a estreptomicina (strepR) calculada a partir de ufc totales o resistentes a kanamicina (kanR). (d) Fracción de células totales que adquieren tanto el plásmido pCRISPR como el oligonucleótido de edición. La cotransformación del plásmido que fija como objetivo pCRISPR produjo más transformados (ensayo t, p=0,004). En todos los casos los valores mostraron la media ± d. e. para tres experimentos independientes.
La Figura 28 ilustra que la transformación de ADN genómico de crR6 conduce a la edición del sitio fijado como objetivo (a) El elemento IS1167 de R6 de S. pneumoniae fue reemplazado por el sitio CRISPR01 de SF370 de S. pyogenes para generar cepa crR6. Este sitio codifica la nucleasa Cas9, una matriz CRISPR con seis espaciadores, el ARNtracr que se requiere para biogénesis de ARNcr y Cas1, Cas2 y Csn2, proteínas no necesarias para fijación como objetivo. La cepa crR6M contiene un sistema CRISPR funcional mínimo sin cas1, cas2 y csn2. El gen aphA-3 codifica resistencia a kanamicina. Se fusionaron protoespaciadores de los bacteriófagos de estreptococos 98232,5 y 9370,1 a un gen de resistencia a cloranfenicol (cat) y se integraron en el gen srtA de cepa R6 para generar las cepas R68232,5 y R6370,1. (b) Panel izquierdo: Transformación de ADN genómico crR6 y crR6M en R682325 y R63701. Como un control de competencia celular también se transformó un gen resistente a estreptomicina. Panel derecho: Análisis PCR de 8 transformados R682325 con ADN genómico crR6. Se usaron cebadores que multiplican el sitio srtA por PCR. 7/8 colonias genotipadas reemplazaron el sitio srtA R68232,5 por el sitio TN del ADN genómico de crR6.
La Figura 29 proporciona cromatogramas de secuencias de ADN de células editadas obtenidas en este estudio. En todos los casos, se indica el protoespaciador natural y mutante y secuencias PAM (o su complemento inverso). Cuando es relevante, se proporciona la secuencia de aminoácidos codificada por el protoespaciador. Para cada experimento de edición, se secuenciaron todas las cepas para las que el análisis PCR y de restricción corroboró la introducción de la modificación deseada. Se muestra un cromatograma representativo. (a) Cromatograma para la introducción de una mutación PAM en el objetivo R682325 (Figura 23d). (b) Cromatogramas para la introducción de las mutaciones R>A y NE>AA en p-galactosidasa (bgaA) (Figura 25c). (c) Cromatograma para la introducción de una supresión de 6.664 pb en ORF (marco de lectura abierta, por sus siglas en inglés) de bgaA (Figuras 25c y 25f). La línea de puntos indica los límites de la supresión. (d) Cromatograma para la introducción de una supresión de 729 pb en ORF de srtA (Figura 25f). La línea de puntos indica los límites de la supresión. (e) Cromatogramas para la generación de un codón de terminación prematuro en ermAM (Figura 33). (f) Edición de rpsL en E. coli (Figura 27).
La Figura 30 ilustra inmunidad de CRISPR contra objetivos de S. pneumoniae aleatorios conteniendo diferentes PAM. (a) Posición de los 10 objetivos aleatorios en el genoma R6 de S. pneumoniae. Los objetivos elegidos presentan diferentes PAM y están en ambas cadenas. (b) Se clonaron los espaciadores correspondientes a los objetivos en una matriz de CRISPR mínima en plásmido pLZ12 y se transformaron en cepa crR6Rc, que suministra la maquinaria de tratamiento y fijación como objetivo en trans. (c) Eficacia de transformación de los diferentes plásmidos en cepa R6 y crR6Rc. No se recuperaron colonias para la transformación de pDB99-108 (T1-T10) en crR6Rc. La línea de puntos representa el límite de detección de la prueba.
La Figura 31 proporciona un esquema general para editar genoma fijado como objetivo. Para facilitar la edición de genoma fijado como objetivo, se logró además que crR6M contuviera ARNtracr, Cas9 y sólo una repetición de la matriz CRISPR seguido por marcador de resistencia a kanamicina (aphA-3), generando cepa crR6Rk. Se usa ADN de esta cepa como una plantilla para PCR con cebadores diseñados para introducir un nuevo espaciador (recuadro verde designado con N). Las PCR izquierda y derecha se reúnen usando el método de Gibson para crear la construcción fijadora de objetivo. Después se transforman las construcciones tanto fijadora de objetivo como de edición en cepa crR6Rc, que es una cepa equivalente a crR6Rk, pero presenta el marcador de resistencia a kanamicina reemplazado por un marcador de resistencia a cloranfenicol (cat). Aproximadamente el 90% de los transformados resistentes a kanamicina contienen la mutación deseada.
La Figura 32 ilustra la distribución de distancias entre las PAM. NGG y CCN que se consideran que son PAM válidas. Se muestran los datos para el genoma R6 de S. pneumoniae, así como para una secuencia aleatoria de
la misma longitud y con el mismo contenido en GC (39,7%). La línea de puntos representa la distancia promedio (12) entre las PAM en el genoma R6.
La Figura 33 ilustra edición mediada por CRISPR del sitio ermAM usando ADN genómico como construcción fijadora de objetivo. Para usar ADN genómico como construcción fijadora de objetivo es necesario evitar autoinmunidad de CRISPR y, por lo tanto, se debe usar un espaciador contra una secuencia no presente en el cromosoma (en este caso el gen de resistencia a eritromicina ermAM). (a) Secuencias de nucleótidos y aminoácidos del gen ermAM natural y mutado (letras rojas). Se muestra el protoespaciador y las secuencias PAM. (b) Un esquema para la edición mediada por CRISPR del sitio ermAM usando a Dn genómico. Una construcción que soporta un espaciador que fija como objetivo ermAM (recuadro azul) se prepara por PCR y ensamblaje de Gibson y se transforma en cepa crR6Rc, que genera cepa JEN37. Después se usó el ADN genómico de JEN37 como una construcción fijadora de objetivo y se cotransformó con la plantilla de edición en JEN38, una cepa en la que el gen srtA fue reemplazado por una copia natural de ermAM. Los transformados resistentes a kanamicina contienen el genotipo editado (JEN43). (c) Número de células resistentes a kanamicina obtenidas después de cotransformación de plantillas de fijación como objetivo y de edición o control. En presencia de la plantilla de control se obtuvieron 5,4*103 ufc/ml y 4,3*105 ufc/ml cuando se usó la plantilla de edición. Esta diferencia indica una eficacia de edición de aproximadamente 99% [(4,3*105-5,4*103)/4,3x105]. (d) Para comprobar la presencia de células editadas se sembraron en estrías siete clones resistentes a kanamicina y JEN38 en placas de agar con eritromicina (erm+) o sin eritromicina (erm-). Sólo el control positivo mostró resistencia a eritromicina. El genotipo mutado de ermAM de uno de estos transformados también fue verificado por secuenciación de ADN (Figura 29e).
La Figura 34 ilustra la introducción secuencial de mutaciones por edición de genomas mediada por CRISPR. (a) Un esquema para introducción secuencial de mutaciones por edición de genomas mediada por CRISPR. Primero, se logra R6 para generar crR6Rk. crR6Rk se co-transforma con una construcción que fija como objetivo srtA fusionado a cat para selección de cloranfenicol de células editadas, junto con una construcción de edición para una supresión dentro del marco de AsrtA. Se genera cepa AsrtA crR6 por selección sobre cloranfenicol. Con posterioridad, se cotransforma la cepa AsrtA con una construcción que fija como objetivo bgaA fusionada a aphA-3 para selección de kanamicina de células editadas y una construcción de edición que contiene una supresión dentro del marco de AbgaA. Finalmente, el sitio de CRISPR logrado puede ser eliminado del cromosoma cotransformando primero ADN de R6 que contiene el sitio IS1167 natural y un plásmido que soporta un protoespaciador bgaA (pDB97) y selección en espectinomicina. (b) Análisis PCR para 8 transformados resistentes a cloranfenicol (Cam) para detectar la supresión en el sitio srtA. (c) Actividad de la p-galactosidasa cuando se mide por la prueba de Miller. En S. pneumoniae, se ancha esta enzima a la pared celular mediante sortasa A. La supresión del gen srtA da como resultado la liberación de p-galactosidasa en el sobrenadante. Los mutantes de AbgaA no muestran actividad. (d) Análisis PCR para 8 transformados resistentes a espectinomicina (Espec) para detectar el reemplazo del sitio CRISPR mediante IS1167 natural.
La Figura 35 ilustra la frecuencia de mutación de fondo de CRISPR en S. pneumoniae. (a) Transformación de las construcciones que fijan como objetivo CRISPR::0 o CRISPR::erm(terminación) en JEN53, con o sin la plantilla de edición de ermAM. La diferencia en UFC de kanR entre CRISPR::0 y CRISPR::erm(terminación) indica que la escisión de Cas9 destruye células no editadas. Los mutantes que escapan a la interferencia de CRISPR en ausencia de plantilla de edición se observan a una frecuencia de 3*10'3 (b) El análisis PCR del sitio CRISPR de escapistas muestra que 7/8 presentan una supresión de espaciador. (c) El escapista #2 soporta una mutación puntual en cas9.
La Figura 36 ilustra que los elementos esenciales del sitio 1 de CRISPR de S. pyogenes se reconstituyen en E. coli usando pCas9. El plásmido contenía ARNtracr, Cas9, así como una secuencia líder que conduce la matriz ARNcr. Los plásmidos pCRISPR contenían el líder y la matriz sólo. Los espaciadores pueden insertarse en la matriz de ARNcr entre los sitios BsaI usando oligonucleótidos hibridados. El diseño de oligonucleótidos se muestra en el fondo. pCas9 soportaba resistencia a cloranfenicol (CmR) y se basa en la cadena principal del plásmido pACYC184 de copia baja. pCRISPR se basa en el plásmido pZE21 de alto número de copias. Se requirieron dos plásmidos debido a que un plásmido pCRISPR que contenía un espaciador que fijada como objetivo el cromosoma de E. coli puede no ser construido usando este organismo como un huésped de clonación si también está presente Cas9 (destruirá el huésped).
La Figura 37 ilustra edición dirigida por CRISPR en MG1655 de E.coli. Un oligonucleótido (W542) que soporta una mutación puntual que tanto confiere resistencia a estreptomicina como suprime la inmunidad de CRISPR, junto con un plásmido que fija como objetivo rpsL (pCRISPR::rpsL) o un plásmido de control (pCRISPR::0) se cotransformaron en MG1655 de la cepa E.coli natural que contenía pCas9. Los transformados se seleccionaron en medios que contenían estreptomicina o kanamicina. La línea de puntos indica el límite de detección de la prueba de transformación.
La Figura 38 ilustra la frecuencia de mutación de fondo de CRISPR en HME63 de E. coli. (a) Transformación de los plásmidos pCRISPR::0 o pCRISPR::rpsL en células competentes HME63. Los mutantes que escapan a la
interferencia de CRISPR fueron observados a una frecuencia de 2,6*10-4. (b) La multiplicación de la matriz CRISPR de escapistas demostró que 8/8 había suprimido el espaciador.
La Figura 39A-D muestra una representación circular del análisis filogenético que revela cinco familias de Cas9s, incluyendo tres grupos de Cas9s grandes (-1.400 aminoácidos) y dos de Cas9s pequeños (— 1.100 aminoácidos). La Figura 40A-F muestra la representación lineal del análisis filogenético que revela cinco familias de Cas9s, incluyendo tres grupos de Cas9s grandes (-1.400 aminoácidos) y dos de Cas9s pequeños (— 1.100 aminoácidos). La Figura 41A-M muestra las secuencias en el caso de que los puntos de mutación estén situados dentro del gen SpCas9.
La Figura 42 muestra una construcción esquemática en la que se fusiona el dominio de activación de la transcripción (VP64) a Cas9 con dos mutaciones en los dominios catalíticos (D10 y H840).
La Figura 43A-D muestra edición de genomas por recombinación homóloga. (a) Esquema de nickasa SpCas9, con mutación D10A en el dominio catalítico RuvC I. (b) Esquema que representa recombinación homóloga (RH) en el sitio EMX1 humano usando oligonucleótido monocatenario sentido o antisentido como plantillas de reparación. La flecha roja arriba indica sitio de escisión de ARNsg; los cebadores PCR para genotipados (Tablas J y K) se indican como flechas en el panel derecho. (c) Secuencia de región modificada por RH. d, prueba SURVEYOR para las inserciones o supresiones medidadas por SpCas9 natural (tn) y nickasa (D10A) en el sitio objetivo 1 de EMX1 (n=3). Las flechas indican las posiciones de tamaños de fragmento esperados.
La Figura 44A-B muestra únicos diseños de vector para SpCas9.
La Figura 45 muestra cuantificación de escisión de construcciones de NLS-Csn1 NLS-Csn1, Csn1, Csn1-NLS, NLS-Csn1-NLS, NLS-Csn1-GFP-NLS y UnTFN.
La Figura 46 muestra la frecuencia del índice de NLS-Cas9, Cas9, Cas9-NLS y NLS-Cas9-NLS.
La Figura 47 muestra un gel que demuestra que SpCas9 con mutaciones de nickasa (individualmente) no inducen roturas de doble cadena.
La Figura 48 muestra un diseño del oligo ADN usado como plantilla de recombinación homóloga (RH) en este experimento y una comparación de la eficacia de la RH inducida por diferentes combinaciones de proteína Cas9 y plantilla de RH.
La Figura 49A muestra el mapa de vectores que fijan como objetivo Cas9, Rosa26 condicional.
La Figura 49B muestra el mapa de vectores que fijan como objetivo Cas9, Rosa26 constitutivo.
La Figura 50A-H muestra las secuencias de cada elemento presentes en los mapas de vectores de las Figuras 49A-B.
La Figura 51 muestra un esquema de los elementos importantes en las construcciones Cas9 constitutiva y condicional.
La Figura 52 muestra la validación funcional de la expresión de las construcciones Cas9 constitutiva y condicional. La Figura 53 muestra la validación de la actividad de la nucleasa Cas9 por Surveyor.
La Figura 54 muestra la cuantificación de actividad de la nucleasa Cas9.
La Figura 55 muestra diseño de construcción y estrategia de recombinación homóloga (RH).
La Figura 56 muestra los resultados de genotipados de PCR genómico para las construcciones constitutiva (derecha) y condicional (izquierda) en dos tiempos de exposición de gel diferentes (fila de arriba durante 3 min y fila del fondo durante 1 min).
La Figura 57 muestra activación de Cas9 en mESCs.
La Figura 58 muestra un esquema de la estrategia usada para mediar la inactivación génica vía NHEJ (recombinación no homóloga, por sus siglas en inglés) usando una versión de nickasa de Cas9 junto con dos ARN guías.
La Figura 59 muestra cómo la reparación de la rotura de doble cadena de ADN (DSB, por sus siglas en inglés) activa la edición de genes. En la ruta de unión de extremos no homólogos propensa a errores (NHEJ), los extremos
de una DSB se tratan mediante maquinarias de reparación de ADN endógeno y se vuelven a unir entre sí, lo que puede dar como resultado mutaciones de inserción/supresión (indel) aleatorias en el sitio de unión. Las mutaciones indel que tienen lugar en la región codificadora de un gen pueden dar como resultado cambio de marco y un codón de terminación prematuro, conduciendo a inactivación génica. Alternativamente, se puede suministrar una plantilla de reparación en la forma de un plásmido u oligodesoxinucleótidos monocatenarios (ODNss) para potenciar la ruta de reparación dirigida por homología (HDR, por sus siglas en inglés), que permite alta fidelidad y edición precisa.
La Figura 60 muestra la cronología y resumen de los experimentos. Las etapas para diseño de reactivo, construcción, validación y expansión de estirpes celulares. Los ARNsg de costumbre (barras azul claro) para cada objetivo, así como cebadores de genotipado, se diseñan in silico vía nuestra herramienta de diseño on-line (disponible en el sitio web genome-engineering.org/tools). Los vectores de expresión ARNsg se clonaron después en un plásmido que contenía Cas9 (PX330) y se verificó por secuenciación de ADN. Los plásmidos completos (los pCRISPR) y las plantillas de reparación opcionales para facilitar la reparación dirigida por homología, se transinfectan después a células y se prueba su capacidad para mediar la escisión fijada como objetivo. Finalmente, las células transinfectadas pueden expandirse de manera clonal para obtener estirpes celulares isogénicas con mutaciones definidas.
La Figura 61A-C muestra selección de objetivo y preparación de reactivo. (a) Para Cas9 de S. pyogene, los objetivos de 20 pb (resaltado en azul) deben ir seguidos por 5'-NGG, que puede tener lugar en cualquier cadena en ADN genómico. Se recomienda usar la herramienta on-line descrita en este protocolo para ayudar a la selección de objetivo (www.genome-engineering.org/tools). (b) Esquema para cotransinfección de plásmido de expresión de Cas9 (PX165) y casete de expresión de ARNsg conducida por U6 multiplicada por PCR. Usar una plantilla de PCR que contenga activador de U6 y un cebador directo fijado (U6 Dir), puede agregarse ADN codificador de ARNsg sobre el cebador inverso de u6 (U6 Inv) y sintetizarse como un oligo ADN extendido (Oligo ultrámeros de IDT). Obsérvese que la secuencia guía (de N azul) en U6 Inv es el complemento inverso de la secuencia objetivo flanqueadora de 5'-NGG. (c) Esquema para clonación sin marca de las oligo secuencias guía en un plásmido que contiene andamio Cas9 y ARNsg (PX330). Los oligos guía (de N azul) contienen salientes para ligadura en el par de sitios de BbsI en PS330, igualándose las orientaciones de las cadenas superior y del fondo a aquéllas del objetivo genómico (es decir, oligo superior es la secuencia de 20 pb que precede 5'-NGG en ADN genómico). La digestión de PX330 con BbsI permite el reemplazo de los sitios de restricción de tipo IIs (trazado azul) con inserción directa de oligos hibridados. Cabe señalar que se puso una G extra antes de la primera base de la secuencia guía. Los solicitantes han encontrado que una G extra delante de la secuencia guía no afecta negativamente a la eficacia de la fijación como objetivo. En los casos en los que la secuencia guía de 20 nucleótidos de elección no empieza con guanina, la guanina extra asegurará que el ARNsg se transcriba de manera eficaz por el activador de U6, que prefiere una guanina en la primera base de la transcripción.
La Figura 62A-D muestra los resultados anticipados para NHEJ múltiplex. (a) Esquema de la prueba SURVEYOR usada para determinar el porcentaje de inserciones o supresiones. Primero, se multiplica por PCR ADN genómico de la población heterogénea de células fijadas como objetivo de Cas9. Se vuelven a hibridar después lentamente los amplicones para generar los heterodúplex. Se escinden los heterodúplex hibridados de nuevo mediante nucleasa SURVEYOR, mientras se dejan intactos los homodúplex. Se calcula la eficacia de escisión mediada por Cas9 (% indel) basándose en la fracción de ADN escindido, cuando se determina por intensidad integrada de bandas de gel. (b) Se diseñan dos ARNsg (barras naranja y azul) para fijar como objetivo los sitios (GRIN2B y DYRK1A humanos. El gel SURVEYOR muestra la modificación en ambos sitios en células transinfectadas. Las flechas coloreadas indicaron los tamaños de fragmento esperados para cada sitio. (c) Se diseña un par de ARNsg (barras azul claro y verde) para escindir un exón (azul oscuro) en el sitio EMX1 humano. Las secuencias objetivo y los PAM (rojo) se muestran en los respectivos colores y los sitios de escisión se indican por triángulo rojo. La unión prevista se muestra a continuación. Los clones individuales aislados de poblaciones de células transinfectadas con ARNsg 3, 4 o ambos se ensayan mediante PCR (OUT Dir, OUT Inv), que refleja una supresión de ~270 pb. Se muestran los clones representativos sin modificación (12/23), modificaciones monoalélicas (10/23) y bi-alélicas (1/23). Se usan cebadores IN Dir e IN Inv para identificar sistemáticamente sucesos de inversión (Fig. 6d). (d) Cuantificación de estirpes clonales con supresiones de exones EMX1. Se usan dos pares de ARNsg (ARNsg de flanqueado izquierdo 3.1, 3.2; ARNsg de flanqueado derecho 4.1,4.2) para mediar las supresiones de tamaños variables alrededor de un exón EMX1. Se aíslan de manera clonal células transinfectadas y se expanden por análisis de genotipado para supresiones y sucesos de inversión. De los 105 clones se detectan sistemáticamente, 51 (49%) y 11 (10%) soportando supresiones heterozigóticas y homozigóticas, respectivamente. Se proporcionan tamaños de supresión aproximados puesto que las uniones pueden ser variables.
La Figura 63A-C muestra la aplicación de ssODN (del inglés, oligodesoxirribonucleótido de cadena sencilla) y vector de fijación como objetivo para mediar la RH con mutante tanto natural como nickasa de Cas9 en células HEK293FT y HUES9 con eficacias que oscilan de 1,0-27%.
La Figura 64 muestra un esquema de un método basado en PCR para fijar como objetivo CRISPR de manera rápida y eficaz en células de mamífero. Un plásmido que contiene el activador U6 de polimerasa III de ARN humano se multiplica por PCR usando un cebador directo específico de U6 y un cebador inverso que soporta el complemento inverso de parte del cebador U6, el andamio ARNsg(+85) con secuencia guía y nucleótidos 7 T para terminación transcripcional. Se purifica el producto PCR resultante y se suministra conjuntamente con un plásmido que soporta Cas9 conducido por el activador CBh.
La Figura 65 muestra estuche de detección de mutación SURVEYOR de resultados transgenómicos para cada ARNg y respectivos controles. Un resultado SURVEYOR positivo es una banda grande que corresponde al PCR genómico y dos bandas más pequeñas que son el producto de la nucleasa SURVEYOR que haga una rotura de doble cadena en el sitio de una mutación. Se validó cada ARNg en la estirpe celular de ratón, Neuro-N2a, por cotransinfección transitoria liposomal con hSpCas9. 72 horas postransinfección se purificó ADN genómico usando ADN QuickExtract de Epicentre. Se realizó PCR para multiplicar el sitio de interés.
La Figura 66 muestra los resultados Surveyor para 38 crías vivas (rutas 1-38) 1 cría muerta (ruta 39) y 1 cría natural para comparación (ruta 40). Se inyectó ARNg Chd8.2 a las crías 1-19 y ARNg Chd8.3 a las crías 20-38. De las 38 crías vivas, 13 fueron positivas para una mutación. Una cría muerta también tuvo una mutación. No se detectó mutación en la muestra natural. La secuenciación PCR genómica fue consistente con los hallazgos de la prueba SURVEYOR.
La Figura 67 muestra un diseño de diferentes construcciones de NLS Cas9. Todos los Cas9 fueron la versión de codón optimizado humano de Sp Cas9. Las secuencias NLS se ligaron al gen cas9 en cualquier N-terminal o C-terminal. Todas las variantes de Cas9 con diferentes diseños NLS se clonaron en un vector de esqueleto que conteniendo eso es conducido por un activador de EF1a. En el mismo vector hay un sitio EMX1 humano que fija como objetivo ARN quimérico conducido por activador U6, formando un sistema de dos componentes.
La Figura 68 muestra la eficacia de la escisión genómica inducida por variantes de Cas9 que soportan diferentes diseños NLS. El porcentaje indica la porción de ADN genómico de EMX1 humano que se escindió por cada construcción. Todos los experimentos son de 3 replicados biológicos. n = 3, el error indica E.E.M.
La Figura 69A muestra un diseño del CRISPR-TF (factor de transcripción) con actividad de activación transcripcional. Se expresa el ARN quimérico por el activador U6, mientras una versión de doble mutante, de codón optimizado humano de la proteína Cas9 (hSpCas9m), ligada de manera operable a NLS triple y un dominio funcional VP64 se expresa por un activador de EF1a. Las dobles mutaciones, D10A y H840A, hacen a la proteína cas9 incapaz de introducir escisión, pero mantiene su capacidad para fijar como objetivo ADN cuando se guía por el ARN quimérico.
La Figura 69B muestra la activación de la transcripción del gen SOX2 humano con sistema CRISPR-TF (ARN quimérico y la proteína de fusión Cas9-NLS-VP64). Se transinfectaron células 293FT con plásmidos que soportaban dos componentes: (1) diferentes ARN quiméricos conducidos por U6 que fijan como objetivo secuencias de 20 pb en o alrededor del sitio genómico SOX2 humano y (2) proteína de fusión hSpCas9m (doble mutante)-NLS-VP64 conducida por EF1a. 96 horas postransinfección, se recogieron las células 293FT y se midió el nivel de activación por la inducción de expresión de ARNm usando una prueba qRT-PCR (del inglés, reacción en cadena de la polimerasa con transcriptasa inversa). Se normalizan todos los niveles de expresión contra el grupo de control (barra gris), que representa los resultados de células transinfectadas con el plásmido de esqueleto de CRISPR-TF sin ARN quimérico. Las sondas de qRT-PCR usadas para detectar el ARNm de SOX2 es la prueba de expresión de genes humanos Taqman (Life Technologies). Todos los experimentos representan datos de 3 replicados biológicos, n=3, las barras de error muestran E.E.M.
La Figura 70 representa la optimización de la arquitectura NLS para SpCas9.
La Figura 71 muestra una representación gráfica de QQ para las secuencias NGGNN.
La Figura 72 muestra un histograma de la densidad de datos con distribución normal fijada (línea negra) y 0,99 de cuantil (línea de puntos).
La Figura 73A-C muestra represión guiada por ARN de expresión de bgaA por ARNdg::cas9**. a. La proteína Cas9 se une al ARNtracr y al precursor de ARN de CRISPR que se trata por RNAseIII para formar el ARNcr. El ARNcr dirige la unión de Cas9 al activador de bgaA y reprime la transcripción. b. Se representan los objetivos usados para dirigir Cas9** al activador de bgaA. El codón putativo -35, -10 así como el codón de partida bgaA están en negrita. c. La actividad de la betagalactosidasa cuando se mide por la prueba de Miller en ausencia de fijación como objetivo y para los cuatro objetivos diferentes.
La Figura 74A-E muestra caracterización de represión mediada por Cas9**. a. El gen gfpmut2 y su activador, incluyendo las señales -35 y -10 se representan junto con la posición de los diferentes sitios objetivo usados en
el estudio. b. Fluorescencia relativa en la fijación como objetivo de la cadena codificadora. c. Fluorescencia relativa en la fijación como objetivo de la cadena no codificadora. d. Método de Northern con sondas B477 y B478 sobre ARN extraído de T5, T10, B10 o una cepa de control sin un objetivo. e. Efecto de un número creciente de mutaciones en el extremo 5' del ARNcr de B1, T5 y B10.
Las figuras en la presente memoria son sólo para fines ilustrativos y no están necesariamente dibujadas a escala.
Descripción detallada de la invención
Los términos "polinucleótido", "nucleótido", "secuencia de nucleótidos", "ácido nucleico" y "oligonucleótido" se usan de forma intercambiable. Se refieren a una forma polimérica de nucleótidos de cualquier longitud, desoxirribonucleótidos o ribonucleótidos o análogos de los mismos. Los polinucleótidos pueden tener cualquier estructura tridimensional y pueden realizar cualquier función, conocida o desconocida. Los siguientes no son ejemplos limitantes de los polinucleótidos: regiones codificadoras o no codificadoras de un gen o fragmento de gen, sitios (sitio) definidos a partir de análisis de unión, exones, intrones, ARN mensajero (ARNm), ARN de transferencia, ARN ribosómico, ARN de interferencia corta (ARNic), ARN de horquilla corta (ARNhc), micro- ARN (ARNmi), ribozimas y ADNc, polinucleótidos recombinantes, polinucleótidos ramificados, plásmidos, vectores, ADN aislado de cualquier secuencia, ARN aislado de cualquier secuencia, sondas de ácidos nucleicos y cebadores. Un polinucleótido puede comprender uno o más nucleótidos modificados, tales como nucleótidos metilados y análogos de nucleótidos. Si hay, se pueden impartir modificaciones a la estructura del nucleótido antes o después del ensamblado del polímero. La secuencia de nucleótidos puede ser interrumpida por componentes no nucleótidos. Un polinucleótido puede ser modificado además después de polimerización, tal como por conjugación con un componente etiquetado.
En aspectos de la descripción, los términos "ARN quimérico", "ARN guía quimérico", "ARN guía", "ARN guía único" y "ARN guía sintético" se usan de forma intercambiable y se refieren a la secuencia de polinucleótidos que comprende la secuencia guía, la secuencia tracr y la secuencia de emparejamiento tracr. El término "secuencia guía" se refiere a la secuencia de aproximadamente 20 pb dentro del ARN guía que especifica el sitio objetivo y se puede usar de manera intercambiable con los términos "guía" o "espaciador". El término "secuencia de emparejamiento tracr" también se puede usar de forma intercambiable con el término "repetición o repeticiones directas".
Como se usa en la presente memoria el término "natural" es un término de la técnica entendido por los expertos y significa la forma típica de un organismo, cepa, gen o característica como se encuentra en la naturaleza como se distingue de formas mutantes o variantes.
Como se usa en la presente memoria, el término "variante" se debería considerar que significa la exhibición de cualidades que presentan un patrón que se desvía de lo que ocurre en la naturaleza.
Los términos "que no se encuentra en la naturaleza" o "logrado" se usan de forma intercambiable e indican la implicación de la mano del hombre. Los términos, cuando se refieren a moléculas de ácido nucleico o polipéptidos significan que la molécula de ácido nucleico o el polipéptido está al menos sustancialmente exento de al menos otro componente con el que se asocian de manera natural en la naturaleza y como se encuentran en la naturaleza.
"Complementariedad" se refiere a la capacidad de un ácido nucleico para formar el enlace o enlaces de hidrógeno con otra secuencia de ácidos nucleicos por emparejamiento de bases de Watson-Crick tradicional u otros tipos no tradicionales. Un porcentaje de complementariedad indica el porcentaje de restos en una molécula de ácido nucleico que puede formar enlaces de hidrógeno (por ej., emparejamiento de bases de Watson-Crick) con una segunda secuencia de ácidos nucleicos (por ej., 5, 6, 7, 8, 9, 10 de un total de 10 siendo complementariedad del 50%, 60%, 70%, 80%, 90% y 100%). "Perfectamente complementario" significa que todos los restos contiguos de una secuencia de ácidos nucleicos formarán enlace de hidrógeno con el mismo número de restos contiguos en una segunda secuencia de ácidos nucleicos. "Sustancialmente complementario" como se usa en la presente memoria se refiere a un grado de complementariedad que es al menos 60%, 65%, 70%, 75%, 80%, 85%, 90%%, 95%, 97%, 98%, 99% o 100% por una región de 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21,22, 23, 24, 25, 30, 35, 40, 45, 50 o más nucleótidos o se refiere a dos ácidos nucleicos que se hibridan en condiciones rigurosas.
Como se usa en la presente memoria, "condiciones rigurosas" para hibridación se refiere a las condiciones en las cuales un ácido nucleico que tiene complementariedad a una secuencia objetivo se hibrida predominantemente con la secuencia objetivo y sustancialmente no se hibrida a las secuencias no objetivo. Las condiciones rigurosas son en general dependientes de la secuencia y varían dependiendo de una serie de factores. En general, cuanto más larga la secuencia, mayor la temperatura a la que se hibrida de manera específica la secuencia a su secuencia objetivo. Se describen con detalle ejemplos no limitantes de condiciones rigurosas en Tijssen (1993), Laboratory Techniques in Biochemistry and Molecular Biology-Hybridization With Nucleic Acid Probes Part I, Segundo Capítulo "Overview of principles of hybridization and the strategy of nucleic acid probe assay", Elsevier, N.Y.
"Hibridación" se refiere a una reacción en la que uno o más polinucleótidos reaccionan para formar un complejo que se estabiliza vía enlace de hidrógeno entre las bases de los restos de nucleótido. El enlace de hidrógeno puede tener lugar por apareamiento de bases de Watson Crick, unión de Hoogstein o de cualquier otra manera específica de la secuencia. El complejo puede comprender dos cadenas que forman una estructura dúplex, tres o más cadenas que forman un complejo multicatenario, una sola cadena que se autohibrida o cualquier combinación de éstas. Una reacción de hibridación puede constituir una etapa en un procedimiento más extenso, tal como la iniciación de PCR o la escisión de un polinucleótido mediante una enzima. Una secuencia capaz de hibridación con una secuencia determinada se refiere como el “complemento” de la secuencia determinada.
Como se usa en la presente memoria, "expresión" se refiere al procedimiento por el que un polinucleótido se transcribe de una plantilla de ADN (tal como en ARNm u otro transcrito de ARN) y/o el procedimiento por el que se traduce con posterioridad un ARNm transcrito en péptidos, polipéptidos o proteínas. Los transcritos y polipéptidos codificados se pueden referir de manera colectiva como “producto génico”. Si el polinucleótido procede de ADN genómico, la expresión puede incluir proceso de corte y empalme del ARNm en una célula eucariota.
Los términos "polipéptido", "péptido" y "proteína" se usan de forma intercambiable en la presente memoria para referirse a polímeros de aminoácidos de cualquier longitud. El polímero puede ser lineal o ramificado y puede comprender aminoácidos modificados y puede ser interrumpido por no aminoácidos. Los términos también incluyen un polímero de aminoácidos que ha sido modificado; por ejemplo, formación de enlace disulfuro, glucosilación, lipidación, acetilación, fosforilación o cualquier otra manipulación, tal como conjugación con un componente etiquetado. Como se usa en la presente memoria el término “aminoácido” incluye aminoácidos naturales y/o no naturales o sintéticos, incluyendo glicina y los dos isómeros ópticos D o L y análogos de aminoácido y peptidomiméticos.
Los términos " individuo," "individual" y "paciente" se usan de forma intercambiable en la presente memoria para referirse a un vertebrado, preferiblemente un mamífero, más preferiblemente un ser humano. Los mamíferos incluyen, pero no están limitados a, murinas, simios, seres humanos, animales de granja, animales deportivos y mascotas. Los tejidos, las células y su progenie de una entidad biológica obtenida in vivo o cultivada in vitro también se incluyen.
Los términos "agente terapéutico", "agente terapéutico capaz" o "agente de tratamiento" se usan de forma intercambiable y se refieren a una molécula o compuesto que confiere algún efecto beneficioso en la administración a un individuo. El efecto beneficioso incluye permitir determinaciones de diagnóstico; alivio de una enfermedad, síntoma, trastorno o afección patológica; reducir o prevenir el comienzo de una enfermedad, síntoma, trastorno o afección y en general contrarrestar una enfermedad, síntoma, trastorno o afección patológica.
Como se usa en la presente memoria, "tratamiento" o " tratar," o "paliar" o "aliviar" se usan de forma intercambiable. Estos términos se refieren a una propuesta para obtener resultados beneficiosos o deseados incluyendo, pero no limitándose a, un beneficio terapéutico y/o un beneficio profiláctico. Por beneficio terapéutico se quiere decir cualquier mejora terapéuticamente relevante en, o efecto sobre, una o más enfermedades, afecciones o síntomas bajo tratamiento. Para beneficio profiláctico, las composiciones se pueden administrar a un individuo con riesgo de desarrollar una enfermedad, afección o síntoma, particular, o a un individuo que indica uno o más de los síntomas fisiológicos de una enfermedad, incluso aunque la enfermedad, afección o síntoma pueda no haberse manifestado aún.
El término "cantidad eficaz" o "cantidad terapéuticamente eficaz" se refiere a la cantidad de un agente que es suficiente para efectuar resultados beneficiosos o deseados. La cantidad terapéuticamente eficaz puede variar dependiendo de uno o más de: el individuo y la enfermedad que se está tratando, el peso y la edad del individuo, la gravedad de la enfermedad, la manera de administración y similares, que puede ser determinado fácilmente por un experto en la materia. El término también se aplica a una dosis que proporcionará una imagen para detección por uno cualquiera de los métodos de formación de imagen descritos en la presente memoria. La dosis específica puede variar dependiendo de uno o más de: el agente particular elegido, el régimen de dosificación que se tenga que seguir, si se administra en asociación con otros compuestos, la sincronización de la administración, el tejido que se tiene que someter a formación de imagen y el sistema de suministro físico en el que se soporte.
La práctica de la presente descripción emplea, a menos que se indique de otro modo, técnicas convencionales de inmunología, bioquímica, química, biología molecular, microbiología, biología celular, ADN genómico y recombinante, que están dentro de la destreza de la técnica. Véase Sambrook, Fritsch y Maniatis, MOLECULAR CLONING: A LABORATORY MANUAL, 2a edición (1989); CURRENT PROTOCOLS IN MOLECULAR BIOLOGY (F. M. Ausubel, et al. eds., (1987)); las series METHODS IN ENZYMOLOGY (Academic Press, Inc.): PCR 2: A PRACTICAL APPROACH (M. J. MacPherson, B. D. Hames y G. R. Taylor eds. (1995)), eds. Harlow and Lane, (1988) ANTIBODIES, A LABORATORY MANUAL y ANIMAL CELL CULTURE (ed. R. I. Freshney, (1987)).
Varios aspectos de la descripción se refieren a sistemas vector que comprenden uno o más vectores o vectores como tales. Se pueden diseñar vectores para expresión de transcritos CRISPR (por ejemplo, transcritos de ácidos nucleicos, proteínas o enzimas) en células procariotas o eucariotas. Por ejemplo, se pueden expresar transcritos de CRISPR en células bacterianas tales como Escherichia coli, células de insecto (usando vectores de expresión de baculovirus), células de levaduras o células de mamíferos. Las células huésped adecuadas se discuten además en Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego; Calif. (1990). Alternativamente, el vector de expresión recombinante se puede transcribir y traducir in vitro, por ejemplo, usando secuencias reguladoras de activador T7 y polimerasa T7.
Se pueden introducir vectores y propagar en una procariota. En algunas realizaciones, se usa una procariota para multiplicar copias de un vector que se tiene que introducir en una célula eucariota o como un vector intermedio en la producción de un vector que se tiene que introducir en una célula eucariota (por ej., multiplicando un plásmido como parte de un sistema de empaquetamiento de vector vírico). En algunas realizaciones, se usa una procariota para multiplicar copias de un vector y expresar uno o más ácidos nucleicos, tal como para proporcionar una fuente de una o más proteínas para suministro a una célula huésped u organismo huésped. La expresión de proteínas en procariotas se lleva a cabo lo más frecuentemente en Escherichia coli con vectores que contienen activadores constitutivos o inducibles que dirigen la expresión de proteínas de fusión o de no fusión. Los vectores de fusión añaden una serie de aminoácidos a una proteína codificada allí, tal como al término amino de la proteína recombinante. Dichos vectores de fusión pueden servir para uno o más fines, tales como: (i) para aumentar la expresión de proteína recombinante; (ii) para aumentar la solubilidad de la proteína recombinante y (iii) para ayudar en la purificación de la proteína recombinante actuando como un ligando en purificación de afinidad. Con frecuencia, en vectores de expresión de fusión, se introduce un sitio de escisión proteolítico en la unión del resto de fusión y la proteína recombinante para permitir la separación de la proteína recombinante del resto de fusión posterior a la purificación de la proteína de fusión. Tales enzimas, y sus secuencias de reconocimiento afines, incluyen Factor Xa, trombina y enterocinasa. Vectores de expresión de fusión de ejemplo incluyen pGEX (Pharmacia Biotech Inc; Smith y Johnson, 1988. Gene 67: 31-40), pMAL (New England Biolabs, Beverly, Mass.) y pRIT5 (Pharmacia, Piscataway, N. J.) que condensan glutationa S-transferasa (GST), proteína de unión de maltosa E o proteína A, respectivamente, a la proteína recombinante objetivo.
Ejemplos de vectores de expresión de E. coli no de fusión, inducibles, adecuados, incluyen pTrc (Amrann et al., (1988) Gene 69: 301-315) y pET 11d (Studier et al., GENE EXPRESSION TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990) 60-89).
En algunas realizaciones, un vector es un vector de expresión de levadura. Ejemplos de vectores para expresión en levadura Saccharomyces cerivisae incluyen pYepSec1 (Baldari, et al., 1987. EMBO J. 6: 229-234), pMFa (Kuijan y Herskowitz, 1982. Cell 30: 933-943), pJRY88 (Schultz et al., 1987. Gene 54: 113-123), pYES2 (Invitrogen Corporation, San Diego, Calif.) y picZ (InVitrogen Corp, San Diego, Calif.).
En algunas realizaciones, un vector conduce la expresión de proteínas en células de insecto usando vectores de expresión de baculovirus. Los vectores baculovirus disponibles para expresión de proteínas en células de insecto cultivadas (por ej., células SF9) incluyen la serie pAc (Smith, et al., 1983. Mol. Cell. Biol. 3: 2.156-2.165) y la serie pVL (Lucklow y Summers, 1989. Virology 170: 31-39).
En algunas realizaciones, un vector es capaz de conducir la expresión de una o más secuencias en células de mamífero usando un vector de expresión de mamífero. Ejemplos de vectores de expresión de mamífero incluyen pCDM8 (Seed, 1987. Nature 329: 840) y pMT2PC (Kaufman, et al., 1987. EMBO J. 6: 187-195). Cuando se usa en células de mamífero, las funciones de control del vector de expresión se proporcionan típicamente por uno o más elementos reguladores. Por ejemplo, los activadores usados comúnmente proceden de polioma, adenovirus 2, citomegalovirus, virus 40 de simio y otros descritos en la presente memoria y conocidos en la técnica. Para otros sistemas de expresión adecuados para células tanto procariotas como eucariotas véanse, por ejemplo, los Capítulos 16 y 17 de Sambrook, et al., MOLECULAR CLONING: A LABORATORY MANUAL. 2a ed., Cold Spring Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N. Y., 1989.
En algunas realizaciones, el vector de expresión de mamíferos recombinante es capaz de dirigir la expresión del ácido nucleico preferentemente en un tipo de célula particular (por ejemplo, se usan elementos reguladores específicos del tejido para expresar el ácido nucleico). Los elementos reguladores específicos del tejido son conocidos en la técnica. Ejemplos no limitantes de activadores específicos del tejido adecuados incluyen el activador de albúmina (específico del hígado; Pinkert, et al., 1987. Genes Dev. 1: 268-277), activadores específicos linfoides (Calame y Eaton, 1988. Adv. Immunol. 43: 235-275), en particular activadores de receptores de células T (Winoto y Baltimore, 1989. EMBO J. 8: 729-733) e inmunoglobulinas (Baneiji, et al., 1983. Cell 33: 729-740; Queen y Baltimore, 1983. Cell 33: 741-748), activadores específicos de las neuronas (por ej., el activador de neurofilamentos; Byrne y Ruddle, 1989. Proc. Natl. Acad Sci. u Sa 86: 5.473-5.477), activadores específicos del páncreas (Edlund, et al., 1985. Science 230: 912-916) y activadores específicos de las glándulas mamarias (por ej., activador de suero de leche; Patente de EE.UU. N° 4. 873. 316 y Publicación de la Solicitud de Patente
Europea N° 264.166). También se incluyen activadores regulados por el desarrollo, por ej., los activadores de hox de murina (Kessel y Gruss, 1990. Science 249: 374-379) y el activador de a-fetoproteína (Campes y Tilghman, 1989. Genes Dev. 3: 537-546).
En algunas realizaciones, un elemento regulador se une de manera operable a uno o más elementos de un sistema CRISPR a fin de conducir la expresión de uno o más elementos del sistema CRISPR. En general, las CRISPR (Repeticiones Palindrómicas Cortas Agrupadas y Regularmente Interespaciadas, por sus siglas en inglés), también conocidas como las SPIDR (Repeticiones Directas Intercaladas Separadas, por sus siglas en inglés), constituyen una familia de sitios de ADN que son normalmente específicos para una especie bacteriana particular. El sitio de CRISPR comprende una clase distinta de repeticiones de secuencias cortas intercaladas (las SSR, por sus siglas en inglés) que se reconocieron en E. coli (Ishino et al., J. Bacteriol., 169: 5.429-5.433 [1987] y Nakata et al., J. Bacteriol., 171: 3.553-3.556 [1989]) y genes asociados. Las SSR intercaladas similares han sido identificadas en Haloferax mediterranei, Streptococcus pyogenes, Anabaena y Mycobacterium tuberculosis (Véase, Groenen et al., Mol. Microbiol., 10: 1.057-1.065 [1993]; Hoe et al., Emerg. Infect. Dis., 5: 254-263 [1999]; Masepohl et al., Biochim. Biophys. Acta 1.307: 26-30 [1996] y Mojica et al., Mol. Microbiol., 17: 85-93 [1995]). El sitio de CRISPR difiere típicamente de las otras SSR por la estructura de las repeticiones, que se han denominado repeticiones espaciadas regularmente cortas (las SRSR, por sus siglas en inglés) (Janssen et al. OMICS J. Integ. Biol., 6: 23-33 [2002] y Mojica et al., Mol. Microbiol., 36: 244-246 [2000]). En general, las repeticiones son elementos cortos que tienen lugar en agrupaciones que están regularmente espaciadas por secuencias de intervención únicas con una longitud sustancialmente constante (Mojica et al., [2000], supra). Aunque las secuencias de repeticiones se conservan mucho entre cepas, el número de repeticiones intercaladas y las secuencias de las regiones espaciadoras difieren típicamente de cepa a cepa (van Embden et al., J. Bacteriol., 182: 2.393-2.401 [2000]). Se ha identificado el sitio de CRISPR en más de 40 procariotas (Véase por ej., Jansen et al., Mol. Microbiol., 43: 1.565-1.575 [2002] y Mojica et al., [2005]) incluyendo, pero no limitado a Aeropyrum, Pyrobaculum, Sulfolobus, Archaeoglobus. Halocarcula, Methanobacterium, Methanococcus, Methanosarcina, Methanopyrus, Pyrococcus, Picrophilus, Thermoplasma, Corynebacterium, Mycobacterium, Streptomyces, Aquifex, Porphyromonas, Chlorobium, Thermus, Bacillus, Listeria, Staphylococcus, Clostridium, Thermoanaerobacter, Mycoplasma, Fusobacterium, Azarcus, Chromobacterium, Neisseria, Nitrosomonas, Desulfovibrio, Geobacter, Myxococcus, Campylobacter, Wolinella. Acinetobacter, Erwinia, Escherichia, Legionella, Methylococcus, Pastaurella, Photobacterium, Salmonella, Xanthomonas, Yersinia, Treponema y Thermotoga.
En general, "sistema CRISPR" se refiere colectivamente a transcritos y otros elementos implicados en la expresión de o dirigidos a la actividad de genes asociados a CRISPR ("Cas"), incluyendo secuencias que codifican un gen Cas, una secuencia tracr (CRISPR trans-activante) (por ej., ARNtracr o un ARNtracr parcial activo), una secuencia de emparejamiento tracr (incluyendo una "repetición directa" y una repetición directa parcial tratada por ARNtracr en el contexto de un sistema CRISPR endógeno), una secuencia guía (también referida como un "espaciador" en el contexto de un sistema CRISPR endógeno) u otras secuencias y transcritos de un sitio CRISPR. En algunas realizaciones, uno o más elementos de un sistema CRISPR proceden de un sistema CRISPR tipo I, tipo II o tipo III. En algunas realizaciones, uno o más elementos de un sistema CRISPR proceden de un organismo particular que comprende un sistema CRISPR endógeno, tal como Streptococcus pyogenes. En general, un sistema CRISPR se caracteriza por elementos que activan la formación de un complejo CRISPR en el sitio de una secuencia objetivo (también referido como un protoespaciador en el contexto de un sistema CRISPR endógeno). En el contexto de formación de un complejo CRISPR, "secuencia objetivo" se refiere a una secuencia para la que se diseña una secuencia guía que presente complementariedad, donde la hibridación entre una secuencia objetivo y una secuencia guía active la formación de un complejo CRISPR. No se requiere necesariamente complementariedad completa, siempre que haya suficiente complementariedad para producir hibridación y activar la formación de un complejo CRISPR. Una secuencia objetivo puede comprender cualquier polinucleótido, tal como polinucleótidos de a Dn o ARN. En algunas realizaciones, se sitúa una secuencia objetivo en el núcleo o citoplasma de una célula. En algunas realizaciones, la secuencia objetivo puede estar dentro de un organelo de una célula eucariota, por ejemplo, mitocondria o cloroplasto. Una secuencia o plantilla que se puede usar para recombinación en el sitio fijado como objetivo que comprende las secuencias objetivo se refiere como una “plantilla de edición” o “polinucleótido de edición” o “secuencia de edición”. En aspectos de la descripción, un polinucleótido de plantilla exógeno se puede referir como una plantilla de edición. En un aspecto de la descripción la recombinación es recombinación homóloga.
Típicamente, en el contexto de un sistema CRISPR endógeno, la formación de un complejo CRISPR (que comprende una secuencia guía hibridada a una secuencia objetivo y complejada con una o más proteínas Cas) da como resultado la escisión de una o más cadenas en o cerca de (por ej., dentro de 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 20, 50 o más pares de bases de) la secuencia objetivo. Sin desear estar limitados por la teoría, la secuencia tracr, que puede comprender o constar de toda o una porción de una secuencia tracr natural (por ej., aproximadamente o más de aproximadamente 20, 26, 32, 45, 48, 54, 63, 67, 85 o más nucleótidos de una secuencia tracr natural), también puede formar parte de un complejo CRISPR, tal como por hibridación a lo largo de al menos una porción de la secuencia tracr a toda o una porción de una secuencia de emparejamiento tracr que esté ligada de manera operable a la secuencia guía. En algunas realizaciones, la secuencia tracr presenta suficiente complementariedad
a una secuencia de emparejamiento traer para hibridar y participar en la formación de un complejo CRISPR. Como con la secuencia objetivo, se cree que no es necesaria la complementariedad completa, siempre que haya suficiente para que sea funcional. En algunas realizaciones, la secuencia tracr presenta al menos 50%, 60%, 70%, 80%, 90%, 95% o 99% de complementariedad de secuencias a lo largo de la longitud de la secuencia de emparejamiento tracr cuando se alinea de manera óptima. En algunas realizaciones, uno o más vectores que conducen la expresión de uno o más elementos de un sistema CRISPR se introducen en una célula huésped de manera que la expresión de los elementos del sistema CRISPR dirija la formación de un complejo CRISPR en uno o más sitios objetivo. Por ejemplo, una enzima Cas, una secuencia guía ligada a una secuencia de emparejamiento tracr y una secuencia tracr podían estar unidas cada una de manera operable a elementos reguladores separados en vectores separados. Alternativamente, dos o más de los elementos expresados de los mismos o diferentes elementos reguladores, se pueden combinar en un vector único, proporcionando uno o más vectores adicionales cualquier componente del sistema CRISPR no incluido en el primer vector. Los elementos del sistema CRISPR que se combinan en un único vector se pueden disponer en cualquier orientación adecuada, tal como un elemento situado en 5' con respecto a ("aguas arriba" de) o 3' con respecto a ("aguas abajo" de) un segundo elemento. La secuencia codificadora de un elemento puede estar situada en la misma cadena u opuesta de la secuencia codificadora de un segundo elemento y orientada en la misma dirección u opuesta. En algunas realizaciones, un activador único conduce la expresión de una transcripción que codifica una enzima CRISPR y una o más de la secuencia guía, la secuencia de emparejamiento tracr (opcionalmente ligada de manera operable a la secuencia guía) y una secuencia tracr embebida dentro de una o más secuencias intrón (por ej., cada una en un intrón diferente, dos o más en al menos un intrón o todas en un solo intrón). En algunas realizaciones, la enzima CRISPR, secuencia guía, secuencia de emparejamiento tracr y secuencia tracr se ligan de manera operable a, y se expresan a partir de, el mismo activador.
En algunas realizaciones, un vector comprende uno o más sitios de inserción, tal como una secuencia de reconocimiento de endonucleasa de restricción (también referido como un “sitio de clonación”). En algunas realizaciones, uno o más sitios de inserción (por ej., aproximadamente o más de aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 o más sitios de inserción) están situados aguas arriba y/o aguas abajo de uno o más elementos de secuencia de uno o más vectores. En algunas realizaciones, un vector comprende un sitio de inserción aguas arriba de una secuencia de emparejamiento tracr y opcionalmente aguas abajo de un elemento regulador ligado de manera operable a la secuencia de emparejamiento tracr, de manera que después de la inserción de una secuencia guía en el sitio de inserción y en la expresión de la secuencia guía dirija la unión específica de la secuencia de un complejo CRISPR a una secuencia objetivo en una célula eucariota. En algunas realizaciones, un vector comprende dos o más sitios de inserción, estando situado cada sitio de inserción entre dos secuencias de emparejamiento tracr de manera que se permita la inserción de una secuencia guía en cada sitio. En dicha disposición, dos o más secuencias guía pueden comprender dos o más copias de una secuencia guía única, dos o más secuencias guía diferentes o combinaciones de éstas. Cuando se usan múltiples secuencias guía diferentes, se puede usar una construcción de expresión única para fijar como objetivo la actividad de CRISPR a múltiples secuencias objetivo correspondientes, diferentes, dentro de una célula. Por ejemplo, un solo vector puede comprender aproximadamente o más de aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20 o más secuencias guía. En algunas realizaciones, se pueden proporcionar aproximadamente o más de aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 o más de tales vectores que contienen secuencia guía y se suministran opcionalmente a una célula.
En algunas realizaciones, un vector comprende un elemento regulador unido de manera operable a una secuencia codificadora de enzima que codifica una enzima CRISPR, tal como una proteína Cas. Ejemplos no limitantes de proteínas Cas incluyen Cas1, Cas1B, Cas2, Cas3, Cas4, Cas5, Cas6, Cas7, Cas8, Cas9 (también conocidas como Csn1 y Csx12), Cas10, Csy1, Csy2, Csy3, Cse1, Cse2, Csc1, Csc2, Csa5, Csn2, Csm2, Csm3, Csm4, Csm5, Csm6, Cmr1, Cmr3, Cmr4, Cmr5, Cmr6, Csb1, Csb2, Csb3, Csx17, Csx14, Csx10, Csx16, CsaX, Csx3, Csx1, Csx15, Csf1, Csf2, Csf3, Csf4, homólogos de las mismas o versiones modificadas de las mismas. Estas enzimas son conocidas; por ejemplo, la secuencia de aminoácidos de proteína Cas9 de S. pyogenes se puede encontrar en la base de datos SwissProt con el número de acceso Q99ZW2. En algunas realizaciones, la enzima CRISPR no modificada presenta actividad de escisión de ADN, tal como Cas9. En algunas realizaciones la enzima CRISPR es Cas9 y puede ser Cas9 de S. pyogenes o S. pneumoniae. En algunas realizaciones, la enzima CRISPR dirige la escisión de una o más cadenas en la posición de una secuencia objetivo, tal como dentro de la secuencia objetivo y/o dentro del complemento de la secuencia objetivo. En algunas realizaciones, la enzima CRISPR dirige la escisión de una o más cadenas dentro de aproximadamente 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 50, 100, 200, 500 o más pares de bases del primer o el último nucleótido de una secuencia objetivo. En algunas realizaciones, un vector codifica una enzima CRISPR que muta con respecto a una enzima natural correspondiente de manera que la enzima CRISPR mutadas carece de la capacidad para escindir una o ambas cadenas de un polinucleótido objetivo que contiene una secuencia objetivo. Por ejemplo, una sustitución de aspartato a alanina (D10A) en el dominio catalítico RuvC I de Cas9 de S. pyogenes convierte Cas9 de una nucleasa que escinde dos cadenas en una nickasa (escinde una única cadena). Otros ejemplos de mutaciones que hacen Cas9 una nickasa incluyen, sin limitación, H840A, N854A y N863A. En algunas realizaciones, se puede usar una nickasa de Cas9 junto con secuencia o secuencias guía, por ejemplo, dos secuencias guía, que fijan como objetivo cadenas transcrita y
complementaria, respectivamente, del objetivo de ADN. Esta combinación permite que ambas cadenas sean nickeadas y usadas para inducir NHEJ. Los solicitantes han demostrado (datos no mostrados) la eficacia de dos objetivos de nickasa (es decir, los ARNsg fijados como objetivo en la misma posición pero para diferentes cadenas de ADN) en la inducción de NHEJ mutagénico. Una única nickasa (Cas9-D10A con un único ARNsg) es incapaz de inducir NHEJ y crear inserciones o supresiones (las indel) pero los Solicitantes han demostrado que la nickasa doble (Cas9-D10A y dos ARNsg fijados como objetivo a diferentes cadenas en la misma posición) lo puede hacer en células madre embrionarias humanas (las hESC, por sus siglas en inglés). La eficacia es aproximadamente 50% de nucleasa (es decir, Cas9 regular sin mutación D10) en las hESC.
Como un ejemplo más, dos o más dominios catalíticos de Cas9 (RuvC I, RuvC II y RuvC III) pueden mutar para producir un Cas9 mutado que carece sustancialmente de toda actividad de escisión de ADN. En algunas realizaciones, una mutación de D10A se combina con una o más de las mutaciones H840A, N854A o N863A para producir una enzima Cas9 que carece sustancialmente de toda actividad de escisión de ADN. En algunas realizaciones, se considera que una enzima CRISPR carece sustancialmente de toda actividad de escisión de ADN cuando la actividad de escisión de ADN de la enzima mutada es menor que aproximadamente 25%, 10%, 5%, 1%, 0,1%, 0,01% o menor con respecto a su forma no mutada. Pueden ser útiles otras mutaciones; en el caso de que la Cas9 u otra enzima CRISPR sea de una especie distinta de S. pyogenes, se pueden realizar mutaciones en los correspondientes aminoácidos para conseguir efectos similares.
En algunas realizaciones, una secuencia codificadora de enzimas que codifica una enzima CRISPR es codón optimizada para expresión en células particulares, tales como células eucariotas. Las células eucariotas pueden ser aquéllas de, o procedentes de, un organismo particular, tal como un mamífero, incluyendo, pero no limitado a, ser humano, ratón, rata, conejo, perro o primate no ser humano. En general, optimización de codón se refiere a un procedimiento para modificar una secuencia de ácidos nucleicos para mejorar la expresión en las células huésped de interés reemplazando al menos un codón (por ej., aproximadamente o más de aproximadamente 1, 2, 3, 4, 5, 10, 15, 20, 25, 50 o más codones) de la secuencia natural con codones que se usan más frecuentemente o lo más frecuentemente en los genes de esa célula huésped al tiempo que se mantiene la secuencia de aminoácidos natural. Diversas especies presentan sesgo particular para ciertos codones de un aminoácido particular. El sesgo de codones (diferencias en el uso de codones entre organismos) con frecuencia se correlaciona con la eficacia de traducción de ARN mensajero (ARNm), que se cree a su vez que depende de, entre otras cosas, las propiedades de los codones que se están traduciendo y la disponibilidad de moléculas de ARN de transferencia (ARNt) particulares. La predominancia de los ARNt seleccionados en una célula es en general un reflejo de los codones usados lo más frecuentemente en síntesis de péptidos. De acuerdo con esto, se pueden adaptar los genes para expresión óptima de los genes en un organismo determinado basándose en optimización de codones. Las tablas de utilización de codones están fácilmente disponibles, por ejemplo, en la “Base de Datos de Uso de Codones” y estas tablas se pueden adaptar de una serie de maneras. Véase Nakamura Y., et al. "Codon usage tabulated from the international DNA sequence databases: status for the year 2000" Nucl. Acids Res. 28: 292 (2000). También están disponibles algoritmos de ordenador para codones optimizando una secuencia particular para expresión en una célula huésped particular, también están disponibles tales como Gene Forge (Aptagen; Jacobus, PA). En algunas realizaciones, uno o más codones (por ej., 1, 2, 3, 4, 5, 10, 15, 20, 25, 50 o más o todos los codones) en una secuencia codificando una enzima CRISPR corresponden al codón usado lo más frecuentemente para un aminoácido particular.
En algunas realizaciones, un vector codifica una enzima CRISPR que comprende una o más secuencias de localización nuclear (las NLS, por sus siglas en inglés), tales como aproximadamente o más de aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 o más NLS. En algunas realizaciones, la enzima CRISPR comprende aproximadamente o más de aproximadamente 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 o más NLS en o cerca del amino terminal, aproximadamente o más de aproximadamente 1,2, 3, 4, 5, 6, 7, 8, 9, 10 o más NLS en o cerca del carboxi terminal o una combinación de éstos (por ejemplo, una o más NLS en el amino terminal y una o más NLS en el carboxi terminal). Cuando está presente más de una NLS, cada una se puede seleccionar independientemente de las otras, de manera que puede estar presente una sola NLS en más de una copia y/o junto con otra u otras NLS más presentes en una o más copias. En una realización preferida de la descripción, la enzima CRISPR comprende a lo sumo 6 NLS. En algunas realizaciones, se considera una NLS cerca del N- o C-terminal cuando el aminoácido más cercano de la NLS está dentro de aproximadamente 1, 2, 3, 4, 5, 10, 15, 20, 25, 30, 40, 50 o más aminoácidos a lo largo de la cadena polipeptídica del N o C-terminal. Típicamente, una NLS consta de una o más secuencias cortas de lisinas o argininas cargadas positivamente expuestas en la superficie de la proteína, pero se conocen otros tipos de NLS. Ejemplos no limitantes de las NLS incluyen una secuencia de NLS procedente de: la NLS del antígeno T grande del virus SV40, que tiene la secuencia de aminoácidos PKKKRKV; la NLS de nucleoplasmina (por ej., la NLS bipartita de nucleoplasmina con la secuencia KRPAATKKAGQAKKKK); la NLS de c-myc que tiene la secuencia de aminoácidos PAAKRVKLD o RQRRNELKRSP; la NLS M9 del hRNPA1 que tiene la secuencia NQSSNFGPMKGGNFGGRSSGPYGGGGQYFAKPRNQGGY; la secuencia RMRIZFKNKGKDTAELRRRRVEVSVELRKAKKDEQILKRRNV del dominio IBB de importina-alfa; las secuencias VSRKRPRP y PPKKARED de la proteína T de mioma; la secuencia POPKKKPL de p53 humano; la secuencia SALIKKKKKMAP de c-abl IV de ratón; las secuencias DRLRR y PKQKKRK del virus de la influenza NS1; la
secuencia RKLKKKIKKL del antígeno delta del virus de la Hepatitis; la secuencia REKKKFLKRR de la proteína Mx1 del ratón; la secuencia KRKGDEVDGVDEVAKKKSKK de la poli(ADP-ribosa) polimerasa humana y la secuencia RKCLQAGMNLEARKTKK del glucocorticoide de receptores de hormonas esteroideas (humano).
En general, una o más NLS son de suficiente fortaleza para conducir la acumulación de la enzima CRISPR en una cantidad detectable en el núcleo de una célula eucariota. En general, la fortaleza de actividad de localización nuclear puede proceder del número de las NLS en la enzima CRISPR, la o las NLS particulares usadas o una combinación de estos factores. La detección de acumulación en el núcleo se puede realizar por cualquier técnica adecuada. Por ejemplo, se puede condensar un marcador detectable a la enzima CRISPR, de manera que se pueda visualizar la localización dentro de una célula, tal como junto con un medio para detectar la localización del núcleo (por ejemplo, una mancha específica para el núcleo tal como DAPI). Ejemplos de marcadores detectables incluyen proteínas fluorescentes (tales como proteínas fluorescentes Verdes o GFP; RFP; CFP) y etiquetas de epítopos (etiqueta HA, etiqueta bandera, etiqueta SNAP). También se pueden aislar los núcleos celulares de las células, los contenidos de los cuales se pueden analizar después por cualquier procedimiento adecuado para detectar proteína, tal como immunohistoquímica, ensayo de Western o ensayo de actividad enzimática. La acumulación en el núcleo también se puede determinar de una manera indirecta, tal como por un ensayo para el efecto de la formación de complejo CRISPR (por ejemplo, ensayo para escisión de ADN o mutación en la secuencia objetivo o ensayo para actividad de expresión de genes modificada afectada por la formación de complejo CRISPR y/o actividad de la enzima CRISPR), cuando se compara con un control no expuesto a la enzima o complejo CRISPR o expuesto a una enzima CRISPR que carece de una o más NLS.
En general, una secuencia guía es cualquier secuencia de polinucleótidos que tiene suficiente complementariedad con una secuencia de polinucleótidos objetivo para hibridación con la secuencia objetivo y dirigir la unión específica de la secuencia de un complejo CRISPR a la secuencia objetivo. En algunas realizaciones, el grado de complementariedad entre una secuencia guía y su correspondiente secuencia objetivo, cuando se alinean de manera óptima usando un algoritmo de alineación adecuado, es aproximadamente o más de aproximadamente 50%, 60%, 75%, 80%, 85%, 90%, 95%, 97,5%, 99% o más. La alineación óptima se puede determinar con el uso de cualquier algoritmo adecuado para alinear secuencias, ejemplo no limitante de lo cual incluye el algoritmo Smith-Waterman, el algoritmo Needleman-Wunsch, los algoritmos basados en la transformación de Burrows-Wheeler (por ej., el Alineador de Burrows Wheeler), ClustalW, Clustal X, BLAT, Novoalign (Novocraft Technologies, ELAND (Illumina, San Diego, CA), SOAP (disponible en soap.genomics.org.cn) y Maq (disponible en maq.sourceforge.net). En algunas realizaciones, una secuencia guía es aproximadamente o más de aproximadamente 5, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 35, 40, 45, 50, 75 o más nucleótidos de longitud. En algunas realizaciones, una secuencia guía es menor que aproximadamente 75, 50, 45, 40, 35, 30, 25, 20, 15, 12 o menos nucleótidos de longitud. La capacidad de una secuencia guía para dirigir la unión específica de la secuencia de un complejo CRISPR a una secuencia objetivo se puede evaluar por cualquier prueba adecuada. Por ejemplo, los componentes de un sistema CRISPR suficientes para formar un complejo CRISPR, incluyendo la secuencia guía que se tiene que ensayar, se pueden proporcionar a una célula huésped que tenga la correspondiente secuencia objetivo, tal como por transinfección con vectores que codifiquen los componentes de la secuencia CRISPR, seguido por una evaluación de la escisión preferente dentro de la secuencia objetivo, tal como mediante ensayo Surveyor como se describe en la presente memoria. De manera similar, la escisión de una secuencia de polinucleótidos objetivo se puede evaluar en un tubo de ensayo proporcionando la secuencia objetivo, los componentes de un complejo CRISPR, incluyendo la secuencia guía que se tiene que ensayar y una secuencia guía de control diferente de la secuencia guía de ensayo y comparando la unión o velocidad de escisión en la secuencia objetivo entre las reacciones de la secuencia guía de ensayo y de control. Son posibles otros ensayos, y tendrán lugar para los expertos en la materia.
Se puede seleccionar una secuencia guía para fijar como objetivo cualquier secuencia objetivo. En algunas realizaciones, la secuencia objetivo es una secuencia dentro de un genoma de una célula. Las secuencias objetivo ejemplares incluyen aquéllas que son únicas en el genoma objetivo. Por ejemplo, para la Cas9 de S. pyogenes, una secuencia objetivo única en un genoma puede incluir un sitio objetivo de Cas9 de la forma MMMMMMMMNNNNNNNNNNNNXGG donde NNNNNNNNNNNNXGG (N es A, G, T o C y X puede ser cualquiera) presenta un solo caso en el genoma. Una única secuencia objetivo en un genoma puede incluir un sitio objetivo de Cas9 de S. pyogenes de la forma MMMMMMMMMNNNNNNNNNNNXGG donde NNNNNNNNNNNXGG (N es A, G, T o C y X puede ser cualquiera) presenta un solo caso en el genoma. Para la Cas9 de CRISPR1 de S. thermophilus, una única secuencia objetivo en un genoma puede incluir un sitio objetivo de Cas9 de la forma MMMMMMMMNNNNNNNNNNNNXXAGAAW donde NNNNNNNNNNNNXXAGAAW (N es A, G, T o C; X puede ser cualquiera y W es A o T) presenta un solo caso en el genoma. Una única secuencia objetivo en un genoma puede incluir un sitio objetivo de Cas9 de CRISPR1 de S. thermophilus de la forma MMMMMMMMMNNNNNNNNNNNXXAGAAW donde NNNNNNNNNNNXXAGAAW (N es A, G, T o C; X puede ser cualquiera y W es A o T) presenta un solo caso en el genoma. Para la Cas9 de S. pyogenes, una sola secuencia objetivo en un genoma puede incluir un sitio objetivo de Cas9 de la forma MMMMMMMMNNNNNNNNNNNNXGGXG donde NNNNNNNNNNNNXGGXG (N es A, G, T o C y X puede ser cualquiera) presenta un solo caso en el genoma. Una única secuencia objetivo en un genoma puede incluir un
sitio objetivo de Cas9 de S. pyogenes de la forma MMMMMMMMNMNNNNNNNNNNXGGXG donde NNNNNNNNNNNXGGXG (N es A, G, T o C y X puede ser cualquiera) presenta un único caso en el genoma. En cada una de estas secuencias "M" puede ser A, G, T o C y requiere que no se considere en la identificación de una secuencia como única.
En algunas realizaciones, se selecciona una secuencia guía para reducir el grado de estructura secundaria dentro de la secuencia guía. La estructura secundaria se puede determinar por cualquier algoritmo de plegamiento de polinucleótidos adecuado. Algunos programas se basan en calcular la energía libre mínima de Gibbs. Un ejemplo de uno de tales algoritmos es mFold, como se describe por Zuker y Stiegler (Nucleic Acids Res. 9 (1981), 133 148). Otro algoritmo de plegamiento de ejemplo es el RNAfold webserver online, desarrollado en el Instituto para Química Teórica de la Universidad de Viena, usando el algoritmo de predicción de estructura centroide (véase por ej., A. R. Gruber et al., 2008, Cell 106 (1): 23-24 y PA Carr y GM Church, 2009, Nature Biotechnology 27 (12): 1.151-62).
En general, una secuencia de emparejamiento tracr incluye cualquier secuencia que presente suficiente complementariedad con una secuencia tracr para activar uno o más de: (1) escisión de una secuencia guía flanqueada por secuencias de emparejamiento tracr en una célula que contiene la correspondiente secuencia tracr y (2) formación de un complejo CRISPr en una secuencia objetivo, en la que el complejo CRISPR comprende la secuencia de emparejamiento tracr hibridada a la secuencia tracr. En general, el grado de complementariedad es con referencia al alineamiento óptimo de la secuencia de emparejamiento tracr y la secuencia tracr, junto con la longitud del acortador de las dos secuencias. El alineamiento óptimo se puede determinar por cualquier algoritmo de alineamiento adecuado y puede justificar además estructuras secundarias, tal como autocomplementareidad dentro de cualquiera, la secuencia tracr o la secuencia de emparejamiento tracr. En algunas realizaciones, el grado de complementariedad entre la secuencia tracr y la secuencia de emparejamiento tracr junto con la longitud del acortador de las dos cuando se alinean de manera óptima es aproximadamente o más de aproximadamente 25%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 95%, 97,5%, 99% o mayor. Las ilustraciones de ejemplo de alineación óptima entre una secuencia tracr y una secuencia de emparejamiento tracr se proporcionan en las Figuras 12B y 13B. En algunas realizaciones, la secuencia tracr es aproximadamente o más de aproximadamente 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 25, 30, 40, 50 o más nucleótidos de longitud. En algunas realizaciones, la secuencia tracr y secuencia de emparejamiento tracr están contenidas dentro de un transcripto único, de manera que la hibridación entre las dos produce un transcripto con una estructura secundaria, tal como una horquilla. Las secuencias formadoras de bucle preferidas para uso en estructuras de horquilla tienen cuatro nucleótidos de longitud y lo más preferiblemente tienen la secuencia GAAA. Sin embargo, se pueden usar secuencias de bucle más largas o más cortas, como se pueden usar secuencias alternativas. Las secuencias incluyen preferiblemente un triplete nucleótido (por ejemplo, AAA) y un nucleótido adicional (por ejemplo, C o G). Ejemplos de secuencias formadoras de bucle incluyen CAAA y AAAG. En una realización de la descripción, el transcripto o secuencia de polinucleótidos transcrita presenta al menos dos o más horquillas. En realizaciones preferidas, el transcripto presenta dos, tres, cuatro o cinco horquillas. En una realización más de la descripción, el transcripto presenta a lo sumo cinco horquillas. En algunas realizaciones, el transcripto único incluye además una secuencia de terminación de la transcripción; preferiblemente esta es una secuencia poliT, por ejemplo, seis nucleótidos T. Una ilustración de ejemplo de dicha estructura de horquilla se proporciona en la porción inferior de la Figura 13B, donde la porción de la secuencia 5' del "N" final y aguas arriba del bucle corresponde a la secuencia de emparejamiento tracr y la porción de la secuencia 3' del bucle corresponde a la secuencia tracr. Ejemplos no limitantes adicionales de polinucleótidos únicos que comprenden una secuencia guía, una secuencia de emparejamiento tracr y una secuencia tracr son como sigue (enumerados 5' a 3'), donde "N" representa una base de una secuencia guía, el primer bloque de letras de la caja inferior representan la secuencia de emparejamiento tracr y el segundo bloque de letras de la caja inferior representa la secuencia tracr y la secuencia poli-T final representa el terminador de la transcripción: (1)
NNNNNNNNNNNNNNNNNNNNgtttttgtactctcaagatttaGAAAtaaatcttgcagaagctacaaagataaggctt
catgccgaaatcaacaccctgtcattttatggcagggtgttttcgttatttaaTTTTTT; (2)
NNNNNNNNNNNNNNNNNNNNgtttttgtactctcaGAAAtgcagaagctacaaagataaggcttcatgccgaaatca
acaccctgtcattttatggcagggtgttttcgttatttaaTTTTTT; (3)
NNNNNNNNNNNNNNNNNNNNgtttttgtactctcaGAAAtgcagaagctacaaagataaggcttcatgccgaaatca
acaccctgtcattttatggcagggtgtTTTTTT; (4)
NNNNNNNNNNNNNNNNNNNNgttttagagctaGAAAtagcaagttaaaataaggctagtccgttatcaacttgaaaa
agtggcaccgagtcggtgcTTTTTT; (5)
NNNNNNNNNNNNNNNNNNNNgttttagagctaGAAATAGcaagttaaaataaggctagtccgttatcaacttgaa
aaagtgTTTTTTT; y (6)
NNNNNNNNNNNNNNNNNNNNgttttagagctagAAATAGcaagttaaaataaggctagtccgttatcaTTTTT
TTT.
En algunas realizaciones, las secuencias (1) a (3) se usan junto con Cas9 de CRISPR1 de S. thermophilus. En algunas realizaciones, las secuencias (4) a (6) se usan junto con Cas9 de S. pyogenes. En algunas realizaciones, la secuencia tracr es un transcrito separado de un transcrito que comprende la secuencia de emparejamiento tracr (tal como se ilustra en la porción superior de la Figura 13B).
En algunas realizaciones, también se proporciona una plantilla de recombinación. Una plantilla de recombinación puede ser un componente de otro vector como se describe en la presente memoria, contenido en un vector separado o proporcionado como un polinucleótido separado. En algunas realizaciones, se diseña una plantilla de recombinación para servir como una plantilla en recombinación homóloga, tal como dentro de o cerca de una secuencia objetivo nickeada o escindida por una enzima CRISPR como parte de un complejo CRISPR. Un polinucleótido de plantilla puede ser de cualquier longitud adecuada, tal como aproximadamente o más aproximadamente 10, 15, 20, 25, 50, 75, 100, 150, 200, 500, 1.000 o más nucleótidos de longitud. En algunas realizaciones, el polinucleótido de plantilla es complementario a una porción de un polinucleótido que comprende la secuencia objetivo. Cuando se alinea de manera óptima, un polinucleótido de plantilla puede solaparse con uno o más nucleótidos de una secuencia objetivo (por ej., aproximadamente o más de aproximadamente 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100 o más nucleótidos). En algunas realizaciones, cuando una secuencia de plantilla y un polinucleótido que comprende una secuencia de plantilla se alinean de manera óptima, el nucleótido más cercano del polinucleótido de plantilla está dentro de aproximadamente 1, 5, 10, 15, 20, 25, 50, 75, 100, 200, 300, 400, 500, 1.000, 5.000, 10.000 o más nucleótidos de la secuencia objetivo.
En algunas realizaciones, la enzima CRISPR es parte de una proteína de fusión que comprende uno o más dominios de proteína heteróloga (por ej., aproximadamente o más de aproximadamente 1,2, 3, 4, 5, 6, 7, 8, 9, 10 o más dominios además de la enzima CRISPR). Una proteína de fusión de enzima CRISPR puede comprender cualquier secuencia de proteínas adicional y opcionalmente una secuencia ligadora entre dos dominios cualesquiera. Ejemplos de dominios de proteínas que se pueden condensar a una enzima CRISPR incluyen, sin limitación, etiquetas de epítopos, secuencias de genes informadores y dominios de proteínas con una o más de las siguientes actividades: actividad de metilasa, actividad de desmetilasa, actividad de activación de la transcripción, actividad de represión de la transcripción, actividad del factor de liberación de la transcripción, actividad de modificación de histona, actividad de escisión de ARN y actividad de unión de ácidos nucleicos. Ejemplos no limitantes de etiquetas de epítopos incluyen etiquetas de histidina (His), etiquetas V5, etiquetas FLAG, etiquetas de hemaglutinina de la influenza (HA), etiquetas Myc, etiquetas VSV-G y etiquetas de tioredoxina (Trx). Los ejemplos de genes informadores incluyen, pero no se limitan a, glutationa-S-transferasa (GST), peroxidasa de rábano (HRP, por sus siglas en inglés), cloramfenicol acetiltransferasa (CAT, por sus siglas en inglés) beta-galactosidasa, beta-glucuronidasa, luciferasa, proteína fluorescente verde (PFV), HcRed, DsRed, proteína fluorescente cian (PFC) proteína fluorescente amarilla (PFAm) y proteínas autofluorescentes incluyendo proteína fluorescente azul (PFA). Una enzima CRISPR se puede condensar a una secuencia de genes que codifique una proteína o un fragmento de una proteína que una moléculas de ADN o una otras moléculas celulares, incluyendo pero no limitándose a (por sus siglas en inglés), proteína de unión de maltosa (MBP), etiqueta S, fusiones de dominios de unión de ADN Lex A (DBD), fusiones de dominios de unión de ADN GAL4 y fusiones de proteínas BP16 de virus del herpes simple (HSV). Los dominios adicionales que pueden formar parte de una proteína de fusión que comprende una enzima CRISPR se describen en la patente de EE.UU. 20110059502. En algunas realizaciones, se usa una enzima CRISPR etiquetada para identificar la posición de una secuencia objetivo.
En algunos aspectos, se describen en la presente memoria métodos que comprenden suministrar uno o más polinucleótidos, tales como o uno o más vectores como se describe en la presente memoria, uno o más transcritos de los mismos y/o una o proteínas transcritas de allí, a una célula huésped. En algunos aspectos, se describen en la presente memoria células producidas por tales métodos y organismos (tales como animales, plantas u hongos) que comprenden o son producidos de tales células. En algunas realizaciones, se suministra una enzima CRISPR junto con (y opcionalmente complejada con) una secuencia guía a una célula. Se pueden usar métodos
de transferencia de genes con base vírica y no vírica convencionales para introducir ácidos nucleicos en células de mamífero o tejidos objetivo. Se pueden usar dichos métodos para administrar componentes que codifican ácidos nucleicos de un sistema CRISPR a células en cultivo o en un organismo huésped. Los sistemas de suministro de vectores no víricos incluyen plásmidos de ADN, ARN (por ej., un transcrito de un vector descrito en la presente memoria), ácido nucleico desnudo y ácido nucleico complejado con un vehículo de suministro, tal como un liposoma. Los sistemas de suministro de vectores víricos incluyen virus de ADN y ARN, que tienen genomas episomales o integrados después de suministro a la célula. Para una Invisión de procedimientos de tratamiento de genes, véase Anderson, Science 256: 808-813 (1992); Nabel & Felgner, TIbTe CH 11: 211-217 (1993) ; Mitani & Caskey, TIBTECH 11: 162-166 (1993); Dillon, TIBTECH 11: 167-175 (1993); Miller, Nature 357: 455-460 (1992); Van Brunt, Biotechnology 6 (10): 1.149-1.154 (1988); Vigne, Restorative Neurology and Neuroscience 8: 35-36 (1995); Kremer & Perricaudet, British Medical Bulletin 51 (1): 31-44 (1995); Haddada et al., en Current Topics in Microbiology and Immunology, Doerfler and Bohm (eds) (1995) y Yu et al., Gene Therapy 1: 13-26 (1994).
Los métodos de suministro no vírico de ácidos nucleicos incluyen lipofección, nucleofección, microinyección, biolística, virosomas, liposomas, inmunoliposomas, conjugados de policatión o lípido:ácido nucleico, ADN desnudo, viriones artificiales y absorción de ADN aumentada por agente. La lipofeción se describe en por ej., las Patentes de EE.UU. N° 5.049.386, 4.946.787 y 4.897.355) y se venden comercialmente los reactivos de lipofección (por ej., Transfectam™ y Lipofectin™). Los lípidos catiónicos y neutros que son adecuados para lipofección de reconocimiento de receptores eficaz de polinucleótidos incluyen aquéllos de Felgner, Patente Internacional WO 91/17424; Patente Internacional WO 91/16024. El suministro puede ser a células (por ej., administración in vitro o ex vivo) o tejidos objetivo (por ej., administración in vivo).
La preparación de complejos de lípido:ácido nucleico, incluyendo liposomas fijados como objetivo tales como complejos de inmunolípidos, es conocida para un experto en la materia (véase, por ej., Crystal, Science 270: 404 410 (1995); Blaese et al., Cancer Gene Ther. 2: 291-297 (1995); Behr et al., Bioconjugate Chem. 5: 382-389 (1994) ; Remy et al., Bioconjugate Chem. 5: 647-654 (1994); Gao et al., Gene Therapy 2: 710-722 (1995); Ahmad et al., Cancer Res. 52: 4.817-4.820 (1992); Patentes de EE.UU. N° 4.186.183; 4.217.344; 4.235.871; 4.261.975; 4.485.054; 4.501.728; 4.774.085; 4.837.028 y 4.946.787).
El uso de sistemas de base vírica de ARN o ADN para el suministro de ácidos nucleicos toma ventaja de los procedimientos altamente desarrollados para fijar como objetivo un virus para células específicas en el cuerpo y traficar la carga útil vírica al núcleo. Se pueden administrar vectores víricos directamente a pacientes (in vivo) o se pueden usar para tratar células in vitro y las células modificadas se pueden administrar opcionalmente a los pacientes (ex vivo). Los sistemas con base vírica convencionales podían incluir vectores retrovirales, de lentivirus, adenovirales, adenoasociados y del virus del herpes simple para transferencia de genes. La integración en el genoma huésped es posible con los métodos de transferencia de genes de retrovirus, lentivirus y virus adenoasociados, dando como resultado con frecuencia expresión a largo plazo del transgén insertado. Adicionalmente, se han observado altas eficacias de transducción en muchos tipos celulares diferentes y tejidos fijados como objetivo.
El tropismo de un retrovirus se puede modificar por incorporación de proteínas de sobre extrañas, expandiendo la población objetivo potencial de células objetivo. Los vectores lentivirales son vectores retrovirales que son capaces de transducir o infectar células no de división y típicamente producen títulos víricos altos. La selección de un sistema de transferencia de genes retrovirales dependería por lo tanto del tejido objetivo. Los vectores retrovirales están constituidos por repeticiones terminales largas que actúan en cis con capacidad de empaquetamiento para hasta 6-10 kb de secuencia extraña. Las LTR mínimas que actúan en cis son suficientes para la replicación y empaquetamiento de los vectores, que se usan después para integrar el gen terapéutico en la célula objetivo para proporcionar expresión de transgenes permanente. Los vectores retrovirales ampliamente usados incluyen los basados en virus de la leucemia murina (MuLV), virus de la leucemia del mono gibón (GaLV), virus de la inmunodeficiencia simia (VIS), virus de la inmunodeficiencia humana (VIH) y combinaciones de los mismos (véase, por ej., Buchscher et al., J. Virol. 66: 2.731-2.739 (1992); Johann et al., J. Virol. 66: 1.635-1.640 (1992); Sommnerfelt et al., Virol. 176: 58-59 (1990); Wilson et al., J. Virol. 63: 2.374-2.378 (1989); Miller et al., J. Virol. 65: 2.220-2.224 (1991); Patente de EE.UU. PCT/US94/05700). En aplicaciones donde se prefiere la expresión transitoria, se pueden usar sistemas a base de adenovirus. Los vectores a base de adenovirus son capaces de una eficacia de transducción muy alta en muchos tipos de células y no requieren división celular. Con tales vectores, se han obtenido altos títulos y niveles de expresión. Este vector se puede producir en grandes cantidades en un sistema relativamente simple. También se pueden usar vectores de virus adenoasociados (“AAV”, por sus siglas en inglés) para transducir células con ácidos nucleicos objetivo, por ejemplo, en la producción in vitro de ácidos nucleicos y péptidos y para procedimientos de tratamiento con genes in vivo y ex vivo (véase, por ej., West et al., Virology 160: 38-47 (1987); la Patente de EE.UU. N° 4.797.368; la patente internacional WO 93/24641; Kotin, Human Gene Therapy 5: 793-801 (1994); Muzyczka, J. Clin. Invest. 94: 1.351 (1994). Se describe la construcción de vectores AAV recombinantes en una serie de publicaciones, incluyendo la Patente de EE.UU. N° 5.173.414; Tratschin et al., Mol. Cell. Biol. 5: 3.251-3.260 (1985); Tratschin, et al., Mol. Cell
Biol. 4: 2.072-2.081 (1984); Hermonat & Muzyczka, PNAS 81: 6.466-6.470 (1984) y Samulski et al., J. Virol. 63: 03822-3828 (1989).
Típicamente se usan células de empaquetamiento para formar partículas de virus que sean capaces de infectar una célula huésped. Tales células incluyen células 293, que empaquetan adenovirus, y células ^2 o células PA317, que empaquetan retrovirus. Los vectores víricos usados en terapia génica se usan generalmente produciendo una estirpe celular que empaqueta un vector de ácidos nucleicos en una partícula vírica. Los vectores contienen típicamente las mínimas secuencias víricas requeridas para empaquetamiento y posterior integración en un huésped, siendo reemplazadas otras secuencias víricas por una casete de expresión para el polinucleótido o los polinucleótidos que se tienen que expresar. Las funciones víricas que faltan se suministran típicamente en trans por la estirpe celular de empaquetamiento. Por ejemplo, los vectores AAV usados en terapia génica típicamente poseen sólo secuencias ITR del genoma AAV que se requieren para empaquetamiento e integración en el genoma huésped. El ADN vírico se empaqueta en una estirpe celular, que contiene un plásmido auxiliar que codifica los otros genes AAV, es decir rep y cap, pero carecen de secuencias ITR. La estirpe celular también puede infectarse con adenovirus como auxiliar. El virus auxiliar activa la replicación del vector AAV y la expresión de genes AAV del plásmido auxiliar. El plásmido auxiliar no se empaqueta en cantidades significativas debido a ausencia de secuencias ITR. La contaminación con adenovirus se puede reducir por, por ejemplo, tratamiento térmico al cual es más sensible el adenovirus que AAV. Los métodos adicionales para el suministro de ácidos nucleicos a células son conocidos para los expertos en la materia. Véase, por ejemplo, la Patente de EE.UU.
20030087817.
En algunas realizaciones, una célula huésped se transinfecta de manera transitoria o de manera no transitoria con uno o más vectores descritos en la presente memoria. En algunas realizaciones, una célula se transinfecta como tiene lugar de manera natural en un individuo. En algunas realizaciones, una célula que se transinfecta es tomada de un individuo. En algunas realizaciones, la célula procede de células tomadas de un individuo, tales como una estirpe celular. Se conoce una amplia variedad de estirpes celulares de cultivos de tejido en la técnica. Ejemplos de estirpes celulares incluyen, pero no se limitan a, C8161, CCRF-CEM, MOLT, mIMCD-3, NHDF, HeLa-S3, Huh1, Huh4, Huh7, HUVEC, HASMC, HEKn, HEKa, MiaPaCell, Panc1, PC-3, TF1, CTLL-2, C1R, Rat6, CV1, RPTE, A10, T24, J82, A375, ARH-77, Calu1, SW480, SW620, SKOV3, SK-UT, CaCo2, P388D1, SEM-K2, WEHI-231, HB56, TIB55, Jurkat, J45.01, LRMB, Bcl-1, BC-3, IC21, DLD2, Raw264.7, NRK, NRK-52E, MRC5, MEF, Hep G2, HeLa B, HeLa T4, COS, COS-1, COS-6, COS-M6A, epitelial de riñón de mono BS-C-1, fibroblasto de embrión de ratón BALB/ 3T3, 3T3 Swiss, 3T3-L1, fibroblastos fetales humanos 132-d5; fibroblastos de ratón 10.1, 293-T, 3T3, 721, 9L, A2780, A2780ADR, A2780cis, A172, A20, A253, A431, A-549, ALC, B16, B35, células BCP-1, BEAS-2B, bEnd.3, BHK-21, BR 293, BxPC3, C3H-10T1/2, C6/36, Cal-27, CHO, CHO-7, CHO-IR, CHO-K1, CHO-K2, CHO-T, CHO Dhfr -/-, COR-L23, COR-L23/CPR, COR-L23/5010, COR-L23/R23, COS-7, COV-434, CML T1, CMT, CT26, D17, DH82, DU145, DuCaP, EL4, EM2, EM3, EMT6/AR1, EMT6/AR10.0, FM3, H1299, H69, HB54, HB55, HCA2, HEK-293, HeLa, Hepa1c1c7, HL-60, HMEC, HT-29, Jurkat, células JY, células K562, Ku812, KCL22, KG1, KYO1, LNCap, Ma-Mel 1-48, MC-38, MCF-7, MCF-10A, MDA-MB-231, MDA-MB-468, MDA-MB-435, MDCK II, MDCK II, MOR/0.2R, MONO-MAC 6, MTD-1A, MyEnd, NCI-H69/CPR, NCI-H69/LX10, NCI-H69/LX20, NCI-H69/LX4, NIH-3T3, NALM-1, NW-145 estirpes celulares OPCN / OPCT, Peer, PNT-1A / PNT 2, RenCa, RIN-5F, RMA/RMAS, células Saos-2, Sf-9, SkBr3, T2, T-47D, T84, estirpe celular THP1, U373, U87, U937, VCaP, células Vero, WM39, WT-49, X63 YAC-1, YAR y variedades transgénicas de los mismos. Las estirpes celulares están disponibles de una variedad de fuentes conocidas por los expertos en la materia (véase, por ej., la Colección de Cultivos de Tipo Americano (ATCC, por sus siglas en inglés) (Manassus, Va.)). En algunas realizaciones, una célula transinfectada con uno o más vectores descritos en la presente memoria se usa para establecer una nueva estirpe celular que comprende una o más secuencias procedentes de vector. En algunas realizaciones, una célula transinfectada de manera transitoria con los componentes de un sistema CRISPR como se describe en la presente memoria (tal como por transinfección transitoria de uno más vectores o transinfección con ARN) y modificada por la actividad de un complejo CRISPR, se usa para establecer una nueva estirpe celular que comprende células que contienen la modificación pero carecen de otra secuencia exógena. En algunas realizaciones, las células transinfectadas de manera transitoria o de manera no transitoria con uno o más vectores descritos en la presente memoria o estirpes celulares procedentes de dichas células se usan en la valoración de uno o más compuestos de ensayo.
En algunas realizaciones, se usa uno o más vectores descritos en la presente memoria para producir un animal transgénico no humano o planta transgénica. En algunas realizaciones, el animal transgénico es un mamífero, tal como un ratón, rata o conejo. En algunas realizaciones, el organismo o individuo es una planta. En algunas realizaciones, el organismo o individuo o planta es un alga. Los métodos para producir plantas y animales transgénicos son conocidos en la técnica y generalmente empiezan con un método de transinfección de células, tal como se describe en la presente memoria.
En un aspecto, se describen en la presente memoria métodos para modificar un polinucleótido objetivo en una célula eucariota. En algunas realizaciones, el método comprende permitir que un complejo CRISPR se una al polinucleótido objetivo para efectuar la escisión de dicho polinucleótido objetivo modificando de ese modo el
polinucleótido objetivo, en la que el complejo CRISPR comprende una enzima CRISPR complejada con una secuencia guía hibridada a una secuencia fijada como objetivo dentro de dicho polinucleótido objetivo, en el que dicha secuencia guía está ligada a una secuencia de emparejamiento tracr que a su vez hibrida a una secuencia tracr.
En un aspecto, se describe en la presente memoria un método para modificar la expresión de un polinucleótido en una célula eucariota. En algunas realizaciones, el método comprende permitir que un complejo CRISPR se una al polinucleótido de manera que dicha unión dé como resultado expresión aumentada o disminuida de dicho polinucleótido; en el que el complejo CRISPR comprende una enzima CRISPR complejada con una secuencia guía hibridada a una secuencia fijada como objetivo dentro de dicho polinucleótido, en el que dicha secuencia guía está ligada a una secuencia de emparejamiento tracr que a su vez hibrida a una secuencia tracr.
Con los recientes avances en la genómica de cultivos, la capacidad para usar sistemas CRISPR-Cas para realizar edición y manipulación de genes eficaz y de coste eficaz permitirá la rápida selección y la comparación de manipulaciones genéticas únicas y multiplexadas para transformar dichos genomas para producción mejorada y cebos mejorados. Con respecto a esto se hace referencia a las Patentes y publicaciones de EE.UU.: Patente de EE.UU. N° 6.603.061 - Agrobacterium-Mediated Plant Transformation Method; la Patente de EE.UU. N° 7.868.149 - Plant Genome Sequences and Uses Thereof y la Patente de EE.UU. 2009/0100536 - Transgenic Plants with Enhanced Agronomic Traits. En la práctica de la descripción, también se hace referencia a los contenidos y la invención de Morrell et al., “Crop genomics: advances and applications Nat Rev Genet., 29 de diciembre de 2011; 13 (2): 85-96. En una realización ventajosa de la invención, el sistema CRISPR/Cas9 se usa para lograr microalgas (Ejemplo 15). De acuerdo con esto, la referencia en la presente memoria a células de animales puede aplicarse también, mutatis mutandis, a células de plantas a menos que sea evidente de otro modo.
En un aspecto, se describen en la presente memoria métodos para modificar un polinucleótido objetivo en una célula eucariota, que puede ser in vivo, ex vivo o in vitro. En algunas realizaciones, el método comprende muestrear una célula o población de células de un animal humano o no humano o planta (incluyendo microalgas) y modificar la célula o células. Puede tener lugar cultivo en cualquier fase ex vivo. La célula o las células pueden ser introducidas de nuevo incluso en el animal no humano o planta (incluyendo microalgas).
En las plantas, los patógenos son con frecuencia específicos del huésped. Por ejemplo, Fusarium oxysporum f. sp. lycopersici produce marchitamiento del tomate pero ataca sólo al tomate y F. oxysporum f. dianthii Puccinia graminis f. sp. tritici ataca sólo al trigo. Las plantas tienen defensas existentes e inducidas para resistir a la mayoría de los patógenos. Las mutaciones y los casos de recombinación a través de generaciones de plantas conducen a variabilidad genética que da lugar a susceptibilidad, especialmente ya que los patógenos se reproducen con más frecuencia que las plantas. En las plantas puede haber resistencia al no huésped, por ej., el huésped y el patógeno son incompatibles. También puede haber Resistencia Horizontal, por ej., resistencia parcial frente a todas las razas de un patógeno, controlado típicamente por muchos genes y Resistencia Vertical, por ej., resistencia completa a algunas razas de un patógeno pero no a otras razas, controlado típicamente por unos genes. En un nivel de Gen por Gen, las plantas y los patógenos evolucionan juntos y los cambios genéticos en uno equilibran los cambios en otro. De acuerdo con eso, usando Variabilidad Natural, los criadores combinan genes lo más útiles para Rendimiento, Calidad, Uniformidad, Dureza, Resistencia. Las fuentes de genes de resistencia incluyen Variedades naturales o extrañas, Variedades Heirloom, Mutaciones relativas a plantas silvestres e inducidas, por ej., tratando material vegetal con agentes mutagénicos. Usando la presente descripción, se les proporciona a los criadores de plantas una nueva herramienta para inducir mutaciones. De acuerdo con esto, un experto en la materia puede analizar el genoma de fuentes de genes de resistencia y en Variedades con características o rasgos deseados se emplea la presente descripción para inducir la elevación de genes de resistencia con más precisión que los agentes mutagénicos previos y por lo tanto se aceleran y mejoran los programas de cultivo de plantas.
En un aspecto, se describen en la presente memoria estuches que contienen uno o más cualesquiera de los elementos descritos en los métodos y composiciones anteriores. En algunas realizaciones, el estuche comprende un sistema vector e instrucciones para usar el estuche. En algunas realizaciones, el sistema vector comprende (a) un primer elemento regulador unido de manera operable a una secuencia de emparejamiento tracr y uno o más sitios de inserción para insertar una secuencia guía aguas arriba de la secuencia de emparejamiento tracr, en el que cuando se expresa, la secuencia guía dirige la unión específica de la secuencia de un complejo CRISPR a una secuencia fijada como objetivo en una célula eucariota, en la que el complejo CRISPR comprende una enzima CRISPR complejada con (1) la secuencia guía que se hibrida a la secuencia objetivo y (2) la secuencia de emparejamiento tracr que se hibrida a la secuencia tracr y/o (b) un segundo elemento regulador unido de manera operable a una secuencia codificadora de enzima que codifica dicha enzima CRISPR que comprende una secuencia de localización nuclear. Los elementos se pueden proporcionar de manera individual o en combinaciones y se pueden proporcionar en cualquier envase adecuado, tal como un vial, un frasco o un tubo. En algunas realizaciones, el estuche incluye instrucciones en uno o más idiomas, por ejemplo, en más de un idioma.
En algunas realizaciones, un estuche comprende uno o más reactivos para uso en un procedimiento utilizando uno o más de los elementos descritos en la presente memoria. Los reactivos se pueden proporcionar en cualquier envase adecuado. Por ejemplo, un estuche puede proporcionar uno o más tampones de reacción o de almacenamiento. Los reactivos se pueden proporcionar en una forma que sea utilizable en un ensayo particular o en una forma que requiera la adición de otro u otros componentes más antes de uso (por ejemplo, en forma concentrada o liofilizada). Un tampón puede ser cualquier tampón, incluyendo, pero no limitándose a un tampón de carbonato de sodio, un tampón de bicarbonato de sodio, un tampón de borato, un tampón de Tris, un tampón de MOPS, un tampón de HEPES y combinaciones de los mismos. En algunas realizaciones, el tampón es alcalino. En algunas realizaciones, el tampón presenta un pH de aproximadamente 7 a aproximadamente 10. En algunas realizaciones, el estuche comprende uno o más oligonucleótidos correspondientes a una secuencia guía para inserción en un vector de manera que se una de manera operable a la secuencia guía y un elemento regulador. En algunas realizaciones, el estuche comprende un polinucleótido de plantilla de recombinación homólogo.
En un aspecto, se describen en la presente memoria métodos para usar uno o más elementos de un sistema CRISPR. El complejo CRISPR que se describe en la presente memoria proporciona un medio eficaz para modificar un polinucleótido objetivo. El complejo CRISPR que se describe en la presente memoria presenta una amplia variedad de utilidad incluyendo modificación (por ej., supresión, inserción, traslocación, inactivación, activación) de un polinucleótido objetivo en una multiplicidad de tipos de células. Como tal el complejo CRISPR que se describe en la presente memoria presenta un amplio espectro de aplicaciones en, por ej., tratamiento génico, investigación de fármacos, diagnóstico de enfermedades y prognosis. Un complejo CRISPR ejemplar comprende una enzima CRISPR complejada con una secuencia guía hibridada a una secuencia objetivo dentro del polinucleótido objetivo. La secuencia guía está ligada a una secuencia de emparejamiento tracr, que a su vez hibrida a una secuencia tracr.
El polinucleótido objetivo de un complejo CRISPR puede ser cualquier polinucleótido endógeno o exógeno a la célula eucariota. Por ejemplo, el polinucleótido objetivo puede ser un polinucleótido que resida en el núcleo de la célula eucariota. El polinucleótido objetivo puede ser una secuencia que codifique un producto génico (por ej., una proteína) o una secuencia no codificadora (por ej., un polinucleótido regulador o un a Dn basura). Sin desear estar limitados por la teoría, se cree que la secuencia objetivo debería estar asociada a un PAM (unidad adyacente protoespaciadora); esto es, una secuencia corta reconocida por el complejo CRISPR. Los requerimientos de secuencia y longitud precisos para el PAM difieren dependiendo de la enzima CRISPR usada, pero los PAM son típicamente secuencias de 2-5 pares de bases adyacentes al protoespaciador (esto es, la secuencia objetivo). Se proporcionan ejemplos de secuencias PAM en la sección de ejemplos a continuación y el experto podrá identificar secuencias PAM adicionales para uso con una enzima CRISPR determinada.
El polinucleótido objetivo de un complejo CRISPR puede incluir una serie de genes y polinucleótidos asociados a enfermedades así como genes y polinucleótidos asociados a rutas bioquímicas de señalización.
Ejemplos de polinucleótidos objetivo incluyen una secuencia asociada a una ruta bioquímica de señalización, por ej., un gen o polinucleótido asociado a una ruta bioquímica de señalización. Ejemplos de polinucleótidos objetivo incluyen un gen o polinucleótido asociado a una enfermedad. Un gen o polinucleótido "asociado a una enfermedad" se refiere a cualquier gen o polinucleótido que esté proporcionando productos de transcripción o traducción en un nivel anormal o en una forma anormal en las células procedentes de tejidos afectados por una enfermedad comparado con tejidos o células de un control no de enfermedad. Puede ser un gen que llegue a expresarse a un nivel anormalmente alto; puede ser un gen que llegue a expresarse a un nivel anormalmente bajo, donde la expresión modificada se correlaciona con la aparición y/o progresión de la enfermedad. Un gen asociado a la enfermedad también se refiere a un gen que posee mutación o mutaciones o variación genética que es directamente responsable o está ligada a desequilibrio con un gen, o genes, que es responsable de la etiología de una enfermedad. Los productos transcritos o traducidos pueden ser conocidos o desconocidos y puede ser a un nivel normal o anormal.
Ejemplos de genes y polinucleótidos asociados a una enfermedad están disponibles en Instituto McKusick-Nathans de Medicina Genética, Universidad de Johns Hopkins (Baltimore, Md.) y Centro Nacional para Información de Biotecnología, Biblioteca Nacional de Medicina (Bethesda, Md.), disponible en la Red Informática Mundial.
Ejemplos de genes y polinucleótidos asociados a una enfermedad se enumeran en las Tablas A y B. La información específica de la enfermedad está disponible en el Instituto McKusick-Nathans de Medicina Genética, Universidad de Johns Hopkins (Baltimore, Md.) y Centro Nacional para Información de Biotecnología, Biblioteca Nacional de Medicina (Bethesda, Md.), disponible en la Red Informática Mundial. Ejemplos de genes y polinucleótido asociados a una ruta bioquímica de señalización se enumeran en la Tabla C.
Las mutaciones en estos genes y rutas pueden dar como resultado la producción de proteínas inapropiadas o proteínas en cantidades inapropiadas que afecten a la función. Tales genes, proteínas y rutas pueden ser el polinucleótido objetivo de un complejo CRISPR.
Tabla A


Tabla B


Tabla C


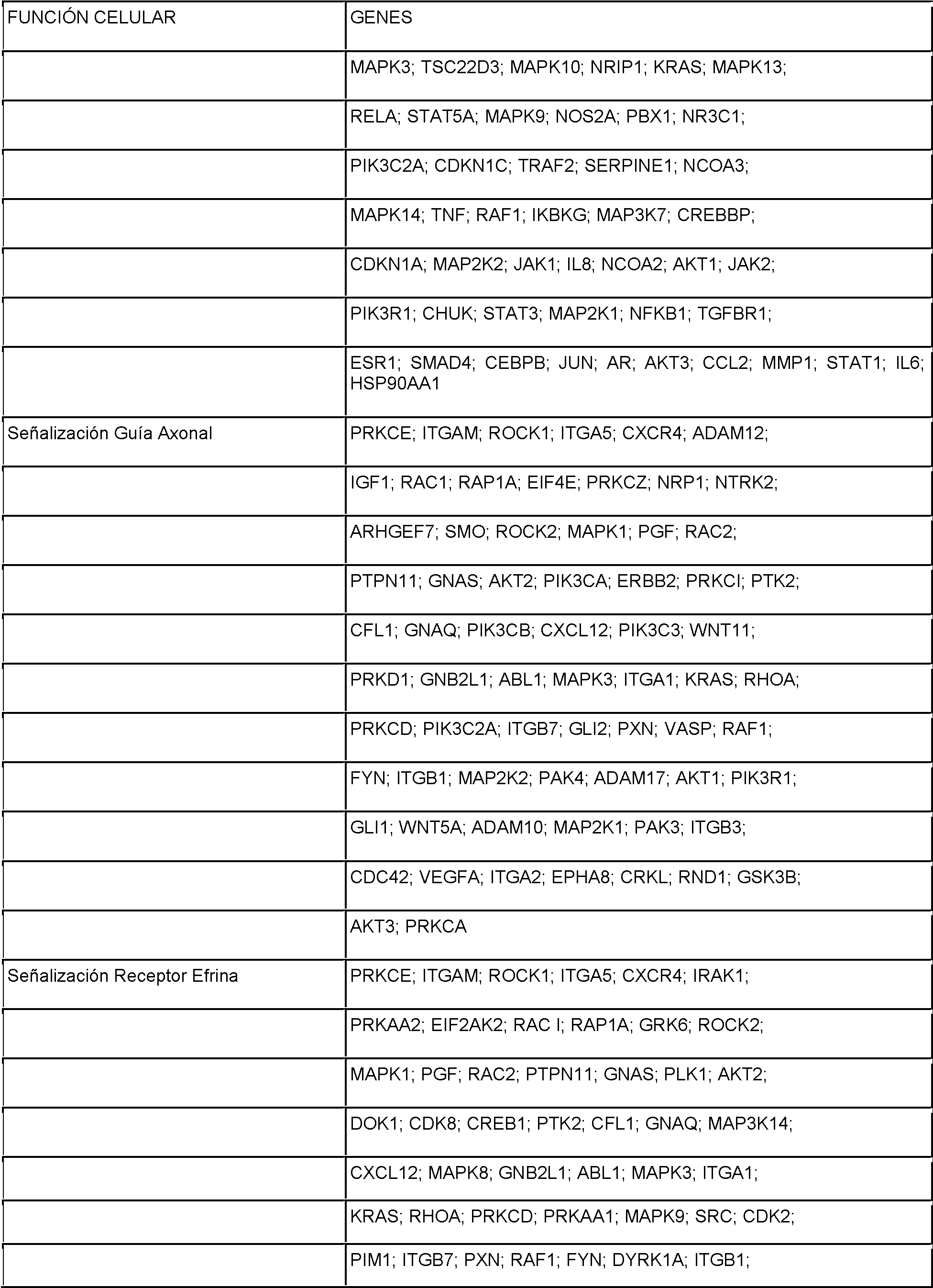











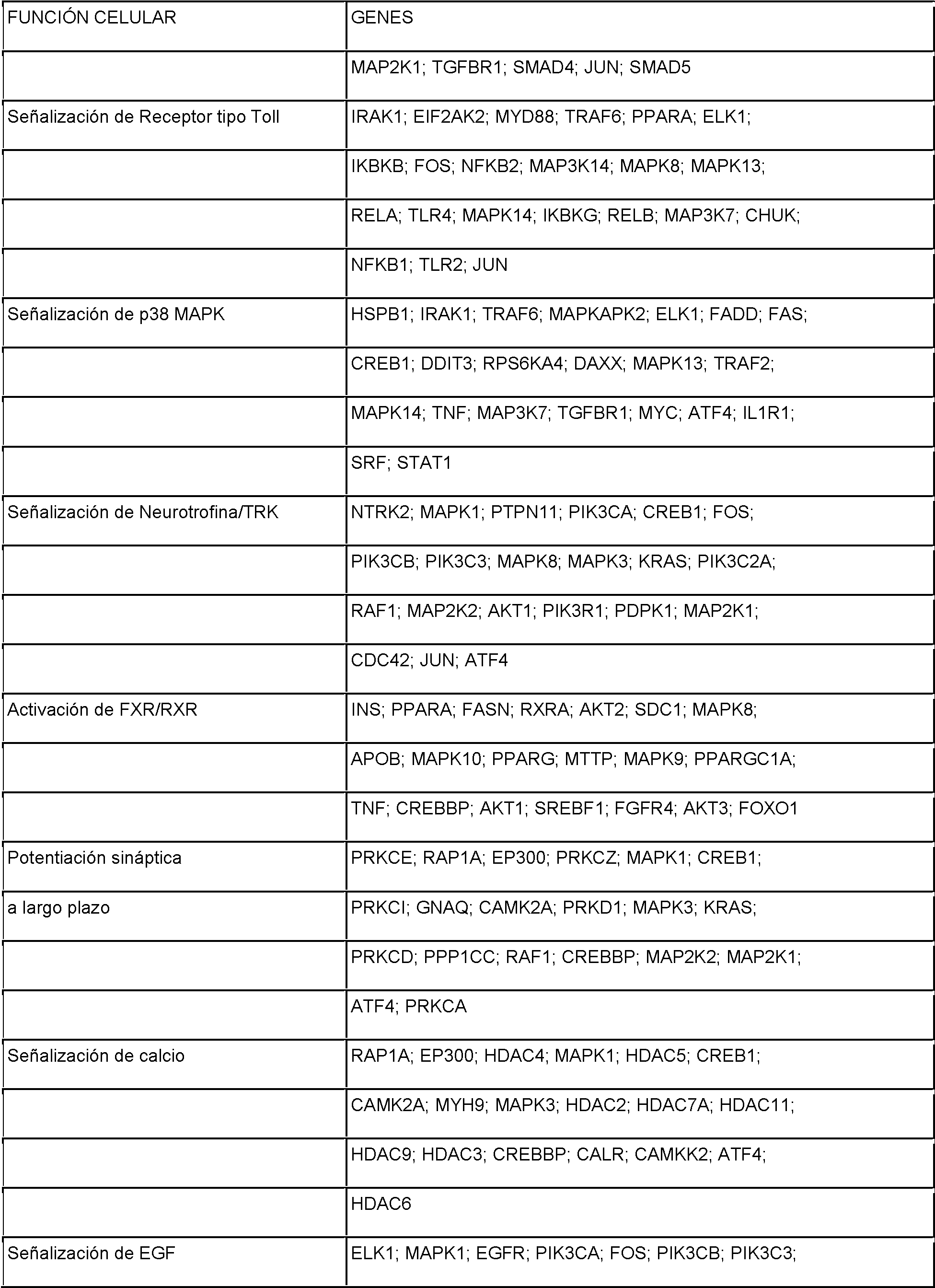






Las realizaciones de la descripción también se refieren a métodos y composiciones relacionadas con eliminación de genes, la multiplicación de genes y reparación de mutaciones particulares asociadas a la inestabilidad de repeticiones de a Dn y trastornos neurológicos (Robert D. Wells, Tetsuo Ashizawa, Genetic Instabilities and Neurological Diseases, Segunda Edición, Academic Press, 13 de octubre de 2011-Medical). Se han encontrado aspectos específicos de secuencias de repetición en tándem responsables de más de veinte enfermedades humanas (Nuevos conocimientos en la inestabilidad de la repetición: función de híbridos de ARN ADN. McIvor EI, Polak U, Napierala M. RNA Biol. Sep-Oct 2010; 7 (5): 551-8). El sistema CRISPR-Cas puede ser aprovechado para corregir estos defectos de inestabilidad genómica.
Un aspecto más de la descripción se refiere a la utilización del sistema CRISPR-Cas para corregir defectos en los genes EMP2A y EMP2B que se ha identificado que están asociados a la enfermedad Lafora. La enfermedad de Lafora es un trastorno autosómico recesivo que se caracteriza por epilepsia mioclónica progresiva que puede empezar como ataques epilépticos en la adolescencia. Algunos casos de la enfermedad pueden estar causados por mutaciones en genes que se tienen que identificar, sin embargo. La enfermedad produce ataques, espasmos musculares, dificultad para caminar, demencia y eventualmente la muerte. En la actualidad no hay tratamiento que se haya demostrado eficaz contra el progreso de la enfermedad. Otras anormalidades genéticas asociadas a la epilepsia también pueden ser fijadas como objetivo mediante el sistema CRISPR-Cas y la genética subyacente se describe además en Genetics of Epilepsy and Genetic Epilepsies, editado por Giuliano Avanzini, Jeffrey L. Noebels, Mariani Foundation Paediatric Neurology: 20; 2009).
En otro aspecto más de la descripción, el sistema CRISPR-Cas puede ser usado para corregir defectos oculares que surgen de mutaciones genéticas diversas descritas además en Genetic Diseases of the Eye, Segunda Edición, editado por Elias I. Traboulsi, Oxford University Press, 2012.
Diversos aspectos adicionales de la descripción se refieren a corregir defectos asociados a un amplio intervalo de enfermedades genéticas que se describen además en la página web del Instituto Nacional para la Salud bajo la subsección tópica Trastornos Genéticos (sitio web en health.nih.gov/topic/GeneticDisorders). Las enfermedades cerebrales genéticas pueden incluir, pero no se limitan a, Adrenoleucodistrofia, Angenesia del Cuerpo Calloso, Síndrome de Aicardi, Enfermedad de Alper, Enfermedad de Alzheimer, Síndrome de Barth, Enfermedad de Batten, CADASIL, Degeneración Cerebelar, Enfermedad de Fabry, Enfermedad de Gerstmann-Straussler-Scheinker, Enfermedad de Huntington y otros Trastornos de Triple Repetición, Enfermedad de Leigh, Síndrome de Lesch-Nyhan, Enfermedad de Menkes, Miopatías Mitocondriales y Colpocefalia de NINDS (por sus siglas en inglés). Estas enfermedades se describen además en el sitio web del Instituto Nacional de la Salud bajo la subsección Trastornos Cerebrales Genéticos.
En algunas realizaciones, la afección puede ser neoplasia. En algunas realizaciones, donde la afección es neoplasia, los genes que se tienen que fijar como objetivo son cualquiera de los enumerados en la Tabla A (en este caso PTEN, etc). En algunas realizaciones, la afección puede ser Degeneración Macular Relacionada con la Edad. En algunas realizaciones, la afección puede ser un Trastorno Esquizofrénico. En algunas realizaciones, la afección puede ser un trastorno de Repetición del Trinucleótido. En algunas realizaciones, la afección puede ser Síndrome Frágil X. En algunas realizaciones, la afección puede ser un Trastorno Relacionado con la Secretasa. En algunas realizaciones, la afección puede ser un trastorno relacionado con el Prión. En algunas realizaciones, la afección puede ser ELA. En algunas realizaciones, la afección puede ser una drogadicción. En algunas realizaciones, la afección pueden ser Autismo. En algunas realizaciones, la afección puede ser Enfermedad de Alzheimer. En algunas realizaciones, la afección puede ser inflamación. En algunas realizaciones, la afección puede ser Enfermedad de Parkinson.
Ejemplos de proteínas asociadas a la enfermedad de Parkinson incluyen, pero no se limitan a, a-sinucleína, DJ-1, LRRK2, PINK1, Parkin, UCHL1, Synphilin-1 y NURR1.
Ejemplos de proteínas relacionadas con la adicción pueden incluir ABAT, por ejemplo.
Ejemplos de proteínas relacionadas con la inflamación pueden incluir la proteína quimioatrayente de monocitos -1 (MCP1) codificada por el gen Ccr2, el receptor de quimiocina C-C tipo 5 (CCR5) codificada por el gen Ccr5, el receptor IIB de IgG (FCGR2b, también denominado CD32) codificado por el gen Fcgr2b o la proteína de R1g Fc épsilon (FCER1g) codificada por el gen Fcer1g, por ejemplo.
Ejemplos de enfermedades cardiovasculares asociadas a proteínas pueden incluir IL1B (interleucina 1, beta), XDH (xantina deshidrogenasa), TP53 (proteína tumoral p53), PTGIS (prostaglandina 12 (prostaciclina) sintasa), MB (mioglobina), IL4 (interleucina 4), ANGPT1 (angiopoyetina 1), ABCG8 (casete de unión a ATP, sub-familia G (WHITE), miembro 8) o CTSK (catepsina K), por ejemplo.
Ejemplos de proteínas asociadas a la enfermedad de Alzheimer pueden incluir la proteína receptora de lipoproteína de densidad muy baja (VLDLR, por sus siglas en inglés) codificada por el gen VLDLR, la enzima activadora de modificador de tipo ubiquitina 1 (UBA1) codificada por el gen UBA1 o la proteína de subunidad catalítica de enzima E1 activadora de NEDD8 (UBE1C) codificada por el gen UBA3, por ejemplo.
Ejemplos de proteínas asociadas a Trastorno por Espectro Autista pueden incluir la proteína 1 asociada a receptor de benzodiazapina (periférica) (BZRAP1) codificada por el gen BZRAP1, la proteína del miembro 2 de la familia AF4/FMR2 (AFF2) codificada por el gen AFF2 (también denominado MFR2), la proteína de homólogo 1 autosómico de retraso mental frágil X (FXR1) codificada por el gen FXR1 o la proteína de homólogo 2 autosómico del retraso mental frágil X (FXR2) codificada por el gen FXR2, por ejemplo.
Los ejemplos de proteínas asociadas a Degeneración Macular pueden incluir la casete de unión de ATP, proteína del miembro 4 (ABCA4) de la sub-familia A (ABC 1) codificada por el gen ABCR, la proteína apolipoproteína E (APOE) codificada por el gen APOE o la proteína de Ligando 2 (CCL2) de quimiocina (unidad C-C) codificada por el gen CCL2, por ejemplo.
Ejemplos de proteínas asociadas a Esquizofrenia pueden incluir NRG1, ErbB4, CPLX1, TPH1, TPH2, NRXN1, GSk3a , BDNF, DISC1, GSK3B y combinaciones de las mismas.
Ejemplos de proteínas implicadas en la supresión de tumores pueden incluir ATM (ataxia telangiectasia mutada), ATR (ataxia telangiectasia y relacionada con Rad3), EGFR (receptor de factor de crecimiento epidérmico), ERBB2 (homólogo 2 de oncogén vírico de leucemia eritroblástica v-erb-b2), ERBB3 (homólogo 3 de oncogén vírico de leucemia eritroblástica v-erb-b2), ERBB4 (homólogo 4 de oncogén vírico de leucemia eritroblástica v-erb-b2), Notch 1, Notch2, Notch 3 o Notch 4, por ejemplo.
Ejemplos de proteínas asociadas a trastorno de la secretas pueden incluir PSENEN (homólogo de estimulador 2 de presenilina (C. elegans)), CTSB (catepsina B), PSEN1 (presenilina 1), APP (proteína precursora de amiloide beta (A4)), APH1B (homólogo B defectuoso anterior de la faringe (C. elegans)), PSEN2 (presenilina 2 (enfermedad de Alzheimer 4)) o BACE1 (enzima 1 de escisión de APP de sitio beta), por ejemplo.
Los ejemplos de proteínas asociadas a Esclerosis Lateral Amiotrófica pueden incluir SOD1 (superóxido dismutasa 1), a LS2 (esclerosis lateral amiotrófica 2), FUS (fusionado en sarcoma), TARDBP (proteína de unión de ADN TAR), VAGFA (factor A de crecimiento endotelial vascular), VAGFB (factor B de crecimiento endotelial vascular) y VAGFC (factor C de crecimiento endotelial vascular) y cualquier combinación de los mismos.
Ejemplos de proteínas asociadas a enfermedades por priones pueden incluir SOD1 (superóxido dismutasa 1), ALS2 (esclerosis lateral amiotrófica 2), FUS (fusionado en sarcoma), TARDBP (proteína de unión de ADN TAR), VAGFA (factor A de crecimiento endotelial vascular), VAGFB (factor B de crecimiento endotelial vascular) y VAGFC (factor C de crecimiento endotelial vascular) y cualquier combinación de los mismos.
Los ejemplos de proteínas relacionadas con enfermedades neurodegenerativas en trastornos por priones pueden incluir A2M (Alfa-2-Macroglobulina), AATF (Factor de transcripción de antagonización de muerte celular programada), ACPP (fosfatasa ácida prostática), ACTA2 (Actina alfa 2 de músculo liso aorta), ADAM22 (dominio de la metalopeptidasa ADAM), ADORA3 (Receptor de adenosina A3) o ADRA1D (Receptor adrenérgico Alfa-1D para adrenoreceptor Alfa-1D), por ejemplo.
Los ejemplos de proteínas asociadas a Inmunodeficiencia pueden incluir A2M [alfa-2-macroglobulina]; AANAT [arilalquilamina N-acetiltransferasa]; ABCA1 [casete de unión a ATP, sub-familia A (ABC1), miembro 1]; ABCA2 [casete de unión a ATP, sub-familia A (ABC1), miembro 2] o ABCA3 [casete de unión a ATP, sub-familia A (ABC1), miembro 3]; por ejemplo.
Ejemplos de proteínas asociadas a Trastornos de Repetición del Trinucleótido incluyen AR (receptor de andrógeno), FMR1 (retraso mental 1 frágil X), HTT (huntingtina) o DMPK (distrofia miotónica-proteína cinasa), FXN (frataxina), AtXn2 (ataxina 2), por ejemplo.
Ejemplos de proteínas asociadas a Trastornos de Neurotransmisión incluyen SST (somatostatina), NOS1 (óxido nítrico sintasa 1 (neuronal)), ADRA2A (receptor alfa-2A- adrenérgico), ADRA2C (receptor alfa-2C- adrenérgico), TACR1 (receptor de taquicinina 1) o HTR2c (receptor 2C de 5-hidroxitriptamina (serotonina)), por ejemplo. Ejemplos de secuencias asociadas al neurodesarrollo incluyen A2BP1 [proteína 1 de unión de ataxina 2], AADAT [aminoadipato aminotransferasa], AANAT [arilalquilamina N-acetiltransferasa], ABAT [4-aminobutirato
aminotransferasa], ABCA1 [casete de unión a ATP, sub-familia A (ABC1), miembro 1] o ABCA13 [casete de unión a ATP, sub-familia A (ABC1), miembro 13], por ejemplo.
Más ejemplos de trastornos preferidos tratables con el presente sistema se pueden seleccionar de: Síndrome de Aicardi-Goutiéres; Enfermedad de Alexander; Síndrome de Allan-Herndon-Dudley; Trastornos Relacionados con POLG; Alfa-Manosidosis (Tipo II y III); Síndrome de Alstrom; Angelman; Síndrome; Ataxia-Telangiectasia; Lipofuscinosis Ceroide Neuronal; Beta-Talasemia; Atrofia Óptica Bilateral y Atrofia Óptica (Infantil) Tipo 1; Retinoblastoma (bilateral); Enfermedad de Canavan; Síndrome 1 Cerebroculofacioesquelético [COFS1]; Xantomatosis Cerebrotendinosa; Síndrome de Cornelia de Lange; Trastornos Relacionados con MAPT; Enfermedades Genéticas por Priones; Síndrome de Dravet; Enfermedad de Alzheimer Familiar de Comienzo Temprano; Ataxia de Friedreich [FRDA]; Síndrome de Fryns; Fucosidosis; Distrofia Muscular Congénita de Fukuyama; Galactosialidosis; Enfermedad de Gaucher; Acidemias Orgánicas; Linfohistiocitosis Hemofagocítica; Síndrome de Progeria de Hutchinson-Gilford; Mucolipidosis II; Enfermedad de Almacenamiento de Ácido Siálico Libre Infantil; Neurodegeneración Asociada a PLA2G6; Síndrome de Jervell y Lange-Nielsen; Epidermolisis Bullosa sobre las Articulaciones; Enfermedad de Huntington; Enfermedad de Krabbe (Infantil); Síndrome de Leigh Asociado al ADN Mitocondrial y NARP; Síndrome de Lesch-Nyhan; Lisencefalia Asociada a LIS1; Síndrome de Lowe; Enfermedad Urinaria de Jarabe de Maple; Síndrome de Duplicación de MECP2; Trastornos de Transporte de Cobre Relacionado con ATP7A; Distrofia Muscular Relacionada con LAMA2; Deficiencia de Arilsulfatasa A; Mucopolisacaridosis Tipos I, II o III; Trastornos de la Biogénesis del Peroxisoma, Espectro del Síndrome de Zellweger; Neurodegeneración con Trastornos de Acumulación de Hierro en el Cerebro; Deficiencia de Esfingomielinasa Ácida; Enfermedad de Niemann-Pick Tipo C; Encefalopatía por Glicina; Trastornos Relacionados con ARX; Trastornos del Ciclo de la Urea; Osteogénesis Imperfecta Relacionada con COL1A1/2; Síndromes de Supresión de ADN Mitocondrial; Trastornos Relacionados con PLP1; Síndrome de Perry; Síndrome de Phelan-McDermid; Enfermedad de Almacenamiento de Glucógeno Tipo II (Enfermedad de Pompe) (Infantil); Trastornos Relacionados con MAPT; Trastornos Relacionados con MECP2; Condrodisplasia Punctata Rizomélica Tipo 1; Síndrome de Roberts; Enfermedad de Sandhoff; Enfermedad de Schindler - Tipo 1; Deficiencia de Adenosina Deaminasa; Síndrome de Smith-Lemli-Opitz; Atrofia Muscular Espinal; Ataxia Espinocerebelar de Comienzo Infantil; Deficiencia de Hexosaminidasa A; Displasia Tanatofórica de Tipo 1; Trastornos Relacionados con el Colágeno Tipo VI; Síndrome de Usher Tipo I; Distrofia Muscular Congénita; Síndrome de Wolf-Hirschhorn; Deficiencia de Lipasa Ácida Lisosomal y Xerodermia Pigmentosa.
Como será evidente, se prevé que el presente sistema se pueda usar para fijar como objetivo cualquier secuencia de polinucleótidos de interés. Algunos ejemplos de afecciones o enfermedades que se podían tratar positivamente usando el presente sistema se incluyen en las Tablas anteriores y también se proporcionan allí ejemplos de genes asociados en la actualidad a esas enfermedades. Sin embargo, los genes ejemplificados no son exhaustivos.
Ejemplos
Los siguientes ejemplos se proporcionan para el fin de ilustración de diversas realizaciones de la descripción y no están destinados a limitar la presente invención de ningún modo. Los presentes ejemplos, junto con los métodos descritos en la presente memoria son representativos en el momento presente de realizaciones preferidas, son ejemplares, y no se destinan como limitaciones del alcance de la invención.
Ejemplo 1: Actividad del complejo CRISPR en el núcleo de una célula eucariota.
Un sistema CRISPR de ejemplo de tipo II es el sitio CRISPR de tipo II de Streptococcus pyogenes SF370, que contiene una agrupación de cuatro genes Cas9, Cas1, Cas2 y Csn1, así como dos elementos de ARN no codificante, ARNtracr y una serie característica de secuencias repetitivas (repeticiones directas) interespaciadas por tramos cortos de secuencias no repetitivas (espaciadores, aproximadamente 30 pb cada uno). En este sistema, se genera rotura de doble cadena (RDC) de ADN fijado como objetivo en cuatro etapas secuenciales (Figura 2A). Primero, dos ARN no codificantes, la matriz pre-ARNcr y ARNtracr, se transcriben del sitio CRISPR. Segundo, ARNtracr se hibrida a las repeticiones directas de pre-ARNcr, que se tratan después en ARNcr maduros que contienen secuencias espaciadoras individuales. Tercero, el complejo ARNcr:ARNtracr maduro dirige Cas9 al ADN objetivo que consiste en el protoespaciador y la correspondiente PAM vía la formación de heterodúplex entre la región espaciadora del ARNcr y el ADN protoespaciador. Finalmente, Cas9 media la escisión de ADN objetivo aguas arriba de PAM para crear una RDC dentro del protoespaciador (Figura 2A). Este ejemplo describe un procedimiento de ejemplo para adaptar este sistema de nucleasa programable de ARN para dirigir la actividad del complejo CRISPR en el núcleo de las células eucariotas.
Cultivo celular y transinfección.
Se mantuvo la estirpe celular HEK 293FT (Life Technologies) de riñón embriónico humano (HEK, por sus siglas en inglés) en Medio Eagle modificado de Dulbecco (DMEM, por sus siglas en inglés) enriquecido con suero fetal bovino al 10% (HyClone), GlutaMAX 2 mM (Life Technologies), 100 U/ml de penicilina y 100 pg/ml de estreptomicina a 37°C con incubación en CO2 al 5%. Se mantuvo la estirpe celular neuro2A (N2A) de ratón (ATCC)
con DMEM enriquecido con suero fetal bovino al 5% (HyClone), GlutaMAX 2 mM (Life Technologies), 100 U/ml de penicilina y 100 pg/ml de estreptomicina a 37°C con CO2 al 5%.
Se sembraron células 293FT de HEK o N2A en placas de 24 pozos (Corning) un día previamente a transinfección a una densidad de 200.000 células por pozo. Se transinfectaron las células usando Lipofectamine 2000 (Life Technologies) siguiendo el protocolo recomendado por el fabricante. Para cada pozo de una placa de 24 pozos, se usó un total de 800 ng de plásmido.
Ensayo surveyor y análisis de secuenciación para modificación de genomas.
Se transinfectaron células 293FT de HEK o N2A con ADN de plásmido como se describió anteriormente. Después de transinfección, se incubaron las células a 37°C durante 72 horas antes de extracción de ADN genómico. Se extrajo ADN genómico usando el estuche de extracción de ADN QuickExtract (Epicentre) siguiendo el protocolo del fabricante. En resumen, se volvieron a suspender las células en disolución QuickExtract y se incubaron a 65°C durante 15 minutos y 98°C durante 10 minutos. El ADN genómico extraído se trató inmediamente o se almacenó a -20°C.
La región genómica que rodea un sitio objetivo de CRISPR para cada gen se multiplicó por PCR y se purificaron los productos usando una columna de QiaQuick Spin (Qiagen) siguiendo el protocolo del fabricante. Se mezcló un total de 400 ng los productos PCR purificados con 2 pl de tampón PCR Taq Polimerasa (Enzymatics) X 10 y agua ultrapura a un volumen final de 20 pl y se sometió a un procedimiento de rehibridación para permitir la formación de heterodúplex: 95°C durante 10 min, 95°C a 85°C descendiendo a -2°C/s, 85°C a 25°C a - 0,25°C/s y 25°C mantenidos durante 1 minuto. Después de rehibridación, se trataron los productos con nucleasa Surveyor y estimulador S Surveyor (Transgenomics) siguiendo el protocolo recomendado por el fabricante y se analizaron sobre geles de poliacrilamida Novex TBE al 4-20% (Life Technologies). Se tiñeron los geles con tinción de ADN SYBR Gold (Life Technologies) durante 30 minutos y se formaron imágenes con un sistema de formación de imágenes de gel Gel Doc (Bio-rad). La cuantificación estuvo basada en intensidades de banda relativas, como una medición de la fracción de ADN escindido. La Figura 8 proporciona una ilustración esquemática de esta prueba Surveyor.
Prueba de polimorfismo de longitud de fragmento de restricción para detección de recombinación homóloga. Se transinfectaron células 293FT de HEK y N2A con ADN de plásmido y se incubaron a 37°C durante 72 horas antes de extracción de ADN genómico como se describió anteriormente. La región genómica objetivo se multiplicó por PCR usando cebadores fuera de las ramas de homología de la plantilla de recombinación homóloga (RH). Los productos PCR se separaron en un gel de agarosa al 1% y se extrajo con Estuche MinElute GelExtraction (Qiagen). Se digirieron los productos purificados con HindIII (Fermentas) y se analizaron en un gel de poliacrilamida Novex TBE al 6% (Life Technologies).
Predicción y análisis de la estructura secundaria de ARN.
Se realizó la predicción de la estructura secundaria de ARN usando el Webserver RNAfold online desarrollado en el Instituto para Química Teórica de la Universidad de Viena, usando el algoritmo de predicción de estructura centroide (véase por ej., A. R. Gruber et al., 2008, Cell 106 (1): 23-24 y PA Carr y GM Church, 2009, Nature Biotechnology 27 (12): 1.151-62).
Prueba de interferencia de transformación de plásmidos bacterianos.
Se reconstituyeron elementos del sitio 1 de CRISPR de S. pyogenes suficientes para actividad de CRISPR en E. coli usando plásmido de pCRISPR (ilustrado de manera esquemática en la Figura 10A). pCRISPR contenía ARNtracr, SpCas9, y una secuencia líder que conducía a la matriz de ARNcr. Se insertaron espaciadores (también referidos como “secuencias guía”) en la matriz de ARNcr entre los si tios BsaI usando oligonucleótidos hibridados, como se ilustra. Se construyeron plásmidos provocadores usados en la prueba de interferencia insertando la secuencia protoespaciadora (también referida como una “secuencia diana”) junto con una secuencia unidad de CRISPR adyacente (PAM) en pUC19 (véase la Figura 10B). El plásmido provocador contenía resistencia a la ampicilina. La figura 10C proporciona una representación esquemática de la prueba de interferencia. Las cepas E. coli químicamente competentes que ya soportaban pCRISPR y el espaciador apropiado se transformaron con el plásmido provocador que contenía la correspondiente secuencia protoespaciador-PAM. Se usó pUC19 para valorar la eficacia de la transformación de cada cepa competente soportando pCRISPR. La catividad CRISPR dio como resultado escisión del plásmido pPSP soportando el protoespaciador, que excluye la resistencia a la ampicilina conferida de otro modo por pUC19 que carece del protoespaciador. La figura 10d ilustra competencia de cada cepa de E. coli que soporta pCRISPR usada en las pruebas ilustradas en la Figura 4C.
Purificación de ARN.
Se mantuvieron células de HEK 293FT y se transinfectaron como se indicó anteriormente. Se recogieron las células por tripsinización seguido por lavado en disolución salina tamponada de fosfato (PBS). Se extrajo ARN de células totales con reactivo TRI (Sigma) siguiendo el protocolo del fabricante. Se cuantificó el ARN total extraido usando Naonodrop (Thermo Scientific) y se normalizó a la misma concentración.
Análisis por el método Northern de expresión de ARNcr y ARNtracr en células de mamífero.
Se mezclaron ARN con volúmenes iguales de tampón de carga X2 (Ambion), se calentó a 95°C durante 5 min., se enfrió en hielo durante 1 min., y después se cargó en geles de poliacrilamida desnaturalizada al 8% (SequaGel, National Diagnostics) después de hacer operar previamente el gel durante al menos 30 minutos. Se sometieron las muestras a electroforesis durante 1,5 horas a un límite de 40 W. Después, se transfirió el ARN a membrana Hybond N+ (GE Healthcare) a 300 mA en un aparato de transferencia semiseco (Bio-rad) a temperatura ambiente durante 1,5 horas. Se reticuló el ARN a la membrana usando un botón de autoreticulación en Reticulador UV Stratagene el Stratalinker (Stratagene). Se hibridó previamente la membrana en Tampón de Hibridación ULTRAhyb-Oligo (Ambion) durante 30 min., con rotación a 42°C y se añadieron después sondas y se hibridaron durante la noche. Se pidieron las sondas de IDT y se etiquetaron con ATP [gamma-32P] (Perkin Elmer) con T4 polinucleótido cinasa (New England Biolabs). Se lavó la membrana una vez con SSC (citrato de sodio - salino, por sus siglas en inglés) x2 precalentado (42°C), SDS (dodecilsulfato de sodio, por sus siglas en inglés) al 0,5% durante 1 min., seguido por dos lavados de 30 minutos a 42°C. Se expuso la membrana a pantalla de fósforo durante una hora o durante la noche a temperatura ambiente y después se escaneó con un fosforimager (Typhoon).
Construcción y evaluación de sistema CRISPR bacteriano.
Los elementos del sitio CRISPR, incluyendo ARNtracr, Cas9, y líder se multiplicaron por PCR de ADN genómico SF370 de Streptococcus pyogenes con ramas de homología de flanqueamiento por Gibson Assembly. Se introdujeron dos sitios Bsal tipo IIS en medio de dos repeticiones directas para facilitar la fácil inserción de espaciadores (Figura 9). Se clonaron los productos PCR en pACYC184 EcoRV-digerido aguas abajo del activador tet usando Mezcla Maestra de Gibson Assembly (NEB). Se omitieron otros elementos del sistema CRISPR endógeno, con la excepción de las últimas 50 pb de Csn2. Se clonaron espaciadores codificadores de oligos (Tecnología de ADN Integrada) con salientes complementarios en el vector Bsal-digerido pDC000 (NEB) y se ligaron después con T7 ligasa (Enzymatics) para generar plásmidos pCRISPR. Se crearon espaciadores que contenían plásmidos provocadores con secuencias de PAM (también referido en la presente memoria como “secuencias de unidades CRISPR”) ligando olig os hibridados soportando salientes compatibles (Tecnología de ADN Integrada) en pUC19 BamHI-digeridos. Se realizó clonación para todas las construcciones en cepa JM109 de E. coli (Zymo Research).
Las células que soportaban pCRISPR se hicieron competentes usando el Estuche de Transformación de E. coli Z-Competente y Ajuste de Tampón (Zymo Research, T3001) según las instrucciones del fabricante. En la prueba de transformación, se descongelaron sobre hielo alícuotas de 50 ul de células competentes que soportaban pCRISPR y se transformaron con 1 ng de plásmido espaciador o pUC19 sobre hielo durante 30 minutos, seguido por 45 segundos de choque térmico a 42°C y 2 minutos sobre hielo. Con posterioridad, se añadieron 250 ul de SOC (Invitrogen) seguido por incubación con agitación a 37°C durante 1 h y se pusieron en placas 100 ul de excrecencia pos-SOC en placas de selección dobles (12,5 ug/ml de cloranfenicol, 100 ug/ml de ampicilina). Para obtener ufc/ng de ADN, los números de colonias totales se multiplicaron por 3.
Para mejorar la expresión de los componentes CRISPR en células de mamífero, se codón-optimizaron dos genes del sitio 1 de SF370 de Streptococcus pyogenes (S. pyogenes), Cas9 (SpCas9) y RNasa III (SpRNasa III). Para facilitar la localización nuclear, se incluyó una señal de localización nuclear (NLS, por sus siglas eninglés) en el término (N) amino o (C) carboxilo de tanto SpCas9 como SpRNasa III (Figura 2B). Para facilitar la visualización de la expresión de proteínas, también se incluyó un marcador de proteína fluorescente en el N- o C-terminal de ambas proteínas (Figura 2B). También se generó una versión de SpCas9 con una NLS unido a tanto N- como C-terminal (2xNLS-SpCas9). Se transinfectaron construcciones que contenían SpCas9 NLS-fusionada y SpRNasa III en células de riñón embriónico humano (HEK) 293FT y se encontró que el posicionamiento relativo de la NLS para SpCas9 y SpRNasa III afectaba a su eficacia de localización nuclear. Mientras la NLS C-terminal fue suficiente para fijar como objetivo SpRNasa III para los núcleos, la unión de una sola copia de estas NLS particulares en cualquiera de los N- o C-terminales de SpCas9 no pudo conseguir la localización nuclear adecuada en este sistema. En este ejemplo, la NLS C-terminal fue la de nucleoplasmina (KRPAATKKAGQAKKKK) y la NLS C-terminal fue la del antígeno T grande SV40 (PKKKRKV). De las versiones de SpCas9 ensayadas, sólo 2xNLS -SpCas9 presentó localización nuclear (Figura 2B).
La ARNtracr del sitio CRISPR de S. pyogenes SF370 presenta dos sitios de comienzo transcripcional, dando lugar a dos transcritos de 89 nucleótidos (nt) y 171 nt que se tratan con posterioridad en ARNtracr maduros de 75 nt
idénticos. Se seleccionó el ARNtracr de 89 nt más corto para expresión en células de mamífero (construcciones de expresión ilustradas en la Figura 7A, con funcionalidad cuando se determina por los resultados de la prueba de Surveryor mostrados en la Figura 7B). Los sitios de comienzo de la transcripción se señalan como 1 y el terminador de la transcripción y la secuencia probada por el ensayo de northern se indican también. La expresión de ARNtracr tratado también se confirmó por ensayo de Northern. La Figura 7C muestra los resultados de un análisis por el método de Northern de ARN total extraído de células 293FT transinfectadas con construcciones de expresión U6 que soportaban ARNtracr largo o corto, así como SpCas9 y DR-EMX1(1)-DR. Los paneles izquierdo y derecho son de células 293FT transinfectadas sin o con SpRNasa III, respectivamente. U6 indica control de carga representado gráficamente con una sonda que fija como objetivo ARNsn U6 humano. La transinfección de la construcción de expresión de ARNtracr corto conduce a niveles abundantes de la forma tratada de ARNtracr (~75 pb). Se detectan cantidades muy bajas de ARNtracr largo en el análisis de Northern.
Para activar la iniciación precisa transcripcional, se seleccionó el activador de U6 basado en ARN polimerasa III para conducir la expresión de ARNtracr (Figura 2C). De manera similar, se desarrolló una construcción a base de activador U6 para expresar una matriz pre-ARNcr que consiste en un único espaciador flanqueado por dos repeticiones directas (las RD, también incluida por el término "secuencias de emparejamiento tracr"; Figura 2C). Se diseñó el espaciador inicial para fijar como objetivo un sitio objetivo de 33 pares de bases (pb) (protoespaciador de 30 pb más una secuencia de unidad (PAM) CRISPR de 3 pb que satisface la unidad de reconocimiento NGG de Cas9) en el sitio EMX1 humano (Figura 2C), un gen clave en el desarrollo de la corteza cerebral.
Para ensayar si la expresión heteróloga del sistema CRISPR (SpCas9, SpRNasa III, ARNtracr y pre-ARNcr) en células de mamífero puede conseguir la escisión fijada como objetivo de cromosomas de mamífero, se transinfectaron células HEK 293FT con combinaciones de componentes CRISPR. Puesto que las RDC en núcleos de mamífero se reparan parcialmente por la ruta de unión de extremos no homólogos (NHEJ, por sus siglas en inglés), que conduce a la formación de inserciones o supresiones, se usó el ensayo Surveyor para detectar potencial actividad de escisión en el sitio EMX1 objetivo (Figura 8) (véase por ej., Guschin et al., 2010, Methods Mol Biol 649: 247). La transinfección conjunta de los cuatro componentes CRISPR pudo inducir una escisión hasta del 5,0% en el protoespaciador (véase la Figura 2D). La transinfección conjunta de todos los componentes CRISPR menos SpRNasa III también indujo hasta 4,7% de inserción o supresión en el protoespaciador, que sugiere que puede haber RNasas de mamífero endógenas que son capaces de ayudar a la maduración de ARNcr, tal como por ejemplo las enzimas Dicer y Drosha relacionadas. Retirar cualquiera de los tres componentes restantes anuló la actividad de escisión del genoma del sistema CRISPR (Figura 2D). La secuenciación Sanger de amplicones que contienen el sitio fijado como objetivo verificó la actividad de escisión: en 43 clones secuenciados, se encontraron 5 alelos mutados (11,6%). Experimentos similares usando una variedad de secuencias guía produjeron porcentajes de inserción o supresión tan altos como 29% (véanse las Figuras 4-7, 12 y 13). Estos resultados definen un sistema de tres componentes para modificación de genomas mediada por CRISPR eficaz en células de mamífero. Para optimizar la eficacia de escisión, los solicitantes también ensayaron si diferentes isoformas de ARNtracr afectaban a la eficacia de la escisión y encontraron que, en este sistema de ejemplo, sólo la forma transcrita corta (89 pb) era capaz de mediar la escisión del sitio genómico EMX1 humano (Figura 7B).
La Figura 14 proporciona un análisis de Northern adicional de tratamiento de ARNcr en células de mamífero. La Figura 14A ilustra un esquema que muestra el vector de expresión para un único espaciador flanqueado por dos repeticiones directas (RD-EMX1(1)- RD). El espaciador de 30 pb que fija como objetivo el protoespaciador 1 del sitio EMX1 humano (véase la Figura 6) y las secuencias de repetición directa se muestran en la secuencia debajo de la Figura 14A. La línea indica la región cuya secuencia de complemento inverso se usó para generar sondas de Northern para detección de ARNcr de e MX1(1). La Figura 14B muestra un análisis Northern de ARN total extraído de células 293FT transinfectadas con construcciones de expresión U6 que soportan RD-EMX1(1)- RD. Los paneles izquierdo y derecho son de células 293FT transinfectadas sin o con SpRNasa III respectivamente. Se trató RD-EMX1(1)- RD en ARNcr maduros sólo en presencia de SpCas9 y ARNtracr corto y no fue dependiente de la presencia de SpRNasa III. El ARNcr maduro detectado de ARN total de 293FT transinfectado es ~33 pb y es más corto que el ARNcr maduro de 39-42 pb de S. pyogenes. Estos resultados demuestran que un sistema CRISPR se puede trasplantar en células eucariotas y reprogramar para facilitar la escisión de polinucleótidos objetivo de mamíferos endógenos.
La Figura 2 ilustra el sistema CRISPR bacteriano descrito en este ejemplo. La Figura 2A ilustra un esquema que muestra el sitio 1 de CRISPR de SF370 de Streptococcus pyogenes y un mecanismo propuesto de escisión de ADN mediada por CRISPR por este sistema. El ARNcr maduro tratado de la matriz de repetición directa -espaciador dirige Cas9 a objetivos genómicos que consisten en protoespaciadores favorables y una unidad adyacente al protoespaciador (PAM). En el emparejamiento de base objetivo-espaciador, Cas9 media una rotura de doble cadena en el ADN objetivo. La Figura 2B ilustra el logro de Cas9 de S. pyogenes (SpCas9) y RNasa III (SpRNasa III) con señales de localización nuclear (las NLS) para permitir la importación al núcleo del mamífero. La Figura 2C ilustra la expresión de mamífero de SpCas9 y SpRNasa III conducida por el activador EF1a constitutivo y ARNtracr y la matriz pre-ARNcr (RD-Espaciador-RD) conducida por el activador U6 de ARN Pol3
para activar iniciación y terminación de transcripción precisa. Se usa un protoespaciador del sitio EMX1 humano con una secuencia PAM satisfactoria como el espaciador en la serie pre-ARNcr. La Figura 2D ilustra ensayo de la nucleasa surveyor para inserciones y supresiones minoritarias mediadas por SpCas9. SpCas9 se expresó con y sin SpRNasa III, ARNtracr y una matriz pre-ARNcr que soporta el espaciador objetivo EMX1. La Figura 2E ilustra una representación esquemática de emparejamiento de bases entre el sitio objetivo y ARNcr que fija como objetivo EMX1, así como un cromatograma de ejemplo que muestra una microsupresión adyacente al sitio de escisión de SpCas9. La Figura 2F ilustra alelos mutados identificados de análisis de secuenciación de 43 amplicones clonales que muestran una variedad de microinserciones y supresiones. Las líneas discontinuas indican bases suprimidas y bases no alineadas o desajustadas indican inserciones o mutaciones. Barra de escala = 10 pm.
Para simplificar además el sistema de tres componentes, se adaptó un diseño híbrido de ARNcr-ARNtracr quimérico, donde un ARNcr maduro (que comprende una secuencia guía) se fusiona a un ARNtracr parcial vía un tallo-bucle para imitar el dúplex ARNcr:ARNtracr natural (Figura 3A). Para aumentar la eficacia de suministro conjunto, se creó un vector de expresión bicistrónico para conducir la coexpresión de un ARN quimérico y SpCas9 en células transinfectadas (Figuras 3A y 8). Paralelamente, se usaron los vectores bicistrónicos para expresar un pre-ARNcr (RD-secuencia guía-RD) con SpCas9, para inducir el progreso en ARNcr con un ARNtracr expresado por separado (compare la Figura 13B superior y del fondo). La Figura 9 proporciona ilustraciones esquemáticas de vectores de expresión bicistrónicos para matriz pre-ARNcr (Figura 9A) o ARNcr quimérico (representado por la línea corta aguas abajo del sitio de inserción de la secuencia guía y aguas arriba del activador EF1a en la Figura 9B) con hSpCas9, que muestra la posición de varios elementos y el punto de inserción de la secuencia guía. La secuencia expandida alrededor de la posición del sitio de inserción de la secuencia guía en la Figura 9B también muestra una secuencia RD parcial (GTTTAGAGCTA) y una secuencia de ARNtracr parcial (TAGCAAGTTAAAATAAGGCTAGTCCGTTt Tt ). Las secuencias guía se pueden insertar entre sitios Bbsl usando oligonucleótidos hibridados. El diseño de las secuencias para los oligonucleótidos se muestra bajo las ilustraciones esquemáticas en la Figura 9, con los adaptadores de ligadura apropiados indicados. WPRE representa el elemento regulador postranscripcional del virus de la hepatitis Woodchuck. Se ensayó la eficacia de la escisión mediada por ARN quimérico fijando como objetivo el mismo sitio EMX1 descrito anteriormente. Usando tanto la prueba Surveyor como la secuenciación de Sanger de amplicones, los solicitantes confirmaron que el diseño de ARN quimérico facilita la escisión del sitio EMX1 humano con aproximadamente una relación de modificación de 4,7% (Figura 4).
Se ensayó el carácter generalizable de la escisión medidad por CRISPR en células eucariotas fijando como objetivo los sitios genómicos adicionales en células tanto humanas como de ratón diseñando múltiples sitios que fijen como objetivo ARN quimérico en el EMX1 y PVALB humanos, así como los sitios Th de ratón. La Figura 15 ilustra la selección de algunos protoespaciadores fijados como objetivo adicionales en PVALB humano (Figura 15A) y sitios Th de ratón (Figura 15B). Se proporcionan esquemas de los sitios génicos y la posición de tres protoespaciadores en el último exón de cada uno. Las secuencias subrayadas incluyen 30 pb de secuencia protoespaciadora y 3 pb en el extremo 3' correspondiente a las secuencias de PAM. Los protoespaciadores en las cadenas codificante y no codificante se indican por encima y por debajo de las secuencias de ADN, respectivamente. Se consiguió una tasa de modificación de 6,3% y 0,75% para los sitios PVALB humano y Th de ratón, respectivamente, que demuestra la amplia aplicabilidad del sistema CRISPR en la modificación de diferentes sitios a través de múltiples organismos (Figuras 3B y 6). Aunque la escisión sólo se detectó con uno de tres espaciadores para cada sitio usando las construcciones quiméricas, todas las secuencias objetivo se escindieron con eficacia de producción de inserción o supresión que alcanzó el 27% cuando se usó la disposición de pre-ARNcr expresada conjuntamente (Figura 6).
La Figura 13 proporciona una ilustración más de que SpCas9 puede ser reprogramado para fijar como objetivo múltiples sitios genómicos en células de mamífero. La Figura 13A proporciona un esquema del sitio EMX1 humano que muestra la posición de cinco protoespaciadores, indicado por las secuencias subrayadas. La Figura 13B proporciona un esquema del complejo pre-ARNcr/ARNtrcr que muestra la hibridación entre la región de repetición directa del pre-ARNcr y ARNtracr (parte superior) y un esquema de un diseño de ARN quimérico que comprende una secuencia guía de 20 pb y secuencias de emparejamiento tracr y tracr que consisten en secuencias de repetición directa parcial y ARNtracr hibridadas en una estructura de horquilla (fondo). Los resultados de un ensayo Surveyor comparando la eficacia de escisión mediada por Cas9 en cinco protoespaciadores en el sitio EMX1 humano se ilustra en la Figura 13C. Cada protoespaciador se fija como objetivo usando complejo pre-ARNcr/ARNtracr tratado (ARNcr) o ARN quimérico (ARNqui).
Puesto que la estructura secundaria de ARN puede ser crucial para interacciones intermoleculares, se usó un algoritmo de predicción de estructuras basado en energía libre mínima y conjunto de estructura pesada de Boltzmann para comparar la estructura secundaria putativa de todas las secuencias guía usadas en nuestro experimento de fijación como objetivo de genoma (Figura 3B) (véase por ej., Gruber et al., 2008, Nucleic Acids Research, 36: W70). El análisis reveló que en la mayoría de los casos, las secuencias guía eficaces en el contexto de ARNcr quimérico estaban sustancialmente exentas de unidades de estructura secundaria, mientras las secuencias guía ineficaces era más probable que formaran estructuras secundarias internas que podían evitar el
emparejamiento de bases con el ADN de protoespaciador objetivo. Así es posible que la variabilidad en la estructura secundaria del espaciador pueda impactar en la eficacia de la interferencia mediada por CRISPR cuando se usa un ARNcr quimérico.
La Figura 3 ilustra vectores de expresión de ejemplo. La Figura 3A proporciona un esquema de un vector bicistrónico para conducir la expresión de una quimera ARNcr-ARNtracr sintética (ARN quimérico) así como SpCas9. El ARN guía quimérico contiene una secuencia guía de 20 pb correspondiendo al protoespaciador en el sitio objetivo genómico. La Figura 3B proporciona un esquema que muestra secuencias guía que fijan como objetivo los sitios EMX1, PVALB humano y Th de ratón, así como sus estructuras secundarias previstas. La eficacia de la modificación en cada sitio objetivo se indica debajo del dibujo de la estructura secundaria del ARN (EMX1, n = 216 lecturas de secuenciación de amplicones; PVALB, n = 224 lecturas; Th, n = 265 lecturas). El algoritmo de plegamiento produjo una salida con cada base coloreada de acuerdo con su probabilidad de asumir la estructura secundaria prevista, como se indica por una escala en arco iris que se reproduce en la Figura 3B en escala de grises. Más diseños de vectores para SpCas9 se presentan en la Figura 44, que ilustra vectores únicos de expresión que incorporan un activador U6 ligado a un sitio de inserción para un oligo guía y un activador Cbh ligado a secuencia de codificación de SpCas9. El vector mostrado en la Figura 44b incluye una secuencia codificadora de ARNtracr ligada a un activador H1.
Para ensayar si los espaciadores que contienen estructuras secundarias son capaces de funcionar en células procariotas en las cuales los CRISPR operan de manera natural, se ensayó interferencia de transformación de plásmidos que soportan protoespaciadores en una cepa E. coli que expresa de manera heteróloga el sitio 1 CRISPR SF370 de S. pyogenes (Figura 10). Se clonó el sitio CRISPR en un vector de expresión E. coli de copia baja y se reemplazó la matriz de ARNcr con un espaciador único flanqueado por un par de las RD (pCRISPR). Se transformaron cepas E. coli que albergaban diferentes plásmidos pCRISPR con plásmidos problema conteniendo el correspondiente protoespaciador y secuencias pAm (Figura 10C). En el ensayo de bacterias, todos los espaciadores facilitaron interferencia de CRISPR eficaz (Figura 4C). Estos resultados sugieren que puede haber factores adicionales que afecten a la eficacia de la actividad de CRISPR en células de mamífero.
Para investigar la especificidad de la escisión mediada por CRISPR, se analizó el efecto de mutaciones de único nucleótido en la secuencia guía sobre la escisión del protoespaciador en el genoma de mamífero usando una serie de ARNcr quiméricos que fijan como objetivo EMX1 con mutaciones de puntos únicos (Figura 4A). La Figura 4B ilustra los resultados de un ensayo de nucleasa Surveyor comparando la eficacia de la escisión de Cas9 cuando se empareja con diferentes ARN quiméricos mutantes. El desajuste de bases únicas hasta 12 pb 5' de la PAM sustancialmente anuló la escisión genómica por SpCas9, mientras los espaciadores con mutaciones en posiciones aguas arriba más lejos retuvieron la actividad frente al objetivo del protoespaciador original (Figura 4B). Además de la PAM, SpCas9 presenta especificidad de base única dentro de las últimas 12 pb del espaciador. Además, CRISPR es capaz de mediar la escisión genómica tan eficazmente como un par de nucleasas TALE (TALEN) fijando como objetivo el mismo protoespaciador EMX1. La Figura 4C proporciona un esquema que muestra el diseño de las TALEN que fijan como objetivo EMX1 y la Figura 4D muestra un gel Surveyor comparando la eficacia de TALEN y Cas9 (n=3).
Teniendo establecida una serie de componentes para conseguir edición de genes mediada por CRISPR en células de mamífero a través del mecanismo NHEJ con predisposición a errores, se ensayó la capacidad de CRISPR para estimular la recombinación homóloga (RH), una ruta de reparación de genes de alta fidelidad para preparar ediciones precisas en el genoma. SpCas9 natural es capaz de mediar las RDC específicas del sitio, que se pueden reparar a través de tanto NHEJ como RH. Además, se logró una sustitución de aspartato a alanina (D10A) en el dominio catalítico RuvC I de SpCas9 para convertir la nucleasa en una nickasa (SpCas9n; ilustrado en la Figura 5A) (véase por ej., Sapranauskas et al., 2011, Nucleic Acids Research, 39: 9.275; Gasiunas et al., 2012, Proc. Natl. Acad. Sci. USA, 109: E2579), de manera que el ADN genómico nickeado experimenta la reparación dirigida por homología de alta fidelidad (HDR, por sus siglas en inglés). La prueba Surveyor confirmó que SpCas9n no generaba inserciones o supresiones en el objetivo del protoespaciador EMX1. Como se ilustra en la Figura 5B, la expresión conjunta de ARNcr quimérico que fija como objetivo EMX1 con SpCas9 produjo inserciones o supresiones en el sitio fijado como objetivo, mientras la expresión conjunta con SpCas9n no lo hizo (n=3). Por otra parte, la secuenciación de 327 amplicones no detectó ninguna inserción o supresión inducida por SpCas9n. Se seleccionó el mismo sitio para ensayar RH mediada por CRISPR por transinfección conjunta de células HEK 293FT con el ARN quimérico que fija como objetivo e MX1, hSpCas9 o hSpCas9n, así como una plantilla de RH para introducir un par de sitios de restricción (HindIII y NheI) cerca del protoespaciador. La Figura 5C proporciona una ilustración en esquema de la estrategia de la Rh , con posiciones relativas de puntos de recombinación y secuencias de hibridación de cebadores (flechas). SpCas9 y SpCas9n por supuesto catalizaron la integración de la plantilla de RH en el sitio EMX1. La multiplicación por PCR de la región fijada como objetivo seguida por digestión de restricción con HindIII reveló productos de escisión correspondiendo a tamaños de fragmentos esperados (flechas en análisis de gel de polimorfismo de longitud del fragmento de restricción mostrado en la Figura 5D), mediando SpCas9 y SpCas9n niveles similares de eficacias de la RH. Los solicitantes verificaron además RH usando secuenciación de Sanger de amplicones genómicos (Figura 5E). Estos resultados demuestran
la utilidad de CRISPR para facilitar la inserción de genes fijados como objetivo en el genoma del mamífero. Dada la especificidad objetivo de 14 pb (12 pb del espaciador y 2 pb de la PAM) de SpCas9 natural, la disponibilidad de una nickasa puede reducir significativamente la probabilidad de modificaciones fuera de lo proyectado, puesto que las roturas de cadena única no son sustratos para la ruta NHEJ con predisposición a errores.
Las construcciones de expresión que imitan la arquitectura natural de sitios CRISPR con espaciadores formados (Figura 2A) se construyeron para ensayar la posibilidad de fijar como objetivo secuencias multiplexadas. Usando una única matriz de CRISPR codificando un par de espaciadores que fijan como objetivo EMX1 y PVALB, se detectó la escisión eficaz en ambos sitios (Figura 4F, que muestra tanto un diseño esquemático de la matriz ARNcr como un análisis Surveyor mostrando la mediación eficaz de la escisión). La supresión fijada como objetivo de regiones genómicas mayores a través de las RDC concurrentes usando espaciadores frente a dos objetivos dentro de EMX1 espaciado por 119 pb también se ensayó y se detectó una eficacia de la supresión del 1,6% (3 de un total de 182 amplicones; Figura 4G). Esto demuestra que el sistema CRISPR puede mediar la edición multiplexada dentro de un único genoma.
Ejemplo 2: Modificaciones y alternativas del sistema CRISPR.
La capacidad para usar ARN para programar escisión de ADN específica de la secuencia define una nueva clase de herramientas de ingeniería del genoma para una variedad de aplicaciones de investigación e industriales. Diversos aspectos del sistema CRISPR se pueden mejorar además para aumentar la eficacia y la versatilidad de la fijación como objetivo de CRISPR. La actividad óptima de Cas9 puede depender de la disponibilidad de Mg2+ libre en niveles mayores que el presente en el núcleo de los mamíferos (véase por ej., Jinek et al., 2012, Science, 337: 816) y la preferencia para una unidad de NGG inmediatamente aguas abajo del protoespaciador restringe la capacidad para fijar como objetivo de promedio cada 12 pb en el genoma humano (Figura 11, evaluando las cadenas positiva y negativa de secuencias de cromosomas humanos). Algunas de estas restricciones se pueden superar explorando la diversidad de los sitios CRISPR a través del metagenoma microbiano (véase por ej., Makarova et al., 2011, Nat Rev Microbiol, 9: 467). Otros sitios CRISPR pueden ser trasplantados en el medio celular de los mamíferos mediante un procedimiento similar al descrito en el Ejemplo 1. Por ejemplo, la Figura 12 ilustra la adaptación del sistema CRISPR de Tipo II de CRISPR 1 de LMD-9 de Streptococcus thermophilus para expresión heteróloga en células de mamífero para conseguir edición de genomas mediada por CRISPR. La Figura 12A proporciona una ilustración esquemática de CRISPR 1 de LMD-9 de S. thermophilus. La Figura 12B ilustra el diseño de un sistema de expresión para el sistema CRISPR de S. thermophilus. Se expresa hStCas9 codónoptimizado humano usando un activador de EF1a constitutivo. Las versiones maduras de ARNtracr y ARNcr se expresan usando el activador U6 para activar la iniciación de transcripción precisa. Se ilustran las secuencias del ARNcr y ARNtracr maduros. Se usa una sola base indicada por el caso inferior “un” en la secuencia de ARNcr para retirar la secuencia poliU, que sirve como un terminador transcripcional de ARN polIII. La Figura 12C proporciona un esquema que muestra secuencias guía que fijan como objetivo el sitio EMX1 humano, así como sus estructuras secundarias previstas. La eficacia de la modificación en cada sitio fijado como objetivo se indica debajo de las estructuras secundarias de ARN. El algoritmo que genera las estructuras colorea cada base de acuerdo con su probabilidad para asumir la estructura secundaria prevista, que se indica por una escala en arco iris reproducida en la Figura 12C en escala gris. La Figura 12D muestra los resultados de la escisión mediada por hStCas9 en el sitio fijado como objetivo usando la prueba Surveyor. Los espaciadores 1 y 2 guía de ARN indujeron 14% y 6,4%, respectivamente. El análisis estadístico de la actividad de escisión a través de réplicas biológicas en estos dos sitios del protoespaciador se proporciona también en la Figura 6. La Figura 16 proporciona un esquema de protoespaciador adicional y los correspondientes objetivos de secuencia PAM del sistema CRISPR de S. thermophilus en el sitio EMX1 humano. Se resaltan dos secuencias protoespaciadoras y se indican sus correspondientes secuencias PAM satisfaciendo la unidad NNAGAAW subrayando 3' con respecto a la correspondiente secuencia resaltada. Los dos protoespaciadores fijan como objetivo la cadena no codificante.
Ejemplo 3: Algoritmo de selección de secuencias objetivo de muestra.
Se diseña un programa informático para identificar secuencias objetivo de CRISPR candidato en ambas cadenas de una secuencia de ADN de entrada basada en longitud deseada de secuencias guía y una secuencia de unidad CRISPR (PAM) para una enzima CRISPR especificada. Por ejemplo, los sitios objetivo para Cas9 de S. pyogenes, con secuencias NGG de PAM, se pueden identificar por investigación para 5'-Nx-NGG-3' ambas en la secuencia de entrada y en el complemento inverso de la entrada. Asimismo, los sitios objetivo para Cas9 de CRISPR1 de S. thermophilus, con secuencia NNAGAAW de PAM, se pueden identificar por investigación para 5'-Nx-NNAGAAW-3' ambos en la secuencia de entrada y en el complemento inverso de la entrada. Asimismo, los sitios objetivo para Cas9 de CRISPR3 de S. thermophilus, con secuencia NGGNG de PAM, se pueden identificar por investigación para 5'-Nx-NGGNG-3' ambos en la secuencia de entrada y en el complemento inverso de la entrada. El valor "x" en Nx puede ser fijado por el programa o especificado por el usuario, tal como 20.
Puesto que múltiples apariciones en el genoma del sitio objetivo de ADN pueden conducir a edición de genoma no especifica, después de identificar todos los sitios potenciales, el programa filtra secuencias basadas en el número de veces que aparecen en el genoma de referencia relevante. Para esas enzimas CRISPR para las cuales se determina la especificidad de secuencia mediante una secuencia "simiente", tal como la 5' de 11-12 pb de la secuencia PAM, incluyendo la propia secuencia PAM, la etapa de filtración puede estar basada en la secuencia simiente. Así, para evitar la edición en sitios genómicos adicionales, los resultados se filtran basándose en el número de apariciones de la secuencia simiente:PAM en el genoma relevante. Se puede permitir que el usuario elija la longitud de la secuencia simiente. También se puede permitir que el usuario especifique el número de apariciones de la secuencia simiente:PAM en un genoma para fines de pase del filtro. El defecto es investigar secuencias únicas. El nivel de filtración se modifica cambiando tanto la longitud de la secuencia simiente como el número de apariciones de la secuencia en el genoma. El programa puede proporcionar además o alternativamente la secuencia de una secuencia guía complementaria a la secuencia o secuencias objetivo indicadas proporcionando el complemento inverso de la secuencia o secuencias objetivo identificadas.
Ejemplo 4: Evaluación de híbridos ARNcr-ARNtracr quiméricos múltiples.
Este ejemplo describe los resultados obtenidos para los ARN quiméricos (los ARNqui; que comprenden una secuencia guía, una secuencia de emparejamiento tracr y una secuencia tracr en un transcrito único) que tienen secuencias tracr que incorporan diferentes longitudes de secuencia ARNtracr natural. La Figura 18a ilustra un esquema de un vector bicistrónico de expresión para ARN quimérico y Cas9. Cas9 es conducido por el activador CBh y el ARN quimérico es conducido por un activador U6. El ARN guía quimérico consta de una secuencia guía de 20 pb (Ns) unida a la secuencia tracr (barriendo desde la primera "U" de la cadena menor al extremo del transcrito), que se trunca en diversas posiciones como se indica. Las secuencias guía y tracr se separan mediante la secuencia de emparejamiento tracr GUUUUAGAGCUA seguido por la secuencia bucle GAAA. Los resultados de los ensayos SURVEYOR para inserciones o supresiones mediadas por Cas9 en los sitios EMX1 y PVALB humanos se ilustran en la Figura 18b y 18c, respectivamente. Las flechas indican los fragmentos SURVEYOR esperados. Los ARNqui se indican por su designación "+n" y ARNcr se refiere a un ARN híbrido donde las secuencias guía y tracr se expresan como transcritos separados. La cuantificación de estos resultados, realizados por triplicado, se ilustran por histograma en las Figuras 19a y 19b, que corresponden a las Figuras 18b y 18c, respectivamente ("N. D." indica no inserciones o supresiones detectadas). Los ID del protoespaciador y su correspondiente objetivo genómico, secuencia del protoespaciador, secuencia PAM y posición de la cadena se proporcionan en la Tabla D. Las secuencias guía se diseñaron para que fueran complementarias a la secuencia completa del protoespaciador en el caso de transcritos separados en el sistema híbrido o sólo a la porción subrayada en el caso de ARN quiméricos.
Tabla D:

Cultivo celular y transinfección.
Se mantuvo la estirpe celular 293FT (Life Technologies) de riñón embriónico humano (HEK) en Medio Eagle modificado de Dulbecco (DMEM) enriquecido con suero fetal bovino al 10% (HyClone), GlutaMAX 2 mM (Life Technologies), 100 U/ml de penicilina y 100 pg/ml de estreptomicina a 37°C con incubación en CO2 al 5%. Se sembraron células 293FT en placas de 24 pozos (Coming) 24 horas previamente a transinfección a una densidad de 150.000 células por pozo. Se transinfectaron las células usando Lipofectamine 2000 (Life Technologies) siguiendo el protocolo recomendado por el fabricante. Para cada pozo de una placa de 24 pozos, se usó un total de 500 ng de plásmido.
Prueba SURVEYOR para modificación de genomas.
Se transinfectaron células 293FT con ADN de plásmido como se describió anteriormente. Se incubaron las células a 37°C durante 72 horas post-transinfección previamente a extracción de ADN genómico. Se extrajo ADN genómico usando la disolución de extracción de ADN QuickExtract (Epicentre) siguiendo el protocolo del fabricante. En resumen, se volvieron a suspender las células del botón en disolución QuickExtract y se incubaron a 65°C durante 15 minutos y 98°C durante 10 minutos. La región genómica que flanquea el sitio objetivo de CRISPR para cada gen se multiplicó por PCR (cebadores enumerados en la Tabla E) y se purificaron los productos usando una columna de QiaQuick Spin (Qiagen) siguiendo el protocolo del fabricante. Se mezclaron 400 ng totales de los productos PCR purificados con 2 pl 10X de tampón PCR Taq ADN Polimerasa (Enzymatics) y agua ultrapura a un volumen final de 20 pl y se sometieron a un procedimiento de rehibridación para permitir la formación de heterodúplex: 95°C durante 10 min, 95°C a 85°C descendiendo a -2°C/s, 85°C a 25°C a - 0,25°C/s y 25°C mantenidos durante 1 minuto. Después de rehibridación, se trataron los productos con nucleasa SURVEYOR y estimulador S SURVEYOR (T ransgenomics) siguiendo el protocolo recomendado por el fabricante y se analizaron sobre geles de poliacrilamida Novex TBE al 4-20% (Life Technologies). Se tiñeron los geles con tinción de ADN SYBR Gold (Life Technologies) durante 30 minutos y se formaron imágenes con un sistema de formación de imágenes de gel Gel Doc (Bio-rad). La cuantificación estuvo basada en intensidades de banda relativas.
Tabla E:
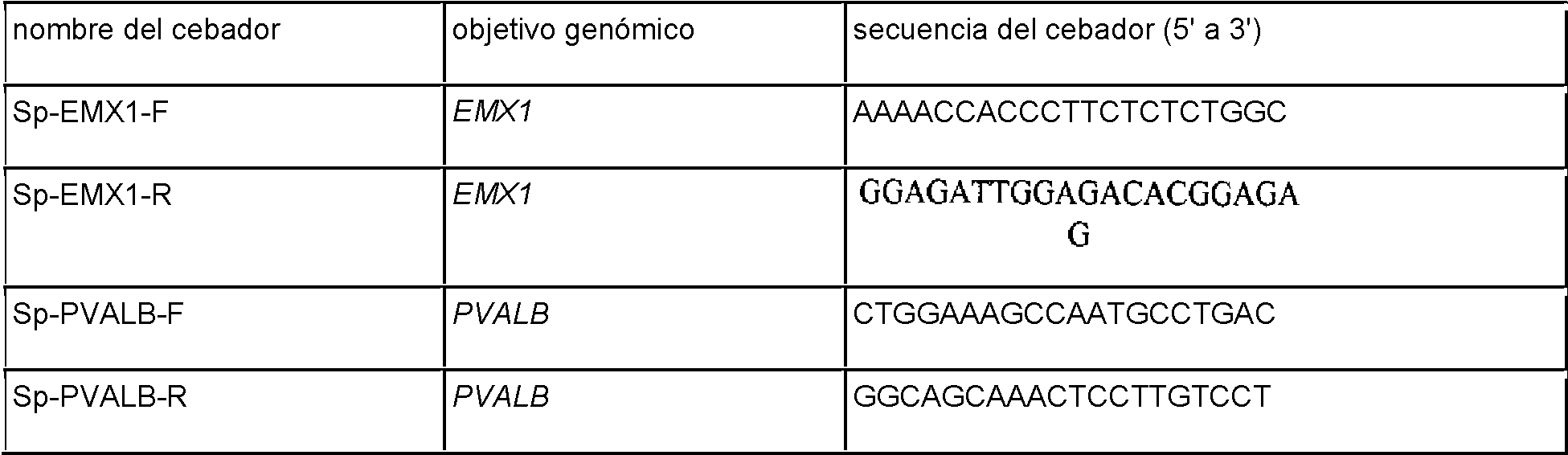
identificación computacional de sitios fijados como objetivo de CRISPR únicos.
Para identificar sitios fijados como objetivo, únicos, para la enzima SF370 Cas9 (SpCas9) de S. pyogenes en genoma de ser humano, ratón, rata, pez cebra, mosca de la fruta y C. elegans, se desarrolló un paquete informático para escanear ambas cadenas de una secuencia de ADN e identificar todos los posibles sitios objetivo de SpCas9. Para este ejemplo, cada sitio objetivo SpCas9 se definió operacionalmente como una secuencia de 20 pb seguido por una secuencia de unidad adyacente del protoespaciador (PAM) de NGG y se identificaron todas las secuencias que satisfacían esta definición de 5'-N20-NGG-3' en todos los cromosomas. Para evitar la edición de genoma no específica, después de identificar todos los sitios potenciales, se filtraron todos los sitios objetivo basándose en el número de veces que aparecían en el genoma de referencia relevante. Para sacar provecho de la especificidad de la secuencia de la actividad de Cas9 conferida por una secuencia "simiente", que puede ser, por ejemplo, secuencia 5' de aproximadamente 11-12 pb de la secuencia PAM, se seleccionaron secuencias 5'-NNNNNNNNNN-NGG-3' para que fueran únicas en el genoma relevante. Todas las secuencias genómicas se descargaron de UCSC Genome Browser (genoma humano hg19, genoma de ratón mm9, genoma de rata rn5, genoma de pez cebra danRer7, genoma dm4 de D. melanogaster y genoma ce10 de C. elegans). Los resultados de la investigación completa están disponibles para navegar usando la información de UCSC Genome Browser. Una visualización de ejemplo de algunos sitios objetivo en el genoma humano se proporciona en la Figura 21.
Inicialmente, se fijaron como objetivo tres sitios dentro del sitio de EMX1 en células HEK 293FT humanas. Se evaluó la eficacia de la modificación del genoma de cada ARNqui usando la prueba de la nucleasa SURVEYOR, que detecta mutaciones que resultan de las roturas de la doble cadena de ADN (las RDC) y su posterior reparación por la ruta de reparación de daños de ADN de unión de extremos no homólogos (NHEJ). Las construcciones designadas ARNqui(+n) indican que hasta el nucleótido n de ARNtracr natural se incluye en la construcción de ARN quimérico, con valores de 48, 54, 67 y 85 usados para n. Los ARN quiméricos que contienen fragmentos mayores de ARNtracr natural (ARNqui (+67) y ARNqui (+85)) mediaron la escisión de ADN en los tres sitios objetivo de EMX1, demostrando ARNqui (+85) en particular niveles significativamente mayores de escisión de ADN que los correspondientes híbridos ARNcr/ARNtracr que expresaron secuencias guía y tracr en transcritos separados (Figuras 18b y 19a). También se fijaron como objetivo dos sitios en el sitio PVALB que no proporcionaron escisión detectable usando el sistema híbrido (secuencia guía y secuencia tracr expresadas como transcritos separados) usando los ARNqui. ARNqui(+67) y ARNqui(+85) pudieron mediar escisión significativa en los dos protoespaciadores PVALB (Figuras 18c y 19b).
Para los cinco objetivos en los sitios EMX1 y PVALB, se observó un incremento consistente en la eficacia de modificación del genoma con longitud creciente de la secuencia tracr. Sin desear estar limitados por ninguna teoría, la estructura secundaria formada por el extremo 3' de la ARNtracr puede desempeñar una función en la activación de la tasa de formación de complejo CRISPR. Una ilustración de las estructuras secundarias previstas para cada uno de los ARN quiméricos usados en este ejemplo se proporciona en la Figura 21. Se predijo la estructura secundaria usando RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi) usando energía libre mínima y algoritmo de función de partición. El pseudocolor para cada base (reproducido en escala de grises) indica la probabilidad de emparejamiento. Debido a que los ARNqui con secuencias tracr más largas eran capaces de escindir objetivos que no eran escindidos por híbridos ARNcr/ARNtracr de CRISPR naturales, es posible que se pueda cargar ARN quimérico en Cas9 más eficazmente que su contrapartida híbrida natural. Para facilitar la aplicación de Cas9 para edición de genoma específica del sitio en células y organismos eucariotas, todos los sitios objetivo únicos previstos para la Cas9 de S. pyogenes se identificaron de manera computacional en los genomas del ser humano, ratón, rata, pez cebra, C. elegans y D. melanogaster. Se pueden diseñar ARN quiméricos para enzimas Cas9 de otros microbios para expandir el espacio fijado como objetivo de las nucleasas programables de ARN de CRISPR.
La Figura 22 ilustra un vector de expresión bicistrónico ejemplar para expresión de ARN quimérico incluyendo hasta el nucleótido 85 de secuencia de ARNtracr natural y SpCas9 con secuencias de localización nuclear. SpCas9 se expresa a partir de un activador CBh y termina con la señal poliA bGH (bGH pA). La secuencia expandida ilustrada inmediatamente debajo del esquema corresponde a la región que rodea al sitio de inserción de la secuencia guía e incluye, desde 5' a 3', la porción 3' del activador U6 (primera región sombreada), sitios de escisión BbsI (flechas), repetición directa parcial (secuencia de emparejamiento tracr GTTTTAGAGCTA, subrayada), secuencia bucle GAAA y secuencia tracr 85 (secuencia subrayada después de la secuencia bucle). Un inserto de la secuencia guía ejemplar se ilustra debajo del sitio de la inserción de la secuencia guía, con nucleótidos de la secuencia guía para un objetivo seleccionado representado por un "N".
Las secuencias descritas en los ejemplos anteriores son como sigue (las secuencias de polinucleótidos son 5' a 3'): ARNtracr corto U6 (Streptococcus pyogenes SF370):
GAGGGCCTATTTCCCATGATTCCTTCATATTTGCATATACGATACAAGGCT GTT AGAGAGAT AATTGGAATT A ATTT GACTGT A A ACAC A AAGAT ATT AGT ACA A A A T ACGT G ACGT AG AAAGT AAT A ATTTCTTGGGT AGTTTGC AGTTTT AAAATT ATGTTTT AAAATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTAT ATATCTTGTGGAAAGGACGAAACACCGGAACCATTCAAAACAGCATAGCAAGTTA AAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTT TTT
(negrita = secuencia ARNtracr; subrayado = secuencia terminadora)
ARNtracr largo U6 (SF370 Streptococcus pyogenes):
GAGGGCCTATTTCCCATGATTCCTTCATATTTGCATATACGATACAAGGCT
GTTAGAGAGATAATTGGAATTAATTTGACTGTAAACACAAAGATATTAGTACAAAA TACGTGACGTAGAAAGTAATAATTTCTTGGGTAGTTTGCAGTTTTAAAATTATGTTTT AAAATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTAT AT AT CTT GTGGA AAGGACG AAAC ACCGGTAGT ATTAAGT ATT GTTTT ATGGCT G AT A AATTT CTTT G AATTT CTCCTT GATT ATTT GTT AT AAAAGTT AT AA AAT AAT CTT GTT G GA ACCATTCA AA ACAGC AT AGC A AGTTA A A AT A AGGCT AGTCCGTT ATCAACTTG A AAAAGTGGCACCGAGTCGGTGCTTTTTTT
U6-DR-Bbsl cadena principal-RD (SF370 Streptococcus pyogenes):
GAGGGCCTATTTCCCATGATTCCTTCATATTTGCATATACGATACAAGGCT GTTAGAGAGATAATTGGAATTAATTTGACTGTAAACACAAAGATATTAGTACAAAA T ACGTGACGT AGAAAGT AATA ATTTCTT GGGT AGTTTGC AGTTTT AAAATT ATGTTTT A A AAT GGACT AT CAT ATGCTT ACCGT A ACTTGA A AGT ATTT CG ATTTCTTGGCTTT AT ATATCTTGTGGAAAGGACGAAACACCGGGTTTTAGAGCTATGCTGTTTTGAATGGTC CCAAAACGGGTCTTCGAGAAGACGTTTTAGAGCTATGCTGTTTTGAATGGTCCCAAA AC
U6-ARN quimérico-Cadena principal BbsI (SF370 Streptococcus pyogenes)
GAGGGCCTATTTCCCATGATTCCTTCATATTTGCATATACGATACAAGGCT
GTTAGAGAGATAATTGGAATTAATTTGACTGTAAACACAAAGATATTAGTACAAAA T ACGT G ACGT AG AAAGT AAT AATTT CTTGGGT AGTTTGC AGTTTT AAAATT AT GTTTT AAAATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTAT ATATCTTGTGGAAAGGACGAAACACCGGGTCTTCGAGAAGACCTGTTTTAGAGCTA GAAATAGCAAGTTAAAATAAGGCTAGTCCG
NLS-SpCas9-EGFP:
MDYKDHDGDYKDHDIDYKDDDDKMAPKKKRKVG1HGVPAADKKYSIGLDI GTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTARR RYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFLVEEDKKHERHPIFGNIVDEVAYHE KYPTIYHLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDVDKLFIQLVQ TYNQLFEENPINASGVDAKAILSARLSKSRRLENFTAQLPGEKKNGFFGNLIALSLGFTPN FKSNFDLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKMLSDA1LLSD1LRVNT EITKAPLSASM1KRYDEHHQDLTFLKAFVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQ EEFYKFIKPTLEKMDGTEELLVKLNREDLLRKQRTFDNGSIPHQTHLGELH AILRRQEDFY PFLKDNREKIEKILTFR1PYYVGPLARGNSRFAWMTRKSEETITPWNFEEVVDKGASAQS FIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIV DLLFKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDN EENEDIFEDIVFTLTFFEDREMIEERLKTYAHLFDDKVMKQLKRRRYTGWGRFSRKLIN
G1RDKQSGKTILDFLKSDGFANRNFMQL1HDDSLTFKEDIQKAQVSGQGDSLHEH1ANLA
GSPAIKKGILQTVKVVDELVKVMGRHKPENIVIEMARENQTTQKGQKNSRERMKRTEEG 1KELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSF LKDDSIDNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKEDNLTKAE RGGLSELDKAGFIKRQLVETRQITKHVAQILDSRMNTKYDENDKLIREVKVITLKSKLVS DFRKDFQFYKVRETNNYHHAHDAYLNAVVGTALIKKYPKLESEFVYGDYKVYDVRKMI AKSEQEIGKATAKYFFYSNIMNFFKTEITLANGETRKRPLIETNGETGETVWDKGRDFATV RKVLSMPQVNIVKKTEVQTGGFSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYS VLVVAKVEKGKSKKLKSVKELLGIT1MERSSFEKNP1DFLEAKGYKEVKKDLIIKJLPKYS LFELENGRKRMLASAGELQKGNELALPSKYVNFLYLASHYEKLKGSPEDNEQKQLFVE QHKH Y LDEIJLEQ1SEFSKR VILADAN LDKVLS A YNKHRDKP1REQ AEN 11E[LFT LTN LG AP AAFKYFDTTIDRKRYTSTKEVLDATL1HQSITGLYETR1DLSQLGGDAAAVSKGEELFTG VVPILVELDGDVNGHKFSVSGEGEGDATYGKLTLKFTCTTGKLPVPWPTLVTTLTYGVQ CFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRTELKGI DFKEDGNTLGHKLEYNYNSFTNVYIMADKQKNGTKVNFKTRFIMEDGSVQLADHYQQNT PIGDGPVLLPDNHYLSTQSALSKDPNEKRDHMVLLEFVTAAGITLGMDELYK
SpCas9-EGFP-NLS:
MDKKYS1GLD1GTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLF
DSGETAEATRLKRTARRRYTRRKNRTCYLQE1FSNEMAKVDDSFFHRLEESFLVEEDKK HERHP1FGNIVDEVAYHEKYPT1YHLRKKLVDSTDKADLRL1YLALAHMIKFRGHFLIEG DLNPDNSDVDKLFIQLVQTYNQLFEENPINASGVDAKAILSARLSKSRRLENLIAQLPGE KKNGLFGNLTALSLGLTPNFKSNFDLAEDAKLQLSKDTYDDDLDNLLAQTGDQYADLFL AAKNLSDAILLSDILRVNTEITKAPLSASMIKRYDEHHQDLTLLKALVRQQLPEKYKEIFF DQSKNGYAGYTDGGASQEEFYKFIKPILEKMDGTEELLVKLNREDLLRKQRTFDNGSIPH
Q1HLGELHA1LRRQEDFYPFLKDNREK1EKILTFR1PYYVGPLARGNSRFAWMTRKSEET1 TPWNFEEVVDKGASAQSFIERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTE GMRKPAFLSGEQKKAIVDLLFKTNRKVTVKQLKEDYFKKIECFDSVE1SGVEDRFNASL GTYHDLLKMKDKDFLDNEENEDILEDIVLTLTLFEDREMIEERLKTYAHLFDDKVMKQL KRRRYTGWGRLSRKLINGJRDKQSGKTILDFLKSDGFANRNFMQL1HDDSLTFKEDIQKA QVSGQGDSLHEHIANLAGSPAIKKG1LQTVKVVDELVKVMGRHKPENIV1EMARENQTT QKGQKNSRERMKRTEEGTKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQEL DINRLSDYDVDHIVPQSFLKDDSIDNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQL
FNAKFITQRKFDNFTKAERGGFSEFDKAGFIKRQLVETRQITKHVAQILDSRMNTKYDE NDKLIREVKVTTLKSKLVSDFRKDFQFYKVREINNYEIHAHDAYLNAVVGTALIKKYPKL ESEFVYGDYKVYDVRKMIAKSEQE1GKATAKYFFYSN1MNFFKTEITLANGEJRKRPL1ET TMGETGEIVWDKGRDFATVRKVLSMPQVNIVKKTEVQTGGFSKESIFPKRNSDKLIARKK DWDPKKYGGFDSPTVAYSVFVVAKVEKGKSKKFKSVKELFGITIMERSSFEKNPIDFLE AKGYKEVKKDEIIKFPKYSFFEFENGRKRMLASAGEFQKGNELALPSKYVNFLYLASHY EKLKGSPEDNEQKQFFVEQHKHYFDEITEQISEFSKRVIFADAIMLDKVLSAYNKFIRDKPI REQAENIIHLFTLTNLGAPAAFKYFDTTIDRtCRYTSTKEVFDATLIHQSITGLYETRJDLSQ LGGDAAAVSKGEELFTGVVPIFVELDGDVNGHKFSVSGEGEGDATYGKLTLKFICTTGK FPVPWPTFVTTFTYGVQCFSRYPDHMKQHDFFKSAMPEGYVQERTIFFK.DDGNYKTRA EVKFEGDTFVNRTELKGIDFKEDGNILGHKLEYNYNSHNVY1MADKQKNGIKVNFIC1RH NIEDGSVQLADHYQQNTPIGDGPVLFPDNHYLSTQSAFSKDPNEKRDHMVLLEFVTAAG ITFGMDELYKKRPAATKKAGQAKKKK NLS-SpCas9-EGFP-NLS:
MDYKDHDGDYKDHDIDYKDDDDKMAPKKKRKVGIHGVPAADKKYSIGLDI
GTNSVGWAVITDEYKVPSKKFKVFGNTDRHSIKKNFIGAFFFDSGETAEATRFKRTARR RYTRRKNRICYFQEIFSNEMAKVDDSFFHRFEESFLVEEDKKHERHPIFGNIVDEVAYHE KYPTIYHFRKKFVDSTDKADFRLIYFAFAHMIKFRGHFFIEGDFNPDNSDVDRLFIQFVQ TYNQFFEENPINASGVDAKAIFSARFSKSRRFENFIAQFPGEKKNGFFGNFIAFSFGFTPN FKSNFDFAEDAKLQLSKDTYDDDFDNLFAQIGDQYADLFLAAKNFSDAIFLSD1LRVNT E1TKAPFSASM1KRYDEHHQDFTFFKAFVRQQFPEKYKEIFFDQSKNGYAGYIDGGASQ EEFYKFIKPIFEKMDGTEEFFVKFNREDFFRKQRTFDNGSIPHQIHFGEFHAIFRRQEDFY PFFKDNREKJEKJFTFR1PYYVGPFARGNSRFAWMTRKSEETITPWNFEEVVDKGASAQS FIERMTNFDKNFPNEKVFPKHSFFYEYFTVYNEFTKVKYVTEGMRKPAFPSGEQKKAIV DFFFKTNRKVTVKQFKEDYFKKIECFDSVEISGVEDRFNASFGTYHDFFKIIKDKDFFDN EENEDIFEDIVFTFTFFEDREM1EERFKTYAHFFDDKVMKQFKRRRYTGWGRFSRKF1N GIRDKQSGKTIFDFFKSDGFANRNFMQFIHDDSFTFKEDTQKAQVSGQGDSFHEHIANFA GSPA1KKG1FQTVKVVDEFVKVMGRHKPEN1VIEMARENQTTQKGQKNSRERMKR1EEG IKEFGSQIFKFHPVENTQFQNEKFYFYYFQNGRDMYVDQEFD1NRFSDYDVDH1VPQSF LKDDSIDNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQFFNAKFITQRKFDNLTKAE RGGFSEFDKAGF1KRQF V ET RQITKH VAQIFDSRMNTKY DENDKFIRE ViCVITFKSKFV S
DFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLESEFVYGDYKVYDVRKMI AKSEQEIGKATAKYFFYSNIMNFFKTEITLANGEIRKRPL1ETNGETGEIVWDKGRDFATV RKVLSMPQVN1VKKTEVQTGGFSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYS VLVVAKVEKGKSRKLKSVKELLGITIMERSSFEKNPIDFLEAKGYKEVKKDLIIKLPKYS LFELENGRKRMLASAGELQKGNELALPSKYVNFLYLASHYEKLKGSPEDNEQKQLFVE QHKHYLDE11EQISEFSKRVILADANLDKVLSAYNKHRDKPIREQAENI1HLFTLTNLGAP AAFKYFDTTIDRKRYTSTKEVLDATLIHQSITGLYETRIDLSQLGGDAAAVSKGEELFTG VVP1LVELDGDVNGHKFSVSGEGEGDATYGKLTLKEICTTGK-LPVPWPTLVTTLTYGVQ CFSRYPDHMKQHDFFKSAMPEGYVQERTIFFKDDGNYKTRAEVKFEGDTLVNRIELKGI DFKEDGNILGHKLEYNYNSHNVYIMADKQKNGIKVNFKIRHNIEDGSVQLADHYQQNT
P1GDGPVLLPDNHYLSTQSALSKDPNERRDHMVLLEFVTAAGITLGMDELYKKRPAATK KAGQAKKKK NLS-SpCas9-NLS:
MDYKDHDGDYKDHDTDYKDDDDKMAPKKKRKVGTHGVPAADKKYSTGLDT
GTNSVGWAVITDEYKVPSKKFKVLGNTDRHSIKKNLIGALLFDSGETAEATRLKRTARR RYTRRKNRICYLQEIFSNEMAKVDDSFFHRLEESFLVEEDKKHERHPIFGNIVDEVAYHE KYPTIYHLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDVDKLFIQLVQ TYNQLFEENPINASGVDAKAILSARESKSRREENEIAQLPGEKKNGLFGNL1ALSLGLTPN FKSNFDLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKNLSDAILLSDILRVNT EITKAPLSASMIKRYDEHHQDLTLLKALVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQ EEFYKF1KJPILEKMDGTEELLVKLNREDLLRKQRTFDNGSIPHQ1HLGELHAILRRQEDFY PFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEWDKGASAQS FTERMTNFDKNLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKATV DLLFKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDN EENEDILEDIVLTLTLFEDREMTEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKL1N GIRDKQSGKTILDFLKSDGFANRNFMQLIHDDSLTFKEDIQKAQVSGQGDSLHEHIANLA GSPA1KKGIEQTVKVVDEEVKVMGRHKPENIV1EMARENQTTQKGQKNSRERMKRIEEG IKELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSF LKDDS1DNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKEITQRKFDNLTKAE RGGLSELDKAGFIKRQLVETRQITKHVAQILDSRMNTKYDENDKLIREVKVITLKSKLVS DFRKDFQFYKVREINNYHHAHDAYLNAVVGTALIKKYPKLESEFVYGDYKVYDVRKMI
AKSEQEIGKATAKYFFYSNIMNFFKTEITLANGEIRKRPLIETNGETGEIVWDKGRDFATV RKVLSMPQVNIVKKTEVQTGGFSKESILPKRNSDKLIARKKDWDPKKYGGFDSPTVAYS VLVVAKVEKGKSKKLKSVKELLGITIMERSSFEKNPTDFLEAKGYKEVKKDLTTKLPKYS LFELENGRKRMLASAGELQKGNELALPSKYVNFLYLASEPFEKLKGSPEDNEQKQLFVE QHKHYLDEIIEQ1SEFSKRVILADANLDKVLSAYNKHRDKPIREQAENIIHLFTLTNLGAP AAFKYFDTTIDRKRYTSTKEVLDATL1HQSITGLYETRIDLSQLGGDKRPAATKKAGQAK KKK NLS-mCherry-SpRNasa3:
MFLFLSLTSFLSSSRTLVSKGEEDNMAIIKEFMRFKVHMEGSVNGHEFEIEGE
GEGRPYEGTQTAKLKVTKGGPLPFAWDILSPQFMYGSKAYVKHPADIPDYLKLSFPEGF KWERVMNFEDGGVVTVTQDSSLQDGEFrYKVKLRGTNFPSDGPVMQKKTMGWEASSE RMYPEDGALKGEIKQRLKLKDGGHYDAEVKTTYKAKKPVQLPGAYNVNIKLDITSHNE DYTIVEQYERAEGRHSTGGMDELYKGSKQLEELLSTSFDIQFNDLTLLETAFTHTSYANE HRLLNVSHNERLEFLGDAVLQLIISEYLFAKYPKKTEGDMSKLLRSM1VREESLAGFSRFC SFDAYIKLGKGEEKSGGRRRDT1LGDLFEAFLGALLLDKG1DAVRRFLKQVMIPQVEKG NFERVKDYKTCLQEFLQTKGDVAIDYQVISEKGPAHAKQFEVSIVVNGAVLSKGLGKSK KLAEQDAAKNALAQLSEV
SpRNasa3-mCherry-NLS:
MKQLEELLSTSFDIQFNDLTLLETAFTHTSYANEHRLLNVSHNERLEFLGDAV
LQLIISEYLFAKYPKKTEGDMSKLRSMIVREESLAGFSRFCSFDAYIKLGKGEEKSGGRR RDT1LGDLFEAFLGALLLDKG1DAVRRFLKQVMIPQVEKGNFERVKDYKTCLQEFLQTK GDVAIDYQVISEKGPAHAKQFEVSIVVNGAVLSKGLGKSKKLAEQDAAKNALAQLSEV GSVSKGEEDNMAIIKEFMRFKVHMEGSVNGHEFEIEGEGEGRPYEGTQTAKLKVTKGGP LPFAWDILSPQFMYGSKAYVKHPADIPDYLKLSFPEGFKWERVMNFEDGGVVTVTQDS SLQDGEFIYKVKLRGTNFPSDGPVMQKKTMGWEASSERMYPEDGALKGE1KQRLKLKD GGHYDAEVKTTYKAKKPVQLPGAYNVNIKLDITSHNEDYT1VEQYERAEGRHSTGGMD ELYKKRPAATKKAGQAKKKK NLS-SpCas9n-NLS (la mutación de nickasa D10A es minúscula):
MDYKDHDGDYKDHDTDYKDDDDKMAPKKKRKVGTHGVPAADKKYSIGLal GTNSVGWAV1TDEYKVPSKXFKVLGNTDRHS1KKNL1GALLFDSGETAEATRLKRTARR RYTRRKNRTCYLQETFSNEMAKVDDSFFHRLEESFLVEEDKKHERHPTFGNIVDEVAYHE
KYPTIYHLRKKLVDSTDKADLRLIYLALAHMIKFRGHFLIEGDLNPDNSDVDKLFIQLVQ TYNQLFEENPINASGVDAKAILSARLSKSRRLENL1AQLPGEKKNGLFGNLIALSLGLTPN FKSNFDLAEDAKLQLSKDTYDDDLDNLLAQIGDQYADLFLAAKNLSDAILLSDILRVNT EITKAPLSASMTKRYDEHHQDLTLLKALVRQQLPEKYKEIFFDQSKNGYAGYIDGGASQ EEFYKFTKPILEKMDGTEELLVKLNREDLLRKQRTFDNGSTPTTQTHLGELÍJAILRRQEDFY PFLKDNREKIEKILTFRIPYYVGPLARGNSRFAWMTRKSEETITPWNFEEVVDKGASAQS FIERMTNFDK.NLPNEKVLPKHSLLYEYFTVYNELTKVKYVTEGMRKPAFLSGEQKKAIV DLLFKTNRKVTVKQLKEDYFKKIECFDSVEISGVEDRFNASLGTYHDLLKIIKDKDFLDN EENEDILEDTVLTLTLFEDREMIEERLKTYAHLFDDKVMKQLKRRRYTGWGRLSRKLIN GIRDKQSGKTILDFLKSDGFANRJMFMQLIHDDSLTFKEDIQKAQVSGQGDSLHEHIANLA GSPAIKKGILQTVKVVDELVKVMGRHKPENIVIEMARENQTTQKGQKNSRERMKRIEEG
1KELGSQILKEHPVENTQLQNEKLYLYYLQNGRDMYVDQELDINRLSDYDVDHIVPQSF LKDDSIDNKVLTRSDKNRGKSDNVPSEEVVKKMKNYWRQLLNAKLITQRKFDNLTKAE RGGLS ELDKAGFIKRQ LVETRQITKHV AQILDSRMNTKYDENDKLIRE VKVITLKSKLV S DFRKDFQFYKVREINNYHHAHDAYLNAWGTALIKKYPKLESEFVYGDYKVYDVRKMI AKSEQEIGKATAKYFFY SNIMNFFKTEITLANGEIRKRPLIETN GETGEIVWDKGRDFATV RKVLSMPQVNIVKKTEVQTGGFSKES1LPKRNSDKL1ARKKDWDPKKYGGFDSPTVAYS VLWAKVEKGKSKKLKSYKELLGITIMERSSFEKNPIDFLEAKGYKEVKKDLIIKLPKYS LFELENGRKRMLASAGELQfCGNELALPSKYVNFLYLASHYEKLKGSPEDNEQRQLFVE QHKHYLDEIIEQISEFSKRVILADANLDKVLSAYNKHRDKPIREQAENHHLFTLTNLGAP AAFKYFDTTIDRKRYTSTKEVLDATLIHQSITGLYETRIDLSQLGGDKRPAATKKAGQAK K K K
hEMX1-Plantilla RH-Hindll-Nhel:
GAATGCTGCCCTCAGACCCGCTTCCTCCCTGTCCTTGTCTGTCCAAGGAGA
ATGAGGTCTCACTGGTGGATTTCGGACTACCCTGAGGAGCTGGCACCTGAGGGACA AGGCCCCCCACCTGCCCAGCTCCAGCCTCTGATGAGGGGTGGGAGAGAGCTACATG AGGTTGCTAAGAAAGCCTCCCCTGAAGGAGACCACACAGTGTGTGAGGTTGGAGTC TCTAGCAGCGGGTTCTGTGCCCCCAGGGATAGTCTGGCTGTCCAGGCACTGCTCTTG ATATAAACACCACCTCCTAGTTATGAAACCATGCCCATTCTGCCTCTCTGTATGGAA AAGAGC ATGGGGCT GGCCCGT GGGGTGGT GT CC ACTTT AGGCCCTGTGGGAG AT CA TGGGAACCCACGCAGTGGGTCATAGGCTCTCTCATTTACTACTCACATCCACTCTGT
GAAGAAGCGATTATGATCTCTCCTCTAGAAACTCGTAGAGTCCCATGTCTGCCGGCT TCCAGAGCCTGCACTCCTCC’ACCTTGGCTTGGCTTTGCTGGGGCTAGAGGAGCTAGG ATGCACAGCAGCTCTGTGACCCTTTGTTTGAGAGGAACAGGAAAACCACCCTTCTCT CTGGCCCACTGTGTCCTCTTCCTGCCCTGCCATCCCCTTCTGTGAATGTTAGACCCAT GGGAGCAGCTGGTCAGAGGGGACCCCGGCCTGGGGCCCCTAACCCTATGTAGCCTC AGTCTTCCCATCAGGCTCTCAGCTCAGCCTGAGTGTTGAGGCCCCAGTGGCTGCTCT GGGGGCCTCCTGAGTTTCTCATCTGTGCCCCTCCCTCCCTGGCCCAGGTGAAGGTGT GGTTCCAGAACCGGAGGACAAAGTACAAACGGCAGAAGCTGGAGGAGGAAGGGCC TGAGTCCGAGCAGAAGAAGAAGGGCTCCCATCACATCAACCGGTGGCGCATTGCCA CGAAGCAGGCCAATGGGGAGGACATCGATGTCACCTCCAATGACaagcttgctagcGGTGG GCAACCACAAACCCACGAGGGCAGAGTGCTGCTTGCTGCTGGCCAGGCCCCTGCGT GGGCCCAAGCTGGACTCTGGCCACTCCCTGGCCAGGCTTTGGGGAGGCCTGGAGTC ATGGCCCCACAGGGCTTGAAGCCCGGGGCCGCCATTGACAGAGGGACAAGCAATGG GCT GGCT GAGGCCT G GG ACC ACTTGGCCTTCTC CT CGGAGAGCCTGCCT GCCTGGGC GGGCCCGCCCGCCACCGCAGCCTCCCAGCTGCTCTCCGTGTCTCCAATCTCCCTTTTG TTTTGATGCATTTCTGTTTTAATTTATTTTCCAGGCACCACTGTAGTTTAGTGATCCCC AGTGTCCCCCTTCCCTATGGGAATAATAAAAGTCTCTCTCTTAATGACACGGGCATC CAGCTCCAGCCCCAGAGCCTGGGGTGGTAGATTCCGGCTCTGAGGGCCAGTGGGGG CTGGTAGAGCAAACGCGTTCAGGGCCTGGGAGCCTGGGGTGGGGTACTGGTGGAGG GGGTCAAGGGTAATTCATTAACTCCTCTCTTTTGTTGGGGGACCCTGGTCTCTACCTC CAGCTCCACAGCAGGAGAAACAGGCTAGACATAGGGAAGGGCCATCCTGTATCTTG AGGGAGGACAGGCCCAGGTCTTTCTTAACGTATTGAGAGGTGGGAATCAGGCCCAG GTAGTTCAATGGGAGAGGGAGAGTGCTTCCCTCTGCCTAGAGACTCTGGTGGCTTCT CCAGTTGAGGAGAAACCAGAGGAAAGGGGAGGATTGGGGTCTGGGGGAGGGAACA CCATTCACAAAGGCTGACGGTTCCAGTCCGAAGTCGTGGGCCCACCAGGATGCTCA CCTGTCCTTGGAGAACCGCTGGGCAGGTTGAGACTGCAGAGACAGGGCTTAAGGCT GAGCCTGCAACCAGTCCCCAGTGACTCAGGGCCTCCTCAGCCCAAGAAAGAGCAAC GT GCC AGGGCCCGCT G A GCTCTTGT GTT C ACCTG NLS-StCsn1-NLS:
MKRPAATKKAGQAKKKKSDLVLGLDIGIGSVGVGILNKVTGEI1HKNSRIFPA
AQAENNLVRRTNRQGRRLARRKKHRRVRLNRLFEESGLITDFTKISTNLNPYQLRVKGL
TDELSNEELFIALKNMVKHRGISYLDDASDDGNSSVGDYAQIVKENSKQLETKTPGQIQL ERYQTYGQLRGDFTVEKDGKKHRLINVFPTSAYRSEALR1LQTQQEFNPQITDEFINRYL EILTGKRKYYHGPGNEKSRTDYGRYRTSGETLDNIFGIL1GKCTFYPDEFRAAKASYTAQ EFNLLNDLNNLTVPTETKKLSKEQKNQIINYVKNEKAMGPAKLFKYÍAKLLSCDVADIK GYRIDKSGKAE1HTFEAYRKMKTLETLD1EQMDRETLDKLAYVLTLNTEREGIQEALEHE FADGSFSQKQVDELVQFRKANSSIFGKGWHNFSVKLMMELIPELYETSEEQMT1LTRLG KQKTTSSSNKTKYIDEKLLTEEIYNPVVAKSVRQAIKIVNAAIKEYGDFDNIVIEMARETN EDDEKKA1QK1QKANKDEKDAAMLKAANQYNGKAELPHSVFHGHRQLATKIRLWHQQ GERCL YT GKTTSIEIDLINN SN QFEVDHILPLSITFDD SLANKYLVY AT AN QEKGQRTP Y Q ALDSMDDAWSFRELKAFVRESKTLSNKKKEYLLTEED1SKFDVRKKFIERNLVDTRYAS RVVLNALQEHFRAHKIDTfCVSVVRGQFTSQLRRHWGIEKTRDTYHHHAVDALIIAASSQ LNLWKKQKNTLVSYSEDQLLDIETGELISDDEYKESVFKAPYQHFVDTLKSKEFEDSILF S Y Q VDSKFNRK1SDATIY ATRQ AKV GKDKADETYVLGKJKDIYTQDGYDAFMK1YKKD KSKFLMYRHDPQTFEKVIEPTLENYPNKQTNEKGKEVPCNPFLKYKEEHGYTRKYSKKGN GPEIKSLKYYDSKLGNHIDTTPKDSNNKVVLQSVSPWRADVYFNKTTGKYETLGLKYAD LQFEKGTGTYKISQEKYNDIKKKEGVDSDSEFKFTLYKNDLLLVKDTETKEQQLFRFLSR TMPKQKHYVELKPYDKQRFEGGEAL1RVLGNVANSGQCKKGLGKSNISIYKVRTDVLG N QHIIKN EG DRPKL DF KRP AATKKAGQ A KK RK
U6-St_ARNtracr(7-97):
GAGGGCCT ATTTCCCAT GATTCCTTCAT ATTTGCAT AT ACGATAC AAGGCT GTT AG AG AG AT AATT GG AATT AATTT G ACT GT AAAC AC AAAG AT ATT AGT AC AAAA T ACGT G ACGT AG AAAGT AAT AATTT CTTGGGTAGTTTGC AGTTTT AAAATTAT GTTTT A A A ATGG ACT AT C AT ATGCTT ACCGT A ACTTG A A AGT ATTTCG ATTTCTT GGCTTT AT AT AT CTT GTGGA A AGGACG AA AC ACCGTT ACTT A A ATCTTGC AGA AGCT ACAAAGA TAAGGCTTCATGCCGAAATCAACACCCTGTCATTTTATGGCAGGGTGTTTTCGTTATT TAA
U6-RD-espaciador-RD (SF370 S. pyogenes)

(subrayado minúsculas = repetición directa; N = secuencia guía; negrita = terminador)
ARN quimérico que contiene 48 ARNtracr (SF370 S. pyogenes)
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattggaattaatttgactgtaaa cacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgc ttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccNNNNNNNNNNNNNNNNNN NN gttttaga acta gaaatagcaagttaaaataaggctagtcc gTTTTTTT
(N = secuencia guía; primer subrayado = secuencia de emparejamiento traer; segundo subrayado = secuencia traer; negrita = terminador)
ARN quimérico que contiene 54 ARNtracr (SF370 S. pyogenes)
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattggaattaatttgactgtaaa cacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgc ttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccNNNNNNNNNNÍNNNNNNNN N N gttttagagctagaaatagcaagttaaaataaggctagtccgttatcaTTTTTTTT
(N = secuencia guía; primer subrayado = secuencia de emparejamiento traer; segundo subrayado = secuencia traer; negrita = terminador)
ARN quimérico que contiene 67 ARNtracr (SF370 S. pyogenes)

(N = secuencia guía; primer subrayado = secuencia de emparejamiento tracr; segundo subrayado = secuencia tracr; negrita = terminador)
ARN quimérico que contiene 85 ARNtracr (SF370 S. pyogenes)
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattggaattaatttgactgtaaa
ttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccNNNNNNNNNNNNNNNNNN NNgttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcTTTTTTT (N = secuencia guía; primer subrayado = secuencia de emparejamiento tracr; segundo subrayado = secuencia tracr; negrita = terminador)
CBh-NLS-SpCas9-NLS CGTTACATAACTTACGGTAAATGGCCCGCCTGGCTGACCGCCCAACGACC CCCGCCCATTGACGTCAATAATGACGTATGTTCCCATAGTAACGCCAATAGGGACTT
TCCATTGACGTCAATGGGTGGAGTATTTACGGTAAACTGCCCACTTGGCAGTACATC AAGTGTATCATATGCCAAGTACGCCCCCTATTGACGTCAATGACGGTAAATGGCCCG CCTGGCATTATGCCCAGTACATGACCTTATGGGACTTTCCTACTTGGCAGTACATCTA CGTATTAGTCATCGCTATTACCATGGTCGAGGTGAGCCCCACGTTCTGCTTCACTCTC CCCATCTCCCCCCCCTCCCCACCCCCAATTTTGTATTTATTTATTTTTTAATTATTTTG TGCAGCGATGGGGGCGGGGGGGGGGGGGGGGCGCGCGCCAGGCGGGGCGGGGCGG GGCGAGGGGCGGGGCGGGGCGAGGCGGAGAGGTGCGGCGGCAGCCAATCAGAGCG GCGCGCTCCGAAAGTTTCCTTTTATGGCGAGGCGGCGGCGGCGGCGGCCCTATAAA AAGCGAAGCGCGCGGCGGGCGGGAGTCGCTGCGACGCTGCCTTCGCCCCGTGCCCC GCTCCGCCGCCGCCTCGCGCCGCCCGCCCCGGCTCTGACTGACCGCGTTACTCCCAC AGGTGAGCGGGCGGGACGGCCCTTCTCCTCCGGGCTGTAATTAGCTGAGCAAGAGG TAAGGGTTTAAGGGATGGTTGGTTGGTGGGGTATTAATGTTTAATTACCTGGAGCAC CTGCCTGAAATCACTmTTTCAGGTTGGaccegteccaccATGGACTATAAGGACCACGA CGGAGACTACAAGGATCATGATATTGATTACAAAGACGATGACGATAAGATGGCCC CAAAGAAGA\GCGGAAGGTCGGTATCCACGGAGTCCCAGCAG( CG\CAAGAAGTA CAGCATCGGCCTGGACATCGGCACCAACTCTGTGGGCTGGGCCGTGATCACCGACG AGTACAAGGTGCCCAGCAAGAAATTCAAGGTGCTGGGCAACACCGACCGGCACAGC ATCAAGAAGAACCTGATCGGAGCCCTGCTGTTCGACAGCGGCGAAACAGCCGAGGC CACCCGGCTGAAGAGAACCGCCAGAAGAAGATACACCAGACGGAAGAACCGGATC TGCTATCTGCAAGAGATCTTCAGCAACGAGATGGCCAAGGTGGACGACAGCTTCTTC CACAGACTGGAAGAGTCCTTCCTGGTGGAAGAGGATAAGAAGCACGAGCGGCACCC CATCTTCGGCAACATCGTGGACGAGGTGGCCTACCACGAGAAGTACCCCACCATCT ACCACCTGAGAAAGAAACTGGTGGACAGCACCGACAAGGCCGACCTGCGGCTGATC TATCTGGCCCTGGCCCACATGATCAAGTTCCGGGGCCACTTCCTGATCGAGGGCGAC CTGAACCCCGACAACAGCGACGTGGACAAGCTGTTCATCCAGCTGGTGCAGACCTA CAACCAGCTGTTCGAGGAAAACCCCATCAACGCCAGCGGCGTGGACGCCAAGGCCA TCCTGTCTGCCAGACTGAGCAAGAGCAGACGGCTGGAAAATCTGATCGCCCAGCTG CCCGGCGAGAAGMGMTCGCriGTTCGGCMCCTGATTCCCCTGAGCCTGGGCCT GACCCCCAACTTCAAGAGCAACTTCGACCTGGCCGAGGATGCCAAACTGCAGCTGA GCAAGGACACCTACGACGACGACCTGGACAACCTGCTGGCCCAGATCGGCGACCAG TACGCCGACCTGTTTCTGGCCGCCAAGAACC'TGTCCGACGCCATCCTGCTGAGCGAC
ATCCTGAGAGTGAACACCGAGATCACCAAGGCCCCCCTGAGCGCCTCTATGATCAA GAGATACGACGAGCACCACCAGGACCTGACCCTGCTGAAAGCTCTCGTGCGGCAGC AGCTGCCTGAGAAGTACAAAGAGATTTTCTTCGACCAGAGCAAGAACGGCT ACGCC GGC TACATTGACGGCGGAGCCAGCCAGGAAGAGTTCTACAAGTTCATCAAGCCCAT CCTGGAAAAGATGGACGGCACCGAGGAACTGCTCGTGAAGCTGAACAGAGAGGAC CTGCTGCGGAAGCAGCGGACCTICGACAACGGCAGCATCCCCCACCAGATCCACCT GGGAGAGCTGCACGCCATTCTGCGGCGGCAGGAAGATTTTTACCCATTCCTGAAGG ACAACCGGGAAAAGATCGAGAAGATCCTGACCTTCCGCATCCCCTACTACGTGGGC C CTCTGGC CAGGGG AAAC AGCAG ATTCGC CT GG AT G ACO AG AAAG AGC G AGGAAA CCATCACCCCCTGGAACTTCGAGGAAGTGGTGGACAAGGGCGCTTCCGCCCAGAGC TTCATCGAGCGGATGACCAACTTCGATAAGAACCTGCCCAACGAGAAGGTGCTGCC CAAGCACAGCCTGCTGTACGAGTACTTCACCGTGTATAACGAGCTGACCAAAGTGA AATACGTGACCGAGGGAATGAGAAAGCCCGCCTTCCTGAGCGGCGAGCAGAAAAA GGCCATCGTGGACCTGCTGTTCAAGACCAACCGGA AAGTGACCGTGAAGCAGCTGA AAGAGGACT ACTTCAAGAAAATCGAGTGCTTCGACTCCGTGGAAATCTCCGGCGTG GAAGATCGGTTCAACGCCTCCCTGGGCACATACCACGATCTGCTGAAAATTATCAAG GACAAGGACTTCCTGGACAATGAGGAAAACGAGGACATTCTGGAAGATATCGTGCT GACCCTGACACTGTTTGAGGACAGAGAGATGATCGAGGAACGGCTGAAAACCTATG CCCACCTGTTCGACGACAAAGTGATGAAGCAGCTGAAGCGGCGGAGATACACCGGC TGGGGCAGGCTGAGCCGGAAGCTGATCAACGGCATCCGGGACAAGCAGTCCGGCAA GACAATCCTGGATTTCCTGAAGTCCGACGGCTTCGCCAACAGAAACTTCATGCAGCT GATCCACGACGACAGCCTGACCTTTAAAGAGGACATCCAGAAAGCCCAGGTGTCCG GCCAGGGCGATAGCCTGCACGAGCACATTGCCAATCTGGCCGGCAGCCCCGCCATT AAGAAGGGCATCCTGCAGACAGTGAAGGTGGTGGACGAGCTCGTGAAAGTGATGGG CCGGCACAAGCCCGAGAACATCGTGATCGAAATGGCCAGAGAGAACCAGACCACCC AGAAGGGACAGAAGAACAGCCGCGAGAGAATGAAGCGGATCGAAGAGGGCATCAA AGAGCTGGGCAGCCAGATCCTGAAAGAACACCCCGTGGAAAACACCCAGCTGCAGA ACGAGAAGCTGTACCTGTACTACCTGCAGAATGGGCGGGATATGTACGTGGACCAG GAACTGGACATCAACCGGCTGTCCGACTACGATGTGGACCATATCGTGCCTCAGAG CTTTCTGAAGGACGACTCCATCGACAACAAGGTGCTGACCAGAAGCGACAAGAACC GGGGCAAGAGCGACAACGTGCCCTCCGAAGAGGTCGTGAAGAAGATGAAGAACTA
CTGGCGGCAGCTGCTGAACGCCAAGCTGATTACCCAGAGAAAGTTCGACAATCTGA
CCAAGGCCGAGAGAGGCGGCCTGAGCGAACTGGATAAGGCCGGCTTCATCAAGAG
ACAGCTGGTGGAAACCCGGCAGATCACAAAGCACGTGGCACAGATCCTGGACTCCC
GGATGAACACTAAGTACGACGAGAATGACAAGCTGATCCGGGAAGTGAAAGTGATC
ACCCTGAAGTCCAAGCTGGTGTCCGATTTCCGGAAGGATTTCCAGTTTTACAAAGTG
CGCGAGATCAACAACT ACCACC ACGCCCACGACGCCT ACCTGAACGCCGTCGTGGG
AACCGCCCTGATCAAAAAGTACCCTAAGCTGGAAAGCGAGTTCGTGTACGGCGACT ACAAGGTGTACGACGTGCGGAAGATGATCGCCAAGAGCGAGCAGGAAATCGGCAA GGCTACCGCC AAGT ACTTCTTCTAC AGC AACAT CAT G AACTTTTT CAAGACCGAGAT TACCCTGGCCAACGGCGAGATCCGGAAGCGGCCTCTGATCGAGACAAACGGCGAAA
CCGGGGAGATCGTGTGGGATAAGGGCCGGGATTTTGCCACCGTGCGGAAAGTGCTG
AGCATGCCCCAAGTGAATATCGTGAAAAAGACCGAGGTGCAGACAGGCGGCTTCAG
CAAAGAGTCTATCCTGCCCAAGAGGAACAGCGATAAGCTGATCGCCAGAAAGAAGG
ACTGGGACCCTAAGAAGTACGGCGGCTTCGACAGCCCCACCGTGGCCTATTCTGTGC
T GGTGGT GGCC AAAGTGGAAAAGGGC AAGTCC AAGAAACT GAAGAGT GT GAAAG A GCTGCTGGGGATCACCATCATGGAAAGAAGCAGCTTCGAGAAGAATCCCATCGACT TTCTGGAAGCCAAGGGCTACAAAGAAGTGAAAAAGGACCTGATCATCAAGCTGCCT AAGTACTCCCTGTTCGAGCTGGAAAACGGCCGGAAGAGAATGCTGGCCTCTGCCGG
CGAACTGCAGAAGGGAAACGAACTGGCCCTGCCCTCCAAATATGTGAACTTCCTGT
ACCTGGCCAGCCACTATGAGAAGCTGAAGGGCTCCCCCGAGGATAATGAGCAGAAA
CAGCTGTTTGTGGAACAGCACAAGCACTACCTGGACGAGATCATCGAGCAGATCAG
CGAGTTCTCCAAGAGAGTGATCCTGGCCGACGCTAATCTGGACAAAGTGCTGTCCGC
CTACAACAAGCACCGGGATAAGCCCATCAGAGAGCAGGCCGAGAATATCATCCACC
TGTTTACCCTGACCAATCTGGGAGCCCCTGCCGCCTTCAAGTACTTTGACACCACCA TCGACCGGAAGAGGTACACCAGCACCAAAGAGGTGCTGGACGCCACCCTGATCCAC
CAGAGCATCACCGGCCTGTACGAGACACGGATCGACCTGTCTCAGCTGGGAGGCGA
CTTTCTTTTTCTTAGCTTGACCAGCTTTCTTAGTAGCAGCAGGACGCTTTAA
(subrayado = NLS-hSpCas9-NLS)
ARN quimérico de ejemplo para Cas9 de CRISPR1 LMD-9 de S. thermophilus (con PAM de NNAGAAW)
NNNNNNNNNNNNNNNNNNNNgtttttgtactctcaagatttaGAAAtaaatcttgcagaagctacaa agataaggcttcatgccgaaatcaacaccctgtcattttatggcagggtgttttcüttatttaaTTTTTT
(N = secuencia guía; primer subrayado = secuencia de emparejamiento traer; segundo subrayado = secuencia traer; negrita = terminador)
ARN quimérico de ejemplo para Cas9 de CRISPR1 LMD-9 de S. thermophilus (con PAM de NNAGAAW)
J^MNMNNNlSnNTNTNNNNNNNNNNgtttttgtactctcaGAAAtgcagaagctacaaagataaggcttca tgccgaaatcaacaccctgtcattttatggcagggtgttttcgttatttaaTTTTTT
(N = secuencia guía; primer subrayado = secuencia de emparejamiento traer; segundo subrayado = secuencia traer; negrita = terminador)
ARN quimérico de ejemplo para Cas9 de CRISPR1 LMD-9 de S. thermophilus (con PAM de NNAGAAW)
NNNNNNNNNNNNNNNNNNNNgtttttgtactctcaGAAAtgcagaagctacaaagataaggcttca tgccgaaatcaacaccctgtcattttatggcagggtgtTTTTTT
(N = secuencia guía; primer subrayado = secuencia de emparejamiento tracr; segundo subrayado = secuencia tracr; negrita = terminador)
ARN quimérico de ejemplo para Cas9 de CRISPR1 LMD-9 de S. thermophilus (con PAM de NNAGAAW)
NNNNNNNNNNNNNNNNNNNNgttattgtactctcaagatttaGAAAtaaatcttgcagaagctacaa agataaggcttcatgccgaaatcaacaccctgtcattttatggcagggtgttttcgttatttaaTTTTTT
(N = secuencia guía; primer subrayado = secuencia de emparejamiento tracr; segundo subrayado = secuencia tracr; negrita = terminador)
ARN quimérico de ejemplo para Cas9 de CRISPR1 LMD-9 de S. thermophilus (con PAM de NNAGAAW)
NNNNNNNNNNNNNNNNNNNNgttattgtactctcaGAAAtgcagaagctacaaagataaggcttca tgccgaaatcaacaccctgtcattttatggcagggtgttttcgttatttaaTTTTTT
(N = secuencia guía; primer subrayado = secuencia de emparejamiento tracr; segundo subrayado = secuencia tracr; negrita = terminador)
ARN quimérico de ejemplo para Cas9 de CRISPR1 LMD-9 de S. thermophilus (con PAM de NNAGAAW)
NNNNISTNNNNNNNNNNNNNNNgttattgtactctcaGAAAtgcagaagctacaaagataaggcttca tgccgaaatcaacaccctgtcattttatggcagggtgtTTTTTT
(N = secuencia guía; primer subrayado = secuencia de emparejamiento tracr; segundo subrayado = secuencia tracr; negrita = terminador)
ARN quimérico de ejemplo para Cas9 CRISPR1 LMD-9 de S. thermophilus (con PAM de NNAGAAW)

(N = secuencia guía; primer subrayado = secuencia de emparejamiento tracr; segundo subrayado = secuencia tracr; negrita = terminador)
ARN quimérico de ejemplo para Cas9 CRISPR1 LMD-9 de S. thermophilus (con PAM de NNAGAAW)
NNNNNNNNNNNNNNNNNNNNgttattgtactctcaGAAAtgcagaagctacaatgataaggcttca tgccgaaatcaacaccctgtcattttatggcagggtgttttcgttatttaaTTTTTT
(N = secuencia guía; primer subrayado = secuencia de emparejamiento tracr; segundo subrayado = secuencia tracr; negrita = terminador)
ARN quimérico de ejemplo para Cas9 CRISPR1 LMD-9 de S. thermophilus (con PAM de NNAGAAW)
^^ >^^^^^MNN7^^^^^MNNNNNNNNNgttattgtactctcaGAAAtgcagaagctacaatgataaggcttca tgccgaaatcaacaccctgtcattttatggcagggtgtTTTTTT
(N = secuencia guía; primer subrayado = secuencia de emparejamiento traer; segundo subrayado = secuencia traer; negrita = terminador)
ARN quimérico de ejemplo para Cas9 CRISPR3 LMD-9 de S. thermophilus (con PAM de NGGNG)
NNNNNNNNNNNNNNNNNNNNgttttagagctgtgGAAAcacagcgagttaaaataaggcttagtc cgtactcaacttgaaaaggtggcaccgattcggtgtTTTTTT
(N = secuencia guía; primer subrayado = secuencia de emparejamiento traer; segundo subrayado = secuencia traer; negrita = terminador)
Versión codón-optimizada de Cas9 de sitio CRISPR3 LMD-9 de S. thermophilus (con una NLS en ambos extremos 5' y 3')
ATGAAAAGGCCGGCGGCCACGAAAAAGGCCGGCCAGGCAAAAAAGAAA
AAGACCAAGCCCTACAGCATCGGCCTGGACATCGGCACCAATAGCGTGGGCTGGGC CGTGACCACCGACAACTACAAGGTGCCCAGCAAGAAAATGAAGGTGCTGGGCAACA CCTCCAAGAAGTACATCAAGAAAAACCTGCTGGGCGTGCTGCTGTTCGACAGCGGC ATTACAGCCGAGGGCAGACGGCTGAAGAGAACCGCCAGACGGCGGTACACCCGGC GGAGAAACAGAATCCTGTATCTGCAAGAGATCTTCAGCACCGAGATGGCTACCCTG GACGACGCCTTCTTCCAGCGGCTGGACGACAGCTTCCTGGTGCCCGACGACAAGCG GGACAGCAAGTACCCCATCTTCGGCAACCTGGTGGAAGAGAAGGCCTACCACGACG AGTTCCCCACCATCTACCACCTGAGAAAGTACCTGGCCGACAGCACCAAGAAGGCC GACCTGAGACTGGTGTATCTGGCCCTGGCCCACATGATCAAGTACCGGGGCCACTTC CTGATCGAGGGCGAGTTCAACAGCAAGAACAACGACATCCAGAAGAACTTCCAGGA
CTTCCTGGACACCTACAACGCCATCTTCGAGAGCGACCTGTCCCTGGAAAACAGCAA GC AGCT GGAAG AGATCGT GAAGG ACAAG AT CAGC AAGCT GG AAAAGAAGG ACCGC ATCCTGAAGCTGTTCCCCGGCGAGAAGAACAGCGGAATCTTCAGCGAGTTTCTGAA GCTGATCGTGGGCAACCAGGCCGACTTCAGAAAGTGCTTCAACCTGGACGAGAAAG CCAGCCTGCACTTCAGCAAAGAGAGCTACGACGAGGACCTGGAAACCCTGCTGGGA TATATCGGCGACGACTACAGCGACGTGTTCCTGAAGGCCAAGAAGCTGTACGACGC TATCCTGCTGAGCGGCTTCCTGACCGTGACCGACAACGAGACAGAGGCCCCACTGA GCAGCGCCATGATTAAGCGGTACAACGAGCACAAAGAGGATCTGGCTCTGCTGAAA GAGTACATCCGGAACATCAGCCTGAAAACCTACAATGAGGTGTTCAAGGACGACAC CAAGAACGGCTACGCCGGCTACATCGACGGCAAGACCAACCAGGAAGATTTCTATG TGTACCTGAAGAAGCTGCTGGCCGAGTTCGAGGGGGCCGACTACTTTCTGGAAAAA ATCGACCGCGAGGATTTCCTGCGGAAGCAGCGGACCTTCGACAACGGCAGCATCCC CTACCAGATCCATCTGCAGGAAATGCGGGCCATCCTGGACAAGCAGGCCAAGTTCT ACCCATTCCTGGCCAAGAACAAAGAGCGGATCGAGAAGATCCTGACCTTCCGCATC CCTTACTACGTGGGCCCCCTGGCCAGAGGCAACAGCGATTTTGCCTGGTCCATCCGG AAGCGCAATGAGAAGATCACCCCCTGGAACTTCGAGGACGTGATCGACAAAGAGTC CAGCGCCGAGGCCTTCATCAACCGGATGACCAGCTTCGACCTGTACCTGCCCGAGG AAAAGGTGCT GCCC A AGC AC AGC CT GCT GT ACGAG AC ATTC AAT GT GT AT A ACG AG CTGACCAAAGTGCGGTTTATCGCCGAGTCTATGCGGGACTACCAGTTCCTGGACTCC AAGCAGAAAAAGGACATCGTGCGGCTGTACTTCAAGGACAAGCGGAAAGTGACCG ATAAGGACATCATCGAGTACCTGCACGCCATCTACGGCTACGATGGCATCGAGCTG AAGGGCATCGAGAAGCAGTTCAACTCCAGCCTGAGCACATACCACGACCTGCTGAA CATTATCAACGACAAAGAATTTCTGGACGACTCCAGCAACGAGGCCATCATCGAAG AGATCATCCACACCCTGACCATCTTTGAGGACCGCGAGATGATCAAGCAGCGGCTG AGCAAGTTCGAGAACATCTTCGACAAGAGCGTGCTGAAAAAGCTGAGCAGACGGCA CTACACCGGCTGGGGCAAGCTGAGCGCCAAGCTGATCAACGGCATCCGGGACGAGA AGTCCGGCAACACAATCCTGGACTACCTGATCGACGACGGCATCAGCAACCGGAAC TTCATGCAGCTGATCCACGACGACGCCCTGAGCTTCAAGAAGAAGATCCAGAAGGC CCAGATCATCGGGGACGAGGACAAGGGCAACATCAAAGAAGTCGTGAAGTCCCTGC CCGGCAGCCCCGCCATCAAGAAGGGAATCCTGCAGAGCATCAAGATCGTGGACGAG CTCGTGAAAGTGATGGGCGGCAGAAAGCCCGAGAGCATCGTGGTGGAAATGGCTAG
AGAGAACCAGTACACCAATCAGGGCAAGAGCAACAGCCAGCAGAGACTGAAGAGA CTGGAAAAGT CCCT G AAAG AGCTGGGC AGC AAG ATT CT G AAA G AG AAT ATCCCTGC CAAGCTGTCCAAGATCGACAACAACGCCCTGCAGAACGACCGGCTGTACCTGTACT ACCTGCAGAATGGCAAGGACATGTATACAGGCGACGACCTGGATATCGACCGCCTG AGCAACTACGACATCGACCATATTATCCCCCAGGCCTTCCTGAAAGACAACAGCATT GACAACAAAGTGCTGGTGTCCTCCGCCAGCAACCGCGGCAAGTCCGATGATGTGCC CAGCCTGGAAGTCGTGAAAAAGAGAAAGACCTTCTGGTATCAGCTGCTGAAAAGCA AGCTGATT AGCC AGAGGAAGTTCG ACAACCT GACCAAGGCCGAGAG AG GCGGC CTG AGCCCTGAAGATAAGGCCGGCTTCATCCAGAGACAGCTGGTGGAAACCCGGCAGAT CACCAAGCACGTGGCCAGACTGCTGGATGAGAAGTTTAACAACAAGAAGGACGAGA ACAACCGGGCCGTGCGGACCGTGAAGATCATCACCCTGAAGTCCACCCTGGTGTCC CAGTTCCGGAAGGACTTCGAGCTGTATAAAGTGCGCGAGATCAATGACTTTCACCAC GCCCACGACGCCTACCTGAATGCCGTGGTGGCTTCCGCCCTGCTGAAGAAGTACCCT AAGCTGGAACCCGAGTTCGTGTACGGCGACTACCCCAAGTACAACTCCTTCAGAGA GCGGAAGTCCGCCACCGAGAAGGTGTACTTCTACTCCAACATCATGAATATCTTTAA GAAGTCCATCTCCCTGGCCGATGGCAGAGTGATCGAGCGGCCCCTGATCGAAGTGA ACGAAGAGACAGGCGAGAGCGTGTGGAACAAAGAAAGCGACCTGGCCACCGTGCG GCGGGTGCTGAGTTATCCTCAAGTGAATGTCGTGAAGAAGGTGGAAGAACAGAACC ACGGCCTGGATCGGGGCAAGCCCAAGGGCCTGTTCAACGCCAACCTGTCCAGCAAG C CT AAGCCCAACT CCAACG AG AATCTCGT GGGGGCCAAAG AGT ACCTGGACCCTAA GAAGTACGGCGGATACGCCGGCATCTCCAATAGCTTCACCGTGCTCGTGAAGGGCA CAATCGAGAAGGGCGCTAAGAAAAAGATCACAAACGTGCTGGAATTTCAGGGGATC TCTATCCTGGACCGGATCAACTACCGGAAGGATAAGCTGAACTTTCTGCTGGAAAAA GGCTACAAGGACATTGAGCTGATTATCGAGCTGCCTAAGTACTCCCTGTTCGAACTG AGCGACGGCTCCAGACGGATGCTGGCCTCCATCCTGTCCACCAACAACAAGCGGGG CGAGATCCACAAGGGAAACCAGATCTTCCTGAGCCAGAAATTTGTGAAACTGCTGT ACC ACGCC AAGCGG ATCT CC AAC ACC AT C AAT G AGAACCACCGGA AAT ACGT GG AA AACCACAAGAAAGAGTTTGAGGAACTGTTCTACTACATCCTGGAGTTCAACGAGAA CTATGTGGGAGCCAAGAAGAACGGCAAACTGCTGAACTCCGCCTTCCAGAGCTGGC AGAACCACAGCATCGACGAGCTGTGCAGCTCCTTCATCGGCCCTACCGGCAGCGAG CGGAAGGGACTGTTTGAGCTGACCTCCAGAGGCTCTGCCGCCGACTTTGAGTTCCTG
GGAGTGAAGATCCCCCGGTACAGAGACTACACCCCCTCTAGTCTGCTGAAGGACGC
CACCCTGATCCACCAGAGCGTGACCGGCCTGTACGAAACCCGGATCGACCTGGCTA
AGCTGGGCGAGGGAAAGCGTCCTGCTGCTACTAAGAAAGCTGGTCAAGCTAAGAAA
AAGAAATAA
Ejemplo 5: Edición guiada de ARN de genomas bacterianos usando sistemas CRISPR-Cas.
Los solicitantes usaron la endonucleasa Cas9 asociada a CRISPR para introducir mutaciones precisas en los genomas de Streptococcus pneumoniae y Escherichia coli. La propuesta se basa en escisión dirigida por Cas9 en el sitio fijado como objetivo para destruir células no mutadas y evitar la necesidad de marcadores seleccionables o sistemas contra la selección. La especificidad de Cas9 se volvió a programar cambiando la secuencia de ARN CRISPR (ARNcr) corto para hacer cambios de nucleótidos únicos y múltiples soportados sobre plantillas de edición. El uso simultáneo de dos ARNcr permitió la mutagénesis múltiplex. En S. pneumoniae, casi el 100% de las células que sobrevivió a la escisión de Cas9 contenía la mutación deseada y el 65% cuando se usó junto con recombinación en E. coli. Los solicitantes analizaron de manera exhaustiva los requerimientos de fijación como objetivo de Cas9 para definir el intervalo de secuencias fijables como objetivo y mostraron las estrategias para editar sitios que no requieren estos requerimientos, sugiriendo la versatilidad de esta técnica para lograr genomas bacterianos.
El entendimiento de la función génica depende de la posibilidad de modificar las secuencias de ADN dentro de la célula de un modo controlado. La mutagénesis específica del sitio en eucariotas se consigue por el uso de nucleasas específicas de la secuencia que activan la recombinación homóloga de un ADN de plantilla que contiene la mutación de interés. Las nucleasas con dedos de cinc (las ZFN, por sus siglas en inglés), las nucleasas efectoras de tipo activador de la transcripción (las TALEN, por sus siglas en inglés) y meganucleasas de direccionamiento específico pueden programarse para escindir genomas en posiciones específicas, pero estas propuestas requieren lograr nuevas enzimas para cada secuencia diana. En organismos procariotas, los métodos de mutagénesis introducen un marcador de selección en el sitio editado o requieren un procedimiento de dos etapas que incluya un sistema contra la selección. Más recientemente, las proteínas de recombinación de fagos se han usado para recombinación, una técnica que activa la recombinación homóloga de ADN lineal u oligonucleótidos. Sin embargo, debido a que no hay selección de mutaciones, la eficacia de la recombinación puede ser relativamente baja (0,1-10% para mutaciones puntuales hasta 10-5-10-6 para mayores modificaciones), requiriendo en muchos casos la detección sistemática de un gran número de colonias. Por lo tanto, aún se requieren nuevas tecnologías que sean a un precio asequible, fáciles de usar y eficaces para ingeniería genética de organismos tanto eucariotas como procariotas.
El trabajo reciente sobre el sistema inmunitario adaptativo de CRISPR (repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas) de procariotas ha conducido a la identificación de nucleasas cuya especificidad de secuencias se programa por ARN pequeños. Los sitios de CRISPR están constituidos por una serie de repeticiones separadas por secuencias “espadadoras” que igualan los genomas de los bacteriófagos y otros elementos genéticos móviles. La matriz repetición-espaciador se transcribe como un gran precursor y se trata dentro de secuencias de repetición para generar ARNcr pequeño que especifique las secuencias diana (también conocidas como protoespaciadores) escindidas por sistemas CRISPR. Es esencial para la escisión la presencia de una unidad de secuencia inmediatamente aguas abajo de la región fijada como objetivo, conocida como la unidad protoespaciadoraadyacente (PAM). Los genes asociados a CRISPR (cas) normalmente flanquean la matriz repetición-espaciador y codifican la maquinaria enzimática responsable de la biogénesis y fijación como objetivo de ARNcr. Cas9 es una endonucleasa de ADNdc que usa una guía de ARNcr para especificar el sitio de escisión. La carga de la guía de ARNcr sobre Cas9 tiene lugar durante el tratamiento del precursor de ARNcr y requiere un ARN complementario pequeño para el precursor, el ARNtracr y RNAsa III. A diferencia de edición de genomas con las ZFN o las TALEN, cambiar la especificidad fijada como objetivo de Cas9 no requiere lograr proteínas sino sólo el diseño de la guía de ARNcr corta.
Los solicitantes mostraron recientemente en S. pneumoniae que la introducción de un sistema CRISPR que fija como objetivo un sitio cromosómico conduce a la destrucción de las células transformadas. Se observó que los supervivientes ocasionales contenían mutaciones en la región fijada como objetivo, que sugiere que la actividad de la endonucleasa de ADNdc de Cas9 contra dianas endógenas podía ser usada para edición de genomas. Los solicitantes mostraron que las mutaciones sin marcador podían introducirse por la transformación de un fragmento de ADN de plantilla que se recombinarán en el genoma y eliminarán el reconocimiento de la diana de Cas9. Dirigir la especificidad de Cas9 con varios ARNcr diferentes permite la introducción de múltiples mutaciones al mismo tiempo. Los solicitantes también caracterizaron con detalle los requerimientos de la secuencia para fijar como objetivo Cas9 y mostrar que la propuesta puede combinarse con recombinación para edición de genomas en E. coli.
Resultados: Edición de genomas por escisión de Cas9 de un objetivo cromosómico.
La cepa crR6 de S. pneumoniae contiene un sistema CRISPR a base de Cas9 escinde una secuencia diana presente en el bacteriófago 98232.5. Esta diana se integró en el sitio cromosómico srtA de una segunda cepa el R682325. Se integró una secuencia diana modificada que contenía una mutación en la región PAM en el sitio srtA de una tercera cepa R63701, haciendo esta cepa “inmune” a escisión de CRISPR (Figura 28a). Los solicitantes transformaron células R68232,5 y R6370,1 con ADN genómico de células crR6, esperando que la transformación exitosa de las células de R682325 conduciría a escisión del sitio fijado como objetivo y la muerte celular. A diferencia de esta previsión, los solicitantes aislaron transformados de R682325, aunque con aproximadamente 10 veces menos eficacia que los transformados de R6370,1 (Figura 28b). El análisis genético de ocho transformados de R682325 (Figura 28) reveló que la gran mayoría son el producto de un caso de doble recombinación que elimina la toxicidad de la fijación como objetivo de Cas9 reemplazando el objetivo 98232,5 con el sitio srtA natural del genoma de crR6, que no contiene el protoespaciador requerido para reconocimiento de Cas9. Estos resultados eran evidencia de que la introducción concurrente de un sistema CRISPR que fija como objetivo un sitio genómico (la construcción fijadora de objetivo) junto con una plantilla para recombinación en el sitio fijado como objetivo (la plantilla de edición) condujo a edición de genoma fijada como objetivo (Figura 23a).
Para crear un sistema simplificado para edición de genomas, los solicitantes modificaron el sitio CRISPR en la cepa crR6 suprimiendo los genes casi, cas2 y csn2, que ha demostrado ser prescindible para fijar como objetivo CRISPR, proporcionando la cepa crR6M (Figura 28a). Esta cepa mantuvo las mismas propiedades de crR6 (Figura 28b). Para aumentar la eficacia de edición a base de Cas9 y demostrar que puede usarse un ADN de plantilla de elección para controlar la mutación introducida, los solicitantes cotransformaron células R682325 con productos PCR del gen srtA natural o el objetivo R63701 mutante, cualquiera de los cuales debería ser resistente a escisión por Cas9. Esto dio como resultado un aumento de 5 a 10 veces de la frecuencia de transformación comparado con ADN de crR6 genómico sólo (Figura 23b). La eficacia de la edición también aumentó sustancialmente, con 8/8 de los transformados ensayados conteniendo una copia de srtA natural y conteniendo 7/8 la mutación PAM presente en el objetivo R63701 (Figura 23b y Figura 29a). Tomados juntos, estos resultados mostraron el potencial de la edición de genomas asistida por Cas9.
Análisis de los requerimientos del objetivo de Cas9: Para introducir cambios específicos en el genoma, se debe usar una plantilla de edición que soporte mutaciones que anulen la escisión mediada por Cas9, evitándose de ese modo la muerte celular. Esto es fácil de conseguir cuando se busque la supresión del objetivo o su reemplazo por otra secuencia (inserción de genes). Cuando el objetivo es producir fusiones de genes o generar mutaciones de nucleótidos solos, la supresión de la actividad de la nucleasa Cas9 sólo será posible introduciendo mutaciones en la plantilla de edición que modifiquen la PAM o las secuencias del protoespaciador. Para determinar las restricciones de la edición mediada por CRISPR, los solicitantes realizaron un análisis exhaustivo de mutaciones de PAM y protoespaciador que anularan la fijación como objetivo de CRISPR.
Los estudios previos propusieron que Cas9 de S. pyogenes requiere una PAM de NGG inmediatamente aguas abajo del protoespaciador. Sin embargo, debido a que se ha descrito hasta ahora sólo un número muy limitado de mutaciones de inactivación de PAM, los solicitantes llevaron a cabo un análisis sistemático para encontrar todas las secuencias de 5 nucleótidos siguiendo al protoespaciador que elimina la escisión de CRISPR. Los solicitantes usaron oligonucleótidos aleatorizados para generar todas las posibles 1.024 secuencias de PAM en un producto PCR heterogéneo que se transformó en células crR6 o R6. Se esperó que se reconocieran las construcciones que soportaban las PAM funcionales y fueran destruidas por Cas9 en células crR6 pero no R6 (Figura 24a). Se mezclaron más de 2*105 colonias para extraer ADN para usar como plantilla para la multiplicación conjunta de todos los objetivos. Se secuenciaron a fondo los productos PCR y se encontró que contenían todas las 1.024 secuencias, con un cubrimiento que oscilaba de 5 a 42.472 lecturas (véase la sección "Analysis of deep sequencing data"). La funcionalidad de cada PAM se estimó por la proporción relativa de sus lecturas en la muestra de crR6 sobre la muestra de R6. El análisis de las primeras tres bases de la PAM, promediando sobre las dos últimas bases, demostró claramente que el patrón de NGG estaba infrarepresentado en transformados de crR6 (Figura 24b). Además, las siguientes dos bases no presentaron un efecto detectabla sobre la PAM de NGG (véase la sección "Analysis of deep sequencing data"), que demuestra que la secuencia NGGNN era suficiente para conceder actividad de Cas9. Se observó fijación como objetivo parcial para secuencias de PAM de NAG (Figura 24b). También el patrón de NNGGN inactivó parcialmente la fijación como objetivo de CRISPR (Tabla G), que indica que la unidad de NGG aún puede ser reconocida por Cas9 con eficacia reducida cuando se desplaza por 1 pb. Estos datos arrojaron luz sobre el mecanismo molecular del reconocimiento de objetivo de Cas9 y revelaron que las secuencias NGG (o CCN en la cadena complementaria) son suficientes para fijar como objetivo Cas9 y que las mutaciones de NGG a NAG o NNGGN en la plantilla de edición deberían evitarse. Debido a la alta frecuencia de estas secuencias de trinucleótidos (una vez cada 8 pb), esto significa que casi cualquier posición del genoma puede editarse. Por lo tanto, los solicitantes ensayaron diez objetivos elegidos de manera aleatoria soportando varias PAM y todas fueron encontradas funcionales (Figura 30).
Otra manera de alterar la escisión mediada por Cas9 es introducir mutaciones en la región protoespaciadora de la plantilla de edición. Se sabe que las mutaciones puntuales en la “secuencia simiente” (los 8 a 10 nucleótidos del protoespaciador inmediatamente adyacentes a la PAM) pueden anular la escisión por nucleasas CRISPR. Sin embargo, no se conoce la longitud de exacta de esta región y no está claro si las mutaciones para cualquier nucleótido en la simiente pueden anular el reconocimiento del objetivo de Cas9. Los solicitantes siguieron la misma propuesta de secuenciación profunda descrita anteriormente para aleatorizar la secuencia completa del protoespaciador implicada en los contactos de pares de bases con el ARNcr y determinar todas las secuencias que alteran la fijación como objetivo. Cada posición de los 20 nucleótidos de igualación (14) en el presente objetivo spcl en células R682325 (Figura 23a) fue aleatorizada y transformada en células crR6 y R6 (Figura 24a). Consistente con la presencia de una secuencia simiente, sólo las mutaciones en los 12 nucleótidos inmediatamente aguas arriba de la PAM anularon la escisión por Cas9 (Figura 24c). Sin embargo, diferentes mutaciones mostraron efectos notablemente diferentes. Las posiciones distales (de la PAM) de la simiente (12 a 7) toleraron la mayoría de las mutaciones y sólo una sustitución de base particular anuló la fijación como objetivo. Por el contrario, las mutaciones en cualquier nucleótido en las posiciones próximas (6 a 1, excepto 3) eliminaron la actividad de Cas9, aunque en diferentes niveles para cada sustitución particular. En la posición 3, sólo dos sustituciones afectaron a la actividad de CRISPR y con diferente fortaleza. Los solicitantes concluyeron que, aunque las mutaciones de la secuencia simiente pueden evitar la fijación como objetivo de CRISPR, hay restricciones teniendo en cuenta los cambios de nucleótidos que pueden realizarse en cada posición de la simiente. Además, estas restricciones lo más probablemente pueden variar para diferentes secuencias espaciadoras. Por lo tanto, los solicitantes creen que las mutaciones en la secuencia PAM, si es posible, deberían ser la estrategia de edición preferida. Alternativamente, se pueden introducir múltiples mutaciones en la secuencia simiente para evitar la actividad de la nucleasa Cas9.
Edición de genomas mediada por Cas9 en S. pneumonía: Para desarrollar un método rápido y eficaz para editar genomas fijados como objetivo, los solicitantes lograron la cepa crR6Rk, una cepa en la que se pueden introducir fácilmente espaciadores por PCR (Figura 33). Los solicitantes decidieron editar el gen de p-galactosidasa (bgaA) de S. pneumoníae, cuya actividad puede medirse fácilmente. Los solicitantes introdujeron sustituciones de alanina de aminoácidos en el sitio activo de esta enzima: mutaciones R481A (R^-A) y N563A, E564A (NE^-AA). Para ilustrar diferentes estrategias de edición, los solicitantes diseñaron mutaciones de tanto la secuencia PAM como la simiente del protoespaciador. En los dos casos, se usó la misma construcción fijadora de objetivo con un ARNcr complementario a una región del gen de p-galactosidasa que es adyacente a una secuencia de PAM TGG (CCA en la cadena complementaria, Figura 26). La plantilla de edi ción de R ^A creó un desajuste de tres nucleótidos en la secuencia simiente del protoespaciador (CGT a GCA, introduciendo también un sitio de restricción de BtgZI). En la plantilla de edición de N E ^ a A, los solicitantes introdujeron simultáneamente una mutac ión sinónima que creó una PAM inactiva (TGG a TTG) junto con mutaciones que son 218 nt aguas abajo de la región protoespaciadora (AAT GAA a GCT GCA, generando también un sitio de restricción de TseI). Esta última estrategia de edición demostró la posibilidad de usar una PAM remota para hacer mutaciones en sitios donde puede ser duro elegir un objetivo apropiado. Por ejemplo, aunque el genoma R6 de S. pneumoníae, que presenta un contenido del 39,7% de GC, contiene de promedio una unidad PAM cada 12 pb, algunas unidades PAM se separan por hasta 194 pb (Figura 33). Además, los solicitantes diseñaron una supresión dentro del marco de AbgaA de 6.664 pb. En los tres casos, la cotransformación de las plantillas de fijación como objetivo y de edición produjo 10 veces más células resistentes a kanamicina que la cotransformación con una plantilla de edición de control que contenía secuencias bgaA naturales (Figura 25b). Los solicitantes genotiparon 24 transformados (8 para cada experimento de edición) y encontraron que todos excepto uno incorporaron el cambio deseado (Figura 25c). La secuenciación de ADN también confirmó no sólo la presencia de las mutaciones introducidas sino también la ausencia de mutaciones secundarias en la región de objetivo (Figura 29b,c). Finalmente, los solicitantes midieron la actividad de la p-galactosidasa para confirmar que todas las células editadas mostraban el fenotipo esperado (Figura 25d).
La edición mediada por Cas9 también puede usarse para generar múltiples mutaciones para el estudio de rutas biológicas. Los solicitantes decidieron ilustrar esto para la ruta dependiente de sortasa que ancla las proteínas superficiales a la envoltura de las bacterias Gram-positivas. Los solicitantes introdujeron una supresión de sortasa por cotransformación de una construcción fijadora como objetivo resistente a cloranfenicol y una plantilla de edición de AsrtA (Figura 33a,b), seguido por una supresión de AbgaA usando una construcción fijadora de objetivo resistente a kanamicina que reemplaza la previa. En S. pneumoníae, la p-galactosidasa está ligada mediante enlaces covalentes a la pared celular mediante sortasa. Por lo tanto, la supresión de srtA da como resultado la liberación de la proteína superficial en el sobrenadante, mientras que la doble supresión no presenta actividad de la p-galactosidasa detectabla (Figura 34c). Dicha selección secuencial puede iterarse tantas veces como se requiera para generar múltiples mutaciones.
Estas dos mutaciones también se pueden introducir al mismo tiempo. Los solicitantes diseñaron una construcción fijadora de objetivo que contenía dos espaciadores, uno igualando srtA y el otro igualando bgaA y cotransformándola con las dos plantillas de edición al mismo tiempo (Figura 25e). Los análisis genéticos de transformados mostraron que la edición tenía lugar en 6/8 de casos (Figura 25f). En particular, los dos clones
restantes contenían cada uno una supresión de AsrtA o una de AbgaA, que sugiere la posibilidad de realizar mutagénesis combinatoria usando Cas9. Finalmente, para eliminar las secuencias CRISPR, los solicitantes introdujeron un plásmido que contenía el objetivo bgaA y un gen de resistencia a espectinomicina junto con ADN genómico de la cepa R6 natural. Los transformados resistentes a espectinomicina que conservan el plásmido eliminaron las secuencias CRISPR (Figura 34a,d).
Mecanismo y eficacia de edición: Para comprender los mecanismos subyacentes a la edición de genomas con Cas9, los solicitantes diseñaron un experimento en el que la eficacia de edición se midió independientemente de escisión de Cas9. Los solicitantes integraron el gen de resistencia a eritromicina ermAM en el sitio srtA e introdujeron un codón de terminación prematuro usando edición mediada por Cas9 (Figura 33). La cepa resultante (JEN53) contiene un alelo ermAM(interrupción) y es sensible a eritromicina. Esta cepa puede usarse para valorar la eficacia con la que se repara el gen ermAM midiendo la fracción de células que restablecen la resistencia a los antibióticos con o sin el uso de escisión de Cas9. Se transformó JEN53 con una plantilla de edición que restablece el alelo natural, junto con construcción CRISPR resistente a kanamicina que fija como objetivo el alelo ermAM(interrupción) (CRISPR::ermAM(interrupción)) o una construcción de control sin un espaciador (CRISPR::0) (Figura 26a,b). En ausencia de selección de kanamicina, la fracción de colonias editadas estuvo en el orden de 10-2 (ufc resistentes a eritromicina/ufc totales) (Figura 26c), que representa la frecuencia de referencia de recombinación sin selección mediada por Cas9 frente a células no editadas. Sin embargo, si se aplicaba selección de kanamicina y se cotransformaba la construcción CRISPR de control, la fracción de colonias editadas aumentó a aproximadamente 10-1 (ufc resistentes a kanamicina y eritromicina/ufc resistentes a kanamicina) (Figura 26c). Este resultado muestra que la selección para la recombinación del sitio CRISPR se coseleccionó para recombinación en el sitio ermAM independientemente de escisión Cas9 del genoma, que sugiere que una subpoblación de células es más propensa a transformación y/o recombinación. La transformación de la construcción CRISPR::ermAM(interrupción) seguido por selección de kanamicina dio como resultado un aumento de la fracción de células editadas, resistentes a eritromicina a 99% (Figura 26c). Para determinar si este aumento se produce por la destrucción de células no editadas, los solicitantes compararon las unidades formadoras de colonias (cfu) resistentes a kanamicina obtenidas después de cotransformación de células JEN53 con las construcciones CRISPR: :ermAM(interrupción) o CRISPR:: 0.
Los solicitantes contaron 5,3 veces menos colonias resistentes a kanamicina después de transformación de la construcción ermAM(interrupción) (2,5*104/4,7*103, Figura 35a), un resultado que sugiere que por supuesto fijar como objetivo un sitio cromosómico por Cas9 conduce a la destrucción de células no editadas. Finalmente, debido a que la introducción de roturas de ADNdc en el cromosoma bacteriano se sabe que provoca mecanismos de reparación que aumentan la tasa de recombinación del ADN dañado, los solicitantes investigaron si la escisión por Cas9 induce la recombinación de la plantilla de edición. Los solicitantes contaron 2,2 veces más colonias después de cotransformación con la construcción CRISPR::erm(interrupción) que con la construcción CRISPR::0 (Figura 26d), que indica que hubo una modesta inducción de recombinación. Tomados juntos, estos resultados mostraron que la coselección de células transformables, inducción de recombinación por escisión mediada por Cas9 y selección contra células no editadas, contribuyeron cada una a la alta eficacia de la edición de genomas en S. pneumoniae.
Como la escisión del genoma por Cas9 debería destruir las células no editadas, no se debería espera recuperar ninguna célula que recibiera la casete de Cas9 que contiene resistencia a kanamicina pero no la plantilla de edición. Sin embargo, en ausencia de la plantilla de edición, los solicitantes recuperaron muchas colonias resistentes a kanamicina después de transformación de la construcción CRISPR::ermAM(interrupción) (Figura 35a). Estas células que “escapan” a la muerte inducida por CRISPR produjeron un fon do que determinó un límite del método. Esta frecuencia del fondo puede calcularse como la relación de ufc de CRISPR::ermAM(interrupción)/CRISPR::0, 2,6*10-3 (7,1x10V2,7x104) en este experimento, que significa que si la frecuencia de recombinación de la plantilla de edición es menor que este valor, la selección de CRISPR puede no recuperar con eficacia los mutantes deseados por encima del fondo. Para entender el origen de estas células, los solicitantes genotiparon 8 colonias de fondo y encontraron que 7 contenían supresiones del espaciador de fijación como objetivo (Figura 35b) y una albergó una mutación presumiblemente inactivante en Cas9 (Figura 35c).
Edición de genomas con Cas9 en E. coli: La activación de fijación como objetivo de Cas9 por la integración cromosómica de un sistema CRISPR-Cas sólo es posible en organismos que sean altamente recombinogénicos. Para desarrollar un método más general que sea aplicable a otros microbios, los solicitantes decidieron realizar edición de genomas en E. coli usando un sistema CRISPR-Cas a base de plásmido. Se construyeron dos plásmidos: un plásmido pCas9 soportando el ARNtracr, Cas9 y una casete de resistencia a cloranfenicol (Figura 36) y un plásmido resistente a kanamicina de pCRISPR soportando la matriz de espaciadores de CRISPR. Para medir la eficacia de la edición independientemente de la selección de CRISPR, los solicitantes buscaron introducir una transversión de A a C en el gen rpsL que confiriera resistencia a estreptomicina. Los solicitantes construyeron un plásmido de pCRISPR::rpsL que albergaba un espaciador que guiaría la escisión de Cas9 del alelo rpsL natural, pero no el mutante (Figura 27b). El plásmido pCas9 fue introducido primero en MG1655 de E. coli y se cotransformó la cepa resultante con el plásmido pCRISPR::rpsL y W542, un oligonucleótido de edición que
contenía la mutación A a C. Sólo se recuperaron colonias resistentes a estreptomicina después de la transformación del plásmido pCRISPR::rpsL, que sugiere que la escisión de Cas9 induce la recombinación del oligonucleótido (Figura 37). Sin embargo, el número de colonias resistentes a estreptomicina fue dos órdenes de magnitud menor que el número de colonias resistentes a kanamicina, que son presumiblemente células que escapan a la escisión por Cas9. Por lo tanto, en estas condiciones, la escisión por Cas9 facilitó la introducción de la mutación, pero con una eficacia que no fue suficiente para seleccionar las células mutantes por encima del fondo de “escapistas”.
Para mejorar la eficacia de edición de genomas en E. coli, los solicitantes aplicaron su sistema CRISPR con recombinación, usando muerte celular inducida por Cas9 para seleccionar las mutaciones deseadas. Se introdujo el plásmido pCas9 en la cepa recombinante HME63 (31), que contenía las funciones Gam, Exo y Beta del fago □ -rojo. Se cotransformó la cepa resultante con el plásmido pCRISPR::rpsL (o un control de pCRISPR::0) y el oligonucleótido W542 (Figura 27a). La eficacia de recombinación fue 5,3*10-5, calculada como la fracción de células totales que llegaba a ser resistente a estreptomicina cuando se usaba el plásmido de control (Figura 27c). Por el contrario, la trasformación con el plásmido pCRISPR::rpsL aumentó el porcentaje de células mutantes a 65 ± 14 % (Figuras 27c y 29f). Los solicitantes observaron que el número de ufc se reducía por aproximadamente tres órdenes de magnitud después de la transformación del plásmido pCRISPR::rpsL que el plásmido de control (4,8*105/5,3*102, Figura 38a), que sugiere que la selección da como resultado muerte inducida por CRISPR de células no editadas. Para medir la tasa a la que se inactivó la escisión de Cas9, un parámetro importante del método de los solicitantes, los solicitantes transformaron células con pCRISPR::rpsL o el plásmido de control sin el oligonucleótido de edición W542 (Figura 38a). Este fondo de “escapistas” de CRISPR, medido como la relación de ufc de pCRISPR::rpsL/pCRISPR::0, fue 2,5*10-4 (1,2*102/4,8*105). Genotipar ocho de estos escapistas reveló que en todos los casos había una supresión del espaciador de fijación como objetivo (Figura 38b). Este fondo fue mayor que la eficacia de recombinación de la mutación rpsL, 5,3*10-5, que sugiere que para obtener el 65% de las células editadas, la escisión de Cas9 debe inducir recombinación de oligonucleótidos. Para confirmar esto, los solicitantes compararon el número de ufc resistentes a kanamicina y estreptomicina después de la transformación de pCRISPR::rpsL o pCRISPR::0 (Figura 27d). Como en el caso para S. pneumoniae, los solicitantes observaron una modesta inducción de recombinación, aproximadamente 6,7 veces (2,0*10-4/3,0*10-5). Tomados juntos, estos resultados indicaron que el sistema CRISPR proporcionaba un método para seleccionar mutaciones introducidas por recombinación.
Los solicitantes mostraron que los sistemas CRISPR-Cas pueden usarse para edición fijada como objetivo de genomas en bacterias por la cointroducción de una construcción de fijación como objetivo que destruye células naturales y una plantilla de edición que tanto eliminaba la escisión de CRISPR como introducía las mutaciones deseadas. Se pueden generar diferentes tipos de mutaciones (inserciones, supresiones o sustituciones de un solo nucleótido sin marca). Se pueden introducir múltiples mutaciones al mismo tiempo. La especificidad y versatilidad de la edición usando el sistema CRISPR se basaba en las diversas propiedades únicas de la endonucleasa Cas9: (i) su especificidad de objetivo puede programarse con un ARN pequeño, sin la necesidad de lograr enzimas, (ii) la especificidad del objetivo fue muy alta, determinada por una interacción ARN-ADN de 20 pb con baja probabilidad de no reconocimiento de objetivo, (iii) casi ninguna secuencia puede fijarse como objetivo, siendo el único requerimiento la presencia de una secuencia NGG adyacente, (iv) casi ninguna mutación en la secuencia NGG, así como mutaciones en la secuencia simiente del protoespaciador, elimina la fijación como objetivo.
Los solicitantes mostraron que la ingeniería genómica usando el sistema CRISPR no sólo funcionaba en bacterias altamente recombinogénicas tales como S. pneumoniae, sino también en E. coli. Los resultados en E. coli sugirieron que el método podía ser aplicable a otros organismos para los cuales se pueden introducir plásmidos. En E. coli, la propuesta complementa la recombinación de oligonucleótidos mutagénicos. Para usar esta metodología en microbios en los que no es posible la recombinación, se puede usar la maquinaria de recombinación homóloga del huésped para proporcionar la plantilla de edición en un plásmido. Además, debido a que la evidencia acumulada indica que la escisión mediada por CRISPR del cromosoma conduce a muerte celular en muchas bacterias y arqueas, es posible prever el uso de sistemas CRISPR-Cas endógenos para fines de edición.
Tanto en S. pneumoniae como en E. coli, los solicitantes observaron que aunque se facilitó la edición por una coselección de células transformables y una pequeña inducción de recombinación en el sitio fijado como objetivo por escisión de Cas9, el mecanismo que contribuyó más a la edición fue la selección contra células no editadas. Por lo tanto, la mayor limitación del método fue la presencia de un fondo de células que escaparan a la muerte celular inducida por CRISPR y ausencia de la mutación deseada. Los solicitantes mostraron que estos “escapistas” surgían principalmente por la supresión del espaciador de fijación como objetivo, presumiblemente después de la recombinación de las secuencias de repetición que flanqueaban el espaciador de fijación como objetivo. Más mejoras pueden centrarse en conseguir secuencias de flanqueamiento que puedan soportar aún la biogénesis de los ARNcr funcionales pero que sean suficientemente diferentes entre sí para eliminar la recombinación. Alternativamente, la trasformación directa de los ARNcr quiméricos puede explorarse. En el caso particular de E. coli, la construcción del sistema CRISPR-Cas no fue posible si este organismo se usaba también como un huésped
de clonación. Los solicitantes resolvieron esta cuestión poniendo Cas9 y el ARNtracr en un plásmido diferente que la matriz de CRISPR. La ingeniería de un sistema inducible también puede evitar esta limitación.
Aunque las nuevas tecnologías de síntesis de ADN proporcionan la capacidad para crear con coste eficaz cualquier secuencia con un alto rendimiento, permanece el reto de integrar ADN sintético en células vivas para crear genomas funcionales. Recientemente, se mostró la estrategia MAGE de coselección para mejorar la eficacia de mutación de recombinación por selección de una subpoblación de células que presenta una probabilidad aumentada para conseguir recombinación en o alrededor de un sitio determinado. En este método, la introducción de mutaciones seleccionables se usa para aumentar los cambios de generación de mutaciones cercanas no seleccionables. Al contrario que la selección indirecta proporcionada por esta estrategia, el uso del sistema CRISPR lo hace posible para seleccionar de manera directa la mutación deseada y recuperarla con una alta eficacia. Estas tecnologías se añaden al conjunto de los logros genéticos y junto con la síntesis de ADN, pueden avanzar sustancialmente tanto la capacidad para decodificar la función genética como para manipular organismos para fines biotecnológicos. Otros dos estudios también se relacionan con la ingeniería asistida por CRISPR de genomas de mamífero. Se espera que estas tecnologías de edición de genomas dirigida por ARNcr puedan ser ampliamente útiles en ciencias básicas y médicas.
Cepas y condiciones de cultivo. Se proporcionó cepa R6 de S. pneumoniae por el Dr. Alexander Tomasz. Se generó cepa crR6 en un estudio previo. Se cultivaron cultivos líquidos de S. pneumoniae en medio THYE (30 g/l de agar Todd-Hewitt, 5 g/l de extracto de levadura). Se cultivaron células en agar de soja tríptica (TSA, por sus siglas en inglés) enriquecido con sangre de oveja desfibrinada al 5%. Cuando fue apropiado, se añadieron antibióticos como sigue: kanamicina (400 pg/ml), cloranfenicol (5 pg/ml), eritromicina (1 pg/ml), estreptomicina (100 pg/ml) o espectinomicina (100 pg/ml). Se realizaron mediciones de actividad de p-galactosidasa usando la prueba de Miller como se describió previamente.
Se proporcionaron cepas MG1655 y HME63 de E. coli (procedentes de MG1655, A(argF-lac) U169 A cI857 AcrobioA galK tyr 145 UAG mutS<>amp) (31) por Jeff Roberts y Donald Court, respectivamente. Se cultivaron cultivos líquidos de E. coli en medio LB (Difco). Cuando fue apropiado, se añadieron antibióticos como sigue: cloranfenicol (25 pg/ml), kanamicina (25 pg/ml) y estreptomicina (50 pg/ml).
Transformación de S. pneumoniae. Se prepararon células competentes como se describió previamente (23). Para todas las transformaciones de edición de genomas, se descongelaron suavemente las células sobre hielo y se volvieron a suspender en 10 volúmenes de medio M2 enriquecido con 100 ng/ml de péptido estimulador de competencia CSP1(40) y seguido por adición de construcciones de edición (se añadieron construcciones de edición a células a una concentración final entre 0,7 ng/pl y 2,5 pg/ul). Se incubaron las células 20 min a 37°C antes de la adición de 2 pl de construcciones de fijación como objetivo y después se incubaron 40 min a 37°C. Se pusieron en placas diluciones en serie de células en el medio apropiado para determinar el recuento de unidades formadoras de colonias (ufc).
Recombinación de lambda-rojo de E. coli. Se usó cepa HME63 para todos los experimentos de recombinación. Se prepararon células de recombinación y se manipularon de acuerdo con un protocolo publicado previamente (6). En pocas palabras, se cultivaron 2 ml de cultivo durante la noche (medio LB) inoculado de una única colonia obtenida de una placa a 30°C. Se diluyó el cultivo durante la noche 100 veces y se cultivó a 30 °C con agitación (21 rad/s (200 rpm)) hasta que la DO600 es de 0,4-0,5 (aproximadamente 3 h). Para inducción de Lambda-rojo, se transfirió el cultivo a un baño de agua a 42 °C para agitar a (21 rad/s (200 rpm)) durante 15 min. Inmediatamente después de inducción, se distribuyó el cultivo en una suspensión de hielo-agua y se enfrió sobre hielo durante 5 10 min. Después se lavaron las células y se tomaron alícuotas según el protocolo. Para electrotransformación, se mezclaron 50 pl de células con 1 mM de oligos sin sal (IDT) o 100-150 ng de ADN de plásmido (preparado por Estuche QIAprep Spin Miniprep, Qiagen). Se electroporaron las células usando una cubeta de 1 mm de Gene Pulser (Bio-rad) a 1,8 kV y se volvieron a suspender inmediatamente en 1 ml de medio LB a temperatura ambiente. Se recuperaron las células a 30 °C durante 1 -2 h antes de que se pusieran en placas sobre agar LB con resistencia apropiada a los antibióticos y se incubaron a 32 °C durante la noche.
Preparación de ADN genómico de S. pneumoniae. Para fines de transformación, se extrajo ADN genómico de S. pneumoniae usando el estuche de purificación de ADN genómico Wizard, siguiendo las instrucciones proporcionadas por el fabricante (Promega). Para fines de genotipado, se granularon 700 ul de cultivos de S. pneumoniae durante la noche, se volvieron a suspender en 60 ul de disolución de lisozima (2 mg/ml) y se incubaron 30 min a 37°C. Se extrajo el ADN genómico usando estuche QIAprep Spin Miniprep (Qiagen).
Construcción de cepas. Todos los cebadores usados en este estudio se proporcionan en la Tabla G. Para generar crR6M de S. pneumoniae, se preparó una cepa intermedia, LAM226. En esta cepa se volvió a poner el gen aphA-3 (proporcionando resistencia a kanamicina) adyacente a la matriz de CRISPR de cepa crR6 de S. pneumoniae por un gen cat (que proporciona resistencia a cloranfenicol). En pocas palabras, se multiplicó ADN genómico de crR6 usando cebadores L448/L444 y L447/L481, respectivamente. El gen cat se multiplicó de plásmido pC194 usando los cebadores L445/L446. Cada producto PCR se purificó en gel y los tres se fusionaron por PCR SOEing
con los cebadores L448/L481. El producto PCR resultante se transformó en células crR6 de S. pneumoniae competentes y se seleccionaron transformados resistentes a cloranfenicol. Para generar ADN genómico de crR6M de S. pneumoniae, crR6 de S. pneumoniae se multiplicó por PCR usando los cebadores L409/L488 y L448/L481, respectivamente. Cada producto PCR se purificó en gel y se fusionaron por PCR SOEing con cebadores L409/L481. El producto PCR resultante se transformó en células LAM226 de S. pneumoniae competentes y se seleccionaron transformados resistentes a kanamicina.
Para generar crR6Rc de S. pneumoniae, se multiplicó ADN genómico de crR6M de S. pneumoniae por PCR usando cebadores L430/W286 y se multiplicó ADN genómico LAM226 de S. pneumoniae por PCR usando cebadores W288/L481. Cada producto PCR se purificó en gel y se fusionaron por PCR SOEing con cebadores L430/L481. El producto PCR resultante se transformó en células crR6M de S. pneumoniae competentes y se seleccionaron transformados resistentes a cloranfenicol.
Para generar crR6Rk de S. pneumoniae, se multiplicó ADN genómico de crR6M de S. pneumoniae por PCR usando cebadores L430/W286 y W287/L481, respectivamente. Cada producto PCR se purificó en gel y se fusionaron por PCR SOEing con cebadores L430/L481. El producto pCr resultante se transformó en células crR6Rc de S. pneumoniae compententes y se seleccionaron transformados resistentes a kanamicina.
Para generar JEN37, se multiplicó ADN genómico de crR6Rk de S. pneumoniae por PCR usando cebadores L430/W356 y W357/L481, respectivamente. Cada producto PCR se purificó en gel y se fusionaron por PCR SOEing con cebadores L430/L481. El producto PCR resultante se transformó en células crR6Rc compententes de S. pneumoniae y transformados resistentes a kanamicina se seleccionaron.
Para generar JEN38, se multiplicó ADN genómico R6 usando cebadores L422/L461 y L459/L426, respectivamente. El gen ermAM (que especifica resistencia a eritromicina) se multiplicó de plásmido pFW1543 usando cebadores L457/L458. Cada producto PCR se purificó en gel y los tres se fusionaron por PCR SOEing con cebadores L422/L426. El producto PCR resultante se transformó en células crR6Rc de S. pneumoniae compententes y se seleccionaron transformados resistentes a eritromicina.
Se generaron JEN53 S. pneumoniae en dos etapas. Primero se construyó JEN43 como se ilustra en la Figura 33. Se generó JEN53 transformando ADN genómico de JEN25 en células JEN43 competentes y seleccionando en tanto cloranfenicol como eritromicina.
Para generar JEN62S. pneumoniae, se multiplicó ADN genómico crR6Rk S. pneumoniae por PCR usando cebadores W256/W365 y W366/L403, respectivamente. Cada producto PCR fue purificado y ligado por ensamblado de Gibson. El producto de ensamblado se transformó en células crR6Rc compententes de S. pneumoniae y se seleccionaron transformados resistentes a kanamicina.
Construcción de plásmido. Se construyó pDB97 por fosforilación e hibridación de oligonucleótidos B296/B297, seguido por ligadura en pLZ12spec digerido por EcoRI/BamHI. Los solicitantes secuenciaron completamente pLZ12spec y depositaron su secuencia en genebank (acceso: KC112384).
Se obtuvo pDB98 después de clonar la secuencia líder de CRISPR se clonó junto con una unidad repeticiónespaciador-repetición en pLZ12spec. Esto se consiguió por multiplicación de ADN de crR6Rc con cebadores B298/B320 y B299/B321, seguido por PCR SOEing de los dos productos y clonación en pLZ12spec con sitios de restricción BamHI/EcoRI. De esta manera se logró que la secuencia espaciadora en pDB98 contuviera dos sitios de restricción BsaI en direcciones opuestas que permitieran la clonación sin marca de nuevos espaciadores. Se construyeron pDB99 a pDB108 por hibridación de oligonucleótidos B300/B301 (pDB99), B302/B303 (pDB100), B304/B305 (pDB101), B306/B307 (pDB102), B308/B309 (pDB103), B310/B311 (pDB104), B312/B313 (pDB105), B314/B315 (pDB106), B315/B317 (pDB107), B318/B319 (pDB108), seguido por ligadura en corte de pDB98 por BsaI.
Se construyó el plásmido pCas9 como sigue. Se multiplicaron elementos CRISPR esenciales de ADN genómico de SF370 Streptococcos pyogenes con brazos de homología de flanqueamiento para ensamblado de Gibson. El ARNtracr y Cas9 se multiplicaron con oligos HC008 y HC010. Las secuencias líder y CRISPR se multiplicaron HC011/HC014 y HC015/HC009, de manera que se introdujeron dos sitios SsaI tipo IIS entre dos repeticiones directas para facilitar la fácil inserción de espaciadores.
Se construyó pCRISPR por subclonación de la matriz CRISPR pCas9 en pZE21 -MCS1 por multiplicación con oligos B298+B299 y restricción con EcoRI y BamHI. Se clonó el espaciador de fijación como objetivo de rpsL por hibridación de oligos B352+B353 y clonando en el corte de BsaI pCRISPR proporcionando pCRISPR::rpsL. Generación de construcciones de fijación como objetivo y edición. Las construcciones de fijación como objetivo usadas para edición de genomas se prepararon por ensamblado de Gibson de los PCR izquierdo y PCR derecho (Tabla G). Las construcciones de edición se prepararon por PCR SOEing fusionando productos PCR A (PCR A),
productos PCR B (PCR B) y productos PCR C (PCR C) cuando fue aplicable (Tabla G). Se generaron las construcciones de fijación como objetivo CRISPR::0 y CRISPR::ermAM(interrupción) por multiplicación PCR de JEN62 y ADN genómico crR6 respectivamente, con oligos L409 y L481.
Generación de objetivos con PAM o secuencias del protoespaciador aleatorizadas. Se aleatorizaron los 5 nucleótidos siguiendo el objetivo espaciador 1 por la multiplicación de ADN genómico de R682325 con cebadores W377/ L426. Este producto PCR se ensambló después con el gen cat y la región aguas arriba de srtA que se multiplicó a partir de la misma matriz con los cebadores L422/W376. Se usaron 80 ng del ADN ensamblado para transformar las cepas R6 y crR6. Se prepararon las muestras para los objetivos aleatorizados usando los siguientes cebadores: B280-B290/L426 para aleatorizar las bases 1-10 del objetivo y B269-B278/L426 para aleatorizar las bases 10-20. Se usaron los cebadores L422/B268 y L422/B279 para multiplicar el gen cat y la región aguas arriba de srtA para ensamblar con los 10 productos PCR primeros y últimos, respectivamente. Se mezclaron juntas las construcciones ensambladas y se transformaron 30 ng en R6 y crR6. Después de la transformación, se pusieron en placas las células en selección de cloranfenicol. Para cada muestra se mezclaron entre sí más de 2*105 células en 1 ml de THYE y se extrajo ADN genómico con el estuche Promega Wizard. Se usaron los cebadores B250/B251 para multiplicar la región fijada como objetivo. Se marcaron los productos PCR y se barrieron sobre una vía de extremos apareados Illumina MiSeq usando 300 ciclos.
Análisis de los datos de secuenciación profunda.
PAM aleatorizada: Para el experimento de PAM aleatorizada se obtuvieron 3.429.406 lecturas para crR6 y 3.253.998 para R6. Se espera que sólo la mitad de ellas corresponda al objetivo PAM mientras la otra mitad secuenciará el otro extremo del producto PCR. 1.623.008 de las lecturas de crR6 reads y 1.537.131 de las lecturas de R6 soportaron una secuencia diana exenta de error. La existencia de cada posible PAM entre estas lecturas se muestra en un archivo suplementario. Para estimar la funcionalidad de una PAM, se cómputo su proporción relativa en la muestra de crR6 sobre la muestra de R6 y se indica rijklm donde I,j,k,l,m son una de las 4 posibles bases. Se construyó el siguiente modelo estadístico:
logOykim) = p b2¡ b3, b4k b2b3y b3b4,.k+ s1]klm,
donde £ es el error residual, b2 es el efecto de la 2a base de la PAM, b3 de la tercera, b4 de la cuarta, b2b3 es la interacción entre las bases segunda y tercera, b3b4 entre las bases tercera y cuarta. Se realizó un análisis de la varianza:
Tabla Anova
Df Suma Cuadrado Media Cuadrado Valor F Pr (>F)
b3 3 151,693 50,564 601,8450 < 2,2e-16
b2 3 90,521 30,174 359,1454 < 2,2e-16
b4 3 1,881 0,627 7,4623 6,070e-05
b3:b2 9 228,940 25,438 302,7738 < 2,2e-16
b3:b4 9 3,010 0,334 3,9809 5,227e-05
Residuos 996 83,680 0,084
Cuando se añade este modelo, no parece que b1 o b5 sean significativos y también se pueden desechar otras interacciones que las incluidas. La lección de modelo se realizó por comparación de sucesivas de modelos más o menos completos usando el método anova en R. Se usó el ensayo de significancia verdadera de Tukey para determinar si eran significativas las diferencias de pares entre efectos.
Los patrones NGGNN eran significativamente diferentes de los otros patrones y soportan el efecto más fuerte (véase la tabla a continuación).
Para demostrar que las posiciones, 1, 4 ó 5 no afectan al patrón NGGNN, los solicitantes miraron estas secuencias solamente. Este efecto parece que está normalmente distribuido (véase la representación gráfica QQ en la Figura 71) y comparaciones de modelo usando el método anova en R muestran que el modelo nulo es el mejor, es decir, no hay una función significativa de b1, b4 y b5.
Comparación de modelo usando el método anova en R para las secuencias NGGNN.
Modelo 1: proporción.log ~ 1
Modelo 2: proporción.log ~ b1 b4 b5
Df Res. RSS Df Suma de Cuadrados F Pr (>F)
1 63 14,579
2 54 11,295 9 3,2836 1,7443 0,1013
Interferencia parcial de los patrones NAGNN y NNGGN
Los patrones NAGNN eran significativamente diferentes de los otros patrones pero soportan un efecto mucho menor que NGGNN (véase el ensayo de significancia verdadera de Tukey a continuación).
Finalmente, los patrones NTGGN y NCGGN son similares y muestran interferencia de CRISPR más significativamente que los patrones NTGHN y NCGHN (donde H es A,T o C), como se muestra por un ensayo de student de pares ajustados de bonferroni.
Comparaciones de pares del efecto de b4 sobre secuencias NYGNN usando ensayos t con DE agrupada.
Datos: b4
A C G
C 1,00 - -G 9,2e-05 2,4e-06 -T 0,31 1,00 1,2e-08
Tomados juntos, estos resultados permiten concluir que los patrones NNGGN producen en general una interferencia completa en el caso de NGGGN, o una interferencia parcial en el caso de NAGGN, NTGGN o NCGGN.
Comparaciones múltiples de Tukey de medias: 95% de nivel de confianza a nivel familiar.
$b2:b3
dif infer super p adj
G:G-A:A -2,76475 -2,94075 -2,58875 <1E -07
G:G-C:A -2,79911 -2,97511 -2,62311 <1E -07
G:G-T:A -2,7809 -2,9569 -2,6049 <1E -07
G:G-A:C -2,81643 -2,99244 -2,64043 <1E -07
$b2:b3
dif infer super p adj
G:G-C:C -2,77903 -2,95504 -2,60303 <1E -07 G:G-G:C -2,64867 -2,82468 -2,47267 <1E -07 G:G-T:C -2,79718 -2,97319 -2,62118 <1E -07 G:G-A:G -2,67068 -2,84668 -2,49468 <1E -07 G:G-C:G -2,73525 -2,91125 -2,55925 <1E -07 G:G-T:G -2,7976 -2,62159 -2,9736 <1E -07 G:G-A:T -2,76727 -2,59127 -2,94328 <1E -07 G:G-C:T -2,84114 -2,66513 -3,01714 <1E -07 G:G-G:T -2,76409 -2,58809 -2,94009 <1E -07 G:G-T:T -2,76781 -2,59181 -2,94381 <1E -07 G:G-G:A -2,13964 -2,31565 -1,96364 <1E -07 G:A-A:A -0,62511 -0,80111 -0,4491 <1E -07
G:A-C:A -0,65947 -0,83547 -0,48346 <1E -07 G:A-T:A -0,64126 -0,46525 -0,81726 <1E -07 G:A-A:C -0,67679 -0,50078 -0,85279 <1E -07 G:A-C:C -0,63939 -0,46339 -0,81539 <1E -07 G:A-G:C -0,50903 -0,33303 -0,68503 <1E -07 G:A-T:C -0,65754 -0,48154 -0,83354 <1E -07 G:A-A:G -0,53104 -0,35503 -0,70704 <1E -07 G:A-C:G -0,59561 -0,4196 -0,77161 <1E -07 G:A-T:G -0,65795 -0,48195 -0,83396 <1E -07 G:A-A:T -0,62763 -0,45163 -0,80363 <1E -07 G:A-C:T -0,70149 -0,52549 -0,8775 <1E -07 G:A-G:T -0,62445 -0,44844 -0,80045 <1E -07 G:A-T:T -0,62817 -0,45216 -0,80417 <1E -07
$b2:b3
dif infer super p adj
$b3:b4
dif infer super p adj
G:G-G:A -0,33532 -0,51133 -0,15932 <1E-07
G:G-G:C -0,18118 -0,35719 -0,00518 0,036087
G:G-G:T -0,31626 -0,14026 -0,49226 <1 E-07
Objetivo aleatorizado
Para el experimento de objetivo aleatorizado se obtuvieron 540.726 lecturas para crR6 y 753.570 para R6. Como antes, se esperó que sólo la mitad de las lecturas secuenciara el extremo de interés del producto pCr . Después de filtrar para las lecturas que soportan un objetivo que está exento de error o con una mutación puntual única, permanecieron 217.656 y 353.141 lecturas para crR6 y R6, respectivamente. Se computó la proporción relativa de cada mutante en la muestra crR6 sobre la muestra R6 (Figura 24c). Todas las mutaciones fuera de la secuencia simiente (13-20 bases lejos de la PAM) muestran interferencia completa. Esas secuencias se usaron con una referencia para determinar si se podía decir que otras mutaciones en el interior de la secuencia simiente alteraban significativamente la interferencia. Se fijó una distribución normal para estas secuencias usando la función fitdistr del paquete MASS R. El cuantil 0,99 de la distribución fijada se muestra como una línea de puntos en la Figura 24c. La Figura 72 muestra un histograma de la densidad de datos con distribución normal fijada (línea negra) y el cuantil 0,99 (línea de puntos).
Tabla F. Abundancia relativa de secuencias PAM en las muestras crR6/R6 promediadas sobre las bases 1 y 5.

Tabla G. Cebadores usados en este estudio.




Tabla H. Diseño de construcciones de fijación como objetivo y edición usadas en este estudio

Ejemplo 6: Optimización del ARN guía para Cas9 de Streptococcus pyogenes (referido como SpCas9).
Los solicitantes mutaron las secuencias ARNtracr y de repetición directa o mutaron el ARN guía quimérico para activar los ARN en las células.
La optimización se basa en la observación de que hay tramos de timinas (Ts) en el ARNtracr y ARN guía, que podían conducir a terminación de la transcripción temprana por el activador pol 3. Por lo tanto, los Solicitantes generaron las siguientes secuencias optimizadas. Se presentan en parejas ARNtracr optimizada y la repetición directa optimizada correspondiente.
ARNtracr optimizado 1 (mutación subrayada):
GG AACC ATT CAt AAC AGCAT AGC AAGTT AtAAT AAGGCTAGTCCGTT ATCA ACTT GAAA AAGT GGC ACCG AGT CGGTGCTTTTT
Repetición directa optimizada 1 (mutación subrayada):
GTTaTAGAGCTATGCTGTTaTGAATGGTCCCAAAAC
ARNtracr optimizado 2 (mutación subrayada):
GGAACCATTCA AtACAGCAT AGCAAGTTAAtAT AAGGCT AGTCCGTT ATCA ACTTGAAAAAGTGGCACCGAGTC'GGTGCTTTTT
Repetición directa optimizada 2 (mutación subrayada):
GTaTTAGAGCTATGCTGTaTTGAATGGTCCCAAAAC
Los solicitantes también optimizaron el ARN guía quimérico para actividad óptima en células eucariotas.
ARN guía original :
NNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTAGAAATAGCAAGTTAAAA TAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTT
Secuencia de ARN guía quimérico optimizado 1:
NNNNNNNNNNNNNNNNNNNNGTATTAGAGCTAGAAATAGCAAGTTAATA TAAGGCTAGTC'CGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTT
Secuencia de ARN guía quimérico optimizado 2:
NNNNNNNNNNNNNNNNNNNNGTTTTAGAGCTATGCTGTTTTGGAAACAA
AACAGCATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCAC C G AGTCGGT GCTTTTTTT
Secuencia de ARN guía quimérico optimizado 3:
NNNNNNNNNNNNNNNNNNNNGTATTAGAGCTATGCTGTATTGGAAACAA
IACAGCATAGCAAGTTAAIAT AAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACC GAGTCGGTGCTTTTTTT
Los solicitantes mostraron que el ARN guía quimérico optimizado funciona mejor como se indica en la Figura 3. El experimento se realizó por cotransinfección de células 293FT con Cas9 y una casete de ADN ARN guía de U6 para expresar una de las cuatro formas de ARN mostradas anteriormente. El objetivo del ARN guía es el mismo sitio objetivo en el lugar Emx1 humano: "GTCACCTCCAATGACTAGGG".
Ejemplo 7: Optimización de Cas9 CRISPR1 LMD-9 de Streptococcus thermophilus (referido como StlCas9).
Los solicitantes diseñaron los ARN quiméricos guía como se muestra en la Figura 4.
Los ARN guía de StlCas9 pueden experimentar el mismo tipo de optimización que para los ARN guía de SpCas9, por rotura de los tramos de politiminas (Ts).
Ejemplo 8: Diversidad de Cas9 y mutaciones.
El sistema CRISPR-Cas es un mecanismo inmunitario adaptativo contra ADN exógeno invasor empleado por diversas especies a través de bacterias y arqueas. El sistema CRISPR-Cas9 de tipo II consiste en una serie de genes que codifica proteínas responsables de la “adquisición” de ADN extraño en el lugar CRISPR, así como una serie de genes que codifica la “ejecución” del mecanismo de escisión de ADN; estos incluyen la ADN nucleasa (Cas9), un ARN-cr de transactivación no codificante (ARNtracr) y una matriz de espaciadores procedentes de ADN extraño flanqueados por repeticiones directas (los ARNcr). En la maduración por Cas9, el dúplex de ARNtracr y ARNcr guía la nucleasa Cas9 para una secuencia de ADN fijada como objetivo especificada por las secuencias guía espaciadoras y media roturas de doble cadena en el ADN cerca de una unidad de secuencia corta en el ADN fijado como objetivo que se requiere para escisión y es específico para cada sistema CRISPR-Cas. Los sistemas CRISPR-Cas de tipo II se encuentran por todo el reino bacteriano y son muy diversos en secuencia y tamaño de la proteína Cas9, secuencia de repetición directa de ARNtracr y ARNcr, organización de genomas de estos elementos y el requerimiento de unidad para escisión fijada como objetivo. Una especie puede presentar múltiples sistemas CRISPR-Cas distintos.
Los solicitantes evaluaron 207 Cas9s putativas de especies bacterianas identificadas basándose en homología de secuencias para conocer Cas9s y estructuras ortólogas para conocer subdominios, incluyendo el dominio de la endonucleasa HNH y los dominios de endonucleasa RuvC [información de the Eugene Koonin and Kira Makarova]. El análisis filogenético basado en la conservación de secuencias de proteínas de este conjunto reveló cinco familias de Cas9s, incluyendo tres grupos de Cas9s grandes (-1.400 aminoácidos) y dos de Cas9s pequeños (~1.100 aminoácidos) (Figuras 39 y 40A-F).
En este ejemplo, los solicitantes muestran que las siguientes mutaciones pueden convertir SpCas9 en una enzima de mellado: D10A, E762A, H840A, N854A, N863A, D986A.
Los solicitantes proporcionan secuencias que muestran donde se sitúan los puntos de mutación dentro del gen SpCas9 (Figura 41). Los solicitantes muestran también que las nickasas aún pueden mediar la recombinación homóloga (Prueba indicada en la Figura 2). Además, los solicitantes muestran que SpCas9 con estas mutaciones (individualmente) no inducen doble rotura de cadena (Figura 47).
Ejemplo 9: Suplemento para especificidad de fijación como objetivo de ADN de la nucleasa Cas9 guiada por ARN. Cultivo celular y transinfección.
Se mantuvo la estirpe celular 293FT (Life Technologies) de riñón embriónico humano (HEK) en medio Eagle modificado de Dulbecco (DMEM) enriquecido con suero fetal bovino al 10% (HyClone), GlutaMAX 2 mM (Life Technologies), 100 U/ml de penicilina y 100 pg/ml de estreptomicina a 37°C con incubación con CO2 al 5%.
Se sembraron células 293FT en placas de 6 pozos, placas de 24 pozos o placas de 96 pozos (Corning) 24 horas previas a transinfección. Se transinfectaron las células usando Lipofectamine 2000 (Life Technologies) a confluencia del 80-90% siguiendo el protocolo recomendado por el fabricante. Para cada pozo de una placa de 6 pozos, se usó un total de 1 ug de plásmido Cas9+ARNsg. Para cada pozo de una placa de 24 pozos, se usó un total de 500 ng de plásmido Cas9+ARNsg a menos que se indicara de otro modo. Para cada pozo de una placa de 96 pozos, se usaron 65 ng de plásmido Cas9 a una relación molar de 1:1 para el producto PCR de U6-ARNsg.
Se mantuvo estirpe de células madre embrionarias humanas HUES9 (núcleo del instituto de células madre de Harvard) en condiciones exentas de alimentador en GelTrex (Life Technologies) en medio mTesR (Stemcell Technologies) enriquecido con 100 ug/ml de Normocina (InvivoGen). Se transinfectaron células HUES9 con estuche Amaxa P3 Primary Cell 4-D Nucleofector (Lonza) siguiendo el protocolo del fabricante.
Prueba de la nucleasa SURVEYOR para modificación de genomas.
Se transinfectaron células 293FT con ADN de plásmido como se describió anteriormente. Se incubaron las células a 37°C durante 72 horas postransinfección previamente a extracción de ADN genómico. Se extrajo ADN genómico usando la disolución de extracción de ADN QuickExtract (Epicentre) siguiendo el protocolo del fabricante. En resumen, se volvieron a suspender las células granuladas en disolución QuickExtract y se incubaron a 65°C durante 15 minutos y 98°C durante 10 minutos.
La región genómica flanqueando el sitio fijado como objetivo de CRISPR para cada gen se multiplicó por PCR (cebadores enumerados en las Tablas J y K) y se purificaron los productos usando Columna QiaQuick Spin (Qiagen) siguiendo el protocolo del fabricante. Se mezclaron 400 ng totales de los productos PCR purificados con 2 pl de tampón PCR de ADN polimerasa 10X Taq (Enzymatics) y agua ultrapura a un volumen final de 20 pl y se sometió a un procedimiento de hibridación de nuevo para permitir la formación de heterodúplex: 95°C durante 10 min, 95°C a 85°C variando a - 2°C/s, 85°C a 25°C a -0,25°C/s y se mantuvieron 25°C durante 1 minuto. Después de hibridar de nuevo, los productos se trataron con nucleasa SURVEYOR y activador S SURVEYOR (Transgenomics) siguiendo el protocolo recomendado por el fabricante y se analizaron en geles de poliacrilamida Novex TBE al 4-20% (Life Technologies). Se tiñeron los geles con
tinte de ADN SYBR Gold (Life Technologies) durante 30 minutos y se formaron imágenes con un sistema de formación de imágenes de gel Doc (Bio-rad). La cuantificación se basó en intensidades de banda relativas.
Análisis de Northern de expresión de ARNtracr en células humanas.
Se realizaron análisis Northern como se describió previamente 1. En resumen, se calentaron los ARN a 95°C durante 5 min antes de la carga en geles de poliacrilamida desnaturalizadantes al 8% (SequaGel, National Diagnostics). Después, se transfirió el ARN a una membrana Hybond N+ prehibridada (GE Healthcare) y se reticuló con reticulador de UV Stratagene (Stratagene). Se etiquetaron las sondas con [gamma-32P] ATP (Perkin Elmer) con T4 polinucleótido cinasa (New England Biolabs). Después de lavado, se expuso la membrana a pantalla de fósforo durante una hora y se escaneó con fosforimager (Typhoon).
Secuenciación de bisulfito para valorar estado de metilación de ADN.
Se transinfectaron células 293FT HEK con Cas9 como se describió anteriormente. Se aisló ADN genómico con el estuche DNeasy Blood & Tissue (Qiagen) y bisulfito convertido con estuche EZ DNA Methylation-Lightning (Zymo Research). Se llevó a cabo PCR con bisulfito usando ADN polimerasa KAPA2G Robust HotStart (KAPA Biosystems) con cebadores diseñados usando el buscador de cebadores de bisulfito (Zymo Research, Tablas J y K). Los amplicones PCR resultantes se purificaron en gel, se digirieron con EcoRI y HindIII y se ligaron en una cadena principal de pUC19 previamente a transformación. Los clones individuales fueron sometidos a secuenciación de Sanger para valorar el estado de metilacion de ADN.
Prueba de transcripción y escisión in vivo.
Se transinfectaron células 293FT HEK con Cas9 como se describió anteriormente. Se prepararon después lisados celulares completos con un tampón de lisis (HEPES 20 mM, KCl 100 mM, MgCl25 mM, DtT 1 mM, glicerol al 5%, Tritón X-100 al 0,1%) enriquecido con cóctel de inhibidores de proteasas (Roche). Se transcribió ARNsg conducido por T7 in vitro usando oligos modificados (Ejemplo 10) y estuche de transcripción in vitro de T7 HiScribe (NEB), siguiendo el protocolo recomendado por el fabricante. Para preparar sitios fijados como objetivo metilados, se metiló plásmido de pUC19 por M.SssI y después se linearizó por NheI. Se realizó la prueba de escisión in vitro como sigue: para una reacción de escisión de 20 ul, se incuban 10 ul de lisado celular con 2 ul de tampón de escisión (HEPES 100 mM, KCl 500 mM, MgCl225 mM, DTT 5 mM, glicerol al 25%), el ARN transcrito in vitro y 300 ng de ADN de plásmido de pUC19.
Secuenciación profunda para valorar la especificidad de la fijación como objetivo.
Se transinfectaron células 293FT HEK en placas de 96 pozos con ADN de plásmido Cas9 y casete PCR de ARN de una sola guía (ARNsg) 72 horas previas a extracción de ADN genómico (Fig. 72). La región genómica que flanquea el sitio fijado como objetivo de CRISPR para cada gen se multiplicó (Fig. 74, Fig. 80, (Ejemplo 10) por un método PCR de fusión para unir los adaptadores Illumina P5 así como códigos de barras específicos de una muestra única para los amplicones fijados como objetivo (esquema descrito en la Fig. 73). Se purificaron los productos PCR usando placas de filtro del 96 pozos EconoSpin (Epoch Life Sciences) siguiendo el protocolo recomendado por el fabricante.
Se cuantificaron las muestras de ADN codificadas por barras y purificadas mediante el estuche de prueba de ADNdc Quant-iT PicoGreen o Fluorómetro Qubit 2.0 (Life Technologies) y se mezclaron en una relación equimolar. Se secuenciaron después en profundidad bibliotecas de secuenciación con el secuenciador personal MiSeq (Life Technologies).
Análisis de datos de secuenciación y detección de Indel.
Se filtraron lecturas de MiSeq requiriendo una calidad promedio Phred (puntuación Q) de al menos 23, así como igualaciones perfectas de secuencias para cebadores directos de códigos de barras y amplicones. Se analizaron las lecturas de los lugares en y fuera del objetivo realizando primero alineamientos de Smith-Waterman frente a secuencias de amplicones que incluían 50 nucleótidos aguas arriba y aguas abajo del sitio fijado como objetivo (un total de 120 pb). Se analizaron los indel en alineamientos, mientras tanto, de 5 nucleótidos aguas arriba a 5 nucleótidos aguas abajo del sitio fijado como objetivo (un total de 30 pb). Se desecharon las regiones fijadas como objetivo analizadas si parte de su alineamiento caía fuera de la propia lectura de MiSeq o si los pares de bases igualados comprendían menos del 85% de su longitud total.
Los controles negativos para cada muestra proporcionaron una medida para la inclusión o exclusión de los indel como casos de corte putativos. Para cada muestra, se contó un indel sólo si su puntuación de calidad excedía de y-o, donde y era la puntuación de calidad media del control negativo que correspondía a esa muestra y o era la desviación estándar del mismo. Esto proporcionó tasas de indel de región de objetivo total para tanto los controles negativos como sus correspondientes muestras. Usando la tasa de error por región de objetivo por lectura del control negativo, q, el recuento de indel observado de la muestra n y su recuento de lectura R, una estimación probablemente máxima para la fracción de lecturas con regiones de objetivo con los indel verdaderos, P, se derivó por aplicación de un modelo de error binomial, como sigue.
Suponiendo que el número (desconocido) de lecturas en una muestra con regiones fijadas como objetivo contadas de manera incorrecta que tiene al menos 1 indel es E, se puede escribir (sin presumir acerca del número de los indel verdaderos)

puesto de R(1-p) es el número de lecturas que tiene regiones fijadas como objetivo sin indel verdadero. Mientras tanto, debido a que el número de lecturas que se observa que presenta indel es n, n=E+Rp , en otras palabras, el número de lecturas que tiene regiones fijadas como objetivo con error, pero no tienen indel verdadero más el número de lecturas cuyas regiones fijadas como objetivo tienen correctamente indel. Se puede volver a escribir entonces lo anterior:

Tomando todos los valores de la frecuencia de regiones fijadas como objetivo con P inserciones o supresiones verdaderas que sean igualmente probables a priori, Prob(n|p) k Prob(p|n). La estimación de probabilidad máxima (EPM) para la frecuencia de regiones fijadas como objetivo con inserciones o supresiones verdaderas se fijó por lo tanto como el valor de P que hizo máxima la Prob(n|p). Esto se evaluó de manera numérica.
Para poner límites de error en las frecuencias de lectura de inserción o supresión verdadera en las propias bibliotecas de secuenciación, se calcularon los intervalos de puntuación de Wilson (2) para cada muestra, proporcionando la estimación de EPM para regiones fijadas como objetivo de inserción o supresión verdadera, RP y el número de lecturas R. De manera explícita, el límite inferior / y el límite superior u se calcularon como:
l =
(flp y - W flpfl - p) rV^/ÍJ?
z 2)

donde z, la puntuación estándar para la confianza requerida de la distribución normal de la varianza 1, se fijó en 1,96, que significa una confianza del 95%. Los límites superiores máximos y los límites inferiores mínimos para cada replicado biológico se enumeran en las Figs. 80-83.
Análisis qRT-PCR de expresión relativa de Cas9 y ARNsg.
Se transinfectaron células 293FT puestas en placas de 24 pozos como se describió anteriormente. 72 horas postransinfección, se recogió ARN total con microestuche miRNeasy (Qiagen). Se realizó síntesis de cadena inversa para los ARNsg con estuche de ADNc qScript Flex (VWR) y cebadores de síntesis de primera cadena modificados (Tablas J y K). Se realizó análisis qPCR con mezclador maestro verde de Fast SYBR (Life Technologies) y cebadores modificados (Tablas J y K), usando GAPDH como un control endógeno. Se calculó la cuantificación relativa por el método AACT.
Tabla I | Secuencias del sitio fijado como objetivo. Sitios fijados como objetivo ensayados para sistema CRISPR de tipo II de S. pyogenes con la PAM requisito. Se transinfectaron células con Cas9 y ARNcr-ARNtracr o ARNsg quimérico para cada objetivo.


Tabla J | Secuencias de cebadores


Tabla K | Secuencias para cebadores para ensayar la arquitectura de ARNsg. Los cebadores se hibridan a la cadena inversa del activador U6 a menos que se indique de otro modo. El sitio para cebar U6 está en cursiva, la secuencia guía se indica como un tramo de N, la secuencia repetida directa se destaca en negrita y subrayada la secuencia de ARNtracr. La estructura secundaria de cada arquitectura de ARNsg se muestra en la Fig. 43.

Tabla L | Sitios fijados como objetivo con las PAM alternas para ensayar la especificidad de PAM de Cas9. Todos los sitios fijados como objetivo para ensayar la especificidad de PAM se encuentran dentro del lugar EMX1 humano.

Ejemplo 10: Secuencias suplementarias
Todas las secuencias están en la dirección 5' a 3'. Para la transcripción de U6, la hilera de T subrayadadas sirven como el terminador de la transcripción.
> ARNtracr corto de U6 (SF370 Streptococcus pyogenes)
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattggaattaatttgactgtaaa cacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgc ttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccGGAACCATTCAAAACAGC ATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGA GTCGGTGCTTTTTTT
(Secuencia ARNtracr en negrita)
>U6-RD-secuencia guía-RD (SF370 Streptococcus pyogenes)
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattggaattaatttgactgtaaa cacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgc ttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgggttttagagctatgctgttttgaatggtccc aaaacNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNgttttagagctatgctgttttgaatggtcccaaaacT
(la secuencia de repetición directa se resalta en gris y la secuencia guía son N en negrita)
> ARNsg que contiene 48 ARNtracr (SF370 Streptococcus pyogenes)
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattggaattaatttgactgtaaa cacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgc ttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccNNNNNNNNNNNNNNNNNN NNgttttagagctugaaatageaagttaaaataaggctagtccgTTTIJTT
(la secuencia guía son N en negrita y el fragmento ARNtracr está en negrita)
> ARNsg que contiene 54 ARNtracr (SF370 Streptococcus pyogenes)
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattggaattaatttgactgtaaa
ttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccNN NNNNNNNNNNNNNNNN NNuttttagagctagaaatagcaagttaaaataaggctagtccgttatcaTTTTTTTT
(la secuencia guía son N en negrita y el fragmento ARNtracr está en negrita)
> ARNsg que contiene 67 ARNtracr (SF370 Streptococcus pyogenes)
gagggcctatttcccatgattccttcatatttgcatatacgatacaaggctgttagagagataattggaattaatttgactgtaaa cacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgggtagtttgcagttttaaaattatgttttaaaatggactatcatatgc ttaccgtaacttgaaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccNNNNNNNNNNNNNNNNNN NNgttttagagctagaaatagcaagttaaaataaggctagtccgttatcaacttgaaaaagtgTTTTTTX
(la secuencia guía son N en negrita y el fragmento ARNtracr está en negrita)
> ARNsg que contiene 85 ARNtracr (SF370 Streptococcus pyogenes)

(la secuencia guía son N en negrita y el fragmento ARNtracr está en negrita)
> CBh-NLS-SpCas9-NLS
CGTTACATAACTTACGGTAAATGGCCCGCCTGGCTGACCGCCCAACGACC CCCGCCCATTGACGTCAATAATGACGTATGTTCCCATAGTAACGCCAATAGGGACTT TCCATTGACGTCAATGGGTGGAGTATTTACGGTAAACTGCCCACTTGGCAGTACATC AAGTGTATCATATGCCAAGTACGCCCCCTATTGACGTCAATGACGGTAAATGGCCCG CCTGGCATTATGCCCAGTACATGACCTTATGGGACTTTCCTACTTGGCAGTACATCTA CGTATTAGTCATCGCTATTACCATGGTCGAGGTGAGCCCCACGTTCTGCTTCACTCTC CCCAT CTCCCCCC CCT CCCC ACCCCC AATTTTGTATTT ATTT ATTTTTT AATT ATTTT G TGCAGCGATGGGGGCGGGGGGGGGGGGGGGGCGCGCGCCAGGCGGGGCGGGGCGG GGCGAGGGGCGGGGCGGGGCGAGGCGGAGAGGTGCGGCGGCAGCCAATCAGAGCG GCGCGCTCCGAAAGTTTCCTTTTATGGCGAGGCGGCGGCGGCGGCGGCCCTATAAA AAGCGAAGCGCGCGGCGGGCGGGAGTCGCTGCGACGCTGCCTTCGCCCCGTGCCCC GCTCCGCCGCCGCCTCGCGCCGCCCGCCCCGGCTCTGACTGACCGCGTTACTCCCAC AGGTGAGCGGGCGGGACGGCCCTTCTCCTCCGGGCTGTAATTAGCTGAGCAAGAGG T A AGGGTTT AAGGGATGGTTGGTTGGTGGGGT ATT AATGTTT AATT ACCTGGAGC AC CTGCCTGAAATCACTTTTTTTCAGGTTGGaccggtgccaccATGGACTATAAGGACCACG ACGGAGACTACAAGGATCATGATATTGATTACAAAGACGATGACGATAAGATG GCCCCAAAGAAGAAGCGGAAGGTCGGTATCCACGGAGTCCCAGCAGCCGACAA GAAGTACAGCATCGGCCTGGACATCGGCACCAACTCTGTGGGCTGGGCCGTGA TCACCGACGAGTACAAGGTGCCCAGCAAGAAATTCAAGGTGCTGGGCAACACC GACCGGCACAGCATCAAGAAGAACCTGATCGGAGCCCTGCTGTTCGACAGCGG CGAAACAGCCGAGGCCACCCGGCTGAAGAGAACCGCCAGAAGAAGATACACCA GACGGAAGAACCGGATCTGCTATCTGCAAGAGATCTTCAGCAACGAGATGGCC AAGGTGGACGACAGCTTCTTCCACAGACTGGAAGAGTCCTTCCTGGTGGAAGA GGATAAGAAGCACGAGCGGCACCCCATCTTCGGCAACATCGTGGACGAGGTGG CCTACCACGAGAAGTACCCCACCATCTACCACCTGAGAAAGAAACTGGTGGAC AGCACCGACAAGGCCGACCTGCGGCTGATCTATCTGGCCCTGGCCCACATGAT CAAGTTCCGGGGCCACTTCCTGATCGAGGGCGACCTGAACCCCGACAACAGCG ACGTGGACAAGCTGTTCATCCAGCTGGTGCAGACCTACAACCAGCTGTTCGAG GAAAACCCCATCAACGCCAGCGGCGTGGACGCCAAGGCCATCCTGTCTGCCAG
ACTGAGCAAGAGCAGACGGCTGGAAAATCTGATCGCCCAGCTGCCCGGCGAGA AGAAGAATGGCCTGTTCGGCAACCTGATTGCCCTGAGCCTGGGCCTGACCCCC AACTTCAAGAGCAACTTCGACCTGGCCGAGGATGCCAAACTGCAGCTGAGCAA GGACACCTACGACGACGACCTGGACAACCTGCTGGCCCAGATCGGCGACCAGT ACGCCGACCTGTTTCTGGCCGCCAAGAACCTGTCCGACGCCATCCTGCTGAGC GACATCCTGAGAGTGAACACCGAGATCACCAAGGCCCCCCTGAGCGCCTCTAT GATCAAGAGATACGACGAGCACCACCAGGACCTGACCCTGCTGAAAGCTCTCG TGCGGCAGCAGCTGCCTGAGAAGTACAAAGAGATTTTCTTCGACCAGAGCAAG AACGGCTACGCCGGCTACATTGACGGCGGAGCCAGCCAGGAAGAGTTCTACAA GTTCATCAAGCCCATCCTGGAAAAGATGGACGGCACCGAGGAACTGCTCGTGA AGCTGAACAGAGAGGACCTGCTGCGGAAGCAGCGGACCTTCGACAACGGCAG CATCCCCCACCAGATCCACCTGGGAGAGCTGCACGCCATTCTGCGGCGGCAGG AAGATTTTTACCCATTCCTGAAGGACAACCGGGAAAAGATCGAGAAGATCCTG ACCTTCCGCATCCCCTACTACGTGGGCCCTCTGGCCAGGGGAAACAGCAGATT CGCCTGGATGACCAGAAAGAGCGAGGAAACCATCACCCCCTGGAACTTCGAGG AAGTGGTGGACAAGGGCGCTTCCGCCCAGAGCTTCATCGAGCGGATGACCAAC TTCGATAAGAACCTGCCCAACGAGAAGGTGCTGCCCAAGCACAGCCTGCTGTA CGAGTACTTCACCGTGTATAACGAGCTGACCAAAGTGAAATACGTGACCGAGG GAATGAGAAAGCCCGCCTTCCTGAGCGGCGAGCAGAAAAAGGCCATCGTGGAC CTGCTGTTCAAGACCAACCGGAAAGTGACCGTGAAGCAGCTGAAAGAGGACTA CTTCAAGAAAATCGAGTGCTTCGACTCCGTGGAAATCTCCGGCGTGGAAGATC GGTTCAACGCCTCCCTGGGCACATACCACGATCTGCTGAAAATTATCAAGGAC AAGG ACTTCCTGG AC AAT GAGG AAAACGAGG AC ATTCTGG A AG AT ATCGT GCT GACCCTGACACTGTTTGAGGACAGAGAGATGATCGAGGAACGGCTGAAAACCT ATGCCCACCTGTTCGACGACAAAGTGATGAAGCAGCTGAAGCGGCGGAGATAC ACCGGCTGGGGCAGGCTGAGCCGGAAGCTGATCAACGGCATCCGGGACAAGC AGTCCGGCAAGACAATCCTGGATTTCCTGAAGTCCGACGGCTTCGCCAACAGA AACTTCATGCAGCTGATCCACGACGACAGCCTGACCTTTAAAGAGGACATCCA GAAAGCCCAGGTGTCCGGCCAGGGCGATAGCCTGCACGAGCACATTGCCAATC TGGCCGGCAGCCCCGCCATTAAGAAGGGCATCCTGCAGACAGTGAAGGTGGTG GACGAGCTCGTGAAAGTGATGGGCCGGCACAAGCCCGAGAACATCGTGATCGA
AATGGCCAGAGAGAACCAGACCACCCAGAAGGGACAGAAGAACAGCCGCGAG AGAATGAAGCGGATCGAAGAGGGCATCAAAGAGCTGGGCAGCCAGATCCTGAA AGAACACCCCGTGGAAAACACCCAGCTGCAGAACGAGAAGCTGTACCTGTACT ACCTGCAGAATGGGCGGGATATGTACGTGGACCAGGAACTGGACATCAACCGG CTGTCCGACTACGATGTGGACCATATCGTGCCTCAGAGCTTTCTGAAGGACGA CTCCATCGACAACAAGGTGCTGACCAGAAGCGACAAGAACCGGGGCAAGAGCG ACAACGTGCCCTCCGAAGAGGTCGTGAAGAAGATGAAGAACTACTGGCGGCAG CTGCTGAACGCCAAGCTGATTACCCAGAGAAAGTTCGACAATCTGACCAAGGC CGAGAGAGGCGGCCTGAGCGAACTGGATAAGGCCGGCTTCATCAAGAGACAG CTGGTGGAAACCCGGCAGATCACAAAGCACGTGGCACAGATCCTGGACTCCCG GATGAACACTAAGTACGACGAGAATGACAAGCTGATCCGGGAAGTGAAAGTGA TCACCCTGAAGTCCAAGCTGGTGTCCGATTTCCGGAAGGATTTCCAGTTTTACA AAGTGCGCGAGATCAACAACTACCACCACGCCCACGACGCCTACCTGAACGCC GTCGTGGGAACCGCCCTGATCAAAAAGTACCCTAAGCTGGAAAGCGAGTTCGT GTACGGCGACTACAAGGTGTACGACGTGCGGAAGATGATCGCCAAGAGCGAGC AGGAAATCGGCAAGGCTACCGCCAAGTACTTCTTCTACAGCAACATCATGAACT TTTTCAAGACCGAGATTACCCTGGCCAACGGCGAGATCCGGAAGCGGCCTCTG ATCGAGACAAACGGCGAAACCGGGGAGATCGTGTGGGATAAGGGCCGGGATT TTGCCACCGTGCGGAAAGTGCTGAGCATGCCCCAAGTGAATATCGTGAAAAAG ACCGAGGTGCAGACAGGCGGCTTCAGCAAAGAGTCTATCCTGCCCAAGAGGAA CAGCGATAAGCTGATCGCCAGAAAGAAGGACTGGGACCCTAAGAAGTACGGCG GCTTCGACAGCCCCACCGTGGCCTATTCTGTGCTGGTGGTGGCCAAAGTGGAA AAGGGCAAGTCCAAGAAACTGAAGAGTGTGAAAGAGCTGCTGGGGATCACCAT CATGGAAAGAAGCAGCTTCGAGAAGAATCCCATCGACTTTCTGGAAGCCAAGG GCTACAAAGAAGTGAAAAAGGACCTGATCATCAAGCTGCCTAAGTACTCCCTG TTCGAGCTGGAAAACGGCCGGAAGAGAATGCTGGCCTCTGCCGGCGAACTGCA GAAGGGAAACGAACTGGCCCTGCCCTCCAAATATGTGAACTTCCTGTACCTGG CCAGCCACTATGAGAAGCTGAAGGGCTCCCCCGAGGATAATGAGCAGAAACAG CTGTTTGTGGAACAGCACAAGCACTACCTGGACGAGATCATCGAGCAGATCAG CGAGTTCTCCAAGAGAGTGATCCTGGCCGACGCTAATCTGGACAAAGTGCTGT CCGCCTACAACAAGCACCGGGATAAGCCCATCAGAGAGCAGGCCGAGAATATC
ATCCACCTGTTTACCCTGACCAATCTGGGAGCCCCTGCCGCCTTCAAGTACTTT GACACCACCATCGACCGGAAGAGGTACACCAGCACCAAAGAGGTGCTGGACGC CACCCTGATCCACCAGAGCATCACCGGCCTGTACGAGACACGGATCGACCTGT CTCAGCTGGGAGGCGACTTTCTTTTTCTTAGCTTGACCAGCTTTCTTAGTAGCA GCAGGACGCTTTAA
(NLS-hSpCas9-NLS se resalta en negrita)
> Amplicón de secuenciación para guías de EMX1 1.1, 1.14, 1.17
CCAATGGGGAGGACATCGATGTCACCTCCAATGACTAGGGTGGGCAACC ACAAACCCACG AGGGCAGAGT GCTGCTTGCT GCT GGCCAGGCCCCT GCGT GGGCCC AAGCTGGACTCTGGCCAC
> Amplicón de secuenciación para guías de EMX1 1.2, 1.16
CGAGC AGAAGAAGAAGGGCTCCCATC AC ATC AACCGGTGGC GC ATTGCC ACGAAGCAGGCCAATGGGGAGGACATCGATGTCACCTCCAATGACTAGGGTGGGCA ACCACAAACCCACGAG
> Amplicón de secuenciación para guías de EMX1 1.3, 1.13, 1.15
GGAGGACAAAGTACAAACGGCAGAAGCTGGAGGAGGAAGGGCCTGAGTC CGAGCAGAAGAAGAAGGGCTCCCATCACATCAACCGGTGGCGCATTGCCACGAAGC AGGCCAATGGGGAGGACATCGAT
> Amplicón de secuenciación para guías de EMX1 1.6
AGAAGCTGGAGGAGGAAGGGCCTGAGTCCGAGCAGAAGAAGAAGGGCTC CCATCACATCAACCGGTGGCGCATTGCCACGAAGCAGGCCAATGGGGAGGACATCG ATGTCACCTCCAATGACTAGGGTGG
> Amplicón de secuenciación para guías de EMX1 1.10
CCTCAGTCTTCCCATCAGGCTCTCAGCTCAGCCTGAGTGTTGAGGCCCCAG TGGCTGCTCTGGGGGCCTCCTGAGTTTCTCATCTGTGCCCCTCCCTCCCTGGCCCAGG TGAAGGTGTGGTTCCA
> Amplicón de secuenciación para guías de EMX1 1.11, 1.12
TCATCTGTGCCCCTCCCTCCCTGGCCCAGGTGAAGGTGTGGTTCCAGAACC GGAGGACAAAGTACAAACGGCAGAAGCTGGAGGAGGAAGGGCCTGAGTCCGAGCA GAAGA AGA AGGGCT CCC AT CACA
> Amplicón de secuenciación para guías de EMX1 1.18, 1.19
CTCCAATGACTAGGGTGGGCAACCACAAACCCACGAGGGCAGAGTGCTG CTTGCTGCTGGCCAGGCCCCTGCGTGGGCCCAAGCTGGACTCTGGCCACTCCCTGGC CAGGCTTTGGGGAGGCCTGGAGT
> Amplicón de secuenciación para guías de EMX1 1.20
CTGCTTGCTGCTGGCCAGGCCCCTGCGTGGGCCCAAGCTGGACTCTGGCC ACTCCCTGGCCAGGCTTTGGGGAGGCCTGGAGTCATGGCCCCACAGGGCTTGAAGC CCGGGGCCGCCATTGACAGAG
> Cebador F de activador T7 para hibridación con cadena fijada como objetivo
>oligo que contiene sitio 1 fijado como objetivo de pUC19 para metilación (T7 inversa)
AAAAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCC TT ATTTT AACTT GCT ATTTCT AGCTCT AAAAC A ACGACGAGCGT GAC ACC ACCCT AT AGT GAGTCGTATTAATTTC
>oligo que contiene sitio 2 fijado como objetivo de pUC19 para metilación (T7 inversa)
AAAAAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCC TTATTTTAACTTGCTATTTCTAGCTCTAAAACGCAACAATTAATAGACTGGACCTATA GTGAGTCGTATT A ATTTC
Ejemplo 11: Recombinación homologa inducida por Cas9 oligo-mediada.
El ensayo de recombinación homóloga de oligonucleótidos es una comparación de eficacia a través de diferentes variantes de Cas9 y diferentes plantillas de RH (oligonucleótido frente a plásmido).
Se usaron células 293FT. SpCas9 = Cas9 natural y SpCas9n = Cas9 de nickasa (D10A). El objetivo de ARN quimérico es el mismo objetivo 1 protoespaciador EMX1 que en los Ejemplos 5, 9 y 10 y oligonucleótidos sintetizados por IDT usando purificación PAGE.
La Figura 44 representa un diseño de oligo-ADN usado como plantilla de recombinación homóloga (RH) en este experimento. Los oligonucleótidos largos contienen homología de 100 pb en el lugar EMX1 y un sitio de restricción HindIII. Se cotransinfectaron células 293FT con: primero, un plásmido que contenía un lugar EMX1 humano de fijación como objetivo de ARN quimérico y proteína cas9 natural y segundo, el oligo-ADN como plantilla de RH. Las muestras son de células 293FT recogidas 96 horas postransinfección con Lipofectamina 2000. Todos los productos se multiplicaron con un cebador de RH EMX1, purificado en gel, seguido por digestión con HindIII para detectar la eficacia de integración de plantilla de RH en el genoma humano.
Las Figuras 45 y 46 representan una comparación de eficacia de RH inducida por diferente combinación de proteína Cas9 y plantilla de RH. La construcción Cas9 usada fue Cas9 natural o la versión de nickasa de Cas9 (Cas9n). La plantilla de RH usada fue: oligo-ADN complementario (oligonucleótido complementario en la figura anterior) u oligo-ADN transcrito (oligonucleótido transcrito en la figura anterior) o plantilla de RH de plásmido (plantilla de RH en la figura anterior). La definición del transcrito/complementario es que la cadena transcrita de manera activa con secuencia correspondiente al ARNm transcrito se define como la cadena transcrita de genoma. Se muestra la eficacia de RH como porcentaje de banda de digestión de HindIII como frente al producto multiplicado por PCR genómico (números del fondo).
Ejemplo 12: Ratón autista
Recientes iniciativas de secuenciación a gran escala han producido un gran número de genes asociados a enfermedad. El descubrimiento de los genes es sólo el comienzo del entendimiento de lo que hace el gen y cómo
conduce a un fenotipo enfermo. Las tecnologías y propuestas actuales para estudiar genes candidatos son lentas y laboriosas. Las reglas de oro, fijación como objetivo de genes e inactivaciones genéticas, requieren una significativa inversión de tiempo y recursos, tanto monetariamente como en términos de personal investigador. Los solicitantes proponen utilizar la nucleasa hSpCas9 para fijar como objetivo muchos genes y haciéndolo con mayor eficacia y menor cambio comparado con cualquier otra tecnología. Debido a la alta eficacia de hSpCas9 los solicitantes pueden realizar inyección de ARN en zigotos de ratón y conseguir inmediatamente animales con genomas modificados sin la necesidad de hacer ninguna fijación como objetivo de genes de manera preliminar en mESC.
La proteína 8 de unión a ADN helicasa con cromodominio (CHD8) es un gen pivotal implicado en el desarrollo y la morfogénesis de los vertebrados inicial. Los ratones que carecen de CHD8 mueren durante el desarrollo embrionario. Las mutaciones en el gen CHD8 se han asociado con un trastorno del espectro autista en seres humanos. Esta asociación se realizó en tres diferentes artículos publicados simultáneamente en Nature. Los mismos tres estudios identificaron una plétora de genes asociados al trastorno del espectro autista. El objeto de los solicitantes fue crear ratones inactivados para los cuatro genes que se encontraron en todos los artículos, Chd8, Katnal2, Kctd13 y Scn2a. Además, los solicitantes eligen otros dos genes asociados al trastorno del espectro autista, esquizofrenia y ADHD, GIT1, CACNA1C y CACNB2. Y finalmente, como control positivo, los solicitantes deciden fijar como objetivo MeCP2. Para cada gen, los solicitantes diseñaron tres ARNg que inactivarían probablemente el gen. Tendría lugar una inactivación después de que la nucleasa hSpCas9 realizara una doble rotura de cadena y la ruta de reparación de ADN propensa a error, unión de extremos no homólogos, corrija la rotura, creando una mutación. El resultado más probable es una mutación que desplace el marco de lectura que inactivaría el gen. La estrategia de fijación como objetivo implicó encontrar protoespaciadores en los exones del gen que tuvieran una secuencia PAM, NGG, y fuera única en el genoma. Se dio preferencia a los protoespaciadores en el primer exón, que sería lo más perjudicial para el gen.
Se validó cada ARNg en la estirpe celular de ratón, Neuro-N2a, por cotransinfección transitoria de liposomas con hSpCas9. 72 horas postransinfección se purificó el ADN genómico usando ADN QuickExtract de Epicentre. Se realizó PCR para multiplicar el lugar de interés. Con posterioridad, se siguió el estuche de detección de mutación SURVEYOR de Transgenomics. Los resultados de SURVEYOR para cada ARNg y respectivos controles se muestran en la Figura A1. Un resultado de SURVEYOR positivo es una banda amplia correspondiendo al PCR genómico y dos bandas más pequeñas que son el producto de la nucleasa SURVEYOR realizando una doble rotura de cadena en el sitio de una mutación. La eficacia de corte promedio de cada ARNg se determinó también para cada ARNg. El ARNg que se eligió para inyección fue el ARNg de más alta eficacia que fue el más único dentro del genoma.
Se inyectó ARN (ARN hSpCas9+ ARNg) en el pronúcleo de un zigoto y se trasplantó más tarde en una madre adoptiva. Se dejó que las madres completarán la gestación y se muestrearon las crías agarrando las colas 10 días posparto. Se extrajo ADN y se usó como una plantilla para PCR, que se trató después por SURVEYOR. Adicionalmente, los productos PCR se enviaron para secuenciación. Los animales que se detectaron como positivos en cualquiera de, el ensayo SURVEYOR o la secuenciación PCR habrían clonado sus productos PCR genómicos en un vector pUC19 y se secuenciaron para determinar mutaciones putativas para cada alelo.
Hasta ahora, las crías de ratón del experimento de fijación como objetivo de Chd8 se han tratado completamente hasta el punto de secuenciación del alelo. Los resultados Surveyor para 38 crías vivas (vías 1-38) 1 cría muerta (vía 39) y 1 cría natural para comparación (vía 40) se muestran en la Figura A2. Se inyectó a las crías 1-19 ARNg Chd8.2 y a las crías 20-38 se inyectó ARNg Chd8.3. De las 38 crías vivas, 13 fueron positivas para una mutación. La cría muerta también tuvo una mutación. No hubo mutación detectada en la muestra natural. La secuenciación por PCR genómica fue consistente con los hallazgos de la prueba SURVEYOR.
Ejemplo 13: Modulación transcripcional mediada por CRISPR/Cas.
La Figura 67 representa un diseño del CRISPR-FT (Factor de Transcripción) con actividad de activación transcripcional. El ARN quimérico se expresa por el activador de U6, mientras una versión de doble mutante, codón optimizada humana de la proteína Cas9 (hSpCas9m), ligada de manera operable a un dominio funcional de NLS triple y uno de VP64 se expresa por un activador de EF1a. Las dobles mutaciones, D10A y H840A, hacen la proteína cas9 incapaz de introducir escisión, pero mantuvieron su capacidad para unirse a un ADN fijado como objetivo cuando se guía por el ARN quimérico.
La Figura 68 representa activación transcripcional del gen SOX2 humano con sistema CRISPR-FT (ARN quimérico y la proteína de fusión Cas9-NLS-VP64). Se transinfectaron células 293FT con plásmidos que soportaban dos componentes: (1) secuencias de 20 pb de fijación como objetivo de ARN quiméricos diferentes conducidos por U6 dentro o alrededor del lugar genómico SOX2 humano y (2) proteína de fusión hSpCas9m (doble mutante)-NLS-VP64 conducida por EF1a. 96 horas postransinfección, se recogieron células 293FT y se midió el nivel de activación por la inducción de expresión de ARNm usando una prueba qRT-PCR. Todos los niveles de expresión se normalizan frente al grupo de control (barra gris), que representa los resultados de células transinfectadas con el plásmido de cadena principal de CRISPR-FT sin ARN quimérico. Las ondas qRT-PCR usadas para detectar el ARNm de SOX2 es prueba
de expresión de genes humanos de Taqman (Life Technologies). Todos los experimentos representan datos de 3 replicados biológicos, n=3, las barras de error muestran e. e. m.
Ejemplo 14: NLS: NLS de Cas9
Se transinfectaron células 293FT con plásmido que contenía dos componentes: (1) activador de EF1a que conducía la expresión de Cas9 (Sp Cas9 codón optimizada, humana, natural) con diferentes diseños NLS (2) activador de U6 que conducía el mismo lugar EMX1 humano que fija como objetivo ARN quimérico.
Se recogieron células en el instante de tiempo de 72 h postransinfección y se extrajeron después con 50 pl de la disolución de extracción de ADN genómico QuickExtract siguiendo el protocolo del fabricante. Se multiplicó por PCR ADN genómico de EMX1 fijado como objetivo y se purificó en gel con gel de agarosa al 1%. Se volvió a hibridar el producto PCR genómico y se sometió a la prueba Surveyor siguiendo el protocolo del fabricante. La eficacia de escisión genómica de diferentes construcciones se midió usando SDS-PAGe en un gel TBE-PAGE al 4-12% (Life Technologies), se analizó y se cuantificó con el programa informático ImageLab (Bio-rad), todo siguiendo el protocolo del fabricante.
La Figura 69 representa un diseño de diferentes construcciones NLS Cas9. Todas las Cas9 eran la versión codón optimizada humana de la Sp Cas9. Se ligaron las secuencias NLS al gen cas9 en el N-terminal o C-terminal. Todas las variantes de Cas9 con diferentes diseños de NLS se clonaron en una cadena principal conteniendo así vector para ser conducido por el activador de EF1a. En el mismo vector hay un lugar EMX1 humano de fijación como objetivo de ARN quimérico conducido por activador de U6, formando juntos un sistema de dos componentes.
Tabla M. Resultados de ensayo de diseño NLS Cas9. Cuantificación de escisión genómica de diferentes construcciones cas9-nls por prueba surveyor.

La Figura 70 representa la eficacia de escisión genómica inducida por variantes de Cas9 que soportan diferentes diseños de NLS. El porcentaje indica la porción de ADN genómico de EMX1 humano que fue escindido por cada construcción. Todos los experimentos son de 3 replicados biológicos. n = 3, error indica E. E. M.
Ejemplo 15: Ingeniería del microalgas usando Cas9
Métodos de suministrar Cas9
Método 1: Los solicitantes suministran Cas9 y ARN guía usando un vector que expresa Cas9 bajo el control de un activador constitutivo tal como Hsp70A-Rbc S2 o Beta2-tubulina.
Método 2: Los solicitantes suministran Cas9 y Polimerasa T7 usando vectores que expresan Cas9 y Polimerasa T7 bajo el control de un activador constitutivo tal como Hsp70A-Rbc S2 o Beta2-tubulina. Se suministró ARN guía usando un vector que contenía activador de T7 que conduce el ARN guía.
Método 3: Los solicitantes suministran ARNm de Cas9 y ARN guía transcrito in vitro a células de algas. El ARN puede ser transcrito in vitro. El ARNm de Cas9 constará de la región codificadora para Cas9, así como 3'UTR de Cop1 para asegurar la estabilización del ARNm de Cas9.
Para recombinación homóloga, los solicitantes proporcionan una plantilla de reparación dirigida por homología adicional.
Secuencia para un casete que conduce la expresión de Cas9 bajo el control de activador de beta-2 tubulina, seguido por 3' UTR de Cop 1.
TCTTTCTTGCGCTATGACACTTCCAGCAAAAGGTAGGGCGGGCTGCGAGA CGGCTTCCCGGCGCTGCATGCAACACCGATGATGCTTCGACCCCCCGAAGCTCCTTC GGGGCTGCATGGGCGCTCCGATGCCGCTCCAGGGCGAGCGCTGTTTAAATAGCCAG GCCCCCGATTGCAAAGACATTATAGCGAGCTACCAAAGCCATATTCAAACACCTAG ATCACTACCACTTCTACACAGGCCACTCGAGCTTGTGATCGCACTCCGCTAAGGGGG CGCCTCTTCCTCTTCGTTTCAGTCACAACCCGCAAACATGTACCCATACGATGTTCCA GATTACGCTTCGCCGAAGAAAAAGCGCAAGGTCGAAGCGTCCGACAAGAAGTACAG CATCGGCCTGGACATCGGCACCAACTCTGTGGGCTGGGCCGTGATCACCGACGAGT ACAAGGTGCCCAGCAAGAAATTCAAGGTGCTGGGCAACACCGACCGGCACAGCATC AAGAAGAACCTGATCGGAGCCCTGCTGTTCGACAGCGGCGAAACAGCCGAGGCCAC CCGGCTGAAGAGAACCGCCAGAAGAAGATACACCAGACGGAAGAACCGGATCTGC TATCTGCAAGAGATCTTCAGCAACGAGATGGCCAAGGTGGACGACAGCTTCTTCCAC AGACTGGAAGAGTCCTTCCTGGTGGAAGAGGATAAGAAGCACGAGCGGCACCCCAT CTTCGGCAACATCGTGGACGAGGTGGCCTACCACGAGAAGTACCCCACCATCTACC ACCTGAGAAAGAAACTGGTGGACAGCACCGACAAGGCCGACCTGCGGCTGATCTAT CTGGCCCTGGCCCACATGATCAAGTTCCGGGGCCACTTCCTGATCGAGGGCGACCTG AACCCCGACAACAGCGACGTGGACAAGCTGTTCATCCAGCTGGTGCAGACCTACAA CCAGCTGTTCGAGGAAAACCCCATCAACGCCAGCGGCGTGGACGCCAAGGCCATCC
TGTCTGCCAGACTGAGCAAGAGCAGACGGCTGGAAAATCTGATCGCCCAGCTGCCC GGCGAGAAGAAGAATGGCCTGTTCGGCAACCTGATTGCCCTGAGCCTGGGCCTGAC CCCCAACTTCAAGAGCAACTTCGACCTGGCCGAGGATGCCAAACTGCAGCTGAGCA AGGACACCTACGACGACGACCTGGACAACCTGCTGGCCCAGATCGGCGACCAGTAC GCCGACCTGTTTCTGGCCGCCAAGAACCTGTCCGACGCCATCCTGCTGAGCGACATC CTGAGAGTGAACACCGAGATCACCAAGGCCCCCCTGAGCGCCTCTATGATCAAGAG ATACGACGAGCACCACCAGGACCTGACCCTGCTGAAAGCTCTCGTGCGGCAGCAGC TGCCTGAGAAGTACAAAGAGATTTTCTTCGACCAGAGCAAGAACGGCTACGCCGGC TACATTGACGGCGGAGCCAGCCAGGAAGAGTTCTACAAGTTCATCAAGCCCATCCT GG AAAAG ATGG ACGGCACCG AGGAACTGCTCGT GA AGCT G AAC AG AGAGG ACCT G CTGCGGAAGCAGCGGACCTTCGACAACGGCAGCATCCCCCACCAGATCCACCTGGG AGAGCTGCACGCCATTCTGCGGCGGCAGGAAGATTTTTACCCATTCCTGAAGGACAA CCGGGAAAAGATCGAGAAGATCCTGACCTTCCGCATCCCCTACTACGTGGGCCCTCT GGCCAGGGGAAACAGCAGATTCGCCTGGATGACCAGAAAGAGCGAGGAAACCATC ACCCCCTGGAACTTCGAGGAAGTGGTGGACAAGGGCGCTTCCGCCCAGAGCTTCAT CGAGCGGATGACCAACTTCGATAAGAACCTGCCCAACGAGAAGGTGCTGCCCAAGC ACAGCCTGCTGTACGAGTACTTCACCGTGTATAACGAGCTGACCAAAGTGAAATAC GTGACCGAGGGAATGAGAAAGCCCGCCTTCCTGAGCGGCGAGCAGAAAAAGGCCA TCGTGGACCTGCTGTTCAAGACCAACCGGAAAGTGACCGTGAAGCAGCTGAAAGAG GACTACTTCAAGAAAATCGAGTGCTTCGACTCCGTGGAAATCTCCGGCGTGGAAGAT CGGTTCAACGCCTCCCTGGGCACATACCACGATCTGCTGAAAATTATCAAGGACAA GGACTTCCTGGACAATGAGGAAAACGAGGACATTCTGGAAGATATCGTGCTGACCC TGACACTGTTTGAGGACAGAGAGATGATCGAGGAACGGCTGAAAACCTATGCCCAC CTGTTCGACGACAAAGTGATGAAGCAGCTGAAGCGGCGGAGATACACCGGCTGGGG CAGGCTGAGCCGGAAGCTGATCAACGGCATCCGGGACAAGCAGTCCGGCAAGACA ATCCTGGATTTCCTGAAGTCCGACGGCTTCGCCAACAGAAACTTCATGCAGCTGATC CACGACGACAGCCTGACCTTTAAAGAGGACATCCAGAAAGCCCAGGTGTCCGGCCA GGGCGATAGCCTGCACGAGCACATTGCCAATCTGGCCGGCAGCCCCGCCATTAAGA AGGGCATCCTGCAGACAGTGAAGGTGGTGGACGAGCTCGTGAAAGTGATGGGCCGG CACAAGCCCGAGAACATCGTGATCGAAATGGCCAGAGAGAACCAGACCACCCAGA AGGGACAGAAGAACAGCCGCGAGAGAATGAAGCGGATCGAAGAGGGCATCAAAGA
GCTGGGCAGCCAGATCCTGAAAGAACACCCCGTGGAAAACACCCAGCTGCAGAACG AGAAGCTGTACCTGTACTACCTGCAGAATGGGCGGGATATGTACGTGGACCAGGAA CTGGACATCAACCGGCTGTCCGACTACGATGTGGACCATATCGTGCCTCAGAGCTTT CTGAAGGACGACTCCATCGACAACAAGGTGCTGACCAGAAGCGACAAGAACCGGG GCAAGAGCGACAACGTGCCCTCCGAAGAGGTCGTGAAGAAGATGAAGAACTACTGG CGGCAGCTGCTGAACGCCAAGCTGATTACCCAGAGAAAGTTCGACAATCTGACCAA GGCCGAGAGAGGCGGCCTGAGCGAACTGGATAAGGCCGGCTTCATCAAGAGACAG CTGGTGGAAACCCGGCAGATCACAAAGCACGTGGCACAGATCCTGGACTCCCGGAT GAACACTAAGTACGACGAGAATGACAAGCTGATCCGGGAAGTGAAAGTGATCACCC TGAAGTCCAAGCTGGTGTCCGATTTCCGGAAGGATTTCCAGTTTTACAAAGTGCGCG AGATCAACAACTACCACCACGCCCACGACGCCTACCTGAACGCCGTCGTGGGAACC GCCCTGATCAAAAAGTACCCTAAGCTGGAAAGCGAGTTCGTGTACGGCGACTACAA GGTGTACGACGTGCGGAAGATGATCGCCAAGAGCGAGCAGGAAATCGGCAAGGCT ACC GCC AAGT AC TTCTTCTACAGC AACATC AT G A ACTTTTT CAAG ACCG AG ATT ACC CTGGCCAACGGCGAGATCCGGAAGCGGCCTCTGATCGAGACAAACGGCGAAACCGG GGAGATCGTGTGGGATAAGGGCCGGGATTTTGCCACCGTGCGGAAAGTGCTGAGCA TGCCCCAAGTGAATATCGTGAAAAAGACCGAGGTGCAGACAGGCGGCTTCAGCAAA GAGTCTATCCTGCCCAAGAGGAACAGCGATAAGCTGATCGCCAGAAAGAAGGACTG GGACCCTAAGAAGTACGGCGGCTTCGACAGCCCCACCGTGGCCTATTCTGTGCTGGT GGTGGCCAAAGTGGAAAAGGGCAAGTCCAAGAAACTGAAGAGTGTGAAAGAGCTG CTGGGGATCACCATCATGGAAAGAAGCAGCTTCGAGAAGAATCCCATCGACTTTCT GG AAGCC AAGGGCT ACAAAG AAGT G AAAAAGG ACCTG ATC AT C AAGCTGCCT AAGT ACTCCCTGTTCGAGCTGGAAAACGGCCGGAAGAGAATGCTGGCCTCTGCCGGCGAA CTGCAGAAGGGAAACGAACTGGCCCTGCCCTCCAAATATGTGAACTTCCTGTACCTG GCCAGCCACTATGAGAAGCTGAAGGGCTCCCCCGAGGATAATGAGCAGAAACAGCT GTTT GTGGAACAGC ACAAGCACT ACCTGGACGAG ATC ATCGAGC AGATCAGCGAGT TCTCCAAGAGAGTGATCCTGGCCGACGCTAATCTGGACAAAGTGCTGTCCGCCTACA ACAAGCACCGGGATAAGCCCATCAGAGAGCAGGCCGAGAATATCATCCACCTGTTT ACCCTGACCAATCTGGGAGCCCCTGCCGCCTTCAAGTACTTTGACACCACCATCGAC CGGAAGAGGTACACCAGCACCAAAGAGGTGCTGGACGCCACCCTGATCCACCAGAG CATCACCGGCCTGTACGAGACACGGATCGACCTGTCTCAGCTGGGAGGCGACAGCC
CCAAGAAGAAGAGAAAGGTGGAGGCCAGCTAAGGATCCGGCAAGACTGGCCCCGC TTGGCAACGCAACAGTGAGCCCCTCCCTAGTGTGTTTGGGGATGTGACTATGTATTC GTGTGTTGGCCAACGGGTCAACCCGAACAGATTGATACCCGCCTTGGCATTTCCTGT C AGAATGTAACGTCA GTTGATGGTACT
Secuencia para un casete que conduce la expresión de polimerasa T7 bajo el control de activador de beta-2 tubulina, seguido por la 3' UTR de Cop1:
TCTTTCTTGCGCTATGACACTTCCAGCAAAAGGTAGGGCGGGCTGCGAGA CGGCTTCCCGGCGCTGCATGCAACACCGATGATGCTTCGACCCCCCGAAGCTCCTTC GGGGCTGCATGGGCGCTCCGATGCCGCTCCAGGGCGAGCGCTGTTTAAATAGCCAG GCCCCCGATTGCAAAGACATTATAGCGAGCTACCAAAGCCATATTCAAACACCTAG ATCACTACCACTTCTACACAGGCCACTCGAGCTTGTGATCGCACTCCGCTAAGGGGG CGCCTCTTCCTCTTCGTTTCAGTCACAACCCGCAAACatgcctaagaagaagaggaaggttaacacgatt aacatcgctaagaacgacttctctgacatcgaactggctgctatcccgttcaacactctggctgaccattacggtgagcgtttagctcgcgaa
aacgctgccgccaagcctctcatcactaccctactccctaagatgattgcacgcatcaacgactggtttgaggaagtgaaagctaagcgcg gcaagcgcccgacagccttccagttcctgcaagaaatcaagccggaagccgtagcgtacatcaccattaagaccactctggcttgcctaac cagtgctgacaatacaaccgttcaggctgtagcaagcgcaatcggtcgggccattgaggacgaggctcgcttcggtcgtatccgtgacctt gaagctaagcacttcaagaaaaacgttgaggaacaactcaacaagcgcgtagggcacgtctacaagaaagcatttatgcaagttgtcgag gctgacatgctctctaagggtctactcggtggcgaggcgtggtcttcgtggcataaggaagactctattcatgtaggagtacgctgcatcgag atgctcattgagtcaaccggaatggttagcttacaccgccaaaatgctggcgtagtaggtcaagactctgagactatcgaactcgcacctga atacgctgaggctatcgcaacccgtgcaggtgcgctggctggcatctctccgatgttccaaccttgcgtagttcctcctaagccgtggactgg cattactggtggtggctattgggctaacggtcgtcgtcctctggcgctggtgcgtactcacagtaagaaagcactgatgcgctacgaagacg tttacatgcctgaggtgtacaaagcgattaacattgcgcaaaacaccgcatggaaaatcaacaagaaagtcctagcggtcgccaacgtaatc
gaggctctcaccgcgtggaaacgtgctgccgctgctgtgtaccgcaaggacaaggctcgcaagtctcgccgtatcagccttgagttcatgc ttgagcaagccaataagtttgctaaccataaggccatctggttcccttacaacatggactggcgcggtcgtgtttacgctgtgtcaatgttcaac
ggtgcaaactgtgcgggtgtcgacaaggttccgttccctgagcgcatcaagttcattgaggaaaaccacgagaacatcatggcttgcgctaa gtctccactggagaacacttggtgggctgagcaagattctccgttctgcttccttgcgttctgctttgagtacgctggggtacagcaccacggc ctgagctataactgctcccttccgctggcgtttgacgggtcttgctctggcatccagcacttctccgcgatgctccgagatgaggtaggtggtc gcgcggttaacttgcttcctagtgaaaccgttcaggacatctacgggattgttgctaagaaagtcaacgagattctacaagcagacgcaatca
atgggaccgataacgaagtagttaccgtgaccgatgagaacactggtgaaatctctgagaaagtcaagctgggcactaaggcactggctg gtcaatggctggcttacggtgttactcgcagtgtgactaagcgttcagtcatgacgctggcttacgggtccaaagagttcggcttccgtcaac aagtgctggaagataccattcagccagctattgattccggcaagggtctgatgttcactcagccgaatcaggctgctggatacatggctaag ctgatttgggaatctgtgagcgtgacggtggtagctgcggttgaagcaatgaactggcttaagtctgctgctaagctgctggctgctgaggtc aaagataagaagactggagagattcttcgcaagcgttgcgctgtgcattgggtaactcctgatggtttccctgtgtggcaggaatacaagaa gcctattcagacgcgcttgaacctgatgttcctcggtcagttccgcttacagcctaccattaacaccaacaaagatagcgagattgatgcaca caaacaggagtctggtatcgctcctaactttgtacacagccaagacggtagccaccttcgtaagactgtagtgtgggcacacgagaagtac ggaatcgaatcttttgcactgattcacgactccttcggtacgattccggctgacgctgcgaacctgttcaaagcagtgcgcgaaactatggttg acacatatgagtcttgtgatgtactggctgatttctacgaccagttcgctgaccagttgcacgagtctcaattggacaaaatgccagcacttcc ggctaaaggtaacttgaacctccgtgacatcttagagtcggacttcgcgttcgcgtaaGGATCCGGCAAGACTGGCCCC GCTTGGCAACGCAACAGTGAGCCCCTCCCTAGTGTGTTTGGGGATGTGACTATGTAT TCGTGTGTTGGCCAACGGGTCAACCCGAACAGATTGATACCCGCCTTGGCATTTCCT GT CAGAATGTAACGTCAGTTGATGGTACT
Secuencia de ARN guía conducida por el activador T7 (activador T7, las N representan secuencia de fijación como objetivo):
gaaatTAATACGACTCACTATANNNNNNNNNNNNNNNNNNNNettttaeaectaG AAAtagcaagttaaaataaggctagtccgttatcaacttgaaaaagtggcaccgagtcggtgcttttttt
Suministro génico:
Se usarán cepas CC-124 y CC-125 de Chlamydomonas reinhardtii del centro de recursos de Chlamydomonas para electroporación. El protocol de electroporación sigue el protocolo recomendado estándar del estuche de ingeniería de Chlamydomonas GeneArt.
También, los solicitantes generan una línea de Chlamydomonas reinhardtii que expresa Cas9 de manera constitutiva. Esto se puede realizar usando pChlamy1 (linearizado usando PvuI) y seleccionando colonias resistentes a higromicina. La secuencia para pChlamy1 que contiene Cas9 está a continuación. De esta manera para conseguir inactivación génica se requiere simplemente suministrar ARN para el ARN guía. Para recombinación homóloga, los solicitantes suministran ARN guía, así como una plantilla de recombinación homóloga linearizada.
pChlamy1-Cas9:
TGCGGTATTTCACACCGCATCAGGTGGCACTTTTCGGGGAAATGTGCGCG G AACCCCTATTT GTTT ATTTTT CT AAAT AC ATTC AAAT AT GT ATCCGCTC AT G AG ATT ATCAAAAAGGATCTTCACCTAGATCCTTTTAAATTAAAAATGAAGTTTTAAATCAAT
CT AAAGT AT AT ATGAGT AA ACTT GGTCTG AC AGTTACCAATGCTT AATC AGTGAGGC ACCT ATCTC AGCGAT CTGTCT ATTTCGTT CAT CCAT AGTTGCCT G ACTCCCCGT CGTG TAGATAACTACGATACGGGAGGGCTTACCATCTGGCCCCAGTGCTGCAATGATACCG CGAGACCCACGCTCACCGGCTCCAGATTTATCAGCAATAAACCAGCCAGCCGGAAG GGCCGAGCGCAGAAGTGGTCCTGCAACTTTATCCGCCTCCATCCAGTCTATTAATTG TTGCCGGGAAGCTAGAGTAAGTAGTTCGCCAGTTAATAGTTTGCGCAACGTTGTTGC CATTGCTACAGGCATCGTGGTGTCACGCTCGTCGTTTGGTATGGCTTCATTCAGCTCC GGTTCCC A ACGATC A AGGCGAGTT AC AT GATCCCCC AT GTT GTGC A A A AA AGCGGTT AGCTCCTTCGGTCCTCCGATCGTTGTCAGAAGTAAGTTGGCCGCAGTGTTATCACTC ATGGTTATGGCAGCACTGCATAATTCTCTTACTGTCATGCCATCCGTAAGATGCTTTT CTGTGACTGGTGAGTACTCAACCAAGTCATTCTGAGAATAGTGTATGCGGCGACCGA GTTGCTCTTGCCCGGCGTCAATACGGGATAATACCGCGCCACATAGCAGAACTTTAA AAGTGCTCATCATTGGAAAACGTTCTTCGGGGCGAAAACTCTCAAGGATCTTACCGC TGTTGAGATCCAGTTCGATGTAACCCACTCGTGCACCCAACTGATCTTCAGCATCTTT TACTTTCACCAGCGTTTCTGGGTGAGCAAAAACAGGAAGGCAAAATGCCGCAAAAA AGGGAAT AAGGGCGACACGGAA ATGTTGAAT ACTCAT ACTCTTCCTTTTTCAAT ATT ATTGAAGCATTTATCAGGGTTATTGTCTCATGACCAAAATCCCTTAACGTGAGTTTTC GTTCCACTGAGCGTCAGACCCCGTAGAAAAGATCAAAGGATCTTCTTGAGATCCTTT TTTTCTGCGCGTAATCTGCTGCTTGCAAACAAAAAAACCACCGCTACCAGCGGTGGT TTGTTTGCCGGATCAAGAGCTACCAACTCTTTTTCCGAAGGTAACTGGCTTCAGCAG AGCGCAGATACCAAATACTGTTCTTCTAGTGTAGCCGTAGTTAGGCCACCACTTCAA GAACTCTGTAGCACCGCCTACATACCTCGCTCTGCTAATCCTGTTACCAGTGGCTGTT GCCAGTGGCGATAAGTCGTGTCTTACCGGGTTGGACTCAAGACGATAGTTACCGGAT AAGGCGCAGCGGTCGGGCTGAACGGGGGGTTCGTGCACACAGCCCAGCTTGGAGCG AACGACCTACACCGAACTGAGATACCTACAGCGTGAGCTATGAGAAAGCGCCACGC TTCCCGAAGGGAGAAAGGCGGACAGGTATCCGGTAAGCGGCAGGGTCGGAACAGG AGAGCGCACGAGGGAGCTTCCAGGGGGAAACGCCTGGTATCTTTATAGTCCTGTCG GGTTTCGCCACCTCTGACTTGAGCGTCGATTTTTGTGATGCTCGTCAGGGGGGCGGA GCCTATGGAAAAACGCCAGCAACGCGGCCTTTTTACGGTTCCTGGCCTTTTGCTGGC CTTTTGCTCACATGTTCTTTCCTGCGTTATCCCCTGATTCTGTGGATAACCGTATTACC GCCTTTGAGTGAGCTGATACCGCTCGCCGCAGCCGAACGACCGAGCGCAGCGAGTC
AGTGAGCGAGGAAGCGGTCGCTGAGGCTTGACATOATTGGTGCGTATGTTTGTATGA AGCTACAGGACTGATTTGGCGGGCTATGAGGGCGGGGGAAGCTCTGGAAGGGCCGC GATGGGGCGCGCGGCGTCCAGAAGGCGCCATACGGCCCGCTGGCGGCACCCATCCG GTATAAAAGCCCGCGACCCCGAACGGTGACCTCCACTTTCAGCGACAAACGAGCAC TTATACATACGCGACTATTCTGCCGCTATACATAACCACTCAGCTAGCTTAAGATCC CATCAAGCTTGCATGCCGGGCGCGCCAGAAGGAGCGCAGCCAAACCAGGATGATGT TTGATGGGGTATTTGAGCACTTGCAACCCTTATCCGGAAGCCCCCTGGCCCACAAAG GCTAGGCGCCAATGCAAGCAGTTCGCATGCAGCCCCTGGAGCGGTGCCCTCCTGAT AAACCGGCCAGGGGGCCTATGTTCTTTACTTTTTTACAAGAGAAGTCACTCAACATC TTAAAATGGCCAGGTGAGTCGACGAGCAAGCCCGGCGGATCAGGCAGCGTGCTTGC AGATTTGACTTGCAACGCCCGCATTGTGTCGACGAAGGCTTTTGGCTCCTCTGTCGCT GTCTCAAGCAGCATCTAACCCTGCGTCGCCGTTTCCATTTGCAGGAGATTCGAGGTA CCATGTACCCATACGATGTTCCAGATTACGCTTCGCCGAAGAAAAAGCGCAAGGTC GAAGCGTCCGACAAGAAGTACAGCATCGGCCTGGACATCGGCACCAACTCTGTGGG CTGGGCCGTGATCACCGACGAGTACAAGGTGCCCAGCAAGAAATTCAAGGTGCTGG GCAACACCGACCGGCACAGCATCAAGAAGAACCTGATCGGAGCCCTGCTGTTCGAC AGCGGCGAAACAGCCGAGGCCACCCGGCTGAAGAGAACCGCCAGAAGAAGATACA CCAGACGGAAGAACCGGATCTGCTATCTGCAAGAGATCTTCAGCAACGAGATGGCC AAGGTGGACGACAGCTTCTTCCACAGACTGGAAGAGTCCTTCCTGGTGGAAGAGGA TAAGAAGCACGAGCGGCACCCCATCTTCGGCAACATCGTGGACGAGGTGGCCTACC ACGAGAAGTACCCCACCATCTACCACCTGAGAAAGAAACTGGTGGACAGCACCGAC AAGGCCGACCTGCGGCTGATCTATCTGGCCCTGGCCCACATGATCAAGTTCCGGGGC CACTTCCTGATCGAGGGCGACCTGAACCCCGACAACAGCGACGTGGACAAGCTGTT CATCCAGCTGGTGCAGACCTACAACCAGCTGTTCGAGGAAAACCCCATCAACGCCA GCGGCGTGGACGCCAAGGCCATCCTGTCTGCCAGACTGAGCAAGAGCAGACGGCTG GAAAATCTGATCGCCCAGCTGCCCGGCGAGAAGAAGAATGGCCTGTTCGGCAACCT GATTGCCCTGAGCCTGGGCCTGACCCCCAACTTCAAGAGCAACTTCGACCTGGCCGA GGATGCCAAACTGCAGCTGAGCAAGGACACCTACGACGACGACCTGGACAACCTGC TGGCCCAGATCGGCGACCAGTACGCCGACCTGTTTCTGGCCGCCAAGAACCTGTCCG ACGCCATCCTGCTGAGCGACATCCTGAGAGTGAACACCGAGATCACCAAGGCCCCC CTGAGCGCCTCTATGATCAAGAGATACGACGAGCACCACCAGGACCTGACCCTGCT
GAAAGCTCTCGTGCGGCAGCAGCTGCCTGAGAAGTACAAAGAGATTTTCTTCGACC AGAGCAAGAACGGCTACGCCGGCTACATTGACGGCGGAGCCAGCCAGGAAGAGTTC TACAAGTTCATCAAGCCCATCCTGGAAAAGATGGACGGCACCGAGGAACTGCTCGT GAAGCTGAACAGAGAGGACCTGCTGCGGAAGCAGCGGACCTTCGACAACGGCAGC AT CCCCC ACCAG AT CC ACCTGGGAG AGC'T GCACGCC ATT CT GCGGCGGC AGGAAGA TTTTTACCCATTCCTGAAGGACAACCGGGAAAAGATCGAGAAGATCCTGACCTTCCG CATCCCCTACTACGTGGGCCCTCTGGCCAGGGGAAACAGCAGATTCGCCTGGATGA CCAGAAAGAGCGAGGAAACCATCACCCCCTGGAACTTCGAGGAAGTGGTGGACAA GGGCGCTTCCGCCCAGAGCTTCATCGAGCGGATGACCAACTTCGATAAGAACCTGC CCAACGAGAAGGTGCTGCCCAAGCACAGCCTGCTGTACGAGTACTTCACCGTGTAT AACGAGCTGACCAAAGTGAAATACGTGACCGAGGGAATGAGAAAGCCCGCCTTCCT GAGCGGCGAGCAGAAAAAGGCCATCGTGGACCTGCTGTTCAAGACCAACCGGAAA GTGACCGTGAAGCAGCTGAAAGAGGACTACTTCAAGAAAATCGAGTGCTTCGACTC CGTGGAAATCTCCGGCGTGGAAGATCGGTTCAACGCCTCCCTGGGCACATACCACG ATCTGCTGAAAATTATCAAGGACAAGGACTTCCTGGACAATGAGGAAAACGAGGAC ATTCTGGAAGATATCGTGCTGACCCTGACACTGTTTGAGGACAGAGAGATGATCGA GG AACGGCT G AA AACCTAT GCCC ACCTGTT C'G ACG AC AAAGT G AT GAAG CAGCT G A AGCGGCGGAGATACACCGGCTGGGGCAGGCTGAGCCGGAAGCTGATCAACGGCATC CGGGACAAGCAGTCCGGCAAGACAATCCTGGATTTCCTGAAGTCCGACGGCTTCGC CAACAGAAACTTCATGCAGCTGATCCACGACGACAGCCTGACCTTTAAAGAGGACA TCCAGAAAGCCCAGGTGTCCGGCCAGGGCGATAGCCTGCACGAGCACATTGCCAAT CTGGCCGGCAGCCCCGCCATTAAGAAGGGCATCCTGCAGACAGTGAAGGTGGTGGA CGAGCTCGTGAAAGTGATGGGCCGGCACAAGCCCGAGAACATCGTGATCGAAATGG CCAGAGAGAACCAGACCACCCAGAAGGGACAGAAGAACAGCCGCGAGAGAATGAA GCGGATCGAAGAGGGCATCAAAGAGCTGGGCAGCCAGATCCTGAAAGAACACCCC GTGGAAAACACCCAGCTGCAGAACGAGAAGCTGTACCTGTACTACCTGCAGAATGG GCGGGATATGTACGTGGACCAGGAACTGGACATCAACCGGCTGTCCGACTACGATG TGGACCATATCGTGCCTCAGAGCTTTCTGAAGGACGACTCCATCGACAACAAGGTGC TGACCAGAAGCGACAAGAACCGGGGCAAGAGCGACAACGTGCCCTCCGAAGAGGT CGTGAAGAAGATGAAGAACTACTGGCGGCAGCTGCTGAACGCCAAGCTGATTACCC AGAGAAAGTTCGACAATCTGACCAAGGCCGAGAGAGGCGGCCTGAGCGAACTGGAT
AAGGCCGGCTTCATCAAGAGACAGCTGGTGGAAACCCGGCAGATCACAAAGCACGT GGCACAGATCCTGGACTCCCGGATGAACACTAAGTACGACGAGAATGACAAGCTGA TCCGGGAAGTGAAAGTGATCACCCTGAAGTCCAAGCTGGTGTCCGATTTCCGGAAG GATTTCCAGTTTTACAAAGTGCGCGAGATCAACAACTACCACCACGCCCACGACGCC TACCTGAACGCCGTCGTGGGAACCGCCCTGATCAAAAAGTACCCTAAGCTGGAAAG CGAGTTCGTGTACGGCGACTACAAGGTGTACGACGTGCGGAAGATGATCGCCAAGA GCGAGCAGGAAATCGGCAAGGCTACCGCCAAGTACTTCTTCTACAGCAACATCATG AACTTTTTCAAGACCGAGATTACCCTGGCCAACGGCGAGATCCGGAAGCGGCCTCT GATCGAGACAAACGGCGAAACCGGGGAGATCGTGTGGGATAAGGGCCGGGATTTTG CCACCGTGCGGAAAGTGCTGAGCATGCCCCAAGTGAATATCGTGAAAAAGACCGAG GTGCAGACAGGCGGCTTCAGCAAAGAGTCTATCCTGCCCAAGAGGAACAGCGATAA GCTGATCGCCAGAAAGAAGGACTGGGACCCTAAGAAGTACGGCGGCTTCGACAGCC CCACCGTGGCCTATTCTGTGCTGGTGGTGGCCAAAGTGGAAAAGGGCAAGTCCAAG AAACTGAAGAGTGTGAAAGAGCTGCTGGGGATCACCATCATGGAAAGAAGCAGCTT CGAGAAGAATCCCATCGACTTTCTGGAAGCCAAGGGCTACAAAGAAGTGAAAAAGG ACCTGATCATCAAGCTGCCTAAGTACTCCCTGTTCGAGCTGGAAAACGGCCGGAAG AGAATGCTGGCCTCTGCCGGCGAACTGCAGAAGGGAAACGAACTGGCCCTGCCCTC CAAATATGTGAACTTCCTGTACCTGGCCAGCCACTATGAGAAGCTGAAGGGCTCCCC CGAGGATAATGAGCAGAAACAGCTGTTTGTGGAACAGCACAAGCACTACCTGGACG AG ATCAT CG AG C A GAT C AGC G AGTTCT C CAAG AG AGTG AT CCTG G CCG ACG CT AAT CTGGACAAAGTGCTGTCCGCCTACAACAAGCACCGGGATAAGCCCATCAGAGAGCA GGCCGAGAATATCATCCACCTGTTTACCCTGACCAATCTGGGAGCCCCTGCCGCCTT C AAGTACTTT G AC ACC ACC ATCGACC GGAAG AGGTAC ACC AGC ACCAAAG AGGTGC TGGACGCCACCCTGATCCACCAGAGCATCACCGGCCTGTACGAGACACGGATCGAC CTGTCTCAGCTGGGAGGCGACAGCCCCAAGAAGAAGAGAAAGGTGGAGGCCAGCT AACATATGATTCGAATGTCTTTCTTGCGCTATGACACTTCCAGCAAAAGGTAGGGCG GGCTGCGAGACGGCTTCCCGGCGCTGCATGCAACACCGATGATGCTTCGACCCCCCG AAGCTCCTTCGGGGCTGCAT GGGCGCTCCGATGCCGCTCCAGGGCGAGCGCTGTTT A AATAGCCAGGCCCCCGATTGCAAAGACATTATAGCGAGCTACCAAAGCCATATTCA AAC ACCT AGAT C ACT ACC ACTTCTAC AC AGGC CACTCGAGCTTGT G ATC G CACTCCG CTAAGGGGGCGCCTCTTCCTCTTCGTTTCAGTCACAACCCGCAAACATGACACAAGA
ATCCCTGTTACTTCTCGACCGTATTGATTCGGATGATTCCTACGCGAGCCTGCGGAA CGACCAGGAATTCTGGGAGGTGAGTCGACGAGCAAGCCCGGCGGATCAGGCAGCGT GCTTGCAGATTTGACTTGCAACGCCCGCATTGTGTCGACGAAGGCTTTTGGCTCCTCT GTCGCTGTCTCAAGCAGCATCTAACCCTGCGTCGCCGTTTCCATTTGCAGCCGCTGG CCCGCCGAGCCCTGGAGGAGCTCGGGCTGCCGGTGCCGCCGGTGCTGCGGGTGCCC GGCGAGAGCACCAACCCCGTACTGGTCGGCGAGCCCGGCCCGGTGATCAAGCTGTT CGGCGAGCACTGGTGCGGTCCGGAGAGCCTCGCGTCGGAGTCGGAGGCGTACGCGG TCCTGGCGGACGCCCCGGTGCCGGTGCCCCGCCTCCTCGGCCGCGGCGAGCTGCGGC CCGGCACCGGAGCCTGGCCGTGGCCCTACCTGGTGATGAGCCGGATGACCGGCACC ACCTGGCGGTCCGCGATGGACGGCACGACCGACCGGAACGCGCTGCTCGCCCTGGC CCGCGAACTCGGCCGGGTGCTCGGCCGGCTGCACAGGGTGCCGCTGACCGGGAACA CCGTGCTCACCCCCCATTCCGAGGTCTTCCCGGAACTGCTGCGGGAACGCCGCGCGG CGACCGTCGAGGACCACCGCGGGTGGGGCTACCTCTCGCCCCGGCTGCTGGACCGC CTGGAGGACTGGCTGCCGGACGTGGACACGCTGCTGGCCGGCCGCGAACCCCGGTT CGTCCACGGCGACCTGCACGGGACCAACATCTTCGTGGACCTGGCCGCGACCGAGG TCACCGGGATCGTCGACTTCACCGACGTCTATGCGGGAGACTCCCGCTACAGCCTGG TGCAACTGCATCTCAACGCCTTCCGGGGCGACCGCGAGATCCTGGCCGCGCTGCTCG ACGGGGCGCAGTGGAAGCGGACCGAGGACTTCGCCCGCGAACTGCTCGCCTTCACC TTCCTGCACGACTTCGAGGTGTTCGAGGAGACCCCGCTGGATCTCTCCGGCTTCACC GATCCGGAGGAACTGGCGCAGTTCCTCTGGGGGCCGCCGGACACCGCCCCCGGCGC CTGATAAGGATCCGGCAAGACTGGCCCCGCTTGGCAACGCAACAGTGAGCCCCTCC CTAGTGTGTTTGGGGATGTGACTATGTATTCGTGTGTTGGCCAACGGGTCAACCCGA ACAG ATT G ATACCCGCCTT GGC ATTTCCTGTC AG AAT GT AACGTC AGTTGAT GGTAC T
Para todas las células de Chlamydomonas reinhardtii, los solicitantes usaron PCR, prueba de la nucleasa SURVEYOR y secuenciación de ADN para verificar la modificación exitosa.
Ejemplo 16: Uso de Cas9 como un represor transcripcional en bacterias.
La capacidad para transcripción de control de manera artificial es esencial tanto para el estudio de la función génica como para la construcción de redes de genes sintéticos con propiedades deseadas. Los solicitantes describen en la presente el uso de la proteína Cas9 guiada por ARN como un represor transcripcional programable.
Los solicitantes han demostrado previamente cómo puede usarse la proteína Cas9 de SF370 de Streptococcus pyogenes para dirigir la edición de genomas en Streptococcus pneumoniae. En este estudio, los solicitantes lograron la cepa crR6Rk que contenía un sistema CRISPR mínimo, que consistía en cas9, el ARNtracr y una repetición. Se introdujeron las mutaciones D10A-H840 en cas9 en esta cepa, proporcionando cepa crR6Rk**. Se clonaron cuatro espaciadores fijando como objetivo diferentes posiciones del activador del gen de p-galactosidasa bgaA en la matriz CRISPR por el plásmido pDB98 descrito previamente. Los solicitantes observaron una reducción de X a Y veces en actividad de la p-galactosidasa dependiendo de la posición fijada como objetivo, que demuestra el potencial de Cas9 como un represor programable (Figura 73).
Para conseguir la represión de Cas9** en Escherichia coli se construyó un plásmido informador de proteína fluorescente verde (g Fp , por sus siglas en inglés) (pDB127) para expresar el gen gfpmut2 de un activador constitutivo.
Se diseñó el activador para que soportara varias PAM de NPP en las dos cadenas, para medir el efecto de la unión de Cas9** en varias posiciones. Los solicitantes introdujeron las mutaciones D10A-H840 en pCas9, un plásmido que se describe que soporta el ARNtracr, cas9 y una matriz de CRISPR mínima diseñada para la clonación fácil de nuevos espaciadores. Se diseñaron veintidós espaciadores diferentes para fijar como objetivo diferentes regiones del activador de gfpmut2 y marco de lectura abierto. Se observó una reducción de fluorescencia de aproximadamente 20 veces en las regiones de fijación como objetivo que solapan o están adyacentes a los elementos activadores -35 y -10 y a la secuencia Shine-Dalgarno. Los objetivos en ambas cadenas mostraron niveles de represión similares. Estos resultados sugieren que la unión de Cas9** a cualquier posición de la región activadora evita la iniciación de la transcripción, presumiblemente por inhibición estérica de la unión de RNAP.
Para determinar si Cas9** podía evitar la extensión de la transcripción, los solicitantes la dirigieron al marco de lectura de gpfmut2. Se observó una reducción en la fluorescencia cuando las cadenas codificante y no codificante eran fijadas como objetivo, que sugiere que la unión de Cas9 es en realidad suficientemente fuerte para representar un obstáculo para el funcionamiento de RNAP. Sin embargo, aunque se observó un 40% de reducción en la expresión cuando la cadena codificante era el objetivo, se observó una reducción de 20 veces para la cadena no codificante (Fig 21b, compare T9, T10 y T11 a B9, B10 y B11). Para determinar directamente los efectos de la unión de Cas9** en la transcripción, los solicitantes extrajeron ARN de cepas que soportaban T5, T10, B10 o una construcción de control que no fija como objetivo pDB127 y la sometieron a análisis Northern usando una sonda que se une antes (B477) o después de (B510) los sitios fijados como objetivo B10 y T10. Consistente con los métodos de fluorescencia de los solicitantes, no se detectó transcripción de gfpmut2 cuando se dirigió Cas9** a la región activadora (objetivo T5) y se observó una transcripción después de la fijación como objetivo de la región T10. Convenientemente, se observó un transcrito más pequeño con la sonda B477. Esta banda corresponde al tamaño esperado de un transcrito que se interrumpiría por Cas9** y es una indicación directa de una terminación transcripcional ocasionada por la unión de ARNdg::Cas9** a la cadena codificante. Sorprendentemente, los solicitantes no detectaron transcrito cuando la cadena no codificante fue fijada como objetivo (B10). Puesto que la unión de Cas9** a la región B10 es improbable que interfiera con la iniciación de la transcripción, este resultado sugiere que se degradó el ARNm. Se mostró que ARNdg::Cas9 se unía a ARNmc in vitro. Los solicitantes especulan que la unión puede provocar la degradación del ARNm por nucleasas huésped. Por supuesto, la inmovilización de los ribosomas puede inducir la escisión en el ARNm traducido en E. coli.
Algunas solicitudes requieren una afinación precisa de la expresión génica en vez de su completa represión. Los solicitantes buscaron conseguir niveles de represión intermedios por la introducción de desajustes que debilitarían las interacciones ARNcr/objetivo. Los solicitantes crearon una serie de espaciadores basados en las construcciones B1, T5 y B10 con números crecientes de mutaciones en el extremo 5' del ARNcr. Hasta 8 mutaciones en B1 y T5 no afectaban al nivel de represión y se observó un progresivo aumento en la fluorescencia para mutaciones adicionales.
La represión observada con sólo una igualación de 8 nt entre el ARNcr y su objetivo plantea la cuestión de efectos inesperados del uso de Cas9** como un regulador transcripcional. Puesto que también se requiere una buena PAM (NGG) para unión de Cas9, el número de nucleótidos para igualar obtener algún nivel de respiración es 10. Una igualación de 10 nt tiene lugar aleatoriamente una vez cada ~1 Mpb y tales sitios es así improbable que se encuentren incluso en genomas bacterianos pequeños. Sin embargo, para reprimir con eficacia la transcripción, se requiere que dicho sitio esté en una región activadora de gen, que hace mucho menos probable el efecto inesperado. Los solicitantes también demostraron que la expresión génica puede verse afectada si la cadena no codificante de un gen se fija como objetivo. Para que esto ocurra, un objetivo aleatorio tendría que estar en la orientación correcta, pero tales casos and the o, dicho sitio es un objetivo aleatorio tendría que estar en la orientación correcta pero que tales casos ocurran relativamente más probablemente. De hecho, durante el transcurso de este estudio, los solicitantes no pudieron construir uno de los espaciadores diseñados en pCas9**. Los solicitantes encontraron más tarde que este espaciador mostraba una igualación de 12 pb próxima a una buena PAM en el gen murC esencial. Dicho efecto inesperado podía evitarse fácilmente mediante un chorro sistemático de los espaciadores diseñados.
Referencias
1. Urnov, F.D., Rebar, E.J., Holmes, M.C., Zhang, H.S. & Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11, 636-646 (2010).
2. Bogdanove, A.J. & Voytas, D.F. TAL effectors: customizable proteins for DNA targeting. Science 333, 1843-1846 (2011).
3. Stoddard, B.L. Homing endonuclease structure and function. Q. Rev. Biophys. 38, 49-95 (2005).
4. Bae, T. & Schneewind, O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55, 58-63 (2006).
5. Sung, C.K., Li, H., Claverys, J.P. & Morrison, D.A. An rpsL cassette, janus, for gene replacement through negative selection in Streptococcus pneumoniae. Appl. Environ. Microbiol. 67, 5190-5196 (2001).
6. Sharan, S.K., Thomason, L.C., Kuznetsov, S.G. & Court, D.L. Recombineering: a homologous recombinationbased method of genetic engineering. Nat. Protoc. 4, 206-223 (2009).
7. Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816-821 (2012).
8. Deveau, H., Garneau, J.E. & Moineau, S. CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 64, 475-493 (2010).
9. Horvath, P. & Barrangou, R. CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167-170 (2010).
10. Terns, M.P. & Terns, R.M. CRISPR-based adaptive immune systems. Curr. Opin. Microbiol. 14, 321-327 (2011).
11. van der Oost, J., Jore, M.M., Westra, E.R., Lundgren, M. & Brouns, S.J. CRISPR-based adaptive and heritable immunity in prokaryotes. Trends. Biochem. Sci. 34, 401-407 (2009).
12. Brouns, S.J. et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960-964 (2008).
13. Carte, J., Wang, R., Li, H., Terns, R.M. & Terns, M.P. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 22, 3489-3496 (2008).
14. Deltcheva, E. et al. CRiSp R RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602-607 (2011).
15. Hatoum-Aslan, A., Maniv, I. & Marraffini, L.A. Mature clustered, regularly interspaced, short palindromic repeats RNA (crRNA) length is measured by a ruler mechanism anchored at the precursor processing site. Proc. Natl. Acad. Sci. U.S.A. 108, 21218-21222 (2011).
16. Haurwitz, R.E., Jinek, M., Wiedenheft, B., Zhou, K. & Doudna, J.A. Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329, 1355-1358 (2010).
17. Deveau, H. et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol.
190, 1390-1400 (2008).
18. Gasiunas, G., Barrangou, R., Horvath, P. & Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A. (2012).
19. Makarova, K.S., Aravind, L., Wolf, Y.I. & Koonin, E.V. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Direct. 6, 38 (2011).
20. Barrangou, R. RNA-mediated programmable DNA cleavage. Nat. Biotechnol. 30, 836-838 (2012).
21. Brouns, S.J. Molecular biology. A Swiss army knife of immunity. Science 337, 808-809 (2012).
22. Carroll, D. A CRISPR Approach to Gene Targeting. Mol. Ther. 20, 1658-1660 (2012).
23. Bikard, D., Hatoum-Aslan, A., Mucida, D. & Marraffini, L.A. CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cel1Host Microbe 12, 177-186 (2012).
24. 24. Sapranauskas, R. et al. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. (2011).
25. Semenova, E. et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U.S.A. (2011).
26. Wiedenheft, B. et al. RNA-guided complex from a bacterial immune system enhances target recognition through seed sequence interactions. Proc. Natl. Acad. Sci. U.S.A. (2011).
27. Zahner, D. & Hakenbeck, R. The Streptococcus pneumoniae beta-galactosidase is a surface protein. J. Bacteriol.
182, 5919-5921 (2000).
28. Marraffini, L.A., Dedent, A.C. & Schneewind, O. Sortases and the art of anchoring proteins to the envelopes of gram-positive bacteria. Microbiol. Mol. Biol. Rev. 70, 192-221 (2006).
29. Motamedi, M.R., Szigety, S.K. & Rosenberg, S.M. Double-strand-break repair recombination in Escherichia coli: physical evidence for a DNA replication mechanism in vivo. Genes Dev. 13, 2889-2903 (1999).
30. Hosaka, T. et al. The novel mutation K87E in ribosomal protein S12 enhances protein synthesis activity during the late growth phase in Escherichia coli. Mol. Genet. Genomics 271, 317-324 (2004).
31. Costantino, N. & Court, D.L. Enhanced levels of lambda Red-mediated recombinants in mismatch repair mutants. Proc. Natl. Acad. Sci. U.S.A. 100, 15748-15753 (2003).
32. Edgar, R. & Qimron, U. The Escherichia coli CRISPR system protects from lambda lysogenization, lysogens, and prophage induction. J. Bacteriol. 192, 6291-6294 (2010).
33. Marraffini, L.A. & Sontheimer, E.J. Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463, 568-571 (2010).
34. Fischer, S. et al. An archaeal immune system can detect multiple Protospacer Adjacent Motifs (PAMs) to target invader DNA. J. Biol. Chem. 287, 33351-33363 (2012).
35. Gudbergsdottir, S. et al. Dynamic properties of the Sulfolobus CRISPR/Cas and CRISPR/Cmr systems when challenged with vector-borne viral and plasmid genes and protospacers. Mol. Microbiol. 79, 35-49 (2011).
36. Wang, H.H. et al. Genome-scale promoter engineering by coselection MAGE. Nat Methods 9, 591-593 (2012).
37. Cong, L. et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science In press (2013).
38. Mali, P. et al. RNA-Guided Human Genome Engineering via Cas9. Science In press (2013).
39. Hoskins, J. et al. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 183, 5709-5717 (2001).
40. Havarstein, L.S., Coomaraswamy, G. & Morrison, D.A. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc. Natl. Acad. Sci. U.S.A. 92, 11140-11144 (1995).
41. Horinouchi, S. & Weisblum, B. Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J. Bacteriol. 150, 815-825 (1982).
42. Horton, R.M. In Vitro Recombination and Mutagenesis of DNA : SOEing Together Tailor-Made Genes. Methods Mol. Biol. 15, 251-261 (1993).
43. Podbielski, A., Spellerberg, B., Woischnik, M., Pohl, B. & Lutticken, R. Novel series of plasmid vectors for gene inactivation and expression analysis in group A streptococci (GAS). Gene 177, 137-147 (1996).
44. Husmann, L.K., Scott, J.R., Lindahl, G. & Stenberg, L. Expression of the Arp protein, a member of the M protein family, is not sufficient to inhibit phagocytosis of Streptococcus pyogenes. Infection and immunity 63, 345-348 (1995).
45. Gibson, D.G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343 345 (2009).
Claims (29)
1. Una composición que comprende un sistema de repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas (CRISPR, por sus siglas en inglés), comprendiendo el sistema CRISPR:
- una proteína Cas9 que comprende una o más secuencias de localización nuclear (NLS);
- una secuencia guía unida a una secuencia de emparejamiento tracr y
- una secuencia tracr con 30 o más nucleótidos de longitud;
en donde la secuencia guía dirige la unión específica de la secuencia de un complejo CRISPR a una secuencia diana en una célula eucariota.
2. La composición según la reivindicación 1, en donde dicha secuencia guía, dicha secuencia de emparejamiento tracr y dicha secuencia tracr están comprendidas en un ARN quimérico.
3. La composición según la reivindicación 1 o 2, en donde la Cas9 dirige la escisión de una o dos cadenas en la posición de la secuencia diana.
4. La composición según la reivindicación 3, en donde la Cas9 muta con respecto a una enzima natural correspondiente de manera que la Cas9 mutada carece de la capacidad para escindir una o ambas cadenas de un polinucleótido diana que contiene una secuencia diana.
5. La composición según la reivindicación 4, en donde la Cas9 comprende una mutación seleccionada del grupo que consiste en D10A, H840A, N854A y N863A con referencia a la numeración de la posición de una proteína Cas9 de Streptococcus pyogenes (SpCas9).
6. La composición según una cualquiera de las reivindicaciones 1 a 5, en donde la Cas9 comprende una o más NLS en el amino terminal, o una o más NLS en el carboxi terminal, o una combinación de estos.
7. La composición según una cualquiera de las reivindicaciones 1 a 6, en donde la composición de Cas9 comprende al menos dos NLS.
8. La composición según una cualquiera de las reivindicaciones 1 a 7, en donde la Cas9 es S. pyogenes o Cas9 de S. thermophilus.
9. La composición según una cualquiera de las reivindicaciones 1 a 8, en donde la secuencia tracr tiene 50 o más nucleótidos de longitud.
10. La composición según una cualquiera de las reivindicaciones 1 a 9, en donde dicha secuencia tracr comprende una secuencia tracr natural.
11. La composición según una cualquiera de las reivindicaciones 1 a 10, en donde la Cas9 incluye uno o más dominios de proteína heteróloga.
12. Un sistema vector (CRISPR-Cas) de repeticiones palindrómicas cortas agrupadas y regularmente interespaciadas (CRISPR) y (Cas) asociado a CRISPR que comprende uno o más vectores que comprenden:
a) un primer elemento regulador ligado de manera operable a uno o más elementos de un sistema CRISPR de tipo II que comprende secuencias que codifican:
1) una secuencia guía ligada a una secuencia de emparejamiento tracr,
2) una secuencia tracr,
b) un segundo elemento regulador unido de manera operable a una secuencia codificadora de proteínas que codifica una proteína Cas9 que comprende una o más secuencias de localización nucleares (las NLS);
en donde los componentes (a) y (b) se sitúan en los mismos vectores del sistema o en diferentes y
en donde la hibridación entre una secuencia diana y una secuencia guía activa la formación de un complejo CRISPR en una célula eucariota.
13. El sistema vector según la reivindicación 12, en donde los componentes (a) y (b) están situados en el mismo vector.
14. El sistema vector según una cualquiera de las reivindicaciones 12 o 13, en donde la proteína Cas9 comprende al menos dos NLS.
15. El sistema vector según una cualquiera de las reivindicaciones 12 a 14, en donde la secuencia tracr tiene 30 o más nucleótidos de longitud, opcionalmente la secuencia tracr tiene 50 o más nucleótidos de longitud.
16. El sistema vector según una cualquiera de las reivindicaciones 12 a 15, en donde la secuencia tracr y la secuencia de emparejamiento tracr están contenidas en un transcripto único, de manera que la hibridación entre las dos produce una estructura secundaria que es una horquilla.
17. El sistema vector según una cualquiera de las reivindicaciones 12 a 16, en donde el sistema vector codifica dos o más secuencias guía, en donde cuando se expresa, cada una de las dos o más secuencias guía dirige la unión específica de la secuencia de un complejo CRISPR a una secuencia diana diferente en una célula eucariota.
18. El sistema vector según una cualquiera de las reivindicaciones 12 a 17, en donde el vector es un vector vírico.
19. El sistema vector según la reivindicación 18, en donde el vector vírico es un retrovirus, un lentivirus, un adenovirus, un virus adenoasociado o un vector vírico de herpes simple.
20. El sistema vector según una cualquiera de las reivindicaciones 12 a 19, que comprende uno o más activadores de pol III.
21. La composición según una cualquiera de las reivindicaciones 1 a 11 o el sistema vector según una cualquiera de las reivindicaciones 12 a 20, para uso en terapia.
22. La composición o el sistema vector para uso según la reivindicación 21, en donde dicha terapia es terapia génica.
23. La composición o el sistema vector según la reivindicación 22, para uso en la modificación de un polinucleótido diana en una célula eucariota, en donde el método comprende permitir que se una un complejo CRISPR al polinucleótido diana para efectuar la escisión de dicho polinucleótido diana modificando de ese modo el polinucleótido diana.
24. Uso de la composición según una cualquiera de las reivindicaciones 1 a 11 o el sistema vector según una cualquiera de las reivindicaciones 12 a 20, en la producción de un animal, distinto de ser humano, modificado de manera genética, siempre que el uso no sea un método para el tratamiento del cuerpo de un animal por terapia.
25. Uso de la composición según una cualquiera de las reivindicaciones 1 a 11 o el sistema vector según una cualquiera de las reivindicaciones 12 a 20, en la producción de un animal, distinto de un ser humano, transgénico o una planta transgénica.
26. Un método para modificar un polinucleótido diana en una célula eucariota, en donde el método comprende permitir que un complejo CRISPR se una a dicho polinucleótido diana para efectuar la escisión de dicho polinucleótido diana, modificando de ese modo dicho polinucleótido diana, en donde el complejo CRISPR comprende una proteína Cas9 que comprende una o más secuencias de localización nuclear (NLS) complejadas con una secuencia guía hibridada a una secuencia diana en dicho polinucleótido diana en donde la secuencia guía se une a una secuencia de emparejamiento tracr que se hibrida, a su vez, a una secuencia tracr que tiene 30 o más nucleótidos de longitud, en donde el método no es un método para tratamiento por terapia del cuerpo de un animal o de un ser humano y en donde el método no es un procedimiento para modificar la identidad genética de línea germinal de seres humanos.
27. Una célula huésped eucariota que comprende uno o más vectores del sistema vector según una cualquiera de las reivindicaciones 12 a 20.
28. Un método ex vivo o in vitro para modificar un polinucleótido diana en una célula eucariota, en donde el método comprende permitir que un complejo CRISPR se una al polinucleótido diana para efectuar la escisión de dicho
polinucleótido diana, modificándose de ese modo el polinucleótido diana, en donde el complejo CRISPR comprende una proteína Cas9 que comprende una o más secuencias de localización nuclear (NLS) complejadas con una secuencia guía hibridada a una secuencia diana en dicho polinucleótido diana en donde la secuencia guía está ligada a una secuencia de emparejamiento tracr que, a su vez, hibrida a una secuencia tracr que tiene 30 o más nucleótidos de longitud, en donde dicho método no es un procedimiento de modificación de la identidad genética de línea germinal de seres humanos.
29. Un complejo CRISPR para uso en la modificación de un polinucleótido diana en una célula eucariota en un método de terapia génica, en donde el método comprende permitir que un complejo CRISPR se una al polinucleótido diana para efectuar la escisión de dicho polinucleótido diana, modificándose de ese modo el polinucleótido diana, en donde el complejo CRISPR comprende una proteína Cas9 que comprende una o más secuencias de localización nuclear (NLS) complejadas con una secuencia guía hibridada a una secuencia diana en dicho polinucleótido diana en donde la secuencia guía está ligada a una secuencia de emparejamiento tracr que, a su vez, hibrida a una secuencia tracr que tiene 30 o más nucleótidos de longitud.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261736527P | 2012-12-12 | 2012-12-12 | |
| US201361748427P | 2013-01-02 | 2013-01-02 | |
| US201361757972P | 2013-01-29 | 2013-01-29 | |
| US201361768959P | 2013-02-25 | 2013-02-25 | |
| US201361791409P | 2013-03-15 | 2013-03-15 | |
| US201361835931P | 2013-06-17 | 2013-06-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2861623T3 true ES2861623T3 (es) | 2021-10-06 |
Family
ID=55306645
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13812430.0T Active ES2635244T3 (es) | 2012-12-12 | 2013-12-12 | Sistemas, métodos y composiciones de componentes de CRISPR-Cas para manipulación de secuencias |
| ES17162030T Active ES2861623T3 (es) | 2012-12-12 | 2013-12-12 | Sistemas, métodos y composiciones de componentes de CRISPR-Cas para manipulación de secuencias |
| ES14188820.6T Active ES2618531T3 (es) | 2012-12-12 | 2013-12-12 | Sistemas, métodos y composiciones de componentes de CRISPR-Cas para manipulación de secuencias |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13812430.0T Active ES2635244T3 (es) | 2012-12-12 | 2013-12-12 | Sistemas, métodos y composiciones de componentes de CRISPR-Cas para manipulación de secuencias |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES14188820.6T Active ES2618531T3 (es) | 2012-12-12 | 2013-12-12 | Sistemas, métodos y composiciones de componentes de CRISPR-Cas para manipulación de secuencias |
Country Status (4)
| Country | Link |
|---|---|
| DK (1) | DK2825654T3 (es) |
| ES (3) | ES2635244T3 (es) |
| HK (2) | HK1208045A1 (es) |
| RU (1) | RU2701662C9 (es) |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013066438A2 (en) | 2011-07-22 | 2013-05-10 | President And Fellows Of Harvard College | Evaluation and improvement of nuclease cleavage specificity |
| US20150044192A1 (en) | 2013-08-09 | 2015-02-12 | President And Fellows Of Harvard College | Methods for identifying a target site of a cas9 nuclease |
| US9359599B2 (en) | 2013-08-22 | 2016-06-07 | President And Fellows Of Harvard College | Engineered transcription activator-like effector (TALE) domains and uses thereof |
| US9322037B2 (en) | 2013-09-06 | 2016-04-26 | President And Fellows Of Harvard College | Cas9-FokI fusion proteins and uses thereof |
| US9526784B2 (en) | 2013-09-06 | 2016-12-27 | President And Fellows Of Harvard College | Delivery system for functional nucleases |
| US9340799B2 (en) | 2013-09-06 | 2016-05-17 | President And Fellows Of Harvard College | MRNA-sensing switchable gRNAs |
| US9068179B1 (en) | 2013-12-12 | 2015-06-30 | President And Fellows Of Harvard College | Methods for correcting presenilin point mutations |
| WO2016022363A2 (en) | 2014-07-30 | 2016-02-11 | President And Fellows Of Harvard College | Cas9 proteins including ligand-dependent inteins |
| IL294014B2 (en) | 2015-10-23 | 2024-07-01 | Harvard College | Nucleobase editors and their uses |
| CA3032699A1 (en) | 2016-08-03 | 2018-02-08 | President And Fellows Of Harvard College | Adenosine nucleobase editors and uses thereof |
| AU2017308889B2 (en) | 2016-08-09 | 2023-11-09 | President And Fellows Of Harvard College | Programmable Cas9-recombinase fusion proteins and uses thereof |
| US11542509B2 (en) | 2016-08-24 | 2023-01-03 | President And Fellows Of Harvard College | Incorporation of unnatural amino acids into proteins using base editing |
| WO2018071868A1 (en) | 2016-10-14 | 2018-04-19 | President And Fellows Of Harvard College | Aav delivery of nucleobase editors |
| US10745677B2 (en) | 2016-12-23 | 2020-08-18 | President And Fellows Of Harvard College | Editing of CCR5 receptor gene to protect against HIV infection |
| EP3592853A1 (en) | 2017-03-09 | 2020-01-15 | President and Fellows of Harvard College | Suppression of pain by gene editing |
| JP2020510439A (ja) | 2017-03-10 | 2020-04-09 | プレジデント アンド フェローズ オブ ハーバード カレッジ | シトシンからグアニンへの塩基編集因子 |
| IL269458B2 (en) | 2017-03-23 | 2024-02-01 | Harvard College | Nucleic base editors that include nucleic acid programmable DNA binding proteins |
| WO2018209320A1 (en) | 2017-05-12 | 2018-11-15 | President And Fellows Of Harvard College | Aptazyme-embedded guide rnas for use with crispr-cas9 in genome editing and transcriptional activation |
| JP2020534795A (ja) | 2017-07-28 | 2020-12-03 | プレジデント アンド フェローズ オブ ハーバード カレッジ | ファージによって支援される連続的進化(pace)を用いて塩基編集因子を進化させるための方法および組成物 |
| US11319532B2 (en) | 2017-08-30 | 2022-05-03 | President And Fellows Of Harvard College | High efficiency base editors comprising Gam |
| US11795443B2 (en) | 2017-10-16 | 2023-10-24 | The Broad Institute, Inc. | Uses of adenosine base editors |
| DE112020001342T5 (de) | 2019-03-19 | 2022-01-13 | President and Fellows of Harvard College | Verfahren und Zusammensetzungen zum Editing von Nukleotidsequenzen |
| DE112021002672T5 (de) | 2020-05-08 | 2023-04-13 | President And Fellows Of Harvard College | Vefahren und zusammensetzungen zum gleichzeitigen editieren beider stränge einer doppelsträngigen nukleotid-zielsequenz |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2003290937A1 (en) * | 2002-11-15 | 2004-06-15 | Trustees Of Boston University | Cis/trans riboregulators |
| WO2007025097A2 (en) * | 2005-08-26 | 2007-03-01 | Danisco A/S | Use |
| US20100076057A1 (en) * | 2008-09-23 | 2010-03-25 | Northwestern University | TARGET DNA INTERFERENCE WITH crRNA |
| US20140113376A1 (en) * | 2011-06-01 | 2014-04-24 | Rotem Sorek | Compositions and methods for downregulating prokaryotic genes |
-
2013
- 2013-12-12 ES ES13812430.0T patent/ES2635244T3/es active Active
- 2013-12-12 RU RU2015128102A patent/RU2701662C9/ru active
- 2013-12-12 ES ES17162030T patent/ES2861623T3/es active Active
- 2013-12-12 ES ES14188820.6T patent/ES2618531T3/es active Active
- 2013-12-12 DK DK13812430.0T patent/DK2825654T3/en active
-
2015
- 2015-08-21 HK HK15108120.9A patent/HK1208045A1/xx unknown
-
2018
- 2018-06-05 HK HK18107321.5A patent/HK1247961A1/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| RU2019127238A (ru) | 2019-09-20 |
| RU2701662C9 (ru) | 2019-10-31 |
| HK1247961A1 (zh) | 2018-10-05 |
| DK2825654T3 (en) | 2017-08-21 |
| ES2618531T3 (es) | 2017-06-21 |
| HK1208045A1 (en) | 2016-02-19 |
| RU2701662C2 (ru) | 2019-09-30 |
| RU2015128102A3 (es) | 2018-12-21 |
| RU2015128102A (ru) | 2018-12-21 |
| ES2635244T3 (es) | 2017-10-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7542681B2 (ja) | 配列操作のためのCRISPR-Cas成分系、方法および組成物 | |
| ES2861623T3 (es) | Sistemas, métodos y composiciones de componentes de CRISPR-Cas para manipulación de secuencias | |
| JP7542595B2 (ja) | 配列操作のための系、方法および最適化ガイド組成物のエンジニアリング | |
| JP7463436B2 (ja) | 遺伝子産物の発現を変更するためのCRISPR-Cas系および方法 | |
| DK2921557T3 (en) | Design of systems, methods and optimized sequence manipulation guide compositions | |
| ES2786193T3 (es) | Modificación por tecnología genética y optimización de sistemas, métodos y composiciones enzimáticas mejorados para la manipulación de secuencias | |
| KR102649341B1 (ko) | H1 프로모터를 사용하여 crispr 가이드 rna를 발현시키기 위한 조성물 및 방법 | |
| ES2777217T3 (es) | Suministro, modificación y optimización de sistemas de guía en tándem, métodos y composiciones para la manipulación de secuencias | |
| WO2014093694A1 (en) | Crispr-cas nickase systems, methods and compositions for sequence manipulation in eukaryotes |