ES2812452T3 - Compuestos de 2-oxo-1,2-dihidropiridin-3-carboxamida y su uso como inhibidores de PDK1 - Google Patents
Compuestos de 2-oxo-1,2-dihidropiridin-3-carboxamida y su uso como inhibidores de PDK1 Download PDFInfo
- Publication number
- ES2812452T3 ES2812452T3 ES16735586T ES16735586T ES2812452T3 ES 2812452 T3 ES2812452 T3 ES 2812452T3 ES 16735586 T ES16735586 T ES 16735586T ES 16735586 T ES16735586 T ES 16735586T ES 2812452 T3 ES2812452 T3 ES 2812452T3
- Authority
- ES
- Spain
- Prior art keywords
- oxo
- dihydropyridine
- carboxamide
- difluorobenzyl
- amino
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 B=C(c1cc(**NC(C2=CC=CN(Cc(cc3F)ccc3F)C2=O)=O)ccc1N1)C1=O Chemical compound B=C(c1cc(**NC(C2=CC=CN(Cc(cc3F)ccc3F)C2=O)=O)ccc1N1)C1=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Plural Heterocyclic Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Un compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida de Fórmula (I) **(Ver fórmula)** en donde m es 0 o 1 n es 0 o 1 A se selecciona del grupo que consiste en (-NH-CO-CH2-), (-NH-CO-CH(Ph)-), (-NH-CO-CH2-CH2-), (-NH-CO-CH2- CH2-CH2-) y (-NH-CO-Ph-); y B es CH-D, donde D se selecciona del grupo que consiste en tienilo, fenilo, imidazolilo, estando D opcionalmente sustituido con un halógeno, o una sal farmacéutica del mismo, para su uso en el tratamiento de patologías que requieren el uso de un inhibidor de la enzima PDK1.
Description
DESCRIPCIÓN
Compuestos de 2-oxo-1,2-dihidropiridin-3-carboxamida y su uso como inhibidores de PDK1
Campo de la invención
La invención trata de compuestos de 2-oxo-1,2-dihidropiridin-3-carboxamida y su uso como inhibidores y/o moduladores de PDK1.
Estado de la técnica
El eje de señalización Pl3K/PDK1/Akt está implicado de forma importante en la inhibición de la apoptosis y la estimulación de la proliferación celular y se ha estimado que al menos 50% de todos los tipos de cáncer está relacionado con la desregulación de esta ruta de señalización.
Debido a su función clave como regulador de la supervivencia y el metabolismo celulares, la desregulación de esta ruta se manifiesta en varias patologías humanas incluyendo cánceres y enfermedades neurodegenerativas. La cinasa dependiente de fosfoinosítido (PDK1) actúa como uno de los principales mediadores de la ruta. La PDK1 es una serina/treonina proteína cinasa que representa un papel clave en la regulación del crecimiento, la proliferación y la supervivencia celulares a través de mecanismos tanto dependientes de Akt como independientes de Akt. La ruta dependiente de Akt se caracteriza por la implicación de proteínas aguas abajo como mTOR, Ras y GSK, todas controladas por Akt. La señal independiente de Akt actúa a través de PLCy1, una fosfolipasa implicada en la metástasis.
La fosforilación y, por lo tanto, la activación de múltiples sustratos que parecen ser constitutivamente activos en tejido tumoral (tales como isoformas de AKT, S6K, SGK, RSK y PKC) puede explicar la influencia de esta cinasa sobre una variedad de procesos celulares incluyendo la proliferación, la migración y la supervivencia.
Por estas razones, la PDK1, también conocida como "cinasa maestra" de las AGC cinasas, ha atraído un interés considerable como un objetivo de fármacos anticancerosos. Sin embargo, aunque se han realizado enormes esfuerzos para descubrir moléculas específicas que se dirijan a PI3K y Akt, la PDK1 se ha pasado bastante por alto. Recientemente, el interés creciente en esta cinasa incitó a muchos grupos de investigación a trabajar en esta dirección, publicando y patentando así varias series de moléculas capaces de inhibir este importante nudo de las Pl3K/PDK1/Akt.
La PDK1 representa un papel pleotrópico en el crecimiento y el desarrollo. Hallazgos recientes revelaron que una activación elevada de PDK1 induce tumorigénesis al potenciar la proliferación celular e inhibir la apoptosis. Además, una evidencia creciente muestra que la PDK1 representa un papel fundamental en la migración celular y la metástasis. Su papel en estos procesos se probó en diferentes tipos de células y organismos incluyendo células endoteliales, células del músculo liso, linfocitos T, neutrófilos y varias líneas de células tumorales tales como cánceres de mama, glioblastoma (Signore, M., et al., Combined PDK1 and CHK1 inhibition is required to kill glioblastoma stem-like cells in vitro and in vivo. Cell death & disease, 2014. 5(5): pág. e1223) y pancreático.
En el documento WO2008/005457 se describen nuevos compuestos de piridinonilo que tienen actividad de inhibición de PDK1 según se demuestra para algunos compuestos preparados in vitro. Los mismos compuestos también se estudiaron sobre líneas celulares y los resultados se presentan en Nagashima, K., et al., Genetic and Pharmacological Inhibition of PDK1 in Cancer Cells CHARACTERIZATION OF A SELECTIVE ALLOSTERIC KINASE INHIBITOR. Journal of Biological Chemistry, 2011.286(8): pág. 6433-6448. Aunque algunos compuestos se reivindicaban como inhibidores de PDK1 en el documento WO2008/005457, no inhiben significativamente el crecimiento celular sobre un cultivo de células de tejido estándar, específicamente líneas de cáncer de próstata PC3.
Existe una gran necesidad de inhibidores eficaces frente a la enzima PDK1.
Compendio de la invención
El objetivo anterior se ha alcanzado mediante un compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida de Fórmula (I)

en donde
m es 0 o 1
n es 0 o 1
B es CH-D, donde D se selecciona del grupo que consiste en tienilo, fenilo, imidazolilo, estando D opcionalmente sustituido con un halógeno; y A se selecciona del grupo que consiste en (-NH-CO-CH2-), (-NH-CO-CH(Ph)-), (-NH-CO-CH2-CH2-), (-NH-CO-CH2-CH2-CH2-) y (-NH-CO-Ph-),
o una de sus sales farmacéuticas para su uso en el tratamiento de patologías que requieran el uso de un inhibidor de la enzima PDK1.
En otro aspecto, la invención trata de un nuevo compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida de Fórmula (II)
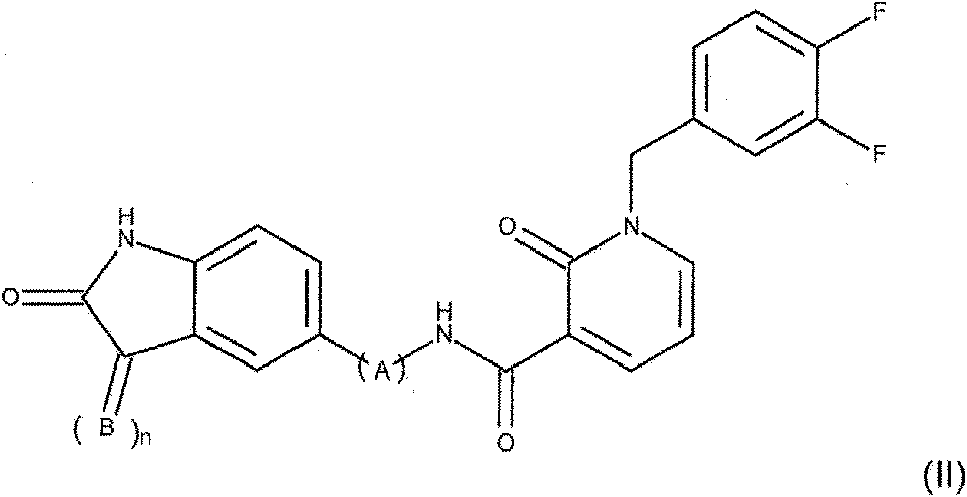
en donde
n es 0 o 1
B es CH-D, donde D se selecciona del grupo que consiste en tienilo, fenilo, imidazolilo, estando D opcionalmente sustituido con un halógeno; y A se selecciona del grupo que consiste en (-NH-CO-CH2-), (-NH-CO-CH(Ph)-) (-NH-CO-CH2-CH2-) (-NH-CO-CH2-CH2-CH2-) y (-NH-CO-Ph-),
o una de sus sales farmacéuticas.
En otro aspecto la invención trata de un compuesto de Fórmula (II) para su uso como medicamento.
En un aspecto adicional la invención trata de una composición farmacéutica que comprende un compuesto de Fórmula (II) y un portador farmacéuticamente aceptable.
En un aspecto adicional más, la invención trata de un compuesto de Fórmula (II) para su uso en el tratamiento de patologías que requieran el uso de un inhibidor de PDK1.
Las patologías que requieren un inhibidor de la enzima PDK1 son diabetes, enfermedades neurodegenerativas tales como enfermedad de Alzheimer y enfermedades priónicas, y tumores tales como cánceres de mama y pancreático y glioblastoma. Preferiblemente, esta patología es un cáncer, más preferiblemente glioblastoma (GBM).
En esta invención, un compuesto de Fórmula (I) o Fórmula (II) puede existir como enantiómeros R y S y como mezcla racémica. Esta invención incluye en su alcance de protección todos los posibles isómeros y mezclas racémicas. Siempre que deban estar presentes centros de simetría adicionales, esta invención incluye todos los posibles diastereoisómeros y también mezclas relativas.
Descripción de las figuras
La Figura 1 ilustra la inhibición de la proliferación de células U87MG después de 24 h de incubación con compuestos de la invención;
la Figura 2 ilustra la inhibición de la proliferación de células U87MG después de 72 h de incubación con compuestos de la invención;
la Figura 3 muestra los efectos de inhibidores de PDK1 de la invención sobre la inducción de la apoptosis de U87MG usando en protocolo de tinción con anexina V;
la Figura 4 muestra los efectos de inhibidores de PDK1 de la invención sobre la inducción de la apoptosis de U87MG como porcentaje de células apoptósicas frente al número total de células;
la Figura 5 muestra los efectos de los compuestos de la invención sobre la actividad de PDK1 en células U87 MG;
la Figura 6 muestra micrografías en la evaluación de efectos de los inhibidores de PDK1 de la invención sobre la migración para la curación de heridas;
la Figura 7 muestra medidas de superficies huecas en la evaluación de efectos de los inhibidores de PDK1 de la invención sobre la migración para la curación de heridas;
la Figura 8 muestra los efectos del tratamiento de los compuestos de la invención sobre la proliferación de GSC después de cuatro días;
la Figura 9 muestra los efectos del tratamiento de los compuestos de la invención sobre la proliferación de GSC después de siete días;
la Figura 10 muestra los efectos de inhibidores de PDK1 de la invención sobre la apoptosis de GSC usando en protocolo de tinción con anexina V;
la Figura 11 muestra los efectos de inhibidores de PDK1 de la invención sobre la apoptosis de GSC como porcentaje de células apoptósicas frente al número total de células; y
la Figura 12 muestra los efectos de inhibidores de PDK1 de la invención sobre GSC mediante ensayos ELISA. Descripción detallada de la invención
Por lo tanto, la invención trata de un compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida de Fórmula (I)

en donde
m es 0 o 1
n es 0 o 1
A se selecciona del grupo que consiste en (-NH-CO-CH2-), (-NH-CO-CH(Ph)-), (-NH-CO-CH2-CH2-), (-NH-CO-CH2-CH2-CH2-) y (-NH-CO-Ph-); y
B es CH-D, donde D se selecciona del grupo que consiste en tienilo, fenilo, imidazolilo, estando D opcionalmente sustituido con un halógeno,
o una de sus sales farmacéuticas, para su uso en el tratamiento de patologías que requieran el uso de un inhibidor de la enzima PDK1.
Cuando m es 0, n es preferiblemente 1.
Cuando m es 1, n puede ser 0 o 1.
Preferiblemente, m es 1 y n es 1.
Cuando m es 1, A se selecciona del grupo que consiste en (-NH-CO-CH2-), (-NH-CO-CH(Ph)-), (-NH-CO-CH2-CH2-), (-NH-CO-CH2-CH2-CH2-) y (-NH-CO-Ph-).
Preferiblemente, A es (-NH-CO-CH2-), (-NH-CO-CH2-CH2-), (-NH-CO-CH2-CH2-CH2-) o (-NH-CO-Ph-).
Cuando n es 1, B es CH-D, donde D se selecciona del grupo que consiste en tienilo, fenilo, imidazolilo, estando D opcionalmente sustituido con un halógeno.
Preferiblemente, D es imidazolilo, preferiblemente 1H-imidazol-5-ilo o 1H-imidazol-2-ilo.
En una realización preferida, A es (-NH-CO-CH(Ph)-) y D es imidazolilo, preferiblemente 1 H-imidazol-5-ilo.
En una realización más preferida, A es (-NH-CO-CH2-) y D es imidazolilo, preferiblemente 1H-imidazol-5-ilo.
En una realización más preferida, A es (-NH-CO-CH2-CH2) y D es imidazolilo, preferiblemente 1 H-imidazol-5-ilo. En una realización más preferida, A es (-NH-CO-CH2-CH2) y D es imidazolilo, preferiblemente 1 H-imidazol-2-ilo. En una realización aún más preferida, A es (-NH-CO-CH2-CH2-CH2-) o (-NH-CO-Ph), más preferiblemente D es imidazolilo, aún más preferiblemente 1 H-imidazol-5-ilo.
El compuesto preferido para su uso como inhibidor de PDK1 es uno de los compuestos presentados en la Tabla a continuación.



Más preferiblemente, el compuesto de Fórmula (I) se selecciona del grupo (Z)-N-(2-((3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1-feniletil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-2-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1 -feniletil)-1 -(3,4 difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)carbamoil)fenil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-N-(4-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-4-oxobutil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida y (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-3-oxopropil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida.
Aún más preferiblemente, el compuesto de Fórmula (I) es (Z)-N-(2-((3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida.
También son compuestos preferidos (Z)-N-(4-((3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-4-oxobutil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida y (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-3-oxopropil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida. En otro aspecto, la invención se refiere a un compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida de Fórmula (II)
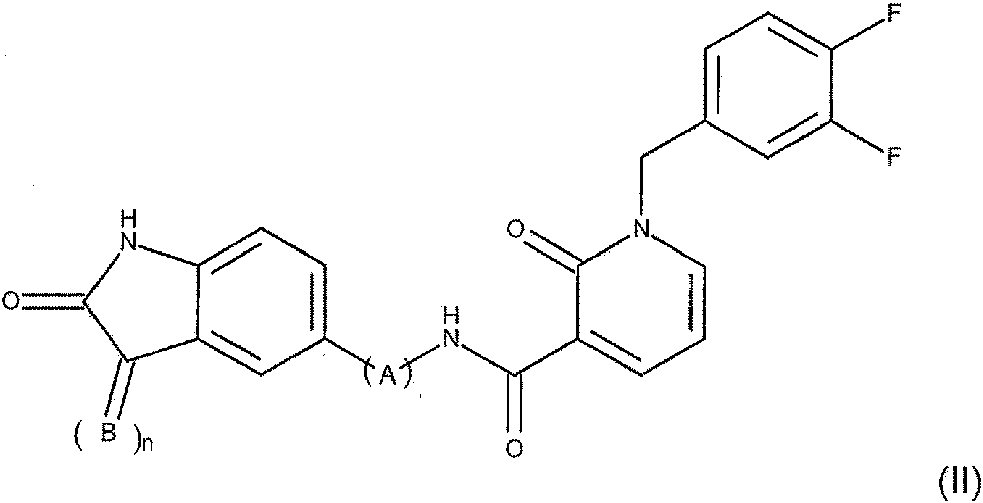
en donde
n es 0 o 1
B es CH-D, donde D se selecciona del grupo que consiste en tienilo, fenilo, imidazolilo, estando D opcionalmente sustituido con un halógeno; y A se selecciona del grupo que consiste en (-NH-CO-CH2-), (-NH-CO-CH(Ph)-) (-NH-CO-CH2-CH2-), (-NH-CO-CH2-CH2-CH2-) y (-NH-CO-Ph-),
o una de sus sales farmacéuticas.
n es 0 o 1, más preferiblemente n es 1.
A se selecciona del grupo que consiste en (-NH-CO-CH2-), (-NH-CO-CH(Ph)-) (-NH-CO-CH2-CH2-), (-NH-CO-CH2-CH2-CH2-) y (-NH-CO-Ph-). Preferiblemente, A es (-NH-CO-CH2-), (-NH-CO-CH2-CH2-CH2-), (-NH-CO-CH2-CH2-) o (-NH-CO-Ph-). En una realización más preferida, n es 1 y A se selecciona del grupo que consiste en (-NH-CO-CH2-), (-NH-CO-CH2-CH2-) (-NH-CO-CH2-CH2-CH2-) y (-NH-CO-Ph-).
Cuando n es 1, B es CH-D, donde D se selecciona del grupo que consiste en tienilo, fenilo, imidazolilo, estando D opcionalmente sustituido con un halógeno.
Preferiblemente, D es imidazolilo.
En una realización preferida, A es (-NH-CO-CH(Ph)-) y D es imidazolilo, preferiblemente 1 H-imidazol-5-ilo.
En una realización más preferida, A es (-NH-CO-CH2-) y D es imidazolilo, preferiblemente 1 H-imidazol-5-ilo.
En una realización más preferida, A es (-NH-CO-CH2-CH2) y D es imidazolilo, preferiblemente 1 H-imidazol-5-ilo. El compuesto preferido es uno de los compuestos presentados en la Tabla a continuación.



Más preferiblemente, el compuesto de Fórmula (II) se selecciona del grupo (Z)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1-feniletil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-2-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1-feniletil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)carbamoil)fenil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-N-(4-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-4-oxobutil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida y (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-3-oxopropil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida.
Aún más preferiblemente, el compuesto de Fórmula (II) es (Z)-N-(2-((3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida.
También son compuestos preferidos (Z)-N-(3-((3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)carbamoil)fenil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida o (Z)-N-(4-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-4-oxobutil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida o (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-3-oxopropil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida. Los compuestos de la invención se pueden preparar al usar procedimientos fáciles de aumentar a escala y evitando etapas de preparación prolongadas y costosas, obteniendo así un alto rendimiento de un compuesto de calidad farmacéutica estable como será evidente a partir de la parte experimental de la presente descripción.
Los compuestos de la invención de Fórmulas (I) y (II) como tales o una de sus sales farmacéuticas se podrían usar en medicina, en particular como un inhibidor de la enzima PDK1.
En otro aspecto, la presente invención proporciona una composición farmacéutica que comprende un compuesto de Fórmula (I) y/o Fórmula (II) y un excipiente, por ejemplo un portador, farmacéuticamente aceptable. La composición farmacéutica también puede comprender un compuesto inhibidor de PDK1 conocido.
Los compuestos de la invención de Fórmula (I) y de Fórmula (II) se pueden administrar solos o se pueden coadministrar al paciente. Se entiende que la coadministración incluye la administración simultánea o secuencial de los compuestos individualmente o en combinación (más de un compuesto). Así, la preparación también se puede combinar, cuando se desee, con otras sustancias activas.
Se podrían usar en combinación con un portador farmacéuticamente aceptable y opcionalmente con excipientes adecuados para obtener composiciones farmacéuticas. El término "portador farmacéuticamente aceptable" significa disolventes, agentes portadores, agentes diluyentes y similares que se usan en la administración de compuestos de la invención.
Tales composiciones farmacéuticas se pueden administrar mediante administración parenteral, oral, bucal, sublingual, nasal, rectal, tópica o transdérmica. Composiciones de la invención adecuadas para la administración oral serán convenientemente unidades discretas tales como comprimidos, cápsulas, sellos, polvos o pellas o como una suspensión líquida.
El comprimido también puede contener excipientes adecuados usados habitualmente en el campo farmacéutico tales como almidón pregelatinizado, celulosa microcristalina, amiloglicolato sódico, talco, lactosa, estearato magnésico, sacarosa, ácido esteárico, manitol. La composiciones para administración parental pueden incluir convenientemente preparaciones estériles.
Una composición para la administración tópica se puede formular convenientemente como cremas, pastas, aceites, pomadas, emulsiones, espumas, geles, gotas, soluciones para pulverización y parches transdérmicos.
Los compuestos de la invención se pueden usar como un medicamento en el tratamiento de patologías que requieren un inhibidor de la enzima PDK1 tales como cánceres, preferiblemente en el tratamiento de glioblastoma (GBM).
Se muestra que los compuestos de Fórmula (I) y Fórmula (II) inhiben la enzima PDK1 con valores de IC50 en el intervalo de nM a pM como será evidente a partir de la parte experimental de la descripción.
La clasificación del valor de IC50 sobre PDK1 recombinante reflejaba la clasificación de afinidad hacia líneas celulares de glioblastoma, confirmando así que la actividad antiproliferativa está mediada por PDK1.
Ventajosamente, los compuestos preferidos (Z)-N-(2-((3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1 -feniletil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-2-il)amino)-2-oxo-1-feniletil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida,(Z)-W-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)carbamoil)fenil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-N-(4-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-4-oxobutil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida y (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-3-oxopropil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida inhibían la actividad constitutiva de PDK1 en células de glioblastoma; como resultado, los compuestos de Fórmula (II) disminuían la viabilidad celular y desencadenaban la apoptosis. Por otra parte, la inhibición de la viabilidad celular era prolongada. En el ensayo de curación de heridas, como se aclarará posteriormente, se mostraba que los inhibidores de PDK1 inhiben significativamente la migración celular, tanto después de 6 h como de 24 h desde la escarificación. Estos resultados confirmaban que una inhibición de PDK1 conduce a una supresión de la migración de células tumorales.
Finalmente, los compuestos de las Fórmulas (I), y preferiblemente de Fórmula (II), aún más preferiblemente los compuestos preferidos (Z)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1 -feniletil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-2-il)amino)-2-oxo-1-feniletil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)carbamoil)fenil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, y (Z)-N-(4-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-4-oxobutil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida se caracterizaban en células progenitoras de glioma (GSC, por sus siglas en inglés) aisladas de células de glioblastoma. Los resultados obtenidos indicaban que los compuestos inducían una inhibición dependiente del tiempo y la concentración de la proliferación y la apoptosis desencadenada de GSC, confirmando así que los inhibidores de PDK1 son capaces de bloquear la proliferación de GSC.
La invención se detallará ahora por medio de los siguientes ejemplos que se refieren a la preparación de algunos compuestos de la invención y a la evaluación de su actividad frente al receptor de PDK1.
Partes experimentales
Preparación de los compuestos de Fórmulas (I) y (II)
Se usan disolventes anhidros de calidad comercial sin secado adicional. Los productos químicos disponibles comercialmente se adquirieron de Sigma-Aldrich o Alfa Aesar y se usaron sin purificación adicional. La evaporación se realizó a vacío (evaporador giratorio). Siempre se usaba Na2SÜ4 anhidro como el agente de secado.
Se realizó cromatografía de desarrollo rápido sobre gel de sílice de calidad de alta pureza de 60 Á de Merck (0,40-63 pm). Las reacciones se siguieron mediante TLC, realizada sobre láminas de aluminio-gel de sílice de Merck (60 F254). Los puntos se observaron bajo una lámpara UV (254 nm) o con la ayuda de ácido fosfomolíbdico al 10% en EtOH. Los puntos de fusión se determinaron con un aparato de banco térmico de Kofler y no estaban corregidos.
Las reacciones de hidrogenación se realizaron a través de un generador de hidrógeno HG 2000 CLAIND®. La reacción de microondas se realizó usando un microondas CEM Discover® LabMateTM. Se usó Celite® 545 como agente filtrante.
Los espectros de RMN de 1H, 13C y 19F se obtuvieron usando un espectrómetro Bruker Avance 400 y se registraron a 400, 101 y 376 MHz, respectivamente. RMN de 1H [t=373K] se registró a 250 MHz en un espectrómetro Bruker Avance II 250.
Los desplazamientos químicos se registran en valores 5 en partes por millón (ppm), con relación a la referencia interna tetrametilsilano (TMS) y con referencia desde la resonancia del disolvente como el estándar interno: deuterocloroformo [5 7,26 (espectros de 1H), 577,16 (espectros de 13C)]; deuterodimetilsulfóxido [52,50 (espectros de 1H), 539,52 (espectros de 13C)]; deuteroacetonitrilo [5 1,94 (espectros de 1H)]. Las constantes de acoplamiento J se presentan en hertzios (Hz). Los espectros de RMN de 19F y 13C están desacoplados de 1H. Los espectros de RMN de 19F no tienen referencia, corregidos a partir de ácido trifluoroacético (TFA, por sus siglas en inglés) como estándar externo (-76,2 ppm).
Los patrones de señal se indican como sigue: singlete (s), doblete (d), triplete (t), doble doblete (dd), doble triplete (dt), multiplete (m), singlete ancho (s a), doblete ancho (d a) y triplete ancho (t a).
Cuando el nitrógeno amídico soporta dos sustituyentes, los espectros de RMN de 1H muestran la presencia de dos rotámeros diferentes en equilibrio.
Las composiciones elementales de los compuestos (C, H, N) coinciden hasta dentro de (±) 0,4% de los valores teóricos. Cuando no se incluye el análisis elemental, los compuestos se usaban en la siguiente etapa sin purificación adicional.
Abreviaturas: TBTU= Tetrafluoroborato de N,N,N',N'-tetrametil-O-(benzotriazol-1-il)uronio; DIPEA= N,N-diisopropiletilamina; DMF= N,N-Dimetilformamida; CDI= N,N'-carbonildiimidazol; TFA= Ácido trifluoroacético; NaH = Hidruro sódico; DCM= diclorometano
Ejemplo 1: Preparación del compuesto JJ31
(Z)-N-(3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)-1-(3.4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida El compuesto JJ31 se preparó al seguir el Esquema 1
Esquema 1

aReactivos y condiciones: I) H2, Pd-C 10%, EtOH absoluto, t.a., 4 h; II) TBTU, DIPEA, DMF seca, 0°C ^ t.a., 12 h; III) 1 H-imidazol-5-carbaldehído, EtOH absoluto, piperidina, 110°C, 4 h.
La hidrogenación catalítica de 5-nitroxindol en EtOH absoluto en presencia de Pd-C 10% daba la amina 2. La condensación del ácido carboxílico 3 con TBTU y DIPEA en DMF seca durante 16 h daba el compuesto SST143. El compuesto 3 se sintetizó a través de la alquilación con bromuro de 3,4-difluorobencilo del ácido 2-oxopiridino-3-carboxílico en presencia de NaH 60%. La condensación posterior del derivado SST143 con 1 H-imidazol-5-carbaldehído y una cantidad catalítica de piperidina daba el correspondiente compuesto del epígrafe. El 2-oxindol sustituido en 3 puede existir como el isómero bien Z o bien E dependiendo de las características de los sustituyentes en la posición C3 del núcleo de 2-oxindol. Las configuraciones de los compuestos se asignaron al analizar sus espectros de RMN de 1H.
Síntesis de 5-amino-1,3-dihidro-2H-indol-2-ona (2).
El 5-nitro-2-oxindol 1 (1,00 g, 5,61 mmol) se hidrogenó en (70 ml) en presencia de Pd al 10%-C (315 mg, 2,97 mmol) durante 4 h a temperatura ambiente. A continuación, el catalizador se filtró a través de Celite, la Celite se enjuagó con EtOH adicional y la disolución se evaporó, para dar 2 como un sólido pardo (698 mg, 4,71 mmol, 84% de rendimiento).
RMN de 1H (400 MHz, DMSO-de): 53,30 (s, 2H, CH2); 4,62 (s a, 2H, NH2); 6,37 (dd, 1H, J = 2,2, 8,2 Hz, Ar); 6,48 6,50 (m, 2H, Ar); 9,90 (s a, 1H, Nh) ppm. Anál. Calc. para C8H8N2O: C, 64,85%; H, 5,44%; N 18,91%; Encontrado: C, 65,03%; H, 5,49%; N, 18,83%.
Síntesis de ácido 1-(3,4-difluorobencil)-2-oxo-1,2-dihidropirídino-3-carboxílico (3).
Una suspensión agitada de dispersión de NaH al 60% en aceite mineral (206 mg, 5,14 mmol), lavada dos veces con n-hexano destilado y una vez con Et2O en 2 ml de DMF anhidra bajo atmósfera de N2, se trató gota a gota con una disolución que contenía el ácido 2-hidroxinicotínico (600 mg, 4,31 mmol) en 5 ml de DMF anhidra. La mezcla se dejó bajo agitación a temperatura ambiente durante 2 h y a continuación se añadió bromuro de 3,4-difluorobencilo (1,06 g, 5,14 mmol) y la mezcla se agitó y se calentó a 50°C durante 16 h. Después, la mezcla se concentró bajo presión reducida y el residuo se trató con agua para dar un sólido, que se recogió mediante filtración a vacío. Posteriormente, el sólido se sometió a reflujo durante 4 h en NaOH ac. al 10% (10 ml) y la mezcla resultante se enfrió y se acidificó con HCl ac. 1 N. El sólido blanco formado se recogió mediante filtración y se lavó con n-hexano y Et2O, dando el derivado 3.9 como un sólido blanco (857 mg, 3,23 mmol, 75% de rendimiento).
RMN de 1H (400 MHz, DMSO-de): 55,30 (s, 2H, CH2); 6,78 (t, 1H, J = 6,9 Hz, Ar); 7,22-7,24 (m, 1H, Ar); 7,41-7,53 (m, 2H, Ar); 8,41 (d, 2H, J = 6,9 Hz, Ar) ppm. Anál. Calc. para C13H9NO3F2: C, 58,87%; H, 3,42%; N, 5,28%; Encontrado: C, 58,99%; H, 3,47%; N, 5,43%.
Síntesis de 1 -(3,4-difluorobencil)-2-oxo-N-(2-oxoindolin-5-il)- 1,2-dihidropiridin-3-carboxamida (SST143).
El derivado carboxílico 3 (300 mg, 1,13 mmol) y la amina 2 (168 mg, 1,13 mmol) se combinaron a través de una reacción de condensación en presencia del agente de condensación TBTU (363 mg, 1,13 mmol). Los disolventes se evaporaron bajo presión reducida y el producto en bruto se purificó mediante cromatografía de desarrollo rápido sobre gel de sílice, usando MeOH al 0 - 4% como un gradiente en CHCh, para obtener SST143 puro como un sólido blanco (210 mg, 0,53 mmol, 47% de rendimiento).
p.f.: 285-288°C. RMN de 1H (400 MHz, DMSO-de): 53,48 (s, 2H, CH2 indol); 5,28 (s, 2H, CH2); 6,68 (dd, 1H, J= 6,8, 7.2 Hz, Ar); 6,77 (d, 1H, J = 8,4 Hz, Ar); 7,20-7,23 (m, 1H, Ar); 7,40-7,52 (m, 3H, Ar); 7,66 (s, 1H, Ar); 8,29 (dd, 1H, J = 2,2, 6,8 Hz, Ar); 8,46 (dd, 1H, J = 2,2, 6,8 Hz, Ar); 10,33 (s, 1H, NH), 11,87 (s, 1H, NH) ppm. RMN de 13C (101 MHz, DMSO-de): 5 176,23, 161,54, 160,73, 150,35, 147,90, 143,78, 143,42, 139,86, 134,02, 132,32, 126,40, 124,90, 120,35, 119,02, 117,74, 117,25, 116,90, 109,10, 107,22, 51,45, 36,02 ppm. RMN de 19F (376 MHz; DMSO-de): 5 -138,17 (d, 1 F, J = 24 Hz); -139,76 (d, 1 F, J = 24 Hz) ppm. Anál. Calc. para C21H15F2N3O3: C, 63,80%; H, 3,82%; N 10,63%; Encontrado: C, 63,72%; H, 3,55%; N, 10,50%.
Síntesis de (Z)-N-(3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida (JJ31)
El producto en bruto se purificó mediante cristalización en EtOH, proporcionando el isómero Z como un sólido amarillo (29 mg, 0,06 mmol, 34% de rendimiento).
p.f.: 285-288 °C. RMN de 1H (400 MHz, DMSO-de): 5 5,31 (s, 2H, CH2); 6,70 (dd, 1H, J = 6,8, 7,2 Hz, Ar); 6,87 (d, 1H, J = 8,4 Hz, Ar); 7,22-7,25 (m, 1H, Ar); 7,42-7,62 (m, 3H, Ar); 7,68 (s, 1H, Ar); 7,94 (s, 1H, CH=); 8,01 (s, 1H, Ar); 8.03 (s, 1H, Ar); 8,32 (d, 1H, J = 7,2 Hz, Ar); 8,50 (d, 1H, J = 6,8 Hz, Ar); 11,00 (s, 1H, NH); 11,99 (s, 1H, NH) ppm. RMN de 13C (101 MHz, DMSO-de): 5 169,05, 161,58, 160,85, 150,36, 147,91, 143,85, 143,55, 139,67, 138,75, 136,10, 134,09, 132,79, 128,13, 124,85, 123,45, 120,29, 119,87, 117,86, 117,69, 117,25, 117,08, 111,39, 109,94, 107,28, 51,53 ppm. RMN de 19F (376 MHz; DMSO-de): - 138,13 (d, 1 F, J = 24 Hz); -139,76 (d, 1 F, J = 24 Hz) ppm. Anál. Calc. para C25H17F2N5O3: C, 63,15%; H, 4,03%; N 14,73%; Encontrado: C, 63,34%; H, 4,17%; N, 14,89%.
Ejemplo 2:
Preparación de los compuestos IB35, IB36, SA16, SA23, DD21-23, DD25, DF8 y DF9
Los compuestos IB35, IB36, SA16, SA23, DD21-23, DD25, DF8 y DF9 se prepararon al seguir el Esquema 2
Esquema 2

Reactivos y condicionesa: I) (Boc)2O, NaOH 1 M/iPrOH 4:3, t.a., 2 h; II) 5-amino-2-oxo-indol, TBTU, DIPEA, DMF seca, t.a., 16 h; III) TFA, DCM, -10°C/-20°C, 3 h; IV) TBTU, DIPEA, DMF seca, t.a., 16 h; V) Carbaldehído apropiado, iPrOH, DMF, piperidina, 110°C, 4 h
(r=0 y R=H IB32, IB35, IB36; r=0 y R=Fenilo SA16 y SA23; r=1 y R= H D21, D22, D23, DD25; r=2 y R=H, DF8, DF9)
Los compuestos de Fórmula (II) se sintetizaron mediante la inserción de un conector para ligar el núcleo de 2-oxindol y el centro de difluorobencilpiridonilo. El procedimiento sintético se llevó a cabo a través de condensación amídica al usar como espaciador aminoácidos, tales como glicina, (R)-(-)-2-fenilglicina o p-alanina.
El aminoácido apropiado se protegió en el grupo amina con (Boc)2O y se condensó con 5-aminoxindol, conduciendo al derivado amídico 5a-d. Posteriormente, la reacción de desprotección con ácido trifluoroacético proporcionaba la amina 6a-d como sal trifluoroacética, que se hizo reaccionar con el ácido carboxílico 3 para dar el derivado IB32 o 7. La reacción de Knovenagel con el carbaldehído apropiado, en presencia de una cantidad catalítica de piperidina, proporcionaba los compuestos deseados finales IB35-36, SA16, SA23, DD21-23, DD25, DF8 y DF9. Según se observaba previamente, los nuevos compuestos se obtenían como forma isómera Z, E o mezcla de E/Z. De nuevo, las configuraciones se asignaron al analizar sus espectros de RMN de 1H.
Procedimiento general para la síntesis de derivados de N-(terc-butoxicarbonil)aminoácido (4a-d)
Se añadió Boc2O (2,9 g, 13,32 mmol) a una disolución del aminoácido apropiado (13,32 mmol) en NaOH 1 M/iPrOH (4:3). La reacción, controlada mediante TLC, se agitó a temperatura ambiente durante 2 h y a continuación se lavó con Et2O, se acidificó hasta pH 3,0 con HCl 1 N y finalmente se extrajo varias veces con AcOEt. La capa orgánica se secó sobre Na2SO4 anhidro y se evaporó bajo presión reducida. El producto 4a-d en bruto se usó para la siguiente etapa sin purificación adicional (1,68 g, 9,59 mmol, 72% de rendimiento).
Ácido 2-((terc-butoxicarbonil)amino)acético (4a) RMN de 1H (400 MHz, CDCta): 51,45 (s, 9H, Boc); 3,90-4,08 (m, 2H, CH2); 5,07 (s a, 1H, NH) ppm. Anál. Calc. para C7H13NO4: C, 47,99%; H, 7,48%; N 8,00%; Encontrado: C, 48,18%; H, 7,36%; N, 8,26%.
Ácido (R)-2-((terc-butoxicarbonil)amino)-2-fenilacético (4b) RMN de 1H (400 MHz, CDCI3): 5 1,21 (s, 6H, Boc); 1,43 (s, 3H, Boc); 5,12-5,51 (m, 1H); 7,29-7,44 (m, 5 H); 7,96 (s a, 1H, NH) ppm. Anál. Calc. para C13H17NO4: C, 62,14%; H, 6,82%; N 5,57%; Encontrado: C, 62,18%; H, 7,06%; N, 5,86%.
Ácido 3-((terc-butoxicarbonil)amino)propanoico (4c) RMN de 1H (400 MHz, CDCh): 5 1,43 (s a, 9H, Boc); 2,52-2,59 (m, 2H); 3,39-3,41 (s a, 2H); 5,05-5,17 (m, 1H); Anál. Calc. para C8H15NO4: C%, 50,78%; H, 7,99; N 7,40; Encontrado% : C, 51,02; H, 8,02; N, 7,54.
Ácido 4-((terc-butoxicarbonil)amino)butanoico (4d): El producto en bruto (921 mg, 4,53 mmol, 50% de rendimiento) se usó para la siguiente etapa sin purificación adicional. RMN de 1H (400 MHz, CDCh): 5 1,44 (s, 9H, CH3); 1,79 1,86 (m, 2H, CH2); 2,40 (t, 2H, J=7,2 Hz, CH2); 3,20-3,18 (m, 2H, CH2) ppm. Anál. Calc. para C9H17NO4: C, 53,19%; H, 8,43%; N, 6,89%; Encontrado: C, 53,07%; H, 8,57%; N, 6,93%.
Procedimiento general para la síntesis de compuestos 5a-d
El derivado N-Boc 4a-d (2,64 mmol) se hizo reaccionar a través de una reacción de condensación con 5-amino-indol-2-ona (391 mg, 2,64 mmol), en presencia del agente de condensación TBTU (848 mg, 2,64 mmol) y DIPEA (5,28 mmol, 0,92 ml) como base.
(2-oxo-2-((2-oxoindolin-5-il)amino)etil)carbamato de terc-butilo (5a)
La amida se obtuvo después de la purificación del producto en bruto mediante cromatografía en columna sobre gel de sílice usando CHCh/MeOH 92:8 como el eluyente (693 mg, 2,27 mmol, 86% de rendimiento). RMN de 1H (400 MHz, DMSO-ds): 5 1,39 (s, 9H, Boc); 3,46 (s, 2H, CH2 indol); 3,68 (d, 2H, J = 6,0 Hz, CH2 glicina); 6,73 (d, 1H, J = 8,2 Hz, Ar); 7,00 (t, 1H, J = 6,0 Hz, NH); 7,32 (dd, 1H, J=2,0, 8,2 Hz, Ar); 7,49 (s, 1H, Ar); 9,74 (s a, 1H, NH); 10,27 (s a, 1H, NH) ppm. Anál. Calc. para C15H19N3O4: C, 59,01%; H, 6,27%; N 13,76%; Encontrado: C, 59,10%; H, 6,19%; N, 13,99%.
(R)-(2-oxo-2-((2-oxoindolin-5-il)amino)-1-feniletil)-carbamato de terc-butilo (5b)
El compuesto final se ha purificado mediante cromatografía en columna sobre gel de sílice usando CHCh/MeOH 92:8 como el eluyente. RMN de 1H (400 MHz, DMSO-d6): 5 1,39 (s, 9H, Boc); 3,44 (s, 2H, CH2 indol); 5,30-5,33 (m, 1H); 6,72 (d, 1H, J = 8,4 Hz, Ar); 7,26-7,36 (m, 4H, Ar); 7,44-7,48 (m, 4H, Ar); 10,09 (s a, 1H, NH); 10,29 (s a, 1H, NH) ppm. Anál. Calc. para C21H23N3O4: C, 66,13%; H, 6,08%; N 11,02%; Encontrado: C, 66,17%; H, 6,23%; N, 11,21%.
3-((terc-butoxicarbonil)-amino)-N-(2-oxo-2,3-dihidro-1H-indol-5-il)propanamida (5c)
El compuesto final se ha purificado mediante cromatografía en columna sobre gel de sílice usando CHCl3/MeOH 92:8 como el eluyente. RMN de 1H (400 MHz, DMSO-ds): 5 1,39 (s, 9H, Boc); 2,42 (t, 2H, J = 7,2 Hz); 3,16-3,22 (m, 2H); 3,45 (s, 2H); 6,71 (d, 1H, J = 8,2 Hz, Ar); 6,84-6,88 (m, 1H); 7,32(d, 1H, J= 8,2Hz); 7,50 (s, 1H, Ar); 9,78 (s a, 1H, nH); 10,28 (s a, 1H, NH) ppm. Anál. Calc. para C16H20N3O4: C, 60,37%; H, 6,33%; N 13,20%; Encontrado: C, 60,17%; H, 6,23%; N, 13,22%.
4-((terc-butoxicarbonil)-amino)-N-(2-oxo-2,3-dihidro-1H-indol-5-il)butanamida (5d)
El compuesto final (726 mg, 1,92 mmol, 95% de rendimiento) se ha purificado mediante cromatografía en columna sobre gel de sílice usando CHCh/MeOH (95:5) como el eluyente. r Mn de 1H (400 MHz, DMSO-d6): 5 1,37 (s, 9H, CH3); 1,65-1,69 (m, 2H, CH2); 2,24 (t, 2H, J = 7,6 Hz, CH2); 2,92-2,97 (m, 2H, CH2); 3,44 (s, 2H, CH2); 6,71 (d, 1H, J = 8,2 Hz, Ar); 6,79 (s a, 1H, NH); 7,30 (d, 1H, J = 8,2 Hz, Ar); 7,49 (s, 1H, Ar); 9,68 (s a, 1H, NH); 10,23 (s a, 1H, NH) ppm. Anál. Calc. para C17H23N3O4: C, 61,25%; H, 6,95%; N, 12,60%; Encontrado: C, 60,98%; H, 6,89%; N, 12,41%. Procedimiento general para la síntesis de compuestos 6a-d
Se añadió TFA (4,54 ml) a una suspensión agitada de 5a-d (2,27 mmol) en DCM (4,54 ml) enfriada a -10°C/- 20°C. La reacción se controló mediante TLC y alcanzaba la finalización en 3 h a la misma temperatura. A continuación, la mezcla de reacción se evaporó hasta sequedad para proporcionar 6a-d como una sal trifluoroacética, usada para la siguiente etapa sin purificaciones adicionales.
2,2,2-trifluoroacetato de 2-oxo-2-((2-oxoindolin-5-il)amino)etanaminio (6a)
RMN de 1H (400 MHz, DMSO-d6): 53,49 (s, 2H, CH2 indol); 3,74 (d, 2H, J = 5,6 Hz, CH2 glicina); 6,78 (d, 1H, J = 8,0 Hz, Ar); 7,33 (dd, 1H, J = 2,2, 8,2 Hz, Ar); 7,47 (s, 1H, Ar); 8,09 (s a, 3H, NH3+); 10,26 (s a, 1H, NH); 10,34 (s a, 1H, NH) ppm.
(R)-2,2,2-trífluoroacetato de 2-oxo-2-((2-oxoindolin-5-il)amino)-1-feniletanaminio (6b)
RMN de 1H (400 MHz, DMSO-d6): 53,47 (s, 2H, CH2); 5,03-5,05 (m, 1H); 6,76 (d, 1H, J = 8,4 Hz, Ar); 7,29 (dd, 1H, J = 2,0, 8,4 Hz, Ar); 7,42-7,50 (m, 3H, Ar); 7,57-7,59 (m, 2H, Ar); 8,72-8,74 (m, 1H, Ar); 10,36 (s a, 1H, NH); 10,47 (s a, 1H, NH) ppm.
3- amino-N- (2-oxo-2,3-dihidro-1 H-indol-5-il)propanamida (6c)
RMN de 1H (400 MHz, DMSO-ds): 52,66 (t, 2H, J= 6,8Hz); 3,06-3,11 (m, 2H); 3,49 (s, 2H, CH2 indol); 6,75 (d, 1H, J = 8,4 Hz, Ar); 7,33 (dd, 1H, J = 2,0, 8,4 Hz, Ar); 7,50 (s, 1H, Ar); 7,79 (s a, 3H, NH3+); 10,02 (s a, 1H, NH); 10,33 (s a, 1H, NH) ppm.
4- amino-N-(2-oxo-2,3-dihidro-1H-indol-5-il)butanamida (6d) RMN de 1H (400 MHz, DMSO-d6): 5 1,82-1,85 (m, 2H, CH2); 2,38 (t, 2H, J = 6,8 Hz, CH2); 2,83-2,85 (m, 2H, CH2); 3,45 (s, 2H, CH2); 6,73 (d, 1H, J = 8,0 Hz, Ar); 7,31 (d, 1H, J= 8,0 Hz, Ar), 7,50 (s, 1H, Ar); 7,76 (s a, 3H, NH3+); 9,84 (s a, 1H, NH); 10,29 (s a, 1H, NH) ppm.
1-(3,4-difluorobencil)-2-oxo-N-(4-oxo-4-((2-oxoindolin-5-il)amino)butil)-1,2-dihidropiridin-3-carboxamida (7)
El ácido carboxílico 3 (480 mg, 1,81 mmol) se hizo reaccionar a través de una reacción de condensación con la sal de amina 6d (480 mg, 1,81 mmol), en presencia de TBTU (581,16 mg, 1,81 mmol). El procedimiento seguido es el mismo descrito para el derivado SST143 en el ejemplo 1. El producto en bruto se purificó mediante cromatografía de desarrollo rápido sobre gel de sílice, usando CHCh/MeOH (92:8) como el eluyente, para obtener 7 puro como un sólido blanco (680 mg, 1,42 mmol, 79% de rendimiento).
p.f.: 199-204°C RMN de 1H (400 MHz, DMSO-d6): 5 1,76-1,84 (m, 2H, CH2); 2,30 (t, 2H, J = 7,6 Hz, CH2); 3,34-3,36 (m, 2H, CH2); 3,43 (s, 2H, CH2); 5,20 (s, 2H, CH2); 6,57 (t, 1H, J = 7,2 Hz, Ar); 6,70 (d, 1H, J = 8,4 Hz, Ar); 7,16-7,19 (m, 1H, Ar); 7,30 (dd, 1H, J = 2,0, 8,4 Hz, Ar); 7,38-7,48 (m, 3H, Ar); 8,21 (dd, 1H, J = 2,2, 7,2 Hz, Ar); 8,34 (dd, 1H, J = 2,2, 7,2 Hz, Ar); 9,66 (t a, 1H, J =5,8 NH); 9,74 (s a, 1H, NH); 10,25 (s a, 1H, NH) ppm. RMN de 13C (101 MHz, DMSO-d6): 5 176,23; 170,15; 162,98; 161,23; 150,31; 147,87; 143,40; 142,92; 139,06; 134,21; 133,38; 125,90; 124,85; 120,49; 118,42; 117,70; 117,20; 116,46; 108,73; 106,60; 51,26; 38,29; 36,01; 33,85; 25,41 ppm. RMN de 19F (376 MHz, DMSO-d6): 5 -138,23 (d, 1 F, J = 22 Hz); - 139,82 (d, 1 F, J = 24 Hz) ppm. Anál. Calc. para C25H22N4O4F2 : C, 62,50%; H, 4,62%; N, 11,66%; Encontrado: C, 62,32%; H, 4,79%; N, 11,81%.
Síntesis de 1-(3,4-difluorobencil)-2-oxo-N-(2-oxo-2-((2-oxoindolin-5-il)amino)etil)-1,2-dihidropiridin-3-carboxamida (IB32).
Partiendo del ácido carboxílico 3 (154 mg, 0,58 mmol) y el 2,2,2-trifluoroacetato de 2-oxo-2-((2-oxoindolin-5-il)amino)etanaminio (0,58 mmol), se sintetizó el derivado amídico, usando TBTU (187 mg, 0,58 mmol) como agente de condensación. El procedimiento seguido es el mismo que se describe para el derivado SST143 en el ejemplo 1 anterior.
El producto en bruto se purificó mediante cromatografía en columna sobre gel de sílice usando CHCh/MeOH 92:8 como el eluyente para obtener el producto como un sólido blanco (63 mg, 0,14 mmol, 25% de rendimiento).
p.f.: 257-262°C. RMN de 1H (400 MHz, D M S O d): 5 3,45 (s, 2H, CH2 indol); 4,13 (d, 2H, J = 5,2 Hz, CH2 glicina); 5,23 (s, 2H, CH2); 6,58 (t, 1H, J = 6,9 Hz, Ar); 6,74 (d, 1H, J = 8,4 Hz, Ar); 7,19-7,22 (m, 1H, Ar); 7,33 (dd, 1H, J= 2,0, 8,4 Hz, Ar); 7,40-7,50 (m, 3H, Ar); 8,25 (dd, 1H, J = 2,1,6,9 Hz, Ar); 8,35 (dd, 1H, J = 2,1, 6,9 Hz, Ar); 9,94 (s a, 1H, NH); 9,99 (t, 1H, J = 5,2 Hz, NH); 10,28 (s a, 1H, NH) ppm. RMN de 13C (101 MHz, DMSO-d6): 5 176,17, 166,69, 163,04, 161,09, 150,22, 147,83, 143,48, 143,19, 139,33, 134,18, 132,82, 126,05, 124,89, 120,20, 118,48, 117,65, 117,23, 116,44, 108,81, 106,45, 51,34, 43,05, 35,97 ppm. RMN de 19F (376 MHz; DMSO-d6): 5 -138,27 (d, 1 F, J = 24 Hz); -139,80 (d, 1F, J = 24 Hz) ppm. Anál. Calc. para C23H18N4O4F2: C, 61,06%; H, 4,01%; N 12,38%; Encontrado: C, 61,22%; H, 4,00%; N, 12,51%.
Procedimiento general para sintetizar los compuestos IB35, IB36, SA16, SA23, DD21-23, DD25, DF8, DF9
Se añadió el carbaldehído apropiado (0,12 mmol) y una cantidad catalítica de piperidina a una disolución del derivado 2-oxoindólico (0,12 mmol) disuelto en iPrOH/DMF o EtOH absoluto (5 ml). La disolución resultante se agitó y se calentó para alcanzar 110°C durante 4 h, y a continuación la disolución se evaporó hasta sequedad. El material residual se purificó mediante cristalización.
(E/Z)-1-(3,4-difluorobencil)-2-oxo-N-(2-oxo-2-((2-oxo-3-(tiofen-2-ilmetilen)indolin-5-il)amino)etil)-1,2-dihidropiridin-3-carboxamida (IB36)
El material residual se purificó mediante cristalización en iPrOH, proporcionando el producto final como un sólido naranja que consistía en una mezcla isómera de E /Z 1:2 (35 mg, 0,06 mmol, 53% de rendimiento). p.f.: 223-227°C. RMN de 1H (400 MHz, DMSO-d6): 54,17 (d, 2H, J = 4,8 Hz, CH2 glicina); 5,24 (s, 2H, CH2); 6,59 (t, 1H, J = 7,0 Hz, Ar); 6,80 (d, 0,66H, J = 8,4 Hz, Ar); 6,85 (d, 0,33H, J = 8,4 Hz, Ar); 7,17-7,23 (m, 2H, Ar); 7,31-7,33 (m, 0,33H, Ar); 7,40-7,50 (m, 2,66H, Ar); 7,78 (s, 0,33H, isómero E, Ar); 7,82 (d, 0,33H, J = 3,6 Hz, Ar, isómero E); 7,89 (d, 0,66H, J = 4,8 Hz, Ar, isómero Z); 7,98-8,00 (m, 1,32H, Ar, isómero Z); 8,03-8,05 (m, 1H, Ar, isómeros E+Z); 8,26-8,28 (m,
1H, Ar, isómeros E+Z); 8,36-8,39 (m, 1H, Ar, isómeros E+Z); 8,58 (s, 0,33H, C4, J = 1,6 Hz Ar, isómero E); 9,99 10,05 (m, 1,66H, NH); 10,11 (s a, 0,33H, NH); 10,55 (s a, 1H, NH) ppm. RMN de 13C (DMSO-afe): 5 169,27, 167,41, 166.95, 166,89 163,12, 161,15, 150,41, 147,90,138,46, 138,16, 137,23, 137,12, 136,58, 136,03, , 134,57, 134,26, 132,88, 132,64, 132,19, 128,73, 128,26, 127,40, 124,93, 124,31, 123,54, 121,69, 121,06, 120,75, 120,29, 117,73, 117,28, 115,08, 111,59, 109,65, 106,54, 51,45, 43,08 ppm. RMN de 19F (376 MHz; DMSO-cfe): 5 -138,24 (d, 1F, J = 24 Hz); -139,76 (d, 0,33F, J = 24 Hz), -139,79 (d, 0,66F, J = 24 Hz) ppm. Anál. Calc. para C28H20N4O4F2S: C, 61,53%; H, 3,69%; N 10,25%; Encontrado: C, 61,62%; H, 3,63%; N, 10,39%.
(Z)-N-(2-((3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropirídin-3-carboxamida (IB35)
El material residual se purificó mediante cristalización en iPrOH, proporcionando el isómero Z como un sólido amarillo (32 mg, 0,06 mmol, 52% de rendimiento). p.f.: 230-235°C. r Mn de 1H (400 MHz, DMSO-d6): 54,17 (d, 2H, J = 4,4 Hz, CH2 glicina); 5,24 (s, 2H, CH2); 6,60 (t, 1H, J = 6,9 Hz, Ar); 6,85 (d, 1H, J = 8,8 Hz, Ar); 7,19-7,21 (m, 2H, Ar); 7,43-7,50 (m, 3H, Ar); 7,71 (s, 1H, Ar); 7,75 (s, 1H, Ar); 8,01 (s, 1H, Ar); 8,05 (s, 1H, Ar); 8,27 (dd, 1H, J = 2,0, 6,9 Hz, Ar); 8,37 (d, 1H, J = 2,0, 6,9 Hz, Ar); 10,61 (s a, 2H, NH); 10,95 (s a, 1H, NH) ppm. RMN de 13C (101 MHz, DMSO-ds): 5 169,05, 166,93, 163,12, 161,16, 150,41, 147,90, 143,57, 143,31, 139,62, 138,85, 135,79, 134,32, 133,20, 128,07, 124,93, 124,47, 122,90, 120,25, 120,10, 119,67, 117,73, 117,23, 111,17, 109,86, 106,52, 51,45, 43,14 ppm. RMN de 19F (376 MHz; DMSO-ds): 5 -138,25 (d, 1F, J = 24 Hz); -139,80 (d, 1F, J = 24 Hz) ppm. Anál. Calc. para C27H20N6O4F2: C, 61,13%; H, 3,80%; N 15,84%; Encontrado: C, 61,15%; H, 4,03%; N, 15,89%.
(Z)-(R)-N-(2-((3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1-feniletil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida (SA16)
El material residual se purificó mediante cristalización en EtOH, proporcionando el isómero Z como un sólido amarillo (41 mg, 0,07 mmol, 57% de rendimiento). p.f.: 248-255°C. RMN de 1H (400 MHz, DMSO-d6): 5 5,20-5,31 (m, 2H, CH2); 5,81 (d, 1H, J = 7,2 Hz, CH2); 6,60 (t, 1H, J = 6,8 Hz, Ar); 6,83 (d, 1H, J = 8,0 Hz, Ar); 7,19-7,21 (m, 2H, Ar); 7,29-7,52 (m, 8H, Ar); 7,71 (s, 1H, Ar); 7,78 (s, 1H, Ar); 8,01 (s, 1H, Ar); 8,04 (s, 1H, Ar); 8,27 (dd, 1H, J = 2,0, 6,8 Hz, Ar); 8,35 (dd, 1H, J = 2,0, 6,8 Hz, Ar); 10,46 (s a, 1H, NH); 10,72 (d, 1H, J = 7,2 Hz, NH); 10,97 (s a, 1H, NH) ppm. RMN de 13C (101 MHz, DMSO-d6): 5 169,03, 167,95, 162,32, 161,30, 150,35, 147,91, 143,82, 143,57, 139,68, 138.95, 138,72, 136,04, 134,24, 132,92, 128,70, 128,07, 127,89, 126,83, 124,87, 124,51, 123,16, 119,90, 119,77, 117,79, 117,15, 111,16, 109,88, 106,68, 57,06, 51,36 ppm. Anál. Calc. para C33H24N6O4F2: C, 65,34%; H, 3,99%; N 13,85%; Encontrado: C, 65,25%; H, 4,03%; N, 13,79%.
(Z)-(R)-N-(2-((3-((1H-imidazol-2-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1-feniletil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida (SA23)
El material residual se purificó mediante cristalización en EtOH, proporcionando el sólido amarillo como el isómero Z, que se interconvierte lentamente en la mezcla E/Z 2:1 (45 mg, 0,07 mmol, 62% de rendimiento). p.f.: 304-307°C. RMN de 1H (400 MHz, DMSO-d6) [t=0]: 55,20-5,30 (m, 2H, CH2); 5,81 (d, 1H, J= 7,2 Hz, CH2); 6,60 (t, 1H, J = 7,0 Hz, Ar); 6,86 (d, 1H, J = 8,4 Hz, Ar); 7,17-7,20 (m, 1H, Ar); 7,21-7,56 (m, 11H, Ar); 7,58 (s, 1H, CH=); 7,86 (d, 1H, J = 2,0 Hz, C4, Ar); 8,26 (dd, J = 2,0, 7,0 Hz, 1H, Ar); 8,36 (dd, J = 2,0, 7,0 Hz, 1H, Ar); 10,45 (s a, 1H, NH); 10,68 (d, 1H, J = 7,2 Hz, NH); 11,11 (s a, 1H, NH) ppm. RMN de 19F (376 MHz; DMSO-d6) [t=0]: 5 - 138,14 (d, 1 F, J = 24 Hz); -139,76 (d, 1 F, J = 24 Hz) ppm. RMN de 13C (101 MHz, DMSO-d6) [t=12 h]: 5 169,50 (isómero Z), 168,89 (isómero E), 168,01, 162,31, 162,25, 161,26, 150,46, 147,44, 143,78 (isómero E), 143,50 (isómero Z), 143,35 (isómero E), 142,89 (isómero Z), 138,92 (isómero Z), 138,86 (isómero Z), 138,56 (isómero E), 136,23 (isómero E), 134,16, 133,09, 132,62, 132,11, 128,68 (isómero E), 128,60 (isómero Z), 127,87 (isómero E), 127,77 (isómero Z), 126,85, 124,85, 124,31, 123,95, 123,42, 122,36, 121,90, 120,91 (isómero Z), 120,79 (isómero E), 119,93 (isómero Z), 119,86 (isómero E), 117,74, 117,16, 111,74, 110,06, 108,85, 106,65, 57,06, 51,28 ppm. RMN de 19F (376 MHz; DMSO-d6) [t=24 h]: 5 -138,14 (d, 0,33F, J= 24 Hz, isómero Z); -138,16 (d, 0,66F, J = 24 Hz, isómero E); -139,76 (d, 0,33F, J = 24 Hz, isómero Z); -139,79 (d, 0,66F, J = 24 Hz, isómero E) ppm. Anál. Calc. para C33H24N6O4F2: C, 65,34%; H, 3,99%; N 13,85%; Encontrado: C, 65,25%; H, 4,03%; N, 13,79%.
(E/Z)-1-(3,4-difluorobencil)-2-oxo-N-(3-oxo-3-((2-oxo-3-(tiofen-2-ilmetilen)indolin-5-il)amino)propil)-1,2-dihidropiridin-3-carboxamida (DD22).
El material residual se purificó mediante cristalización en iPrOH. RMN de 1H (400 MHz, DMSO-d6): 5 2,57-2,59 (m, 2H, CH2); 3,57-3,61 (m, 2H, CH2); 5,22 (s, 2H, CH2 bencílico); 6,57 (t, 1H, J = 6,8Hz, Ar); 6,78 (d, 0,75H, J = 8,4 Hz, Ar); 6,81 (d, 0,25H, J = 8,4 Hz, Ar); 7,15-7,22 (m, 3H, Ar); 7,31-7,44 (m, 2 H, Ar); 7,76 (s, 0,25H, isómero E, Ar); 7,89 (d, 0,90H, J = 3,2 Hz, Ar, isómero Z); 7,96-8,00 (m, 3H, Ar, isómero E+Z); 8,19-8,21 (m, 1H, Ar, isómero E+Z); 8,35 8,38 (m, 1H, Ar, isómero E+Z); 8,58 (s, 0,11H, C4, J = 1,6 Hz Ar, isómero E); 9,79 (s a, 1H, NH); 9,87 (m, 0,80H, NH); 9,95 (m, 0,20H, NH); 10,52 (s a, 0,35H, NH); 10,55 (s a, 1H, NH) ppm.
(Z/E)-N-(3-((3-((1H-imidazol-2-il)metilen)-2-oxoindolin-5-il)amino)-3-oxopropil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropirídin-3-carboxamida(mezcla 88:12=Z:E) (DD25)
RMN de 1H (400 MHz, DMSO-d6): 52,58 (t, 2H, J = 6,4Hz, CH2); 3,59 (q, 2H, J = 6,4 Hz, CH2); 4,17 (d, 2H, J=4,4 Hz, CH2); 5,20 (s, 2H, CH2); 6,57 (t, 1H, J=6,8 Hz, Ar); 6,82 (d, 1H, J=8,4 Hz, Ar); 7,12-7,14 (m, 1H, Ar); 7,33-7,43
(m, 4H, Ar); 7,52 (s, 1H, Ar); 7,56 (s, 1H, Ar); 7,92 (s, 1H, Ar); 8,19 (dd, 1H, J =2,0, 6,8 Hz, Ar); 8,35 (dd, 1H, J =2,0, 6,8 Hz, Ar); 9,81 (s a, 1H, NH); 9,91 (s, 1H, NH); 11,08 (s a, 1H, NH) ppm.
(Z)-N-(3-((3-((1H-imidazoi-5-ii)metiien)-2-oxoindoiin-5-ii)amino)-3-oxopropii)-1-(3,4-difiuorobencii)-2-oxo-1,2-dihidropirídin-3-carboxamida (DD21)
RMN de 1H (400 MHz, DMSO-d6): 5 2,59 (m a, 2H, CH2); 3,57-3,61 (m, 2H, CH2); 5,19 (s, 2H, CH2); 6,57 (t, 1H, J=6,8 Hz, Ar); 6,82 (d, 1H, J=8,0 Hz, Ar); 7,14-7,21 (m, 2H, Ar); 7,33-7,45 (m, 2H, Ar); 7,71 (s, 1H, Ar); 8,01 (s, 1H, Ar); 8,19 (d, 1H, J =6,0 Hz, Ar); 8,36 (d, 1H, J =6,0 Hz, Ar); 9,80 (s a, 1H, NH); 9,90 (s a, 1H, NH); 10,93 (s a, 1H, NH) ppm.
(Z)-1-(3,4-difiuorobencii)-N-(3-((3-(4-fiuorobenciiiden)-2-oxoindoiin-5-ii)amino)-3-oxopropii)-2-oxo-1,2-dihidropiridin-3-carboxamida (DD23)
RMN de 1H (400 MHz, DMSO-de): 5 2,50-2,53 (m, 2H, CH2); 3,53-3,55 (m, 2H, CH2); 5,18 (s, 2H, CH2 bencílico); 6,56 (t, 1H, J = 7,2 Hz, Ar); 6,80 (d, 1H, J = 8,4 Hz, Ar); 7,10-7,16 (m, 1H, Ar); 7,32-7,44 (m, 5H, Ar); 7,58 (s, 1H, CH=); 7,76-7,80 (m, 2H, Ar); 7,98 (s, 1H, C4 Ar); 8,20 (d, 1H, J = 6,4 Hz, Ar); 8,34 (d, 1H, J = 6,4 Hz, Ar); 9,78-9,82 (m, 2H, NH); 10,53 (s a, 1H, NH) ppm.
(Z)-N-(4-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-4-oxobutil)-5-(3,4-difluorobencil)-6-oxocidohexa-1,3-dieno-1-carboxamida (DF8). Se añadió el 4-imidazolcarbaldehído (14 mg, 0,15 mmol) y una cantidad catalítica de piperidina a una disolución del derivado 2-oxoindólico 7 (70 mg, 0,15 mmol) disuelto en iPrOH/DMF. El procedimiento seguido es el mismo descrito para el derivado IB35. El material residual se purificó mediante cristalización en iPrOH, proporcionando el isómero Z como un sólido naranja (44 mg, 0,08 mmol, 66% de rendimiento). p.f.: 240-244° C. RMN de 1H (400 MHz, DMSO-de): 5: 1,80-1,85 (m, 2H, CH2); 2,34 (t, 2H, J = 7,2 Hz, CH2); 3,35-3,39 (m, 2H, CH2); 5,19 (s, 2H, CH2); 6,57 (t, 1H, J = 7,0 Hz, Ar); 6,80 (d, 1H, J= 8,4 Hz, Ar); 7,18-7,21 (m, 2H, Ar); 7,38-7,46 (m, 2H, Ar); 7,65-7,75 (m, 2H, Ar); 7,95-8,05 (m, 2H, Ar); 8,20 (dd, 1H, J = 1,8, 7,0 Hz, Ar); 8,35 (dd, 1H, J = 1,8, 7,0 Hz, Ar); 9,68 (t a, 1H, J = 5,6 Hz, NH); 9,84 (s a, 1H, NH); 10,89 (s a, 1H, NH) ppm. RMN de 13C (101 MHz, DMSO-d6): 5 170,33; 169,04; 162,99; 161,24; 150,05; 147,87; 143,41; 142,91; 139,52; 138,73; 134,20; 133,68; 124,85; 124,30; 122,70; 120,50; 120,20; 119,77; 117,70; 117,20; 111,22; 109,70; 106,60; 51,27; 38,31; 33,79; 25,38 ppm. RMN de 19F (376 MHz, DMSO-de): 5: -138,22 (d, 1 F, J = 24 Hz); - 139,81 (d, 1 F, J = 24 Hz) ppm. Anál. Calc. para C29H24N6O4F2: C, 62,36%; H, 4,33%; N, 15,05%; Encontrado: C, 62,66%; H, 4,21%; N, 15,39%.
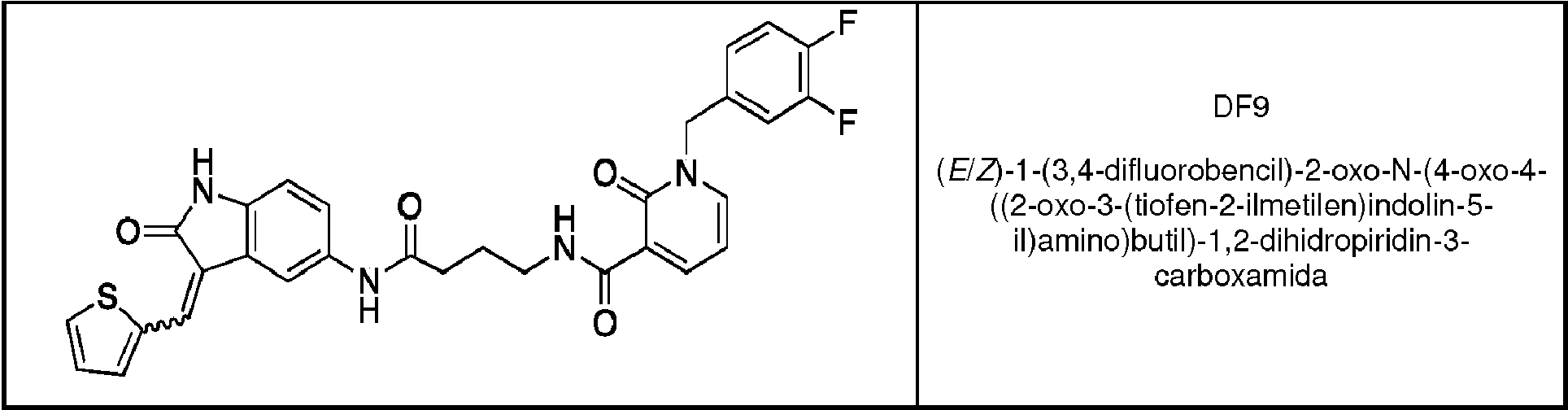
(E/Z)-1-(3,4-difiuorobencii)-2-oxo-N-(4-oxo-4-((2-oxo-3-(tiofen-2-iimetiien)indoiin-5-ii)amino)butii)-1,2-dihidropiridin-3-carboxamida (DF9). Se añadió el 2-tiofenocarbaldehído (17 mg, 0,15 mmol) y una cantidad catalítica de piperidina a una disolución del derivado 2-oxoindólico 7 (70 mg, 0,15 mmol) disuelto en iPrOH/DMF. El procedimiento seguido es el mismo descrito para el derivado IB35. El material residual se purificó mediante cristalización en iPrOH, proporcionando el producto final como un sólido naranja que consiste en una mezcla isómera E/Z 40:60 (52 mg, 0,09 mmol, 76% de rendimiento). p.f.: 189-194°C. RMN de 1H (400 MHz, DMSO- de): 5 1,80-1,87 (m, 2H, CH2); 2,32-2,36 (m, 2H, CH2); 3,35-3,39 (m, 2H, CH2); 5,19 (m, 2H, CH2); 6,54-6,59 (m, 1H, Ar); 6,77 (d, 0,60 H, J = 8,0 Hz, Ar, isómero Z); 6,81 (d, 0,40 H, J = 8,4 Hz, Ar, isómero E); 7,18-7,23 (m, 2,20 H, Ar); 7,31-7,32 (m, 0,40 H, Ar, isómero E); 7,40-7,47 (m, 2,40 H, Ar); 7,76 (s, 0,40 H, Ar, isómero E); 7,81 -7,88 (m, 1H, Ar, isómero E/Z); 7,96-7,99 (m, 1,60 H, Ar), 8,03 (m, 0,60H, Ar, isómero Z); 8,20-8,22 (m, 1H, Ar); 8,33-8,37 (m, 1H, Ar); 8,61 (m, 0,40H, Ar, isómero E); 9,67-9,69 (m a, 1H, NH); 9,82 (s a, 0,60H, NH, isómero Z); 9,90 (s a, 0,40H, NH, isómero E); 10,51 (s a, 1H, NH) ppm. RMN de 13C (101 MHz, DMSO-de): 5 170,32 (isómero Z); 169,31 (isómero E); 167,43; 163,02; 161,26; 150,34; 148,09; 143,45 (isómero Z); 142,97 (isómero E); 138,19 (isómero E); 138,04 (isómero Z); 137,23 (isómero Z); 137,18 (isómero E); 136,39 (isómero Z); 135,89 (isómero E); 134,48; 134,23; 133,39 (isómero E); 133,14 (isómero Z); 132,17; 128,70 (isómero Z); 128,08 (isómero E); 127,41 (isómero Z); 127,19 (isómero E); 124,88; 124,17 (isómero Z); 123,63 (isómero E); 121,81; 120,95; 120,65; 120,50; 120,44; 117,74; 117,23; 115,14; 111,65; 109,68 (isómero E); 109,34 (isómero Z); 106,50; 51,31; 38,34; 33,96 (isómero E); 33,77 (isómero Z); 25,42 ppm. RMN de 19F (376 MHz, DMSO- de): 5: - 139,22 (d, 1F, J = 24 Hz) ; -139,81 (d, 1F, J = 24 Hz) ppm. Anál. Calc. para C30H24N4O4F2S: C, 62,71%; H, 4,21%; N, 9,75%; Encontrado: C, 63,89%; H, 4,01%; N, 9,55%.
Preparación de los compuestos DD40 y DD41
Los compuestos DD40 y DD41 se prepararon al seguir el Esquema 3
Esquema 3

Reactivos y condiciones: I. SOCI2, 82°C, 12 h; II. Piridina, 3 h; III. H2, Pd/C, EtOH; IV. TBTU, DIPEA, DMF seca, t.a., 16 h; V. Carboxialdehído, Piperidina, EtOH, reflujo 110°C, 4h.
Cloruro de 3-nitrobenzoílo. Se hicieron reaccionar ácido 3-nitrobenzoico (600 mg, 3,59 mmol) y cloruro de tionilo (0,26 ml, 3,59 mmol) bajo atmósfera de N2 a 82°C durante 16 h. Una vez que la TLC verificaba la desaparición del precursor, la disolución se evaporaba hasta sequedad y el producto en bruto se usaba inmediatamente/directamente para la siguiente etapa sin purificación adicional.
3-nitro-N-(2-oxoindolin-5-il)benzamida (4e) Se añadió el 5-amino-2-oxindol (379 mg, 2,56 mmol) a una mezcla de cloruro de nitrobenzoílo (3,59 mmol) en piridina. La mezcla se agitó a temperatura ambiente durante 3 h y se vertió en agua de hielo. El precipitado se filtró, se lavó con agua y se secó a vacío para dar el producto final como un polvo blanco (544,1 mg, 1,83 mmol, 71% de rendimiento). RMN de 1H (400 MHz, DMSO-ds): 53,51 (s, 2H, CH2); 6,81 (d, 1H, J = 8,4 Hz, Ar); 7,52 (d, 1H, J = 8,4 Hz, Ar); 7,66 (s, 1H, Ar); 7,83 (t, 1H, J = 8,0 Hz, Ar); 8,37-8,43 (m, 2H, Ar); 8,77 (s, 1H, Ar); 10,35 (s a, 1H, NH); 10,45 (s a, 1H, NH) ppm. Anál. Calc. para C15H11N3O4: C, 60,61%; H, 3,73%; N, 14,14%; Encontrado: C, 60,78%; H, 3,86%; N, 14,05%.
Cloruro de 3-((2-oxoindolin-5-il)carbamoil)bencenaminio (5e). El derivado 4e (544 mg, 1,83 mmol) disuelto en EtOH absoluto (70 ml) se hidrogenó en presencia de Pd al 10%-C durante 4 h a t.a. A continuación, el catalizador se separó por filtración a través de Celite, la Celite se enjuagó con EtOH adicional y la disolución se evaporó, para dar 5e como un sólido pardo. El producto en bruto se transformó en la sal de hidrocloruro correspondiente (171,5 mg, 0,57 mmol, 31% de rendimiento). RMN de 1H (400 MHz, CD3OD): 53,55 (s, 2H, CH2); 6,90 (d, 1H, J = 8,4 Hz, Ar); 7,49 (dd, 1H, J = 1,8, 8,4 Hz, Ar); 7,59-7,62 (m, 2H, Ar); 7,70 (t, 1H, J = 8,0 Hz, Ar); 7,95 (d, 1H, J = 1,8, Ar); 8,05 (d, 1H, J = 8,0 Hz, Ar) ppm. Anál. Calc. para C15HmN3O2CI: C, 59,51%; H, 4,33%; N, 13,88%; Encontrado: C, 59,78%; H, 4,47%; N, 13,90%.
1 -(3,4-difluorobendl)-2-oxo-N-(3-((2-oxoindorm-5-il)carbamoil)fenil)- 1,2-dihidropiridin-3-carboxamida (8). El ácido carboxílico 3 (175 mg, 0,66 mmol) se hizo reaccionar con el derivado amínico 5e (200 mg, 0,66 mmol) en DMF seca (4 ml). La reacción se efectuó en presencia de TBTU (212 mg, 0,66 mmol) y DIPEA (4 ml) según se presenta previamente para el compuesto SST143. A continuación, el disolvente orgánico se evaporó bajo vacío y el producto en bruto se purificó mediante cromatografía de desarrollo rápido sobre gel de sílice, usando CHCh/MeOH (97:3) como el eluyente, dando 8 puro como un sólido blanco (80 mg, 0,16 mmol, 24% de rendimiento). RMN de 1H (400 MHz, DMSO-ds): 53,50 (s, 2H, CH2); 5,31 (s, 2H, CH2); 6,71 (t, 1H, J = 7,0 Hz, Ar); 6,79 (d, 1H, J = 8,4 Hz, Ar); 7,22-7,24 (m, 1H, Ar); 7,41-7,52 (m, 4H); 7,66-7,68 (m, 2H, Ar); 8,00 (d, 1H, J = 8,4 Hz, Ar); 8,13 (s, 1H, Ar); 8,33 (dd, 1H, J = 2,0, 7,0 Hz, Ar); 8,51 (dd, 1H, J = 2,0, 7,0 Hz, Ar); 10,15 (s a, 1H, NH); 10,32 (s a, 1H, NH); 12,14 (s a, 1H, NH) ppm. RMN de 13C (101 MHz, DMSO-ds): 5 176,30, 164,75, 161,60, 161,45, 150,37, 147,92, 144,29, 143,91, 139,77, 138,28, 135,92, 134,02, 133,03, 129,07, 125,90, 124,84, 122,94, 122,48, 120,01, 118,82, 117,83, 117,67, 117,27, 117,09, 108,74, 107,34, 51,52, 36,03 ppm. RMN de 19F (376 MHz, DMSO-ds): 5 -138,12 (d, 1F, J = 24 Hz, Ar); -139,73 (d, 1F, J = 24 Hz, Ar) ppm. Anál. Calc. para C28H20N4O4F2: C, 65,37 %; H, 3,92%; N, 10,89%; Encontrado: C, 65,69%; H, 3,79%; N, 10,93%.
(Z)-N-(3-((3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)carbamoil)fenil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida (DD40). Se añadió el 4-imidazolcarbaldehído (8 mg, 0,08 mmol) y una cantidad catalítica de piperidina a una disolución del derivado 8 (40 mg, 0,08 mmol) en iPrOH/DMF. El procedimiento seguido es el mismo descrito para el derivado IB35. El material residual se purificó mediante cristalización en iPrOH proporcionando el sólido amarillo como el isómero Z (15 mg, 0,03 mmol, 37% de rendimiento). p.f.: 326-330°C; Rm N de 1H (400 MHz, DMSO-ds): 55,31 (s, 2H, CH2); 6,71 (t, 1H, J = 6,8 Hz, Ar); 6,89 (d, 1H, J = 8,4 Hz, Ar); 7,22-7,24 (m, 1H, Ar); 7,41 -7,54 (m, 4H, Ar); 7,71 (d, 1H, J = 8,0, Ar); 7,75-7,77 (m, 2H, Ar); 8,01-8,03 (m, 2H, Ar); 8,10 (s, 1H, Ar); 8,18 (s, 1H,
Ar); 8,33 (dd, 1H, J = 2,0, 6,8 Hz, Ar); 8,52 (dd, 1H, J = 2,0, 6,8 Hz, Ar); 10,25 (s a, 1H, NH); 12,16 (s a, 1H, NH) ppm. RMN de 13C (101 MHz, DMSO-ds): 5 164,84, 161,60, 161,46, 150,36, 147,85, 144,29, 143,91, 138,30, 136,21, 135,78, 134,02, 133,24, 129,11, 128,07, 124,81, 124,35, 122,94, 122,55, 121,36, 120,02, 118,82, 117,75, 117,16, 112,80, 109,65, 107,34, 51,52 ppm. RMN de 19F (376 MHz, DMSO-ds): 5 -138,12 (d, 1F, J = 24 Hz, Ar); -139,73 (d,
1F, J = 24 Hz, Ar) ppm. Anál. Calc. para C32H22N6O4F2: C, 64,86 %; H, 3,74%; N, 14,18%; Encontrado: C, 65,08%;
H, 3,77%; N, 14,39%.
(E/Z)-1-(3,4-difluorobencil)-2-oxo-N-(3-((2-oxo-3-(tiofen-2-ilmetilen)indolin-5-il)carbamoil)fenil)-1,2-dihidropiridin-3-carboxamida (DD41). Se añadió el 2-tiofenocarbaldehído (9 mg, 0,08 mmol) y una cantidad catalítica de piperidina a una disolución del derivado 2-oxoindólico 8 (40 mg, 0,08 mmol) disuelto en iPrOH/DMF. El procedimiento seguido es el mismo descrito para el derivado IB35. El material residual se purificó mediante cristalización en iPrOH, proporcionando el producto final (12 mg, 0,02 mmol, 25% de rendimiento) como un sólido naranja que consiste en una mezcla de isómeros E /Z (1:4). p.f.: 295-298°C; RMN de 1H (400 MHz, DMSO-d6): 55,32 (s, 2H, CH2); 6,71-6,73
(m, 1H, Ar); 6,86-6,88 (m, 1H, Ar); 7,23-7,34 (m, 2H, Ar); 7,44-7,52 (m, 3,75H, Ar); 7,62-8,20 (m, 7H, Ar); 8,34 (s, 1H,
Ar); 8,53 (s, 1H, Ar); 8,78 (s, 0,25H, Ar, isómero E); 10,25-10,33 (m a, 1H, NH); 10,60 (s a, 1H, NH); 12,17 (s a, 1H, nH) ppm. RMN de 13C (101 MHz, DMSO-d6): 5 169,35 (isómero E), 167,45 (isómero Z), 164,97 (isómero E), 164,80 (isómero Z), 161,62, 161,47, 150,37, 147,93, 144,30, 143,92, 138,86 (isómero E), 138,32 (isómero Z), 137,96 (isómero E), 137,25 (isómero Z), 137,01 (isómero E), 135,91, 134,48, 134,01, 132,94 (isómero E), 132,70 (isómero
Z), 132,25 (isómero E), 129,12 (isómero Z), 128,69 (isómero Z), 128,20 (isómero E), 127,43 (isóm 124,22, 123,59 (isómero E), 123,02 (isómero E), 122,93 (isómero Z), 122,66 (isómero E), 122,55 (isómero Z), 122,04 (isómero Z), 121,74 (isómero Z), 120,64 (isómero E), 120,04 (isómero Z), 118,92 (isómero E), 118,83 (isómero Z), 117,78, 117,18, 116,57 (isómero E), 113,26 (isómero Z), 109,68 (isómero E), 109,27 (isómero Z), 106,50, 51,54 ppm.
RMN de 19F (376 MHz, DMSO-d6): 5 -138,12 (d, 1F, J = 24 Hz, Ar); -139,73 (d, 1F, J = 24 Hz, Ar) ppm. Anál. Calc.
para C33H22N4O4F2S: C, 65,12 %; H, 3,64%; N, 9,21%; Encontrado: C, 65,38%; H, 3,69%; N, 9,28%.
Evaluación de los compuestos de Fórmulas (I) y (II)
La actividad de los compuestos inhibidores se puede ensayar utilizando métodos conocidos en la técnica y/o métodos presentados en esta memoria.
Los compuestos de Fórmula (I) y (II) se probaron en la prueba biológica denominada ensayo Z'-LYTE® específico de cinasa (Invitrogen Corporation, Life Technologies). De ahí que los compuestos sintetizados se sometieran a ensayo
Z'-Lyte basado en FRET frente a cinasa directa PDK1 para evaluar las actividades inhibidoras de cinasa (Invitrogen).
Los datos indican que los compuestos JJ31, IB32, IB35, SA23, SA16, DD22, DD21, DD40, DF8 y DF9 exhibían los mejores efectos sobre PDK1 (>50% de inhibición frente a PDK1 a 10 pM). Los valores de IC50 y el porcentaje de inhibición se presentan en la siguiente tabla.




El compuesto IB35 que mostraba una inhibición óptima de PDK1 también se probó con respecto a la selectividad al comparar su capacidad para inhibir otras cinasas en la ruta de Akt. Los resultados se presentan a continuación.
Características de IB35 con cinasas a 100 nM
Conc. % Compuesto % Inhibición % Inhibición Inhibición IB35 (nM) ATP Cinasa Tecnología 1 2 promedio 100 Km ap CDK7/ciclina H/MNAT1 Adapta 2 -1 1 100 Km ap CDK9/ciclina T1 Adapta 7 5 6 100 Km ap CHUK (IKK alfa) Adapta -2 7 3 100 Km ap PI4KB (PI4K beta) Adapta -3 12 4 100 Km ap PIK3CA/PIK3R1 (p110 Adapta 16 24 20 alfa/p85 alfa)
100 Km ap PIK3CD/PIK3R1 (p110 Adapta 13 21 17 delta/p85 alfa)
100 Km ap PIK3CG (p110 gamma) Adapta 6 -1 2 100 Km ap ABL1 ZLYTE 17 12 14 100 Km ap AKT1 (PKB alfa) ZLYTE -6 -9 -8 100 Km ap AKT2 (PKB beta) ZLYTE 0 -9 -4 100 Km ap AKT3 (PKB gamma) ZLYTE 3 1 2 100 Km ap AMPK A1/B1/G1 ZLYTE 8 7 7
Km ap AMPK A2/B1/G1 ZLYTE 10 11 10 Km ap AURKA (Aurora A) ZLYTE 37 38 38 100 BRAF ZLYTE 16 17 17 Km ap BTK ZLYTE -4 -2 -3 Km ap CAMK2A (CaMKII alfa) ZLYTE -1 2 0 Km ap CDC42 BPA (MRCKA) ZLYTE -1 -6 -3 Km ap CDK1/ciclina B ZLYTE 10 3 6 Km ap CDK2/ciclina A ZLYTE -1 8 4 Km ap CDK5/p25 ZLYTE -7 -10 -8 Km ap CDK5/p35 ZLYTE -2 6 2 Km ap CHEK1 (CHK1) ZLYTE 10 2 6 Km ap CHEK2 (CHK2) ZLYTE 1 8 4 Km ap CLK1 ZLYTE 4 -7 -2 Km ap CSNK1A1 (CK1 alfa 1) ZLYTE -3 -2 -2 Km ap CSNK1G1 (CK1 gamma ZLYTE 3 2 3 1)
Km ap CSNK2A2 (CK2 alfa 2) ZLYTE 4 3 3 Km ap FRAP1 (mTOR) ZLYTE -10 -18 -14 Km ap GRK5 ZLYTE -7 0 -4 Km ap GSK3A (GSK3 alfa) ZLYTE 43 43 43 Km ap GSK3B (GSK3 beta) ZLYTE 23 25 24 Km ap IRAK4 ZLYTE 3 -14 -6 Km ap ITK ZLYTE 17 12 14 Km ap JAK1 ZLYTE -10 -6 -8 Km ap LCK ZLYTE 10 8 9 100 MAP2K1 (MEK1) ZLYTE -20 -6 -13 100 MAP2K2 (MEK2) ZLYTE 13 6 9 Km ap MAPK1 (ERK2) ZLYTE 5 6 6 Km ap MAPK14 (p38 alfa) ZLYTE 2 4 3 Directa
Km ap MAPK3 (ERK1) ZLYTE -8 1 -3 100 MAPK8 (JNK1) ZLYTE 4 21 12 100 MAPK9 (JNK2) ZLYTE 3 -5 -1 Km ap MAPKAPK2 ZLYTE -2 2 0 Km ap MARK1 (MARK) ZLYTE 20 13 17 Km ap PAK6 ZLYTE 1 -9 -4 Km ap PRKACA (PKA) ZLYTE -5 -5 -5
100 Km ap PRKCA (PKC alfa) ZLYTE -2 2 0
100 Km ap PRKCB1 (PKC beta I) ZLYTE -10 -10 -10
100 Km ap PRKCD (PKC delta) ZLYTE -2 -12 -7
100 Km ap PRKCE (PKC épsilon) ZLYTE 4 3 3
100 Km ap PRKCZ (PKC zeta) ZLYTE -3 -2 -3
100 100 RAF1 (cRAF) Y340D ZLYTE 10 11 10 Y341D
100 Km ap ROCK1 ZLYTE -9 -6 -7
100 Km ap ROCK2 ZLYTE 3 1 2
100 Km ap RPS6KA1 (RSK1) ZLYTE -3 7 2
100 Km ap RPS6KB1 (p70S6K) ZLYTE -3 -10 -6
100 Km ap SRC ZLYTE 8 6 7
Como es evidente a partir de los datos presentados, no existía actividad sobre 58 cinasas implicadas en la ruta de Akt, con la única excepción de Aurora A y GSK3 que resultaba que eran inhibidas con valores porcentuales de 38%, 43% a 100 nM del compuesto probado.
A fin de estudiar más en profundidad las propiedades biológicas de los nuevos compuestos y correlacionar la actividad de inhibición de PDK1 con las propiedades antiproliferativas in vitro, los compuestos IB35, SA23 y SA16 se investigaron adicionalmente en una serie de ensayos biológicos in vitro sobre líneas celulares U87MG y células precursoras aisladas de células U87MG.
Cultivo de la línea celular GBM y aislamiento de GSC. La línea celular U87MG se obtuvo de the National Institute for Cancer Research de Génova (Italia) y se cultivó en DMEM-FBS al 10% y glutamina 2 mM. Para aislar GSC, aproximadamente 2,0 x 106 células se suspendieron en 1 ml de un medio para células precursoras neurales (NSC, por sus siglas en inglés) libre de suero definido (Daniele, S., et al., Modulation of A1 and A2B adenosine receptor activity: a new strategy to sensitise glioblastoma stem cells to chemotherapy. Cell death & disease, 2014. 5(11): pág. e1539). Después de 3-4 días de cultivo, las neuroesferas se recogieron, se suspendieron en medio NSC, se disociaron en células individuales y se sembraron para los ensayos. Para el tratamiento a largo plazo de las células, el NSC o medio completo que contenía fármacos se reemplazó cada de dos a tres días.
Ensayos de proliferación celular de células GBM y GSC. Las células U87MG o GSC humanas se sembraron en una densidad de 3 x 103 células/pocillo. Después de 24 h, las células se trataron de uno a siete días con medio de crecimiento reciente que contenía diferentes concentraciones de SA16, SA23 o IB35. Tras el período de tratamiento, se determinó la proliferación celular usando el ensayo de MTS según las instrucciones del fabricante. La absorbancia de formazano a 490 nM se midió en un ensayo colorimétrico con un lector de placas automatizado (Victor Wallac 2, Perkin Elmer). Los resultados se calcularon al sustraer el fondo medio de los valores obtenidos a partir de cada evaluación y se expresaron como el porcentaje del control (células no tratadas). Se generaba una curva sigmoidea de respuesta a la dosis, de la que se derivaban los valores de IC50.
Tinción con anexina V y 7-AAD en células U87MG y en GSC. Se realizó una tinción doble con anexina V conjugada a isotiocianato de fluoresceína (FITC, por sus siglas en inglés) y 7-aminoactinomicina (7-AAD) usando el estuche disponible comercialmente (Muse Annexin V and Dead Cell Kit; Merck KGaA, Darmstadt, Alemania). Las células U87MG o las GSC se trataron con DMSO (control), SA16, SA23 o IB35 durante 72 h o siete días, respectivamente. Al final de los períodos de tratamiento, los porcentajes de células vivas, apoptósicas y muertas se adquirieron y se analizaron mediante el analizador celular Muse™ según se describe previamente (Daniele, S., et al., Combined inhibition of AKT/mTOR and MDM2 enhances Glioblastoma Multiforme cell apoptosis and differentiation of cancer stem cells. Sci. Rep., 2015. 5).
Actividad de PDK1 en células U87MG y en GSC. Se cultivaron células U87MG en microplacas de 96 pocillos (5.000 células/pocillo) y se trataron durante diferentes tiempos con SA16, SA23 o IB35 10 pM. Al final del período de tratamiento, las GSC se centrifugaron a 500 x g durante 3 minutos; las células se lavaron dos veces usando solución salina reciente y se fijaron rápidamente con formaldehído al 4% (para células U87MG adherentes) o al 8% (para GSC en suspensión) para conservar la activación de una modificación de proteína específica. La actividad de PDK1 se determinó midiendo los niveles de AKT fosforilada sobre Thr308, usando anticuerpos primarios específicos. La incubación posterior con un anticuerpo conjugado a HRP secundario y la disolución de revelado permitía la cuantificación colorimétrica de los niveles de proteína fosforilada. A continuación, el número relativo de células en cada pocillo se determinó usando el ensayo del violeta cristal. Los resultados se calcularon al sustraer el valor de
fondo medio de los valores obtenidos bajo cada condición de prueba: los valores se normalizaron con respecto al número de células en cada pocillo y se expresaron como los porcentajes de los valores de control (células no tratadas).
Ensayo de migración para curación de heridas Se sembraron células U87MG (1x105) sobre placas de seis pocillos hasta formar una monocapa confluente, y se trataron durante 72 h con SA16, SA23 o IB35 10 pM. A continuación, la monocapa celular se escarificó en una línea recta para realizar una 'herida por escarificación' con una punta de pipeta de 0,2 ml y el residuo celular se retiró al lavar las células con solución salina tamponada con fosfato. Se añadió medio RPMl complementado con FBS al 1% y se capturaron imágenes del cierre de la escarificación a las 0, 6 y 24 h.
Análisis estadístico. Se usó el programa de ajuste de curvas no lineal polivalente GraphPad Prism (GraphPad Software Inc., San Diego, CA) para el análisis de datos y las presentaciones gráficas. Todos los datos se presentan como la media ± EEM. El análisis estadístico se realizó mediante análisis de varianza (ANOVA) unidireccional con la prueba de la t corregida de Bonferroni para comparaciones emparejadas retrospectivas. P<0,05 se consideraba estadísticamente significativa.
Resultados
Efectos de los inhibidores de PDK1 sobre la proliferación celular de U87MG.
Como línea celular GBM representativa, se usaron las células U87MG anteriores; esta línea celular humana daba como resultado un modelo apropiado para estudiar los efectos de la inhibición de PDK1, debido a que es deficiente para el supresor tumoral fosfatasa y homólogo de tensina (PTEN), un regulador negativo de la ruta PI3K/AKT (Wang, S.I., et al., Somatic mutations of PTEN in glioblastoma multiforme. Cancer research, 1997. 57(19): pág. 4183-4186). Para examinar los efectos de SA16, SA23 e IB35 sobre el crecimiento/la supervivencia celulares de GBM, se incubaron células U87MG con diferentes concentraciones de los nuevos compuestos (1 nM-10 pM) durante 24 y 72 h. Específicamente, las células U87MG se trataron en medio completo con diferentes concentraciones de SA16, SA23 o IB35 (1 nM-10 pM) durante 24 h (A) o 72 h (B). Al final del tratamiento, la proliferación celular se midió usando el ensayo de MTS. Los datos se expresan como un porcentaje con respecto al de células no tratadas (control), que se fijaba al 100%, y son los valores medios ± EEM de tres experimentos independientes, cada uno realizado por duplicado. La significación de las diferencias presentadas en las figuras 1 y 2 se determinó con un ANOVA unidireccional con prueba a posteriori de Bonferroni (*p<0,05, **p<0,01, *** p<0,001 frente a células de control).
Después de 24 h (Figura 1), los tres compuestos inducían solo una inhibición moderada la proliferación de células U87MG; el mayor efecto se observó con IB35 (41% de inhibición, figura 1). En contraste, después de 72 h, todos los compuestos disminuían significativamente la proliferación de células U87MG, de un modo dependiente de la concentración, dando valores de IC50 de 449,7 ± 46,2 nM (IB35), 2,05 ± 0,19 pM (SA16) y 1,17 ± 0,10 pM (SA23) (Figura 2). La clasificación de los valores de IC50 de los nuevos compuestos sobre células U87MG reflejaba la clasificación de afinidad hacia PDK1 recombinante, confirmando así que las actividades antiproliferativas están mediadas por la inhibición de PDK1.
Efecto de inhibidores de PDK1 sobre la inducción de apoptosis de U87MG.
A continuación, se investigó si la reducción en la proliferación celular provocada por los inhibidores de PDK1 se podía asociar con la apoptosis. Se trataron células U87MG durante 72 h con DMSO (control), o SA16, SA23 o IB35 10 pM. Al final del período de tratamiento, las células se recogieron y el nivel de externalización de fosfatidilserina se evaluó usando el protocolo de tinción con anexina V, según se describe anteriormente (Figura 3). En la Figura 4, los datos se expresan como el porcentaje de células apoptósicas (los datos para las células apoptósicas de la fase temprana mostrados en blanco, y los datos para las células apoptósicas/necróticas de fase tardía mostrados en gris) frente al número total de células. Los datos mostrados representaban la media ± EEM de tres experimentos diferentes. La significación de las diferencias se determinó con un ANOVA unidireccional con prueba a posteriori de Bonferroni (***p<0,001 frente al control).
El tratamiento de células U87MG con SA16, SA23 e IB35 (10 pM) durante 72 h inducía una externalización de fosfatidilserina significativa, en ausencia (apoptosis temprana) o en presencia de 7-aminoactinomicina que se une a ADN (apoptosis/muerte tardía) (Figuras 3 y 4). De acuerdo con los datos obtenidos en el ensayo de MTS, IB35 mostraba el mayor grado de apoptosis de GBM (porcentaje de células apoptósicas totales: 23,0 ± 1,9).
Efectos de SA16, SA23 e IB35 sobre la actividad de PDK1 en células U87 MG.
Para valorar si SA16, SA23 e IB35 podían inhibir eficazmente la ruta AKT/PDK1 en células U87MG, se trataron U87MG durante diferentes tiempos con 10 pM de cada inhibidor; después de la incubación, la actividad de PDK1 se determinó evaluando la fosforilación de AKT en Thr308. Según se presenta en la bibliografía, las células U87MG exhibían un alto nivel constitutivo de AKT fosforilada en los sitios reguladores tanto T308 como Ser473, en línea con un perfil celular de células cancerosas deficientes en PTEN (González-Angulo, A.M., et al., SU2C Phase Ib Study of Paclitaxel and MK-2206 in Advanced Solid Tumors and Metastatic Breast Cancer. Journal of the National Cancer
Institute, 2015. 107(3): p. dju493; Zinda, M.J., C.J. Vlahos, y M.T. Lai, Ceramide induces the dephosphorylation and inhibition of constitutively activated Akt in PTEN negative U87mg cells. Biochemical and biophysical research communications, 2001. 280(4): pág. 1107-1115). De ahí que U87MG se trataran durante los tiempos indicados (0 120 min) con medio completo que contenía DMSO (control) o SA16, SA23 o IB35 10 pM. Al final de los períodos de tratamiento, los niveles de fosforilación de (T308)AKT se evaluaron usando un estuche de ELISA, según se describe anteriormente. Los datos presentados en la Figura 5 se expresaron como el porcentaje de (T308)AKT fosforilada con relación a los de células no tratadas (control), que se fijaron al 100%, y eran los valores medios ± EEM de tres experimentos independientes realizados por triplicado. Según se representa en la Figura 5, SA23 e IB35, probados a 10 pM, inhibían significativamente la fosforilación de AKT en Thr308, de un modo dependiente del tiempo, con un máximo de inhibición después de 120 min, confirmando que los compuestos eran capaces de bloquear la actividad de PDK1. En contraste, SA16 provocaba una cinética bifásica de inhibición de (T308)AKT, mostrando máximos después de 5 min y 120 min de tratamiento (Figura 5).
Efectos de inhibidores de PDK1 sobre la migración celular.
Para valorar si la inhibición de la actividad de PDK1 se podía asociar con un bloqueo de la migración celular, se realizó una prueba de herida por escarificación. Imágenes microscópicas representativas se muestran en la Figura 6. Las células U87MG se trataron durante 72 h con DMSO (control) o SA16, SA23 e IB35 10 pM. A continuación, la monocapa celular se escarificó (momento inicial) y las células se dejaron crecer en medio reciente. Se tomaron micrografías representativas después de 6 h y 24 h desde la escarificación (Figura 6). La superficie hueca (Figura 7) se midió después de 6 h y 24 h desde la escarificación; los recuentos eran los valores medios ± EEM de tres experimentos independientes. La significación de las diferencias se determinó usando un ANOVA unidireccional con la prueba a posteriori de Bonferroni (**P<0,05, ***P<0,001 frente al control; ###P<0,001 frente a la superficie hueca respectiva a las 6 h desde la escarificación).
El análisis cuantitativo de la superficie hueca (Figura 7) demostraba que el tratamiento de células U87MG con inhibidores de PDK1 inhibe significativamente la migración celular, después tanto de 6 h como de 24 h desde la escarificación. Estos resultados confirmaban que una inhibición de PDK1 conduce a una supresión de la migración de células tumorales.
Efectos del tratamiento con SA16, SA23 e IB35 sobre la proliferación de GSC
La inhibición de PDK1 se ha presentado como un enfoque terapéutico potencialmente eficaz para reducir la autorrenovación y la propagación de GSC (Signore, M., etal., Combined PDK1 and CHK1 inhibition is required to kill glioblastoma stem-like cells in vitro and in vivo. Cell death & disease, 2014. 5(5): pág. e1223). Sobre esta base, se evaluaron los efectos de SA16, SA23 e IB35 sobre GSC, aisladas de células U87MG según se presenta previamente (Daniele, S., et al., Modulation of A1 and A2B adenosine receptor activity: a new strategy to sensitise glioblastoma stem cells to chemotherapy. Cell death & disease, 2014. 5(11): pág. e1539). Se incubaron GSC derivadas de U87MG con las concentraciones indicadas de IB35, SA23 o SA16 durante cuatro días (Figura 8) o siete días (Figura 9). Al final de los períodos de tratamiento, se midió la proliferación celular usando el ensayo de MTS. Los datos se expresaron como un porcentaje con respecto al de células no tratadas (control), que se fijó hasta 100%, y son los valores medios ± EEM de tres experimentos independientes, cada uno realizado por duplicado. Los valores de IC50 después de siete días de tratamiento se calcularon a partir de la curva sigmoidea de respuesta a la dosis (Figura 9). La significación de las diferencias se determinó con un ANOVA unidireccional con prueba a posteriori de Bonferroni (** p<0,01, *** p<0,001 frente al control).
Cuando las GSC se incubaban con IB35 y SA23, se producía una inhibición significativa de la proliferación celular, partiendo de cuatro días de tratamiento (Figura 8). Después de siete días de tratamiento, los compuestos inducían una inhibición dependiente de la concentración de la proliferación de GSC, dando valores de IC50 de 3,36 ± 0,40 nM (IB35), 0,30 ± 0,04 nM (SA23) y 5,47 ± 0,10 nM (SA16) (Figura 9). Estos datos confirmaban que los inhibidores de PDK1 son capaces de bloquear la proliferación de GSC.
Efectos de inhibidores de PDK1 sobre la apoptosis de GSC.
Se investigó si la reducción de proliferación celular provocada por los dos compuestos se podía asociar a la apoptosis celular. Las GSC se trataron durante 7 días con medio NSC que contenía DMSO (control) o 10 pM de IB35, SA16 o SA23 durante siete días. Al final de los tratamientos, las células se recogieron y el grado de externalización de fosfatidilserina se evaluó usando el protocolo de anexina V, según se describe anteriormente (figura 10). Los datos se expresaron como el porcentaje de células apoptósicas (apoptósicas tempranas en blanco, apoptósicas/necróticas tardías en gris) con relación al número total de células y se representaron en la Figura 11. Los datos eran los valores medios ± EEM de tres experimentos diferentes. La significación de las diferencias se determinó con un ANOVA unidireccional con prueba a posteriori de Bonferroni (*** p<0,001 frente al control).
Cuando se medía la tinción con anexina V después de 7 días de tratamiento de GSC, todos los compuestos inducían una externalización de fosfatidilserina significativa, en ausencia (apoptosis temprana) o en presencia de 7-aminoactinomicina 11 que se une a ADN (apoptosis/muerte tardía) (Figuras 10 y 11). Como en células U87MG, IB35 mostraba el mayor grado de apoptosis de GBM (porcentaje de células apoptósicas totales: 38,5 ± 3,9).
Efectos de SA16, SA23 e IB35 sobre la actividad de PDK1 en GSC.
Los efectos de los nuevos compuestos sobre la actividad de PDK1 se investigaron mediante ensayos ELISA también en GSC. GSC derivadas de U87MG se trataron durante los tiempos indicados (0-120 min) con medio NSC que contenía DMSO (control) o IB35, SA23 o SA16 10 qM. Al final de los períodos de tratamiento, se evaluaron los niveles de fosforilación de AKT sobre Thr308 usando un ensayo ELISA, según se describe anteriormente. Los datos se expresaron como el porcentaje de AKT fosforilada con relación a los de células no tratadas (control), que se fijaron a 100%, y eran los valores medios ± EEM de tres experimentos realizados por triplicado. Los datos se representan en la Figura 12. La significación de las diferencias se realizó usando un ANOVA unidireccional con prueba a posteriori de Bonferroni (* p< 0,05, ** p< 0,01, *** p<0,001 frente al control). De acuerdo con los datos obtenidos en células U87MG, SA23 e IB35, probados a 10 qM, inhibían significativamente la fosforilación de AKT en Thr308, de un modo dependiente del tiempo, con un máximo de inhibición después de 120 min. (Figura 12). En contraste, según se evidencia en células U87MG, SA16 provocaba una cinética bifásica de inhibición de AKT, que mostraba máximos después de 5 min y 120 min de tratamiento. Se debe apreciar que los porcentajes de inhibición de PDK1 eran significativamente superiores en GSC con respecto a células U87MG (máximo de inhibición para IB35: 54,0 ± 5,2 en células U87MG; 80,4 ± 3,7 en GSC; máximo de inhibición para SA16: 33,3 ± 4,5 en células U87MG; 74,8 ± 3,0 en GSC; máximo de inhibición para SA23: 48,1 ± 5,0 en células U87MG; 69,0 ± 4,5 en GSC.
Comparación de actividades antiproliferativas con un fármaco conocido en GBM y GSC
Los compuestos de la invención IB35, SA16 y SA23 se compararon con everolimus (dihidroxi-12-[(2R)-1-[(1S,3fí,4fí)-4-(2-hidroxietoxi)-3-metoxiciclohexil]propan-2-il]-19,30-dimetoxi-15,17,21,23,29,35-hexametil-11,36-dioxa-4-azatriciclo[30.3.1.0-hexatriaconta-16,24,26,28-tetraeno-2,3,10,14,20-pentona), un fármaco de referencia de Novartis para la terapia dirigida (ahora en fase I de estudios clínicos para GBM) (Daniele, S., et al., Combined inhibition of AKT/mTOR and MDM2 enhances Glioblastoma Multiforme cell apoptosis and differentiation of cancer stem cells. Sci. Rep., 2015. 5).
Los resultados se presentan en la tabla a continuación.

Los datos de la tabla muestran que aunque el resulte que IB35 SA16 y SA23 inhiban el crecimiento celular sobre la línea celular U87, la potencia es inferior que everolimus. Por el contrario, la actividad antiproliferativa frente a GSC es de 2 a 50 veces superior que everolimus, indicando que IB35, SA16 y SA23 son más capaces de acelerar la inducción de la apoptosis y así potenciar la inhibición de la viabilidad de GSC con respecto a everolimus.
A partir de la parte biológica experimental presentada anteriormente, se muestra que los compuestos JJ31, IB32, IB35, SA16 y SA23 inhiben la enzima PDK1 con valores de IC50 en el intervalo de nM a qM.
La clasificación del valor de IC50 sobre PDK1 recombinante reflejaba la clasificación de afinidad hacia líneas celulares U87MG, confirmando así que la actividad antiproliferativa estaba medidas por PDK1. IB35, SA16 y SA23 inhibían la actividad constitutiva de PDK1 en células U87MG; como resultado, los nuevos compuestos disminuían la viabilidad celular y desencadenaban la apoptosis. Por otra parte, la inhibición de la viabilidad celular era de larga duración (72 h). En el ensayo de escarificación con herida, se mostraba que los inhibidores de PDK1 inhiben significativamente la migración celular, después tanto de 6 h como de 24 h desde la escarificación. Estos resultados confirmaban que una inhibición de PDK1 conduce a una supresión de la migración de células tumorales.
Finalmente, los nuevos compuestos se caracterizaban en células precursoras de glioma (GSC) aisladas de células U87MG. Los resultados obtenidos indicaban que los nuevos compuestos inducían una inhibición dependiente de la concentración y el tiempo de la proliferación de GSC y desencadenaban la apoptosis, confirmando así que los inhibidores de PDK1 eran capaces de bloquear la proliferación de GSC.
Claims (22)
1. Un compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida de Fórmula (I)

en donde
m es 0 o 1
n es 0 o 1
A se selecciona del grupo que consiste en (-NH-CO-CH2-), (-NH-CO-CH(Ph)-), (-NH-CO-CH2-CH2-), (-NH-CO-CH2-CH2-CH2-) y (-NH-CO-Ph-); y
B es CH-D, donde D se selecciona del grupo que consiste en tienilo, fenilo, imidazolilo, estando D opcionalmente sustituido con un halógeno,
o una sal farmacéutica del mismo, para su uso en el tratamiento de patologías que requieren el uso de un inhibidor de la enzima PDK1.
2. El compuesto para el uso según la reivindicación 1, en donde m es 0 y n es 1.
3. El compuesto para el uso según la reivindicación 1, en donde m es 1 y n es 1.
4. El compuesto para el uso según la reivindicación 1, en donde A es (-NH-CO-CH2-), (-NH-CO-CH2-CH2-CH2-), (-NH-CO-CH2-CH2), o (-NH-CO-Ph-) o (-NH-CO-CH(Ph)-).
5. El compuesto para el uso según la reivindicación 4, en donde A es (-NH-CO-CH2-) o (-NH-CO-CH2-CH2).
6. El compuesto para el uso según una cualquiera de las reivindicaciones 1, 4-5, en donde n es 1 y B es CH-D, donde D es imidazolilo, preferiblemente 1 H-imidazol-5-ilo.
7. El compuesto para el uso según la reivindicación 1, en donde dicho compuesto se selecciona del grupo que consiste en (Z)-N-(3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, 1-(3,4-difluorobencil)-2-oxo-N-(2-oxo-2-((2-oxoindolin-5-il)amino)etil)-1,2-dihidropiridin-3-carboxamida, (E/Z)-1-(3,4-difluorobencil)-2-oxo-N-(2-oxo-2-((2-oxo-3-(tiofen-2-ilmetilen)indolin-5-il)amino)etil)-1,2-dihidropiridin-3-carboxamida, (Z)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1-feniletil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-2-il)amino)-2-oxo-1 -feniletil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (E/Z)-1 -(3,4-difluorobencil)-2-oxo-N-(3-oxo-3-((2-oxo-3-(tiofen-2-ilmetilen)indolin-5-il)amino)propil)-1,2-dihidropiridin-3-carboxamida, (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-3-oxopropil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (E/Z)-1 -(3,4-difluorobencil)-N-(3-((3-(4-fluorobenciliden)-2-oxoindolin-5-il)amino)-3-oxopropil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z/E)-N-(3-((3-((1 H-imidazol-2-il)metilen)-2-oxoindolin-5-il)amino)-3-oxopropil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)carbamoil)fenil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (E/Z)-1-(3,4-difluorobencil)-2-oxo-N-(3-((2-oxo-3-(tiofen-2-ilmetilen)indolin-5-il)carbamoil)fenil)-1,2-dihidropiridin-3-carboxamida, (Z)-N-(4-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-4-oxobutil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida y (E/Z)-1-(3,4-difluorobencil)-2-oxo-N-(4-oxo-4-((2-oxo-3-(tiofen-2-ilmetilen)indolin-5-il)amino)butil)-1,2-dihidropiridin-3-carboxamida.
8. El compuesto para el uso según la reivindicación 7, en donde dicho compuesto se selecciona del grupo (Z)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (z )-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1 -feniletil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-2-il)amino)-2-oxo-1 -feniletil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)carbamoil)fenil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida y (Z)-N-(4
((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-4-oxobutil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida.
9. El compuesto para el uso según la reivindicación 8, en donde el compuesto es (Z)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida.
10. El compuesto para el uso según una cualquiera de las reivindicaciones 1-9, en donde la patología que requiere un inhibidor de PDK1 es un cáncer, preferiblemente glioblastoma.
11. Un compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida de Fórmula (II)

en donde
n es 0 o 1
B es CH-D, donde D se selecciona del grupo que consiste en tienilo, fenilo, imidazolilo, estando D opcionalmente sustituido con un halógeno; y
A se selecciona del grupo que consiste en (-NH-CO-CH2-), (-NH-CO-CH(Ph)-), (-NH-CO-CH2-CH2-), (-NH-CO-CH2-CH2-CH2-) y (-NH-CO-Ph-)
o una sal farmacéutica del mismo.
12. El compuesto según la reivindicación 11, en donde A se selecciona del grupo que consiste en (-NH-CO-CH2-), (-NH-CO-CH2-CH2-), (-NH-CO-CH2-CH2-CH2-) y (-NH-CO-Ph-).
13. El compuesto según la reivindicación 12, en donde A es (-NH-CO-CH2-).
14. El compuesto según una cualquiera de las reivindicaciones 11-13, en donde n es 1 y B es CH-D, donde D es imidazolilo, preferiblemente 1 H-imidazol-5-ilo.
15. El compuesto según la reivindicación 11, en donde dicho compuesto se selecciona del grupo que consiste en (E/Z)-1-(3,4-difluorobencil)-2-oxo-N-(2-oxo-2-((2-oxo-3-(tiofen-2-ilmetilen)indolin-5-il)amino)etil)-1,2-dihidropiridin-3-carboxamida, (Z)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1-feniletil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-2-il)amino)-2-oxo-1 -feniletil)-1 -(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (E/Z)-1-(3,4-difluorobencil)-2-oxo-N-(3-oxo-3-((2-oxo-3-(tiofen-2-ilmetilen)indolin-5-il)amino)propil)-1,2-dihidropiridin-3-carboxamida, (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-3-oxopropil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (E/Z)-1-(3,4-difluorobencil)-N-(3-((3-(4-fluorobenciliden)-2-oxoindolin-5-il)amino)-3-oxopropil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z/E)-N-(3-((3-((1 H-imidazol-2-il)metilen)-2-oxoindolin-5-il)amino)-3-oxopropil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)carbamoil)fenil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (E/Z)-1-(3,4-difluorobencil)-2-oxo-N-(3-((2-oxo-3-(tiofen-2-ilmetilen)indolin-5-il)carbamoil)fenil)-1,2-dihidropiridin-3-carboxamida, (Z)-N-(4-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-4-oxobutil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida y (E/Z)-1 -(3,4-difluorobencil)-2-oxo-N-(4-oxo-4-((2-oxo-3-(tiofen-2-ilmetilen)indolin-5-il)amino)butil)-1,2-dihidropiridin-3-carboxamida.
16. El compuesto según la reivindicación 15, en donde dicho compuesto se selecciona del grupo (Z)-N-(2-((3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-(R)-N-(2-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1-feniletil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida y (Z)-(R)-N-(2-((3-((1 H-imidazol-2-il)metilen)-2-oxoindolin-5-il)amino)-2-oxo-1 -feniletil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, (Z)-N-(3-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)carbamoil)fenil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida, y (Z)-N-(4-((3-((1 H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-4-oxobutil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida.
17. El compuesto según la reivindicación 14, en donde el compuesto es (Z)-N-(2-((3-((1H-imidazol-5-il)metilen)-2-oxoindolin-5-il)amino)-2-oxoetil)-1-(3,4-difluorobencil)-2-oxo-1,2-dihidropiridin-3-carboxamida.
18. Un compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida según una cualquiera de las reivindicaciones 11-17, para su uso como medicamento.
19. Una composición farmacéutica que comprende un compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida según una cualquiera de las reivindicaciones 11-18 y un portador farmacéuticamente aceptable.
20. Un compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida según una cualquiera de las reivindicaciones 11-17, para su uso en el tratamiento de patologías que requieren un inhibidor de la enzima PDK1.
21. El compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida para el uso según la reivindicación 20, en donde la patología se selecciona del grupo que consiste en diabetes, enfermedades neurodegenerativas tales como enfermedades de Alzheimer y priónicas, y tumores tales como cánceres de mama y pancreático y glioblastoma.
22. El compuesto de 2-oxo-1,2-dihidropiridin-3-carboxamida para el uso según la reivindicación 21, en donde la patología es un cáncer, preferiblemente glioblastoma (GBM).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ITUB20150999 | 2015-06-11 | ||
| PCT/EP2016/063293 WO2016198597A1 (en) | 2015-06-11 | 2016-06-10 | 2-oxo-1,2-dihydropyridine-3-carboxamide compounds and their use as inhibitors of pdk1 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2812452T3 true ES2812452T3 (es) | 2021-03-17 |
Family
ID=53901023
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16735586T Active ES2812452T3 (es) | 2015-06-11 | 2016-06-10 | Compuestos de 2-oxo-1,2-dihidropiridin-3-carboxamida y su uso como inhibidores de PDK1 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US10351548B2 (es) |
| EP (1) | EP3307726B1 (es) |
| ES (1) | ES2812452T3 (es) |
| WO (1) | WO2016198597A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ITUA20164278A1 (it) * | 2016-06-10 | 2017-12-10 | International Soc For Drug Development S R L | COMPOSTI 2-OXO-1,2-DIIDROPIRIDIN-3-CARBOSSAMMIDE E LORO USO COME INIBITORI DUALI DI PDK1/AurA |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8778977B2 (en) | 2006-06-30 | 2014-07-15 | Sunesis Pharmaceuticals, Inc. | Pyridinonyl PDK1 inhibitors |
| US8552019B2 (en) * | 2008-12-05 | 2013-10-08 | Merck Sharp & Dohme Corp. | Inhibitors of phosphoinositide dependent kinase 1 (PDK1) |
-
2016
- 2016-06-10 US US15/735,468 patent/US10351548B2/en active Active
- 2016-06-10 ES ES16735586T patent/ES2812452T3/es active Active
- 2016-06-10 WO PCT/EP2016/063293 patent/WO2016198597A1/en active Application Filing
- 2016-06-10 EP EP16735586.6A patent/EP3307726B1/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| US10351548B2 (en) | 2019-07-16 |
| WO2016198597A1 (en) | 2016-12-15 |
| EP3307726A1 (en) | 2018-04-18 |
| EP3307726B1 (en) | 2020-05-20 |
| US20180155320A1 (en) | 2018-06-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10351514B2 (en) | Benzimidazole inhibitors of the sodium channel | |
| JP6869947B2 (ja) | 置換キナゾリン化合物ならびにそのg12c変異kras、hrasおよび/またはnrasタンパク質の阻害剤としての使用 | |
| ES2289349T3 (es) | Derivados de heteroaril-pirimidina como inhibidores de jak. | |
| RU2715708C2 (ru) | Ингибиторы fgfr4 | |
| DK2396307T3 (en) | NOVEL AMINO-azaheterocyclic carboxamides | |
| CN103797002B (zh) | Dyrk1抑制剂及其用途 | |
| ES2824814T3 (es) | Compuestos farmacéuticos | |
| JP6912486B2 (ja) | Rsk阻害剤として有用なカルボキサミド誘導体 | |
| JP2013510824A (ja) | セリン/トレオニンキナーゼを調節または制御するための化合物、その調製のための方法、医薬組成物、化合物の使用、方法ならびにセリン/トレオニンキナーゼ調節剤 | |
| BR112013012488B1 (pt) | Azetidinas quinazolina carboxamida, seus usos e seu processo de fabricação, composição farmacêutica, e kit | |
| Du et al. | Discovery of phosphonamidate IDO1 inhibitors for the treatment of non-small cell lung cancer | |
| Kardile et al. | Design, synthesis, and biological evaluation of novel quinoline derivatives as small molecule mutant EGFR inhibitors targeting resistance in NSCLC: In vitro screening and ADME predictions | |
| WO2015189799A1 (en) | Compounds comprising 1,1',2,5'-tetrahydrospiro[indole-3,2'-pyrrole]-2,5'-dione system as inhibitors p53-mdm2 protein-protein interaction | |
| AU2017368286B2 (en) | 3-amino-1,5-dihydro-pyrazolo[3,4-d]pyrimidin-4-ones as cyclin dependent kinase inhibitors | |
| CA3172987A1 (en) | Small molecule inhibitors of oncogenic chd1l with preclinical activity against colorectal cancer | |
| WO2021074251A1 (en) | Pyrrolo[2,3-d]pyrimidine derivatives and their use in the treatment of cancer | |
| WO2018214796A1 (zh) | 一类异吲哚酮-酰亚胺环-1,3-二酮-2-烯化合物、其组合物和用途 | |
| Li et al. | Design, synthesis and biological evaluation of aminopyrimidine derivatives bearing a 4, 5, 6, 7-tetrahydrothieno [3, 2-c] pyridine as potent EGFR inhibitors | |
| CA3000565C (en) | 2-aminoquinazoline derivatives as p70s6 kinase inhibitors | |
| ES2812452T3 (es) | Compuestos de 2-oxo-1,2-dihidropiridin-3-carboxamida y su uso como inhibidores de PDK1 | |
| JP6987125B2 (ja) | 新規2,4,6−三置換s−トリアジン化合物並びにその製造方法および使用 | |
| WO2019157959A1 (zh) | 一种嘧啶类化合物、其制备方法及其医药用途 | |
| Xi et al. | Modification of osimertinib to discover new potent EGFRC797S-TK inhibitors | |
| Dimitrov et al. | Design and Optimization of Novel Benzimidazole-and Imidazo [4, 5-b] pyridine-Based ATM Kinase Inhibitors with Subnanomolar Activities | |
| JP2021500350A (ja) | 変異体egfrファミリーチロシンキナーゼの阻害剤 |