ES2769031T3 - Agentes citotóxicos bifuncionales que contienen el farmacóforo cti - Google Patents
Agentes citotóxicos bifuncionales que contienen el farmacóforo cti Download PDFInfo
- Publication number
- ES2769031T3 ES2769031T3 ES16714004T ES16714004T ES2769031T3 ES 2769031 T3 ES2769031 T3 ES 2769031T3 ES 16714004 T ES16714004 T ES 16714004T ES 16714004 T ES16714004 T ES 16714004T ES 2769031 T3 ES2769031 T3 ES 2769031T3
- Authority
- ES
- Spain
- Prior art keywords
- alkyl
- antibody
- methyl
- dihydro
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 230000001588 bifunctional effect Effects 0.000 title description 15
- 229940127089 cytotoxic agent Drugs 0.000 title description 10
- 231100000599 cytotoxic agent Toxicity 0.000 title description 5
- 239000002254 cytotoxic agent Substances 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 183
- 125000002252 acyl group Chemical group 0.000 claims abstract description 25
- 150000003839 salts Chemical class 0.000 claims abstract description 25
- 239000012453 solvate Substances 0.000 claims abstract description 20
- 125000003147 glycosyl group Chemical group 0.000 claims abstract description 11
- -1 PO 3 H 2 Chemical group 0.000 claims description 216
- 206010028980 Neoplasm Diseases 0.000 claims description 98
- 201000011510 cancer Diseases 0.000 claims description 75
- 229940049595 antibody-drug conjugate Drugs 0.000 claims description 56
- 239000000611 antibody drug conjugate Substances 0.000 claims description 55
- 238000011282 treatment Methods 0.000 claims description 48
- 239000008194 pharmaceutical composition Substances 0.000 claims description 15
- 229960000575 trastuzumab Drugs 0.000 claims description 7
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 6
- 206010006187 Breast cancer Diseases 0.000 claims description 3
- 208000026310 Breast neoplasm Diseases 0.000 claims description 3
- 229960005395 cetuximab Drugs 0.000 claims description 3
- 210000001072 colon Anatomy 0.000 claims description 3
- 206010017758 gastric cancer Diseases 0.000 claims description 3
- 102100022337 Integrin alpha-V Human genes 0.000 claims description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 2
- 206010033128 Ovarian cancer Diseases 0.000 claims description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 2
- 108010048673 Vitronectin Receptors Proteins 0.000 claims description 2
- 229960000548 alemtuzumab Drugs 0.000 claims description 2
- 229960000397 bevacizumab Drugs 0.000 claims description 2
- 229960001776 edrecolomab Drugs 0.000 claims description 2
- 229960000578 gemtuzumab Drugs 0.000 claims description 2
- 229950000518 labetuzumab Drugs 0.000 claims description 2
- 201000005202 lung cancer Diseases 0.000 claims description 2
- 208000020816 lung neoplasm Diseases 0.000 claims description 2
- 229960002450 ofatumumab Drugs 0.000 claims description 2
- 229950007283 oregovomab Drugs 0.000 claims description 2
- 229960001972 panitumumab Drugs 0.000 claims description 2
- 229960004641 rituximab Drugs 0.000 claims description 2
- 201000011549 stomach cancer Diseases 0.000 claims description 2
- 229960005267 tositumomab Drugs 0.000 claims description 2
- 206010005003 Bladder cancer Diseases 0.000 claims 1
- 206010008342 Cervix carcinoma Diseases 0.000 claims 1
- 206010014733 Endometrial cancer Diseases 0.000 claims 1
- 206010014759 Endometrial neoplasm Diseases 0.000 claims 1
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims 1
- 208000008839 Kidney Neoplasms Diseases 0.000 claims 1
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims 1
- 206010060862 Prostate cancer Diseases 0.000 claims 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims 1
- 206010038389 Renal cancer Diseases 0.000 claims 1
- 208000000453 Skin Neoplasms Diseases 0.000 claims 1
- 208000024313 Testicular Neoplasms Diseases 0.000 claims 1
- 206010057644 Testis cancer Diseases 0.000 claims 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims 1
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims 1
- 201000010881 cervical cancer Diseases 0.000 claims 1
- 201000004101 esophageal cancer Diseases 0.000 claims 1
- 230000002496 gastric effect Effects 0.000 claims 1
- 201000010982 kidney cancer Diseases 0.000 claims 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims 1
- 201000002528 pancreatic cancer Diseases 0.000 claims 1
- 208000008443 pancreatic carcinoma Diseases 0.000 claims 1
- 201000000849 skin cancer Diseases 0.000 claims 1
- 201000003120 testicular cancer Diseases 0.000 claims 1
- 201000003073 testicular leukemia Diseases 0.000 claims 1
- 201000002814 testicular lymphoma Diseases 0.000 claims 1
- 201000005112 urinary bladder cancer Diseases 0.000 claims 1
- 229910018828 PO3H2 Inorganic materials 0.000 abstract 1
- 125000004122 cyclic group Chemical group 0.000 description 141
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 134
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 130
- 239000000203 mixture Substances 0.000 description 124
- 239000000243 solution Substances 0.000 description 106
- 238000006243 chemical reaction Methods 0.000 description 91
- 238000003786 synthesis reaction Methods 0.000 description 89
- 230000015572 biosynthetic process Effects 0.000 description 88
- 238000000034 method Methods 0.000 description 86
- 125000000217 alkyl group Chemical group 0.000 description 84
- 210000004027 cell Anatomy 0.000 description 82
- 239000007787 solid Substances 0.000 description 77
- 108090000765 processed proteins & peptides Proteins 0.000 description 74
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 66
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 63
- 125000005647 linker group Chemical group 0.000 description 59
- 230000014759 maintenance of location Effects 0.000 description 56
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 55
- 125000000623 heterocyclic group Chemical group 0.000 description 53
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 51
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 51
- 239000003814 drug Substances 0.000 description 50
- 229940079593 drug Drugs 0.000 description 49
- 239000000427 antigen Substances 0.000 description 47
- 108091007433 antigens Proteins 0.000 description 47
- 102000036639 antigens Human genes 0.000 description 47
- 239000000047 product Substances 0.000 description 46
- 241000282414 Homo sapiens Species 0.000 description 45
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 45
- 229910052739 hydrogen Inorganic materials 0.000 description 45
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 45
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 45
- 235000001014 amino acid Nutrition 0.000 description 44
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 43
- 102000004196 processed proteins & peptides Human genes 0.000 description 42
- 125000001424 substituent group Chemical group 0.000 description 42
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 40
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 39
- 229910052757 nitrogen Inorganic materials 0.000 description 39
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 38
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 38
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 37
- 230000027455 binding Effects 0.000 description 37
- 229920001184 polypeptide Polymers 0.000 description 37
- 238000002360 preparation method Methods 0.000 description 37
- 125000001475 halogen functional group Chemical group 0.000 description 36
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 36
- 229940024606 amino acid Drugs 0.000 description 34
- 150000001413 amino acids Chemical class 0.000 description 34
- 239000012071 phase Substances 0.000 description 33
- 239000002904 solvent Substances 0.000 description 33
- 229910052760 oxygen Inorganic materials 0.000 description 32
- 239000000562 conjugate Substances 0.000 description 30
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 30
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 29
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 28
- 238000003756 stirring Methods 0.000 description 28
- 229910052796 boron Inorganic materials 0.000 description 26
- 230000021615 conjugation Effects 0.000 description 25
- 239000011541 reaction mixture Substances 0.000 description 25
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical group CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 24
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 24
- 125000004432 carbon atom Chemical group C* 0.000 description 23
- 238000004587 chromatography analysis Methods 0.000 description 23
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 23
- 125000005842 heteroatom Chemical group 0.000 description 23
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 22
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 22
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 22
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 21
- 125000004404 heteroalkyl group Chemical group 0.000 description 21
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 21
- 238000005160 1H NMR spectroscopy Methods 0.000 description 20
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 20
- 125000003275 alpha amino acid group Chemical group 0.000 description 20
- 125000001072 heteroaryl group Chemical group 0.000 description 20
- 239000000463 material Substances 0.000 description 20
- 230000000269 nucleophilic effect Effects 0.000 description 20
- 238000000746 purification Methods 0.000 description 19
- 0 C[C@](C(*=CI=*1)C1=C)C(C(C)=O)=C* Chemical compound C[C@](C(*=CI=*1)C1=C)C(C(C)=O)=C* 0.000 description 18
- 108020004414 DNA Proteins 0.000 description 18
- 238000007792 addition Methods 0.000 description 18
- 125000003118 aryl group Chemical group 0.000 description 18
- 235000019439 ethyl acetate Nutrition 0.000 description 18
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 18
- 239000002609 medium Substances 0.000 description 18
- 108090000623 proteins and genes Proteins 0.000 description 18
- 239000000741 silica gel Substances 0.000 description 18
- 229910002027 silica gel Inorganic materials 0.000 description 18
- 210000004881 tumor cell Anatomy 0.000 description 18
- 125000002947 alkylene group Chemical group 0.000 description 17
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 17
- 229910052717 sulfur Inorganic materials 0.000 description 17
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 16
- 125000005843 halogen group Chemical group 0.000 description 16
- 239000000651 prodrug Substances 0.000 description 16
- 229940002612 prodrug Drugs 0.000 description 16
- 235000018102 proteins Nutrition 0.000 description 16
- 102000004169 proteins and genes Human genes 0.000 description 16
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 15
- 239000002253 acid Substances 0.000 description 15
- 229910052799 carbon Inorganic materials 0.000 description 15
- 239000012043 crude product Substances 0.000 description 15
- 239000000539 dimer Substances 0.000 description 15
- 238000004007 reversed phase HPLC Methods 0.000 description 15
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 14
- 239000002246 antineoplastic agent Substances 0.000 description 14
- 208000035475 disorder Diseases 0.000 description 14
- 239000002953 phosphate buffered saline Substances 0.000 description 14
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 13
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 13
- 102000035195 Peptidases Human genes 0.000 description 13
- 108091005804 Peptidases Proteins 0.000 description 13
- 239000004365 Protease Substances 0.000 description 13
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 13
- 125000003710 aryl alkyl group Chemical group 0.000 description 13
- 239000000872 buffer Substances 0.000 description 13
- 239000012634 fragment Substances 0.000 description 13
- 238000004128 high performance liquid chromatography Methods 0.000 description 13
- 238000006467 substitution reaction Methods 0.000 description 13
- 238000012360 testing method Methods 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 12
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 12
- 150000001720 carbohydrates Chemical class 0.000 description 12
- 235000014633 carbohydrates Nutrition 0.000 description 12
- 238000002347 injection Methods 0.000 description 12
- 239000007924 injection Substances 0.000 description 12
- 241000894007 species Species 0.000 description 12
- 239000000126 substance Substances 0.000 description 12
- FONKWHRXTPJODV-DNQXCXABSA-N 1,3-bis[2-[(8s)-8-(chloromethyl)-4-hydroxy-1-methyl-7,8-dihydro-3h-pyrrolo[3,2-e]indole-6-carbonyl]-1h-indol-5-yl]urea Chemical compound C1([C@H](CCl)CN2C(=O)C=3NC4=CC=C(C=C4C=3)NC(=O)NC=3C=C4C=C(NC4=CC=3)C(=O)N3C4=CC(O)=C5NC=C(C5=C4[C@H](CCl)C3)C)=C2C=C(O)C2=C1C(C)=CN2 FONKWHRXTPJODV-DNQXCXABSA-N 0.000 description 11
- 102000005600 Cathepsins Human genes 0.000 description 11
- 108010084457 Cathepsins Proteins 0.000 description 11
- 108060003951 Immunoglobulin Proteins 0.000 description 11
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 11
- 235000018417 cysteine Nutrition 0.000 description 11
- 230000001472 cytotoxic effect Effects 0.000 description 11
- 239000012039 electrophile Substances 0.000 description 11
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 11
- 102000018358 immunoglobulin Human genes 0.000 description 11
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 11
- 125000003396 thiol group Chemical group [H]S* 0.000 description 11
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 10
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 10
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 10
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 10
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 10
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 10
- 229910019142 PO4 Inorganic materials 0.000 description 10
- 229960002433 cysteine Drugs 0.000 description 10
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 10
- 229960002743 glutamine Drugs 0.000 description 10
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 10
- 235000021317 phosphate Nutrition 0.000 description 10
- 230000002829 reductive effect Effects 0.000 description 10
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 10
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 9
- 241001465754 Metazoa Species 0.000 description 9
- 229950006844 bizelesin Drugs 0.000 description 9
- 230000037396 body weight Effects 0.000 description 9
- GTCAXTIRRLKXRU-UHFFFAOYSA-N carbamic acid methyl ester Natural products COC(N)=O GTCAXTIRRLKXRU-UHFFFAOYSA-N 0.000 description 9
- 239000013058 crude material Substances 0.000 description 9
- 231100000433 cytotoxic Toxicity 0.000 description 9
- 229910052805 deuterium Inorganic materials 0.000 description 9
- 201000010099 disease Diseases 0.000 description 9
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 9
- 125000000717 hydrazino group Chemical group [H]N([*])N([H])[H] 0.000 description 9
- 239000007788 liquid Substances 0.000 description 9
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 9
- 239000010452 phosphate Substances 0.000 description 9
- 238000001542 size-exclusion chromatography Methods 0.000 description 9
- 239000011780 sodium chloride Substances 0.000 description 9
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 9
- 239000003981 vehicle Substances 0.000 description 9
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 8
- 102000002274 Matrix Metalloproteinases Human genes 0.000 description 8
- 108010000684 Matrix Metalloproteinases Proteins 0.000 description 8
- 108060008539 Transglutaminase Proteins 0.000 description 8
- 125000004429 atom Chemical group 0.000 description 8
- 230000004071 biological effect Effects 0.000 description 8
- 238000001514 detection method Methods 0.000 description 8
- 229960005501 duocarmycin Drugs 0.000 description 8
- 229930184221 duocarmycin Natural products 0.000 description 8
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 8
- 125000004474 heteroalkylene group Chemical group 0.000 description 8
- 230000004048 modification Effects 0.000 description 8
- 238000012986 modification Methods 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- 239000000843 powder Substances 0.000 description 8
- 125000006239 protecting group Chemical group 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- 102000003601 transglutaminase Human genes 0.000 description 8
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 7
- 125000003277 amino group Chemical group 0.000 description 7
- 239000001257 hydrogen Substances 0.000 description 7
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 7
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 7
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 7
- 229940127121 immunoconjugate Drugs 0.000 description 7
- 230000003834 intracellular effect Effects 0.000 description 7
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 7
- 230000003389 potentiating effect Effects 0.000 description 7
- 238000001959 radiotherapy Methods 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- ILJSQTXMGCGYMG-UHFFFAOYSA-N triacetic acid Chemical compound CC(=O)CC(=O)CC(O)=O ILJSQTXMGCGYMG-UHFFFAOYSA-N 0.000 description 7
- 239000003643 water by type Substances 0.000 description 7
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 6
- DJQYYYCQOZMCRC-UHFFFAOYSA-N 2-aminopropane-1,3-dithiol Chemical compound SCC(N)CS DJQYYYCQOZMCRC-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 6
- 241001529936 Murinae Species 0.000 description 6
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 6
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 6
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 6
- VZTDIZULWFCMLS-UHFFFAOYSA-N ammonium formate Chemical compound [NH4+].[O-]C=O VZTDIZULWFCMLS-UHFFFAOYSA-N 0.000 description 6
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 238000004132 cross linking Methods 0.000 description 6
- XLJMAIOERFSOGZ-UHFFFAOYSA-M cyanate Chemical compound [O-]C#N XLJMAIOERFSOGZ-UHFFFAOYSA-M 0.000 description 6
- 239000006260 foam Substances 0.000 description 6
- 229910052736 halogen Inorganic materials 0.000 description 6
- 238000004191 hydrophobic interaction chromatography Methods 0.000 description 6
- NONOKGVFTBWRLD-UHFFFAOYSA-N isocyanatosulfanylimino(oxo)methane Chemical compound O=C=NSN=C=O NONOKGVFTBWRLD-UHFFFAOYSA-N 0.000 description 6
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 6
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 229910052763 palladium Inorganic materials 0.000 description 6
- 239000000377 silicon dioxide Substances 0.000 description 6
- 229910052938 sodium sulfate Inorganic materials 0.000 description 6
- 235000011152 sodium sulphate Nutrition 0.000 description 6
- NXLNNXIXOYSCMB-UHFFFAOYSA-N (4-nitrophenyl) carbonochloridate Chemical compound [O-][N+](=O)C1=CC=C(OC(Cl)=O)C=C1 NXLNNXIXOYSCMB-UHFFFAOYSA-N 0.000 description 5
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 5
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 5
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 5
- 108091007491 NSP3 Papain-like protease domains Proteins 0.000 description 5
- 229920005654 Sephadex Polymers 0.000 description 5
- 239000012507 Sephadex™ Substances 0.000 description 5
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 5
- 239000012346 acetyl chloride Substances 0.000 description 5
- 150000001299 aldehydes Chemical class 0.000 description 5
- 125000003545 alkoxy group Chemical group 0.000 description 5
- 239000002168 alkylating agent Substances 0.000 description 5
- 230000029936 alkylation Effects 0.000 description 5
- 238000005804 alkylation reaction Methods 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 5
- 231100000263 cytotoxicity test Toxicity 0.000 description 5
- 238000013461 design Methods 0.000 description 5
- 239000003937 drug carrier Substances 0.000 description 5
- 238000005516 engineering process Methods 0.000 description 5
- 125000005549 heteroarylene group Chemical group 0.000 description 5
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 5
- 238000001802 infusion Methods 0.000 description 5
- 239000012948 isocyanate Substances 0.000 description 5
- 150000002513 isocyanates Chemical class 0.000 description 5
- 239000011159 matrix material Substances 0.000 description 5
- 239000002207 metabolite Substances 0.000 description 5
- 230000035772 mutation Effects 0.000 description 5
- YUOCYTRGANSSRY-UHFFFAOYSA-N pyrrolo[2,3-i][1,2]benzodiazepine Chemical compound C1=CN=NC2=C3C=CN=C3C=CC2=C1 YUOCYTRGANSSRY-UHFFFAOYSA-N 0.000 description 5
- 125000002653 sulfanylmethyl group Chemical group [H]SC([H])([H])[*] 0.000 description 5
- 208000024891 symptom Diseases 0.000 description 5
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical class CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 5
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 4
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 4
- ZZOKVYOCRSMTSS-UHFFFAOYSA-N 9h-fluoren-9-ylmethyl carbamate Chemical compound C1=CC=C2C(COC(=O)N)C3=CC=CC=C3C2=C1 ZZOKVYOCRSMTSS-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 208000023275 Autoimmune disease Diseases 0.000 description 4
- 239000004971 Cross linker Substances 0.000 description 4
- 230000007118 DNA alkylation Effects 0.000 description 4
- 230000004568 DNA-binding Effects 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 4
- YGRDBLHSNXKCES-VIFPVBQESA-N [(8R)-8-(chloromethyl)-2-methyl-7,8-dihydro-6H-thieno[2,3-e]indol-4-yl] acetate Chemical compound C(C)(=O)OC1=C2C(=C3[C@H](CNC3=C1)CCl)SC(=C2)C YGRDBLHSNXKCES-VIFPVBQESA-N 0.000 description 4
- 125000000539 amino acid group Chemical group 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- 150000001721 carbon Chemical class 0.000 description 4
- 238000003776 cleavage reaction Methods 0.000 description 4
- 229940125782 compound 2 Drugs 0.000 description 4
- 238000013270 controlled release Methods 0.000 description 4
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 4
- 238000002784 cytotoxicity assay Methods 0.000 description 4
- HPYNZHMRTTWQTB-UHFFFAOYSA-N dimethylpyridine Natural products CC1=CC=CN=C1C HPYNZHMRTTWQTB-UHFFFAOYSA-N 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 238000003818 flash chromatography Methods 0.000 description 4
- 238000013467 fragmentation Methods 0.000 description 4
- 238000006062 fragmentation reaction Methods 0.000 description 4
- 238000004108 freeze drying Methods 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 239000011521 glass Substances 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 208000032839 leukemia Diseases 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- 238000004949 mass spectrometry Methods 0.000 description 4
- 239000000178 monomer Substances 0.000 description 4
- 239000001301 oxygen Substances 0.000 description 4
- 230000001575 pathological effect Effects 0.000 description 4
- 229910052698 phosphorus Inorganic materials 0.000 description 4
- 230000009257 reactivity Effects 0.000 description 4
- 229920006395 saturated elastomer Polymers 0.000 description 4
- 230000007017 scission Effects 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 4
- 239000011550 stock solution Substances 0.000 description 4
- 238000001356 surgical procedure Methods 0.000 description 4
- 239000013077 target material Substances 0.000 description 4
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 4
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 4
- 229910052721 tungsten Inorganic materials 0.000 description 4
- ZJLNQOIFTYLCHC-VXKWHMMOSA-N (2s)-5-(carbamoylamino)-2-[[(2s)-2-(9h-fluoren-9-ylmethoxycarbonylamino)-3-methylbutanoyl]amino]pentanoic acid Chemical compound C1=CC=C2C(COC(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=O)C(O)=O)C3=CC=CC=C3C2=C1 ZJLNQOIFTYLCHC-VXKWHMMOSA-N 0.000 description 3
- LOVPHSMOAVXQIH-UHFFFAOYSA-N (4-nitrophenyl) hydrogen carbonate Chemical compound OC(=O)OC1=CC=C([N+]([O-])=O)C=C1 LOVPHSMOAVXQIH-UHFFFAOYSA-N 0.000 description 3
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 3
- 125000001140 1,4-phenylene group Chemical group [H]C1=C([H])C([*:2])=C([H])C([H])=C1[*:1] 0.000 description 3
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 3
- NEFYNJFWPKOSAZ-UHFFFAOYSA-N 2-[carboxy(methyl)amino]ethyl-methylcarbamic acid Chemical compound OC(=O)N(C)CCN(C)C(O)=O NEFYNJFWPKOSAZ-UHFFFAOYSA-N 0.000 description 3
- ALYNCZNDIQEVRV-UHFFFAOYSA-N 4-aminobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C=C1 ALYNCZNDIQEVRV-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 208000035473 Communicable disease Diseases 0.000 description 3
- 230000000970 DNA cross-linking effect Effects 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Divinylene sulfide Natural products C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 3
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 102000005741 Metalloproteases Human genes 0.000 description 3
- 108010006035 Metalloproteases Proteins 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- 241000699660 Mus musculus Species 0.000 description 3
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 3
- 229910004013 NO 2 Inorganic materials 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 230000021736 acetylation Effects 0.000 description 3
- 238000006640 acetylation reaction Methods 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 229940100198 alkylating agent Drugs 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 229940125644 antibody drug Drugs 0.000 description 3
- 230000006907 apoptotic process Effects 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 125000002619 bicyclic group Chemical group 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 3
- 125000004452 carbocyclyl group Chemical group 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- 230000003833 cell viability Effects 0.000 description 3
- 238000002512 chemotherapy Methods 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000001085 cytostatic effect Effects 0.000 description 3
- 230000003013 cytotoxicity Effects 0.000 description 3
- 231100000135 cytotoxicity Toxicity 0.000 description 3
- 239000002619 cytotoxin Substances 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- FONOSWYYBCBQGN-UHFFFAOYSA-N ethylene dione Chemical group O=C=C=O FONOSWYYBCBQGN-UHFFFAOYSA-N 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 238000002474 experimental method Methods 0.000 description 3
- 239000012065 filter cake Substances 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 150000002430 hydrocarbons Chemical class 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 230000002401 inhibitory effect Effects 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 238000011068 loading method Methods 0.000 description 3
- 235000018977 lysine Nutrition 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- JCAZSWWHFJVFPP-UHFFFAOYSA-N methyl 5-chloro-5-oxopentanoate Chemical compound COC(=O)CCCC(Cl)=O JCAZSWWHFJVFPP-UHFFFAOYSA-N 0.000 description 3
- 230000000813 microbial effect Effects 0.000 description 3
- 239000002808 molecular sieve Substances 0.000 description 3
- OWIUPIRUAQMTTK-UHFFFAOYSA-M n-aminocarbamate Chemical compound NNC([O-])=O OWIUPIRUAQMTTK-UHFFFAOYSA-M 0.000 description 3
- 229930014626 natural product Natural products 0.000 description 3
- 239000002773 nucleotide Substances 0.000 description 3
- 125000003729 nucleotide group Chemical group 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 239000003960 organic solvent Substances 0.000 description 3
- 150000002923 oximes Chemical class 0.000 description 3
- YVOFTMXWTWHRBH-UHFFFAOYSA-N pentanedioyl dichloride Chemical compound ClC(=O)CCCC(Cl)=O YVOFTMXWTWHRBH-UHFFFAOYSA-N 0.000 description 3
- 210000002307 prostate Anatomy 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- UOWVMDUEMSNCAV-WYENRQIDSA-N rachelmycin Chemical class C1([C@]23C[C@@H]2CN1C(=O)C=1NC=2C(OC)=C(O)C4=C(C=2C=1)CCN4C(=O)C1=CC=2C=4CCN(C=4C(O)=C(C=2N1)OC)C(N)=O)=CC(=O)C1=C3C(C)=CN1 UOWVMDUEMSNCAV-WYENRQIDSA-N 0.000 description 3
- 102000005962 receptors Human genes 0.000 description 3
- 108020003175 receptors Proteins 0.000 description 3
- 230000010076 replication Effects 0.000 description 3
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 3
- 239000011593 sulfur Substances 0.000 description 3
- 125000004434 sulfur atom Chemical group 0.000 description 3
- 230000009885 systemic effect Effects 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- SRVJKTDHMYAMHA-WUXMJOGZSA-N thioacetazone Chemical compound CC(=O)NC1=CC=C(\C=N\NC(N)=S)C=C1 SRVJKTDHMYAMHA-WUXMJOGZSA-N 0.000 description 3
- 238000011830 transgenic mouse model Methods 0.000 description 3
- PYHOFAHZHOBVGV-UHFFFAOYSA-N triazane Chemical compound NNN PYHOFAHZHOBVGV-UHFFFAOYSA-N 0.000 description 3
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- CIQYBTAUACRWKP-UHFFFAOYSA-N (2,3,4,5,6-pentafluorophenyl) 6-(2,5-dioxopyrrol-1-yl)hexanoate Chemical compound FC1=C(F)C(F)=C(F)C(F)=C1OC(=O)CCCCCN1C(=O)C=CC1=O CIQYBTAUACRWKP-UHFFFAOYSA-N 0.000 description 2
- TZCPCKNHXULUIY-RGULYWFUSA-N 1,2-distearoyl-sn-glycero-3-phosphoserine Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@H](COP(O)(=O)OC[C@H](N)C(O)=O)OC(=O)CCCCCCCCCCCCCCCCC TZCPCKNHXULUIY-RGULYWFUSA-N 0.000 description 2
- GHSXIZYVEWSKER-UHFFFAOYSA-N 1-(chloromethyl)-2,3-dihydro-1h-benzo[e]indol-5-ol Chemical compound C12=CC=CC=C2C(O)=CC2=C1C(CCl)CN2 GHSXIZYVEWSKER-UHFFFAOYSA-N 0.000 description 2
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 description 2
- KWQMJXZUGBNSOY-UHFFFAOYSA-N 1h-thieno[2,3-g]indole Chemical compound C1=C2SC=CC2=C2NC=CC2=C1 KWQMJXZUGBNSOY-UHFFFAOYSA-N 0.000 description 2
- HUHXLHLWASNVDB-UHFFFAOYSA-N 2-(oxan-2-yloxy)oxane Chemical class O1CCCCC1OC1OCCCC1 HUHXLHLWASNVDB-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- CDAWCLOXVUBKRW-UHFFFAOYSA-N 2-aminophenol Chemical compound NC1=CC=CC=C1O CDAWCLOXVUBKRW-UHFFFAOYSA-N 0.000 description 2
- ANDCDPBTSFSYLB-UHFFFAOYSA-N 2-cyclopropylpyrrolo[2,3-e]indole Chemical class C1CC1C(C=C1C=C2)=NC1=C1C2=NC=C1 ANDCDPBTSFSYLB-UHFFFAOYSA-N 0.000 description 2
- XASOHFCUIQARJT-UHFFFAOYSA-N 8-methoxy-6-[7-(2-morpholin-4-ylethoxy)imidazo[1,2-a]pyridin-3-yl]-2-(2,2,2-trifluoroethyl)-3,4-dihydroisoquinolin-1-one Chemical compound C(N1C(=O)C2=C(OC)C=C(C=3N4C(=NC=3)C=C(C=C4)OCCN3CCOCC3)C=C2CC1)C(F)(F)F XASOHFCUIQARJT-UHFFFAOYSA-N 0.000 description 2
- 102000052030 Aldehyde Dehydrogenase 1 Family Human genes 0.000 description 2
- 101710196131 Aldehyde dehydrogenase 1 Proteins 0.000 description 2
- 108010083359 Antigen Receptors Proteins 0.000 description 2
- 102000006306 Antigen Receptors Human genes 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- 101100240517 Caenorhabditis elegans nhr-11 gene Proteins 0.000 description 2
- 239000004215 Carbon black (E152) Substances 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- 208000001333 Colorectal Neoplasms Diseases 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 2
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 2
- 102000001390 Fructose-Bisphosphate Aldolase Human genes 0.000 description 2
- 108010068561 Fructose-Bisphosphate Aldolase Proteins 0.000 description 2
- ZWZWYGMENQVNFU-UHFFFAOYSA-N Glycerophosphorylserin Natural products OC(=O)C(N)COP(O)(=O)OCC(O)CO ZWZWYGMENQVNFU-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 229910004373 HOAc Inorganic materials 0.000 description 2
- 208000017604 Hodgkin disease Diseases 0.000 description 2
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- 229930182816 L-glutamine Natural products 0.000 description 2
- 108090001090 Lectins Proteins 0.000 description 2
- 102000004856 Lectins Human genes 0.000 description 2
- 206010025323 Lymphomas Diseases 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 206010027476 Metastases Diseases 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical class CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- 101001043818 Mus musculus Interleukin-31 receptor subunit alpha Proteins 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical class C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- 102000007079 Peptide Fragments Human genes 0.000 description 2
- 108010033276 Peptide Fragments Proteins 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 2
- 102000004338 Transferrin Human genes 0.000 description 2
- 108090000901 Transferrin Proteins 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 101800003344 Vaccinia growth factor Proteins 0.000 description 2
- ASTSAPGHWBBDFY-NHCUHLMSSA-N [(8S)-6-[5-[(8S)-4-acetyloxy-8-(chloromethyl)-2-methyl-7,8-dihydrothieno[2,3-e]indol-6-yl]-5-oxopentanoyl]-8-(chloromethyl)-2-methyl-7,8-dihydrothieno[2,3-e]indol-4-yl] acetate Chemical compound C(C)(=O)OC1=C2C(=C3[C@@H](CN(C3=C1)C(CCCC(=O)N1C[C@H](C3=C4C(=C(C=C13)OC(C)=O)C=C(S4)C)CCl)=O)CCl)SC(=C2)C ASTSAPGHWBBDFY-NHCUHLMSSA-N 0.000 description 2
- 150000001242 acetic acid derivatives Chemical class 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 239000003463 adsorbent Substances 0.000 description 2
- 239000003708 ampul Substances 0.000 description 2
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical class C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 2
- 230000000890 antigenic effect Effects 0.000 description 2
- 229940041181 antineoplastic drug Drugs 0.000 description 2
- 125000000732 arylene group Chemical group 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 230000001363 autoimmune Effects 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical class OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 2
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 2
- 238000012925 biological evaluation Methods 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000002837 carbocyclic group Chemical group 0.000 description 2
- 125000000837 carbohydrate group Chemical group 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 238000007385 chemical modification Methods 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 238000010367 cloning Methods 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 239000003431 cross linking reagent Substances 0.000 description 2
- 238000009295 crossflow filtration Methods 0.000 description 2
- 239000000824 cytostatic agent Substances 0.000 description 2
- 238000012217 deletion Methods 0.000 description 2
- 230000037430 deletion Effects 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- 238000011026 diafiltration Methods 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 238000000502 dialysis Methods 0.000 description 2
- HDFFVHSMHLDSLO-UHFFFAOYSA-M dibenzyl phosphate Chemical compound C=1C=CC=CC=1COP(=O)([O-])OCC1=CC=CC=C1 HDFFVHSMHLDSLO-UHFFFAOYSA-M 0.000 description 2
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical class C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 238000009510 drug design Methods 0.000 description 2
- 239000003640 drug residue Substances 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 238000001962 electrophoresis Methods 0.000 description 2
- BJXYHBKEQFQVES-NWDGAFQWSA-N enpatoran Chemical compound N[C@H]1CN(C[C@H](C1)C(F)(F)F)C1=C2C=CC=NC2=C(C=C1)C#N BJXYHBKEQFQVES-NWDGAFQWSA-N 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 125000002541 furyl group Chemical group 0.000 description 2
- 125000000404 glutamine group Chemical group N[C@@H](CCC(N)=O)C(=O)* 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 230000013595 glycosylation Effects 0.000 description 2
- 238000006206 glycosylation reaction Methods 0.000 description 2
- 150000002367 halogens Chemical class 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 125000004475 heteroaralkyl group Chemical group 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 238000004896 high resolution mass spectrometry Methods 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 125000002883 imidazolyl group Chemical group 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 238000011065 in-situ storage Methods 0.000 description 2
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 2
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 2
- 125000001041 indolyl group Chemical group 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- 238000000322 laser mass spectrometry Methods 0.000 description 2
- 239000002523 lectin Substances 0.000 description 2
- 239000007791 liquid phase Substances 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 238000007477 logistic regression Methods 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- 229960003646 lysine Drugs 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 230000009401 metastasis Effects 0.000 description 2
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 108020004707 nucleic acids Proteins 0.000 description 2
- 102000039446 nucleic acids Human genes 0.000 description 2
- 150000007523 nucleic acids Chemical class 0.000 description 2
- 239000012038 nucleophile Substances 0.000 description 2
- 150000002894 organic compounds Chemical class 0.000 description 2
- RQKYHDHLEMEVDR-UHFFFAOYSA-N oxo-bis(phenylmethoxy)phosphanium Chemical compound C=1C=CC=CC=1CO[P+](=O)OCC1=CC=CC=C1 RQKYHDHLEMEVDR-UHFFFAOYSA-N 0.000 description 2
- 210000000496 pancreas Anatomy 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 229960005190 phenylalanine Drugs 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 239000002504 physiological saline solution Substances 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 238000000159 protein binding assay Methods 0.000 description 2
- 230000006337 proteolytic cleavage Effects 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- 125000003226 pyrazolyl group Chemical group 0.000 description 2
- 125000002098 pyridazinyl group Chemical group 0.000 description 2
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 2
- 239000002994 raw material Substances 0.000 description 2
- 238000003259 recombinant expression Methods 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 125000006413 ring segment Chemical group 0.000 description 2
- 229930195734 saturated hydrocarbon Natural products 0.000 description 2
- 238000012163 sequencing technique Methods 0.000 description 2
- 229940054269 sodium pyruvate Drugs 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 210000000130 stem cell Anatomy 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 150000003457 sulfones Chemical class 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 125000003831 tetrazolyl group Chemical group 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- ATGUDZODTABURZ-UHFFFAOYSA-N thiolan-2-ylideneazanium;chloride Chemical compound Cl.N=C1CCCS1 ATGUDZODTABURZ-UHFFFAOYSA-N 0.000 description 2
- 229930192474 thiophene Natural products 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 239000012581 transferrin Substances 0.000 description 2
- 230000005945 translocation Effects 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- 125000001425 triazolyl group Chemical group 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 229940124543 ultraviolet light absorber Drugs 0.000 description 2
- 230000035899 viability Effects 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- FLCQLSRLQIPNLM-UHFFFAOYSA-N (2,5-dioxopyrrolidin-1-yl) 2-acetylsulfanylacetate Chemical compound CC(=O)SCC(=O)ON1C(=O)CCC1=O FLCQLSRLQIPNLM-UHFFFAOYSA-N 0.000 description 1
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 1
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 1
- VEGGTWZUZGZKHY-GJZGRUSLSA-N (2s)-2-[[(2s)-2-amino-3-methylbutanoyl]amino]-5-(carbamoylamino)-n-[4-(hydroxymethyl)phenyl]pentanamide Chemical compound NC(=O)NCCC[C@H](NC(=O)[C@@H](N)C(C)C)C(=O)NC1=CC=C(CO)C=C1 VEGGTWZUZGZKHY-GJZGRUSLSA-N 0.000 description 1
- LOVPHSMOAVXQIH-UHFFFAOYSA-M (4-nitrophenyl) carbonate Chemical compound [O-]C(=O)OC1=CC=C([N+]([O-])=O)C=C1 LOVPHSMOAVXQIH-UHFFFAOYSA-M 0.000 description 1
- STPKWKPURVSAJF-LJEWAXOPSA-N (4r,5r)-5-[4-[[4-(1-aza-4-azoniabicyclo[2.2.2]octan-4-ylmethyl)phenyl]methoxy]phenyl]-3,3-dibutyl-7-(dimethylamino)-1,1-dioxo-4,5-dihydro-2h-1$l^{6}-benzothiepin-4-ol Chemical compound O[C@H]1C(CCCC)(CCCC)CS(=O)(=O)C2=CC=C(N(C)C)C=C2[C@H]1C(C=C1)=CC=C1OCC(C=C1)=CC=C1C[N+]1(CC2)CCN2CC1 STPKWKPURVSAJF-LJEWAXOPSA-N 0.000 description 1
- 125000006832 (C1-C10) alkylene group Chemical group 0.000 description 1
- 125000003837 (C1-C20) alkyl group Chemical group 0.000 description 1
- 125000006735 (C1-C20) heteroalkyl group Chemical group 0.000 description 1
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- 125000006694 (C2-C10) heterocyclyl group Chemical group 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- VYEWZWBILJHHCU-OMQUDAQFSA-N (e)-n-[(2s,3r,4r,5r,6r)-2-[(2r,3r,4s,5s,6s)-3-acetamido-5-amino-4-hydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-[2-[(2r,3s,4r,5r)-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]-2-hydroxyethyl]-4,5-dihydroxyoxan-3-yl]-5-methylhex-2-enamide Chemical compound N1([C@@H]2O[C@@H]([C@H]([C@H]2O)O)C(O)C[C@@H]2[C@H](O)[C@H](O)[C@H]([C@@H](O2)O[C@@H]2[C@@H]([C@@H](O)[C@H](N)[C@@H](CO)O2)NC(C)=O)NC(=O)/C=C/CC(C)C)C=CC(=O)NC1=O VYEWZWBILJHHCU-OMQUDAQFSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical group CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- QIDUHGHFWAMMPV-UHFFFAOYSA-N 1,1-diphenylethylbenzene Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(C)C1=CC=CC=C1 QIDUHGHFWAMMPV-UHFFFAOYSA-N 0.000 description 1
- 125000004607 1,2,3,4-tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- RHKRZQHJRZVTBP-UHFFFAOYSA-N 1-(chloromethyl)-2,3-dihydro-1h-benzo[e]indole Chemical class C1=CC=CC2=C3C(CCl)CNC3=CC=C21 RHKRZQHJRZVTBP-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical group O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 1
- GPAAEZIXSQCCES-UHFFFAOYSA-N 1-methoxy-2-(2-methoxyethoxymethoxymethoxy)ethane Chemical compound COCCOCOCOCCOC GPAAEZIXSQCCES-UHFFFAOYSA-N 0.000 description 1
- SDTORDSXCYSNTD-UHFFFAOYSA-N 1-methoxy-4-[(4-methoxyphenyl)methoxymethyl]benzene Chemical compound C1=CC(OC)=CC=C1COCC1=CC=C(OC)C=C1 SDTORDSXCYSNTD-UHFFFAOYSA-N 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N 1-nonene Chemical group CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 125000006023 1-pentenyl group Chemical group 0.000 description 1
- 125000006069 2,3-dimethyl-2-butenyl group Chemical group 0.000 description 1
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 1
- QEBYEVQKHRUYPE-UHFFFAOYSA-N 2-(2-chlorophenyl)-5-[(1-methylpyrazol-3-yl)methyl]-4-[[methyl(pyridin-3-ylmethyl)amino]methyl]-1h-pyrazolo[4,3-c]pyridine-3,6-dione Chemical compound C1=CN(C)N=C1CN1C(=O)C=C2NN(C=3C(=CC=CC=3)Cl)C(=O)C2=C1CN(C)CC1=CC=CN=C1 QEBYEVQKHRUYPE-UHFFFAOYSA-N 0.000 description 1
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 1
- NOIRDLRUNWIUMX-UHFFFAOYSA-N 2-amino-3,7-dihydropurin-6-one;6-amino-1h-pyrimidin-2-one Chemical compound NC=1C=CNC(=O)N=1.O=C1NC(N)=NC2=C1NC=N2 NOIRDLRUNWIUMX-UHFFFAOYSA-N 0.000 description 1
- KQLXBKWUVBMXEM-UHFFFAOYSA-N 2-amino-3,7-dihydropurin-6-one;7h-purin-6-amine Chemical compound NC1=NC=NC2=C1NC=N2.O=C1NC(N)=NC2=C1NC=N2 KQLXBKWUVBMXEM-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- 125000006029 2-methyl-2-butenyl group Chemical group 0.000 description 1
- 125000006024 2-pentenyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- HQXYNMLKJGNCSV-PHXRZEHOSA-N 3-[(8S)-4-bis(phenylmethoxy)phosphoryloxy-8-(chloromethyl)-1-methyl-7,8-dihydrothieno[3,2-e]indole-6-carbonyl]bicyclo[1.1.1]pentane-1-carboxylic acid Chemical compound C(C1=CC=CC=C1)OP(=O)(OCC1=CC=CC=C1)OC1=C2C(=C3[C@@H](CN(C3=C1)C(=O)C13CC(C1)(C3)C(=O)O)CCl)C(=CS2)C HQXYNMLKJGNCSV-PHXRZEHOSA-N 0.000 description 1
- PBVAJRFEEOIAGW-UHFFFAOYSA-N 3-[bis(2-carboxyethyl)phosphanyl]propanoic acid;hydrochloride Chemical compound Cl.OC(=O)CCP(CCC(O)=O)CCC(O)=O PBVAJRFEEOIAGW-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- UJZHYIMESNWEQA-UHFFFAOYSA-N 3-methoxycarbonylbicyclo[1.1.1]pentane-1-carboxylic acid Chemical compound C1C2(C(O)=O)CC1(C(=O)OC)C2 UJZHYIMESNWEQA-UHFFFAOYSA-N 0.000 description 1
- 125000006027 3-methyl-1-butenyl group Chemical group 0.000 description 1
- QFVHZQCOUORWEI-UHFFFAOYSA-N 4-[(4-anilino-5-sulfonaphthalen-1-yl)diazenyl]-5-hydroxynaphthalene-2,7-disulfonic acid Chemical compound C=12C(O)=CC(S(O)(=O)=O)=CC2=CC(S(O)(=O)=O)=CC=1N=NC(C1=CC=CC(=C11)S(O)(=O)=O)=CC=C1NC1=CC=CC=C1 QFVHZQCOUORWEI-UHFFFAOYSA-N 0.000 description 1
- VZHNAVSRNGLHRD-UHFFFAOYSA-N 5-[(2-methylpropan-2-yl)oxy]-5-oxopentanoic acid Chemical compound CC(C)(C)OC(=O)CCCC(O)=O VZHNAVSRNGLHRD-UHFFFAOYSA-N 0.000 description 1
- QCLYBSPPEGJKJC-UHFFFAOYSA-N 5-thia-10-azatetracyclo[7.4.0.01,12.02,6]trideca-2,8,11-trien-13-one Chemical compound C1C2SCC=C2C23C(=O)C3=CNC2=C1 QCLYBSPPEGJKJC-UHFFFAOYSA-N 0.000 description 1
- BYQPXOBDTLDUHS-UHFFFAOYSA-N 5H-pyrrolo[3,2-e]indol-4-one Chemical compound C1=CN=C2C1=C1C=CN=C1CC2=O BYQPXOBDTLDUHS-UHFFFAOYSA-N 0.000 description 1
- 125000004008 6 membered carbocyclic group Chemical group 0.000 description 1
- GDUANFXPOZTYKS-UHFFFAOYSA-N 6-bromo-8-[(2,6-difluoro-4-methoxybenzoyl)amino]-4-oxochromene-2-carboxylic acid Chemical compound FC1=CC(OC)=CC(F)=C1C(=O)NC1=CC(Br)=CC2=C1OC(C(O)=O)=CC2=O GDUANFXPOZTYKS-UHFFFAOYSA-N 0.000 description 1
- UEZYJDITXKYWHK-UHFFFAOYSA-N 8-(chloromethyl)-1-methyl-3,6,7,8-tetrahydropyrrolo[3,2-e]indol-4-ol Chemical compound C1=C2NCC(CCl)C2=C2C(C)=CNC2=C1O UEZYJDITXKYWHK-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 102100033350 ATP-dependent translocase ABCB1 Human genes 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 102000000412 Annexin Human genes 0.000 description 1
- 108050008874 Annexin Proteins 0.000 description 1
- 108090000672 Annexin A5 Proteins 0.000 description 1
- 102000004121 Annexin A5 Human genes 0.000 description 1
- 108010083590 Apoproteins Proteins 0.000 description 1
- 102000006410 Apoproteins Human genes 0.000 description 1
- 101100096578 Arabidopsis thaliana SQD2 gene Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 108010051479 Bombesin Proteins 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 102100022595 Broad substrate specificity ATP-binding cassette transporter ABCG2 Human genes 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- 125000004418 C2-C8 unsaturated alkyl group Chemical group 0.000 description 1
- AWYXVAVNCXKNTD-UHFFFAOYSA-N CC(C(C1)=CC2=C1C=C[IH]C=C2)=O Chemical compound CC(C(C1)=CC2=C1C=C[IH]C=C2)=O AWYXVAVNCXKNTD-UHFFFAOYSA-N 0.000 description 1
- VPMFSMYHGIUWQV-JLNHVWKISA-N CC(Oc1c(cc(C)[s]2)c2c([C@@H](CCl)CN2C(C(C3)(C4)CC34C(N3c4cc(OC(C)=O)c(cc(C)[s]5)c5c4[C@@H](CCl)C3)=O)=O)c2c1)=O Chemical compound CC(Oc1c(cc(C)[s]2)c2c([C@@H](CCl)CN2C(C(C3)(C4)CC34C(N3c4cc(OC(C)=O)c(cc(C)[s]5)c5c4[C@@H](CCl)C3)=O)=O)c2c1)=O VPMFSMYHGIUWQV-JLNHVWKISA-N 0.000 description 1
- HWNNFUANVUYGDB-UHFFFAOYSA-N CC(c1cc(C=C(C2)N)c2cc1)=O Chemical compound CC(c1cc(C=C(C2)N)c2cc1)=O HWNNFUANVUYGDB-UHFFFAOYSA-N 0.000 description 1
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 201000009030 Carcinoma Diseases 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 238000003734 CellTiter-Glo Luminescent Cell Viability Assay Methods 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- 244000110556 Cyclopia subternata Species 0.000 description 1
- DSLZVSRJTYRBFB-LLEIAEIESA-N D-glucaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O DSLZVSRJTYRBFB-LLEIAEIESA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- OFDNQWIFNXBECV-UHFFFAOYSA-N Dolastatin 10 Natural products CC(C)C(N(C)C)C(=O)NC(C(C)C)C(=O)N(C)C(C(C)CC)C(OC)CC(=O)N1CCCC1C(OC)C(C)C(=O)NC(C=1SC=CN=1)CC1=CC=CC=C1 OFDNQWIFNXBECV-UHFFFAOYSA-N 0.000 description 1
- VQNATVDKACXKTF-UHFFFAOYSA-N Duocarmycin SA Natural products COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C(C64CC6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-UHFFFAOYSA-N 0.000 description 1
- GKQLYSROISKDLL-UHFFFAOYSA-N EEDQ Chemical compound C1=CC=C2N(C(=O)OCC)C(OCC)C=CC2=C1 GKQLYSROISKDLL-UHFFFAOYSA-N 0.000 description 1
- 108010066486 EGF Family of Proteins Proteins 0.000 description 1
- 102000018386 EGF Family of Proteins Human genes 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 102400001368 Epidermal growth factor Human genes 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 102400000921 Gastrin Human genes 0.000 description 1
- 102000004862 Gastrin releasing peptide Human genes 0.000 description 1
- 108090001053 Gastrin releasing peptide Proteins 0.000 description 1
- 102100036519 Gastrin-releasing peptide Human genes 0.000 description 1
- 108010052343 Gastrins Proteins 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical group NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000823298 Homo sapiens Broad substrate specificity ATP-binding cassette transporter ABCG2 Proteins 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- 102100026120 IgG receptor FcRn large subunit p51 Human genes 0.000 description 1
- 101710177940 IgG receptor FcRn large subunit p51 Proteins 0.000 description 1
- 102000009786 Immunoglobulin Constant Regions Human genes 0.000 description 1
- 108010009817 Immunoglobulin Constant Regions Proteins 0.000 description 1
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 1
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 102000004889 Interleukin-6 Human genes 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- 102000007330 LDL Lipoproteins Human genes 0.000 description 1
- 108010007622 LDL Lipoproteins Proteins 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 241000721701 Lynx Species 0.000 description 1
- 231100000070 MTS assay Toxicity 0.000 description 1
- 238000000719 MTS assay Methods 0.000 description 1
- 229930126263 Maytansine Natural products 0.000 description 1
- 108010047230 Member 1 Subfamily B ATP Binding Cassette Transporter Proteins 0.000 description 1
- 102000014842 Multidrug resistance proteins Human genes 0.000 description 1
- 108050005144 Multidrug resistance proteins Proteins 0.000 description 1
- AVYVHIKSFXVDBG-UHFFFAOYSA-N N-benzyl-N-hydroxy-2,2-dimethylbutanamide Chemical compound C(C1=CC=CC=C1)N(C(C(CC)(C)C)=O)O AVYVHIKSFXVDBG-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- UJCYQVOGBMCBSI-UHFFFAOYSA-N O=C(C(C1)(C2)CC12C(Cl)=O)Cl Chemical compound O=C(C(C1)(C2)CC12C(Cl)=O)Cl UJCYQVOGBMCBSI-UHFFFAOYSA-N 0.000 description 1
- WCNARHWVZDVGFB-UHFFFAOYSA-N O=P(OCC1=CC=CC=C1)(OCC1=CC=CC=C1)OC1=C(C=CN2)C2=CC=C1 Chemical compound O=P(OCC1=CC=CC=C1)(OCC1=CC=CC=C1)OC1=C(C=CN2)C2=CC=C1 WCNARHWVZDVGFB-UHFFFAOYSA-N 0.000 description 1
- 101100510696 Oncidium hybrid cultivar LCY-E gene Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 101710160107 Outer membrane protein A Proteins 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 101100272976 Panax ginseng CYP716A53v2 gene Proteins 0.000 description 1
- 241000009328 Perro Species 0.000 description 1
- 108010038512 Platelet-Derived Growth Factor Proteins 0.000 description 1
- 102000010780 Platelet-Derived Growth Factor Human genes 0.000 description 1
- 208000034255 Primary dystonia, DYT2 type Diseases 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 239000012979 RPMI medium Substances 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 229910006069 SO3H Inorganic materials 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 239000004268 Sodium erythorbin Substances 0.000 description 1
- 102000005157 Somatostatin Human genes 0.000 description 1
- 108010056088 Somatostatin Proteins 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 239000012505 Superdex™ Substances 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- 239000003490 Thiodipropionic acid Substances 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 108010009583 Transforming Growth Factors Proteins 0.000 description 1
- 102000009618 Transforming Growth Factors Human genes 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- YJQCOFNZVFGCAF-UHFFFAOYSA-N Tunicamycin II Natural products O1C(CC(O)C2C(C(O)C(O2)N2C(NC(=O)C=C2)=O)O)C(O)C(O)C(NC(=O)C=CCCCCCCCCC(C)C)C1OC1OC(CO)C(O)C(O)C1NC(C)=O YJQCOFNZVFGCAF-UHFFFAOYSA-N 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 1
- WHXNNQCQEWTPOP-PUAPYOPTSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[3-[2-[2-[2-[2-[2-[2-(2,5-dioxopyrrol-1-yl)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl (4-nitrophenyl) carbonate Chemical compound CC(C)[C@H](NC(=O)CCOCCOCCOCCOCCOCCOCCN1C(=O)C=CC1=O)C(=O)N[C@@H](CCCNC(N)=O)C(=O)Nc1ccc(COC(=O)Oc2ccc(cc2)[N+]([O-])=O)cc1 WHXNNQCQEWTPOP-PUAPYOPTSA-N 0.000 description 1
- YGNJVEMJXJNRIE-UHFFFAOYSA-N [6-acetyloxy-8-(chloromethyl)-1-methyl-7,8-dihydrothieno[3,2-e]indol-4-yl] acetate Chemical compound C(C)(=O)ON1CC(C2=C3C(=C(C=C12)OC(C)=O)SC=C3C)CCl YGNJVEMJXJNRIE-UHFFFAOYSA-N 0.000 description 1
- SORGEQQSQGNZFI-UHFFFAOYSA-N [azido(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(N=[N+]=[N-])OC1=CC=CC=C1 SORGEQQSQGNZFI-UHFFFAOYSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- WEVYAHXRMPXWCK-FIBGUPNXSA-N acetonitrile-d3 Chemical compound [2H]C([2H])([2H])C#N WEVYAHXRMPXWCK-FIBGUPNXSA-N 0.000 description 1
- XBJFCYDKBDVADW-UHFFFAOYSA-N acetonitrile;formic acid Chemical compound CC#N.OC=O XBJFCYDKBDVADW-UHFFFAOYSA-N 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- BYRVKDUQDLJUBX-JJCDCTGGSA-N adozelesin Chemical compound C1=CC=C2OC(C(=O)NC=3C=C4C=C(NC4=CC=3)C(=O)N3C[C@H]4C[C@]44C5=C(C(C=C43)=O)NC=C5C)=CC2=C1 BYRVKDUQDLJUBX-JJCDCTGGSA-N 0.000 description 1
- 229950004955 adozelesin Drugs 0.000 description 1
- 208000037844 advanced solid tumor Diseases 0.000 description 1
- 229960003767 alanine Drugs 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 238000006668 aldol addition reaction Methods 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 229960004050 aminobenzoic acid Drugs 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 1
- 235000011130 ammonium sulphate Nutrition 0.000 description 1
- 230000002491 angiogenic effect Effects 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000002494 anti-cea effect Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- UOWVMDUEMSNCAV-UHFFFAOYSA-N antibiotic cc 1065 Chemical compound N1C=2C(OC)=C(O)C=3N(C(N)=O)CCC=3C=2C=C1C(=O)N1CCC(C=2C=3)=C1C(O)=C(OC)C=2NC=3C(=O)N1CC2CC22C1=CC(=O)C1=C2C(C)=CN1 UOWVMDUEMSNCAV-UHFFFAOYSA-N 0.000 description 1
- 238000011230 antibody-based therapy Methods 0.000 description 1
- 230000005888 antibody-dependent cellular phagocytosis Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 238000003782 apoptosis assay Methods 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 230000003140 astrocytic effect Effects 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 239000012752 auxiliary agent Substances 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 235000015278 beef Nutrition 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 150000001555 benzenes Chemical class 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 108010085672 beta-lactamase PSE-2 Proteins 0.000 description 1
- SBLRPOGZAJTJEG-UHFFFAOYSA-N bicyclo[1.1.1]pentane-1,3-dicarboxylic acid Chemical compound C1C2(C(O)=O)CC1(C(=O)O)C2 SBLRPOGZAJTJEG-UHFFFAOYSA-N 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Chemical class 0.000 description 1
- SIPUZPBQZHNSDW-UHFFFAOYSA-N bis(2-methylpropyl)aluminum Chemical compound CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 1
- ACBQROXDOHKANW-UHFFFAOYSA-N bis(4-nitrophenyl) carbonate Chemical compound C1=CC([N+](=O)[O-])=CC=C1OC(=O)OC1=CC=C([N+]([O-])=O)C=C1 ACBQROXDOHKANW-UHFFFAOYSA-N 0.000 description 1
- 210000003443 bladder cell Anatomy 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- DNDCVAGJPBKION-DOPDSADYSA-N bombesin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC=1NC2=CC=CC=C2C=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1NC(=O)CC1)C(C)C)C1=CN=CN1 DNDCVAGJPBKION-DOPDSADYSA-N 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 210000002798 bone marrow cell Anatomy 0.000 description 1
- 201000008275 breast carcinoma Diseases 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000001354 calcium citrate Substances 0.000 description 1
- 229940112129 campath Drugs 0.000 description 1
- 230000009702 cancer cell proliferation Effects 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 229910002091 carbon monoxide Inorganic materials 0.000 description 1
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 238000001516 cell proliferation assay Methods 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- AOXOCDRNSPFDPE-UKEONUMOSA-N chembl413654 Chemical compound C([C@H](C(=O)NCC(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CCSC)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](C)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@@H](N)CCC(O)=O)C1=CC=C(O)C=C1 AOXOCDRNSPFDPE-UKEONUMOSA-N 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000000451 chemical ionisation Methods 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 230000010428 chromatin condensation Effects 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 150000001860 citric acid derivatives Chemical class 0.000 description 1
- 239000011247 coating layer Substances 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 230000004540 complement-dependent cytotoxicity Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125807 compound 37 Drugs 0.000 description 1
- 229940127271 compound 49 Drugs 0.000 description 1
- 229940127113 compound 57 Drugs 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- TXWRERCHRDBNLG-UHFFFAOYSA-N cubane Chemical class C12C3C4C1C1C4C3C12 TXWRERCHRDBNLG-UHFFFAOYSA-N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000058 cyclopentadienyl group Chemical group C1(=CC=CC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 150000001945 cysteines Chemical class 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- OESHPIGALOBJLM-REOHCLBHSA-N dehydroascorbate Chemical compound OC[C@H](O)[C-]1OC(=O)C(=O)C1=O OESHPIGALOBJLM-REOHCLBHSA-N 0.000 description 1
- 235000020960 dehydroascorbic acid Nutrition 0.000 description 1
- 239000011615 dehydroascorbic acid Substances 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000010612 desalination reaction Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- URIILRBPLOOVJY-OAQYLSRUSA-N dibenzyl [(8S)-8-(chloromethyl)-1-methyl-7,8-dihydro-6H-thieno[3,2-e]indol-4-yl] phosphate Chemical compound P(=O)(OCC1=CC=CC=C1)(OCC1=CC=CC=C1)OC1=C2C(=C3[C@@H](CNC3=C1)CCl)C(=CS2)C URIILRBPLOOVJY-OAQYLSRUSA-N 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- RXKJFZQQPQGTFL-UHFFFAOYSA-N dihydroxyacetone Chemical compound OCC(=O)CO RXKJFZQQPQGTFL-UHFFFAOYSA-N 0.000 description 1
- 230000010339 dilation Effects 0.000 description 1
- KUMNEOGIHFCNQW-UHFFFAOYSA-N diphenyl phosphite Chemical compound C=1C=CC=CC=1OP([O-])OC1=CC=CC=C1 KUMNEOGIHFCNQW-UHFFFAOYSA-N 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 1
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000002224 dissection Methods 0.000 description 1
- 239000012738 dissolution medium Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- OFDNQWIFNXBECV-VFSYNPLYSA-N dolastatin 10 Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C=1SC=CN=1)CC1=CC=CC=C1 OFDNQWIFNXBECV-VFSYNPLYSA-N 0.000 description 1
- 108010045524 dolastatin 10 Proteins 0.000 description 1
- 229960005510 duocarmycin SA Drugs 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- ZSWFCLXCOIISFI-UHFFFAOYSA-N endo-cyclopentadiene Natural products C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 1
- 230000002357 endometrial effect Effects 0.000 description 1
- 210000004696 endometrium Anatomy 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- 229940082789 erbitux Drugs 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 125000005519 fluorenylmethyloxycarbonyl group Chemical group 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- HQVFCQRVQFYGRJ-UHFFFAOYSA-N formic acid;hydrate Chemical compound O.OC=O HQVFCQRVQFYGRJ-UHFFFAOYSA-N 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 230000022244 formylation Effects 0.000 description 1
- 238000006170 formylation reaction Methods 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 208000010749 gastric carcinoma Diseases 0.000 description 1
- PUBCCFNQJQKCNC-XKNFJVFFSA-N gastrin-releasingpeptide Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)CNC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)C(C)C)[C@@H](C)O)C(C)C)[C@@H](C)O)C(C)C)C1=CNC=N1 PUBCCFNQJQKCNC-XKNFJVFFSA-N 0.000 description 1
- 238000005227 gel permeation chromatography Methods 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 239000003168 generic drug Substances 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 230000002518 glial effect Effects 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 239000003966 growth inhibitor Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 201000010536 head and neck cancer Diseases 0.000 description 1
- 208000014829 head and neck neoplasm Diseases 0.000 description 1
- 201000005787 hematologic cancer Diseases 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- UQEAIHBTYFGYIE-UHFFFAOYSA-N hexamethyldisiloxane Chemical compound C[Si](C)(C)O[Si](C)(C)C UQEAIHBTYFGYIE-UHFFFAOYSA-N 0.000 description 1
- 235000014304 histidine Nutrition 0.000 description 1
- 229960002885 histidine Drugs 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 210000004408 hybridoma Anatomy 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- 230000002267 hypothalamic effect Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000005462 in vivo assay Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- KCKIKSJECTZLJA-UHFFFAOYSA-N indol-4-one Chemical compound O=C1C=CC=C2N=CC=C12 KCKIKSJECTZLJA-UHFFFAOYSA-N 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000010189 intracellular transport Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 230000002601 intratumoral effect Effects 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 229960005386 ipilimumab Drugs 0.000 description 1
- IQPQWNKOIGAROB-UHFFFAOYSA-N isocyanate group Chemical group [N-]=C=O IQPQWNKOIGAROB-UHFFFAOYSA-N 0.000 description 1
- 235000014705 isoleucine Nutrition 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 235000005772 leucine Nutrition 0.000 description 1
- 229960003136 leucine Drugs 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 238000001972 liquid chromatography-electrospray ionisation mass spectrometry Methods 0.000 description 1
- 239000003589 local anesthetic agent Substances 0.000 description 1
- 208000019420 lymphoid neoplasm Diseases 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 239000004337 magnesium citrate Substances 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- 235000006109 methionine Nutrition 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 229960004452 methionine Drugs 0.000 description 1
- NSPJNIDYTSSIIY-UHFFFAOYSA-N methoxy(methoxymethoxy)methane Chemical compound COCOCOC NSPJNIDYTSSIIY-UHFFFAOYSA-N 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- SZRWOLOVRDQPFQ-UHFFFAOYSA-N methyl n-[2-(methylamino)ethyl]carbamate Chemical compound CNCCNC(=O)OC SZRWOLOVRDQPFQ-UHFFFAOYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 238000000302 molecular modelling Methods 0.000 description 1
- IBMRTYCHDPMBFN-UHFFFAOYSA-N monomethyl glutaric acid Chemical compound COC(=O)CCCC(O)=O IBMRTYCHDPMBFN-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003136 n-heptyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000001613 neoplastic effect Effects 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 239000000346 nonvolatile oil Substances 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000003431 oxalo group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003585 oxepinyl group Chemical group 0.000 description 1
- AHHWIHXENZJRFG-UHFFFAOYSA-N oxetane Chemical compound C1COC1 AHHWIHXENZJRFG-UHFFFAOYSA-N 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 239000005022 packaging material Substances 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000006320 pegylation Effects 0.000 description 1
- 125000000538 pentafluorophenyl group Chemical group FC1=C(F)C(F)=C(*)C(F)=C1F 0.000 description 1
- 239000000863 peptide conjugate Substances 0.000 description 1
- 125000005062 perfluorophenyl group Chemical group FC1=C(C(=C(C(=C1F)F)F)F)* 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 239000002831 pharmacologic agent Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 125000005547 pivalate group Chemical class 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 239000004331 potassium propionate Substances 0.000 description 1
- 230000001855 preneoplastic effect Effects 0.000 description 1
- 238000004262 preparative liquid chromatography Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000005522 programmed cell death Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 229960002429 proline Drugs 0.000 description 1
- 235000013930 proline Nutrition 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 230000009145 protein modification Effects 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000005494 pyridonyl group Chemical group 0.000 description 1
- 125000001348 pyrrole-2,5-diyl group Chemical group N1C(=CC=C1*)* 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 230000010837 receptor-mediated endocytosis Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 102200160920 rs35304565 Human genes 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 210000003079 salivary gland Anatomy 0.000 description 1
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 1
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 229960001153 serine Drugs 0.000 description 1
- 235000004400 serine Nutrition 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 229910052709 silver Inorganic materials 0.000 description 1
- 239000004332 silver Substances 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 1
- 229960000553 somatostatin Drugs 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000008227 sterile water for injection Substances 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 201000000498 stomach carcinoma Diseases 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 230000003319 supportive effect Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- VMKKXJFHURYIIA-IOPIWRGFSA-N tert-butyl N-[2-[[4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[3-[2-[2-[2-[2-[2-[2-(2,5-dioxopyrrol-1-yl)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methoxycarbonyl-methylamino]ethyl]-N-methylcarbamate Chemical compound CC(C)[C@H](NC(=O)CCOCCOCCOCCOCCOCCOCCN1C(=O)C=CC1=O)C(=O)N[C@@H](CCCNC(N)=O)C(=O)Nc1ccc(COC(=O)N(C)CCN(C)C(=O)OC(C)(C)C)cc1 VMKKXJFHURYIIA-IOPIWRGFSA-N 0.000 description 1
- 238000010998 test method Methods 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 235000008521 threonine Nutrition 0.000 description 1
- 229960002898 threonine Drugs 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 201000003353 torsion dystonia 2 Diseases 0.000 description 1
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 231100000440 toxicity profile Toxicity 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- LGSAOJLQTXCYHF-UHFFFAOYSA-N tri(propan-2-yl)-tri(propan-2-yl)silyloxysilane Chemical compound CC(C)[Si](C(C)C)(C(C)C)O[Si](C(C)C)(C(C)C)C(C)C LGSAOJLQTXCYHF-UHFFFAOYSA-N 0.000 description 1
- 239000001393 triammonium citrate Substances 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- WILBTFWIBAOWLN-UHFFFAOYSA-N triethyl(triethylsilyloxy)silane Chemical compound CC[Si](CC)(CC)O[Si](CC)(CC)CC WILBTFWIBAOWLN-UHFFFAOYSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229960004799 tryptophan Drugs 0.000 description 1
- MEYZYGMYMLNUHJ-UHFFFAOYSA-N tunicamycin Natural products CC(C)CCCCCCCCCC=CC(=O)NC1C(O)C(O)C(CC(O)C2OC(C(O)C2O)N3C=CC(=O)NC3=O)OC1OC4OC(CO)C(O)C(O)C4NC(=O)C MEYZYGMYMLNUHJ-UHFFFAOYSA-N 0.000 description 1
- 229960004441 tyrosine Drugs 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 210000003606 umbilical vein Anatomy 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- 125000005500 uronium group Chemical group 0.000 description 1
- 235000014393 valine Nutrition 0.000 description 1
- 229960004295 valine Drugs 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 230000007332 vesicle formation Effects 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 101150061972 zur gene Proteins 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39558—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against tumor tissues, cells, antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6807—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug or compound being a sugar, nucleoside, nucleotide, nucleic acid, e.g. RNA antisense
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6863—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from stomach or intestines cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6865—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from skin, nerves or brain cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6869—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from a cell of the reproductive system: ovaria, uterus, testes, prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6871—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting an enzyme
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2866—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against receptors for cytokines, lymphokines, interferons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
- C07K5/06052—Val-amino acid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0815—Tripeptides with the first amino acid being basic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Cell Biology (AREA)
- Oncology (AREA)
- Biomedical Technology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Reproductive Health (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Un compuesto, o una sal o solvato farmacéuticamente aceptable del mismo, seleccionado entre: **(Ver fórmula)** donde Pg es H, acilo, PO3H2 o glicosilo
Description
DESCRIPCIÓN
Agentes citotóxicos bifuncionales que contienen el farmacóforo CTI
Campo de la invención
La presente invención está dirigida a nuevos dímeros bifuncionales CTI-CTI útiles para el tratamiento de enfermedades proliferativas. Los dímeros de la invención pueden funcionar como medicamentos independientes, cargas útiles en conjugados anticuerpo-fármaco (CAF) y compuestos de carga útil de enlace útiles en conexión con la producción o administración de tales CAF. La presente invención se refiere además a composiciones que incluyen los dímeros mencionados anteriormente, cargas útiles de enlace y CAF, y procedimientos para el uso de estos dímeros, cargas útiles de enlace y CAF, para el tratamiento de afecciones patológicas, incluido el cáncer.
Antecedentes
Los análogos bifuncionales que contienen dos motivos de alquilación de ADN activos (es decir, un CBI o un CPI) contienen dos alquilaciones (por ejemplo, dos motivos de CPI) fusionados. Debido a la presencia de dos motivos de alquilación reactivos, estos compuestos son reticuladores de ADN activos, mientras que los compuestos con un solo motivo de alquilación (por ejemplo, duocarmicinas) son mono-alquiladores de ADN.

Los compuestos mostrados anteriormente son ejemplos representativos de las referencias y se ha documentado que son citotoxinas potentes: A ("Glycosidic Prodrugs of Highly Potent Bifunctional Duocarmycin Derivatives for Selective Treatment of Cancer", Angew. Chem. Int. Ed. 2010, 49, 7336 -7339; "Duocarmycin Analogues Target Aldehyde Dehydrogenase 1 in LungCancer Cells", Angew. Chem. Int. Ed. 2012, 51,2874 -2877; "Bifunctional prodrugs and drugs", WO 2011/054837, DE 102009 051 799; "The Two Faces of Potent Antitumor Duocarmycin-Based Drugs: A Structural Dissection Reveals Disparate Motifs for DNA versus Aldehyde Dehydrogenase 1 Affinity", Angew. Chem. Int. Ed. 2013, 52, 1-6 ; B ("Interstrand DNA Cross-linking with Dimers of the Spirocyclopropyl Alkylating Moiety of C C -1065", J. Am. Chem. SOC. 1989, 11 1, 6428-6429; "CC-1065 analogs having two CPI subunits useful as antitumor agents and ultraviolet light absorbers", Eur. Pat. Appl. (1990), EP 359454, también para los compuestos C y D; C ("Synthesis and DNA Cross-Linking by a Rigid CPI Dimer", J. Am. Chem. SO C. 1991, 113, 8994-8995; "Nucleotide Preferences for DNA Interstrand Cross-Linking Induced by the Cyclopropylpyrroloindole Analogue U-77,779", Biochemistry 1993, 32, 2592-2600; "Determination of the Structural Role of the Internal Guanine-Cytosine Base Pair in Recognition of a Seven-Base-Pair Sequence Cross-Linked by Bizelesin", Biochemistry 1995, 34, 11005- 11016; "Analysis of the Monoalkylation and Cross-Linking Sequence Specificity of Bizelesin, a Bifunctional Alkylation AgentRelated to (+)-C C -1065", J. Am. Chem. SO C. 1993, 115, 5925-5933; "Mapping of DNA Alkylation Sites Induced by Adozelesin and Bizelesin in Human Cells by Ligation-Mediated Polymerase Chain Reaction", Biochemistry 1994, 33, 6024-6030; "DNA Interstrand Cross-Links Induced by the Cyclopropylpyrroloindole Antitumor Agent Bizelesin Are Reversible upon Exposure to Alkali", Biochemistry 1993, 32, 9108-9114; "Replacement of the Bizelesin Ureadiyl Linkage by a Guanidinium Moiety Retards Translocation from Monoalkylation to Cross-Linking Sites on DNA", J. Am. Chem. Soc. 1997, 119, 3434-3442; "DNA interstrand cross-linking, DNA sequence specificity, and induced conformational changes produced by a dimeric analog of (+)-C C -1065", Anti-Cancer Drug Design (1991), 6 , 427-452; "A phase I study of bizelesin, a highly potent and selective DNAinteractive agent, in patients with advanced solid malignancies", Ann Oncol. mayo de 2003;14(5):775-782; "A Phase I study of bizelesin (NSC 615291) in patients with advanced solid tumors", Clin Cancer Res. 2002, 3, 712-717; "Solution conformation of a bizelesin A-tract duplex adduct: DNA-DNA cross-linking of an A-tract straightens out bent DNA", J Mol Biol. 1995, 252, 86- 101; "Preclinical pharmacology of bizelesin, a potent bifunctional analog of the DNA-binding antibiotic C C -1065", Cancer Chemother Pharmacol. 1994, 34, 317-322; y D ("CC-1065 analogs having two CPI subunits useful as antitumor agents and
ultraviolet light absorbers", Eur. Pat. Appl. (1990), EP 359454. El motivo de alquilación de ADN activo puede, en principio, existir en forma de profármaco que se convierte en el fármaco activo en el medio biológico, o en su estado activo que no requiere conversión adicional. La conversión de profármaco a fármaco activo para los reticuladores bifuncionales se ejemplifica en el dímero CBI que se muestra a continuación:

Se lleva a cabo una conversión correspondiente para todos los reticuladores bifuncionales que existen en sus estados de profármaco.
Se han documentado otros reticuladores bifuncionales relacionados. ("Chemical and Biological Explorations of the Family of C C -1065 and the Duocarmycin Natural Products", Current Topics in Medicinal Chemistry, 2009, 9, 1494 1524; "DNA interstrand cross-linking agents and their chemotherapeutic potential", Curr Med Chem. 2012, 19, 364 385; "Design and Synthesis of a Novel DNA-DNA Interstrand Adenine-Guanine Cross-Linking Agent", J. Am. Chem. Soc. 2001, 123, 4865-4866; "Effect of base sequence on the DNA cross-linking properties of pyrrolobenzodiazepine (PBD) dimers", Nucleic Acids Res. 2011, 39, 5800- 5812; "Sequence-selective recognition of duplex DNA through covalent interstrand cross-linking: kinetic and molecular modeling studies with pyrrolobenzodiazepine dimers", Biochemistry. 2003, 42, 8232- 8239; "Bifunctional alkylating agents derived from duocarmycin SA: potent antitumor activity with altered sequence selectivity", Bioorg Med Chem Lett. 2000, 10, 495-498; "Design, Synthesis and Cytotoxicity Evaluation of 1-Chloromethyl-5-hydroxy-1,2-dihydro-3H-benz[e]indole (seco-CBI) Dimers", Bioorganic & Medicinal Chemistry 2000, 8, 1607- 1617.
Zhao y col. describieron una estrategia de profármacos de fosfato para citotoxinas que contienen seco-CBI monomérico. ("Synthesis and biological evaluation of antibody conjugates of phosphate prodrugs of cytotoxic DNA alkylators for the targeted treatment of cancer", J. Med. Chem. 2012, 55, 766- 782) y Zhang y col. ("Immunoconjugates containing phosphate-prodrugged DNA minor groove binding agents, compositions containing them, and methods of making them and their use for treating cancer", WO 2012/162482).
Recientemente se han descrito ciertos dímeros de CBI como cargas útiles de CAF ("1-(Chloromethyl)-2 ,3-Dihydro-1H-Benzo[e]indole Dimer Antibody-Drug Conjugate Compounds, and Methods of Use and Treatment", WO2015/023355).
La conjugación de fármacos con anticuerpos, ya sea directamente o mediante enlazadores, implica una consideración de una variedad de factores, incluyendo la identidad y ubicación del grupo químico para la conjugación del fármaco, el mecanismo de liberación del fármaco, los elementos estructurales que proporcionan la liberación del fármaco y la modificación estructural del fármaco libre liberado. Además, si el fármaco se va a liberar después de la internalización de anticuerpos, el mecanismo de liberación del fármaco debe estar en consonancia con el tráfico intracelular del conjugado.
Si bien se han probado una serie de diferentes clases de medicamentos para su administración mediante anticuerpos, solo unas pocas clases de fármacos han demostrado ser eficaces como conjugados de fármacos de anticuerpos, manteniendo un perfil de toxicidad adecuado. Una de esas clases es la de las auristatinas, derivados del producto natural dolastatina 10. Las auristatinas representativas incluyen (N-metilvalina-valinadolaisoleuina-dolaproinanorefedrina) y (N-metilvalina-valina-dolaisoleuina-dolaproina-fenilalanina). Otros agentes de unión a la tubulina relacionados incluyen las maytansinas (por ejemplo, véase "Cell-binding agent-maytansinoid conjugates linked via a noncleavable linker, preparation methods, and methods using them for targeting specific cell populations" publicado como el documento W o 2005/037992). Otros fármacos citotóxicos que se han empleado en la unión con anticuerpos incluyen fármacos que se unen al ADN, tales como la caliqueamicina, que causa la escisión del ADN de doble cadena específica de la secuencia. Otra clase de fármacos citotóxicos que se unen al ADN empleados en los CAF incluye pirrolobenzodiacepinas diméricas (por ejemplo, véase "Preparation of unsymmetrical pyrrolobenzodiazepines dimers for inclusion in targeted conjugates" publicado como el documento WO2013/041606). Otra clase de fármaco de este tipo en el que se ha intentado la administración por anticuerpos son los agentes alquilantes de unión al ADN, tales como el análogo de duocarmicina C C -1065 (véase "Preparation of C C -1065 analogs and their conjugates for treatment of cancer" publicado como el documento WO2010/062171) y compuestos relacionados (véase "Antibody-drug peptide conjugates for use as cytotoxins in cancer treatment" publicado como el documento WO 2007/038658, y "Immunoconjugates containing phosphate-prodrugged DNA minor groove binding agents, compositions containing them, and methods of making them and their use for treating cancer" publicado como el documento WO2012/162482). Sin embargo, todos estos fármacos tienen limitaciones relacionadas con las indicaciones de la enfermedad y el perfil de tratamiento, y por lo tanto, sigue existiendo la necesidad de fármacos adicionales con propiedades mejoradas que se puedan administrar mediante la conjugación con anticuerpos. Por consiguiente, la presente invención proporciona
nuevos CAF con dímeros como cargas útiles.
Otro heterociclo que se sabe que es un potente motivo de alquilación de ADN está representado por el grupo "CTI" (CTI: 1,2 ,8,8a-tetrahidro-4H-ciclopropa[c]tieno[3,2-e]indol-4-ona, "Fundamental relationships between structure, reactivity, and biological activity for the duocarmycins and C C -1065", J. Med. Chem. 2009, 52(19), 5771-5780, "Rational Design, Synthesis, and Evaluation of Key Analogues of C C -1065 and the Duocarmycins", J. a M. CHEM. SO C. 2007, 129, 14092- 14099). Los monómeros de duocarmicina que contienen CTI se han descrito como cargas útiles de CAF ("Synthesis of functionalized thieno-indole derivs. optionally containing a peptidic residue for the treatment of cancer and their use in the preparation of conjugates", WO 2013/ 149946, "Preparation of new functionalized alkylating agents containing a thieno-indole moiety linked to a DNA-binding moiety for treating cancers and their use in the prepn. of conjugates", WO 2013/149948). Las diferencias estructurales entre "CBI", "CPI" y "CTI" se representan en el siguiente dibujo:

CBI, CPI y CTI difieren en el primer anillo aromático, teniendo el CBI un fenilo, el CPI un pirrol y el CTI un heterociclo de tiofeno, respectivamente. El dibujo también muestra los profármacos de cloruro. Estos profármacos se convierten en el fármaco activo en el medio biológico bajo pérdida de cloruro de hidrógeno. Por lo tanto, tanto el profármaco de cloruro como la especie de ciclopropilo activo deben considerarse equivalentes con respecto a su actividad biológica. El concepto de los profármacos de cloruro para CBI, CPI y los grupos relacionados han sido bien documentados en las referencias ("Design, Synthesis, and Evaluation of Duocarmycin O-Amino Phenol Prodrugs Subject", J. Med. Chem.
2010, 53, 7731-7738 y la referencia 8 citada en el presente documento).
Con respecto al grupo CTI, dos variaciones estructurales son de particular interés, a saber, los grupos Me-CTI e iso-Me-CTI que se muestran en el siguiente dibujo:
Ambos grupos Me-CTI e /so-Me-CTI se han descrito en el contexto de duocarmicinas ("Chemical and biological explorations of the family of C C -1065 and the duocarmycin natural products", Curr Top Med Chem. 2009, 9(16), 1494 1524).
Sumario de la invención
La invención describe nuevos análogos de dímeros estructurales basados en el motivo CTI. La invención también describe elementos espaciadores para los correspondientes dímeros de CTI. Se desvelan estructuras mixtas CBI-CTI. Los términos CBI y CTI se usan tanto para la versión de profármacos de cloruro como para las especies de ciclopropilo activas. Además, se describe la sustitución del enlazador a estas especies diméricas, así como la preparación de conjugados anticuerpos-fármacos.
No se conocen dímeros que contengan el motivo CTI y el uso de dímeros CTI en la presente invención da como resultado cambios significativos en la reactividad química y las propiedades biológicas en comparación con las especies de dímeros descritas anteriormente. Por lo tanto, los dímeros preparados con especies CTI representan entidades farmacológicas distintivas. En la presente invención, los presentes inventores describen nuevos dímeros que contienen dos grupos CTI. Además, los presentes inventores desvelan cómo estas especies de dímeros están conectadas a moléculas enlazadoras adecuadas para la unión a anticuerpos.
De manera más específica, la presente invención está dirigida a dímeros citotóxicos que comprenden dímeros a base de CTI/CTI (incluyendo las formas seco de CTI, tal como se detalla en el presente documento), a conjugados de anticuerpos-fármacos que comprenden tales dímeros, y a procedimientos para el uso de los mismos para tratar el cáncer y otras indicaciones. Tanto las estructuras CTI como CBI se pueden representar por su forma seco y puede sustituirse y derivatizarse tal como se detalla en el presente documento. Además, la función fenol en las formas seco puede derivatizarse con grupos acetato. Las funciones de acetato fenólico son funcionalmente equivalentes al fenol ya que los grupos acetato se hidrolizan fácilmente para dar los fenoles libres en medio biológico.
Por lo tanto, la presente invención se refiere a compuestos y composiciones farmacéuticas que los contienen, a su preparación, y a los usos de los compuestos, principalmente, pero no exclusivamente, agentes anticancerígenos. De acuerdo con un aspecto, la presente invención se refiere a compuestos de "carga útil" de la reivindicación 1.
También se desvelan compuestos de Fórmula I:
F1-L1-T-L2-F2 (Fórmula I)
o una sal farmacéuticamente aceptable o solvato de los mismos, en la que: cada uno de F1 y F2 se selecciona independientemente entre los sistemas de anillo A, B, C, D, E, F, G y H:


(Sistema de Anillo C)


(Sistema de Anillo H)
donde:
está presente al menos uno de los sistemas de anillo A, B, C y D;
cada R se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C20, -alquenilo C2-C6, -alquinilo C2-C6, halo, deuterio, hidroxilo, alcoxi, -NH2, -NH(alquilo C1-C8), -N(alquilo C1-C8)2, -NO2, -arilo C6-C14 y -heteroarilo C6-C14, en los que dos o más R se unen opcionalmente para formar un anillo o anillos, y en los que dicho -arilo C6-C14 y -heteroarilo C6-C14 están opcionalmente sustituidos con 1 a 5 sustituyentes seleccionados independientemente entre -alquilo C1-C10, -alcoxi C1-C10, -halo, -alquiltio C1-C10, -trifluorometilo, -NH2, -NH(alquilo C1-C8), -N(alquilo C1-C8)2, -alquil C1-C10-N(alquilo C1-C8)2, -alquiltio C1-C3, -NO2 o -heterociclilo C1-C10, para cada sistema de anillo en el que aparece R;
cada V 1 es independientemente un enlace, O, N(R) o S, para cada sistema de anillo en el que aparece V 1; cada V 2 es independientemente O, N(R) o S, para cada sistema de anillo en el que aparece V 2;
cada uno de W 1 y W 2 es independientemente H, -alquilo C1-C5, -fenilo, -C(O)OR, -C(O)SR, -C(O)NHN(R)2 o -C(O)N(R)2 para cada sistema de anillo en el que aparecen W 1 y W 2;
cada X es independientemente -OH, -O-acilo, azido, halo, cianato, tiocianato, isocianato, tioisocianato, o

para cada sistema de anillo en el que aparece X;
cada Y se selecciona independientemente entre el grupo que consiste en H, -alquil C i -C 6-R a, -C(O)RA, -C(S)R A, -C(O)ORA, -S(O)2ORa, -C(O)N(Ra)2, -C(S)N(Ra)2, un carbohidrato, glicosilo, -NO2, -PO(ORA)2, un aminoácido, y un péptido (por ejemplo, un péptido que se escinde con proteasas tales como catepsinas y metaloproteinasas de matriz) para cada sistema de anillo en el que aparece Y, en el que cada RA se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C20, -heteroalquilo C1-C8, -arilo C6-C14, aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8 y -alquil C1-C20-N(R)2,
en el que dichos alquilo -C 1-C 20, -heteroalquilo C 1-C 8, -arilo C 6-C 14, aralquilo, -heterociclilo C 1-C 10, -carbociclilo C 3-s y -alquil C1-C20-N(R)2 están opcionalmente sustituidos con 1 a 3 sustituyentes seleccionados independientemente entre R;
cada Z se selecciona independientemente entre el grupo que consiste en H, -alquilo Ci-Cs, -heteroalquilo Ci-Cs, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-CS, -C(O)Oalquilo C i-Cs, -C(O)N(alquilo Ci-Cs)2, -C(O)OH, -C(O)NHNH2 y -C(O)-halo, y en el que dichos alquilo C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-CS, -C(O)Oalquilo C1-Cs, -C(O)N(alquilo C1-Cs)2, -C(O)OH, -C(O)NHNH2 y -C(O)-halo están cada uno opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R, para cada sistema de anillo en el que aparece Z;
cada uno de L1 y L2 se selecciona independientemente entre un enlace directo, carbonilo o un grupo carbonil acilo unido a F1 o F2 en el resto acilo, donde el grupo carbonil acilo se selecciona entre el grupo que consiste en:

en el que
U1 se selecciona entre H, -CH3, -OH, -OCH3, -NO2, -NH2, -NHNHAc, -NHNHC(O)CH3, -NHC(O)fenilo o -halo,
U2 es H, -OH o -OCH3,
U3 es H, -CH3 o -C2H5,
U4 es H o CH3S-,
cada uno de U5 y U6 se selecciona independientemente entre H, -halo, -alquilo C1-C4, -alcoxi C1-C3, -dialquilamino
C1-C6, -NO2 -NHC(O)alquilo C1-C10, -OH, -NH2, -NHC(O)NH2, -NHC(O)CH3 o -NHC(O)fenilo,
Q1 es -O-, -S - o -NH-, y
cada uno de Q2 y Q3 es independientemente -C H - o -N-;
T se selecciona entre:
- NHC(O)-,
- C(O)NH-,
- C(O)O-,
- OC(O)-,
- NRB-T 1-N RC- donde cada uno de RB y RC es independientemente H o -alquilo C1-C8, o RB y RC juntos se unen
para formar un anillo y juntos son (CH2)2-3, donde T1 se selecciona entre el grupo que consiste en -C(O)-, -C(O)(CH2)nC(O)- donde n es un número entero de 0 a 50 y -C(O)PhC(O)- donde Ph es 1,3- o 1,4-fenileno, y donde T 1 está opcionalmente sustituido con 1-2 R,
- C(O)hetC(O)- en el que het es un heteroarilo mono-, bi- o tricíclico de 5 a 12 miembros, que contiene uno, dos o tres heteroátomos seleccionados independientemente entre O, N, S, P y B, en el que het está opcionalmente sustituido con 1 a 8 sustituyentes, cada uno seleccionado independientemente entre el grupo que consiste en
-alquilo C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, NHRD y -NO2, y dichos sustituyentes opcionales en het están opcionalmente sustituidos con RE,
en el que cada RD se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -C(O)-alquilo
C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2 y -C(O)-halo, opcionalmente sustituido con RE,
en el que cada RE se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2 y -C(O)-halo, y en el que cada RE está opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R,
-C(A1)X1-T 2-X 1C(B1)-, donde T2 es:

en el que cada X 1 es independientemente un enlace, -N RE-, -O - o -S-, en el que cada uno de A1 y B1 es independientemente =O o =S, en el que cada uno de R1, R2, R3 y R4 es independientemente RE o R1 y R2 forman un sistema de anillo, o R3 y R4 forman un sistema de anillo, o ambos R1 y R2, y R3 y R4, forman cada uno independientemente sistemas de anillo, o R1 y R3 forman un sistema de anillo, o R2 y R4 forman un sistema de anillo, o ambos R1 y R3, y R2 y R4, forman cada uno independientemente sistemas de anillo,
donde dichos sistemas de anillo se seleccionan independientemente entre -heterociclilo C1-C10 o -carbociclilo
C3-C8, o cada uno de R1, R2, R3 y R4 es un enlace a diferentes carbonos en D, en el que cada uno de g y j es independientemente un número entero de 0 a 50 y m es un número entero de 1 a 50, y en el que D es un enlace o se selecciona entre el grupo que consiste en -S -, -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heteroalquileno C1-C8-, -aralquileno, -heterociclo C1-C10 y -carbociclo C3-C8, donde dichos -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heteroalquileno C1-C8-, -aralquileno, -heterociclo C1-C10 y --carbociclo
C3-C8 están opcionalmente sustituidos con -R E, -C(O )RE, -C(O)ORE, -N(RE)2, -N(R)C(O)RE o -N(R)C(O)ORE, y
D está opcionalmente sustituido adicionalmente con 1 a 2 R, y -G 1-T 2-G 2-, donde cada uno de G 1 y G 2 es independientemente -S(O )X1- o -S(O)2X1-.
En realizaciones de los compuestos desvelados, la variable n es de 0 a 50, preferentemente de 0 a 25, preferentemente de 0 a 10, y preferentemente 1- 5. Preferentemente, la variable n puede ser 0, 1, 2 , 3, 4 o 5.
En otras realizaciones de los compuestos desvelados, la variable -Y - es C(O)N(RA)2 o C(S)N(RA)2 donde un RA es
hidrógeno o -alquilo C1-C20 y el otro RA es -alquil C i -C 20-N(R)2, de tal forma que la estructura:

se forma, donde A es oxígeno o azufre.
Como se ha indicado anteriormente, realizaciones de los presentes compuestos desvelados incluyen aquellas en las que cada uno de R1, R2, R3 y R4 se une a diferentes carbonos en D. Cuando D es un anillo carbocíclico de 6 miembros (en negrita, a continuación), esta realización puede tomar la forma de un cubano:

También son posibles otras formas de cubanos (por ejemplo, formas sustituidas como las que se resumen en el presente documento) y no cubanos y se incluyen dentro de la invención.
De acuerdo con otro aspecto de la invención, se proporciona un compuesto "enlazador-carga útil" de la reivindicación 2.
También se desvelan compuestos "enlazador-carga útil" de Fórmula IIA:
L-P (Fórmula IIA)
o una sal farmacéuticamente aceptable o solvato de los mismos, en la que:
P es:
F1- L1-T -L2- F2
en la que:
cada uno de F1 y F2 se selecciona independientemente entre los sistemas de anillo A, B, C, D, E, F, G y H:


(Sistema de Anillo C)
(Sistema de Anillo D)


(Sistema de Anillo H)
donde:
al menos uno de los sistemas de anillo A, B, C y D está presente;
cada R se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C20, -alquenilo C2-C6, -alquinilo C2-C6, halo, deuterio, hidroxilo, alcoxi, -NH2, -NH(alquilo C1-C8), -N(alquilo C1-C8)2, -NO2, -arilo C6-C14 y -heteroarilo C6-C14, en los que dos o más R se unen opcionalmente para formar un anillo o anillos, y en los que dicho -arilo C6-C14 y -heteroarilo C6-C14 están opcionalmente sustituidos con 1 a 5 sustituyentes seleccionados independientemente entre -alquilo C1-C10, -alcoxi C1-C10, -halo, -alquiltio C1-C10, -trifluorometilo, -NH2, -NH(alquilo C1-C8), -N(alquilo C1-C8)2, -alquil C1-C10-N(alquilo C1-C8)2, -alquiltio C1-C3, -NO2 o -heterociclilo C1-C10, para cada sistema de anillo en el que aparece R;
cada V 1 es independientemente un enlace, O, N(R) o S, para cada sistema de anillo en el que aparece V 1; cada V 2 es independientemente O, N(R) o S, para cada sistema de anillo en el que aparece V 2;
cada uno de W 1 y W 2 es independientemente H, -alquilo C1-C5, -fenilo, -C(O)OR, -C(O)SR, -C(O)NHN(R)2 o -C(O)N(R)2 para cada sistema de anillo en el que aparecen W 1 y W2;
cada X se selecciona independientemente entre -OH, -O-acilo, azido, halo, cianato, tiocianato, isocianato, tioisocianato, o

para cada sistema de anillo en el que aparece X;
cada Y se selecciona independientemente entre un enlace, H, -C(O)RA, -C(S)R A, -C(O)ORA, S(O)2ORA, -C(O)N(Ra)2, -C(S)N(Ra)2, un carbohidrato tal como glicosilo, -NO2, -P(O)(ORa)2, un aminoácido y un péptido (en particular un péptido que se escinde con proteasas tales como catepsinas y metaloproteinasas de matriz) para cada sistema de anillo en el que aparece Y, en el que cada RA se selecciona independientemente entre H, -alquilo
C1-C20, -heteroalquilo C1-C8, -arilo C6-C14, aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -alquil C1-C20N(R)2, -alquileno C1-C20, -heteroalquileno C1-C8, -arileno C6-C14, aralquileno, -heterociclo C1-C10, -carbociclo C3-C8y -alquil
C1-C20N(R)-, y RF donde dicho RA está opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R, y en el que un Y es divalente y está unido a L,
RF es -N(R6)QN(R5)C(O)- y está unido a L en el carbonilo adyacente N(R5), en el que cada uno de R5 y R6 se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -heteroalquilo C1-C8, -arilo C 6-C14, -aralquilo, -heterociclilo C1-C10 y -carbociclilo C3-C8, o R5 o R6 se unen con un carbono sustituido en Q para formar un anillo -heterocíclico C1-C10 o -heteroarilo C6-C14, o R5 y R6 se unen para formar un sistema de anillo -heterocíclico C1-C10 o -heteroarilo C6-C14, y donde Q es -alquileno C1-C8-, -heteroalquileno C1-C8-, -arileno C6-C14-, -aralquileno-, -heterociclo C1-C10- o -carbociclo C3-C8-, en el que cada uno de Q, R5 y R6 está opcionalmente sustituido independientemente con 1 a 3 sustituyentes seleccionados independientemente entre R;
cada Z se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C C(O)OH, -C(O)NHNH2 y -C(O)-halo, y en el que dichos alquilo C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo CrC8)2, -C(O)OH, -C(O)NHNH2 y -C(O)-halo están cada uno opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R, para cada sistema de anillo en el que aparece Z;
cada uno de L1 y L2 se selecciona independientemente entre un enlace directo, carbonilo o un grupo carbonil acilo unido a F1 o F2 en el resto acilo, donde el grupo carbonil acilo se selecciona entre el grupo que consiste en:

en el que
U1 se selecciona entre H, -CH3, -OH, -OCH3, -NO2, -NH2, -NHNHAc, -NHNHC(O)CH3, -NHC(O)fenilo o -halo,
U2 es H, -OH o -OCH3,
U3 es H, -CH3 o -C2H5,
U4 es H o CH3S-,
cada uno de U5 y U6 se selecciona independientemente entre H, -halo, -alquilo C1-C4, -alcoxi C1-C3, -dialquilamino C1-C6, -NO2, -NHC(O)alquilo C1-C10, -OH, -NH2, -NHC(O)NH2, -NHC(O)CH3 o -NHC(O)fenilo,
Q 1 es -O-, -S - o -NH-, y
cada uno de Q2 y Q3 es independientemente -C H - o -N-;
T se selecciona entre:
-NHC(O)-,
-C(O)NH-,
-C(O)O-,
-OC(O)-,
-N RB-T 1-N RC- donde cada uno de RB y RC es independientemente H o -alquilo C1-C8, o R B y RCjuntos se unen para formar un anillo y juntos son (CH2)2-3, donde T 1 se selecciona entre el grupo que consiste en -C(O)-, -C(O)(CH2)nC(O)- donde n es un número entero de 0 a 50 y -C(O)PhC(O)- donde Ph es 1,3- o 1,4-fenileno, y donde T 1 está opcionalmente sustituido con 1-2 R,
-C(O)hetC(O)- en el que het es un heteroarilo mono-, bi- o tricíclico de 5 a 12 miembros, que contiene uno, dos o tres heteroátomos seleccionados independientemente entre O, N, S, P y B, donde het está opcionalmente sustituido con 1 a 8 sustituyentes, cada uno seleccionado independientemente entre el grupo que consiste en -alquilo C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -NH2, -NHRD y -NO2, y dichos sustituyentes opcionales en het están opcionalmente sustituidos con RE,
en el que cada R D se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -C(O)-alquilo C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2 y -C(O)-halo, opcionalmente sustituido con RE,
en el que cada RE se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -heteroalquilo C1-C8, -arilo, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2 y -C(O)-halo, y en el que cada RE está opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R,
-C(A1 )X1 -T 2-X 1C(B1)-, donde T2 es:

en el que cada X 1 es independientemente un enlace, -N RE-, -O - o -S-, en el que cada uno de A1 y B1 es independientemente =O o =S, en el que cada uno de R1, R2, R3 y R4 es independientemente RE o R1 y R2 forman un sistema de anillo, o R3 y R4 forman un sistema de anillo, o ambos R1 y R2, y R3 y R4, forman cada uno independientemente sistemas de anillo, o R1 y R3 forman un sistema de anillo, o R2 y R4 forman un sistema de anillo, o ambos R1 y R3, y R2 y R4, forman cada uno independientemente sistemas de anillo, donde dichos sistemas de anillo se seleccionan independientemente entre -heterociclilo C1-C10 o -carbociclilo C3-C8, o cada uno de R1, R2, R3 y R4 es un enlace a diferentes carbonos en D, en el que cada uno de g y j es independientemente un número entero de 0 a 50 y m es un número entero de 1 a 50, y en el que D es un enlace o se selecciona entre el grupo que consiste en -S -, -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heteroalquileno C1-C8-, -aralquileno, -heterociclo C1-C10 y -carbociclo C3-C8, donde dichos -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heteroalquileno C1-C8-, -aralquileno, -heterociclo C1-C10 y -carbociclo C3-C8 están opcionalmente sustituidos con -R E, -C(O )RE, -C(O)ORE, -N(RE)2, -N(R)C(O)RE o -N(R)C(O)ORE, y D está opcionalmente sustituido adicionalmente con 1 a 2 R, y
-G 1-T 2-G 2-, donde cada uno de G 1 y G 2 es independientemente -S(O )X1- o -S(O)2X1-;
L es La-L b-(Lc)1-3, en el que LA se selecciona entre el grupo que consiste en -halo, -N(R)2, -CON(R)2, -S-arilo opcionalmente sustituido con -NO2 o -CON(R)2, -S-heteroarilo opcionalmente sustituido con -NO2, alquil-SO2-heteroarilo, arilSO2-heteroaril-,

Lb es l b1-LB2-LB3 en el que LB1 está ausente o es uno o más componentes seleccionados entre el grupo que consiste en -C(O)-, -C(S)-, -C(O)NR-, -C(O)alquil C1-C6-, -C(O)NRalquil C1-C6-, -alquil C i -C6(OCH2CH2)i -6-, -C(O)alquil C i -C 6NRC(O)-, -C(O)alquil C i -C6(OCH2CH2)i -6-, -alquil C i-C6(OCH2CH2)i -6-C(O)-, -alquil C1-C6-S-S-alquil C i -C 6NRC(O)CH2-, -alquil C i -C6(OCH2CH2)i -6NRC(O)CH2-, -C(O)alquil C i-C 6-NRC(O)alquil C1-6-, -N=CR-fenil-O-alquil C1-C6-, -N=CR-fenil-O-alquil C i-C 6-C(O)-, -C(O)-alquil C i -C6(OCH2CH2)i -6NRC(O)-, -C(O)alquil C i-C 6-fenil(NR-C(O)alquil Ci-C6)i-4-, -C(O)alquil Ci-C6(OCH2CH2)i-6-NRC(O)alquil C1-C6-, -alquil C i-C 6-, -S-, -C(O)-CH(NR-C(O)alquil Ci-Ce)-alquil C i-C 6- y (-CH2-CH2-O-K20,
en el que LB2 es AA0-12, en el que AA es un aminoácido natural, un aminoácido no natural o -(C R 15)o-S-S-(CR15)p donde cada uno de o y p es independientemente un número entero de i a 20,
LB3 es -PABA-, -PABC-, -C(O)(CH2)nC(O)- o está ausente;
Lc está ausente o se selecciona independientemente entre el grupo que consiste en -alquileno C1-C6-, -NRheterociclil C3-C8NR-, -NRcarbociclil C3-C8NR-, -NRalquil C1-C6NR-, -NRalquileno C1-C6-, -S-, -NR-, -N RNR-, -O(CR2)i-4S-S(CR2)i-4N(R)-, -NRalquilenofenileno C1-C6NR-, -NRalquilenofenileno C1-C6SO2NR-, -Oalquil C i-C 6S-Salquil C i -C 6C(CO O r )Nr -, -NRC(COOR)alquil C i-C 6S-Salquil C1-C6O-,
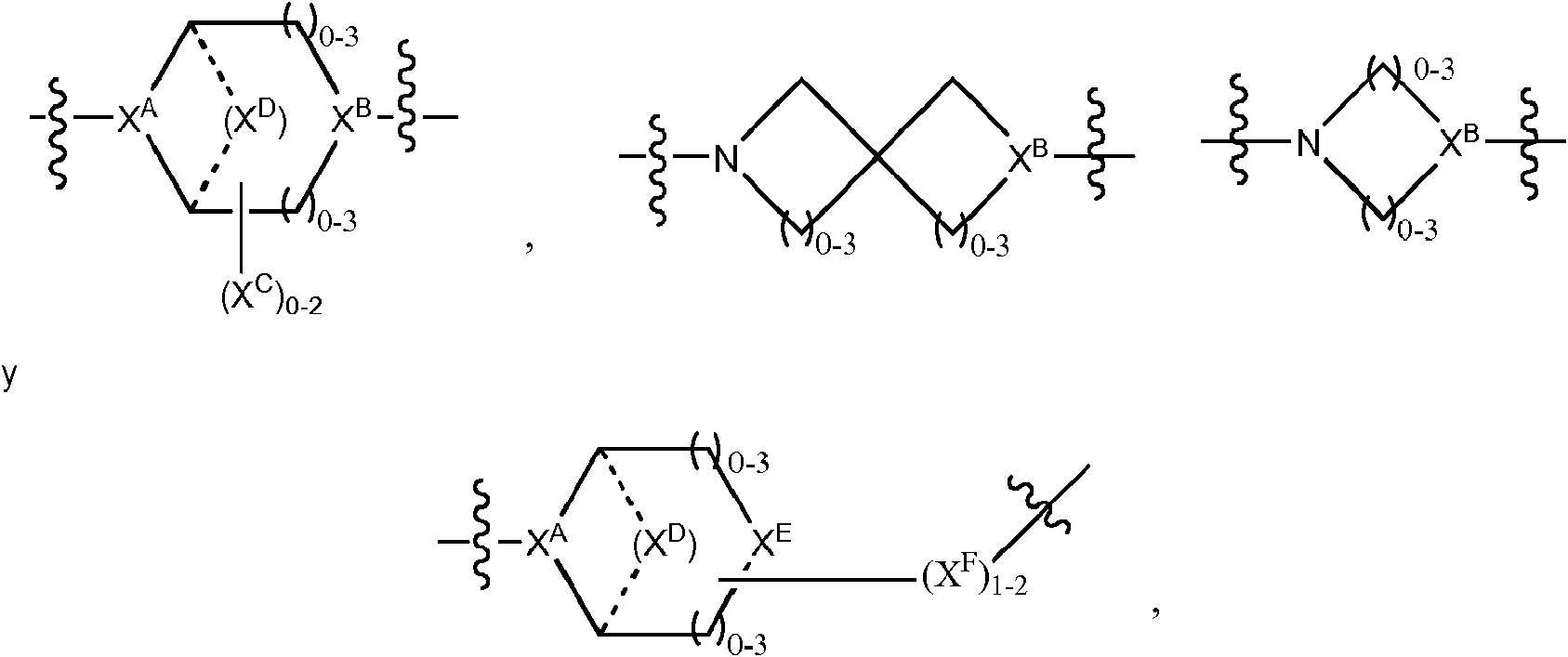
en el que
X a es C R o N,
X B es CH, CR(C(R)2)i -3NR, CR(C(R)2)i -30, CR(C(R)2)i -3C(0 )NR, CR-(C(R)2)i-3C(O)NRNR, CR(C(R)2)i -3SO2NR, CR(C(R)2)i -3NRNR, CR(C(R)2)i -3NRC(0 ) o N,
cada X C es R,
cada X D es -(CH2)i -5-, o está ausente;
X E es O, S, C(R)2, C(R)(C(R)2)i -3-NR2 o NR y cada X F es (C(R)2)i -3-NR o C(R)2-(C(R)2)i -3-0.
En otras realizaciones de los compuestos desvelados, la variable -Y - es C(O)N(RA)2 o C(S)N(Ra)2 donde un RA es hidrógeno o -alquilo C1-C20 y el otro RA es -alquil C i-C 20-N(R)-, de tal forma que la estructura:
se forma, donde cada A es independientemente oxígeno o azufre.
De acuerdo con otro aspecto más de la invención, se proporciona un compuesto conjugado anticuerpo-fármaco de la reivindicación 3.
También se desvela un conjugado anticuerpo-fármaco de Fórmula IIIA:
AB-(L-P)i -20 (Fórmula IIIA)
o una sal farmacéuticamente aceptable o solvato de los mismos, en la que:
AB es un anticuerpo;
P es:
f 1-l 1-t -l 2-f2
en la que:
cada uno de F1 y F2 se selecciona independientemente entre los sistemas de anillo A, B, C, D, E, F, G y H:


(Sistema de Anillo D)

(Sistema de Anillo E)

donde:
al menos uno de los sistemas de anillo A, B, C y D está presente;
cada R se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C20, -alquenilo C2-C3, -alquinilo C2-C3, halo, deuterio, hidroxilo, alcoxi, -NH2, -NH(alquilo C1-C8), -N(alquilo C1-C8)2, -NO2, -arilo C6-C14 y -heteroarilo C6-C14, en los que dos o más R se unen opcionalmente para formar un anillo o anillos, y en los que dicho -arilo C6-C14 y -heteroarilo C6-C14 están opcionalmente sustituidos con 1 a 5 sustituyentes seleccionados independientemente entre -alquilo C1-C10, -alcoxi C1-C10, -halo, -alquiltio C1-C10, -trifluorometilo, -NH2, -NH(alquilo C1-C8), -N(alquilo C1-C8)2, -alquil C1-C10-N(alquilo C1-C8)2, -alquiltio C1-C3, -NO2 o -heterociclilo C1-C10, para cada sistema de anillo en el que aparece R;
cada V 1 es independientemente un enlace, O, N(R) o S, para cada sistema de anillo en el que aparece V 1; cada V2 es independientemente O, N(R) o S, para cada sistema de anillo en el que aparece V2;
cada uno de W 1 y W 2 es independientemente H, -alquilo C1-C5, -fenilo, -C(O)OR, -C(O)SR, -C(O)NHN(R)2 o -C(O)N(R)2 para cada sistema de anillo en el que aparecen W 1 y W 2;
cada X se selecciona independientemente entre -OH, -O-acilo, azido, halo, cianato, tiocianato, isocianato, tioisocianato, o
O
í 11
f — O -S -R
* 11
O ,
para cada sistema de anillo en el que aparece X;
cada Y se selecciona independientemente entre un enlace, H, -C(O)RA, -C(S)R A, -C(O)ORA, -S(O)2ORA, -C(O)N(Ra)2, -C(S)N(Ra)2, un carbohidrato tal como glicosilo, -NO2, -P(O)(ORA)2, un aminoácido y un péptido (en particular un péptido que se escinde con proteasas tales como catepsinas y metaloproteinasas de matriz) para cada sistema de anillo en el que aparece Y, en el que cada RA se selecciona independientemente entre H, -alquilo C1-C20, -heteroalquilo C1-C8, -arilo C6-C14, aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -alquil C1-C20N(R)2, -alquileno C1-C20, -heteroalquileno C1-C8, -arileno C6-C14, aralquileno, -heterociclo C1-C10, -carbociclo C3-C8 y -alquil C1-C20N(R)-, y RF donde dicho RA está opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R, y en el que un Y es divalente y está unido a L, RF es -N(R6)QN(R5)C(O)- y está unido a L en el carbonilo adyacente N(R5), en el que cada uno de R5 y R6 se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -heteroalquilo C1-C8, -arilo C 6
C14, -aralquilo, -heterociclilo C1-C10 y -carbociclilo C3-C8, o R5 o R6 se unen con un carbono sustituido en Q para formar un anillo -heterocíclico C1-C10 o -heteroarilo C6-C14, o R5 y R6 se unen para formar un sistema de anillo -heterocíclico C1-C10 o -heteroarilo C6-C14, y donde Q es -alquileno C1-C8-, -heteroalquileno C1-C8-, -arileno Ca-C14-, -aralquileno-, -heterociclo C1-C10- o -carbociclo C3-C8-, en el que cada uno de Q, R5 y R6 está opcionalmente sustituido independientemente con 1 a 3 sustituyentes seleccionados independientemente entre R;
cada Z se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -heteroalquilo C1-C 8, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2, -C(O)OH, -C(O)NHNH2 y -C(O)-halo, y en el que dichos alquilo C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2, -C(O)OH, -C(O)NHNH2 y -C(O)-halo están cada uno opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R, para cada sistema de anillo en el que aparece Z;
cada uno de L1 y L2 se selecciona independientemente entre un enlace directo, carbonilo o un grupo carbonil acilo unido a F1 o F2 en el resto acilo, donde el grupo carbonil acilo se selecciona entre el grupo que consiste en:

en el que
U1 se selecciona entre H, -CH3, -OH, -OCH3, -NO2, -NH2, -NHNHAc, -NHNHC(O)CH3, -NHC(O)fenilo o -halo, U2 es H, -OH o -OCH3,
U3 es H, -CH3 o -C2H5,
U4 es H o CH3S-,
cada uno de U5 y U6 se selecciona independientemente entre H, -halo, -alquilo C1-C4, -alcoxi C1-C3, -dialquilamino C1-C6, -NO2, -NHC(O)alquilo C1-C10, -OH, -NH2, -NHC(O)NH2, -NHC(O)CH3 o -NHC(O)fenilo, Q1 es -O-, -S - o -NH-,
cada uno de Q2 y Q3 es independientemente -C H - o -N-;
T se selecciona entre:
-NHC(O)-,
-C(O)NH-,
-C(O)O-,
-OC(O)-,
-N R B-T 1-N RC- donde cada uno de RB y RC es independientemente H o -alquilo C1-C8, o RB y RC juntos se unen para formar un anillo y juntos son (CH2)2-3, donde T 1 se selecciona entre el grupo que consiste en -C(O)-, -C(O)(CH2)nC(O)- donde n es un número entero de 0 a 50 y -C(O)PhC(O)- donde Ph es 1,3-o 1,4-fenileno, y donde T 1 está opcionalmente sustituido con 1-2 R,
-C(O)hetC(O)- en el que het es un heteroarilo mono-, bi- o tricíclico de 5 a 12 miembros, que contiene uno, dos o tres heteroátomos seleccionados independientemente entre O, N, S, P y B, donde het está opcionalmente sustituido con 1 a 8 sustituyentes, cada uno seleccionado independientemente entre el grupo que consiste en -alquilo C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -NH2, -N HRD y -NO2, y dichos sustituyentes opcionales en het están opcionalmente sustituidos con RE,
en el que cada RD se selecciona independientemente entre el grupo que consiste en H o -alquilo C1-C8, -C(O)-alquilo C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C 8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2 y -C(O)-halo, opcionalmente sustituido con RE,
en el que cada RE se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -heteroalquilo C1-C8, -arilo, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2 y -C(O)-halo, y en el que cada RE está opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R,
-C (A 1)X1-T 2-X 1C(B1)-, donde T2 es:

en el que cada X 1 es independientemente un enlace, -N RE-, -O - o -S-, en el que cada uno de A1 y B1 es independientemente =O o =S, en el que cada uno de R1, R2, R3 y R4 es independientemente RE o R1 y R2 forman un sistema de anillo, o R3 y R4 forman un sistema de anillo, o ambos R1 y R2, y R3 y R4, forman cada uno independientemente sistemas de anillo, o R1 y R3 forman un sistema de anillo, o R2 y R4 forman un sistema de anillo, o ambos R1 y R3, y R2 y R4, forman cada uno independientemente sistemas de anillo, donde dichos sistemas de anillo se seleccionan independientemente entre -heterociclilo C1-C10 o -carbociclilo C3-C8, o cada uno de R1, R2, R3 y R4 es un enlace a diferentes carbonos en D, en el que cada uno de g y j es independientemente un número entero de 0 a 50 y m es un número entero de 1 a 50, y en el que D es un enlace o se selecciona entre el grupo que consiste en -S-, -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heteroalquileno C1-C8-, -aralquileno, -heterociclo C1-C10 y -carbociclo C3-C8, donde dichos -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heteroalquileno C1-C8-, -aralquileno, -heterociclo C1-C10 y --carbociclo C3-C8 están opcionalmente sustituidos con -R E, -C(O )RE, -C(O)ORE, -N(RE)2, -N(R)C(O)RE o -N(R)C(O)ORE, y D está opcionalmente sustituido adicionalmente con 1 a 2 R, y
-G 1-T 2-G 2-, donde cada uno de G 1 y G 2 es independientemente -S(O )X1- o -S(O)2X1-;
L es La-L b-(Lc)1-3;
LA se selecciona entre: un enlace a AB, -NR-(enlace a AB), -heteroaril-(enlace a AB),

LB1-LB2-LB3 en el que LB1 está ausente o es uno o más componentes seleccionados entre el grupo que consiste en -C(O)-, -C(S)-, -C(O)NR-, -C(O)alquil C1-C6-, -C(O)NRalquil C1-C6-, -alquil C1
C6(OCH2CH2)i -6-, -C(O)alquil Ci-CaNRC(O)-, -C(O)alquil Ci-Ca(OCH2CH2)i-a-, -alquil C i-Ca(OCH2CH2)i-6-C(O)-, -alquil Ci-Ca-S-S-alquil Ci-CaNRC(O)CH2-, -alquil Ci-Ca(OCH2CH2)iaNRC(O)CH2-, -C(O)alquil C i-C a-
NRC(O)alquil Ci-a-, -N=CR-fenil-O-alquil Ci-Ca-, -N=CR-fenil-O-alquil Ci-Ca-C(O)-, -C(O)-alquil C i-Ca(OCH2CH2)i-aNRC(O)-,
-C(O)alquil Ci-Ca-fenil(NR-C(O)alquil Ci-Ca)i-4-, -C(O)alquil Ci-Ca(OCH2CH2)i-a-NRC(O)alquil Ci-Ca-,
-alquil Ci-Ca-, -S -, -C(O)-CH(NR-C(O)alquil Ci-Ca)-alquil C i-C a - y (-CH2-CH2-O -)i -20;
LB2 es AAo-i2, en el que AA es un aminoácido natural, un aminoácido no natural o -(C R i5)o-S-S-(CR i5)p donde cada uno de o y p es independientemente un número entero de 1 a 20,
LB3 es -PABA-, -PABC-, -C(O)(CH2)nC(O)- o está ausente;
Lc está ausente o se selecciona independientemente entre el grupo que consiste en -alquileno Ci-Ca-, -NRheterociclil C3-C8NR-, -NRcarbociclil C3-C8NR-, -NRalquil C i-CaNR-, -NRalquileno Ci-Ca-, -S-, -NR-, -NRNR-, - 0 (CR2)i -4S -S (C R 2)i -4N(R)-, -NR-alquilenofenileno Ci-CaNR-, -NRalquilenofenileno C i-CaSO2NR-, -Oalquil Ci-CaS-Salquil Ci-CaC(COOR)N R-, -NRC(COOR)alquil Ci-CaS-Salquil Ci-CaO-,

en el que
X a es CR o N,
X B es CH, CR(C(R)2)í 43NR, CR(C(R)2)i -30, CR(C(R)2)i -3C(0 )NR, CR-(C(R)2)i -3C(O)NRNR, CR(C(R)2)i -3S 02NR, CR(C(R)2)i -3NRNR, CR(C(R)2)i-3NRC(O) o N, cada X C es R;
cada X D es -(CH2)i -5-, o está ausente;
X E es O, S, C(R)2, C(R)(C(R)2)i -3-N R2 o NR, y
cada X F es (C(R)2)i -3-NR o C(R)2-(C(R)2)i -3-0.
También se desvela un compuesto "enlazador-carga útil" de Fórmula IIB:
F ^ - T - lA F 2
l
L
(Fórmula IIB)
o una sal farmacéuticamente aceptable o solvato de los mismos, en la que:
cada uno de F 1 y F2 se selecciona independientemente entre los sistemas de anillo A, B, C, D, E, F, G y H:
i8

(Sistema de Anillo A)

(Sistema de Anillo B)

(Sistema de Anillo D)
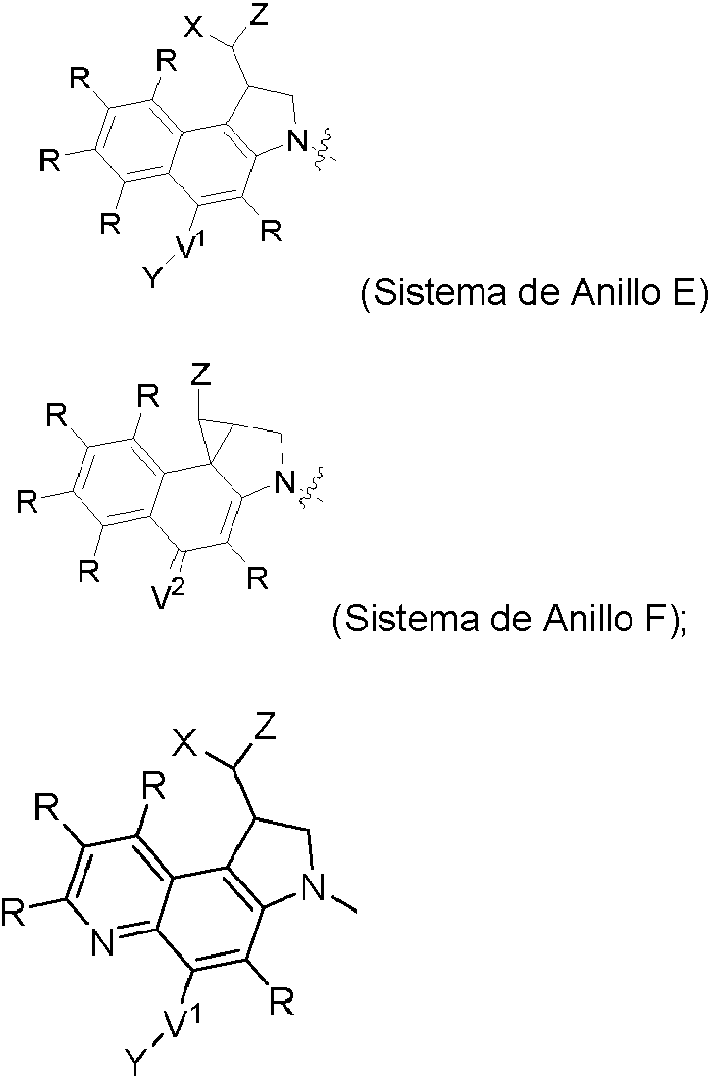
(Sistema de Anillo G)

donde:
al menos uno de los sistemas de anillo A, B, C y D está presente;
cada R se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C20, -alquenilo C2-C6, -alquinilo C2-C3, halo, deuterio, hidroxilo, alcoxi, -NH2, -NH(alquilo C1-C8), -N(alquilo C1-C8)2, -NO2, -arilo C6-C14 y -heteroarilo C6-C14, en los que dos o más R se unen opcionalmente para formar un anillo o anillos, y en los que dicho -arilo C6-C14 y -heteroarilo C6-C14 están opcionalmente sustituidos con 1 a 5 sustituyentes seleccionados independientemente entre -alquilo C1-C10, -alcoxi C1-C10, -halo, -alquiltio C1-C10, -trifluorometilo, -NH2, -NH(alquilo C1-C8), -N(alquilo C1-C8)2, -alquil C1-C10-N(alquilo C1-C8)2, -alquiltio C1-C3, -NO2 o -heterociclilo C1-C10, para cada sistema de anillo en el que aparece R;
cada V 1 es independientemente un enlace, O, N(R) o S, para cada sistema de anillo en el que aparece V 1; cada V 2 es independientemente O, N(R) o S, para cada sistema de anillo en el que aparece V 2;
cada uno de W 1 y W 2 es independientemente H, -alquilo C1-C5, -fenilo, -C(O)OR, -C(O)SR, -C(O)NHN(R)2 o -C(O)N(R)2 para cada sistema de anillo en el que aparecen W 1 y W 2;
cada X es independientemente -OH, -O-acilo, azido, halo, cianato, tiocianato, isocianato, tioisocianato, o
O
s
f—O - S - R ' 11
■ O
para cada sistema de anillo en el que aparece X;
cada Y se selecciona independientemente entre el grupo que consiste en H, -alquil C1-C6-RA -C(O)RA, -C(S)R A, -C(O)ORA, -S(O)2ORa, -C(O)N(Ra)2, -C(S)N(Ra)2, un carbohidrato tal como glicosilo, -NO2, -PO(ORA)2, un aminoácido y un péptido (en particular un péptido que se escinde con proteasas tales como catepsinas y metaloproteinasas de matriz) para cada sistema de anillo en el que aparece Y, en el que cada RA se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C20, -heteroalquilo C1-C8, -arilo C6-C14, aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8 y -alquil C1-C20N(R)2, en el que dichos alquilo -C1-C20, -heteroalquilo C1-C 8, -arilo C6-C14, aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8 y -alquil C1-C20-N(R)2 están opcionalmente sustituidos con 1 a 3 sustituyentes seleccionados independientemente entre R;
cada Z se selecciona independientemente entre el grupo que consiste en H, -alquilo C 1-C 8, -heteroalquilo C 1-C 8, -arilo C 6-C 14, -aralquilo, -heterociclilo C 1-C 10, -carbociclilo C 3-C 8, -C(O)Oalquilo C 1-C 8, -C(O)N(alquilo C 1-C 8)2, -C(O)OH, -C(O)Nh Nh 2 y -C(O)-halo, y en el que dichos alquilo C 1-C 8, -heteroalquilo C 1-C 8, -arilo C 6-C 14, -aralquilo, -heterociclilo C 1-C 10, -carbociclilo C 3-C 8, -C(O)Oalquilo C 1-C 8, -C(O)N(alquilo C 1-C 8)2, -C(O)OH, -C(O)NHNH2 y -C(O)-halo están cada uno opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R, para cada sistema de anillo en el que aparece Z;
cada uno de L1 y L2 se selecciona independientemente entre un enlace directo, carbonilo o un grupo carbonil acilo unido a F 1 o F2 en el resto acilo, donde el grupo carbonil acilo se selecciona entre el grupo que consiste en:

en el que
U1 se selecciona entre H, -CH3, -OH, -OCH3, -NO2, -NH2, -NHNHAc, -NHNHC(O)CH3, -NH-C(O)fenilo o -halo,
U2 es H, -OH o -OCH3,
U3 es H, -CH3 o -C2H5,
U4 es H o CH3S-,
cada uno de U5y U6 se selecciona independientemente entre H, -halo, -alquilo C1-C4, -alcoxi C1-C3, -dialquilamino
C1-C6, -NO2, -NHC(O)alquilo C1-C10, -OH, -NH2, -NHC(O)NH2, -N H C (O )C H o -NHC(O)fenilo,
Q1 es -O-, -S - o -NH-, y
cada uno de Q2 y Q3 es independientemente -C H - o -N-;
T se selecciona entre:
-C(A 1)X1-T 2-X 1C(B1)-, donde T2 es:

en el que cada X 1 es independientemente un enlace, -N R E-, -O - o -S-, en el que cada uno de A1 and B1 es independientemente =O o =S, en el que cada uno de R1, R2, R3, y R4 es independientemente RE, o R1 y R2 forman un sistema de anillo, o R3 y R4 forman un sistema de anillo, o ambos R1 y R2, uno independientemente sistemas de anillo, o R1 y R3 forman un sistema de anillo, o R2 y R4 forman un sistema de anillo, o ambos R1 y R3, y R2 y R4 forman cada uno sistemas de anillo, donde los sistemas de anillo se seleccionan independientemente entre -heterociclilo C1-C10 o -carbociclilo C3-C8, o cada uno de R1, R2, R3 y R4 es un enlace a diferentes carbonos en D, en el que cada g y j es independientemente un número entero de 0 a
50 y m es un número entero de 1 a 50, y en el que D se selecciona entre el grupo que consiste en -alquileno
C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heteroalquileno C1-C8-, -aralquileno, -heterociclo C1-C10 y -carbociclo C3-C8, donde dichos -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heteroalquileno C1-C 8-, -aralquileno, -heterociclo C1-C10 y -carbociclo C3-C8 están sustituidos con un miembro del grupo seleccionado entre N(RE)C(O)- donde el carbonilo está unido a L, y -C(O)- donde el carbonilo está unido a L, y opcionalmente sustituidos adicionalmente con 1 a 2 R;
donde cada RE se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -heteroalquilo C1-C8, -arilo, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2 y -C(O)-halo, y en el que cada RE está opcionalmente sustituido con 1 a 3 sustituyentes
seleccionados independientemente entre R;
L es LA-L B-(LC)i-a;
LA se selecciona entre -halo, -N(R)2, -CON(R)2, -S-arilo opcionalmente sustituido con -NO2 o -CONR2, -S -heteroarilo opcionalmente sustituido con -NO2, alquil-SO2-heteroarilo, arilSO2-heteroaril-,

LB es LB1-LB2-LB3
en el que LB1 está ausente o es uno o más componentes seleccionados entre el grupo que consiste en -C(O)-, -C(S)-, -C(O)NR-, -C(O)alquil C1-C6-, -C(O)NRalquil C1-C6-, -alquil C1-Cs(OCH2CH2)1-6-, -C(O)alquil C1-CsNRC(O)-, -C(O)alquil C1-C6(OCH2CH2)1-6-, -alquil C1-C6(OCH2CH2)1-6-C(O)-, -alquil C1-C6-S-S-alquil C1-C6NRC(O)CH2-, -alquil C1-C6(OCH2CH2)1-6NRC(O)CH2-, -C(O)alquil C1-C6-NRC(O)alquil C1-6-, -N=CR-fenil-O-alquil C1-C6-, -N=CR-fenil-O-alquil C1-C6-C(O)-, -C(O)-alquil C1-C6(OCH2CH2)1-6NRC(O)-, -C(O)alquil C1-C6-fenil(NR-C(O)alquil C1-C6)1-4-, -C(O)alquil C1-C6(OCH2CH2)1-6-NRC(O)alquil C1-C6-, -alquil C1-C6-, -S-, -C(O)-CH(NR-C(O)alquil C1-C6)-alquil C1-C6- y (-CH2-CH2-O-)1-20;
LB2 es AA0-12, en el que AA es un aminoácido natural, un aminoácido no natural o -(C R 15)o-S-S-(CR15)p donde cada uno de o y p es independientemente un número entero de 1 a 20,
LB3 es -PABA-, -PABC-, -C(O)(CH2)„C(O)- o está ausente;
LC está ausente o se selecciona independientemente entre el grupo que consiste en -alquileno C1-C6-, -NRheterociclil C3-C8NR-, -NRcarbociclil C3-C8NR-, -NRalquil C1-C6NR-, -NRalquileno C1-C6-, -S-, -NR-, -N RNR-, -O(CR2)1-4S-S(CR2)1-4N(R)-, -NRalquilenofenileno C1-C6NR-, -NRalquilenofenileno C1-C6SO2NR-, -Oalquil C1-C 6S-Salquil C1-C6C(CO O r )Nr -, -NRC(COOR)alquil C1-C6S-Salquil C1-C6O-,

en el que
X a es C R o N,
X B es CH, CR(C(R)2)1-3NR, CR(C(R)2)1-3O, CR(C(R)2)1-3C(O)NR, CR-(C(R)2)1-3C(O)NRNR, CR(C(R)2)1-3SO2NR, CR(C(R)2)1-3NRNR, CR(C(R)2)1-3NRC(O) o N;
cada X C es R;
cada X D es -(CH2)1-5-, o está ausente;
X E es O, S, C(R)2, C(R)(C(R)2)1-3-NR2 o NR, y
cada X F es (C(R)2)1-3-NR o C(R)2-(C(R)2)1-3-O.
También se desvela un compuesto conjugado anticuerpo-fármaco de Fórmula IIIB:

(Fórmula IIIB)
a sal farmacéuticamente aceptable o solvato de los mismos, en la que:
AB es un anticuerpo;
cada uno de F 1 y F2 se selecciona independientemente entre los sistemas de anillo A, B, C, D, E, F, G y H:

(Sistema de Anillo A)

(Sistema de Anillo B)
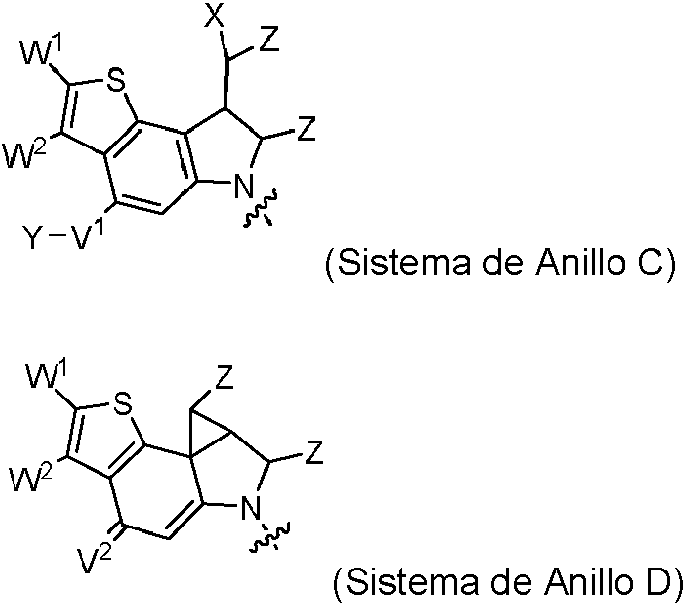

(Sistema de Anillo F);

s ema e n o
donde:
al menos uno de los sistemas de anillo A, B, C y D está presente;
cada R se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C20, -alquenilo C2-C6, -alquinilo C2-C6, halo, deuterio, hidroxilo, alcoxi, -NH2, -NH(alquilo C1-C8), -N(alquilo C1-C8)2, -NO2, -arilo C6-C14 y -heteroarilo C6-C14, en los que dos o más R se unen opcionalmente para formar un anillo o anillos, y en los que dicho -arilo C6-C14 y -heteroarilo C6-C14 están opcionalmente sustituidos con 1 a 5 sustituyentes seleccionados independientemente entre -alquilo C1-C10, -alcoxi C1-C10, -halo, -alquiltio C1-C10, -trifluorometilo, -NH2, -NH(alquilo C1-C8), -N(alquilo C1-C8)2, -alquil C1-C10-N(alquilo C1-C8)2, -alquiltio C1-C3, -NO2 o -heterociclilo C1-C10, para cada sistema de anillo en el que aparece R;
cada V 1 es independientemente un enlace, O, N(R) o S, para cada sistema de anillo en el que aparece V 1;
cada V 2 es independientemente O, N(R) o S, para cada sistema de anillo en el que aparece V2;
cada uno de W 1 y W 2 es independientemente H, -alquilo C1-C5, -fenilo, -C(O)OR, -C(O)SR, -C(O)NHN(R)2 o -C(O)N(R)2 para cada sistema de anillo en el que aparecen W 1 y W 2;
cada X es independientemente -OH, -O-acilo, azido, halo, cianato, tiocianato, isocianato, tioisocianato, o
O
i <1
I—O - S - R ' 11
O
para cada sistema de anillo en el que aparece X;
cada Y se selecciona independientemente entre el grupo que consiste en H, -alquil C1-C8-RA-C(O)RA, -C(S)R A, -C(O)ORA, -S(O)2ORa, -C(O)N(Ra)2, -C(S)N(Ra)2, un carbohidrato tal como glicosilo, -NO, -PO(ORA)2, un aminoácido y un péptido (en particular un péptido que se escinde con proteasas tales como catepsinas y metaloproteinasas de matriz), para cada sistema de anillo en el que aparece Y, en el que cada RA se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C20, -heteroalquilo C1-C8, -arilo C6-C14, aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8 y -alquil C1-C20N(R)2, en el que dichos alquilo -C1-C20, -heteroalquilo C1-C8, -arilo C6-C14, aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8 y -alquil C1-C20N(R)2 están opcionalmente sustituidos con 1 a 3 sustituyentes seleccionados independientemente entre R;
cada Z se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -heteroalquilo C1-C 8, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2, -C(O)OH, -C(O)NHNH2 y -C(O)-halo, y en el que dichos alquilo C1-C8, -heteroalquilo C1-C8, -arilo C6-C14, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2, -C(O)OH, -C(O)NHNH2 y -C(O)-halo están cada uno opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R, para cada sistema de anillo en el que aparece Z;
cada uno de L1 y L2 se selecciona independientemente entre un enlace directo, carbonilo o un grupo carbonil acilo unido a F1 o F2 en el resto acilo, donde el grupo carbonil acilo se selecciona entre el grupo que consiste en:

en el que
U1 se selecciona entre H, -CH3, -OH, -OCH3, -NO2, -NH2, -NHNHAc, -NHNHC(O)CH3, -NH-C(O)fenilo o -halo,
U2 es H, -OH o -OCH3,
U3 es H, -CH3 o -C2H5,
U4 es H o CH3S-,
cada uno de U5 y U6 se selecciona independientemente entre H, -halo, -alquilo C1-C4, -alcoxi C1-C3, -dialquilamino C1-C8,
-NO2, -NHC(O)alquilo C1-C10, -OH, -NH2, -NHC(O)NH2, -NHC(O)CH3 o -NHC(O)fenilo,
Q1 es -O-, -S - o -NH-, y
cada uno de Q2 y Q3 es independientemente -C H - o -N-;
T se selecciona entre:
-C(A 1)X1-T 2-X 1C(B1)-, donde T2 es:

en el que cada X 1 es independientemente un enlace, -N R E-, -O - o -S-, en el que cada uno de A1 y B1 es independientemente =O o =S, en el que cada uno de R1, R2, R3, y R4 es independientemente RE, o R1 y R2 forman un sistema de anillo, o R3 y R4 forman un sistema de anillo, o ambos R1 y R2, y R3 y R4 forman cada uno independientemente sistemas de anillo, o R1 y R3 forman un sistema de anillo, o R2 y R4 forman un
sistema de anillo, o ambos R1 y R3, y R2 y R4 forman cada uno sistemas de anillo, donde los sistemas de anillo se seleccionan independientemente entre -heterociclilo C1-C10 o -carbociclilo C3-C8, o cada uno de R1, R2, R3 y R4 es un enlace a diferentes carbonos en D, en el que cada g y j es independientemente un número entero de 0 a 50 y m es un número entero de 1 a 50, y en el que D se selecciona entre el grupo que consiste en -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heteroalquileno C1-C8-, -aralquileno, -heterociclo C1-C10 y -carbociclo C3-C8, donde dichos -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heteroalquileno C1-C8-, -aralquileno, -heterociclo C1-C10 y -carbociclo C3-C8 están sustituidos con un miembro del grupo seleccionado entre N(RE)C(O)- donde el carbonilo está unido a L, y -C(O)- donde el carbonilo está unido a L, y opcionalmente sustituidos adicionalmente con 1 a 2 R; donde cada RE se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8, -heteroalquilo C1-C8, -arilo, -aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -C(O)Oalquilo C1-C8, -C(O)N(alquilo C1-C8)2 y -C(O)-halo, y en el que cada RE está opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R;
L es La-L b-(Lc)1-3;
LA se selecciona entre: un enlace a AB, -NR-(enlace a AB), -heteroaril-(enlace a AB),

en el que LB1 está ausente o es uno o más componentes seleccionados entre el grupo que consiste en -C(O)-, -C(S)-, -C(O)NR-, -C(O)alquil C1-C6-, -C(O)NRalquil C1-C6-, -alquil C1-Ca(OCH2CH2)1-6-, -C(O)alquil C r CaNRC(O)-, -C(O)alquil C1-Ca(OCH2CH2)1-6-, -alquil C1-Ca(OCH2CH2)1-a-C(O)-, -alquil C1-Ca-S-S-alquil C1-CaNRC(O)CH2-, -alquil C1-Ca(OCH2CH2)1-aNRC(O)CH2-, -C(O)alquil C^Ca-NRC(O)alquil C1.6-, -N=CR-fenil-O-alquil C1-C6-, -N=CR-fenil-O-alquil C1-Ca-C(O)-, -C(O)-alquil C1-Ca(OCH2CH2)1-6NRC(O)-, -C(O)alquil C1-Ca-fenil(NR-C(O)alquil C1-Ca)1-4-, -C(O)alquil C1-Ca(OCH2CH2)1-a-NRC(O)alquil C1-C6-, -alquil C1-C6-, -S-, -C(O)-CH(NR-C(O)alquil C1-Ca)-alquil C 1-C a-y (-CH2-C H 2-O -)1-20;
LB2 es AA0-12, en el que AA es un aminoácido natural, un aminoácido no natural o -(C R 15)o-S-S-(CR15)p donde cada uno de o y p es independientemente un número entero de 1 a 20,
LB3 es -PABA-, -PABC-, -C(O)(CH2)nC(O)- o está ausente;
LC está ausente o se selecciona independientemente entre el grupo que consiste en -alquileno C1-Ca-, -NRheterociclil C3-C8NR-, -NRcarbociclil C3-C8NR-, -NRalquil C1-CaNR-, -NRalquileno C1-Ca-, -S-, -NR-, -NRNR-, -O(CR2)1-4S-S(CR2)1-4N(R)-, -NRalquilenofenileno C1-CaNR-, -NRalquilenofenileno C1-CaSO2NR-, -Oalquil C1-CaS-Salquil C1-CaC(COOR)NR-, -NRC(COOR)alquil C1-CaS-Salquil C1-CaO-,

en el que
X a es CR o N,
2a
X B es CH, CR(C(R)2)i-aNR, CR(C(R)2)i-3O, CR(C(R)2)i-3C(O)NR, CR-(C(R)2)i-3C(O)NRNR, CR(C(R)2)i-3SO2NR, CR(C(R)2)i-aNRNR, CR(C(R)2)i-3NRC(O) o N;
cada X C es R;
cada X D es -(CH2)i-5-, o está ausente;
X E es O, S, C(R)2, C(R)(C(R)2)i-3-N R2 o NR, y
cada X F es (C(R)2)i-3-NR o C(R)2-(C(R)2)i-3-O.
Aspectos adicionales de la invención incluyen compuestos tales como los mencionados en el presente documento, donde
cada R se selecciona independientemente entre el grupo que consiste en H, deuterio, -alquilo C1-C20 y -NH2; cada V 1 es independientemente O o N(R) para cada sistema de anillo en el que aparece V 1;
cada V2 es independientemente O o N(R) para cada sistema de anillo en el que aparece V2;
cada uno de W 1 y W 2 es independientemente H, -alquilo C1-C5, -C(O)OR o -C(O)NR2 para cada sistema de anillo en el que aparecen W i y W 2;
cada X es independientemente halo, para cada sistema de anillo en el que aparece X;
cada Y se selecciona independientemente entre el grupo que consiste en H, -C(O)RA, -C(O)N(RA)2, un carbohidrato tal como glicosilo, -NO2, -PO(ORa)2, un aminoácido y un péptido (en particular un péptido que se escinde con proteasas tales como catepsinas y metaloproteinasas de matriz) para cada sistema de anillo en el que aparece Y, en el que cada RA se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C20, -heteroalquilo C1-C8, -carbociclilo C3-C8 y -alquil Ci-C20-N(R)2, en el que dichos alquilo -C1-C20, -heteroalquilo C1-C8, -carbociclilo C3-C8 y -alquil Ci-C20-N(R)2 están opcionalmente sustituidos con 1 a 3 sustituyentes seleccionados independientemente entre R;
cada uno de L1 y L2 se selecciona independientemente entre un enlace directo y carbonilo; y
T se selecciona entre:
-N R B-T 1-N RC- donde cada uno de RB y RC es independientemente H o -alquilo C1-C8, -C(O)hetC(O)- en el que het es un heteroarilo monocíclico de 5 a 12 miembros, que contiene uno o dos heteroátomos seleccionados independientemente entre O, N y S, en el que het está opcionalmente sustituido con 1 a 8 sustituyentes, cada uno seleccionado independientemente entre el grupo que consiste en -alquilo C1-C8, -NH2 y -NH2, y dichos sustituyentes opcionales en het están opcionalmente sustituidos con -alquilo C1-C8, y
-C (A 1)X1-T 2-X 1C(B1)-, donde T2 es:

en el que cada X 1 es un enlace, en el que cada uno de A1 y B1 es independientemente =0, en el que cada uno de R1, R2, R3 y R4 es independientemente H o R1 y R2 forman un sistema de anillo, o R3 y R4 forman un sistema de anillo, o ambos R1 y R2, y R3 y R4, forman cada uno independientemente sistemas de anillo, o R1 y R3 forman un sistema de anillo, o R2 y R4 forman un sistema de anillo, o ambos R1 y R3, y R2 y R4, forman cada uno independientemente sistemas de anillo, donde dichos sistemas de anillo se seleccionan independientemente entre -heterociclilo C1-C10 o -carbociclilo C3-C8, y en el que D es un enlace o se selecciona entre el grupo que consiste en -S -, -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heterociclo C1-C10 y -carbociclo C3-C8, donde dichos -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heterociclo C1-C10 y --carbociclo C3-C8 están opcionalmente sustituidos con -NH2, -N(R)C(O)H o -N(R)C(O)OH.
Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde dos o más R se unen opcionalmente para formar un anillo o anillos.
Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde
cada R se selecciona independientemente entre el grupo que consiste en H, deuterio, -alquilo C1-C20 y -NH2; cada V 1 es independientemente O o N(R) para cada sistema de anillo en el que aparece V 1;
cada V2 es independientemente O o N(R) para cada sistema de anillo en el que aparece V2;
cada uno de W 1 y W 2 es independientemente H, -alquilo C1-C5, -C(O)OR o -C(O)NR2 para cada sistema de anillo en el que aparecen W 1 y W 2;
cada X es independientemente halo, para cada sistema de anillo en el que aparece X;
cada Y se selecciona independientemente entre un enlace, H, -C(O)RA, -C(S)R A, -C(O)ORA, -S(O)2ORA, -C(O)N(RA)2, -C(S)N(Ra)2, un carbohidrato tal como glicosilo, -NO2, -P(O)(ORA)2, un aminoácido y un péptido (en particular un péptido que se escinde con proteasas tales como catepsinas y metaloproteinasas de matriz) para cada sistema de anillo en el que aparece Y, en el que cada RA se selecciona independientemente entre H, -alquilo C1-C20, -heteroalquilo C1-C8, -arilo C6-C14, aralquilo, -heterociclilo C1-C10, -carbociclilo C3-C8, -alquil Ci-C20N(R)2, -alquileno C1-C20,
heteroalquileno C1-C8, -arileno C b-C-m, aralquileno, -heterociclo C1-C10, -carbociclo C3-C8 y -alquil C i -C2oN(R)-, y RF donde dicho RA está opcionalmente sustituido con 1 a 3 sustituyentes seleccionados independientemente entre R, y en el que un Y es divalente y está unido a L,
RF es -N(R6)QN(R5)C(O)- y está unido a L en el carbonilo adyacente N(R5), en el que cada uno de R5 y R6 se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C8 y -heteroalquilo C1-C8, o R5 o R6 se unen con un carbono sustituido en Q para formar un anillo -heterocíclico C1-C10 o -heteroarilo C b-C i 4, o R5 y R6 se unen para formar un sistema de anillo -heterocíclico C1-C10 o -heteroarilo C6-C14, y donde Q es -alquileno C1-C8-, -arileno C6-C14- o -carbociclo C3-C8-, en el que cada uno de Q, R5 y R6 está opcionalmente sustituido independientemente con 1 a 3 sustituyentes seleccionados independientemente entre R;
cada uno de L1 y L2 se selecciona independientemente entre un enlace directo y carbonilo; y
T se selecciona entre:
- NRB-T 1-N RC- donde cada uno de RB y RC es independientemente H o -alquilo C1-C8,
- C(O)hetC(O)- en el que het es un heteroarilo monocíclico de 5 a 12 miembros, que contiene uno o dos heteroátomos seleccionados independientemente entre O, N y S, en el que het está opcionalmente sustituido con 1 a 8 sustituyentes, cada uno seleccionado independientemente entre el grupo que consiste en -alquilo C1-C8, -NH2 y -NH2, y dichos sustituyentes opcionales en het están opcionalmente sustituidos con -alquilo C1-C8, y
- C(A1)X1-T 2-X 1C(B1)-, donde T2 es:

en el que cada X 1 es un enlace, en el que cada uno de A1 y B1 es independientemente =0, en el que cada uno de R1, R2, R3 y R4 es independientemente H o R1 y R2 forman un sistema de anillo, o R3 y R4 forman un sistema de anillo, o ambos R1 y R2, y R3 y R4, forman cada uno independientemente sistemas de anillo, o R1 y R3 forman un sistema de anillo, o R2 y R4 forman un sistema de anillo, o ambos R1 y R3, y R2 y R4, forman cada uno independientemente sistemas de anillo, donde dichos sistemas de anillo se seleccionan independientemente entre -heterociclilo C1-C10 o -carbociclilo C3-C8, y en el que D es un enlace o se selecciona entre el grupo que consiste en -S -, -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heterociclo C1-C10 y -carbociclo C3-C8, donde dichos -alquileno C 1-C 8-, -arileno C 6-C 14-, -heteroarileno C 6-C 14-, -heterociclo C 1-C 10 y --carbociclo C 3-C 8 están opcionalmente sustituidos con -NH2, -N(R)C(O)H o -N(R)C(O)OH.
Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde
cada R se selecciona independientemente entre el grupo que consiste en H, deuterio, -alquilo C1-C20 y -NH2; cada V 1 es independientemente O o N(R) para cada sistema de anillo en el que aparece V 1;
cada V2 es independientemente O o N(R) para cada sistema de anillo en el que aparece V2;
cada uno de W 1 y W 2 es independientemente H, -alquilo C1-C5, -C(O)OR o -C(O)NR2 para cada sistema de anillo en el que aparecen W 1 y W 2;
cada X es independientemente halo, para cada sistema de anillo en el que aparece X;
cada Y se selecciona independientemente entre el grupo que consiste en H, -C(O )RA, -C(O)N(RA)2, un carbohidrato tal como glicosilo, -NO2, -PO(ORa)2, un aminoácido y un péptido (en particular un péptido que se escinde con proteasas tales como catepsinas y metaloproteinasas de matriz) para cada sistema de anillo en el que aparece Y, en el que cada RA se selecciona independientemente entre el grupo que consiste en H, -alquilo C1-C20, -heteroalquilo C1-C8, -carbociclilo C3-C8 y -alquil C i -C2o-N(R)2, en el que dichos alquilo -C1-C20, -heteroalquilo C1-C8, -carbociclilo C3-C8 y -alquil C i -C2o-N(R)2 están opcionalmente sustituidos con 1 a 3 sustituyentes seleccionados independientemente entre R;
cada uno de L1 y L2 se selecciona independientemente entre un enlace directo y carbonilo; y
T es -C(A 1)X1-T 2-X 1C(B1)-, donde T2 es:

en el que cada X 1 es un enlace, en el que cada uno de A1 y B1 es independientemente =0, en el que cada uno de R1,
R2, R3 y R4 es independientemente H o R1 y R2 forman un sistema de anillo, o R3 y R4 forman un sistema de anillo, o
ambos R1 y R2, y R3 y R4, forman cada uno independientemente sistemas de anillo, o R1 y R3 forman un sistema de
anillo, o R2 y R4 forman un sistema de anillo, o ambos R1 y R3, y R2 y sistemas de anillo, donde dichos sistemas de anillo se seleccionan independientemente entre -heterociclilo C1-C10 o -carbociclilo C3-C8, y en el que D es un enlace o se selecciona entre el grupo que consiste en -S-, -alquileno C1-C8-, -arileno C6-C14-, -heteroarileno C6-C14-, -heterociclo C r C ^ y -carbociclo C3-C8, donde dichos -alquileno C1-C8-, -arileno
C6-C14-, -heteroarileno C6-C14-, -heterociclo C1-C10 y --carbociclo C3-C8 están opcionalmente sustituidos con -NH2, -N(R)C(O)H o -N(R)C(O)OH.
Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente
documento donde LA se selecciona entre el grupo que consiste en -halo, -N(R)2, -CON(R)2, -S-arilo opcionalmente
sustituido con -NO2 o -CON(R)2, -S-heteroarilo opcionalmente sustituido con -NO2, alquil-SO2-heteroarilo, arilSO2-heteroaril-, y

Lb es l B1-L B2-LB3 en el que LB1 está ausente o es uno o más componentes seleccionados entre el grupo que
consiste en -C(O)-, -C(S)-, -C(O)NR-, -C(O)alquil C1-C6-, -C(O)NRalquil C1-C6-, -alquil C1-C6(OCH2CH2)1-6-, -C(O)alquil C1-C6NRC(O)-, -C(O)alquil C1-C6(OCH2CH2)1-6-, -alquil C1-C6)(OCH2CH2)1-6-C(O)-, -alquil C1-C6-S-S-alquil C1-C6NRC(O)CH2-, -alquil C1-C6(OCH2CH2)1-6NRC(O)CH2-, -C(O)alquil C1-C6-NRC(O)alquil C1-6-, -C(O)-alquil C1-C6(OCH2CH2)1-6NRC(O)-, -C(O)alquil C1-C6-fenil(NR-C(O)alquil C r C6)1-4-, -C(O)alquil C r C6(OCH2CH2)1-6-NRC(O)alquil C1-C6-, -alquil C1-C6-, -S-, -C(O)-CH(NR-C(O)alquil C1-C6)-alquil C1-C6- y (-CH2-CH2-O-)1-20, en el
que LB2 es AA0-12, en el que AA es un aminoácido natural, un aminoácido no natural o -(C R 15)o-S-S-(CR15)p donde
cada uno de o y p es independientemente un número entero de 1 a 20, y LB3 es -PABA-, -PABC-, -C(O)(CH2)nC(O)-o está ausente; y LC está ausente.
Aspectos adicionales de los compuestos desvelados incluyen conjugados anticuerpo-fármaco tales como los
mencionados en el presente documento donde LA se selecciona entre: un enlace a AB, -NR-(enlace a AB), -heteroaril-(enlace a AB),

LB es lb1-L B2-L B3 en el que Lb1 está ausente o es uno o más componentes seleccionados entre el grupo que consiste
en -C(O)-, -C(S)-, -C(O)NR-, -C(O)alquil C1-C6-, -C(O)NRalquil C1-C6-, -alquil C1-C6(OCH2CH2)1-6-, -C(O)alquil C1-C 6NRC(O)-, -C(O)alquil C1-C6(OCH2CH2)1-6-, -alquil C1-C6(OCH2CH2)1-6-C(O)-, -alquil C1-C6-S-S-alquil C1-C6NRC(O)CH2-, -alquil C r C6(OCH2CH2)1-6NRC(O)CH2-, -C(O)alquil C r C 6-NRC(O)alquil C1.6-, -C(O)-alquil C1-C6(OCH2CH2)1-6NRC(O)-, -C(O)alquil C1-Carfenil(NR-C(O)alquil C1-C6)1-4-, -C(O)alquil C1-C6(OCH2CH2)1-6-NRC(O)alquil C1-C6-, -alquil C1-C6-, -S-, -C(O)-CH(NR-C(O)alquil C1-C6)-alquil C1-C6- y (-CH2-CH2-O-)1-20, en el que
LB2 es AA0-12, en el que Aa es un aminoácido natural, un aminoácido no natural o -(C R 15)o-S-S-(CR15)p donde cada
uno de o y p es independientemente un número entero de 1 a 20, y LB3 es -PABA-, -PABC-, -C(O)(CH2)nC(O)- o está
ausente.
Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde RF se selecciona entre:

Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde uno o más W es alquilo C1-C3.
Aspectos adicionales de los í compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde X es cloro.
Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde un Y es H o -C(O)alquilo C1-C10.
Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde uno o más Z es H.
Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde T se selecciona entre una amida o amino-unión-amino de fórmula -NH-C(O)-NH- o -NH-C(O)-het-C(O)-NH-.
Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde la amida es -C(O)NH- o -NHC(O)-.
Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde het es un heteroarilo seleccionado entre pirrol-2-,5-diil-, fur-2 ,5-diil-, indol-2 ,5-diilo, benzofuran-2 ,5-diilo y 3, 6-dihidrobenzo[1,2-b:4 ,3-b]dipirrol-2 ,7-diilo.
Aspectos adicionales de los compuestos desvelados incluyen compuestos tales como los mencionados en el presente documento donde L1 y L2 se seleccionan entre carbonilo, 2-carbonilindolo-5-ilo, 2-carbonil-6-hidroxi-7-metoxiindol-5-ilo, 2-carbonil-1,2 ,3,6-tetrahidrobenzo[1,2-b:4 ,3-b ]dipirrol-7-ilo, 2-carbonil-4-hidroxi-5-metoxi-1,2 ,3,6-tetrahidrobenzo[1,2-b:4 ,3-b']dipirrol-7-ilo, y 2-carbonil-4-hidroxi-5-metoxi-1,2 ,3,6-tetrahidrobenzo[1,2-b:4 ,3-b']dipirrol-7-ilo.
Aspectos adicionales de los compuestos desvelados son aquellos compuestos enumerados en el presente documento donde se aplican uno o más de los siguientes: W es metilo; X es un halógeno; Y es hidrógeno o -CO R donde R es alquilo C1-C10; y Z es hidrógeno.
Los compuestos desvelados también incluyen compuestos como los descritos en el presente documento donde T se selecciona entre una amida (es decir, -C(O)NH- o -NHC(O)-); o un amino-unión-amino de fórmula -NH-T'-NH donde T' es carbonilo o -C-(O)-het-C(O)-. Cuando T es un amino-unión-amino de fórmula NH-T'-NH, T' puede ser carbonilo (es decir, -C-(O)-) o -C(O)-het-C(O)- donde het es un heteroarilo seleccionado entre pirrol-2 ,5-diil-; fur-2 ,5-diil-; indol-2 ,5-diilo; benzofuran-2 ,5-diilo; o 3, 6-dihidrobenzo[1,2-b:4 ,3-b]dipirrol-2 ,7-diilo.
También se incluyen en realizaciones de los compuestos desvelados aquellos compuestos que se han descrito en el presente documento donde L1 y L2 se seleccionan entre 2-carbonilindolo-5-ilo; 2-carbonil-6-hidroxi-7-metoxiindol-5-ilo; 2-carbonil-1,2 , 3, 6-tetrahidrobenzo[1,2-b:4 ,3-b]dipirrol-7-ilo; 2-carbonil-4-hidroxi-5-metoxi-1,2 ,3,6-tetrahidrobenzo[1,2-b:4 ,3-b']dipirrol-7-ilo; y 2-carbonil-4-hidroxi-5-metoxi-1,2 ,3,6-tetrahidrobenzo[1,2-b:4 ,3-b']dipirrol-7-ilo.
Otro aspecto de los compuestos desvelados incluye compuestos como los descritos en el presente documento donde LA es

La invención incluye, también, compuestos enlazador-carga útil o conjugados anticuerpo-fármaco que comprenden un radical de los compuestos carga útil descritos en el presente documento.
Notablemente, la invención incluye composiciones farmacéuticas de los compuestos, y cualquier sal farmacéuticamente aceptables o solvato de los mismos, descritos en el presente documento, donde la composición farmacéutica incluye un excipiente farmacéuticamente aceptable.
La invención también se refiere a compuestos de la invención, o a una composición o composiciones farmacéuticas que comprenden uno o más de estos compuestos, para su uso en el tratamiento del cáncer.
Algunos compuestos, incluyendo cargas útiles, compuestos enlazador-carga útil y ADC representados en el presente documento, se muestran en una forma estereoisomérica específica. La invención, sin embargo, pretende incluir todas las formas estereoisoméricas de estos compuestos. Por ejemplo, un compuesto con dos centros estereoisoméricos puede representarse como la forma R,S del compuesto, pero la invención refleja todas la formas estereoisoméricas, por ejemplo, R,R; R,S; S,R y S,S.
Descripción detallada
La presente invención se refiere a compuestos bifuncionales citotóxicos, a conjugados anticuerpo-fármaco (ADC) que comprenden dichos compuestos bifuncionales citotóxicos y a los mismos para su uso para tratar el cáncer y otras afecciones patológicas. También se desvelan procedimientos de uso de dichos compuestos y/o conjugados in vitro, in situ e in vivo para la detección, diagnóstico y tratamiento de células de mamíferos, afecciones patológicas asociadas.
Definiciones y abreviaturas
A menos que se indique otra cosa, los siguientes términos y frases, tal como se usan en el presente documento, tienen el siguiente significado. Cuando se utilizan marcas comerciales en el presente documento, la marca comercial incluye la formulación del producto, el fármaco genérico y el ingrediente o ingredientes farmacéuticos activos del producto de marca comercial, salvo que se indique otra cosa por el contexto.
El término "anticuerpo" (o "Ab") en el presente documento se usa en el sentido más amplio y abarca específicamente anticuerpos monoclonales intactos, anticuerpos policlonales, anticuerpos monoespecíficos, anticuerpos multiespecíficos (por ejemplo, anticuerpos biespecíficos) y fragmentos de anticuerpos que presentan la actividad biológica deseada. Un anticuerpo intacto tiene principalmente dos regiones: una región variable y una región constante. La región variable se une e interactúa con un antígeno diana. La región variable incluye una región determinante de complementariedad (CDR) que reconoce y se une a un sitio específico de unión a un antígeno particular. La región constante puede ser reconocida e interactuar con el sistema inmunológico (véase, por ejemplo, Janeway y col., 2001, Immuno. Biology, 5a Ed., Garland Publishing, Nueva York). Un anticuerpo puede ser de cualquier tipo o clase (por ejemplo, IgG, IgE, IgM, IgD e IgA) o subclase (por ejemplo, IgG1, IgG2 , IgG3, IgG4 , IgA1 y IgA2). El anticuerpo puede provenir de cualquier especie adecuada. En algunas realizaciones, el anticuerpo es de origen humano o murino. Un anticuerpo puede ser, por ejemplo, humano, humanizado o quimérico.
Las expresiones "se une específicamente" y "unión específica" se refieren a la unión del anticuerpo a un antígeno predeterminado. Por lo general, el anticuerpo se une con una afinidad de al menos aproximadamente 1x107 M-1, y se une al antígeno predeterminado con una afinidad que es al menos dos veces mayor que su afinidad por la unión a un antígeno no específico (por ejemplo, BSA, caseína) que no sea el antígeno predeterminado o un antígeno estrechamente relacionado.
La expresión "anticuerpo monoclonal" tal como se utiliza en el presente documento, se refiere a un anticuerpo obtenido a partir de una población de anticuerpos sustancialmente homogéneos, es decir, los anticuerpos individuales que comprenden la población son idénticos excepto para posibles mutaciones de origen natural que pueden estar presentes en cantidades menores. Los anticuerpos monoclonales son altamente específicos, estando dirigidos contra un solo sitio antigénico. El modificador "monoclonal" indica el carácter del anticuerpo como obtenido de una población de anticuerpos sustancialmente homogénea, y no debe interpretarse como que requiere la producción del anticuerpo por ningún procedimiento particular.
La expresión "anticuerpos monoclonales" incluye específicamente anticuerpos "quiméricos" en los que una porción de la cadena pesada y/o ligera es idéntica a u homóloga con la correspondiente secuencia de anticuerpos derivados de una especie particular o que pertenecen a una clase o subclase particular de anticuerpos, mientras que el resto de la(s) cadena(s) es idéntica a u homóloga con las correspondientes secuencias de anticuerpos derivados de otra
especie o que pertenecen a otra clase o subclase de anticuerpos, así como fragmentos de tales anticuerpos, siempre que presenten la actividad biológica y la especificidad deseada.
Un "anticuerpo intacto" es uno que comprende una región variable de unión a antígeno, así como un dominio constante de cadena ligera (C l) y los dominios constantes de cadena pesada, C h1, C h2, C h3 y C h4, según corresponda para la clase de anticuerpos. Los dominios constantes pueden ser dominios constantes de secuencia natural (por ejemplo, dominios constantes de secuencia natural humana) o variantes de secuencia de aminoácidos de los mismos.
Un anticuerpo intacto puede tener una o más "funciones efectoras", que se refiere a aquellas actividades biológicas atribuibles a la región Fc (por ejemplo, una región Fc de secuencia natural o una región Fc de variante de secuencia de aminoácidos) de un anticuerpo. Los ejemplos de funciones efectoras de anticuerpos incluyen citotoxicidad dependiente del complemento, citotoxicidad celular dependiente de anticuerpos (ADCC, por sus siglas en inglés) y fagocitosis celular dependiente de anticuerpos.
Un "fragmento de anticuerpo" comprende una porción de un anticuerpo intacto, que preferentemente comprende la unión a antígeno o a la región variable de la misma. Los ejemplos de fragmentos de anticuerpos incluyen los fragmentos Fab, Fab', F(ab')2 y Fv, diacuerpos, triacuerpos, tetracuerpos, anticuerpos lineales, moléculas de anticuerpos de cadena sencilla, scFv, scFv-Fc, fragmentos de anticuerpos multiespecíficos formados a partir de fragmentos de anticuerpos, uno o varios fragmentos producidos por una biblioteca de expresión Fab, o un fragmento de unión al epítopo de cualquiera de los anteriores que se unen inmunoespecíficamente a un antígeno diana (por ejemplo, un antígeno de células cancerosas, un antígeno vírico o un antígeno microbiano).
La expresión "variable" en el contexto de un anticuerpo se refiere a ciertas porciones de los dominios variables del anticuerpo que difieren ampliamente en secuencia y se usan en la unión y especificidad de cada anticuerpo particular para su antígeno particular. Esta variabilidad se concentra en tres segmentos llamados "regiones hipervariables" en los dominios variables de la cadena ligera y la cadena pesada. Las partes más altamente conservadas de los dominios variables se denominan regiones marco (FR, por sus siglas en inglés). Los dominios variables de las cadenas pesadas y ligeras naturales comprenden cada uno cuatro FR conectadas por tres regiones hipervariables.
El término "región hipervariable" cuando se usa en el presente documento se refiere a los restos de aminoácidos de un anticuerpo que son responsables de la unión al antígeno. La región hipervariable generalmente comprende restos de aminoácidos de una "región determinante de complementariedad" o "CDR" (por ejemplo, los restos 24-34 (L1), 50 56 (L2) y 89-97 (L3) en el dominio variable de cadena ligera y 31-35 (H1), 50-65 (H2) y 95-102 (L3) en el dominio variable de cadena pesada; Kabat y col. (Sequences of Proteins of Immunological Interest, 5a Ed. Public Health Service, National Institutes of Health, Bethesda, Md. (1991)) y/o los restos de un "bucle hipervariable" (por ejemplo los restos 26-32 (L1), 50-52 (L2) y 91-96 (L3) en el dominio variable de cadena ligera y 26-32 (H1), 53-55 (142) y 96-101 (H3) en el dominio variable de cadena pesada; Chothia y Lesk, 1987, J. Mol. Biol. 196:901- 917). Los restos de FR son aquellos restos de dominio variable distintos de los restos de la región hipervariable tal como se define en el presente documento.
Un fragmento de anticuerpo "Fv de cadena simple" o "scFv" comprende los dominios VH y VL de un anticuerpo, en el que estos dominios están presentes en una sola cadena polipeptídica. Por lo general, el polipéptido de Fv comprende además un enlazador de polipéptido entre los dominios V.sub.H y V.sub.L, lo que permite que scFv forme la estructura deseada para la unión al antígeno. Para una revisión de scFv, véase Pluckthun en The Pharmacology of Monoclonal Antibodies, vol. 113, Rosenburg y Moore eds., Springer-Verlag, Nueva York, págs. 269-315 (1994).
El término "diacuerpo" se refiere a fragmentos de anticuerpos pequeños con dos sitios de unión a antígeno, cuyos fragmentos comprenden un dominio pesado variable (Vh) conectado a un dominio ligero variable (Vl) en la misma cadena polipeptídica. Usando un enlazador que sea demasiado corto para permitir el emparejamiento entre los dos dominios de la misma cadena, se fuerza a los dominios a emparejarse con los dominios complementarios de otra cadena y crear dos sitios de unión a antígeno. Los diacuerpos se describen más completamente en, por ejemplo, el documento EP 0404097; el documento WO 93/11161; y Hollinger y col., 1993, Proc. Natl. Acad. Sci. e E.UU. 90:6444-6448.
Las formas "humanizadas" de anticuerpos no humanos (por ejemplo, roedores) son anticuerpos quiméricos que contienen una secuencia mínima derivada de la inmunoglobulina no humana. En su mayoría, los anticuerpos humanizados son inmunoglobulinas humanas (anticuerpo receptor) en la que los restos de una región hipervariable del receptor son sustituidos por restos de una región hipervariable de una especie no humana (anticuerpo donante), tal como ratón, rata, conejo o primate no humano que tiene la especificidad, afinidad y capacidad deseadas. En algunos casos, los restos de la región marco (FR) de la inmunoglobulina humana están sustituidos por los correspondientes restos no humanos. Además, los anticuerpos humanizados pueden comprender restos que no se encuentran en el anticuerpo receptor o en el anticuerpo donante. Estas modificaciones se efectúan para refinar adicionalmente el funcionamiento del anticuerpo. En general, el anticuerpo humanizado comprenderá sustancialmente la totalidad de al menos uno, y normalmente dos, dominios variables, en los que todos o sustancialmente todos los bucles hipervariables se corresponden con los de una inmunoglobulina no humana y todas o sustancialmente todas las FR son las de una secuencia de inmunoglobulina humana. El anticuerpo humanizado comprenderá también opcionalmente al menos una parte de una región constante de la inmunoglobulina (Fc), normalmente de una inmunoglobulina humana. Para más
detalles, véase Jones y col., 1986, Nature 321:522-525; Riechmann y col., 1988, Nature 332:323-329; y Presta, 1992, Curr. Op. Struct. Biol. 2:593-596.
tal como se usa en el presente documento, "aislado" significa separado de otros componentes de (a) una fuente natural, tal como una célula vegetal o animal o un cultivo celular, o (b) una mezcla de reacción química orgánica sintética. Tal como se usa en el presente documento, "purificado" significa que cuando está aislado, el aislado contiene al menos el 95 % , y en otro aspecto al menos el 98 % , de un compuesto (por ejemplo, un conjugado) en peso del aislado.
Un anticuerpo "aislado" es uno que se ha identificado y separado y/o recuperado a partir de un componente de su ambiente natural. Los componentes contaminantes de su ambiente natural son materiales que podrían interferir con los usos diagnósticos o terapéuticos del anticuerpo y pueden incluir enzimas, hormonas y otros solutos proteicos o no proteicos. En las realizaciones preferidas, el anticuerpo se purificará (1) a más del 95 % en peso de anticuerpo según lo determinado por el procedimiento de Lowry, y lo más preferentemente, a más del 99 % en peso, (2) hasta un grado suficiente para obtener al menos 15 restos de secuencia de aminoácidos N-terminal o interna mediante el uso de un secuenciador de copa giratoria o (3) hasta homogeneidad mediante SD S-P A G E en condiciones reductoras o no reductoras usando azul de Coomassie o, preferentemente, tinción de plata. El anticuerpo aislado incluye el anticuerpo in situ en el interior de células recombinantes ya que al menos un componente del entorno natural del anticuerpo no estará presente. Habitualmente, sin embargo, el anticuerpo aislado se preparará mediante al menos una etapa de purificación.
Un anticuerpo que "induce apoptosis" es uno que induce la muerte celular programada según lo determinado por la unión de anexina V, fragmentación de ADN, contracción celular, dilatación del retículo endoplásmico, fragmentación celular y/o formación de vesículas de membrana (llamadas cuerpos apoptóticos). La célula es una célula tumoral, por ejemplo, una célula de mama, de ovario, de estómago, de endometrio, de glándula salival, de pulmón, de riñón, de colon, de tiroides, de páncreas o de vejiga. Hay varios procedimientos disponibles para evaluar los eventos celulares asociados con la apoptosis. Por ejemplo, la translocación de fosfatidil serina (PS) se puede medir mediante la unión de anexina; la fragmentación del a Dn se puede evaluar a través de marcadores de ADN; y la condensación nuclear/cromatina junto con la fragmentación del ADN pueden evaluarse mediante cualquier aumento en las células hipodiploides.
El término "cantidad terapéuticamente eficaz" se refiere a una cantidad de un fármaco eficaz para tratar una enfermedad o trastorno en un mamífero. En el caso del cáncer, la cantidad terapéuticamente eficaz del medicamento puede reducir la cantidad de células cancerosas; reducir el tamaño del tumor; inhibir (ralentizar hasta cierto punto y preferentemente detener) la infiltración de células cancerosas en los órganos periféricos; inhibir (ralentizar hasta cierto punto y preferentemente detener) la metástasis tumoral; inhibir, hasta cierto punto, el crecimiento tumoral; y/o aliviar hasta cierto punto uno o más de los síntomas asociados con el cáncer. En la medida en que el medicamento pueda inhibir el crecimiento y/o eliminar las células cancerosas existentes, puede ser citostático y/o citotóxico. Para el tratamiento del cáncer, la eficacia puede, por ejemplo, medirse evaluando el tiempo hasta la progresión de la enfermedad (TTP, del inglés time to disease progression) y/o determinando la tasa de respuesta (RR, del inglés response rate).
La expresión "cantidad sustancial" se refiere a una mayoría, es decir, más del 50 % de una población, de una mezcla o una muestra.
La expresión "metabolito intracelular" se refiere a un compuesto que es resultado de un proceso metabólico o reacción dentro de una célula en un conjugado anticuerpo-fármaco (CAF). El proceso o reacción metabólica puede ser un proceso enzimático tal como la escisión proteolítica de un enlazador peptídico del CAF. Los metabolitos intracelulares incluyen, aunque sin limitación, anticuerpos y fármaco libre que se han sometido a escisión intracelular después de la entrada, difusión, captación o transporte a una célula.
Los términos "intracelularmente escindido" y "escisión intracelular" se refieren a un proceso metabólico o reacción dentro de una célula en un CAF o similar, por el cual la unión covalente, por ejemplo, el enlazador, entre el resto del fármaco y el anticuerpo se rompe, dando como resultado el fármaco libre u otro metabolito del conjugado disociado del anticuerpo dentro de la célula. Los restos escindidos del CAF son, por lo tanto, metabolitos intracelulares.
El término "biodisponibilidad" se refiere a la disponibilidad sistémica (es decir, niveles en sangre/plasma) de una cantidad dada de un fármaco administrado a un paciente. La biodisponibilidad es un término absoluto que indica la medición tanto del tiempo (tasa) como de la cantidad total (extensión) del medicamento que alcanza la circulación general desde una forma de dosificación administrada.
La expresión "actividad citotóxica" se refiere a una muerte celular, un efecto citostático o antiproliferativo de un CAF o un metabolito intracelular de dicho CAF. La actividad citotóxica puede expresarse como el valor de CI50 , que es la concentración (molar o de masa) por unidad de volumen a la que sobreviven la mitad de las células.
Un "trastorno" es cualquier afección que se beneficiaría del tratamiento con un fármaco o conjugado de anticuerpofármaco. Esto incluye trastornos crónicos y agudos o enfermedades que incluyen dichas afecciones patológicas que predisponen al mamífero al trastorno en cuestión. Los ejemplos no limitantes de trastornos a tratar en el presente
documento incluyen cánceres benignos y malignos; leucemia y neoplasias linfoides, trastornos neuronales, de la glía, astrocíticos, hipotalámicos y de otras glándulas, macrofágicos, epiteliales, estromáticos y blastocélicos; y trastornos inflamatorios, angiogénicos e inmunológicos.
Los términos "cáncer" y "canceroso" se refieren a o describen la afección fisiológica o el trastorno en mamíferos que se caracteriza típicamente por un crecimiento celular no regulado. Un "tumor" comprende una o más células cancerosas.
Los ejemplos de un "paciente" incluyen, aunque sin limitación, un ser humano, rata, ratón, cobaya, mono, cerdo, cabra, vaca, caballo, perro, gato, pájaros y aves. En una realización ejemplar, el paciente es un ser humano.
Los términos "tratar" o "tratamiento", salvo que se indique otra cosa por el contexto, se refieren al tratamiento terapéutico y medidas profilácticas para evitar la recaída, en el que el objeto es inhibir o ralentizar (disminuir) un cambio o trastorno fisiológico no deseado, como el desarrollo o la propagación del cáncer. Para los fines de la presente invención, los resultados clínicos beneficiosos o deseados incluyen, aunque sin limitación, el alivio de síntomas, la disminución del grado de la enfermedad, el estado patológico estabilizado (es decir, sin empeoramiento), el retraso o la ralentización de la progresión de la enfermedad, la mejora o la atenuación del estado patológico y la remisión (bien total o parcial), ya sea detectable o indetectable. "Tratamiento" también significa prolongar la supervivencia en comparación con la supervivencia esperada si no recibiera tratamiento. Aquellos que necesitan tratamiento incluyen aquellos que ya tienen la afección o trastorno, así como aquellos propensos a tener la afección o trastorno.
En el contexto del cáncer, el término "tratar" incluye cualquiera o todos los inhibidores del crecimiento de células tumorales, células cancerosas o de un tumor; la inhibición de la replicación de células tumorales o células cancerosas, la disminución de la carga tumoral general o la disminución del número de células cancerosas, y la mejora de uno o más síntomas asociados con la enfermedad.
En el contexto de una enfermedad autoinmunitaria, el término "tratar" incluye cualquiera o todos de la inhibición de la replicación de células asociadas con un estado patológico autoinmune que incluye, aunque no de forma limitativa, células que producen un anticuerpo autoinmune, la disminución de la carga de anticuerpos autoinmunes y la mejora de uno o más síntomas de una enfermedad autoinmune.
En el contexto de una enfermedad infecciosa, el término "tratar" incluye cualquiera o todos de: la inhibición del crecimiento, la multiplicación o la replicación del patógeno que causa la enfermedad infecciosa y la mejora de uno o más síntomas de una enfermedad infecciosa.
El término "prospecto" se utiliza para referirse a las instrucciones habitualmente incluidas en los paquetes comerciales de productos terapéuticos, que contienen información sobre las indicaciones, el uso, la dosificación, la administración, las contraindicaciones y/o las advertencias sobre el uso de dichos productos terapéuticos.
Tal como se usa en el presente documento, los términos "célula", "línea celular", y "cultivo celular" se usan indistintamente y todas esas designaciones incluyen la progenie. Las palabras "transformantes" y "células transformadas" incluyen la célula principal y los cultivos o la progenie que proviene de la misma sin tener en cuenta el número de transferencias. También se entiende que toda la progenie puede no ser exactamente idéntica en contenido de ADN, debido a mutaciones deliberadas o inadvertidas. Se incluye la progenie mutante que tiene la misma función o actividad biológica que la seleccionada en la célula transformada originalmente. Cuando se pretenda hacer designaciones distintas, será claro por el contexto.
Tal como se usa en el presente documento, CBI se refiere a 1,2,9,9a-tetrahidro-4H-benzo[e]ciclopropa[c]indol-4-ona, o una forma sustituida o derivada de la misma. CBI también puede referirse a la forma seco de CBI, o seco-CBI, que también se conoce como 1-(clorometil)-2,3-dihidro-1H-benzo[e]indol-5-ol, o una forma (o formas) sustituida o derivatizada de las mismas.
Tal como se usa en el presente documento, CPI se refiere a 1,2,8,8a-tetrahidrociclopropa[c]pirrolo[3,2-e]indol-4(5H)-ona o una forma sustituida o derivatizada de la misma. CPI también puede referirse a la forma seco de CPI, o seco-CPI, que también se conoce como 8-(clorometil)-1-metil-3,6,7,8-tetrahidropirrolo[3,2-e]indol-4-ol, o una forma (o formas) sustituidas o derivadas de las mismas.
A menos que se indique lo contrario, el término "alquilo", por sí mismo o como parte de otro término, se refiere a un hidrocarburo saturado, de cadena lineal o ramificada, que tiene el número indicado de átomos de carbono (por ejemplo, alquilo "C-i-Ce" se refiere a un grupo alquilo que tiene de 1 a 8 átomos de carbono). Típicamente, los grupos alquilo contienen de 1 a 20 átomos de carbono, preferentemente de 1 a 8 átomos de carbono, y más preferentemente de 1 a 4 átomos de carbono. Cuando no se indica el número de átomos de carbono, el grupo alquilo tiene de 1 a 8 átomos de carbono. Los alquilos C-i-Ce de cadena lineal representativos incluyen, pero sin limitación, metilo, etilo, n-propilo, nbutilo, n-pentilo, n-hexilo, n-heptilo y n-octilo; mientras que los alquilos C-i-Ce ramificados incluyen, pero sin limitación, -isopropilo, -sec-butilo, -isobutilo, -terc-butilo, -isopentilo y -2-metilbutilo; los alquilos C 2-C 8 insaturados incluyen, pero sin limitación, vinilo, alilo, 1 -butenilo, 2-butenilo, isobutilenilo, 1-pentenilo, 2-pentenilo, 3-metil-1-butenilo, 2-metil-2-butenilo, 2,3-dimetil-2-butenilo, 1-hexilo, 2-hexilo, 3-hexilo, acetilenilo, propinilo, 1 -butinilo, 2-butinilo, 1 -pentinilo, 2pentinilo y 3-metil-1-butinilo. La referencia a "alquilo" en el presente documento se refiere a restos sin sustituir y sustituidos, como se han descrito anteriormente.
A menos que se indique otra cosa, "alquileno", por sí mismo o como parte de otro término, se refiere a un radical de hidrocarburo saturado de cadena lineal o ramificada o cíclico del número indicado de átomos de carbono, típicamente 1-18 átomos de carbono, y que tiene dos centros de radicales monovalentes derivados mediante la eliminación de dos átomos de hidrógeno de los mismos o dos átomos de carbono diferentes de un alcano original.) Los grupos alquileno comprenden típicamente de 1 a 18 átomos de carbono, preferentemente de 1 a 10 átomos de carbono, más preferentemente de 1 a 8 átomos de carbono, y lo más preferentemente de 1 a 4 átomos de carbono. Los radicales alquileno típicos incluyen, pero sin limitación: metileno (-CH 2 -), 1,2-etileno (-CH 2 CH 2-), 1,3-propileno (-CH 2 CH 2 CH 2 -), 1,4-butileno (-CH 2 CH 2 CH 2 CH 2 -) y similares. Un alquileno "C1 -C 10 " de cadena lineal es un grupo hidrocarburo saturado, de cadena lineal, de fórmula -( ^ 2 )1 -10 -. Los ejemplos de un alquileno C 1 -C 10 incluyen metileno, etileno, propileno, butileno, pentileno, hexileno, heptileno, ocitileno, nonileno y decaleno. En el presente documento, cualquier mención a "alquileno" se refiere a restos sin sustituir y sustituidos como se han descrito anteriormente.
A menos que se indique otra cosa, el término "heteroalquilo", por sí mismo o junto con otro término, significa, a menos que se indique otra cosa, un hidrocarbono estable de cadena lineal o ramificada, o sus combinaciones, totalmente saturado o que contiene de 1 a 3 grados de insaturación, que consiste en el número indicado de átomos de carbono y de uno a tres heteroátomos seleccionados entre el grupo que consiste en O, N, Si y S, y en el que los átomos de nitrógeno y azufre pueden estar opcionalmente oxidados y el heteroátomo de nitrógeno puede estar opcionalmente cuaternizado. El heteroátomo o heteroátomos O, N y S pueden estar situados en cualquier posición interior del grupo heteroalquilo. El heteroátomo Si puede estar situado en cualquier posición del grupo heteroalquilo, incluyendo la posición en la que el grupo alquilo está unido al resto de la molécula. Hasta dos heteroátomos pueden ser consecutivos. Los grupos heteroalquilo comprenden típicamente de 1 a 15 átomos de carbono, preferentemente de 1 a 12 átomos de carbono, más preferentemente de 1 a 8 átomos de carbono, y lo más preferentemente de 1 a 4 átomos de carbono. Cualquier mención a "heteroalquilo" en el presente documento se refiere a restos sin sustituir y sustituidos como se ha descrito anteriormente.
A menos que se indique otra cosa, el término "heteroalquileno", por sí mismo o como parte de otro sustituyente, significa un grupo divalente obtenido a partir de heteroalquilo (como se ha tratado anteriormente). Para los grupos heteroalquileno, los heteroátomos también pueden ocupar cualquiera o ambos extremos de la cadena. Cualquier mención a "heteroalquileno" en el presente documento se refiere a restos sin sustituir y sustituidos como se ha descrito anteriormente.
Típicamente, el término H o hidrógeno en el presente documento se refiere a un átomo de hidrógeno que comprende un solo protón y ningún neutrón, pero también incluye el isótopo de hidrógeno conocido como deuterio que comprende un solo protón y un solo neutrón.
A menos que se indique otra cosa, "arilo", por sí mismo o una parte de otro término, significa un radical de hidrocarburo aromático, carbocíclico, monovalente, sustituido o sin sustituir, de 5-20, preferentemente 5-14 o 6-14, átomos de carbono, obtenido a partir de la eliminación de un átomo de hidrógeno de un solo átomo de carbono de un sistema de anillo aromático parental. Los grupos arilo típicos incluyen, pero sin limitación, radicales derivados de benceno, benceno sustituido, naftaleno, antraceno, bifenilo y similares. Un grupo aromático carbocíclico sustituido (por ejemplo, un grupo arilo) puede estar sustituido con uno o más, preferentemente de 1 a 5, de los siguientes grupos: alquilo C 1 -C b, -O-(alquilo CrCa), -C(O )R9, -OC(O)R9, -C(O)OR9, -C(O)NH2 , -C(O)NHR', -C(O)N(R')2 , -NHC(O)R', -S(O)2 R', -S(O)R', -OH, halógeno, -N 3 , -NH 2 , -NH(R9), -N(R9 )2 y -CN; en los que cada R9 se selecciona independientemente entre -H, alquilo C 1 -C 8 y arilo sin sustituir. En algunas realizaciones, un grupo aromático carbocíclico sustituido puede incluir adicionalmente uno o más de: -NHC(=NH)NH2 , -NHCONH 2 , -S(=O)2 R9 y -S R 9. "Arileno" es el resto divalente correspondiente.
"Alquilo sustituido" (o "alquileno sustituido", "heteroalquilo sustituido" o "heteroalquileno sustituido") significa un grupo o radical que contiene alquilo adecuado como se ha analizado anteriormente en el que uno o más átomos de hidrógeno se reemplazan, cada uno independientemente, por un sustituyente. Los sustituyentes típicos incluyen, pero sin limitación, -X, -R 10, -O-, -O R 10, -S R 10, -S-, -N R 102 , -N R 103 , =NR10, -C X 3 , -CN, -OCN, -SCN , -N=C=O, -N CS, -NO, -NO 2 , =N2, -N3, -N R10C(=O)R10R10 -C(=O)NR102, -SO3-, -SO3H -S(=O)2R10 , -OS(=O)2OR10, -S(=O)2NR10, -S(=O)R10, -OP(=O)(OR10)2, -P(=O)(OR10)2, -PO32- PO 3 H2 , -AsO 2 H2 , -C ( O)R10, -C( O)X, -C(=S)R 10, -C O 2 R 10 , -C O 2 -, -C(=S)O R10, -C(=O )SR10, -C (=S)SR 10, -C(=O)NR10 -C(=S)N R 102 o -C(=N R10)N R1 , en los que cada X es independientemente un halógeno: -F, -CI, -Br o -I; y cada R10 es independientemente -H, alquilo C 1 -C 20 , heteroalquilo C 1 -C 20 , arilo C 6-C 20 , heterociclilo C 1 -C 10 , un grupo protector o un resto de profármaco. Los grupos arilo, alquileno y heteroalquileno como se ha descrito anteriormente también pueden estar sustituidos de manera similar.
A menos que se indique otra cosa, "aralquilo", por sí mismo o como parte de otro término, significa un grupo alquilo, como se ha definido anteriormente, sustituido con un grupo arilo, como se ha definido anteriormente.
A menos que se indique otra cosa, "heterociclilo C 3-C 10 ", por sí mismo o como parte de otro término, se refiere a un sistema de anillo monocíclico, bicíclico o tricíclico, aromático o no aromático, sustituido o sin sustituir, monovalente, que tiene de 2 a 10, de 2 a 14 o 2-20 átomos de carbono, preferentemente de 3 a 8, átomos de carbono (también
denominados miembros de anillo) y de uno a cuatro miembros de anillo heteroátomos seleccionados independientemente entre N, O, P o S, y derivados por la eliminación de un átomo de hidrógeno de un átomo del anillo de un sistema de anillo parental. Uno o más de los átomos de N, C o S en el heterociclilo se pueden oxidar. El anillo que incluye el heteroátomo puede ser aromático o no aromático. En el presente documento, los heterociclos aromáticos se denominan, en algunas ocasiones, heteroarilos. A menos que se indique otra cosa, el heterociclilo está unido a su grupo colgante en cualquier heteroátomo o átomo de carbono que dé como resultado una estructura estable. Los ejemplos representativos de un heterociclilo C 2 -C 10 incluyen, pero sin limitación, tetrahidrofuranilo, oxetanilo, piranilo, pirrolidinilo, piperidinilo, piperazinilo, benzofuranilo, benzotiofeno, benzotiazolilo, indolilo, benzopirazolilo, pirrolilo, tiofenilo (tiofeno), furanilo, tiazolilo, imidazolilo, pirazolilo, triazolilo, quinolinilo, incluyendo restos tales como 1,2,3,4-tetrahidro-quinolinilo, pirimidinilo, piridinilo, piridonilo, pirazinilo, piridazinilo, isotiazolilo, isoxazolilo, tetrazolilo, epóxido, oxetano y BODIPY (sustituido o sin sustituir). Un heterociclilo C 2-C 10 puede estar sustituido hasta con siete grupos incluyendo, pero sin limitación, alquilo C 1 -C 8 , heteroalquilo C 1 -C 8 , -O R 11, arilo, -C(O )R11, -OC(O)R11, -C(O)OR11, -C(O)NH2 , -C(O)NHR11, -C(O)N(R11)2 , -NHC(O)R11, -S(=O)2 R11, -S(O )R11, halógeno, -N 3 , -NH 2 , -NH(R11), -N(R11)2 y -CN; en los que cada R11 se selecciona independientemente entre -H, alquilo C 1 -C 8 , heteroalquilo C 1 -C 8 y arilo. En algunas realizaciones, un heterociclilo sustituido también puede incluir uno o más de: -NHC(=NH)NH2 , -NHCONH2 , -S(=O)2 R11 y -S R 11. Heterociclo o heterociclo C 2 -C 10 es el resto divalente correspondiente. En el presente documento, los heterociclos aromáticos divalentes se denominan, en algunas ocasiones, heteroarileno o heteroarileno C 2 -C 10.
Como se ha indicado anteriormente, En el presente documento, los heterociclos aromáticos se denominan, en algunas ocasiones, heteroarilos, y preferentemente contienen 5-14, 6-14, o 6-20 átomos de carbono además de heteroátomos. Los heteroarilos pueden ser sistemas de anillo monocíclicos, bicíclicos o tricíclicos. Los heteroarilos representativos incluyen, pero sin limitación, triazolilo, tetrazolilo, oxadiazolilo, piridilo, furilo, benzofuranilo, tiofenilo, benzotiofenilo, quinolinilo, pirrolilo, indolilo, oxazolilo, benzoxazolilo, imidazolilo, benzoimidazolilo, tiazolilo, benzotiazolilo, isoxazolilo, pirazolilo, isotiazolilo, piridazinilo, pirimidinilo, pirazinilo, triazinilo, cinnolinilo, ftalazinilo, quinazolinilo, pirimidilo, azepinilo, oxepinilo y quinoxalinilo. Los heteroarilos están opcionalmente sustituidos. Los sustituyentes típicos incluyen, pero sin limitación, -X, -R h, -O-, -O R h, -S R h, -S-, -N R h2 , -N R h3 , =NRh, -C X 3 , -CN, -OCN, -SCN , -N=C=O, -NCS, -NO, -NO 2 , =N2 , -N 3 , -N R hC(=O)Rh, -C(=O)NR h2 , -S O 3-, -SO 3 H, -S(=O)2 Rh, -OS(=O)2 O Rh, -S(=O)2 NRh, -S(=O )Rh, -OP(=O)(ORh)2 , -P(=O)(ORh)2 , -PO 32-, PO3 H2 , -AsO 2 H2 , -C(=O )Rh, -C(=O)X, -C(=S)R h, -C O 2 Rh, -C O 2 -, -C(=S)O Rh, -C(=O )SRh, -C (=S)SR h, -C(=O)NRh2 , -C(=S)N R h2 , -C(=NR)NRh2 , heteroalquilo C 1 -C 20 , arilo C 6-C 20 , heterociclilo C 3-C 8 , un grupo protector o un resto de profármaco, en los que cada X es independientemente un halógeno: -F, -Cl, -Br o -I; y cada Rh es independientemente -H o alquilo C 1 -C 8. En el presente documento, los heterociclos aromáticos divalentes se denominan, en algunas ocasiones, heteroarilenos o heteroarilenos C 1 -C 10.
A menos que se indique otra cosa, "heteroaralquilo", por sí mismo o como parte de otro término, significa un grupo alquilo, como se ha definido anteriormente, sustituido con un grupo heterociclilo aromático, como se ha definido anteriormente. Heteroaralquilo es el resto divalente correspondiente.
A menos que se indique otra cosa, "carbociclilo C 3-C 8", por sí mismo o como parte de otro término, es un anillo carbocíclico, monocíclico o bicíclico, no aromático, saturado o insaturado, sustituido o sin sustituir, monovalente, de 3, 4, 5, 6, 7 u 8 miembros, derivado mediante la eliminación de un átomo de hidrógeno de un átomo del anillo de un sistema de anillo parental. Los grupos carbociclilo C 3-C 8 representativos incluyen, pero sin limitación, ciclopropilo, ciclobutilo, ciclopentilo, ciclopentadienilo, ciclohexilo, ciclohexenilo, 1,3-ciclohexadienilo, 1,4-ciclohexadienilo, cicloheptilo, 1,3-cicloheptadienilo, 1,3,5-cicloheptatrienilo, ciclooctilo, ciclooctadienilo, biciclo(1.1.1.)pentano y biciclo(2.2.2.)octano. Un grupo carbociclilo C 3-C 8 puede estar sin sustituir o sustituido hasta con siete grupos, incluyendo, pero sin limitación, alquilo C 1 -C 8 , heteroalquilo C 1 -C 8 , -O R 11, arilo, -C(O )R11, -O C(O )R11, -C(O)OR11, -C(O)NH2 , -C(O)NHR11, -C(O)N(R11)2 , -NHC(O)R11, -S(=O)2 R11, -S(=O )R11, -OH, -halógeno, -N 3 , -NH2 , -NH(R11), -N(R11)2 y -CN; donde cada R11 se selecciona independientemente entre -H, alquilo C 1 -C 8, heteroalquilo C 1 -C 8 y arilo. "carbociclo C 3-C 8" es el resto divalente correspondiente.
Como se usa en el presente documento, un sustituyente azido se refiere a -N=N=N; un sustituyente cianato se refiere a -O-CN; un sustituyente tiocianato se refiere a -S-CN ; un sustituyente isocianato se refiere a -N=C=O; y un sustituyente tioisocianato se refiere a -S-N=C=O .
El término "quiral" se refiere a moléculas que tienen la propiedad de no superposición del compañero de imagen especular, mientras que el término "aquiral" se refiere a moléculas que pueden superponerse sobre su compañero de imagen especular.
El término "estereoisómeros" se refiere a compuestos que tienen idéntica constitución química, pero se diferencian con respecto a la disposición de los átomos o grupos en el espacio.
"Diastereómero" se refiere a un estereoisómero con dos o más centros de quiralidad y cuyas moléculas no son imágenes especulares entre sí. Los diastereómeros tienen propiedades físicas diferentes, por ejemplo, puntos de fusión, puntos de ebullición, propiedades espectrales y reactividades. Las mezclas de diastereómeros pueden separase en procedimientos analíticos de alta resolución tales como electroforesis y cromatografía.
"Glicosilo" se refiere a la estructura:

o formas sustituidas de la misma, por  de referencia sustituidas para formar estructuras tales como:
de referencia sustituidas para formar estructuras tales como:

y muchas otras.
Las definiciones y convenciones estereoquímicas usadas en el presente documento generalmente siguen S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms, McGraw-Hill Book Company, Nueva York (1984); y Eliel and Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., Nueva York (1994). Muchos compuestos orgánicos existen en formas ópticamente activas, es decir, tienen la capacidad de rotar el plano de luz polarizada en el plano. A la hora de describir un compuesto ópticamente activo, los prefijos D y L o R y S se usan para indicar la configuración absoluta de la molécula alrededor de su centro o centros quirales. Los prefijos d y I o (+) y (-) se emplean para designar el signo de rotación del plano de luz polarizada por el compuesto, de forma que (-) o 1 significa que el compuesto es levógiro. Un compuesto con el prefijo (+) o d es dextrógiro. Para una estructura química dada, estos estereoisómeros son idénticos, con la excepción de que son imágenes especulares entre sí. Un estereoisómero específico también puede denominarse un enantiómero, y una mezcla de dichos isómeros normalmente se denomina mezcla enantiomérica. Una mezcla 50:50 de enantiómeros se denomina mezcla racémica o racemato, que puede aparecer cuando no ha habido estereoselección o estereoespecificidad en una reacción o procedimiento químico. Los términos "mezcla racémica" y "racemato" se refieren a una mezcla equimolar de dos especies enantioméricas, desprovista de actividad óptica.
Como se usa en el presente documento, "-PABA-" o "PABA" se refiere al ácido p-aminobenzoico y restos derivados del mismo, por ejemplo, la estructura:

o variantes de la misma.
Como se usa en el presente documento, "-PABC-" o "PABC" se refiere a p-aminobenciloxicarbonilo y restos derivados del mismo, por ejemplo, la estructura:

o variantes de la misma.
Un "derivado" aminoacídico incluye un aminoácido que tiene sustituciones o modificaciones por unión covalente de un aminoácido parental, tal como, por ejemplo, por alquilación, glicosilación, acetilación, fosforilación y similares. También se incluyen dentro de la definición de "derivado", por ejemplo, uno o más análogos de aminoácido con engarces sustituidos, así como otras modificaciones conocidas en la técnica.
Un "aminoácido natural" se refiere a arginina, glutamina, fenilalanina, tirosina, triptófano, lisina, glicina, alanina, histidina, serina, prolina, ácido glutámico, ácido aspártico, treonina, cisteína, metionina, leucina, asparagina, isoleucina y valina, a menos que se indique otra cosa por el contexto.
"Grupo protector" se refiere a un resto que, cuando se une a un grupo reactivo en una molécula, enmascara, reduce o previene esa reactividad. Pueden encontrarse ejemplos de grupos protectores en T. W. Greene y P. G. M. Wuts, Protective Groups in Organic Synthesis, 3a edición, John Wiley & Sons, Nueva York, 1999, y Harrison and Harrison y col., Compendium of Synthetic Organic Methods, vol. 1-8 (John Wiley y Sons, 1971-1996). Los grupos protectores de hidroxi representativos incluyen grupos acilo, bencilo y éteres de tritilo, éteres de tetrahidropiranilo, éteres de trialquilsililo y éteres de alilo. Los grupos protectores de amino representativos incluyen, formilo, acetilo, trifluoroacetilo, bencilo, benciloxicarbonilo (CBZ), ferc-butoxicarbonilo (Boc), trimetil sililo (TMS), 2-trimetilsilil-etanosulfonilo (SES), grupos tritilo y tritilo sustituidos, aliloxicarbonilo, 9-fluorenilmetiloxicarbonilo (FMOC), nitro-veratriloxicarbonilo (NVOC) y similares.
Los ejemplos de un "grupo protector de hidroxilo" incluyen, pero sin limitación, metoximetil éter, 2-metoxietoximetil éter, tetrahidropiranil éter, bencil éter, p-metoxibencil éter, trimetilsilil éter, trietilsilil éter, triisopropil silil éter, tbutildimetil silil éter, trifenilmetil silil éter, éster de acetato, ésteres de acetato sustituidos, pivaloato, benzoato, metanosulfonato y p-toluenosulfonato.
"Grupo saliente" se refiere a un grupo funcional que puede estar sustituido con otro grupo funcional. Dichos grupos salientes son bien conocidos en la técnica, y sus ejemplos incluyen, pero sin limitación, un haluro (por ejemplo, cloruro, bromuro, yoduro), metanosulfonilo (mesilo), p-toluenosulfonilo (tosilo), trifluorometilsulfonilo (triflato) y trifluorometilsulfonato.
La expresión "sal farmacéuticamente aceptable", como se usa en el presente documento, se refiere a sales orgánicas o inorgánicas farmacéuticamente aceptables de un compuesto. El compuesto típicamente contiene al menos un grupo amino y, por consiguiente, pueden formarse sales de adición de ácidos con este grupo amino. Las sales ejemplares incluyen, pero sin limitación, sales sulfato, citrato, acetato, oxalato, cloruro, bromuro, yoduro, nitrato, bisulfato, fosfato, fosfato ácido, isonicotinato, lactato, salicilato, citrato ácido, tartrato, oleato, tanato, pantotenato, bitartrato, ascorbato, succinato, maleato, malato, gentisinato, fumarato, gluconato, glucuronato, sacarato, formiato, benzoato, glutamato, metanosulfonato, etanosulfonato, bencenosulfonato, p-toluenosulfonato y pamoato (es decir, 1,1'-metileno-bis-(2-hidroxi-3-naftoato)). Una sal farmacéuticamente aceptable puede implicar la inclusión de otra molécula, tal como un ion acetato, un ion succinato u otro contraión. El contraión puede ser cualquier resto orgánico o inorgánico que estabilice la carga en el compuesto parental. Además, una sal farmacéuticamente aceptable puede tener más de un átomo cargado en su estructura. Los casos en los que múltiples átomos cargados son parte de la sal farmacéuticamente aceptable pueden tener múltiples contraiones. Por lo tanto, una sal farmacéuticamente aceptable puede tener uno o más átomos cargados y/o uno o más contraiones.
"Solvato farmacéuticamente aceptable" o "solvato" se refieren a una asociación de una o más moléculas de disolvente y un compuesto o conjugado de la invención. Los ejemplos de disolventes que forman solvatos farmacéuticamente aceptables incluyen, pero sin limitación, agua, isopropanol, etanol, metanol, DMSO, acetato de etilo, ácido acético y etanolamina.
Los términos "carga" o "carga de fármaco" o "carga útil" representan o se refieren al número promedio de cargas útiles ("carga útil" y "cargas útiles" se usan de forma intercambiable en el presente documento con "fármaco" y "fármacos") por anticuerpo en una molécula de CAF. La carga de fármaco puede variar de 1 a 20 fármacos por anticuerpo. Esto a veces se cita como el DAR, o la proporción de fármaco frente a anticuerpo (por sus siglas en inglés). Las composiciones de los CAF descritos en el presente documento típicamente tienen DAR de 1-20, y en ciertas realizaciones de 1-8, de 2-8, de 2-6, de 2-5 y de 2-4. Los valores típicos de DAR son 2, 4, 6 y 8. El número promedio de fármacos por anticuerpo, o valor DAR, puede caracterizarse por medios convencionales tales como la espectroscopia UV/visible, espectrometría de masas, ensayo ELISA y HPLC. El valor DAR cuantitativo también se puede determinar. En algunos casos, la separación, la purificación y la caracterización de CAF homogéneos que tienen un valor DAR particular se puede lograr por medios tales como HpLC de fase inversa o electroforesis. El d A r puede estar limitado por el número de sitios de unión en el anticuerpo. Por ejemplo, cuando la unión es un tiol de cisteína, un anticuerpo puede tener solo uno o varios grupos tiol de cisteína, o puede tener solo uno o varios grupos tiol suficientemente reactivos a través de los cuales se puede unir una unidad enlazadora. En algunas realizaciones, el tiol de cisteína es un grupo tiol de un resto de cisteína que forma un enlace disulfuro intercatenario. En algunas realizaciones, el tiol de cisteína es un grupo tiol de un resto de cisteína que no forma un enlace disulfuro intercatenario. Por lo general, menos que el máximo teórico de restos de fármaco se conjugan con un anticuerpo durante una reacción de conjugación. Un anticuerpo puede contener, por ejemplo, muchos restos de lisina que no reaccionan con un enlazador o un intermediario de enlazador. Solo los grupos de lisina más reactivos pueden reaccionar con un reactivo enlazador que es reactivo.
Generalmente, los anticuerpos no contienen muchos, en caso de haberlos, grupos tiol de cisteína libres y reactivos que pueden estar unidos a un fármaco a través de un enlazador. La mayoría de los restos de tiol de cisteína en los anticuerpos existen como puentes disulfuro y deben reducirse con un agente reductor tal como el ditiotreitol (DTT). El anticuerpo puede someterse a condiciones desnaturalizantes para revelar grupos nucleofílicos reactivos tales como lisina o cisteína. La carga (proporción fármaco/anticuerpo) de un CAF puede controlarse de varias maneras diferentes, que incluyen: (i) limitar el exceso molar de fármaco-enlazador en relación con el anticuerpo, (ii) limitar el tiempo o temperatura de reacción de conjugación, y (iii) condiciones reductoras parciales o limitantes para la modificación del tiol de cisteína. Cuando más de un grupo nucleófilo reacciona con un fármaco-enlazador, el producto resultante es
una mezcla de CAF con una distribución de uno o más restos de fármacos por anticuerpo. El número promedio de fármacos por anticuerpo puede calcularse a partir de la mezcla mediante, por ejemplo, doble ensayo de anticuerpos ELISA, específico para anticuerpo y específico para el fármaco. Los CAF individuales pueden identificarse en la mezcla por espectroscopía de masas y separarse por HPLC, por ejemplo, cromatografía de interacción hidrofóbica.
A continuación se muestra una lista de abreviaturas y definiciones que de otra manera no se podrían definir o describir en la presente solicitud: DMSO (se refiere a dimetilsulfóxido), HRMS (se refiere a espectrometría de masas de alta resolución), DAD (se refiere a la detección de matriz de diodos), TFA (se refiere al ácido 2,2,2-trifluoroacético o ácido trifluoroacético), TFF (se refiere a filtración por flujo tangencial), EtOH (se refiere a etanol), PM (se refiere al peso molecular), HPLC (se refiere a la cromatografía líquida de alto rendimiento), HPLC de preparación (se refiere a cromatografía líquida preparativa de alto rendimiento), etc. (se refiere a y así sucesivamente), tritilo (se refiere a 1,1',1"-etano-1,1,1-triiltribenceno), THF (se refiere a tetrahidrofurano), NHS (se refiere a 1-hidroxi-2,5-pirrolidinediona), Cbz (se refiere a carboxibencilo), eq. (se refiere al equivalente), n-BuLi (se refiere a n-butil litio), OAc (se refiere a acetato), MeOH (se refiere a metanol), i-P r (se refiere a isopropilo o propan-2-ilo), NMM (se refiere a 4-metilmorfolina) y "-" (en una tabla se refiere a que no hay datos disponibles en este momento).
Los restos y sustituyentes divalentes usados en el presente documento pretenden referirse a dichos restos o sustituyentes unidos o enlazados en cualquier dirección o en ambas direcciones. Por ejemplo, el resto -C(O)NR- (en la definición de LB1, y en otros lugares) está destinado a transmitir -C(O)NR- así como -NRC(O)-, el resto -C(O)-alquilo C 1 -C 6- está destinado a transmitir -C(O)C-alquilo C 1 -C 6, así como -alquil-C 1 -C 6-C(O)-, y así sucesivamente. Más generalmente, una descripción de un resto divalente no simétrico enlazado en sus lados "izquierdo" y "derecho" pretende transmitir tanto el resto como se presenta (lado izquierdo del resto enlazado en el lado izquierdo tal como está escrito, lado derecho del resto enlazado en el lado derecho tal como está escrito) y el inverso del resto tal como se presenta (lado izquierdo del resto enlazado en el lado derecho tal como está escrito, el lado derecho del resto enlazado en el lado izquierdo tal como está escrito).
Los términos "enlace" y "ausente" se usan en el presente documento para describir una variable que no incluye un átomo o átomos. Por lo tanto, cuando se dice que una variable divalente está "ausente" se entiende que los restos adyacentes están unidos entre sí. Por ejemplo, si LB2 está ausente se entiende que LB1 puede estar unido a LB3; o si LB1 y yoB2 ambos están ausentes se entiende que LA puede estar unido a LB3. De manera similar, si una variable divalente se define como un "enlace", se entiende que esto significa que no hay átomos presentes y que los restos adyacentes están unidos entre sí. Por lo tanto, por ejemplo, cuando la variable "D" se define como un enlace, se aprecia que los carbonos adyacentes a D (en la estructura que define T2) están unidos entre sí. Se entiende que una variable monovalente ausente es un hidrógeno o un par de electrones capaz de una unión covalente adicional.
La unidad de anticuerpo (A, Ab o AB)
Como se indica anteriormente, el término "anticuerpo" (o "A", "Ab" o "AB") se usa en el presente documento en el sentido más amplio y abarca específicamente anticuerpos monoclonales intactos, anticuerpos policlonales, anticuerpos monoespecíficos, anticuerpos multiespecíficos (por ejemplo, anticuerpos biespecíficos) y fragmentos de anticuerpos que presentan la actividad biológica deseada. Además, mientras que ciertos aspectos de la invención descritos en el presente documento se refieren a conjugados de anticuerpo-fármaco, se prevé además que la porción de anticuerpo del conjugado podría reemplazarse con cualquier cosa que se una específicamente o que se asocie o forme complejos reactivamente con un receptor, antígeno u otro resto receptivo asociado con una población de células diana dada. Por ejemplo, en lugar de contener un anticuerpo, los conjugados de la invención podrían contener una molécula de direccionamiento que se une a, forma complejos con, o reacciona con un receptor, antígeno u otro resto receptivo de una población celular que se busca que sea terapéuticamente o biológicamente modificada. Los ejemplos de tales moléculas incluyen proteínas de menor peso molecular, polipéptido o péptidos, lectinas, glucoproteínas, no péptidos, vitaminas, moléculas de transporte de nutrientes (tales como, pero no limitado a transferrina), o cualquier otra molécula o sustancia de unión celular. En determinados aspectos, el anticuerpo u otra molécula dirigida de este tipo actúa para administrar un fármaco a la población de células diana particular con la que interactúa el anticuerpo u otra molécula de direccionamiento.
En otro aspecto, la presente invención se refiere a un compuesto de conjugado de anticuerpo-fármaco en el que el anticuerpo AB se selecciona de: trastuzumab, mutantes de trastuzumab (por ejemplo, los mutantes de trastuzumab desvelados en el presente documento o en la solicitud de patente internacional PCT/IB2012/056234), oregovomab, edrecolomab, cetuximab, un anticuerpo monoclonal humanizado para el receptor de vitronectina (ayp3 ), alemtuzumab, anticuerpos anti-HLA-DR que incluyen un anticuerpo anti-HLA-DR humanizado para el tratamiento del linfoma no de Hodgkin, 131l Lym-1, anticuerpos anti-HLA-Dr10 que incluyen un anticuerpo anti-HLA-Dr10 de murino para el tratamiento del linfoma no de Hodgkin, anticuerpos anti-cd33, anticuerpos anti-cd22 que incluyen un mAb anti-CD22 humanizado para el tratamiento de la enfermedad de Hodgkin o el linfoma no de Hodgkin, labetuzumab, bevacizumab, ibritumomab tiuxetán, ofatumumab, panitumumab, rituximab, tositumomab, ipilimumab y gemtuzumab.
Los heteroátomos que puedan estar presentes en una unidad de anticuerpos incluyen azufre (en una realización, de un grupo sulfhidrilo de un anticuerpo), oxígeno (en una realización, de un grupo carbonilo, carboxilo o hidroxilo de un anticuerpo) y nitrógeno (en una realización, de un grupo amino primario o secundario de un anticuerpo). Estos heteroátomos pueden estar presentes en el anticuerpo en el estado natural del anticuerpo, por ejemplo, un anticuerpo
natural, o puede introducirse en el anticuerpo mediante modificación química.
En una realización, una unidad de anticuerpo tiene un grupo sulfhidrilo y la unidad de anticuerpo se une a través del átomo de azufre del grupo sulfhidrilo.
En otra realización, el anticuerpo tiene restos de lisina que pueden reaccionar con los ésteres activados (tales ésteres incluyen, entre otros, N-hidroxisuccinimida, pentafluorofenilo y ésteres de p-nitrofenilo) y, por lo tanto, forman un enlace amida que consiste en el átomo de nitrógeno de la unidad de anticuerpo y un carbonilo.
En otro aspecto más, la unidad de anticuerpo tiene uno o más restos de lisina que pueden modificarse químicamente para introducir uno o más grupos sulfhidrilo. Los reactivos que se pueden usar para modificar lisinas incluyen, aunque sin limitación, N-succinimidil S-acetiltioacetato (SATA) y clorhidrato de 2-iminotiolano (reactivo de Traut).
En otra realización, la unidad de anticuerpos puede tener uno o más grupos de carbohidratos que pueden modificarse químicamente para tener uno o más grupos sulfhidrilo.
En otra realización más, la unidad de anticuerpos puede tener uno o más grupos de carbohidratos que pueden oxidarse para proporcionar un grupo aldehído (véase, por ejemplo, Laguzza y col., 1989, J. Med. Chem. 32(3):548-55). El correspondiente aldehído puede formar un enlace con un sitio reactivo tal como, por ejemplo, hidrazina e hidroxilamina. Otros protocolos para la modificación de proteínas para la unión o asociación de fármacos se describen en Coligan y col., Current Protocols in Protein Science, vol. 2, John Wiley y Sons (2002).
Cuando los conjugados comprenden proteínas no inmunorreactivas, polipéptidos, o unidades peptídicas en lugar de un anticuerpo, la proteína no inmunorreactiva útil, el polipéptido, o las unidades peptídicas incluyen, aunque sin limitación, transferrina, factores de crecimiento epidérmico ("e G f "), bombesina, gastrina, péptido liberador de gastrina, factor de crecimiento derivado de plaquetas, IL-2, IL-6, factores de crecimiento transformadores ("TOP"), tales como T G F -a y TGF-p, factor de crecimiento de vaccinia ("VGF"), factores de crecimiento de insulina y de tipo insulina I y II, somatostatina, lectinas y apoproteína de lipoproteína de baja densidad.
Los anticuerpos policlonales útiles son poblaciones heterogéneas de moléculas de anticuerpos derivadas de los sueros de animales inmunizados. Los anticuerpos monoclonales útiles son poblaciones homogéneas de anticuerpos para un determinante antigénico particular (por ejemplo, un antígeno de células cancerosas, un antígeno vírico, un antígeno microbiano, una proteína, un péptido, un carbohidrato, un compuesto químico, ácido nucleico, o fragmentos de los mismos). Se puede preparar un anticuerpo monoclonal (mAb) contra un antígeno de interés utilizando cualquier técnica conocida en la técnica que proporcione la producción de moléculas de anticuerpo mediante líneas celulares continuas en cultivo.
Los anticuerpos monoclonales útiles incluyen, aunque sin limitación, anticuerpos monoclonales humanos, anticuerpos monoclonales humanizados, fragmentos de anticuerpos o anticuerpos monoclonales quiméricos. Los anticuerpos monoclonales humanos se pueden generar mediante cualquiera de las numerosas técnicas conocidas en la técnica (por ejemplo, Teng y col., 1983, Proc. Natl. Acad. Sci. E E .u U., 80:7308-7312; Kozbor y col., 1983, Immunology Today 4:72-79; y Olsson y col., 1982, Meth. Enzymol. 92:3-16).
El anticuerpo también puede ser un anticuerpo biespecífico. Los procedimientos para generar anticuerpos biespecíficos son conocidos en la técnica y se discuten más adelante.
El anticuerpo puede ser un fragmento funcionalmente activo, derivado o análogo de un anticuerpo que se une inmunoespecíficamente a las células diana (por ejemplo, antígenos de células cancerosas, antígenos víricos, o antígenos microbianos) u otros anticuerpos que se unen a las células tumorales o matriz. En este sentido, "funcionalmente activo" significa que el fragmento, derivado o análogo es capaz de provocar anticuerpos anti-antiidiotipo que reconocen el mismo antígeno que el anticuerpo del que se deriva el fragmento, derivado o análogo. De manera específica, En una realización ejemplar, la antigenicidad del idiotipo de la molécula de inmunoglobulina puede potenciarse mediante la eliminación de las secuencias marco y CDR que están en C-terminal a la secuencia de CDR que reconoce específicamente el antígeno. Para determinar qué secuencias de CDR se unen al antígeno, los péptidos sintéticos que contienen las secuencias de CDR pueden usarse en ensayos de unión con el antígeno mediante cualquier procedimiento de ensayo de unión conocido en la técnica (por ejemplo, el ensayo BIA core) (para la ubicación de las secuencias de CDR, véase, por ejemplo, Kabat y col., 1991, Sequences of Proteins of Immunological Interest, Quinta edición, National Institute of Health, Bethesda, Md.; Kabat E y col., 1980, J. Immunology 125(3):961-969).
Otros anticuerpos útiles incluyen fragmentos de anticuerpos tales como, aunque no de forma limitativa, fragmentos F(ab')2 , fragmentos Fab, Fvs, anticuerpos de cadena simple, diacuerpos, triacuerpos, tetracuerpos, scFv, scFv-FV, o cualquier otra molécula con la misma especificidad que el anticuerpo.
Además, los anticuerpos recombinantes, tales como los anticuerpos monoclonales quiméricos y humanizados, que comprenden porciones tanto humanas como no humanas, que se pueden generar usando técnicas convencionales de ADN recombinante, son anticuerpos útiles. Un anticuerpo quimérico es una molécula en la que diferentes porciones proceden de diferentes especies animales, tales como, por ejemplo, aquellos que tienen una región variable derivada de un monoclonal de murino y regiones constantes de inmunoglobulina humana. (Véase, por ejemplo, la patente de
EE.UU. N.° 4.816.567; y la patente de EE.UU. N.° 4.816.397.) Los anticuerpos humanizados son moléculas de anticuerpos de especies no humanas que tienen una o más regiones determinantes de complementariedad (CDR) de las especies no humanas y una región marco de una molécula de inmunoglobulina humana. (Véase, por ejemplo, la patente de EE.UU. N.° 5.585.089). Tales anticuerpos monoclonales humanizados y quiméricos se pueden producir mediante técnicas de ADN recombinante conocidas en la materia, por ejemplo, utilizando los procedimientos descritos en la Publicación Internacional N.° WO 87/02671; la Publicación de Patente Europea N.° 01 8 4 187; la Publicación de Patente Europea N.° 0171 496; la Publicación de Patente Europea N.° 0173 494; la Publicación internacional N.° WO 86/01533; la Patente de EE.UU. N.° 4.816.567; la Publicación de Patente Europea N.° 012 023; Berter y col., 1988, Science 240:1041-1043; Liu y col., 1987, Proc. Natl. Acad. Sci. EE.UU. 84:3439-3443; Liu y col., 1987, J. Immunol.
139:3521-3526; Sun y col., 1987, Proc. Natl. Acad. Sci. EE.UU. 84:214-218; Nishimura y col., 1987, Cancer. Res.
47:999-1005; Wood y col., 1985, Nature 314:446-449; y Shaw y col., 1988, J. Natl. Cancer Inst. 80:1553-1559; Morrison, 1985, Science 229:1202-1207; Oi y col., 1986, BioTechniques 4:214; Patente de EE.UU. N.° 5.225.539; Jones y col., 1986, Nature 321:552-525; Verhoeyan y col., 1988, Science 239:1534; y Beidler y col., 1988, J. Immunol.
141:4053-4060.
Los anticuerpos completamente humanos son particularmente deseables y pueden producirse usando ratones transgénicos que son incapaces de expresar genes de cadenas pesadas y ligeras de inmunoglobulina endógena, pero que pueden expresar genes humanos de cadena pesada y ligera. Los ratones transgénicos se inmunizan de manera normal con un antígeno seleccionado, por ejemplo, todo o una porción de un polipéptido de la invención. Los anticuerpos monoclonales dirigidos contra el antígeno pueden obtenerse usando tecnología de hibridoma convencional. Los transgenes de inmunoglobulina humana albergados por los ratones transgénicos se reorganizan durante la diferenciación de los linfocitos B y, posteriormente, se someten a cambio de clase y a mutación somática. Por lo tanto, utilizando dicha técnica, es posible producir anticuerpos IgG, IgA, IgM e IgE terapéuticamente útiles. Para una descripción general de esta tecnología para producir anticuerpos humanos, véase Lonberg y Huszar, 1995, Int. Rev. Immunol. 13:65-93. Para una discusión detallada de esta tecnología para producir anticuerpos humanos y anticuerpos monoclonales humanos y los protocolos para producir dichos anticuerpos, véase, por ejemplo, las Pat. de EE.UU. N.° 5.625.126; 5.633.425; 5.569.825; 5.661.016; 5.545.806. Otros anticuerpos humanos se pueden obtener comercialmente de, por ejemplo, Abgenix, Inc. (ahora Amgen, Freemont, Calif.) y Medarex (Princeton, N.J.).
Los anticuerpos completamente humanos que reconocen un epítopo seleccionado se pueden generar utilizando una técnica denominada "selección guiada". En este enfoque, un anticuerpo monoclonal no humano seleccionado, por ejemplo, un anticuerpo de ratón, se usa para guiar la selección de un anticuerpo completamente humano que reconoce el mismo epítopo. (Véase, por ejemplo, Jespers y col., 1994, Biotechnology 12:899-903). Los anticuerpos humanos también se pueden producir usando diversas técnicas conocidas en la materia, incluidas las bibliotecas de presentación en fago (véase, por ejemplo, Hoogenboom y Winter, 1991, J. Mol. Biol. 227:381; Marks y col., 1991, J. Mol. Biol. 222:581; Quan y Carter, 2002, The rise of monoclonal antibodies as therapeutics, In Anti-lgE and Allergic Disease, Jardieu y Fick, eds., Marcel Dekker, Nueva York, N.Y., Capítulo 20, págs. 427-469).
En otras realizaciones, el anticuerpo es una proteína de fusión de un anticuerpo, o un fragmento funcionalmente activo del mismo, por ejemplo, en el que el anticuerpo se fusiona a través de un enlace covalente (por ejemplo, un enlace peptídico), ya sea en el extremo N-terminal o en el extremo C-terminal para una secuencia de aminoácidos de otra proteína (o una porción de la misma, preferentemente) al menos una porción de 10, 20 o 50 aminoácidos de la proteína) que no es de un anticuerpo. Preferentemente, el anticuerpo o fragmento del mismo está unido covalentemente a la otra proteína en el extremo N-terminal del dominio constante.
Los anticuerpos incluyen análogos y derivados que se modifican, es decir, mediante la unión covalente de cualquier tipo de molécula siempre que dicha unión covalente permita que el anticuerpo conserve su inmunoespecificidad de unión al antígeno. Por ejemplo, pero no a modo de limitación, los derivados y análogos de los anticuerpos incluyen aquellos que se han modificado adicionalmente, por ejemplo, mediante glucosilación, acetilación, pegilación, fosforilación, amidación, derivatización mediante grupos protectores/bloqueadores conocidos, escisión proteolítica, enlace a una unidad de anticuerpo celular u otra proteína, etc. Cualquiera de las numerosas modificaciones químicas puede llevarse a cabo mediante técnicas conocidas que incluyen, aunque no de forma limitativa, escisión química específica, acetilación, formilación, síntesis metabólica en presencia de tunicamicina, etc. Además, el análogo o derivado puede contener uno o más aminoácidos no naturales.
Los anticuerpos pueden tener modificaciones (por ejemplo, sustituciones, deleciones o adiciones) en restos de aminoácidos que interactúan con los receptores de Fc. En particular, los anticuerpos pueden tener modificaciones en los restos de aminoácidos identificados como implicados en la interacción entre el anti-dominio Fc y el receptor FcRn (véase, por ejemplo, la Publicación Internacional N.° WO 97/34631).
Los anticuerpos inmunoespecíficos para un antígeno de células cancerosas pueden obtenerse comercialmente o se pueden producir mediante cualquier procedimiento conocido por un experto en la técnica, tal como, por ejemplo, la síntesis química o las técnicas de expresión recombinante. Se puede obtener la secuencia de nucleótidos que codifica los anticuerpos inmunoespecíficos para un antígeno de células cancerosas, por ejemplo, de la base de datos GenBank o de una base de datos similar, de publicaciones de referencias, o por clonación y secuenciación convencional.
En una realización específica, se pueden usar anticuerpos conocidos para el tratamiento del cáncer. Los anticuerpos
inmunoespecíficos para un antígeno de células cancerosas pueden obtenerse comercialmente o se pueden producir mediante cualquier procedimiento conocido por un experto en la técnica, tal como, por ejemplo, mediante técnicas de expresión recombinante. Se puede obtener la secuencia de nucleótidos que codifica los anticuerpos inmunoespecíficos para un antígeno de células cancerosas, por ejemplo, de la base de datos GenBank o de una base de datos similar, las publicaciones de referencias, o por clonación y secuenciación convencional. Los ejemplos de anticuerpos disponibles para el tratamiento del cáncer incluyen, aunque sin limitación, OVAREX, que es un anticuerpo murino para el tratamiento del cáncer de ovario; PAN O REX (Glaxo Wellcome, NC) que es un anticuerpo IgGna2 a de murino para el tratamiento del cáncer colorrectal; Cetuximab ERBITUX (Imclone Systems Inc., n Y), que es un anticuerpo quimérico IgG anti-EGFR para el tratamiento de cánceres positivos del factor de crecimiento epidérmico, como el cáncer de cabeza y cuello; Vitaxin (Medlmmune, Inc., MD) que es un anticuerpo humanizado para el tratamiento del sarcoma; CAMPATH I/H (Leukosite, MA) que es un anticuerpo humanizado IgG1 para el tratamiento de leucemia linfocítica crónica (LLC); SMART ID10 (Protein Design Labs, Inc., CA) que es un anticuerpo humanizado anti-HLA-DR para el tratamiento del linfoma no de Hodgkin; ONCOLYM (Techniclone, Inc., CA), que es un anticuerpo anti-HLA-Dr10 de murino radiomarcado para el tratamiento del linfoma no de Hodgkin; ALLOMUNE (BioTransplant, CA), que es un mAb anti-CD2 humanizado para el tratamiento de la enfermedad de Hodgkin o el linfoma no de Hodgkin; y CEA CID E (Immunomedics, NJ), que es un anticuerpo anti-CEA humanizado para el tratamiento del cáncer colorrectal.
En un intento por descubrir dianas celulares eficaces para el diagnóstico y la terapia del cáncer, los investigadores han tratado de identificar polipéptidos transmembrana o de otro tipo asociados a tumores que se expresan específicamente en la superficie de uno o más tipos particulares de células cancerosas en comparación con una o más células no cancerosas normales. A menudo, dichos polipéptidos asociados a tumores se expresan más abundantemente en la superficie de las células cancerosas en comparación con la superficie de las células no cancerosas. La identificación de dichos polipéptidos de antígeno de superficie celular asociados a tumores ha dado lugar a la capacidad de atacar específicamente a las células cancerosas para su destrucción a través de terapias basadas en anticuerpos.
La unidad enlazadora (L)
Un enlazador (a veces denominado "[enlazador]" en el presente documento) es un compuesto bifuncional que puede usarse para enlazar un fármaco y un anticuerpo para formar un conjugado de anticuerpo-fármaco (c A f ). Tales conjugados son útiles, por ejemplo, en la formación de inmunoconjugados dirigidos contra antígenos asociados a tumores. Dichos conjugados permiten el suministro selectivo de fármacos citotóxicos a las células tumorales.
En un CAF, el enlazador sirve para unir la carga útil al anticuerpo.
En un aspecto, se introduce una segunda sección de la unidad enlazadora que tiene un segundo sitio reactivo, por ejemplo, un grupo electrófilo que es reactivo con un grupo nucleófilo presente en una unidad de anticuerpo (por ejemplo, un anticuerpo). Los grupos nucleófilos útiles en un anticuerpo incluyen, pero sin limitación, los grupos sulfhidrilo, hidroxilo y amino. El heteroátomo del grupo nucleófilo de un anticuerpo es reactivo con un grupo electrófilo en una unidad enlazadora y forma un enlace covalente con una unidad enlazadora. Los grupos electrófilos útiles incluyen, aunque sin limitación, los grupos maleimida y haloacetamida. El grupo electrófilo proporciona un sitio conveniente para la unión de los anticuerpos.
En otra realización, una unidad enlazadora tiene un sitio reactivo que tiene un grupo nucleófilo que es reactivo con un grupo electrófilo presente en un anticuerpo. Los grupos electrófilos útiles en un anticuerpo incluyen, aunque sin limitación, grupos aldehído y cetona carbonilo. El heteroátomo de un grupo nucleófilo de una unidad enlazadora puede reaccionar con un grupo electrofílico en un anticuerpo y formar un enlace covalente con el anticuerpo. Los grupos nucleófilos útiles en una unidad enlazadora incluyen, aunque sin limitación, hidrazida, oxima, amino, hidrazina, tiosemicarbazona, carboxilato de hidrazina y arilhidrazida. El grupo electrófilo en un anticuerpo proporciona un sitio conveniente para la unión con una unidad enlazadora.
Los grupos amino funcionales también son sitios reactivos útiles para una unidad enlazadora porque pueden reaccionar con ácido carboxílico o ésteres activados de un compuesto para formar un enlace amida. Por lo general, los compuestos a base de péptidos de la invención pueden prepararse formando un enlace peptídico entre dos o más aminoácidos y/o fragmentos de péptidos. Tales enlaces peptídicos se pueden preparar, por ejemplo, de acuerdo con el procedimiento de síntesis en fase líquida (véase, por ejemplo, Schroder y Lubke, "The Peptides", volumen 1, págs 76-136, 1965, Academic Press) que es bien conocido en el campo de la química de péptidos.
En el contexto de la invención y de otros compuestos desvelados, particularmente pero no limitado a componentes enlazadores tales como L-i, L2 (incluyendo L2 A, L2 B y L2 C) y L3 , la expresión "seleccionado de uno o más de" o "uno o más de" indica que múltiples componentes, que pueden ser el mismo o diferentes, están o se pueden organizar secuencialmente. Por lo tanto, por ejemplo, L3 puede ser -alquil C 1 -6 -, -N R- u otros componentes enumerados individualmente, pero también -alquil C 1 -6 -NR-, o cualquier otra combinación de 2 o más componentes listados. En otra realización, una unidad enlazadora tiene un sitio reactivo que puede reaccionar con nucleófilos de anticuerpos, tales como las cisteínas. El sitio reactivo está compuesto por un heterociclo que está sustituido con una sulfona. La
sulfona se reemplaza luego por el nucleófilo del anticuerpo (es decir, la cisteína) y el enlace recién formado entre el anticuerpo y el heterociclo conecta el anticuerpo con el enlazador. Véase, el documento WO 2014/144878.
Síntesis de compuestos y conjugados de anticuerpo-fármaco de los mismos
Los compuestos y conjugados de la invención y otros compuestos desvelados se pueden preparar usando los procedimientos de síntesis desvelados a continuación en la Ejemplificación. Tal como se describe con más detalle a continuación, los compuestos y conjugados de la invención pueden prepararse usando una sección de una unidad enlazadora que tiene un sitio reactivo para la unión al compuesto. En un aspecto, se introduce una segunda sección de la unidad enlazadora que tiene un segundo sitio reactivo, por ejemplo, un grupo electrófilo que es reactivo con un grupo nucleófilo presente en una unidad de anticuerpo (por ejemplo, un anticuerpo). Los grupos nucleófilos útiles en un anticuerpo incluyen, pero sin limitación, los grupos sulfhidrilo, hidroxilo y amino. El heteroátomo del grupo nucleófilo de un anticuerpo es reactivo con un grupo electrófilo en una unidad enlazadora y forma un enlace covalente con una unidad enlazadora. Los grupos electrófilos útiles incluyen, aunque sin limitación, los grupos maleimida y haloacetamida. El grupo electrófilo proporciona un sitio conveniente para la unión de los anticuerpos.
En otra realización, una unidad enlazadora tiene un sitio reactivo que tiene un grupo nucleófilo que es reactivo con un grupo electrófilo presente en un anticuerpo. Los grupos electrófilos útiles en un anticuerpo incluyen, aunque sin limitación, grupos aldehído y cetona carbonilo. El heteroátomo de un grupo nucleófilo de una unidad enlazadora puede reaccionar con un grupo electrofílico en un anticuerpo y formar un enlace covalente con el anticuerpo. Los grupos nucleófilos útiles en una unidad enlazadora incluyen, aunque sin limitación, hidrazida, oxima, amino, hidrazina, tiosemicarbazona, carboxilato de hidrazina y arilhidrazida. El grupo electrófilo en un anticuerpo proporciona un sitio conveniente para la unión con una unidad enlazadora.
Los grupos amino funcionales también son sitios reactivos útiles para una unidad enlazadora porque pueden reaccionar con ácido carboxílico o ésteres activados de un compuesto para formar un enlace amida. Por lo general, los compuestos a base de péptidos de la invención pueden prepararse formando un enlace peptídico entre dos o más aminoácidos y/o fragmentos de péptidos. Tales enlaces peptídicos se pueden preparar, por ejemplo, de acuerdo con el procedimiento de síntesis en fase líquida (véase, por ejemplo, Schroder y Lubke, "The Peptides", volumen 1, págs 76-136, 1965, Academic Press) que es bien conocido en el campo de la química de péptidos.
Tal como se describe con más detalle a continuación, los conjugados pueden prepararse usando una sección del enlazador que tiene un sitio reactivo para la unión a un compuesto de la invención e introducir otra sección de la unidad enlazadora que tiene un sitio reactivo para un anticuerpo. En un aspecto, una unidad enlazadora tiene un sitio reactivo que tiene un grupo electrófilo que es reactivo con un grupo nucleófilo presente en una unidad de anticuerpos, tal como un anticuerpo. El grupo electrófilo proporciona un sitio conveniente para la unión de los anticuerpos. Los grupos nucleófilos útiles en un anticuerpo incluyen, pero sin limitación, los grupos sulfhidrilo, hidroxilo y amino. El heteroátomo del grupo nucleófilo de un anticuerpo es reactivo con un grupo electrófilo en una unidad enlazadora y forma un enlace covalente con una unidad enlazadora. Los grupos electrófilos útiles incluyen, aunque sin limitación, los grupos maleimida y haloacetamida.
En otra realización, una unidad enlazadora tiene un sitio reactivo que tiene un grupo nucleófilo que es reactivo con un grupo electrófilo presente en una unidad de anticuerpo. El grupo electrófilo en un anticuerpo proporciona un sitio conveniente para la unión con una unidad enlazadora. Los grupos electrófilos útiles en un anticuerpo incluyen, aunque sin limitación, grupos aldehído y cetona carbonilo. El heteroátomo de un grupo nucleófilo de una unidad enlazadora puede reaccionar con un grupo electrofílico en un anticuerpo y formar un enlace covalente con el anticuerpo. Los grupos nucleófilos útiles en una unidad enlazadora incluyen, aunque sin limitación, hidrazida, oxima, amino, hidrazina, tiosemicarbazona, carboxilato de hidrazina y arilhidrazida.
En otra realización de la invención, el enlace de la presente invención emplea un polipéptido de dominio constante de anticuerpo diseñado genéticamente, o una porción del mismo, en la que el dominio constante diseñado genéticamente comprende al menos una sustitución de aminoácidos para introducir un resto de cisteína útil para la conjugación, y en el que el polipéptido de dominio constante es un polipéptido del dominio constante (Cy) de cadena pesada de IgG humana diseñada genéticamente, o una porción del mismo, que comprende al menos una sustitución de aminoácidos seleccionada del grupo que consiste en K246, D249, D265, S267, D270, N276, Y278, E283, R292, E293, E294, Y300, V302, V303, L314, N315, E318, K320, I332, E333, K334, 1336, E345, Q347, S354, R355, M358, K360, Q362, K370, Y373, D376, A378, E380, E382, Q386, E388, N390, K392, T393, D401, F404, T 411, D413, K414, R416, Q418, Q419, N421, M428, A431, L432, T437, Q438, K439, L443 y S444, según el índice EU de Kabat.
En otra realización de la invención, el enlace de la presente invención emplea un polipéptido de dominio constante de anticuerpo diseñado genéticamente, o una porción del mismo, en el que el dominio constante diseñado genéticamente comprende al menos una sustitución de aminoácidos para introducir un resto de cisteína útil para la conjugación, y en el que el polipéptido de dominio constante es un polipéptido de dominio constante de cadena ligera (CA) lambda humana diseñada genéticamente, o una porción del mismo, que comprende al menos una sustitución de aminoácidos seleccionada del grupo que consiste en K110, A 111, L125, K149C, V155, G158, T 161, Q185, S188, H189, S191, T197, V205, E206, k 207, T208 y A210, según la numeración de Kabat.
En otra realización más de la invención, el enlace de la presente invención emplea un polipéptido de dominio constante de anticuerpo modificado, o una porción del mismo, en el que el dominio constante diseñado genéticamente comprende al menos una sustitución de aminoácidos para introducir un resto de cisteína útil para la conjugación, y en el que el polipéptido de dominio constante es un polipéptido de dominio constante de cadena ligera kappa (CK) humana diseñada genéticamente, o una porción del mismo, que comprende al menos una sustitución de aminoácidos seleccionada del grupo que consiste en A 111, K183 y n 210, según la numeración de Kabat.
En otra realización más de la invención, el enlace de la presente invención emplea un polipéptido de dominio constante de anticuerpo modificado, o una porción del mismo, en el que el dominio constante diseñado genéticamente comprende al menos una sustitución de aminoácidos para introducir un resto de cisteína útil para la conjugación, y en el que el polipéptido de dominio constante es un polipéptido Cy diseñado genéticamente, o una porción del mismo, que comprende al menos una secuencia de aminoácidos seleccionada del grupo que consiste en una secuencia de aminoácidos de las SEQ ID NO: 97-100, 102, 104, 107-127 y 129-163, tal como se desvela en el documento WO2013/093809.
En otra realización más de la invención, el enlace de la presente invención emplea un polipéptido de dominio constante de anticuerpo diseñado genéticamente, o una porción del mismo, en la que el dominio constante modificado comprende al menos una sustitución de aminoácidos para introducir un resto de cisteína útil para la conjugación, y en el que el polipéptido de dominio constante es un polipéptido CK diseñado genéticamente, o una porción del mismo, que comprende al menos una secuencia de aminoácidos seleccionada del grupo que consiste en una secuencia de aminoácidos de las SEQ ID NO: 90, 92, 95, 164, 166 y 169, tal como se desvela en el documento WO2013/093809. En otra realización más de la invención, el enlace de la presente invención emplea un polipéptido de dominio constante de anticuerpo diseñado genéticamente, o una porción del mismo, en la que el dominio constante modificado comprende al menos una sustitución de aminoácidos para introducir un resto de cisteína útil para la conjugación, y en el que el polipéptido de dominio constante es un polipéptido CA diseñado genéticamente, o una porción del mismo, que comprende al menos una secuencia de aminoácidos seleccionada del grupo que consiste en una secuencia de aminoácidos de las SEQ ID NO: 172-186, tal como se desvela en el documento WO2013/093809.
En otra realización de la invención, el polipéptido Cy diseñado genéticamente anteriormente mencionado comprende además al menos una mutación seleccionada del grupo que consiste en una mutación en la posición de aminoácido 284, 287, A327, N384, L398 y V422, según el índice EU de Kabat.
Conjugación con transglutaminasa
En ciertas realizaciones, un compuesto de la invención puede reticularse covalentemente con un polipéptido diseñado genéticamente que contiene Fc o Fab con un marcador que contiene glutamina donadora de acilo (por ejemplo, marcadores de péptido que contienen Gln o marcadores Q) o una glutamina endógena reactiva (es decir, la capacidad de formar un enlace covalente como donador de acilo en presencia de una amina y una transglutaminasa) mediante diseño genético de polipéptidos (por ejemplo, mediante deleción, inserción, sustitución, mutación de aminoácidos o cualquier combinación de los mismos en el polipéptido), en presencia de transglutaminasa, siempre que el compuesto de la invención comprenda un agente donador de amina (por ejemplo, una molécula pequeña que comprende o está unida a una amina reactiva), formando de este modo una población estable y homogénea de un conjugado de polipéptido que contiene Fc diseñado genéticamente con un agente donador de amina que está conjugado específicamente en el sitio con el polipéptido que contiene Fc o que contiene Fab a través del marcador que contiene glutamina donadora de acilo o la glutamina endógena expuesta/accesible/reactiva. Por ejemplo, los compuestos de la invención pueden conjugarse tal como se describe en la Solicitud de Patente Internacional de N.° de serie PCT/IB2011/054899. En ciertas realizaciones, para facilitar la conjugación del compuesto de la invención a un polipéptido que contiene Fc o Fab diseñado genéticamente con un marcador que contiene glutamina donante de acilo o una glutamina endógena hecha reactiva por diseño genético de polipéptidos en presencia de transglutaminasa, Z es NH2.
Conjugación con la región constante del dom inio kappa de la cadena ligera humana
En ciertas realizaciones, un compuesto de la invención puede unirse covalentemente a la cadena lateral de K188 de la región constante del dominio kappa de la cadena ligera humana (CLk) (numeración de la cadena ligera completa según Kabat). Por ejemplo, los compuestos de la invención pueden conjugarse tal como se describe en la Solicitud de Patente de los Estados Unidos de Número de Serie 13/180.204. En ciertas realizaciones, para facilitar la conjugación con K188 CLk, Z es

R7 se selecciona independientemente cada vez que está presente entre el grupo que consiste en F, Cl, I, Br, NO2 , CN
y C F 3 ; y h es 1, 2, 3, 4 o 5.
En ciertas realizaciones, la invención proporciona una composición que comprende un compuesto de la invención conjugado covalentemente con un anticuerpo (o una porción de unión a antígeno del mismo), en el que al menos aproximadamente el 50 % , o al menos aproximadamente el 60 % , o al menos aproximadamente el 70 % , o al menos aproximadamente el 80 % , o al menos aproximadamente el 90 % del compuesto de la invención en la composición se conjuga con el anticuerpo o la porción de unión antígeno del mismo en K188 CLk.
En ciertas realizaciones, los compuestos de la invención pueden conjugarse con el sitio de combinación de un anticuerpo catalítico, tal como anticuerpos de aldolasa, o la porción de unión a antígeno de los mismos. Los anticuerpos de aldolasa contienen porciones de sitio de combinación que, cuando no están comprometidas (por ejemplo, por conjugación), catalizan una reacción de adición de aldol entre un donante de cetona alifática y un aceptor de aldehído. El contenido de la Publicación de Solicitud de Patente de EE. UU. N.° US 2006/205670, en particular las páginas 78 118 que describen enlazadores, y los párrafos [0153]-[0233] que describen anticuerpos, fragmentos útiles, variantes y modificaciones de los mismos, h38C2, sitios de combinación y regiones determinantes de complementariedad (CDR), y tecnología de anticuerpos relacionada. La expresión "sitio de combinación" generalmente incluye las CDR y los restos de la región marco adyacentes que están implicados en la unión al antígeno.
Composiciones y procedimientos de administración
En otras realizaciones, otro aspecto de la invención se refiere a composiciones farmacéuticas que incluyen una cantidad eficaz de un compuesto de la invención y/o un conjugado de anticuerpo-fármaco del mismo y un vehículo o excipiente farmacéuticamente aceptable. En ciertas realizaciones, las composiciones son adecuadas para la administración veterinaria o humana.
Las presentes composiciones farmacéuticas pueden estar en cualquier forma que permita que la composición se administre a un paciente. Por ejemplo, la composición puede estar en forma de un sólido o de un líquido. Las vías de administración típicas incluyen, aunque sin limitación, la administración parenteral, ocular e intratumoral. La administración parenteral incluye inyecciones subcutáneas, intravenosas, la inyección intramuscular o intraesternal o técnicas de infusión. En un aspecto, las composiciones se administran por vía parenteral. En una realización específica, las composiciones se administran por vía intravenosa.
Las composiciones farmacéuticas pueden formularse para permitir que un compuesto de la invención y/o un conjugado de anticuerpo-fármaco del mismo esté biodisponible tras la administración de la composición a un paciente. Las composiciones pueden tomar la forma de una o más unidades de dosificación, en las que, por ejemplo, un comprimido puede ser una unidad de dosificación única, y un recipiente de un compuesto de la invención y/o un conjugado de anticuerpo-fármaco del mismo en forma líquida puede contener una pluralidad de unidades de dosificación.
Los materiales utilizados en la preparación de las composiciones farmacéuticas pueden ser no tóxicos en las cantidades utilizadas. Será evidente para los expertos en la materia que la dosificación óptima del principio o los principios activos en la composición farmacéutica dependerá de una variedad de factores. Los factores relevantes incluyen, aunque sin limitación, el tipo de animal (por ejemplo, el ser humano), la forma particular del compuesto de la invención y/o el conjugado del anticuerpo-fármaco del mismo, el modo de administración y la composición empleada. El vehículo o excipiente farmacéuticamente aceptable puede ser sólido o particulado, para que las composiciones estén, por ejemplo, en forma de comprimidos o en polvo. El o los vehículos pueden ser líquidos. Además, el o los vehículos pueden ser particulados.
La composición puede estar en forma de un líquido, por ejemplo, una solución, emulsión o suspensión. En una composición para administración por inyección, uno o más de un tensioactivo, conservante, agente humectante, agente dispersante, agente de suspensión, tampón, estabilizante y agente isotónico también se pueden incluir. Las composiciones líquidas, tanto si son soluciones, como suspensiones u otra forma similar, también pueden incluir uno o más de los siguientes: diluyentes estériles tales como agua para inyección, solución salina, preferentemente, solución salina fisiológica, solución de Ringer, cloruro de sodio isotónico, aceites fijos tales como mono o diglicéridos sintéticos que pueden servir como disolvente o medio de suspensión, polietilenglicoles, glicerina, ciclodextrina, propilenglicol u otros disolventes; agentes antibacterianos tales como alcohol bencílico o metilparabenos; antioxidantes tales como ácido ascórbico o bisulfito de sodio; agentes quelantes tales como ácido etilendiaminotetraacético; tampones tales como acetatos, citratos, fosfatos o aminoácidos, y agentes para el ajuste de la tonicidad tales como cloruro de sodio o dextrosa. Una composición parenteral puede estar encapsulada en una ampolla, una jeringa desechable o un frasco de dosis múltiples hecho de vidrio, plástico u otro material. La solución salina fisiológica es un adyuvante ejemplar. Una composición inyectable es preferentemente estéril.
La cantidad de un compuesto de la invención y/o conjugado de anticuerpo-fármaco del mismo que es eficaz en el tratamiento de un trastorno o afección particular dependerá de la naturaleza del trastorno o afección, y puede determinarse mediante técnicas clínicas convencionales. Además, los ensayos in vitro o in vivo se pueden emplear opcionalmente para ayudar a identificar intervalos de dosificación óptimos. La dosis precisa que se empleará en las composiciones también dependerá de la vía de administración y la gravedad de la enfermedad o trastorno, y debe
decidirse de acuerdo con el criterio del médico y las circunstancias de cada paciente.
Las composiciones comprenden una cantidad eficaz de un compuesto de la invención y/o un conjugado de anticuerpofármaco del mismo de modo que se obtenga una dosificación adecuada. Por lo general, esta cantidad es al menos aproximadamente el 0,01 % de un compuesto de la invención y/o de un conjugado de anticuerpo-fármaco del mismo en peso de la composición. En una realización ejemplar, las composiciones farmacéuticas se preparan de manera que una unidad de dosificación parenteral contenga de aproximadamente el 0,01 % a aproximadamente el 2 % en peso de la cantidad de un compuesto de la invención y/o un conjugado de anticuerpo-fármaco del mismo.
Para la administración intravenosa, la composición puede comprender de aproximadamente 0,01 a aproximadamente 100 mg de un compuesto de la invención y/o conjugado de anticuerpo-fármaco del mismo por kg del peso corporal del paciente. En un aspecto, la composición puede incluir de aproximadamente 1 a aproximadamente 100 mg de un compuesto de la invención y/o conjugado de anticuerpo-fármaco del mismo por kg del peso corporal del paciente. En otro aspecto, la cantidad administrada estará en el intervalo de aproximadamente 0,1 a aproximadamente 25 mg/kg de peso corporal de un compuesto de la invención y/o conjugado de anticuerpo-fármaco del mismo.
Generalmente, la dosificación de un compuesto de la invención y/o conjugado de anticuerpo-fármaco del mismo administrado a un paciente es típicamente de aproximadamente 0,01 mg/kg a aproximadamente 20 mg/kg del peso corporal del paciente. En un aspecto, la dosis administrada a un paciente está entre aproximadamente 0,01 mg/kg y aproximadamente 10 mg/kg del peso corporal del paciente. En otro aspecto, la dosis administrada a un paciente está entre aproximadamente 0,1 mg/kg y aproximadamente 10 mg/kg del peso corporal del paciente. En otro aspecto más, la dosis administrada a un paciente está entre aproximadamente 0,1 mg/kg y aproximadamente 5 mg/kg del peso corporal del paciente. En otro aspecto más, la dosis administrada está entre aproximadamente 0,1 mg/kg y aproximadamente 3 mg/kg del peso corporal del paciente. En otro aspecto más, la dosis administrada es de aproximadamente 1 mg/kg a aproximadamente 3 mg/kg del peso corporal del paciente.
Un compuesto de la invención y/o un conjugado de anticuerpo-fármaco del mismo pueden administrarse por cualquier vía conveniente, por ejemplo por infusión o inyección en bolo. La administración puede ser sistémica o local. Se conocen varios sistemas de administración, por ejemplo, encapsulación en liposomas, mieropartículas, microcápsulas, cápsulas, etc., y puede usarse para administrar un compuesto de la invención y/o un conjugado de anticuerpo-fármaco del mismo. En ciertas realizaciones, se administra a un paciente más de un compuesto de la invención y/o conjugado de anticuerpo-fármaco del mismo.
En realizaciones específicas, puede ser deseable administrar uno o más compuestos de la invención y/o conjugados de anticuerpo-fármaco de los mismos localmente al área que necesita tratamiento. Esto se puede lograr, por ejemplo, y no a modo de limitación, por infusión local durante la cirugía; aplicación tópica, por ejemplo, junto con un apósito para heridas después de la cirugía; por inyección; por medio de un catéter; o por medio de un implante, siendo el implante de un material poroso, no poroso o gelatinoso, incluyendo membranas, tales como membranas sialásticas o fibras. En una realización, la administración puede ser por inyección directa en el sitio (o en el sitio anterior) de un cáncer, tumor o tejido neoplásico o preneoplásico. En otra realización, la administración puede ser por inyección directa en el sitio (o en el sitio anterior) de una manifestación de una enfermedad autoinmune.
En otra realización más, el compuesto de la invención y/o el conjugado de anticuerpo-fármaco del mismo se puede administrar en un sistema de liberación controlada, tal como, pero sin limitación, una bomba, o se pueden usar diversos materiales poliméricos. En otra realización más, se puede colocar un sistema de liberación controlada cerca de la diana del compuesto de la invención y/o del conjugado de anticuerpo-fármaco del mismo, por ejemplo, el hígado, requiriendo por lo tanto solo una parte de la dosis sistémica (véase, por ejemplo, Goodson, en Medical Applications of Controlled Release, citado anteriormente, vol. 2, págs. 115 -138 (1984)). Se pueden utilizar otros sistemas de liberación controlada tratados en la revisión de Langer (Science 249:1527-1533 (1990)).
El término "vehículo" se refiere a un diluyente, adyuvante o excipiente, con el que se administra un compuesto o conjugado de anticuerpo-fármaco del mismo. Dichos vehículos farmacéuticos pueden ser líquidos, tales como agua y aceites, incluidos los de origen del petróleo, animal, vegetal o sintético. Los vehículos pueden ser solución salina, y similares. Además, se pueden usar agentes auxiliares, estabilizantes y otros agentes. En una realización, cuando se administra a un paciente, el compuesto o conjugado y los vehículos farmacéuticamente aceptables son estériles. El agua es un vehículo ejemplar cuando el compuesto o el conjugado se administran por vía intravenosa. Se pueden emplear también soluciones salinas y soluciones acuosas de dextrosa y glicerol como vehículos líquidos, particularmente para soluciones inyectables. Las presentes composiciones, si se desea, también puede contener cantidades menores de agentes humectantes o emulsionantes, o agentes tamponantes del pH.
Las presentes composiciones pueden tomar la forma de soluciones, microgránulos, polvos, formulaciones de liberación sostenida, o cualquier otra forma adecuada para su uso. Otros ejemplos de vehículos farmacéuticos adecuados se describen en "Remington's Pharmaceutical Sciences" de E. W. Martin.
En una realización, el compuesto de la invención y/o el conjugado de anticuerpo-fármaco del mismo se formulan de acuerdo con procedimientos habituales tales como una composición farmacéutica adaptada para administración intravenosa a animales, particularmente seres humanos. Por lo general, los vehículos o excipientes para la
administración intravenosa son soluciones tampón acuosas isotónicas estériles. Cuando sea necesario, las composiciones también pueden incluir un agente solubilizante. Las composiciones para la administración intravenosa pueden comprender opcionalmente un anestésico local tal como lignocaína para aliviar el dolor en el sitio de la inyección. Generalmente, los ingredientes se suministran por separado o se mezclan en forma de dosificación unitaria, por ejemplo, como polvo liofilizado seco o concentrado libre de agua en un recipiente sellado herméticamente, tal como una ampolla o bolsita que indica la cantidad de agente activo. Cuando un compuesto de la invención y/o un conjugado de anticuerpo-fármaco del mismo se va a administrar por infusión, se puede dispensar, por ejemplo, con una botella de infusión que contenga agua estéril de grado farmacéutico o solución salina. Cuando el compuesto de la invención y/o el conjugado de anticuerpo-fármaco del mismo se administra mediante inyección, se puede proporcionar una ampolla de agua estéril para inyección o solución salina para que los ingredientes se puedan mezclar antes de la administración.
La composición puede incluir diversos materiales que modifican la forma física de una unidad de dosificación sólida o líquida. Por ejemplo, la composición puede incluir materiales que forman una capa de recubrimiento alrededor de los principios activos. Los materiales que forman la cubierta del revestimiento son típicamente inertes y pueden seleccionarse de, por ejemplo, azúcar, goma laca y otros agentes de recubrimiento entérico. Como alternativa, los principios activos se pueden encerrar en una cápsula de gelatina.
Ya sea en forma sólida o líquida, las presentes composiciones pueden incluir un agente farmacológico utilizado en el tratamiento del cáncer.
Usos terapéuticos de compuestos y conjugados de anticuerpo-fármaco de los mismos
Otro aspecto de la invención se relaciona con los compuestos de la invención y sus conjugados de anticuerpo-fármaco para su uso en el tratamiento del cáncer.
Los compuestos de la invención y/o sus conjugados de anticuerpo-fármaco son útiles para inhibir la multiplicación de una célula tumoral o célula cancerosa, causando la apoptosis en una célula tumoral o cancerosa, o para tratar el cáncer en un paciente. Los compuestos de la invención y/o sus conjugados de anticuerpo-fármaco pueden usarse en consecuencia en una variedad de entornos para el tratamiento de cánceres de animales. Dichos conjugados se pueden usar para administrar un compuesto de la invención a una célula tumoral o a una célula cancerosa. Sin quedar ligados a teoría alguna, en una realización, el anticuerpo del conjugado se une o se asocia con una célula cancerosa o un antígeno asociado a células tumorales, y el conjugado puede ser absorbido (internalizado) dentro de una célula tumoral o célula cancerosa a través de la endocitosis mediada por receptor u otro mecanismo de internalización. El antígeno puede unirse a una célula tumoral o célula cancerosa o puede ser una proteína de matriz extracelular asociada con la célula tumoral o célula cancerosa. En ciertas realizaciones, una vez dentro de la célula, una o más secuencias peptídicas específicas se cortan enzimática o hidrolíticamente por una o más proteasas asociadas a células tumorales o cancerosas, dando como resultado la liberación de un compuesto de la invención del conjugado. El compuesto de la invención liberado es libre de migrar dentro de la célula e inducir actividades citotóxicas o citostáticas. El conjugado también se puede escindir mediante una proteasa intracelular para liberar un compuesto de la invención. En una realización alternativa, el compuesto de la invención se escinde del conjugado fuera de la célula tumoral o la célula cancerosa, y el compuesto de la invención penetra posteriormente en la célula.
En ciertas realizaciones, los conjugados proporcionan un tumor específico de conjugación o un fármaco dirigido contra el cáncer, reduciendo así la toxicidad general de los compuestos de la invención.
En otra realización, la unidad de anticuerpo se une a la célula tumoral o a la célula cancerosa.
En otra realización, la unidad de anticuerpo se une a una célula tumoral o al antígeno de células cancerosas que está en la superficie de la célula tumoral o de la célula cancerosa.
En otra realización, la unidad de anticuerpo se une a una célula tumoral o al antígeno de células cancerosas que es una proteína de matriz extracelular asociada con la célula tumoral o con la célula cancerosa.
La especificidad de la unidad de anticuerpo para una célula tumoral o célula cancerosa particular puede ser importante para determinar aquellos tumores o cánceres que se tratan con mayor eficacia.
Los tipos particulares de cánceres que pueden tratarse con un compuesto de la invención y/o conjugado de anticuerpofármaco del mismo, incluyen pero no se limitan a, carcinomas de vejiga, mama, cuello del útero, colon, endometrio, riñón, pulmón, esófago, ovario, próstata, páncreas, piel, estómago y testículos; y cánceres de sangre que incluyen, pero no se limitan a, leucemias y linfomas.
Terapia multimodal para el cáncer. Los cánceres, incluyendo, aunque no de forma limitativa, un tumor, metástasis u otra enfermedad o trastorno caracterizado por un crecimiento celular descontrolado, se pueden tratar o inhibir mediante la administración de un compuesto de la invención y/o de un conjugado de anticuerpo-fármaco del mismo.
En otras realizaciones, se proporcionan compuestos de la invención y conjugados de anticuerpo-fármaco para su uso en el tratamiento del cáncer, incluyendo la administración a un paciente que lo necesite de una cantidad eficaz de un
compuesto de la invención y/o un conjugado de anticuerpo-fármaco del mismo y un agente quimioterapéutico. En una realización, el agente quimioterapéutico es aquel con el que no se ha descubierto que el tratamiento del cáncer sea refractario. En otra realización, el agente quimioterapéutico es aquel con el que se ha descubierto que el tratamiento del cáncer es refractario. Un compuesto de la invención y/o el conjugado de anticuerpo-fármaco del mismo se puede administrar a un paciente que también se ha sometido a cirugía como tratamiento para el cáncer.
En algunas realizaciones, el paciente también recibe un tratamiento adicional, tal como radioterapia. En una realización específica, el compuesto de la invención y/o el conjugado de anticuerpo-fármaco del mismo se administra simultáneamente con el agente quimioterapéutico o con radioterapia. En otra realización específica, el agente quimioterapéutico o la radioterapia se administran antes o después de la administración de un compuesto de la invención y/o del conjugado de anticuerpo-fármaco del mismo.
Se puede administrar un agente quimioterapéutico durante una serie de sesiones. Cualquiera o una combinación de los agentes quimioterapéuticos, tales agentes quimioterapéuticos de tratamiento habitual, se pueden administrar. Además, un compuesto de la invención y/o un conjugado de anticuerpo-fármaco del mismo para su uso en el tratamiento del cáncer se proporcionan como una alternativa a la quimioterapia o radioterapia en la que la quimioterapia o la radioterapia han demostrado o pueden demostrar ser demasiado tóxicas, por ejemplo, resultados de efectos secundarios inaceptables o insoportables, para el sujeto que se está tratando. El paciente que está siendo tratado puede, opcionalmente, ser tratado con otro tratamiento contra el cáncer tal como cirugía, radioterapia o quimioterapia, dependiendo de qué tratamiento se considere aceptable o soportable.
Los compuestos de la invención y/o sus conjugados de anticuerpo-fármaco también se pueden usar de forma in vitro o ex vivo, tal como para el tratamiento de ciertos tipos de cáncer, incluyendo, pero sin limitación, leucemias y linfomas, implicando dicho tratamiento los trasplantes autólogos de células madre. Esto puede implicar un procedimiento de varias etapas en el que las células madre hematopoyéticas autólogas del animal se recolectan y se purgan de todas las células cancerosas, la población de células de médula ósea restante del animal se erradica a través de la administración de una dosis alta de un compuesto de la invención y/o de un conjugado de anticuerpo-fármaco del mismo con o sin radioterapia de dosis alta, y el injerto de células madre se infunde nuevamente en el animal. Luego se brinda atención de apoyo mientras se restaura la función de la médula ósea y el paciente se recupera.
La invención se describe adicionalmente en los siguientes ejemplos y ejemplos de referencia.
Procedimientos generales
Procedimientos sintéticos experimentales:
Los experimentos se realizaron generalmente en una atmósfera inerte (nitrógeno o argón), especialmente en los casos en los que se emplearon reactivos o intermedios sensibles al oxígeno o a la humedad. Generalmente, se usaron disolventes y reactivos comerciales sin purificación adicional, incluyendo disolventes anhidros, cuando era apropiado (generalmente productos Sure-Seal™ de la Aldrich Chemical Company, Milwaukee, Wisconsin). Los datos de espectrometría de masas se indican a partir de cromatografía líquida-espectrometría de masas (CL-EM) o de ionización química a presión atmosférica (IQPA). Los desplazamientos químicos en los datos de resonancia magnética nuclear (r Mn ) se expresan en partes por millón (ppm, 8) referidos a los picos residuales de los disolventes deuterados utilizados.
Para las síntesis que hagan referencia a los procedimientos de otros Ejemplos o Procedimientos, el protocolo de reacción (duración de reacción y temperatura) pueden variar. En general, las reacciones se siguieron por cromatografía de capa fina, CL-EM o HPLC, y se sometieron a tratamiento cuando era apropiado. Las purificaciones pueden variar entre experimentos: por lo general, los disolventes y las proporciones de disolventes usadas para eluyentes/gradientes se seleccionaron para proporcionar tiempos de retención apropiados. A menos que se especifique otra cosa, las fracciones de HpLC de fase inversa se concentraron por liofilización/secado por congelación. Los compuestos intermedios y finales se almacenaron a (0 °C) o a temperatura ambiente en viales o matraces cerrados en una atmósfera de nitrógeno. Los nombres de los compuestos se generaron con software de ACD Labs.
Las abreviaturas para los disolventes y/o reactivos se basan en las directrices de la American Chemical Society y se destacan a continuación:
Ac = Acetilo
Boc = W-terc-butoxicarbonilo
CDI = W,W'-Carbonildiimidazol
D CC = 1,3-Diciclohexilcarbodiimida
DCE = Dicloroetano
DCM = Diclorometano
DEA = W,W-Dietilamina
DEAD = Azodicarboxilato de dietilo
DIAD = Azodicarboxilato de diisopropilo
DIBAL-H = Hidruro de diisobutilaluminio
DIPEA (o) Base de Hunig = N,N-Diisopropiletilamina
DMA = Dimetilacetamida
DMAP = 4-Dimetilaminopiridina
DME = Dimetoxietano
DMF = N,N-Dimetilformamida
DMSO = Dimetilsulfóxido
DPPA = Difenilfosforil azida
EDCI = 1-Etil-3-(3-dimetilaminopropil)carbodiimida
EtOAc = Acetato de etilo
Fmoc = Fluorenilmetiloxicarbonilo
h = hora
HATU = Hexafluorofosfato de o-(7-azabenzotriazol-1-il)-N,N,N',N'-te-trametiluronio
HBTU = Hexafluorofosfato de N,N,N',N'-Tetrametil-O-(1H-benzotriazol-1-il)uronio
HOAc = Ácido acético
HOAt = 1-Hidroxi-7-azabenzotriazol
HOBt = 1-Hidroxibenzotriazol hidrato
LDA = Diisopropilamida de litio
Me = Metilo
MS = Tamices moleculares
MTBE = Metil terc-butil éter
n-BuLi = n-Butil litio
NBS = W-Bromosuccinimida
NMM = N-metil morfolina
Ph = Fenilo
PPTS = p-Toluenosulfonato de piridinio
p-TsOH = Ácido p-toluenosulfónico
ta = temperatura ambiente
TBAI = Yoduro de tetrabutilamonio
TEA = Trietilamina
Tf = Trifluorometanosulfonato
TFA = TFA
THF = Tetrahidrofurano
TPTU = Tetrafluoroborato de O-(2-oxo-1(2H)piridil)-N,N,N,'N'-tetrametiluronio
Condiciones de HPLC y CL-EM usadas para el análisis
Protocolo A: Columna: Waters Acquity UPLC H SS T3, 2,1 mm x 50 mm, C18, 1,7 pm; Fase móvil A: :ácido fórmico al 0,1 % en agua (v/v); Fase móvil B: ácido fórmico al 0,1 % en acetonitrilo (v/v); Gradiente: B al 5 % durante 0,1 minutos, B del 5 % al 95 % durante 2,5 minutos, B al 95 % durante 0,35 minutos; Caudal: 1,25 ml/minuto. Temperatura: 60 °C; Detección: 200-450 nm; intervalo de EM (+) 100-2000 daltons; Volumen de inyección: 5 pl; Instrumento: Waters Acquity.
Protocolo B: Columna: Waters Acquity UPLC HSS T3, C18, 2,1 x 50 mm, 1,7 pm; Fase móvil A: ácido fórmico al 0,1 % en agua (v/v); Fase móvil B: ácido fórmico al 0,1 % en acetonitrilo (v/v); Gradiente: B al 5 % durante 0,1 minutos, B del 5 % al 95 % durante 1,5 minutos, B al 95 % durante 0,35 minutos; Caudal: 1,25 ml/minuto. Temperatura: 60 °C; Detección: 200-450 nm; intervalo de EM (+) 100-2000 daltons; Volumen de inyección: 5 pl; Instrumento: Waters Acquity.
Condiciones de HPLC usadas para la purificación
Procedimiento A: Columna: Phenomenex Luna Fenilhexil 150 x 21,2 mm, 5 pm; Fase móvil A: TFA al 0,02 % en agua (v/v); Fase móvil B: TFA al 0,02 % en acetonitrilo (v/v); Gradiente: B al 20 % durante 1,5 minutos, de B al 2 0 % a B al 100 % durante 8,5 minutos, después B al 100 % durante 2,0 minutos; Caudal: 27 ml/minuto. Temperatura: no controlada; Detección: DAD 210-360 nm; intervalo de EM (+) 150-2000 daltons; Instrumento: 305 RP Waters Fractional Lynx LCMS
Procedimiento B: Columna: Phenomenex Luna C18, 100 x 30 mm, 5 pm; Fase móvil A: TFA al 0,02 % en agua (v/v); Fase móvil B: TFA al 0,02 % en acetonitrilo (v/v); Gradiente: B al 20 % durante 1,5 minutos, de B al 20 % a B al 100 % durante 8,5 minutos, después B al 95 % durante 2,0 minutos; Caudal: 30 ml/minuto. Temperatura: no controlada; Detección: UV 215 nm; Instrumento: Gilson
Procedimiento C : Phenomenex Luna C18, 100 x 30 mm, 5 pm; Fase móvil A: TFA al 0,02 % en agua (v/v); Fase móvil B: TFA al 0,02 % en acetonitrilo (v/v); Gradiente: variable, gradiente en aumento de B en A durante 10 a 20 minutos. Caudal: de 27 a 30 ml/minuto. Temperatura: no controlada; Detección: UV 215 nm; Instrumento: Gilson Procedimientos generales:
Procedimiento general A: A una solución en agitación del mono o diácido, en THF, diclorometano o una mezcla de ambos a 0 °C, se le añadió cloruro de oxalilo (1-2,5 equiv.) seguido de una cantidad catalítica de DMF. La reacción se dejó en agitación a 0 °C durante varios minutos antes de que se dejara calentar a temperatura ambiente, y después se agitó a temperatura ambiente durante 30 minutos a varias horas. Después, la reacción se concentró al vacío. En algunos casos, el material en bruto después se destiló azeotrópicamente de una a varias veces con heptano, u otro disolvente o disolventes adecuados. Después, el material en bruto se secó a alto vacío antes de que se usara en la siguiente etapa.
Procedimiento general B: A una solución en agitación de la amina (2-2,5 equiv.) en THF, diclorometano o una mezcla de ambos a 0 °C (o en algunos casos otro disolvente o disolventes adecuados), se le añadió el cloruro de ácido, o dicloruro de ácido, seguido de piridina (3-6 equiv.), trietilamina (3-6 equiv.) u otra base adecuada (3-6 equiv.). La reacción se dejó en agitación a 0 °C durante unos segundos a varios minutos antes de que se dejara calentar a temperatura ambiente, y después se agitó a temperatura ambiente durante 10 minutos a varias horas. Después, la reacción se concentró al vacío. En algunos casos, el material en bruto después se destiló azeotrópicamente de una a varias veces con heptano, u otro disolvente o disolventes adecuados. En la mayoría de los casos, el material en bruto se purificó después por un procedimiento descrito tal como cromatografía sobre sílice o cromatografía de fase inversa C 18 a media presión.
Ejemplos:
Preparación de acetato de (S)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilo (4).

Etapa 1: Síntesis de (S)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de terc-butilo (2). A una solución en agitación de (8S)-4-(benciloxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de terc-butilo (1) [preparado como se describe en J. Am. Chem. Soc., 2007, 129, 14092-14099.] (100 mg, 0,225 mmol) en THF (1,5 ml) se le añadió Pd al 10 % /C (33 mg) seguido de una solución acuosa al 25 % de formiato amónico (0,15 ml). La solución se agitó durante 2 h. La solución se diluyó con éter (6 ml) y se añadió sulfato sódico. La mezcla se filtró a través de Celite y el disolvente se retiró al vacío, proporcionando el producto deseado 2 en forma de un sólido de color blanco (79 mg, 100 %).
Etapa 2: Síntesis de (S)-4-acetoxi-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de tercbutilo (3). Se disolvió (S)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de tercbutilo (2,69 mg, 0,19 mmol) en CH 2 C l2 (2 ml). Piridina (46 mg, 0,585 mmol). Se añadió cloruro de acetilo (16 ml, 0,234 mmol) y la reacción se agitó durante 2 h. El disolvente se retiró al vacío. El residuo en bruto se purificó por cromatografía ultrarrápida (EtOAc al 0-100 %/heptano) para proporcionar el producto deseado (45 mg, 58 %). Etapa 3: Síntesis de acetato de (S)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilo (4): Se recogió (S)-4-acetoxi-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de terc-butilo (3) (45 mg, 0,11 mmol) en dioxano (1 ml). Se añadió HCI 4 N en dioxano (1 ml) y la solución se agitó durante 2 h. El disolvente se retiró para proporcionar el material diana en bruto con rendimiento cuantitativo y el material se usó inmediatamente tal cual.
Preparación de cloruro del ácido biciclo[1.1.1]-1,3-dicarboxílico (6)

Se puso ácido biciclo[1.1.1]pentano-1,3-dicarboxílico (50 mg) en un vial, se recogió en THF (1 ml) y se añadió una gota de DMF. Se añadió lentamente dicloruro de oxalilo (122 mg, 0,961 mmol, 0,0825 ml) y la solución se burbujeó rápidamente. Después de 2 h, el disolvente se retiró y el dicloruro de ácido 6 se usó tal cual en reacciones posteriores. Preparación de diacetato de (8S,8'S)-(biciclo[1.1.1]pentano-1,3-dicarbonil)bis(8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6,4-diilo) (7).

Etapa 1: diacetato de (8S,8'S)-(biciclo[1,1,1 ]pentano-1,3-dicarbonil)bis(8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6,4-diilo) (7). : El compuesto del título se preparó siguiendo el procedimiento general B usando 4 (20,0 mg, 0,060 mmol), 6 (5,81 mg, 0,0301 mmol, 0,0301 ml, 1 M en THF), piridina (14,3 0,181 mmol) y THF (1,0 ml), y la purificación usando el Material se purificó por cromatografía C18 de fase inversa a media presión (Gradiente: acetonitrilo del 0 % al 80 % en agua con TFA al 0,02 % en cada fase) (proporcionando el producto deseado 7. (8,0 mg, 20 % ). CL-EM (Protocolo B): m /z 711,3 [M+H]+, tiempo de retención = 1,12 minutos
Preparación de acetato de (R)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilo (11).

Etapa 1: Síntesis de (R)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de terc-butilo (9). A una solución en agitación de (8R)-4-(benciloxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de terc-butilo (8) [preparado como se describe en J. Am. Chem. Soc., 2007, 129, 14092-14099.] (100 mg, 0,225 mmol) en THF (1,5 ml) se le añadió Pd al 10 % /C (33 mg) seguido de una solución acuosa al 25 % de formiato amónico (0,15 ml). La solución se agitó durante 2 h. La solución se diluyó con éter (6 ml) y se añadió sulfato sódico. La mezcla se filtró a través de Celite y el disolvente se retiró al vacío, proporcionando el producto deseado 9 en forma de un sólido de color blanco (79 mg, 100 %).
Etapa 2: Síntesis de (R)-4-acetoxi-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de tercbutilo (10): Se disolvió (R)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de tercbutilo (2) (69 mg, 0,19 mmol) en CH 2 O 2 (2 ml). Se añadió piridina (46 mg, 0,585 mmol). Se añadió cloruro de acetilo (16 ml, 0,234 mmol) y la reacción se agitó durante 2 h. El disolvente se retiró al vacío. El residuo en bruto se purificó por cromatografía ultrarrápida (EtOAc al 0-100 %/heptano) para proporcionar el producto deseado 10 (53 mg, 69 % ).
Etapa 3: Síntesis de acetato de (R)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilo (11): Se recogió (R)-4-acetoxi-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de terc-butilo (10) (53 mg, 0,13 mmol) en dioxano (1 ml). Se añadió HCI 4 N en dioxano (1 ml) y la solución se agitó durante 2 h. El disolvente se retiró para proporcionar el material diana en bruto con rendimiento cuantitativo y el material se usó inmediatamente tal cual en la preparación de diacetato de (8R,8'R)-(biciclo[1.1.1]pentano-1,3-dicarbonil)bis(8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6,4-diilo) (11).
Preparación de diacetato de (8R,8'R)-(biciclo[1.1.1]pentano-1,3-dicarbonil)bis(8-(dorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6,4-diilo) (12)

Etapa 1: Síntesis de diacetato de (8R,8'R)-(biciclo[1.1.1]pentano-1,3-dicarbonil)bis(8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6,4-diilo) (12).
El compuesto del título se preparó siguiendo el procedimiento general B usando 11(20,0 mg, 0,060 mmol) y 6 (5,81 mg, 0,0301 mmol, 0,0301 ml, 1 M en THF), piridina (14,3 0,181 mmol) y THF (1,0 ml) y se purificó usando cromatografía de fase inversa C 18 a media presión (Gradiente: acetonitrilo del 0 % al 80 % en agua con TFA al 0,02 % en cada fase), proporcionando el producto deseado 12. (5,9 mg, 14 % ). CL-EM (Protocolo B): m /z 711,2 [M+H]+, tiempo de retención
= 1,12 minutos
Preparación de acetato de (S)-8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indol-4-ilo (16):

Etapa 1: Síntesis de (S)-8-(dorometil)-4-hidroxi-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6-carboxilato de terc-butilo (14): (S)-4-(benciloxi)-8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6-carboxilato de terc-butilo (13) [preparado como se describe en J. Am. Chem. Soc., 2007, 129, 14092-14099.] (250 mg, 0,563 mmol) se recogió en THF (5,63 ml). Se añadió Pd al 10 % /C (80 mg). Se añadió una sal de amonio del ácido fórmico acuosa al 25 % recién preparada (500 mg, 2 mmol, 0,5 ml) y la reacción se agitó durante 30 min. La mezcla de reacción se diluyó con éter (15 ml) y se añadió Na2 SO 4. La solución se filtró a través de Celite y el disolvente se retiró al vacío para proporcionar el compuesto del título 14 (191 mg, 95 % )
Etapa 2: Síntesis de (S)-4-acetoxi-8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6-carboxilato de tercbutilo (15): Se disolvió (S)-8-(clorometil)-4-hidroxi-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6-carboxilato de tercbutilo (14, 171 mg, 0,483 mmol) en CH 2CI2 (5,0 ml). Se añadió piridina (213 mg, 2,69 mmol) seguido de cloruro de acetilo (114 mg, 1,45 mmol). La reacción se agitó durante una noche y la solución se volvió de color naranja/pardo. El disolvente se retiró al vacío, dejando un sólido en bruto de color naranja. El residuo en bruto se purificó por cromatografía ultrarrápida (EtOAc al 0-100 %/heptano) para proporcionar, después de la retirada del disolvente, el producto deseado 15 (171,0 mg, 89,4 % )
Etapa 3: Síntesis de acetato de (S)-8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indol-4-ilo (16): Se recogió (S)-4-acetoxi-8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6-carboxilato de terc-butilo (15, 20 mg, 0,051 mmol) en HCI 4 N en dioxano (1 ml). La reacción se dejó en reposo durante una noche y el disolvente se retiró para proporcionar el material diana en bruto 16 con rendimiento cuantitativo y el material se usó inmediatamente
Preparación de diacetato de biddo[1.1.1]pentano-1,3-diNbis[carboml(8S)-8-(dorometN)-2-metM-7,8-dihidro-6H-tieno[2,3-e]indolo-6,4-diilo] (17)

Etapa 1: Síntesis de diacetato de biciclo[1.1.1]pentano-1,3-diilbis[carbonil(8S)-8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6,4-diilo] (17): El compuesto del título se preparó siguiendo el procedimiento general B usando 16, (17 mg, 0,051) y 6 (4,94 mg, 0,026 mmol, 0,0301 ml, 1 M en THF), piridina (14,3 0,181 mmol) y THF (1,0 ml) y se purificó usando cromatografía de fase inversa C 18 a media presión (Gradiente: acetonitrilo del 0 % al 80 % en agua con TFA al 0,02 % en cada fase), proporcionando el producto deseado 17 (10,0 mg, 27 % ) CL-EM (Protocolo B): m/z 711,1 [M+H]+, tiempo de retención = 1,12 minutos
Preparación de acetato de (R)-8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indol-4-ilo (21)
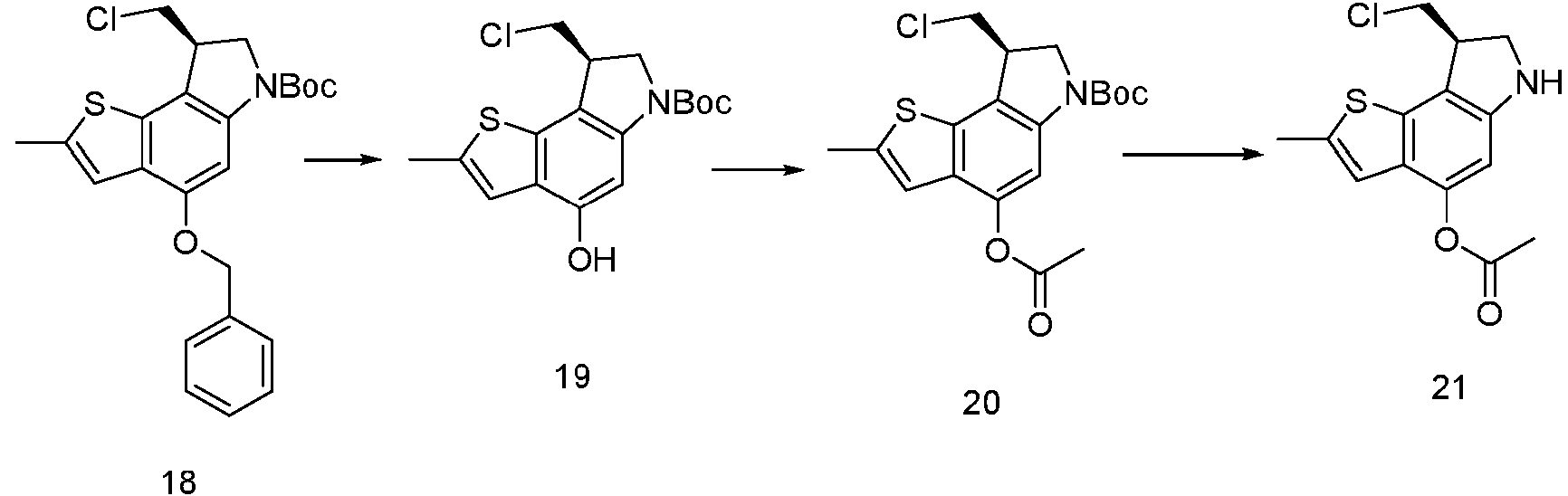
Etapa 1: Síntesis de (R)-8-(dorometil)-4-hidroxi-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6-carboxilato de terc-butilo (19): (R)-4-(benciloxi)-8-(dorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6-carboxilato de terc-butilo (18) [preparado como se describe en J. Am. Chem. Soc., 2007, 129, 14092-14099.] (250 mg, 0,563 mmol) se recogió en THF (5,63 ml). Se añadió Pd al 10 % /C (80 mg). Se añadió una sal de amonio del ácido fórmico acuosa al 25 % recién preparada (500 mg, 2 mmol, 0,5 ml) y la reacción se agitó durante 30 min. La mezcla de reacción se diluyó con éter (15 ml) y se añadió Na2SO4. La solución se filtró a través de Celite y el disolvente se retiró al vacío, dejando un sólido de color blanco de 19 en bruto (156 mg, 78 % )
Etapa 2: Síntesis de (R)-4-acetoxi-8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6-carboxilato de tercbutilo (20): Se disolvió (R)-8-(clorometil)-4-hidroxi-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6-carboxilato de tercbutilo (19, 136 mg, 0,384 mmol) en DCM (5,0 ml). Se añadió piridina (213 mg, 2,69 mmol) seguido de cloruro de acetilo (114 mg, 1,45 mmol). La reacción se agitó durante una noche y la solución se volvió de color naranja/pardo. El disolvente se retiró al vacío, dejando un sólido en bruto de color naranja. El residuo en bruto se purificó por cromatografía ultrarrápida (EtOAc al 0-100 %/heptano), proporcionando, después de la retirada del disolvente, el producto deseado 20 (113,0 mg, 74 % )
Etapa 3: Síntesis de acetato de (R)-8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indol-4-ilo (21): Se recogió (R)-4-acetoxi-8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6-carboxilato de terc-butilo (20, 25 mg, 0,051 mmol) en HCI 4 N en dioxano (1 ml). La reacción se dejó en reposo durante una noche y el disolvente se retiró para proporcionar el material diana en bruto 21 con rendimiento cuantitativo y el material se usó inmediatamente
Preparación de diacetato de biddo[1.1.1]pentano-1,3-diMbis[carboml(8R)-8-(dorometM)-2-metM-7,8-dihidro-6H-tieno[2,3-e]indolo-6,4-diilo] (22)

Etapa 1: Síntesis de diacetato de biciclo[1.1.1]pentano-1,3-diilbis[carbonil(8R)-8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6,4-diilo] (22): El compuesto del título se preparó siguiendo el procedimiento general B usando 21, (17 mg, 0,051 mmol) y 6 (4,94 mg, 0,026 mmol, 0,0301 ml, 1 M en THF), piridina (14,3 0,181 mmol) y THF (1,0 ml) y se purificó usando cromatografía de fase inversa C 18 a media presión (Gradiente: acetonitrilo del 0 % al 80 % en agua con TFA al 0,02 % en cada fase), proporcionando el producto deseado 22 (8,6 mg, 19 % ) CL-EM (Protocolo B): m/z 711,1 [M+H]+, tiempo de retención = 1,12 minutos
Preparación de diacetato de (8S,8'S)-glutaroilbis(8-(dorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6,4-diilo) (24)

Etapa 1: Síntesis de diacetato de (8S,8'S)-glutaroilbis(8-(dorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6,4-diilo) (24): Una solución de 3 (10 mg, 0,025 mmol) se trató con HCI 4 M (0,5 ml en dioxano) a ta durante 1 h y se concentró al vacío. El residuo se disolvió en THF (2 ml) y se añadió cloruro de glutarilo (2,13 mg, 0,0125 mmol) seguido de DIPEA (13 ul, 0,074 mmol). La mezcla resultante se agitó a ta durante 30 min. La mezcla de reacción se concentró y el residuo se purificó por cromatografía sobre gel de sílice usando MeOH/DCM (0 -10 % ) para dar el producto en forma de un sólido de color blanquecino, que se trató con MeOH y se filtró para dar el producto 24 en forma de un sólido de color blanquecino, 6 mg (70 % ). CL-EM (Protocolo B): m/z 687,1 [M+H]+, tiempo de retención = 1,11 minutos.
1H RMN (400 MHz, CDCla) 8 = 8,24 (s, 2H), 7 ,13 (s, 2H), 4,36 (d, J = 10,5 Hz, 2H), 4 ,17 (m, 2H), 4,06 (m, 2H), 3,74 (d, J = 10,9 Hz, 2H), 3,39 (t, J = 10,9 Hz, 2H), 2,80 (m, 2H), 2,66 (m, 2H), 2,55 (s, 6H), 2,37 (s, 6H), 2,22 (m, 2H).
Preparación de diacetato de (8R,8'R)-glutaroilbis(8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]mdolo-6,4-diilo) (25):

Etapa 1: Síntesis de diacetato de (8R,8'R)-glutaroilbis(8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6,4-diilo) (25):
Una solución de 10 (10 mg, 0,025 mmol) se trató con HCI 4 M (0,5 ml en dioxano) a ta durante 1 h y se concentró al vacío. El residuo se disolvió en THF (2 ml) y se añadió cloruro de glutarilo (2,13 mg, 0,0125 mmol) seguido de piridina (12 mg, 0,15 mmol). La mezcla resultante se agitó a ta durante 4 h, se concentró al vacío y el residuo se purificó por HPLC de fase inversa (Procedimiento C) para dar el producto 25 en forma de un sólido de color blanquecino, 1,5 mg (17 % ). CL-EM (Protocolo B): m/z 687,1 [M+H]+, tiempo de retención = 1,11
1H RMN (400 MHz, CDCla) 8 = 8,24 (s, 2H), 7 ,13 (s, 2H), 4,37 (d, J = 10,5 Hz, 2H), 4,18 (m, 2H), 4,06 (m, 2H), 3,73 (d, J = 10,9 Hz, 2H), 3,39 (t, J = 10,9 Hz, 2H), 2,77 (m, 2H), 2,66 (m, 2H), 2,55 (s, 6H), 2,37 (s, 6H), 2,22 (m, 2H).
Preparación de diacetato de (8S,8'S)-glutaroilbis(8-(clorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6,4-diilo) (26)
Etapa 1: diacetato de (8S,8'S)-glutaroilbis(8-(dorometil)-2-metil-7,8-dihidro-6H-tieno[2,3-e]indolo-6,4-diilo) (26): Una solución de 15 (12 mg, 0,03 mmol) se trató con HCI 4 M (0,5 ml en dioxano) a ta durante 1 h y se concentró al vacío. El residuo se disolvió en THF (2 ml) y se añadió cloruro de glutarilo (2,5 mg, 0,015 mmol) seguido de DIPEA (16 ul, 0,09 mmol). La mezcla resultante se agitó a ta durante 30 min, se concentró al vacío y el residuo se purificó por ISCO usando MeOH/DCM (0 - 10 % ) para dar el producto 26 en forma de un sólido de color blanquecino, 6 mg (60 % ): C L-EM (Protocolo B): m /z 687,1 [M+H]+, tiempo de retención = 1,11.
1H RMN (400 MHz, CDCla) 8 = 8,12 (s, 2H), 6,84 (s, 2H), 4,32 (m, 2H), 4,2 (m, 2H), 4,05 (d, J = 10,9 Hz, 2H), 3,93 (m, 2H), 3,62 (m, 2H), 2,68 (s a, 4H), 2,56 (s, 6H), 2,37 (s, 6H), 2,19 (s a, 2H).
Preparación de 4-nitrofenil carbonato de (8S)-8-(clorometM)-6-[(3-{[(8S)-8-(clorometM)-1-metN-4-(fosfonooxi)-7,8-dihidro-6H-tieno[3,2-e]indol-6-il]carbonil}biciclo[1.1.1]pent-1 -il)carbonil]-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilo (33).

Etapa 1: Síntesis de (8S)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de terc-butilo (2). A una solución en agitación de (8S)-4-(benciloxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de terc-butilo 1 [preparado como se describe en J. Am. Chem. Soc., 2007, 129, 14092-14099.] (575 mg, 1,30 mmol) en 18 ml de THF a 0 °C, se le añadió paladio al 10 % en peso sobre carbono (250 mg) seguido de la lenta adición gota a gota de 2,0 ml de formiato amónico al 25 % en agua. La reacción se dejó en agitación a 0 °C durante ~90 minutos. La reacción se diluyó con éter seguido de la adición de sulfato sódico. La reacción se filtró a través de un lecho fino de celite, que después se lavó dos veces con éter. Los extractos orgánicos se combinaron y después se redujeron antes de ponerse al vacío, produciendo 2 (458 mg, cuantitativo) en forma de un sólido de color blanquecino. CL-EM (Protocolo A): m/z 352,2 [M-H], tiempo de retención = 1,96 minutos.
Etapa 2: Síntesis de acetato de (8S)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilo (4). A una solución en agitación de 2 (209 mg, 0,591 mmol) en 8 ml de diclorometano a 0 °C se le añadió cloruro de acetilo (0,0462 ml, 0,650 mmol) seguido inmediatamente de piridina (0,0714 ml, 0,886 mmol). La reacción se dejó en agitación a 0 °C durante ~10 minutos. La reacción se redujo sobre sílice. Después, se realizó cromatografía sobre sílice (gradiente: acetona al 0 % - 15 % en heptanos). Los tubos de ensayo apropiados se concentraron y se pusieron a alto vacío para producir un sólido de color blanco. A este sólido de color blanco se le añadió HCI 4 M en dioxano (10 ml, 40 mmol) y la reacción se dejó en agitación a temperatura ambiente durante ~90 minutos. La reacción se diluyó con heptano, se concentró al vacío y después se puso a alto vacío para producir 4 (170 mg, rendimiento del 80 % , 2 etapas) en forma de un sólido de color naranja claro. CL-EM (Protocolo A): m/z 296,1 [M+H]+, tiempo de retención = 1,56 minutos. 1H RMN (400 MHz, DMSO-d6): 87,53 (s, 1H), 7,05 (s, 1H), 4,27-4,19 (m, 1H), 3,93-3,86 (m, 1H), 3,80-3,75 (m, 1H), 3,72-3,64 (m, 2H), 2,54 (s, 3H), 2,37 (s, 3H).
Etapa 3: Síntesis de 3-(clorocarbonil)biciclo[1,1,1]pentano-1-carboxilato de metilo (28). Siguiendo el procedimiento general A y usando ácido 3-(metoxicarbonil)biciclo[1.1.1]pentano-1-carboxílico 27 (86,8 mg, 0,510 mmol), cloruro
de oxalilo (0,0525 ml, 0,612 mmol), THF (6 ml) y 1 gota de DMF, se preparó 28 en forma de un sólido de color blanco (97 mg, cuant.). El producto 28 en bruto se usó tal cual sin purificación adicional.
Etapa 4: Síntesis de (8S)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il fosfato de dibencilo (29). A una solución en agitación de 2 (4,5 g, 13,4 mmol) en 8 ml de THF y 8 ml de acetonitrilo se le añadió tetracloruro de carbono (0,997 ml, 10,3 mmol) seguido de base de Hunig (0,512 ml, 2,94 mmol), dibencilfosfito (0,974 ml, 4,41 mmol) y DMAP (18 mg, 0,147 mmol). La reacción se dejó en agitación a temperatura ambiente durante ~10 minutos. La reacción se redujo sobre sílice. Después, se realizó cromatografía sobre sílice (gradiente: acetona al 0 % -25 % en heptanos). Los tubos de ensayo apropiados se concentraron y se pusieron a alto vacío para producir un sólido de color blanco. El material en bruto se disolvió en 5 ml de diclorometano seguido de la adición de ácido trifluoroacético (5 ml, 70 mmol). La reacción se dejó en agitación a temperatura ambiente durante 60 segundos, se concentró inmediatamente al vacío, y después se puso a alto vacío, proporcionando 29 (321 mg, rendimiento del 70 % , 2 etapas) en forma de una mezcla transparente de aceite y sólido de color blanco. CL-EM (Protocolo A): m/z 514,1 [M+H]+, tiempo de retención = 2,14 minutos.
Etapa 5: Síntesis de ácido 3-{[(8S)-4-{[bis(benciloxi)fosforil]oxi}-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il]carbonil}biciclo[1.1.1]pentano-1-carboxílico (30). Siguiendo el procedimiento general B y usando 29 (315 mg, 0,502 mmol), 28 (94,6 mg, 0,502 mmol), trietilamina (0,210 ml, 1,50 mmol) y THF (20 ml) y purificación usando cromatografía sobre gel de sílice (Gradiente: acetona del 0 % al 35 % en heptano). Los tubos de ensayo apropiados se combinaron y se concentraron al vacío, produciendo un sólido de color blanco. El material se disolvió en THF (10 ml) seguido de la adición de hidróxido de litio disuelto en 2,5 ml de agua. La reacción se dejó en agitación a temperatura ambiente durante ~45 minutos. La reacción se diluyó con diclorometano y se interrumpió a través de la adición de HCl 1 N (ac.). La reacción se transfirió a un embudo de decantación. La fase orgánica se separó y la fase acuosa se lavó dos veces con diclorometano. Las fases orgánicas se combinaron, se lavaron una vez con salmuera, se lavaron una vez con agua, se secaron sobre sulfato sódico, se filtraron, se concentraron al vacío y después se pusieron a alto vacío, produciendo 30 (178 mg, rendimiento del 57 % , 2 etapas). CL-EM (Protocolo A): m/z 652,2 [M+H]+, tiempo de retención = 1,97 minutos.
Etapa 6: Síntesis de (8S)-6-{[3-(clorocarbonil)biciclo[1.1.1jpent-1-il]carbonil}-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il fosfato de dibencilo (31). Siguiendo el procedimiento general A y usando 30 (174 mg, 0,267 mmol), cloruro de oxalilo (0,0298 ml, 0,347 mmol), THF (5 ml), diclorometano (1 ml) y 1 gota de DMF, se preparó 31 en forma de un sólido de color blanco (182 mg, cuant.). El producto 31 en bruto se usó tal cual sin purificación adicional.
Etapa 7: Síntesis de acetato de (8S)-6-[(3-{[(8S)-4-{[bis(benciloxi)fosforil]oxi}-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il]carbonil}biciclo[1.1.1]pent-1-il)carbonil]-8-(clorometil)-1-metil-7,8-dihidro-6H -tieno[3,2-e]indol-4-ilo (32). Siguiendo el procedimiento general B y usando 31 (167 mg, 0,249 mmol), 4 (99,3 mg, 0,299 mmol), trietilamina (0,104 ml, 0,747 mmol) y THF (20 ml) y purificación usando cromatografía sobre gel de sílice (Gradiente: acetona del 0 % al 50 % en heptano). Los tubos de ensayo apropiados se combinaron y se concentraron al vacío, produciendo 32 (103 mg, 45 % ) en forma de un sólido de color blanco. CL-EM (Protocolo A): m /z 929,3 [M+H]+, tiempo de retención = 2,44 minutos. 1H RMN (400 MHz, DMSO-d6): 88,37 (s, 1H), 8,09 (s, 1H), 7,54-7,50 (m, 2H), 7,39-7,33 (m, 10H), 5,23-5,14 (m, 4H), 4,53-4,46 (m, 2H), 4,38-4,25 (m, 4H), 4,02-3,95 (m, 2H), 3,78-3,69 (m, 2H), 2,63 (s, 6H), 2,57-2,53 (m, 6H), 2,39 (s, 3H).
Etapa 8: Síntesis de 4-nitrofenil carbonato de (8S)-8-(clorometil)-6-[(3-{[(8S)-8-(clorometil)-1-metil-4-(fosfonooxi)-7,8-dihidro-6H-tieno[3,2-e]indol-6-il]carbonil}biciclo[1.1.1]pent-1 -il)carbonil]-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il (33). A una solución en agitación de 32 (99 mg, 0,11 mmol) en 6 ml de metanol, se le añadió HCI 4 M en dioxano (6,0 ml, 20 mmol). La reacción se dejó en agitación a temperatura ambiente durante ~ 15 minutos. La reacción se redujo y después se puso a alto vacío. A una solución en agitación del material en bruto en 6 ml de diclorometano y 6 ml de THF a 0 °C se le añadió cloroformiato de p-nitrofenilo (38,6 mg, 0,192 mmol) seguido inmediatamente de trietilamina (0,0742 ml, 0,532 mmol). La reacción se dejó en agitación a 0 °C durante ~5 minutos y después se dejó calentar a temperatura ambiente mientras se agitaba. La reacción se dejó en agitación a temperatura ambiente durante ~10 minutos. La reacción se redujo. A una solución en agitación del material en bruto en 3 ml de diclorometano se le añadió una solución de TFA (3,0 ml, 39 mmol) en 3 ml de diclorometano seguido de la adición de tiofenol (0,109 ml, 1,06 mmol). La reacción se dejó en agitación a temperatura ambiente durante ~6 horas. La reacción se redujo. El material en bruto se diluyó con unos mililitros de DMSO y después se inyectó en una precolumna C 18 de 25 g (que se había equilibrado previamente con acetonitrilo y después con agua, con TFA al 0,02 % en cada fase). El material se purificó por cromatografía de fase inversa C 18 a media presión (Gradiente: acetonitrilo del 20 % al 65 % en agua con TFA al 0,02 % en cada fase) concentrando los tubos de ensayo apropiados usando un genevac, produciendo 33 (37 mg, 40 % , 3 etapas) en forma de un sólido de color blanco. C L -E m (Protocolo A): m /z 872,3 [M+H]+, tiempo de retención = 1,86 minutos. 1H RMN (400 MHz, DMSO-d6): 88,44 (s, 1H), 8,40-8,33 (m, 3H), 7,78-7,72 (m, 2H), 7,59 (m, 1H), 7,47 (m, 1H), 4,53-4,44 (m, 2H), 4,38-4,22 (m, 4H), 4,04-3,94 (m, 2H), 3,81-3,75 (m, 1H), 3,72-3,65 (m, 1H), 2,62 (s, 6H), 2,58 (s, 3H), 2,54 (s, 3H).
Preparación de N-[1-(2,5-dioxo-2,5-dihidro-1H-pirroM-M)-21-oxo-3,6,9,12,15,18-hexaoxahemcosan-21-M]-L-valil-N~5~carbamoil-N-{4-[({metil[2-(metilamino)etil]carbamoil}oxi)metil]fenil}-L-omitinamida (40).

Etapa 1: Síntesis de N-[1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-21-oxo-3,6,9,12,15,18-hexaoxahenicosan-21-il]-L-valil-N~5~carbamoil-N-[4-(hidroximetil)fenil]-L-omitinamida (36). A un matraz de fondo redondo que contenía ácido 1-(2,5-dioxo-2,5-dihidro-lH-pirrol-1-il)-3,6,9,12,15,18-hexaoxahenicosan-21-oico se le añadieron 34 (628 mg, 1,45 mmol), 20 ml de diclorometano, 2 ml de DMF, HATU (501 mg, 1,32 mmol) y base de Hunig (0,92 ml, 5,3 mmol). La reacción se dejó en agitación a temperatura ambiente durante 2 minutos antes de la adición de L-valil-N~5~-carbamoil-N-[4-(hidroximetil)fenil]-L-ornitinamida, 35 (500 mg, 1,32 mmol). La reacción se dejó en agitación a temperatura ambiente durante ~90 minutos antes de que se interrumpiera mediante la adición de TFA. La reacción se concentró a un volumen menor, se diluyó con unos ml de DMSO y después se inyectó en una precolumna C 18 de 25 g (que se había equilibrado previamente con acetonitrilo y después con agua, con TFA al 0,02 % en cada fase). El material se purificó por cromatografía de fase inversa C18 a media presión (Gradiente: acetonitrilo del 5 % al 40 % en agua con TFA al 0,02 % en cada fase) concentrando los tubos de ensayo apropiados usando un genevac, produciendo 36 (514 mg, 49 % ) en forma de un sólido transparente. CL-EM (Protocolo A): m/z 795,5 [M+H]+, tiempo de retención = 1,01 minutos.
Etapa 2: Síntesis de N-[1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-21-oxo-3,6,9,12,15,18-hexaoxahenicosan-21-il]-L-valil-N~5~-carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (37). A una solución en agitación de 36 (210 mg, 0,264 mmol) y carbonato de bis(4-nitrofenilo) (161 mg, 0,528 mmol) en 4 ml de DMF se le añadió base de Hunig (0,096 ml, 0,554 mmol). La reacción se dejó en agitación a temperatura ambiente durante ~2 horas. La reacción se inyectó en una precolumna C 18 de 25 g (que se había equilibrado previamente con acetonitrilo y después con agua, con TFA al 0,02 % en cada fase). El material se purificó por cromatografía de fase inversa C 18 a media presión (Gradiente: acetonitrilo del 5 % al 55 % en agua con TFA al 0,02 % en cada fase) concentrando los tubos de ensayo apropiados usando un genevac, produciendo 37 (180 mg, 71 % ) en forma de un sólido. C L-EM (Protocolo A): m /z 960,5 [M+H]+, tiempo de retención = 1,48 minutos.
Etapa 3: Síntesis de N-[1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-21-oxo-3,6,9,12,15,18-hexaoxahenicosan-21-il]-L-valil-N~5~-carbamoil-N-[4-(4,7,10,10-tetrametil-3,8-dioxo-2,9-dioxa-4,7-diazaundec-1-il)fenil]-L-ornitinamida (39). A una solución en agitación de 37 (640 mg, 0,667 mmol) y 38 [preparado como se describe en J. Med. Chem.
1992, 33, 559-567] (127 mg, 0,674 mmol) en 6 ml de DMA se le añadió 2,6-Lutidina (0,154 ml, 1,33 mmol) seguido de base de Hunig (0,232 ml, 1,33 mmol) y HOAT (9,1 mg, 0,67 mmol). La reacción se dejó en agitación a temperatura ambiente durante ~15 minutos. La reacción se inyectó en una precolumna C 18 de 25 g (que se había equilibrado previamente con acetonitrilo y después con agua, con TFA al 0,02 % en cada fase). El material se purificó por cromatografía de fase inversa C 18 a media presión (Gradiente: acetonitrilo del 5 % al 40 % en agua con TFA al 0,02 % en cada fase) concentrando los tubos de ensayo apropiados usando un genevac, produciendo 39 (564 mg, 84 % ) en forma de un sólido de tipo cera de color blanco. CL-EM (Protocolo A): m/z 1009,7 [M+H]+, tiempo de retención = 1,43 minutos.
Etapa 4: Síntesis de N-[1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-21-oxo-3,6,9,12,15,18-hexaoxahenicosan-21-il]-L-valil-N~5~-carbamoil-N-{4-[({metil[2-(metilamino)etil]carbamoil}oxi)metil]fenil}-L-ornitinamida (40). A una mezcla en agitación de 39 (470 mg, 0,466 mmol) en 6 ml de diclorometano se le añadió TFA (3,0 ml, 40 mmol). La reacción se dejó en agitación a temperatura ambiente durante ~10 minutos. La reacción se redujo. El residuo se purificó por cromatografía de fase inversa C 18 a media presión (Gradiente: acetonitrilo del 5 % al 30 % en agua con TFA al 0,02 % en cada fase) concentrando los tubos de ensayo apropiados usando un genevac, produciendo 40 (326 mg, 68 % ) en forma de una mezcla de aceite/sólido de color blanco. CL-EM (Protocolo A): m/z 909,8 [M+H]+, tiempo de retención = 0,91 minutos.
Preparación de N-[1-(2,5-dioxo-2,5-dihidro-1H-pirroM-M)-21-oxo-3,6,9,12,15,18-hexaoxahemcosan-21-M]-L-valil-N~5~-carbamoil-N-[4-({[(2-{[({(8S)-8-(cloromethil)-6-[(3-{[(8S)-8-(clorometil)-1-metil-4-(fosfonooxi)-7,8-dihidro-6H-tieno[3,2-e]mdol-6-N]carbonM)biddo[1.1.1]pent-1-N)carboml]-1-metN-7,8-dihidro-6H-tieno[3,2-e]indol-4-il}oxi)carbonil](m etil)am ino}etil)(m etil)carbamoil]oxi}metil)fenil]-L-om itinam ida (41).

Etapa 1: Síntesis de N-[1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-21-oxo-3,6,9,12,15,18-hexaoxahenicosan-21-il]-L-valil-N~5~-carbamoil-N-[4-({[(2-{[({(8S)-8-(clorometil)-6-[(3-{[(8S)-8-(clorometil)-1-metil-4-(fosfonooxi)-7,8-dihidro-6H-tieno[3,2-e]indol-6-il]carbonil}biciclo[1.1.1]pent-1-il)carbonil]-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il}oxi)carbonil](metil)amino}etil)(metil)carbamoil]oxi}metil)fenil]-L-ornitinamida (41). A un vial de 2 dram que contenía 33 (8,0 mg, 0,0092 mmol) y 40 (10,3 mg, 0,0101 mmol) se le añadió 1,0 ml de DmA seguido de base de Hunig (0,00639 ml, 0,0367 mmol), 2,6-Lutidina (0,00425 ml, 0,0367 mmol) y HOAT (1,25 mg, 0,0367 mmol). La reacción se dejó en agitación a temperatura ambiente durante ~5 minutos. La reacción en bruto se inyectó en una precolumna C 18 de 4 g (que se había equilibrado previamente con acetonitrilo y después con agua, con TFA al 0,02 % en cada fase). El material se purificó por cromatografía de fase inversa C 18 a media presión (Gradiente: acetonitrilo del 15 % al 50 % en agua con TFA al 0,02 % en cada fase) seguido de una segunda purificación por el procedimiento A, concentrando los tubos de ensayo apropiados usando un genevac, produciendo 41 (8,9 mg, 59 % ) en forma de un sólido de color blanco. CL-EM (Protocolo A): m/z 1642,9 [M+2H]+, tiempo de retención = 1,61 minutos.
Preparación de LP: ácido (2S,3S,4S,5R,6S)-6-(((S)-8-(dorometN)-6-(5-((S)-8-(dorometM)-4-(((2-((((4-((23S,26S)-1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-23-isopropil-21,24-dioxo-26-(3-ureidopropil)-3,6,9,12,15,18-hexaoxa-22,25-diazaheptacosan-27-amido)bencil)oxi)carbonil)(metil)amino)etil)(metil)carbamoil)oxi)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-3,4,5-trihidroxitetrahidro-2H-piran-2-carboxílico (49)
O oruro de oxa 10
tT T 7 < io r r O y 1

Etapa 1: Síntesis de 5-cloro-5-oxopentanoato de ferc-butilo (44): A una solución de ácido 5-(ferc-butoxi)-5-oxopentanoico (110 mg, 0,58 mmol) en THF (3 ml) se le añadió cloruro de oxalilo (0,58 ml, 1,2 mmol, 2 M en DCM) a 0 °C , seguido de 1 gota de DMF. La mezcla se agitó a 0 °C durante 30 min y se concentró al vacío para dar el cloruro de ácido 44 correspondiente en forma de un sólido de color blanco.
Etapa 2. Síntesis de triacetato de (2S,3R,4S,5S,6S)-2-(((S)-6-(ferc-butoxicarbonil)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-6-(metoxicarbonil)tetrahidro-2H-piran-3,4,5-triilo (43)
A una solución de 2 (226 mg, 0,64 mmol) en DCM (23 ml) se le añadieron tamices moleculares de 4 A (1,17 g, en polvo, <5 micrómetros, activados) y la mezcla se agitó a temperatura ambiente durante 30 min. A la mezcla de reacción se le añadió 1-2,2,2-tricloroetanimidato de 2,3,4-triacetato del éster metílico de alfa-D-glucurónido (42, 367 mg, 0,77 mmol) y la mezcla se enfrió a -25 °C. Se añadió lentamente una solución de BF3 -Et2 O (0,13 ml, 0,32 mmol) en DCM (10 ml) y la mezcla se agitó por debajo de -20 °C durante 1 h. La mezcla de reacción se filtró y la solución se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice usando un gradiente de EtOAc al 0-60 % en Heptanos para dar el producto 43 en forma de un sólido de color amarillo, 261 mg (61 %). CL-EM (Protocolo B): m/z 692,1 (M+Na), tiempo de retención = 1,09 min. 1H RMN (400 MHz, CLOROFORMO-d) 8 = 7,29 (s, 1H), 7 ,13 (s, 1H), 5,40 (s a, 3H), 4,30 (d, J = 7,4 Hz, 2H), 4,06 - 3,92 (m, 2H), 3,85 - 3,73 (m, 4H), 3,69 (d, J = 10,5 Hz, 1H), 3,36 (t, J = 10,5 Hz, 1H), 2,56 (s, 3H), 2,10 (s, 6H), 2,08 (s, 3H), 1,62 (s, 9H).
Etapa 2. Síntesis de triacetato de (2S,3R,4S,5S,6S)-2-(((S)-6-(5-(ferc-butoxi)-5-oxopentanoil)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-6-(metoxicarbonil)tetrahidro-2H-piran-3,4,5-triilo (45)
Una solución de 43 (261 mg, 0,39 mmol) se trató con HCI 4 M en dioxano (3 ml) durante 1 h. Se concentró al vacío, el residuo se disolvió en THF (3,0 ml) y se añadió una solución de cloruro de ácido 44 (0,58 mmol) en THF (3,0 ml) seguido de TEA (0,163 ml, 1,2 mmol). La mezcla se agitó a 0 °C durante 30 min. La mezcla se concentró y el residuo se purificó por cromatografía sobre gel de sílice usando un gradiente de EtOAc (0-70 % ) en heptanos para dar el producto en forma de un sólido de color blanquecino, 224 mg (78 %). CL-EM (Protocolo B): m/z 740,2 [M+H]+, tiempo de retención = 1,07 minutos. 1H RMN (400 MHz, CLOROFORMO-d) 8 = 8,26 (s a, 1H), 7 ,13 (s, 1H), 5,45 -5,25 (m, 4H), 4,32 (d, J = 9,0 Hz, 2H), 4 ,12 (t, J = 8,8 Hz, 1H), 4,05 (d, J = 8,6 Hz, 1H), 3,79 - 3,66 (m, 5H), 3,33 (t, J = 10,9 Hz, 1H), 2,63 (d, J = 7,0 Hz, 1H), 2,58 -2 ,47 (m, 4H), 2,45 -2 ,35 (m, 2H), 2,06 (m, 11H), 1,47 (s, 9H).
Etapa 3. Síntesis de triacetato de (2S,3R,4S,5S,6S)-2-(((S)-8-(clorometil)-6-(5-((S)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-6-(metoxicarbonil)tetrahidro-2H-piran-3,4,5-triilo (46)
Una solución de 45 (116 mg, 0,16 mmol) se trató con DCM (1,5 ml) y TFA (1 ml) a ta durante 1 h, se concentró al vacío, el residuo se disolvió en THF a 0 °C y se añadieron cloruro de oxalilo (0,16 ml, 0,32 mmol, 2 M en DCM) y DMF (1 gota). La mezcla se agitó a 0 °C durante 30 min y se concentró al vacío para dar el cloruro de ácido correspondiente.
En un vial separado, el compuesto 2 (83 mg, 0,24 mmol) se trató con HCI 4 M (2 ml) en dioxano a ta durante 1 h, se concentró al vacío, el residuo se disolvió en THF (5 ml) a 0 °C y se añadió Et3N (100 ul, 0,78 mmol), seguido
de una solución del cloruro de ácido anterior en THF (5 ml). La mezcla se agitó a 0 °C durante 20 min. La mezcla se concentró y el residuo se purificó por cromatografía sobre gel de sílice usando un gradiente de EtOAc (0 -100 % ) en heptanos para dar el producto 46 en forma de un sólido de color blanquecino, 114 mg (79 % ). CL-EM (Protocolo B): m/z 919,1 [M+H]+, tiempo de retención = 1,05 minutos, 1H RMN (400 MHz, DMSO-d6) 8 = 10,41 (s, 1H), 8,22 (s, 1H), 7,93 (s, 1H), 7,47 (s, 1H), 7,38 (s, 1H), 5,81 -5 ,70 (m, 4H), 5,63 -5 ,52 (m, 1H), 5,19 (t, J = 8,6 Hz, 1H), 5,12 (t, J = 9,8 Hz, 1H), 4,76 (d, J = 9,8 Hz, 1H), 4,30 - 4,16 (m, 3H), 3,86 (m, 2H), 3,68 (s, 3H), 3,64 - 3,51 (m, 2H), 2,73 (s a, 1H), 2,62 (m, 2H), 2,50 (s, 6H), 2,10-2,02 (m, 12H).
Etapa 4. Síntesis de triacetato de (2S,3R,4S,5S,6S)-2-(((S)-6-(5-((S)-4-(((2-((tercbutoxicarbonil)(metil)amino)etil)(metil)carbamoil)oxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-6-(metoxicarbonil)tetrahidro-2H-piran-3,4,5-triilo (47): A una solución de 46 (50 mg, 0,054 mmol) en THF (3 ml) a 0 °C se le añadió una solución de cloroformiato de 4-nitrofenilo (22 mg, 0,10 mmol) en DCM (0,5 ml), seguido de DIPEA (57 ul, 0,33 mmol) y la mezcla se agitó a 0 °C durante 1 h. A la mezcla anterior se le añadió una solución de 38 [preparado como se describe en J. Med. Chem. 1992, 33, 559-567] (31 mg, 0,16 mmol) en THF (0,5 ml), la mezcla se agitó a 0 °C durante 30 min y se concentró al vacío y el residuo se purificó se purificó por cromatografía sobre gel de sílice usando un gradiente de EtOAc (0 -100 % ) en heptanos para dar el producto 47 en forma de un sólido de color blanco, 56 mg (91 %). CL-EM (Protocolo B): m/z 1150,2 [M+NH4], tiempo de retención = 1,14 minutos
Etapa 5: Síntesis de ácido (2S,3S,4S,5R,6S)-6-(((S)-6-(5-((S)-4-(((2-((tercbutoxicarbonil)(metil)amino)etil)(metil)carbamoil)oxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-3,4,5-trihidroxitetrahidro-2H-piran-2-carboxílico (48): A una solución de 47 (56 mg, 0,049 mmol) en THF/MeOH (1/1, 6 ml) a 0 °C se le añadió una solución de L O H H 2 O (21 mg, 0,49 mmol) en agua (0,5 ml). La mezcla se agitó a 0 °C durante 1 h. Se añadió ácido acético (50 mg) y la reacción se concentró al vacío para dar el producto en bruto 48 en forma de un sólido de color blanco, 45 mg (90 % ). CL-EM (Protocolo B): m/z 1014,9 [M+Na], tiempo de retención = 1,0 minutos
Etapa 6: Síntesis de ácido (2S,3S,4S,5R,6S)-6-(((S)-8-(clorometil)-6-(5-((S)-8-(clorometil)-4-(((2-((((4-((23S,26S)-1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-23-isopropil-21,24-dioxo-26-(3-ureidopropil)-3,6,9,12,15,18-hexaoxa-22,25-diazaheptacosan-27-amido)bencil)oxi)carbonil)(metil)amino)etil)(metil)carbamoil)oxi)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-3,4,5-trihidroxitetrahidro-2H-piran-2-carboxílico (49) y ácido (8S)-8-(clorometil)-6-{5-[(8S)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il]-5-oxopentanoil}-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilbeta-D-glucopiranosidurónico (57): El compuesto 48 (23 mg, 0,02 mmol) se trató con TFA enfriado previamente (1 ml) a 0 °C durante 5 min y se concentró al vacío. El residuo se disolvió en DMF (2 ml), se añadieron el compuesto 37(19 mg, 0,02 mmol), lutidina (14 ul, 0,12 mmol), DIPEA (21 ul, 0,12 mmol) y HOAt (2,7 mg, 0,02 mmol) y la mezcla se agitó a 30 °C durante 1 h La mezcla se purificó por HPLC de fase inversa (Procedimiento C) para dar el compuesto 49 en forma de un sólido de color blanquecino, 8 mg (20 % ). CL-EM (Protocolo B): m/z 1714,6 [M+H]+, tiempo de retención = 0,89 minutos y el compuesto 57 en forma de una goma 7 mg (50 %): CL-EM (Protocolo B): m/z 779,1 [M+H]+, tiempo de retención = 0,90 minutos
Preparación de: (4-((23S,26S)-1-(2,5-dioxo-2,5-dihidro-1H-pirroM-M)-23-isopropN-21,24-dioxo-26-(3-ureidopropM)-3,6,9,12,15,18-hexaoxa-22,25-diazaheptacosan-27-amido)bencil)etano-1,2-diilbis(metilcarbamato) de (S)-8-(dorometN)-6-(5-((S)-8-(dorometN)-1-metM-4-(fosfonooxi)-7,8-dihidro-6H-tieno[3,2-e]mdol-6-M)-5-oxopentanoN)-1-metM-7,8-dihidro-6H-tieno[3,2-e]mdol-4-No (56)

Etapa 1: Síntesis de 5-cloro-5-oxopentanoato de metilo (51): Se disolvió ácido 5-metoxi-5-oxopentanoico (128 mg, 0,88 mmol) en THF (5 ml) a 0 °C, se añadieron cloruro de oxalilo (0,9 ml, 1,8 mmol, 2 M en DCM) y DMF (1 gota) y la mezcla se agitó a 0 °C durante 30 min. Se concentró al vacío para dar el cloruro de ácido 51 correspondiente en forma de un sólido de color blanco que se usó sin purificación adicional.
Etapa 2. Síntesis de (S)-4-((bis(benciloxi)fosforil)oxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de ferc-butilo (50)
A una solución de 2 (260 mg, 0,74 mmol) en ACN (8 ml) y THF (8 ml) se le añadieron CCU (1 ml), DIPEA (0,52 ml, 2,94 mmol), difenilfosfito (1,03 ml, 4,41 mmol) y DMAP (18 mg). La mezcla se agitó a ta durante 15 min, se concentró al vacío y el residuo se purificó por cromatografía sobre gel de sílice usando un gradiente (0-60 % ) de EtOAc en heptanos para dar el producto 50 en forma de un aceite incoloro, 365 mg (81 % ). CL-EM (Protocolo B): m /z 631,2 [M+H]+, tiempo de retención = 1,18 minutos
Etapa 3. Síntesis de (S)-5-(4-((bis(benciloxi)fosforil)oxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoato de metilo (52)
A una solución de 50 (365 mg, 0,58 mmol) en DCM (3 ml) se le añadió TFA (3 ml) y la mezcla se agitó a ta durante 2 min y se concentró al vacío. El residuo se disolvió en THF (5,0 ml), se añadió una solución del cloruro de ácido 51 (0,88 mmol) en THF (5,0 ml) seguido de Et3 N (0,37 ml, 0,24 mmol) y la mezcla se agitó a 0 °C durante 30 min. La reacción se concentró y el residuo se purificó por cromatografía sobre gel de sílice usando un gradiente (0 80 % ) de EtOAc en heptanos para dar el producto 52 en forma de un aceite incoloro, 240 mg (64 % ). CL-EM (Protocolo B): m/z 642,1 [M+H]+, tiempo de retención = 1,09 minutos
Etapa 4. Síntesis de ácido (S)-5-(4-((bis(benciloxi)fosforil)oxi)-8-(clorometil)-1-meti-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoico (53)
A una solución de 52 (235 mg, 0,37 mmol) en THF (10 ml) a 0 °C se le añadió una solución de LO H/H 2 O (155 mg, 3,7 mmol) en agua (2,5 ml) y la mezcla se agitó a 0 °C durante 1,5 h. La mezcla se diluyó con DCM, se acidificó
con HCl 1 M, la fase orgánica se separó y la fase acuosa se extrajo dos veces con DCM. Las fases orgánicas combinadas se secaron sobre MgSO4. La mezcla se concentró al vacío y el residuo que se purificó por HPLC de fase inversa (Procedimiento C) para dar el producto 53 en forma de una espuma de color blanquecino, 27 mg (12 % ). C L -e M (Protocolo B): m/z 628,1 [M+H]+, tiempo de retención = 1,01 minutos
Etapa 5. Síntesis de ((S)-8-(clorometil)-6-(5-((S)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)fosfato de dibencilo (54)
A una solución de 53 (27 mg, 0,043 mmol) en THF (5 ml) a 0 °C se le añadieron cloruro de oxalilo (0,043 ml, 0,086 mmol, 2 M en DCM) y DMF (1 gota). La mezcla se agitó a 0 °C durante 30 min y se concentró al vacío para dar el cloruro de ácido correspondiente en forma de una espuma de color amarillo.
En un vial separado, el compuesto 2 (23 mg, 0,064 mmol) se trató con HCI 4 M (1 ml) a ta durante 1 h y se concentró al vacío. El residuo se disolvió en THF (5 ml), se enfrió a 0 °C y se añadió a una solución del cloruro de ácido anterior en THF (5 ml) y Et3 N (0,018 ml, 0,13 mmol). La mezcla se concentró al vacío y el residuo se purificó por HPLC de fase inversa (Procedimiento C) para dar el producto 54 en forma de un sólido de color blanquecino, 28 mg (75 % ). CL-EM (Protocolo B): m/z 863,2 [M+H]+, tiempo de retención = 1,16 minutos
Etapa 6. Síntesis de (4-nitrofenil)carbonato de (S)-8-(clorometil)-6-(5-((S)-8-(clorometil)-1-metil-4-(fosfonooxi)-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilo (55) y dihidrogenofosfato de (8S)-8-(clorometil)-6-{5-[(8S)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il]-5-oxopentanoil}-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilo (61)
A una solución de 54 (40 mg, 0,046 mmol) en THF (5 ml) a 0 °C se le añadió una solución de cloroformiato de 4-nitrofenilo (19,4 mg, 0,092 mmol) en Dc M (0,5 ml) y DIPEA (0,049 ml, 0,28 mmol). La mezcla se agitó a 0 °C durante 30 min. La reacción se concentró al vacío y el residuo se purificó por cromatografía sobre gel de sílice usando un gradiente (0-60 % ) de acetona en heptanos para dar el intermedio PNP carbonato en forma de un sólido de color blanco, 48 mg. Se disolvió en DCM (2 ml), se añadieron TFA (2 ml) y tiofenol (0,047 ml, 0,46 mmol) y la mezcla se agitó a ta durante 3 h. La reacción se concentró al vacío y el residuo se purificó por HPLC de fase inversa (Procedimiento C) para dar 17,8 mg (46 % ) del producto 55 en forma de un sólido de color blanco (17,8 mg, 46 % ). C l -EM (Protocolo B): m/z 848,2 [M+H]+, tiempo de retención = 1,09 minutos y 5,2 mg (17 % ) de producto 61 en forma de una goma CL-EM (Protocolo B): m/z 683,2 [M+H]+, tiempo de retención = 0,93 minutos Etapa 7. Síntesis de (4-((23S,26S)-1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-23-isopropil-21,24-dioxo-26-(3-ureidopropil)-3,6,9,12,15,18-hexaoxa-22,25-diazaheptacosan-27-amido)bencil) etano-1,2-diilbis(metilcarbamato) de (S)-8-(clorometil)-6-(5-((S)-8-(clorometil)-1-metil-4-(fosfonooxi)-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilo (56)
A una solución de 55 (10,5 mg, 0,012 mmol) en DMF (1 ml) se le añadieron 40 (13,9 mg, 0,014 mmol), lutidina (0,009 ml, 0,074 mmol), DIPEA (0,013 ml, 0,074 mmol) y HOAt (1,7 mg, 0,012 mmol). La mezcla se agitó a ta durante 20 min, se concentró al vacío y el residuo se purificó por HPLC de fase inversa (Procedimiento C) para dar el producto 56 en forma de una espuma de color blanco, 12 mg (60 % ). CL-EM (Protocolo B): m/z 1619,6 [M+2H]+, tiempo de retención = 0,95 minutos
Preparación de diacetato de pentaddo[4.2.0,0~2,5~.0~3,8~.0~4,7~]octano-1,4-diNbis[carboml(8S)-8-(dorometM)-1-metN-7,8-dihidro-6H-tieno[3,2-e]mdolo-6,4-diNo] (60)

Etapa 1: Síntesis de diacetato de pentaciclo[4.2.0,0~2,5~.0~3,8~.0~4,7~]octano-1,4-diilbis[carbonil(8S)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6,4-diilo] (60): Una solución de 3 (47 mg, 0,12 mmol) se trató con HCI 4 M (1,5 ml en dioxano) a ta durante 90 min, se concentró al vacío, el residuo se disolvió en DCM (4 ml) y TEA (0,025 ml), se añadió a una solución de ácido 1,4-cubanodicarboxílico (11,5 mg, 0,06 mmol, 72) y se disolvió en 1 ml de diclorometano anhidro seguido de la adición de HATU (47 mg, 0,12 mmol). La reacción se agitó a temperatura ambiente durante 16 h. La mezcla se purificó con una columna de HPLC de fase inversa: (Columna Phenomenex Luna C 18 5u 150 x 21,5 mm, Gradiente A cC n al 20 % -90 % / Agua (con AcOH al 0,02 % en cada una) durante 20 min (más un tiempo isocrático inicial de 6 minutos a AcCN al 20 %/agua) con 0,02 % para proporcionar 3,1 mg de 60 (sólido de
color blanco, 7 % ). CL-EM (Protocolo A): m/z: 747,1 (M+H)+, tiempo de retención = 2,39 min
Preparación de dihidrogenofosfato de (S)-8-(dorometM)-6-(5-((S)-1-(dorometM)-5-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanamido)-3-metilbutanamido)propanamido)-1,2-dihidro-3H-benzo[e]indol-3-il)-5-oxopentanoM)-1-metN-7,8-dihidro-6H-tieno[3,2-e]mdol-4-No (62)

Etapa 1: Síntesis de (S)-4-(benciloxi)-8-(dorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol (63): A una solución agitada del compuesto 1 (300 mg, 0,676 mmol) en DCM seco (5 ml) se le añadió gota a gota HCI 4,0 M en EtOAc (5 ml) a 0 °C. Después de la adición, la mezcla se agitó a ta durante 1,5 h. La mezcla se concentró al vacío y después se co-evaporó una vez con DCM para dar el producto 63 (260 mg, 100 % ) que se usó tal cual en la siguiente etapa.
Etapa 2: Síntesis de (S)-5-(4-(benciloxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoato de ferc-butilo (63)
A un matraz de fondo redondo que contenía (S)-4-(benciloxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indolo-6-carboxilato de ferc-butilo (1) (500 mg, 1,13 mmol) se le añadió TFA al 25 % en DCM (10 ml). La reacción se agitó durante 30 min. La reacción se concentró y se puso al vacío durante 30 min para dar el residuo de-boc 63 que se recogió en 5 ml de DCM y se usó en la siguiente etapa.
A un matraz de fondo redondo purgado con N2 , que contenía ácido 5-(ferc-butoxi)-5-oxopentanoico (212 mg, 1,13 mmol) en 5 ml de DCM anhidro, se le añadió cloruro de oxalilo (0,101 ml, 1, 13 mmol). A esta solución se le añadió 1 gota de DMF y el sistema se agitó durante 3 horas. Se observó inmediatamente formación de gas. La reacción se concentró al vacío para dar el cloruro de ácido 44 en bruto, que se recogió en DCM y se añadió a un matraz de fondo redondo que contenía el residuo desprotegido 63 descrito anteriormente y TEA (0,144 ml). La reacción se agitó a temperatura ambiente durante 2 horas. La mezcla de reacción en bruto se concentró al vacío, se recogió en 25 ml de DCM y se transfirió a un embudo de decantación. La fase orgánica se lavó con HCl 1 M (3 x), agua (3 x) y salmuera (2 x). La fase orgánica se secó sobre sulfato sódico y se filtró y el filtrado se concentró para dar un sólido en bruto. El producto en bruto se purificó por cromatografía sobre gel de sílice (Gradiente: MeOH del 0 % al 10 % en DCM) para dar 64 en forma de un sólido de color amarillo (525 mg, 90 % ). CL-EM (Protocolo A): m /z 514 [M+H]+, tiempo de retención = 2,43 minutos.
Etapa 3: Síntesis de (S)-5-((S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)propanamido)-1-(clorometil)-1,2-dihidro-3H-benzo[e]indolo-3-carboxilato de ferc-butilo (66).
A un matraz de fondo redondo con (S)-5-amino-1-(clorometil)-1,2-dihidro-3H-benzo[e]indolo-3-carboxilato de fercbutilo (1000 mg, 3,0 mmol, 65) (preparado como se describe en el documento W O2015023355) en 15 ml de DCM se le añadió (S)-(1-cloro-1-oxopropan-2-il)carbamato de (9H-fluoren-9-il)metilo (991 mg, 3,0 mmol seguido de 1,2 ml de base de Hunig. La reacción se agitó durante 1 hora y se concentró para dar un vidrio en bruto. La mezcla de reacción en bruto se purificó por cromatografía sobre gel de sílice (gradiente: MeOH del 0 % al 10 % en DCM) para dar 66 en forma de un sólido de color blanco (1529 mg, 80 % ). CL-EM (Protocolo A): m/ z 626 [M+H]+, tiempo de retención = 2,32 minutos.
Etapa 4: Síntesis de ((S)-1-(((S)-3-(5-((S)-4-(benciloxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-(clorometil)-2,3-dihidro-1H-benzo[e]indol-5-il)amino)-1-oxopropan-2-il)carbamato de (9H-fluoren-9-il)metilo (67).
A una solución en agitación de (S)-5-(4-(benciloxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoato de ferc-butilo (500 mg, 0,973 mmol, 64) en 10 ml de DCM se le añadieron 2,5 ml de TFA y la reacción se agitó durante 3 horas. Después de que se completara, la reacción se concentró al vacío para dar un sólido de color blanco pálido. Después, el sólido se recogió en 5 ml de DCM anhidro y cloruro de oxalilo (0,32 ml, 0,93 mmol). La reacción se agitó durante 3 horas y se concentró al vacío para dar un sólido de color blanco. Se recogió (S)-5-((S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)propanamido)-1-(clorometil)-1,2-dihidro-3H-benzo[e]indolo-3-carboxilato de ferc-butilo (0,973 mmol, 66) en TFA al 25 % en DCM (5 ml) y se agitó durante 30 min. La reacción se concentró al vacío y se recogió de nuevo en 5 ml de DCM. Se añadió base de Hunig (0,32 ml) seguido del cloruro de ácido descrito previamente. La reacción se agitó durante 2 horas. Después de que se completara, la mezcla de reacción se concentró al vacío. El producto en bruto se purificó por cromatografía sobre gel de sílice (Gradiente: MeOH del 0 % al 10 % en DCM) para dar 67 en forma de un sólido de color blanco (510 mg, 54 % ). % ). CL-EM (Protocolo A): m/ z 965 [M+H]+, tiempo de retención = 2,61 minutos.
Etapa 5: Síntesis de ((S)-1-(((S)-1-(((S)-3-(5-((S)-4-(benciloxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-(clorometil)-2,3-dihidro-1H-benzo[e]indol-5-il)amino)-1-oxopropan-2-il)amino)-3-metil-1-oxobutan-2-il)carbamato de ferc-butilo (68).
A una solución en agitación de ((S)-1-(((S)-3-(5-((S)-4-(benciloxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-(clorometil)-2,3-dihidro-1H-benzo[e]indol-5-il)amino)-1-oxopropan-2-il)carbamato de (9H-fluoren-9-il)metilo (500 mg, 0,518 mmol, 67) en 5 ml de DCM se le añadieron 5 ml de dietilamina. La reacción se agitó durante 3 horas y se concentró al vacío para dar un sólido de color amarillo. El sólido en bruto de color amarillo se recogió en 10 ml de THF anhidro seguido de (ferc-butoxicarbonil)-L-valinato de 2,5-dioxopirrolidin-1-ilo (163 mg, 0,518 mmol) seguido de TEA (0,2 ml). La reacción se agitó a 70 grados Celsius durante 4 horas. Después de que se completara, la mezcla de reacción se concentró al vacío. El producto en bruto se purificó por cromatografía sobre gel de sílice (Gradiente: MeOH del 0 % al 10 % en DCM) para dar 68 en forma de un sólido de color blanco (198 mg, 40 % ). % ). CL-EM (Protocolo A): m/ z 942 [M+H]+, tiempo de retención = 2,49 minutos.
Etapa 6: Síntesis de ((S)-1-(((S)-1-(((S)-1-(clorometil)-3-(5-((S)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-2,3-dihidro-1H-benzo[e]indol-5-il)amino)-1-oxopropan-2-il)amino)-3-metil-1-oxobutan-2-il)carbamato de ferc-butilo (69).
Una solución en agitación de ((S)-1-(((S)-1-(((S)-3-(5-((S)-4-(benciloxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-(clorometil)-2,3-dihidro-1H-benzo[e]indol-5-il)amino)-1-oxopropan-2-il)amino)-3-metil-1-oxobutan-2-il)carbamato de ferc-butilo (325 mg, 0,345 mmol, 68) en 7 ml de THF en una atmósfera de nitrógeno se enfrió a 0 C usando un baño de hielo. Después, se añadió paladio al 10 % en peso sobre carbono activado (10 mg) seguido de la lenta adición gota a gota de 0,5 ml de formiato amónico al 25 % en agua. La reacción se dejó en agitación a 0 C. durante 1 hora. Después de que se completara, la mezcla de reacción se filtró a través de una capa de celite y el filtrado se concentró al vacío. El producto en bruto se purificó por cromatografía sobre gel de sílice (gradiente: MeOH del 0 % al 10 % en DCM) para dar 69 en forma de un sólido de color amarillo (181 mg, 61 % ). CL-EM (Protocolo A): m/ z 852 [M+H]+, tiempo de retención = 2,18 minutos.
Etapa 7: Síntesis de ((S)-1-(((S)-1-(((S)-3-(5-((S)-4-((bis(benciloxi)fosforil)oxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-(clorometil)-2,3-dihidro-1H-benzo[e]indol-5-il)amino)-1-oxopropan-2-il)amino)-3-metil-1-oxobutan-2-il)carbamato de ferc-butilo (70).
A una solución en agitación de ((S)-1-(((S)-1-(((S)-1-(clorometil)-3-(5-((S)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-2,3-dihidro-1H-benzo[e]indol-5-il)amino)-1-oxopropan-2-il)amino)-3-metil-1-oxobutan-2-il)carbamato de ferc-butilo (165 mg, 0,193 mmol, 69) en 10 ml de Th F y 10 ml de AcCN se le añadió tetracloruro de carbono (2,04 ml, 21,0 mmol) seguido de base de Hunig (1,12 ml, 6,45 mmol), dibencilfosfito (320 mg, 1,16 mmol) y DMAP (catalítico). La reacción se dejó en agitación a temperatura ambiente durante 20 minutos. La reacción se concentró para dar un vidrio en bruto. La mezcla de reacción en bruto se purificó por cromatografía sobre gel de sílice (gradiente: MeOH del 0 % al 10 % en DCM) para dar 70 en forma de un vidrio de color blanco (51 mg, 24 %). CL-e M (Protocolo A): m/ z 1113 [M-H]-, tiempo de retención = 2,50 minutos.
Etapa 8: Síntesis de dihidrogenofosfato de (S)-8-(clorometil)-6-(5-((S)-1-(clorometil)-5-((S)-2-((S)-2-(6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanamido)-3-metilbutanamido)propanamido)-1,2-dihidro-3H-benzo[e]indol-3-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilo (62).
En un matraz de fondo redondo equipado con una barra de agitación, se recogió ((S)-1-(((S)-1-(((S)-3-(5-((S)-4-((bis(benciloxi)fosforil)oxi)-8-(dorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-(dorometil)-2,3-dihidro-1H-benzo[e]indol-5-il)amino)-1-oxopropan-2-il)amino)-3-metil-1-oxobutan-2-il)carbamato de ferc-butilo (40 mg 0,036 mmol, 70) en 5 ml de DCM. Se añadieron TFA (2,5 ml) y 2 gotas de tiofenol y la reacción se agitó durante 6 horas. El material en bruto se concentró al vacío. El residuo en bruto se recogió en 3 ml de DMF y se añadió 6-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)hexanoato de pentafluorofenilo (71) (13,6 mg, 0,036 mmol) seguido de TEA (0,036 mmol). La reacción se agitó durante 1 hora. La reacción se concentró al vacío y se purificó por el Procedimiento de HPLC para dar 62 en forma de un sólido de color blanco (25 mg, 68 % ), tiempo de retención = 7 ,115 minutos. CL-EM (Protocolo A): m/ z 1025 [M+H]+, tiempo de retención = 2,01 minutos
Preparación de N~2~-acetN-N~6~-[(9H-fluoren-9-Nmetoxi)carbonM]-L-NsN-L-valN-N~5~-carbamoN-N-{4-[({metil[2-(metilam ino)etil)carbamoil}oxi)metil)fenil}-L-omitinamida (74):

Etapa 1: Síntesis de N~2~-acetil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N~5~-carbamoil-N-[4-(4,7,10,10-tetrametil-3,8-dioxo-2,9-dioxa-4,7-diazaundec-1-il)fenil]-L-ornitinamida (72A): A una solución agitada de N~2~-acetil-N~6~-[(9H-fluoren-9-ilmetoxi)carbonilj-L-lisil-L-valil-N-5--carbamoil-N-[4-({[(4-nitrofenoxi)carbonil]oxi}metil)fenil]-L-ornitinamida (72) (preparada como se describe en el documento US 9169264) (1,00 g, 1,07 mmol) en DMF (20 ml) se le añadió 38 [preparado como se describe en J. Med. Chem. 1992, 33, 559 567] (241 mg, 1,28 mmol) a 0 °C en un globo de N2. La mezcla resultante se agitó a 0 °C durante 30 min. La mezcla se vertió en TBME (200 ml). La suspensión de color blanco resultante se filtró y se lavó con TBME (200 ml) para proporcionar el compuesto del título 72A (750 mg, 71,3 % ) en forma de un sólido de color amarillo
Etapa 2: Síntesis de N~2~-acetil-N-6--[(9H-fluoren-9-ilmetoxi)carbonil]-L-lisil-L-valil-N~5~-carbamoil-N-{4-[({metil[2-(metilamino)etil]carbamoil}oxi)metil]fenil}-L-ornitinamida (74): A una suspensión agitada del compuesto 72a (10,00 g, 10,1 mmol) en DCM (60,0 ml) se le añadió TFA (50,0 ml) a 0 °C. La solución resultante se agitó a 0 °C durante 40 min. La suspensión resultante se filtró. La torta de filtro se lavó con TBME (200 ml) y se secó al vacío a sequedad para proporcionar el producto en bruto. El producto en bruto se purificó por HPLC preparativa (Columna: Phenomenex Synergi Max-RP 250X50 mm, 10 um, Gradiente: acetonitrilo del 25 % al 55 % en agua con TFA al 0,1 % en cada fase durante 17,5 min y mantenido durante 8 min a acetonitrilo al 100 % en agua que contenía TFA al 0,1 % , Caudal: 80 ml/min) para dar el compuesto del título 74 (5,2 g, rendimiento del 51,3 % ) en forma de un sólido de color blanco
1H RMN (400 MHz, DMSO-d6) □ 10,03 (s, 1H), 8,47 (s a, 2H), 8,10 (d, J = 7,0 Hz, 1H), 8,03 (d, J = 7,8 Hz, 1H), 7,89 (d, J = 7,5 Hz, 2H), 7,69 (d, J = 7,3 Hz, 3H), 7,60 (d, J = 8,5 Hz, 2H), 7,45 - 7,38 (m, 2H), 7,36 - 7,29 (m, 4H), 7,26 (t, J = 5,4 Hz, 1H), 6,02 (s a, 1H), 5,01 (s, 2H), 4,43 -4 ,33 (m, 1H), 4,32 -4 ,16 (m, 5H), 3,49 (t, J = 6,0 Hz, 2H), 3,06-2,92 (m, 6H), 2,86 (s, 3H), 2,62 -2 ,53 (m, 3H), 1,99 (dd, J = 6,5, 13,3 Hz, 1H), 1,85 (s, 3H), 1,76 - 1,27 (m, 10H), 0,83 (d, J = 6,8 Hz, 3H), 0,86 (d, J = 6,8 Hz, 3H)
Preparación de LP: ácido (2S,3S,4S,5R,6S)-6-(((S)-6-(5-((S)-4-(((2-((((4-((S)-2-((S)-2-((S)-2-acetamido-6-aminohexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencil)oxi)carbonil)(metil)amino)etil)(metil)carbamoil)oxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-3,4,5-trihidroxitetrahidro-2H-piran-2-carboxílico (76)

Etapa 1: Síntesis de triacetato de (2S,3R,4S,5S,6S)-2-(((S)-8-(clorometil)-6-(5-((S)-8-(clorometil)-1-metil-4-(((4-nitrofenoxi)carbonil)oxi)-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-6-(metoxicarbonil)tetrahidro-2H-piran-3,4,5-triilo (73)
A una solución de 46 (120 mg, 0,13 mmol) en THF (5 ml) a 0 °C se le añadió una solución de cloroformiato de 4-nitrofenilo (55 mg, 0,26 mmol) en DCM (0,5 ml), seguido de Et3 N (109 ul, 0,78 mmol) y la mezcla se agitó a 0 °C durante 30 min. La mezcla se concentró al vacío y el residuo se purificó por HPLC de fase inversa (Procedimiento C) para dar el producto 73 en forma de un sólido de color blanco, 109 mg (77 % ). CL-EM (Protocolo B): m/z 1086,5 (m+H), tiempo de retención = 1,24 min Etapa 2: Síntesis de triacetato de (2s ,3R,4S,5S,6S)-2-(((S)-6-(5-((S)-4-(((2-((((4-((9S,12S,15S)-9-acetamido-1-(9H-fluoren-9-il)-12-isopropil-3,10,13-trioxo-15-(3-ureidopropil)-2-oxa-4,11,14-triazahexadecan-16-amido)bencil)oxi)carbonil)(metil)amino)etil)(metil)carbamoil)oxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-6-(metoxicarbonil)tetrahidro-2H-piran-3,4,5-triilo (75)
El compuesto 73 (50 mg, 0,046 mmol) en DMF (2 ml) y se le añadieron metil(2-(metilamino)etil)carbamato de 4-((9S,12S,15S)-9-acetamido-1-(9H-fluoren-9-il)-12-isopropil-3,10,13-trioxo-15-(3-ureidopropil)-2-oxa-4,11,14-triazahexadecan-16-amido)bencilo (74, 55 mg, 0,055 mmol), lutidina (0,021 ml, 0,18 mmol), DIPEA (0,032 ml, 0,18 mmol) y HOAt (6 mg, 0,046 mmol). La mezcla se agitó a ta durante 1 h. La mezcla se concentró al vacío y el residuo se purificó por HPLC de fase inversa (Procedimiento C) para dar el producto 75 en forma de un polvo de color blanco después de la liofilización, 72 mg (85 %). CL-EM (Protocolo B): m/z 1833,3 (M+H) tiempo de retención = 1,17 min 1H RMN (400 MHz, DMSO-d6) 8 = 10,02 (s a, 1H), 8,22 (s, 1H), 8,19 -8 ,13 (m, 1H), 8,10 (d, J = 6,6 Hz, 1H), 8,02 (d, J = 7,8 Hz, 1H), 7,89 (d, J = 7,4 Hz, 2H), 7,69 (d, J = 7,0 Hz, 3H), 7,57 (d, J = 7,0 Hz, 2H), 7,54 - 7,38 (m, 5H), 7,37 -7 ,30 (m, 3H), 7,26 (s a, 3H), 5,99 (s a, 1H), 5,74 (d, J = 7,0 Hz, 1H), 5,64 -5 ,49 (m, 1H), 5,20 (t, J = 8,8 Hz, 1H), 5 ,13 (t, J = 9,6 Hz, 1H), 5,04 (s a, 1H), 5,00 (s a, 1H), 4,81 -4 ,70 (m, 1H), 4,39 (s a, 2H), 4,32 -4 ,11 (m, 14H), 3,95 - 3,80 (m, 3H), 3,62 (s a, 1H), 3,56 (s a, 1H), 3,48 (s a, 2H), 3 ,12 (s a, 1H), 3,03 (s a, 2H), 3,00 -2,91 (m, 6H), 2,88 (s a, 3H), 2,82 - 2,67 (m, 4H), 2,67 - 2,55 (m, 7H), 2,10 (s, 1H), 2,08 - 1,91 (m, 14H), 1,85 (s, 3H), 1,79 - 1,64 (m, 2H), 1,61 (s a, 2H), 1,54 - 1,32 (m, 5H), 1,31 - 1,23 (m, 2H), 1,23 - 1,12 (m, 2H), 0,84 (d, J = 6,6 Hz, 3H), 0,87 (d, J = 6,6 Hz, 3H).
Etapa 3. Preparación de LP: Se disolvió ácido (2S,3S,4S,5R,6S)-6-(((S)-6-(5-((S)-4-(((2-((((4-((S)-2-((S)-2-((S)-2-acetamido-6-aminohexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencil)oxi)carbonil)(metil)amino)etil)(metil)carbamoil)oxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-3,4,5-trihidroxitetrahidro-2H-piran-2-carboxílico (76) compuesto 75 (44 mg, 0,024 mmol) en DMF (1 ml), se añadieron THF (5 ml) y MeOH (1 ml), enfriado a 0 °C , L O H H 2 O (10 mg, 0,24 mmol) y la mezcla se agitó a 0 °C durante 60 min. La mezcla se acidificó mediante la adición de HOAc (30 ul) y se concentró al vacío. El residuo se purificó por HPLC de fase inversa (Procedimiento C) para dar 25,0 mg (66 % ) del producto 76 en forma de un polvo de color blanco.
CL-EM (Protocolo B): m/z 1471,1 (M+2H)+ tiempo de retención = 0,75 min 1H RMN (500 MHz, DMSO-d6) 8 = 8,09
(s a, 2H), 7,97 (d, J = 8,1 Hz, 1H), 7,68 (s a, 2H), 7,48 (d, J = 7,6 Hz, 1H), 7,44 - 7,39 (m, 1H), 7,39 - 7,31 (m, 2H), 7,22 - 7 ,12 (m, 1H), 5,97 (s a, 1H), 5,45 - 5,32 (m, 2H), 5,18 (s a, 1H), 5,06 (d, J = 5,6 Hz, 1H), 4,98 (s a, 1H), 4,92 (s a, 1H), 4,31 (s a, 1H), 4,26 -4,05 (m, 8H), 3,82 (d, J = 10,0 Hz, 1H), 3,76 (d, J = 10,5 Hz, 1H), 3,70 (d, J = 7,6 Hz, 1H), 3,66 - 3,57 (m, 1H), 3,57 - 3,44 (m, 5H), 3,43 - 3,36 (m, 2H), 3,36 - 3,28 (m, 3H), 3 ,12 - 3,07 (m, 2H), 3,05 (s a, 1H), 3,00 - 2,91 (m, 2H), 2,88 (d, J = 9,8 Hz, 3H), 2,80 (s, 2H), 2,72 - 2,59 (m, 4H), 2,59 - 2,37 (m, 22H), 2,02 (s, 2H), 1,97 - 1,83 (m, 3H), 1,78 (s, 3H), 1,69 (td, J = 3,2, 6,4 Hz, 2H), 1,66 - 1,48 (m, 3H), 1,47 - 1,32 (m, 4H), 1,32 -1,14 (m, 4H), 0,76 (d, J = 6,4 Hz, 3H), 0,79 (d, J = 5,9 Hz, 3H).
Preparación de ((S)-8-(dorometM)-6-(5-((S)-8-(dorometM)-1-metN-4-(((2S,3R,4S,5R,6R)-3,4,5-trihidroxi-6-(hidroximetil)tetrahidro-2H-piran-2-il)oxi)-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)etano-1,2-diilbis(metilcarbamato) de 4-((S)-2-((S)-2-((S)-2-acetamido-6-aminohexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (77)

Etapa 1. Síntesis de triacetato de (2R,3S,4S,5R,6S)-2-(acetoximetN)-6-(((S)-6-(terc-butoxicarbonil)-8-(clorometil)-1 -metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)tetrahidro-2H-piran-3,4,5-triilo (78)
El compuesto 2 (300 mg, 0,85 mmol) se disolvió en DCM (30 ml), se añadieron tamices moleculares de 4 A (1,5 g, en polvo, <5 micrómetros, activados) y la mezcla se agitó a temperatura ambiente durante 30 min. A la mezcla de reacción se le añadió alfa-D-galactopanosa, 1-2,2,2-tricloroetanimidato de 2,3,4,6-tetraacetato (449 mg, 93 % , 0,85 mmol), la mezcla se enfrió a -15 °C, se añadió lentamente una solución de BF3 Et2 O (0,052 ml, 0,42 mmol) en DCM (5 ml) y la mezcla de reacción se agitó a -15 °C - -20 °C durante 1 h. La mezcla de reacción se retiró por filtración a través de una capa de Celite, se lavó con acetona y se concentró para dar un residuo. El residuo se trató con MeOH y se concentró al vacío para dar 580 mg (100 % ) del producto 78 en forma de un sólido de color blanquecino C L -E m (Protocolo B): m/z: 706,4 (M+Na) tiempo de retención = 1,09 min
Etapa 2. Síntesis de triacetato de (2R,3S,4S,5R,6S)-2-(acetoximetil)-6-(((S)-8-(clorometil)-1-metil-7,8-dihidro-6Htieno[3,2-e]indol-4-il)oxi)tetrahidro-2H-piran-3,4,5-triilo (79)
El compuesto 78 (580 mg, 0,85 mmol) se trató con una solución de HCI 4 M en dioxano (5 ml) durante 30 min y la mezcla se concentró al vacío para dar el producto en bruto 79 en forma de un sólido de color verde que se usó sin purificación adicional CL-EM (Protocolo B): m/z 584,3 (M+H), tiempo de retención = 0,94 min. 1H r Mn (400 MHz, METANOL-d4) 8 = 7,53 (s, 1H), 7,22 (s, 1H), 5,57 - 5,46 (m, 3H), 5,34 (dd, J = 2,0, 9,0 Hz, 1H), 4,53 (d, J = 6,6 Hz, 2H), 4,42 (t, J = 6,4 Hz, 1H), 4,25 (dt, J = 6,4, 11,2 Hz, 2H), 4,18 - 4,01 (m, 5H), 3,74 (dd, J = 8,6, 11,7 Hz, 2H), 3,69 (s, 14H), 2,68 - 2,58 (m, 4H), 2,23 (s, 3H), 2,19 - 2,14 (m, 2H), 2,14 - 2,07 (m, 7H), 2,07 - 1,94 (m, 6H), 1,33 (s, 3H).
Etapa 3. Síntesis de triacetato de (2R,3S,4S,5R,6S)-2-(acetoximetil)-6-(((S)-6-(5-(terc-butoxi)-5-oxopentanoil)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)tetrahidro-2H-piran-3,4,5-triilo (80)
Una solución de ácido 5-(terc-butoxi)-5-oxopentanoico (44, 191 mg, 1,02 mmol) en THF (10 ml) se enfrió a 0 °C y se añadió cloruro de oxalilo (1,02 ml, 2 M en DCM) seguido de 1 gota de DMF. La mezcla se agitó a 0 °C durante 30 min y se concentró al vacío para dar el cloruro de ácido correspondiente en forma de una cera. Este se disolvió en THF (10 ml) y se añadió a una solución de 79 (526 mg, 0,85 mmol) en THF (10 ml), seguido de Et3 N (0,354 ml, 2,54 mmol). La mezcla se agitó a 0 °C durante 30 min. La mezcla se concentró y el residuo se purificó por cromatografía sobre gel de sílice usando un gradiente de acetato de etilo del 0 % al 70 % en heptanos para dar 448 mg (70 % ) del producto 80 en forma de un sólido de color blanquecino
CL-EM (Protocolo B): m/z 754,4 (M+H), tiempo de retención = 1,06 min
Etapa 4. Síntesis de triacetato de (2R,3S,4S,5R,6S)-2-(acetoximetil)-6-(((S)-8-(clorometil)-6-(5-((S)-8-(clorometil)-4-hidroxi-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)tetrahidro-2H-piran-3,4,5-triilo (84)
Una solución del compuesto 80 (448 mg, 0,59 mmol) se trató con DCM (3 ml) y TFA (2 ml) a ta durante 1 h y la mezcla se concentró al vacío para dar el ácido libre 81 en forma de un sólido de color verde, 212 mg. El ácido 81 (167 mg, 0,24 mmol) se disolvió en THF (5 ml), la solución se enfrió a 0 °C y se añadió cloruro de oxalilo (0,24 ml, 2 M en DCM), seguido de DMF (1 gota). La mezcla se agitó a 0 °C durante 30 min y se concentró al vacío para dar el cloruro de ácido 82 correspondiente que se usó sin purificación adicional en la siguiente etapa. Una solución del compuesto 2 (102 mg, 0,28 mmol) en HCI 4 M (2 ml) en dioxano se agitó a ta durante 1 h y la mezcla se concentró al vacío para dar el compuesto 83 en forma de un sólido de color verde que se disolvió en THF (5 ml) y se enfrió a 0 °C . Se añadió TEA (0,2 ml, 1,44 mmol) seguido de una solución del cloruro de ácido 82 en bruto en THF (5 ml) y la mezcla se agitó a 0 °C durante 20 min. La mezcla se concentró y el residuo se purificó por cromatografía sobre gel de sílice usando un gradiente de acetona del 0 % al 70 % en heptanos para dar 196 mg (88 % ) del producto 84 en forma de un sólido de color amarillo
CL-EM (Protocolo B): m/z 933,5 (M+H), tiempo de retención = 1,05 min. 1H RMN (400 MHz, DMSO-d6) 8 = 8,26 (s, 1H), 7,94 (s, 1H), 7,46 (s, 1H), 7,39 (s, 1H), 5,51 (d, J = 7,8 Hz, 1H), 5,42 - 5,26 (m, 3H), 4,59 - 4,46 (m, 1H), 4,33 - 4,23 (m, 2H), 4,23 - 4,15 (m, 4H), 4,15 - 4,07 (m, 2H), 4,04 (s, 4H), 3,87 (dd, J = 10,5, 18,0 Hz, 2H), 3,67 (t, J = 9,8 Hz, 1H), 3,57 (t, J = 10,1 Hz, 1H), 3,33 (s, 1H), 3,31 (s, 1H), 2,83 -2 ,67 (m, 2H), 2,60 (dd, J = 6,4, 16,2 Hz, 3H), 2,19 (s, 3H), 2 ,15 -2 ,09 (m, 4H), 2,07 (s, 4H), 1,97 (s, 6H).
Etapa 5: Síntesis de triacetato de (2R,3S,4S,5R,6S)-2-(acetoximetil)-6-(((S)-8-(clorometil)-6-(5-((S)-8-(clorometil)-1-metil-4-(((4-nitrofenoxi)carbonil)oxi)-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)tetrahidro-2H-piran-3,4,5-triilo (85)
A una solución de 84 (100 mg, 0,107 mmol) en THF (5 ml) a 0 °C se le añadió cloroformiato de 4-nitrofenilo (45 mg, 0,21 mmol) seguido de Et3 N (0,060 ml, 0,43 mmol). La mezcla se agitó a 0 °C durante 60 min. La mezcla se concentró al vacío para dar el producto en bruto en forma de una espuma de color amarillo, 142 mg, que se purificó por HPLC de fase inversa (Procedimiento C) para dar 50 mg (43 % ) del producto 85 en forma de un sólido de color blanquecino. CL-EM (Protocolo B): m/z 1098,5 (M+H), tiempo de retención = 1,05 min
Etapa 6: Síntesis de triacetato de (2S,3R,4S,5S,6R)-2-(((S)-6-(5-((S)-4-(((2-((((4-((9S,12S,15S)-9-acetamido-1-(9H-fluoren-9-il)-12-isopropil-3,10,13-trioxo-15-(3-ureidopropil)-2-oxa-4,11,14-triazahexadecan-16-amido)bencil)oxi)carbon-il)(metil)amino)etil)(metil)carbamoil)oxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-6-(acetoximetil)tetrahidro-2H-piran-3,4,5-triilo (86): A una solución de 85 (50 mg, 0,045 mmol) en DMF (2 ml) se le añadieron metil(2-(metilamino)etil)carbamato de 4-((9S,12S,15S)-9-acetamido-1-(9H-fluoren-9-il)-12-isopropil-3,10,13-trioxo-15-(3-ureidopropil)-2-oxa-4,11,14-triazahexadecan-16-amido)bencilo (7445 mg, 0,045 mmol) y DIPEA (0,024 ml, 0,136 mmol). La mezcla se agitó a ta durante 60 min y se purificó usando HPLC de fase inversa (Procedimiento C) para dar 26 mg (31 % ) del producto 86 en forma de un sólido de color blanco: CL-EM (Protocolo B): m/z 1847,1 (M+H), tiempo de retención = 1,06 min 1H RMN (400 MHz, DMSO-d6) 8 = 10,01 (s a, 1H), 8,25 (s, 1H), 8,20 - 8,12 (m, 1H), 8,09 (d, J = 6,6 Hz, 1H), 8,02 (d, J = 8,2 Hz, 1H), 7,89 (d, J = 7,8 Hz, 2H), 7,69 (d, J = 7,0 Hz, 3H), 7,57 (d, J = 7,4 Hz, 2H), 7,47 - 7,37 (m, 4H), 7,37 - 7,30 (m, 3H), 7,26 (s a, 3H), 5,98 (s a, 1H), 5,50 (d, J = 7,0 Hz, 1H), 5,46 - 5,27 (m, 5H), 5,02 (d, J = 19,1 Hz, 2H), 4,52 (s a, 1H), 4,39 (s a, 1H), 4,32 - 4,14 (m, 13H), 4,14 - 4,03 (m, 1H), 3,95 - 3,81 (m, 2H), 3,75 - 3,59 (m, 6H), 3,56 (s a, 2H), 3,48 (s a, 2H), 3,12 (s a, 1H), 3,03 (s a, 2H), 2,94 (s, 3H), 2,97 (s, 2H), 2,87 (s a, 2H), 2,83 - 2,67 (m, 3H), 2,66 - 2,54 (m, 7H), 2,19 (s, 4H), 2 ,12 (s, 3H), 2,09 -2 ,03 (m, 4H), 2,03 - 1,90 (m, 7H), 1,85 (s, 3H), 1,78 (t, J = 6,4 Hz, 1H), 1,68 (d, J = 7,8 Hz, 1H), 1,61 (s a, 2H), 1,53 - 1,33 (m, 5H), 1,32 - 1, 18 (m, 2H), 0,84 (d, J = 7,0 Hz, 3H), 0,87 (d, J = 6,6 Hz, 3H).
Etapa 7. Síntesis de LP: etano-1,2-diilbis(metilcarbamato) de 4-((S)-2-((S)-2-((S)-2-acetamido-6-aminohexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo ((S)-8-(clorometil)-6-(5-((S)-8-(clorometil)-1-metil-4-(((2S,3R,4S,5R,6R)-3,4,5-trihidroxi-6-(hidroximetil)tetrahidro-2H-piran-2-il)oxi)-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-ilo) (77): A una solución de 86 (24 mg, 0,013 mmol) en THF (2 ml) y MeOH (2 ml), a 0 °C se le añadió a una solución de LiOH (1 M, 0,26 ml, 0 , 2 6 mmol) en agua y la mezcla se agitó a 0 °C a ta durante 2 h. Se añadió ácido acético (20 ul), la mezcla se concentró y el residuo se purificó por HPLC de fase inversa (Procedimiento C) para dar 14 mg (69 % ) del producto 77 en forma de una espuma de color blanco: CL-EM (Protocolo B): m/z 1456,9 (M+2H)+, tiempo de retención = 0,75 min 1H RMN (500 MHz, DMSO-d6 ) 8 = 10,07 - 9,96 (m, 1H), 8,29 - 8,18 (m, 1H), 8,18 - 8,09 (m, 2H), 8,06 (d, J = 8,1 Hz, 1H), 7,73 (d, J = 8 , 6 Hz, 1H), 7,66 (s a, 3H), 7,56 (d, J = 7,8 Hz, 2H), 7,50 (d, J = 9,3 Hz, 1H), 7,47 - 7,39 (m, 2H), 7,35 (d, J = 7,6 Hz, 1H), 7,32 -7 ,25 (m, 2H), 6,04 (s a, 1H), 5,11 (d, J = 6,4 Hz, 1H), 5,05 (s a, 1H), 5,01 (s a, 1H), 4,40 (s a, 1H), 4,36 -4 ,16 (m, 10H), 4,10 (d, J = 11,7 Hz, 3H), 4,00 (s a, 3H), 3,96 -3 ,80 (m, 10H), 3,77 (s a, 4H), 3,73 - 3,55 (m, 11H), 3,52 (s a, 1H), 3,48 (s a, 3H), 3 ,12 (s a, 1H), 3,05 (s a, 2H), 3,01 - 2,91 (m, 4H), 2,88 (s a, 2H), 2,86 -2 ,72 (m, 4H), 2,72 -2 ,53 (m, 13H), 2,45 (t, J = 7,5 Hz, 1H), 2,06 - 1,92 (m, 3H), 1,86 (s, 4H), 1,71 -1,56 (m, 3H), 1,56 - 1,42 (m, 4H), 1,42 - 1,23 (m, 4H), 0,85 (dd, J = 6 ,6 , 14,7 Hz, 6 H).
Preparación de metil(2-(metilamino)etil)carbamato de 4-((S)-2-((S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-metilbutanamido)-5-ureidopentanamido)bencilo (96)

Etapa 1. Síntesis de (((9H-fluoren-9-il)metoxi)carbonil)-L-valinato de 2,5-dioxopirrolidin-1-ilo (88)
A una solución del compuesto 87 (150 g, 0,442 mol) y HOSu (56 g, 0,487 mol) en THF (1800 ml) se le añadió en porciones D CC (100 g, 0,487 mol) en un baño de hielo. Después de la adición, la reacción se agitó a temperatura ambiente durante una noche. La reacción se enfrió a -5 °C , se filtró y se lavó con THF frío, el filtrado se concentró al vacío y el residuo se recristalizó en MTBE para dar el compuesto 88 en forma de un sólido de color blanco, 175 g (91 %).
Etapa 2. Síntesis de ácido (S)-2-((S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-metilbutanamido)-5-ureidopentanoico (90)
A una solución del compuesto 89 (40,14 g, 0,229 mol) y NaHCO 3 (19,23 g, 0,229 mol) en agua (750 ml) se le añadió gota a gota la solución del compuesto 88 (100 g, 0,229 mol) en DME (750 ml) en un baño de hielo. Durante la adición, se formó una suspensión de color blanco. Se añadió más cantidad de THF (400 ml) para mejorar la solubilidad. Después de la adición, la solución se agitó a 25 - 30 °C durante 2 días. A la reacción se le añadió K2 CO 3 ac. saturado para ajustar el pH a 8 - 9, y después se extrajo con EtOAc (500 ml x 5). La fase acuosa se ajustó a pH 3 - 4 con ácido cítrico ac. Se formó un material gelatinoso y se filtró. La torta húmedas se disolvió en THF (1,5 l). Se añadió metanol hasta que el sólido se disolvió. La solución se concentró al vacío para retirar el 30 % del disolvente y después se enfrió a temperatura ambiente. A la solución se le añadió TBME (2 l) y la mezcla se agitó a temperatura ambiente durante una noche. La mezcla se filtró y la torta húmeda se secó al vacío para dar el compuesto 90 en forma de un sólido de
color blanco, 60 g (53 %).
Etapa 3. Síntesis de ((S)-1-(((S)-1-((4-(hidroximetil)fenil)amino)-1-oxo-5-ureidopentan-2-il)amino)-3-metil-1-oxobutan-2-il)carbamato de (9H-fluoren-9-il)metilo (92)
A la suspensión del compuesto 90 (70 g, 0,141 mol) en DCM/MeOH (1 L/500 ml) se le añadió el compuesto 91 (34,7 g, 0,282 mol) seguido de EEDQ (69,7 g, 0,282 mol). La mezcla se agitó a 40 °C durante una noche. La mezcla de reacción se filtró y la torta húmeda se suspendió en EtOAc/TBME (500 ml/200 ml), se agitó durante 30 min y después se filtró. El sólido se lavó con EtOAc/TBME para proporcionar el compuesto 92 en forma de un sólido de color blanquecino, 65 g (77 % ). 1H RMN (400 MHz, DMSO-d6) 8 = 9,98 (s, 1H), 8,11 (d, 1H), 7,87 (d, 2H), 7,77 (m, 2H), 7,52 (d, 2H), 7,39 (m, 3H), 7,30 (m, 2H), 7,21 (d, 2H), 5,97 (m, 1H), 5,41 (s, 2H), 5,10 (m, 1H), 4,42 (m, 3H), 4,22 (m, 3H), 3,90 (m, 1H), 2,93 (m, 2H), 1,98 (m, 1H), 1,50 (m, 2H), 1,30 (m, 2H), 0,84 (m, 6H).
Etapa 4. Síntesis de ((S)-3-metil-1-(((S)-1-((4-((((4-nitrofenoxi)carbonil)oxi)metil)fenil)amino)-1-oxo-5-ureidopentan-2-il)amino)-1-oxobutan-2-il)carbamato de (9H-fluoren-9-il)metilo (94)
A una solución de ((S)-1-(((S)-1-((4-(hidroximetil)fenil)amino)-1-oxo-5-ureidopentan-2-il)amino)-3-metil-1-oxobutan-2-il)carbamato de (9H-fluoren-9-il)metilo (3,74 g, 8,31 mmol, 92) en DMF anhidra (120 ml) se le añadió en porciones carbonato de bis(4-nitrofenilo) (3,78 g, 12,4 mmol, 93), seguido gota a gota de DIPEA (1,21 g, 9,32 mmol) a 0 °C. La mezcla de reacción se agitó a temperatura ambiente durante una noche. El análisis por TLC (MeOH: CH 2 O 2 = 1:10) mostró que la reacción se había completado. La mezcla de reacción se añadió gota a gota a MTBE (2,5 l) con agitación. El producto en bruto se recogió por filtración. La torta de filtro se lavó con MTBE y se secó a alto vacío para proporcionar el compuesto 94 en forma de un sólido de color pardo, 2,7 g (57 %).
Etapa 5. Síntesis de etano-1,2-diilbis(metilcarbamato) de 4-((S)-2-((S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-metilbutanamido)-5-ureidopentanamido)bencil tere-butilo (95)
A una solución del compuesto 94 (1,7 g, 2 ,217 mmol) en DMA (15 ml) se le añadieron HOAT (332 mg, 2,44 mmol) y 2,6-lutidina (950 mg, 8,87 mmol) a 0 °C. La mezcla se agitó a 0 °C durante 5 minutos. Después, a la mezcla se le añadió metil(2-(metilamino)etil)carbamato de tere-butilo (38, 459 mg, 2,44 mmol) a 0 °C seguido de DIPEA (860 mg, 6,65 mmol) a 0 °C. La mezcla se agitó a ta durante 1 h, se vertió en TBME (800 ml), se agitó durante 30 min, se filtró y la torta de filtro se concentró al vacío para dar el producto en bruto (1,06 g) en forma de un sólido de color amarillo. El producto en bruto se purificó por cromatografía sobre gel de sílice (Gradiente: DCM:MeOH = 100 :1-10 :1) para dar el producto 95 en forma de un sólido de color blanco, 600 mg (33 %).
Etapa 6. Síntesis de metil(2-(metilamino)etil)carbamato de 4-((S)-2-((S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-metilbutanamido)-5-ureidopentanamido)bencilo (96)
A una solución agitada del compuesto 95 (600 mg, 0,735 mmol) en DCM seco (10 ml) se le añadió TFA (5 ml) a 0 °C y se agitó a 0 °C durante 1 h. La mezcla se concentró al vacío para dar el compuesto del título 96 en forma de un sólido de color blanco, 550 mg (100 % ). 1H RMN (400 MHz, DMSO-d6) 8 = 10,09 (a, 1H), 8,40 (s, 2H), 8,14 (d, 1H), 7,90 (d, 2H), 7,74 (m, 2H), 7,61 (d, 2H), 7,43 (m, 3H), 7,33 (m, 4H), 6,00 (s, 1H), 5,43 (s, 2H), 5,02 (s, 2H), 4,42-4,23 (m, 4H), 3,93 (m, 1H), 3,50 (m, 2H), 3,08-2,94 (m, 4H), 2,88 (s, 3H), 2,59 (m, 3H), 1,99 (m, 1H), 1,69-1,59 (m, 2H), 1,46-1,39 (m, 2H), 0,90-0,85 (m, 6H).
Preparación alternativa de ácido (2S,3S,4S,5R,6S)-6-(((S)-8-(dorometM)-6-(5-((S)-8-(dorometM)-4-(((2-((((4-((23S,26S)-1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1 -il)-23-isopropil-21,24-dioxo-26-(3-ureidopropil)-3,6,9,12,15,18-hexaoxa-22,25-diazaheptacosan-27-amido)bencil)oxi)carbonil)(metil)amino)-etil)(metil)carbamoil)oxi)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-3,4,5-trihidroxitetrahidro-2H-piran-2-carboxílico (49)

Etapa 1: Síntesis de triacetato de (2S,3R,4S,5S,6S)-2-(((S)-6-(5-((S)-4-(((2-((((4-((S)-2-((S)-2-((((9H-fluoren-9-il)metoxi)carbonil)amino)-3-metilbutanamido)-5-ureidopentanamido)bencil)oxi)carbonil)(metil)amino)etil)(metil)carbamoil)oxi)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-6-(metoxicarbonil)tetrahidro-2H-piran-3,4,5-triilo (97)
A una solución del compuesto 73 (26 mg, 0,024 mmol) en DMF (1,5 ml) se le añadieron el enlazador Fmoc-VCPABC-DM EA 96 (24 mg, 0,029 mmol), 2,6-lutidina (0,0167 ml, 0,14 mmol), DIPEA (0,0253 ml, 0,14 mmol) y HOAt (3,3 mg, 0,024 mmol). La mezcla se agitó a ta durante 30 min y se concentró al vacío para dar un residuo. El producto en bruto se sometió a purificación por HPLC (Procedimiento C) para dar el producto 97 en forma de
un sólido de color blanco, 33 mg (83 % ). CL-EM (Protocolo B): m/z 1660,9 (M+H), tiempo de retención = 1,11 min.
Etapa 2: Síntesis de ácido (2S,3S,4S,5R,6S)-6-(((S)-6-(5-((S)-4-(((2-((((4-((S)-2-((S)-2-amino-3-metilbutanamido)-5-ureidopentanamido)bencil)oxi)carbonil)(metil)amino)-etil)(metil)carbamoil)oxi)-8-(dorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-8-(clorometil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-3,4,5-trihidroxitetrahidro-2H-piran-2-carboxílico (98): A una solución de 97 (46 mg, 0,028 mmol) en THF (3 ml) y MeOH (3 ml), enfriada a 0 °C, se le añadió una solución de L O H H 2 O (12 mg, 0,28 mmol) en agua (0,5 ml). La mezcla se agitó a 0 °C durante 1 h. Se añadió AcOH (0,03 ml) para neutralizar la mezcla y se concentró al vacío para dar un residuo sólido de color blanco. Se purificó por HPLC (Procedimiento C) para dar el producto 98 en forma de un sólido de color blanquecino, 28 mg (72 % ). CL-EM (Protocolo B): m/z 1300,6 (M+H), tiempo de retención = 0,78 min. 1H RMN (400 MHz, DMF) 8 = 10,33 - 10,21 (m, 1H), 8,84 (d, J = 7,4 Hz, 1H), 8,63 (s a, 3H), 8,41 - 8,28 (m, 2H), 8,05 (s, 1H), 7,81 - 7,63 (m, 2H), 7,57 - 7,41 (m, 3H), 7,38 (s a, 2H), 6,52 (s a, 1H), 5,39 (d, J = 6,6 Hz, 1H), 5 ,17 (s a, 1H), 5 ,12 (s a, 1H), 4,74 (s a, 1H), 4,48 -4 ,28 (m, 5H), 4,24 (s a, 1H), 4,09 (s, 4H), 4,04 - 3,89 (m, 2H), 3,84 - 3,67 (m, 6H), 3,67 - 3,52 (m, 4H), 3,32 (s, 9H), 3,26 (s a, 2H), 3,19 (s a, 1H), 3,14 - 3,04 (m, 4H), 3,00 (s, 2H), 2,94 (s a, 2H), 2,85 (s a, 2H), 2,79 - 2,68 (m, 5H), 2,62 (s, 4H), 2,64 (s, 3H), 2,42 - 2,29 (m, 1H), 2,23 - 2,05 (m, 11H), 1,92 (s a, 1H), 1,78 (s a, 1H), 1,60 (s a, 2H), 1,15 (s a, 6H).
Etapa 3: Síntesis de ácido (2S,3S,4S,5R,6S)-6-(((S)-8-(clorometil)-6-(5-((S)-8-(clorometil)-4-(((2-((((4-((23S,26S)-1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-23-isopropil-21,24-dioxo-26-(3-ureidopropil)-3,6,9,12,15,18-hexaoxa-22,25-diazaheptacosan-27-amido)bencil)oxi)carbonil)(metil)amino)etil)(metil)carbamoil)oxi)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)oxi)-3,4,5-trihidroxitetrahidro-2H-piran-2-carboxílico (49): Una solución de 98 (10 mg, 0,007 mmol) se disolvió en DMF (1 ml) y se le añadieron 1-(2,5-dioxo-2,5-dihidro-1H-pirrol-1-il)-3,6,9,12,15,18-hexaoxahenicosan-21-oato de perfluorofenilo (5,6 mg, 0,009 mmol, 99) y DIPEA (0,007 ml, 0,042 mmol). La mezcla se agitó a ta durante 30 min. La mezcla se purificó por HPLC (Procedimiento C) para dar el producto 49 en forma de una espuma de color blanco, 10 mg (82 % ). CL-EM (Protocolo B): m/z 1715 ,7 (M+H), tiempo de retención = 0,89 min. 1H RMN (400 MHz, ACETONITRILO-d3) 8 = 8,21 (s a, 2H), 7,56 (d, J = 7,4 Hz, 1H), 7,31 -7 ,10 (m, 4H), 6,78 (s, 2H), 5,25 (s a, 1H), 5,11 (s a, 1H), 5,02 (s a, 1H), 4,50 (s a, 1H), 4,18 (s a, 2H), 3,77 (s a, 2H), 3,71 (s a, 3H), 3,66 -3 ,42 (m), 3,37 -3 ,24 (m, 4H), 3,16 (s a, 1H), 3 ,12 - 2,95 (m, 4H), 2,93 (s, 2H), 2,77 - 2,47 (m, H), 2,22 - 2,02 (m, 4H), 1,51 (s a, 2H), 0,95 (s a, 6H).
Preparación de ((S)-8-(dorometM)-6-(5-((S)-8-(dorometM)-1-metN-4-(fosfonooxi)-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-methy)-7,8-dihidro-6H-tieno[3,2-e]indo)-4-il)etano-1,2-diilbis(metilcarbamato) de 4-((S)-2-((S)-2-((S)-2-acetamido-6-aminohexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (100)

Etapa 1: Síntesis de ((S)-8-(clorometil)-6-(5-((S)-8-(clorometil)-1-metil-4-(fosfonooxi)-7,8-dihidro-6H-tieno[3,2-e]indol-6-il)-5-oxopentanoil)-1-metil-7,8-dihidro-6H-tieno[3,2-e]indol-4-il)etano-1,2-diilbis(metilcarbamato) de 4-((S)-2-((S)-2-((S)-2-acetamido-6-aminohexanamido)-3-metilbutanamido)-5-ureidopentanamido)bencilo (100): A una solución de 55 (35 mg, 0,041 mmol) en DMF (3 ml) se le añadieron 74 (45 mg, 0,045 mmol), lutidina (0,019 ml, 0,16 mmol), DIPEA (0,029 ml, 0,16 mmol) y HOAt (5,6 mg, 0,041 mmol). La mezcla se agitó a ta durante 3 h. A la mezcla resultante se le añadió piperidina (0,3 ml, 3 mmol) y se agitó a ta durante 20 min. La mezcla se concentró al vacío y el residuo se purificó por HPLC (Procedimiento C) para dar el producto 100 en forma de un polvo de color blanco después de la liofilización, 35 mg (62 % ). CL-EM (Protocolo B): m/z 1374,8 (M+H), tiempo de retención = 0,92 min. 1H RMN (400 MHz, DMSO-d6) 8 = 8,45 (d, J = 13,7 Hz, 1H), 8,14 (s, 1H), 8,17 (s, 1H), 8,05 (d, J = 7,4 Hz, 1H), 7,76 (s a, 3H), 7,58 (d, J = 8,2 Hz, 1H), 7,52 - 7,39 (m, 2H), 7,30 - 7,16 (m, 1H), 6,09 (s a, 1H), 5,15 - 4,94 (m, 2H), 4,40 (s a, 1H), 4,35 -4,08 (m, 8H), 3,95 -3 ,79 (m, 2H), 3,69 (s a, 1H), 3,60 (d, J = 10,5 Hz, 3H), 3,49 (s a, 3H), 3,13 (s a, 1H), 3,06 (s a, 1H), 3,03 - 2,91 (m, 4H), 2,88 (s, 2H), 2,76 (s a, 4H), 2,61 (d, J = 6,6 Hz, 2H), 2,56 (s, 6H), 1,98 (m, 3H), 1,86 (s, 3H), 1,65 (m, 3H), 1,58 - 1,49 (m, 3H), 1,46 (m, 2H), 1,40 - 1,20 (m, 3H), 0,85 (m, 6H).
Tal como se indica en el presente documento, los compuestos descritos en las Tablas 1 y 2 pueden existir en su etapa de profármaco de acetato o fenol, que se convierten ambos rápidamente en la especie de fármaco activo bis ciclopropilo. Esta interconversión tiene lugar rápidamente en el medio de ensayo. Por lo tanto, las tres formas del compuesto son funcionalmente equivalentes y dan como resultado los mismos valores inhibidores del crecimiento en los ensayos de proliferación de células cancerosas. Por lo tanto, describir compuestos como "bis-acetatos" también implica la descripción como sus especies de fenol y bis ciclopropilo funcionalmente equivalentes.

Se prepararon las siguientes cargas útiles o cargas de referencia. Todas las cargas útiles o de referencia se prepararon utilizando los procedimientos generales A y B o mediante los procedimientos indicados
Tabla 1: Ejemplos de cargas útiles o cargas útiles de referencia

continuación

continuación
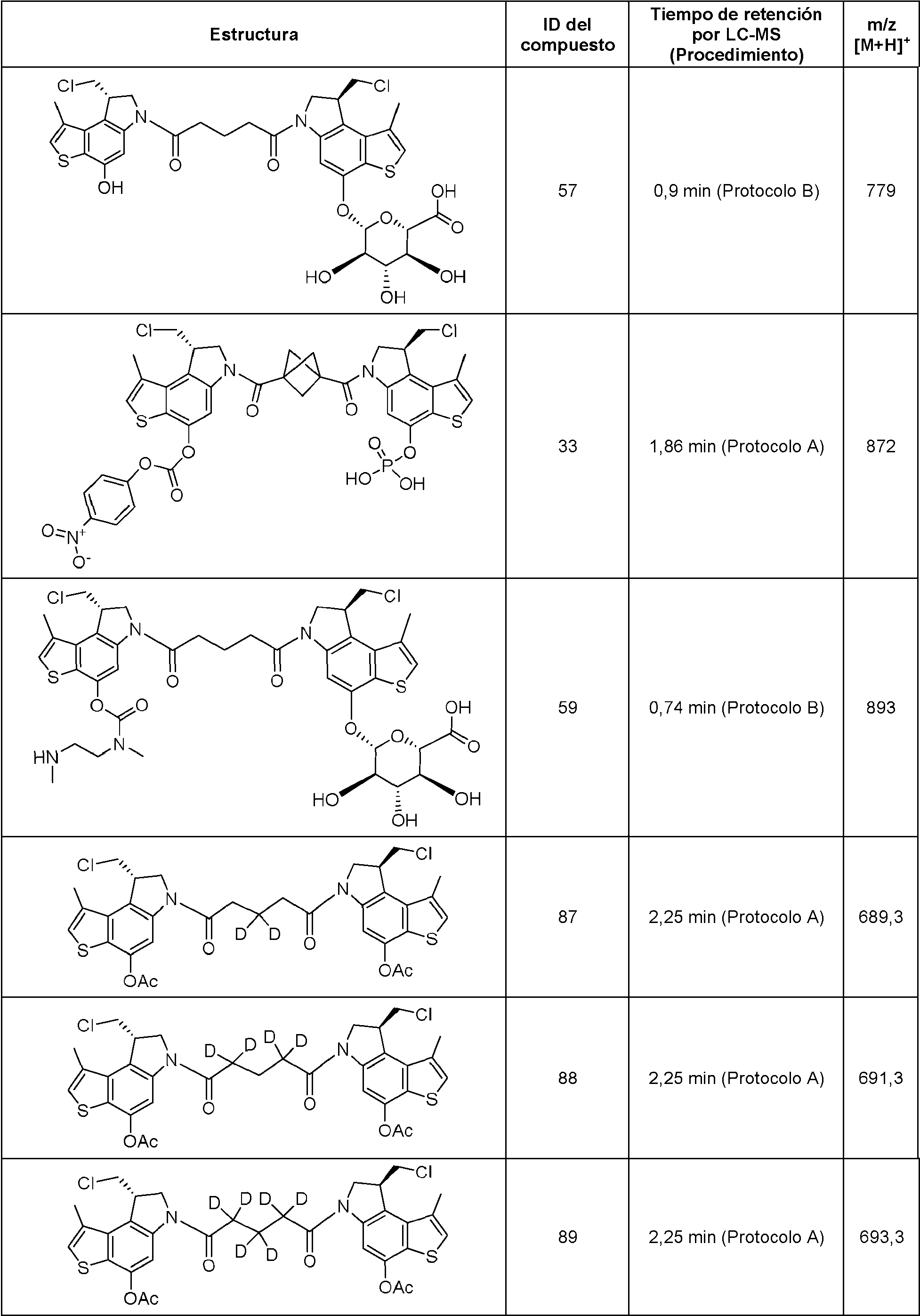
Tabla 2:_ Las siguientes cargas útiles o cargas de referencia se preparan utilizando los procedimientos sintéticos generales A y B.
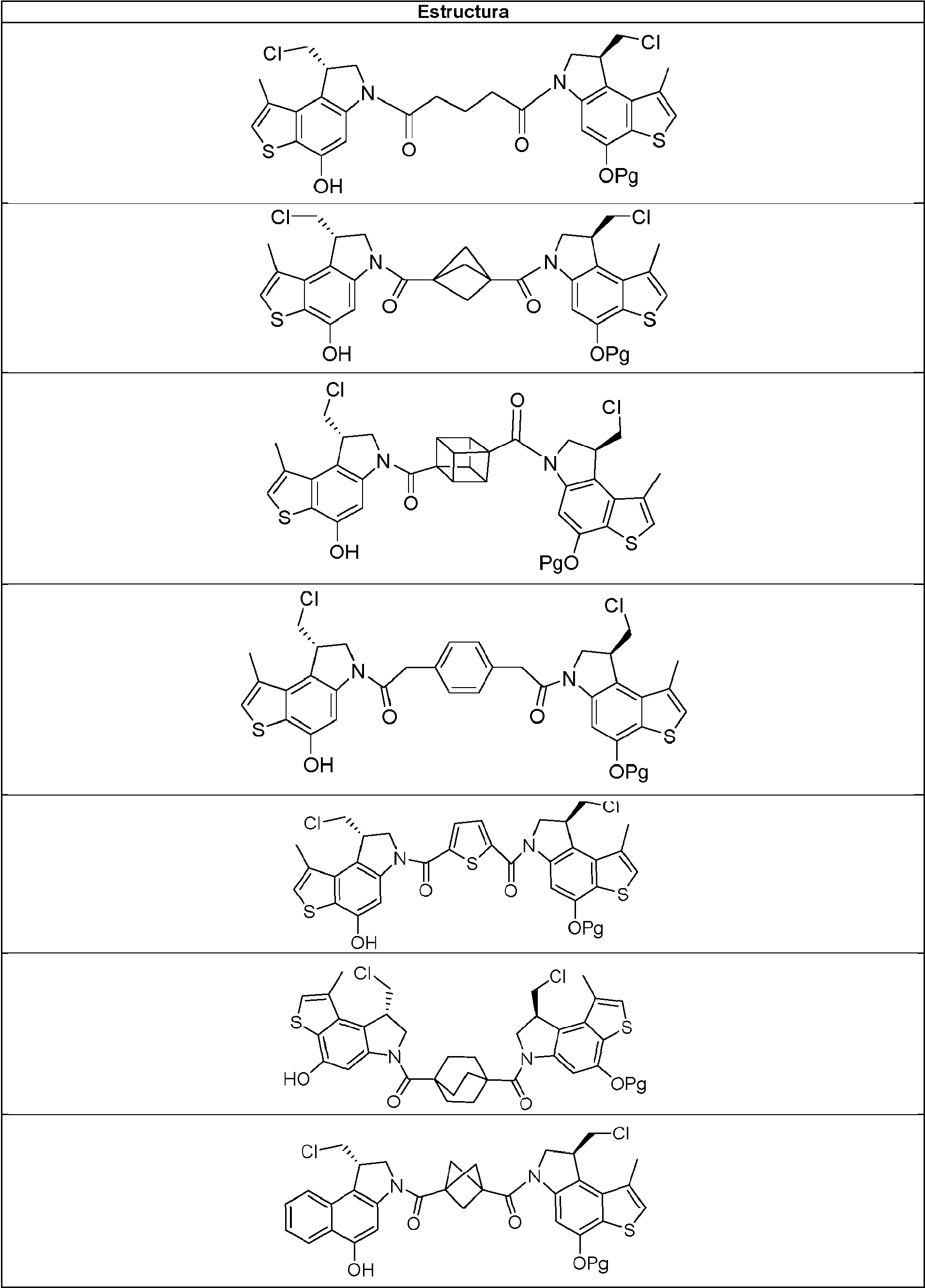
continuación

continuación


continuación

En los ejemplos anteriores, la variable Pg es H, acilo, fosfato PO 3 H2 , un carbohidrato, un aminoácido o un péptido (y en particular un péptido que es escindido por proteasas, tales como catepsinas y metaloproteinasas de matriz). Se prepararon los siguientes enlazadores/cargas útiles o enlazadores de referencia/cargas útiles:
Tabla 3. Enlazador-carga útil preparados o enlazador-carga útil de referencia

Tabla 4: Los siguientes conjuntos de enlazador/carga útil o enlazador/carga útil de referencia se preparan utilizando el procedimiento descrito anteriormente para la preparación de análogos de enlazador/carga útil que se muestran en la Tabla 3.
Estructuras de enlazador para la Tabla 4

en donde
P representa el punto de unión a dicha carga útil, R5 *7 es independientemente H o -alquilo C 1 -C 20 ,
R8 *10es -alquilo C 1 -C 20 , -arilo C 6-C 14 o -heteroarilo C 6-C 14 ,
n = 0-20, y
m = 0-20.
Tabla 4. Enlazador/carga útil adicional o enlazador/car útil de referencia

continuación

continuación

En la Tabla 4, Pg es H, acilo, fosfato PO3 H2 , un carbohidrato, un aminoácido o un péptido (en particular un péptido que se escinde por proteasas, tales como catepsinas y metaloproteinasas de matriz).
Estructuras de enlazador para la Tabla 5
de:

en donde
P representa el punto de unión a dicha carga útil, R7 es independientemente H o -alquilo C 1 -C 20 , R8 es -alquilo C 1 -C 20 , -arilo C 6-C 14 o -heteroarilo C 6-C 14 ,
n = 0-20, y
m = 0-20.

continuación

continuación

En la Tabla 5, Pg es H, acilo, fosfato PO 3 H, un carbohidrato, un aminoácido o un péptido (en particular un péptido que se escinde por proteasas, tales como catepsinas y metaloproteinasas de matriz).
Procedimientos experimentales para la evaluación biológica de cargas útiles y conjugados de anticuerpofármaco
Líneas celulares para ensayos de viabilidad de carga útil
Se obtuvieron líneas celulares de cáncer de A TCC (Manassas, VA). N87 (carcinoma gástrico humano derivado del sitio metastásico del hígado). HL60 (leucemia) y Md A-MB-361 Dy T2 (carcinoma de mama humano MDA-MB-361) se cultivaron en medio RPMI 1640. Todos los medios se suplementaron con suero bovino fetal al 10 % , piruvato de sodio al 1 % y L-glutamina al 1 % (Invitrogen, Grand Island, NY). Las células endoteliales de la vena umbilical humana (HUVEC, por sus siglas en inglés) se obtuvieron de Lonza (Allendale, NJ) y se mantuvieron en medio EGM2 suplementado con EGM -2 SingleQuots (N.° de Lonza CC-4176). Todas las células se mantuvieron en una incubadora humidificada (37 °C, CO 2 al 5 %).
Procedimiento de ensayo de citotoxicidad para cargas útiles
Las células en 100 |jl de medio se cultivaron en una placa de 96 pocillos. Las líneas celulares de cáncer se trataron con los compuestos indicados mediante la adición de 50 j l de solución madre a 3X por duplicado a 10 concentraciones. Las células se incubaron con los compuestos durante cuatro días, luego se añadieron a las células 30 j l de solución CellTiter® 96 AQueous One MTS (N.° de cat. de Promega G3582), se incubaron 1,5 horas a 37 °C, después se midió la absorbancia a 490 nm en un lector de placas Victor (Perkin Elmer, Waltham, MA). La viabilidad celular relativa se determinó como un porcentaje de los pocillos de control no tratados. Los valores de CI50 se calcularon utilizando el modelo logístico de cuatro parámetros N.° 203 con XLfit v4.2 (IDBS, Guildford, Surry, Reino Unido).
Procedimiento de ensayo de citotoxicidad para CAF de anti-IL13Ra2
Líneas celulares utilizadas: A375 (melanoma), PC3MM2 (próstata), PC3 (próstata)
Las células se sembraron en una placa de 96 pocilios durante la noche antes de exponerlas a concentraciones crecientes de CAF de anti-IL13Ra2 o de CAF de control. Después de cuatro días, se evaluó la viabilidad de cada cultivo frente a las células de control. Se calcularon los valores de CI50 por regresión logística no lineal.
Procedimiento de ensayos de citotoxicidad para CAF de anti-CD33
Las líneas celulares de LMA humana (HL60, NB4, HEL92.1.7 y Raji) se obtuvieron de la American Type Culture Collection (Manassas, VA). Las células se cultivaron en medio RPMI, suplementado con FBS al 10 % , mezcla de Pen/Estrep y L-glutamina al 1 % , piruvato de sodio 1 mM, H EPES 10 mM y glucosa al 0,27 % (Life Technology, Carlsbad, c A). Se analizaron las líneas celulares para determinar sus niveles de actividad endógena de glucoproteína P, la proteína de resistencia a múltiples fármacos y la proteína de resistencia al cáncer de mama se determinaron utilizando un kit comercial de citometría de flujo (eFLUXX-ID; ENZO Life Sciences, Plymouth Meeting, PA).
Para ensayos de citotoxicidad, las células se sembraron en placas opacas de 96 pocillos (Corning) y se trataron con concentraciones variables de compuestos durante 4 días. La viabilidad se determinó utilizando un kit de ensayo de viabilidad de células luminiscentes CellTiter Glo (Promega, Madison, WI) y se midió con un lector de placas Victor X3 (Perkin Elmer Waltham, MA). Los datos se normalizaron al grupo de control (DMSO o PBS). Los valores de CI50 se definieron como la concentración que causa una inhibición del crecimiento del 50 % . Los valores de CI50 se calcularon utilizando una regresión logística no lineal, modelo n.° 203 con XL fit v4.2 (IDBS, Guldford, Surry, Reino Unido). Todos los puntos experimentales se configuraron en dos pocillos replicados y se realizaron de forma independiente por duplicado. Ensayos de citotoxicidad para CAF de anti-Trastuzumab
Las células que expresan la diana N87 (cáncer gástrico), MDA-MB-361-DYT2 (cáncer de mama) las que no la expresan (HT-29) se colocaron en placas de cultivo celular de 96 pocillos durante 24 horas antes del tratamiento. Las células se trataron con conjugados de anticuerpo-fármaco diluidos 3 veces de forma seriada. La viabilidad celular se determinó mediante el ensayo CellTiter 96® AQ.ueousOne Solution Cell Proliferation MTS (Promega, Madison WI) 96 horas después del tratamiento. La viabilidad celular relativa se determinó como porcentaje del control no tratado. Los valores de CI50 se calcularon utilizando un modelo logístico de cuatro parámetros N.° 203 con XLfit v4. 2 (IDBS, Guildford, Surry, Reino Unido).
Los resultados de citotoxicidad de las cargas útiles, las cargas útiles de referencia y los CAF y los CAF de referencia se muestran en las Tablas 6 y 10
Tabla 6: Potencias de carga útil libre del ejemplo:

Ejemplificación de conjugados de anticuerpo-fármaco
Procedimiento D: Procedimiento general para la conjugación de anticuerpos con enlazador-carga útil a través de d isulfuros internos
Se redujo una solución de anticuerpo terapéutico en solución salina tamponada con fosfato de Dulbecco (PBS, Lonza pH 7,4) mediante la adición de 3,0-3,5 equivalentes de clorhidrato de tris(2-carboxietil)fosfina (TCEP, solución de 5 mM en PBS). La reacción se incubó a 37 °C durante 1-2 horas y luego se dejó enfriar a temperatura ambiente. La conjugación se realizó mediante la adición de 7 equivalentes de enlazador-carga útil [solución de 10 mM en N, N
dimetilacetamida (DMA)]. Se añadió DMA adicional a la mezcla de reacción para lograr un 15 % (v/v) de componente de disolvente orgánico total en la mezcla de reacción final. La reacción se incubó durante 1 hora a temperatura ambiente. Después de 1 hora a temperatura ambiente, a la reacción cruda se le intercambió el tampón con PBS mediante columnas de desalación G E Sephadex o el exceso de enlazador-carga útil se inactivó mediante la adición de 10 equivalentes de cisteína (solución de 20 mM en PBS). En cualquier caso, el material bruto se purificó luego por cromatografía de exclusión por tamaño (SEC, Protocolo C). Las fracciones monoméricas se agruparon y se concentraron si fuera necesario para dar el CAF.
Procedimiento E: Conjugación específica del sitio del enlazador-carga útil con un anticuerpo trastuzumab que contiene restos de cisteína diseñados genéticamente
Se preparó una solución de anticuerpo terapéutico que contenía un resto de cisteína diseñado genéticamente (numeración Kabat, véase el documento WO2013093809) en tampón fosfato 50 mM, a pH 7,4. Se añadió PBS, EDTA (solución madre de 0,5 M) y T C E P (solución madre de 0,5 M) de modo que la concentración final de proteína fue de ~10 mg/ml, la concentración final de EDTA fue de ~20 mM, y la concentración final de T C EP fue de aproximadamente ~6,6 mM (100 eq. molares). Se permitió que la reacción permaneciera a temperatura ambiente durante 2-48 h y luego se intercambió el tampón en PBS usando columnas G E PD-10 Sephadex g 25 según las instrucciones del fabricante. Los procedimientos alternativos tales como la diafiltración o la diálisis también son útiles en circunstancias particulares. La solución resultante se trató con aproximadamente 50 equivalentes de deshidroascorbato (solución madre de 50 mM en EtOH/agua a 1:1). El anticuerpo se dejó en reposo a 4 °C durante la noche y posteriormente se intercambió el tampón en PBS usando columnas G E PD-10 Sephadex G25 según las instrucciones del fabricante. De nuevo, los procedimientos alternativos tales como la diafiltración o la diálisis también son útiles en circunstancias particulares. La conjugación con el anticuerpo así preparado se realizó mediante la adición de 10 equivalentes de enlazador-carga útil [solución de 10 mM en N,N-dimetilacetamida (DMA)]. Se añadió DMA adicional a la mezcla de reacción para lograr un 15 % (v/v) de componente de disolvente orgánico total en la mezcla de reacción final. En algunos casos, se añadió PBS adicional para lograr la concentración final de anticuerpos totales en la reacción de 5-10 mg/ml. Después de 1-2 horas a temperatura ambiente, a la mezcla de reacción se le intercambió el tampón en PBS (según lo anterior) y se purificó por cromatografía de exclusión por tamaño (SEC, Protocolo D). Las fracciones monoméricas se agruparon y se concentraron si fuera necesario para dar el CAF. En algunos casos, el CAF se incubó posteriormente después de la purificación por S E C con 0,5-1,0 g de adsorbente Bio-Beads SM-2 (Bio-Rad) a 23-37 °C durante 4-18 h, se filtró y luego se concentró si fuese necesario.
Procedimiento F: Conjugación mediada por enzimas con anticuerpos que llevan restos reactivos de glutamina: El anticuerpo terapéutico que lleva restos de glutamina reactivos a la enzima transglutamina se dializó en agua estéril (Lonza). La conjugación mediada por transglutaminasa se llevó a cabo mezclando 5,0-15 mg/ml de anticuerpo que contiene glutamina reactiva con transglutaminasa en agua con fosfato de potasio 30 mM a pH 7,4 concentrado de medio de disolución DILUT-IT (J.T. Baker), cloruro de sodio 150 mM (NaCl), exceso molar de 15,0-20,0 veces de enlazador aminoalquil que transporta carga útil (solución de 10-30 mM en DMSO) y 30-60 mg/ml de polvo de enzima transglutaminasa (Ajinomoto Activa TI). Además, se añadió DMSO a la mezcla de reacción para lograr un 5-10 % (v/v) de componente de disolvente orgánico total en la mezcla de reacción final. En algunos casos, se añadió agua adicional para lograr la concentración final de anticuerpos totales en la reacción de 5-10 mg/ml. La mezcla resultante se agitó a temperatura ambiente durante 5-6 horas. A continuación, se añadieron otros 30-60 mg/ml de transglutaminasa sólida y la mezcla se incubó adicionalmente a temperatura ambiente durante 18 horas adicionales. Después de toda la noche, a la reacción cruda se le intercambió el tampón con solución salina tamponada con fosfato de Dulbecco (PBS, a pH7,4, Lonza) utilizando columnas de intercambio de tampones Sephadex de G E Healthcare según las instrucciones del fabricante. El material crudo se purificó por cromatografía de exclusión por tamaño (SEC, Protocolo D) o por cromatografía de interacción hidrófoba (HIC, Protocolo G). Después de la purificación, las fracciones monoméricas se agruparon y se concentraron si fuera necesario para dar el CAF. En ejemplos en los que el CA F se purificó mediante HIC, a las fracciones agrupadas se les intercambió el tampón con solución salina tamponada con fosfato de Dulbecco (PBS, a pH7,4, Lonza) utilizando columnas de intercambio de tampones Sephadex de G E Healthcare según las instrucciones del fabricante. En algunos casos, el CA F se incubó posteriormente después de la purificación con 0,5 1,0 g de adsorbente Bio-Beads SM-2 (Bio-Rad) a 23-37 °C durante 4-18 h, se filtró y luego se concentró si fuera necesario.
El ADC se caracterizó mediante cromatografía de exclusión por tamaño (SEC) y cromatografía de interacción hidrofóbica (HIC) para la pureza, y se hizo una cromatografía líquida de espectrometría de masas en tándem de ionización por electroaspersión (LC-ESI MS) para calcular la proporción de fármaco-anticuerpo (DAR, carga).
Protocolo C: Columna: Agilent Poroshell 300SB-C8, 75 x 2,1 mm, 2,6 pm; Fase móvil A: Ácido fórmico al 0,1 % en agua (v/v); Fase móvil B: Ácido fórmico al 0 ,1 % en acetonitrilo (v/v); Gradiente: Condiciones iniciales: 20 % de B al 45 % de B durante 4 minutos; Caudal: 1,0 ml/minuto. Temperatura: 60 °C; Detección: 220 nm; MS (+) intervalo de 400-2000Da; Volumen de inyección: 10 pl; Equipo: Agilent 1100 LC, Waters MicromassZQ MS. La desconvolución se realizó utilizando MaxEnt1.
Protocolo D: Columna: G E Superdex 200 (10/300 GL); Fase móvil: Solución salina tamponada con fosfato (PBS,
1X, a pH 7,4); Isocrático; Caudal: 1,0 ml/minuto. Temperatura: temperatura ambiente; Equipo: G E Akta Explorer.
Protocolo E : Columna = Waters BEH300-C4, 2,1 x 100 mm (P/N = 186004496); Equipo = Acquity UPLC con un detector de especificación de masa SQD2; Caudal = 0,7 ml/min; Temperatura = 80 °C ; Tampón A = agua ácido fórmico al 0,1 % ; Tampón B = acetonitrilo ácido fórmico al 0,1 % . El gradiente va del 3 % de B al 95 % de B durante 2 minutos, se mantiene al 95 % de B durante 0,75 min, y luego se reequilibra al 3 % de B. La muestra se reduce con T C EP o DTT inmediatamente antes de la inyección. El eluato se controla por LCMS (400-2000 daltons) y el pico de proteína se desconvoluciona usando MaxEnt1. El DAR se documenta como una carga promedio en peso como se ha descrito anteriormente.
Protocolo F: Columna: TSKGel Butyl NPR, 4,6 mm x 3,5 cm (P/N = S0557-835); Tampón A = sulfato de amonio 1,5 M que contiene fosfato 10 mM, a pH 7; Tampón B = fosfato 10 mM, a pH 7 alcohol isopropílico al 20 % ; Caudal = 0,8 ml/min; Temperatura = ambiente; Gradiente = 0 % de B al 100 % B durante 12 minutos, mantener al 100 % de B durante 2 minutos, luego reequilibrar al 100 % de A; Equipo: Agilent 1100 HPLC.
Protocolo G: Columna: G E HiTrap Butyl HP, 5 ml; Fases móviles: Fosfato de potasio 1 M, Tris-HCl 50 mM, pH7 (tampón A); Tris-HCl 50 mM, pH7 (tampón B); Gradiente de elución: 0-100 % de tampón B sobre 10-20 volúmenes de columna; Caudal: 5,0 ml/minuto. Temperatura: temperatura ambiente; Equipo: G E Akta Explorer.
Tabla 7: Estructura del CAF y CAF de referencia y enlazador-carga útil utilizados para prepararlos

X = En la tabla anterior, "X" indica el anticuerpo utilizado para la conjugación tal como se indica en la Tabla 5.
Tabla 8: Procedimiento general para la preparación de CAF y CAF de referencia

Tabla 9: Procedimiento de purificación y caracterización analítica de CAF y CAF de referencia

Tabla 10: Datos de citotoxicidad in vitro para los CAF N.° 1-6 en líneas celulares de Antígeno ve y Antígeno
-ve
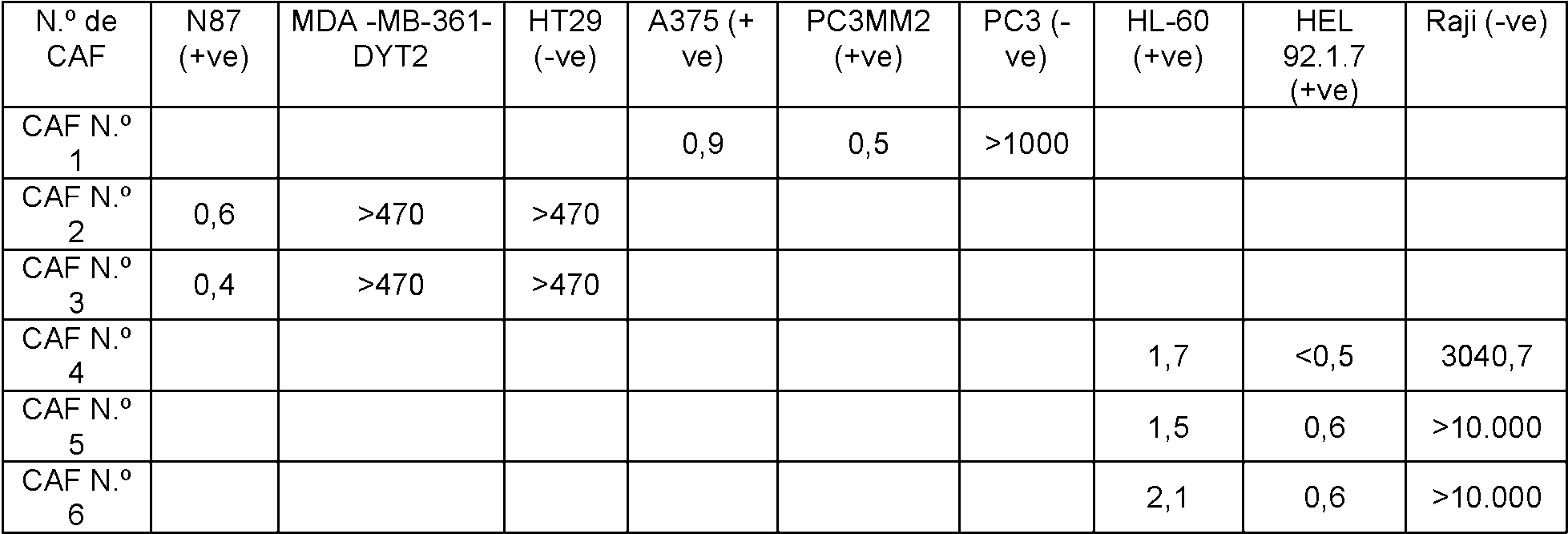
Se preparan los siguientes CAF y CAF de referencia, incluyendo el enlazador/carga útil que se muestra en las Tablas 4 y 5 conjugado con anticuerpos de interés utilizando el procedimiento de conjugación apropiado:
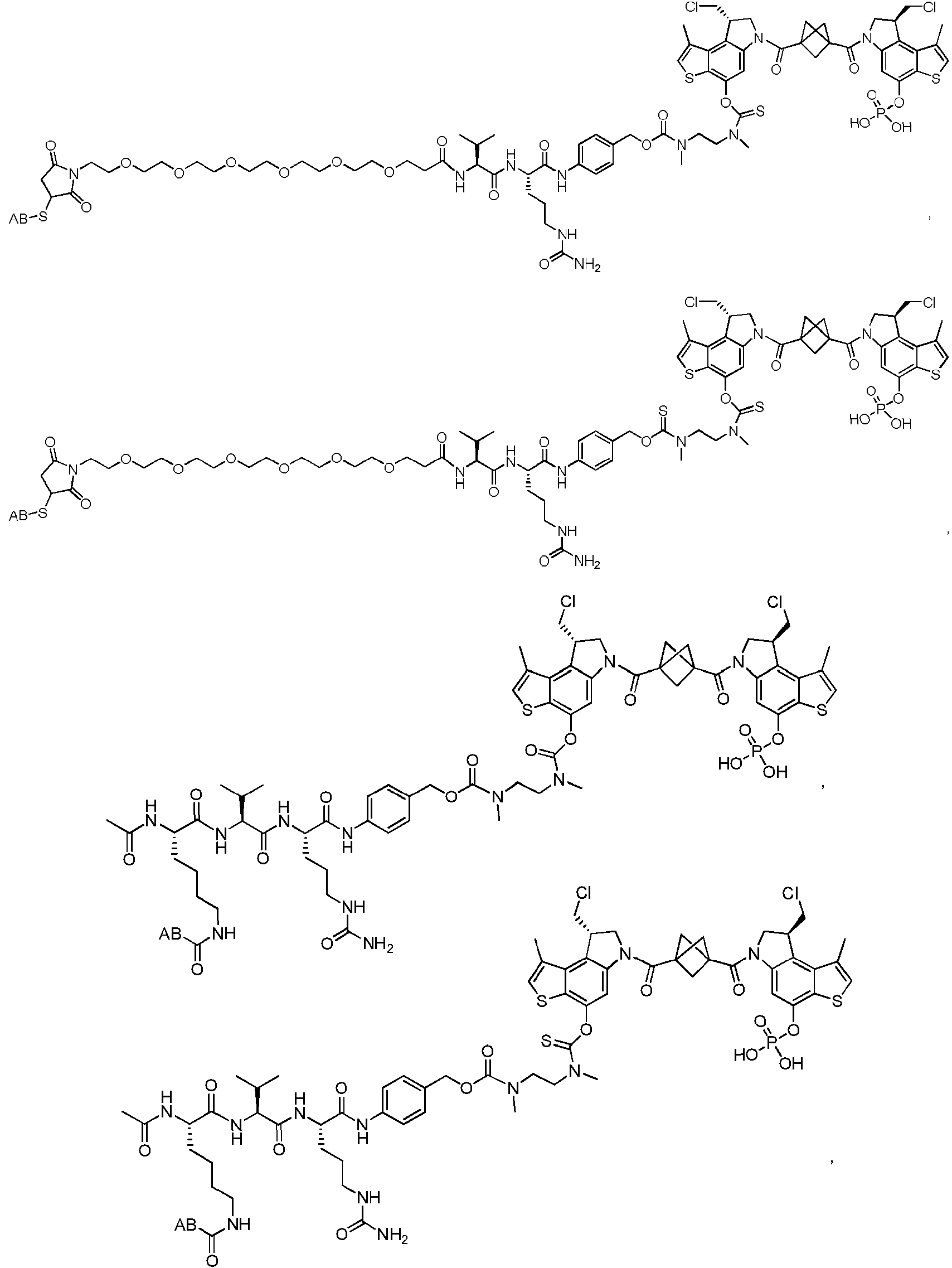


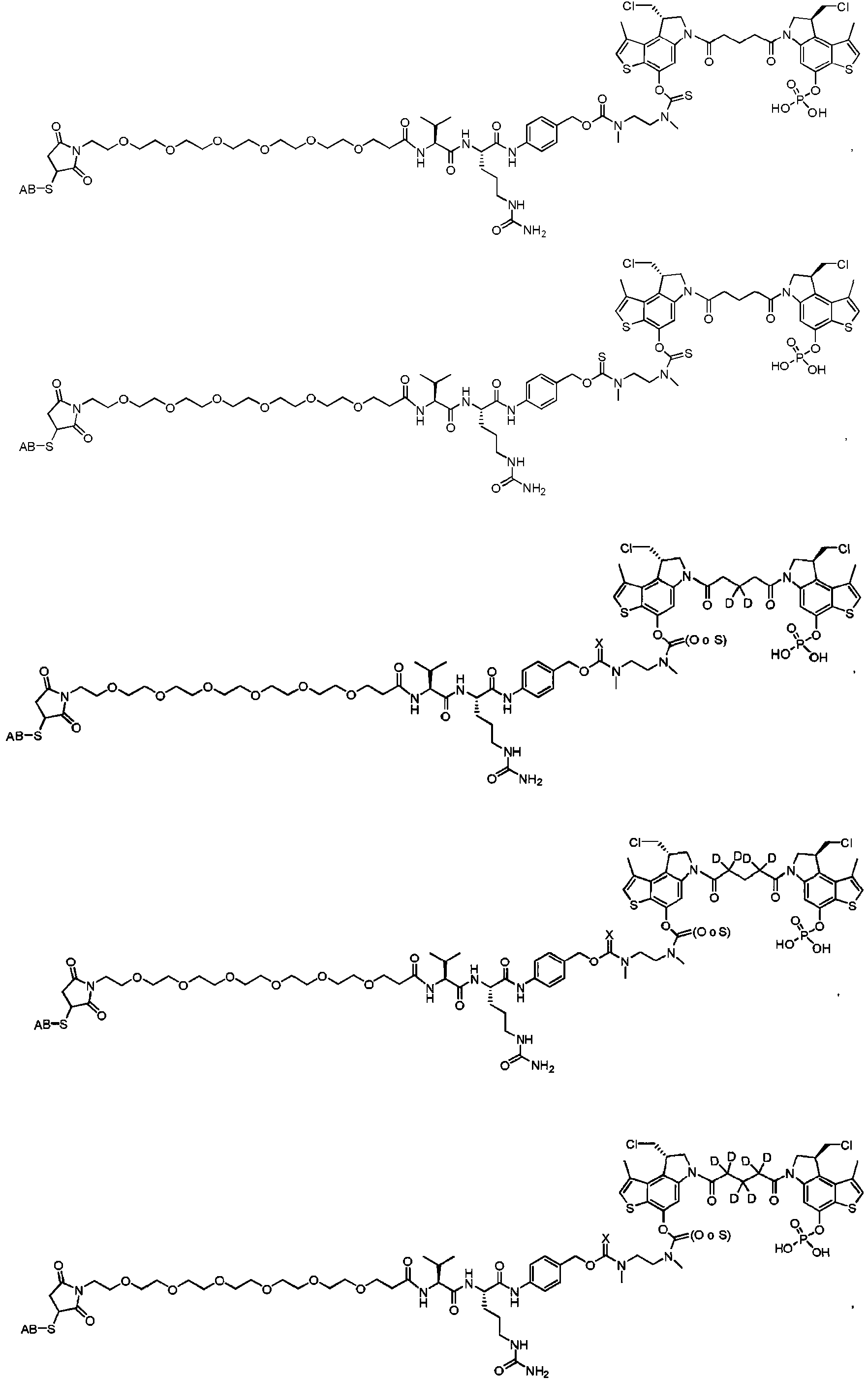








Claims (10)
1. Un compuesto, o una sal o solvato farmacéuticamente aceptable del mismo, seleccionado entre:


donde Pg es H, acilo, PO 3 H2 o glicosilo.
2. Un compuesto, o una sal o solvato farmacéuticamente aceptable del mismo, seleccionado entre:

3. Un compuesto, o una sal o solvato farmacéuticamente aceptable del mismo, seleccionado entre:
Ċ





4. El compuesto de la reivindicación 3, o una sal o solvato farmacéuticamente aceptable del mismo, en el que dicho anticuerpo AB se selecciona entre trastuzumab, mutantes de trastuzumab, oregovomab, edrecolomab, cetuximab, un anticuerpo monoclonal humanizado para el receptor de vitronectina (avp3), alemtuzumab, anticuerpos anti-HLA-DR, 1311 Lym-1, anticuerpos anti-HLA-Dr10, anticuerpos anti-cd33, anticuerpos anti-cd22, labetuzumab, bevacizumab, ibritumomab tiuxetán, ofatumumab, panitumumab, rituximab, tositumomab, ipilimumab y gemtuzumab.
5. Un compuesto de acuerdo con la reivindicación 3, o una sal o solvato farmacéuticamente aceptable del mismo, seleccionado entre:

en la que X es un anticuerpo seleccionado entre el grupo que consiste en IL13Ra2-AB08-v1.0, C D 33 -11A 1-v 1417 -hG1-(C), C D 33-11A 1 -v1417-K334C-K392C-hG 1-(C334+C392), y C D 33-11A 1-v1417-K 334 C -h G 1-(C 334 ).
6. Una carga útil de enlazador o un conjugado anticuerpo-fármaco, o una sal o solvato farmacéuticamente aceptable del mismo, que comprende un radical de un compuesto de la reivindicación 1.
7. Un conjugado anticuerpo-fármaco, o una sal o solvato farmacéuticamente aceptable del mismo, que comprende un radical de un compuesto de la reivindicación 2.
8. Una composición farmacéutica que comprende un compuesto de una cualquiera de las reivindicaciones 1 a 7, o una sal o solvato farmacéuticamente aceptable del mismo, y un excipiente farmacéuticamente aceptable.
9. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 7, o una sal o solvato farmacéuticamente aceptable del mismo, o una composición farmacéutica de la reivindicación 8, para su uso en el tratamiento de cáncer.
10. Un compuesto, o una sal o solvato farmacéuticamente aceptable del mismo, o una composición farmacéutica para su uso de acuerdo con la reivindicación 9, en el que dicho cáncer es cáncer de vejiga, cáncer de mama, cáncer de cuello de útero, cáncer de colon, cáncer de endometrio, cáncer de riñón, cáncer de pulmón, cáncer de esófago, cáncer de ovarios, cáncer de próstata, cáncer pancreático, cáncer de piel, cáncer de estómago (gástrico), cáncer testicular, leucemias y linfomas.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562136223P | 2015-03-20 | 2015-03-20 | |
| US201562138505P | 2015-03-26 | 2015-03-26 | |
| PCT/IB2016/051465 WO2016151432A1 (en) | 2015-03-20 | 2016-03-15 | Bifunctional cytotoxic agents containing the cti pharmacophore |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2769031T3 true ES2769031T3 (es) | 2020-06-24 |
Family
ID=60675468
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES16714004T Active ES2769031T3 (es) | 2015-03-20 | 2016-03-15 | Agentes citotóxicos bifuncionales que contienen el farmacóforo cti |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP3271365B1 (es) |
| CN (1) | CN107660208B (es) |
| DK (1) | DK3271365T5 (es) |
| ES (1) | ES2769031T3 (es) |
| HK (1) | HK1245252A1 (es) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MX2022003671A (es) * | 2019-09-27 | 2022-04-25 | Univ Texas | Inhibidores de proteina cinasa i de interaccion con receptores para el tratamiento de enfermedades. |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090118349A1 (en) * | 2007-08-16 | 2009-05-07 | Onco-Pharmakon Incorporated | Bis-alkylating agents and their use in cancer therapy |
| DE102009051799B4 (de) * | 2009-11-03 | 2021-07-01 | Georg-August-Universität Göttingen Stiftung Öffentlichen Rechts | Bifunktionale Prodrugs und Drugs |
| RU2632199C2 (ru) * | 2012-04-05 | 2017-10-03 | НЕРВИАНО МЕДИКАЛ САЙЕНСИЗ С.р.л. | Функционализированные производные тиеноиндола для лечения ракового заболевания |
-
2016
- 2016-03-15 EP EP16714004.5A patent/EP3271365B1/en active Active
- 2016-03-15 ES ES16714004T patent/ES2769031T3/es active Active
- 2016-03-15 CN CN201680029088.7A patent/CN107660208B/zh active Active
- 2016-03-15 DK DK16714004.5T patent/DK3271365T5/da active
-
2018
- 2018-04-04 HK HK18104498.9A patent/HK1245252A1/zh unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CN107660208A (zh) | 2018-02-02 |
| DK3271365T3 (da) | 2020-02-24 |
| HK1245252A1 (zh) | 2018-08-24 |
| EP3271365A1 (en) | 2018-01-24 |
| EP3271365B1 (en) | 2019-12-18 |
| CN107660208B (zh) | 2020-06-30 |
| DK3271365T5 (da) | 2020-03-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101871088B1 (ko) | 이관능성 세포독성제 | |
| US11365263B2 (en) | Bifunctional cytotoxic agents containing the CTI pharmacophore | |
| JP7555998B2 (ja) | ヘテロアリールスルホンをベースとするコンジュゲーションハンドル、これらの調製のための方法、および抗体薬物コンジュゲートの合成におけるこれらの使用 | |
| ES2769031T3 (es) | Agentes citotóxicos bifuncionales que contienen el farmacóforo cti |