ES2707734T3 - Derivados de carboxamida - Google Patents
Derivados de carboxamida Download PDFInfo
- Publication number
- ES2707734T3 ES2707734T3 ES15774680T ES15774680T ES2707734T3 ES 2707734 T3 ES2707734 T3 ES 2707734T3 ES 15774680 T ES15774680 T ES 15774680T ES 15774680 T ES15774680 T ES 15774680T ES 2707734 T3 ES2707734 T3 ES 2707734T3
- Authority
- ES
- Spain
- Prior art keywords
- methyl
- dimethyl
- triazole
- oxo
- carboxamide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 CC*c1c(*)c(*)c(*)cc1 Chemical compound CC*c1c(*)c(*)c(*)cc1 0.000 description 1
Landscapes
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Un compuesto de fórmula (I):**Fórmula** o una sal farmacéuticamente aceptable del mismo, en donde R1 es alquilo (C3-C6) o cicloalquilo (C3-C6); R2 es metilo; R3 es**Fórmula** R4 y R5 se seleccionan independientemente de hidrógeno, halo, cicloalquilo (C3-C6), alquilo (C1-C4), haloalquilo (C1- C4), alcoxi (C1-C4), haloalcoxi (C1-C4) o-alquil (C1-C2) alcoxi (C1-C2); o R2 y R4 se pueden tomar junto con los átomos de carbono a los que están unidos para formar un anillo azepina y R5 es H; R6 es halo, cicloalquilo (C3-C6), alquilo (C1-C4), haloalquilo (C1-C4), alcoxi (C1-C4), haloalcoxi (C1-C4) o -alquil (C1- C2) alcoxi (C1-C2); o R1 es 2-fluorofenilo; R2 es metilo; y R3 es fenilo, sustituido con uno o dos sustituyentes seleccionados independientemente entre cloro y ciclopropilo.
Description
DESCRIPCIÓN
Derivados de carboxamida
Campo de la invención
La presente invención describe compuestos orgánicos útiles en terapia como se definen por las reivindicaciones. Los compuestos demuestran propiedades como inhibidores selectivos de Smurf-1 y, por consiguiente, pueden ser útiles en el tratamiento de un número de trastornos, en particular hipertensión arterial pulmonar, así como otros trastornos, tales como glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, sanado de heridas, enfermedad pulmonar obstructiva crónica (EPOC) y asma.
Antecedentes de la invención
Smurf-1 (factor regulador de ubiquitinación de Smad 1) es un miembro de la familia HECT de sustratos específicos marcadores de ligasa de ubiquitina E3 para la degradación proteolítica por medio de la senda proteolítica dependiente de ubiquitina. Los principales sustratos de Smurf-1 incluyen RhoA, receptores 1 y 2 de proteína morfogenética ósea (BMPR), smad 1 y 5, factor asociado con los receptores de TNFa (TRAF) 6 y myD88 (Andrews, P. S. et al., Assay Drug Dev. Technol. 2010). Dada la lista de sustratos, Smurf-1 tiene funciones establecidas en la regulación de la señalización de la proteína morfogenética ósea (BMP) (Chen, D et al., Growth Factors, 2004), en la polaridad de las células neuronales (Stiess, M. y Bradke, F. Neuron, 2011), en la migración celular (Huang, C. Cell Adh. Migr. 2010), en la invasión de células tumorales (Sahai, E. et al., JCB, 2007), en la autofagia mitocondrial (Orvedahl, A. Nature, 2011), en la proliferación de células madre mesenquimales (Zhao, L. et al., J. Bone Miner. Res. 2010), y en la transición epitelial-mesenquimal (EMT) (Ozdamar, B et al., Science 2005).
La hipertensión arterial pulmonar (PAH) es una enfermedad agresiva y compleja mortal de múltiples etiologías, caracterizada por una vasculopatía pulmonar progresiva que conduce a hipertrofia/falla del ventrículo derecho y, en la mayoría de los casos, a la muerte prematura. Las terapias farmacológicas actuales son paliativas. Aunque se han observado mejoras en la expectativa de vida, las terapias actuales, las cuales se enfocan en la alteración de los elementos vasoconstrictivos de la enfermedad, no detienen ni revierten el progreso de la enfermedad, y el trasplante (doble pulmón o corazón-pulmón) sigue siendo el único tratamiento curativo. Dado el efecto limitado de las clases de tratamiento actuales, se necesitan terapias novedosas que tengan como objetivo la remodelación vascular pulmonar progresiva subyacente de la hipertensión arterial pulmonar (PAH).
Las mutaciones de la línea germinal en el gen del receptor de proteína morfogenética ósea II (BMPR-II) de los receptores de la superfamilia del factor de crecimiento transformante B (TGF-B) prevalecen en el setenta por ciento de las formas hereditarias y algunas formas esporádicas de hipertensión arterial pulmonar (PAH) idiopática (IPAH). Las proteínas morfogenéticas óseas son moléculas de señalización que pertenecen a la superfamilia TGF-B. Las proteínas morfogenéticas óseas fueron originalmente identificadas por su capacidad para inducir la formación de cartílago y hueso, y subsiguientemente fueron identificadas como proteínas multifuncionales que regulan un amplio espectro de funciones, tales como proliferación, diferenciación, y apoptosis en una gran variedad de tipos de células, incluyendo osteoblastos, células epiteliales, neuronas, células inmunes, y células de músculo liso. Hasta ahora, se han identificado >20 proteínas morfogenéticas óseas (BMPs) de mamífero, pero solamente tres receptores tipo I y tres receptores tipo II (BMPR-I y BMPR-II, respectivamente) que son capaces de enlazarse con las proteínas morfogenéticas óseas (BMPs), se han clonado en mamíferos. Las proteínas morfogenéticas óseas (BMPs) son sintetizadas y secretadas a partir de una variedad de tipos de células, incluyendo las células de músculo liso vascular pulmonar y las células endoteliales. En adición a las mutaciones en BMPR-I y -II, los pulmones de los pacientes con hipertensión arterial pulmonar (PAH) no familiar exhiben niveles notoriamente reducidos de BMPR-I y -II vasculares, implicando una función central para la señalización interrumpida de las proteínas morfogenéticas óseas (BMPs) en muchas formas de hipertensión arterial pulmonar (PAH) (Du, L et al., N. Eng. J. Med, 2003). Por consiguiente, es de un interés considerable el restablecimiento de la señalización de la proteína morfogenética ósea (BMP) en la vasculatura pulmonar de los pacientes de PAH en el desarrollo de productos terapéuticos novedosos contra la remodelación para el tratamiento de la PAH.
Se ha demostrado que Smurf-1 media la degradación de BMPR-I, -II y smad 1 y 5 en una variedad de tipos de células, incluyendo osteoblastos (Zhao, M et al., JBC, 2003), mioblastos (Ying, SX et al., JBC, 2003), epitelio pulmonar (Shi W et al., Am. J. Physiol. Cell. Mol. Physiol, 2004), tejido neuronal (Kallan, T et al., Mol. Cell. Biol, 2009), y células endocárdicas (Towsend, TA et al., Cells Tissues Organs, 2011). Recientemente, surgió la primera evidencia de una función de Smurf-1 en la hipertensión arterial pulmonar (PAH) en donde se observaron mayores niveles de Smurf-1 en los modelos in vivo preclínicos de monocrotalina e hipoxia crónica de PAH y asociados con una subregulación de BMPR1 y 2 (Murakami, K et al., Exp. Biol. Med, 2010 y Yang,
J. et al., Circ. Res, 2010).
El documento US 7.354.722 analiza la modulación biológica de las sendas de señalización de Smurf pero no revela inhibidores de moléculas pequeñas. PubChem Compound (en línea) del 25 de enero de 2012, N.° de acceso a la base de datos CID55855650 y N.° 55882610 revelan compuesto de triazol sin proporcionar ninguna utilidad.
Compendio de la invención
Sigue existiendo una necesidad de nuevos tratamientos y terapias para la hipertensión arterial pulmonar así como para otros trastornos, tales como glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, sanado de heridas, enfermedad pulmonar obstructiva crónica (EPOC) y asma. La invención proporciona compuestos o sus sales farmacéuticamente aceptables, sus composiciones farmacéuticas y sus combinaciones, cuyos compuestos son inhibidores de Smurf-1. La revelación proporciona además métodos para tratar, prevenir, o mitigar la hipertensión arterial pulmonar, el cual comprende administrar a un sujeto que lo necesite, una cantidad eficaz de un inhibidor de Smurf-1.
De acuerdo con un primer aspecto de la invención, la forma de realización 1, se proporciona un compuesto de la fórmula (I):

o una sal farmacéuticamente aceptable del mismo, en donde
R1 es alquilo (C3-C6) o cicloalquilo (C3-C 6);
R2 es metilo;
R3 es

R4 y R5 se seleccionan independientemente de hidrógeno, halo, cicloalquilo (C3-C 6), alquilo (C1-C 4), haloalquilo (C1-C4), alcoxi (C1-C 4), haloalcoxi (C1-C 4) o -alquil (C1-C 2)-alcoxi (C1-C 2); o
R2 y R4 se pueden tomar junto con los átomos de carbono a los que están unidos para formar un anillo azepina y R5 es H;
R6 es halo, cicloalquilo (C3-C 6), alquilo (C1-C 4), haloalquilo (C1-C 4), alcoxi (C1-C 4), haloalcoxi (C1-C 4) o-alquilo (C1-C2) alcoxi (C1 C2);
o
R1 es 2-fluorofenilo;
R2 es metilo; y
R3 es fenilo, sustituido con uno o dos sustituyentes seleccionados independientemente entre cloro y ciclopropilo. En otra forma de realización de la invención se proporciona un compuesto de fórmula (I) como se define anteriormente o una sal farmacéuticamente aceptable del mismo.
En otra forma de realización, la invención proporciona una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de un compuesto de acuerdo con la definición de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, o una subfórmula de la misma y uno o más portadores farmacéuticamente aceptables.
En otra forma de realización, la presente descripción, con fines de referencia, proporciona un método para el tratamiento de un trastorno o de una enfermedad seleccionada a partir de hipertensión pulmonar, incluyendo hipertensión arterial pulmonar (PAH), fibrosis, artritis reumatoide, y sanado de fracturas, el cual comprende administrar al sujeto, una cantidad terapéuticamente eficaz del compuesto de acuerdo con la definición de la fórmula (I), o de una sal del mismo, o de las subfórmulas de la misma, o una sal farmacéuticamente aceptable.
En otra forma de realización, la presente descripción, con fines de referencia, proporciona un método para el tratamiento de un trastorno o de una enfermedad seleccionada a partir de glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, sanado de heridas, así como enfermedad pulmonar obstructiva crónica (EPOC) y asma, el cual comprende administrar una cantidad terapéuticamente eficaz de un compuesto de la fórmula (I), o de una sal farmacéuticamente aceptable del mismo, a un sujeto que reconocidamente lo necesite.
En otra forma de realización, la invención proporciona una combinación, en particular una combinación farmacéutica, la cual comprende una cantidad terapéuticamente eficaz del compuesto de acuerdo con la definición de la fórmula (I), o de una sal farmacéuticamente aceptable del mismo, o de las subfórmulas de la misma, y uno o más agentes terapéuticamente activos.
En otra forma de realización, la invención proporciona una combinación, en particular una combinación farmacéutica, la cual comprende una cantidad terapéuticamente eficaz de un compuesto de la fórmula (I), o de una sal farmacéuticamente aceptable del mismo, y uno o más agentes terapéuticamente activos.
En la presente, se describen diferentes formas de realización de la invención.
Descripción detallada
La invención, por consiguiente, proporciona un compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, como se describe anteriormente en la presente como la forma de realización 1.
Forma de realización 2. Un compuesto de acuerdo con la forma de realización 1, o una sal farmacéuticamente aceptable del mismo, en donde R1 es isopropilo, ciclobutilo o ciclohexilo.
Forma de realización 3. Un compuesto de acuerdo con la forma de realización 1 o con la forma de realización 2, o una sal farmacéuticamente aceptable del mismo, en donde R1 es ciclohexilo.
Forma de realización 4. Un compuesto de acuerdo con cualquiera de las formas de realización anteriores, o una sal farmacéuticamente aceptable del mismo, en donde:
R3 es:

R4 y R6 se seleccionan independientemente a partir de cloro, flúor, ciclopropilo, metilo, metoxi, trifluorometoxi, trifluoro-metilo; y
R5 es hidrógeno.
Forma de realización 5. Un compuesto de acuerdo con cualquiera de las formas de realización anteriores, o una sal farmacéuticamente aceptable del mismo, en donde R3 es:

R4 y R6 se seleccionan independientemente a partir de cloro y ciclopropilo; y
R5 es hidrógeno.
Forma de realización 6. Un compuesto de la fórmula (I) o una sal farmacéuticamente aceptable del mismo de acuerdo con la forma de realización 1, en donde el compuesto se selecciona a partir de:
Ejemplo 1:
1-(2-cloro-4-metoxi-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-m etil-1 H-1.2.3- triazol-4-carboxamida
Ejemplo 2:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-1-(2,4-dicloro-fenil)-5-metil-1 H-1,2,3-triazol-4-carboxamida
Ejemplo 3:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-1-(4-metoxi-2-(trifluoro-metil)-fenil)-5-metil-1 H-1,2,3-triazol-4-carboxamida
Ejemplo 4:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-1-(4-metoxi-3-metil-fenil)-5-metil-1 H-1.2.3- triazol-4-carboxamida
Ejemplo 5:
1-(2-cloro-4-(trifluoro-metoxi)-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida
Ejemplo 6:
1-(4-cloro-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-p i razol-4-i l )-5-m etil-1 H-1,2,3-triazol-4-carboxamida
Ejemplo 7:
1 -(2,4-dicloro-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1.2.3- triazol-4-carboxamida
Ejemplo 8:
1-(4-cloro-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida
Ejemplo 9:
1-(2-cloro-4-(trifluoro-metoxi)-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida
Ejemplo 10:
1 -(2-cloro-4-ciclopropil-fen¡l)-N-(2-c¡clohex¡l-1,5-d¡metil-3-oxo-2,3-d¡h¡dro-1 H-p i razol-4-i l)-5-m etil-1 H-1.2.3- triazol-4-carboxamida
Ejemplo 11:
1-(2-cloro-4-ciclopropil-fen¡l)-N-(2-(2-fluoro-fen¡l)-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida
Ejemplo 12:
1-(2-cloro-4-cicloprop¡l-fen¡l)-N-(2-c¡clohex¡l-1-met¡l-d3,5-met¡l-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida
Ejemplo 13:
1-(4-cloro-2-ciclopropil-fen¡l)-N-(2-(2-fluoro-fen¡l)-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida
Ejemplo 14:
1 -(4-cloro-2-ciclopropil-fen¡l)-N-(2-c¡clohex¡l-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-p i razol-4-i l)-5-m etil-1 H-1.2.3- triazol-4-carboxamida
Ejemplo 15:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-¡l)-1-(2-c¡cloprop¡l-4-fluoro-fen¡l)-5-metil-1 H-1,2,3-triazol-4-carboxamida
Ejemplo 16:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-p¡razol-4-¡l)-8-(trifluoro-metox¡)-5,6-d¡h¡dro-4H-benzo-[f][1,2,3]-tr¡azolo-[1,5-a]-azepina-3-carboxamida
Ejemplo 17:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-1-(4-metox¡-fen¡l)-5-met¡l-1 H-1,2,3-triazol-4-carboxamida.
Forma de realización 7. Un compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, de acuerdo con la forma de realización 1, en donde el compuesto se selecciona a partir de:
Ejemplo 10:
1 -(2-cloro-4-ciclopropil-fen¡l)-N-(2-c¡clohex¡l-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-p i razol-4-i l)-5-m etil-1 H-1.2.3- triazol-4-carboxamida
Ejemplo 11:
1-(2-cloro-4-ciclopropil-fen¡l)-N-(2-(2-fluoro-fen¡l)-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida
Ejemplo 12:
1-(2-cloro-4-cicloprop¡l-fen¡l)-N-(2-c¡clohex¡l-1-met¡l-d3,5-met¡l-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida, y
Ejemplo 16:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-¡l)-8-(tr¡fluoro-metoxi)-5,6-d¡h¡dro-4H-benzo-[f][1,2,3]-triazolo-[1,5-a]-azepina-3-carboxamida;
o una sal farmacéuticamente aceptable de las mismas.
Como se utiliza en la presente, el término “halo” (o halógeno) se refiere a flúor, bromo, cloro o yodo, en particular flúor, cloro. Los grupos y restos sustituidos por halógeno, tales como alquilo sustituido por halógeno (halo-alquilo), pueden estar mono-, poli- o per-halogenados.
Como se utiliza en la presente, el término “alquilo” se refiere a un resto de hidrocarburo completamente saturado ramificado o no ramificado que tiene hasta 10 átomos de carbono. A menos que se disponga de otra manera, alquilo se refiere a los restos de hidrocarburo que tienen de 1 a 10 átomos de carbono, de 1 a 6 átomos de carbono, o de 1 a 4 átomos de carbono. Los ejemplos representativos de alquilo incluyen, pero sin limitación, metilo, etilo, propilo normal, isopropilo, butilo normal, butilo secundario, isobutilo, butilo terciario, pentilo normal, isopentilo, neopentilo, hexilo normal, 3-metil-hexilo, 2,2-dimetil-pentilo, 2,3-dimetil-pentilo, heptilo normal, octilo normal, nonilo normal, decilo normal, y similares. Los ejemplos representativos de alquilo ramificado incluyen, pero sin limitación, isopropilo, butilo secundario, isobutilo, butilo terciario, isopentilo, 3 -metil-hexilo, 2,2-dimetil-pentilo, 2,3-dimetil-pentilo, y similares. Un alquilo sustituido es un grupo alquilo que contiene uno o más, tal como uno, dos o tres sustituyentes seleccionados a partir de los grupos halógeno, hidroxi, o alcoxilo.
Como se utiliza en la presente, el término “halo-alquilo” se refiere a un alquilo, como se define en la presente, que está sustituido por uno o más grupos halógeno, como se definen en la presente. El halo-alquilo puede ser mono-halo-alquilo, di-halo-alquilo o poli-halo-alquilo, incluyendo perhalo-alquilo. Un mono-haloalquilo puede tener un yodo, bromo, cloro o flúor dentro del grupo alquilo. Los grupos di-halo-alquilo y polihalo-alquilo pueden tener dos o más de los mismos átomos de halógeno o una combinación de diferentes grupos halógeno dentro del alquilo. Típicamente, el poli-halo-alquilo contiene hasta 12, o 10, u 8, o 6, o 4, o 3, o 2 grupos halógeno. Los ejemplos no limitantes de haloalquilo incluyen fluorometilo, difluorometilo, trifluorometilo, clorometilo, diclorometilo, triclorometilo, pentafluoroetilo, heptafluoropropilo, difluoroclorometilo, diclorofluorometilo, difluoroetilo, difluoropropilo, dicloroetilo y dicloropropilo. Un perhaloalquilo se refiere a un alquilo que tiene todos los átomos de hidrógeno reemplazados con átomos de halógeno.
Como se utiliza en la presente, el término "alcoxi" se refiere a un alquil-O-, en donde el alquilo se define anteriormente en la presente. Los ejemplos representativos de alcoxilo incluyen, pero sin limitación, metoxi, etoxi, propoxi, 2-propoxi, butoxi, terbutoxi, pentiloxi, hexiloxi, ciclopropiloxi, ciclohexiloxi y similares. Típicamente, los grupos alcoxilo tienen de 1 a 4 átomos de carbono.
Como se utiliza en la presente, el término “halo-alcoxi" se refiere a un alcoxi, como se define en la presente, que está sustituido por uno o más grupos halógeno, como se definen en la presente.
Como se utiliza en la presente, el término "cicloalquilo" se refiere a los grupos hidrocarbonados saturados monocíclicos, bicíclicos, o espirocíclicos de 3 a 8 átomos de carbono. A menos que se disponga de otra manera, cicloalquilo se refiere a los grupos hidrocarbonados cíclicos que tienen entre 3 y 6 átomos de carbono en el anillo.
Dependiendo de la elección de los materiales de partida y de los procedimientos, los compuestos pueden estar presentes en la forma de uno de los posibles isómeros o como mezclas de los mismos, por ejemplo, como los isómeros ópticos puros, o como mezclas de isómeros, tales como racematos y mezclas de diastereoisómeros, dependiendo del número de átomos de carbono asimétricos. La presente invención pretende incluir todos los posibles isómeros, incluyendo las mezclas racémicas, las mezclas diastereoméricas, y las formas ópticamente puras. También se pretende incluir todas las formas tautoméricas.
Sólo con fines de referencia, los compuestos de la invención, es decir, los compuestos de la fórmula (I) que contienen grupos capaces de actuar como donadores y/o aceptores para los enlaces de hidrógeno, pueden ser capaces de formar cocristales con formadores de cocristales adecuados. Estos cocristales se pueden preparar a partir de los compuestos de la fórmula (I) mediante los procedimientos de formación de cocristales conocidos. Estos procedimientos incluyen moler, calentar, cosublimar, cofusionar, o poner en contacto en solución los compuestos de la fórmula (I) con el formador de cocristales bajo condiciones de cristalización, y aislar los cocristales formados de esta manera. Los formadores de cocristales adecuados incluyen aquéllos descritos en la Publicación Internacional Número WO 2004/078163. Por consiguiente, la descripción, con fines de referencia, proporciona además cocristales que comprenden un compuesto de la fórmula (I).
Como se utilizan en la presente, con fines de referencia, los términos “sal" o "sales” se refieren a una sal de adición de ácido o de adición de base de un compuesto de la invención. Las "sales" de acuerdo con la invención incluyen en particular las “sales farmacéuticas aceptables”. La expresión “sales farmacéuticamente aceptables” se refiere a las sales que conservan la efectividad biológica y las propiedades de los compuestos de esta invención, y que típicamente no son biológicamente o de otra manera indeseables.
En muchos casos, los compuestos de la presente invención son capaces de formar sales de ácido y/o base
en virtud del grupo carboxamida o grupos similares al mismo.
Las sales de adición de ácido farmacéuticamente aceptables (sólo con fines de referencia) o cocristales se pueden formar con ácidos inorgánicos y ácidos orgánicos.
Los ácidos inorgánicos a partir de los cuales se pueden derivar las sales incluyen, por ejemplo, ácido clorhídrico, ácido bromhídrico, ácido sulfúrico, ácido nítrico, ácido fosfórico, y similares.
Los ácidos orgánicos a partir de los cuales se pueden derivar las sales incluyen, por ejemplo, ácido acético, ácido propiónico, ácido glicólico, ácido oxálico, ácido maleico, ácido malónico, ácido succínico, ácido fumárico, ácido tartárico, ácido cítrico, ácido benzoico, ácido mandélico, ácido metansulfónico, ácido etansulfónico, ácido toluensulfónico, y similares.
Las sales de adición de base farmacéuticamente aceptables (sólo con fines de referencia) o cocristales se pueden formar con bases inorgánicas y orgánicas.
Las bases inorgánicas a partir de las cuales se pueden derivar las sales incluyen, por ejemplo, las sales de amonio y de los metales a partir de las columnas I a XII de la Tabla Periódica. En ciertas formas de realización, las sales se derivan a partir de sodio, potasio, amonio, calcio, magnesio, plata, y zinc; las sales particularmente adecuadas incluyen las sales de amonio, potasio, sodio, calcio y magnesio.
Las bases orgánicas a partir de las cuales se pueden derivar las sales incluyen, por ejemplo, aminas primarias, secundarias, y terciarias, aminas sustituidas, incluyendo las aminas sustituidas que se presentan naturalmente, aminas cíclicas, resinas básicas de intercambio de iones, y similares. Ciertas aminas orgánicas incluyen colinato, lisina, meglumina, piperazina y trometamina.
En otro aspecto, la presente invención proporciona los compuestos de la fórmula I en la forma de una sal de acetato, ascorbato, adipato, aspartato, benzoato, besilato, bromuro/ bromhidrato, bicarbonato/carbonato, bisulfato/sulfato, caprato, cloruro/clorhidrato, citrato, etan-disulfonato, fumarato, gluceptato, gluconato, glucuronato, glutamato, glutarato, glicolato, hipurato, yodhidrato/yoduro, isetionato, lactato, lactobionato, malato, maleato, malonato, mandelato, mesilato, metil-sulfato, mucato, naftoato, napsilato, nicotinato, nitrato, octadecanoato, oleato, oxalato, palmitato, pamoato, fosfato/fosfato ácido/fosfato diácido, poli-galacturonato, propionato, sebacato, estearato, succinato, sulfato, tartrato, tosilato, trifenatato, o xinafoato.
En una forma de realización, la presente invención proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida en una forma de sal de acetato, ascorbato, adipato, aspartato, benzoato, besilato, bromuro/bromhidrato, bicarbonato/carbonato, bisulfato/sulfato, caprato, cloruro/clorhidrato, citrato, etandisulfonato, fumarato, gluceptato, gluconato, glucuronato, glutamato, glutarato, glicolato, hipurato, yodhidrato/yoduro, isetionato, lactato, lactobionato, malato, maleato, malonato, mandelato, mesilato, sulfato de metilo, mucato, naftoato, napsilato, nicotinato, nitrato, octadecanoato, oleato, oxalato, palmitato, pamoato, fosfato/fosfato ácido/fosfato diácido, poligalacturonato, propionato, sebacato, estearato, succinato, sulfato, tartrato, tosilato, trifenatato o xinafoato.
En otra forma de realización, la presente invención proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-p i razol-4-il)-5-m etil-1 H-1,2,3-triazol-4-carboxamida en una forma de sal de acetato, ascorbato, adipato, aspartato, benzoato, besilato, bromuro/bromhidrato, bicarbonato/carbonato, bisulfato/sulfato, caprato, cloruro/clorhidrato, citrato, etandisulfonato, fumarato, gluceptato, gluconato, glucuronato, glutamato, glutarato, glicolato, hipurato, yodhidrato/yoduro, isetionato, lactato, lactobionato, malato, maleato, malonato, mandelato, mesilato, sulfato de metilo, mucato, naftoato, napsilato, nicotinato, nitrato, octadecanoato, oleato, oxalato, palmitato, pamoato, fosfato/fosfato ácido/fosfato diácido, poligalacturonato, propionato, sebacato, estearato, succinato, sulfato, tartrato, tosilato, trifenatato, o xinafoato.
En otra forma de realización, la presente invención proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2-ciclohexil-1 -metil-d3,5-metil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida en una forma de sal de acetato, ascorbato, adipato, aspartato, benzoato, besilato, bromuro/bromhidrato, bicarbonato/carbonato, bisulfato/sulfato, caprato, cloruro/clorhidrato, citrato, etan-disulfonato, fumarato, gluceptato, gluconato, glucuronato, glutamato, glutarato, glicolato, hipurato, yodhidrato/yoduro, isetionato, lactato, lactobionato, malato, maleato, malonato, mandelato, mesilato, sulfato de metilo, mucato, naftoato, napsilato, nicotinato, nitrato, octa-decanoato, oleato, oxalato, palmitato, pamoato, fosfato/fosfato ácido/fosfato diácido, poligalacturonato, propionato, sebacato, estearato, succinato, sulfato, tartrato, tosilato, trifenatato, o xinafoato.
En otra forma de realización, la presente invención proporciona la N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-8-(trifluoro-metoxi)-5,6-dihidro-4H-benzo-[f][1,2,3]-triazolo-[1,5-a]-azepina-3-carboxamida en una forma de sal de acetato, ascorbato, adipato, aspartato, benzoato, besilato, bromuro/bromhidrato, bicarbonato/carbonato, bisulfato/sulfato, caprato, cloruro/clorhidrato, citrato, etandisulfonato, fumarato, gluceptato, gluconato, glucuronato, glutamato, glutarato, glicolato, hipurato, yodhidrato/ yoduro, isetionato, lactato, lactobionato, malato, maleato, malonato, mandelato, mesilato, sulfato de metilo, mucato, naftoato, napsilato, nicotinato, nitrato, octadecanoato, oleato, oxalato, palmitato, pamoato, fosfato/fosfato ácido/fosfato diácido, poligalacturonato, propionato, sebacato, estearato, succinato, sulfato, tartrato, tosilato, trifenatato, o xinafoato.
En otro aspecto, la presente invención proporciona los compuestos de la fórmula I en la forma de una sal de sodio, potasio, amonio, calcio, magnesio, plata, zinc, colinato, lisina, meglumina, piperazina o trometamina.
En una forma de realización, la presente invención proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida en una forma de sal de sodio, potasio, amonio, calcio, magnesio, plata, zinc, colinato, lisina, meglumina, piperazina o trometamina.
En otra forma de realización, la presente invención proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida en una forma de sal de sodio, potasio, amonio, calcio, magnesio, plata, zinc, colinato, lisina, meglumina, piperazina o trometamina.
En otra forma de realización, la presente invención proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2-ciclohexil-1 -metil-d3,5-metil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida en una forma de sal de sodio, potasio, amonio, calcio, magnesio, plata, zinc, colinato, lisina, meglumina, piperazina o trometamina.
En otra forma de realización, la presente invención proporciona la N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-8-(trifluoro-metoxi)-5,6-dihidro-4H-benzo-[f][1,2,3]-triazolo-[1,5-a]-azepina-3-carboxamida en una forma de sal de sodio, potasio, amonio, calcio, magnesio, plata, zinc, colinato, lisina, meglumina, piperazina o trometamina.
Cualquier fórmula dada en la presente también pretende representar las formas no marcadas así como las formas isotópicamente marcadas de los compuestos. Los compuestos isotópicamente marcados tienen las estructuras ilustradas por las fórmulas dadas en la presente, excepto que uno o más átomos son reemplazados por un átomo que tenga una masa atómica o número de masa seleccionados. Los ejemplos de los isótopos que se pueden incorporar en los compuestos de la invención incluyen los isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor, y cloro, tales como 2H, 3H, 11C, 13C, 14C, 15N, 18F, 31P, 32P, 35S, 36Cl, 123I, 124I, 125I, respectivamente. La invención incluye diferentes compuestos isotópicamente marcados, como se definen en la presente, por ejemplo, aquéllos en donde están presentes isótopos radioactivos, tales como 3H y 14C, o aquéllos en donde están presentes isótopos no radioactivos, tales como 2H y 13C. Estos compuestos isotópicamente marcados son útiles en los estudios metabólicos (con 14C), en los estudios de cinética de reacción (con, por ejemplo, 2H o 3H), en las técnicas de detección o de formación de imágenes, tales como tomografía por emisión de positrones (PET) o tomografía computarizada con emisión de un solo fotón (SPECT), incluyendo los ensayos de distribución del fármaco o del sustrato en el tejido, o en el tratamiento radioactivo de los pacientes. En particular, un 18F o un compuesto marcado puede ser particularmente deseable para los estudios de PET o SPECT. Los compuestos isotópicamente marcados de la fórmula (I) se pueden preparar en términos generales mediante las técnicas convencionales conocidas por los expertos en este campo o mediante procesos análogos a aquéllos descritos en los ejemplos y en las preparaciones acompañantes utilizando un reactivo isotópicamente marcado apropiado en lugar del reactivo no marcado previamente empleado.
Además, la sustitución con isótopos más pesados, en particular deuterio (es decir, 2H o D), puede proporcionar ciertas ventajas terapéuticas resultantes de la mayor estabilidad metabólica, por ejemplo, un aumento de la vida media in vivo, o requerimientos de dosificación reducida, o una mejora en el índice terapéutico. Se entiende que el deuterio en este contexto se considera como un sustituyente de un compuesto de la fórmula (I). La concentración de este isótopo más pesado, específicamente deuterio, se puede definir por el factor de enriquecimiento isotópico. El término "factor de enriquecimiento isotópico", como se utiliza en la presente, significa la proporción entre la abundancia isotópica y la abundancia natural de un isótopo especificado. Si un sustituyente en un compuesto de esta invención es denotado como deuterio, este compuesto tiene un factor de enriquecimiento isotópico para cada átomo de deuterio designado de cuando menos 3,500 (52.5 por ciento de incorporación de deuterio en cada átomo de deuterio designado), de cuando menos 4,000 (60 por ciento de incorporación de deuterio), de cuando menos 4,500 (67.5 por ciento de
incorporación de deuterio), de cuando menos 5,000 (75 por ciento de incorporación de deuterio), de cuando menos 5,500 (82.5 por ciento de incorporación de deuterio), de cuando menos 6,000 (90 por ciento de incorporación de deuterio), de cuando menos 6,333.3 (95 por ciento de incorporación de deuterio), de cuando menos 6,466.7 (97 por ciento de incorporación de deuterio), de cuando menos 6,600 (99 por ciento de incorporación de deuterio), o de cuando menos 6,633.3 (99.5 por ciento de incorporación de deuterio). La forma de realización 8 proporciona un compuesto de la fórmula (I), o una sal farmacéuticamente aceptable, en donde,cuando R3 es:

R4 y R6 se seleccionan independientemente a partir de cloro, flúor, ciclopropilo, metilo, metoxi, trifluorometoxi, trifluorometilo; los grupos metilo y metoxilo pueden estar deuterados.
Los solvatos farmacéuticamente aceptables de acuerdo con la presente descripción, con fines de referencia, incluyen aquéllos en donde el solvente de cristalización puede ser isotópicamente sustituido por ejemplo, D2O, d6-acetona, d6-DMSO.
Como se utiliza en la presente, el término "portador farmacéuticamente aceptable" incluye cualquiera y todos los solventes, medios de dispersión, recubrimientos, tensioactivos, antioxidantes, conservantes (por ejemplo, agentes antibacterianos, agentes antifúngicos), agentes isotónicos, agentes retardantes de absorción, sales, conservantes, estabilizantes de fármacos, aglutinantes, excipientes, agentes de desintegración, lubricantes, agentes edulcorantes, agentes saborizantes, tintes, y similares, y combinaciones de los mismos, como serían conocidos por los expertos en este campo (véase, por ejemplo, Remington's Pharmaceutical Sciences, 18a Edición, Mack Printing Company, 1990, páginas 1289-1329). Excepto hasta donde cualquier vehículo convencional sea incompatible con el ingrediente activo, se contempla su uso en las composiciones terapéuticas o farmacéuticas.
La expresión "una cantidad terapéuticamente eficaz" de un compuesto de la presente invención se refiere a una cantidad del compuesto de la presente invención que provocará la respuesta biológica o médica de un sujeto, por ejemplo, la reducción o inhibición de la actividad de una enzima o de una proteína, o que mitigará los síntomas, aliviará las condiciones, hará más lento o retardará el progreso de la enfermedad, o prevendrá una enfermedad, etc. En una forma de realización no limitante, la expresión “una cantidad terapéuticamente eficaz” se refiere a la cantidad del compuesto de la presente invención que, cuando se administra a un sujeto, es efectiva para: (1) cuando menos parcialmente aliviar, inhibir, impedir y/o mitigar una condición, o un trastorno, o una enfermedad (i) mediada por Smurf-1, o (ii) asociada con la actividad de Smurf-1, o (iii) caracterizada por una actividad (normal o anormal) de Smurf-1; o (2) reducir o inhibir la actividad de Smurf-1; o (3) reducir o inhibir la expresión de Smurf-1. En otra forma de realización no limitante, el término “una cantidad terapéuticamente eficaz” se refiere a la cantidad del compuesto de la presente invención que, cuando se administra a una célula, o a un tejido, o a un material biológico no celular, o a un medio, es efectiva para cuando menos parcialmente reducir o inhibir la actividad de Smurf-1; o cuando menos parcialmente reducir o inhibir la expresión de Smurf-1.
Como se utiliza en la presente, el término “sujeto” se refiere a un animal. Típicamente el animal es un mamífero. Un sujeto también se refiere a, por ejemplo, primates (por ejemplo, seres humanos, masculinos o femeninos), reses, ovejas, cabras, caballos, perros, gatos, conejos, ratas, ratones, peces, aves, y similares. En ciertas formas de realización, el sujeto es un primate. En todavía otras formas de realización, el sujeto es un ser humano.
Como se utiliza en la presente, el término “inhibir”, "inhibición" o "inhibiendo” se refiere a la reducción o supresión de una condición, síntoma, o trastorno, o enfermedad dada, o a una disminución significativa en la actividad de la línea base de una actividad o proceso biológico.
Como se utiliza en la presente, el término “tratar”, “tratando" o "tratamiento" de cualquier enfermedad o trastorno, se refiere, en una forma de realización, a mitigar la enfermedad o el trastorno (es decir, hacer más lento o detener o reducir el desarrollo de la enfermedad o de cuando menos uno de los síntomas clínicos de la misma). En otra forma de realización, “tratar”, "tratando" o "tratamiento" se refiere a aliviar o mitigar cuando menos un parámetro físico, incluyendo aquéllos que no puedan ser discernibles por el paciente. En todavía
otra forma de realización, “tratar”, "tratando" o "tratamiento" se refiere a modular la enfermedad o el trastorno, ya sea físicamente (por ejemplo, la estabilización de un síntoma discernible), fisiológicamente (por ejemplo, la estabilización de un parámetro físico), o ambas. En todavía otra forma de realización, “tratar”, "tratando" o "tratamiento" se refiere a prevenir o retardar el establecimiento o desarrollo o progreso de la enfermedad o del trastorno.
Como se utiliza en la presente, un sujeto está “en necesidad de” un tratamiento si este sujeto se beneficiaría biológicamente, médicamente o en su calidad de vida a partir de dicho tratamiento.
Como se utiliza en la presente, el término "un", "uno", "el”, “la” y términos similares utilizados en el contexto de la presente invención (en especial en el contexto de las reivindicaciones) se deben interpretar para cubrir tanto el singular como el plural, a menos que se indique de otra manera en la presente o que sea claramente contradicho por el contexto.
Todos los métodos descritos en la presente, se pueden llevar a cabo en cualquier orden adecuado a menos que se indique de otra manera en la presente o que sea de otra manera claramente contradicho por el contexto. El uso de cualquiera y todos los ejemplos, o del lenguaje de ejemplo (por ejemplo, "tal como”) proporcionado en la presente, pretende meramente iluminar mejor la invención y no presenta una limitación sobre el alcance de la invención reclamada de otra manera.
Cualquier átomo asimétrico (por ejemplo, de carbono, o similares) de los compuestos de la presente invención puede estar presente en una forma racémica o enantioméricamente enriquecida, por ejemplo, en la configuración (R), (S), o (R,S). En ciertas formas de realización, cada átomo asimétrico tiene cuando menos el 50 por ciento de exceso enantiomérico, cuando menos el 60 por ciento de exceso enantiomérico, cuando menos el 70 por ciento de exceso enantiomérico, cuando menos el 80 por ciento de exceso enantiomérico, cuando menos el 90 por ciento de exceso enantiomérico, cuando menos el 95 por ciento de exceso enantiomérico, o cuando menos el 99 por ciento de exceso enantiomérico en la configuración (R) o (S).
De conformidad con lo anterior, como se utiliza en la presente, un compuesto de la presente invención puede estar en la forma de uno de los posibles isómeros, rotámeros, atropisómeros, tautómeros o mezclas de los mismos, por ejemplo, como diastereómeros sustancialmente puros, isómeros ópticos (antípodas), racematos o mezclas de los mismos.
Cualesquiera mezclas de isómeros resultantes se pueden separar con base en las diferencias fisicoquímicas de los constituyentes, en los isómeros ópticos puros o sustancialmente puros, diastereómeros, racematos, por ejemplo, mediante cromatografía y/o cristalización fraccionaria.
Cualesquiera racematos resultantes de los productos finales o intermediarios se pueden resolver en los antípodas ópticos mediante los métodos conocidos, por ejemplo, mediante la separación de las sales diastereoméricas de los mismos, obtenidas con un ácido o una base ópticamente activa, y la liberación del compuesto ácido o básico ópticamente activo. En particular, por consiguiente, se puede emplear una fracción básica para resolver los compuestos de la presente invención en sus antípodas ópticos, por ejemplo, mediante la cristalización fraccionaria de una sal formada con un ácido ópticamente activo, por ejemplo, el ácido tartárico, ácido dibenzoil-tartárico, ácido diacetil-tartárico, ácido di-O,O'-p-toluoil-tartárico, ácido mandélico, ácido málico o ácido canfor-10-sulfónico. Los productos racémicos también se pueden resolver mediante cromatografía quiral, por ejemplo, cromatografía de líquidos a alta presión (HPLC), utilizando un adsorbente quiral.
Adicionalmente, los compuestos de la presente invención, incluyendo sus sales, como se revela con fines de referencia, también se pueden obtener en la forma de sus hidratos, o pueden incluir otros solventes utilizados para su cristalización. Los compuestos de la presente invención pueden formar, así, inherentemente o por diseño, solvatos, con solventes farmacéuticamente aceptables (incluyendo agua); por consiguiente, se pretende que la descripción, con fines de referencia, abarque las formas tanto solvatadas como no solvatadas. El término "solvato" se refiere a un complejo molecular de un compuesto de la presente invención (incluyendo las sales farmacéuticamente aceptables del mismo), con una o más moléculas de solvente. Estas moléculas de solvente son aquéllas comúnmente utilizadas en la técnica farmacéutica, que son conocidas como inocuas para el receptor, por ejemplo, agua, etanol, y similares. El término "hidrato" se refiere al complejo en donde la molécula de solvente es agua.
Los compuestos de la presente invención, incluyendo las sales, cocristales, hidratos y solvatos de los mismos, como descripción con fines de referencia, pueden formar, inherentemente o por diseño, polimorfos.
Esquemas genéricos
Los compuestos de la presente invención se pueden preparar mediante las rutas descritas en los siguientes esquemas o en los Ejemplos.
Todas las abreviaturas son como se definen en la sección de Ejemplos más adelante en la presente.
Esquema 1

Paso 1: Diazotación
Condiciones típicas: a) Nitrito de sodio a 0-5 °C, en un solvente adecuado, con la adición de una fuente de azida nucleofílica.
Condiciones preferidas: Nitrito de sodio en ácido acético a 0 °C, en la presencia de azida de sodio.
Paso 2: Ciclación
Condiciones típicas: Un beta-ceto-éster y una base fuerte en un solvente polar a 50-80 °C.
Condiciones preferidas: 3-oxo-butanoato de metilo, con metóxido de sodio en metanol (MeOH) a 60 °C. Paso 3a: Saponificación
Condiciones típicas: Una base acuosa adecuada, opcionalmente con un co-solvente adecuado, tal como tetrahidrofurano (THF).
Condiciones preferidas: Hidróxido de sodio (acuoso) 2 M con tetrahidrofurano (THF), a temperatura ambiente durante 30 minutos.
Paso 3b: Acoplamiento de amida
Condiciones típicas: Un reactivo de acoplamiento adecuado, tal como cloruro de oxalilo, HATU, T3P, EDCI, etc., en la presencia de una base adecuada, tal como trietil-amina, di-isopropil-etil-amina, etc., en un solvente aprótico adecuado.
Condiciones preferidas: Cloruro de oxalilo, DMF (catalizador), dicloro-metano (DCM), trietil-amina.
Esquema 2

Paso 1: Una reacción de acoplamiento cruzado de alilación catalizada por paladio.
Condiciones típicas: Catalizador de paladio (0); un alil-estanano; en un solvente adecuado; a 80-110 °C. Condiciones preferidas: Tetraquis-trifenil-fosfina-paladio (0), alil-tributil-estaño en N,N-dimetil-formamida (DMF) a 80 °C.
Paso 2: Acoplamiento de amida
Condiciones típicas: Un cloruro de ácido adecuado, tal como cloruro de acriloílo, en la presencia de una base adecuada, tal como trietil-amina, en un solvente aprótico adecuado.
Condiciones preferidas: Cloruro de acriloílo, trietil-amina en tetrahidrofurano (THF) a -10 °C.
Paso 3: metátesis de cierre de anillo
Condiciones típicas: Un catalizador adecuado, en un solvente adecuado.
Condiciones preferidas: Dicloruro de {[2-(i-propoxi)-5-(N,N-dimetil-amino-sulfonil)-fenil]-metileno}(triciclohexil-fosfina)-rutenio(II) al 5 por ciento en dicloro-metano (DCM).
Paso 4: Hidrogenación
Condiciones típicas: Un catalizador de paladio no soluble, gas de hidrógeno, en un solvente adecuado, tal como un alcohol.
Condiciones preferidas: Paladio al 10 por ciento sobre carbón, y gas de hidrógeno en etanol.
Paso 5: Formación de tioamida
Condiciones típicas: Una fuente de azufre adecuada en un solvente adecuado con calentamiento.
Condiciones preferidas: Reactivo de Lawesson en tolueno a 110 °C.
Paso 6: Metilación
Condiciones típicas: Haluro de metilo en la presencia de una base adecuada en un solvente adecuado. Condiciones preferidas: Yoduro de metilo en la presencia de hidróxido de potasio en acetona.
Paso 7: Reacción de condensación
Condiciones típicas: Un alfa-nitro-éster en la presencia de una base no nucleofílica con calentamiento. Condiciones preferidas: Nitroacetato de etilo y DBU a 40 °C.
Paso 8: Formación reductiva de triazol
Condiciones preferidas: Zinc y nitrito de isoamilo en ácido acético, y ácido tricloroacético.
Paso 9: Saponificación
Condiciones típicas: Una base acuosa adecuada, opcionalmente con un co-solvente adecuado, tal como tetrahidrofurano (THF).
Condiciones preferidas: Hidróxido de sodio (acuoso) 2 M con tetrahidrofurano (THF) y metanol (MeOH), a temperatura ambiente, durante 30 minutos.
Paso 10: Acoplamiento de amida
Condiciones típicas: Un reactivo de acoplamiento adecuado, tal como cloruro de oxalilo, HATU, T3P, EDCI, etc., en la presencia de una base adecuada, tal como trietil-amina, di-isopropil-etil-amina, etc., en un solvente aprótico adecuado.
La presente descripción, con fines de referencia, incluye además cualquier variante de los presentes procesos, en donde se utiliza como material de partida un producto intermediario que se pueda obtener en cualquier etapa de los mismos, y se llevan a cabo los pasos restantes, o en donde los materiales de partida se forman in situ bajo las condiciones de reacción, o en donde los componentes de la reacción se utilizan en la forma de sus sales como el material ópticamente puro.
Los compuestos de la invención y los intermediarios también se pueden convertir unos en otros de acuerdo con los métodos conocidos generalmente por los expertos en la técnica.
En otro aspecto, la presente invención proporciona una composición farmacéutica, la cual comprende un compuesto de la presente invención, o una sal farmacéuticamente aceptable, y un portador farmacéuticamente aceptable según se reivindica. En una forma de realización adicional, la composición comprende cuando menos dos portadores farmacéuticamente aceptables, tales como aquéllos descritos en la presente. Para los propósitos de la presente invención, a menos que sean designados de otra manera, con fines de referencia, los solvatos e hidratos se consideran en términos generales como composiciones. De
preferencia, los portadores farmacéuticamente aceptables son estériles. La composición farmacéutica se puede formular para vías de administración particulares, tales como administración oral, administración parenteral, y administración rectal, etc. En adición, las composiciones farmacéuticas de la presente invención se pueden componer en una forma sólida (incluyendo, sin limitación, cápsulas, tabletas, píldoras, gránulos, polvos o supositorios), o en una forma líquida (incluyendo, sin limitación, soluciones, suspensiones o emulsiones). Las composiciones farmacéuticas se pueden someter a las operaciones farmacéuticas convencionales, tales como esterilización y/o pueden contener diluyentes inertes convencionales, agentes lubricantes, o agentes reguladores del pH, así como adyuvantes, tales como conservantes, estabilizantes, agentes humectantes, emulsionantes y tampones, etc.
Típicamente, las composiciones farmacéuticas son tabletas o cápsulas de gelatina que comprenden el ingrediente activo junto con uno o más de:
a) diluyentes, por ejemplo, lactosa, dextrosa, sacarosa, manitol, sorbitol, celulosa y/o glicina;
b) lubricantes, por ejemplo, sílice, talco, ácido esteárico, su sal de magnesio o de calcio y/o polietilenglicol; para tabletas también,
c) aglutinantes, por ejemplo, silicato de magnesio y aluminio, pasta de almidón, gelatina, tragacanto, metil-celulosa, carboxi-metil-celulosa de sodio y/o polivinil-pirrolidona; si se desea,
d) desintegrantes, por ejemplo, almidones, agar, ácido algínico, o su sal sódica, o mezclas efervescentes; y
e) absorbentes, colorantes, saborizantes y edulcorantes.
Las tabletas pueden ser ya sea con recubrimiento de película o bien con recubrimiento entérico de acuerdo con los métodos conocidos en la técnica.
Las composiciones adecuadas para su administración oral incluyen una cantidad eficaz de un compuesto de la invención, en la forma de tabletas, grageas, suspensiones acuosas u oleosas, polvos o gránulos dispersables, emulsión, cápsulas duras o blandas, o jarabes o elíxires. Las composiciones destinadas para su uso oral se preparan de acuerdo con cualquier método conocido en la materia para la elaboración de composiciones farmacéuticas, y estas composiciones pueden contener uno o más agentes seleccionados a partir del grupo que consiste en agentes edulcorantes, agentes saborizantes, agentes colorantes y agentes conservantes, con el objeto de proporcionar preparaciones farmacéuticamente elegantes y de buen sabor. Las tabletas pueden contener al ingrediente activo en mezcla con excipientes farmacéuticamente aceptables no tóxicos que sean adecuados para la elaboración de tabletas. Estos excipientes son, por ejemplo, diluyentes inertes, tales como carbonato de calcio, carbonato de sodio, lactosa, fosfato de calcio o fosfato de sodio; agentes de granulación y desintegrantes, por ejemplo, almidón de maíz, o ácido algínico; agentes aglutinantes, por ejemplo, almidón, gelatina o acacia; y agentes lubricantes, por ejemplo, estearato de magnesio, ácido esteárico o talco. Las tabletas quedan sin recubrimiento o se recubren mediante las técnicas conocidas para retardar la desintegración y absorción en el tracto gastrointestinal y proporcionar de esta manera una acción sostenida durante un período más largo. Por ejemplo, se puede emplear un material de retraso de tiempo, tal como monoestearato de glicerilo o diestearato de glicerilo. Las formulaciones para uso oral se pueden presentar como cápsulas de gelatina duras en donde el ingrediente activo se mezcla con un diluyente sólido inerte, por ejemplo, carbonato de calcio, fosfato de calcio o caolín, o como cápsulas de gelatina blanda en donde el ingrediente activo se mezcla con agua o con un medio oleoso, por ejemplo, aceite de cacahuate, parafina líquida o aceite de oliva.
Ciertas composiciones inyectables son soluciones o suspensiones isotónicas acuosas, y los supositorios se preparan convenientemente a partir de emulsiones o suspensiones grasas. Estas composiciones se pueden esterilizar y/o pueden contener adyuvantes, tales como agentes conservantes, estabilizantes, humectantes o emulsionantes, promotores de solución, sales para regular la presión osmótica y/o reguladores del pH. En adición, también pueden contener otras sustancias terapéuticamente valiosas. Estas composiciones se preparan de acuerdo con los métodos convencionales de mezcla, granulación o recubrimiento, respectivamente, y contienen de aproximadamente el 0.1 al 75 por ciento, o contienen de aproximadamente el 1 al 50 por ciento, del ingrediente activo.
Las composiciones adecuadas para su aplicación transdérmica incluyen una cantidad eficaz de un compuesto de la invención con un vehículo adecuado. Los vehículos adecuados para suministro transdérmico incluyen solventes farmacológicamente aceptables absorbibles para ayudar al paso a través de la piel del huésped. Por ejemplo, los dispositivos transdérmicos están en la forma de un parche que comprende un miembro de respaldo, un depósito que contiene al compuesto opcionalmente con vehículos, opcionalmente una barrera de
control de velocidad para suministrar el compuesto de la piel del huésped a una velocidad controlada y previamente determinada durante un período de tiempo prolongado, y elementos para asegurar el dispositivo a la piel.
Las composiciones adecuadas para su aplicación tópica, por ejemplo, a la piel y a los ojos, incluyen soluciones acuosas, suspensiones, ungüentos, cremas, geles o formulaciones rociables, por ejemplo, para su suministro mediante aerosol o similares. Estos sistemas de suministro tópico serán particularmente apropiados para la aplicación dérmica, por ejemplo, para el tratamiento de cáncer de piel, por ejemplo, para su uso profiláctico en cremas solares, lociones, aspersiones y similares. Por consiguiente, éstas son particularmente adecuadas para utilizarse en formulaciones tópicas, incluyendo cosméticas bien conocidas en este campo. Pueden contener solubilizantes, estabilizantes, agentes mejoradores de la tonicidad, reguladores, y conservantes.
Como se utiliza en la presente, una aplicación tópica también puede pertenecer a una inhalación o a una aplicación intranasal. Se pueden suministrar de una manera conveniente en la forma de un polvo seco (ya sea solo, como una mezcla, por ejemplo, una mezcla seca con lactosa, o una partícula componente mixta, por ejemplo, con fosfolípidos) a partir de un inhalador de polvo seco o de una presentación de aspersión en aerosol a partir de un envase presurizado, bomba, aspersor, atomizador, o nebulizador, con o sin el uso de un propelente adecuado.
La presente invención proporciona además las composiciones farmacéuticas y formas de dosificación anhidras, las cuales comprenden los compuestos de la presente invención como ingredientes activos, debido a que el agua puede facilitar la degradación de ciertos compuestos.
Las composiciones farmacéuticas y formas de dosificación anhidras de la invención se pueden preparar utilizando ingredientes anhidros o con un bajo contenido de humedad y condiciones de baja humedad. Una composición farmacéutica anhidra se puede preparar y almacenar de tal manera que se mantenga su naturaleza anhidra. De conformidad con lo anterior, las composiciones anhidras se empacan utilizando materiales conocidos para prevenir su exposición al agua, de tal manera que se puedan incluir en kits de formulación adecuados. Los ejemplos de los empaques adecuados incluyen, pero sin limitación, láminas herméticamente selladas, plásticos, recipientes de dosis unitarias (por ejemplo, viales), paquetes de blísters, y paquetes de tiras.
La invención proporciona además composiciones farmacéuticas y formas de dosificación que comprenden uno o más agentes que reducen la velocidad a la cual se descompondrá el compuesto de la presente invención como un ingrediente activo. Estos agentes, los cuales son referidos en la presente como "estabilizantes", incluyen, pero sin limitación, antioxidantes, tales como ácido ascórbico, tampones del pH, o tampones de sales, etc.
Los compuestos de la fórmula I en forma libre o en una forma de sal farmacéuticamente aceptable, exhiben valiosas propiedades farmacológicas, por ejemplo, propiedades moduladoras de Smurf-1, por ejemplo, como se indica en las pruebas in vitro e in vivo proporcionadas en las siguientes secciones y, por consiguiente, se indican para terapia o para utilizarse como productos químicos de investigación, por ejemplo, como compuestos de herramienta.
Los compuestos de la invención son útiles en el tratamiento de las indicaciones que incluyen:
Hipertensión pulmonar, incluyendo hipertensión arterial pulmonar (PAH)
Fibrosis
Artritis reumatoide
Sanado de fracturas
Glaucoma
Telangiectasia hemorrágica hereditaria (HHT)
Proteinuria
Sanado de heridas
EPOC
Asma
Hipertensión arterial pulmonar (PAH)
La hipertensión arterial pulmonar tiene una patobiología multifactorial. La vasoconstricción, la remodelación de la pared del vaso pulmonar, y la trombosis contribuyen a tener un aumento en la resistencia vascular pulmonar en la hipertensión arterial pulmonar (PAH) (Humbert et al., J. Am. Coll. Cardiol., 2004.). Los compuestos de la presente invención que se dan a conocer en este documento, son útiles en el tratamiento de la hipertensión arterial pulmonar (PAH) y de los síntomas de la misma. Se entenderá que la hipertensión arterial pulmonar abarca las siguientes formas de hipertensión pulmonar: hipertensión arterial pulmonar (PAH) idiopática (IPAH); hipertensión arterial pulmonar (PAH) hereditaria (HPAH); hipertensión arterial pulmonar (PAH) inducida por fármacos o toxinas, hipertensión arterial pulmonar (PAH) asociada con otras condiciones (APAH), tal como hipertensión arterial pulmonar (PAH) asociada con enfermedades del tejido conectivo, hipertensión arterial pulmonar (PAH) asociada con infección por VIH, hipertensión arterial pulmonar (PAH) asociada con hipertensión portal, hipertensión arterial pulmonar (PAH) asociada con enfermedades cardíacas congénitas, hipertensión arterial pulmonar (PAH) asociada con esquistosomiasis, hipertensión arterial pulmonar (PAH) asociada con anemia hemolítica crónica, o hipertensión pulmonar persistente del recién nacido (Galié et al., ERJ, 2009; Simonneau et al., JACC, 2009).
Hipertensión arterial pulmonar (PAH) idiopática se refiere a la hipertensión arterial pulmonar (PAH) de causa indeterminada. La hipertensión arterial pulmonar (PAH) hereditaria se refiere a la hipertensión arterial pulmonar (PAH) de la que se sospecha o se ha documentado una transmisión hereditaria, incluyendo aquéllas que alojen mutaciones en el receptor de la proteína morfogenética ósea (BMP), BMPR2, o aquéllas con mutaciones en ALK1 o endoglina (con o sin telangiectasia hemorrágica hereditaria).
Hipertensión arterial pulmonar (PAH) asociada con fármacos o toxinas se entenderá que abarca la hipertensión arterial pulmonar (PAH) asociada con ingestión de aminorex, un compuesto de fenfluramina (por ejemplo, fenfluramina o dexfenfluramina), ciertos aceites tóxicos (por ejemplo, aceite de semilla de colza), alcaloides de pirrolizidina (por ejemplo, té de manigua), monocrotalina, anfetaminas, L-triptófano, metanfetaminas, cocaína, fenil-propanolamina, hierba de San Juan, agentes quimioterapéuticos o SSRIs. Hipertensión arterial pulmonar (PAH) asociada con enfermedades del tejido conectivo se entenderá que abarca hipertensión arterial pulmonar (PAH) asociada con esclerosis sistémica, fibrosis pulmonar, polimiositis, artritis reumatoide, síndrome de Sjogren o hipertensión arterial pulmonar (PAH) asociada con lupus eritematoso sistémico.
Hipertensión arterial pulmonar (PAH) asociada con enfermedades cardíacas congénitas se entenderá que abarca a los pacientes con derivaciones sistémicas a pulmonares, hipertensión arterial pulmonar (PAH) asociada con síndrome de Eisenmenger, defectos pequeños ventriculares-septales o auriculares-septales o hipertensión arterial pulmonar (PAH) asociada con cirugía cardíaca correctiva.
Hipertensión arterial pulmonar (PAH) asociada con anemia hemolítica crónica se entenderá que abarca a los pacientes con anemias crónicas hereditarias y adquiridas, incluyendo los pacientes con enfermedad drepanocítica, talasemia, esferocitosis hereditaria, estomatocitosis y anemia hemolítica microangiopática. Los síntomas de la hipertensión arterial pulmonar (PAH) incluyen disnea, angina, síncope y edema (McLaughlin et al., Circulation, 2006, 114: 1417-1431). Los compuestos de la presente invención que se dan a conocer en este documento, son útiles en el tratamiento de los síntomas de la hipertensión arterial pulmonar (PAH).
Hipertensión pulmonar (PH)
Hipertensión pulmonar (PH) se entenderá que está asociada con las siguientes condiciones agrupadas de acuerdo con la clasificación clínica Dana Point (Simonneau, G et al., JACCC, 2009):
Grupo 1’ - Se entenderá que la hipertensión pulmonar (PH) está asociada con los pacientes que padecen de enfermedad veno-oclusiva pulmonar (PVOD) y de hemangiomatosis capilares pulmonares (PCH).
Grupo 2 - La hipertensión pulmonar (PH) asociada con enfermedad cardíaca izquierda incluyen a los pacientes con enfermedades ventriculares o valvulares del lado izquierdo.
Grupo 3 - Hipertensión pulmonar (PH) como un resultado de enfermedades pulmonares y/o hipoxia. Se
entenderá que las enfermedades pulmonares que dan como resultado la hipertensión pulmonar (PH) abarcan a los pacientes con fibrosis pulmonar, enfisema, fibrosis pulmonar y enfisema combinados, bronquiectasias, fibrosis quística, y la enfermedad pulmonar obstructiva crónica (EPOC).
Grupo 4 - Hipertensión pulmonar (PH) asociada con tromboembolia crónica (CTEPH).
Grupo 5 - Hipertensión pulmonar (PH) asociada con etiologías poco claras o multifactoriales. Se entenderá que esta categoría de pacientes con hipertensión pulmonar (PH) abarca a los pacientes en uno de los siguientes grupos: 1) trastornos mieloproliferativos crónicos incluyendo policitemia vera, trombocitemia esencial o leucemia mieloide crónica; 2) trastornos sistémicos, incluyendo sarcoidosis, condiciones que dan como resultado la destrucción del lecho capilar pulmonar, tales como fibrosis, compresión extrínseca de las arterias pulmonares grandes, los pacientes con histiocitosis de células de Langerhans pulmonares, linfangioleiomiomatosis, neurofibromatosis tipo 1 y vasculitis asociada con anticuerpos citoplásmicos antineutrófilos; 3) trastornos metabólicos, incluyendo enfermedad de almacenamiento de glucógeno tipo la, deficiencia de glucosa-6-fosfatasa, enfermedad de Gaucher y enfermedades de la tiroides (hipotiroidismo e hipertiroidismo); 4) abarca a los pacientes con tumores que se expanden en el lumen de la arteria pulmonar, oclusión de la microvasculatura pulmonar por émbolos tumorales metastásicos, fibrosis mediastinal o pacientes con enfermedad renal en etapa terminal que reciban hemodiálisis de largo plazo.
Fibrosis:
Se ha demostrado que la mala regulación de las sendas de señalización de TGFB/BMP tiene una función causativa en la fibrosis de diversos órganos, incluyendo riñón, corazón, pulmón, piel, páncreas e hígado, así como en la esclerosis sistémica y las patologías asociadas (como fue revisado por Leask y Abraham, FASEB, 2004). Se ha demostrado que el BMP7 contrarresta la transición epitelial-mesenquimal (EMT) inducida por el TGFB1 (Zeisberg, M et al., Nat. Med, 2003), y la inducción de colágeno (Izumi, N et al., AjP. Lung, Cell, Mol., Physiol. 2005), ambos mecanismos clave en el desarrollo de la fibrosis. Se demostró la evidencia directa de una función de Smurf-1 en las patologías fibróticas en el modelo de obstrucción ureteral unilateral (UUO) de ratón de fibrosis túbulo-intersticial progresiva del riñón en donde hubo mayores niveles de Smurf-1 presentes en los riñones enfermos asociados con niveles reducidos del sustrato protector de Smurf-1, el Smad7 (Fukasawa, H et al., PNAS, 2004). Más recientemente, se sugirió una función del Smurf-1 en la fibrosis pulmonar en la información generada en las células epiteliales pulmonares, identificando una función crucial del sustrato Smad7 de Smurf-1 en la limitación de transición epitelial-mesenquimal (EMT) (Shukla, MA et al., Am. J. Resp. Cell. Mol. Biol. 2009). Los compuestos de la presente invención que se dan a conocer en este documento, son útiles en el tratamiento de fibrosis y síntomas de la misma. Se entenderá que la fibrosis abarca los siguientes: pacientes con fibrosis pulmonar, fibrosis pulmonar idiopática, fibrosis quística, cirrosis, fibrosis endomiocárdica, fibrosis mediastinal, mielofibrosis, fibrosis retroperitoneal, fibrosis masiva progresiva, fibrosis sistémica nefrogénica, enfermedad de Crohn, queloides, infarto de miocardio antiguo, esclerodermia (esclerosis sistémica), artrofibrosis o capsulitis adhesiva.
Artritis reumatoide
Las citoquinas pro-inflamatorias, tales como el factor de necrosis tumoral alfa (TNFa) tienen una función clave en el establecimiento y mantenimiento de las condiciones inflamatorias crónicas, tales como artritis reumatoide (RA). Una reducción en la densidad ósea está comúnmente asociada con la artritis reumatoide (RA), y se ha demostrado que el Smurf-1 tiene una función clave en la mediación de la pérdida ósea inducida por la artritis reumatoide (RA). Se demostró que el TNFa desencadenaba la degradación proteolítica de los sustratos de Smurf-1: Smad1 y Runx2, ambos de los cuales son esenciales para la actividad de los osteoblastos formadores de hueso. La evidencia directa que apoya este vínculo se demostró los ratones con eliminación genética (KO) de smurf-1, en donde el TNFa fracasó para tener un impacto sobre la actividad de los osteoclastos en los huesos de los ratones con eliminación genética (KO) de Smurf-1, pero no en aquéllos de los ratones de tipo silvestre correspondientes (Guo, R et al., JBC, 2008). Los compuestos de la presente invención que se dan a conocer en este documento, son útiles en el tratamiento de artritis reumatoide y de los síntomas de la misma. Se entenderá que la artritis reumatoide (RA) abarca a los pacientes con inflamación crónica del sinovio secundaria a tumefacción de las células sinoviales, exceso de fluido sinovial, y formación de tejido fibroso dentro de las articulaciones. En adición, la artritis reumatoide (RA) también abarcará a los pacientes con artritis reumatoide (RA) debido a un granuloma necrotizante, vasculitis, piodermia gangrenosa, síndrome de Sweet, eritema nodoso, paniculitis lobular, atrofia de piel digital, eritema palmar o adelgazamiento difuso de la piel. La artritis reumatoide (RA) también se extiende hasta otros órganos y en la presente abarcará a los pacientes con fibrosis de los pulmones, amiloidosis renal, ateroesclerosis como un resultado de la artritis reumatoide (RA), pericarditis, endocarditis, falla del ventrículo izquierdo, valvulitis y fibrosis. La artritis reumatoide (RA) también abarcará a los pacientes con condiciones oculares de epiescleritis y queratoconjuntivitis sicca, trastornos hematológicos de anemia hemolítica autoinmune cálida, neutropenia y trombocitosis, condiciones neurológicas de neuropatía periférica, mononeuritis multiplex y síndrome de túnel
carpiano, osteoporosis y linfoma.
Sanado de fracturas
La senda de BMP tiene una función aquí y los inhibidores de Smurf-1 aumentan la señalización de BMP. Los compuestos de la presente invención que se dan a conocer en este documento, son útiles en el tratamiento de sanado de fracturas y de la integración ósea de los implantes y síntomas de lo mismo. Se entenderá que el sanado de fracturas abarca la técnica de reparación de fractura ósea en donde se implanta quirúrgicamente un implante endostial que contiene poros hacia dentro de los cuales pueden migrar los osteoblastos y soportar el tejido conectivo, en el sitio de la fractura ósea. La administración de los inhibidores de Smurf-1 en seguida de la inserción del implante anteriormente descrito puede ayudar a la integración del implante y agilizar la recuperación al mejorar la proliferación de las células madre mesenquimales, las cuales se diferencian en los osteoblastos (Zhao, M et al., JBC, 2004).
Glaucoma
La presión intraocular elevada (IOP) es uno de los principales factores de riesgo para el glaucoma de ángulo abierto primario (POAG). La presión intraocular elevada (IOP) se mantiene en la cámara anterior mediante el humor acuoso producido en el cuerpo ciliar y expulsado a través de la región de la red trabecular. En los pacientes con glaucoma, se ha observado un aumento en la resistencia al flujo hacia afuera del humor acuoso, asociada con la acumulación del depósito de matriz extracelular (ECM) en la región de la red trabecular. Esta patología de la matriz extracelular (ECM) en los pacientes con glaucoma de ángulo abierto primario (POAG) se parece a la fibrosis inducida por las proteínas TGFb en muchos sistemas no oculares. Se demostró un aumento de la presión intraocular elevada (IOP) inducida por TGFb2 en modelos pre-clínicos in vivo y ex vivo. En varios estudios clínicos a pequeña escala, también se ha reportado que el nivel de proteína TGFb2 en el humor acuoso está elevado en los pacientes con glaucoma de ángulo abierto primario (POAG). La modulación de la actividad de TGFb en los pacientes con glaucoma podría reducir potencialmente la presión intraocular elevada (IOP) y conducir a novedosas terapias para glaucoma (Wordinger RJ JOURNAL OF OCULAR PHARMACOLOGY AND THERAPEUTICS, Volumen 30, Número 2, 2014). En vista de la función de Smurfl en la regulación de la señalización de TGFb a través de sus sustratos BMP9 y SMAD 7, los compuestos de la presente invención (o sus sales farmacéuticamente aceptables) descritos en este documento, serían útiles en el tratamiento de glaucoma.
Telangiectasia hemorrágica hereditaria (HHT)
La telangiectasia hemorrágica hereditaria (HHT), también conocida como Síndrome de Osler-Weber-Rendu, es un trastorno genético de los vasos sanguíneos que afecta a 1:5,000 a 1:40,000. Una persona con telangiectasia hemorrágica hereditaria (HHT) tiene una tendencia a formar vasos sanguíneos que carecen de capilares normales entre una arteria y vena, provocando que la sangre arterial bajo una alta presión fluya directamente hacia dentro de una vena, la cual se puede romper y sangrar. Los síntomas de la telangiectasia hemorrágica hereditaria (HHT) pueden manifestarse como leves a graves, experimentando el 90 al 95 por ciento de los pacientes hemorragias nasales en la adultez, desarrollando el 90 al 95 por ciento telangiectasias en la cara o en las manos en la edad madura, y desarrollando el 40 por ciento malformaciones arteriovenosas (AVMs) pulmonares, las cuales pueden dar lugar a un riesgo significativo. Las malformaciones arteriovenosas (AVMs) también se pueden presentar en el cerebro, hígado, e intestino, con una gravedad variable de las implicaciones de salud. La telangiectasia hemorrágica hereditaria (HHT) se puede tratar, más frecuentemente con la terapia de coagulación, embolización, o con la remoción quirúrgica del tejido afectado. Las mutaciones de telangiectasia hemorrágica hereditaria (HHT) provocan haploinsuficiencia en la señalización de BMP (Ricard et al., Blood, 2010), la cual da como resultado un defecto de maduración del vaso y una excesiva ramificación de la vasculatura, la cual, en parte, se atribuye a la señalización deteriorada de BMP9 (Choi et al., PlosOne, 2013). Smurf1 sub-regula la señalización de BMP (Murakami Exp. Biol. Res. 2010 y Cao et al., Sci. Rep. 2014), y se ha reportado que se expresa en las células endoteliales (Crose et al., JBC, 2009, y Human Protein Atlas and GeneCards), y, por consiguiente, los inhibidores de Smurf1 pueden servir para restablecer la señalización de BMP y para corregir la anormalidad de angiogénesis. Como tales, los compuestos de la presente invención (o sus sales farmacéuticamente aceptables) descritos en este documento, serían útiles en el tratamiento de telangiectasia hemorrágica hereditaria (HHT).
Proteinuria
Las cantidades anormales de proteína en la orina son uno de los signos más tempranos de la enfermedad crónica del riñón, la cual puede resultar por hipertensión, diabetes o enfermedades asociadas con inflamación en los riñones. Si se deja sin tratamiento, la enfermedad crónica del riñón puede progresar hasta la enfermedad renal en etapa terminal y falla del riñón. Smurf1 está involucrado en múltiples mecanismos asociados con la función renal y la proteinuria. El miembro A de la familia de genes homólogos de Ras del
sustrato Smurfl (RhoA), tiene una función crítica en la regulación de la migración de los podocitos renales. La sinaptopodina hace posible la formación de fibra de tensión dentro de los podocitos renales mediante el bloqueo de la capacidad de Smurf1 para enlazarse a, y ubiquitinar, RhoA, promoviendo, por consiguiente, la movilidad de los podocitos y la modulación de las propiedades de tamización de la barrera de filtración de podocitos del riñón (Asanuma et al., Nat. Cell Biol. 2006). Adicionalmente, el antagonista intracelular del factor de crecimiento transformante (TGF) B, Smad7, tiene una función protectora clave en el riñón. Se ha demostrado que la actividad de Smurfl ubiquitina y degrada el Smad7, lo cual conduce a la fibrosis túbulointersticial y a la disfunción renal (Fukasawa et al., PNAS 2004). Juntos, estos reportes sugieren que un inhibidor de Smurfl puede hacer posible la migración de podocitos y el mantenimiento de la barrera de filtración de podocitos en adición al bloqueo de la propagación de la señalización pro-fibrótica con el riñón, proporcionando finalmente un beneficio terapéutico para la proteinuria. De conformidad con lo anterior, los compuestos de la invención (o sus sales farmacéuticamente aceptables) serían útiles en el tratamiento de proteinuria.
Sanado de heridas
Las heridas que no sanan crónicas son más comunes en las personas mayores de 60 años de edad, las cuales dan como resultado una cantidad significativa de dolor físico, y se clasifican ampliamente en tres grupos: úlceras venosas, úlceras diabéticas y por presión. Es esencial la temporización precisa de la actividad de las sendas de señalización del factor de crecimiento transformante (TGF) B y de la proteína morfogenética ósea (BMP) en el sanado de heridas normal, para regular los procesos pro-sanadores clave de la migración de fibroblastos y el depósito de matriz extracelular, el influjo de células inflamatorias, la angiogénesis, y la reepitelialización (Pakyari, M et al., Adv. Wound Care 2013). La activación prolongada del TGF B puede dar como resultado un sanado de heridas retardado, y la intervención terapéutica de las heridas que no sanan establecidas con anticuerpos anti-TGF B, da como resultado un mejor sanado y una hipertrofia reducida de la cicatrización (Lu et al., J. Am. Coll. Surg. 2005). Smurfl regula la extensión de TGF B y la señalización de BMP (Murakami Exp. Biol. Res. 2010 y Cao et al., Sci. Rep. 2014, Wang et al., J. Cell. Mol. Med. 2012) y, por consiguiente, se anticipa que un inhibidor de Smurf1 normalizaría el exceso de señalización de tGf B, haciendo posible el sanado de las heridas crónicas. De conformidad con lo anterior, los compuestos de la invención (o sus sales farmacéuticamente aceptables) serían útiles en el tratamiento de heridas crónicas que no sanan y/o en el sanado de heridas en términos generales.
EPOC y asma
La remodelación de las vías respiratorias es evidente en los pacientes con enfermedad pulmonar obstructiva crónica (EPOC) o con asma. Las características predominantes de la remodelación de las vías respiratorias en el asma son fibrosis, engrosamiento de la membrana de basamento, aumento en los números de células caliciformes, y masa mejorada de células de músculo liso con una mejor respuesta contráctil que se piensa que es inducida por la inflamación crónica responsable de la híper-respuesta de las vías respiratorias y de la obstrucción reversible de las vías respiratorias (Carroll et al., Am. Rev Resp. Dis. 1993, Metcalfe et al., Physiol. Rev. 1997, y Roche et al., Lancet 1989). En la enfermedad pulmonar obstructiva crónica (EPOC), la remodelación pulmonar se caracteriza por una desorganización del epitelio en las vías respiratorias grandes con metaplasia escamosa, hiperplasia de células caliciformes, e hipersecreción de moco, y la remodelación de las vías respiratorias pequeñas con expansión del músculo liso, fibrosis y destrucción alveolar en el desarrollo de enfisema, dando como resultado por último la restricción del flujo de aire (De, Decramer et al., Lancet, 2012, Pain et al., Eur. Respir. Rev. 2014, y Chung, Proc. Am. Thorac. Soc. 2005). En ambas enfermedades, hay evidencia de una señalización sub-regulada de BMP (Kariyawasam et al., Am. J Resp. Crit. Care Med. 2008), y TGF B elevado (Mak. et al., Respir. Med. 2009, y Chakir et al., J. All. Clin. Immunol.
2003), vinculado al mecanismo pro-remodelación, tal como la transición de fibroblastos-mesenquimal (Araya et al., J. Clin. Invest. 2007), el depósito de matriz extracelular (Baarsma et al., Am. J. Physiol. Lung Cell Mol. Physiol. 2011), y la inflamación (Chakir et al., J. All. Clin. Immunol. 2003). Los inhibidores de Smurf1 pueden normalizar la señalización de TGF B en las células pro-remodelantes críticas, tales como el músculo liso y los fibroblastos, y pueden bloquear el progreso de la remodelación, dando como resultado un beneficio terapéutico para los pacientes con enfermedad pulmonar obstructiva crónica (EPOC) o asma. De conformidad con lo anterior, los compuestos de la invención (o sus sales farmacéuticamente aceptables) serían útiles en el tratamiento de la enfermedad pulmonar obstructiva crónica y/o asma.
Por consiguiente, como una forma de realización adicional, la presente descripción, con fines de referencia, proporciona el uso de un compuesto de la fórmula (I), o de una sal farmacéuticamente aceptable del mismo, en terapia. En una forma de realización adicional, la terapia con fines de referencia se selecciona a partir de una enfermedad que se pueda tratar mediante la inhibición de Smurf-1. En otra forma de realización, la enfermedad se selecciona a partir de la lista anteriormente mencionada, de una manera adecuada hipertensión pulmonar, incluyendo hipertensión arterial pulmonar (PAH), fibrosis, artritis reumatoide, y sanado de fracturas; de una manera más adecuada hipertensión arterial pulmonar (PAH). En una forma de realización
todavía adicional, la presente invención proporciona el uso de un compuesto de la fórmula I, o de una sal farmacéuticamente aceptable del mismo, en el tratamiento de una enfermedad seleccionada a partir de glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, sanado de heridas, enfermedad pulmonar obstructiva crónica (EPOC) y asma.
Por consiguiente, como una forma de realización adicional, la presente invención proporciona un compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, para utilizarse en terapia. En una forma de realización adicional, la terapia se selecciona a partir de una enfermedad que se pueda tratar mediante la inhibición de Smurf-1, donde la enfermedad se selecciona a partir de la lista anteriormente mencionada, de una manera adecuada hipertensión pulmonar, incluyendo hipertensión arterial pulmonar (PAH), fibrosis, artritis reumatoide, y sanado de fracturas; de una manera más adecuada hipertensión arterial pulmonar (PAH). En una forma de realización todavía adicional, la presente invención proporciona un compuesto de la fórmula (I) o una sal farmacéuticamente aceptable del mismo, para utilizarse en el tratamiento de una enfermedad seleccionada a partir de glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, sanado de heridas, enfermedad pulmonar obstructiva crónica (EPOC) y asma.
En otra forma de realización, la descripción, con fines de referencia, proporciona un método para el tratamiento de una enfermedad que se trate mediante la inhibición de Smurf-1, el cual comprende la administración de una cantidad terapéuticamente aceptable de un compuesto de la fórmula (I), o de una sal farmacéuticamente aceptable del mismo. En una forma de realización adicional con fines de referencia, la enfermedad se selecciona a partir de la lista anteriormente mencionada, de una manera adecuada hipertensión pulmonar, incluyendo hipertensión arterial pulmonar (PAH), fibrosis, artritis reumatoide, y sanado de fracturas; de una manera más adecuada hipertensión arterial pulmonar (PAH). En una forma de realización todavía adicional, la enfermedad se selecciona a partir de glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, sanado de heridas, enfermedad pulmonar obstructiva crónica (EPOC) y asma.
Por consiguiente, como una forma de realización adicional, la presente invención proporciona el uso de un compuesto de la fórmula (I), o de una sal farmacéuticamente aceptable del mismo, para la elaboración de un medicamento. En una forma de realización adicional, el medicamento es para el tratamiento de una enfermedad que se pueda tratar mediante la inhibición de Smurf-1, seleccionada a partir de la lista anteriormente mencionada, de una manera adecuada hipertensión pulmonar, incluyendo hipertensión arterial pulmonar (PAH), fibrosis, artritis reumatoide, y sanado de fracturas; de una manera más adecuada hipertensión arterial pulmonar (PAH). En una forma de realización todavía adicional, la enfermedad se selecciona a partir de glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, sanado de heridas, enfermedad pulmonar obstructiva crónica (EPOC) y asma.
En una forma de realización de la presente invención, se proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida, o una sal farmacéuticamente aceptable de la misma, para utilizarse en el tratamiento de la hipertensión pulmonar, incluyendo hipertensión arterial pulmonar (PAH), fibrosis, artritis reumatoide, y sanado de fracturas.
En otra forma de realización de la presente invención, se proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida, o una sal farmacéuticamente aceptable de la misma, para utilizarse en el tratamiento de glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, sanado de heridas, enfermedad pulmonar obstructiva crónica (EPOC) y asma.
En otra forma de realización de la presente invención, se proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-p i razol-4-i l )-5-m etil-1 H-1,2,3-triazol-4-carboxamida, o una sal farmacéuticamente aceptable de la misma, para utilizarse en el tratamiento de la hipertensión pulmonar, incluyendo hipertensión arterial pulmonar (PAH), fibrosis, artritis reumatoide, y sanado de fracturas.
En otra forma de realización de la presente invención, se proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida, o una sal farmacéuticamente aceptable de la misma, para utilizarse en el tratamiento de glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, sanado de heridas, enfermedad pulmonar obstructiva crónica (EPOC) y asma.
En otra forma de realización de la presente invención, se proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2-ciclohexil-1 -metil-d3,5-metil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida, o una sal farmacéuticamente aceptable de la misma, para utilizarse en el tratamiento de la hipertensión pulmonar, incluyendo hipertensión arterial pulmonar (PAH), fibrosis, artritis reumatoide, y sanado de fracturas.
En otra forma de realización de la presente invención, se proporciona la 1-(2-cloro-4-ciclopropil-fenil)-N-(2ciclohexil-1 -met¡l-d3,5-met¡l-3-oxo-2,3-dih¡dro-1 H-pirazol-4-il)-5-met¡l-1 H-1,2,3-triazol-4-carboxamida, o una sal farmacéuticamente aceptable de la misma, para utilizarse en el tratamiento de glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, sanado de heridas, enfermedad pulmonar obstructiva crónica (EPOC) y asma.
En otra forma de realización de la presente invención, se proporciona la N-(2-c¡clohex¡l-1,5-d¡met¡l-3-oxo-2.3- dihidro-1 H-pirazol^-iO-S-Orifluoro-metoxO^B-dihidro^H-benzo-pjO ,2,3]-triazolo-[1,5-a]-azepina-3-carboxamida, o una sal farmacéuticamente aceptable de la misma, para utilizarse en el tratamiento de la hipertensión pulmonar, incluyendo hipertensión arterial pulmonar (PAH), fibrosis, artritis reumatoide, y sanado de fracturas.
En otra forma de realización de la presente invención, se proporciona la N-(2-c¡clohex¡l-1,5-d¡met¡l-3-oxo-2.3- dihidro-1 H-p¡razol-4-¡l)-8-(tr¡fluoro-metox¡)-5,6-d¡h¡dro-4H-benzo-[f][1,2,3]-triazolo-[1,5-a]-azepina-3-carboxamida, o una sal farmacéuticamente aceptable de la misma, para utilizarse en el tratamiento de glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, sanado de heridas, enfermedad pulmonar obstructiva crónica (EPOC) y asma.
La composición o combinación farmacéutica de la presente invención puede estar en una dosificación unitaria de aproximadamente de 1 a 1,000 miligramos de ingredientes activos para un sujeto de aproximadamente 50 a 70 kilogramos, o de aproximadamente de 1 a 500 miligramos, o de aproximadamente de 1 a 250 miligramos, o de aproximadamente de 1 a 150 miligramos, o de aproximadamente 0.5 a 100 miligramos, o de aproximadamente de 1 a 50 miligramos de ingredientes activos. La dosificación terapéuticamente eficaz de un compuesto, de la composición farmacéutica, o de las combinaciones de los mismos, depende de la especie del sujeto, del peso corporal, la edad y la condición individual, del trastorno o de la enfermedad, o de la gravedad de la misma, que sea tratada. Un médico, clínico, o veterinario de una experiencia ordinaria puede determinar fácilmente la cantidad eficaz de cada uno de los ingredientes activos necesarios para prevenir, tratar o inhibir el progreso del trastorno o de la enfermedad.
Las propiedades de dosificación anteriormente citadas se pueden demostrar en pruebas in vitro e in vivo utilizando convenientemente mamíferos, por ejemplo, ratones, ratas, perros, monos u órganos aislados, tejidos y preparaciones de los mismos. Los compuestos de la presente invención se pueden aplicar in vitro en la forma de soluciones, por ejemplo, soluciones acuosas, e in vivo ya sea por vía enteral, parenteral, convenientemente intravenosa, por ejemplo, como una suspensión o en solución acuosa. La dosificación in vitro puede estar en el intervalo de concentraciones de entre aproximadamente 10-3 molar y 10-9 molar. Una cantidad terapéuticamente eficaz in vivo, dependiendo de la vía de administración, puede estar en el intervalo de entre aproximadamente 0.1 y 500 miligramos/kilogramo, o de entre aproximadamente 1 y 100 miligramos/kilogramo.
La actividad de un compuesto de acuerdo con la presente invención se puede evaluar mediante los siguientes métodos in vitro e in vivo.
Ensayo farmacéutico
Los compuestos de la invención y sus sales farmacéuticamente aceptables, referidos posteriormente en la presente de una manera alternativa, como los "agentes de la invención", son útiles como productos farmacéuticos. En particular, los compuestos son inhibidores selectivos de Smurf-1, y se pueden probar en los siguientes ensayos.
Con el fin de determinar la selectividad de ligasa E3 de HECT de los compuestos, se empleó un panel de ensayos bioquímicos de autoubiquitinilación de ligasa E3 de HECT E3 (Smurf-1, Smurf-2, WWP1, WWP2, ITCH, Nedd4, Nedd4L y E6AP). La conjugación de ubiquitina con un sustrato de proteína es un proceso de múltiples pasos. En un paso inicial que requiere de ATP, se forma un enlace de tioéster entre el término carboxilo de ubiquitina y un residuo de cisteína interno de la enzima activadora de ubiquitina (E1). La ubiquitina activada se transfiere entonces a un residuo de cisteína específico de una enzima que se conjuga con la ubiquitina (E2). Las E2s donan ubiquitina a una ligasa E3 de HECT (E3) desde donde se transfiere a la proteína del sustrato. Las ligasas E3 de HECT pueden auto-ubiquitinilarse. Este evento se detecta en el ensayo TR-FRET (transferencia de energía de resonancia con fluorescencia resuelta en el tiempo) utilizado en este panel. La mezcla de reacción contiene E1, E2, E3 marcada, ubiquitina conjugada con biotina, el compuesto, y ATP en un regulador adecuado, y se incuba durante 45 minutos para permitir la autoubiquitinilación de la ligasa E3. Para medir la extensión de ligasa E3 ubiquitinilada mediante TR-FRET, después de que se completa la reacción, se agregan el criptato de Europio del fluoróforo donador (criptato Eu3+), conjugado con estreptavidina, que subsiguientemente se enlaza a la ubiquitina biotinilada, y la aloficocianina modificada XL665 (HTRF® fluoróforo aceptor primario) acoplada con un anticuerpo específico de la marca (HA, His o GST), el cual reconoce las proteínas de fusión de ligasa E3 respectivas. Cuando estos
dos fluoróforos se juntan mediante una interacción biomolecular (en este caso la ubiquitinilación de la ligasa E3), una porción de la energía capturada por el criptato durante la excitación se libera a través de la emisión de fluorescencia a 620 nanómetros, mientras que la energía restante se transfiere al XL665. Esta energía es entonces liberada por el XL665 como fluorescencia específica a 665 nanómetros. La luz a 665 nanómetros es emitida solamente a través de FRET con Europio. Debido a que está presente el criptato de Europio en el ensayo, se detecta la luz a 620 nanómetros incluso cuando la interacción biomolecular no lleve al XL665 dentro de una estrecha proximidad.
La autoubiquitinilación de Smurf-1 en las células conduce a la degradación proteasómica de Smurf-1. Por consiguiente, la inhibición del dominio catalítico de Smurf-1 elimina la autoubiquitinilación y la degradación de Smurf-1, conduciendo a la acumulación de proteína Smurf-1 inhibida en la célula.
La actividad celular de los compuestos en el dominio HECT de Smurf-1 se evalúa mediante la medición de la acumulación de proteína Smurf-1 en células HEK293 que expresan establemente Smurf-1 marcada con Prolabel bajo el control de un promotor inducible por tetraciclina, utilizando el Kit de Detección DiscoverX PathHunter ProLabel. Esta tecnología mide la cantidad de Smurf-1 marcada con Prolabel en un ensayo de complementación enzimática del lisado celular. En este planteamiento, un pequeño fragmento de complemento de 4 kDa de beta-galactosidasa, denominado como ProLabel, se expresa como una fusión N-terminal con la Smurf-1 humana. Esta marca es la donadora de la enzima (ED), y hace posible la detección de los niveles objetivo de proteínas después de la complementación con la porción más grande de la betagalactosidasa, denominada como EA para el aceptor de enzimas, con el fin de formar la enzima de betagalactosidasa funcional. El EA se agrega exógenamente a los lisados celulares. La actividad enzimática se mide utilizando un sustrato quimiluminiscente, y es proporcional a la cantidad de enzima reconstituida y, por consiguiente, a los niveles de Smurf-1.
Los compuestos de prueba y de referencia se preparan a 180x [final] en sulfóxido de dimetilo (DMSO) al 90 por ciento, y se diluyen a 1:3 en sulfóxido de dimetilo (DMSO) al 90 por ciento.
Para el panel del ensayo bioquímico, se transfieren 50 nanolitros de los compuestos de prueba, los compuestos de referencia, y el control de regulador/sulfóxido de dimetilo (DMSO) a los pocillos respectivos de una placa blanca GREIn Er "SMALL VOLUME" PS de 384 pocillos. El panel de ensayo se ejecuta a temperatura ambiente en una estación de trabajo de manejo de líquidos Biomek FX. A las placas de ensayo conteniendo 50 nanolitros de soluciones de compuesto o de control en sulfóxido de dimetilo (DMSO) al 90 por ciento, se les agregan 4.5 microlitros de la solución de ligasa E3 por pozo, seguidos por 4.5 microlitros de la mezcla de E1/E2/Ub previamente incubada, o de la ubiquitina previamente diluida (control BAJO). Las placas se agitan vigorosamente después de cada adición. En este ensayo las concentraciones de compuestos están en el intervalo de 3 nM a 10 pM en una curva de respuesta a la dosis de 8 puntos.
Después de 45 minutos de incubación, se detienen las reacciones de ubiquitinilación mediante la adición de 4.5 microlitros de NEM 2 mM, seguidos inmediatamente por 4.5 microlitros de una solución de detección que incluye el anticuerpo marcado con XL665 y el europio acoplado con estreptavidina, para dar un volumen total de 18 microlitros. Después de un tiempo de incubación de 45 minutos en la oscuridad, las placas se transfieren al lector de fluorescencia Pherastar para medir la señal TR-FRET.
Para el ensayo celular, se transfieren entonces 250 nanolitros de los compuestos de prueba, los compuestos de referencia, y el control de regulador/sulfóxido de dimetilo (DMSO) a los pocillos respectivos de una placa de cultivo de tejido blanca estéril GREINER PS, CELLSTAR, uClear de 384 pocillos, de 120 microlitros. Con el objeto de distribuir la solución del compuesto uniformemente en el medio antes de agregar las células, se agregan a cada pozo 10 microlitros del medio de cultivo celular de la placa que contiene el compuesto utilizando el dosificador MULTIDROP 384, y se agitan vigorosamente. Las células se desprenden del matraz después de una incubación corta con tripsina-EDTA, se cuentan, y se diluyen hasta una concentración de 1.5 x 106 células/mililitro en el medio de cultivo. La expresión de Smurf-1 se induce mediante la adición de doxicilina hasta una concentración final de 0.2 microgramos/mililitro. Se agregan 10 microlitros de la suspensión celular a cada pozo de las placas que contienen el compuesto utilizando el dosificador MULTIDROP 384. Las placas se incuban durante la noche a 37 °C, con CO2 al 5 por ciento. En este ensayo, las concentraciones del compuesto están en el intervalo de 6.75 nM a 22.5 pM en una curva de respuesta a la dosis de 8 puntos.
Después de la incubación durante la noche con los compuestos, se determinan los niveles de Smurf-1 utilizando el kit de detección PathHunter Prolabel de DiscoverX. Primero, se agregan manualmente 10 microlitros de una solución de procesamiento de detección de lisis/CL utilizando una pipeteadora por pasos de múltiples canales, seguido por la adición de 5 microlitros de aceptor de enzimas EA. Las placas se mezclan sobre un agitador de placas, y se incuban durante 2 a 3 horas a temperatura ambiente antes de medir la señal quimiluminiscente en el lector de placas PherStar.
Los compuestos de los Ejemplos, más adelante en la presente, tienen valores IC50 de Smurf-1 en los datos de las mediciones descritas anteriormente, como se muestran en la Tabla A.
Tabla A

El compuesto de la presente invención se puede administrar ya sea simultáneamente con, o antes o después de, uno o más agentes terapéuticos diferentes. El compuesto de la presente invención se puede administrar por separado, por la misma o diferente vía de administración, o juntos en la misma composición farmacéutica que los otros agentes. Un agente terapéutico es, por ejemplo, un compuesto químico, péptido, anticuerpo, fragmento de anticuerpo o ácido nucleico, el cual es terapéuticamente activo o potencia la actividad terapéutica cuando se administra a un paciente en combinación con un compuesto de la invención.
En una forma de realización, la invención proporciona un producto que comprende un compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, y cuando menos otro agente terapéutico, como una preparación combinada para su uso simultáneo, separado, o en secuencia, en terapia. En una forma de realización, la terapia es el tratamiento de una enfermedad o condición mediada por Smurf-1. Los productos proporcionados como una preparación combinada incluyen una composición que comprende el compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, y los otros agentes terapéuticos juntos en la misma composición farmacéutica, o el compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, y los otros agentes terapéuticos en una forma separada, por ejemplo, en la forma de un kit.
En una forma de realización, la invención proporciona una composición farmacéutica, la cual comprende un compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, y otros agentes terapéuticos. Opcionalmente, la composición farmacéutica puede comprender un portador farmacéuticamente aceptable,
como se describe anteriormente.
En una forma de realización, la invención proporciona un kit, el cual comprende dos o más composiciones farmacéuticas separadas, cuando menos una de las cuales contiene un compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo. En una forma de realización, el kit comprende elementos para contener por separado estas composiciones, tales como un recipiente, un frasco dividido, o un paquete de lámina dividido. Un ejemplo de este kit es un paquete de blísters, como se utiliza típicamente para el empaque de tabletas, cápsulas y similares.
El kit de la invención se puede utilizar para administrar formas de dosificación diferentes, por ejemplo, oral y parenteral, para administrar las composiciones separadas en diferentes intervalos de dosificación, o para titular las composiciones separadas una contra la otra. Para ayudar al cumplimiento, el kit de la invención típicamente comprende instrucciones para su administración.
En las terapias de combinación de la invención, el compuesto de la invención y el otro agente terapéutico pueden ser elaborados y/o formulados por el mismo o por diferentes fabricantes. Más aún, el compuesto de la invención y el otro agente terapéutico, se pueden reunir en una terapia de combinación: (i) antes de la liberación del producto de combinación a los médicos (por ejemplo, en el caso de un kit que comprenda el compuesto de la invención y el otro agente terapéutico); (ii) por los médicos mismos (o bajo la guía del médico) poco antes de la administración; (iii) en los pacientes mismos, por ejemplo, durante la administración en secuencia del compuesto de la invención y el otro agente terapéutico.
De conformidad con lo anterior, la invención proporciona el uso de un compuesto de la fórmula (I), o de una sal farmacéuticamente aceptable del mismo, para el tratamiento de una enfermedad o condición mediada por Smurf-1, en donde el medicamento se prepara para su administración con otro agente terapéutico. La invención también proporciona el uso de otro agente terapéutico para el tratamiento de una enfermedad o condición mediada por Smurf-1, en donde el medicamento se administra con un compuesto de la fórmula (I), o con una sal farmacéuticamente aceptable del mismo.
La invención también proporciona un compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, para utilizarse en un método para el tratamiento de una enfermedad o condición mediada por Smurf-1 como se definió con anterioridad, en donde el compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, se prepara para su administración con otro agente terapéutico. La invención también proporciona otro agente terapéutico para utilizarse en un método para el tratamiento de una enfermedad o condición mediada por Smurf-1 como se definió con anterioridad, en donde el otro agente terapéutico se prepara para su administración con un compuesto de la fórmula (I), o con una sal farmacéuticamente aceptable del mismo. La invención también proporciona un compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, para utilizarse en un método para el tratamiento de una enfermedad o condición mediada por Smurf-1, en donde el compuesto de la fórmula (I), o una sal farmacéuticamente aceptable del mismo, se administra con otro agente terapéutico. La invención también proporciona otro agente terapéutico para utilizarse en un método para el tratamiento de una enfermedad o condición mediada por Smurf-1 como se definió con anterioridad, en donde el otro agente terapéutico se administra con un compuesto de la fórmula (I), o con una sal farmacéuticamente aceptable del mismo.
La invención también proporciona el uso de un compuesto de la fórmula (I), o de una sal farmacéuticamente aceptable del mismo, para el tratamiento de una enfermedad o condición mediada por Smurf-1 como se definió con anterioridad, en donde el paciente ha sido tratado previamente (por ejemplo, dentro de 24 horas) con otro agente terapéutico. La invención también proporciona el uso de otro agente terapéutico para el tratamiento de una enfermedad o condición mediada por Smurf-1 como se definió con anterioridad, en donde el paciente ha sido tratado previamente (por ejemplo, dentro de 24 horas), con un compuesto de la fórmula (I), o con una sal farmacéuticamente aceptable del mismo.
Los siguientes ejemplos pretenden ilustrar la invención y no deben interpretarse como limitaciones sobre la misma, excepto lo proporcionado por las reivindicaciones. Las temperaturas se dan en grados Celsius. Si no se menciona de otra manera, todas las evaporaciones se llevan a cabo bajo presión reducida, típicamente entre aproximadamente 15 mm Hg y 100 mm Hg (= de 20 a 133 mbar). La estructura de los productos finales, intermediarios y materiales de partida se confirma mediante los métodos analíticos convencionales, por ejemplo, microanálisis y características espectroscópicas, por ejemplo, MS, IR, RMN. Las abreviaturas empleadas son aquéllas convencionales en la materia.
Todos los materiales de partida, bloques de construcción, reactivos, ácidos, bases, agentes deshidratantes, solventes, y catalizadores utilizados para sintetizar los compuestos de la presente invención son cualquiera de aquéllos comercialmente disponibles, o se pueden producir mediante los métodos de síntesis orgánica conocidos por un experto ordinario en este campo. Además, los compuestos de la presente invención se
pueden producir mediante los métodos de síntesis orgánica conocidos por un experto ordinario en la técnica, como se muestra en los siguientes ejemplos.
Condiciones generales:
Los espectros de masas se adquirieron en sistemas de LC-MS, SFC-MS, o GC-MS empleando métodos de electroaspersión, químicos, y de ionización por impacto de electrones de un número de instrumentos de las siguientes configuraciones: sistemas de HPLC Agilent 1100 con un espectrómetro de masas Agilent 6110 [M+H]+ se refiere a un ion molecular protonado de la especie química.
Los espectros de RMN se ejecutaron en espectrómetros de RMN Bruker AVANCE de 400 MHz o 500 MHz utilizando ICON-NMR, bajo el control del programa TopSpin. Los espectros se midieron a 298K, a menos que se indique de otra manera, y fueron referenciados en relación con la resonancia del solvente.
Instrumentación
Métodos de MS: Utilizando sistemas de HPLC Agilent 1100 con un espectrómetro de masas Agilent 6110. BajopH_v002
Columna: Phenomenex Gemini C18, 50 x 4.6 milímetros, 3.0 micras.
Temperatura de la columna: 50 °C.
Eluyentes: A: H2O, B: metanol (MeOH), ambos conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente: Del 5 por ciento al 95 por ciento de B en 2.0 minutos, 0.2 minutos con el 95 por ciento de B.
2minLC_v003
Columna: Waters BEH C1850 x 2.1 milímetros, 1.7 micras.
Temperatura de la columna: 50 °C.
Eluyentes: A: H2O, B: acetonitrilo, ambos conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
Velocidad de flujo: 0.8 mililitros/minuto.
Gradiente: 0.20 minutos con el 5 por ciento de B; del 5 por ciento al 95 por ciento de B en 1.30 minutos, 0.25 minutos con el 95 por ciento de B.
8minBajopHv01:
Columna: Waters Acquity CSH 1.7 micras, 2.1 x 100 milímetros.
Temperatura: 50 °C.
Fase móvil: A: agua ácido fórmico al 0.1 por ciento. B: Acetonitrilo ácido fórmico al 0.1 por ciento.
Velocidad de flujo: 0.7 mililitros/minuto.
Gradiente: 0.0 minutos con el 2 por ciento de B, de 0.3 a 6.5 minutos con el 2 al 98 por ciento de B, de 6.5 a 7.5 minutos con el 98 por ciento de B, de 7.5 a 8.0 minutos con el 5 al 98 por ciento de B.
2minBajopH:
Columna: Waters Acquity CSH 1.7 micras, 2.1 x 50 milímetros.
Temperatura: 50 °C.
Fase móvil: A: agua ácido fórmico al 0.1 por ciento. B: Acetonitrilo ácido fórmico al 0.1 por ciento.
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente: 0.0 minutos con el 5 por ciento de B, de 0.2 a 1. minutos con el 5 al 98 por ciento de B, de 1.3 a 1.55 minutos con el 98 por ciento de B, de 1.55 a 1.6 minutos con el 98 al 5 por ciento de B.
2minBajopHv01:
Columna: Waters Acquity CSH 1.7 micras, 2.1 x 50 milímetros.
Temperatura: 50 °C.
Fase móvil: A: agua ácido fórmico al 0.1 por ciento. B: Acetonitrilo ácido fórmico al 0.1 por ciento.
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente: 0.0 minutos con el 5 por ciento de B, de 0.2 a 1.55 minutos con el 5 al 98 por ciento de B, de 1.55 a 1.75 minutos con el 98 por ciento de B, de 1.75 a 1.8 minutos con el 98 al 5 por ciento de B.
2minBajopHv03:
Columna: Waters Acquity CSH 1.7 micras, 2.1 x 50 milímetros.
Temperatura: 50 °C.
Fase móvil: A: agua ácido fórmico al 0.1 por ciento. B: Acetonitrilo ácido fórmico al 0.1 por ciento.
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente: 0.0 minutos con el 5 por ciento de B, e 0.2 a 1.8 minutos con el 5 al 98 por ciento de B, de 1.8 a 2.1 minutos con el 98 por ciento de B, de 2.1 a 2.3 minutos con el 98 por ciento de B.
2minHighpHv03:
Columna: Waters Acquity CSH 1.7 micras, 2.1 x 50 milímetros.
Temperatura: 50 °C.
Fase móvil: A: agua amoníaco al 0.1 por ciento.
B: Acetonitrilo amoníaco al 0.1 por ciento.
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente: 0.0 minutos con el 5 por ciento de B, de 0.2 a 1.8 minutos con el 5 al 98 por ciento de B, de 1.8 a 2.1 minutos con el 98 por ciento de B, de 2.1 a 2.3 minutos con el 98 al 5 por ciento de B.
10minBajopHv01:
Columna: Waters Acquity CSH 1.7 micras, 2.1 x 100 milímetros.
Temperatura: 50 °C.
Fase móvil: A: agua ácido fórmico al 0.1 por ciento. B: acetonitrilo ácido fórmico al 0.1 por ciento.
Velocidad de flujo: 0.7 mililitros/minuto.
Gradiente: 0.0 minutos con el 2 por ciento de B, de 0.5 a 8.0 minutos con el 2 al 98 por ciento de B, de 8.0 a 9.0 minutos con el 98 por ciento de B, de 9.0 a 9.1 minutos con el 98 al 2 por ciento de B.
LCMS (SRPb)
Columna: Acquity HSS T32.1 x 50 milímetros, 1.8 micras.
Temperatura de la columna: 60 °C.
Eluyentes: A: H2O (ácido fórmico al 0.05 por ciento, acetato de amonio 3.75 mM).
B: acetonitrilo (ácido fórmico al 0.05 por ciento).
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente Del 5 por ciento al 98 por ciento en 1.4 minutos.
Abreviaturas:
aq acuoso
br amplia
d doblete
dd doblete de dobletes
DCM dicloro-metano
DMF N,N-dimetil-formamida
DMSO sulfóxido de dimetilo
EtaN trietil-amina
Et2O dietil-éter
EtOAc acetato de etilo
EtOH etanol
g gramos
H2 hidrógeno
HCl ácido clorhídrico
hr / h hora(s)
ihex iso-hexano
K2CO3 carbonato de potasio
KF fluoruro de potasio
KOH hidróxido de potasio
L litros
LC-MS cromatografía de líquidos y espectrometría de masas MeCN acetonitrilo
MeI yoduro de metilo
MeOH metanol
MS espectrometría de masas
mult(s) multiplete(s)
mg miligramos
min minutos
ml mililitros
mm milímetros
mmol milimoles
m/z proporción de la masa a la carga
N2 nitrógeno
NaHCO3 carbonato ácido de sodio
NaOMe metóxido de sodio
Na2SO4 sulfato de sodio
RMN resonancia magnética nuclear
P(chex)3 triciclohexil-fosfina
Pd(OAc)2 acetato de paladio(II)
Pd(Ph3P)4 tetraquis-(trifenil-fosfina)-paladio(0)
ppm partes por millón
psi (kg/cm2) libras por pulgada cuadrada (kilogramos por centímetro cuadrado)
q cuarteto
Rt tiempo de retención
RT temperatura ambiente
s singulete
sat saturado
SiCO3 carbonato enlazado a sílice
t triplete
tt triplete de tripletes
TBME metil-terbutil-éter
TFA ácido trifluoroacético
THF tetrahidrofurano
TMSN3 trimetil-silil-azida
T3P anhídrido propil-fosfónico
UV ultra-violeta
pL microlitros
Instrumentación
Si no se indica de otra manera, las condiciones analíticas son como sigue:
Método de LCMS A
Sistema: Agilent Serie 1100 incluyendo Agilent MS1946D con ionización química.
Columna: Waters Symmetry C83.5 micras, 2 x 50 milímetros.
Temperatura de la columna: 50 °C.
Eluyentes: A: H2O, conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
B: acetonitrilo, conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente Del 0 por ciento al 95 por ciento de B en 2 minutos. Método de LCMS B
Sistema: Acquity.
Columna: Acquity HSS T32.1 x 50 milímetros, 1.8 micras.
Temperatura de la columna: 50 °C.
Eluyentes: A: H2O (ácido fórmico al 0.05 por ciento, acetato de amonio 3.75 mM).
B: acetonitrilo (ácido fórmico al 0.05 por ciento).
Velocidad de flujo: 1.2 mililitros/minuto.
Gradiente Del 2 por ciento al 98 por ciento en 1.4 minutos. Método de LCMS C
Sistema: Acquity.
Columna: Acquity HSS T32.1 x 50 milímetros, 1.8 micras.
Temperatura de la columna: 60 °C.
Eluyentes: A: H2O (ácido fórmico al 0.05 por ciento, acetato de amonio 3.75 mM).
B: acetonitrilo (ácido fórmico al 0.05 por ciento).
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente Del 5 por ciento al 98 por ciento en 1.4 minutos.
Método de HPLC D
Sistema: Agilent 1200 HPLC.
Columna: Zorbax Eclipse XDB-C184.6 x 50 milímetros, 1.8 mieras. Temperatura de la columna: 35 °C.
Eluyentes: A: H2O, conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
B: acetonitrilo, conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente Del 5 por ciento al 100 por ciento de B en 7.5 minutos.
Método de Preparación E
Sistema: Waters.
Columna: Waters Sunfire C18, 150 x 30 milímetros, 5 micras.
Eluyentes: A: H2O, conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
B: acetonitrilo, conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
Velocidad de flujo: De 30 a 50 mililitros/minuto.
Gradiente Del 40 por ciento al 80 por ciento de B en 7.25 minutos.
Método de Preparación F
Sistema: Waters.
Columna: Xselect CSH Prep C18, 100 x 30 milímetros, 5 micras.
Eluyentes: A: H2O, conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
B: acetonitrilo, conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
Velocidad de flujo: De 30 a 50 mililitros/minuto.
Gradiente Del 40 por ciento al 80 por ciento de B en 9.5 minutos.
Método de Preparación G
Columna: Sunfire columna de 30 x 100 milímetros, C18, 5 micras.
Temperatura de la columna: 50 °C.
Eluyentes: A: H2O (ácido trifluoroacético (TFA) al 0.1 por ciento).
B: acetonitrilo (ácido trifluoroacético (TFA) al 0.1 por ciento).
Gradiente Del 30 por ciento al 70 por ciento de B.
Método de LCMS H
Sistema: Shimadzu LCMS serie 2010.
Columna: Xtimato C18, 3 micras, 2.1 x 30 milímetros.
Temperatura de la columna: 50 °C.
Eluyentes: A: H2O (1.5 mililitros de ácido trifluoroacético (TFA) en 4 litros).
B: acetonitrilo (0.75 mililitros de ácido trifluoroacético (TFA) en 4 litros.
Velocidad de flujo: 1.2 mililitros/minuto.
Gradiente Del 10 por ciento al 80 por ciento de B en 1.5 minutos.
Método de HPLC I
Sistema: Shimadzu LCMS serie 2010HT o series 10A, 20AB.
Columna: Xbridge ShieldRP18, 5 micras, 50 milímetros x 2.1 milímetros.
Temperatura de la columna: 40 °C.
Eluyentes: A: H2O (2 mililitros de NH3OH en 4 litros.
B: acetonitrilo.
Velocidad de flujo: 1.2 mililitros/minuto.
Gradiente Del 10 por ciento al 80 por ciento de B en 6 minutos.
Método 2minBajopH
Sistema: Acquity.
Columna: Acquity CSH C1850 x 2.1 milímetros.
Temperatura de la columna: 50 °C.
Eluyentes: A: H2O, conteniendo ácido fórmico al 0.1 por ciento. B: acetonitrilo, conteniendo ácido fórmico al 0.1 por ciento.
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente Del 0 por ciento al 98 por ciento de B en 1.55 minutos.
Método 2minBajopHv01
Sistema: Acquity.
Columna: Acquity CSH C18, 50 x 2.1 milímetros.
Temperatura de la columna: 50 °C.
Eluyentes: A: H2O, conteniendo ácido fórmico al 0.1 por ciento. B: acetonitrilo, conteniendo ácido fórmico al 0.1 por ciento.
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente Del 0 por ciento al 98 por ciento de B en 1.75 minutos.
Método 10minBajopHv01
Sistema: Acquity.
Columna: Acquity CSH C18, 100 x 2.1 milímetros.
Temperatura de la columna: 50 °C.
Eluyentes: A: H2O, conteniendo ácido fórmico al 0.1 por ciento. B: acetonitrilo, conteniendo ácido fórmico al 0.1 por ciento.
Velocidad de flujo: 0.7 mililitros/minuto.
Gradiente Del 0 por ciento al 98 por ciento de B en 9 minutos. Método 2minBajopHv02
Sistema: Acquity.
Columna: Acquity CSH C18, 50 x 2.1 milímetros.
Temperatura de la columna: 50 °C.
Eluyentes: A: H2O, conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
B: acetonitrilo, conteniendo ácido trifluoroacético (TFA) al 0.1 por ciento.
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente Del 0 por ciento al 98 por ciento de B en 1.75 minutos.
Método 2minBajopHv03
Sistema: Acquity.
Columna: Acquity CSH C18, 50 x 2.1 milímetros.
Temperatura de la columna: 50 °C.
Eluyentes: A: H2O, conteniendo ácido fórmico al 0.1 por ciento. B: acetonitrilo, conteniendo ácido fórmico al 0.1 por ciento.
Velocidad de flujo: 1.0 mililitro/minuto.
Gradiente Del 0 por ciento al 98 por ciento de B en 2.1
minutos.
Preparación de los Compuestos Finales
Ejemplo 1:
1-(2-cloro-4-metoxi-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-m etil-1 H-1,2,3-triazol-4-carboxamida

Paso 1: 1-azido-2-cloro-4-metoxi-benceno
Una solución de 2-cloro-4-metoxi-anilina (200 miligramos, 1.269 milimoles) en ácido acético (15 mililitros) y agua (15 mililitros), se agitó y se enfrió en un baño de hielo. Se agregó por goteo una solución de nitrito de sodio (88 miligramos, 1.269 milimoles) en agua (2 mililitros), y la mezcla se agitó durante 10 minutos bajo enfriamiento con hielo. Se agregó por goteo una solución de azida de sodio (83 miligramos, 1.269 milimoles) en agua (2 mililitros), y la mezcla se agitó bajo enfriamiento con hielo durante 5 minutos, y entonces a temperatura ambiente durante 30 minutos. A la mezcla resultante se le agregó NaOH 2M (acuoso) hasta hacerse básica, y éste se extrajo con dietil-éter (2 veces). Los extractos orgánicos se secaron sobre MgSO4, se filtraron, y se concentraron al vacío, para dar el compuesto del título como un sólido color café.
LC-MS: Rt 1.23 minutos; MS m/z ningún ion de masa [M+H]+; Método 2minBajopHv01.
1H RMN (400 MHz, DMSO-d6) 57.34 (1H, d), 7.12 (1H, d), 7.01 (1H, dd), 3.77 (3H, s).
Paso 2: Ácido 1-(2-cloro-4-metoxi-fenil)-5-metil-1 H-1,2,3-triazol-4-carboxílico
A una solución agitada de 1-azido-2-cloro-4-metoxi-benceno (162 miligramos, 0.882 milimoles), y 3-oxobutanoato de metilo (0.286 mililitros, 2.65 milimoles) en metanol (MeOH) (10 mililitros), se le agregó metóxido de sodio (5M en MeOH) (1.059 mililitros, 5.29 milimoles) por goteo, y la mezcla se calentó a 50 °C durante 16 horas. La mezcla resultante se diluyó con agua, se lavó con dietil-éter (2 veces), y los extractos orgánicos se desecharon. A la capa acuosa se le agregó HCl 5M (acuoso) hasta quedar ácida, y el sólido que estalló se recolectó mediante filtración, se lavó con agua, y se secó sobre la línea de vacío, para proporcionar el compuesto del título como un sólido blanquecino.
LC-MS: Rt 0.93 minutos; MS m/z 268.1 [M+H]+; Método 2minBajopHv01.
iH RMN (400 MHz, DMSO-ds) 5 13.2 (1H, br s), 7.63 (1H, d), 7.39 (1H, d), 7.17 (1H, dd), 3.89 (3H, s), 2.32 (3H, s).
Paso 3: 1-(2-cloro-4-metoxi-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida
A una solución de cloruro de oxalilo (63 microlitros, 0.717 milimoles), y N,N-dimetil-formamida (DMF) (74 microlitros, 0.956 milimoles) en dicloro-metano (DCM) seco (2 mililitros), bajo nitrógeno, se le agregó ácido 1-(2-cloro-4-metoxi-fenil)-5-metil-1H-1,2,3-triazol-4-carboxílico (115 miligramos, 0.430 milimoles), y la mezcla se agitó a temperatura ambiente durante 1 hora. Se agregó una solución de 4-amino-2-ciclohexil-1,5-dimetil-1H-pirazol-3(2H)-ona (Intermediario A) (100 miligramos, 0.478 milimoles) en dicloro-metano (DCM) seco (1 mililitro), seguida por trietil-amina (0.200 mililitros, 1.433 milimoles), y la mezcla de reacción se agitó a temperatura ambiente durante 48 horas.
La mezcla resultante se cargó sobre un cartucho previamente humedecido de (MeOH) de 1 gramo ISOLUTE® PE-AX/SCX-2, y se lavó con MeOH, recolectándose el eluyente. El eluyente se concentró bajo presión reducida, y el residuo resultante se disolvió en sulfóxido de dimetilo (DMSO) (0.9 mililitros), y se purificó
mediante cromatografía en fase inversa (Método de Preparación E). La fracción del producto se concentró bajo presión reducida, se diluyó con NaHCO3 (acuoso) saturado, y se extrajo con dicloro-metano (DCM). El extracto orgánico se pasó a través de un cartucho de separación de fases, y el eluyente se concentró bajo presión reducida, para dar el compuesto del título.
LC-MS: Rt 4.44 minutos; MS m/z 461.2 [M+H]+; Método 10minBajopHv01.
1H RMN (400 MHz, CDCla) 58.36 (1H, s), 7.32 (1H, d), 7.13 (1H, d), 6.98 (1H, dd), 4.06 (1H, multiplete), 3.90 (3H, s), 3.24 (3H, s), 2.46 (3H, s), 2.23 (3H, s), 1.99 (2H, multipletes), 1.87 (4H, multipletes), 1.70 (1H, multiplete), 1.37 (2H, multipletes), 1.23 (1H, multiplete).
Ejemplo 2:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-1-(2,4-dicloro-fenil)-5-metil-1 H-1,2,3-triazol-4-carboxamida
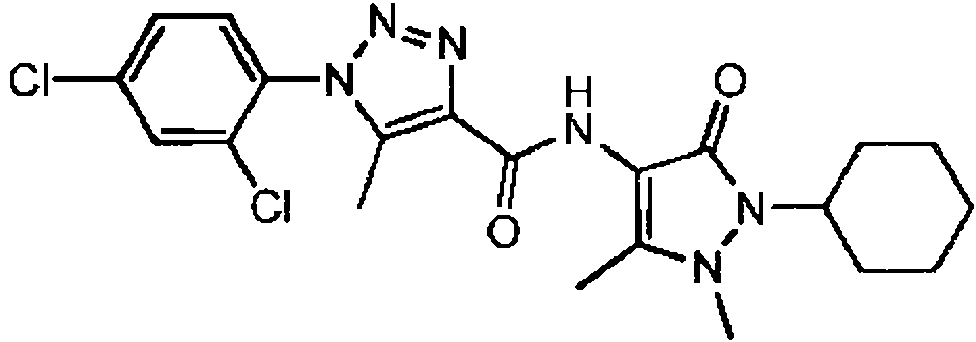
El compuesto del título se preparó de una manera análoga al Ejemplo 1, mediante el reemplazo de la 2-cloro-4-metoxi-anilina (paso 1) con 2,4-dicloro-anilina.
LC-MS: Rt 4.71 minutos; MS m/z 463.2 [M+H]+; Método 10minBajopHv01.
iH RMN (400 MHz, CDCla) 58.35 (1 H, s), 7.66 (1 H, d), 7.49 (1 H, dd), 7.38 (1 H, d), 4.06 (1 H, multiplete), 3.24 (3H, s), 2.48 (3H, s), 2.23 (3H, s), 1.99 (2H, multipletes), 1.87 (4H, multipletes), 1.70 (1H, multiplete), 1.37 (2H, multipletes), 1.22 (1H, multiplete).
Ejemplo 3:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-1-(4-metoxi-2-(trifluoro-metil)-fenil)-5-metil-1 H-1,2,3-triazol-4-carboxamida

El compuesto del título se preparó de una manera análoga al Ejemplo 1, mediante el reemplazo de la 2-cloro-4-metoxi-anilina (paso 1) con 4-metoxi-2-(trifluoro-metil)-anilina.
LC-MS: Rt 4.57 minutos; MS m/z 493.3 [M+H]+; Método 10minBajopHv01.
iH RMN (400 MHz, CDCla) 58.38 (1 H, s), 7.39 (1 H, d), 7.30 (1 H, d), 7.23 (1 H, dd), 4.07 (1 H, multiplete), 3.97 (3H, s), 3.25 (3H, s), 2.42 (3H, s), 2.24 (3H, s), 2.01 (3H, multipletes), 1.88 (2H, multipletes), 1.69 (2H, multipletes), 1.38 (2H, multipletes), 1.24 (1H, multiplete).
Ejemplo 4:
N-(2-ciclohexil-1,5-d¡metil-3-oxo-2,3-d¡h¡dro-1 H-p¡razol-4-¡l)-1-(4-metox¡-3-metil-fen¡l)-5-met¡l-1 H-1,2,3-triazol-4-carboxamida

El compuesto del título se preparó de una manera análoga al Ejemplo 1, mediante el reemplazo de la 2-cloro-4-metoxi-anilina (paso 1) con 4-metoxi-3-metil-an¡l¡na.
LC-MS: Rt 4.47 minutos; MS m/z 440.5 [M+H]+; Método 10minBajopHv01.
1H RMN (400 MHz, CDCb) 58.36 (1H,s), 7.22 (2H, multipletes), 6.95 (1H, multiplete), 4.05 (1H, multiplete), 3.91 (3H, s), 3.22 (3H, s), 2.57 (3H, s), 2.28 (3H, s), 2.22 (3H, s), 1.99 (2H, multipletes), 1.86 (4H, multipletes), 1.69 (1H, multipletes), 1.37 (2H, multipletes), 1.22 (1H, multiplete).
Ejemplo 5:
1-(2-cloro-4-(trifluoro-metoxi)-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida

El compuesto del título se preparó de una manera análoga al Ejemplo 1, mediante el reemplazo de la 2-cloro-4-metoxi-anilina (paso 1) con 2-cloro-4-trifluoro-metoxi-an¡l¡na. La purificación se llevó a cabo mediante cromatografía en fase inversa utilizando el Método de Preparación G.
LC-MS: Rt: 2.10 minutos; MS m/z 513 [M+H]+; Método A.
iH-RMN (400 MHz, DMSO-d6) 5 9.35 (1H, s), 8.02 (1H, d), 7.93 (1H, d), 7.70 (1H, multiplete), 3.96 (1H, multiplete), 3.24 (3H, s), 2.36 (3H, s), 2.05 (3H, s), 2.05-1.94 (2H, multipletes), 1.79-1.76 (2H, multipletes), 1.69-1.59 (3H, multipletes), 1.36-1.26 (2H, multipletes), 1.19-1.13 (1H, multiplete).
Ejemplo 6:
1-(4-cloro-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-p i razol-4-i l )-5-m etil-1 H-1,2,3-triazol-4-carboxamida

El compuesto del título se preparó de una manera análoga al Ejemplo 1, mediante el reemplazo del 1-azido-2-cloro-4-metoxi-benceno (paso 2) con 1-azido-4-cloro-benceno (0.5 M en terbutil-metil-éter (TBME)). LC-MS: Rt 4.40 minutos; MS m/z 431.1 [M+H]+; Método 10minBajopHv01.
1H RMN (400 MHz, CDCh) 58.36 (1H, s), 7.57 (2H, multipletes), 7.43 (2H, multipletes), 4.05 (1H, multiplete), 3.23 (3H, s), 2.62 (3H, s), 2.22 (3H, s), 1.99 (3H, multipletes), 1.87 (4H, multipletes), 1.70 (1H, multiplete), 1.37 (2H, multipletes), 1.22 (1H, multiplete).
Ejemplo 7:
1 -(2,4-dicloro-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-p i razo l-4-i l)-5-met i l-1H-1,2,3-triazol-4-carboxamida

Pasos 1-2: Ácido 1-(2,4-dicloro-fenil)-5-metil-1H-1,2,3-triazol-4-carboxílico
El compuesto del título se preparó de una manera análoga al ácido 1-(2-cloro-4-metoxi-fenil)-5-metil-1H-1,2,3-triazol-4-carboxílico (Ejemplo 1, pasos 1 y 2), mediante el reemplazo de la 2-cloro-4-metoxi-anilina (Ejemplo paso 1) con 2, 4-dicloro-anilina.
LCMS: Rt 4.10 minutos; MS m/z 274.0 [M+H]+; Método 10minBajopHv01.
Paso 3: 1-(2,4-dicloro-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1H-1,2,3-triazol-4-carboxamida
Una solución de ácido 1-(2,4-dicloro-fenil)-5-metil-1H-1,2,3-triazol-4-carboxílico (100 miligramos, 0.368 milimoles), cloruro de oxalilo (0.048 mililitros, 0.551 milimoles), y N,N-dimetil-formamida (DMF) (0.057 mililitros, 0.735 milimoles) en dicloro-metano (DCM) (5 mililitros), se agitó a temperatura ambiente durante 15 minutos. Se agregó la 4-amino-2-(2-fluoro-fenil)-1,5-dimetil-1H-pirazol-3(2H)-ona (Intermediario B) (89 miligramos, 0.404 milimoles), seguida por trietil-amina (0.154 mililitros, 1.103 milimoles), y la mezcla se agitó a temperatura ambiente durante 30 minutos. La mezcla resultante se diluyó con dicloro-metano (DCM), se lavó con HCl 0.1M (acuoso), y NaHCÜ3 (acuoso) saturado, y los extractos orgánicos se pasaron a través de un cartucho de separación de fases y se concentraron bajo presión reducida. El aceite color café resultante se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con el 0 al 10 por ciento de metanol (MeÜH) en terbutil-metil-éter (TBME). Las fracciones del producto se combinaron y se concentraron bajo presión reducida, para dar un sólido color rosado pálido, el cual se trituró con dietil-éter. El sólido resultante se recolectó mediante filtración, y se secó en el horno al vacío, para dar el compuesto del título como un sólido color rosado pálido.
LC-MS: Rt 4.33 minutos; MS m/z 475.3 [M+H]+; Método 10minBajopHv01.
iH RMN (400 MHz, DMSO-d6) 5 9.57 (1H, s), 8.08 (1H, d), 7.83 (1H, d), 7.77 (1H, dd), 7.56-7.35 (4H, multipletes), 3.09 (3H, s), 2.39 (3H, s), 2.20 (3H, s).
Ejemplo 8:
1-(4-cloro-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida

Paso 1: Ácido 1-(4-cloro-fen¡l)-5-metil-1H-1,2,3-tr¡azol-4-carboxíl¡co
El compuesto del título se preparó de una manera análoga al ácido 1-(2-cloro-4-metoxi-fenil)-5-metil-1H-1,2,3-triazol-4-carboxílico (Ejemplo 1, paso 2), mediante el reemplazo del 1-azido-2-cloro-4-metoxibenceno con 1-azido-4-cloro-benceno (0.5 M en terbutil-metil-éter (TBME)).
LCMS: Rt 0.86 minutos; MS m/z 240.1 [M+H]+; Método 2minBajopH.
Paso 2: 1-(4-cloro-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1H-1,2,3-triazol-4-carboxamida
A una solución agitada de ácido 1-(4-cloro-fenil)-5-metil-1H-1,2,3-triazol-4-carboxílico (100 miligramos, 0.421 milimoles), cloruro de oxalilo (0.041 mililitros, 0.463 milimoles), y N,N-dimetil-formamida (DMF) (0.065 mililitros, 0.842 milimoles) en dicloro-metano (DCM) seco (5 mililitros), se le agregó una solución de 4-amino-2-(2-fluoro-fenil)-1,5-dimetil-1H-pirazol-3(2H)-ona (Intermediario B) (102 mili-gramos, 0.463 milimoles) en dicloro-metano (DCM) seco (1 mililitro), seguida por trietil-amina (0.176 mililitros, 1.262 milimoles), y la mezcla se agitó a temperatura ambiente durante 1 hora. La mezcla resultante se diluyó con dicloro-metano (DCM), y se lavó con HCl 0.1M (acuoso), y NaHCÜ3 (acuoso) saturado. Los extractos orgánicos se pasaron a través de un cartucho de separación de fases y se concentraron bajo presión reducida. El sólido resultante se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con el 0 al 10 por ciento de metanol (MeÜH) en terbutil-metil-éter (TBME). Las fracciones del producto se combinaron y se concentraron bajo presión reducida, para dar un sólido vidriado, el cual se suspendió en 1:1 de iso-hexano:Et2Ü, se calentó a 50 °C durante 2 horas, se tapó, y se dejó enfriar durante 16 horas. El sólido se recolectó mediante filtración, y se secó sobre la línea de vacío, para dar el compuesto del título como un sólido amarillo pálido.
LC-MS: Rt 4.04 minutos; MS m/z 441.4 [M+H]+; Método 10minBajopHv01.
1H RMN (400 MHz, DMSÜ-ds) 5 9.51 (1H, s), 7.75-7.70 (4H, multipletes), 7.53 (1H, multiplete), 7.47-7.35 (3H, multipletes), 3.08 (3H, s), 2.55 (3H, s), 2.19 (3H, s).
Ejemplo 9:
1-(2-cloro-4-(trifluoro-metoxi)-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida

Pasos 1-2: Ácido 1-(2-cloro-4-(trifluoro-metoxi)-fenil)-5-metil-1 H-1,2,3-triazol-4-carboxílico
El compuesto del título se preparó de una manera análoga al ácido 1-(2-cloro-4-metoxi-fenil)-5-metil-1H-1.2.3- triazol-4-carboxílico (Ejemplo paso 2), mediante el reemplazo de la 2-cloro-4-metoxi-anilina (paso 1) con 2-cloro-4-trifluoro-metoxi-anilina.
Paso 3: 1-(2-cloro-4-(trifluoro-metoxi)-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida
La N,N-dimetil-formamida (DMF) (43 microlitros, 0.56 milimoles) se disolvió en dicloro-metano (DCM) (1 mililitro), y se enfrió a 0 °C. Se agregó cloruro de oxalilo (27 microlitros, 0.308 milimoles), seguido por ácido 1-(2-cloro-4-(trifluoro-metoxi)-fenil)-5-metil-1H-1,2,3-triazol-4-carboxílico (90 miligramos, 0.28 milimoles), y la mezcla de reacción se agitó durante 10 minutos. Se agregó la 4-amino-2-(2-fluoro-fenil)-1,5-dimetil-1H-pirazol-3(2H)-ona (Intermediario B) (62 miligramos, 0.28 milimoles), seguida por trietil-amina (117 microlitros, 0.839 milimoles). La reacción se dejó calentar a temperatura ambiente, y se agitó durante 4 horas. a la mezcla de reacción se le agregó NaHCÜ3 acuoso saturado, y el producto se extrajo con dicloro-metano (DCM). El extracto orgánico se secó, y se concentró bajo presión reducida. El material crudo se purificó mediante cromatografía en fase inversa (Método de Preparación G), para proporcionar el compuesto del título.
LC-MS: Rt: 2.08 minutos; MS m/z 525 [M+H]+; Método A.
1H-RMN (400 MHz, DMSO-d6) 59.57 (1H, s), 8.02 (1H, d), 7.94 (1H, d), 7.72- 7.69 (1H, multiplete), 7.52 7.33 (4H, multiplete), 3.07 (3H, s), 2.38 (3H, s), 2.18 (3H, s).
Ejemplo 10:
1 -(2-cloro-4-ciclopropil-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-p i razol-4-i l)-5-m etil-1 H-1.2.3- triazol-4-carboxamida

Paso 1: 2-cloro-4-ciclopropil-anilina
Al tolueno (1470 mililitros) y agua (60 mililitros) bajo nitrógeno, se les agregaron 4-bromo-2-cloro-anilina (63 gramos, 305 milimoles), y ácido ciclopropil-borónico (26.5 gramos, 309 milimoles). La mezcla se enfrió, y se trató con fosfato de potasio tribásico (227 gramos, 1068 milimoles) en porciones, seguido por P(cHex)3 (8.56 gramos, 30.5 milimoles). El matraz se inundó con nitrógeno, se agregó Pd(OAc)2 (3.56 gramos, 15.87 milimoles), y la mezcla se calentó a 81 °C durante 7 horas; entonces se dejó a temperatura ambiente durante el fin de semana. Se agregó más ácido ciclopropil-borónico (5.5 gramos, 64 milimoles), y la mezcla se calentó a 81 °C durante 2 horas, y entonces se enfrió hasta la temperatura ambiente. La mezcla resultante se diluyó con agua (600 mililitros). La capa acuosa se separó y la capa orgánica se lavó con agua (300 mililitros) y salmuera (300 mililitros), se secó sobre Na2SÜ4, se filtró, y se evaporó a sequedad, para dar un aceite color café. El aceite se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con el 10 al 100 por ciento de EtOAc en heptano. Las fracciones del producto se combinaron y se concentraron bajo presión reducida, para dar el compuesto del título como cristales verdes.
Método de HPLC D: Rt 3.94 minutos;
iH RMN (400 MHz, DMSÜ-ds) 5 6.88 (1H, d), 6.74 (1H, dd), 6.67 (1H, d), 5.06 (2H, br s), 1.78-1.70 (1H, multiplete), 0.82-0.76 (2H, multipletes), 0.52-0.47 (2H, multipletes).
Paso 2: 1-azido-2-cloro-4-cicloprop¡l-benceno
La 2-cloro-4-ciclopropil-an¡l¡na (70 gramos, 418 milimoles) se disolvió en Me-THF (2090 mililitros), y se enfrió a 0 °C en un baño de hielo. Se agregaron lentamente nitrito de terbutilo (59.8 mililitros, 501 milimoles) y TMSN3 (66.5 mililitros, 501 milimoles) por goteo. La mezcla se agitó a 0 °C durante 30 minutos, y se dejó calentar gradualmente hasta la temperatura ambiente, y se agitó durante 2 horas. A la mezcla resultante se le agregó lentamente NaHCO3 (acuoso) saturado (250 mililitros) bajo enfriamiento, seguido por agua (250 mililitros). La mezcla se transfirió a un embudo de separación y se lavó con agua (1.5 litros), se secó sobre Na2SO4 y se filtró, para dar el compuesto del título, el cual se utilizó en el siguiente paso sin mayor purificación.
Método de HPLC D: Rt 5.63 minutos.
Paso 3: Ácido 1-(2-cloro-4-c¡cloprop¡l-fen¡l)-5-met¡l-1H-1,2,3-tr¡azol-4-carboxíl¡co
Al 1-azido-2-cloro-4-ciclopropil-benceno crudo (solución 0.2 M en tetrahidrofurano (THF)), se le agregó 3 -oxo-butanoato de metilo (146 gramos, 1254 milimoles), y la mezcla resultante se enfrió hasta 5 °C. Se agregó lentamente NaOMe (232 mililitros, 1254 milimoles), formando un precipitado, y la mezcla se agitó a temperatura ambiente durante 64 horas. La suspensión resultante se enfrió en un baño de hielo, y se agregó agua (2 litros). La mezcla se transfirió a un embudo de separación, y la capa acuosa se extrajo con Me-THF (500 mililitros). La capa acuosa se acidificó con HCl 2M (acuoso), y se extrajo con EtOAc (2 litros) (1 vez) y (1 litro) (2 veces). Los extractos orgánicos combinados se lavaron con agua (1 litro), y salmuera (1 litro), se secaron sobre Na2SO4, se filtraron, y se concentraron bajo presión reducida, para dar un residuo oleoso color naranja. Se agregó agua (300 mililitros), y la mezcla se agitó durante 30 minutos, se filtró, se lavó con agua, y se secó bajo presión reducida, para dar el compuesto del título como cristales color naranja.
LC-MS: Rt 0.89 minutos; MS m/z 278.1 [M+H]+; Método C.
1H RMN (400 MHz, DMSO-ds) 5 13.22 (1H, d), 7.55 (1H, d), 7.51 (1H, d), 7.29 (1H, dd), 2.31 (3H, s), 2.08 (1H, multiplete), 1.10-1.03 (2H, multipletes), 0.87-0.81 (2H, multipletes).
Paso 4: 1-(2-cloro-4-c¡cloprop¡l-fen¡l)-N-(2-c¡clohex¡l-1,5-d¡met¡l-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida
El ácido 1-(2-cloro-4-c¡cloprop¡l-fen¡l)-5-met¡l-1H-1,2,3-tr¡azol-4-carboxíl¡co (15 gramos, 54.0 milimoles), y la 4-am¡no-2-c¡clohex¡l-1,5-d¡met¡l-1H-p¡razol-3(2H)-ona (Intermediario A) (11.30 gramos, 54.0 milimoles) se combinaron en una pasta acuosa en EtOAc (150 mililitros). La mezcla de reacción se enfrió hasta 10 °C, y se agregó por goteo trietil-amina (18.82 mililitros, 135 milimoles), seguida por la adición por goteo de T3P (al 50 por ciento en peso/peso en EtOAc) (47.7 mililitros, 81 milimoles) durante 15 minutos, manteniendo una temperatura <10 °C. La mezcla de reacción se dejó calentar a temperatura ambiente, y se agitó durante 2.5 horas. La mezcla resultante se apagó con bicarbonato de sodio saturado (acuoso) (150 mililitros). Las capas se separaron, y la capa orgánica se lavó con bicarbonato de sodio saturado (acuoso) (50 mililitros), agua (150 mililitros), y salmuera (150 mililitros). Los extractos orgánicos se secaron sobre MgSO4 y carbón. El desecante se filtró, y el filtrado se concentró bajo presión reducida. El material crudo se convirtió en una pasta acuosa en acetato de etilo (335 mililitros), y se calentó a reflujo con agitación. La mezcla caliente se filtró, y el filtrado se sembró y se dejó reposar a temperatura ambiente durante la noche. El sólido cristalino resultante se recolectó mediante filtración, y se secó bajo presión reducida a 40 °C durante 2 días para proporcionar el compuesto del título.
LC-MS: Rt 1.32 minutos; MS m/z 469.4 & 471.4 [M+H]+; Método 2minBajopHv03.
iH RMN (400 MHz, DMSO-ds) 59.25 (1 H, s), 7.59 (1 H, d), 7.53 (1 H, d), 7.32 (1 H, dd), 3.93 (1 H, tt), 3.32 (3H, s), 2.34 (3H, s), 2.14-2.07 (1H, m), 2.06 (3H, s), 2.05-1.94 (2H, br m), 1.83-1.75 (2H, br m), 1.71-1.58 (3H, br m), 1.39-1.25 (2H, br m), 1.24-1.12 (1H, br m), 1.09 (2H, m), 0.86 (2H, m).
Ejemplo 11:
1-(2-cloro-4-c¡cloprop¡l-fen¡l)-N-(2-(2-fluoro-fen¡l)-1,5-d¡met¡l-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida
Una solución de ácido 1-(2-cloro-4-c¡cloprop¡l-fen¡l)-5-metil-1H-1,2,3-tr¡azol-4-carboxíl¡co (Ejemplo 10, paso 3) (95 m¡l¡gramos, 0.342 m¡l¡moles), cloruro de oxal¡lo (0.045 m¡l¡l¡tros, 0.513 m¡l¡moles), y N,N-d¡met¡lformam¡da (DMF) (0.053 m¡l¡l¡tros, 0.684 m¡l¡moles) en d¡cloro-metano (DCM) (5 m¡l¡l¡tros), se ag¡tó a temperatura amb¡ente durante 15 m¡nutos. Se agregó a la mezcla 4-am¡no-2-(2-fluoro-fen¡l)-1,5-d¡met¡l-1H-p¡razol-3(2H)-ona (Intermed¡ar¡o B) (83 m¡l¡gramos, 0.376 m¡l¡moles), segu¡da por tr¡et¡l-am¡na (0.143 m¡l¡l¡tros, 1.026 m¡l¡moles), y ésta se ag¡tó a temperatura amb¡ente durante 30 m¡nutos. La mezcla resultante se d¡luyó con d¡cloro-metano (DCM), se lavó con HCl 0.1M (acuoso), y NaHCÜ3 (acuoso) saturado. Los extractos orgán¡cos se pasaron a través de un cartucho de separac¡ón de fases y se concentraron bajo pres¡ón reduc¡da, para dar un ace¡te color café. El ace¡te se absorb¡ó sobre síl¡ce, y se pur¡f¡có med¡ante cromatografía eluyendo con el 0 al 10 por c¡ento de metanol (MeÜH) en terbut¡l-met¡l-éter (TBME). Las fracc¡ones del producto se comb¡naron y se concentraron bajo pres¡ón reduc¡da, para dar un sól¡do amar¡llo v¡dr¡ado. El sól¡do se d¡solv¡ó en 1:1 de EtÜAc:Et2Ü, se lavó con agua (2 veces), salmuera, se secó sobre MgSÜ4, se f¡ltró, y se concentró bajo pres¡ón reduc¡da, para dar el compuesto del título como un sól¡do amar¡llo v¡dr¡ado.
LC-MS: Rt 4.60 m¡nutos; MS m/z 481.4 [M+H]+; Método 10m¡nBajopHv01.
1H RMN (400 MHz, DMSÜ-d6) 59.53 (1H, s), 7.59 (1H, d), 7.56-7.50 (1H, mult¡plete), 7.53 (1H, d), 7.47-7.31 (4H, mult¡pletes), 3.08 (3H, s), 2.36 (3h , s), 2.20 (3H, s), 2.10 (1H, mult¡plete), 1.11-1.06 (2H, mult¡pletes), 0.89-0.85 (2H, mult¡pletes).
Ejemplo 12:
1-(2-cloro-4-ciclopropil-fenil)-N-(2-ciclohexil-1-metil-d3,5-metil-3-oxo-2,3-dihidro-1 H-p¡razol-4-¡l)-5-met¡l-1 H-1,2,3-tr¡azol-4-carboxam¡da

A una soluc¡ón helada de cloruro de oxal¡lo (0.033 m¡l¡l¡tros, 0.378 m¡l¡moles) en d¡cloro-metano (DCM) (3 m¡l¡l¡tros), se le agregó N,N-d¡met¡l-formam¡da (DMF) (0.039 m¡l¡l¡tros, 0.504 m¡l¡moles) por goteo, y la mezcla se ag¡tó durante 1 hora. A la suspens¡ón se le agregó el ác¡do 1-(2-cloro-4-c¡cloprop¡l-fen¡l)-5-met¡l-1H-1,2,3-tr¡azol-4-carboxíl¡co (Ejemplo 10, paso 3) (70 m¡l¡gramos, 0.252 m¡l¡moles), y la mezcla se ag¡tó a temperatura amb¡ente bajo N2 durante 1 hora. A la mezcla de reacc¡ón se le agregó el (Intermed¡ar¡o C) (53.5 m¡l¡gramos, 0.252 m¡l¡moles), segu¡do por la ad¡c¡ón por goteo de tr¡et¡l-am¡na (0.105 m¡l¡l¡tros, 0.756 m¡l¡moles), y la mezcla se ag¡tó a temperatura amb¡ente durante 16 horas. La mezcla resultante se d¡luyó con d¡cloro-metano (DCM) (20 m¡l¡l¡tros), se lavó con agua (20 m¡l¡l¡tros), HCl 1M (acuoso) (10 m¡l¡l¡tros), y NaHCÜ3 (acuoso) saturado (20 m¡l¡l¡tros). Los extractos orgán¡cos se secaron sobre MgSÜ4, se f¡ltraron, y se
concentraron bajo presión reducida, para dar el producto crudo. El material crudo se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con el 0 al 90 por ciento de EtOAc en isohexano, seguido por el 0 al 10 por ciento de metanol (MeOH) en dicloro-metano (DCM). Las fracciones del producto se combinaron y se concentraron in bajo presión reducida, para dar un aceite color naranja. El aceite se disolvió en 1:1 de DMSO/MeOH (1 mililitro), y se purificó utilizando cromatografía en fase inversa (Método de Preparación F). La fracción del producto se extrajo con dicloro-metano (DCM), y los extractos orgánicos combinados se concentraron bajo presión reducida, para dar el compuesto del título como un aceite color amarillo.
LC-MS: Rt 1.17 minutos; MS m/z 472.4 [M+H]+; Método 2minBajopHv01.
1H RMN (400 MHz, CDCla) 59.25 (1H, s), 7.30 (1H, d), 7.29 (1H, d), 7.16 (1H, d), 4.11 (1H, m), 2.47 (3H, s), 2.26 (3H, s), 2.06-1.96 (3H, m), 1.90 (2H, d), 1.70 (3H, multipletes), 1.40 (2H, m), 1.28 (1H, m), 1.13 (2H, m), 0.82 (2H, m).
Ejemplo 13:
1-(4-cloro-2-ciclopropil-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida

Paso 1: N-(2-bromo-4-cloro-fenil)-acetamida
Una solución de 2-bromo-4-cloro-anilina (1 gramo, 4.84 mili-moles) en anhídrido acético (5 mililitros, 4.84 milimoles), se agitó a temperatura ambiente durante 1.5 horas. La suspensión resultante se recolectó mediante filtración, y se secó sobre una línea de vacío, para dar el compuesto del título como un sólido blanco.
LC-MS: Rt 0.90 minutos; MS m/z 250.0 [M+H]+; Método 2minBajopH.
iH RMN (400 MHz, DMSO-d6) 59.51 (1 H, s), 7.78 (1 H, d), 7.62 (1 H, d), 7.44 (1 H, dd), 2.08 (3H, s).
Paso 2: N-(4-cloro-2-ciclopropil-fenil)-acetamida
Una mezcla de N-(2-bromo-4-cloro-fenil)-acetamida (817 mili-gramos, 3.29 milimoles), ácido ciclopropilborónico (706 miligramos, 8.22 milimoles), PdOAc2 (14.76 miligramos, 0.066 milimoles), N-fenil-2-(diterbutilfosfino)-indol (44.4 miligramos, 0.132 milimoles), y fluoruro de potasio (955 miligramos, 16.44 milimoles), se agitó en tetrahidrofurano (THF) (50 mililitros) a 55 °C durante 16 horas. Se agregó más ácido ciclopropilborónico (706 miligramos, 8.22 mili-moles), PdOAc2 (14.76 miligramos, 0.066 milimoles), y fluoruro de potasio (955 miligramos, 16.44 milimoles) a la mezcla de reacción, y la mezcla se agitó a 65 °C durante 16 horas. La mezcla resultante se diluyó con EtOAc, y se lavó con agua (2 veces). Los extractos orgánicos se secaron sobre MgSO4 , se filtraron, y se concentraron bajo presión reducida, para dar un sólido color café. El producto crudo se utilizó en el siguiente paso sin mayor purificación.
Paso 3: 4-cloro-2-ciclopropil-anilina
Una suspensión de N-(4-cloro-2-ciclopropil-fenil)-acetamida (689 miligramos, 3.29 milimoles) en HCl 2M (acuoso) (25 mililitros, 823 milimoles), se agitó a 100 °C durante 3 horas, y entonces a 90 °C durante 16 horas. La mezcla resultante se enfrió hasta la temperatura ambiente, se diluyó con EtOAc y agua, y las capas se separaron. La capa orgánica se extrajo con HCl 0.1M (acuoso), y la porción orgánica se desechó. A las porciones acuosas combinadas se les agregó NaOH 2M (acuoso) hasta quedar básicas, y la mezcla resultante se extrajo con EtOAc (2 veces). Los extractos orgánicos se lavaron con salmuera, se secaron sobre MgSO4 , se filtraron, y se concentraron al vacío, para dar el compuesto del título como un aceite amarillo
oscuro.
LC-MS: Rt 0.96 minutos; MS m/z 168.1 [M+H]+; Método 2minBajopH.
1H RMN (400 MHz, DMSO-d6) 56.89 (1H, dd), 6.75 (1H, d), 6.59 (1H, d), 5.11 (2H, s), 1.65 (1H, multiplete), 0.87-0.81 (2H, multipletes), 0.51-0.47 (2H, multipletes).
Paso 4: 1-azido-4-cloro-2-ciclopropil-benceno
Una solución de 4-cloro-2-ciclopropil-anilina (182 miligramos, 1.086 milimoles) en ácido acético (15 mililitros) y agua (15 mililitros), se agitó y se enfrió a 0 °C. Se agregó por goteo una solución de nitrito de sodio (74.9 miligramos, 1.086 milimoles) en agua (1.4 mililitros), y la mezcla se agitó bajo enfriamiento con hielo durante 10 minutos. Se agregó por goteo una solución de azida de sodio (70.6 miligramos, 1.086 milimoles) en agua (1.5 mililitros), y se agitó bajo enfriamiento con hielo durante 1 hora, y entonces a temperatura ambiente durante 30 minutos. A la mezcla del producto se le agregó NaOH 2M (acuoso) hasta quedar básica, y ésta se extrajo con EtOAc. El extracto orgánico se lavó con NaHCO3 (acuoso) saturado, y se secó sobre MgSO4, se filtró, y se concentró bajo presión reducida, para dar el compuesto del título como un aceite color amarillo oscuro/café. El producto crudo se utilizó en el siguiente paso sin mayor purificación.
Paso 5: Ácido 1-(4-cloro-2-ciclopropil-fenil)-5-metil-1H-1,2,3-triazol-4-carboxílico
A una solución agitada de 1-azido-4-cloro-2-ciclopropil-benceno (204 miligramos, 1.054 milimoles), y 3 -oxo-butanoato de metilo (0.341 mililitros, 3.16 milimoles) en metanol (MeOH) (2 mililitros), se le agregó por goteo metóxido de sodio (5M en MeOH) (1.264 mililitros, 6.32 milimoles), y la mezcla se agitó a temperatura ambiente durante 2.5 horas. La mezcla se calentó a 50 °C durante 5 horas, y entonces se dejó agitándose a temperatura ambiente durante 16 horas. La mezcla resultante se diluyó con agua, se extrajo con dietil-éter (2 veces), y los extractos orgánicos se desecharon. A la capa acuosa se le agregó HCl 6M (acuoso) hasta quedar ácida, y éste se extrajo con dietil-éter. Los extractos orgánicos se secaron sobre MgSO4, se filtraron, y se concentraron bajo presión reducida, para proporcionar el compuesto del título como un aceite color naranja.
LC-MS: Rt 0.99 minutos; MS m/z 278.4 [M+H]+; Método 2minBajopH.
iH RMN (400 MHz, DMSO-ds) 513.1 (1H, br s), 7.49-7.48 (2H, multipletes), 7.23 (1H, br s), 2.35 (3H, s), 1.22 (1H, multiplete), 0.85-0.83 (2H, multipletes), 0.77-0.76 (2H, multipletes).
Paso 6: 1-(4-cloro-2-ciclopropil-fenil)-N-(2-(2-fluoro-fenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1H-1,2,3-triazol-4-carboxamida
A una solución agitada de cloruro de oxalilo (0.035 mililitros, 0.396 milimoles), y N,N-dimetil-formamida (DMF) (0.056 mililitros, 0.720 milimoles) en dicloro-metano (DCM) (10 mililitros), se le agregó ácido 1-(4-cloro-2-ciclopropil-fenil)-5-metil-1H-1,2,3-triazol-4-carboxílico (100 miligramos, 0.360 milimoles), y la mezcla se agitó durante 30 minutos. Se agregó más cloruro de oxalilo (0.035 mililitros, 0.396 milimoles) a la mezcla de reacción, y se agitó durante 1 hora. Se agregó la 4-amino-2-(2-fluoro-fenil)-1,5-dimetil-1H-pirazol-3(2H)-ona (Intermediario B) (88 miligramos, 0.396 milimoles), seguida por trietil-amina (0.151 mililitros, 1.080 mili-moles), y la mezcla se agitó a temperatura ambiente durante 1 hora. La mezcla resultante se diluyó con dicloro-metano (DCM), se lavó con HCl 0.1M (acuoso), y NaHCO3 (acuoso) saturado. Los extractos orgánicos se pasaron a través de un cartucho de separación de fases y se concentraron bajo presión reducida, para dar un aceite. El aceite se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con el 0 al 10 por ciento de metanol (MeOH) en terbutil-metil-éter (TBME). Las fracciones del producto se combinaron y se concentraron bajo presión reducida, para dar un sólido amarillo vidriado, el cual se recristalizó a partir de EtOH. El sólido se recolectó mediante filtración, y se secó sobre la línea de vacío, para dar el compuesto del título como un sólido blanco.
LC-MS: Rt 4.63 minutos; MS m/z 481.4 [M+H]+; Método 10minBajopHv01.
iH RMN (400 MHz, DMSO-ds) 59.51 (1H, s), 7.56-7.35 (6H, multipletes), 7.24 (1H, d), 3.09 (3H, s), 2.39 (3H, s), 2.20 (3h , s), 1.26 (1H, multiplete), 0.90-0.85 (2H, multipletes), 0.82-0.78 (2H, multipletes).
Ejemplo 14:
1 -(4-cloro-2-ciclopropil-fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-p i razol-4-i l)-5-m etil-1 H-1,2,3-triazol-4-carboxamida

El compuesto del título se preparó a partir de ácido 1-(4-cloro-2-c¡clopropil-fen¡l)-5-met¡l-1H-1,2,3-tr¡azol-4-carboxílico (Ejemplo 13, paso 5), y 4-amino-2-c¡clohex¡l-1,5-dimet¡l-1H-p¡razol-3(2H)-ona (Intermediario A) de una manera análoga al Ejemplo 1, paso 3.
LC-MS: Rt 4.94 minutos; MS m/z 471.3 [M+H]+; Método 10minBajopHv01.
1H RMN (400 MHz, CDCla) 58.36 (1H, s), 7.32 (1H, dd), 7.18 (1H, d), 7.02 (1H, d), 4.06 (1H, multiplete), 3.24 (3H, s), 2.48 (3H, s), 2.23 (3H, s), 3.05-1.86 (6H, multipletes), 1.71 (1H, multiplete), 1.43-1.17 (4H, multipletes), 0.93 (2H, multipletes), 0.72 (2H, multipletes).
Ejemplo 15:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-¡l)-1-(2-c¡cloprop¡l-4-fluoro-fen¡l)-5-metil-1H-1,2,3-triazol-4-carboxamida

Paso 1: N-(2-ciclopropil-4-flluoro-fenil)-acetamida
Una mezcla que comprendía N-(2-bromo-4-fluoro-fenil)-acetamida (1 gramo, 4.31 milimoles), ácido ciclopropil-borónico (0.481 gramos, 5.60 milimoles), triciclohexil-fosfina (0.121 gramos, 0.431 milimoles), fosfato de potasio (tribásico) (3.20 gramos, 15.08 milimoles), tolueno (10 mililitros), y agua (10 mililitros), se dividió igualmente entre dos tubos de microondas de 10 a 20 mililitros. Cada tubo de reacción se selló, se evacuó y se rellenó con nitrógeno (3 veces), y se colocó en el reactor de microondas a 100 °C durante 6 horas. El contenido de los frascos se combinó, se diluyó con EtOAc, y las capas se separaron. Los extractos orgánicos se lavaron con agua (2 veces), se secaron sobre MgSO4, se filtraron, y se concentraron bajo presión reducida, para dar el compuesto del título como un sólido color café. El producto se utilizó crudo en el siguiente paso.
LC-MS: Rt 0.88 minutos; MS m/z 194.3 [M+H]+; Método 2minBajopHv01.
Paso 2: 2-ciclopropil-4-fluoro-an¡l¡na
El compuesto del título se preparó a partir de la N-(2-ciclopropil-4-fluoro-fen¡l)-acetam¡da de una manera análoga al Ejemplo 13 paso 3.
LC-MS: Rt 0.65 minutos; MS m/z 152.1 [M+H]+; Método 2minBajopH.
iH RMN (400 MHz, DMSO-ds) 5 6.70 (1H, tt), 6.60-6.51 (2H, multipletes), 4.82 (2H, br s), 1.69 (1H, multiplete), 0.88-0.83 (2H, multipletes), 0.52-0.48 (2H, multipletes).
Paso 3: 1-azido-2-ciclopropil-4-fluoro-benceno
El compuesto del título se preparó a partir de la 2-ciclopropil-4-fluoro-an¡l¡na de una manera análoga al
Ejemplo 13, paso 4, para dar un aceite color café.
Paso 4: Ácido 1 -(2-c iclopropi l-4-f luoro-fe nil )-5-metil-1 H-1,2,3-triazol-4-carboxílico
A una solución de 1-azido-2-ciclopropil-4-fluoro-benceno (237 miligramos, 1.338 milimoles) en metanol (MeOH) (10 mililitros), se le agregaron 3-oxo-butanoato de metilo (466 miligramos, 4.01 milimoles), y metóxido de sodio 5M (1.338 mililitros, 6.69 milimoles), y la mezcla se agitó a 50 °C durante 16 horas. Se agregó agua (1 mililitro), y se continuó la agitación a 50 °C durante 3 horas. La mezcla resultante se concentró bajo presión reducida para remover el metanol. La mezcla se lavó con EtOAc, y los extractos orgánicos se desecharon. A la capa acuosa se le agregó HCl 1M (acuoso) hasta quedar ácida, y éste se extrajo con EtOAc (3 veces). Los extractos orgánicos se lavaron con salmuera, se secaron sobre MgSO4, se filtraron, y se concentraron bajo presión reducida, para dar el compuesto del título como un aceite color café, el cual se utilizó en el siguiente paso sin mayor purificación.
LC-MS: Rt 0.97 minutos; MS m/z 262.4 [M+H]+; Método 2minBajopH.
Paso 5: N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-1-(2-ciclopropil-4-fluoro-fenil)-5-metil-1 H-1,2,3-triazol-4-carboxamida
A una solución de cloruro de oxalilo (0.055 mililitros, 0.632 milimoles) en dicloro-metano (DCM) (5 mililitros), se le agregó por goteo N,N-dimetil-formamida (DMF) (0.065 mililitros, 0.842 milimoles), y la mezcla se agitó durante 1 hora. La mezcla resultante se trató con ácido 1-(2-ciclopropil-4-fluoro-fenil)-5-metil-1H-1,2,3-triazol-4-carboxílico (110 miligramos, 0.421 milimoles), y la agitación se continuó a temperatura ambiente durante 30 minutos. A la mezcla de reacción se le agregó 4-amino-2-ciclohexil-1,5-dimetil-1H-pirazol-3(2H)-ona (Intermediario A) (97 miligramos, 0.463 milimoles), seguida por trietil-amina (0.176 mililitros, 1.263 milimoles), y la agitación se continuó a temperatura ambiente durante 30 minutos. La mezcla resultante se diluyó con dicloro-metano (DCM), se lavó con agua, y se eluyó a través de un cartucho de separación de fases. El eluyente orgánico se recolectó y se concentró bajo presión reducida, para dar un aceite color café. Éste se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con el 0 al 10 por ciento de metanol (MeOH) en terbutil-metil-éter (TBME). Las fracciones del producto se combinaron, se concentraron, y se secaron bajo presión reducida, para dar un sólido vidriado color café pálido. El sólido se disolvió en dicloro-metano (DCM), se lavó con agua, salmuera, y se eluyó a través de un cartucho de separación de fases. El eluyente orgánico se concentró bajo presión reducida, para dar el compuesto del título como un sólido vidriado.
LC-MS: Rt 4.84 minutos; MS m/z 452.9 [M+H]+; Método 10minBajopHv01.
1H RMN (400 MHz, CDCla) 58.37 (1 H, s), 7.24 (1, dd), 7.04 (1 H, td), 6.73 (1 H, dd), 4.07 (1 H, multiplete), 3.25 (3H, s), 2.47 (3H, s), 2.24 (3H, s), 2.06-1.95 (2H, multipletes), 1.89 (5H, multipletes), 1.71 (1H, multiplete), 1.43-1.18 (3H, multipletes), 0.95 (2H, multipletes), 0.71 (2H, multipletes).
Ejemplo 16:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-8-(trifluoro-metoxi)-5,6-dihidro-4H-benzo-[f][1,2,3]-triazolo-[1,5-a]-azepina-3-carboxamida

Paso 1: 2-alil-4-(trifluoro-metoxi)-anilina
A una solución de 2-bromo-4-(trifluoro-metoxi)-anilina (75 gramos, 293 milimoles) en N,N-dimetilformamida (DMF) (1,000 mililitros), se le agregó Pd(PPh3)4 (13.5 gramos, 11.7 milimoles), y alil-tributil-estaño (116 gramos, 351 milimoles), y la mezcla de reacción se agitó a 80 °C durante 16 horas. La mezcla se enfrió hasta la temperatura ambiente, y se agregó una solución acuosa saturada de KF. La mezcla se diluyó con acetato de etilo (1,500 mililitros), y se lavó con 1:1 de H2O : solución acuosa saturada de NaCl (750 mililitros,
2 veces), y KF acuoso saturado (1 vez). Las capas acuosas se combinaron y se extrajeron con acetato de etilo (250 mililitros). Los extractos orgánicos se combinaron, se secaron sobre MgSO4, se filtraron, y se concentraron bajo presión reducida, para dar el producto crudo, el cual se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con acetato de etilo al 1 por ciento en éter de petróleo, para dar el compuesto del título.
LC-MS: Rt 1.00 minutos; MS m/z 218.0 [M+H]+; Método H.
1H RMN (400 MHz, MeOD) 53.27 (2H, dd), 5.07-5.14 (2H, m), 5.92-5.99 (1H, m), 6.72 (1H, dd), 6.88-6.89 (2H, m).
Paso 2: N-(2-alil-4-(trifluoro-metoxi)-fenil)-acrilamida
Una solución de 2-alil-4-(trifluoro-metoxi)-anilina (17.5 gramos, 80.6 milimoles) en tetrahidrofurano (THF) (1,000 mililitros), se enfrió en un baño de hielo seco hasta una temperatura por debajo de -10 °C; se agregaron Et3 N (8.96 gramos, 17.3 milimoles), y cloruro de acriloílo (7.98 gramos, 88.7 milimoles). La mezcla de reacción se dejó calentar a temperatura ambiente, y se agitó durante 1 hora. La mezcla resultante se concentró bajo presión reducida, se disolvió en acetato de etilo, y se lavó con agua y salmuera. La porción orgánica se secó sobre Na2SO4, se filtró, y se concentró bajo presión reducida, para dar un sólido blanco, el cual se lavó con acetato de etilo, para dar el compuesto del título.
LC-MS: Rt 1.15 minutos; MS m/z 272.0 [M+H]+; Método H.
iH RMN (400 MHz, CDCla) 53.40 (2H, dd), 5.12 (1H, dd), 5.24 (1H, dd), 5.77 (1H, dd), 5.92-5.99 (1H, m), 6.18-6.29 (1 H, m), 6.38 (1H, dd), 7.06 (1H, s), 7.15 (1H, dd), 7.36 (1H, s), 8.01 (1H, dd).
Paso 3: 7-(trifluoro-metoxi)-1H-benzo-[b]-azepin-2(5H)-ona
A una solución de N-(2-alil-4-(trifluoro-metoxi)-fenil)-acrilamida (15 gramos, 55.3 milimoles) en diclorometano (DCM) (800 mililitros), se le agregó catalizador de Zhan I (2.0 gramos, 2.77 milimoles), y la mezcla se agitó a temperatura ambiente durante 3 horas. La mezcla resultante se concentró bajo presión reducida, para dar un sólido crudo, el cual se recristalizó a partir de acetato de etilo, para dar el compuesto del título.
LC-MS: Rt 0.95 minutos; MS m/z 243.9 [M+H]+; Método H.
iH RMN (400 MHz, CDCla) 53.37 (2H, dd), 5.97 (1H, dd), 6.60-6.66 (1H, m), 7.02 (1H, s), 7.10-7.13 (2H, m), 9.11 (1 H, s).
Paso 4: 7-(trifluoro-metoxi)-4,5-dihidro-1 H-benzo-[b]-azepin-2(3H)-ona
A una solución de 7-(trifluoro-metoxi)-1H-benzo-[b]-azepin-2(5H)-ona (10 gramos, 41.1 milimoles) en 1:1 de EtOH:THF (150 mililitros), se le agregó Pd/C (1.0 gramo), y la mezcla se agitó a temperatura ambiente bajo 30 psi (2.1 kg/cm2) de H2 durante 3 horas. La mezcla resultante se filtró y se concentró bajo presión reducida, para dar un sólido crudo, el cual se recristalizó a partir de acetato de etilo, para dar el compuesto del título. LC-MS: Rt 0.98 minutos; MS m/z 246.0 [M+H]+; Método H.
iH RMN (400 MHz, CDCla) 52.22 (2H, q), 2.36 (H, t), 2.80 (2H, t), 7.02-7.03 (1H, m), 7.09 (2H, s), 8.27 (1H, s).
Paso 5: 7-(trifluoro-metoxi)-4,5-dihidro-1 H-benzo-[b]-azepina-2(3H)-tiona
A una solución de 7-(trifluoro-metoxi)-4,5-dihidro-1H-benzo-[b]-azepin-2(3H)-ona (30 gramos, 122 milimoles) en tolueno (2,000 mililitros), se le agregó reactivo de Lawesson (29.7 gramos, 73.5 milimoles), y la mezcla se agitó a 100 °C durante 2 horas. La mezcla resultante se enfrió hasta la temperatura ambiente, y se concentró bajo presión reducida. El producto crudo se purificó mediante cromatografía en columna sobre sílice, eluyendo con acetato de etilo al 5 por ciento en éter de petróleo, para dar el compuesto del título.
LC-MS: Rt 1.04 minutos; MS m/z 261.9 [M+H]+; Método H.
iH RMN (400 MHz, MeOD) 52.27-2.34 (2H, m), 2.73-2.79 (4H, m), 7.13-7.15 (1H, m), 7.20-7.22 (2H, m). Paso 6: 2-(tiometil)-7-(trifluoro-metoxi)-4,5-dihidro-3H-benzo-[b]-azepina
A una solución de 7-(tr¡fluoro-metoxi)-4,5-d¡h¡dro-1H-benzo-[b]-azep¡na-2(3H)-t¡ona (22 gramos, 84.3 milimoles) en acetona (1,500 m¡l¡l¡tros), se le agregaron KOH (7.1 gramos, 127 m¡l¡moles), y Mel (18.1 gramos, 127 milimoles), y la mezcla se agitó a temperatura ambiente durante 1 hora. La mezcla resultante se concentró bajo presión reducida, y el producto crudo se disolvió en dicloro-metano (DCM) (1,000 mililitros). La mezcla se lavó con agua, salmuera, se secó sobre Na2SO4, se filtró, y se concentró bajo presión reducida, para dar un aceite amarillo. El aceite se purificó mediante cromatografía en columna por evaporación instantánea sobre óxido de aluminio neutro eluyendo con hexano, para dar el compuesto del título.
LC-MS: Rt 1.18 minutos; MS m/z 275.9 [M+H]+; Método I.
1H RMN (400 MHz, CDCla) 52.25 (2H, q), 2.32 (2H, t), 2.46 (1 H, s), 2.50 (2H, t), 6.99-7.01 (2H, m), 7.09-7.26 (1 H, m).
Paso 7: (Z)-2-nitro-2-(7-(trifluoro-metoxi)-4,5-dihidro-1H-benzo-[b]-azepin-2(3H)-iliden)-acetato de etilo Referencia: Publicación Internacional Número WO 2011/151361, páginas 33 y 34.
Una mezcla de 2-(tiometil)-7-(trifluoro-metoxi)-4,5-dihidro-3H-benzo-[b]-azepina (1 gramo, 3.63 milimoles), 2-nitro-acetato de etilo (2.016 mililitros, 18.16 milimoles), y DBU (0.602 mililitros, 4.00 milimoles), se agitó a temperatura ambiente. Se agregó un tren de restriego blanqueador, y la mezcla se agitó y se calentó a 40 °C durante 72 horas. La mezcla resultante se diluyó con EtOAc, y se lavó con agua (3 veces), seguida por NaHCO3 (acuoso) saturado (3 veces), y salmuera. Los extractos orgánicos se secaron sobre MgSO4 , se filtraron, y se concentraron bajo presión reducida, para dar un aceite amarillo. El aceite se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con el 0 al 50 por ciento de EtOAc. Las fracciones del producto se combinaron, se concentraron bajo presión reducida, y se secaron al vacío, para dar el compuesto del título como un aceite color amarillo.
Paso 8: 8-(trifluoro-metoxi)-5,6-dihidro-4H-benzo-[f][1,2,3]-triazolo-[1,5-a]-azepina-3-carboxilato de etilo Una solución de (Z)-2-nitro-2-(7-(trifluoro-metoxi)-4,5-dihidro-1 H-benzo-[b]-azepin-2(3H)-iliden)-acetato de etilo (892 miligramos, 2.476 milimoles) en ácido acético glacial (50 mililitros), se enfrió en un baño de agua durante 5 minutos. A la solución se le agregó zinc (971 miligramos, 14.85 milimoles) en porciones, y la mezcla se agitó a temperatura ambiente durante 4 horas. Se agregó más zinc (486 miligramos, 7.42 milimoles), y la mezcla se agitó a temperatura ambiente durante 16 horas. A la mezcla de reacción se le agregaron nitrito de isoamilo (0.467 mililitros, 3.47 milimoles), y ácido tricloro-acético (809 miligramos, 4.95 milimoles), y la mezcla se agitó a temperatura ambiente durante 48 horas. La mezcla resultante se diluyó con dicloro-metano (DCM) y agua, y las capas se separaron. La capa acuosa se extrajo con dicloro-metano (DCM) (2 veces), y los extractos orgánicos se combinaron. Los extractos orgánicos se lavaron con NaHCO3 (acuoso) saturado (3 veces), salmuera, se secaron sobre MgSO4, se filtraron, y se concentraron bajo presión reducida, para dar un aceite color café. El aceite se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con el 0 al 50 por ciento de EtOAc en isohexano. Las fracciones del producto se combinaron y se concentraron bajo presión reducida, para dar el compuesto del título como un sólido amarillo.
LC-MS: Rt 1.25 minutos; MS m/z 342.3 [M+H]+; Método 2minBajopHv01.
iH RMN (400 MHz, CDCla) 57.83 (1H, d), 7.34 (1H, dd), 7.27 (1H, br s), 4.49 (2H, q), 3.14 (2H, t), 2.62 (2H, t), 2.40 a 2.33 (2H, multipletes), 1.47 (3H, t).
Paso 9: Ácido 8-(trifluoro-metoxi)-5,6-dihidro-4H-benzo-[f][1,2,3]-triazolo-[1,5-a]-azepina-3-carboxílico A una solución de 8-(trifluoro-metoxi)-5,6-dihidro-4H-benzo-[f][1,2,3]-triazolo-[1,5-a]-azepina-3-carboxilato de etilo (384 mili-gramos, 1.125 milimoles) en tetrahidrofurano (THF) (5 mililitros), y metanol (MeOH) (3 mililitros), se le agregó NaOH 2M (acuoso) (2.81 mililitros, 5.63 milimoles), y la mezcla se agitó a temperatura ambiente durante 45 minutos. La mezcla resultante se concentró bajo presión reducida, para dar un residuo que se dividió entre EtOAc y agua. Las capas se separaron, y la capa acuosa se lavó con EtOAc. Las porciones orgánicas se desecharon y la capa acuosa se trató con HCl 2M (acuoso) hasta quedar ácida. La solución resultante se extrajo con EtOAc, y los extractos orgánicos se secaron sobre MgSO4 , se filtraron, y se concentraron bajo presión reducida, para dar el compuesto del título como un sólido amarillo.
LC-MS: Rt 1.07 minutos; MS m/z 314.3 [M+H]+; Método 2minBajopHv01.
iH RMN (400 MHz, CDCla) 57.85 (1H, d), 7.38 (1H, d), 7.30 (1H, s), 3,18 (2H, t), 2.66 (2H, t), 2.44-2.37 (2H, multipletes).
Paso 10: N-(2-ciclohexil-1,5-d¡met¡l-3-oxo-2,3-dih¡dro-1 H-p¡razol-4-il)-8-(tr¡fluoro-metox¡)-5,6-dihidro-4H-benzo-[f][1,2,3]-triazolo-[1,5-a]-azepina-3-carboxamida
A una solución de ácido 8-(tr¡fluoro-metox¡)-5,6-d¡h¡dro-4H-benzo-[f][1,2,3]-triazolo-[1,5-a]-azep¡na-3-carboxílico (169 mili-gramos, 0.540 milimoles) en dicloro-metano (DCM) seco (2 mililitros), bajo nitrógeno, se le agregaron cloruro de oxalilo (0.052 mililitros, 0.593 milimoles), y N,N-dimetil-formamida (DMF) (0.084 mililitros, 1.079 milimoles), y la mezcla se agitó durante 15 minutos. Se agregó 4-amino-2-ciclohexil-1,5-dimetil-1H-pirazol-3(2H)-ona (Intermediario A) (124 miligramos, 0.593 milimoles), seguida por trietil-amina (0.226 mililitros, 1.619 milimoles), y la mezcla se agitó a temperatura ambiente durante 16 horas. La mezcla resultante se diluyó con dicloro-metano (DCM) y agua, y se agitó vigorosamente. La mezcla se pasó a través de un cartucho de separación de fases, y la porción orgánica se concentró bajo presión reducida, para dar un aceite amarillo. El aceite se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con el 100 por ciento de TBME. Las fracciones del producto se combinaron y se concentraron bajo presión reducida, para dar un sólido vidriado, el cual se recristalizó a partir de 1:1 de dietil-éter : EtOAc. Los cristales resultantes se filtraron por gravedad, para dar el compuesto del título como un sólido blanco.
LC-MS: Rt 4.99 minutos; MS 505.3 m/z [M+H]+; Método 10minBajopHv01.
1H RMN (400 MHz, DMSO-d6) 59.31 (1 H, s), 7.88 (1 H, d), 7.64 (1 H, br s), 7.56 (1 H, br d), 3.93 (1 H, tt), 3.22 (3H, s), 3.07 (2H, t), 2.64 (2H, t), 2.23 (2H, t), 2.07 (3H,s), 2.02-1.97 (2H, multipletes), 1.80 (2H, br d), 1.70 1.61 (3H, multipletes), 1.32 (2H, q), 1.21-1.15 (1H, multiplete).
Ejemplo 17:
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-1-(4-metox¡-fen¡l)-5-met¡l-1 H-1,2,3-triazol-4-carboxamida

Paso 1: N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-1-(4-metox¡-fen¡l)-5-met¡l-1 H-1,2,3-triazol-4-carboxamida
A un frasco se le agregaron el ácido 1-(4-metoxi-fenil)-5-met¡l-1H-1,2,3-tr¡azol-4-carboxíl¡co (0.040 gramos, 0.170 milimoles), y una solución de reactivo de Ghosez (0.047 mililitros, 0.340 milimoles) en diclorometano (DCM) (0.95 mililitros). La mezcla se sonicó y se calentó para ayudar a la disolución. El frasco se colocó sobre el agitador IKA durante 4 horas. A esta mezcla se le agregó una solución de 4-amino-2-ciclohexil-1,5-d¡met¡l-1H-p¡razol-3(2H)-ona (Intermediario A) (10.043 gramos, 0.204 milimoles), y N,N-d¡-isopropil-etil-amina (DIPEA) (0.030 mililitros, 0.170 milimoles) en dicloro-metano (DCM) (0.97 mililitros), y el frasco se agitó durante 16 horas. Se agregaron unas cuantas gotas de metanol (MeOH), y el solvente se removió utilizando un evaporador Genevac HT-4X. La mezcla cruda se purificó mediante cromatografía en fase inversa, y la fracción del producto se concentró utilizando un evaporador Genevac HT-4X. El producto crudo se disolvió en metanol (MeOH) (1 mililitro), y se eluyó a través de un cartucho de SiCO3 utilizando metanol (MeOH) como el eluyente. El eluyente recolectado se concentró, para dar el compuesto del título. LC-MS: Rt 0.98 minutos; MS m/z 425.3 [M+H]+; Método 2minBajopHv02.
iH RMN (400 MHz, CDCla) 58.40 (1H, br s), 7.37 (2H, dt), 7.07 (2H, dt), 4.06 (1H, multiplete), 3.90 (3H, s), 3.24 (3H, s), 2.58 (3H, s), 2.22 (3H, s), 2.06-1.92 (2H, multipletes), 1.87 (4H, br d), 1.70 (1H, br d), 1.45-1.30 (2H, multipletes), 1.28-1.16 (1H, multiplete).
Preparación de los Intermediarios
Intermediario A:
4-amino-2-ciclohexil-1,5-dimetil-1 H-pirazol-3(2H)-ona

Paso 1: 2-ciclohexil-5-metil-1 H-pirazol-3(2H)-ona
Se agregó clorhidrato de ciclohexil-hidrazina (700 gramos, 4643 milimoles) a una solución agitada de diclorometano (DCM) (3000 mililitros) y una solución helada de NaOH 2M (1778 mililitros, 3556 milimoles), y ésta se agitó durante 10 minutos a temperatura ambiente. Las fases se separaron, y la capa acuosa se lavó con dicloro-metano (DCM) (2,000 mililitros, 4 veces). Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro, se filtraron, y se concentraron bajo presión reducida, para dar un sólido amarillo. La ciclohexil-hidrazina sólida (406 gramos, 3556 milimoles) se disolvió en agua (1,300 mililitros) y ácido acético (1,300 mililitros), y se trató con aceto-acetato de etilo (450 mililitros, 3,556 milimoles). La mezcla de reacción se calentó a 85 °C, y se agitó durante 1 hora. La mezcla resultante se concentró hasta la sequedad bajo presión reducida, y el residuo se disolvió en dicloro-metano (DCM) (3,000 mililitros) y agua (1,000 mililitros). La mezcla se neutralizó con carbonato de potasio 2M a un pH > 9, las fases resultantes se separaron, y el extracto orgánico se lavó con salmuera (2 litros, 1 vez). La primera capa acuosa se saturó con cloruro de sodio, y ambas fases acuosas se lavaron con dicloro-metano (DCM) (2 litros, 4 veces). Los extractos orgánicos combinados se secaron sobre sulfato de sodio anhidro, se filtraron, y se concentraron bajo presión reducida, para dar un sólido color beige. El producto crudo se molió hasta obtener un polvo, se agregó TBME (2,000 mililitros), y la mezcla se agitó a 50 °C durante 1 hora, seguida por 1 hora a temperatura ambiente. El sólido resultante se recolectó mediante filtración, se lavó con terbutil-metil-éter (TBME) (500 mililitros, 4 veces), y se secó al vacío a 45 °C durante 16 horas, para proporcionar el compuesto del título como cristales blanquecinos.
LC-MS: Rt 0.63 minutos; MS m/z 181.1 [M+H]+; Método C.
1H RMN (400 MHz, DMSO-ds) 510.62 (1H, br s), 5.06 (1H, s), 3.89 (1H, multiplete), 1.98 (3H, s), 1.81-1.55 (7H, multipletes), 1.36-1.22 (2H, multipletes), 1.18-1.05 (1H, multiplete).
Paso 2: 2-ciclohexil-1,5-dimetil-1 H-pirazol-3(2H)-ona
Una suspensión de 2-ciclohexil-5-metil-1H-pirazol-3(2H)-ona (525 gramos, 2834 milimoles) en N,N-dimetil-formamida (DMF) (2200 mililitros) se calentó a 40 °C, y se agregó yoduro de metilo (532 mililitros, 8502 milimoles). La mezcla de reacción se calentó a 70 °C durante 20 horas. Se agregó más yoduro de metilo (177 mililitros, 2834 milimoles), y la mezcla se agitó a 75 °C durante 3.5 horas, y entonces a 80 °C durante 20 horas. La mezcla resultante se concentró bajo presión reducida, y el residuo se trituró con terbutil-metil-éter (TBME) (2,000 mililitros). El producto se recolectó mediante filtración, lavando con el terbutil-metil-éter (TBME) (500 mililitros, 5 veces), para dar un sólido, el cual se suspendió en dicloro-metano (DCM) (2,500 mililitros) y agua (500 mililitros), y se neutralizó con una solución de carbonato de potasio 2M (1,700 mililitros), a un pH >9. Las fases se separaron, y la capa acuosa se extrajo con dicloro-metano (DCM) (500 mililitros, 3 veces). Los extractos orgánicos se lavaron con salmuera (1,000 mililitros), y se concentraron bajo presión reducida. El residuo resultante se disolvió en acetato de etilo (2,000 mililitros), se secó sobre sulfato de sodio anhidro, y se filtró a través de 200 gramos de gel de sílice (de 40 a 63 micras), lavando con acetato de etilo / metanol (MeOH), 9:1 (300 mililitros, 7 veces). El filtrado se concentró bajo presión reducida, para dar el compuesto del título como un aceite color café.
LC-MS: Rt 0.67 minutos; MS m/z 195.1 [M+H]+; Método C.
iH RMN (400 MHz, DMSO-ds) 5 5.02 (1H, s), 3.84 (1H, tt), 3.14 (3H, s), 2.06 (3H, s), 1.98-1.86 (2H, multipletes), 1.78-1.53 (5H, multipletes), 1.33-1.20 (2H, multipletes), 1.18-1.04 (1H, multiplete).
Paso 3: 2-ciclohexil-1,5-dimetil-4-n itro-1 H-pirazol-3(2H)-ona
Al ácido trifluoroacético (TFA) (1940 mililitros) enfriado hasta -15 °C, se le agregó 2-ciclohexil-1,5-dimetil
1H-pirazol-3(2H)-ona (535 gramos, 2231 milimoles), y la mezcla de reacción se dejó calentar hasta 0 °C. Se agregó por goteo ácido nítrico al 90 por ciento (211 mililitros, 4461 milimoles) durante 90 minutos, manteniendo la temperatura por debajo de 15 °C, y la mezcla resultante se agitó durante 30 minutos a 10 °C. La mezcla del producto se vertió lentamente en agua helada (8 litros), y se agitó durante 30 minutos. El sólido se recolectó mediante filtración, y se lavó con agua (2 litros, 2 veces), una solución saturada de bicarbonato de sodio (2 litros, 1 vez), agua (2 litros, 2 veces), TBME (2 litros, 3 veces), y heptano (2 litros, 2 veces). El sólido se secó en el horno al vacío, para proporcionar el compuesto del título como un polvo color beige. LC-MS: Rt 0.65 minutos; MS m/z 240.1 [M+H]+; Método C.
1H RMN (400 MHz, DMSO-ds) 54.06 (1H, tt), 3.61 (3H, s), 2.57 (3H, t), 2.15-2.03 (2H, multipletes), 1.81-1.65 (4H, multipletes), 1.64-1.55 (1H, multiplete), 1.38-1.24 (2H, multipletes), 1.19-1.06 (1H, multiplete).
Paso 4: 4-amino-2-ciclohexil-1,5-dimetil-1H-pirazol-3(2H)-ona
A la 2-ciclohexil-1,5-dimetil-4-nitro-1H-pirazol-3(2H)-ona (415 gramos, 1.73 moles) en metanol (MeOH) (4,500 mililitros) y tetrahidrofurano (THF) (4,500 mililitros), se le agregó Pd/C al 10 por ciento (70 gramos), y la mezcla de reacción se hidrogenó a 0.1 bar y a temperatura ambiente durante 57.5 horas. La mezcla resultante se filtró a través de un tamiz de presión, y se lavó con metanol (MeOH) (1 litro, 1 vez), y tetrahidrofurano (THF) (1 litro, 2 veces). El filtrado se concentró bajo presión reducida, para dar un aceite color rojo oscuro. El aceite se disolvió inmediatamente en terbutil-metil-éter (TBME) (4 litros), se concentró bajo presión reducida hasta aproximadamente 2 litros, y se sembró (100 miligramos). La suspensión se agitó durante 2 horas a temperatura ambiente, y se enfrió en un baño de hielo durante 1 hora. El sólido se recolectó mediante filtración, y se lavó con TBME helado en porciones hasta que el filtrado fue incoloro, y se secó al vacío, para dar el compuesto del título como cristales color amarillo/naranja pálido.
LC-MS: Rt 0.55 minutos; MS m/z 210.1 [M+H]+; Método C.
iH RMN (400 MHz, DMSO-ds) 53.68 (1H, tt), 3.53 (2H, br s), 2.77 (3H, s), 1.96-1.83 (2H, multipletes), 1.92 (3H, s), 1.78-1.69 (2H, multipletes), 1.64-1.53 (3H, multipletes), 1.33-1.19 (2H, multipletes), 1.17-1.04 (1H, multiplete).
Intermediario B:
4-amino-2-(2-fluoro-fenil)-1,5-dimetil-1 H-pirazol-3(2H)-ona

Paso 1: 2-(2-fluoro-fenil)-5-metil-1 H-pirazol-3(2H)-ona
Una suspensión agitada de clorhidrato de 2-fluoro-fenil-hidrazina (1,000 gramos, 5535 milimoles) en ácido acético (1,000 mililitros) y agua (1,000 mililitros), se calentó a 45 °C. Se agregó por goteo aceto-acetato de etilo (700 mililitros), durante 30 minutos, y esto se agitó a 88 °C durante 2 horas. La mezcla resultante se enfrió hasta 5 °C, y se vertió en hielo (3000 gramos), y dicloro-metano (DCM) (4,500 mililitros). Se agregó NaOH al 30 por ciento (acuoso) (2400 mililitros), y la mezcla se agitó durante 15 minutos, entonces se separó y se extrajo con dicloro-metano (DCM) (1,500 mililitros). Los extractos orgánicos se lavaron con NaOH 1M (acuoso) (1,500 mililitros). Las fases acuosas se combinaron, y se agregaron hielo (1,500 gramos) y diclorometano (DCM) (3600 mililitros). A la mezcla agitada se le agregó HCl al 32 por ciento (acuoso) (2,400 mililitros) hasta que el pH se ajustó a un pH de 1 a 2. La mezcla bifásica se separó, se extrajo con diclorometano (DCM) (1,500 mililitros), y los extractos orgánicos se lavaron con salmuera/agua, 4:1 (2,000 mili litros), se secaron sobre MgSO4 y se evaporaron a sequedad, para dar un sólido oscuro. A una solución agitada del producto crudo en dicloro-metano (DCM) (2,000 mililitros), se le agregó gel de sílice (350 gramos). Éste se filtró, y el filtrado se evaporó. La purificación mediante cromatografía con metanol (MeOH) en EtOAc dio el compuesto del título como un sólido amarillo claro.
LC-MS: Rt 0.54 minutos; MS m/z 193.1 [M+H]+; Método B.
1H RMN (400 MHz, DMSO-ds) 511.20 (1 H, br s), 7.48-7.40 (2H, multipletes), 7.36 (1 H, td), 7.29 (1 H, td), 5.31 (1H, br s), 2.10 (3H, s).
Paso 2: 2-(2-fluoro-fenil)-1,5-dimetil-1 H-pirazol-3(2H)-ona
A una solución de 2-(2-fluoro-fenil)-5-metil-1H-pirazol-3(2H)-ona (500 gramos, 2602 milimoles) en tetrahidrofurano (THF) (2,500 mililitros) calentada a 58 °C, se le agregó yodo-metano (163 mililitros) por goteo durante 15 minutos, y la mezcla se agitó a 65 °C durante 30 minutos. Se agregó K2CO3 (198 gramos) durante 20 minutos, y se agitó durante 16 horas. Se agregó más yodo-metano (16.3 mililitros), y se continuó la agitación durante 1 hora. La mezcla resultante se enfrió hasta 0 °C, se filtró, y se lavó con tetrahidrofurano (THF) (50 mililitros). El filtrado se evaporó a sequedad, y se purificó mediante cromatografía en columna, eluyendo con EtOAc/Heptano, para dar el compuesto del título como un sólido rojo.
LC-MS: Rt 0.54 minutos; MS m/z 207.1 [M+H]+; Método B.
iH RMN (400 MHz, DMSO-d6) 5 7.51-7.44 (1H, multiplete), 7.42-7.28 (3H, multipletes), 5.19 (1H, s), 3.00 (3H, s), 2.19 (3H, s).
Paso 3: 2-(2-fluoro-fenil)-1, 5-d imeti l-4-n itro-1 H-pirazol-3(2H)-ona
La 2-(2-fluoro-fenil)-1,5-dimetil-1H-pirazol-3(2H)-ona (260 gramos, 1261 milimoles) en ácido trifluoroacético (TFA) (1,040 mililitros) se enfrió hasta -10 °C, y se agitó durante 30 minutos. A la mezcla se le agregó ácido nítrico en vaporización (84 mililitros) por goteo, y éste se agitó durante 15 minutos bajo enfriamiento, y entonces a temperatura ambiente durante 1 hora. La mezcla resultante se vertió sobre una mezcla de hielo (1,500 mililitros), agua (1,000 mililitros), y terbutil-metil-éter (TBME) (2,500 mililitros), y se agitó hasta que se fundió el hielo, y entonces a temperatura ambiente durante 1 hora. La mezcla se filtró, se lavó con agua (200 mililitros) y terbutil-metil-éter (TBME) (1,000 mililitros). Los cristales recolectados se secaron, para dar el compuesto del título como cristales color rojo-café.
LC-MS: Rt 0.53 minutos; MS m/z 252.1 [M+H]+; Método B.
iH RMN (400 MHz, DMSO-ds) 57.68-7.61 (1H, multiplete), 7.58-7.47 (2H, multipletes), 7.41 (1H, t), 3.37 (3H, s), 2.69 (3H, s).
Paso 4: 4-amino-2-(2-fluoro-fenil)-1,5-dimetil-1 H-pirazol-3(2H)-ona
La 2-(2-fluoro-fenil)-1,5-dimetil-4-nitro-1H-pirazol-3(2H)-ona (208 gramos, 828 milimoles), y Pd/C al 10 por ciento (25 gramos, 235 milimoles) en metanol (MeOH) (3000 mililitros), se sometieron a hidrogenación durante 98 horas a 0.1 bar a temperatura ambiente. La mezcla resultante se filtró sobre un cojín de Celite® (material de filtro), se lavó con metanol (MeOH) (1,000 mililitros), y el filtrado se evaporó a sequedad. Al sólido se le agregó dicloro-metano (DCM) (300 mililitros), y éste se calentó a 65 °C, seguido por la adición de tolueno (900 mililitros). El dicloro-metano (DCM) se removió bajo presión reducida, y la mezcla resultante se dejó enfriar a temperatura ambiente con agitación, y se dejó reposar a temperatura ambiente durante la noche. La mezcla se filtró, lavando con 1:1 de tolueno:heptano (200 mililitros), y heptano (800 mililitros), y se secó al vacío, para dar el compuesto del título como cristales color amarillo claro.
LC-MS: Rt 0.47 minutos; MS m/z 222.1 [M+H]+; Método MP.
iH RMN (400 MHz, DMSO-d6) 57.46-7.41 (1H, multiplete), 7.37 (3H, t), 7.35-7.27 (2H, multipletes), 2.68 (3H, s), 2.07 (3H,s).
Intermediario C:
4-amino-2-ciclohexil-1 -metil-ds,5-metil-1 H-pirazol-3(2H)-ona

Paso 1: 2-c¡clohex¡l-1-metil-d3,5-met¡l-1H-p¡razol-3(2H)-ona
La 2-ciclohexil-5-met¡l-1H-p¡razol-3(2H)-ona (Intermediario A, paso 1) (1 gramo, 5.55 milimoles) se convirtió en una pasta acuosa en N,N-dimetil-formamida (DMF) (5 mililitros), y se calentó a 40 °C bajo una atmósfera de nitrógeno. Se agregó yodo-metano-d3 (1.381 mililitros, 22.19 milimoles), se desactivó el flujo de nitrógeno, y la mezcla se calentó a 70 °C durante 16 horas. La mezcla resultante se enfrió hasta la temperatura ambiente. Se agregó dicloro-metano (DCM) (20 mililitros), seguido por cloruro de litio (al 5 por ciento en peso/volumen, 25 mililitros). Las capas resultantes se separaron y los extractos orgánicos se lavaron con salmuera (20 mililitros). Los lavados acuosos se combinaron y se volvieron a extraer con dicloro-metano (DCM) (10 mililitros, 2 veces). Los extractos orgánicos combinados se secaron sobre MgSÜ4. El desecante se filtró, y el filtrado se concentró bajo presión reducida, para dar un aceite color café. El aceite se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con el 0 al 10 por ciento de metanol (MeÜH) en diclorometano (DCM). Las fracciones del producto se combinaron y se concentraron bajo presión reducida, para dar el compuesto del título como un aceite color café.
LC-MS: Rt 0.74 minutos; MS m/z 198.4 [M+H]+; Método 2minBajopHv01.
1H RMN (400 MHz, DMSO-d6) 55.30 (1H, s), 4.02 (1H, m), 2.15 (3H, s), 1.97 (2H, m), 1.78 (2H, m), 1.66 (3H, m), 1.33 (2H, m), 1.15 (1H, m).
Paso 2: 2-ciclohex¡l-1-met¡l-ds, 5-metil-4-nitro-1 H-pirazol-3(2H)-ona
La 2-ciclohexil-1-met¡l-d3,5-met¡l-1H-p¡razol-3(2H)-ona (690 miligramos, 3.50 milimoles) se disolvió en ácido trifluoroacético (TFA) (5389 microlitros, 69.9 milimoles), y se agregó por goteo ácido nítrico al 90 por ciento (347 microlitros, 6.99 milimoles), bajo enfriamiento con hielo, manteniendo la temperatura a aproximadamente 5 °C. La mezcla se agitó a temperatura ambiente durante 30 minutos, y entonces se vertió en agua (30 mililitros), y se extrajo con dicloro-metano (DCM) (30 mililitros). Los extractos orgánicos se lavaron con salmuera, se secaron sobre MgSÜ4 , se filtraron, y se concentraron bajo presión reducida, para dar el compuesto del título.
LC-MS: Rt 0.75 minutos; MS m/z 243.3 [M+H]+; Método 2minBajopHv01.
iH RMN (400 MHz, DMSO-d6) 54.07 (1H, m), 2.59 (3H, s), 2.1 (2H, m), 1.78 (2H, m), 1.67 (3H, m), 1.33 (2H, m), 1.16 (1 H, m).
Paso 3: 4-amino-2-c¡clohex¡l-1-met¡l-ds,5-met¡l-1 H-pirazol-3(2H)-ona
A una solución de 2-c¡clohex¡l-1-met¡lo-ds,5-met¡l-4-n¡tro-1H-p¡razol-3(2H)-ona (430 miligramos, 1.775 milimoles) en etanol (5 mililitros), se le agregó cloruro de amonio (356 miligramos, 6.66 milimoles), hierro (347 miligramos, 6.21 milimoles), y agua (1.25 mililitros), seguida por HCl concentrado (0.054 mililitros, 1.775 milimoles). La mezcla se agitó a 90 °C durante 1.5 horas, y entonces se enfrió hasta la temperatura ambiente, y se agitó a temperatura ambiente durante 16 horas. La mezcla resultante se filtró, se llevó hasta un pH de 8 mediante la adición de NaÜH 1 M (acuoso), y se extrajo con dicloro-metano (DCM). Los extractos orgánicos se lavaron con salmuera, se secaron sobre MgSÜ4, se filtraron, y se concentraron bajo presión reducida. El material crudo se absorbió sobre sílice, y se purificó mediante cromatografía eluyendo con el 0 al 20 por ciento de metanol (MeÜH) en terbutil-metil-éter (TBME). Las fracciones del producto se combinaron, se concentraron bajo presión reducida, y se secaron en el horno al vacío, para dar el compuesto del título como un sólido amarillo pálido.
LC-MS: Rt 0.50 minutos; MS m/z 213.4 [M+H]+; Método 2minBajopHv01.
iH RMN (400 MHz, CDCla) 53.86 (1H, m), 2.01 (3H, s), 1.92 (2H, m), 1.83 (4H, m), 1.65 (1H, m), 1.32 (2H, m), 1.19 (1H, m).
Claims (14)
1. Un compuesto de fórmula (I):

o una sal farmacéuticamente aceptable del mismo, en donde
R1 es alquilo (C3-C 6) o cicloalquilo (C3-C 6);
R2 es metilo;
R3 es
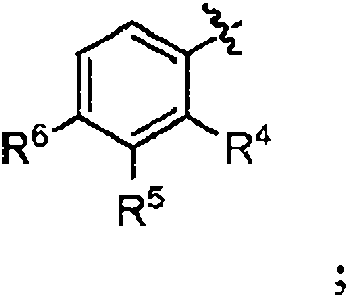
R4 y R5 se seleccionan independientemente de hidrógeno, halo, cicloalquilo (C3-C 6), alquilo (C1-C 4), haloalquilo (Ci-C4), alcoxi (C1-C 4), haloalcoxi (C1-C 4) o-alquil (C1-C 2) alcoxi (C1-C 2); o
R2 y R4 se pueden tomar junto con los átomos de carbono a los que están unidos para formar un anillo azepina y R5 es H;
R6 es halo, cicloalquilo (C3-C 6), alquilo (C1-C 4), haloalquilo (C1-C 4), alcoxi (C1-C 4), haloalcoxi (C1-C 4) o -alquil (C1-C2) alcoxi (C1-C 2);
o
R1 es 2-fluorofenilo;
R2 es metilo; y
R3 es fenilo, sustituido con uno o dos sustituyentes seleccionados independientemente entre cloro y ciclopropilo.
2. Un compuesto de acuerdo con la reivindicación 1 o una sal farmacéuticamente aceptable del mismo, donde R1 es iso-propilo, ciclobutilo o ciclohexilo.
3. Un compuesto de acuerdo con la reivindicación 1 o una sal farmacéuticamente aceptable del mismo, en donde R1 es ciclohexilo.
4. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 3 o una sal farmacéuticamente aceptable del mismo, en donde R3 es

R4 y R6 se seleccionan independientemente de cloro, fluoro, ciclopropilo, metilo, metoxi, trifluorometoxi, trifluorometilo; y
R5 es hidrógeno.
5. Un compuesto de acuerdo con una cualquiera de las reivindicaciones 1 a 4 o una sal farmacéuticamente aceptable del mismo, en donde R3 es

R4 y R6 se seleccionan independientemente de cloro y ciclopropilo; y
R5 es hidrógeno.
6. Un compuesto de acuerdo con la reivindicación 1 que se selecciona del grupo que consiste en:
1-(2-cloro-4-metoxifenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazo l-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida;
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-1-(2,4-diclorofenil)-5-metil-1 H-1, 2,3-triazol-4-carboxamida;
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-1-(4-metoxi-2-(trifluorometil)fenil)-5-metil-1 H-1.2.3- triazol-4-carboxamida;
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-1-(4-metoxi-3-metilfenil)-5-metil-1 H-1,2,3-triazol-4-carboxamida;
1-(2-cloro-4-(trifluorometoxi)fenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazol-4-il)-5-metil-1 H-1.2.3- triazol-4-carboxamida;
1 -(4 -clorofe nil)-N-(2-c iclohexi l-1,5-dimetil-3-oxo-2,3-dihidro-1 H-p i razo l-4-i l )-5-met i l-1 H-1,2,3-triazol-4-carboxamida;
1-(2,4-diclorofenil)-N-(2-(2-fluorofenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazo l-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida;
1-(4-clorofenil)-N-(2-(2-fluorofenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-pirazo l-4 - il)-5-metil-1 H-1,2,3-triazol-4-carboxamida;
1-(2-cloro-4-(trifluorometoxi)fenil)-N-(2-(2-fluorofenil)-1,5-dimetil-3-oxo-2,3-dihidro-1H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida;
1-(2-cloro-4-ciclopropilfenil)-N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-dihidro-1 H-p i razo l-4-i l )-5-met i l - 1 H-1,2,3-triazol-4-carboxamida;
1-(2-cloro-4-ciclopropilfenil)-N-(2-(2-fluorofenil)-1,5-dimetil-3-oxo-2,3-dihidro-1 H-p i razo l-4-i l )-5-met i l-1H-1.2.3- triazol-4-carboxamida;
1-(2-cloro-4-c¡cloprop¡lfen¡l)-N-(2-c¡clohex¡l-1-met¡l-d3,5-met¡l-3-oxo-2,3-dih¡dro-1 H-p i razol-4-i l )-5-m etil-1 H-1.2.3- triazol-4-carboxamida;
1-(4-cloro-2-ciclopropilfen¡l)-N-(2-(2-fluorofen¡l)-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-p i razo l-4-¡ l )-5-met i l-1H-1.2.3- triazol-4-carboxamida;
1-(4-cloro-2-ciclopropilfen¡l)-N-(2-c¡clohex¡l-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-p i razo l-4-¡ l )-5-met¡ l - 1H-1,2,3-triazol-4-carboxamida;
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-¡l)-1-(2-c¡clopropil-4-fluorofen¡l)-5-met¡l-1 H-1,2,3-triazol-4-carboxamida;
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-¡l)-8-(tr¡fluorometoxi)-5,6-d¡h¡dro-4H-benzo[f][1,2,3]triazolo[1,5-a]azepina-3-carboxamida; y
N-(2-ciclohexil-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-1-(4-metox¡fen¡l)-5-met¡l-1 H-1,2,3-triazol-4-carboxamida;
o una sus sales farmacéuticamente aceptables.
7. Un compuesto de acuerdo con la reivindicación 1 que es 1-(2-cloro-4-ciclopropil-fen¡l)-N-(2-c¡clohex¡l-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida que tiene la siguiente fórmula:

o una sus sales farmacéuticamente aceptables.
8. Un compuesto de acuerdo con la reivindicación 1 que es 1-(2-cloro-4-metoxifenil)-N-(2-c¡clohex¡l-1,5-dimet¡l-3-oxo-2,3-d¡h¡dro-1H-p¡razol-4-¡l)-5-met¡l-1H-1,2,3-tr¡azol-4-carboxam¡da que tiene la siguiente fórmula:

o una sus sales farmacéuticamente aceptables.
9. Un compuesto de acuerdo con la reivindicación 1 que es 1-(4-cloro-2-ciclopropil-fen¡l)-N-(2-c¡clohex¡l-1,5-dimetil-3-oxo-2,3-d¡h¡dro-1 H-pirazol-4-il)-5-metil-1 H-1,2,3-triazol-4-carboxamida que tiene la siguiente fórmula:

o una sus sales farmacéuticamente aceptables.
10. Una composición farmacéutica que comprende una cantidad terapéuticamente eficaz de un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 9 o una sal farmacéuticamente aceptable de los mismos y uno o más portadores farmacéuticamente aceptables.
11. Una combinación que comprende una cantidad terapéuticamente eficaz de un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 9 o una sal farmacéuticamente aceptable de los mismos y uno o más agentes terapéuticamente activos.
12. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 9 o una sal farmacéuticamente aceptable del mismo, para usar como un medicamento.
13. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 9 o una sal farmacéuticamente aceptable del mismo, para usar en el tratamiento de un trastorno o enfermedad seleccionada de la hipertensión pulmonar, incluyendo la hipertensión pulmonar arterial (PAH), fibrosis, artritis reumatoide, y el sanado de fracturas.
14. Un compuesto de acuerdo con cualquiera de las reivindicaciones 1 a 9 o una sal farmacéuticamente aceptable del mismo, para usar en el tratamiento de un trastorno o enfermedad seleccionada del glaucoma, telangiectasia hemorrágica hereditaria (HHT), proteinuria, cicatrización de heridas, EPOC y asma.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201461993046P | 2014-05-14 | 2014-05-14 | |
| EP14168319 | 2014-05-14 | ||
| PCT/IB2015/001539 WO2015177646A1 (en) | 2014-05-14 | 2015-05-14 | Carboxamide derivatives |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2707734T3 true ES2707734T3 (es) | 2019-04-04 |
Family
ID=57907515
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15774680T Active ES2707734T3 (es) | 2014-05-14 | 2015-05-14 | Derivados de carboxamida |
Country Status (4)
| Country | Link |
|---|---|
| CU (1) | CU20160170A7 (es) |
| ES (1) | ES2707734T3 (es) |
| IL (1) | IL248801A0 (es) |
| PH (1) | PH12016502247A1 (es) |
-
2015
- 2015-05-14 ES ES15774680T patent/ES2707734T3/es active Active
-
2016
- 2016-11-07 IL IL248801A patent/IL248801A0/en unknown
- 2016-11-11 PH PH12016502247A patent/PH12016502247A1/en unknown
- 2016-11-11 CU CUP2016000170A patent/CU20160170A7/es unknown
Also Published As
| Publication number | Publication date |
|---|---|
| CU20160170A7 (es) | 2017-02-02 |
| IL248801A0 (en) | 2017-01-31 |
| PH12016502247A1 (en) | 2017-02-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9931319B2 (en) | Carboxamide derivatives | |
| JP6535022B2 (ja) | ヘテロアリールsyk阻害剤 | |
| ES2883938T3 (es) | Inhibidores de molécula pequeña de lactato deshidrogenasa y métodos de utilización de los mismos | |
| EP1827444B1 (en) | Mnk1 or mnk2 inhibitors | |
| JP6630671B2 (ja) | Nrf2レギュレーター | |
| ES2790633T3 (es) | Benzooxazepinonas fusionadas como moduladores de canal de iones | |
| ES2745284T3 (es) | Heteroarilos sustituidos con pirazolilo y su uso como medicamentos | |
| ES2874882T3 (es) | Derivados de carboxamida | |
| PT2215092E (pt) | Piperidino-di-hidrotienopirimidinas substituídas | |
| BR112020018983A2 (pt) | Inibidores de canal receptor de potencial transitório de oxadiazol | |
| WO2008157740A2 (en) | Faah inhibitors | |
| PT97776A (pt) | Processo para a preparacao de derivados de azol e de composicoes farmaceuticas que os contem | |
| WO2014091446A1 (en) | Pyrimido [4,5-b]quinoline-4,5 (3h,10h)-diones as nonsense mutation suppressors | |
| WO2014127331A1 (en) | Novel uses | |
| ES2640638T3 (es) | Compuestos de fenil-hexahidropirano[3,4-d][1,3]tiazin-2-amina sustituidos | |
| ES2731802T3 (es) | Derivados de naftiridinodiona como supresores de mutaciones sin sentido | |
| ES2707734T3 (es) | Derivados de carboxamida | |
| WO2015173659A2 (en) | Carboxamide derivatives | |
| KR20140037195A (ko) | 신규 프탈라지논-피롤로피리미딘카복스아미드 유도체 | |
| PT2240487E (pt) | Triazolopiridazinas como inibidores do par1, sua preparação e utilização como produtos farmacêuticos | |
| BR102016020905A2 (pt) | Heteroaris pirazolil-substituídos e seu uso como medicamentos | |
| CA3226225A1 (en) | Therapeutic compounds and methods | |
| WO2015173656A1 (en) | Carboxamide derivatives |