ES2703851T3 - Indazoles - Google Patents
Indazoles Download PDFInfo
- Publication number
- ES2703851T3 ES2703851T3 ES15744882T ES15744882T ES2703851T3 ES 2703851 T3 ES2703851 T3 ES 2703851T3 ES 15744882 T ES15744882 T ES 15744882T ES 15744882 T ES15744882 T ES 15744882T ES 2703851 T3 ES2703851 T3 ES 2703851T3
- Authority
- ES
- Spain
- Prior art keywords
- phenyl
- methanone
- methyl
- pyrrolidin
- pyrazolo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000002473 indoazoles Chemical class 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 134
- 125000004429 atom Chemical group 0.000 claims abstract description 35
- 125000004432 carbon atom Chemical group C* 0.000 claims abstract description 28
- 125000004433 nitrogen atom Chemical group N* 0.000 claims abstract description 24
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 17
- 125000003118 aryl group Chemical group 0.000 claims abstract description 14
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 14
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 13
- 229910052794 bromium Inorganic materials 0.000 claims abstract description 9
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims abstract description 9
- 229910052740 iodine Inorganic materials 0.000 claims abstract description 9
- 125000000217 alkyl group Chemical group 0.000 claims abstract description 8
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 8
- 229920006395 saturated elastomer Polymers 0.000 claims abstract description 8
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims abstract description 7
- 125000000753 cycloalkyl group Chemical group 0.000 claims abstract description 7
- 125000001931 aliphatic group Chemical group 0.000 claims abstract description 5
- 125000006367 bivalent amino carbonyl group Chemical group [H]N([*:1])C([*:2])=O 0.000 claims abstract description 3
- 125000004430 oxygen atom Chemical group O* 0.000 claims abstract description 3
- 239000000203 mixture Substances 0.000 claims description 112
- -1 / -propyl Chemical group 0.000 claims description 99
- 238000000034 method Methods 0.000 claims description 84
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 37
- 150000003839 salts Chemical class 0.000 claims description 34
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 22
- 206010028980 Neoplasm Diseases 0.000 claims description 18
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 claims description 17
- 201000011510 cancer Diseases 0.000 claims description 14
- 239000008194 pharmaceutical composition Substances 0.000 claims description 14
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 13
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 claims description 12
- 238000002360 preparation method Methods 0.000 claims description 12
- 238000011282 treatment Methods 0.000 claims description 12
- 239000003795 chemical substances by application Substances 0.000 claims description 11
- 239000004480 active ingredient Substances 0.000 claims description 10
- FRZVFHFLYODQCE-SFHVURJKSA-N BrC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)C Chemical compound BrC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)C FRZVFHFLYODQCE-SFHVURJKSA-N 0.000 claims description 8
- KMCPIJPJZCDCMY-FQEVSTJZSA-N BrC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CCC Chemical compound BrC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CCC KMCPIJPJZCDCMY-FQEVSTJZSA-N 0.000 claims description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 8
- 239000003814 drug Substances 0.000 claims description 8
- 230000003463 hyperproliferative effect Effects 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 8
- JJIQBYGSBIHLSR-INIZCTEOSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C1=CC=C2C(=N1)C(=NN2)C Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C1=CC=C2C(=N1)C(=NN2)C JJIQBYGSBIHLSR-INIZCTEOSA-N 0.000 claims description 6
- VMSOGMBHDNROTJ-NRFANRHFSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CC(C)C Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CC(C)C VMSOGMBHDNROTJ-NRFANRHFSA-N 0.000 claims description 6
- CVZAKPYVWFEYFW-SFHVURJKSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1F)C Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1F)C CVZAKPYVWFEYFW-SFHVURJKSA-N 0.000 claims description 6
- MONHZYRUJIRNID-AWEZNQCLSA-N NC1=NNC2=NC=C(C=C21)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Br Chemical compound NC1=NNC2=NC=C(C=C21)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Br MONHZYRUJIRNID-AWEZNQCLSA-N 0.000 claims description 6
- 201000010099 disease Diseases 0.000 claims description 6
- 230000002757 inflammatory effect Effects 0.000 claims description 6
- 201000001441 melanoma Diseases 0.000 claims description 6
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 5
- QTXWFJYVYZUQAX-UHFFFAOYSA-N ClC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C1CC1 Chemical compound ClC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C1CC1 QTXWFJYVYZUQAX-UHFFFAOYSA-N 0.000 claims description 5
- ATELZSLYYPHJDP-INIZCTEOSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)F Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)F ATELZSLYYPHJDP-INIZCTEOSA-N 0.000 claims description 5
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 5
- PAUXJRUOCRBJHR-INIZCTEOSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Cl Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Cl PAUXJRUOCRBJHR-INIZCTEOSA-N 0.000 claims description 5
- 210000000481 breast Anatomy 0.000 claims description 5
- 210000001072 colon Anatomy 0.000 claims description 5
- 229940079593 drug Drugs 0.000 claims description 5
- 229910052731 fluorine Inorganic materials 0.000 claims description 5
- 208000027866 inflammatory disease Diseases 0.000 claims description 5
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 5
- KMCPIJPJZCDCMY-UHFFFAOYSA-N BrC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CCC Chemical compound BrC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CCC KMCPIJPJZCDCMY-UHFFFAOYSA-N 0.000 claims description 4
- YFRGYWOOWLDSGP-UHFFFAOYSA-N ClC1=C(C=C(C=C1)C1N(CCC1)C(=O)C1=CC=C2C(=N1)C(=NN2)C)F Chemical compound ClC1=C(C=C(C=C1)C1N(CCC1)C(=O)C1=CC=C2C(=N1)C(=NN2)C)F YFRGYWOOWLDSGP-UHFFFAOYSA-N 0.000 claims description 4
- QLSTWYIPABBKKG-UHFFFAOYSA-N ClC1=CC=C(C=C1)C1N(CCN(C1)C)C(=O)C=1C=C2C(=NNC2=CC=1)C Chemical compound ClC1=CC=C(C=C1)C1N(CCN(C1)C)C(=O)C=1C=C2C(=NNC2=CC=1)C QLSTWYIPABBKKG-UHFFFAOYSA-N 0.000 claims description 4
- BCXLYZDDXDFXDU-JKSUJKDBSA-N ClC1=CC=C(C=C1)[C@@H]1CN(C[C@H]1O)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC1=CC=C(C=C1)[C@@H]1CN(C[C@H]1O)C(=O)C=1C=C2C(=NC=1)NN=C2C BCXLYZDDXDFXDU-JKSUJKDBSA-N 0.000 claims description 4
- GHDVGYOWJVANGE-HNNXBMFYSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2OC Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2OC GHDVGYOWJVANGE-HNNXBMFYSA-N 0.000 claims description 4
- BKZFMJSHBSIEIJ-KRWDZBQOSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)SC Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)SC BKZFMJSHBSIEIJ-KRWDZBQOSA-N 0.000 claims description 4
- PAUXJRUOCRBJHR-UHFFFAOYSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCC1)C1=CC=C(C=C1)Cl Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCC1)C1=CC=C(C=C1)Cl PAUXJRUOCRBJHR-UHFFFAOYSA-N 0.000 claims description 4
- 230000003213 activating effect Effects 0.000 claims description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 4
- 210000003734 kidney Anatomy 0.000 claims description 4
- 210000004072 lung Anatomy 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 4
- 230000008569 process Effects 0.000 claims description 4
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 3
- ZFWBUPMSFPAUMM-INIZCTEOSA-N CC1=NNC=2C1=NC(=CC=2)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)C(F)(F)F Chemical compound CC1=NNC=2C1=NC(=CC=2)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)C(F)(F)F ZFWBUPMSFPAUMM-INIZCTEOSA-N 0.000 claims description 3
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 3
- ODRITQGYYWHQGM-UHFFFAOYSA-N ClC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C ODRITQGYYWHQGM-UHFFFAOYSA-N 0.000 claims description 3
- BXPXFPCETCYHHL-UHFFFAOYSA-N ClC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C(F)(F)F Chemical compound ClC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C(F)(F)F BXPXFPCETCYHHL-UHFFFAOYSA-N 0.000 claims description 3
- FLEYJCJMMRLIBV-KRWDZBQOSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=CN=1)NN=C2C Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=CN=1)NN=C2C FLEYJCJMMRLIBV-KRWDZBQOSA-N 0.000 claims description 3
- 208000007766 Kaposi sarcoma Diseases 0.000 claims description 3
- 208000034578 Multiple myelomas Diseases 0.000 claims description 3
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 3
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 3
- 208000024770 Thyroid neoplasm Diseases 0.000 claims description 3
- 210000004556 brain Anatomy 0.000 claims description 3
- 230000002496 gastric effect Effects 0.000 claims description 3
- 210000004185 liver Anatomy 0.000 claims description 3
- 230000002611 ovarian Effects 0.000 claims description 3
- 210000002307 prostate Anatomy 0.000 claims description 3
- 230000002381 testicular Effects 0.000 claims description 3
- 201000002510 thyroid cancer Diseases 0.000 claims description 3
- KLSJWNVTNUYHDU-UHFFFAOYSA-N Amitrole Chemical compound NC1=NC=NN1 KLSJWNVTNUYHDU-UHFFFAOYSA-N 0.000 claims description 2
- VZPGERSMGVRRCU-UHFFFAOYSA-N ClC1=CC=C(C=C1)C1N(CCOC1)C(=O)C=1C=C2C(=NNC2=CC=1)C Chemical compound ClC1=CC=C(C=C1)C1N(CCOC1)C(=O)C=1C=C2C(=NNC2=CC=1)C VZPGERSMGVRRCU-UHFFFAOYSA-N 0.000 claims description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 2
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 claims description 2
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 claims description 2
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 claims description 2
- 125000004199 4-trifluoromethylphenyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1*)C(F)(F)F 0.000 claims 3
- VSIKBMSMNYEKJE-UHFFFAOYSA-N (3-amino-1H-indazol-5-yl)-[3-(4-chlorophenyl)pyrrolidin-1-yl]methanone Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1CC(CC1)C1=CC=C(C=C1)Cl VSIKBMSMNYEKJE-UHFFFAOYSA-N 0.000 claims 1
- YARWEXGAKZFIGU-INIZCTEOSA-N (3-methyl-2H-pyrazolo[3,4-b]pyridin-5-yl)-[(2S)-2-[4-(trifluoromethyl)phenyl]pyrrolidin-1-yl]methanone Chemical compound CC1=NNC2=NC=C(C=C21)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)C(F)(F)F YARWEXGAKZFIGU-INIZCTEOSA-N 0.000 claims 1
- YARWEXGAKZFIGU-UHFFFAOYSA-N (3-methyl-2H-pyrazolo[3,4-b]pyridin-5-yl)-[2-[4-(trifluoromethyl)phenyl]pyrrolidin-1-yl]methanone Chemical compound CC1=NNC2=NC=C(C=C21)C(=O)N1C(CCC1)C1=CC=C(C=C1)C(F)(F)F YARWEXGAKZFIGU-UHFFFAOYSA-N 0.000 claims 1
- 206010000830 Acute leukaemia Diseases 0.000 claims 1
- LIOIIZNXBDBOIN-UHFFFAOYSA-N BrC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound BrC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C LIOIIZNXBDBOIN-UHFFFAOYSA-N 0.000 claims 1
- IHDYOFIEDGOEQN-UHFFFAOYSA-N BrC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C=NNC2=CC=1 Chemical compound BrC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C=NNC2=CC=1 IHDYOFIEDGOEQN-UHFFFAOYSA-N 0.000 claims 1
- JPZJYEGCEUVLHB-INIZCTEOSA-N BrC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C1=CC=C2C(=N1)C(=NN2)C Chemical compound BrC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C1=CC=C2C(=N1)C(=NN2)C JPZJYEGCEUVLHB-INIZCTEOSA-N 0.000 claims 1
- LIOIIZNXBDBOIN-INIZCTEOSA-N BrC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound BrC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C LIOIIZNXBDBOIN-INIZCTEOSA-N 0.000 claims 1
- IHDYOFIEDGOEQN-KRWDZBQOSA-N BrC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C=NNC2=CC=1 Chemical compound BrC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C=NNC2=CC=1 IHDYOFIEDGOEQN-KRWDZBQOSA-N 0.000 claims 1
- XXIISIAMUYKNPZ-UHFFFAOYSA-N CC1=NNC2=NC=C(C=C21)C(=O)N1C(CCC1)C1=CC=C(C=C1)C Chemical compound CC1=NNC2=NC=C(C=C21)C(=O)N1C(CCC1)C1=CC=C(C=C1)C XXIISIAMUYKNPZ-UHFFFAOYSA-N 0.000 claims 1
- YDXXNUGJCKRMGL-UHFFFAOYSA-N CC1=NNC2=NC=C(C=C21)C(=O)N1C(CCC1)C1=CC=CC=C1 Chemical compound CC1=NNC2=NC=C(C=C21)C(=O)N1C(CCC1)C1=CC=CC=C1 YDXXNUGJCKRMGL-UHFFFAOYSA-N 0.000 claims 1
- LQLNUJNDEOQFCG-UHFFFAOYSA-N CC1=NNC2=NC=C(C=C21)C(=O)N1C(CCCC1)C1=CC=CC=C1 Chemical compound CC1=NNC2=NC=C(C=C21)C(=O)N1C(CCCC1)C1=CC=CC=C1 LQLNUJNDEOQFCG-UHFFFAOYSA-N 0.000 claims 1
- ZUBYHCUPXICHOR-UHFFFAOYSA-N CC1=NNC2=NC=C(C=C21)C(=O)N1CC(CC1)C1=CC=CC=C1 Chemical compound CC1=NNC2=NC=C(C=C21)C(=O)N1CC(CC1)C1=CC=CC=C1 ZUBYHCUPXICHOR-UHFFFAOYSA-N 0.000 claims 1
- ZUBYHCUPXICHOR-CQSZACIVSA-N CC1=NNC2=NC=C(C=C21)C(=O)N1C[C@@H](CC1)C1=CC=CC=C1 Chemical compound CC1=NNC2=NC=C(C=C21)C(=O)N1C[C@@H](CC1)C1=CC=CC=C1 ZUBYHCUPXICHOR-CQSZACIVSA-N 0.000 claims 1
- XXIISIAMUYKNPZ-KRWDZBQOSA-N CC1=NNC2=NC=C(C=C21)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)C Chemical compound CC1=NNC2=NC=C(C=C21)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)C XXIISIAMUYKNPZ-KRWDZBQOSA-N 0.000 claims 1
- YDXXNUGJCKRMGL-INIZCTEOSA-N CC1=NNC2=NC=C(C=C21)C(=O)N1[C@@H](CCC1)C1=CC=CC=C1 Chemical compound CC1=NNC2=NC=C(C=C21)C(=O)N1[C@@H](CCC1)C1=CC=CC=C1 YDXXNUGJCKRMGL-INIZCTEOSA-N 0.000 claims 1
- LQLNUJNDEOQFCG-KRWDZBQOSA-N CC1=NNC2=NC=C(C=C21)C(=O)N1[C@@H](CCCC1)C1=CC=CC=C1 Chemical compound CC1=NNC2=NC=C(C=C21)C(=O)N1[C@@H](CCCC1)C1=CC=CC=C1 LQLNUJNDEOQFCG-KRWDZBQOSA-N 0.000 claims 1
- GQUJOPPZESFRMO-INIZCTEOSA-N COC1=C(C=CC=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound COC1=C(C=CC=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C GQUJOPPZESFRMO-INIZCTEOSA-N 0.000 claims 1
- ULTCWZLKVMRIRC-KRWDZBQOSA-N COC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound COC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C ULTCWZLKVMRIRC-KRWDZBQOSA-N 0.000 claims 1
- XMDMIQFMXUZYSH-UHFFFAOYSA-N ClC1=C(C=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C)F Chemical compound ClC1=C(C=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C)F XMDMIQFMXUZYSH-UHFFFAOYSA-N 0.000 claims 1
- XMDMIQFMXUZYSH-INIZCTEOSA-N ClC1=C(C=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C)F Chemical compound ClC1=C(C=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C)F XMDMIQFMXUZYSH-INIZCTEOSA-N 0.000 claims 1
- GDHUJMZVXRCWBQ-UHFFFAOYSA-N ClC1=C(C=CC=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC1=C(C=CC=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C GDHUJMZVXRCWBQ-UHFFFAOYSA-N 0.000 claims 1
- GDHUJMZVXRCWBQ-INIZCTEOSA-N ClC1=C(C=CC=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC1=C(C=CC=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C GDHUJMZVXRCWBQ-INIZCTEOSA-N 0.000 claims 1
- FPYXHNVSYBMHBN-INIZCTEOSA-N ClC1=CC(=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C)F Chemical compound ClC1=CC(=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C)F FPYXHNVSYBMHBN-INIZCTEOSA-N 0.000 claims 1
- VDLBYRKEWHDMNC-UHFFFAOYSA-N ClC1=CC=C(C=C1)C1CN(CC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC1=CC=C(C=C1)C1CN(CC1)C(=O)C=1C=C2C(=NC=1)NN=C2C VDLBYRKEWHDMNC-UHFFFAOYSA-N 0.000 claims 1
- JTDVBPIICLGREC-UHFFFAOYSA-N ClC1=CC=C(C=C1)C1N(CC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC1=CC=C(C=C1)C1N(CC1)C(=O)C=1C=C2C(=NC=1)NN=C2C JTDVBPIICLGREC-UHFFFAOYSA-N 0.000 claims 1
- PJFOQIYHWXOOHR-UHFFFAOYSA-N ClC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CCC Chemical compound ClC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CCC PJFOQIYHWXOOHR-UHFFFAOYSA-N 0.000 claims 1
- JTDVBPIICLGREC-HNNXBMFYSA-N ClC1=CC=C(C=C1)[C@H]1N(CC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CC1)C(=O)C=1C=C2C(=NC=1)NN=C2C JTDVBPIICLGREC-HNNXBMFYSA-N 0.000 claims 1
- ODRITQGYYWHQGM-INIZCTEOSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C ODRITQGYYWHQGM-INIZCTEOSA-N 0.000 claims 1
- BXPXFPCETCYHHL-AWEZNQCLSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C(F)(F)F Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C(F)(F)F BXPXFPCETCYHHL-AWEZNQCLSA-N 0.000 claims 1
- QTXWFJYVYZUQAX-KRWDZBQOSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C1CC1 Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C1CC1 QTXWFJYVYZUQAX-KRWDZBQOSA-N 0.000 claims 1
- PJFOQIYHWXOOHR-FQEVSTJZSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CCC Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CCC PJFOQIYHWXOOHR-FQEVSTJZSA-N 0.000 claims 1
- CUQBBHUITNFLKO-KRWDZBQOSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)NC Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)NC CUQBBHUITNFLKO-KRWDZBQOSA-N 0.000 claims 1
- GONWDEVAERSSRA-UHFFFAOYSA-N ClC1=NNC2=CC=C(C=C12)C(=O)N1CC(CC1)C1=CC=C(C=C1)Cl Chemical compound ClC1=NNC2=CC=C(C=C12)C(=O)N1CC(CC1)C1=CC=C(C=C1)Cl GONWDEVAERSSRA-UHFFFAOYSA-N 0.000 claims 1
- BSHIEDSBWFUQKU-INIZCTEOSA-N ClC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Cl Chemical compound ClC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Cl BSHIEDSBWFUQKU-INIZCTEOSA-N 0.000 claims 1
- VDABZMQOFGGGFG-UHFFFAOYSA-N ClC=1C=C(C=CC=1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC=1C=C(C=CC=1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C VDABZMQOFGGGFG-UHFFFAOYSA-N 0.000 claims 1
- VDABZMQOFGGGFG-INIZCTEOSA-N ClC=1C=C(C=CC=1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC=1C=C(C=CC=1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C VDABZMQOFGGGFG-INIZCTEOSA-N 0.000 claims 1
- HYZZDAUMDQGNIM-UHFFFAOYSA-N FC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound FC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C HYZZDAUMDQGNIM-UHFFFAOYSA-N 0.000 claims 1
- WFEXSSOVFFHIHV-UHFFFAOYSA-N FC1=CC=C(C=C1)C1N(CCCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound FC1=CC=C(C=C1)C1N(CCCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C WFEXSSOVFFHIHV-UHFFFAOYSA-N 0.000 claims 1
- ZAUVINNJTQBQLS-HNNXBMFYSA-N FC1=CC=C(C=C1)[C@H]1N(CC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound FC1=CC=C(C=C1)[C@H]1N(CC1)C(=O)C=1C=C2C(=NC=1)NN=C2C ZAUVINNJTQBQLS-HNNXBMFYSA-N 0.000 claims 1
- HSVXCKKYKUBIOX-INIZCTEOSA-N FC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C1=CC=C2C(=N1)C(=NN2)C Chemical compound FC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C1=CC=C2C(=N1)C(=NN2)C HSVXCKKYKUBIOX-INIZCTEOSA-N 0.000 claims 1
- WFEXSSOVFFHIHV-KRWDZBQOSA-N FC1=CC=C(C=C1)[C@H]1N(CCCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound FC1=CC=C(C=C1)[C@H]1N(CCCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C WFEXSSOVFFHIHV-KRWDZBQOSA-N 0.000 claims 1
- LNRSURRWPWPDMK-UHFFFAOYSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1C(CC1)C1=CC=C(C=C1)Cl Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1C(CC1)C1=CC=C(C=C1)Cl LNRSURRWPWPDMK-UHFFFAOYSA-N 0.000 claims 1
- GHYABPDMZQFTSA-UHFFFAOYSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1C(CC1)C1=CC=C(C=C1)F Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1C(CC1)C1=CC=C(C=C1)F GHYABPDMZQFTSA-UHFFFAOYSA-N 0.000 claims 1
- PEUOHMPBTLQKMK-UHFFFAOYSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCC1)C1=CC=C(C=C1)Br Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCC1)C1=CC=C(C=C1)Br PEUOHMPBTLQKMK-UHFFFAOYSA-N 0.000 claims 1
- ZWGIAHTXMSBBML-UHFFFAOYSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCC1)C1=CC=C(C=C1)C(C)C Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCC1)C1=CC=C(C=C1)C(C)C ZWGIAHTXMSBBML-UHFFFAOYSA-N 0.000 claims 1
- ZOKLUCHKTUZIJY-UHFFFAOYSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCC1)C1=CC=C(C=C1)F Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCC1)C1=CC=C(C=C1)F ZOKLUCHKTUZIJY-UHFFFAOYSA-N 0.000 claims 1
- HCNADAUGSSOTGC-UHFFFAOYSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCC1)C1=CC=CC=C1 Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCC1)C1=CC=CC=C1 HCNADAUGSSOTGC-UHFFFAOYSA-N 0.000 claims 1
- RPAQNJIGNBXHSR-UHFFFAOYSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCCC1)C1=CC=CC=C1 Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1C(CCCC1)C1=CC=CC=C1 RPAQNJIGNBXHSR-UHFFFAOYSA-N 0.000 claims 1
- GHYABPDMZQFTSA-HNNXBMFYSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CC1)C1=CC=C(C=C1)F Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CC1)C1=CC=C(C=C1)F GHYABPDMZQFTSA-HNNXBMFYSA-N 0.000 claims 1
- PEUOHMPBTLQKMK-INIZCTEOSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Br Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Br PEUOHMPBTLQKMK-INIZCTEOSA-N 0.000 claims 1
- ZWGIAHTXMSBBML-IBGZPJMESA-N NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)C(C)C Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)C(C)C ZWGIAHTXMSBBML-IBGZPJMESA-N 0.000 claims 1
- XIRFWQBNOWOYCK-INIZCTEOSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)C(F)(F)F Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)C(F)(F)F XIRFWQBNOWOYCK-INIZCTEOSA-N 0.000 claims 1
- ZOKLUCHKTUZIJY-INIZCTEOSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)F Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)F ZOKLUCHKTUZIJY-INIZCTEOSA-N 0.000 claims 1
- VXYMGUGNDZARSZ-KRWDZBQOSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCCC1)C1=CC=C(C=C1)F Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCCC1)C1=CC=C(C=C1)F VXYMGUGNDZARSZ-KRWDZBQOSA-N 0.000 claims 1
- RPAQNJIGNBXHSR-KRWDZBQOSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCCC1)C1=CC=CC=C1 Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCCC1)C1=CC=CC=C1 RPAQNJIGNBXHSR-KRWDZBQOSA-N 0.000 claims 1
- MGZYAAMHPOFEEJ-INIZCTEOSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCCC1)CC1=CC=CC=C1 Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1[C@@H](CCCC1)CC1=CC=CC=C1 MGZYAAMHPOFEEJ-INIZCTEOSA-N 0.000 claims 1
- RPAQNJIGNBXHSR-QGZVFWFLSA-N NC1=NNC2=CC=C(C=C12)C(=O)N1[C@H](CCCC1)C1=CC=CC=C1 Chemical compound NC1=NNC2=CC=C(C=C12)C(=O)N1[C@H](CCCC1)C1=CC=CC=C1 RPAQNJIGNBXHSR-QGZVFWFLSA-N 0.000 claims 1
- RDAKOBUFMLFFML-AWEZNQCLSA-N NC1=NNC2=NC=C(C=C21)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Cl Chemical compound NC1=NNC2=NC=C(C=C21)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Cl RDAKOBUFMLFFML-AWEZNQCLSA-N 0.000 claims 1
- QXNLDTWVURNIRW-KRWDZBQOSA-N [(2S)-2-(4-chlorophenyl)azetidin-1-yl]-(3-methyl-2H-indazol-5-yl)methanone Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CC1)C(=O)C=1C=C2C(=NNC2=CC=1)C QXNLDTWVURNIRW-KRWDZBQOSA-N 0.000 claims 1
- QJIOPOJQZXHHEW-SFHVURJKSA-N [(2S)-2-(4-chlorophenyl)pyrrolidin-1-yl]-(3-methyl-2H-indazol-5-yl)methanone Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)C QJIOPOJQZXHHEW-SFHVURJKSA-N 0.000 claims 1
- HYZZDAUMDQGNIM-INIZCTEOSA-N [(2S)-2-(4-fluorophenyl)pyrrolidin-1-yl]-(3-methyl-2H-pyrazolo[3,4-b]pyridin-5-yl)methanone Chemical compound FC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C HYZZDAUMDQGNIM-INIZCTEOSA-N 0.000 claims 1
- 210000003128 head Anatomy 0.000 claims 1
- 210000003739 neck Anatomy 0.000 claims 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 162
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 135
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 132
- 239000000243 solution Substances 0.000 description 102
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 83
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 78
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 75
- 239000007787 solid Substances 0.000 description 64
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 57
- 239000002904 solvent Substances 0.000 description 41
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 33
- 239000011541 reaction mixture Substances 0.000 description 33
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 29
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 28
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 27
- 239000012044 organic layer Substances 0.000 description 27
- 238000004128 high performance liquid chromatography Methods 0.000 description 24
- 238000001914 filtration Methods 0.000 description 22
- 239000000741 silica gel Substances 0.000 description 22
- 229910002027 silica gel Inorganic materials 0.000 description 22
- 102100024456 Cyclin-dependent kinase 8 Human genes 0.000 description 21
- 101000980937 Homo sapiens Cyclin-dependent kinase 8 Proteins 0.000 description 21
- 238000005481 NMR spectroscopy Methods 0.000 description 21
- 235000019441 ethanol Nutrition 0.000 description 21
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 21
- 239000013078 crystal Substances 0.000 description 20
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 20
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 18
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 18
- 239000012043 crude product Substances 0.000 description 18
- 239000003921 oil Substances 0.000 description 18
- 235000019198 oils Nutrition 0.000 description 18
- 238000006243 chemical reaction Methods 0.000 description 17
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 17
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 17
- 238000002953 preparative HPLC Methods 0.000 description 17
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 16
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 14
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 13
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 12
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 11
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 11
- 239000007821 HATU Substances 0.000 description 11
- 239000003208 petroleum Substances 0.000 description 11
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 10
- ODRITQGYYWHQGM-MRXNPFEDSA-N ClC1=CC=C(C=C1)[C@@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC1=CC=C(C=C1)[C@@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C ODRITQGYYWHQGM-MRXNPFEDSA-N 0.000 description 10
- 241000124008 Mammalia Species 0.000 description 10
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 10
- 235000019253 formic acid Nutrition 0.000 description 10
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 10
- 239000012071 phase Substances 0.000 description 10
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 10
- 239000002244 precipitate Substances 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 229910052721 tungsten Inorganic materials 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 9
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 9
- 210000004027 cell Anatomy 0.000 description 9
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 9
- 239000012453 solvate Substances 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 239000002253 acid Substances 0.000 description 8
- 239000012298 atmosphere Substances 0.000 description 8
- 239000000460 chlorine Substances 0.000 description 8
- 238000003818 flash chromatography Methods 0.000 description 8
- 239000010410 layer Substances 0.000 description 8
- 239000007788 liquid Substances 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L sodium carbonate Substances [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 7
- 238000004440 column chromatography Methods 0.000 description 7
- 238000001816 cooling Methods 0.000 description 7
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 7
- PJUPKRYGDFTMTM-UHFFFAOYSA-N 1-hydroxybenzotriazole;hydrate Chemical compound O.C1=CC=C2N(O)N=NC2=C1 PJUPKRYGDFTMTM-UHFFFAOYSA-N 0.000 description 6
- CIHHGGKKRPPWSU-UHFFFAOYSA-N 2-(4-chlorophenyl)pyrrolidine Chemical compound C1=CC(Cl)=CC=C1C1NCCC1 CIHHGGKKRPPWSU-UHFFFAOYSA-N 0.000 description 6
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- VMSOGMBHDNROTJ-OAQYLSRUSA-N ClC1=CC=C(C=C1)[C@@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CC(C)C Chemical compound ClC1=CC=C(C=C1)[C@@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)CC(C)C VMSOGMBHDNROTJ-OAQYLSRUSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- 102000013814 Wnt Human genes 0.000 description 6
- 108050003627 Wnt Proteins 0.000 description 6
- 150000007513 acids Chemical class 0.000 description 6
- 150000001412 amines Chemical class 0.000 description 6
- 238000004296 chiral HPLC Methods 0.000 description 6
- 208000029742 colonic neoplasm Diseases 0.000 description 6
- 238000001514 detection method Methods 0.000 description 6
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 6
- 238000010828 elution Methods 0.000 description 6
- 239000003112 inhibitor Substances 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 230000037361 pathway Effects 0.000 description 6
- 108090000623 proteins and genes Proteins 0.000 description 6
- 238000000926 separation method Methods 0.000 description 6
- 229910052938 sodium sulfate Inorganic materials 0.000 description 6
- 235000011152 sodium sulphate Nutrition 0.000 description 6
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 6
- QHQLWLFLPDYQHI-UHFFFAOYSA-N 3-methyl-1-tritylpyrazolo[4,3-b]pyridine Chemical compound C12=CC=CN=C2C(C)=NN1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 QHQLWLFLPDYQHI-UHFFFAOYSA-N 0.000 description 5
- BGNZTRUIXUSPDY-UHFFFAOYSA-N 3-methyl-2h-indazole-5-carboxylic acid Chemical compound C1=C(C(O)=O)C=C2C(C)=NNC2=C1 BGNZTRUIXUSPDY-UHFFFAOYSA-N 0.000 description 5
- JZKKDADXPOJWMB-UHFFFAOYSA-N 3-methyl-2h-pyrazolo[4,3-b]pyridine-5-carboxylic acid Chemical compound C1=CC(C(O)=O)=NC2=C(C)NN=C21 JZKKDADXPOJWMB-UHFFFAOYSA-N 0.000 description 5
- KMCPIJPJZCDCMY-HXUWFJFHSA-N CCCC1=NNC2=C1C=C(C=C2)C(=O)N1CCC[C@@H]1C1=CC=C(Br)C=C1 Chemical compound CCCC1=NNC2=C1C=C(C=C2)C(=O)N1CCC[C@@H]1C1=CC=C(Br)C=C1 KMCPIJPJZCDCMY-HXUWFJFHSA-N 0.000 description 5
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 5
- 230000002159 abnormal effect Effects 0.000 description 5
- 238000003556 assay Methods 0.000 description 5
- 239000003480 eluent Substances 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 239000000706 filtrate Substances 0.000 description 5
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- 239000012299 nitrogen atmosphere Substances 0.000 description 5
- 229940002612 prodrug Drugs 0.000 description 5
- 239000000651 prodrug Substances 0.000 description 5
- 239000011734 sodium Substances 0.000 description 5
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 5
- 235000017557 sodium bicarbonate Nutrition 0.000 description 5
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 4
- LXSNZLZWYNZHEH-UHFFFAOYSA-N 3-methyl-2h-pyrazolo[3,4-b]pyridine-5-carboxylic acid Chemical compound N1=CC(C(O)=O)=CC2=C(C)NN=C21 LXSNZLZWYNZHEH-UHFFFAOYSA-N 0.000 description 4
- JJLBTVYCYWUHCH-UHFFFAOYSA-N 3-methyl-2h-pyrazolo[4,3-b]pyridine Chemical compound C1=CN=C2C(C)=NNC2=C1 JJLBTVYCYWUHCH-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- OPJNRVAWKYSENX-UHFFFAOYSA-N 5-bromo-3-methyl-2h-pyrazolo[3,4-c]pyridine Chemical compound N1=C(Br)C=C2C(C)=NNC2=C1 OPJNRVAWKYSENX-UHFFFAOYSA-N 0.000 description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 4
- KQXRAIUTSSRSOB-KRWDZBQOSA-N ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)S(=O)(=O)C Chemical compound ClC1=CC=C(C=C1)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NNC2=CC=1)S(=O)(=O)C KQXRAIUTSSRSOB-KRWDZBQOSA-N 0.000 description 4
- 206010009944 Colon cancer Diseases 0.000 description 4
- UOGPBMKOXQEYSJ-UHFFFAOYSA-N FC1=C(C=C2C(=NNC2=C1)C)C(=O)O Chemical compound FC1=C(C=C2C(=NNC2=C1)C)C(=O)O UOGPBMKOXQEYSJ-UHFFFAOYSA-N 0.000 description 4
- SFZOIFKOCQUMQH-AWEZNQCLSA-N NC1=NNC=2C1=NC(=CC=2)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Cl Chemical compound NC1=NNC=2C1=NC(=CC=2)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)Cl SFZOIFKOCQUMQH-AWEZNQCLSA-N 0.000 description 4
- 229960000583 acetic acid Drugs 0.000 description 4
- 239000002775 capsule Substances 0.000 description 4
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 230000008020 evaporation Effects 0.000 description 4
- 239000001257 hydrogen Substances 0.000 description 4
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 4
- 235000011056 potassium acetate Nutrition 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 238000001959 radiotherapy Methods 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- CIHHGGKKRPPWSU-JTQLQIEISA-N (2s)-2-(4-chlorophenyl)pyrrolidine Chemical compound C1=CC(Cl)=CC=C1[C@H]1NCCC1 CIHHGGKKRPPWSU-JTQLQIEISA-N 0.000 description 3
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- NLQQIWGMFAJKIH-UHFFFAOYSA-N 1-(2-bromo-5-fluoropyridin-4-yl)ethanone Chemical compound CC(=O)C1=CC(Br)=NC=C1F NLQQIWGMFAJKIH-UHFFFAOYSA-N 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- HIJZBROSVFKSCP-UHFFFAOYSA-N 2-(4-bromophenyl)pyrrolidine Chemical compound C1=CC(Br)=CC=C1C1NCCC1 HIJZBROSVFKSCP-UHFFFAOYSA-N 0.000 description 3
- SOIMDMPMUKPBOC-UHFFFAOYSA-N 3-(2-methylpropyl)-2H-indazole-5-carboxylic acid Chemical compound CC(C)CC1=NNC2=C1C=C(C=C2)C(O)=O SOIMDMPMUKPBOC-UHFFFAOYSA-N 0.000 description 3
- VXJHBBSBNCHTOF-UHFFFAOYSA-N 3-amino-2h-pyrazolo[3,4-b]pyridine-5-carboxylic acid Chemical compound N1=CC(C(O)=O)=CC2=C(N)NN=C21 VXJHBBSBNCHTOF-UHFFFAOYSA-N 0.000 description 3
- DYZNTIADYAHDQK-UHFFFAOYSA-N 3-iodo-2h-indazole-5-carboxylic acid Chemical compound OC(=O)C1=CC=C2NN=C(I)C2=C1 DYZNTIADYAHDQK-UHFFFAOYSA-N 0.000 description 3
- QGWLKUFLLVCCTP-UHFFFAOYSA-N 3-methyl-2h-pyrazolo[3,4-c]pyridine-5-carboxylic acid Chemical compound C1=NC(C(O)=O)=CC2=C(C)NN=C21 QGWLKUFLLVCCTP-UHFFFAOYSA-N 0.000 description 3
- NIYSJLLWAMUQNS-UHFFFAOYSA-N 3-methyl-4-oxido-1-tritylpyrazolo[4,3-b]pyridin-4-ium Chemical compound C12=CC=C[N+]([O-])=C2C(C)=NN1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 NIYSJLLWAMUQNS-UHFFFAOYSA-N 0.000 description 3
- XMIIGOLPHOKFCH-UHFFFAOYSA-N 3-phenylpropionic acid Chemical compound OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 3
- SSNUTEUZXZIYTB-UHFFFAOYSA-N 5-bromo-2h-pyrazolo[3,4-b]pyridin-3-amine Chemical compound N1=CC(Br)=CC2=C(N)NN=C21 SSNUTEUZXZIYTB-UHFFFAOYSA-N 0.000 description 3
- XDJNHYAQZWCIAH-UHFFFAOYSA-N 5-bromo-3-methyl-2h-indazole Chemical compound C1=CC(Br)=CC2=C(C)NN=C21 XDJNHYAQZWCIAH-UHFFFAOYSA-N 0.000 description 3
- LAHMHTLOIHUMFQ-UHFFFAOYSA-N 5-bromo-6-fluoro-3-methyl-2h-indazole Chemical compound FC1=C(Br)C=C2C(C)=NNC2=C1 LAHMHTLOIHUMFQ-UHFFFAOYSA-N 0.000 description 3
- FQKVEOICHOSVAB-UHFFFAOYSA-N 5-chloro-3-methyl-2h-pyrazolo[4,3-b]pyridine Chemical compound C1=C(Cl)N=C2C(C)=NNC2=C1 FQKVEOICHOSVAB-UHFFFAOYSA-N 0.000 description 3
- SAXJIWTWKKIZMB-UHFFFAOYSA-N BrC=1C=C2C(=CN=1)NN=C2N Chemical compound BrC=1C=C2C(=CN=1)NN=C2N SAXJIWTWKKIZMB-UHFFFAOYSA-N 0.000 description 3
- SABJBUGQMHTTSE-UHFFFAOYSA-N CC(=CC1=NNC2=CC=C(C=C12)C(=O)O)C Chemical compound CC(=CC1=NNC2=CC=C(C=C12)C(=O)O)C SABJBUGQMHTTSE-UHFFFAOYSA-N 0.000 description 3
- IJWOGSNXXMILTM-UHFFFAOYSA-N CSC1=NNC2=CC=C(C=C12)C(=O)O Chemical compound CSC1=NNC2=CC=C(C=C12)C(=O)O IJWOGSNXXMILTM-UHFFFAOYSA-N 0.000 description 3
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 3
- ZQODSTYMINEEDI-UHFFFAOYSA-N ClC1=CC=C(C=C1)C1C(CNC1)O Chemical compound ClC1=CC=C(C=C1)C1C(CNC1)O ZQODSTYMINEEDI-UHFFFAOYSA-N 0.000 description 3
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 3
- FXUBOLKTMDGVMH-UHFFFAOYSA-N NC1=NNC2=CN=C(C=C21)C(=O)O Chemical compound NC1=NNC2=CN=C(C=C21)C(=O)O FXUBOLKTMDGVMH-UHFFFAOYSA-N 0.000 description 3
- 101100030361 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) pph-3 gene Proteins 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 229910000831 Steel Inorganic materials 0.000 description 3
- 235000011054 acetic acid Nutrition 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- 239000012131 assay buffer Substances 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 229910002091 carbon monoxide Inorganic materials 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 239000012230 colorless oil Substances 0.000 description 3
- 239000013058 crude material Substances 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 239000011737 fluorine Substances 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 239000000543 intermediate Substances 0.000 description 3
- 230000002503 metabolic effect Effects 0.000 description 3
- JLMLNYIMUVBZFM-UHFFFAOYSA-N methyl 3-amino-2h-pyrazolo[3,4-b]pyridine-5-carboxylate Chemical compound C1=C(C(=O)OC)C=NC2=NNC(N)=C21 JLMLNYIMUVBZFM-UHFFFAOYSA-N 0.000 description 3
- 210000000066 myeloid cell Anatomy 0.000 description 3
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000005855 radiation Effects 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 239000010959 steel Substances 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 3
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 2
- BVEQCCJUWYROBR-PPHPATTJSA-N (2s)-2-(4-chlorophenyl)pyrrolidine;hydrochloride Chemical compound Cl.C1=CC(Cl)=CC=C1[C@H]1NCCC1 BVEQCCJUWYROBR-PPHPATTJSA-N 0.000 description 2
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 2
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 2
- KGJFRZOATIXYPW-UHFFFAOYSA-N 1,3-dimethyl-2,4-dioxopyrimidine-5-carbaldehyde Chemical compound CN1C=C(C=O)C(=O)N(C)C1=O KGJFRZOATIXYPW-UHFFFAOYSA-N 0.000 description 2
- CCZMVVFNNQQTFC-UHFFFAOYSA-N 1-(3-fluoropyridin-2-yl)ethanone Chemical compound CC(=O)C1=NC=CC=C1F CCZMVVFNNQQTFC-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- MAVGBUDLHOOROM-UHFFFAOYSA-N 1h-indazole-5-carboxylic acid Chemical compound OC(=O)C1=CC=C2NN=CC2=C1 MAVGBUDLHOOROM-UHFFFAOYSA-N 0.000 description 2
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 2
- BVEQCCJUWYROBR-UHFFFAOYSA-N 2-(4-chlorophenyl)pyrrolidine;hydrochloride Chemical compound Cl.C1=CC(Cl)=CC=C1C1NCCC1 BVEQCCJUWYROBR-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- YEGQMUOUKUEJRM-UHFFFAOYSA-N 3-amino-1h-indazole-5-carboxylic acid Chemical compound C1=C(C(O)=O)C=C2C(N)=NNC2=C1 YEGQMUOUKUEJRM-UHFFFAOYSA-N 0.000 description 2
- FGBHCSVATHXMLX-UHFFFAOYSA-N 3-methyl-2h-indazole-5-carbaldehyde Chemical compound C1=C(C=O)C=C2C(C)=NNC2=C1 FGBHCSVATHXMLX-UHFFFAOYSA-N 0.000 description 2
- NYATZKHOQZWOJM-UHFFFAOYSA-N 3-propyl-2h-indazole-5-carboxylic acid Chemical compound C1=C(C(O)=O)C=C2C(CCC)=NNC2=C1 NYATZKHOQZWOJM-UHFFFAOYSA-N 0.000 description 2
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 2
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 2
- SLHQBVVOZYFUSS-UHFFFAOYSA-N 5-pyrrolidin-2-yl-1h-indazole Chemical compound C1CCNC1C1=CC=C(NN=C2)C2=C1 SLHQBVVOZYFUSS-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- DOIZQBMCPUAUBV-UHFFFAOYSA-N BrC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2 Chemical compound BrC1=CC=C(C=C1)C1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2 DOIZQBMCPUAUBV-UHFFFAOYSA-N 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- VNJXXCQDVVQYDQ-UHFFFAOYSA-N CC1=NN(C=2C1=NC(=CC=2)C#N)C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound CC1=NN(C=2C1=NC(=CC=2)C#N)C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1 VNJXXCQDVVQYDQ-UHFFFAOYSA-N 0.000 description 2
- NGHQNXITCKBSBU-XIFFEERXSA-N CC1=NN(C=2C1=NC(=CC=2)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)C(F)(F)F)C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound CC1=NN(C=2C1=NC(=CC=2)C(=O)N1[C@@H](CCC1)C1=CC=C(C=C1)C(F)(F)F)C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1 NGHQNXITCKBSBU-XIFFEERXSA-N 0.000 description 2
- KWUWGJYXFDVVDR-UHFFFAOYSA-N CC1=NN(C=2C1=NC(=CC=2)C(=O)O)C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1 Chemical compound CC1=NN(C=2C1=NC(=CC=2)C(=O)O)C(C1=CC=CC=C1)(C1=CC=CC=C1)C1=CC=CC=C1 KWUWGJYXFDVVDR-UHFFFAOYSA-N 0.000 description 2
- XSARCVOGXVUPKL-UHFFFAOYSA-N CC1=NNC2=CN=C(C=C21)C(=O)OC Chemical compound CC1=NNC2=CN=C(C=C21)C(=O)OC XSARCVOGXVUPKL-UHFFFAOYSA-N 0.000 description 2
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 2
- CUFCDNXOIUSLMJ-UHFFFAOYSA-N CN(C(=O)NC)C(=O)C=1C=C2C(=NC=1)NN=C2C(F)(F)F Chemical compound CN(C(=O)NC)C(=O)C=1C=C2C(=NC=1)NN=C2C(F)(F)F CUFCDNXOIUSLMJ-UHFFFAOYSA-N 0.000 description 2
- BFOLECOSTUQXKU-UHFFFAOYSA-N COC1=NNC2=NC=C(C=C21)C(=O)O Chemical compound COC1=NNC2=NC=C(C=C21)C(=O)O BFOLECOSTUQXKU-UHFFFAOYSA-N 0.000 description 2
- JBPBKTPOJBSQKT-LBAUFKAWSA-N ClC1=CC=C(C=C1)C1C[C@@H](CN1C(=O)C=1C=C2C(=NC=1)NN=C2C)O Chemical compound ClC1=CC=C(C=C1)C1C[C@@H](CN1C(=O)C=1C=C2C(=NC=1)NN=C2C)O JBPBKTPOJBSQKT-LBAUFKAWSA-N 0.000 description 2
- BCXLYZDDXDFXDU-CVEARBPZSA-N ClC1=CC=C(C=C1)[C@H]1CN(C[C@@H]1O)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC1=CC=C(C=C1)[C@H]1CN(C[C@@H]1O)C(=O)C=1C=C2C(=NC=1)NN=C2C BCXLYZDDXDFXDU-CVEARBPZSA-N 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- 102100024170 Cyclin-C Human genes 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- BKHAACFXJMDLRV-UHFFFAOYSA-N FC(C1=NNC2=NC=C(C=C21)C(=O)O)(F)F Chemical compound FC(C1=NNC2=NC=C(C=C21)C(=O)O)(F)F BKHAACFXJMDLRV-UHFFFAOYSA-N 0.000 description 2
- VSKKSLJHPOZGGI-UHFFFAOYSA-N FC1=NNC2=CC=C(C=C12)C(=O)O Chemical compound FC1=NNC2=CC=C(C=C12)C(=O)O VSKKSLJHPOZGGI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 102100034221 Growth-regulated alpha protein Human genes 0.000 description 2
- 101000980770 Homo sapiens Cyclin-C Proteins 0.000 description 2
- 101001069921 Homo sapiens Growth-regulated alpha protein Proteins 0.000 description 2
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- 108010078049 Interferon alpha-2 Proteins 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- HZXRHFFZIBJQGB-UHFFFAOYSA-N NC1=NNC2=CN=C(C=C21)C(=O)OC Chemical compound NC1=NNC2=CN=C(C=C21)C(=O)OC HZXRHFFZIBJQGB-UHFFFAOYSA-N 0.000 description 2
- 0 O=C(c1cc(cn[n]2)c2nc1)N(CCC1)*1c(cc1)ccc1Br Chemical compound O=C(c1cc(cn[n]2)c2nc1)N(CCC1)*1c(cc1)ccc1Br 0.000 description 2
- 108700020796 Oncogene Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 102000007374 Smad Proteins Human genes 0.000 description 2
- 108010007945 Smad Proteins Proteins 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 108700012920 TNF Proteins 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- SNRCKKQHDUIRIY-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloromethane;dichloropalladium;iron(2+) Chemical compound [Fe+2].ClCCl.Cl[Pd]Cl.C1=C[CH-]C(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.C1=C[CH-]C(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 SNRCKKQHDUIRIY-UHFFFAOYSA-L 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 210000001671 embryonic stem cell Anatomy 0.000 description 2
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 201000005787 hematologic cancer Diseases 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- TWBYWOBDOCUKOW-UHFFFAOYSA-N isonicotinic acid Chemical compound OC(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-N 0.000 description 2
- 239000004310 lactic acid Substances 0.000 description 2
- 235000014655 lactic acid Nutrition 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000001630 malic acid Substances 0.000 description 2
- 235000011090 malic acid Nutrition 0.000 description 2
- 229960004961 mechlorethamine Drugs 0.000 description 2
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 2
- 238000012961 medicinal therapy Methods 0.000 description 2
- HWZPYTDUHRCVLH-UHFFFAOYSA-N methyl 3-methyl-2h-indazole-5-carboxylate Chemical compound COC(=O)C1=CC=C2NN=C(C)C2=C1 HWZPYTDUHRCVLH-UHFFFAOYSA-N 0.000 description 2
- JGHZIRKYYYUZAF-UHFFFAOYSA-N methyl 3-methyl-2h-pyrazolo[4,3-b]pyridine-5-carboxylate Chemical compound N1=C(C(=O)OC)C=CC2=NNC(C)=C21 JGHZIRKYYYUZAF-UHFFFAOYSA-N 0.000 description 2
- RJOLBIDETVZLGH-UHFFFAOYSA-N methyl 6-fluoro-3-methyl-2h-indazole-5-carboxylate Chemical compound C1=C(F)C(C(=O)OC)=CC2=C(C)NN=C21 RJOLBIDETVZLGH-UHFFFAOYSA-N 0.000 description 2
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 239000006186 oral dosage form Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 239000003960 organic solvent Substances 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- WLJVNTCWHIRURA-UHFFFAOYSA-N pimelic acid Chemical compound OC(=O)CCCCCC(O)=O WLJVNTCWHIRURA-UHFFFAOYSA-N 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- 238000004237 preparative chromatography Methods 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-O pyridinium Chemical compound C1=CC=[NH+]C=C1 JUJWROOIHBZHMG-UHFFFAOYSA-O 0.000 description 2
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 2
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 2
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 description 2
- 125000004943 pyrimidin-6-yl group Chemical group N1=CN=CC=C1* 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 230000001235 sensitizing effect Effects 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 230000004936 stimulating effect Effects 0.000 description 2
- 238000003860 storage Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- 238000003419 tautomerization reaction Methods 0.000 description 2
- 229960003433 thalidomide Drugs 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 230000000699 topical effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000002103 transcriptional effect Effects 0.000 description 2
- 229960000575 trastuzumab Drugs 0.000 description 2
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- LWDBMUAJGMXQAY-GSEQGPDBSA-L (1r,2r)-cyclohexane-1,2-diamine;platinum(2+);tetradecanoate;hydrate Chemical compound O.[Pt+2].N[C@@H]1CCCC[C@H]1N.CCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCC([O-])=O LWDBMUAJGMXQAY-GSEQGPDBSA-L 0.000 description 1
- WCWUXEGQKLTGDX-LLVKDONJSA-N (2R)-1-[[4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-5-methyl-6-pyrrolo[2,1-f][1,2,4]triazinyl]oxy]-2-propanol Chemical compound C1=C2NC(C)=CC2=C(F)C(OC2=NC=NN3C=C(C(=C32)C)OC[C@H](O)C)=C1 WCWUXEGQKLTGDX-LLVKDONJSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- FWFGIHPGRQZWIW-SQNIBIBYSA-N (2S)-2-[[(2R)-2-[(1S)-1-hydroxy-2-(hydroxyamino)-2-oxoethyl]-4-methyl-1-oxopentyl]amino]-2-phenylacetic acid cyclopentyl ester Chemical compound O=C([C@@H](NC(=O)[C@@H]([C@H](O)C(=O)NO)CC(C)C)C=1C=CC=CC=1)OC1CCCC1 FWFGIHPGRQZWIW-SQNIBIBYSA-N 0.000 description 1
- UUBHZHZSIKRVIV-KCXSXWJSSA-N (2e,6e,10e)-3,7,11,15-tetramethylhexadeca-2,4,6,10,14-pentaenoic acid Chemical compound CC(C)=CCC\C(C)=C\CC\C(C)=C\C=C\C(\C)=C\C(O)=O UUBHZHZSIKRVIV-KCXSXWJSSA-N 0.000 description 1
- DNISEZBAYYIQFB-PHDIDXHHSA-N (2r,3r)-2,3-diacetyloxybutanedioic acid Chemical compound CC(=O)O[C@@H](C(O)=O)[C@H](C(O)=O)OC(C)=O DNISEZBAYYIQFB-PHDIDXHHSA-N 0.000 description 1
- WDQLRUYAYXDIFW-RWKIJVEZSA-N (2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-4-[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-6-[[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl]oxy-6-(hydroxymethyl)oxane-2,3,5-triol Chemical compound O[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)O1 WDQLRUYAYXDIFW-RWKIJVEZSA-N 0.000 description 1
- JUSWZYFYLXTMLJ-JTQLQIEISA-N (2s)-1-(benzenesulfonyl)pyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1S(=O)(=O)C1=CC=CC=C1 JUSWZYFYLXTMLJ-JTQLQIEISA-N 0.000 description 1
- RQYKQWFHJOBBAO-JTQLQIEISA-N (2s)-1-benzoylpyrrolidine-2-carboxylic acid Chemical compound OC(=O)[C@@H]1CCCN1C(=O)C1=CC=CC=C1 RQYKQWFHJOBBAO-JTQLQIEISA-N 0.000 description 1
- PSVUJBVBCOISSP-SPFKKGSWSA-N (2s,3r,4s,5s,6r)-2-bis(2-chloroethylamino)phosphoryloxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound OC[C@H]1O[C@@H](OP(=O)(NCCCl)NCCCl)[C@H](O)[C@@H](O)[C@@H]1O PSVUJBVBCOISSP-SPFKKGSWSA-N 0.000 description 1
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 1
- KCOYQXZDFIIGCY-CZIZESTLSA-N (3e)-4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1,3-dihydrobenzimidazol-2-ylidene]quinolin-2-one Chemical compound C1CN(C)CCN1C1=CC=C(N\C(N2)=C/3C(=C4C(F)=CC=CC4=NC\3=O)N)C2=C1 KCOYQXZDFIIGCY-CZIZESTLSA-N 0.000 description 1
- DEQANNDTNATYII-OULOTJBUSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-19-[[(2r)-2-amino-3-phenylpropanoyl]amino]-16-benzyl-n-[(2r,3r)-1,3-dihydroxybutan-2-yl]-7-[(1r)-1-hydroxyethyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicosane-4-carboxa Chemical compound C([C@@H](N)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC=CC=2)NC1=O)C(=O)N[C@H](CO)[C@H](O)C)C1=CC=CC=C1 DEQANNDTNATYII-OULOTJBUSA-N 0.000 description 1
- MWTUOSWPJOUADP-XDJHFCHBSA-N (5z)-5-(4-hydroxy-6-oxo-3-propan-2-ylcyclohexa-2,4-dien-1-ylidene)-4-(1-methylindol-5-yl)-1,2,4-triazolidin-3-one Chemical compound O=C1C=C(O)C(C(C)C)=C\C1=C\1N(C=2C=C3C=CN(C)C3=CC=2)C(=O)NN/1 MWTUOSWPJOUADP-XDJHFCHBSA-N 0.000 description 1
- FPVKHBSQESCIEP-UHFFFAOYSA-N (8S)-3-(2-deoxy-beta-D-erythro-pentofuranosyl)-3,6,7,8-tetrahydroimidazo[4,5-d][1,3]diazepin-8-ol Natural products C1C(O)C(CO)OC1N1C(NC=NCC2O)=C2N=C1 FPVKHBSQESCIEP-UHFFFAOYSA-N 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- 125000004509 1,3,4-oxadiazol-2-yl group Chemical group O1C(=NN=C1)* 0.000 description 1
- KXZHDRBQPYKHKS-UHFFFAOYSA-N 1-(5-bromo-2,4-difluorophenyl)ethanone Chemical compound CC(=O)C1=CC(Br)=C(F)C=C1F KXZHDRBQPYKHKS-UHFFFAOYSA-N 0.000 description 1
- XNRQIHIOKXQSPG-UHFFFAOYSA-N 1-(5-bromo-2-fluorophenyl)ethanone Chemical compound CC(=O)C1=CC(Br)=CC=C1F XNRQIHIOKXQSPG-UHFFFAOYSA-N 0.000 description 1
- ZQGDSZPGKPJABN-UHFFFAOYSA-N 1-(chloromethyl)-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane Chemical compound C1C[N+]2(CCl)CC[N+]1(F)CC2 ZQGDSZPGKPJABN-UHFFFAOYSA-N 0.000 description 1
- SPMVMDHWKHCIDT-UHFFFAOYSA-N 1-[2-chloro-4-[(6,7-dimethoxy-4-quinolinyl)oxy]phenyl]-3-(5-methyl-3-isoxazolyl)urea Chemical compound C=12C=C(OC)C(OC)=CC2=NC=CC=1OC(C=C1Cl)=CC=C1NC(=O)NC=1C=C(C)ON=1 SPMVMDHWKHCIDT-UHFFFAOYSA-N 0.000 description 1
- PVCULFYROUOVGJ-UHFFFAOYSA-N 1-[2-chloroethyl(methylsulfonyl)amino]-3-methyl-1-methylsulfonylurea Chemical compound CNC(=O)N(S(C)(=O)=O)N(S(C)(=O)=O)CCCl PVCULFYROUOVGJ-UHFFFAOYSA-N 0.000 description 1
- NHDODQWIKUYWMW-UHFFFAOYSA-N 1-bromo-4-chlorobenzene Chemical compound ClC1=CC=C(Br)C=C1 NHDODQWIKUYWMW-UHFFFAOYSA-N 0.000 description 1
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 1
- MFKFCHQDGBJUIU-UHFFFAOYSA-N 2-(4-bromophenyl)pyrrolidine;hydron;chloride Chemical compound Cl.C1=CC(Br)=CC=C1C1NCCC1 MFKFCHQDGBJUIU-UHFFFAOYSA-N 0.000 description 1
- UCOPFPGOAGRDCX-UHFFFAOYSA-N 2-(4-chloro-3-fluorophenyl)pyrrolidine Chemical compound C1=C(Cl)C(F)=CC(C2NCCC2)=C1 UCOPFPGOAGRDCX-UHFFFAOYSA-N 0.000 description 1
- OTOVNNDSINVUBR-UHFFFAOYSA-N 2-(4-chlorophenyl)piperazine Chemical compound C1=CC(Cl)=CC=C1C1NCCNC1 OTOVNNDSINVUBR-UHFFFAOYSA-N 0.000 description 1
- UZYQSNQJLWTICD-UHFFFAOYSA-N 2-(n-benzoylanilino)-2,2-dinitroacetic acid Chemical compound C=1C=CC=CC=1N(C(C(=O)O)([N+]([O-])=O)[N+]([O-])=O)C(=O)C1=CC=CC=C1 UZYQSNQJLWTICD-UHFFFAOYSA-N 0.000 description 1
- OXQGTIUCKGYOAA-UHFFFAOYSA-N 2-Ethylbutanoic acid Chemical compound CCC(CC)C(O)=O OXQGTIUCKGYOAA-UHFFFAOYSA-N 0.000 description 1
- PIMQWRZWLQKKBJ-SFHVURJKSA-N 2-[(2S)-1-[3-ethyl-7-[(1-oxido-3-pyridin-1-iumyl)methylamino]-5-pyrazolo[1,5-a]pyrimidinyl]-2-piperidinyl]ethanol Chemical compound C=1C(N2[C@@H](CCCC2)CCO)=NC2=C(CC)C=NN2C=1NCC1=CC=C[N+]([O-])=C1 PIMQWRZWLQKKBJ-SFHVURJKSA-N 0.000 description 1
- PDWUPXJEEYOOTR-UHFFFAOYSA-N 2-[(3-iodophenyl)methyl]guanidine Chemical compound NC(=N)NCC1=CC=CC(I)=C1 PDWUPXJEEYOOTR-UHFFFAOYSA-N 0.000 description 1
- QXLQZLBNPTZMRK-UHFFFAOYSA-N 2-[(dimethylamino)methyl]-1-(2,4-dimethylphenyl)prop-2-en-1-one Chemical compound CN(C)CC(=C)C(=O)C1=CC=C(C)C=C1C QXLQZLBNPTZMRK-UHFFFAOYSA-N 0.000 description 1
- IEQAICDLOKRSRL-UHFFFAOYSA-N 2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-dodecoxyethoxy)ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO IEQAICDLOKRSRL-UHFFFAOYSA-N 0.000 description 1
- RZHKDBRREKOZEW-AAXZNHDCSA-N 2-[4-[2-[[(2r)-1-[[(4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-4-[[(2r,3r)-1,3-dihydroxybutan-2-yl]carbamoyl]-7-[(1r)-1-hydroxyethyl]-16-[(4-hydroxyphenyl)methyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicos-19-yl] Chemical compound C([C@H](C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC1=O)C(=O)N[C@H](CO)[C@H](O)C)NC(=O)CN1CCN(CC(O)=O)CCN(CC(O)=O)CCN(CC(O)=O)CC1)C1=CC=CC=C1 RZHKDBRREKOZEW-AAXZNHDCSA-N 0.000 description 1
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 1
- KMGUEILFFWDGFV-UHFFFAOYSA-N 2-benzoyl-2-benzoyloxy-3-hydroxybutanedioic acid Chemical compound C=1C=CC=CC=1C(=O)C(C(C(O)=O)O)(C(O)=O)OC(=O)C1=CC=CC=C1 KMGUEILFFWDGFV-UHFFFAOYSA-N 0.000 description 1
- UODINHBLNPPDPD-UHFFFAOYSA-N 2-bromo-5-fluoropyridine Chemical compound FC1=CC=C(Br)N=C1 UODINHBLNPPDPD-UHFFFAOYSA-N 0.000 description 1
- GLUXDIYRUHBWMX-UHFFFAOYSA-N 2-bromo-5-fluoropyridine-4-carbonitrile Chemical compound FC1=CN=C(Br)C=C1C#N GLUXDIYRUHBWMX-UHFFFAOYSA-N 0.000 description 1
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000389 2-pyrrolyl group Chemical group [H]N1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- NDMPLJNOPCLANR-UHFFFAOYSA-N 3,4-dihydroxy-15-(4-hydroxy-18-methoxycarbonyl-5,18-seco-ibogamin-18-yl)-16-methoxy-1-methyl-6,7-didehydro-aspidospermidine-3-carboxylic acid methyl ester Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 NDMPLJNOPCLANR-UHFFFAOYSA-N 0.000 description 1
- AXRCEOKUDYDWLF-UHFFFAOYSA-N 3-(1-methyl-3-indolyl)-4-[1-[1-(2-pyridinylmethyl)-4-piperidinyl]-3-indolyl]pyrrole-2,5-dione Chemical compound C12=CC=CC=C2N(C)C=C1C(C(NC1=O)=O)=C1C(C1=CC=CC=C11)=CN1C(CC1)CCN1CC1=CC=CC=N1 AXRCEOKUDYDWLF-UHFFFAOYSA-N 0.000 description 1
- VFKOTIALXPYKGA-UHFFFAOYSA-N 3-(4-chlorophenyl)-1-methylpiperazine Chemical compound C1N(C)CCNC1C1=CC=C(Cl)C=C1 VFKOTIALXPYKGA-UHFFFAOYSA-N 0.000 description 1
- VKUBCLCJPLUOTA-UHFFFAOYSA-N 3-(4-chlorophenyl)morpholine;hydrochloride Chemical compound Cl.C1=CC(Cl)=CC=C1C1NCCOC1 VKUBCLCJPLUOTA-UHFFFAOYSA-N 0.000 description 1
- ZZUBHVMHNVYXRR-UHFFFAOYSA-N 3-(4-hydroxyphenyl)-2h-chromen-7-ol Chemical compound C1=CC(O)=CC=C1C1=CC2=CC=C(O)C=C2OC1 ZZUBHVMHNVYXRR-UHFFFAOYSA-N 0.000 description 1
- NHFDRBXTEDBWCZ-ZROIWOOFSA-N 3-[2,4-dimethyl-5-[(z)-(2-oxo-1h-indol-3-ylidene)methyl]-1h-pyrrol-3-yl]propanoic acid Chemical compound OC(=O)CCC1=C(C)NC(\C=C/2C3=CC=CC=C3NC\2=O)=C1C NHFDRBXTEDBWCZ-ZROIWOOFSA-N 0.000 description 1
- JMHGATOBRPWPBZ-UHFFFAOYSA-N 3-cyano-4-fluorobenzoic acid Chemical compound OC(=O)C1=CC=C(F)C(C#N)=C1 JMHGATOBRPWPBZ-UHFFFAOYSA-N 0.000 description 1
- YIRTUZMXPMVWAY-UHFFFAOYSA-N 3-cyclopropyl-2H-indazole-5-carboxylic acid Chemical compound C1(CC1)C1=NNC2=CC=C(C=C12)C(=O)O YIRTUZMXPMVWAY-UHFFFAOYSA-N 0.000 description 1
- ITMZYFZDCWKKBQ-UHFFFAOYSA-N 3-cyclopropyl-2h-pyrazolo[3,4-b]pyridine-5-carboxylic acid Chemical compound C=12C=C(C(=O)O)C=NC2=NNC=1C1CC1 ITMZYFZDCWKKBQ-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- SBQXRRPYYHWLOK-UHFFFAOYSA-N 3-methoxy-1H-pyrazolo[3,4-b]pyridine-5-carbonitrile Chemical compound COC1=NNC2=NC=C(C=C21)C#N SBQXRRPYYHWLOK-UHFFFAOYSA-N 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- CLPFFLWZZBQMAO-UHFFFAOYSA-N 4-(5,6,7,8-tetrahydroimidazo[1,5-a]pyridin-5-yl)benzonitrile Chemical compound C1=CC(C#N)=CC=C1C1N2C=NC=C2CCC1 CLPFFLWZZBQMAO-UHFFFAOYSA-N 0.000 description 1
- XXJWYDDUDKYVKI-UHFFFAOYSA-N 4-[(4-fluoro-2-methyl-1H-indol-5-yl)oxy]-6-methoxy-7-[3-(1-pyrrolidinyl)propoxy]quinazoline Chemical compound COC1=CC2=C(OC=3C(=C4C=C(C)NC4=CC=3)F)N=CN=C2C=C1OCCCN1CCCC1 XXJWYDDUDKYVKI-UHFFFAOYSA-N 0.000 description 1
- ZLHFILGSQDJULK-UHFFFAOYSA-N 4-[[9-chloro-7-(2-fluoro-6-methoxyphenyl)-5H-pyrimido[5,4-d][2]benzazepin-2-yl]amino]-2-methoxybenzoic acid Chemical compound C1=C(C(O)=O)C(OC)=CC(NC=2N=C3C4=CC=C(Cl)C=C4C(=NCC3=CN=2)C=2C(=CC=CC=2F)OC)=C1 ZLHFILGSQDJULK-UHFFFAOYSA-N 0.000 description 1
- AGYWDGVTLKNTBS-UHFFFAOYSA-N 4-bromo-1-chloro-2-fluorobenzene Chemical compound FC1=CC(Br)=CC=C1Cl AGYWDGVTLKNTBS-UHFFFAOYSA-N 0.000 description 1
- MDOJTZQKHMAPBK-UHFFFAOYSA-N 4-iodo-3-nitrobenzamide Chemical compound NC(=O)C1=CC=C(I)C([N+]([O-])=O)=C1 MDOJTZQKHMAPBK-UHFFFAOYSA-N 0.000 description 1
- KDDQRKBRJSGMQE-UHFFFAOYSA-N 4-thiazolyl Chemical compound [C]1=CSC=N1 KDDQRKBRJSGMQE-UHFFFAOYSA-N 0.000 description 1
- WVORIWCOSAWJJE-UHFFFAOYSA-N 5-(trifluoromethyl)-1h-pyrazol-3-amine Chemical compound NC1=CC(C(F)(F)F)=NN1 WVORIWCOSAWJJE-UHFFFAOYSA-N 0.000 description 1
- IDPUKCWIGUEADI-UHFFFAOYSA-N 5-[bis(2-chloroethyl)amino]uracil Chemical compound ClCCN(CCCl)C1=CNC(=O)NC1=O IDPUKCWIGUEADI-UHFFFAOYSA-N 0.000 description 1
- UPALIKSFLSVKIS-UHFFFAOYSA-N 5-amino-2-[2-(dimethylamino)ethyl]benzo[de]isoquinoline-1,3-dione Chemical compound NC1=CC(C(N(CCN(C)C)C2=O)=O)=C3C2=CC=CC3=C1 UPALIKSFLSVKIS-UHFFFAOYSA-N 0.000 description 1
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 1
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 1
- STVHMYNPQCLUNJ-UHFFFAOYSA-N 5-bromo-1h-indazole Chemical compound BrC1=CC=C2NN=CC2=C1 STVHMYNPQCLUNJ-UHFFFAOYSA-N 0.000 description 1
- MQOHJAYYYVQBSH-UHFFFAOYSA-N 5-bromo-2-chloropyridine-3-carbonitrile Chemical compound ClC1=NC=C(Br)C=C1C#N MQOHJAYYYVQBSH-UHFFFAOYSA-N 0.000 description 1
- JFJIFJQOTNHZRK-UHFFFAOYSA-N 5-bromo-3-methoxy-1h-pyrazolo[3,4-b]pyridine Chemical compound C1=C(Br)C=C2C(OC)=NNC2=N1 JFJIFJQOTNHZRK-UHFFFAOYSA-N 0.000 description 1
- MXVAGCQKBDMKPG-UHFFFAOYSA-N 5-cyclopropyl-1h-pyrazol-3-amine Chemical compound N1C(N)=CC(C2CC2)=N1 MXVAGCQKBDMKPG-UHFFFAOYSA-N 0.000 description 1
- 125000004938 5-pyridyl group Chemical group N1=CC=CC(=C1)* 0.000 description 1
- CWDWFSXUQODZGW-UHFFFAOYSA-N 5-thiazolyl Chemical group [C]1=CN=CS1 CWDWFSXUQODZGW-UHFFFAOYSA-N 0.000 description 1
- WYWHKKSPHMUBEB-UHFFFAOYSA-N 6-Mercaptoguanine Natural products N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 1
- OZPFIJIOIVJZMN-SFHVURJKSA-N 6-[(7s)-7-hydroxy-5,6-dihydropyrrolo[1,2-c]imidazol-7-yl]-n-methylnaphthalene-2-carboxamide Chemical compound C1=CC2=CC(C(=O)NC)=CC=C2C=C1[C@]1(O)C2=CN=CN2CC1 OZPFIJIOIVJZMN-SFHVURJKSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- SHGAZHPCJJPHSC-ZVCIMWCZSA-N 9-cis-retinoic acid Chemical compound OC(=O)/C=C(\C)/C=C/C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-ZVCIMWCZSA-N 0.000 description 1
- BUROJSBIWGDYCN-GAUTUEMISA-N AP 23573 Chemical compound C1C[C@@H](OP(C)(C)=O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 BUROJSBIWGDYCN-GAUTUEMISA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- MXPOCMVWFLDDLZ-NSCUHMNNSA-N Apaziquone Chemical compound CN1C(\C=C\CO)=C(CO)C(C2=O)=C1C(=O)C=C2N1CC1 MXPOCMVWFLDDLZ-NSCUHMNNSA-N 0.000 description 1
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 102000015790 Asparaginase Human genes 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- VGGGPCQERPFHOB-MCIONIFRSA-N Bestatin Chemical compound CC(C)C[C@H](C(O)=O)NC(=O)[C@@H](O)[C@H](N)CC1=CC=CC=C1 VGGGPCQERPFHOB-MCIONIFRSA-N 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 241000167854 Bourreria succulenta Species 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 108010037003 Buserelin Proteins 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 102100039398 C-X-C motif chemokine 2 Human genes 0.000 description 1
- IVQJSAQLEPPLTG-UHFFFAOYSA-N C1(CC1)C1=NNC2=CC=C(C=C12)C(=O)N(C(=O)NC)C Chemical compound C1(CC1)C1=NNC2=CC=C(C=C12)C(=O)N(C(=O)NC)C IVQJSAQLEPPLTG-UHFFFAOYSA-N 0.000 description 1
- CXCABFWIYFRVNF-UHFFFAOYSA-N C1(CC1)C1=NNC2=NC=C(C=C21)C(=O)N(C(=O)NC)C Chemical compound C1(CC1)C1=NNC2=NC=C(C=C21)C(=O)N(C(=O)NC)C CXCABFWIYFRVNF-UHFFFAOYSA-N 0.000 description 1
- VSEIDZLLWQQJGK-CHOZPQDDSA-N CCC1=C(C)C2=N\C\1=C/C1=C(C)C(C(O)=O)=C(N1)\C(CC(=O)N[C@@H](CC(O)=O)C(O)=O)=C1/N=C(/C=C3\N/C(=C\2)C(C=C)=C3C)[C@@H](C)[C@@H]1CCC(O)=O Chemical compound CCC1=C(C)C2=N\C\1=C/C1=C(C)C(C(O)=O)=C(N1)\C(CC(=O)N[C@@H](CC(O)=O)C(O)=O)=C1/N=C(/C=C3\N/C(=C\2)C(C=C)=C3C)[C@@H](C)[C@@H]1CCC(O)=O VSEIDZLLWQQJGK-CHOZPQDDSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 102000005701 Calcium-Binding Proteins Human genes 0.000 description 1
- 108010045403 Calcium-Binding Proteins Proteins 0.000 description 1
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 1
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- AOCCBINRVIKJHY-UHFFFAOYSA-N Carmofur Chemical compound CCCCCCNC(=O)N1C=C(F)C(=O)NC1=O AOCCBINRVIKJHY-UHFFFAOYSA-N 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 101150090188 Cdk8 gene Proteins 0.000 description 1
- 229920002284 Cellulose triacetate Polymers 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- NYRRKGWPJFGFDI-UHFFFAOYSA-N Cl.Fc1cc(ccc1Cl)C1CCCN1 Chemical compound Cl.Fc1cc(ccc1Cl)C1CCCN1 NYRRKGWPJFGFDI-UHFFFAOYSA-N 0.000 description 1
- VKIRLSJJSBMVHT-WBVHZDCISA-N ClC1=CC=C(C=C1)[C@H]1N(C[C@@H](C1)NC)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound ClC1=CC=C(C=C1)[C@H]1N(C[C@@H](C1)NC)C(=O)C=1C=C2C(=NC=1)NN=C2C VKIRLSJJSBMVHT-WBVHZDCISA-N 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- 108010024986 Cyclin-Dependent Kinase 2 Proteins 0.000 description 1
- 108010025464 Cyclin-Dependent Kinase 4 Proteins 0.000 description 1
- 102100032857 Cyclin-dependent kinase 1 Human genes 0.000 description 1
- 101710106279 Cyclin-dependent kinase 1 Proteins 0.000 description 1
- 102100033145 Cyclin-dependent kinase 19 Human genes 0.000 description 1
- 102100036239 Cyclin-dependent kinase 2 Human genes 0.000 description 1
- 102100036252 Cyclin-dependent kinase 4 Human genes 0.000 description 1
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 1
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 1
- GJKXGJCSJWBJEZ-XRSSZCMZSA-N Deslorelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CNC2=CC=CC=C12 GJKXGJCSJWBJEZ-XRSSZCMZSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- XXPXYPLPSDPERN-UHFFFAOYSA-N Ecteinascidin 743 Natural products COc1cc2C(NCCc2cc1O)C(=O)OCC3N4C(O)C5Cc6cc(C)c(OC)c(O)c6C(C4C(S)c7c(OC(=O)C)c(C)c8OCOc8c37)N5C XXPXYPLPSDPERN-UHFFFAOYSA-N 0.000 description 1
- 108700038672 Edotreotide Proteins 0.000 description 1
- 241000854350 Enicospilus group Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- OBMLHUPNRURLOK-XGRAFVIBSA-N Epitiostanol Chemical compound C1[C@@H]2S[C@@H]2C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 OBMLHUPNRURLOK-XGRAFVIBSA-N 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 229910052693 Europium Inorganic materials 0.000 description 1
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 1
- 108010069236 Goserelin Proteins 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000889128 Homo sapiens C-X-C motif chemokine 2 Proteins 0.000 description 1
- 101000944345 Homo sapiens Cyclin-dependent kinase 19 Proteins 0.000 description 1
- 101000904152 Homo sapiens Transcription factor E2F1 Proteins 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 108010023610 IL13-PE38 Proteins 0.000 description 1
- MPBVHIBUJCELCL-UHFFFAOYSA-N Ibandronate Chemical compound CCCCCN(C)CCC(O)(P(O)(O)=O)P(O)(O)=O MPBVHIBUJCELCL-UHFFFAOYSA-N 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 102100030694 Interleukin-11 Human genes 0.000 description 1
- 102000002791 Interleukin-8B Receptors Human genes 0.000 description 1
- 108010018951 Interleukin-8B Receptors Proteins 0.000 description 1
- BKCJZNIZRWYHBN-UHFFFAOYSA-N Isophosphamide mustard Chemical compound ClCCNP(=O)(O)NCCCl BKCJZNIZRWYHBN-UHFFFAOYSA-N 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 1
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 1
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 1
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 1
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 1
- 239000005536 L01XE08 - Nilotinib Substances 0.000 description 1
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 1
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 1
- 239000002145 L01XE14 - Bosutinib Substances 0.000 description 1
- 239000002146 L01XE16 - Crizotinib Substances 0.000 description 1
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 description 1
- 239000002138 L01XE21 - Regorafenib Substances 0.000 description 1
- 239000002139 L01XE22 - Masitinib Substances 0.000 description 1
- 239000002137 L01XE24 - Ponatinib Substances 0.000 description 1
- 239000002177 L01XE27 - Ibrutinib Substances 0.000 description 1
- UCEQXRCJXIVODC-PMACEKPBSA-N LSM-1131 Chemical compound C1CCC2=CC=CC3=C2N1C=C3[C@@H]1C(=O)NC(=O)[C@H]1C1=CNC2=CC=CC=C12 UCEQXRCJXIVODC-PMACEKPBSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229920001491 Lentinan Polymers 0.000 description 1
- 108010000817 Leuprolide Proteins 0.000 description 1
- HLFSDGLLUJUHTE-SNVBAGLBSA-N Levamisole Chemical compound C1([C@H]2CN3CCSC3=N2)=CC=CC=C1 HLFSDGLLUJUHTE-SNVBAGLBSA-N 0.000 description 1
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 1
- 108010080991 Mediator Complex Proteins 0.000 description 1
- 102000000490 Mediator Complex Human genes 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- QXKHYNVANLEOEG-UHFFFAOYSA-N Methoxsalen Chemical compound C1=CC(=O)OC2=C1C=C1C=COC1=C2OC QXKHYNVANLEOEG-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 102000029749 Microtubule Human genes 0.000 description 1
- 108091022875 Microtubule Proteins 0.000 description 1
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 1
- VIUAUNHCRHHYNE-JTQLQIEISA-N N-[(2S)-2,3-dihydroxypropyl]-3-(2-fluoro-4-iodoanilino)-4-pyridinecarboxamide Chemical compound OC[C@@H](O)CNC(=O)C1=CC=NC=C1NC1=CC=C(I)C=C1F VIUAUNHCRHHYNE-JTQLQIEISA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- LYPFDBRUNKHDGX-SOGSVHMOSA-N N1C2=CC=C1\C(=C1\C=CC(=N1)\C(=C1\C=C/C(/N1)=C(/C1=N/C(/CC1)=C2/C1=CC(O)=CC=C1)C1=CC(O)=CC=C1)\C1=CC(O)=CC=C1)C1=CC(O)=CC=C1 Chemical compound N1C2=CC=C1\C(=C1\C=CC(=N1)\C(=C1\C=C/C(/N1)=C(/C1=N/C(/CC1)=C2/C1=CC(O)=CC=C1)C1=CC(O)=CC=C1)\C1=CC(O)=CC=C1)C1=CC(O)=CC=C1 LYPFDBRUNKHDGX-SOGSVHMOSA-N 0.000 description 1
- GHVQKBGARCXIOU-KRWDZBQOSA-N N1N=CC2=CC(=CC=C12)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C Chemical compound N1N=CC2=CC(=CC=C12)[C@H]1N(CCC1)C(=O)C=1C=C2C(=NC=1)NN=C2C GHVQKBGARCXIOU-KRWDZBQOSA-N 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 108010021717 Nafarelin Proteins 0.000 description 1
- PAUXJRUOCRBJHR-MRXNPFEDSA-N Nc1n[nH]c(cc2)c1cc2C(N(CCC1)[C@H]1c(cc1)ccc1Cl)=O Chemical compound Nc1n[nH]c(cc2)c1cc2C(N(CCC1)[C@H]1c(cc1)ccc1Cl)=O PAUXJRUOCRBJHR-MRXNPFEDSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- DOIZQBMCPUAUBV-OAHLLOKOSA-N O=C(c1cc(cn[nH]2)c2nc1)N(CCC1)[C@H]1c(cc1)ccc1Br Chemical compound O=C(c1cc(cn[nH]2)c2nc1)N(CCC1)[C@H]1c(cc1)ccc1Br DOIZQBMCPUAUBV-OAHLLOKOSA-N 0.000 description 1
- 239000012425 OXONE® Substances 0.000 description 1
- 108010016076 Octreotide Proteins 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 108010057150 Peplomycin Proteins 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- KMSKQZKKOZQFFG-HSUXVGOQSA-N Pirarubicin Chemical compound O([C@H]1[C@@H](N)C[C@@H](O[C@H]1C)O[C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1CCCCO1 KMSKQZKKOZQFFG-HSUXVGOQSA-N 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- HFVNWDWLWUCIHC-GUPDPFMOSA-N Prednimustine Chemical compound O=C([C@@]1(O)CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)[C@@H](O)C[C@@]21C)COC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 HFVNWDWLWUCIHC-GUPDPFMOSA-N 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 239000005464 Radotinib Substances 0.000 description 1
- AHHFEZNOXOZZQA-ZEBDFXRSSA-N Ranimustine Chemical compound CO[C@H]1O[C@H](CNC(=O)N(CCCl)N=O)[C@@H](O)[C@H](O)[C@H]1O AHHFEZNOXOZZQA-ZEBDFXRSSA-N 0.000 description 1
- YJDYDFNKCBANTM-QCWCSKBGSA-N SDZ PSC 833 Chemical compound C\C=C\C[C@@H](C)C(=O)[C@@H]1N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C(=O)[C@H](C(C)C)NC1=O YJDYDFNKCBANTM-QCWCSKBGSA-N 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 229920000519 Sizofiran Polymers 0.000 description 1
- OCOKWVBYZHBHLU-UHFFFAOYSA-N Sobuzoxane Chemical compound C1C(=O)N(COC(=O)OCC(C)C)C(=O)CN1CCN1CC(=O)N(COC(=O)OCC(C)C)C(=O)C1 OCOKWVBYZHBHLU-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 102000004183 Synaptosomal-Associated Protein 25 Human genes 0.000 description 1
- 108010057722 Synaptosomal-Associated Protein 25 Proteins 0.000 description 1
- NAVMQTYZDKMPEU-UHFFFAOYSA-N Targretin Chemical compound CC1=CC(C(CCC2(C)C)(C)C)=C2C=C1C(=C)C1=CC=C(C(O)=O)C=C1 NAVMQTYZDKMPEU-UHFFFAOYSA-N 0.000 description 1
- GKLVYJBZJHMRIY-OUBTZVSYSA-N Technetium-99 Chemical compound [99Tc] GKLVYJBZJHMRIY-OUBTZVSYSA-N 0.000 description 1
- NCLGDOBQAWBXRA-PGRDOPGGSA-N Telotristat Chemical compound N1=C(C)C=CN1C1=CC(Cl)=CC=C1[C@H](C(F)(F)F)OC1=CC(C=2C=CC(C[C@H](N)C(O)=O)=CC=2)=NC(N)=N1 NCLGDOBQAWBXRA-PGRDOPGGSA-N 0.000 description 1
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 1
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- 108010078233 Thymalfasin Proteins 0.000 description 1
- 108010066702 Thyrotropin Alfa Proteins 0.000 description 1
- IVTVGDXNLFLDRM-HNNXBMFYSA-N Tomudex Chemical compound C=1C=C2NC(C)=NC(=O)C2=CC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)S1 IVTVGDXNLFLDRM-HNNXBMFYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- 102100024026 Transcription factor E2F1 Human genes 0.000 description 1
- 102000006747 Transforming Growth Factor alpha Human genes 0.000 description 1
- 101800004564 Transforming growth factor alpha Proteins 0.000 description 1
- YCPOZVAOBBQLRI-WDSKDSINSA-N Treosulfan Chemical compound CS(=O)(=O)OC[C@H](O)[C@@H](O)COS(C)(=O)=O YCPOZVAOBBQLRI-WDSKDSINSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 108010050144 Triptorelin Pamoate Proteins 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- PCWZKQSKUXXDDJ-UHFFFAOYSA-N Xanthotoxin Natural products COCc1c2OC(=O)C=Cc2cc3ccoc13 PCWZKQSKUXXDDJ-UHFFFAOYSA-N 0.000 description 1
- NNLVGZFZQQXQNW-ADJNRHBOSA-N [(2r,3r,4s,5r,6s)-4,5-diacetyloxy-3-[(2s,3r,4s,5r,6r)-3,4,5-triacetyloxy-6-(acetyloxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6s)-4,5,6-triacetyloxy-2-(acetyloxymethyl)oxan-3-yl]oxyoxan-2-yl]methyl acetate Chemical compound O([C@@H]1O[C@@H]([C@H]([C@H](OC(C)=O)[C@H]1OC(C)=O)O[C@H]1[C@@H]([C@@H](OC(C)=O)[C@H](OC(C)=O)[C@@H](COC(C)=O)O1)OC(C)=O)COC(=O)C)[C@@H]1[C@@H](COC(C)=O)O[C@@H](OC(C)=O)[C@H](OC(C)=O)[C@H]1OC(C)=O NNLVGZFZQQXQNW-ADJNRHBOSA-N 0.000 description 1
- XJXKGUZINMNEDK-GPJOBVNKSA-L [(4r,5r)-5-(aminomethyl)-2-propan-2-yl-1,3-dioxolan-4-yl]methanamine;platinum(2+);propanedioate Chemical compound [Pt+2].[O-]C(=O)CC([O-])=O.CC(C)C1O[C@H](CN)[C@@H](CN)O1 XJXKGUZINMNEDK-GPJOBVNKSA-L 0.000 description 1
- ZWGMJLNXIVRFRJ-UHFFFAOYSA-N [1-[(2-methylpropan-2-yl)oxycarbonyl]pyrrol-2-yl]boronic acid Chemical compound CC(C)(C)OC(=O)N1C=CC=C1B(O)O ZWGMJLNXIVRFRJ-UHFFFAOYSA-N 0.000 description 1
- XSMVECZRZBFTIZ-UHFFFAOYSA-M [2-(aminomethyl)cyclobutyl]methanamine;2-oxidopropanoate;platinum(4+) Chemical compound [Pt+4].CC([O-])C([O-])=O.NCC1CCC1CN XSMVECZRZBFTIZ-UHFFFAOYSA-M 0.000 description 1
- CCYVYPOBDYXDKY-UHFFFAOYSA-N [Br].ClC1=CC=C(C=C1)[Mg] Chemical compound [Br].ClC1=CC=C(C=C1)[Mg] CCYVYPOBDYXDKY-UHFFFAOYSA-N 0.000 description 1
- DGYIJVNZSDYBOE-UHFFFAOYSA-N [CH2]C1=CC=NC=C1 Chemical group [CH2]C1=CC=NC=C1 DGYIJVNZSDYBOE-UHFFFAOYSA-N 0.000 description 1
- SJZAPSHKTOTRBQ-UHFFFAOYSA-N [dimethylamino(triazolo[4,5-b]pyridin-1-yl)methylidene]-dimethylazanium Chemical compound C1=CC=C2[N+](=C(N(C)C)N(C)C)N=NC2=N1 SJZAPSHKTOTRBQ-UHFFFAOYSA-N 0.000 description 1
- 229960002184 abarelix Drugs 0.000 description 1
- AIWRTTMUVOZGPW-HSPKUQOVSA-N abarelix Chemical compound C([C@@H](C(=O)N[C@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)N(C)C(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(O)C=C1 AIWRTTMUVOZGPW-HSPKUQOVSA-N 0.000 description 1
- 108010023617 abarelix Proteins 0.000 description 1
- 229960000853 abiraterone Drugs 0.000 description 1
- GZOSMCIZMLWJML-VJLLXTKPSA-N abiraterone Chemical compound C([C@H]1[C@H]2[C@@H]([C@]3(CC[C@H](O)CC3=CC2)C)CC[C@@]11C)C=C1C1=CC=CN=C1 GZOSMCIZMLWJML-VJLLXTKPSA-N 0.000 description 1
- JLFVIEQMRKMAIT-UHFFFAOYSA-N ac1l9mnz Chemical compound O.O.O JLFVIEQMRKMAIT-UHFFFAOYSA-N 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 108010052004 acetyl-2-naphthylalanyl-3-chlorophenylalanyl-1-oxohexadecyl-seryl-4-aminophenylalanyl(hydroorotyl)-4-aminophenylalanyl(carbamoyl)-leucyl-ILys-prolyl-alaninamide Proteins 0.000 description 1
- USZYSDMBJDPRIF-SVEJIMAYSA-N aclacinomycin A Chemical compound O([C@H]1[C@@H](O)C[C@@H](O[C@H]1C)O[C@H]1[C@H](C[C@@H](O[C@H]1C)O[C@H]1C[C@]([C@@H](C2=CC=3C(=O)C4=CC=CC(O)=C4C(=O)C=3C(O)=C21)C(=O)OC)(O)CC)N(C)C)[C@H]1CCC(=O)[C@H](C)O1 USZYSDMBJDPRIF-SVEJIMAYSA-N 0.000 description 1
- 229960004176 aclarubicin Drugs 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 229960001686 afatinib Drugs 0.000 description 1
- ULXXDDBFHOBEHA-CWDCEQMOSA-N afatinib Chemical compound N1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-CWDCEQMOSA-N 0.000 description 1
- 229960002833 aflibercept Drugs 0.000 description 1
- 108010081667 aflibercept Proteins 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- QNAYBMKLOCPYGJ-UHFFFAOYSA-M alaninate Chemical compound CC(N)C([O-])=O QNAYBMKLOCPYGJ-UHFFFAOYSA-M 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 108700025316 aldesleukin Proteins 0.000 description 1
- 229960005310 aldesleukin Drugs 0.000 description 1
- 229960000548 alemtuzumab Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229950009447 alisertib Drugs 0.000 description 1
- 229960001445 alitretinoin Drugs 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229960004701 amonafide Drugs 0.000 description 1
- 229960002550 amrubicin Drugs 0.000 description 1
- VJZITPJGSQKZMX-XDPRQOKASA-N amrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC=C4C(=O)C=3C(O)=C21)(N)C(=O)C)[C@H]1C[C@H](O)[C@H](O)CO1 VJZITPJGSQKZMX-XDPRQOKASA-N 0.000 description 1
- 229960001220 amsacrine Drugs 0.000 description 1
- XCPGHVQEEXUHNC-UHFFFAOYSA-N amsacrine Chemical compound COC1=CC(NS(C)(=O)=O)=CC=C1NC1=C(C=CC=C2)C2=NC2=CC=CC=C12 XCPGHVQEEXUHNC-UHFFFAOYSA-N 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 1
- 238000005349 anion exchange Methods 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 238000011394 anticancer treatment Methods 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940045985 antineoplastic platinum compound Drugs 0.000 description 1
- 229960003982 apatinib Drugs 0.000 description 1
- 229950002465 apaziquone Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 239000003886 aromatase inhibitor Substances 0.000 description 1
- 229940046844 aromatase inhibitors Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 229960003005 axitinib Drugs 0.000 description 1
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 description 1
- 229960002756 azacitidine Drugs 0.000 description 1
- KLNFSAOEKUDMFA-UHFFFAOYSA-N azanide;2-hydroxyacetic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OCC(O)=O KLNFSAOEKUDMFA-UHFFFAOYSA-N 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 125000003725 azepanyl group Chemical group 0.000 description 1
- 125000004566 azetidin-1-yl group Chemical group N1(CCC1)* 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000004266 aziridin-1-yl group Chemical group [H]C1([H])N(*)C1([H])[H] 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 229950011276 belotecan Drugs 0.000 description 1
- LNHWXBUNXOXMRL-VWLOTQADSA-N belotecan Chemical compound C1=CC=C2C(CCNC(C)C)=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 LNHWXBUNXOXMRL-VWLOTQADSA-N 0.000 description 1
- 229960002707 bendamustine Drugs 0.000 description 1
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229950010559 besilesomab Drugs 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 229960002938 bexarotene Drugs 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 238000010256 biochemical assay Methods 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229950008548 bisantrene Drugs 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 229960001467 bortezomib Drugs 0.000 description 1
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 1
- 229960003736 bosutinib Drugs 0.000 description 1
- UBPYILGKFZZVDX-UHFFFAOYSA-N bosutinib Chemical compound C1=C(Cl)C(OC)=CC(NC=2C3=CC(OC)=C(OCCCN4CCN(C)CC4)C=C3N=CC=2C#N)=C1Cl UBPYILGKFZZVDX-UHFFFAOYSA-N 0.000 description 1
- 229960000455 brentuximab vedotin Drugs 0.000 description 1
- CUWODFFVMXJOKD-UVLQAERKSA-N buserelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](COC(C)(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 CUWODFFVMXJOKD-UVLQAERKSA-N 0.000 description 1
- 229960002719 buserelin Drugs 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 229960005069 calcium Drugs 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- IVFYLRMMHVYGJH-PVPPCFLZSA-N calusterone Chemical compound C1C[C@]2(C)[C@](O)(C)CC[C@H]2[C@@H]2[C@@H](C)CC3=CC(=O)CC[C@]3(C)[C@H]21 IVFYLRMMHVYGJH-PVPPCFLZSA-N 0.000 description 1
- 229950009823 calusterone Drugs 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical class C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 229960004117 capecitabine Drugs 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 150000001719 carbohydrate derivatives Chemical class 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 229960002438 carfilzomib Drugs 0.000 description 1
- BLMPQMFVWMYDKT-NZTKNTHTSA-N carfilzomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)[C@]1(C)OC1)NC(=O)CN1CCOCC1)CC1=CC=CC=C1 BLMPQMFVWMYDKT-NZTKNTHTSA-N 0.000 description 1
- 108010021331 carfilzomib Proteins 0.000 description 1
- 229960003261 carmofur Drugs 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 238000005341 cation exchange Methods 0.000 description 1
- 229960000419 catumaxomab Drugs 0.000 description 1
- 229960002412 cediranib Drugs 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- 229940044683 chemotherapy drug Drugs 0.000 description 1
- 235000019693 cherries Nutrition 0.000 description 1
- 239000007958 cherry flavor Substances 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- 229960002559 chlorotrianisene Drugs 0.000 description 1
- BFPSDSIWYFKGBC-UHFFFAOYSA-N chlorotrianisene Chemical compound C1=CC(OC)=CC=C1C(Cl)=C(C=1C=CC(OC)=CC=1)C1=CC=C(OC)C=C1 BFPSDSIWYFKGBC-UHFFFAOYSA-N 0.000 description 1
- 208000037976 chronic inflammation Diseases 0.000 description 1
- 230000006020 chronic inflammation Effects 0.000 description 1
- 229950009003 cilengitide Drugs 0.000 description 1
- AMLYAMJWYAIXIA-VWNVYAMZSA-N cilengitide Chemical compound N1C(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](C(C)C)N(C)C(=O)[C@H]1CC1=CC=CC=C1 AMLYAMJWYAIXIA-VWNVYAMZSA-N 0.000 description 1
- 229950010810 cintredekin besudotox Drugs 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960002436 cladribine Drugs 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229960000928 clofarabine Drugs 0.000 description 1
- WDDPHFBMKLOVOX-AYQXTPAHSA-N clofarabine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1F WDDPHFBMKLOVOX-AYQXTPAHSA-N 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 201000011024 colonic benign neoplasm Diseases 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229960005527 combretastatin A-4 phosphate Drugs 0.000 description 1
- WDOGQTQEKVLZIJ-WAYWQWQTSA-N combretastatin a-4 phosphate Chemical compound C1=C(OP(O)(O)=O)C(OC)=CC=C1\C=C/C1=CC(OC)=C(OC)C(OC)=C1 WDOGQTQEKVLZIJ-WAYWQWQTSA-N 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 229940125936 compound 42 Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 229960005061 crizotinib Drugs 0.000 description 1
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 210000000172 cytosol Anatomy 0.000 description 1
- 229960002465 dabrafenib Drugs 0.000 description 1
- BFSMGDJOXZAERB-UHFFFAOYSA-N dabrafenib Chemical compound S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 BFSMGDJOXZAERB-UHFFFAOYSA-N 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229950002205 dacomitinib Drugs 0.000 description 1
- LVXJQMNHJWSHET-AATRIKPKSA-N dacomitinib Chemical compound C=12C=C(NC(=O)\C=C\CN3CCCCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 LVXJQMNHJWSHET-AATRIKPKSA-N 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 229960002482 dalotuzumab Drugs 0.000 description 1
- 229960000766 danazol Drugs 0.000 description 1
- POZRVZJJTULAOH-LHZXLZLDSA-N danazol Chemical compound C1[C@]2(C)[C@H]3CC[C@](C)([C@](CC4)(O)C#C)[C@@H]4[C@@H]3CCC2=CC2=C1C=NO2 POZRVZJJTULAOH-LHZXLZLDSA-N 0.000 description 1
- 229960002448 dasatinib Drugs 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- 229960002272 degarelix Drugs 0.000 description 1
- MEUCPCLKGZSHTA-XYAYPHGZSA-N degarelix Chemical compound C([C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)NC(=O)[C@H](CC=1C=CC(NC(=O)[C@H]2NC(=O)NC(=O)C2)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(NC(N)=O)C=C1 MEUCPCLKGZSHTA-XYAYPHGZSA-N 0.000 description 1
- 230000003412 degenerative effect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 229960002923 denileukin diftitox Drugs 0.000 description 1
- 108010017271 denileukin diftitox Proteins 0.000 description 1
- 229960001251 denosumab Drugs 0.000 description 1
- 229960005408 deslorelin Drugs 0.000 description 1
- 108700025485 deslorelin Proteins 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- RGLYKWWBQGJZGM-ISLYRVAYSA-N diethylstilbestrol Chemical compound C=1C=C(O)C=CC=1C(/CC)=C(\CC)C1=CC=C(O)C=C1 RGLYKWWBQGJZGM-ISLYRVAYSA-N 0.000 description 1
- 229960000452 diethylstilbestrol Drugs 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 229950009859 dinaciclib Drugs 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229950005778 dovitinib Drugs 0.000 description 1
- ZWAOHEXOSAUJHY-ZIYNGMLESA-N doxifluridine Chemical compound O[C@@H]1[C@H](O)[C@@H](C)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ZWAOHEXOSAUJHY-ZIYNGMLESA-N 0.000 description 1
- 229950005454 doxifluridine Drugs 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 229950006595 edotreotide Drugs 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 229960004137 elotuzumab Drugs 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 150000002085 enols Chemical group 0.000 description 1
- INVTYAOGFAGBOE-UHFFFAOYSA-N entinostat Chemical compound NC1=CC=CC=C1NC(=O)C(C=C1)=CC=C1CNC(=O)OCC1=CC=CN=C1 INVTYAOGFAGBOE-UHFFFAOYSA-N 0.000 description 1
- 229950005837 entinostat Drugs 0.000 description 1
- 229960004671 enzalutamide Drugs 0.000 description 1
- WXCXUHSOUPDCQV-UHFFFAOYSA-N enzalutamide Chemical compound C1=C(F)C(C(=O)NC)=CC=C1N1C(C)(C)C(=O)N(C=2C=C(C(C#N)=CC=2)C(F)(F)F)C1=S WXCXUHSOUPDCQV-UHFFFAOYSA-N 0.000 description 1
- 229950002189 enzastaurin Drugs 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 229950002973 epitiostanol Drugs 0.000 description 1
- 229950009760 epratuzumab Drugs 0.000 description 1
- 229950006835 eptaplatin Drugs 0.000 description 1
- 229960003649 eribulin Drugs 0.000 description 1
- UFNVPOGXISZXJD-XJPMSQCNSA-N eribulin Chemical compound C([C@H]1CC[C@@H]2O[C@@H]3[C@H]4O[C@H]5C[C@](O[C@H]4[C@H]2O1)(O[C@@H]53)CC[C@@H]1O[C@H](C(C1)=C)CC1)C(=O)C[C@@H]2[C@@H](OC)[C@@H](C[C@H](O)CN)O[C@H]2C[C@@H]2C(=C)[C@H](C)C[C@H]1O2 UFNVPOGXISZXJD-XJPMSQCNSA-N 0.000 description 1
- 229960001433 erlotinib Drugs 0.000 description 1
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- 229960005309 estradiol Drugs 0.000 description 1
- 229930182833 estradiol Natural products 0.000 description 1
- 229960001842 estramustine Drugs 0.000 description 1
- FRPJXPJMRWBBIH-RBRWEJTLSA-N estramustine Chemical compound ClCCN(CCCl)C(=O)OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 FRPJXPJMRWBBIH-RBRWEJTLSA-N 0.000 description 1
- 229950006566 etanidazole Drugs 0.000 description 1
- WCDWBPCFGJXFJZ-UHFFFAOYSA-N etanidazole Chemical compound OCCNC(=O)CN1C=CN=C1[N+]([O-])=O WCDWBPCFGJXFJZ-UHFFFAOYSA-N 0.000 description 1
- 125000001033 ether group Chemical group 0.000 description 1
- HHFAWKCIHAUFRX-UHFFFAOYSA-N ethoxide Chemical compound CC[O-] HHFAWKCIHAUFRX-UHFFFAOYSA-N 0.000 description 1
- ZULBYKANHBUAOW-UHFFFAOYSA-N ethyl 3-iodo-2h-indazole-5-carboxylate Chemical compound CCOC(=O)C1=CC=C2NN=C(I)C2=C1 ZULBYKANHBUAOW-UHFFFAOYSA-N 0.000 description 1
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005167 everolimus Drugs 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 229960000255 exemestane Drugs 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 229950011548 fadrozole Drugs 0.000 description 1
- 229950009929 farletuzumab Drugs 0.000 description 1
- 239000010685 fatty oil Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- ODKNJVUHOIMIIZ-RRKCRQDMSA-N floxuridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(F)=C1 ODKNJVUHOIMIIZ-RRKCRQDMSA-N 0.000 description 1
- 229960000961 floxuridine Drugs 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- GAKMQHDJQHZUTJ-ULHLPKEOSA-N fluocortolone Chemical compound C1([C@@H](F)C2)=CC(=O)C=C[C@]1(C)[C@@H]1[C@@H]2[C@@H]2C[C@@H](C)[C@H](C(=O)CO)[C@@]2(C)C[C@@H]1O GAKMQHDJQHZUTJ-ULHLPKEOSA-N 0.000 description 1
- 229960003973 fluocortolone Drugs 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 1
- 229960002074 flutamide Drugs 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 229960004421 formestane Drugs 0.000 description 1
- OSVMTWJCGUFAOD-KZQROQTASA-N formestane Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1O OSVMTWJCGUFAOD-KZQROQTASA-N 0.000 description 1
- 229960004783 fotemustine Drugs 0.000 description 1
- YAKWPXVTIGTRJH-UHFFFAOYSA-N fotemustine Chemical compound CCOP(=O)(OCC)C(C)NC(=O)N(CCCl)N=O YAKWPXVTIGTRJH-UHFFFAOYSA-N 0.000 description 1
- 229960002258 fulvestrant Drugs 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 229950004161 ganetespib Drugs 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- 229960003297 gemtuzumab ozogamicin Drugs 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 229950011595 glufosfamide Drugs 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 229960002913 goserelin Drugs 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- HHXHVIJIIXKSOE-QILQGKCVSA-N histrelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC(N=C1)=CN1CC1=CC=CC=C1 HHXHVIJIIXKSOE-QILQGKCVSA-N 0.000 description 1
- 108700020746 histrelin Proteins 0.000 description 1
- 229960002193 histrelin Drugs 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 229960005236 ibandronic acid Drugs 0.000 description 1
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 1
- 229960001507 ibrutinib Drugs 0.000 description 1
- XYFPWWZEPKGCCK-GOSISDBHSA-N ibrutinib Chemical compound C1=2C(N)=NC=NC=2N([C@H]2CN(CCC2)C(=O)C=C)N=C1C(C=C1)=CC=C1OC1=CC=CC=C1 XYFPWWZEPKGCCK-GOSISDBHSA-N 0.000 description 1
- 229950007440 icotinib Drugs 0.000 description 1
- QQLKULDARVNMAL-UHFFFAOYSA-N icotinib Chemical compound C#CC1=CC=CC(NC=2C3=CC=4OCCOCCOCCOC=4C=C3N=CN=2)=C1 QQLKULDARVNMAL-UHFFFAOYSA-N 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229950008268 idronoxil Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960002411 imatinib Drugs 0.000 description 1
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 1
- 229960002751 imiquimod Drugs 0.000 description 1
- DOUYETYNHWVLEO-UHFFFAOYSA-N imiquimod Chemical compound C1=CC=CC2=C3N(CC(C)C)C=NC3=C(N)N=C21 DOUYETYNHWVLEO-UHFFFAOYSA-N 0.000 description 1
- 229950008097 improsulfan Drugs 0.000 description 1
- DBIGHPPNXATHOF-UHFFFAOYSA-N improsulfan Chemical compound CS(=O)(=O)OCCCNCCCOS(C)(=O)=O DBIGHPPNXATHOF-UHFFFAOYSA-N 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 229940060367 inert ingredients Drugs 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 229950002133 iniparib Drugs 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229950004101 inotuzumab ozogamicin Drugs 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 229950000038 interferon alfa Drugs 0.000 description 1
- 229960003521 interferon alfa-2a Drugs 0.000 description 1
- 229960003507 interferon alfa-2b Drugs 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 229960003795 iobenguane (123i) Drugs 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229960005386 ipilimumab Drugs 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229960002014 ixabepilone Drugs 0.000 description 1
- FABUFPQFXZVHFB-CFWQTKTJSA-N ixabepilone Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@H](C)C(=O)C(C)(C)[C@H](O)CC(=O)N1)O)C)=C\C1=CSC(C)=N1 FABUFPQFXZVHFB-CFWQTKTJSA-N 0.000 description 1
- 229960003648 ixazomib Drugs 0.000 description 1
- MXAYKZJJDUDWDS-LBPRGKRZSA-N ixazomib Chemical compound CC(C)C[C@@H](B(O)O)NC(=O)CNC(=O)C1=CC(Cl)=CC=C1Cl MXAYKZJJDUDWDS-LBPRGKRZSA-N 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960004891 lapatinib Drugs 0.000 description 1
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 1
- 229950000340 laromustine Drugs 0.000 description 1
- 229960004942 lenalidomide Drugs 0.000 description 1
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 1
- 229940115286 lentinan Drugs 0.000 description 1
- 229960003784 lenvatinib Drugs 0.000 description 1
- WOSKHXYHFSIKNG-UHFFFAOYSA-N lenvatinib Chemical compound C=12C=C(C(N)=O)C(OC)=CC2=NC=CC=1OC(C=C1Cl)=CC=C1NC(=O)NC1CC1 WOSKHXYHFSIKNG-UHFFFAOYSA-N 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 1
- 229960004338 leuprorelin Drugs 0.000 description 1
- 229960001614 levamisole Drugs 0.000 description 1
- XMGQYMWWDOXHJM-UHFFFAOYSA-N limonene Chemical compound CC(=C)C1CCC(C)=CC1 XMGQYMWWDOXHJM-UHFFFAOYSA-N 0.000 description 1
- 229950002216 linifanib Drugs 0.000 description 1
- MPVGZUGXCQEXTM-UHFFFAOYSA-N linifanib Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C(N)=NNC=3C=CC=2)=C1 MPVGZUGXCQEXTM-UHFFFAOYSA-N 0.000 description 1
- 229950001762 linsitinib Drugs 0.000 description 1
- PKCDDUHJAFVJJB-VLZXCDOPSA-N linsitinib Chemical compound C1[C@](C)(O)C[C@@H]1C1=NC(C=2C=C3N=C(C=CC3=CC=2)C=2C=CC=CC=2)=C2N1C=CN=C2N PKCDDUHJAFVJJB-VLZXCDOPSA-N 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- DLEDOFVPSDKWEF-UHFFFAOYSA-N lithium butane Chemical compound [Li+].CCC[CH2-] DLEDOFVPSDKWEF-UHFFFAOYSA-N 0.000 description 1
- 229950008991 lobaplatin Drugs 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 229960003538 lonidamine Drugs 0.000 description 1
- WDRYRZXSPDWGEB-UHFFFAOYSA-N lonidamine Chemical compound C12=CC=CC=C2C(C(=O)O)=NN1CC1=CC=C(Cl)C=C1Cl WDRYRZXSPDWGEB-UHFFFAOYSA-N 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 208000037841 lung tumor Diseases 0.000 description 1
- 206010025135 lupus erythematosus Diseases 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 229960004655 masitinib Drugs 0.000 description 1
- WJEOLQLKVOPQFV-UHFFFAOYSA-N masitinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3SC=C(N=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 WJEOLQLKVOPQFV-UHFFFAOYSA-N 0.000 description 1
- 229950008001 matuzumab Drugs 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229960001786 megestrol Drugs 0.000 description 1
- JBVNBBXAMBZTMQ-CEGNMAFCSA-N megestrol Chemical compound C1=CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(=O)C)(O)[C@@]1(C)CC2 JBVNBBXAMBZTMQ-CEGNMAFCSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- NBTOZLQBSIZIKS-UHFFFAOYSA-N methoxide Chemical compound [O-]C NBTOZLQBSIZIKS-UHFFFAOYSA-N 0.000 description 1
- 229960004469 methoxsalen Drugs 0.000 description 1
- YJXJLESRRZVQPW-UHFFFAOYSA-N methyl 3-methyl-2h-pyrazolo[3,4-b]pyridine-5-carboxylate Chemical compound C1=C(C(=O)OC)C=NC2=NNC(C)=C21 YJXJLESRRZVQPW-UHFFFAOYSA-N 0.000 description 1
- YNBADRVTZLEFNH-UHFFFAOYSA-N methyl nicotinate Chemical compound COC(=O)C1=CC=CN=C1 YNBADRVTZLEFNH-UHFFFAOYSA-N 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 210000004688 microtubule Anatomy 0.000 description 1
- 229950010895 midostaurin Drugs 0.000 description 1
- BMGQWWVMWDBQGC-IIFHNQTCSA-N midostaurin Chemical compound CN([C@H]1[C@H]([C@]2(C)O[C@@H](N3C4=CC=CC=C4C4=C5C(=O)NCC5=C5C6=CC=CC=C6N2C5=C43)C1)OC)C(=O)C1=CC=CC=C1 BMGQWWVMWDBQGC-IIFHNQTCSA-N 0.000 description 1
- 229960005225 mifamurtide Drugs 0.000 description 1
- 108700007621 mifamurtide Proteins 0.000 description 1
- 229960003775 miltefosine Drugs 0.000 description 1
- PQLXHQMOHUQAKB-UHFFFAOYSA-N miltefosine Chemical compound CCCCCCCCCCCCCCCCOP([O-])(=O)OCC[N+](C)(C)C PQLXHQMOHUQAKB-UHFFFAOYSA-N 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 229960005485 mitobronitol Drugs 0.000 description 1
- VFKZTMPDYBFSTM-GUCUJZIJSA-N mitolactol Chemical compound BrC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-GUCUJZIJSA-N 0.000 description 1
- 229950010913 mitolactol Drugs 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 229950007699 mogamulizumab Drugs 0.000 description 1
- 229950003968 motesanib Drugs 0.000 description 1
- RAHBGWKEPAQNFF-UHFFFAOYSA-N motesanib Chemical compound C=1C=C2C(C)(C)CNC2=CC=1NC(=O)C1=CC=CN=C1NCC1=CC=NC=C1 RAHBGWKEPAQNFF-UHFFFAOYSA-N 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- LNOPIUAQISRISI-UHFFFAOYSA-N n'-hydroxy-2-propan-2-ylsulfonylethanimidamide Chemical compound CC(C)S(=O)(=O)CC(N)=NO LNOPIUAQISRISI-UHFFFAOYSA-N 0.000 description 1
- MZRVEZGGRBJDDB-UHFFFAOYSA-N n-Butyllithium Substances [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 1
- NJSMWLQOCQIOPE-OCHFTUDZSA-N n-[(e)-[10-[(e)-(4,5-dihydro-1h-imidazol-2-ylhydrazinylidene)methyl]anthracen-9-yl]methylideneamino]-4,5-dihydro-1h-imidazol-2-amine Chemical compound N1CCN=C1N\N=C\C(C1=CC=CC=C11)=C(C=CC=C2)C2=C1\C=N\NC1=NCCN1 NJSMWLQOCQIOPE-OCHFTUDZSA-N 0.000 description 1
- XNSAINXGIQZQOO-UHFFFAOYSA-N n-[1-(2-carbamoylpyrrolidin-1-yl)-3-(1h-imidazol-5-yl)-1-oxopropan-2-yl]-5-oxopyrrolidine-2-carboxamide Chemical compound NC(=O)C1CCCN1C(=O)C(NC(=O)C1NC(=O)CC1)CC1=CN=CN1 XNSAINXGIQZQOO-UHFFFAOYSA-N 0.000 description 1
- WPEWQEMJFLWMLV-UHFFFAOYSA-N n-[4-(1-cyanocyclopentyl)phenyl]-2-(pyridin-4-ylmethylamino)pyridine-3-carboxamide Chemical compound C=1C=CN=C(NCC=2C=CN=CC=2)C=1C(=O)NC(C=C1)=CC=C1C1(C#N)CCCC1 WPEWQEMJFLWMLV-UHFFFAOYSA-N 0.000 description 1
- RWHUEXWOYVBUCI-ITQXDASVSA-N nafarelin Chemical compound C([C@@H](C(=O)N[C@H](CC=1C=C2C=CC=CC2=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 RWHUEXWOYVBUCI-ITQXDASVSA-N 0.000 description 1
- 229960002333 nafarelin Drugs 0.000 description 1
- 229960004719 nandrolone Drugs 0.000 description 1
- NPAGDVCDWIYMMC-IZPLOLCNSA-N nandrolone Chemical compound O=C1CC[C@@H]2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 NPAGDVCDWIYMMC-IZPLOLCNSA-N 0.000 description 1
- YZMHQCWXYHARLS-UHFFFAOYSA-N naphthalene-1,2-disulfonic acid Chemical class C1=CC=CC2=C(S(O)(=O)=O)C(S(=O)(=O)O)=CC=C21 YZMHQCWXYHARLS-UHFFFAOYSA-N 0.000 description 1
- 229950009793 naptumomab estafenatox Drugs 0.000 description 1
- 229960000513 necitumumab Drugs 0.000 description 1
- 229950007221 nedaplatin Drugs 0.000 description 1
- IXOXBSCIXZEQEQ-UHTZMRCNSA-N nelarabine Chemical compound C1=NC=2C(OC)=NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O IXOXBSCIXZEQEQ-UHTZMRCNSA-N 0.000 description 1
- 229960000801 nelarabine Drugs 0.000 description 1
- QZGIWPZCWHMVQL-UIYAJPBUSA-N neocarzinostatin chromophore Chemical compound O1[C@H](C)[C@H](O)[C@H](O)[C@@H](NC)[C@H]1O[C@@H]1C/2=C/C#C[C@H]3O[C@@]3([C@@H]3OC(=O)OC3)C#CC\2=C[C@H]1OC(=O)C1=C(O)C=CC2=C(C)C=C(OC)C=C12 QZGIWPZCWHMVQL-UIYAJPBUSA-N 0.000 description 1
- 229950008835 neratinib Drugs 0.000 description 1
- JWNPDZNEKVCWMY-VQHVLOKHSA-N neratinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 JWNPDZNEKVCWMY-VQHVLOKHSA-N 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 description 1
- 229960001346 nilotinib Drugs 0.000 description 1
- XWXYUMMDTVBTOU-UHFFFAOYSA-N nilutamide Chemical compound O=C1C(C)(C)NC(=O)N1C1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 XWXYUMMDTVBTOU-UHFFFAOYSA-N 0.000 description 1
- 229960002653 nilutamide Drugs 0.000 description 1
- MDJFHRLTPRPZLY-UHFFFAOYSA-N nimorazole Chemical compound [O-][N+](=O)C1=CN=CN1CCN1CCOCC1 MDJFHRLTPRPZLY-UHFFFAOYSA-N 0.000 description 1
- 229960004918 nimorazole Drugs 0.000 description 1
- 229950010203 nimotuzumab Drugs 0.000 description 1
- 229960001420 nimustine Drugs 0.000 description 1
- VFEDRRNHLBGPNN-UHFFFAOYSA-N nimustine Chemical compound CC1=NC=C(CNC(=O)N(CCCl)N=O)C(N)=N1 VFEDRRNHLBGPNN-UHFFFAOYSA-N 0.000 description 1
- 229960004378 nintedanib Drugs 0.000 description 1
- XZXHXSATPCNXJR-ZIADKAODSA-N nintedanib Chemical compound O=C1NC2=CC(C(=O)OC)=CC=C2\C1=C(C=1C=CC=CC=1)\NC(C=C1)=CC=C1N(C)C(=O)CN1CCN(C)CC1 XZXHXSATPCNXJR-ZIADKAODSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 229960003347 obinutuzumab Drugs 0.000 description 1
- 229950009090 ocaratuzumab Drugs 0.000 description 1
- 229960002700 octreotide Drugs 0.000 description 1
- 229960002450 ofatumumab Drugs 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229950000846 onartuzumab Drugs 0.000 description 1
- 231100000590 oncogenic Toxicity 0.000 description 1
- 230000002246 oncogenic effect Effects 0.000 description 1
- 229950009057 oportuzumab monatox Drugs 0.000 description 1
- 108010046821 oprelvekin Proteins 0.000 description 1
- 229960001840 oprelvekin Drugs 0.000 description 1
- 239000007968 orange flavor Substances 0.000 description 1
- 229950006354 orantinib Drugs 0.000 description 1
- 229950007283 oregovomab Drugs 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 229950004023 orteronel Drugs 0.000 description 1
- 201000008482 osteoarthritis Diseases 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 229950000755 palifosfamide Drugs 0.000 description 1
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 1
- 229960003978 pamidronic acid Drugs 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- 229960005184 panobinostat Drugs 0.000 description 1
- FPOHNWQLNRZRFC-ZHACJKMWSA-N panobinostat Chemical compound CC=1NC2=CC=CC=C2C=1CCNCC1=CC=C(\C=C\C(=O)NO)C=C1 FPOHNWQLNRZRFC-ZHACJKMWSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 229960000639 pazopanib Drugs 0.000 description 1
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 1
- 229960001744 pegaspargase Drugs 0.000 description 1
- 108010001564 pegaspargase Proteins 0.000 description 1
- 229960005079 pemetrexed Drugs 0.000 description 1
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 1
- 229960002340 pentostatin Drugs 0.000 description 1
- FPVKHBSQESCIEP-JQCXWYLXSA-N pentostatin Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(N=CNC[C@H]2O)=C2N=C1 FPVKHBSQESCIEP-JQCXWYLXSA-N 0.000 description 1
- QIMGFXOHTOXMQP-GFAGFCTOSA-N peplomycin Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCCN[C@@H](C)C=1C=CC=CC=1)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1NC=NC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C QIMGFXOHTOXMQP-GFAGFCTOSA-N 0.000 description 1
- 229950003180 peplomycin Drugs 0.000 description 1
- 238000010647 peptide synthesis reaction Methods 0.000 description 1
- 229950010307 peretinoin Drugs 0.000 description 1
- SZFPYBIJACMNJV-UHFFFAOYSA-N perifosine Chemical compound CCCCCCCCCCCCCCCCCCOP([O-])(=O)OC1CC[N+](C)(C)CC1 SZFPYBIJACMNJV-UHFFFAOYSA-N 0.000 description 1
- 229950010632 perifosine Drugs 0.000 description 1
- 229960002087 pertuzumab Drugs 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 239000003504 photosensitizing agent Substances 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 229950005566 picoplatin Drugs 0.000 description 1
- IIMIOEBMYPRQGU-UHFFFAOYSA-L picoplatin Chemical compound N.[Cl-].[Cl-].[Pt+2].CC1=CC=CC=N1 IIMIOEBMYPRQGU-UHFFFAOYSA-L 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229950002592 pimasertib Drugs 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229960000952 pipobroman Drugs 0.000 description 1
- NJBFOOCLYDNZJN-UHFFFAOYSA-N pipobroman Chemical compound BrCCC(=O)N1CCN(C(=O)CCBr)CC1 NJBFOOCLYDNZJN-UHFFFAOYSA-N 0.000 description 1
- 229960001221 pirarubicin Drugs 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-N pivalic acid Chemical compound CC(C)(C)C(O)=O IUGYQRQAERSCNH-UHFFFAOYSA-N 0.000 description 1
- 229960004403 pixantrone Drugs 0.000 description 1
- PEZPMAYDXJQYRV-UHFFFAOYSA-N pixantrone Chemical compound O=C1C2=CN=CC=C2C(=O)C2=C1C(NCCN)=CC=C2NCCN PEZPMAYDXJQYRV-UHFFFAOYSA-N 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 229950008499 plitidepsin Drugs 0.000 description 1
- UUSZLLQJYRSZIS-LXNNNBEUSA-N plitidepsin Chemical compound CN([C@H](CC(C)C)C(=O)N[C@@H]1C(=O)N[C@@H]([C@H](CC(=O)O[C@H](C(=O)[C@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N2CCC[C@H]2C(=O)N(C)[C@@H](CC=2C=CC(OC)=CC=2)C(=O)O[C@@H]1C)C(C)C)O)[C@@H](C)CC)C(=O)[C@@H]1CCCN1C(=O)C(C)=O UUSZLLQJYRSZIS-LXNNNBEUSA-N 0.000 description 1
- 108010049948 plitidepsin Proteins 0.000 description 1
- 210000001778 pluripotent stem cell Anatomy 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229960000688 pomalidomide Drugs 0.000 description 1
- UVSMNLNDYGZFPF-UHFFFAOYSA-N pomalidomide Chemical compound O=C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O UVSMNLNDYGZFPF-UHFFFAOYSA-N 0.000 description 1
- 229960001131 ponatinib Drugs 0.000 description 1
- PHXJVRSECIGDHY-UHFFFAOYSA-N ponatinib Chemical compound C1CN(C)CCN1CC(C(=C1)C(F)(F)F)=CC=C1NC(=O)C1=CC=C(C)C(C#CC=2N3N=CC=CC3=NC=2)=C1 PHXJVRSECIGDHY-UHFFFAOYSA-N 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- OKBMCNHOEMXPTM-UHFFFAOYSA-M potassium peroxymonosulfate Chemical compound [K+].OOS([O-])(=O)=O OKBMCNHOEMXPTM-UHFFFAOYSA-M 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- OGSBUKJUDHAQEA-WMCAAGNKSA-N pralatrexate Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CC(CC#C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OGSBUKJUDHAQEA-WMCAAGNKSA-N 0.000 description 1
- 229960000214 pralatrexate Drugs 0.000 description 1
- 229960004694 prednimustine Drugs 0.000 description 1
- OIGNJSKKLXVSLS-VWUMJDOOSA-N prednisolone Chemical compound O=C1C=C[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OIGNJSKKLXVSLS-VWUMJDOOSA-N 0.000 description 1
- 229960005205 prednisolone Drugs 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- XYWJNTOURDMTPI-UHFFFAOYSA-N procodazole Chemical compound C1=CC=C2NC(CCC(=O)O)=NC2=C1 XYWJNTOURDMTPI-UHFFFAOYSA-N 0.000 description 1
- 229950000989 procodazole Drugs 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- NSETWVJZUWGCKE-UHFFFAOYSA-N propylphosphonic acid Chemical compound CCCP(O)(O)=O NSETWVJZUWGCKE-UHFFFAOYSA-N 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 1
- 125000004944 pyrazin-3-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 1
- 125000002206 pyridazin-3-yl group Chemical group [H]C1=C([H])C([H])=C(*)N=N1 0.000 description 1
- 125000004940 pyridazin-4-yl group Chemical group N1=NC=C(C=C1)* 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 229950011613 racotumomab Drugs 0.000 description 1
- 229950004043 radotinib Drugs 0.000 description 1
- DUPWHXBITIZIKZ-UHFFFAOYSA-N radotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3N=CC=NC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 DUPWHXBITIZIKZ-UHFFFAOYSA-N 0.000 description 1
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 1
- 229960004622 raloxifene Drugs 0.000 description 1
- 229960004432 raltitrexed Drugs 0.000 description 1
- 229960002633 ramucirumab Drugs 0.000 description 1
- 229960002185 ranimustine Drugs 0.000 description 1
- BMKDZUISNHGIBY-UHFFFAOYSA-N razoxane Chemical compound C1C(=O)NC(=O)CN1C(C)CN1CC(=O)NC(=O)C1 BMKDZUISNHGIBY-UHFFFAOYSA-N 0.000 description 1
- 229960000460 razoxane Drugs 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 229960004836 regorafenib Drugs 0.000 description 1
- FNHKPVJBJVTLMP-UHFFFAOYSA-N regorafenib Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=C(F)C(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 FNHKPVJBJVTLMP-UHFFFAOYSA-N 0.000 description 1
- OIRUWDYJGMHDHJ-AFXVCOSJSA-N retaspimycin hydrochloride Chemical compound Cl.N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(O)C1=CC(O)=C2NCC=C OIRUWDYJGMHDHJ-AFXVCOSJSA-N 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229960001302 ridaforolimus Drugs 0.000 description 1
- 229950006764 rigosertib Drugs 0.000 description 1
- OWBFCJROIKNMGD-BQYQJAHWSA-N rigosertib Chemical compound COC1=CC(OC)=CC(OC)=C1\C=C\S(=O)(=O)CC1=CC=C(OC)C(NCC(O)=O)=C1 OWBFCJROIKNMGD-BQYQJAHWSA-N 0.000 description 1
- 229950003238 rilotumumab Drugs 0.000 description 1
- 229960005560 rindopepimut Drugs 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- OHRURASPPZQGQM-GCCNXGTGSA-N romidepsin Chemical compound O1C(=O)[C@H](C(C)C)NC(=O)C(=C/C)/NC(=O)[C@H]2CSSCC\C=C\[C@@H]1CC(=O)N[C@H](C(C)C)C(=O)N2 OHRURASPPZQGQM-GCCNXGTGSA-N 0.000 description 1
- 229960003452 romidepsin Drugs 0.000 description 1
- 108010091666 romidepsin Proteins 0.000 description 1
- OHRURASPPZQGQM-UHFFFAOYSA-N romidepsin Natural products O1C(=O)C(C(C)C)NC(=O)C(=CC)NC(=O)C2CSSCCC=CC1CC(=O)NC(C(C)C)C(=O)N2 OHRURASPPZQGQM-UHFFFAOYSA-N 0.000 description 1
- 229960000215 ruxolitinib Drugs 0.000 description 1
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229950006896 sapacitabine Drugs 0.000 description 1
- LBGFKUUHOPIEMA-PEARBKPGSA-N sapacitabine Chemical compound O=C1N=C(NC(=O)CCCCCCCCCCCCCCC)C=CN1[C@H]1[C@@H](C#N)[C@H](O)[C@@H](CO)O1 LBGFKUUHOPIEMA-PEARBKPGSA-N 0.000 description 1
- 229960005399 satraplatin Drugs 0.000 description 1
- 190014017285 satraplatin Chemical compound 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 229960003323 siltuximab Drugs 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 229950001403 sizofiran Drugs 0.000 description 1
- 229950010372 sobuzoxane Drugs 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- NGIYLSFJGRLEMI-MHTUOZSYSA-M sodium 2-[[(2S)-2-[[(4R)-4-[[(2S)-2-[[(2R)-2-[(2R,3R,4R,5R)-2-acetamido-4,5,6-trihydroxy-1-oxohexan-3-yl]oxypropanoyl]amino]propanoyl]amino]-5-amino-5-oxopentanoyl]amino]propanoyl]amino]ethyl [(2R)-2,3-di(hexadecanoyloxy)propyl] phosphate hydrate Chemical compound O.[Na+].CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCCNC(=O)[C@H](C)NC(=O)CC[C@@H](NC(=O)[C@H](C)NC(=O)[C@@H](C)O[C@@H]([C@H](O)[C@H](O)CO)[C@@H](NC(C)=O)C=O)C(N)=O)OC(=O)CCCCCCCCCCCCCCC NGIYLSFJGRLEMI-MHTUOZSYSA-M 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- RMBAVIFYHOYIFM-UHFFFAOYSA-M sodium methanethiolate Chemical compound [Na+].[S-]C RMBAVIFYHOYIFM-UHFFFAOYSA-M 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- MIXCUJKCXRNYFM-UHFFFAOYSA-M sodium;diiodomethanesulfonate;n-propyl-n-[2-(2,4,6-trichlorophenoxy)ethyl]imidazole-1-carboxamide Chemical compound [Na+].[O-]S(=O)(=O)C(I)I.C1=CN=CN1C(=O)N(CCC)CCOC1=C(Cl)C=C(Cl)C=C1Cl MIXCUJKCXRNYFM-UHFFFAOYSA-M 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 239000000126 substance Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 229950010265 tabalumab Drugs 0.000 description 1
- 229950010924 talaporfin Drugs 0.000 description 1
- MUTNCGKQJGXKEM-UHFFFAOYSA-N tamibarotene Chemical compound C=1C=C2C(C)(C)CCC(C)(C)C2=CC=1NC(=O)C1=CC=C(C(O)=O)C=C1 MUTNCGKQJGXKEM-UHFFFAOYSA-N 0.000 description 1
- 229950010130 tamibarotene Drugs 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 238000002626 targeted therapy Methods 0.000 description 1
- 229960003102 tasonermin Drugs 0.000 description 1
- 229950001899 tasquinimod Drugs 0.000 description 1
- ONDYALNGTUAJDX-UHFFFAOYSA-N tasquinimod Chemical compound OC=1C=2C(OC)=CC=CC=2N(C)C(=O)C=1C(=O)N(C)C1=CC=C(C(F)(F)F)C=C1 ONDYALNGTUAJDX-UHFFFAOYSA-N 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229940059294 technetium (99mtc) arcitumomab Drugs 0.000 description 1
- 229960001674 tegafur Drugs 0.000 description 1
- WFWLQNSHRPWKFK-ZCFIWIBFSA-N tegafur Chemical compound O=C1NC(=O)C(F)=CN1[C@@H]1OCCC1 WFWLQNSHRPWKFK-ZCFIWIBFSA-N 0.000 description 1
- 229950002246 telotristat Drugs 0.000 description 1
- 229960002197 temoporfin Drugs 0.000 description 1
- 229960004964 temozolomide Drugs 0.000 description 1
- 229960000235 temsirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-UHFFFAOYSA-N temsirolimus Natural products C1CC(O)C(OC)CC1CC(C)C1OC(=O)C2CCCCN2C(=O)C(=O)C(O)(O2)C(C)CCC2CC(OC)C(C)=CC=CC=CC(C)CC(C)C(=O)C(OC)C(O)C(C)=CC(C)C(=O)C1 QFJCIRLUMZQUOT-UHFFFAOYSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- MODVSQKJJIBWPZ-VLLPJHQWSA-N tesetaxel Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3CC[C@@]2(C)[C@H]2[C@@H](C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C(=CC=CN=4)F)C[C@]1(O)C3(C)C)O[C@H](O2)CN(C)C)C(=O)C1=CC=CC=C1 MODVSQKJJIBWPZ-VLLPJHQWSA-N 0.000 description 1
- 229950009016 tesetaxel Drugs 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- NZVYCXVTEHPMHE-ZSUJOUNUSA-N thymalfasin Chemical compound CC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O NZVYCXVTEHPMHE-ZSUJOUNUSA-N 0.000 description 1
- 229960004231 thymalfasin Drugs 0.000 description 1
- 229960000902 thyrotropin alfa Drugs 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- MNRILEROXIRVNJ-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=NC=N[C]21 MNRILEROXIRVNJ-UHFFFAOYSA-N 0.000 description 1
- 229950009158 tipifarnib Drugs 0.000 description 1
- PLHJCIYEEKOWNM-HHHXNRCGSA-N tipifarnib Chemical compound CN1C=NC=C1[C@](N)(C=1C=C2C(C=3C=C(Cl)C=CC=3)=CC(=O)N(C)C2=CC=1)C1=CC=C(Cl)C=C1 PLHJCIYEEKOWNM-HHHXNRCGSA-N 0.000 description 1
- 229950002376 tirapazamine Drugs 0.000 description 1
- QVMPZNRFXAKISM-UHFFFAOYSA-N tirapazamine Chemical compound C1=CC=C2[N+]([O-])=NC(=N)N(O)C2=C1 QVMPZNRFXAKISM-UHFFFAOYSA-N 0.000 description 1
- 229950005976 tivantinib Drugs 0.000 description 1
- 229960000940 tivozanib Drugs 0.000 description 1
- 229960003989 tocilizumab Drugs 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 229960000303 topotecan Drugs 0.000 description 1
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 1
- 229960005026 toremifene Drugs 0.000 description 1
- XFCLJVABOIYOMF-QPLCGJKRSA-N toremifene Chemical compound C1=CC(OCCN(C)C)=CC=C1C(\C=1C=CC=CC=1)=C(\CCCl)C1=CC=CC=C1 XFCLJVABOIYOMF-QPLCGJKRSA-N 0.000 description 1
- 229950005801 tosedostat Drugs 0.000 description 1
- 229960005267 tositumomab Drugs 0.000 description 1
- 229960000977 trabectedin Drugs 0.000 description 1
- PKVRCIRHQMSYJX-AIFWHQITSA-N trabectedin Chemical compound C([C@@]1(C(OC2)=O)NCCC3=C1C=C(C(=C3)O)OC)S[C@@H]1C3=C(OC(C)=O)C(C)=C4OCOC4=C3[C@H]2N2[C@@H](O)[C@H](CC=3C4=C(O)C(OC)=C(C)C=3)N(C)[C@H]4[C@@H]21 PKVRCIRHQMSYJX-AIFWHQITSA-N 0.000 description 1
- 229950002824 trabedersen Drugs 0.000 description 1
- FNCMIJWGZNHSBF-UHFFFAOYSA-N trabedersen Chemical compound CC1=CN(C2CC(O)C(COP(=O)(S)OC3CC(OC3COP(=O)(S)OC4CC(OC4COP(=O)(S)OC5CC(OC5COP(=O)(S)OC6CC(OC6COP(=O)(S)OC7CC(OC7COP(=O)(S)OC8CC(OC8COP(=O)(S)OC9CC(OC9COP(=O)(S)OC%10CC(OC%10COP(=O)(S)OC%11CC(OC%11COP(=O)(S)OC%12CC(OC%12COP(=O)(S)OC%13CC(OC%13COP(=O)(S)OC%14CC(OC%14COP(=O)(S)OC%15CC(OC%15CO)N%16C=CC(=NC%16=O)N)n%17cnc%18C(=O)NC(=Nc%17%18)N)n%19cnc%20C(=O)NC(=Nc%19%20)N)N%21C=CC(=NC%21=O)N)n%22cnc%23c(N)ncnc%22%23)N%24C=C(C)C(=O)NC%24=O)n%25cnc%26C(=O)NC(=Nc%25%26)N)N%27C=C(C)C(=O)NC%27=O)N%28C=CC(=NC%28=O)N)N%29C=C(C)C(=O)NC%29=O)n%30cnc%31c(N)ncnc%30%31)N%32C=C(C)C(=O)NC%32=O)N%33C=C(C)C(=O)NC%33=O)O2)C(=O)NC1=O.CC%34=CN(C%35CC(OP(=O)(S)OCC%36OC(CC%36OP(=O)(S)OCC%37OC(CC%37OP(=O)(S)OCC%38OC(CC%38O)n%39cnc%40c(N)ncnc%39%40)N%41C=C(C)C(=O)NC%41=O)n%42cnc%43C(=O)NC(=Nc%42%43)N)C(COP(=O)S)O%35)C(=O)NC%34=O FNCMIJWGZNHSBF-UHFFFAOYSA-N 0.000 description 1
- 229960004066 trametinib Drugs 0.000 description 1
- LIRYPHYGHXZJBZ-UHFFFAOYSA-N trametinib Chemical compound CC(=O)NC1=CC=CC(N2C(N(C3CC3)C(=O)C3=C(NC=4C(=CC(I)=CC=4)F)N(C)C(=O)C(C)=C32)=O)=C1 LIRYPHYGHXZJBZ-UHFFFAOYSA-N 0.000 description 1
- 108010075758 trebananib Proteins 0.000 description 1
- 229950001210 trebananib Drugs 0.000 description 1
- 229960003181 treosulfan Drugs 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- KVJXBPDAXMEYOA-CXANFOAXSA-N trilostane Chemical compound OC1=C(C#N)C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@@]32O[C@@H]31 KVJXBPDAXMEYOA-CXANFOAXSA-N 0.000 description 1
- 229960001670 trilostane Drugs 0.000 description 1
- LEIMLDGFXIOXMT-UHFFFAOYSA-N trimethylsilyl cyanide Chemical compound C[Si](C)(C)C#N LEIMLDGFXIOXMT-UHFFFAOYSA-N 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- FIQMHBFVRAXMOP-UHFFFAOYSA-N triphenylphosphane oxide Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)(=O)C1=CC=CC=C1 FIQMHBFVRAXMOP-UHFFFAOYSA-N 0.000 description 1
- VXKHXGOKWPXYNA-PGBVPBMZSA-N triptorelin Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 VXKHXGOKWPXYNA-PGBVPBMZSA-N 0.000 description 1
- 229960004824 triptorelin Drugs 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 229960000875 trofosfamide Drugs 0.000 description 1
- UMKFEPPTGMDVMI-UHFFFAOYSA-N trofosfamide Chemical compound ClCCN(CCCl)P1(=O)OCCCN1CCCl UMKFEPPTGMDVMI-UHFFFAOYSA-N 0.000 description 1
- 229950009811 ubenimex Drugs 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 238000000825 ultraviolet detection Methods 0.000 description 1
- 230000003827 upregulation Effects 0.000 description 1
- 229960001055 uracil mustard Drugs 0.000 description 1
- 229960005486 vaccine Drugs 0.000 description 1
- 229960000653 valrubicin Drugs 0.000 description 1
- ZOCKGBMQLCSHFP-KQRAQHLDSA-N valrubicin Chemical compound O([C@H]1C[C@](CC2=C(O)C=3C(=O)C4=CC=CC(OC)=C4C(=O)C=3C(O)=C21)(O)C(=O)COC(=O)CCCC)[C@H]1C[C@H](NC(=O)C(F)(F)F)[C@H](O)[C@H](C)O1 ZOCKGBMQLCSHFP-KQRAQHLDSA-N 0.000 description 1
- 229950010938 valspodar Drugs 0.000 description 1
- 108010082372 valspodar Proteins 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 229960000241 vandetanib Drugs 0.000 description 1
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 229960003862 vemurafenib Drugs 0.000 description 1
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- HHJUWIANJFBDHT-KOTLKJBCSA-N vindesine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(N)=O)N4C)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 HHJUWIANJFBDHT-KOTLKJBCSA-N 0.000 description 1
- 229960004355 vindesine Drugs 0.000 description 1
- NMDYYWFGPIMTKO-HBVLKOHWSA-N vinflunine Chemical compound C([C@@](C1=C(C2=CC=CC=C2N1)C1)(C2=C(OC)C=C3N(C)[C@@H]4[C@@]5(C3=C2)CCN2CC=C[C@]([C@@H]52)([C@H]([C@]4(O)C(=O)OC)OC(C)=O)CC)C(=O)OC)[C@H]2C[C@@H](C(C)(F)F)CN1C2 NMDYYWFGPIMTKO-HBVLKOHWSA-N 0.000 description 1
- 229960000922 vinflunine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- 229960002360 vintafolide Drugs 0.000 description 1
- KUZYSQSABONDME-QRLOMCMNSA-N vintafolide Chemical compound C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)NNC(=O)OCCSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(O)=O)NC(=O)CC[C@H](NC(=O)C=4C=CC(NCC=5N=C6C(=O)NC(N)=NC6=NC=5)=CC=4)C(O)=O)C(O)=O)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 KUZYSQSABONDME-QRLOMCMNSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229960004449 vismodegib Drugs 0.000 description 1
- BPQMGSKTAYIVFO-UHFFFAOYSA-N vismodegib Chemical compound ClC1=CC(S(=O)(=O)C)=CC=C1C(=O)NC1=CC=C(Cl)C(C=2N=CC=CC=2)=C1 BPQMGSKTAYIVFO-UHFFFAOYSA-N 0.000 description 1
- 229960000237 vorinostat Drugs 0.000 description 1
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical compound ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 1
- XZAFZXJXZHRNAQ-STQMWFEESA-N vosaroxin Chemical compound C1[C@H](OC)[C@@H](NC)CN1C1=CC=C2C(=O)C(C(O)=O)=CN(C=3SC=CN=3)C2=N1 XZAFZXJXZHRNAQ-STQMWFEESA-N 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
- 229950008250 zalutumumab Drugs 0.000 description 1
- 229950009002 zanolimumab Drugs 0.000 description 1
- 229950009268 zinostatin Drugs 0.000 description 1
- FBTUMDXHSRTGRV-ALTNURHMSA-N zorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(\C)=N\NC(=O)C=1C=CC=CC=1)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 FBTUMDXHSRTGRV-ALTNURHMSA-N 0.000 description 1
- 229960000641 zorubicin Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/4545—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring hetero atom, e.g. pipamperone, anabasine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Hematology (AREA)
- Hospice & Palliative Care (AREA)
- Oncology (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Un compuesto de Fórmula (I)**Fórmula** en la que: X, Y son independientemente CH o N, Z es CH, C-Hal o N, R1 es H, LA, CA, NH2, NH(LA) o (LA)NH(LA), Hal, -S(LA), -SO2(LA), O(LA), Cyc es un heterociclo alifático de 3, 4, 5, 6 o 7 miembros que tiene 1 o 2 átomos de N, o 1 átomo de N y 1 átomo de O, R2 es -LA-Ar o Ar, que está en la posición 2 o 3 con respecto al átomo de N de Cyc, R3 es H, OH, NH2, COO(LA), CONH2, CONH(LA) NHCO(LA), (LA)OH, NH(LA) o LA, que está en cualquier posición de Cyc, Ar es un homo o heterociclo, mono o binuclear, alifático o aromático de 3, 4, 5, 6, 7, 8, 9 o 10 miembros, que tiene 0, 1, 2, 3 o 4 átomos de N, O y/o S, que puede estar sin sustituir o mono o disustituido independientemente con Hal, OH, CN, LA, O(LA), S(LA), LA es alquilo sin ramificar o ramificado, que tiene 1, 2, 3, 4 o 5 átomos de carbono, que puede estar saturado o parcialmente insaturado, en el que 1, 2 o 3 átomos de H están reemplazados por Hal, CA es cicloalquilo que tiene 3, 4, 5 o 6 átomos de carbono, o cicloalquil alquilo que tiene 3, 4, 5 o 6 átomos de carbono en el anillo y 1 o 2 átomos de carbono no anulares, Hal es F, CI, Br o I.
Description
DESCRIPCIÓN
Indazoles
Campo de la invención
La invención se refiere a una serie de compuestos de indazol sustituidos novedosos que son útiles en el tratamiento de enfermedades hiperproliferativas tales como cáncer, así como enfermedades inflamatorias o degenerativas, en mamíferos. También quedan abarcados por la presente invención tales compuestos para su uso en el tratamiento de enfermedades hiperproliferativas, inflamatorias o degenerativas en mamíferos, especialmente seres humanos y composiciones farmacéuticas que contienen tales compuestos.
Sumario de la técnica relacionada
CDK8, junto con su isoforma estrechamente relacionada CDK19, es una quinasa reguladora de la transcripción oncogénica. En contraste a miembros mejor conocidos de la familia de la CDK (tales como CDK1, CDK2 y CDK4/6), CDK8 no juega un papel directo en la progresión del ciclo celular. La CDK8 desactivada en células madre embrionarias evita el desarrollo embrionario, debido a su papel esencial en el fenotipo de célula madre pluripotente pero el agotamiento de CDK8 no inhibe el crecimiento de células normales.
El papel de la CDK8 en el cáncer se debe a su función única como regulador de varios programas transcripcionales implicados en la carcinogénesis. La CDK8 se ha identificado como un oncogén en cáncer de melanoma y de colon, amplificándose el gen de la CDK8 en aproximadamente el 50 % de los últimos cánceres. Se ha asociado una expresión superior de CDK8 con una peor prognosis en cáncer de colon, de mama y de ovario. Las actividades relevantes de cáncer conocidas de la CDK8 incluyen regulación positiva de la vía Wnt/p-catenina, transcripción inducida por el factor de crecimiento y señalización de TGFa. La CDK8 también demostró mantener el fenotipo pluripotente de las células madre embrionarias y se ha asociado con el fenotipo de célula madre cancerosa. Los fármacos quimioterapéuticos que dañan el ADN inducen TNFa, un activador del factor de transcripción de NFkB, en células endoteliales y en otros elementos estromales asociados con cáncer. La TNFa derivado del estroma actúa sobre células tumorales, en las que induce una producción mediada por NFkB de citoquinas relacionadas promotoras de tumores CXCL1 y CXCL2. CXCL1/2 atraen células mieloides al tumor, uniéndose al receptor CXCR2 sobre la superficie de células mieloides. Las células mieloides secretan entonces pequeñas proteínas de unión de calcio 5100A8 y A9 que se asocian con inflamación crónica y cáncer. 5100A8/9 actúa sobre células tumorales, estimulando tanto su metástasis como su supervivencia de quimioterapia.
CDK8 es una quinasa dependiente de ciclina que tiene una función conservada en la transcripción como parte del complejo Mediador. Taatjes, D. J., Trends Biochem Sci 35, 315-322 (2010); Conaway, R. C. y Conaway, J. W., Curr Opin Genet Dev 21, 225-230 (2011). Más recientemente, Se ha dado a conocer que la CDK8 como un oncogén en tanto cáncer de colon (Firestein R. y col., Nature 455:547-51 (2008); Morris E. J. y col., Nature 455:552-6 (2008); Starr T. K. y col., Science 323:1747-50 (2009)) como de melanoma (Kapoor A. y col., Nature 468:1105-9 (2010)). c Dk 8 se regula positivamente y amplifica en un subconjunto de tumores de colon de seres humanos. La CDK8 transforma células inmortalizadas y se requiere para la proliferación de cáncer de colon in vitro (Firestein, R. y col., Nature 455, 547-551 (2008)). También se ha encontrado que la CDK8 está sobreexpresada y es esencial para la proliferación en melanoma (Kapoor, A. y col., Nature 468, 1105-1109 (2010)). La CDK8 ha demostrado regular varias vías de señalización que son reguladores clave de tanto pluripotencia de ES como cáncer. La CDK8 activa la vía de Wnt estimulando la expresión de genes diana de p-Catenina (Firestein, R. y col., Nature 455, 547-551 (2008)) o inhibiendo E2F1, un potente inhibidor de la actividad transcripcional de la p-Catenina (Morris, E. J. y col., Nature 455, 552-556 (2008) ). La CDK8 estimula la expresión del gen diana de Notch mediante la fosforilación del dominio intracelular de Notch, activando los complejos potenciadores de Notch en genes diana (Fryer C. J. et al., Mol Cell 16:509-20 (2004)). Por último, la fosforilación de CDK8 de proteínas SMAD lleva a la activación de genes diana TGFp/BMP seguido por la degradación de las proteínas SMAD para limitar la expresión del gen diana (Alarcon, C. y col., Cell 139, 757-769 (2009) ).
Otros compuestos que se centran en la vía de Wnt se desvelan en, es decir, los documentos WO 2010/041054, WO 2013/110433, WO 2014/063778 o WO 2014/086453.
Los documentos US 2011/0034441 y US 6.107.305 desvelan inhibidores de la CDK.
Sin embargo, como un terapéutico dirigido a la vía de Wnt y, en particular, a CDK8/19, aún se tiene que comercializar, aún existe una necesidad médica insatisfecha significante, de modo que deben identificarse y desarrollarse más inhibidores de la vía de Wnt prometedores.
Descripción de la invención
Es, por lo tanto, el objeto de la presente invención proporcionar inhibidores de CDK8/19 novedosos útiles en el tratamiento de enfermedades inflamatorias o proliferativas, tales como cáncer en mamíferos, con propiedades farmacológicas superiores tanto con respecto a sus actividades como a su solubilidad, eliminación metabólica y características de biodisponibilidad.
Como resultado, la presente invención proporciona compuestos de indazol sustituidos novedosos o sus estereoisómeros o tautómeros, o sales farmacéuticamente aceptables, que son inhibidores de CDK8/19 y útiles como medicamentos, especialmente en el tratamiento de las enfermedades mencionadas anteriormente y a continuación.
Los compuestos se definen mediante la Fórmula (I):

en la que:
X, Y son independientemente CH o N,
Z es CH, C(Hal) o N,
R1 es H, LA, CA, NH2 , NH(LA) o (LA)NH(LA), Hal, -S(LA), -SO2(LA), O(LA),
Cyc es un heterociclo alifático de 3, 4, 5, 6 o 7 miembros que tiene 1 o 2 átomos de N, o 1 átomo de N y 1 átomo de O,
R2 es -LA-Ar o Ar, que está en la posición 2 o 3 con respecto al átomo de N de Cyc,
R3 es H, OH, NH2 , COO(LA), CONH2 , CONH(LA), NHCO(LA), (LA)OH, NH(LA) o LA, que está en cualquier posición de Cyc,
Ar es un homo o heterociclo, mono o binuclear, alifático o aromático de 3, 4, 5, 6, 7, 8, 9 o 10 miembros, que tiene 0, 1, 2, 3 o 4 átomos de N, O y/o S, que puede estar sin sustituir o mono o disustituido independientemente con Hal, OH, CN, LA, O(LA), S(LA),
LA es alquilo sin ramificar o ramificado, que tiene 1,2, 3, 4 o 5 átomos de carbono, que puede estar saturado o parcialmente insaturado, en el que 1, 2 o 3 átomos de H están reemplazados por Hal,
CA es cicloalquilo que tiene 3, 4, 5 o 6 átomos de carbono, o cicloalquil alquilo que tiene 3, 4, 5 o 6 átomos de carbono en el anillo y 1 o 2 átomos de carbono no anulares,
Hal es F, CI, Br o I.
En general, todos los residuos que aparecen más de una vez pueden ser idénticos o diferentes, es decir, son independientes unos de otros. Anteriormente y más adelante, los restos y parámetros tienen los significados indicados para la Fórmula (I), a menos que se indique expresamente lo contrario.
Por consiguiente, la invención se refiere, en particular, a los compuestos de la Fórmula (I) en la que al menos uno dichos restos tiene uno de los significados preferidos indicados más adelante.
Hal representa flúor, cloro, bromo o yodo, en particular flúor o cloro, y preferiblemente cloro.
"LA" representa por ejemplo metilo, etilo, trifluorometilo, difluorometilo, 1,1,1 -trifluoroetilo, propilo, isopropilo, butilo, isobutilo, sec-butilo o ferc-butilo, isopropenilo, etenilo, etinilo o prop-1-inilo.
"CA" representa por ejemplo ciclopropilo, (ciclopropil)metilo, ciclobutilo o (ciclopentil)etilo.
"Cyc" representa, por ejemplo aziridinilo, azetidinilo, pirrolidinilo, piperidinilo, azepanilo, preferiblemente aziridin-1-ilo, azetidin-1-ilo, pirrolidin-1-ilo, piperidin-1-ilo, azepan-1-ilo.
"Ar" representa, por ejemplo, fenilo, 2- o 3-furilo, 2- o 3-tienilo, 1-, 2- o 3-pirrolilo, 1 -, 2, 4- o 5-imidazolilo, 1 -, 3-, 4- o 5 pirazolilo, 2-, 4- o 5-oxazolilo, 3-, 4- o 5-isoxazolilo, 2-, 4- o 5-tiazolilo, 3-, 4- o 5-isotiazolilo, 2-, 3- o 4-piridilo, 2-, 4-, 5-o 6-pirimidinilo, 2-, 3-, 5-, o 6-pirazin-1 - o 4-ilo, más preferiblemente 1,2,3-triazol-1 -, -4- o -5-ilo, 1,2,4- triazol-1 -, -3- o 5-ilo, 1- o 5-tetrazolilo, 1,2,3-oxadiazol-4- o -5-ilo, 1,2,4-oxadiazol-3- o -5-ilo, 1,3,4-oxadiazol-2-ilo, 1,3,4-tiadiazol-2- o
-5-ilo, 1,2,4-tiadiazol-3- o -5-ilo, 1,2,3- tiadiazol-4- o -5-ilo, 3- o 4-piridazinilo, 1 -, 2-, 3-, 4-, 5-, 6- o 7-indolilo, 2-, 3-, 4-o 5-iso-indolilo, 2-, 6, - u 8-purinilo, 1-, 2-, 4- o 5-benzoimidazolilo, 1-, 3-, 4-, 5-, 6- o 7-benzopirazolilo, 2-, 4-, 5-, 6- o 7-benzoxazolilo, 3-, 4-, 5-, 6- o 7-benzoisoxazolilo, 2-, 4-, 5-, 6- o 7-benzotiazolilo, 2-, 4-, 5-, 6- o 7-benzoisotiazolilo, 4-, 5-, 6- o 7-benz-2,1,3-oxadiazolilo, 1 -, 3-, 4-, 5-, 6-, 7- u 8-isoquinolinilo, 3-, 4-, 5-, 6-, 7- u 8-quinolinilo, 2-, 4-, 5-, 6 , 7- u 8-quinazolinilo, quinoxalin-2-, 3-, 4- o 5-ilo, 4-, 5- o 6-ftalazinilo, 2-, 3-, 5-, 6-, 7- u 8-2H-benzo-1,4-oxazinilo. Preferiblemente, "Ar" representa 2-, 3- o -4-piridilo, fenilo, 2-, 4-, 5- o 6-pirimidinilo, 2- o 3-pirazinilo.
"LA-Ar" representa, por ejemplo, 2-, 3- o 4-fenilmetilo, 2-, 3- o 4-feniletilo, 2-, 3- o 4-piridilmetilo, 2-, 3- o 4-piridiletilo,
En una realización preferida los compuestos de la invención están de acuerdo con la Fórmula (I) como se ha mostrado anteriormente, en la que:
X, Y, Z son independientemente CH o N,
R1 es H, LA, CA, NH2 , NH(LA) o (LA)NH(LA),
Cyc es un heterociclo alifático de 3, 4, 5, 6 o 7 miembros que tiene 1 átomo de N,
R2 es -LA-Ar o Ar, que está en la posición 2 o 3 con respecto al átomo de N de Cyc,
R3 es H, OH, NH2 , COO(LA), CONH2 , CONH(LA) o LA, que está en cualquier posición de Cyc,
Ar es un homo o heterociclo, mono o binuclear, alifático o aromático de 3, 4, 5, 6, 7, 8, 9 o 10 miembros, que tiene 0, 1, 2, 3 o 4 átomos de N, O y/o S, que puede estar sin sustituir o mono o disustituido independientemente con Hal, OH, CN, LA, O(LA), S(LA),
LA es alquilo sin ramificar o ramificado, que tiene 1, 2, 3, 4 o 5 átomos de carbono, que puede estar saturado o parcialmente insaturado, en el que 1, 2 o 3 átomos de H están reemplazados por Hal,
CA es cicloalquilo que tiene 3, 4, 5 o 6 átomos de carbono, o cicloalquil alquilo que tiene 3, 4, 5 o 6 átomos de carbono en el anillo y 1 o 2 átomos de carbono no anulares,
Hal es F, CI, Br o I.
En otra realización preferida, los compuestos de la invención están de acuerdo con la Fórmula (I) como se ha mostrado anteriormente, en la que:
X, Y son independientemente CH o N,
Z es CH,
R1 es H, LA, CA, NH2 , NH(LA) o (LA)NH(LA),
Cyc es un heterociclo alifático de 3, 4, 5, 6 o 7 miembros que tiene 1 átomo de N,
R2 es -LA-Ar o Ar, que está en la posición 2 o 3 con respecto al átomo de N de Cyc,
R3 es H, NH2 o LA, que está en cualquier posición de Cyc,
Ar es un homo o heterociclo mononuclear, aromático, de 6 miembros, que tiene 0, 1 o 2 átomos de N, que puede estar sin sustituir o mono o disustituido independientemente con Hal, OH, CN, LA, O(LA), S(LA), LA es un alquilo sin ramificar o ramificado, que tiene 1, 2, 3, 4 o 5 átomos de carbono, que puede estar saturado o parcialmente insaturado, en el que 1, 2 o 3 átomos de H están reemplazados por Hal,
CA es cicloalquilo que tiene 3, 4, 5 o 6 átomos de carbono, o cicloalquil alquilo que tiene 3, 4, 5 o 6 átomos de carbono en el anillo y 1 o 2 átomos de carbono no anulares,
Hal es F, CI, Br o I.
En una realización más preferida, los compuestos de la invención están de acuerdo con las Fórmulas (lla) o (IIb),

en las que todos los sustituyentes tienen los significados según se han indicado para la Fórmula (I).
En una realización preferida adicional, los compuestos de la invención están de acuerdo con las Subfórmulas 1 a 26 de la Fórmula (I) (lla) o (IIb), en la que
en la Subfórmula 1
Ar es un homo o heterociclo mononuclear, aromático, de 6 miembros, que tiene 0, 1 o 2 átomos de N, que está sin sustituir o monosustituido con Hal, LA u O(LA),
en la Subfórmula 2
X es N,
en la Subfórmula 3
X es CH,
en la Subfórmula 4
Y es CH,
en la Subfórmula 5
Y es N,
en la Subfórmula 6
Z es CH,
en la Subfórmula 7
X es CH,
Y es N,
Z es CH,
en la Subfórmula 8
X es N,
Y es CH,
Z es CH,
en la Subfórmula 9
Ar es un homo o heterociclo mononuclear, aromático, de 6 miembros, que tiene 0, 1 o 2 átomos de N, que puede estar sin sustituir o mono o disustituido independientemente con Hal, OH, CN, LA, O(LA), S(LA),
X es CH,
Y es N,
Z es CH,
en la Subfórmula 10
Ar es un homo o heterociclo mononuclear, aromático, de 6 miembros, que tiene 0, 1 o 2 átomos de N, que puede estar sin sustituir o mono o disustituido independientemente con Hal, OH, CN, LA, O(LA), S(LA),
X es N,
Y es CH,
Z es CH,
en la Subfórmula 11
R1 es LA o NH2 , NHLA
en la Subfórmula 12
Cyc tiene 4, 5 o 6 átomos en el anillo,
en la Subfórmula 13
R3 es H, OH, NH2 o metilo,
en la Subfórmula 14
R3 es H,
en la Subfórmula 15
R2 es bencilo,
R3 es H, NH2 o metilo,
Cyc tiene 6 átomos en el anillo,
en la Subfórmula 16
Cyc tiene 5 o 6 átomos en el anillo,
R2 es fenilo, que está sin sustituir, o mono o disustituido independientemente con Hal o LA,
en la Subfórmula 17
Z es CH,
R2 es bencilo o fenilo, fenilo que está sin sustituir, o mono o disustituido independientemente con Hal o LA,
en la Subfórmula 18
X es N o CH,
Y es CH,
Z es CH,
R1 es metilo, n-propilo, /-propilo, ciclopropilo, NH2 , NHLA
en la Subfórmula 19
Z es CH,
Cyc tiene 5 átomos en el anillo,
en la Subfórmula 20
Z es CH,
R2 es fenilo, que está sin sustituir o monosustituido con Br, CI, metilo o CF3 ,
R3 es H,
en la Subfórmula 21
Z es CH,
R2 es fenilo, que está sin sustituir o monosustituido con Br, CI, metilo o CF3 ,
R3 es H,
en la Subfórmula 22
Z es CH,
R2 es fenilo, que está sin sustituir o para-sustituido con Br, CI, metilo o CF3 ,
R3 es H,
en la Subfórmula 23
Y es CH,
Z es CH,
R1 es metilo, n-propilo, /-propilo, ciclopropilo o NH2 ,
en la Subfórmula 24
X es N o CH,
Y es CH,
Z es CH,
R1 es metilo, n-propilo, /-propilo, ciclopropilo o NH2 ,
Cyc tiene 4, 5 o 6 átomos en el anillo,
en la Subfórmula 25
X es N o CH,
Y es CH,
Z es CH,
R1 es metilo, n-propilo, /-propilo, ciclopropilo o NH2 ,
Cyc tiene 4, 5 o 6 átomos en el anillo,
R2 es fenilo, que está sin sustituir o para-sustituido con Br, CI, metilo o CF3 ,
R3 es H,
en la Subfórmula 26
X es N o CH,
Y es CH,
Z es CH,
R1 es metilo, n-propilo, /-propilo, ciclopropilo o NH2 ,
Cyc tiene 5 átomos en el anillo,
R2 es fenilo, que está sin sustituir o para-sustituido con Br, CI, metilo o CF3 ,
R3 es H,
en la Subfórmula 27
R1 es metilo, n-propilo, /-propilo, ciclopropilo, metilsulfanilo, metanosulfonilo, metoxi, F o NH2 ,
en la Subfórmula 28
R3 es H, OH, NH2 , metilo, acetamido, 2-hidroxietilo o metilamino.
en la Subfórmula 29
Z es CH o C(Hal),
Cyc tiene 4, 5 o 6 átomos en el anillo, de los cuales 1 átomo es N los otros átomos son C,
R2 es fenilo, que está sin sustituir o para-sustituido con Br, CI, F, metilo, metoxi, isopropilo o CF3 , o están independientemente meta- / para-disustituidos con CI y F,
R3 es H.
en la Subfórmula 30
Z es CH o C(Hal),
Cyc tiene 4 o 5 átomos en el anillo, de los cuales 1 átomo es N los otros átomos son C,
R2 es fenilo, que está sin sustituir o para-sustituido con Br, CI, F, metilo, metoxi, isopropilo o CF3 , o meta-sustituido con F y para-sustituido con CI,
R3 es H.
y los restos restantes tienen el significado según se ha indicado para la Fórmula (I).
Los compuestos de la Fórmula (I) pueden tener uno o más centros de quiralidad. En consecuencia, pueden aparecer en diversas formas enantioméricas y estar en forma racémica u ópticamente activa. La invención, por lo tanto, también se refiere a las formas ópticamente activas, enantiómeros, racematos, diastereómeros, en conjunto: estereoisómeros, de estos compuestos.
Puesto que la actividad farmacéutica de los racematos o estereoisómeros de los compuestos de acuerdo con la invención puede diferir, puede ser deseable usar los enantiómeros. En estos casos, el producto final o incluso los intermedios pueden separarse en compuestos enantioméricos por medidas químicas o físicas conocidas para la persona experta en la materia o incluso emplearse como tales en las síntesis.
En el caso de aminas racémicas, se forman diastereómeros de la mezcla por reacción con un agente de resolución ópticamente activo. Son ejemplos de agentes de resolución adecuados, ácidos ópticamente activos, tales como las formas R y S de ácido tartárico, ácido diacetiltartárico, ácido dibenzoiltartárico, ácido mandélico, ácido málico, ácido láctico, adecuadamente aminoácidos N-protegidos (por ejemplo N-benzoilprolina o N-bencenosulfonilprolina), o los diversos ácidos alcanforsulfónicos ópticamente activos. También es ventajosa la resolución de enantiómeros cromatográfica con la ayuda de un agente de resolución ópticamente activo (por ejemplo dinitrobenzoilfenilglicina, celulosa triacetato u otros derivados de carbohidratos o polímeros de metacrilato derivatizados quiralmente inmobilizados sobre gel de sílice). Los eluyentes adecuados para este propósito son mezclas de disolventes acuosos o alcohólicos, tales como, por ejemplo, hexano/isopropanol/ acetonitrilo, por ejemplo en la proporción 82:15:3.
Un procedimiento elegante para la resolución de racematos que contienen grupos éster (por ejemplo acetil ésteres) es el uso de enzimas, en particular esterasas.
Es bien conocido que algunos los átomos pueden tener masas atómicas o números másicos que difieren de las masas atómicas o números másicos de los átomos que normalmente aparecen en la naturaleza. Los ejemplos de isótopos que están fácilmente disponibles en el mercado y que pueden incorporarse en un compuesto de la presente invención por métodos bien conocidos incluyen isótopos de hidrógeno, carbono, nitrógeno, oxígeno, fósforo, flúor y cloro, por ejemplo 2H, 3H, 13C, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F y 36CI, respectivamente. La incorporación de isótopos más pesados, especialmente deuterio (2H), en un compuesto de la invención tiene ventajas terapéuticas debido a la mayor estabilidad metabólica de este compuesto marcado con isótopo. Una estabilidad metabólica mayor se traduce directamente en una semivida in vivo aumentada o dosificaciones menores. Por lo tanto, estos isótopos están incluidos en la definición de átomos H, C, N etc., según se usan en los compuestos químicos de esta invención.
También se desvela que los compuestos de la presente invención pueden estar en forma de un compuesto profármaco. "Compuesto profármaco" significa un derivado que se convierte en un compuesto biológicamente activo de acuerdo con la presente invención en condiciones fisiológicas el organismo vivo, por ejemplo, por oxidación, reducción, hidrólisis o similares, cada uno de los cuales se realiza enzimáticamente o sin implicación enzimática. Son ejemplos de profármacos, compuestos, en los que el grupo amino en un compuesto de la presente invención se acila, alquila o fosforila, por ejemplo, eicosanoilamino, alanilamino, pivaloiloximetilamino o en el que el grupo hidroxilo se acila, alquila, fosforila o convierte en el borato, por ejemplo acetiloxi, palmitoiloxi, pivaloiloxi, succiniloxi, fumariloxi, alaniloxi o en el que el grupo carboxilo se esterifica o amida, o en el que un grupo sulfhidrilo forma un puente disulfuro con una molécula portadora, por ejemplo un péptido, que libera el fármaco selectivamente a una diana y/o al citosol de una célula. Estos compuestos pueden producirse a partir de compuestos de la presente invención de acuerdo con procedimientos bien conocidos. Otros ejemplos de profármacos son compuestos, en los que el carboxilato en un compuesto de la presente invención se convierte, por ejemplo, en un alquil-, aril-, colina-, amino, aciloximetiléster, linolenoil-éster.
Donde puede suceder tautomerismo, por ejemplo, tautomerismo ceto-enol, de compuestos de la presente invención o sus profármacos, las formas individuales, por ejemplo, la forma ceto o la enol, se reivindican por separado y junto como mezclas en cualquier proporción. Lo mismo se aplica para estereoisómeros, por ejemplo, enantiómeros, isómeros cis/trans, confórmeros y similares.
Si se desea, los isómeros pueden separarse por procedimientos bien conocidos en la técnica, por ejemplo por cromatografía líquida. Lo mismo se aplica para enantiómeros, por ejemplo, usando fases quirales estacionarias. Además, los enantiómeros pueden aislarse convirtiéndolos en diastereómeros, es decir, acoplando con un compuesto auxiliar enantioméricamente puro, separación posterior de los diastereómeros resultantes y escisión del residuo auxiliar. Como alternativa, cualquier enantiómero de un compuesto de la presente invención puede obtenerse a partir de síntesis estereoselectiva usando materiales de partida ópticamente puros.
Los compuestos de la presente invención pueden estar en forma de una sal farmacéuticamente aceptable, un solvato farmacéuticamente aceptable, o un solvato farmacéuticamente aceptable de una sal farmacéuticamente aceptable. La expresión "sales farmacéuticamente aceptables" se refiere a sales preparadas a partir de bases o ácidos farmacéuticamente aceptables, incluyendo bases o ácidos inorgánicos y bases o ácidos orgánicos. En casos donde los compuestos de la presente invención contienen uno o más grupos ácidos o básicos, la invención también comprende sus sales farmacéuticamente aceptables correspondientes. Por lo tanto, los compuestos de la presente invención que contienen grupos ácidos pueden estar presentes en forma de sal, y pueden usarse de acuerdo con la invención, por ejemplo, como sales de metal alcalino, sales de metal alcalinotérreo o sales de amonio. Algunos ejemplos más precisos de dichas sales incluyen sales de sodio, sales de potasio, sales de calcio, sales de magnesio o sales con amoniaco o aminas orgánicas, tales como, por ejemplo, etilamina, etanolamina, trietanolamina o aminoácidos. Los compuestos de la presente invención que contienen uno o más grupos básicos, es decir grupos que pueden estar protonados, pueden estar presentes en forma de sal, y pueden usarse de acuerdo con la invención en forma de sus sales de adición con ácidos inorgánicos u orgánicos. Los ejemplos de ácidos adecuados incluyen cloruro de hidrógeno, bromuro de hidrógeno, ácido fosfórico, ácido sulfúrico, ácido nítrico, ácido metanosulfónico, ácido ptoluenosulfónico, ácidos naftalenodisulfónicos, ácido oxálico, ácido acético, ácido tartárico, ácido láctico, ácido salicílico, ácido benzoico, ácido fórmico, ácido propiónico, ácido piválico, ácido dietilacético, ácido malónico, ácido succínico, ácido pimélico, ácido fumárico, ácido maleico, ácido málico, ácido sulfámínico, ácido fenilpropiónico, ácido glucónico, ácido ascórbico, ácido isonicotínico, ácido cítrico, ácido adípico y otros ácidos conocidos para la persona experta en la materia. Si los compuestos de la presente invención contienen simultáneamente grupos ácidos y básicos en la molécula, la invención también incluye, además de las formas de sal mencionadas, sales internas o betaínas (zwitteriones). Las sales respectivas pueden obtenerse por procedimientos que son conocidos para una persona experta en la materia, por ejemplo poniendo en contacto estos con un ácido o base orgánica o inorgánica en un disolvente o dispersante, o mediante intercambio aniónico o intercambio catiónico con otras sales. La presente invención también incluye todas las sales de los compuestos de la presente invención que, debido a su baja compatibilidad fisiológica, no son directamente adecuadas para su uso en productos farmacéuticos, pero que pueden usarse, por ejemplo, como intermedios para reacciones químicas o para la preparación de sales farmacéuticamente aceptables.
La expresión ''solvatos farmacéuticamente aceptables" significa formas de adición con disolventes farmacéuticamente aceptables que contienen cantidades estequiométricas o no estequiométricas de disolvente. Algunos compuestos tienen una tendencia a atrapar una proporción molar fija de moléculas de disolvente en el estado sólido cristalino, formando de este modo un solvato. Si el disolvente es agua, el solvato formado es un hidrato, por ejemplo un monoo dihidrato. Si el disolvente es alcohol, el solvato formado es un alcoholato, por ejemplo, un metanolato o etanolato. Si el disolvente es un éter, el solvato formado es un eterato, por ejemplo, dietil eterato.
Por lo tanto, los siguientes artículos también están de acuerdo con la invención:
a) todos los estereoisómeros o tautómeros de los compuestos, incluyendo mezclas de los mismos en todas las proporciones,
b) sales farmacéuticamente aceptables de los compuestos y del artículo mencionado bajo (a),
c) solvatos farmacéuticamente aceptables de los compuestos y de los artículos mencionados bajo (a) y (b).
Debe entenderse que todas las referencias a compuestos anteriores y más adelante están destinadas a incluir estos artículos, en particular solvatos farmacéuticamente aceptables de los compuestos, o solvatos farmacéuticamente aceptables de sus sales farmacéuticamente aceptables.
Asimismo, la presente invención se refiere a composiciones farmacéuticas que comprenden un compuesto de la presente invención, o sus estereoisómeros o tautómeros, o sales farmacéuticamente aceptables de cada uno de los anteriores, incluyendo mezclas de los mismos en todas las proporciones, como principio activo, junto con un vehículo farmacéuticamente aceptable.
"Composición farmacéutica" se refiere a uno o más principios activos y a uno o más ingredientes inertes que conforman el portador, así como cualquier producto que sea el resultado, directa o indirectamente, de la combinación, formación de complejos o agregación de dos o más ingredientes cualesquiera, o de la disociación de uno o más de los ingredientes, o de otros tipos de reacciones o interacciones de uno o más de los ingredientes. En consecuencia, las composiciones farmacéuticas de la presente invención abarcan cualquier composición preparada premezclando un compuesto de la presente invención y un portador farmacéuticamente aceptable.
Una composición farmacéutica de la presente invención puede comprender adicionalmente uno o más otros compuestos como ingredientes activos, tales como una o más compuestos adicionales de la presente invención u otros inhibidores de la vía de Wnt.
Las composiciones farmacéuticas incluyen composiciones adecuadas para administración oral, rectal, tópica, parenteral (incluyendo subcutánea, intramuscular e intravenosa), ocular (oftálmica), pulmonar (inhalación nasal o bucal) o nasal, aunque la vía más adecuada en cualquier caso dado dependerá de la naturaleza y gravedad de las afecciones que se estén tratando y de la naturaleza del principio activo. Pueden presentarse convenientemente en forma monodosis y prepararse por cualquiera de los métodos bien conocidos en la técnica de farmacia.
En una realización, dichos compuestos y composición farmacéutica son para el tratamiento de cáncer tal como de cerebro, pulmón, colon, epidermoide, células escamosas, vejiga, gástrico, pancreático, mama, cabeza y cuello, renal, riñón, el hígado, ovárico, próstata, uterino, esofágico, testicular, ginecológico, cáncer de tiroides, melanoma, así como tumores hematológicos malignos tales como leucemia mielógena aguda, mieloma múltiple, leucemia mielógena crónica, leucemia mieloide aguda, sarcoma de Kaposi o cualquier otro tipo de tumores sólidos o líquidos. Preferentemente, el cáncer a tratar se escoge entre de colon, pulmón, mama y tipor de tumor hematológico.
Además, dichos compuestos y composición farmacéutica son para el tratamiento de enfermedades inflamatorias tales como esclerosis múltiple, artritis reumatoide, lupus sistémico, enfermedades de intestinales inflamatorias o enfermedades degenerativas tales como osteoartritis y enfermedad de Alzheimer.
El tratamiento anticanceroso definido anteriormente y a continuación puede aplicarse como monoterapia o puede implicar, además de los compuestos desvelados en el presente documento de Fórmula (I), terapia de cirugía convencional o radioterapia o medicinal. Tal terapia medicinal, por ejemplo, una quimioterapia o una terapia dirigida, puede incluir uno o más, pero preferentemente uno, de los siguientes agentes antitumorales:
- Agentes alquilantes, tales como altretamina, bendamustina, busulfán, carmustina, clorambucilo, clormetina, ciclofosfamida, dacarbazina, ifosfamida, tosilato de improsulfano, lomustina, melfalán, mitobronitol, mitolactol, nimustina, ranimustina, temozolomida, tiotepa, treosulfano, mecloretamina, carbocuona, apaziquona, fotemustina, glufosfamida, palifosfamida, pipobromano, trofosfamida, uramustina;
- Compuestos de platino, tales como carboplatino, cisplatino, eptaplatino, hidrato de miriplatino, oxaliplatino, lobaplatino, nedaplatino, picoplatino, satraplatino;
- Agentes alteradores de ADN, tales como amrubicina, bisantreno, decitabina, mitoxantrona, procarbazina, trabectedina, clofarabina, amsacrina, brostalicina, pixantrona, laromustina;
- Inhibidores de topoisomerasas, tal como etopósido, irinotecán, razoxano, sobuzoxano, tenipósido, topotecán, amonafida, belotecan, acetato de eliptinio, voreloxin;
- Modificadores de microtúbulos, tales como cabacitaxel, docetaxel, eribulina, ixabepilona, paclitaxel, vinblastina, vincristina, vinorelbina, vindesina, vinflunina, fosbretabulina, tesetaxel:
- Antimetabolitos, tales como asparaginasa, azacitidina, levofolionato de calcio, capecitabina, cladribina, citarabina, enocitabina, floxuridina, fludarabina, fluorouracilo, gemcitabina, mercaptopurina, metotrexato, nelarabina, pemetrexed, pralatrexato, azatioprina, tioguanina, carmofur, doxifluridina, elacitarabina, raltitrexed, sapacitabina, tegafur, trimetrexato;
- Antibióticos anticancerosos, tales como bleomicina, dactinomicina, doxorrubicina, epirrubicina, idarrubicina, levamisol, miltefosina, mitomicina C, romidepsin, estreptozocina, valrubicina, zinostatina, zorrubicina, daunurobicina, plicamicina, aclarubicina, peplomicina, pirarrubicina;
- Hormonas/Antagonistas, tales como abarelix, abiraterona, bicalutamida, buserelina, calusterona, clorotrianiseno, degarelix, dexametasona, estradiol, fluocortolona, fluoximasterona, flutamida, fulvestrant, goserelina, histrelina, leuprorelina, megestrol, mitotano, nafarelina, nandrolona, nilutamida, octreotida, prednisolona, raloxifeno, tamoxifeno, tirotropina alfa, toremifeno, trilostano, triptorelina, dietilestilbestrol, acolbifeno, danazol, deslorelina, epitiostanol, orteronel, enzalutamida;
- Inhibidores de la aromatasa, tales como aminoglutehimida, anastrozol, exemestano, fadrozol, letrozol, testolactona, formestano;
- Moléculas pequeñas inhibidoras de quinasa, tales como crizotinib, dasatinib, erlotinib, imatinib, lapatinib, nilotinib, pazopanib, regorafenib, ruxolitinib, sorafenib, sunitinib, vandetanib, vemurafenib, bosutinib, gefitinib, axitinib, afatinib, alisertib, dabrafenib, dacomitinib, dinaciclib, dovitinib, enzastaurina, nintedanib, lenvatinib, linifanib, linsitinib, masitinib, midostaurina, motesanib, neratinib, orantinib, perifosina, ponatinib, radotinib, rigosertib, tipifarnib, tivantinib, tivozanib, trametinib, pimasertib, alaninato de brivanib, cediranib, apatinib, S-malato de cabozantinib, carfilzomib, ibrutinib, icotinib;
- Fotosensibilizadores, tales como metoxsaleno, porfimer sódico, talaporfina, temoporfina;
- Anticuerpos, tales como alemtuzumab, besilesomab, brentuximab vedotina, cetuximab, denosumab, ipilimumab, ofatumumab, panitumumab, rituximab, tositumomab, trastuzumab, bevacizumab, catumaxomab, elotuzumab, epratuzumab, farletuzumab, mogamulizumab, necitumumab, nimotuzumab, obinutuzumab, ocaratuzumab, oregovomab, ramucirumab, rilotumumab, siltuximab, tocilizumab, zalutumumab, zanolimumab, matuzumab, dalotuzumab, onartuzumab, pertuzumab, racotumomab, tabalumab;
- Citoquinas, tales como aldesleuquina, interferón alfa, interferón alfa2a, interferón alfa2b, tasonermina, teceleuquina, oprelvekina;
- Conjugados de fármacos, tales como diftitox de denileuquina, ibritumomab tiuxetan, iobenguano I123, prednimustina, emtansina de trastuzumab, estramustina, gemtuzumab ozogamicina, aflibercept, besudotox de cintredequina, edotreotida, inotuzumab ozogamicina, naptumomab estafenatox, oportuzumab monatox, tecnetio (99mTc) arcitumomab, vintafolida;
- Vacunas, tales como sipuleucel, vitespen, emepepimut-S, oncoVAX, rindopepimut, troVax, stimuvax;
- Agentes miscelaneos, tales como alitretinoina, bexaroteno, bortezomib, everolimus, ácido ibandronico, imiquimod, lenalidomida, lentinan, metirosina, mifamurtida, ácido pamidrónico, pegaspargasa, pentostatina, sipuleucel, sizofiran, tamibaroteno, temsirolimus, talidomida, tretinoína, vismodegib, ácido zolendrónico, talidomida, vorinostat, celecoxib, cilengitida, entinostat, etanidazol, ganetespib, idronoxil, iniparib, ixazomib, lonidamina, nimorazol, panobinostat, peretinoina, plitidepsina, pomalidomida, procodazol, ridaforolimus, tasquinimod, telotristat, timalfasina, tirapazamina, tosedostat, trabedersen, ubenimex, valspodar, gendicina, picibanil, reolisina, clorhidrato de retaspimicina, trebananib, virulizina.
También se desvela un método para inhibir el crecimiento celular anormal en un mamífero o para tratar un trastorno hiperproliferativo que comprende la administración al mamífero de una cantidad de un compuesto de la presente invención o composición farmacéutica, en combinación con radioterapia, en la que las cantidades del compuesto o composición farmacéutica, son en combinación con la terapia de radiación eficaces en la inhibición del crecimiento celular anormal o el tratamiento del trastorno hiperproliferativo en el mamífero. Se conocen técnicas para la administración de terapia de radiación en la técnica y estas técnicas pueden usarse en la terapia de combinación que se describe en el presente documento. La administración de un compuesto de la invención o una composición farmacéutica, en esta terapia de combinación puede determinarse tal como se describe en el presente documento. Se cree que los compuestos de la presente invención pueden volver a las células anormales más sensibles al tratamiento con radiación para los fines de destruir y/o inhibir el crecimiento de tales células.
En consecuencia, también se desvela un método para sensibilizar células anormales en un mamífero para su tratamiento con radiación que comprende la administración al mamífero de una cantidad de un compuesto de la presente invención o composición farmacéutica, cuya cantidad es eficaz en la sensibilización de las células anormales para el tratamiento con radiación. La cantidad del compuesto en este método puede determinarse de acuerdo con los medios para determinar cantidades eficaces de tales compuestos descritos en el presente documento.
En su uso práctico, los compuestos de la presente invención se pueden combinar como el principio activo en mezcla íntima con un portador farmacéutico de acuerdo con técnicas convencionales de composición farmacéutica. El portador puede adoptar una amplia diversidad de formas dependiendo de la forma de preparación deseada para su administración, por ejemplo, oral o parenteral (incluyendo intravenosa). En la preparación de las composiciones para formas orales de dosificación, puede emplearse cualquiera de los medios farmacéuticos habituales, tales como, por ejemplo, agua, glicoles, aceites, alcoholes, agentes aromatizantes, conservantes, agentes colorantes y similares. En el caso de preparaciones líquidas orales, puede emplearse cualquiera de los medios farmacéuticos habituales, tales como, por ejemplo, suspensiones, elixires y soluciones; o portadores tales como almidones, azúcares, celulosa microcristalina, diluyentes, agentes de granulación, lubricantes, aglutinantes, agentes disgregantes y similares. En el caso de preparaciones sólidas orales la composición puede tomar formas tales como, por ejemplo, polvos, cápsulas duras y blandas y comprimidos, prefiriéndose las preparaciones sólidas orales sobre las preparaciones líquidas. Debido a su facilidad de administración, los comprimidos y cápsulas representan la forma monodosis oral más ventajosa, en cuyo caso se emplean, obviamente, portadores farmacéuticos sólidos. Si se desea, los comprimidos pueden recubrirse mediante técnicas convencionales acuosas o no acuosas. Dichas composiciones y preparaciones deberían contener al menos un 0,1 por ciento de compuesto activo. El porcentaje de compuesto activo en estas composiciones puede, por supuesto, variarse y puede ser de forma conveniente entre aproximadamente el 2 por ciento a aproximadamente el 60 por ciento del peso de la unidad. La cantidad de compuesto activo en tales composiciones terapéuticamente útiles es tal que se obtendrá una dosificación eficaz. Los compuestos activos también pueden administrarse por vía intranasal, por ejemplo, gotas líquidas o aerosol. Los comprimidos, píldoras, cápsulas y similares también pueden contener un aglutinante tal como goma tragacanto, goma arábiga, almidón de maíz o gelatina; excipientes tales como fosfato de dicalcio; un agente disgregante, tal como almidón de maíz, almidón de patata, ácido algínico; un lubricante, tal como estearato de magnesio, y un agente edulcorante, tal como sacarosa, lactosa o sacarina. Cuando una forma farmacéutica unitaria es una cápsula, puede contener, además de los materiales del tipo anterior, un portador líquido, tal como un aceite graso.
Puede haber diversos otros materiales presentes como recubrimientos o para modificar la forma física de la unidad de dosificación. Por ejemplo, los comprimidos pueden estar recubiertos con goma laca, azúcar o ambos. Un jarabe o elixir puede contener, además del principio activo, sacarosa como agente edulcorante, metilo y propilparabenos como conservantes, un colorante y un aromatizante como sabor a cereza o naranja.
Los compuestos de la presente invención también pueden administrarse por vía parenteral. Se pueden preparar soluciones o suspensiones de estos compuestos activos en agua mezclada de forma adecuada con un tensioactivo tal como hidroxipropilcelulosa. También se pueden preparar dispersiones en glicerol, polietilenglicoles líquidos y mezclas de los mismos y en aceites. En condiciones normales de almacenamiento y uso, estos preparados contienen un conservante para impedir el crecimiento de microorganismos.
Las formas farmacéuticas adecuadas para uso inyectable incluyen soluciones o dispersiones acuosas estériles y polvos estériles para la preparación extemporánea de soluciones o dispersiones inyectables estériles. En todos los casos, la forma debe ser estéril y debe ser fluida hasta el punto de que pueda inyectarse fácilmente. Debe ser estable en las condiciones de fabricación y almacenamiento y ha de preservarse frente a la acción contaminante de microorganismos, tales como bacterias y hongos. El portador puede ser un disolvente o medio de dispersión que contiene, por ejemplo, agua, etanol, poliol (por ejemplo, glicerol, propilenglicol y polietilenglicol líquido), mezclas adecuadas de los mismos, y aceites vegetales.
Puede emplearse cualquier vía de administración adecuada para proporcionar a un mamífero, especialmente un ser humano, una dosis eficaz de un compuesto de la presente invención. Por ejemplo, oral, rectal, tópica, administración parenteral, ocular, pulmonar, nasal y similares, se pueden emplear. Las formas de dosificación incluyen comprimidos, trociscos, dispersiones, suspensiones, soluciones, cápsulas, cremas, ungüentos, aerosoles y similares. Preferentemente, los compuestos de la presente invención se administran por vía oral.
La dosificación eficaz del principio activo empleado puede variar dependiendo del compuesto particular empleado, el modo de administración, la afección a tratar y la gravedad de la afección a tratar. Tal dosificación puede confirmarse fácilmente por un experto en la materia.
Cuando se tratan enfermedades inflamatorias, degenerativas o hiperproliferativas para las cuales se han indicado los compuestos de la presente invención, se obtienen resultados generalmente satisfactorios cuando los compuestos de la presente invención se administran a una dosificación diaria de aproximadamente 0,01 miligramos a aproximadamente 100 miligramos por kilogramo de peso corporal, preferentemente proporcionado como una dosificación diaria única. Para la mayoría de los grandes mamíferos, la dosificación diaria total es de aproximadamente 0,1 miligramos a aproximadamente 1000 miligramos, preferentemente de aproximadamente 0,2 miligramos a aproximadamente 50 miligramos. En el caso de un ser humano adulto de 70 kg, la dosificación diaria total será generalmente de aproximadamente 0,2 miligramos a aproximadamente 200 miligramos. Esta dosificación puede ajustarse para proporcionar la respuesta terapéutica óptima.
La invención también se refiere a un conjunto (kit) que consiste en envases separados de
a) una cantidad eficaz de un compuesto según la invención o sus estereoisómeros o tautómeros o sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas las mezclas de los mismos en todas las relaciones, y
b) una cantidad eficaz de un principio activo de medicamento adicional.
El conjunto comprende recipientes adecuados, tales como cajas, botellas individuales, bolsas o ampollas.
A modo de ejemplo, el conjunto puede comprender ampollas separadas, que contiene cada una, una cantidad eficaz de un compuesto según la invención y una cantidad eficaz de un principio activo de medicamento adicional en forma disuelta o liofilizada.
Sección Experimental
Algunas abreviaturas que pueden aparecer en la presente solicitud son como se indica a continuación:
Abreviaturas


Los compuestos de la presente invención pueden prepararse de acuerdo con los procedimientos de los siguientes Esquemas y Ejemplos, usando materiales adecuados y se ilustran adicionalmente mediante los siguientes ejemplos específicos.
Además, utilizando los procedimientos descritos en el presente documento, junto con habilidades habituales en la técnica, pueden prepararse fácilmente compuestos adicionales de la presente invención reivindicada en el presente documento. Los compuestos ilustrados en los ejemplos no deben, sin embargo, interpretarse como que forman el único género que se considera como la invención. Los ejemplos ilustran adicionalmente detalles para la preparación de los compuestos de la presente invención. Los expertos en la materia entenderán rápidamente que pueden usarse variaciones conocidas de las condiciones y procesos de los siguientes procedimientos preparativos para preparar estos compuestos.
Los presentes compuestos generalmente se aíslan en forma de sus sales farmacéuticamente aceptables, tales como las descritas anteriormente. Las de amina correspondientes a las sales aisladas pueden generarse mediante neutralización con una base adecuada, tal como hidrogenocarbonato sódico acuoso, carbonato sódico, hidróxido sódico e hidróxido potásico, y extracción de la base libre de amina liberada en un disolvente orgánico, seguido de evaporación. La base libre de amina, aislada de esta manera, puede convertirse adicionalmente en otra sal farmacéuticamente aceptable mediante disolución en un disolvente orgánico, seguido de la adición del ácido adecuado y posterior evaporación, precipitación o cristalización.
La invención se ilustrará, aunque sin limitación, por referencia a realizaciones específicas descritas en los siguientes ejemplos. A menos que se indique otra cosa en los esquemas, las variables tienen el mismo significado que se ha descrito anteriormente.
Salvo que se especifique otra cosa, todos los materiales de partida se obtienen de proveedores comerciales y se usan sin purificaciones adicionales. Salvo que se especifique otra cosa, todas las temperaturas se expresan en °C y todas las reacciones se realizan a TA. Los compuestos se purificaron por cromatografía de gel de sílice o HPLC preparativa.
La presente invención también se refiere a un proceso para la fabricación de compuestos de Fórmula (I), en los que un compuesto de Fórmula (IV)

se hace reaccionar, opcionalmente en presencia de un agente de activación, con un compuesto de Fórmula (III),

para producir
Los expertos en la materia están al corriente de una diversidad de agentes de activación, usados normalmente en síntesis de péptidos para acelerar la reacción, tales como carbodiimidas y triazoles. Son ejemplos específicos de estos agentes: DCC, DIC, HBTU, HATU, HCTU, TBTU, DAPECI, PyBOP, HOBt o HOAt.
Ejemplos
Los ejemplos funcionales presentados más adelante están destinados a ilustrar realizaciones particularmente preferidas de la invención, y no están destinados a limitar el ámbito de la memoria descriptiva o las reivindicaciones de ningún modo.
Procedimientos HPLC
Procedimiento A (Procedimiento de HPLC/EM, polar)
Disolvente A: agua 0,05 % de ácido fórmico
Disolvente B: acetonitrilo 0,04 % de ácido fórmico
Flujo: 2,4 ml/min, longitud de onda: 220 nm
Gradiente: 0,00 min 5 % de B
2.80 min 100 % de B
3.30 min 100 % de B
3.40 min 5 % de B
Columna: Chromolith® Speed ROD RP18e 50-4,6 mm
Procedimiento B (procedimiento de HPLC/EM)
Disolvente A: aguas 0,1 % de TFA
Disolvente B: acetonitrilo 0,1 % de TFA
Flujo: 2 ml/min, longitud de onda: 220 nm
Gradiente: 0,00 min 1 % de B
0,20 min 1 % de B
3.80 min 100 % de B
4.20 min 100 % de B
4.30 min 1 % de B
Columna: Chromolith® Performance RP18e 100-3 mm
Procedimiento C (procedimiento de HPLC/EM)
Disolvente A: agua 0,1 % de ácido fórmico
Disolvente B: acetonitrilo 0,08 % de ácido fórmico
Flujo: 0,9 ml/min, longitud de onda: 220 nm
Gradiente: 0,00 min 5 % de B
1.00 min 100 % de B
1,2 min 100 % de B
1.40 min 5 % de B
Columna: ACQUITY UPLC® BEH C18 1,7 pm
Procedimiento D (procedimiento de HPLC/EM)
Disolvente A: agua 0,05 % de ácido fórmico
Disolvente B: acetonitrilo 0,05 % de ácido fórmico
Flujo: 2,5 ml/min, longitud de onda: 220 nm
Gradiente: 0,00 min 0 % de B
1.40 min 100 % de B
2.00 min 100 % de B
2.20 min 0 % de B
Columna: Chromolith® Performance RP18e 100-3 mm
Procedimiento E (procedimiento de HPLC/EM)
Disolvente A: agua NH4HCO35 mM
Disolvente B: acetonitrilo
Flujo: 1,5 ml/min, longitud de onda: 190-400 nm
Gradiente: 0,01 min 10 % de B
4.20 min 70 % de B
5.20 min 70 % de B
5.30 min 10 % de B
5,60 min, parada
Columna: XBridge BEH C183,5 pM 50-4,6 mm
Temp. de columna: 40 °C
Procedimiento F (procedimiento de HPLC/EM)
Disolvente A: agua 0,05 % de TFA
Disolvente B: acetonitrilo 0,05 % de TFA
Flujo: 1 ml/min, longitud de onda: 220 nm;
Gradiente: 0,01 min 5 % de B
3.00 min 50 % de B
5.00 min 50 % de B
5.20 min 5 % de B
5.60 min, parada
Columna: Shim-pack VP-ODS 50-3 mm
Temp. de columna: 40 °C
Procedimiento G (procedimiento de HPLC/EM)
Disolvente A: aguas 0,05 % de TFA
Disolvente B: acetonitrilo 0,05 % de TFA
Flujo: 1 ml/min, longitud de onda: 220 nm
Gradiente: 0,01 min 5 % de B
2.20 min 100 % de B
3.20 min 100 % de B
3.30 min 5 % de B
3.60 min, parada
Columna: Shim-pack VP-ODS 50-3 mm
Temp. de columna: 40 °C
Procedimiento H (procedimiento de HPLC/EM)
Disolvente A: agua NH4HCO35 mM
Disolvente B: acetonitrilo
Flujo: 1,5 ml/min, longitud de onda: 190-400 nm
Gradiente: 0,01 min 5 % de B
2.20 min 95 % de B
3.20 min 95 % de B
3.30 min 5 % de B
3.60 min, parada
Columna: XBridge BEH C183,5 |uM 50-4,6 mm
Temp. de columna: 40 °C
Procedimiento I (procedimiento de HPLC/EM)
Disolvente A: aguas 0,05 % de TFA
Disolvente B: acetonitrilo 0,05 % de TFA
Flujo: 1 ml/min, longitud de onda: 220 nm
Gradiente: 0,01 min 5 % de B
4.20 min 100 % de B
5.20 min 100 % de B
5.30 min 5 % de B
5.60 min, parada
Columna: Shim-pack VP-ODS 50-3 mm
Temp. de columna: 40 °C
Procedimiento J (procedimiento de HPLC/EM)
Disolvente A: aguas 0,1 % de TFA
Disolvente B: acetonitrilo 0,1 % de TFA
Flujo: 1,5 ml/min, longitud de onda: 220 nm
Gradiente: 0,01 min 10 % de B
2.00 min 95 % de B
2.60 min 95 % de B
2,70 min 10 % de B
3.00 min, parada
Columna: ACE UltraCore 2,5 Super C18, 50-3 mm
Temp. de columna: 40 °C
Procedimiento K de CLEM:
La separación analítica se realizó a 40 °C en una columna Merck Purospher STAR (RP-18e, 30 x 4 mm) usando un caudal de 3 ml/min en una elución en gradiente de 2 minutos con detección a 254 nm. La fase móvil fue una mezcla de metanol (disolvente A) y agua (disolvente B), conteniendo ambos 0,1 % de ácido fórmico. La elusión en gradiente fue de la siguiente manera: 1:9 (A/B) a 9:1 (A/B) durante 1,25 min, 9:1 (A/B) durante 0,5 min, y después vuelta a 1:9 (A/B) durante 0,15 min, finalmente 1:9 (A/B) durante 0,1 min
Procedimiento L de CLEM:
La separación analítica se realizó a 30 °C en una columna Merck Purospher STAR (RP-18e, 30 x 4 mm) usando un caudal de 1,5 ml/min en una elución en gradiente de 4 minutos con detección a 254 nm. La fase móvil fue una mezcla de metanol (disolvente A) y agua (disolvente B), conteniendo ambos 0,1 % de ácido fórmico. La elusión en gradiente fue de la siguiente manera: 1:9 (A/B) a 9:1 (A/B) durante 2,5 min, 9:1 (A/B) durante 1 min, y después vuelta a 1:9 (A/B) durante 0,3 min, finalmente 1:9 (A/B) durante 0,2 min.
Procedimiento M de CLEM:
La separación analítica se realizó a 30 °C en una columna Phenomenex Kinetex XB-C18 (30 x 2,1 mm, 1,7u, 100A) usando un caudal de 0,5 ml/min en una elución en gradiente de 2 minutos con detección a 254 nm. La fase móvil fue una mezcla de metanol (disolvente A) y agua que contenía ácido fórmico a 0,1 % (disolvente B). La elusión en gradiente fue de la siguiente manera: 1:9 (A/B) a 9:1 (A/B) durante 1,25 min, 9:1 (A/B) durante 0,5 min, y después vuelta a 1:9 (A/B) durante 0,15 min, finalmente 1:9 (A/B) durante 0,1 min.
Síntesis químicas
En esta sección experimental se proporcionan detalles para una diversidad de compuestos de Ejemplo de acuerdo con la Fórmula (I) e intermedios sintéticos de los mismos.
1. [(S)-2-(4-Bromo-fenil)-pirrolidin-1-il]-(3-propil-1H-indazol-5-il)-metanona 5 y [(R)-2-(4-bromo-fenil)-pirrolidin-1 -il]-(3-propil-1 H-indazol-5-il)-metanona 6

1.1 [2-(4-Bromo-fenil)-pirrolidin-1-il]-(3-propil-1 H-indazol-5-il)-metanona 4

A una solución de ácido 3-propil-1H-indazol-5-carboxílico (50,0 mg, 0,24 mmol), 2-(4-bromo-fenil)-pirrolidina, 97 % (110 mg, 0,47 mmol) y 4-metilmorfolina (0,064 ml, 0,58 mmol) en N,N-dimetil-formamida (2 ml), se añadieron tetrafluoroborato de 2-(1H-benzotriazol-1-il)-1,1,3,3-tetrametilaminio (TBTU, 83,0 mg, 0,26 mmol) e hidrato de 1-hidroxibenzotriazol (9,00 mg, 0,068 mmol) y se agitaron a temperatura ambiente durante una noche. Se añadió agua y el precipitado formado se retiró por filtración y se secó al vacío. El producto en bruto se purificó por cromatografía ultrarrápida (diclorometano/metanol) para producir 52,0 mg (52 %) del compuesto del título en forma de cristales de color amarillo.
Síntesis alternativa:
En un recipiente de cristal con tapón de rosca, se disolvió ácido 3-propil-1H-indazol-5-carboxílico (50,0 mg, 0,24 mmol), 2-(4-bromo-fenil)-pirrolidina, 97 % (72,0 mg, 0,31 mmol) en diclorometano (5 ml), seguido de N,N-diisopropiletilamina (0,12 ml, 0,73 mmol) y anhídrido cíclico del ácido 1-propilfosfónico (50 % en acetato de etilo, 0,28 ml, 0,47 mmol). La solución se agitó a temperatura ambiente durante una noche. La solución se trituró con acetato de etilo, se lavó con agua, una solución de NaHCÜ3 y salmuera. La capa orgánica se secó sobre sulfato sódico, se filtró y se evaporó a sequedad. El producto en bruto se purificó por cromatografía ultrarrápida (ciclohexano/acetato de etilo/metanol) para producir 7,30 mg (8 %) del compuesto del título en forma de cristales de color amarillo.
1.2. [(S)-2-(4-Bromo-fenil)-pirrolidin-1-il]-(3-propil-1H-indazol-5-il)-metanona 5 y [(R)-2-(4-bromo-fenil)-pirrolidin-1-il]-(3-propil-1 H-indazol-5-il)-metanona 6

Se separaron 52,0 mg (0,13 mmol) de la mezcla racémica disueltos en metanol (1 ml) en los materiales enantioméricamente puros contenidos por HPLC quiral en porciones de 25 pl/ciclo para producir 19,9 mg (38 %) de cristales de color amarillo claro como 5 y 19,4 mg (37 %) de cristales de color amarillo claro como 6. HPLC/EM (quiral): Tr 2,40 min (procedimiento posterior, 5), Tr 3,32 min (procedimiento posterior, 6).
Instrumento: SFC Berger Minigram, columna: ChiralPak AD-H, eluyente: CÜ2/metanol 0,5 % de dietilamina 60:40, isocrático, flujo: 5 ml/min, detección: 220 nm.
De acuerdo con estos procedimientos, se sintetizaron los compuestos 8, 9, 11 y 12.
2. (S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1H-pirazolo[3,4-b]piridin-5-il)- metanona 13 y [(R)-2-(4-cloro-fenil)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona 14

2.1. Ácido 3-metil-1 H-pirazolo[3,4-b]piridin-5-carboxílico
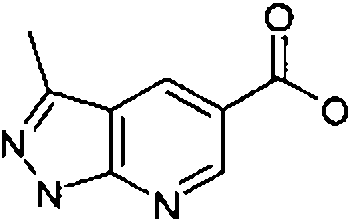
Se disolvió 3-metil-1H-pirazolo[3,4-b]piridin-5-carboxilato de metilo (500 mg, 2,54 mmol) en tetrahidrofurano (100 ml). Se añadió hidróxido de litio (180 mg, 7,60 mmol) disuelto en agua (50 ml) y se agitó a temperatura ambiente durante una noche. La mezcla de reacción se acidificó a pH 5 usando HCI 1 N y el precipitado se retiró por filtración, se lavó con metil ferc-butil éter y se secó al vacío para dar como resultado 377 mg (83 %) del compuesto del título en forma de cristales de color blanco. HPLC: (pureza) 99 %. HPLC EM: Temperatura ambiente 1,21 min (procedimiento A).
2.2. [2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)- metanona 10
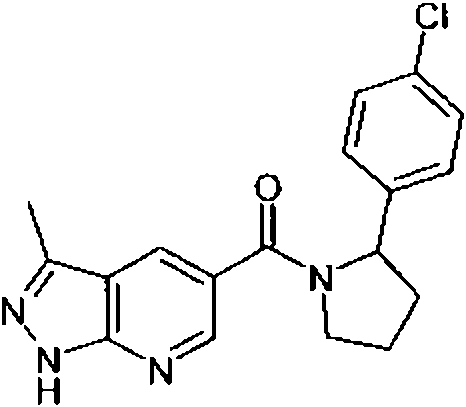
A una solución de ácido 3-metil-1H-pirazolo[3,4-b]piridin-5-carboxílico (510 mg, 2,74 mmol), clorhidrato de 2-(4-clorofenil)-pirrolidina (1,00 g, 4,56 mmol) y 4-metilmorfolina (0,064 ml, 0,58 mmol) en N,N-dimetilformamida (2 ml) se añadió tetrafluoroborato de 2-(1H-benzotriazol-1-il)-1,1,3,3-tetrametilaminio (TBTU, 83,0 mg, 0,26 mmol) e hidrato de 1 hidroxibenzotriazol (9,00 mg, 0,068 mmol) a temperatura ambiente. Se agitó a temperatura ambiente durante una noche. Se añadió agua a la mezcla y la capa acuosa se extrajo con acetato de etilo. El producto se encontró en la fase acuosa. Se evaporó a sequedad y el producto en bruto se purificó por cromatografía ultrarrápida (diclormetano/metanol) para producir 667 mg (71 %) del compuesto del título en forma de cristales de color amarillo.
2.3. (S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1H-pirazolo[3,4-b]piridin-5-il)- metanona 13 y [(R)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona 14
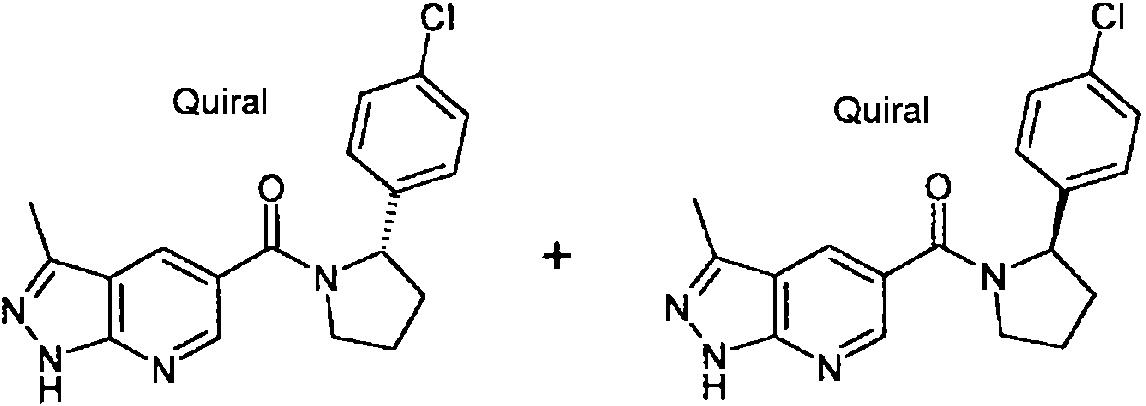
Se separaron 100 mg (0,56 mmol) de la mezcla racémica disueltos en metanol (1 ml) en los materiales enantioméricamente puros contenidos por HPLC quiral en porciones de 60 pl/ciclo para producir 39,1 mg (39 %) de cristales de color amarillo claro como 13 y 40,2 mg (40 %) de cristales de color amarillo claro como 14. HPLC/EM (quiral): Tr 3,84 min (procedimiento posterior, 13), Tr 5,96 min (procedimiento posterior, 14).
Instrumento: SFC Berger Minigram, columna: ChiralPak AD-H, eluyente: CÜ2/metanol 0,5 % de dietilamina 60:40, isocrático, flujo: 5 ml/min, detección: 220 nm.
De acuerdo con estos procedimientos se sintetizaron los compuestos 1-3, 7, 15-30, 32-34, 36-41 y 43-46, 55-69, 94 95, 121-123, 125, 127, 129, 130, 136-139, 141-146, 149-150, 157-160, 162 y 170.
3. [(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-isobutil-1H-indazol-5-il)-metanona 35 y [(R)-2-(4-cloro-fenil)-pirrolidin-1-il]-(3-isobutil-1 H-indazol-5-il)- metanona 31

3.1 Ácido 3-yodo-1 H-indazol-5-carboxílico

En un matraz de fondo redondo de 25 ml se puso ácido 1H-indazol-5-carboxílico (200 mg, 1,23 mmol), N,N-dimetilformamida (3 ml), hidróxido potásico (138 mg, 2,46 mmol) y yodo (470 mg, 1,85 mmol). La solución se agitó durante 3 h a 25 °C. La reacción se interrumpió mediante la adición de 10 ml de Na2S2Ü3. El valor del pH de la solución se ajustó a 6 con una solución de cloruro de hidrógeno (10 %). Los sólidos se recogieron por filtración. Esto dio como resultado 300 mg (84 %) de ácido 3-yodo-1 H-indazol-5-carboxílico en forma de un sólido de color blanco.
3.2. Ácido 3-(2-metilprop-1 -en-1 -il)-1 H-indazol-5-carboxílico

En un matraz de fondo redondo de 250 ml purgado y mantenido con una atmósfera inerte de nitrógeno, se puso ácido 3-yodo-1H-indazol-5-carboxílico (2,00 g, 6,94 mmol), 4,4,5,5-tetrametil-2-(2-metilprop-1-en-1-il)-1,3,2-dioxaborolano (1,90 g, 10,4 mmol), Pd(PPh3)4 (800 mg, 0,69 mmol), carbonato potásico (2,86 g, 20,7 mmol), dioxano (40 ml) y agua (10 ml). La solución se agitó durante 3 h a 90 °C. El valor del pH de la solución se ajustó a 6 con una solución de cloruro de hidrógeno (10 %). Los sólidos se recogieron por filtración. Esto dio como resultado 1,30 g (87 %) de ácido 3-(2-metilprop-1 -en-1 -il)-1 H-indazol-5-carboxílico en forma de un sólido de color amarillo.
3.3. Ácido 3-(2-metilpropil)-1H-indazol-5-carboxílico

En un tubo cerrado herméticamente de 2000 ml se pusieron ácido 3-(2-metilprop-1-en-1-il)-1H-indazol-5-carboxílico (1,20 g, 5,55 mmol) y paladio sobre carbono (2,00 g, 18,8 mmol). Esto se siguió de la adición de metanol (800 ml). En la mezcla anterior se introdujo hidrógeno y se añadió una solución de cloruro de hidrógeno (4 ml, 10 %). La solución se agitó durante 4 h a 60 °C. Los sólidos se retiraron por filtración. La mezcla se concentró al vacío. Esto dio como resultado 1,00 g (83 %) de ácido 3-(2-metilpropil)-1 H-indazol-5-carboxílico en forma de un sólido de color amarillo.
3.4. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-isobutil-1 H-indazol-5-il)-metanona 35 y [(R)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-isobutil-1 H-indazol-5-il)- metanona 31

En un matraz de fondo redondo de 25 ml, se puso ácido 3-(2-metilpropil)-1H-indazol-5-carboxílico (120 mg, 0,55 mmol), 2-(4-clorofenil)pirrolidina (150 mg, 0,83 mmol), hexafluorofosfato de 3-óxido de 1-[bis(dimetilamino)metileno]-1 H-1,2,3-triazolo[4,5-¿>]piridinio (HATU, 208 mg, 0,55 mmol), N,N-diisopropiletilamina
(212 mg, 1,64 mmol) y N,N-dimetilformamida (2 ml). La solución se agitó durante 1 h a temperatura ambiente. La mezcla se concentró al vacío. El resto se purificó por HPLC prep. para dar como resultado 80 mg (38 %) de material racémico. La mezcla racémica se purificó por HPLC prep. quiral (columna: ChiralPak IC, 2*25 cm, 5 gm, fase móvil: hexano/isopropanol (parada a 50 % de isopropanol en 25 min), Detector: UV 254/220 nm). Se obtuvieron 20 mg (10 %) de 5-[[(2S)-2-(4-clorofenil)pirrolidin-1 -il]carbonil]-3-(2-metilpropil)-1 H-indazol 35 en forma de un sólido de color blanco y 20 mg (10 %) de 5-[[(2R)-2-(4-clorofenil)pirrolidin-1 -il]carbonil]-3-(2-metilpropil)-1 H-indazol 31 en forma de un sólido de color blanco. De acuerdo con estos procedimientos generales, se sintetizó el compuesto 42.
4. (3-Amino-1 H-indazol-5-il)-[2-(4-cloro-fenil)-pirrolidin-1 -il]-metanona 78

4.1. Ácido 3-amino-1H-indazol-5-carboxílico

Se disolvieron ácido 3-ciano-4-fluoro-benzoico (2,00 g, 12,1 mmol) e hidróxido de hidrazinio (0,60 ml, 12,1 mmol) en 1-butanol (40 ml) y se agitaron a 110 °C durante una noche. Los cristales formados después de refrigeración a temperatura ambiente se retiraron por filtración, se lavaron con metil ferc-butil éter y se secaron al vacío a 40 °C para producir 1,16 g (66 %) del compuesto del título en forma de cristales de color beige. Cl/EM: Tr 0,99 min (procedimiento A).
4.2. (3-Amino-1 H-indazol-5-il)-[2-(4-cloro-fenil)-pirrolidin-1 -il]-metanona

A una solución de ácido 3-amino-1H-indazol-5-carboxílico (50,0 mg, 0,28 mmol), clorhidrato de 2-(4-cloro-fenil)-pirrolidina (123 mg, 0,56 mmol) y 4-metilmorfolina (0,064 ml, 0,58 mmol) en N,N-dimetilformamida (2 ml), se añadieron tetrafluoroborato de 2-(1H-benzotriazol-1-il)- 1,1,3,3-tetrametilaminio (TBTU, 83,0 mg, 0,26 mmol) e hidrato de 1-hidroxibenzotriazol (9,00 mg, 0,068 mmol) y se agitaron a temperatura ambiente durante una noche. Se añadió agua a la mezcla, el precipitado se retiró por filtración y se secó al vacío. El producto en bruto se purificó por HPLC preparativa para producir 48,0 mg (50 %) del compuesto del título en forma de un sólido de color blanquecino.
4.3. (3-Amino-1H-indazol-5-il)-[(S)-2-(4-cloro-fenil)-pirrolidin-1-il]-metanona 79 y (3-amino-1H-indazol-5-il)-[(R)-2-(4-cloro-fenil)-pirrolidin-1 -il]-metanona 80

Se separaron 45,0 mg (0,13 mmol) de la mezcla racémica disueltos en metanol (1 ml) en los materiales enantioméricamente puros contenidos por HPLC quiral en porciones de 90 pl/ciclo para producir 22,1 mg (49 %) de cristales de color amarillo claro como 79 y 20,0 mg (44 %) de cristales de color amarillo claro como 80. HPLC/EM (quiral): Tr 3,47 min (procedimiento posterior, 79), Tr 6,45 min (procedimiento posterior, 80).
Instrumento: SFC Berger Minigram, columna: ChiralPak AD-H, eluyente: CÜ2/metanol 0,5 % de dietilamina 60:40, isocrático, flujo: 5 ml/min, Detección UV: 220 nm.
De acuerdo con estos procedimientos generales, se sintetizaron los compuestos 74-77, 81-93,104-118, 120, 124, 131 135, 147, 148 y 167.
5. [2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-ciclopropil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona 70

5.1. 1 -(3-Ciclopropil-1 H-indazol-5-carbonil)-1,3-dimetil-urea

Una mezcla de 3-amino-5-ciclopropil-1H-pirazol (250 mg, 2,03 mmol) y 1,3-dimetil-2,4-dioxo-1,2,3,4-tetrahidropirimidina-5-carbaldehído (360 mg, 2,03 mmol) en ácido acético glacial (20 ml) se calentó a reflujo durante 6 h. Después de un periodo de refrigeración, el disolvente se evaporó a presión reducida. El residuo se disolvió en metanol y la solución se dejó en reposo durante una noche. La mezcla se diluyó con éter dietílico, los cristales formados se retiraron por filtración y se secaron al vacío para obtener 246 mg (44 %) del compuesto del título en forma de cristales de color beige claro.
5.2. Ácido 3-ciclopropil-1H-indazol-5-carboxílico

Una mezcla de 1-(3-ciclopropil-1H-pirazolo[3,4-b]piridin-5-carbonil)-1,3-dimetil-urea (246 mg, 0,89 mmol) y una solución de hidróxido sódico, 10 % (8 ml) se calentó y se agitó durante 2 h a 60 °C. Después de un periodo de
refrigeración, la mezcla de reacción se filtró y el filtrado se acidificó con HCI diluido. La mezcla se extrajo con diclorometano. La fase orgánica se dejó en reposo durante una noche para formar una suspensión. Los cristales se retiraron por filtración, se lavaron con agua y se secaron al vacío para producir 133 mg (74 %) del compuesto del título en forma de cristales de color blanco.
5.3. [2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-ciclopropil-1H-pirazolo[3,4-b]piridin-5-il)- metanona 70

A una solución de ácido 3-ciclopropil-1H-pirazolo[3,4-b]piridin-5-carboxílico (100 mg, 0,49 mmol), 2-(4-cloro-fenil)-pirrolidina (152 mg, 0,84 mmol) y 4-metilmorfolina (0,064 ml, 0,58 mmol) en N,N-dimetilformamida (2 ml), se añadieron tetrafluoroborato de O-(1H-benzotriazol-1-il)- N,N,N',N'-tetrametiluronio (TBTU, 83,0 mg, 0,26 mmol) e hidrato de 1 hidroxibenzotriazol (9,00 mg, 0,07 mmol) y se agitaron a temperatura ambiente durante una noche. Se añadió agua a la mezcla, se extrajo dos veces con acetato de etilo y las fases orgánicas combinadas se secaron con Na2SO4, se filtraron y se evaporaron a sequedad. El producto en bruto se purificó por HPLC preparativa (acetonitrilo/agua). Las fracciones combinadas se hicieron alcalinas con NaOH 1 N, se extrajeron dos veces con diclorometano, las fases orgánicas combinadas se secaron sobre Na2SO4, se filtraron y se evaporaron a sequedad para producir 92,3 mg del compuesto del título en forma de cristales de color blanco. De acuerdo con estos procedimientos generales, se sintetizaron los compuestos 71-73, 152-153 y 168-169.
6. (S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1H-pirazolo[3,4-b]piridin-5-il)-metanona 155 y [(R)-2-(4-cloro-fenil)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona 156

6.1. [2-(4-Bromo-fenil)-pirrolidin-1-il]-(1H-pirazolo[3,4-b]piridin-5-il)-metanona 154

A una solución de ácido 1H-lndazol-5-carboxílico (100 mg, 0,60 mmol), clorhidrato de 2-(4-bromo-fenil)-pirrolidina (280 mg, 1,02 mmol) y 4-metilmorfolina (0,064 ml, 0,58 mmol) en N,N-dimetilformamida (2 ml) se añadió tetrafluoroborato de 2-(1H-benzotriazol-1-il)-1,1,3,3-tetrametilaminio (TBTU, 83,0 mg, 0,26 mmol) e hidrato de 1 hidroxibenzotriazol (9,00 mg, 0,068 mmol) a temperatura ambiente. Se agitó a temperatura ambiente durante una noche. Se añadió agua a la mezcla y la capa acuosa se extrajo con acetato de etilo. El producto se encontró en la fase acuosa. Se evaporó a sequedad y el producto en bruto se purificó por cromatografía ultrarrápida (diclormetano/metanol) para producir 41,1 mg (19 %) del compuesto del título en forma de cristales de color blanco.
6.2. (S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1H-pirazolo[3,4-b]piridin-5-il)- metanona 155 y [(R)-2-(4-cloro-fenil)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona 156

Se separaron 35,5 mg (0,096 mmol) de la mezcla racémica disueltos en metanol (0,5 ml) en los materiales enantioméricamente puros contenidos por HPLC quiral en porciones de 60 pl/ciclo para producir 13,8 mg (39 %) de cristales de color amarillo claro como 155 y 14,2 mg (40 %) de cristales de color amarillo claro como 156. HPLC/EM (quiral): Tr 4,66 min (procedimiento posterior, 155), Tr 7,33 min (procedimiento posterior, 156).
Instrumento: SFC Berger Minigram, columna: ChiralPak AD-H, eluyente: CÜ2/metanol 0,5 % de dietilamina 60:40, isocrático, flujo: 5 ml/min, detección: 220 nm.
7. [(S)-2-(4-Bromo-fenil)-pirrolidin-1 -il]-(3-metil-1 H-indazol-5-il)-metanona 49

7.1. 5-Bromo-3-metil-1H-indazol

En un tubo de 50 ml cerrado herméticamente, purgado y mantenido con una atmósfera inerte de nitrógeno, se añadieron 1-(5-bromo-2-fluorofenil)etan-1-ona (6,00 g, 27,7 mmol) e hidrato de hidrazina (30 ml). La solución se agitó durante 14 h a 117 °C. La mezcla de reacción se concentró al vacío. El residuo se disolvió en agua y se extrajo con 3 veces con 20 ml de acetato de etilo. Las capas orgánicas se combinaron, se secaron sobre sulfato sódico anhidro y se concentraron. El residuo se purificó mediante una columna de gel de sílice, eluyendo con éter de petróleo:acetato de etilo (5:1). Las fracciones recogidas se combinaron y se concentraron al vacío. Esto dio como resultado 3,00 g (51 %) de 5-bromo-3-metil-1 H-indazol en forma de un sólido de color amarillo.
7.2. Éster metílico del ácido 3-metil-1 H-indazol-5-carboxílico

En un reactor de tanque de acero de 50 ml purgado y mantenido con una atmósfera de monóxido de carbono (5 atm), se añadieron 5-bromo-3-metil-1H-indazol (1,00 g, 4,74 mmol), Pd(dppf)Cl2.CH2Cl2 (400 mg, 0,49 mmol), acetato potásico (1,40 g, 14,3 mmol), N,N-dimetilformamida (5 ml) y metanol (25 ml). La solución se agitó durante 14 h a 80 °C. La mezcla de reacción se concentró al vacío. El residuo se disolvió en agua y se extrajo 3 veces con 20 ml de acetato de etilo. Las capas orgánicas se combinaron, se secaron sobre sulfato sódico anhidro y se concentraron. El residuo se purificó mediante una columna de gel de sílice eluyendo con acetato de etilo/éter de petróleo (3:7). Esto dio como
resultado 750 mg (83 %) de éster metílico del ácido 3-metil-1H-¡ndazol-5-carboxílico en forma de un sólido de color amarillo.
7.3. Ácido 3-metil-1H-indazol-5-carboxílico

En un matraz de fondo redondo de 100 ml se añadieron éster metílico del ácido de 3-metil-1H-indazol-5-carboxílico (500 mg, 2,63 mmol), hidróxido sódico (210 mg, 5,25 mmol), tetrahidrofurano (50 ml) y agua (13 ml). La solución se agitó durante 2 h a 50 °C. La mezcla de reacción se concentró al vacío. El residuo se disolvió en 30 ml de agua. El valor del pH de la solución se ajustó a 3 con una solución de HCI (1 M). El precipitado se recogió por filtración. Esto dio como resultado 300 mg (65 %) de ácido 3-metil-1 H-indazol-5-carboxílico en forma de un sólido de color amarillo.
7.4. [(S)-2-(4-Bromo-fenil)-pirrolidin-1 -il]-(3-metil-1 H-indazol-5-il)-metanona 49

En un matraz de fondo redondo de 25 ml, se añadieron ácido 3-metil-1H-indazol-5-carboxílico (80,0 mg, 0,45 mmol), 2-(4-clorofenil)pirrolidina (124 mg, 0,68 mmol), diisopropiletilamina (176 mg, 1,36 mmol), hexafluorofosfato de 3-óxido de 1-[bis(dimetilamino)metileno]-1H-1,2,3-triazolo[4,5-b]piridinio (HÁTU, 207 mg, 0,54 mmol) y N,N-dimetilformamida (5 ml). La solución se agitó durante 3 h a 25 °C. La mezcla de reacción se concentró al vacío. El residuo se disolvió en agua y se extrajo 3 veces con 10 ml de acetato de etilo. Las capas orgánicas se combinaron. El producto en bruto (200 mg) se purificó por HPLC prep. (acetonitrilo/agua). El producto racémico (150 mg, 87 %) se purificó adicionalmente por HPLC quiral preparativa con las siguientes condiciones: Columna: Phenomenex Lux5 g Cellulose-4, 250*21,2 mm, 5 gm; fase móvil: hexano y etanol (20 % de etanol en hexano isocrático en 21 min); Detector, UV 254/220 nm. Esto dio como resultado 80,0 mg (46 %) de 5-[[(2S)-2-(4-bromofenil)pirrolidin-1-il]carbonil]-3-metil-1H-indazol 49 en forma de un sólido de color blanco.
De acuerdo con estos procedimientos generales, se sintetizaron los compuestos 47, 48 y 50.
8. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[4,3-b]piridin-5-il)- metanona 51

8.1. 3-Metil-1 H-pirazolo[4,3-b]piridina

En un tubo cerrado herméticamente de 50 ml se añadieron 1-(3-fluoropiridin-2-il)etan-1-ona (2,00 g, 14,4 mmol) e hidrato de hidrazina (25 ml). La solución se agitó durante 3 h a 130 °C. La mezcla de reacción se lavó 3 veces con 50 ml de acetato de etilo, se secó sobre sulfato sódico anhidro y se concentró. El residuo se purificó mediante una columna de gel de sílice eluyendo con acetato de etilo/éter de petróleo (2:3). Esto dio como resultado 900 mg (47 %) de 3-metil-1 H-pirazolo[4,3-b]piridina en forma de un sólido de color amarillo.
8.2. 3-Metil-1 -tritil-1 H-pirazolo[4,3-b]piridina

En un matraz de fondo redondo de 100 ml purgado y mantenido con una atmósfera inerte de nitrógeno se añadieron 3-metil-1H-pirazolo[4,3-b]piridina (2,43 g, 18,3 mmol) y N,N-dimetilformamida (20 ml) a 0 °C. Después, se añadió hidruro sódico (660 mg, 27,5 mmol) y después de 30 min (clorodifenilmetil)benceno (5,60 g, 20,1 mmol) a la mezcla de reacción. La solución se agitó durante 2 h a 25 °C. Después, la reacción se interrumpió mediante la adición de 5 ml de solución de NH4CI. La mezcla se concentró al vacío. El residuo se disolvió en 20 ml de acetato de etilo, se lavó 3 veces con 30 ml de agua, se secó sobre sulfato sódico anhidro y se concentró. El residuo se purificó mediante una columna de gel de sílice eluyendo con éter de petróleo:acetato de etilo (3:1). Esto dio como resultado 3,00 g (44 %) de 3-metil-1 -(trifenilmetil)-1 H-pirazolo[4,3-b]piridina en forma de un sólido de color amarillo.
8.3. 4-Óxido de 3-metil-1 -tritil-1 H-pirazolo[4,3-b]piridina

En un matraz de fondo redondo de 50 ml se añadieron 3-metil-1-(trifenilmetil)-1H-pirazolo[4,3-b]piridina (3,00 g, 7,99 mmol) y diclorometano (20 ml) a 0 °C. Después, se añadió ácido mefa-cloroperoxibenzoico (1,50 g, 8,69 mmol). La solución se agitó durante 2 h a 25 °C. La mezcla de reacción se lavó 3 veces con 20 ml de solución de carbonato sódico, se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó mediante una columna de gel de sílice eluyendo con diclorometano/acetato de etilo (1:1). Esto dio como resultado 2,70 g (86 %) de 4-óxido de 3-metil-1 -tritil-1 H-pirazolo[4,3-b]piridina en forma de un sólido de color blanquecino.
8.4. 5-Cloro-3-metil-1 H-pirazolo[4,3-b]piridina

En un tubo cerrado herméticamente de 50 ml, se añadieron 4-óxido de 3-metil-1 -tritil-1 H-pirazolo[4,3-b]piridina (2,84 g, 7,25 mmol), cloruro de fosforilo (20 ml). La solución se agitó durante 1 h a 130 °C. Después, la reacción se interrumpió mediante la adición de 20 ml de suspensión de agua/hielo. Se usó una solución de hidróxido sódico (6 M) para ajustar el pH de la mezcla a 7. La solución se extrajo 5 veces con 200 ml de diclorometano y las capas orgánicas se combinaron, se secaron sobre sulfato sódico anhidro y se concentraron. El residuo se purificó mediante una columna de gel de sílice eluyendo con diclorometano/metanol (20:1). Esto dio como resultado 1,40 g (98 %) de 5-cloro-3-metil-1 H-pirazolo[4,3-b]piridina en forma de un sólido de color blanquecino.
8.5. Éster metílico del ácido 3-metil-1 H-p¡razolo[4,3-b]p¡r¡d¡n-5-carboxílico

En un reactor de tanque de acero de 250 ml purgado y mantenido con una atmósfera de monóxido de carbono (5 atm), se añadieron 5-cloro-3-metil-1 H-pirazolo[4,3-b]piridina (1,20 g, 7,16 mmol), metanol (160 ml), acetato potásico (2,10 g, 21,4 mmol) y Pd(dppf)CÍ2.CH2Cl2 (585 mg, 0,72 mmol). La solución se agitó durante 14 h a 80 °C. La mezcla de reacción se concentró al vacío. El residuo se disolvió en 50 ml de agua. La solución se extrajo 3 veces con 60 ml de acetato de etilo y las capas orgánicas se combinaron y se concentraron. El residuo se purificó mediante una columna de gel de sílice eluyendo con acetato de etilo/éter de petróleo (3:2). Esto dio como resultado 720 mg (53 %) de éster metílico del ácido 3-metil-1H-pirazolo[4,3-b]piridin-5-carboxílico en forma de un sólido de color blanquecino.
8.6. Ácido 3-metil-1H-pirazolo[4,3-b]piridin-5-carboxílico

En un matraz de fondo redondo de 50 ml, se añadieron éster metílico del ácido 3-metil-1H-pirazolo[4,3-b]piridin-5-carboxílico (720 mg, 3,77 mmol), tetrahidrofurano (10 ml), agua (2 ml) e hidróxido sódico (226 mg, 5,65 mmol). La solución se agitó durante 2 h a 50 °C. La mezcla de reacción se concentró al vacío. El residuo se disolvió en 20 ml de agua. El sólido formado se retiró por filtración. Se añadió una solución de HCI (1 M) para ajustar el pH a 7. El precipitado se recogió por filtración. Esto dio como resultado 350 mg (52 %) de ácido 3-metil-1 H-pirazolo[4,3-b]piridin-5-carboxílico en forma de un sólido de color blanquecino.
8.7. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[4,3-b]piridin-5-il)- metanona 51

En un matraz de fondo redondo de 25 ml, se añadieron ácido 3-metil-1H-pirazolo[4,3-b]piridin-5-carboxílico (170 mg, 0,96 mmol), 2-(4-clorofenil)pirrolidina (261 mg, 1,44 mmol), N,N-dimetilformamida (4 ml), hexafluorofosfato de 3-óxido de 1-[bis(dimetilamino)metileno]-1H-1,2,3-triazolo[4,5-b]piridinio (HATU, 401 mg, 1,05 mmol) y diisopropiletilamina (372 mg, 2,88 mmol). La solución se agitó durante 1 h a 25 °C. La mezcla se concentró al vacío. El residuo se purificó por HPLC preparativa (acetonitrilo/agua). El producto racémico (80,0 mg, 24 %) se purificó adicionalmente por HPLC quiral preparativa con las siguientes condiciones: Columna: Phenomenex Lux 5u Cellulose-4, 250*21,2 mm, 5 gm; fase móvil: hexano y metanol (20 % de metanol en hexano isocrático en 12 min); Detector, UV 254/220 nm. Esto dio como resultado 35 mg (11 %) de (2S)-2-(4-clorofenil)-1-([3-metil-1H-pirazolo[4,3-b]piridin-5-il]carbonil)pirrolidina en forma de un sólido de color blanco.
De acuerdo con estos procedimientos generales, se sintetizaron los compuestos 52, 53 y 54.
9. (3-Amino-1 H-pirazolo[3,4-b]piridin-5-il)-[(S)-2-(4-bromo-fenil)-pirrolidin-1 -il]- metanona 98

9.1. 5-Bromo-1 H-pirazolo[3,4-b]piridin-3-ilamina

En un tubo cerrado herméticamente de 100 ml, se añadió 5-bromo-2-cloropiridin-3-carbonitrilo (2,50 g, 11,5 mmol) e hidrato de hidrazina (30 ml). La solución se agitó durante 3 h a 130 °C. La mezcla de reacción se concentró al vacío. El residuo se disolvió en 30 ml de agua. Sucedió la precipitación. Los precipitados se recogieron por filtración. Esto dio como resultado 2,00 g (82 %) de 5-bromo-1 H-pirazolo[3,4-b]piridin-3-amina en forma de un sólido de color amarillo.
9.2. Éster metílico del ácido 3-amino-1 H-pirazolo[3,4-b]piridin-5-carboxílico

En un reactor de tanque de acero de 30 ml purgado y mantenido con una atmósfera de monóxido de carbono (5 atm), se puso 5-bromo-1H-pirazolo[3,4-b]piridin-3-amina (500 mg, 2,35 mmol), Pd(dppf)Cl2.CH2Cl2 (192 mg, 0,24 mmol), acetato potásico (691 mg, 7,04 mmol), N,N-dimetilformamida (5 ml) y metanol (5 ml). La mezcla se agitó durante 14 h a 80 °C y después se concentró al vacío. La solución se extrajo 3 veces con 20 ml de acetato de etilo y las capas orgánicas se combinaron. El residuo se purificó mediante una columna de gel de sílice eluyendo con acetato de etilo/éter de petróleo (7:3). Esto dio como resultado 200 mg (44 %) de 3-amino-1H-pirazolo[3,4-b]piridin-5-carboxilato de metilo en forma de un sólido de color pardo claro.
9.3. Ácido 3-amino-1H-pirazolo[3,4-b]piridin-5-carboxílico

En un matraz de fondo redondo de 25 ml se añadió 3-amino-1 H-pirazolo[3,4-b]piridin-5-carboxilato de metilo (200 mg, 1,04 mmol), tetrahidrofurano (8 ml), agua (2 ml) e hidróxido sódico (84,0 mg, 2,10 mmol). La solución se agitó durante 3 h a 50 °C. La mezcla se concentró al vacío. El residuo se disolvió en 10 ml de agua. El valor del pH de la solución se ajustó a 3 con cloruro de hidrógeno (1 mol/l). Los precipitados se recogieron por filtración. Esto dio como resultado 100 mg (54 %) de ácido 3-amino-1 H-pirazolo[3,4-b]piridin-5-carboxílico en forma de un sólido de color amarillo.
9.4. (3-Amino-1 H-pirazolo[3,4-b]piridin-5-il)-[(S)-2-(4-bromo-fenil)-pirrolidin-1 -il]- metanona 98

En un matraz de fondo redondo de 25 ml, se puso ácido 3-amino-1H-pirazolo[3,4-b]piridin-5-carboxílico (40 mg, 0,22 mmol), 2-(4-bromofenil)pirrolidina (76,0 mg, 0,34 mmol), N,N-dimetilformamida (5 ml), hexafluorofosfato de 3-óxido de 1 -[bis(dimetilamino)metileno]-1 H-1 ,2,3-triazolo[4,5-b]piridinio (HATU, 94 mg, 0,25 mmol), diisopropiletilamina (87,0 mg, 0,67 mmol). La solución se agitó durante 1 h a 25 °C. La mezcla se concentró al vacío. El residuo se disolvió en 10 ml de acetato de etilo. La mezcla se lavó tres veces con 10 ml de agua. El producto en bruto se purificó por HPLC preparativa (acetonitrilo/agua). El producto en bruto (50 mg, 59 %) se purificó por HPLC preparativa quiral con las siguientes condiciones: Columna: Chiralpak IC, 2*25cm, 5 gm; fase móvil: hexano-HPLC y etanol-HPLC (parada 50 % de etanol-HPLC en 20 min); Detector, UV 254/220 nm. Esto dio como resultado 19 mg (21 %) de 5-[[(2S)-2-(4-bromofenil)pirrolidin-1 -il]carbonil]-1 H-pirazolo[3,4-b]piridin-3-amina en forma de un sólido de color blanquecino. De acuerdo con estos procedimientos generales, se sintetizaron los compuestos 96, 97 y 99.
10. [2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-trifluorometil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona (177)

10.1. 1,3-Dimetil-1-(3-trifluorometil-1 H-pirazolo[3,4-b]piridin-5-carbonil)-urea

Una mezcla de 5-trifluorometil-2H-pirazol-3-ilamina (500 mg, 3,31 mmol) y 1,3-dimetil-2,4-dioxo-1,2,3,4-tetrahidropirimidina-5-carbaldehído (586 mg, 3,31 mmol) en ácido acético (glacial, 40 ml) se calentó a reflujo durante 6 h. Después de un periodo de refrigeración, el disolvente se evaporó a presión reducida. El residuo se disolvió en metanol, la solución se dejó en reposo durante una noche, los cristales precipitados se suspendieron en éter dietílico, se retiraron por filtración y se secaron. El producto en bruto se purificó por cromatografía ultrarrápida (diclorometano/metanol) para producir 630 mg (49 %) del compuesto del título en forma de un sólido incoloro. HPLC (pureza) 77 %. HPLC/EM: Tr 1,66 min (procedimiento A).
10.2. Ácido 3-trifluorometil-1 H-pirazolo[3,4-b]piridin-5-carboxílico

Una mezcla de 1,3-dimetil-1-(3-trifluorometil-1H-pirazolo[3,4-b]piridin-5-carbonil)- urea (630 mg, 1,61 mmol) y una solución de hidróxido sódico en agua (10 %, 16 ml) se calentó y se agitó durante 2 h a 60 °C. Después de un periodo de refrigeración, la mezcla de reacción se filtró y el filtrado se acidificó con HCI diluido. El precipitado formado se retiró por filtración, se lavó con agua y se secó para producir 206 mg (51 %) del compuesto del título en forma de un sólido de color beige. HPLC (pureza) 92 %. HPLC/eM: Tr 1,58 min.
10.3. [2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-trifluorometil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona

A una solución de ácido 3-trifluorometil-1 H-pirazolo[3,4-b]piridin-5-carboxílico (100 mg, 0,40 mmol), 2-(4-cloro-fenil)-pirrolidina (115 mg, 0,60 mmol) y 4-metilmorfolina (0,064 ml, 0,58 mmol) en W,W-dimetilformamida (2 ml) se añadió a TA clorhidrato de W-(^3-dimetilaminopropil)-W'-etilcarbodiimida, DAPECl (92,0 mg, 0,48 mmol) e hidrato de 1-hidroxibenzotriazol (9,00 mg, 0,07 mmol) y la mezcla de reacción se agitó a TA durante una noche. La mezcla se vertió en agua, el precipitado se retiró por filtración y se secó al vacío. El producto en bruto se purificó por cromatografía ultrarrápida (n-heptano/acetato de etilo) para producir 69,0 mg (44 %) del compuesto del título en forma de un sólido de color blanco. Rm N 1H (400 MHz, DMSO-d6) ppm = 8,90, 8,55, 8,49, 7,84 (4 x s, 2H, proporción = 2:3 mezcla de rotámeros), 7,46, 7,38, 7,23, 7,02 (2 x d, J = 8,2 Hz, 2 x d, J = 7,7 Hz, 4H, proporción = 2:3 mezcla de rotámeros), 5,23 - 5,10, 4,98 - 4,87 (2x m, 1H, proporción = 2:3 mezcla de rotámeros), 3,97 - 3,76, 3,66 - 3,24 (1x m, 1x m Hd O, 2H), 2,46 - 2,30 (m, 1H), 2,03 - 1,68 (m, 3H).
Se separaron 62,7 mg de la mezcla racémica por HPLC prep. (SFC Berger Minigram, columna: ChiralPak AD-H, disolvente: CO2/Metanol 85:15, caudal: 5 ml/min, longitud de onda: 220 nm) para producir los compuestos 180 (26,6 mg, 42 %) y 181 (26,3 mg, 42 %) en forma de sólidos de color blanco.
De manera análoga a estos procedimientos, se sintetizaron los comps. 183 y 184.
11. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-c]piridin-5-il)- metanona (190)

11.1. 1 -(2-Bromo-5-fluoropiridin-4-il)etan-1 -ona

En un matraz de fondo redondo de tres bocas y 250 ml, purgado y mantenido con una atmósfera inerte de nitrógeno, se puso una solución de LDA (35,0 ml, 258 mmol) en tetrahidrofurano (30 ml). Esto se siguió de la adición de una solución de 2-bromo-5-fluoropiridina (5,00 g, 28,4 mmol) en tetrahidrofurano (20 ml) a -78 °C. La mezcla se agitó durante 2 h a -78 °C. A la mezcla se le añadió una solución de W-metoxi-W-metilacetamida (6,40 g, 62,1 mmol) en tetrahidrofurano (20 ml) a -78 °C. La solución se calentó hasta TA y se agitó durante 3 h a TA. Después, la mezcla se inactivó mediante la adición de 100 ml de agua. La solución se extrajo con 200 ml de acetato de etilo y la capa orgánica se secó sobre sulfato sódico anhidro y se concentró al vacío. El residuo se purificó sobre una columna de gel de sílice con éter de petróleo/acetato de etilo (50:1). Esto dio como resultado 4,00 g (65 %) de 1-(2-bromo-5-fluoropiridin-4
il)etan-1 -ona en forma de un aceite de color amarillo.
11.2. 5-Bromo-3-metil-1 H-pirazolo[3,4-c]piridina

En un tubo cerrado herméticamente de 30 ml, se puso 1-(2-bromo-5-fluoropiridin-4-il)etan-1-ona (2,00 g, 9,17 mmol), NH2NH2*agua (500 mg, 9,99 mmol) y etano-1,2-diol (10 ml). La solución se agitó durante 4 h a 165 °C en un baño de aceite. La mezcla se diluyó con 100 ml de agua. Los sólidos se recogieron por filtración. Esto dio como resultado 1,70 g (87 %) de 5-bromo-3-metil-1 H-pirazolo[3,4-c]piridina en forma de un sólido de color amarillo.
11.3. 3-Metil-1 H-pirazolo[3,4-c]piridin-5-carboxilato de metilo

En un tubo cerrado herméticamente de 30 ml se puso 5-bromo-3-metil-1 H-pirazolo[3,4-c]piridina (900 mg, 4,24 mmol), Pd(dppf)CÍ2 (315 mg, 0,43 mmol), KOAc (1,25 g, 12,7 mmol) y W,W-dimetilformamida (10 ml). Esto se siguió de la adición de metanol (10 ml) y gas de CO. La solución se agitó durante 1 h durante una noche a 80 °C en un baño de aceite. La mezcla se concentró al vacío. El residuo se purificó sobre una columna de gel de sílice con diclorometano/metanol (20:1). Esto dio como resultado 0,60 g (74 %) de 3-metil-1 H-pirazolo[3,4-c]piridin-5-carboxilato de metilo en forma de un sólido de color pardo.
11.4. Ácido 3-metil-1 H-pirazolo[3,4-c]piridin-5-carboxílico

En un matraz de fondo redondo de 50 ml se puso 3-metil-1 H-pirazolo[3,4-c]piridin-5-carboxilato de metilo (300 mg, I , 57 mmol), LiOH*H2O (328 mg, 7,82 mmol), tetrahidrofurano (20 ml) y agua (5 ml). La solución se agitó durante 5 h a 60 °C en un baño de aceite. La mezcla se concentró al vacío. El pH de la solución se ajustó a 4 con cloruro de hidrógeno (1 M). Los sólidos se recogieron por filtración. Esto dio como resultado 190 mg (68 %) de ácido 3-metil-1H-pirazolo[3,4-c]piridin-5-carboxílico en forma de un sólido de color amarillo.
I I .5. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-c]piridin-5-il)- metanona
En un matraz de fondo redondo de 25 ml se puso ácido 3-metil-1 H-pirazolo[3,4-c]piridin-5-carboxílico (130 mg, 0,73 mmol), (S)-2-(4-clorofenil)pirrolidina (172 mg, 0,95 mmol), HATU (277 mg, 0,73 mmol), diacetato de etilo (282 mg, 2,18 mmol) y W,W-dimetilformamida (4 ml). La solución se agitó durante 3 h a TA. La mezcla se concentró al vacío. El producto en bruto se purificó por HPLC prep. (acetonitrilo/agua). Esto dio como resultado 20,0 mg (8 %) de (2S)-2-(4-clorofenil)-1-([3-metil-1H-pirazolo[3,4-c]piridin-5-il]carbonil)pirrolidina en forma de un sólido de color blanco. RMN 1H (300 MHz, DMSO-d6) ppm = 13,40 (m, 1H), 9,00 (s, 0,67H), 8,72 (s, 0,33H), 8,14 (s, 0,67H), 7,83 (s, 0,33H), 7,40-7,32 (m, 3H), 7,15-6,98 (m, 1H), 5,84-5,82 (m, 0.33H), 5,26-5,21 (m, 0,67H), 4,10-4,02 (m, 0.67H), 3,87-3,70 (m, 1.33H), 2,55 (s, 2H), 2,41 (s, 1H), 2,38-2,27 (m, 1H), 1,87-1,68 (m, 3H).
De manera análoga a estos procedimientos, se sintetizó 189.

12. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(6-fluoro-3-metil-1 H-indazol-5-il)- metanona (206)

12.1. 5-bromo-6-fluoro-3-metil-1 H-indazol

En un matraz de fondo redondo de 50 ml se puso etano-1,2-diol (30 ml), 1-(5-bromo-2,4-difluorofenil)etan-1-ona (5,95 g, 25,3 mmol), NH2NH2*H2Ü (1,90 g, 38,0 mmol). La solución se agitó durante 20 h a 120 °C en un baño de aceite. La mezcla se diluyó con 200 ml de agua y se extrajo tres veces con 70 ml de acetato de etilo. Las capas orgánicas combinadas se concentraron al vacío. Esto dio como resultado 4,45 g (77 %) de 5-bromo-6-fluoro-3-metil-1 H-indazol en forma de un sólido de color amarillo.
12.2. 6-Fluoro-3-metil-1 H-indazol-5-carboxilato de metilo

En un tubo cerrado herméticamente de 250 ml se puso metanol (180 ml), H,H-dimetilformamida (10 ml), 5-bromo-6-fluoro-3-metil-1H-indazol (4,00 g, 17,5 mmol), Pd(dppf)CÍ2*diclorometano (2,55 g, 3,12 mmol) y acetato potásico (5,15 g, 52,4 mmol). La solución se agitó durante 20 h a 80 °C en un baño de aceite. La mezcla se concentró al vacío. El residuo se diluyó con 100 ml de agua. La solución se extrajo tres veces con 50 ml de acetato de etilo y las capas orgánicas combinadas se evaporaron a sequedad. El residuo se purificó sobre una columna de gel de sílice con acetato de etilo/hexano (1:3). Esto dio como resultado 2,64 g (73 %) de 6-fluoro-3-metil-1H-indazol-5-carboxilato de metilo en forma de un sólido de color amarillo.
12.3. Ácido 6-fluoro-3-metil-1 H-indazol-5-carboxílico

En un matraz de fondo redondo de 100 ml se puso tetrahidrofurano (50 ml), 6-fluoro-3-metil-1 H-indazol-5-carboxilato de metilo (2,64 g, 12,7 mmol), agua (10 ml) y LiOH (1,52 g, 63,47 mmol). La solución se agitó durante 4 h a 60 °C en un baño de aceite. La mezcla se concentró al vacío. El pH de la solución se ajustó a 4 con cloruro de hidrógeno (12 M). Los sólidos se recogieron por filtración y se descartaron. El filtrado se concentró al vacío para dar como resultado 2,21 g (90 %) de ácido 6-fluoro-3-metil-1 H-indazol-5-carboxílico en forma de un sólido de color amarillo claro.
12.4. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(6-fluoro-3-metil-1 H-indazol-5-il)-metanona (206)

En un matraz de fondo redondo de 25 ml se puso W,W-dimetilformamida (5 ml), ácido 6-fluoro-3-metil-1 H-indazol-5-carboxílico (200 mg, 1,03 mmol), (2S)-2-(4-clorofenil)pirrolidina (243,261 mg, 1,34 mmol), diacetato de etilo (399 mg, 3,09 mmol) y HATU (392 mg, 1,03 mmol). La solución se agitó durante 1 h a 25 °C. Los sólidos se retiraron por filtración y el producto en bruto se purificó por HPLC prep. (acetonitrilo/agua) para dar como resultado 60 mg (16 %) de 5-[[(2S)-2-(4-clorofenil)pirrolidin-1 -il]carbonil]-6-fluoro-3-metil-1 H-indazol en forma de un sólido de color blanco. r Mn 1H (300 MHz, CDCI3) ppm = 7,78 (d, 0,65H), 7,33-7,12 (m, 2.33H), 7,09-7,06 (m, 1.90H), 6,93-6,78 (m, 1.17H), 5,36-5,32 (m, 0.69H), 4,72-4,68 (m, 0.38H), 4,00-3,90 (m, 0.75H), 3,70-3,66 (m, 0.65H), 3,49-3,45 (m, 0.65H), 2,57-2,45 (s, 2H), 2,43-2,39 (m, 1.02H), 2,38-2,36 (s, 1.18H), 2,07-2,00 (m, 1.14H), 1,96-1,85 (m, 2.14H).
De manera análoga a estos procedimientos, se sintetizó 203
13. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-metanosulfonil-1 H-indazol-5-il)-metanona (220)

13.1. Ácido 3-metilsulfanil-1 H-indazol-5-carboxílico

En un vial para microondas de 2 ml, se pesaron y disolvieron éster etílico del ácido 3-yodo-1 H-indazol-5-carboxílico (100 mg, 0,32 mmol), yoduro de cobre (l) (6,02 mg, 0,03 mmol) y metanotiolato sódico (123 pl, 1,58 mmol) en dimetilsulfóxido seco (4 ml) y agua (0,6 ml). La mezcla se hizo reaccionar a 120 °C durante 2 h en irradiación de microondas. La mezcla se diluyó con una solución 0,5 N de HCI (25 ml) y se extrajo dos veces con acetato de etilo (50 ml). Las capas orgánicas combinadas se evaporaron a sequedad y se purificaron por cromatografía ultrarrápida (acetato de etilo/metanol) para producir ácido 3-metilsulfanil-1 H-indazol-5-carboxílico (62,0 mg, 77 %) en forma de un aceite incoloro.
13.2. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-metilsulfanil-1 H-indazol-5-il)- metanona (210)

Se pesaron ácido 3-metilsulfanil-1 H-indazol-5-carboxílico (62,0 mg, 0,24 mmol), clorhidrato de (S)-2-(4-cloro-fenil)-pirrolidina (63,8 mg, 0,29 mmol) y tetrafluorborato de O-(1H-benzotriazol-1-il)-N,N,N',N'-tetrametiluronio (TBTU, 157 mg, 0,49 mmol) y se disolvieron en W,W-dimetilformamida (2 ml) y 4-metilmorfolina (82,1 gl, 0,73 mmol). La solución transparente se agitó a TA durante 30 min. La mezcla de reacción se vertió en una solución sat. de cloruro de amonio (30 ml). El precipitado se retiró por filtración, se lavó con agua (10 ml) y se secó a 70 °C al vacío durante una noche para dar [(S)-2-(4-cloro-fenil)-pirrolidin-1-il]-(3-metilsulfanil-1H-indazol-5-il)-metanona (26,2 mg, 27 %) en forma de un sólido de color blanquecino.
13.3. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-metanosulfonil-1 H-indazol-5-il)- metanona (220)
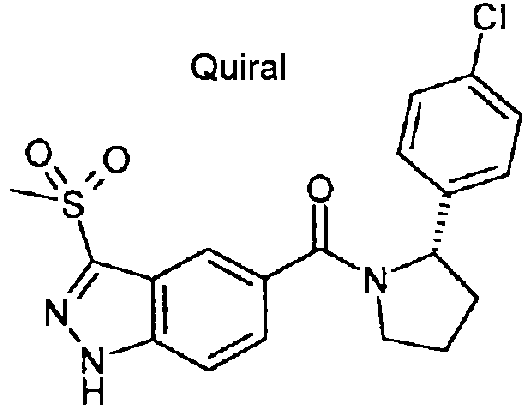
En un tarro con tapón de rosca de 12 ml, se pesaron [(S)-2-(4-cloro-fenil)-pirrolidin-1-il]-(3-metilsulfanil-1H-indazol-5-il)-metanona (17,5 mg, 0,04 mmol) y Oxone®, monopersulfato (Peroximonosulfato potásico, 54,1 mg, 0,09 mmol) y se suspendieron en W,W-dimetilformamida (1 ml). La mezcla se agitó a TA durante 3 días. La suspensión se inactivó con una solución de NaHSO3 (3 ml, ~39 % en agua). La mezcla se diluyó con agua y se neutralizó con NaHCO3 sólido. Se extrajo dos veces con acetato de etilo (30 ml). Las capas orgánicas combinadas se secaron, se filtraron, se evaporaron a sequedad y se purificaron adicionalmente por cromatografía ultrarrápida (diclorometano/metanol) para producir 12,4 mg (71 %) de [(S)-2-(4-cloro-fenil)-pirrolidin-1 -il]-(3-metanosulfonil-1 H-indazol-5-il)- metanona en forma de un sólido de color blanco.
14. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-fluoro-1 H-indazol-5-il)-metanona (216)

14.1. Ácido 3-fluoro-1 H-indazol-5-carboxílico

Un frasco con tapón de rosca se cargó con ácido 1 H-indazol-5-carboxílico (64,0 mg, 0,38 mmol) y se suspendió en
acetonitrilo ( 8 ml) y ácido acético (glacial, 1 ml). A esta suspensión turbia se añadió gota a gota ditetrafluoroborato de 1-(clorometil)-4-fluoro-1,4-diazoniabiciclo[2,2,2]octano (214 mg, 0,57 mmol) disuelto en 1 ml de acetonitrilo y la mezcla se agitó durante una noche a 60 °C y durante 4 días a 80 °C. La mezcla se evaporó a sequedad, el residuo se disolvió en acetato de etilo y se lavó dos veces con agua y una vez con salmuera. La fase orgánica se secó con sulfato sódico y la fase orgánica se evaporó a sequedad. El residuo se separó por cromatografía preparativa (acetonitrilo/agua) para producir 16 mg (23 %) de un sólido de color blanco.
14.2. [(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-fluoro-1 H-indazol-5-il)-metanona (216)

A una solución de ácido 3-fluoro-1 H-indazol-5-carboxílico (16,0 mg, 0,09 mmol) en W,W-dimetilformamida (3 ml), se añadieron clorhidrato de (S)-2-(4-cloro-fenil)-pirrolidina (23,0 mg, 0,11 mmol), 4-metilmorfolina (0,03 ml, 0,26 mmol) y tetrafluoroborato de [(benzotriazol-1-iloxi)-dimetilamino-metileno]-dimetilamonio (TBTU), 56,5 mg, 0,18 mmol) y la mezcla se agitó durante 5 min a 60 °C. La mezcla se diluyó con acetato de etilo, se lavó una vez con una solución 1 N de NaOH, con agua y salmuera. La capa orgánica se separó y se secó con sulfato sódico, se filtró y se evaporó a sequedad. El residuo se purificó por cromatografía preparativa (acetonitrilo/agua) para producir 17 mg (55 %) del compuesto del título en forma de un sólido de color blanquecino. RMN 1H (400 m Hz, DMSo-d6 , 90 °C) ppm = 12,43 (s, 1H), 7,78 (s, 1H), 7,54 - 7,39 (m, 2H), 7,34 - 7,18 (m, 4H), 5,12 (t, J = 6,7 Hz, 1H), 3,86 - 3,76 (m, 1H), 3,69 - 3,58 (m, 1H), 2,43 - 2,33 (m, 1H), 1,94-1,83 (m, 2H), 1,83 - 1,72 (m, 1H).
15. [(S)-2-(1 H-lndazol-5-il)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona (245) y [(R)-2-(1 H-lndazol-5-il)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona (246)

15.1. 2-(1 H-Indazol-5-il)-1 H-pirrol-1 -carboxilato de ferc-butilo

En un matraz de fondo redondo de 250 ml, purgado y mantenido con una atmósfera inerte de nitrógeno, se puso carbonato sódico (1,08 g, 10,1 mmol), agua (20 ml), dimetil éter de etilenglicol (100 ml), 5-bromo-1 H-indazol (1,00 g, 5,08 mmol), ácido [1-[(ferc-butoxi)carbon¡l]-1H-p¡rrol-2-¡l]borónico (1,29 g, 6,09 mmol) y Pd(PPh3)4 (586 mg, 0,51 mmol). La solución se agitó durante 4 h a 95 °C en un baño de aceite y se diluyó con 50 ml de agua después de refrigeración. La solución se extrajo dos veces con 50 ml de diclorometano y las capas orgánicas combinadas se concentraron a sequedad. El residuo se purificó sobre una columna de gel de sílice con acetato de etilo/éter de petróleo
(1:10). Esto dio como resultado 1 g (70 %) de 2-(1 H-indazol-5-il)-1 H-pirrol-1 -carboxilato de ferc-butilo en forma de un aceite de color amarillo.
15.2. 5-(Pirrolidin-2-il)-1 H-indazol

En un reactor de tanque de presión de 20 ml, purgado y mantenido con una atmósfera de hidrógeno, se puso 2-(1 H-indazol-5-il)-1 H-pirrol-1 -carboxilato de ferc-butilo (500 mg, 1,76 mmol), etanol (5,00 ml), cloruro de hidrógeno (1 N, 0,50 ml) y PtÜ2 (40,1 mg, 0,18 mmol). La solución se agitó durante 5 días a 60 °C en un baño de aceite. La mezcla se concentró al vacío. El residuo se purificó sobre una columna de gel de sílice con metanol/agua (1:99). Esto dio como resultado 200 mg (61 %) de 5-(pirrolidin-2-il)-1 H-indazol en forma de un aceite de color amarillo.
15.3. Se prepararon [(S)-2-(1H-indazol-5-il)-pirrolidin-1-il]-(3-metil-1H-pirazolo[3,4-b]piridin-5-il)- metanona (245) y [(fí)-2-(1H-indazol-5-il)-pirrolidin-1-il]-(3-metil-1H-pirazolo[3,4-b]piridin-5-il)-metanona (246) de acuerdo con los procedimientos generales para producir 29 mg (7 %) del compuesto del título en forma de sólidos de color blanco. RMN 1H (245, 300 Hz, DMSO-ds) ppm = 13,11 (S, 1H), 12,76 (s, 1H), 8,52 (s, 1H), 8,40-8,09 (m, 1H), 7,95 (s, 1H), 7,62-7,50 (m, 1H), 7,50-7,35 (m, 1H), 7,22 (s, 1H), 5,24 (s, 1H), 3,92-3,90 (m, 1H), 3,79-3,72 (m, 1H), 2,42-2,31 (m, 4H), 1,97-1,90 (m, 3H).
16. [(2S,4R)-2-(4-Cloro-fenil)-4-metilamino-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona (254)

16.1

En un matraz de fondo redondo de 250 ml se puso (3S)-4-cloro-3-hidroxibutanonitrilo (5,00 g, 41,8 mmol), N,N- dimetilformamida (80 ml), TBSCI (8,10 g, 53,74 mmol), imidazol (5,60 g, 82,3 mmol). La mezcla de reacción se agitó durante 1 h por la noche a TA. La solución se diluyó con 200 ml de acetato de etilo. La mezcla se concentró al vacío. El residuo se purificó sobre una columna de gel de sílice con acetato de etilo/éter de petróleo (1:10). Esto dio como resultado 7,80 g (80 %) de (3S)-3-[(ferc-butildimetilsilil)oxi]-4-clorobutanonitrilo en forma de un líquido incoloro.
16.2. (3S)-3-[(ferc-Butildimetilsilil)oxi]-5-(4-clorofenil)-3,4-dihidro-2H-pirrol

En un matraz de fondo redondo de 3 bocas y 50 ml, purgado y mantenido con una atmósfera inerte de nitrógeno, se puso (3S)-3-[(ferc-butildimetilsilil)oxi]-4-clorobutanonitrilo (300 mg, 1,28 mmol) en metil ferc-butil éter (5 ml). Esto se siguió de la adición de bromo(4-clorofenil)magnesio (3,80 ml, 17,6 mmol) a 0 °C. La mezcla se agitó durante 3 h a 25 °C. A la mezcla se le añadió dimetil éter de etilenglicol (5 ml) y la solución se agitó durante 2 h a TA. La solución se diluyó con 50 ml de acetato de etilo. La mezcla se concentró al vacío. El residuo se purificó sobre una columna de gel de sílice con éter de petróleo/acetato de etilo (50:1). Esto dio como resultado 240 mg (60 %) de (3S)-3-[(fercbutildimetilsilil)oxi]-5-(4-clorofenil)-3,4-dihidro-2H-pirrol en forma de un aceite de color pardo.
16.3. (2RS,4S)-4-[(ferc-Butildimetilsilil)oxi]-2-(4-clorofenil)pirrolidina

En un matraz de fondo redondo de 50 ml se puso (3S)-3-[(ferc-butildimetilsilil)oxi]-5-(4-clorofenil)-3,4-dihidro-2H-pirrol (300 mg, 0,97 mmol) en etanol ( 6 ml). Esto se siguió de la adición de PtÜ2 (30 mg, 0,13 mmol). La solución se agitó en una atmósfera de hidrógeno durante 1 h, durante una noche a TA. Los sólidos se retiraron por filtración y se descartaron. El filtrado se concentró al vacío para dar como resultado 300 mg (99 %) de (2RS,4S)-4-[(fercbutildimetilsilil)oxi]-2-(4-clorofenil)pirrolidina en forma de un aceite de color pardo.
16.4. (2RS,4S)-4-[(ferc-Butildimetilsilil)oxi]-2-(4-clorofenil)-1-((3-metil-1 H-pirazolo[3,4-b]piridin-5-il|carbonil)pirrolidina

En un matraz de fondo redondo de 50 ml se puso ácido 3-metil-1 H-pirazolo[3,4-b]piridin-5-carboxílico (300 mg, 1,69 mmol), (2RS,4S)-4-[(ferc-butildimetilsilil)oxi]-2-(4-clorofenil|pirrolidina (631 mg, 2,02 mmol), HATU (640 mg, 1,68 mmol), diacetato de etilo (652 mg, 5,04 mmol) y W,W-dimetilformamida (5 ml). La solución se agitó durante 3 h a TA. La mezcla se concentró al vacío. El residuo se purificó sobre una columna de gel de sílice con diclorometano/metanol (50:1). Esto dio como resultado 250 mg (31 %) de (2RS,4S)-4-[(ferc-butildimetilsilil)oxi]-2-(4-clorofenil)-1 -([3-metil-1 H-pirazolo[3,4-b]piridin-5-il]carbonil)pirrolidina en forma de un producto de color amarillo.
16.5. (3 S ,5 R S )-5 - (4 -C lo ro fe n il) -1 - ({3 -m e til-1 H -p ira z o lo [3 ,4 -b ]p ir id in -5 - i l |c a rb o n il)p ir ro l id in -3 -o l

En un matraz de fondo redondo de 25 ml se puso (2RS,4S)-4-[(ferc-butildimetilsilil)oxi]-2-(4-clorofenil)-1-([3-metil-1H-pirazolo[3,4-b]piridin-5-il]carbonil)pirrolidina (200 mg, 0,42 mmol) en tetrahidrofurano (5 ml). Se añadió TBAF (2 ml, 7,65 mmol) a 0 °C y la solución se agitó durante 30 min a 0 °C en un baño de agua/hielo. La mezcla se concentró al vacío. El residuo se aplicó en una columna de gel de sílice con diclorometano/metanol (20:1). Esto dio como resultado 140 mg (92 %) de (3S,5RS)-5-(4-clorofenil)-1-([3-metil-1 H-pirazolo[3,4-b]piridin-5-il]carbonil)pirrolidin-3-ol en forma de un sólido de color amarillo.
16.6. Metanosulfonato de (3S,5RS)-5-(4-clorofenil)-1 -({3-metil-1 H-pirazolo[3,4-b]piridin-5-il|carbonil)pirrolidin-3-ilo

En un matraz de fondo redondo de 25 ml se puso (3S,5RS)-5-(4-clorofenil)-1-([3-metil-1H-pirazolo[3,4-b]piridin-5-il]carbonil)pirrolidin-3-ol (140 mg, 0,39 mmol), diclorometano (10 ml), trietilamina (119 mg, 1,18 mmol), cloruro de mesilo (58,0 mg, 0,51 mmol). La solución se agitó durante 2 h a 0 °C en un baño de agua/hielo. La solución se diluyó con 50 ml de diclorometano. La mezcla se secó con sulfato sódico y se concentró al vacío. Esto dio como resultado 140 mg (82 %) de metanosulfonato de (3S,5RS)-5-(4-clorofenil)-1-([3-metil-1H-pirazolo[3,4-b]piridin-5-il]carbonil)pirrolidin-3-ilo en forma de un aceite de color amarillo.
16.7. [(2S,4R)-2-(4-Cloro-fenil)-4-metilamino-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona

En un tubo cerrado herméticamente de 30 ml, se puso metanosulfonato de (3S,5RS)-5-(4-clorofenil)-1-([3-metil-1H-pirazolo[3,4-b]piridin-5-il]carbonil)pirrolidin-3-ilo (140 mg, 0,32 mmol), metilamina (434 mg, 13,97 mmol) y etanol ( 8 ml). La solución se agitó durante 1 por la noche a 100 °C en un baño de aceite. La mezcla se concentró al vacío. El producto en bruto se purificó por HPLC prep. (acetonitrilo/agua) para producir 5,00 mg (4 %) de [(2S,4R)-2-(4-clorofenil)-4-metilamino-pirrolidin-1-il]-(3-metil-1H-pirazolo[3,4-b]piridin-5-il)-metanona en forma de un sólido de color blanco. RMN 1H (400 MHz, DMSO-d6) ppm = 13,38 (s, 1H), 8,69 (s, 1H), 8,52 (s, 1H), 7,46 (d, J = 8 Hz, 2H), 7,38 (d, J = 8 Hz, 2H), 5,20 (m, 1H), 4,11-4,07 (m, 1H), 3,46-3,41 (m, 2H), 2,59 (s, 3H), 2,37-2,15 (m, 5H), 1,84 (m, 2H).
17. [(3S,4R)-3-(4-Cloro-fenil)-4-hidroxi-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona (256)

17.1.3-(4-Clorofenil)-4-hidroxipirrolidin-1 -carboxilato de ferc-butilo

En un matraz de fondo redondo de tres bocas y 100 ml, purgado y mantenido con una atmósfera inerte de nitrógeno, se puso 1-bromo-4-clorobenceno (2,33 g, 12,2 mmol) en tetrahidrofurano (20 ml). A esta solución se añadió gota a gota n-BuLi (5,40 ml, 2,5mol/l) con agitación a -80 °C durante 30 min. Después, a la mezcla se añadió gota a gota BF3*Et2Ü (1,80 ml, 14,2 mmol) con agitación a -80 °C en 10 min. Se añadió gota a gota una solución de 6-oxa-3-azabiciclo[3,1,0]hexano-3-carboxilato de ferc-butilo (1,00 g, 5,40 mmol) en tetrahidrofurano (3 ml) con agitación de nuevo a -80 °C. La solución se agitó durante 3 h a -80 °C. Después, la reacción se interrumpió mediante la adición de 20 ml de bicarbonato sódico acuoso. La mezcla se extrajo dos veces con 20 ml de tetrahidrofurano/acetato de etilo (1:1) y las capas orgánicas combinadas se concentraron al vacío. El residuo se purificó sobre una columna de gel de sílice con éter de petróleo/acetato de etilo (5:2). Esto dio como resultado 600 mg (37 %) de 3-(4-clorofenil)-4-hidroxipirrolidin-1 -carboxilato de ferc-butilo en forma de un aceite incoloro.
17.2. 4-(4-Clorofenil)pirrolidin-3-ol

En un matraz de fondo redondo de 50 ml se puso 3-(4-clorofenil)-4-hidroxipirrolidin-1 -carboxilato de ferc-butilo (600 mg, 2,01 mmol) en diclorometano (15 ml) y se añadió ácido trifluoroacético (3 ml) a TA. La solución se agitó durante 2 h a TA. La mezcla se concentró al vacío. El residuo se purificó sobre una columna de gel de sílice con metanol/agua (1:99). Esto dio como resultado 300 mg (75 %) de 4-(4-clorofenil)pirrolidin-3-ol en forma de un aceite incoloro.
17.3. [(3S,4R)-3-(4-Cloro-fenil)-4-hidroxi-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona (256) y [(3R,4S)-3-(4-Cloro-fenil)-4-hidroxi-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona (255)

En un matraz de fondo redondo de 25 ml se puso ácido 3-metil-1H-pirazolo[3,4-b]piridin-5-carboxílico (150 mg, 0,85 mmol), 4-(4-clorofenil)pirrolidin-3-ol (201 mg, 1,02 mmol), N,N-dimetilformamida (5 ml), EDCI (325 mg, 1,69 mmol) y 4-dimetilaminopiridina (155 mg, 1,27 mmol). La solución se agitó durante 3 h a TA. El residuo se purificó sobre una columna de gel de sílice con metanol/diclorometano (3:10). El producto en bruto se purificó por HPLC prep. (acetonitrilo/agua). Esto dio como resultado 30 mg (10 %) de trans-[3-(4-cloro-fenil)-4-hidroxi-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona racémica en forma de un sólido de color blanco. Después, la mezcla racémica se purificó por HpLC quiral (Chiralpak IB4,6*250 mm, 5 gm, 100 % de metanol (0,1 % de dietilamina) para producir [(3S,4R)-3-(4-Cloro-fenil)-4-hidroxi-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona (256) y [(3R,4S)-3-(4-Cloro-fenil)-4-hidroxi-pirrolidin-1-il]-(3-metil-1H-pirazolo[3,4-b]piridin-5-il)-metanona (255). RMN 1H (256, 300 Hz, DMSO-ds) ppm = 13,15 (s, 1H), 8,65-8,64 (m, 1H), 8,36-8,35 (m, 1H), 7,38-7,33 (m, 4H), 5,30-5,00 (m, 1H), 4,27-4,25 (m, 1H), 4,04-3,98 (m, 1H), 3,86-3,80 (m, 1H), 3,65-3,59 (m, 1H), 3,46-3,40 (m, 1H), 3,32-3,24 (m, 1H), 2,52 (s, 3H).
18. 3-(4-Clorofenil)morfolino)(3-metil-1 H-indazol-5-il)metanona (178)

A ácido 3-metil-1 H-indazol-5-carboxílico (40 mg, 0,23 mmol) en /V,N-dimetil formamida (1,9 ml) se añadió HATU (104 mg, 0,272 mmol), clorhidrato de 3-(4-clorofenil)morfolina (53 mg, 0,23 mmol) y DIp Ea (87 gl, 0,50 mmol). La mezcla de reacción se agitó a TA durante una noche, se concentró y el material en bruto se purificó mediante cromatografía en columna Biotage (diclorometano/etanol 99/1 a 93/7) y por TLC prep. (1 mm, diclorometano/etanol, 98/2). Después, el producto se filtró en una columna SCX- 2 y se liberó con NH31 N en metanol para dar el compuesto del título (5 mg, rendimiento del 6 %). RMN 1H (500 MHz, CD3OD) ppm = 7,86-7,85 (m, 1H), 7,60-7,51 (m, 3H), 7,47 7,40 (m, 3H), 5,70-5,40 (sa, 1H), 4,49 (d, J = 12,4 Hz, 1H), 4,00-4,64 (sa, 1H), 4,00 (dd, J = 12,4, 3,6 Hz, 1H), 3,94 3,83 (m, 1 h ), 3,73-3,64 (m, 1 H), 3,39-3,30 (m, 1 H), 2,56 (s, 3H). CL - EN (Ie N, m/z, procedimiento l) Tr = 2,71 min -356/358 (M+H)+. IEN-HRMS: Observado: 356,1153 calculado para C1gH1935CIN3O2 (M+H)+: 356.1160.
19. (2-(4-Clorofenil)-4-metilpiperazin-1 -il)(3-metil-1 H-indazol-5-il)metanona (244)

19.1

A una solución de 2-(4-clorofenil)piperazina (200 mg, 1,02 mmol) y trietilamina (1,08 ml, 7,73 mmol) en acetona (3,2 ml) se añadió gota a gota yoduro de metilo (70 gl, 1,1 mmol) a 02C. La reacción se agitó a TA durante una noche. Se añadieron 1 equivalente más de yoduro de metilo y 1 ml de acetona. La reacción se agitó a TA durante 5 h más. Se añadieron una solución saturada de NH4CI y acetato de etilo, la capa acuosa se extrajo tres veces con acetato de etilo y la capa orgánica se secó sobre MgSÜ4 , se filtró y se concentró. La mezcla en bruto se usó en la siguiente etapa sin purificación.
19. 2. (2-(4-Clorofenil)-4-metilpiperazin-1 -il)(3-metil-1 H-indazol-5-il)metanona

A ácido 3-metil-1 H-indazol-5-carboxílico (92 mg, 0,52 mmol), HATU (238 mg, 0,626 mmol) y 3-(4-clorofenil)-1-metilpiperazina (110 mg, 0,522 mmol) se añadieron DIPEA (200 gl, 1,15 mmol) y N,N-dimetil formamida (4,4 ml). La mezcla de reacción se agitó a TA durante 2 h y el disolvente se evaporó (V10). El producto en bruto se purificó mediante cromatografía en columna Biotage (diclorometano/etanol 99:1 a 80:20) seguido de HPLC prep. El producto se filtró en una columna SCX y se liberó con NH31 N en metanol para dar el compuesto del título (30 mg, rendimiento del 16 %). RMN 1H (500 MHz, CD3OD) ppm = 7,84 (s, 1H), 7,55-7,34 (m, 6 H), 5,64 (s a, 1H), 3,92 (s a, 1H), 3,51-3,44 (m, 1H), 3,27-3,18 (m, 1H), 2,78-2,72 (m, 1H), 2,54 (s, 3H), 2,53 - 2,48 (m, 1H), 2,29 (s, 3H), 2,20-2,14 (m, 1H). CL -EN (IEN, m/z, procedimiento l) Tr = 0,96 min - 369/371 (m+H)+. IEN-HRMS: Observado: 369,1471 calculado para C2üH2235CIN4O (M+H)+: 369.1477.
20. (S)-(2-(4-Clorofenil)pirrolidin-1 -il)(3-metoxi-1 H-pirazolo[3,4-b]piridin-5-il)metanona (243)

20.1. 3 -M e to x i-1 H -p ira z o lo [3 ,4 -b ]p ir id in -5 -c a rb o n it r i lo

Se cargaron 5-bromo-3-metoxi-1 H-pirazolo[3,4-b]piridina (300 mg, 1,32 mmol), Pd(PPh3)4 (152 mg, 0,132 mmol) y cianuro de cinc (170 mg, 1,45 mmol) en un vial para microondas y se añadió W,W-dimetilformamida (8,7 ml). La mezcla de reacción se agitó a 60 °C durante una noche. El disolvente se evaporó y la mezcla en bruto se purificó mediante cromatografía en columna Biotage (diclorometano/etanol 99:1 a 96:4) para dar el compuesto del título (160 mg que contenía 17 % de óxido de trifenilfosfina, rendimiento corregido 58 %). RMN 1H (500 MHz, CD3OD) ppm = 8,72 (d, J = 2,0 Hz, 1H), 8,55 (d, J = 2,0 Hz, 1H), 4,11 (s, 3H). CL - EN (IEN, m/z, procedimiento M) Tr = 0,96 min -175 (M+H)+.
20.2. Ácido 3-metoxi-1H-pirazolo[3,4-b]piridin-5-carboxílico
A 3-metoxi-1 H-pirazolo[3,4-b]piridin-5-carbonitrilo (160 mg, 0,919 mmol) en etanol (7,1 ml) se añadió NaOH 2 M (6,9 ml, 13,78 mmol). La reacción se calentó a 100 °C durante 12 h. Se añadió HCI en dioxano (13,8 ml, 13,78 mmol) a la mezcla de reacción y la mezcla de reacción se concentró al vacío. El producto en bruto se usó en la siguiente etapa sin purificación.
20.3. (S)-(2-(4-Clorofenil)pirrolidin-1 -il)(3-metoxi-1 H-pirazolo[3,4-b]piridin-5-il)metanona

A ácido 3-metoxi-1 H-pirazolo[3,4-b]piridin-5-carboxílico (59 mg, 0,31 mmol), clorhidrato de (S)-2-(4-clorofenil)pirrolidina (67 mg, 0,31 mmol) y HATU (139 mg, 0,367 mmol) se añadieron N,N-dimetil formamida (2,5 ml) y DIPEA (117 pl, 0,672 mmol). La mezcla de reacción se agitó a TA durante 1 h 30 y después se concentró (V10), se cargó en seco sobre sílice y se purificó por cromatografía en columna Biotage (SNAP 25 g, diclorometano/etanol 99:1 a 95:5) y por TLC prep. (0,5 mm, diclorometano/etanol 98:2) para dar el compuesto del título (18 mg, rendimiento del 16 %). RMN 1H (500 MHz, CD3OD, 2 rotámeros) ppm = 8,72 (s, 0,6H), 8,39 (s, 0.6H), 8,26 (s, 0.4H), 7,86 (s, 0.4H), 7,37 (d, J = 8,1Hz, 1.2H), 7,32 (d, J = 8,1Hz, 1.2H), 7,17 (d, J = 8,0 Hz, 0,8H), 6,97 (d, J = 8,0 Hz, 0,8H), 5,21 (t, J = 7,3 Hz, 0,6H), 5,02-4,96 (m, 0.4H), 4,09 (s, 1.8H), 4,01 (s, 1.2H), 4,00-3,85 (m, 1.4H), 3,75-3,68 (m, 0.6H), 2,50-2,40 (m, 1H), 2,08-1,98 (m, 1.4H), 1,96 - 1,83 (m, 1.6 H). CL - En (IEN, m/z, procedimiento l) Tr = 2,67 min - 357/359 (M+H)+. IEN-HRMS: Observado: 357,1111 calculado para C1bH1b35CIN402 (M+H)+: 357.1113.
21. (S ) - (2 - (4 - f lu o ro fe n il)p ir ro lid in -1 - il) (3 -m e til-1 H -p ira z o lo [4 ,3 -b ]p ir id in -5 - i l)m e ta n o n a (242 )

A ácido 3-metil-1 H-pirazolo[4,3-b)piridin-5-carboxílico (40 mg, 0,23 mmol), HATU (103 mg, 0,271 mmol) y clorhidrato de (S)-2-(4-fluorofenil)pirrolidina (45,5 mg, 0,226 mmol) se añadieron DIPEA (87 pl, 0,50 mmol) y N,N- dimetilformamida (1,9 ml). La mezcla de reacción se agitó a TA durante 1 h. Después, la mezcla de reacción se concentró (V10), se cargó en seco sobre sílice y se purificó mediante cromatografía en columna Biotage (diclorometano/etanol 99:1 a 85:15) y por TLC prep. (0,5 mm, diclorometano/etanol 98:2) para dar el compuesto del título (16 mg, rendimiento del 22 %). RMN 1H (500 m Hz , CDCI3 , 2 rotámeros) ppm = 7,97 (d, J = 8,7 Hz, 0,45H), 7,79 (d, J = 8,7 Hz, 0,45H), 7,66 (d, J = 8,7 Hz, 0.55H), 7,59 (d, J = 8,7 Hz, 0,55H), 7,32-7,26 (m, 0.90H), 7,01-6,93 (m, 2H), 6,82-6,76 (m, 1.1H), 5,89-5,84 (m, 0.55H), 5,42-5,37 (m, 0.45H), 4,39 (dt, J = 12,3, 6 , 8 Hz, 0,45H), 4,11 (dt, J = 12,3, 6 , 8 Hz, 0,45H), 4,06 - 3,94 (m, 1.1H), 2,73 (s, 1.35H), 2,48 (s, 1.65H), 2,52-2,37 (m, 1.1H), 2,10 - 1,85 (m, 2.9H). RMN 19F (470 MHz, CDCI3) ppm = -116,35. CL - EN (Ie N, m/z, procedimiento l) Tr = 2,55 min - 325 (M+H)+. IEN-Hr MS: Observado: 325,1456 calculado para C18H18FN4O (M+H)+: 325.1459.
22. (2-(4-Cloro-3-fluorofenil)pirrolidin-1 -il)(3-metil-1 H-pirazolo[4,3-b]piridin-5-il)metanona (260)

22.1.2-(4-Cloro-3-fluorofenil)-1 H-pirrol-1 -carboxilato de terc-butilo

Se cargaron ácido (1-(terc-butoxicarbonil)-1H-pirrol-2-il)borónico (0,625 g, 2,96 mmol), carbonato sódico (1,05 g, 9,87 mmol) y Pd(PPh3)4 (0,285 g, 0,247 mmol) en un matraz y después se añadieron d Me (50 ml), agua (10 ml) y 4-bromo-1-cloro-2-fluorobenceno (0,300 ml, 2,47 mmol). La mezcla de reacción se agitó a 95 °C durante 3 h 30. Se añadieron agua y diclorometano y las capas se separaron. La capa acuosa se extrajo con diclorometano y las capas orgánicas se secaron y se concentraron. El producto en bruto se purificó por cromatografía en columna Biotage (ciclohexano/diclorometano 100/0 a 90/10) para dar el compuesto del título ( 6 00 mg, rendimiento del 82 %). RMN 1H (500 MHz, CDCI3) ppm = 7,38-7,34 (m, 2H), 7,15 (dd, J = 10,0, 2,0 Hz, 1H), 7,11-7,08 (m, 1H), 6,24-6,20 (m, 2H), 1,43 (s, 9H). CL - EM (IEN, m/z. procedimiento K) Tr = 1,67 min - 196/198 (M-Boc+H)+.
22.2. 2-(4-Cloro-3-fluorofenil)pirrolidina

A 2-(4-cloro-3-fluorofenil)-1 H-pirrol-1 -carboxilato de ferc-butilo (600 mg, 2,03 mmol) en etanol (11,9 ml) se añadió HCI (867 pl, 9,13 mmol) y PtÜ2 (60 mg). Después de 27 h a 13 °C, la mezcla de reacción se filtró sobre celite y el filtrado se concentró. Se añadieron metanol (2 ml) y HCI 4 N en dioxano (2 ml) al residuo obtenido. Después de 2 h a TA, la mezcla de reacción se concentró. La mezcla en bruto se filtró en una columna SCX2 y el producto se liberó con NH3 1 N en metanol y se usó en la siguiente etapa sin purificación adicional. RMN 1H (500 MHz, CDCI3) ppm = 7,30 (dd, J = 8,2, 7,6 Hz, 1H), 7,22-7,15 (m, 1H), 7,11-7,04 (m, 1H), 4,11 (t, J = 7,7 Hz, 1H), 3,16 (ddd, J = 10,1,7,7, 5,3 Hz, 1H), 3,02 (ddd, J = 10,1, 8,2, 6,7 Hz, 1H), 2,23-2,12 (m, 1H), 1,96-1,77 (m, 2H), 1,63-1,55 (m, 1H). CL - EN (IEN, m/z, procedimiento K) Tr = 0,69 min - 200/202 (M H)+.
22.3. (2-(4-Cloro-3-fluorofenil)pirrolidin-1-il)(3-metil-1 H-pirazolo[4,3-b]piridin-5-il)metanona

Se mezcló ácido 3-metil-1 H-pirazolo[4,3-b]piridin-5-carboxílico (80 mg, 0,45 mmol) con /V,A/-dimetilformamida (2,3 ml) y trietilamina (126 pl, 0,903 mmol). La suspensión se enfrió en un baño de hielo/acetona. Se añadió cloroformiato de etilo (43 pl, 0,45 mmol) a la solución fría y la reacción se agitó durante 15 min. Se añadió una solución de clorhidrato de 2-(4-cloro-3-fluorofenil)pirrolidina (107 mg, 0,452 mmol) en W,W-dimetilformamida (2 ml) a la mezcla de reacción, que se agitó durante 30 min más en el baño de refrigeración. La reacción se agitó a TA durante una noche. Se añadieron agua y diclorometano a la mezcla de reacción. Las capas se separaron y la capa acuosa se extrajo tres veces con acetato de etilo. Las capas orgánicas se secaron sobre MgSCL y se concentraron al vacío. El material en bruto se disolvió en metanol (3 ml) y se añadió carbonato de cesio (147 mg, 0,452 mmol). La mezcla de reacción se agitó a 40 °C durante 1 h. Después de la evaporación del disolvente, el material en bruto se purificó mediante cromatografía en columna Biotage (diclorometano/etanol, 98/2 a 94/6) y por filtración en una columna SCX para dar el compuesfo del fífulo (18 mg, rendimiento del 11 %). RMN 1H (500 MHz, CDCI3 , 2 rotámeros) ppm = 7,98 (d, J = 8 , 8 Hz, 0,5H), 7,85 (dd, J = 8 ,8 , 5,5 Hz, 1H), 7,71 (d, J = 8 , 8 Hz, 0.5H), 7,29 (t, J = 8,1 Hz, 0.5H), 7,17 (dd, J = 8,3, 7,4 Hz, 0,5H), 7,09 (dd, J = 10,0, 2,0 Hz, 0,5H), 7,05 (dd, J = 8 ,1 ,2,1Hz, 0.5H), 6 , 8 8 (dd, J = 10,0, 2,0 Hz, 0.5H), 6,81 (dd, J = 8,3, 2,1Hz, 0.5H), 5,92 (dd, J = 7,5, 2,7 Hz, 0.5H), 5,34 (dd, J = 7,8, 5,3 Hz, 0,5H), 4,38 (dt, J= 11,7, 6,9 Hz, 0,5H), 4,11 (dt, J= 11,7, 6,9 Hz, 0,5H), 4,02 - 3,94 (m, 1H), 2,75 (s, 1.5H), 2,49 - 2,34 (m, 2.5H), 2,10 - 1,82 (m, 3H). CL - EN (IEN, m/z, procedimiento K) Tr = 1,40 min - 359/361 (M+H)+. IEN-HRMS: Observado: 359,1086 calculado para C1bH1735CIFN4C (M+H)+: 359.1069.
23. (S )-(3 -M e tiI-1 H -p ira z o lo [4 ,3 -b ]p ir id in -5 - il) (2 - (4 - ( t r if lu o ro m e t il ) fe n il)p ir ro lid in -1 - i l)m e ta n o n a (257 )

23.1

Una solución de 1 -(3-fluoropiridin-2-il)etanona (1,00 g, 7,19 mmol) en monohidrato de hidrazina (12 ml, 247,00 mmol) se calentó a 130 °C durante 3 h en irradiación de microondas. Se añadió agua (50 ml) y la mezcla se extrajo con acetato de etilo (3 x 25 ml). Las capas orgánicas combinadas se secaron (MgSO4), se filtraron y se concentraron al vacío. Se purificó por Biotage (columna SNAP 50 g, acetato de etilo/etanol 95/5 -> 70/30) para dar un sólido de color crema (575 mg, 60 %). RMN 1H (500 MHz, DMSO-da) 5 12,87 (s a, 1H), 8,46 (dd, J = 4,3, 1,4 Hz, 1H), 7,91 (dd, J = 8,5, 1,4 Hz, 1H), 7,33 (dd, J = 8,5, 4,3 Hz, 1H), 2,53 (s, 3H). RMN 13C (126 MHz, DMSO-ds) 5144,51, 142,04, 139,94, 133,42, 121,12, 118,49, 11,24. CLEM (Procedimiento M) Tr 0,52 min, [M+H]+ 134; HRMS calc. para C7H8N3 : 134,0718, observado: 134.0726.
23.2. 3-Metil-1 -tritil-1 H-pirazolo[4,3-b]piridina

A una solución de 3-metil-1H-pirazolo[4,3-b]piridina (497 mg, 3,73 mmol) en DMF (10 ml) a 0 °C se añadió NaH (al 60 % en aceite mineral; 300 mg, 7,50 mmol) y la mezcla se dejó calentar a TA y se agitó durante 20 min. Después, se añadió cloruro de tritilo (1,35 g, 4,84 mmol) y la mezcla se agitó a TA durante 2,5 h. La reacción se interrumpió con NH4CI ac. sat. (5 ml) y la mezcla se concentró al vacío. El residuo se recogió en acetato de etilo (75 ml) y se lavó con agua (3 x 50 ml). La capa orgánica se secó (MgSO4), se filtró, se concentró al vacío y el residuo se purificó por Biotage (columna SNAP 50 g, ciclohexano/diclorometano 100/0 -> 50/50) para dar un sólido de color blanquecino (1,086 g, 77 %). RMN 1H (500 MHz, CDCI3) 58,47 (dd, J = 4,4, 1,3 Hz, 1H), 7,32-7,27 (m, 15H), 6,90 (dd, J = 8,7, 4,4 Hz, 1H), 6,59 (dd, J = 8,7, 1,3 Hz, 1H), 2,69 (s, 3H). RMN 13C (126 MHz, CDCI3) 5 144,43, 142,72 (3 x ArC), 142,66, 135,23, 130,46, 130,18 ( 6 x ArCH), 127,73 ( 6 x ArCH), 127,52 (3 x ArCH), 121,35, 119,81,78,48, 11,32. CLEM (Procedimiento M) Tr 1,67 min, [M+H]+ 376; h Rm S calc. para C26H22N3 : 376,1814, observado: 376.1816.
23.3. 4-Óxido de 3-metil-1 -tritil-1 H-pirazolo[4,3-b]piridina

A una solución de 3-metil-1 -tritil-1 H-pirazolo[4,3-b]piridina (108 mg, 0,29 mmol) en diclorometano (0,8 ml) a 0 °C se añadió mCPBA (77 %; 80 mg, 0,36 mmol). Después, la mezcla se dejó calentar a TA y se agitó durante 1,5 h. Después, la mezcla de reacción se diluyó con diclorometano (10 ml) y se lavó con NaHCO3 ac. sat. (3 x 10 ml). La capa orgánica se filtró a través de un separador de fases y se concentró al vacío. El residuo resultante se purificó por Biotage (columna SNAP 10 g, diclorometano/acetato de etilo 100/0 -> 70/30) para dar un sólido de color blanco (94 mg, 83 %). RMN 1H (500 MHz, CDCIa) 58,03 (d, J = 6,0 Hz, 1H), 7,35-7,29 (m, 9H), 7,21-7,18 (m, 6H), 6,75 (dd, J = 8,8, 6,0 Hz, 1H), 6,19 (d, J = 8,8 Hz, 1H), 2,85 (s, 3H). RMN 13C (126 MHz, CDCIa) 5 142,01 (3xArCH), 139,70, 138,83, 132,14, 130,26, 130,10 (6xArCH), 127,82 (6 x ArCH), 120,97, 113,41, 79,14, 14,26. CLEM (Procedimiento K) Tr 1,54 min, [M+H]+ 392; HRMS calc. para C26H22Nao: 392,1757, observado: 392.1744.
23.4. 3-Metil-1 -tritil-1 H-pirazolo[4,3-b]piridin-5-carbonitrilo

A una suspensión de 4-óxido de 3-metil-1 -tritil-1 H-pirazolo[4,3-b]piridina (94 mg, 0,24 mmol) y trietilamina (0,05 ml, 0,36 mmol) en una mezcla de acetonitrilo (1 ml) y diclorometano (0,2 ml) se añadió TMS-CN (0,07 ml, 0,52 mmol). La mezcla resultante se calentó a reflujo durante 18 h. Después, la mezcla se dejó enfriar a TA, se concentró al vacío y el residuo se purificó por Biotage (columna SNAP 10 g, diclorometano/acetato de etilo 100/0 -> 70/30) para dar un aceite incoloro (81 mg, 84 %). RMN 1H (500 MHz, CDCb) 57,35 - 7,29 (m, 9H), 7,25 - 7,20 (m, 7H), 6,59 (d, J = 8,8 Hz, 1H), 2,67 (s, 3H). CLEM (Procedimiento M) Tr 1,14 min (masa no encontrada; únicamente masa para tritilo).
23.5. Ácido 3-metil-1 -tritil-1 H-pirazolo[4,3-b]piridin-5-carboxílico

Se recogió 3-metil-1 -tritil-1 H-pirazolo[4,3-b]piridin-5-carbonitrilo (112 mg, 0,28 mmol) en etanol (1,5 ml) y se añadió NaOH 2 M (1 ml, 2,00 mmol). La mezcla resultante se calentó a 130 °C durante 1 h en irradiación de microondas. Después, el pH se ajustó a 3 con HCI 2 M y se extrajo con diclorometano (3 x 20 ml). Las capas orgánicas combinadas se filtraron a través de un separador de fases, se concentraron al vacío y el residuo se purificó por Biotage (columna SNAP 10 g, diclorometano/acetato de etilo 100/0 -> 75/25) para dar un sólido de color blanco (67 mg, 57 %). RMN 1H (500 MHz, CDCb) 57,85 (d, J = 8,9 Hz, 1H), 7,39-7,26 (m, 9H), 7,26-7,12 (m, 6H), 6,72 (d, J = 8,9 Hz, 1H), 2,68 (s, 3H). CLEM (Procedimiento M) Tr 1,54 min, [M+Na]+ 442; HRMS calc. para C27H21Nao2Na: 442,1531, observado: 442.1542.
23.6. (S)-(3-Metil-1 -tritil-1 H-pirazolo[4,3-b]piridin-5-il)(2-(4-(trifluorometil)fenil)pirrolidin-1 -il)metanona

A una mezcla de ácido 3-metil-1 -tritil-1 H-pirazolo[4,3-b]piridin-5-carboxílico (67 mg, 0,16 mmol) y clorhidrato de (S)-2-(4-(trifluorometil)fenil)pirrolidina (70 mg, 0,28 mmol) en DMF (0,8 ml) se añadió Ha Tu (95 mg, 0,25 mmol) y DIPEA (0,3 ml, 1,72 mmol) y la mezcla resultante se agitó a TA durante 18 h. Después, la mezcla se concentró al vacío y el residuo se purificó por Biotage (columna SNAP 10 g, ciclohexano/acetato de etilo 100/0 -> 70/30) para dar el compuesto del título en forma de una mezcla de rotámeros (78 mg). CLEM (Procedimiento M) Tr 1,71 min, [M+H]+ 617; HRMS calc. para C38H32F3N40 : 617,2528, observado: 617.2556.
23.7. (S)-(3-Metil-1 H-pirazolo[4,3-b]piridin-5-il)(2-(4-(trifluorometil)fenil)pirrolidin-1 -il}metanona (257)

A una solución de (S)-(3-metil-1-tritil-1H-pirazolo[4,3-b]piridin-5-il)(2-(4-(trifluorometil)fenil)pirrolidin-1-il)metanona (78 mg, 0,13 mmol) en diclorometano (0,8 ml) se añadió t Fa (0,2 ml, 2,60 mmol) y la mezcla resultante se agitó a TA durante 45 min. Después, la reacción se interrumpió con NaHCO3 ac. sat. ( 6 ml) y se extrajo con acetato de etilo (3 x 10 ml). Las capas orgánicas combinadas se secaron (MgSO4), se filtraron y se concentraron al vacío. El residuo se purificó por Biotage (columna SNAP 10 g, ciclohexano/acetato de etilo 100/0 -> 0/100) para dar el compuesto del título en forma de una mezcla de rotámeros (46 mg, 97 %). RMN 1H (500 MHz, CDCI3) 5 7,98 (d, J = 8,7 Hz, 1H), 7,82 -7,74 (m, 2H), 8,00 - 7,55 (m, 3H), 7,47 - 7,42 (m, 4H), 7,21 (d, J = 8,0 Hz, 2H), 6,02 - 5,97 (m, 1H), 5,47 (dd a, J = 7,8, 5,2 Hz, 1H), 4,45 (dt, J = 12,0, 6,9 Hz, 1H), 4,19 (dt, J = 12,4, 6 , 8 Hz, 1H), 4,08 - 4,01 (m, 2H), 2,72 (s, 3H), 2,51 - 2,42 (m, 2H), 2,32 (s, 3H), 2,10 - 1,98 (m, 4H), 1,96 - 1,89 (m, 2H). RMN 13C (126 MHz, CDCI3) 5 167,01, 166,48, 148,93, 148,40, 148,00, 147,45 (2 x C), 144,98, 144,60, 138,60, 138,10, 133,43, 133,12, 129,10, 128,84, 128,59, 125,91 (4 x C), 125,51 (c, J = 3,8 Hz, 2 x C), 125,8 (c, J = 3,8 Hz, 2 x C), 123,14, 122,99, 122,26, 121,97, 117,95, 117,80, 65,88, 63,12, 62,14, 51,04, 48,52, 36,40, 34,15, 25,13, 21,35, 14,13, 10,93, 10,51. CLEM (Procedimiento M) Tr 1,31 min, [M+H]+ 375; HRMS calc. para C19H18F3N40 : 375,1433, observado: 375.1429.
24. (3-Amino-1 H-pirazolo[4,3-b]piridin-5-il)-[(S)-2-(4-cloro-fenil)-pirrolidin-1 -il]- metanona (265)

24.1. 5-Bromo-1 H-pirazolo[3,4-c]piridin-3-ilamina

En un tubo de 10 ml cerrado herméticamente, se puso 2-bromo-5-fluoropiridin-4-carbonitrilo (700 mg, 3,13 mmol, 90 %), DMSO (5 ml) y NH2NH2.H2O (2,5 ml). La mezcla se agitó durante 3 h a 120 °C. La mezcla de reacción se diluyó con 50 ml de agua. La capa orgánica se aisló y la capa acuosa se extrajo con 50 ml de acetato de etilo. Las capas orgánicas combinadas se lavaron dos veces con 2 0 ml de cloruro sódico, se secaron sobre Na2SO4 , se filtraron y se evaporaron a sequedad. Esto dio como resultado 600 mg (81 %) de 5-bromo-1H-pirazolo[3,4-c]piridin-3-amina en forma de un sólido de color amarillo.
24.2. Éster metílico del ácido 3-amino-1 H-pirazolo[3,4-clpiridin-5-carboxílico

En un tanque a presión de 20 ml purgado y mantenido con una atmósfera de CO(g), se puso 5-bromo-1 H-pirazolo[3,4-c]piridin-3-amina (600 mg, 2,53 mmol, 90 %), Pd(dppf)CÍ2.CH2CÍ2 (104 mg, 0,13 mmol), AcOK (498 mg, 5,07 mmol), metanol (5 ml) y N,N-dimetilformamida (5 ml). La solución resultante se agitó durante una noche a 80 °C. La solución resultante se diluyó con 10 ml de agua y se extrajo con 50 ml de acetato de etilo. La capa orgánica se evaporó a sequedad. Esto dio como resultado 500 mg (72 %) de 3-amino-1H-pirazolo[3,4-c]piridin-5-carboxilato de metilo en forma de un sólido de color naranja.
24.3. Ácido 3-amino-1H-pirazolo[3,4-c]piridin-5-carboxílico

En un matraz de fondo redondo de 50 ml, se puso LiOH (99,7 mg, 4,16 mmol), agua (3 ml), tetrahidrofurano (10 ml) y 3-amino-1H-pirazolo[3,4-c]piridin-5-carboxilato de metilo (500 mg, 2,08 mmol, 80 %). La solución se agitó durante 20 h a 50 °C. La mezcla se concentró por evaporación y se añadieron 10 ml de agua. El valor del pH de la solución se ajustó a 2 con una solución de cloruro de hidrógeno (2 N). La mezcla se extrajo dos veces con 30 ml de acetato de etilo y las capas orgánicas se combinaron. La fase orgánica se lavó dos veces con 20 ml de agua, se secó sobre sulfato sódico y después de filtración se evaporó a sequedad. Esto dio como resultado 400 mg (76 %) de ácido 3 amino-1 H-pirazolo[3,4-c]piridin-5-carboxílico en forma de un sólido de color amarillo.
24.4. (3-Amino-1 H-pirazolo[4,3-b]piridin-5-il)-[(S)-2-(4-cloro-fenil)-pirrolidin-1 -il]- metanona (265)

En un matraz de fondo redondo de 10 ml, se puso ácido 3-amino-1H-pirazolo[3,4-c]piridin-5-carboxílico (100 mg, 0,39 mmol, 70 %), 2-(4-clorofenil)pirrolidina (112 mg, 0,59 mmol), 4-dimetilaminopiridina (82,3 mg, 0,67 mmol), EDCI (215 mg, 1,12 mmol) y N,N-dimetilformamida (5 ml). La mezcla de reacción se agitó durante 3 h a TA. La mezcla se concentró al vacío. El residuo se aplicó a una columna de gel de sílice con metanol:diclorometano (3:10). El producto racémico en bruto se purificó por HPLC prep. (acetonitrilo/agua), seguido de una separación de HPLC prep. quiral
(Chiralpak IB4, 6*250 mm, 5 pM, 100 % de metanol (0,1 % de dietilamina) para producir 13 mg (9 %) de (3-amino-1 H-pirazolo[4,3-b]piridin-5-il)-[(S)-2-(4-cloro-fenil)-pirrolidin-1 -il]-metanona en forma de un sólido de color blanco. RMN 1H (300 Hz, DMSO-d6) ppm = 11,84 (s, 1H), 8,65-8,64 (m, 1H), 8,13 (s, 1H), 7,27 (s, 4H), 5,60-5,20 (m, 3H), 4,12-3,90 (m, 1H), 3,90-3,70 (m, 1H), 2,41-2,29 (m, 1H), 1,99-1,77 (m, 4H). CLEM (Procedimiento J) Tr 1,17 min, [M+H]+ 342.
Los compuestos restantes se sintetizaron a partir de los ácidos carboxílicos descritos o disponibles en el mercado con aminas descritas o disponibles en el mercado como se describe en 2.2., seguido parcialmente de una separación quiral como se describe en 2.3 dado como resultado 2 o más enantiómeros distintos. De este modo, se sintetizaron los comps. 171-176, 179, 182, 185-188, 191 -202, 204-205, 207-209, 211-215, 217-219, 221-241,247-253, 258-259 y 261-264.
Actividad biológica
Ensayo bioquímico de las actividades inhibitorias de la CDK8
Ensayo de competición de unión de Lanthascreen basado en FRET: Una sonda competitiva ATP marcada con colorante servida como aceptador de FRET cuando se une a CDK8 marcada con un quelato de Eu de estreptavidina (a través de un anticuerpo anti His biotinilatado). El resultado fue una señal de fluorescencia en 647 nm. En caso esta sonda se sometió a competición mediante un inhibidor, tal señal ya no puede generarse más. La CDK8 usada para este ensayo puede una proteína co-expresada con CycC.
El procedimiento de ensayo para un ensayo en una placa de 1536 pocillos se realizó según lo que sigue: 2 pl de CDK8 / biotina-anti-His Ab / mezcla de SA-Eu en tampón de ensayo se pipetearon en los pocillos de una microplaca. Se añadió 1 pl del compuesto en 20 mM de tampón de Hepes / 5 % de DMSO. La placa se agitó durante 30 seg y se incubó durante 20 min a TA.
Se añadieron 2 pl de sonda de Alexa647 en tampón de ensayo. La placa se agitó durante 30 seg de nuevo y se incubó durante 60 min a TA en la oscuridad.
A continuación, la placa se leyó sobre un Perkin Elmer Envision (modo LANCE/TRF, excitación 340 nm emisión 650 nm).
El tampón de ensayo fue de 50 mM Hepes, pH 7,5 (Merck n.° 1,10110), 10 mM de MgCl2 (Merck n.° 1,05833), 1 mM de EGTA (Merck n .21,08435), 0,01 % de Brij-35 (Pierce n .228316).
Las concentraciones finales de los componentes de reacción en 5 pl de volumen de ensayo total fueron: 1 % de DMSO (Merck n.° 1,02950), 5 nM de CDK8 (CDK8 /CycC Invitrogen n.° PV4402), 2 nM de biotina-a-His Ab (Invitrogen n.° PV6089), 2 nM de SA-Europium (Invitrogen n.° PV5899), 10 nM de indicador de Alexa647 (Invitrogen n.° PV5592). Las diluciones del compuesto para las curvas de respuesta a la dosis fueron de 30, 10, 3,2, 1, 0,32, 0,2, 0,032, 0,01, 0,0032, 0,001 pM (final) u óptima (como dilución 1:5) 30, 6 , 1,2, 0,24, 0,048, 0,0096, 0,0019, 0,0004, 8e-5, 2e-5 pM. Las lecturas se ajustaron a curvas CI50 usando Genedata Condoseo.
Para evaluar el potencial inhibitorio de los compuestos sobre la CDK8 , se determinaron los valores IC50, como se muestra en la Tabla 1 a continuación, mediante la cual se usa la siguiente clasificación:
CI50 < 10 nM "A'
10 nM < CI50 < 0,1 pM "B'
0,1 pM < CI50 < 1 pM "C
1 < < CI50 < 10 pM "D1
Tabla 1





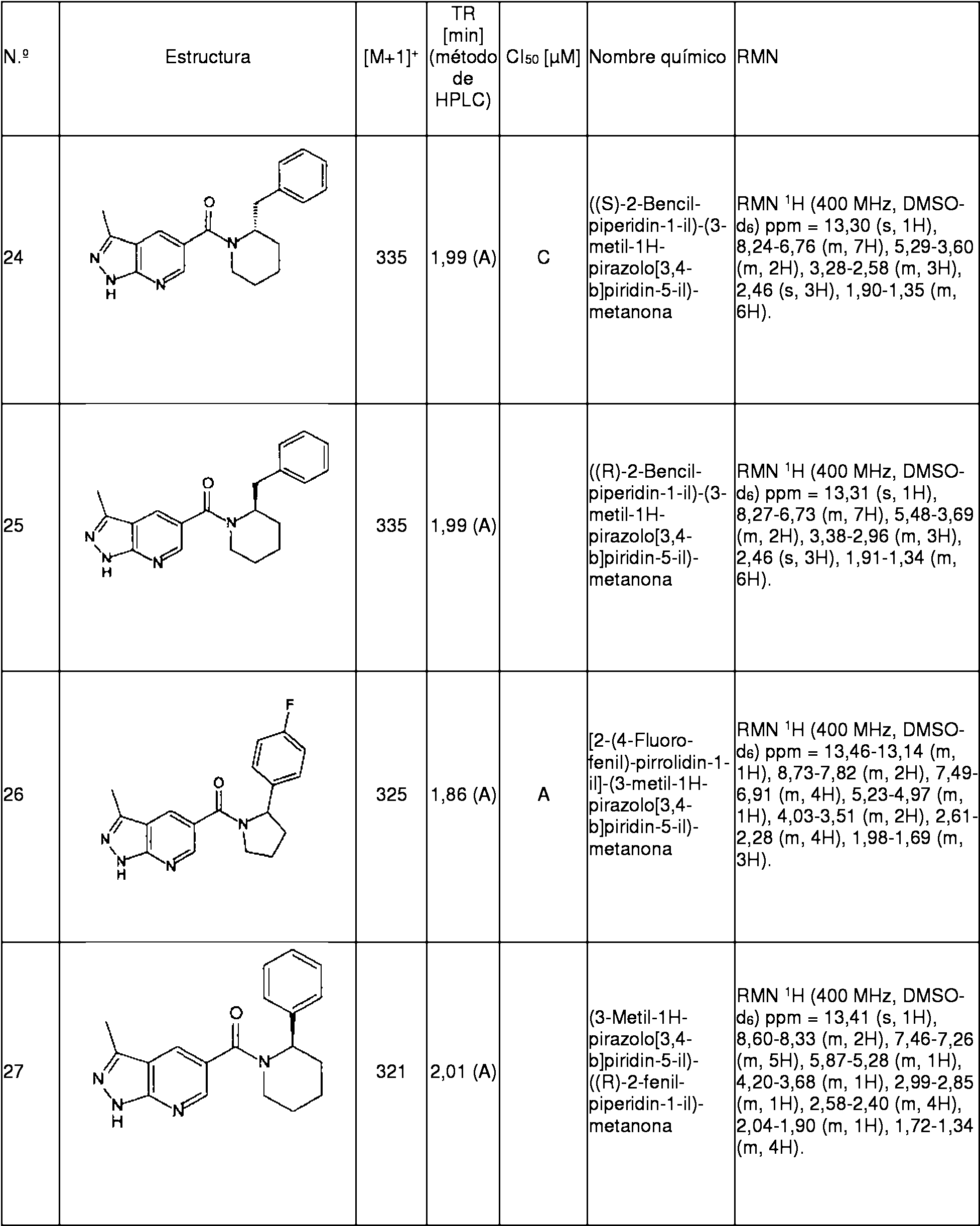




















,90
,50
,90 ,50
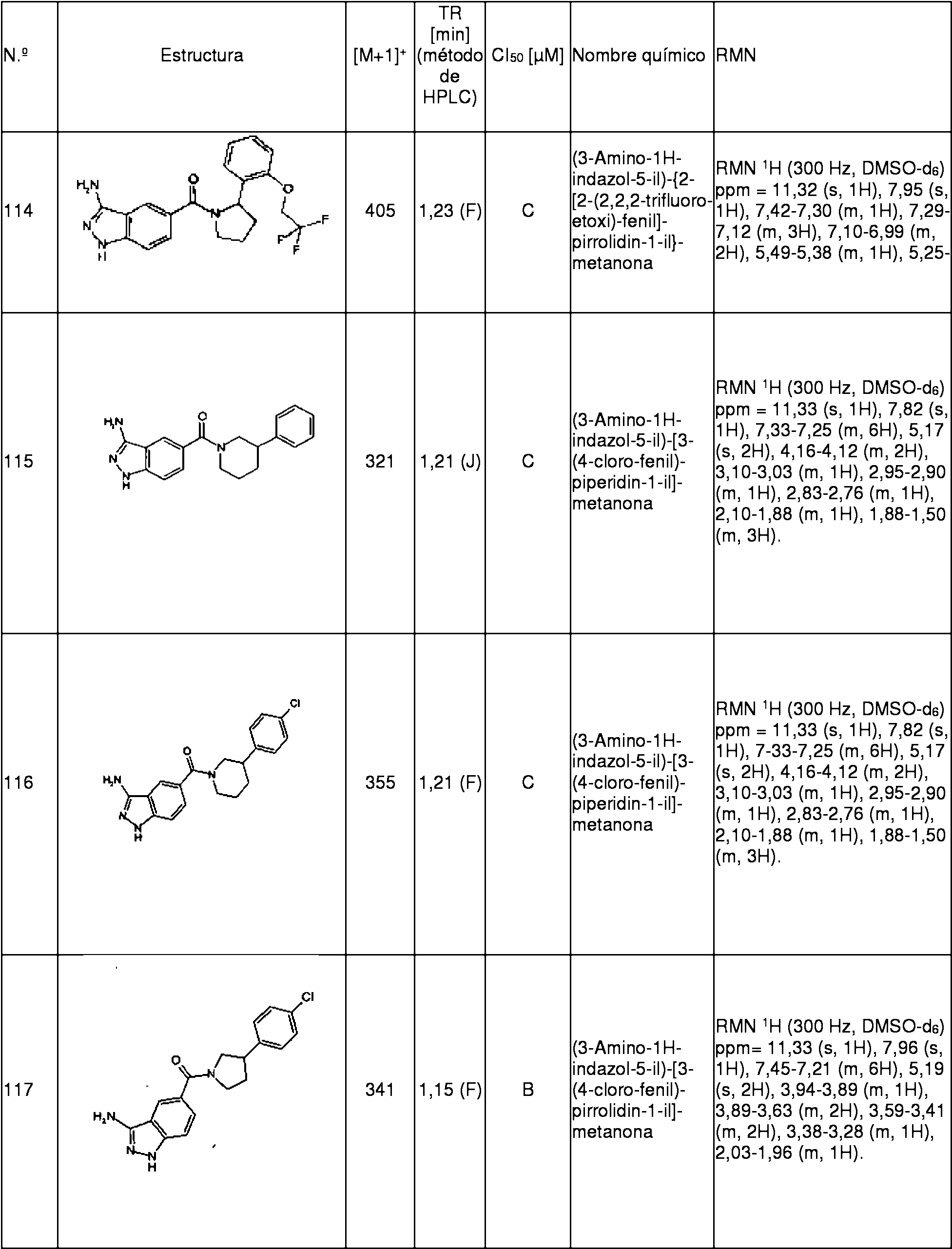










,86




,90






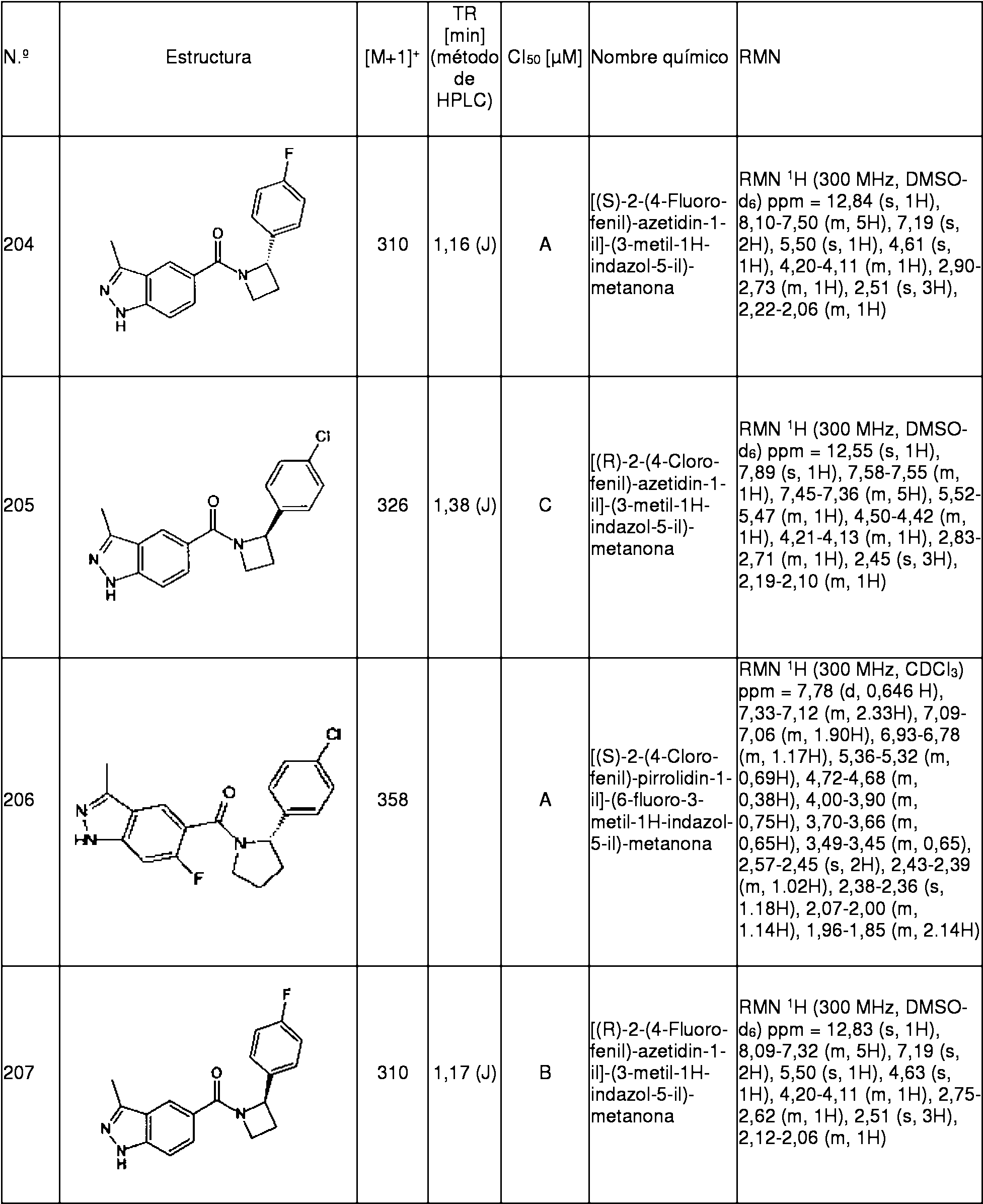
,56













Los números de ejemplo 7, 8, 42, 75, 119, 126, 128, 140, 146, 151, 161, 163, 187, 201, 212, 213, 233 y 234 se omitieron intencionadamente.
Claims (15)
1. Un compuesto de Fórmula (I)

en la que:
X, Y son independientemente CH o N,
Z es CH, C-Hal o N,
R1 es H, LA, CA, NH2 , NH(LA) o (LA)NH(LA), Hal, -S(LA), -SO2(LA), O(LA),
Cyc es un heterociclo alifático de 3, 4, 5, 6 o 7 miembros que tiene 1 o 2 átomos de N, o 1 átomo de N y 1 átomo de O,
R2 es -LA-Ar o Ar, que está en la posición 2 o 3 con respecto al átomo de N de Cyc,
R3 es H, OH, NH2 , COO(LA), CONH2 , CONH(LA) NHCO(LA), (LA)OH, NH(LA) o LA, que está en cualquier posición de Cyc,
Ar es un homo o heterociclo, mono o binuclear, alifático o aromático de 3, 4, 5, 6, 7, 8, 9 o 10 miembros, que tiene 0, 1, 2, 3 o 4 átomos de N, O y/o S, que puede estar sin sustituir o mono o disustituido independientemente con Hal, OH, CN, LA, O(LA), S(LA),
LA es alquilo sin ramificar o ramificado, que tiene 1,2, 3, 4 o 5 átomos de carbono, que puede estar saturado o parcialmente insaturado, en el que 1, 2 o 3 átomos de H están reemplazados por Hal,
CA es cicloalquilo que tiene 3, 4, 5 o 6 átomos de carbono, o cicloalquil alquilo que tiene 3, 4, 5 o 6 átomos de carbono en el anillo y 1 o 2 átomos de carbono no anulares,
Hal es F, CI, Br o I.
2. El compuesto de acuerdo con la reivindicación 1, o sus estereoisómeros o tautómeros, o sales farmacéuticamente aceptables de cada uno de los anteriores, incluyendo mezclas de los mismos en todas las proporciones, en la que X, Y, Z son independientemente CH o N,
R1 es H, LA, CA, NH2 , NH(LA) o (LA)NH(LA),
Cyc es un heterociclo alifático de 3, 4, 5, 6 o 7 miembros que tiene 1 átomo de N,
R2 es -LA-Ar o Ar, que está en la posición 2 o 3 con respecto al átomo de N de Cyc,
R3 es H, OH, NH2 , COO(LA), CONH2 , CONH(LA) o LA, que está en cualquier posición de Cyc,
Ar es un homo o heterociclo, mono o binuclear, alifático o aromático de 3, 4, 5, 6, 7, 8, 9 o 10 miembros, que tiene 0, 1, 2, 3 o 4 átomos de N, O y/o S, que puede estar sin sustituir o mono o disustituido independientemente con Hal, OH, CN, LA, O(LA), S(LA),
LA es alquilo sin ramificar o ramificado, que tiene 1,2, 3, 4 o 5 átomos de carbono, que puede estar saturado o parcialmente insaturado, en el que 1, 2 o 3 átomos de H están reemplazados por Hal,
CA es cicloalquilo que tiene 3, 4, 5 o 6 átomos de carbono, o cicloalquil alquilo que tiene 3, 4, 5 o 6 átomos de carbono en el anillo y 1 o 2 átomos de carbono no anulares,
Hal es F, CI, Br o I.
3. El compuesto de acuerdo con la reivindicación 2, o sus estereoisómeros o tautómeros, o sales farmacéuticamente aceptables de cada uno de los anteriores, incluyendo mezclas de los mismos en todas las proporciones, en la que X, Y son independientemente CH o N,
Z es CH,
R1 es H, LA, CA, NH2 , NH(LA) o (LA)NH(LA),
Cyc es un heterociclo alifático de 3, 4, 5, 6 o 7 miembros que tiene 1 átomo de N,
R2 es -LA-Ar o Ar, que está en la posición 2 o 3 con respecto al átomo de N de Cyc,
R3 es H, NH2 o LA, que está en cualquier posición de Cyc,
Ar es un homo o heterociclo mononuclear, aromático, de 6 miembros, que tiene 0, 1 o 2 átomos de N, que puede estar sin sustituir o mono o disustituido independientemente con Hal, OH, CN, LA, O(LA), S(LA),
LA es alquilo sin ramificar o ramificado, que tiene 1, 2, 3, 4 o 5 átomos de carbono, que puede estar saturado o parcialmente insaturado, en el que 1, 2 o 3 átomos de H están reemplazados por Hal,
CA es cicloalquilo que tiene 3, 4, 5 o 6 átomos de carbono, o cicloalquil alquilo que tiene 3, 4, 5 o 6 átomos de carbono en el anillo y 1 o 2 átomos de carbono no anulares,
Hal es F, CI, Br o I.
4. El compuesto de acuerdo con la reivindicación 1, 2 o 3, que está de acuerdo con las Fórmulas (lla) o (IIb),

o sus estereoisómeros o tautómeros, o sales farmacéuticamente aceptables de cada uno de los anteriores, incluyendo mezclas de los mismos en todas las proporciones.
5. El compuesto de acuerdo con la reivindicación 1 o 4, o sus estereoisómeros o tautómeros, o sales farmacéuticamente aceptables de cada uno de los anteriores, incluyendo mezclas de los mismos en todas las proporciones, en el que los restos no designados con mayor detalle tienen el significado indicado en la reivindicación 1, pero en los que
en la Subfórmula 1
Ar es un homo o heterociclo mononuclear, aromático, de 6 miembros, que tiene 0, 1 o 2 átomos de N, que está sin sustituir o monosustituido con Hal, LA u O(LA),
en la Subfórmula 2
X es N,
en la Subfórmula 3
X es CH,
en la Subfórmula 4
Y es CH,
en la Subfórmula 5
Y es N,
en la Subfórmula 6
Z es CH,
en la Subfórmula 7
X es CH,
Y es N,
Z es CH,
en la Subfórmula 8
X es N,
Y es CH,
Z es CH,
en la Subfórmula 9
Ar es un homo o heterociclo mononuclear, aromático, de 6 miembros, que tiene 0, 1 o 2 átomos de N, que puede estar sin sustituir o mono o disustituido independientemente con Hal, OH, CN, LA, O(LA), S(LA), X es CH, Y es N,
Z es CH,
en la Subfórmula 10
Ar es un homo o heterociclo mononuclear, aromático, de 6 miembros, que tiene 0, 1 o 2 átomos de N, que puede estar sin sustituir o mono o disustituido independientemente con Hal, OH, CN, LA, O(LA), S(LA), X es N, Y es CH,
Z es CH,
en la Subfórmula 11
R1 es LA o NH2 , NHLA
en la Subfórmula 12
Cyc tiene 4, 5 o 6 átomos en el anillo,
en la Subfórmula 13
R3 es H, OH, NH2 o metilo,
en la Subfórmula 14
R3 es H,
en la Subfórmula 15
R2 es bencilo,
R3 es H, NH2 o metilo,
Cyc tiene 6 átomos en el anillo,
en la Subfórmula 16
Cyc tiene 5 o 6 átomos en el anillo,
R2 es fenilo, que está sin sustituir, o mono o disustituido independientemente con Hal o LA,
en la Subfórmula 17
Z es CH,
R2 es bencilo o fenilo, fenilo que está sin sustituir, o mono o disustituido independientemente con Hal o LA,
en la Subfórmula 18
X es N o CH,
Y es CH,
Z es CH,
R1 es metilo, n-propilo, /-propilo, ciclopropilo, NH2 , NHLA
en la Subfórmula 19
Z es CH,
Cyc tiene 5 átomos en el anillo,
en la Subfórmula 20
Z es CH,
R2 es fenilo, que está sin sustituir o monosustituido con Br, CI, metilo o CF3 ,
R3 es H,
en la Subfórmula 21
Z es CH,
R2 es fenilo, que está sin sustituir o monosustituido con Br, CI, metilo o CF3 ,
R3 es H,
en la Subfórmula 22
Z es CH,
R2 es fenilo, que está sin sustituir o para-sustituido con Br, CI, metilo o CF3 ,
R3 es H,
en la Subfórmula 23
Y es CH,
Z es CH,
R1 es metilo, n-propilo, /-propilo, ciclopropilo o NH2 ,
en la Subfórmula 24
X es N o CH,
Y es CH,
Z es CH,
R1 es metilo, n-propilo, /-propilo, ciclopropilo o NH2 ,
Cyc tiene 4, 5 o 6 átomos en el anillo,
en la Subfórmula 25
X es N o CH,
Y es CH,
Z es CH,
R1 es metilo, n-propilo, /-propilo, ciclopropilo o NH2 ,
Cyc tiene 4, 5 o 6 átomos en el anillo,
R2 es fenilo, que está sin sustituir o para-sustituido con Br, CI, metilo o CF3 ,
R3 es H,
en la Subfórmula 26
X es N o CH,
Y es CH,
Z es CH,
R1 es metilo, n-propilo, /-propilo, ciclopropilo o NH2 ,
Cyc tiene 5 átomos en el anillo,
R2 es fenilo, que está sin sustituir o para-sustituido con Br, CI, metilo o CF3 ,
R3 es H.
en la Subfórmula 27
R1 es metilo, n-propilo, /-propilo, ciclopropilo, metilsulfanilo, metanosulfonilo, metoxi, F o NH2 ,
en la Subfórmula 28
R3 es H, OH, NH2 , metilo, acetamido, 2-hidroxietilo o metilamino.
en la Subfórmula 29
Z es CH o C(Hal),
Cyc tiene 4, 5 o 6 átomos en el anillo, de los cuales 1 átomo es N los otros átomos son C,
R2 es fenilo, que está sin sustituir o para-sustituido con Br, CI, F, metilo, metoxi, isopropilo o CF3 , o independientemente meta-/ para- disustituido con CI y F,
R3 es H.
en la Subfórmula 30
Z es CH o C(Hal),
Cyc tiene 4 o 5 átomos en el anillo, de los cuales 1 átomo es N los otros átomos son C,
R2 es fenilo, que está sin sustituir o para-sustituido con Br, CI, F, metilo, metoxi, isopropilo o CF3 , o meta-sustituido con F y para-sustituido con CI,
R3 es H.
6. El compuesto de acuerdo con la reivindicación 1, en el que el compuesto se selecciona entre el grupo que consiste en:
[2-(4-Bromo-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(S)-2-(4-Bromo-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[2-(4-Bromo-fenil)-pirrolidin-1-il]-(3-propil-1 H-indazol-5-il)-metanona,
[(S)-2-(4-Bromo-fenil)-pirrolidin-1-il]-(3-propil-1 H-indazol-5-il)-metanona,
[2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-propil-1 H-indazol-5-il)-metanona,
[2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-propil-1 H-indazol-5-il)-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[2-(4-Fluoro-fenil)-piperidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(S)-2-(4-Fluoro-fenil)-piperidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-(2-fenil-piperidin-1-il)-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-(2-fenil-pirrolidin-1-il)-metanona,
[2-(3-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-(3-fenil-pirrolidin-1-il)-metanona,
[2-(2-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[2-(4-Fluoro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-((S)-2-fenil-piperidin-1-il)-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1 -il]-(3-isobutil-1 H-indazol-5-il)-metanona,
[(S)-2-(2-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-((S)-2-fenil-pirrolidin-1-il)-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-((S)-3-fenil-pirrolidin-1-il)-metanona,
[(S)-2-(4-Fluoro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(S)-2-(3-Cloro-fenil)-pirrolidin-1 -il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1 H-indazol-5-il)-metanona,
[(S)-2-(4-Bromo-fenil)-pirrolidin-1-il]-(3-metil-1 H-indazol-5-il)-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[4,3-b]piridin-5-il)-metanona,
[(S)-2-(4-Bromo-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[4,3-b]piridin-5-il)-metanona,
[2-(4-Cloro-fenil)-azetidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-(2-p-tolil-pirrolidin-1-il)-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-[2-(4-trifluorometil-fenil)-pirrolidin-1-il]-metanona,
[3-(4-Cloro-fenil)-piperidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[3-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-ciclopropil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
(3-Amino-1 H-indazol-5-il)-[2-(4-bromo-fenil)-pirrolidin-1-il]-metanona,
(3-Amino-1 H-indazol-5-il)-[(S)-2-(4-bromo-fenil)-pirrolidin-1-il]-metanona,
(3-Amino-1 H-indazol-5-il)-[2-(4-cloro-fenil)-pirrolidin-1-il]-metanona,
(3-Amino-1 H-indazol-5-il)-[(S)-2-(4-cloro-fenil)-pirrolidin-1-il]-metanona,
(3-Amino-1 H-indazol-5-il)-(2-fenil-piperidin-1 -il)-metanona,
(3-Amino-1 H-indazol-5-il)-(2-fenil-pirrolidin-1 -il)-metanona,
(3-Amino-1 H-indazol-5-il)-[2-(4-fluoro-fenil)-pirrolidin-1-il]-metanona,
(3-Amino-1 H-indazol-5-il)-((S)-2-bencil-piperidin-1-il)-metanona,
(3-Amino-1 H-indazol-5-il)-((S)-2-fenil-piperidin-1-il)-metanona,
(3-Amino-1 H-indazol-5-il)-((R)-2-fenil-piperidin-1-il)-metanona,
(3-Amino-1 H-indazol-5-il)-[(S)-2-(4-fluoro-fenil)-piperidin-1-il]-metanona,
(3-Amino-1 H-pirazolo[3,4-b]piridin-5-il)-[(S)-2-(4-cloro-fenil)-pirrolidin-1-il]-metanona,
(3-Amino-1 H-pirazolo[3,4-b]piridin-5-il)-[(S)-2-(4-bromo-fenil)-pirrolidin-1-il]-metanona,
(3-Amino-1 H-indazol-5-il)-[2-(4-cloro-fenil)-azetidin-1-il]-metanona,
(3-Amino-1 H-indazol-5-il)-[2-(4-fluoro-fenil)-azetidin-1-il]-metanona,
(3-Amino-1 H-indazol-5-il)-[3-(4-cloro-fenil)-pirrolidin-1-il]-metanona,
(3-Amino-1 H-indazol-5-il)-[(S)-2-(4-fluoro-fenil)-pirrolidin-1-il]-metanona,
(3-Amino-1 H-indazol-5-il)-[(S)-2-(4-fluoro-fenil)-azetidin-1-il]-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-((S)-2-p-tolil-pirrolidin-1-il)-metanona,
[(S)-2-(2-Metoxi-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(2R,3R)-3-Amino-2-(4-fluoro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-ciclopropil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[2-(4-Bromo-fenil)-pirrolidin-1-il]-(1 H-indazol-5-il)-metanona,
[(S)-2-(4-Bromo-fenil)-pirrolidin-1-il]-(1 H-indazol-5-il)-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-[(S)-2-(4-trifluorometil-fenil)-pirrolidin-1-il]-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-[3-metil-2-(4-trifluorometil-fenil)-pirrolidin-1-il]-metanona,
N-4-(4-Cloro-fenil)-1-(3-metil-1H-pirazolo[3,4-b]piridin-5-carbonil)-pirrolidin-3-il]-acetamida,
(3-Amino-1 H-indazol-5-ilH2-(4-trifluorometil-fenil)-pirrolidin-1-il]-metanona,
[(S)-2-(1 H-lndazol-6-il)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
(3-Cloro-1 H-indazol-5-il)-[(S)-2-(4-cloro-fenil)-pirrolidin-1-il]-metanona,
[(S)-2-(4-Fluoro-fenil)-azetidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metoxi-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
(3-Cloro-1 H-indazol-5-il)-[3-(4-cloro-fenil)-pirrolidin-1-il]-metanona,
[2-(4-Cloro-fenil)-4-metil-piperazin-1-il]-(3-metil-1 H-indazol-5-il)-metanona,
[2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-trifluorometil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
(3-Amino-1 H-indazol-5-il)-[(S)-2-(4-trifluorometil-fenil)-pirrolidin-1-il]-metanona,
(3-Amino-1 H-indazol-5-il)-[2-(4-isopropil-fenil)-pirrolidin-1 -il]-metanona,
(3-Cloro-1 H-indazol-5-ilH2-(4-trifluorometil-fenil)-pirrolidin-1-il]-metanona,
[(S)-2-(4-Cloro-fenil)-azetidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(s)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-c]piridin-5-il)-metanona,
[2-(4-Cloro-3-fluoro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(S)-2-(4-Cloro-2-fluoro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[3-(4-Cloro-fenil)-morfolin-4-il]-(3-metil-1 H-indazol-5-il)-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-fluoro-1 H-indazol-5-il)-metanona,
[(S)-2-(4-Metoxi-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
(3-Metil-1 H-pirazolo[3,4-b]piridin-5-il)-[(2S,3R)-3-metil-2-(4-trifluorometil-fenil)- pirrolidin-1-il]-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-trifluorometil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metilsulfanil-1 H-indazol-5-il)-metanona,
[2-(4-lsopropil-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
Trans-[3-(4-Cloro-fenil)-4-hidroxi-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
(3-Amino-1 H-indazol-5-il)-[(S)-2-(4-isopropil-fenil)-pirrolidin-1-il]-metanona,
[(S)-2-(4-Fluoro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[4,3-b]piridin-5-il)-metanona,
(3-Metil-1 H-pirazolo[4,3-b]piridin-5-il)-[(S)-2-(4-trifluorometil-fenil)-pirrolidin-1-il]-metanona,
[(S)-2-(4-Cloro-3-fluoro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[2-(4-Cloro-3-fluoro-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[4,3-b]piridin-5-il)-metanona,
[(S)-2-(4-lsopropil-fenil)-pirrolidin-1-il]-(3-metil-1 H-pirazolo[3,4-b]piridin-5-il)-metanona,
[(S)-2-(4-Fluoro-fenil)-azetidin-1-il]-(3-metil-1 H-indazol-5-il)-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(3-metilamino-1 H-indazol-5-il)-metanona,
[(S)-2-(4-Cloro-fenil)-pirrolidin-1-il]-(6-fluoro-3-metil-1 H-indazol-5-il)-metanona,
[(S)-2-(4-Cloro-fenil)-azetidin-1-il]-(3-metil-1 H-indazol-5-il)-metanona.
7. Una composición farmacéutica que comprenden un compuesto según una o más de las reivindicaciones 1 a 6, o sus estereoisómeros o tautómeros o sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas las mezclas de los mismos en todas las relaciones, como principio activo, junto con un vehículo farmacéuticamente aceptable.
8. Un compuesto según una o más de las reivindicaciones 1 a 6, o sus estereoisómeros o tautómeros o sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas las mezclas de los mismos en todas las relaciones, para su uso en el tratamiento de una enfermedad hiperproliferativa, inflamatorias o degenerativa.
9. El compuesto para su uso según la reivindicación 8, o sus estereoisómeros o tautómeros o sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas las mezclas de los mismos en todas las relaciones, en donde la enfermedad hiperproliferativa es cáncer.
10. El compuesto para su uso según la reivindicación 9, o sus estereoisómeros o tautómeros o sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas las mezclas de los mismos en todas las relaciones, en donde el cáncer se selecciona entre el grupo que consiste en cerebro, pulmón, colon, epidermoide, células escamosas, vejiga, gástrico, pancreático, mama, cabeza y cuello, renal, riñón, el hígado, ovárico, próstata, uterino, esofágico, testicular, ginecológico, cáncer de tiroides, melanoma, leucemia mielógena aguda, mieloma múltiple, leucemia mielógena crónica, leucemia mieloide aguda, sarcoma de Kaposi.
11. Uso de un compuesto de una o más de las reivindicaciones 1 a 6, o sus estereoisómeros o tautómeros o sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas las mezclas de los mismos en todas las relaciones, para la preparación de un medicamento para el tratamiento de una enfermedad hiperproliferativa, inflamatorias o degenerativa.
12. Uso según la reivindicación 11 en donde la enfermedad es cáncer.
13. Uso según la reivindicación 12 en donde el cáncer se selecciona entre el grupo que consiste en cerebro, pulmón, colon, epidermoide, células escamosas, vejiga, gástrico, pancreático, mama, cabeza y cuello, renal, riñón, el hígado, ovárico, próstata, uterino, esofágico, testicular, ginecológico, cáncer de tiroides, melanoma, leucemia mielógena aguda, mieloma múltiple, leucemia mielógena crónica, leucemia mieloide aguda, sarcoma de Kaposi.
14. Conjunto (kit) que consiste en envases separados de
a) una cantidad eficaz de un compuesto según una o más de las reivindicaciones 1 a 6, o sus estereoisómeros o tautómeros o sales farmacéuticamente aceptables de cada uno de los anteriores, incluidas las mezclas de los mismos en todas las relaciones, y
b) una cantidad eficaz de un principio activo de medicamento adicional.
15. Procedimiento para la fabricación de compuestos de Fórmula (I) como se define en la reivindicación 1, en el que un compuesto de Fórmula (IV)

se hace reaccionar, opcionalmente en presencia de un agente de activación, con un compuesto de Fórmula (III),

para producir un compuesto de Fórmula (I).
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP14002929 | 2014-08-22 | ||
| PCT/EP2015/001570 WO2016026549A1 (en) | 2014-08-22 | 2015-07-30 | Indazoles |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2703851T3 true ES2703851T3 (es) | 2019-03-12 |
Family
ID=51690171
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES15744882T Active ES2703851T3 (es) | 2014-08-22 | 2015-07-30 | Indazoles |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US9862717B2 (es) |
| EP (1) | EP3183239B1 (es) |
| JP (1) | JP6434614B2 (es) |
| CN (1) | CN106573932B (es) |
| AU (1) | AU2015306458B2 (es) |
| CA (1) | CA2958508A1 (es) |
| ES (1) | ES2703851T3 (es) |
| IL (1) | IL250306B (es) |
| WO (1) | WO2016026549A1 (es) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101731624B1 (ko) * | 2014-07-01 | 2017-05-04 | 광주과학기술원 | 세포 리프로그래밍 유도용 조성물 |
| MX2020001531A (es) * | 2017-08-07 | 2020-03-20 | Biocad Joint Stock Co | Nuevos compuestos hetorociclicos como inhibidores de cdk8/19. |
| US11261184B2 (en) | 2017-10-02 | 2022-03-01 | Boehringer Ingelheim International Gmbh | [1,6]naphthyridine compounds and derivatives as CDK8/CDK19 inhibitors |
| EP3990432A1 (en) * | 2019-06-27 | 2022-05-04 | Biogen MA Inc. | 2h-indazole derivatives and their use in the treatment of disease |
| EP4255904A2 (en) * | 2020-12-03 | 2023-10-11 | Domain Therapeutics | Novel par-2 inhibitors |
| AR124369A1 (es) | 2020-12-16 | 2023-03-22 | Amgen Inc | Inhibidores de prmt5 novedosos |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE69837282T2 (de) | 1997-12-13 | 2007-11-08 | Bristol-Myers Squibb Co. | VERWENDUNG VON PYRAZOLO Ä 3,4-bÜ PYRIDIN ALS CYCLIN-ABHÄNGIGE KINASE HEMMER |
| ES2236217T3 (es) * | 2000-04-25 | 2005-07-16 | Bristol-Myers Squibb Company | Uso de 5.tio.,sulfinil- y sulfonilpirazolo 3,4-b)-piridinas como inhibidores de la quinasa dependiente de la ciclina. |
| CA2644910C (en) * | 2006-03-31 | 2014-01-28 | Abbott Laboratories | Indazole compounds |
| WO2011000566A2 (en) * | 2009-06-30 | 2011-01-06 | Savira Pharmaceuticals Gmbh | Compounds and pharmaceutical compositions for the treatment of negative-sense ssrna virus infections |
| EP2464231A4 (en) * | 2009-08-10 | 2013-02-06 | Samumed Llc | INDAZOLE AS WNT / B-CATENINE SIGNALING PATHWASHER AND THERAPEUTIC APPLICATIONS THEREOF |
| CA2885392A1 (en) * | 2012-12-10 | 2014-06-19 | Zhanling CHENG | Novel bi-ring phenyl-pyridines/pyrazines for the treatment of cancer |
-
2015
- 2015-07-30 EP EP15744882.0A patent/EP3183239B1/en active Active
- 2015-07-30 JP JP2017510567A patent/JP6434614B2/ja not_active Expired - Fee Related
- 2015-07-30 ES ES15744882T patent/ES2703851T3/es active Active
- 2015-07-30 AU AU2015306458A patent/AU2015306458B2/en not_active Ceased
- 2015-07-30 US US15/505,345 patent/US9862717B2/en active Active
- 2015-07-30 CN CN201580045223.2A patent/CN106573932B/zh active Active
- 2015-07-30 CA CA2958508A patent/CA2958508A1/en not_active Abandoned
- 2015-07-30 WO PCT/EP2015/001570 patent/WO2016026549A1/en active Application Filing
-
2017
- 2017-01-26 IL IL250306A patent/IL250306B/en active IP Right Grant
Also Published As
| Publication number | Publication date |
|---|---|
| US20170267675A1 (en) | 2017-09-21 |
| US9862717B2 (en) | 2018-01-09 |
| IL250306B (en) | 2019-01-31 |
| EP3183239B1 (en) | 2018-09-26 |
| WO2016026549A1 (en) | 2016-02-25 |
| CN106573932B (zh) | 2019-07-30 |
| AU2015306458B2 (en) | 2019-05-02 |
| EP3183239A1 (en) | 2017-06-28 |
| AU2015306458A1 (en) | 2017-04-06 |
| CN106573932A (zh) | 2017-04-19 |
| JP2017525717A (ja) | 2017-09-07 |
| IL250306A0 (en) | 2017-03-30 |
| CA2958508A1 (en) | 2016-02-25 |
| JP6434614B2 (ja) | 2018-12-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2703851T3 (es) | Indazoles | |
| US10428080B2 (en) | TBK/IKK inhibitor compounds and uses thereof | |
| AU2015349899B9 (en) | Heteroaryl compounds as IRAK inhibitors and uses thereof | |
| JP7249950B2 (ja) | ヘテロ環化合物 | |
| AU2016215185B2 (en) | Macrocyclic compounds as IRAK 1/4 inhibitors and uses thereof | |
| US20160016951A1 (en) | Novel naphthyridines and isoquinolines and their use as cdk8/19 inhibitors | |
| WO2016041618A1 (en) | Substituted indazoles and related heterocycles | |
| ES2720324T3 (es) | Piridil piperidinas | |
| ES2698960T3 (es) | Compuestos de 2-aminopiridina | |
| KR20200051626A (ko) | Cdk8/19 저해제로서 새로운 헤테로사이클릭 화합물 | |
| AU2018352699A1 (en) | Pyrimidine TBK/IKKE inhibitor compounds and uses thereof | |
| WO2016128140A1 (en) | Pyrimidine derivatives for use in the treatment of cancer | |
| ES2656096T3 (es) | Nuevos derivados de imidazol-piperidinilo como moduladores de la actividad de quinasa | |
| JP7417540B2 (ja) | シクロアルカン-1,3-ジアミン誘導体 |